Область техники
Настоящее изобретение относится к депротеинизированному препарату из телячьей крови для применения в предотвращении или лечении болезни периферических артерий. Более конкретно, настоящее изобретение относится к депротеинизированному препарату из телячьей крови для применения в предотвращении или лечении болезни периферических артерий, где депротеинизированный препарат из телячьей крови вводят по меньшей мере один раз парентерально, предпочтительно внутривенно, с последующим периодом энтерального введения, предпочтительно - перорального введения. В предпочтительном варианте осуществления настоящего изобретения депротеинизированный препарат из телячьей крови представляет собой лекарственное средство Актовегин®.
Уровень техники
Болезнь периферических артерий (PAD, БПА), иначе называемая "окклюзионная болезнь периферических артерий", является проявлением системного атеросклероза и определяется прогрессирующим стенозом или окклюзией в артериях нижних конечностей [1]. БПА затрагивает приблизительно 202 миллиона взрослых во всем мире [2]. Распространенность БПА возрастает с возрастом и количеством факторов сосудистого риска (например, диабет, курение, гипертония, гиперхолестеринемия, возраст, пол, семейная история) [1, 3].
Что еще более важно, она является маркером атеросклеротической болезни и ассоциирована с повышением смертности от сердечно-сосудистых и цереброваскулярных причин [1, 3].
Снижение притока крови к ногам, вызванное БПА, может быть умеренным или тяжелым и обуславливает широкий спектр симптомов. Пациенты могут не ощущать заметных симптомов со стороны конечностей, но могут и испытывать перемежающуюся хромоту (ПХ, IC), или у них могут проявляться симптомы тяжелой ишемии конечностей. ПХ, наиболее распространенный симптом БПА, определяется как усталость, судороги или явная боль мышц ягодиц, бедер или голеней, которые регулярно провоцируются физическими упражнениями и воспроизводимо купируются отдыхом.
Пациенты с ПХ часто ограничены в своей повседневной деятельности из-за нарушений ходьбы и, в связи с этим, испытывают снижение качества жизни. При постоянном воздействии факторов риска атеросклероза БПА может прогрессировать до критической ишемии конечностей, что предвещает тяжелое снижение качества жизни и ассоциировано с высокой вероятностью ампутации и значительным увеличением краткосрочной смертности. Таким образом, БПА является распространенным проявлением атеросклероза, ассоциированным с рядом симптомов, различным воздействием на качество жизни и повышенным риском сердечно-сосудистых ишемических событий [1].
Актовегин® представляет собой депротеинизированный гемодериват из телячьей крови, который был очищен от пирогенов и антигенов в процессе производства с применением ступенчатой ультрафильтрации.
Актовегин® содержит физиологические компоненты крови, включая витамины, аминокислоты, липиды, олигосахариды и олигопептиды, нуклеозиды, промежуточные продукты метаболизма углеводов и жирных кислот, а также компоненты клеточных мембран, такие как гликосфинголипиды. Он обладает плейотропными метаболическими, нейропротекторными и регенеративными свойствами [4]. Экспериментальные исследования показали, что Актовегин® улучшает использование и усвоение кислорода, а также энергетический обмен и усвоение глюкозы [5-7]. Показано, что Актовегин® обладает нейропротекторными эффектами и увеличивает выживаемость нейронов в условиях ишемии [8]. Он также уменьшает апоптоз, вызванный бета-амилоидным белком, наряду с уменьшением образования активных форм кислорода [9], модулирует активность ядерного фактора kappa B [10] и ингибирует ядерный фермент поли(АДФ-рибозу) полимеразу, что также может частично объяснять его нейропротекторные свойства [11]. Актовегин®оказывает положительное влияние на микроциркуляцию и эндотелий микрососудов, увеличивая скорость капиллярного кровотока и уменьшая перикапиллярную зону и артериовенозное шунтирование кровотока [12].
Данные по клинической эффективности Актовегина® свидетельствуют о возможности лечения нарушений мозгового кровообращения, включая деменцию [13, 14] и постинсультное когнитивное расстройство [15]. Для этих показаний было проведено несколько рандомизированных контролируемых испытаний и много неконтролируемых исследований. Имеющиеся клинические данные также поддерживают применение Актовегина® для лечения диабетической полинейропатии [16]. Эффективность Актовегина® для этих показаний дополнительно подтверждается неклиническими фармакодинамическими исследованиями. Данные по эффективности также свидетельствуют в пользу возможности лечения периферических нарушений перфузии свидетельствуя о положительных результатах исследований в отношении максимальной дистанции безболевой ходьбы при БПА и времени заживления при венозной язве [17-21].
Актовегин® впервые был одобрен в Федеративной Республике Германии в 1976 году и остается на рынке более чем в 20 странах (в том числе, в нескольких странах Содружества Независимых Государств и в нескольких странах Европы и Азии). Он представлен в разных препаратах для перорального, внутривенного (в/в) и внутримышечного применения. Актовегин® был одобрен для следующих показаний (которые варьируются в разных странах): симптоматическое лечение когнитивных нарушений, включая постинсультные когнитивные нарушения и деменцию; симптоматическое лечение нарушений периферической перфузии и их последствий; и симптоматическое лечение диабетической полинейропатии.
Актовегин® используется в клинической практике более 40 лет. Благодаря тому, что он состоит из биологических компонентов, присутствующих в нормальных физиологических условиях, и строго контролируемому процессу производства препарат обладает превосходным профилем безопасности, что отражает клинический опыт, эквивалентный экспозиции препарата, соответствующей более 1 200 000 пациентогодам (Периодический отчет по безопасности [PSUR] 2014, представленный Австрийскому агентству по здравоохранению и безопасности пищевых продуктов). Профиль нежелательных лекарственных реакций (НЛР) был стабильным по всем PSUR, не показывая токсичности в отношении конкретных органов-мишеней.
Актовегин® используется с 1970-х годов для лечения БПА различной степени тяжести. В нескольких исследованиях оценивали эффективность Актовегина® (в/в) у пациентов с БПА преимущественно II и III стадией по Фонтейну.
Было проведено рандомизированное, открытое, 8-недельное исследование для исследования влияния Актовегина® на ишемический синдром нижних конечностей у пациентов с диабетом типа 1 и типа 2 по сравнению с сулодексидом [17]. Пациенты были рандомизированы для получения либо в/в инфузий Актовегина® (2000 мг один раз в день, n=12), либо сулодексида (1200 липопротеинлипазных единиц [LSU] в день, n=14) в течение 2 недель с последующим пероральным введением обоих средств в течение еще 6 недель (1200 мг/день и 1000 LSU в день соответственно). Оба средства увеличивали максимальную продолжительность безболевой ходьбы после 8 недель лечения (p <0,05). Но лечение Актовегином® значимо увеличивало максимальную продолжительность безболевой ходьбы после 2 недель лечения по сравнению с исходным уровнем (p <0,05), в отличие от лечения сулодексидом. Кроме того, относительное увеличение времени безболевой ходьбы было больше в группе Актовегина® по сравнению с группой сулодексида (95,1% по сравнению с 38,1% соответственно, p <0,05).
Было проведено рандомизированное, двойное слепое, плацебо-контролируемое, исследование в параллельных группах, включающее 60 пациентов с II стадией БПА [18]. Все пациенты принимали участие в программе регулярных физических тренировок продолжительностью 12 недель, по меньшей мере за 4 недели до рандомизации. Затем пациенты получали ежедневно в/в инфузии Актовегина® 20% (250 мл/день) или плацебо (физиологический раствор 250 мл/день) в течение 4 недель. Первичная конечная точка, дистанция безболевой ходьбы на беговой дорожке, увеличилась на 27% (с 73 до 93 метров) в группе Актовегина® и уменьшилась в группе плацебо на 12% (на 10 метров). Разница между двумя группами была статистически значимой (р <0,001). Аналогичный результат был получен для максимальной дистанции ходьбы (+25% по сравнению с -10,4% для Актовегина® и плацебо, соответственно, p <0,01).
Horsch et al. провели рандомизированное, двойное слепое плацебо-контролируемое исследование, включающее 138 пациентов с БПА II стадии [19]. Актовегин® 20% (250 мл) и подходящее плацебо вводили в виде ежедневных внутривенных инфузий в течение 3-недельного периода. Эффективность оценивали с помощью анализа числа ответивших пациентов, где ответ определялся как увеличение дистанции безболевой ходьбы на 35% от исходного уровня, измеренное на беговой дорожке. В группе Актовегина® было 57% ответивших по сравнению с 33% в группе плацебо (p=0,01).
В другом двойном слепом исследовании Müller-Bühl et al. оценивали эффективность и совместимость инфузии Актовегина® 20% (250 мл) по сравнению с плацебо (0,9% физиологический раствор 250 мл) у пациентов с БПА II стадии [20]. В этом исследовании Актовегин® и плацебо вводили внутриартериально в бедренную артерию ноги, ограничивающей дистанцию ходьбы; пациенты должны были иметь стабильную дистанцию ходьбы до рандомизации для получения либо Актовегина® (n=40), либо плацебо (n=40).
Инфузии вводили ежедневно, за исключением выходных, в течение 4 недель, а тренировки прекращали с началом фазы лечения. В результате терапии Актовегином® наблюдалось увеличение дистанции безболевой ходьбы от 112 до 162 метров (49%) и максимальной дистанции от 171 до 266 метров (59%). В группе плацебо дистанция безболевой ходьбы увеличилась с 114 до 135 метров (23%), а максимальная дистанция - от 176 до 201 метров (17%). Различия между двумя группами были значимыми для обоих параметров (р <0,05). После прекращения лечения Актовегином® и плацебо 8-недельное наблюдение показало, что дистанция ходьбы заметно снизилась в группе плацебо по сравнению с пациентами, которых ранее лечили Актовегином®.
Также было проведено рандомизированное односторонне слепое исследование для сравнения эффективности Актовегина® и бенциклана при БПА III стадии [21]. Пятьдесят восемь пациентов были рандомизированы для получения либо инфузии Актовегина® 20% (20 внутриартериальных инфузий), либо 250 мг бенциклана в 250 мл физиологического раствора 0,9% в течение 4 недель. Анализ конечных точек показал, что через 4 недели из пациентов, получавших Актовегин®, 23,1% испытывали боль в ночное время, по сравнению с 61,5% пациентов, получавших бенциклан (р <0,05). Ни один из пациентов, получавших Актовегин®, не сообщал о постоянной боли через 4 недели, тогда как 16% пациентов, получавших бенциклан, жаловались на нее (p <0,05). Применение обезболивающих в течение 4 недель также было ниже у пациентов, получавших Актовегин®, по сравнению с пациентами, получавшими бенциклан (28,7% по сравнению с 42,3% соответственно, p <0,05). Наконец, оба средства увеличивали дистанцию безболевой ходьбы (29,2 ± 14,6 м и 25,8 ± 18,7 м для групп Актовегина® и бенциклана соответственно), но статистически значимой разницы между двумя группами не было.
Таким образом, было показано, что в/в лечение Актовегином® увеличивает начальную ходьбы дистанцию и облегчает симптомы боли у пациентов с БПА в небольших двойных слепых контролируемых исследованиях. Однако существует настоятельная потребность в новой и эффективной схеме лечения БПА.
Краткое описание изобретения
Настоящее изобретение обеспечивает в первом аспекте депротеинизированный препарат из телячьей крови, такой как лекарственное средство Актовегин®, для применения в предотвращении или лечении болезни периферических артерий, где депротеинизированный препарат из телячьей крови вводят по меньшей мере один раз парентерально, предпочтительно внутривенно, с последующим периодом энтерального введения, предпочтительно - перорального.
В другом аспекте настоящее изобретение относится к применению депротеинизированного препарата из телячьей крови, такого как лекарственное средство Актовегин®, для профилактики или лечения болезни периферических артерий, где указанный депротеинизированный препарат из телячьей крови вводят по меньшей мере один раз парентерально, предпочтительно внутривенно, с последующим периодом энтерального введения, предпочтительно - перорального.
В другом аспекте настоящее изобретение относится к способу профилактики или лечения болезни периферических артерий у пациента, который в этом нуждается, причем указанный способ включает следующие этапы: а) введение по меньшей мере один раз парентерально, предпочтительно внутривенно, указанному пациенту терапевтически эффективного количества депротеинизированного препарата из телячьей крови, такого как лекарственное средство Актовегин®, с последующим b) введением по меньшей мере один раз энтерально, предпочтительно перорально, указанному пациенту терапевтически эффективного количества указанного депротеинизированного препарата из телячьей крови.
Настоящее изобретение может быть дополнительно кратко описано в следующих пунктах:
1. Депротеинизированный препарат из телячьей крови, такой как лекарственное средство Актовегин®, для применения в предотвращении или лечении болезни периферических артерий, где указанный депротеинизированный препарат из телячьей крови вводят по меньшей мере один раз парентерально, предпочтительно внутривенно, с последующим периодом энтерального введения, предпочтительно - перорального.
2. Депротеинизированный препарат из телячьей крови для применения по п. 1, где указанный депротеинизированный препарат из телячьей крови вводят парентерально один раз в день.
3. Депротеинизированный препарат из телячьей крови для применения по п. 1 или 2, где указанный депротеинизированный препарат из телячьей крови вводят парентерально в течение по меньшей мере примерно 10 последовательных дней.
4. Депротеинизированный препарат из телячьей крови для применения по п. 1 или 2, где указанный депротеинизированный препарат из телячьей крови вводят парентерально в течение от примерно 10 до примерно 18 последовательных дней.
5. Депротеинизированный препарат из телячьей крови для применения по любому из пп. 1-4, где указанный депротеинизированный препарат из телячьей крови вводят парентерально в течение примерно 14 дней.
6. Депротеинизированный препарат из телячьей крови для применения по любому из пп. 1-5, где парентеральное введение представляет собой внутривенное введение.
7. Депротеинизированный препарат из телячьей крови для применения по п. 6, где указанный депротеинизированный препарат из телячьей крови вводят внутривенно путем инфузии.
8. Депротеинизированный препарат из телячьей крови для применения по любому из пп. 1-7, где общая суточная доза парентерально вводимого депротеинизированного препарата из телячьей крови находится в диапазоне от примерно 1000 мг до примерно 1400 мг.
9. Депротеинизированный препарат из телячьей крови для применения по п. 8, где общая суточная доза парентерально вводимого депротеинизированного препарата из телячьей крови составляет примерно 1200 мг.
10. Депротеинизированный препарат из телячьей крови для применения по любому из пп. 1-9, где указанный депротеинизированный препарат из телячьей крови вводят один раз в день внутривенно в дозе в диапазоне от примерно 1000 мг до примерно 1400 мг.
11. Депротеинизированный препарат из телячьей крови для применения по п. 10, где указанный депротеинизированный препарат из телячьей крови вводят один раз в день внутривенно в дозе примерно 1200 мг.
12. Депротеинизированный препарат из телячьей крови для применения по любому из пп. 1-11, где указанный депротеинизированный препарат из телячьей крови вводят энтерально по меньшей мере один раз в день.
13. Депротеинизированный препарат из телячьей крови для применения по любому из пп. 1-12, где депротеинизированный препарат из телячьей крови вводят энтерально по меньшей мере два раза в день.
14. Депротеинизированный препарат из телячьей крови для применения по любому из пп. с 1 по 13, где депротеинизированный препарат из телячьей крови вводят энтерально три раза в день.
15. Депротеинизированный препарат из телячьей крови для применения по любому из пп. 1-14, где депротеинизированный препарат из телячьей крови вводят энтерально в течение по меньшей мере примерно 63 последовательных дней.
16. Депротеинизированный препарат из телячьей крови для применения по любому из пп. 1-15, где депротеинизированный препарат из телячьей крови вводят энтерально в течение периода от примерно 63 до примерно 77 последовательных дней.
17. Депротеинизированный препарат из телячьей крови для применения по любому из пп. 1-16, где депротеинизированный препарат из телячьей крови вводят энтерально в течение примерно 70 последовательных дней.
18. Депротеинизированный препарат из телячьей крови для применения по любому из пп. 1-17, где энтеральное введение представляет собой пероральное введение.
19. Депротеинизированный препарат из телячьей крови для применения по п. 18, где депротеинизированный препарат из телячьей крови вводят перорально в форме таблетки.
20. Депротеинизированный препарат из телячьей крови для применения по любому из пп. 1-19, где общая суточная доза энтерально вводимого депротеинизированного препарата из телячьей крови находится в диапазоне от примерно 400 мг до примерно 2000 мг.
21. Депротеинизированный препарат из телячьей крови для применения по любому из пп. 1-20, где общая суточная доза энтерально вводимого депротеинизированного препарата из телячьей крови составляет примерно 1200 мг.
22. Депротеинизированный препарат из телячьей крови для применения по любому из пп. 1-19, где депротеинизированный препарат из телячьей крови вводят три раза в день перорально каждый раз в дозе в диапазоне от примерно 300 до примерно 500 мг.
23. Депротеинизированный препарат из телячьей крови для применения по п. 22, где депротеинизированный препарат из телячьей крови вводят три раза в день перорально каждый раз в дозе примерно 400 мг.
24. Депротеинизированный препарат из телячьей крови для применения по п. 23, где депротеинизированный препарат из телячьей крови вводят перорально в форме таблетки, содержащей примерно 200 мг указанного депротеинизированного препарата из телячьей крови.
25. Депротеинизированный препарат из телячьей крови для применения по любому из пп. с 1 по 24, где указанный депротеинизированный препарат из телячьей крови является лекарственным средством Актовегин®.
26. Депротеинизированный препарат из телячьей крови для применения по любому из пп. 1-25, где указанная болезнь периферических артерий является болезнью переферических артерий стадии II по Фонтейну.
27. Депротеинизированный препарат из телячьей крови для применения по п. 26, в котором указанная болезнь периферических артерий является болезнью периферических артерий стадии IIB по Фонтейну, например, подтвержденным ультразвуковым цветовым дуплексным сканированием.
28. Депротеинизированный препарат из телячьей крови для применения по любому из пп. с 1 по 27, где пациентом является человек, например, человек, пораженный или страдающий сахарным диабетом.
29. Депротеинизированный препарат из телячьей крови для применения по п. 28, где пациент-человек имеет допплеровский лодыжечно-плечевой индекс в покое ≤ 0,9.
30. Депротеинизированный препарат из телячьей крови для применения по пунктам 28 или 29, где у пациента-человека наблюдается перемежающаяся хромота с ICD <200 метров.
31. Депротеинизированный препарат из телячьей крови для применения по любому из пп. 28-30, в котором возраст пациента-человека составляет по меньшей мере примерно 40 лет.
32. Депротеинизированный препарат из телячьей крови для применения по любому из пп. 28-31, где пациент-человек является мужчиной.
33. Депротеинизированный препарат из телячьей крови для применения по любому из пп. 28-32, где пациент-человек является женщиной.
34. Применение депротеинизированного препарата из телячьей крови в предотвращении или лечении болезни периферических артерий, где депротеинизированный препарат из телячьей крови вводят по меньшей мере один раз парентерально, предпочтительно внутривенно, с последующим периодом энтерального введения, предпочтительно перорального введения.
35. Применение по п. 34, где депротеинизированный препарат из телячьей крови вводят парентерально один раз в день.
36. Применение по пунктам 34 или 35, где депротеинизированный препарат из телячьей крови вводят парентерально в течение по меньшей мере примерно 10 последовательных дней.
37. Применение по пунктам 34 или 35, в котором депротеинизированный препарат из телячьей крови вводят парентерально в течение периода от примерно 10 до примерно 18 последовательных дней.
38. Применение по любому из пп. 34-37, где депротеинизированный препарат из телячьей крови вводят парентерально в течение примерно 14 последовательных дней.
37. Применение по любому из пп. 34-36, где парентеральное введение представляет собой внутривенное введение.
38. Применение по п. 37, где депротеинизированный препарат из телячьей крови вводят внутривенно путем инфузии.
39. Применение по любому из пп. 34-38, где общая суточная доза парентерально вводимого депротеинизированного препарата из телячьей крови находится в диапазоне от примерно 1000 мг до примерно 1400 мг.
40. Применение по п. 39, где общая суточная доза парентерально вводимого депротеинизированного препарата из телячьей крови составляет примерно 1200 мг.
41. Применение по любому из пп.34-40, где депротеинизированный препарат из телячьей крови вводят один раз в день внутривенно в дозе от примерно 1000 мг до примерно 1400 мг.
42. Применение по п. 41, где депротеинизированный препарат из телячьей крови вводят один раз в день внутривенно в дозе примерно 1200 мг.
43. Применение по любому из пп. 34-42, где депротеинизированный препарат из телячьей крови вводят энтерально по меньшей мере один раз в день.
44. Применение по любому из пп. 34-43, где депротеинизированный препарат из телячьей крови вводят энтерально по меньшей мере два раза в день.
45. Применение по любому из пп. 34-44, где депротеинизированный препарат из телячьей крови вводят энтерально три раза в день.
46. Применение по любому из пп. 34-45, где депротеинизированный препарат из телячьей крови вводят энтерально в течение по меньшей мере примерно 63 последовательных дней.
47. Применение по любому из пп. 34-46, где депротеинизированный препарат из телячьей крови вводят энтерально в течение от 63 до 77 последовательных дней.
48. Применение по любому из пп. 34-47, где депротеинизированный препарат из телячьей крови вводят энтерально в течение примерно 70 последовательных дней.
49. Применение по любому из пп. 34-48, где энтеральное введение представляет собой пероральное введение.
50. Применение по п. 49, в котором депротеинизированный препарат из телячьей крови вводят перорально в форме таблетки.
51. Применение по любому из пп. 34-50, где общая суточная доза энтерально вводимого депротеинизированного препарата из телячьей крови находится в диапазоне от примерно 400 мг до примерно 2000 мг.
52. Применение по любому из пп. 34-51, где общая суточная доза вводимого энтерально депротеинизированного препарата из телячьей крови составляет примерно 1200 мг.
53. Применение по любому из пп.34-50, где депротеинизированный препарат из телячьей крови вводят три раза в день перорально в дозе от примерно 300 до примерно 500 мг каждый раз.
54. Применение по п. 53, где депротеинизированный препарат из телячьей крови вводят три раза в день перорально в дозе примерно 400 мг каждый раз.
55. Применение по п. 54, где депротеинизированный препарат из телячьей крови вводят перорально в форме таблетки, содержащей примерно 200 мг указанного депротеинизированного препарата из телячьей крови.
56. Применение по любому из пп. 34-55, где указанный депротеинизированный препарат из телячьей крови является лекарственным средством Актовегин®.
57. Применение по любому из пп. 34-56, где указанная болезнь периферических артерий является болезнью периферических артерий стадии II по Фонтейну.
58. Применение по п. 57, где указанная болезнь периферических артерий является болезнью периферических артерий стадии IIB по Фонтейну, например, подтвержденным ультразвуковым цветовым дуплексным сканированием.
59. Применение по любому из пп. 34-58, где пациентом является человек, такой как человек, пораженный или страдающий сахарным диабетом.
60. Применение по п. 59, где пациент-человек имеет допплеровский лодыжечно-плечевой индекс в покое ≤ 0,9.
61. Применение по пунктам 59 или 60, где у пациента-человека наблюдается перемежающаяся хромота с ICD <200 метров.
62. Применение по любому из пп. 59-61, где возраст пациента-человека составляет по меньшей мере примерно 40 лет.
63. Применение по любому из пп. 59-62, где пациент-человек является мужчиной.
64. Применение по любому из пп. 59-62, где пациент-человек является женщиной.
65. В другом аспекте настоящее изобретение относится к способу профилактики или лечения болезни периферических артерий у пациента, который в этом нуждается, причем указанный способ включает в себя следующие этапы: а) введение по меньшей мере один раз парентерально, предпочтительно внутривенно, указанному пациенту терапевтически эффективного количества депротеинизированного препарата из телячьей крови, такого как лекарственное средство Актовегин®, с последующим b) введением по меньшей мере один раз энтерально, предпочтительно перорально, указанному пациенту терапевтически эффективного количества указанного депротеинизированного препарата из телячьей крови.
66. Способ по п. 65, где депротеинизированный препарат из телячьей крови вводят парентерально один раз в день.
67. Способ по пунктам 65 или 66, где депротеинизированный препарат из телячьей крови вводят парентерально в течение по меньшей мере примерно 10 последовательных дней.
68. Способ по пунктам 65 или 66, где депротеинизированный препарат из телячьей крови вводят парентерально в течение от примерно 10 до примерно 18 последовательных дней.
69. Способ по любому из пп. 65-68, где депротеинизированный препарат из телячьей крови вводят парентерально в течение примерно 14 последовательных дней.
70. Способ по любому из пп. 65-69, где парентеральное введение представляет собой внутривенное введение.
71. Способ по п. 70, где депротеинизированный препарат из телячьей крови вводят внутривенно путем инфузии.
72. Способ по любому из пп. 65-71, где общая суточная доза парентерально вводимого депротеинизированного препарата из телячьей крови находится в диапазоне от примерно 1000 мг до примерно 1400 мг.
73. Способ по п. 72, где общая суточная доза парентерально вводимого депротеинизированного препарата из телячьей крови составляет примерно 1200 мг.
74. Способ по любому из пп. 65-71, где депротеинизированный препарат из телячьей крови вводят один раз в день внутривенно в дозе от примерно 1000 мг до примерно 1400 мг.
75. Способ по п. 74, где депротеинизированный препарат из телячьей крови вводят один раз в день внутривенно в дозе примерно 1200 мг.
76. Способ по любому из пп. 65-75, где депротеинизированный препарат из телячьей крови вводят энтерально по меньшей мере один раз в день.
77. Способ по любому из пп. 65-76, где депротеинизированный препарат из телячьей крови вводят энтерально по меньшей мере два раза в день.
78. Способ по любому из пп. 65-77, где депротеинизированный препарат из телячьей крови вводят энтерально три раза в день.
79. Способ по любому из пп. 65-78, где депротеинизированный препарат из телячьей крови вводят энтерально в течение периода по меньшей мере примерно 63 последовательных дней.
80. Способ по любому из пп. 65-79, где депротеинизированный препарат из телячьей крови вводят энтерально в течение от примерно 63 до примерно 77 последовательных дней.
81. Способ по любому из пп. 65-80, где депротеинизированный препарат из телячьей крови вводят энтерально в течение периода приблизительно 70 последовательных дней.
82. Способ по любому из пп. 65-81, где энтеральное введение представляет собой пероральное введение.
83. Способ по п. 82, где депротеинизированный препарат из телячьей крови вводят перорально в форме таблетки.
84. Способ по любому из пп. с 65 по 83, где общая суточная доза вводимого энтерально депротеинизированного препарата из телячьей крови находится в диапазоне от примерно 400 мг до примерно 2000 мг.
85. Способ по любому из пп. 65-84, где общая суточная доза вводимого энтерально депротеинизированного препарата из телячьей крови составляет примерно 1200 мг.
86. Способ по любому из пп.65-83, где депротеинизированный препарат из телячьей крови вводят три раза в день каждый раз перорально в дозе от примерно 300 до примерно 500 мг.
87. Способ по п. 86, где депротеинизированный препарат из телячьей крови вводят три раза в день каждый раз перорально в дозе примерно 400 мг.
88. Способ по п. 87, где депротеинизированный препарат из телячьей крови вводят перорально в форме таблетки, содержащей примерно 200 мг указанного депротеинизированного препарата из телячьей крови.
89. Способ по любому из пп. 65-88, где указанный депротеинизированный препарат из телячьей крови является лекарственным средством Актовегин®.
90. Способ по любому из пп. 65-89, где указанная болезнь периферических артерий является болезнью периферических артерий стадии II по Фонтейну.
91. Способ по п. 90, где указанная болезнь периферических артерий является болезнью периферических артерий стадии IIB по Фонтейну, например подтвержденной ультразвуковым цветовым дуплексным сканированием.
92. Способ по любому из пп. 65-191, где пациентом является человек, такой как человек, пораженный или страдающий сахарным диабетом.
93. Способ по п. 92, где у пациента-человека наблюдается допплеровский лодыжечно-плечевой индекс в покое ≤ 0,9.
94. Способ по п. 92 или 93, где у пациента-человека наблюдается перемежающаяся хромота с ICD <200 метров.
95. Способ по любому из пп. 92-94, где возраст пациента-человека составляет по меньшей мере 40 лет.
96. Способ по любому из пп. 92-95, где пациент-человек является мужчиной.
97. Способ по любому из пп. с 92-95, где пациент-человек является женщиной.
Краткое описание чертежей
Фигура 1: Схема дизайна исследования
Подробное описание изобретения
За исключением специально определенных в данном документе случаев, все используемые технические и научные термины имеют то же значение, которое обычно понимается квалифицированным специалистом в области медицины и фармацевтики.
Все способы и материалы, аналогичные или эквивалентные описанным здесь, могут быть использованы при осуществлении или тестировании настоящего изобретения; подходящие для способы и материалы описаны в настоящем документе. Все публикации, патентные заявки, патенты и другие источники, упомянутые в настоящем документе, полностью включены него посредством ссылок. В случае конфликта настоящее описание, включая определения, будет иметь преимущественную силу. Кроме того, материалы, способы и примеры являются только иллюстративными и не имеют ограничительного характера, если не указано иное.
В случаях, когда здесь указывается числовой предел или диапазон, крайние точки также считаются включенными. Кроме того, все значения и поддиапазоны в пределах любого числового предела или диапазона считаются специально включенными, как если бы они были явно выписаны.
Ниже настоящее изобретение будет описано более подробно.
В соответствии с одним аспектом в настоящем изобретении предложен депротеинизированный препарат из телячьей крови для применения в предотвращении или лечении болезни периферических артерий (БПА).
Болезнь периферических артерий (БПА), также известная как "окклюзионная болезнь периферических артерий", является хорошо известным проявлением системного атеросклероза, определяющимся прогрессирующим стенозом или окклюзией. Болезнь периферических артерий чаще всего поражает артерии нижних конечностей, но также могут быть затронуты другие артерии. Классическим симптомом является боль в ногах при ходьбе, которая проходит при отдыхе, известная как перемежающаяся хромота. В пораженной ноге могут возникать и другие симптомы, включая кожные язвы, синюшную кожу, холодную кожу или плохой рост ногтей и волос. Осложнения могут включать инфекцию или омертвение ткани, которое может потребовать ампутации; ишемическую болезнь сердца или инсульт. До 50% случаев БПА являются бессимптомными. Общеизвестные факторы риска включают курение, сахарный диабет, дислипидемию и гипертонию. Риск БПА также возрастает у лиц старше 50 лет, мужчин, лиц, страдающих ожирением, сердечными приступами или после инсульта или с семейной историей сосудистых заболеваний.
Более конкретно, в настоящем изобретении предложен депротеинизированный препарат из телячьей крови, такой как лекарственное средство Актовегин®, для применения в предотвращении или лечении болезни периферических артерий, где указанный депротеинизированный препарат из телячьей крови вводят по меньшей мере один раз парентерально, предпочтительно внутривенно, с последующим периодом энтерального введения, предпочтительно перорального введения.
В настоящем изобретении также предложено применение депротеинизированного препарата из телячьей крови, такого как лекарственное средство Актовегин®, в предотвращении или лечении болезни периферических артерий, при этом депротеинизированный препарат из телячьей крови вводят по меньшей мере один раз парентерально, предпочтительно внутривенно, с последующим периодом энтерального введения, предпочтительно перорального введения.
В настоящем изобретении также предложен способ профилактики или лечения болезни периферических артерий у пациента, нуждающегося в этом, причем указанный способ включает следующие этапы: а) введение по меньшей мере один раз парентерально, предпочтительно внутривенно, указанному пациенту терапевтически эффективного количества депротеинизированного препарата из телячьей крови, такого как лекарственное средство Актовегин®, с последующим б) введением по меньшей мере один раз энтерально, предпочтительно перорально, указанному пациенту терапевтически эффективного количества указанного депротеинизированного препарата из телячьей крови.
В настоящем документе термин "парентеральный" или "парентерально" относится к пути введения лекарственного средства, которое не затрагивает желудочно-кишечный тракт (ЖКТ), и включает любую соответствующую форму введения, такую как внутривенное (инъекция в вену), подкожное (инъекция под кожу), внутримышечное (инъекция в мышцу), ингаляция (инфузия через легкие) или чрескожное (абсорбция через неповрежденную кожу).
В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят парентерально один раз в день.
В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят парентерально в течение по меньшей мере примерно 10 последовательных дней, например, в течение периода, равного от примерно 10 до примерно 18, от примерно 11 до примерно 17, примерно от 12 до примерно 16 или от 13 до 15 последовательных дней.
В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят парентерально в течение периода от примерно 10 до примерно 18 последовательных дней. В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят парентерально в течение периода от примерно 11 до примерно 17 последовательных дней. В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят парентерально в течение периода от примерно 12 до примерно 16 последовательных дней. В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят парентерально в течение периода от примерно 13 до примерно 15 последовательных дней.
В соответствии с некоторыми вариантами осуществления настоящего изобретения, депротеинизированный препарат из телячьей крови вводят парентерально в течение примерно 14 последовательных дней.
В соответствии с некоторыми вариантами осуществления настоящего изобретения парентеральное введение представляет собой внутривенное введение, например, путем инфузии.
Обычно вводят объем инфузии от примерно 100 мл до примерно 500 мл, содержащий депротеинизированный препарат из телячьей крови, например примерно 200 мл, 250 мл, 280 мл, 300 мл, 350 мл, 400 мл или 450 мл. В соответствии с некоторыми вариантами осуществления вводят объем инфузии от 200 до 400 мл, содержащий депротеинизированный препарат из телячьей крови. В соответствии с некоторыми вариантами осуществления вводят объем инфузии от примерно 250 до примерно 300 мл, содержащий депротеинизированный препарат из телячьей крови. В соответствии с некоторыми вариантами осуществления вводят объем инфузии примерно 280 мл, содержащий депротеинизированный препарат из телячьей крови.
Рекомендуемая общая суточная доза для парентерально вводимого депротеинизированного препарата из телячьей крови, такого как лекарственное средство Актовегин®, находится в диапазоне от примерно 800 до примерно 2000 мг. Таким образом, в соответствии с некоторыми вариантами осуществления настоящего изобретения общая суточная доза парентерально вводимого депротеинизированного препарата из телячьей крови находится в диапазоне от примерно 800 до примерно 2000 мг.
В соответствии с некоторыми вариантами осуществления настоящего изобретения общая суточная доза парентерально вводимого депротеинизированного препарата из телячьей крови находится в диапазоне от примерно 900 мг до примерно 1800 мг. В соответствии с некоторыми вариантами осуществления настоящего изобретения общая суточная доза парентерально вводимого депротеинизированного препарата из телячьей крови находится в диапазоне от примерно 1000 мг до примерно 1400 мг. В соответствии с некоторыми вариантами осуществления настоящего изобретения общая суточная доза парентерально вводимого депротеинизированного препарата из телячьей крови находится в диапазоне от примерно 1100 до примерно 1300 мг.
В соответствии с некоторыми вариантами осуществления настоящего изобретения, общая суточная доза вводимого парентерально депротеинизированного препарата из телячьей крови составляет примерно 1200 мг.
В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят один раз в день внутривенно, например, путем инфузии, в дозе от примерно 1000 мг до примерно 1400 мг.
В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят один раз в день внутривенно, например, путем инфузии, в дозе примерно 1200 мг.
Как описано выше, за периодом парентерального введения следует период энтерального введения.
В настоящем документе термин "энтеральный" или "энтерально" относится к пути введения лекарственного средства через желудочно-кишечный тракт и включает любую соответствующую форму введения, такую как оральное, сублингвальное (растворение лекарственного средства под языком) или ректальное.
В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят энтерально по меньшей мере один раз в день. В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят энтерально по меньшей мере два раза в день.
В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят энтерально три раза в день.
В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят энтерально в течение по меньшей мере примерно 63 последовательных дней, например, в течение периода от примерно 65 до примерно 75, от примерно 66 до примерно 74, от примерно 67 до примерно 73, от 68 до 72 или от 69 до 71 дня подряд.
В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят энтерально в течение периода от 63 до 77 последовательных дней. В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят энтерально в течение периода от 65 до 75 последовательных дней. В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят энтерально в течение периода от примерно 66 до примерно 74 последовательных дней. В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят энтерально в течение периода от 67 до 73 последовательных дней. В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят энтерально в течение периода от примерно 68 до примерно 72 последовательных дней. В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят энтерально в течение периода от 69 до 71 дня подряд.
В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят энтерально в течение примерно 70 последовательных дней.
В соответствии с некоторыми вариантами осуществления настоящего изобретения энтеральное введение представляет собой пероральное введение.
В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят перорально в форме таблетки.
Рекомендуемая общая суточная доза для энтерально вводимого депротеинизированного препарата из телячьей крови, такого как лекарственное средство Актовегин®, находится в диапазоне от примерно 400 мг до примерно 2000 мг. Таким образом, в соответствии с некоторыми вариантами осуществления настоящего изобретения общая суточная доза парентерально вводимого депротеинизированного препарата из телячьей крови находится в диапазоне от примерно 400 мг до примерно 2000 мг.
В соответствии с некоторыми вариантами осуществления настоящего изобретения общая суточная доза энтерально вводимого депротеинизированного препарата из телячьей крови находится в диапазоне от примерно 600 мг до примерно 1800 мг. В соответствии с некоторыми вариантами осуществления настоящего изобретения общая суточная доза энтерально вводимого депротеинизированного препарата из телячьей крови находится в диапазоне от примерно 800 до примерно 1600 мг. В соответствии с некоторыми вариантами осуществления настоящего изобретения общая суточная доза энтерально вводимого депротеинизированного препарата из телячьей крови находится в диапазоне от примерно 1000 мг до примерно 1400 мг. В соответствии с некоторыми вариантами осуществления настоящего изобретения общая суточная доза энтерально вводимого депротеинизированного препарата из телячьей крови находится в диапазоне от примерно 1100 мг до примерно 1300 мг.
В соответствии с некоторыми вариантами осуществления настоящего изобретения общая суточная доза энтерально вводимого депротеинизированного препарата из телячьей крови составляет примерно 1200 мг.
В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят три раза в день перорально в дозе в диапазоне от примерно 300 до примерно 500 мг каждый раз.
В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят три раза в день перорально в дозе примерно 400 мг каждый раз.
В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови вводят перорально в форме таблетки, содержащей примерно 200 мг указанного депротеинизированного препарата из телячьей крови.
Депротеинизированные препараты из телячьей крови хорошо известны специалистам в данной области. Примеры приготовления таких препаратов подробно описаны в Примерах 1-3 Международной заявки на патент WO 2010/034315, содержание которой включено в настоящее описание посредством конкретной ссылки. Предпочтительный препарат из телячьей крови доступен на рынке в виде лекарственного средства Актовегин®.
В соответствии с некоторыми вариантами осуществления настоящего изобретения депротеинизированный препарат из телячьей крови является лекарственным средством Актовегин®.
В соответствии с некоторыми вариантами осуществления настоящего изобретения болезнь периферических артерий является болезнью периферических артерий стадии II по Фонтейну.
В соответствии с некоторыми вариантами осуществления настоящего изобретения болезнь периферических артерий является болезнью периферических артерий стадии IIB по Фонтейну.
Болезнь периферических артерий II стадии по Фонтейну, и более конкретно, болезнь периферических артерий стадии IIB по Фонтейну, может быть подтверждена с помощью ультразвукового цветового дуплексного сканирования.
В соответствии с некоторыми вариантами осуществления настоящего изобретения пациент, подлежащий лечению, является человеком.
В соответствии с некоторыми вариантами осуществления настоящего изобретения пациент-человек является мужчиной.
В соответствии с некоторыми вариантами осуществления настоящего изобретения пациент-человек является женщиной.
В соответствии с определенными вариантами осуществления настоящего изобретения у пациента-человека наблюдается допплеровский лодыжечно-плечевой индекс ≤ 0,9.
В соответствии с некоторыми вариантами осуществления настоящего изобретения у пациента-человека наблюдается перемежающаяся хромота с ICD <200 метров.
В соответствии с некоторыми вариантами осуществления настоящего изобретения пациентом-человеком является взрослый человек.
В соответствии с некоторыми вариантами осуществления настоящего изобретения возраст пациента-человека составляет по меньшей мере 40 лет. В соответствии с некоторыми вариантами осуществления, возраст пациента-человека составляет по меньшей мере 45 лет. В соответствии с некоторыми вариантами осуществления возраст пациента-человека составляет не менее 50 лет. В соответствии с некоторыми вариантами осуществления, возраст пациента-человека составляет по меньшей мере 55 лет. В соответствии с некоторыми вариантами осуществления, возраст пациента-человека составляет по меньшей мере 60 лет. В соответствии с некоторыми вариантами осуществления, возраст пациента-человека составляет по меньшей мере 65 лет. В соответствии с некоторыми вариантами осуществления возраст пациента-человека составляет по меньшей мере 70 лет.
В соответствии с некоторыми вариантами осуществления настоящего изобретения возраст пациента-человека составляет от 40 до 75 лет. В соответствии с некоторыми вариантами осуществления возраст пациента-человека составляет от 45 до 75 лет. В соответствии с некоторыми вариантами осуществления, возраст пациента-человека составляет примерно от 50 до 75 лет. В соответствии с некоторыми вариантами осуществления, возраст пациента-человека составляет примерно от 55 до 75 лет. В соответствии с некоторыми вариантами осуществления, возраст пациента-человека составляет от 60 до 75 лет. В соответствии с некоторыми вариантами осуществления возраст пациента-человека составляет примерно 65-75 лет.
В соответствии с некоторыми вариантами осуществления пациент-человек страдает сахарным диабетом, таким как сахарный диабет 1-го типа или сахарный диабет типа 2. В соответствии с некоторыми вариантами осуществления пациент-человек страдает сахарным диабетом 1-го типа. В соответствии с некоторыми вариантами осуществления пациент-человек страдает сахарным диабетом 2-го типа.
В соответствии с определенными вариантами осуществления возраст пациента-человека составляет по меньшей мере 40 лет, например, по меньшей мере 50 лет, и страдает сахарным диабетом.
В соответствии с определенными вариантами осуществления возраст пациента-человека составляет по меньшей мере 40 лет, например, по меньшей мере 50 лет, и он страдает сахарным диабетом и имеет по меньшей мере один сердечно-сосудистый фактор риска, такой как курение.
В соответствии с некоторыми вариантами осуществления возраст пациента-человека составляет по меньшей мере 40 лет, например, по меньшей мере 50 лет, и он страдает сахарным диабетом и имеет по меньшей мере еще один фактор риска атеросклероза, например другой фактор риска атеросклероза, выбранный из группы, включающей курение, дислипидемию, гипертониию или гипергомоцистеинемию.
Список сокращений
ACD - абсолютная дистанция, проходимая при перемежающейся хромоте
НЛР - нежелательная лекарственная реакция
НЯ - нежелательное явление
АЛТ - аланинаминотрансфераза
АСТ - аспартатаминотрансфераза
CPMP - Комитет по патентованным лекарственным средствам
ЭКГ - электрокардиограмма
eCRF - электронная индивидуальная регистрационная карта
EMA - Европейское агентство по лекарственным средствам
ФСГ - фолликулостимулирующий гормон
GCP - Правила проведения качественных клинических исследований
HbA1c - гликозилированный гемоглобин
ХГЧ - хорионический гонадотропин человека
IC- перемежающаяся хромота
ICD - начальная дистанция, проходимая при перемежающейся хромоте
ICH - Международная конференция по гармонизации
ИЛП - исследуемый лекарственный препарат
ИП - исследуемый продукт
IRT - интерактивная технология связи
в/в - внутривенно
LFT - функциональная проба печени
LSU - липопротеинлипазная единица
MedDRA - Медицинский словарь регуляторной деятельности
Med ID - идентификационный номер лекарственного средства
PAD - болезнь периферических артерий
PSUR - периодический отчет по безопасности лекарственного средства
PTE - нежелательное явление, возникшее до начала лечения
СНЯ- серьезное нежелательное явление
SAP - план статистического анализа
SF-36 - краткий опросник по оценке состояния здоровья из 36 пунктов
SUSAR - Предполагаемая непредвиденная серьезная нежелательная реакция
TID - 3 раза в день
ВГН - верхняя граница нормы
С учетом общего описания этого изобретения, дополнительное понимание может быть получено при помощи ссылки на некоторые конкретные примеры, которые приведены здесь только для иллюстрации и не имеют ограничительного характера, если не указано иное.
Примеры
Пример 1 - Рандомизированное международное многоцентровое, в параллельных группах, двойное слепое, плацебоконтролируемое исследование для оценки эффективности и безопасности 12-недельного лечения препаратом Актовегин®, вводимым сперва внутривенно и в дальнейшем перорально у пациентов с окклюзионным болезнью периферических артерий стадии IIB по Фонтейну
1. ЦЕЛИ И КОНЕЧНЫЕ ТОЧКИ ИССЛЕДОВАНИЯ
1.1 Цели
1.1.1 Основная цель
Основная цель - исследование эффективности Актовегина® для симптоматического лечения БПА стадии IIB по Фонтейну.
1.1.2 Вторичные цели
Изучение влияния Актовегина® на устойчивое улучшение дистанции, проходимой при хромоте, у пациентов с БПА стадии IIB по Фонтейну.
Изучение влияния Актовегина® на качество жизни пациентов.
1.1.3 Цель по безопасности
Оценка безопасности Актовегина® по сравнению с плацебо.
1.2 Конечные точки
1.2.1 Основная конечная точка
Первичная конечная точка представляет собой процентное изменение начальной дистанции, проходимой при хромоте (ICD), от исходного уровня до 12 недель лечения.
1.2.2 Вторичные конечные точки
Процентное изменение ICD от исходного уровня до 2 и 24 недель после рандомизации.
Изменение абсолютной дистанции при перемежающейся хромоте (ACD) от исходного уровня до 2, 12 и 24 недель после рандомизации.
Доля пациентов с болью в состоянии покоя через 12 и 24 недель после рандомизации.
Доля пациентов, подвергнутых процедурам реваскуляризации, через 24 недель после рандомизации.
Изменение в баллах краткого опросника по оценке состояния здоровья из 36 пунктов (SF-36) через 12 и 24 недель после рандомизации.
1.2.3 Дополнительные конечные точки: безопасность
Безопасность и переносимость Актовегина® будут оцениваться путем оценки нежелательных явлений (НЯ), клинических лабораторных тестов безопасности, жизненно важных показателей, веса, электрокардиограммы (ЭКГ) и физического обследования.
2. ДИЗАЙН И ОПИСАНИЕ ИССЛЕДОВАНИЯ
2.1 Дизайн исследования
Это рандомизированное многоцентровое, в параллельных группах, двойное слепое, плацебоконтролируемое исследование фазы 3b для оценки эффективности и безопасности 12-недельного лечения Актовегином®, вводимого сперва внутривенно и в дальнейшем перорально у пациентов с БПА стадии IIB по Фонтейну.
В исследование будут зачислены в общей сложности 366 пациентов с БПА стадии IIB по Фонтейну в примерно 20-25 центрах в 5 странах (в России, Украине, Белоруссии, Казахстане и Грузии).
Исследование будет состоять из 1-2-недельного периода скрининга, рандомизации, 12-недельного периода лечения и 12-недельного периода последующего наблюдения. Общая продолжительность исследования составит от 25 до 26 недель.
Пациенты будут проходить 1-2-недельный период период скрининга, в течение которого будет проверена стабильность состояния пациента, будет подтвержден диагноз БПА, и будут выявлены и исключены пациенты с высокой нестабильностью проходимой дистанции. Для этой цели будут выполняться 2 теста на беговой дорожке с промежутком ≥1 недели и ≤2 недель. Пациенты, показавшие изменение в ACD более чем на 25% в течение скринингового периода, будут исключены из исследования.
Право на включение в исследование будут иметь пациенты с историей перемежающейся хромотой продолжительностью не менее 6 месяцев до включения. Диагноз БПА (ICD <200 метров) будет подтвержден с помощью ультразвукового цветового дуплексного сканирования и теста на беговой дорожке.
Пригодные пациенты будут рандомизированы в соотношении 1:1 для получения либо Актовегина®, либо плацебо. Период лечения будет включать 2 недели в/в инфузий Актовегина® (депротеинизированный гемо-дериват) в дозе 1200 мг/сут, а затем 10 недель перорального приема в таблетках в дозе 1200 мг/день (две таблетки по 200 мг три раза ежедневно [TID]). На протяжении всего периода лечения для поддержания ослепления будет использоваться соответствующее по виду плацебо (в ампулах и таблетках). Общая продолжительность лечения составит 12 недель.
За 12-недельным периодом лечения последует 12-недельный период наблюдения без лечения исследуемым лекарственным средством (ИЛП), предназначенный для изучения стойкой эффективности после лечения, а также безопасности после прекращения лечения Актовегином®.
Схема дизайна исследования приведена на рисунке 1. График оценок приведен в таблице 1.
Таблица 1: График процедур исследования
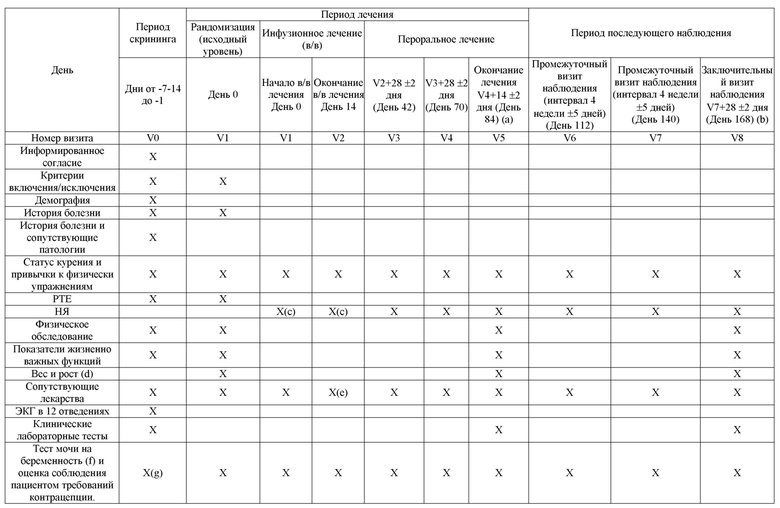
Сноски расшифрованы на последней странице таблицы.
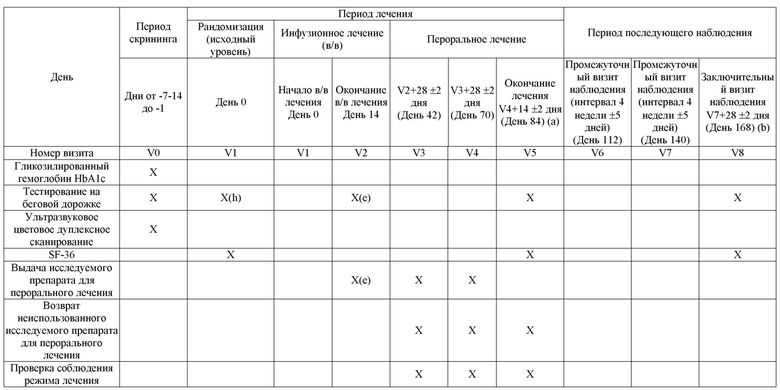
V= визит.
(a) Визит досрочного прекращения для пациентов, которые прекращают участие в исследовании до завершения периода перорального лечения.
(b) Визит досрочного прекращения для пациентов, которые прекращают участие в исследовании в течение периода последующего наблюдения.
(c) В День 0, Визит 1, НЯ следует проверять после инфузии. Начиная с Дня 1, НЯ следует проверять до инфузии [НЯ с момента последнего визита] и после инфузии.
(d) Высота и вес будут измеряться на визите V1. На визитах V5 и V8 будет измеряться только вес.
(e) После заключительной инфузии.
(f) только у женщин, способных к деторождению.
(g) Оценка соблюдения требований контрацепции на этом визите не требуется.
(h) Промежуток времени от первого теста должен составлять ≥ 1 недели и ≤2 недель.
2.2 Обоснование дизайна исследования, дозы и конечных точек
Данное рандомизированное, в параллельных группах, двойное слепое, плацебоконтролируемое исследование согласовано с рекомендациями CPMP/EWP/714/98 "Примечание к руководству по клиническим исследованиям лекарственных средств для лечения окклюзионной болезни периферических артерий".
Схема применения для этого исследования соответствует инструкции по применению лекарственного средства, за исключением того, что пероральное лечение будет более длительным, чем указано в инструкции. Рекомендуемая доза для Актовегина® составляет от 800 до 2000 мг (20-50 мл) внутривенно в течение 4 недель. Рекомендуемая доза для таблеток составляет от 1 до 2 таблеток (каждая таблетка, содержащая 200 мг Актовегина®) TID со средней продолжительностью лечения от 4 до 6 недель. Пероральная схема в данном исследовании - 2 таблетки (200 мг) TID (1200 мг/день) в течение 10 недель.
Эффективность Актовегина® 1000 мг в/в была продемонстрирована в сравнительном рандомизированном исследовании у пациентов с БПА стадии IIB по Фонтейну. Максимальная дистанция ходьбы увеличилось после 4 недель лечения на 100,7% в группе Актовегина® по сравнению с 67% и 8,1% в группах нафтидрофурила и витамина B, соответственно [22]. В другом открытом контролируемом исследовании Актовегина® назначался по 1000 мг в/в ежедневно в течение 10 дней и показал увеличение дистанции безболевой ходьбы, сравнимое с группой пентоксифиллина. Анализ микроциркуляции в коже с использованием лазерной допплеровской флоуметрии показал, что Актовегин® имеет ряд значительных эффектов, более выраженных по сравнению с группой пентоксифиллина [23].
Такой длительный период перорального лечения продемонстрировал свою безопасность и эффективность в предыдущих испытаниях Актовегина®.
В качестве основной конечной точки изменения ICD от исходного уровня была выбрана перемежающаяся хромота, так как она является ключевым симптомом II этапа БПА и рекомендована в руководстве Европейского агентства по лекарственным средствам (EMA) "Примечание к руководству по клиническим исследованиям лекарственных средств для лечения окклюзионного болезни периферических артерий".
3. ОТБОР И ПРЕКРАЩЕНИЕ ЛЕЧЕНИЯ/ПРЕКРАЩЕНИЕ УЧАСТИЯ ПАЦИЕНТОВ
Все критерии включения, включая результаты тестов, должны быть подтверждены до рандомизации или первой дозы.
3.1 Критерии включения
Пригодность пациента для участия определяется в соответствии со следующими критериями до вступления в исследование:
1. Пациент - мужчина или женщина и в возрасте от 40 до 75 лет включительно.
2. В анамнезе стабильная перемежающаяся хромота, которая длится более 6 месяцев до скрининга.
3. Диагноз БПА (код I70.2 в соответствии с Международной классификацией болезней 10-го пересмотра) стадии IIB по Фонтейну подтвержден с помощью ультразвукового цветового дуплексного сканирования.
4. Впервые диагностирована БПА или в анамнезе стабильная терапия БПА (включая статус курения, привычки к физическим упражнениям и лекарства) в течение как минимум 3 месяцев до рандомизации.
5. Доплеровский лодыжечно-плечевой индекс=0,9.
6. Перемежающаяся хромота с ICD <200 метров.
7. По мнению исследователя, пациент способен понимать и соблюдать требования протокола.
8. Пациент или, в соответствующих случаях, законный представитель пациента подписывает и датирует письменную форму информированного согласия и любое необходимое разрешение на обработку личных данных до начала любых процедур исследования.
9. Пациент-мужчина, который не стерилизован и сексуально активен с партнершей, способной к деторождению, соглашается использовать барьерный метод контрацепции (например, презерватив со спермицидом или без него) с момента подписания информированного согласия на протяжении всего исследования или в течение 30 дней после последней дозы в случае преждевременного прекращения лечения. Партнерше пациента-мужчины также следует рекомендовать использовать высокоэффективный/эффективный метод контрацепции на протяжении всего исследования или в течение 30 дней после последней дозы в случае преждевременного прекращения.
10. Пациент-женщина, способная к деторождению, которая сексуально активна с нестерилизованным партнером-мужчиной, соглашается использовать эффективный или высокоэффективный метод контрацепции с момента подписания информированного согласия на протяжении всего исследования или в течение 30 дней после последней дозы в случае преждевременного прекращения. Определения эффективных/высокоэффективных методов контрацепции приведены в разделе 5.1.11, а ответственность по информированию определена в разделе 5.1.12.
3.2 Критерии исключения
Любой пациент, отвечающий любому из следующих критериев после периода скрининга, непригоден для участия в исследования:
1. БПА стадии III или IV по Фонтейну (боль в состоянии покоя, незаживающее изъязвление или гангрена).
2. Признаки неатеросклеротической БПА.
3. Вариабельность ACD > 25% по данным теста на беговой дорожке в течение периода скрининга.
4. Реконструкция артерий нижней конечности (хирургическая или эндоваскулярная) или симпатэктомии в течение 3 месяцев до скрининга.
5. Кандидат на реваскуляризацию или ангиопластику.
6. У пациента был инфаркт миокарда или хирургическое вмешательство на сердце в течение 3 месяцев до скрининга.
7. Застойная сердечная недостаточность (класс III/IV по классификации Нью-йоркской кардиологической ассоциации).
8. Неконтролируемый сахарный диабет (гликозилированный гемоглобин [HbA1c> 9%]) или диабетическая полинейропатия.
9. Какая-либо другая болезнь, которая значительно ограничивает способность к нагрузке, или другое медицинское состояние, включая любое психическое расстройство, которое ограничивает участие (по мнению исследователя).
10. Прием любого запрещенного лекарства (раздел 7.3) в течение 14 дней до рандомизации (день 0).
11. Прием любого экспериментального соединения в течение 30 дней до скрининга.
12. Прием Актовегина® в качестве терапевтического агента в течение 30 дней до начала скрининга.
13. Пациент является непосредственным членом семьи сотрудника, сотрудником центра исследования, родственником сотрудника центра, который участвует в проведении этого исследования (например, является супругом, родителем, ребенком, родным братом) или может согласиться на участие по принуждению.
14. Клинически значимые по мнению исследователя аномальные гематологические параметры гемоглобина, гематокрита или эритроцитов при скрининге.
15. В анамнезе гиперчувствительность или аллергия на Актовегин® или аналогичные препараты или вспомогательные вещества.
16. В анамнезе злоупотребление веществами (определяемое как любое употребление запрещенных средств) или злоупотребление алкоголем в течение 6 месяцев до скрининга.
17. Для пациентов-женщин, беременность или кормление грудью, или намерение забеременеть до участия в этом исследовании и в ходе исследования или намерение стать донором яйцеклетки в течение этого периода времени.
18. Участие пациента в другом клиническом исследовании в течение 30 дней до скрининга.
3.3 Исключенные лекарства и терапевтические процедуры
Сопутствующие и предшествующие лекарства следует тщательно проверять и документировать в электронной индивидуальной регистрационной карте (eCRF).
Ниже перечислены лекарства, которые запрещены в ходе исследования:
Алпростадил и другие простагландины
Ангиогенные факторы роста
Хроническое применение нестероидных противовоспалительных средств
Цилостазол
Экстракт гинкго билоба
L-карнитин
L-лизин эсцинат
Нафтидрофурил
Никотиновая кислота
Пентоксифиллин
Сулодексид
Винкамин
Гипербарическая оксигенация и физиотерапия любого типа, которые могут потенциально влиять на периферическое кровообращение, в ходе исследования не допускаются.
Применение сопутствующего лекарства для лечения любого другого острого или хронического заболевания может быть начато или продолжено в соответствии с назначением врача. Пациентов следует проинструктировать не принимать какие-либо лекарства, в том числе безрецептурные, без предварительной консультации с исследователем. Дополнительная информация о сопутствующей терапии БПА представлена в разделе 4.1.1.2.
3.4 Рацион, потребление жидкости, контроль активности, лечебные учреждения и общие рекомендации и ограничения
Все пациенты получат рекомендации по изменению образа жизни, чтобы свести к минимуму факторы риска для сосудов (например, прекращение курения и регулярные физические упражнения). Курение и привычка к физическим упражнениям должны быть задокументированы и стабильны как минимум за 3 месяца до рандомизации.
Кроме того, на БПА также могут влиять лечение и статус гликемического контроля у пациентов с диабетом, а также нерегулярное использование нестероидных противовоспалительных агентов. Эти препараты следует проанализировать и задокументировать в eCRF.
Во время периода в/в лечения требуется госпитализация или дневной стационар, чтобы обеспечить выполнение процедур лечения.
3.5 Критерии прекращения лечения или участия пациента
Основная причина прекращения лечения исследуемым препаратом или участия пациента в исследовании должна быть записана в eCRF, с использованием следующих категорий, перечисленных ниже. Случаи пациентов, не прошедших скрининг обсуждаются в Разделе 5.1.14.
1. Нежелательное явление до начала лечения (PTE) или НЯ. Пациент испытал PTE или НЯ, требующие досрочного прекращения участия, поскольку продолжение участия налагает неприемлемый риск для здоровья пациента или пациент не желает продолжать участие из-за PTE или НЯ.
PTE определяется как любое неблагоприятное медицинское явление у пациента клинического исследования, который подписал информированное согласие на участие в исследовании, но до введения любого исследуемого препарата; оно не обязательно должно иметь причинно-следственную связь с участием в исследовании.
НЯ определяется как любое неблагоприятное медицинское явление у пациента клинического исследования, которому вводили лекарственное средство; оно не обязательно должно иметь причинно-следственную связь с этим лечением.
Таким образом, НЯ представлять собой любой неблагоприятный и непреднамеренный показатель (например, клинически значимая аномальная лабораторная величина), симптом или заболевание, связанные по времени с применением лекарственного средства независимо от того, сочтено ли оно обусловленным лекарственным средством.
Аномальные показатели функции печени (LFT)
Прием исследуемого препарата следует немедленно прекратить с соответствующим клиническим наблюдением (включая повторные лабораторные анализы, пока лабораторный профиль пациента не вернется к нормальному/исходному состоянию, см. Раздел 5.1.10), при наступлении в любое время во время лечения исследуемым препаратом следующих условий:
- аланинаминотрансфераза (АЛТ) или аспартатаминотрансфераза (АСТ)> 8 × верхняя граница нормы (ВГН) или
- АЛТ или АСТ> 5 × ВГН и сохраняется более 2 недель, или
- АЛТ или АСТ> 3 × ВГН в сочетании с повышенным общим билирубином > 2 × ВГН или
- АЛТ или АСТ> 3 × ВГН с появлением по меньшей мере 1 из следующих симптомов: усталость, тошнота, рвота, боль или болезненность в правом верхнем квадранте, лихорадка, сыпь и/или эозинофилия (> 5%).
2. Значительное отклонение от протокола. Обнаружение после рандомизации, что пациент не соответствовал критериям включения согласно протоколу или не соблюдал требования протокола, и продолжение участия несет неприемлемый риск для здоровья пациента.
3. Невозможность последующего наблюдения из-за утраты связи с пациентом. Пациент не вернулся в клинику, и попытки связаться с ним не увенчались успехом. Попытки связаться с пациентом должны быть задокументированы в исходных документах пациента.
4. Добровольное прекращение участия. Пациент (или законный представитель пациента) желает выйти из исследования. Причина прекращения участия, если она предоставлена, должна быть записана в eCRF.
Примечание: необходимо предпринять все попытки определить первопричину выхода и, по возможности, зафиксировать определяющую причину (т.е. выход из-за НЯ не следует регистрировать в категории "добровольного выхода". Аналогичным образом, отсутствие эффективности не следует регистрировать в категории "добровольного выхода").
5. Прекращение исследования. Спонсор, комитет по этике или регулирующее агентство прекращают исследование.
6. Беременность. Обнаружение беременности у пациента.
Примечание. Если у пациента обнаружена беременность, пациент должен быть немедленно выведен из исследования. Процедура описана в разделе 5.1.12.
7. Прогрессирование заболевания. Выявлены клинические признаки критической ишемии конечностей (боль в состоянии покоя, образование язв и гангрена), подтверждаемые инструментальными методами, требующие процедуры реваскуляризации.
8. Другое.
Примечание. Конкретные причины должны быть указаны в поле "Уточнить" в eCRF.
3.6 Процедуры прекращения лечения или участия пациента
Исследователь может прекратить участие в исследовании пациента в любое время в ходе исследования, если пациент соответствует критериям прекращения участия в исследовании, описанным в Разделе 3.5. Кроме того, пациент может прекратить свое участие без объяснения причин в любое время в ходе исследования. В случае прекращения участия пациента основной критерий прекращения должен быть зарегистрирован исследователем. Кроме того, необходимо предпринять усилия для выполнения всех процедур, запланированных для Визита досрочного прекращения. Прекратившие лечение или участие пациенты замене не подлежат.
4. Управление материальными ресурсами клинического исследования
Этот раздел содержит информацию обо всех лекарственных средствах и материалах, предоставляемых непосредственно спонсором, и/или другими способами, которые требуются по протоколу исследования, включая важные разделы, описывающие управление материалами исследования.
4.1 Лекарственные препараты и материалы исследования
4.1.1 Лекарственная форма, производитель, упаковка и маркировка
В данном протоколе термин "исследуемый препарат" относится к активному средству Актовегин® (400 мг/10 мл и 200 мг) и соответствующему плацебо, как описано ниже. Исследуемые препараты будут упакованы в слепом режиме.
4.1.1.1 Исследуемый препарат
Актовегин®, ампулы (400 мг/10 мл) и таблетки (200 мг)
Исследуемый препарат (Актовегин®) будет поставляться в виде ампул (400 мг/10 мл) и таблеток (200 мг). Актовегин® (депротеинизированный гемодериват) доступен на рынке как в ампулах, так и в таблетках. Доступные на рынке ампулы и таблетки Актовегина® будут упакованы и маркированы в слепом режиме контрактной производственной организацией (Fisher Clinical Services, Швейцария), и таким образом оба эти продукта считаются исследуемым лекарственным препаратом.
Takeda GmbH, Австрия, является оригинальным производителем ампул Актовегина® 400 мг/10 мл. Одна ампула содержит 400 мг Актовегина® в 10 мл воды для инъекций. Раствор прозрачный, желтоватый и почти не содержит частиц. Цвет может меняться до более интенсивного желтого цвета в течение срока годности. Принимая во внимание цвет раствора Актовегина® и различную конструкцию ампул, в исследовательскую группу центра будет включен фармацевт, от которого не будет скрыта информация о препарате, который будет отвечать за приготовление исследуемого продукта (ИП) в течение периода в/в лечения.
Ампулы будут упаковываться в наборы InPatient ("стационар") из расчета на 7 дней ((21 ампула на 1 неделю, плюс 3 ампулы на 1 день). Дозирование включает 3 ампулы в день.
Каждая ампула будет снабжена многоязычной брошюрой-инструкцией или одноязычной инструкцией, которая включает соответствующую информацию об исследовании. Каждый набор из ампул 400 мг/10 мл Актовегина® будет снабжен многоязычной брошюрой-инструкцией или одноязычной инструкцией, которая включает соответствующую информацию об исследовании вместе с предупреждением о мерах предосторожности при использовании.
Основным производителем таблеток Актовегина® 200 мг является Takeda GmbH, Германия. Таблетки круглые, двояковыпуклые, яркого зеленовато-желтого цвета, с пленочным покрытием, блестящие без гравировки.
Таблетки будут упаковываться расфасованы в бутылки с защитой от детей, содержащие по 50 таблеток. Дозирование включает 6 таблеток в день.
Каждая бутылка таблеток 200 мг Актовегина® будет снабжена многоязычной брошюрой-инструкцией или одноязычной инструкцией, которая включает соответствующую информацию об исследовании вместе с предупреждением. Каждый набор бутылок также будет снабжен многоязычной брошюрой-инструкцией или одноязычной инструкцией, которая включает соответствующую информацию об исследовании вместе с предупреждением о мерах предосторожности при использовании.
Инструкции будут на соответствующем языке, в зависимости от страны выдачи исследуемого препарата.
Ампулы и таблетки плацебо
Ампулы плацебо, имитирующие 400 мг ампулы Актовегина® представляют собой доступные на рынке ампулы, содержащие 10 мл 0,9% раствора NaCl для инфузий.
Оригинальным производителем ампул плацебо является Fresenius Kabi, Испания. Ампулы будут поставляться, упаковываться и маркироваться Fisher Clinical Services, Швейцария.
Ампулы плацебо будут упакованы в 7-дневные наборы In-Patient (21 ампула на 1 неделю, плюс 3 ампулы на 1 день). Дозирование включает 3 ампулы в день.
Каждая ампула будет снабжена многоязычной брошюрой-инструкцией или одноязычной инструкцией, которая включает соответствующую информацию об исследовании. Каждый набор ампул плацебо будет снабжен многоязычной брошюрой-инструкцией или одноязычной инструкцией, которая включает соответствующую информацию об исследовании вместе с предупреждением о мерах предосторожности при использовании.
Таблетки плацебо, имитирующие таблетки Актовегина® 200 мг, будут изготовлены Globopharm Pharmazeutische Produktions- und Handelsgesellschaft mbH, Австрия, а затем упакованы и маркированы в Fisher Clinical Services, Швейцария.
Таблетки будут упаковываться расфасованы в бутылки с защитой от детей, содержащие по 50 таблеток. Дозирование включает 6 таблеток в день.
Каждая бутылка таблеток плацебо будет снабжена многоязычной брошюрой-инструкцией или одноязычной инструкцией, которая включает соответствующую информацию об исследовании вместе с предупреждением. Каждый набор бутылок также будет снабжен многоязычной брошюрой-инструкцией или одноязычной инструкцией, которая включает соответствующую информацию об исследовании вместе с предупреждением о мерах предосторожности при использовании.
Инструкции будут на соответствующем языке, в зависимости от страны выдачи исследуемого препарата.
4.1.1.2 Сопутствующие лекарства
Сопутствующая терапия БПА
Пациенты могут продолжить свое текущее лечение БПА. Эти виды лечения должны быть оптимизированы и стабильны в течение как минимум 3 месяцев на момент рандомизации. Статус курения, привычки к физическим упражнениям и лекарства будут оцениваться и документироваться. Эти виды лечения должны поддерживаться на стабильном уровне в течение всего исследования.
Терапия для лечения БПА включает:
- Антитромбоцитарные препараты и антикоагулянты.
- Средства, снижающие уровень липидов.
- Антигипертензивные препараты.
- Физические упражнения.
- Отказ от курения.
В дополнение к перечисленным лекарствам от БПА, на БПА также могут влиять лечение и состояние гликемического контроля у пациентов с диабетом и нерегулярное использование нестероидных противовоспалительных средств. Эти препараты следует проанализировать и задокументировать в eCRF.
4.1.1.3 Вспомогательные материалы
Спонсор также будет снабжать центры инфузионными мешками с 250 мл 0,9% раствора NaCl, защитными мешками для инфузионных мешков и вспомогательными этикетками для прикрепления к защитным мешкам в центрах исследования.
Оригинальным производителем 0,9% раствора NaCl для инфузионных мешков, 250 мл является Fresenius Kabi, Германия.
4.1.1.4 Поставляемый спонсором препарат
Спонсор будет поставлять Актовегин® в ампулах (400 мг/10 мл) и таблетках (200 мг), ампулы плацебо и таблетки плацебо. Кроме того, спонсор будет поставлять инфузионные мешки, содержащие 250 мл 0,9% раствора NaCl для инфузии, покровные защитные мешки и вспомогательные этикетки для инфузионных мешков.
4.1.2 Хранение
Исследуемый препарат, т.е. ампулы Актовегина® (400 мг/10 мл), таблетки Актовегина® (200 мг), ампулы плацебо, таблетки плацебо и вспомогательные материалы т.е. 0,9% раствор NaCl для инфузионных мешков, защитные мешки и вспомогательные этикетки должны храниться в надлежащем, защищенном месте с ограниченным доступом до его использования или возвращения спонсору или назначенному лицу для уничтожения. Исследуемый препарат и вспомогательные материалы должны храниться в условиях, указанных на этикетке (от 15°C до 25°C), в защищенном от света месте и оставаться в оригинальной упаковке до выдачи. Дополнительная информация приведена в фармацевтическом руководстве. Ежедневный температурный журнал зоны хранения препаратов должен заполняться каждый рабочий день.
4.1.3 Доза и схема
4.1.3.1 Период в/в лечения (недели 0-2)
Каждый пациент, который удовлетворяет критериям отбора, будет рандомизирован для приема либо Актовегина®, либо плацебо с помощью интерактивной онлайновой системы связи. После рандомизации (визит 1, день 0) пациенты получат 3 ампулы либо Актовегина® 400 мг/10 мл, либо плацебо/10 мл в 250 мл 0,9% NaCl (общий объем инфузии 280 мл) в виде единственной инфузии. Скорость инфузии будет составлять примерно 2 мл в минуту (от 40 до 50 капель в минуту). Пациенты будут получать 1 инфузию в день в течение 14 дней. Исследователь или назначенное лицо должны обеспечить, чтобы препарат, поставляемый спонсором, был приготовлен и введен в соответствии с Фармацевтическим руководством.
Инфузии ИП должны проводиться в центре исследования под наблюдением врача. В течение первых 15 минут первой инфузии исследователь (или назначенный врач) должен осуществлять контроль для выявления возможных аллергических реакций и принятия неотложных мер для их предотвращения или лечения.
4.1.3.2 Период пероральной терапии (недели 2-12)
По окончании периода в/в лечения (визит 2) пациентам будет выдана коробка с 4 бутылками, содержащая 200 таблеток Актовегина® 200 мг или плацебо. Пациенты будут проинструктированы принимать 2 таблетки Актовегина® 200 мг или плацебо TID, всего 6 таблеток в день. Пациенты получат исследуемый препарат на 4 недели лечения, и им напомнят о необходимости принести с собой бутылку с исследуемым препаратом (включая неиспользованные таблетки) на следующий визит.
На третьем визите (визит 3) пациенту будет выдана еще одна коробка с 4 бутылками, содержащая в общей сложности 200 таблеток Актовегина® 200 мг или плацебо, рассчитанная еще на 4 недели лечения. Пациенты будут проинструктированы принимать 2 таблетки Актовегина® 200 мг или плацебо TID, всего 6 таблеток в день. Пациенту также напомнят о необходимости принести бутылку с исследуемым препаратом (включая неиспользованные таблетки) на следующий визит.
На четвертом визите (визит 4), пациенту будет выдана последняя коробка, содержащая 2 бутылки, в общей сложности 100 таблеток Актовегина® 200 мг или плацебо, рассчитанная на 2 недели лечения. Пациенты будут проинструктированы принимать 2 таблетки Актовегина® 200 мг или плацебо TID, всего 6 таблеток в день. Пациенту также напомнят о необходимости принести бутылку с исследуемым препаратом (включая неиспользованные таблетки) на следующий визит (визит 5).
После этого пациенты перейдут в фазу последующего наблюдения, где они не будут получать никакого исследуемого препарата.
Таблица 2 описывает дозу и количество ампул/таблеток, которые будут предоставлены каждой группе.
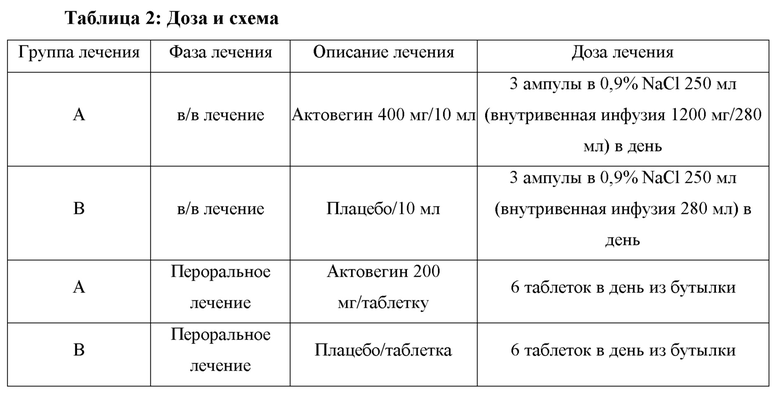
4.1.4 Передозировка
Передозировка определяется как известное преднамеренное или случайное введение исследуемого препарата, пациенту исследования или пациентом исследования, в дозе, превышающей дозу, назначенную этому конкретному пациенту в соответствии с протоколом исследования.
Все случаи передозировки (со связанными НЯ или без таковых) будут задокументированы на странице передозировки eCRF, с целью последовательно фиксировать эту важную информацию по безопасности в базе данных.
Случаи передозировки без явных признаков или симптомов не считаются НЯ. НЯ, связанные с передозировкой, будут задокументированы в разделе НЯ eCRF.
Серьезные нежелательные явления (СНЯ), связанные с передозировкой, подлежат обязательной отчетности.
Опыта передозировки Актовегина® не имеется. Согласно данным неклинического исследования, Актовегин® не проявляет токсического эффекта при дозах, до 30-40 раз превышающих дозу, рекомендуемую для применения у человека.
Специфических инструкций по лечению передозировки не имеется. Исходя из фармакологии препарата, нежелательных эффектов не ожидается.
4.2 Процедуры назначения и дозирования препарата
Исследователь или назначенное исследователем лицо заходят в интерактивную систему связи (IRT) при скрининге, для получения идентификационного номера пациента в данном исследовании ("номер пациента").
Исследователь или назначенное исследователем лицо будут использовать систему IRT для рандомизации пациента в исследовании. Во время этого сеанса связи исследователь или назначенное лицо предоставят необходимую информацию для идентификации пациента, включая номер пациента, назначенный при скрининге.
Во время визитов с выдачей препарата исследователь или назначенное лицо снова свяжутся с системой IRT для регистрации визитов. На визитах 1, 2, 3 и 4 для всех пациентов в обеих группах лечения системой IRT будет назначен идентификационный номер препарата (Med ID), который будет предоставлен фармацевту/медсестре центра, от которых не маскируют информацию, по электронной почте.
Для поддержания ослепления, в системе IRT предусмотрено, чтобы исследователь или назначенное лицо не знали о присвоенных идентификаторах Med ID.
Если препарат, поставляемый спонсором, потерян или поврежден, фармацевт /медсестра центра, от которого не маскируют информацию, может запросить по системе IRT замену.
4.3 Создание и хранение кода рандомизации
Сотрудник Takeda, ответственный за рандомизацию, или назначенное лицо сгенерирует график рандомизации для исследования; для рандомизации пациентов и назначения исследуемого препарата будет централизованно использоваться система IRT.
Вся информация о рандомизации будет храниться в охраняемой зоне, доступной только уполномоченному персоналу.
4.4 Поддержание маскировки исследуемого препарата
Маскировка исследуемого препарата будет поддерживаться с использованием системы IRT. Главный исследователь в каждом центре получит инструкции о получении назначения лекарств через систему IRT.
От исследователей, сотрудников клинических центров, участвующих в уходе за пациентами или клинических обследованиях, статистической группы, группы управления данными и пациентов, информация относительно типа терапии будет скрыта (маскирована) до момента блокировки базы данных. От сотрудников аптечного пункта (медсестры, фармацевта) информацию маскировать не будут. Все мешки и инфузионные системы для инфузий будут в закрытом виде, и запечатанный материал не позволит выполнить несанкционированное открытие, для поддержания маскировки.
Персонал исследования, назначенный центром, будет хранить информацию о лекарственных препаратах в слепом режиме.
Во время регулярных запланированных контрольных визитов монитор, от которого информация не маскирована, от спонсора или назначенного лица будет выполнять инвентаризацию коробок/бутылок нераспределенного и распределенного исследуемого препарата. Все нераспределенные и распределенные упаковки будут сверены и возвращены спонсору или назначенному лицу до прекращения исследования.
4.5 Процедура демаскировки
Маскировка исследуемого препарата не должна нарушаться исследователем, кроме случаев, когда информация об исследуемом препарате требуется для лечения пациента. По возможности, спонсора/назначенное лицо следует уведомить до демаскировки препарата. При возникновении чрезвычайной ситуации, требующей немедленной демаскировки, исследователь (или назначенное лицо) в центре свяжется со спонсором или назначенным лицом, чтобы оценить необходимость демаскировки препарата.
Для демаскировки информации о назначенном конкретному пациенту препарате, ее можно будет получить, обратившись к системе IRT. При демаскировке препарата следует немедленно уведомить спонсора /назначенное лицо. Дата, время и причина демаскировки должны быть занесены в исходные документы и в соответствующий раздел eCRF.
Если какой-либо сотрудник центра получил демаскированную информацию о назначенном конкретному пациенту препарате, прем исследуемого препарата быть немедленно прекращен, и данный пациент должен быть выведен из исследования. Причина вывода должна быть записана как "Значительное отклонение от протокола".
4.6 Отчетность и уничтожение поставляемых спонсором препаратов
Препараты подлежат учету и сверке в центре до возвращения спонсору или назначенному лицу.
Исследователь или назначенное лицо должны обеспечить, чтобы препарат, предоставленный спонсором (ампулы Актовегина®/плацебо, таблетки Актовегина®/плацебо), и вспомогательный материал (0,9% раствор NaCl для инфузии, защитные мешки и вспомогательные этикетки), использовались в соответствии с протоколом и выдавались только пациентам, включенным в исследование. Для документирования надлежащего использования лекарственного средства, предоставленного спонсором, исследователь или назначенное лицо должны вести учет всех поставок препарата, предоставленного спонсором, в центр, инвентаризацию центра, выдачу и использование по каждому пациенту и осуществлять возврат спонсору или назначенному лицу.
После получения лекарственного препарата, предоставленного спонсором, исследователь или назначенное лицо должны сверить содержимое поставки с упаковочным листом. Проверяющее лицо должен убедиться, что количество совпадает, и лекарство находится в хорошем состоянии. Если количество и состояние приемлемы, исследователь или назначенное лицо должны подтвердить получение груза, подписав нижнюю половину упаковочного листа и отправив по электронной почте/факсу в соответствии с инструкциями, представленными в форме/путем регистрации в системе IRT. При наличии каких-либо расхождений между упаковочным листом и фактическим полученным продуктом, для решения проблемы необходимо связаться с Takeda или назначенным лицом. Упаковочный лист должен быть внесен в основной файл документов исследователя.
Исследователь или назначенное лицо должны вести 100-процентную отчетность по всем полученным от спонсора и выданным за время всего его участия в исследовании лекарствам. Надлежащая отчетность лекарственных средств включает, не ограничиваясь этим:
Постоянный мониторинг сроков годности (через систему IRT).
Регулярная сверка фактических запасов с документами.
Проверка заполнения журнала с указанием Идентификатора/других данных лекарства, используемого для приготовления каждой дозы.
Проверка правильности документирования всех используемых контейнеров в журнале.
Проверка правильности и читаемости заполненных полей.
Представитель центра, от которого информация не маскируется, или монитор, от которого информация не маскируется, будет проверять график рандомизации и журнал дозирования пациентов для обеспечения того, чтобы все пациенты получили надлежащую дозу исследуемого препарата.
При обнаружении каких-либо ошибок или несоответствий, следует немедленно уведомить спонсора.
Система IRT будет содержать всю необходимую информацию в виде отдельной записи для каждого пациента, которому выдано лекарство, предоставляемое спонсором.
Исследователь или назначенное лицо должны фиксировать текущие запасы всех лекарств, поставляемых спонсором, в одобренном спонсором журнале отчетности о препарате. Как минимум, будет фиксироваться следующая информация: номер и название протокола, имя исследователя, идентификатор и номер центра, описание поставляемых спонсором лекарств, срок годности и выданное количество, включая инициалы, печать или подпись лица, отпускающего лекарство, а также дату и количество, возвращенные в центр пациентом, включая инициалы, печать или подпись лица, получившего лекарство, поставляемое спонсором. Журнал должен содержать всю необходимую информацию в виде отдельной записи для каждого пациента (по идентификатору пациента), которому выдан препарат, поставляемый спонсором.
Исследователь или назначенный сотрудник центра, который вводит исследуемый препарат (инфузия Актовегина®/плацебо), должен заполнять журнал отчетности по отдельным пациентам, для документирования того, выполнена ли инфузия или не выполнена, и лекарственные препараты были возвращены в аптеку, включая дату и количество, возвращенное в аптеку, включая инициалы, печать или подпись лица, выполняющего инфузию.
Все случаи невозвращения исследуемого препарата в центр пациентом должны быть расследованы в центре и надлежащим образом документированы в журнале отчетности препарата.
Перед закрытием центра или через определенные промежутки времени представитель от спонсора или его назначенное лицо будут выполнять отчет и сверку поставляемого спонсором препарата до возвращения препарата спонсору или назначенному лицу для уничтожения. Исследователь или назначенное лицо будут сохранять копию документации, касающейся отчетности, возвращения и/или уничтожения поставляемого спонсором препарата, а оригиналы будут отправлены спонсору или назначенному им лицу.
Во время проведения исследования исследователь будет уведомлен о сроке годности лекарственного препарата, предоставленного спонсором. По получении уведомления об истечении срока годности от спонсора или назначенного лица центр должен выполнить все инструкции, указанные в уведомлении, включая изоляцию препарата с истекающим сроком годности, предоставленного спонсором, для возвращения спонсору или назначенному им лицу для уничтожения.
5. ПЛАН ИССЛЕДОВАНИЯ
5.1 Процедуры исследования
В следующих разделах описаны процедуры исследования и данные, которые необходимо будет собрать. Для каждой процедуры пациенты должны оцениваться, по возможности, одним и тем же исследователем или сотрудником центра. График процедур исследования приведен в таблице 1 выше.
5.1.1 Процедура информированного согласия
Информированное согласие должно быть получено до включения пациента в исследование и до выполнения каких-либо процедур по протоколу.
Каждому пациенту будет присвоен уникальный идентификационный номер пациента (номер пациента) в момент получения информированного согласия; этот номер пациента будет использоваться на протяжении всего исследования.
5.1.2 Процедура сбора демографических данных, истории болезни и лекарственного анамнеза
Собираемая демографическая информация будет включать дату рождения, пол, расу, сообщенную пациентом, рост, вес и статус курения пациента при скрининге.
Собираемая история болезни будет включать определение наличия у пациента каких-либо существенных состояний или болезней, имеющих значение для исследуемого заболеванию, которые разрешились на момент или до подписания информированного согласия, включая психиатрическую историю (любые психиатрические расстройства являются критериями исключения). Текущие патологии считаются сопутствующими патологиями (раздел 5.1.9).
Собираемая информация в отношении лекарственного анамнеза включает любые лекарства, имеющие значение для критериев отбора (раздел 4.1.1.2), прекращенные за 14 дней до рандомизации (день 0).
5.1.3 Процедура физического обследования
Исходное физическое обследование (определяемое как оценка до первой дозы исследуемого препарата) будет включать следующие системы организма: (1) глаза; (2) уши, нос, горло; (3) сердечно-сосудистая система; (4) дыхательная система; (5) желудочно-кишечная система; (6) дерматологическая система; (7) конечности; (8) костно-мышечная система; (9) нервная система; (10) лимфатические узлы; и (11) другое. Все последующие физические обследования должны оценивать клинически значимые изменения по сравнению с оценкой до первой дозы.
5.1.4 Вес и рост
Вес и рост пациентов следует измерять в комнатной одежде и без обуви. Значения должны указываться с 1 знаком после запятой.
5.1.5 Статус курения
Статус курения будет классифицироваться следующим образом:
1. Никогда не курил.
2. Активный курильщик (собирается информация о числе лет курения и текущем количестве сигарет в день).
3. Бывший курильщик (собирается информация о числе лет курения, примерном количестве сигарет, ранее выкуриваемых в день, и дате прекращения курения).
Для целей исследования, при определении истории курения, использование электронной сигареты учитывать не следует. Один грамм табака=1 сигарета.
Статус курения будет проверяться на каждом визите.
5.1.6 Процедура определения показателей жизненно важных функций
Показатели жизненно важных функций включают артериальное давление (систолическое и диастолическое) и пульс (ударов/минуту) после отдыха не менее 5 минут в сидячем положении. Когда сбор показателей жизненно важных функций назначен одновременно с забором крови, забор крови будет приоритетным, а сбор показателей жизненно важных функций должен выполняться за 0,5 часа до или после запланированного забора крови.
5.1.7 Измерения эффективности
5.1.7.1 Тестирование на беговой дорожке
Беговая дорожка - это устройство, используемое для ходьбы или бега на месте. В этом исследовании данный метод будет использоваться для определения дистанций, пройденных при хромоте. Дистанция, пройденная при хромоте, будет измеряться с использованием протокола постоянной рабочей нагрузки (постоянная скорость 3,0 км/ч и наклон 10% на беговой дорожке).
С момента подписания информированного согласия тест на беговой дорожке будет проводиться дважды. Первое тестирование должно быть выполнено в течение периода скрининга. Второе тестирование на беговой дорожке должно быть повторено с промежутком от =1 до =2 недель и должно быть выполнено в День 0 до рандомизации.
Пациенты с высокой вариабельностью в величины ACD [> 25% для максимальной пройденной дистанции при хромоте] будут исключены. Если пациент рандомизирован для получения исследуемой терапии, результаты тестирования в День 0 будут использоваться в качестве исходного значения.
5.1.7.2 SF-36
SF-36 - это инструмент для измерения со слов пациента связанного со здоровьем качества жизни из 36 пунктов. SF-36 ранее был подробно оценен и протестирован с различными популяциями и позволяет различать группы с различным статусом качества жизни в клинических испытаниях [24, 25]. SF-36 состоит из 8 разделов (жизненная активность, физическое функционирование, интенсивность боли, общее состояние здоровья, ролевое физическое функционирование, ролевое эмоциональное функционирование, ролевое социальное функционирование и психическое здоровье), которые являются взвешенными суммами вопросов в каждом домене здоровья. Каждая шкала напрямую преобразуется в шкалу от 0 до 100, исходя из предположения, что каждый вопрос несет равный вес. Чем ниже оценка, тем больше инвалидизация, и чем выше оценка, тем меньше инвалидизация, т.е. оценка 0 эквивалентна максимальной инвалидизации, а оценка 100 эквивалентна отсутствию инвалидизации.
Для каждой участвующей страны будет доступна валидированная версия SF-36 на соответствующем языке; каждому пациенту должна быть предоставлена версия SF-36 на его родном языке. Пациенты должны заполнять SF-36 на визитах 1, 5 и 8 (или визите окончания лечения) до проведения любых связанных с исследованием процедур. Исследователь должен проверить, что заполнены ответы на все вопросы.
5.1.8 Документирование сопутствующих процедур и лекарств
Сопутствующим лекарством является любой препарат, назначаемый в дополнение к исследуемому препарату. Они могут быть назначены врачом или приобретены пациентом без рецепта. Сопутствующее лекарство не предоставляется спонсором. Во время каждого визита пациентам будет задан вопрос о том, принимали ли они какие-либо лекарства помимо исследуемого препарата (используемые с момента подписания информированного согласия и вплоть до конца исследования), и все лекарства, включая витаминные добавки, безрецептурные лекарства и пероральные травяные препараты должны быть записаны в eCRF. Кроме того, будут учитываться и регистрироваться в eCRF все сопутствующие процедуры.
5.1.9 Документирование сопутствующих медицинских патологий
Сопутствующими патологиями являются те значительные текущие состояния или заболевания, которые присутствуют при подписании информированного согласия. Это включает клинически значимые аномалии лабораторных показателей, ЭКГ или физического обследования, отмеченные на скрининговом обследовании, согласно решению исследователя.
Ультразвуковое цветовое дуплексное сканирование будет использоваться при скрининге для проверки диагноза БПА и исключения другой патологии, которая может вести к окклюзии периферических артерий. Также будет идентифицировано местонахождение сужения и окклюзии артерии, аортально-подвздошное или бедренно-подколенное поражение будет задокументировано в индивидуальной регистрационной карте. Это обследование будет проводиться на сертифицированном оборудовании квалифицированным специалистом по ультразвуковой диагностике в соответствии с общепринятой методологией. Это обследование будет проводиться в соответствии с местной рутинной практикой. Результаты (как минимум, заключение в письменном виде) должны храниться вместе с другими исходными документами.
5.1.10 Процедуры для клинических лабораторных образцов
Все образцы будут забираться натощак и в соответствии с приемлемыми местными лабораторными процедурами. Максимальный объем крови, собранной на каждом визите, будет составлять приблизительно 10 мл, а приблизительный общий объем собранной крови в ходе исследования - 30 мл.
Сроки сбора образцов для клинических лабораторных тестов представлены в таблице 1 и таблице 3.
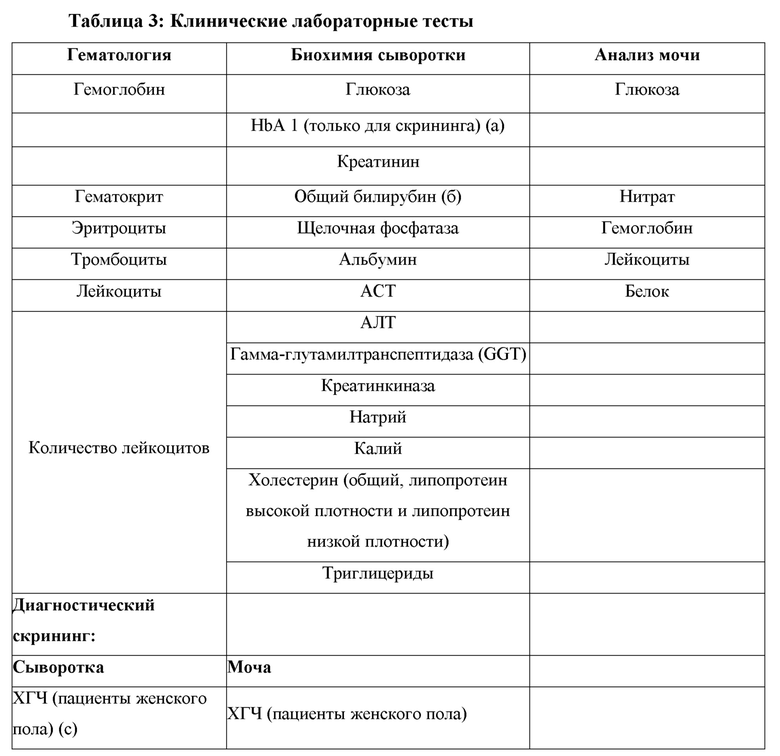
(a) HbA1c при скрининге для оценки права на участие в исследовании.
(b) Если значения выше нормального диапазона, необходимо определение прямого и свободного билирубина.
(c) При скрининге.
Лабораторные тесты на гематологию, биохимию сыворотки и анализ мочи будет выполнять местная лаборатория. Результаты лабораторных тестов будут возвращаться исследователю, который отвечает за проверку и архивирование этих результатов.
При выявлении у пациента АЛТ или АСТ> 3 × ВГН, должны быть проведены дополнительные лабораторные тесты (как минимум, сывороточная щелочная фосфатаза, АЛТ, АСТ, общий билирубин и гамма-ГТФ) в течение максимум 7 дней и предпочтительно в течение 48-72 часов после выявления аномалии (см. в разделе 3.5 соответствующее руководство по отчетности об анормальных результатах тестов функции печени).
Исследователь должен постоянно иметь копию аккредитации лаборатории и диапазоны нормальных значений показателей для используемой лаборатории.
5.1.11 Процедура контрацепции и предотвращения беременности
5.1.11.1 Пациенты-мужчины и их партнерши
С момента подписания информированного согласия и на протяжении всего исследования или в течение 30 дней после последней дозы исследуемого препарата (в случае досрочного прекращения исследования) нестерилизованные пациенты мужского пола, сексуально активные с партнершей, способной к деторождению, должны использовать барьерную контрацепцию (например, презерватив с спермицидным кремом или гелем или без них). Кроме того, им не следует быть донорами спермы в течение этого периода. Женщинам, способным к деторождению, которые являются партнершами пациентов-мужчин, также рекомендуется использовать дополнительную контрацепцию, согласно перечню высокоэффективной/эффективной контрацепции (раздел 5.1.11.3).
5.1.11.2 Женщины-пациенты и их партнеры-мужчины
С момента подписания информированного согласия и на протяжении всего исследования или в течение 30 дней после последней дозы исследуемого препарата (в случае досрочного прекращения исследования) женщины, способные к деторождению, которые сексуально активны с нестерилизованным партнером-мужчиной, должны использовать высокоэффективный/эффективный метод контрацепции (раздел 5.1.11.3).
Кроме того, им не следует быть донорами яйцеклетки в течение этого периода.
5.1.11.3 Определения и процедуры по контрацепции и предотвращению беременности
Следующие определения применяются для процедур контрацепции и предотвращения беременности.
Состояние постменопаузы определяется как отсутствие менструаций в течение 12 месяцев без альтернативной медицинской причины. Для подтверждения состояния постменопаузы у молодых женщин (например, тех, кто моложе 45 лет) или женщин, не использующих гормональную контрацепцию или гормональную заместительную терапию может использоваться высокий уровень фолликулостимулирующего гормона (ФСГ) в постменопаузальном диапазоне (ФСГ> 40 МЕ/л).
Однако, при отсутствии 12 месяцев аменореи, одно измерение ФСГ является недостаточным.
У стерилизованных мужчин должен пройти как минимум 1 год после двусторонней вазэктомии, с документальным подтверждением отсутствия спермы в эякуляте, либо они должны подвергнуться двусторонней орхидэктомии.
Следующие процедуры применяются для контрацепции и предотвращения беременности.
1. Высокоэффективные методы контрацепции определяются как методы, которые, по отдельности или в комбинации, обеспечивают низкий показатель неэффективности (то есть менее 1% зачатий в год при постоянном и правильном использовании). В этом исследовании, включающем лекарства и устройства, содержащие гормоны, приемлемыми высокоэффективными методами контрацепции являются:
Негормональные методы:
o Внутриматочное устройство.
o Двусторонняя окклюзия трубы.
o Вазэктомированный партнер (при условии, что этот партнер является единственным сексуальным партнером участника исследования и что этот вазэктомированный партнер получил медицинское заключение об успехе операции).
Гормональные методы:
Комбинированная (эстроген и прогестоген) гормональная контрацепция, ассоциированная с ингибированием овуляции, начатая по меньшей мере за 3 месяца до первой дозы исследуемого препарата ИЛИ в сочетании с барьерным методом (мужской презерватив, женский презерватив или диафрагма), при более коротком сроке гормональной контрацепции, пока этот срок не достигнет 3 месяцев:
- Пероральная
- Внутривагинальная (например, кольцо)
- Трансдермальная
Гормональная контрацепция, содержащая только прогестоген, ассоциированная с подавлением овуляции, начатая по меньшей мере за 3 месяца до первой дозы исследуемого препарата ИЛИ в сочетании с барьерным методом (мужской презерватив, женский презерватив или диафрагма), при более коротком сроке гормональной контрацепции, пока этот срок не достигнет 3 месяцев:
- Пероральная
- Инъекционная
- Имплантируемая
2. Эффективные методы контрацепции определяются как методы, обеспечивающие низкий показатель неэффективности, который может быть выше, чем 1% зачатий. В этом исследовании, включающем лекарства и устройства, содержащие гормоны, приемлемыми эффективными методами контрацепции являются:
Двухбарьерный метод (контрацептивная губка, диафрагма или шеечный колпачок с спермицидными гелями или кремами ПЛЮС мужской презерватив).
Гормональная контрацепция, содержащая только прогестоген, где ингибирование овуляции не является основным способом действия ПЛЮС презерватив со спермицидом или без него.
3. Неприемлемыми методами контрацепции являются:
Периодическое воздержание (например, календарный, овуляционный, симптомотермический, постовуляционный методы).
Только спермициды.
Прерванный половой акт.
Отсутствие какого-либо метода.
Совместное использование женских и мужских презервативов.
Колпачок/диафрагма/губка без спермицида и без презерватива.
Половое воздержание НЕ является приемлемым методом контрацепции.
4. Пациентам будет предоставлена информация о высокоэффективных и эффективных методах контрацепции в процессе информированного согласия пациента, и им будет предложено подписать форму согласия, в которой говорится, что они понимают требования к предотвращению беременности, донорству яйцеклеток и пожертвованию спермы в ходе исследования.
5. В ходе исследования будут проводиться регулярные тесты на беременность на ХГЧ мочи, только у женщин, способных к деторождению, а все пациенты (мужчины и женщины) будут получать в рамках процедур исследования руководство-напоминание для предотвращения беременности и донорства спермы. Такое руководство должно включать напоминание о следующем:
а. Требования исследования к контрацепции.
b. Оценка соблюдения пациентом этих требований, с помощью следующих вопросов:
I. Вы использовали контрацепцию последовательно и должным образом со времени последнего визита?
II. Вы забывали использовать контрацепцию со времени последнего визита?
III. Есть ли у вас задержка месячных? (если ответ "да", даже у женщин с нерегулярными или замедленными менструальными циклами должен быть выполнен тест на беременность)
IV. Есть ли вероятность, что вы можете быть беременной?
6. В дополнение к отрицательному тесту сыворотки на беременность на ХГЧ при скрининге женщины пациенты, способные к деторождению, также должны подтвердить менструацию в течение месяца перед первым дозированием (отсутствие задержки менструации) и показать отрицательный тест на беременность на ХГЧ на визите рандомизации (исходном визите) до получения любой дозы исследуемого препарата.
5.1.12 Беременность
При обнаружении беременности у любой пациентки в ходе исследования, ее участие в исследовании и прием любого препарата, поставляемого спонсором, следует немедленно прекратить. Кроме того, любые беременности у партнерши мужчины-пациента в ходе исследования или в течение 30 дней после последней дозы (в случае досрочного прекращения исследования) также должны отражаться в отчетности после получения разрешения от партнерши пациента.
Если беременность возникает во время приема исследуемого препарата, например, после Визита 1 или в течение 30 дней после последней дозы исследуемого препарата, о беременности следует сообщить немедленно, используя форму уведомления о беременности.
Если беременность возникает во время или после применения ослепленного препарата, исследователь должен сообщить пациенту о его праве на получение информации о полученном лечении. Если пациент решает получить информацию о демаскированном лечении, исследователь должен раскрыть лечение.
Если женщина-пациент и/или партнерша мужчины-пациента согласны с информированием своего основного лечащего врача, исследователь должен уведомить лечащего врача о том, что женщина-пациент или партнерша мужчины-пациента участвовала в клиническом исследовании на момент наступления беременности и предоставить ему подробную информацию об исследуемом препарате, полученном пациентом (ослепленную или открытую, в зависимости от обстоятельств).
Все беременности, в том числе беременности партнерш мужчины-пациента, у пациентов, получавших активный лекарственный препарат, будут отслеживаться до их исхода с использованием формы беременности. Беременности будут оставаться ослепленной для исследовательской группы. Исход беременности, включая любое досрочное прекращение, должен быть сообщен спонсору. Также будет проведена оценка после рождения ребенка.
5.1.13 Процедура ЭКГ
Будет регистрироваться стандартная ЭКГ в 12 отведениях. Исследователь (или квалифицированный наблюдатель в центре исследования) будет интерпретировать ЭКГ, согласно одной из следующих категорий: в нормальных пределах, анормальная, но не клинически значимая, либо анормальная и клинически значимая. Копия ЭКГ должна храниться с исходной документацией исследования данного пациента.
5.1.14 Документирование не прошедших скрининг
Исследователи должны учитывать всех пациентов, которые подписали информированное согласие. Если пациент исключен на скрининговом визите, исследователь должен заполнить eCRF.
Данные, которые должны собираться в eCRF для не прошедших скрининг как минимум включают: номер и инициалы пациента, дату информированного согласия, демографические данные, критерии включения/исключения, PTE/СНЯ, дата непрохождения скрининга.
Основная причина непрохождения скрининга записывается в eCRF, используя следующие категории::
• PTE/НЯ.
• Несоответствие критериям включения или соответствие критериям исключения <указать причину>.
• Значительное отклонение от протокола.
• Невозможность последующего наблюдения из-за утраты связи с пациентом.
• Добровольный выход < указать причину >.
• Прекращение исследования.
• Другое <указать причину>.
Идентификационные номера пациентов, присвоенные не прошедшим скрининг, не должны использоваться повторно.
5.1.15 Документирование рандомизации
Только пациенты, которые соответствуют всем критериям включения и ни одному из критериев исключения, могут участвовать в рандомизации для лечения.
Если признано, что пациент не подходит для рандомизации, исследователь должен записать основную причину отказа в соответствующем разделе eCRF.
5.2 Мониторинг соблюдения пациентом режима лечения
Во время периода в/в лечения ежедневные внутривенные инфузии будут регистрироваться в журнале инфузий. Будут фиксироваться следующие данные:
• Данные набора инфузии.
• Дата инфузии.
• Время начала.
• Время прекращения.
• Инициалы лица, вводившего ИЛП.
• Инфузия успешно завершена; да или нет.
• Комментарии.
В течение периода перорального лечения исследователь должен проверять соблюдение пациентом режима лечения, подсчитывая количество возвращенных таблеток в моменты времени, указанные в обзоре клинического исследования. Пациенты должны будут приносить выданные бутылки с исследуемым лекарственным средством на каждый визит.
Соблюдение режима лечения будет рассчитываться исследователем на Визитах 3, 4, и 5, используя формулу:
Соблюдение режима в %=
(число таблеток, принятых с момента последнего визита/число таблеток, которые должны быть приняты с момента последнего визита)*100
Эти данные впоследствии будут занесены в eCRF персоналом центра.
В случае отклонения от режима лечения, пациента следует повторно проинструктировать относительно требований дозирования во время контактов с исследователем. Уполномоченный персонал исследования, проводящий повторный инструктаж, должен задокументировать этот процесс в исходных записях.
Если соблюдение режима лечения находится вне диапазона 70%-130% за период лечения с момента последнего визита, его следует включить в отчетность как отклонение от протокола.
Если пациент постоянно не соблюдает режим лечения исследуемым препаратом (например, соблюдение для двух визитов подряд вне диапазона 70% до 130%), может быть целесообразным (на основании решения исследователя) вывести пациента из исследования.
5.3 График наблюдений и процедур
График всех связанных с исследованием процедур для всех оценок приведен в таблице 1. Оценки должны быть выполнены в назначенный визит/точки времени.
5.3.1 Период скрининга
Скрининг считается начатым с момента подписания ICF. Пациенты должны пройти скрининг в течение 1-2 недель до рандомизации. Пациенты будут проходить скрининг в соответствии с предопределенными критериями включения и исключения, как описано в разделе 3. Процедуры документирования не прошедших скрининг см. в Разделе 5.1.14.
Следующие процедуры предусмотрены при скрининге (Визит 0):
• Информированное согласие.
• Демографические данные.
• Анамнез и сопутствующие патологии.
• Лекарственный анамнез.
• Сопутствующие лекарства.
• PTE.
• Физическое обследование.
• Показатели жизненно важных функций.
• Статус курения и привычки к физически упражнениям.
• ЭКГ в 12 отведениях.
• Клинические лабораторные тесты.
• Тест на беременность мочи (только для женщин, способных к деторождению), и устное подтверждение менструаций в течение месяца перед первым дозированием (без задержки менструации).
• Ультразвуковое цветовое дуплексное сканирование.
• Тестирование на беговой дорожке (первое тестирование за период скрининга).
• Критерии включения/исключения.
5.3.2 Рандомизация (исходный уровень), День 0, Визит 1
Рандомизация состоится в День 0.
Следующие процедуры предусмотрены при рандомизации (Визит 1):
• Критерии включения/исключения.
• SF-36 (до всех процедур).
• PTE.
• Статус курения и привычки к физически упражнениям.
• Тестирование на беговой дорожке (второе тестирование за период скрининга, чтобы проверить, что результаты обоих тестов не отличаются больше, чем на 25%) (разделы 3.2 и 5.1.7.1, таблица 1).
• Тест мочи на беременность (только для женщин, способных к деторождению).
• Оценка соответствия пациента требованиям контрацепции.
• Физическое обследование.
• Показатели жизненно важных функций.
• Вес и рост.
• Сопутствующие лекарства.
• Лекарственный анамнез.
Если пациент соответствует всем критериям включения и ни одному из критериев исключения для рандомизации, пациент следует рандомизировать с использованием системы IRT, как описано в разделе 4.2.
5.3.3 Период лечения инфузиями (в/в)
После рандомизации пациенты вступают в период в/в лечения (День 0, Визит 1 - День 14, Визит 2), в течение которого они получают ежедневные инфузии Актовегина® или плацебо (до 14 инфузий).
Инфузии ИП должны выполняться в центре исследования под наблюдением врача. В течение первых 15 минут первой инфузии такое наблюдение должно проводиться Исследователем (или назначенным врачом), чтобы немедленно выявить возможные аллергические реакции и принять срочные меры для их предотвращения или лечения.
Следующие данные будут собраны записаны в течение периода в/в лечения:
• НЯ (в день 0 после первой инфузии). Начиная с Дня 1, НЯ следует проверять до (НЯ с момента последнего визита) и после инфузии.
• Сопутствующие лекарства.
• Статус курения и привычки к физически упражнениям.
• Тест мочи на беременность (только для женщин, способных к деторождению).
• Оценка соблюдения пациентом требований контрацепции.
5.3.4 Окончание в/в лечения, День 14, Визит 2
Окончание в/в лечения определяется как день последней инфузии (День 14) и до начала перорального лечения. Оценки, требуемые на этом визите, должны проводиться после последной инфузии.
Визит окончания в/в лечения и последующий переход к пероральному лечению могут быть выполнены либо пока пациент еще госпитализирован, либо уже после выписки, в зависимости от потребности пациента.
Следующие процедуры предусмотрены на Визите окончания в/в лечения (Визит 2):
• НЯ (до инфузии [НЯ с момента последнего визита] и после инфузии).
• Сопутствующие лекарства.
• Статус курения и привычки к физически упражнениям.
• Тестирование на беговой дорожке.
• Тест мочи на беременность (только для женщин, способных к деторождению).
• Оценка соблюдения пациентом требований контрацепции.
• Выдача исследуемого препарата для перорального лечения.
5.3.5 Период перорального лечения
Период перорального лечения определяется как период от дня после последней инфузии до 12 недель после рандомизации (дни 15-84). В течение периода перорального лечения пациент должен посетить центр в День 42 (± 2 дня), Визит 3 и День 70 (± 2 дня), Визит 4.
Следующие процедуры предусмотрены в течение периода перорального лечения (Визиты 3 и 4):
НЯ (с момента последнего визита).
Сопутствующие лекарства.
Статус курения и привычки к физически упражнениям.
Тест мочи на беременность (только для женщин, способных к деторождению).
Оценка соблюдения пациентом требований контрацепции.
Выдача исследуемого препарата для перорального лечения.
Возврат неиспользованного исследуемого препарата для перорального лечения.
Проверка соответствия соблюдения пациентом режима лечения исследуемым препаратом.
5.3.6 Окончание перорального лечения, День 84, Визит 5/Визит досрочного прекращения
Визит окончания перорального лечения пройдет в последний день перорального введения через 12 недель (День 84 ± 2 дня). Это также будет Визитом досрочного прекращения лечения для пациентов, которые прекращают участие в исследовании до завершения периода перорального лечения.
Следующие процедуры предусмотрены на Визите окончания перорального лечения (Визит 5):
• НЯ (с момента последнего визита).
• Физическое обследование.
• Показатели жизненно важных функций.
• Вес.
• Сопутствующие лекарства.
• Статус курения и привычки к физически упражнениям.
• SF-36 (перед всеми процедурами).
• Клинические лабораторные тесты.
• Тест мочи на беременность (только для женщин, способных к деторождению).
• Оценка соблюдения пациентом требований контрацепции.
• Тестирование на беговой дорожке.
• Возврат неиспользованного исследуемого препарата для перорального лечения.
• Проверка соответствия соблюдения пациентом режима лечения исследуемым препаратом.
5.3.7 Период последующего наблюдения
12-недельный период последующего наблюдения начнется в первый день после визита окончания перорального лечения (Визит 5). В течение этого периода пациенты будут посещать центр каждые 4 недели, в день 112 (± 5 дней), Визит 6 и день 140 (± 5 дней), Визит 7.
Во время каждого визита последующего наблюдения будут собираться следующие данные (Визиты 6 и 7):
НЯ (с момента последнего визита).
Сопутствующие лекарства.
Статус курения и привычки к физически упражнениям.
Тест мочи на беременность (только для женщин, способных к деторождению).
Оценка соблюдения пациентом требований контрацепции.
5.3.8 Заключительный визит последующего наблюдения/Визит досрочного прекращения
Заключительный визит последующего наблюдения пройдет в последний день периода наблюдения (День 168 ± 2 дня). Это также будет визит досрочного прекращения для пациентов, которые прекращают участие в исследовании в течение периода последующего наблюдения.
Во время заключительного визита последующего наблюдения (Визит 8) предусмотрено следующее:
НЯ (с момента последнего визита).
Физическое обследование.
Показатели жизненно важных функций.
Вес.
Сопутствующие лекарства.
Статус курения и привычки к физически упражнениям.
Клинические лабораторные тесты.
Тест мочи на беременность (только для женщин, способных к деторождению).
Оценка соблюдения пациентом требований контрацепции.
Тестирование на беговой дорожке.
SF-36 (перед всеми процедурами).
Для всех пациентов, получавших исследуемый препарат, исследователь должен заполнить страницу окончания исследования в eCRF.
5.3.9 Терапия после исследования
Исследуемый препарат не будет доступен после завершения участия пациента в исследовании. Пациент должен быть возвращен для лечения своему врачу, с продолжением стандартных методов лечения по мере необходимости.
6 СТАТИСТИЧЕСКИЕ МЕТОДЫ
6.1 Статистический и аналитический планы
План статистического анализа (SAP) будет подготовлен и завершен до демаскировки назначенного пациентам лечения. В этом документе будут представлены дополнительные сведения в отношении определения переменных анализа и методологии анализа для решения всех задач исследования.
Анализ маскированных данных будет проведен до демаскировки назначенного пациентам лечения. В этом обзоре будет дана оценка точности и полноты базы данных исследования, возможности оценки пациентов и адекватности планируемых статистических методов.
6.1.1 Популяции анализа
Популяция полного анализа (FAS) будет включать всех пациентов, которые были рандомизированы, получили по меньшей мере 1 дозу исследуемого препарата и имеют по меньшей мере 1 достоверное значение после исходного для оценки основной конечной точки. В сводках и анализах эффективности FAS пациенты будут анализироваться в соответствии с видом лечения, на который они были рандомизированы.
Популяция оценки безопасности будет включать всех пациентов, которые были рандомизированы и получили по меньшей мере 1 дозу исследуемого препарата в двойном слепом режиме. В сводках безопасности пациенты будут анализироваться в соответствии с фактически полученным ими лечением. В случае, если пациент получит более одного вида лечения, фактическое лечение будет определяться как наиболее часто используемое. Если два наиболее распространенных вида лечения используются с равной частотой, то в качестве фактического лечения будет использоваться рандомизированное лечение.
6.1.2 Анализ демографии и других исходных характеристик
Исходные характеристики будут сведены в список и обобщены по демографии (пол, возраст и раса), данным физического обследования и истории болезни, включая психиатрический анамнез, историю курения, диабетический статус и сопутствующие лекарства.
Исходные значения для параметров эффективности и безопасности будут приведены в стандартных таблицах, обобщающих данные по визитам; однако исходные данные по эффективности будут также представлены отдельно с учетом всех рандомизированных пациентов.
Для непрерывных переменных сопоставимость групп лечения будет оцениваться с использованием анализа дисперсии, с лечением и центром исследования в качестве факторов. Для дискретных переменных сопоставимость будет оцениваться с использованием общего критерия взаимосвязи Кохрана-Мантеля-Гензеля, стратифицированного по центрам. P-значения будут приведены в качестве описательной статистики сопоставимости.
6.1.3 Анализ эффективности
Основные и вторичные конечные точки будут проанализированы у всех рандомизированных пациентов, получавших по меньшей мере одну дозу исследуемого препарата.
Процентное изменение ICD относительно исходного уровня и изменение балла SF-36 относительно исходного уровня будет проанализировано с использованием смешанной модели для ковариационного анализа повторных измерений, с лечением, центром, неделей, взаимодействием вида лечения от недели, взаимодействием исходного значения от недели в качестве фиксированных эффектов, исходным значением в качестве ковариаты, и полностью неструктурированной ковариационной матрицей. Сравнения между Актовегином® и плацебо будут проводиться во всех точках оценки. Исходя из предположения о случайных пропусках, этот анализ будет проводиться с использованием данных только наблюдаемых случаев. Эффект в каждый точке времени для каждого вида лечения может свободно варьироваться, причем предполагается неструктурированная ковариационная матрица.
Доля пациентов с болью в покое и доля пациентов, подвергнутых процедуре реваскуляризации, будет анализироваться во всех точках времени путем логистической регрессии с поправкой на лечение.
6.1.4 Анализ безопасности
6.1.4.1 НЯ
Отчеты о НЯ будут представляться на протяжении всего исследования.
Определение НЯ, возникающих при лечении, будет предоставлено в SAP. НЯ будут кодироваться с использованием MedDRA и обобщаться по классам систем и органов и по предпочтительному термину в течение основного периода лечения и всего исследования.
НЯ, неоднократно выявленные у пациента в течение одного периода, будут засчитаны только один раз для этого пациента и периода с максимальной степенью тяжести.
6.1.4.2 Клинические оценки
Абсолютные значения и изменения по сравнению со скринингом/исходным уровнем в клинических лабораторных тестах безопасности, показатели жизненно важных функций и вес будут обобщаться для каждой группы лечения с использованием дескриптивных методов. Значения вне нормальных диапазонов и потенциально клинически значимые величины будут специально помечены и сведены в таблицу. Результаты физического обследования также будут обобщены для каждой группы лечения.
6.2 Промежуточный анализ и критерии досрочного прекращения
Промежуточный анализ не планируется.
6.3 Определение размера выборки
Предполагая стандартное отклонение 85% для процентного изменения ICD от исходного уровня и 20%-ный уровень отсева, достаточно в общей сложности 366 пациентов (по 183 на каждую группу лечения) для достижения минимум 80% мощности для обнаружения разницы в 28% в процентном изменении ICD от исходного уровня между Актовегином® и плацебо с помощью двухвыборочного t-критерия с двусторонним уровнем значимости 0,05.
Обоснование размера выборки базируется на опубликованных данных для ICD для нафтадрофурила, согласно которым разница в процентном изменении ICD от исходного уровня между нафтидрофурилом и плацебо составляет приблизительно 28%, тогда как соответствующее стандартное отклонение составляет примерно 85% [26].
Список некоторых документов, цитируемых в описании
1. Bordeaux L, Reich L, Hirsch A. The epidemiology and natural history of peripheral arterial disease. In: Coffman JD, Eberhardt RT, editors. Contemporary Cardiology: Peripheral Arterial Disease: Diagnosis and Treatment.. Totowa, NJ: Humana Press Inc.; 2010, p. 21-34.
2. Fowkes FG, Rudan D, Rudan I, Aboyans V, Denenberg JO, McDermott MM, et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: a systematic review and analysis. Lancet 2013;382(9901):1329-40.
3. Dhaliwal G, Mukherjee D. Peripheral arterial disease: epidemiology, natural history, diagnosis and treatment. Int J Angiol 2007;16(2):36-44.
4. Buchmayer F, Pleiner J, Elmlinger MW, Lauer G, Nell G, Sitte HH. Actovegin®: a biological drug for more than 5 decades. Wien Med Wochenschr 2011;161(3-4):80-8.
5. Kuninaka T, Senga Y, Senga H, Weiner M. Nature of enhanced mitochondrial oxidative metabolism by a calf blood extract. J Cell Physiol 1991;146(1):148-55.
6. Bachmann W, Forster H, Mehnert H. Experimental studies in animals on the effect of a protein-free blood extract on the metabolism of glucose [in German]. ArzneimittelForschung/ Drug Research 1968;18:1023-7.
7. Jacob S, Dietze GJ, Machicao F, Kuntz G, Augustin HJ. Improvement of glucose metabolism in patients with type II diabetes after treatment with a hemodialysate. Arzneimittelforschung 1996;46(3):269-72.
8. Meilin S, Machicao F, Elmlinger M. Treatment with Actovegin® improves spatial learning and memory in rats following transient forebrain ischaemia. J Cell Mol Med 2014;18(8):1623-30.
9. Elmlinger MW, Kriebel M, Ziegler D. Neuroprotective and anti-oxidative effects of the hemodialysate Actovegin® on primary rat neurons in vitro. Neuromolecular Med 2011;13(4):266-74.
10. Machicao F, Muresanu DF, Hundsberger H, Pfluger M, Guekht A. Pleiotropic neuroprotective and metabolic effects of Actovegin®'s mode of action. J Neurol Sci 2012;322(1-2):222-7.
11. Dieckmann A, Kriebel M, Andriambeloson E, Ziegler D, Elmlinger M. Treatment with Actovegin® improves sensory nerve function and pathology in streptozotocin-diabetic rats via mechanisms involving inhibition of PARP activation. Exp Clin Endocrinol Diabetes 2012;120(3):132-8.
12. Fedorovich AA. Non-invasive evaluation of vasomotor and metabolic functions of microvascular endothelium in human skin. Microvasc Res 2012;84(1):86-93.
13. Herrmann WM, Bohn-Olszewsky WJ, Kuntz G. Actovegin infusion treatment in patients with primarily degenerative dementia of the Alzheimer type and multi-infarct dementia 46-55. Zeitschrift für Geriatrie 1992;5:46-55.
14. Kanowski S, Kinzler E, Lehmann E, Schweizer A, Kuntz G. Confirmed clinical efficacy of Actovegin® in elderly patients with organic brain syndrome. Pharmacopsychiatry 1995;28(4):125-33.
15. Guekht A, Skoog I, Edmundson S, Zakharov V, Korczyn AD. ARTEMIDA Trial (A Randomized Trial of Efficacy, 12 Months International Double-Blind Actovegin®): A Randomized Controlled Trial to Assess the Efficacy of Actovegin® in Poststroke Cognitive Impairment. Stroke 2017;48(5):1262-70.
16. Ziegler D, Movsesyan L, Mankovsky B, Gurieva I, Abylaiuly Z, Strokov I. Treatment of symptomatic polyneuropathy with Actovegin® in type 2 diabetic patients. Diabetes Care 2009;32(8):1479-84.
17. Gurieva IV, Begma IV, Kuzina IV, et al. [Actovegin pathogenetic treatment of neuroischemic pain syndrome in the lower extremities in diabetes mellitus. (In Russ.)]. RMJ 2009;17:8-11.
18. Angelkort B, Blume J, de la Haye R, Kuntz G. Metabolically active calves blood extract in welltrained patients with AVD in stage II. A placebo-controlled doubleblind study. VASA - J Vasc Dis 1988(Suppl 23):141-3.
19. Horsch S, Claeys L, Diehm C, Rieger H, Cachovan M, Baitsch C. [Assessment of the clinical effectiveness of actihaemyl in Fontaine stage IIb intermittent claudication]. Vasa Suppl 1992;37:64-5.
20. Muller-Buhl U, Ruhlmann KU, Meister K, Diehm C, Kuntz G. Increase of walking distance by intra-arterial infusions of hemodialysate. Therapiewoche 1991;41:188-96.
21. Angelkort B, Ruhlmann KU, de la Haye R, Kuntz G. Influence of deproteinized hemodialysate on rest pain and walking distance in the presence of peripheral chronic arterial occlusive disease. Angiology 1992;43(1):47-58.
22. Blume J. Peripheral arterial occlusive disease - Intravenous and intraarterial infusion therapy with Actovegin® (In German). Therapiewoche 1986;36:5355-58.
23. Uchkin IG, Zudin AM, Bagdasarian AG, Fedorovich AA. Effect of drug therapy for chronic obliterating diseases of lower-limb arteries on the state of the microcirculatory bed. Angiol Sosud Khir 2014;20(2):27-36.
24. Garratt AM, Ruta DA, Abdalla MI, Buckingham JK, Russell IT. The SF36 health survey questionnaire: an outcome measure suitable for routine use within the NHS? BMJ 1993;306(6890):1440-4.
25. Ware J, Kosinski M. Improvements in the content and scoring of the SF-36 Health Survey Version 2. Accessed 16 July 2014. Available at: http://www.sf-36.org/.
26. Adhoute G, Bacourt F, Barral M, Cardon JM, Chevalier JM, Cuny A, et al. Naftidrofuryl in chronic arterial disease. Results of a six month controlled multicenter study using Naftidrofuryl tablets 200 mg. Angiology 1986;37(3 Pt 1):160-7.
| название | год | авторы | номер документа |
|---|---|---|---|
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ, КОМПОЗИЦИИ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2731000C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ, ИХ КОМПОЗИЦИИ И ПРИМЕНЕНИЯ | 2017 |
|
RU2766155C2 |
| ТРЕХФАЗНЫЙ ПЕРОРАЛЬНЫЙ КОНТРАЦЕПТИВ | 1999 |
|
RU2225207C2 |
| СПОСОБ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ РЕЦИДИВА ВЕНОЗНОГО ТРОМБОЗА ПОСЛЕ ПЕРЕНЕСЕННОЙ НОВОЙ КОРОНАВИРУСНОЙ ИНФЕКЦИИ | 2022 |
|
RU2791373C1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ДГЛК, И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2714323C2 |
| ТЕРАПИЯ С ИНТЕРВАЛАМИ ДЛЯ ЛЕЧЕНИЯ ТИННИТУСА | 2008 |
|
RU2446794C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ПАЦИЕНТОВ С ГЕТЕРОЗИГОТНОЙ СЕМЕЙНОЙ ГИПЕРХОЛЕСТЕРИНЕМИЕЙ (HEFH) | 2015 |
|
RU2735521C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ 15-HEPE, ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ ГЕМАТОЛОГИЧЕСКИХ НАРУШЕНИЙ И/ИЛИ СВЯЗАННЫХ ЗАБОЛЕВАНИЙ | 2020 |
|
RU2833560C1 |
| Способ лечения пациентов с тяжелыми формами реперфузионного синдрома | 2023 |
|
RU2807877C1 |
| Способ лечения пациентов с тяжелыми формами реперфузионного синдрома, осложненными послеоперационной лимфореей | 2023 |
|
RU2808921C1 |

Группа изобретений относится к медицине, а именно к применению депротеинизированного препарата из телячьей крови для предотвращения или лечения болезни периферических артерий стадии IIB по Фонтейну и способу лечения. Предлагается применение депротеинизированного препарата из телячьей крови для предотвращения или лечения болезни периферических артерий стадии IIB по Фонтейну, где указанный депротеинизированный препарат из телячьей крови вводят по меньшей мере один раз внутривенно посредством инфузии в дозе 1200 мг один раз в день в течение периода 14 последовательных дней, с последующим периодом перорального введения дозы в форме двух таблеток по 200 мг, принимаемых три раза в день в течение периода 70 последовательных дней. Способ профилактики или лечения болезни периферических артерий стадии IIB по Фонтейну у пациента, который в этом нуждается, включает определенные стадии. Вышеописанная группа позволяет эффективно профилактировать и лечить болезни периферических артерий стадии IIB по Фонтейну у пациента. 2 н. и 16 з.п. ф-лы, 1 ил., 3 табл., 1 пр.
1. Применение депротеинизированного препарата из телячьей крови для предотвращения или лечения болезни периферических артерий стадии IIB по Фонтейну, где указанный депротеинизированный препарат из телячьей крови вводят по меньшей мере один раз внутривенно посредством инфузии в дозе 1200 мг один раз в день в течение периода 14 последовательных дней, с последующим периодом перорального введения дозы в форме двух таблеток по 200 мг, принимаемых три раза в день в течение периода 70 последовательных дней.
2. Применение по п. 1, где депротеинизированным препаратом из телячьей крови является лекарственное средство «Актовегин®».
3. Применение по п. 1 или 2, в котором указанная болезнь периферических артерий является болезнью периферических артерий стадии IIB по Фонтейну, например, подтвержденной ультразвуковым цветовым дуплексным сканированием.
4. Применение по любому из пп. 1-3, где пациентом является человек, например человек, пораженный или страдающий сахарным диабетом.
5. Применение по п. 4, где пациент-человек имеет допплеровский лодыжечно-плечевой индекс в покое ≤ 0,9.
6. Применение по п. 4 или 5, где у пациента-человека наблюдается перемежающаяся хромота с ICD < 200 метров.
7. Применение по любому из пп. 4-6, в котором возраст пациента-человека составляет по меньшей мере примерно 40 лет.
8. Применение по любому из пп. 4-7, где пациент-человек является мужчиной.
9. Применение по любому из пп. 4-7, где пациент-человек является женщиной.
10. Способ профилактики или лечения болезни периферических артерий стадии IIB по Фонтейну у пациента, который в этом нуждается, включающий следующие стадии: а) введение по меньшей мере один раз внутривенно посредством инфузии указанному пациенту терапевтически эффективного количества депротеинизированного препарата из телячьей крови в дозе 1200 мг один раз в день в течение периода 14 последовательных дней, с последующим b) введением по меньшей мере один раз перорально указанному пациенту терапевтически эффективного количества указанного депротеинизированного препарата из телячьей крови в дозе в форме двух таблеток по 200 мг, принимаемых три раза в день в течение периода 70 последовательных дней.
11. Способ по п. 10, где указанный депротеинизированный препарат из телячьей крови является лекарственным средством Актовегин®.
12. Способ по п. 10 или 11, где указанная болезнь периферических артерий стадии IIB по Фонтейну, например, подтверждена ультразвуковым цветовым дуплексным сканированием.
13. Способ по любому из пп. 10-12, где пациентом является человек, такой как человек, пораженный или страдающий сахарным диабетом.
14. Способ по п. 13, где у пациента-человека наблюдается допплеровский лодыжечно-плечевой индекс в покое ≤ 0,9.
15. Способ по п. 13 или 14, где у пациента-человека наблюдается перемежающаяся хромота с ICD < 200 метров.
16. Способ по любому из пп. 13-15, где возраст пациента-человека составляет по меньшей мере 40 лет.
17. Способ по любому из пп. 13-16, где пациент-человек является мужчиной.
18. Способ по любому из пп. 13-16, где пациент-человек является женщиной.
| КУДЫКИН М.Н | |||
| Применение препарата Актовегин® у пациентов с облитерирующими заболеваниями периферических артерий нижних конечностей // СПРАВОЧНИК ПОЛИКЛИНИЧЕСКОГО ВРАЧА | |||
| N | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Способ запрессовки не выдержавших гидравлической пробы отливок | 1923 |
|
SU51A1 |
| Гурьева И.В | |||
| Актовегин® в комплексном подходе к лечению и профилактике поздних осложнений сахарного диабета // Эффективная фармакотерапия | |||
| Способ образования коричневых окрасок на волокне из кашу кубической и подобных производных кашевого ряда | 1922 |
|
SU32A1 |
| Способ изготовления звездочек для французской бороны-катка | 1922 |
|
SU46A1 |

