Область техники
Настоящее изобретение относится к соединениям N-замещенного диоксоциклобутениламино-3-гидроксипиколинамида, которые ингибируют CC-хемокиновый рецептор 6 (CCR6), фармацевтическим композициям, содержащим эти соединения, и применению этих соединений для лечения или профилактики заболеваний, состояний и расстройств, ослабляемых ингибированием CCR6.
Уровень техники
Хемокиновые рецепторы представляют собой большое подсемейство из семи трансмембранных белков клеточной поверхности. Согласно функциям их можно разделить на две основные группы: хемокиновые рецепторы, сопряженные с G-белком, которые опосредуют транспорт лейкоцитов, и «атипичные хемокиновые рецепторы», которые могут передавать сигналы через механизмы, не сопряженные с G-белком, и функционировать в качестве «мусорщиков» хемокинов, подавляя воспаление или формируя градиенты хемокинов [Bachelerie, 2014; Murphy, 2002]. Хемокины, естественные лиганды хемокиновых рецепторов, представляют собой низкомолекулярные белки, которые стимулируют рекрутмент лейкоцитов. На основании присутствия и относительного положения NH2-концевых остатков Cys, хемокины структурно сгруппированы в хемокины CC, CXC, CX3C и C [White, 2013]. У человека хемокины и хемокиновые рецепторы образуют фармакологически сложную систему. Во многих случаях распознавание одного хемокина разными рецепторами, а также связывание разных хемокинов с одним и тем же рецептором, приводит к разной передаче сигналов и транспорту [Kufareva, 2016]. Физиологически члены семейства хемокинов индуцируют рекрутмент четко определенных субпопуляций лейкоцитов и играют важную роль в развитии иммунитета и аутоиммунных заболеваний.
СС-хемокиновый рецептор 6 (CCR6) экспрессируется на разных ключевых иммунных клеток, включая незрелые дендритные клетки, B-клетки, Т-клетки памяти (включая все клетки Th17), нейтрофилы и субпопуляцию Treg. CCR6 является единственным известным рецептором хемокина CCL20 (альтернативно называемого LARC или MIP-3a). CCL20 продуцируется синовиоцитами, эпителиальными клетками толстого кишечника, различными клетками кожи (например, кератиноцитами и дермальными фибробластами) и альвеолярными эпителиальными клетками. Пара лиганд-рецептор CCL20-CCR6 является ответственной за миграцию незрелых дендритных клеток и эффекторных Т-клеток/Т-клеток памяти на коже и поверхности слизистых оболочек в условиях гомеостаза и воспаления, а также при аутоиммунных заболеваниях, таких как псориаз и воспалительные заболевания кишечника [Liao, 1999; Schutyser, 2003].
CCL20 представляет собой индуцибельный хемокин, который высоко активируется в очагах воспаления при различных аутоиммунных заболеваниях, включая псориаз, язвенный колит, болезнь Крона, псориатический артрит и ревматоидный артрит. Повышенное количество CCR6-положительных Т-клеток и дендритных клеток было обнаружено в очагах поражения, локализованных вместе с экспрессией CCL20. Эти кластеры Т-клетки-дендритные клетки, поддерживаемые взаимодействием CCR6-CCL20, имеют ключевое значение для инициации и сохранения заболевания. У мышей как нокаут CCR6, так и нейтрализующие антитела против CCL20 имели протективный эффект на модели индуцированного IL-23 воспаления кожи, предполагая, что блокирование CCR6-CCL20-опосредованного рекрутмента иммунных клеток представляет собой привлекательный механизм для новой низкомолекулярной терапии при аутоиммунных и воспалительных заболеваниях [Homey, 2000; Kim, 2014; Kwon, 2002; Shen, 2010].
Хемокиновый рецептор C-X-C типа 1 (CXCR1) и хемокиновый рецептор C-X-C типа 2 (CXCR2) представляют собой хемокиновые рецепторы, экспрессируемые на нейтрофилах. Оба рецептора связывают хемокин IL-8 (CXCL8) с высокой аффинностью. Напротив, CXCL1 (GRO альфа) и CXCL2 (GRO бета) являются специфическими лигандами для CXCR2 с 90% гомологией последовательностей. В клинических исследованиях двойные антагонисты CXCR1/2, такие как навариксин, показали обратимое снижение абсолютного количества нейтрофилов у пациентов [Hastrup, 2015]. Пациенты со сниженным уровнем нейтрофилов могут подвергаться повышенному риску инфекций. Следовательно, можно предположить, что антагонисты CCR6 с пониженной активностью в отношении антагонизма к рецептору CXCR2 могут иметь улучшенный профиль безопасности по сравнению с недифференцирующими антагонистами. Генетические данные для человека и результаты исследования с нокаутом на мышах предполагают, что выход нейтрофилов из костного мозга зависит от функции CXCR2 и опосредуется специфическими для CXCR2 лигандами CXCL1 (GRO alpha) и CXCL2 (GRO beta) [Auer, 2014; Eash, 2010]. Таким образом, чтобы понять фармакологию ингибиторов CXCR2 в отношении мобилизации нейтрофилов из костного мозга, важно использовать соответствующий лиганд CXCR2 (CXCL1 или CXCL2) в системе первичного анализа нейтрофилов человека.
Auer P. L. et. al. Nature Genetics (2014) 46, 629-634.
Bachelerie F. et. al. Pharmacol. Rev. (2014) 66, 1-79.
Eash K. J. et. al. J. Clin. Invest. (2010) 120, 2423-2431.
Hastrup N. et. al. Cytokine (2015) 72, 197-203.
Homey B. et. al. J. Immunol. (2000) 164, 6621-6632.
Kim T.-G. et. al. J. Invest. Dermatol. (2014) 134, 1462-1465.
Kufareva I. Curr. Opin. Pharmacol. (2016) 30, 27-37.
Kwon J. H. et. al. Gut (2002) 51, 818-826.
Liao, F. et. al. J. Immunol. (1999) 162, 186-194.
Murphy P. M. Pharmacol. Rev. (2002) 54, 227-229.
Schutyser E.; Struyf S.; Van Damme J. Cytokine Growth Factor Rev. (2003) 14, 409-426.
Shen H.; Goodall J. C.; Gaston J. S. H. J. Rheumatol. (2010) 37, 2096-2099.
White G. E.; Iqbal A. J.; Greaves D. R. Pharmacol. Rev. (2013) 65, 47-89.
Сущность изобретения
Настоящее изобретение относится к соединениям формул (IA) и (IB), которые ингибируют CCR6 и пригодны для лечения или профилактики расстройств, ослабляемых ингибированием CCR6, у людей:
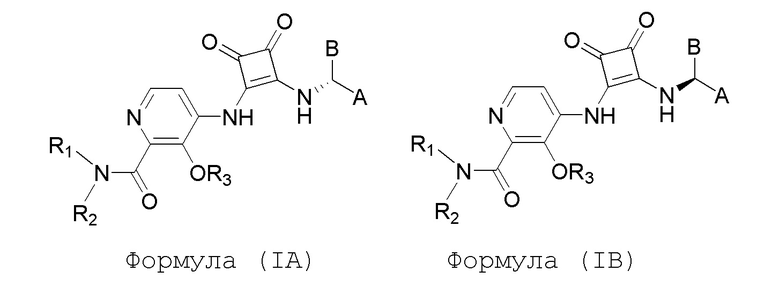
или его фармацевтически приемлемой соли или гидрату, где:
R1 и R2 независимо представляют собой H или (C1-C6)алкил, или R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют 4-, 5- или 6-членный гетероцикл, содержащий один гетероатом N и необязательно 1 или 2 дополнительных гетероатома, выбранных из группы, состоящей из O, N и S, где гетероцикл необязательно замещен 1, 2 или 3 (C1-C4)алкильными группами;
R3 представляет собой H, (C1-C6)алкил, (C1-C4)алкилкарбонил, -C(=O)CH=CHCO2H, -SO2NH2, -CH2OC(=O)(C1-C4)алкил, -CH2OP(=O)(OH)2 или -C(=O)NRARB, где (C1-C4)алкилкарбонил необязательно замещен -CO2H или -NH2, где -CH2OC(=O)(C1-C4)алкил необязательно замещен -NH2, и где RA и RB независимо представляют собой H или (C1-C6)алкил;
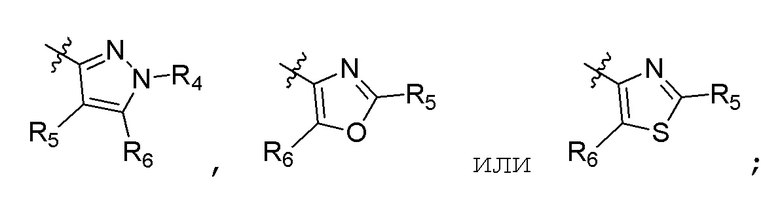
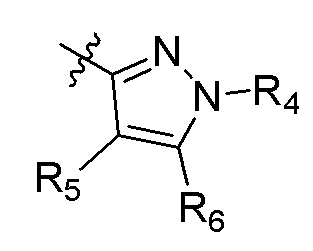
А представляет собой:
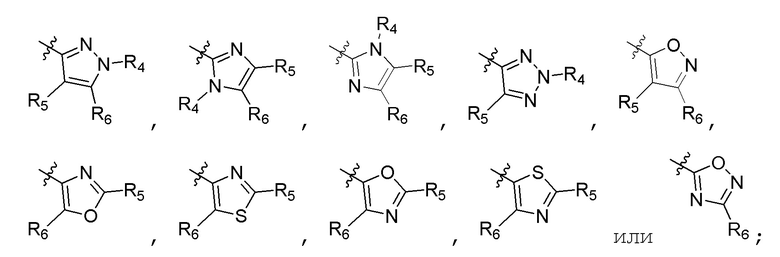
R4 представляет собой H, (C1-C4)алкил, (C3-C4)циклоалкил, (C3-C4)циклоалкил(C1-C4)алкил или галоген(C1-C4)алкил;
R5 и R6 независимо представляют собой H, дейтерий, (C2-C4)алкенил, (C1-C4)алкокси, (C1-C4)алкил, (C1-C4)алкил-d1-9, (C3-C4)циклоалкил, (C3-C4)циклоалкил(C1-C4)алкил, циано, атом галогена, галоген(C1-C4)алкокси, галоген(C1-C4)алкил или гидрокси(C1-C4)алкил;
B представляет собой:
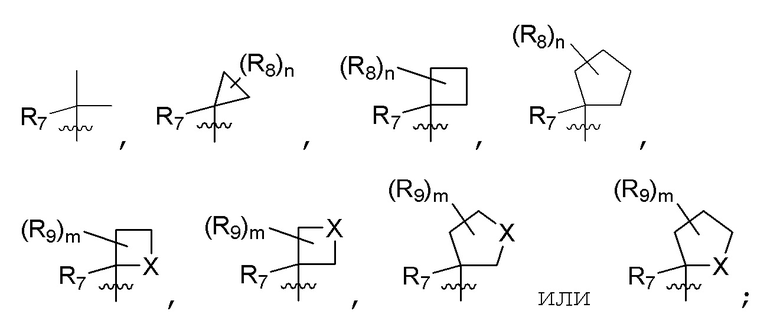
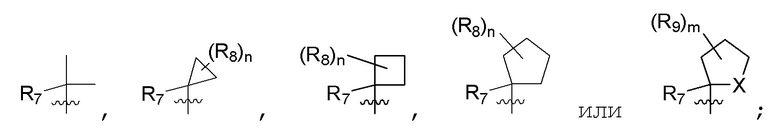
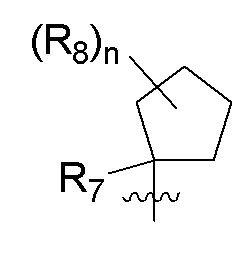
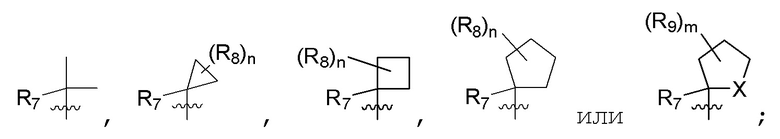
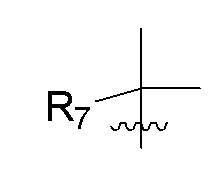
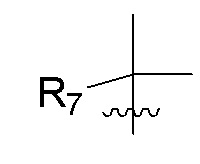
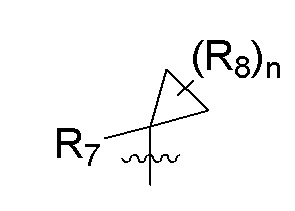
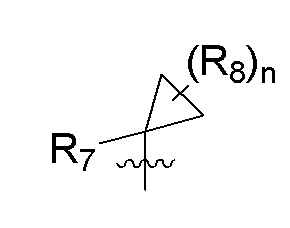
R7 представляет собой -F, -CN, (C1-C3)алкил, (C1-C3)алкил-d1-7 или галоген(C1-C3)алкил;
R8 в каждом случае независимо представляет собой дейтерий, -F, -Cl, -Br или -I, или два R8, присоединенные к одному и тому же атому углерода, образуют (C3-C5)циклоалкильную группу;
n равно 0, 1, 2, 3 или 4;
R9 в каждом случае независимо представляет собой дейтерий, -F, -Cl, -Br или -I, или два R9, присоединенные к одному и тому же атому углерода, образуют (C3-C5) циклоалкильную группу;
m равно 1, 2, 3 или 4; и
X представляет собой O, S или NRC, где RC представляет собой H или (C1-C4)алкил.
В другом варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IA) или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IB) или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (IA) или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (IB) или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к соединению формулы (IA) или его фармацевтически приемлемой соли или гидрату для применения в качестве лекарственного средства.
В еще одном варианте осуществления настоящее изобретение относится к соединению формулы (IB) или его фармацевтически приемлемой соли или гидрату для применения в качестве лекарственного средства.
В еще одном варианте осуществления настоящее изобретение относится к соединению формулы (IA) или его фармацевтически приемлемой соли или гидрату для применения в лечении или профилактике заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к соединению формулы (IB) или его фармацевтически приемлемой соли или гидрату для применения в лечении или профилактике заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (IA) или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (IB) или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (IA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (IB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей соединение формулы (IA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей соединение формулы (IB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей соединение формулы (IA) или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей соединение формулы (IB) или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей соединение формулы (IA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей соединение формулы (IB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (IA) или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (IB) или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
Краткое описание фигур
На фиг. 1 представлена полученная с помощью рентгеноструктурного анализа структура (ORTEP-диаграмма) кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
На фиг. 2 представлена полученная с помощью рентгеноструктурного анализа структура (ORTEP-диаграмма) кристаллического (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
На фиг. 3 представлена полученная с помощью рентгеноструктурного анализа структура (ORTEP-диаграмма) кристаллической (S)-4,4-дифтор-2-метилтетрагидрофуран-2-карбоновой кислоты ((1R,4aS,10aR)-7-изопропил-1,4a-диметил-1,2,3,4,4a, 9,10,10a-октагидрофенантрен-1-ил) метанаминовой соли (пример 36F).
На фиг. 4 представлены результаты анализа порошковой рентгеновской дифракцией кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
На фиг. 5 представлены результаты анализа дифференциальной сканирующей калориметрией кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
На фиг. 6 представлены результаты термогравиметрического анализа кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
На фиг. 7 представлены результаты анализа порошковой рентгеновской дифракцией кристаллического (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
На фиг. 8 представлены результаты анализа дифференциальной сканирующей калорметрией кристаллического (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида.
На фиг. 9 представлены результаты термогравиметрического анализа кристаллического (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
На фиг. 10 представлена полученная с помощью рентгеноструктурного анализа структура (ORTEP-диаграмма) кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата.
На фиг. 11 представлена полученная с помощью рентгеноструктурного анализа структура (ORTEP-диаграмма) кристаллического (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата.
На фиг. 12 представлены результаты анализа порошковой рентгеновской дифракцией кристаллического (R)-4-((2- ((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата.
Подробное описание изобретения
В еще одном варианте осуществления настоящее изобретение относится к соединениям формул (IA) и (IB):
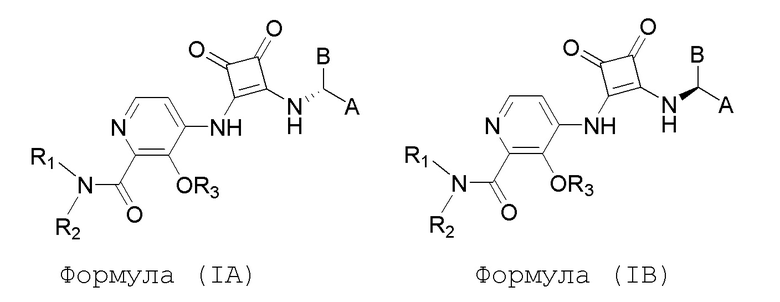
или их фармацевтически приемлемой соли или гидрату, где R1 и R2 независимо представляют собой H или (C1-C6)алкил, или R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют 4-, 5- или 6-членный гетероцикл, содержащий один гетероатом N и необязательно 1 или 2 дополнительных гетероатома, выбранных из группы, состоящей из O, N и S, необязательно замещенный (C1-C4)алкилом; R3 представляет собой H, (C1-C6)алкил, (C1-C4)алкилкарбонил, -C(=O)CH=CHCO2H, -SO2NH2, -CH2OC(=O)(C1-C4)алкил, -CH2OP(=O)(OH)2 или -C(=O)NRARB, где (C1-C4)алкилкарбонил необязательно замещен -CO2H или -NH2, где -CH2OC(=O)(C1-C4)алкил необязательно замещен -NH2, и где RA и RB независимо представляют собой H или (C1-C6)алкил;
А представляет собой:
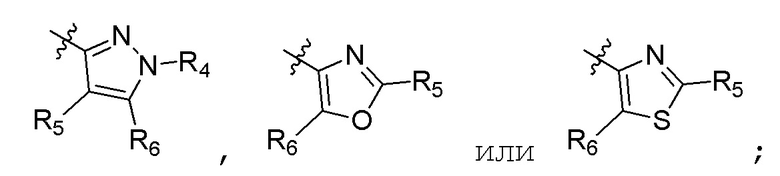
R4 представляет собой (С1-С4)алкил; R5 и R6 независимо представляют собой H, дейтерий, (C1-C4)алкокси, (C1-C4)алкил, (C1-C4)алкил-d1-9, (C3-C4)циклоалкил, циано, атом галогена, галоген(C1-C4)алкокси, галоген(C1-C4)алкил или гидрокси(C1-C4)алкил; B представляет собой:
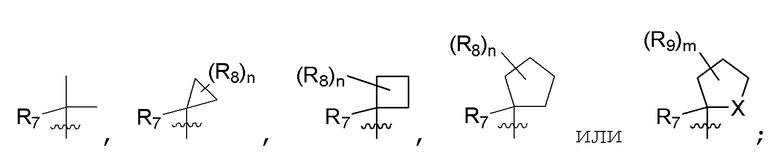
R7 представляет собой (C1-C3)алкил или (C1-C3)алкил-d1-7; R8 представляет собой дейтерий или два R8, присоединенные к одному и тому же атому углерода, образуют (C3-C5)циклоалкильную группу; n равно 0 или 2; R9 в каждом случае представляет собой F; m равно 1, 2, 3 или 4; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IA) и (IB) или их фармацевтически приемлемой соли или гидрату, где R1 и R2 независимо представляют собой (C1-C6)алкил или R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют 4-, 5- или 6-членный гетероцикл, содержащий один гетероатом N и необязательно 1 или 2 дополнительных гетероатома, выбранных из группы, состоящей из O, N и S, необязательно замещенный (C1-C4) алкилом; R3 представляет собой H; А представляет собой:
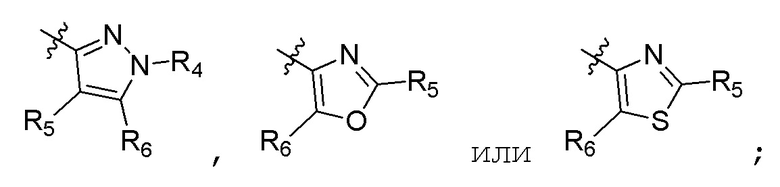
R4 представляет собой (C1-C4)алкил; R5 и R6 независимо представляют собой H, дейтерий, (C1-C4)алкокси, (C1-C4)алкил, (C1-C4)алкил-d1-9, (C3-C4)циклоалкил, циано, атом галогена, галоген(C1-C4)алкокси, галоген(C1-C4)алкил или гидрокси(C1-C4)алкил; B представляет собой:
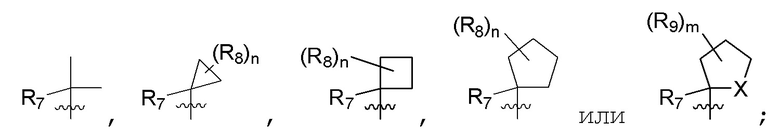
R7 представляет собой (C1-C3)алкил или (C1-C3)алкил-d1-7; R8 представляет собой дейтерий или два R8, присоединенные к одному и тому же атому углерода, образуют (C3-C5)циклоалкильную группу; n равно 0 или 2; R9 в каждом случае представляет собой F; m равно 2; и X представляет собой О.
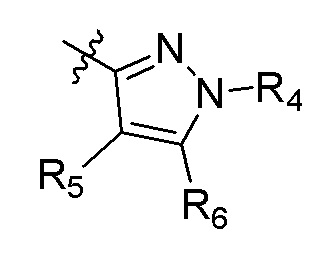
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IA) и (IB) или их фармацевтически приемлемой соли или гидрату, где R1 и R2 независимо представляют собой метил, этил или изопропил, или R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или пиперазин, где пиперазин замещен метилом; R3 представляет собой H; А представляет собой:
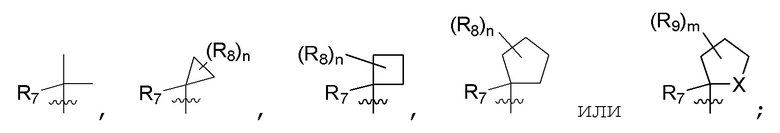
R4 представляет собой метил; R5 представляет собой метил, метил-d3, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой H или метил; B представляет собой:
R7 представляет собой метил; R8 представляет собой дейтерий или два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; n равно 0 или 2; R9 в каждом случае представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формул (IA) и (IB) или их фармацевтически приемлемой соли или гидрату, где R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; А представляет собой:
R4 представляет собой метил; R5 представляет собой метил, метил-d3, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
R7 представляет собой метил; R8 представляет собой дейтерий или два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; n равно 0 или 2; R9 в каждом случае представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формул (IA) и (IB) или их фармацевтически приемлемой соли или гидрату, где R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; А представляет собой:
R4 представляет собой метил; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIA):
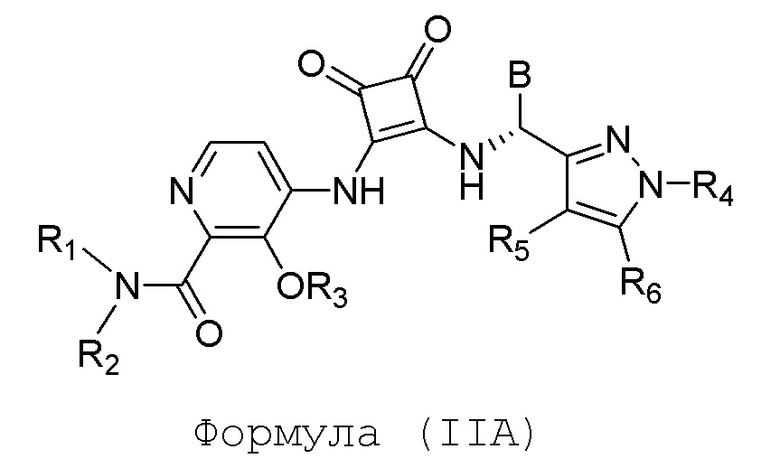
или их фармацевтически приемлемой соли или гидрату, где R1 и R2 независимо представляют собой (C1-C6)алкил или R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют 4-, 5- или 6-членный гетероцикл, содержащий один гетероатом N и необязательно 1 или 2 дополнительных гетероатома, выбранных из группы, состоящей из О, N и S, необязательно замещенный (C1-C4)алкилом; R3 представляет собой H, (C1-C6)алкил, (C1-C4)алкилкарбонил, -C(=O)CH=CHCO2H, -SO2NH2, -CH2OC(=O)(C1-C4)алкил, -CH2OP(=O)(OH)2 или -C(=O)NRARB, где (C1-C4)алкилкарбонил необязательно замещен -CO2H или -NH2, где -CH2OC(=O)(C1-C4)алкил необязательно замещен -NH2, и где RA и RB независимо представляют собой H или (C1-C6)алкил; R4 представляет собой (C1-C4)алкил; R5 и R6 независимо представляют собой H, дейтерий, (C1-C4)алкокси, (C1-C4)алкил, (C1-C4)алкил-d1-9, (C3-C4)циклоалкил, циано, атом галогена, галоген(C1-C4)алкокси или галоген(C1-C4)алкил; B представляет собой:
R7 представляет собой (C1-C3)алкил или (C1-C3)алкил-d1-7; R8 представляет собой дейтерий или два R8, присоединенные к одному и тому же атому углерода, образуют (C3-C5)циклоалкильную группу; n равно 0 или 2; R9 в каждом случае представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIA) или их фармацевтически приемлемой соли или гидрату, где R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
R7 представляет собой метил.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIA) или их фармацевтически приемлемой соли или гидрату, где R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
R7 представляет собой метил.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIA) или их фармацевтически приемлемой соли или гидрату, где R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIA) или их фармацевтически приемлемой соли или гидрату, где R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, метил-d3, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIA) или их фармацевтически приемлемой соли или гидрату, где R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, метил-d3, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
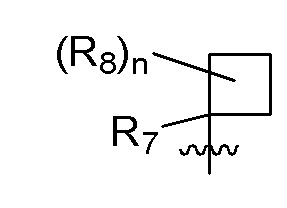
R7 представляет собой метил; два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; и n равно 0 или 2.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIA) или их фармацевтически приемлемой соли или гидрату, где R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
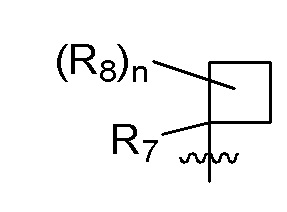
R7 представляет собой метил; два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; и n равно 0 или 2.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIA) или их фармацевтически приемлемой соли или гидрату, где R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, метил-d3, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
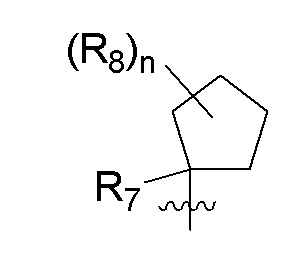
R7 представляет собой метил; R8 представляет собой дейтерий; и n равно 0 или 2.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIA) или их фармацевтически приемлемой соли или гидрату, где R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
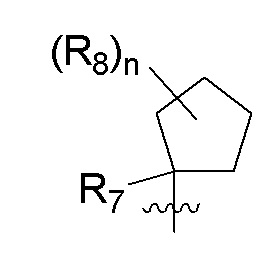
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIA) или их фармацевтически приемлемой соли или гидрату, где R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
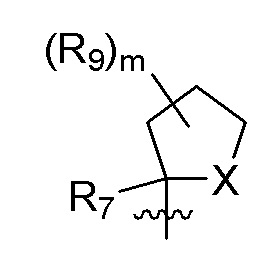
R7 представляет собой метил; R9 представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIA) или их фармацевтически приемлемой соли или гидрату, где R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
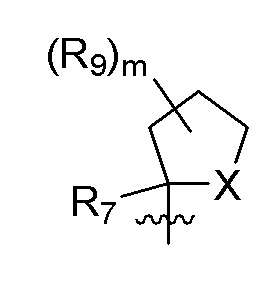
R7 представляет собой метил; R9 представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления соединение формулы (IIA) представляет собой (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид или его фармацевтически приемлемую соль или гидрат.
В еще одном варианте осуществления соединение формулы (IIA) представляет собой (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид.
В еще одном варианте осуществления настоящее изобретение относится к соединению формулы (IIA) или его фармацевтически приемлемой соли или гидрату для применения в качестве лекарственного средства.
В еще одном варианте осуществления настоящее изобретение относится к соединению формулы (IIA) или его фармацевтически приемлемой соли или гидрату для применения в лечении или профилактике заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IIA) или ее фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IIA) или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительных заболеваний, состояний или расстройств у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IIA) или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунных заболеваний, состояний или расстройств у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IIA) или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IIA) или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества пролекарства (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N -диметилпиколинамида.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (IIA) или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (IIA) или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (IIA) или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (IIA) или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (IIA) или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению пролекарства (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей пролекарство (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино))-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей пролекарство (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино))-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей пролекарство ((R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино))-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата, и по меньшей мере один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей пролекарство (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата, и по меньшей мере один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат, и по меньшей мере один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, включающей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей пролекарство (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (IIA) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино))-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей пролекарство (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино))-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата, и по меньшей мере один противовоспалительный агент, в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино))-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики 18,7±0,2, 19,1±0,2 и 20,2±0,2 градусов 2-тета.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему рентгеновскую порошковую дифрактограмму, одержащую дифракционные пики 17,6±0,2, 18,4±0,2, 18,7± 0,2, 19,1±0,2, и 20,2±0,2 градусов 2-тета.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики 11,4±0,2, 15,5±0,2, 17,6±0,2, 18,4±0,2, 18,7±0,2, 19,1±0,2, 20,2±0,2 и 24,3±0,2 градусов 2-тета.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики 11,4±0,2, 12,4±0,2, 15,5±0,2, 17,6 ±0,2, 18,4±0,2, 18,7±0,2, 19,1±0,2, 20,2±0,2, 24,3±0,2, 26,8± 0,2 и 30,5±0,2 градусов 2-тета.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую 3-10 дифракционных пиков, выбранных из группы, состоящей из 11,4±0,2, 12,4±0,2 , 15,5±0,2, 17,6±0,2, 18,4±0,2, 18,7±0,2, 19,1±0,2, 20,2±0,2, 24,3±0,2, 26,8±0,2 и 30,5± 0,2 градусов 2-тета.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую 3-10 дифракционных пиков, приведенных в таблице 7.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики, приведенные в таблице 7.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики, показанные на фиг. 4.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему результаты анализа дифференциальной сканирующей калориметрией, показанные на фиг. 5.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему результаты термогравиметрического анализа, показанные на фиг. 6.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату для применения в качестве лекарственного средства.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату для применения в лечении или профилактике заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительных заболеваний, состояний или расстройств человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунных заболеваний, состояний или расстройств человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического (R)-4- ((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата, в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита, или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6, у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере одно, противовоспалительное средство.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройствя у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду гемисоли кальция, моногидрату.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду гемисоли кальция, моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики 7,8±0,2, 10,3±0,2 и 10,7±0,2 градусов 2-тета.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду гемисоли кальция, моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики 7,8±0,2, 10,3± 0,2, 10,7±0,2, 15,5±0,2 и 18,4±0,2 градусов 2-тета.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду гемисоли кальция, моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики 7,7±0,2, 7,8±0,2, 10,3±0,2, 10,7±0,2, 15,5±0,2, 17,0±0,2, 18,4± 0,2, 20,8±0,2 и 21,0±0,2 градусов 2-тета.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксо-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду гемисоли кальция, моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики 7,7±0,2, 7,8±0,2, 10,3±0,2, 10,7± 0,2, 15,5±0,2, 17,0±0,2, 18,4±0,2, 20,5±0,2, 20,8±0,2, 21,0± 0,2, 24,0±0,2 и 25,6±0,2 градусов 2-тета.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоцикло-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколин гемисоли кальция, моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики 7,7±0,2, 7,8±0,2, 10,3±0,2, 10,7± 0,2, 11,9±0,2, 13,6±0,2, 15,5±0,2, 16,6±0,2, 17,0±0,2, 18,4± 0,2, 20,5±0,2, 20,8±0,2, 21,0±0,2, 22,3±0,2, 24,0±0,2, 24,9± 0,2, 25,6±0,2, 26,1±0,2, 31,3±0,2 и 31,4±0,2 градусов 2-тета.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду гемисоли кальция, моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую 3-10 дифракционных пиков, приведенных в таблице 10.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4- диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду гемисоли кальция, моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики, приведенные в таблице 10.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоцикло-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду гемисоли кальция, моногидрату, имеющему рентгеновскую порошковую дифрактограмму, показанную на фиг. 12.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду гемисоли кальция, моногидрату для применения в качестве лекарственного средства.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду гемисоли кальция, моногидрату для применения в лечении или профилактике заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического ((R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопен)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительных заболеваний, состояний или расстройств человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунных заболеваний, состояний или расстройств человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилцикло)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического (R)-4-((2-((( 1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса T-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата, в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата, в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата, в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата, в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид, гемисоль кальция, моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один противовоспалительный агент.
В другом варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения ревматоидного артрита в человеке.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду.
В еще одном варианте осуществления настоящее изобретение относится к (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики 18,5±0,2, 18,8±0,2 и 19,2±0,2 градусов 2-тета.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики 8,4±0,2, 11,5± 0,2, 12,5±0,2, 18,5±0,2, 18,8±0,2 и 19,2±0,2 градусов 2-тета.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики 8,4±0,2, 9,3±0,2, 11,5±0,2, 12,5± 0,2, 18,5±0,2, 18,8±0,2, 19,2±0,2 и 20,4±0,2 градусов 2-тета.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую 3-10 дифракционных пиков, выбранных из группы, состоящей из 8,4±0,2, 9,3±0,2, 11,5±0,2, 12,5±0,2, 18,5±0,2, 18,8±0,2, 19,2±0,2, 20,4±0,2, 24,5±0,2, 25,1±0,2 и 26,7±0,2 градусов 2-тета.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую 3-10 дифракционных пиков, приведенных в таблице 8.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики, приведенные в таблице 8.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему рентгеновскую порошковую дифрактограмму, показанную на фиг. 7.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему результаты анализа дифференциальной сканирующей калориметрией, показанные на фиг. 8.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату, имеющему результаты термогравиметрического анализа показанные на фиг. 9.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату для применения в качестве лекарственного средства.
В еще одном варианте осуществления настоящее изобретение относится к кристаллическому (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду моногидрату для применения в лечении или профилактике заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительных заболеваний, состояний или расстройств человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунных заболеваний, состояний или расстройств человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества кристаллического (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатрического артрита или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению кристаллического (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино))-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей кристаллический (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино))-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино))-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и по меньшей мере один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения заболевания, состояния или расстройства, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения заболевания, состояния или расстройства, ослабляемых ингибированием хемотаксиса Т-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей кристаллический (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид моногидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIB):
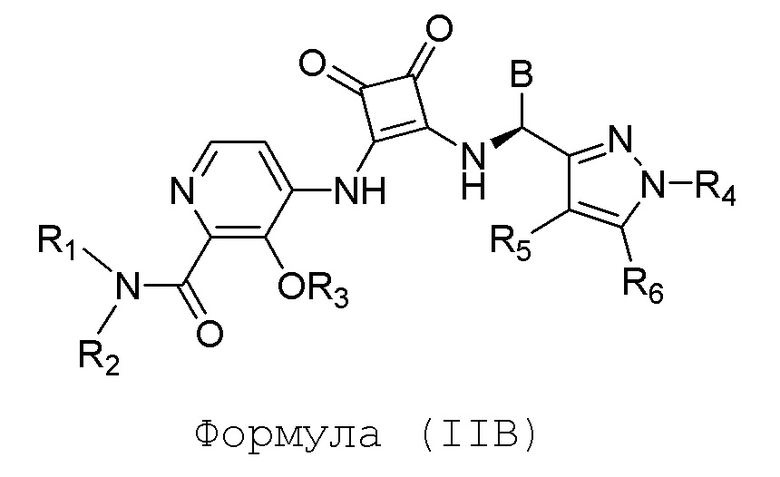
или их фармацевтически приемлемой соли или гидрату, где R1 и R2 независимо представляют собой (C1-C6)алкил или R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют 4-, 5- или 6-членный гетероцикл, содержащий один гетероатом N и необязательно 1 или 2 дополнительных гетероатома, выбранных из группы, состоящей из O, N и S, необязательно замещенный (C1-C4)алкилом; R3 представляет собой H, (C1-C6)алкил, (C1-C4)алкилкарбонил, -C(=O)CH=CHCO2H, -SO2NH2, -CH2OC(=O)(C1-C4)алкил, -CH2OP(=O)(OH)2 или -C(=O)NRARB, где (C1-C4)алкилкарбонил необязательно замещен -CO2H или -NH2, где -CH2OC(= O)(C1-C4)алкил необязательно замещен -NH2, и где RA и RB независимо представляют собой H или (C1-C6)алкил; R4 представляет собой (C1-C4)алкил; R5 и R6 независимо представляют собой H, дейтерий, (C1-C4)алкокси, (C1-C4)алкил, (C1-C4)алкил-d1-9, (C3-C4)циклоалкил, циано, атом галогена, галоген(C1-C4)алкокси или галоген(C1-C4)алкил; B представляет собой:
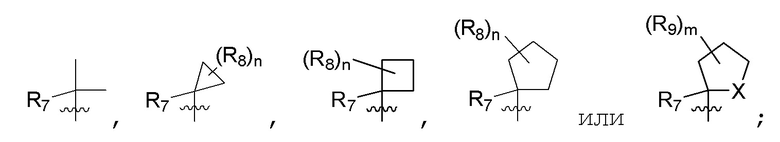
R7 представляет собой (C1-C3)алкил или (C1-C3)алкил-d1-7; R8 представляет собой дейтерий или два R8, присоединенные к одному и тому же атому углерода, образуют (C3-C5)циклоалкильную группу; n равно 0 или 2; R9 в каждом случае представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIB) или их фармацевтически приемлемой соли или гидрату, где R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
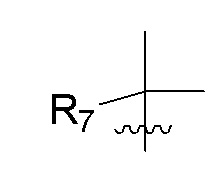
R7 представляет собой метил.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIB) или их фармацевтически приемлемой соли или гидрату, где R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
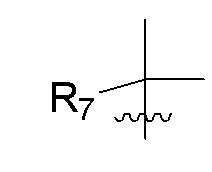
R7 представляет собой метил.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIB) или их фармацевтически приемлемой соли или гидрату, где R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
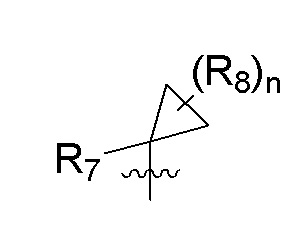
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIB) или их фармацевтически приемлемой соли или гидрату, где R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, метил-d3, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIB) или их фармацевтически приемлемой соли или гидрату, где R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, метил-d3, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
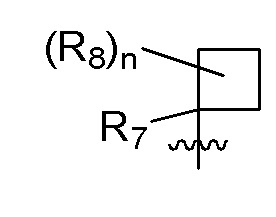
R7 представляет собой метил; два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; и n равно 0 или 2.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIB) или их фармацевтически приемлемой соли или гидрату, где R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
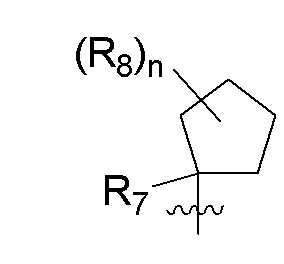
R7 представляет собой метил; два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIB) или их фармацевтически приемлемой соли или гидрату, где R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
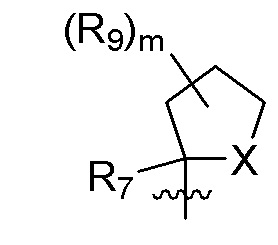
R7 представляет собой метил; R8 представляет собой дейтерий; и n равно 0 или 2.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIB) или их фармацевтически приемлемой соли или гидрату, где R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
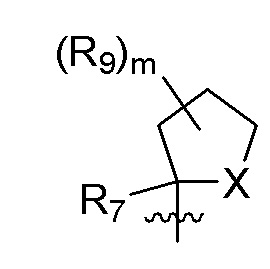
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIB) или их фармацевтически приемлемой соли или гидрату, где R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
R7 представляет собой метил; R9 представляет собой F; m равно 2, и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIB) или их фармацевтически приемлемой соли или гидрату, где R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R4 представляет собой метил; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил; R6 представляет собой H; B представляет собой:
R7 представляет собой метил; R9 представляет собой F; m равно 2, и X представляет собой О.
В еще одном варианте осуществления соединение формулы (IIA) представляет собой (S)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид или его фармацевтически приемлемую соль или гидрат.
В еще одном варианте осуществления соединение формулы (IIA) представляет собой (S)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид.
В еще одном варианте осуществления настоящее изобретение относится к соединению формулы (IIB) или его фармацевтически приемлемой соли или гидрату для применения в качестве лекарственного средства.
В еще одном варианте осуществления настоящее изобретение относится к соединению формулы (IIB) или его фармацевтически приемлемой соли или гидрату для применения в лечении или профилактике заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IIB) или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IIB) или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительных заболеваний, состояний или расстройств у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IIB) или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунных заболеваний, состояний или расстройств у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IIB) или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IIB) или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения (S)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения пролекарства (S)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (IIB) или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (IIB) или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (IIB) или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (IIB) или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (IIB) или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению (S)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению пролекарства (S)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей (S)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей пролекарство (S)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино))-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (IIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей (S)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей пролекарство (S)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей (S)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино))-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей пролекарство (S)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата, и, по меньшей мере, один фармацевтически приемлемый эксиципент, разбавитель или носитель, в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей (S)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей пролекарство (S)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино))-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата, и по меньшей мере один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, включающей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей (S)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей пролекарство (S)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (IIB) или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей (S)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино))-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид или его фармацевтически приемлемую соль или гидрат и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей пролекарство (S)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида или его фармацевтически приемлемой соли или гидрата, и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA):
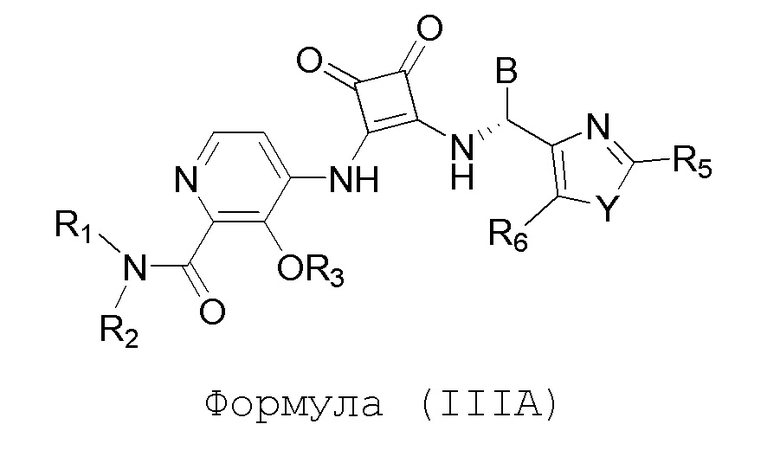
или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2 независимо представляют собой (C1-C6)алкил или R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют 4-, 5- или 6-членный гетероцикл, содержащий один гетероатом N и необязательно 1 или 2 дополнительных гетероатома, выбранных из группы, состоящей из O, N и S, необязательно замещенный (C1-C4)алкилом; R3 представляет собой H, (C1-C6)алкил, (C1-C4)алкилкарбонил, -C(=O)CH=CHCO2H, -SO2NH2, -CH2OC(=O)(C1-C4)алкил, -CH2OP(=O)(OH)2 или -C(=O)NRARB, где (C1-C4)алкилкарбонил необязательно замещен -CO2H или -NH2, где -CH2OC(=O)(C1-C4)алкил необязательно замещен -NH2, и где RA и RB независимо представляют собой H или (C1-C6)алкил; R5 и R6 независимо представляют собой H, дейтерий, (C1-C4)алкокси, (C1-C4) алкил, (C1-C4)алкил-d1-9, (C3-C4)циклоалкил, циано, атом галогена, галоген(C1-C4)алкокси или галоген(C1-C4)алкил; B представляет собой:
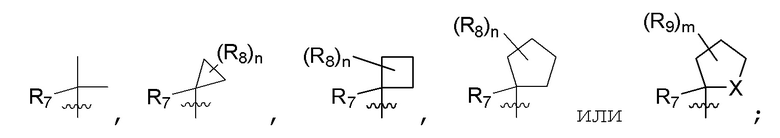
R7 представляет собой (C1-C3)алкил или (C1-C3)алкил-d1-7; R8 представляет собой дейтерий или два R8, присоединенные к одному и тому же атому углерода, образуют (C3-C5)циклоалкильную группу; n равно 0 или 2; R9 в каждом случае представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой О или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
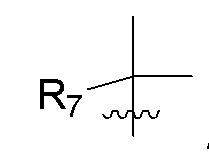
R7 представляет собой метил.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
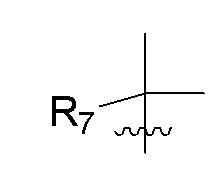
R7 представляет собой метил.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
R7 представляет собой метил.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
R7 представляет собой метил.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой О или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
R7 представляет собой метил.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой О или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
R7 представляет собой метил.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
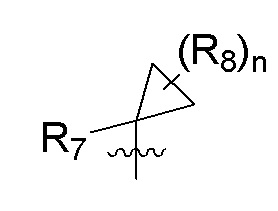
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
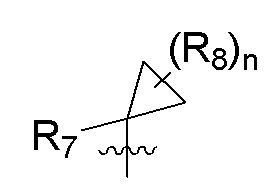
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
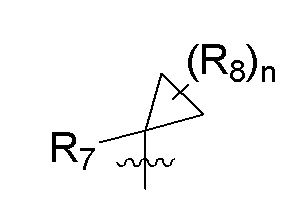
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
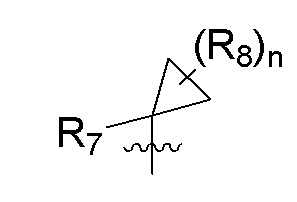
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
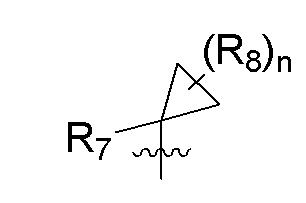
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
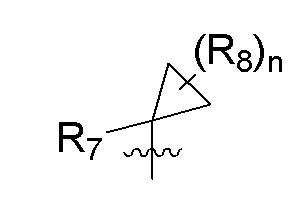
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
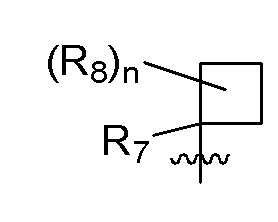
R7 представляет собой метил; два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; и n равно 0 или 2.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
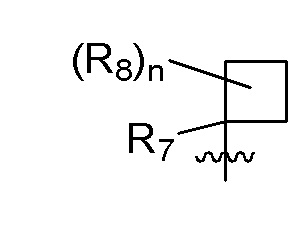
R7 представляет собой метил; два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; и n равно 0 или 2.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
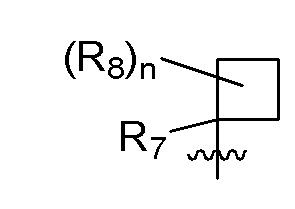
R7 представляет собой метил; два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; и n равно 0 или 2.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
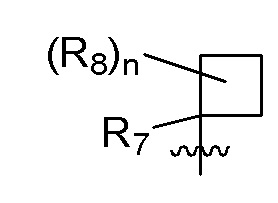
R7 представляет собой метил; два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; и n равно 0 или 2.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
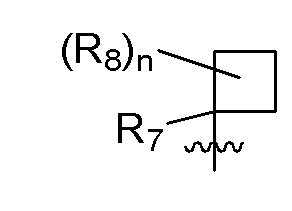
R7 представляет собой метил; два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; и n равно 0 или 2.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
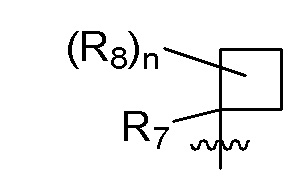
R7 представляет собой метил; два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; и n равно 0 или 2.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
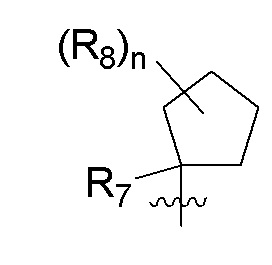
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
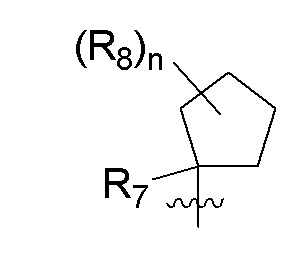
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
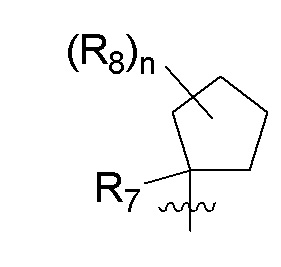
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
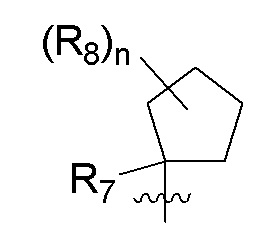
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
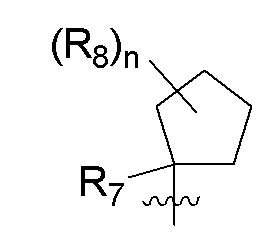
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
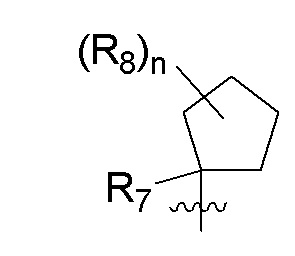
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
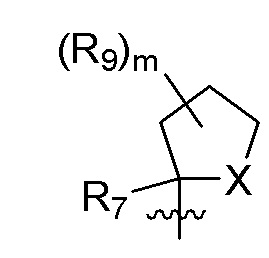
R7 представляет собой метил; R9 представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
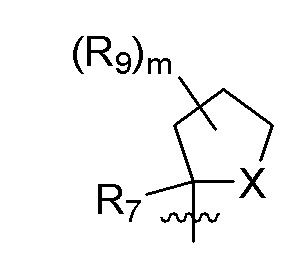
R7 представляет собой метил; R9 представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
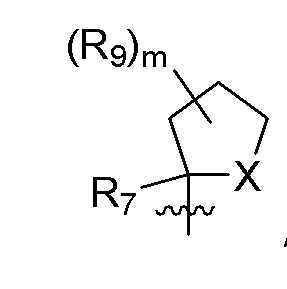
R7 представляет собой метил; R9 представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:

R7 представляет собой метил; R9 представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
R7 представляет собой метил; R9 представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIA) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
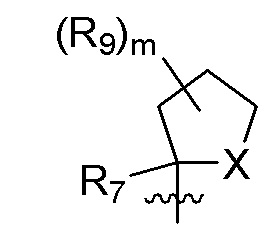
R7 представляет собой метил; R9 представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB):
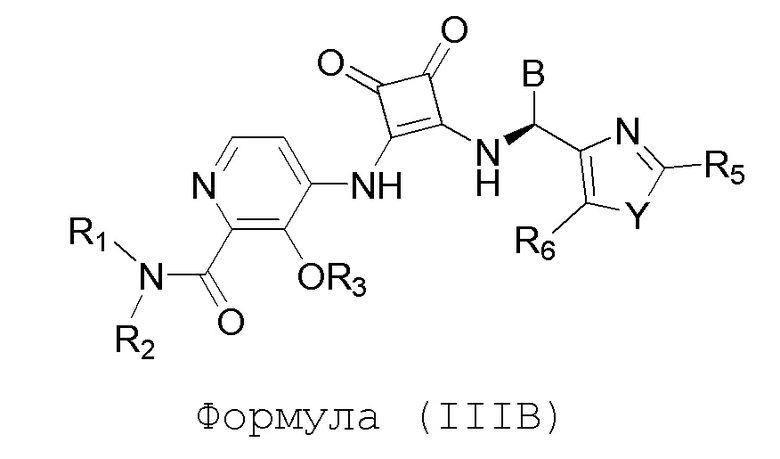
или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2 независимо представляют собой (C1-C6)алкил или R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют 4-, 5- или 6-членный гетероцикл, содержащий один гетероатом N и необязательно 1 или 2 дополнительных гетероатома, выбранных из группы, состоящей из O, N и S, необязательно замещенный (C1-C4)алкилом; R3 представляет собой H, (C1-C6)алкил, (C1-C4)алкилкарбонил, -C(=O)CH=CHCO2H, -SO2NH2, -CH2OC(=O)(C1-C4)алкил, -CH2OP(=O)(OH)2 или -C(=O)NRARB, где (C1-C4)алкилкарбонил необязательно замещен -CO2H или -NH2, где -CH2OC(=O)(C1-C4)алкил необязательно замещен -NH2, и где RA и RB независимо представляют собой H или (C1-C6)алкил; R5 и R6 независимо представляют собой H, дейтерий, (C1-C4)алкокси, (C1-C4)алкил, (C1-C4)алкил-d1-9, (C3-C4)циклоалкил, циано, атом галогена, галоген(C1-C)алкокси или галоген(C1-C4)алкил; B представляет собой:
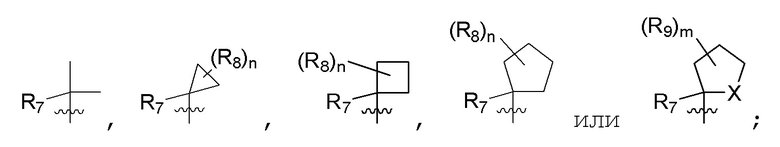
R7 представляет собой (C1-C3)алкил или (C1-C3)алкил-d1-7; R8 представляет собой дейтерий или два R8, присоединенные к одному и тому же атому углерода, образуют (C3-C5)циклоалкильную группу; n равно 0 или 2; R9 в каждом случае представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
R7 представляет собой метил.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
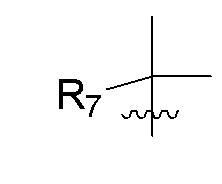
R7 представляет собой метил.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
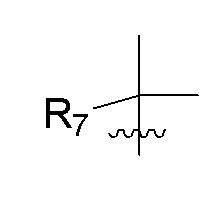
R7 представляет собой метил.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
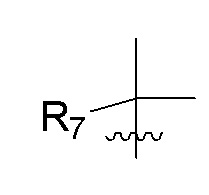
R7 представляет собой метил.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
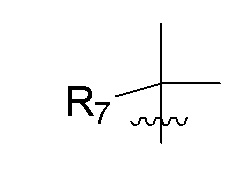
R7 представляет собой метил.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
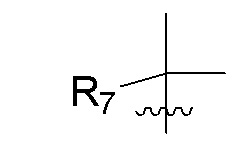
R7 представляет собой метил.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
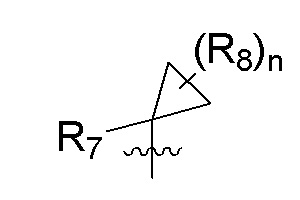
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
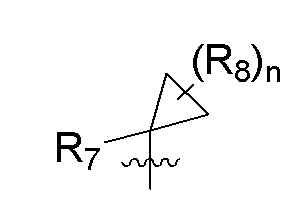
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
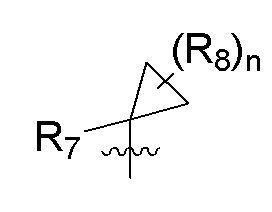
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
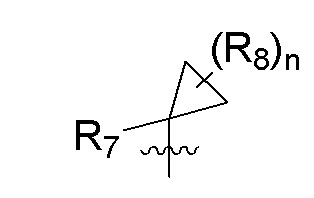
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
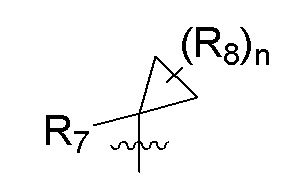
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
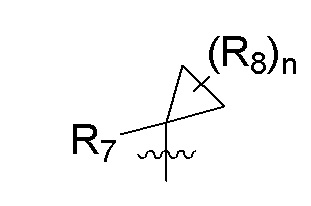
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
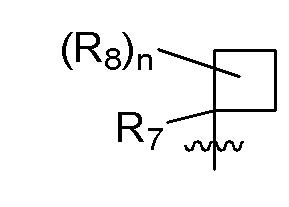
R7 представляет собой метил; два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; и n равно 0 или 2.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
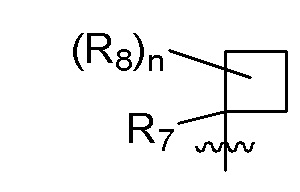
R7 представляет собой метил; два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; и n равно 0 или 2.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
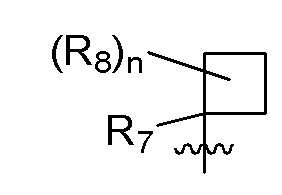
R7 представляет собой метил; два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; и n равно 0 или 2.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
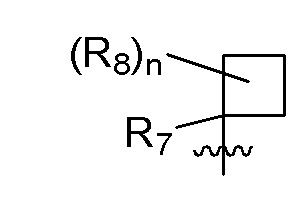
R7 представляет собой метил; два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; и n равно 0 или 2.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:

R7 представляет собой метил; два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; и n равно 0 или 2.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
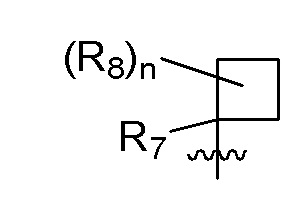
R7 представляет собой метил; два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил; и n равно 0 или 2.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
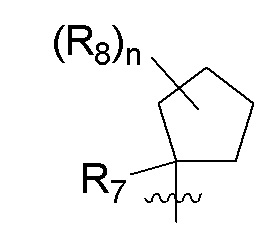
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
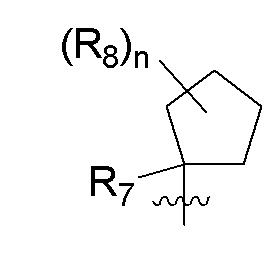
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
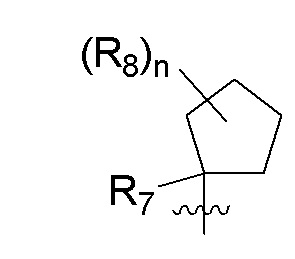
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
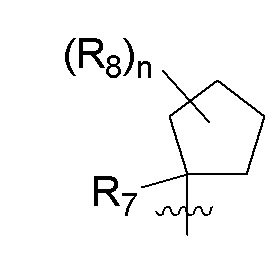
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
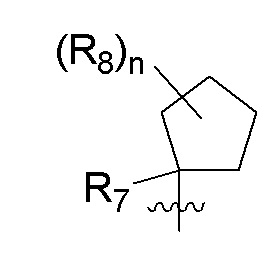
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
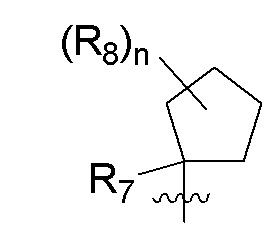
R7 представляет собой метил; и n равно 0.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
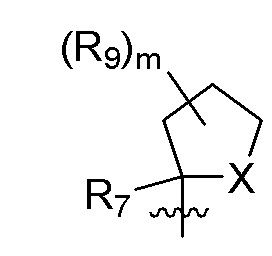
R7 представляет собой метил; R9 представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
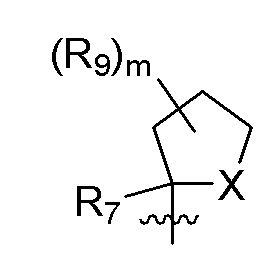
R7 представляет собой метил; R9 представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 представляет собой метил; R2 представляет собой метил, этил или изопропил; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
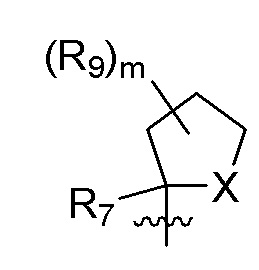
R7 представляет собой метил; R9 представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 и R6 независимо представляют собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; B представляет собой:
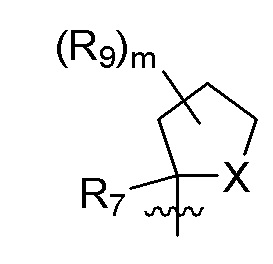
R7 представляет собой метил; R9 представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано, циклопропил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
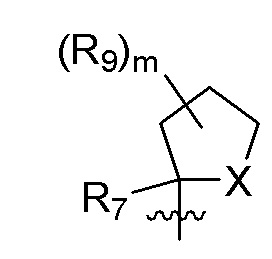
R7 представляет собой метил; R9 представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (IIIB) или их фармацевтически приемлемой соли или гидрату, где Y представляет собой O или S; R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин; R3 представляет собой H; R5 представляет собой метил или гидроксиметил; R6 представляет собой метил или метокси; B представляет собой:
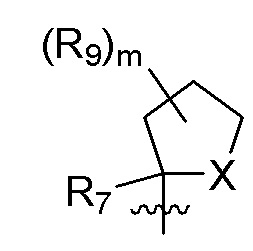
R7 представляет собой метил; R9 представляет собой F; m равно 2; и X представляет собой О.
В еще одном варианте осуществления настоящее изобретение относится к соединению формулы (IIIA) или (IIIB) или его фармацевтически приемлемой соли или гидрату для применения в качестве лекарственного средства.
В еще одном варианте осуществления настоящее изобретение относится к соединению формулы (IIIA) или (IIIB), или его фармацевтически приемлемой соли или гидрату, для применения в лечении или профилактике заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IIIA) или (IIIB), или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IIIA) или (IIIB), или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительных заболеваний, состояний или расстройств у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IIIA) или (IIIB), или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунных заболеваний, состояний или расстройств у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IIIA) или (IIIB), или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IIIA) или (IIIB) или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (IIIA) или (IIIB) или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (IIIA) или (IIIB) или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (IIIA) или (IIIB) или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (IIIA) или (IIIB) или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения формулы (IIIA) или (IIIB) или его фармацевтически приемлемой соли или гидрата в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической композиции, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один фармацевтически приемлемый эксципиент, разбавитель или носитель, в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтической комбинации, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунного заболевания, состояния или расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической комбинации, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием CCR6 у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения заболеваний, состояний или расстройств, ослабляемых ингибированием хемотаксиса Т-клеток у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения воспалительного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения аутоиммунного заболевания, состояния или расстройства у человека.
В еще одном варианте осуществления настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, и, по меньшей мере, один противовоспалительный агент, в производстве лекарственного средства для лечения псориаза, язвенного колита, болезни Крона, воспалительного заболевания кишечника, псориатического артрита или ревматоидного артрита у человека.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики иммунного расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB), или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики аутоиммунного расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB), или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики нейродегенеративного или нейровоспалительного расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB), или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики ревматоидного артрита, ювенильного артрита, болезни Стилла, ювенильного ревматоидного артрита, ревматоидного артрита с системным началом, пауциартикулярного ревматоидного артрита, пауциартикулярного юношеского ревматоидного артрита, полиартикулярного ревматоидного артрита, энтеропатического артрита, ювенильного синдрома Рейтера, анкилозирующего спондилита, ювенильного анкилозирующего спондилита, SEA- синдрома, реактивного артрита (реактивной артропатии), псориатической артропатии, ювенильного энтеропатического артрита, ревматической полимиалгии, энтеропатического спондилита, ювенильного идиопатического артрита (JIT), ювенильного псориатрического артрита, ювенильного ревматоидного артрита, ювенильного ревматоидного артрита с системным началом, гигантоклеточного артериита или вторичного остеоартрита в результате воспалительных заболеваний у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IA), (IB), (IIA), ( IIB), (IIIA), или (IIIB), или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики волчанки, системной красной волчанки, ювенильной системной красной волчанки, волчаночного нефрита, синдрома Шегрена, склеродермии (системного склероза), феномена Рейно, ювенильной склеродермии, полимиозита, дерматомиозита, полимиозита-дерматомиозита, смешанного заболевания соединительной ткани, саркоидоза, фибромиалгии, микроскопического полиангиита, системного васкулита, васкулита, эозинофильного гранулематоза с полиангиитом (ранее известного как синдром Черджа-Штрауса), гранулематоза с полиангиитом (ранее известного как гранулематоз Вегенера), узелкового полиартериита, пурпуры Геноха-Шенлейна, идиопатической тромбоцитопенической тромботической пурпуры, ювенильного васкулита, узелкового полиартериита (также известного как узелковый панартериит, узелковый периартериит, болезнь Куссмауля, болезнь Куссмауля-Майера или PAN), сывороточной болезни, миастении гравис, артериита Такаясу, синдрома Бехчета, болезни Кавасаки (синдрома кожнослизистых лимфоузлов), болезни Бюргера (облитерирующего тромбангиита), синдрома Фогта-Коянаги-Харада, болезни Аддисона, тиреоидита Хашимото, склерозирующего холангита, мембранозной гломерулопатии, полимиозита, миозита, атеросклероза, аутоиммунной гемолитической анемии, аутоиммунного орхита или болезни Гудпасчера у человека, включащего введение терапевтически эффективного количества соединения формулы (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB) или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики воспалительных заболеваний кишечника, болезни Крона, язвенного колита, глютеновой энтеропатии, глютеновых болезней, проктита, эозинофильного гастроэнтерита, аутоиммунного атрофического гастрита при пернициозной анемии или мастоцитоза у человека, включающему введение человеку, нуждающегося в таком лечении, терапевтически эффективного количества соединения формулы (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB), или его фармацевтически приемлемой соли или его гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечение псориаза, атопического дерматита, экзематозного дерматита, дерматита, зуда, алопеции, аутоиммунной алопеции, витилиго, эпидермальной гиперплазии, ювенильного дерматомиозита или дерматомиозита. В некоторых других вариатах осуществления псориаз представляет собой бляшечный псориаз, гуттатный псориаз, псориатическую эпидермальную гиперплазию, обратный псориаз, пустулезный псориаз, эритродермический псориаз у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB), или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики неалкогольного стеатогепатита, неалкогольной жировой болезни печени, аутоиммунного гепатита, хронического агрессивного гепатита или первичного билиарного склероза у человека, включающему введение человеку, нуждающемуся в таком лечение, терапевтически эффективного количества соединения формулы (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB) или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики рассеянного склероза, бокового амиотрофического склероза, болезни Гийена-Барре, аутоиммунного энцефаломиелита, болезни Альцгеймера, большого депрессивного расстройства, черепно-мозговой травмы, эпилепсии, болезни Паркинсона или биполярного расстройства у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB), или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики болезни Грейвса, неинфекционного увеита, синдрома сухого глаза, симпатической офтальмии, синдрома Когана, кератоконъюнктивита, весеннего конъюнктивита, увеита, включая увеит, ассоциированный с болезнью Бехчета, и увеит, вызванный линзами, кератита, герпетического кератита, конического кератита, дистрофии эпителия роговицы, кератолейкомы, пемфигуса конъюктивы глаза, язвы Мурена, склерита, сухого кератоконъюнктивита (сухой глаз), фликтенулы, иридоциклита, саркоидоза, эндокринной офтальмопатии, симпатической офтальмии, аллергического конъюктивита, окулярной неоваскуляризации или пролиферативной диабетической ретинопатии у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB), или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики асмы, аллергии, хронического обструктивного заболевания легких или острого респираторного заболевания у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формула (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB) или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики рака органов пищеварительного/желудочно-кишечного тракта, рака толстого кишечника, рака печени, рака кожи, включая опухоль из тучных клеток и плоскоклеточную капциному, рака молочной железы, рака яичника, рака предстательной железы, лейкоза, диффузной В-крупноклеточной лимфомы, кожной Т-клеточной лимфомы, неходжкинской лимфомы, рака почки, рака легкого, опухолей мышечной ткани, рака костей, рака мочевого пузыря, рака головного мозга, меланомы, включая оральную и метастатическую меланому, саркомы Капоши, множественной миеломы, миелопролиферативных расстройств, глиобластомы, олигодендроглиомы, рака поджелудочной железы, опухолей головного мозга или глиом, включая астроцитому. В некоторых других вариантах осуществления лейкоз представляет собой острый миелоидный лейкоз, Т-клеточный острый лимфобластный лейкоз или Т-клеточный лейкоз взрослых у человека, включающеиу введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (IA) , (IB), (IIA), (IIB), (IIIA) или (IIIB), или его фармацевтически приемлемой соли или гидрата.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения или профилактики сахарного диабета I типа, сахарного диабета II типа или ювенильного диабета у человека, включающему введение человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения. формулы (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB), или его фармацевтически приемлемой соли или гидрата.
Определения
Как используется в описании и в прилагаемой формуле изобретения, следующие термины имеют следующие значения.
Как здесь используется, термин «(C2-C4)алкенил» означает углеводород с прямой или разветвленной цепью, содержащий от 2 до 4 атомов углерода и содержащий одну двойную углерод-углеродную связь. Репрезентативные примеры (C2-C4)алкенила включают, не ограничиваясь этим, этенил, 2-пропенил, 2-метил-2-пропенил и 3-бутенил.
Как здесь используется, термин «(С1-С4)алкокси», означает (С1-С4)алкильную группу, как здесь определено, присоединенную к исходному молекулярному фрагменту через атом кислорода. Репрезентативные примеры (C1-C4)алкокси включают, не ограничиваясь этим, метокси, этокси, пропокси, 2-пропокси, бутокси и трет-бутокси.
Как здесь используется, термин «(C1-C3)алкил» означает углеводород с прямой или разветвленной цепью, содержащий от 1 до 3 атомов углерода. Репрезентативные примеры (C1-C3)алкила включают метил, этил, н-пропил и изопропил.
Как здесь используется, термин «(C1-C3)алкил-d1-7» означает углеводород с прямой или разветвленной цепью, содержащий от 1 до 3 атомов углерода, в котором от одного до семи атомов водорода заменены на дейтерий (2H или D). Репрезентативные примеры (C1-C3) алкил-d1-7 включают метил-d3, этил-d5 и этил-2,2,2-d3.
Как здесь используется, термин «(C1-C4)алкил» означает углеводород с прямой или разветвленной цепью, содержащий от 1 до 4 атомов углерода. Репрезентативные примеры (C1-C4)алкила включают метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил и трет-бутил.
Как здесь используется, термин «(C1-C4)алкил-d1-9» означает углеводород с прямой или разветвленной цепью, содержащий от 1 до 4 атомов углерода, в котором от 1 до 9 атомов водорода заменены на дейтерий (2H или D). Репрезентативные примеры (C1-C4)алкил-d1-9 включают метил-d3, этил-d5 и этил-2,2,2-d3.
Как здесь используется, термин «(C1-C6)алкил» означает углеводород с прямой или разветвленной цепью, содержащий от 1 до 6 атомов углерода. Репрезентативные примеры (C1-C6)алкила включают, не ограничиваясь этим, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, изопентил, неопентил и н-гексил.
Как здесь используется, термин «(С1-С4)алкилкарбонил» означает (С1-С4)алкильную группу, как здесь определено, присоединенную к исходному молекулярному фрагменту через карбонильную группу, как здесь определено. Репрезентативные примеры (C1-C4) алкилкарбонила включают, не ограничиваясь этим, ацетил, 1-оксопропил и 2,2-диметил-1-оксопропил.
Как здесь используется, термин «карбонил» означает группу -C(=O)-.
Как здесь используется, термин «циано» означает группу -CN.
Как здесь используется, термин «(C3-C4)циклоалкил» означает насыщенную циклическую углеводородную группу, содержащую от 3 до 4 атомов углерода, примеры (C3-C4)циклоалкила включают циклопропил и циклобутил. (C3-C4)циклоалкильные группы по изобретению необязательно замещены 1, 2 или 3 заместителями, которые независимо представляют собой (C1-C4)алкил, CN, атом галогена или OH.
Как здесь используется, термин «(C3-C4)циклоалкил(C1-C4)алкил» означает (C3-C4)циклоалкил, как здесь определено, присоединенный к исходному молекулярному фрагменту через (C1-C4) алкильную группу, как здесь определено. Репрезентативные примеры (C3-C4)циклоалкил(C1-C4)алкила включают циклопропилметил, 2-циклопропилэтил, 2-циклопропилпропил, 3-циклопропилпропил, циклобутилметил, 2-циклобутилэтил, 2-циклобутилпропил и 3-циклобутил.
Как здесь используется, термин «(C3-C5)циклоалкил» означает насыщенную циклическую углеводородную группу, содержащую от 3 до 5 атомов углерода, примеры (C3-C5)циклоалкила включают циклопропил, циклобутил и циклопентил. (C3-C5)циклоалкильные группы по изобретению необязательно замещены 1, 2 или 3 заместителями, которые независимо представляют собой (C1-C4) алкил, CN, атом галогена или OH.
Как здесь используется, термин «атом галогена» или «галоген» означает -Cl, -Br, -I или -F.
Как здесь используется, «галоген(С1-С4)алкокси» означает, по меньшей мере, один галоген, как здесь определено, присоединенный к исходному молекулярному фрагменту через (С1-С4)алкоксигруппу, как здесь определено. Типичные примеры галоген(C1-C4)алкокси включают, не ограничиваясь этим, хлорметокси, дифторметокси, 2-фторэтокси, трифторметокси и пентафторэтокси.
Как здесь используется, термин «галоген(C1-C3)алкил» означает, по меньшей мере, один галоген, как здесь определено, присоединенный к исходному молекулярному фрагменту через (C1-C3) алкильную группу, как здесь определено. Репрезентативные примеры галоген(C1-C3)алкила включают, не ограничиваясь этим, хлорметил, дифторметил, 2-фторэтил, трифторметил и пентафторэтил.
Как здесь используется, термин «галоген(C1-C4)алкил» означает, по меньшей мере, один галоген, как здесь определено, присоединенный к исходному молекулярному фрагменту через (C1-C4) алкильную группу, как здесь определено. Репрезентативные примеры галоген(C1-C4)алкила включают, не ограничиваясь этим, хлорметил, 2-фторэтил, трифторметил и пентафторэтил.
Как здесь используется, термин «гетероцикл» или «гетероциклический» означает 4-, 5- или 6-членное кольцо, содержащее один гетероатом N и необязательно 1 или 2 дополнительных гетероатома, выбранных из группы, состоящей из O, N и S. Репрезентативные примеры гетероцикла включают, не ограничиваясь этим, азетидинил, имидазолидинил, морфолинил, оксадиазолидинил, оксазолидинил, пиперазинил, пиперидинил, пиразолидинил, пирролидинил, тиадиазолидинил, тиазолидинил и тиоморфолинил. Гетероциклические группы по изобретению необязательно замещены 1, 2 или 3 (C1-C4) алкильными группами.
Как здесь используется, термин «гидрокси(C1-C4)алкил» означает, по меньшей мере, одну гидроксигруппу, как здесь определено, присоединенную к исходному молекулярному фрагменту через (C1-C4)алкильную группу, как здесь определено. Репрезентативные примеры гидрокси(C1-C4)алкила включают, не ограничиваясь этим, гидроксиметил, 2-гидроксиэтил, 3-гидроксипропил, 2,4-дигидроксибутил и 2,3-дигидроксипропил.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтическим комбинациям для местного введения, содержащим соединение формулы (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, комбинации с другим фармацевтическим агентом для лечения заболеваний, состояний и/или расстройств, описанных здесь. Подходящие фармацевтические агенты, которые можно использовать в комбинации с соединениями по настоящему изобретению для местного введения, включают, не ограничиваясь этим: второе соединение формулы (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат; ингибитор изофермента PDE4, включая, не ограничиваясь этим, ампремиласт, рофлумиласт, ролипрам, пикламиласт, кризаборол, PF-07038124, PF-07091905, PF-07090414, PF-07062087, PF-07062077 и PF-07566; кортикостероид, включая, не ограничиваясь этим, флуоцинонид, дезоксиметазон, мометазон, триамцинолон, бетаметазон, алклометазон, десонид, гидрокортизон, LEO-134310A и мапракорат; ингибитор кальциневрина, включая, не ограничиваясь этим, такролимус, пимекролимус и циклоспорин; ингибитор JAK, включая, не ограничиваясь этим, тофацитиниб, JTE-052, барицитиниб, упадацитиниб, PF-04965842, PF-06651600 и PF-06700841; ингибиторы TYK, включая, не ограничиваясь этим, PF-06826647 и BMS-986165; ингибитор ITK, включая, не ограничиваясь этим, JTE-051; ингибитор SYK, включая, не ограничиваясь этим, фостаматиниб, цердулатиниб, энтосплетиниб, TAK-659, ASN-002 и GS-9876; ингибитор тирозинкиназы, включая, не ограничиваясь этим, цердулатиниб; ингибитор IRAK4, включая, не ограничиваясь этим, 1-(((2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил)метокси)-7-метоксиизохинолин-6-карбоксамид; противовоспалительное средство, включая, не ограничиваясь этим, WBI-1001 и MRX-6; производные ретиноевой кислоты, включая, не ограничиваясь этим, алитретиноин; селективный агонист Х-рецепторов печени (LXR), включая, не ограничиваясь этим, VTP-38543; антагонисты рецептора H4, включая, не ограничиваясь этим, ZPL-389; антагонисты рецептора NKI, включая, не ограничиваясь этим, апрепитант и традипитант; антагонисты рецептора CRTH2, включая, не ограничиваясь этим, февипипрант и OC-459; ингибиторы химазы, включая, не ограничиваясь этим, SUN 13834; ингибиторы GATA-3, включая, не ограничиваясь этим, SB-011; и обратный агонист RORC2, включая, не ограничиваясь этим, PF-06763809, ESR-114, VTP-43742, ARN6039, TAK-828, RTA-1701, BOS-172767, AUR-101 и JTE-451.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтическим комбинациям для перорального введения, содержащим соединение формулы (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат, в комбинации с другим фармацевтическим агентом для лечения заболеваний, состояний и/или расстройств, описанных здесь. Подходящие фармацевтические средства, которые можно использовать в комбинации с соединениями по настоящему изобретению для перорального введения, включают, не ограничиваясь этим: второе соединение формулы (IA), (IB), (IIA), (IIB), (IIIA), или (IIIB), или его фармацевтически приемлемую соль или гидрат; противовоспалительные средства для перорального введения, включая, не ограничиваясь этим, аспирин, ибупрофен, напроксен, сулиндак, индометацин, мефенамат, дроксикам, диклофенак, сульфинпиразон, пироксикам, CELEBREX® и их фармацевтически приемлемые соли или пролекарства; производные ретиноевой кислоты для перорального введения, включая, не ограничиваясь этим, алитретиноин; селективные агонисты Х-рецептора печени (LXR) для перорального введения, включая, не ограничиваясь этим, VTP-38543; антагонисты рецептора H4 для перорального введения, включая, не ограничиваясь этим, ZPL-389; антагонисты рецептора NKI для перорального введения, включая, не ограничиваясь этим, апрепитант и традипитант; антагонисты рецептора CRTH2 для перорального введения, включая, не ограничиваясь этим, февипипрант и ОС-459; ингибиторы химазы для перорального введения, включая, не ограничиваясь этим, SUN 13834; ингибиторы GATA-3 для перорального введения, включая, не ограничиваясь, SB-011; обратные агонисты RORC2 для перорального введения, включая, не ограничиваясь этим, ESR-114, VTP-43742, ARN6039, TAK-828, RTA-1701, BOS-172767, AUR-101 и JTE-451; пероральные ингибиторы JAK, включая, не ограничиваясь этим, тофацитиниб, JTE-052, барицитиниб, упадацитиниб, PF-04965842, PF-06651600 и PF-06700841; ингибиторы TYK для перорального введения, включая, не ограничиваясь этим, PF-06826647 и BMS-986165; ингибиторы ITK для перорального введения, включая, не ограничиваясь этим, JTE-051; ингибиторы SYK для перорального введения, включая, не ограничиваясь этим, фостаматиниб, цердулатиниб, энтосплетиниб, TAK-659, ASN-002 и GS-9876; модуляторы рецептора S1P для перорального введения, включая, не ограничиваясь этим, озанимод и этразимод; и ингибиторы IRAK4 для перорального введения, включая, не ограничиваясь этим, 1-(((2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил)метокси)-7-метоксиизохинолин-6-карбоксамид и BAY 1830839.
В еще одном варианте осуществления настоящее изобретение относится к фармацевтическим комбинацим для инъекционного введения, содержащим соединение формулы (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB), или его фармацевтически приемлемую соль или гидрат в комбинации с другим фармацевтическим агентом для лечения заболеваний, состояний и/или расстройств, описанных здесь. Подходящие фармацевтические средства, которые можно использовать в комбинации с соединениями по настоящему изобретению для инъекционного введения, включают, не ограничиваясь этим: ингибиторы TNF, включая, не ограничиваясь этим, инфликсимаб, адалимумаб, голимумаб и цертолизумаб пэгол; анти-MAdCAM, включая, не ограничиваясь этим, SHP647; анти-IL-12 P40, включая, не ограничиваясь этим, устекинумаб; анти-IL-23 P19, включая, не ограничиваясь этим, рисанкизумаб, мирикизумаб, бразикумаб, гуселкумаб и тидракизумаб; анти-IL-17, включая, не ограничиваясь этим, секукинумаб, бродалумаб и иксекизумаб; ингибиторы интегрина, включая, не ограничиваясь этим, натализумаб, ведолизумаб и этролизумаб.
Комбинированная терапия включает введение двух или более терапевтических средств одновременно или последовательно. Средства можно вводить в любом порядке. Альтернативно, несколько терапевтических средств можно объединить в одной композиции, которую можно вводить пациенту. Например, одна фармацевтическая композиция может содержать соединение или его фармацевтически приемлемую соль, гидрат, сложный эфир или пролекарство в соответствии с формулой (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB), другой терапевтический агент или его фармацевтически приемлемую соль, гидрат, сложный эфир или пролекарство и, по меньшей мере, один фармацевтически приемлемый эксципиент или носитель.
Как здесь используется, термин «фармацевтически приемлемая соль» или «соль» относится к солям, которые хорошо известны в данной области. Например, S.M Berge et al. подробно описывают фармацевтически приемлемые соли в публикации в J. Pharmaceutical Sciences, 66: 1-19 (1977), которая включена здесь посредством ссылки. Примерами фармацевтически приемлемых нетоксичных кислотно-аддитивных солей являются соли по аминогруппе, образованные с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и хлорная кислота, или с органическими кислотами, такими как уксусная кислота, щавелевая кислота, малеиновая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота, или с использованием других методов, используемых в данной области, таких как ионный обмен. Другие фармацевтически приемлемые соли включают нитрат, бисульфат, борат, формиат, бутират, валерат, 3-фенилпропионат, камфорат, адипат, бензоат, безилат, олеат, пальмитат, стеарат, лаурат, лактат, фумарат, аскорбат, аспартат, никотинат, п-толуолсульфонат, камфорсульфонат, метансульфонат, 2-гидроксиэтансульфонат, глюконат, глюкогептонат, лактобионат, глицерофосфат, пектинат, лаурилсульфат и тому подобное, соли металлов, такие как соли натрия, калия, магния или кальция, или аминосоли, такие как соли аммония, триэтиламина, и подобные, которые все можно получить обычными способами.
Основно-аддитивные соли можно получить in situ во время конечного выделения и очистки соединений по настоящему изобретению взаимодействием гидроксигруппы в положении 3 N, N-диметилпиколинамидного фрагмента с подходящим основанием, таким как гидроксид, карбонат или бикарбонат фармацевтически приемлемого катиона металла или аммонием, или органическим первичным, вторичным или третичным амином. Фармацевтически приемлемые соли включают, не ограничиваясь этим, катионы на основе щелочных металлов или щелочноземельных металлов, такие как соли лития, натрия, калия, кальция, магния и алюминия и т.п., и нетоксичные катионы четвертичного аммония и аминов, включая аммоний, тетраметиламмоний, тетраэтиламмоний, метиламмоний, диметиламмоний, триметиламмоний, триэтиламмоний, диэтиламмоний, этиламмоний и тому подобное. Другие репрезентативные органические амины, пригодные для образования основно-аддитивных солей, включают этилендиамин, этаноламин, диэтаноламин, пиперидин, пиперазин и тому подобное.
Настоящее изобретение также включает фармацевтически приемлемые соли пролекарств соединений по настоящему изобретению. Репрезентативные примеры включают, не ограничиваясь этим, соединения, приведенные ниже, где R+ представляет собой литий, натрий, калий, аммоний, тетраметиламмоний, тетраэтиламмоний, метиламмоний, диметиламмоний, триметиламмоний, триэтиламмоний, диэтиламмоний, этиламмоний и т.п., и где R2+ представляет собой кальций, магний, алюминий и тому подобное.
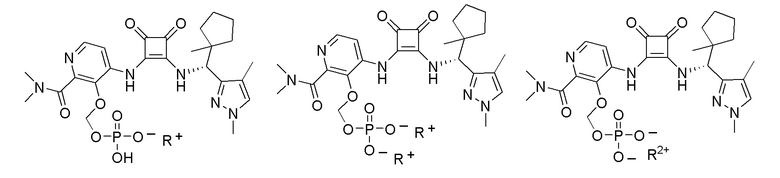
Соединения по настоящему изобретению можно выделить и использовать сами по себе в форме их фармацевтически приемлемых солей, включая соли пролекарства. В соответствии с настоящим изобретением соединения с несколькими основными атомами азота могут образовывать соли с различным числом эквивалентов («экв») кислоты. Соединения с несколькими кислотными группами могут образовывать основно-аддитивные соли с различным числом эквивалентов («экв») основной аддитивной молекулы или положительного противоиона. Практикующим специалистам будет понятно, что все такие соли входят в объем настоящего изобретения.
Настоящее изобретение охватывает соединения формулы (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB), полученные способами синтеза или метаболическими процессами. Получение соединений изобретения с помощью метаболических процессов, включают процессы, происходящие в организме человека или животного (in vivo), или процессы, происходящие in vitro.
Настоящее изобретение также предусматривает фармацевтически активные метаболиты, образованные биотрансформацией in vivo соединений формулы (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB). Как здесь используется, термин «фармацевтически активный метаболит» относится к соединению, образовавшемуся in vivo в результате биотрансформации соединений формулы (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB). Подробное обсуждение биотрансформации представлено в монографии (Goodman and Gilman’s, The Pharmacological Basis of Therapeutics, седьмое издание, MacMillan Publishing Company, New York, NY, (1985)).
При использовании в вышеуказанных или других способах лечения терапевтически эффективное количество одного из соединений по настоящему изобретению можно использовать в чистой форме или, если такие формы существуют, в форме фармацевтически приемлемой соли, сложного эфира, амида или пролекарства. Альтернативно, соединение можно вводить в виде фармацевтической композиции, содержащей соединение, представляющее интерес, в комбинации с одним или более фармацевтически приемлемыми носителями. Выражение «терапевтически эффективное количество» соединения по настоящему изобретению означает количество соединения, достаточное для лечения заболеваний, состояний или расстройств, указанных здесь, при разумном соотношении польза/риск, применимом к любому медицинскому лечению. Конкретный терапевтически эффективный уровень дозы для любого конкретного пациента будет зависеть от многих факторов, включая заболевание, которое лечат, и тяжесть расстройства; активность конкретного используемого соединения; конкретную используемую композицию; возраст, массу тела, общее состояние здоровья, пол и диету пациента; время введения, путь введения и скорость выведения конкретного применяемого соединения; продолжительность лечения; лекарственные средства, применяемые в комбинации или одновременно с конкретным используемым соединением; и подобные факторы, хорошо известные в области медицины.
Фармацевтические композиции и составы
В еще одном варианте осуществления настоящее изобретение относится к фармацевтическим композициям или составам, содержащим терапевтически эффективное количество соединения по настоящему изобретению и фармацевтически приемлемый эксципиент, разбавитель или носитель. Фармацевтические композиции или составы по настоящему изобретению можно вводить людям и другим млекопитающим местно, перорально, парентерально, интрацистернально, интравагинально, внутрибрюшинно, буккально, в виде перорального спрея, в виде назального спрея, ректально в виде суппозитория или в форме липосомы.
Типичную фармацевтическую композицию или состав получают смешиванием соединения по настоящему изобретению и носителя, разбавителя или эксципиента. Подходящие носители, разбавители и эксципиенты включают такие вещества, как углеводы, воска, водорастворимые и/или набухающие полимеры, гидрофильные или гидрофобные вещества, желатин, масла, растворители, воду и тому подобное. Конкретный используемый носитель, разбавитель или эксципиент будет зависеть от средств и цели, для которой применяется соединение по настоящему изобретению. Подходящие водные растворители включают воду, этанол, пропиленгликоль, полиэтиленгликоли (например, PEG400, PEG300) и т.д. и их смеси. Составы могут также включать одно или более из буферов, стабилизаторующих агентов, поверхностно-активных веществ, смачивающих агентов, смазывающих веществ, эмульгаторов, суспендирующих агентов, консервантов, антиоксидантов, замутнителей, скользящих веществ, технологических добавок, красителей, подсластителей, отдушек, ароматизаторов и других известных добавок для обеспечения приятного лекарственного средства (т.е. соединения по настоящему изобретению или его фармацевтической композиции) или помощи в производстве фармацевтического продукта (т.е. для использования в приготовлении лекарственного средства).
Составы можно приготовить с использованием обычных процедур растворения и смешивания. Например, насыпную лекарственную субстанцию (т.е. соединение по настоящему изобретению или стабилизированную форму соединения (например, комплекс с производным циклодекстрина или другим известным комплексообразующим агентом)) растворяют в подходящем растворителе в присутствии одного или более из эксципиентов, описанных выше. Степень растворения плохо растворимых в воде соединений можно повысить за счет использования высушенных распылением дисперсий, таких как описанные в публикации Takeuchi, H., et al. в «Enhancement of the dissolution rate of a poorly water-soluble drug (tolbutamide) by a spray-drying solvent deposition method and disintegrants» J. Pharm. Pharmacol., 39, 769-773 (1987); and EP0901786 B1 (US2002/009494), которая включена здесь посредством ссылки. Соединение по настоящему изобретению обычно готовят в виде фармацевтических лекарственных форм для обеспечения легко контролируемой дозы лекарственного средства и предоставления пациенту приятного и легко используемого продукта.
Фармацевтическая композиция или состав для применения могут быть упакованы различными способами в зависимости от метода, используемого для введения лекарственного средства. Обычно изделие для распространения включает контейнер, в котором помещен фармацевтический состав в соответствующей форме. Подходящие контейнеры включают в себя такие материалы, как бутыли (пластиковые и стеклянные), пакетики, ампулы, пластиковые пакеты, металлические цилиндры и тому подобное. Контейнер может также включать в себя защищенную от неосторожного обращения сборку для предотвращения нетребуемого доступа к содержимому упаковки. Кроме того, на контейнере наносится этикетка, на которой описано содержимое контейнера. Этикетка может также включать соответствующие предупреждения.
Термин «фармацевтически приемлемый носитель» относится к носителю-среде, который обеспечивает соответствующую доставку эффективного количества активного агента, как здесь определено, не оказывает влияния на эффективность проявления биологической активности активного агента и является достаточно нетоксичным для хозяина или пациента. Репрезентативные носители включают воду, масла, как растительные, так и минеральные, основы для кремов, основы для лосьонов, основы для мазей и т.п. Эти основы включают суспендирующие агенты, загустители, усилители проникновения и т.п. Дополнительную информацию о носителях можно найти в монографии Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams & Wilkins (2005), которая включена здесь посредством ссылки. Другими примерами веществ, которые могут служить фармацевтически приемлемыми носителями, являются сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлоза и ее производные, такие как натрий карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; порошкообразный трагакант; солод; желатин; тальк; эксципиенты, такие как масло какао и воска для суппозиториев; масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные агенты, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический физиологический раствор; раствор Рингера; этиловый спирт и фосфатные буферные растворы, а также другие нетоксичные совместимые смазочные вещества, такие как лаурилсульфат натрия и стеарат магния, а также красители, антиадгезионные средства, покрывающие вещества, подсластители, ароматизаторы и отдушки, консерванты и антиоксиданты также могут присутствовать в композиции согласно мнению специалиста, занимающегося составлением рецептуры.
Термин «фармацевтически приемлемый носитель для местного введения» относится к фармацевтически приемлемым носителям, как здесь описано выше, подходящим для местного введения. Неактивный жидкий или кремообразный носитель, способный суспендировать или растворять активный агент (агенты) и не обладающий свойствами индукции токсичности и воспаления при нанесении на кожу, ноготь, волосы, коготь или копыто является примером фармацевтически приемлемого носителя для местного введения. Данный термин специально предназначен для охвата веществ носителя, одобренных для применения в косметике для местного применения.
Термин «местное введение» относится к нанесению фармацевтического агента на наружную поверхность кожи, ногтя, волос, когтя или копыта, таким образом, что агент проникает через наружную поверхность кожи, ногтя, волос, когтя или копыта и попадает в нижележащие ткани. Местное введение включает нанесение композиции на неповрежденную кожу, ноготь, волосы, коготь или копыто или на нарушенную, необработанную или открытую рану кожи, ногтя, волос, когтя или копыта. Местное введение фармацевтического агента может привести к ограниченному распределению агента на коже и окружающих тканях или, когда агент удаляется из области лечения кровотоком, что может привести к системному распределению агента.
Лекарственные формы для местного или трансдермального введения соединения по настоящему изобретению включают мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, ингалянты или пластыри. Активный компонент смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами или буферами, которые могут потребоваться. Для летучих соединений может потребоваться смешивание со специальными формулирующими агентами или специальными упаковочными материалами, чтобы обеспечить правильную доставку доз. Кроме того, для соединений по настоящему изобретению, которые имеют плохую проницаемость через кожу человека, может потребоваться включение одного или более усилителей проникновения, тогда как соединения, быстро всасывающиеся через кожу, могут потребовать формуляции с агентами, замедляющими всасывание или барьеры.
Мази, пасты, кремы, лосьоны, гели, порошки и растворы для местного введения могут содержать, помимо активного соединения по настоящему изобретению, фармацевтически приемлемые эксципиенты, такие как животные и растительные жиры, масла, воска, парафины, крахмал, трагакант, производные целлюлозы, полиэтиленгликоли, силиконы, бентониты, кремниевую кислоту, тальк, оксид цинка, консерванты, антиоксиданты, отдушки, эмульгаторы, красители, инертные наполнители, вещества, снимающие раздражение, вещества, повышающие клейкость, отдушки, замутнители, антиоксиданты, гелеобразователи, поверхностно-активные вещества, смягчающие вещества, красители, консерванты, буферные агенты, усилители проникновения или их смеси. Вспомогательные вещества для местного применения не должны влиять на эффективность биологической активности активного агента и не быть вредными для эпителиальных клеток или их функции.
Термины «усилитель проникновения» или «усилитель проницаемости» относятся к агентам, которые повышают проницаемость кожи, ногтей, волос, когтей или копыт для лекарственного средства, для повышения скорости, с которой лекарственное средство проникает через кожу, ноготь, волосы, коготь или копыто. Усиление проникновения, достигаемое за счет применения таких усилителей, можно наблюдать, например, измерением скорости диффузии лекарственного средства через кожу, ноготь, волосы, коготь или копыто животного или человека с использованием прибора для оценки диффузии в клетки. Прибор для оценки диффузии в клетки описан Merritt et al. Diffusion Apparatus for Skin Penetration, J. of Controlled Release, 1 (1984) pp. 161-162. Термин «усилитель проникновения» или «усилитель проницаемости» означает вещество или смесь веществ, которые, по отдельности или в комбинации, действуют, увеличивая проницаемость кожи, ногтей, волос или копыт для лекарственного средства.
Термин «трансдермальная доставка» относится к диффузии агента через барьер кожи, ногтей, волос, когтей или копыт в результате местного введения или другого применения композиции. Роговой слой функционирует в качестве барьера, и немногие фармацевтические агенты способны проникать через неповрежденную кожу. Напротив, эпидермис и дерма проницаемы для многих растворенных веществ, и поэтому всасывание лекарственных препаратов легче происходит через кожу, ноготь, волосы, коготь или копыто, которые истираются или с иным образом удаленным роговым слоем, обнажая при этом эпидермис. Трансдермальная доставка включает инъекцию или другую доставку через любую часть кожи, ногтя, волос, когтя, копыта или слизистой оболочки и всасывание или проникновение через остальную часть. Всасывание через неповрежденную кожу, ноготь, волосы, коготь или копыто может быть усилено помещением активного агента в соответствующий фармацевтически приемлемый носитель перед нанесением на кожу, ноготь, волосы, коготь или копыто. Пассивное местное введение может состоять из нанесения активного агента непосредственно на область лечения в комбинации со смягчающими средствами или усилителями проникновения. В контексте настоящего описания под трансдермальной доставкой подразумевается доставка посредством проникновения через или сквозь кожный покров, то есть кожу, ноготь, волосы, коготь или копыто.
Порошки и спреи могут содержать, помимо соединений по настоящему изобретению, лактозу, тальк, кремниевую кислоту, гидроксид алюминия, силикаты кальция и порошкообразный полиамид или смеси этих веществ. Спреи могут дополнительно содержать обычные пропелленты, такие как хлорфторуглеводороды.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение смешано, по меньшей мере, с одним инертным фармацевтически приемлемым носителем, таким как цитрат натрия или фосфат кальция, и/или а) наполнители или разбавители, такие как крахмалы, лактоза, сахароза, глюкоза, маннит и салициловая кислота; b) связующие вещества, такие как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидинон, сахароза и гуммиарабик; c) увлажнители, такие как глицерин; d) дезинтегрирующие агенты, такие как агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые силикаты и карбонат натрия; e) средства, замедляющие растворение, такие как парафин; f) ускорители всасывания, такие как соединения четвертичного аммония; g) смачивающие агенты, такие как цетиловый спирт и моностеарат глицерина; h) абсорбенты, такие как каолин и бентонитовая глина; и i) смазывающие вещества, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси. В случае капсул, таблеток и пилюль лекарственная форма может также содержать буферные агенты.
Твердые композиции подобного типа также могут использоваться в качестве наполнителей в мягких и твердых желатиновых капсулах с использованием лактозы или молочного сахара, а также высокомолекулярных полиэтиленгликолей и т.п.
Твердые лекарственные формы, включающие таблетки, драже, капсулы, пилюли и гранулы, можно приготовить с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные в области фармацевтических препаратов. Они могут необязательно содержать замутнители, а также могут представлять собой композицию, которая высвобождает активный ингредиент(ы) только или предпочтительно в определенной части кишечного тракта замедленным образом. Примеры композиций для включения, которые можно использовать, включают полимерные вещества и воска.
Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активным соединениям жидкие лекарственные формы могут содержать инертные разбавители, обычно используемые в данной области, такие как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, масло зародышей пшеницы, оливковое, касторовое и кунжутное), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот сорбитана и их смесей.
Помимо инертных разбавителей, оральные композиции могут также включать адъюванты, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, подсластители, ароматизаторы и отдушки.
Как здесь используется, термин «парентерально» относится к способам введения, которые включают внутривенное, внутримышечное, внутрибрюшинное, интрастернальное, подкожное, внутрисуставное введение и инфузию. Фармацевтические композиции по настоящему изобретению для парентеральной инъекции включают фармацевтически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии и стерильные порошки для восстановления в стерильные инъекционные растворы или дисперсии. Примеры подходящих водных и неводных носителей, разбавителей, растворителей или эксципиентов включают воду, этанол, полиолы (пропиленгликоль, полиэтиленгликоль, глицерин и т.п.), их подходящие смеси, растительные масла (такие как оливковое масло) и органические сложные эфиры для инъекций, такие как этилолеат. Надлежащую текучесть можно поддерживать, например, посредством использования покрытия, такого как лецитин, путем поддержания требуемого размера частиц в случае дисперсий и путем использования поверхностно-активных веществ.
Инъекционные формы-депо получают посредством формирования микрокапсулированных матриц лекарственного средства в биоразлагаемых полимерах, таких как полилактид-полигликолид. В зависимости от соотношения лекарственного средства к полимеру и природы конкретного используемого полимера скорость высвобождения лекарственного средства можно контролировать. Примеры других биоразлагаемых полимеров включают поли(ортоэфиры) и поли(ангидриды). Инъекционные препараты-депо также получают посредством включения лекарственного средства в липосомы или микроэмульсии, которые совместимы с тканями организма.
Инъекционные составы можно стерилизовать, например, фильтрованием через фильтр, задерживающий бактерии, или включением стерилизующих агентов в форме стерильных твердых композиций, которые можно растворить или диспергировать в стерильной воде или другой стерильной среде для инъекций непосредственно перед применением.
Инъекционные препараты, например стерильные водные или масляные суспензии для инъекций, можно составить с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильный инъекционный препарат может также представлять собой стерильный раствор, суспензию или эмульсию для инъекций в нетоксичном, приемлемом для парентерального введения разбавителе или растворителе, таком как раствор в 1,3-бутандиоле. Среди приемлемых носителей и растворителей, которые можно использовать, можно назвать воду, раствор Рингера, U.S.P. и изотонический раствор натрия хлорида. Кроме того, стерильные нелетучие масла обычно используются в качестве растворителя или суспендирующей среды. Для этой цели можно использовать любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, при приготовлении инъекционных препаратов используются жирные кислоты, такие как олеиновая кислота.
Фармацевтические композиции или составы для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые можно приготовить смешиванием соединений по настоящему изобретению с подходящими нераздражающими носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при температуре окружающей среды, но становятся жидкими при температуре тела и, следовательно, плавятся в прямой кишке или полости влагалища и высвобождают активное соединение.
Соединения по настоящему изобретению также можно вводить в форме липосом. Липосомы обычно получают из фосфолипидов или других липидных соединений, и они образованы моно- или многослойными гидратированными жидкими кристаллами, которые диспергированы в водной среде. Можно использовать любой нетоксичный, физиологически приемлемый и метаболизируемый липид, способный образовывать липосомы. Настоящие композиции в липосомной форме могут содержать, помимо соединений по настоящему изобретению, стабилизаторы, консерванты и тому подобное. Предпочтительными липидами являются природные и синтетические фосфолипиды и фосфатидилхолины (лецитины), используемые отдельно или вместе. Способы образования липосом известны в данной области. См., Например, Prescott, Ed., Methods in Cell Biology, Volume XIV, Academic Press, New York, N. Y., (1976), p 33 et seq.
Фармацевтические композиции или составы по настоящему изобретению могут также содержать адъюванты, такие как консерванты, смачивающие агенты, эмульгирующие агенты и диспергирующие агенты. Предупреждение действия микроорганизмов может быть обеспечено различными антибактериальными и противогрибковыми средствами, например парабенами, хлорбутанолом, фенолом, сорбиновой кислотой и т.п. Также может быть желательным включить изотонические агенты, например сахара, хлорид натрия и тому подобное. Продолжительное всасывание инъекционной фармацевтической формы может быть достигнуто за счет использования агентов, замедляющих абсорбцию, например моностеарата алюминия и желатина.
Фармацевтические композиции или составы по изобретению могут представлять собой суспензии. Суспензии, помимо активных соединений, могут содержать суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбитол и сложные эфиры сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар, трагакант и их смеси.
Фактические уровни дозировок активных ингредиентов в фармацевтических композициях по настоящему изобретению могут варьироваться для получения количества активного(ых) соединения(й), которое является эффективным для достижения желаемого терапевтического ответа для конкретного пациента, композиций и способа введения. Выбранный уровень дозировок будет зависеть от активности конкретного соединения, пути введения, тяжести состояния, которое лечат, а также состояния и предшествующей истории болезни пациента, который подвергается лечению. Однако специалист в данной области техники может начинать дозы соединения с уровней ниже, чем требуется для достижения желаемого терапевтического эффекта, и постепенно увеличивать дозу до тех пор, пока желаемый эффект не будет достигнут.
Общая суточная доза соединений по настоящему изобретению, вводимая человеку или низшему животному, может находиться в диапазоне примерно от 0,0001 до примерно 15 мг/кг/день. Для перорального введения более предпочтительные дозы могут находиться в диапазоне примерно от 0,01 до примерно 5 мг/кг/ день. Для местного применения более предпочтительные дозы могут находиться в диапазоне от 0,001 мг/кг/день до примерно 5 мг/кг/ день. При желании эффективную суточную дозу можно разделить на несколько доз для введения, например от двух до четырех отдельных доз в день.
Фармацевтические композиции также включают сольваты и гидраты соединений по настоящему изобретению. Термин «сольват» относится к молекулярному комплексу соединения, представленного формулой (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB), включая его фармацевтически приемлемые соли или гидраты, с одной или более молекулами растворителя. Такие молекулы растворителя обычно используются в фармацевтике, которые, как известно, безвредны для реципиента, например вода, этанол, этиленгликоль, (S)-пропиленгликоль, (R)-пропиленгликоль и т.п. Термин «гидрат» относится к комплексу, в котором молекулой растворителя является вода. Сольваты и/или гидраты предпочтительно находятся в кристаллической форме. Другие растворители можно использовать в качестве промежуточных сольватов при получении более желательных сольватов. Промежуточные растворители включают, не ограничиваясь этим, метанол, метил-трет-бутиловый эфир, этилацетат, метилацетат, 1,4-бутиндиол и тому подобное.
Соединения по настоящему изобретению могут находиться в более чем одной кристаллической форме. Полиморфы соединений формул (IA), (IB), (IIA), (IIB), (IIIA) или (IIIB) и их солей (включая сольваты и гидраты) составляют часть данного изобретения и могут быть получены кристаллизацией соединения по настоящему изобретению в различных условиях. Например, используют разные растворители или смеси разных растворителей для перекристаллизации; кристаллизацию при разных температурах; различные режимы охлаждения, от очень быстрого до очень медленного охлаждения во время кристаллизации. Полиморфы также можно получить нагреванием или плавлением соединения настоящего изобретения с последующим постепенным или быстрым охлаждением. Присутствие полиморфов можно определить с помощью твердотельной спектроскопии ядерного магнитного резонанса (ЯМР), инфракрасной (ИК) спектроскопии, дифференциальной сканирующей калориметрии, порошковой дифракции рентгеновских лучей или других подобных методов.
Настоящее изобретение также включает меченые изотопами соединения, которые идентичны соединениям, описанным формулами (IA), (IB), (IIA), (IIB), (IIIA) и (IIIB), но с тем отличием, что один или более атомов заменяются атомом с атомной массой или массовым числом, отличным от атомной массы или массового числа, обычно встречающихся в природе. Примеры изотопов, которые могут быть включены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, серы и фтора, такие как 2H, 3H, 13C, 14C, 15N, 18O, 17O, 35S, 36Cl, 125I, 129I и 18F соответственно. Некоторые меченные изотопами соединения по настоящему изобретению, например те, в которые включены радиоактивные изотопы, такие как 3H и 14C, пригодны для использования в исследованиях распределения лекарственных препаратов и/или субстратов в тканях. Изотопы, содержащие тритий (т.е. 3H) и углерод-14 (т.е. 14C), особенно предпочтительны за счет простоты их получения и детектирования. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий (т.е. 2H), может обеспечить определенные терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличенный период полураспада in vivo или снижение необходимости введения дозы, и, следовательно, может быть предпочтительным в некоторых обстоятельствах. Меченые изотопами соединения по настоящему изобретению, как правило, можно получить, выполняя процедуры, раскрытые на схемах и/или в примерах ниже, путем замены легко доступного изотопно меченного реагента на реагент, не меченный изотопами.
В частности, настоящее изобретение включает дейтерированные соединения формул (IA), (IB), (IIA), (IIB), (IIIA) и (IIIB). Любой из атомов водорода, содержащихся в соединениях настоящего изобретения, может быть заменен на дейтерий, включая атомы водорода пиридина, как показано ниже.
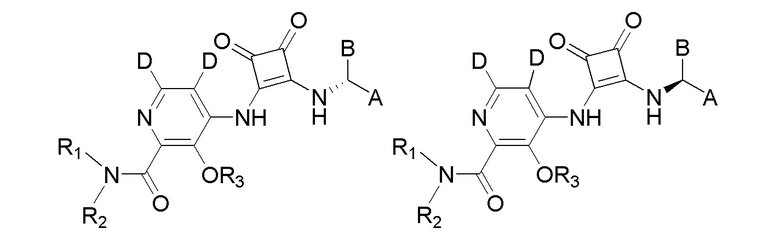
Репрезентативные примеры дейтерированных соединений по настоящему изобретению включают, но не ограничиваются ими, примеры 39 и 40.
Все цитированные патенты и публикации США (включая все технические бюллетени, на которые есть ссылки в примерах) полностью включены здесь посредством ссылки.
Соединения по настоящему изобретению или их фармацевтически приемлемые соли или гидраты можно получить способами, описанными ниже, вместе с методами синтеза, известными в области органической химии, или модификациями и дериватизациями, которые знакомы обычным специалистам в данной области техники. Используемые здесь исходные вещества являются коммерчески доступными или их можно получить обычными способами, известными в данной области (такими как способы, раскрытые в стандартных справочниках, включая Compendium of Organic Synthetic Methods, Vol. I-VI, опубликованный Wiley-Interscience). Предпочтительные способы включают, не ограничиваясь этим, способы, описанные ниже.
Во время любой из следующих синтетических последовательностей может быть необходимо и/или желательно защитить чувствительные или реактивные группы на любой из рассматриваемых молекул. Это может быть достигнуто с помощью обычных защитных групп, таких как описанные в монографиях T. W. Greene, Protective Groups in Organic Chemistry, John Wiley & Sons, 1981; T. W. Greene и P. G.M. Wuts, Protective Groups in Organic Chemistry, John Wiley & Sons, 1991, и T. W. Greene, P. G. M. Wuts, Protective Groups in Organic Chemistry, John Wiley & Sons, 1999, которые включены здесь посредством ссылки.
Соединения по настоящему изобретению можно получить в виде одного энантиомера или в виде смеси отдельных энантиомеров, которая включает рацемические смеси. Способы получения предпочтительно одного энантиомера из смеси отдельных энантиомеров или рацемической смеси хорошо известны специалистам в области органической химии. Такие методы включают, не ограничиваясь этим, предпочтительную кристаллизацию диастереомерных солей (например, тартрата или камфорсульфоната), ковалентную дериватизацию хиральным нерацемическим реагентом с последующим разделением полученных диастереомеров обычными методами (например, кристаллизацией, хроматографическим разделением или дистилляцией) и химическое превращение в скалемическое соединение, или жидкостная хроматография высокого/среднего давления или сверхкритическая жидкостная хроматография с использованием хиральной неподвижной фазы. Данные методы можно выполнить с конечными соединениями по изобретению или с любыми промежуточными продуктами соединений по изобретению, которые несут стереогенный центр. Кроме того, для облегчения разделения любым из способов, описанных выше, соединения по изобретению или любые промежуточные соединения для соединений по изобретению, которые несут стереогенный центр, можно временно подвергнуть взаимодействию с ахиральным реагентом, разделить и затем превратить в скалемическое соединение с использованием обычных синтетических методик.
Стереоизомеры обозначаются (R) или (S) в зависимости от конфигурации заместителей вокруг хирального атома углерода. Термины (R) и (S), используемые здесь, являются конфигурациями, как определено в Рекомендациях IUPAC 1974 г. Section E, Fundamental Stereochemistry, Pure Appl. Chem., (1976), 45: 13-30. В частности, стереохимия в точке присоединения переменных «А» и «В»:
в формулах (IA), (IB), (IIA), (IIB), (IIIA) и (IIIB) может независимо представлять (R) или (S). Энантиомеры по настоящему изобретению, обозначенные (R), (S) или *, по существу не содержат другого энантиомера. «По существу не содержат» означает, что энантиомерный избыток составляет примерно более 90%, предпочтительно примерно более 95% и более предпочтительно примерно более 99%. В контексте энантиомерного избытка термин «примерно» означает ± 1,0%. Символ * обозначает хиральный атом углерода либо как (R), либо как (S) стереохимию в зависимости от конфигурации заместителей вокруг хирального атома углерода.
Соединения по настоящему изобретению, не обозначенные (R), (S) или *, могут находиться в виде рацематов (т.е. 50% (R) и 50% (S)) или в виде смеси двух энантиомеров, где один энантиомер находится в избытке. Например, энантиомерные смеси могут включать (R) энантиомер в количестве 51% и (S) энантиомер в количестве 49% или наоборот, или любую комбинацию (R) и (S), отличную от рацемической смеси 50% (R) и 50% (S). Настоящее изобретение включает рацематы и энантиомерные смеси соединений по настоящему изобретению.
Соединения по настоящему изобретению могут находиться в различных стабильных конформационных формах, которые можно разделить. Торсионная асимметрия за счет ограниченного вращения вокруг асимметричной одинарной связи, например, за счет стерических затруднений или деформации кольца, может допускать разделение различных конформеров. Соединения по настоящему изобретению дополнительно включают каждый конформационный изомер соединений формулы (IA), (IB), (IIA), (IIB), (IIIA), (IIIB) и их смеси.
Таутомеры могут присутствовать в соединениях по настоящему изобретению и конкретно включаются в объем настоящего изобретения. Термин «таутомер» в контексте настоящего описания означает сдвиг протона от одного атома молекулы к другому атому той же самой молекулы, где два или более структурно различных соединения находятся в равновесии друг с другом. Соединения по настоящему изобретению могут находиться в виде таутомеров. Настоящее изобретение рассматривает таутомеры за счет сдвигов протонов от одного атома к другому атому одной и той же молекулы, с образованием двух или более различных соединений, находящихся в равновесии друг с другом. В частности, фрагмент бис(амино)циклобут-3-ен-1,2-диона, входящий в состав соединений по настоящему изобретению, может таутомеризоваться, как показано ниже, и они включаются в объем настоящего изобретения.
Соединения по настоящему изобретению или их фармацевтически приемлемые соли можно получить в соответствии со схемами и примерами реакций, раскрытыми здесь ниже. Если не указано иное, то R1, R2, R3, R4, R5, R6, R7, R8, R9, m, n, A, B, X, RA, RB и RC на схемах реакций имеют значения, определенные в формулах (IA) и (IB) в разделе «Сущность изобретения» выше. Специалисту в данной области техники должно быть понятно, что различные символы, надстрочные и подстрочные индексы, используемые на схемах реакций, способах и примерах, используются для удобства представления и/или для отражения порядка, в котором они вводятся на схемах, и не предназначены для обязательного соответствия символам, надстрочным или подстрочным индексам в прилагаемой формуле изобретения. На схемах реакций представлены способы, используемые в синтезе соединений по настоящему изобретению. Они никоим образом не ограничивают объем изобретения. Выделение и очистка продуктов выполняются стандартными процедурами, известными специалисту с обычной квалификаией в данной области.
Схема реакций А
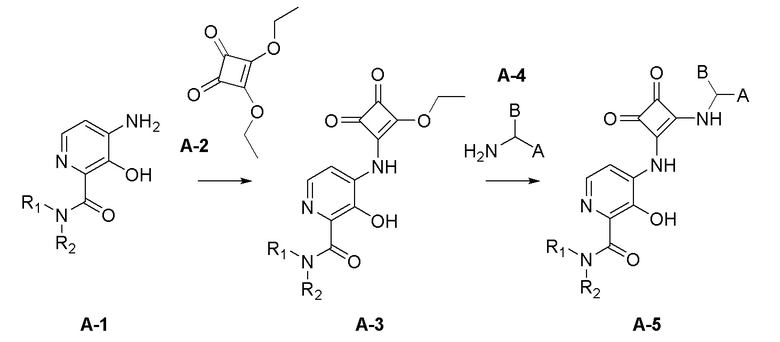
Соединения формулы (IA) и (IB) можно получить, как показано на схеме реакций A. Нуклеофильное ароматическое замещение между аминопиридином A-1 (полученным, как показано на схеме реакций B) и коммерчески доступным 3,4-диэтоксициклобут-3-ен-1,2-дионом (A-2) дает соединения формулы A-3. Второе нуклеофильное ароматическое замещение между A-3 и амином A-4 (полученным, как показано на схемах реакций C-G) дает соединения формулы A-5.
Схема реакций В
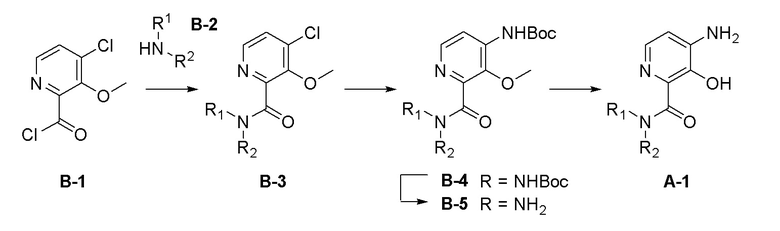
Соединения формулы A-1, используемые на схеме реакций A, можно получить, как показано на схеме реакций B. Амидирование коммерчески доступного 4-хлор-3-метоксипиколиноилхлорида (B-1) и коммерчески доступного амина B-2 в щелочных условиях дает соединения формулы В-3. Сочетание Бухвальда между B-3 и трет-бутилкарбаматом дает соединения формулы B-4, которые можно подвергнуть воздействию кислотных условий для удаления Boc-группы и получить соединения формулы B-5. Деметилирование B-5 приводит к получению соединений формулы A-1.
Схема реакций С
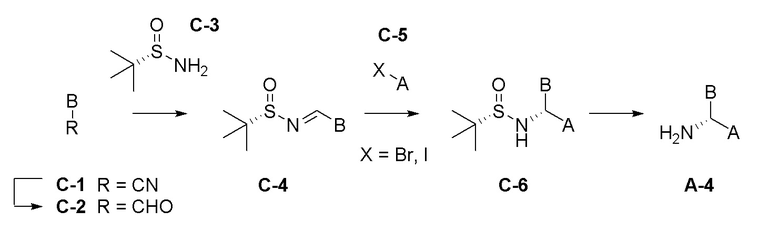
Соединения формулы A-4, используемые на схеме реакций A, где амины энантиомерно обогащены, можно получить, как показано на схеме реакций C. Опосредованная этоксидом титана (IV) конденсация альдегида C-2 (либо коммерчески доступного, либо полученного восстановлением соответствующего нитрила C-1, коммерчески доступного или полученного способами, известными в данной области техники) с (S)-2-метилпропан-2-сульфинамидом (C-3) обеспечивает соединения формулы C-4. Металлирование гетероарилгалогенидов C-5, где X представляет собой йод или бром, обеспечивает высоко диастереоселективное присоединение к соединению C-4 с получением сульфонамидов формулы C-6 в качестве основного диастереомера. Последующее удаление хирального вспомогательного соединения в кислых условиях приводит к образованию соединений формулы A-4. Абсолютную стереохимию аминов А-4 можно установить методами, известными специалистам в данной области, включая анализ амидов Мошера и рентгеновскую кристаллографию монокристаллов, или определить по аналогии с известными соединениями.
Схема реакций D
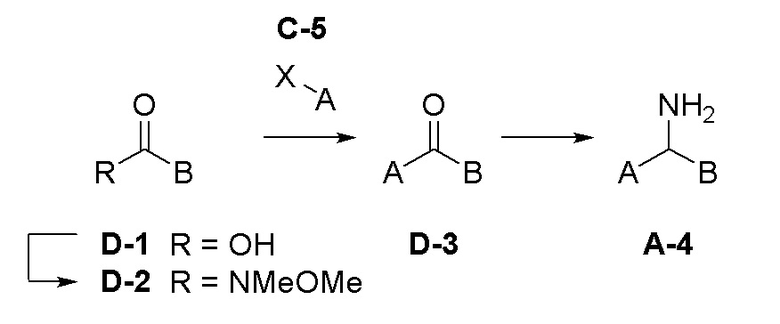
Соединения формулы A-4, используемые на схеме реакций A, где амины представляют собой рацемические смеси или диастереомерные смеси, можно получить, как показано на схеме реакций D. Карбоновые кислоты формулы D-1 (либо коммерчески доступные, либо полученные, как описано здесь ниже), можно превратить в соответствующий амид Вайнреба D-2 посредством HATU-опосредованной конденсации с метоксиметиламином. Металлирование гетероарилгалогенидов C-5, где X представляет собой йод или бром, с последующей реакцией с D-2 дает кетоны формулы D-3. Восстановительное аминирование D-3 с раствором аммиака в метаноле дает соединения формулы A-4 в виде рацемической или диастереомерной смеси.
Схема реакций E

Альтернативно, соединения формулы A-4, где переменная A формулы (IA) или (IB) определена как замещенный пиразол, можно получить из 3-галоген-1-алкилпиразолов E-1 (R4=алкил и X=бром или йод), как показано на схеме реакций E. Металлирование E-1 и его присоединение к сульфинамину C-4 или к амиду Вайнреба D-2 дает продукты присоединения формулы E-2 или E-4 соответственно. Введение заместителя R5 можно провести электрофильным ароматическим замещением или реакцией с NIS, и затем опосредованной металлом реакции перекрестного сочетания образующегося иодида с получением соединений E-3 или E-5. Затем хиральный вспомогательный компонент E-3 можно удалить в кислых условиях с получением соединений формулы A-4. Восстановительное аминирование E-5 с использованием раствора аммиака в метаноле дает соединения формулы A-4.
Схема реакций F
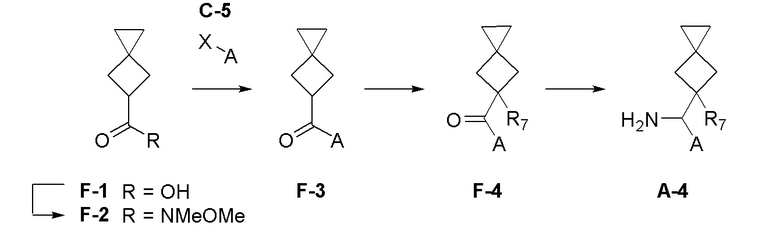
Соединения формулы A-4, используемые на схеме A, где переменная B формулы (IA) или (IB) определяется как 5-метилспиро [2,3]гексан-5-ил, можно получить, как показано на схеме реакций F. Коммерчески доступная спиро[2.3]гексан-5-карбоновая кислота (F-1) может быть преобразована в соответствующий амид Вейнреба F-2 посредством HATU-опосредованной конденсации с метоксиметиламином. Металлирование гетероарилгалогенидов C-5, где X представляет собой йод или бром, с последующей реакцией с F-2 дает кетоны формулы F-3. Последующее алкилирование F-3 с использованием алкилгалогенида (такого как йодметан или йодэтан) и подходящего основания с последующим восстановительным аминированием с использованием раствора аммиака в метаноле дает соединения формулы A-4 в виде рацемической смеси.
Схема реакций G
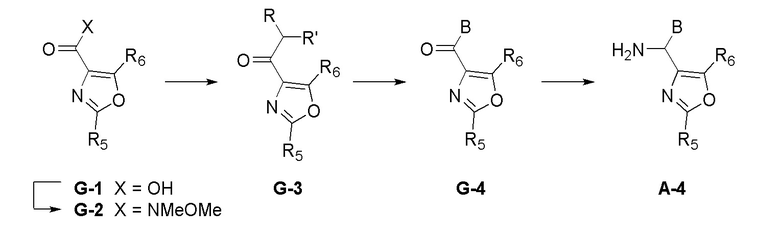
Соединения формулы A-4, используемые на схеме A, где переменная A формулы (IA) или (IB) определяется как 2,5-дизамещенный оксазол, можно получить, как показано на схеме реакций G. Карбоновые кислоты формулы G-1, коммерчески доступные или полученные способами, известными в данной области, можно конденсировать с метоксиметиламином в присутствии HATU с получением амидов Вайнреба формулы G-2. Соединение G-2 можно подвергнуть взаимодействию с реагентом Гриньяра (таким как бромид изопропилмагния или бромид циклопентилмагния) с образованием кетонов формулы G-3, где R и R' представляют собой метил или R и R', взятые вместе с атомом углерода, к которому они присоединены, образуют C3-5циклоалкановое кольцо. Последующее алкилирование G-3 с использованием алкилгалогенида (такого как йодметан или йодэтан) и подходящего основания с последующим восстановительным аминированием с использованием раствора аммиака в метаноле дает соединения формулы A-4 в виде рацемической смеси.
Схема реакций H
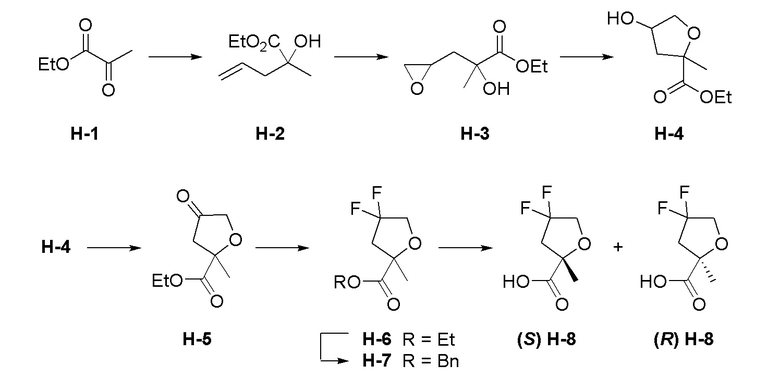
Конкретную карбоновую кислоту D-1, используемую на схеме D, где переменная B формулы (IA) или (IB) определяется как (S) или (R)-4,4-дифтор-2-метилтетрагидрофуран-2-ил, можно получить, как показано на схеме реакций Н. Опосредованное хлоридом титана (IV) аллилирование коммерчески доступного этил 2-оксопропаноата (H-1) с аллилтриметилсиланом дает этил 2-гидрокси-2-метилпент-4-еноат (H-2). Эпоксидирование концевого алкена H-2 с использованием 3-хлорпербензойной кислоты с последующей внутримолекулярной циклизацией, опосредованной бромидом магния, дает этил 4-гидрокси-2-метилтетрагидрофуран-2-карбоксилат (H-4). Последующее окисление вторичного спирта с использованием хлорхромата пиридиния с последующим фторированием с использованием трифторида диэтиламиносеры дает этил-4,4-дифтор-2-метилтетрагидрофуран-2-карбоксилат (H-6). Сложный эфир H-6 переэтерифицировали до бензилового эфира H-7, который затем разделяли с помощью хиральной ВЭЖХ. Последующее омыление сложного эфира дает индивидуальные энантиомеры H-8, которые можно использовать на схеме реакций D. Абсолютную стереохимию H-8 определяли с помощью рентгеновской кристаллографии монокристаллов (см. фиг. 3).
Схема реакций I
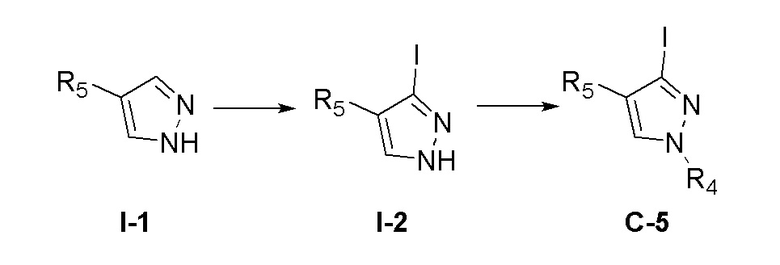
1,4-Дизамещенные-3-иодопиразолы, которые можно использовать в синтетических методологиях, показанных на схемах C, D и F, можно получить, как показано на схеме реакций I. Иодирование коммерчески доступных 4-замещенных пиразолов (I-1) с использованием NIS обеспечивает соединения формулы I-2. Последующее N-алкилирование I-2 алкилгалогенидами (например, йодметаном, йодэтаном) с последующим разделением региоизомеров хроматографией дает соединения формулы C-5, которые можно использовать в синтетических методологиях, показанных на вышеприведенных схемах.
Схема реакций J
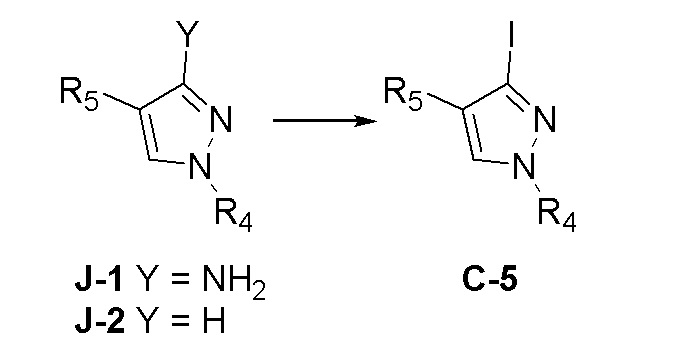
Альтернативно 1,4-дизамещенные-3-иодопиразолы, которые можно использовать в синтетических методологиях, показанных на схемах C, D и F, можно получить, как показано на схеме реакций J. 1,4-дизамещенные-3-аминопиразолы (J-1) можно преобразовать в соединения формулы C-5 посредством реакции Зандмейера с использованием нитрита натрия и иодида калия. Соединения формулы C-5 также можно получить прямым йодированием коммерчески доступных 1,4-дизамещенных пиразолов J-2 с использованием NIS.
Схема реакций K
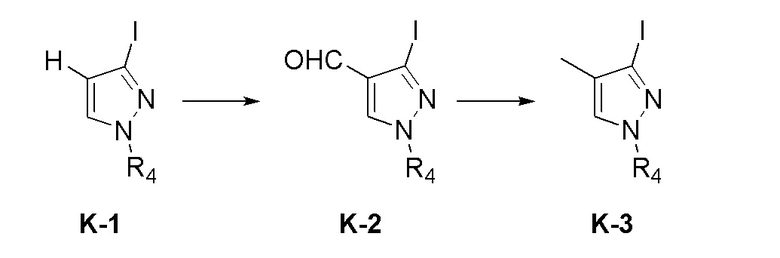
В еще одном варианте 1,4-дизамещенные 3-иодопиразолы, используемые на схемах реакций C-D и схеме реакций F, можно получить, как показано на схеме реакций K, в частности, где R4=R5=метил. 3-Иод-1-метил-1H-пиразол, K-1, можно подвергнуть региоселективному формилированию с помощью реагента Вильсмайера с образованием 3-иод-1-метил-1H-пиразол-4-карбальдегида (K-2), который, в свою очередь, можно восстановить (ТФК, триэтилсилан) с получением 3-иод-1,4-диметил-1H-пиразола, K-3.
Схема реакций L
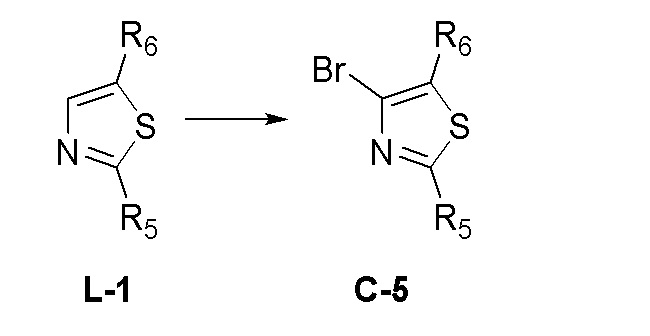
2,5-Дизамещенные 4-бромтиазолы, используемые на схемах реакций C-D и схеме реакций F, можно получить, как показано на схеме реакций L. Бромирование коммерчески доступных 2,5-дизамещенных тиазолов L-1 с использованием NBS дает соединения формулы C-5.
Схема реакций М
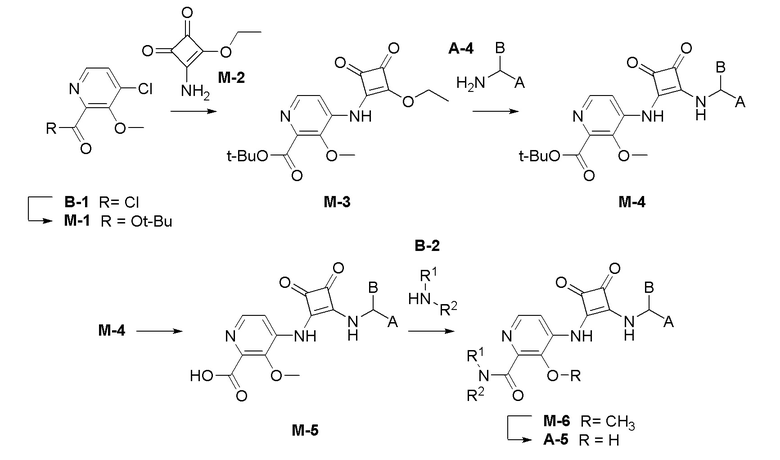
Альтернативно, соединения формулы (IA) и (IB) можно получить, как показано на схеме реакций М. Этерификация коммерчески доступного 4-хлор-3-метоксипиколиноилхлорида (B-1) в щелочных условиях дает трет-бутил 4-хлор-3-метоксипиколинат (М-1). Кросс-сочетание Бухвальда между M-1 и коммерчески доступным 3-амино-4-этоксициклобут-3-ен-1,2-дионом (M-2) с последующим нуклеофильным ароматическим замещением с аминами A-4 приводит к образованию соединений M-4. Сложноэфирный гидролиз в кислых условиях с последующим HATU-опосредованным амидированием аминами B-2 и O-деметилированием с использованием бромида магния дает соединения формулы A-5.
Схема реакций N
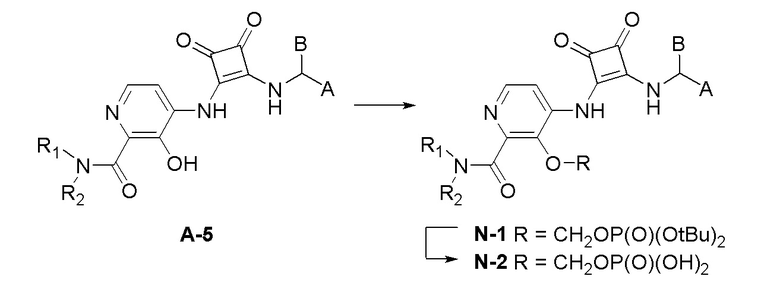
Соединения формулы (IA) и (IB), где R3 представляет собой -CH2OP(O)(OH)2, можно получить, как показано на схеме реакций N. Алкилирование соединений формулы A-5 коммерчески доступным ди-трет-бутил(хлорметилом)фосфатом в щелочных условиях обеспечивает соединение N-1. Последующее отщепление защитных групп трет-бутилового эфира в кислотных или нейтральных условиях приводит к получению соединения N-2.
Примеры
Настоящее изобретение будет легче понять при обращении к следующим примерам, которые включены только в целях иллюстрации определенных аспектов и вариантов осуществления настоящего изобретения и не предназначены для ограничения изобретения. Следующее описание иллюстрирует синтез различных соединений по настоящему изобретению. Дополнительные соединения по настоящему изобретению можно получить с использованием способов, описанных в этих примерах и на схемах, либо по отдельности, либо в сочетании с методами, обычно известными в данной области.
Эксперименты обычно проводили в инертной атмосфере (атмосфере азота или аргона), особенно в случаях, когда применялись реагенты или промежуточные соединения, чувствительные к кислороду или влаге. Коммерчески доступные растворители и реагенты обычно использовали без дополнительной очистки, включая безводные растворители, где это необходимо. Данные масс-спектрометрии получали с использованием приборов для жидкостной хроматографии-масс-спектрометрии (LCMS), химической ионизации при атмосферном давлении (APCI) или газовой хроматографии-масс-спектрометрии (GCMS). Химические сдвиги для данных ядерного магнитного резонанса (ЯМР) выражены в частях на миллион (ppm) относительно остаточных пиков от используемых дейтерированных растворителей. Константы спин-спинового взаимодействия (значения J) указаны в герцах. Для синтезов, с ссылкой на процедуры в других примерах, условия реакции (продолжительность реакции и температура) могут варьироваться. Обычно за ходом реакций следили с помощью тонкослойной хроматографии или масс-спектрометрии и при необходимости подвергали очистке. Степень очистки может варьироваться в зависимости от эксперимента: как правило, растворители и соотношения растворителей, используемые для элюентов/градиентов, были выбраны для обеспечения подходящего Rf или времени удерживания (RetT).
Соединения по настоящему изобретению были названы согласно Chemdraw Professional версии 16.0 или им были даны названия, которые соответствовали номенклатуре Chemdraw.
В данном документе используются следующие сокращения: DCM: дихлорметан; DAST: (диэтиламино)трифторид серы; DEA, диэтиламин; DIPEA: диизопропилэтиламин; ДМФА: диметилформамид; EtOAc: этилацетат; EtOH: этанол; HATU: 1-[бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиний-3-оксид гексафторфосфат; IPA: изопропиловый спирт; ВЭЖХ: жидкостная хроматография высокого давления; LHMDS: литий гексаметилдисилазид; МеОН: метанол; MTBE: метил трет-бутиловый эфир; NMM: N-метилморфолин; NIS: N-иодосукцинимид; PCC: хлорхромат пиридиния; PE: петролейный эфир; SFC: сверхкритическая жидкостная хроматография; TBAI: иодид тетрабутиламмония; TEA: триэтиламин; ТФК: трифторуксусная кислота; и ТГФ: тетрагидрофуран.
Использовали следующие методы SFC. Метод SFC A: Chiral Tech OD-H 250 мм × 4,6 мм, 5 мкм, от 5 до 60% 0,2% NH4+ (7 Н в MeOH) в EtOH, 3,0 мл/мин. Метод SFC B: Chiralpak AD-3 50 мм × 3 мм, 3 мкм, от 5 до 40% 0,05% DEA в EtOH, 2,5 мл/мин, 4°C. Метод SFC C: Chiralcel OD 250 мм × 4,6 мм, 5 мкм, от 5 до 60% 0,2% NH4+ (7 M в MeOH) в EtOH, 3,0 мл/мин. Метод SFC D: Chiralpak AD-3 150 мм × 4,6 мм, 3 мкм, от 5 до 40% 0,05% DEA в EtOH, 2,5 мл/мин, 40°C. Метод SFC E: Chiralcel OJ-H 150 мм 4,6 мм × 5 мкм, от 5 до 40% 0,05% DEA в EtOH, 2,5 мл/мин, 40°C. Метод SFC F: Chiralcel OD-3 100 мм × 4,6 мм, 3 мкм, от 5 до 40% 0,05% DEA в EtOH, 2,8 мл/мин, 40°C. Метод SFC G: Chiralpak AD-3 150 мм × 4,6 мм, 3 мкм, от 5 до 40% 0,05% DEA в IPA, 2,5 мл/мин, 40°C. Метод SFC H: Chiralcel OD-3 150 × 4,6 мм, внутренний диаметр 3 мкм, от 5 до 40% 0,05% DEA в EtOH, 2,5 мл/мин, 40°C). Метод SFC I: REGIS (s, s) WHELK-O1 250 мм × 30 мм, 5 мкм 40% 0,05% DEA в EtOH, 2,5 мл/мин, 35°C. Метод SFC J: Chiralpak AS-3 150 × 4,6 мм, 3 мкм, от 5 до 40% 0,05% DEA в EtOH, 2,5 мл/мин, 35°C. Метод SFC K: Chiralpak AS-3 100 × 4,6 мм, 3 мкм, от 5 до 40% 0,05% DEA в EtOH, 2,8 мл/мин, 40°C. Метод SFC L. Lux Amylose W-1, 250 мм × 4,6 мм, 5 мкм, от 5 до 60% 0,2% NH3 в EtOH, 3 мл/мин. Метод SFC M. Lux Cellulose, 150 мм × 4,6 мм, 3 мкм, от 5 до 40% MeOH, 2 мл/мин. Метод SFC N: Chiralpak AD-3 150 мм × 4,6 мм, 3 мкм, от 5 до 40% 0,1% этаноламина в EtOH, 2,5 мл/мин.
Использовали следующие методы ВЭЖХ. Метод ВЭЖХ A: Chiralcel OD-RH, 150 мм × 4,6 мм, 5 мкм, от 10 до 80% MeCN в 0,069% ТФК в H2O, 0,8 мл/мин, 30°C. ВЭЖХ, метод B: Chiralpak AS-RH, 150 мм × 4,6 мм × 5 мкм, от 10 до 80% MeCN в 0,069% ТФК в H2O, 0,8 мл/мин, 30°C.
Пример 1
(R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
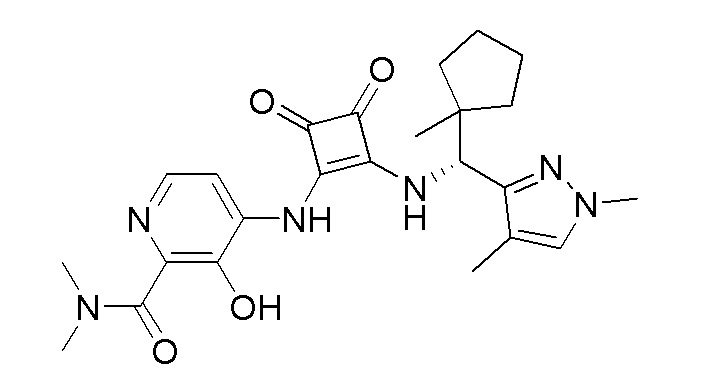
Стадия 1А. 1-метилциклопентан-1-карбонитрил
К раствору LHMDS (280 мл, 280 ммоль, 1 М раствор в ТГФ) при -70°C добавляли по каплям раствор циклопентанкарбонитрила (26,67 г, 280,3 ммоль) в ТГФ (20 мл) в течение 15 мин. После перемешивания в течение 30 мин по каплям добавляли йодметан (59,7 г, 26,2 мл, 420 ммоль), и реакционной смеси давали подогреться до температуры окружающей среды и перемешивали в течение 16 ч. Полученный желтый раствор охлаждали до 0°C и гасили насыщенным водным раствором NH4Cl (200 мл) и водой (100 мл). Смесь экстрагировали МТВЕ (2,5 л × 2), и объединенные органические экстракты промывали насыщенным раствором соли (1 л), сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме. Неочищенный продукт дважды очищали с помощью колоночной хроматографии на силикагеле (100% петролейный эфир), с получением 60 г (65%) указанного в заголовке соединения в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 2,20-2,10 (м, 2H), 1,90-1,70 (м, 4H), 1,65-1,55 (м, 2H), 1,41 (с, 3H).
Стадия 1В. 1-Метилциклопентан-1-карбальдегид
К раствору DIBAL-H (824 мл, 824 ммоль, 1 M раствор в толуоле) при -65°C добавляли по каплям раствор 1-метилциклопентан-1-карбонитрила (30 г, 275 ммоль) в DCM (30 мл). Смесь перемешивали при той же температуре в течение 30 мин. Реакционную смесь гасили насыщенным водным раствором NH4Cl (1 л) при -40°C и интенсивно перемешивали при 25°C в течение 10 мин. Смесь разбавляли DCM (1 л), затем фильтровали и твердые частицы промывали DCM (500 мл × 3). Объединенный фильтрат промывали насыщенным раствором соли (1 л), сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде раствора в DCM/толуоле (3/2, 2 л). Раствор непосредственно использовали без дополнительной очистки, допуская количественный выход. 1H ЯМР (400 МГц, CDCl3) δ 9,59 (с, 1H), 2,15-2,01 (м, 2H), 1,80-1,75 (м, 4H), 1,55-1,45 (м, 2H), 1,25 (с, 3H).
Стадия 1С. (S, E)-2-Метил-N-((1-метилциклопентил)метилен) пропан-2-сульфинамид
К раствору 1-метилциклопентан-1-карбальдегида (30,8 г, 275 ммоль) в смеси DCM/толуоле (3/2, 2,0 л) при 20°C добавляли этоксид титана (IV)(163 г, 717 ммоль). Реакционную смесь перемешивали в течение 20 мин, и затем добавляли (S)-2-метилпропан-2-сульфинамид (33,3 г, 275 ммоль). Полученную смесь перемешивали при температуре окружающей среды в течение 16 ч. Реакциоггую смесь гасили водой (250 мл). Смесь фильтровали, и твердые частицы промывали ТГФ (3 л × 3). Объединенные органические слои концентрировали в вакууме. Неочищенный продукт очищали хроматографией на колонке с силикагелем (100% петролейный эфир) с получением 44,89 г (38%) указанного в заголовке соединения в виде светло-желтого масла. ЖХМС m/z 216,3 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,94 (с, 1H), 1,93-1,85 (м, 2H), 1,80-1,60 (м, 4H), 1,55-1,45 (м, 2H), 1,21 (с, 3H), 1,20 (с, 9H).
Стадия 1D. 3-Иод-1,4-диметил-1H-пиразол
Путь A
3-Иод-1,4-диметил-1H-пиразол. К раствору 1,4-диметил-1H-пиразол-3-амина (1,0 г, 8,6 ммоль) в конц. HCl (7,15 мл) при 0°C добавляли раствор нитрита натрия (1,22 г, 17 ммоль) в воде (1,78 мл) в течение 5 мин. Затем в течение 5 мин по каплям добавляли раствор иодида калия (3,57 г, 21,5 ммоль) в воде (3,6 мл). Смесь перемешивали при 0°C в течение 30 мин, и затем подогревали до температуры окружающей среды и перемешивали в течение 2 ч. Реакционную смесь разбавляли ТГФ (8 мл) и водой (8 мл) и экстрагировали EtOAc (30 мл × 4). Объединенные органические экстракты промывали насыщенным водным раствором Na2S2O3 (30 мл × 2), затем водой (30 мл) и насыщенным раствором соли (30 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали хроматографией на колонке с силикагелем (от 0 до 60% EtOAc в гептане) с получением 988 мг (52%) указанного в заголовке соединения в виде белого твердого вещества. ЖХМС m/z 223,0 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,05 (с, 1H), 3,87 (с, 3H), 1,98 (с, 3H).
Путь B
Стадия 1. 3-иод-1-метил-1H-пиразол-4-карбальдегид. POСl3 (45,0 мл, 481 ммоль) добавляли к раствору 3-иод-1-метил-1H-пиразола (25 г, 120,2 ммоль) в ДМФА (150 мл) при 0°C. Через 10 мин реакционную смесь нагревали до 65°C в течение 18 ч. К реакционной смеси медленно добавляли раствор NaH2PO4 (50 г в 200 мл), обеспечивая поддержание температуры в пределах 25-35°C и pH не выше 4. После добавления реакционную смесь перемешивали при комнатной температуре в течение 45 мин и затем подщелачивали осторожным добавлением насыщенного раствора Na2CO3. Водную смесь экстрагировали EtOAc. Органический слой промывали насыщенным раствором соли, сушили и фильтровали. Фильтрат концентрировали. Полученное желтое масло перекристаллизовывали из смеси EtOAc/ гептана с получением 28,4 г (88%) указанного в заголовке соединения в виде рыжевато-коричневого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 9,67 (с, 1H), 7,84 (с, 1H), 4,00 (с, 4H).
Стадия 2. 3-Иод-1,4-диметил-1H-пиразол. Триэтилсилан (5,08 мл, 0,32 ммоль) и ТФК (1,36 мл, 0,18 ммоль) добавляли к смеси 3-иод-1-метил-1H-пиразол-4-карбальдегида (1,0 г, 0,042 ммоль) в хлорбензоле (10 мл). Смесь нагревали до 50°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры и добавляли насыщенный водный раствор NaHCO3. Смесь экстрагировали EtOAc. Органический слой промывали насыщенным раствором соли, сушили (Na2SO4) и фильтровали. Фильтрат концентрировали, и остаток очищали хроматографией на колонке с силикагелем (смесь EtOAc/ гептан) с получением 0,64 г (68%) указанного в заголовке соединения. 1H ЯМР (400 МГц, CDCl3) δ 7,08 (с, 1H), 3,90 (с, 3H), 2,01 (с, 3H).
Путь C
Стадия 1: 3-иод-4-метил-1H-пиразол. NIS (2196,0 г, 9,76 моль) добавляли порциями к раствору 4-метил-1H-пиразола (800,0 г, 9,76 моль) в ДМФА (5 л), поддерживая температуру ниже 25°C. Смесь перемешивали при 25°C в течение 20 ч. Две партии объединяли. Объединенные смеси выливали в воду (20 л) и затем экстрагировали MTBE (5 л × 5). Объединенные органические слои промывали насыщенным раствором соли (5 л × 3), сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме и остаток растирали со смесью петролейный эфир/EtOAc (5 л, 10:1). Суспензию перемешивали при 16°C в течение 2 ч, и затем фильтровали. Полученное твердое вещество сушили в вакууме с получением 1280,0 г (32%) указанного в заголовке соединения в виде светло-желтого твердого вещества. 1H ЯМР (CDCl3,400 МГц) δ 7,42 (с, 1H), 2,07 (с, 3H).
Стадия 2. 3-иод-1,4-диметил-1H-пиразол. К суспензии 60% NaH в минеральном масле (165,0 г, 4,12 моль) в безводном ТГФ (4,5 л) добавляли смесь 3-иод-4-метил-1H-пиразола (710,0 г, 3,41 моль) в безводном ТГФ (1,5 л) по каплям при 0°C. Смесь перемешивали при 10°C в течение 1 ч. К смеси по каплям добавляли йодметан (496,0 г, 3,49 моль) при 0°C. Полученную смесь перемешивали при 20°C в течение 16 ч. Реакционную смесь гасили водой (4,5 л) и экстрагировали EtOAc (2,5 л × 3). Объединенные органические слои сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (смесь петролейный эфир:EtOAc=от 10:1 до 1:1) с получением двух фракций. Первую фракцию (150 г) разбавляли петролейным эфиром (200 мл) и перемешивали при 10°C в течение 30 мин. Суспензию фильтровали. Твердое вещество промывали петролейным эфиром (100 мл) и сушили в вакууме с получением 100,0 г указанного в заголовке соединения в виде белого твердого вещества. Фильтрат концентрировали в вакууме, объединяли со второй фракцией (1300 г) и очищали колоночной хроматографией на силикагеле (смесь петролейный эфир:EtOAc=от 10:1 до 1:1) с получением дополнительного количества желаемого соединения (500,0 г). Это соединение разбавляли петролейным эфиром (800 мл) и перемешивали при 10°C в течение 30 мин. Суспензию фильтровали. Осадок на фильтре промывали петролейным эфиром (500 мл) и сушили в вакууме с получением 440,0 г указанного в заголовке соединения в виде белого твердого вещества. Общий выход составил 540 г (35,6%). ЖХМС m/z 222,8 [M+H]+. 1H ЯМР (CDCl3,400 МГц) δ 7,04 (с, 1 H), 3,86 (с, 3H), 1,97 (с, 3 H).
Стадия 1E. (S)-N-((R)-(1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)-2-метилпропан-2-сульфинамид
3-Йод-1,4-диметил-1H-пиразол (33 г, 148,6 ммоль) в ТГФ (50 мл) добавляли в течение 1 ч к раствору 1,0 M iPrMgCl⋅LiCl в ТГФ (189 мл, 189 ммоль), поддерживая внутреннюю температуру на уровне 0-5°C в атмосфере азота. Через 1 ч добавляли (S, E)-2-метил-N-((1-метилциклопентил)метилен)пропан-2-сульфинамид (20 г, 92,85 ммоль) в ТГФ (50 мл) и смесь перемешивали при 25°C в течение 18 ч. Реакционную смесь охлаждали до 0°C и добавляли 10% уксусную кислоту. Органический слой отделяли и частично концентрировали. Добавляли MTBE и воду. Смесь перемешивали в течение 10 мин и слои разделяли. Органический слой концентрировали с получением указанного в заголовке соединения (чистота 66%), которое использовали на следующей стадии без дополнительной очистки.
Стадия 1F. (R)-(1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метанамин
Неочищенный (S)-N-((R)-(1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)-2-метилпропан-2-сульфинамид (231 г) суспендировали в MTBE (300 мл), охлаждали до 10°C и концентрировали. Добавляли HCl (38,4 мл, 2 экв). Смесь перемешивали при 10-20°C в течение 1 ч, и затем разбавляли водой (300 мл). Слои разделяли, и органический слой отбрасывали. Водный слой подщелачивали 20% NaOH до pH 11-13 и экстрагировали МТВЕ (2 × 300 мл). Объединенные органические слои концентрировали с получением 120 г указанного в заголовке соединения в виде масла. Абсолютную стереохимию указанного в заголовке соединения определяли на основании анализа открытого переходного состояния, как описано в литературе (Robak M.T., Herbage M. A. Ellman, J. A. Chem. Rev. 2010, 110, 3600), и окончательно подтверждали анализом амида Мошера.
Стадия 1G. (R)-(1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метанамин соль L-пироглутаминовой кислоты
L-пироглутаминовую кислоту (77,7 г, 0,6 моль) добавляли к раствору (R)-(1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил) метанамина (100 г, 0,48 моль) в ТГФ (1,3 л) при 10-20°C. Смесь нагревали до 50°C и перемешивали в течение 2 ч, затем охлаждали до 25°C в течение 18 ч. Твердое вещество отфильтровывали и промывали ТГФ (890 мл). Твердое вещество сушили в вакууме при 45°C в течение 6 ч с получением 259,6 г (88%) указанного в заголовке соединения. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,58 (шир. с, 1H), 7,40 (с, 1H), 3,93 (с, 1H), 3,87-3,84 (м, 1H), 3,77 (с, 3H), 2,27-2,14 (м, 1H), 2,12-2,02 (м, 2H), 1,98 (с, 3H), 1,96-1,84 (м, 1H), 1,70-1,48 (м, 6H), 1,42-1,31 (м, 1H), 1,08-1,02 (м, 1H), 0,97 (с, 3H). Хиральная SFC (метод SFC M) RT=3,81 мин, 99% ее.
Стадия 1H. 4-((2-Этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
4-((2-Этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид получали, как описано в WO/2010/131145.
Стадия 1I. (R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
Смесь 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида (180 г, 590 ммоль), (R)-(1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метанамина, соли L-пироглутаминовой кислоты (209 г, 625 ммоль), EtOH (1 л) и DIPEA (205 мл, 1180 ммоль) перемешивали при 20-30°C в течение 2 ч. Добавляли HOAc (23,8 мл, 416 ммоль) для доведения pH до 6-7. Затем реакционную смесь концентрировали при пониженном давлении примерно до половины объема. Смесь разбавляли водой (3,2 л) и перемешивали в течение 1,5 ч. Полученное твердое вещество фильтровали, промывали водой и затем сушили в вакууме при 40-50°C в течение 20 ч с получением 256 г (93%) указанного в заголовке соединения в виде твердого вещества желтого цвета. ЖХМС m/z 467,4 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,59 (шир. с, 1H), 9,92 (шир. с, 1H), 9,16 (с, 1H), 8,03-8,00 (м, 2H), 7,43 (с, 1H), 5,33 (д, J=10,0 Гц, 1H), 3,80 (с, 3H), 3,18 (с, 3H), 3,05 (с, 3H), 2,00 (с, 3H), 1,80-1,50 (м, 6H), 1,20-1,15 (м, 1H), 1,14-1,09 (м, 1H), 1,08 (с, 3H). [α]20D = -78,4 (c=1,0, MeOH). Хиральная SFC (метод SFC N) RT=4,77 мин, 98,5% ее. Абсолютную конфигурацию устанавливали рентгеноструктурным анализом монокристалла (фиг. 1).
Пример 2
(S)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
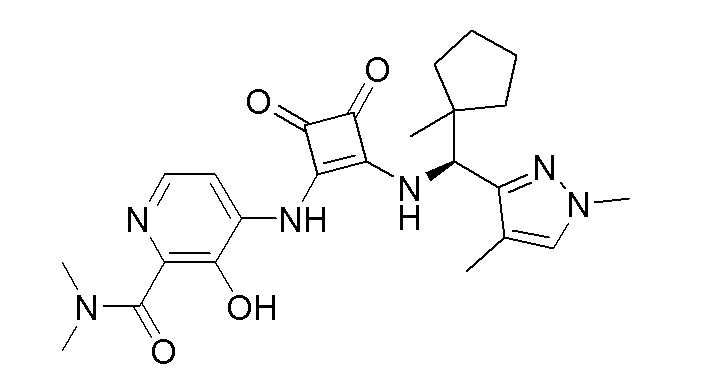
Указанное в заголовке соединение получали аналогично тому, как описано в примере 1, с использованием (R)-2-метилпропан-2-сульфинамида. ЖХМС m/z 467,4 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,18 (шир. с, 1H), 8,12-7,85 (м, 2H), 7,43 (с, 1H), 5,34 (д, J=10,0 Гц, 1H), 3,80 (с, 3H), 3,15 (шир. с, 3H), 3,05 (шир. с, 3H), 1,98 (с, 3H), 1,74-1,52 (м, 6H), 1,41-1,27 (м, 1H) 1,23-1,15 (м, 1H), 1,08 (с, 3H). [α]29D = +88,49 (c=0,5, MeOH). Хиральная SFC (метод SFC F) RT=5,45 мин, 100% ее.
Пример 3
(R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N-изопропил-N-метилпиколинамид
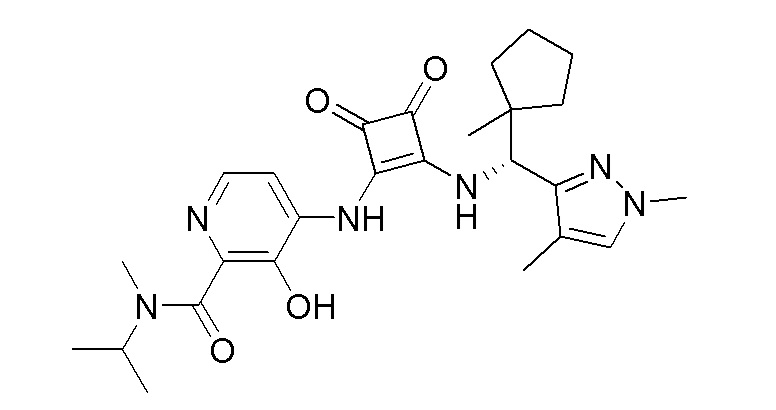
Стадия 3А. 4-Хлор-3-метоксипиколиноилхлорид
К раствору 4-хлор-3-метоксипиколиновой кислоты (5,0 г, 30 ммоль) в DCM (59 мл) при 0°C по каплям добавляли оксалилхлорид (8,46 г, 5,71 мл, 66,6 ммоль). Полученную смесь подогревали до температуры окружающей среды и перемешивали в течение 2 ч. Реакционную смесь концентрировали в вакууме с получением 5,49 г (100%) указанного в заголовке соединения в виде светло-желтого твердого вещества. Неочищенный продукт непосредственно использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 8,43 (д, J=5,1 Гц, 1H), 7,71-7,51 (м, 1H), 4,01 (с, 3H).
Стадия 3В. 4-Хлор-N-изопропил-3-метокси-N-метилпиколинамид
К раствору 4-хлор-3-метоксипиколиноилхлорида (175 г, 0,85 моль) в DCM (500 мл) добавляли Et3N (172 г, 1,7 моль) при 0°C. Затем добавляли N-метилпропан-2-амин (62 г, 0,85 моль), поддерживая внутреннюю температуру ниже 10°C. Смесь перемешивали при 15°C в течение 16 ч. Добавляли воду (500 мл) и органический слой отделяли. Водный слой экстрагировали DCM (200 мл × 2). Объединенные органические экстракты промывали насыщенным раствором соли (500 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением 153 г (74%) указанного в заголовке соединения в виде коричневого масла. Неочищенный продукт непосредственно использовали без дополнительной очистки.
Стадия 3C. трет-Бутил (2-(изопропил(метил)карбамоил)-3-метоксипиридин-4-ил)карбамат
К раствору 4-хлор-N-изопропил-3-метокси-N-метилпиколинамида (153 г, 0,63 моль) в диоксане (1 л) добавляли NH2Boc (88,5 г, 0,76 моль) и K2CO3 (130 г, 0,95 моль). Смесь продували N2 и добавляли Pd(OAc)2 (11,3 г, 0,05 моль) и ксантфос (36,4 г, 0,063 моль). Смесь нагревали до 120°C и перемешивали в течение 16 ч. Реакционную смесь охлаждали до 20°C и фильтровали через целит. Фильтрат концентрировали в вакууме. Остаток распределяли между EtOAc (500 мл) и водой (500 мл). Органический слой отделяли и промывали насыщенным раствором соли (500 мл), сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме и неочищенный продукт очищали хроматографией на колонке с силикагелем (от 50 до 66% EtOAc в петролейном эфире) с получением 160 г (78%) указанного в заголовке соединения в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,24-8,20 (м, 1H), 8,07-8,04 (м, 1H), 7,24 (с, 1H), 5,03-5,00 (м, 0,4H), 3,90 (с, 3H) , 3,66-3,63 (м, 0,6H), 2,98 (с, 1,7H), 2,69 (с, 1,3H), 1,54 (с, 9H), 1,25-1,20 (м, 3H), 1,16-1,13 (м, 3H).
Стадия 3D. 4-Амино-N-изопропил-3-метокси-N-метилпиколинамид
К раствору трет-бутил (2-(изопропил(метил)карбамоил)-3-метоксипиридин-4-ил)карбамата (160 г, 0,50 моль) в EtOAc (320 мл) добавляли смесь HCl/EtOAc (4,0 M, 750 мл). Смесь перемешивали при 20°C в течение 4 ч. Реакционную смесь концентрировали в вакууме с получением 150 г (>100%) указанного в заголовке соединения в виде коричневого твердого вещества. Неочищенный продукт непосредственно использовали без дополнительной очистки.
Стадия 3E. 4-Амино-3-гидрокси-N-изопропил-N-метилпиколинамид
К раствору 4-амино-N-изопропил-3-метокси-N-метилпиколинамида (150 г, 0,67 моль) в DCM (1,5 л) добавляли TBAI (168 г, 0,47 моль). Смесь охлаждали до 0°C и по каплям добавляли раствор BBr3 (420 г, 1,68 моль) в DCM (500 мл), поддерживая внутреннюю температуру ниже 10°C. Реакционную смесь перемешивали при 20°C в течение 16 ч. Реакцию гасили насыщенным водным раствором NaHCO3 (2,5 л) и доводили pH до 6-7. Органический слой отделяли и промывали водой (500 мл). Объединенные водные слои подвергали обратной экстракции DCM (1 л × 2). Объединенные органические экстракты концентрировали в вакууме, и остаток обрабатывали смесью DCM:MeOH=10:1 (2 л) в течение 2 ч. Смесь фильтровали, и фильтрат концентрировали в вакууме. Остаток обрабатывали смесью DCM:MeOH=10:1 (600 мл) в течение 1 ч и смесь фильтровали. Фильтрат концентрировали в вакууме с получением 140 г (99%) указанного в заголовке соединения в виде розового твердого вещества. Неочищенный продукт непосредственно использовали без дополнительной очистки.
Стадия 3F. 4-((2-Этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N-изопропил-N-метилпиколинамид
К раствору 4-амино-3-гидрокси-N-изопропил-N-метилпиколинамида (140 г, 0,67 моль) в EtOH (1,4 л) добавляли DIPEA (147 г, 1,14 моль) и 3,4-диэтоксициклобут-3-ен-1,2-дион (159 г, 0,94 моль). Смесь нагревали до 35°C и перемешивали в течение 16 ч. Реакционную смесь фильтровали, и фильтрат сохраняли для дальнейшего использования. Осадок на фильтре растворяли в воде (500 мл), доводили pH до 6 с помощью HCl (1,0 М водный раствор) и экстрагировали DCM (500 мл × 2). Объединенные экстракты DCM промывали насыщенным раствором соли (1 л), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток растирали с МТВЕ (500 мл) в течение 2 ч, и затем фильтровали с получением 73 г (33%) указанного в заголовке соединения в виде желтого твердого вещества. Маточный раствор объединяли с ранее выделенным фильтратом и концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 25 до 50% EtOAc в петролейном эфире) с получением дополнительных 30 г (13%) указанного в заголовке соединения в виде желтого твердого вещества. ЖХМС m/z 334,2 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 13,30 (с, 1H), 8,09 (с, 1H), 8,01-7,90 (м, 2H), 5,85-5,80 (м, 0,5H), 4,95-4,88 (м, 2,5H), 3,50-3,31 (м, 1,5H), 3,03-2,95 (м, 1,5H), 1,55-1,50 (м, 6H), 1,42-1,32 (м, 3H).
Стадия 3G. (S)-N-((R)-(1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)-2-метилпропан-2-сульфинамид
В высушенную в печи круглодонную колбу загружали i-PrMgCl·LiCl (67 мл, 87,1 ммоль, 1,3 М раствор в ТГФ). Раствор охлаждали до 0°C, и затем через капельную воронку в течение 15 мин по каплям добавляли раствор 3-йод-1,4-диметил-1H-пиразола (15,5 г, 69,7 ммоль) в ТГФ (90 мл). Реакционную смесь нагревали до температуры окружающей среды и перемешивали в течение 1 ч. Смесь снова охлаждали до 0°C и добавляли (S, E)-2-метил-N-((1-метилциклопентил)метилен)пропан-2-сульфинамид (препаративная стадия 1C)(10,0 г, 46,4 ммоль) по каплям в течение 5 мин. Смесь подогревали до температуры окружающей среды и перемешивали в течение 18 ч. Реакционную смесь медленно выливали в насыщенный водный раствор NH4Cl (300 мл) при 0°C. Затем смесь экстрагировали EtOAc (350 мл × 2). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме. Неочищенное масло растворяли в МТВЕ (50 мл) и гептане (100 мл) с получением гомогенного раствора, который концентрировали в вакууме с получением твердого вещества. Твердое вещество суспендировали в гептане (50 мл), и затем концентрировали в вакууме. Этот процесс повторяли дважды, и конечную суспензию в гептане (50 мл) охлаждали до 0°C, чтобы дать возможность образоваться большему количеству твердого вещества. Суспензию фильтровали с получением 11,5 г (80%) указанного в заголовке соединения в виде белого твердого вещества. ЖХМС m/z 312,2 [M+H]+. 1H ЯМР (500 МГц, ДМСО-d6) δ 7,34 (с, 1H), 4,37 (д, J=6,5 Гц, 1H), 4,10 (д, J=6,4 Гц, 1H), 3,72 (с, 3H), 1,96 (с, 3H), 1,72-1,51 (м, 6H), 1,44-1,35 (м, 1H), 1,16-1,03 (м, 1H), 1,01 (д, J=1,8 Гц, 12H).
Стадия 3H. (R)-(1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метанамина гидрохлорид
К раствору (S)-N-((R)-(1,4-диметил-1H-пиразол)-3-ил)(1-метилциклопентил)метил)-2-метилпропан-2-сульфинамида (28,7 г, 92,14 ммоль) в MeOH (150 мл) при 5°C, добавляли HCl (50 мл, 200 ммоль, 4,0 M раствор в 1,4-диоксане). После перемешивания в течение 3 ч при 20°C раствор концентрировали в вакууме с получением указанного в заголовке соединения в виде соли моно-HCl, предполагая количественный выход. Неочищенный продукт непосредственно использовали без дополнительной очистки. ЖХМС m/z 191,2 [M-NH2]+. 1H ЯМР (400 МГц, CD3OD) δ 7,39 (с, 1H), 4,25 (с, 1H), 3,86 (с, 3H), 2,07 (с, 3H), 1,75-1,54 (м, 7H), 1,21-1,20 (м, 1H), 1,14 (с, 3H).
Стадия 3I. (R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N-изопропил-N-метилпиколинамид
К раствору (R)-(1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метанамина гидрохлорида (3,50 г, 14,36 ммоль) в EtOH (50 мл) при 10°C добавляли DIEA (3,10 г, 4,31 мл, 24,0 ммоль). После перемешивания в течение 20 мин добавляли желтую суспензию 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N-изопропил-N-метилпиколинамида (4,0 г, 12,0 ммоль) в EtOH (100 мл), и полученный коричневый раствор перемешивали при той же температуре в течение 16 ч. Реакционную смесь концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 80% до 100% EtOAc в петролейном эфире). Неочищенный продукт подвергали азеотропной перегонке с EtOH (50 мл × 2) и полученную желтую суспензию концентрировали до примерно 30 мл. Добавляли дополнительное количество EtOH (10 мл) и суспензию перемешивали при 15°C в течение 20 мин. Смесь фильтровали и осадок на фильтре промывали EtOH (10 мл). Осадок на фильтре собирали и сушили с получением 2,62 г (44%) указанного в заголовке соединения в виде светло-желтого твердого вещества. ЖХМС m/z 495,5 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,35 (с, 0,3H), 10,85 (с, 0,7H), 9,88 (с, 1H), 9,11 (д, J=9,8 Гц, 1H), 8,05-7,90 (м, 2H), 7,43 (с, 1H), 5,33 (д, J=10,0 Гц, 1H), 4,81 (с, 0,3H), 4,20 (с, 0,7H), 3,80 (с, 3H), 2,88 (с , 3H), 1,98 (с, 3H), 1,75-1,51 (м, 6H), 1,36-1,32 (м, 1H), 1,25-1,10 (м, 7H), 1,08 (с, 3H). [α]20D = -49,8 (с=0,26, МеОН). Условия хиральной SFC (метод SFC D) RT=4,62 мин, 100% ее.
Пример 4
(R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N-этил-3-гидрокси-N-метилпиколинамид
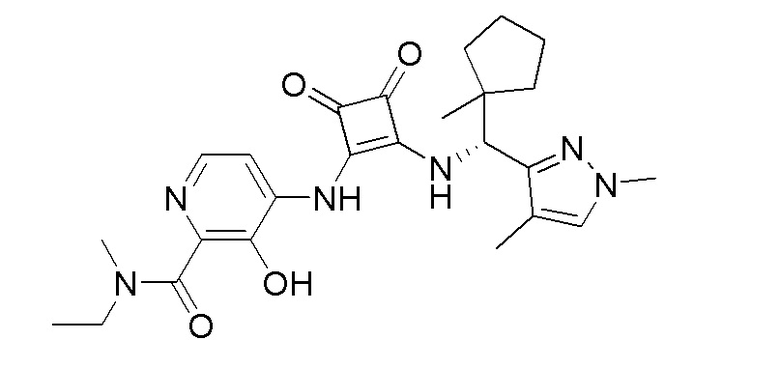
Стадия 4А. 4-Хлор-N-этил-3-метокси-N-метилпиколинамид
N-Метилэтанамин (138 г, 233 ммоль) и триэтиламин (32,3 мл, 233 ммоль) добавляли к раствору 4-хлор-3-метоксипиколиноилхлорида (препаративная стадия 3A)(32 г, 155 ммоль) в DCM (150 мл). Смесь перемешивали при температуре окружающей среды в течение 48 ч, и затем концентрировали. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 100% EtOAc в петролейном эфире) с получением 22 г (62%) указанного в заголовке соединения в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 8,25 (дд, J=5,3, 6,8 Гц, 1H), 7,35 (д, J=5,3 Гц, 1H), 3,94 (д, J=1,8 Гц, 3H), 3,66-3,55 (м, 1H), 3,23-3,12 (м, 1H), 3,12-2,72 (м, 3H), 1,35-1,05 (м, 3H).
Стадия 4В. трет-Бутил (2-(этил(метил)карбамоил)-3-метоксипиридин-4-ил)карбамат
К раствору 4-хлор-N-этил-3-метокси-N-метилпиколинамида (21,3 г, 93 ммоль) в диоксане (120 мл) добавляли NH2Boc (21,8 г, 186 ммоль) и K2CO3 (25,7 г, 186 ммоль). Смесь защищали N2 и добавляли Pd(OAc)2 (1,05 г, 4,7 ммоль) и ксантфос (2,7 г, 4,7 ммоль). Смесь нагревали до 120°C и перемешивали в течение 16 ч. Реакционную смесь охлаждали до 20°C и фильтровали через целит. Фильтрат концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 100% EtOAc в петролейном эфире) с получением 24 г (83%) указанного в заголовке соединения в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,25 (ддд, J=5,5, 3,8, 0,6 Гц, 1H), 8,09 (дд, J=5,5, 1,6 Гц, 1H), 7,26 (д, J=5,6 Гц, 1H) , 3,92 (д, J=1,0 Гц, 3H), 3,64 (кв, J=7,2 Гц, 1H), 3,18 (кв, J=7,1 Гц, 1H), 3,13 (с, 1,5H), 2,86 (с, 1,5 H), 1,56 (с, 9H), 1,28 (т, J=7,2 Гц, 1,5H), 1,13 (т, J=7,1 Гц, 1,5H).
Стадия 4C. 4-Амино-N-этил-3-метокси-N-метилпиколинамид
Раствор HCl/EtOAc (4,0 M, 40 мл) добавляли к трет-бутил (2- (этил(метил)карбамоил)-3-метоксипиридин-4-ил)карбамату (16 г, 51,7 ммоль), и смесь перемешивали при 20°C в течение 18 ч. Смесь концентрировали в вакууме с получением 12,71 г (100%) указанного в заголовке соединения в виде желтого твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 8,00 (дд, J=6,9, 1,5 Гц, 1H), 7,02 (дд, J=6,8, 0,7 Гц, 1H), 3,86 (д, J=2,7 Гц, 3H), 3,35 -3,28 (м, 2H), 3,16 (с, 1,5H), 3,00 (с, 1,5), 1,33-1,17 (м, 3H).
Стадия 4D. 4-Амино-N-этил-3-гидрокси-N-метилпиколинамид
К раствору 4-амино-N-этил-3-метокси-N-метилпиколинамида (5 г, 24 ммоль) в DCM (100 мл) добавляли TBAI (5,7 г, 15,5 ммоль). Смесь охлаждали до 0°C и по каплям добавляли раствор BBr3 (23,9 г, 95,6 ммоль) в DCM (100 мл), поддерживая внутреннюю температуру ниже 10°C. Реакционную смесь перемешивали при 10°C в течение 15 ч. Реакционную смесь гасили водным раствором NaOH/MeOH (200 мл MeOH, 11,5 г NaOH, 50 мл воды), поддерживая внутреннюю температуру на уровне 0°C. Смесь концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 3 до 9% MeOH в DCM) с получением 3,1 г (78%) указанного в заголовке соединения в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,81 (д, J=6,3 Гц, 1H), 7,39 (шир. с, 3H), 6,85 (д, J=6,2 Гц, 1H), 3,52-3,18 (м , 2H), 2,91 (шир. с, 3H), 1,37-0,80 (м, 3H).
Стадия 4E. 4-((2-Этокси-3,4-диоксоциклобут-1-ен-1-ил)амино) -N-этил-3-гидрокси-N-метилпиколинамид
К раствору 4-амино-N-этил-3-гидрокси-N-метилпиколинамида (7,01 г, 35,9 ммоль) в EtOH (150 мл) добавляли K2CO3 (4,96 г, 35,9 ммоль) и 3,4-диэтоксициклобут-3-ен-1,2-дион (9,17 г, 53,9 ммоль). Смесь нагревали до 50°C и перемешивали в течение 16 ч. Реакционную смесь фильтровали через целит, и фильтрат концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 10 до 100% DCM в петролейном эфире, от 0 до 20% MeOH в DCM) с получением 4,77 г (42%) указанного в заголовке соединения в виде коричневого масла. 1H ЯМР (400 МГц, CDCl3) δ 13,62 (шир. с, 0,5H), 13,50 (шир. с, 0,5H), 8,07 (д, J=5,2 Гц, 1H), 7,94 (с, 1H), 7,88 (с, 1H), 4,90 (кв., J=7,1 Гц, 2H), 4,16 (шир. с, 1H), 3,62 (шир. с, 3H), 3,14 (шир. с, 1H), 1,55 (т, J=7,1 Гц, 3H) 1,45-1,20 (м, 3H).
Стадия 4F. (R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N-этил-3-гидрокси-N-метилпиколинамид
Раствор 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-N-этил-3-гидрокси-N-метилпиколинамида (5,92 г, 18,54 ммоль) в EtOH (140 мл) добавляли к (R)-(1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метанамину соли L-пироглутаминовой кислоты (препаративная стадия 1G)(7,48 г, 22,2 ммоль) и DIPEA (4,79 г, 37,1 ммоль) в EtOH (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, и затем реакционную смесь концентрировали. Остаток очищали хроматографией на колонке с силикагелем (смесь петролейный эфир/EtOAc, от 1:5 до 0:1) с получением 4,4 г (49%) указанного в заголовке соединения в виде желтого твердого вещества. ЖХМС m/z 481,1 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ 8,22 (шир. с, 1H), 7,94 (шир.с, 1H), 7,28 (с, 1H), 5,44 (с, 1H), 3,83 (с, 3H), 3,65 (шир. с, 2H), 3,28-3,06 (м, 3H), 2,09 (с, 3H), 1,92-1,58 (м, 6H), 1,53-1,42 (м, 1H), 1,38-1,21 (м, 4H), 1,17 (с, 3H). [α]26D = -77,88 (c=0,5, MeOH). Хиральная SFC (метод SFC F) RT=3,52 мин, 99% ее.
Пример 5
(S)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N-этил-3-гидрокси-N-метилпиколинамид
Указанное в заголовке соединение получали аналогично тому, как описано в примере 4, с использованием (S)-(1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метанамина (пример 2). ЖХМС m/z 481,4 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,70 (шир. с, 1H), 9,90 (шир. с, 1H), 9,15 (шир. с, 1H), 8,00 (шир. с, 2H) , 7,43 (с, 1H), 5,34 (д, J=10,0 Гц, 1H), 3,80 (с, 3H), 3,54 (шир. с, 2H), 3,17 (шир. с, 1H), 3,02 (шир. с, 2H), 1,98 (с, 3H), 1,75-1,52 (м, 6H), 1,40-1,28 (м, 1H), 1,25-1,10 (4H), 1,08 (с, 3H). [α]29D = +80,8 (c=0,5, MeOH). Хиральная SFC (метод SFC K) RT=3,11 мин, 99% ее.
Пример 6
(R)-3-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-4-((3-гидрокси-2-(морфолин-4-карбонил)пиридин-4-ил)амино)циклобут-3-ен-1,2-дион
Стадия 6А. трет-Бутил 4-хлор-3-метоксипиколинат
К раствору пиридина (7,91 г, 8,05 мл, 100 ммоль) и t-BuOH (7,90 г, 10,1 мл, 107 ммоль) в DCM (32 мл) добавляли по каплям при 0°C раствор 4-хлор-3-метоксипиколиноилхлорида (препаративная стадия 3A)(5,49 г, 26,7 ммоль) в DCM (26,7 мл). Реакционную смесь перемешивали при 0°C в течение 15 мин, подогревали до температуры окружающей среды и затем нагревали с обратным холодильником в течение 4 ч. Растворитель удаляли в вакууме и остаток растворяли в EtOAc. Раствор промывали 1 Н водным раствором NaOH и насыщенным раствором соли, фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (от 0 до 20% EtOAc в DCM) с получением 3,10 г (48%) указанного в заголовке соединения в виде жидкости. ЖХМС m/z 188,1 [M-tBu]+. 1H ЯМР (400 МГц, CDCl3) δ 8,30 (д, J=5,1 Гц, 1H), 7,42 (д, J=5,1 Гц, 1H), 3,98 (с, 3H), 1,65 (с, 9H).
Стадия 6В. трет-Бутил 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-метоксипиколинат
К смеси 3-амино-4-этоксициклобут-3-ен-1,2-диона (169,2 г, 1,2 моль) и Na2CO3 (67,84 г, 0,64 моль) в ТГФ (1,8 л) добавляли трет-бутил-4-хлор-3-метоксипиколинат (97,2 г, 0,4 моль) при 25°C. Смесь дегазировали и 3 раза продували N2. Добавляли tBuXPhos-Pd-G3 (15,9 г, 0,02 моль) и tBuXPhos (8,48 г, 0,02 моль). Реакционную смесь дегазировали и 3 раза продували N2. Реакционную смесь нагревали до 80°C и перемешивали в течение 16 ч. Смесь охлаждали до 25°C и затем фильтровали через целит. Фильтрат концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 50% DCM в петролейном эфире, затем от 0 до 2,5% ТГФ в DCM). Полученное масло растирали с гептаном (250 мл) и перемешивали при 25°C в течение 3 ч. Смесь фильтровали с получением 84,7 г, 60% указанного в заголовке соединения в виде светло-желтого твердого вещества. ЖХМС m/z 349,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 8,38 (д, J=5,4 Гц, 1H), 7,93 (шир. с, 2H), 4,95 (кв, J=7,1 Гц, 2H), 4,01 (с, 3H), 1,68 (с, 9H), 1,59 (т, J=7,2 Гц, 3H).
Стадия 6C. трет-Бутил (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-метоксипиколинат
DIEA (6,0 г, 46,4 ммоль) добавляли к раствору (R)-(1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метанамина гидрохлорида (препаративная стадия 3H)(4,70 г, 19,3 ммоль) в EtOH (70 мл). Смесь перемешивали в течение 10 мин, и затем добавляли трет-бутил 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-метоксипиколинат (4,0 г, 11,5 ммоль). Полученную смесь перемешивали при 20°C в течение 70 ч, и затем концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 80% EtOAc в петролейном эфире) с получением 5,85 г (100%) указанного в заголовке соединения в виде желтого твердого вещества. ЖХМС m/z 510,1 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,97 (с, 1H), 9,06-9,02 (м, 1H), 8,20-8,18 (м, 1H), 8,09-8,07 (м, 1H), 7,44 (с, 1H), 5,36-5,33 (м, 1H), 3,84 (с, 3H), 3,80 (с, 3H), 1,98 (с, 3H), 1,72-1,53 (м, 15H), 1,35-1,30 (м, 1H), 1,24-1,15 (м, 1H), 1,09 (с, 3H).
Стадия 6D. (R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-метоксипиколиновая кислота
ТФК (25 г, 16,3 мл, 219,3 ммоль) добавляли к раствору трет-бутил (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-метоксипиколината (6,60 г, 13,0 ммоль) в DCM (50 мл). Полученный раствор перемешивали при температуре окружающей среды в течение 44 ч. Смесь концентрировали в вакууме, и pH доводили до 8, используя насыщенный водный раствор NaHCO3. Смесь экстрагировали DCM (20 мл). Водный слой подкисляли примерно до pH 3 с помощью 1 Н водного раствора HCl, и затем экстрагировали DCM (100 мл × 5). Объединенные органические экстракты концентрировали в вакууме с получением 5,8 г (99%) указанного в заголовке соединения в виде желтого твердого вещества. ЖХМС m/z 454,4 [M+H]+.
Стадия 6E. (R)-3-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-4-((3-метокси-2-(морфолин-4-карбонил)пиридин-4-ил)амино)циклобут-3-ен-1,2-дион
HATU (377 мг, 0,99 ммоль) добавляли к раствору (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-метоксипиколиновой кислоты (300 мг, 0,66 ммоль), морфолина (57,6 мг, 0,66 ммоль) и TEA (134 мг, 1,32 ммоль) в DCM (5 мл) при 0°C. Полученную смесь перемешивали при 25°C в течение 16 ч, и затем концентрировали в вакууме. Остаток растворяли в EtOAc (10 мл) и промывали насыщенным водным раствором KHCO2 (10 мл). Водный слой экстрагировали EtOAc (40 мл × 3). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде желтого твердого вещества. Неочищенный продукт непосредственно использовали без дополнительной очистки, допуская количественный выход. ЖХМС m/z 523,2 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ 8,28-8,26 (м, 1H), 8,21-8,19 (м, 1H), 7,29 (с, 1H), 5,45 (с, 1H), 3,94 (с, 3H), 3,90- 3,75 (м, 5H), 3,68-3,64 (м, 2H), 3,01-2,96 (м, 4H), 2,09 (с, 3H), 1,90-1,65 (м, 7H), 1,50-1,42 (м, 1H), 1,18 (с, 3H).
Стадия 6F. (R)-3-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-4-((3-гидрокси-2-(морфолин-4-карбонил)пиридин-4-ил)амино)циклобут-3-ен-1,2-дион
К раствору (R)-3-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-4-((3-метокси-2-(морфолин-4-карбонил)пиридин-4-ил)амино)циклобут-3-ен-1,2-диона (600 мг, 1,15 ммоль) в 1,4-диоксане (5 мл) при 15°C добавляли MgBr2 (634 мг, 3,44 ммоль). Смесь нагревали при 120°C в течение 2 ч. Реакционную смесь фильтровали, и фильтрат концентрировали в вакууме. Остаток очищали с помощью препаративной ВЭЖХ (Agela Durashell C18 150 мм × 25 мм × 5 мкм, от 30 до 50% MeCN в 0,225% муравьиной кислоте в воде, 25 мл/мин, 11 мин) с получением 66 мг (11%) указанного в заголовке соединение в виде желтого твердого вещества. ЖХМС m/z 509,4 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ 8,22 (д, J=5,5 Гц, 1H), 7,91 (шир. с, 1H), 7,28 (с, 1H), 5,44 (с, 1H), 3,90-3,70 (м, 11H), 2,09 (с, 3H), 1,90-1,65 (м, 6H), 1,50-1,43 (м, 1H), 1,35-1,26 (м, 1H), 1,17 (с, 3H). [α]20D = -65,6 (c=0,17, MeOH). Условия хиральной SFC (метод SFC K) RT=3,47 мин, 100% ее.
Пример 7
(R)-3-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-4-((3-гидрокси-2-(4-метилпиперазин-1-карбонил)пиридин-4-ил)амино)циклобут-3-ен-1,2-дион
Указанное в заголовке соединение получали, следуя процедуре, аналогичной получению (R)-3-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-4-((3-гидрокси-2-(морфолин-4-карбонил)пиридин-4-ил)амино)циклобут-3-ен-1,2-диона (пример 6) с использованием 1-метилпиперазина с выходом 19,7 мг (9%) указанного в заголовке соединения в виде желтого твердого вещества. ЖХМС m/z 522,5 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ 8,35 (с, 1H), 8,24 (д, J=5,6 Гц, 1H), 7,90 (д, J=5,6 Гц, 1H), 7,28 (с, 1H), 5,44 (с , 1H), 3,92 (шир. с, 4H), 3,82 (с, 3H), 2,80 (шир. с, 4H), 2,53 (с, 3H), 2,09 (с, 3H), 1,90-1,65 (м, 6H), 1,51-1,42 (м, 1H), 1,35-1,25 (м, 1H), 1,17 (с, 3H). Хиральная SFC (метод SFC F) RT=3,66 мин, 98,6% ее.
Пример 8
(R)-4-((2-(((4-Хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
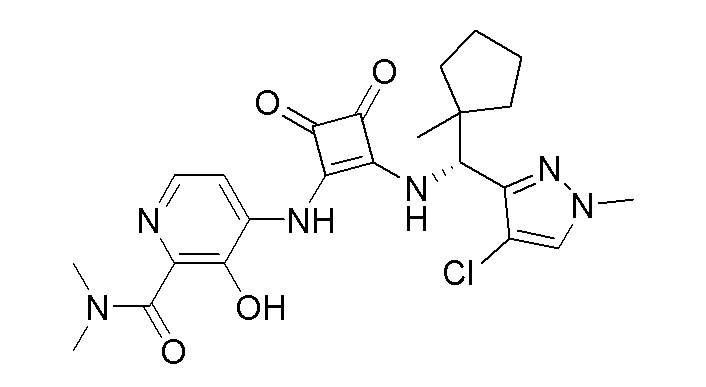
Стадия 8А. (R)-2-Метил-N-((R)-(1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)пропан-2-сульфинамид
К раствору i-PrMgCl⋅LiCl (20,1 мл, 26,2 ммоль, 1,3 М раствор в ТГФ) при -30°C добавляли раствор 3-иод-1-метил-1H-пиразола (4,35 г, 20,9 ммоль) в ТГФ (20 мл). Полученный желтый раствор перемешивали при 30-40°C в течение 2 ч. Реакционную смесь охлаждали до -30°C и добавляли по каплям раствор (S, E)-2-метил-N-((1-метилциклопентил)метилен)пропан-2-сульфинамида (препаративная стадия 1C)(3,0 г, 13,93 ммоль) в ТГФ (5 мл). Реакционную смесь нагревали до 30°C и перемешивали в течение 16 ч. Реакционную смесь медленно выливали в насыщенный водный раствор NH4Cl (100 мл) при 5°C и разбавляли водой (20 мл). Затем смесь экстрагировали EtOAc (50 мл × 2). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 80% EtOAc в петролейном эфире) с получением 3,48 г (84%) указанного в заголовке соединения в виде желтого масла. ЖХМС m/z 297,9 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,26 (д, J=2,1 Гц, 1H), 6,08 (д, J=2,2 Гц, 1H), 4,34 (д, J=3,4 Гц, 1H), 3,86 (с, 3H), 3,59-3,55 (м, 1H), 1,80-1,55 (м, 6H), 1,50-1,43 (с, 1H), 1,18 (с, 9H), 1,18-1,12 (м, 1H), 0,98 (с, 3H).
Стадия 8В. (R)-(1-Метил-1H-пиразол-3-ил)(1-метилциклопентил) метанамин
HCl (80 мл, 4,0 М раствор в MeOH) добавляли к раствору (R)-2-метил-N-((R)-(1-метил-1H-пиразол-3-ил)(1-метилциклопентил)мет)пропан-2-сульфинамида (6,59 г, 22,15 ммоль) в MeOH (50 мл). Полученный раствор перемешивали при температуре окружающей среды в течение 3 ч, и затем концентрировали в вакууме. Указанное в заголовке соединение выделяли в виде моносоли HCl, допуская количественный выход, и непосредственно использовали без дополнительной очистки. ЖХМС m/z 193,8 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ 7,63 (д, J=2,3 Гц, 1H), 6,33 (д, J=2,3 Гц, 1H), 4,25 (с, 1H), 3,92 (с, 3H), 1,85-1,65 (м, 6H), 1,60-1,55 (м, 1H), 1,31-1,25 (м, 1H), 1,05 (с, 3H). Абсолютную стереохимию указанного в заголовке соединения определяли по аналогии с препаративным примером 1F.
Стадия 8C. трет-Бутил (R)-((1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)карбамат
К раствору (R)-(1-метил-1H-пиразол-3-ил)(1-метилциклопентил) метанамина (5,09 г, 22,2 ммоль) в MeOH (150 мл) при 15°C добавляли TEA (9,25 мл, 66,5 ммоль) и (Boc)2O (7,25 г, 33,2 ммоль). Полученную смесь перемешивали в течение 16 ч. Реакционную смесь концентрировали в вакууме и остаток очищали хроматографией на колонке с силикагелем (от 0 до 20% EtOAc в петролейном эфире) с получением 5,72 г (88%) указанного в заголовке соединения в виде светло-желтого масла. ЖХМС m/z 176,8 [M-BocNH]+. 1H ЯМР (400 МГц, CDCl3) δ 7,24 (д, J=2,2 Гц, 1H), 6,07 (д, J=2,2 Гц, 1H), 5,39-5,30 (м, 1H), 4,67 (д, J=9,5 Гц, 1H), 3,87 (с, 3H), 1,79-1,61 (м, 6H), 1,44 (с, 9H), 1,38-1,17 (м, 2H), 0,94 (с, 3H).
Стадия 8D. трет-Бутил (R)-((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)карбамат
NCS (3,12 г, 23,4 ммоль) порциями добавляли к раствору трет-бутил (R)-((1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил) карбамата (5,72 г, 19,5 ммоль) в ДМФА (100 мл). Смесь нагревали при 50°C в течение 16 ч. После охлаждения до температуры окружающей среды реакционную смесь выливали в 3% водный раствор LiCl (150 мл) и затем экстрагировали EtOAc (70 мл × 2). Объединенные органические экстракты концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (от 0 до 15% EtOAc в петролейном эфире) с получением 6,0 г (94%) указанного в заголовке соединения в виде бледно-желтого масла. ЖХМС m/z 210,7 [M-BocNH]+. 1H ЯМР (400 МГц, CDCl3) δ 7,25 (с, 1H), 5,32-5,29 (м, 1H), 4,79 (д, J=9,7 Гц, 1H), 3,82 (с, 3H), 1,80-1,55 (м, 6H), 1,42 (с, 9H), 1,40-1,30 (м, 1H), 1,21-1,12 (м, 1H), 1,00 (с, 3H).
Стадия 8E. (R)-(4-Хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метанамин
К смеси трет-бутил (R)-((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)карбамата (6,0 г, 18,3 ммоль) в MeOH (20 мл) при 15°C добавляли HCl (150 мл, 600 ммоль, 4,0 M в MeOH). Смесь перемешивали в течение 5 ч, и затем концентрировали в вакууме. Указанное в заголовке соединение выделяли в виде моносоли HCl, допуская количественный выход, и непосредственно использовали без дополнительной очистки. ЖХМС m/z 228,0 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ 7,77 (с, 1H), 4,32 (с, 1H), 3,92 (с, 3H), 1,90-1,55 (м, 7H), 1,30-1,22 (м, 1H), 1,15 с, 3Н).
Стадия 8F. (R)-4-((2-(((4-Хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
DIEA (5,0 г, 38,7 ммоль) добавляли к суспензии (R)-(4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метанамина (5,90 г, 22,3 ммоль) в EtOH (100 мл). Смесь перемешивали в течение 30 мин, и затем добавляли 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид (препаративная стадия 1H)(5,0 г, 16,4 ммоль). Полученную смесь перемешивали при 20°C в течение 16 ч. Реакционную смесь фильтровали, и твердые частицы промывали EtOH (10 мл × 3). Твердые частицы суспендировали в воде (30 мл) и нагревали при 50°C в течение 3 ч. Суспензию фильтровали. Твердые частицы промывали водой (10 мл × 3) и сушили с получением 5,24 г (66%) указанного в заголовке соединения в виде твердого вещества желтого цвета. ЖХМС m/z 487,3 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,67 (шир. с, 1H), 9,95 (шир. с, 1H), 9,18 (с, 1H), 8,01 (с, 2H), 7,95 (с, 1H), 5,44 (д, J=10,0 Гц, 1H), 3,86 (с, 3H), 3,18 (шир. с, 3H), 3,05 (шир. с, 3H), 1,78-1,58 (м, 6H), 1,39-1,31 (м, 1H) 1,21-1,10 (м, 1H), 1,07 (с, 3H). [α]24D = -145,151 (c=0,50, MeOH). Хиральная SFC (метод SFC F) RT=3,85 мин, 100% ее. Абсолютную конфигурацию устанавливали рентгеноструктурным анализом монокристалла (фиг. 2).
Пример 9
(S)-4-((2-(((4-Хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
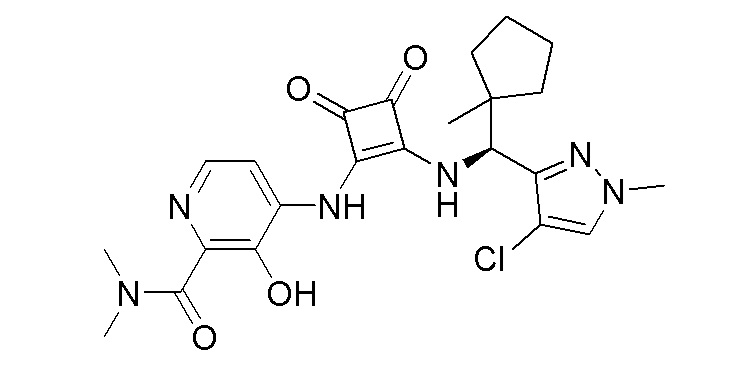
Указанное в заголовке соединение получали аналогично тому, как описано в примере 8, с использованием (R)-2-метилпропан-2-сульфинамида. ЖХМС m/z 487,4 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,68 (шир. с, 1H), 9,92 (шир. с, 1H), 9,16 (шир. с, 1H), 8,02-7,98 (м, 2H), 7,94 (с, 1H), 5,37 (д, J=10,2 Гц, 1H), 3,86 (с, 3H), 3,18 (шир. с, 3H), 3,05 (шир. с, 3H), 1,74-1,55 ( м, 6H), 1,37-1,30 (м, 1H), 1,21-1,14 (м, 1H), 1,07 (с, 3H). [α]20D = +107,3 [c=1, MeOH). Хиральная SFC (метод SFC L) RT=6,28 мин, 98% ее.
Пример 10
(R)-4-((2-(((4-Хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N-изопропил-N-метилпиколинамид
Указанное в заголовке соединение получали из 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N-изопропил-N-метилпиколинамида (препаративная стадия 3F) и (R)-(4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метанамина (препаративная стадия 8E), следуя той же процедуре, описанной для получения примера 8. Желтое твердое вещество, 64 мг (41%). ЖХМС m/z 515,3 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ 8,21 (д, J=5,1 Гц, 1H), 7,93 (шир. с, 1H), 7,64 (с, 1H), 5,55 (с, 1H), 4,70-4,55 (м, 0,4 H), 4,40-4,20 (м, 0,6H), 3,88 (с, 3H), 3,01 (с, 3H), 1,95-1,62 (м, 6H), 1,51-1,42 (м, 1H), 1,35-1,20 (м , 7H), 1,18 (с, 3H). [α]20D = -75,5 (c=0,23, MeOH). Хиральная SFC (метод SFC B) RT=1,59 мин, 100% ее.
Пример 11
(R)-3-Гидрокси-N-изопропил-4-((2-((((4-метокси-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N-метилпиколинамид
Стадия 11А. 3-Иод-4-метокси-1-метил-1H-пиразол
NIS (4,41 г, 19,6 ммоль) добавляли к раствору 4-метокси-1-метил-1H-пиразола (2,20 г, 19,6 ммоль) в ДМФА (20 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 18 ч. Смесь разбавляли водой (100 мл) и насыщенным раствором соли (20 мл) и экстрагировали EtOAc (50 мл × 5). Объединенные органические экстракты концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 16% EtOAc в петролейном эфире) с получением 310 мг (7%) указанного в заголовке соединения в виде коричневого твердого вещества. ЖХМС m/z 238,8 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 6,91 (с, 1H), 3,86 (с, 3H), 3,78 (с, 3H).
Стадия 11В. (R)-N-((R)-(4-Метокси-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)-2-метилпропан-2-сульфинамид
К раствору 3-иод-4-метокси-1-метил-1H-пиразола (399 мг, 1,68 ммоль) в свежеперегнанном ТГФ (4 мл) при -40°C добавляли i-PrMgCl⋅LiCl (3 мл, 3,90 ммоль, 1,3 М раствор в ТГФ). Полученную смесь перемешивали при температуре окружающей среды в течение 2 ч, и затем охлаждали до -40°C. Добавляли раствор (S, E)-2-метил-N -((1-метилциклопентил)метилен)пропан-2-сульфинамида (препаративная стадия 1C)(250 мг, 1,16 ммоль) в свежеперегнанном ТГФ (1 мл). Реакционную смесь подогревали до температуры окружающей среды и перемешивали в течение 16 ч. Реакционную смесь гасили насыщенным водным раствором NH4Cl (1 мл) при 0°C и экстрагировали EtOAc. Объединенные органические экстракты концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 100% EtOAc в петролейном эфире) с получением 150 мг, 33% указанного в заголовке соединения в виде желтого масла. ЖХМС m/z 327,9 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 6,95 (с, 1H), 4,31-4,29 (м, 1H), 4,15-4,13 (м, 1H), 3,77 (с, 3H), 3,69 (с, 3H), 1,80- 1,70 (м, 2H), 1,70-1,55 (м, 4H), 1,50-1,42 (м, 2H), 1,11 (с, 9H), 1,02 (с, 3H).
Стадия 11C. (R)-(4-Метокси-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метанамин
HCl (2,5 мл, 4,0 М раствор в 1,4-диоксане) добавляли к раствору (R)-N-((R)-(4-метокси-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)-2-метилпропан-2-сульфинамида (150 мг, 0,458 ммоль) в MeOH (15 мл). Полученный раствор перемешивали в течение 4 ч, и затем концентрировали в вакууме. Указанное в заголовке соединение выделяли в виде моносоли HCl, допуская количественный выход, и непосредственно использовали без дополнительной очистки. ЖХМС m/z 207,3 [M-NH2]+. 1H ЯМР (400 МГц, CD3OD) δ 7,40 (с, 1H), 4,19 (с, 1H), 3,82 (с, 3H), 3,75 (с, 3H), 1,80-1,61 (м, 6H), 1,51-1,45 (м, 1H), 1,25-1,15 (м, 1H), 1,09 (с, 3H). Абсолютную стереохимию указанного в заголовке соединения определяли по аналогии с препаративным примером 1F.
Стадия 11D. (R)-3-Гидрокси-N-изопропил-4-((2-((((4-метокси-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N-метилпиколинамид
К суспензии (R)-(4-метокси-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метанамина (2,43 г , 9,35 ммоль) в EtOH (60 мл) при 5°C добавляли DIEA (11,8 г, 91,5 ммоль). После перемешивания в течение 10 мин добавляли 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N-изопропил-N-метилпиколинамид (препаративная стадия 3F)(3,05 мг , 9,15 ммоль). Полученный раствор перемешивали в течение 3 суток. Смесь концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 20 до 100% EtOAc в петролейном эфире). Неочищенный продукт растворяли в EtOH (30 мл) и перемешивали при 20°C в течение 16 ч. Суспензию фильтровали с получением 3,26 г (70%) указанного в заголовке соединения в виде желтого твердого вещества. ЖХМС m/z 511,4 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,34 (с, 0,3H), 10,84 (с, 0,7H), 9,94 (с, 1H), 9,13 (д, J=9,8 Гц, 1H), 7,99-7,94 (м, 2H), 7,48 (с, 1H), 5,34 (д, J=10,3 Гц, 1H), 4,82 (шир. с, 0,3H), 4,18 (шир. с, 0,7H), 3,78 (с, 3H), 3,67 (с, 3H), 2,95-2,84 (м, 3H), 1,72-1,50 (м, 6H), 1,35-1,24 (м, 1H), 1,21-1,10 (м, 7H), 1,02 (с, 3H). [α]24D = -114,4 (c=0,50, MeOH). Хиральная SFC (метод SFC B) RT=1,52 мин, 100% ее.
Пример 12
(R)-3-Гидрокси-4-((2-(((4-метокси-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N, N-диметилпиколинамид
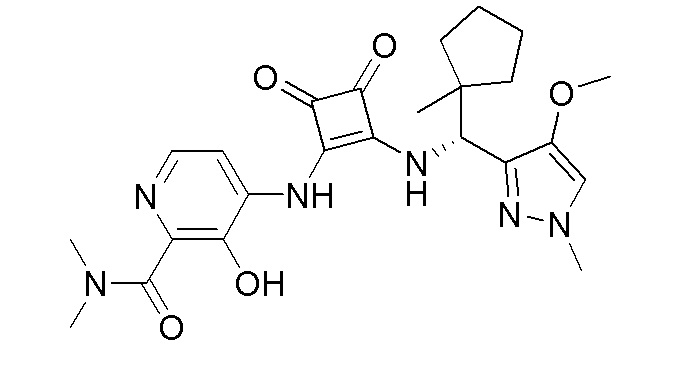
Указанное в заголовке соединение получали из 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида (препаративная стадия 1H) и (R)-(4-метокси-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метанамина (препаративная стадия 11C) в соответствии с процедурой, аналогичной получению примера 11. Желтое твердое вещество, 45 мг (56%). ЖХМС m/z 483,4 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ 8,21 (д, J=5,5 Гц, 1H), 7,90 (д, J=5,5 Гц, 1H), 7,30 (с, 1H), 5,43 (с, 1H), 3,78 (с , 3H), 3,74 (с, 3H), 3,24 (с, 3H), 3,17 (шир. С, 3H), 1,91-1,74 (м, 2H), 1,70-1,60 (м, 4H), 1,45-1,35 (м , 1H), 1,30-1,20 (м, 1H), 1,11 (с, 3H). [α]24D = -87,5 (c=0,50, MeOH). Хиральная SFC (метод SFC F) RT=4,86 мин, 97,4% ее.
Пример 13
(R)-3-Гидрокси-4-((2-((1-(4-метокси-1-метил-1H-пиразол-3-ил)-2,2-диметилпропил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N, N-диметилпиколинамид
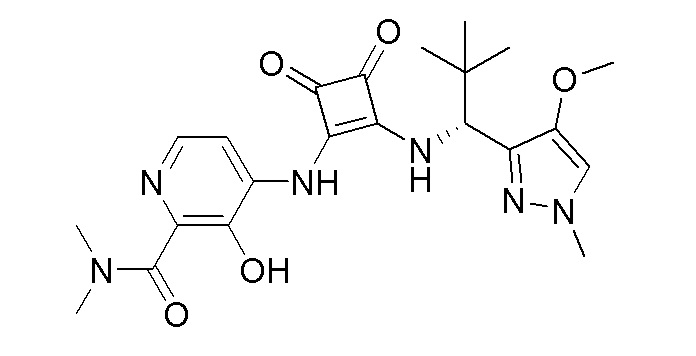
Стадия 13А. (S, E)-N-(2,2-Диметилпропилиден)-2-метилпропан-2-сульфинамид
Этоксид титана (IV)(68,9 г, 302 ммоль) добавляли к раствору пивальдегида (10,0 г, 116 ммоль) в DCM. (830 мл). Смесь перемешивали в течение 20 мин, и затем добавляли (S)-2-метилпропан-2-сульфинамид (14,1 г, 116 ммоль). Полученную смесь перемешивали при температуре окружающей среды в течение 16 ч, а затем гасили водой (250 мл). Смесь фильтровали, и твердые частицы промывали ТГФ (500 мл × 2). Объединенные органические слои концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 5% EtOAc в петролейном эфире) с получением 24,4 г (75%) указанного в заголовке соединения в виде бесцветного масла. ЖХМС m/z 190,2 [M+H]+. 1H ЯМР (400 МГц, CDCl3) 7,92 (с, 1H), 1,20 (с, 9H), 1,16 (с, 9H).
Стадия 13В. (R)-N-((R)-1-(4-Метокси-1-метил-1H-пиразол-3-ил)-2,2-диметилпропил)-2-метилпропан-2-сульфинамид
К раствору 3-йод-4-метокси-1-метил-1H-пиразола (препаративная стадия 11A)(250 мг, 1,05 ммоль) в свежеперегнанном ТГФ (0,7 мл) при -40°C, добавляли i-PrMgCl·LiCl (1,7 мл, 2,22 ммоль), 1,3 М раствор в ТГФ). Полученную смесь перемешивали при 15°C в течение 1 ч, и затем охлаждали до -40°C. Добавляли раствор (S, E)-N-(2,2-диметилпропилиден)-2-метилпропан-2-сульфинамида (140 мг, 0,74 ммоль) в свежеперегнанном ТГФ (0,5 мл). Смесь нагревали до 15°C и перемешивали в течение 16 ч. Реакционную смесь гасили насыщенным водным раствором NH4Cl (1 мл) при 0°C и экстрагировали EtOAc. Объединенные органические экстракты концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 100% EtOAc в петролейном эфире) с получением 80 мг (36%) указанного в заголовке соединения в виде желтого масла. ЖХМС m/z 301,9 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 6,97 (с, 1H), 5,32-5,31 (м, 1H), 4,20-4,15 (м, 1H), 3,83 (с, 3H), 3,71 (с, 3H), 1,11 ( с, 9H), 1,01 (с, 9H).
Стадия 13C. (R)-1-(4-Метокси-1-метил-1H-пиразол-3-ил)-2,2-диметилпропан-1-амин
HCl (1,2 мл, 4,0 М в MeOH) добавляли к раствору (R)-N-((R)-1-(4-метокси-1-метил-1H-пиразол-3-ил)-2,2-диметилпропил)-2-метилпропан-2-сульфинамида (80 мг, 0,27 ммоль) в МеОН (7 мл). Смесь перемешивали при 15°C в течение 4 ч, и затем концентрировали в вакууме. Указанное в заголовке соединение выделяли в виде моносоли HCl, допуская количественный выход, и непосредственно использовали без дополнительной очистки. ЖХМС m/z 180,8 [M-NH2]+. 1H ЯМР (400 МГц, CD3OD) δ 7,43 (с, 1H), 4,10 (с, 1H), 3,84 (с, 3H), 3,78 (с, 3H), 1,05 (с, 9H). Хиральная SFC (метод SFC J) RT=1,85 мин, 93,7% ее. Абсолютную стереохимию указанного в заголовке соединения определяли по аналогии с препаративным примером 1F.
Стадия 13D. (R)-3-Гидрокси-4-((2-((1-(4-метокси-1-метил-1H-пиразол-3-ил)-2,2-диметилпропил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N, N-диметилпиколинамид
К раствору (R)-1-(4-метокси-1-метил-1H-пиразол-3-ил)-2,2-диметилпропан-1-амина (60 мг, 0,26 ммоль) в EtOH (3 мл) при 15°C добавляли DIEA (63,5 мг, 0,49 ммоль). После перемешивания в течение 10 мин добавляли 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид (препаративная стадия 1H)(50 мг, 0,16 ммоль). Полученную смесь перемешивали при температуре окружающей среды в течение 16 ч, и затем концентрировали в вакууме. Остаток очищали с помощью препаративной ВЭЖХ (Agela Durashell C18 150 мм × 25 мм, 5 мкм, от 14 до 54% CH3CN в 0,225% муравьиной кислоте в воде, 25 мл/мин, 11 мин) с получением 42,1 мг (56%) указанного в заголовке соединения в виде желтого твердого вещества. ЖХМС m/z 457,4 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,64 (шир. с, 1H), 9,99 (с, 1H), 9,12 (д, J=8,8 Гц, 1H), 8,01 (с, 2H), 7,49 (с, 1H), 5,22 (д, J=10,3 Гц, 1H), 3,79 (с, 3H), 3,67 (с, 3H), 3,19 (шир. с, 3H), 3,06 (шир. с, 3H), 0,94 (с, 9H). [α]24D = -110,8 (c=0,33, MeOH). Хиральная SFC (метод SFC J) RT=3,95 мин, 100% ее.
Пример 14
(R)-3-Гидрокси-N-изопропил-4-((2-((1-(4-метокси-1-метил-1H-пиразол-3-ил)-2,2-диметилпропил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N-метилпиколинамид
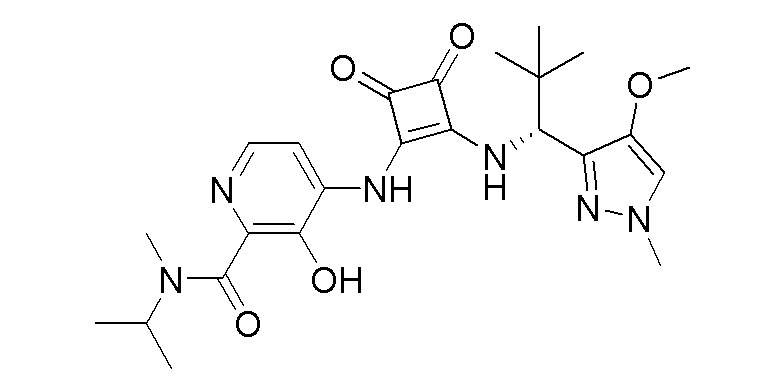
Указанное в заголовке соединение получали из 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N-изопропил-N-метилпиколинамида (препаративная стадия 3F) и (R)-1-(4-метокси-1-метил-1H-пиразол-3-ил)-2,2-диметилпропан-1-амина (препаративная стадия 13C) в соответствии с процедурами, аналогичными получению примера 13. Желтое твердое вещество, 18 мг (25%). ЖХМС m/z 485,4 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ 8,18 (шир. с, 1H), 7,94 (шир. с, 1H), 7,33 (с, 1H), 5,35 (с, 1H), 4,64 (шир. с, 0,3H), 4,30 (шир. с, 0,7H), 3,81 (с, 3H), 3,76 (с, 3H), 3,02 (с, 3H), 1,33-1,20 (м, 6H), 1,04 (с, 9H). Хиральная SFC (метод SFC K) RT=2,78 мин, 98,7% ее.
Пример 15
(R)-4-((2-(((4-(Дифторметокси)-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-метилпиколинамид
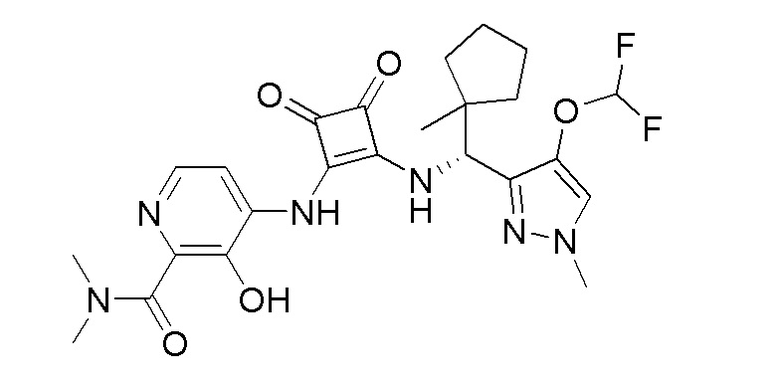
Стадия 15А. 4-(Дифторметокси)-1-метил-1H-пиразол
К раствору 1-метил-1H-пиразол-4-ола (1,0 г, 10,2 ммоль) в IPA (10 мл) при 15°C добавляли КОН (2,86 г, 51,0 ммоль), и затем хлордифторметан (8,81 г, 102 ммоль). Реакция была экзотермической. Смесь перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь выливали в воду и экстрагировали EtOAc. Объединенные органические экстракты промывали водой и насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (33% EtOAc в петролейном эфире) с получением 700 мг (46%) указанного в заголовке соединения в виде желтого масла. ЖХМС m/z 148,9 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,37 (с, 1H), 7,32 (с, 1H), 6,34 (т, J=73,03 Гц, 1H), 3,98-3,78 (с, 3H). 19F ЯМР (376 МГц, CDCl3) δ -83,6.
Стадия 15В. 4-(Дифторметокси)-3-иод-1-метил-1H-пиразол
NIS (6,38 г, 28,4 ммоль) добавляли к раствору 4- (дифторметокси)-1-метил-1H-пиразола (1,40 г, 9,45 ммоль) в ацетонитриле (25 мл). Полученную смесь нагревали при 50°C в течение 84 ч. Реакционную смесь разбавляли водой (15 мл) и экстрагировали EtOAc (20 мл × 3). Объединенные органические экстракты промывали насыщенным водным раствором Na2S2O3 и насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью препаративной ТСХ на силикагеле (50% EtOAc в петролейном эфире) с получением 260 мг (10%) указанного в заголовке соединения в виде желтого твердого вещества. ЖХМС m/z 275,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,27 (с, 1H), 6,41 (т, J=73,03 Гц, 1H), 3,92 (с, 3H). 19F ЯМР (376 МГц, CDCl3) δ -83,6.
Стадия 15C. (R)-N-((R)-(4-(Дифторметокси)-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)-2-метилпропан-2-сульфинамид
К раствору 4-(дифторметокси)-3-иод-1-метил-1H-пиразола (66,2 мг, 1,3 ммоль) в ТГФ (5 мл) при -40°C по каплям добавляли i-PrMgCl⋅LiCl (0,43 мл, 0,557 ммоль, 1,3 М раствор в ТГФ). Полученную смесь перемешивали при -10°C в течение 1 ч, и затем охлаждали до -40°C. Добавляли раствор (S, E)-2-метил-N-((1-метилциклопентил)метилен)пропан-2-сульфинамида (препаративная стадия 1С)(40 мг, 0,19 ммоль) в ТГФ (3 мл). Реакционную смесь нагревали до 50°C и перемешивали в течение ночи. Смесь гасили насыщенным водным раствором NH4Cl при 10°C и экстрагировали EtOAc. Объединенные органические экстракты концентрировали в вакууме. Остаток очищали с помощью препаративной ТСХ на силикагеле (80% EtOAc в петролейном эфире) с получением 70 мг (46%) указанного в заголовке соединения в виде желтого масла. ЖХМС m/z 363,9 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,25 (с, 1H), 6,37 (т, J=73,03 Гц, 1H), 4,33 (д, J=6,8 Гц, 1H), 3,90-3,71 (м, 4H), 1,77 -1,55 (м, 6H), 1,54-1,42 (м, 1H), 1,28-1,13 (м, 10H), 1,08-0,98 (м, 3H). 19F ЯМР (376 МГц, CDCl3) δ -83,2.
Стадия 15D. (R)-(4-(Дифторметокси)-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метанамин
HCl (1 мл, 4,0 М раствор в MeOH) добавляли к раствору (R)-N- ((R)-(4-(дифторметокси)-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)-2-метилпропан-2-сульфинамида (35 мг, 0,096 ммоль) в MeOH (1 мл) при 5°C. Смесь перемешивали при 20°C в течение 3 ч, и затем концентрировали в вакууме. Указанное в заголовке соединение выделяли в виде моносоли HCl, допуская количественный выход, и непосредственно использовали без дополнительной очистки. Абсолютную стереохимию указанного в заголовке соединения определяли по аналогии с препаративным примером 1F. ЖХМС m/z 242,8 [M-NH2]+.
Стадия 15E. (R)-4-((2-(((4-(Дифторметокси)-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
К раствору (R)-(4-(дифторметокси)-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метанамина (28 мг, 0,11 ммоль) в EtOH (2 мл) при 20°C добавляли DIEA (140 мг, 1,08 ммоль). После перемешивания в течение 5 мин добавляли 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид (препаративная стадия 1H)(30 мг, 0,108 ммоль). Полученную смесь перемешивали при 30°C в течение 6 ч, и затем концентрировали в вакууме. Остаток очищали с помощью препаративной ВЭЖХ (Xbridge 150 мм × 30 мм, 10 мкм, от 15 до 55% CH3CN в 0,225% муравьиной кислоте в воде, 25 мл/мин, 10 мин) с получением 52 мг (55%) указанного в заголовке соединения в виде желтого твердого вещества. ЖХМС m/z 519,3 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ 8,22 (д, J=5,5 Гц, 1H), 7,92 (шир. с, 1H), 7,57 (с, 1H), 6,65 (дд, J=75,5, 72,9 Гц, 1H), 5,46 (с, 1H), 3,86 (с, 3H), 3,24 (с, 3H), 3,17 (с, 3H), 1,87-1,62 (м, 6H), 1,51-1,43 (м, 1H), 1,33- 1,25 (м, 1H), 1,15 (с, 3H). 19F ЯМР (376 МГц, CDCl3) δ -85,1. Хиральная SFC (метод SFC B) RT=1,34 мин, 100% ее.
Пример 16
(R)-4-((2-(((4-Циано-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
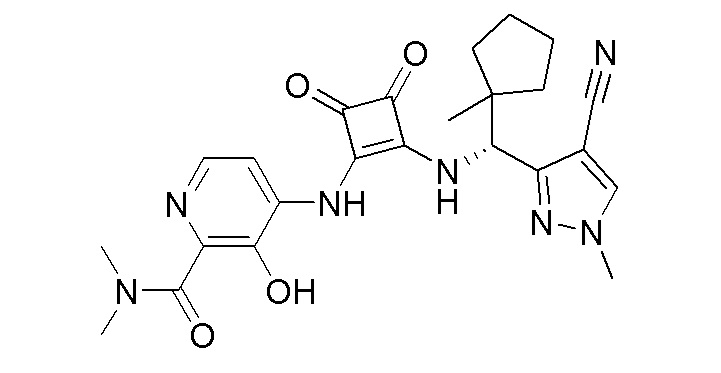
Стадия 16А. 3-Амино-1-метил-1H-пиразол-4-карбонитрил
К раствору 3-амино-1H-пиразол-4-карбонитрила (15,0 г, 139 ммоль) в ДМФА (700 мл) при 0°C добавляли K2CO3 (23,0 г, 167 ммоль). Смесь перемешивали в течение 45 мин, и затем добавляли MeI (23,6 г, 167 ммоль). Полученную смесь нагревали при 90°C в течение 16 ч. Реакционную смесь выливали в ледяную воду (200 мл) и экстрагировали EtOAc (400 мл × 8). Объединенные органические экстракты концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 20% EtOAc в петролейном эфире) с получением 3,30 г (20%) указанного в заголовке соединения в виде белого твердого вещества. ЖХМС m/z 122,8 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,02 (с, 1H), 5,53 (с, 2H), 3,61 (с, 3H).
Стадия 16В. 3-Иод-1-метил-1H-пиразол-4-карбонитрил
К перемешиваемому раствору 3-амино-1-метил-1H-пиразол-4-карбонитрила (1,0 г, 8,19 ммоль) и p-TsOH·вода (3,55 г, 18,7 ммоль) в MeCN (30 мл) при 0°C по каплям добавляли раствор NaNO2 (1,29 г, 18,7 ммоль) и KI (3,1 г, 18,7 ммоль) в воде (4,0 мл). Полученную смесь перемешивали при 15°C в течение 3 суток. Реакционную смесь концентрировали в вакууме. Остаток разбавляли водой (10 мл) и насыщенным водным раствором Na2SO3 (20 мл), и затем нейтрализовали NaOH (1,0 Н водный раствор) до pH=8. Смесь экстрагировали EtOAc (100 мл × 4) и объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 100% EtOAc в петролейном эфире) с получением 398 мг (21%) указанного в заголовке соединения в виде желтого твердого вещества. ЖХМС m/z 234,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 8,48 (с, 1H), 3,94 (с, 3H).
Стадия 16C. (R)-N-((R)-(4-Циано-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)-2-метилпропан-2-сульфинамид
К раствору 3-йод-1-метил-1H-пиразол-4-карбонитрила (398 мг, 1,71 ммоль) в свежеперегнанном ТГФ (12 мл) при -65°C по каплям добавляли i-PrMgCl·LiCl (2,60 мл, 3,38 ммоль, 1,3 М раствор в ТГФ). Полученную смесь перемешивали при -40°C в течение 1 ч, и затем охлаждали до -65°C. Добавляли по каплям раствор (S, E)-2-метил-N-((1-метилциклопентил)метилен)пропан-2-сульфинамида (препаративная стадия 1C)(260 мг, 1,21 ммоль) в свежеперегнанном ТГФ (3 мл). Смесь подогревали до 15°C и перемешивали в течение 3 суток. Реакционную смесь выливали в 25% водный раствор NH4Cl (20 мл) при 15°C и экстрагировали EtOAc (50 мл × 3). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 100% EtOAc в петролейном эфире) с получением 84 мг (17%) указанного в заголовке соединения в виде желтого твердого вещества. ЖХМС m/z 322,9 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,76 (с, 1H), 4,48-4,46 (м, 1H), 3,93 (с, 3H), 3,82-3,75 (м, 1H), 1,85-1,50 (м, 8H), 1,29 (с, 9H), 1,08 (с, 3H).
Стадия 16D. (R)-3-(Амино(1-метилциклопентил)метил)-1-метил-1H-пиразол-4-карбонитрил
К раствору (R)-N-((R)-(4-циано-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)-2-метилпропан-2-сульфинамида (84 мг, 0,26 ммоль) в DCM (6 мл) при 0°C добавляли HCl (2 мл, 4,0 мл в EtOAc). Смесь подогревали до 15°C и перемешивали в течение 2 ч. Затем реакционную смесь концентрировали в вакууме. Указанное в заголовке соединение выделяли в виде моносоли HCl, допуская количественный выход, и непосредственно использовали без дополнительной очистки. ЖХМС m/z 219,3 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ 8,31 (с, 1H), 4,40 (с, 1H), 4,00 (с, 3H), 1,90-1,50 (м, 8H), 1,11 (с, 3H). Абсолютную стереохимию указанного в заголовке соединения определяли по аналогии с препаративным примером 1F.
Стадия 16E. (R)-4-((2-(((4-Циано-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
К раствору (R)-3-(амино(1-метилциклопентил)метил)-1-метил-1H-пиразол-4-карбонитрила (24 мг, 0,094 ммоль) в EtOH (3 мл) при 0°C добавляли DIEA (59,3 мг, 0,459 ммоль) и 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид (препаративная стадия 1H)(28 мг, 0,092 ммоль). Полученный раствор перемешивали при 20°C в течение 16 ч. Реакционную смесь концентрировали в вакууме. Остаток очищали с помощью препаративной ВЭЖХ (Agela Durashell C18 150 мм × 25 мм, 5 мкм, от 18 до 58% MeCN в 0,225% муравьиной кислоте в воде, 25 мл/мин, 12 мин) с получением 13 мг (18%) указанного в заголовке соединения в виде желтого твердого вещества. ЖХМС m/z 478,4 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ 8,22 (д, J=5,5 Гц, 1H), 8,19 (с, 1H), 7,95-7,85 (м, 1H), 5,52 (с, 1H), 3,95 (с, 3H) , 3,24 (шир. с, 3H), 3,17 (шир. с, 3H), 1,95-1,65 (м, 6H), 1,55-1,45 (м, 1H), 1,44-1,35 (м, 1H), 1,15 (с, 3H). Хиральная ВЭЖХ (метод ВЭЖХ A) RT=8,65 мин, 98,0% ее.
Пример 17
(R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(5-метилспиро[2.3] гексан-5-ил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
Пример 17А. N-Метокси-N-метилспиро[2.3]гексан-5-карбоксамид
NMM (1,09 г, 1,18 мл, 10,8 ммоль) добавляли к раствору N, O-диметилгидроксиламина · HCl (300 мг, 3,07 ммоль) в ДМФА (30,7 мл). Раствор перемешивали в течение 5 мин, и затем добавляли спиро[2,3]гексан-5-карбоновую кислоту (465 мг, 3,69 ммоль) и HATU (1,75 г, 4,61 ммоль). Полученную смесь перемешивали в течение ночи. Реакционную смесь гасили насыщенным водным раствором NH4Cl и экстрагировали EtOAc. Объединенные органические экстракты промывали водой (100 мл × 2) и насыщенным раствором соли (100 мл) и концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 100% EtOAc в гептане) с получением 440 мг (85%) указанного в заголовке соединения в виде масла. ГХМС m/z 169,1 [M]. 1H ЯМР (400 МГц, CDCl3) δ 3,67 (с, 3H), 3,20 (с, 3H), 2,65-2,42 (м, 2H), 2,26-2,00 (м, 2H), 0,61-0,23 (м, 4H).
Пример 17В. (1,4-Диметил-1H-пиразол-3-ил)(спиро[2.3]гексан-5-ил)метанон
К раствору 3-иод-1,4-диметил-1H-пиразола (препаративная стадия 1D)(304 мг, 1,37 ммоль) в ТГФ (2,36 мл) по каплям добавляли i-PrMgCl⋅LiCl (1,82 мл, 2,36 ммоль, 1,3 M раствор в THF). Полученный желтый раствор подогревали до температуры окружающей среды и перемешивали в течение 1 ч. Затем по каплям добавляли раствор N-метокси-N-метилспиро[2.3]гексан-5-карбоксамида (200 мг, 1,18 ммоль) в ТГФ (1 мл) и смесь перемешивали в течение 16 ч. Реакционную смесь выливали в насыщенный водный раствор NH4Cl. Смесь экстрагировали DCM, и объединенные органические экстракты концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 100% EtOAc в гептане) с получением 220 мг (91%) указанного в заголовке соединения в виде твердого вещества. ЖХМС m/z 205,2 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,15 (с, 1H), 4,31 (тт, J=9,2, 7,6 Гц, 1H), 3,89 (с, 3H), 2,67-2,48 (м, 2H), 2,32 (с, 3H), 2,26-2,21 (м, 2H), 0,59-0,28 (м, 4H).
Пример 17C. (1,4-Диметил-1H-пиразол-3-ил)(5-метилспиро[2.3]гексан-5-ил)метанон
NaH (47,0 мг, 1,17 ммоль, 60 мас.% в минеральном масле) и йодметан (208 мг, 1,47 ммоль) добавляли к раствору (1,4-диметил-1H-пиразол-3-ил)(спиро[2.3]гексан-5-ил)метанона (72 мг, 0,37 ммоль) в ТГФ (9,79 мл). Полученную смесь перемешивали при температуре окружающей среды в течение 24 ч. Реакционную смесь гасили насыщенным водным раствором NH4Cl и экстрагировали EtOAc. Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 100% EtOAc в гептане) с получением 40 мг (19%) указанного в заголовке соединения в виде твердого вещества. ЖХМС m/z 219,2 [M+H]+.
Пример 17D. (1,4-Диметил-1H-пиразол-3-ил)(5-метилспиро[2.3] гексан-5-ил)метанамин
Этоксид титана (IV)(157 мг, 0,144 мл, 0,688 ммоль) добавляли к раствору (1,4-диметил-1H-пиразол-3-ил)(5-метилспиро[2.3]гексан-5-ил)метанона (50 мг, 0,23 ммоль) в метанольном растворе NH3 (0,983 мл, 7,0 M в MeOH). Полученную смесь перемешивали при 40°C в течение ночи. Реакционную смесь охлаждали до температуры окружающей среды, разбавляли MeOH (2 мл) и добавляли NaBH4 (13 мг, 0,344 ммоль). Смесь перемешивали в течение 4 ч. Реакционную смесь гасили насыщенным водным раствором NH4Cl и фильтровали через целит. Осадок на фильтре промывали 20% MeOH в DCM. Органический слой фильтрата отделяли, и водный слой подкисляли до pH 2, используя 1,0 М раствор HCl. Водный слой экстрагировали DCM (30 мл). Затем водный слой подщелачивали до pH 12, используя NaOH, и экстрагировали DCM (20 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения и непосредственно использовали на следующей стадии без дополнительной очистки, допуская количественный выход.
Пример 17Е. (R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(5-метилспиро[2.3]гексан-5-ил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
К смеси (1,4-диметил-1H-пиразол-3-ил)(5-метилспиро[2.3] гексан-5-ил)метанамина (52 мг, 0,236 ммоль) в EtOH (1,97 мл) при 20°C добавляли DIEA (381 мг, 0,529 мл, 2,95 ммоль). Полученный раствор перемешивали в течение 20 мин, и затем добавляли 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид (препаративная стадия 1H)(60 мг, 0,20 ммоль). Полученную суспензию перемешивали при 20°C в течение 4 суток, и затем концентрировали в вакууме. Остаток очищали в условиях хиральной SFC (Chiral Tech OD-H 21,2 × 21,2 мм, внутренний диаметр 5 мкм, 45% (0,2% 7 Н аммиак в MeOH) в EtOH, 80 мл/мин). Выделение первого элюированного изомера дает 21,6 мг (23%) указанного в заголовке соединения, и второй элюированный изомер дает соответствующий энантиомер (24 мг, 26%). ЖХМС m/z 479,8 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,93 (шир. с, 1H), 9,18 (шир. С, 1H), 8,04-8,00 (м, 2H), 7,44 (с, 1H), 5,53 (д, J=10,1 , 1H), 3,79 (с, 3H), 3,20 (шир. с, 3H), 3,07 (шир. с, 3H), 2,28 (д, J=11,7, 1H), 2,08 (д, J=11,3, 1H), 2,01 (с, 3H), 1,73 (д, J=11,3 Гц, 1H), 1,66 (д, J=10,5 Гц, 1H), 1,35 (с, 3H), 0,45-0,28 (м, 4H). Условия хиральной SFC (Chiral Tech OD-H 21,2 × 21,2 мм, внутренний диаметр 5 мкм, 45% (0,2% 7 Н аммиак в MeOH) в EtOH, 80 мл/мин) RT=5,39 мин, 98,8% ee (указанное в заголовке соединение) и RT=5,98 мин, 96% ее (энантиомер).
Соединения 1-14
Указанные в заголовке соединения в таблице 1 получали аналогично получению (R)-(1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил) метанамина (препаративная стадия 1F) из соответствующего альдегида и гетероарилгалогенида.
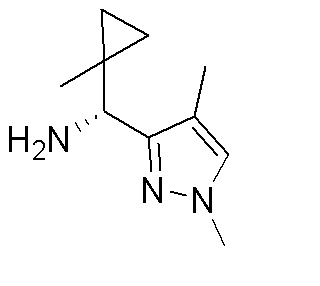
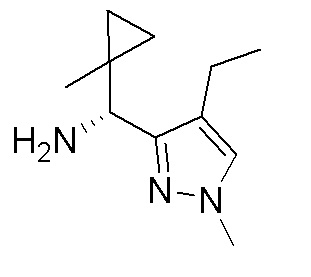
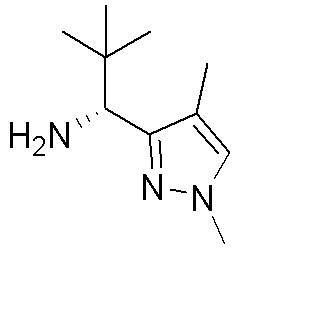
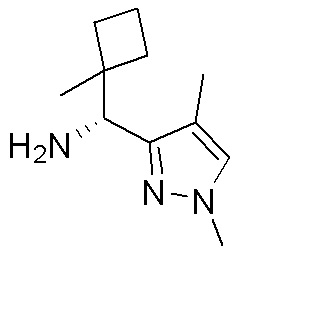
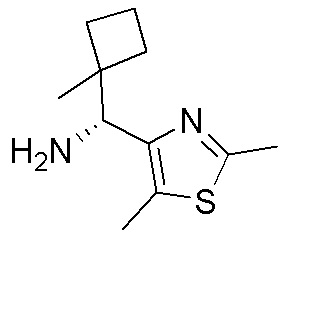
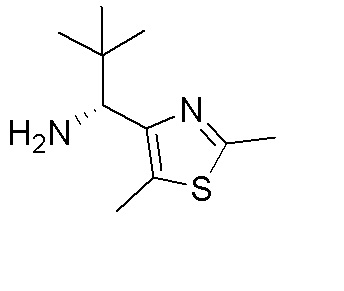
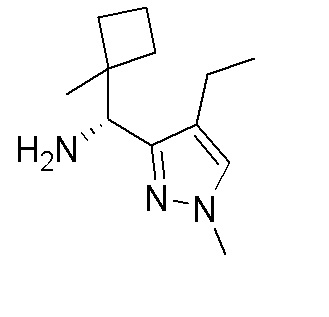
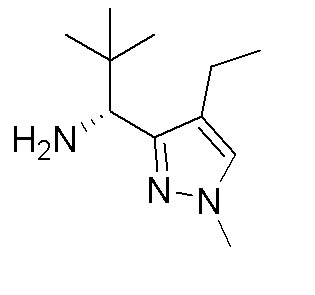
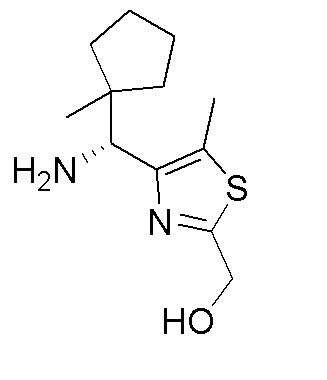
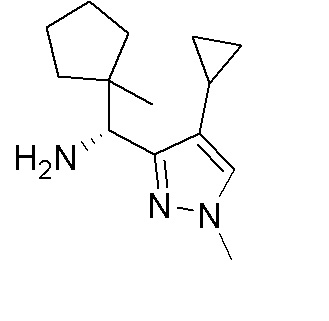
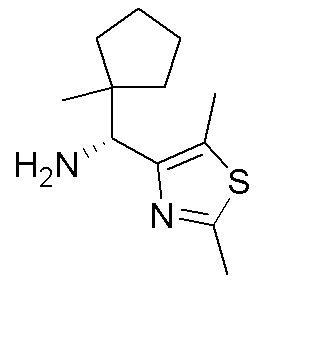
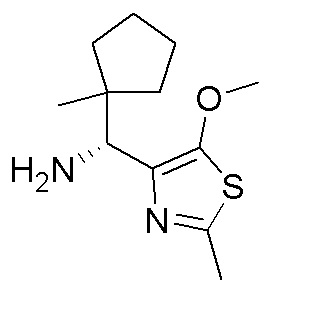
Соединения 18-33
Указанные в заголовке соединения в таблице 2 получали аналогично получению (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида (препаративная стадия 1I) из соответствующего амина и 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диалкилпиколинамида.
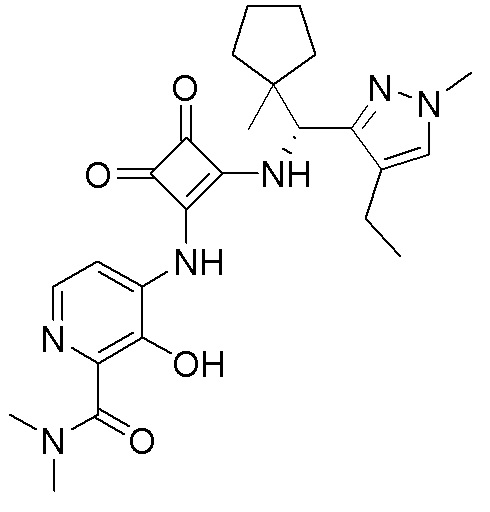

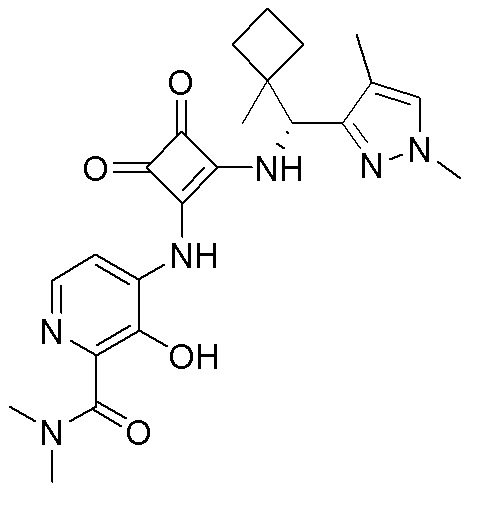
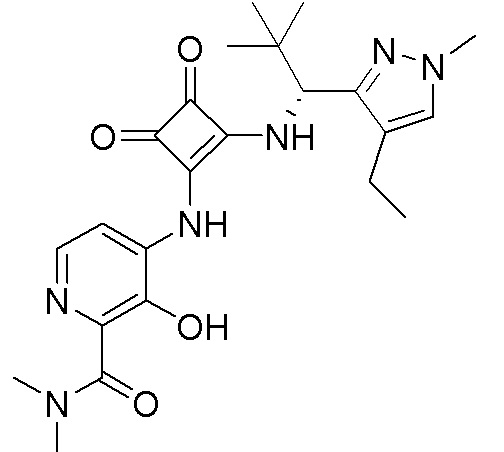
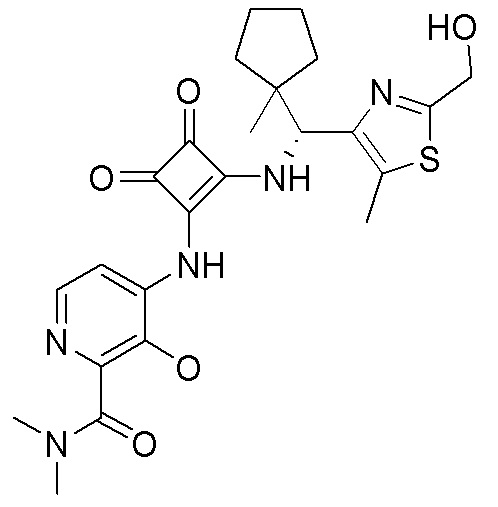

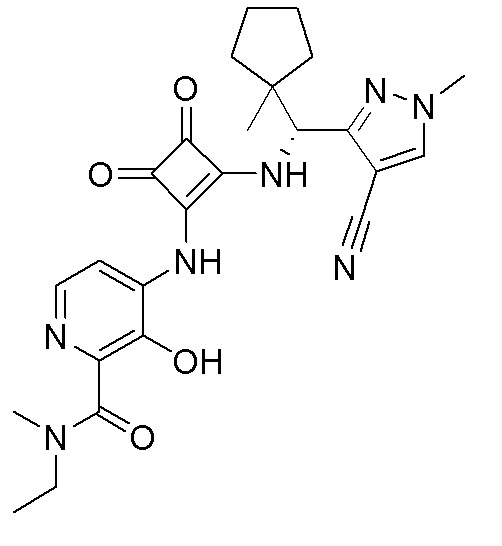
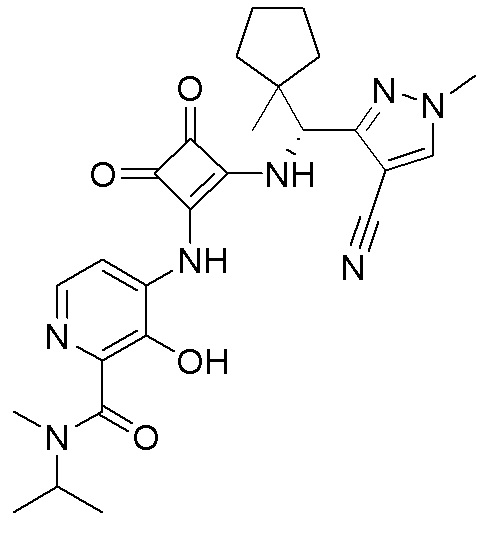
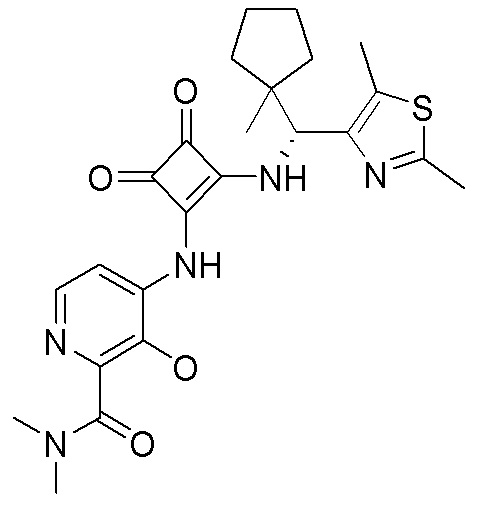

Пример 34
(R)-4-((2-(((2,5-Диметилоксазол-4-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
Стадия 34А. (2,5-Диметилоксазол-4-ил)(1-метилциклопентил) метанамин
Указанное в заголовке соединение получали аналогично тому, как описано для (1,4-диметил-1H-пиразол-3-ил)(5-метилспиро[2.3] гексан-5-ил)метанамина (препаративная стадия 17D) с использованием 2,5-диметилоксазол-4-карбоновой кислоты и бромида циклопентилмагния. ЖХМС m/z 192,2 [M-NH2]+.
Стадия 34В. (R)-4-((2-(((2,5-Диметилоксазол-4-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
К раствору (2,5-диметилоксазол-4-ил)(1-метилциклопентил) метанамина (45 мг, 0,22 ммоль) в EtOH (1,6 мл) добавляли 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида (препаративная стадия 1H)(50 мг, 0,16 ммоль) и DIEA (318 мг, 2,46 ммоль). Полученный раствор перемешивали при 40°C в течение ночи. Реакционную смесь концентрировали в вакууме. Остаток подвергали разделению энантиомеров в условиях хиральной SFC (Chiralcel OD, 250 мм × 21 мм × 5 мкм, от 5 до 60% 0,2% NH4 + (7 Н в MeOH) в MeOH, 80 мл/ мин). Выделение первого элюированного изомера дает 16 мг (21%) указанного в заголовке соединения, и второй элюированный изомер дает 23 мг (30%) соответствующего энантиомера. ЖХМС m/z 468,6 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,23-9,10 (м, 1H), 7,98-7,97 (м, 1H), 7,91 (шир. с, 1H), 5,17-5,15 (с, 1H), 3,17 (с, 3H), 3,04 (с, 3H), 2,37 (с, 3H), 2,25 (с, 3H), 1,78-1,47 (м, 7H), 1,37-1,10 (м, 3H), 1,05 (с, 3H). RT=5,58 мин, 100% ее (метод SFC C)
Пример 35
(R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-этилциклобутил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
Стадия 35А. (1,4-Диметил-1H-пиразол-3-ил)(1-этилциклобутил) метанамин
Указанное в заголовке соединение получали аналогично тому, как описано для (1,4-диметил-1H-пиразол-3-ил)(5-метилспиро[2.3] гексан-5-ил)метанамина (препаративная стадия 17D) с использованием 1-этилциклобутан-1-карбоновой кислоты и 3-иод-1,4-диметил-1H-пиразола. ЖХМС m/z 190,8 [M-NH2]+.
Стадия 35В. (R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-этилциклобутил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
К раствору (1,4-диметил-1H-пиразол-3-ил)(1-этилциклобутил) метанамина (800 мг, 3,28 ммоль) в EtOH (15 мл) добавляли 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид (препаративная стадия 1H)(120 мг, 0,39 ммоль) и DIEA (152 мг, 1,18 ммоль). Полученный раствор перемешивали при 30°C в течение ночи. Реакционную смесь концентрировали в вакууме. Остаток очищали с помощью препаративной ВЭЖХ (Agela Durashell C18 150 мм × 25 мм × 5 мкм, от 13 до 53% CH3CN в 0,05% NH4OH в воде, 25 мл/мин, 11 мин) с получением 100 мг рацемического продукта. Рацемическую смесь подвергали разделению энантиомеров с использованием условий хиральной SFC (REGIS (s, s) WHELK-O1 250 мм × 30 мм, 5 мкм, 45% 0,1% NH4OH в EtOH, 60 мл/мин). Выделение первого элюированногго изомера дает 33 мг (18%) указанного в заголовке соединения в виде желтого твердого вещества, и второй элюированный изомер дает 34 мг (19%) соответствующего энантиомера в виде желтого твердого вещества. ЖХМС m/z 467,4 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ 8,27 (д, J=5,6 Гц, 1H), 7,94 (д, J=5,7 Гц, 1H), 7,31 (с, 1H), 5,51 (с, 1H), 3,84 (с , 3H), 3,30-3,10 (м, 6H), 2,46-2,38 (м, 1H), 2,15-2,05 (м, 4H), 1,95-1,60 (м, 6H), 1,00 (т, J=7,4, 3H). Хиральная SFC (метод SFC I) RT=10,44 мин, 97,2% ее (указанное в заголовке соединение) и RT=14,45 мин, 91,9% ее (энантиомер).
Пример 36
4-((2-(((S)-((S)-4,4-Дифтор-2-метилтетрагидрофуран-2-ил)(1,4-диметил-1H-пиразол-3-ил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
Стадия 36А. Этил 2-гидрокси-2-метилпент-4-еноат
К раствору этил 2-оксопропаноата (76 г, 654,5 ммоль) в DCM (380 мл) при -78°C добавляли хлорид титана (IV)(124 г, 655 ммоль). Полученную смесь перемешивали при той же температуре в течение 30 мин, пока она не превратилась в желтую суспензию. Затем добавляли аллилтриметилсилан (97,2 г, 851 ммоль) и реакционную смесь перемешивали в течение 2 ч. Реакционную смесь нагревали до 0°C, гасили насыщенным водным раствором Na2CO3 (1,5 л) и экстрагировали DCM (3,0 л × 2). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 5 до 10% EtOAc в петролейном эфире) с получением 134 г (63%) указанного в заголовке соединения в виде светло-желтого масла. ЖХМС m/z 158,8 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 5,80-5,70 (м, 1H), 5,12 (с, 1H), 5,10-5,07 (м, 1H), 4,25-4,15 (м, 2H), 3,19 (с, 1H), 2,53-2,45 (м, 1H), 2,41-2,38 (м, 1H), 1,41 (с, 3H), 1,32-1,25 (м, 3H).
Стадия 36В. Этил 2-гидрокси-2-метил-3-(оксиран-2-ил) пропаноат
К раствору этил 2-гидрокси-2-метилпент-4-еноата (46 г, 290,8 ммоль) в DCM (1,45 л) при 0°С добавляли порциями м-CPBA (81,5 г, 378 ммоль, чистота 80%) в течение 20 мин. Реакционную смесь перемешивали при 10°C в течение 1 ч, и затем нагревали до 35°C и перемешивали в течение 16 ч. Смесь охлаждали до 0°C и гасили насыщенным водным раствором Na2S2O3. Органический слой отделяли и промывали насыщенным водным раствором Na2CO3, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем с получением 81 г (80%) указанного в заголовке соединения в виде светло-желтого масла, состоящего из смеси диастереомеров. 1H ЯМР (400 МГц, CDCl3) δ 4,35-4,20 (м, 2H), 3,50 (с, 0,5H), 3,35 (с, 0,5H), 3,15-3,05 (м, 1H), 2,80-2,70 (м, 1H), 2,50-2,42 (м, 1H), 2,01-1,85 (м, 2H), 1,48 (с, 1,5H), 1,45 (с, 1,5H), 1,35-1,30 (м, 3H).
Стадия 36C. Этил 2-метил-4-оксотетрагидрофуран-2-карбоксилат
Бромид магния (12 г, 65,4 ммоль) добавляли к раствору этил 2-гидрокси-2-метил-3-(оксиран-2-ил)пропаноата (76 г, 436 г). ммоль) в ТГФ (957 мл). Смесь нагревали при 80°C в течение 16 ч. Затем реакционную смесь концентрировали в вакууме. Остаток растворяли в воде (100 мл) и экстрагировали DCM (500 мл × 2). Объединенные органические экстракты концентрировали в вакууме. Остаток растворяли в DCM (2080 мл) и охлаждали до 0°C. Добавляли силикагель (78,6 г, 1310 ммоль) и PCC (122 г, 567 ммоль), и смесь перемешивали при 10°C в течение 16 ч. Добавляли дополнительно PCC (18,8 г, 87,3 ммоль) и смесь перемешивали при 30°C в течение 4 ч. Реакционную смесь фильтровали, и твердые частицы промывали DCM (500 мл × 2). Органический фильтрат сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 10 до 15% EtOAc в петролейном эфире) с получением 24,3 г (32%) указанного в заголовке соединения в виде светло-желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 4,26-4,20 (м, 2H), 4,14 (с, 2H), 2,90-2,85 (м, 1H), 2,47-2,41 (м, 1H), 1,63 (с, 3H), 1,32-1,28 (м, 3H).
Стадия 36D. Этил 4,4-дифтор-2-метилтетрагидрофуран-2-карбоксилат
DAST (37,4 г, 232 ммоль) добавляли по каплям к чистому образцу этил 2-метил-4-оксотетрагидрофуран-2-карбоксилата (20,0 г, 116,2 ммоль) при 10°С. Полученную смесь перемешивали при той же температуре в течение 15 ч. Затем смесь разбавляли DCM (50 мл) и выливали в насыщенный водный раствор NaHCO3 (500 мл) при 0°C. Смесь экстрагировали DCM (500 мл × 2), и объединенные органические экстракты промывали насыщенным раствором соли (100 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде желтого масла. Неочищенный продукт непосредственно использовали без дополнительной очистки, допуская количественный выход. 1H ЯМР (400 МГц, CDCl3) δ 4,30-4,20 (м, 2H), 4,20-4,05 (м, 2H), 2,95-2,85 (м, 1H), 2,40-2,25 (м, 1H), 1,57 (с, 3H), 1,35-1,25 (м, 3H). 19F ЯМР (376 МГц, CD3OD) δ -101, -98,0.
Стадия 36E. Бензил (S)-4,4-дифтор-2-метилтетрагидрофуран-2-карбоксилат
К раствору этил 4,4-дифтор-2-метилтетрагидрофуран-2-карбоксилата (36,5 г, 140 ммоль) в ТГФ (1,5 л) при 15°С добавляли 1,0 М водный раствор LiOH (214 мл, 214 ммоль). Смесь перемешивали при 15°C в течение 2 ч, и затем концентрировали в вакууме. Остаток подкисляли 1,0 М водным раствором HCl до тех пор, пока pH раствора не достигал 3, и затем экстрагировали DCM (600 мл × 3) и DCM/MeOH (10/1, 600 мл × 2). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток растворяли в ацетонитриле (500 мл) и порциями добавляли K2CO3 (25 г, 181 ммоль). Затем по каплям добавляли бензилбромид (25 г, 146 ммоль). Полученную суспензию перемешивали при 60°C в течение 16 ч. Реакционную смесь гасили водой (500 мл) и экстрагировали EtOAc (500 мл × 3). Объединенные органические экстракты концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 25% EtOAc в петролейном эфире) с получением рацемического продукта в виде желтого масла (24,7 г, 80%). Часть рацемического продукта (5,0 г) подвергали хиральному разделению с использованием условий хиральной хроматографии с нормальной фазой (Phenomenex Lux 10 мкм Cellulose-3 45% EtOH в гексане, 20 мл/мин, 12 мин). Выделение второго пика элюирования давало 2,56 г указанного в заголовке соединения в виде бесцветного масла. ЖХМС m/z 274,2 [M+вода]. 1H ЯМР (400 МГц, CDCl3) δ 7,39-7,34 (м, 5H), 5,21 (с, 2H), 4,15-4,04 (м, 2H), 2,96-2,84 (м, 1H), 2,40-2,24 (м, 1H), 1,59 (с, 3H). 19F ЯМР (376 МГц, CDCl3) δ -101, -98,0.
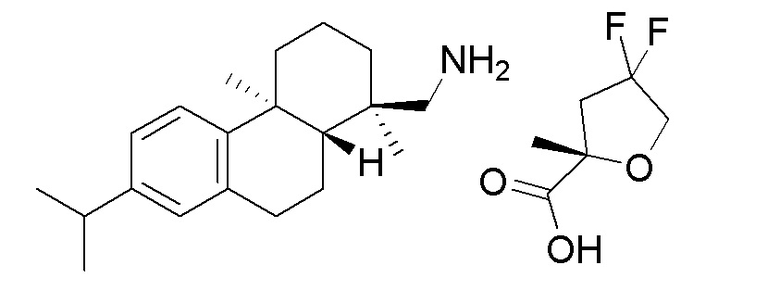
Стадия 36F. (S)-4,4-дифтор-2-метилтетрагидрофуран-2-карбоновая кислота ((1R,4aS,10aR)-7-изопропил-1,4a-диметил-1,2,3,4,4a,9,10,10a-октагидрофенантрен-1-ил) соль метанамина
Абсолютную стереохимию указанного в заголовке соединения определяли с помощью рентгеновской кристаллографии монокристаллов (фиг. 3). Бензил (S)-4,4-дифтор-2-метилтетрагидрофуран-2-карбоксилат гидролизовали до соответствующей кислоты и кристаллизовали с ((1R,4aS,10aR)-7-изопропил-1,4a-диметил-1,2, 3,4,4a, 9,10,10a-октагидрофенантрен-1-ил)метанамином из этанола.
Стадия 36G. (S)-(4,4-Дифтор-2-метилтетрагидрофуран-2-ил)(1,4-диметил-1H-пиразол-3-ил)метанон
К раствору 3-иод-1,4-диметил-1H-пиразола (препаративная стадия 1D)(394 мг, 1,77 ммоль) в ТГФ (3,23 мл) при 0°C по каплям добавляли i-PrMgCl⋅LiCl (1,37 мл, 1,77 ммоль, 1,3 М раствор в ТГФ). Полученный желтый раствор перемешивали при той же температуре в течение 2 ч, и затем нагревали до температуры окружающей среды и перемешивали в течение 30 мин. Смесь охлаждали до 0°C и добавляли бензил (S)-4,4-дифтор-2-метилтетрагидрофуран-2-карбоксилат (413 мг, 1,61 ммоль). Смесь перемешивали при 0°C в течение 30 мин, и затем подогревали до температуры окружающей среды и перемешивали в течение >12 ч. Реакционную смесь гасили насыщенным водным раствором NH4Cl и смесь экстрагировали DCM. Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 100% EtOAc в DCM) с получением 351 мг (89%) указанного в заголовке соединения в виде масла. ЖХМС m/z 245,3 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,16 (с, 1H), 4,20-4,05 (м, 2H), 3,90 (с, 3H), 3,34-3,23 (м, 1H), 2,62-2,51 (м, 1H), 2,29 (с, 3H), 1,78 (с, 3H).
Стадия 36H. (S, R)-((S)-4,4-Дифтор-2-метилтетрагидрофуран-2-ил)(1,4-диметил-1H-пиразол-3-ил)метанамин
К раствору (S)-(4,4-дифтор-2-метилтетрагидрофуран-2-ил)(1,4-диметил-1H-пиразол-3-ил)метанона (352 мг, 1,44 ммоль) в метанольном растворе аммиака (1,23 г, 10,3 мл, 72,1 ммоль, 7,0 M в MeOH) добавляли этоксид титана (IV)(1,97 г, 1,81 мл, 8,65 ммоль). Полученную суспензию герметично закрывали и перемешивали при 60°C в течение ночи. Реакционную смесь охлаждали до 0°C и добавляли NaBH4 (224 мг, 5,91 ммоль). Смесь перемешивали при 0°C в течение 30 мин, и затем при температуре окружающей среды в течение 1 ч. Реакционную смесь концентрировали в вакууме. К остатку добавляли HCl (1,0 Н водный раствор) до тех пор, пока смесь не стаовилась кислой, и затем экстрагировали Et2O. Водный слой нейтрализовали NaOH (1,0 Н водный раствор), и затем экстрагировали DCM (×2). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток растворяли в DCM (2 мл) и добавляли HCl (4 мл, 4,0 M раствор в MeOH). Раствор перемешивали в течение 10 мин и затем концентрировали в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества в виде соли моно-HCl. Продукт выделяли в виде смеси двух диастереомеров и непосредственно использовали без дополнительной очистки.
Стадия 36I. 4-((2-(((S)-((S)-4,4-дифтор-2-метилтетрагидрофуран-2-ил)(1,4-диметил-1H-пиразол-3-ил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
DIEA (131 мг, 1,02 ммоль) и (S, R)-((S)-4,4-дифтор-2-метилтетрагидрофуран-2-ил)(1,4-диметил-1H-пиразол-3-ил) метанамин (292 мг, 1,19 ммоль) добавляли к 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамиду (препаративная стадия 1G)( 330 мг, 1,08 ммоль) в EtOH (1,9 мл). Смесь перемешивали при комнатной температуре в течение 18 ч, и затем концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (от 0 до 10% MeOH в DCM). Полученную диастереомерную смесь разделяли с помощью хиральной SFC-хроматографии (Chiral Tech OD-H, 250 мм × 30 мм, 5 мкм, 25% этанол, содержащий 0,2% 7 Н раствора аммиак в метаноле, 80 мл/мин). Выделение первого элюированного пика давало 299 мг (55%) указанного в заголовке соединения в виде желтого твердого вещества. ЖХМС m/z 505,4 [M+H]+. 1H ЯМР (600 МГц, ДМСО-d6) 10,48 (шир. с, 2 H), 9,23 (шир. С, 1H), 7,99 (д, J=5,3 Гц, 1H), 7,91 (шир.с, 1H), 7,49 (с, 1H), 5,56 (д, J=8,4 Гц, 1H), 4,10-3,96 (м, 1H), 3,93-3,82 (м, 1H), 3,80 (с, 3H), 3,14 (шир. с, 3H), 3,03 (шир. с, 3H), 2,55-2,45 (м, 1H), 2,34-2,22 (м, 1H), 1,99 (с, 3H), 1,49 (с, 3H). Хиральная SFC (метод SFC A) RT=5,26 мин, 100% ее.
Примеры 37 и 38
Указанные в заголовке соединения в таблице 3 получали аналогично тому, как получали 4-((2-(((S)-(S)-4,4-дифтор-2-метилтетрагидрофуран-2-ил)(1,4-диметил-1H-пиразол-3-ил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид (пример 36) с использованием соответствующего гетероарилгалогенида.
Пример 39
(R)-3-гидрокси-N, N-диметил-4-((2-((((1-метил-4-(метил-d3)-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)пиколинамид
Стадия 39А. 4,4,5,5-Тетраметил-2-(метил-d3)-1,3,2-диоксаборолан
В высушенную и заполненную азотом колбу, снабженную холодильником и капельной воронкой, добавляли металлический Mg (1,26 г, 51,7 ммоль) и хлопья йода. Дно колбы нагревали с помощью теплового пистолета до тех пор, пока пурпурный газ не распространился по поверхности Mg. Добавляли Et2O (5 мл). Раствор перемешивали до исчезновения йодной окраски. Когда раствор стал бесцветным, медленно добавляли раствор йодметана-d3 (5,0 г, 34,49 ммоль) в Et2O (45 мл). После завершения добавления реакционную смесь кипятили с обратным холодильником в течение 20 мин. Образовавшийся реактив Гриньяра добавляли по каплям к раствору 2-метокси-4,4,5,5-тетраметил-1,3,2-диоксаборолана (5,45 г, 5,65 мл, 34,5 ммоль) в Et2O (20 мл) при температуре -60°C. Затем реакционной смеси давали подогреться до 20°C и перемешивали в течение 1 ч. Реакционную смесь гасили насыщенным NH4Cl и слои разделяли. Органический слой сушили (Na2SO4) и фильтровали. Фильтрат концентрировали с получением указанного в заголовке соединения, которое использовали без дополнительной очистки.
Стадия 39В. N-Метокси-N,1-диметилциклопентан-1-карбоксамид
К раствору 1-метилциклопентан-1-карбоновой кислоты (60 г, 470 ммоль) в DCM (1,5 л) добавляли HN(Me)(OMe)⋅HCl (50,2 г, 515 ммоль), TEA (194 г, 1,92 моль) и HATU (267 г, 702 ммоль) при 0°C. Смесь перемешивали при 25°C в течение 15 ч, и затем концентрировали. Остаток добавляли к насыщенному водному раствору Na2CO3 (1 л) и экстрагировали EtOAc (2 × 1 л). Объединенные органические экстракты сушили (Na2SO4) и фильтровали. Фильтрат концентрировали и остаток очищали перегонкой с получением 62,1 г (77%) указанного в заголовке соединения в виде бесцветного масла. ЖХМС 193,2 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 3,69 (с, 3H), 3,20 (с, 3H), 2,15-2,01 (м, 2H), 1,73-1,54 (м, 6H), 1,26 (с, 3H).
Стадия 39C. (1-Метил-1H-пиразол-3-ил)(1-метилциклопентил)метанон
3-Иод-1-метил-1H-пиразол (2,19 г, 10,5 ммоль) добавляли по каплям к iPrMgCl·LiCl (10,8 мл, 14,0 ммоль, 1,3 М раствор в ТГФ) и ТГФ (14 мл) охлаждали до 0°С в атмосфере азота. Смесь оставляли перемешиваться при комнатной температуре в течение 1 ч. Затем по каплям добавляли N-метокси-N,1-диметилциклопентан-1-карбоксамид (1,2 г, 7,0 ммоль). Смесь перемешивали при комнатной температуре в течение 18 ч, и затем гасили насыщенным водным NH4Cl. Реакционную смесь экстрагировали EtOAc. Органический экстракт сушили (Na2SO4) и фильтровали. Фильтрат концентрировали и остаток очищали хроматографией на колонке с силикагелем (10-50% EtOAc/ гептан) с получением 992 мг (74%) указанного в заголовке соединения. ЖХМС 172,4 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,34 (д, J=2,3 Гц, 1H), 6,81 (д, J=2,3 Гц, 1H), 3,98 (с, 3H), 2,46-2,33 (м, 2H), 1,79 -1,63 (м, 6H), 1,49 (с, 3H).
Стадия 39D. (4-Иод-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метанон
NIS (702 мг, 3,12 ммоль) добавляли к раствору (1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метанона (400 мг, 2,08 ммоль) в уксусной кислоте (4,2 мл), и смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь концентрировали и остаток очищали хроматографией на колонке с силикагелем (0-20% EtOAc/гептан) с получением 595 мг (90%) указанного в заголовке соединения в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,47 (с, 1H), 3,98 (с, 3H), 2,43-2,32 (м, 2H), 1,80-1,60 (м, 6H), 1,47 (с, 3H).
Стадия 39E. (1-Метил-4-(метил-d3)-1H-пиразол-3-ил)(1-метилциклопентил)метанон
Флакон, содержащий (4-иод-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метанон (532 мг, 1,67 ммоль), 4,4,5,5-тетраметил-2-(метил-d3)-1,3,2-диоксаборолан (1,7 г, 11,7 ммоль), диоксан (8 мл) и водный раствор K3PO4 (3,55 г, 16,7 ммоль, 8,36 мл, 2,0 М) продували газообразным аргоном в течение 5 мин. Добавляли P(tBu)3 Pd-G2 (177 мг, 0,33 ммоль) и смесь нагревали до 100°C в течение 4 ч. Реакционную смесь разделяли с EtOAc. Органический слой сушили (Na2SO4) и фильтровали. Фильтрат концентрировали и остаток очищали хроматографией на колонке с силикагелем (смесь 50% DCM/гептан) с получением 185 мг (53%) указанного в заголовке соединения. 1H ЯМР (400 МГц, CDCl3) δ 7,14 (с, 1H), 3,90 (с, 3H), 2,43-2,32 (м, 2H), 1,78-1,61, (м, 6H), 1,49 (с, 3H).
Стадия 39F. (1-Метил-4-(метил-d3)-1H-пиразол-3-ил)(1-метилциклопентил)метаноноксим
Флакон, содержащий NaOAc (129 мг, 1,58 ммоль), гидроксиламина гидрохлорид (219 мг, 3,15 ммоль), (1-метил-4-(метил-d3)-1H-пиразол-3-ил)(1-метилциклопентил)метанон (165 мг, 0,788 ммоль) и EtOH (3,94 мл) нагревали при 95°C в течение 2 ч и затем охлаждали до комнатной температуры. Растворитель удаляли и остаток распределяли между DCM и насыщенным водным раствором NaCl. Слои разделяли, и водный слой экстрагировали DCM (2 × 20 мл). Объединенные органические экстракты сушили (Na2SO4) и фильтровали. Фильтрат концентрировали с получением 212 мг указанного в заголовке соединения в виде твердого вещества. ЖХМС m/z 225,2 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,21 (с, 1H), 3,89 (с, 3H), 2,11-1,99 (м, 2H), 1,77-1,60 (м, 4H), 1,53-1,40 (м, 2H), 1,25 (с, 3H).
Стадия 39G. (1-Метил-4-(метил-d3)-1H-пиразол-3-ил)(1-метилциклопентил)метанамин
HOAc (3 мл), и затем Zn (311 мг, 4,76 ммоль) добавляли в колбу, содержащую (1-метил-4-(метил-d3)-1H-пиразол-3-ил)(1-метилциклопентил)метаноноксим (178 мг, 0,79 ммоль). Смесь нагревали при 60°C в течение ночи. После охлаждения до комнатной температуры реакционную смесь фильтровали через целит и промывали HOAc. Фильтрат концентрировали и подвергали азеотропной перегонке с толуолом (3 раза). Остаток разбавляли DCM и Et2O, и pH доводили примерно до pH 14 с помощью 50% водного раствора NaOH. Слои разделяли, и водный слой дважды экстрагировали смесью DCM/Et2O (2:8). Объединенные органические экстракты сушили (Na2SO4) и фильтровали. Фильтрат концентрировали с получением 145 мг (87%) указанного в заголовке соединения. ЖХМС m/z 194,2 [M-NH2]+. 1H ЯМР (400 МГц, CDCl3) 7,05 (с, 1H), 3,81 (с, 3H), 3,55-3,47 (м, 1H), 1,81-1,14 (м, 8H), 1,05 (с, 3H).
Стадия 39H. (R)-3-Гидрокси-N, N-диметил-4-((2-(((1-метил-4- (метил-d3)-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)пиколинамид
EtOH (3,0 мл), 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид (препаративная стадия 1H)(842 мг, 0,7 ммоль) и DIEA (267 мг, 0,36 мл, 2,07 ммоль) добавляли в колбу, содержащую (1-метил-4-(метил-d3)-1H-пиразол-3-ил)(1-метилциклопентил)метанамин (145 мг, 0,69 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали и фильтрат концентрировали. Остаток очищали хроматографией на колонке с силикагелем (6% MeOH/DCM) с получением рацемического продукта. Рацемат разделяли с помощью хиральной SFC-хроматографии (Chiralcel OD, 250 × 30 мм, 5 мкм, 80 мл/мин, 25% MeOH, содержащий 0,2% 7 Н аммиак/MeOH). Выделение первого элюированного энантиомера давало 70 мг указанного в заголовке соединения. ЖХМС m/z 470,5 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,70 (с, 1H), 9,92 (с, 1H), 9,13 (д, J=10,1 Гц, 1H), 8,02 (с, 2H), 7,44 (с, 1H), 5,35 (д, J=10,1 Гц, 1H), 3,81 (с, 3H), 3,22-3,07 (м, 6H), 1,72-1,60 (м, 6H), 1,39-1,12 (м, 2H), 1,10 (с, 3Н). Второй элюированный энантиомер выделяли с получением 70 мг соответствующего (S)-энантиомера.
Пример 40
4-((2-(((1R)-(1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил-3,4-d2)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
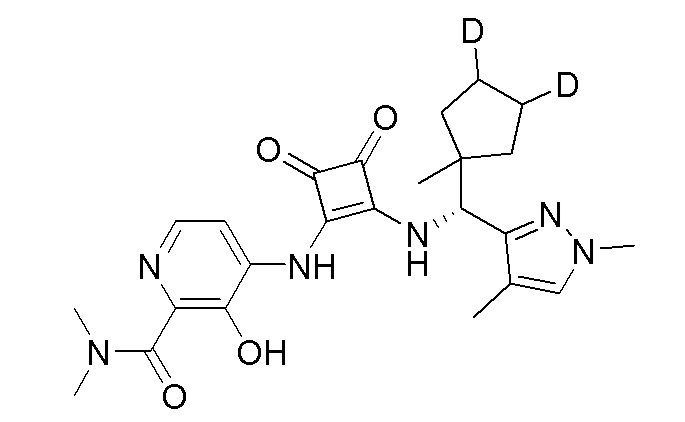
Стадия 40А. Циклопент-3-ен-1-карбоновая кислота
К раствору метил 1-метилциклопент-3-ен-1-карбоксилата (50 г, 396 ммоль) в ТГФ:H2O (500 мл:125 мл) при 25°C добавляли LiOH (19 г, 793 ммоль). Через 15 ч реакционную смесь экстрагировали EtOAc. Затем водный слой подкисляли до pH 3 с использованием 1 Н HCl и экстрагировали EtOAc (3 × 1 л). Объединенные органические экстракты объединяли, сушили (Na2SO4) и фильтровали. Фильтрат концентрировали с получением 40 г (90%) указанного в заголовке соединения в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 5,68 (с, 2H), 3,29-3,03 (м, 1H), 2,82 2,53 (м, 4H).
Стадия 40В. 1-Метилциклопент-3-ен-1-карбоновая кислота
К раствору LDA (2M в смесм ТГФ/гептан/этилбензол, 446 мл) в ТГФ (800 мл) по каплям добавляли раствор циклопент-3-ен-1-карбоновой кислоты (40 г, 357 ммоль) в ТГФ (200 мл), поддерживая температуру при -30°C. Смесь нагревали до 25°C и перемешивали в течение 15 ч. Затем смесь охлаждали до -30°C и добавляли йодметан (50,6 г, 22,2 мл, 357 ммоль). Через 2 ч при 25°C смесь гасили 3 М HCl и экстрагировали EtOAc (3 × 1 л). Объединенные органические экстракты сушили (Na2SO4) и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем (EtOAc) с получением 45 г (100%) указанного в заголовке соединения в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 5,64 (с, 2H), 2,98 (д, J=14,7 Гц, 2H), 2,28 (д, J=14,7 Гц, 2H), 1,36 (с, 3H).
Стадия 40C. N-Метокси-N,1-диметилциклопент-3-ен-1-карбоксамид
Раствор 1-метилциклопент-3-ен-1-карбоновой кислоты (45 г, 357 ммоль) в DCM (1 л) при 0°C добавляли HN(Me)(OMe) ⋅ HCl (38,3 г, 392 ммоль), TEA (148 г, 1,46 моль) и HATU (203 г, 535 ммоль). После перемешивания при комнатной температуре в течение 24 ч реакционную смесь концентрировали при пониженном давлении. Остаток разбавляли EtOAc (1 л) и промывали насыщенным водным Na2CO3. Водный слой дополнительно экстрагировали EtOAc (1 л). Объединенные органические экстракты сушили (Na2SO4) и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем (смесь PE:EtOAc, от 20:1 до 5:1) с получением 23,4 г (39%) указанного в заголовке соединения в виде светло-желтого масла. ЖХ/МС m/z 170,3 [M+H]+; 1H ЯМР (400 МГц, CDCl3) δ 5,59 (с, 2H), 3,69 (с, 3H), 3,20 (с, 3H), 2,88 (д, J=15,1 Гц, 2H), 2,23 (д, J=15,1 Гц, 2H), 1,27 (с, 3H).
Стадия 40D. (1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопент-3-ен-1-ил)метанон
Круглодонную колбу объемом 125 мл с мешалкой и перегородкой снова заполняли азотом три раза, после чего добавляли ТГФ (7,0 мл), и затем 3-иод-1,4-диметил-1H-пиразол (препаративная стадия 1D)(917 мг, 4,13 ммоль). К этому раствору по каплям добавляли 1,3 М раствор iPrMgCl⋅LiCl в ТГФ (5,45 мл) и смесь перемешивали в течение 1 ч при комнатной температуре. По каплям добавляли N-метокси-N,1-диметилциклопент-3-ен-1-карбоксамид (600 мг, 3,55 ммоль) в ТГФ (5 мл). После перемешивания в течение 2 ч реакционную смесь гасили насыщенным водным NH4Cl (20 мл) и экстрагировали DCM (3 × 25 мл). Объединенные экстракты сушили (Na2SO4) и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (EtOAc в гептане, 0-100%) с получением 500 мг (69%) указанного в заголовке соединения в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 7,14 (с, 1H), 5,70-5,64 (м, 2H), 3,90 (с, 3H), 3,20-3,12 (м, 2H), 2,44-2,36 (м, 2H), 2,30 (с, 3H), 1,52 (с, 3H).
Стадия 40E. N-((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопент-3-ен-1-ил)метил)формамид
В колбу добавляли формамид (10,5 мл) и муравьиную кислоту (6 мл), содержащую (1,4-диметил-1H-пиразол-3-ил)(1-метилциклопент-3-ен-1-ил)метанон (1,1 г, 5,4 ммоль). Смесь нагревали при 135°C в течение 20 ч, и затем охлаждали до комнатной температуры. Добавляли воду (60 мл) и смесь экстрагировали МТВЕ (3 × 50 мл). Объединенные органические экстракты сушили (Na2SO4) и фильтровали. Фильтрат концентрировали с получением указанного в заголовке соединения, которое использовали на следующей стадии без очистки. ЖХ/МС m/z=234,0 [M+H]+.
Стадия 40F. (1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопент-3-ен-1-ил)метанамин
1 Н водный раствор HCl (24 мл) добавляли в колбу, содержащую N-((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопент-3-ен-1-ил)метил)формамид (1,26 г, 5,4 ммоль). Смесь нагревали при 100°C в течение 2 ч, и затем охлаждали до комнатной температуры. pH раствора доводили примерно до pH 11 твердым NaOH. Смесь экстрагировали МТВЕ (3 × 30 мл). Объединенные органические экстракты сушили (Na2SO4) и фильтровали. Фильтрат концентрировали с получением 1,1 г (100%) указанного в заголовке соединения. ЖХМС m/z 189,1 [M-NH2]+. 1H ЯМР (400 МГц, CD3OD) δ 7,27 (с, 1H), 5,66-5,58 (м, 2H), 3,86 (с, 1H), 3,82 (с, 3H), 2,67-2,51 (м, 2H), 2,09- 1,97 (м, 4H), 1,93-1,83 (м, 1H), 1,10 (с, 3H).
Стадия 40G. 4-((2-(((R)-(1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопент-3-ен-1-ил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
Смесь 4-((2-этокси-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид (препаративная стадия 1H)(300 мг, 0,983 ммоль), (1,4-диметил-1H-пиразол-3-ил)(1-метилциклопент-3-ен-1-ил)метанамин ( 242 мг, 1,18 ммоль), этанол (9,83 мл) и диизопропилэтиламин (2,65 мл, 14,7 ммоль) перемешивали при 40°C в течение ночи. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (0-20% MeOH в DCM) с получением 450 мг рацемического продукта. Энантиомеры разделяли с помощью хиральной SFC (Chiral Tech OD-H, 250 мм × 21,2 мм, 5 мкм, 22,5% метанола, содержащего 0,2% 7 Н аммиак/МеОН, 80 мл/ мин). Выделение первого элюированного энантиомера давало 80 мг указанного в заголовке соединения. ЖХМС m/z 465,4 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 12,84 (шир. с, 1H), 8,27 (шир. с, 1H), 8,02 (д, J=5,1 Гц, 1H), 7,97 (шир. с, 1H) , 7,22 (шир. с, 1H), 7,11 (шир. с, 1H), 5,63 (шир. с, 2H), 5,50 (шир. с, 1H), 3,81 (с, 3H), 3,61 (шир. с, 3H), 3,19 (шир. с, 3H), 2,78-2,64 (м, 2H), 2,15-2,02 (м, 5H), 1,21 (с, 3H). Второй элюированный энантиомер выделяли с получением 100 мг соответствующего (S)-энантиомера.
Стадия 40H. 4-((2-(((1R)-(1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил-3,4-d2)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид
В реактор Парра с мешалкой добавляли 4-((2-(((R)-(1,4-диметил-1H-пиразол-3-ил)(1-метилциклопент-3-ен-1-ил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид (20 мг, 0,043 ммоль), 10% Pd/C (6,05 мг) и MeOH (10 мл). Реактор дегазировали азотом и затем загружали газообразным D2 до 10 фунтов/кв. дюйм. Реакционную смесь перемешивали в течение 16 ч, и затем фильтровали через целит. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем (0-20% MeOH/ DCM) с получением 16 мг (80%) указанного в заголовке соединения в виде желтого твердого вещества. ЖХМС m/z 469,4 [M+H]+. 1H ЯМР (400 МГц, ДМСО d6) δ 11,75 (шир. с, 1H), 9,96 (шир. с, 1H), 9,17 (шир. с, 1H), 8,05-7,89 (м, 2H), 7,44 (с, 1H), 5,35 (д, J=10,2 Гц, 1H), 3,81 (с, 3H), 3,20 (шир. с, 3H), 3,07 (шир. с, 3H), 2,00 (с, 3H), 1,72-1,55 (м, 4H), 1,37-1,31 (м, 1H), 1,25-1,19 (м, 1H), 1,10 (с, 3H).
Пример 41
(R)-((4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-2-(диметилкарбамоил)пиридин-3-ил)окси)метилдигидрофосфат
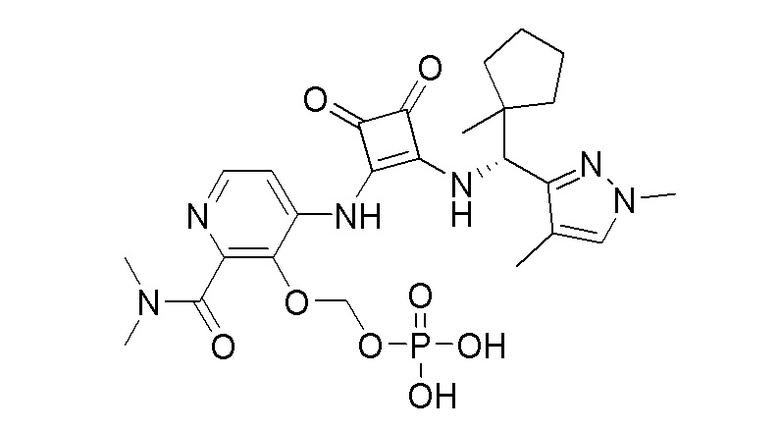
Стадия 41А. (R)-ди-трет-Бутил (((4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут -1-ен-1-ил)амино)-2-(диметилкарбамоил)пиридин-3-ил)окси)метил) фосфат
K2CO3 (89 мг, 0,643 ммоль), TBAI (119 мг, 0,322 ммоль) и ди-трет-бутил (хлорметил)фосфат (0,65 мл, 0,75 ммоль) при комнатной температуре добавляли к раствору (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид (пример 1)(250 мг, 0,536 ммоль) в ДМФА. Реакционную смесь перемешивали при комнатной температуре в течение 2 суток, и затем добавляли смесь толуол:H2O (4 мл:2 мл). Слои разделяли, и органический слой промывали насыщенным раствором соли, H3PO4 (1 М раствор) и 10% Na2CO3. Органический слой концентрировали. Остаток помещали в смесь толуол:МТВЕ (1 мл:20 мл) и помещали в холодильник. Твердый осадок собирали и фильтрат концентрировали. К фильтрату добавляли гептан для осаждения твердого вещества, которое отфильтровывали, собирали и сушили в высоком вакууме с получением 140 мг (38%) указанного в заголовке соединения. ЖХМС 689,3 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 10,08 (с, 1H), 8,43-8,08 (м, 3H), 6,95 (с, 1H), 5,57 (с, 2H), 5,39 (д, J=10,2 Гц, 1H) , 3,68 (с, 3H), 3,06 (с, 3H), 2,75 (с, 3H), 2,00 (с, 3H), 1,82-1,53 (м, 6H), 1,45 (с, 9H), 1,43 (с, 9H)), 1,41-1,18 (м, 2H), 1,09 (с, 3H).
Стадия 41В. (R)-((4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-2-(диметилкарбамоил)пиридин-3-ил)окси)метилдигидрофосфат
К раствору (R)-ди-трет-бутил (((4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-2-(диметилкарбамоил)пиридин-3-ил)окси)метил)фосфата (80 мг, 0,12 ммоль) в диоксане (1 мл) добавляли 4 Н HCl в диоксане (3 мл). После перемешивания в течение 45 ч растворитель удаляли с получением 73 мг (100%) указанного в заголовке соединения в виде соли HCl. ЖХМС 577,5 [M+H]+. 1H ЯМР (400 МГц, CD3OD) δ 8,86 (д, J=7,0 Гц, 1H), 8,49 (д, J=6,6 Гц, 1H), 7,78 (с, 1H), 5,75-5,63 (м, 1H), 5,52 -5,40 (м, 2H), 4,01 (с, 3H), 3,22 (с, 3H), 3,04 (с, 3H), 2,23 (с, 3H), 1,84-1,74 (м, 6H), 1,66-1,60 (м , 1H), 1,50-1,41 (м, 1H), 1,22 (с, 3H).
Пример 42
(R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид гемисоль кальция, моногидрат
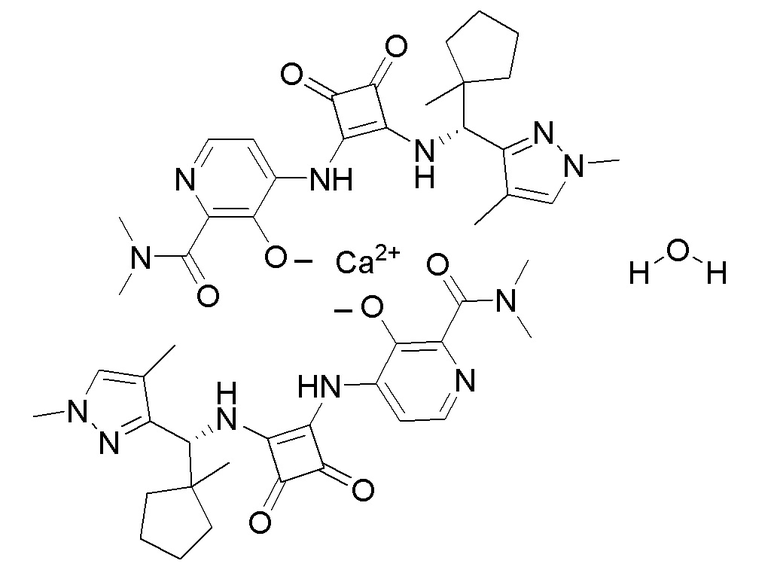
Метод A. Гидроксид кальция (7,94 мг, 0,107 ммоль) добавляли во флакон на 7 мл с мешалкой, содержащий (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид (пример 1)(100 мг, 0,214 ммоль) и МеОН (0,75 мл). Смесь нагревали при 55°C в течение 2 ч, и затем охлаждали до комнатной температуры и перемешивали в течение 72 ч. Полученную суспензию фильтровали с получением указанного в заголовке соединения в виде желтых кристаллов, имеющих дифрактограмму PXRD, соответствующую дифрактограмме PXRD, приведенной на фиг. 12. Если желаемая дифрактограмма PXRD не была получена, то полученную соль помещали в MeOH (1 г вещества/10 мл MeOH) и суспендировали до тех пор, пока не наблюдали дифрактограмму PXRD, соответствующую приведенной на фиг. 12.
Метод B. (R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид (пример 1)(6,0 г, 12,86 ммоль), МеОН (45 мл) и воду (3,6 мл), и затем гидроксид кальция (476 мг, 6,43 ммоль) добавляли в круглодонную колбу на 250 мл с мешалкой. Суспензию перемешивали при 45°C в течение 4 ч, и затем при комнатной температуре в течение 4 суток. Суспензию фильтровали. Затем твердые частицы добавляли в круглодонную колбу емкостью 125 мл с мешалкой и суспендировали в МеОН (50 мл). В смесь вносили затравку 1 мг указанного в заголовке соединения, имеющего дифрактограмму PXRD, соответствующую приведенной на фигуре 12. Смесь перемешивали при комнатной температуре в течение 20 ч, и затем фильтровали с получением 4,21 г указанного в заголовке соединения в виде твердого вещества слегка желтого цвета, которое, как было установлено, имело рентгенограмму, приведенную на фиг. 12. ЖХМС m/z 467,5 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,19 (шир. д, J=8,2 Гц, 1H), 7,66 (д, J=4,7 Гц, 1H), 7,40 (с, 1H), 7,35 (м, 1H), 5,37 (д, J=9,8 Гц, 1 H), 3,77 (с, 3H), 3,06 (с, 3H), 2,00 (с, 3H), 1,99 (с, 3H), 1,72-1,59 (м, 6H), 1,22-1,20 (м, 1H), 1,19-1,18 (м, 1H), 1,09 (с, 3H).
Рентгеноструктурный анализ монокристаллов кристаллического (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобута-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата
Кристалл, подходящий для рентгеноструктурного анализа, получали перекристаллизацией из этанола.
Сбор данных проводили на дифрактометре Bruker D8 Venture при комнатной температуре. Сбор данных состоял из омега- и фи- сканирования. Структуру расшифровали методом внутреннего фазирования с использованием программного пакета SHELX в тетрагональной пространственной группе класса P4(3). В дальнейшем структуру уточняли полноматричным методом наименьших квадратов. Все неводородные атомы были найдены и уточнены с использованием параметров анизотропного смещения. Атомы водорода, расположенные на азоте и кислороде, были найдены из карты разностной электронной плотности метода Фурье и уточнены с ограниченными расстояниями. Остальные атомы водорода были размещены в расчетных положениях и позволили им перемещаться на своих атомах-носителях. Конечное уточнение включало параметры изотропного смещения для всех атомов водорода.
Анализ абсолютной структуры с использованием методов правдоподобия (Hooft, 2008) выполняли с использованием PLATON (Spek, 2010). Полагая, что представленный образец является энантиочистым, результаты показывают, что абсолютная структура была определена правильно. Итоговый R-индекс составил 4,3%. Конечное различие Фурье не выявило отсутствующей или смещенной электронной плотности.
На фиг. 1 представлена полученная с помощью рентгеноструктурного анализа структура (ORTEP-диаграмма) кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино))-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата. Данные кристаллической структуры приведены в таблице 4.
Кристалографические данные и уточнение кристаллической структуры
Рентгеноструктурный анализ монокристаллов кристаллического (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата
Кристалл, подходящий для рентгеновского анализа, был выращен из смеси гептана, этилацетата, ацетона и DCM посредством медленного испарения в течение 48 ч.
Сбор данных проводили на дифрактометре Bruker D8 Venture при комнатной температуре. Сбор данных состоял из омега- и фи- сканирования. Структуру расшифровали методом внутреннего фазирования с использованием программного пакета SHELX в тетрагональной пространственной группе класса P43. В дальнейшем структура была уточнена полноматричным методом наименьших квадратов. Все неводородные атомы были найдены и уточнены с использованием параметров анизотропного смещения. Вода была обнаружена из карты разностной электронной плотности во время уточнения, моделированной с заполнением 0,33. Атомы водорода, связанные с водой, не были включены в уточнение. Атомы водорода, расположенные на азоте и кислороде, были найдены из карты разностной электронной плотности метода Фурье и уточнены с ограниченными расстояниями. Остальные атомы водорода были размещены в расчетных положениях и позволили им перемещаться на своих атомах-носителях. Конечное уточнение включало параметры изотропного смещения для всех атомов водорода.
Анализ абсолютной структуры с использованием методов правдоподобия (Hooft, 2008) выполняли с использованием PLATON (Spek, 2010). Полагая, что представленный образец является энантиочистым, результаты показывают, что абсолютная структура была определена правильно. Итоговый R-индекс составил 6,4%. Конечное различие Фурье не выявило отсутствующей или смещенной электронной плотности.
На фиг. 2 представлена полученная с помощью рентгеноструктурного анализа структура (ORTEP-диаграмма) кристаллического (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил))амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата. Данные кристаллической структуры приведены в таблице 5.
Кристалографические данные и уточнение кристаллической структуры
Рентгеноструктурный анализ монокристаллов кристаллической (S)-4,4-дифтор-2-метилтетрагидрофуран-2-карбоновой кислоты ((1R, 4aS,10aR)-7-изопропил-1,4a-диметил-1,2,3,4,4a,9,10,10a-октагидрофенантрен-1-ил)метанамина соли (пример 36F)
Кристалл, подходящий для рентгеноструктурного анализа, получали перекристаллизацией из этанола.
Сбор данных проводили на дифрактометре Bruker D8 Venture при комнатной температуре. Сбор данных состоял из омега- и фи- сканирования. Структуру расшифровали методом внутреннего фазирования с использованием программного пакета SHELX в моноклинной пространственной группе класса P21. В дальнейшем структура была уточнена полноматричным методом наименьших квадратов. Все неводородные атомы были найдены и уточнены с использованием параметров анизотропного смещения. Атомы водорода, расположенные на азоте и кислороде, были найдены из карты разностной электронной плотности метода Фурье и уточнены с ограниченными расстояниями. Остальные атомы водорода были размещены в расчетных положениях и позволили им перемещаться на своих атомах-носителях. Конечное уточнение включало параметры изотропного смещения для всех атомов водорода.
Стереохимия основана на известных хиральных конфигурациях центров C4(-R), C7(-R) и C8(-S) основания. Конечный R-индекс составил 7,6%. Конечное различие Фурье не выявило отсутствующей или смещенной электронной плотности.
На фиг. 3 представлена полученная с помощью рентгеноструктурного анализа структура (ORTEP-диаграмма) кристаллической (S)-4,4-дифтор-2-метилтетрагидрофуран-2-карбоновой кислоты ((1R,4aS,10aR)-7-изопропил-1,4a-диметил-1,2,3,4,4a,9,10,10a-октагидрофенантрен-1-ил)метанаминовой соли. Данные кристаллической структуры приведены в таблице 6.
Кристалографические данные и уточнение кристаллической структуры
Анализ порошковой рентгеновской дифракцией кристаллического (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобута-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата
Анализ порошковой рентгеновской дифракцией проводили с использованием дифрактометра Bruker AXS D8 Advance, снабженного источником излучения Cu. Дифрагированное излучение регистрировали на детекторе LYNXEYE_EX с моторизованными щелями. Как первичный, так и вторичный детекторы имели щели Соллера 2,5. Напряжение и сила тока рентгеновской трубки были установлены на 40 кВ и 40 мА соответственно. Данные собирали в гониометре Theta-Theta при сканировании заблокированной пары при длине волны Cu K-альфа от 3,0 до 40,0 градусов 2-тета с шагом 0,01 градуса, с использованием скорости сканирования 1,0 с на шаг. Образцы готовили их помещением в кремниевый низкофоновый держатель образцов (номер Bruker: C79298A3244B261). Данные собирали и анализировали с помощью программного обеспечения Bruker DIFFRAC Plus, и анализ выполняли с помощью программного обеспечения EVA Diffrac Plus (v4.2.1.10). Обычно для предварительного определения пиков использовали пороговое значение 1 и значение ширины 0,3. Для обеспечения достоверности поправки вносили вручную; результаты автоматических назначений проверяли визуально, и положения пиков доводили до максимума. Обычно выбирали пики с относительной интенсивностью ≥ 3%. Пики, которые не были разрешены или соответствовали шуму, не выбирались. Типичная ошибка, связанная с положением пика кристаллического вещества, из PXRD, указанная в USP, составляет до +/- 0,2° 2-тета (USP-941).
На фиг. 4 представлена полученная рентгеновская порошковая дифрактограмма кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата, и в таблице 7 приведены дифракционные пики в значениях 2-тета.
2-тета
(градусы)
(%)
Анализ дифференциальной сканирующей калориметрией кристаллического (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата
Измерения с помощью дифференциального сканирующего калориметра (DSC) выполняли с помощью прибора Discovery DSC (ТА instruments), оснащенного охлаждающим устройством. Все эксперименты проводили в алюминиевых тиглях standard/Tzero. Константу ячейки определяли с использованием индия, и калибровку температуры выполняли с использованием индия и олова в качестве стандартов. Все измерения проводили при непрерывной продувке сухим азотом (50 мл/мин). Навеску твердого образца примерно 1-5 мг помещали в алюминиевый тигль Tzero, герметично закрывали и нагревали от 25°C до, по меньшей мере, 250°C со скоростью нагревания 10°C/мин. Экспериментальные данные анализировали с использованием комерчески доступного программного обеспечения (программное обеспечение TA Universal Analysis 2000/Trios, TA Instruments).
На фиг. 5 представлены полученные результаты анализа дифференциальной сканирующей калориметриетрией для кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
Термогравиметрический анализ кристаллического (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
Термогравиметрический анализ проводили на термогравиметрическом анализаторе Discovery TGA (TA instruments). Навеску образца примерно 10 мг помещали в алюминиевые тигли и нагревали от температуры окружающей среды до, по меньшей мере, 250°C со скоростью нагревания 10°C/мин при продувке азотом (10 мл/мин для камеры для образцов и весов).
На фиг. 6 представлены полученные результаты термогравиметрического анализа кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
Анализ порошковой рентгеновской дифракцией кристаллического (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата
Анализ порошковой рентгеновской дифракцией проводили, как описано выше. На фиг. 7 представлена полученная рентгеновская порошковая дифрактограмма кристаллического (R)-4-(2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино))-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата, и в таблице 8 приведены дифракционные пики в значениях 2-тета.
(%)
Анализ дифференциальной сканирующей калориметрией кристаллического (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата
Измерения DSC выполняли, как описано выше.
На фиг. 8 представлены полученные результаты анализа дифференциальной сканирующей калориметрией кристаллического (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
Термогравиметрический анализ кристаллического (R)-4-((2-(((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата
Термогравиметрический анализ выполняли, как описано выше.
На фиг. 9 представлены полученные результаты термогравиметрического анализа кристаллического (R)-4-((2-((((4-хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида моногидрата.
Рентгеноструктурный анализ монокристаллов кристаллического (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобута-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата
Твердый гидроксид кальция (0,978 мг, 0,0132 ммоль, 0,50 экв) вносили во флакон для ВЭЖХ с мешалкой. Раствор (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида (пример 1)(0,504 мл, 0,0265 ммоль, 1,00 экв, 25,4 мг/мл) в метаноле вносили пипеткой во флакон. Смесь перемешивали при 60°C в течение 3 ч. За это время образовалось желтое твердое вещество. Перемешивание останавливали, и смесь медленно охлаждали до 21°C со скоростью -0,1°C/мин. Желтое твердое вещество (порошок) все еще присутствовало после охлаждения до 21°C. Твердому веществу давали отстояться в маточном растворе в течение нескольких недель при комнатной температуре до образования подходящих кристаллов.
Сбор данных проводили на дифрактометре Bruker D8 Venture при комнатной температуре. Сбор данных состоял из омега- и фи- сканирования. Структуру расшифровали методом внутреннего фазирования с использованием программного пакета SHELX в пространственной группе Р21212. В дальнейшем структуру уточняли полноматричным методом наименьших квадратов. Все неводородные атомы были найдены и уточнены с использованием параметров анизотропного смещения. Атомы водорода, расположенные на азоте и кислороде, были найдены из карты разностной электронной плотности метода Фурье и уточнены с ограниченными расстояниями. Остальные атомы водорода были размещены в расчетных положениях и позволили им перемещаться на своих атомах-носителях. Конечное уточнение включало параметры изотропного смещения для всех атомов водорода.
Анализ абсолютной структуры с использованием методов правдоподобия (Hooft, 2008) выполняли с использованием PLATON (Spek, 2010). Полагая, что представленный образец является энантиочистым, результаты показывают, что абсолютная структура была определена правильно. Итоговый R-индекс составил 6,92%. Конечное различие Фурье не выявило отсутствующей или смещенной электронной плотности.
На фиг. 10 представлена полученная с помощью рентгеноструктурного анализа структура (ORTEP-диаграмма) кристаллического (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино))-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата. На фиг. 11 представлена полученная с помощью рентгеноструктурного анализа структура (ORTEP-диаграмма), соответствующая элементарному кристаллу. Данные кристаллической структуры приведены в таблице 9.
Кристалографические данные и уточнение кристаллической структуры
Анализ порошковой рентгеновской дифракцией монокристаллов кристаллического (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобута-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата
Анализ порошковой рентгеновской дифракцией проводили с использованием дифрактометра Bruker AXS D4 Endeavour, снабженного источником излучения Cu. Щель расходимости устанавливали на 0,6 мм, в то время как во вторичной оптике использовали переменные щели. Дифрагированное излучение регистрировали на детекторе PSD-Lynx Eye. Напряжение и сила тока рентгеновской трубки были установлены на 40 кВ и 40 мА соответственно. Данные собирали в гониометре Theta-2Theta при Cu (среднее k-альфа) от 3,0 до 40,0 градусов 2-тета с использованием размера шага 0,01 градуса и времени 1,0 с на шаг. Образцы готовили их помещением в кремниевый низкофоновый держатель образцов и вращали во время сбора. Данные собирали с помощью Bruker DIFFRAC Plus XRD Commander (версия 2.6.1), и анализ выполняли с помощью программного обеспечения EVA diffract plus (версия 3.1). Файл данных PXRD не обрабатывался до поиска пика. Используя алгоритм поиска пиков в программном обеспечении EVA, пики, выбранные с пороговым значением 1, использовали для предварительного отнесения пиков. Для обеспечения достоверности поправки вносили вручную. Результаты автоматических назначений проверяли визуально, и положения пиков доводили до максимума. Обычно выбирали пики с относительной интенсивностью ≥ 3%. Пики, которые не были разрешены или соответствовали шуму, не выбирались. Типичная ошибка, связанная с положением пика кристаллического вещества, из PXRD, указанная в USP, составляет до +/- 0,2° 2-тета (USP-941).
На фиг. 12 представлена полученная рентгеновская порошковая дифрактограмма кристаллического кристаллического (R)-4-((2-((((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамида гемисоли кальция, моногидрата, и в таблице 10 приведены дифракционные пики в значениях 2-тета.
2-тета
(градусы)
(%)
Биологические данные
Анализ хемотаксиса CCR6+ Т-клеток
CD4+ CCR6+ CXCR3- T-клетки человека выделяли из лейкопака от здоровых доноров с использованием набора EasySep™ Human Th17 Cell Enrichment Kit (StemCell Technologies, 18162). Для получения больших количеств клеток, CCR6+ Т-клетки активировали с использованием магнитных частиц Dynabeads Human T-activator (номер по каталогу 11132D, Gibco) при плотности 1×106 клеток/мл в культуральной среде (среда RPMI1640 с 10% сывороткой, 4 нг/мл IL-2) с соотношением клеток к частицам 1:1,5. На сутки 4 после активации частицы Dynabeads удаляли из культуры. Активированные Т-клетки поддерживали на уровне 1-2×106 клеток/мл в течение 15 суток подпиткой свежей культуральной среды, когда это было необходимо.
Анализ хемотаксиса CCR6+ Т-клеток проводили на сутки 12-15 после активации Т-клеток с использованием 96-луночной одноразовой системы для оценки хемотаксиса ChemoTx® (Neuroprobe 101-5) в соответствии с протоколом изготовителя. После одного промывания аналитическим буфером (1× HBSS, содержащий 20 мМ HEPES и 0,25% BSA) клетки инкубировали с тестируемыми соединениями в течение 30 мин при комнатной температуре до начала хемотаксиса. Для определения IC50 верхняя и нижняя части камеры для хемотаксиса содержали одинаковую концентрацию соединения. Содержание ДМСО поддерживали на постоянном уровне 0,1% (об/об) во всех лунках. Конечная концентрация CCL20 (Peprotech 300-29A) в нижней камере составляла 0,5 нМ. Полностью собранный планшет для хемотаксиса помещали в инкубатор для клеточных культур при 37°C, 5% CO2 на 1 ч. После инкубации верхний фильтр удаляли, и затем нижнюю камеру быстро замораживали при -80°C в течение 1 ч. Мигрировавшие клетки в нижней камере окрашивали красителем CyQUANT (Life Technologies, C7026) для определения количества клеток.
Значения IC50 для соединений по настоящему изобретению (таблица 11) определяли с помощью нелинейного регрессионного анализа кривых доза-ответ.
Анализ хемотаксиса нейтрофилов человека
Человеческие нейтрофилы выделяли из свежей человеческой цельной крови иммуномагнитной отрицательной селекцией с использованием набора EasySepTM Direct Human Neutrophil Isolation Kit (StemCell Technologies, # 19666) в соответствии с инструкциями изготовителя.
Анализ хемотаксиса нейтрофилов человека проводили с 96-луночными планшетами Corning FluoroBlok cell insert system (Corning # 351164). Выделенные нейтрофилы ресуспендировали в буфере для хемотаксиса (1× HBSS, содержащий 25 мМ HEPES, pH 7,4 и 0,25% BSA) до рабочего титра 2×106 клеток/мл и инкубировали в течение 30 мин при 37°C с кальцеином AM для окрашивания клеток. Через 30 мин меченые нейтрофилы дважды промывали буфером для хемотаксиса, ресуспендировали, как описано выше, и немедленно использовали. Для анализа тестируемые соединения анализировали в формате зависимости доза-ответ для определения IC50. Меченые нейтрофилы (50 мкл) предварительно инкубировали с разбавленным тестируемым соединением (50 мкл) в течение 30 мин в 96-луночном планшете (Greiner). Затем 100 мкл разбавленного соединения смешивали со 100 мкл GRO (Peprotech # 300-11, 2 нМ) в нижней части камеры для хемотаксиса. Затем собирали вместе верхнюю часть (вкладыш) системы хемотаксиса, содержащую пористый мембранный вкладыш FluoroBlok. 50 мкл предварительно инкубированных нейтрофилов наносили на мембранный вкладыш FluoroBlok (верхняя часть), и хемотаксис измеряли по способности нейтрофилов мигрировать через мембрану к специфическому лиганду CXCR2, GRO. Для оценки изменений флуоресцентного сигнала (флуорофор кальцеин AM) при длине волны 485/535 нм, генерируемого мечеными нейтрофилами, прошедшими через пористую мембрану, использовали планшетный ридер Envision multilabel (Perkin Elmer) и/или лазерный сканер для флуоресцентного анализа Typhoon Fluorescent imager (GE).
Значения IC50 для соединений по настоящему изобретению (таблица 12) определяли с помощью нелинейного регрессионного анализа кривых доза-ответ. Навариксин ((R)-2-гидрокси-N, N-диметил-3-((2-((1-(5-метилфуран-2-ил)пропил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)бензамид) использовали в качестве контрольного стандартного образца в анализе (IC50=16,8, n=52).
Мышиная модель in vivo IL-23-индуцированного псориазоподобного кожного воспаления
Эффективность антагониста CCR6 по настоящему изобретению оценивали на мышиной модели псориазоподобного воспаления кожи in vivo. Рекомбинантный мышиный IL-23 вводили внутрикожными инъекциями в левое ухо самкам мышей C57BL/6 в возрасте 8-10 недель. Это приводило к развитию симптомов псориазоподобного заболевания, включая утолщение и эритему кожи левого уха. Мышам, обработанным IL-23, вводили с суток 0 по сутки 11 ежедневные (QD) пероральные дозы антагониста CCR6 или два раза в сутки (BID) дозы антагониста CCR6. Кроме того, группу мышей обрабатывали анти-IL-17 антителами в качестве положительного контроля эффективности. Репрезентативные группы по дозам в исследования включали следующее:
группа, получавшая перорально два раза в сутки (BID) носитель, группа отрицательного контроля,
группа, получавшая внутрибрюшинную инъекцию два раза в неделю анти-IL-17 Ab (5 мг/кг), группа положительного контроля,
группа, получавшая перорально соединение А в дозе 3-100 мг/кг один раз в сутки (QD) или два раза в сутки (BID),
необработанная, контрольная группа.
Десять мышей набирали в группу каждого вида обработки, за исключением контрольной группы, которая включала пять мышей. Сутки 0 в исследовании принимали как первый день обработки, и измерения толщины уха проводили ежедневно с помощью инженерного микрометра (Mitutoyo, Aurora, IL, USA). Показатели измерения отека ушей для каждой мыши сравнивали с исходным уровнем и выражали в микронах.
Соединения по настоящему изобретению избирательно ингибируют хемотаксис Т-клеток по сравнению с хемотаксисом нейтрофилов (см. таблицу 13). Такая селективность делает соединения по настоящему изобретению перспективными терапевтическими агентами для лечения воспалительных, иммунных, аутоиммунных, нейродегенеративных и нейровоспалительных заболеваний, состояний или расстройств у людей с пониженным риском проявления нейтропении.
** Отсутствие селективности для хемотаксиса Т-клеток по сравнению с хемотаксисом нейтрофилов.
Авторы изобретения неожиданно установили, что ингибирование хемотаксиса Т-клеток по сравнению с ингибированием хемотаксиса нейтрофилов усиливается заменой водорода на метильную группу альфа в точке присоединения гетероарильного кольца «А» (см. таблицу 14).
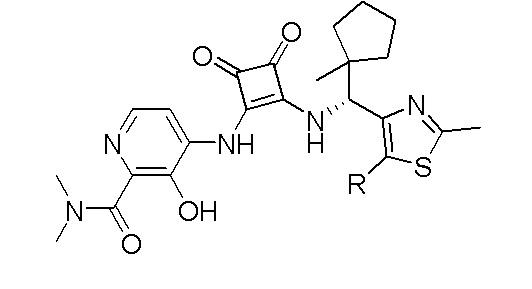
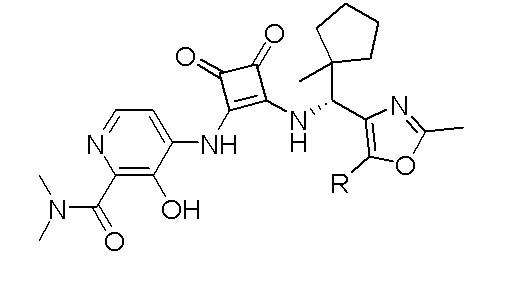
В WO 2010/131145 раскрываются примеры 112 и 120.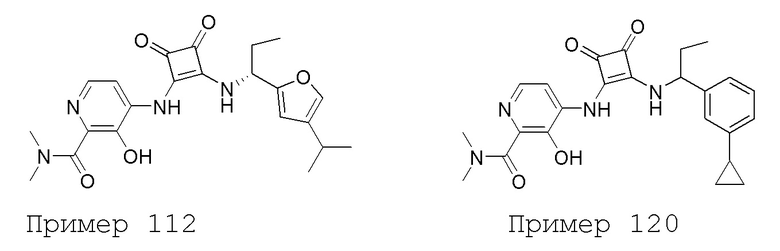
Авторы изобретения предоставили сравнительные данные по хемотаксису в таблице 15 для соединений WO 2010/131145. Данные в таблице 15 демонстрируют, что примеры 112 и 120 более эффективны в отношении ингибирования миграции нейтрофилов, чем соединения по настоящему изобретению, и менее эффективны в отношении ингибирования миграции Т-клеток, чем соединения по настоящему изобретению. Соединения по WO 2010/131145 являются функционально селективными в отношении CXCR2 по сравнению с CCR6 в противоположность соединениям по настоящему изобретению. Эти данные предполагают, что соединения WO 2010/131145 менее привлекательны в качестве агентов для лечения заболеваний, состояний или расстройств, ослабляемых за счет снижения хемотаксиса Т-клеток посредством ингибирования CCR6, и что соединения по WO 2010/131145 также имеют больший риск развития нейтропении у пациентов по сравнению с соединениями по настоящему изобретению.
В WO 2013/061005 раскрываются примеры 22, 50 и 53 и приводятся функциональные данные CXCR2 и CCR6 для каждого соединения, которые воспроизведены из WO 2013/061005 в таблице 16 ниже. Активность антагониста CXCR2 определяли по ингибированию рекрутмента β-аррестина после активации с помощью CXCL8 в клетках Path-Hunter HEK293. Активность антагониста CCR6 определяли по ингибированию потока Ca2+ в клетках после активации CCR6 на платформе FLIPR TETRA®. Об активирующем агенте CCR6 не сообщалось.
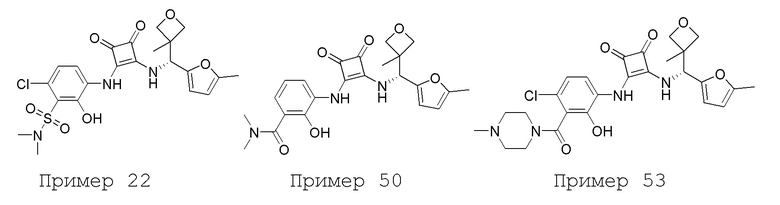
IC50 (нМ)
IC50 (нМ)
Авторы изобретения предоставляют следующие сравнительные данные по хемотаксису в таблице 17 для соединений в WO 2013/061005. Данные в таблице 17 демонстрируют, что пример 22 является эффективным в ингибировании хемотаксиса Т-клеток и нейтрофилов с несколько большей активностью (1,5×) для ингибирования CCR6. Высокая аффинность к обоим рецепторам в сочетании с низкой селективностью в отношении CCR6 делает это соединение менее привлекательным в качестве противовоспалительного агента с пониженным потенциалом индукции нейтропении, чем соединения по настоящему изобретению. Пример 50 имеет низкую аффинность к CCR6 и ингибирует хемотаксис нейтрофилов лучше, чем хемотаксис Т-клеток. Профиль хемотаксиса примера 50 менее привлекателен для его разработки в качестве противовоспалительного средства, чем соединения по настоящему изобретению. Пример 53 не является сильным ингибитором хемотаксиса Т-клеток или хемотаксиса нейтрофилов. Отсутствие функциональной активности на CCR6 делает пример 53 менее привлекательным для лечения воспалительных заболеваний, чем соединения по настоящему изобретению.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ НАФТИРИДИНА В КАЧЕСТВЕ АНТАГОНИСТОВ АЛЬФА V БЕТА 6 ИНТЕГРИНА ДЛЯ ЛЕЧЕНИЯ, В ЧАСТНОСТИ, ФИБРОЗНЫХ ЗАБОЛЕВАНИЙ | 2015 |
|
RU2692775C2 |
| СОЕДИНЕНИЯ ПИРАЗОЛО[3,4-b]ПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ТАМ И МЕТ | 2019 |
|
RU2773611C1 |
| АНТИПРОЛИФЕРАТИВНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ PAH | 2020 |
|
RU2786588C1 |
| ПИРИДОПИРИМИДИНОВЫЕ ИНГИБИТОРЫ CDK2/4/6 | 2017 |
|
RU2726115C1 |
| 3-КАРБОНИЛАМИНО-5-ЦИКЛОПЕНТИЛ-1H-ПИРАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ ИНГИБИТОРОВ CDK2 | 2020 |
|
RU2797889C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛАМИНОПИРИМИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ОБОГАЩЕННОЙ ЛЕЙЦИНОВЫМИ ПОВТОРАМИ КИНАЗЫ 2 | 2013 |
|
RU2637947C2 |
| ТРИЦИКЛИЧЕСКИЕ КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ПИРИДИН-2-ОНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ BRD4 | 2016 |
|
RU2734959C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PRMT5 | 2018 |
|
RU2797822C2 |
| ЗАМЕЩЕННЫЕ 1,2-ДИГИДРО-3Н-ПИРАЗОЛО[3,4-D]ПИРИМИДИН-3-ОНЫ | 2019 |
|
RU2812726C2 |
| ГИДРОКСИПИРИДОКСАЗЕПИНЫ В КАЧЕСТВЕ АКТИВАТОРОВ NRF2 | 2020 |
|
RU2812931C2 |
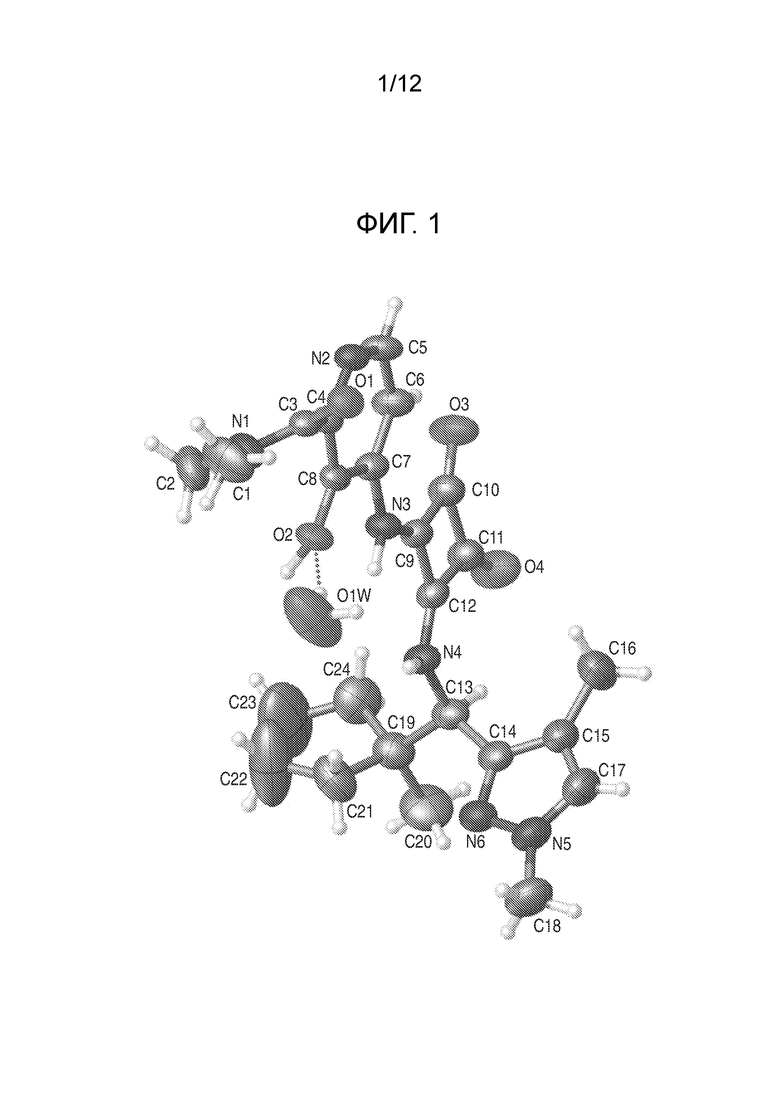
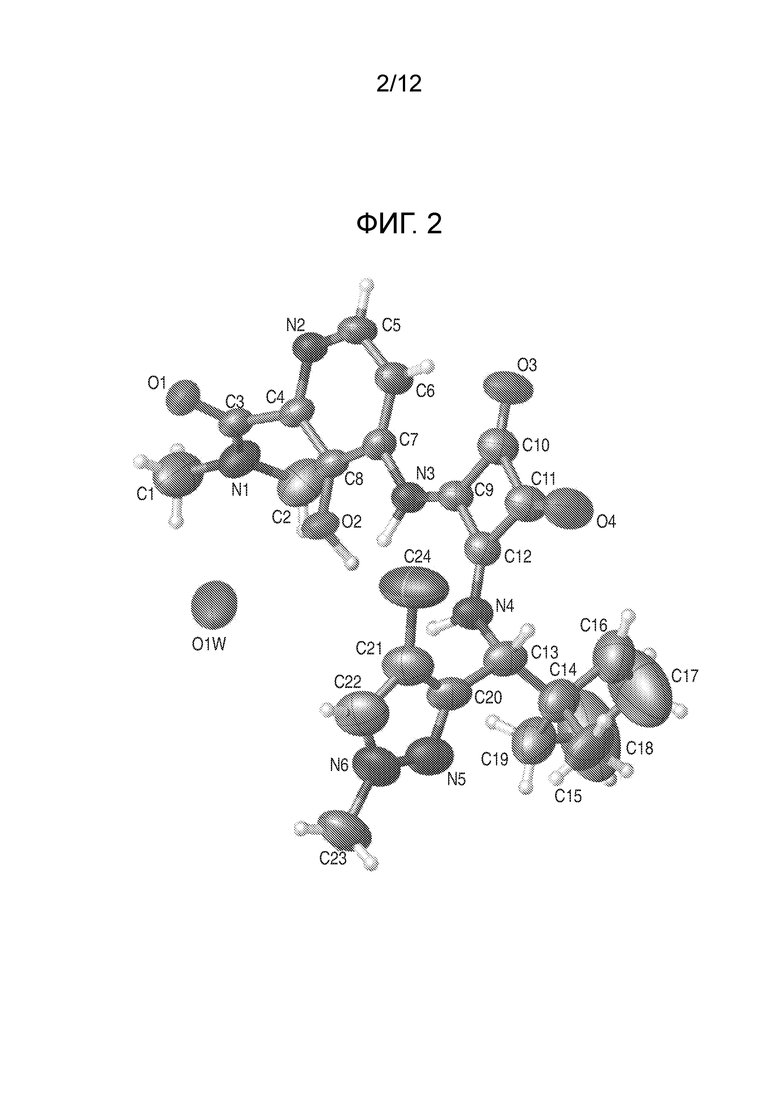
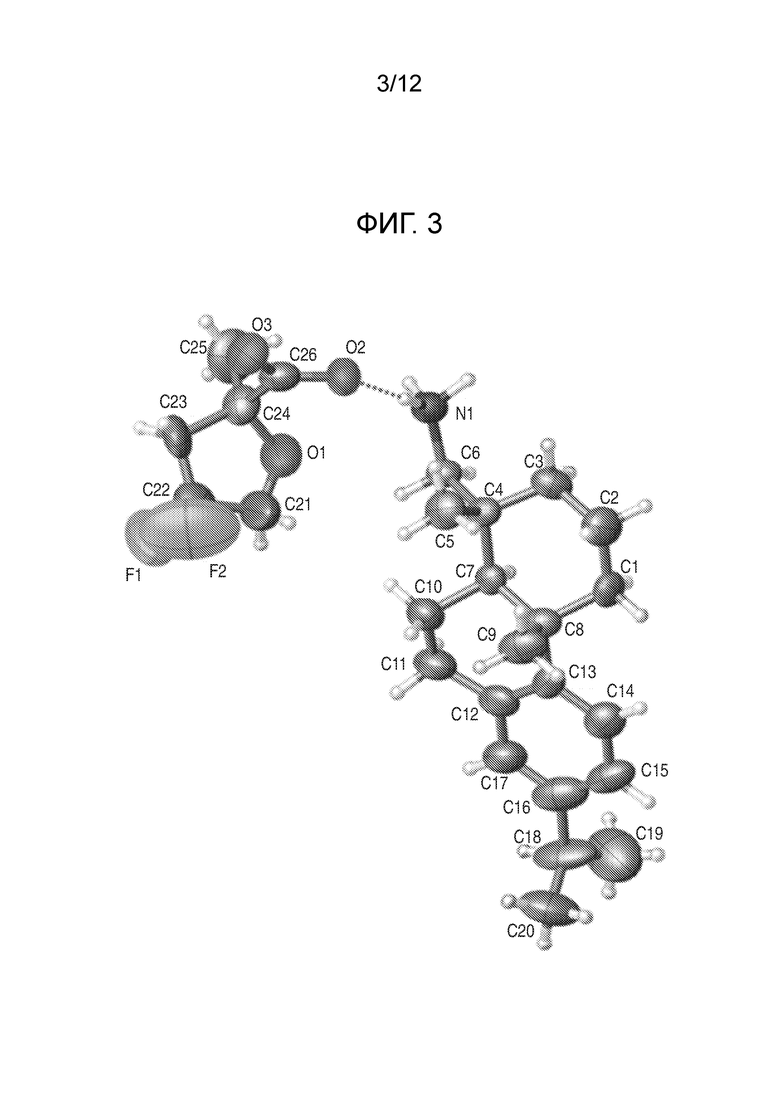
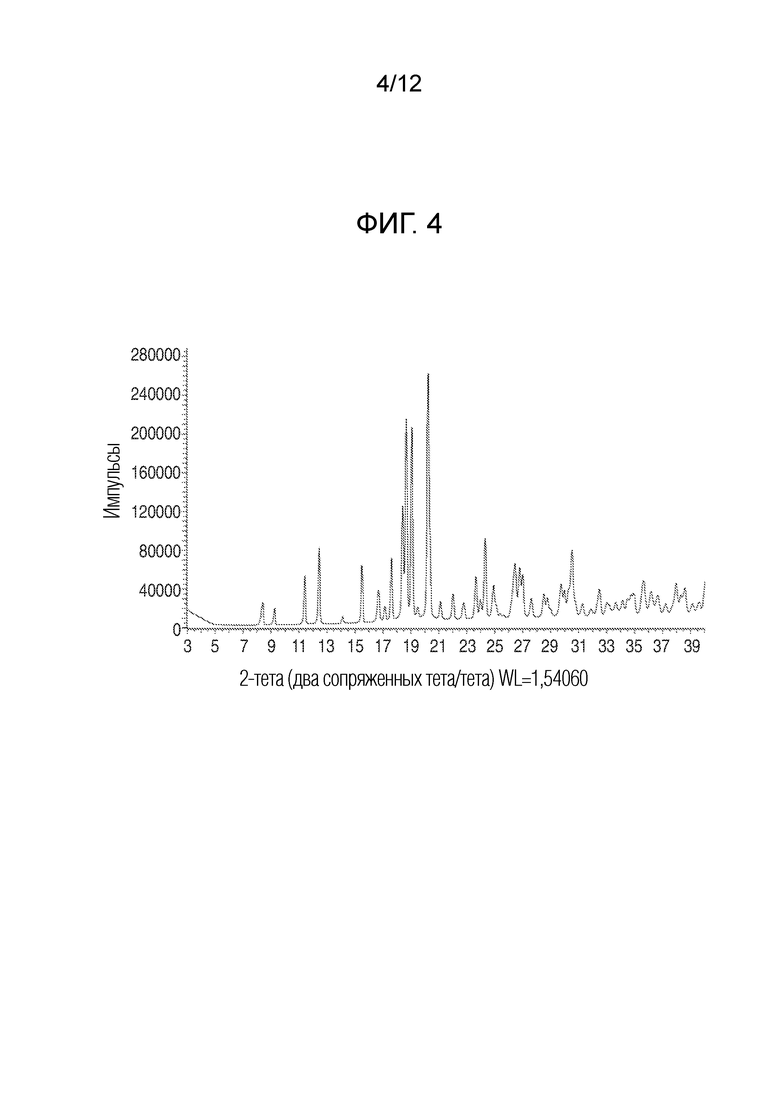
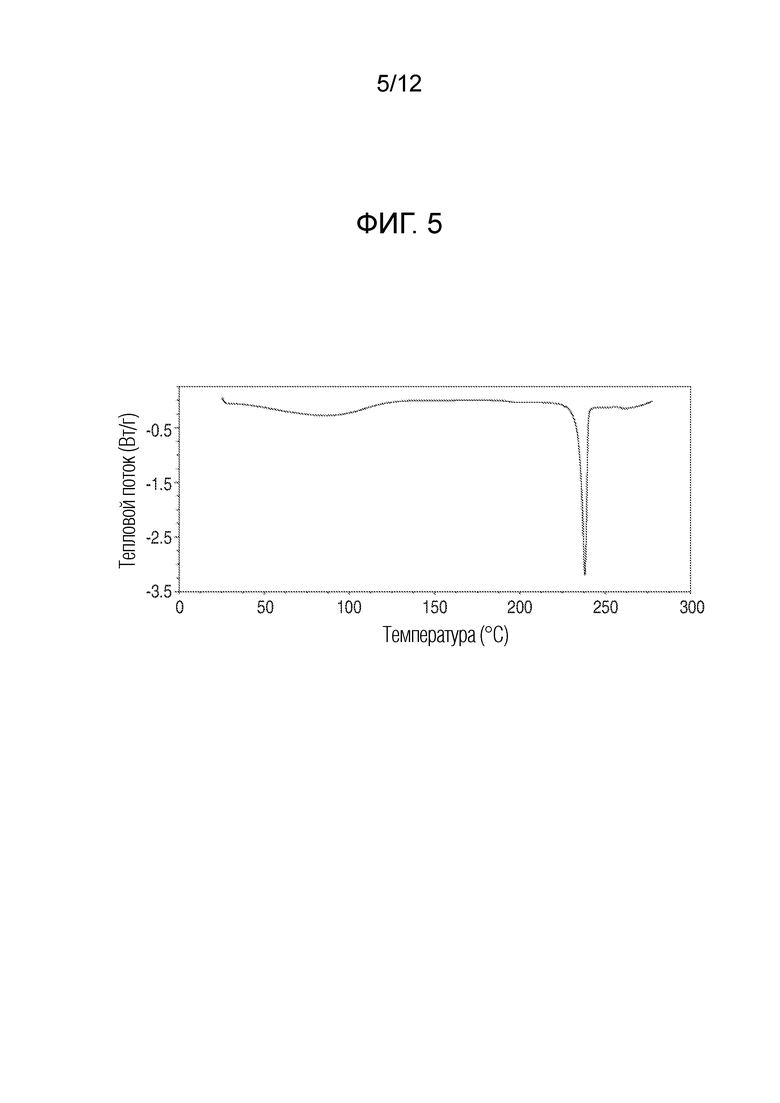
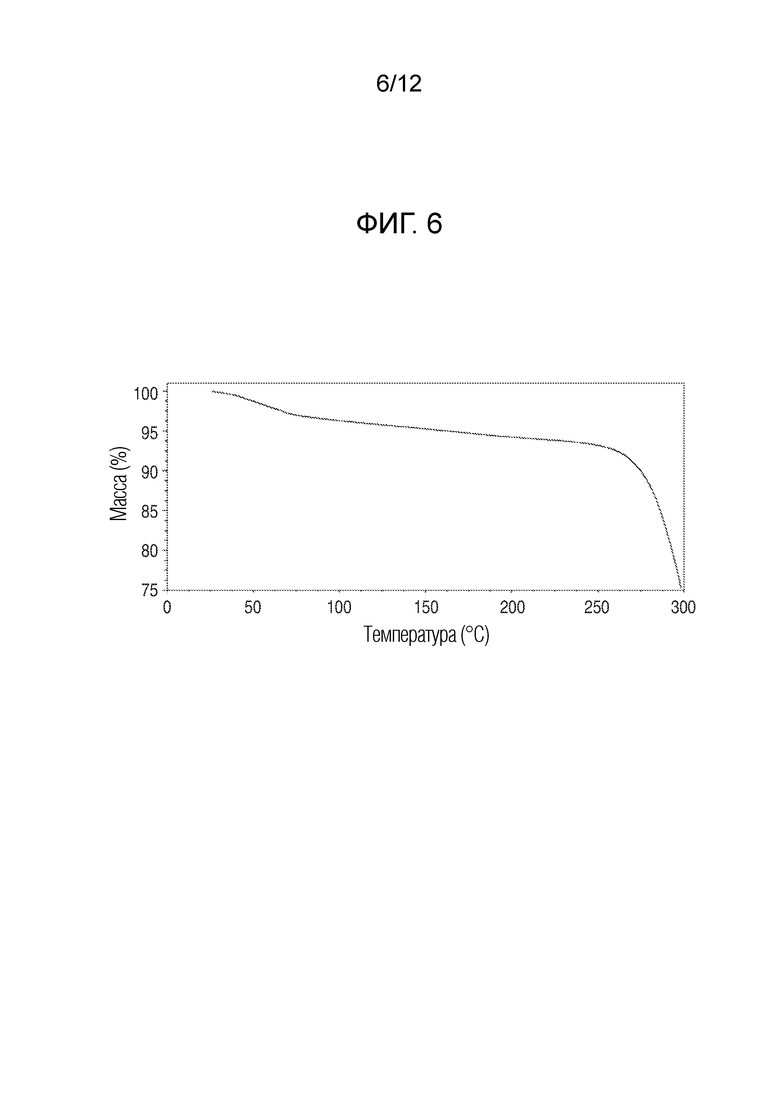
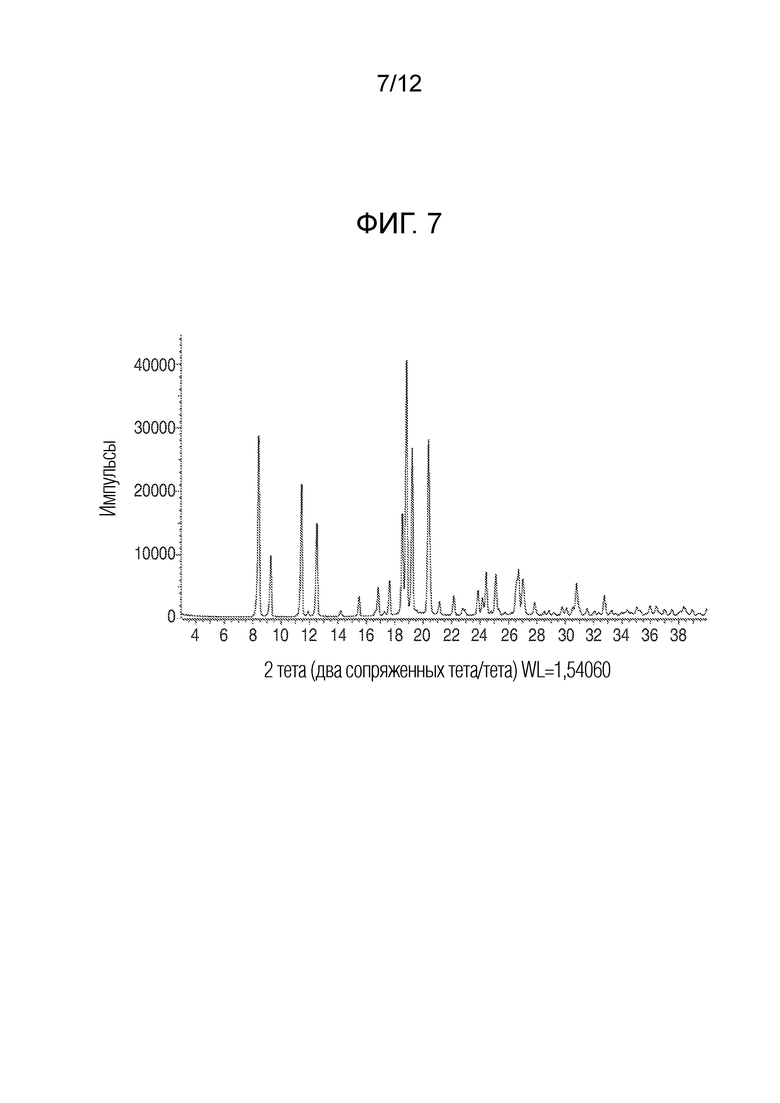
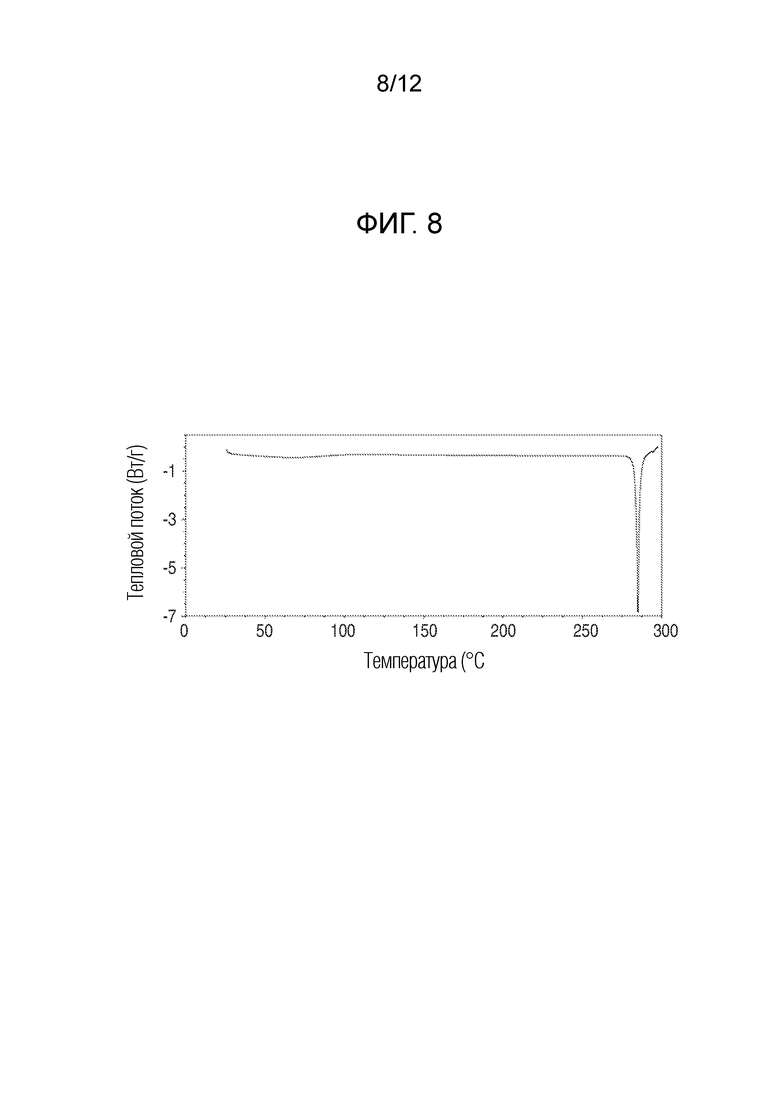
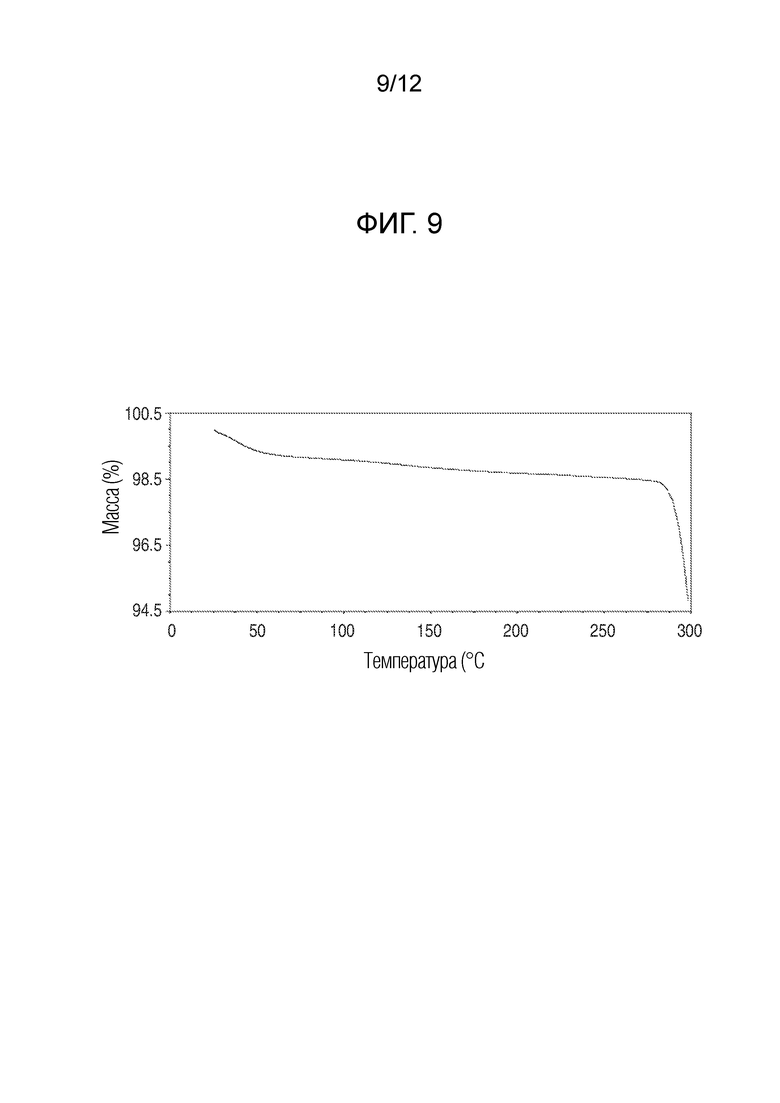
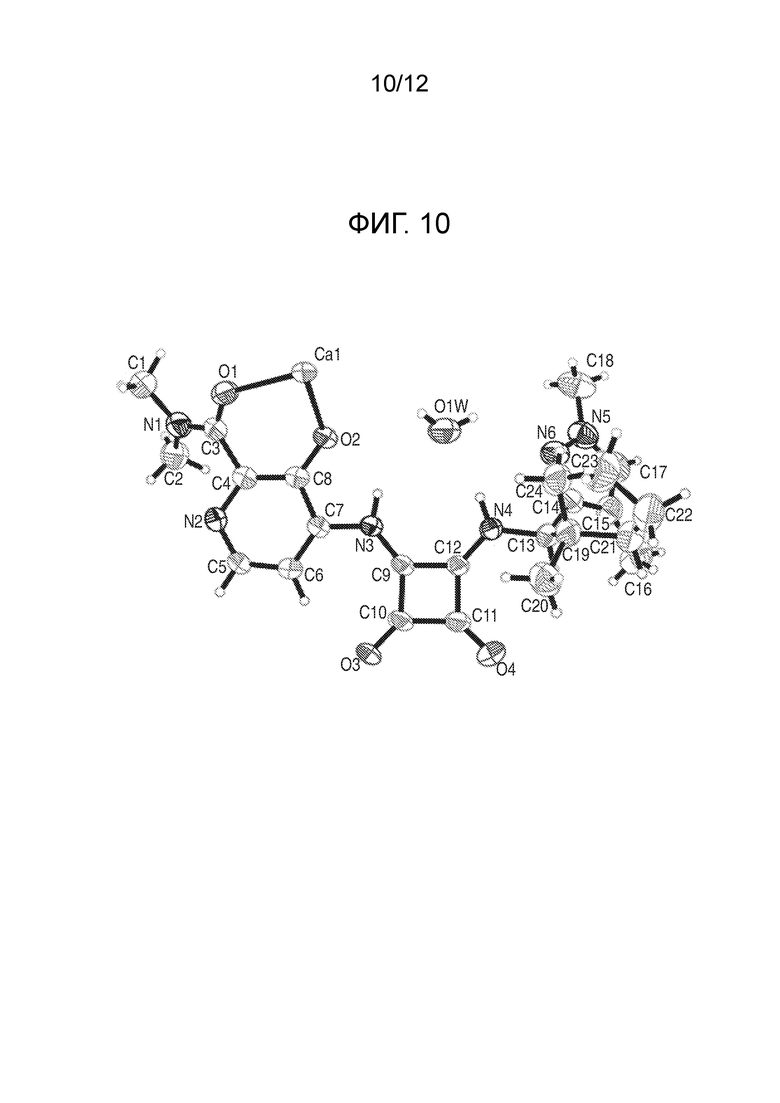
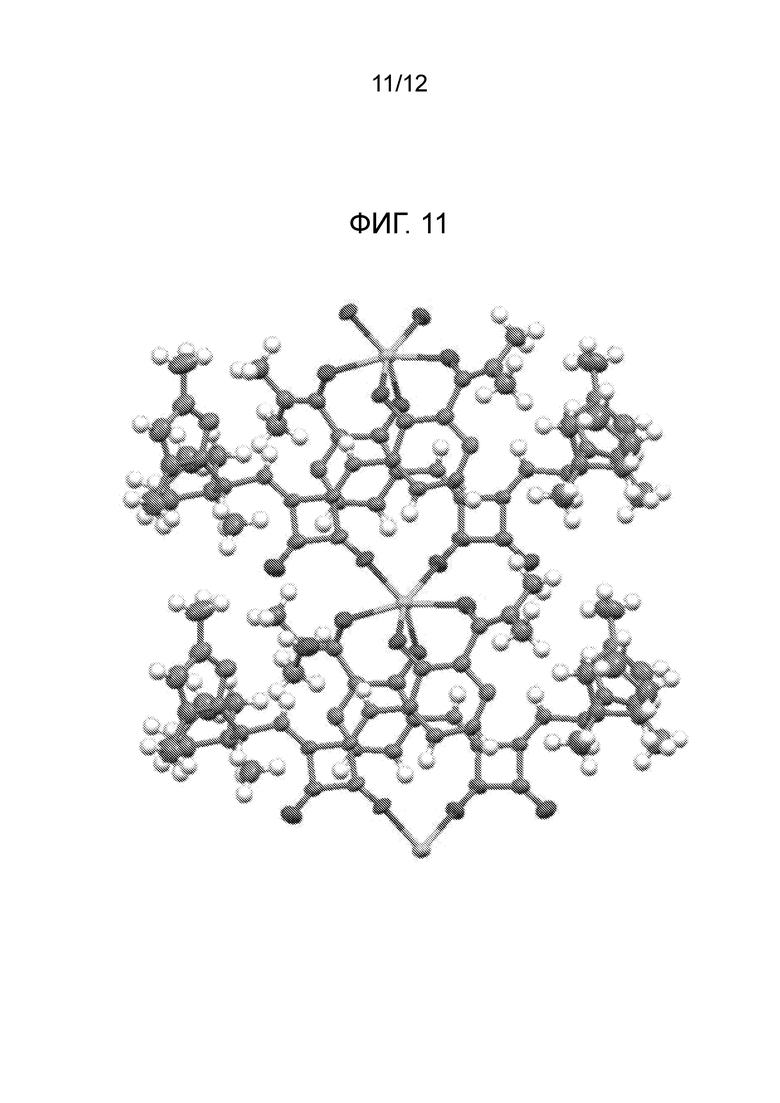
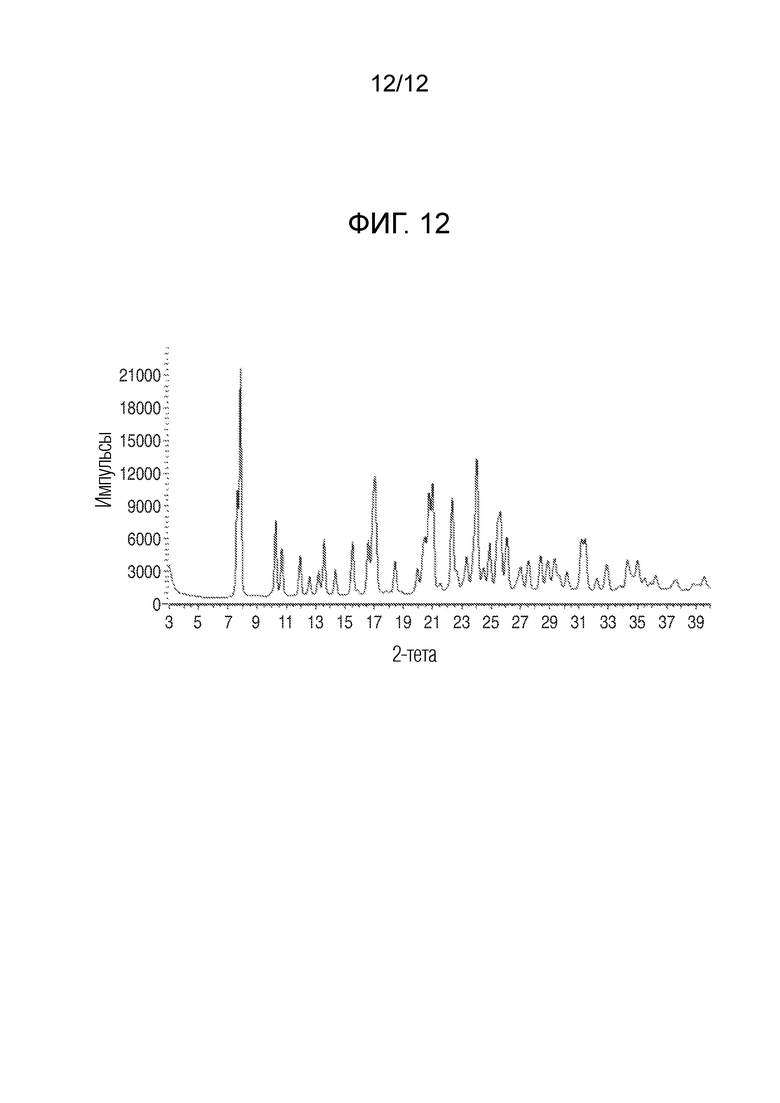
Изобретение относится к области органической химии, а именно к соединениям N-замещенного диоксоциклобутениламино-3-гидроксипиколинамида формул (IA и IB), или его фармацевтически приемлемая соль, где R1 и R2 независимо представляют собой (C1-C6)алкил, или R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют 6-членный гетероцикл, содержащий один гетероатом N и 1 дополнительный гетероатом, выбранный из группы, состоящей из O и N, где гетероцикл необязательно замещен одной (C1-C4)алкильной группой; R3 представляет собой H; А представляет собой 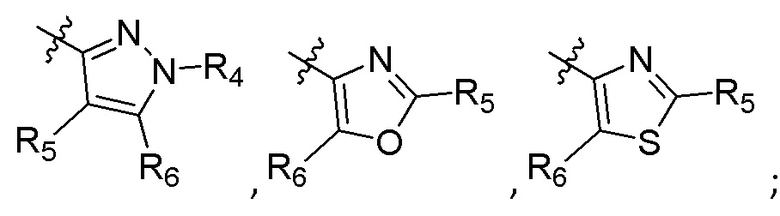 R4 представляет собой (C1-C4)алкил; R5 и R6 независимо представляют собой H, (C1-C4)алкокси, (C1-C4)алкил, (C1-C4)алкил-d1-9, (C3-C4)циклоалкил, циано, атом галогена, галоген(C1-C4)алкокси или гидрокси(C1-C4)алкил; B представляет собой
R4 представляет собой (C1-C4)алкил; R5 и R6 независимо представляют собой H, (C1-C4)алкокси, (C1-C4)алкил, (C1-C4)алкил-d1-9, (C3-C4)циклоалкил, циано, атом галогена, галоген(C1-C4)алкокси или гидрокси(C1-C4)алкил; B представляет собой 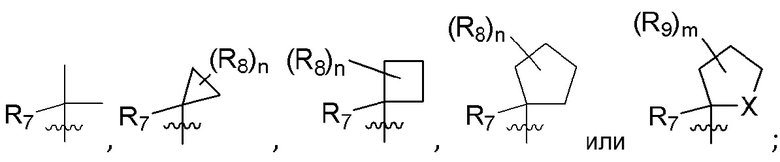 R7 представляет собой (C1-C3)алкил; R8 в каждом случае независимо представляет собой дейтерий, или два R8, присоединенные к одному и тому же атому углерода, образуют (C3-C5)циклоалкильную группу; n равно 0 или 2; R9 в каждом случае независимо представляет собой дейтерий или –F; m равно 2; и X представляет собой O. Также изобретение относится к конкретным соединениям N-замещенного диоксоциклобутениламино-3-гидроксипиколинамида формул (IA и IB). Технический результат: получены соединения, которые ингибируют СС-хемокиновый рецептор 6 (ССR6), полезные при лечении или профилактики состояний или заболеваний, ослабляемых ингибированием ССR6. 8 н. и 5 з.п. ф-лы, 12 ил., 17 табл., 42 пр.
R7 представляет собой (C1-C3)алкил; R8 в каждом случае независимо представляет собой дейтерий, или два R8, присоединенные к одному и тому же атому углерода, образуют (C3-C5)циклоалкильную группу; n равно 0 или 2; R9 в каждом случае независимо представляет собой дейтерий или –F; m равно 2; и X представляет собой O. Также изобретение относится к конкретным соединениям N-замещенного диоксоциклобутениламино-3-гидроксипиколинамида формул (IA и IB). Технический результат: получены соединения, которые ингибируют СС-хемокиновый рецептор 6 (ССR6), полезные при лечении или профилактики состояний или заболеваний, ослабляемых ингибированием ССR6. 8 н. и 5 з.п. ф-лы, 12 ил., 17 табл., 42 пр.
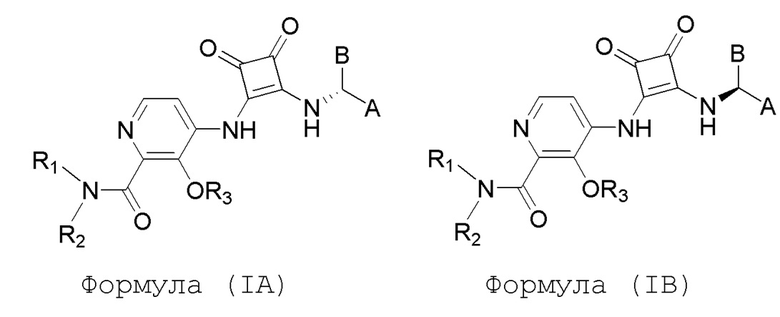
1. Соединение формулы (IA) или (IB):
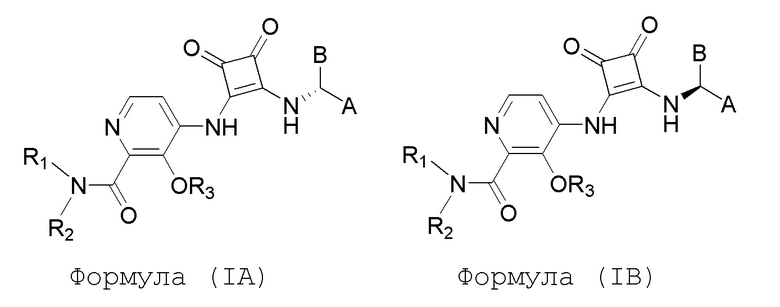
или его фармацевтически приемлемая соль, где:
R1 и R2 независимо представляют собой (C1-C6)алкил, или R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют 6-членный гетероцикл, содержащий один гетероатом N и 1 дополнительный гетероатом, выбранный из группы, состоящей из O и N, где гетероцикл необязательно замещен одной (C1-C4)алкильной группой;
R3 представляет собой H;
А представляет собой:
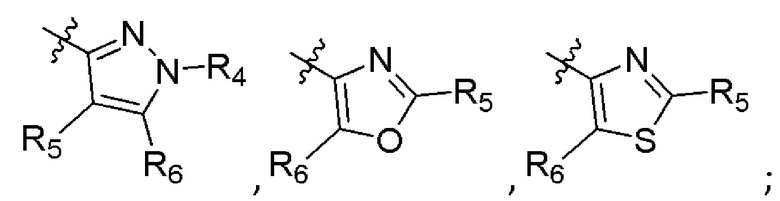
R4 представляет собой (C1-C4)алкил;
R5 и R6 независимо представляют собой H, (C1-C4)алкокси, (C1-C4)алкил, (C1-C4)алкил-d1-9, (C3-C4)циклоалкил, циано, атом галогена, галоген(C1-C4)алкокси или гидрокси(C1-C4)алкил;
B представляет собой:
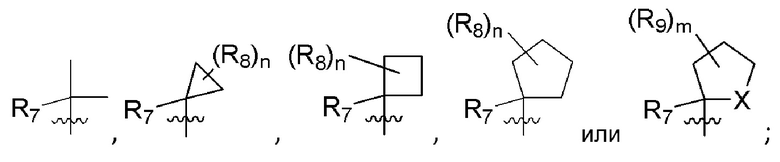
R7 представляет собой (C1-C3)алкил;
R8 в каждом случае независимо представляет собой дейтерий, или два R8, присоединенные к одному и тому же атому углерода, образуют (C3-C5)циклоалкильную группу;
n равно 0 или 2;
R9 в каждом случае независимо представляет собой дейтерий или –F;
m равно 2; и
X представляет собой O.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где:
R1 представляет собой метил;
R2 представляет собой метил, этил, изопропил;
А представляет собой:
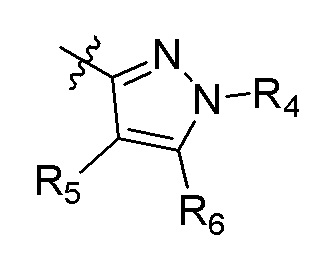
R4 представляет собой метил;
R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил;
R6 представляет собой H;
R7 представляет собой метил;
два R8, присоединенные к одному и тому же атому углерода, образуют циклопропил;
n равно 0 или 2;
R9 в каждом случае представляет собой F;
m равно 2; и
X представляет собой О.
3. Соединение по п.1 или его фармацевтически приемлемая соль, где:
R1 представляет собой метил;
R2 представляет собой метил, этил, изопропил;
А представляет собой:
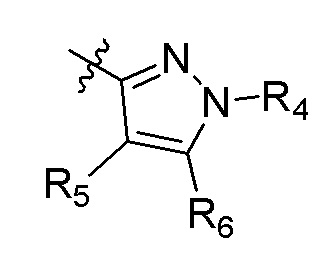
R4 представляет собой метил;
R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил;
R6 представляет собой H;
B представляет собой:
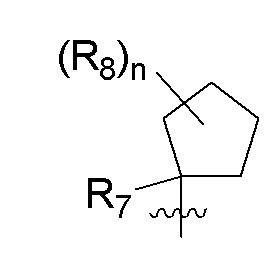
R7 представляет собой метил; и
n равно 0.
4. Соединение по п.1 формулы (IIA):
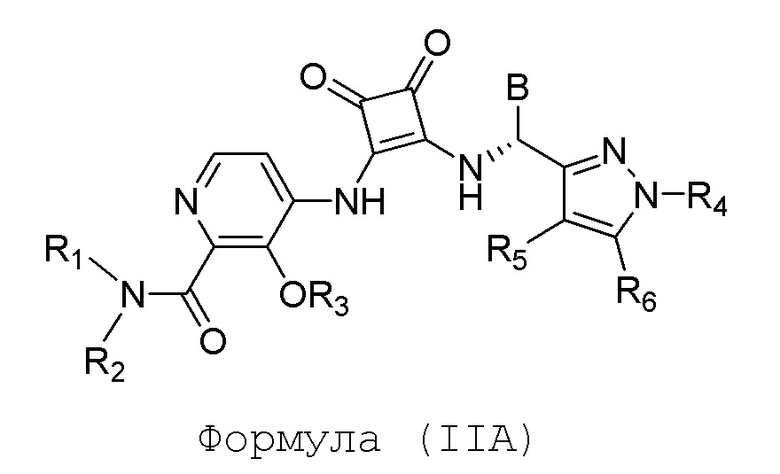
или его фармацевтически приемлемая соль.
5. Соединение по п.4 или его фармацевтически приемлемая соль, где:
R1 представляет собой метил;
R2 представляет собой метил, этил или изопропил;
R4 представляет собой метил;
R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил;
R6 представляет собой H;
B представляет собой:
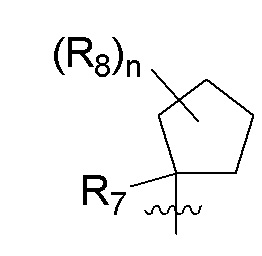
R7 представляет собой метил; и
n равно 0.
6. Соединение по п.4 или его фармацевтически приемлемая соль, где:
R1 и R2, взятые вместе с атомом азота, к которому они присоединены, образуют морфолин или 4-метилпиперазин;
R4 представляет собой метил;
R5 представляет собой метил, этил, метокси, Cl, дифторметокси, циано или циклопропил;
R6 представляет собой H;
B представляет собой:
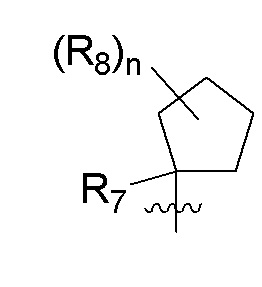
R7 представляет собой метил; и
n равно 0.
7. Соединение, выбранное из группы, включающей:
(R)-4-((2-(((4-Хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопен)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
(R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N-изопропил-N-метилпиколинамид;
(R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(5-метилспиро[2.3]гексан-5-ил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
4-((2-(((S)-((S)-4,4-Дифтор-2-метилтетрагидрофуран-2-ил)(4-этил-1-метил-1H-пиразол-3-ил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
(R)-4-((2-(((2,5-Диметилтиазол-4-ил)(1-метилциклобутил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
(S)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
(R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N-этил-3-гидрокси-N-метилпиколинамид;
(S)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N-этил-3-гидрокси-N-метилпиколинамид;
(R)-3-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-4-((3-гидрокси-2-(морфолин-4-карбонил)пиридин-4-ил)амино)циклобут-3-ен-1,2-дион;
(R)-3-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-4-((3-гидрокси-2-(4-метилпиперазин-1-карбонил)пиридин-4-ил)амино)циклобут-3-ен-1,2-диoн;
(S)-4-((2-(((4-Хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
(R)-4-((2-(((4-Хлор-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N-изопропил-N-метилпиколинамид;
(R)-3-Гидрокси-N-изопропил-4-((2-(((4-метокси-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N-метилпиколинамид;
(R)-3-Гидрокси-4-((2-(((4-метокси-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N,N-диметилпиколинамид;
(R)-3-Гидрокси-4-((2-((1-(4-метокси-1-метил-1H-пиразол-3-ил)-2,2-диметилпропил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N, N-диметилпиколинамид;
(R)-3-Гидрокси-N-изопропил-4-((2-((1-(4-метокси-1-метил-1H-пиразол-3-ил)-2,2-диметилпропил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N-метилпиколинамид;
(R)-4-((2-(((4-(Дифторметокси)-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N,N-диметилпиколинамид;
(R)-4-((2-(((4-Циано-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
(R)-4-((2-(((2,5-Диметилтиазол-4-ил)(1-метилциклопропил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
(R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопропил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
(R)-4-((2-(((4-Этил-1-метил-1H-пиразол-3-ил)(1-метилциклопропил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
(R)-4-((2-((1-(1,4-Диметил-1H-пиразол-3-ил)-2,2-диметилпропил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
(R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклобутил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
(R)-4-((2-((1-(2,5-Диметилтиазол-4-ил)-2,2-диметилпропил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
(R)-4-((2-(((4-Этил-1-метил-1H-пиразол-3-ил)(1-метилциклобутил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
(R)-4-((2-((1-(4-Этил-1-метил-1H-пиразол-3-ил)-2,2-диметилпропил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
(R)-3-Гидрокси-4-((2-(((2-(гидроксиметил)-5-метилтиазол-4-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N,N-диметилпиколинамид;
(R)-4-((2-(((4-Циклопропил-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N,N-диметилпиколинамид;
(R)-4-((2-(((4-Циано-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N-этил-3-гидрокси-N-метилпиколинамид;
(R)-4-((2-(((4-Циано-1-метил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N-изопропил-N-метилпиколинамид;
(R)-4-((2-(((2,5-Диметилтиазол-4-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
(R)-3-Гидрокси-4-((2-(((5-метокси-2-метилтиазол-4-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-N,N-диметилпиколинамид;
(R)-4-((2-(((2,5-Диметилоксазол-4-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
(R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-этилциклобутил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
4-((2-(((S)-((S)-4,4-Дифтор-2-метилтетрагидрофуран-2-ил)(1,4-диметил-1H-пиразол-3-ил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
(R)-3-Гидрокси-N,N-диметил-4-((2-(((1-метил-4-(метил-d3)-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)пиколинамид;
4-((2-(((1R)-(1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил-3,4-d2)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид; и
4-((2-(((S)-((S)-4,4-Дифтор-2-метилтетрагидрофуран-2-ил)(2,5-диметилтиазол-4-ил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N, N-диметилпиколинамид;
или его фармацевтически приемлемая соль.
8. (R)-4-((2-(((1,4-Диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N,N-диметилпиколинамид или его фармацевтически приемлемая соль.
9. Соединение структуры:
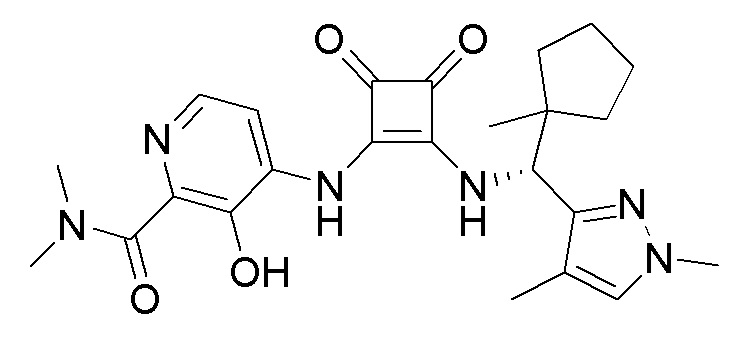 .
.
10. Кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N,N-диметилпиколинамид моногидрат, имеющий рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики 18,8±0,2, 19,2±0,2 и 20,4±0,2 градусов 2-тета.
11. Кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N,N-диметилпиколинамид моногидрат, имеющий рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики 17,6±0,2, 18,5±0,2, 18,8±0,2, 19,2±0,2 и 20,4±0,2 градусов 2-тета.
12. Кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N,N-диметилпиколинамид моногидрат, имеющий рентгеновскую порошковую дифрактограмму, содержащую дифракционные пики 11,5±0,2, 15,5±0,2, 17,6±0,2, 18,5±0,2, 18,8±0,2, 19,2±0,2, 20,4±0,2 и 24,5±0,2 градусов 2-тета.
13. Кристаллический (R)-4-((2-(((1,4-диметил-1H-пиразол-3-ил)(1-метилциклопентил)метил)амино)-3,4-диоксоциклобут-1-ен-1-ил)амино)-3-гидрокси-N,N-диметилпиколинамид моногидрат, имеющий рентгеновскую порошковую дифрактограмму, приведенную на фиг. 4.
| WO 2013061005 A1, 02.05.2013 | |||
| WO 2010131145 A1, 18.11.2010 | |||
| 3,4-ДИЗАМЕЩЕННЫЕ ЦИКЛОБУТЕН-1,2-ДИОНЫ КАК ЛИГАНДЫ СХС-ХЕМОКИНОВОГО РЕЦЕПТОРА | 2002 |
|
RU2344123C9 |
| Приспособление для профилирования дорожного полотна | 1932 |
|
SU28509A1 |

