ОБЛАСТЬ ТЕХНИКИ
[0001] Настоящее изобретение относится к производному хинолина и способу его получения. Более конкретно, настоящее изобретение относится к производному хинолина высокой чистоты и способу его получения для эффективного получения производного хинолина.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
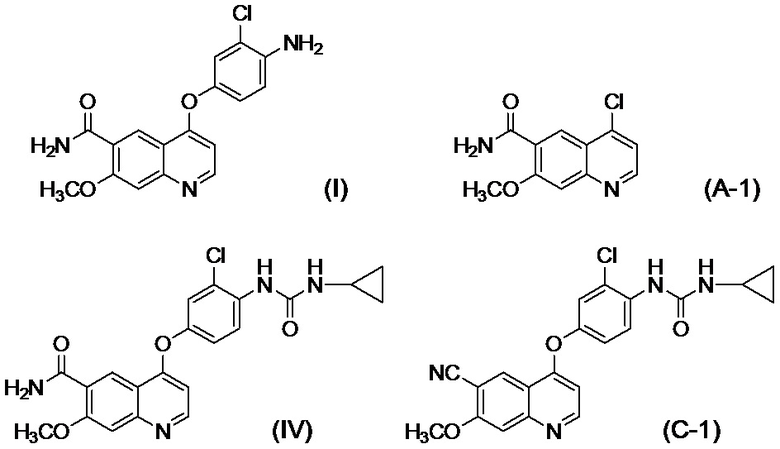
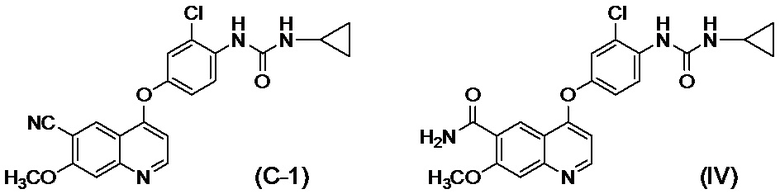
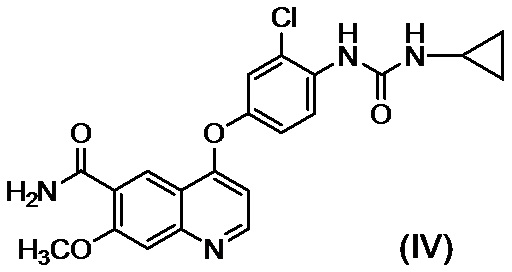
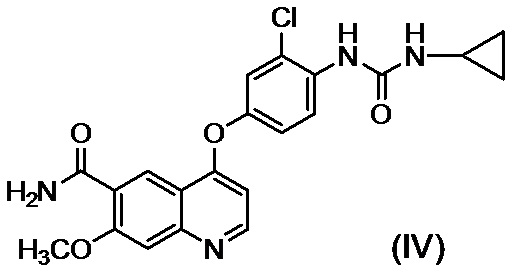
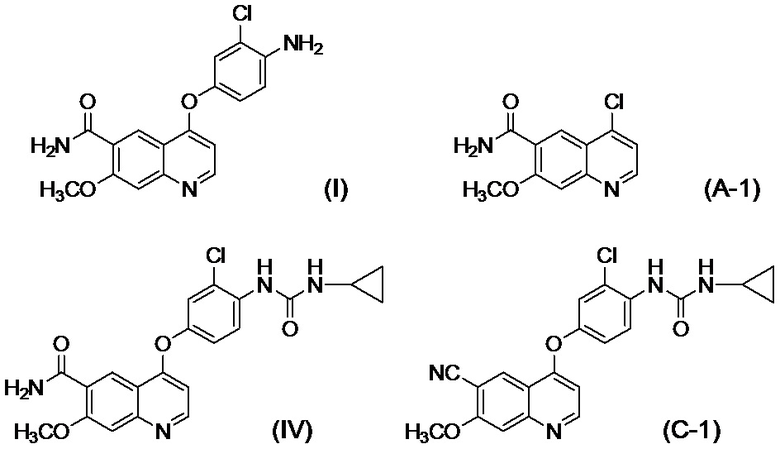
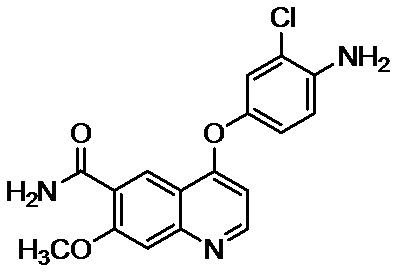
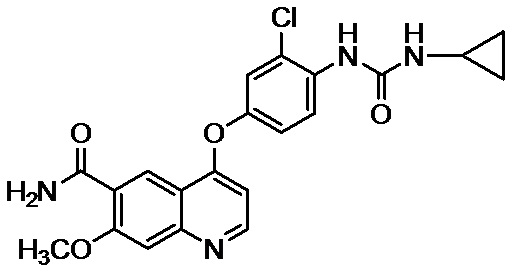
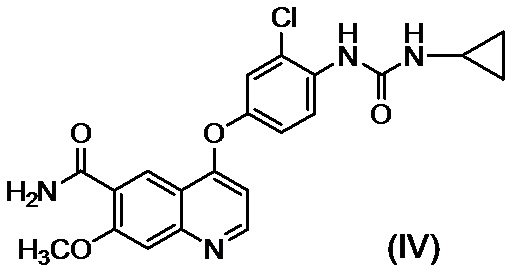
[0002] Известно, что производные хинолина, представленные соединением (IV):
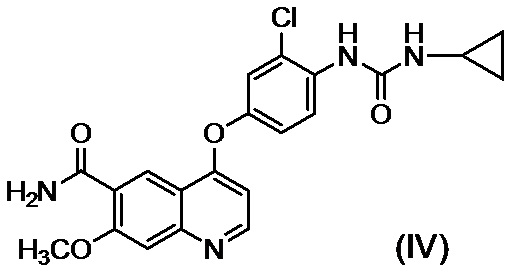 ,
,
проявляют превосходную противоопухолевую активность (PTL 1).
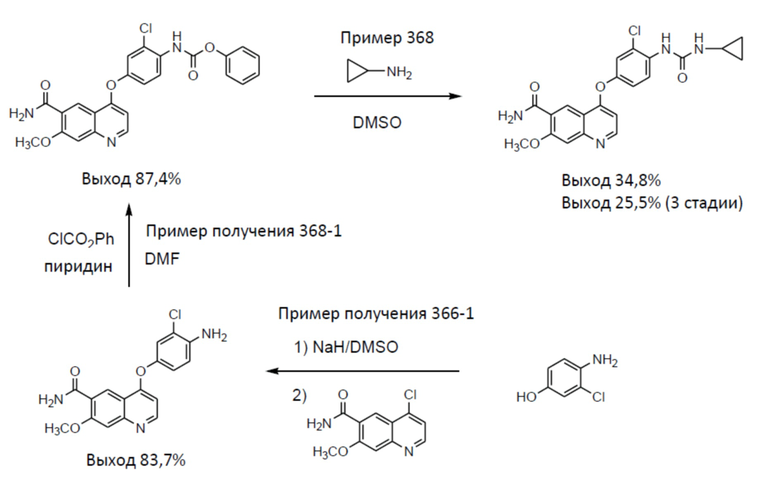
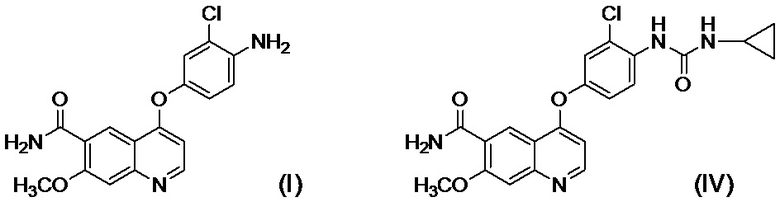
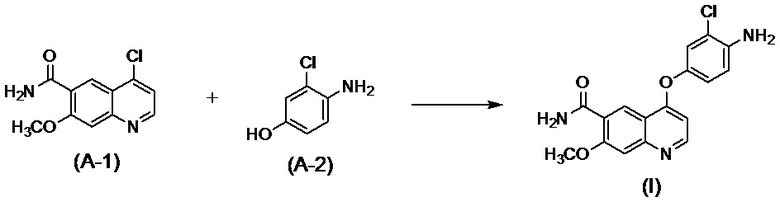
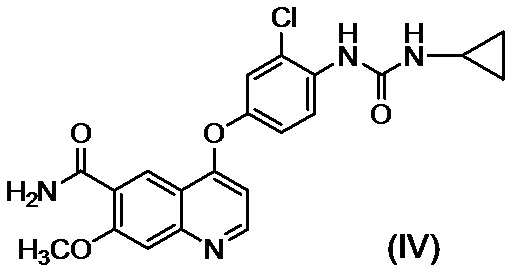
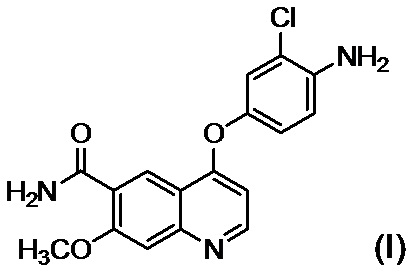
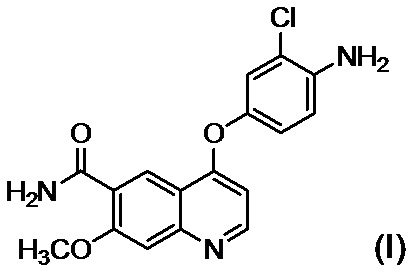
[0003] В PTL 1, 2, 3, 4 и 5 раскрываются способы получения таких производных хинолина. В частности, в способе получения по PTL 1 (таком как описан в примере 368), 4-амино-3-хлорфенол гидрохлорид вводят в реакцию с 4-хлор-7-метоксихинолин-6-карбоксамидом (стадия A), фенилхлорформиат вводят в реакцию с полученным 4-(4-амино-3-хлорфенокси)-7-метоксихинолин-6-карбоксамидом и полученный в результате фенил-N-{4-(6-карбамоил-7-метокси-4-хинолил)окси-2-хлорфенил}карбамат (стадия B) выделяют, а затем циклопропиламин дополнительно вводят в реакцию с карбаматом (стадия C) с получением целевого соединения 4-[3-хлор-4-(циклопропиламинокарбонил)аминофенокси]-7-метокси-6-хинолинкарбоксамида (здесь и далее называемого "соединение (IV)"), с общим выходом 25,5% для трех стадий.
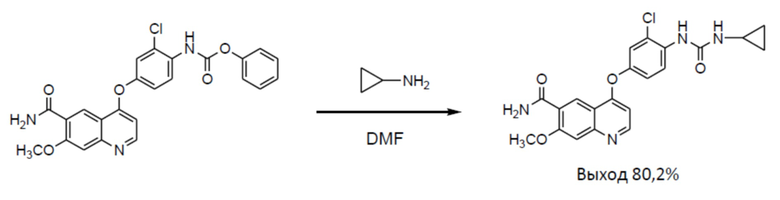
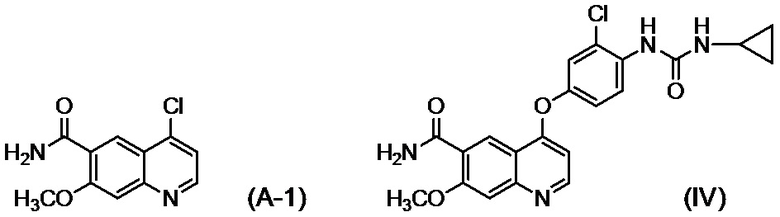
[0004] В способах получения, описанных в PTL 2 (сравнительный пример 1) и PTL 4 (пример получения 1), циклопропиламин вводят в реакцию с фенил N-{4-(6-карбамоил-7-метокси-4-хинолил)окси-2-хлорфенил}карбаматом с получением соединения (IV), с выходом 80,2%.
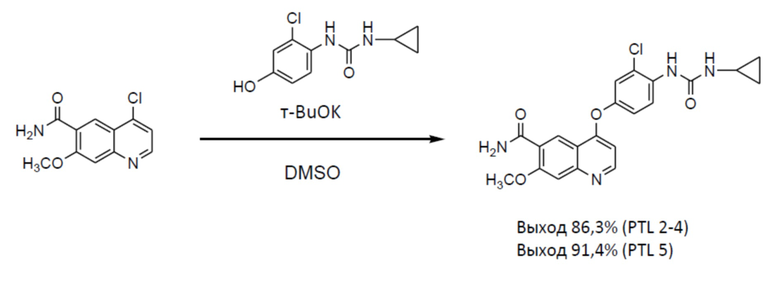
[0005] В способах получения, описанных в PTL 2 (сравнительный пример 3), PTL 3 (пример 4), PTL 4 (пример получения 3) и PTL 5 (пример 1a), целевое соединение (IV) получают с помощью одной стадии из 4-хлор-7-метоксихинолин-6-карбоксамида, с выходом 86,3% в PTL 2-4 и выходом 91,4% в PTL 5.
[0006] Далее будут конкретно описаны способы получения, описанные в PTL 1-5. Способ получения, описанный в PTL 1 (пример 368 и т. п.), является таким, как описано в следующих формулах.
[0007] Схема реакции для способа получения в PTL 2 (сравнительный пример 1) и PTL 4 (пример получения 1) является следующей.
[0008] Способ получения в PTL 2 (сравнительный пример 3), PTL 3 (пример 4), PTL 4 (пример получения 3) и PTL 5 (пример 1a) характеризуется следующей схемой реакции.
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
ПАТЕНТНАЯ ЛИТЕРАТУРА
[0009] [PTL 1] US2004/0053908
[PTL 2] US2007/0004773
[PTL 3] US2007/0037849
[PTL 4] US2007/0078159
[PTL 5] US2007/0117842
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ЗАДАЧА
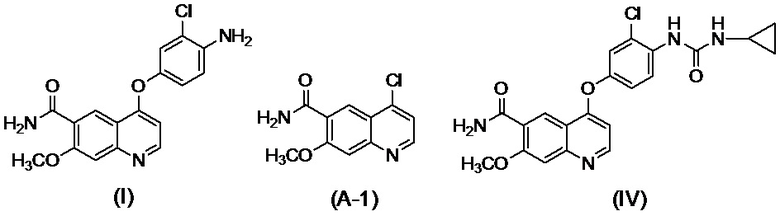
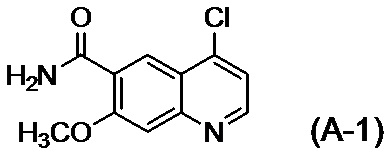
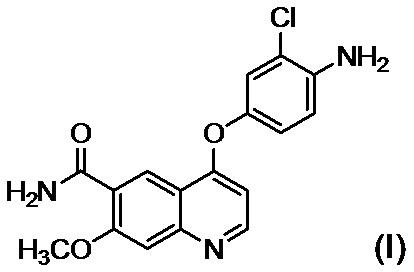
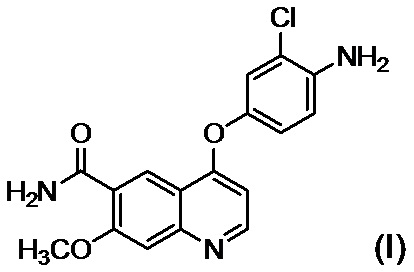
[0010] Авторы настоящего изобретения обнаружили, что в случае если соединение, представленное формулой (IV), или его соль получают путем применения способов получения, описанных в PTL 1-5, при этом продукт содержит соединение, представленное формулой (I), соединение, представленное формулой (A-1), соединение, представленное формулой (C-1), и им подобные в качестве примесей, и что такие примеси сложно удалять с помощью обычного способа очистки, такого как хроматография и кристаллизация.
[0011] Таким образом, целью настоящего изобретения является обеспечение производного хинолина высокой чистоты с небольшим количеством примесей. Другой целью настоящего изобретения является обеспечение способа получения, подходящего для крупномасштабного производства с целью получения производного хинолина высокой чистоты с высоким выходом.
РЕШЕНИЕ ЗАДАЧИ
[0012] Авторы настоящего изобретения, в результате тщательного изучения при рассмотрении вышеописанной ситуации, обнаружили новый способ получения вышеописанного производного хинолина, завершив таким образом настоящее изобретение. Таким образом, настоящее изобретение предусматривает следующие [1]-[27].
[1] Соединение, представленное формулой (IV), или его соль, где содержание соединения, представленного формулой (I), составляет 350 ppm по массе или менее.
[2] Соединение, представленное формулой (IV), или его соль, где содержание соединения, представленного формулой (I), составляет 183 ppm по массе или менее.
[3] Соединение, представленное формулой (IV), или его соль, где содержание соединения, представленного формулой (A-1), составляет 60 ppm по массе или менее.
[4] Соединение, представленное формулой (IV), или его соль, где содержание соединения, представленного формулой (I), составляет 350 ppm по массе или менее, и содержание соединения, представленного формулой (A-1), составляет 60 ppm по массе или менее.
[5] Соединение, представленное формулой (IV), или его соль, где содержание соединения, представленного формулой (I), составляет 183 ppm по массе или менее, и содержание соединения, представленного формулой (A-1), составляет 60 ppm по массе или менее.
[6] Соединение представленное формулой (IV), или его соль, где содержание соединения, представленного формулой (C-1) составляет 0,10% по массе или менее.
[7] Соединение, представленное формулой (IV), или его соль, в соответствии с любым из [1]-[6], где содержание соединения, представленного формулой (IV), составляет 98,0% по массе или более.
[8] Композиция, где содержание соединения, представленного формулой (IV), или его соли, составляет 98,0% по массе или более, и содержание соединения, представленного формулой (I), или его соли, составляет 350 ppm по массе или менее.
[9] Композиция, где содержание соединения, представленного формулой (IV), или его соли, составляет 98,0% по массе или более, и содержание соединения, представленного формулой (I), или его соли, составляет 183 ppm по массе или менее.
[10] Композиция, где содержание соединения, представленного формулой (IV), или его соли, составляет 98,0% по массе или более, и содержание соединения, представленного формулой (A-1), или его соли, составляет 60 ppm по массе или менее.
[11] Композиция, где содержание соединения, представленного формулой (IV), или его соли, составляет 98,0% по массе или более, содержание соединения представленного формулой (I), или его соли, составляет 350 ppm по массе или менее, и содержание соединения, представленного формулой (A-1), или его соли, составляет 60 ppm по массе или менее.
[12] Композиция, где содержание соединения, представленного формулой (IV), или его соли, составляет 98,0% по массе или более, содержание соединения, представленного формулой (I), или его соли, составляет 183 ppm по массе или менее, и содержание соединения, представленного формулой (A-1), или его соли, составляет 60 ppm по массе или менее.
[13] Композиция, где содержание соединения, представленного формулой (IV), или его соли, составляет 98,0% по массе или более, и содержание соединения, представленного формулой (C-1), или его соли, составляет 0,10% по массе или менее.
[14] Фармацевтический препарат, содержащий соединение в соответствии с любым из [1]-[7] или его соль в качестве активного ингредиента.
[15] Фармацевтический препарат, содержащий композицию в соответствии с любым из [8]-[13] в качестве активного ингредиента.
[16] Фармацевтическая композиция с использованием соединения или его соли в соответствии с любым из [1]-[7] в качестве активного ингредиента, где фармацевтическая композиция дополнительно содержит фармацевтически приемлемый носитель.
[17] Фармацевтическая композиция, содержащая композицию в соответствии с любым из [8]-[13] в качестве активного ингредиента, где фармацевтическая композиция дополнительно включает фармацевтически приемлемый носитель.
[18] Твердый состав для перорального применения, содержащий соединение или его соль в соответствии с [4] в качестве активного ингредиента, где твердый состав для перорального применения дополнительно включает фармацевтически приемлемый носитель, и содержание соединения, представленного формулой (I), составляет 0,06% по массе или менее.
[19] Твердый состав для перорального применения, содержащий композицию в соответствии с [8] или [11] в качестве активного ингредиента, где твердый состав для перорального применения дополнительно содержит фармацевтически приемлемый носитель, и содержание соединения, представленного формулой (I), составляет 0,06% по массе или менее.
[20] Твердый состав для перорального применения, содержащий соединение или его соль в соответствии с [5] в качестве активного ингредиента, где твердый состав для перорального применения дополнительно включает фармацевтически приемлемый носитель, и содержание соединения, представленного формулой (I), составляет 0,040% по массе или менее.
[21] Твердый состав для перорального применения, содержащий композицию в соответствии с [9] или [12] в качестве активного ингредиента, при этом твердый состав для перорального применения дополнительно содержит фармацевтически приемлемый носитель, и содержание соединения, представленного формулой (I), составляет 0,040% по массе или менее.
[22] Способ получения соединения, представленного формулой (IV):
 ,
,
или его соли, включающий:
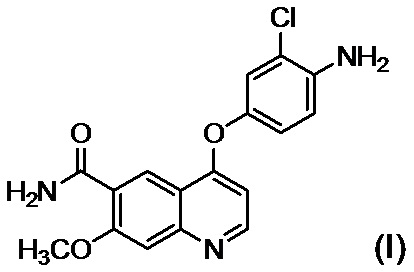
стадию B обеспечения реакции соединения, представленного формулой (I):
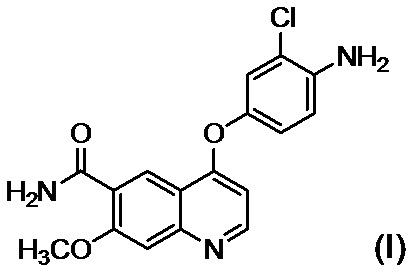 ,
,
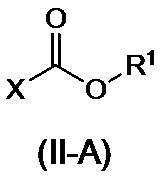
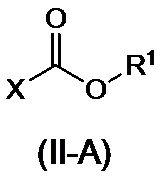
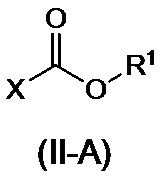
или его соли, с соединением, представленным формулой (II-A) или формулой (II-B):
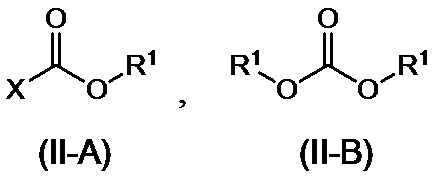 ,
,
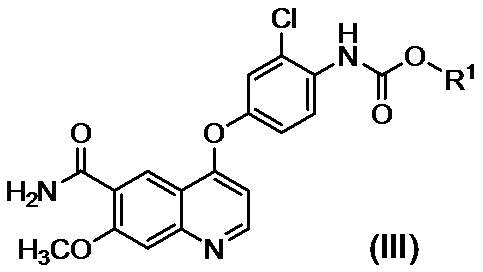
где R1 представляет собой C1-6алкильную группу, C2-6алкенильную группу, C6-10арильную группу или C7-11аралкильную группу, где C1-6алкильная группа или C2-6алкенильная группа могут иметь от одного до трех заместителей, которые могут быть идентичными или разными и являются выбранными из группы, состоящей из атома галогена и метоксигруппы, и где C6-10арильная группа или C7-11аралкильная группа могут иметь от одного до трех заместителей, которые могут быть идентичными или разными и являются выбранными из группы, состоящей из атома галогена, метильной группы, метоксигруппы и нитро-группы; и X представляет собой атом галогена,
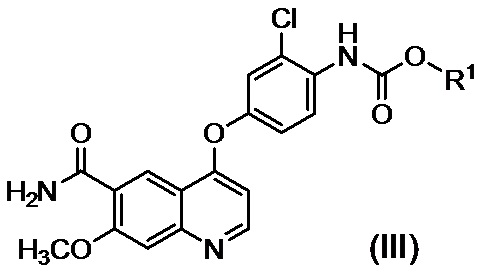
в присутствии основания, с получением таким образом соединения, представленного формулой (III):
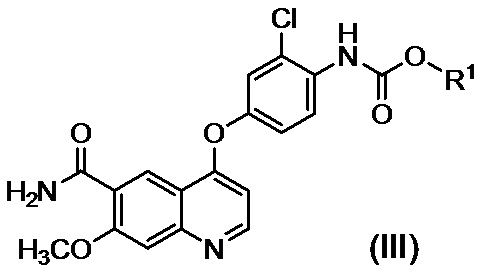 ,
,
где R1 представляет собой такую же группу, как описано выше, и
стадию C, на которой после обеспечения реакции соединения, представленного формулой (III), полученного на стадии B без выделения, с циклопропиламином, осуществляют осаждение и выделение соединения, представленного формулой (IV):
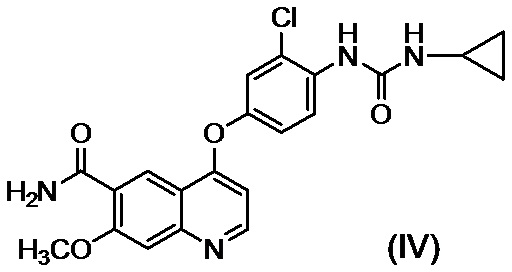 ,
,
или его соли путем введения в реакционный раствор водного органического растворителя.
[23] Способ получения соединения, представленного формулой (IV):
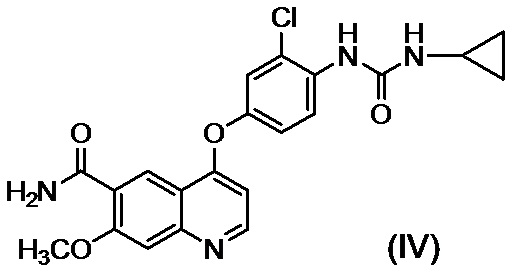 ,
,
или его соли, включающий:
стадию A, на которой после обеспечения реакции соединения, представленного формулой (A-1):
 ,
,
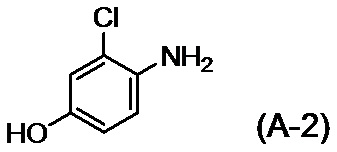
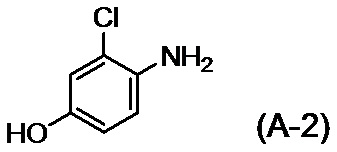
с соединением, представленным формулой (A-2):
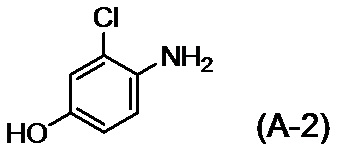 ,
,
или его солью в присутствии основания, осуществляют осаждение и выделение соединения, представленного формулой (I):
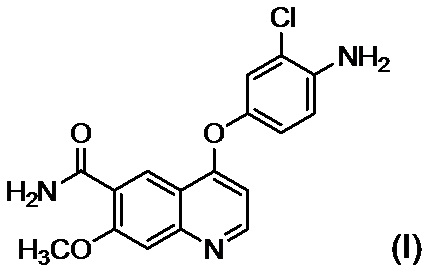 ,
,
или его соли из реакционного раствора путем введения в реакционный раствор водного органического растворителя,
стадию B обеспечения реакции соединения, представленного формулой (I):
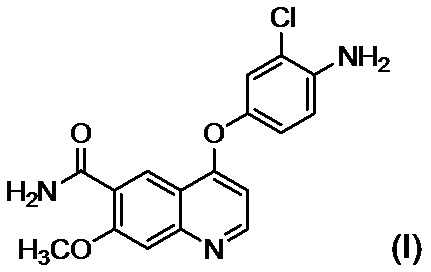 ,
,
или его соли, полученных на стадии A, с соединением, представленным формулой (II-A) или формулой (II-B):
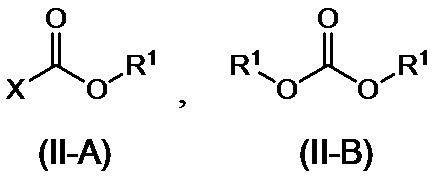 ,
,
где R1 представляет собой C1-6алкильную группу, C2-6алкенильную группу, C6-10арильную группу или C7-11аралкильную группу, где C1-6алкильная группа или C2-6алкенильная группа могут иметь от одного до трех заместителей, которые могут быть идентичными или разными и являются выбранными из группы, состоящей из атома галогена и метоксигруппы, и где C6-10арильная группа или C7-11аралкильная группа могут иметь от одного до трех заместителей, которые могут быть идентичными или разными и являются выбранными из группы, состоящей из атома галогена, метильной группы, метоксигруппы и нитро-группы; и X представляет собой атом галогена,
в присутствии основания, с получением таким образом соединения, представленного формулой (III):
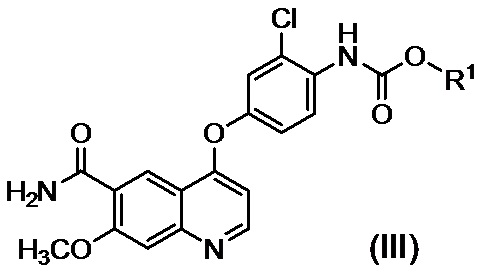 ,
,
где R1 представляет собой такую же группу, как описано выше, и
стадию C, на которой после обеспечения реакции соединения, представленного формулой (III), полученного на стадии B без выделения, с циклопропиламином, осуществляют осаждение и выделение соединения, представленного формулой (IV):
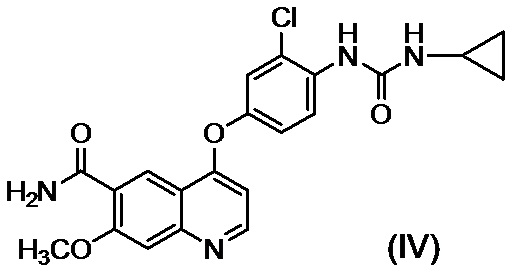 ,
,
или его соли путем введения в реакционный раствор водного органического растворителя.
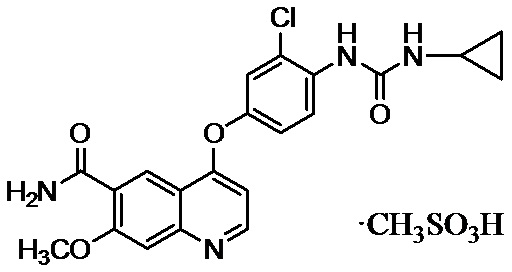
[24] Способ в соответствии с [22] или [23], дополнительно включающий стадию D превращения соединения, представленного формулой (IV), полученного на стадии C, в соль соединения, представленного формулой (IV).
[25] Способ в соответствии с [24], где соль, полученная на стадии D, представляет собой метансульфонат.
[26] Способ в соответствии с любым из [22]-[25], где стадия B представляет собой стадию обеспечения реакции соединения, представленного формулой (I):
 ,
,
или его соли, с соединением, представленным формулой (II-A):
 ,
,
где R1 представляет собой C6-10арильную группу, которая может иметь от одного до трех заместителей, которые могут быть идентичными или разными и являются выбранными из группы, состоящей из атома галогена, метильной группы, метоксигруппы и нитро-группы; и X представляет собой атом галогена,
в присутствии основания, с получением таким образом соединения, представленного формулой (III):
 ,
,
где R1 представляет собой такую же группу, как описано выше.
[27] Способ в соответствии с любым из [22]-[26], где соединение, представленное формулой (II-A), представляет собой фенилхлорформиат.
ПОЛЕЗНЫЕ ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
[0013] В соответствии с настоящим изобретением может быть обеспечено соединение (IV) с высоким выходом и высокой чистотой.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
[0014] Далее будут объяснены обозначения и термины, применяемые по всему настоящему описанию.
[0015] В настоящем описании ангидраты, гидраты и сольваты включены в понятие "соединение". Также в настоящем описании каждое из понятий "соединение (I)" и ему подобное означают то же соединение, что и "соединение, представленное формулой (I)" и т. п.
[0016] В настоящем описании "соединение или его соль" относится к соединению или его соли, которые содержат 90% по массе или более соединения и могут содержать исходный материал или побочный продукт, которые могут образовываться в виде примесей. Например, "соединение, представленное формулой (IV), или его соль" содержит 90% по массе или более соединения (IV) или его соли, и может содержать исходный материал, такой как соединение (I), соединение (A-1), а также побочный продукт, такой как соединение (C-1), который может образовываться на каждой стадии получения. Соответственно "соединение или его соль" в настоящем описании, которое может содержать побочный продукт и т. п. в качестве примесей, имеет вид "композиции". В случае выражения в данном документе содержания примесей, таких как соединение (I), соединение (A-1) и соединение (C-1), содержание основано на общей массе соединения (IV) или его соли.
[0017] В настоящем описании понятие "фармацевтическая композиция" относится к композиции, содержащей соединение, которое обладает фармакологическим эффектом, или его соль, и фармацевтически приемлемый носитель. Примером соединения, обладающего фармакологическим эффектом, или его соли, является соединение (IV) или его соль. В качестве альтернативы понятие "состав" означает составы, которые были подвергнуты обработке (такой как стерилизация и таблетирование), переводящей их в состояние, в котором их можно при необходимости вводить нуждающемуся в этом субъекту, по сравнению с фармацевтическими композициями. В качестве альтернативы понятие "фармацевтический препарат" представляет собой препарат, применяемый для терапии или профилактики заболевания и включает любые необязательные формы.
[0018] Кроме того, термин "C1-6алкильная группа", используемый в данном документе, означает одновалентную группу, образованную путем удаления любого одного атома углерода из C1-6алифатического насыщенного углеводорода, и при этом она представляет собой C1-6заместитель с линейной цепью или разветвленной цепью. Примеры C1-6алкильных групп включают метильную, этильную, 1-пропильную, 2-пропильную, 2-метил-1-пропильную, 2-метил-2-пропильную, 1-бутильную, 2-бутильную, 1-пентильную, 2-пентильную, 3-пентильную, 1-гексильную, 2-гексильную и 3-гексильную группы, при этом метильная, этильная и 1-пропильная группы являются предпочтительными.
[0019] Термин "C1-6алкенильная группа", используемый в данном документе, означает одновалентную группу, образованную путем удаления любого одного атома водорода из C1-6алифатического углеводорода с ненасыщенной связью, и при этом она представляет собой C1-6заместитель с линейной цепью или разветвленной цепью. Примеры C1-6алкенильных групп включают 2-пропенильную, 2-бутенильную, 2-пентенильную, 3-пентенильную, 2-гексенильную, 3-гексенильную и 4-гексенильную группы, при этом 2-пропенильная группа является предпочтительной.
[0020] Термин "C6-10арильная группа", используемый в данном документе, относится к C6-10ароматической циклической углеводородной группе. Примеры C6-10арильных групп включают фенильную, 1-нафтильную и 2-нафтильную группы, при этом фенильная группа является предпочтительной.
[0021] Термин "C7-11аралкильная группа", используемый в данном документе, относится к C7-11аралкильной группе. Примеры C7-11аралкильных групп включают бензильную и нафтилметильную группы, при этом бензильная группа является предпочтительной.
[0022] Термин "атом галогена", используемый в данном документе, относится к атомам фтора, хлора, брома или йода, а предпочтительно к атому хлора.
[0023] Термин "основание", используемый в данном документе, может относится к неорганическому основанию, такому как гидроксид лития, гидроксид натрия, гидроксид калия, карбонат лития, карбонат натрия, карбонат калия, трет-бутоксид калия, трет-бутоксид натрия, гидрокарбонат натрия, гидрокарбонат калия или карбонат цезия; органометаллическому реагенту, такому как бутиллитий, метиллитий, бистриметилсилиламид лития, бистриметилсилиламид натрия или бистриметилсилиламид калия; гидриду, такому как гидрид лития, гидрид натрия или гидрид калия; гетероциклическому соединению, такому как имидазол, пиридин, диметилпиридин, триметилпиридин или 4-диметиламинопиридин; или органическому амину, такому как триэтиламин, N,N-диизопропилэтиламин или диазабициклоундецен.
[0024] Соединение (I) или его соль могут представлять собой ангидрид, гидрат или сольват, при этом примером сольвата является сольват диметилсульфоксида.
[0025] Не существует конкретного ограничения относительно солей соединения (I), и при этом примеры солей соединения (I) включают соли неорганических кислот, соли органических кислот и соли кислых аминокислот.
[0026] Также не существует конкретных ограничений относительно солей соединения (IV), и при этом примеры солей соединения (IV) включают соли неорганических кислот, соли органических кислот и соли кислых аминокислот.
[0027] Предпочтительные примеры солей неорганических кислот включают соли хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, азотной кислоты и фосфорной кислоты.
[0028] Предпочтительные примеры солей органических кислот включают соли уксусной кислоты, янтарной кислоты, фумаровой кислоты, малеиновой кислоты, винной кислоты, лимонной кислоты, молочной кислоты, стеариновой кислоты, бензойной кислоты, метансульфоновой кислоты, этансульфоновой кислоты и п-толуолсульфоновой кислоты, при этом метансульфоновые соли являются предпочтительными.
[0029] Предпочтительные примеры солей кислых аминокислот включают соли аспарагиновой кислоты и глутаминовой кислоты.
[0030] Не существует конкретных ограничений относительно солей соединений, представленных формулой (A-2), и при этом примеры включают соли неорганических кислот, таких как хлористоводородная кислота и бромистоводородная кислота.
[0031] Далее способ получения по настоящему изобретению будет описан более подробно.
[0032]
Способ получения 1: способ получения соединения (I) или его соли (стадия A)
[0033] Стадия A представляет собой стадию, в которой соединение (A-2) или его соль вводят в реакцию с соединением (A-1) с получением соединения (I) или его соли.
[0034] Реакционный раствор конкретно не ограничен, поскольку он растворяет исходный материал и не вступает в реакцию и, например, он может представлять собой диметилсульфоксид, N,N-диметилформамид, N,N-диметилацетамид, 1,3-диметил-2-имидазолидинон или т. п., при этом диметилсульфоксид является предпочтительным.
[0035] Соединение (A-2) или его соль можно применять при 1,0-2,0 эквивалентах относительно количества молей соединения (A-1).
[0036] Основание конкретно не ограничено и, например, оно может представлять собой основание, такое как карбонат цезия, т-бутоксид калия или гидроксид калия, при этом гидроксид калия является предпочтительным. Основание можно применять при 1,5-2,0 эквивалентах относительно количества молей соединения (A-2) или его соли, применяемых в реакции.
[0037] Как правило, время реакции также будет отличаться, в зависимости от исходных материалов, растворителя и других реагентов, применяемых в реакции, однако оно предпочтительно составляет от 5 до 100 часов и более предпочтительно от 10 до 30 часов.
[0038] Как правило, температура реакции будет подобным образом отличаться, в зависимости от исходных материалов, растворителя и других реагентов, применяемых в реакции, однако предпочтительно она составляет от комнатной температуры до температуры кипения растворителя с обратным холодильником, более предпочтительно от 60°C до 80°C и еще более предпочтительно от 65°C до 75°C.
[0039] По завершении реакции содержащий воду органический растворитель можно вводить в реакционную смесь для осаждения и выделения соединения (I) или его соли. Количество вводимого содержащего воду органического растворителя может составлять 10-20-кратный (об./вес) объем относительно массы соединения (A-1). Кроме того, применяемый органический растворитель, содержащий воду, может представлять собой, например, смесь вода/ацетон (объемное отношение: от 50/50 до 80/20).
[0040] Соединение (I) или его соль могут быть получены в виде ангидрида, гидрата или сольвата путем изменения условий высушивания, т. е. условий, таких как температура и степень снижения давления.
[0041]
Способ получения 2: способ получения соединения (IV) или его соли (стадии B и C)
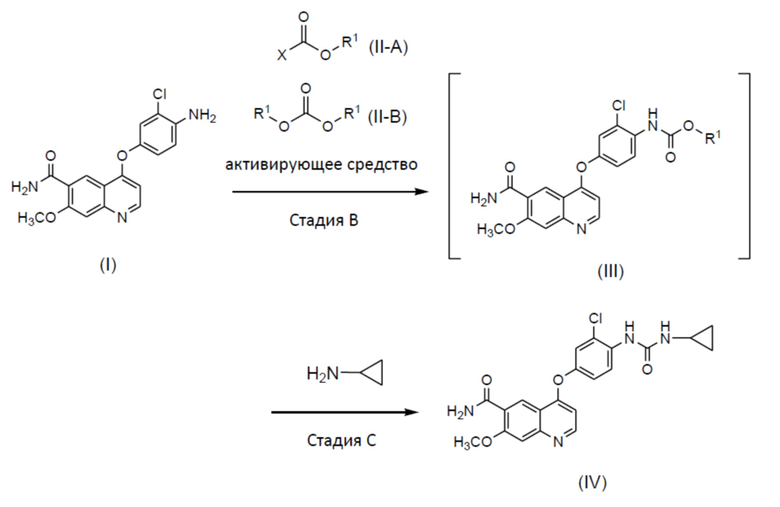
[0042] Данный способ включает стадию, в которой соединение (I) или его соль, полученные в вышеописанном способе получения 1, вводят в реакцию с соединением (II) с получением соединения (III) (стадия B), и стадию, в которой соединение (III) в виде активированной формы соединения (I) вводят в реакцию с циклопропиламином без выделения, с получением соединения (IV) или его соли (стадия C). Термин "соединение (II)" представляет собой общий термин, относящийся к реагенту для преобразования соединения (I) в соединение (III) в качестве его активированной формы, и он представляет собой соединение (II-A), соединение (II-B) или другой активирующий реагент.
[0043] Реакционный раствор конкретно не ограничен, поскольку он не ингибирует реакцию и можно применять, например, N,N-диметилформамид, 1-метил-2-пирролидон, 1,3-диметил-2-имидазолидинон, диметилсульфоксид, тетрагидрофуран, ацетонитрил или им подобные, при этом N,N-диметилформамид является предпочтительным.
[0044] В соединении, представленном формулой (II-A) или формулой (II-B):
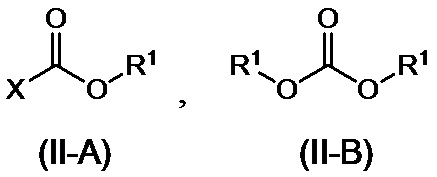
R1 представляет собой C1-6алкильную, C1-6алкенильную, C6-10арильную или C7-11аралкильную группы, при этом C1-6алкильная группа или C1-6алкенильная группа необязательно имеют 1-3 идентичных или разных заместителей, выбранных из группы, состоящей из атомов галогена и метокси-групп, и C6-10арильная группа или C7-11аралкильная группа необязательно имеют 1-3 идентичных или разных заместителей, выбранных из атомов галогена, метила, метокси и нитро-групп, и X представляет собой атом галогена. Кроме того, две R1 группы в формуле (II-B) могут вместе составлять циклический сложный эфир карбоновой кислоты с алкиленовой группой, такой как этиленовая группа.
[0045] Примеры соединения (II-A) включают метилхлорформиат, этилхлорформиат, изопропилхлорформиат, 2-метоксиэтилхлорформиат, 1-хлорэтилхлорформиат, изобутилхлорформиат, 2,2,2-трихлорэтилхлорформиат, пропилхлорформиат, 2-хлорэтилхлорформиат, фенилхлорформиат, 2-нафтилхлорформиат, бензилхлорформиат, 4-хлорфенилхлорформиат и 4-нитрофенилхлорформиат, и примеры соединения (II-B) включают диметилкарбонат, диэтилкарбонат, трифосген, бис(2-хлорэтил)карбонат, диаллилкарбонат, дифенилкарбонат, дибензилкарбонат и этиленкарбонат. В качестве других активированных реагентов вместо соединения (II-A) или соединения (II-B) можно применять сложные эфиры дикарбоновой кислоты, такие как ди-т-бутилдикарбонат или 1,1'-карбонилдиимидазол. Соединение (II) предпочтительно представляет собой фенилхлорформиат.
[0046] Соединение (II) можно применять при 1,0-3,0 эквивалентах относительно количества молей соединения (I).
[0047] Относительно основания конкретных ограничений не существует, и можно применять, например, пиридин, триметилпиридин, диметилпиридин, гидроксид калия, карбонат калия, гидрокарбонат натрия, триэтиламин, N,N-диизопропилэтиламин или им подобные, при этом пиридин является предпочтительным.
[0048] Основание можно применять при 1,0-3,0 эквивалентах относительно количества молей соединения (I).
[0049] К реакционному раствору можно добавлять предпочтительно 0,5-2,0 эквивалента, более предпочтительно 1,0-1,5 эквивалента и особенно предпочтительно 1,0 эквивалент воды относительно количества молей соединения (I).
[0050] Как правило, время реакции для стадии B также будет отличаться в зависимости от исходных материалов, растворителя и других реагентов, применяемых в реакции, но предпочтительно оно составляет от 15 минут до 24 часа.
[0051] Как правило, температура реакции B также будет отличаться в зависимости от исходных материалов, растворителя и других реагентов, применяемых в реакции, но предпочтительно она составляет от -50°C до комнатной температуры и более предпочтительно от -30°C до 0°C.
[0052] Соединение (III) подают на стадию C без выделения из реакционной смеси на стадии B. Циклопропиламин применяют при 1,0-7,2 эквивалентах относительно количества молей соединения (II).
[0053] Реакция на стадии C будет происходить с циклопропиламином отдельно, однако она будет происходить также при совместном присутствии как циклопропиламина, так и другого основания. Не существует конкретных ограничений относительно других оснований, которые могут представлять собой третичные амины, такие как триэтиламин, N,N-диизопропилэтиламин или трибутиламин, или гетероциклические соединения, такие как пиридин. В данном случае циклопропиламин можно применять при 1,0-5,0 эквивалентах относительно количества молей соединения (II), и другие основания можно применять при 1,0-5,0 эквивалентах относительно количества молей соединения (II).
[0054] Как правило, время реакции для стадии C также будет отличаться в зависимости от исходных материалов, растворителя и других реагентов, применяемых в реакции, но предпочтительно оно составляет от 30 минут до 90 часов.
[0055] Как правило, температура реакции C также будет отличаться в зависимости от исходных материалов, растворителя и других реагентов, применяемых в реакции, но предпочтительно она составляет от -20°C до 40°C и более предпочтительно от 0°C до 20°C.
[0056] После завершения реакции соединение (IV) или его соль можно осаждать и выделять посредством введения в реакционный раствор водного органического растворителя. Количество водного органического растворителя, подлежащего введению, может быть установлено в объеме от 10- до 20-кратного количества (об./вес) относительно массы соединения (I). Примеры органического растворителя, которые можно применять в качестве водного органического растворителя, включают без конкретного ограничения ацетон, изопропилацетат, этанол, 1-пропанол, 2-пропанол и N,N-диметилформамид. Примерами водного органического растворителя предпочтительно являются: вода/ацетон (объемное отношение 3/100-1/20), вода/изопропилацетат (объемное отношение 1/20) и вода/этанол (объемное отношение 1/1), и более предпочтительно вода/ацетон (объемное отношение 1/20). Следует отметить, что при необходимости могут быть добавлены затравочные кристаллы, в случае введения водного органического растворителя. В качестве альтернативы соединение (IV) или его соль также можно осаждать и выделять путем введения воды в реакционный раствор после завершения реакции.
[0057] Полученные кристаллы можно прополаскивать с помощью растворителя, такого как вода или ацетон, с получением кристаллов соединения (IV) (неочищенный продукт). Кристаллы (неочищенный продукт) можно затем кристаллизовать с помощью растворителя для очистки, такого как 1,3-диметил-2-имидазолидинон, N,N-диметилформамид, диметилсульфоксид, 2-пропанол или изопропилацетат.
[0058] Стадия D представляет собой стадию, в которой соединение (IV), полученное на стадии C, превращают в соль. Соль соединения (IV) предпочтительно представляет собой соль метансульфоновой кислоты.
[0059] Кристаллы соли, такие как 4-[3-хлор-4-(циклопропиламинокарбонил)аминофенокси]-7-метокси-6-хинолинкарбоксамида метансульфонат, могут образовываться посредством способа, описанного в PTL 4.
[0060] Более конкретно, в случае получения 4-[3-хлор-4-(циклопропиламинокарбонил)аминофенокси]-7-метокси-6-хинолинкарбоксамида метансульфоната, например, метансульфонат (кристаллы (C), описанный в патентной литературе 4) может быть получен после смешивания 4-[3-хлор-4-(циклопропиламинокарбонил)аминофенокси]-7-метокси-6-хинолинкарбоксамида, уксусной кислоты и метансульфоновой кислоты для растворения 4-[3-хлор-4-(циклопропиламинокарбонил)аминофенокси]-7-метокси-6-хинолинкарбоксамида с помощью добавления 1-пропанола в качестве слабого растворителя и постепенного охлаждения данного раствора. Следует отметить, что предпочтительно, чтобы кристаллы метансульфоната (C) в качестве затравочных кристаллов добавляли вместе со слабым растворителем, и чтобы для облегчения осаждения добавляли изопропилацетат. В качестве затравочных кристаллов можно использовать кристаллы метансульфоната (C), полученные в соответствии со способом, описанным в патентной литературе 4, или со способом, раскрытым в настоящем описании.
[0061] Количество добавляемой уксусной кислоты конкретно не ограничивается, однако предпочтительно можно применять 5-10-кратное количество и более предпочтительно 6-8-кратное количество относительно массы соединения (IV).
[0062] В качестве количества добавляемой метансульфоновой кислоты можно применять 1,00-1,50 эквивалента, предпочтительно 1,05-1.30 эквивалента, более предпочтительно 1,05-1,22 эквивалента и особенно предпочтительно 1,20 эквивалента относительно количества молей соединения (IV).
[0063] Метансульфоновую кислоту можно смешивать с соединением (IV) сразу или частями, и после применения предпочтительно от 1,00 эквивалента до 1,10 эквивалента и более предпочтительно 1,05 эквивалента, предпочтительно, чтобы дополнительно применять предпочтительно от 0,10 эквивалента до 0,20 эквивалента и более предпочтительно дополнительно 0,15 эквивалента относительно количества молей соединения (IV).
[0064] В случае, если получают соль 4-[3-хлор-4-(циклопропиламинокарбонил)аминофенокси]-7-метокси-6-хинолинкарбоксамида и другой кислоты, требуемую кислоту можно применять вместо метансульфоновой кислоты. Количество добавляемой кислоты должно корректироваться соответствующим образом, ссылаясь на количество добавляемой метансульфоновой кислоты.
[0065] Температура реакции на стадии D обычно отличается, в зависимости от исходных материалов, растворителей и других реагентов, применяемых в реакции, и предпочтительно составляет от 20 до 40°C и более предпочтительно от 25 до 35°C.
[0066] В качестве слабого растворителя можно применять метанол, этанол, 1-пропанол, 2-пропанол и им подобные, предпочтительно можно применять 1-пропанол.
[0067] Количество слабого растворителя конкретно не ограничивается, однако предпочтительно применяют 2-15-кратное количество и более предпочтительно 8-10-кратное количество относительно массы соединения (IV).
[0068] В случае, если добавляют изопропилацетат, количество конкретно не ограничивается, однако предпочтительно применяют 2-10-кратное количество и более предпочтительно 5-кратное количество относительно массы соединения (IV).
[0069] Температура охлаждения конкретно не ограничивается, однако она предпочтительно составляет от 15 до 25°C.
[0070] Кристаллы, полученные путем фильтрации, перемешивают в этаноле. Количество этанола, подлежащее применению, конкретно не ограничивается, однако предпочтительно применяют 5-10-кратное количество и более предпочтительно 7,5-кратное количество относительно массы соединения (IV).
[0071] Полученные кристаллы перемешивают в этаноле предпочтительно при 20-60°C в течение 2-5 часов и предпочтительно в течение 3 часов.
[0072] В соответствии с вышеприведенным способом получения, в метансульфонате соединения (IV) содержание соединения (A-1), соединения (I) и соединения (C-1) может быть установлено до 60 ppm по массе или менее, 350 ppm по массе или менее и 0,10% по массе или менее, соответственно.
[0073] В частности, содержание соединения (I) в метансульфонате соединения (IV) может быть снижено до 183 ppm по массе или менее при применении в избытке циклопропиламина на стадии C, или путем осуществления перекристаллизации соединения (IV) перед синтезом метансульфоната соединения (IV).
[0074] Соединение (A-1) представляет собой исходный материал стадии A, однако его растворимость в органических растворителях является низкой. Соответственно, удаление соединения (A-1) из соединения (IV) или его соли путем перекристаллизации является сложным. Однако в соответствии со способом получения по настоящему изобретению, содержание соединения (A-1) в соединении (IV) или его соли может быть снижено путем подвергания нескольким стадиям пути синтеза из стадии A через стадию B к стадии C. В частности, в соответствии с рассмотрением авторов настоящего изобретения, поскольку существует возможность того, что соединение (A-1) демонстрирует генотоксичность, то важно снижать содержание соединения (A-1) в соединении (IV) или его соли.
[0075] Предпочтительно, чтобы содержание соединения (A-1) в соединении (IV) или его соли составляло 60 ppm по массе или менее на основании Уровней предельно допустимого токсикологического воздействия (TTC), указанных в "Руководстве по предельным содержаниям генотоксичных примесей" ("Guideline on the Limits of Genotoxic Impurities"), выданном Европейским агентством по лекарственным средствам.
[Выражение 1]

[0076] Соединение (I) представляет собой исходный материал стадии B, а не вступившее в реакцию соединение (I) остается в качестве примеси в соединении (III) или образовано путем разложения соединения (III) или соединения (IV) или его соли на стадии B. В частности, если метансульфонат соединения (IV) нагревают после растворения в растворителе, то соединение (I) образуется в качестве продукта разложения соединения (IV) и т. п. В соответствии со способом получения по настоящему изобретению содержание соединения (I) в соединении (IV) или его соли может быть дополнительно снижено при использовании циклопропиламина в избытке на стадии C, или путем разделения минимального необходимого количества метансульфоновой кислоты и смешивания данного количества с соединением (IV), если соль соединения (IV) синтезируют на стадии D. Кроме того, содержание соединения (I) в соли соединения (IV) может быть дополнительно снижено путем осуществления перекристаллизации соединения (IV) для снижения содержания соединения (I) в соединении (IV) перед синтезом соли соединения (IV). В частности, соединение (I) представляет собой химическое вещество, размещенное на сайте Безопасности труда "Chemical substances on which strong mutagenicity was recognized" Министерства здравоохранения, труда и социального обеспечения Японии (Публичное извещение № 166 Министерства здравоохранения, труда и социального обеспечения от 27 марта 2012 г.), и важным является снижение содержания соединения (I) в соединении (IV).
[0077] Поскольку сложно постоянно контролировать то, является ли содержание соединения (I) в соединении (IV) или его соли равным или ниже TTC, то предпочтительно, чтобы содержание было на практически допустимом низшем уровне (ALARP), т. е. составляло 350 ppm по массе или менее на основании средних измеренных значений партий продукции 1-8 и верхнего предела доверительного интервала. В соответствии с одним вариантом осуществления способа получения по настоящему изобретению, как показано в таблице 1, содержание соединения (I), содержащегося в метансульфонате соединения (IV), может быть снижено до 350 ppm по массе или менее. В частности, содержание соединения (I) может быть снижено до 350 ppm по массе или менее путем соответствующего комбинирования с использованием гидроксида калия в качестве основания на стадии A в партиях 5-8, дополнительного выделения соединения (I) в виде кристаллов его ангидрида после стадии A и добавления воды в реакционный раствор на стадии B в партиях 6-8 с использованием циклопропиламина в избытке на стадии C и осуществления перекристаллизации соединения (IV) перед стадией D в партиях 5-8, и т. п.
[Таблица 1]
aНижний предел количественного определения (нижний предел) составляет 7 ppm по массе.
bВерхний предел доверительного интервала=трехкратное стандартное отклонение данных анализа партии
[0078] Поскольку сложно постоянно контролировать то, является ли содержание соединения (I) в соединении (IV) или его соли равным или ниже TTC, то предпочтительно, чтобы содержание было на практически допустимом низшем уровне (ALARP), т. е. составляло 183 ppm по массе или менее на основании среднего измеренного значения партий продукции 5-10 и верхнего предела доверительного интервала. В частности, содержание соединения (I), содержащегося в метансульфонате соединения (IV), может быть дополнительно снижено до 183 ppm по массе или менее, как показано в таблице 2, путем соответствующего комбинирования с использованием гидроксида калия в качестве основания на стадии A в партиях 5-10, дополнительного выделения соединения (I) в виде кристаллов его ангидрида после стадии A и добавления воды в реакционный раствор на стадии B в партиях 6-10 с использованием избыточного количества циклопропиламина на стадии C и осуществления перекристаллизации соединения (IV) перед стадией D в партиях 5-10, а также разделения метансульфоновой кислоты и смешивания ее с соединением (IV) на стадии D в партиях 9-10, и т. п.
[Таблица 2]
aНижний предел количественного определения (нижний предел) составляет 7 ppm по массе.
bВерхний предел доверительного интервала=трехкратное стандартное отклонение данных анализа партии
[0079] Соединение (C-1) представляет собой побочный продукт, образованный главным образом на стадии B. На стадии B образование соединения (C-1) может быть снижено более эффективно путем дополнительного добавления в реакционный раствор одного эквивалента воды. Следует отметить, что в случае, если соединение (I)⋅моногидрат применяют в качестве исходного материала, то образование соединения (C-1) может быть снижено без добавления одного эквивалента воды.
[0080] Предпочтительно, чтобы содержание соединения (C-1) в соединении (IV) или его соли составляло 0,10% по массе или менее, в соответствии с руководствами ICH Q3A.
[0081] Предпочтительно, чтобы чистота соединения (IV) или его соли составляла 98,0% по массе или более с учетом данных анализа партии, теста стабильности и аналитической изменчивости.
[0082] В случае, если составляют соединение (IV) или его соль, как правило, применяют фармацевтическую композицию, содержащую соединение (IV) или его соль и соответствующую добавку в виде фармацевтически приемлемого носителя. Однако приведенное выше описание не предназначено, чтобы воспрепятствовать тому, что составление осуществляют при использовании только соединения (IV) или его соли.
[0083] В качестве вышеуказанной добавки можно применять вспомогательное средство, связующее, смазывающее средство, разрыхлитель и им подобные, которые можно обычно применять в области фармацевтики. В качестве вышеуказанной добавки данные средства можно соответствующим образом применять также в комбинации.
[0084] Примеры вышеуказанного вспомогательного средства включают лактозу, сахарозу, глюкозу, маннит, прежелатинизированный крахмал и кристаллическую целлюлозу.
[0085] Примеры вышеуказанного связующего включают метилцеллюлозу, гидроксипропилметилцеллюлозу и гидроксипропилцеллюлозу.
[0086] Примеры вышеуказанного смазывающего средства включают стеарат магния, тальк, полиэтиленгликоль и коллоидный диоксид кремния.
[0087] Примеры вышеуказанного разрыхлителя включают кристаллическую целлюлозу, агар, желатин, карбонат кальция и гидрокарбонат натрия.
[0088] Кроме того, примеры вышеуказанного состава включают твердые составы для перорального применения, такие как таблетки, порошки, гранулы, капсулы, сиропы, пастилки и средства для ингаляции. Составы, полученные путем составления соединения (IV) или его соли, или содержащая их фармацевтическая композиция обычно предоставляются в соответствующей первичной упаковке (контейнере или пакете) и транспортируются как фармацевтический препарат. В качестве первичной упаковки можно применять упаковку в форме, подходящей для применения каждого состава.
[0089] Вышеуказанный твердый состав для перорального применения составляют путем комбинирования соответствующим образом вышеуказанных добавок. Следует отметить, что при необходимости на поверхность твердого состава для перорального применения может быть нанесено покрытие.
[0090] Твердый состав для перорального применения может быть получен в соответствии с описанием, например в WO 2006/030826 или WO 2011/021597. В случае, если 5% водный раствор (вес/вес) получают для стабилизации соединения (IV) или его соли, то в качестве фармацевтически приемлемого носителя предпочтительно применять соединение, значение pH которого становится 8 или более. В качестве альтернативы, для стабилизации соединения (IV) или его соли можно применять карбонат щелочноземельного металла в качестве фармацевтически приемлемого носителя.
[0091] Первичная упаковка для твердого состава для перорального применения представляет собой, например, стеклянный или пластмассовый флакон или баночку. Пластмасса в данном документе означает полимеры, такие как полиэтилен высокой плотности (HDPE). Кроме того, в случае предоставления твердого состава для перорального применения в бутылке, с приведенным составом может быть инкапсулировано высушивающее средство, такое как силикагель.
[0092] Один вариант осуществления вышеуказанного фармацевтического средства представляет собой флакон из HDPE, в котором инкапсулированы таблетки или капсулы, содержащие соединение (IV) или его соль и силикагель. В частности, примером является флакон из HDPE, в котором инкапсулированы приблизительно 30 капсул, содержащих соединение (IV) или его соль и приблизительно 2 г силикагеля.
[0093] Другой пример первичной упаковки для твердого состава для перорального применения представляет собой блистерную упаковку. Пример блистерной упаковки представляет собой выдавливаемую упаковку (PTP). PTP состоит из формовочных материалов, покрывных материалов и т. п.
[0094] Примеры компонентов вышеуказанных формовочных материалов включают металлы, такие как алюминий, и пластмассы, такие как поливинилхлорид (PVC), поливинилиденхлорид (PVDC), циклические полиолефины, полиамиды и полипропилен (PP). Формовочный материал может представлять собой однослойный материал, состоящий из одного компонента, или может представлять собой слоистый материал, состоящий из множества компонентов, такой как алюминиевая слоистая пленка. Покрывной материал состоит из материала-основы, такого как алюминий или пластмасса, и при необходимости средства для горячей закупорки и т. п.
[0095] Один вариант осуществления PTP представляет собой например PTP, состоящий из формовочного материала из алюминиевой слоистой пленки и покрывного материала из алюминия, или PTP, состоящего из формовочного материала, изготовленного из пластмассы и покрывного материала из алюминия. Для такого PTP в случае необходимости можно применять вторичную упаковку (упаковку-вкладыш) с использованием полиэтилена или алюминия. Кроме того, в упаковке-вкладыше с PTP можно применять высушивающее средство.
[0096] Один вариант осуществления вышеуказанного фармацевтического препарата представляет собой PTP, в которой предоставляются таблетки или капсулы, содержащие соединение (IV) или его соль, где PTP состоит из алюминиевой слоистой пленки и алюминия.
[0097] Вышеуказанный флакон или вышеуказанная PTP могут предоставляться, в качестве окончательной формы упаковки, с листком-вкладышем в упаковке фармацевтического препарата в коробке и т. п.
[0098] В твердом составе для перорального применения, содержащем соединение (IV) или его соль, соединение (I) увеличивается не более чем на 0,02% на протяжении периода хранения в испытании на воздействие ускорения, как показано в примерах, описанных ниже. Другими словами, как показано в таблице 1, если твердый состав для перорального применения, содержащий соединение (IV) или его соль, в котором содержание соединения (I) составляет 350 ppm по массе или менее, хранят в условиях хранения теста на воздействие ускорения, описанного ниже, или на протяжении периода хранения при комнатной температуре в течение трех лет, содержание соединения (I) может сохранять значение 0,06% по массе или менее в твердом составе для перорального применения.
[0099] Соответственно, один аспект настоящего изобретения представляет собой твердый состав для перорального применения, который содержит соединение (IV) или его соль и фармацевтически приемлемый носитель, и в котором содержание соединения (I) составляет 0,06% по массе или менее.
[0100] В качестве альтернативы, как показано в таблице 2, если твердый состав для перорального применения, содержащий соединение (IV) или его соль, в которой содержание соединения (I) составляет 183 ppm по массе или менее, хранят в условиях хранения теста на воздействие ускорения, описанного ниже, или на протяжении периода хранения при комнатной температуре в течение трех лет, содержание соединения (I) может сохранять значение 0,04% по массе или менее, или 0,040% по массе или менее в твердом составе для перорального применения.
[0101] Соответственно один аспект настоящего изобретения представляет собой твердый состав для перорального применения, который содержит соединение (IV) или его соль и фармацевтически приемлемый носитель, и в котором содержание соединения (I) составляет 0,04% по массе или менее или 0,040% по массе или менее.
[0102] В случае применения соединения (IV) или его соли для получения фармацевтического препарата, применяемое количество варьирует в зависимости от симптомов, возраста и форм введения, но обычно взрослому вводят от 100 мкг до 10 г один раз в сутки, или применяют частями несколько раз в сутки.
ПРИМЕРЫ
[0103] Теперь настоящее изобретение будет дополнительно объяснено с помощью примеров, при этом следует понимать, что настоящее изобретение не ограничено данными примерами.
[0104]
Пример 1: 4-(4-амино-3-хлорфенокси)-7-метоксихинолин-6-карбоксамид
Смесь 43,5 кг 4-амино-3-хлорфенола гидрохлорида, 53,8 кг 48,5 вес/вес % водного раствора гидроксида калия, 44,0 кг 4-хлор-7-метоксихинолин-6-карбоксамида и 396 л диметилсульфоксида перемешивали при 70°C в течение 20 часов в атмосфере азота. После добавления в реакционную смесь при 55°C содержащего воду ацетона (ацетон: 220 л, очищенная вода: 440 л), смесь охлаждали до 8°C и осажденный осадок фильтровали. Осадок прополаскивали водным раствором ацетона и полученное твердое вещество высушивали при сниженном давлении с получением 59,3 кг 4-(4-амино-3-хлорфенокси)-7-метоксихинолин-6-карбоксамида (выход: 93%).
[0105]
Пример 2: 4-[3-хлор-4-(циклопропиламинокарбонил)аминофенокси]-7-метокси-6-хинолинкарбоксамид
К смеси 26,0 кг 4-(4-амино-3-хлорфенокси)-7-метоксихинолин-6-карбоксамида, 13,2 кг пиридина, 1,36 кг воды и 196,0 л N,N-диметилформамида добавляли 26,6 кг фенилхлорформиата при -20°C в атмосфере азота и смесь перемешивали в течение 3 часов. Далее, 19,4 кг циклопропиламина дополнительно добавляли при 8°C и смесь перемешивали в течение 15 часов. После добавления в реакционную смесь 13,0 л воды и 261,0 л ацетона осажденный осадок фильтровали. Осадок прополаскивали ацетоном и полученное твердое вещество высушивали при сниженном давлении с получением 28,7 кг неочищенного продукта указанного в заголовке соединения (89% выход). Полученное кристаллизовали из 359,6 л 1,3-диметил-2-имидазолидинона и 575,0 л 2-пропанола с получением 25,7 кг соединения (IV) (90% выход).
[0106] В примерах 1 и 2 общий выход составлял 83% в двух стадиях с получением неочищенного продукта соединения (IV) относительно исходного материала соединения (I), и он представлял собой высокий выход по сравнению с выходом в способе получения PTL 1 (три стадии, 25,5%). Кроме того, кристаллизация соединения (IV) обеспечивала получение высокой чистоты соединения (IV) с выходом 90%.
[0107]
Пример 3: 4-[3-хлор-4-(циклопропиламинокарбонил)аминофенокси]-7-метокси-6-хинолинкарбоксамида метансульфонат
В смешанном растворе метансульфоновой кислоты (5,44 кг) и уксусной кислоты (150 л) растворяли 4-[3-хлор-4-(циклопропиламинокарбонил)аминофенокси]-7-метокси-6-хинолинкарбоксамид (23,0 кг) при температуре от 20°C до 35°C. Дополнительно добавляли метансульфоновую кислоту (777 г), раствор фильтровали при температуре 35°C или менее, и фильтровальную бумагу промывали уксусной кислотой (11,5 л). К фильтрату добавляли 1-пропанол (46,0 л) и затравочные кристаллы (230 г) при температуре от 25°C до 45°C, и дополнительно по каплям добавляли 1-пропанол (161 л) и изопропилацетат (115 л) при температуре от 25°C до 45°C. Смешанный раствор охлаждали до температуры от 15°C до 25°C, а затем осажденные кристаллы фильтровали и промывали смешанным раствором 1-пропанола и изопропилацетата (концентрация 1-пропанола: 33 об./об. %). К полученным влажным кристаллам добавляли этанол (173 л) и перемешивали при температуре от 20°C до 60°C в течение трех часов. После того, как кристаллы были собраны с помощью фильтрации и промыты этанолом, кристаллы высушивали при сниженном давлении при температуре 80°C или менее с получением таким образом 4-[3-хлор-4-(циклопропиламинокарбонил)аминофенокси]-7-метокси-6-хинолинкарбоксамида метансульфоната (27,5 кг, выход: 94%).
[0108]
Пример 4: 4-(4-амино-3-хлорфенокси)-7-метоксихинолин-6-карбоксамида⋅моногидрат
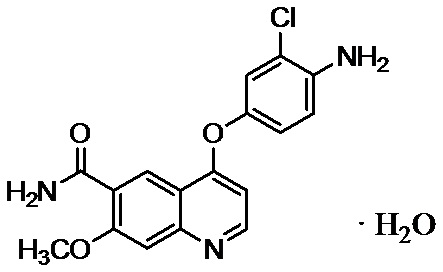
Смесь 4-амино-3-хлорфенола гидрохлорида (593,4 г), 48,7 вес/вес % водного раствора гидроксида калия (730,6 г), 4-хлор-7-метоксихинолин-6-карбоксамида (600,0 г) и диметилсульфоксида (5,4 л) перемешивали в атмосфере азота при 70°C в течение 21 часа. После введения в реакционный раствор 3,0 г затравочных кристаллов добавляли водный ацетон (ацетон: 3 л, очищенная вода: 6 л) при 55°C и охлаждали до 8°C, и осажденный осадок фильтровали. Осадок промывали водным ацетоном и твердое вещество, полученное с использованием роторного испарителя, высушивали при 60°C при сниженном давлении с получением таким образом 4-(4-амино-3-хлорфенокси)-7-метоксихинолин-6-карбоксамида⋅моногидрата (862,7 г, выход: 94%).
[0109]
Пример 5: 4-[3-хлор-4-(циклопропиламинокарбонил)аминофенокси]-7-метокси-6-хинолинкарбоксамид
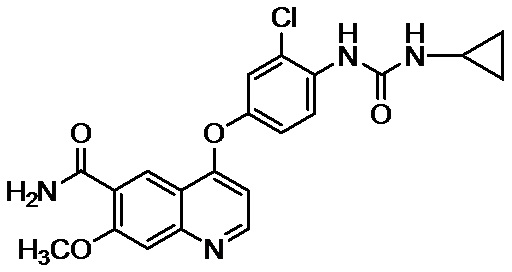
К смеси 4-(4-амино-3-хлорфенокси)-7-метоксихинолин-6-карбоксамида⋅моногидрата (800 г), пиридина (524,8 г) и N,N-диметилформамида (8 л) добавляли фенилхлорформиат (865,6 г) в атмосфере азота при -20°C и перемешивали в течение одного часа. Кроме того, добавляли циклопропиламин (757,6 г) и перемешивали при 8°C в течение 18 часов. К реакционному раствору добавляли воду (8 л) и осажденный осадок фильтровали. Осадок промывали водным N,N-диметилформамидом и этанолом, и полученное в результате твердое вещество высушивали при сниженном давлении с получением таким образом неочищенного продукта указанного в заголовке соединения (910 г, выход: 96%). Пятьсот грамм неочищенного продукта кристаллизовали из 1,3-диметил-2-имидазолидинона (6250 мл) и 2-пропанола (10 л) с получением таким образом соединения (IV) (450 г, выход: 90%).
[0110] Тест чистоты 1
В отношении осажденного неочищенного продукта соединения (IV), полученного в примере 2, соединения (IV), полученного в соответствии со способом получения, описанным в патентной литературе 1, и осажденного неочищенного продукта соединения (IV), полученного в соответствии со способами получения, описанными в патентной литературе 2, 3, 4 и 5, значения их чистоты анализировали посредством жидкостной хроматографии и каждое из них сравнивали. Как показано в таблице 3, содержание соединения (IV), полученного в примере 2, было выше, чем содержание соединения (IV), полученного в соответствии со способами получения, описанными в патентной литературе 1-5, и общее содержание примесей было ниже.
[0111] Результаты приведены в таблице 3.
[Таблица 3]
bИзмерено с использованием соединения (IV), полученного посредством способа получения, описанного в патентной литературе 1 (пример 368)
cИзмерено с использованием осажденного неочищенного продукта соединений (IV), полученных посредством способов получения, описанных в патентной литературе 2 (сравнительный пример 3), патентной литературе 3 (пример 4), патентной литературе 4 (пример получения 3) и патентной литературе 5 (пример 1a)
[0112] Расчет площади % в таблице 3 осуществляли следующим образом. Рассчитывали площадь пиков для пиков, полученных из образца на хроматограмме, полученной при нижеследующих условиях измерения, площадь пиков для каждого пика делили на общее число, для получения таким образом общего числа фигур пиков, соответствующих примесям, в виде общего содержания примесей и фигуры, соответствующей соединению (IV) в виде содержания соединения (IV).
[0113] Кроме того, расчет % по массе в таблице 3 осуществляли следующим образом. Вначале, в отношении содержания соединения (IV), при использовании стандарта соединения (IV), полученного путем кристаллизации, в качестве внешнего контроля и сравнении площади пиков для каждого пика, соответствующего соединению (IV) в стандарте и в образце, рассчитывали содержание соединения (IV) в образце. Затем, с целью компенсации разности поглощения каждой примеси на единицу массы, впоследствии определяли каждую примесь в соответствии с процедурой, описанной в тесте чистоты 2, и синтезировали образец каждой примеси, при этом поглощение (коэффициент чистоты) каждой примеси определяли, если поглощение соединения (IV) принимали за 1. Затем, путем применения площадей пиков и коэффициентов чувствительности примесей в образце, рассчитывали массу каждой примеси (%) и общее количество обнаруженных примесей, которое превышало 0,05% по массе, принимали за общее содержание примесей.
[0114]
Условия измерения при жидкостной хроматографии:
детектор: ультрафиолетовый абсорбциометр (длина волны при измерениях: 252 нм);
колонка: YMC-Pack ProC18 (YMC Inc.), внутренний диаметр: 4,6 мм, длина: 15 см, диаметр частиц наполнителя: 3 мкм;
температура колонки: постоянная температура около 25°C;
подвижная фаза: раствор A и раствор B, содержащие нижеследующие композиции, элюировали с линейным градиентом, показанным в таблице 2;
раствор A: смесь вода/ацетонитрил/70% перхлорной кислоты (990:10:1, об./об./об.);
раствор B: смесь вода/ацетонитрил/70% перхлорной кислоты (100:900:1, об./об./об.);
скорость потока: 1,0 мл/мин;
скорость впрыска: 10 мкл;
температура штатива для проб: постоянная температура около 15°C;
диапазон измерения площади: 45 минут.
[0115] [таблица 4]
(мин.)
[0116] Следует отметить, что каждый из пределов количественного определения (нижних пределов) соединения (A-1), соединения (I) и соединения (C-1) в условиях измерения теста чистоты 1 составлял 0,0020% по массе (20 ppm по массе), 0,0020% по массе (20 ppm по массе), и 0,0022% по массе (22 ppm по массе).
[0117] Тест чистоты 2
В условиях измерения теста чистоты 1 сравнивали каждое значение времени удерживания соединения (A-1), соединения (I), соединения (C-1) и соединения (IV). "Относительное время удерживания", показанное в таблице 5, означает относительное время удерживания соединения (A-1), соединения (I) и соединения (C-1) относительно соединения (IV). То есть значение, полученное при делении времени удерживания пика, который получен от каждого соединения на хроматограмме, полученной в условиях измерения теста чистоты 1, на время удерживания пика, полученного при впрыскивании соединения (IV), описывали как "относительное время удерживания".
[0118] [таблица 5]
[0119] В вышеуказанных условиях измерения каждое соединение идентифицировали с помощью того факта, что время его элюирования в HPLC соответствовало времени элюирования образца. Следует отметить, что образцы каждого соединения синтезировали отдельно, и каждую из химических структур определяли на основании их 1H-ЯМР и MS спектров.
[0120]
Соединение (C-1): 1-{2-хлор-4-[(6-циано-7-метоксихинолин-4-ил)окси]фенил}-3-циклопропилмочевина
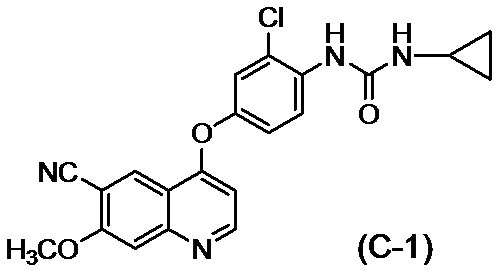
1H-ЯМР (600 МГц, DMSO-d6) δ (ppm): 0,42 (2H,m), 0,66 (2H,m), 2,57 (1H,dtt,J=3,4,7 Гц), 4,05 (3H,s), 6,58 (1H,d,J=5 Гц), 7,20 (1H,d,J=3 Гц), 7,25 (1H,dd,J=3,9 Гц), 7,49 (1H,d,J=3 Гц), 7,58 (1H,s), 7,98 (1H,s), 8,28 (1H,d,J=9 Гц), 8,72 (1H,s), 8,73 (1H,d,J=5 Гц).
[0121] Затем, в отношении соединения (IV), полученного в примере 2, и соединения (IV), полученного как описано в патентной литературе 2, 3, 4 и 5, измеряли содержание соединения (A-1) посредством жидкостной хроматографии. В результате, как показано в таблице 6, содержание соединения (A-1) составляло 1311 ppm по массе в соединении (IV), полученном посредством способов получения, описанных в патентной литературе 2, 3, 4 и 5, тогда как содержание снижалось до 20 ppm по массе или менее в соединении (IV), полученном в примере 2.
[0122] [таблица 6]
**Способы получения, описанные в патентной литературе 2 (сравнительный пример 3), патентной литературе 3 (пример 4), патентной литературе 4 (пример получения 3) и патентной литературе 5 (пример 1a)
[0123] В отношении соединения (IV), полученного в примере 2, и соединения (IV), полученного посредством способа получения, описанного в патентной литературе 1, содержание соединения (C-1) измеряли посредством жидкостной хроматографии. В результате, как показано в таблице 7, содержание соединения (C-1) составляло 3,37% по массе в соединении (IV), полученном посредством способа получения, описанного в патентной литературе 1, тогда как содержание соединения (C-1) снижалось до 0,05% по массе или менее в соединении (IV), полученном в примере 2.
[0124] [таблица 7]
**Измерено с использованием соединения (IV), полученного посредством способа получения, описанного в патентной литературе 1 (пример 368)
[0125] Тест чистоты 3
В отношении метансульфоната соединения (IV), полученного в примере 3, определяли каждое из соединения (C-1) при нижеследующих условиях измерения A, а также соединения (A-1) и соединения (I) при нижеследующих условиях измерения B. В частности, в отношении соединения (A-1) и соединения (I) измерение можно было осуществлять с хорошей чувствительностью посредством способа внешнего стандарта, в котором применяли стандартные растворы, полученные из этих стандартов, в следующих условиях. Следует отметить, что чистота метансульфоната соединения (IV), полученного в примере 3, составляла 99,3% по массе.
[0126]
Условия измерения при жидкостной хроматографии:
детектор: ультрафиолетовый абсорбциометр (длина волны при измерениях: 252 нм);
колонка: YMC-Pack ProC18 (YMC Inc.), внутренний диаметр: 4,6 мм, длина: 7,5 см, диаметр частицы наполнителя: 3 мкм;
температура колонки: постоянная температура около 40°C;
подвижная фаза: раствор A и раствор B, содержащие нижеследующие композиции, элюировали с линейным градиентом, показанным в таблице 8;
раствор A: смесь вода/ацетонитрил/70% перхлорной кислоты (990:10:1, об./об./об.);
раствор B: смесь вода/ацетонитрил/70% перхлорной кислоты (100:900:1, об./об./об.);
скорость потока: 1,0 мл/мин;
скорость впрыска: 10 мкл;
температура штатива для проб: постоянная температура около 15°C;
диапазон измерения площади: 30 минут.
[0127] [таблица 8]
(мин.)
(об. %)
[0128] Следует отметить, что предел количественного определения (нижний предел) соединения (C-1) в вышеуказанных условиях измерения A в тесте чистоты 3 составлял 0,01% по массе.
[0129]
Условия измерения при жидкостной хроматографии:
детектор: ультрафиолетовый абсорбциометр (длина волны при измерениях: 252 нм);
колонка: YMC-Pack ProC18 (YMC Inc.), внутренний диаметр: 4,6 мм, длина: 7,5 см, диаметр частицы наполнителя: 3 мкм;
температура колонки: постоянная температура около 40°C;
подвижная фаза: раствор A и раствор B, содержащие нижеследующие композиции, элюировали с линейным градиентом, показанным в таблице 9;
раствор A: смесь вода/ацетонитрил/70% перхлорной кислоты (990:10:1, об./об./об.);
раствор B: смесь вода/ацетонитрил/70% перхлорной кислоты (100:900:1, об./об./об.);
скорость потока: 1,0 мл/мин;
скорость впрыска: 5 мкл;
температура штатива для проб: постоянная температура около 15°C;
диапазон измерения площади: 13 минут.
[Таблица 9]
(мин.)
(об. %)
[0130] Следует отметить, что пределы количественного определения (нижние пределы) соединения (I) и соединения (A-1) составляли соответственно 7 ppm по массе и 12 ppm по массе при условиях измерения B.
[0131] Содержания каждого полученного соединения, показаны в таблице 10.
[Таблица 10]
[0132] Пример 6
Капсулы из 4-мг капсул и 10-мг капсул получали с использованием метансульфоната соединения (IV), показанного в таблице 1 или таблице 2, и с использованием D-маннита, осажденного карбоната кальция, гидроксипропилцеллюлозы с низкой степенью замещения, кристаллической целлюлозы, гидроксипропилцеллюлозы, талька и т. п. Следует отметить, что "4-мг капсула" означает капсулу, содержащую 4 мг соединения (IV) в капсуле. Масса гранул, которые являются содержимым капсулы, составляет 100 мг на капсулу. Содержания соединения (I) (% по массе) относительно общей массы капсулы во время получения капсулы (также называемые "исходным содержанием") показаны в таблице 11.
[Таблица 11]
(% по массе)
Капсула
Капсула
[0133] С помощью 4-мг и 10-мг капсул соединения (IV), полученного с мезилатом соединения (IV) в партии 5, 6 или 7 (масса гранул, которые являются содержимым капсулы, составляет 100 мг на капсулу), осуществляли тест на воздействие ускорения (40°C/75% RH, PTP (формовочный материал: алюминиевая слоистая пленка (полиамид/алюминий/поливинилхлорид), покрывной материал: алюминиевая фольга)) и тест с длительным хранением (25°C/60% RH, PTP (формовочный материал: алюминиевая слоистая пленка (полиамид/алюминий/поливинилхлорид), покрывной материал: алюминиевая фольга)).
[0134] В тесте на воздействие ускорения на 4-мг и 10-мг капсулы, содержание соединения (I) повышалось не более чем на 0,02% по массе и 0,01% по массе соответственно, по сравнению с исходным содержанием. Кроме того, содержание соединения (I) в тесте с длительным хранением в течение 24 месяцев повышалось незначительно, по сравнению с исходным содержанием. Повышение содержания соединения (I) в тесте с длительным хранением было меньшим, чем эффективная фигура предела количественного определения, и конкретно составляло от 0,003% по массе до 0,004% по массе. Измерение содержания соединения (I) в данных капсулах осуществляли посредством жидкостной хроматографии (предел обнаружения (нижний предел): 0,0020% по массе), и при этом предел количественного определения (нижний предел) составлял 0,01% по массе.
Изобретение относится к области органической химии, а именно к способу получения (варианты) метансульфонатной соли соединения формулы (IV), включающему стадию A приведения во взаимодействие соединения формулы (A-1) с соединением формулы (A-2) в присутствии подходящего основания с последующим осаждением и выделением соединения формулы (I) из реакционного раствора путем введения в реакционный раствор водного органического растворителя, стадию B приведения во взаимодействие соединения формулы (I), полученного на стадии A, с соединением формулы (II-A), где R1 представляет собой C1-6алкильную группу или C6арильную группу и X представляет собой атом галогена, в присутствии подходящего основания и воды, с получением таким образом соединения формулы (III), где R1 представляет собой такую же группу, как описано выше, стадию C приведения во взаимодействие соединения формулы (III), полученного на стадии B без выделения, с циклопропиламином с последующим осаждением и выделением соединения формулы (IV) путем введения в реакционный раствор водного органического растворителя и стадию D превращения соединения формулы (IV), полученного на стадии C, в метансульфонатную соль соединения формулы (IV). Технический результат: разработан способ получения метансульфонатной соли соединения формулы IV, проявляющего противоопухолевую активность, обеспечивающий высокий выход и высокую чистоту целевого соединения. 2 н. и 1 з.п. ф-лы, 11 табл., 6 пр.
 ,
,  ,
,  ,
,  ,
,  ,
, 
1. Способ получения метансульфонатной соли соединения, представленного формулой (IV)
 ,
,
включающий:
стадию B приведения во взаимодействие соединения, представленного формулой (I)
 ,
,
с соединением, представленным формулой (II-A)
 ,
,
где R1 представляет собой C1-6алкильную группу или C6арильную группу и X представляет собой атом галогена,
в присутствии подходящего основания и воды с получением таким образом соединения, представленного формулой (III)
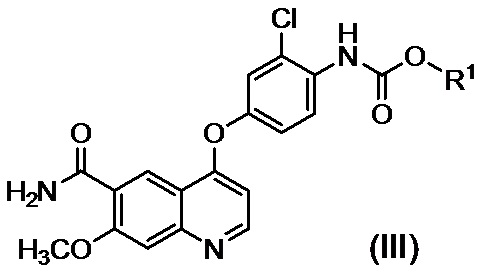 ,
,
где R1 представляет собой такую же группу, как описано выше, и
стадию C приведения во взаимодействие соединения, представленного формулой (III), полученного на стадии B без выделения, с циклопропиламином с последующим осаждением и выделением соединения, представленного формулой (IV)
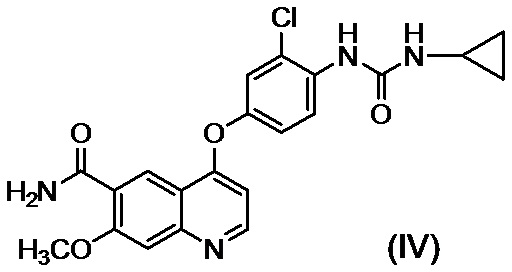 ,
,
путем введения в реакционный раствор водного органического растворителя, и
стадию D превращения соединения, представленного формулой (IV), полученного на стадии C, в метансульфонатную соль соединения, представленного формулой (IV).
2. Способ получения метансульфонатной соли соединения, представленного формулой (IV)
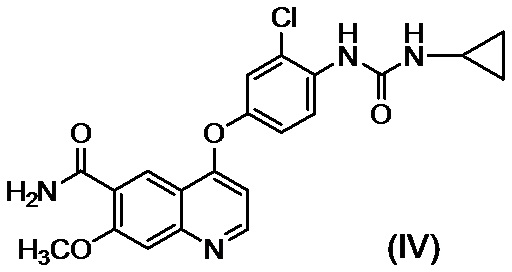 ,
,
включающий:
стадию A приведения во взаимодействие соединения, представленного формулой (A-1)
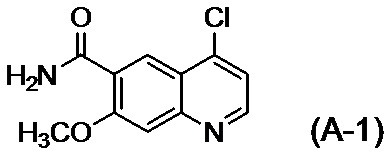 ,
,
с соединением, представленным формулой (A-2)
 ,
,
в присутствии подходящего основания с последующим осаждением и выделением соединения, представленного формулой (I)
 ,
,
из реакционного раствора путем введения в реакционный раствор водного органического растворителя,
стадию B приведения во взаимодействие соединения, представленного формулой (I)
 ,
,
полученного на стадии A, с соединением, представленным формулой (II-A)
,
где R1 представляет собой C1-6алкильную группу или C6арильную группу и X представляет собой атом галогена,
в присутствии подходящего основания и воды с получением таким образом соединения, представленного формулой (III)
,
где R1 представляет собой такую же группу, как описано выше,
стадию C приведения во взаимодействие соединения, представленного формулой (III), полученного на стадии B без выделения, с циклопропиламином с последующим осаждением и выделением соединения, представленного формулой (IV)
,
путем введения в реакционный раствор водного органического растворителя, и
стадию D превращения соединения, представленного формулой (IV), полученного на стадии C, в метансульфонатную соль соединения, представленного формулой (IV).
3. Способ по любому из пп. 1, 2, где соединение, представленное формулой (II-A), представляет собой фенилхлорформиат.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| ЕР 1894918 А1, 06.03.2008 | |||
| Устройство для горизонтального вытяжения позвоночника | 1990 |
|
SU1797881A1 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА СОЛИ 4-(3-ХЛОР-4-(ЦИКЛОПРОПИЛАМИНОКАРБОНИЛ)АМИНОФЕНОКСИ)-7-МЕТОКСИ-6-ХИНОЛИНКАРБОКСАМИДА ИЛИ СОЛЬВАТА ЭТОЙ СОЛИ И СПОСОБЫ ЕЕ ПОЛУЧЕНИЯ | 2004 |
|
RU2328489C2 |
| Разборный домашний спортивный комплекс | 1986 |
|
SU1683785A1 |
| АЗОТСОДЕРЖАЩИЕ АРОМАТИЧЕСКИЕ ПРОИЗВОДНЫЕ, ИХ ПРИМЕНЕНИЕ, ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 2001 |
|
RU2264389C2 |

