Область техники, к которой относится изобретение
Настоящее изобретение относится к способам лечения кожных заболеваний, обусловленных базофилами и тучными клетками, таких как хроническая спонтанная крапивница (CSU), с применением ингибитора тирозинкиназы Брутона.
Предпосылки изобретения
Крапивница представляет собой гетерогенную группу заболеваний, характеризующихся зудящей крапивной сыпью и/или ангионевротическим отеком. Хроническая крапивница определяется как крапивница, которая присутствует постоянно или периодически в течение более 6 недель (Maurer M et al. (2013) Revisions to the international guidelines on the diagnosis and therapy of chronic urticaria. J Dtsch Dermatol Ges.; Bernstein JA, Lang DM, Khan DA, et al (2014) The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol; 133(5):1270-7). Хроническую крапивницу далее дополнительно делят на две подгруппы: хроническую спонтанную крапивницу (CSU) и индуцибельную крапивницу (IU), при этом последняя включает физическую крапивницу, как например тепловая, холодовая или крапивница вследствие давления, а также особые варианты, как например холинергическая крапивница. CSU определяется как спонтанное появление зудящих волдырей, ангионевротического отека или того и другого на протяжении более 6 недель по известным или неизвестным причинам (Zuberbier T, et al. (2018) The EAACI/GA(2) LEN/EDF/WAO Guideline for the definition, classification, diagnosis, and management of urticaria: the 2017 revision and update. Allergy; 73(7):1393-1414). Возможна комбинация CSU с индуцируемой формой крапивницы, такая как часто наблюдаемая комбинация симптоматической дерматографической крапивницы и CSU.
Ранее все формы хронической крапивницы без известного триггера назывались "хронической идиопатической крапивницей" (CIU). Благодаря прогрессу в области медицины в настоящее время стало известно, что при некоторых из ранее считавшихся "идиопатическими" формах крапивницы в действительности могут быть обнаружены аутоантитела. Однако, ежедневное нестабильное проявление симптомов при такой хронической крапивнице с наличием аутоантител все еще остается непредсказуемым и не провоцируется очевидным триггером, вследствие чего симптомы проявляются спонтанно. Чтобы надлежащим образом отразить в терминологии тот факт, что при некоторых из ранее считавшихся "идиопатическими" формах в действительности могут иметься поддающиеся выявлению аутоантитела, эту популяцию теперь называют хронической спонтанной крапивницей (CSU) в соответствии с Международным руководством (Maurer M et al. (2013) Revisions to the international guidelines on the diagnosis and therapy of chronic urticaria. J Dtsch Dermatol Ges.; Zuberbier T, et al. (2018) The EAACI/GA(2) LEN/EDF/WAO Guideline for the definition, classification, diagnosis, and management of urticaria: the 2017 revision and update. Allergy; 73(7):1393-1414). Использование выражения "хроническая идиопатическая крапивница" в медицинской практике больше не рекомендуется. Однако данное новое соглашение о наименовании реализовано не во всех частях мира, и в таких странах, как США, популяцию пациентов с хронической крапивницей неспецифической этиологии или с неизвестными триггерами все еще характеризуют как пациентов с хронической идиопатической крапивницей (CIU). Согласно Международному руководству эта нозологическая единица в целях единообразия в данном документе называется CSU.
Риск заболеть CSU в течение жизни составляет примерно 1,8%, а у 20% пациентов с CSU заболевание все еще может иметься по прошествии 20 лет (Greaves M (2000) Chronic urticaria. J Allergy Clin Immunol; 105(4):664-72; Zuberbier T, Balke M, Worm M, et al (2010) Epidemiology of urticaria: a representative cross-sectional population survey. Clin Exp Dermatol; 35(8):869-73). У болеющих пациентов часто наблюдается зудящая крапивная сыпь с сопутствующей эритемой и/или эпизодами ангионевротического отека. Сообщается, что ангионевротический отек ассоциирован примерно с 33-67% случаев CSU (Juhlin L (1981) Recurrent urticaria: clinical investigation of 330 patients. Br J Dermatol; 104(4):369-81 ; Toubi E, Kessel A, Avshovich N, et al (2004) Clinical and laboratory parameters in predicting chronic urticaria duration: a prospective study of 139 patients. Allergy; 59(8):869-73; Zuberbier T, Balke M, Worm M, et al (2010) Epidemiology of urticaria: a representative cross-sectional population survey. Clin Exp Dermatol; 35(8):869-73; Maurer M, Weller K, Bindslev-Jensen C, et al (2011) Unmet clinical needs in chronic spontaneous urticaria. A GA²LEN task force report. Allergy; 66(3):317-30). Классическое поражение кожи при крапивнице представляет собой волдырь и покраснение с бледным приподнятым участком поражения и окружающей эритемой размером от нескольких миллиметров до нескольких сантиметров в поперечнике, которые обычно возникают группами и часто сливаются с образованием больших сливных поражений. CSU ассоциируется с сильным зудом и оказывает большое влияние на самочувствие и качество жизни пациента, которое, как представляется, сравнимо с влиянием тяжелой ишемической болезни сердца (Greaves MW (2003) Chronic idiopathic urticaria. Curr Opin Allergy Clin Immunol; 3(5):363-8. Review; Powell RJ, Du Toit GL, Siddique N, et al (2007) BSACI guidelines for the management of chronic urticaria and angio-oedema. Clin Exp Allergy; 37(5):631-50). Симптомы крапивницы и ангионевротического отека, ассоциированного с крапивницей, отрицательно влияют на повседневную активность и сон (O'Donnell BF, Lawlor F, Simpson J, et al (1997). The impact of chronic urticaria on the quality of life. Br J Dermatol; 136(2):197-201). Таким образом, при ведении пациентов с крапивницей результаты с точки зрения пациента (например, DLQI) являются важными показателями лечения (Kaplan A., et al. (2013) Omalizumab in patients with symptomatic chronic idiopathic/spontaneous urticaria despite standard combination therapy. J Allergy Clin Immunol; 132(1):101-9; Maurer M et al. (2013) Revisions to the international guidelines on the diagnosis and therapy of chronic urticaria. J Dtsch Dermatol Ges; Zuberbier T, Aberer W, Asero R, et al (2018) The EAACI/GA(2) LEN/EDF/WAO Guideline for the definition, classification, diagnosis, and management of urticaria: the 2017 revision and update. Allergy; 73(7):1393-1414).
Патогенез CSU не до конца выяснен. До 50% случаев CSU связаны с индуцирующими высвобождение гистамина аутоантителами ко множеству антигенов, включая высокоаффинный рецептор IgE (FcεRI) или антитела класса IgE; при этом клиническое значение этих аутоантител четко не установлено, хотя есть предположения, что они могут быть вовлечены в патогенез заболевания (Kaplan AP (2002) Chronic urticaria--new concepts regarding pathogenesis and treatment. Curr Allergy Asthma Rep; 2(4):263-4; Sabroe RA, Greaves MW (2006) Chronic idiopathic urticaria with functional autoantibodies: 12 years on. Br J Dermatol; 154(5):813-9. Review). Было также высказано предположение, что базофилы пациентов с CSU могут иметь отчетливые изменения в опосредованной FcεRIα дегрануляции, независимо от какой-либо потенциальной роли аутоантител (Eckman JA, et al. (2008) Basophil phenotypes in chronic idiopathic urticaria in relation to disease activity and autoantibodies. J Invest Dermatol; 128(8):1956-63).
Лечение CSU является сложной задачей, а не обладающие седативным действием (относящиеся ко второму поколению) H1-антигистаминные средства (H1-AH) являются основой симптоматической терапии CSU. Хотя H1-AH в одобренных дозах приносит облегчение некоторым пациентам, у более 50% пациентов отсутствует ответ на H1-AH при стандартных дозах. Даже при четырехкратном повышении дозы по сравнению с одобренной дозой, осуществляемом в соответствии со вторым этапом алгоритма лечения согласно действующему Международному руководству (Zuberbier T, et al. (2018) The EAACI/GA(2) LEN/EDF/WAO Guideline for the definition, classification, diagnosis, and management of urticaria: the 2017 revision and update. Allergy; 73(7):1393-1414), у значительной части пациентов не наблюдается контроля симптомов крапивницы (Maurer M, Weller K, Bindslev-Jensen C, et al (2011) Unmet clinical needs in chronic spontaneous urticaria. A GA²LEN task force report. Allergy; 66(3):317-30; Marrouche N, Grattan C (2014) Update and insights into treatment options for chronic spontaneous urticaria. Expert Rev Clin Immunol; 10(3):397-403). Для пациентов, у которых не удается обеспечить контроль заболевания при использовании четырехкратных доз H1-AH, третий этап алгоритма лечения согласно Международному руководству предусматривает добавление омализумаба, а при недостаточном ответе на омализумаб может использоваться циклоспорин в качестве терапии последней линии.
Убедительность экспериментальных доказательств эффективности антагонистов лейкотриеновых рецепторов (LTRA) при крапивнице является низким. С появлением омализумаба (для применения вне показаний) LTRA больше не рекомендуются для лечения CSU, не поддающейся лечению H1-антигистаминными средствами (Zuberbier 2018). К схемам лечения третьего уровня могут быть добавлены короткие курсы (макс. 10 дней) системных кортикостероидов, если это требуется в связи с обострениями. Ввиду нежелательных эффектов, связанных с хроническим системным воздействием кортикостероидов, более длительный курс лечения не рекомендуется. Другие варианты лечения, которые использовались ранее, такие как иммуноглобулин G для внутривенного введения, дапсон, гидроксихлорохин, H2-антигистаминные средства (H2-AH), метотрексат и циклофосфамид, характеризуются неблагоприятным соотношением риска и пользы или профилем со значительными побочными эффектами и больше не рекомендуются для терапии CSU (Kaplan AP (2002) Chronic urticaria--new concepts regarding pathogenesis and treatment. Curr Allergy Asthma Rep; 2(4):263-4 ; Powell RJ, Du Toit GL, Siddique N, et al (2007) BSACI guidelines for the management of chronic urticaria and angio-oedema. Clin Exp Allergy; 37(5):631-50; Zuberbier T, et al. (2018) The EAACI/GA(2) LEN/EDF/WAO Guideline for the definition, classification, diagnosis, and management of urticaria: the 2017 revision and update. Allergy; 73(7):1393-1414).
Омализумаб является одобренным терапевтическим средством для лечения CSU, невосприимчивой к стандартному лечению, и демонстрирует благоприятное соотношение польза-риск. Это рекомбинантное гуманизированное моноклональное антитело подкласса IgG1, которое связывается с IgE-специфическими эпитопами в пределах области C3 (FcεRI-связывающей) молекулы IgE, и его применение показано во многих странах для лечения плохо поддающейся контролю умеренной или тяжелой астмы и CSU, невосприимчивой к стандартной терапии. Точный механизм действия омализумаба у пациентов с CSU неизвестен. Пациентам, страдающим CSU, омализумаб вводят в виде раствора для инъекций.
Несмотря на наличие средств лечения CSU, в области медицины остается высокая потребность в новых вариантах лечения для субъектов с CSU. Менее 40% субъектов с CSU, которых лечили с помощью H1-антигистаминных средств второго поколения, не отвечают на лечение должным образом (Guillén-Aguinaga et al 2016, Br J Dermatol; 175(6):1153-65). Кроме того, менее чем у 50% субъектов, которых лечат с помощью омализумаба, удается достичь полного контроля признаков и симптомов CSU (Kaplan et al. 2016, J Allergy Clin Immunol; 137(2):474-81).
КРАТКОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является предоставление нового способа лечения или предупреждения кожных заболеваний, обусловленных базофилами и тучными клетками, таких как хроническая спонтанная крапивница и атопический дерматит, у нуждающегося в таком лечении субъекта, предусматривающего введение указанному субъекту терапевтически эффективного количества N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида.
Следовательно, в данном документе раскрыты способы лечения хронической спонтанной крапивницы (CSU), предусматривающие введение нуждающемуся в таком лечении пациенту суточной дозы, составляющей от приблизительно 0,5 мг до приблизительно 600 мг, предпочтительно от приблизительно 10 мг до приблизительно 200 мг, или более предпочтительно в суточной дозе, составляющей от приблизительно 10 мг до приблизительно 100 мг, N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида, который представляет собой соединение формулы (I)
 (I),
(I),
или его фармацевтически приемлемую соль.
Также раскрывается N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид; или его фармацевтически приемлемая соль для применения в лечении хронической спонтанной крапивницы (CSU), где N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид или его фармацевтически приемлемая соль вводятся в суточной дозе, составляющей от приблизительно 0,5 мг до приблизительно 600 мг, предпочтительно в суточной дозе, составляющей от приблизительно 10 мг до приблизительно 200 мг, и наиболее предпочтительно в суточной дозе, составляющей от приблизительно 10 до приблизительно 100 мг.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фигура 1. Ингибирующие эффекты соединения формулы (I) при обратной пассивной реакции Артюса.
Фигура 2. Занятость BTK в селезенке через 5 часов после введения дозы.
Фигура 3. Соединение формулы (I) ингибирует PCA после сенсибилизации низкими дозами IgE.
Фигура 4. Занятость BTK в селезенке при сенсибилизации низкой дозой IgE.
Фигура 5. Концентрация в крови в зависимости от времени для соединения (I) после однократных нарастающих доз в диапазоне 0,5 мг - 600 мг.
Фигура 6. Концентрация в крови в зависимости от времени для соединения (I) после многократных нарастающих доз в диапазоне 10 мг - 400 мг при введении дозы q.d.
Фигура 7. Концентрация в крови в зависимости от времени для соединения (I) после многократных нарастающих доз в диапазоне от 100 мг b.i.d. до 200 мг b.i.d.
Фигура 8. Влияние приема пищи, наблюдаемое после однократной пероральной дозы 60 мг соединения формулы (I).
Фигура 9. Среднее арифметическое (SD) процентного значения степени занятости BTK в периферической крови после однократной дозы соединения формулы (I).
Фигура 10. Медиана процентного значения ингибирования активации базофилов в зависимости от общей суточной дозой соединения формулы (I) в день 12 введения многократных нарастающих доз соединения формулы (I).
Фигура 11. Уменьшение размера волдырей в кожном прик-тесте при многократных нарастающих дозах.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Тирозинкиназа Брутона (BTK) является цитоплазматической тирозинкиназой и членом семейства киназ TEC. BTK экспрессируется в выбранных клетках адаптивной и врожденной иммунной системы, включая В-клетки, макрофаги, тучные клетки/базофилы и тромбоциты. BTK необходима для передачи сигналов через рецептор Fc-эпсилон (FcεR1 для IgE) и активирующие Fc-гамма-рецепторы (FcγR для IgG), а также через B-клеточный рецептор антигена (BCR) и ингибиторы BTK. Ингибиторы BTK, такие как ибрутиниб, одобрены для лечения B-клеточных злокачественных новообразований (Hendriks et al. 2014). Недавно было продемонстрировано, что ингибирование BTK приводит к ингибированию активации/дегрануляции тучных клеток и базофилов in vitro и к уменьшению размеров волдырей в кожных прик-тестах у пациентов, страдающих от IgE-опосредованной аллергии (Smiljkovic et al. 2017; Regan et al. 2017; Dispenza et al. 2018). Следовательно, ингибирование BTK является привлекательной терапевтической концепцией для лечения различных аутоиммунных и хронических воспалительных заболеваний, включая ревматоидный артрит, рассеянный склероз, системную красную волчанку, хроническую крапивницу, атопический дерматит, астму и первичный синдром Шегрена (Tan et al 2013; Whang и Chang 2014).
N-(3-(6-Амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид описан в заявке WO2015/079417, поданной 4 июня 2015 г. (номер дела патентного поверенного PAT056021-WO-PCT). Это соединение является селективным, сильным, необратимым ковалентным ингибитором тирозинкиназы Брутона (BTK).
Согласно настоящему изобретению авторы настоящего изобретения продемонстрировали, что N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид эффективно ингибирует активацию базофилов у здоровых добровольцев и у субъектов с атопией, у которых наблюдается сходный с CSU патомеханизм, что измеряется по уровню ингибирования повышения экспрессии CD63. Кроме того, авторы настоящего изобретения продемонстрировали, что N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид обеспечивает уменьшение размеров волдырей в кожном прик-тесте.
Соответственно, в настоящее время авторы настоящего изобретения разработали схемы введения доз для лечения пациентов с CSU с помощью соединения, представляющего собой N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид или его фармацевтически приемлемую соль.
Определения
Для целей толкования данного описания будут применяться следующие определения, и при необходимости термины, используемые в единственном числе, будут также включать множественное число, и наоборот.
Используемая в данном документе фраза "фармацевтически приемлемый" относится к тем соединениям, материалам, композициям и/или лекарственным формам, которые в рамках здравого медицинского суждения являются подходящими для применения в контакте с тканями людей и животных без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений в соответствии с приемлемым соотношением пользы и риска.
Любая формула, приведенная в данном документе, также предназначена для представления немеченых форм, а также меченых изотопом форм соединений. Меченые изотопами соединения имеют структуры, изображенные посредством формул, приведенных в данном документе, за исключением того, что один или несколько атомов заменены атомом, характеризующимся выбранными атомной массой или массовым числом. Изотопы, которые могут быть включены в соединения по настоящему изобретению, включают, например, изотопы водорода, углерода, азота, кислорода, фтора, йода и хлора, такие как 3H, 11C, 13C, 14C, 15N, 18F и 36Cl. Соответственно, следует понимать, что настоящее изобретение предусматривает соединения, в которые включены один или несколько любых из вышеуказанных изотопов, в том числе, например, радиоактивных изотопов, таких как 3H и 14C, или соединения, в которых присутствуют нерадиоактивные изотопы, такие как 2H и 13C. Такие меченые изотопами соединения применимы в исследованиях метаболизма (с использованием 14C), исследованиях кинетики реакций (с использованием, например, 2H или 3H), методиках выявления или визуализации, таких как позитронно-эмиссионная томография (PET) или однофотонная эмиссионная компьютерная томография (SPECT), в том числе в анализах распределения лекарственного средства или субстрата в тканях, или в радиоизотопном лечении пациентов. В частности, меченое 18F соединение может быть особенно пригодным для исследований с помощью PET или SPECT. Изотопно меченые соединения, как правило, можно получать с помощью традиционных методик, известных специалистам в данной области техники, например с использованием подходящих изотопно меченых реагентов вместо немеченого реагента, используемого ранее.
Термин "фармацевтическая комбинация", используемый в данном документе, означает продукт, который получают в результате смешивания или объединения более чем одного активного ингредиента. Следует понимать, что фармацевтическая комбинация, используемая в данном документе, включает как фиксированные, так и нефиксированные комбинации активных ингредиентов. Термин "фиксированная комбинация" означает, что активные ингредиенты, например соединение формулы (I) или его фармацевтически приемлемая соль и один или несколько партнеров по комбинации, вводятся пациенту одновременно в форме одного препарата или лекарственной формы. Термин в таком случае относится к комбинации с фиксированной дозой в одной стандартной лекарственной форме (например, капсуле, таблетке или саше). Оба термина "нефиксированная комбинация" или "набор частей" означают, что активные ингредиенты, например соединение по настоящему изобретению и один или несколько партнеров по комбинации и/или один или несколько дополнительных средств, вводят или совместно вводят пациенту независимо в виде отдельных препаратов либо одновременно, либо параллельно, либо последовательно без конкретных временных ограничений, где такое введение обеспечивает терапевтически эффективные уровни двух соединений в организме пациента, особенно если эти временные интервалы позволяют партнерам по комбинации продемонстрировать совместный, например аддитивный или синергический, эффект. Термин "нефиксированная комбинация" также применяется в отношении "коктейльной терапии", например к введению трех или больше активных ингредиентов. Термин "нефиксированная комбинация", таким образом, определяет в частности введение, применение, композицию или состав в том смысле, что дозы соединений, описанных в данном документе, можно вводить независимо друг от друга, то есть одновременно или в разные моменты времени. Следует понимать, что термин "нефиксированная комбинация" также охватывает использование отдельного средства вместе с одним или несколькими фиксированными комбинированными продуктами, при этом каждый независимый состав имеет различные количества содержащихся в нем активных ингредиентов. Также следует понимать, что комбинированные продукты, описанные в данном документе, а также термин "нефиксированные комбинации" охватывают активные ингредиенты (включая соединения, описанные в данном документе), где партнеры по комбинации вводятся в виде полностью отдельных фармацевтических лекарственных форм или в виде фармацевтических составов, которые также продаются независимо друг от друга. Инструкции по применению нефиксированной комбинации предоставлены или могут быть предоставлены в упаковке, например в виде листка-вкладыша и т. п., или в виде другой информации, которая предоставляется врачам и/или медицинскому персоналу. Независимые составы или части состава, продукты или композиции затем можно вводить одновременно или через определенные промежутки времени, что означает, что каждую отдельную часть из набора частей можно вводить в различные моменты времени и/или с одинаковыми или различными временными интервалами в случае любой части из набора частей. В частности, временные интервалы для введения доз выбирают таким образом, чтобы эффект на заболевание, в отношении которого осуществляют лечение, при комбинированном применении частей был больше/сильнее, чем эффект, получаемый при применении только соединения формулы (I) или его фармацевтически приемлемой соли; таким образом, соединения, применяемые в фармацевтической комбинации, описанной в данном документе, являются совместно активными. Соотношение общих количеств соединения формулы (I) или его фармацевтически приемлемой соли и второго средства, подлежащих введению в виде фармацевтической комбинации, можно варьировать или корректировать для обеспечения лучшего соответствия потребностям определенной субпопуляции пациентов, подлежащей лечению, или потребностям одного пациента, при этом различные потребности могут быть связаны с возрастом, полом, весом тела и т. д. пациентов.
Термины "совместное введение", или "комбинированное введение", или подобные таковым, используемые в данном документе, следует рассматривать как охватывающие введение одного или нескольких соединений, описанных в данном документе, совместно с выбранным партнером по комбинации одному нуждающемуся в этом субъекту (например, пациенту или субъекту), и предполагают включение схем лечения, в которых соединения необязательно вводят одним и тем же путем введения и/или в одно и то же время.
Термин "фармацевтическая композиция", определяемый в данном документе, относится к смеси (например, раствору или эмульсии), содержащей по меньшей мере один активный ингредиент или терапевтическое средство, подлежащие введению теплокровному животному, например млекопитающему или человеку, для того, чтобы предупредить или лечить конкретное заболевание или состояние, поражающее теплокровное животное.
Термин "терапевтически эффективное количество" соединения (т. е. соединения формулы (I) или его фармацевтически приемлемой соли) по настоящему изобретению относится к количеству соединения по настоящему изобретению, которое будет вызывать биологический или медицинский ответ у субъекта (пациента или субъекта), например снижение или ингибирование активности фермента или белка, или ослаблять симптомы, облегчать состояния, замедлять или сдерживать прогрессирование заболевания, или предупреждать заболевание и т. д. Терапевтически эффективная доза соединения, фармацевтической композиции или их комбинаций зависит от биологического вида пациента, веса тела, возраста, пола и индивидуального состояния, нарушения или заболевания, лечение которых осуществляют, или их тяжести. Врач, клиницист или ветеринар средней квалификации может легко определить эффективное количество каждого из активных ингредиентов, необходимое для предупреждения, лечения или подавления прогрессирования нарушения или заболевания.
Частоту введения дозы можно варьировать в зависимости от используемого соединения и конкретного состояния, подлежащего лечению или предупреждению. В целом, применение минимальной дозы, достаточной для обеспечения эффективной терапии, является предпочтительным. Мониторинг терапевтической эффективности у пациентов, как правило, можно осуществлять с применением анализов, подходящих для состояния, подлежащего лечению или предупреждению, что будет известно специалистам в данной области техники.
Используемые в данном документе термины "носитель" или "фармацевтически приемлемый носитель" включают все возможные растворители, дисперсионные среды, покрытия, поверхностно-активные вещества, антиоксиданты, консерванты (например, антибактериальные средства, противогрибковые средства), средства для обеспечения изотоничности, замедляющие абсорбцию средства, соли, консерванты, лекарственные средства, стабилизаторы лекарственных средств, связующие средства, вспомогательные вещества, разрыхляющие средства, смазывающие вещества, подслащивающие вещества, ароматизирующие средства, красители и т. п., а также их комбинации, которые будут известны специалистам в данной области техники (см., например, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289-1329). За исключением случаев, когда традиционный носитель является несовместимым с активным ингредиентом, предполагается его применение в терапевтических или фармацевтических композициях.
Используемый в данном документе термин "субъект" относится к животному. Как правило, животное является млекопитающим. Субъект также относится, например, к приматам (например, людям, мужчинам или женщинам), коровам, овцам, козам, лошадям, собакам, кошкам, кроликам, крысам, мышам, рыбам, птицам и т. п. В определенных вариантах осуществления субъектом является примат. В предпочтительном варианте осуществления субъектом является человек. Термин "субъект" используется взаимозаменяемо с "пациентом", если он относится к человеку.
Как используется в данном документе, субъект является "нуждающимся в" лечении, если в результате такого лечения такой субъект получит пользу с биологической, медицинской точки зрения, или улучшится качество его жизни.
Используемая в данном документе фраза "популяция пациентов" означает группу пациентов.
Термин "содержащий" охватывает "включающий", а также "состоящий", например композиция, "содержащая" X, может состоять исключительно из X или может включать что-либо дополнительное, например X+Y.
Термин "приблизительно" по отношению к числовому значению х означает, например, +/-10%. В случае использования перед числовым диапазоном или перечнем чисел термин "приблизительно" применяется к каждому числу в серии, например, фразу "приблизительно 1-5" следует интерпретировать как "приблизительно 1 - приблизительно 5", или, например, фразу "приблизительно 1, 2, 3, 4" следует интерпретировать как "приблизительно 1, приблизительно 2, приблизительно 3, приблизительно 4 и т. д.".
Термин "лечение" или "лечить" в данном документе определяется как применение или введение соединения в соответствии с настоящим изобретением (соединения формулы (I), или его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей указанное соединение, субъекту или в выделенную ткань или линию клеток от субъекта, где у субъекта имеется конкретное заболевание (например, CSU), симптом, связанный с заболеванием (например, CSU), или предрасположенность к развитию заболевания (например, CSU) (если применимо), при этом целью является излечение (если применимо), задержка начала развития, уменьшение тяжести, облегчение, ослабление одного или нескольких симптомов заболевания, улучшение течения заболевания, уменьшение или снижение проявления любых связанных симптомов заболевания или предрасположенности к развитию заболевания. Термин "лечение" или "лечить" включает лечение пациента с подозрением на наличие заболевания, а также больных пациентов и пациентов, которые были диагностированы, как страдающие заболеванием или медицинским состоянием, и включает подавление клинического рецидива.
Используемые в данном документе фразы "ранее не получавший лечение с помощью системного средства лечения CSU" или "не подвергавшийся лечению" относятся к пациенту с CSU, который ранее не получал лечение с помощью системного средства, например циклоспорина А, монтелукаста, H1-антигистаминных средств (H1-AH), H2-антигистаминных средств (H2-AH) и антагониста лейкотриеновых рецепторов (LTRA), биологического средства (например, омализумаба) и т. д., для лечения CSU. Системные средства (т. е. средства, вводимые перорально, с помощью инъекции и т. д.) отличаются от локально действующих средств (например, местных средств и фототерапии) тем, что системные средства обладают системным (охватывающим весь организм) эффектом при доставке пациенту. В некоторых вариантах осуществления раскрытых способов, схем, вариантов применения, наборов и фармацевтических композиций пациенту ранее не вводили системное средство лечения CSU.
Используемая в данном документе фраза "ранее получавший лечение с помощью системного средства для лечения CSU" используется для обозначения пациента, который ранее подвергался лечению CSU с использованием системного средства. Такие пациенты включают пациентов, ранее получавших лечение с помощью H1-антигистаминных средств или биологических средств, таких как омализумаб, и пациентов, ранее получавших лечение с помощью небиологических средств, таких как циклоспорин. В некоторых вариантах осуществления настоящего изобретения пациенту ранее вводили системное средство для лечения CSU. В некоторых вариантах осуществления пациенту ранее вводили системное средство для лечения CSU (например, циклоспорин), однако при этом пациенту ранее не вводили системное биологическое лекарственное средство (т. е. лекарственное средство, продуцируемое живым организмом, например, антитела, рецепторы-ловушки и т. д.) для лечения CSU (например, омализумаб). В данном случае пациента называют "не получавшим лечение с помощью биологического средства". В некоторых вариантах осуществления пациент является не получавшим лечение с помощью биологического средства.
Как используется в данном документе, термины "осуществление отбора" и "отобранный", используемые в отношении пациента, означают, что конкретный пациент специально выбран из большей группы пациентов на основании того (вследствие того), что конкретный пациент отвечает заранее определенным критериям. Подобным образом, "селективное лечение" относится к обеспечению лечением пациента, имеющего конкретное заболевание, при этом данный пациент специально выбран из большей группы пациентов на основании того, что конкретный пациент отвечает заранее определенному критерию. Подобным образом, "селективное введение" относится к введению лекарственного средства пациенту, который специально выбран из большей группы пациентов на основании того (вследствие того), что конкретный пациент отвечает заранее определенному критерию. Под "осуществлением отбора", "селективным лечением" и "селективным введением" подразумевают, что пациенту предоставляется персонализированная терапия, основанная на личном анамнезе пациента (например, предшествующие терапевтические вмешательства, например предшествующее лечение биологическими средствами), биологии пациента (например, конкретные генетические маркеры) и/или проявлении у пациента (например, несоответствие определенным диагностическим критериям), вместо применения стандартной схемы лечения, основанной исключительно на принадлежности пациента к большей группе. Отбор, применительно к способу лечения в контексте данного документа, не относится к случайному лечению пациента, отвечающего конкретному критерию, но скорее относится ко взвешенному решению, принимаемому в отношении введения лекарственного средства пациенту на основании того, что пациент отвечает конкретному критерию. Таким образом, селективное лечение/введение отличается от стандартного лечения/введения, в ходе которого обеспечивается доставка конкретного лекарственного средства всем пациентам, имеющим конкретное заболевание, независимо от их личного анамнеза, проявлений заболевания и/или биологических особенностей. В некоторых вариантах осуществления пациент был выбран для лечения на основании наличия у него CSU.
Варианты осуществления настоящего изобретения
Хроническая крапивница и эффективность лечения в соответствии с настоящим изобретением
Раскрытый ингибитор BTK, т. е. соединение формулы (I) или его фармацевтически приемлемую соль, можно применять in vitro, ex vivo, или включать в фармацевтические композиции и вводить in vivo для лечения пациентов с CSU (например, пациентов-людей).
Крапивница представляет собой гетерогенную группу заболеваний, характеризующихся зудящей крапивной сыпью и/или ангионевротическим отеком.
Хроническая крапивница определяется как крапивница, которая постоянно или периодически присутствует в течение более 6 недель (Maurer, et al 2013, Bernstein, et al 2014). Хроническую крапивницу далее дополнительно делят на две подгруппы: хроническую спонтанную крапивницу (CSU) и индуцибельную крапивницу (IU), при этом последняя включает физическую крапивницу, как например тепловая, холодовая или крапивница вследствие давления, а также особые варианты, как например холинергическая крапивница. CSU определяется как спонтанное появление зудящих волдырей, ангионевротического отека или и того, и другого в течение ≥ 6 недель по известным или неизвестным причинам (Zuberbier, et al. 2018). Возможна комбинация CSU с индуцируемой формой крапивницы, такая как часто наблюдаемая комбинация бессимптомной дерматографической крапивницы и CSU.
Эффективность лечения CSU оценивают с использованием различных известных способов и инструментов, с помощью которых осуществляют измерения для определения состояния заболевания CSU и/или клинического ответа при CSU. Некоторые примеры включают, например, ежедневно заполняемый дневник пациента с крапивницей (UPDD), оценку активности ангионевротического отека (AAS), еженедельную оценку тяжести крапивной сыпи (HSS7), еженедельную оценку тяжести зуда (ISS7), еженедельную оценку активности крапивницы (UAS7) и улучшение связанного со здоровьем качества жизни, измеряемого с помощью дерматологического индекса качества жизни (DLQI).
Ежедневно заполняемый дневник пациента с крапивницей (UPDD)
UPDD предусматривает оценку активности крапивницы (UAS), в рамках которой дважды в день оценивают тяжесть зуда и количество элементов крапивной сыпи, использование лекарственных препаратов неотложной помощи, влияние на сон и активность, частота возникновения ангионевротического отека, его лечение, и в нем записывают звонки специалисту здравоохранения (HCP).
Компоненты представлены в таблице 1 и соответствующие еженедельные оценки
описаны ниже.
Таблица 1. UPDD
- Был ли у пациента эпизод
- Если у пациента был эпизод, как происходило его лечение?
В некоторых вариантах осуществления, когда популяцию пациентов с CSU лечат в соответствии с раскрытыми способами, пациент достигает улучшенной оценки UPDD.
Еженедельная оценка тяжести крапивной сыпи (HSS7)
Оценка тяжести крапивной сыпи (волдырей), определяемая количеством элементов крапивной сыпи, записывается субъектом дважды в день в своем электронном дневнике по шкале от 0 (отсутствие) до 3 (> 12 элементов крапивной сыпи/12 часов; таблица 2). Еженедельную оценку (HSS7) получали путем суммирования усредненных ежедневных оценок за 7 дней, предыдущих визиту. Таким образом, возможный диапазон еженедельных оценок составлял от 0 до 21.
Таблица 2. Оценка тяжести крапивной сыпи
В некоторых вариантах осуществления, если популяцию пациентов с CSU лечат в соответствии с раскрытыми способами, оценка тяжести крапивной сыпи (HSS7) улучшается на по меньшей мере 5 баллов. Более того, по сравнению с группой плацебо разница между группой лечения и группой плацебо составляет по меньшей мере 4 балла, предпочтительно по меньшей мере 5 баллов. В одном варианте осуществления, если пациента лечат в соответствии с раскрытыми способами, оценка тяжести крапивной сыпи (HSS7) составляет менее 6, предпочтительно менее 4, предпочтительно менее 2 баллов, и наиболее предпочтительно оценка HSS7 составляет 0 баллов.
Еженедельная оценка тяжести зуда (ISS7)
Степень интенсивности зуда записывается субъектом два раза в сутки в своем электронном дневнике по шкале от
0 (отсутствие) до 3 (сильный) (таблица 3). Еженедельную оценку (ISS7) получали путем суммирования усредненных ежедневных оценок за 7 дней, предшествующих визиту. Таким образом, возможный диапазон еженедельных оценок составлял от 0 до 21.
Таблица 3. Оценка тяжести зуда
В некоторых вариантах осуществления, если популяцию пациентов с CSU лечат в соответствии с раскрытыми способами, оценка тяжести зуда (ISS7) улучшается на по меньшей мере 5 баллов. Более того, по сравнению с группой плацебо разница между группой лечения и группой плацебо составляет по меньшей мере 4 балла, предпочтительно по меньшей мере 5 баллов. В одном варианте осуществления, если пациента лечат в соответствии с раскрытыми способами, оценка тяжести зуда (ISS7) составляет менее 6, предпочтительно менее 4, предпочтительно менее 2 баллов, и наиболее предпочтительно оценка ISS7 составляет 0 баллов.
Еженедельная оценка активности крапивницы (UAS7)
Показатель UAS7 представляет собой сумму оценок по шкалам HSS7 и ISS7. Возможный диапазон еженедельной
оценки UAS7 составляет от 0 до 42 (максимальная активность).
В некоторых вариантах осуществления у пациента с CSU достигают улучшенной оценки UAS7 в ответ на лечение с помощью соединения формулы (I) или его фармацевтически приемлемой соли.
В предпочтительном варианте осуществления при лечении способом по настоящему изобретению у пациента с CSU наблюдается уменьшение количества элементов крапивной сыпи и уменьшение зуда, характеризующиеся оценкой UAS7≤6, к неделе 4 или к неделе 12.
В наиболее предпочтительном варианте осуществления при лечении способом по настоящему изобретению у пациента с CSU наблюдается полное отсутствие элементов крапивной сыпи и зуда к неделе 12, что оценивается как UAS7=0.
Более того, по сравнению с группой плацебо разница между группой лечения и группой плацебо составляет по меньшей мере 8 баллов, предпочтительно по меньшей мере 10 баллов.
Еженедельная оценка влияния на сон
Влияние на сон оценивается субъектом один раз в сутки утром в электронном дневнике. Оно
оценивается по шкале от 0 до 3. Еженедельная оценка варьирует от 0 до 21 (таблица 4).
Таблица 4. Оценка влияния на сон
В некоторых вариантах осуществления, если популяцию пациентов с CSU лечат в соответствии с раскрытыми способами, оценка влияния на сон улучшается на по меньшей мере 5 баллов. В предпочтительном варианте осуществления, если пациента лечат в соответствии с раскрытыми способами, оценка влияния на сон составляет менее 6, предпочтительно менее 4, предпочтительно менее 2 баллов, и наиболее предпочтительно оценка влияния на сон составляет 0 баллов.
Еженедельная оценка влияния на активность
Влияние на повседневную активность оценивается субъектами по шкале от 0 до 3 один раз в сутки вечером в электронном дневнике. Виды повседневной активности могут включать работу, учебу, спорт, хобби и виды активности, осуществляемой совместно с друзьями и семьей. Еженедельная оценка влияния на активность варьирует от 0 до 21 (таблица 5).
Таблица 5. Оценка влияния на повседневную активность
В некоторых вариантах осуществления, если популяцию пациентов с CSU лечат в соответствии с раскрытыми способами, оценка влияния на активность улучшается на по меньшей мере 5 баллов. В предпочтительном варианте осуществления, если пациента лечат в соответствии с раскрытыми способами, еженедельная оценка влияния на активность составляет менее 6, предпочтительно менее 4, предпочтительно менее 2 баллов, и наиболее предпочтительно еженедельная оценка влияния на активность составляет 0 баллов.
Применение H1-антигистаминных лекарственных препаратов неотложной помощи
Количество таблеток лекарственного препарата неотложной помощи, принятых за последние 24 часа для контроля таких состояний, как зуд или крапивная сыпь, записывается субъектом один раз в сутки вечером в электронном дневнике. Суточная доза
лекарственного препарата неотложной помощи рассчитывается как количество таблеток в день, умноженное на дозу
каждой таблетки, затем недельная доза лекарственного препарата неотложной помощи рассчитывается как сумма суточной дозы за 7 дней.
В некоторых вариантах осуществления, если популяцию пациентов с CSU лечат в соответствии с раскрытыми способами, количество доз в неделю лекарственного препарата неотложной помощи уменьшается. В одном аспекте данного варианта осуществления больше нет необходимости в использовании лекарственного препарата неотложной помощи.
Количество осуществленных телефонных звонков врачу или медсестре
Количество осуществленных телефонных звонков врачу, медсестре или практикующей медсестре из-за состояния кожи субъекта
записывается субъектом один раз в сутки в электронном дневнике.
Оценка активности ангионевротического отека (AAS)
AAS записывается субъектом один раз в сутки вечером в электронном дневнике. Данный валидированный инструмент позволяет оценить частоту возникновения и тяжесть эпизодов ангионевротического отека (Weller et al (2013), Allergy 68(9): 1185-92). Частота возникновения ангионевротического отека записывается субъектом один раз в сутки вечером в электронном дневнике. Действия и/или виды лечения, связанные с такими случаями возникновения ангионевротического отека, также записываются в электронном дневнике следующим образом (возможно несколько ответов):
Ничего не делал
Принимал лекарственные препараты, отпускаемые по рецепту или без рецепта
Позвонил своему врачу, медсестре или практикующей медсестре
Посетил своего врача, медсестру или практикующую медсестру
Отправился в отделение неотложной помощи в больнице
Был госпитализирован
Если субъекты отвечают на вводный вопрос "нет", оценка AAS для этого дня равна 0. Если ответом на вводный вопрос является "да", субъект продолжает отвечать на вопросы о продолжительности, тяжести, влиянии на повседневное функционирование и возникновении ангионевротического отека.
Каждому полю для ответа присваивается оценка от 0 до 3. Оценка AAS в этом исследовании
представлена в виде еженедельной оценки AAS (AAS7). Минимальный и максимальный возможные баллы по шкале AAS7 составляют 0 и 105 соответственно.
Более высокий балл означает более высокую степень тяжести.
В некоторых вариантах осуществления, если популяцию пациентов с CSU лечат в соответствии с раскрытыми способами, у пациентов достигают снижения оценки AAS7, предпочтительно до 0 баллов по шкале AAS7, к неделе 12 лечения. В другом аспекте этого варианта осуществления у пациента достигается ноль по шкале AAS7 в течение нескольких недель, например в течение по меньшей мере 4 недель лечения, по меньшей мере 8 недель лечения или в течение всех 12 недель лечения.
В некоторых вариантах осуществления, если популяцию пациентов с CSU лечат в соответствии с раскрытыми способами, у пациентов достигается показатель ≥95,5% дней без ангионевротического отека (AAS=0) с недели 4 по неделю 12.
Более того, при сравнении с группой плацебо разница между группой лечения и группой плацебо составляет по меньшей мере 6%.
Дерматологический индекс качества жизни (DLQI)
Дерматологический индекс качества жизни (DLQI) представляет собой показатель качества жизни (QoL), связанный с заболеваниями кожи (Finlay et al 1994). DLQI был валидирован для пациентов в возрасте 16 лет и старше. Субъекты оценивают свои дерматологические симптомы, а также влияние состояния их кожи на различные аспекты своей жизни, учитывая предыдущие 7 дней.
Рассчитывают общий балл, и он варьирует от 0 до 30 (более высокий балл означает худшее QoL, связанное с заболеванием). Оценки по сферам рассчитывают для следующего: симптомы и чувства (0-6), повседневная активность
(0-6), досуг (0-6), работа и школа (0-3), личные отношения (0-6), лечение (0-3).
Общий диапазон оценок DLQI был разделен на конкретные зоны оценок (Hongbo et al 2005) и их валидировали в отношении
их значения/значимости для пациентов следующим образом.
Таблица 6. Зоны оценок DLQI и их влияние на жизнь пациента
Зона DLQI Значение оценки
Оценка DLQI > 10 соответствует очень большому влиянию на жизнь пациентов и является обоснованием для назначения биологического средства, например как при псориазе (Kaplan et al. 2005). Опросники по DLQI заполняются во время рандомизации (день 1), на неделе 4 (день 29) и неделе 12 (день 85) в электронном дневнике.
Оценку DLQI следует выполнять перед любой другой оценкой и перед введением соединения формулы (I) или его фармацевтически приемлемой соли.
В некоторых вариантах осуществления у пациента с CSU достигают улучшения оценки DLQI в ответ на лечение с помощью соединения формулы (I) или его фармацевтически приемлемой соли. В некоторых вариантах осуществления у пациента с CSU достигают оценки DLQI, составляющей 0 или 1 балл, на неделе 4 или на неделе 12 лечения.
Индекс хронической крапивницы (индекс CU)
CU-Index® представляет собой коммерчески доступный анализ высвобождения гистамина из базофилов in vitro, в котором сыворотка крови пациента смешивается с донорскими базофилами, и уровни высвобожденного гистамина измеряются с помощью количественного иммуноферментного анализа. Значение индекса CU, составляющее 10 или больше, указывает на то, что у пациента имеется либо аутоиммунная основа своего заболевания (аутоантитела к IgE, FcεRI или антитела к FcεRII; положительный результат не позволяет определить, какое именно аутоантитело), либо альтернативный фактор высвобождения гистамина (Biagtan MJ, Viswanathan RK, Evans MD, et al (2011) Clinical utility of the Chronic Urticaria Index. J Allergy Clin Immunol; 127(6):1626-7).
В некоторых вариантах осуществления у пациента с CSU в ответ на лечение с помощью соединения формулы (I) или его фармацевтически приемлемой соли наблюдается снижение титров антител, связанных с патогенезом CSU (например, аутоантител к IgE, FcεRI или антител к FcεRII).
В другом варианте осуществления у пациента с CSU в ответ на лечение с помощью соединения формулы (I) или его фармацевтически приемлемой соли наблюдается снижение значения индекса CU. В некоторых вариантах осуществления пациент с CSU достигают снижения индекса CU до менее 10 на неделе 4 или на неделе 12 лечения.
В некоторых вариантах осуществления пациент подвергается лечению CSU в соответствии с заявленными способами в течение по меньшей мере 4 недель, по меньшей мере 12 недель, по меньшей мере 16 недель, по меньшей мере 48 недель или по меньшей мере 2 лет.
В предпочтительном варианте осуществления у пациента ранее наблюдался недостаточный ответ на традиционную системную терапию в отношении CSU (например, H1-антигистаминными препаратами второго поколения).
В других предпочтительных вариантах осуществления пациент представляет собой взрослого пациента (возрастом > 18 лет), имеющего CSU со степенью тяжести от умеренной до тяжелой. CSU со степенью тяжести от умеренной до тяжелой определяется как CSU, при которой у пациента имеется 7-дневная оценка активности крапивницы (UAS7), составляющая ≥16, и/или 7-дневная оценка тяжести крапивной сыпи (HSS7), составляющая ≥8.
В некоторых вариантах осуществления в ответ на лечение в соответствии с заявленными способами у пациента наблюдается быстрое уменьшение зуда при крапивной сыпи, измеряемое с помощью оценки UAS7, уже через 4 недели после введения начальной дозы или через 12 недель после введения начальной дозы. В предпочтительном варианте осуществления у пациента наблюдается уменьшение количества элементов крапивной сыпи и зуда с достижением показателя UAS7 ≤6 на неделе 4 или неделе 12. В наиболее предпочтительном варианте осуществления у пациента наблюдается полное отсутствие элементов крапивной сыпи и зуда (UAS7=0) на неделе 4 или неделе 12.
В некоторых вариантах осуществления в ответ на лечение в соответствии с заявленным способами у пациента наблюдается быстрое уменьшение частоты возникновения и тяжести ангионевротического отека, измеряемое с помощью оценки AAS7, уже через 4 недели после введения начальной дозы или через 12 недель после введения начальной дозы. В одном варианте осуществления у пациента наблюдается полное отсутствие ангионевротического отека в течение по меньшей мере 8 недель из 12 недель лечения (измеряемое как оценка AAS7, составляющая ноль в течение периода по меньшей мере 8 недель из 12 недель лечения). В предпочтительном варианте осуществления у пациента наблюдается полное отсутствие ангионевротического отека в течение всего периода лечения (измеряемое как оценка AAS7, равная нулю, в течение периода 12 недель).
В некоторых вариантах осуществления в ответ на лечение заявленными способами у пациента наблюдается снижение титров антител, связанных с патогенезом CSU (аутоантител к IgE, FcεRI или антител к FcεRII), и/или снижение значения индекса CU до менее 10.
Фармацевтическая композиция
Ингибитор BTK, т. е. соединение формулы (I) или его фармацевтически приемлемую соль, можно использовать в качестве фармацевтической композиции в комбинации с фармацевтически приемлемым носителем. Такая композиция может содержать, в дополнение к соединению формулы (I) или его фармацевтически приемлемой соли, носители, различные разбавители, наполнители, соли, буферы, стабилизаторы, солюбилизаторы и другие материалы, известные из уровня техники. Характеристики носителя зависят от пути введения. Фармацевтические композиции для применения в раскрытых способах также могут содержать дополнительные терапевтические средства для лечения конкретного целевого нарушения. Например, фармацевтическая композиция может также включать противовоспалительные и противозудные средства. Такие дополнительные факторы и/или средства могут быть включены в фармацевтическую композицию для получения синергического эффекта с соединением формулы (I) или его фармацевтически приемлемой солью, или для минимизации побочных эффектов, вызванных соединением формулы (I) или его фармацевтически приемлемой солью. В предпочтительных вариантах осуществления фармацевтическая композиция для применения в раскрытых способах содержит соединение формулы (I) или его фармацевтически приемлемую соль в дозе приблизительно 5 мг, приблизительно 10 мг, приблизительно 20 мг, приблизительно 25 мг, приблизительно 50 мг или приблизительно 100 мг.
Подходящие композиции для перорального введения включают эффективное количество соединения по настоящему изобретению в форме таблеток, пастилок для рассасывания, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул, или сиропов, или настоек. Композиции, предназначенные для перорального применения, получают в соответствии с любым способом, известным из уровня техники, предназначенным для изготовления фармацевтических композиций, и такие композиции могут содержать одно или несколько средств, выбранных из группы, состоящей из подслащивающих веществ, ароматизирующих средств, красящих средств и консервантов, с целью получения препаратов, которые являются фармацевтически эстетичными и приятными на вкус. Таблетки могут содержать активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми вспомогательными веществами, которые являются подходящими для изготовления таблеток. Такие вспомогательные вещества представляют собой, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и разрыхляющие средства, например кукурузный крахмал или альгиновая кислота; связующие средства, например крахмал, желатин или аравийская камедь; и смазывающие средства, например стеарат магния, стеариновая кислота или тальк. Таблетки являются непокрытыми или покрытыми посредством известных методик для замедления распада и абсорбции в желудочно-кишечном тракте и обеспечения таким образом устойчивого действия в течение более длительного периода. Например, можно использовать материал для обеспечения замедленного действия, такой как глицерилмоностеарат или глицерилдистеарат. Составы для перорального применения могут быть представлены в виде твердых желатиновых капсул, где активный ингредиент смешан с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где активный ингредиент смешан с водой или масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом.
Фармацевтические композиции для применения в раскрытых способах можно изготавливать обычным способом. В одном варианте осуществления фармацевтическая композиция предусмотрена для перорального введения. Например, фармацевтические композиции представляют собой таблетки или желатиновые капсулы, содержащие активный ингредиент вместе с
a) разбавителями, например лактозой, декстрозой, сахарозой, маннитом, сорбитом, целлюлозой и/или глицином;
b) смазывающими веществами, например диоксидом кремния, тальком, стеариновой кислотой, ее магниевой или кальциевой солью и/или полиэтиленгликолем; в случае таблеток также со
c) связующими средствами, например алюмосиликатом магния, крахмальной пастой, желатином, трагакантом, метилцеллюлозой, натрия карбоксиметилцеллюлозой и/или поливинилпирролидоном; при необходимости
d) разрыхлителями, например видами крахмала, агаром, альгиновой кислотой или ее натриевой солью или шипучими смесями; и/или
e) абсорбентами, красящими веществами, ароматизаторами и подсластителями.
Таблетки могут быть покрыты либо пленочной оболочкой, либо кишечнорастворимой оболочкой в соответствии со способами, известными из уровня техники.
Комбинации
При практическом применении некоторых способов лечения или вариантов применения настоящего изобретения терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли вводится пациенту, например млекопитающему (например, человеку). Хотя понятно, что раскрытые способы предусматривают лечение пациентов с CSU с применением соединения формулы (I) или его фармацевтически приемлемой соли, терапия не обязательно является монотерапией. Действительно, если пациент выбран для лечения с помощью соединения формулы (I) или его фармацевтически приемлемой соли, то соединение формулы (I) или его фармацевтически приемлемую соль можно вводить в соответствии со способами по настоящему изобретению либо отдельно, либо в комбинации с другими средствами и терапевтическими средствами для лечения пациентов с CSU, например в комбинации с по меньшей мере одним дополнительным средством для лечения CSU. При совместном введении с одним или несколькими дополнительными средствами для лечения CSU соединение формулы (I) или его фармацевтически приемлемую соль можно вводить либо одновременно с другим средством, либо последовательно. При последовательном введении лечащий врач примет решение относительно соответствующей последовательности введения соединения формулы (I) в комбинации с другими средствами и соответствующих дозировок для совместной доставки.
В ходе лечения CSU различные терапевтические средства могут быть объединены с раскрытым соединением формулы (I), с обеспечением благоприятного эффекта. Такие терапевтические средства включают местные средства для лечения (кремы [нестероидные или стероидные], жидкости для промывания, антисептики), системные средства для лечения (например, с использованием биологических препаратов, антибиотиков или химических веществ) и антисептики, фотодинамическую терапию и хирургическое вмешательство (воздействие лазером, дренирование или разрезание, иссечение).
Неограничивающие примеры местных средств для лечения CSU для применения с раскрытыми соединением формулы (I) или его фармацевтически приемлемой солью, включают бензоилпероксид, местные стероидные кремы, местные антибиотики аминогликозидной группы, такие как клиндамицин, гентамицин и эритромицин, резорциновый крем, скрабы с йодом и хлоргексидин.
Неограничивающие примеры средств для лечения CSU, используемых в системном лечении, предусматриваемых для применения с раскрытыми соединением формулы (I) или его фармацевтически приемлемой солью, включают антагонисты IgE (омализумаб, лигезумаб).
Дополнительные средства для лечения CSU, предусматриваемые для применения в комбинации с раскрытым соединением формулы (I) при лечении CSU, включают циклоспорин и кортикостероиды (инъекционные или пероральные).
Квалифицированный специалист в данной области техники сможет определить соответствующие дозы вышеуказанных средств для лечения CSU для совместной доставки с раскрытыми соединением формулы (I) или его фармацевтически приемлемой солью.
Наборы по настоящему изобретению
Настоящее изобретение также предусматривает наборы для лечения CSU. Такие наборы содержат ингибитор BTK, например соединение формулы (I) или фармацевтическую композицию на его основе. В одном варианте осуществления набор содержит две или больше отдельных фармацевтических композиций, по меньшей мере одна из которых содержит соединение формулы (I) или его фармацевтически приемлемую соль. В одном варианте осуществления набор содержит средства для раздельного содержания указанных композиций, такие как контейнер, разделенная бутылка или разделенный пакет из фольги. Примером такого набора является блистерная упаковка, как правило, применяемая для упаковывания таблеток, капсул и т. п.
Набор по настоящему изобретению можно применять для введения различных лекарственных форм, например для перорального и парентерального применения, для введения отдельных композиций с различными интервалами между введениями доз или для титрования отдельных композиций относительно друг друга. В целях содействия соблюдению режима лечения набор по настоящему изобретению, как правило, содержит инструкции по введению.
В видах комбинированной терапии по настоящему изобретению соединение формулы (I) или его фармацевтически приемлемая соль и другое средство для лечения CSU (как определено в данном документе) могут быть изготовлены и/или составлены одними и теми же или различными производителями. Более того, соединение формулы (I) или его фармацевтически приемлемая соль и другое средство для лечения CSU могут быть объединены с получением средства комбинированной терапии: (i) до того, как комбинированный продукт попадает к врачам (например, в случае набора, содержащего соединение формулы (I) или его фармацевтически приемлемую соль и другое средство для лечения CSU); (ii) самими врачами (или под наблюдением врача) незадолго до введения; (iii) в организме пациента, например, при последовательном введении соединения формулы (I) или его фармацевтически приемлемой соли и другого средства для лечения CSU.
Дополнительные варианты осуществления
Соединение формулы (I) или его фармацевтически приемлемую соль в целях удобства вводят пациенту (предпочтительно перорально) в дозе, составляющей от приблизительно 10 мг до приблизительно 200 мг в сутки.
Соединение формулы (I) или его фармацевтически приемлемую соль в целях удобства вводят пациенту (предпочтительно перорально) в суточной дозе, составляющей от приблизительно 10 мг до приблизительно 200 мг в сутки.
В некоторых вариантах осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в суточной дозе, составляющей от приблизительно 10 мг до приблизительно 100 мг.
В некоторых вариантах осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в суточной дозе приблизительно 10 мг, приблизительно 20 мг, приблизительно 25 мг, приблизительно 35 мг, приблизительно 50 мг, приблизительно 100 мг или приблизительно 200 мг.
В других вариантах осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в суточной дозе приблизительно 100 мг.
В других вариантах осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в суточной дозе приблизительно 50 мг.
В других вариантах осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в суточной дозе приблизительно 35 мг.
В других вариантах осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в суточной дозе приблизительно 25 мг.
В других вариантах осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят в суточной дозе приблизительно 20 мг.
В одном варианте осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят один раз в сутки в дозе приблизительно 10 мг, приблизительно 35 мг, приблизительно 50 мг или приблизительно 100 мг.
В другом варианте осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят два раза в сутки в дозе приблизительно 10 мг, приблизительно 25 мг, приблизительно 50 мг или приблизительно 100 мг.
Следует понимать, что для некоторых пациентов может потребоваться повышение дозы, например для пациентов с CSU, у которых наблюдается недостаточный ответ (например, при измерении с помощью любой из систем оценки CSU, раскрытых в данном документе, например с помощью способа по любому из предыдущих пунктов, где у указанного пациента достигается устойчивый ответ, измеряемый в виде полного ответа (оценка тяжести крапивной сыпи и зуда UAS7) и в виде дерматологического индекса качества жизни (DLQI) и т. д.), на лечение с помощью соединения формулы (I) или его фармацевтически приемлемой соли к неделе 4 или неделе 12 лечения. Также будет понятно, что снижение дозы также может потребоваться для определенных пациентов, например пациентов с CSU, у которых проявляются нежелательные явления или нежелательная реакция на лечение соединением формулы (I) или его фармацевтически приемлемой солью. Таким образом, дозировки соединения формулы (I) или его фармацевтически приемлемой соли могут составлять менее приблизительно 10 мг, приблизительно 20 мг, приблизительно 25 мг, приблизительно 50 мг или приблизительно 100 мг.
Временные рамки введения доз, как правило, отмеряют от дня введения первой дозы соединения формулы (I) или его фармацевтически приемлемой соли (что также известно как "исходный уровень"). Временные рамки введения доз, как правило, отмеряют от дня введения первой дозы соединения формулы (I) или его фармацевтически приемлемой соли (что также известно как "исходный уровень").
Однако медицинские работники зачастую используют разные способы наименования для идентификации графиков введения доз. Для обеспечения ясности, как описано в данном документе, первый день введения доз называется день 1. Однако квалифицированному специалисту в данной области техники будет понятно, что этот способ наименования используется просто для обеспечения соответствия и не должен рассматриваться как ограничивающий, т. е. ежедневное введение доз представляет собой обеспечение ежедневной дозы соединения формулы (I) или его фармацевтически приемлемой соли, и врач может ссылаться на конкретный день как на "день 0" или "день 1".
В данном документе раскрыты способы лечения хронической спонтанной крапивницы (CSU), предусматривающие введение нуждающемуся в этом пациенту соединения формулы (I) или его фармацевтически приемлемой соли в дозе, составляющей от приблизительно 10 мг до приблизительно 200 мг.
В данном документе также раскрыты способы лечения хронической спонтанной крапивницы (CSU), предусматривающие введение нуждающемуся в этом пациенту соединения формулы (I) или его фармацевтически приемлемой соли в суточной дозе, составляющей от приблизительно 10 мг до приблизительно 200 мг.
В данном документе также раскрыто соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении CSU, где суточная доза соединения составляет от приблизительно 10 мг до приблизительно 200 мг.
В одном варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемую соль вводят в суточной дозе, составляющей от приблизительно 10 мг до приблизительно 100 мг.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемую соль вводят в суточной дозе приблизительно 10 мг, приблизительно 20 мг, приблизительно 25 мг, приблизительно 35 мг, приблизительно 50 мг, приблизительно 100 мг или приблизительно 200 мг.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемую соль вводят в суточной дозе приблизительно 100 мг.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемую соль вводят в суточной дозе приблизительно 50 мг.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемую соль вводят в суточной дозе приблизительно 35 мг.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемую соль вводят в суточной дозе приблизительно 25 мг.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемую соль вводят в суточной дозе приблизительно 20 мг.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемую соль вводят один раз в сутки в дозе приблизительно 10 мг, приблизительно 35 мг, приблизительно 50 мг или приблизительно 100 мг.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемую соль вводят в дозе приблизительно 10 мг, приблизительно 25 мг, приблизительно 50 мг или приблизительно 100 мг два раза в сутки.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов до лечения с помощью соединения формулы (I) или его фармацевтически приемлемой соли пациент ранее получал лечение с помощью системного средства для лечения CSU. В одном аспекте данного варианта осуществления системное средство выбрано из группы, состоящей из H1-антигистаминных средств (H1-AH), H2-антигистаминных средств (H2-AH) и антагониста лейкотриеновых рецепторов (LTRA) и их комбинаций.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов до лечения с помощью соединения формулы (I) или его фармацевтически приемлемой соли пациент ранее не получал лечение с помощью системного средства для лечения CSU.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов до лечения с помощью соединения формулы (I) или его фармацевтически приемлемой соли у пациента имеется CSU со степенью тяжести от умеренной до тяжелой; т. е. пациент имеет оценку UAS7 ≥16 и/или оценку HSS7 ≥8.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов до лечения с помощью соединения формулы (I) или его фармацевтически приемлемой соли у пациента имеется либо аутоиммунная основа своего заболевания (аутоантитела к IgE, FcεRI или антитела к FcεRII) или альтернативный фактор высвобождения гистамина.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов до лечения с помощью соединения формулы (I) или его фармацевтически приемлемой соли пациент имеет индекс CU, составляющий 10 или больше. В конкретном аспекте данного варианта осуществления к неделе 4 или неделе 12 лечения у пациента достигается снижение индекса CU до менее 10.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов пациент выбирается в соответствии с по меньшей мере одним из следующих критериев:
а) до лечения с помощью соединения формулы (I) или его фармацевтически приемлемой соли пациент имел оценку UAS7 ≥16;
b) до лечения с помощью соединения формулы (I) или его фармацевтически приемлемой соли пациент имел оценку HSS7 ≥8.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов пациент является взрослым.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов к неделе 4 или неделе 12 лечения у пациента достигается по меньшей мере одно из следующего:
а) уменьшение количества элементов крапивной сыпи и уменьшение зуда, измеряемые как UAS≤6, или полное отсутствие крапивной сыпи и зуда (UAS7=0);
b) дерматологический индекс качества жизни (DLQI), составляющий 0-1;
c) отсутствие ангионевротического отека, измеряемое как оценка активности ангионевротического отека (AAS7), составляющая ноль.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов у пациента достигается устойчивый ответ, измеренный в виде полного ответа, в котором учитывается крапивная сыпь и зуд ([UAS7]=0), и/или в виде дерматологического индекса качества жизни (DLQI), составляющий 0-1, и/или в виде продолжающегося отсутствия ангионевротического отека (AAS7=0) к неделе 4 после завершения лечения.
Дополнительные пронумерованные варианты осуществления
1. Способ лечения хронической спонтанной крапивницы (CSU), включающий введение нуждающемуся в этом субъекту суточной дозы, составляющей от приблизительно 10 мг до приблизительно 200 мг N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида или его фармацевтически приемлемой соли.
2. Способ в соответствии с вариантом осуществления 1, где суточная доза N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида или его фармацевтически приемлемой соли составляет от приблизительно 10 мг до приблизительно 100 мг.
3. Способ в соответствии с вариантом осуществления 1, где суточная доза N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида или его фармацевтически приемлемой соли составляет приблизительно 100 мг.
4. Способ в соответствии с вариантом осуществления 1, где суточная доза N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида или его фармацевтически приемлемой соли составляет приблизительно 50 мг.
5. Способ в соответствии с вариантом осуществления 1, где суточная доза N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида или его фармацевтически приемлемой соли составляет приблизительно 35 мг.
6. Способ в соответствии с вариантом осуществления 1, где суточная доза N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида или его фармацевтически приемлемой соли составляет приблизительно 25 мг.
7. Способ в соответствии с вариантом осуществления 1, где суточная доза N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида или его фармацевтически приемлемой соли составляет приблизительно 20 мг.
8. Способ в соответствии с вариантом осуществления 1, где N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид или его фармацевтически приемлемую соль вводят один раз в сутки в дозе приблизительно 10 мг, приблизительно 35 мг, приблизительно 50 мг или приблизительно 100 мг.
9. Способ в соответствии с вариантом осуществления 1, где N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид или его фармацевтически приемлемую соль вводят в дозе приблизительно 10 мг, приблизительно 25 мг, приблизительно 50 мг или приблизительно 100 мг два раза в сутки.
10. Способ в соответствии с любым из предыдущих вариантов осуществления, где до лечения субъект ранее получал лечение с помощью системного средства для лечения CSU.
11. Способ в соответствии с вариантом осуществления 10, где системное средство выбрано из группы, состоящей из H1-антигистаминных средств (H1-AH), H2-антигистаминных средств (H2-AH) и антагониста лейкотриеновых рецепторов (LTRA) и их комбинаций.
12. Способ в соответствии с любым из вариантов осуществления 1-9, где до лечения субъект ранее не получал лечение с помощью системного средства для лечения CSU.
13. Способ в соответствии с любым из предыдущих вариантов осуществления, где у субъекта имеется CSU со степенью тяжести от умеренной до тяжелой.
14. Способ в соответствии с любым из вариантов осуществления 1-13, где субъект выбран в соответствии с по меньшей мере одним из следующих критериев:
а) до лечения с помощью соединения формулы (I) или его фармацевтически приемлемой соли субъект имеет оценку UAS7 ≥16;
b) до лечения с помощью соединения формулы (I) или его фармацевтически приемлемой соли субъект имеет оценку HSS7 ≥8;
15. Способ в соответствии с любым из предыдущих вариантов осуществления, где субъект является взрослым.
16. Способ в соответствии с любым из предыдущих вариантов осуществления, где к неделе 4 или неделе 12 лечения у указанного субъекта достигается по меньшей мере одно из следующего:
а) уменьшение количества элементов крапивной сыпи и уменьшение зуда, измеряемые как UAS≤6, или полное отсутствие крапивной сыпи и зуда (UAS7=0); или
b) дерматологический индекс качества жизни (DLQI), составляющий 0-1;
c) отсутствие ангионевротического отека, измеряемое как оценка активности ангионевротического отека (AAS7), составляющая ноль.
17. Способ в соответствии с любым из предыдущих вариантов осуществления, где у указанного субъекта достигается устойчивый ответ, измеренный в виде полного ответа, в котором учитывается крапивная сыпь и зуд ([UAS7]=0), и/или в виде дерматологического индекса качества жизни (DLQI)=0-1, и/или продолжающегося отсутствия ангионевротического отека (AAS7=0) к неделе 4 после завершения лечения.
18. Способ в соответствии с любым из предыдущих вариантов осуществления, где N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид или его фармацевтически приемлемая соль помещены в фармацевтический состав, где указанный фармацевтический состав дополнительно содержит фармацевтически приемлемые носители.
19. Способ в соответствии с любым из вариантов осуществления 1-18, где N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид или его фармацевтически приемлемая соль характеризуются значением Tmax, составляющим приблизительно 0,5-3 часа.
СОКРАЩЕНИЯ
AE нежелательный эффект
AUC площадь под кривой
AUCinf площадь под кривой зависимости концентрации в плазме крови (или сыворотке крови или крови) от времени,
с момента времени ноль до бесконечности (масса x время/объем)
AUClast площадь под кривой зависимости концентрации в плазме крови (или сыворотке крови или крови) от времени,
с момента времени ноль до момента времени, в котором концентрация поддается количественному определению (масса x время/объем)
AUCtau площадь под кривой зависимости концентрации в плазме крови (или сыворотки крови или крови) от времени,
с момента времени ноль до окончания интервала введения доз тау (масса x время/объем)
b.i.d. два раза в сутки
BMI индекс массы тела
CL/F кажущийся системный (или общий организменный) клиренс из плазмы крови (или сыворотки крови, или крови)
после введения (масса/объем)
Cmax максимальная концентрация после введения лекарственного средства
CSU хроническая спонтанная крапивница
ECG электрокардиограмма
Emax максимальное изменение эффекта по сравнению с плацебо
FcγR Fc-гамма-рецептор
FcεR Fc-альфа-рецептор
MRT среднее время удержания
PK фармакокинетика
PD фармакодинамика
PRO результаты лечения, сообщаемые пациентами
QoL качество жизни
q.d. один раз в сутки
QTcF интервал QT, скорректированный по формуле Фредерика
SAE серьезный нежелательный эффект
Tmax предельная продолжительность периода времени до достижения максимальной концентрации после введения лекарственного средства
T1/2 конечный период полувыведения
Tlast время последней измеряемой концентрации в профиле PK
Vz/F кажущийся объем распределения во время конечной фазы выведения
после введения (объем)
Пример 1. Доклинические исследования
Пример 1a. Взаимосвязь занятости BTK и PK/PD в доклинических исследованиях
PD-эффекты in vivo необратимого ингибитора BTK, такого как соединение (I), определяются степенью и продолжительностью ковалентной занятости BTK ингибитором. Занятость BTK после обработки соединением формулы (I) (также называемым соединением (I)) измеряли с помощью иммунологического анализа ex vivo. Фракцию незанятого белка BTK анализировали после инкубации in vitro с ковалентным биотинилированным зондом BTK, поскольку соединение (I) и зонд связываются с BTK взаимоисключающим образом. Незанятый белок BTK, а также относительные уровни общего белка BTK определяли в лизатах выбранных тканей, и уровни незанятого белка BTK нормализовали к уровням общего белка BTK в тех же образцах.
У самок крыс однократная пероральная доза 3 мг/кг соединения (I) приводила к полной занятости BTK селезенки, доза 1 мг/кг приводила к занятости 76%-81%, тогда как после однократной дозы 0,3 мг/кг была достигнута только частичная занятость 30%. Занятость BTK в крови достигло уровней, соответствующих тем, которые наблюдались в селезенке. Из экспериментальных данных было очевидно, что кратковременного присутствия соединения (I) в системном кровотоке достаточно для достижения полной занятости BTK в нескольких тканях при низких пероральных дозах 1-3 мг/кг. Содержание соединения (I) в крови после дозы 1 мг/кг достигало 49,1 нМ через 0,5 часа и составляло 5,6 нМ через 5 часов после введения дозы. Такое кратковременное присутствие в системном кровотоке в очень низком количестве согласуется с моделью PK/PD, типичной для необратимых ингибиторов.
Продолжительность занятости BTK определяли у крыс и мышей после однократной пероральной дозы соединения (I) для селезенки, крови, лимфатических узлов и легких. У крыс для BTK в занятом состоянии был продемонстрирован длинный период полужизни в крови, составляющий примерно 87 часов. Расчетный период полужизни BTK в занятом состоянии в селезенке крысы значительно короче, чем в крови, всего примерно 5 часов. Различная скорость метаболизма может отражать тот факт, что В-клетки и моноциты в периферической крови, экспрессирующие BTK, находятся в состоянии покоя и метаболически относительно неактивны по сравнению с таковыми в селезенке. Ранее сообщалось о более длительной персистенции BTK в занятом состоянии в крови (Advani et al 2013, J Clin Onc; 31(1):88-94). Все другие проанализированные ткани (легкие и лимфатические узлы) показали метаболизм и период полужизни BTK в занятом состоянии, подобные таковым в селезенке.
Пример 1B. Эффективность in vivo в моделях острой кожной гиперчувствительности на мышах
Эффекты соединения (I) в отношении FcγR- и FcεR-индуцированной гиперчувствительности оценивали в двух моделях острых кожных реакций на мышах. Из-за менее благоприятной PK соединения (I) у мышей дозу соединения вводили два раза в сутки.
Продолжительность PD-эффекта в коже после однократной дозы оценивали на модели обратной пассивной реакции Артюса (RPA) воспаления, опосредованного FcγRIII тучных клеток. В этой модели RPA поликлональное антитело IgG вводят локально в дерму, в то время как растворимый антиген вводят системно путем внутривенной инъекции. В этой модели на мышах FcγRIII тучных клеток играет доминирующую роль с незначительным вкладом системы комплемента (Hazenbos et al. 1998, journal of immunology, 161(6), pp. 3026; Hazenbos et al., Immunity, 1996, 5(2), pp. 181; Köhl & Gessner 1999, Molecular immunology, 36(13-14), pp. 893; Sylvestre & Ravetch 1996, Immunity, 5(4), pp. 387; Sylvestre & Ravetch 1994, Science, 265(5175), pp. 1095). Было показано, что мыши с генетическим дефицитом BTK защищены от пассивной кожной реакции Артюса (Fiedler et al. 2011, blood, 117(4), pp. 1329).
Пероральная обработка соединением (I) в однократной дозе 3, 10, 30 и 100 мг/кг, проведенная за 2 часа до индукции ответа на RPA, уменьшала отек кожи дозозависимым образом. Максимальные эффекты наблюдали при 30 и 100 мг/кг (уровень ингибирования составлял 73,0 и 61,2% соответственно), а частичные эффекты наблюдали при 3 и 10 мг/кг (уровень ингибирования составлял 22,9 и 29,2% соответственно) (фигура 1). Занятость BTK в селезенке, наблюдаемая в полной мере через 5 часов после введения дозы, коррелировала с эффективностью в отношении отека кожи со следующими значениями: 68,1% (3 мг/кг), 82,1% (10 мг/кг), 91,3% (30 мг/кг), 99,3% (100 мг/кг) (фигура 2).
В этой модели ингибирование отека кожи было максимальным, когда соединение (I) вводили за 2 часа до индуцирования реакции Артюса. Эффект постепенно уменьшался и достигал исходного уровня, когда реакцию Артюса запускали через 45 часов или позже после введения дозы соединения (I). Это говорит о том, что занятость BTK в коже характеризуется такой же динамикой, как и в селезенке, легких и лимфатических узлах.
Эффекты соединения (I) в отношении острой кожной гиперчувствительности также оценивали после двух пероральных доз соединения (I) на мышах в модели пассивной кожной анафилаксии (PCA) (Ovary, 1958). Модель PCA на мышах использовали при низкой дозе сенсибилизирующих антител IgE для минимизации комплемент-зависимых анафилатоксинов (Schäfer et al. 2013, The journal of allergy and clinical immunology, 131(2), pp. 541; Hata et al. 1998, Journal of Experimental Medicine, 187(8), pp. 1235). Две пероральные дозы при каждом уровне из 3, 10, 30 или 100 мг/кг соединения (I), введенные за 14 и 2 часа до индуцирования кожной анафилаксии с помощью гаптена, показали дозозависимое ингибирование отека кожи, измеренное путем определения проницаемости сосудов с помощью красителя синего Эванса. Ингибирование отека кожи, измеренное путем определения проницаемости сосудов с помощью красителя синего Эванса, находилось в диапазоне от 60,7% (3 мг/кг), 66,9% (10 мг/кг), 69,2% (30 мг/кг) до 87,4% (100 мг/кг) (фигура 3). Занятость в селезенке через 2,5 часа после последней дозы находилась в диапазоне от 89,1 до 99,7% (что соответствует максимальной занятости) (фигура 4).
Исследования в примерах 1a и 1b позволяют предположить, что соединение (I) достигает своей мишени в коже и эффективно ингибирует кожные воспалительные реакции.
В этих доклинических фармакологических исследованиях занятость BTK и соответствующие фармакологические данные показали сильную корреляцию. Таким образом, занятость BTK и реакции кожной гиперчувствительности, опосредованные тучными клетками, являются подходящим биомаркером PD для применения в клинических исследованиях и, следовательно, их использовали в клинических исследованиях фазы 1.
Пример 2. Клиническое испытание фазы 1
Исследование первого применения у человека проводили для оценки безопасности и переносимости, фармакокинетики (PK) и фармакодинамики (PD) однократной и многократных доз соединения (I) при пероральном введении как один раз в сутки (qd), так и два раза в сутки (bid), у здоровых добровольцев и пациентов с атопическим диатезом для поддержки дальнейшей клинической разработки соединения (I) при аутоиммунных заболеваниях. В этом исследовании также изучали влияние приема пищи.
Исследование первого применения у человека с участием примерно 168 здоровых добровольцев (HV), из которых 64 (в частях 2 и 4) имели бессимптомный атопический диатез.
Часть 1 представляла собой двойное слепое (для субъекта и исследователя данные были замаскированы, для спонсора - нет) плацебо-контролируемое исследование с повышением дозы с использованием однократных нарастающих доз (SAD) с участием 10 когорт (N=80).
Часть 2 представляла собой двойное слепое (для субъекта и исследователя данные были замаскированы, для спонсора - нет), плацебо-контролируемое исследование с повышением дозы с использованием многократных нарастающих доз (MAD) (13 доз в течение 12 дней) с введением дозы один раз в сутки в 6 когортах здоровых добровольцев с бессимптомным атопическим диатезом (N=48).
Часть 3 представляла собой открытое перекрестное исследование эффектов приема пищи при однократной дозе у 12 HV.
Часть 4 представляла собой двойное слепое (для субъекта и исследователя данные были замаскированы, для спонсора - нет), плацебо-контролируемое исследование с использованием многократных доз (25 доз в течение 12 дней) с введением дозы два раз в сутки в 2 когортах здоровых добровольцев с бессимптомным атопическим диатезом (N=16).
Часть SAD (часть 1) предусматривала десять уровней доз, а части MAD (части 2 и 4) предусматривали восемь уровней доз (6 когорт с введением дозы один раз в сутки в части 2 и 2 когорты с введением дозы два раза в сутки в части 4). В каждую когорту случайным образом распределяли по восемь субъектов для получения либо соединения (I), либо соответствующего плацебо в соотношении 6:2 (активное вещество:плацебо) в частях SAD и MAD. В рамках части SAD, дозы, примерно в 4 раза превышающие расчетную фармакологически активную дозу (PAD), должны были подвергаться оценке до того, как была начата часть исследования MAD, при условии, что до того времени от части SAD не поступало сигнала безопасности. Общая суточная доза соединения (I), используемая в части 2 (схема MAD qd) и части 4 (схема многократных доз bid), не превышала наивысшего исследованного уровня дозы в части SAD. Более того, общая суточная доза в части 4 не превышала общую суточную дозу в части 2.
В части 1 (SAD) для первого введения на каждом уровне дозы должны были применять стратегию дозорного введения доз следующим образом. Первым двум субъектам дозу вводили в первый день (одному с активным лекарственным средством, одному с плацебо). После 48-часового периода наблюдения дозы вводили остальным 6 субъектам когорты (пяти с активным лекарственным средством, одному с плацебо).
Во всех частях исследования использовали стандартный мониторинг безопасности. Была включена специальная оценка возможных случаев возникновения синяков на коже. Для каждой когорты перед повышением дозы необходимо было проводить слепой обзор всех показателей жизненно важных функций, физикального обследования и анамнеза пациента, ЭКГ, нежелательных явлений и лабораторных параметров безопасности (биохимический анализ крови, гематологическое исследование и анализ мочи) до 96 часов после последней дозы, а также данных PK от группы предыдущей дозы (если таковые доступны) до 48 часов после последней дозы. Сводные отчеты о безопасности по сообщенным нежелательным явлениям, лабораторным параметрам клинической безопасности, QTc и частоте сердечных сокращений были предоставлены после завершения каждого уровня дозы.
В частях 1, 2 и 4 каждый субъект участвовал в 28-дневном скрининговом периоде (от дня -29 до дня -2), периоде определения исходного уровня, периоде лечения и периоде последующего наблюдения, который включал оценку в конце исследования.
В части 1 субъекты поступали в центр исследования в день -2 или -1 для оценки безопасности на исходном уровне и подтверждения соответствия критериям пригодности для включения в исследование. Пригодные для включения в исследование субъекты получали однократную дозу соединения (I) или плацебо натощак в день 1. Их госпитализировали с дня -1 до утра дня 5 (96 часов после последнего введения лекарственного средства).
В частях 2 и 4 субъекты поступали в день -2 или -1 для оценки безопасности на исходном уровне и подтверждения соответствия критериям пригодности для включения в исследование. Пригодные для включения в исследование субъекты получали первую дозу соединения (I) натощак в день 1 и продолжали принимать исследуемый лекарственный препарат натощак до дня 12 включительно. Субъектов госпитализировали со дня -2 или -1 до утра дня 16, который соответствовал 96 часам после приема последней дозы соединения (I). В частях 2 и 4 исследуемый лекарственный препарат давали один раз в сутки и два раза в сутки соответственно (подробности см. в графике проведения оценок).
Часть 3 представляла собой открытое рандомизированное двустороннее перекрестное исследование однократной дозы для оценки влияния приема пищи. В части 3 каждый субъект участвовал в 28-дневном скрининговом периоде (от дня -29 до дня -2), в 2 периодах определения исходного уровня (день -1) и 2 периодах лечения, каждый из которых состоял из введения однократной дозы в день 1 с последующей оценкой безопасности и PK до дня 5. Период лечения 2 состоял из визита последующего наблюдения и оценки в конце исследования в дни 22 и 40 соответственно. Два периода лечения были разделены периодом вымывания продолжительностью по меньшей мере 18 дней (+/- 1 день).
Основная(-ые) цель(-и)
Включена специальная оценка частоты возникновения синяков на коже.
Второстепенные цели
Исследовательские цели
Ответы на известный аллерген в кожном прик-тесте (части 2 и 4)
Клеточные биомаркеры PD ex vivo, которые обеспечивали дополнительные средства измерения ответа на лекарственное средство для соединения (I) за счет ингибирования дегрануляции базофилов периферической крови (FcεR-индуцированная экспрессия CD63 и CD203c)
Ключевые критерии включения
1. Здоровые субъекты мужского и женского пола в возрасте от 18 до 65 лет (включительно), имеющие хорошее состояние здоровья, как определено на основании анамнеза, физикального обследования, показателей жизненно важных функций, электрокардиограммы и лабораторных тестов при скрининге. Здоровые субъекты участвовали в части 2 или части 4 с атопическим диатезом согласно критериям пригодности для включения в исследование в этих конкретных частях исследования. У здоровых добровольцев с атопией при скрининге должен был наблюдаться положительный результат прик-теста на известный аллерген (атопический диатез), но они должны были быть клинически бессимптомными и не должны были нуждаться в приеме каких-либо системных лекарственных препаратов.
2. Субъекты должны были иметь массу тела по меньшей мере 50 кг и индекс массы тела (BMI) в диапазоне 18-30 кг/м2 (включительно). BMI=вес тела (кг)/[рост (м)]2
3. При скрининге и первом определении исходного уровня показатели жизненно важных функций (температура тела, систолическое и диастолическое кровяное давление и пульс) оценивали в положении сидя после пребывания субъекта в состоянии покоя в течение по меньшей мере 3 минут и еще раз (при необходимости) после 3 минут в положении стоя. Показатели жизненно важных функций в положении сидя должны были находиться в пределах следующих диапазонов (включительно):
температура тела при измерении в полости рта 35,0-37,5°C
систолическое кровяное давление 90-139 мм рт. ст.
диастолическое кровяное давление 50-89 мм рт. ст.
пульс 50-90 уд./мин.
Ключевые критерии исключения
1. В анамнезе повышенная чувствительность к любому из исследуемых лекарственных средств или к лекарственным средствам аналогичных химических классов.
2. В анамнезе клинически значимые отклонения ЭКГ или любые из следующих отклонений ЭКГ при скрининге и/или перед лечением:
интервал PR > 200 мс
комплекс QRS > 120 мс
QTcF > 450 мс (мужчины)
QTcF > 460 мс (женщины)
3. Уровни гемоглобина ниже 12,0 г/дл при скрининге или первом определении исходного уровня.
4. Количество тромбоцитов выходит за пределы диапазона нормальных значений (ниже 150×109/л или выше 450×109) при скрининге или первом определении исходного уровня.
5. Любые клинически значимые отклонения в любом из стандартных исследований коагуляции, включая протромбиновое время (PT), частичное тромбопластиновое время (PTT) или международное нормализованное отношение (INR) при скрининге и/или определении исходного уровня.
6. Наличие тромботических или тромбоэмболических событий или наличие таковых в анамнезе, или повышенный риск развития тромботических или тромбоэмболических событий.
Вводимые средства для лечения
Часть 1 (SAD)
Субъектов распределяли в одну из следующих 10 когорт. В каждую когорту случайным образом распределяли по 8 субъектов для получения либо соединения (I), либо соответствующего плацебо при общем соотношении 6:2. Первую субкогорту случайным образом распределяли в соотношении 1:1: одного субъекта для получения соединения (I) и одного субъекта для получения соответствующего плацебо. Остальных 6 субъектов каждой когорты, которым вводили дозу после 48-часового периода наблюдения за 2 субъектами, получившими дозу ранее, случайным образом распределяли в соотношении 5:1.
Когорта 1: однократная пероральная доза 0,5 мг соединения (I) или соответствующего плацебо.
Когорта 2: однократная пероральная доза 1,5 мг соединения (I) или соответствующего плацебо.
Когорта 3: однократная пероральная доза 5 мг соединения (I) или соответствующего плацебо.
Когорта 4: однократная пероральная доза 15 мг соединения (I) или соответствующего плацебо.
Когорта 5: однократная пероральная доза 30 мг соединения (I) или соответствующего плацебо.
Когорта 6: однократная пероральная доза 60 мг соединения (I) или соответствующего плацебо.
Когорта 7: однократная пероральная доза 100 мг соединения (I) или соответствующего плацебо.
Когорта 8: однократная пероральная доза 200 мг соединения (I) или соответствующего плацебо.
Когорта 9: однократная пероральная доза 400 мг соединения (I) или соответствующего плацебо.
Когорта 10: однократная пероральная доза 600 мг соединения (I) или соответствующего плацебо.
Часть 2 (MAD, схема qd)
Субъектов распределяли в одну из следующих 6 когорт. В каждой когорте по 8 субъектов случайным образом распределяли либо для получения соединения (I), либо для получения соответствующего плацебо в соотношении 6:2.
Когорта 1: многократные пероральные дозы 10 мг соединения (I) или соответствующего плацебо.
Когорта 2: многократные пероральные дозы 25 мг соединения (I) или соответствующего плацебо.
Когорта 3: многократные пероральные дозы 50 мг соединения (I) или соответствующего плацебо.
Когорта 4: многократные пероральные дозы 100 мг соединения (I) или соответствующего плацебо.
Когорта 5: многократные пероральные дозы 400 мг соединения (I) или соответствующего плацебо.
Когорта 6: многократные пероральные дозы до 600 мг соединения (I) или соответствующего плацебо.
Часть 3 (влияние приема пищи)
Субъектов случайным образом распределяли в одну из 2 последовательностей лечения (таблица 9) в соотношении 1:1.
Определение последовательности лечения для части 3.
Часть 4 (MAD, схема bid)
Субъектов распределяли в одну из следующих когорт. В каждой когорте по 8 субъектов случайным образом распределяли либо для получения соединения (I), либо для получения соответствующего плацебо в соотношении 6:2.
Когорта 1: многократные пероральные дозы 100 мг соединения (I) или соответствующего плацебо в схеме bid.
Когорта 2: многократные пероральные дозы 200 мг соединения (I) или соответствующего плацебо в схеме bid.
Данные фармакокинетики
Биоаналитические методы:
Фармакокинетические образцы получали из крови и оценивали при всех уровнях дозы у всех субъектов. Образцы от субъектов в группах плацебо не анализировали. Образцы для оценки PK от субъектов собирали в моменты времени, определенные в исследовании. Концентрации соединения (I) определяли в крови с помощью утвержденного метода LC-MS/MS.
Фармакокинетика однократных нарастающих доз 0,5 мг - 600 мг
Средняя концентрация соединения (I) в крови в зависимости от времени после однократных нарастающих доз показана на фигуре 5.
Соединение (I) быстро абсорбировалось, при этом время достижения Cmax составляло приблизительно 1-1,5 часа при всех дозах. Фаза абсорбции характеризовалась единственным отчетливым пиком абсорбции у большинства субъектов. Для распределения лекарственного средства было показано двухфазное снижение. Большая часть лекарственного средства была выведена в фазе первоначального распределения, что позволяет предположить, что по сути клиренс лекарственного средства может происходить до достижения равновесия в тканях всего тела. Кажущаяся конечная фаза выведения наступала только через 12 часов после введения дозы и могла быть измерена только у субъектов, получавших дозы 100 мг и выше. Измеряемые конечные периоды полужизни находились в диапазоне от 4 часов (100 мг) до 18 часов (600 мг), что приводило к тому, что среднее время удержания (MRT) в кровотоке составляло от 1 часа до 5 часов (MRT≈ T1/2/ln2). Фаза распределения продемонстрировала доминирующий независимый от дозы T1/2, составляющий ~1 ч. Расчетное среднее геометрическое значение клиренса из крови после перорального введения однократной дозы (CL/F) находилось в диапазоне от 250 до 506 л/ ч. в когортах SAD, с оценочным значением, равным приблизительно 383 л/ ч., во всех когортах.
Фармакокинетика многократных пероральных доз
Средняя концентрация соединения (I) в крови в зависимости от времени после многократных нарастающих доз 10 мг - 400 мг показана на фигуре 6.
Геометрическое среднее значение кажущегося клиренса в равновесном состоянии после перорального введения дозы (CLss/F, день 12 MAD, qd) находилось в диапазоне от 246 л/ ч. до 414 л/ ч. в разных когортах. В целом более низкий клиренс наблюдали в равновесном состоянии по сравнению с днем 1, но эта разница почти исчезала при дозах 100 мг и выше (таблица 8-1 (день 1) и таблица 8-2 (день 12)). Причиной такого поведения, вероятно, является ковалентное связывание мишени (BTK), способствующее первоначальному клиренсу соединения (I). Этот эффект наиболее заметен в день 1, так как в последующие дни оставшаяся мишень в занятом состоянии при минимальном уровне снижает долю вклада связывания мишени в клиренс (CLss/F). Естественно, эта разница уменьшается с увеличением дозы, когда занятость мишени при минимальном уровне является почти полной. Следовательно, обнаружили, что содержание лекарственного средства (AUC, Cmax) было выше в день 12 по сравнению с днем 1, о чем свидетельствует (внутрисубъектно) коэффициент накопления лекарственного средства (Racc), который находился в диапазоне от 5 (низкая доза) до 1,2 (высокая доза) и, как правило, был выше для AUC, чем для Cmax, что подтверждает возможность влияния на системный клиренс.
Таблица 8-1. Сводная информация о PK-параметрах соединения (I) при многократных нарастающих дозах от 10 до 600 мг, введение дозы q.d.
Профиль: день 1
N=6
N=6
N=6
N=6
N=6
N=6
Медиана (мин.-макс.) [n]
CV%=коэффициент вариации (%)=SD/среднее значение*100
Для Tmax и T1/2 представлены только значения медианы (мин.-макс.) [n].
Таблица 8-2.
Профиль: день 12
N=6
N=6
N=6
N=6
N=6
N=6
Медиана (мин.-макс.) [n]
CV%=коэффициент вариации (%)=SD/среднее значение*100
Для Tmax и T1/2 представлены только значения медианы (мин.-макс.) [n].
В целом, концентрации в крови через 24 часа после последней дозы обычно были ниже 1 нг/мл, за исключением нескольких субъектов, принимавших дозы 100 мг и выше, что указывало на почти полное вымывание соединения (I) за две последовательные дозы. Последнее также предполагает, что равновесное состояние достигается за несколько доз.
Из-за более быстрого метаболизма BTK в тканях также исследовали введение дозы b.i.d. Профили средней концентрации в крови с течением времени, полученные после многократных нарастающих доз 100 мг и 200 мг два раза в сутки, показаны на фигуре 7. В соответствии с результатами других когорт, быстрая абсорбция с Tmax примерно 1 ч. наблюдалась для доз после схемы b.i.d. Наблюдаемый коэффициент накопления (Racc) составлял 1,5 (100 мг) и 2,0 (200 мг) для AUC и приблизительно 1,65 для Cmax (обе дозы). Пропорциональное дозе увеличение AUCtau наблюдали в день 12, тогда как для Cmax обнаружили лишь небольшое увеличение (в 1,33 раза). В заключение, введение дозы соединения (I) b.i.d обеспечивает возможность более быстрого ресинтеза мишени в тканях во время интервала дозирования без ущерба для общего PK-профиля и необходимости лечения высокими дозами q.d.
Влияние приема пищи: результат части 3
Данные PK от когорты, в которой определяли влияние приема пищи, обобщенные в таблице 9 ниже, указывают на сниженную скорость абсорбции, о чем свидетельствует снижение Cmax в 1,25 раза, и более полную общую абсорбцию, о чем свидетельствует повышение значения AUC0-24 в 1,4 раза. Что наиболее важно, среднее значение Tmax смещалось с 1 ч. (натощак) до >3 ч. (после приема пищи) (фигура 8).
Таблица 9. Сводные данные по PK-параметрам: влияние приема пищи на соединение (I) после однократной дозы 60 мг.

CV=коэффициент вариации (%)=SD/среднее значение*100
Фармакодинамика
Определение фармакодинамических (PD) характеристик осуществляли путем оценки степени занятости мишени и ингибирования в дистальном участке пути. Измеренные значения степени занятости BTK в цельной крови человека (полученной в виде соотношения свободной и общей BTK) служили прямым маркером терапевтического воздействия на мишень.
Взаимосвязь между степенью занятости BTK, дозой, содержанием соединения в системном кровотоке и эффективностью в отношении сложного пути in vivo и данными по болезни установили на доклинических моделях для соединения формулы (I) (например, пример 1).
Соединение формулы (I) является необратимым ингибитором BTK, определяли степень и продолжительность занятости BTK. PD-эффект соединения (I) оценивали путем измерения содержания как свободной BTK (не связанной), так и общей BTK, в цельной крови с помощью твердофазного иммуноферментного анализа (ELISA) на платформе Meso Scale Diagnostics (MSD) в двух отдельных анализах.
Взаимосвязь между дозой и фармакодинамическими показателями характеризовали с помощью измеренных значений степени занятости BTK в крови человека (полученной в виде соотношения свободной и общей BTK), прямого маркера терапевтического воздействия на мишень. Степень занятости BTK определялась для однократных нарастающих доз в диапазоне от 0,5 до 400 мг, для многократных нарастающих доз q.d. в диапазоне от 10 до 400 мг и для многократных нарастающих доз b.i.d. в диапазоне от 100 до 200 мг.
Соединение формулы (I) продемонстрировало явное дозозависимое увеличение как степени, так и продолжительности занятости BTK в периферической крови. Пиковую степень занятости мишени обычно наблюдали уже через 0,5 часа после введения дозы, что указывает на быстрое начало действия без значимого гистерезиса действия лекарственного средства по сравнению с пиковым содержанием лекарственного средства. Исходя из его способности ковалентно связывать BTK, занятость мишени сохранялась еще долго после его удаления из системного кровообращения, что указывает на неравновесную взаимосвязь PK-PD. Соответственно, делают вывод, что продолжительность занятости BTK определяется скоростью синтеза BTK de novo.
В отличие от когорт с более низкими дозами (0,5-1,5 мг), однократные дозы 15 мг соединения (I) и выше обеспечивали установление максимальной степени занятости мишени, приближающейся к 100%, почти у всех субъектов, которая оставалась выше 80% через 24 ч. В то время как ответ значительно варьировал среди субъектов при дозе 15 мг, дозы 30 мг и выше обеспечивали устойчивую (> 24 ч.) и почти полную (>90%) занятость у всех субъектов с явно уменьшенной межсубъектной вариабельностью. Время обновления пула белка BTK до уровней до введения дозы составляло приблизительно 10 дней, что соответствует медиане T1/2 метаболизма приблизительно 48 часов (фигура 9).
После многократных доз соединения (I) уже при использовании 10 мг соединения (I) q.d. достигалась остаточная степень занятости BTK в крови >96% перед введением дозы в день 12.
Кроме того, ингибирование ex vivo активации базофилов (отслеживаемое по поверхностной экспрессии CD63 и CD203c) использовали в качестве дистального механистического биомаркера для исследования последующих PD-эффектов соединения (I). Для определения PD-эффекта соединения (I) в отношении активации базофилов цельную кровь стимулировали ex vivo с помощью антител к IgE. Дегрануляцию оценивали по процентному содержанию базофилов CD63+и CD203+с помощью проточной цитометрии.
После однократных нарастающих доз соединения (I) данные указывают на дозозависимое ингибирование FcεR1-опосредованной активации базофилов. Активация базофилов крови ex vivo, измеренная с помощью CD63, почти полностью ингибировалась (>89%) при дозах 60 мг и достигала почти 100% ингибирования при более высоких дозах через 24 ч. после введения дозы. Напротив, максимальное ингибирование CD203c через 24 ч. после однократной дозы соединения (I) (примерно 50% ингибирование) было достигнуто только при использовании 200 мг соединения (I).
В день 12, через 8 ч. после введения MAD q.d. или b.i.d. соединения формулы (I), уже наиболее низкая испытанная доза соединения (I) (10 мг q.d.) приводила к >90% ингибированию повышения экспрессии CD63, и остаточный уровень ингибирования CD63 составлял >90% при дозах соединения (I) ≥50 мг q.d. (фигура 10). Максимальное остаточное ингибирование активации CD203c в день 12 было неизменно выше, чем после однократной дозы соединения (I), и достигалось только при введении b.i.d. 100 мг и 200 мг соединения (I).
Способность соединения (I) ингибировать ответ на определенный аллерген оценивали с помощью кожного прик-теста (SPT) у здоровых субъектов с атопией в части исследования с использованием MAD в исследовании первого применения у человека. SPT выполняли перед введением дозы (при скрининге, на исходном уровне и перед введением дозы в день 1) и в различные моменты времени после первой дозы (день 1) и через 11 дней после введения дозы один раз в сутки (день 12).
Подобно ингибированию активации базофилов ex vivo, дозозависимый эффект в отношении диаметра волдыря был ярко выражен в когортах многократных нарастающих доз, на что указывало уменьшение среднего размера волдырей после введения дозы по сравнению с исходным уровнем (фигура 11). Эффект начал выходить на плато примерно при 100 мг соединения (I) q.d.
Обоснование выбора дозы/заключение
Здоровых добровольцев подвергали воздействию соединения (I) в клинических исследованиях фазы 1 с диапазоном доз от 0,5 до 600 мг, которые вводили в виде однократной дозы или вводили в течение вплоть до 18 дней один или два раза в сутки. Соединение (I) хорошо переносилось, и не было серьезных или тяжелых нежелательных явлений, связанных с приемом соединения (I). В клиническом исследовании наблюдаемые нежелательные явления (AE), по-видимому, не зависели от дозы, большинство из них были единичными и, как правило, носили умеренный характер. Таким образом, информация о клинической безопасности свидетельствует в пользу доз, выбранных для данного исследования фазы 2b.
Уровни доз по настоящему изобретению были получены в следующих анализах: степень занятости BTK, ингибирование активации базофилов (отслеживаемое по повышению экспрессии CD63 и CD203c) у здоровых добровольцев; и влияние на кожные прик-тесты (SPT) у здоровых добровольцев с бессимптомной атопией - непрямой показатель ингибирования тучных клеток и базофилов в коже.
В данном вышеописанном клиническом испытании введение 10 мг соединения (I) q.d. приводило к почти полной занятости BTK в крови, >90% снижению повышения экспрессии CD63 (через 8 ч. после введения СОЕДИНЕНИЯ (I) в равновесном состоянии), и минимальному ингибированию размера волдырей в SPT. Таким образом, 10 мг соединения (I) q.d соответствует началу биологической активности. При дозе 100 мг соединения формулы (I) среднее уменьшение размера волдырей в SPT начинало выходить на плато. Следовательно, 100 мг соединения соответствует максимальному эффекту СОЕДИНЕНИЯ (I). Средняя доза соединения (I), составляющая тридцать пять мг q.d., хорошо подходит для точного описания кривой доза-ответ СОЕДИНЕНИЯ (I) при введении q.d.
Соединение формулы (I) ингибирует BTK за счет ковалентного связывания. В то время как продолжительность занятости BTK в крови составляет >24 часа (ч.), быстрый метаболизм BTK в ткани (например, примерно 5 часов в селезенке грызунов) может потребовать введения соединения (I) b.i.d. для достижения максимальной эффективности. Дозы 10, 25 и 100 мг соединения (I) b.i.d. соответственно точно описывают кривые доза-ответ для соединения (I) при приеме два раза в сутки.
Безопасность для человека
Для анализа нежелательных эффектов субъектов, получавших плацебо, из всех когорт с использованием SAD и MAD (по 2 на когорту) и разделенные на част с использованием SAD и MAD, объединяли в одну группу плацебо (n=20 для SAD и n=16 для MAD), для сравнения с каждой отдельной группой дозы соединения (I) (n=6 в каждой) и с общей группой соединения (I) (n=60 для SAD, и n=48 для MAD). Не наблюдали очевидных серьезных различий в демографических данных между группой с использованием плацебо и группами с использованием активного ингредиента как для популяции SAD, так и для популяции MAD. В результате оценки безопасности в рамках исследования FIH на здоровых добровольцах не было выявлено значимых проблем с безопасностью при введении дозы вплоть до 600 мг.
Пример 3. Данные относительно эффективности и безопасности у пациентов с CSU, у которых симптомы сохраняются, несмотря на стандартное лечение
Фаза 2b проводится в виде многоцентрового рандомизированного двойного слепого плацебо-контролируемого исследования с параллельными группами для изучения безопасности, переносимости и эффективности шести групп доз перорально вводимого соединения формулы (I) у субъектов с недостаточно контролируемой CSU, несмотря на лечение с помощью H1-антигистаминных препаратов (второго поколения).
На протяжении всего исследования (т. е. с дня -14 до дня 113) субъекты находились на стабильной схеме лечения H1-антигистаминным лекарственным препаратом второго поколения в утвержденной на местном уровне лицензированной схеме введения доз ("фоновый лекарственный препарат").
Субъекты могут принимать дополнительный H1-антигистаминный лекарственный препарат второго поколения, который выводится в первую очередь посредством почечной экскреции (например, цетиризин, левоцетиризин или биластин), в качестве препарата неотложной помощи. H1-антигистаминный препарат неотложной помощи должен отличаться от фонового H1-антигистаминного препарата и может применяться только для лечения невыносимых симптомов (зуда) CSU на ежедневной основе на протяжении всего исследования (с дня -14 до дня 113).
Все другие виды терапии CSU (включая H1-антигистаминные препараты в дозах, превышающих утвержденные) запрещены (см. таблицу 10).
Схема плана исследования включает три периода:
скрининговый период (10-14 дней до рандомизации): в течение скринингового периода субъектов, предоставивших информированное согласие, оценивают на соответствие критериям пригодности для включения в исследование;
период лечения (с дня 1 по день 85): после скрининга пригодных для включения в исследование субъектов случайным образом распределяют в одну из следующих групп лечения в соотношении 1:1:1:1:1:1:1:
10 мг соединения (I) один раз в сутки;
35 мг соединения (I) один раз в сутки;
100 мг соединения (I) один раз в сутки;
10 мг соединения (I) два раза в сутки;
25 мг соединения (I) два раза в сутки;
100 мг соединения (I) два раза в сутки;
плацебо;
промежуточный анализ проводят для оценки зависимости доза-ответ соединения формулы (I) после того, как все субъекты осуществили свое посещение на неделе 4 (первичная конечная точка);
период последующего наблюдения (с дня 86 по день 113): за субъектами проводят последующее наблюдение для оценки устойчивости ответа после отмены лечения и для дальнейшей оценки безопасности.
Таблица 10. Запрещенные лекарственные препараты
Примечание: с дня 1 (исходный уровень) до недели 12 (период окончания лечения) использование системных кортикостероидов вообще запрещено.
Резюме протокола
Оценка эффективности соединения (I) по сравнению с плацебо в отношении изменения значения UAS7 от исходного уровня с течением времени.
Оценка эффективности соединения (I) по сравнению с плацебо в отношении достижения полного клинического ответа (UAS7=0) с течением времени.
Оценка эффективности соединения (I) по сравнению с плацебо в отношении достижения контроля заболевания (UAS7≤ 6) с течением времени.
Оценка влияния соединения (I) на ангионевротический отек (AAS7) в отношении количества недель с ответом AAS7=0 от исходного уровня до недели 12.
Оценка влияния соединения (I) на связанное с заболеванием качество жизни в отношении достижения оценки DLQI, равной 0 или 1 на неделе 4 и неделе 12.
Оценка влияния соединения (I) на связанное с CSU качество жизни в отношении изменения DLQI от исходного уровня до недели 4 и недели 12.
Оценка фармакокинетики (PK) соединения (I), полученной в результате перорального введения дозы (отбор образцов для исследования PK проводят на неделе 4 и неделе 12).
Оценка безопасности и переносимости соединения (I) у субъектов с CSU.
Диагноз CSU, поставленный за ≥ 6 месяцев перед скринингом.
Диагноз CSU, недостаточно контролируемой H1-антигистаминными препаратами второго поколения, определяемый следующим:
наличие зуда и крапивной сыпи в течение ≥6 недель подряд перед скринингом, несмотря на использование неседативных H1-антигистаминных препаратов в соответствии с местными руководствами по лечению в течение этого периода времени;
оценка UAS7 (диапазон 0-42) ≥16 и оценка HSS7 (диапазон 0-21) ≥8 в течение 7 дней до рандомизации (день 1);
наличие крапивной сыпи должно быть задокументировано в течение трех месяцев до рандомизации (либо при скрининге и/или рандомизации, либо задокументировано в истории болезни субъекта).
Другие заболевания с симптомами крапивницы или ангионевротического отека, включая без ограничения уртикарный васкулит, пигментную крапивницу, многоформную эритему, мастоцитоз, наследственную крапивницу или приобретенную/лекарственную крапивницу.
Любое другое кожное заболевание, связанное с хроническим зудом, которое, по мнению исследователей, может повлиять на оценки и результаты исследования, например атопический дерматит, буллезный пемфигоид, герпетиформный дерматит, старческий зуд или псориаз.
Наличие на момент исследования или в анамнезе диагностированных отклонений ЭКГ от нормы, указывающих на значительный риск безопасности для субъектов, участвующих в исследовании, таких как следующие:
• сопутствующие клинически значимые формы нарушения сердечного ритма, например устойчивая желудочковая тахикардия и клинически значимая AV-блокада второй или третьей степени без кардиостимулятора;
• наличие синдрома наследственного удлиненного интервала QT в анамнезе или известный семейный анамнез двунаправленной веретенообразной желудочковой тахикардии;
• частота сердечных сокращений в покое (медицинское обследование или ЭКГ в 12 отведениях) < 60 уд./мин.;
• QTcF в состоянии покоя ≥450 мс (мужчины) или ≥460 мс (женщины) до лечения [при скрининге] или невозможность определения интервала QTcF;
• применение средств, которые, как известно, обеспечивают удлинение интервала QT, если только они не могут быть отменены на весь период исследования.
Значительный риск кровотечения или нарушения свертываемости крови.
Известное или предполагаемое текущее, хроническое или рецидивирующее инфекционное заболевание в анамнезе, включая без ограничения оппортунистические инфекции (например, туберкулез, атипичные микобактериозы, листериоз или аспергиллез), HIV, гепатит B/C.
Ежедневно заполняемый дневник пациента с крапивницей (UPDD), включая следующее:
оценка активности крапивницы (UAS);
оценка активности ангионевротического отека (AAS);
оценка влияния CSU на сон и повседневную активность;
дерматологический индекс качества жизни (DLQI).
Физикальное обследование
Показатели жизненно важных функций
Мониторинг лабораторных маркеров в крови и моче
Мониторинг ЭКГ
Первичные конечные точки
Первичной переменной для исследования является изменение значения UAS7 от исходного уровня до недели 4. UAS7 представляет собой сумму усредненных ежедневных оценок UAS за 7 дней. Следует обратить внимание, что еженедельная оценка рассчитывается с использованием последних 7 дней до визита.
Вторичные конечные точки
Эффективность настоящего способа анализируется по группам лечения и визитам (если применимо) с использованием описательной статистики, которая включает абсолютные и относительные частоты для категориальных переменных и среднее арифметическое, стандартное отклонение, минимум, максимум, медиану и первый и третий квартили для непрерывных переменных. Для вторичного анализа UAS7, определения полного клинического ответа, контролируемости заболевания и AAS7=0 проводят попарные сравнения каждой дозы соединения (I) с плацебо.
UAS7
Обобщенная статистика абсолютного и процентного изменения значения UAS7 от исходного уровня анализируется по группам лечения и визитам в периоды лечения и последующего наблюдения.
Полный клинический ответ
Полный клинический ответ, т. е. отсутствие крапивной сыпи и зуда, определяется как субъекты, у которых достигается UAS7=0.
Количество субъектов с UAS7=0 в зависимости от группы лечения и визита в периоды лечения и последующего наблюдения.
Попарные сравнения между группами лечения (отдельные группы соединения (I) по сравнению с плацебо).
Контролируемое заболевание (UAS7≤ 6)
Количество субъектов с UAS7≤ 6 в зависимости от группы лечения и визита в периоды лечения и последующего наблюдения.
Попарные сравнения между группами лечения (отдельные группы соединения (I) по сравнению с плацебо).
Отсутствие ангионевротического отека (AAS7=0)
Суммарное количество недель с ответом AAS7=0 между исходным уровнем и неделей 12.
Показатель получают на основе электронного дневника AAS. Еженедельную оценку AAS7 получали путем суммирования ежедневных оценок за 7 дней, предыдущих визиту, и она находится в диапазоне от 0 до 105. Если оценка AAS7 отсутствует, это рассматривается как отсутствие ответа в течение суммарного количества недель, в течение которых субъекты достигают ответа AAS7=0. Суммарное количество недель, в течение которых достигается ответ AAS7=0 между исходным уровнем и неделей 12, моделируется с использованием модели отрицательной биномиальной регрессии с логарифмической связью, с использованием группы лечения, страт рандомизации и состояния AAS7=0 на исходном уровне.
DLQI
Семь оценок получают из DLQI: оценку каждого из шести параметров, а также общую оценку DLQI рассчитывают на основе правил разработчиков. Для каждой из этих семи оценок также получают изменение от исходного уровня и процентное изменение от исходного уровня. Обобщенную статистику рассчитывают для абсолютных значений, а также для изменения и процентного изменения с разбивкой по визитам и группам лечения.
Количество субъектов с оценкой DLQI 0 или 1 в зависимости от группы лечения и визита.
Конечные точки безопасности
Все конечные точки безопасности (т. е. AE, лабораторные данные, показатели жизненно важных функций и ЭКГ, а также потенциальные риски, определенные в плане профилирования безопасности) обобщаются в зависимости от лечения для всех субъектов выборки безопасности. Все данные включаются в анализ независимо от того, использовались ли лекарственные препараты неотложной помощи.
Данные, полученные в клиническом исследовании, а также доклинические данные, представленные выше, демонстрируют, что соединение (I) при суточных дозах от 10 мг до 200 мг является безопасным и фармакологически активным в отношении лечения кожных заболеваний, обусловленных базофилами и тучными клетками.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ЛЕЧЕНИЯ ХРОНИЧЕСКОЙ СПОНТАННОЙ КРАПИВНИЦЫ С ПРИМЕНЕНИЕМ ЛИГЕЛИЗУМАБА | 2019 |
|
RU2783540C2 |
| СПОСОБЫ ЛЕЧЕНИЯ СИНДРОМА ШЕГРЕНА С ПРИМЕНЕНИЕМ ИНГИБИТОРА ТИРОЗИНКИНАЗЫ БРУТОНА | 2020 |
|
RU2824354C2 |
| ИНГИБИТОРЫ LTA4H ДЛЯ ЛЕЧЕНИЯ ГНОЙНОГО ГИДРАДЕНИТА | 2020 |
|
RU2821362C2 |
| КОМБИНАЦИЯ ИНГИБИТОРА PI3 КИНАЗЫ С ПАКЛИТАКСЕЛОМ ДЛЯ ИСПОЛЬЗОВАНИЯ ПРИ ЛЕЧЕНИИ ИЛИ ПРЕДОТВРАЩЕНИИ РАКА ГОЛОВЫ И ШЕИ | 2014 |
|
RU2672555C2 |
| Применение антагониста рецептора NK-1 серлопитанта при зуде | 2014 |
|
RU2666219C2 |
| ПРИМЕНЕНИЕ ИНГИБИТОРОВ BTK ПРИ ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ | 2021 |
|
RU2837046C1 |
| СПОСОБЫ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С РЕЦЕПТОРОМ S1P1 | 2021 |
|
RU2821032C1 |
| Фармацевтические комбинации | 2017 |
|
RU2759669C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА BTK | 2020 |
|
RU2828460C2 |
| ЛЕЧЕНИЕ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ С ИСПОЛЬЗОВАНИЕМ МОДУЛЯТОРОВ ИЗОФОРМ PIЗ-КИНАЗЫ | 2014 |
|
RU2705204C2 |
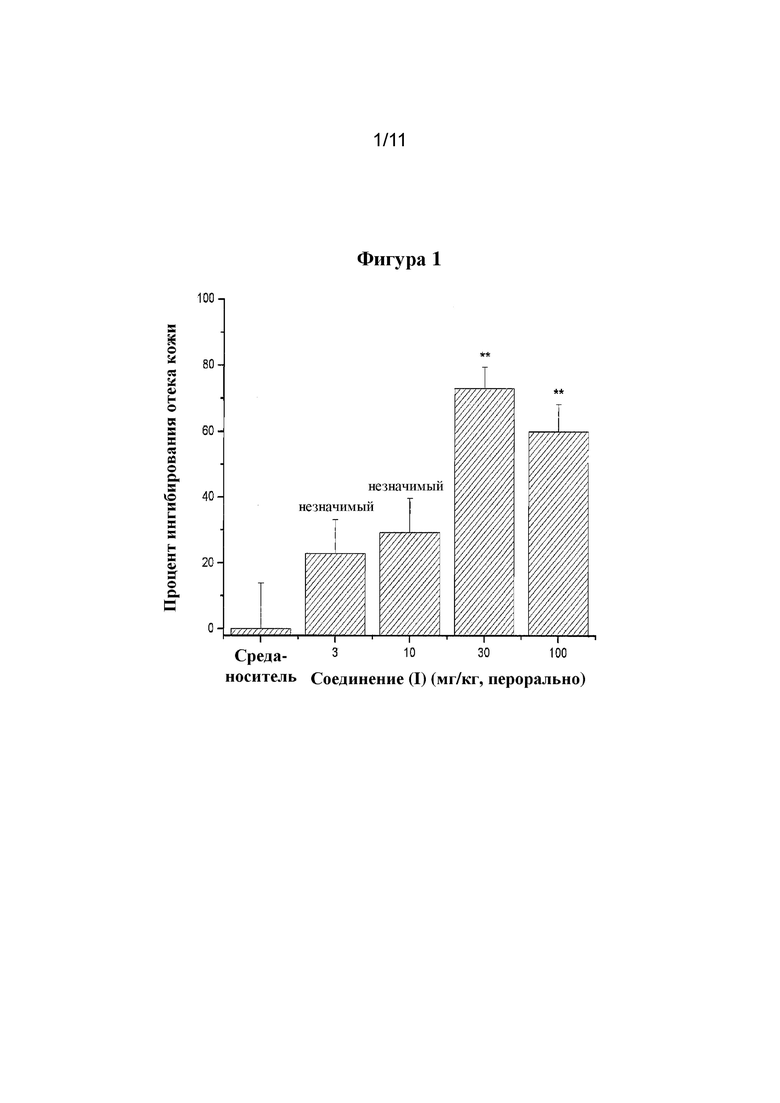
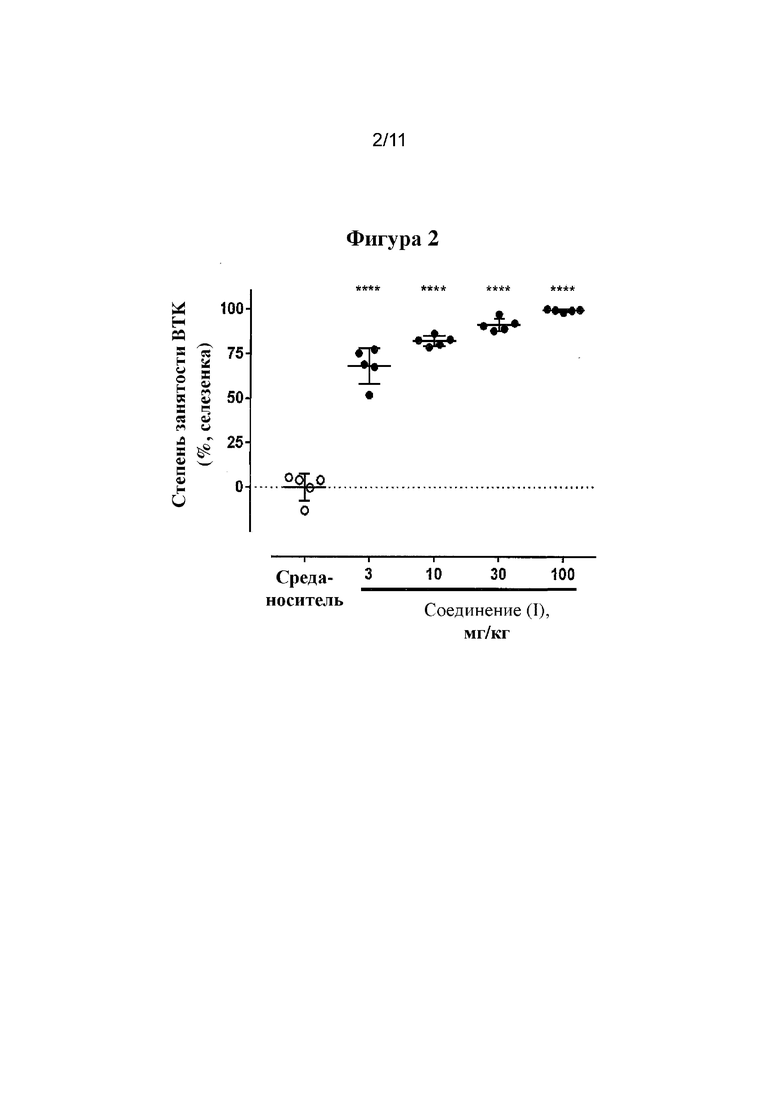









Изобретение относится к способам лечения кожных заболеваний. Раскрыт способ лечения хронической спонтанной крапивницы (CSU), включающий введение нуждающемуся в этом субъекту суточной дозы от приблизительно 10 мг до приблизительно 100 мг соединения формулы (I)
 (I),
(I),
где термин «приблизительно» означает +/-10%. Изобретение обеспечивает безопасное и эффективное лечение спонтанной хронической крапивницы. 17 з.п. ф-лы, 11 ил., 10 табл., 3 пр.
1. Способ лечения хронической спонтанной крапивницы (CSU), включающий введение нуждающемуся в этом субъекту суточной дозы от приблизительно 10 мг до приблизительно 100 мг соединения формулы (I)
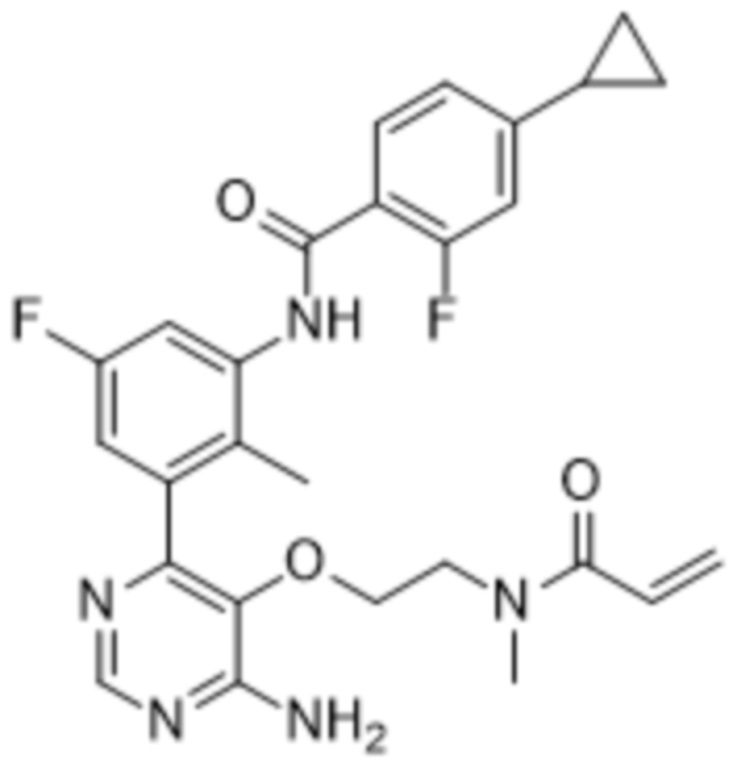 (I),
(I),
где термин «приблизительно» означает +/-10%.
2. Способ по п. 1, где суточная доза составляет 100 мг +/-10%.
3. Способ по п. 1, где суточная доза составляет 50 мг +/-10%.
4. Способ по п. 1, где суточная доза составляет 35 мг +/-10%.
5. Способ по п. 1, где суточная доза составляет 25 мг +/-10%.
6. Способ по п. 1, где суточная доза составляет 20 мг +/-10%.
7. Способ по п. 1, где соединение формулы (I) вводят один раз в сутки в дозе приблизительно 10 мг, приблизительно 35 мг, приблизительно 50 мг или приблизительно 100 мг и где термин «приблизительно» означает +/-10%.
8. Способ по п. 1, где соединение формулы (I) вводят в дозе приблизительно 10 мг, приблизительно 25 мг или приблизительно 50 мг два раза в сутки и где термин «приблизительно» означает +/-10%.
9. Способ по любому из предыдущих пунктов, где перед лечением с помощью соединения формулы (I) субъект ранее получал лечение с помощью системного средства для лечения CSU.
10. Способ по п. 9, где системное средство выбрано из группы, состоящей из H1-антигистаминных средств (H1-AH), H2-антигистаминных средств (H2-AH) и антагониста лейкотриеновых рецепторов (LTRA) и их комбинаций.
11. Способ по любому из пп. 1-8, где перед лечением с помощью соединения формулы (I) субъект ранее не получал лечения с помощью системного средства для лечения CSU.
12. Способ по любому из предыдущих пунктов, где субъект имеет CSU со степенью тяжести от умеренной до тяжелой.
13. Способ по любому из пп. 1-11, где субъект выбран в соответствии с по меньшей мере одним из следующих критериев:
а) до лечения с помощью соединения формулы (I) субъект имеет оценку UAS7, которая больше или равняется 16;
b) до лечения с помощью соединения формулы (I) субъект имеет оценку HSS7, которая больше или равняется 8.
14. Способ по любому из предыдущих пунктов, где субъект является взрослым.
15. Способ по любому из предыдущих пунктов, где у указанного субъекта к неделе 4 или неделе 12 лечения достигается по меньшей мере одно из следующего:
а) уменьшение количества элементов крапивной сыпи и зуда, измеряемое как значение UAS, которое меньше или равняется 6, или полное отсутствие крапивной сыпи и зуда (значение UAS7, равняющееся 0); или
b) дерматологический индекс качества жизни (DLQI), составляющий 0-1;
c) отсутствие ангионевротического отека, измеряемое как оценка активности ангионевротического отека (AAS7), составляющая ноль.
16. Способ по любому из предыдущих пунктов, где у указанного субъекта достигается устойчивый ответ, измеряемый в виде полного ответа, в котором учитывается крапивная сыпь и зуд ([UAS7] равняется 0), и/или в виде дерматологического индекса качества жизни (DLQI), равняющегося 0-1, и/или продолжающегося отсутствия ангионевротического отека (AAS7 равняется 0) к неделе 4 после завершения лечения.
17. Способ по любому из предыдущих пунктов, где соединение формулы (I) помещено в фармацевтический состав и где указанный фармацевтический состав дополнительно содержит фармацевтически приемлемые носители.
18. Способ по любому из пп. 1-16, где предельная продолжительность периода времени достижения соединением формулы (I) максимальной концентрации после введения составляет приблизительно 0,5-3 часа и где термин «приблизительно» означает +/-10%.
| US 20150152068 A1, 04.06.2015 | |||
| WO 2015105926 A1, 16.07.2015 | |||
| KIRAN G | |||
| et al | |||
| Position statement for the use of omalizumab in the management of chronic spontaneous urticaria in Indian patients // Indian Dermatology Online Journal, 2016, V | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| FERRER M | |||
| et al | |||
| Predicting Chronic Spontaneous Urticaria Symptom Return After Omalizumab | |||

