ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Область техники, к которой относится изобретение
Настоящее изобретение направлено на кристаллические солевые формы соединения–ингибитора JAK, полезные для лечения респираторных и других заболеваний. Настоящее изобретение также направлено на фармацевтические композиции, содержащие такое соединение, способы применения солевых форм для лечения, например, респираторных и глазных заболеваний и способы и промежуточные соединения, пригодные для получения таких солевых форм.
Предшествующий уровень техники
Цитокины являются межклеточными сигнальными молекулами, которые включают хемокины, интерфероны, интерлейкины, лимфокины и фактор некроза опухоли. Цитокины имеют решающее значение для нормального роста клеток и иммунорегуляции, но также стимулируют иммуноопосредованные заболевания и способствуют росту злокачественных клеток. Повышенные уровни многих цитокинов вовлечены в патологию большого количества различных заболеваний или состояний, особенно тех заболеваний, которые характеризуются воспалением. Многие из цитокинов, связанных с заболеванием, действуют через сигнальные пути, зависящие от Janus семейства тирозинкиназ (JAK), которые передают сигналы через семейство транскрипционных факторов переносчиков сигнала и активаторов транскрипции (STAT).
Семейство JAK включает четыре члена: JAK1, JAK2, JAK3 и тирозинкиназа 2 (TYK2). Связывание цитокина с JAK–зависимым цитокиновым рецептором индуцирует димеризацию рецептора, что приводит к фосфорилированию тирозиновых остатков на JAK–киназе, вызывая активацию JAK. Фосфорилированные JAK, в свою очередь, связывают и фосфорилируют различные STAT белки, которые димеризуются, интернализуются в ядре клетки и непосредственно модулируют транскрипцию генов, приводя, среди других эффектов, к нисходящим эффектам, ассоциированным с воспалительным заболеванием. JAK обычно ассоциированы в паре с цитокиновыми рецепторами в виде гомодимеров или гетеродимеров. Конкретные цитокины ассоциированы с конкретными JAK–спариваниями. Каждый из четырех членов семейства JAK участвует в передаче сигналов по меньшей мере одного из цитокинов, связанных с воспалением.
Астма представляет собой хроническое заболевание дыхательных путей, для которого не существует никакой профилактики или лечения. Заболевание характеризуется воспалением, фиброзом, гиперчувствительностью и ремоделированием дыхательных путей, все это способствует ограничению воздушного потока. По оценкам, в мире от астмы страдают 300 миллионов человек, и, согласно оценкам, к 2025 году число людей, страдающих астмой, вырастет более чем на 100 миллионов. Хотя большинство пациентов могут контролировать симптомы астмы с использованием ингаляционных кортикостероидов, которые можно комбинировать с модификатором лейкотриенов и/или бета–агонистом длительного действия, остается подгруппа пациентов с тяжелой формой астмы, у которых заболевание не контролируется традиционными терапиями. Цитокины, связанные с воспалением при астме, которые передают сигналы через путь JAK–STAT, включают IL–2, IL–3, IL–4, IL–5, IL–6, IL–9, IL–11, IL–13, IL–23, IL–31, IL–27, тимусный стромальный лимфопоэтин (TSLP), интерферон–γ (IFNγ) и гранулоцитарно–макрофагальный колониестимулирующий фактор (GM–CSF). Воспаление дыхательных путей характерно и для других респираторных заболеваний, помимо астмы. Хроническая обструктивная болезнь легких (COPD), кистозный фиброз (CF), пневмония, интерстициальное заболевание легких (включая идиопатический легочный фиброз), острое повреждение легких, синдром острой дыхательной недостаточности, бронхит, эмфизема, и облитерирующий бронхиолит также являются заболеваниями дыхательных путей, патофизиология которых, как считают, связана с JAK–сигнальными цитокинами.
Воспаление играет значимую роль при многих глазных заболеваниях, включая увеит, диабетическую ретинопатию, диабетический макулярный отек, синдром сухого глаза, возрастную дегенерацию желтого пятна и атопический кератоконъюнктивит. Увеит охватывает множество внутриглазных воспалительных состояний и часто является аутоиммунным, возникающим без известного инфекционного возбудителя. По оценкам, этим состоянием страдают около 2 миллионов пациентов в США. У некоторых пациентов хроническое воспаление, ассоциированное с увеитом, приводит к разрушению ткани, и он является пятой основной причиной слепоты в США. Цитокины, уровень которых повышен в глазах пациентов с увеитом, которые передают сигналы через JAK–STAT путь, включают IL–2, IL–4, IL–5, IL–6, IL–10, IL–23 и IFN–γ. (Horai and Caspi, J Interferon Cytokine Res, 2011, 31, 733–744; Ooi et al, Clinical Medicine and Research, 2006, 4, 294–309). Существующие терапии увеита часто недостаточно эффективны, и у многих пациентов заболевание плохо контролируется. Стероиды, хотя часто эффективны, связаны с катарактами и повышенным внутриглазным давлением/глаукомой.
Причиной диабетической ретинопатии (DR) является повреждение кровеносных сосудов в сетчатке. Это наиболее распространенная причина потери зрения среди людей с диабетом. Ангиогенные, а также воспалительные пути играют важную роль в заболевании. Часто DR прогрессирует до диабетического макулярного отека (DME), наиболее частой причины потери зрения у пациентов с диабетом. По оценкам, только в США от этого заболевания страдают около 1,5 миллиона пациентов, из которых примерно у 20% заболеванием поражены оба глаза. Считают, что цитокины, которые передают сигналы по пути JAK–STAT, такие как IL–6, а также другие цитокины, такие как IP–10 и MCP–1 (альтернативно называемые CCL2), продукция которых частично управляется передачей сигналов по пути JAK–STAT, играют роль в воспалении, ассоциированном с DR/DME (Abcouwer, J Clin Cell Immunol, 2013, Suppl 1, 1–12; Sohn et al., American Journal of Opthalmology, 2011, 152, 686–694; Owen and Hartnett, Curr Diab Rep, 2013, 13, 476–480; Cheung et al, Molecular Vision, 2012, 18, 830–837; Dong et al, Molecular Vision, 2013, 19, 1734–1746; Funatsu et al, Ophthalmology, 2009, 116, 73–79). Существующие методы лечения DME недостаточно эффективны: интравитреальные анти–VEGF лечения эффективны только у части пациентов, а стероиды связаны с катарактой и повышенным внутриглазным давлением.
Синдром сухого глаза (DED) представляет собой многофакторное заболевание, которым страдают около 5 миллионов пациентов в США. Считается, что воспаление поверхности глаза играет важную роль в развитии и распространении этого заболевания. Повышенные уровни цитокинов, таких как IL–1, IL–2, IL–4, IL–5, IL–6 и IFN–γ, были отмечены в глазных жидкостях пациентов с DED (Stevenson et al., Arch Ophthalmol, 2012, 130, 90–100), и уровни часто коррелируют с тяжестью заболевания. Также считается, что возрастная дегенерация желтого пятна и атопический кератоконъюнктивит связаны с JAK–зависимыми цитокинами.
Принадлежащая тому же правообладателю заявка США с серийным № 15/341226, поданная 02 ноября 2016 года, раскрывает диамино соединения, полезные в качестве ингибиторов JAK. В частности, соединение 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенол (соединение 1)
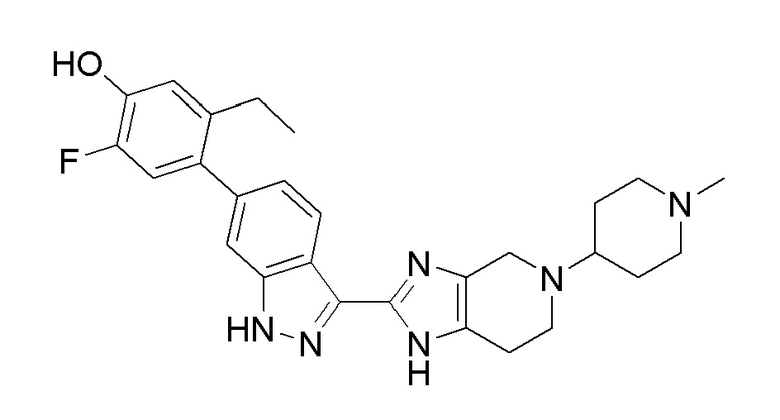
1
специально раскрыто в заявке как сильный ингибитор пан–JAK.
Для эффективного применения этого соединения в качестве терапевтического средства было бы желательно иметь солевую форму в кристаллическом состоянии. Например, было бы весьма желательно иметь физическую форму, которая термостабильна при достаточно высокой температуре, облегчая таким образом обработку и хранение вещества. Кристаллические твердые вещества, как правило, предпочтительнее аморфных форм для повышения чистоты и стабильности производимого продукта. Однако образование кристаллических форм органических соединений слишком непредсказуемо. Не существует никаких надежных способов для определения какая, если вообще таковая имеется, форма органического соединения будет кристаллической. Кроме того, не существует никаких способов для предсказания какая, если вообще таковая имеется, кристаллическая форма будет иметь физические свойства, желаемые для использования в качестве фармацевтических средств.
Никакие кристаллические солевые формы соединения 1 не были описаны ранее. Соответственно, существует потребность в кристаллических солевых формах соединения 1.
Сущность изобретения
Настоящее изобретение предоставляет кристаллогидраты оксалатной и сукцинатной солей 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола (1).
Было обнаружено, что кристаллогидрат оксалатной соли 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола имеет температуру плавления в диапазоне от около 266°C до около 276°C и демонстрирует общее поглощение влаги около 1% в условиях относительной влажности от около 30% до около 90% при комнатной температуре.
Было обнаружено, что кристаллогидрат сукцинатной соли 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола имеет температуру плавления в диапазоне от около 180°C до около 190°C и демонстрирует общее поглощение влаги около 2% в условиях относительной влажности от около 5% до около 90% при комнатной температуре.
Помимо прочих применений, кристаллические твердые формы по изобретению, как ожидают, будут полезны для получения фармацевтических композиций для лечения или облегчения заболевания, которое можно лечить ингибитором JAK, в частности респираторного заболевания. Соответственно, в еще одном из его аспектов, относящихся к композициям, изобретение предоставляет фармацевтическую композицию, содержащую фармацевтически приемлемый носитель и активное вещество, выбранное из кристаллогидрата оксалатной соли 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола и кристаллогидрата сукцинатной соли 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола.
Изобретение также предоставляет способ лечения респираторного заболевания, в частности астмы, у млекопитающего, включающий введение млекопитающему кристаллической твердой формы или фармацевтической композиции по изобретению. В отдельных и особых аспектах изобретение также предоставляет способы синтеза, пригодные для получения кристаллических форм по изобретению.
Изобретение также предоставляет способ лечения глазного воспалительного заболевания у млекопитающего, включающий введение в глаз млекопитающего кристаллической твердой формы или фармацевтической композиции по изобретению.
Изобретение также предоставляет кристаллическую твердую форму по изобретению, описанную в настоящей заявке, для применения в медицинском лечении, а также применение кристаллической твердой формы по изобретению для получения композиции или лекарственного средства для лечения респираторного заболевания у млекопитающего.
Краткое описание чертежей
Различные аспекты настоящего изобретения проиллюстрированы со ссылкой на прилагаемые чертежи.
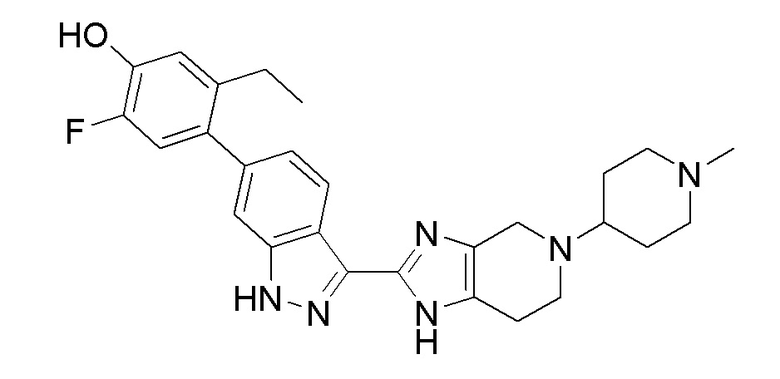
Фиг. 1 показывает порошковую рентгеновскую дифрактограмму (PXRD) кристаллогидрата оксалатной соли 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола (далее 'гидрат оксалата').
Фиг. 2 показывает термограмму, полученную методом дифференциальной сканирующей калориметрии (DSC), гидрата оксалата по изобретению.
Фиг. 3 показывает график термогравиметрического анализа (TGA) гидрата оксалата по изобретению.
Фиг. 4 показывает изотерму динамической сорбции влаги (DMS) гидрата оксалата по изобретению, наблюдаемую при температуре около 25°C.
Фиг. 5 показывает порошковую рентгеновскую дифрактограмму (PXRD) кристаллогидрата сукцината 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола (далее 'гидрат сукцината').
Фиг. 6 показывает термограмму, полученную методом дифференциальной сканирующей калориметрии (DSC), гидрата сукцината по изобретению.
Фиг. 7 показывает график термогравиметрического анализа (TGA) гидрата сукцината по изобретению.
Фиг. 8 показывает изотерму динамической сорбции влаги (DMS) гидрата сукцината по изобретению, наблюдаемую при температуре около 25°C.
Подробное описание изобретения
Определения
При описании настоящего изобретения, включая его различные аспекты и варианты осуществления, следующие термины имеют следующие значения, если не указано иное.
Термин “терапевтически эффективное количество” означает количество, достаточное для осуществления лечения при введении пациенту, нуждающемуся в лечении.
Термин “лечащий” или “лечение” означает профилактику, улучшение или подавление медицинского состояния, заболевания или расстройства, подлежащего лечению (например, респираторного заболевания) у пациента (в частности, человека); или облегчение симптомов медицинского состояния, заболевания или расстройства.
Термин "гидрат" означает комплекс или агрегат, обычно в кристаллической форме, образованный молекулами воды и соединения по изобретению, где отношение молекул воды к молекулам соединения может быть меньше чем 1:1 или больше чем 1:1.
Термин “около” означает ± 5 процентов от указанного значения.
Следует отметить, что, как используется в описании и в прилагаемой формуле изобретения, форма единственного числа, определяемая при помощи “a”, “an”, “one” и “the”, может включать соответствующие термины во множественном числе, если контекст явно не диктует иное.
Способ наименования
Соединение 1 определено как 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенол в соответствии с номенклатурой IUPAC, реализованной в программе ChemDraw (PerkinElmer, Inc., Cambridge, MA).
Кроме того, имидазо часть тетрагидроимидазопиридиновой группы в структуре соединения 1 существует в таутомерных формах, проиллюстрированных ниже для фрагмента соединения Примера 1
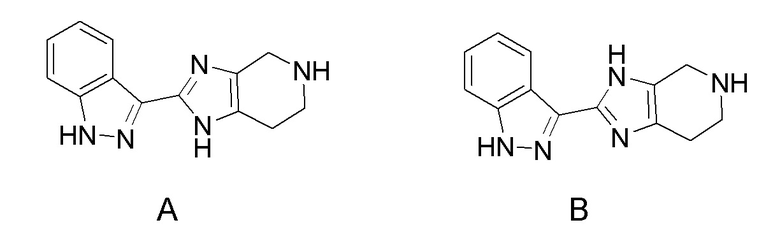
В соответствии с номенклатурой IUPAC, представленные структуры приводят к разной нумерации атомов имидазольной части: 2–(1H–индазол–3–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин (структура A) vs. 2–(1H–индазол–3–ил)–4,5,6,7–тетрагидро–3H–имидазо[4,5–c]пиридин (структура B). Должно быть понятно, что, хотя структуры показаны или названы в конкретной форме, изобретение также включает их таутомер.
Кристаллические формы по изобретению
В одном аспекте изобретение предоставляет кристаллогидрат оксалатной соли 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола (1).
В одном аспекте кристаллогидрат оксалата характеризуется порошковой рентгеновской дифрактограммой (PXRD), имеющей значимые дифракционные пики, среди других пиков, при 2θ значениях 6,77±0,20, 12,13±0,20, 13,54±0,20, 17,23±0,20 и 18,00±0,20. Кристаллогидрат оксалата может, кроме того, характеризоваться PXRD дифрактограммой, имеющей два или более дополнительных дифракционных пиков, в том числе три или более дополнительных дифракционных пиков при 2θ значениях, выбранных из 11,56±0,20, 14,29±0,20, 19,51±0,20, 21,38±0,20 и 23,63±0,20. В другом аспекте кристаллогидрат оксалата характеризуется PXRD дифрактограммой, имеющей дифракционные пики при 2θ значениях 6,77±0,20, 11,56±0,20, 12,13±0,20, 13,54±0,20, 14,29±0,20, 17,23±0,20, 18,00±0,20, 19,51±0,20, 21,38±0,20 и 23,63±0,20.
Как хорошо известно в области порошковой рентгеновской дифракции, положения пиков PXRD спектров сравнительно менее чувствительны к деталям эксперимента, таким как детали подготовки образцов и геометрия инструмента, чем относительная высота пиков. Такм образом, в одном аспекте кристаллогидрат оксалата характеризуется порошковой рентгеновской дифрактограммой, на которой положения пиков по существу соответствуют тем, которые показаны на Фиг. 1.
В другом аспекте кристаллогидрат оксалата характеризуется поведением под воздействием высокой температуры. Как продемонстрировано на Фиг. 2, кривая дифференциальной сканирующей калориметрии (DSC), полученная при скорости нагрева 10°C в минуту, демонстрирует эндотерму десольватации с началом при около 59°C и пиком при около 97°C и пиком в эндотермическом тепловом потоке, идентифицированным как переход в расплав, в диапазоне от около 266°C до около 276°C, в том числе от около 268°C до около 273°C. Кривая термогравиметрического анализа (TGA) на Фиг. 3 показывает начало десольватации при температуре около 26°C и начало разложения при температуре от около 250°C. Взятые вместе, кривые DSC и TGA указывают на то, что переход в расплав сопровождается разложением. TGA профиль показывает потерю массы около 5,5% между около 25°C и около 75°C.
Было продемонстрировано, что кристаллогидрат оксалата по изобретению имеет обратимый профиль сорбции/десорбции с небольшой склонностью к гигроскопичности. Гидрат оксалата показал общее поглощение влаги около 1% в условиях относительной влажности в диапазоне от около 30% до около 90% при комнатной температуре, как показанно на Фиг. 4. Обратимый переход гидратация/дегидратация наблюдали при относительной влажности между около 0% и 15%. Никакого гистерезиса не наблюдали в двух циклах сорбции и десорбции.
В другом аспекте изобретение предоставляет кристаллогидрат сукцинатной соли 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола (1)
В одном аспекте кристаллогидрат сукцината характеризуется порошковой рентгеновской дифрактограммой (PXRD), имеющей значимые дифракционные пики, среди других пиков, при 2θ значениях 4,81±0,20, 9,66±0,20, 14,93±0,20 и 16,78±0,20. Кристаллогидрат сукцината может, кроме того, характеризоваться PXRD дифрактограммой, имеющей два или более дополнительных дифракционных пиков, в том числе три или более дополнительных дифракционных пиков при 2θ значениях, выбранных из 10,46±0,20, 16,21±0,20, 17,45±0,20, 22,87±0,20 и 24,77±0,20. В другом аспекте кристаллогидрат оксалата характеризуется PXRD дифрактограммой, имеющей дифракционные пики при 2θ значениях 4,81±0,20, 9,66±0,20, 10,46±0,20, 14,93±0,20, 16,21±0,20, 16,78±0,20, 17,45±0,20, 22,87±0,20 и 24,77±0,20. Еще в одном аспекте кристаллогидрат сукцината характеризуется порошковой рентгеновской дифрактограммой, на которой положения пиков по существу соответствуют тем, которые показаны на Фиг. 5.
Кристаллогидрат сукцината также характеризуется поведением под воздействием высокой температуры. Как продемонстрировано на Фиг. 6, кривая дифференциальной сканирующей калориметрии (DSC), полученная при скорости нагрева 10°C в минуту, демонстрирует две эндотермы десольватации: одна с началом при около 20°C и пиком при около 50°C, а вторая эндотерма десольватации с началом при около 103°C и пиком при около 129°C. Кривая DSC также демонстрирует пик в эндотермическом тепловом потоке, идентифицированный как переход в расплав, в диапазоне от около 180°C до около 190°C, в том числе от около 183°C до около 188°C. Кривая термогравиметрического анализа (TGA) на Фиг. 7 показывает начало разложения при температуре около 200°C.
Было продемонстрировано, что кристаллогидрат сукцината имеет обратимый профиль сорбции/десорбции с небольшой склонностью к гигроскопичности. Гидрат сукцината показал общее поглощение влаги около 2% в условиях относительной влажности в диапазоне от около 5% до около 90% при комнатной температуре, как показанно на Фиг. 8. Никакого гистерезиса не наблюдали в двух циклах сорбции и десорбции.
Процедуры синтеза
Соединение 1 можно получить из легко доступных исходных веществ с использованием процедур, описанных в Примерах ниже, или с использованием процедур, описанных в принадлежащей тому же заявителю заявке США, указанной в разделе “Предпосылки создания изобретения” настоящей заявки.
Кристаллогидрат оксалата по изобретению удобно получают путем растворения эквимолярной смеси соединения 1 и щавелевой кислоты в 1:1 смеси тетрагидрофурана и воды при комнатной температуре, с последующим добавлением 1:1:2 смеси тетрагидрофуран:вода:ацетонитрил в качестве антирастворителя с получением суспензии. Полученную реакционную смесь перемешивают примерно в течение одного дня при комнатной температуре, промывают ацетонитрилом и сушат с получением формы кристаллогидрата.
Кристаллогидрат сукцината по изобретению можно получить трехстадийным способом. Во–первых, эквимолярную смесь соединения 1 и янтарной кислоты суспендируют в изопропаноле и перемешивают примерно в течение одного дня при комнатной температуре. Полученные твердые частицы фильтруют, промывают изопропанолом и сушат с получением первого промежуточного кристаллического твердого вещества. Во–вторых, выделенное первое промежуточное кристаллическое твердое вещество сушат при около 150°C примерно 30 минут с получением второго промежуточного кристаллического твердого вещества. В–третьих, второе промежуточное твердое вещество уравновешивают в условиях относительной влажности около 80%–90% примерно в течение одного дня при комнатной температуре с получением формы кристаллогидрата сукцината.
Соответственно, в аспекте, относящемся к способу, изобретение предоставляет способ получения кристаллогидрата оксалатной соли 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола, включающий (a) растворение 1:1 смеси 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола:щавелевой кислоты в 1:1 смеси тетрагидрофурана:воды при комнатной температуре, (b) добавление 1:1:2 смеси тетрагидрофурана:воды:ацетонитрила с получением суспензии, (c) перемешивание суспензии примерно в течение одного дня и (d) выделение кристаллогидрата оксалата из суспензии.
Фармацевтические композиции
Кристаллические твердые формы по изобретению обычно используют в форме фармацевтической композиции или состава. Такие фармацевтические композиции могут быть эффективны при введении пациенту путем ингаляции. Кроме того, фармацевтические композиции можно вводить любым приемлемым способом введения, включая, но не ограничиваясь этим, пероральный, местный (включая чрескожный), ректальный, назальный и парентеральный пути введения.
Соответственно, в одном из его аспектов, относящимся к композициям, изобретение направлено на фармацевтическую композицию, содержащую фармацевтически приемлемый носитель или эксципиент и кристаллогидрат оксалата или кристаллогидрат сукцината соединения 1. Необязательно, такие фармацевтические композиции могут содержать другие терапевтические средства и/или рецептурные добавки, если желательно. При обсуждении композиций и их применений кристаллические твердые формы по изобретению также могут быть указаны в настоящей заявке как "активное вещество".
Фармацевтические композиции по изобретению типично содержат терапевтически эффективное количество кристаллических форм по настоящему изобретению. Специалистам в данной области, однако, должно быть понятно, что фармацевтическая композиция может содержать больше, чем терапевтически эффективное количество, то есть нерасфасованные композиции, или меньше, чем терапевтически эффективное количество, то есть отдельные единичные дозы, предназначенные для многократного введения для достижения терапевтически эффективного количества.
Как правило, такие фармацевтические композиции будут содержать от около 0,01 до около 95% по массе активного вещества; включая, например, от около 0,05 до около 30% по массе; и от около 0,1% до около 10% по массе активного вещества.
В фармацевтических композициях по настоящему изобретению можно использовать любой обычный носитель или эксципиент. Выбор конкретного носителя или эксципиента, или комбинаций носителей или эксципиентов, будет зависеть от способа введения, используемого для лечения конкретного пациента, или от типа медицинского состояния или болезненного состояния. В этом отношении, получение подходящей фармацевтической композиции для конкретного способа введения находится в пределах компетенции специалистов в области фармацевтики. Кроме того, носители или эксципиенты, используемые в фармацевтических композициях по настоящему изобретению, являются коммерчески доступными. В качестве дополнительной иллюстрации, традиционные методы формулирования описаны в Remington: The Science and Practice of Pharmacy, 20th Edition, Lippincott Williams & White, Baltimore, Maryland (2000); и H.C. Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems, 7th Edition, Lippincott Williams & White, Baltimore, Maryland (1999).
Репрезентативные примеры веществ, которые могут служить в качестве фармацевтически приемлемых носителей, включают, но не ограничиваются этим, следующие: сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлоза, такая как микрокристаллическая целлюлоза, и ее производные, такие как натрий карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; порошкообразный трагакант; солод; желатин; тальк; эксципиенты, такие как масло какао и воски для суппозиториев; масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль; полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные агенты, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический солевой раствор; раствор Рингера; этиловый спирт; фосфатно–буферные растворы; и другие нетоксичные совместимые вещества, используемые в фармацевтических композициях.
Фармацевтические композиции обычно получают путем тщательного и тесного смешивания или вымешивания активного вещества с фармацевтически приемлемым носителем и одним или несколькими необязательными ингредиентами. Полученную однородно смешанную смесь затем можно формовать или загружать в таблетки, капсулы, пилюли и т.п., используя обычные процедуры и оборудование.
В одном аспекте фармацевтическая композиция является подходящей для ингаляционного введения. Фармацевтические композиции для ингаляционного введения обычно находятся в форме аэрозоля или порошка. Такие композиции обычно вводят с использованием устройств доставки ингаляционных препаратов, таких как ингалятор сухого порошка (DPI), дозирующий ингалятор (MDI), ингалятор–небулайзер или аналогичное устройство доставки.
В конкретном варианте осуществления фармацевтическую композицию вводят путем ингаляции, используя ингалятор сухого порошка. Такие ингаляторы сухого порошка, как правило, вводят фармацевтическую композицию в виде свободнотекучего порошка, который диспергируется в потоке воздуха при вдыхании пациентом. Чтобы получить композицию свободнотекучего порошка, терапевтическое средство обычно формулируют в композицию с подходящим эксципиентом, таким как лактоза, крахмал, маннит, декстроза, полимолочная кислота (PLA), полилактид–со–гликолид (PLGA) или их комбинации. Как правило, терапевтическое средство тонко измельчают и объединяют с подходящим носителем для образования композиции, подходящей для ингаляции.
Типичная фармацевтическая композиция для применения в ингаляторе сухого порошка включает лактозу и кристаллическую твердую форму по изобретению в микронизированной форме. Такая сухая порошковая композиция может быть получена, например, путем объединения сухой измельченной лактозы с терапевтическим средством и затем сухого смешивания компонентов. Затем композицию обычно загружают в дозатор сухого порошка или в ингаляционные картриджи или капсулы для использования с устройством доставки сухого порошка.
Устройства доставки в виде ингаляторов сухого порошка, подходящие для введения путем ингаляции, описаны в данной области техники, и примеры таких устройств являются коммерчески доступными. Например, типичные устройства доставки или продукты для ингаляции сухого порошка включают Aeolizer (Novartis); Airmax (IVAX); ClickHaler (Innovata Biomed); Diskhaler (GlaxoSmithKline); Diskus/Accuhaler (GlaxoSmithKline); Ellipta (GlaxoSmithKline); Easyhaler (Orion Pharma); Eclipse (Aventis); FlowCaps (Hovione); Handihaler (Boehringer Ingelheim); Pulvinal (Chiesi); Rotahaler (GlaxoSmithKline); SkyeHaler/Certihaler (SkyePharma); Twisthaler (Schering–Plough); Turbuhaler (AstraZeneca); Ultrahaler (Aventis); и т.п.
В другом конкретном варианте осуществления фармацевтическую композицию вводят путем ингаляции, используя дозирующий ингалятор. Такие дозирующие ингаляторы, как правило, выпускают отмеренное количество терапевтического средства, используя сжатый газ–пропеллент. Соответственно, фармацевтические композиции, вводимые с использованием дозирующего ингалятора, как правило, содержат раствор или суспензию терапевтического средства в сжиженном пропелленте. Можно использовать любой подходящий сжиженный пропеллент, включая гидрофторалканы (HFA), такие как 1,1,1,2–тетрафторэтан (HFA 134a) и 1,1,1,2,3,3,3–гептафтор–н–пропан, (HFA 227); и хлорфторуглероды, такие как CCl3F. В конкретном варианте осуществления пропеллент включает гидрофторалканы. В некоторых вариантах осуществления композиция гидрофторалканов содержит со–растворитель, такой как этанол или пентан, и/или поверхностно–активное вещество, такое как сорбитантриолеат, олеиновая кислота, лецитин и глицерин.
Типичная фармацевтическая композиция для применения в дозирующем ингаляторе содержит от около 0,01% до около 5% по массе соединения по настоящему изобретению; от около 0% до около 20% по массе этанола; и от около 0% до около 5% по массе поверхностно–активного вещества; остальное количество составляет HFA пропеллент. Такие композиции, как правило, получают путем добавления охлажденного или находящегося под давлением гидрофторалкана в подходящий контейнер, содержащий терапевтическое средство, этанол (если присутствует) и поверхностно–активное вещество (если присутствует). Для получения суспензии терапевтическое средство тонко измельчают, а затем объединяют с пропеллентом. Затем композицию загружают в аэрозольный баллончик, который обычно является частью дозирующего ингаляционного устройства.
Дозирующие ингаляторы, подходящие для введения терапевтических средств путем ингаляции, описаны в данной области техники, и примеры таких устройств являются коммерчески доступными. Например, типичные дозирующие ингаляторы или продукты включают ингаляционную систему AeroBid (Forest Pharmaceuticals); ингаляционный аэрозоль Atrovent (Boehringer Ingelheim); Flovent (GlaxoSmithKline); ингалятор Maxair (3M); ингалятор Proventil (Schering); ингаляционный аэрозоль Serevent (GlaxoSmithKline); и т.п.
В другом конкретном аспекте фармацевтическую композицию вводят путем ингаляции с использованием ингалятора–небулайзера. Такие небулайзеры, как правило, обычно производят поток воздуха с высокой скоростью, который заставляет фармацевтическую композицию распыляться в виде тумана, который переносится в дыхательные пути пациента. Соответственно, при формулировании для использования в ингаляторе–небулайзере терапевтическое средство можно растворить в подходящем носителе для образования раствора. Альтернативно, терапевтическое средство можно подвергнуть микронизации или измельчению до наночастиц и объединить с подходящим носителем для образования суспензии.
Типичная фармацевтическая композиция для применения в ингаляторе–небулайзере включает раствор или суспензию, содержащую от около 0,05 мкг/мл до около 20 мг/мл соединения по настоящему изобретению и эксципиенты, совместимые с распыляемыми составами. В одном варианте осуществления раствор имеет pH от около 3 до около 8.
Небулайзеры, подходящие для введения терапевтических средств путем ингаляции, описаны в данной области техники, и примеры таких устройств являются коммерчески доступными. Например, типичные распыляющие устройства или продукты включают ингалятор Respimat Softmist (Boehringer Ingelheim); систему доставки в легкие AERx (Aradigm Corp.); многоразовый небулайзер PARI LC Plus (Pari GmbH); и т.п.
В еще одном аспекте фармацевтические композиции по настоящему изобретению альтернативно можно получить в лекарственной форме, предназначенной для перорального введения. Подходящие фармацевтические композиции для перорального введения могут быть в форме капсул, таблеток, пилюль, пастилок, саше, драже, порошков, гранул; или в виде раствора или суспензии в водной или неводной жидкости; или в виде жидкой эмульсии масло–в–воде или вода–в–масле; или в виде эликсира или сиропа; и т.п.; при этом каждая из этих форм содержит заранее определенное количество соединения по настоящему изобретению в качестве активного ингредиента.
Если они предназначены для перорального введения в твердой лекарственной форме, фармацевтические композиции по настоящему изобретению, как правило, будут включать активное вещество и один или несколько фармацевтически приемлемых носителей, таких как цитрат натрия или дикальций фосфат. Необязательно или альтернативно, такие твердые лекарственные формы также могут включать: наполнители или добавки для увеличения объема, связующие, увлажнители, замедляющие растворение вещества, ускорители абсорбции, смачивающие вещества, абсорбенты, смазывающие вещества, красители и буферные агенты. В фармацевтических композициях по настоящему изобретению также могут присутствовать агенты, облегчающие высвобождение, смачивающие агенты, агенты покрытия, подсластители, отдушки и ароматизаторы, консерванты и антиоксиданты.
Кристаллические твердые формы также могут быть сформулированы в виде стерильной водной суспензии или раствора для глазного введения. Полезные эксципиенты, которые могут быть включены в такую водную композицию, включают полисорбат 80, карбоксиметилцеллюлозу, хлорид калия, хлорид кальция, хлорид натрия, хлорид магния, ацетат натрия, цитрат натрия, гистидин, дигидрат α–трегалозы, сахарозу, полисорбат 20, гидроксипропил–β–циклодекстрин и фосфат натрия. Бензиловый спирт может служить в качестве консерванта, а хлорид натрия может быть включен для регулирования тоничности. Кроме того, к раствору можно добавить хлористоводородную кислоту и/или гидроксид натрия для регулирования рН. Могут быть получены водные композиции для внутриглазных инъекций, не содержащие консервантов.
Альтернативные лекарственные формы также могут включать лекарственные формы с контролируемым высвобождением, жидкие лекарственные формы для перорального введения, трансдермальные пластыри и парентеральные лекарственные формы. Обычные эксципиенты и способы получения таких альтернативных лекарственных форм описаны, например, в ссылочном документе Remington, указанном выше.
Следующие неограничивающие примеры иллюстрируют типичные фармацевтические композиции по настоящему изобретению.
Сухая порошковая композиция
Микронизированную твердую форму по изобретению (1 г) смешивают с измельченной лактозой (25 г). Эту перемешанную смесь затем загружают в отдельные блистеры отслаиваемой блистерной упаковки в количестве, достаточном для обеспечения от около 0,1 до около 4 мг соединения формулы I на дозу. Содержимое блистеров вводят с использованием ингалятора сухого порошка.
Сухая порошковая композиция
Микронизированную твердую форму по изобретению (1 г) смешивают с измельченной лактозой (20 г) с образованием сыпучей композиции, имеющей массовое отношение соединения к измельченной лактозе 1:20. Смешанную композицию упаковывают в устройство для ингаляции сухого порошка, способное доставлять от около 0,1 до около 4 мг соединения формулы I на дозу.
Композиция для дозирующего ингалятора
Микронизированную твердую форму по изобретению (10 г) диспергируют в растворе, полученном путем растворения лецитина (0,2 г) в деминерализованной воде (200 мл). Полученную суспензию сушат распылением и затем измельчают с образованием микронизированной композиции, содержащей частицы, имеющие средний диаметр менее чем около 1,5 мкм. Микронизированную композицию затем загружают в картриджи дозирующего ингалятора, содержащие 1,1,1,2–тетрафторэтан под давлением, в количестве, достаточном для обеспечения от около 0,1 мг до около 4 мг соединения формулы I на дозу при введении при помощи дозирующего ингалятора.
Композиция для небулайзера
Твердую форму по изобретению (25 мг) растворяют в растворе, содержащем 1,5–2,5 эквивалента хлористоводородной кислоты, с последующим добавлением гидроксида натрия для доведения рН до 3,5–5,5 и 3% по массе глицерина. Раствор тщательно перемешивают до растворения всех компонентов. Раствор вводят с использованием небулайзера, который обеспечивает от около 0,1 мг до около 4 мг соединения формулы I на дозу.
Водная композиция для глазной инъекции
Каждый мл стерильной водной суспензии включает от 5 мг до 50 мг твердой формы по изобретению, хлорид натрия для тоничности, 0,99% (масс/об) бензилового спирта в качестве консерванта, 0,75% натрий карбоксиметилцеллюлозы и 0,04% полисорбата. Можно включить гидроксид натрия или хлористоводородную кислоту для доведения pH до 5–7,5.
Водная композиция для глазной инъекции
Стерильная не содержащая консервантов водная суспензия включает от 5 мг/мл до 50 мг/мл твердой формы по изобретению в 10 мМ фосфата натрия, 40 мМ хлорида натрия, 0,03% полисорбата 20 и 5% сахарозы.
Полезность
Было показано, что соединение по изобретению, 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенол, (соединение 1), является сильным ингибитором JAK семейства ферментов: JAK1, JAK2, JAK3 и TYK2.
Респираторные заболевания
Кроме того, как описано в анализах ниже, соединение 1 продемонстрировало сильное ингибирование провоспалительных и профибротических цитокинов, связанных с астмой и другими респираторными заболеваниями. Абсорбцию и дистрибуцию соединения определяли в доклинических анализах. У мышей соединение показало экспозицию в легких приверно в 55 раз больше, чем экспозиция в плазме. Важно, что концентрация соединения 1 в легких мыши, как было показано, коррелирует с предсказанным фармакодинамическим эффектом ингибирования фермента JAK. В частности, было показано, что соединения по изобретению ингибируют эффект провоспалительного цитокина IL–13 в ткани легкого мыши. В частности, соединение продемонстрировало ингибирование IL–13–индуцированного фосфорилирования STAT6 в ткани легкого, что свидетельствует о локальном связывании с JAK мишенью в легком in vivo. Этот эффект наблюдался при введении провоспалительного цитокина IL–13 через 4 часа после введения испытываемого соединения, что является дополнительным доказательством значительного удерживания в легком.
Противовоспалительная активность ингибиторов JAK была четко продемонстрирована на доклинических моделях астмы (Malaviya et al., Int Immunopharmacol, 2010, 10, 829,–836; Matsunaga et al., Biochem and Biophys Res Commun, 2011, 404, 261–267; Kudlacz et al., Eur J Pharmacol, 2008, 582, 154–161.) Соответственно, ожидается, что соединения по изобретению будут полезны для лечения воспалительных заболеваний дыхательных путей, в частности астмы. Воспаление и фиброз легких характерны для других респираторных заболеваний, помимо астмы, таких как хроническая обструктивная болезнь легких (COPD), кистозный фиброз (CF), пневмонит, интерстициальное заболевание легких (включая идиопатический легочный фиброз), острое повреждение легких, синдром острой дыхательной недостаточности, бронхит, эмфизема и облитерирующий бронхиолит. Следовательно, ожидают, что соединения по настоящему изобретению также будут полезны для лечения хронической обструктивной болезни легких, кистозного фиброза, пневмонита, интерстициального заболевания легких (включая идиопатический легочный фиброз), острого повреждения легких, острого респираторного дистресс–синдрома, бронхита, эмфиземы и облитерирующего бронхиолита. Как описано выше, для лечения респираторных заболеваний твердые формы особенно полезны для введения путем ингаляции.
Поэтому в одном аспекте настоящее изобретение предоставляет способ лечения респираторного заболевания у млекопитающего (например, человека), при этом способ включает введение млекопитающему терапевтически эффективного количества соединения по настоящему изобретению или фармацевтической композиции, содержащей фармацевтически приемлемый носитель и твердую форму по настоящему изобретению.
В одном аспекте респираторное заболевание представляет собой астму, хроническую обструктивную болезнь легких, кистозный фиброз, пневмонию, хроническую обструктивную болезнь легких (COPD), кистозный фиброз (CF), пневмонит, интерстициальное заболевание легких (включая идиопатический легочный фиброз), острое повреждение легких, острый респираторный дистресс–синдром, бронхит, эмфизему и облитерирующий бронхиолит. В другом аспекте респираторное заболевание представляет собой астму или хроническую обструктивную болезнь легких. В другом аспекте твердые формы по изобретению вводят путем ингаляции.
Настоящее изобретение также предоставляет способ лечения астмы у млекопитающего, при этом способ включает введение млекопитающему твердой формы по изобретению или фармацевтической композиции, содержащей фармацевтически приемлемый носитель и твердую форму по изобретению.
При использовании для лечения астмы соединения по изобретению обычно вводят в однократной суточной дозе или в виде нескольких доз в сутки, хотя можно использовать другие формы введения. Количество вводимого активного вещества на дозу или общее количество, вводимое в день, обычно определяется лечащим врачом с учетом соответствующих обстоятельств, включая состояние, подлежащее лечению, выбранный путь введения, конкретное вводимое соединение и его относительную активность, возраст, массу тела и ответ конкретного пациента, тяжесть симптомов у пациента и т.п.
В дополнение к продемонстрированному сильному ингибированию цитокинов, связанных с воспалением, соединение 1 продемонстрировало ингибирование T–клеточной активации и активность в анализах легочной эозинофилии и нейтрофилии у грызунов. Поэтому считают, что твердые формы по изобретению будут полезными для лечения дополнительных респираторных заболеваний.
Дополнительные респираторные заболевания включают легочные инфекции, гельминтоз, легочную артериальную гипертензию, саркоидоз, лимфангиолейомиоматоз, бронхоэктаз и инфильтративное легочное заболевание. Считают, что твердые формы по изобретению будут полезны для лечения лекарственного пневмонита, грибкового пневмонита, аллергического бронхолегочного аспергиллеза, гиперчувствительного пневмонита, эозинофильного грануломатоза с полиангиитом, идиопатической острой эозинофильной пневмонии, идиопатической хронической эозинофильной пневмонии, гиперэозинофильного синдрома, синдрома Леффлера, облитерирующего бронхиолита с организующейся пневмонией или индуцированного ингибитором иммунных контрольных точек пневмонита.
Цитокины, передающие сигналы через JAK, также играют большую роль в активации T–клеток, подтипа иммунных клеток, который имеет важное значение для многих иммунных процессов. Патологическая активация Т–клеток имеет решающее значение в этиологии множества респираторных заболеваний. Аутореактивные Т–клетки играют роль в облитерирующем бронхиолите с организующейся пневмонией (также называемом COS). Подобно COS, этиология отторжения трансплантата легкого связана с аберрантной T–клеточной активацией Т–клеток реципиентов трансплантированным легким донора. Отторжения трансплантата легких могут произойти раньше в виде первичной дисфункции трансплантата (PGD), организующейся пневмонии (OP), острого отторжения (AR) или лимфоцитарного бронхиолита (LB), или они могут произойти спустя годы после трансплантации легкого в виде хронической дисфункции аллотрансплантата легкого (CLAD). CLAD ранее был известен как облитерирующий бронхиолит (BO), но теперь считается синдромом, который может иметь различные патологические проявления, включая BO, рестриктивную CLAD (rCLAD или RAS) и нейтрофильную дисфункцию аллотрансплантата. Хроническая дисфункция аллотрансплантата легкого (CLAD) является серьезной проблемой в тактике долгосрочного лечения реципиентов с легочным трансплантатом, поскольку заставляет трансплантированное легкое постепенно терять функциональность (Gauthier et al., Curr Transplant Rep., 2016, 3(3), 185–191). CLAD плохо реагирует на лечение, и, следовательно, остается потребность в эффективных соединениях, способных к предотвращению или лечению этого состояния. Некоторые JAK–зависимые цитокины, такие как IFNγ и IL–5, активируются при CLAD и отторжении трансплантата легкого (Berastegui et al, Clin Transplant. 2017, 31, e12898). Кроме того, высокий уровень хемокинов CXCR3 в легких, таких как CXCL9 и CXCL10, которые находятся на нисходящем JAK–зависимом IFN сигнальном пути, связаны с худшими исходами у пациентов с трансплантированным легким (Shino et al, PLOS One, 2017, 12 (7), e0180281). Было показано, что системное ингибирование JAK эффективно при отторжении трансплантата почки (Vicenti et al., American Journal of Transplantation, 2012, 12, 2446–56). Поэтому ингибиторы JAK могут быть эффективными при лечении или профилактике отторжения трансплантата легких и CLAD. Подобные события активации Т–клеток, описанные в качестве основы для отторжения трансплантата легкого, также считают основным фактором, вызывающим заболевание легких, ассоциированное с реакцией трансплантат–против–хозяина (GVHD), которая может происходить после трансплантации гемопоэтических стволовых клеток. Как и в случае с CLAD, GVHD легкого является хроническим прогрессирующим заболеванием с крайне плохими исходами, и в настоящее время нет одобренного лечения. Ретроспективное многоцентровое обзорное исследование 95 пациентов с стероид–резистентной острой или хронической GVHD, которые получали системный ингибитор JAK руксолитиниб в качестве консервативной терапии, продемонстрировало полный или частичный ответ на руксолитиниб у большинства пациентов, включая пациентов с GVHD легких (Zeiser et al, Leukemia, 2015, 29, 10, 2062–68). Поскольку системное ингибирование JAK связывают с неблагоприятными явлениями и низким терапевтическим индексом, остается потребность в ингаляционном, направленно действующем на легкие, несистемном ингибиторе JAK для профилактики и/или лечения отторжения трансплантата легкого или GVHD легкого.
Соответственно, изобретение также предоставляет способ лечения дополнительных респираторных состояний, описанных выше, у млекопитающего, включающий введение млекопитающему твердой формы по изобретению или фармацевтической композиции, содержащей фармацевтически приемлемый носитель и твердую форму по изобретению.
Глазные заболевания
Было показано, что многие глазные заболевания связаны с повышением уровня провоспалительных цитокинов, которые зависят от пути JAK–STAT. Поскольку соединение по изобретению демонстрирует сильное ингибирование всех четырех ферментов JAK, ожидается, что оно должно сильно ингибировать передачу сигналов и патогенные эффекты многочисленных цитокинов (таких как IL–6, IL–2 и IFN–γ), которые передают сигналы через JAK, а также препятствовать повышению уровней других цитокинов (таких как MCP–1 и IP–10), продукция которых зависит от сигнального пути JAK–STAT.
В частности, соединение 1 показало pIC50 значения 6,7 или больше (IC50 значения 200 нМ или меньше) для ингибирования передачи сигналов IL–2, IL–4, IL–6 и IFNγ в клеточных анализах, описанных в Анализах 3–7, включая анализы, регистрирующие ингибирование нисходящих эффектов повышения уровня цитокинов.
Фармакокинетическое исследование в Анализе 12 продемонстрировало длительную экспозицию в глазных тканях кролика после одной интравитреальной инъекции суспензии кристаллического соединения 1 примера 2 и концентрацию в плазме по меньшей мере на три порядка ниже, чем концентрация, наблюдаемая в стекловидной ткани. Анализы 13 и 14 продемонстрировали фармакодинамический эффект соединения у крыс и кроликов.
Поэтому ожидают, что твердые формы по изобретению будут полезны при различных глазных заболеваниях, которые включают, но не ограничиваются этим, увеит, диабетическую ретинопатию, диабетический макулярный отек, синдром сухого глаза, возрастную дегенерацию желтого пятна и атопический кератоконъюнктивит.
В частности, увеит (Horai and Caspi, J Interferon Cytokine Res, 2011, 31, 733–744), диабетическая ретинопатия (Abcouwer, J Clin Cell Immunol, 2013, Suppl 1, 1–12), диабетический макулярный отек (Sohn et al., American Journal of Opthalmology, 2011, 152, 686–694), синдром сухого глаза (Stevenson et al, Arch Ophthalmol, 2012, 130, 90–100) и возрастная дегенерация желтого пятна (Knickelbein et al, Int Ophthalmol Clin, 2015, 55(3), 63–78) характеризуются повышением уровней некоторых провоспалительных цитокинов, которые передают сигналы через JAK–STAT путь. Соответственно, твердые формы по изобретению могут облегчать ассоциированное глазное воспаление и вызывать регрессию прогрессирующего заболевания или обеспечивать облегчение симптомов.
Окклюзия вен сетчатки (RVO) является широко распространенным глазным инвалидизирующим заболеванием. Обструкция кровотока сетчатки может привести к повреждению сосудистой сети сетчатки, кровоизлиянию и ишемии тканей. Хотя причины RVO являются многофакторными, было показано, что важное значение имеют как сосудистые, так и воспалительные медиаторы (Deobhakta et al., International Journal of Inflammation, 2013, article ID 438412). Повышенные уровни цитокинов, которые передают сигналы через путь JAK–STAT, таких как IL–6 и IL–13, а также других цитокинов, таких как MCP–1, продукция которых частично управляется передачей сигналов JAK–STAT, были обнаружены в тканях глаз пациентов с RVO (Shchuko et al, Indian Journal of Ophthalmology, 2015, 63(12), 905–911). Соответственно, твердые формы по изобретению могут облегчать ассоциированное глазное воспаление и вызывать регрессию прогрессирующего заболевания или обеспечивать облегчение симптомов при этом заболевании. Хотя многих пациентов с RVO лечат фотокоагуляцией, это по своей сути деструктивная терапия. Анти–VEGF средства также используют, но они эффективны только у части пациентов. Стероидные препараты, которые снижают уровень воспаления в глазу (триамцинолон ацетонид и дексаметазоновые имплантаты), также, как было показано, дают полезные результаты для пациентов с некоторыми формами RVO, но также было показано, что они вызывают катаракты и повышенное внутриглазное давление/глаукому.
Поэтому в одном аспекте изобретение предоставляет способ лечения глазного заболевания у млекопитающего, включающий введение твердой формы по изобретению в глаз млекопитающего. В одном аспекте глазное заболевание представляет собой увеит, диабетическую ретинопатию, диабетический макулярный отек, синдром сухого глаза, возрастную дегенерацию желтого пятна или атопический кератоконъюнктивит. В одном аспекте глазное заболевание представляет собой окклюзию вен сетчатки.
ПРИМЕРЫ
Следующие примеры синтеза и биологические примеры предложены для иллюстрации изобретения, и никоим образом не должны рассматриваться как ограничивающие объем изобретения. В приведенных ниже примерах следующие аббревиатуры имеют следующие значения, если не указано иное. Аббревиатуры, не определенные ниже, имеют общепринятые значения.
ACN=ацетонитрил
CPME=циклопентилметиловый эфир
DCM=дихлорметан
DCC=дициклогексилкарбодиимид
DIPEA=N, N–диизопропилэтиламин
DMAc=диметилацетамид
DIPEA=N, N–диизопиропилэтиламин
DMAc=диметилацетамид
DMF=N, N–диметилформамид
EtOAc=этилацетат
ч=час(часы)
IPAc=изопиропилацетат
KOAc=ацетат калия
MeOH=метанол
MeTHF=2–метилтетрагидрофуран
мин=минута(минуты)
MTBE=метил трет–бутиловый эфир
NMP=N–метил–2–пирролидон
Pd(amphos)2Cl2=бис(ди–трет–бутил(4–диметиламинофенил)–фосфин)дихлорпалладий(II)
Pd(dppf)Cl2=дихлор(1,1’–бис(дифенилфосфино)–ферроцен)дипалладий(II)
Pd(PPh3)4=тетракис(трифенилфосфин)палладий(0)
Pd(t–Bu3P)2=бис(три–трет–бутилфосфин)палладий(0)
RT=комнатная температура
TEA=триэтиламин
TFA=трифторуксусная кислота
THF=тетрагидрофуран
бис(пинаколато)дибор=4,4,5,5,4',4',5',5'–октаметил–[2,2']би[[1,3,2]диоксабороланил]
Реагенты и растворители приобретали у коммерческих поставщиков (Aldrich, Fluka, Sigma и т.д.) и использовали без дополнительной очистки. Реакции отслеживали при помощи тонкослойной хроматографии (ТСХ), аналитической высокоэффективной жидкостной хроматографии (анал. ВЭЖХ) и масс–спектрометрии. Реакционные смеси обрабатывали, как описано конкретно для каждой реакции; обычно их очищали экстракцией и другими методами очистки, такими как температура– и растворитель–зависимая кристаллизация, а также преципитация. Кроме того, реакционные смеси обычно очищали колоночной хроматографией или препаративной ВЭЖХ, обычно с использованием колонок с насадкой C18 или BDS и традиционных элюентов. Типичные условия препаративной ВЭЖХ описаны ниже.
Продукты реакции обычно охарактеризовывали методом масс– и 1H–ЯМР спектрометрии. Для ЯМР анализа образцы растворяли в дейтерированном растворителе (таком, как CD3OD, CDCl3 или d6–DMSO) и спектры 1H–ЯМР получали с использованием устройства Varian Gemini 2000 (400 МГц) в стандартных условиях наблюдения. Масс–спектрометрическую идентификацию соединений осуществляли методом электрораспылительной ионизации (ESMS) с использованием устройства Applied Biosystems (Foster City, CA) модель API 150 EX или устройства Waters (Milford, MA) 3100, соединенных с системами автоочистки.
Условия препаративной ВЭЖХ
B=ACN+0,05% TFA
Неочищенные соединения растворяли в смеси 1:1 вода:уксусная кислота при около 50 мг/мл. 4–минутный цикл в аналитическом масштабе осуществляли, используя колонку C18 2,1×50 мм, с последующим 15– или 20–минутным циклом в препаративном масштабе, используя объем вводимой пробы 100 мкл, с градиентом, основанном на удерживании % B эксперимента в аналитическом масштабе. Точные градиенты зависели от образца. Образцы с близко элюируемыми примесями проверяли на колонке C18 21×250 мм и/или колонке C14 21×150 мм для лучшего разделения. Фракции, содержащие желаемый продукт, идентифицировали при помощи масс–спектрометрического анализа.
Условия аналитической ВЭЖХ
Метод А
B=ACN:TFA (99,95:0,05)
Метод В
B=ACN:Вода:TFA (90:10:0,1)
Метод С
B=ACN:Вода:TFA (90:10:0,1)
Получение 1: 1–бензил–4–имино–1,4–дигидропиридин–3–амин
Смесь пиридин–3,4–диамина (445 г, 4,078 моль) и ACN (11,0 л) перемешивали в течение 80 мин при температуре от 25°C до 15°C. Добавляли бензилбромид (485 мл, 4,078 моль) в течение 20 мин и реакционную смесь перемешивали при 20°C в течение ночи. Реакционную смесь охлаждали до 10°C и фильтровали. В реактор добавляли ACN (3 л), который охлаждали до 10°C. Лепешку промывали раствором для промывки реактора и снова промывали ACN (3 л), нагретым до 25°C. Твердое вещество сушили на фильтре в течение 24 ч в атмосфере азота, при 55°C в вакууме в течение 2 ч и затем при комнатной температуре в течение ночи и в течение 4 дней с получением HBr соли указанного в заголовке соединения (1102,2 г, 3,934 моль, выход 96%). ВЭЖХ Метод A Время удерживания 4,12 мин.
Получение 2: 5–Бензил–2–(6–бром–1H–индазол–3–ил)–5H–имидазо[4,5–c]пиридин
(a) 5–Бензил–2–(6–бром–1H–индазол–3–ил)–5H–имидазо[4,5–c]пиридин
Раствор 6–бром–1H–индазол–3–карбальдегида (550 г, 2,444 моль), 1–бензил–4–имино–1,4–дигидропиридин–3–амина HBr (721 г, 2,333 моль) и DMAc (2,65 л) перемешивали в течение 60 мин и добавляли бисульфит натрия (257 г, 2,468 моль). Реакционную смесь нагревали до 135°C и поддерживали в течение 3 ч и давали охладиться до 20°C и поддерживали при 20°C в течение ночи. Добавляли ацетонитрил (8 л) и реакционную смесь перемешивали в течение 4 ч при 15°C. Суспензию фильтровали на фильтре, работающем под давлением, при средней скорости фильтрации. В реактор добавляли ACN (1 л). Лепешку промывали раствором ACN для промывки реактора и сушили в атмосфере азота в течение ночи и затем в вакууме при 50°C в течение 24 ч с получением HBr сол указанного в заголовке соединения (1264 г, 2,444 моль, выход 100%, чистота 94%) в виде плотного мокрого бежевого/коричневого твердого вещества. ВЭЖХ Метод A Время удерживания 8,77 мин.
Смесь продукта с предыдущей стадии (1264 г, 2,444 моль), MeTHF (6 л) и воды (2,75 л) нагревали до 65°C и добавляли гидроксид натрия 50% масс. (254 г, 3,177 моль) в течение 5 мин и реакционную смесь перемешивали при 65°C в течение 1 ч, охлаждали до комнатной температуры, затем до 5°C и поддерживали в течение 2 ч. Суспензию фильтровали и реактор и лепешку промывали MeTHF (1 л). Полученное твердое вещество бежевого до желтого цвета сушили на фильтре в атмосфере азота в течение 3 дней с получением указанного в заголовке соединения (475 г, 1,175 ммоль, выход 48%) в виде бежевого/желтого твердого вещества. Маточный раствор (около 8 л) концентрировали до около 2 л, при этом начинали выделяться твердые частицы. Суспензию нагревали до 50°C, поддерживали в течение 2 ч, охлаждали до 5°C в течение 2 ч, перемешивали в течение ночи и фильтровали. Лепешку промывали MeTHF (100 мл) и сушили в течение ночи в вакууме при 40°C с получением дополнительного количества указанного в заголовке соединения (140 г, 0,346 моль, выход 14%).
Смесь всего продукта с предыдущей стадии, объединенного с продуктом второй партии в том же масштабе (1500 г, 3,710 моль) и MeTHF (4 л), перемешивали при 20°C в течение 2 ч и фильтровали. Реактор и лепешку промывали MeTHF (1,5 л). Полученное твердое вещество бежевого–желтого цвета сушили в атмосфере азота в течение 3 дней с получением указанного в заголовке соединения в виде бежевого/желтого твердого вещества (1325 г, 3,184 моль, выход 86% (общий выход 68%), чистота 97%). ВЭЖХ Метод A Время удерживания 8,77 мин.
Получение 3: 5–бензил–2–(6–бром–1H–индазол–3–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин
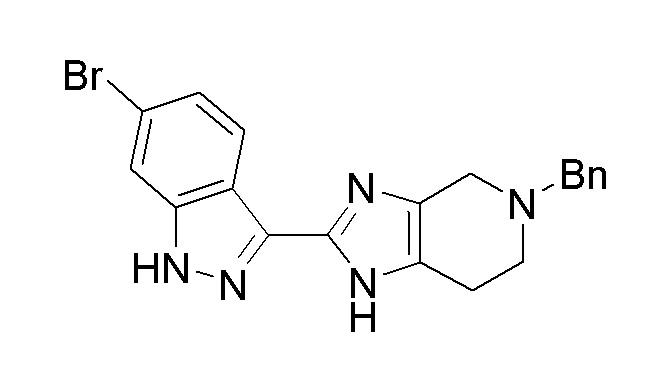
В 15–л колбу добавляли 5–бензил–2–(6–бром–1H–индазол–3–ил)–5H–имидазо[4,5–c]пиридин (440 г, 1,088 моль) с последующим добавлением MeTHF (4,5 л), метанола (2,25 л) и воды (1,125 л). Суспензию охлаждали до 20°C, перемешивали в течение 1 ч и добавляли NaBH4 (247 г, 6,530 моль). Реакционную смесь перемешивали при 25°C в течение 18 ч. Добавляли воду (1,125 л) с последующим добавлением 20% масс. раствора хлорида натрия (1,125 л) и смесь перемешивали в течение 30 мин и давали слоям разделиться. Водный слой сливали. Добавляли предварительно смешанный раствор NaOH (522 г) и воды (5 л) и реакционную смесь перемешивали в течение 60 мин; давали слоям разделиться и водный слой сливали. Получали две дополнительные партии в одинаковом масштабе.
Органический слой из одной партии концентрировали при пониженном давлении в 15–л снабженном рубашкой реакторе с установленной температурой рубашки 50°C, внутренней температурой 20°C. Дополнительные партии поочередно добавляли в реактор и концентрировали, что приводило к образованию суспензии объемом около 6 л. Суспензию нагревали до 50°C, добавляли IPAc (6 л) и смесь поддерживали при 60°C в течение 1,5 ч, охлаждали до 20°C в течение 10 ч, нагревали до 60°C в течение 50 ч, охлаждали до 20°C через 5 ч, затем охлаждали до 5°C и поддерживали в течение 3 ч. Смесь фильтровали и реактор и лепешку промывали предварительно смешанным раствором IPAc (1 л) и MeTHF (1 л), предварительно охлажденным до 5°C. Твердые вещества сушили в атмосфере азота на фильтре при 40°C в течение 3 дней с получением указанного в заголовке соединения (1059 г, 2,589 моль, выход 79%) в виде не совсем белого твердого вещества. Это вещество затем сушили в вакуумной печи при 50–60°C в течение 8 ч и при 27°C в течение 2 дней с получением указанного в заголовке соединения (1043 г, 2,526 моль, выход 77%, чистота 99%). ВЭЖХ Метод A Время удерживания 6,73 мин.
Получение 4: (4–(Бензилокси)–2–этил–5–фторфенил)трифторборат калия
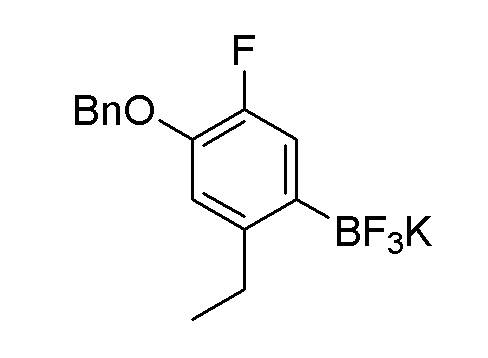
(a) 2–(4–(Бензилокси)–2–этил–5–фторфенил)–4,4,5,5–тетраметил–1,3,2–диоксаборолан
Смесь 1–(бензилокси)–4–бром–5–этил–2–фторбензола (520 г, 1682 ммоль) и диоксане (5193 мл) продували азотом и затем добавляли бис(пинаколато)дибор (641 г, 2523 ммоль) с последующим добавлением ацетата калия (495 г, 5046 ммоль). Реакционную смесь продували азотом; добавляли Pd(dppf)Cl2 (41,2 г, 50,5 ммоль); реакционную смесь продували азотом, нагревали при 103°C в атмосфере азота в течение 5 ч; и охлаждали до комнатной температуры. Реакционную смесь концентрировали вакуумной дистилляцией и распределяли между этилацетатом (5204 мл) и водой (5212 мл). Реакционную смесь фильтровали через Целит; органический слой промывали насыщенным солевым раствором (2606 мл), затем растворитель удаляли вакуумной дистилляцией с получением неочищенного продукта в виде густого черного масла (~800 г).
Неочищенный продукт растворяли в DCM (1289 мл) и очищали хроматографией на силикагеле (2627 г диоксида кремния, предварительно суспендированного в гексане, элюировали при помощи 20% этилацетата в гексане (10,35 л)). Растворитель удаляли вакуумной дистилляцией с получением светло–желтого масла (600 г). ВЭЖХ Метод B Время удерживания 33,74 мин.
(b) (4–(бензилокси)–2–этил–5–фторфенил)трифторборат калия
Продукт с предыдущей стадии (200 г, 561 ммоль) смешивали с ацетоном (1011 мл) до полного растворения и добавляли метанол (999 мл) с последующим добавлением 3 M гидродифторида калия (307 г, 3930 ммоль), растворенного в воде (1310 мл). Реакционную смесь перемешивали в течение 3,5 ч. Бóльшую часть органического растворителя удаляли вакуумной дистилляцией. Добавляли воду (759 мл) и полученную густую суспензию перемешивали в течение 30 мин и фильтровали. Лепешку промывали водой (506 мл) и твердые частицы сушили на фильтре в течение 30 мин. Твердые частицы суспендировали в ацетоне (1237 мл) и перемешивали в течение 1 ч. Полученную суспензию фильтровали и твердые частицы промывали ацетоном (247 мл). Ацетоновый раствор концентрировали путем вакуумной дистилляции и постоянный объем (2 л) поддерживали путем медленного добавления толуола (2983 мл) до полной отгонки ацетона и воды. Толуольный раствор подвергали дистилляции на роторном испарителе до получения густой желтой суспензии, в течение этого времени продукты осаждались в виде белых твердых частиц. К смеси добавляли дополнительную порцию толуола (477 мл) и перемешивали в течение 1 ч. Смесь затем фильтровали и промывали толуолом (179 мл) и сушили в вакууме при 50°C в течение 24 ч с получением указанного в заголовке соединения (104 г, 310 ммоль, выход 55%) в виде свободнотекучего рыхлого слегка беловатого твердого вещества. ВЭЖХ Метод B Время удерживания 27,71 мин.
Получение 5: 5–Бензил–2–(6–(4–(бензилокси)–2–этил–5–фторфенил)–1H–индазол–3–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин
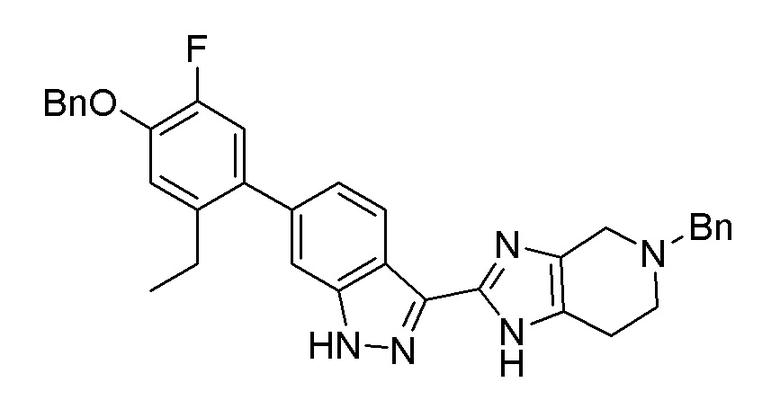
(a) 5–Бензил–2–(6–(4–(бензилокси)–2–этил–5–фторфенил)–1H–индазол–3–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин
Смесь бис(пинаколато)дибора (250 г, 984 ммоль) и IPA (1,88 л) перемешивали до растворения и затем добавляли по порциям раствор гидродифторида калия (538 г, 6,891 моль) в воде (2,31 л) в течение 10 мин. Реакционную смесь перемешивали в течение 1 ч и фильтровали. Гелеобразные твердые вещества суспендировали в воде (1,33 л) до образования прозрачного гидрогеля, а затем еще в течение 45 мин. Полученные твердые вещества/гель фильтровали, затем снова суспендировали в ацетоне (1,08 л), фильтровали, подвергали воздушной сушке на фильтре в течение 30 мин и сушили в течение ночи с получением рыхлого белого твердого вещества (196,7 г).
В 5–л колбу добавляли 5–бензил–2–(6–бром–1H–индазол–3–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин (135 г, 331 ммоль), (4–(бензилокси)–2–этил–5–фторфенил)–трифторборат калия (133 г, 397 ммоль) и белый твердый продукт с предыдущей стадии (40,5 г) с последующим добавлением MeTHF (1,23 л) и MeOH (1,75 л). Полученную суспензию дегазировали три раза азотом. К суспензии добавляли дегазированный раствор карбоната цезия (431 г, 1,323 моль) в воде (1,35 л). Суспензию дегазировали два раза, добавляли Pd (amphos)2Cl2 (11,71 г, 16,53 ммоль), суспензию снова дегазировали два раза и реакционную смесь перемешивали при 67°C в течение ночи и охлаждали до 20°C. Слои разделяли и подвергали обратной экстракции при помощи MeTHF (550 мл). Органические слои объединяли и концентрировали на роторном испарителе до осаждения твердых веществ. Добавляли MeTHF (700 мл) и реакционную смесь перемешивали при 65°C. Слои разделяли и водную фазу подвергали обратной экстракции при помощи MeTHF (135 мл). Органические фазы объединяли и концентрировали до около 300 мл, получая густую оранжевую суспензию. К суспензии добавляли MeOH (270 мл) с последующим добавлением 1M HCl (1,325 л) при 20°C при быстром перемешивании. Реакционную смесь перемешивали в течение 5 мин и добавляли воду (1 л) и полученную суспензию перемешивали в течение 1 ч. Твердые частицы фильтровали, промывали водой (150 мл), сушили на фильтре в течение 10 мин и при 45°C в атмосфере азота в течение 16 ч с получением 2 HCl соли указанного в заголовке соединения (221,1 г, 351 ммоль, чистота 92,2%) в виде светло–желтого твердого вещества. ВЭЖХ Метод B время удерживания 23,41 мин.
Получение 6: 5–этил–2–фтор–4–(3–(4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенол
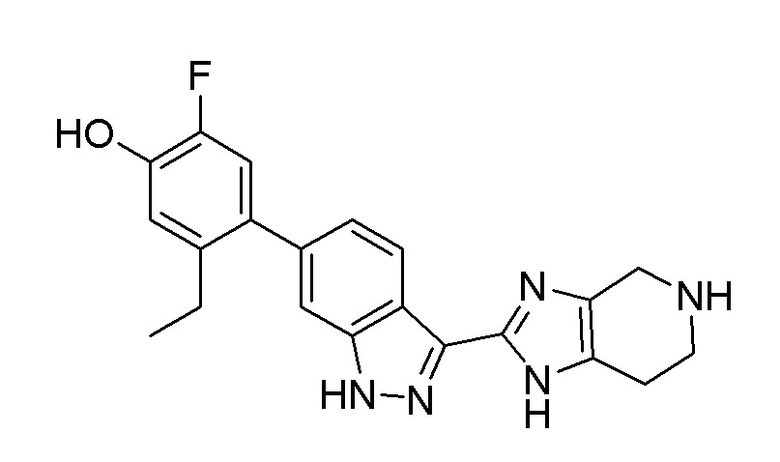
В 1–л колбу добавляли 5–бензил–2–(6–(4–(бензилокси)–2–этил–5–фторфенил)–1H–индазол–3–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин, 2 HCl (40 г, 63,4 ммоль) в виде суспензии в этаноле (348 мл) и 1,25 M HCl в MeOH (101 мл) и воде (17,14 мл). Реакционную смесь дегазировали азотом в течение 5 мин и добавляли 10% масс.Pd/C, 50% масс. H2O (4,05 г, 1,903 ммоль). Реактор герметично закрывали, продували при помощи H2, повышали давление до 1–2 ф/дюйм2(0,07–0,14 кг/см2), нагревали до 50°C и реакционную смесь перемешивали в течение ночи и фильтровали через Целит. Реактор и фильтр промывали метанолом (100 мл).
Отфильтрованный раствор объединяли с продуктом второй партии в масштабе 98 ммоль и концентрировали до 390 г. Медленно добавляли EtOAc (2,04 л) при перемешивании и затем раствор охлаждали до 5°C при перемешивании. Твердые вещества фильтровали, промывали при помощи EtOAc (510 мл) и сушили в течение ночи при 45°C в атмосфере азота с получением 2 HCl соли указанного в заголовке соединения (58 г, выход 80%) в виде не совсем белого твердого вещества. ВЭЖХ Метод B время удерживания 12,83 мин.
Пример 1: 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенол гидрат
В 125–мл колбу добавляли NMP (19,23 мл) и 5–этил–2–фтор–4–(3–(4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенол 2 HCl (6 г, 13,32 ммоль) при перемешивании с последующим добавлением NMP (19,23 мл). Добавляли уксусную кислоту (2,52 мл) и затем добавляли 1–метилпиперидин–4–он (3,28 мл, 26,6 ммоль) одной порцией и реакционную смесь перемешивали при 25°C в течение 30 мин и охлаждали до 15°C. Добавляли триацетоксиборогидрид натрия (7,91 г, 37,3 ммоль) и устанавливали температуру внешнего кожуха при 20°C через 20 мин. Через 3,5 ч общий объем раствора составлял 35 мл. Реактор промывали метанолом (5 мл). Половину раствора (17,5 мл), затем половину метанольной промывки (2,5 мл) добавляли к предварительно смешанному раствору метанола (28 мл), гидроксида аммония (17 мл, 270 ммоль) и воды (9 мл), поддерживая температуру ниже 5°C. Твердые вещества осаждались через 10 мин. Суспензию перемешивали в течение 30 мин, добавляли ACN (60 мл) медленно в течение 30 мин и суспензию перемешивали в течение 2 ч при 0°C, фильтровали и промывали при помощи ACN. Твердые вещества сушили при 50°C в течение 12 ч с получением указанного в заголовке соединения (2,95 г, выход 93,2%, (выход 85,2%, скорректированный на содержание воды)) в виде не совсем белого твердого вещества. ВЭЖХ Метод C время удерживания 12,11 мин
Пример 2: Кристаллогидрат 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола
К раствору 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола (10 г, 21,07 ммоль), полученному как в Примере A, в DMSO (19,99 мл) добавляли этанол (19,93 мл). Нерастворенные твердые частицы удаляли фильтрованием и половину DMSO раствора добавляли к перемешиваемому раствору 20% воды в метаноле (30 мл). Через 10 мин образовывалась суспензия, которую перемешивали при комнатной температуре в течение 4 ч и фильтровали. Полученное желтое твердое вещество сушили в течение 3 ч при 50°C в атмосфере азота. Твердое вещество суспендировали в растворе 20% воды в ацетоне (110 мл) при 45°C при перемешивании в течение 35 ч, фильтровали и промывали раствором 15% воды в ацетоне и сушили в течение ночи с получением указанного в заголовке соединения (4,40 г, выход 88%) в виде светло–желтого твердого вещества.
Пример 3: Кристаллогидрат оксалата 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола
В 20–мл стеклянном флаконе кристаллогидрат Соединения 1 (248,5 мг), продукт Примера 2 и безводную щавелевую кислоту (48,0 мг) растворяли в 1:1 смеси тетрагидрофуран:вода (5 мл). Добавляли ацетонитрил (5 мл) с получением суспензии. Полученную реакционную смесь перемешивали в течение одного дня при комнатной температуре, фильтровали, промывали ацетонитрилом (2 мл) и сушили в условиях окружающей среды в течение ночи с получением указанного в заголовке соединения.
Пример 4: Кристаллогидрат сукцината 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола
В 4–мл стеклянном флаконе кристаллогидрат Соединения 1 (40 мг) и янтарную кислоту (10 мг) суспендировали в изопропаноле (1 мл). Реакционную суспензию перемешивали в течение семи дней при комнатной температуре. Твердые вещества фильтровали, промывали изопропанолом (0,5 мл) и сушили в условиях окружающей среды в течение ночи с получением кристаллического сольвата сукцината. Выделенный сольват сукцината в виде твердого вещества сушили при 150°C в течение 30 мин в вакуумной печи с получением второй твердой формы, которую уравновешивали при 80%–90% относительной влажности в течение одного дня при комнатной температуре с получением указанного в заголовке соединения.
Примеры 5–7: Свойства твердых форм по изобретению
Образцы кристаллогидрата оксалатной соли 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола Примера 3 и кристаллогидрата сукцинатной соли 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола Примера 4 анализировали методами порошковой рентгеновской дифракции (PXRD), дифференциальной сканирующей калориметрии (DSC), термогравиметрического анализа (TGA) и динамической сорбции влаги (DMS).
Пример 5 Порошковая рентгеновская дифракция
Рентгеновские порошковые дифрактограммы, показанные на Фиг. 1, получали с использованием рентгеновского дифрактометра Bruker D8–Advance с Cu–Kα излучением (λ = 1,54051 Å), напряжением на выходе 45 кВ и током 40 мА. Устройство работало в геометрии Брэгга–Бретано с входной щелью, щелью расходимости и щелью рассеивания, установленных для максимизации интенсивности на образце. Для измерения небольшое количество порошка (5–25 мг) осторожно спрессовывали на держателе образца с получением ровной поверхности и подвергали рентгеновскому облучению. Образцы сканировали в режиме 2θ–2θ от 2° до 40° в 2θ с размером шага 0,02° и скоростью сканирования 0,30° секунд на шаг. Сбор данных контролировали с использованием программы измерений Bruker DiffracSuite и анализировали с использованием программы Jade (версия 7.5.1). Устройство калибровали с использованием корундового стандарта, в пределах ±0,02° угла два–тета. Наблюдаемые положения пиков PXRD два–тета и межатомные расстояния показаны в Таблицах 1 и 2 для кристаллогидрата оксалата и кристаллогидрата сукцината по изобретению, соответственно.
PXRD данные для кристаллогидрата оксалата
PXRD данные для кристаллогидрата сукцината
Пример 6: Термический анализ
Дифференциальную сканирующую калориметрию (DSC) осуществляли с использованием модуля TA Instruments Model Q–100 с контроллером Thermal Analyst. Данные собирали и анализировали с использованием программы TA Instruments Thermal Analysis. Образец каждой кристаллической формы точно отвешивали в закрываемую алюминиевую чашу. После 5–мин периода изотермического уравновешивания при 5°C образец нагревали с использованием линейного увеличения температуры 10°C/мин от 0°C до 250°C. Репрезентативная DSC термограмма кристаллогидрата оксалата и кристаллогидрата сукцината по изобретению показана на Фиг. 2 и 6, соответственно.
Термогравиметрический анализ (TGA) осуществляли с использованием модуля TA Instruments Model Q–50 с высокой разрешающей способностью. Данные собирали с использованием контроллера TA Instruments Thermal Analyst и анализировали с использованием программы TA Instruments Universal Analysis. Взвешенный образец помещали на платиновую чашу и сканировали со скоростью нагрева 10°C от температуры окружающей среды до 300°C. Камеры весов и печи продували азотом в процессе использования. Репрезентативные TGA кривые кристаллогидрата оксалата и кристаллогидрата сукцината по изобретению показаны на Фиг. 3 и 7, соответственно.
Пример 7: Метод динамической сорбции влаги
Анализ динамической сорбции влаги (DMS) осуществляли с использованием микровесов VTI при атмосферном давлении, SGA–100 системы (VTI Corp., Hialeah, FL 33016). Использовали взвешенный образец, и в начале анализа влажность имела самое низкое возможное значение (близкое к 0% RH). DMS анализ состоял из начальной стадии сушки (~0%RH) в течение 120 минут, с последующими двумя циклами сорбции и десорбции, со скоростью сканирования 5% RH/шаг в диапазоне влажности от 5% RH до 90% RH. DMS анализ осуществляли изотермически при 25°C. Репрезентативные DMS кривые для кристаллогидрата оксалата и кристаллогидрата сукцината по изобретению показаны на Фиг. 4 и 8, соответственно.
Биологические анализы
5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенол (соединение 1) было охарактеризовано в следующих биологических анализах.
Анализ 1: Биохимические анализы JAK киназ
Панель из четырех LanthaScreen биохимических анализов JAK (JAK1, 2, 3 и Tyk2) осуществляли в обычном буфере для киназной реакции (50 мМ HEPES, pH 7,5, 0,01% Brij–35, 10 мМ MgCl2 и 1 мМ EGTA). Рекомбинантные GST–меченные JAK ферменты и GFP–меченный STAT1 пептидный субстрат получали от Life Technologies.
Серийно разведенное соединение предварительно инкубировали с каждым из четырех JAK ферментов и субстратом в белых 384–луночных микропланшетах (Corning) при температуре окружающей среды в течение 1 часа. Затем добавляли АТФ для инициирования киназных реакций в общем объеме 10 мкл, с 1% DMSO. Конечные концентрации ферментов для JAK1, 2, 3 и Tyk2 составляли 4,2 нМ, 0,1 нМ, 1 нМ и 0,25 нМ, соответственно; соответствующие используемые Km АТФ концентрации составляли 25 мкМ, 3 мкМ, 1,6 мкМ и 10 мкМ; при этом концентрация субстрата составляла 200 нМ для всех четырех анализов. Киназным реакциям давали осуществиться в течение 1 часа при температуре окружающей среды, затем добавляли 10 мкл препарата EDTA (конечная концентрация 10 мМ) и Tb–анти–pSTAT1 (pTyr701) антитело (Life Technologies, 2 нМ конечная концентрация) в TR–FRET буфере для разведения (Life Technologies). Планшеты оставляли для инкубации при температуре окружающей среды в течение 1 часа, затем считывали на планшет–ридере EnVision (Perkin Elmer). Сигналы эмиссии (520нм/495нм) регистрировали и использовали для расчета процента ингибирования на основании DMSO и фоновых контролей.
Для анализа доза–ответ проценты ингибирования наносили на график против концентраций соединения и IC50 значения определяли из 4–параметрической робастной модели подгонки с использованием программы Prism (GraphPad Software). Результаты выражали как pIC50 (отрицательный логарифм IC50) и затем преобразовывали в pKi (отрицательный логарифм константы диссоциации, Ki) с использованием уравнения Ченга–Прусоффа.
Соединение 1 показало следующую активность в отношении ферментов.
pKi
pKi
pKi
pKi
Анализ 2: Клеточный анализ активности в отношении JAK: Ингибирование IL–13
AlphaScreen клеточный анализ активности в отношении JAKI осуществляли путем измерения интерлейкин–13 (IL–13, R&D Systems)–индуцированного фосфорилирования STAT6 в эпителиальных клетках легкого человека BEAS–2B (ATCC). Анти–STAT6 антитело (Cell Signaling Technologies) конъюгировали с AlphaScreen акцепторными шариками (Perkin Elmer), тогда как анти–pSTAT6 (pTyr641) антитело (Cell Signaling Technologies) биотинилировали с использованием Сульфо–NHS–Биотина EZ–Link (Thermo Scientific).
BEAS–2B клетки выращивали при 37°C в 5% CO2 увлажненном инкубаторе в среде 50% DMEM/50% F–12 (Life Technologies), дополненной 10% FBS (Hyclone), 100 Ед./мл пенициллина, 100 мкг/мл стрептомицина (Life Technologies) и 2 мМ GlutaMAX (Life Technologies). В день 1 анализа клетки высевали при плотности 7500 клеток/лунка в белые покрытые поли–D–лизином 384–луночные планшеты (Corning) с 25 мкл среды и оставляли в инкубаторе в течение ночи для адгезии. В день 2 анализа среду удаляли и заменяли на 12 мкл аналитического буфера (сбалансированный солевой раствор Хэнкса/HBSS, 25 мМ HEPES и 1 мг/мл бычьего сывороточного альбумин/BSA), содержащего доза–ответы испытываемых соединений. Соединение серийно разводили в DMSO и затем снова разбавляли 1000–кратно в среде для доведения конечной концентрации DMSO до 0,1%. Клетки инкубировали с испытываемыми соединениями при 37°C в течение 1 часа и затем добавляли 12 мкл предварительно нагретого IL–13 (80 нг/мл в аналитическом буфере) для стимуляции. После инкубации при 37°C в течение 30 минут аналитический буфер (содержащий соединение и IL–13) удаляли и добавляли 10 мкл буфера для лизиса клеток (25 мМ HEPES, 0,1% SDS, 1% NP–40, 5 мМ MgCl2, 1,3 мМ EDTA, 1 мМ EGTA и дополненный ингибиторами протеаз Complete Ultra mini и PhosSTOP от Roche Diagnostics). Планшеты встряхивали при температуре окружающей среды в течение 30 минут перед добавленим реагентов для детекции. Сначала добавляли смесь биотин–анти–pSTAT6 и анти–STAT6–конъюгированных акцепторных шариков и инкубировали при температуре окружающей среды в течение 2 часов, с последующим добавлением стрептавидин–конъюгированных донорных шариков (Perkin Elmer). После минимум 2 часов инкубации аналитические планшеты считывали на планшет–ридере EnVision. Сигналы люминесценции AlphaScreen регистрировали и использовали для расчета процента ингибирования на основании DMSO и фоновых контролей.
Для анализа доза–ответ процент ингибирования наносили на график против концентраций соединения и определяли IC50 значения из 4–параметрической робастной модели подгонки с использованием программы Prism. Результаты также могут быть выражены как отрицательный логарифм IC50 значения, pIC50. В этом анализе соединение 1 показало pIC50 значение 8,2.
Анализ 3: Клеточный анализ активности в отношении JAK: Ингибирование IL–2/анти–CD3–стимулированного IFNγ в человеческих PBMC
Активность испытываемого соединения для ингибирования интерлейкин–2 (IL–2)/анти–CD3–стимулированного интерферона гамма (IFNγ) измеряли в мононуклеарных клетках периферической крови человека (PBMC), выделенных из человеческой цельной крови (Stanford Blood Center). Поскольку IL–2 передает сигналы через JAK, этот анализ обеспечивает определение активности в отношении JAK в клетках.
(1) Мононуклеарные клетки периферической крови человека (PBMC) выделяли из человеческой цельной крови здоровых доноров с использованием градиента фиколла. Клетки культивировали в 37°C, 5% CO2 увлажненном инкубаторе в RPMI (Life Technologies), дополненной 10% термоинактивированной фетальной бычьей сыворотки (FBS, Life Technologies), 2 мМ Glutamax (Life Technologies), 25 мМ HEPES (Life Technologies) и 1X пенициллина/стрептомицина (Life Technologies). Клетки высевали при плотности 200000 клеток/лунка в среду (50 мкл) и культивировали в течение 1 часа. Соединение серийно разводили в DMSO и затем еще разбавляли 500–кратно (до 2х конечной анализируемой концентрации) в среде. Испытываемые соединения (100 мкл/лунка) добавляли к клеткам и инкубировали при 37°C, 5% CO2 в течение 1 часа с последующим добавлением IL–2 (R&D Systems; конечная концентрация 100 нг/мл) и анти–CD3 (BD Biosciences; конечная концентрация 1 мкг/мл) в предварительно нагретой среде для количественного определения (50 мкл) в течение 24 часов.
(2) После стимуляции цитокинами клетки центрифугировали при 500 g в течение 5 минут и супернатанты удаляли и замораживали при –80°C. Для определения ингибиторной активности испытываемых соединений в ответ на IL–2/анти–CD3 измеряли концентрации IFNγ в супернатанте методом ELISA (R&D Systems). IC50 значения определяли из анализа кривых ингибирования, представляющих концентрацию IFNγ против концентрации соединения. Данные выражены как pIC50 (отрицательный десятичный логарифм IC50) значения. В этом анализе соединение 1 показало pIC50 значение около 7,3.
Анализ 4: Клеточный анализ активности в отношении JAK: Ингибирование IL–2–стимулированного pSTAT5 в CD4+ T–клетках
Активность испытываемого соединения для ингибирования интерлейкин–2 (IL–2)/анти–CD3–стимулированного фосфорилирования STAT5 измеряли в CD4–положительных (CD4+) T–клетках в мононуклеарных клетках периферической крови человека (PBMC), выделенных из человеческой цельной крови (Stanford Blood Center), с использованием проточной цитометрии. Поскольку IL–2 передает сигналы через JAK, этот анализ обеспечивает определение активности в отношении JAK в клетках.
CD4+ T–клетки идентифицировали с использованием фикоэритробилин (PE)–конъюгированного анти–CD4 антитела (Клон RPA–T4, BD Biosciences), тогда как Alexa Fluor 647–конъюгированное анти–pSTAT5 антитело (pY694, Клон 47, BD Biosciences) использовали для детекции фосфорилирования STAT5.
(1) Следовали протоколу Анализа 3, параграф (1), за исключением того, что стимуляцию цитокином с анти–CD3 осуществляли в течение 30 минут вместо 24 часов.
(2) После стимуляции цитокинами клетки фиксировали предварительно нагретым фиксирующим раствором (200 мкл; BD Biosciences) в течение 10 минут при 37°C, 5% CO2, промывали два раза DPBS буфером (1 мл, Life Technologies) и ресуспендировали в ледяном Perm Буфере III (1000 мкл, BD Biosciences) в течение 30 минут при 4°C. Клетки промывали два раза 2% FBS в DPBS (FACS буфер) и затем ресуспендировали в FACS буфере (100 мкл), содержащем анти–CD4 PE (1:50 разведение) и анти–CD3 анти–CD3Alexa Fluor 647 (1:5 разведение), в течение 60 минут при комнатной температуре в темноте. После инкубации клетки промывали два раза в FACS буфере, затем анализировали с использованием проточного цитометра LSRII (BD Biosciences). Для определения ингибиторной активности испытываемого соединения в ответ на IL–2/анти–CD3 измеряли среднюю интенсивность флуоресценции (MFI) pSTAT5 в CD4+ T–клетках. IC50 значения определяли из анализа кривых ингибирования, представляющих MFI против концентрации соединения. Данные выражены как pIC50 (отрицательный десятичный логарифм IC50) значения. В этом анализе соединение 1 показало pIC50 значение около 7,7.
Анализ 5: Клеточный анализ активности в отношении JAK: Ингибирование IL–4–стимулированного pSTAT6 в CD3+ T клетках
Эффективность ингибирования испытываемым соединением интерлейкин–4(IL–4)–стимулированного фосфорилирования STAT6 измеряли в CD3–положительных (CD3+) Т–клетках в мононуклеарных клетках периферической крови человека (РВМС), выделенных из цельной крови человека (Stanford Blood Center), с использованием проточной цитометрии. Поскольку IL–4 передает сигналы через JAK, этот анализ обеспечивает показатель эффективности в клетках в отношении JAK.
CD3+ T–клетки идентифицировали, используя конъюгированное с фикоэритробилином (PE) анти–CD3 антитело (Клон UCHT1, BD Biosciences), при этом конъюгированное с Alexa Fluor 647 анти–pSTAT6 антитело (pY641, Клон 18/P, BD Biosciences) использовали для детекции фосфорилирования STAT6.
Мононуклеарные клетки периферической крови человека (РВМС) выделяли из цельной крови человека здоровых доноров, как в Анализах 3 и 4. Клетки высевали при 250000 клеток/лунка в среду (200 мкл) и культивировали в течение 1 часа и затем ресуспендировали в среде для анализа (50 мкл) (RPMI, дополненная 0,1% бычьим сывороточным альбумином (Sigma), 2мМ Glutamax, 25мМ HEPES и 1X ПенСтреп), содержащей различные концентрации испытываемых соединений. Соединения серийно разводили в DMSO и затем еще 500–кратно разводили (до 2х конечной анализируемой концентрации) в среде для анализа. Испытываемые соединения (50 мкл) инкубировали с клетками при 37°C, 5% CO2 в течение 1 часа, с последующим добавлением IL–4 (50 мкл) (R&D Systems; конечная концентрация 20 нг/мл) в предварительно нагретой среде для анализа в течение 30 мин. После стимуляции цитокинами клетки фиксировали предварительно нагретым фиксирующим раствором (100 мкл) (BD Biosciences) в течение 10 минут при 37°C, 5% CO2, дважды промывали FACS буфером (1 мл) (2% FBS в DPBS) и ресуспендировали в охлажденном льдом Perm Буфере III (1000 мкл) (BD Biosciences) в течение 30 минут при 4°C. Клетки дважды промывали FACS буфером и затем ресуспендировали в FACS буфере (100 мкл), содержащем анти–CD3 PE (1:50 разведение) и анти–pSTAT6 Alexa Fluor 647 (1:5 разведение), в течение 60 минут при комнатной температуре в темноте. После инкубации клетки дважды промывали в FACS буфере перед анализом с использованием проточного цитометра LSRII (BD Biosciences).
Для определения ингибирующей активности испытываемого соединения в ответ на IL–4, среднюю интенсивность флуоресценции (MFI) pSTAT6 измеряли в CD3+ T–клетках. Значения IC50 определяли из анализа кривых ингибирования, представляющих MFI против концентрации соединения. Данные выражены в виде значений pIC50 (отрицательный десятичный логарифм IC50). В этом анализе соединение 1 показало pIC50 значение 8,1.
Анализ 6: Клеточный анализ активности в отношении JAK: ингибирование IL–6–стимулированного pSTAT3 в CD3+ T–клетках
Использовали протокол, аналогичный протоколу Анализа 5, для определения эффективности ингибирования испытываемым соединением интерлейкин–6(IL–6)–стимулированного фосфорилирования STAT3. Конъюгированное с Alexa Fluor 647 анти–pSTAT3 антитело (pY705, Клон 4/P, BD Biosciences) использовали для детекции фосфорилирования STAT3.
В этом анализе соединение 1 показало pIC50 значение 7,4.
Анализ 7: Клеточный анализ активности в отношении JAK: Ингибирование IFNγ–индуцированного pSTAT1
Активность испытываемого соединения для ингибирования интерферон гамма (IFNγ)–стимулированного фосфорилирования STAT1 измеряли в CD14–положительных (CD14+) моноцитах, выделенных из человеческой цельной крови (Stanford Blood Center), с использованием проточной цитометрии. Поскольку IFNγ передает сигналы через JAK, этот анализ обеспечивает определение активности в отношении JAK в клетках.
Моноциты идентифицировали с использованием флуоресцеинизотиоцианат (FITC)–конъюгированного анти–CD14 антитела (Клон RM052, Beckman Coulter), а Alexa Fluor 647–конъюгированное анти–pSTAT1 антитело (pY701, Клон 4a, BD Biosciences) использовали для детекции фосфорилирования STAT1.
Мононуклеарные клетки периферической крови человека (PBMC) выделяли из человеческой цельной крови здоровых доноров с использованием градиента фиколла. Клетки культивировали в 37°C, 5% CO2 увлажненном инкубаторе в RPMI (Life Technologies), дополненной 10% фетальной бычьей сыворотки (FBS, Life Technologies), 2 мМ Glutamax (Life Technologies), 25 мМ HEPES (Life Technologies) и 1X пенициллином/стрептомицином (Life Technologies). Клетки высевали при плотности 250000 клеток/лунка в среду (200 мкл), культивировали в течение 2 часов и ресуспендировали в среде для количественного определения (50 мкл) (RPMI, дополненной 0,1% бычьего сывороточного альбумина (Sigma), 2 мМ Glutamax, 25 мМ HEPES и 1X ПенСтреп), содержащей различные концентрации испытываемых соединений. Соединение серийно разводили в DMSO и затем снова разбавляли 1000–кратно в среде для доведения конечной концентрации DMSO до 0,1%. Разведения испытываемого соединения инкубировали с клетками при 37°C, 5% CO2, в течение 1 часа с последующим добавлением предварительно нагретого IFNγ (R&D Systems) в среде (50 мкл) при конечной концентрации 0,6 нг/мл в течение 30 минут. После стимуляции цитокинами клетки фиксировали предварительно нагретым фиксирующим раствором (100 мкл) (BD Biosciences) в течение 10 минут при 37°C, 5% CO2, промывали два раза FACS буфером (1 мл) (1% BSA в PBS), ресуспендировали в смеси 1:10 анти–CD14 FITC:FACS буфер (100 мкл) и инкубировали при 4°C в течение 15 минут. Клетки промывали один раз и затем ресуспендировали в ледяном Perm Буфере III (BD Biosciences) (100 мкл) в течение 30 минут при 4°C. Клетки промывали два раза FACS буфером и затем ресуспендировали в смеси 1:10 анти–pSTAT1 Alexa Fluor 647:FACS буфер (100 мкл) в течение 30 минут при комнатной температуре в темноте, промывали два раза в FACS буфере и анализировали с использованием проточного цитометра LSRII (BD Biosciences).
Для определения ингибиторной активности испытываемого соединения среднюю интенсивность флуоресценции (MFI) pSTAT1 измеряли в CD14+ моноцитах. IC50 значения определяли из анализа кривых ингибирования, представляющих MFI против концентрации соединения. Данные выражены как pIC50 (отрицательный десятичный логарифм IC50) значения. В этом анализе соединение 1 показало pIC50 значение около 7,5.
Анализ 8: Фармакокинетика в плазме и легком мыши
Уровни испытываемого соединения в плазме и легких и их соотношения определяли следующим образом. В анализе использовали мышей BALB/c от Charles River Laboratories. Испытываемые соединения отдельно формулировали в 20% растворе пропиленгликоля в pH 4 цитратном буфере при концентрации 0,2 мг/мл и 50 мкл раствора вводили в трахею мыши путем оральной аспирации. В различных временных точках (типично 0,167, 2, 6, 24ч) после введения дозы брали образцы крови путем сердечной пункции и у мышей вырезали интактные легкие. Образцы крови центрифугировали (центрифуга Эппендорфа, 5804R) в течение 4 минут при приблизительно 12000 об/мин при 4°C для сбора плазмы. Легкие сушили, промакивая тампоном, взвешивали и гомогенизировали при разведении 1:3 в стерильной воде. Уровни испытываемого соединения в плазме и легких определяли анализом ЖХ–МС против аналитических стандартов, построенных в виде стандартной кривой в тестируемой матрице. Отношение легкие:плазма определяли как отношение AUC в мкг×ч/г в легких к AUC мкг×ч/мл в плазме, где AUC обычно определяют как площадь под кривой концентрации испытываемого соединения в зависимости от времени.
Соединение по изобретению показало экспозицию в легких мыши примерно в 55 раз больше, чем экспозиция в плазме.
Анализ 9: Мышиная (мышь) модель IL–13–индуцированной индукции pSTAT6 в ткани легкого
IL–13 является важным цитокином, лежащим в основе патофизиологии астмы (Kudlacz et al. Eur. J. Pharmacol, 2008, 582,154–161). IL–13 связывается с рецепторами клеточной поверхности, активируя членов семейства Janus киназ (JAK), которые затем фосфорилируют STAT6, и затем активирует дальнейшие пути транскрипции. В описанной модели дозу IL–13 доставляли локально в легкие мышей для индукции фосфорилирования STAT6 (pSTAT6), которое затем измеряли в качестве конечной точки.
В анализе использовали взрослых мышей balb/c от Harlan. В день исследования животных слегка анестезировали изофлураном и вводили либо носитель, либо испытываемое соединение (0,5 мг/мл, общий объем 50 мкл за несколько вдохов) посредством оральной аспирации. Животных помещали в боковое лежачее положение после введения дозы и проверяли на полное восстановление после анестезии перед возвращением в их клетку. Через четыре часа животных снова ненадолго анестезировали и вводили либо носитель, либо IL–13 (общая доставленная доза 0,03 мкг, общий объем 50 мкл) посредством оральной аспирации, а затем проверяли на восстановление от анестезии и возвращали в их клетки. Через один час после введения носителя или IL–13 легкие собирали для определения pSTAT6 с использованием анти–pSTAT6 ELISA (кроличье mAb иммобилизованное/покрывающее антитело; мышиное mAb детекторное/репортерное антитело: анти–pSTAT6–pY641; вторичное антитело: анти–мышиное IgG–HRP) и анализировали на общую концентрацию лекарственного средства, как описано выше в Анализе 12.
Об активности в модели свидетельствует снижение уровня pSTAT6, присутствующего в легких обработанных животных через 5 часов, по сравнению с обработанными носителем IL–13–индуцированными контрольными животными. Разница между контрольными животными, которые были обработаны носителем и IL–13–индуцированы, и контрольными животными, которые были обработаны носителем и индуцированы носителем, определяла ингибирующий эффект 0% и 100%, соответственно, в любом данном эксперименте. Соединение 1 показало примерно 60% ингибирование фосфорилирования STAT6 через 4 часа после IL–13 стимуляции.
Анализ 10: Мышиная модель Alternaria alternata–индуцированного эозинофильного воспаления легких
Эозинофилия дыхательных путей является отличительным признаком астмы человека. Alternaria alternata является грибковым аэроаллергеном, который может усугублять астму у людей и вызывать эозинофильное воспаление в легких мышей (Havaux et al. Clin Exp Immunol. 2005; 139(2):179–88). На мышах было продемонстрировано, что альтернария опосредованно активирует присутствующие в ткани легких врожденные лимфоидные клетки типа 2, которые отвечают на (например, IL–2 и IL–7) и высвобождают JAK–зависимые цитокины (например, IL–5 и IL–13) и координируют эозинофильное воспаление (Bartemes et al. J Immunol. 2012; 188(3):1503–13).
В исследовании использовали самцов мышей C57 в возрасте от семи до девяти недель от Taconic. В день исследования животных слегка анестезировали изофлураном и вводили либо носитель, либо испытываемое соединение (0,1–1,0 мг/мл, общий объем 50 мкл за несколько вдохов) посредством орофарингеальной аспирации. Животных помещали в боковое лежачее положение после введения дозы и проверяли на полное восстановление от анестезии перед их возвращением в клетку. Через один час животных снова ненадолго анестезировали и индуцировали либо носителем, либо экстрактом альтернарии (общее доставляемое количество экстракта 200 мкг, общий объем 50 мкл) посредством орофарингеальной аспирации, а затем проверяли на полное восстановление от анестезии и возвращали в их клетки. Через сорок восемь часов после введения альтернарии собирали жидкость бронхоальвеолярного лаважа (BALF) и подсчитывали эозинофилы в BALF с использованием гематологической системы Advia 120 (Siemens).
Активность в модели подтверждается снижением уровня эозинофилов, присутствующих в BALF обработанных животных через сорок восемь часов, по сравнению с обработанными носителем, зараженными альтернарией контрольными животными. Данные выражены как процент ингибирования ответа обработанных носителем альтернария–индуцированных эозинофилов в BALF. Для расчета процента ингибирования количество эозинофилов в BALF для каждого состояния преобразуют в процент от среднего числа обработанных носителем альтернария–индуцированных эозинофилов в BALF и вычитают из ста процентов. Соединение 1 показало примерно 88% ингибирование количества эозинофилов в BALF через сорок восемь часов после заражения альтернарией.
Анализ 11: Мышиная модель LPS/G–CSF/IL–6/IFNγ коктейль–индуцированного нейтрофильного воспаления дыхательных путей модели легких
Нейтрофилия дыхательных путей является отличительным признаком ряда респираторных заболеваний человека. Соединение 1 испытывали в модели нейтрофильного воспаления дыхательных путей с использованием LPS/G–CSF/IL–6/IFNγ коктейля для индукции нейтрофилии дыхательных путей.
В анализе использовали семи–девятинедельных самцов мышей Balb/C (дикого типа) от Jackson Laboratory. В день исследования животных слегка анестезировали изофлураном и вводили либо носитель, либо испытываемое соединение (1,0 мг/мл, общий объем 50 мкл за несколько вдохов) посредством орофаригеальной аспирации. Животных помещали в боковое лежачее положение после введения дозы и проверяли на полное восстановление после анестезии перед возвращением в их клетку. Через один час животных снова ненадолго анестезировали и вводили либо носитель, либо LPS; 0,01 мг/кг/G–CSF; 5 мкг/IL–6; 5 мкг/IFNγ; 5 мкг (общий объем 100 мкл) посредством орофаригеальной аспирации (OA). Через двадцать четыре часа после введения LPS/G–CSF/IL–6/IFNγ коктейля собирали жидкость бронхоальвеолярного лаважа (BALF) и подсчитывали количество нейтрофилов.
После OA обработки соединением 1 наблюдали статистически значимое уменьшение нейтрофилов в дыхательных путях (84% по сравнению с мышами, обработанными носителем), указывающее на то, что блокада JAK–зависимой передачи сигналов имеет эффекты на нейтрофильное воспаление дыхательных путей.
Анализ 12: Фармакокинетика в глазных тканях кроликов
Целью этого анализа было определение фармакокинетики испытываемого соединения в глазных тканях кролика.
Композиция раствора
Соединение по изобретению 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенол (1) растворяли либо в 10% растворе 2–гидроксипропил–β–циклодекстрина до достижения целевой концентрации 4 мг/мл, либо в очищенной воде до достижения целевой концентрации 1 мг/мл. Билатеральную интравитреальную инъекцию (50 мкл/глаз) раствора испытываемого соединения вводили новозеландским белым кроликам в двух дозовых группах, 200 мкг/глаз и 50 мкг/глаз, соответственно, для композиций в циклодекстрине и воде, соответственно. Концентрацию испытываемого соединения измеряли в глазных тканях: стекловидном теле, внутриглазной жидкости, сетчатке/сосудистой оболочке и иридо–цилиарной зоне в заранее определенных точках времени после инъекции (30 минут, 4 ч, 1 д, 3 д, 7 д, 14 д). Введение осуществляли двум кроликам (четыре глаза) для каждой временной точки. В ткани стекловидного тела соединение 1 показало двухфазное снижение концентрации, характеризующееся начальным снижением концентрации с периодом полужизни примерно 12 часов и в завершение с конечным периодом полужизни примерно 3,6 дня. Было обнаружено, что соединение также быстро распределяется в сетчатке и в сосудистой оболочке и показывает такой же фармакокинетический профиль, как в ткани стекловидного тела.
Композиция суспензии
Композицию суспензии получали путем объединения кристаллического соединения 1 Примера 2 с раствором 0,5% гидроксипропилметилцеллюлозы (HPMC E5) + 0,02% Tween 80 до достижения целевой концентрации 10 мг/мл. Билатеральную интравитреальную инъекцию (50 мкл/глаз) суспензии испытываемого соединения вводили новозеландским белым кроликам (500 мкг/глаз). Концентрацию испытываемого соединения измеряли в глазных тканях, как в анализе композиции суспензии, через 30 минут, 2 недели, 4 недели, 6 недель и 8 недель после инъекции. Соединение показало линейное снижение концентрации лекарственного средства в ткани стекловидного тела от 30 минут до 6 недель при скорости выведения лекарственного средства из организма примерно 3 мкг/мл/день. Такое поведение согласуется с растворимостью соединения 1 в носителе и фармакокинетикой в глазных тканях при использовании композиции раствора. Измеряли концентрацию лекарственного средства в плазме, и было найдено, что она по меньшей мере на 3 порядка величины ниже, чем концентрация в ткани стекловидного тела.
Анализ 13: Фармакодинамический анализ: Ингибирование IL6–индуцированного pSTAT3 у крыс
Способность испытываемого соединения при разовом интравитреальном введении ингибировать IL–6–индуцированный pSTAT3 измеряли в гомогенатах сетчатки/сосудистой оболочки глаз крыс.
Композиции суспензии получали путем объединения кристаллического соединения 1 Примера 2 с 0,5% гидроксипропилметилцеллюлозы (HPMC E5 LV), 0,02% Tween 80 и 0,9% хлорида натрия в очищенной воде до достижения целевой концентрации 3 мг/мл и 10 мг/мл.
Самкам крыс Lewis интравитреально (и/в) вводили (5 мкл/глаз) композиции суспензии или носитель лекарственного средства. Через три дня интравитреально вводили IL–6 (Peprotech; 0,1 мг/мл; 5 мкл/глаз) или носитель для индукции pSTAT3. Глазные ткани иссекали через один час после второй и/в инъекции IL–6. Ткани сетчатки/сосудистой оболочки гомогенизировали и уровни pSTAT3 измеряли с использованием ELISA (Cell Signaling Technology). Процент ингибирования IL–6–индуцированного pSTAT3 рассчитывали по сравнению с группами носитель/носитель и носитель/IL–6. Ингибирование больше чем 100% отражает снижение уровней pSTAT3 до ниже тех, которые наблюдали в группе носитель/носитель.
При предварительном лечении за 3 дня до IL–6–стимуляции доза 15 мкг и 50 мкг соединения по изобретению, вводимая с использованием композиции суспензии, ингибировала IL–6–индуцированный pSTAT3 на 33% и 109%, соответственно, в ткани сетчатки/сосудистой оболочки.
Анализ 14: Фармакодинамический анализ: Ингибирование IFNγ–индуцированного IP–10 у кроликов
Способность испытываемого соединения при разовом интравитреальном введении ингибировать уровни интерферон–гамма (IFNγ)–индуцированного IP–10 белка измеряли в стекловидном теле и тканях сетчатки/сосудистой оболочки глаз кролика.
Композиции растворов при концентрациях 1 мг/мл и 4 мг/мл соединения 1 Примера 2 получали как в Анализе 12. Композицию суспензии получали путем объединения кристаллического соединения 1 Примера 2 с раствором 0,5% гидроксипропилметилцеллюлозы (HPMC E5), 0,02% Tween 80 и 9 мг/мл хлорида натрия в очищенной воде до достижения целевой концентрации 20 мг/мл.
Для исследований использовали самцов новозеландских белых кроликов (Liveon Biolabs, India). Животным давали акклиматизироваться после их доставки в лабораторию для проведения исследований (Jubilant Biosys Ltd., India). Каждому кролику делали две интравитреальные (и/в) инъекции с общим объемом дозы 50 мкл/глаз. Первая и/в инъекция (45 мкл/глаз) обеспечивала доставку испытываемого соединения или носителя в определенной временной точке (т.е. 24 часа для композиций растворов или 1 неделя для композиции суспензии). Вторая и/в инъекция (5 мкл/глаз) обеспечивала доставку IFNγ (1 мкг/глаз; исходный раствор 1 мг/мл; Kingfisher Biotech) или носителя для индукции IP–10. Вкратце, в день введения инъекций кроликов анестезировали внутримышечной инъекцией кетамина (35 мг/кг) и ксилазина (5 мг/кг). При достижении глубокой анестезии каждый глаз промывали стерильным физиологическим раствором и вводили и/в инъекции с использованием 0,5 мл инсулинового шприца (50 единиц=0,5 мл) с иглой 31 калибра на надназальной стороне обоих глаз, отмечая положение при помощи фиксированного штангенциркуля Браунштейна (2 3/4”) на расстоянии 3,5 мм от прямой мышцы и 4 мм от лимба.
Ткани собирали через 24 часа после второй и/в инъекции IFNγ. Ткани стекловидного тела (VH) и ткани сетчатки/сосудистой оболочки (R/C) собирали и гомогенизировали, и уровни белка IP–10 измеряли с использованием набора для ELISA CXCL10 (IP–10) кролика (Kingfisher Biotech). Процент ингибирования IFNγ–индуцированного IP–10 рассчитывали по сравнению с группами носитель/носитель и носитель/IFNγ.
При предварительном лечении за 24 часа до IFNγ–стимуляции композиция раствора, содержащая 45 мкг соединения 1, ингибировала IFNγ–индуцированный IP–10 на 70% и 86% в стекловидном теле и тканях сетчатки/сосудистой оболочки, соответственно, тогда как композиция, содержащая 180 мкг соединения, ингибировала IFNγ–индуцированный IP–10 на 91% и 100% в стекловидном теле и тканях сетчатки/сосудистой оболочки, соответственно.
При предварительном лечении за 1 неделю до IFNγ–стимуляции композиция в форме кристаллической суспензии соединения 1 ингибировала IFNγ–индуцированный IP–10 на 100% как в стекловидном теле, так и в тканях сетчатки/сосудистой оболочки.
Анализ 15: Ингибирование IFNγ– и IL–27–индуцированных хемокинов CXCL9 и CXCL10 в 3D культурах ткани дыхательных путей человека
Тканевые культуры эпителия дыхательных путей получали от Mattek (AIR–100). Культуры были получены от астматических доноров. Во вставке для культивирования клеток человеческие эпителиальные клетки трахеи/бронхов выращивали и дифференцировали на подложке из пористой мембраны, обеспечивая границу раздела воздушной и жидкой среды с нагретой культуральной средой ниже клеток и газообразной атмосферой испытания выше. Ткани культивировали в поддерживающей среде (Mattek, AIR–100–MM) в 37°С, 5% СО2 увлажненном инкубаторе. Были протестированы четыре донора. В день 0 культуры тканей обрабатывали испытываемыми соединениями при 10 мкМ, 1 мкМ и/или 0,1 мкМ. Соединения разбавляли в диметилсульфоксиде (DMSO, Sigma) до конечной концентрации 0,1%. DMSO при концентрации 0,1% использовали в качестве контроля. Испытываемые соединения инкубировали с культурами в течение 1 часа при 37°C, 5% CO2, с последующим добавлением предварительно нагретой среды, содержащей IFNγ (R&D Systems) или IL–27 (R&D Systems) в конечной концентрации при 100 нг/мл. Тканевые культуры поддерживали в течение 8 дней. Среду заменяли каждые 2 дня свежей средой, содержащей соединения и IFNγ или IL–27. В день 8 культуры тканей и супернатанты собирали для анализа. Образцы супернатантов анализировали на CXCL10 (IP–10) и CXCL9 (MIG) с использованием luminex анализа (EMD Millipore). Данные выражены как % ингибирования +/– стандартное отклонение (± STDV). Процент ингибирования определяли на основании ингибирующей активности соединения против IFNγ– или IL–27–индуцированной секреции CXCL10 или CXCL9 по сравнению с клетками, обработанными носителем. Данные представляли собой среднее значение от 3 или 4 доноров. Соединение 1 было способно ингибировать IFNγ–индуцированную секрецию CXCL10 на 99%±2,0 (при 10мкМ), 71%±19 (при мкМ) и 17%±12 (при 0,1мкМ) при сравнении с носителем, используемым в качестве контроля. Соединение 1 было способно ингибировать IFNγ–индуцированную секрецию CXCL9 на 100%±0,3 (при 10мкМ), 99%±0,9 (при 1мкМ) и 74%±17 (при 0,1мкМ) при сравнении с носителем. Соединение 1 было способно ингибировать IL–27–индуцированную секрецию CXCL10 на 108%±11 (при 10мкМ), 98%±10 (при 1мкМ) и 73%±8,5 (при 0,1мкМ) при сравнении с носителем, используемым в качестве контроля. Соединение 1 было способно ингибировать IL–27–индуцированную секрецию CXCL9 на 100%±0 (при 10мкМ), 95%±3,7 (при 1мкМ) и 75%±3,5 (при 0,1мкМ) при сравнении с носителем, используемым в качестве контроля.
Анализ 16: Анализ IL–5–опосредованного выживания эозинофилов
Активность испытываемого соединения в отношении IL–5–опосредованного выживания эозинофилов измеряли в эозинофилах человека, выделенных из цельной крови человека (AllCells). Поскольку IL–5 передает сигналы через JAK, этот анализ обеспечивает измерение активности в клетках в отношении JAK.
Эозинофилы человека выделяли из свежей цельной крови человека (AllCells) от здоровых доноров. Кровь смешивали с 4,5% декстраном (Sigma–Aldrich) в 0,9% растворе хлорида натрия (Sigma–Aldrich). Эритроциты оставляли для осаждения в течение 35 минут. Верхний слой, богатый лейкоцитами, отделяли и наслаивали на Ficoll–Paque (GE Healthcare) и центрифугировали при 600g в течение 30 минут. Слои плазмы и мононуклеарных клеток удаляли до того, как слой гранулоцитов лизировали водой, для удаления любых загрязняющих эритроцитов. Эозинофилы дополнительно очищали с использованием набора для выделения эозинофилов человека (Miltenyi Biotec). Фракцию очищенных эозинофилов инкубировали с анти–CD16 FITC (Miltenyi Biotec) в течение 10 минут при 4°C в темноте. Чистоту анализировали с использованием проточного цитометра LSRII (BD Biosciences).
Клетки культивировали в 37°С, 5% СО2 увлажненном инкубаторе в RPMI 1640 (Life Technologies), дополненной 10% термоинактивированной фетальной бычьей сыворотки (FBS, Life Technologies), 2 мМ Glutamax (Life Technologies), 25 мМ HEPES (Life Technologies) и 1X Пенициллина/Стрептомицина (Life Technologies). Клетки высевали при 10000 клеток/лунка в среду (50 мкл). Планшет центрифугировали при 300 g в течение 5 минут и супернатанты удаляли. Соединения серийно разводили в DMSO, а затем еще 500–кратно разбавляли до 2х конечной анализируемой концентрации в среде. Испытываемые соединения (50 мкл/лунка) добавляли к клеткам и инкубировали при 37°С, 5% СО2 в течение 1 часа с последующим добавлением IL–5 (R&D Systems; конечные концентрации 1 нг/мл и 10 пг/мл) в предварительно нагретой среде для анализа (50 мкл) в течение 72 часов.
После стимуляции цитокинами клетки центрифугировали при 300 g в течение 5 минут и дважды промывали холодным DPBS (Life Technologies). Для достижения жизнеспособности и апоптоза клетки инкубировали с пропидий йодидом (Thermo Fisher Scientific) и APC аннексином V (BD Biosciences) и анализировали с использованием проточного цитометра LSRII (BD Biosciences). Значения IC50 определяли из анализа кривых жизнеспособности, представляющих процент жизнеспособных клеток в зависимости от концентрации соединения. Данные выражены в виде значений pIC50 (отрицательный десятичный логарифм IC50). Соединение 1 показало pIC50 значение 7,9±0,5 в присутствии 10 пг/мл IL–5 и pIC50 значение 6,5±0,2 в присутствии 1 нг/мл IL–5.
Хотя настоящее изобретение было описано со ссылкой на конкретные аспекты или варианты его осуществления, специалистам в данной области техники должно быть понятно, что возможны различные изменения, или можно использовать заменяющие эквиваленты без отступления от сущности и объема изобретения. Кроме того, в той степени, в которой это допускается применимыми патентными законами и положениями, все публикации, патенты и патентные заявки, процитированные в настоящей заявке, включены в настоящую заявку посредством ссылки во всей их полноте в той степени, как если бы каждый документ был отдельно включен в настоящую заявку посредством ссылки.
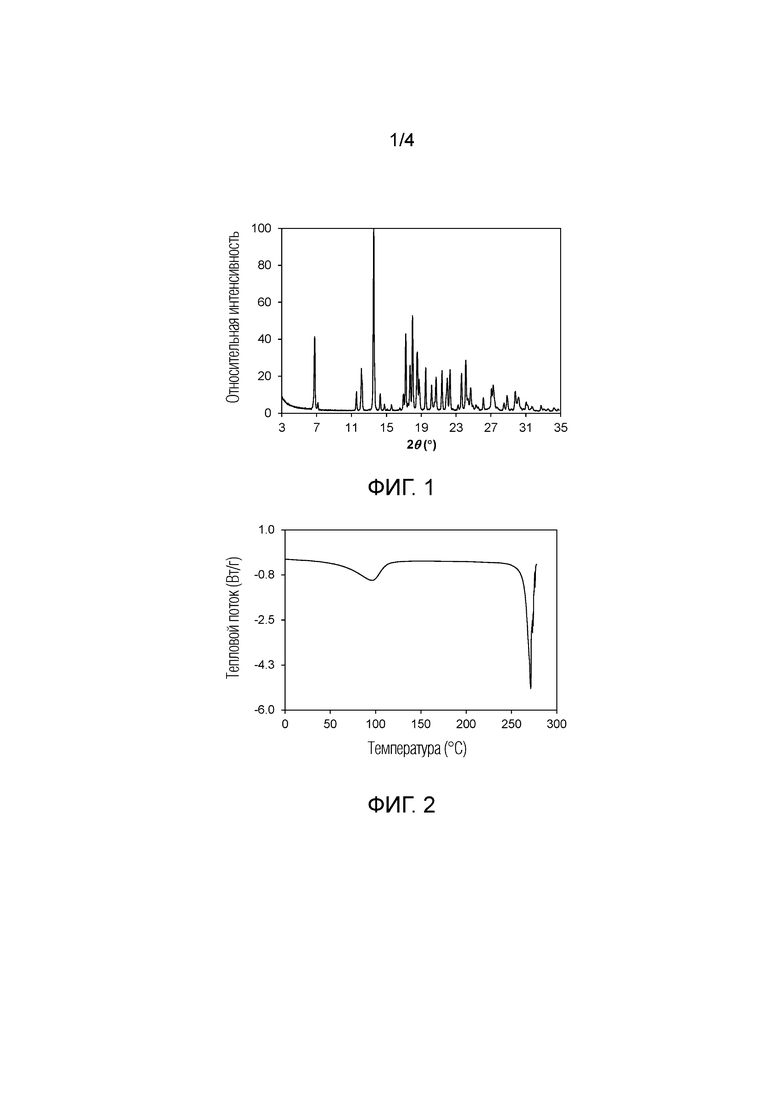
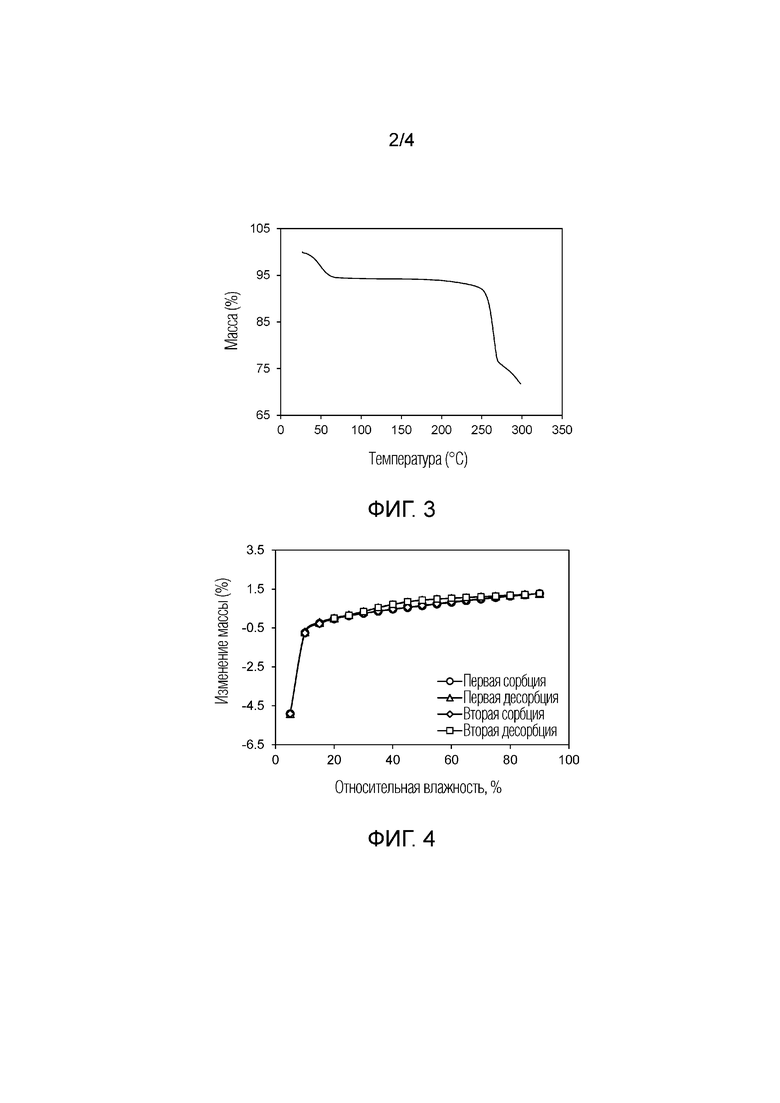
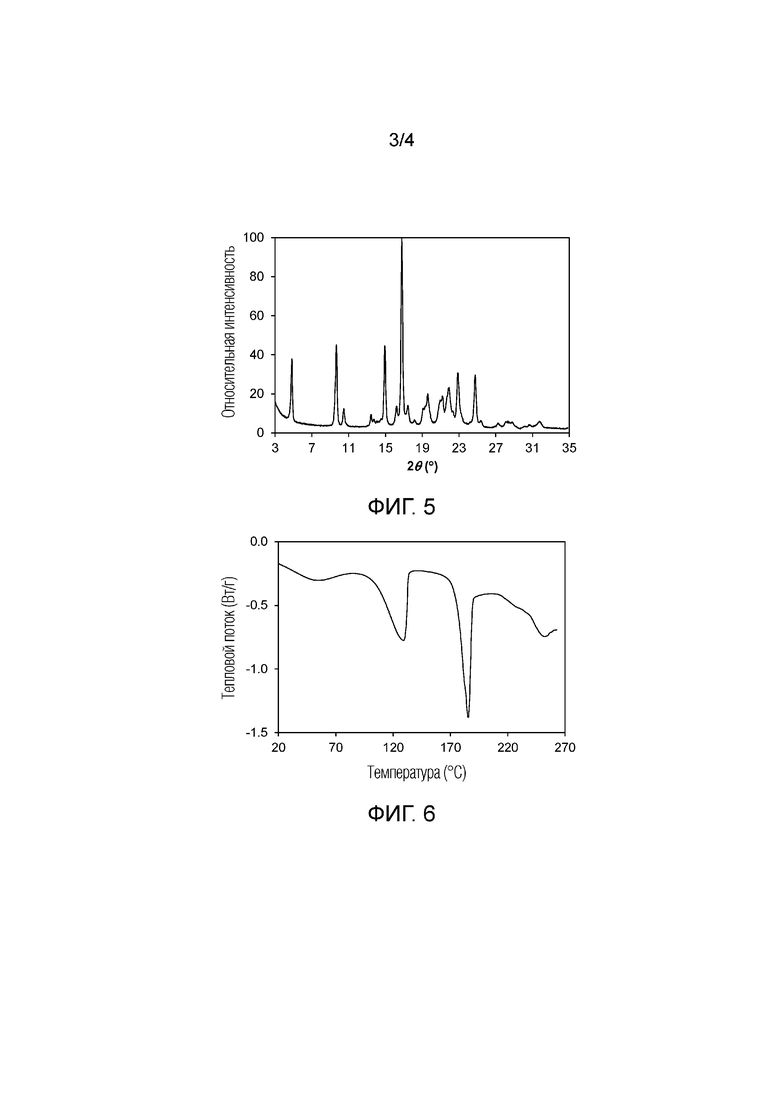
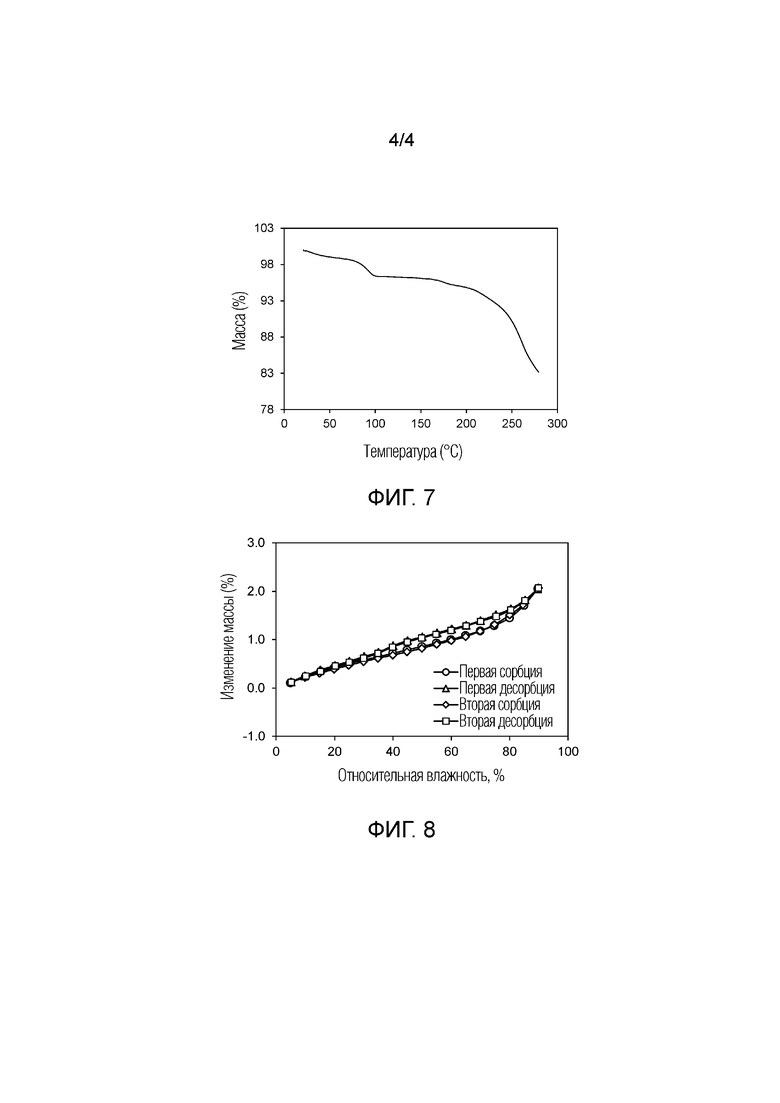
Изобретение относится к области органической химии, а именно к кристаллогидрату оксалатной соли 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола. Также изобретение относится к фармацевтической композиции на основе указанного кристаллогидрата оксалата, способу лечения воспаления, ассоциированного с респираторным заболеванием или глазным заболеванием, применению указанного кристаллогидрата оксалата. Технический результат: ингибирующая активность в отношении ферментов JAK1, JAK2, JAK3 и Tyk2 кристаллогидратом оксалата 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола, что полезно при лечении воспаления, ассоциированного с респираторными и глазными заболеваниями. 6 н. и 14 з.п. ф-лы, 8 ил., 3 табл., 7 пр.
1. Кристаллогидрат оксалатной соли 5–этил–2–фтор–4–(3–(5–(1–метилпиперидин–4–ил)–4,5,6,7–тетрагидро–1H–имидазо[4,5–c]пиридин–2–ил)–1H–индазол–6–ил)фенола.
2. Кристаллогидрат оксалата по п. 1, где кристаллогидрат оксалата характеризуется порошковой рентгеновской дифрактограммой, содержащей дифракционные пики при значениях 2θ 6,77±0,20, 12,13±0,20, 13,54±0,20, 17,23±0,20 и 18,00±0,20.
3. Кристаллогидрат оксалата по п. 2, где порошковая рентгеновская дифрактограмма дополнительно характеризуется тем, что имеет два или более дополнительных дифракционных пиков при значениях 2θ, выбранных из 11,56±0,20, 14,29±0,20, 19,51±0,20, 21,38±0,20 и 23,63±0,20.
4. Кристаллогидрат оксалата по п. 1, где кристаллогидрат оксалата характеризуется порошковой рентгеновской дифрактограммой, на которой положения пиков по существу соответствуют положениям пиков на дифрактограмме, показанной на фиг. 1.
5. Кристаллогидрат оксалата по п. 1, где кристаллогидрат оксалата характеризуется кривой дифференциальной сканирующей калориметрии, полученной при скорости нагрева 10°C в минуту, которая показывает максимум в эндотермическом тепловом потоке при температуре между 266°C и 276°C.
6. Кристаллогидрат оксалата по п. 1, где кристаллогидрат оксалата характеризуется кривой дифференциальной сканирующей калориметрии по существу в соответствии с той, которая показана на фиг. 2.
7. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении ферментов JAK1, JAK2, JAK3 и Tyk2, содержащая терапевтически эффективное количество кристаллогидрата оксалата по любому из пп. 1–6 и фармацевтически приемлемый носитель.
8. Способ лечения воспаления, ассоциированного с респираторным заболеванием, у млекопитающего, включающий введение млекопитающему фармацевтической композиции, содержащей кристаллогидрат оксалата по любому из пп. 1–6 и фармацевтически приемлемый носитель.
9. Способ по п. 8, где респираторное заболевание представляет собой астму, хроническую обструктивную болезнь легких, кистозный фиброз, пневмонит, идиопатический легочный фиброз, острое повреждение легкого, острый респираторный дистресс–синдром, бронхит, эмфизему и облитерирующий бронхиолит.
10. Способ по п. 8, где респираторное заболевание представляет собой облитерирующий бронхиолит.
11. Способ по п. 8, где респираторное заболевание представляет собой астму или хроническую обструктивную болезнь легких.
12. Способ по п. 11, где респираторное заболевание представляет собой астму.
13. Способ по п. 8, где фармацевтическую композицию вводят путем ингаляции.
14. Способ по п. 8, где респираторное заболевание представляет собой легочную инфекцию, гельминтоз, легочную артериальную гипертензию, саркоидоз, лимфангиолейомиоматоз, бронхоэктаз и инфильтративное легочное заболевание.
15. Способ по п. 8, где респираторное заболевание представляет собой лекарственный пневмонит, грибковый пневмонит, аллергический бронхолегочный аспергиллез, гиперчувствительный пневмонит, эозинофильный грануломатоз с полиангиитом, идиопатическую острую эозинофильную пневмонию, идиопатическую хроническую эозинофильную пневмонию, гиперэозинофильный синдром, синдром Леффлера, облитерирующий бронхиолит с организующейся пневмонией или индуцированный ингибитором иммунных контрольных точек пневмонит.
16. Кристаллогидрат оксалата по любому из пп. 1–6 для применения в лечении воспаления, ассоциированного с респираторным заболеванием, у млекопитающего.
17. Применение кристаллогидрата оксалата по любому из пп. 1–6 для получения лекарственного средства для лечения воспаления, ассоциированного с респираторным заболеванием, у млекопитающего.
18. Способ лечения воспаления, ассоциированного с глазным заболеванием, у млекопитающего, включающий введение в глаз млекопитающего фармацевтической композиции, содержащей кристаллогидрат оксалата по любому из пп. 1–6 и фармацевтически приемлемый носитель.
19. Кристаллогидрат оксалата по любому из пп. 1–6 для применения в лечении воспаления, ассоциированного с глазным заболеванием, у млекопитающего.
20. Применение кристаллогидрата оксалата по любому из пп. 1–6 для получения лекарственного средства для лечения воспаления, ассоциированного с глазным заболеванием, у млекопитающего.
| WO 2013014567 A1, 31.01.2013 | |||
| B | |||
| ABELSON MARK и др | |||
| "Sorting Out the Stats from the Jaks", REVIEW OF OPHTALMOLOGY, 05.04.2013,с.84-88 | |||
| ТРИАЗОЛПИРИДИНОВЫЕ СОЕДИНЕНИЯ, ИНГИБИРУЮЩИЕ JAK, И СПОСОБЫ | 2009 |
|
RU2560153C2 |
| Приспособление для подвода массы на сетку бумагоделательной машины | 1932 |
|
SU34914A1 |

