Область техники, к которой относится изобретение
Настоящее изобретение относится к области биотехнологии, медицины, в частности, онкологии и фармацевтики, и направлено на лекарственные формы лекарственного препарата для комбинированной ген-направленной энзиматической пролекарственной терапии - иммунной терапии солидных злокачественных новообразований.
Предложенные лекарственные формы могут быть использованы для лечения различных солидных злокачественных новообразований у человека. Предложенные лекарственные формы демонстрируют высокую стабильность и сохраняют терапевтическую эффективность на протяжение по крайней мере 18 месяцев.
Уровень техники
Рак (злокачественные новообразования) представляет собой одну из наиболее серьезных проблем медицины: он занимает второе место по смертности в мире, и онкологическая заболеваемость демонстрирует тенденцию к росту.
Онкологические заболевания сочетают в себе как сложность клеточной организации, так и свойства сложной растущей системы, способной приспосабливаться к внешним воздействиям, в том числе и лекарственным, благодаря микрогетерогенности внутри опухолей. Гетерогенность раковых опухолей заключается в том, что каждая раковая клетка отличается от всех других клеток этой же опухоли, а опухоль одного пациента отличается от опухоли того же типа у другого. Поэтому среди множества клеток в опухоли всегда существуют клетки, устойчивые к терапевтическому воздействию и способные дать рост нового, устойчивого к данному воздействию, клона опухоли. В результате этого становится очевидным, что достаточно сложно однозначно идентифицировать какие-либо универсальные ключевые гены/генные продукты, которые могут быть использованы в качестве мишеней для противоопухолевой терапии.
Классическая химиотерапия в качестве стандартного лечения онкологических больных, созданная в течение нескольких десятилетий, доказала свою эффективность в замедлении развития различных типов злокачественных новообразований. Однако терапевтический индекс классической химиотерапии часто является низким по причинам отсутствия специфичности лекарственных средств к раковым клеткам по сравнению с нормальными клетками и трудности достижения терапевтических интратуморальных концентраций в отсутствие системной токсичности. Данные проблемы особенно актуальны для солидных опухолей, так как плохая неоваскуляризация и связанные с ней некротические области часто делают опухоли относительно устойчивыми к эффективному поглощению системно доставляемых лекарственных средств. В связи со сложившейся ситуацией во всем мире идет поиск и внедрение новейших стратегий терапии рака.
Одной из таких стратегий является ген-направленная энзиматическая пролекарственная терапии (ГНЭПТ, Gene-directed enzyme prodrug therapy, (GDEPT)) (Sverdlov E.D. Curr. Gene Ther. 2011, 11(6), p. 501-531). Данный подход также известен под названиями «суицидальная генная терапия», или «генная хирургия». Подход генной хирургии заключается в доставке в раковые клетки-мишени генов, кодирующих фермент, который в клетках, где он экспрессируется, способен превращать свой субстрат, вводимый системно, в высокотоксичный агент. Данная стратегия имеет сходство с химиотерапией, однако, в отличие от нее, в случае генной хирургии токсин образуется внутри раковой клетки, поэтому токсическое воздействие на нормальные клетки организма сводится к минимуму.
Подход ГНЭПТ позволяет убивать раковые клетки независимо от их генезиза и свойств. Еще одним важным преимуществом генной хирургии является то, что даже небольшого количества клеток, экспрессирующих трансген, достаточно для того, чтобы вызвать гибель большого числа опухолевых клеток. Токсин, образовавшийся в клетках, экспрессирующих трансген, может высвобождаться из них и проникать в соседние клетки, не получившие трансген, вызывая их гибель. Данное явление получило название «эффект свидетеля» (bystander effect).
Наиболее изученной в настоящее время системой для проведения ГНЭПТ является комбинация «тимидинкиназа вируса простого герпеса (HSVtk)/ганцикловир (GCV)», достигшая поздних стадий клинических испытаний, в ходе которых была показана ее эффективность и безопасность (Immonen A. et al. Mol. Ther. 2004, 10(5), p. 967-972; Westphal M. et al. Lancet Oncol. 2013, 14(9), p. 823-833). Ганцикловир является зарегистрированным лекарственным средством и коммерчески доступен, например, как препарат «Цимевен»). Доставка терапевтического гена, кодирующего фермент тимидинкиназу, например, тимидинкиназу вируса простого герпеса (HSVtk), в составе той или иной экспрессионной конструкции в опухолевые клетки обеспечивает продукцию фермента HSVtk в указанных клетках. Ганцикловир является аналогом гуанозина и представляет собой пролекарство, которое вводят системно. Под действием фермента тимидинкиназы в опухолевой клетке, экспрессирующей HSVtk, происходит фосфорилирование ганцикловира до ганцикловирмонофосфата. Затем под действием внутриклеточных киназ ганцикловирмонофосфат последовательно превращается в ганцикловирдифосфат и ганцикловиртрифосфат. Действуя как субстрат и встраиваясь в ДНК, ганцикловиртрифосфат конкурентно ингибирует ДНК-полимеразу, что приводит к подавлению синтеза ДНК за счет ингибирования элонгации цепи ДНК и к одиночным разрывам ДНК и, в конечном итоге, к гибели опухолевых клеток. Кроме того, вновь образуемый ганцикловиртрифосфат может выходить из опухолевых клеток, в которых он образовался, и проникать в соседние опухолевые клетки, приводя к их гибели (упомянутый выше «эффект свидетеля»). Гибель клеток, опосредованная комбинацией HSVtk/ганцикловир, может быть вызвана различными механизмами: апоптозом и некрозом, а также иммуногенной гибелью клеток (ICD) с высвобождением опухолеспецифических антигенов и молекулярных фрагментов, ассоциированных с повреждениями (DAMP), что приводит к инфильтрации опухоли различными антиген-презентирующими клетками, включая дендритные и Т-клетки (Sverdlov E.D. Curr. Gene Ther. 2011, 11(6), p. 501-531).
В связи с тем, что часть клеток опухоли (или опухоль в целом) может быть устойчива к воздействию некоторых токсических агентов, используемых в системах ГНЭПТ, исследователями разрабатываются подходы к повышению эффективности генной хирургии. Одним из таких подходов является комбинирование генной хирургии с иммунотерапией. Такой подход иногда называют «ген-иммунной терапией».
Известно, что клетки опухоли на ранних этапах канцерогенеза по своему молекулярному составу практически неотличимы от клеток окружающей ткани, они не могут быть узнаны и уничтожены иммунной системой, являясь неиммуногенными. По мере роста опухоли за счёт накопления мутаций в ДНК происходит образование так называемых неоантигенов, которые могут быть презентированы дендритными клетками Т-клеткам хелперам, являющимся активаторами специфического иммунитета к опухолям. Появление неоантигенов происходит с задержкой, что позволяет клеткам опухоли реализовать механизмы защиты от иммунной системы, в частности, создать микроокружение, которое ингибирует специфический противоопухолевый иммунитет.
Многие представители семейства цитокинов способны мобилизовать иммунную систему пациентов, способствуя развитию специфического противоопухолевого ответа. Например, в генотерапевтических противоопухолевых препаратах Imlygic (Amgen Inc., США) для усиления презентации опухолевых антигенов дендритными клетками использован подход, основанный на увеличении продукции цитокина гранулоцитарно-макрофагального колониестимулирующего фактора (GM-CSF). В исследованиях показано, что клеточные вакцины, содержащие GM-CSF, обеспечивают развитие у животных более сильного противоопухолевого ответа, чем вакцины, включающие другие цитокины (Dranoff G, Jaffee Ε et al. (1993) Proc. Natl. Acad. Sci. USA 90:3539-43).
Гранулоцитарно-макрофагальный колониестимулирующий фактор (GM-CSF) принадлежит к семейству гемопоэтических цитокинов. Биологическая активность GM-CSF опосредуется связыванием GM-CSF с рецептором-гетеромером, который экспрессируется на моноцитах, макрофагах, гранулоцитах, лимфоцитах, эндотелиальных клетках и клетках альвеолярного эпителия. После специфического связывания GM-CSF с рецептором происходит передача сигнала по JAK2 пути, в результате чего развивается функциональный ответ. GM-CSF стимулирует пролиферацию и дифференцировку гранулоцитов и моноцитов, усиливает фагоцитарную и цитотоксическую активность гранулоцитов и эозинофилов, стимулирует пролиферацию, созревание и дифференцировку дендритных клеток, играет важную роль в рекрутинге и активации антиген-презентирующих клеток.
Известно, что экспрессия GM-CSF в клетках опухоли повышает ее иммуногенность, тем самым способствуя ее элиминации и обеспечивая развитие противоопухолевого ответа. В исследованиях in vivo продемонстрировано, что эффективность системы HSVtk/GCV резко возрастает, если в клетки опухоли вводить ген HSVtk совместно с геном GM-CSF (Jones R.K., Pope I.M., et al. (2000) Cancer Gene Ther. 7:1519-28; Finocchiaro L.M., Fiszman G.L., et al. (2008) Cancer Gene Ther. 15:165-72)).
В исследованиях ex vivo и in vivo продемонстрировано превосходство комбинированной терапии, сочетающей введение GM-CSF и системы HSVtk/GCV, над генной хирургией, включающей только введение системы HSVtk/GCV.
Полагают, что противоопухолевый эффект GM-CSF в сочетании с системой HSVtk/GCV обусловлен следующим механизмом: при одновременном попадании в раковые клетки генов HSVtk и GM-CSF будут происходить гибель раковых клеток за счет работы системы HSVtk/GCV и высвобождение из них опухолевых антигенов. Опухолевые антигены будут эффективно представляться GM-CSF-активированными антиген-презентирующими клетками Т-клеткам иммунной системы, обеспечивая активацию специфического противоопухолевого иммунитета, в результате чего возрастает гибель опухолевых клеток и снижается вероятность возникновения метастазов. Привлечение к опухоли антиген-презентирующих клеток и их активация происходит посредством синтезированного GM-CSF. В результате происходит эффективное представление опухолевых антигенов GM-CSF-активированными антиген-презентирующими клетками Т-клеткам иммунной системы и, как следствие, активация специфического противоопухолевого иммунитета.
Одним из важных элементов успеха генной терапии рака является адекватная система доставки и экспрессии терапевтических генов, поскольку данные гены должны работать в раковых клетках и не работать в нормальных клетках организма. Для доставки терапевтических конструкций в клетки опухоли пациента используют вирусные и невирусные системы. Основным недостатком невирусных систем доставки, по сравнению с вирусными, является их более низкая трансфекционная эффективность. При этом невирусные системы доставки обладают рядом преимуществ, такими как высокая пакующая емкость, низкая иммуногенность, высокая безопасность, возможность экономичного производства (Yue Y. and Wu С. (2013) Biomaterials Science 1:152-170).
В настоящее время в качестве невирусных систем доставки ДНК при генной терапии широкое применение находят поликатионы. Комплексы ДНК с поликатионами, или полиплексы, способны компактизовать ДНК и защищать ее от воздействия внешних факторов. В силу особенностей химической структуры полиплексы предоставляют возможность модификации, в частности, путем ковалентного присоединения молекул, обеспечивающих направленное (таргетное) взаимодействие с мишенью. Полиплексы способны преодолевать эндосомные мембраны и выходить в цитозоль, они непатогенны, и практически неиммуногенны, и нетоксичны. Относительно недорогая стоимость производства делает их весьма привлекательными для фармакологических компаний, а воспроизводимость производства полиплексов с ковалентно присоединенными лигандами достаточно высока (Kang Н.С., Lee Μ., et al. (2005) Crit. Rev. Eukaryot. Gene Expr. 15:317-42). Полиплексы можно использовать как для системного, так и для локального введения - интратуморально в легкие, различные опухоли, мозг, молочные железы и яйцеводы. Полиплексы оказались эффективными для генотерапии гепатоцеллюлярной карциномы (Iwai Μ, Harada Y et al. (2002) Biochem. Biophys. Res. Commun. 291:48-54) и диссеминированного рака поджелудочной железы (Aoki К, Furuhata S et al. (2001) Gene Ther. 8:508-14).
Указанный подход комбинированной ГНЭПТ - иммунной терапии с использованием поликатионной системы доставки был успешно реализован, например, в фармацевтической композиции, описанной в RU 2 575 077. Фармацевтическая композиция включает следующие активные вещества: 1) нуклеиновую кислоту, кодирующую два терапевтических гена (ген-убийца и иммуномодулирующий ген) и регуляторные элементы, необходимые для их экспрессии; 2) полимерный поликатионный носитель, который состоит из трех компонентов - полиэтиленгликоля (ПЭГ), полиэтиленимина (ПЭИ) и ТАТ-пептида (ТАТ). Нуклеиновая кислота представляет собой высокоочищенную сверхскрученную форму плазмиды общей формулы pCMV-HSVtk-hGM-CSF и кодирует тимидинкиназу вируса простого герпеса (HSVtk) и гранулоцитарно-макрофагальный колониестимулирующий фактор человека (GM-CSF) под контролем промотора (управляющего участка ДНК) цитомегаловируса (CMV). Поликатионный носитель представляет собой блок-сополимер (БС) ПЭИ и ПЭГ с коньюгированным ТАТ-пептидом (все вместе ППТ). ТАТ-пептид, характеризующийся последовательностью GRKKKRRQRC, представляет собой фрагмент ТАТ-белка и повышает способность блок-сополимера, в состав которого он входит, проникать в клетки и обеспечивать доставку плазмиды в клетки опухоли (Ulasov AV, Khramtsov YV et al. (2011) Mol. Ther. 19:103-12; Rudolph C, Plank С et al. (2003) J. Biol. Chem. 278:11411-8).
Рекомбинантная плазмидная ДНК, описанная в RU 2 575 077, состоит из следующих компонентов: фрагмента промоторного участка (581 нуклеотидная пара), который определяет транскрипцию генов; гена HSVtk, при экспрессии которого синтезируется форма HSVtk, состоящая из 374 аминокислотных остатков; участка внутренней посадки рибосомы (IRES), обеспечивающего экспрессию обоих терапевтических генов с одного вектора (Mizuguchi, Xu et al. (2000) Mol. Ther. 1:376-82); гена hGM-CSF или mGM-CSF, при экспрессии которого синтезируется форма hGM-CSF (человеческая) или mGM-CSF (мышиная), соответственно; сигнала полиаденилирования и терминатора транскрипции SV40, обеспечивающих синтез зрелой РНК генов. Поскольку белок гранулоцитарно-макрофагального колониестимулирующего фактора является видоспецифичным (Shi Y, Liu СН et al. (2006) Cell Res. 16:126-33), а для тестирования эффективности генотерапевтических конструкций наиболее часто применяют животных с привитыми опухолями (ксенотрансплантаты), в RU 2 575 077 описаны два варианта конструкции, один из которых содержит ген GM-CSF мыши и предназначен для модельных экспериментов на мышах, а другой содержит ген GM-CSF человека и предназначен для проведения клинических испытаний и непосредственного использования на пациентах. Кроме того рекомбинантная плазмидная ДНК, описанная в RU 2 575 077, дополнительно содержит вспомогательные области, необходимые для эффективного биосинтеза плазмидной ДНК в промышленном масштабе в клетках штамма-продуцента E. coli. К указанным вспомогательным областям относятся, в частности, f1 ori, ColE1 ori, селективный маркер устойчивости к ампициллину AmpR.
При проникновении молекул плазмиды pCMV-HSVtk-hGM-CSF внутрь опухолевых клеток происходит синтез фермента HSVtk, способного фосфорилировать нуклеозидный аналог ганцикловир, вводимый системно, до ганцикловирмонофосфата, который затем фосфорилируется эндогенными клеточными киназами до токсичной формы - ганцикловиртрифосфата. Ганцикловиртрифосфат при клеточном делении включается во вновь синтезированную цепь ДНК и обрывает ее дальнейший синтез, вызывая гибель клетки. Также при проникновении молекул плазмиды внутрь опухолевых клеток происходит синтез белка GM-CSF, который секретируется во внеклеточное пространство и стимулирует рост, развитие и дифференцировку гранулоцитов и антиген-презентирующих клеток с привлечением их к зоне роста опухоли, обеспечивая индукцию специфичного противоопухолевого иммунного ответа. Механизм индукции противоопухолевого иммунитета с участием GM-CSF включает прямое вовлечение натуральных киллеров и антиген-презентирующих клеток, например, дендритных клеток в зоне роста опухоли, что приводит к активации Т-клеток. Таким образом, за счет работы тимидинкиназы вируса простого герпеса вводимый ганцикловир вызывает гибель опухолевых клеток, из которых высвобождаются опухолевые антигены, которые антиген-презентирующие клетки, активированные посредством GM-CSF, представляют Т-клеткам иммунной системы. Активация адаптивного иммунного ответа, вызываемого препаратом, может обеспечивать локальное и системное иммуноопосредованное разрушение отдаленных опухолей, тем самым обеспечивая дополнительный эффект «противоопухолевой вакцинации», и приводить к развитию иммунологической памяти, а также к долгосрочному контролю роста опухоли с помощью иммунного надзора.
Фармацевтическая композиция, описанная в RU 2 575 077, содержит следующие вспомогательные вещества: 5 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота), рН 7,4, 12,5 мМ боратный буфер, рН 7,5, и 5% глюкозу.
В модельных экспериментах на мышах в RU 2 575 077 показана высокая эффективность комбинированной системы ГНЭПТ - иммунной терапии, основанной на использовании плазмиды pCMV-HSVtk-GM-CSF в сочетании с системным введением ганцикловира, против широкого спектра солидных злокачественных новообразований. В частности, эффективность указанной системы продемонстрирована в отношении саркомы, аденокарциномы толстой кишки, рака шейки матки, карциномы гортани, фибросаркомы.
Одной из основных проблем при использовании генотерапевтических препаратов в медицине остается их стабильность при хранении. Как уже упоминалось выше, используемые в клинической практике генотерапевтические препараты можно разделить по способу доставки на вирусные и невирусные. Хотя невирусные генотерапевтические препараты обладают целым рядом преимуществ по сравнению с вирусными, таких как низкие токсичность, патогенность и иммуногенность, меньшая стоимость производства, большая простота в изготовлении и/или модификации определённым образом для достижения высокой специфичности, и те, и другие препараты не стабильны при хранении. Стабильность таких препаратов достигается только при отрицательных температурах и зачастую требует хранения при -70°С, при этом нарушение температуры хранения может приводить к полной потере активности препарата.
Готовые лекарственные формы генотерапевтических препаратов должны отвечать определенным требованиям, в частности:
1) процесс производства должен быть технологичным;
2) готовая лекарственная форма должна обладать стабильностью при хранении и транспортировке;
3) желательно, чтобы срок хранения был продолжительным;
4) готовая лекарственная форма должна характеризоваться удобством подготовки к применению и удобством самого применения.
Один из подходов при разработке лекарственных форм для генной терапии состоит в получении лиофилизированных препаратов. Так, в WO 2009/021017 описана успешная разработка лиофилизированной композиции, содержащей вспомогательное вещество - наполнитель, нуклеиновую кислоту и катионный липополимер Указанный липополимер содержит каркас на основе катионного полимера с присоединенными к нему ковалентно молекулами холестерола и молекулами полиэтиленгликоля. Однако данный подход, как правило, не подходит для лиофилизации комплексов ДНК с синтетическими векторами, поскольку при использовании данного подхода наблюдается изменение физико-химических свойств, что приводит к агрегации и потере способности к трансфекции при восстановлении лиофилизированной композиции при помощи растворителя. Применение криопротекторов, в частности, таких как различные сахара, декстраны или полиэтиленгликоль, может приводить к улучшению стабильности, однако достигаемый эффект варьирует и зависит не только от конкретно используемого криопротектора, но и от типа используемого синтетического вектора.
Еще одним немаловажным фактором, влияющим на возможность использования лиофилизации для получения лекарственных форм для генной терапии, является то, что при использовании даже тех сахаров, которые являются наиболее эффективными лиопротекторами, требуется очень высокое мольное соотношение сахар/ДНК (обычно выше 1000:1) для обеспечения стабильности. В результате лиофилизированный препарат зачастую требует очень высокой степени разведения при восстановлении для достижения приемлемой осмолярности (в идеале - изотоничности), что приводит к драматическому уменьшению концентрации ДНК в восстановленном препарате по сравнению с исходной композицией, подвергаемой лиофилизации. Для многих препаратов, в которых в качестве носителя используются катионные векторы, конечная концентрация ДНК может составлять приблизительно 0,1-0,2 мг/мл и ниже, что создает определенные трудности при клиническом применении, поскольку для введения оптимальных доз могут потребоваться такие объемы, которые несовместимы с местным путем введения. В WO 2009/021017 сообщается, что указанное фармацевтическое ограничение является одним из основных факторов, обусловливающих субоптимальную эффективность синтетических систем доставки генов при клинических испытаниях на людях.
Таким образом, в данной области техники существует потребность в разработке новых лекарственных форм генотерапевтических препаратов, в частности, лекарственного препарата для комбинированной ГНЭПТ - иммунной терапии на основе терапевтической плазмиды pCMV-HSVtk-GM-CSF, сочетающих в себе возможность длительного хранения в относительно нестрогих условиях, технологичность процесса производства, удобство подготовки препарата к введению субъекту, нуждающемуся в лечении, и удобство самого введения.
Раскрытие сущности изобретения
Настоящее изобретение относится к двухкомпонентному лекарственному препарату для комбинированной ген-направленной энзиматической пролекарственной терапии - иммунной терапии солидных злокачественных новообразований, причем готовый раствор для инъекций, предназначенный для введения пациенту, получают в результате смешивания первого компонента со вторым компонентом. Первый компонент представляет собой водный раствор, хранящийся до использования в замороженном виде, который содержит в качестве активного вещества сверхскрученную плазмидную ДНК pCMV-HSVtk-hGM-CSF, полученную биосинтезом в клетках E. coli и обладающую непосредственно лечебным эффектом. Первый компонент содержит активное вещество в терапевтически эффективном количестве. Второй компонент представляет собой водный раствор, хранящийся до использования в замороженном виде, который содержит в качестве активного вещества химически синтезированный в водной фазе ковалентносвязанный блок-сополимер полиэтиленимин-полиэтиленгликоль-ТАТ пептид (БС ППТ), который обеспечивает проникновение плазмидной ДНК в клетку. Второй компонент содержит активное вещество в терапевтически эффективном количестве.
Настоящее изобретение также относится к двухкомпонентному лекарственному препарату для комбинированной ген-направленной энзиматической пролекарственной терапии - иммунной терапии солидных злокачественных новообразований, причем готовый раствор для инъекций, предназначенный для введения пациенту, получают в результате восстановления первого компонента путем добавления воды и последующего смешивания со вторым компонентом. Первый компонент представляет собой лиофилизат, хранящийся при температуре не выше 4°С, полученный из водного раствора, содержащего в качестве активного вещества сверхскрученную плазмидную ДНК pCMV-HSVtk-hGM-CSF, полученную биосинтезом в клетках E. coli и обладающую непосредственно лечебным эффектом. Первый компонент содержит активное вещество в терапевтически эффективном количестве. Второй компонент представляет собой водный раствор, хранящийся до использования в замороженном виде, который содержит в качестве активного вещества химически синтезированный в водной фазе ковалентносвязанный БС ППТ, который обеспечивает проникновение плазмидной ДНК в клетку. Второй компонент содержит активное вещество в терапевтически эффективном количестве.
В обоих вариантах лекарственного препарата готовый раствордля инъекций, предназначенный для введения пациенту, готовят ex tempore, т.е. непосредственно перед введением субъекту, нуждающемуся в лечении солидного злокачественного новообразования.
В основе механизма действия заявленных лекарственных препаратов лежит доставка терапевтических генов HSVtk и hGM-CSF в опухолевые клетки с помощью невирусного вектора, представляющего собой блок-сополимер ПЭГ-ПЭИ-ТАТ (ППТ), посредством инъекции, предпочтительно - путем интратуморального введения с целью обеспечения продукции фермента HSVtk и цитокина hGM-CSF.
Технический результат, обеспечиваемый изобретением, состоит, в частности, в том, что предложенные лекарственные формы демонстрируют высокую стабильность и сохраняют терапевтическую эффективность на протяжение по крайней мере 18 месяцев.
Таким образом, в своем первом аспекте заявленное изобретение относится к лекарственному препарату для лечения солидных злокачественных новообразований методом комбинированной ген-направленной энзиматической пролекарственной терапии - иммунной терапии, содержащему две герметически укупоренные емкости, причем:
первая емкость содержит первый компонент, содержащий сверхскрученную плазмидную ДНК pCMV-HSVtk-hGM-CSF в терапевтически эффективном количестве, по меньшей мере одну фармацевтически приемлемую добавку и воду,
вторая емкость содержит второй компонент, содержащий:
блок-сополимер ПЭГ-ПЭИ-ТАТ пептид в терапевтически эффективном количестве, по меньшей мере одну фармацевтически приемлемую добавку и воду,
причем содержимое каждой из емкостей является стерильным, и причем емкости хранят при температуре не выше -18°С.
В одном из воплощений лекарственный препарат характеризуется тем, что:
первый компонент содержит:
сверхскрученную плазмидную ДНК pCMV-HSVtk-hGM-CSF - приблизительно 150 - 170 мкг,
декстрозу - приблизительно 100 мг,
HEPES - приблизительно 2,38 мг,
воду - до 1,0 мл,
второй компонент содержит:
блок-сополимер ПЭГ-ПЭИ-ТАТ пептид - приблизительно 0,63 - 0,74 мг (в расчете на ПЭИ),
тетраборат натрия - приблизительно 2,38 мг,
воду - до 1,0 мл.
В одном из воплощений лекарственного препарата готовый раствор для инъекций получают путем размораживания содержимого указанных первой и второй емкостей и их смешивания.
В еще одном из воплощений лекарственного препарата готовый раствор для инъекций вводят интратуморально.
В следующем воплощении солидные злокачественные новообразования выбраны из группы, включающей: злокачественные новообразования легкого, злокачественные новообразования поджелудочной железы, меланому, фибросаркому, саркому, злокачественные новообразования головы и шеи.
В одном из воплощений лекарственного препарата блок-сополимер ПЭГ-ПЭИ-ТАТ пептид характеризуется мольным соотношением ПЭГ/ПЭИ приблизительно от 1,0 до 3,0 и мольным соотношением ТАТ/ПЭИ приблизительно от 0,8 до 1,5. В предпочтительном воплощении блок-сополимер ПЭГ-ПЭИ-ТАТ пептид характеризуется мольным соотношением ПЭГ/ПЭИ приблизительно от 2,0 до 2,5.
В еще одном из воплощений лекарственный препарат характеризуется тем, что:
первый компонент содержит:
сверхскрученную плазмидную ДНК pCMV-HSVtk-hGM-CSF - приблизительно 160 мкг,
декстрозу - приблизительно 100 мг,
HEPES - приблизительно 2,38 мг,
воду - до 1,0 мл,
а второй компонент содержит:
блок-сополимер ПЭГ-ПЭИ-ТАТ пептид - приблизительно 0,69 мг (в расчете на ПЭИ);
тетраборат натрия - приблизительно 2,38 мг,
воду - до 1,0 мл,
причем блок-сополимер ПЭГ-ПЭИ-ТАТ пептид характеризуется мольным соотношением ПЭГ/ПЭИ приблизительно 2,0.
В своем втором аспекте настоящее изобретение относится к лекарственному препарату для лечения солидных злокачественных новообразований методом комбинированной ген-направленной энзиматической пролекарственной терапии - иммунной терапии, содержащему две герметически укупоренные емкости, причем:
первая емкость содержит первый компонент, представляющий собой лиофилизат, содержащий сверхскрученную плазмидную ДНК pCMV-HSVtk-hGM-CSF в терапевтически эффективном количестве и по меньшей мере одну фармацевтически приемлемую добавку,
а вторая емкость содержит второй компонент, содержащий блок-сополимер ПЭГ-ПЭИ-ТАТ пептид в терапевтически эффективном количестве, по меньшей мере одну фармацевтически приемлемую добавку и воду,
причем содержимое каждой из емкостей является стерильным, причем первую емкость хранят при температуре не выше 4°С, а вторую емкость хранят при температуре не выше -18°С.
В одном из воплощений лекарственный препарат характеризуется тем, что: первый компонент, представляющий собой лиофилизат, получен путем лиофилизации раствора, содержащего:
сверхскрученную плазмидную ДНК pCMV-HSVtk-hGM-CSF - приблизительно 150 - 170 мкг,
декстрозу- приблизительно 100 мг,
HEPES - приблизительно 2,38 мг,
воду - до 1,0 мл,
второй компонент содержит:
блок-сополимер ПЭГ-ПЭИ-ТАТ пептид - приблизительно 0,63 - 0,74 мг (в расчете на ПЭИ);
тетраборат натрия - приблизительно 2,38 мг,
воду - до 1,0 мл.
В одном из воплощений лекарственного препарата готовый раствор для инъекций получают путем восстановления содержимого первой емкости путем добавления 1,0 мл воды, размораживания содержимого второй емкости и их смешивания.
В следующем воплощении лекарственного препарата готовый раствор для инъекций вводят интратуморально.
В еще одном из воплощений солидные злокачественные новообразования выбраны из группы, включающей: злокачественные новообразования легкого, злокачественные новообразования поджелудочной железы, меланому, фибросаркому, саркому, злокачественные новообразования головы и шеи.
В следующем воплощении лекарственного препарата блок-сополимер ПЭГ-ПЭИ-ТАТ пептид характеризуется мольным соотношением ПЭГ/ПЭИ приблизительно от 1,0 до 3,0 и мольным соотношением ТАТ/ПЭИ приблизительно от 0,8 до 1,5. В предпочтительном воплощении блок-сополимер ПЭГ-ПЭИ-ТАТ пептид характеризуется мольным соотношением ПЭГ/ПЭИ приблизительно от 2,0 до 2,5.
В еще одном из воплощений лекарственный препарат характеризуется тем, что:
первый компонент, представляющий собой лиофилизат, получен путем лиофилизации раствора, содержащего:
сверхскрученную плазмидную ДНК pCMV-HSVtk-hGM-CSF - приблизительно 160 мкг,
декстрозу - приблизительно 100 мг,
HEPES - приблизительно 2,38 мг,
воду - до 1,0 мл,
а второйкомпонент содержит:
блок-сополимер ПЭГ-ПЭИ-ТАТ пептид - приблизительно 0,69 мг (в расчете на ПЭИ);
тетраборат натрия - приблизительно 2,38 мг,
воду - до 1,0 мл,
причем блок-сополимер ПЭГ-ПЭИ-ТАТ пептид характеризуется мольным соотношением ПЭГ/ПЭИ приблизительно 2,0.
Краткое описание чертежей
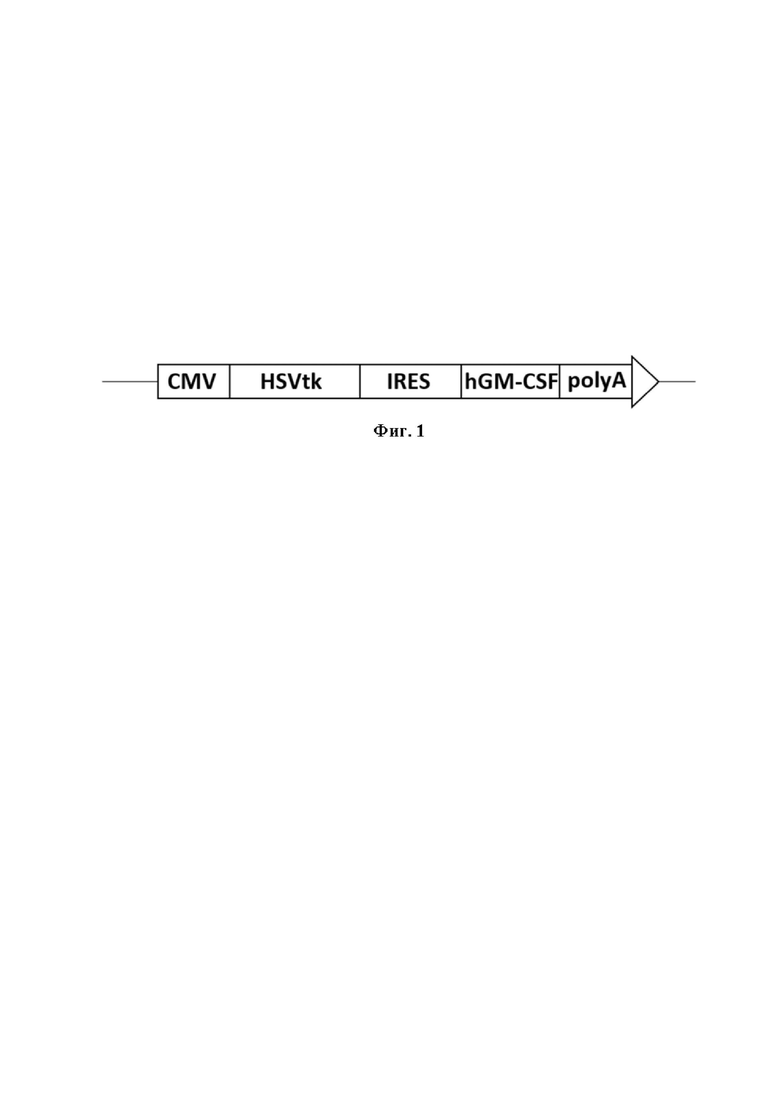
Фиг. 1 - Cтруктура экспрессионной кассеты cверхскрученной плазмидной ДНК pCMV-HSVtk-hGM-CSF. CMV - промотор цитомегаловируса, HSVtk - ген тимидинкиназы вируса простого герпеса, hGM-CSF - ген гранулоцитарно-макрофагального колониестимулирующего фактора человека, IRES - сайт внутренней посадки рибосом вируса энцефаломиокардита, polyA SV40 - сигнал полиаденилирования вируса SV40.
Осуществление изобретения
В многочисленных предварительных экспериментах изобретатели настоящего изобретения исследовали различные лекарственные формы лекарственного препарата для комбинированной ген-направленной энзиматической пролекарственной терапии - иммунной терапии солидных злокачественных новообразований. Исследования включали как жидкие (в виде однокомпонентных или двухкомпонентных систем), так и лиофилизированные (как полностью, так и частично) лекарственные формы. На основании результатов проведенных исследований изобретатели пришли к заключению о необходимости использования двукомпонентных систем, поскольку однокомпонентные системы (как жидкие, так и лиофилизированные) не обеспечивали необходимой стабильности лекарственного препарата. Кроме того, использование лиофилизации в отношении одного из активных веществ лекарственного препарата, а именно, ППТ, неизбежно приводило к неудовлетворительным результатам. Вследствие этого для целей дальнейшей разработки лекарственных форм лекарственного препарата для комбинированной ген-направленной энзиматической пролекарственной терапии - иммунной терапии солидных злокачественных новообразований изобретатели остановили свой выбор на двухкомпонентных системах, в которых первый компонент, содержащий в качестве активного вещества терапевтическую плазмиду, может представлять собой либо замороженный раствор, либо лиофилизат, в то время как второй компонент, содержащий в качестве активного вещества ППТ, должен в обязательном порядке представлять собой замороженный раствор.
Таким образом, в первом варианте осуществления изобретения лекарственный препарат для комбинированной ГНЭПТ - иммунной терапии солидных злокачественных новообразований содержит две герметически укупоренные емкости, причем содержимое первой емкости (первый компонент) представляет собой замороженный раствор, содержащий в качестве активного вещества высокоочищенную сверхскрученную плазмидную ДНК (плДНК) pCMV-HSVtk-hGM-CSF.
Содержимое второй емкости (второй компонент) представляет собой замороженный раствор, содержащий в качестве активного вещества ППТ. Блок-сополимер ППТ - химически синтезированный в водной фазе ковалентносвязанный сополимер, в состав которого входят: ПЭГ - модифицированный полиэтиленгликоль MAL-dPEG®₂₄-NHS эфир, ПЭИ - полиэтиленимин линейный 25000 и ТАТ-пептид. Готовый раствор для инъекций, предпочтительно осуществляемых интратуморально, получают ex tempore путем смешивания первого компонента со вторым компонентом после их предварительного размораживания. В результате объединения двух растворов происходит образование полиплексов (комплексов плазмидной ДНК pCMV-HSVtk-hGM-CSF с блок-сополимером ППТ), способных проникать в опухолевые клетки и индуцировать в них синтез двух терапевтических белков HSVtk и hGM-CSF.
Во втором варианте осуществления изобретения лекарственный препарат для комбинированной ГНЭПТ - иммунной терапии солидных злокачественных новообразований содержит две герметически укупоренные емкости, причем содержимое первой емкости (первый компонент) представляет собой лиофилизат, содержащий в качестве активного вещества высокоочищенную сверхскрученную плазмидную ДНК (плДНК) pCMV-HSVtk-hGM-CSF.
Содержимое второй емкости (второй компонент) представляет собой замороженный раствор, содержащий в качестве активного вещества ППТ.
Во втором варианте осуществления изобретения готовый раствор для инъекций, предпочтительно осуществляемых интратуморально, получают ex tempore путем восстановления первого компонента путем добавления воды до первоначального объема и последующего смешивания со вторым компонентом, после его предварительного размораживания. В результате объединения двух растворов происходит образование полиплексов (комплексов плазмидной ДНК pCMV-HSVtk-hGM-CSF с блок-сополимером ППТ), способных проникать в опухолевые клетки и индуцировать в них синтез двух терапевтических белков HSVtk и hGM-CSF.
Структура и свойства плазмидной ДНК pCMV-HSVtk-hGM-CSF
Плазмида pCMV-HSVtk-hGM-CSF содержит экспрессионную кассету, состоящую из двух терапевтических генов - гена тимидинкиназы вируса простого герпеса HSVtk и гена гранулоцитарно-макрофагального колониестимулирующего фактора человека hGM-CSF, связанных через IRES-элемент, под контролем цитомегаловирусного промотора CMV (Фиг. 1).
Сверхскрученную плДНК pCMV-HSVtk-hGM-CSF получают биосинтезом в клетках-хозяевах Escherichia coli. В качестве клетки-хозяина может быть использован любой подходящий штамм E. coli, как хорошо известно специалисту в данной области техники. В настоящем изобретении предпочтительно используют штамм E. coli DH-5α, трансформированный плазмидой pCMV-HSVtk-hGM-CSF, конкретно, E. coli DH-5α/pCMV-HSVtk-hGM-CSF.
При проникновении молекул плазмиды pCMV-HSVtk-hGM-CSF внутрь опухолевых клеток происходит синтез фермента HSVtk, способного фосфорилировать нуклеозидный аналог ганцикловир, вводимый системно, до ганцикловирмонофосфата, который затем фосфорилируется клеточными киназами до токсичной формы - ганцикловиртрифосфата. Также при проникновении молекул плазмиды внутрь опухолевых клеток происходит синтез белка GM-CSF человека, который секретируется во внеклеточное пространство и стимулирует рост, развитие и дифференцировку гранулоцитов и антиген-представляющих клеток с привлечением их к зоне роста опухоли, обеспечивая индукцию специфичного противоопухолевого иммунного ответа.
В лекарственном препарате настоящего изобретения сверхскрученная плазмидная ДНК pCMV-HSVtk-hGM-CSF содержится в количестве приблизительно 150 - 170 мкг.
Структура и свойства блок-сополимера ППТ (БС ППТ)
При получении БС ППТ в настоящем изобретении используют модифицированный полиэтиленгликоль MAL-dPEG®24-NHS эфир (ПЭГ):
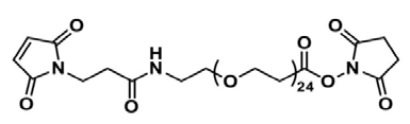 .
.
Эмпирическая формула: C62H111N3O31, молекулярная масса: 1394,55 Да.
При получении БС ППТ в настоящем изобретении используют полиэтиленимин линейный 25000 (ПЭИ):
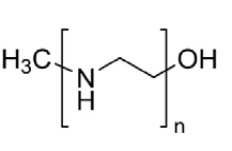 .
.
Эмпирическая формула (CH2CH2NH)n, молекулярная масса: 25000 Да.
При получении БС ППТ в настоящем изобретении используют ТАТ-пептид (ТАТ), имеющий следующую аминокислотную последовательность:
Gly-Arg-Lys-Lys-Lys-Arg-Arg-Gln-Arg-Cys.
Эмпирическая формула: C52H102N26O12S1, молекулярная масса: 1315,6 Да.
Блок-сополимер ППТ представляет собой химически синтезированный в водной фазе высокомолекулярный биополимер, положительно заряженный при рН 7,4 - 8,3. Основу биополимера составляют молекулы линейного ПЭИ с молекулярной массой 25 кДа. ПЭИ соединяет в себе высокую способность к дестабилизации клеточных мембран с высокой способностью конденсировать ДНК, предохраняя ее от ферментативного разрушения и увеличивая вероятность попадания интактной ДНК в ядро. К первичным аминам ПЭИ с помощью химической реакции ковалентно присоединены молекулы ПЭГ с молекулярной массой приблизительно 1395 Да, а к ПЭГ присоединен ТАТ-пептид. Модификация ПЭИ с помощью ПЭГ обеспечивает нейтральный поверхностный заряд, уменьшая тем самым агрегацию частиц, снижает токсическое действие биополимера, повышает гидрофильность и эффективность проникновения плДНК в ядра трансфицируемых опухолевых клеток. TAT-пептид, несущий сигнал ядерной локализации, обеспечивает транспорт полиплекса (комплекс плазмидной ДНК и ППТ) в ядро независимо от стадии клеточного цикла, способствует проникновению всей конструкции в клетку.
В водных растворах положительно заряженные молекулы блок-сополимера ППТ образуют нековалентные сферические комплексы (полиплексы) с молекулами отрицательно заряженной плДНК за счет электростатического взаимодействия. Эти комплексы взаимодействуют с клеточной мембраной и впоследствии поглощаются путем эндоцитоза. Комплекс ППТ-плДНК в составе эндосомы способен буферизовать эндосомную везикулу, что приводит к ее набуханию и лизису, с последующим выходом плДНК в цитоплазму. Далее комплексы ППТ-плДНК по цитоскелетным путям транспортируются к ядру. В ядре полиплекс деконденсируется с отделением плДНК от ППТ. Гены, содержащиеся в плДНК pCMV-HSVtk-hGM-CSF, транскрибируются и транслируются, что приводит к синтезу в клетке двух терапевтических белков: тимидинкиназы вируса простого герпеса и гранулоцитарно-макрофагального колониестимулирующего фактора человека.
В предпочтительных воплощениях настоящего изобретения БС ППТ характеризуется мольным соотношением ПЭГ/ПЭИ приблизительно от 1,0 до 3,0 и мольным соотношением ТАТ/ПЭИ приблизительно от 0,8 до 1,5. В наиболее предпочтительных воплощениях настоящего изобретения мольное соотношение ПЭГ/ПЭИ составляет приблизительно от 2,0 до 2,5.
Для эффективной трансфекции опухолевых клеток экспериментально подобрано оптимальное соотношение молекул ППТ и плДНК в полиплексе, выражающееся в соотношении N (количество молекул азота) к Р (количество молекул фосфора), равное приблизительно от 20 до 40, предпочтительно - приблизительно 30. Для количественного определения ДНК проводят определение неорганического фосфора в соответствии с ГФ 14, ОФС.1.7.2.0018.15 (Определение нуклеиновых кислот по методу Спирина в биологических лекарственных препаратах).
В лекарственном препарате настоящего изобретения блок-сополимер ПЭГ-ПЭИ-ТАТ пептид содержится в количестве приблизительно 0,63 - 0,74 мг (в расчете на ПЭИ).
Фармацевтически приемлемые добавки.
В качестве по меньшей мере одной фармацевтически приемлемой добавки при изготовлении первого компонента, содержащего плазмидную ДНК pCMV-HSVtk-hGM-CSF, используют фармацевтически приемлемый буфер, который обеспечивает физиологически приемлемое значение рН раствора в диапазоне от 7,0 до 8,0. Предпочтительно в качестве буфера использовать буфер HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота), который в сравнении с другими буферными системами (фосфатные, карбонатные) значительно лучше поддерживает заданное значение рН при минимальной цитотоксичности. HEPES включен в Государственную фармакопею Российской Федерации XIV издания (ГФ 14) в качестве компонента буферного раствора (ОФС.1.3.0003.15 Буферные растворы, Буферный (HEPES) раствор рН 7,5, т. 1, стр. 1808). Согласно ГФ 14, буферные растворы применяются в том числе: (1) для достижения изотоничности при приготовлении жидких лекарственных форм; (2) для поддержания стабильности дозированных лекарственных форм (Государственная фармакопея Российской Федерации XIV издание, 2018).
Также для предотвращения потери трансфекционной активности ДНК перед замораживанием раствора или процессом лиофилизации к ДНК добавляют криопротекторы, такие как моно- и дисахариды, например лактозу, глюкозу или сахарозу. Предпочтительно для изготовления первого компонента, содержащего плазмидную ДНК pCMV-HSVtk-hGM-CSF, используют глюкозу (декстрозу). Глюкоза может быть использована как в безводной форме, так и в виде моногидрата, причем содержание глюкозы должно составлять в препарате приблизительно 100 мг в расчете на безводную форму.
В качестве по меньшей мере одной фармацевтически приемлемой добавки при изготовлении второго компонента, содержащего БС ППТ, используют фармацевтически приемлемый буфер, который обеспечивает физиологически приемлемое значение рН раствора в диапазоне от 7,1 до 8,2. Предпочтительно в качестве буфера используют боратный буфер, для изготовления которого применяют тетраборат натрия. Тетраборат натрия может быть представлен как безводной формой, так и любыми его известными кристаллогидратными формами, например, декагидратом (тетраборат натрия десятиводный). При этом содержание тетрабората натрия должно составлять в препарате приблизительно 2,38 мг в расчете на безводную форму.
Настоящее изобретение обеспечивает лекарственный препарат для лечения солидных злокачественных новообразований. Структура ткани солидных опухолей содержит взаимозависимые тканевые компартменты, включая паренхиму (раковые клетки) и поддерживающие стромальные клетки, которые могут обеспечивать поддерживающее микроокружение, среди которых распределены раковые клетки. Солидные злокачественные новообразования включают, но не ограничиваются перечисленным, саркомы, карциномы и лимфомы, включая рак легких, рак груди, рак простаты, рак толстого кишечника, рак прямой кишки, и рак мочевого пузыря. В некоторых воплощениях рак выбран из рака легкого, рака поджелудочной железы, меланомы, фибросаркомы, саркомы, рака головы и шеи (включая, например, плоскоклеточную карциному головы и шеи (HNSCC).
Ниже приведены примеры осуществления, которые служат для иллюстрации заявленного изобретения, но не предназначены для ограничения объема правовой охраны. Объем правовой охраны определяется исключительно прилагаемой формулой изобретения.
Пример 1.
Выделение, сбор и хранение бактериальной биомассы
Биомассу, т.е. осадок клеток, отделяют от культуральной жидкости в напольной высокоскоростной центрифуге периодического действия с угловым ротором. Для этого в стаканы центрифуги при помощи мерного цилиндра вносят порции по 800 мл культуральной суспензии. Стаканы поочередно взвешивают на весах, тарируют весы по массе самого тяжелого из 6 стаканов и поочередно доводят разницу в массе самого тяжелого заполненного стакана и остальных до (0,0±0,5) г внесением культуральной суспензии пастеровской пипеткой. Стаканы герметично закрывают крышками и помещают в ротор. Проводят центрифугирование в течение 15 мин при установленной скорости 10 тыс. об./мин. и температуре рабочей камеры центрифуги 4°C. Супернатант культуральной жидкости сливают в емкость и передают на обеззараживание автоклавированием. Стаканы с осадком повторно заполняют культуральной суспензией и повторяют центрифугирование. Во время последнего цикла центрифугирования для доведения массы стаканов при нехватке культуральной суспензии используют воду очищенную. По окончании последнего цикла центрифугирования стаканы ставят горловиной вниз на лист фильтровальной бумаги и выдерживают (5±1) мин. для удаления остатков жидкости. Отбирают металлическим шпателем пробу биомассы на контроль подлинности методом электрофореза в агарозном геле. Полученную биомассу при помощи стального шпателя переносят в предварительно взвешенный на весах контейнер с крышкой, заполненный контейнер закрывают, повторно взвешивают, наклеивают этикетку установленного образца, помещают в защитный пакет полиэтиленовый с застежкой «Зип-лок» и хранят в низкотемпературном холодильнике при температуре ниже минус 65°C.
Пример 2. Получение осветленного лизата бактериальной биомассы
Процедура получения осветленного лизата состоит из суспендирования бактериальных клеток, щелочного лизиса клеток и осветления лизата.
Получение осветленного лизата осуществляют за 3 цикла. Каждый цикл включает: суспендирование биомассы, лизис биомассы, и осветление лизата.
Суспендирование биомассы
На переработку 600 г биомассы требуется 3 цикла лизиса по 200 г биомассы каждый. Для ресуспендирования каждых 200 г биомассы расходуется 2 л раствора I. Раствор I включает трис-(гидроксиметил)-аминометан, этилендиаминтетраацетат натрия 2-замещенный, моногидрат глюкозы.
Контейнер с биомассой размораживают (при необходимости) при температуре +5±3°С до полной разморозки содержимого. Отвешивают 200 г размороженной биомассы на весах,
добавляют 2,0 л раствора I, ставят перемешивать на магнитную мешалку до получения однородной суспензии без видимых сгустков клеток
Лизис клеток
Для лизиса каждых 200 г биомассы суспензии клеток, полученной на стадии суспендирования, добавляют 2 л раствора II. В емкость с завинчивающейся крышкой переливают полученную суспензию клеток и вливают 2,0 л раствора II, перемешивают переворачиванием 1 минуту, инкубируют раствор 5 минут.
Раствор II готовится следующим образом. В ёмкость наливают 1,5 л ВДИ, вносят 16,00 гидроксида натрия 20,00 г додецилсульфата натрия, перемешивают с помощью магнитной мешалки до полного растворения реактивов. Доводят объём раствор до 2,0 л водой для инъекций (ВДИ).
Осветление лизата
Для осветления лизата из 200 г биомассы расходуют 2 л раствора III.
В ёмкость с лизатом, полученным на стадии лизиса, вливают 2,0 л раствора III и перемешивают интенсивным переворачиванием несколько раз. Раствор III готовят следующим образом. В ёмкость наливают 0.9 л ВДИ, вносят 588,9 г ацетата калия, перемешивают с помощью магнитной мешалки до полного растворения реактивов. Доводят pH до значения 5,5 внесением 0,3 л раствора уксусной кислоты. Доводят объём раствор до 2,0 л ВДИ. Фильтруют буферный раствор в ёмкость, используя фильтр. Затем инкубируют раствор 10 минут. Клеточный дебрис убирают центрифугированием при 15 000 об./мин в течение 30 мин или фильтрованием через плотный бумажный фильтр. Осветленный клеточный лизат концентрируют с помощью ультрафильтрации с использованием стекловолоконного фильтра.
Пример 3. Выделение плазмидной ДНК из биомассы
В качестве первичной хроматографической очистки сверхскрученной плазмидной ДНК используют стадию гель-фильтрационной хроматографии, которая позволяет избавить раствор плазмиды от примеси рибонуклеиновой кислоты. Очистку проводят на колонне, заполненной сорбентом Sepharose 6FF, с помощью хроматографической системы Akta Pilot. Для получения необходимого количества конечного продукта и очистки всего объема сконцентрированного осветленного лизата требуется приблизительно 4 цикла гель-фильтрационной очистки: по 2 цикла на каждый цикл ультрафильтрации. После каждого цикла проводят переуравновешивание сорбента, а каждые два цикла проводят его регенерацию с последующим переуравновешиванием.
Для очистки сверхскрученной кольцевой формы целевой ДНК от релаксированной используется стадия аффинной хроматографии. Очистку проводят на колонне, заполненной сорбентом Plasmid Select Xtra, с помощью хроматографической системы Akta Pilot.
Для очистки целевой объединенной фракции, полученной с одной стадии гельфильтрации, требуется 2-3 цикла очистки на аффинном сорбенте. Для очистки всего объема фракции пДНК со стадии гель-фильтрации и получения необходимого количества целевого вещества суммарно требуется приблизительно 6 циклов аффинной хроматографии. После каждого цикла проводят регенерацию и переуравновешивание сорбента.
Для окончательной очистки сверхскрученной кольцевой формы ДНК от примесей используется стадия анионообменной хроматографии. Очистку проводят на колонне, заполненной сорбентом Source 30Q, с помощью хроматографической системы Akta Pilot. После каждого цикла проводят регенерацию, перед каждым циклом сорбент депирогенизируют и переуравновешивают. Для переработки всего объема целевого продукта требуется приблизительно 2 цикла хроматографической очистки.
Пример 4. Розлив во флаконы и укупорка раствора дезоксирибонуклеиновой кислоты плазмидной со вспомогательными веществами (первый компонент)
Производят розлив препарата в соответствии с рассчитанной дозой розлива (100-102,5%) в помещении класса чистоты В зоне А при включенном ламинарном потоке воздуха во флаконы 2R (в кассетах) из светлого стекла первого гидролитического класса, которые поступают в помещение розлива и укупорки через проходной сухожаровой шкаф. На первых (3±1) флаконах устанавливают дозу (объем) розлива. Розлив ведут посредством перистальтического дозирующего насоса и стерильного силиконового шланга для розлива с внутренним диаметром 3,2 мм. Контроль дозы осуществляют по массе в начале, середине, конце розлива. Содержимое первых 3-х флаконов (соответствует примерно 0,0036 л), заполненных не полностью или с ошибкой дозы, передают на утилизацию или переработку. Наполнение проверяют весовым методом, не менее 5 раз за время работы. Проводят розлив 0,594 л раствора. В линии (шлангах) остаётся 0,015 л раствора, который сливают в стерильный сборник и затем передают на утилизацию или переработку.
Пример 5. Розлив во флаконы и укупорка раствора Блок-сополимер ППТ со вспомогательными веществами (второй компонент)
Производят розлив препарата в соответствии с рассчитанной дозой розлива (100-102,5%) в помещении класса чистоты В зоне А при включенном ламинарном потоке воздуха во флаконы 2R (в кассетах) из светлого стекла первого гидролитического класса, которые поступают в помещение розлива и укупорки через проходной сухожаровой шкаф На первых (3±1) флаконах устанавливают дозу (объем) розлива 1,1-1,12. Розлив ведут посредством перистальтического дозирующего насоса и стерильного силиконового шланга для розлива с внутренним диаметром 3,2 мм. Контроль дозы осуществляют по массе в начале, середине, конце розлива.
Содержимое первых 3-х флаконов (соответствует примерно 0,0033 л), заполненных не полностью или с ошибкой дозы, передают на утилизацию или переработку. Наполнение проверяют весовым методом, не менее 5 раз за время работы.
На протяжении всего розлива флакон мгновенно накрывают пробкой.
Во время работы следят за качеством укупорки флаконов пробками (визуально).
В линии (шлангах) остаётся 0,0015 л раствора, который сливают в стерильный сборник и затем передают на утилизацию или переработку.
Наполненные и укупоренные флаконы помещают в кассету и передают на стадию обкатки флаконов флипами.
Пример 6. Лекарственный препарат для комбинированной ген-направленной энзиматической пролекарственной терапии - иммунной терапии солидных злокачественных новообразований, 2 мл (вариант 1)
При производстве лекарственного препарата используют сырье, прошедшее входной контроль на соответствие нормативно-технической документации:
- субстанцию Дезоксирибонуклеиновая кислота плазмидная
- субстанцию Блок-сополимер ППТ
- Тетраборат натрия десятиводный
- HEPES 1М раствор
- Декстрозы моногидрат 40% раствор (в расчете на безводную глюкозу).
Первый компонент - раствор плДНК pCMV-HSVtk-hGM-CSF для приготовления раствора для инъекций, 1 мл.
Действующее вещество: Дезоксирибонуклеиновая кислота плазмидная 160,0 мкг
Вспомогательные вещества: Декстроза 100 мг
HEPES 2,38 мг
Вода для инъекций до 1,0 мл
Описание. Раствор замороженный. Представляет собой плотную затвердевшую массу беловатого цвета. После размораживания: бесцветный прозрачный раствор без видимых включений.
Хранение. При температуре не выше минус 18°С. Не допускается хранение размороженного препарата. Повторное замораживание не допускается.
Второй компонент - раствор блок-сополимера полиэтиленгликоль-полиэтиленимин-ТАТ-пептид, для приготовления раствора для инъекций, 1 мл
Действующее вещество: Блок-сополимер ППТ 0,69 мг
Вспомогательные вещества: Тетраборат натрия 2,38 мг
Вода для инъекций до 1,0 мл
Описание. Раствор замороженный. Представляет собой плотную затвердевшую массу беловатого цвета. После размораживания: бесцветный прозрачный раствор без видимых включений.
Хранение. При температуре не выше минус 18°С. Не допускается хранение размороженного препарата. Повторное замораживание не допускается.
Приготовление готовой лекарственной формы препарата (вариант 1)
Для приготовления готовой лекарственной формы «раствор для инъекций» лекарственного препарата необходимо разморозить содержимое флаконов, содержащих первый и второй компоненты. Затем необходимо осторожно перемешать содержимое флаконов, совершая круговые движения и осторожно покачивая их. При приготовлении готовой лекарственной формы препарата следует избегать энергичного перемешивания или встряхивания. После размораживания и перемешивания содержимое флакона, содержащего первый компонент, переносят во флакон со вторым компонентом и перемешивают покачиванием. Смесь выдерживают при комнатной температуре 30 минут. Готовый препарат должен быть использован в течение 2 часов с момента приготовления. Следует соблюдать осторожность для обеспечения стерильности подготовленного раствора, так как препарат не содержит консервантов или бактериостатических веществ.
Пример 7. Получение лиофилизированной формы первого компонента, содержащего плазмидную ДНК.
Флаконы, содержащие первый компонент лекарственного препарата, как описано в Примере 6, подвергают лиофилизации в лиофильной сушке в соответствии с программой, приведенной в Таблице 1.
После окончания программы сушки флаконы герметично укупоривают в атмосфере аргона в лиофильной сушке. Флаконы хранят в защищенном от света месте при температуре не выше 4°С.
Пример 8. Лекарственный препарат для комбинированной ген-направленной энзиматической пролекарственной терапии - иммунной терапии солидных злокачественных новообразований, 2 мл (вариант 2)
Состав первого компонента, лимофилизированная плазмидная ДНК pCMV-HSVtk-hGM-CSF для приготовления раствора для инъекций
Получение первого компонента. Раствор плазмидной ДНК pCMV-HSVtk-hGM-CSF, полученный, как описано в Примере 3, подвергают лиофилизации, как описано в Примере 7.
Описание. Лиофилизат. Представляет собой массу беловатого цвета. После восстановления путем добавления 1,0 мл воды для инъекций: бесцветный прозрачный раствор без видимых включений.
Хранение. При температуре не выше 4°С
Состав второго компонента, раствор ППТ, 1 мл для приготовления раствора для инъекций
Приготовление готовой лекарственной формы препарата (вариант 2)
Для приготовления готовой лекарственной формы «раствор для инъекций» лекарственного препарата необходимо разморозить содержимое флакона, содержащего второй компонент. Во флакон с первым компонентом необходимо добавить 1,1 мл воды для инъекций, и осторожно перемешать до полного растворения лиофилизата. Затем необходимо осторожно перемешать содержимое флаконов, совершая круговые движения и осторожно покачивая их. При приготовлении готовой лекарственной формы препарата следует избегать энергичного перемешивания или встряхивания. После размораживания и перемешивания содержимое флакона, содержащего первый компонент, переносят во флакон со вторым компонентом и перемешивают покачиванием. Смесь выдерживают при комнатной температуре 30 минут. Готовый препарат должен быть использован в течение 2 часов с момента приготовления. Следует соблюдать осторожность для обеспечения стерильности подготовленного раствора, так как препарат не содержит консервантов или бактериостатических веществ.
Состав готовой лекарственной формы «раствор для инъекций» лекарственного препарата после смешивания первого и второго компонентов (вариант 2).
Описание. Прозрачная бесцветная жидкость без посторонних включений.
Пример 9. Цитотоксический тест для определения специфической активности препарата
Специфическая активность проверяется по образованию комплекса плазмидной ДНК pCMV-HSVtk-hGM-CSF (первый компонент) с блок-сополимером ППТ (второй компонент) в цитотоксическом тесте на культуре клеток линии С-26 (колоректальная аденокарцинома мыши CT26.WT, ATCC® CRL-2638™) in vitro. Метод оценки активности препарата основан на способности препарата вызывать гибель раковых клеток линии С-26, трансфицированных препаратом, в присутствии ганцикловира. Количество жизнеспособных клеток не должно быть более 50% или 20% при концентрации ганцикловира 2 мкМ или 12,5 мкМ в ростовой среде, соответственно.
Все работы выполняются в стерильных условиях под ламинарным потоком и с использованием стерильных пробирок и пипеток.
Клетки наращивают в питательной среде RPMI 1640 (Gibco, США). Клетки выращивают в СО2-инкубаторе при 37°С в атмосфере, содержащей 5% СО2. При достижении конфлюэнтности монослоя 90% (оценивают визуально) клетки пересевают в питательную среду RPMI1640/антибиотик в соотношении 1:10. Для теста используют клетки на 3 - 10 пассаже.
За 20 - 24 часа до теста клетки С-26 пересевают на 12-луночный планшет.
Проведение трансфекции клеток линии С-26.
В лунки 12-луночного планшета (например, Corning-353043, Corning, США) вносят 2 - 3×105 клеток в 2 мл среды RPMI 1640 (без антибиотика) и инкубируют 24 ч в СО2-инкубаторе. Перед добавлением растворов комплексов для трансфекции и испытуемого образца препарата среду из лунок планшета удаляют пипеткой, не касаясь дна и стенок лунки. После добавления растворов комплексов и испытуемого образца препарата планшет инкубируют 48 часов в СО2-инкубаторе.
Определение количества жизнеспособных клеток.
Вынимают планшет из СО2-инкубатора, отбирают пипеткой среду из всех лунок и промывают лунки 2 мл фосфатного буфера PBS. В каждую лунку добавляют по 0,2 мл раствора трипсин-ЭДТА и инкубируют 2 минуты в СО2-инкубаторе. Добавляют 1 мл среды RPMI 1640/антибиотик, ресуспендируют клетки пипеткой и переносят в стерильные микропробирки.
Определяют концентрацию жизнеспособных клеток с помощью трипанового синего.
Определение количества флуоресцирующих клеток.
Для определения эффективности трансфекции клеток линии С-26, трансфицированных с помощью Липофектамина 3000 и ППТ, содержащих индикаторную плазмиду pEGFP-N1, проводят подсчет трансфицированных клеток с использованием проточного цитофлуориметра. Пробу образцов Ккл и КGFP (клетки, трансфицированные комплексами pEGFP-N1 с Липофектамином и pEGFP-N1 с ППТ, соответственно) в количестве 50 мкл вносят в микропробирки, содержащие 50 мкл буфера для измерений, содержащего 10 мМ фосфатный буфер рН 7,4 с 2,5 % БСА и 0,1% азидом натрия.
Эффективность трансфекции рассчитывают как процент клеток с флуоресценцией, превышающей уровень аутофлуоресценции нетрансфицированных клеток.
После проведения трансфекции по 100 мкл суспензии трансфицированных и нетрансфицированных клеток вносят в две 96-луночные плашки. Плашки с клеточными суспензиями инкубируют 24 часа в СО2-инкубаторе. Далее готовят раствор ганцикловира в концентрации 0, 0,4, 4,0, 25,0 и 100,0 мкМ. После инкубации клеточных суспензий в лунки вносят по 100 мкл ганцикловира в различной концентрации.
Плашки с клеточными суспензиями инкубируют 72 часа в СО2-инкубаторе.
По окончании инкубации из лунок отбирают среду и вносят по 200 мкл ганцикловира в различной концентрации.
Плашки инкубируют в СО2-инкубаторе до достижения клеточного монослоя в лунках с концентрацией ганцикловира 0 мкМ. Оценивают визуально.
По окончании инкубации отбирают по 100 мкл среды из каждой лунки. Во все лунки вносят по 20 мкл MTS-реагента (Cell Titer 96® AQueous One Solution Cell Proliferation Assay (MTS), кат №: G3582, Promega). Содержимое лунок перемешивают на шейкере и инкубируют 30 мин в СО2-инкубаторе.
Измеряют оптическую плотность АN в лунках при длине волны 492 нм на планшетном фотометре. Рассчитывают значение оптической плотности AабсN для каждой лунки с учетом фонового значения по формуле
AабсN = АN - Афон
Рассчитывают среднее значение AабсNср для трех повторностей для каждого образца трансфицированных клеток линии С-26.
Рассчитывают % жизнеспособных клеток в разведениях ганцикловира для всех опытных и контрольных образцов трансфицированных клеток по формуле:
% жизн. клеток = AабсNср × СN × 100/ Aабс0ср, где
AабсNср - среднее значение оптической плотности из трех повторностей для каждого образца трансфицированных клеток при разных разведениях ганцикловира,
СN - концентрация в лунках ганцикловира,
Aабс0ср - среднее значение оптической плотности из трех повторностей для каждого образца трансфицированных клеток при концентрации ганцикловира 0.
Тест считается значимым, если:
- % жизнеспособных клеток в отрицательных контролях Ккл и КGFP и нетрансфицированных клетках превышает 90%,
Серия препарата считается пригодной для применения, если % жизнеспособных клеток в 3-х образцах препарата из серии не превышает 20 % в лунках с концентрацией ганцикловира 12,5 мкМ.
Пример 10. Определение характеристик ППТ (второй компонент)
Измерение содержания ПЭИ в полученном ППТ проводят спектрофотометрически, например, на планшетном ридере Synergy 4 (BioTek Instruments, США, или аналогичном) при длине волны 620 нм. Для построения калибровки и измерения концентраций конъюгатов в каждую ячейку 96-луночного планшета добавляют 50 мкл соответствующего образца ПЭИ или воды, 100 мкл 40 мМ Cu(CH3COO)2⋅H2O и 50 мкл воды. Измерение калибровочных растворов проводят в трёх повторах. Из полученного среднего значения оптической плотности вычитают оптическую плотность 20 мМ Cu(CH3COO)2⋅H2O. Поглощение образующегося комплекса с медью при 620 нм линейно зависит от концентрации ПЭИ (СПЭИ = а1⋅А620, где СПЭИ - концентрация ПЭИ в калибровочном образце в мкМ, а A620 - средняя оптическая плотность калибровочного образца за вычетом оптической плотности образца без ПЭИ, а1 - коэффициент пропорциональности). Концентрацию ППТ принимают равной концентрации ПЭИ в растворе ППТ.
Измерение содержания ПЭГ в полученном ППТ проводят спектрофотометрически, например, на планшетном ридере Synergy 4 (BioTek Instruments, США), или аналогичном, при длине волны 540 нм. Для этого используют свежеприготовленные калибровочные растворы ПЭГ с мол. массой 1500 Да: 200 мкМ, 160 мкМ, 120 мкМ, 80 мкМ, 40 мкМ и 20 мкМ ПЭГ, а также 5% раствор хлорида бария и раствор 12,3 г/л I2 в 2% KI. В лунки 96-луночного планшета добавляют по 10 мкл раствора ПЭГ с известной концентрацией или измеряемого образца (предварительно разведенного до концентрации 20 мкМ ПЭИ), по 10 мкл 20 мкМ раствора ПЭИ или воды для измеряемого неизвестного образца, по 180 мкл воды, по 50 мкл раствора хлорида бария и по 25 мкл раствора йода в йодиде калия и через 10-13 минут измеряют поглощение растворов при 540 нм. Время отсчитывают от момента добавления раствора йода. Измерения производят в трёх повторах. Поглощение при 540 нм линейно зависит от концентрации ПЭИ (СПЭГ/СПЭИ = а2⋅А540, где СПЭГ/СПЭИ - мольное соотношение ПЭГ/ПЭИ в калибровочном образце в мкМ, а A540 - средняя оптическая плотность калибровочного образца за вычетом оптической плотности образца без ПЭГ, а2 - коэффициент пропорциональности).
Измерение содержания ТАТ-пептида в полученном ППТ проводят флуориметрически по содержанию аддукта, образуемого аминогруппами ТАТ-петида в присутствии флуорескамина. Для этого готовят раствор флуорескамина в ацетоне (1 мг/мл) и 0,25 М фосфатный буфер, рН 9,3. Разводят 6 мг/мл раствор ТАТ-пептида до концентрации 100 мкМ. Готовят 3 мл смеси ТАТ-пептида с флуорескамином, так, чтобы конечная концентрация ТАТ-пептида была 0,5 мкМ: 15 мкл 100 мкМ раствора ТАТ-пептида, 60 мкл - раствора флуорескамина с концентрацией 1 мг/мл и 2925 мкл 0,25 М фосфатного буфера, рН 9,3. Готовят 3 мл смеси блок-сополимера ПЭИ-ПЭГ-ТАТ с флуорескамином, так, чтобы конечная концентрация ПЭИ в блок-сополимере была 1 мкМ. Измерения флуоресценции проводят в стеклянной кювете при длине волны возбуждения флуоресценции 395 нм и длине волны флуоресценции 490 нм. Значения флуоресценции в каждой точке измеряют восемь раз и показания усредняют. Эффективность синтеза считают оптимальной, когда мольное соотношение ТАТ/ПЭИ составляет приблизительно от 0,8 до 1,5.
В ходе предварительных экспериментов были определены соотношения ПЭГ/ПЭИ, при которых полиплексы обеспечивают высокую эффективность доставки плДНК в раковые клетки. Для всех исследованных линий клеток наибольшая эффективность трансфекции достигалась при мольном соотношении ПЭГ/ПЭИ приблизительно от 1,0 до 3,0, более предпочтительно при соотношении приблизительно от 2,0 до 2,5, наиболее предпочтительно при соотношении, равном приблизительно 2,0.
Пример 11. Стабильность лекарственной формы лекарственного препарата (вариант 1 - первый и второй компоненты в виде замороженных растворов)
Целью исследования было изучение стабильности физических, химических и биологических характеристик лекарственного препарата настоящего изобретения в условиях хранения при температуре - 20°С ± 2°С в период предполагаемого срока годности 18 месяцев.
Первый и второй компоненты хранили при температуре -20°С ± 2°С упакованными во флаконы бесцветные гидролитического 1 класса, герметично укупоренные резиновыми пробками и обкатанные алюминиевыми колпачками.
Стабильность компонентов и готовой лекарственной формы препарата оценивалась по ряду параметров, в том числе: описание, подлинность (рестриктазный анализ), подлинность (ПЦР со специфическими праймерами), прозрачность, цветность, механические включения, остаточные белки штамма-продуцента, бактериальные эндотоксины, аномальная токсичность, стерильность, специфическая активность. Результаты, полученные в рамках изучения стабильности приведены в Таблицах 2-4.
Таким образом, при исследовании стабильности установлено, что условия хранения при температуре - 20°С ± 2°С обеспечивают стабильность показателей качества первого и второго компонентов, а также готового к применению раствора для инъекций, получаемого после смешивания первого и второго компонентов, в течение срока хранения 18 месяцев. Установленный срок годности препарата составляет не менее 18 месяцев.
Пример 12. Стабильность лекарственной формы лекарственного препарата (вариант 2 - первый компонент в виде лиофилизата, второй компонент в виде замороженного раствора)
Целью исследования было изучение стабильности физических, химических и биологических характеристик лекарственного препарата в условиях хранения первого компонента при температуре 4°С и второго компонента при температуре - 20°С ± 2°С в период предполагаемого срока годности 24 месяца.
Первый компонент хранили при температуре 4°С упакованным во флаконы бесцветные гидролитического 1 класса, герметично укупоренные резиновыми пробками и обкатанные алюминиевыми колпачками. Второй компонент хранили при температуре -20°С±2°С упакованным во флаконы бесцветные гидролитического 1 класса, герметично укупоренные резиновыми пробками и обкатанные алюминиевыми колпачками.
Готовую лекарственную форму получали, как описано выше в Примере 8.
Исследование стабильности проводили посредством измерения ряда параметров, в том числе специфической активности готовой лекарственной формы препарата (вариант 2) через 1 день, 2 недели, 3 месяца, 6 месяцев, 12 месяцев и 18 месяцев после начала эксперимента. Измерение специфической активности проводили, как описано выше в Примере 9 (раздел «Цитотоксический тест для определения специфической активности»). В результате проведенных экспериментов было показано, что лекарственный препарат (вариант 2) сохраняет биологическую активность на протяжении по меньшей мере 18 месяцев.
Пример 13. Исследование противоопухолевой активности лекарственного препарата
Противоопухолевое действие лекарственного препарата исследовали на моделях ксенотрансплантатов опухолей человека у иммунодефицитных мышей nude, полученных путем инокуляции опухолевых клеток человека: карциномы слизистой оболочки полости рта КВ, карциномы гортаноглотки человека НЕр2, колоректальной аденокарциномы HT29 и аденокарциномы шейки матки HeLa. Лечение животных (самки, n=9-12 животных на группу) проводили с использованием эффективного режима. Лечение начинали на 11-е (КВ, НТ29, НЕр2) и 13-е (HeLa) сутки роста опухоли, когда объем опухоли составлял приблизительно 100±10мм3.
Обобщенные результаты исследования представлены в Таблице 5.
Примечание: Показатели ТРО (торможение роста опухоли, %) и Т/С (степень торможения опухоли, %) приведены на 28-й день после окончания лечения (для КВ), на 30-й день (для HeLa и НЕр2), на 38-й день (для НТ29). * - высокая противоопухолевая активность (критерии эффективности: ТРО ≥70%, Т/С ≤ 15%, УПЖ ≥50% (увеличение продолжительности жизни, %). **- отличие от группы животных, которым вводили PBS (контрольная группа), статистически значимо (p<0,05; t-критерий Стьюдента). Т-С - задержка роста опухоли.
Как следует из результатов, представленных в Таблице 5, высокий (по критериям эффективности) противоопухолевый эффект выявлен у мышей nude с карциномой КВ слизистой оболочки полости рта человека: ТРО возрастало по мере увеличения срока наблюдения и составило 66-82% на 46-60 сутки после инокуляции опухолевого материала, задержка роста опухоли - 32 дня, медианы выживаемости животных в опытной и контрольной группах различались на 24 суток - 82 и 58 суток, соответственно.
Значимый противоопухолевый эффект наблюдался и в отношении аденокарциномы HT29 толстой кишки человека: ТРО - более 70% в течение 32-67 суток наблюдения, причем максимальное ТРО (89%) отмечено на наиболее поздний срок - 67 сутки; также наблюдали задержку роста опухоли, которая составляла 49 суток. Медиана выживаемости животных в опытной группе превышает таковую животных контрольной группы на 14 суток (89 и 75 суток, соответственно).
Эффективность комбинации «лекарственный препарат настоящего изобретения + ганцикловир» у мышей с ксенографтом HeLa была существенно ниже, чем на моделях опухолей НТ29 и КВ. На модели ксенографта НЕр2 у иммунодефицитных животных была отмечена наименьшая противоопухолевая эффективность лечения. При этом, однако, медианы выживаемости животных в опытной и контрольной группах достоверно различались на 13,6 суток (74,8 и 61,2 суток, соответственно).
Результаты исследований in vivo с использованием животных моделей ряда солидных злокачественных новообразований человека выявили, что наиболее чувствительными являются карцинома слизистой оболочки полости рта КВ и колоректальная карцинома НТ29 человека.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАКОЛОГИЧЕСКАЯ КОМБИНАЦИЯ ПОЛИКАТИОННОГО НОСИТЕЛЯ ПЭГ-ПЭИ-ТАТ С ЗАКЛЮЧЕННОЙ В НЕМ ПЛАЗМИДОЙ, НЕСУЩЕЙ ТЕРАПЕВТИЧЕСКИЕ ГЕНЫ HSVtk И GM-CSF ДЛЯ ЦЕЛЕЙ ГЕНОТЕРАПИИ ОПУХОЛЕВЫХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2575077C2 |
| МНОГОПРОФИЛЬНЫЙ ПРОМОТОР, ЭКСПРЕССИРУЮЩИЙ ВЕКТОР И СПОСОБ ИЗБИРАТЕЛЬНОГО УБИЙСТВА РАКОВЫХ КЛЕТОК С ИХ ИСПОЛЬЗОВАНИЕМ | 2012 |
|
RU2476596C1 |
| УНИВЕРСАЛЬНЫЕ РАКОВОСПЕЦИФИЧНЫЕ ПРОМОТОРЫ И ИХ ИСПОЛЬЗОВАНИЕ В ПРОТИВООПУХОЛЕВОЙ ТЕРАПИИ | 2013 |
|
RU2539764C2 |
| Генотерапевтический препарат FCU1-BsFm/PP для лечения солидных опухолей | 2023 |
|
RU2824977C1 |
| Гистоны и биодеградируемые липиды как средство для доставки нуклеиновых кислот в клетки эукариот | 2015 |
|
RU2637371C2 |
| УНИВЕРСАЛЬНЫЙ ПРОМОТОР ДЛЯ ЭКСПРЕССИИ ТЕРАПЕВТИЧЕСКИХ ГЕНОВ В КЛЕТКАХ МЛЕКОПИТАЮЩИХ | 2013 |
|
RU2551784C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ ДЛЯ ИНДУКЦИИ РАЗВИТИЯ КРОВЕНОСНЫХ СОСУДОВ В ТКАНЯХ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПОЛУЧЕННАЯ ЭТИМ СПОСОБОМ, И СПОСОБ ЛЕЧЕНИЯ ИШЕМИИ ТКАНЕЙ И/ИЛИ ОРГАНОВ ЧЕЛОВЕКА | 2012 |
|
RU2542385C2 |
| МОЛЕКУЛЯРНЫЙ КОМПЛЕКС ДЛЯ ТРАНСФЕКЦИИ КЛЕТОК МЛЕКОПИТАЮЩИХ, СОДЕРЖАЩИЙ ПЛАЗМИДНУЮ ДНК, МОДИФИЦИРОВАННЫЙ ПОЛИЭТИЛЕНИМИН И МОЛЕКУЛЫ-ЛИГАНДЫ | 2005 |
|
RU2303064C2 |
| КОМПОЗИЦИЯ ДНК ДЛЯ ВЫЗОВА ИММУННОГО ОТВЕТА ПРОТИВ ОПУХОЛЕАССОЦИИРОВАННЫХ МАКРОФАГОВ | 2007 |
|
RU2459631C2 |
| ИММУНОИНДУЦИРУЮЩЕЕ СРЕДСТВО | 2017 |
|
RU2733841C2 |
Настоящее изобретение относится к области биотехнологии, медицины, в частности, онкологии и фармацевтики. Описаны две фармацевтические комбинации для приготовления раствора для инъекций для лечения солидных злокачественных новообразований методом ген-иммунной терапии. В первом варианте фармацевтическая комбинация содержит два компонента, каждый из которых укупорен в емкость, причем: первый компонент укупорен в первую емкость и представляет собой замороженный стерильный раствор, содержащий сверхскрученную плазмидную ДНК pCMV-HSVtk-hGM-CSF в терапевтически эффективном количестве, по меньшей мере, одну фармацевтически приемлемую добавку и воду, второй компонент укупорен во вторую емкость и представляет собой замороженный стерильный раствор, содержащий блок-сополимер ПЭГ-ПЭИ-ТАТ пептид в терапевтически эффективном количестве, по меньшей мере, одну фармацевтически приемлемую добавку и воду. Во втором варианте в состав композиции входят два компонента, каждый из которых укупорен в емкость, причем: первый компонент укупорен в первую емкость и представляет собой стерильный лиофилизат, содержащий сверхскрученную плазмидную ДНК pCMV-HSVtk-hGM-CSF в терапевтически эффективном количестве и, по меньшей мере, одну фармацевтически приемлемую добавку, второй компонент укупорен во вторую емкость и представляет собой замороженный стерильный раствор, содержащий блок-сополимер ПЭГ-ПЭИ-ТАТ пептид в терапевтически эффективном количестве, по меньшей мере, одну фармацевтически приемлемую добавку и воду. Готовый раствор для инъекций, предназначенный для введения пациенту, готовят ex tempore путем восстановления первого компонента посредством добавления воды, размораживания второго компонента и их смешивания. Описанные технические решения демонстрируют высокую стабильность и сохраняют терапевтическую эффективность на протяжении по крайней мере 18 месяцев для лечения солидных злокачественных новообразований человека. 2 н. и 10 з.п. ф-лы, 1 ил., 5 табл., 13 пр.
1. Фармацевтическая комбинация для приготовления раствора для инъекций для лечения солидных злокачественных новообразований методом ген-иммунной терапии, содержащая два компонента, каждый из которых укупорен в емкость, причем:
первый компонент укупорен в первую емкость и представляет собой замороженный стерильный раствор, содержащий сверхскрученную плазмидную ДНК pCMV-HSVtk-hGM-CSF в терапевтически эффективном количестве, по меньшей мере, одну фармацевтически приемлемую добавку и воду,
второй компонент укупорен во вторую емкость и представляет собой замороженный стерильный раствор, содержащий блок-сополимер ПЭГ-ПЭИ-ТАТ пептид в терапевтически эффективном количестве, по меньшей мере, одну фармацевтически приемлемую добавку и воду.
2. Фармацевтическая комбинация по п. 1, в которой:
первый компонент содержит:
сверхскрученную плазмидную ДНК pCMV-HSVtk-hGM-CSF – приблизительно 150–170 мкг,
декстрозу – приблизительно 100 мг,
HEPES – приблизительно 2,38 мг,
воду – до 1,0 мл,
второй компонент содержит:
блок-сополимер ПЭГ-ПЭИ-ТАТ пептид – приблизительно 0,63–0,74 мг (в расчете на ПЭИ),
тетраборат натрия – приблизительно 2,38 мг,
воду – до 1,0 мл.
3. Фармацевтическая комбинация по п. 1, где солидные злокачественные новообразования выбраны из группы, включающей: злокачественные новообразования легкого, меланому, фибросаркому, саркому, злокачественные новообразования головы и шеи.
4. Фармацевтическая комбинация по п. 1, в которой блок-сополимер ПЭГ-ПЭИ-ТАТ пептид характеризуется мольным соотношением ПЭГ/ПЭИ приблизительно от 1,0 до 3,0 и мольным соотношением ТАТ/ПЭИ приблизительно от 0,8 до 1,5.
5. Фармацевтическая комбинация по п. 4, в которой блок-сополимер ПЭГ-ПЭИ-ТАТ пептид характеризуется мольным соотношением ПЭГ/ПЭИ приблизительно от 2,0 до 2,5.
6. Фармацевтическая комбинация по любому из пп. 1-5, в которой:
первый компонент содержит:
сверхскрученную плазмидную ДНК pCMV-HSVtk-hGM-CSF – приблизительно 160 мкг,
декстрозу – приблизительно 100 мг,
HEPES – приблизительно 2,38 мг,
воду – до 1,0 мл,
второй компонент содержит:
блок-сополимер ПЭГ-ПЭИ-ТАТ пептид – приблизительно 0,69 мг (в расчете на ПЭИ),
тетраборат натрия – приблизительно 2,38 мг,
воду – до 1,0 мл,
причем блок-сополимер ПЭГ-ПЭИ-ТАТ пептид характеризуется мольным соотношением ПЭГ/ПЭИ приблизительно 2,0.
7. Фармацевтическая комбинация для приготовления раствора для инъекций для лечения солидных злокачественных новообразований методом ген-иммунной терапии, содержащая два компонента, каждый из которых укупорен в емкость, причем:
первый компонент укупорен в первую емкость и представляет собой стерильный лиофилизат, содержащий сверхскрученную плазмидную ДНК pCMV-HSVtk-hGM-CSF в терапевтически эффективном количестве и, по меньшей мере, одну фармацевтически приемлемую добавку,
второй компонент укупорен во вторую емкость и представляет собой замороженный стерильный раствор, содержащий блок-сополимер ПЭГ-ПЭИ-ТАТ пептид в терапевтически эффективном количестве, по меньшей мере, одну фармацевтически приемлемую добавку и воду.
8. Фармацевтическая комбинация по п. 7, в которой:
первый компонент, представляющий собой лиофилизат, получен путем лиофилизации раствора, содержащего:
сверхскрученную плазмидную ДНК pCMV-HSVtk-hGM-CSF – приблизительно 150–170 мкг,
декстрозу – приблизительно 100 мг,
HEPES – приблизительно 2,38 мг,
воду – до 1,0 мл,
второй компонент содержит:
блок-сополимер ПЭГ-ПЭИ-ТАТ пептид – приблизительно 0,63–0,74 мг (в расчете на ПЭИ);
тетраборат натрия – приблизительно 2,38 мг,
воду – до 1,0 мл.
9. Фармацевтическая комбинация по п. 7, где солидные злокачественные новообразования выбраны из группы, включающей: злокачественные новообразования легкого, меланому, фибросаркому, саркому, злокачественные новообразования головы и шеи.
10. Фармацевтическая комбинация по п. 7, в которой блок-сополимер ПЭГ-ПЭИ-ТАТ пептид характеризуется мольным соотношением ПЭГ/ПЭИ приблизительно от 1,0 до 3,0 и мольным соотношением ТАТ/ПЭИ приблизительно от 0,8 до 1,5.
11. Фармацевтическая комбинация по п. 10, в которой блок-сополимер ПЭГ-ПЭИ-ТАТ пептид характеризуется мольным соотношением ПЭГ/ПЭИ приблизительно от 2,0 до 2,5.
12. Фармацевтическая комбинация по любому из пп. 7-11, в которой:
первый компонент, представляющий собой лиофилизат, получен путем лиофилизации раствора, содержащего:
сверхскрученную плазмидную ДНК pCMV-HSVtk-hGM-CSF – приблизительно 160 мкг,
декстрозу – приблизительно 100 мг,
HEPES – приблизительно 2,38 мг,
воду – до 1,0 мл,
второй компонент содержит:
блок-сополимер ПЭГ-ПЭИ-ТАТ пептид – приблизительно 0,69 мг (в расчете на ПЭИ);
тетраборат натрия – приблизительно 2,38 мг,
воду – до 1,0 мл,
причем блок-сополимер ПЭГ-ПЭИ-ТАТ пептид характеризуется мольным соотношением ПЭГ/ПЭИ приблизительно 2,0.
| ФАРМАКОЛОГИЧЕСКАЯ КОМБИНАЦИЯ ПОЛИКАТИОННОГО НОСИТЕЛЯ ПЭГ-ПЭИ-ТАТ С ЗАКЛЮЧЕННОЙ В НЕМ ПЛАЗМИДОЙ, НЕСУЩЕЙ ТЕРАПЕВТИЧЕСКИЕ ГЕНЫ HSVtk И GM-CSF ДЛЯ ЦЕЛЕЙ ГЕНОТЕРАПИИ ОПУХОЛЕВЫХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2575077C2 |
| СПОСОБ ХРАНЕНИЯ ДНК | 2007 |
|
RU2352636C1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| А.Ю | |||
| СИДУЛЛИН, В.Н | |||
| ОСЛОПОВ, Фармацевтическая несовместимость при комбинации различных лекарств в инфузионной терапии, ПРАКТИЧЕСКАЯ МЕДИЦИНА, 5 (74) октябрь 2013, стр.46 | |||
| Организация работы лабораторий, использующих методы | |||

