Настоящее изобретение относится к катализатору на основе TiO2 и/или ZrO2 для получения алкилмеркаптана и к способу его получения. Настоящее изобретение также относится к способу получения алкилмеркаптана посредством реакции алкилового спирта с сероводородом в присутствии катализатора на подложке в соответствии с настоящим изобретением или в присутствии катализатора, полученного посредством способа его получения в соответствии с настоящим изобретением.
Алкилмеркаптаны являются промышленно важными промежуточными соединениями для получения продуктов с высокой экономической ценностью. В частности, метилмеркаптан (CH3SH) является промышленно важным промежуточным соединением, например, в синтезе метионина, диметилсульфоксида, диметилсульфона или метансульфоновой кислоты. В прошлом было разработано множество различных путей синтеза, например, гидрогенизация карбонилсульфида (COS) или дисульфида углерода (CS2), тиолирование метанола с помощью CS2 в присутствии и в отсутствие H2 и тиолирование метанола сероводородом (H2S) в качестве тиолирующего средства в присутствии катализатора на основе оксида алюминия. Последний способ представляет собой текущий промышленный способ из уровня техники, который, как правило, осуществляют в газовой фазе при значениях температуры в диапазоне от 300°C до 500°C и давлении в диапазоне от 1 до 25 бар.
Полученная таким образом реакционная смесь содержит необходимый продукт в виде метилмеркаптана вместе с непрореагировавшими исходными материалами и побочными продуктами, например, диметилсульфидом и диметиловым эфиром, а также газами, инертными в отношении реакции, например, метаном, монооксидом углерода, водородом и азотом. Следовательно, образовавшийся метилмеркаптан необходимо отделить от этой реакционной смеси.
Отделение метилмеркаптана от газообразной реакционной смеси, как правило, осуществляют путем конденсации метилмеркаптана. В этой части значимым фактором, влияющим на стоимость, является энергопотребление, необходимое для охлаждения реакционной смеси. Таким образом, для обеспечения надлежащей экономической эффективности способа образование метилмеркаптана должно протекать с высокой степенью превращения и высокой селективностью в целях поддержания энергозатрат и инвестиционных затрат на минимально возможном уровне.
Улучшения активности и селективности можно достичь путем увеличения молярного соотношения сероводорода и метанола. Обычно применяют молярные соотношения от 1 до 10. Однако высокое молярное соотношение также подразумевает большой избыток сероводорода в реакционной смеси и, соответственно, необходимость циркуляции больших количеств газа. Поэтому для снижения расхода потребляемой для этого энергии соотношение сероводорода и метанола должно лишь незначительно отклоняться от 1.
Для увеличения активности и селективности были разработаны специфические катализаторы на основе Al2O3. Принято считать, что оксид алюминия, в частности γ-Al2O3, в принципе является катализатором для тиолирования метанола с образованием метилмеркаптана. Однако активность оксида алюминия слишком высока и, соответственно, катализируемая реакция не останавливается при получении необходимого продукта в виде метилмеркаптана. Вместо этого очень активный оксид алюминия также катализирует дополнительную реакцию превращения метилмеркаптана в диметилсульфид. Таким образом, оксид алюминия, как правило, смешивают с вольфраматом щелочного металла, например, с вольфраматом калия или вольфраматом цезия, для снижения его активности, что приводит к увеличению селективности образования метилмеркаптана и снижению селективности образования побочных продуктов, таких как диметилсульфид. В полученных таким образом катализаторах оксид алюминия также называют подложкой или носителем катализатора, а вольфрамат щелочного металла называют промотором. Доля вольфрамата в пересчете на общий вес катализатора, как правило, составляет не более приблизительно 20 вес. %, как описано, например, в патенте США №2820062. Катализатор, раскрытый в данном документе, обладает хорошими показателями активности и селективности в получении алкантиолов при температуре реакции, составляющей 400°C, и молярном соотношении сероводорода и алкилового спирта, составляющем 2:1.
В относительно сложном способе получения, который включает многократную пропитку материала подложки раствором вольфрамата щелочного металла, долю вольфрамата щелочного металла в катализаторе можно увеличить до 25 вес. % и больше. В патенте США №5852219 раскрыты преимущества применения вольфрамата цезия (Cs2WO4) в качестве промотора вместо вольфрамата калия (K2WO4). Таким образом, может быть достигнута увеличенная активность одновременно с хорошей селективностью. Согласно Mashkina et al. (React. Kinet. Catal. Lett., vol. 36, No. 1, 159-164 (1988)) наилучшая селективность образования алкилмеркаптана из алкиловых спиртов и сероводорода достигается с помощью катализаторов, в которых соотношение щелочной металл/вольфрам составляет 2:1. Пропитка носителя на основе оксида алюминия раствором, содержащим цезий и вольфрам в стехиометрическом соотношении 2:1, позволяет достигать нагрузки промотора, составляющей не более 40 вес. % в пересчете на общий вес катализатора, как описано в патенте США №5 852 219. Увеличение концентрации вольфрамата щелочного металла до 25 вес. % или больше приводит к увеличению селективности образования метилмеркаптана. Однако недостатком является то, что при этом снижается активность катализатора.
Многократная пропитка материала подложки раствором, содержащим цезий и вольфрам в нестехиометрическом соотношении, составляющем менее 2:1, как описано в заявке на патент США № 2009/306432 A1, позволяет увеличить нагрузку катализатора промотором до значений, составляющих 25 вес. % или больше в пересчете на общий вес катализатора. Хотя нагрузку оксида алюминия промотором можно увеличить до более чем 35% по весу путем применения нестехиометрического соотношения цезия и вольфрама в пропиточном растворе, при таких высоких нагрузках больше не наблюдается значительного увеличения активности или селективности. В частности, активность и селективность даже снижаются при нагрузках выше 45% по весу в пересчете на общий вес катализатора. Дополнительным недостатком является то, что в катализаторе, полученном посредством способа многократной пропитки, распределение цезия и вольфрама во всем теле катализатора является неоднородным. Однако для достижения высокой активности и высокой селективности в катализируемых реакциях считается необходимым однородное распределение каталитически активных компонентов во всем теле катализатора.
В заявке на патент США № 2014/357897 A1 раскрыты катализаторы с однородным распределением щелочного металла и вольфрама во всем формованном теле катализатора. Эти катализаторы получают путем смешивания материала подложки с материалом на основе оксидного соединения вольфрама, таким как вольфрамовая кислота, и по меньшей мере одним отдельным соединением щелочного металла, таким как гидроксид цезия, с получением массы катализатора и последующим формованием указанной массы катализатора. В полученных таким образом катализаторах нагрузка промотора составляет 45 вес. % или больше в пересчете на общий вес катализатора. Это позволяет дополнительно повысить выход и селективность образования метилмеркаптана. Однако высокий выход и селективность указанных катализаторов недолговечны, поэтому они не подходят для применения в промышленных способах.
В документе WO 2017/114858 A1 раскрыт другой способ получения катализаторов для получения алкантиолов. Этот способ получения представляет собой комбинацию общепринятой процедуры пропитки и способа смешивания или формования, описанного в заявке на патент США №2014/357897 A1. Активность и селективность в отношении метилмеркаптана катализаторов, полученных таким образом, сопоставимы с таковыми других катализаторов. Однако довольно сложный способ получения не приводит к увеличению производительности.
Как описано выше, были предприняты значительные усилия для повышения активности и селективности катализатора тиолирования метанола. Однако оказалось, что каталитическая система не обеспечивает никаких дополнительных улучшений при тиолировании метанола. Следовательно, необходимо было решить проблему, чтобы обеспечить улучшенный катализатор тиолирования метанола, который обладает улучшенной селективностью по сравнению с катализаторами из предшествующего уровня техники.
Неожиданно было обнаружено, что эта проблема решается путем применения подложки катализатора, отличной от подложки из предшествующего уровня техники. На эту конкретную подложку катализатора нагружают оксид щелочного металла.
Таким образом, целью настоящего изобретения является катализатор, содержащий или состоящий из подложки и от 5 до 20 вес. % промотора в пересчете на общий вес катализатора, где подложка содержит или состоит из диоксида титана, диоксида циркония и/или их смеси, и промотор представляет собой оксид щелочного металла.
Отклонения от содержания, составляющего от 5 до 20 вес. % промотора в пересчете на общий вес катализатора, также находятся в пределах объема настоящего изобретения при условии, что они также приводят к эффектам настоящего изобретения.
В соответствии с настоящим изобретением подложка катализатора содержит или состоит из диоксида титана, диоксида циркония и/или их смеси. Таким образом, подложка необязательно должна состоять из диоксида титана, диоксида циркония и/или их смеси. При этом учитываются случаи, когда материалы также содержат связующие, наполнители или любые другие компоненты их производства. Однако независимо от этого основное количество материала должно представлять собой диоксид титана, диоксид циркония и/или их смесь. Предпочтительно подложка катализатора содержит по меньшей мере 50 вес. %, в частности от 55 до 100 вес. %, от 60 до 100 вес. %, от 65 до 100 вес. %, от 70 до 100 вес. %. от 75 до 100 вес. %, от 80 до 100 вес. %, от 85 до 100 вес. %, от 90 до 100 вес. % или от 95 до 100 вес. % диоксида титана, диоксида циркония и/или их смесей. В крайнем случае подложка состоит из диоксида титана, диоксида циркония и/или их смеси.
Материалы подложек в соответствии с настоящим изобретением отличаются от материалов из предшествующего уровня техники не только элементным составом, но и структурой. Они содержат или состоят из тетрагональной фазы. В то же время, чистый γ-Al2O3 в качестве подложки катализаторов в предшествующем уровне техники характеризуется кубической фазой.
В одном варианте осуществления по меньшей мере часть подложки катализатора в соответствии с настоящим изобретением характеризуется тетрагональной фазой. Предпочтительно подложка катализатора в соответствии с настоящим изобретением состоит из тетрагональной фазы.
Катализаторы из предшествующего уровня техники, как правило, содержат вольфрамат щелочного металла в качестве промотора. Однако было обнаружено, что для обеспечения катализатора с необходимой селективностью образования алкилмеркаптана в реакции тиолирования алкилового спирта не обязательно нужен вольфрамат щелочного металла. Напротив, достаточно даже применять оксид щелочного металла в качестве промотора. Во время фазы запуска катализатора оксид щелочного металла сульфидируют.
В принципе, настоящее изобретение не ограничено в отношении выбора конкретного щелочного металла для промотора. Таким образом, щелочной металл может представлять собой любой известный щелочной металл, предпочтительно натрий, калий, цезий или рубидий. Однако цезий является щелочным металлом, который в наибольшей степени улучшает селективность катализатора.
Таким образом, в дополнительном варианте осуществления способа получения катализатора в соответствии с настоящим изобретением щелочной металл представляет собой натрий, калий, цезий или рубидий.
В другом варианте осуществления катализатора в соответствии с настоящим изобретением промотор представляет собой оксид цезия.
Нагруженные Cs катализаторы в соответствии с настоящим изобретением обеспечивают значительное увеличение селективности в отношении метилмеркаптана по сравнению с катализаторами из предшествующего уровня техники. Для катализатора на основе ZrO2 с нагрузкой 10 вес. % Cs селективность в отношении метилмеркаптана увеличивалась до диапазона от SCH3SH, 300°C = 99,9% до SCH3SH, 360°C = 99,1%. Единственным обнаруженным побочным продуктом являлся диметилсульфид, селективность в отношении которого составляла от SDMS, 300°C = 0,1% до SDMS, 360°C = 0,9%. Увеличение нагрузки Cs до 20 вес. % приводило к еще большей селективности, составляющей от SCH3SH, 300°C = 99,9% до SCH3SH, 360°C = 99,4%. Аналогичные результаты были получены для катализаторов на основе TiO2: при нагрузке 10 вес. % Cs селективность в отношении метилмеркаптана увеличивалась до диапазона от SCH3SH, 300°C = 99,9% до SCH3SH, 360°C = 99,4%. И в этом случае единственным обнаруженным побочным продуктом являлся диметилсульфид, селективность в отношении которого составляла от SDMS, 300°C = 0,1% до SDMS, 360°C = 0,6%. Увеличение нагрузки Cs до 20 вес. % и в этом случае приводило к еще большей селективности, составляющей от SCH3SH, 300°C = 99,9% до SCH3SH, 360°C = 99,5%. Селективность в отношении метилмеркаптана, достигаемая с помощью катализатора в соответствии с настоящим изобретением, в абсолютном выражении более чем на 2% выше, чем селективность в отношении метилмеркаптана, достигаемая с помощью катализаторов из предшествующего уровня техники. Для сравнения, лучшие катализаторы, описанные в документе WO 2013/092129 A1, обеспечивают селективность в отношении метилмеркаптана, составляющую в лучшем случае 97,9%.
Катализатор в соответствии с настоящим изобретением содержит от 5 до 20 вес. % промотора в пересчете на общий вес катализатора. В случае, если катализатор представляет собой оболочечный катализатор, количество от 5 до 20 вес. % относится к составу оболочки.
В принципе, катализатор в соответствии с настоящим изобретением не ограничен в отношении своей формы. В своей простейшей форме указанный катализатор представляет собой катализатор на подложке, где щелочной металл в окисленной и/или сульфидной форме нанесен на подложку, содержащую диоксид циркония и/или диоксид титана. В этом случае тело подложки непосредственно пропитывают водным пропиточным раствором, содержащим соединение с щелочным металлом в окисленной и/или сульфидной форме, с получением катализатора в форме катализатора на подложке. Тело подложки не ограничено в отношении конкретного размера частиц. Например, оно может присутствовать в виде порошка с размером частиц, составляющим менее 1000 мкм, менее 500 мкм, не более 250 мкм, например, от 125 до 250 мкм, или не более 125 мкм, например, от 25 до 125 мкм, определяемым во влажной дисперсии путем лазерного рассеяния в соответствии с международным стандартом ISO 13320 (2009). Таким образом, катализатор в соответствии с настоящим изобретением также может присутствовать в форме экструдатов или пеллет, получаемых путем пропитки порошкообразной подложки водным раствором, содержащим соединение, содержащее щелочной металл в окисленной и/или сульфидной форме, с последующим высушиванием и прокаливанием, где прокаливание превращает щелочной металл в окисленной и/или сульфидной форме в оксид щелочного металла. Полученную таким образом каталитическую массу смешивают со связующим и подвергают формованию с получением цельного катализатора. Как правило, полученный таким образом катализатор снова подвергают прокаливанию, при котором выжигают любые связующие, с необязательной последующей стадией термообработки при температуре от 100°C до 200°C.
В дополнительном варианте осуществления катализатор в соответствии с настоящим изобретением представляет собой цельный катализатор.
При получении катализатора типа «ядро-оболочка» порошкообразную подложку, как упомянуто выше, пропитывают водным раствором, содержащим соединение, содержащее щелочной металл в окисленной и/или сульфидной форме. Полученную таким образом смесь необязательно прокаливают, смешивают со связующим и наносят на инертное ядро подложки в форме сферы, например, из керамического материала, с последующим прокаливанием и необязательно термообработкой с получением катализатора типа «ядро-оболочка».
В другом варианте осуществления катализатор в соответствии с настоящим изобретением представляет собой катализатор типа «ядро-оболочка».
Катализатор в соответствии с настоящим изобретением требует только присутствия щелочного металла в окисленной и/или сульфидной форме на подложке, но не обязательно присутствия соединения с вольфрамом в окисленной форме, такого как вольфрамовая кислота или вольфрамат. Это также значительно упрощает получение катализатора по сравнению со способами из предшествующего уровня техники, где необходимо было соблюдать конкретное соотношение щелочного металла и вольфрама.
Другой целью настоящего изобретения является способ получения катализатора в соответствии с настоящим изобретением, включающий стадии:
a) пропитки подложки, содержащей или состоящей из диоксида титана, диоксида циркония и/или их смеси, водным раствором, содержащим растворимое соединение щелочного металла, с получением пропитанной подложки,
b) высушивания пропитанной подложки, полученной на стадии a), и
c) прокаливания высушенной пропитанной подложки, полученной на стадии b), с получением катализатора.
Для нанесения пропиточного раствора на подложку можно применять различные техники пропитки, такие как пропитка погружением, пропитка распылением, вакуумная пропитка и пропитка с заполнением объема пор. Это также позволяет проводить пропитку более одного раза. В случае формованных изделий выбранный способ пропитки должен позволять наносить необходимое количество промотора с хорошей однородностью его распределения по всему поперечному сечению формованных изделий. Пропиточный раствор предпочтительно наносят на формованные изделия в одну или две стадии путем пропитки распылением или вакуумной пропитки. При пропитке распылением на тела подложек распыляют водный пропиточный раствор. При вакуумной пропитке создают пониженное давление с помощью вакуумного насоса в сосуде, заполненном формованными изделиями. При открытии соединительного элемента для подачи водного пропиточного раствора раствор всасывается в сосуд до тех пор, пока не покроет всю загрузку формованных изделий. По завершении периода пропитки, составляющего от 0,2 до 2 часов, не впитанный материалом раствор выгружают или сливают. Первоначальный градиент концентрации по поперечному сечению формованных изделий можно практически полностью уравновесить путем их предварительной сушки при комнатной температуре в течение периода времени от 1 до 10 часов. Таким образом улучшается однородность пропитки по поперечному сечению частиц катализатора. Полученные таким образом предшественники катализатора предпочтительно высушивают в течение ночи, например, в течение периода времени от 1 до 10 часов при температуре от 50°C до 100°C, предпочтительно от 60°C до 80°C, для удаления остаточной влаги. Затем их прокаливают в течение периода времени от 1 до 20 часов, предпочтительно от 1 до 5 часов, при температуре от 300 до 600°С, предпочтительно от 420 до 480°С. В результате, щелочной металл в окисленной и/или сульфидной форме из пропиточного раствора переходит в оксид щелочного металла в качестве промотора, при этом указанный промотор фиксируется на подложке, а содержащийся в пропиточном растворе анион разрушается и удаляется. Во время сушки и прокаливания необязательно можно пропускать поток газа через загрузку тел подложек для предшественников катализатора, что улучшает удаление остаточной влаги и газообразных продуктов разложения. Отклонения от явно упомянутых значений периода времени и температуры прокаливания находятся в пределах объема настоящего изобретения при условии, что они приводят к тому же качеству эффекта, что и явно упомянутые значения.
Способ в соответствии с настоящим изобретением не ограничен в отношении выбора соединения щелочного металла для пропиточного раствора. Единственным требованием является то, что соединение щелочного металла должно обладать достаточной растворимостью в воде, чтобы нагрузить на подложку необходимую концентрацию щелочного металла, и что анион должен легко разлагаться во время стадии прокаливания. Таким образом, предпочтительно применять гидроксид щелочного металла, ацетат щелочного металла, карбонат щелочного металла или нитрат щелочного металла.
Несмотря на вышесказанное, также можно применять соединение щелочного металла с относительно низкой растворимостью в воде. Если низкая растворимость соединения щелочного металла в воде не позволяет получить необходимую нагрузку щелочного металла за одну стадию пропитки, пропитка подложки также может проводиться в несколько стадий, в частности в две стадии. Например, пропиточный раствор, применяемый на первой стадии, содержит от одной до двух третей от общего количества соединения, содержащего щелочной металл в окисленной и/или сульфидной форме, а оставшееся количество наносят на подложку на второй или любой дополнительной стадии. В случае многостадийной, например, двухстадийной процедуры, промежуточный продукт, полученный на первой стадии, необязательно не кальцинируют. Кроме того, на второй стадии выполняют ту же программу пропитки, высушивания и прокаливания, которая описана для одностадийного способа.
В одном варианте осуществления способа получения катализатора в соответствии с настоящим изобретением стадии от a) до b) или от a) до c) повторяют по меньшей мере один раз.
Катализатор, полученный таким образом, в частности, на стадии с), можно смешивать со связующим и затем подвергать способу формования, такому как экструзия или гранулирование, с получением цельного катализатора. Полученные таким образом экструдаты или пеллеты подвергают окончательному прокаливанию и необязательно термообработке.
В другом варианте осуществления способ получения катализатора дополнительно включает стадию
d1) формования катализатора, полученного на стадии c) способа получения катализатора, с получением цельного катализатора.
В качестве альтернативы также можно наносить катализатор со стадии с) на ядро с получением катализатора типа «ядро-оболочка». С этой целью катализатор, полученный на стадии c), суспендируют в растворителе, предпочтительно в воде, смешивают со связующим, и полученную таким образом смесь наносят на инертное ядро, например, изготовленное из керамического материала, например, путем высушивания распылением с последующим прокаливанием, чтобы удалить растворитель и выжечь связующее, и необязательно термообработкой.
В альтернативном варианте осуществления способ получения катализатора дополнительно включает стадию
d2) нанесения катализатора, полученного на стадии c) способа получения катализатора, на ядро с получением катализатора типа «ядро-оболочка».
Катализаторы в соответствии с настоящим изобретением, а также катализаторы, полученные посредством способа в соответствии с настоящим изобретением, подходят для катализируемой реакции алкиловых спиртов с сероводородом с образованием алкилмеркаптанов, также известной как тиолирование алкиловых спиртов.
Таким образом, дополнительной целью настоящего изобретения является способ получения алкилмеркаптана, где алкиловый спирт реагирует с сероводородом в присутствии катализатора в соответствии с настоящим изобретением или катализатора, полученного посредством способа в соответствии с настоящим изобретением.
В принципе, способ тиолирования в соответствии с настоящим изобретением не ограничен применением конкретного алкилового спирта или получением конкретного алкилмеркаптана. Однако экономически наиболее релевантным алкиловым спиртом является метилмеркаптан.
Таким образом, в варианте осуществления способа тиолирования в соответствии с настоящим изобретением алкиловый спирт, который вступает в реакцию, представляет собой метанол, и получаемый алкилмеркаптан представляет собой метилмеркаптан.
Настоящее изобретение дополнительно проиллюстрировано следующими пунктами.
1. Катализатор, содержащий подложку и промотор, где подложка содержит диоксид титана, диоксид циркония и/или их смесь, и промотор представляет собой щелочной металл в окисленной и/или сульфидной форме.
2. Катализатор по пункту 1, где по меньшей мере часть подложки имеет тетрагональную фазу.
3. Катализатор по пункту 1 или 2, где промотор представляет собой оксид щелочного металла и/или сульфид щелочного металла.
4. Катализатор по любому из пунктов 1-3, где щелочной металл представляет собой натрий, калий, цезий или рубидий.
5. Катализатор по любому из пунктов 1-4, где катализатор содержит не более 25 вес. % промотора в пересчете на общий вес катализатора.
6. Катализатор по любому из пунктов 1-5, где катализатор содержит от 5 до 20 вес. % промотора в пересчете на общий вес катализатора.
7. Катализатор по любому из пунктов 1-6, где промотор представляет собой оксид цезия и/или сульфид цезия, а катализатор содержит от 5 до 20 вес. % указанного промотора в пересчете на общий вес катализатора.
8. Катализатор по любому из пунктов 1-7, где катализатор представляет собой цельный катализатор.
9. Катализатор по любому из пунктов 1-8, где катализатор представляет собой катализатор типа «ядро-оболочка».
10. Способ получения катализатора на подложке по любому из пунктов 1-7, включающий стадии
a) пропитки подложки, содержащей диоксид титана, диоксид циркония и/или их смесь, водным раствором, который содержит растворимое соединение щелочного металла,
b) высушивания пропитанной подложки, полученной на стадии a), и
c) прокаливания высушенной пропитанной подложки из стадии b) с получением катализатора.
11. Способ по пункту 10, где стадии от a) до c) повторяют по меньшей мере один раз.
Способ по пункту 10 или 11, дополнительно включающий стадию
d1) формования катализатора, полученного на стадии c), с получением цельного катализатора.
13. Способ по пункту 10 или 11, дополнительно включающий стадию
d2) нанесения катализатора, полученного на стадии c), на ядро с получением катализатора типа «ядро-оболочка».
14. Способ получения алкилмеркаптана, где алкиловый спирт реагирует с сероводородом в присутствии катализатора по любому из пунктов 1-9 или катализатора, полученного посредством способа по любому из пунктов 10-13.
15. Способ по пункту 14, где алкиловый спирт, который вступает в реакцию, представляет собой метанол, и получаемый алкилмеркаптан представляет собой метилмеркаптан.
Описание графических материалов
На фигуре 1 показаны рентгенограммы XRD чистых оксидов металлов (a) и оксидов металлов, нагруженных 5 вес. % Cs (b), 10 вес. % Cs (c), 15 вес. % Cs (d) и 20 вес. % Cs (e), где γ указывает на характеристические сигналы чистого γ-Al2O3, t указывает на тетрагональный ZrO2, A указывает на анатаз (TiO2), и Cs указывает на Cs2CO3.
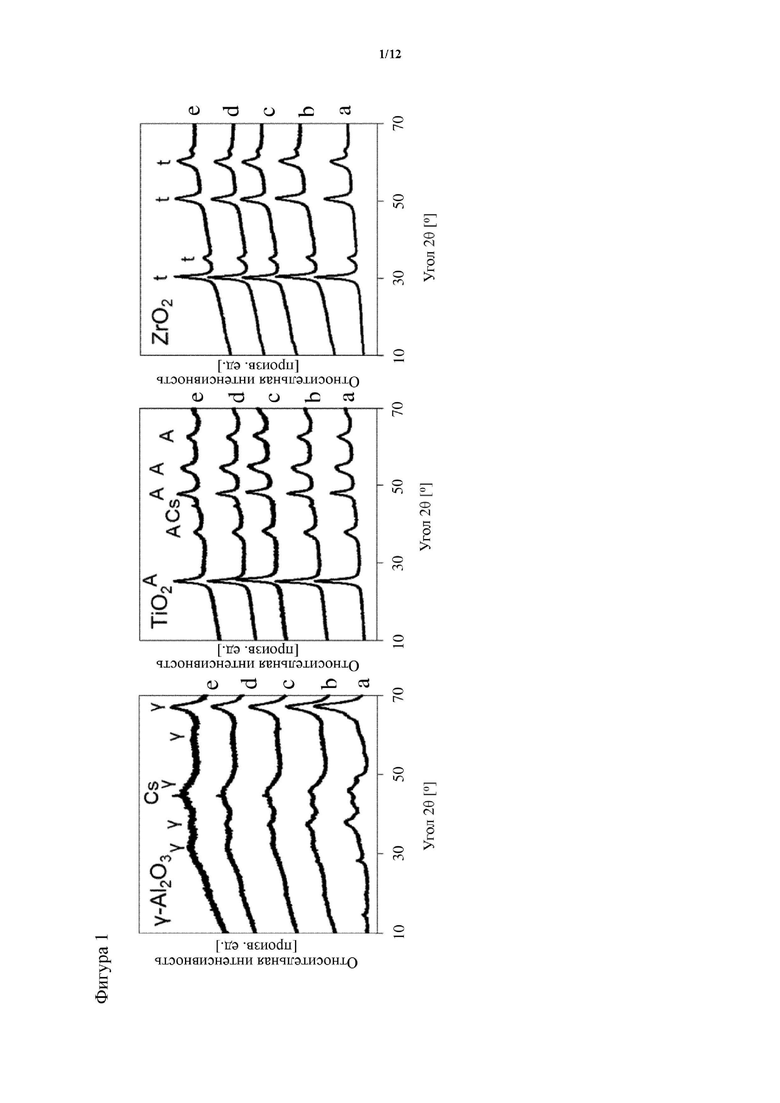
На фигуре 2 показаны вычтенные ИК-спектры пиридина, адсорбированного на чистых оксидах металлов при 50°C, где сплошная линия представляет собой ИК-спектр, снятый при парциальном давлении пиридина, составляющем 0,1 мбар, а пунктирная линия представляет собой ИК-спектр, снятый после вакуумирования при 10-7 мбар.
На фигуре 3 показаны разностные спектры области колебаний OH-групп γ-Al2O3, ZrO2 и TiO2, где сплошная линия представляет собой ИК-спектр, снятый при парциальном давлении пиридина, составляющем 0,1 мбар, а пунктирная линия представляет собой ИК-спектр, снятый после вакуумирования при 10-7 мбар.
На фигуре 4 показаны вычтенные ИК-спектры оксидов металлов с нагрузкой Cs, составляющей 10 или 20 вес. % при 50°C, где сплошная линия представляет собой ИК-спектр, снятый при парциальном давлении пиридина, составляющем 0,1 мбар, а пунктирная линия представляет собой ИК-спектр, снятый после вакуумирования при 10-7 мбар.
На фигуре 5 показаны ИК-спектры CO, адсорбированного на γ-Al2O3 (слева), ZrO2 (посередине) и TiO2 (справа) при парциальном давлении CO, составляющем 5 мбар, и при -150°C, где (a) указывает на чистые оксиды металлов, (b) указывает на нагрузку Cs, составляющую 10 вес. %, а (c) указывает на нагрузку Cs, составляющую 20 вес. %.
На фигуре 6 показаны ИК-спектры метанола, адсорбированного на Al2O3 (слева), ZrO2 (посередине) и TiO2 (справа) при парциальном давлении метанола, составляющем 0,1 мбар, и при 50°C, где (a) указывает на чистые оксиды металлов, (b) указывает на нагрузку Cs, составляющую 10 вес. %, а (c) указывает на нагрузку Cs, составляющую 20 вес. %.
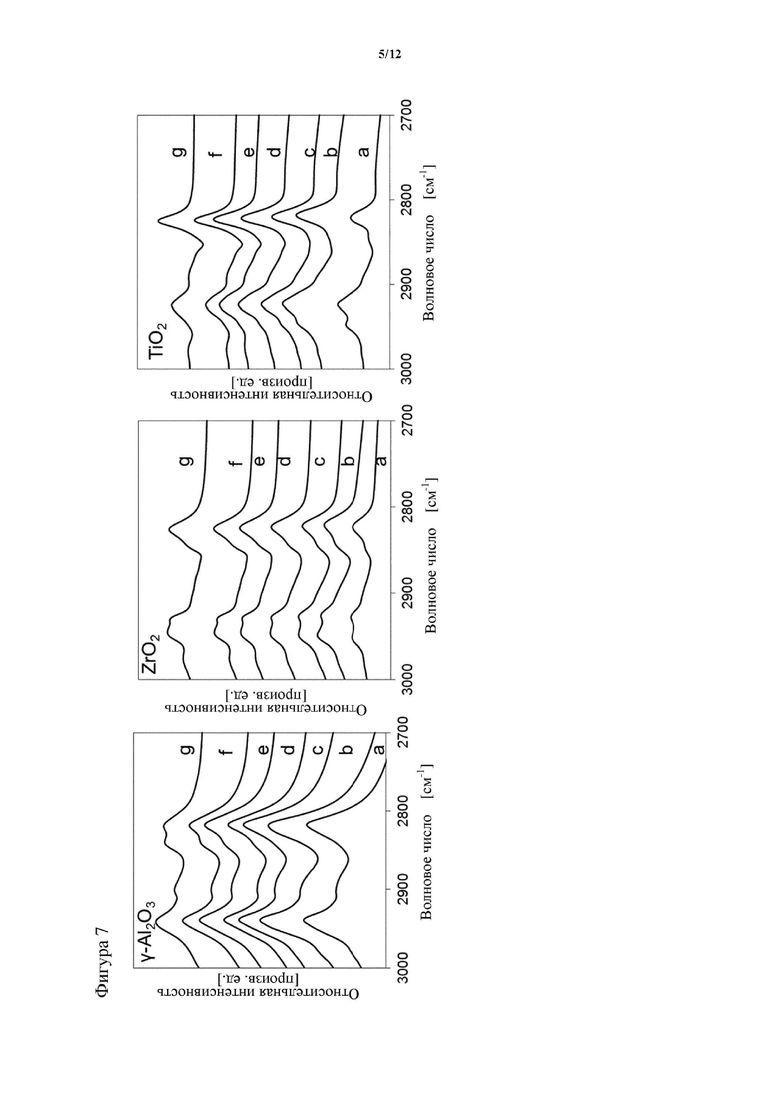
На фигуре 7 показаны ИК-спектры метанола, адсорбированного на чистых оксидах металлов γ-Al2O3 (слева), ZrO2 (посередине) и TiO2 (справа) при значениях температуры и парциального давления метанола, составляющих (a) 50°C и 0,1 мбар, (b) 50°C и 1 мбар, (c) 100°C и 1 мбар, (d) 150°C и 1 мбар, (e) 200°C и 1 мбар, (f) 250°C и 1 мбар и (g) 300°C и 1 мбар.
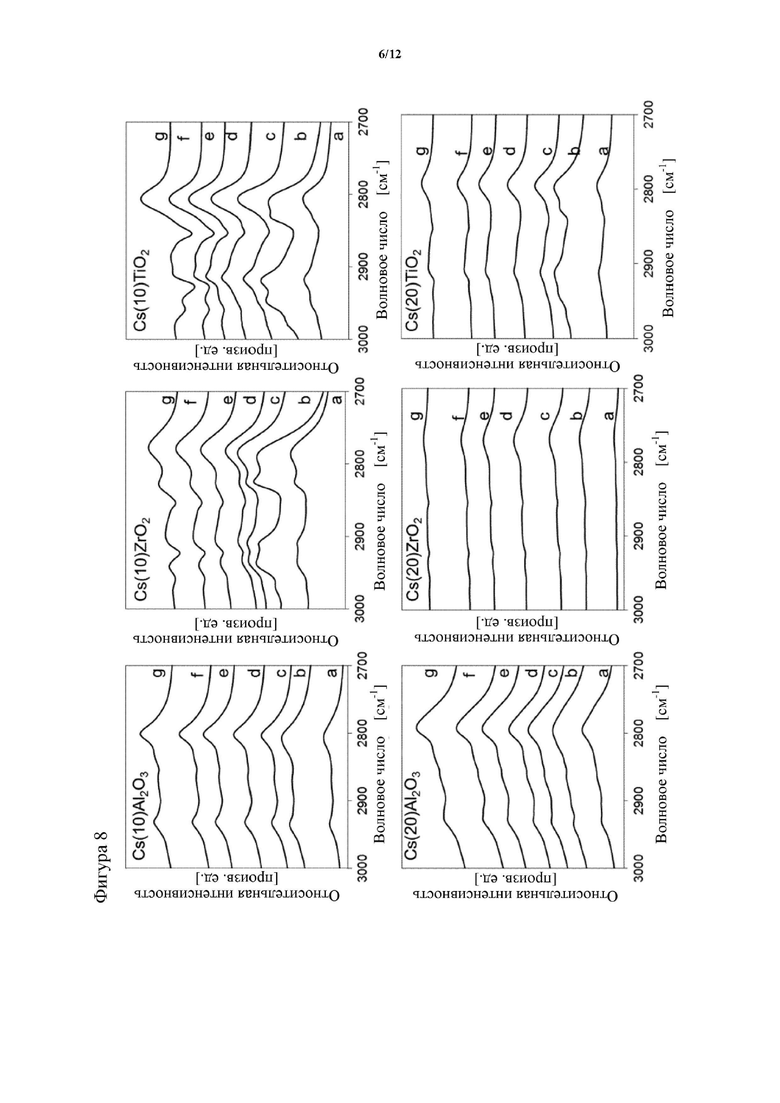
На фигуре 8 показаны ИК-спектры метанола на γ-Al2O3 c 10 и 20 вес. % Cs (слева), ZrO2 c 10 и 20 вес. % Cs (посередине) и TiO2 c 10 и 20 вес. % Cs (справа) при значениях температуры и парциального давления метанола, составляющих (a) 50°C и 0,1 мбар, (b) 50°C и 1 мбар, (c) 100°C и 1 мбар, (d) 150°C и 1 мбар, (e) 200°C и 1 мбар, (f) 250°C и 1 мбар и (g) 300°C и 1 мбар.
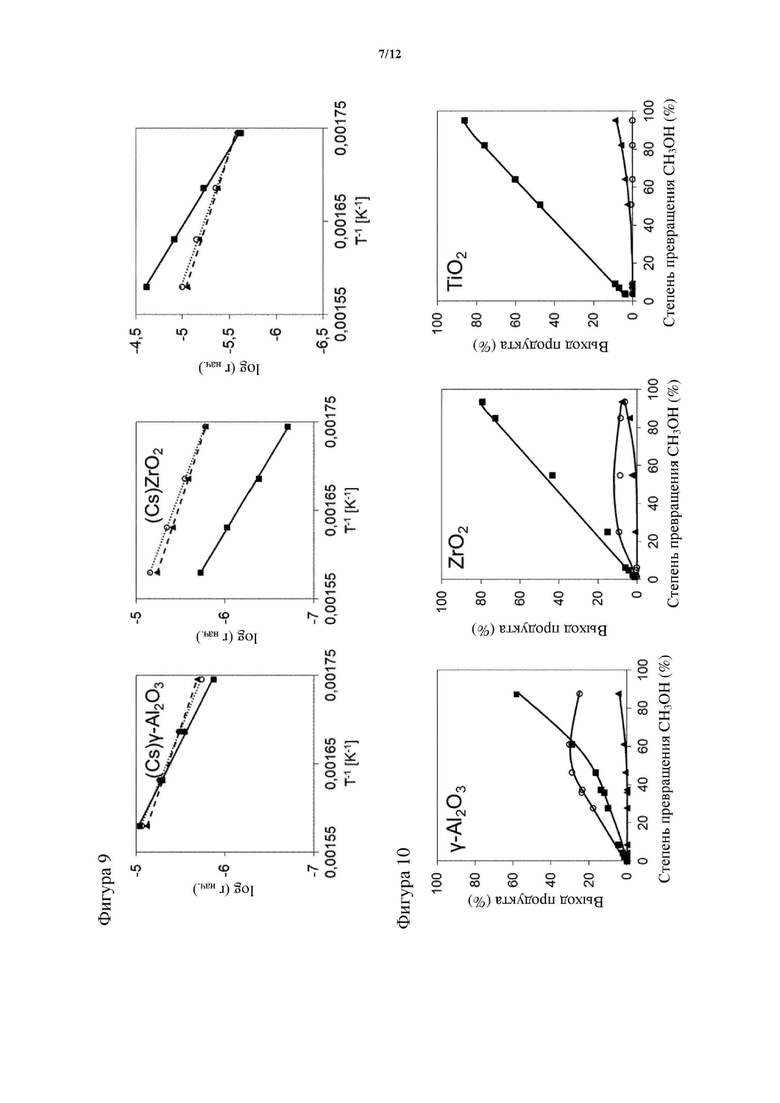
На фигуре 9 показаны начальные скорости образования метилмеркаптана на γ-Al2O3 (слева), ZrO2 (посередине) и TiO2 (справа) при температуре от 300°C до 360°C для чистых оксидов металлов (сплошная линия с кубиками), нагрузки Cs, составляющей 10 вес. % (точечная линия с кружками), и нагрузки Cs, составляющей 20 вес. % (пунктирная линия с треугольниками).
На фигуре 10 показаны выходы метилмеркаптана (кубики), диметилового эфира (кружки) и диметилсульфида (треугольники) в зависимости от степени превращения метанола на чистых оксидах металлов γ-Al2O3 (слева), ZrO2 (посередине) и TiO2 (справа) при 360°C.
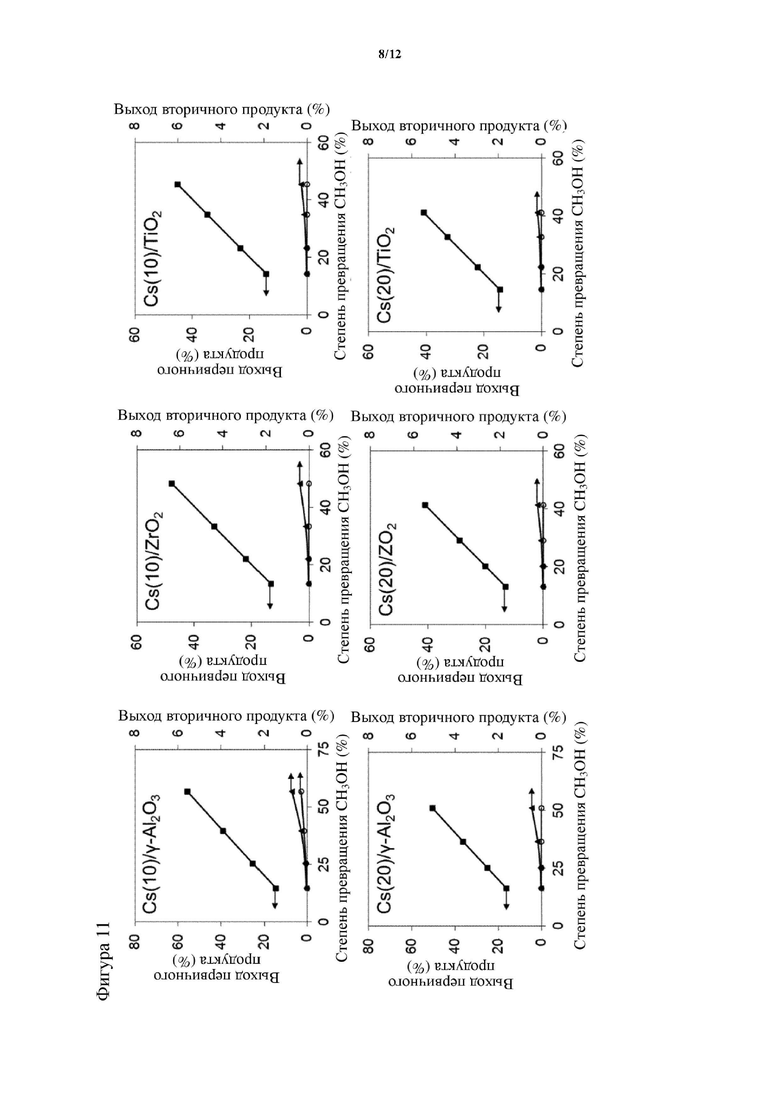
На фигуре 11 показаны значения выхода метилмеркаптана (кубики), диметилового эфира (кружки) и диметилсульфида (треугольники) в зависимости от степени превращения метанола на γ-Al2O3 с 10 и 20 вес. % Cs (слева), ZrO2 c 10 и 20 вес. % Cs (посередине) и TiO2 c 10 и 20 вес. % Cs (справа) при значениях температуры от 300°C до 360°C.
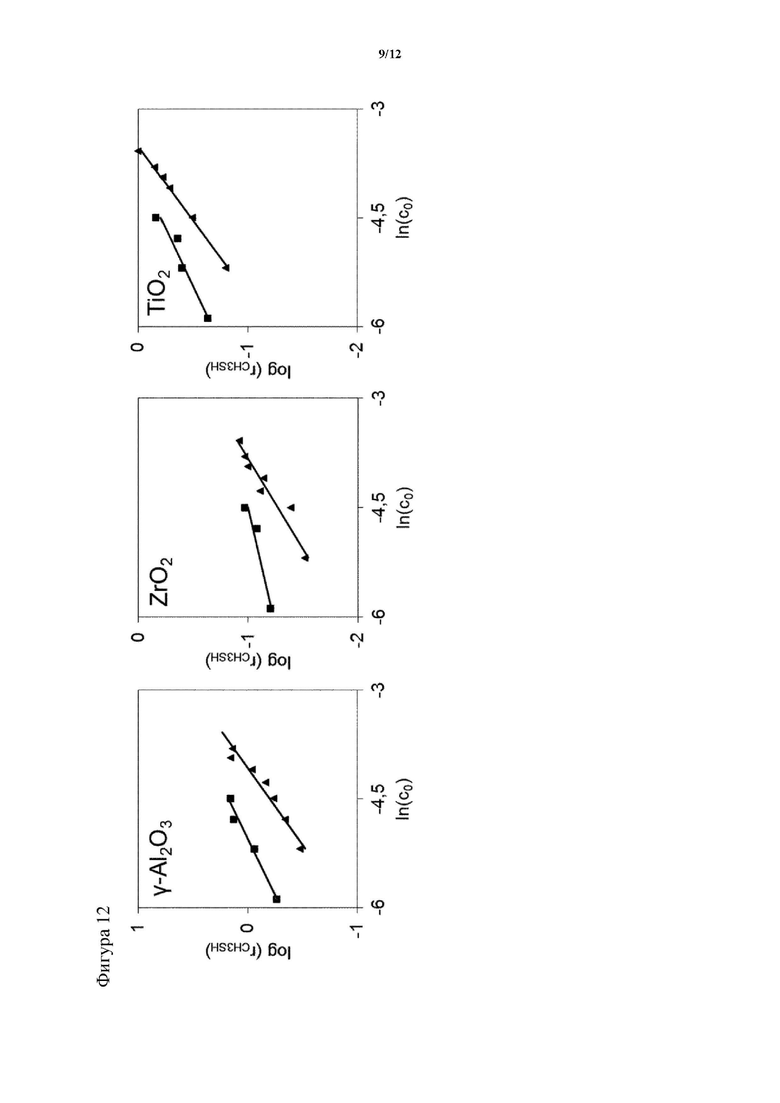
На фигуре 12 показана зависимость скорости образования метилмеркаптана на γ-Al2O3 в метаноле (кубики; y = 0,3x + 1,7) и сероводороде (треугольники; y = 0,5x + 1,9), ZrO2 в метаноле (кубики; y = 0,2x - 0,3) и сероводороде (треугольники; y = 0,4x - 0,6) и TiO2 в метаноле (кубики; y = 0,3x + 1,2) и сероводороде (треугольники; y = 0,5x + 1,7), концентрация в моль/л.
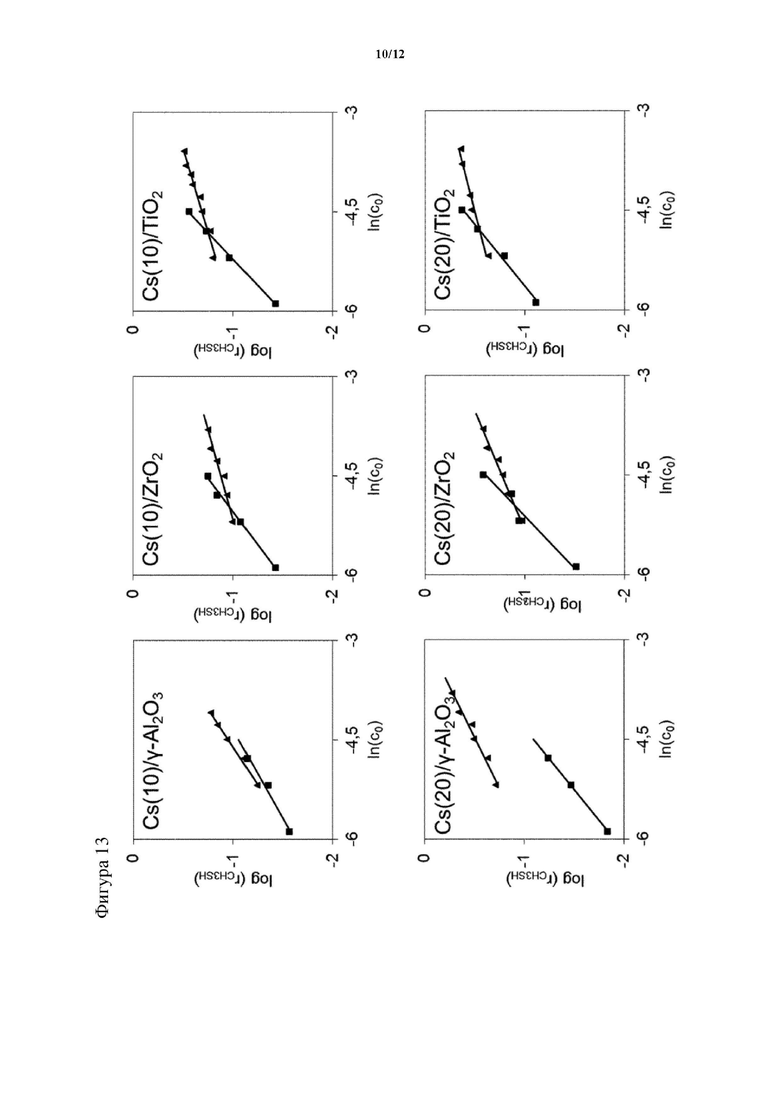
На фигуре 13 показана зависимость скорости образования метилмеркаптана на катализаторах с разными нагрузками цезия: 10 вес. % Cs (первый ряд) в метаноле, обозначенном кубиками (γ-Al2O3: y = 0,4x + 1,0; ZrO2: y = 0,5x + 1,5; и TiO2: y = 0,6x + 2,3), и сероводороде, обозначенном треугольниками (γ-Al2O3: y = 0,4x + 0,6; ZrO2: y = 0,3x - 0,01; и TiO2: y = 0,2x + 0,2). 20 вес. % Cs (второй ряд) в метаноле, обозначенном кубиками (γ-Al2O3: y = 0,3x + 1,0; ZrO2: 0,6x + 2,3; и TiO2: y = 0,5x + 2,0), и сероводороде, обозначенном треугольниками (γ-Al2O3: y = 0,5x + 1,3; ZrO2: y = 0,3x + 0,5; и TiO2: y = 0,2x + 0,3), концентрация в моль/л.
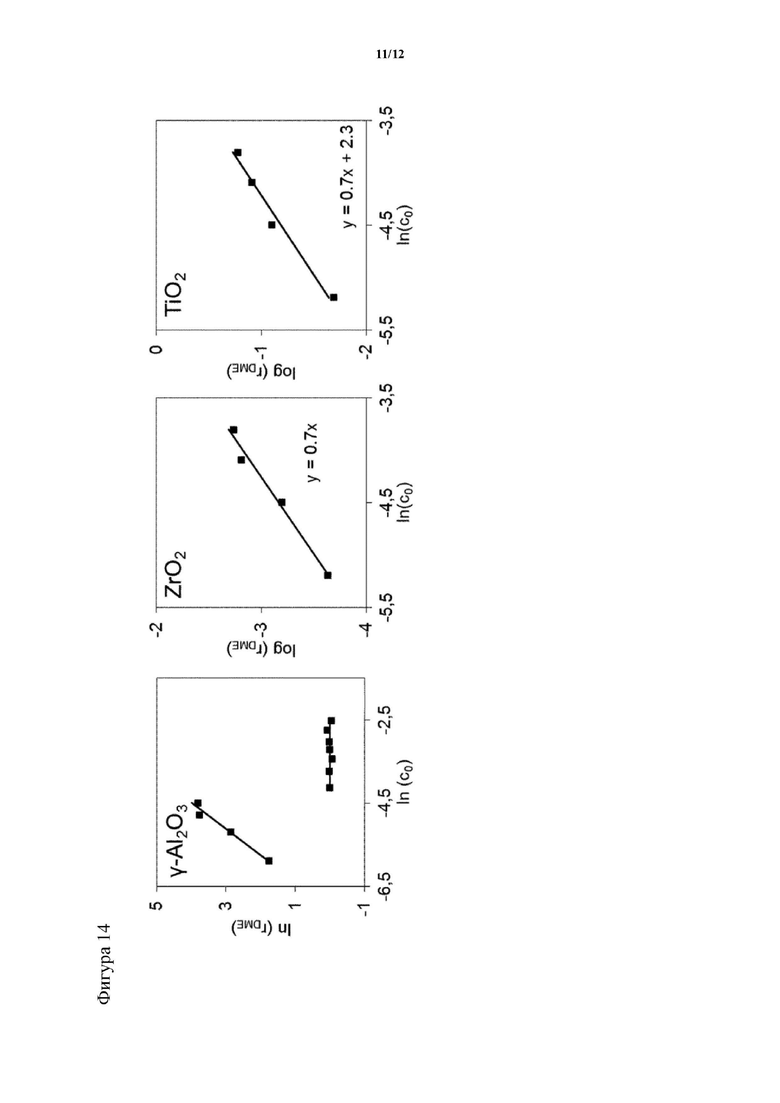
На фигуре 14 показана зависимость скорости образования диметилового эфира на чистом γ-Al2O3 в метаноле (обозначенном кубиками; y = 1,5x + 11,0) и сероводороде (обозначенном треугольниками; y = 0,0x), на чистом ZrO2 в метаноле (обозначенном кубиками; y = 0,7x) и на чистом TiO2 (обозначенном кубиками; y = 0,7x + 2,3), концентрация в моль/л.
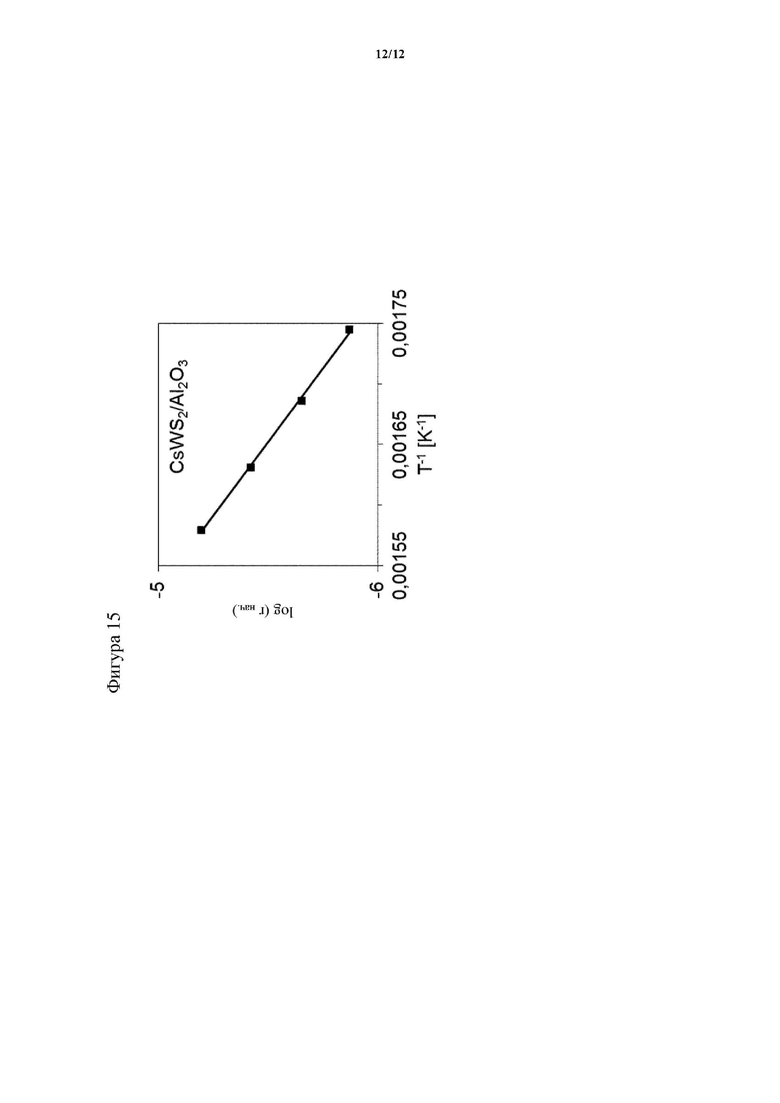
На фигуре 15 показаны начальные скорости образования метилмеркаптана на γ-Al2O3, нагруженном CsWS2 (с содержанием вольфрама 5,1 вес. % и содержанием цезия 20,6 вес. %) при температурах 300°C, 320°C, 340°C и 360°C (закрашенные квадратики).
Примеры
1. Получение нагруженных Cs оксидов металлов в соответствии с настоящим изобретением
Катализаторы с нагрузкой Cs, составляющей 5, 10, 15 и 20 вес. % в пересчете на общий вес катализаторов, получали путем капиллярной пропитки коммерчески доступных оксидов металлов γ-Al2O3 (Spheralite 101, Axens), TiO2 (Hombikat 100 UV, Sachtleben) и ZrO2 (SZ 61152, Norpro), каждый из которых имеет размер зерна 0,125-0,25 мм, водным раствором ацетата цезия, добавляемым по каплям к перемешиваемому твердому веществу. Для каждой нагрузки Cs получали разные пропиточные растворы, содержащие необходимое количество ацетата цезия для обеспечения необходимой нагрузки Cs. 76 мг ацетата цезия (Sigma Aldrich, ≥ 99,99%) растворяли в 0,5 мл H2O на 1 г подложки с получением нагрузки Cs, составляющей 5 вес. %, соответственно растворяли 160,5 мг ацетата цезия с получением нагрузки Cs, составляющей 10 вес. %, 255 мг ацетата цезия с получением нагрузки Cs, составляющей 15 вес. %, и 361,0 мг ацетата цезия с получением нагрузки Cs, составляющей 20 вес. %. Пропитанные оксиды металлов высушивали в течение ночи при 70°C с последующим прокаливанием в потоке синтетического воздуха со скоростью потока 100 мл/мин. и при температуре 400°C в течение 2 ч., достигаемой при скорости изменения температуры 0,5°C/мин. Перед применением в каталитических испытаниях все образцы активировали путем обработки в H2S со скоростью потока 20 мл/мин. при температуре 360°C в течение 2 часов.
2. Определение характеристик полученных катализаторов
2.1 Определение элементного состава и площади поверхности
Элементный состав полученных катализаторов в соответствии с настоящим изобретением определяли с помощью атомно-абсорбционной спектроскопии (AAS). Измерения проводили на АА-спектрометре UNICAM 939. Для определения текстурных характеристик проводили физическую адсорбцию N2 на сорбтометре Porous Materials Inc. BET-121. После активации при 250°C в течение 2 ч. в вакууме проводили адсорбцию N2 при температуре 77,4°К. Площадь поверхности рассчитывали по методу BET. Результаты элементного анализа и определения площади поверхности всех полученных катализаторов обобщены в таблице 1 ниже.
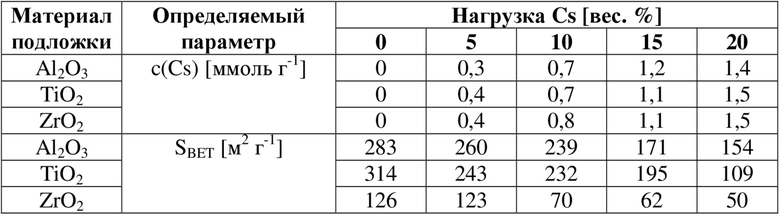
Таблица 1. Результаты элементного анализа и определения площади поверхности для всех полученных катализаторов
Результаты показывают, что сопоставимые нагрузки Cs достигались для каждого из трех типов материалов подложки. Как правило, удельная площадь поверхности полученных катализаторов на подложке уменьшается с увеличением нагрузки Cs. Это может быть связано с увеличенными плотностью катализатора и покрытием поверхности с помощью Cs, что приводит к потере площади поверхности.
В таблице 2 ниже обобщены различные значения массы ацетата цезия в различных пропиточных растворах, применяемые при получении катализаторов, значения массы Cs+ в этих пропиточных растворах (весом ацетатного противоиона пренебрегали), значения массы катализаторов (подложка + Cs+), теоретическая концентрация (CTh(M(цезий)/m(катализатор)) Cs+ в полученных катализаторах и концентрация (CEA (M(цезий)/m(катализатор)) Cs+ в полученных катализаторах, найденные методом элементного анализа.
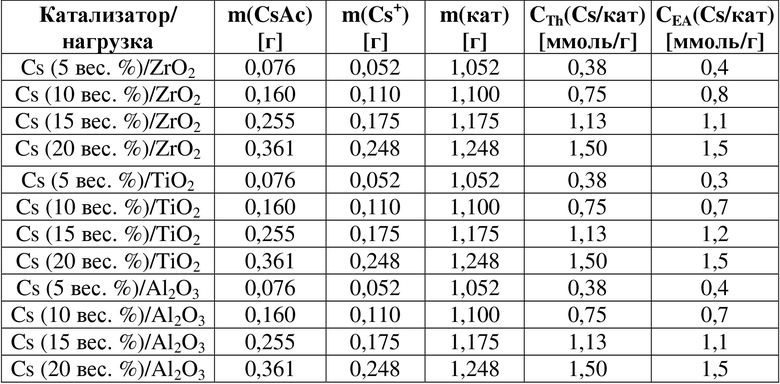
Таблица 2. Обзор полученных катализаторов и концентрации цезия в них.
2.2 Кристаллическая структура
Кристаллическую структуру всех подложек и всех катализаторов определяли методом порошковой дифракционной рентгенографии. Рентгенограммы XRD получали с помощью дифрактометра Philips X’Pert (Cu-Kα-излучение, 0,1542 нм), работающего при 45 кВ/40 мА, с применением никелевого Kβ-фильтра и твердотельного детектора (X’Celerator). Измерения проводили с размером шага 0,017 и временем сканирования 0,31 с на шаг.
Материалы подложки обеспечивали получение ожидаемых дифракционных картин, являясь фазово-чистыми в γ-Al2O3, анатазом в TiO2 и тетрагональным диоксидом циркония в ZrO2. Рентгенограммы XRD показаны на фигуре 1. При добавлении Cs кристаллическая структура материала подложки не изменялась, а дифракционная картина оставалась такой же. Дополнительный дифракционный пик наблюдался на рентгенограмме γ-Al2O3 и TiO2 при 45°C, что указывает на Cs2CO3. Считается, что карбонат образуется в результате реакции частиц Cs на поверхности с атмосферным CO2. При сульфидировании карбонат и его пики исчезали, что приводило к образованию оксианионов серы, которые не были обнаружены методом дифракционной рентгенографии. Других отражений не было. Таким образом, можно сделать вывод, что активные частицы Cs являются аморфными согласно XRD.
2.3 Определение характеристик кислотно-основных свойств
Поглощение СО и пиридина на чистых оксидах металлов и полученных катализаторах контролировали с помощью ИК-спектроскопии в режиме пропускания и поглощения (образцы прессовали в самонесущие пластины) для измерения кислотности по Льюису. Перед адсорбцией образцы нагревали до 360°C со скоростью нагрева 10°C в минуту в потоке гелия 10 мл в минуту. Затем образцы сульфидировали в течение 0,5 ч. при 360°C в потоке 10 мл в минуту 10 об. % сероводорода в азоте. Для удаления физически адсорбированного сероводорода образец продували потоком He со скоростью 10 мл в минуту в течение еще 15 мин., после чего вакуумировали до 10-7 мбар и охлаждали до 50°C. Для адсорбции пиридина ячейку охлаждали до 50°C и образец подвергали воздействию пиридина при парциальном давлении пиридина, составляющем 1 мбар, с последующим уменьшением парциального давления пиридина. Дальнейшее вакуумирование до 10-5 мбар не приводило к адсорбции пиридина на образцах, содержащих Cs. Таким образом, спектры разных катализаторов сравнивали при 0,1 мбар перед вакуумированием. Концентрации координирующего пиридина рассчитывали с помощью интегрального молярного коэффициента экстинкции 0,96 см на мкмоль, определенного для характеристической полосы при 1450 см-1. Адсорбцию CO осуществляли путем охлаждения ИК-ячейки до -150°C с помощью жидкого азота. Спектры записывали при парциальном давлении CO, составляющем 5 мбар.
Адсорбцию метанола проводили при 50°C, постепенно увеличивая парциальное давление метанола (0,1 мбар, 0,5 мбар, 1 мбар и 5 мбар) с последующим повышением температуры до 300°C. Все спектры записывали с помощью спектрометра Nicolet 6700 FTIR (собирали 64 сканирования для получения каждого спектра). Из всех спектров вычитали фон и нормировали к массе пластины.
2.4 Поглощение пиридина
Кислотность оксидов металлов измеряли с помощью адсорбированного пиридина методом ИК-спектроскопии, фигура 2. На чистом γ-Al2O3 наблюдались восемь полос при 1621, 1612, 1591, 1577, 1450 и 1440 см-1. Полосы при 1621 и 1612 см-1 относятся к колебательной моде 8a пиридина, координационно связанного с кислотными центрами Льюиса (LAS) кислот различной силы (волновое число увеличивается с силой кислоты), тогда как полоса при 1579 см-1 относится к колебательной моде 8b. Полоса при 1591 см-1 относится к колебательной моде 8a H-связанного пиридина, образованного путем взаимодействия пиридина со слабыми кислотными поверхностными гидроксильными группами. Сигнал при 1450 см-1 относится к колебанию 9b пиридина на LAS, тогда как полоса при 1440 см-1 снова относится к пиридину, Н-связанному на гидроксильных группах. Центры, которые относятся к пиридину, координационно связанному с LAS (1450 см-1, 1612-1620 см-1), были устойчивы к вакуумированию, тогда как полосы Н-связанного пиридина (1440 и 1593 см-1) исчезали после вакуумирования из-за слабого взаимодействия с молекулярным зондом. Это соответствует высвобождению групп ОН, что приводит к уменьшению отрицательной полосы ОН около области 3700 см-1 по мере десорбции Н-связанного пиридина (фигура 3).
С помощью ИК-спектроскопии пиридина, адсорбированного на ZrO2 и TiO2, получали полосы при 1604, 1593, 1573 и 1445 см-1. Полоса при 1604 см-1 относится к колебательной моде 8a пиридина, связанного с кислотными центрами Льюиса (LAS) ZrO2 и TiO2, тогда как полоса при 1573 см-1 относится к колебательной моде 8a. Полоса при 1593 см-1 относится к колебательной моде 8a H-связанного пиридина, образованного путем взаимодействия пиридина со слабыми кислотными поверхностными гидроксильными группами. Как и в случае с γ-оксидом алюминия, этот сигнал пропадал после вакуумирования. Сигнал при 1445 см-1 относится к колебанию 9b пиридина на LAS. При интегрировании полосы при 1450 см-1 концентрация LAS на оксиде металла составляла 454 мкмоль г-1 на γ-Al2O3, 220 мкмоль г-1 на ZrO2 и 749 мкмоль г-1 на TiO2. Сдвиг сигналов пиридина в область более низких волновых чисел от γ-Al2O3 (1450 см-1) до ZrO2 и TiO2 (1445 см-1) показывает более высокую силу кислоты Льюиса первого, чем остальных двух.
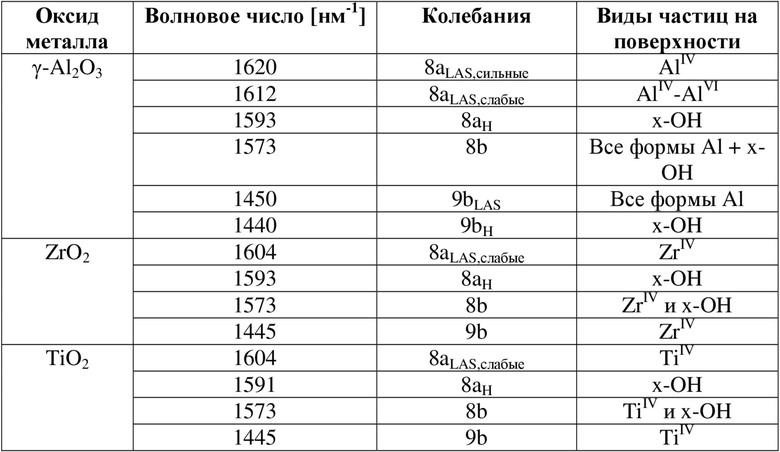
Таблица 3. Краткое описание всех описанных сигналов металла и их отнесений.
Добавление Cs к оксидам металлов приводило к изменению ИК-спектров адсорбированного пиридина (фигура 4). На среднем допированном γ-Al2O3, Cs(10)/γ-Al2O3, полосы, которые относятся к колебательной моде 8a пиридина, координационно связанного с сильными (1612 см-1) и слабыми (1609 см-1) LAS, больше не обнаруживались, как и сигнал Н-связанного пиридина. Появилась новая полоса при 1583 см-1, соответствующая колебательной моде 8a пиридина, координационно связанного с ионом щелочного металла в качестве слабой кислоты Льюиса, т.е. с Cs+ с более низкой силой кислоты Льюиса, чем измеренные на γ-Al2O3. При добавлении Cs на ZrO2 и TiO2, Cs(10)/ZrO2 и Cs(10)/TiO2 не наблюдались полосы, которые относятся к LAS подложки. Как и в случае Cs/γ-Al2O3, появлялись новые полосы при 1600 и 1583 см-1, что соответствует колебательной моде обертона колебаний 1 + 6a и 8a пиридина на Cs, соответственно. После вакуумирования сигналы пиридина на центрах Cs исчезали во всех трех образцах, частично оставаясь на Cs(10)/γ-Al2O3 и Cs(10)/TiO2.
Дополнительное количество Cs, Cs(20)/γ-Al2O3 приводило к уменьшению полосы при 1612 см-1. Наблюдался новый сигнал при 1600 см-1, который относили к обертону колебаний 1 + 6a пиридина на Cs. Уже упоминалось колебание 8a пиридина на центрах Cs при 1583 см-1 и колебание 8b пиридина на LAS и Cs при 1573 см-1. Таким образом, постепенное добавление Cs на поверхность γ-Al2O3 приводило к замене сильных LAS γ-Al2O3 на более слабые LAS Cs. Адсорбция пиридина на катализаторах Cs(20)/ZrO2 и Cs(20)/TiO2 приводила только к получению пиридина, координационно связанного с центрами Cs+ (8a, 8b и 1+6a). Все частицы пиридина, адсорбированные на ZrO2 и TiO2, допированных Cs, десорбировали в вакууме; при этом оставался незначительный сигнал LAS на Cs(20)/γ-Al2O3.
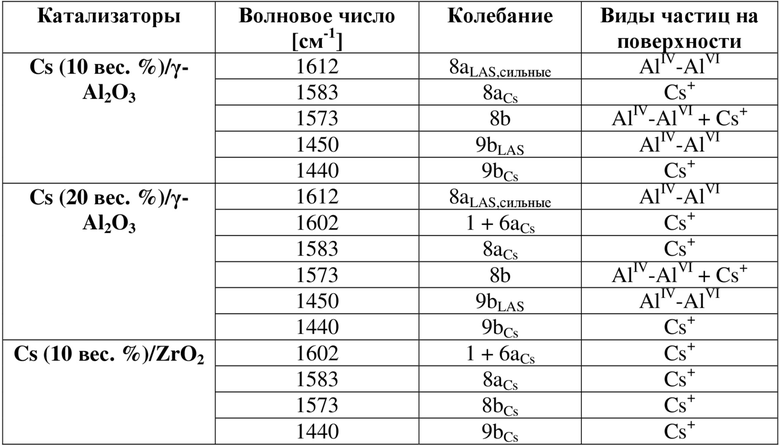
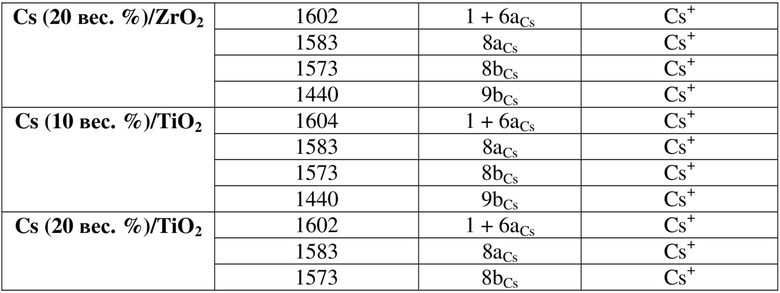
Таблица 4. Отнесение полос поглощения пиридина на нагруженных Cs оксидах металлов.
Титрование кислотных центров пиридином указывает на высокую гетерогенность центров LAS в γ-Al2O3, который характеризуется двумя типами LAS, тогда как TiO2 и ZrO2 характеризуются только одним типом LAS с одинаковой силой на обоих материалах, что совпадает с отмеченными в литературе. Эффект осаждения Cs можно объяснить следующим образом: при средней нагрузке Cs, Cs+ модифицирует центры на поверхности оксида металла путем прямого взаимодействия с увеличением основности поверхности за счет более низкой электроотрицательности Сандерсона. Это прямое взаимодействие осуществляется путем обмена поверхностных протонов на катионы Cs+.
При высокой нагрузке Cs на поверхности преобладает Cs. Как предполагается для калия на TiO2, высокая нагрузка щелочного металла обеспечивает полное покрытие поверхности оксида металла, что приводит к свойствам поверхности, аналогичным свойствам основного щелочного материала.
2.5 Поглощение CO
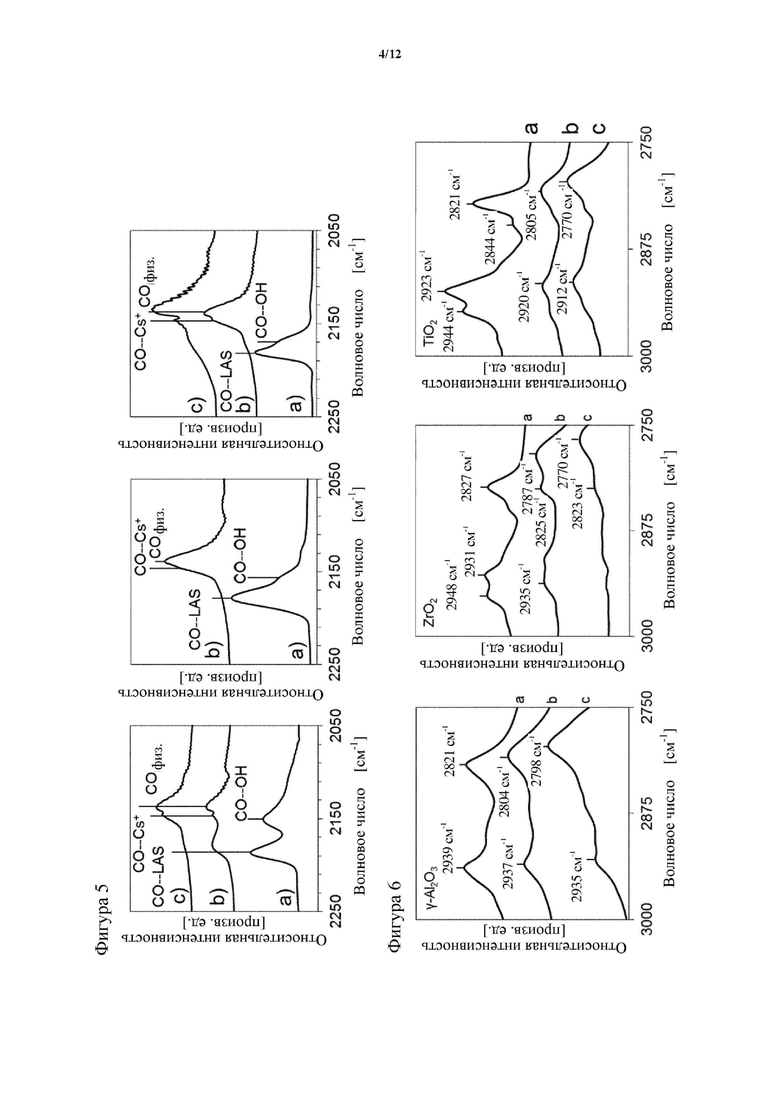
Данные ИК-спектроскопии по адсорбции CO на катализаторах показаны на фигуре 5. Отнесение различных полос поглощения CO приведено в таблице 4. Аналогичные полосы ИК-спектра получали при адсорбции СО на различных оксидах металлов. Полосы между 2180-2190 см-1 относятся к поглощению CO на LAS, тогда как полосы около 2150 см-1 относятся к поверхностному гидроксилу. Валентное колебание CO в γ-Al2O3 (2188 см-1) характеризовалось более высоким волновым числом, чем в ZrO2 (2177 см-1) и TiO2 (2181 см-1), что указывает на более сильное возмущение связи CO. Эта тенденция такая же, как и наблюдаемая для пиридина, что подразумевает более высокую силу кислотных центров Льюиса γ-Al2O3.
Добавление 10 вес. % Cs на три подложки приводило к уменьшению валентного колебания CO на LAS до более низких волновых чисел (2138-2136 см-1), что соответствует адсорбции CO на ионах Cs+. В случае Cs(10)/γ-Al2O3 появляется дополнительная полоса при 2179 см-1, соответствующая LAS в подложке γ-Al2O3, замененным катионом щелочного металла. Для СО, адсорбированного на группах ОН, полосы не наблюдались. В образцах с высокой нагрузкой Cs, равной 20 вес. %, регистрировали только сигналы CO на катионах Cs и физически адсорбированного CO. CO не адсорбировался на образце Cs(20)/ZrO2.
Красный сдвиг валентного колебания CO на LAS с Cs на поверхности γ-Al2O3 связан с увеличением основности и уменьшением электроотрицательности Сандерсона. Результаты для TiO2 и ZrO2 соответствуют результатам адсорбции пиридина, при этом LAS на этих материалах недоступны при нагрузке 10 вес. % Cs. Как и в случае адсорбции пиридина, Cs+ является единственной частицей, доступной для адсорбции CO на материалах, сильнодопированных Cs. Данные ИК-спектроскопии по адсорбции CO соответствуют данным по адсорбции пиридина; кислотные центры Льюиса на подложке отсутствуют при нагрузке 10 вес. % Cs, за исключением Cs(10)/γ-Al2O3.
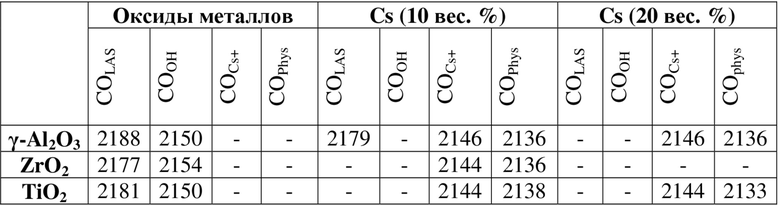
Таблица 5. Отнесение полос поглощения CO на чистых и нагруженных Cs оксидах металлов.
2.6 Поглощение метанола
ИК-спектры метанола, адсорбированного на оксидах металлов и допированных Cs оксидах металлов, показаны на фигуре 6 и демонстрируют полосы в области 3000-2750 см-1 (колебания алкильной группы (sp3) C-H). Область между 3000-2900 см-1 относится к асимметричным валентным колебаниям (υas(CH3)) или их резонансу Ферми с деформационными колебаниями CH3 (2δs(CH3)), тогда как менее интенсивные полосы относятся к симметричным валентным колебаниям (υs(CH3)). Разная интенсивность наблюдалась для полос ИК-спектра, которые относятся к адсорбции метанола на сильных кислотных центрах Льюиса и сильных основных центрах Льюиса, как для υas, так и для υs при 50°C (фигуры 6-8). Первый центр приводил к образованию мостикового метоксида, известного как частица I, с полосами ИК-спектра, которые характеризуются более высоким волновым числом как для υas (2943, 2948 и 2944 см-1 для γ-Al2O3, ZrO2 и TiO2), так и для υs (2845, 2852 и 2844 см-1 для γ-Al2O3, ZrO2 и TiO2). Второй центр приводил к образованию алкоголята (диссоциация группы O-H), известного как частица II, как для υas (2939, 2931 и 2923 см-1 для γ-Al2O3, ZrO2 и TiO2), так и для υs (2821, 2827 и 2821 см-1 для γ-Al2O3, ZrO2 и TiO2). На ZrO2 наблюдалась относительно более высокая концентрация диссоциированного метанола, которая еще дополнительно увеличивалась для TiO2. В случае γ-Al2O3 нагревание ИК-ячейки приводило к увеличению интенсивности мостиковых метоксидов (частица I). В двух других подложках при нагревании значительных изменений не наблюдалось. Относительные интенсивности метанола на поверхностных частицах непосредственно приводят к выводу, что общий кислотный характер оксида металла уменьшается до более основного в порядке γ-Al2O3 > ZrO2 ~ TiO2.
3. Каталитические испытания катализаторов на подложке в соответствии с настоящим изобретением
Проводили каталитическое тиолирование метанола в реакционной трубке объемом 25 мл. Перед реакцией 125,0 мг катализатора (125-250 мкм), разбавленного в 1 г SiC, сульфидировали в потоке 20 мл мин-1 H2S при 360°C и 9 бар. Объем катализатора был почти незначительным по сравнению с объемом пустого реактора идеального вытеснения (20 мл). Это приводило к относительно низкой среднечасовой скорости подачи жидкости (LHSV) в пересчете на жидкий метанол (CH3OH), составляющей всего 0,054 ч-1. Среднечасовая скорость подачи газа (GHSV) в пересчете на полную подачу (H2S, CH3OH и N2) составляла 150 ч-1 (при стандартных условиях при 0°C и 1,013 бар в соответствии с DIN 1343). Для определения значений энергии активации реакцию проводили с потоком газообразного CH3OH (10 мл мин-1), смешанного с H2S (20 мл мин-1) и N2 (20 мл мин-1), при давлении потока подаваемого материала, составляющем 9 бар, со значениями парциального давления, составляющими 3,6 бар для N2, 3,6 бар для H2S и 1,8 бар для метанола. Реакционную трубку нагревали до температур от 300°C до 360°C с помощью теплоносителя, пропускаемого через рубашку.
Стандартные расчеты модуля Вайса-Пратера показали, что он составлял <1 для всех катализаторов при всех условиях, и, следовательно, можно сделать вывод, что эффекты внутреннего переноса массы не повлияли на кинетические результаты. Онлайн-анализ потока продукта проводили с помощью Shimadzu GC-2014, оборудованного колонкой HP-PLOT Q (2,7 м, внутренний диаметр 2,0 мм) с применением детектора TCD. Константы скорости реакции рассчитывали с применением интегрального кинетического уравнения реакции 0,5 порядка в CH3OH и H2S для CH3SH. Во всем диапазоне степени превращения для изучения распределения продукта время пребывания регулировали с поддержанием парциального давления CH3OH на уровне 2,2 бар, а N2 и H2S на уровне 3,3 бар при 360°C.
Порядок реакций определяли при 360°C. Для порядков реакции по H2S парциальное давление метанола поддерживали постоянным на уровне 2,2 бар, тогда как парциальное давление H2S варьировали от 1,1 до 5,6 бар. Чтобы измерить порядки реакции метанола парциальное давление H2S устанавливали на уровне 4,5 бар, а парциальное давление CH3OH варьировали от 0,6 мбар до 2,2 бар газообразного CH3OH. Поток газа N2 регулировали так, чтобы компенсировать изменения объемного потока и поддерживать постоянный общий объемный поток на уровне 80 мл/мин. Количество катализатора, применяемое в каждом эксперименте, регулировали соответственно, чтобы обеспечить степень превращения CH3OH ниже 10%. Порядки реакции для материалов, модифицированных цезием, измеряли с применением 10,0 мг катализаторов, тогда как для TiO2 и ZrO2 было достаточно 5,0 мг, а для γ-Al2O3 было достаточно 1,0 мг. В случае γ-Al2O3 катализатор физически смешивали с SiO2, который, как известно, неактивен в исследуемой реакции, в соотношении 1:9, чтобы избежать эффектов каналирования.
3.1 Каталитическая активность
Начальные скорости образования метилмеркаптана (CH3SH) показаны на фигуре 9. Наибольшая скорость тиолирования метанола наблюдалась для TiO2 (0,17-1,4⋅10-6 мольCH3SH с-1 гкат-1), после него для γ-Al2O3 (0,13-9,2·10-6 мольCH3SH с-1 гкат-1) и ZrO2 (0,02-0,2⋅10-6 мольCH3SH с-1 гкат-1). Для систем, допированных Cs, скорости образования CH3SH уменьшались в порядке Cs (10 вес. %)/γ-Al2O3 (1,8 - 8,7⋅10-6 мольCH3SH с-1 гкат-1) > Cs (10 вес. %)/ZrO2 (1,7 - 7,1·10-6 мольCH3SH с-1 гкат-1гкат) > Cs (10 вес. %)/TiO2 (1,8 - 6,6·10-6 мольCH3SH с-1 гкат-1). Более высокая нагрузка Cs, равная 20 вес. %, не приводила к более активным катализаторам, напротив, активность была слегка ниже для Cs (20 вес. %)/γ-Al2O3 (2,0 - 7,6⋅10-6 мольCH3SH с-1 гкат-1), Cs (20 вес. %)/ZrO2 (1,7 - 7,1·10-6 мольCH3SH с-1 гкат-1) и Cs (20 вес. %)/TiO2 (1,8 - 5,8·10-6 мольCH3SH с-1 гкат-1). Хотя существует одно различие в величине скорости образования CH3SH с применением разных оксидов металлов, активность систем с Cs продемонстрировала лишь незначительные различия. Это указывает на то, что общая активность определяется частицами Cs на поверхности, которые, по-видимому, одинаковы на всех трех подложках на основе оксидов металлов. Действительно, скорость образования CH3SH слегка снизилась для всех трех систем.
Выходы метилмеркаптана, диметилсульфида (DMS) и диметилового эфира (DME) измеряли в зависимости от степени превращения метанола при 360°C для всех трех оксидов металлов (фигура 10). На γ-Al2O3 в качестве первичных продуктов получали CH3SH и (DME), при этом доля первичного продукта DME была наиболее высокой при степени превращения метанола, составляющей до 60%. При преодолении 60% степени превращения, выход DME снижался до 20% при степени превращения 90%, при этом основным продуктом являлся CH3SH. Такое поведение объясняется повторной адсорбцией DME на катализаторе, подвергающегося вторичной реакции с образованием CH3SH. Аналогичные результаты наблюдались для ZrO2, при этом основным первичным продуктом являлся CH3SH, а выход диметилового эфира составлял менее 10%. Стоит отметить, что DME не образовывался при степени превращения ниже 10% на ZrO2. Как на γ-Al2O3, так и на ZrO2 диметилсульфид обнаруживали при более высокой степени превращения, при этом он являлся вторичным продуктом образования CH3SH. На TiO2 не наблюдалось диметилового эфира, при этом единственным побочным продуктом являлся диметилсульфид. Проведение реакции без H2S на ZrO2 и TiO2 приводило к образованию DME (фигура 10), что указывает на конкуренцию между реагентами на ZrO2 и TiO2. Выходы CH3SH возрастают в порядке γ-Al2O3 < ZrO2 < TiO2. Выходы CH3SH, DMS и DME измеряли в зависимости от степени превращения CH3OH при 360°C для всех трех оксидов металлов с нагрузкой Cs, составляющей 10 и 20 вес. % (фигура 11). Для всех систем, содержащих Cs, наблюдалась общая тенденция: CH3SH получали в качестве основного продукта, тогда как единственным катализатором, обеспечивающим выход DME, являлся Cs (10 вес. %)/Al2O3, при этом выход DME составлял 0,3% при 360°C. Основным побочным продуктом являлся DMS, при этом его максимальный выход составлял 0,7% на Cs (10 вес. %)/Al2O3 при 360°C. Отсутствие DME в присутствии Cs на поверхности объяснялось отсутствием сильных LAS. Эти результаты подтверждаются данными ИК-спектроскопии по адсорбции пиридина и CO, что демонстрирует резкое снижение кислотности Льюиса за счет допирования Cs.
3.2 Кинетика
a) Образование метилмеркаптана
Зависимость скоростей образования метилмеркаптана в метаноле и сероводороде на чистых оксидах металлов и оксидах металлов с нагрузкой 10 и 20 вес. % Cs показана на фигурах 12 и 13. Порядки реакции образования метилмеркаптана относительно CH3OH и H2S приведены в таблице 6.
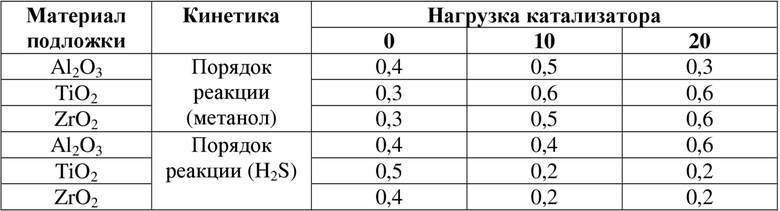
Таблица 6. Определенный порядок реакции образования метилмеркаптана по H2S и CH3OH на всех испытанных системах.
На всех оксидах металлов порядок реакции как по H2S, так и по CH3OH при образовании CH3SH, составляющий 0,5, указывает на диссоциацию обоих реагентов перед протеканием реакции по бимолекулярному механизму Ленгмюра-Хиншельвуда. Уравнение скорости образования метилмеркаптана выглядит следующим образом:
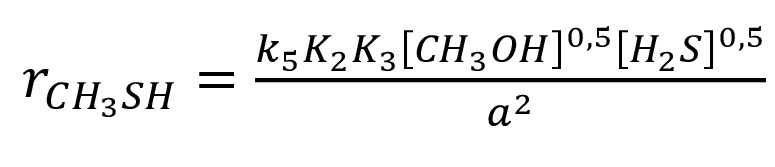 ,
,
где a = (1 + K20,5[CH3OH]0,5 + K30,5[H2S]0,5 + [CH3SH]0,5/K60,5 + [H2O]0,5/K70,5).
Уравнение скорости образования диметилового эфира выглядит следующим образом:
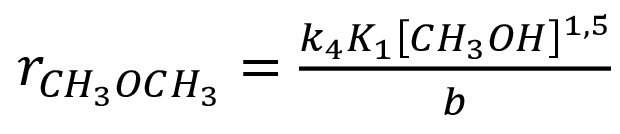 ,
,
где b = (1 + K10,5[CH3OH]0,5 + [H2O]0,5/K80,5).
Известно, что сероводород диссоциативно адсорбируется на поверхности оксидов металлов, тогда как метанол также диссоциативно адсорбируется с образованием метанолята на кислотно-основных парах Льюиса поверхностных оксидов. Таким образом, считается, что оба субстрата диссоциируют на основных центрах одного и того же типа. Можно предположить, что уменьшение порядка реакции с парциальным давлением приведет к тому, что субстраты будут конкурировать за адсорбцию на поверхности. Однако в случае оксидов металлов это не наблюдается.
Было установлено, что кажущаяся энергия активации образования метилмеркаптана составляет приблизительно 112 кДж моль-1 на γ-Al2O3, 115 кДж моль-1 на ZrO2 и 107 кДж моль-1 на TiO2. Она представляет собой кажущуюся энергию активации метилмеркаптана, образованного на активных центрах оксидов металлов, поскольку Cs отсутствует.
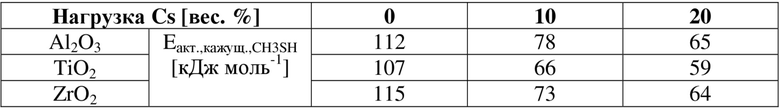
Таблица 7. Определенная кажущаяся энергия активации образования метилмеркаптана.
Добавление Cs (10 вес. %) приводило к порядку реакции по обоим реагентам, близкому к 0,5, что указывает на такой же диссоциативный механизм реакции, как предлагаемый для чистых оксидов металлов. Однако порядок реакции по H2S, составляющий 0,2, предполагает, что катализаторы Cs/TiO2 и Cs/ZrO2 работают при частичном покрытии H2S. Кажущаяся энергия активации снижалась до значений от 66 до 78 кДж моль-1. Более низкая энергия активации этих катализаторов относительно оксидов металлов связана с увеличением основности. Присутствие катиона Cs+ на подложке указывает на покрытие ее поверхностных гидроксилов, аналогично тому, что наблюдается в случае натрия и калия на оксиде алюминия. Это подтверждалось отсутствием полос ОН при исследовании адсорбции пиридина и СО с помощью ИК-спектроскопии. На всех материалах, сильнодопированных Cs (20 вес. %), получали значения порядка реакции, аналогичные 10 вес. % Cs, что также указывает на диссоциативный механизм. Для трех сильнодопированных материалов кажущийся активационный барьер находился в пределах 65-59 кДж моль-1. Снижение кажущейся энергии активации можно объяснить полной модификацией поверхности. Как показывает адсорбция пиридина, единственными поверхностными частицами, доступными при адсорбции пиридина, являлись частицы Cs, подавляющие химические свойства оксидов металлов и выступающие в качестве очень слабых LAS. Кроме того, отсутствие сильных кислотных центров Льюиса приводило только к образованию поверхностного метанолята, наблюдаемого при исследовании адсорбции метанола на катализаторах, сильнодопированных Cs, с помощью ИК-спектроскопии.
Порядки реакции образования DME по метанолу и сероводороду определяли для всех чистых оксидов металлов (таблица 8 и фигура 14). Порядок реакции образования диметилового эфира составлял 1,5 по метанолу и 0 по H2S на γ-Al2O3. Нулевой порядок в H2S показывает, что H2S не конкурирует с метанолом на центрах образования DME. Порядок реакции по метанолу, составляющий 1,5, объясняется частичным покрытием поверхности катализатора метанолом. На γ-Al2O3 адсорбция метанола, по-видимому, более благоприятна по сравнению с H2S, что приводит к образованию DME и нулевому порядку по H2S. Было установлено, что на ZrO2 и TiO2 порядок реакции образования DME (без присутствия H2S) составлял 0,7. Считается, что на этих материалах покрытие поверхности метанолом выше по сравнению с γ-Al2O3. Более низкая кажущаяся энергия активации на γ-Al2O3 по сравнению с двумя другими материалами объясняется более высокой силой кислоты Льюиса, как показывает адсорбция CO и пиридина, что способствует разрыву связи CO в CH3OH.
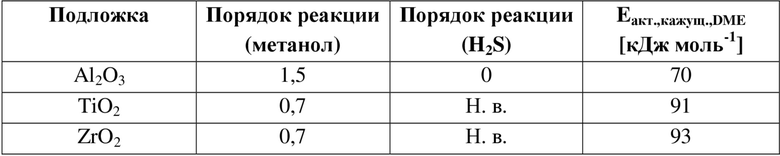
Таблица 8. Порядки реакции по метанолу и сероводороду и кажущаяся энергия активации образования диметилового эфира.
3.3 Каталитическая селективность
Для катализаторов на основе чистых оксидов металлов γ-Al2O3 обладал самой низкой селективностью образования метилмеркаптана, при этом диметиловый эфир являлся основным продуктом в диапазоне температур от 300°C до 320°C (с SDME, 300°C = 71,2% и SDME, 320°C = 63,4%). С повышением температуры селективность в отношении диметилового эфира снижалась до SDME, 340°C = 49,8% и в конце до SDME, 360°C = 28,6%. Селективность в отношении метилмеркаптана увеличивалась от 28,5% при 300°C до 71,2% при 360°C по мере снижения селективности в отношении диметилового эфира. Селективность образования диметилсульфида составляла менее 5%.
На чистом ZrO2 cелективность образования метилмеркаптана составляла 53,1% при 300°C и 60% при 360°C. И в этом случае основным побочным продуктом являлся диметиловый эфир, однако при этом селективность снижалась от 46,7% при 300°C до 36,6% при 360°C. Как и для γ-Al2O3, селективность в отношении диметилсульфида составляла менее 5%.
Из всех чистых оксидов металлов TiO2 обеспечивает наивысшую селективность образования метилмеркаптана: 96% при 300°C, при этом селективность снижалась при более высоких температурах (SDME, 360°C = 79,2%). В отличие от двух других оксидов металлов, селективность в отношении диметилового эфира увеличивалась с повышением температуры от SDME, 300°C = 4,0% до SDME, 360°C = 16,6%). Диметилсульфид получали с селективностью менее 4%.
По сравнению с катализаторами на основе чистых оксидов металлов все нагруженные Cs катализаторы демонстрировали резкое увеличение селективности в отношении CH3SH. Для 10 вес. % Cs на γ-Al2O3 селективность образования CH3SH увеличивалась до диапазона от SCH3SH, 300°C = 99,7% до SCH3SH, 360°C = 98,3%. Селективность в отношении побочного продукта увеличивалась с повышением температуры: DME образовывался с селективностью от SDME, 300°C = 0,1% до SDME, 360°C = 0,5%, а DMS образовывался с селективностью от SDMS, 300°C = 0,2% до SDMS, 360°C = 1,2%. Дальнейшее увеличение нагрузки Cs до 20 вес. % также увеличивало селективность в отношении CH3SH до селективности от SCH3SH, 300°C = 99,9% до SCH3SH, 360°C = 99,1%. В этом случае единственным обнаруженным побочным продуктом являлся DMS, селективность в отношении которого составляла от SDMS, 300°C = 0,1% до SDMS, 360°C = 0,9%.
Для катализаторов на основе ZrO2 селективность в отношении CH3SH увеличивалась до диапазона от SCH3SH, 300°C = 99,9% до SCH3SH, 360°C = 99,1%. И в этом случае единственным обнаруженным побочным продуктом являлся DMS, селективность в отношении которого составляла от SDMS, 300°C = 0,1% до SDMS, 360°C = 0,9%. Увеличение нагрузки Cs до 20 вес. % приводило к еще большей селективности, составляющей от SCH3SH, 300°C = 99,9% до SCH3SH, 360°C = 99,4%.
Аналогичные результаты были получены для катализаторов на основе TiO2: при нагрузке 10 вес. % Cs селективность в отношении CH3SH увеличивалась до диапазона от SCH3SH, 300°C = 99,9% до SCH3SH, 360°C = 99,4%. И в этом случае единственным обнаруженным побочным продуктом являлся диметилсульфид, селективность в отношении которого составляла от SDMS, 300°C = 0,1% до SDMS, 360°C = 0,6%. Увеличение нагрузки Cs до 20 вес. % и в этом случае приводило к еще большей селективности, составляющей от SCH3SH, 300°C = 99,9% до SCH3SH, 360°C = 99,5%.
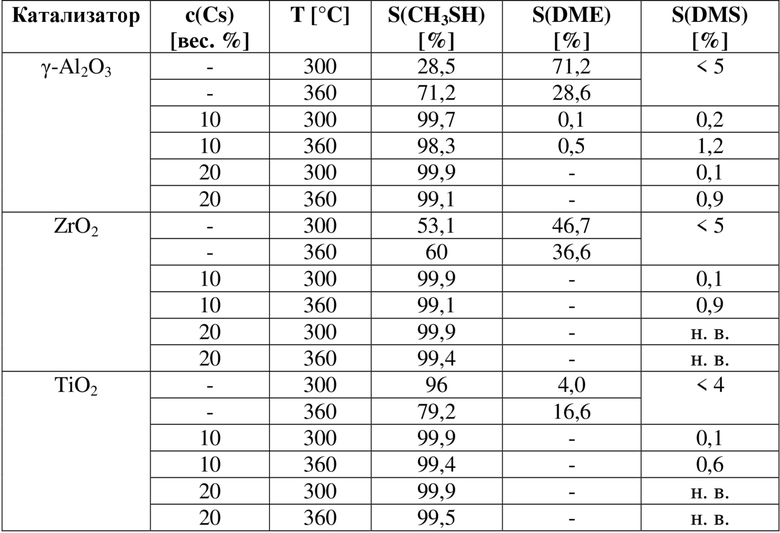
Таблица 9. Краткое описание результатов по селективности полученных катализаторов в отношении продуктов (н. в. = не выявлено)
4. Сравнительные примеры
Сравнительные примеры выполняли с применением катализатора, содержащего Cs2WS4 на γ-Al2O3. Указанный катализатор получали с помощью двухстадийного способа капиллярной пропитки. Сначала 5,0 г γ-Al2O3 (аналог SPH 509 Axens, размер зерна 150-250 мкм) пропитывали с помощью 0,64 г ацетата цезия (Sigma Aldrich, ≥ 99,99%), растворенного в 1,6 мл H2O. Образец высушивали при комнатной температуре в течение ночи с получением Cs/Al2O3. Затем синтезировали систему Cs2WS4/Al2O3 следующим образом: обеспечивали образование кристаллов Cs2WS4 посредством осаждения при смешивании растворов 350 мг (NH4)2WS4 в 20 мл H2O и 325 мг Cs2CO3 в 20 мл H2O. Образовывался желтый осадок. Эти твердые вещества фильтровали, промывали ледяной водой и 1-пропанолом. Из-за низкой растворимости Cs2WS4 растворяли 450 мг этих кристаллов в 150 мл воды. Затем к раствору добавляли 2 г Cs/Al2O3. Воду удаляли путем выпаривания при непрерывном вращении с осаждением кристаллов Cs2WS4 на твердом образце. Образец высушивали при комнатной температуре в течение ночи. После высушивания образец прокаливали при 455°C в течение 4 ч. со скоростью нагрева 5°C/мин. Полученный катализатор характеризовался содержанием вольфрама, составляющим 5,1 вес. %, содержанием цезия, составляющим 20,6 вес. %, объемом пор, составляющим 0,20 см3 г-1, и площадью поверхности по методу BET, составляющей 141 м2 г-1, которые измеряли, как описано выше. Адсорбцию с последующей температурно-программируемой десорбцией H2S проводили импульсным методом на аппарате проточного типа, оснащенном масс-спектрометром (QME 200, Pfeiffer Vacuum). Образец катализатора загружали в кварцевый реактор и активировали in situ в атмосфере 4,2 об. % H2S/He со скоростью потока 6 мл/мин. при 360°C в течение 2 ч. Для адсорбции H2S температуру устанавливали на уровне 360°C и образец продували He в течение 1 часа перед адсорбцией. Каждые 30 мин. вводили импульсы 4,4 об. % H2S в He (5,0 мкмоль/мин. H2S). Общую концентрацию адсорбированного газа рассчитывали как сумму поглощения за импульс.
Полученный таким образом катализатор испытывали при тех же условиях реакции и в той же реакционной трубке, как в примере 3. Перед применением в испытаниях катализатор активировали путем обработки H2S со скоростью потока 20 мл/мин. при температуре 360°C в течение 2 часов.
Степень превращения MeOH, значения выходов CH3SH, DME и DMS, а также значения селективности в отношении CH3SH, DME и DMS при температурах 300°C, 320°C, 340°C и 360°C обобщены в таблице 10.
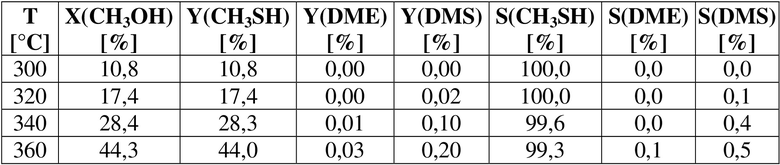
Таблица 10. Краткое описание результатов сравнительных примеров
Начальные скорости образования CH3SH показаны на фигуре 15. Наибольшая скорость тиолирования метанола наблюдалась при температуре 300°C (1,34⋅10-6 мольCH3SH с-1 гкат.-1) со следующими скоростями при более высоких температурах и наименьшей скоростью при температуре 360°C (6,38·10-6 мольCH3SH с-1 гкат.-1).
| название | год | авторы | номер документа |
|---|---|---|---|
| СОДЕРЖАЩИЕ ВОЛЬФРАМАТ КАТАЛИЗАТОРЫ СИНТЕЗА АЛКИЛМЕРКАПТАНА И СПОСОБ ИХ ПРИГОТОВЛЕНИЯ | 2005 |
|
RU2387476C2 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТИЛМЕРКАПТАНА ИЗ ДИАЛКИЛСУЛЬФИДОВ И ДИАЛКИЛПОЛИСУЛЬФИДОВ | 2008 |
|
RU2490255C2 |
| КАТАЛИЗАТОРЫ, КОТОРЫЕ СОДЕРЖАТ ГАЛОГЕНИДСОДЕРЖАЩИЕ ВОЛЬФРАМАТЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ, ДЛЯ СИНТЕЗА АЛКИЛМЕРКАПТАНОВ И СПОСОБ ИХ ПРИГОТОВЛЕНИЯ | 2005 |
|
RU2384364C2 |
| МОЛИБДЕНСОДЕРЖАЩИЙ КАТАЛИЗАТОР, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ МЕТИЛМЕРКАПТАНА | 2007 |
|
RU2436626C2 |
| КАТАЛИЗАТОР СИНТЕЗА АЛКИЛМЕРКАПТАНА И СПОСОБ ПРИГОТОВЛЕНИЯ ТАКОГО КАТАЛИЗАТОРА | 2004 |
|
RU2342992C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ ФОРМУЛЫ RSH ПУТЕМ ГИДРОСУЛЬФУРИЗАЦИИ | 2020 |
|
RU2805660C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛМЕРКАПТАНОВ В МНОГОЗОННОМ РЕАКТОРЕ С НЕПОДВИЖНЫМ СЛОЕМ | 2007 |
|
RU2443686C2 |
| КАТАЛИЗАТОР ДЛЯ ПОЛУЧЕНИЯ МЕТИЛМЕРКАПТАНА | 2008 |
|
RU2497588C2 |
| МЕМБРАНА НА ПОДЛОЖКЕ, ФУНКЦИОНАЛИЗОВАННАЯ ГЕКСА- И ОКТАЦИАНОМЕТАЛЛАТАМИ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И СПОСОБ РАЗДЕЛЕНИЯ С ПРИМЕНЕНИЕМ ЭТОЙ МЕМБРАНЫ | 2013 |
|
RU2645989C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛМЕРКАПТАНОВ | 2015 |
|
RU2712218C2 |

Настоящее изобретение относится к способу получения алкилмеркаптана, согласно которому алкиловый спирт подвергают реакции с сероводородом в присутствии катализатора, содержащего или состоящего из подложки и от 5 до 20 вес.% промотора в пересчете на общий вес катализатора. Подложка содержит или состоит из диоксида титана, диоксида циркония и/или их смеси. Промотор представляет собой оксид щелочного металла. Во время фазы запуска катализатора оксид щелочного металла сульфидируют. Технический результат заключается в повышении активности и селективности катализатора тиолирования метанола. 10 з.п. ф-лы, 15 ил., 10 табл., 4 пр.
1. Способ получения алкилмеркаптана, где алкиловый спирт подвергают реакции с сероводородом в присутствии катализатора, содержащего или состоящего из подложки и от 5 до 20 вес.% промотора в пересчете на общий вес катализатора, где подложка содержит или состоит из диоксида титана, диоксида циркония и/или их смеси и промотор представляет собой оксид щелочного металла и где во время фазы запуска катализатора оксид щелочного металла сульфидируют.
2. Способ по п. 1, где по меньшей мере часть подложки катализатора характеризуется тетрагональной фазой.
3. Способ по п. 1 или 2, где щелочной металл катализатора представляет собой натрий, калий, цезий или рубидий.
4. Способ по любому из пп. 1-3, где промотор катализатора представляет собой оксид цезия.
5. Способ по любому из пп. 1-4, где катализатор представляет собой цельный катализатор.
6. Способ по любому из пп. 1-5, где катализатор представляет собой катализатор типа «ядро-оболочка».
7. Способ по любому из пп. 1-6, где катализатор получают с помощью способа, включающего стадии:
a) пропитки подложки, содержащей или состоящей из диоксида титана, диоксида циркония и/или их смеси, водным раствором, содержащим растворимое соединение щелочного металла,
b) высушивания пропитанной подложки, полученной на стадии а), и
c) прокаливания высушенной пропитанной подложки из стадии b) с получением катализатора.
8. Способ по п. 7, где стадии от а) до с) способа, с помощью которого получают катализатор, повторяют по меньшей мере один раз.
9. Способ по п. 7 или 8, где способ, с помощью которого получают катализатор, дополнительно включает стадию
d1) формования катализатора, полученного на стадии с), с получением цельного катализатора.
10. Способ по п. 7 или 8, где способ, с помощью которого получают катализатор, дополнительно включает стадию
d2) нанесения катализатора, полученного на стадии с), на ядро с получением катализатора типа «ядро-оболочка».
11. Способ по любому из пп. 1-10, где алкиловый спирт, который подвергают реакции, представляет собой метанол и получаемый алкилмеркаптан представляет собой метилмеркаптан.
| СПОСОБ ПОЛУЧЕНИЯ МЕТИЛМЕРКАПТАНА ИЗ ДИАЛКИЛСУЛЬФИДОВ И ДИАЛКИЛПОЛИСУЛЬФИДОВ | 2008 |
|
RU2490255C2 |
| US 9024090 B2, 05.05.2015 | |||
| US 2017080388 A1, 23.03.2017 | |||
| СПОСОБ СНИЖЕНИЯ ОБЩЕГО СОДЕРЖАНИЯ СЕРЫ В ГАЗАХ, ВКЛЮЧАЮЩИХ СЕРОВОДОРОД И ДРУГИЕ СОДЕРЖАЩИЕ СЕРУ КОМПОНЕНТЫ | 1997 |
|
RU2177361C2 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| МАГНИТНОЕ УСТРОЙСТВО ЭЛЕКТРИЧЕСКОЙ МАШИНЫ С ТРУБОПРОВОДОМ ОХЛАДИТЕЛЯ | 2009 |
|
RU2491698C2 |
| 0 |
|
SU352674A1 | |
| КАТАЛИЗАТОР СИНТЕЗА АЛКИЛМЕРКАПТАНА И СПОСОБ ПРИГОТОВЛЕНИЯ ТАКОГО КАТАЛИЗАТОРА | 2004 |
|
RU2342992C2 |

