ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] Данная заявка претендует на приоритет по предварительной заявке на патент США №62/201510, поданной 5 августа 2015 г. Эта заявка включена в настоящее описание путем ссылки.
УРОВЕНЬ ТЕХНИКИ
[0002] Область техники
[0003] Варианты осуществления могут относиться к получению по существу диастереомерно чистых активированных мономеров, которые представляют собой морфолино-фосфорамидохлоридатные субъединицы. Варианты осуществления могут также относиться к применению по существу диастереомерно чистых активированных фосфорилированных морфолиновых мономеров для получения молекул путем стереоспецифичных реакций сочетания.
[0004] Уровень техники
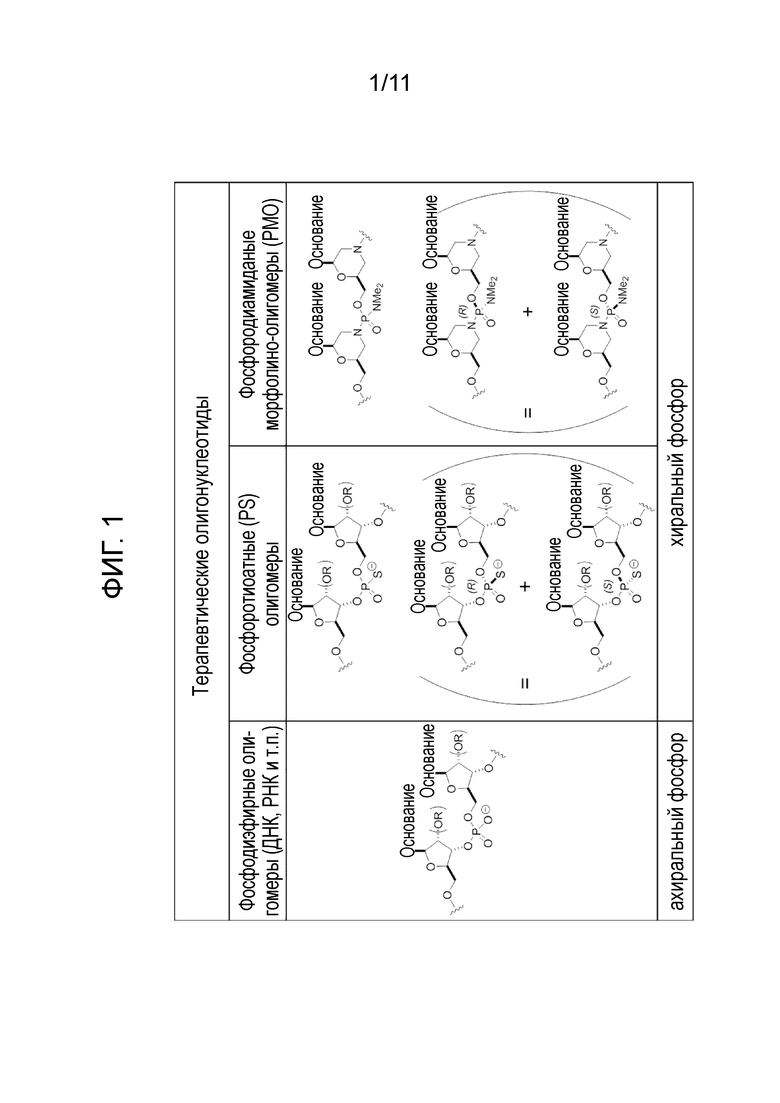
[0005] Синтез диастереомерно чистых фосфородиамидатных олигонуклеотидов существенно осложняется наличием хиральных фосфорных связей. Это контрастирует, например, с фосфодиэфирными связями, которые не имеют хирального фосфора. Примеры приведены на фиг.1, на которой который сравниваются фосфодиэфир, фосфоротиоат (который также включает в себя хиральный фосфор) и фосфородиамидат.
[0006] Присутствие хирального фосфора представляет значительные трудности при синтезе, который включает соединение серии фосфородиамидатных нуклеотидов. Отсутствие стереохимически чистых реагентов (матриц, субъединиц, структурных звеньев), которые позволяют стереоспецифичное образование фосфородиамидатных связей, приводит к реакции на стереоцентре, когда невозможно контролировать хиральность фосфора получаемого соединения.
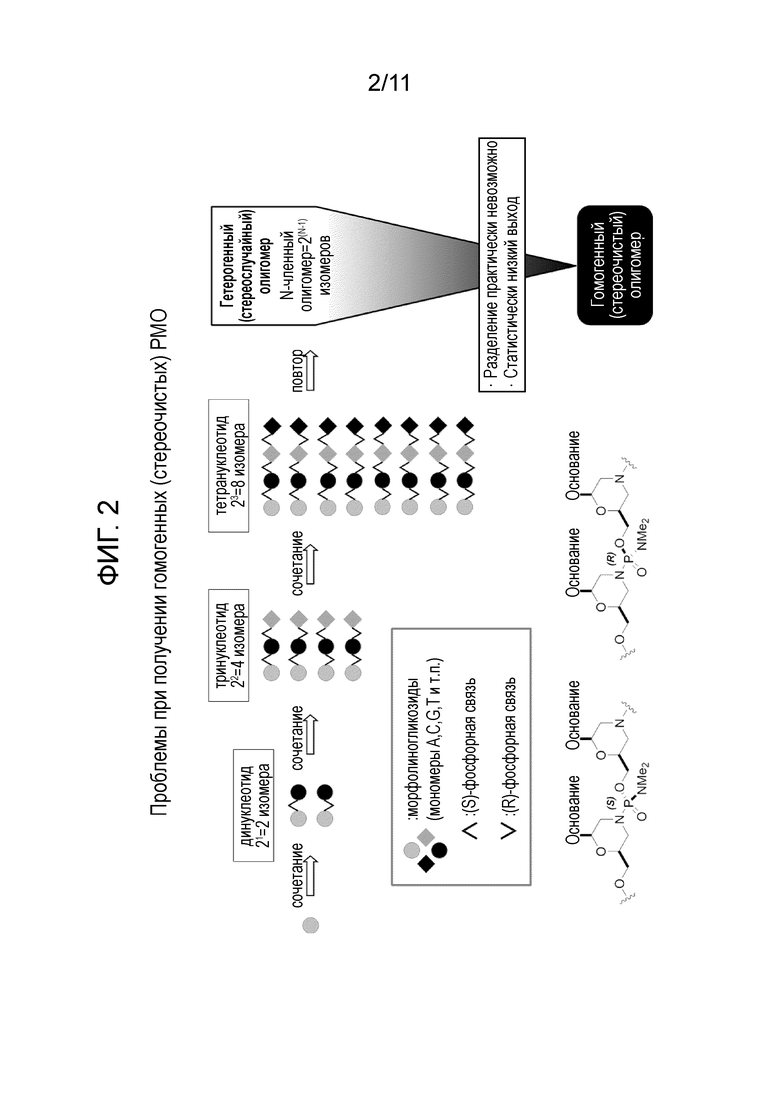
[0007] Как показано графически на фиг.2, использование диастереомерных смесей нуклеотидов для стереохимически неконтролируемой реакции сочетания, чтобы получить олигонуклеотид какой-либо значительной длины и последовательности, дает гетерогенную смесь множества диастереомеров. Количество диастереомеров теоретически составляет 2(n-1), где n представляет собой число нуклеотидов, которые соединяются с образованием олигонуклеотида. Как показано на фиг.2, получение даже простейшего четырехнуклеотидного олигонуклеотида (тетрануклеотида) может привести к образованию смеси из восьми отдельных диастереомеров.
[0008] Образование значительного количества диастереомеров может создать необходимость в чувствительных методах разделения после синтеза. Выход целевого продукта может отрицательно сказаться на использовании сырья вследствие получения множества диастереомеров, которые не являются целевыми.
[0009] Было бы полезно иметь возможность выбрать конкретный диастереомер перед синтезом, а затем синтезировать выбранный диастереомер в стереохимически чистой или по существу чистой форме.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
[0010] В вариантах осуществления предложены один или несколько стереохимически чистых или по существу стереохимически чистых соединений, указанных в таблице 1. В дополнительных вариантах осуществления также предлагаются энантиомеры соединений из таблицы 1. Как правило, стереохимия этих энантиомеров отличается от таковой для соединений из таблицы 1, путем изменения стереохимии морфолинового кольца.
[0011] ТАБЛИЦА 1
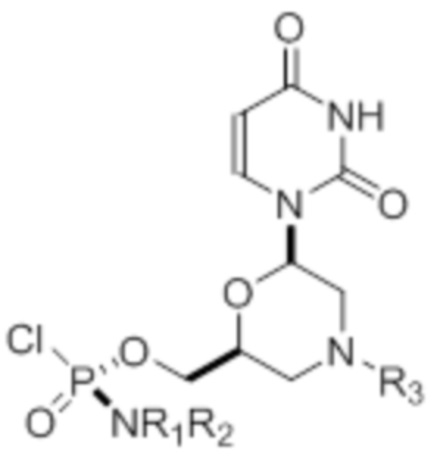
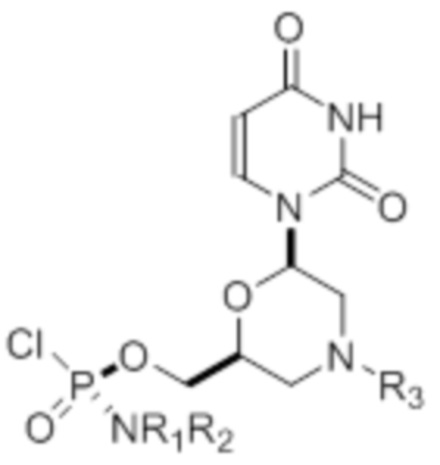
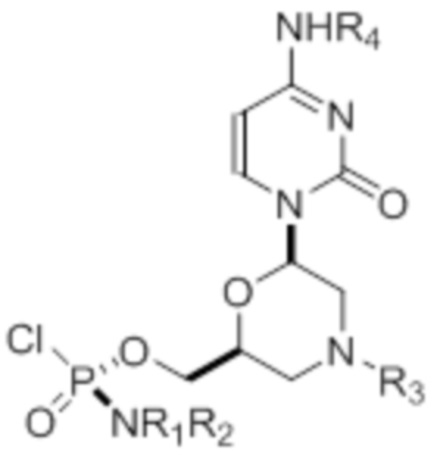
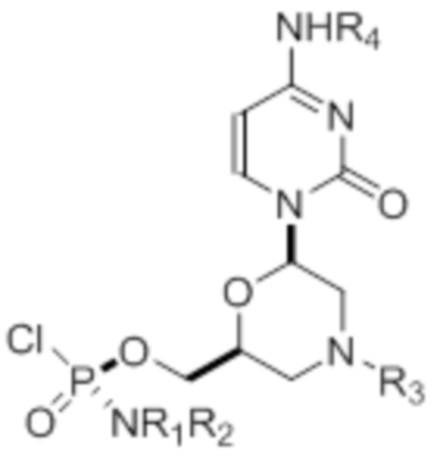
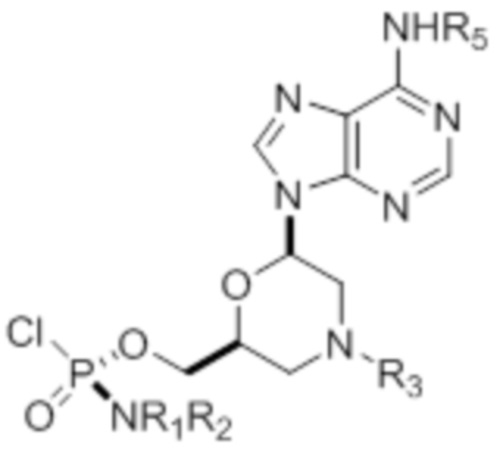
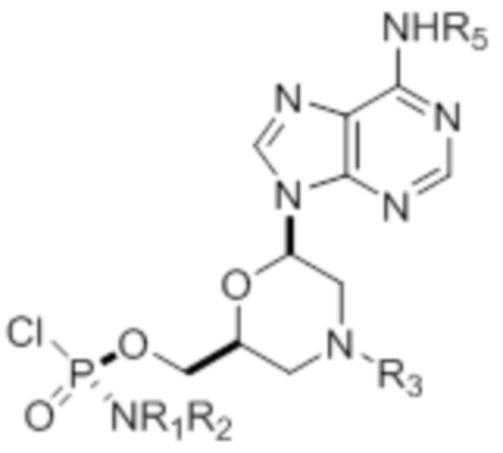
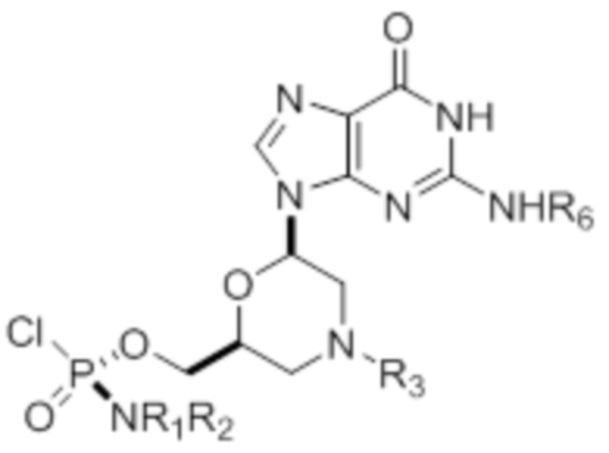
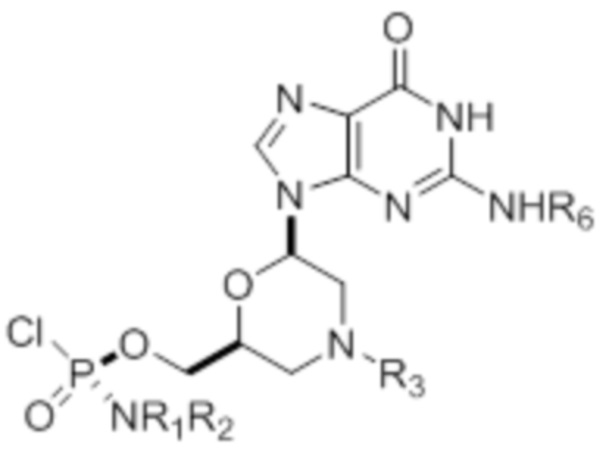
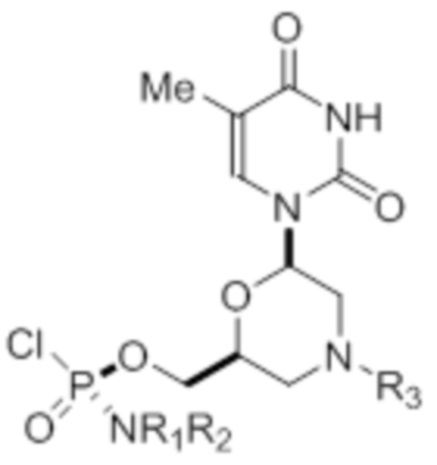
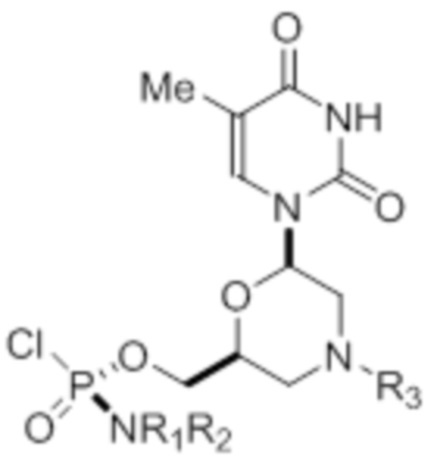
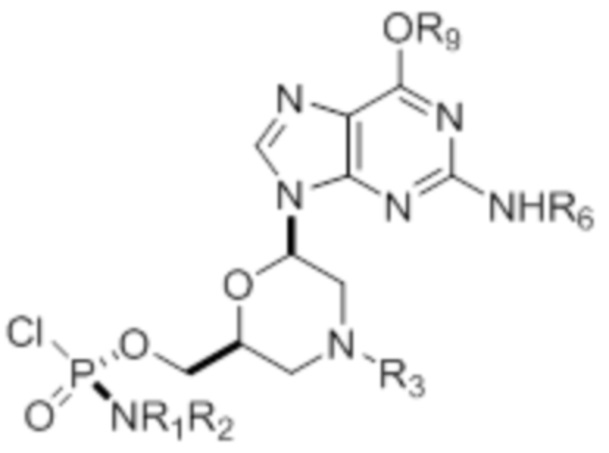

[0012] R1 и R2 могут быть одинаковыми или различными, и могут представлять собой -Н, необязательно замещенный С1-С3-алкил, необязательно замещенный фенил, необязательно замещенный нафтил, или же с атомом азота, к которому они присоединены, они образуют необязательно замещенный гетероцикл, который может представлять собой, например, пирролидин, пиперазин или морфолин.
[0013] Необязательно замещенные группы могут быть замещены одним или несколькими из метила, этила, галогена, нитро-группы, метокси-группы или циано-группы.
[0014] R3 может представлять собой тритил (Tr), который может являться замещенным тритилом, включая, но не ограничиваясь этим, например, MMTr (п-метоксифенилдифенилметил), необязательно замещенный бензил, 4-метоксибензил (PMB, MPM), 3,4-диметоксибензил, дифенилметил (Dpm) или сульфонил, который может являться расщепляемым сульфонилом. В некоторых вариантах осуществления изобретения сульфонил представляет собой 2-нитробензолсульфонил, 4-нитробензолсульфонил или 2,4-динитробензолсульфонил.
[0015] R4, R5, R6 могут представлять собой -H, -C(O)R7 или -C(O)OR7, где R7 представляет собой алкил (метил, этил, изопропил или другой С1-С6-алкил), бензил, 2,2,2-трихлорэтил или арил (включая, но не ограничиваясь ими, фенил, 4-метоксифенил, 4-бромфенил и 4-нитрофенил). R9 может являться, необязательно, замещенным алкилом, цианоэтилом, ацилом, карбонатом, карбаматом, необязательно замещенным бензилом, 4-пивалоилоксибензилом и силилом.
[0016] В дополнительных вариантах осуществления изобретения морфолиновые нуклеозиды в дополнение к приведенным в таблице 1 могут быть получены в диастереомерно чистой или по существу диастереомерно чистой форме.
[0017] В вариантах осуществления могут также быть предложены способы разделения диастереомерных смесей раскрытых выше соединений на стереохимически чистые или по существу стереохимически чистые соединения. В дополнительных вариантах осуществления могут быть предложены фармацевтические композиции, содержащие стереохимически чистые или по существу стереохимически чистые соединения, как описанные в настоящем документе. В других вариантах осуществления могут быть предложены фармацевтические композиции, содержащие фармацевтически приемлемые соли стереохимически чистых или по существу стереохимически чистых соединений, как описанные в настоящем документе. Фармацевтические композиции могут быть введены в эффективном количестве пациентам, нуждающимся в лечении. Фармацевтические композиции могут дополнительно включать фармацевтически приемлемые носители.
[0018] В некоторых вариантах осуществления следующая группа в каждом из соединений из таблицы 1 может быть замещена в положениях a, b и е одной или двумя метильными группами, и может быть замещена в положениях с и d одной метильной группой. В каждом случае метильная группа может находиться на любой стороне от плоскости морфолинового кольца. В других вариантах осуществления изобретения дополнительный метилен, необязательно замещенный одной или несколькими метильными группами, может быть вставлен рядом с атомом азота в морфолиновой группе, чтобы обеспечить возможность расширения до семичленного кольца.
[0019] В вариантах осуществления изобретения дополнительно предложено получение стереохимически чистых олигонуклеотидов путем стереоспецифичного сочетания активированных мономеров. Дополнительные варианты осуществления относятся к по существу диастереомерно чистому олигомеру, изготовленному стереоспецифичным сочетанием активированных мономеров. Другие варианты осуществления относятся к по существу диастереомерно чистой композиции, содержащей по существу диастереомерно чистые соединения, описанные в настоящем документе.
ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0020] На фиг. 1 показаны фосфодиэфирная, фосфоротиоатная и фосфородиамидатная олигонуклеотидные связи.
[0021] На фиг. 2 показаны R- и S-фосфорные связи в фосфородиамидатном морфолино-олигомере (PMO). На фиг. 2 также показана увеличение количества диастереомеров, которое, как правило, возникает, когда диастереомерные смеси предшественников фосфородиамидатного олигомера используются для синтеза динуклеотидов (2-членных), тринуклеотидов (3-членных), тетрануклеотидов (4-членных) и N-членных олигомеров.
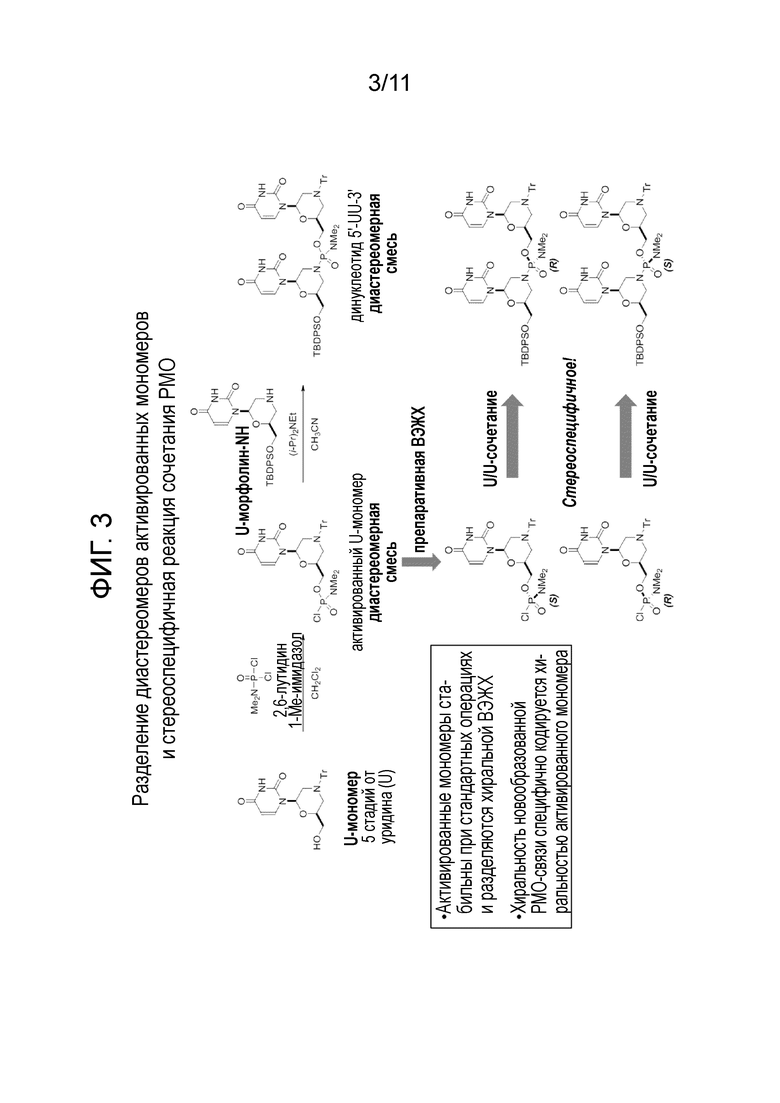
[0022] На фиг. 3 показано получение диастереомерно чистых динуклеотидов как с помощью диастереомерно чистых фосфорамидохлоридатов, так и с использованием диастереомерной смеси фосфорамидохлоридатов.
[0023] На фиг. 4A и фиг. 4В показаны общие диастереомерные смеси фосфорамидохлоридатов, которые могут быть полезны для получения диастереомерно чистых или по существу диастереомерно чистых диастереомерных субъединиц.
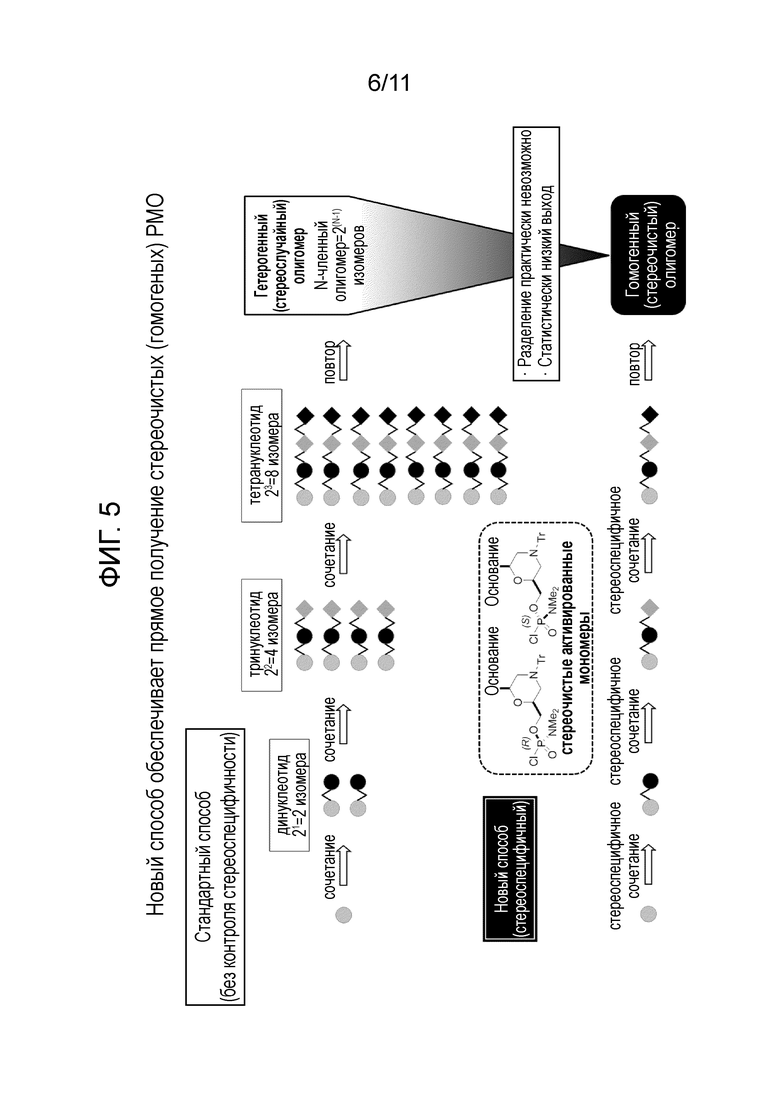
[0024] На фиг. 5 показано получение диастереомерно чистых (гомогенных) олигонуклеотидов с использованием диастереомерно чистых фосфорамидохлоридатов, описанных в настоящем документе.
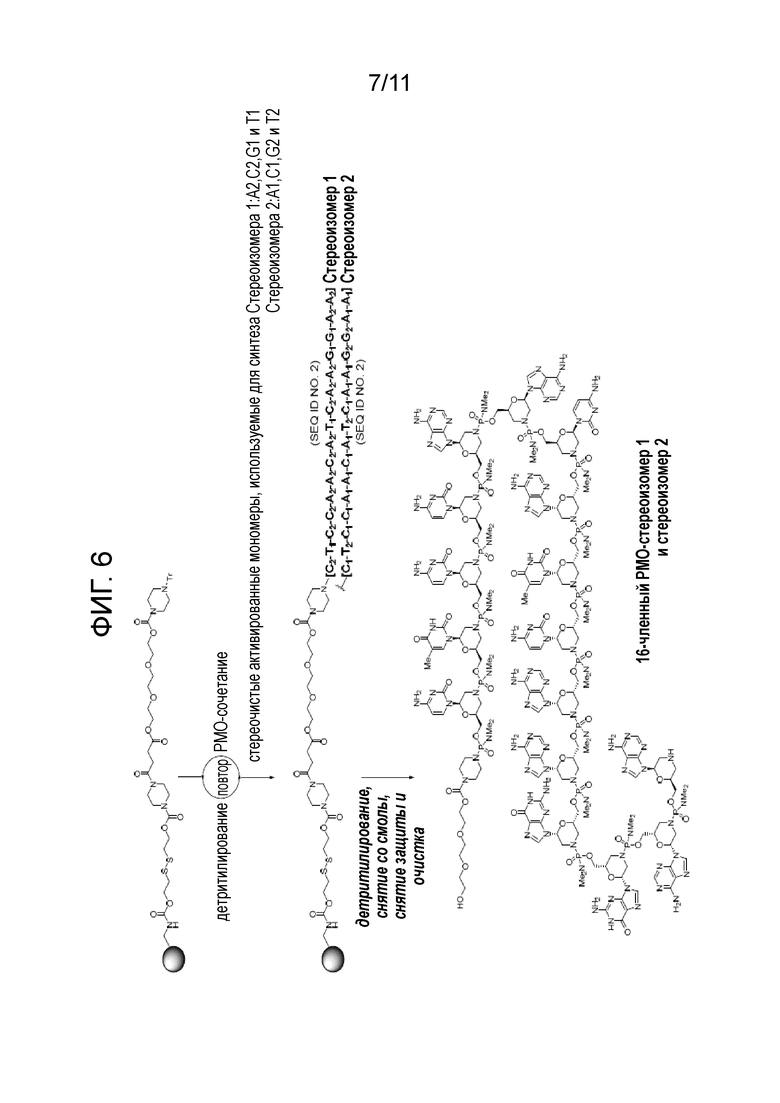
[0025] На фиг. 6 показана схема стереоспецифичного синтеза 16-членного PMO и дифференциация стереоизомеров с помощью биофизического анализа.
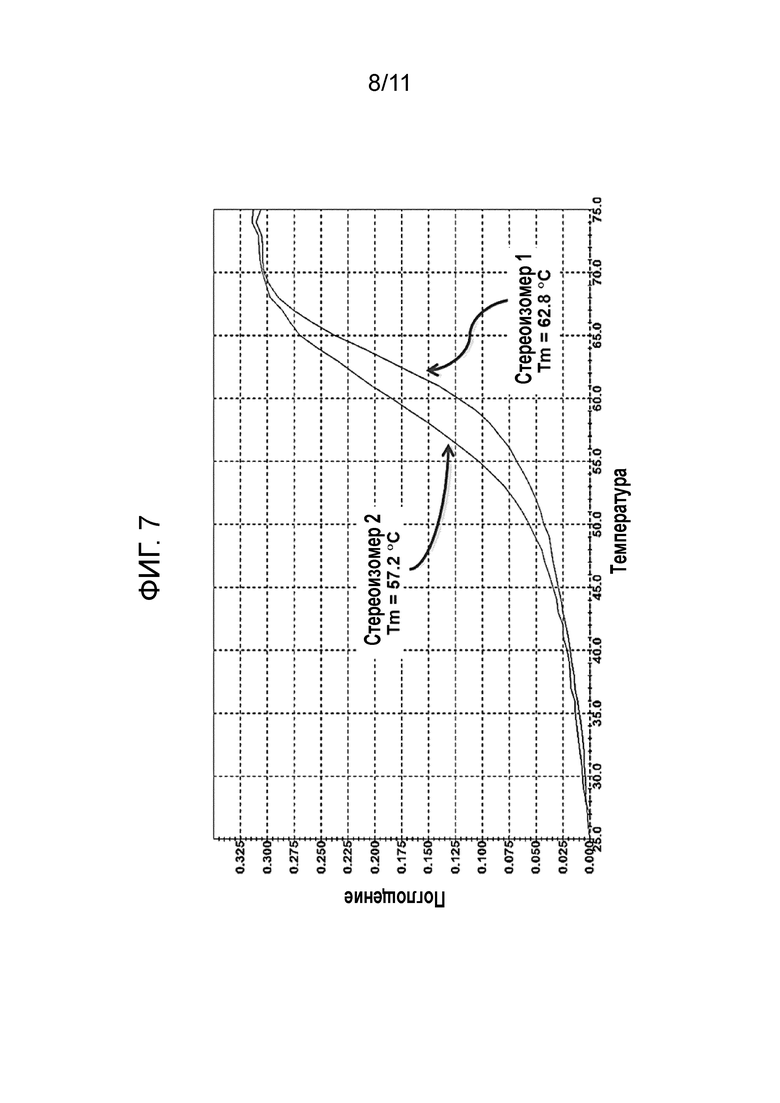
[0026] На фиг. 7 показаны температуры плавления для стереоизомеров 1 и 2 из примера, показывающего стереоспецифичный синтез 16-членного PMO и дифференциацию стереоизомеров с помощью биофизического анализа, как описано ниже.
[0027] На фиг. 8 показаны ORTEP-диаграммы кристаллического соединения 100, описанного ниже.
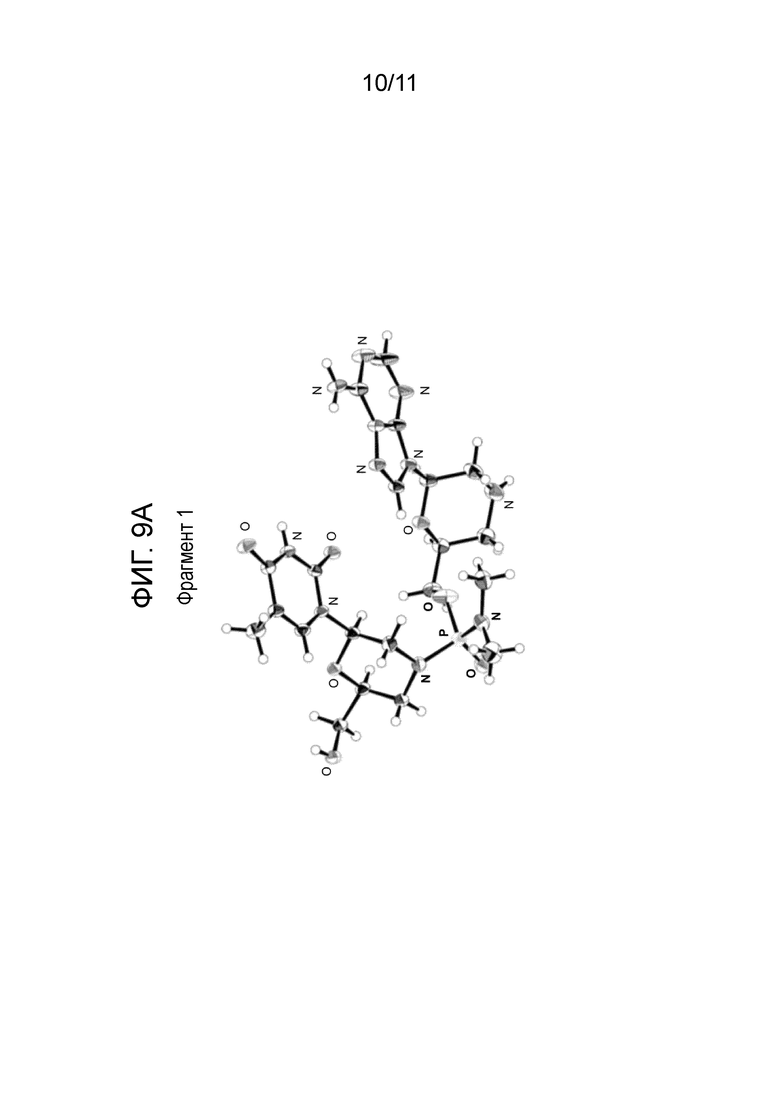
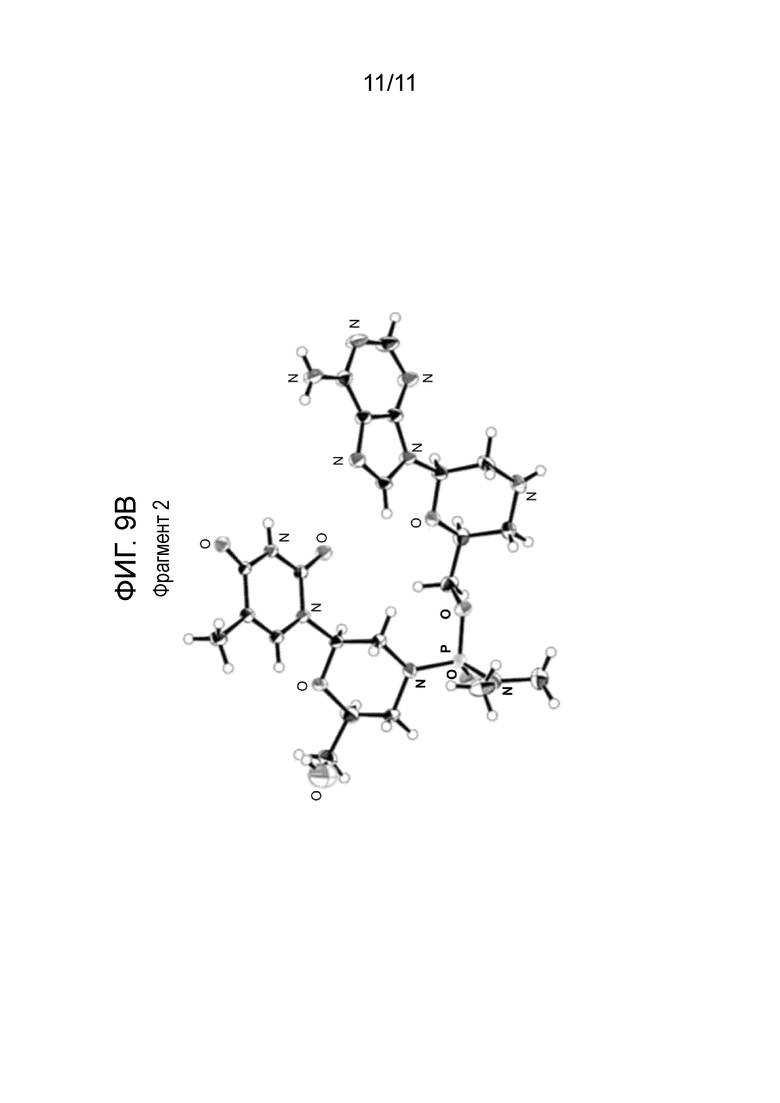
[0028] На фиг. 9А и фиг. 9B показаны ORTEP-диаграммы двух фрагментов соединения 100, описанного ниже.
ПОДРОБНОЕ ОПИСАНИЕ
[0029] Авторы изобретения обнаружили, что два диастереомера активированных морфолиновых субъединиц могут быть разделены по своим физическим свойствам, что позволяет получение диастереомерно чистых изомеров. Это позволяет получение стереохимически чистых или по существу стереохимически чистых PMO при контролируемых условиях реакции, которые затем могут быть использованы для селективного получения олигонуклеотидов с желаемой стереохимией.
[0030] Варианты осуществления могут также относиться к способам разделения диастереомерных смесей раскрытых выше соединений на стереохимически чистые или по существу стереохимически чистые соединения. После разделения чистые диастереомеры из предшествующей диастереомерной смеси могут быть использованы для получения диастереомерно чистых соединений посредством стереоспецифичных реакций сочетания.
[0031] Диастереомерно чистые соединения и по существу диастереомерно чистые соединения, полученные, как описано в настоящем документе, может представлять собой диастереомерно чистые фосфородиамидатные олигонуклеотиды. Эти диастереомерно чистые и по существу диастереомерно чистые фосфородиамидатные олигонуклеотиды могут иметь широкий спектр вариантов применения. Например, они могут быть полезны в качестве лекарственных средств. Они могут быть выбраны из-за свойств, которые потенциально превосходят таковые у гетерогенных (стереослучайных) смесей диастереомеров фосфородиамидатных олигонуклеотидов. Например, они могут быть выбраны благодаря отличиям в реакционоспособности, эффективности, стабильности, безопасности и специфичности. Диастереомерно чистые и по существу диастереомерно чистые олигомеры могут иметь физические, химические и биологические свойства, которые отличаются от таковых у стереохимически гетерогенных смесей олигомеров.
[0032] «Стереоизомеры» относится к изомерам, которые отличаются только расположением атомов в пространстве.
[0033] «Диастереомеры» относится к стереоизомерам, которые не являются зеркальными отражениями друг друга.
[0034] «Энантиомеры» относится к стереоизомерам, которые являются несовпадающими при наложении зеркальными изображениями друг друга. Энантиомеры включают «энантиомерно чистые изомеры», которые содержат по существу единственный энантиомер, например, более чем или 90%, 92%, 95%, 98% или 99%, либо 100% одного энантиомера.
[0035] «Активированный мономер» относится к 5'-О-фосфорилированным морфолино-субъединицам, которые имеют реакционноспособный фосфор, имеющий уходящие группы, включая, но не ограничиваясь ими, хлоридные и галогенидные уходящие группы, которые подвергаются реакции замещения нуклеофилами, включая, но не ограничиваясь ими, амины и спирты.
[0036] «R» и «S» в качестве терминов, описывающих изомеры, представляют собой идентификаторы стереохимической конфигурации при асимметрично замещенных атомах, включая, но не ограничиваясь ими: углерод, серу, фосфор и азот в аммониевой группе. Обозначение асимметрично замещенных атомов как «R» или «S» осуществляется с помощью применения правил приоритета Кана-Ингольда-Прелога, как хорошо известно специалистам в данной области техники и описано в Правилах Международного союза теоретической и прикладной химии (IUPAC) для номенклатуры органической химии. Раздел E, Стереохимия.
[0037] Энантиомер может быть охарактеризован направлением, в котором он вращает плоскость плоскополяризованного света, как хорошо известно специалистам в области химии. Если он поворачивает свет по часовой стрелке (как видно наблюдателю, к которому движется свет), то энантиомер обозначают (+) и называют правовращающим. Его зеркальное изображение будет вращать плоскополяризованный свет против часовой стрелки, и будет обозначаться как (-) или левовращающее. Направление вращения плоскополяризованного света энантиомерно чистым соединением, называемое знаком оптического вращения, может быть легко измерено в стандартном устройстве, известном как поляриметр.
[0038] «Рацемический» относится к смеси, содержащей равные части отдельных энантиомеров.
[0039] «Нерацемический» относится к смеси, содержащей неравные части отдельных энантиомеров. Нерацемическая смесь может быть обогащена по R- или S-конфигурации, включая, без ограничения, смеси в соотношении примерно 50/50, примерно 60/40 и примерно 70/30 R- относительно S-энантиомера или S- относительно R-энантиомера.
[0040] Термины «по существу стереохимически чистый» и «значительная стереохимическая чистота» относятся к энантиомерам или диастереомерам, которые находятся в энантиомерном избытке или диастереомерном избытке, соответственно, равном или больше 80%. В некоторых вариантах осуществления «по существу стереохимически чистый» и «значительная стереохимическая чистота» относятся к энантиомерам или диастереомерам, которые находятся в энантиомерном избытке или диастереомерном избытке, соответственно, равном или больше 87%, равном или больше 90%, равном или больше 95%, равном или больше 96%, равном или больше 97%, равном или больше 98% или равном или больше 99%. «По существу диастереомерно чистые» относится к диастереомерам, которые находятся в диастереомерном избытке, равном или больше 87%, равном или больше 90%, равном или больше 95%, равном или больше 96%, равном или больше 97%, равном или больше 98% или равном или больше 99%.
[0041] «Энантиомерный избыток» (ЭИ) энантиомера представляет собой [(мольная доля основного энантиомера) минус (мольная доля минорного энантиомера)]×100. Диастереомерный избыток (ДЭ) диастереомера в смеси двух диастереомеров определяется аналогично.
[0042] Термин «фармацевтически приемлемая соль», используемый в данном описании, относится к солям присоединения кислот или солям присоединения оснований для соединений в настоящем описании. Фармацевтически приемлемая соль представляет собой любую соль, которая сохраняет активность исходного соединения и не оказывает чрезмерно вредного или нежелательного эффекта на субъекта, которому ее вводят, и в том контексте, в котором ее вводят. Фармацевтически приемлемые соли включают, но не ограничиваются ими, комплексы металлов и соли как неорганических, так и карбоновых кислот. Фармацевтически приемлемые соли также включают соли металлов, такие как алюминиевые, кальциевые, железные, магниевые, марганцевые и комплексные соли. Кроме того, фармацевтически приемлемые соли включают, но не ограничиваются ими, соли кислот, таких как уксусная, аспарагиновая, алкилсульфоновая, арилсульфоновая, аксетильная, бензолсульфоновая, бензойная, бикарбоновая, бисульфитная, битартариновая, масляная, эдетат кальция, камзиловая, карбоновая, хлорбензойная, лимонная, этилендиаминтетрауксусная, эдисиловая, эстоловая, эзиловая, эзиловая, муравьиная, фумаровая, глюцептиновая, глюконовая, глутаминовая, гликолевая, гликолиларсаниловая, гексамическая, гексилрезорциновая, гидрабамовая, бромистоводородная, хлористоводородная, йодистоводородная, гидроксинафтоевая, изетионовая, молочная, лактобионовая, малеиновая, яблочная, малоновая, миндальная, метансульфоновая, метилазотная, метилсерная, муциновая, муконовая, напсиловая, азотная, щавелевая, п-нитрометансульфоновая, памовая, пантотеновая, фосфорная, моногидрофосфорная, дигидрофосфорная, фталевая, полигалактуроновая, пропионовая, салициловая, стеариновая, янтарная, сульфаминовая, сульфаниловая, сульфоновая, серная, дубильная, винная, теоклиновая, толуолсульфоновая и т.п.
[0043] «Эффективное количество» комбинации терапевтических агентов (например, соединения 1 и ингибитора CDK4/6) представляет собой количество, достаточное, чтобы обеспечить наблюдаемый терапевтический эффект по сравнению с HCC или IHCC, не подвергающихся лечению у субъекта или пациента.
[0044] Действующие агенты, описанные в настоящем документе, могут быть объединены с фармацевтически приемлемым носителем, чтобы получить их фармацевтические составы. Конкретный выбор носителя и состава будет зависеть от конкретного способа введения, для которого предназначена композиция.
[0045] Термин «фармацевтически приемлемый носитель», используемый в настоящем документе, относится к нетоксичному носителю, адъюванту или средству доставки, которые не нарушают фармакологическую активность соединения, с которым их объединяют в составе. Фармацевтически приемлемые носители, адъюванты или средства доставки, которые могут быть использованы в композициях по настоящему изобретению, включают, но не ограничиваются ими, сорбиновую кислоту, сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, гидрофосфат динатрия, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натриевую карбоксиметилцеллюлозу, полиакрилаты, воски, полиэтиленгликоль и ланолин.
[0046] Композиции по настоящему изобретению могут быть пригодны для парентерального, перорального, ингаляционно-распылительного, местного, ректального, назального, буккального, вагинального введения или введения с помощью имплантированного резервуара и т.д. В некоторых вариантах осуществления изобретения состав содержит ингредиенты, которые имеют природное и неприродное происхождение. В некоторых вариантах осуществления состав или носитель может быть предложены в стерильной форме. Неограничивающие примеры стерильного носителя включают свободную от эндотоксинов воду или апирогенную воду.
[0047] Термин «парентеральный», используемый в настоящем документе, включает методики подкожной, внутривенной, внутримышечной, внутрисуставной, внутриартериальной, интрасиновиальной, интрастернальной, интратекальной, внутрипеченочной, интралезиональной и внутричерепной инъекции или инфузии. В конкретных вариантах осуществления соединения вводят внутривенно, перорально, подкожно или путем внутримышечного введения. Стерильные инъекционные формы композиций по настоящему изобретению могут представлять собой водную или масляную суспензии. Эти суспензии могут быть изготовлены в соответствии с методиками, известными в данной области техники, с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильный инъекционный препарат может также представлять собой стерильный инъекционный раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе. Из приемлемых носителей и растворителей, которые могут быть использованы, можно упомянуть воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные нелетучие масла.
[0048] Варианты осуществления настоящего изобретения относятся к получению стереохимически чистых изомеров или по существу стереохимически чистых изомеров с последующим использованием чистых изомеров для стереоспецифичного получения диастереомерно чистых фосфородиамидатных морфолиновых олигомеров (PMO). Получение может выполнено путем разделения диастереомерной смеси фосфорамидохлоридатных нуклеотидов. Разделение может быть выполнено, например, с помощью хроматографии, например, высокоэффективной жидкостной хроматографии или «ВЭЖХ». Разделение также может быть осуществлено путем кристаллизации.
[0049] Разделенные мономеры могут быть указаны как «активные» мономеры. «Активный» означает, что мономеры включают фосфорамидохлоридатную группу, которая является реакционноспособной в отношении различных нуклеофилов, которые включают, но не ограничиваются ими: амины, спирты/алкоксиды, тиол/тиолят, алкиллитий и реактивы Гриньяра.
[0050] I. Получение диастереомерных изомеров
[0051] В одном варианте осуществления изобретения стереохимически чистые или по существу стереохимически чистые активированные мономеры могут быть получены разделением диастереомерной смеси мономеров. Разделение может быть осуществлено способами, которые позволяют различить стереоизомеры с использованием физических свойств. Например, разделение может быть выполнено с помощью хроматографии или кристаллизации. Подходящие типы хроматографии, например, включают, но не ограничиваются ими, высокоэффективную жидкостную хроматографию (ВЭЖХ), хроматографию в псевдодвижущемся слое, хроматографию в противотоке и другие типы разделяющей хроматографии. Например, диастереомерная смесь может быть подвергнута ВЭЖХ с элюцией быстро движущейся фракции и медленно движущейся фракции. Каждая из этих фракций является отдельным стереохимически чистым или по существу стереохимически чистым количеством мономера. Как описано ниже, эти мономеры могут быть использованы для получения олигомеров с желаемой стереохимией путем стереоспецифичного сочетания с использованием контролируемых условий реакции.
[0052] Авторы изобретения дополнительно установили, что после разделения стереохимически чистые активированные мономеры имеют достаточную стабильность для использования в дальнейших химических реакциях. Кроме того, авторы изобретения пришли к выводу, что стереохимически чистые активированные мономеры могут участвовать в стереоспецифичных химических реакциях. Таким образом, как описано более подробно ниже, эти стереохимически чистые активированные мономеры могут быть использованы для стереоспецифичных реакций сочетания для получения стереохимически чистых продуктов.
[0053] Как указано выше, варианты осуществления изобретения могут относиться к одному или нескольким стереохимически чистым или по существу стереохимически чистым соединениям из таблицы 1, которые могут быть получены с использованием различных физических свойств стереоизомеров. Другие варианты осуществления могут также относиться к энантиомерам соединений из таблицы 1. Как правило, стереохимия этих энантиомеров отличается от таковой для соединений из таблицы 1 путем изменения стереохимии морфолинового кольца.
[0054] ТАБЛИЦА 1
[0055] Где R3 представляет собой необязательно замещенный трифенилметил (также указывается как «тритил»), необязательно замещенный бензил, или сульфонил. R4, R5 и R6 могут представлять собой -C(O)R7 или -C(O)OR8, где R7 представляет собой метил, этил или фенил, а R8 представляет собой бензил или 2,2,2-трихлорэтил. R9 может представлять собой необязательно замещенный алкил, цианоэтил (для использования в качестве защитной группы см., например, публикацию заявки на патент США №US2013/0197220), ацил, сульфонил, ацеталь/кеталь, карбонат, карбамат, необязательно замещенный бензил, 4-пивалоилоксибензил и силил.
[0056] В некоторых вариантах осуществления изобретения необязательно замещенный бензил представляет собой 4-метоксибензил (РМВ, МРМ). В некоторых вариантах осуществления изобретения сульфонил представляет собой расщепляемый сульфонил. В некоторых вариантах осуществления изобретения сульфонил представляет собой 2-нитробензолсульфонил, 4-нитробензолсульфонил или 2,4-динитробензолсульфонил.
[0057] R1 и R2 могут быть одинаковыми или различными, и могут представлять собой -Н, необязательно замещенный С1-С3-алкил, необязательно замещенный фенил, необязательно замещенный нафтил, или же с атомом азота, к которому они присоединены, они образуют необязательно замещенный гетероцикл, который может представлять собой, например, пирролидин, пиперазин или морфолин.
[0058] Необязательно замещенные группы могут быть замещены одним или несколькими из метила, этила, галогена, нитро-группы или циано-группы.
[0059] R3 может представлять собой тритил (Tr), который может являться замещенным тритилом, включая, но не ограничиваясь этим, например, MMTr (п-метоксифенилдифенилметил), бензил, 4-метоксибензил (PMB, MPM) и 3,4-диметоксибензил, дифенилметил (Dpm).
[0060] R4, R5, R6 могут представлять собой -H, -C(O)R7 или -C(O)OR7, где R7 представляет собой алкил (метил, этил, изопропил или другой С1-С6-алкил) или арил (включая, но не ограничиваясь ими, фенил, 4-метоксифенил, 4-бромфенил и 4-нитрофенил).
[0061] II. Стереоспецифичное сочетание
[0062] В дополнение к определению того, что технология разделения может быть использована для получения по существу стереохимически чистых количеств активированного мономера, авторы изобретения установили, что эти активированные мономеры могут при определенных условиях реакции использоваться для выполнения стереоспецифичного сочетания для получения стереохимически чистых динуклеотидов, стереохимически чистых тринуклеотидов и более длинных стереохимически чистых олигомеров. С использованием способов, описанных в настоящем документе, хиральность новообразованной РМО-связи может специально кодироваться хиральностью стереохимически чистых активных мономеров, используемых для образования олигомера.
[0063] Типичные условия реакции для стереоспецифичного сочетания включают реакцию в апротонных растворителях. Этими растворителями могут, например, являться, но не ограничиваются ими, ацетонитрил, тетрагидрофуран (THF), 1,3-диметилимидазолидинон (DMI), диметилформамид (DMF), N-метил-2-пирролидон (NMP), диметилацетамид (DMAc), дихлорметан (DCM), 1,2-дихлорэтан (DCE), хлороформ, 1,4-диоксан, этилацетат, 2-метилтетрагидрофуран и изопропилацетат. Реакции сочетания могут проводиться в присутствии ненуклеофильных оснований третичных аминов и ароматических оснований. Подходящие основания включают, но не ограничиваются ими, диизопропилэтиламин, триэтиламин, 2,6-лутидин, триметилпиридины (коллидины) и N-этилморфолин. Температура реакции может находиться в диапазоне от комнатной температуры (около 20°C) до 50°С. В некоторых случаях может использоваться обработка ультразвуком для растворения субстрата(ов).
[0064] Для того, чтобы продемонстрировать возможность стереоспецифического сочетания, по существу множество стереохимически чистых РМО-динуклеотидов были получены путем стереоспецифического РМО-сочетания медленно и быстро элюирующихся по существу стереохимически чистых активных мономеров (фосфорамидохлоридатов). В таблице 2 ниже приведены ВЭЖХ-профили удерживания этих по существу стереохимически чистых РМО-динуклеотидов. В таблице сравниваются времена удерживания для динуклеотидов, которые были получены из комбинаций 5'-концевых мономеров, перечисленных в левой части таблицы, с обоими быстро элюирующимися и медленнее элюирующимися изомерами 3'-концевых мономеров, перечисленных в верхней части таблицы. В таблице продемонстрировано, что стереоспецифичное сочетание по существу стереохимически чистых активных мономеров дает различные диастереомерно чистые динуклеотиды, имеющие разные физические свойства.
ТАБЛИЦА 2:
b Аминогруппа гуанина защищена изобутирильной группой
c Аминогруппа цитозина защищена: (1) ацетильной группой при сочетании с U, C, A и G, (2) бензоильной группой при сочетании с T
d Аминогруппа аденина защищена бензоильной группой
[0065] Условия аналитической ВЭЖХ для получения профилей по существу стереохимически чистых РМО-динуклеотидов из таблицы 2 приведены ниже:
[0066] ПРИМЕРЫ
[0067] III. Примеры диастереомерного разделения активированных мономеров
[0068] В нижеследующих примерах показано диастереомерное разделение активированных мономеров в соответствии с некоторыми вариантами осуществления, предложенными в настоящем документе.
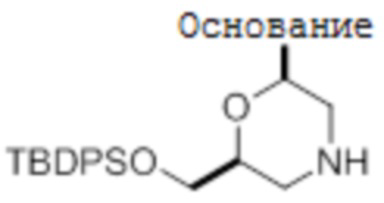
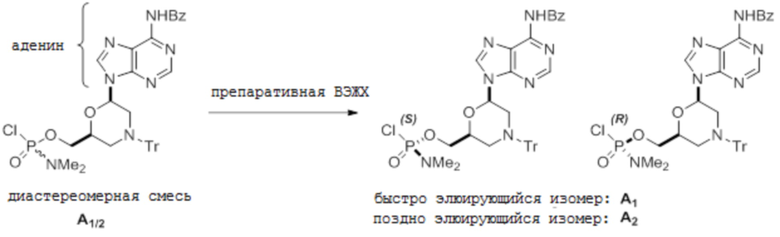
[0069] A. U-мономеры
[0070] Условия аналитической ВЭЖХ для активированных мономеров U:
[0071] Условия препаративной ВЭЖХ для активированных мономеров U:
[0072] Chiralpak IC, 21×250 мм, 5 мкм; элюция с колонки при 11 мл/мин этилацетатом при комнатной температуре; детекция при 260 нм.
[0073] [1Н-ЯМР данные для U1]
[0074] 1Н-ЯМР (400 МГц, CDCl3) δ 8,18 (шир., 1H), 7,45 (м, 6H), 7,15-7,32 (м, 10H), 6,12 (дд, 1H, J=2,0 и 9,6 Гц), 5,62 (д, 1H, J=8,0 Гц), 4,39 (м, 1H), 4,11 (м, 2H), 3,39 (д, 1Н, J=11 Гц), 3,15 (д, 1Н, J=11 Гц), 2,65 (с, 3H), 2,62 (с, 3H), 1,49 (т, 1H, J=11 Гц), 1,39 (т, 1H, J=11 Гц).
[0075] [1Н-ЯМР данные для U2]
[0076] 1Н-ЯМР (400 МГц, CDCl3) δ 8,07 (шир., 1H), 7,44 (м, 6H), 7,14-7,34 (м, 10 Н), 6,12 (дд, 1H, J=2 и 9 Гц), 5,61 (д, 1H, 8,0 Гц), 4,39 (м, 1H), 4,08 (м, 2H), 3,39 (д, 1H, J=12 Гц), 3,15 (д, 1H, J=12 Гц), 2,66 (с, 3H), 2,62 (с, 3H), 1,46 (т, 1H, J=11 Гц), 1,38 (т, 1H, J=11 Гц).
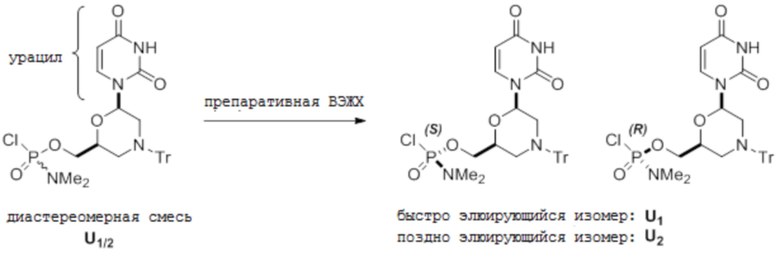
[0077] B. A-мономеры
[0078] Условия аналитической ВЭЖХ для активированных мономеров А:
[0079] Условия препаративной ВЭЖХ для активированных мономеров А:
[0080] Chiralpak IC, 21×250 мм, 5 мкм; элюция с колонки при 11 мл/мин 100%-м этилацетатом при комнатной температуре; УФ-детекция при 260 нм.
[0081] [1Н-ЯМР данные для A1]
[0082] 1H-ЯМР (400 МГц, CDCl3) δ 9,01 (шир., 1H), 8,79 (с, 1H), 8,00 (м, 3H), 7,58 (м, 1H), 7,4-7,6 (м, 8Н), 7,2-7,4 (м, 10H), 6,42 (д, 1H, J=8,4 Гц), 4,51 (м, 1H), 4,12 (м, 3H), 3,54 (д, 1H, J=12 Гц), 3,25 (д, 1H, J=12 Гц), 2,62 (с, 3H), 2,59 (с, 3H), 1,81 (т, 1H, J=11 Гц), 1,62 (т, 1H, J=11 Гц).
[0083] [1Н-ЯМР данные для А2]
[0084] 1Н-ЯМР (400 МГц, CDCl3) δ 9,04 (шир., 1H), 8,79 (с, 1H), 8,00 (м, 3H), 7,56 (м, 1H), 7,4-7,6 (м, 8Н), 7,2-7,4 (м, 10H), 6,41 (д, 1H, J=8,4 Гц), 4,51 (м, 1H), 4,12 (м, 3H), 3,54 (д, 1H, J=12 Гц), 3,25 (д, 1H, J=12 Гц), 2,64 (с, 3H), 2,61 (с, 3H), 1,82 (т, 1H, J=11 Гц), 1,63 (т, 1H, J=11 Гц).
[0085] С. С-мономеры
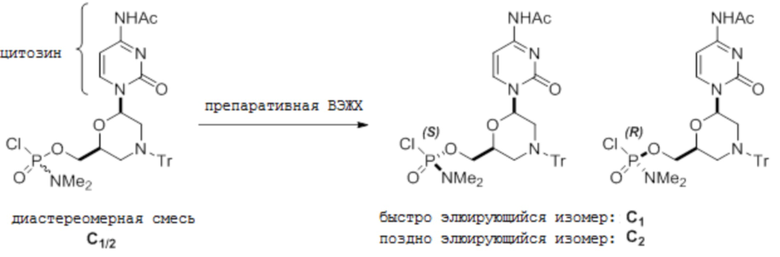
[0086] Условия аналитической ВЭЖХ для активированных мономеров C:
[0087] Условия препаративной ВЭЖХ для активированных мономеров C:
[0088] Колонку Chiralpak IC элюируют при 15 мл/мин 75%-м этилацетатом и 25%-м н-гептаном. Температура комнатная и УФ-детекция при 260 нм.
[0089] [1Н-ЯМР данные для C1]
[0090] 1H ЯМР (400 МГц, CDCl3) δ 7,66 (д, 1H, J=7,8 Гц), 7:43 (м, 6H), 7:33 (д, 1H, J=7,4 Гц), 7:15 до 7:32 (м, 9H), 6,18 (дд, 1H, J=2,2 и 9,2 Гц), 4,42 (м, 1H), 4,08 до 4,16 (м, 2H), 3:54 (д, 1H, J=11 Гц), 3,14 (д, 1H, J=12 Гц), 2,64 (с, 3H), 2,60 (с, 3H), 2,23 (с, 3H), 1,51 (т, 1H, J=11 Гц), 1,25 (м, 1H).
[0091] [1Н-ЯМР данные для С2]
[0092] 1H ЯМР (400 МГц, CDCl3) δ 7,64 (д, 1H, J=7,8 Гц), 7:43 (м, 6H), 7:32 (д, 1H, J=7,4 Гц), 7:15 до 7:32 (м, 9H), 6,19 (дд, 1H, J=2,1 и 9,2 Гц), 4,41 (м, 1H), 4,06 до 4,15 (м, 2H), 3:54 (д, 1H, J=11 Гц), 3,15 (д, 1H, J=12 Гц), 2,64 (с, 3H), 2,61 (с, 3H), 2,22 (с, 3H), 1:49 (т, 1H, J=11 Гц), 1,25 (м, 1H).
D. G-мономеры (монозащищенный гуанин)
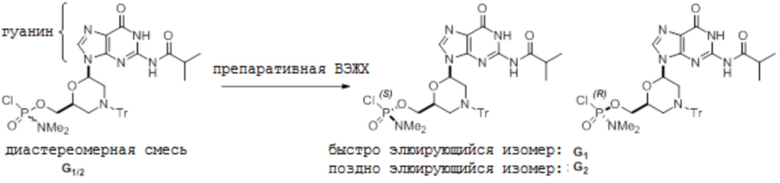
[0094] Условия аналитической ВЭЖХ для активированных мономеров G:
[0095] Условия препаративной ВЭЖХ для активированных мономеров G:
[0096] Колонку Chiralpak IC элюируют при 15 мл/мин 100%-м этилацетатом. Температура комнатная и УФ-детекция при 260 нм.
[0097] E. Т-мономеры
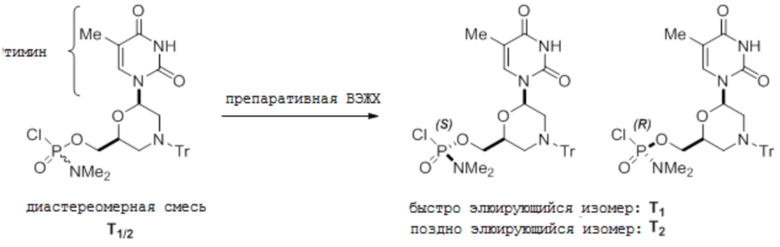
[0098] Условия аналитической ВЭЖХ для активированных мономеров Т:
[0099] Условия препаративной ВЭЖХ для активированных мономеров Т:
Chiralpak IC, 50×500 мм, 20 мкм. Элюция колонки при 60 мл/мин этилацетатом, при комнатной температуре, детекция при 260 нм. Время удерживания 25 и 40 минут.
[0100] [1H-ЯМР данные для T1]
[0101] 1H ЯМР (400 МГц, CDCl3) δ 7,4-7,5 (м, 5H), 7,26-7,33 (м, 6H), 7,16-7,22 (м, 3H), 7,04 (д, 1H, J=1 Гц), 6,12 (дд, 1H, J=2 и 10 Гц), 4,39 (м, 1H), 4,12 (м, 2H), 3,37 (д, 1H, J=12 Гц), 3,15 (д, 1Н, J=12 Гц), 2,66 (с, 3H), 2,63 (с, 3H), 1,83 (д, 1H, J=1 Гц), 1,49 (т, 1H, J=11 Гц), 1,41 (т, 1H, J=11 Гц).
[0102] [1Н-ЯМР данные для Т2]
[0103] 1H-ЯМР (400 МГц, CDCl3) δ 7,4-7,5 (м, 6Н), 7,24-7,35 (м, 6H), 7,14-7,22 (м, 3H), 7,03 (с, 1H), 6,12 (дд, 1Н, J=2 и 10 Гц), 4,39 (м, 1H), 4,09 (м, 2H), 3,37 (д, 1Н, J=11 Гц), 3,15 (д, 1Н, J=11 Гц), 2,66 (с, 3H), 2,62 (с, 3H), 1,82 (с, 3H), 1,48 (т, 1H, J=11 Гц), 1,40 (т, 1H, J=11 Гц).
[0104] F. C-мономеры (NBz)
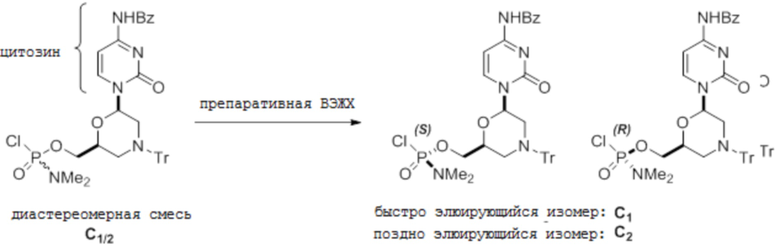
[0105] Условия аналитической ВЭЖХ для активированных мономеров C (NBz):
[0106] Условия препаративной ВЭЖХ для активированных мономеров C (NBz):
Колонку Chiralpak IВ 20×250 мм элюируют при 9 мл/мин 100%-м ацетонитрилом. Температура комнатная и УФ-детекция при 260 нм. Время удерживания 13 и 16 минут.
G. G-мономеры (гуанин с двумя защитными группами)
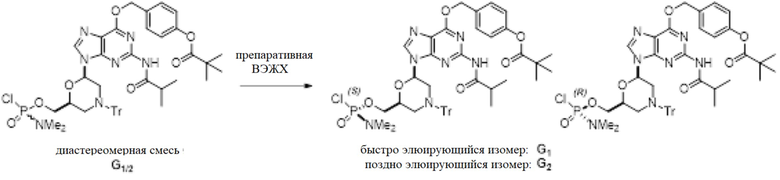
[0108] Условия аналитической ВЭЖХ для активированных мономеров G (гуанин с двумя защитными группами):
[0109] Условия препаративной ВЭЖХ для активированных мономеров G (гуанин с двумя защитными группами):
[0110] Колонку Chiralpak IA 50×500 мм, элюировали при 60 мл/мин 100%-м этилацетатом. Температура комнатная и УФ-детекция при 260 нм. Время удерживания 20 и 24 минуты.
[0111] [1Н-ЯМР данные для G1 (гуанин с двумя защитными группами)]
[0112] 1H ЯМР (400 МГц, CDCl3) δ 7,76 (с, 2H), 7,50 (д, 2H, J=9 Гц), 7,4-7,5 (м, 6H), 7,26-7,32 (м, 6H), 7,16-7,22 (м, 3H), 7,02 (д, 2H, J=9 Гц), 6,24 (дд, 1H, J=2 и 10 Гц), 5,61 (д, 1H, J=12 Гц), 5,56 (д, 1H, J=12 Гц), 4,48 (м, 1H), 4,1 (м, 2H), 3,47 (д, 1Н, J=11 Гц), 3,23 (д, 1H, J=12 Гц), 3,2 (м, 1H), 2,62 (с, 3H), 2,59 (с, 3H), 1,75 (т, 1H, J=11 Гц), 1,57 (т, 1H, J=12 Гц), 1,33 (с, 9H), 1,33 (т, 6Н, J=7 Гц).
[0113] [1Н-ЯМР данные для G2 (гуанин с двумя защитными группами)]
[0114] 1H ЯМР (400 МГц, CDCl3) δ 7,78 (с, 1H), 7,77 (с, 1H), 7,50 (д, 2H, J=9 Гц), 7,4-7,5 (м, 6H), 7,26-7,33 (м, 6H), 7,15-7,22 (м, 3H), 7,02 (д, 2H, J=9 Гц), 6,23 (дд, 1H, J=2 и 10 Гц), 5,61 (д, 1Н, J=12 Гц), 5,56 (д, 1H, J=12 Гц), 4,47 (м, 1H), 4,1 (м, 2H), 3,47 (д, 1Н, J=11 Гц), 3,22 (д, 1Н, J=12 Гц), 3,2 (м, 1H), 2,64 (с, 3H), 2,60 (с, 3H), 1,75 (т, 1H, J=11 Гц), 1,58 (т, 1H, J=11 Гц), 1,33 (с, 9H), 1,33 (т, 6Н, J=7 Гц).
[0115] IV. Примеры стереоспецифичного РМО-сочетания с диастереомерно чистыми активированными мономерами.
[0116] В последующих примерах описано использование стереоспецифичного сочетания для получения стереохимически гомогенных продуктов.
[0117] A. Активированные U-мономеры (U1 & U2)+U-морфолин-NH (1)
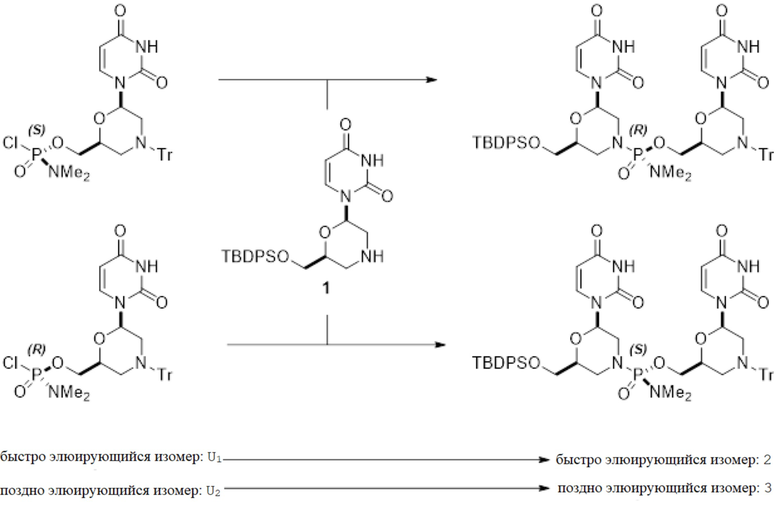
[0118] U1 (11 мг, 0,018 ммоль, 1 экв., 99,0% ДИ (диастереоизомерный избыток)) растворяли в ацетонитриле (0,11 мл) и смешивали с диизопропилэтиламином (8 мкл, 0,05 ммоль, 2,5 экв.). Добавляли U-морфолин-NH (14 мг, 0,030 ммоль, 1,6 экв.) и использовали обработку ультразвуком для облегчения растворения. Через 0,5 ч перемешивания небольшую аликвоту реакционной смеси разбавляли CDCl3 и анализировали с помощью 1H-ЯМР. Всю остальную реакционную смесь разбавляли ацетонитрилом (8 мл) для ВЭЖХ-анализа и хранили в морозильной камере. Стереоспецифичное образование соединения 2 было подтверждено ВЭЖХ-анализом (99,4% ДИ). Вышеуказанный протокол также использовали для реакции сочетания U2 (95,6% ДИ) для стереоспецифичного получения соединения 3 (96,0% ДИ).
[0119] Условия аналитической ВЭЖХ U/U-сочетания:
(99,0% ДИ)
(99,4% ДИ)
(95,6% ДИ)
(96,0% ДИ)
[0120] [1Н-ЯМР данные для соединения 2]
[0121] 1H-ЯМР (400 МГц, CDCl3) δ 7,6 (м, 4H), 7,2-7,5 (м, 20H), 7,1-7,2 (м, 3Η), 6,15 (д, 1H, J=8,0 Гц), 5,73 (д, 1H, J=8,0 Гц), 5,66 (д, 1H, J=8,0 Гц), 5,54 (д, 1H, J=8,0 Гц), 4,40 (м, 1H), 3,93 (м, 2H), 3,81 (м, 1H), 3,70 (м, 2H), 3,41 (м, 2H), 3,40 (м, 3H), 3,11 (д, 1H, J=12 Гц), 2,78 (м, 1H), 2,56 (с, 3H, Me), 2,54 (с, 3H, NMe), 2,48 (м, 1H), 1,47 (т, 1H, J=11 Гц), 1,35 (т, 1H, J=11 Гц), 1,04 (с, 9H).
[0122] [1Н-ЯМР данные для соединения 3]
[0123] 1Н-ЯМР (400 МГц, CDCl3) δ 7,6 (м, 4H), 7,3-7,5 (м, 1 1H), 7,2-7,3 (м, 9H), 7,1 (м, 3H), 6,12 (дд, 1H, J=2,0 и 9,6 Гц), 5,71 (д, 1H, J=8,4 Гц), 5,70 (д, 1H, J=8,0 Гц), 5,47 (дд, 1H, J=2,0 и 10,4 Гц), 4,31 (м, 1H), 3,97 (м, 1H), 3,85 (м, 1H), 3,73 (м, 2H), 3,65 (м, 1H), 3,31 (м, 2H), 3,24 (м, 1H), 3,07 (д, 1H, J=12 Гц), 2,68 (м, 1H), 2,65 (с, 3H, NMe), 2,62 (с, 3H, NMe), 2,26 (м, 1H), 1,45 (т, 1H, J=12 Гц), 1,29 (т, 1H, J=11 Гц), 1,04 (с, 9H).
[0124] B. Активированные C-мономеры (C1 & C2)+С-морфолин-NH (4)

[0125] C1 (20 мг, 0,031 ммоль, 1 экв., 93,5% ДИ) растворяли/суспендировали в THF (0,40 мл) и смешивали с диизопропилэтиламином (12 мкл, 0,069 ммоль, 2,3 экв.). Добавляли морфолино-цитозин (4; 16 мг, 0,035 ммоль, 1,1 экв.) в THF (0,20 мл). После перемешивания в течение 1,0-2,0 ч небольшую аликвоту реакционной смеси разбавляли в ацетонитриле и анализировали с помощью ЖХ/МС. Аликвоту (30-50 мкл) реакционной смеси разбавляли дихлорметаном (0,6 мл) для ВЭЖХ-анализа. Стереоспецифичное образование соединения 6 было подтверждено с помощью ВЭЖХ-анализа (94,3% ДИ). Реакционную смесь напрямую наносили на колонку с силикагелем и элюировали градиентом подвижной фазы 0-15% метанола в этилацетате. Вышеуказанный протокол также использовали для С/C-сочетания С2 (90,2% ДИ), чтобы стереоспецифично получить соединение 5 (90,0% ДИ).
[0126] Условия аналитической ВЭЖХ для C/C-сочетания:
(93,5% ДИ)
(94,3% ДИ)
(90,2% ДИ)
(90,0% ДИ)
[0127] [1Н-ЯМР данные для соединения 5]
[0128] 1Н-ЯМР (400 МГц, CDCl3) δ 10,9 (шир., 1H), 7,69 (д, 1H, J=7,4 Гц), 7,62 (м, 5H), 7,35-7,44 (м, 13H), 7,21-7,35 (м, 6H), 7,15 (м, 4H), 6,14 (шир.д, 1H, J=7,8 Гц), 5,58 (дд, 1H, J=2,4 и 9,4 Гц), 5,53 (шир., 1H), 4,51 (дд, 1H, J=8,6 и 10 Гц), 4,09 (м, 1H), 3,70-3,80 (м, 4H), 3,60 (дд, 1H, J=6,3 и 10 Гц), 3,56 (д, 1Н, J=11 Гц), 3,28 (м, 1H), 2,96 (д, 1Н, J=11 Гц), 2,69 (с, 3H, NMe), 2,67 (с, 3H, NMe), 2,65 (м, 1H), 2,25 (м, 1H), 2,07 (с, 3H), 1,31 (т, 1H, J=11 Гц), 1,13 (т, 1H, J=11 Гц), 1,04 (с, 9H).
[0129] [[1Н-ЯМР данные для соединения 6]
[0130] 1H-ЯМР (400 МГц, CDCl3) δ 9,57 (шир., 1H), 7,62-7,70 (м, 7H), 7,35-7,50 (м, 14H), 7,23-7,35 (м, 4H), 7,12 (м, 4H), 6,31 (м, 1H), 5,79 (м, 1H), 5,70 (м, 1H), 4,61 (м, 1H), 4,03 (м, 1H), 3,80-3,90 (м, 2H), 3,72 (м, 2H), 3,58 (м, 1H), 3,48 (м, 1H), 3,09 (м, 1H), 2,75 (м, 1H), 2,58 (с, 3H, NMe), 2,55 (с, 3H, NMe), 2,53 (м, 1H), 2,38 (м, 1H), 2,21 (с, 3H), 1,47 (т, 1H, J=10 Гц), 1,22 (т, 1H, J=10 Гц), 1,06 (с, 9H).
[0131] С. Активированные А-мономеры (A1 & A2)+U-морфолин-NH (1)

[0132] А1 (5,9 мг, 0,008 ммоль) ресуспендировали в ацетонитриле (118 мкл). Добавляли диизопропилэтиламин (5 мкл, 0,03 ммоль), после чего добавляли морфолино-урацил (1; 4,6 мг, 0,01 ммоль). Ультразвук использовали в течение 1 мин, и полученную гомогенную смесь перемешивали при комнатной температуре. После перемешивания в течение ночи смесь (густую белую пасту) разбавляли смесью ацетонитрила (5,0 мл) и метанола (0,30 мл), получая гомогенный прозрачный раствор. Небольшую аликвоту напрямую анализировали с помощью ВЭЖХ без дальнейшего разбавления.
[0133] А2 (F2; 5,0 мг, 0,007 ммоль) ресуспендировали в ацетонитриле (100 мкл). Добавляли диизопропилэтиламин (4 мкл, 0,02 ммоль), после чего добавляли морфолино-урацил (4,1 мг, 0,009 ммоль). Ультразвук использовали в течение 1 мин, и полученную густую суспензию перемешивали при комнатной температуре. После перемешивания в течение ночи добавляли ацетонитрил (5,0 мл) и использовали обработку ультразвуком, получая гомогенный прозрачный раствор. Небольшую аликвоту напрямую анализировали с помощью ВЭЖХ без дальнейшего разбавления.
[0134] Условия аналитической ВЭЖХ для U/A-сочетания:
(97,8% ДИ)
(R-изомер, 96,2% ДИ)
(98,4% ДИ)
(S-изомер, 98,3% ДИ)
[0135] D. Активированные G-мономеры (G1 & G2)+U-морфолин-NH (1)
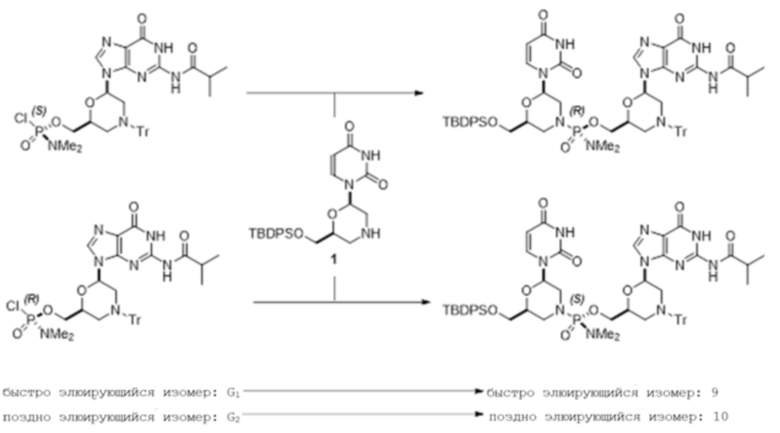
[0136] G1 (6,5 мг, 0,009 ммоль, 1 экв., 99,9% ДИ) растворяли/суспендировали в THF (0,13 мл) и смешивали с диизопропилэтиламином (3,6 мкл, 0,02 ммоль, 2,2 экв.). Добавляли морфолино-урацил (1; 4,7 мг, 0,010 ммоль, 1,1 экв.), растворенный в THF (0,07 мл). После перемешивания в течение 1,0-2,0 ч небольшую аликвоту реакционной смеси разбавляли ацетонитрилом и анализировали с помощью ЖХ/МС. Аликвоту (100 мкл) из реакционной смеси разбавляли дихлорметаном (0,4 мл) для ВЭЖХ-анализа. Стереоспецифичное образование соединения 9 было подтверждено с помощью ВЭЖХ-анализа (99,9% ДИ).
[0137] условия аналитической ВЭЖХ для U/G-сочетания:
(99,9% ДИ)
(99,9% ДИ)
[0138] Диастереомерно по существу чистые соединения, описанные выше, могут использоваться для получения стереохимически чистых олигонуклеотидов и других соединений. Примеры возможных олигонуклеотидов приведены, например, в Summerton, J (1999). "Morpholino Antisense Oligomers: The Case for an RNase-H Independent Structural Type.". Biochimica et Biophysica Acta 1489 (1): 141-58; и в Summerton, J; Weller D. (1997). "Morpholino Antisense Oligomers: Design, Preparation and Properties". Antisense & Nucleic Acid Drug Development 7 (3): 187-95. Оба эти документа включены в настоящее описание посредством ссылки.
[0139] V. Пример стереоспецифичного синтеза 16-членных РМО и разделение стереоизомеров биофизическим методом
[0140] В этом примере описан синтез, целью которого является пара стереочистых 16-членных РМО, путем стереоспецифичного сочетания с использованием активированных мономеров. Эти РМО имеют противоположные стереохимические характеристики своих фосфорных связей.
Целевая последовательность:
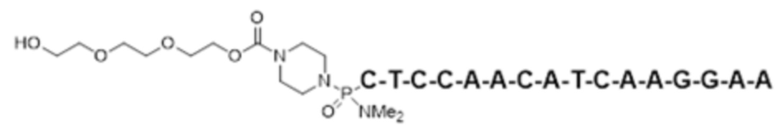
(SEQ ID NO: 2)
Стереочистые активные мономеры (структурные звенья):
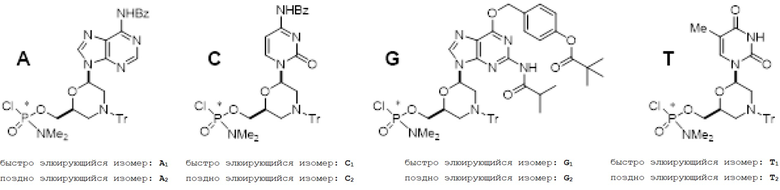
[0141] Схема стереоспецифичного синтеза 16-членных РМО и разделения стереоизомеров биофизическим методом показана на фиг. 6. 16-членные РМО-стереоизомеры 1 и 2 получены вручную методом твердофазного синтеза на аминометилполистиролдисульфидной смоле (нагрузка ~300 мкмоль/г, см. публикацию заявки на патент США №20090131624A1, которая включена в данное описание посредством ссылки) в масштабе 50 мг (начальный вес смолы).
[0142] Исходные растворы для твердофазного синтеза:
*0,11 M N-этилморфолин в 1,3-диметилимидазолидиноне (DMI)
[0143] Операционный цикл для каждого РМО-сочетания:
*40°С в течение 3 часов или комнатная температура в течение 12 часов.
[0144] Снятие со смолы и удаление защиты:
[0145] К связанному со смолой 16-членному олигонуклеотиду (после детритилирования) добавляли 1:3 (об./об.) 28%-го водного раствора аммиака в этаноле (~5 мл). Смесь герметично закрывали и нагревали при температуре 45°С в течение 20 часов. После охлаждения до комнатной температуры смесь фильтровали и промывали метанолом. Фильтрат концентрировали и проводили диафильтрацию против 15 мМ триэтиламмонийного (ТЕАА) буфера, рН 7,0. Используемым устройством была перемешиваемая ячейка Amicon (50 мл) с UF-мембраной Ultracel 1 кДа. Образцы диафильтровали путем разбавления/концентрации до тех пор, пока концентрация исходного растворителя не снижалась до 1% от исходной концентрации (приблизительно за 5 циклов), а затем очищали с помощью препаративной обращенно-фазовой ВЭЖХ.
[0146] Способ очистки РМО путем обращенно-фазовой препаративной ВЭЖХ:
Растворитель B: ацетонитрил+10% MeOH
[0147] ЖХ/МС-способ для оценки качества РМО:
Растворитель B: ацетонитрил/метанол 1/1 об./об. с 50 мM ацетатом аммония
[0148] Материалы и условия для измерения температуры плавления (Tm)
Смеси затем нагревали до 95°С, а затем охлаждали до 25°С для отжига перед измерением Tm.
Анализ Tm (УФ 260 нм) проводили от 25°С до 105°С при 0,5°С/мин (температуру возвращали к исходным условиям после каждого прогона) и затем повторяли при тех же условиях.
Для вычисления Tm использовали аналитическую программу (Shimadzu) с использованием «усредняющей функции».
[0149] Результаты термического плавления комплексов стереохимически отличающихся РМО с комплементарной РНК:
[0150] Температуры плавления для стереоизомеров 1 и 2 показаны на фиг. 7. Исходя из разных температур плавления можно сделать вывод, что были получены определенные количества по существу чистых стереоизомеров.
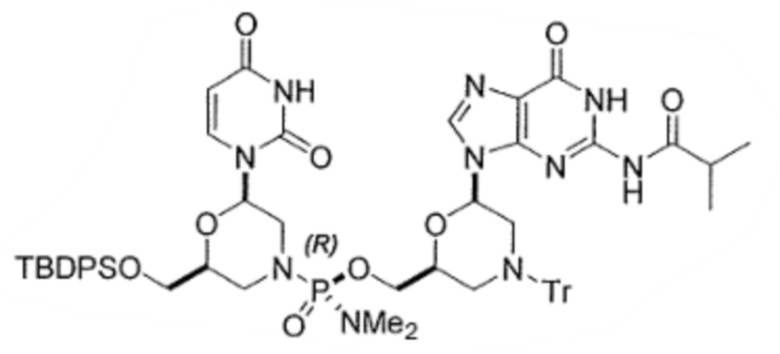
[0151] VI. Пример стереоспецифичного синтеза и абсолютного стереохимического определения стереочистого РМО-динуклеотидного соединения 100 (5'-ΤΑ2-3')
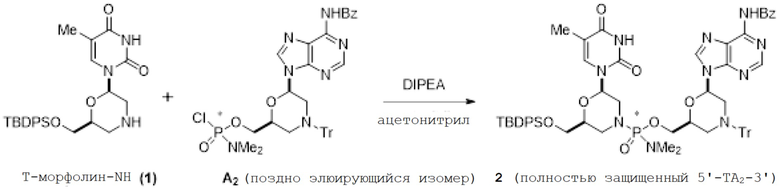
[0152] Поздно элюирующийся активный мономер А (А2; 200 мг, 0,277 ммоль, 1 экв.) растворяли в смеси ацетонитрила (2,0 мл) и DIPEA (0,12 мл, 0,69 ммоль, 2,5 экв.). Затем добавляли Т-морфолин-NH (1; 146 мг, 0,305 ммоль, 1,1 экв), и полученную суспензию обрабатывали ультразвуком в течение нескольких минут до получения прозрачного раствора. Реакционную смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции по данным ЖХ/МС смесь концентрировали и хроматографировали на колонке (в 3%-м метаноле в DCM, Biotage SnapUltra 10 г SiО2). Чистые фракции, содержащие продукт, объединяли и концентрировали в вакууме, получая полностью защищенный стереочистый 5'-ТА-3' динуклеотид 2 в виде белого твердого вещества (240 мг, 0,206 ммоль, выход 74%).
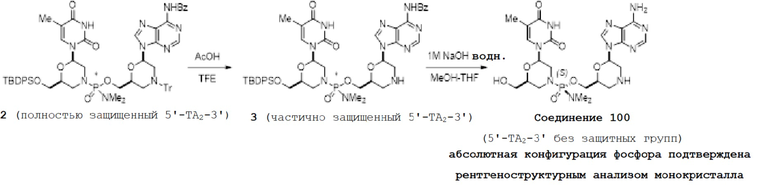
[0153] К полностью защищенному динуклеотиду 2 (500 мг, 0,429 ммоль) в 25-мл колбе добавили 2,2,2-трифторэтанол (TFE, 4,0 мл) и уксусную кислоту (1,0 мл) при комнатной температуре. Полученную смесь перемешивали при комнатной температуре и прохождение реакции контролировали с помощью ЖХ/МС. Через 30 минут реакционную смесь гасили насыщенным водным раствором NaHCО3 и DCM. После разделения двух слоев водный слой повторно экстрагировали. Все органические слои объединяли, промывали наполовину насыщенным маточным раствором, высушивали над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного продукта в виде белой пены. Неочищенный продукт очищали с помощью колоночной хроматографии (в 20% MeOH в ацетоне на картридже Biotage Snap Ultra 25 г SiО2) с получением частично защищенного динуклеотида 3 в виде стеклообразного твердого вещества (300 мг, 0,325 ммоль, выход 76%).
[0154] Частично защищенный динуклеотид 3 (250 мг, 0,271 ммоль) растворяли в смеси метанола (12,5 мл) и THF (12,5 мл) и обрабатывали 1 М NaOH (10,8 мл) при комнатной температуре. После перемешивания при комнатной температуре в течение 22 ч (прогресс мониторили с помощью ЖХ/МС), смесь нейтрализовали 1 М HCl (10,8 мл), чтобы подвести рН до 8, а затем концентрировали в вакууме досуха. Остаток растворяли в воде (5 мл) и промывали EtOAc (5 мл). Водный слой концентрировали в вакууме досуха с получением неочищенного продукта в виде белого твердого вещества (480 мг). Неочищенный продукт очищали с помощью гель-хроматографии (на Sephadex® LH-20 в смеси метанол/вода 4:1) с получением соединения 100 (динуклеотида с полностью удаленными защитными группами) в виде белого твердого вещества (137 мг, 0,236 ммоль, выход 87%).
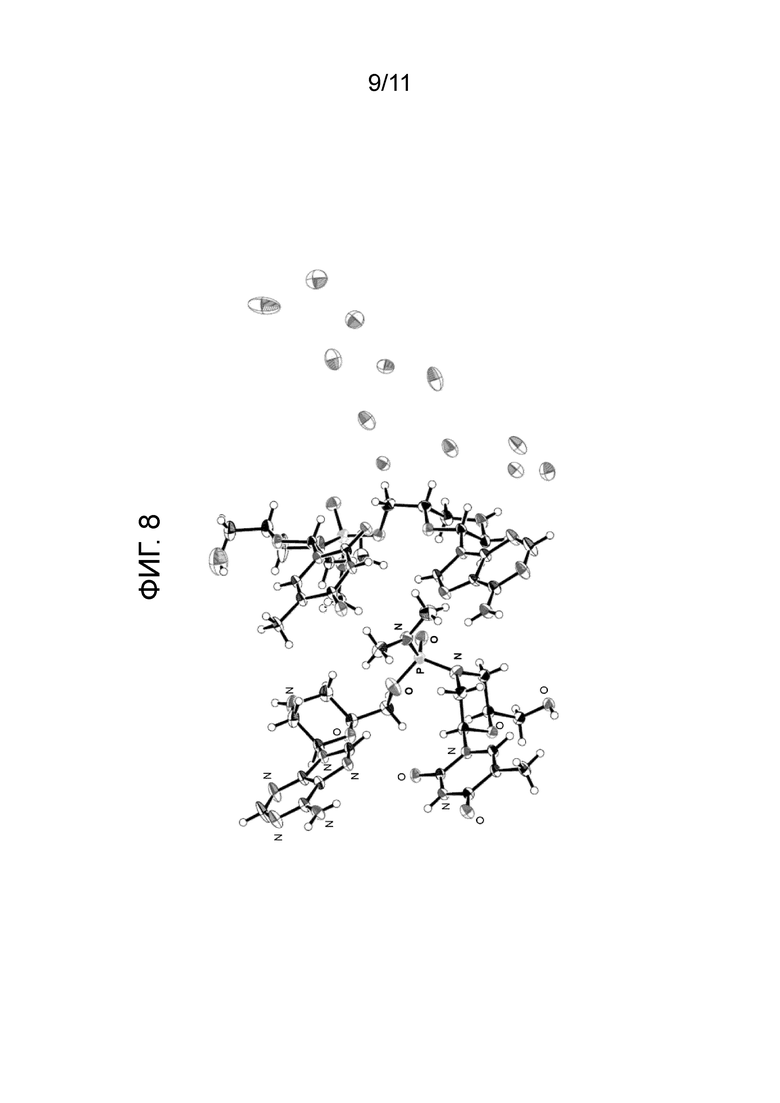
[0155] Каплю водного раствора соединения 100 (200 мг/мл) запечатывали в лунке с чистой водой на один день для выращивания монокристаллов. Рентгеноструктурный анализ монокристалла подтвердил абсолютную конфигурацию фосфорной связи как S. Эта рентгеновская структура показана в виде ORTEP-диаграммы на фиг. 8. ORTEP-диаграммы отдельных фрагментов показаны на фиг. 9А и фиг. 9В. Рентгеноструктурные данные были получены, как описано ниже.
[0156] Получение рентгеноструктурных данных
[0157] Монокристалл соединения 100 (C22H33N10O7P) был установлен на стекловолокне. Все измерения проводились на дифрактометре с использованием монохроматического излучения (Cu-Kα, графитовый монохроматизатор).
[0158] Параметры ячейки и матрица ориентации для данных были получены из уточнения методом наименьших квадратов с использованием установочных углов 36473 хорошо центрированных отражений в диапазоне 7,75 <2θ <147,10°, и соответствовали C-центрированной моноклинной ячейке с размерами:
а=33,3523(2) 
b=13,80020(11) β=96,8075(6)°
с=14,19956(10)
V=6489,53(8) 3
[0159] Для Z=4 и FW=580,54, расчетная плотность составляет 0,594 г/см3. Исходя из условий отражения:
hkl: h+k=2n
упаковки, статистического анализа распределения интенсивности и успешного решения и уточнения структуры, пространственная группа была определена как:
С2(#5)
[0160] Данные были получены при температуре 23±1°C с использованием метода сканирования ω-2θ до максимального значения 2θ 147,7°. Омега-сканирования нескольких интенсивных отражений, сделанных до сбора данных, имели среднюю ширину на полувысоте 0,00° с углом отражения 6,0°. Сканы (0,00+0,00 тангенс θ)° были сделаны со скоростью 0,0°/мин (в ω).
[0161] Сокращение количества данных
[0162] Было собрано 50795 отражений, из которых 12008 были уникальными (Rint=0,0453). Данные были собраны и обработаны с использованием CrysAlisPro (Rigaku Oxford Diffraction). (CrysAlisPro: Data Collection and Processing Software, Rigaku Corporation (2015) Tokyo 196-8666, Japan.). Коррекция на распад не применялась.
[0163] Коэффициент линейного поглощения, μ, для Cu-Κα-излучения составляет 6,011 см-1. Применяли эмпирическую коррекцию поглощения, что давало факторы в пределах от 0,341 до 1,000. Данные были скорректированы на эффекты Лоренца и поляризации.
[0164] Решение и уточнение структуры
[0165] Структура была решена прямыми методами (SHELXT Version 2014/5: Sheldrick, G. M. (2014). Acta Cryst. A70, C1437) и расширенными с использованием Фурье-методов. Неводородные атомы были уточнены анизотропно. Атомы водорода были уточнены с использованием модели наездника. Окончательный цикл уточнения полноматричным методом наименьших квадратов (с использованием минимизированной функции наименьших квадратов: (SHELXL Version 2014/7); Σw(Fо2-Fс2)2, где w=вес наименьших квадратов) на F2 был основан на 12008 наблюдаемых отражениях и 849 переменных параметрах и сходился (наибольший сдвиг параметра составлял 0,00 от его оценочного значения среднеквадратичного отклонения) с невзвешенными и взвешенными факторами согласия:
R1=Σ||Fo|-|Fc||/Σ|Fo|=0,0522
wR2=[Σ(w(Fo2-Fc2)2)/Σw(Fo2)2]1/2=0,1632
[0166] Степень согласия составляла 1,45. Степень согласия определяется следующим образом: [Σw(Fo2-Fc2)2/(No-Nv)]1/2, где: NО=число наблюдений и Nv=число переменных.
[0167] Использовали удельный вес. Максимальные и минимальные пики на конечной разностной карте Фурье соответствовали 1,79 и -0,69 е-/Å3, соответственно. Конечный параметр Флека равнялся 0,029(7), что свидетельствует о том, что структура является двойником инверсии. (Parsons, S. and Flack, H. (2004), Acta Cryst. A60, s61; Flack, H.D. and Bernardinelli (2000), J. Appl. Cryst. 33, 114-1148).
[0168] Факторы рассеяния нейтральных атомов были взяты из Международных таблиц для кристаллографии (IT), том С, таблица 6.1.1.4. (International Tables for Crystallography, Vol.C (1992). Ed. A.J.C. Wilson, Kluwer Academic Publishers, Dordrecht, Netherlands, Table 6.1.1.4, pp. 572). Эффекты аномальной дисперсии были включены в Fcalc (Ibers, J. A. & Hamilton, W. C.; Acta Crystallogr., 17, 781 (1964)); значения для Δf' и Δf" были были взяты из справочника Creagh и McAuley. (Creagh, D. C. & McAuley, W.J.; "International Tables for Crystallography", Vol C, (A.J.C. Wilson, ed.), Kluwer Academic Publishers, Boston, Table 4.2.6.8, pages 219-222 (1992)). Значения коэффициентов затухания масс были взяты из Creagh и Hubbell. (Creagh, D. C. & Hubbell, J.H..; "International Tables for Crystallography", Vol C, (A.J.C. Wilson, ed.), Kluwer Academic Publishers, Boston, Table 4.2.4.3, pages 200-206 (1992)). Все расчеты проводились с использованием пакета кристаллографического программного обеспечения CrystalStructure, за исключением уточнения, которое было выполнено с использованием SHELXL Version 2014/7. (CrystalStructure 4.2: Crystal Structure Analysis Package, Rigaku Corporation (2000-2015). Tokyo 196-8666, Japan; SHELXL Version 2014/7: Sheldrick, G. M. (2008). Acta Cryst. A64, 112-122).
[0169] Кристаллические данные, измерения интенсивности и решение и уточнение структуры приведены ниже:
A. Кристаллические данные
3
B. Измерения интенсивности
)
С. Решение и уточнение структуры
33
[0170] [1H-ЯМР данные для соединения 100]
[0171] 1H-ЯМР (400 МГц, D2О) δ 8,25 (с, 1H), 8,15 (с, 1H), 7,40 (с, 1H), 5,85 (д, 1H), 5,45 (д, 1H), 4,25 (м, 2H), 4,05 (м, 1H), 3,85 (м, 1H), 3,6 (м, 2H), 3,4 (м, 4H), 2,90 (м, 4H), 2,60 (д, 6H), 1,8 (с, 3H).
[0172] Все документы, указанные в настоящей заявке, включены в нее путем ссылки. Если присутствует какое-либо расхождение между включенным документом и данным документом, то данный документ превалирует.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ХИНОЛИНОВЫЕ СОЕДИНЕНИЯ | 2018 |
|
RU2810113C2 |
| НОВЫЕ СОЕДИНЕНИЯ - АНТАГОНИСТЫ РЕЦЕПТОРА НЕЙРОКИНИНА 1 | 2013 |
|
RU2631319C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2437886C2 |
| АТРОПОИЗОМЕРЫ ПРОИЗВОДНЫХ 3-ЗАМЕЩЕННОГО-4-АРИЛХИНОЛИН-2-ОНА | 2003 |
|
RU2352563C2 |
| ПРОИЗВОДНЫЕ 2-ПИРИДОНА | 2010 |
|
RU2551847C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5-БИФЕНИЛ-4-АМИНО-2-МЕТИЛПЕНТАНОВОЙ КИСЛОТЫ | 2008 |
|
RU2530900C2 |
| СОЕДИНЕНИЯ | 2016 |
|
RU2725616C2 |
| ЭНАНТИОМЕРНО ЧИСТЫЕ ОСНОВНЫЕ ЭФИРЫ АРИЛ-ЦИКЛОАЛКИЛГИДРОКСИКАРБОНОВЫХ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В ЛЕКАРСТВЕННЫХ СРЕДСТВАХ | 1997 |
|
RU2238936C2 |
| СОЕДИНЕНИЯ ИМИДАЗОТРИАЗИНОНА | 2011 |
|
RU2603140C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2419619C2 |


Настоящее изобретение относится к способу получения по существу диастереомерно чистого фосфородиамидатного морфолино-олигомера, содержащего хиральные фосфорные связи, который используют для получения стереохимически чистого олигонуклеотида. Предлагаемый способ включает следующие стадии: выбор по существу стереохимически чистых фосфорамидохлоридатных морфолино-мономеров; и синтез по существу диастереомерно чистого фосфородиамидатного морфолино-олигомера путем стереоспецифичного сочетания выбранных по существу стереохимически чистых фосфорамидохлоридатных морфолино-мономеров, где указанное стереоспецифическое сочетание проводят в апротонном растворителе в присутствии ненуклеофильного третичного амина или ароматического основания при температуре от 20°C до 50°C. При этом по существу стереохимически чистые фосфорамидохлоридатные морфолино-мономеры получают путем разделения диастереомерной смеси фосфорамидохлоридатных морфолино-мономеров на по существу стереохимически чистые фосфорамидохлоридатные морфолино-мономеры. По существу стереохимически чистый в данном случае относятся к энантиомерам или диастереомерам, которые находятся в энантиомерном или диастереомерном избытке, соответственно, равном или больше 87%, а по существу диастереомерно чистый относится к диастереомерам, которые находятся в диастереомерном избытке, равном или больше 87%. 2 н. и 4 з.п. ф-лы, 11 ил., 2 табл., 4 пр.
1. Способ получения по существу диастереомерно чистого фосфородиамидатного морфолино-олигомера, содержащего хиральные фосфорные связи, включающий:
выбор по существу стереохимически чистых фосфорамидохлоридатных морфолино-мономеров; и
синтез по существу диастереомерно чистого фосфородиамидатного морфолино-олигомера путем стереоспецифичного сочетания выбранных по существу стереохимически чистых фосфорамидохлоридатных морфолино-мономеров, где указанное стереоспецифическое сочетание проводят в апротонном растворителе в присутствии ненуклеофильного третичного амина или ароматического основания при температуре от 20°C до 50°C;
где по существу стереохимически чистые фосфорамидохлоридатные морфолино-мономеры получают путем разделения диастереомерной смеси фосфорамидохлоридатных морфолино-мономеров на по существу стереохимически чистые фосфорамидохлоридатные морфолино-мономеры, и
где по существу стереохимически чистый относятся к энантиомерам или диастереомерам, которые находятся в энантиомерном или диастереомерном избытке, соответственно, равном или больше 87%, и
по существу диастереомерно чистый относится к диастереомерам, которые находятся в диастереомерном избытке, равном или больше 87%.
2. Способ по п.1, где разделение диастереомерной смеси происходит по меньшей мере с помощью одного метода из группы, состоящей из хроматографии и кристаллизации.
3. Способ по п.2, где хроматографию выбирают из группы, состоящей из высокоэффективной жидкостной хроматографии (ВЭЖХ), хроматографии в псевдодвижущемся слое и хроматографии в противотоке.
4. Способ по любому из пп.1-3, где по существу стереохимически чистые фосфорамидохлоридатные морфолино-мономеры выбирают из группы, состоящей из соединения формулы 20, соединения формулы 21, соединения формулы 22, соединения формулы 23, соединения формулы 24, соединения формулы 25, соединения формулы 26, соединения формулы 27, соединения формулы 28, соединения формулы 29, соединения формулы 30 и соединения формулы 31,
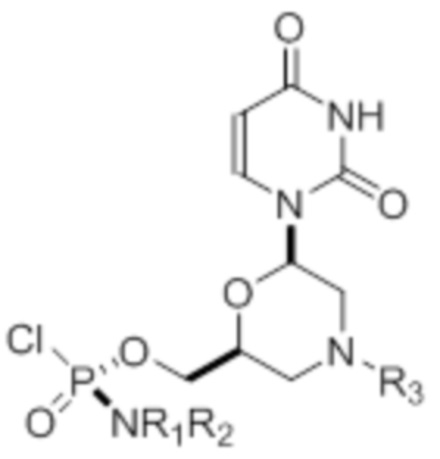
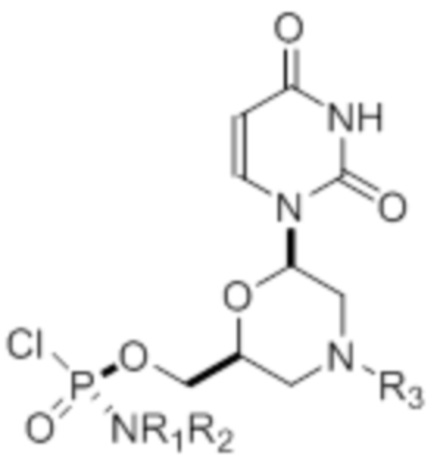
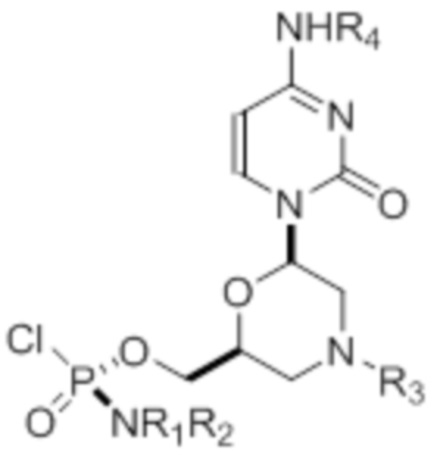
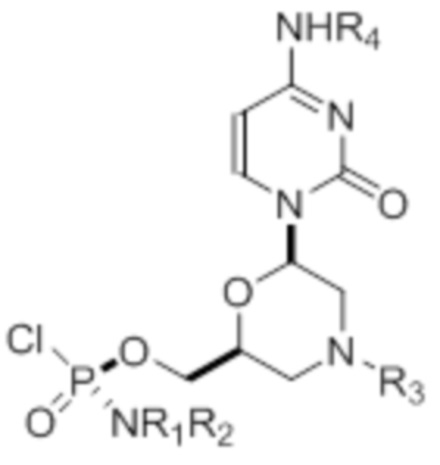
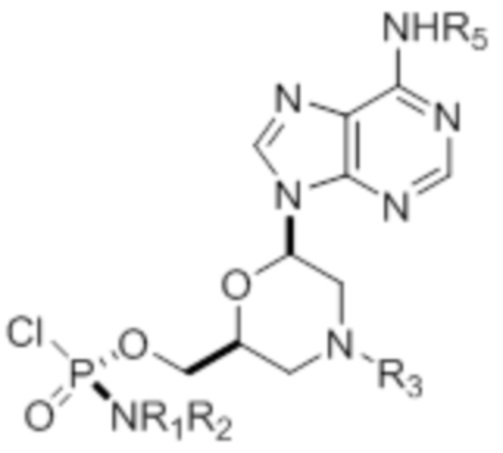
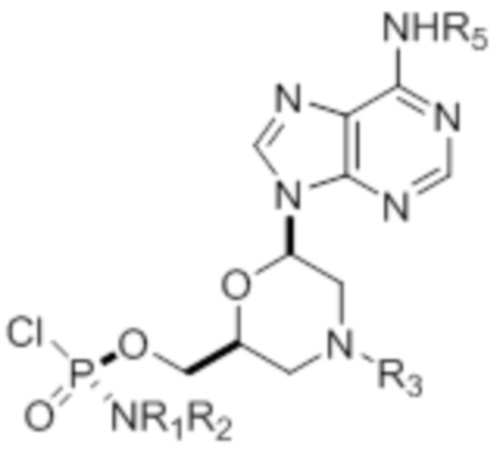
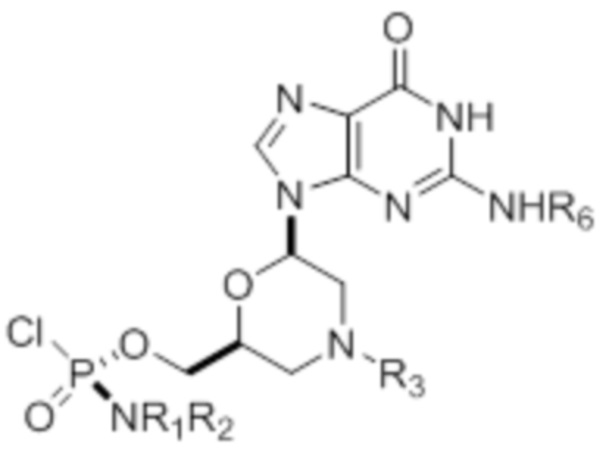

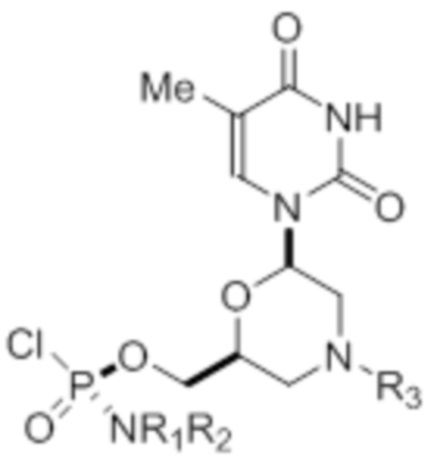
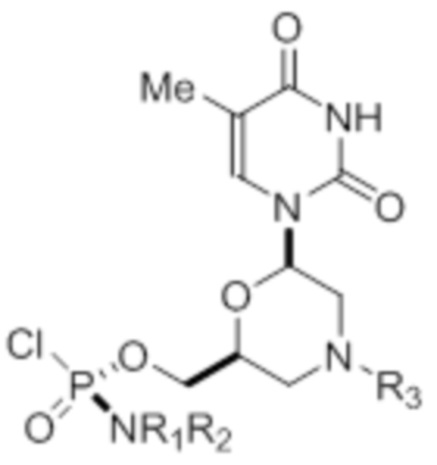
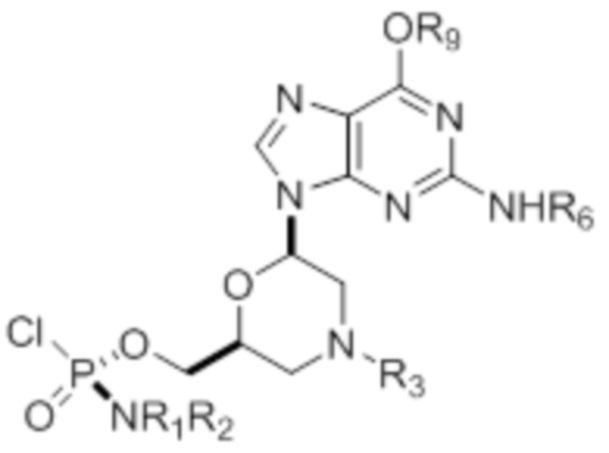
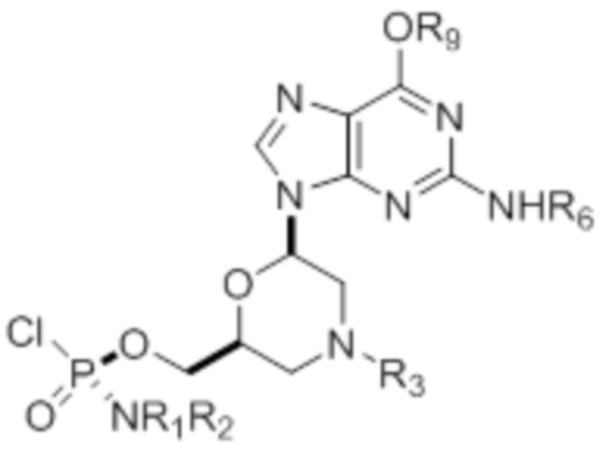
где R1 и R2 могут быть одинаковыми или различными и выбраны из группы, состоящей из -Н, С1-С3-алкила, фенила, нафтила, или же с атомом азота, к которому они присоединены, они образуют замещенный гетероцикл;
где R3 выбран из группы, состоящей из тритила (Tr), замещенного тритила, бензила, 4-метоксибензила (PMB, MPM), 3,4-диметоксибензила, дифенилметила (Dpm) и сульфонила;
R4, R5 и R6 выбраны из группы, состоящей из -H, -C(O)R7 и -C(O)OR7, где R7 представляет собой C1-C6-алкил, бензил, 2,2,2-трихлорэтил и арильную группу, выбранную из фенила, 4-метоксифенила, 4-бромфенила и 4-нитрофенила;
R9 выбран из группы, состоящей из алкила, цианоэтила, ацила, карбоната, карбамата, бензила, 4-пивалоилоксибензила и силила,
и их энантиомеров.
5. Способ по п.4, где сульфонил представляет собой расщепляемый сульфонил, выбранный из группы, состоящей из 2-нитробензолсульфонила, 4-нитробензолсульфонила и 2,4- динитробензолсульфонила.
6. По существу диастереомерно чистый фосфородиамидатный морфолино-олигомер, содержащий хиральные фосфорные связи, полученный способом по любому из пп.1-5, для получения стереохимически чистого олигонуклеотида, где указанный олигонуклеотид выбирают из группы, состоящей из:
 ;
;
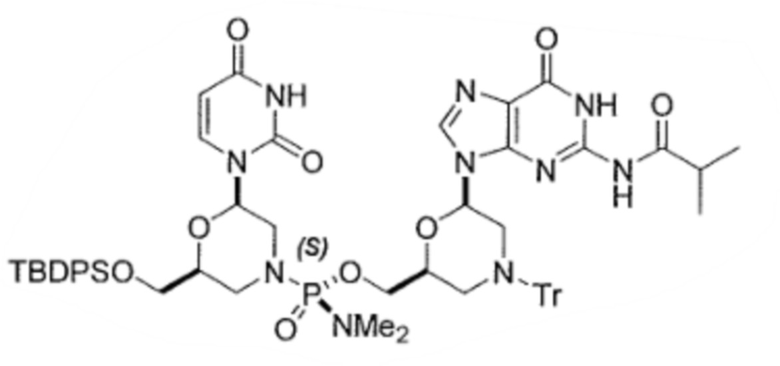 ;
;
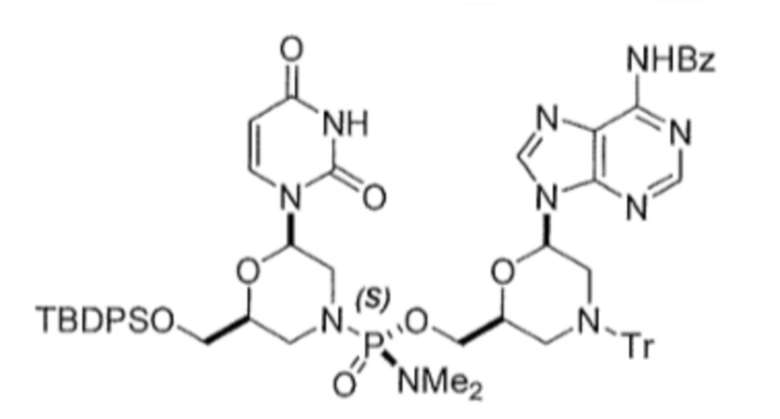 ;
;
;
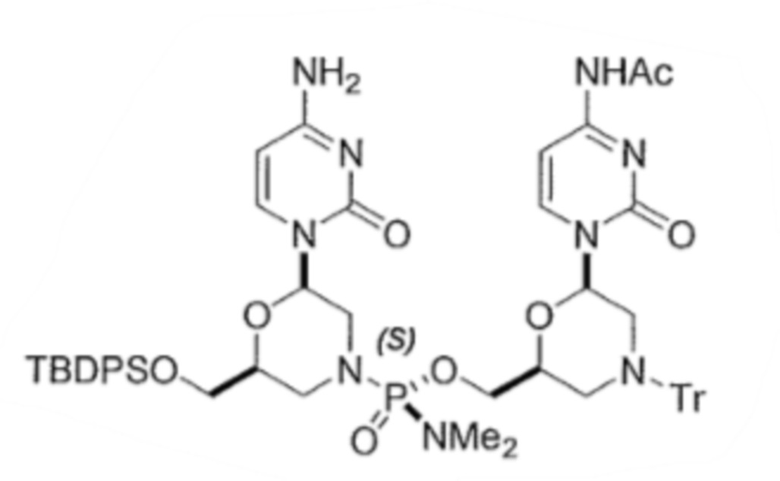 ;
;
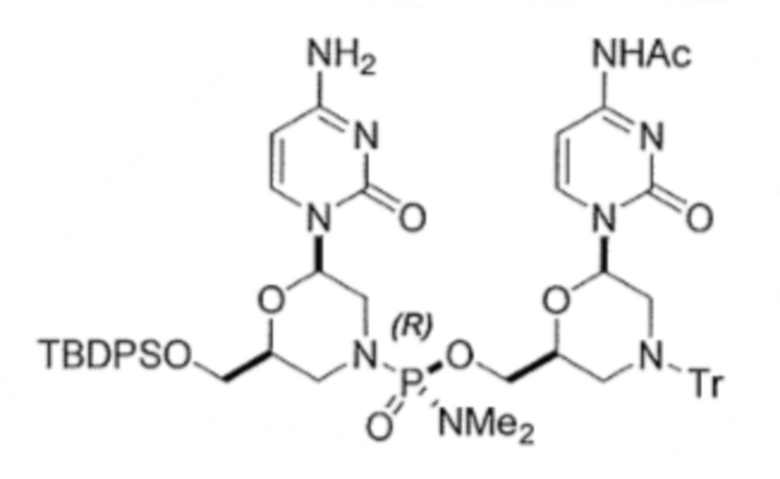 ;
;
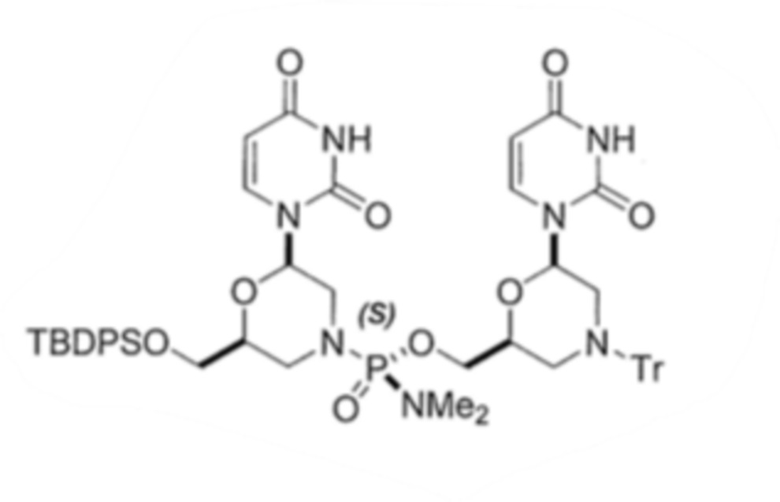 ;
;
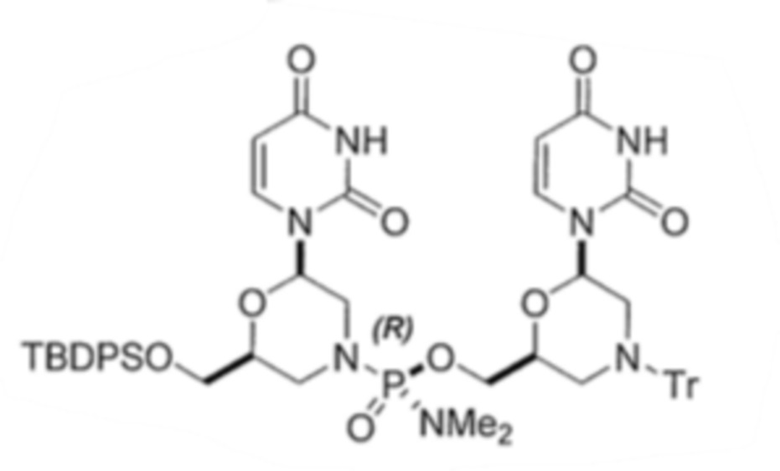 .
.
| WO 9825944 A1, 18.06.1998 | |||
| Обеспыливающий теплообменник вращающейся печи | 1984 |
|
SU1176151A1 |
| СПОСОБ ПСИХОТЕРАПИИ НЕВРОТИЧЕСКИХ РАССТРОЙСТВ | 2008 |
|
RU2357764C1 |
| WO 9938878 A1, 05.08.1999 | |||
| WO 2009064471 A1, 22.05.2009 | |||
| CN 102702265 A, 03.10.2012 | |||
| RU 2004102903, 10.07.2005. | |||

