Настоящее изобретение относится к новым пиразолпиримидинам формулы I
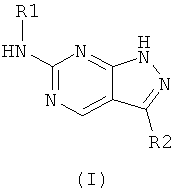
Новые пиразолпиримидины обладают способностью ингибировать активность циклин-зависимых киназ, наиболее предпочтительно, циклин-зависимой киназы 1 (Cdk1), циклин-зависимой киназы 2 (Cdk2) и циклин-зависимой киназы 4 (Cdk4). Эти соединения и их фармацевтически приемлемые соли и сложные эфиры проявляют антипролиферативную активность и используются, среди прочих, при лечении или контролировании раковых, в частности, твердых опухолей. Настоящее изобретение предлагает также фармацевтические композиции, содержащие такие соединения, и способы лечения или контролирования рака, наиболее предпочтительно, лечения или контролирования опухолей грудной железы, легкого, толстой кишки и простаты.
Неконтролируемая клеточная пролиферация является отличительной особенностью рака. Клетки раковой опухоли, как правило, имеют некоторые поврежденные формы генов, которые прямо или косвенно регулируют цикл клеточного деления.
Прохождение клеток через различные фазы клеточного цикла регулируется рядом мультиферментных комплексов, включающих регуляторный белок, циклин и киназу. Эти киназы носят название циклин-зависимых киназ (Cdks). Cdks экспрессируют во всем клеточном цикле, в то время, как уровни циклинов варьируются в зависимости от фазы клеточного цикла.
Четыре основные фазы регуляции клеточного цикла принято обозначать как G1, S, G2 и М. Оказалось, что некоторыми незаменимыми ферментами для регуляции клеточного цикла являются циклин D/Cdк4, циклин D/Cdк6, циклин E/Cdк2, циклин A/Cdк2 и циклин B/Cdк1 (известный также, как Cdc2/циклин В). Циклин D/Cdк4, циклин D/Cdк6, циклин E/Cdк2 контролируют прохождение через G1-фазу и переход G1-фазы в S-фазу посредством фосфорилирования белка ретинобластомы - pRb. Циклин A/Cdк2 регулирует прохождение через S-фазу, а циклин B/Cdк1 контролирует G2 критическую точку и регулирует вход в М-фазу (митоз).
Прохождение клеточного цикла регулируется Cdk1 (cdc2) и Cdк2 до начала фазы G1, когда клетки переходят к цитокинезу. Вследствие этого лекарственное ингибирование этих Cdкs, по-видимому, не только тормозит клеточную пролиферацию, но также запускает апоптоз клеток. Если клетки прошли G1 критическую точку и вступили в S-фазу, они становятся независимыми от стимуляции фактором роста для продолжения прохождения клеточного цикла.
Вслед за завершением ДНК репликации клетки входят в G2-фазу клеточного цикла и готовы к вхождению в М-фазу и цитокинезу. Cdk1, как было показано, регулирует прохождение клеток через эти более поздние фазы клеточного цикла совместно с обоими циклинами А и В. Полная активация Cdk1 требует как связывания циклина, так и специфического форфорилирования (Morgan, D.О., De Bondt, H.L., Curr. Opin. Cell. Biol., 1994, 6, 239-246). После активации Cdk1/циклиновый комплекс готовит клетку к делению в М-фазе.
Переход из G1-фазы в S-фазу, как установлено выше, регулируется комплексом Cdk4 с циклином D и комплексом Cdk2 с циклином Е. Эти комплексы фосфорилируют клеточный супрессорный белок ретинобластому (pRb), реализуя транскрипционный фактор E2F и запуская экспрессию генов, требуемую в S-фазе (Nevins, J.R., Science, 1992, 258, 424-429; Lavia, P. BioEssays, 1999, 21, 221-230). Блокирование активности Cdk4/циклин D и Cdk2/циклин Е комплексов тормозят клеточный цикл в G1-фазе. Например, белки INK4 семейства, включая рINK4a, которые блокируют киназную активность Cdk4/циклин D комплекса, вызывают торможение в фазе G1, (Sherr, С.J., Science, 1996, 274, 1672-1677). Специальная литература рассмотрена в (Vidal, A.Gene, 2000, 247, 1-15).
Недавно проведенные эксперименты показали, что комплекс Cdk4 с циклином D3 также играет роль в клеточном цикле при прохождении через G2-фазу. Ингибирование этого комплекса либо посредством р16, либо с использованием доминантного негативного Cdk4, приводит к торможению в G2-фазе в клетках, которые не экспрессируют pRb (Gabrielli В.G. et al., J.Biol. Chem., 1999, 274, 13961-13969).
Многочисленные дефекты в ходе метаболизма pRb, как было показано, включаются в различные виды раковых опухолей. Например, сверхэкспрессия Cdk4 наблюдается в случаях наследственной меланомы (Webster, К.R., Ехр. Opin. Invest. Drugs, 1988, 7, 865-887); циклин D сверхэкспрессирует во многих видах раковых опухолей человека (Sherr, С.J., Science, 1996, 274, 1672-1677); р16 мутирует или делецирует во многих опухолях (Webster, К.R., Ехр. Opin. Invest. Drugs, 1988, 7, 865-887); и pRb-функция теряется из-за мутации или делеции во многих видах раковых опухолей (Weinberg R.A., Cell, 1995, 81, 323-330). Дефекты в таком метаболизме, как было показано, могут быть использованы для прогнозирования болезни. Например, отсутствие р16 связано с плохим прогнозом относительно немелкоклеточной карциномы легких (NSCLC) и злокачественной меланомы (Tsihlias, J. et al., Annu. Rev. Med., 1999, 50, 401-423). Дефекты циклина D1 и/или pRb в гене и/или уровень экспрессии присутствуют в более чем 90% случаев образцов немелкоклеточной карциномы легких, означая, что циклин D1 и/или pRb представляют важную ступень в легочном опухолевом генезисе (Marchetti, A., et al., Int. J.Cancer, 1998, 75, 573-582). В 49 из 50 случаев карциномы поджелудочной железы (98%) pRb/p16 метаболический путь отменялся исключительно благодаря инактивации р16 гена и связанного циклина D (Shutte, M., et al.,Cancer Res., 1998, 57, 3126-3134). Обзор по связи между экспрессией pRb и циклин/циклин-зависимым киназам в ряде тканей представлен в Teicher, В.A., Cancer Chemother. Pharmacol., 2000, 46, 293-304.
Из-за вовлечения Сdr4/циклин D/pRb метаболизма в раковые заболевания человека через его роль в регуляции протекания клеточного цикла от G1 до S-фазы и потенциального положительного терапевтического эффекта от модулирования этого метаболизма, возникает значительный интерес к агентам, которые ингибируют или промотируют элементы этого метаболизма. Например, воздействие на раковые клетки осуществляется при использовании антител, антисмысловых олигонуклеотидов и сверхэкспрессии или добавления белков, включенных в метаболизм. См., например, Lukas, J. et al., Nanure, 1995, 79, 573-582; Nevins J.R., Science, 1992, 258, 424-429; Lim I.K. et al., Molecular Carcinogenesis, 1998, 23, 25-35; Tam, S.W. et al., Oncogene, 1994, 9, 2663-2674; Driscoll, В. et al., Am. J. Physiol., 1997, 273 (Lung Cell. Mol. Physiol,), 941-949; и Sang, J. et al., Chin. Sci. Bull., 1999, 44, 541-544).
Роль Cdks в регуляции клеточной пролиферации, таким образом, твердо установлена. Например, как показано выше, существует обширная часть литературы, подтверждающая применение соединений, ингибирующих мишени в Cdk4, Cdk2 и Cdk1 метболизме, в качестве антипролиферативных терапевтических агентов. Ингибиторы клеточной пролиферации тем самым действуют, как обратимые цитостатические агенты, используемые в лечении болезней, особенностью которых является отклоняющийся от нормы клеточный рост, таких, как рак и другие клеточные пролиферативные заболевания, включающие, например, воспаление (как, например, доброкачественная гипертрофия простаты, семейный аденоматоз, полипоз, нейрофиброматоз, атеросклероз, легочный фиброз, артрит, псориаз, воспалительная болезнь кишечника, инфекционное отторжение трансплантата), вирусные инфекции (включая, но не лимитируя, вирус герпеса, вирус оспы, вирус Эпштейна-Барра), аутоиммунные заболевания (например, волчанка, ревматоидный артрит, псориаз, воспалительная болезнь кишечника), нейродегенеративные заболевания (включая, но не лимитируя, болезнь Альцгеймера) и нейродегенеративные болезни (например, болезнь Паркинсона, амиотрофический латеральный склероз, пигментное воспаление сетчатки, спинная мускульная атрофия и церебральная дегенерация).
Несколько различных классов небольших молекул идентифицированы, как ингибиторы Cdks: оломуцин и другие аналоги пурина, флавопиридол, стауроспорин, UCN-01 и другие индолокарбазолы, 9-гидроксиэллиптицин, индирубин, пауллоны, диарилмочевины, хиназолины, индопиразолы, [2,3-d]пиридопиримидины, фаскаплизин, аминотиазолы, диаминотиазолы, птеридиноны и пиразолы или пример (Carlson et al., Cancer Res., 1996, 56, 2973-2978; De Azevedo et al., Eur. J. Biochem., 1997, 243, 518-526; Bridges, A.J., Exp. Opin. Ther. Patents, 1955, 5, 124512257; Reinhold Cancer Res., 1998, 58, 704-710, 3803-3807; Kakeya, H. et al., Cancer Res., 1998, 58, 704-710; Harper, J.W., Cancer Surveys, 1997, 29, 91-107; Harrington, E.A. et al., Proc. Natl. Acad. Sci. USA, 1998, 95, 11945-11950; Meijer, L., Eur. J. Biochem., 2000, 267, 1-13; Garrett, M.D. et al., Current Opin. Genetics Develop., 1999, 9, 104-111; Mgbonyebi, O, P. et al., Cancer Res., 1999, 59, 1903-1910; Hoessel en al., Nanure Cell Biology., 1999, 1, 60-67; Zaherevitz et al., Cancer Res., 1999, 59, 2466-2569; Honma, Т. et al., 221St National ACS Meeting, 2001: Medi 136; Sielecki, N.V. et al., Bioorg. Med. Chem. Lett., 2001, 11, 1157-1160; Nugiel, D., J. Med. Chem., 2001, 44, 1334-1336; Fry, D.W. et al., J. Biol. Chem., 2001, 276, 16617-15523; Sony, R., et al., Biochem. Biophys. Res. Commun., 2000, 275, 877; Ryu, C-K., et al., Bioorg. Med. Chem. Lett., 2000, 10, 461; Jeong, H-W., et al., Bioorg. Med. Chem. Lett., 2000, 10, 1819; Toogood et al., J. Med. Chem., 2000, 43, 4606-4616; Chong, W., Fischer, Current Opin. In Drug Discov. and Develop., 2001, 4, 623-634, WO 0009921845, Toogood P., WO 0119825, Toogood P., WO 0138315, Reich S.H., WO 0179198, Webster, K. US 6262096.
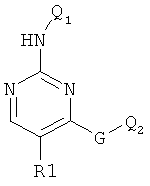
Класс диаминопиримидинов представлен соединениями формулы
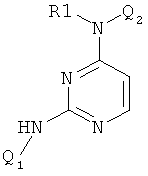
ингибирующими, как установлено, Cdk4 и FAK3. См. WO 0012485 (фирма Astra Zeneca).
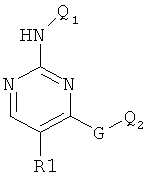
Заявка WO 9118887 (фирма Smith Kline Beecham) относится к диаминопиримидинам формулы
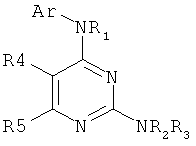
ингибирующим желудочную секрецию.
Заявка WO 0039101 (фирма Astra Zeneca) относится к пиримидиновым соединениям формулы
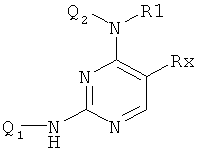
действующим как противораковые агенты.
Заявка WO 0164653 (фирма Astra Zeneca) относится к пиримидиновым соединениям формулы
действующим как Cdk ингибиторы и FAK ингибиторы.
Заявка WO 0164654 (фирма Astra Zeneca) относится к пиримидиновым соединениям формулы
действующим как Cdk ингибиторы и FAK ингибиторы.
Кроме того, заявка WO 0164656 (фирма Astra Zeneca) относится к пиримидиновым соединениям формулы
действующим также как Cdk ингибиторы и FAK ингибиторы.
Обзор соединений, ингибирующих метаболический путь Cdk4/циклин D представлен в: Yarris, W. и Wilkinson, S., Emerging Drugs., 2000, 5, 287-297; Dumas, J., Exp. Opin. Ther., Patents, 2000, 11, 405-429; Sielecki T.J. Med. Chem., 2000, 43, 1-18.
Настоящее изобретение предлагает новые пиразолпиримидины, имеющие формулу (I):
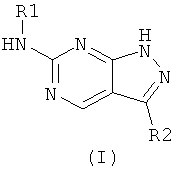
или их фармацевтически приемлемые соли или сложные эфиры, где
R1 выбирают из группы, включающей:
(а) гетероцикл, который может содержать до четырех заместителей, независимо выбранных из группы, включающей:
(i) низший алкил, который может быть замещен ОН, CO2R3, COR4, CONR5R6, NR5R6, OR7 или S(O)nR8; и
(ii) CO2R3, COR4, CONR5R6, NR5R6, OR7 или S(O)nR8;
(б) арил, который может содержать до четырех заместителей, независимо выбранных из группы, включающей:
(i) S(O)nR8, NR5R6, низший алкил, OR7, галоген или низший алкил, который может быть замещен ОН, CO2R3, COR4, CONR5R6, NR5R6 или OR7;
(ii) COOH; и
(iii) карбонил, замещенный низшим алкилом, OR7 или NR5R6;
(в) циклоалкил, который может быть замещен OR7, NR5R6 или S(O)nR8; и
(г) низший алкил, который может быть замещен:
(i) OR7, NR5R6, S(O)nR8, NHS(O)nR8 или CO2R3;
(ii) гетероциклом, который может быть замещен низшим алкилом, CO2R3 или S(O)nR8;
(iii) гетероарилом, который может быть замещен низшим алкилом, CO2R3 или S(O)nR8;
и
(iv) арилом, который может быть замещен низшим алкилом, CO2R3, галогеном, COR4, или NR5R6;
R2 выбирают из группы, включающей:
(i) H;
(ii) низший алкил или низший алкил, который может быть замещен ОН, CO2R3, COR4, CONR5R6, NR5R6, OR7;
(iii) арил, который может быть замещен галогеном, NO2, CN, NR5R6, низшим алкилом, низшей алкоксигруппой и низшим алкилом, замещенным галогеном или OR7;
(iv) гетероарил, который может быть замещен низшим алкилом, CO2R3, COR4, CONR5R6, NR5R6, галогеном и низшим алкилом, который может быть замещен CO2R3, COR4, CONR5R6, NR5R6, OR7;
R3 выбирают из группы, включающей:
(i) H;
(ii) низший алкил, который может быть замещен OR7, COR4, NR5R6 или CONR5R6;
(iii) арил, который может содержать до трех заместителей, независимо выбранных из группы, включающей низший алкил, галоген и NR5R6; и
(iv) циклоалкил, который может быть замещен ОН или NH2;
R4 выбирают из группы, включающей:
(i) H; и
(ii) низший алкил, который может быть замещен OR7 или NR5R6;
R5 и R6 независимо друг от друга выбирают из группы, включающей:
(i) H;
(ii) низший алкил, который может быть замещен ОН, СО2R3, CONR10R11, SO2R3, OR7, NR10R11, гетероциклом или гетероарилом;
(iii) циклоалкил, который может быть замещен CO2R3, CONR10R11, SO2R4, OR7 или NR10R11;
(iv) арил, который может быть замещен СО2R3, CONR10R11, SO2R3, OR7, NR10R11, галогеном, низшим алкилом и низшим алкилом, замещенным галогеном, CO2R3, CONR10R11, OR7, NR10R11, или ОН;
(v) SO2R3,
(vi) CO2R3, и
(vii) COR3, или
альтернативно, NR5R6 может образовать кольцо с общим числом кольцевых атомов от 3 до 7, включающих в дополнение к атому азота, к которому присоединены R5 и R6, углеродные кольцевые атомы, при этом названные углеродные кольцевые атомы необязательно заменены одним или более дополнительными N или О кольцевыми атомами или группой SO2, и названные кольцевые атомы необязательно при этом замещены ОН, оксогруппой, NR5R6, низшим алкилом и низшим алкилом, замещенным OR7;
R7 выбирают из группы, включающей Н и низший алкил, необязательно замещенный NR5R6 или OR9;
R8 выбирают из группы, включающей:
(i) арил, который может быть замещен CO2R3, CONR5R6, SO2R3, OR7, NR5R6, галогеном, низшим алкилом или низшим алкилом, замещенным галогеном, CO2R3, CONR5R6, OR7, NR5R6 и ОН;
(ii) гетероарил, который может быть замещен CO2R3, CONR5R6, SO2R3, OR7, NR5R6, галогеном, низшим алкилом или низшим алкилом, замещенным галогеном, CO2R3, CONR5R6, OR7, NR5R6 или ОН;
(iii) NR5R6,
(iv) низший алкил, который может быть замещен галогеном, CO2R3, CONR5R6, OR7, NR5R6 или ОН; и
(v) гетероцикл, который может быть замещен CO2R3, COR3, SO2R3, CONR5R6, OR7 или NR5R6;
R9 выбирают из группы, включающей Н, низший алкил, и низший алкил, замещенный ОН или галогеном;
R10 и R11 независимо друг от друга выбирают из группы, включающей:
(i) H;
(ii) низший алкил, который может быть замещен ОН, CO2R3, CONR5R6, SO2R3, OR7, NR5R6, гетероциклом или гетероарилом;
(iii) циклоалкил, который может быть замещен СО2R3, CONR5R6, SO2R3, OR7 или NR5R6;
(iv) арил, который может быть замещен СО2R3, CONR5R6, SO2R3, OR7, NR5R6, галогеном, низшим алкилом или низшим алкилом, замещенным галогеном, СО2R3, CONR5R6, OR7, NR5R6, или ОН;
(v) SO2R3,
(vi) CO2R3, и
(vii) COR3, или
альтернативно, NR10R11 может образовать кольцо с общим числом кольцевых атомов от 3 до 7, включающих в дополнение к атому азота, к которому присоединены R10 и R11, углеродные кольцевые атомы, при этом названные углеродные кольцевые атомы необязательно заменены одним или более дополнительными N или О кольцевыми атомами или группой SO2, и названные кольцевые атомы при этом необязательно замещены ОН, оксогруппой, NR5R6, низшим алкилом и низшим алкилом, замещенным OR7;
и n=1 или 2.
Эти соединения ингибируют циклин-зависимые киназы, наиболее предпочтительно, циклин-зависимую киназу 1 (Cdk1), циклин-зависимую киназу 2 (Cdk2) и циклин-зависимую киназу 4 (Cdk4). Эти соединения и их фармацевтически приемлемые соли и сложные эфиры проявляют антипролиферативную активность и используются, среди прочих, при лечении или контролировании рака, в частности твердых опухолей.
Настоящее изобретение предлагает также фармацевтические композиции, включающие одно или более соединений по изобретению или их фармацевтически приемлемые соли или сложные эфиры, и фармацевтически приемлемый носитель или наполнитель.
Настоящее изобретение предлагает далее способ лечения или контролирования рака, более предпочтительно лечения или контролирования твердых опухолей, наиболее предпочтительно, лечения или контролирование опухолей грудной железы, легкого, толстой кишки и простаты, посредством введения пациенту, нуждающемуся в таком лечении, эффективного количества соединения формулы I или его фармацевтически приемлемых соли или сложного эфира.
Наконец, настоящее изобретение относится к новым промежуточным соединениям, применяемым при получении соединения формулы I.
В настоящем описании используются термины, имеющие следующие значения.
«Арил» означает моновалентный, моноциклический или бициклический ароматический карбоциклический углеводородный радикал, предпочтительно, 6-10-членную ароматическую кольцевую систему. Предпочтительные арильные группы включают, не лимитируя, фенил, нафтил, толил и ксилил.
«Карбонил» означает радикал С=O.
«Циклоалкил» означает неароматический, частично или полностью насыщенный моновалентный циклический углеводородный радикал, содержащий от 3 до 8 атомов. Примеры циклоалкильных групп включают циклопропил, циклобутил, циклопентил и циклогексил.
«Эффективное количество» означает количество, являющееся эффективным для профилактики, облегчения или улучшения симптомов болезни или продления жизни субъекта, подлежащего лечению.
«Галоген» обозначает фтор, хлор, бром или йод, предпочтительно фтор или хлор, более предпочтительно фтор.
«Гетероатом» означает атом, выбранный из N, О или S.
«Гетероарил» означает ароматическую гетероциклическую кольцевую систему, содержащую до двух колец. Предпочтительные гетероароматические группы включают, не лимитируя, тиенил, фурил, индолил, пирролил, пиридинил, пиразинил, оксазолил, тиаксолил, хинолинил, пиримидинил, имидазолил, бензофуранил и тетразолил.
«Гетероцикл» или «гетероциклил» означают насыщенный или частично ненасыщенный неароматический циклический радикал, содержащий от 3 до 8 циклических атомов, в котором от 1 до 3 циклических атомов являются гетероатомами, выбранными из азота, кислорода, S(O)n (где n обозначает целое число от 0 до 2), или их комбинацию, при этом оставшиеся кольцевые атомы являются атомами углерода. Примерами предпочтительных гетероциклов являются пиперидин, пиперазин, пирролидин, морфолин, индолин, тетрагидропиран, тиоморфолин, пентаметиленсульфид и пентаметиленсульфон.
«IC50» относится к концентрации отдельного соединения по изобретению, требуемой для ингибирования 50% специфической измеренной активности. Величина IC50 может быть измерена, среди прочих, как описано в примерах.
«КI» относится к измерению термодинамического связывания лиганд/ингибитор (то есть соединения по изобретению) с белком-мишенью. KI можно измерить, среди прочего, как описано в примерах.
«Низший алкил» отдельно, или в сочетании с другим термином, например низший алкил-гетероцикл, означает линейный или разветвленный насыщенный алифатический углеводород, содержащий от 1 до 6, предпочтительно, от 1 до 4 атомов углерода. Типичные низшие алкильные группы включают метил, этил, пропил, изопропил, бутил, трет-бутил, 2-бутил, пентил, гексил и им подобные.
«Низшая алкоксигруппа» отдельно, или в сочетании с другим термином, например низшая алкоксигруппа-гетероцикл, означает линейный или разветвленный насыщенный алифатический алканол, содержащий от 1 до 6, предпочтительно, от 1 до 4 атомов углерода. Типичные низшие алкоксигруппы включают метоксигруппу, этоксигруппу, пропоксигруппу, изопропоксигруппу, бутоксигруппу, трет-бутоксигруппу, 2-бутоксигруппу, пентоксигруппу, гексоксигруппу и им подобные.
«Оксогруппа» означает = O.
Под «фармацевтически приемлемым сложным эфиром» подразумевают стандартно получаемые сложные эфиры соединения формулы I, имеющего карбоксильную группу, сохраняющие биологическую активность и свойства соединения формулы I и расщепляющиеся in vivo (в организме) до соответствующей карбоновой кислоты. Дополнительная информация относительно примеров и применения сложных эфиров для производства фармацевтических соединений имеется в Design of Prodrugs. Bundgaard H ed. (Elsevier, 1985), а также в H. Ansel et. al., Pharmaceutical Dosage Forms и Drug Delivery Systems (6th Ed. 1995), стр.108-109; Krogsgaard-Larsen, et. al., Textbook of Drug Design and Development (2d Ed. 1996) стр.152-191.
Термин «фармацевтически приемлемая соль» относится к стандартным кислотно-аддитивным или основно-аддитивным солям, которые сохраняют биологическую эффективность и свойства соединений формулы I и образуются из соответствующих нетоксичных органических и неорганических кислот или органических или неорганических оснований. Примеры кислотно-аддитивных солей включают соли, полученные из неорганических кислот таких, как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, сульфаминовая кислота, фосфорная кислота и азотная кислота, и соли, полученные из органических кислот таких, как п-толуолсульфокислота, салициловая кислота, метансульфоновая кислота, щавелевая кислота, янтарная кислота, лимонная кислота, яблочная кислота, молочная кислота, фумаровая кислота, и подобных им. Примеры основно-аддитивных солей включают соли, полученные из гидроксидов аммония, калия, натрия и четвертичного аммониевого основания такого, как, например, гидроксид тетраметиламмония. Технология химической модификации фармацевтических соединений (например, лекарств) в соли для получения соединений с улучшенной физической и химической стабильностью, гигроскопичностью, сыпучестью и растворимостью хорошо известна химикам-фармацевтам. См., например, Н. Ansel et. al., Pharmaceutical Dosage Forms and Drug Delivery Systems (6th Ed. 1995) стр.196 и 1456-1457.
Термин «фармацевтически приемлемый» как, например, фармацевтически приемлемый носитель, наполнитель и т.п., подразумевает, что они фармакологически приемлемы и практически нетоксичны по отношению к субъекту, которому вводится данное соединение.
Термин «замещенный» подразумевает, что замещение может иметь место в одном или более положениях и, если не указано иначе, заместители на каждом участке замещения независимо выбраны из определенных вариантов.
Термин «терапевтически эффективное количество» подразумевает количество, по крайней мере, одного определенного соединения формулы I или его фармацевтически приемлемых соли или сложного эфира, которое существенно ингибирует пролиферацию и/или предотвращает дифференцировку опухолевой человеческой клетки, включая линии клеток человеческой опухоли.
Настоящее изобретение предлагает соединения формулы (I):
или их фармацевтически приемлемые соли или сложные эфиры, где
R1 выбирают из группы, включающей:
(а) гетероцикл, который может содержать до четырех заместителей, независимо выбранных из группы, включающей:
(i) низший алкил, который может быть замещен ОН, CO2R3, COR4, CONR5R6, NR5R6, OR7 или S(O)nR8; и
(ii) CO2R3, COR4, CONR5R6, NR5R6, OR7 или S(O)nR8;
(б) арил, который может содержать до четырех заместителей, независимо выбранных из группы, включающей:
(i) S(O)nR8, NR5R6, низший алкил, OR7, галоген или низший алкил, который может быть замещен ОН, CO2R3, COR4, CONR5R6, NR5R6 или OR7;
(ii) COOH; и
(iii) карбонил, замещенный низшим алкилом, OR7 или NR5R6;
(в) циклоалкилы, которые могут быть замещены OR7, NR5R6 или S(O)nR8; и
(г) низшие алкилы, которые могут быть замещены:
(i) OR7, NR5R6, S(O)nR8, NHS(O)nR8 или CO2R3;
(ii) гетероциклами, которые могут быть замещены низшим алкилом, CO2R3 или S(O)nR8;
(iii) гетероарилами, которые могут быть замещены низшим алкилом, CO2R3 или S(O)nR8; и
(iv) арилами, которые могут быть замещены низшим алкилом, CO2R3, COR4, галогеном или NR5R6.
Предпочтительно, R1 обозначает гетероцикл, выбранный из группы, включающей пиперидин, пиперазин или пирролидин; или арил, выбранный из группы, включающей фенил, толил и ксилил; или циклоалкил, выбранный из группы, включающей циклопропил, циклобутил, циклопентил и циклогексил; или низший алкил, выбранный из группы, включающей метил, этил, пропил, изопропил, бутил, трет-бутил, 2-бутил, пентил или гексил. Более предпочтительно, R1 выбирают из группы, включающей замещенный или незамещенный пиперидин, фенил и
С2-С5алкильные группы. Более предпочтительно, R1 замещен SО2СН3, СН3, СООСН2СН3, SO2NH2, F, ОСН3, ОН, NH2, N(СН3).
R2 выбирают из группы, включающей:
H;
низший алкил или низший алкил, который может быть замещен ОН, CO2R3, COR4, CONR5R6;
арил, который может быть замещен галогеном, NO2, CN, NR5R6, низшим алкилом, низшей алкоксигруппой и низшим алкилом, замещенным галогеном или OR7;
гетероарил, который может быть замещен низшим алкилом, CO2R3, COR4, CONR5R6, NR5R6, галогеном и низшим алкилом, который может быть замещен CO2R3, COR4, CONR5R6, NR5R6, OR7.
Предпочтительно, R2 обозначает водород; или низший алкил, выбранный из группы, включающей метил, этил, пропил, изопропил, бутил, трет-бутил, 2-бутил, пентил и гексил; или арил, выбранный из группы, включающей фенил, толил и ксилил. Более предпочтительно, R2 выбирают из группы, включающей водород, метил и фенил, который может быть замещен фтором или метоксигруппой.
R3 выбирают из группы, включающей:
Н;
низший алкил, который может быть замещен OR7, COR4, NR5R6 или CONR5R6;
арил, который может содержать до трех заместителей, независимо выбранных из группы, включающей низший алкил, галоген и NR5R6; и
циклоалкил, который может быть замещен ОН или NH2.
Предпочтительно, R3 обозначает водород или низший алкил, выбранный из группы, включающей метил, этил, пропил, изопропил, бутил, трет-бутил, 2-бутил, пентил и гексил. Более предпочтительно, R3 обозначает водород, метил, этил, пропил, или изопропил.
R4 выбирают из группы, включающей:
Н; и
низший алкил, который может быть замещен OR7 или NR5R6.
Предпочтительно, R4 обозначает водород или низший алкил, выбранный из группы, включающей метил, этил, пропил, изопропил, бутил, трет-бутил, 2-бутил, пентил и гексил. Более предпочтительно, R4 обозначает водород, метил, этил, пропил, или изопропил.
R5 и R6 независимо друг от друга выбирают из группы, включающей:
Н;
низший алкил, который может быть замещен ОН, CO2R3, CONR10R11, SO2R3, OR7, NR10R11, гетероциклом или гетероарилом;
циклоалкил, который может быть замещен CO2R3, CONR10R11, SO2R4, OR7 или NР10R11;
арил, который может быть замещен CO2R3, CONR10R11, SO2R3, OR7, NR10R11, галогеном, низшим алкилом и низшим алкилом, замещенным галогеном, CO2R3, CONR10R11, OR7, NR10R11 или ОН;
SO2R3,
CO2R3, и
COR3.
Альтернативно, NR5R6 может образовать кольцо с общим числом кольцевых атомов от 3 до 7, включающих в дополнение к атому азота, к которому присоединены R5 и R6, углеродные кольцевые атомы, при этом названные углеродные кольцевые атомы необязательно заменены одним или более дополнительными N или О кольцевыми атомами или группой SO2, и названные кольцевые атомы необязательно при этом замещены ОН, оксогруппой, NR5R6, низшим алкилом и низшим алкилом, замещенным OR7.
Предпочтительно, R5 и R6 независимо друг от друга выбирают из группы, включающей водород или низший алкил, выбранный из группы, включающей метил, этил, пропил, изопропил, бутил, трет-бутил, 2-бутил, пентил и гексил. Более предпочтительно, R5 и R6 обозначают водород, метил, этил, пропил или изопропил.
R7 выбирают из группы, включающей Н и низший алкил, необязательно замещенный NR5R6 или OR9.
Предпочтительно, R7 обозначает водород или низший алкил, выбранный из группы, включающей метил, этил, пропил, изопропил, бутил, трет-бутил, 2-бутил, пентил и гексил. Более предпочтительно, R7 обозначает водород, метил, этил, пропил, или изопропил.
R8 выбирают из группы, включающей:
арил, который может быть замещен CO2R3, CONR5R6, SO2R3, OR7, NR5R6, галогеном, низшим алкилом или низшим алкилом, замещенным галогеном, CO2R3, CONR5R6, OR7, NR5R6 и ОН;
гетероарил, который может быть замещен CO2R3, CONR5R6, SO2R3, OR7, NR5R6, галогеном, низшим алкилом или низшим алкилом, замещенным галогеном, CO2R3, CONR5R6, OR7, NR5R6 или ОН;
NR5R6,
низший алкил, который может быть замещен галогеном, CO2R3, CONR5R6, OR7, NR5R6 или ОН; и
гетероцикл, который может быть замещен CO2R3, COR3, SO2R3, CONR5R6, OR7 или NR5R6.
Предпочтительно, R8 обозначает водород или низший алкил, выбранный из группы, включающей метил, этил, пропил, изопропил, бутил, трет-бутил, 2-бутил, пентил и гексил. Более предпочтительно, R8 обозначает водород, метил, этил, пропил, или изопропил.
R9 выбирают из группы, включающей Н, низший алкил, и низший алкил, замещенный ОН или галогеном.
Предпочтительно, R9 обозначает водород или низший алкил, выбранный из группы, включающей метил, этил, пропил, изопропил, бутил, трет-бутил, 2-бутил, пентил и гексил. Более предпочтительно, R9 обозначает водород, метил, этил, пропил, или изопропил.
R10 и R11 независимо друг от друга выбирают из группы, включающей:
Н;
низший алкил, который может быть замещен ОН, СО2R3, CONR5R6, SO2R3, OR7, NR5R6, гетероциклами или гетероарилами;
циклоалкил, который может быть замещен СО2R3, CONR5R6, SO2R3, OR7 или NR5R6;
арил, который может быть замещен СО2R3, CONR5R6, SO2R3, OR7, NR5R6, галогеном, низшим алкилом или низшим алкилом, замещенным галогеном, CO2R3, CONR5R6, OR7, NR5R6 или ОН;
SO2R3,
CO2R3, или
COR3.
Альтернативно, NR10R11 может образовать кольцо с общим числом кольцевых атомов от 3 до 7, включающих в дополнение к атому азота, к которому присоединены R10 и R11, углеродные кольцевые атомы, при этом названные углеродные кольцевые атомы необязательно заменены одним или более дополнительными N или О кольцевыми атомами или группой SO2, и названные кольцевые атомы при этом необязательно замещены ОН, оксогруппой, NR5R6, низшим алкилом и низшим алкилом, замещенным OR7.
Предпочтительно, R10 и R11 независимо друг от друга выбирают из группы, включающей водород или низший алкил, выбранный из группы, включающей метил, этил, пропил, изопропил, бутил, трет-бутил, 2-бутил, пентил и гексил. Более предпочтительно, R10 и R11 обозначают водород, метил, этил, пропил или изопропил.
Примеры соединений формулы (I) включают:
[3-(2,3-дифтор-6-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин (пример 16);
(1-метансульфонилпиперидин-4-ил)-(1Н-пиразоло[3,4-d]пиримидин-6-ил)амин (пример 19);
(1-метилпиперидин-4-ил)-(3-метил-1Н-пиразоло[3,4-d]пиримидин-6-ил)амин (пример 23);
[3-(5-фтор-2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин (пример 25);
[3-(5-фтор-2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин (пример 26);
[3-(5-фтор-2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин (пример 27);
(1-метансульфонилпиперидин-4-ил)-[3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидин-6-ил]амин; соединение с трифторуксусной кислотой (пример 28);
[3-(2,6-дифторфенил)-1H-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин (пример 29);
(1-метансульфонилпиперидин-4-ил)-(3-фенил-1H-пиразоло[3,4-d]пиримидин-6-ил)амин (пример 30);
[3-(3-фторфенил)-1H-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин (пример 31);
4-[3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидин-6-иламино]бензолсульфонамид; соединение с трифторуксусной кислотой (пример 32);
[3-(2,6-дифторфенил)-1H-пиразоло[3,4-d]пиримидин-6-ил]-(4-фторфенил)амин (пример 33);
2-[3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидин-6-иламино]этанол (пример 34);
[3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидин-6-ил]пентан-1,5-диамин трифторуксусной кислоты (пример 35);
N'-[3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидин-6-ил]-N,N-диметилпропан-1,3-диамин (пример 36);
N-[3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидин-6-ил]-N,N-диметилпентан-1,5-диамин трифторуксусной кислоты (пример 37);
[3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидин-6-ил]фениламин (пример 38);
(4-метоксифенил)-[3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидин-6-ил]амин трифторуксусной кислоты (пример 39);
[3-(2,6-дифторфенил)-1H-пиразоло[3,4-d]пиримидин-6-ил]-(2-метоксифенил)амин трифторуксусной кислоты (пример 40);
4-(3-метил-1H-пиразоло[3,4-d]пиримидин-6-иламино]бензолсульфонамид трифторуксусной кислоты (пример 41);
2-(3-метил)-1H-пиразоло[3,4-d]пиримидин-6-иламино]этанол; соединение с трифторуксусной кислотой (пример 42);
этиловый эфир 4-[3-(3-фторфенил)-1H-пиразоло[3,4-d]пиримидин-6-иламино]пиперидин-1-карбоновой кислоты; соединение с трифторуксусной кислотой (пример 43);
(1-метилпиперидин-4-ил)-(3-метил-1H-пиразоло[3,4-d]пиримидин-6-ил)амин (пример 44).
Предпочтительные примеры соединений формулы (I) включают:
[3-(2,3-дифтор-6-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метан сульфонилпиперидин-4-ил)амин (пример 16);
(1-метансульфонилпиперидин-4-ил)-(1Н-пиразоло[3,4-d]пиримидин-6-ил)амин (пример 19);
[3-(5-фтор-2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин (пример 25);
[3-(5-фтор-2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин (пример 27);
(1-метансульфонилпиперидин-4-ил)-[3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидин-6-ил]амин; соединение с трифторуксусной кислотой (пример 28);
[3-(3-фторфенил)-1H-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин (пример 31);
4-[3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидин-6-иламино]бензолсульфонамид; соединение с трифторуксусной кислотой (пример 32);
Соединения, раскрытые в данном описании и соответствующие формуле (I), представленной выше, могут проявлять таутомерную или структурную изомерию. Следует иметь в виду, что изобретение включает в себя любую таутомерную или структурную изомерную форму этих соединений, или смеси таких форм и не ограничивается одной таутомерией или структурной изомерной формой, изображенной в формуле выше.
Соединения по настоящему изобретению могут быть получены любыми стандартными методами. Подходящие методы синтеза этих соединений приведены в примерах. В общем, соединения формулы (I) могут быть получены согласно любой из описанных ниже синтетических схем.
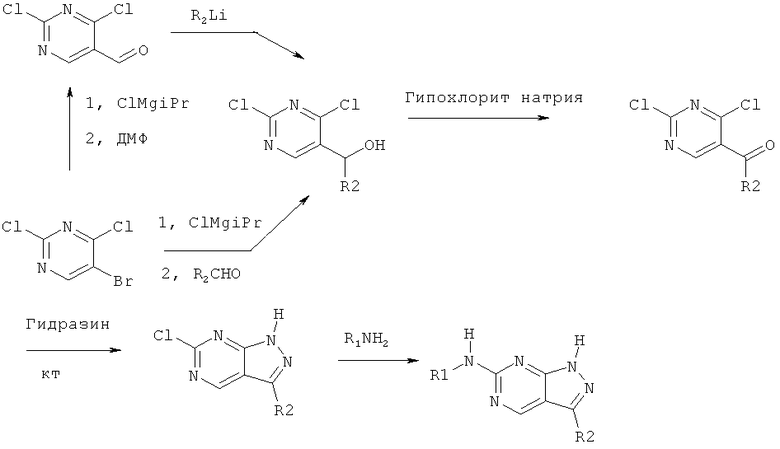
В общем, эти соединения могут быть получены в соответствии с синтетическими схемами, приведенными выше. Соответствующие методики для получения этих соединений приведены в примерах.
Необязательное разделение изомерных структур формулы (I) может быть проведено согласно известным методам таким, как, например, растворение или хиральная высокоэффективная жидкостная хроматография (известная также, как ВЭЖХ). Методы разделения хорошо известны и суммированы в «Enantiomers, Racemates and Resolutions» (Jacques, J. et al., John Willey and Sons, NY, 1981). Методы хиральной ВЭЖХ также хорошо известны и суммированы в «Separation of Enantiomers by Liquid Chromatographic Methods» (Pirkle, W.H. and Finn, J. In «Asymmetric Synthesis», vol.1, Morrison, J.D., Academic Press, Inc., NY 1983, pp.87-124).
Необязательное превращение соединения формулы (I), имеющего основной азот, в фармацевтически приемлемую кислотно-аддитивную соль может быть осуществлено с помощью стандартных методов. Например, соединение обрабатывают неорганической кислотой такой, как, например, хлористоводородная кислота, бромистоводородная кислота, серная кислота, фосфорная кислота, азотная кислота, или соответствующей органической кислотой такой, как уксусная кислота, лимонная кислота, винная кислота, метансульфоновая кислота, п-толуолсульфокислота и подобными им.
Необязательное превращение соединения формулы (I), имеющего карбоксильную группу, в фармацевтически приемлемую соль щелочного металла может быть осуществлено стандартными методами. Например, соединение обрабатывают неорганическим основанием таким, как гидроксид лития, гидроксид натрия, гидроксид калия и подобными им.
Необязательное превращение соединения формулы (I), имеющего карбоксильную группу, в фармацевтически приемлемый сложный эфир может быть осуществлено стандартными методами. Условия образования сложного эфира зависит от стабильности других функциональных групп в молекуле в реакционных условиях. Если другие части молекулы стабильны в кислых условиях, сложный эфир обычно получают нагреванием в растворе минеральной кислоты (например, серной кислоты) в спирте. Другие методы получения сложного эфира, которые могут быть подходящими, если молекулы нестабильны в кислых условиях, включают обработку соединения спиртом в присутствии конденсирующего агента и при необязательном присутствии дополнительных агентов, ускоряющих реакцию. Большое число таких конденсирующих агентов известно любому специалисту в области органической химии. Двумя примерами являются дициклогексилкарбодиимид и трифенилфосфин/диэтилазодикарбоксилат. В случае, когда в качестве конденсирующего агента используется дициклогексилкарбодиимид, реакцию обычно проводят с применением кислоты в спирте при необязательном присутствии каталитического количества (0-10 мол.%) N,N-диметиламинопиридина, в инертном растворителе типа галогенированного углеводорода (например, дихлорметана), при температуре от 0°С до комнатной температуры, предпочтительно, при комнатной температуре. В случае, когда в качестве конденсирующего агента используется трифенилфосфин/диэтилазодикарбоксилат, реакцию обычно проводят с применением кислоты в спирте, трифенилфосфина и диэтилазодикарбоксилата, в инертном растворителе типа эфира (например, тетрагидрофурана) или ароматического углеводорода (например, бензола), при температуре от 0°С до комнатной температуры, предпочтительно, около 0°С.
В альтернативном варианте настоящее изобретение включает фармацевтические композиции, содержащие по меньшей мере одно соединение формулы (I) или его фармацевтически приемлемые соль или сложный эфир и фармацевтически приемлемые наполнитель и/или носитель.
Эти фармацевтические композиции могут быть введены орально, например, в форме таблеток, таблеток в оболочке, драже, твердых или мягких желатиновых капсул, растворов, эмульсий или суспензий. Они могут быть также введены ректально, например, в форме суппозиториев, или парентерально, например, в форме инъекционных растворов. Фармацевтические композиции по настоящему изобретению, включающие соединения формулы (I) и/или их соли или сложные эфиры, могут быть получены методами, известными из уровня техники, то есть посредством смешения, инкапсуляции, растворения, гранулирования, эмульгирования, включения в гель, дражирования или лиофилизации. Эти фармацевтические препараты могут быть сформированы с помощью терапевтически инертных, неорганических или органических носителей. При этом в качестве носителей для таблеток, таблеток в оболочке, драже и твердых или мягких желатиновых капсул могут быть использованы лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или ее соли. Подходящими носителями для мягких желатиновых капсул служат растительные масла, воски и жиры. В зависимости от природы активной субстанции обычно не требуется никаких носителей в случае твердых желатиновых капсул. Подходящими носителями для приготовления растворов и сиропов являются вода, полиолы, сахароза, инвертный сахар и глюкоза. Подходящими носителями для инъекций служат вода, спирты, полиолы, глицерин, растительные масла, фосфолипиды и поверхностно-активные вещества. Подходящими носителями для суппозиториев являются натуральные или гидрированные масла, воски, жиры и полужидкие полиолы.
Фармацевтические препараты могут также содержать консервирующие агенты солюбилизирующие агенты, стабилизаторы, поверхностно-активные вещества, эмульгаторы, подсластители, красители, вкусовые агенты, соли для изменения осмотического давления, буферы, глазировочные агенты или антиоксиданты. Они могут также содержать другие терапевтически ценные субстанции, включая дополнительные активные ингредиенты, иные, чем соединения формулы (I).
Как отмечено выше, соединения по настоящему изобретению, включающие соединения формулы (I), применяются при лечении и контролировании нарушений клеточной пролиферации, включая химиопрофилактику рака. Химиопрофилактика подразумевает ингибирование развития начальной стадии рака посредством либо блокирования инициирования мутагенного случая, либо блокирования развития предраковых клеток, которые уже испытали травму от рецидива ингибирующей опухоли. Эти соединения и композиции, содержащие названные соединения, применяются, в частности, при лечении или контролировании твердых опухолей таких, как, например, опухоли грудной железы, толстой кишки, легкого и простаты.
Терапевтически эффективное количество соединения по настоящему изобретению обозначает количество соединения, являющееся эффективным для профилактики, облегчения или улучшения симптомов болезни или продления жизни субъекта, подлежащего лечению. Определение терапевтически эффективного количества находится в компетенции специалиста в данной области техники.
Терапевтически эффективное количество или дозирование соединения по настоящему изобретению может варьироваться в широких пределах и устанавливаться методом, известным из уровня техники. Такое дозирование должно регулироваться согласно индивидуальным требованиям в каждом особом случае, включая специфику соединения(ий), подлежащего введению, способ ведения, состояние, требующее лечения, а также пациента, подлежащего лечению. В общем, в случае орального или парентерального введения взрослому человеку с весом около 70 кг, суточная доза составляет приблизительно от 10 мг до 10000 мг, предпочтительно приблизительно от 200 мг до 1000 мг, хотя верхний предел при необходимости может быть превышен. Суточная доза может быть введена в виде разовой дозы, а в случае парентерального введения может применяться длительное внутривенное вливание.
Соединения по настоящему изобретению могут применяться в комбинации (совместное или раздельное введение) с известными противораковыми методами лечения такими, как радиационная терапия, или с цитостатическими или цитотоксическими агентами такими, как, например, не лимитируя, ДНК интерактивные агенты, как, например, цисплатин или доксорубицин; ингибиторы топоизомеразы II, как, например, этопозид; ингибиторы топоизомеразы I, как, например, СРТ-11 или топотекан; тубулин интерактивирующие агенты, как, например, паклитаксель, доцетаксель или эпотионы; гормональные агенты, как, например, тамоксифен; ингибиторы тимидилатсинтазы, как например, 5-фторурацил; и антиметаболиты такие, как метотрексат. Соединения формулы (I) могут быть также использованы в комбинации с модуляторами р53 трансактивации.
При формировании смешанной дозы описанная выше комбинация препаратов включает соединения по настоящему изобретению в пределах диапазона дозирования, описанного выше, и другой фармацевтически активный агент или лечение в пределах принятой для него дозы. Например, ранее описанный Cdk1 ингибитор оломуцин, совместно с хорошо известными цитотоксическими агентами индуцирует апоптоз. (J. Cell Sci., 1995, 108, 2829-2904). Соединения формулы (I) могут быть также введены последовательно с известными противораковыми или цитотоксическими агентами, когда сопутствующее введение или комбинация неуместны. Настоящее изобретение не лимитируется последовательностью введения: соединения формулы (I) могут быть введены либо до, либо после введения противоракового или цитотоксического агента. Тогда как, например, цитотоксическая активность Cdk ингибитора флавопиридола зависит от последовательности введения его с антираковым агентом (Cancer Research, 1997, 57, 3375).
Примеры
Следующие примеры иллюстрируют предпочтительные методы синтеза и применения соединений и препаратов по настоящему изобретению. Эти примеры и препараты служат иллюстрацией и не ограничивают изобретения. Следует понимать, что могут существовать другие варианты, которые входят в объем и соответствуют сущности изобретения, как определено в прилагающейся формуле изобретения.
Пример 1: (2,4-дихлорпиримидин-5-ил)-(2,3-дифтор-6-метоксифенил)метанол
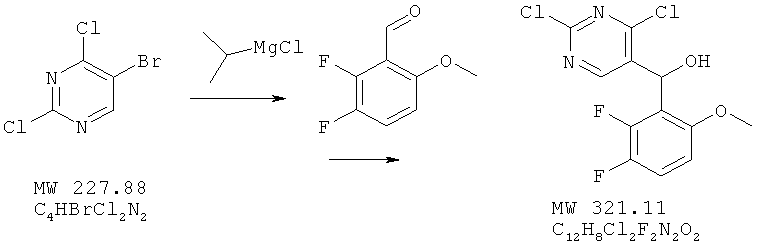
В перемешиваемый раствор 5-бром-2,4-дихлорпиримидина (фирма Aldrich, 3,02 г, 13,25 ммолей) в 20 мл ТГФ при -30°С вносят по каплям изопропилмагнийхлорид (фирма Aldrich, 2-молярный раствор в ТГФ, 7,12 мл, 13,25 ммолей) и перемешивают смесь в течение 20 мин. Затем добавляют 2,3-дифтор-6-метоксибензальдегид (фирма Matrix, 2,28 г, 13,25 ммолей), смесь нагревают до 0°С и перемешивают в течение 40 мин. Реакцию останавливают насыщенным раствором хлорида аммония и образовавшуюся смесь экстрагируют этилацетатом. Экстракт высушивают над сульфатом натрия. Растворитель удаляют на роторном испарителе, а твердое вещество обрабатывают гексаном (8 мл), отфильтровывают и высушивают, получая почти белое твердое вещество. Выход 3,42 г (80,5%). МС (М+Н)+, 322.
Пример 2: (2,4-дихлорпиримидин-5-ил)-(2,3-дифтор-6-метоксифенил)метанон
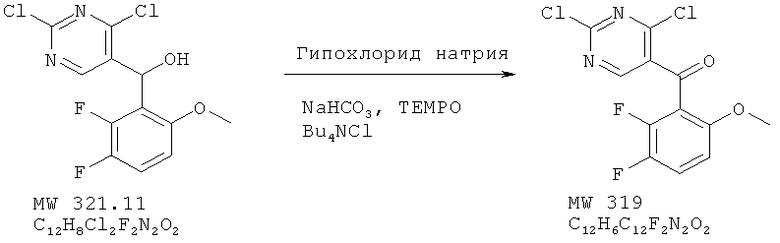
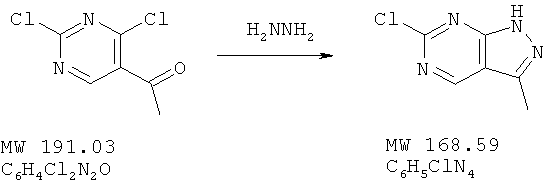
Спирт (2,00 г, 6,23 ммолей) растворяют в метиленхлориде (20 мл) и при перемешивании добавляют воду (2 мл), за которой следует гидрокарбонат натрия (235 мг, 2,8 ммолей), тетрабутиламмонийхлорид (фирма Aldrich, 60 мг, 0,18 ммолей) и TEMPO (фирма Aldrich, 10 мг, 0,062 ммоля). Смесь охлаждают затем до 0°С и медленно добавляют гипохлорит натрия (фирма Aldrich, активный CI2, 5,8%, 8,78 мл), а затем переносят в воду и экстрагируют метиленхлоридом. Экстракт высушивают над сульфатом натрия. Удаление растворителя дает продукт в виде бледно-желтого твердого вещества. Выход 1,84 г (92%). МС (М+Н)+, 319.
Пример 3: 6-хлор-3-(2,3-дифтор-6-метоксифенил)-1Н пиразоло[3,4-d]пиримидин
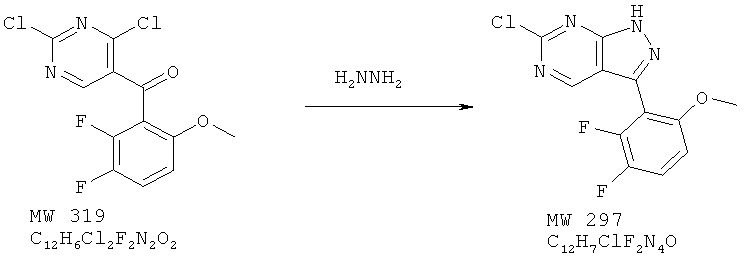
В перемешиваемый раствор кетона (638 мг, 2 ммоля) в ТГФ вносят гидразин (фирма Aldrich, 80 мг, 2,5 ммоля) и смесь перемешивают в течение 30 мин при КТ, а затем при 50°С в течение 1 ч. Растворитель удаляют, а твердое вещество промывают водой и высушивают, получая желтое твердое вещество. Выход 580 мг (97%). МС (М+Н)+, 297.
Пример 4: (2,4-дихдорпиримидин-5-ил)-(2-метоксифенил)метанол
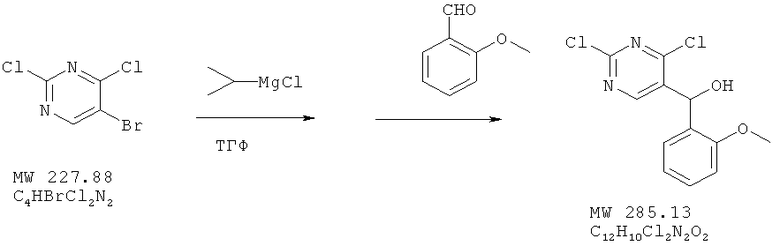
Соединение получают из 5-бром-2,4-дихлорпиримидина (фирма Aldrich) и 2-метоксибензальдегида (фирма Aldrich) с 77%-ным выходом методом, аналогичным описанному в примере 1.
ЯМР (300 МГц, CDCl3) δ 8,75 (s, 1H), 7,34 (d, t 1H, J=2 Гц, 8 Гц), 7,16 (d, d, 1H, J=1,8 Гц, 7,3 Гц), 6,96 (d, 1H, J=7,3 Гц), 6,95 (d, 1H, J=8 Гц), 6,21 (s, 1H), 3,85 (s, 3Н), 3,148 (расширенный s, 1H).
Пример 5: (2,4-дихлорпиримидин-5-ил)-(2-метоксифенил)метанон
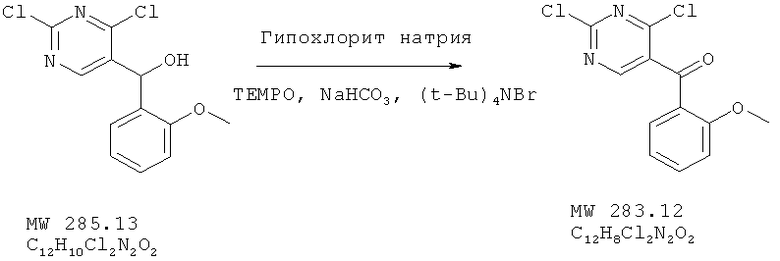
Соединение получено из (2,4-дихлорпиримидин-5-ил)-(2-метоксифенил)метанола (пример 4) с 96%-ным выходом методом, аналогичным описанному в примере 2.
ЯМР (300 МГц, CDCl3) δ 8,60 (s, 1H), 7,84 (d, d 1H, J=2 Гц, 8 Гц), 7,60 (d, d, 1H, J=2 Гц, 7,3 Гц), 7,12 (d, 1H, J=7,3 Гц), 6,95 (d, 1H, J=8 Гц), 3,67 (s, 3Н).
Пример 6: 6-хлор-3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин
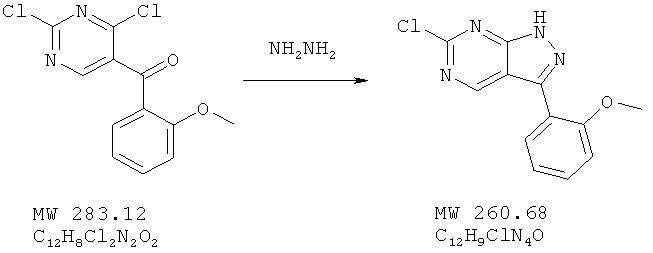
Соединение получено из (2,4-дихлорпиримидин-5-ил)-(2-метоксифенил)метанона (пример 5) с 35%-ным выходом методом, аналогичным описанному в примере 3.
MC(M+H)+, 261,11.
Пример 7: (2,4-дихлорпиримидин-5-ил)-(3-фторфенил)метанол
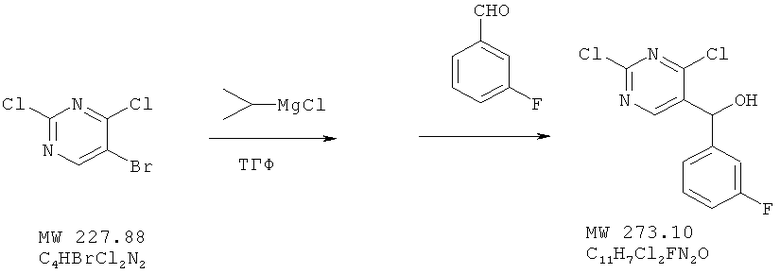
Соединение получено из 5-бром-2,4-дихлорпиримидина (фирма Aldrich) и 3-фторбензальдегида (фирма Aldrich) с 79%-ным выходом методом, аналогичным описанному в примере 1.
ЯМР (300 МГц, CDCl3) δ 8,85 (s, 1Н), 7,35 (m, 1H), 7,14 (m, 3H), 7,14 (m, 3H), 6,077 (s, 1H).
Пример 8: (2,4-дихлорпиримидин-5-ил)-(3-фторфенил)метанон
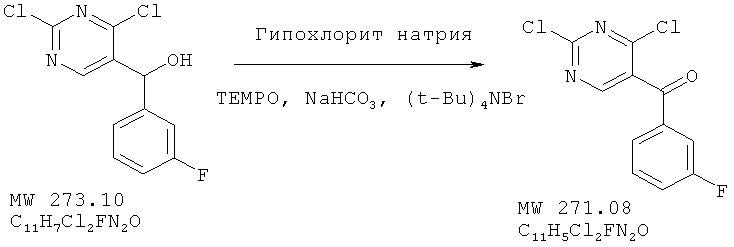
Соединение получено из (2,4-дихлорпиримидин-5-ил)-(3-фторфенил)метанола (пример 7) с 99%-ным выходом методом, аналогичным описанному в примере 2.
ЯМР (300 МГц, CDCl3) δ 8,632 (s, 1H), 7,52 (m, 4H), 7,43 (m, 1H), 5,303 (s, 1H).
Пример 9: 6-хлор-3-(3-фторфенил)-1Н-пиразоло[3,4-d]пиримидин
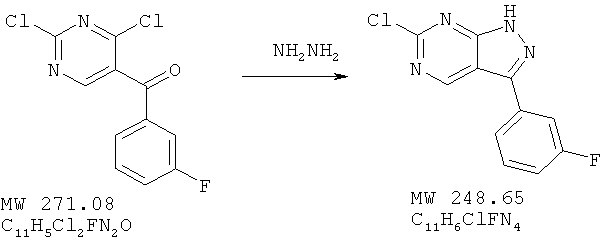
Соединение получено из (2,4-дихлорпиримидин-5-ил)-(3-фторфенил)метанона (пример 8) с 75%-ным выходом методом, аналогичным описанному в примере 3. (М+Н)+, 261 МС (М+Н)+, 261.
Пример 10: (2,4-дихлорпиримидин-5-ил)-(2,6-дифторфенил)метанол
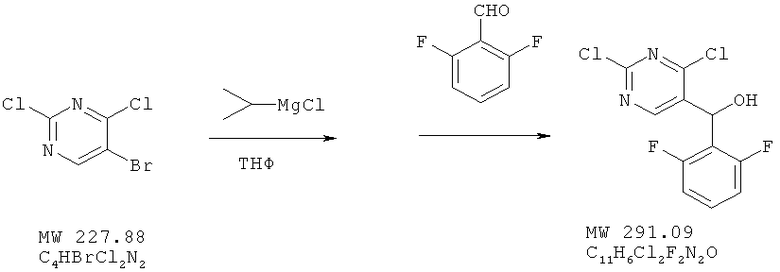
Соединение получено из 5-бром-2,4-дихлорпиримидина (фирма Aldrich) и 2,6-дифторбензальдегида (фирма Aldrich) с 88%-ным выходом методом, аналогичным описанному в примере 1.
ЯМР (300 МГц, CDCl3) δ 9,09 (s, 1Н), 7,35 (m, 1H), 7,92 (m, 2H), 6,35 (s, 1H), 2,67 (расширенный s, 1H).
Пример 11: (2,4-дихлорпиримидин-5-ил)-(2,6-дифторфенил)метанон
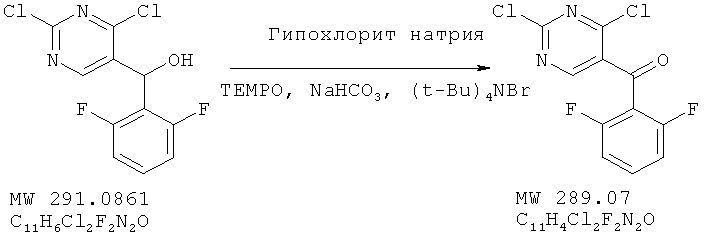
Соединение получено из (2,4-дихлорпиримидин-5-ил)-(2,6-дифторфенил)метанола (пример 10) с 98%-ным выходом методом, аналогичным описанному в примере 2.
ЯМР (300 МГц, CDCl3) δ 8,79 (s, 1Н), 7,56 (m, 1H), 7,04 (m, 2H).
Пример 12: 6-хлор-3-(2,6-дифторфенил)-1Н-пиразоло[3,4-d]пиримидин
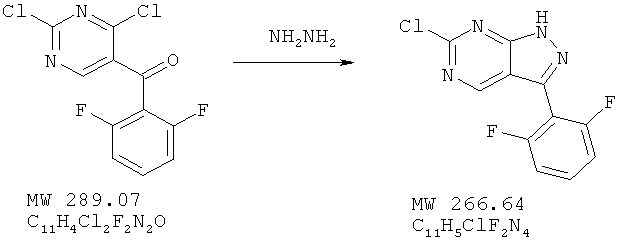
Соединение получено из (2,4-дихлорпиримидин-5-ил)-(2,6-дифторфенил)метанона (пример 11) с 71%-ным выходом методом, аналогичным описанному в примере 3. МС (М+Н)+, 267,08.
Пример 13: (2,4-дихлорпиримидин-5-ил)фенилметанол
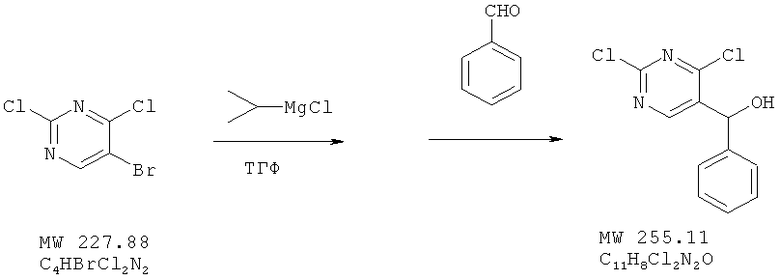
Соединение получено из 5-бром-2,4-дихлорпиримидина (фирма Aldrich) и бензальдегида (фирма Aldrich) с 79%-ным выходом методом, аналогичным описанному в примере 1.
ЯМР (300 МГц, CDCl3) δ 8,91 (s, 1H), 7,36 (m, 5H), 6,06 (d, 1H, J=3,0 Гц), 2,48 (расширенный m, 1H).
Пример 14: (2,4-дихлорпиримидин-5-ил)фенилметанон
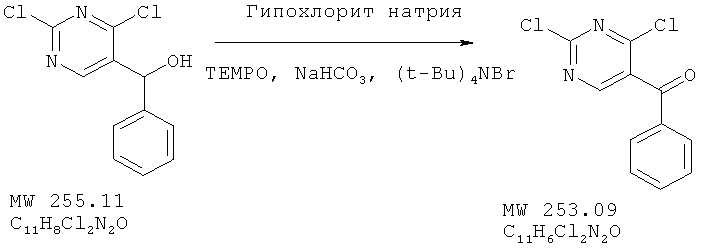
Соединение получено из (2,4-дихлорпиримидин-5-ил)фенилметанола (пример 13)
с 98%-ным выходом методом, аналогичным описанному в примере 2.
ЯМР (300 МГц, CDCl3) δ 8,62 (s, 1Н), 7,79 (m, 2H), 7,70 (m, 1H), 7,54 (m, 2H).
Пример 15: 6-хлор-3-фенил-1Н-пиразоло[3,4-d]пиримидин
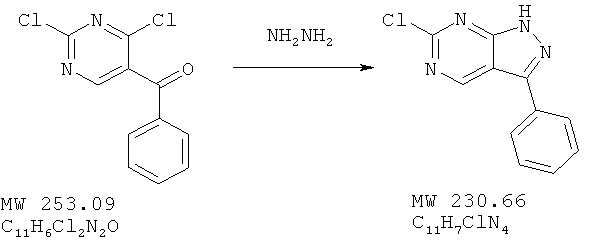
Соединение получено из (2,4-дихлорпиримидин-5-ил)фенил)метанона (пример 14) с 99%-ным выходом методом, аналогичным описанному в примере 3. MC(M+H)+, 231,08.
Пример 16: [3-(2,3-дифтор-6-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин
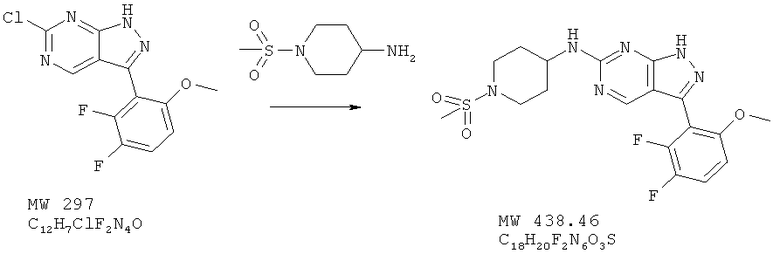
В раствор 6-хлор-3-(2,3-дифтор-6-метоксифенил)-1Н пиразоло[3,4-d]пиpимидинa (пример 3, 148 мг, 0,50 ммоля) в ДМФ (2,5 мл) вносят при перемешивании бикарбонат натрия (100 мг) и 1-метансульфонилпиперидин-4-иламин (125 мг, 0,70 ммоля) и перемешивают смесь при 100°С в течение 4 ч. Растворитель удаляют при пониженном давлении, а остаток обрабатывают водой. Твердое вещество отфильтровывают и высушивают. Перекристаллизация из смеси 2% метилового спирта/дихлорметана дает 92 мг (42%) почти белого вещества. МС (М+Н)+, 439.
Пример 17: 2,4-дихлорпиримидин-5-карбальдегид
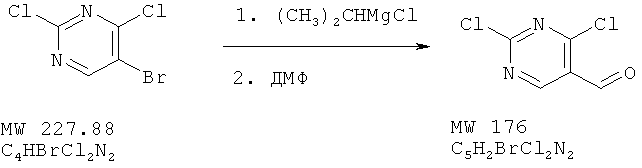
В перемешиваемый раствор 5-бром-2,4-дихлорпиримидина (фирма Aldrich, 454 мг, 2 ммоля) в ТГФ при -78°С вносят диизопропилмагнийхлорид (2-молярный раствор в ТГФ, 1,05 мл, 2,1 ммоля) и перемешивают смесь в течение 20 мин при -35°С. Затем раствор охлаждают до -78°С и добавляют 1 мл сухого ДМФ. Смесь перемешивают в течение 120 мин при -78°С, а затем останавливают реакцию с помощью 1-нормального раствора хлористоводородной кислоты. Смесь экстрагируют этилацетатом и высушивают над сульфатом натрия. Растворитель удаляют, а остаток хроматографируют, получая 102 мг (28%) коричневого твердого вещества.
ЯМР (300 МГц, CDCl3) δ (м.д.) 9,0 (s, 1H), 10,4 (s, 1H).
Пример 18: 6-хлор-1Н-пиразоло[3,4-d]пиримидин
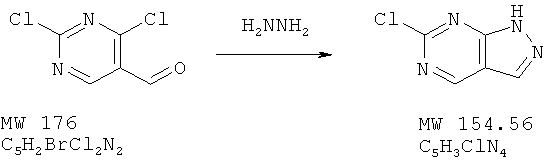
В перемешиваемый раствор гидразина (фирма Aldrich, 62 мг, 2 ммоля) в ТГФ (5 мл) вносят 2,4-дихлорпиримидин-5-карбальдегид (пример 17, 176 мг, 1 ммол) и перемешивают смесь при КТ в течение 30 мин. Затем смесь выливают в воду и экстрагируют этилацетатом. Экстракт высушивают над сульфатом натрия, растворитель удаляют, получая 128 мг (82%) оранжевого твердого вещества.
МС (М+Н)+ 155.
Пример 19: (1-метансульфонилпиперидин-4-ил)-(1Н-пиразоло [3,4-d]пиримидинамин
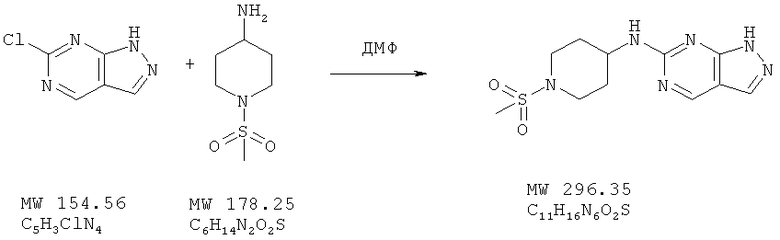
К перемешиваемому раствору 6-хлор-1Н-пиразоло[3,4-d] пиримидина (пример 18, 61 мг, 0,40 ммоля) в ДМФ (2,5 мл) добавляют бикарбонат натрия (60 мг) и 1-метансульфонилпиперидин-4-иламин (85 мг, 0,48 ммолей) и перемешивают смесь при 100°С в течение 15 ч. Растворитель удаляют при пониженном давлении, а остаток обрабатывают водой и экстрагируют этилацетатом. Экстракт высушивают над сульфатом натрия и концентрируют, получая твердое вещество, которое после очистки с помощью ВЭЖХ с обращенной фазой представляет собой почти белое твердое вещество. Выход 29 мг (29%). МС (М+Н)+, 297.
Пример 20: 1-(2,4-дихлорпиримидин-5-ил)этанол
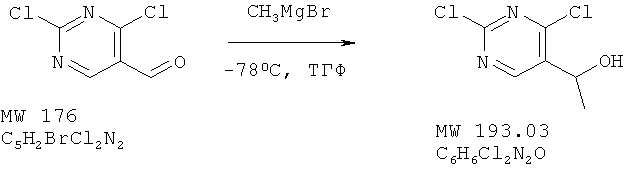
К перемешиваемому раствору 2,4-дихлорпиримидин-5-карбальдегида (пример 17, 1,01 г, 5,7 ммоля) в ТГФ (10 мл) при -78°С медленно добавляют метилмагнийбромид (фирма Aldrich, 3-молярный раствор в эфире, 6,27 ммолей, 2,1 мл) и дополнительно перемешивают смесь в течение 2 ч при -78°С. Затем останавливают реакцию с помощью 1-нормального раствора хлористоводородной кислоты (10 мл). Образовавшуюся смесь экстрагируют этилацетатом и высушивают над сульфатом натрия. Растворитель удаляют, получая 1,07 г (97%) коричневого твердого вещества. МС (М+Н)+, 193.
Пример 21: 1-(2,4-дихлорпиримидин-5-ил)этанон

К перемешиваемой суспензии 1-(2,4-дихлорпиримидин-5-ил)этанола (пример 20, 1,00 г, 5,2 ммоля) в смеси с метиленхлоридом (30 мл), ТГФ (8 мл) и водой (2 мл) добавляют тетрабутиламмонийхлорид (фирма Aldrich, 50 мг, 0,15 ммол), ТЕМПО (фирма Aldrich, 9 мг, 0,05 ммол) и бикарбонат натрия (208 мг, 2,48 ммолей). Смесь охлаждают до 0°С и при перемешивании добавляют colorex (фирма Aldrich, активный Cl2, 10-13%, 3,75 мл), а затем перемешивают в течение 2 ч при 0°С. Добавляют воду (10 мл) отделяют органический слой и высушивают над сульфатом натрия. После удаления растворителя остается масло, которое очищают хроматографией на силикагеле (1% MeOH/CH2Cl2), получая бесцветное масло. Выход 232 г (23%).
MC(M+H)+, 191.
Пример 22: 6-хлор-1-Н-пиразоло[3,4-d] пиримидин
К перемешиваемому раствору гидразина (фирма Aldrich, 102,4 мг, 3,2 ммоля) в ТГФ (5 мл) добавляют 1-(2,4-дихлорпиримидин-5-ил)этанон (пример 21, 191 г, 1 ммоль) и перемешивают смесь при КТ в течение 1 ч. Затем смесь выливают в воду (5 мл) и экстрагируют (трижды по 5 мл) смесью этилацетат/ТГФ в соотношении 1:1. Экстракт высушивают над сульфатом натрия и концентрируют, получая твердое вещество. Выход 126 мг (75%).
МС (М+Н)+, 169.
Пример 23: (1-метилпиперидин-4-ил)-(3-метил-1H-пиразоло [3,4-d]пиримидин-6-ил)амин
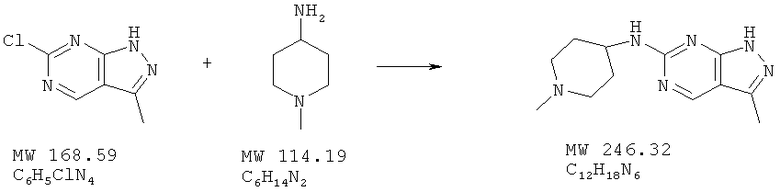
К перемешиваемому раствору 6-хлор-3-метил-1Н-пиразоло[3,4-d]пиримидина (пример 22, 56 мг, 0,3 ммоля) в ДМФ (1,5 мл) добавляют 1-метилпиперидин-4-иламин (46 мг, 0,40 ммолей) и перемешивают смесь при 100°С в течение 2 ч. Растворитель удаляют, а остаток хроматографируют (5% 7-нормального раствора аммиака в MeOH/CH2Cl2), получая 45 мг (61%) белого твердого вещества.
МС (М+Н)+, 247.
Пример 24: 6-хлор-3-(5-фтор-2-метоксифенил)-1Н-пиразоло[3,4-d] пиримидин
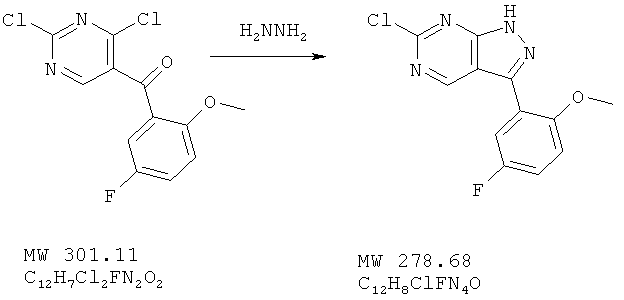
К перемешиваемому раствору (2,4-дихлорпиримидин-5-ил)-(5-фтор-2-метоксифенил)метанона (602 мг, 2 ммоля) в ТГФ (8 мл) добавляют гидразин (фирма Aldrich, 98%-ный, 64 мг, 2 ммоля) и перемешивают смесь в течение 20 мин, после чего прибавляют вторичную порцию гидразина (0,1 мл). Реакция заканчивается через 5 мин. Растворитель удаляют, твердое вещество промывают смесью вода/ацетонитрил в соотношении 3:1, получая 456 г (82%) желтого твердого вещества.
МС (М+Н)+, 278.
Пример 25: [3-(5-фтор-2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин
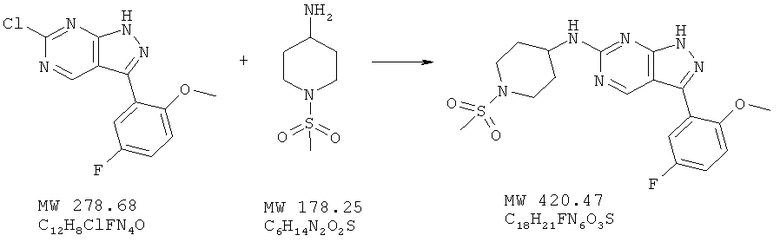
В перемешиваемый раствор 6-хлор-3-(5-фтор-2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидина (пример 24; 97 мг, 0,35 ммоля) в ДМФ (4 мл) вносят бикарбонат натрия (60 мг) и 1-метансульфонилпиперидин-4-иламин (93 мг, 0,52 ммоля) и перемешивают смесь при 100°С в течение 4 ч. Реакционную смесь охлаждают, и переносят в воду (60 мл). Твердое вещество отфильтровывают и высушивают, получая желтое твердое вещество, которое после очистки с помощью ВЭЖХ с обращенной фазой, представляет собой слегка желтое твердое вещество. Выход 52 мг (35%).
MC (M+Н)+, 421.
Пример 26: [3-(5-фтор-2-метоксифенил)-1Н-пиразоло [3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин
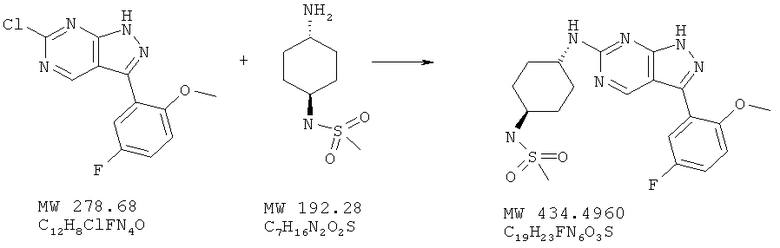
Тем же методом, описанном в примере 25, исходя из 6-хлор-3-(5-фтор-2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидина (пример 24; 97 мг, 0,35 ммоля) и N-(4-аминоциклогексил)метансульфонамида (101 мг, 0,52 ммоля), получают 70 мг (46%) желтого твердого вещества.
МС (М+Н)+, 435.
Пример 27: [3-(5-фтор-2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин
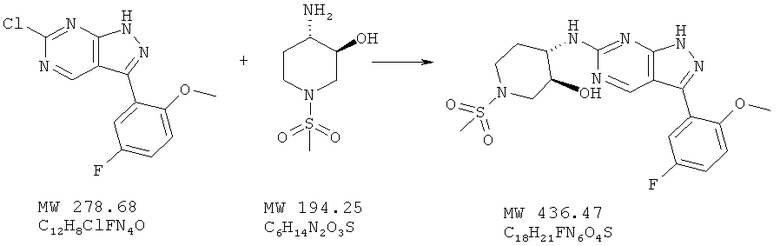
Тем же методом, описанном в примере 25, исходя из 6-хлор-3-(5-фтор-2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидина (пример 24; 103 мг, 0,37 ммоля) и (3S,4S)-4-амино-1-метансульфонилпиперидин-3-ола (115 мг, 0,59 ммоля), получают 70 мг (46%) желтого твердого вещества.
МС (М+Н)+, 437.
Пример 28: 1-(метансульфонилпиперидин-4-ил)-[3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]амин; соединение с трифторуксусной кислотой
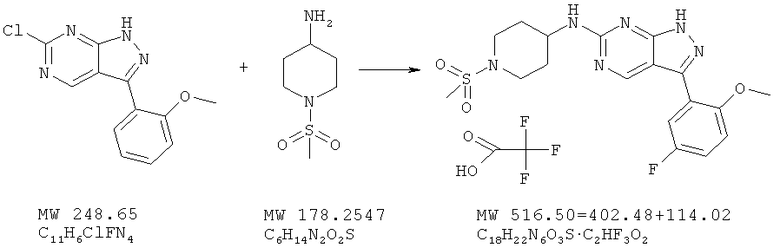
Соединение получают из 6-хлор-3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидина (пример 6) и 1-метансульфонилпиперидин-4-иламина, соединение с трифторуксусной кислотой получают аналогично методу, описанному в примере 4, и очищают с помощью ВЭЖХ, получая соль трифторуксусной кислоты (36%).
МС (М+Н)+, 403,24.
Пример 29: [3-(2,6-дифторфенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-1-(метансульфонилпиперидин-4-ил)амин
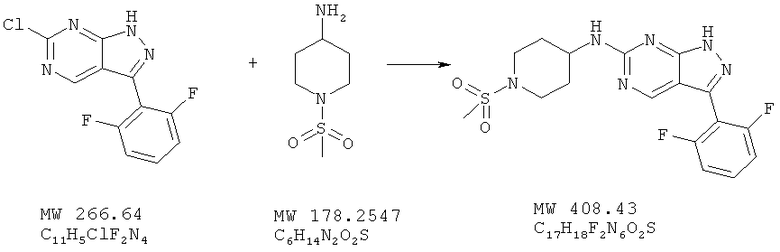
Соединение получают из 6-хлор-3-(2,6-дифторфенил)-1H-пиразоло[3,4-d]пиримидина (пример 8) и 1-метансульфонилпиперидин-4-иламина; соединение с трифторуксусной кислотой получают аналогично методу, описанному в примере 4, и очищают с помощью ВЭЖХ (Gilson), получая свободное основание (34,5%). МС (М+Н)+, 409,22.
Пример 30: 1-(метансульфонилпиперидин-4-ил)-(3-фенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]амин
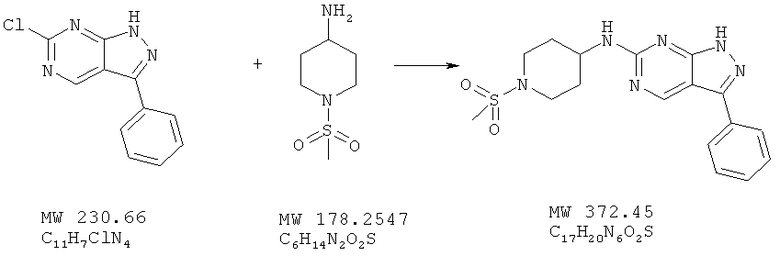
Соединение получают из 6-хлор-3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидина (пример 14) и 1-метансульфонилпиперидин-4-иламина; соединение с трифторуксусной кислотой получают аналогично методу, описанному в примере 4, и очищают с помощью ВЭЖХ (Gilson), получая свободное основание (21%).
МС (M+H)+, 373,24.
Пример 31: [3-(3-фторфенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-1-(метансульфонилпиперидин-4-ил)амин
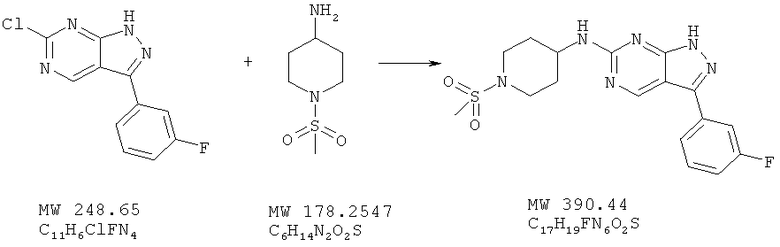
Соединение получают из 6-хлор-3-(3-фторфенил)-1H-пиразоло[3,4-d]пиримидина (пример 9) и 1-метансульфонилпиперидин-4-иламина; соединение с трифторуксусной кислотой получают аналогично методу, описанному в примере 4, и очищают с помощью ВЭЖХ (Gilson), получая свободное основание (6%).
МС (М+Н)+, 391,25.
Пример 32: 4-[3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-иламино]бензосульфонамид; соединение с трифторуксусной кислотой
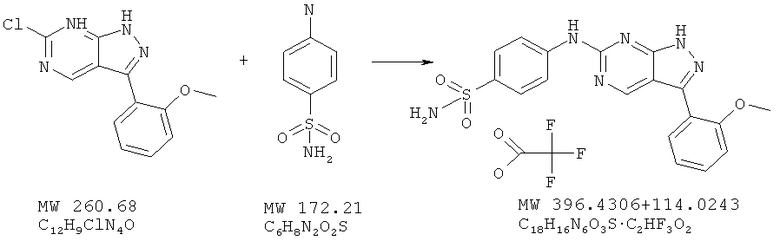
6-Хлор-3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидин (пример 6; 50 мг, 0,192 ммоля) и сульфаниламид (фирма Aldrich, 47,6 мг, 0,276 ммоля) соединяют с 2-пропанолом (1 мл) в микроволновой трубке и нагревают при 170°С в течение 10 мин в микроволновой печи (Smith Synthesizer, Personal Chemistry). Затем смесь охлаждают, отфильтровывают суспензию, а твердое вещество собирают и промывают холодным 2-пропанолом. Очистка с помощью ВЭЖХ дает 40,1 мг (41%) вещества в виде соли трифторуксусной кислоты. МС (М+Н)+, 397,1.
Пример 33: [3-(2,6-дифторфенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(4-фторфенил)амин
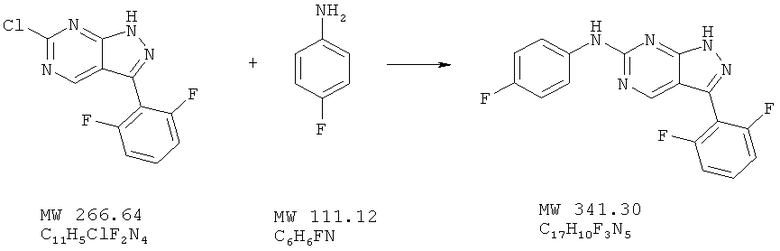
Соединение получают из 6-хлор-3-(2,6-дифторфенил)-1H-пиразоло[3,4-d]пиримидина (пример 12) и 4-фторанилина (фирма Aldrich) аналогично методу, описанному в примере 32 (160°С). Фильтрование и промывка реакционной смеси 2-пропанолом дает названное в заголовке соединение (59%). МС (М+Н)+, 342,1.
Пример 34: 2-[3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-иламино]этанол
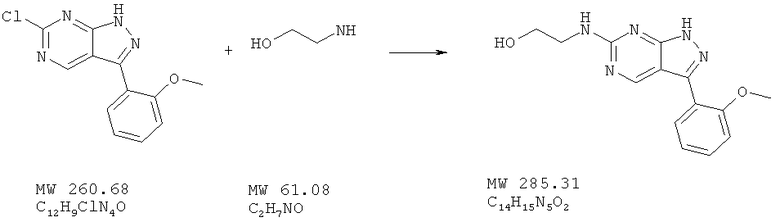
Соединение получают из 6-хлор-3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидина (пример 6) и этаноламина (фирма AnaLar) аналогично методу, описанному в примере 32. Очистка с помощью ВЭЖХ дает названное в заголовке соединение с выходом 24 мг (37%). МС (М+Н)+, 286,2.
Пример 35: [3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]пентан-1,5-диамин; соединение с трифторуксусной кислотой
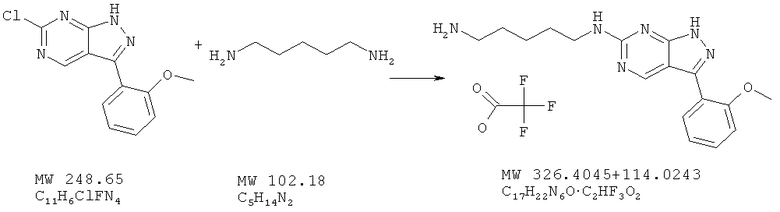
Соединение получают из 6-хлор-3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидина (пример 6) и 1,5-диаминопентана (фирма ACROS) аналогично методу, описанному в примере 32 (микроволновый диапазон при 120°С). Очистка с помощью ВЭЖХ дает соль трифторуксусной кислоты (68%). МС (М+Н)+, 327,3.
Пример 36: N'-[3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-N,N-диметилпропан-1,3-диамин
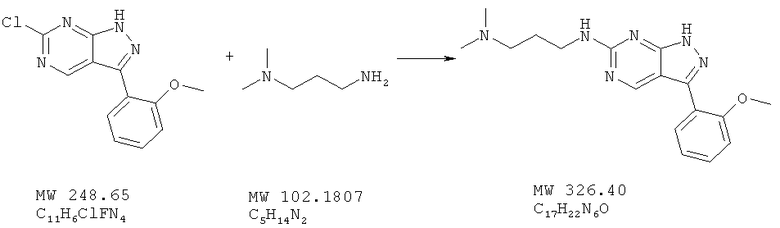
Соединение получают из 6-хлор-3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидина (пример 6) и 3-диамино(пропиламина) (фирма Aldrich) аналогично методу, описанному в примере 32 (120°С). Получают 9,2 мг (12%) свободного основания, а очистка с помощью ВЭЖХ маточного раствора дает соль трифторуксусной кислоты (50 мг, 49%). МС (М+Н)+, 327,2.
Пример 37: N-[3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-N,N-диметилпентан-1,5-диамин; соединение с трифторуксусной кислотой
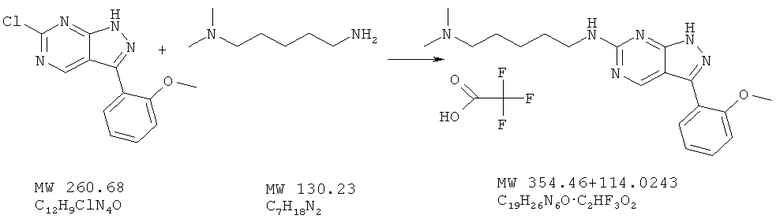
Соединение получают из 6-хлор-3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидина (пример 6) и 5-(диметиламино)амиламина (фирма Matrix) аналогично методу, описанному в примере 32 (120°С). Очистка с помощью ВЭЖХ дает соль трифторуксусной кислоты (59%). МС (М+Н)+, 355,25.
Пример 38: [3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]фениламин
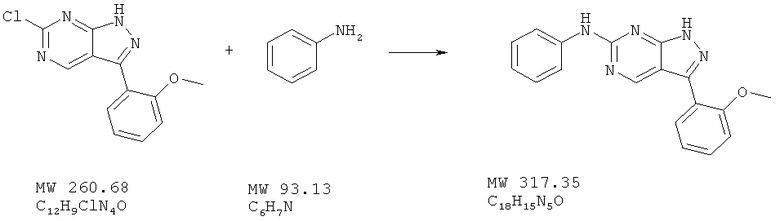
Соединение получают из 6-хлор-3-(2-метоксифенил)-1H-пиразоло[3,4-d]пиримидина (пример 6) и анилина (фирма Aldrich) аналогично методу, описанному в примере 32 (175°С). Названное в заголовке соединение получено с выходом 69% МС (М+Н)+, 318,2.
Пример 39: (4-метоксифенил)-[3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-иламино]бензосульфонамид; соединение с трифторуксусной кислотой
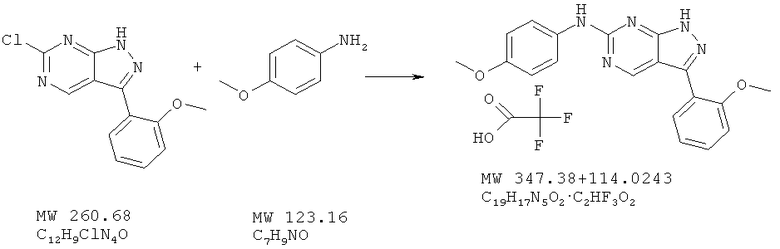
Соединение получают из 6-хлор-3-(2-метоксифенил)-1H-пиразоло[3,4-d] пиримидина (пример 6) и п-анизидина (фирма Aldrich) аналогично методу, описанному в примере 32 (175°С). Очистка с помощью ВЭЖХ дает названное в заголовке соединение в виде соли трифторуксусной кислоты с выходом 59%. МС (М+Н)+, 348,2.
Пример 40: [3-(2,6-дифторфенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(2-метоксифенил)амин; соединение с трифторуксусной кислотой
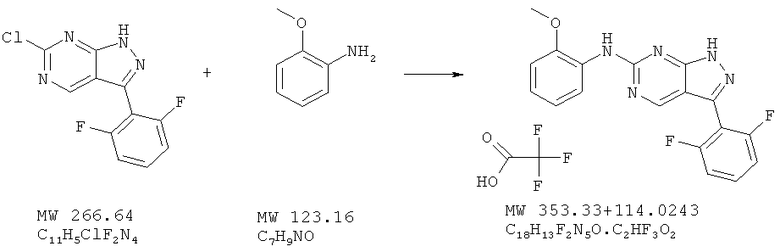
Соединение получают из 6-хлор-3-(2,6-дифторфенил)-1H-пиразоло[3,4-d]пиримидина (пример 12) и о-анизидина (фирма Aldrich) аналогично методу, описанному в примере 32 (160°С). Очистка с помощью ВЭЖХ дает соль трифторуксусной кислоты с выходом 42%. МС (М+Н)+, 354,1.
Пример 41: 4-(3-метил-1Н-пиразоло[3,4-d]пиримидин-6-иламино]бензосульфонамид; соединение с трифторуксусной кислотой
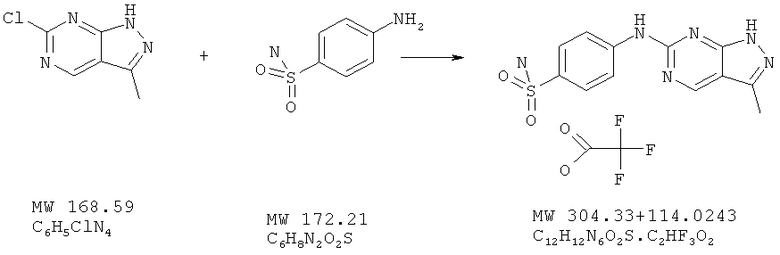
Соединение получают из 6-хлор-3-метил-1H-пиразоло[3,4-d]пиримидина (пример 22) и сульфаниламида (фирма Aldrich) аналогично методу, описанному в примере 32 (175°С). Очистка с помощью ВЭЖХ дает названное в заголовке соединение в виде соли трифторуксусной кислоты с выходом 13%. МС (М+Н)+, 305,0.
Пример 42: 2-(3-метил-1Н-пиразоло[3,4-d]пиримидин-6-иламино]этанол; соединение с трифторуксусной кислотой
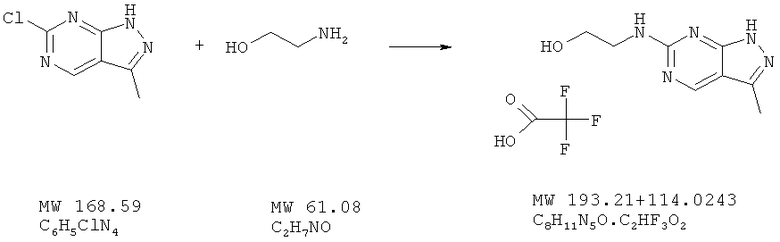
Соединение получают из 6-хлор-3-метил-1H-пиразоло[3,4-d]пиримидина (пример 22) и этаноламина (фирма AnaLar) аналогично методу, описанному в примере 32 (120°С). Очистка с помощью ВЭЖХ дает соль трифторуксусной кислоты с выходом 56%. МС (М+Н)+, 193,9.
Пример 43: этиловый эфир 4-[3-(3-фторфенил)-1Н-пиразоло[3,4-d]пиpимидин-6-илaминo]пипepидин-1-кapбoнoвoй кислоты: соединение с трифторуксусной кислотой
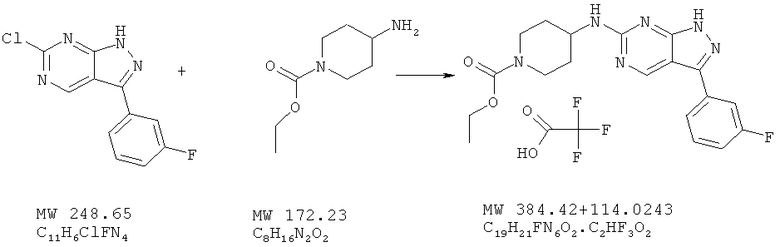
Соединение получают из 6-хлор-3-(3-фторфенил)-1H-пиразоло[3,4-d]пиримидина (пример 9) и этил-4-амино-1-пиперидинкарбоксилата (фирма Aldrich) аналогично методу, описанному в примере 32 (160°С). Очистка с помощью ВЭЖХ дает соединение в виде соли трифторуксусной кислоты с выходом 18%. МС (М+Н)+, 385,1.
Пример 44: 1-(метилпиперидин-4-ил)-(3-метил-1Н-пиразоло[3,4-d]пиримидин-6-ил] амин
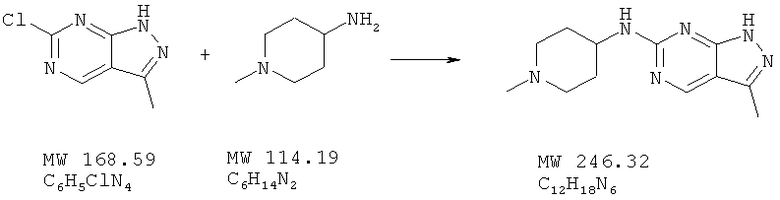
Соединение получают из 6-хлор-3-метил-1H-пиразоло[3,4-d]пиримидина (пример 22) и 4-амино-N-метилпиперидина (фирма Aldrich) аналогично методу, описанному в примере 32 (175°С). МС (М+Н)+, 247.
Пример 45
Фармакологические свойства соединений по настоящему изобретению подтверждаются рядом фармакологических анализов. Приведенные далее в качестве примеров фармакологические анализы проводят с соединениями по изобретению и их солями. Соединения по изобретению проявляют Cdk4/циклин D активность с величинами IC50 и Ki, равными менее 1,0 мкМ. Кроме того, антипролиферативная активность некоторых соединений по изобретению, протестированная на линии клеток НСТ116 в тканях толстой кишки человека, показала величины IC50, полученные в ММТ анализе, составляющие менее 30 мкМ, предпочтительно, менее 5 мкМ.
Анализы киназ
Для определения ингибирования Cdk4, Cdk2 и Cdk1 активности анализы киназ проводят с использованием FlashPlateТМ анализов (NENТМ-Life Science Products). FlashPlate анализы осуществляют, используя рекомбинантные человеческий циклин B-Cdk1, человеческий циклин E-Cdk2 или человеческий циклин D1-Cdk4 комплексы. GST-циклин Е (GST-cycE), Cdk2, GST-циклин В (GST-cycB), Cdk1, GST-Cdk4 и циклин D1 (cycD1) кДНК клоны в бакуловирусных векторах предоставлены Dr. W. Harper из Baylor College of Medicine, Houston, TX. Белки соэкспрессируют в High FiveТМ клетках насекомых и комплекс очищают с использованием глутатионовой Sepharose смолы (Pharmacia, Piscataway, NJ), как ранее описано (J.W.Harper et al., Cell, 1993, 75, 805-816). 6х-Гистидин-меченые укороченные формы белка ретинобластомы (Rb) (аминокислота 386-928) используются в качестве субстрата для cycD1-Cdk4, cycB-Cdk1 и cycE-Cdk2 анализов (экспрессионная плазмида предоставлена Dr. Veronica Sillivan, Departament of Molecular Virology, Roshe Research Centre, Welwyn Garden City, United Kingdom). Rb белок является натуральным субстратом для фосфорилирования с помощью Cdk4, Cdk2 и Cdk1 (см. Herwig and Strauss, Eur. J.Biochem., vol.246 (1997), pp.581-601 и приведенные там ссылки).
Экспрессию 62Kd белка контролируют IPTG индуцируемым промотором в M15 линии E.coli. Клетки лизируют ультразвуком и проводят очистку лизатов при рН 8,0 через наполненную Ni-хелированной агарозой колонку, предварительно обработанную 1 мМ имидазола. Смолу затем несколько раз промывают буфером с постепенно уменьшающимися значениями рН до 6,0 и элютируют 500 мл имидазола. Элютированный белок подвергается диализу с использование 20 мМ HEPES рН 7,5, 30% глицерина, 200 мМ NaCl и 1 мМ DTT. В очищенном Rb слитом белке количественно оценивалась концентрация белка, он разделялся на аликвоты и хранился при -70°С.
Для всех трех анализов киназ, приведенных в данном описании, 96-ячеистые Flash планшеты покрывают Rb белком при 10 мкг/мл, используя 100 мкл на ячейку. Планшеты инкубируют при 4°С в течение ночи или в течение 3 ч при комнатной температуре на вибраторе. Чтобы контролировать неспецифическое фосфорилирование один ряд ячеек покрывают 100 мкл/ячейку буфером для покрытия (20 мМ HEPES, 0,2 М NaCl). Планшеты затем дважды отмывают буфером для отмывания (0,01% Tween 20 в фосфат-буферированном рассоле). Соединения, подлежащие тестированию, («тестируемые соединения») добавляют в ячейки с пятикратной конечной концентрацией. Реакции инициируют немедленным добавлением 40 мкл реакционной смеси (25 мМ HEPES, 20 мМ MgCl2, 0,002% Tween 20, 2 мМ DTT, 1 мкМ АТР, 4 нМ 33Р-АТР), а добавление достаточного количества фермента дает импульсы, по крайней мере, десятикратно превышающие фоновые значения. Планшеты инкубируют при комнатной температуре на вибраторе в течение 30 мин, а затем четыре раза отмывают буфером для отмывания, запаивают и считывают на TopCount сцинтилляционном счетчике (Packard Instrument Co., Downers Grove, IL). Процент ингибирования Rb фосфорилирования, являющийся мерой ингибирования Cdk активности, определяют согласно следующей формуле:
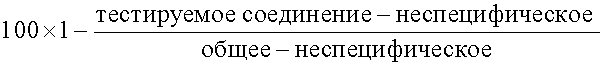
где «тестируемое соединение» относится к среднему числу импульсов в мин тестируемых дублетов, «неспецифическое» относится к среднему числу импульсов в мин, когда циклин D/Cdk и другие не были добавлены, и «общее» относится к среднему числу импульсов в мин, когда никакое соединение не было добавлено. Величина IС50 обозначает концентрацию тестируемого соединения, которая на 50% уменьшает индуцируемое протеинкиназой внедрение радиометок при описанных условиях тестирования.
Результаты проведенных в условиях in vitro экспериментов представлены ниже в таблице 1.
Клеточные анализы (анализ пролиферации с помощью красителя тиазолил голубой)
Пролиферация оценивалась с помощью анализа окрашивания красителем тиазолил голубой согласно методу, предложенному Denizot и Lang (Denizot, F. and Lang, R., J.Immunol. Methods, 1986, 89, 271-277). Была использована клеточная линия НСТ116, клеточная линия колоректальной карциномы получена из Американской коллекции типичных клеточных культур (АТСС; Rockville, MD). Клетки выращены в McCoy's 5A среде, снабженной 10% FCS (плодная телячья сыворотка) и L-глутамином. Клетки помещают в планшеты с соответствующей высеваемой плотностью, дающие десятикратный логарифмический рост по сравнению с анализом в 96-ячеистом планшете с тканевой культурой. Планшеты инкубируют в течение ночи при 37°С во влажном инкубаторе с 5% углекислого газа. На следующий день тестируемые соединения по сериям 4 раза разбавлялись до конечной концентрации в соответствующей среде, содержащей 1,2% ДМСО. Одна четвертая часть каждого финального объема каждого разбавления добавлялась в дублете в планшеты, содержащие клетки. Такой же объем 1,2% ДМСО в среде был добавлен в ряд «контрольных ячеек» так, что финальная концентрация ДМСО в каждой ячейке составляла 0,3%. Эксперимент, в котором в ячейки не были добавлены клетки, служил «слепым опытом». Эксперимент, в котором в ячейки не был добавлен ингибитор, служил «контрольным без ингибитора». Планшеты были возвращены в инкубатор и при установлении временных точек (определенных по кривым роста) планшеты анализировались, как описано ниже.
3-(4,5-Диметилтиазол-2-ил)-2,5-дифенил-2Н-тетразолийбромид (тиазолил голубой; МТТ; фирма SIGMA) добавляют в каждую ячейку до конечной концентрации 1 мг/мл. Планшеты возвращают в инкубатор на 2,5-3 ч при 37°С. МТТ-содержащую среду удаляют и образовавшийся формазан метаболит растворяют в 100%-ном этаноле при встряхивании в течение 15 мин при КТ. Считывание оптической плотности проводят в титрационном микропланшете-ридере (динамическое и молекулярное устройства планшет-ридеров взаимозаменяемы) при длине волны 570 нм относительно 650 нм. Процент ингибирования (% INH) вычисляют вычитанием оптической плотности слепого опыта из всех ячеек, затем вычитанием отношения средней оптической плотности каждого тестируемого дубликата (SAVE) к среднему контролю (CAVE) из 1,00. Конечное число затем умножают на 100 (% INH=(1,00-SAVE/CAVE)×100). Концентрация, при которой получено 50%-ное ингибирование клеточной пролиферации, определяют из линейной регрессии графика концентрации по отношению к проценту ингибирования (в логарифмическом масштабе). Величины IС50 также представлены ниже в таблице 1.
В данной таблице представлены величины IС50 соединений примеров при Cdk4, Cdk2 и Cdk1 анализах киназы, а также величины IС50 при клеточных анализах (МТТ)
IС50 (мкМ)
IС50 (мкМ)
IС50 (мкМ)
IС50 (мкМ)
Пример 46
Изготовление таблеток
Метод изготовления:
Смешивают номера 1, 2 и 3 в подходящем смесителе в течение 15 мин.
Гранулируют порошкообразную смесь из стадии 1 с 20% раствором Повидона К30 (номер 4).
Высушивают гранулят из стадии 2 при 50°С.
Пропускают гранулят из стадии 3 через подходящее помольное оборудование.
Добавляют номер 5 к размельченному грануляту из стадии 4 и смешивают в течение 3 мин.
Продавливают гранулят из стадии 5 через подходящий пресс.
Пример 47
Изготовление капсул
Метод изготовления:
Смешивают номера 1, 2 и 3 в подходящем смесителе в течение 15 мин.
Добавляют номера 4 и 5 и смешивают в течение 3 мин.
Загружают в подходящую капсулу.
Пример 48
Приготовление инъекционного раствора/эмульсии
Метод приготовления:
Растворить номер 1 в номере 2.
Добавить номера 3, 4, и 5 к номеру 6 и перемешать до образования дисперсии, затем гомогенизировать до образования полупрозрачной дисперсии.
Через стерильный фильтр с порами 0,2 мкм отфильтровать и заполнить в пузырьки.
Пример 49
Приготовление инъекционного раствора/эмульсии
Метод приготовления:
Растворить номер 1 в номере 2.
Добавить номера 3, 4, и 5 к номеру 6 и перемешать до образования дисперсии, затем гомогенизировать.
Добавить раствор из стадии 1 к смеси стадии 2 и гомогенизировать до образования полупрозрачной дисперсии.
Через стерильный фильтр с порами 0,2 мкм отфильтровать и заполнить в пузырьки.
Хотя изобретение проиллюстрировано ссылками на специфические и предпочтительные варианты, специалистам в данной области техники должно быть очевидным, что варианты и модификации могут осуществляться, не выходя за рамки экспериментирования и практики данного изобретения. Таким образом, изобретение не ограничивается вышеприведенным описанием, но определяется формулой изобретения и ее эквивалентами.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ 2,6-ДИАМИНОПИРИДИН-3-ОНА | 2005 |
|
RU2385866C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 2,4-ДИАМИНОТИАЗОЛ-5-ОНА | 2005 |
|
RU2395501C2 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛО [4,5-d]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ ПУРИНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2002 |
|
RU2317084C2 |
| ПРОИЗВОДНЫЕ N-(ИМИДАЗОПИРИМИДИН-7-ИЛ)-ГЕТЕРОАРИЛАМИДОВ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE10A | 2011 |
|
RU2562066C2 |
| ЛЕЧЕНИЕ ЗАВИСИМОСТИ И РАССТРОЙСТВА ПОБУЖДЕНИЙ С ПРИМЕНЕНИЕМ ИНГИБИТОРОВ ФДЭ7 | 2011 |
|
RU2661410C2 |
| ПРОИЗВОДНЫЕ N-ФЕНИЛ-2-ПИРИМИДИНАМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ИНГИБИРОВАНИЯ (ЛЕЧЕНИЯ) ОПУХОЛИ | 1994 |
|
RU2135491C1 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ОПОСРЕДОВАНИЯ АКТИВНОСТИ ТИРОЗИНКИНАЗЫ 2 | 2020 |
|
RU2826012C2 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2539568C2 |
| 2-ЦИАНО-4-АМИНОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ С ИНГИБИРУЮЩИМ ДЕЙСТВИЕМ НА КАТЕПСИН К, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 2002 |
|
RU2326119C2 |
Изобретение относится к новым пиразол[3,4-d]пиримидинам формулы (I), которые обладают способностью ингибировать активность циклин-зависимых киназ, наиболее предпочтительно, циклин-зависимой киназы 1 (Cdk1), циклин-зависимой киназы 2 (Cdk2) и циклин-зависимой киназы 4 (Cdk4). Соединения могут найти применение для лечения опухолевых заболеваний, таких как твердые опухоли, опухоли грудной железы, легкого, толстой кишки или простаты. В соединении формулы (I):
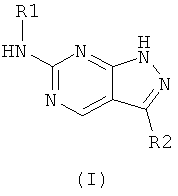
R1 выбирают из группы, включающей: (а) насыщенный циклический радикал, содержащий 3-8 кольцевых атомов, из которых от 1 до 3 атомов представляют собой N, который может содержать до четырех заместителей, независимо выбранных из группы, включающей: (i) низший алкил; и (ii) CO2R3, OR7 или S(O)nR8; (б) С6-С10арил, который может содержать до четырех заместителей, независимо выбранных из группы, включающей: (i) S(O)nR8, низший алкил, OR7 и галоген; (в) С3-С8циклоалкил, который может быть замещен NR5R6; и (г) низший алкил, который может быть замещен: (i) OR7, NR5R6; R2 выбирают из группы, включающей: (i) Н; (ii) низший алкил; (iii) С6-С10арил, который может быть замещен галогеном, низшим алкилом, низшей алкоксигруппой; R3 выбирают из группы, включающей: (i) H; (ii) низший алкил; (iv) С3-С8циклоалкил; R5 и R6 независимо друг от друга выбирают из группы, включающей: (i) Н; (ii) низший алкил; (iii) С3-С8циклоалкил; (v) SO2R3; и (vi) CO2R3; R7 выбирают из группы, включающей Н и низший алкил; R8 выбирают из группы, включающей: (iii) NR5R6; (iv) низший алкил; и n равно 1 или 2. 2 н. и 18 з.п. ф-лы, 1 табл.
1. Соединение формулы (I):
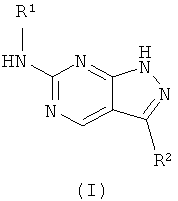
или его фармацевтически приемлемые соли, где
R1 выбирают из группы, включающей:
(а) насыщенный циклический радикал, содержащий 3-8 кольцевых атомов, из которых от 1 до 3 атомов представляют собой N, который может содержать до четырех заместителей, независимо выбранных из группы, включающей:
(i) низший алкил; и
(ii) CO2R3, OR7 или S(O)nR8;
(б) С6-С10арил, который может содержать до четырех заместителей, независимо выбранных из группы, включающей:
(i) S(O)nR8, низший алкил, OR7 и галоген;
(в) С3-С8циклоалкил, который может быть замещен NR5R6; и
(г) низший алкил, который может быть замещен:
(i) OR7, NR5R6;
R2 выбирают из группы, включающей:
(i) H;
(ii) низший алкил;
(iii) С6-С10арил, который может быть замещен галогеном, низшим алкилом, низшей алкоксигруппой;
R3 выбирают из группы, включающей:
(i) Н;
(ii) низший алкил;
(iv) С3-С8циклоалкил;
R5 и R6 независимо друг от друга выбирают из группы, включающей:
(i) Н;
(ii) низший алкил;
(iii) С3-С8циклоалкил;
(v) SO2R3; и
(vi) CO2R3;
R7 выбирают из группы, включающей Н и низший алкил;
R8 выбирают из группы, включающей:
(iii) NR5R6;
(iv) низший алкил; и
n равно 1 или 2.
2. Соединение по п.1, где R1 выбирают из группы, включающей пиперидин, пиперазин, пирролидин, фенил, толил, ксилил, метил, этил, пропил, изопропил, бутил, трет-бутил, 2-бутил, пентил и гексил.
3. Соединение по п.2, где R1 выбирают из группы, включающей пиперидин, фенил, этил, пропил, изопропил, бутил, трет-бутил, 2-бутил и пентил.
4. Соединение по п.1, где R1 представляет собой пиперидин, замещенный SO2СН3, СН3, СООСН2СН3 или ОН; фенил; фенил, замещенный SO2NH2, F или ОСН3; или низший алкил, замещенный ОН, NH2 или N(СН3)2.
5. Соединение по п.1, где R2 выбирают из группы, включающей Н, метил, этил, пропил, изопропил, бутил, трет-бутил, 2-бутил, пентил, гексил, фенил, толил и ксилил.
6. Соединение по п.1, где R2 обозначает Н, метил или фенил, который может быть замещен фтором или метоксигруппой.
7. Соединение по п.1, выбранное из группы, включающей
[3-(2,3-дифтор-6-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин;
(1-метансульфонилпиперидин-4-ил)-(1Н-пиразоло[3,4-d]пиримидин-6-ил)амин;
(1-метилпиперидин-4-ил)-(3-метил-1Н-пиразоло[3,4-d]пиримидин-6-ил)амин;
[3-(5-фтор-2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин;
(1-метансульфонилпиперидин-4-ил)-[3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]амин; соединение с трифторуксусной кислотой;
[3-(2,6-дифторфенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин;
(1-метансульфонилпиперидин-4-ил)-(3-фенил-1Н-пиразоло[3,4-d]пиримидин-6-ил)амин;
[3-(3-фторфенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин.
8. Соединение по п.1, выбранное из группы, включающей
4-[3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-иламино]бензолсульфонамид; соединение с трифторуксусной кислотой;
[3-(2,6-дифторфенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(4-фторфенил)амин;
2-[3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-иламино]этанол;
[3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиpимидин-6-ил]пентан-1,5-диамин; соединение с трифторуксусной кислотой;
N'-[3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-N,N-диметилпропан-1,3-диамин;
N-[3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-N,N-диметилпентан-1,5-диамин; соединение с трифторуксусной кислотой;
[3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]фениламин;
(4-метоксифенил)-[3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]амин; соединение с трифторуксусной кислотой;
[3-(2,6-дифторфенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(2-метоксифенил)амин; соединение с трифторуксусной кислотой;
4-(3-метил-1Н-пиразоло[3,4-d]пиримидин-6-иламино]бензолсульфонамид; соединение с трифторуксусной кислотой.
9. Соединение по п.1, выбранное из группы, включающей
2-(3-метил)-1Н-пиразоло[3,4-d]пиримидин-6-иламино]этанол; соединение с трифторуксусной кислотой;
этиловый эфир 4-[3-(3-фторфенил)-1Н-пиразоло[3,4-d]пиримидин-6-иламино]пиперидин-1-карбоновой кислоты; соединение с трифторуксусной кислотой;
(1-метилпиперидин-4-ил)-(3-метил-1Н-пиразоло[3,4-d]пиримидин-6-ил)амин.
10. Соединение по п.1, представляющее собой [3-(2,3-дифтор-6-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин.
11. Соединение по п.1, представляющее собой (1-метансульфонилпиперидин-4-ил)-(1Н-пиразоло[3,4-d]пиримидин-6-ил)амин.
12. Соединение по п.1, представляющее собой [3-(5-фтор-2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин.
13. Соединение по п.1, представляющее собой [3-(5-фтор-2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин.
14. Соединение по п.1, представляющее собой (1-метансульфонилпиперидин-4-ил)-[3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]амин; соединение с трифторуксусной кислотой.
15. Соединение по п.1, представляющее собой [3-(3-фторфенил)-1Н-пиразоло[3,4-d]пиримидин-6-ил]-(1-метансульфонилпиперидин-4-ил)амин.
16. Соединение по п.1, представляющее собой 4-[3-(2-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-6-иламино]бензолсульфонамид; соединение с трифторуксусной кислотой.
17. Фармацевтическая композиция, обладающая антипролиферативной активностью, включающая эффективное количество соединения формулы (I)
или его фармацевтически приемлемых солей, где значения R1 и R2 определены в п.1, и фармацевтически приемлемый наполнитель.
18. Фармацевтическая композиция по п.17, предназначенная для приготовления лекарственного средства для лечения рака.
19. Фармацевтическая композиция по п.17 для лечения твердых опухолей.
20. Фармацевтическая композиция по п.17, предназначенная для приготовления лекарственного средства для лечения опухолей грудной железы, легкого, толстой кишки или простаты.
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| СПОСОБЫ ИНГИБИРОВАНИЯ ТИРОЗИНКИНАЗЫ РЕЦЕПТОРА ЭПИДЕРМАЛЬНОГО ФАКТОРА РОСТА, БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ПИРИМИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ИНГИБИРУЮЩЕЙ ТИРОЗИНКИНАЗУ РЕЦЕПТОРА ЭПИДЕРМАЛЬНОГО ФАКТОРА РОСТА АКТИВНОСТЬЮ, И КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ КОНТРАЦЕПТИВНЫМ ДЕЙСТВИЕМ | 1995 |
|
RU2174980C2 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| US 6107305 A, 22.08.2000. | |||

