ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001]
Настоящее изобретение относится к соединению, обладающему ингибирующей активностью в отношении моноацилглицеринацилтрансферазы 2 (здесь и далее в настоящем документе называемой «MGAT2»), или к его фармацевтически приемлемой соли, и к содержащей его фармацевтической композиции.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
[0002]
Ожирение определяют как чрезмерно высокое количество жира или жировой ткани организма в сравнении с мышечной массой тела и рассматривают как главный фактор риска возникновения проблем со здоровьем. Индекс массы тела (BMI) представляет собой простое соотношение веса и роста, которое обычно используют для классификации избыточного веса и ожирения во взрослых (в возрасте 15 лет и старше) группах населения и у отдельных лиц. Его определяют как отношение веса в килограммах к росту в метрах в квадрате (кг/м2). Всемирная организация здравоохранения определяет «избыточный вес» при BMI, равном 25 кг/м2 или более, и «ожирение» при BMI, равном 30 кг/м2 или более. С другой стороны, Японское общество по изучению ожирения определяет «ожирение» при BMI, равном 25 кг/м2 или более. Это связано с тем, что согласно BMI количество связанных с ожирением нарушений, включая диабет и дислипидемию, возрастает, и при BMI, равном 25 кг/м2, среднее число связанных с ожирением нарушений составляет 1,0 или более. Всемирной организацией здравоохранения сообщалось, что в 2005 году приблизительно 1600 миллионов, и по меньшей мере 400 миллионов, человек во всем мире имеют избыточный вес и ожирение, соответственно. Ожирение, как правило, вызвано потреблением большего количества калорий, чем используется при физической активности и в повседневной жизни. Количество людей с ожирением увеличилось за счет употребления большего количества пищи с высоким содержанием жира и/или сахара, и по оценкам 700 миллионов человек или более будут страдать от ожирения во всем мире в 2015 году. Для лечения ожирения применяют диетотерапию, терапию физической нагрузкой, лекарственную терапию и т.п. При лекарственной терапии используют лекарства, включая орлистат, мазиндол и сибутрамин. Тем не менее, они не удовлетворяют требованиям согласно двум аспектам: эффективности и побочным эффектам.
Одной из причин ожирения является чрезмерное потребление нейтрального жира. Нейтральный жир (триглицерид), потребляемый с пищей, расщепляется под действием панкреатической липазы в желудочно-кишечном тракте на 2-моноацилглицерин и свободные жирные кислоты, которые всасываются эпителиальными клетками тонкого кишечника. Ацильная группа переносится со свободных жирных кислот на 2-моноацилглицерин под действием моноацилглицеринацилтрансферазы (MGAT). Образовавшийся диацилглицерин далее преобразуется в нейтральный жир под действием диацилглицеринацилтрансферазы (DGAT).
Были определены три изоформы MGAT, а именно MGAT1, MGAT2 и MGAT3. Среди них MGAT2 и MGAT3 характеризуются высоким уровнем экспрессии в тонком кишечнике, и, как считается, вовлечены в процесс всасывания жира в тонком кишечнике.
Сообщалось, что в эксперименте с нокаутными по MGAT2 мышами было продемонстрировано, что диета с высоким содержанием жира усиливает экспрессию MGAT2 в тонком кишечнике с увеличением активности MGAT (непатентный документ 1). Кроме того, у нокаутных по MGAT2 мышей было обнаружено снижение набора веса, вызванное рационом с высоким содержанием жиров, супрессия индукции резистентности к инсулину, снижение повышенного уровня холестерина в крови, предотвращение жировой трансформации печени, и т.п., и усиление энергозатрат (непатентный документ 2).
Хотя ранее сообщалось о соединениях, обладающих ингибирующей активностью в отношении MGAT2 (патентные документы 1-19, непатентные документы 3-13), описанные ниже соединения согласно настоящему изобретению не были раскрыты.
ССЫЛКИ НА ИЗВЕСТНЫЙ УРОВЕНЬ ТЕХНИКИ
Патентные документы
[0003]
[Патентный документ 1] Международная публикация WO 2010/095767 A
[Патентный документ 2] Международная публикация WO 2012/091010 A
[Патентный документ 3] Международная публикация WO 2012/124744 A
[Патентный документ 4] Международная публикация WO 2013/082345 A
[Патентный документ 5] Международная публикация WO 2013/112323 A
[Патентный документ 6] Международная публикация WO 2013/116065 A
[Патентный документ 7] Международная публикация WO 2013/116075 A
[Патентный документ 8] Международная публикация WO 2014/074365 A
[Патентный документ 9] Международная публикация WO 2014/133134 A
[Патентный документ 10] Международная публикация WO 2014/193884 A
[Патентный документ 11] JP 2014-5245 A
[Патентный документ 12] JP 2014-9165 A
[Патентный документ 13] Международная публикация WO 2015/112465 A
[Патентный документ 14] Международная публикация WO 2015/129845 A
[Патентный документ 15] Международная публикация WO 2015/134699 A
[Патентный документ 16] Международная публикация WO 2015/134701 A
[Патентный документ 17] Международная публикация WO 2015/191681 A
[Патентный документ 18] Международная публикация WO 2016/121782 A
[Патентный документ 19] Международная публикация WO 2017/069224 A
Непатентные документы
[0004]
[Непатентный документ 1] Journal of Biological Chemistry (2004), 279, 18878-18886
[Непатентный документ 2] Nature Medicine (2009), 15, (4), 442-446
[Непатентный документ 3] Bioorganic & Medicinal Chemistry Letter (2013), 23, 2721-2726
[Непатентный документ 4] Med. Chem. Commun (2013), 4, 1305-1311
[Непатентный документ 5] Bioorganic & Medicinal Chemistry Letter (2015), 23, 5922-5931
[Непатентный документ 6] Bioorganic & Medicinal Chemistry Letter (2015), 23, 4544-4560
[Непатентный документ 7] Journal of Lipid Research 2015, 56, 747-753
[Непатентный документ 8] European Journal of Pharmacology, 2015, 758, 72-81
[Непатентный документ 9] Journal of Medicinal Chemistry (2015), 58, 3892-3909
[Непатентный документ 10] HETEROCYCLES 2016, 92, 470-484
[Непатентный документ 11] Chemical and Pharmaceutical Bulletin, 2016, 64, 228-238
[Непатентный документ 12] European Journal of Pharmacology, 2016, 791, 569-577
[Непатентный документ 13] Analytical Biochemistry, 2016, 501, 48-55
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Задачи, подлежащие решению настоящим изобретением
[0005]
Цель настоящего изобретения заключается в предоставлении соединения, обладающего ингибирующей активностью в отношении MGAT2, или его фармацевтически приемлемой соли и содержащей его фармацевтической композиции.
Средства решения задачи
[0006]
Авторы настоящего изобретения провели активные исследования и преуспели в синтезе превосходных соединений, обладающих ингибирующей активностью в отношении MGAT2. Настоящее изобретение включает следующее:
[0007]
[1] Соединение, представленное формулой (I):
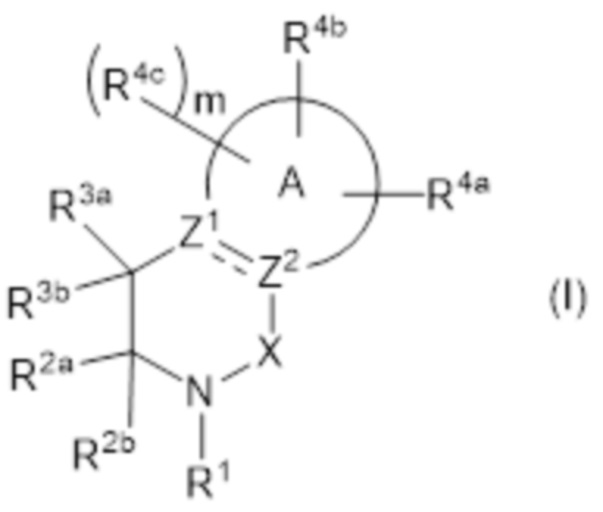
где
фрагмент, представленный формулой:

представляет собой
 ;
;
где X представляет собой C(=O);
R1 представляет собой водород;
R2a представлен формулой:

где кольцо C представляет собой бензол или пиридин;
каждый R5 независимо представляет собой галоген, галогеналкил, галогеналкилокси, неароматический карбоциклил, необязательно замещенный галогеном или галогеналкилом, или неароматический гетероциклил, необязательно замещенный галогеном или галогеналкилом, и
n равен целому числу от 1 до 3,
R2b представляет собой алкил или галогеналкил, или
необязательно, R2a и R2b объединены вместе со смежным атомом углерода с формированием кольца B,
кольцо B представлено формулой:

где каждый R6 независимо представляет собой галоген, галогеналкил, галогеналкилокси, неароматический карбоциклил, необязательно замещенный галогеном или галогеналкилом, или неароматический гетероциклил, необязательно замещенный галогеном или галогеналкилом,
каждый R14a и R14b независимо представляет собой водород или галоген, и
n равен целому числу от 1 до 3;
R3a представляет собой водород,
R3b представляет собой водород;
R4a представляет собой карбокси или представлен формулой:
 ;
;
L3 представляет собой одинарную связь или алкилен,
R7 представляет собой водород, галоген, алкилсульфонил, неароматический гетероциклил, необязательно замещенный оксо, или неароматический карбоциклилсульфонил, необязательно замещенный алкилом, или представлен формулой: -S(=O)(=N-H)-RS1,
R4b представляет собой алкил, необязательно замещенный заместителем группы α, ароматический карбоциклил, необязательно замещенный заместителем группы β, или ароматический гетероциклил, необязательно замещенный заместителем группы β,
RS1 представляет собой алкил,
заместитель группы α представляет собой галоген, галогеналкилокси и неароматический карбоциклил, и
заместитель группы β представляет собой галоген, циано, алкил, галогеналкил и алкилокси,
или его фармацевтически приемлемая соль.
[2] Соединение или его фармацевтически приемлемая соль в соответствии с пунктом [1], где R2a и R2b объединены вместе со смежным атомом углерода с формированием кольца B, и
кольцо B представлено любой из формул:

где значение каждого символа описано в пункте 1.
[3] Соединение или его фармацевтически приемлемая соль в соответствии с пунктом [1], где R2a представлен формулой:

где значение каждого символа описано в пункте 1, и
R2b представляет собой алкил или галогеналкил.
[4] Соединение или его фармацевтически приемлемая соль в соответствии с пунктом [1], где соединение выбирают из группы, состоящей из соединений I-8, I-10, I-12, I-23, I-24, I-34, I-67, I-170, I-190, I-212, I-236, I-253, I-275, I-276, II-93, II-94, II-103, II-121, II-151, II-168, II-174, II-203, II-225, II-233, II-270, II-276 и II-295.
[5] Соединение или его фармацевтически приемлемая соль в соответствии с пунктом [1], где соединение выбирают из группы, состоящей из соединений I-9, I-72, I-97, I-100, I-104, I-110, I-122, I-131, I-133, I-163, I-167, I-168, I-186, I-193, I-200, I-242, I-280, I-284, II-67, II-72, II-87, II-137, II-147, II-201, II-202, II-214, II-238, II-262, II-273 и II-274.
[6] Фармацевтическая композиция, содержащая соединение или его фармацевтически приемлемую соль в соответствии с любым из пунктов [1] - [5].
[7] Фармацевтическая композиция в соответствии с пунктом [6], обладающая ингибирующей активностью в отношении MGAT2.
[8] Фармацевтическая композиция в соответствии с пунктом [6] или [7] для применения при лечении или профилактике MGAT2-опосредованного заболевания.
[9] Фармацевтическая композиция в соответствии с пунктом [8] для применения при лечении или профилактике ожирения, метаболического синдрома, гиперлипидемии, гипертриглицеридемии, гипер-ЛПОНП-триглицеридемии, гиперлипоацидемии, сахарного диабета или артериосклероза.
[10] Способ лечения и профилактики MGAT2-опосредованного заболевания, включающий введение соединения или его фармацевтически приемлемой соли в соответствии с любым из пунктов [1] - [5].
[11] Соединение или его фармацевтически приемлемая соль в соответствии с любым из пунктов [1] - [5] для лечения и профилактики MGAT2-опосредованного заболевания.
[12] Применение соединения в соответствии с любым из пунктов [1] - [5] или его фармацевтически приемлемой соли для лечения и профилактики MGAT2-опосредованного заболевания.
ЭФФЕКТ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
[0008]
Соединения согласно настоящему изобретению обладают ингибирующей активностью в отношении MGAT2 и применимы в качестве профилактического средства и/или терапевтического средства, например, при ожирении, метаболическом синдроме, гиперлипидемии, гипертриглицеридемии, гипер-ЛПОНП-триглицеридемии, гиперлипоацидемии, сахарном диабете или артериосклерозе.
ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
[0009]
Ниже пояснены термины, использованные в настоящем описании. Если не указано иное, то каждый термин имеет одно и то же значения, если он используется по отдельности или вместе с другими терминами.
Термин «состоящий из» означает содержание только указанных компонентов.
Термин «содержащий» означает отсутствие ограничения указанными компонентами без исключения неописанных признаков.
[0010]
Термин «галоген» включает атом фтора, атом хлора, атом брома и атом йода. В частности, предпочтительными являются атом фтора и атом хлора.
[0011]
Термин «алкил» включает C1-C15, предпочтительно C1-C10, более предпочтительно C1-C6 и более предпочтительно C1-C4 линейную или разветвленную углеводородную группу. Примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил, изогексил, н-гептил, изогептил, н-октил, изооктил, н-нонил и н-децил.
Предпочтительным вариантом осуществления «алкила» является метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил или н-пентил. Более предпочтительным вариантом осуществления является метил, этил, н-пропил, изопропил или трет-бутил.
[0012]
Термин «алкилен» включает C1-C15, предпочтительно C1C10, более предпочтительно C1-C6 и более предпочтительно C1-C4 линейную или разветвленную двухвалентную углеводородную группу. Примеры включают метилен, этилен, пропилен, тетраметилен, пентаметилен и гексаметилен.
[0013]
Термин «ароматический карбоциклил» означает циклическую ароматическую углеводородную группу, которая представляет собой моноциклическую группу или полициклическую группу, содержащую два или более колец. Примеры включают фенил, нафтил, антрил и фенантрил.
Предпочтительным вариантом осуществления «ароматического карбоциклила» является фенил.
[0014]
Термин «неароматический карбоциклил» означает циклическую насыщенную углеводородную группу или циклическую ненасыщенную неароматическую углеводородную группу, которая представляет собой моноциклическую группу или полициклическую группу, содержащую два или более колец. «Неароматический карбоциклил», который представляет собой полициклическую группу, содержащую два или более колец, включает конденсированную кольцевую группу, в которой неароматический карбоциклил, который является моноциклической группой или полициклической группой, содержащей два или более колец, конденсирован с кольцом описанного выше «ароматического карбоциклила».
Кроме того, примеры «неароматического карбоциклила» также включают группу, содержащую мостиковую связь, или группу, формирующую следующее спирокольцо:

Неароматический карбоциклил, который представляет собой моноциклическую группу, предпочтительно представляет собой C3-C16 карбоциклил, более предпочтительно C3-C12 карбоциклил и более предпочтительно C4-C8 карбоциклил. Примеры включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил, циклодецил, циклопропенил, циклобутенил, циклопентенил, циклогексенил, циклогептенил и циклогексадиенил.
Неароматический карбоциклил, который представляет собой полициклическую группу, содержащую два или более колец, предпочтительно представляет собой C8C20 карбоциклил и более предпочтительно C8-C16 карбоциклил. Примеры включают инданил, инденил, аценафтил, тетрагидронафтил и флуоренил.
[0015]
Термин «ароматический гетероциклил» означает ароматический циклил, который представляет собой моноциклическую группу или полициклическую группу, содержащую два или более колец, содержащий один или несколько одинаковых или разных гетероатомов, независимо выбранных из O, S и N. «Ароматический гетероциклил», который представляет собой полициклическую группу, содержащую два или более колец, включает конденсированную кольцевую группу, в которой ароматический гетероциклил, который представляет собой моноциклическую группу или полициклическую группу, содержащую два или более колец, конденсирован с кольцом описанного выше «ароматического карбоциклила».
Ароматический гетероциклил, который представляет собой моноциклическую группу, предпочтительно представляет собой 5-8-членное кольцо и более предпочтительно 5-6-членное кольцо. Примеры включают пирролил, имидазолил, пиразолил, пиридил, пиридазинил, пиримидинил, пиразинил, триазолил, триазинил, тетразолил, фурил, тиенил, изоксазолил, оксазолил, оксадиазолил, изотиазолил, тиазолил и тиадиазолил.
Ароматический гетероциклил, который представляет собой бициклическую группу, предпочтительно представляет собой 8-10-членное кольцо и более предпочтительно 9- или 10-членное кольцо. Примеры включают индолил, изоиндолил, индазолил, индолизинил, хинолинил, изохинолинил, циннолинил, фталазинил, хиназолинил, нафтиридинил, хиноксалинил, пуринил, птеридинил, бензимидазолил, бензизоксазолил, бензоксазолил, бензоксадиазолил, бензизотиазолил, бензотиазолил, бензотиадиазолил, бензофурил, изобензофурил, бензотиенил, бензотриазолил, имидазопиридил, триазолопиридил, имидазотиазолил, пиразинопиридазинил, оксазолопиридил и тиазолопиридил.
Примеры ароматического гетероциклила, который представляет собой полициклическую группу, содержащую три или более колец, включают карбазолил, акридинил, ксантенил, фенотиазинил, феноксатиинил, феноксазинил и дибензофурил.
[0016]
Термин «неароматический гетероциклил» означает неароматический циклил, который представляет собой моноциклическую группу или полициклическую группу, содержащую два или более колец, содержащий один или несколько одинаковых или разных гетероатомов, независимо выбранных из O, S и N. «Неароматический гетероциклил», который представляет собой полициклическую группу, содержащую два или более колец, включает конденсированную кольцевую группу, в которой неароматический гетероциклил, который представляет собой моноциклическую группу или полициклическую группу, содержащую два или более колец, конденсирован с кольцом описанного выше «ароматического карбоциклила», «неароматического карбоциклила» и/или «ароматического гетероциклила», и включает конденсированную кольцевую группу, в которой кольцо описанного выше «ароматического гетероциклила» конденсировано с описанным выше «неароматическим карбоциклилом», который представляет собой моноциклическую группу или полициклическую группу, содержащую два или более колец.
Кроме того, примеры «неароматического гетероциклила» также включают группу, содержащую мостиковую связь, или группу, формирующую следующее спирокольцо:

Неароматический гетероциклил, который представляет собой моноциклическую группу, предпочтительно является 3-8-членным и более предпочтительно 5-6-членным. Примеры включают диоксанил, тииранил, оксиранил, оксетанил, оксатиоланил, азетидинил, тианил, тиазолидинил, пирролидинил, пирролинил, имидазолидинил, имидазолинил, пиразодидинил, пиразолинил, пиперидил, пиперазинил, морфолинил, морфолино, тиоморфолинил, тиоморфолино, дигидропиридил, тетрагидропиридил, тетрагидрофурил, тетрагидропиранил, дигидротиазолил, тетрагидроизотиазолил, дигидрооксазинил, гексагидроазепинил, тетрагидродиазепинил, тетрагидропиридазинил, гексагидропиримидинил, диоксоланил, диоксазинил, азиридинил, диоксолинил, оксепанил, тиоланил, тиинил и тиазинил.
Неароматический гетероциклил, который представляет собой полициклическую группу, содержащую два или более колец, предпочтительно является 8-20-членным и более предпочтительно 8-10-членным. Примеры включают индолинил, изоиндолинил, хроманил и изохроманил.
[0017]
Термин «необязательно замещенный заместитель группы α» означает, что атом углерода, атом азота или атом серы в любом положении может быть связан с одной или несколькими группами, выбранными из заместителя группы α.
Здесь и далее в тексте, то же самое относится к термину «необязательно замещенный заместитель группы β».
[0018]
Характерной чертой соединения согласно настоящему изобретению является то, что оно обладает ингибирующей активностью в отношении MGAT2 посредством конденсирования кольца A с моноциклом, таким как дигидропиридон формулы (I). Другой характерной чертой является то, что кольцо A конденсировано с моноциклом, таким как дигидропиридон, посредством чего еноновая структура может быть исключена, и токсичность может быть супрессирована.
[0019]
Соединения, представленные формулой (I), не ограничиваются определенными изомерами, а включают все возможные изомеры (например, кетоенольные изомеры, иминенаминовые изомеры, диастереоизомеры, энантиомеры, ротамеры, и т.п.), рацематы и их смеси.
[0020]
Один или несколько атомов водорода, углерода и/или других атомов в соединениях, представленных формулой (I) могут быть заменены изотопами атомов водорода, углерода и/или других атомов, соответственно. Примеры изотопов включает изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, йода и хлора, такие как 2H, 3H, 11C, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F, 123I и 36Cl, соответственно. Соединения, представленные формулой (I), включают соединения с заменой указанными изотопами. Соединения с заменой указанными изотопами применимы в качестве лекарственных средств и включают все меченые радиоактивными метками соединения, представленные формулой (I). «Способ мечения радиоактивными изотопами» при производстве «меченых радиоактивными изотопами соединений» охватывается настоящим изобретением, и «меченые радиоактивными изотопами соединения» применимы для исследований фармакокинетики метаболизируемого соединения, для анализа связывания и/или в качестве диагностических средств.
[0021]
Меченые радиоактивными изотопами соединения, представленные формулой (I), могут быть получены с использованием способов, хорошо известных в данной области техники. Например, меченое тритием соединение, представленное формулой (I), может быть получено путем введения трития в определенное соединение, представленное формулой (I), посредством каталитической реакции дегалогенирования с использованием трития. Данный способ включает осуществление взаимодействия галогенированного соответствующим образом предшественника соединения, представленного формулой (I), с газообразным тритием в присутствии соответствующего катализатора, такого как Pd/C, и в присутствии или в отсутствие основания. За другим подходящим способом получения меченого тритием соединения можно обратиться к "Isotopes in the Physical and Biomedical Sciences, Vol. 1, Labeled Compounds (Part A), Chapter 6 (1987)". 14C-меченое соединение может быть получено путем использования исходного вещества, содержащего 14C.
[0022]
Фармацевтически приемлемые соли соединений, представленных формулой (I), включают, например, соли соединений, представленных формулой (I), с щелочным металлом (например, литий, натрий, калий, и т.п.), щелочно-земельным металлом (например, кальций, барий, и т.п.), магнием, металлом переходной группы (например, цинк, железо, и т.п.), аммонием, органическими основаниями (например, триметиламин, триэтиламин, дициклогексиламин, этаноламин, диэтаноламин, триэтаноламин, меглумин, этилендиамин, пиридин, пиколин, хинолин, и т.п.) или аминокислотами, или соли с неорганическими кислотами (например, соляная кислота, серная кислота, азотная кислота, угольная кислота, бромистоводородная кислота, фосфорная кислота, йодистоводородная кислота, и т.п.) или органическими кислотами (например, муравьиная кислота, уксусная кислота, пропионовая кислота, трифторуксусная кислота, лимонная кислота, молочная кислота, винная кислота, щавелевая кислота, малеиновая кислота, фумаровая кислота, миндальная кислота, глутаровая кислота, яблочная кислота, бензойная кислота, фталевая кислота, аскорбиновая кислота, бензолсульфоновая кислота, пара-толуолсульфоновая кислота, метансульфоновая кислота, этансульфоновая кислота, и т.п.). В первую очередь, включены соли с соляной кислотой, серной кислотой, фосфорной кислотой, винной кислотой, метансульфоновой кислотой, и т.п. Указанные соли могут быть получены обычными способами.
[0023]
Соединения, представленные формулой (I), или их фармацевтически приемлемые соли согласно настоящему изобретению могут формировать сольваты (например, гидраты, и т.п.), сокристаллы и/или кристаллические полиморфы. Настоящее изобретение охватывает указанные различные сольваты, сокристаллы и кристаллические полиморфы. «Сольваты» могут представлять собой сольваты, в которых любое число молекул растворителя (например, молекул воды, и т.п.) находится в координационной связи с соединениями, представленными формулой (I). Если соединения, представленные формулой (I), или их фармацевтически приемлемые соли оставляют отстаиваться в атмосфере, то соединения могут поглощать воду, что приводит к присоединению поглощенной воды и формированию гидратов. Путем перекристаллизации соединений, представленных формулой (I), или их фармацевтически приемлемых солей можно получать кристаллические полиморфы. Термин «сокристалл» означает, что соединение, представленное формулой (I), или его соль и контрмолекула присутствуют в одной и той же кристаллической решетке, и может формироваться сокристалл с любым числом контрмолекул.
[0024]
Соединения, представленные формулой (I), или их фармацевтически приемлемые соли могут формировать пролекарства. Настоящее изобретение также охватывает такие различные пролекарства. Пролекарства являются производными соединений согласно настоящему изобретению, которые содержат химически или метаболически разлагаемые группы, и соединениями, которые преобразуются до фармацевтически активных соединений согласно настоящему изобретению посредством сольволиза или в физиологических условиях in vivo. Пролекарства включают соединения, которые преобразуются до соединений, представленных формулой (I), посредством ферментативного окисления, восстановления, гидролиза, и т.п. в физиологических условиях и in vivo, соединения, которые преобразуются до соединений, представленных формулой (I), посредством гидролиза желудочным соком, и т.д., и т.п. Способы выбора и получения подходящих пролекарственных производных описаны, например, в "Design of Prodrugs, Elsevier, Amsterdam, 1985". Пролекарства сами по себе могут обладать некоторой активностью.
[0025]
Если соединения, представленные формулой (I), или их фармацевтически приемлемые соли содержат гидроксигруппу(ы), то пролекарства включают ацилоксипроизводные и сульфонилоксипроизводные, которые получают, например, путем осуществления взаимодействия содержащих гидроксигруппу(ы) соединений с подходящим ацилгалогенидом, подходящим ангидридом кислоты, подходящим сульфонилхлоридом, подходящим сульфонилангидридом и смешанным ангидридом или с конденсирующим агентом. Примеры включают CH3COO-, C2H5COO-, трет-BuCOO-, C15H31COO-, PhCOO-, (m-NaOOCPh)COO-, NaOOCCH2CH2COO-, CH3CH(NH2)COO-, CH2N(CH3)2COO-, CH3SO3-, CH3CH2SO3-, CF3SO3-, CH2FSO3-, CF3CH2SO3-, пара-CH3O-PhSO3-, PhSO3- и пара-CH3PhSO3-.
[0026]
Способ получения соединения согласно настоящему изобретению
Например, соединения, представленные формулой (I), согласно настоящему изобретению могут быть получены в соответствии с общими методиками, описанными ниже. При проведении экстрагирования, очистки и других операций, может применяться любой процесс, обычно проводимый для этого в экспериментальной органической химии.
Соединение согласно настоящему изобретению может быть синтезировано в соответствии со способом, известным в данной области техники.
[0027]
Общая методика синтеза 1
Соединение согласно настоящему изобретению, которое представлено формулой (I) (a5 ниже), может быть получено, например, посредством следующего способа получения.

где X1 представляет собой хлор, бром, йод или трифторметансульфонат; X2 представляет собой хлор, бром или йод; и значение каждого из других символов описано выше.
Стадия 1
Соединение a3 может быть получено путем осуществления взаимодействия соединения a2 и основания с соединением a1.
В качестве примеров основания служат триэтиламин, диизопропилэтиламин, бикарбонат натрия, карбонат цезия и бикарбонат калия.
Температура реакции составляет от 0°C до 100°C.
Продолжительность реакции составляет от 1 часа до 10 часов.
В качестве примеров реакционного растворителя служат метанол, этанол и тетрагидрофуран.
Стадия 2
Соединение a4 может быть получено путем осуществления взаимодействия соединения a3 с галогенирующим агентом, таким как хлорид меди или бромид меди, кислотой и водным раствором нитрита натрия, и т.п.
В качестве примеров кислоты служат соляная кислота и уксусная кислота.
Температура реакции составляет от 0°C до 100°C.
Продолжительность реакции составляет от 1 часа до 10 часов.
Стадия 3
Соединение a5 может быть получено путем осуществления взаимодействия соединения a4 с бороновой кислотой, боронатным эфиром, триалкилстаннаном и т.п., в присутствии металлсодержащего катализатора и основания.
В качестве примеров металлсодержащего катализатора служат ацетат палладия, бис(дибензилиденацетон)палладий, тетракис(трифенилфосфин)палладий, дихлорид бис(трифенилфосфин)палладия (II), бис(три-трет-бутилфосфин)палладий и т.п., и может быть использовано от 0,001 до 0,5 мол. эквивалентов металлсодержащего катализатора относительно соединения a4.
В качестве примеров основания служат гидроксид лития, гидроксид натрия, гидроксид калия, трет-бутоксид калия, трет-бутоксид натрия, карбонат натрия, бикарбонат калия, бикарбонат натрия, фосфат натрия, гидрофосфат натрия, фосфат калия, гидрофосфат калия и т.п., и может быть использовано от 1 до 10 мол. эквивалентов основания относительно соединения a4.
Может быть использовано от 1 до 10 мол. эквивалентов бороновой кислоты, боронатного эфира, триалкилстаннана, и т.п., относительно соединения a4.
Температура реакции может составлять от 20°C до температуры возгонки растворителя, при необходимости, с использованием микроволнового облучения.
Продолжительность реакции составляет от 0,1 до 48 часов, предпочтительно от 0,5 до 12 часов.
В качестве примеров реакционного растворителя служат тетрагидрофуран, толуол, DMF, диоксан и вода, и может быть использован один из указанных растворителей или смесь из них.
Соединение a5, содержащее в R4a амин, имин или циан и т.п., может быть синтезировано путем осуществления взаимодействия соединения a4 с амином, имином, цианидом калия, и т.п.
Если R4a представляет собой карбониламино, сульфониламино и т.п., то синтез может проводиться путем добавления различных функциональных групп к соединению a3.
[0028]
Альтернативный способ
Соединение a3, описанное выше, может быть получено, например, посредством следующего способа получения.

где значение каждого из символов описано выше.
Стадия 1
Соединение a6 может быть получено путем осуществления взаимодействия гидразингидрата с соединением a1.
Температура реакции составляет от 0°C до 100°C.
Продолжительность реакции составляет от 1 часа до 20 часов.
В качестве примеров реакционного растворителя служат метанол, этанол и тетрагидрофуран.
Стадия 2
Соединение a3 может быть получено путем осуществления взаимодействия основания, галогенида и т.п., с соединением a6.
В качестве примеров основания служат бикарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия, триэтиламин, диизопропилэтиламин и гидрид натрия.
Температура реакции составляет от 0°C до 100°C.
Продолжительность реакции составляет от 1 часа до 10 часов.
В качестве примеров реакционного растворителя служат тетрагидрофуран, дихлорметан и диметилформамид.
[0029]
Соединения согласно настоящему изобретению обладают ингибирующей активностью в отношении MGAT2 и применимы в качестве профилактического средства и/или терапевтического средства, например, при ожирении, метаболическом синдроме, гиперлипидемии, гипертриглицеридемии, гипер-ЛПОНП-триглицеридемии, гиперлипоацидемии, сахарном диабете или артериосклерозе.
Соединения согласно настоящему изобретению обладают не только ингибирующей активностью в отношении MGAT2, но также применимы в качестве лекарственного средства, и обладают какими-либо отдельными или всеми из следующих превосходных характеристик:
a) наличием высокой метаболической стабильности,
b) проявлением высокой растворимости,
c) наличием сниженного риска фототоксичноси,
d) наличием сниженного риска гепатотоксичности,
e) наличием сниженного риска почечной токсичности,
f) наличием сниженного риска сердечно-сосудистой токсичности,
g) наличием сниженного риска желудочно-кишечных нарушений,
h) наличием сниженного риска взаимодействия лекарств,
i) наличием высокой пероральной всасываемости,
j) наличием низкого клиренса,
k) наличием высокого распределения в ткани-мишени,
l) наличием интенсивной ферментативной активности,
m) сниженной индукцией метаболизирующего лекарство фермента,
n) наличием выраженной эффективности,
o) наличие высокой селективности ингибирующей активности в отношении MGAT2 и
p) наличием высокой химической стабильности.
[0030]
Фармацевтическая композиция согласно настоящему изобретению может быть введена перорально или парентерально. Способы парентерального введения включают кожное, подкожное, внутривенное, интраартериальное, внутримышечное, интраперитонеальное, чресслизистое, ингаляционное, трансназальное, офтальмическое введение, введение во внутреннее ухо или вагинальное введение.
[0031]
В случае перорального введения, могут быть приготовлены в соответствии с обычным способом и введены любые обычно используемые формы, такие как пероральные твердые составы (например, таблетки, порошки, гранулы, капсулы, пилюли или пленки) и пероральные жидкие составы (например, суспензия, эмульсия, эликсир, сироп, лимонад, спиртовой раствор, ароматизированная вода, экстракт, отвар или настойка). Таблетки могут представлять собой таблетки с сахарным покрытием, таблетки с пленочным покрытием, таблетки с кишечнорастворимым покрытием, таблетки с замедленным высвобождением, таблетки-пастилки, подъязычные таблетки, буккальные таблетки, жевательные таблетки или таблетки, диспергируемые в полости рта. Порошки и гранулы могут находиться в виде сухого сиропа. Капсулы могут представлять собой мягкие капсулы, микрокапсулы или капсулы с замедленным высвобождением.
[0032]
В случае парентерального введения, могут быть предпочтительно введены любые обычно используемые формы, такие как инъекции, капельницы и препараты для наружного применения (например, глазные капли, капли в нос, ушные капли, аэрозоли, ингаляции, лосьон, инфузия, линимент, ополаскиватель для ротовой полости, клизма, мазь, гипс, желе, крем, пластырь, горячий компресс, порошок для наружного применения или суппозиторий). Инъекции могут представлять собой эмульсии типа «масло/вода», «вода/масло», «масло/вода/масло», «вода/масло/вода», и т.п.
[0033]
Фармацевтическая композиция может быть приготовлена путем смешивания эффективного количества соединения согласно настоящему изобретению с различными фармацевтическими добавками, подходящими для приготовления состава, такими как наполнители, связующие вещества, разрыхлители, смазки и разбавители. Кроме того, фармацевтическая композиция может быть адаптирована для педиатрических пациентов, гериатрических пациентов, серьезных случаев или оперативных вмешательств путем соответствующего изменения эффективного количества соединения согласно настоящему изобретению, состава и/или различных фармацевтических добавок. Педиатрические фармацевтические композиции предпочтительно вводят пациентам младше 12 или 15 лет. Кроме того, педиатрические фармацевтические композиции можно вводить пациентам в возрасте менее 27 суток от рождения, от 28 суток до 23 месяцев после рождения, от 2 до 11 лет, от 12 до 16 лет или 18 лет. Гериатрические фармацевтические композиции предпочтительно вводят пациентам в возрасте от 65 лет и старше.
[0034]
Несмотря на то, что дозировку фармацевтической композиции согласно настоящему изобретению следует определять с учетом возраста и массы тела пациента, типа и степени тяжести заболеваний, пути введения и т.п., обычная пероральная дозировка составляет от 0,05 до 100, и предпочтительно от 0,1 до 10 мг/кг в сутки. Для парентерального введения, хотя дозировка сильно варьирует в зависимости от путей введения, обычная дозировка составляет от 0,005 до 10 мг/кг в сутки, и предпочтительно от 0,01 до 1 мг/кг в сутки. Дозировка может быть введена однократно или несколькими порциями в течение суток.
[0035]
Доза для совместно вводимых лекарств может быть подходящим образом выбрана с учетом клинической дозы. Рецептурное соотношение соединений согласно настоящему изобретению и совместно вводимых лекарств может быть подходящим образом выбрано в зависимости от подлежащего лечению субъекта, пути введения, подлежащего лечению заболевания, симптомов, сочетания лекарств и т.п. Для введения людям, например, 1 часть по массе соединений согласно настоящему изобретению может быть использована в сочетании с 0,01-100 частями по массе совместно вводимых лекарств.
[0036]
Фармацевтическая композиция согласно настоящему изобретению также эффективна при ожирении (однако, только в том случае, если имеется и диабет 2 типа, и дислипидемия, и BMI составляет 25 кг/м2 или более, даже при наличии диетотерапии/терапии физической нагрузкой).
[0037]
Фармацевтическая композиция согласно настоящему изобретению также эффективна при тяжелом ожирении, для которого эффект предварительно применявшейся диетотерапии и терапии физической нагрузкой является недостаточен.
[0038]
Фармацевтическая композиция согласно настоящему изобретению может быть использована в сочетании с другим(и) средством(ами) против ожирения (фармацевтическая композиция, содержащая соединения, обладающие эффектом против ожирения, или медицинское средство от ожирения или для регулирования веса при ожирении). Например, для профилактики и/или лечения ожирения или для регулирования веса при ожирении может быть использовано сочетанное лечение фармацевтической композицией, содержащей соединение, обладающее эффектом против ожирения, и соединением согласно настоящему изобретению. Сочетанное лечение фармацевтической композицией, содержащей соединение согласно настоящему изобретению и фармацевтической(ими) композицией(ями), содержащей(ими) соединение, обладающее эффектом против ожирения, может быть использовано для профилактики и/или лечения ожирения или для регулирования веса при ожирении. Кроме того, способ лечения путем введения фармацевтической композиции согласно настоящему изобретению может быть использован в сочетании с диетотерапией, лекарственной терапией, физической нагрузкой и т.п.
[0039]
В настоящем описании, значения каждого сокращения являются следующими:
DIEA: N,N-диизопропилэтиламин
DMA: диметилацетамид
DMF: N,N-диметилформамид
DMSO: диметилсульфоксид
HATU: O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат
ПРИМЕРЫ
[0040]
Настоящее изобретение будет более подробно описано со ссылкой, но без ограничения, на следующие Примеры и Тестовые примеры.
[0041]
Пример 1

Стадия 1
Известное соединение 1 (WO2017069224A1) (54,7 мг, 0,142 ммоль) растворяли в тетрагидрофуране (1,1 мл), при охлаждении на льду добавляли фенилгидразин (28 мкл, 0,28 ммоль), и перемешивали смесь в течение 20 минут. К реакционному раствору при охлаждении на льду добавляли DIEA (50 мкл, 0,28 ммоль), смесь перемешивали при комнатной температуре в течение 3 часов, и дополнительно перемешивали при 70°C в течение 1 часа. Растворитель реакционного раствора выпаривали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии (хлороформ-метанол) с получением соединения 2 (59,4 мг, выход 92%).
1H-ЯМР (CDCl3) δ: 2,01-2,10 (2H, м), 2,17 (1H, дт, J=12,8, 8,3 Гц), 2,24-2,39 (2H, м), 2,49 (1H, ддд, J=12,8, 7,4, 3,8 Гц), 2,83-3,01 (2H, м), 2,93 (1H, д, J=15,9 Гц), 3,06 (1H, д, J=15,9 Гц), 4,02 (2H, т, J=6,0 Гц), 5,12 (2H, с), 5,37 (1H, с), 6,78 (1H, с), 6,79 (1H, д, J=7,5 Гц), 7,29 (1H, д, J=7,5 Гц), 7,39 (1H, т, J=7,3 Гц), 7,51 (2H, т, J=7,8 Гц), 7,57 (2H, д, J=7,5 Гц).
Стадия 2
Соединение 2 (44,9 мг, 98,4 мкмоль), 2-метансульфонилуксусную кислоту (54 мг, 0,40 ммоль) и N,N-дициклогексилкарбодиимид (82 мг, 0,40 ммоль) суспендировали в дихлорметане (2 мл) и перемешивали при комнатной температуре в течение 25 часов. к реакционному раствору добавляли N,N-диметиламино-4-пиридин (2,4 мг, 20 мкмоль), и дополнительно перемешивали смесь при комнатной температуре в течение 4 часов. К реакционному раствору добавляли 2-метансульфонилуксусную кислоту (27 мг, 0,20 ммоль) и N,N-дициклогексилкарбодиимид (41 мг, 0,20 ммоль), и дополнительно перемешивали смесь при комнатной температуре в течение 24 часов. Реакционный раствор фильтровали, и выпаривали растворитель фильтрата в условиях пониженного давления. Полученный остаток очищали методом хроматографии на силикагеле (хлороформ-метанол). Затем, полученное твердое вещество промывали диизопропиловым эфиром с получением соединения I-34 (36,3 мг, выход 64%).
1H-ЯМР (CDCl3) δ: 1,95-2,06 (2H, м), 2,19-2,32 (3H, м), 2,44-2,54 (1H, м), 2,90 (3H, с), 2,91-3,04 (2H, м), 3,09 (1H, д, J=15,8 Гц), 3,18 (1H, д, J=15,8 Гц), 3,88-3,97 (2H, м), 4,02 (1H, д, J=14,3 Гц), 4,22 (1H, д, J=14,3 Гц), 6,12 (1H, с), 6,66-6,72 (1H, м), 6,76-6,79 (1H, м), 7,18 (1H, д, J=8,3 Гц), 7,35-7,52 (5H, м), 9,66 (1H, с).
[0042]
Пример 2

Стадия 1
Соединение 1 (1,0 г, 2,60 ммоль) растворяли в этаноле (15 мл), и добавляли гидразина моногидрат (0,65 мл, 13,0 ммоль). Смесь перемешивали при температуре от комнатной до 80°C в течение 18 часов. При комнатной температуре, к реакционному раствору добавляли водный бикарбонат натрия, а затем экстрагировали хлороформом, и сушили продукт над сульфатом натрия. Растворитель выпаривали в условиях пониженного давления, и сушили путем суспендирования (этилацетат/гексан) с получением соединения 3 (890 мг, выход 90%).
1H-ЯМР (CDCl3) δ: 2,01-2,18 (м, 3H), 2,25-2,42 (м, 3H), 2,81-3,02 (м, 4H), 4,00 (т, J=5,9 Гц, 2H), 5,49 (с, 1H), 6,75-6,77 (м, 2H), 7,22 (д, J=9,0 Гц, 1H).
Стадия 2
Соединение 3 (100 мг, 0,24 ммоль) растворяли в дихлорметане (1,0 мл), добавляли 1-бром-2-метоксиэтан (66,7 мг, 0,48 ммоль) и карбонат цезия (313 мг, 0,96 ммоль), и перемешивали 90°C в течение 2 часов. При комнатной температуре, к реакционному раствору добавляли воду, а затем экстрагировали этилацетатом, и сушили продукт над сульфатом натрия. Растворитель выпаривали в условиях пониженного давления с получением соединения 4 в виде неочищенного продукта.
Стадия 3
Неочищенное соединение 4 (0,24 ммоль) растворяли в толуоле (1 мл), при комнатной температуре добавляли 2-метансульфонилуксусную кислоту (66,2 мг, 0,48 ммоль) и N,N′-дициклогексилкарбодиимид (99,0 мг, 0,48 ммоль), и перемешивали в течение 1 часа. При комнатной температуре, к реакционному раствору добавляли воду, а затем экстрагировали этилацетатом, и сушили продукт над сульфатом натрия. Растворитель выпаривали в условиях пониженного давления, и очищали полученный остаток методами колоночной хроматографии на силикагеле (гексан-этилацетат) и обращенно-фазовой хроматографии с получением соединения I-55 (30,0 мг, выход в две стадии 22%).
1H-ЯМР (CDCl3) δ: 2,01-2,20 (м, 3H), 2,25-2,44 (м, 3H), 2,85-2,97 (м, 3H), 3,09 (д, J=15,8 Гц, 1H), 3,30 (ушир. с, 3H), 3,39 (с, 3H), 3,72 (т, J=5,0 Гц, 2H), 3,99 (т, J=5,9 Гц, 2H), 4,13 (ушир. с, 2H), 4,28 (ушир. с, 2H), 5,68 (с, 1H), 6,77 (ушир. с, 2H), 7,20-7,22 (м, 1H).
[0043]
Пример 3

Стадия 1
Соединение 3 (719 мг, 1,89 ммоль) растворяли в DMF (7 мл), и добавляли 2-циклопропилэтилметансульфонат (466 мг, 2,84 ммоль) и карбонат калия (1,31 г, 9,45 ммоль). Смесь перемешивали при 80°C в течение 5 часов. При комнатной температуре, к реакционному раствору добавляли воду, а затем экстрагировали хлороформом, и сушили продукт над сульфатом натрия. Растворитель выпаривали в условиях пониженного давления, и очищали полученный остаток методом колоночной хроматографии (этилацетат) с получением соединения 5 (358 мг, выход 42%).
1H-ЯМР (CDCl3) δ: 0,06-0,10 (м, 2H), 0,46-0,51 (м, 2H), 0,63-0,72 (м, 1H), 1,69-1,75 (м, 2H), 2,03-2,15 (дт, J=30,7, 9,3 Гц, 3H), 2,28-2,43 (м, 3H), 2,82-3,00 (м, 4H), 3,93-4,02 (м, 4H), 4,78 (ушир. с, 2H), 5,23 (с, 1H), 6,76-9,78 (м, 2H), 7,23-7,25 (м, 1H).
Стадия 2
Соединение 5 (2,30 г, 5,13 ммоль) и дихлорид меди (1,38 г, 10,3 ммоль) растворяли в уксусной кислоте (7 мл) и концентрированной соляной кислоте (11 мл), при охлаждении на льду по каплям добавляли водный раствор (1 мл) нитрита натрия (460 мг, 6,67 ммоль), и перемешивали в течение 45 минут при охлаждении на льду. Реакционный раствор экстрагировали хлороформом, органический слой промывали насыщенным водным раствором хлорида аммония и 2M водным раствором карбоната натрия, а затем сушили над сульфатом натрия. Растворитель выпаривали в условиях пониженного давления, и очищали полученный остаток методом колоночной хроматографии (хлороформ-этилацетат) с получением соединения 6 (1,24 г, выход 52%).
1H-ЯМР (CDCl3) δ: -0,04-0,05 (2H, м), 0,40-0,51 (2H, м), 0,61-0,72 (1H, м), 1,69-1,82 (2H, м), 2,01-2,10 (2H, м), 2,14 (1H, дт, J=13,1, 8,3 Гц), 2,25-2,42 (3H, м), 2,82-3,00 (2H, м), 2,94 (1H, д, J=15,8 Гц), 3,08 (1H, д, J=15,8 Гц), 4,01 (2H, т, J=5,9 Гц), 4,24 (2H, т, J=7,0 Гц), 5,59 (1H, с), 6,76-6,80 (2H, м), 7,22 (1H, д, J=9,0 Гц).
Стадия 3
Соединение 6 (1,57 г, 3,36 ммоль) растворяли в DMA (16 мл), добавляли цианид калия (678 мг, 10,4 ммоль), и перемешивали смесь при 200°C в течение 3 часов в условиях обработки микроволновым излучением. После того, как полученный реакционный раствор оставляли охладиться до комнатной температуры, к нему добавляли насыщенный водный раствор бикарбоната натрия и воду, а затем экстрагировали водный слой смесью гексан-этилацетат (2:1). Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над сульфатом натрия. Растворитель выпаривали в условиях пониженного давления с получением неочищенного соединения 7 (1,46 г).
Стадия 4
Неочищенное соединение 7 (212 мг), азид натрия (150 мг, 2,31 ммоль) и хлорид аммония (124 мг, 2,31 ммоль) суспендировали в DMF (4,2 мл) и воде (0,4 мл) и перемешивали при 140°C в течение 3 часов. После того, как полученный реакционный раствор оставляли охладиться до комнатной температуры, к нему добавляли водный раствор лимонной кислоты, а затем экстрагировали водный слой этилацетатом. Органический слой сушили над сульфатом натрия, и выпаривали растворитель в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (хлороформ-метанол) с получением соединения I-24 (162 мг, выход в две стадии 66%).
1H-ЯМР (CDCl3) δ: 0,01-0,06 (2H, м), 0,36-0,42 (2H, м), 0,73-0,82 (1H, м), 1,88 (2H, кв, J=7,1 Гц), 2,02-2,11 (2H, м), 2,18 (1H, с), 2,20 (1H, дт, J=13,3, 8,2 Гц), 2,26-2,39 (2H, м), 2,45 (1H, ддд, J=13,0, 7,7, 4,5 Гц), 2,88-3,06 (2H, м), 3,12 (1H, д, J=15,9 Гц), 3,22 (1H, д, J=15,9 Гц), 4,03 (2H, т, J=6,0 Гц), 4,99-5,13 (2H, м), 5,97 (1H, с), 6,79-6,83 (2H, м), 7,24 (1H, д, J=9,3 Гц).
[0044]
Пример 4
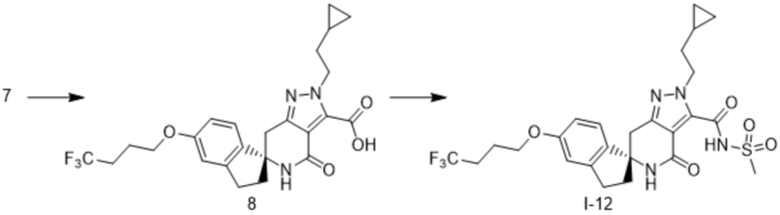
Стадия 1
Соединение 7 (1,46 г, 3,18 ммоль) растворяли в тетрагидрофуране (6 мл) и метаноле (6 мл), и добавляли 5н водный раствор гидроксида натрия (6,4 мл,3,2 ммоль). Смесь перемешивали при 120°C в течение 75 минут в условиях обработки микроволновым излучением. После того, как полученный реакционный раствор оставляли охладиться до комнатной температуры, добавляли 2н соляную кислоту (30 мл), а затем экстрагировали водный слой хлороформом, и сушили органический слой над сульфатом натрия. Растворитель выпаривали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (хлороформ-метанол) с получением соединения 2 (1,12 г, выход 73%).
1H-ЯМР (CDCl3) δ: -0,02-0,02 (2H, м), 0,39-0,45 (2H, м), 0,69-0,77 (1H, м), 1,78 (2H, кв, J=7,2 Гц), 2,02-2,11 (2H, м), 2,21 (1H, дт, J=13,2, 8,2 Гц), 2,26-2,39 (2H, м), 2,45 (1H, ддд, J=13,0, 7,7, 4,5 Гц), 2,93 (1H, дт, J=16,2, 7,7 Гц), 3,01 (1H, ддд, J=16,5, 8,6, 4,3 Гц), 3,09 (1H, д, J=16,2 Гц), 3,19 (1H, д, J=16,1 Гц), 4,03 (2H, т, J=6,0 Гц), 4,76-4,89 (2H, м), 6,01 (1H, с), 6,79-6,84 (2H, м), 7,19-7,23 (1H, м), 15,66 (1H, с).
Стадия 2
Соединение 2 (486 мг, 1,02 ммоль) и карбонилдиимидазол (495 мг, 3,05 ммоль) растворяли в тетрагидрофуране (5 мл) и перемешивали при 70°C в течение 100 минут. После того, как полученный реакционный раствор оставляли охладиться до комнатной температуры, добавляли амид метансульфоновой кислоты (484 мг, 5,09 ммоль) и диазабициклоундецен (0,77 мл, 5,1 ммоль), и перемешивали смесь при комнатной температуре в течение 14 часов. После того, как растворитель выпаривали в условиях пониженного давления, к остатку добавляли 2н соляную кислоту (20 мл), а затем экстрагировали водный слой хлороформом, и сушили органический слой над сульфатом натрия. Растворитель выпаривали в условиях пониженного давления, и очищали полученный остаток путем отверждения (диизопропиловый эфир-хлороформ) с получением соединения I-12 (281 мг, выход 50%).
1H-ЯМР (CDCl3) δ: 0,00-0,04 (2H, м), 0,41-0,47 (2H, м), 0,67-0,76 (1H, м), 1,78(2H, кв.д, J=7,2, 1,4 Гц), 2,02-2,11 (2H, м), 2,18 (1H, дт, J=13,2, 8,2 Гц), 2,26-2,36 (2H, м), 2,40 (1H, ддд, J=13,1, 7,7, 4,4 Гц), 2,91 (1H, дт, J=16,4, 8,1 Гц), 2,99 (1H, ддд, J=16,5, 8,6, 4,5 Гц), 3,07 (1H, д, J=15,8 Гц), 3,18 (1H, д, J=15,8 Гц), 3,38 (3H, с), 4,02 (2H, т, J=6,0 Гц), 4,75-4,87 (2H, м), 5,95 (1H, с), 6,79-6,83 (2H, м), 7,20-7,23 (1H, м), 14,99 (1H, с).
[0045]
Пример 5

Стадия 1
Соединение 7 (23,1 мг, 50,4 мкмоль) и 5% палладированный уголь (23 мг) суспендировали в метаноле (1,5 мл) и концентрированной соляной кислоте (0,2 мл), и перемешивали при комнатной температуре в течение 4 часов в атмосфере водорода. После фильтрования реакционного раствора, растворитель выпаривали в условиях пониженного давления с получением соединения 9 (24,9 мг, выход 99%).
1H-ЯМР (DMSO-d6) δ: -0,08-0,04 (2H, м), 0,34-0,41 (2H, м), 0,58-0,67 (1H, м), 1,59-1,67 (2H, м), 1,91 (2H, дт, J=15,6, 6,4 Гц), 2,14-2,28 (2H, м), 2,36-2,45 (2H, м), 2,90 (2H, т, J=6,7 Гц), 2,94 (1H, д, J=15,8 Гц), 3,01 (1H, д, J=15,8 Гц), 4,01 (2H, т, J=6,1 Гц), 4,22 (2H, т, J=6,7 Гц), 4,38 (1H, д, J=15,2 Гц), 4,43 (1H, д, J=15,2 Гц), 6,68 (1H, дд, J=8,4, 2,3 Гц), 6,85 (1H, д, J=2,3 Гц), 7,01 (1H, д, J=8,4 Гц), 8,42 (1H, с), 8,73 (3H, с).
Стадия 2
Соединение 9 (6,0 мг, 12 мкмоль) суспендировали в тетрагидрофуране (0,7 мл), добавляли уксусный ангидрид (6 мкл, 0,06 ммоль) и триэтиламин (17 мкл, 0,12 ммоль), и перемешивали смесь при комнатной температуре в течение 20 минут. После добавления метанола (2 мл), растворитель выпаривали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (хлороформ-метанол) с получением соединения I-10 (4,6 мг, выход 76%).
1H-ЯМР (CDCl3) δ: -0,05-0,05 (2H, м), 0,39-0,48 (2H, м), 0,60-0,72 (1H, м), 1,75 (2H, кв, J=7,0 Гц), 2,02 (3H, с), 2,03-2,10 (2H, м), 2,14 (1H, дт, J=13,1, 8,0 Гц), 2,25-2,36 (2H, м), 2,40 (1H, ддд, J=12,9, 7,7, 4,2 Гц), 2,83-2,97 (2H, м), 2,96 (1H, д, J=15,8 Гц), 3,07 (1H, д, J=15,8 Гц), 4,02 (2H, т, J=6,0 Гц), 4,27 (2H, т, J=7,2 Гц), 4,55 (1H, дд, J=16,1, 4,8 Гц), 4,76 (1H, дд, J=16,1, 6,0 Гц), 5,73 (1H, с), 6,76-6,81 (2H, м), 7,19-7,24 (1H, м), 8,65-8,72 (1H, м).
[0046]
Пример 11
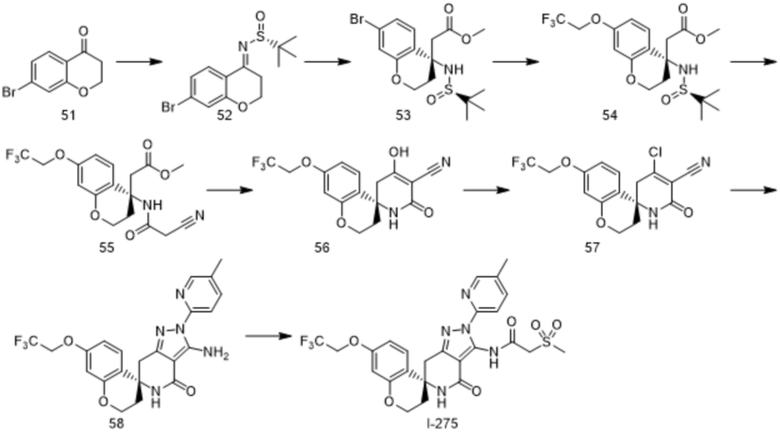
Стадия 1
Раствор тетраэтоксититана (141 мл, 666 ммоль), раствора соединения 51 (100 г, 444 ммоль) в тетрагидрофуране (250 мл) и толуола (250 мл) добавляли к раствору (R)-2-метилпропан-2-сульфинамида (134 г, 1,11 моль) в толуоле (500 мл), и перемешивали смесь при 90°C в течение 12 часов. После добавления к реакционному раствору при комнатной температуре насыщенного раствора хлорида аммония, экстрагировали этилацетатом. Органический слой промывали насыщенным раствором хлорида аммония, а затем сушили над безводным сульфатом натрия. Растворитель выпаривали в условиях пониженного давления, и очищали полученный остаток методом колоночной хроматографии на силикагеле (гексан-этилацетат) с получением соединения 52 (82 г, выход 56%).
1H-ЯМР (CDCl3) δ: 1,32 (с, 9H), 3,24-3,31 (м, 1H), 3,45-3,53 (м, 1H), 4,27-4,41 (м, 2H), 7,09-7,12 (м, 2H), 7,83 (д, 1H, J=8,31 Гц).
Стадия 2
К 1М раствору гексаметилдисилазида лития в тетрагидрофуране (334 мл, 333 ммоль), при 78°C добавляли метилацетат (26,5 мл, 333 ммоль), и перемешивали смесь в течение 1 часа. К реакционному раствору при 78°C по каплям добавляли раствор триизопропоксида хлорида титана (110 г, 424 ммоль) в тетрагидрофуране (250 мл), и перемешивали смесь в течение 2 часов. К реакционному раствору при 78°C по каплям добавляли раствор соединения 52 (50,0 г, 151 ммоль) в тетрагидрофуране (250 мл), и перемешивали смесь в течение 30 минут. Добавляли насыщенный раствор хлорида аммония и сегнетову соль, смесь нагревали до комнатной температуры и перемешивали в течение 12 часов. После этого, проводили экстрагирование этилацетатом, органический слой промывали водой, а затем сушили над безводным сульфатом натрия. Растворитель выпаривали в условиях пониженного давления, и очищали полученный остаток методом колоночной хроматографии на силикагеле (гексан-этилацетат) с получением соединения 53 (35 г, выход 57%).
1H-ЯМР (400 МГц, DMSO-d6) δ: 1,09 (с, 9H), 2,26-2,36 (м, 2H), 3,04 (кв, 2H, J=5,6 Гц), 3,53 (с, 3H), 4,22-4,26 (м, 1H), 4,34-4,37 (м, 1H), 5,45 (с, 1H), 7,01-7,07 (м, 2H), 7,28 (д, 1H, J=8,56 Гц).
Стадия 3
К раствору соединения 53 (1,4 г, 3,46 ммоль) в диоксане (15 мл) добавляли бис(пинаколато)дибор (1,32 г, 5,19 ммоль), комплекс дихлорида [1,1'-бис(дифенилфосфино)ферроцен]-палладия (II) и дихлорметана (1:1) (0,141 г, 0,17 ммоль) и ацетат калия (1,02 г, 10,4 ммоль), и перемешивали смесь при 100°C в течение 4 часов. К реакционному раствору при комнатной температуре добавляли 30% водный пероксид водорода (1,96 мл, 17,3 ммоль), и перемешивали смесь в течение 2 часов. К реакционному раствору добавляли воду, а затем экстрагировали этилацетатом, органический слой промывали 10% водным раствором тиосульфата натрия, а затем сушили над безводным сульфатом натрия, и выпаривали растворитель в условиях пониженного давления. Полученный остаток растворяли в DMF (10 мл), и добавляли карбонат цезия (1,69 г, 5,19 ммоль) и 2,2,2-трифторэтиловый эфир трифторметансульфоновой кислоты (597 мл, 4,16 ммоль). Смесь перемешивали при комнатной температуре в течение 12 часов. К реакционному раствору добавляли 10% водный раствор лимонной кислоты, а затем экстрагировали этилацетатом. После промывки насыщенным водным бикарбонатом натрия и насыщенным водным раствором хлорида натрия, проводили сушку над безводным сульфатом натрия, и выпаривали растворитель в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (гексан-этилацетат) с получением соединения 54 (1,23 г, выход 84%).
1H-ЯМР (CDCl3) δ: 1,22 (с, 9H), 2,21-2,28 (м, 1H), 2,52-2,57 (м, 1H), 2,77 (д, J=15,7 Гц, 1H), 2,96 (д, J=15,7 Гц, 1H), 3,71 (с, 3H), 4,19-4,25 (м, 1H), 4,31 (кв, J=8,1 Гц, 2H), 4,45-4,51 (м, 1H), 5,13 (с, 1H), 6,40 (д, J=2,6 Гц, 1H), 6,56 (дд, J=8,8, 2,6 Гц, 1H), 7,17 (д, J=8,8 Гц, 1H).
Стадия 4
К раствору соединения 54 (545 мг, 1,29 ммоль) в метаноле (1,1 мл) добавляли 4М раствор хлороводорода в диоксане (0,64 мл, 2,57 ммоль), и перемешивали смесь при комнатной температуре в течение 1 часа. После выпаривания растворителя из реакционного раствора в условиях пониженного давления, полученный остаток растворяли в тетрагидрофуране (4,5 мл), и добавляли 2-цианоуксусную кислоту (219 мг, 2,57 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид (493 мг, 2,57 ммоль) и триэтиламин (0,71 мл, 5,15 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов. К реакционному раствору добавляли воду, а затем экстрагировали этилацетатом. После промывки насыщенным водным бикарбонатом натрия и насыщенным водным раствором хлорида натрия, проводили сушку над безводным сульфатом натрия, и выпаривали растворитель в условиях пониженного давления с получением соединения 55 (483 мг, выход 97%).
1H-ЯМР (CDCl3) δ: 2,14-2,20 (м, 1H), 2,81-2,88 (м, 1H), 3,02 (д, J=15,1 Гц, 1H), 3,17 (д, J=15,1 Гц, 1H), 3,33 (дд, J=20,2, 18,9 Гц, 2H), 3,70 (с, 3H), 4,19-4,32 (м, 4H), 6,40 (д, J=2,8 Гц, 1H), 6,54 (дд, J=8,8, 2,8 Гц, 1H), 7,15(с, 1H), 7,20 (д, J=8,8 Гц, 1H).
Стадия 5
К раствору соединения 55 (497 мг, 1,29 ммоль) в метаноле (2,5 мл) добавляли 28% раствор метоксида натрия в метаноле (993 мг, 5,15 ммоль), и перемешивали смесь при комнатной температуре в течение 1 часа. К реакционному раствору добавляли 2М водный раствор соляной кислоты (6 мл) и воду, а затем экстрагировали этилацетатом. Органический слой промывали водой, а затем сушили над безводным сульфатом магния. После концентрирования, полученный остаток растворяли в этилацетате и обратно экстрагировали насыщенным водным бикарбонатом натрия. После этого, к водному слою при охлаждении на льду добавляли концентрированную соляную кислоту, и собирали выпавшее в осадок твердое вещество путем фильтрования с получением соединения 56 (340 мг, 75%).
1H-ЯМР (DMSO-d6) δ: 1,99-2,15 (м, 2H), 2,67 (д, J=17,3 Гц, 1H), 3,21 (д, J=17,3 Гц, 1H), 4,09-4,15 (м, 1H), 4,20-4,25 (м, 1H), 4,72 (кв, J=8,9 Гц, 2H), 6,49 (д, J=2,6 Гц, 1H), 6,64 (дд, J=8,8, 2,6 Гц, 1H), 7,41 (д, J=8,8 Гц, 1H), 7,89 (с, 1H).
Стадия 6
Раствор фосфорилхлорида (0,152 мл, 1,69 ммоль) и DMF (0,136 мл, 1,70 ммоль) в дихлорэтане (5 мл) добавляли к раствору соединения 56 (500 мг, 1,41 ммоль) в дихлорэтане (5 мл), и перемешивали смесь при комнатной температуре в течение 75 часов. К реакционному раствору добавляли воду, а затем экстрагировали этилацетатом. После промывки насыщенным водным бикарбонатом натрия и насыщенным водным раствором хлорида натрия, проводили сушку над безводным сульфатом магния, и выпаривали растворитель в условиях пониженного давления с получением соединения 57 (486 мг, выход 92%).
1H-ЯМР (CDCl3) δ: 2,24-2,27 (м, 2H), 3,00 (д, J=18,8 Гц, 1H), 3,43 (д, J=18,8 Гц, 1H), 4,17-4,35 (м, 4H), 5,92 (с, 1H), 6,42 (д, J=2,5 Гц, 1H), 6,63 (дд, J=8,8, 2,5 Гц, 1H), 7,33 (д, J=8,8 Гц, 1H).
Стадия 7
К раствору соединения 57 (100 мг, 0,27 ммоль) в этаноле (2 мл) добавляли 2-гидразинил-5-метилпиридина гидрохлорид (51,4 мг, 0,32 ммоль) и бикарбонат натрия (56,3 мг, 0,67 ммоль), и перемешивали смесь при 70°C в течение 7 часов. Затем, добавляли воду, а затем экстрагировали хлороформом. После промывки насыщенным водным раствором хлорида натрия, проводили сушку над безводным сульфатом магния, и выпаривали растворитель в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (хлороформ-метанол) с получением соединения 58 (77,6 мг, выход 63%).
1H-ЯМР (CDCl3) δ: 2,15-2,26 (м, 2H), 2,36 (с, 3H), 3,01 (д, J=16,2 Гц, 1H), 3,27 (д, J=16,2 Гц, 1H), 4,21-4,24 (м, 2H), 4,32 (кв, J=8,1 Гц, 2H), 5,20 (с, 1H), 6,42 (д, J=2,6 Гц, 1H), 6,61 (дд, J=8,8, 2,6 Гц, 1H), 7,18 (с, 2H), 7,50 (д, J=8,8 Гц, 1H), 7,63 (дд, J=8,6, 2,2 Гц, 1H), 7,77 (д, J=8,4 Гц, 1H), 8,19 (с, 1H).
Стадия 8
К раствору соединения 58 (430 мг, 0,94 ммоль) в дихлорметане (5 мл), при 0°C добавляли раствор 2-(метансульфонил)ацетилхлорида (3,74 ммоль) и пиридина (0,303 мл, 3,74 ммоль) в дихлорметане (3 мл), и перемешивали смесь в течение 1,5 часов. При комнатной температуре, добавляли воду, а затем экстрагировали этилацетатом, и продукт сушили над сульфатом магния. Растворитель выпаривали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (хлороформ-метанол, амино-силикагель), и измельчали в порошок со смесью гексан-этилацетат с получением соединения I-275 (320 мг, 59%).
1H-ЯМР (CDCl3) δ: 2,19-2,31 (м, 2H), 2,40 (с, 3H), 3,11 (д, J=16,3 Гц, 1H), 3,22 (с, 3H), 3,41 (д, J=16,3 Гц, 1H), 4,14 (с, 2H), 4,23-4,25 (м, 2H), 4,33 (кв, J=8,0 Гц, 2H), 5,59 (с, 1H), 6,44 (д, J=2,6 Гц, 1H), 6,62 (дд, J=8,8, 2,6 Гц, 1H), 7,49 (д, J=8,8 Гц, 1H), 7,69-7,72 (м, 1H), 7,81 (д, J=8,3 Гц, 1H), 8,30 (ушир. с, 1H), 11,19 (с, 1H).
[0047]
Пример 12

Стадия 1
К соединению 51 (1,75 г, 7,69 ммоль) добавляли (S)-2-метилпропан-2-сульфинамид (1,03 г, 8,45 ммоль) и тетраэтоксититан (3,22 мл, 6 ммоль), и перемешивали смесь при 100°C в течение 2,5 часов. Реакционный раствор разбавляли этилацетатом и водой, а затем экстрагировали этилацетатом. После промывки органического слоя насыщенным водным раствором хлорида натрия, растворитель выпаривали в условиях пониженного давления, и очищали полученный остаток методом колоночной хроматографии на силикагеле (гексан-этилацетат) с получением соединения 59 (1,86 г, выход 73%).
1H-ЯМР (CDCl3) δ: 1,32 (с, 9H), 3,24-3,31 (м, 2H), 3,46-3,53 (м, 2H), 4,28-4,41 (м, 2H), 7,09-7,13 (м, 2H), 7,84 (д, J=8,4 Гц, 1H).
Стадия 2
К 1М раствору гексаметилдисилазида лития в тетрагидрофуране (9,01 мл, 9,01 ммоль), при 78°C добавляли тетрагидрофуран (15 мл) и метилацетат (0,72 мл, 9,01 ммоль), и перемешивали в течение 30 минут. К реакционному раствору при 78°C по каплям добавляли 1М раствор триизопропоксида хлорида титана (11,3 мл, 11,3 ммоль) в гексане, и перемешивали смесь в течение 20 минут. После этого, при 78°C по каплям добавляли раствор соединения 59 (1,86 г, 5,63 ммоль) в тетрагидрофуране (16 мл), и перемешивали смесь в течение 1 часа. Добавляли насыщенный водный раствор хлорида натрия, а затем экстрагировали этилацетатом, и после промывки органического слоя водой выпаривали растворитель в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (гексан-этилацетат) с получением соединения 60 (2,07 г, выход 77%).
1H-ЯМР (CDCl3) δ: 1,22 (с, 9H), 2,22-2,29 (м, 1H), 2,56-2,61 (м, 2H), 2,78 (д, J=15,8 Гц, 1H), 2,93 (д, J=15,8 Гц, 1H), 3,71 (с, 3H), 4,20-4,25 (м, 1H), 4,43-4,49 (м, 1H), 5,17 (с, 1H), 7,02-7,05 (м, 2H), 7,11 (д, J=8,3 Гц, 1H).
Стадия 3
Бис(пинаколато)дибор (341 мг, 1,34 ммоль), ацетат калия (359 мг, 3,66 ммоль) и аддукт дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия (II) и дихлорметана (19,9 мг, 0,024 ммоль) добавляли к раствору соединения 60 (512 мг, 1,22 ммоль) в 1,4-диоксане (5 мл), и после трех повторений снижения давления/замещения атмосферы азотом, продолжали нагревание с обратным холодильником в течение 3 часов. После охлаждения до комнатной температуры, остаток, полученный путем фильтрования нерастворимого вещества через Celite и выпаривания растворителя в условиях пониженного давления, разбавляли тетрагидрофураном (5 мл), и добавляли 30% водный раствор пероксида водорода (277 мг, 2,44 ммоль) и 1М водный раствор гидроксида натрия (0,244 мл, 0,244 ммоль). Смесь перемешивали при комнатной температуре в течение 19,5 часов. Реакционный раствор обрабатывали пентагидратом тиосульфата натрия (606 мг, 2,44 ммоль), а затем экстрагировали этилацетатом. После промывки органического слоя водой, растворитель выпаривали в условиях пониженного давления, и измельчали полученный остаток в порошок со смесью гексан-этилацетат с получением соединения 61 (391 мг, выход 94%).
1H-ЯМР (CDCl3) δ: 1,23 (с, 9H), 2,21-2,28 (м, 1H), 2,45-2,50 (м, 1H), 2,78 (д, J=15,9 Гц, 1H), 2,99 (д, J=15,9 Гц, 1H), 3,71 (с, 3H), 4,17-4,22 (м, 1H), 4,43-4,50 (м, 1H), 5,10 (с, 1H), 6,23 (с, 1H), 6,34 (с, 1H), 6,40 (д, J=8,3 Гц, 1H), 7,05 (д, J=8,6 Гц, 1H).
Стадия 4
К раствору соединения 61 (380 мг, 1,11 ммоль) в DMF (1,5 мл) добавляли 2,2,2-трифторэтилтрифторметансульфонат (310 мг, 1,34 ммоль) и карбонат калия (185 мг, 1,34 ммоль), и после перемешивания смеси при комнатной температуре в течение 14 часов, смесь нагревали до 60°C и дополнительно перемешивали в течение 2 часов. Реакционный раствор разбавляли этилацетатом и водой, и после промывки органического слоя водой выпаривали растворитель в условиях пониженного давления. К раствору полученного остатка в метаноле (2 мл) добавляли 4М раствор хлороводорода в диоксане (0,557 мл, 2,23 ммоль), и перемешивали смесь при комнатной температуре в течение 1 часа. Растворитель реакционного раствора выпаривали в условиях пониженного давления, и дополнительно добавляли толуол для выпаривания растворителя в условиях пониженного давления. К раствору полученного остатка в дихлорметане (4 мл), добавляли 2-цианоуксусную кислоту (161 мг, 1,89 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (363 мг, 1,89 ммоль) и триэтиламин (0,463 мл, 3,34 ммоль), и перемешивали при комнатной температуре в течение 19 часов. Растворитель реакционного раствора выпаривали в условиях пониженного давления, и разбавляли этилацетатом и водой. Органический слой промывали водой, и выпаривали растворитель в условиях пониженного давления с получением неочищенного соединения 62.
Стадия 5
К раствору неочищенного соединения 62 в метаноле (4 мл) добавляли 60% гидрид натрия (270 мг, 6,75 ммоль), и перемешивали смесь при комнатной температуре в течение 5 часов. После добавления 2М водного раствора соляной кислоты (3,34 мл, 6,68 ммоль), растворитель выпаривали в условиях пониженного давления, и разбавляли этилацетатом и водой. После промывки органического слоя водой, растворитель выпаривали в условиях пониженного давления, и измельчали полученный остаток в порошок со смесью гексан-этилацетат с получением соединения 63 (324 мг, выход 82%).
1H-ЯМР (DMSO-d6) δ: 1,99-2,13 (м, 2H), 2,65 (д, J=17,1 Гц, 1H), 3,19 (д, J=17,1 Гц, 1H), 4,09-4,17 (м, 1H), 4,20-4,25 (м, 1H), 4,72 (кв, J=8,9 Гц, 2H), 6,48 (д, J=2,3 Гц, 1H), 6,64 (дд, J=8,8, 2,3 Гц, 1H), 7,41 (д, J=8,8 Гц, 1H), 7,84 (с, 1H).
Стадия 6
К суспензии соединения 63 (308 мг, 0,869 ммоль) в 1,2-дихлорэтане (6,2 мл) добавляли N,N-диметилформамид (63,5 мг, 0,869 ммоль) и оксихлорид фосфора (133 мг, 0,869 ммоль), и перемешивали смесь при комнатной температуре в течение 18,5 часов. К реакционному раствору добавляли воду, и собирали выпавшее в осадок твердое вещество путем фильтрования с получением соединения 64 (312 мг, выход 96%).
1H-ЯМР (CDCl3) δ: 2,26 (2H, т, J=5,5 Гц), 3,00 (1H, д, J=18,8 Гц), 3,44 (1H, д, J=18,8 Гц), 4,15-4,29 (2H, м), 4,30 (1H, д, J=7,9 Гц), 4,34 (1H, д, J=7,9 Гц), 5,83 (1H, с), 6,42 (1H, д, J=2,5 Гц), 6,63 (1H, дд, J=8,8, 2,4 Гц), 7,33 (1H, д, J=8,8 Гц).
Стадия 7
К раствору соединения 64 (500 мг, 1,34 ммоль) в тетрагидрофуране (4 мл) добавляли гидразина моногидрат (225 мг, 3,35 ммоль), и перемешивали смесь при комнатной температуре в течение 1 часа. К реакционному раствору добавляли воду, а затем экстрагировали этилацетатом. Растворитель выпаривали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (хлороформ-метанол) с получением соединения 65 (432 мг, выход 87%).
[M+H]=369,00, условия проведения измерений C
Стадия 8
Соединение 66 (324 мг, 1,09 ммоль) и карбонат цезия (397 мг, 1,22 ммоль) добавляли к раствору соединения 65 (333 мг, 0,91 ммоль) в ацетонитриле (2 мл), и перемешивали смесь при 80°C в течение 1 часа. К реакционному раствору добавляли воду, а затем экстрагировали этилацетатом. Растворитель выпаривали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (хлороформ-метанол) с получением соединения 67 (137 мг, выход 31%).
1H-ЯМР (CDCl3) δ: 2,14-2,26 (м, 4H), 2,93 (д, J=16,1 Гц, 1H), 3,21 (д, J=16,1 Гц, 1H), 3,97-4,03 (м, 4H), 4,19-4,21 (м, 2H), 4,32 (кв, J=8,1 Гц, 2H), 4,83 (с, 2H), 5,19 (с, 1H), 6,41 (д, J=2,5 Гц, 1H), 6,60 (дд, J=8,8, 2,5 Гц, 1H), 7,46 (д, J=8,8 Гц, 1H).
Стадия 9
К раствору соединения 67 (137 мг, 0,28 ммоль) в толуоле (2 мл) при комнатной температуре добавляли 2-(метансульфонил)уксусную кислоту (191 мг, 1,38 ммоль) и N,N'-диизопропилкарбодиимид (0,22 мл, 1,38 ммоль), и перемешивали смесь в течение 3 часов. При комнатной температуре добавляли воду, а затем экстрагировали этилацетатом. После промывки органического слоя насыщенным водным бикарбонатом натрия, растворитель выпаривали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (хлороформ-метанол) и измельчали в порошок со смесью метанол-вода с получением соединения II-121 (82,4 мг, выход 49%).
1H-ЯМР (CDCl3) δ: 2,09-2,18 (м, 2H), 2,29-2,36 (м, 2H), 3,03 (д, J=16,3 Гц, 1H), 3,21 (с, 3H), 3,31 (д, J=16,3 Гц, 1H), 3,99-4,35 (м, 10H), 5,92 (с, 1H), 6,40 (д, J=2,4 Гц, 1H), 6,60 (дд, J=8,8, 2,4 Гц, 1H), 7,41 (д, J=8,8 Гц, 1H), 9,56 (с, 1H).
[0048]
Пример 13

Стадия 1
К раствору соединения 53 (5,50 г, 13,6 ммоль) в метаноле (60 мл) добавляли 4М раствор хлороводорода в диоксане (3,40 мл, 13,6 ммоль), и перемешивали смесь при комнатной температуре в течение 1 часа. Затем, после выпаривания растворителя в условиях пониженного давления, полученный остаток растворяли в тетрагидрофуране (60 мл), и добавляли 2-цианоуксусную кислоту (2,31 г, 27,2 ммоль), 1-этил-3-(3-диметиламинопропил)-карбодиимида гидрохлорид (5,22 г, 27,2 ммоль) и триэтиламин (7,54 мл, 54,4 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли насыщенный раствор хлорида аммония, а затем экстрагировали этилацетатом. После промывки водой, проводили сушку над безводным сульфатом натрия, и выпаривали растворитель в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (гексан-этилацетат) с получением соединения 68 (4,80 г, выход 96%).
Стадия 2
К соединению 68 (4,80 г, 13,1 ммоль) добавляли 1М раствор метоксида натрия в метаноле (26,1 мл, 26,1 ммоль), и перемешивали смесь при комнатной температуре в течение 3 часов. К реакционному раствору добавляли 1М водный раствор соляной кислоты (150 мл), а затем экстрагировали этилацетатом. Органический слой промывали водой, а затем сушили над безводным сульфатом натрия. После концентрирования, полученный остаток измельчали в порошок со смесью гексан-этилацетат с получением соединения 69 (3,75 г, 86%).
1H-ЯМР (DMSO-d6) δ: 2,06-2,12 (2H, м), 2,71 (1H, д, J=17,5 Гц), 3,19 (1H, д, J=17,0 Гц), 4,12-4,17 (1H, м), 4,22-4,27 (1H, м), 7,01 (1H, д, J=2,0 Гц), 7,11 (1H, дд, J=8,5, 2,0 Гц), 7,42 (1H, д, J=8,5 Гц), 7,91 (1H, с).
Стадия 3
К раствору соединения 69 (2,00 г, 5,97 ммоль) в дихлорэтане (20 мл) добавляли DMF (4,64 мл, 59,7 ммоль) и фосфорилхлорид (0,61 мл, 6,56 ммоль), и перемешивали смесь при комнатной температуре в течение 19 часов. К реакционному раствору добавляли воду, а затем экстрагировали хлороформом. После промывки насыщенным водным бикарбонатом натрия и насыщенным водным раствором хлорида натрия, проводили сушку над безводным сульфатом магния, и выпаривали растворитель в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (гексан-этилацетат) и измельчали в порошок со смесью гексан-этилацетат с получением соединения 70 (1,03 г, выход 49%).
1H-ЯМР (CDCl3) δ: 2,27 (ушир. с, 2H), 3,03 (д, J=18,6 Гц, 1H), 3,41 (д, J=18,8 Гц, 1H), 4,17-4,31 (м, 2H), 5,96 (с, 1H), 7,07-7,27 (м, 3H).
Стадия 4
К раствору соединения 70 (250 мг, 0,71 ммоль) в этаноле (5 мл) добавляли 2-гидразинил-5-метилпиридина гидрохлорид (135 мг, 0,85 ммоль) и бикарбонат натрия (148 мг, 1,77 ммоль), и перемешивали смесь при 70°C в течение 1 часа. К реакционному раствору добавляли воду, экстрагировали хлороформом, сушили над безводным сульфатом магния, и выпаривали растворитель в условиях пониженного давления. К полученному остатку для измельчения добавляли диизопропиловый эфир с получением тем самым соединения 71 (237 мг, выход 76%).
1H-ЯМР (CDCl3) δ: 2,15-2,29 (м, 2H), 2,36 (с, 3H), 3,03 (д, J=16,1 Гц, 1H), 3,24 (д, J=16,1 Гц, 1H), 4,22-4,24 (м, 2H), 5,23 (с, 1H), 7,04 (д, J=2,0 Гц, 1H), 7,10 (дд, J=8,4, 2,0 Гц, 1H), 7,19 (ушир. с, 2H), 7,42 (д, J=8,4 Гц, 1H), 7,61-7,64 (м, 1H), 7,76 (д, J=8,5 Гц, 1H), 8,19 (ушир. с, 1H).
Стадия 5
К раствору соединения 71 (100 мг, 0,23 ммоль) в толуоле (2 мл) и воде (0,4 мл) добавляли циклопропилтрифторборат калия (101 мг, 0,68 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)-ферроцен]палладия (II) (0,148 г, 0,227 ммоль) и карбонат цезия (0,222 г, 0,681 ммоль), и перемешивали при 100°C в течение 3 часов. К реакционному раствору добавляли воду, а затем смесь экстрагировали этилацетатом, органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Растворитель выпаривали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (гексан-этилацетат) и измельчали в порошок со смесью гексан-этилацетат с получением соединения 72 (20,4 мг, выход 22%).
Стадия 6
К раствору соединения 72 (20,4 мг, 0,051 ммоль) в дихлорметане (1 мл), при 0°C добавляли раствор 2-(метансульфонил)ацетилхлорида (0,20 ммоль) и пиридина (0,016 мл, 0,20 ммоль) в дихлорметане (0,3 мл), и перемешивали смесь в течение 3 часов. При комнатной температуре к реакционному раствору добавляли воду, а затем экстрагировали этилацетатом, и сушили продукт над сульфатом магния. Растворитель выпаривали в условиях пониженного давления, и очищали полученный остаток методом колоночной хроматографии на силикагеле (хлороформ-метанол, амино-силикагель) с получением соединения II-168 (14,2 мг, 54%).
1H-ЯМР (CDCl3) δ: 0,68-0,72 (м, 2H), 0,95-1,00 (м, 2H), 1,81-1,88 (м, 1H), 2,19-2,28 (м, 2H), 2,40 (с, 3H), 3,11 (д, J=16,3 Гц, 1H), 3,23 (с, 3H), 3,42 (д, J=16,3 Гц, 1H), 4,14 (с, 2H), 4,20-4,22 (м, 2H), 5,61 (с, 1H), 6,57 (д, J=1,8 Гц, 1H), 6,73 (дд, J=8,2, 1,8 Гц, 1H), 7,42 (д, J=8,2 Гц, 1H), 7,70 (дд, J=8,4, 1,8 Гц, 1H), 7,81 (д, J=8,4 Гц, 1H), 8,30 (с, 1H), 11,17(с, 1H).
[0049]
Пример 14

Стадия 1
К раствору (R)-2-метилпропан-2-сульфинамида (631 мг, 5,21 ммоль) в толуоле (2,5 мл) добавляли тетраэтоксититан (0,654 мл, 3,12 ммоль), и добавляли раствор соединения 73 (384 мг, 2,08 ммоль) в толуоле (2,5 мл). Смесь перемешивали при 100°C в течение 3 часов. К реакционному раствору добавляли при комнатной температуре водный раствор лимонной кислоты и этилацетат, нерастворимые вещества фильтровали, а затем экстрагировали этилацетатом. Органический слой промывали насыщенным раствором хлорида натрия, а затем сушили над безводным сульфатом магния. Растворитель выпаривали в условиях пониженного давления, и очищали полученный остаток методом колоночной хроматографии на силикагеле (гексан-этилацетат) с получением соединения 74 (428 мг, выход 72%).
1H-ЯМР (CDCl3) δ: 1,32 (с, 9H), 3,31-3,38 (м, 1H), 3,45-3,52 (м, 1H), 4,27-4,33 (м, 1H), 4,36-4,41 (м, 1H), 6,43-6,49 (м, 2H).
Стадия 2
К 1М раствору гексаметилдисилазида лития в тетрагидрофуране (2,23 мл, 2,23 ммоль) при 78°C добавляли тетрагидрофуран (1 мл) и метилацетат (0,178 мл, 2,23 ммоль), и перемешивали в течение 1 часа. После этого, при 78°C по каплям добавляли 1М раствор триизопропоксида хлорида натрия в гексане (2,98 мл, 2,98 ммоль), и перемешивали смесь в течение 30 минут. После этого, при 78°C по каплям добавляли раствор соединения 74 (428 мг, 1,49 ммоль) в тетрагидрофуране (1 мл), и перемешивали смесь в течение 4 часов. К реакционному раствору добавляли 10% водный раствор лимонной кислоты и сегнетову соль, смесь нагревали до комнатной температуры и перемешивали в течение 12 часов, а затем экстрагировали этилацетатом. Органический слой промывали насыщенным водным бикарбонатом натрия и насыщенным водным раствором хлорида натрия, а затем сушили над безводным сульфатом магния. Растворитель выпаривали в условиях пониженного давления, и очищали полученный остаток методом колоночной хроматографии на силикагеле (гексан-этилацетат) с получением соединения 75 (305 мг, выход 57%).
1H-ЯМР (CDCl3) δ: 1,22 (с, 9H), 2,19-2,27 (м, 1H), 2,45-2,49 (м, 1H), 2,74 (д, J=15,9 Гц, 1H), 3,40 (д, J=15,9 Гц, 1H), 3,70 (с, 3H), 4,19-4,24 (м, 1H), 4,51-4,57 (м, 1H), 5,02(с, 1H), 6,36-6,47 (м, 2H).
Стадия 3
К раствору соединения 75 (305 мг, 0,85 ммоль) в метаноле (2,6 мл) добавляли 4М раствор хлороводорода в диоксане (0,44 мл, 1,77 ммоль), и перемешивали смесь при 0°C в течение 1 часа. После выпаривания растворителя из реакционного раствора в условиях пониженного давления, полученный остаток растворяли в тетрагидрофуране (2,5 мл), и добавляли 2-цианоуксусную кислоту (108 мг, 1,27 ммоль), HATU (482 мг, 1,27 ммоль) и триэтиламин (0,29 мл, 2,11 ммоль). Смесь перемешивали при комнатной температуре в течение 4 часов. К реакционному раствору добавляли воду, а затем экстрагировали этилацетатом. После промывки насыщенным водным бикарбонатом натрия и насыщенным водным раствором хлорида натрия, проводили сушку над безводным сульфатом натрия, и выпаривали растворитель в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (гексан-этилацетат) и измельчали в порошок со смесью гексан-этилацетат с получением соединения 76 (170 мг, выход 62%).
1H-ЯМР (CDCl3) δ: 2,08-2,14 (м, 1H), 2,83-2,90 (м, 1H), 2,96 (д, J=14,7 Гц, 1H), 3,26-3,33 (м, 3H), 3,75 (с, 3H), 4,13-4,18 (м, 1H), 4,27-4,33 (м, 1H), 6,35-6,44 (м, 2H), 7,51 (с, 1H).
Стадия 4
К раствору соединения 76 (393 мг, 1,21 ммоль) в метаноле (4 мл) и тетрагидрофурану (1 мл) добавляли 1М раствор метоксида натрия в метаноле (2,6 мл, 2,6 ммоль), и перемешивали смесь при комнатной температуре в течение 7 часов. К реакционному раствору добавляли 2М водный раствор соляной кислоты (1,3 мл), и добавляли метанол к полученному после концентрирования остатку. Нерастворимое вещество удаляли путем фильтрования, и выпаривали растворитель в условиях пониженного давления. К полученному остатку для измельчения добавляли этилацетат с получением тем самым соединения 77 (213 мг, 60%).
1H-ЯМР (DMSO-d6) δ: 2,07-2,17 (м, 2H), 2,72 (д, J=17,7 Гц, 1H), 3,19 (д, J=17,7 Гц, 1H), 4,10-4,22 (м, 2H), 6,61 (ушир. д, J=10,2 Гц, 1H), 6,78-6,84 (м, 1H), 7,89 (с, 1H).
Стадия 5
К раствору соединения 77 (1,0 г, 3,42 ммоль) в дихлорэтане (10 мл) добавляли DMF (2,66 мл, 34,2 ммоль) и фосфорилхлорид (0,35 мл, 3,76 ммоль), и перемешивали смесь при комнатной температуре в течение 20 часов. К реакционному раствору добавляли воду, а затем экстрагировали хлороформом. После промывки насыщенным водным бикарбонатом натрия и насыщенным водным раствором хлорида натрия, проводили сушку над безводным сульфатом магния, и выпаривали растворитель в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (гексан-этилацетат) с получением соединения 78 (413 мг, выход 39%).
[M+H]=310,95, условия проведения измерения C
Стадия 6
К раствору соединения 78 (50 мг, 0,16 ммоль) в этаноле (1 мл) добавляли 2-гидразинил-5-метилпиридина гидрохлорид (30,8 мг, 0,19 ммоль) и бикарбонат натрия (34 мг, 0,40 ммоль), и перемешивали смесь при 80°C в течение 2 часов. К реакционному раствору добавляли воду, а затем экстрагировали этилацетатом, и после промывки насыщенным водным раствором хлорида натрия проводили сушку над безводным сульфатом магния. Растворитель выпаривали в условиях пониженного давления. Полученный остаток растворяли в дихлорметане (1,0 мл), и при 0°C добавляли раствор 2-(метансульфонил)ацетилхлорида (0,64 ммоль) в дихлорметане (1,0 мл) и пиридин (0,052 мл, 0,64 ммоль). Смесь перемешивали в течение 3 часов. При комнатной температуре, добавляли воду, а затем экстрагировали этилацетатом, и сушили продукт над сульфатом магния. Растворитель выпаривали в условиях пониженного давления. Полученный остаток очищали методом колоночной хроматографии на силикагеле (хлороформ-метанол, амино-силикагель) и измельчали в порошок гексан-этилацетат с получением соединения II-203 (52,7 мг, 41%).
1H-ЯМР (CDCl3) δ: 2,19-2,26 (м, 1H), 2,32-2,40 (м, 4H), 2,99 (д, J=16,2 Гц, 1H), 3,22 (с, 3H), 3,81 (д, J=16,2 Гц, 1H), 4,15-4,20 (м, 4H), 5,77 (с, 1H), 6,47-6,52 (м, 2H), 7,69-7,71 (м, 1H), 7,81 (д, J=8,3 Гц, 1H), 8,30 (ушир. с, 1H).
[0050]
Следующие соединения были синтезированы аналогичным образом.
[0051]
Таблица 1

[0052]
Таблица 2

[0053]
Таблица 3
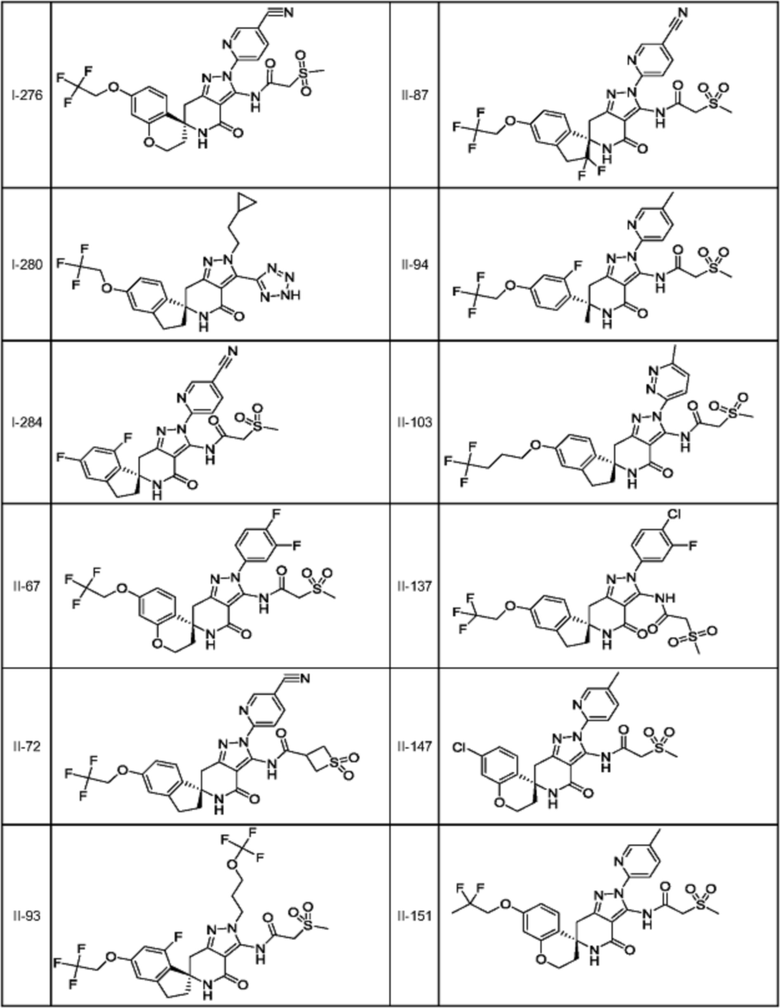
[0054]
Таблица 4

[0055]
Таблица 5

[0056]
Хотя стереохимическая структура не определена, II-276 представляет собой оптически активное соединение с различной стереохимией сульфоксимина.
[0057]
Ниже представлены физические характеристики соединений.
В таблицах, «RT» означает время удерживания (в минутах) согласно LC/MS (жидкостной хроматографии/масс-спектрометрии), «MS» означает массу (M+H) согласно LC/MS, и «Способ LCMS» означает любые из следующих условий проведения LC/MS-измерений.
Условия проведения измерений A
Колонка: ACQUITY UPLC (зарегистрированный товарный знак) BEH C18 (1,7 мкм, внутр. диам. 2,1×50 мм) (Waters)
Скорость потока: 0,8 мл/мин
Длина волны УФ-обнаружения: 254 нм
Подвижная фаза: [A] представляет собой 0,1% раствор муравьиной кислоты в воде, и [B] представляет собой 0,1% раствор муравьиной кислоты в ацетонитриле.
Проводят линейный градиент от 5% до 100% растворителя [B] в течение 3,5 минут, и выдерживают при 100% растворителя [B] в течение 0,5 минуты.
Условия проведения измерений B
Колонка: Shim-pack XR-ODS (2,2 мкм, внутр. диам. 50×3,0 мм) (Shimadzu)
Скорость потока: 1,6 мл/мин
Длина волны УФ-обнаружения: 254 нм
Подвижная фаза: [A] представляет собой 0,1% раствор муравьиной кислоты в воде, и [B] представляет собой 0,1% раствор муравьиной кислоты в ацетонитриле.
Градиент: Проводят линейный градиент от 10% до 100% растворителя [B] в течение 3 минут, и выдерживают при 100% растворителя [B] в течение 0,5 минуты.
Условия проведения измерений C
Колонка: ACQUITY UPLC (зарегистрированный товарный знак) BEH C18 (1,7 мкм, внутр. диам. 2,1×50 мм) (Waters)
Скорость потока: 0,55 мл/мин
Длина волны УФ-обнаружения: 254 нм
Подвижная фаза: [A] представляет собой 0,1% раствор муравьиной кислоты в воде, и [B] представляет собой 0,1% раствор муравьиной кислоты в ацетонитриле.
Градиент: Проводят линейный градиент от 5% до 100% растворителя [B] в течение 3 минут, и выдерживают при 100% растворителя [B] в течение 0,5 минуты.
[0058]
Таблица 6

[0059]
ЯМР-анализ каждого Примера проводили при 300 МГц с использованием DMSO-d6 или CDCl3.
[0060]
Таблица 7
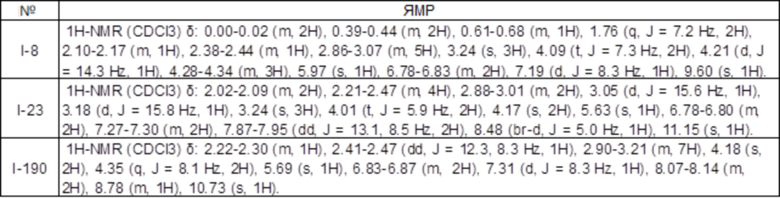
[0061]
Ниже описаны биологические тестовые примеры для соединений согласно настоящему изобретению.
[0062]
Подготовительный пример 1: Получение рекомбинантной MGAT2 человека
Полноразмерный ген MGAT2 человека, к которому на N-конце была добавлена метка Flag, встраивали в pFastBac (производства Invitrogen). Рекомбинантный бакуловирус получали в соответствии с протоколом для бакуловирусной экспрессирующей системы Bac-to-Bac (производства Invitrogen), и инфицировали им клетки Sf-9. Клетки собирали и обрабатывали ультразвуком, а затем собирали мембранную фракцию посредством центрифугирования. Для мембранной фракции проводили вестерн-блоттинг с анти-Flag-антителами для подтверждения экспрессии, и использовали мембранную фракцию в качестве раствора рекомбинантного фермента MGAT2 человека.
[0063]
ТЕСТОВЫЙ ПРИМЕР 1: Измерение ингибирующей активности в отношении MGAT2 человека
Каждый из растворов соединений согласно настоящему изобретению в DMSO разделяли на аликвоты по 0,2 мкл и помещали в 384-луночный полистироловый микропланшет производства Corning Incorporated, и добавляли к ним 5 мкл раствора фермента, приготовленного с аналитическим буфером (100 ммоль/л фосфатный буфер (pH 7,4), содержащий 2 ммоль/л DTT) и 5 мкл раствора субстрата (100 ммоль/л фосфатный буфер (рН 7,4), 30 мкмоль/л 2-олеоилглицерин, 10 мкмоль/л олеоил-КоА), полученную смесь перемешивали, центрифугировали и инкубировали во влажной камере при комнатной температуре в течение 1 часа. После ферментативной реакции, для остановки реакции добавляли 50 мкл раствора для гашения (содержащего 0,2 мкмоль/л Диолеин-d5, 0,4% муравьиную кислоту и 50% изопропанол), содержащего внутренний стандарт (IS), конечную смесь помещали в планшет производства Shimadzu GLC Ltd., а затем перемешивали и центрифугировали, и проводили измерения на Q-TOF масс-спектрометрах RapidFire360 и Agilent 6550 с ионизацией электрораспылением. Определяли диолеин как продукт реакции (Р) 2-олеиоилглицерина в качестве субстрата, и аддукт иона аммония IS, и вычисляли соотношение интенсивностей пиков P/IS по высоте пиков для оценки ингибирующей активности. Значения ингибирующей активности без добавления/с добавлением фермента определяли как Контроль(+)/Контроль(-), соответственно, и определяли соответствующее ингибирование (в %) как ингибирование на 0% и ингибирование на 100%. Ингибирующую активность рассчитывали по формуле, представленной ниже, с помощью TIBCO Spotfire (производства TIBCO Software Inc.):
Ингибирующая активность (в %) = [1-{Образец-Контроль()}/{Контроль(+) - Контроль(-)}] * 100,
где Образец означает соотношение пиков интенсивности P/IS при добавлении соединения согласно настоящему изобретению.
Результаты ингибирующей активности соединений согласно настоящему изобретению представлены в следующей таблице. Значения IC50 (нМ) в таблицах указывает на концентрацию, демонстрирующую 50% ингибирование фермента.
[0064]
[Таблица 8]

[0065]
ТЕСТОВЫЙ ПРИМЕР 2: Тест на метаболическую стабильность
С использованием коммерчески доступных объединенных микросом печени человека соединение согласно настоящему изобретению приводили во взаимодействие в течение фиксированного времени, рассчитывали оставшуюся долю путем сравнения прореагировавшего образца и не прореагировавшего образца, оценивая тем самым степень метаболизма соединения согласно настоящему изобретению.
[0066]
Реакцию (реакцию окисления) проводили при температуре 37°C в течение 0 минут или 30 минут в присутствии 1 ммоль/л NADPH в 0,2 мл буфера (50 ммоль/л Tris-HCl pH 7,4, 150 ммоль/л хлорида калия, 10 ммоль/л хлорида магния), содержащего 0,5 мг белка/мл микросом печени человека. После реакции, 50 мкл реакционного раствора добавляли к 100 мкл смеси метанол/ацетонитрил (1/1 (об./об.)), перемешивали и центрифугировали при 3000 об./мин. в течение 15 минут. Соединение согласно настоящему изобретению в надосадочной жидкости оценивали количественно методом LC/MS/MS, и рассчитывали оставшееся после реакции количество соединения согласно настоящему изобретению, принимая за 100% количество соединения в момент времени 0 минут.
(Результат) Оставшаяся доля представлена для концентрации тестируемого соединения 0,5 мкмоль/л.
[Таблица 9]

[0067]
ТЕСТОВЫЙ ПРИМЕР 3: Тест на растворимость
Растворимость соединения согласно настоящему изобретению определяли в условиях добавления 1% DMSO. Приготавливали 10 ммоль/л раствор соединения в DMSO, и добавляли 6 мкл раствора соединения согласно настоящему изобретению к 594 мкл искусственного кишечного сока с рН 6,8 (118 мл 0,2 моль/л тестового раствора NaOH и воду добавляли к 250 мл 0,2 моль/л тестового раствора дигидрофосфата калия до получения 1000 мл). Смесь оставляли отстаиваться в течение 16 часов при температуре 25°C, и проводили вакуумное фильтрование смеси. Фильтрат в два раза разбавляли смесью метанол/вода (1/1 (об./об.)), и измеряли концентрацию соединения в фильтрате методом HPLC или LC/MS/MS с использованием метода градуировочного графика по абсолютным значениям.
(Результат)
[Таблица 10]

[0068]
ТЕСТОВЫЙ ПРИМЕР 4: Тест на фототоксичность
Тест на фотогемолиз эритроцитов (Wolfgang J. W. Pepe et al., ATLA29, 145-162, 2001), который представляет собой способ оценки с использованием в качестве показателей эффекта на биомембраны и фотопероксидирование, проводили в качестве теста на фототоксичность in vitro. В этом способе приготавливали раствор соединения согласно настоящему изобретению с диметилсульфоксидом в качестве среды, к которому добавляли раствор эритроцитов овцы в соотношении 2,5% (об./об.) относительно приготовленного раствора, и использовали полученный тем самым смешанный раствор (концентрация: от 0,1 до 0,0008%). Смешанный раствор добавляли в два микропланшета, один из приготовленных микропланшетов облучали светом UVA и UVB спектра (10 Дж/см2, от 290 до 400 нм) с использованием ультрафиолетовой флуоресцентной лампы (лампа GL20SE, SANKYO DENKI Co., Ltd., и лампа FL20S-BLB, Panasonic Corporation), и подвергали центрифугированию вместе с микропланшетом, который не облучали светом, а затем измеряли оптическую плотность надосадочной жидкости (540 нм или 630 нм). Для определения двух показателей (эффекта на биомембраны и фотопероксидирование) для оценки фототоксичности значение оптической плотности среды вычитали из значения оптической плотности, полученного для соединения согласно настоящему изобретению в каждом из случаев с облучением и без облучения светом, и использовали вычисленные таким образом значения в последующих расчетах. Применительно к эффекту на биомембраны, степень фотогемолиза определяли по разности значений оптической плотности (540 нм) в случае с облучением светом и без облучения светом, и применительно к фотопероксидированию, определяли разность значений оптической плотности (630 нм) в случае с облучением светом и без облучения светом. При вычислении степени фотогемолиза оптическую плотность (540 нм), полученную от 2,5% (об./об.) раствора эритроцитов овцы, подвергнутого принудительному гемолизу дистиллированной водой, принимали за 100% степень гемолиза и использовали в качестве точки отсчета. Результат считали (-), если степень фотогемолиза составляла менее чем 10%, и разность значений оптической плотности при 630 нм составляла менее чем 0,05. Результат считали (+), если степень фотогемолиза составляла 10% или более, и разность значений оптической плотности при 630 нм составляла 0,05 или более.
(Результат)
Соединение I-34: (-)
[0069]
Тестовый Пример 5: Тест на цитотоксичность
Для оценки цитотоксичности соединения согласно настоящему изобретению, клетки после обработки соединением автоматически подсчитывали с использованием анализатора изображений клеток Toxinsight (Thermo Fisher Scientific).
Клетки HepG2 (полученные из злокачественных клеток печени человека) высевали на 384-луночный планшет в количестве 60000 клеток/мл, и добавляли раствор соединения в каждую лунку через 24 часа. Раствор соединения представлял собой раствор соединения согласно настоящему изобретению в DMSO (выполненное 5 раз 2-кратное разведение от максимальной концентрации в 50 мкмоль/л до минимальной концентрации приблизительно 3,1 мкмоль/л), и использовали раствор, состоящий только из DMSO, в качестве отрицательного контроля, а раствор камптотецина использовали в качестве положительного контроля. Раствор соединения согласно настоящему изобретению в DMSO, раствор отрицательного контроля или раствор положительного контроля добавляли в каждую лунку. Через 71 час в каждую лунку добавляли раствор Hoechst 33342, разбавленный фосфатным буферным раствором Дульбекко (D-PBS) до конечной концентрации 1 мкг/мл, и проводили окрашивание ядер в инкубаторе при температуре 37°C и 5% CO2 в течение 1 часа. После окрашивания результат фиксировали 4% параформальдегидом в инкубаторе CO2 при температуре 37°C в течение 20 минут. В завершение, лунки три раза промывали D-PBS, и подсчитывали в каждой лунке ядра с испусканием флуоресценции с использованием Toxinsight (Thermo Fisher Scientific). Для одной концентрации отводили четыре лунки, и вычисляли среднее значение и стандартное отклонение (SD) для количества ядер (количества клеток, для которых не была обнаружена токсичность) в четырех лунках. Проводили сравнение с группой отрицательного контроля, и рассчитывали экспозиционную концентрацию соединения (IC50), при которой среднее значение было снижено до менее чем 50% от среднего значения для отрицательного контроля. Меньшее значение IC50 оценивали как более высокий риск цитотоксичности.
[0070]
Тестовый Пример 6: Тест на эффект против ожирения
Эффект соединения согласно настоящему изобретению против ожирения тестировали с использованием мышей C57BL/6j (мышей DIO), обеспечиваемых пищей с высоким содержанием жиров (TestDiet; 58Y1).
Приобретали самцов мышей C57BL/6j в возрасте пяти недель (CLEA Japan, Inc.) и растили их с использованием пищи с высоким содержанием жира, в 12-часовых циклах смены дня и ночи в течение 4 недель с получением мышей DIO. Среду (0,5% HPMC) вводили дважды в сутки, начиная за 3 недели до введения соединения. Применяли рандомизацию для деления на группы (n=7) на основании массы тела и изменения в потреблении пищи в течение периода введения для подготовки. Принудительное пероральное введение тестируемого соединения или среды (0,5% HPMC) осуществляли дважды в сутки с 1 по 28 сутки. Массу тела и потребление пищи измеряли каждый день. На 28 сутки выполняли вскрытие, и проводили измерение массы эпидидимального жира и биохимический анализ собранной крови.
[0071]
ТЕСТОВЫЙ ПРИМЕР 7: Тест на ингибирование CYP
Используя коммерчески доступные смешанные микросомы печени человека и применяя в качестве маркеров О-деэтилирование 7-этоксирезоруфина (CYP1A2), метилгидроксилирование толбутамида (CYP2C9), 4'-гидроксилирование мефенитоина (CYP2C19), O-деметилирование декстрометорфана (CYP2D6) и гидроксилирование терфенадина (CYP3A4) в качестве типичных реакций метаболизма субстратов основных пяти форм фермента CYP человека (CYP1A2, 2C9, 2C19, 2D6, 3A4), оценивали степень ингибирования продуцируемого количества каждого метаболита соединением согласно настоящему изобретению.
Условия реакции были следующими: субстрат - 0,5 мкмоль/л этоксирезоруфина (CYP1A2), 100 мкмоль/л толбутамида (CYP2C9), 50 мкмоль/л S-мефенитоина (CYP2C19), 5 мкмоль/л декстрометорфана (CYP2D6), 1 мкмоль/л терфенедина (CYP3A4); время реакции - 15 минут; температура реакции - 37°C; фермент - смешанные микросомы печени человека, 0,2 мг белка/мл; концентрации соединения согласно настоящему изобретению - 1, 5, 10, 20 мкмоль/л (четыре точки).
Каждые пять видов субстратов, микросомы печени человека или соединение согласно настоящему изобретению в 50 ммоль/л буфере Hepes в качестве реакционного раствора добавляли в 96-луночный планшет в композиции, описанной выше, и добавляли NADPH в качестве кофактора для инициирования метаболических реакций. После инкубирования при температуре 37°C в течение 15 минут, добавляли смесь метанол/ацетонитрил (1/1 об./об.) для остановки реакции. После центрифугирования при 3000 об./мин. в течение 15 минут, в надосадочной жидкости количественно оценивали резоруфин (метаболит CYP1A2) посредством многоканального флуоресцентного счетчика или LC/MS/MS, гидроксид толбутамида (метаболит CYP2C9), 4'-гидроксид мефенитоина (метаболит CYP2C19), декстрорфан (метаболит CYP2D6) и терфенадиновый спирт (метаболит CYP3A4) количественно оценивали посредством LC/MS/MS.
Добавление в реакционную систему только DMSO, который является растворителем для растворения лекарства, принимали в качестве контроля (100%). Рассчитывали остаточную активность (%). Значение IC50 рассчитывали путем обратного предположения с помощью логистической модели с использованием концентрации и степени ингибирования.
(Результат)
Соединение II-103: пять видов > 20 мкмоль/л
[0072]
Тестовый пример 8: Тест на биодоступность (ВА)
Материалы и методы для экспериментов по оценке всасывания при пероральном введении
(1) Животные: использовали мышей или крыс SD.
(2) Условия содержания: мышам или крысам SD позволяли свободно принимать твердую пищу и стерилизованную водопроводную воду.
(3) Условия дозирования и деления на группы: Пероральное введение и внутривенное введение проводили в предопределенной дозировке. Деление на группы проводили как указано ниже (дозировка менялась для соединения).
Пероральное введение: от 1 до 30 мг/кг (n=2-3)
Внутривенное введение: от 0,5 до 10 мг/кг (n=2-3)
(4) Приготовление вводимого раствора: Пероральное введение осуществляли в форме суспензии или раствора. Внутривенное введение осуществляли после солюбилизации.
(5) Пути введения: Пероральное введение осуществляли принудительно в желудок посредством перорального зонда. Внутривенное введение осуществляли через хвостовую вену или бедренную вену посредством шприцов с иглой.
(6) Параметры оценки: забор крови выполняли в динамике, и измеряли концентрацию соединения согласно настоящему изобретению в плазме посредством LC/MS/MS.
(7) Статистический анализ: с учетом изменения концентрации соединения согласно настоящему изобретению в плазме, рассчитывали площадь под кривой зависимости концентрации в плазме от времени (AUC) с помощью программы нелинейного анализа по методу наименьших квадратов WinNonlin (зарегистрированная торговая марка), и рассчитывали исходя из AUC биодоступность соединения согласно настоящему изобретению в группе перорального введения и в группе внутривенного введения.
(Результат)
Соединение II-94: 73%
[0073]
Тестовый пример 9: Тест на необратимое ингибирование (MBI) CYP3A4 (MDZ)
Тест на MBI CYP3A4 (MDZ) представляет собой тест на исследование потенциала необратимого ингибирования в отношении CYP3A4 по усилению степени ингибирования метаболической реакции, вызванной соединением согласно настоящему изобретению. Ингибирование CYP3A4 оценивали с использованием смешанных микросом печени человека в реакции 1-гидроксилирования мидазолама (MDZ) в качестве маркера реакции.
Условия реакции были следующими: субстрат - 10 мкмоль/л MDZ; время предварительной реакции - 0 или 30 минут; время взаимодействия с субстратом - 2 минуты; температура реакции - 37°C; содержание белка в смешанных печеночных микросомах человека: во время предварительной реакции - 0,5 мг/мл, в реакционное время - 0,05 мг/мл (при 10-кратном разбавлении); концентрации соединения согласно настоящему изобретению во время предварительной реакции - 1, 5, 10, 20 мкмоль/л (четыре точки).
Смешанные микросомы печени человека и раствор соединения согласно настоящему изобретению в K-Pi буфере (pH 7,4) в качестве раствора для предварительной реакции добавляли в 96-луночный планшет в композиции для предварительной реакции. Часть раствора для предварительной реакции переносили в другой 96-луночный планшет и разбавляли 1/10 K-Pi буфером, содержащим субстрат. Для инициирования реакции добавляли NADPH в качестве ко-фактора маркерной реакции (вариант без предварительной реакции). После предопределенного времени реакции, добавляли смесь метанол/ацетонитрил (1/1, об./об.) для остановки реакции. Кроме того, в оставшийся раствор для предварительной реакции добавляли NADPH для инициирования предварительной реакции (вариант с предварительной реакцией). После предопределенного времени предварительной реакции, часть переносили в другой планшет и разбавляли 1/10 K-Pi буфером, содержащим субстрат, для инициирования реакции, в качестве маркерной реакции. После предопределенного времени реакции, добавляли раствор метанол/ацетонитрил (1/1, об./об.) для остановки реакции. Планшет, в котором осуществляли каждую показательную реакцию, центрифугировали при 3000 об./мин. в течение 15 минут, а затем количественно оценивали в надосадочной жидкости 1-гидроксилированный мидазолам посредством LC/MS/MS.
Образец с добавлением в реакционную систему DMSO в качестве растворителя вместо раствора для растворения соединения согласно настоящему изобретению принимали за контроль (100%). Остаточную активность (%) рассчитывали для каждой концентрации соединения согласно настоящему изобретению в сравнении с контролем, и рассчитывали значение IC путем обратного предположения с помощью логистической модели с использованием концентрации и степени ингибирования. Отношение [IC для преинкубации в течение 0 минут/IC для преинкубации в течение 30 минут] определяли как значение смещенного IC, и пример, когда значение смещенного IC составляло 1,5 или более, рассматривали как «положительный», а пример, когда значение смещенного IC составляло 1,0 или менее, рассматривали как «отрицательный».
(Результат)
Соединение II-103: «отрицательный»
[0074]
ТЕСТОВЫЙ ПРИМЕР 10: Тест на растворимость порошка
Соответствующее количество соединения согласно настоящему изобретению помещали в подходящие контейнеры. В каждый контейнер независимо добавляли 200 мкл JP-1 раствора (к 2,0 г хлорида натрия и 7,0 мл соляной кислоты добавляли воду до достижения 1000 мл), 200 мкл JP-2 раствора (500 мл воды добавляли к 500 мл фосфатного буфера (pH 6,8)) или 20 ммоль/л раствора таурохолата натрия (TCA)/JP-2 (раствор JP-2 добавляли к 1,08 г TCA до достижения 100 мл). Когда все количество растворялось после добавления тестируемого реагента, соответствующим образом добавляли соединение согласно настоящему изобретению. После герметизации и перемешивания встряхиванием при температуре 37°C в течение 1 часа, раствор фильтровали, и добавляли 100 мкл метанола к 100 мкл каждого фильтрата для двукратного разведения. При необходимости, степень разбавления меняли. После проверки на отсутствие пузырьков и осадка, контейнер герметизировали и перемешивали встряхиванием. Соединение согласно настоящему изобретению измеряли методом HPLC с использованием метода градуировочного графика по абсолютным значениям.
(Результат)
Соединение II-94: раствор JP-2: 3,4 мкмоль/л
[0075]
Тестовый пример 11: Флуктуационный тест Эймса
Оценивали мутагенность соединения согласно настоящему изобретению.
В 10 мл жидкой питательной среды (2,5% питательный бульон Oxoid №2) вносили 20 мкл замороженных бацилл тифа крыс (штамм Salmonella typhimurium TA98, штамм TA100) и культивировали, после чего перемешивали при температуре 37°C в течение 10 часов. Для удаления культурального раствора 8,0 мл бактериального раствора TA98 штамма центрифугировали (2000 об./мин., 10 минут). Бактерии суспендировали в 8,0 мл буфера Micro F (K2HPO4: 3,5 г/л, KH2PO4: 1 г/л, (NH4)2SO4: 1 г/л, тринатрий цитрата дигидрат: 0,25 г/л, MgSO4·7H2O: 0,1 г/л), добавляли суспензию в 120 мл экспозиционной среды (буфер Micro F, содержащий 8 мкг/мл биотина, 0,2 мкг/мг гистидина, 8 мг/мл глюкозы). Для приготовления тестируемого бактериального раствора, к 120 мл экспозиционной среды добавляли штамма TA100 в количестве 3,1 мл бактериального раствора. Смешивали 12 мкл каждого раствора соединения согласно настоящему изобретению (несколько 2-3-кратных разведений от максимальной дозы 50 мг/мл) в DMSO, DMSO в качестве отрицательного контроля, и 50 мкг/мл раствора 4-нитрохинолин-1-оксида в DMSO для штамма TA98, 0,25 мкг/мл раствора 2-(2-фурил)-3-(5-нитро-2-фурил)акриламида в DMSO для штамма TA100 в условиях отсутствия активации метаболизма, 40 мкг/мл раствора 2-аминоантрацена в DMSO для штамма TA98, 20 мкг/мл раствора 2-аминоантрацена в DMSO для штамма TA100 в условиях активации метаболизма, в качестве положительного контроля, и 588 мкл тестируемого бактериального раствора (смешанный раствор 498 мкл тестируемого бактериального раствора и 90 мкл смеси S9 в условиях активации метаболизма), и культивировали эту смесь с перемешиванием при температуре 37°C в течение 90 минут. 230 мкл бактериального раствора, обработанного соединением согласно настоящему изобретению, смешивали с 1150 мкл индикаторной среды (буфер Micro F, содержащий 8 мкг/мл биотина, 0,2 мкг/мл гистидина, 8 мг/мл глюкозы, 37,5 мкг/мл бромокрезола красного), каждые 50 мкл переносили на микропланшет в 48 лунок на дозу, и подвергали стационарному культивированию при температуре 37°C в течение 3 суток. Поскольку в лунке, содержащей бактерию, которая приобрела способность к пролиферации посредством мутации гена фермента, синтезирующего аминокислоту (гистидин), цвет изменяется из красного в желтый вследствие изменения рН, подсчитывали лунки с пролиферацией бактерий, которые цвет на менялся на желтый в 48 лунках на дозу, и оценивали в сравнении с группой отрицательного контроля. (-) и (+) означают отрицательную и положительную мутагенность, соответственно.
(Результат)
Соединение I-24: (-)
[0076]
Тестовый пример 12: Тест с hERG
Для оценки риска удлинения QT интервала электрокардиограммы соединением согласно настоящему изобретению, с использованием клеток CHO, экспрессирующих ген альфа-субъединицы калиевого канала человека (hERG) исследовали эффекты соединения согласно настоящему изобретению на калиевый ток замедленного выпрямления (IKr), который играет важную роль в процессе реполяризации желудочка.
После фиксирования клетки с мембранным потенциалом -80 мВ методом пэтч-клампа в конфигурации «целая клетка» с использованием автоматической пэтч-кламп системы (QPatch; Sophion BIoscience A/S) регистрировали IKr, индуцированный воздействием потенциала утечки в -50 мВ, с последующей деполяризационной импульсной стимуляцией в +20 мВ в течение 2 секунд, а затем реполяризационной импульсной стимуляцией в -50 мВ в течение 2 секунд. После стабилизации генерируемого тока, клетку обрабатывали при комнатной температуре в течение 10 минут внеклеточным раствором (NaCl: 145 ммоль/л, KCl: 4 ммоль/л, CaCl2: 2 ммоль/л, MgCl2: 1 ммоль/л, глюкоза: 10 ммоль/л, HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота): 10 ммоль/л, pH=7,4), в котором было растворено соединение согласно настоящему изобретению в целевой концентрации. По результатам регистрации IKr с использованием аналитического программного обеспечения (Falster Patch; Sophion Bioscience A/S) измеряли абсолютное значение пикового следового тока на основании значения тока потенциала покоя мембраны. Кроме того, для оценки влияния соединения согласно настоящему изобретению на IKr вычисляли ингибирования (в %) пикового следового тока соединением согласно настоящему изобретению в сравнении с пиковым следовым током после обработки носителем.
(Результат) Ингибирование (в %) представлено для 10 мкмоль/л тестируемого соединения.
Соединение I-253: 6,9%
[0077]
ПРИМЕР СОСТАВА
Соединение согласно настоящему изобретению может быть введено в виде фармацевтической композиции посредством любого общепринятого пути, в частности, энтерально, например, перорально, например, в форме таблеток или капсул, или парентерально, например, в форме инъекционных растворов или суспензий, местно, например, в форме лосьонов, гелей, мазей или кремов, или в назальной форме или в форме суппозитория. Фармацевтические композиции, содержащие соединение согласно настоящему изобретению в свободной форме или в форме фармацевтически приемлемой соли, в сочетании, по меньшей мере, с одним фармацевтически приемлемым носителем или разбавителем, могут быть приготовлены общепринятым образом посредством способов смешивания, гранулирования или нанесения покрытий. Например, пероральные композиции могут представлять собой таблетки, гранулы или капсулы, содержащие наполнители, разрыхлители, связующие вещества, смазки, и т.п., и активные ингредиенты. Композиции для инъекций могут представлять собой растворы или суспензию, могут быть стерилизованы и могут содержать консерванты, стабилизаторы, буферные средства, и т.п.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
[0078]
Поскольку соединения согласно настоящему изобретению обладают ингибирующей активностью в отношении MGAT2, они применимы в качестве лекарственного средства против MGAT2-опосредованных заболеваний, включая ожирение, метаболический синдром, гиперлипидемию, гипертриглицеридемию, гипер-ЛПОНП-триглицеридемию, гиперлипоацидемию, сахарный диабет или артериосклероз.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ДИГИДРОПИРАЗОЛОПИРАЗИНОНА, ОБЛАДАЮЩЕЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ MGAT-2 | 2020 |
|
RU2803743C2 |
| ЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДЫ ДЛЯ ИНГИБИРОВАНИЯ ГАММА-СЕКРЕТАЗЫ | 2004 |
|
RU2334743C2 |
| ТРИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИНГИБИТОРЫ JAK | 2012 |
|
RU2632870C2 |
| 6-ЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2016 |
|
RU2683245C1 |
| АМИДНОЕ ПРОИЗВОДНОЕ, ИМЕЮЩЕЕ ПРОТИВОВИРУСНУЮ АКТИВНОСТЬ | 2021 |
|
RU2836730C1 |
| МОНОЦИКЛИЧЕСКОЕ ПИРИДИНОВОЕ ПРОИЗВОДНОЕ | 2014 |
|
RU2645352C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА ИЛИ ИМИДАЗОПИРИДИНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ БОЛЕЗНЕЙ, АССОЦИИРОВАННЫХ С АСС2 | 2016 |
|
RU2702637C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2009 |
|
RU2490257C2 |
| ПРОИЗВОДНЫЕ 4-АМИНОМЕТИЛБЕНЗОЙНОЙ КИСЛОТЫ | 2014 |
|
RU2673245C2 |
| ПРОИЗВОДНЫЕ АМИНОДИГИДРОТИАЗИНА, ЗАМЕЩЕННЫЕ ЦИКЛИЧЕСКОЙ ГРУППОЙ | 2008 |
|
RU2476430C2 |
Изобретение относится к соединению, представленному формулой (I), или к его фармацевтически приемлемой соли, где фрагмент, представленный формулой:
 представляет собой
представляет собой ; где X представляет собой C(=O);
; где X представляет собой C(=O);
R1 представляет собой водород; R2a представлен формулой:  , где кольцо C представляет собой бензол; каждый R5 независимо представляет собой галоген или галогенC1-C4алкилокси, и n равен целому числу от 1 до 2, R2b представляет собой C1-C4алкил или галогенC1-C4алкил, или необязательно, R2a и R2b объединены вместе со смежным атомом углерода с формированием кольца B, кольцо B представлено формулой:
, где кольцо C представляет собой бензол; каждый R5 независимо представляет собой галоген или галогенC1-C4алкилокси, и n равен целому числу от 1 до 2, R2b представляет собой C1-C4алкил или галогенC1-C4алкил, или необязательно, R2a и R2b объединены вместе со смежным атомом углерода с формированием кольца B, кольцо B представлено формулой:
 , где каждый R6 независимо представляет собой галоген, галогенC1-C4алкил, галогенC1-C4алкилокси, неароматический C3-C6карбоциклил или неароматический гетероциклил, который представляет собой 4-членную моноциклическую группу, содержащую один гетероатом, выбранный из N, и необязательно замещенный галогеном или галогенC1-C4алкилом, каждый R14a и R14b независимо представляют собой водород или галоген, и n равен целому числу от 1 до 2; R3a представляет собой водород, R3b представляет собой водород; R4a представляет собой карбокси или представлен формулой:
, где каждый R6 независимо представляет собой галоген, галогенC1-C4алкил, галогенC1-C4алкилокси, неароматический C3-C6карбоциклил или неароматический гетероциклил, который представляет собой 4-членную моноциклическую группу, содержащую один гетероатом, выбранный из N, и необязательно замещенный галогеном или галогенC1-C4алкилом, каждый R14a и R14b независимо представляют собой водород или галоген, и n равен целому числу от 1 до 2; R3a представляет собой водород, R3b представляет собой водород; R4a представляет собой карбокси или представлен формулой:
 ; L3 представляет собой одинарную связь или C1-C4алкилен, R7 представляет собой водород, C1-C4алкилсульфонил, неароматический гетероциклил, который представляет собой 4-членную моноциклическую группу, содержащую один гетероатом, выбранный из S, и необязательно замещенный оксо, или неароматический C3-C6карбоциклилсульфонил, необязательно замещенный C1-C4алкилом, или представлен формулой: -S(=O)(=N-H)-RS1, R4b представляет собой C1-C4алкил, необязательно замещенный заместителем группы α, фенил, необязательно замещенный заместителем группы β, или ароматический гетероциклил, который представляет собой 5-6-членное кольцо, содержащее один или два одинаковых или разных гетероатома, выбранных из O и N, и необязательно замещенный заместителем группы β, RS1 представляет собой C1-C4алкил, заместитель группы α представляет собой галоген, галогенC1-C4алкилокси и неароматический C3-C6карбоциклил, и заместитель группы β представляет собой галоген, циано, C1-C4алкил, галогенC1-C4алкил и C1-C4алкилокси. Изобретение также относится к фармацевтической композиции, обладающей ингибирующей активностью в отношении MGAT2, на основе указанных соединений формулы (I). Технический результат – получение новых соединений и фармацевтической композиции на их основе, которые могут найти применение в медицине для лечения или профилактики ожирения, метаболического синдрома, гиперлипидемии, гипертриглицеридемии, гипер-ЛПОНП-триглицеридемии, гиперлипоацидемии, сахарного диабета или артериосклероза. 9 н. и 6 з.п. ф-лы, 10 табл., 14 пр.
; L3 представляет собой одинарную связь или C1-C4алкилен, R7 представляет собой водород, C1-C4алкилсульфонил, неароматический гетероциклил, который представляет собой 4-членную моноциклическую группу, содержащую один гетероатом, выбранный из S, и необязательно замещенный оксо, или неароматический C3-C6карбоциклилсульфонил, необязательно замещенный C1-C4алкилом, или представлен формулой: -S(=O)(=N-H)-RS1, R4b представляет собой C1-C4алкил, необязательно замещенный заместителем группы α, фенил, необязательно замещенный заместителем группы β, или ароматический гетероциклил, который представляет собой 5-6-членное кольцо, содержащее один или два одинаковых или разных гетероатома, выбранных из O и N, и необязательно замещенный заместителем группы β, RS1 представляет собой C1-C4алкил, заместитель группы α представляет собой галоген, галогенC1-C4алкилокси и неароматический C3-C6карбоциклил, и заместитель группы β представляет собой галоген, циано, C1-C4алкил, галогенC1-C4алкил и C1-C4алкилокси. Изобретение также относится к фармацевтической композиции, обладающей ингибирующей активностью в отношении MGAT2, на основе указанных соединений формулы (I). Технический результат – получение новых соединений и фармацевтической композиции на их основе, которые могут найти применение в медицине для лечения или профилактики ожирения, метаболического синдрома, гиперлипидемии, гипертриглицеридемии, гипер-ЛПОНП-триглицеридемии, гиперлипоацидемии, сахарного диабета или артериосклероза. 9 н. и 6 з.п. ф-лы, 10 табл., 14 пр.

1. Соединение, представленное формулой (I):
 ,
,
где
фрагмент, представленный формулой:

представляет собой
 ;
;
где X представляет собой C(=O);
R1 представляет собой водород;
R2a представлен формулой:
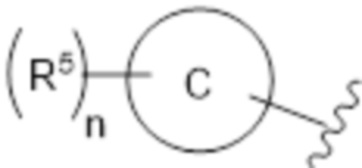 ,
,
где кольцо C представляет собой бензол;
каждый R5 независимо представляет собой галоген или галогенC1-C4алкилокси, и
n равен целому числу от 1 до 2,
R2b представляет собой C1-C4алкил или галогенC1-C4алкил, или
необязательно, R2a и R2b объединены вместе со смежным атомом углерода с формированием кольца B,
кольцо B представлено формулой:
 ,
,
где каждый R6 независимо представляет собой галоген, галогенC1-C4алкил, галогенC1-C4алкилокси, неароматический C3-C6карбоциклил или неароматический гетероциклил, который представляет собой 4-членную моноциклическую группу, содержащую один гетероатом, выбранный из N, и необязательно замещенный галогеном или галогенC1-C4алкилом,
каждый R14a и R14b независимо представляют собой водород или галоген, и
n равен целому числу от 1 до 2;
R3a представляет собой водород,
R3b представляет собой водород;
R4a представляет собой карбокси или представлен формулой:
 ;
;
L3 представляет собой одинарную связь или C1-C4алкилен,
R7 представляет собой водород, C1-C4алкилсульфонил, неароматический гетероциклил, который представляет собой 4-членную моноциклическую группу, содержащую один гетероатом, выбранный из S, и необязательно замещенный оксо, или неароматический C3-C6карбоциклилсульфонил, необязательно замещенный C1-C4алкилом, или представлен формулой: -S(=O)(=N-H)-RS1,
R4b представляет собой C1-C4алкил, необязательно замещенный заместителем группы α, фенил, необязательно замещенный заместителем группы β, или ароматический гетероциклил, который представляет собой 5-6-членное кольцо, содержащее один или два одинаковых или разных гетероатома, выбранных из O и N, и необязательно замещенный заместителем группы β,
RS1 представляет собой C1-C4алкил,
заместитель группы α представляет собой галоген, галогенC1-C4алкилокси и неароматический C3-C6карбоциклил, и
заместитель группы β представляет собой галоген, циано, C1-C4алкил, галогенC1-C4алкил и C1-C4алкилокси,
или его фармацевтически приемлемая соль.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где R2a и R2b объединены вместе со смежным атомом углерода с формированием кольца B и
кольцо B представлено любой из формул:
 ,
,
где значение каждого символа описано в п. 1.
3. Соединение или его фармацевтически приемлемая соль по п. 1, где R2a представлен формулой:
 ,
,
где значение каждого символа описано в п. 1 и
R2b представляет собой C1-C4алкил или галогенC1-C4алкил.
4. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, где R2a и R2b объединены вместе со смежным атомом углерода с формированием кольца B и
кольцо B представлено любой из формул:
 ,
,
где каждый R6 независимо представляет собой галоген, галогенC1-C4алкил или галогенC1-C4алкилокси,
каждый R14a и R14b независимо представляют собой водород, и
n равен целому числу от 1 до 2;
R4a представлен формулой:
 ;
;
L3 представляет собой C1-C4алкилен,
R7 представляет собой C1-C4алкилсульфонил,
R4b представляет собой C1-C4алкил, необязательно замещенный заместителем группы α, фенил, необязательно замещенный заместителем группы β, или ароматический гетероциклил, который представляет собой 5-6-членное кольцо, содержащее один или два одинаковых или разных гетероатома, выбранных из O и N, и необязательно замещенный заместителем группы β,
заместитель группы α представляет собой галогенC1-C4алкилокси, и
заместитель группы β представляет собой галоген или C1-C4алкил, или его фармацевтически приемлемая соль.
5. Соединение или его фармацевтически приемлемая соль по п. 1, где соединение выбирают из группы, состоящей из следующих соединений:


 .
.
6. Соединение или его фармацевтически приемлемая соль, где соединение представлено формулой:

I-253.
7. Соединение или его фармацевтически приемлемая соль, где соединение представлено формулой:
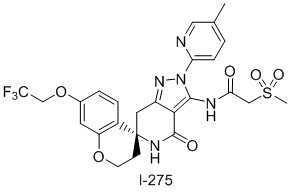 .
.
8. Соединение или его фармацевтически приемлемая соль, где соединение представлено формулой:
 .
.
9. Соединение или его фармацевтически приемлемая соль, где соединение представлено формулой:
 .
.
10. Соединение или его фармацевтически приемлемая соль, где соединение представлено формулой:
 .
.
11. Соединение или его фармацевтически приемлемая соль, где соединение представлено формулой:
 .
.
12. Соединение или его фармацевтически приемлемая соль, где соединение представлено формулой:
 .
.
13. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении MGAT2, содержащая эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп. 1-12.
14. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-12 для лечения или профилактики MGAT2-опосредованного заболевания.
15. Соединение или его фармацевтически приемлемая соль по п. 14 для лечения или профилактики ожирения, метаболического синдрома, гиперлипидемии, гипертриглицеридемии, гипер-ЛПОНП-триглицеридемии, гиперлипоацидемии, сахарного диабета или артериосклероза.
| WO 2017069224 A1, 27.04.2017 | |||
| WO 2014133134 A1, 04.09.2014 | |||
| JP 2014009165 A, 20.01.2014 | |||
| WO 2010095767 A1, 26.08.2010 | |||
| WO 2009126584 A1, 15.10.2009 | |||
| WO 2015073767 A1, 21.05.2015 | |||
| EA 22649 B1, 29.02.2016. |

