Область техники, к которой относится изобретение
Настоящее изобретение относится к биотехнологии, в частности генной инженерии, и может быть использовано для лечения новообразований различной природы.
Рак представляет собой в настоящее время наиболее серьезную проблему медицины. Наиболее обычные типы рака - эпителиальные, в наименьшей степени поддаются традиционному лечению. Даже когда эпителиальная опухоль на начальном периоде поддается лечению, в конечном счете, опухоль возвращается. Открытия различных генетических детерминант рака, происходившие на протяжении многих лет, оказали только незначительное влияние на клиническую эффективность лечения. Болезнь очевидно сложна. Она широко распространена и занимает второе место по смертности в мире и, вероятно, перейдет на первое место в ближайшем будущем. Несмотря на громадные федеральные и индустриальные вложения и громадное число фундаментальных открытий, рак обычно рассматривается, в лучшем случае, как только минимально поддающийся контролю средствами современной медицины, особенно когда его сравнивают с другими распространенными болезнями. Действительно, смертность от рака в 21 веке такая же, как и 50 лет назад, тогда как смертность от сердечных, церебрососудистых и инфекционных болезней уменьшилась за это время на 2/3 (Varmus H (2006) Science 312:1162-5). Недавние статистические исследования показывают, что смертность от рака в США в последнее время снизилась (Winer E and Gralow J et al. (2009) J Clin Oncol 27:812-26) (Jemal A and Siegel R et al. (2009) CA Cancer J Clin 59:225-49); (Malvezzi M and Arfe A et al. (2011) Ann Oncol). Подобное снижение отмечается и в странах ЕС (La Vecchia С and Bosetti С et al. (2010) Ann Oncol 21:1323-60); (Malvezzi M and Arfe A et al. (2011) Ann Oncol); (Gondos A and Bray F et al. (2009) Ann Oncol 20:564-73). Однако причиной этого снижения являются главным образом профилактические мероприятия: резкое снижение курения и потребления алкоголя, улучшение диагностики и увеличения общих осмотров населения. Только незначительную долю в этом снижении играют улучшение терапевтических результатов в лечении отдельных видов рака, особенно рака, возникающего в детском возрасте (Brennan R and Federico S et al. (2010) Oncotarget 1:77-83).
Методы неиммунной противораковой терапии могут быть разделены (Hellerstein MK (2008) Metab Eng 10:1-9) на две очень широкие категории: традиционные методы химиотерапии (XT) и появившиеся относительно недавно методы молекулярной таргетной терапии, в которых действие лекарства направлено на определенную молекулярную мишень - (таргет), которая предположительно является существенной для развития рака. Следует при этом заметить, что каждое лекарство имеет свою мишень, поэтому термин таргетная терапия является неопределенным (Druker BJ (2004) Cell Cycle 3:833-5) (Hait WNHambley TW (2009) Cancer Res 69:1263-7; discussion 1267) и для целей точности в данном описании используются следующие термины (согласующиеся с часто используемыми в литературе): (1) "молекулярная таргетная терапия" (МТТ) и (2) "терапия, нацеленная на пролиферирующие клетки" (ТНПК).
Первый термин относится к новой генерации противораковых средств, нацеленных на a priory идентифицированное молекулярное звено, как правило, белок, который предположительно играет важную роль в росте и прогрессии опухоли. Этот подход противоположен по логике более ранним традиционным подходам, использовавшимся при идентификации цитотоксических средств химиотерапии, которые продолжают оставаться основным средством противораковой терапии. Здесь эти средства определены как ТНПК.
В свою очередь МТТ делится на две субкатегории:
(i) МТТ, направленная на звено, непосредственное повреждение которого служит одной из причин рака. Такая терапия может быть определена как генетическая таргетная (Druker BJ (2004) Cell Cycle 3:833-5) (МТТГ). Наиболее известным агентом такого типа является низкомолекулярный ингибитор протеинкиназ Иматиниб, который ингибирует химерную киназу BCR-ABL, образующуюся при хромосомной перестройке, известной как филадельфийская хромосома (NCI (2010)). BCR-ABL - это единственная молекулярная мутация, которая вызывает клеточную пролиферацию в клетках, содержащих Филадельфийскую хромосому, в хронической миелоидной лейкемии (CML) (NCI (2010)) (Druker BJ (2004) Cell Cycle 3:833-5).
(ii) Молекулярная таргетная терапия, нацеленная на звено, не являющееся непосредственной причиной рака, но расположенное в той же сигнальной цепочке (pathway), что и причинное звено (Sawyers С (2004) Nature 432:294-7) (МТТП).
Поскольку оба подхода МТТ направлены на определенные звенья в клеточных сигнальных системах, то полагали, что они будут менее токсичны и более эффективны для организма, чем ТНПК.
В противоположность агентам МТТ, агенты ТНПК идентифицируют по способности оказывать цитотоксический эффект на линии раковых клеток без предварительного знания мишени (Hait WN and Hambley TW (2009) Cancer Res 69:1263-7). Фактически в существенном большинстве случаев ТНПК агенты действуют на быстро делящиеся клетки (хотя есть исключения). При этом ингибирование нацелено не на отдельное молекулярное звено, а на системы, вовлеченные в репликацию ДНК. Обычные агенты этого типа относятся к нескольким группам:
(i) Алкилирующие агенты вносят повреждения непосредственно в ДНК и ингибируют ее репликацию.
(ii) Ингибиторы топоизомераз, препятствующие расплетанию ДНК.
(iii) Таксаны и винка-алкалоиды, препяствующие образованию микротрубочек, необходимых для митотического деления клеток.
(iv) Антиметаболиты, которые блокируют образование и использование предшественников нуклеиновых кислот, важных для репликации ДНК (Dy GK and Adjei AA (2008) Cancer 113:1857-87) (DeVita VT and Chu E (2008) Cancer Res 68:8643-53); (Morrison WB (2010) J Vet Intern Med 24:1249-62).
Таким образом, мишени в этом походе существуют, но они не являются отдельными молекулами, а представляют собой системы.
Кроме раковых клеток, агенты ТНПК, которые вводят в организм больного, поражают также все относительно быстро делящиеся клетки (например, клетки желудочно-кишечного эпителия, клетки костного мозга и др.) и поэтому эти агенты являются высокотоксичными (Gerber DE (2008) Am Fam Physician 77:311-9; Hait WN and Hambley TW (2009) Cancer Res 69:1263-7; discussion 1267).
В отличие от них агенты МТТ, действуя на определенные молекулярные мишени, являются менее токсичными.
МТТ агенты для терапии солидных опухолей вышли на рынок или в клинически испытания в последние несколько лет (Bria E and Di Maio M et al. (2009) J Exp Clin Cancer Res 28:66).
При использовании этой стратегии были достигнуты некоторые успехи. Часто в качестве ее парадигмы используют Иматиниб (Гливек), низкомолекулярный ингибитор протеин киназы, образующейся в раковых клетках в результате перегруппировки хромосом, приводящей к гибридному белку Bcr:abl с киназной активностью. Этот ингибитор показал хорошие результаты при лечении хронической миелоидной лейкемии и гастроинтестинальных стромальных опухолей. Однако сегодня есть много оснований полагать, что это скорее исключение, чем правило, и что это исключение показывает неверное общее направление в выборе правильных стратегий лечения рака (Hambley TW and Hait WN (2009) Cancer Res 69:1259-62; Murdoch DSager J (2008) Curr Opin Oncol 20:104-11). Другой пример относительно успешного применения таргетного агента представляет собой Герцептин (Trastuzumab) (Hait WN and Hambley TW (2009) Cancer Res 69:1263-7; discussion 1267). Это моноклональное антитело, направленное против рецептора HER-2/Neu, содержание которого повышено на поверхности раковых клеток некоторых пациентов, больных раком молочной железы. Однако он применим только примерно к 20% таких пациентов, имеющих нужный рецептор-мишень на поверхности опухолевых клеток (Ross JS and Slodkowska EA et al. (2009) Oncologist 14:320-68), и приводит к среднему увеличению продолжительности жизни примерно 5 месяцев (Bria E and Di Maio M et al. (2009) J Exp Clin Cancer Res 28:66). Серьезным препятствием перед методами молекулярной таргетной терапии является быстрое возникновение устойчивости к обработке, что наблюдается также и в случае иматиниба. Подавляющее большинство из агентов МТТ оказались малоэффективными (Bria E and Di Maio M et al. (2009) J Exp Clin Cancer Res 28:66; Duenas-Gonzalez A and Garcia-Lopez P et al. (2008) Mol Cancer 7:82; Hambley TW and Hait WN (2009) Cancer Res 69:1259-62; Hellerstein MK (2008) Metab Eng 10:1-9).
Все это привело к определенному кризису в выработке стратегий лечения рака путем молекулярной таргетной терапии, что выражается в замедлении появления новых агентов этого типа (Hambley TW and Hait WN (2009) Cancer Res 69:1259-62). Это вполне ожидаемый кризис ввиду особенностей рака как болезни, которая сочетает в себе сложность клеточной организации, присущую и другим болезням, со сложностью растущей эвольвирующей системы, способной адаптироваться к воздействиям лекарствами путем существования микрогетерогенности внутри опухоли (Merlo LM and Pepper JW et al. (2006) Nat Rev Cancer 6:924-35). Эта гетерогенность вызывается делением раковых клеток при росте опухоли. При каждом делении клетки две дочерние клетки получают мутации так, что они отличаются и друг от друга, и от исходной клетки. Таким образом, в принципе каждая клетка данной раковой опухоли отличается от всех других клеток этой же опухоли, а опухоль одного пациента отличается от опухоли того же типа у другого пациента (Glazier A (2007) www.curecancerproject.org/). В этом отношении опухоль подобна другим сложным многоклеточным эволюционирующим системам (Sverdlov ED (2009) Biochemistry (Mosc) 74:939-44; Veitia RA (2005) J Biosci 30:21-30). Тем более отличаются друг от друга опухоли разных типов. Среди множества клеток в этой микрогетерогенной популяции всегда находятся такие, которые устойчивы к воздействиям, не погибают в результате них и дают рост нового, устойчивого к данному воздействию клона опухоли. Дополнительные сложности в лечение опухолей вносит то, что опухоль активно взаимодействует с окружением, модифицирует его и метастазирует в другие органы организма (Merlo LM and Pepper JW et al. (2006) Nat Rev Cancer 6:924-35).
Процесс эволюции опухоли начинается с первой мутации, которая (придерживаясь гипотезы раковых стволовых клеток - РСК (Bjerkvig R and Tysnes BB et al. (2005) Nat Rev Cancer 5:899-904; Reya Т and Morrison SJ et al. (2001) Nature 414:105-11)) превращает нормальную стволовую клетку в раковую. Вновь образованная раковая стволовая клетка становится таковой не вследствие любой мутации, а тех мутаций (которые получили название драйверов), которые дают ей преимущества в скорости делений, доли симметричных делений, которые увеличивают долю стволовых клеток в популяции, или какие-то еще преимущества и, кроме того, запрещают или затрудняют нормальную дифференцировку стволовых клеток. РСК самообновляется, т.е. постоянно реплицируется, и в силу этого постоянно получает мутации. Большинство этих мутаций безвредно или почти безвредно. Но существует вероятность того, что какая-то из РСК-потомков первичной РСК получит среди массы этих мутаций еще одну, дающую еще большие преимущества в росте, и, кроме того, еще какие-либо преимущества, допустим, способность избегать апоптоза. Это повторяется при каждом клеточном делении. Происходит типичная соматическая эволюция, в процессе которой РСК постепенно накапливают мутации внутри одних и тех же клеток, и РСК с накопленными мутациями становятся все более приспособленными с точки зрения борьбы за ресурсы и за пространство существования. Портрет (т.е. распределение по геному) мутаций отличается для разных клеток данной опухоли на разных этапах ее развития. Естественно, что мутационный портрет метастазов данной опухоли отличается от портрета исходной опухоли, и разные метастазы одной опухоли имеют разные портреты (Park SY and Gonen M et al. (2010) J Clin Invest 120:636-44) (Langley RR and Fidler IJ (2007) Endocr Rev 28:297-321) (Campbell PJ and Yachida S et al. (2010) Nature 467:1109-13); (Li J and Wang К et al. (2010) BMC Res Notes 3:321).
Меж- и внутриопухолевая гетерогенность еще увеличивается благодаря эпигенетической гетерогенности (Jones PA and Baylin SB (2007) Cell 128:683-92).
Практические выводы из существования громадной меж- и внутриопухолевой гетерогенности неблагоприятны для МТТ. Поскольку множество раковых генов участвуют в развитие рака только в малой части клеток раковой опухоли, то их трудно идентифицировать для использования в качестве мишеней (Stratton MR and Campbell PJ et al. (2009) Nature 458:719-24); (Salk J and Fox E et al. (2010) Annu. Rev. Pathol. Mech. Dis. 5:51-75).
Одна из основных гипотез, которая направлена на облегчение поиска таких мишеней, заключается в том, что не все гены вносят равный вклад в прогрессию опухоли. Наиболее важными являются так называемые ведущие (драйверные) мутации, которые придают раковой клетке преимущества в росте. Эти мутации можно рассматривать как причинные в раковом росте и можно ожидать их позитивной селекции в процессе эволюции опухоли (Salk J and Fox E et al. (2010) Annu. Rev. Pathol. Mech. Dis. 5:51-75) (Stratton MR and Campbell PJ et al. (2009) Nature 458:719-24) (Wood LD and Parsons DW et al. (2007) Science 318:1108-13). Все другие мутации являются случайными пассажирами ("passengers") в процессе эволюции и могут не приниматься в расчет при поиске мишени. Предполагалось, что количество таких драйверных мутаций должно быть ограничено (Armitage P and Doll R (2004) Br J Cancer 91:1983-9) и что набор драйверных мутаций затрагивает определенный ограниченный спектр раковых генов (для обзора см. (Salk J and Fox E et al. (2010) Annu. Rev. Pathol. Mech. Dis. 5:51-75) (Stratton MR and Campbell PJ et al. (2009) Nature 458:719-24)). Если это так, то драйверные мутации могли бы быть идентифицированы полногеномным секвенированием нескольких индивидуальных опухолей, как систематически повторяющиеся от опухоли к опухоли мутации (Wood LD and Parsons DW et al. (2007) Science 318:1108-13); (Parmigiani G and Boca S et al. (2009) Genomics 93:17-21) (обзоры (Salk J and Fox E et al. (2010) Annu. Rev. Pathol. Mech. Dis. 5:51-75) (Stratton MR and Campbell PJ et al. (2009) Nature 458:719-24)).
Первый же почти полногеномный анализ подобного рода (Wood LD and Parsons DW et al. (2007) Science 318:1108-13), результаты которого были представлены в виде трехмерного ландшафта, показывающего позиции генов в горизонтальной плоскости и частоты их встречаемости в индивидуальных опухолях в третьем измерении, показал, что мутации только в очень немногих генах появляются с относительно высокими частотами, образуя так называемые горы на ландшафте. Подавляющее большинство генов редко оказывались мутантными даже хотя бы в двух сравниваемых опухолях. Такие редко повторяющиеся гены образовывали на ландшафте так называемые холмы. Вывод заключался в том, что большое число мутаций-холмов (а не только «горы»), каждая с незначительным вкладом в прогрессию опухоли, являются драйверами и движущей силой эволюции опухоли. Позже этот вывод был подтвержден многими полногеномными секвенированиями разных опухолей (Copeland NG and Jenkins NA (2009) Trends Genet 25:455-62) (Salk J and Fox E et al. (2010) Annu. Rev. Pathol. Mech. Dis. 5:51-75) (Stratton MR and Campbell PJ et al. (2009) Nature 458:719-24)).
Такое разнообразие и редкость драйверов делают их обнаружение чрезвычайно трудной задачей. Теоретическое рассмотрение и экспериментальные данные показывают, что трудно, если вообще возможно, получить разумное число лекарств МТТ, которые позволяли бы осуществлять лечение различных видов рака (Salk J and Fox E et al. (2010) Annu. Rev. Pathol. Mech. Dis. 5:51-75).
Положение, возможно, может облегчаться тем, что множество драйверных мутаций располагаются в меньшем числе сигнальных путей, и тогда одно лекарство, направленное на ингибирование сигнального пути, содержащего несколько драйверных мутаций, могло бы подавлять их все. Такой МТТП подход широко обсуждается (см. (Salk J and Fox E et al. (2010) Annu. Rev. Pathol. Mech. Dis. 5:51-75)), однако, хотя определяют шесть основных характерных изменений в физиологии клетки при ее превращении в раковую (которые теперь широко известны как обязательные признаки, hallmarks, раковой клетки (Hanahan D and Weinberg RA (2000) Cell 100:57-70)), за ними стоит громадное разнообразие генотипов раковых клеток, постоянно меняющихся в процессе развития и метастазирования опухолей. Недавние результаты показывают, что в типе рака может быть вовлечено до 12 различных путей передачи информации. К тому же большинство биологических процессов имеет альтернативные пути передачи сигнала, развитые в процессе эволюционных приспособлений. В результате, если удается ингибировать один из них, то другой может активироваться и компенсировать отсутствие сигнала от первого (Hambley TW and Hait WN (2009) Cancer Res 69:1259-62), (Bria E and Di Maio M et al. (2009) J Exp Clin Cancer Res 28:66).
Это сразу ставит вопрос о принципиальной трудности или невозможности развития эффективной молекулярной таргетной терапии, которой сейчас уделяется очень большое внимание как потенциальному средству лечения опухолей (Hait WN and Hambley TW (2009) Cancer Res 69:1263-7) (Hambley TW and Hait WN (2009) Cancer Res 69:1259-62) (Bria E and Di Maio M et al. (2009) J Exp Clin Cancer Res 28:66; Dy GK and Adjei AA (2008) Cancer 113:1857-87). При этом помимо принципиальных проблем существуют еще и труднопреодолимые технические проблемы, а именно проблемы идентификации подходящих молекулярных мишеней (Hambley TW and Hait WN (2009) Cancer Res 69:1259-62).
Общий принцип генной терапии (ГТ) заключается в доставке регулируемого генетического материала в раковые клетки, где в результате производятся продукты, способные к уничтожению раковых клеток. Здесь следует отметить, что подходы ГТ, могут быть разделены на две широкие категории (Sverdlov E (2009) Molecular Genetics, Microbiology and Virology English version 24:93-113). В первой используют стратегию таргетной терапии. При этом в роли таргетного агента выступает продукт гена, вводимого каким-либо способом в клетку опухоли, который является ингибитором того или иного продукта, концентрация которого в раковой клетке повышена, и это является одной из причин ракового процесса. К этой же категории можно отнести обратную технологию - доставка в опухоль гена, продукт которого компенсирует недостаток определенного белка в раковых клетках. В обоих вариантах мишенью воздействия является определенное звено в сигнальных системах клетки, которое меняется при раковом перерождении и способствует ему. Варианты ГТ, основанные на таргетном принципе, страдают от тех же недостатков, что и МТТ. Тем не менее, ГТ расширяет возможности таргетной терапии. Прежде всего, гены, вводимые в клетку, могут придавать этой клетке новые фенотипические признаки. Эта возможность широко используется в таких вариантах генной терапии, как генная иммунотерапия рака (Loisel-Meyer S and Foley R et al. (2008) Front Biosci 13:3202-14; Seth P (2005) Cancer Biol Ther 4:512-7) (Collins SA and Guinn BA et al. (2008) Curr Gene Ther 8:66-78). ГТ позволяет доставлять к клетки гены онкосупрессоров, которые были инактивированы в процессе канцерогенеза, например ген, кодирующий супрессор, который часто инактивирован в раковых клетках (Fang В and Roth JA (2003) Cancer Biol Ther 2:8115-21); (Räty JK (2008) Current Molecular Pharmacology 1:13-23); (Huang CL and Yokomise H et al. (2007) Future Oncol 3:83-93). Возможно вызывать апоптоз в раковых клетках путем доставки про-апоптических генов (Lumniczky К and Safrany G (2006) Pathol Oncol Res 12:118-24; Seth P (2005) Cancer Biol Ther 4:512-7). ГТ позволяет также блокировать одновременно несколько сигнальных путей с помощью РНК интерференции (RNAi) (Wagner E (2007) J Buon 12 Suppl 1:S77-82) (Wang SL and Yao HH et al. (2009) Expert Opin Biol Ther 9:1357-68). Клинические испытания ГТ препаратов показывают их безопасность и положительные клинические отклики (Fang В and Roth JA (2003) Cancer Biol Ther 2:S115-21); (Huang CL and Yokomise H et al. (2007) Future Oncol 3:83-93; Räty JK (2008) Current Molecular Pharmacology 1:13-23; Xue W and Zender L et al. (2007) Nature 445:656-60).
Первый генно-терапевтический вирус для терапии рака головы и шеи был одобрен для клинического применения в Китае в 2003 г. под названием Гендицин (Gendicine). Вирус содержит в качестве терапевтического гена р53 (Peng Z (2005) Hum Gene Ther 16:1016-27); (Wilson JM (2005) Hum Gene Ther 16:1014-5). В сочетании с радиотерапией Гендицин вызывал полную регрессию опухоли у 64% пациентов и частичную - у 29%, тогда как одна радиотерапия давала полную регрессию у 19% и частичную у 60%, что по-видимому показывает заметное улучшение результата при комбинированной терапии (Peng Z (2005) Hum Gene Ther 16:1016-27). Но эти результаты показывают также, что только 64% откликаются на обработку, и дальнейшее поведение их болезни неизвестно. Есть основания ожидать появления вторичного роста опухоли, поскольку было показано, что при введении р53 в опухоли, в которых он исходно поврежден, вызывает появление устойчивых к р53 вариантов (Martins CP and Brown-Swigart L et al. (2006) Cell 127:1323-34). Полученные данные с использованием ГТ в качестве молекулярной таргетной терапии предполагают, что также как и классическая МТТ и вследствие тех же причин, она едва ли будет высокоэффективной или универсальной.
В последнее время широко обсуждаются так называемые онколитические вирусы (OB), которые, обладая способностью селективно экспрессироваться в опухолевых клетках и лизировать их, заражают и лизируют также соседние опухолевые клетки (Hall К and Blair Zajdel ME et al. (2010) Biochem J 431:321-36; Liu TC and Kim D (2008) Gene Ther 15:877-84). Этот подход обладает целым рядом преимуществ, главным из которых является цепная реакция распространения вируса в опухоли и связанное с этим быстрое уничтожение тех опухолевых клеток, которые чувствительны к вирусу. Однако при этом остаются клетки, резистентные к вирусу, которые с высокой вероятностью присутствуют в гетерогенной опухолевой популяции, в результате чего после периода уменьшения размеров опухоли наступает ее вторичный рост. Вновь образуемая опухоль устойчива к обработке вирусом. В целом онколитические вирусы также представляют собой варианты МТТ. Но, кроме того, они дороги в производстве и иммуногенны, что затрудняет повторное их введение в организм.
Вторая стратегия генной терапии, направленная на уничтожение опухолевых клеток, как таковых, путем использования их свойств, которые характерны для всех раковых клеток, например повышенная скорость митотических делений, в этом отношении подобна хемотерапии, или ТНПК. Однако, в отличие от последней, токсин, убивающий раковые клетки путем ингибирования систем репликации, образуется внутри них, так что свойственная ТНПК токсичность в данном случае резко снижается. Этот подход известен, как ген-направленная энзиматическая пролекарственная терапия, ГНЭПТ (Gene-directed enzyme prodrug therapy, GDEPT) или генная терапия с использованием генов самоубийства опухоли (suicide gene therapy) (Altaner С (2008) Cancer Lett 270:191-201; Fillat С and Carrio M et al. (2003) Curr Gene Ther 3:13-26; Portsmouth D and Hlavaty J et al. (2007) Mol Aspects Med 28:4-41; Seth P (2005) Cancer Biol Ther 4:512-7). Подход не является молекулярно таргетированным и потому избегает всех недостатков МТТ. Он основан на введении в опухолевые клетки гена, превращающего внутри них нетоксичный про-агент в токсичный агент.
Все клетки опухоли и ее метастаз, как бы гетерогенны они ни были, имеют одно общее фундаментальное свойство - они все непрерывно пролиферируют, реплицируя ДНК. Это же общее свойство клеток разных опухолей. Вот то общее, что может быть универсальной мишенью в любой опухоли и в ее метастазах. В версии ГНЭПТ, которая использована авторами, заложены два фундаментальных принципа:
1. Терапевтическое воздействие должно осуществляться на реплицирующуюся ДНК, наиболее универсальный и главный элементу репликации клеток, или на системы, непосредственно участвующие в репликации.
2. Клетка, где осуществляется воздействие, должна распространять его на окружающие клетки.
Схематически этот подход изображен на фиг.4 на примере системы, включающей ген, кодирующий фермент тимидинкиназу вируса простого герпеса (HSV-tk) и пролекарство - известный малотоксичный противогерпетический препарат ганцикловир (GCV). Ген HSV-tk вводят в раковые клетки, где он работает и синтезирует фермент - вирусную тимидинкиназу. Затем пациент системно получает GCV. Вирусная тимидинкиназа, в отличие от клеточных ферментов, фосфорилирует GCV, превращая его в монофосфат, который затем последовательно превращается клеточными киназами в ди- и трифосфаты ганцикловира. Трифосфат включается в реплицирующуюся ДНК и обрывает синтез растущих цепей. Раковая клетка погибает. Превращение нетоксичного GCV в токсичный трифосфат происходит внутри раковой клетки и поэтому не оказывает токсичного эффекта на здоровые клетки. Таким образом, осуществляется принцип 1, сформулированный выше.
Принцип 2 осуществляется за счет так называемого эффекта байстэндер. Фосфорилированный ганцикловир выходит из клеток опухоли и проникает в соседние клетки. Там он превращается в трифосфат и, если это реплицирующиеся клетки, то включается в ДНК и обрывает ее синтез. Соседние клетки погибают, хотя в них может не быть гена HSV-tk. По имеющимся данным, достаточно, чтобы ген попал всего в 10% опухолевых клеток, чтобы были уничтожены все клетки опухоли (Mesnil MYamasaki H (2000) Cancer Res 60:3989-99). Это чрезвычайно важно, поскольку практически невозможно доставить ген во все опухолевые клетки. Таким образом, антиметаболит, ингибирующий репликацию ДНК, образуется внутри клетки и не может проявлять токсичность на клетках, удаленных от опухоли.
Аналогичная система использует ген цитозиндезаминазы бактерий или дрожжей. Это иллюстрируется фиг.5. В данном случае внутри клетки из нетоксичного фторцитозина образуется антиметаболит фторурацил.
Использование генов убийц является одним из наиболее перспективных направлений, оно приводит к сочетанию сильных свойств хемотерапии с сильными свойствами и низкой токсичностью таргетной терапии.
В настоящее время проводятся клинические испытания, которые позволяют надеяться, что этот принцип будет доведен до реального способа лечения рака (www.wiley.co.uk/genetherapy/clinical/), (Portsmouth D and Hlavaty J et al. (2007) Mol Aspects Med 28:4-41).
Однако результаты клинических испытаний на пациентах дают пока значительно худшие результаты, чем удается получить на животных. В значительной степени это связано с недостаточно высокой специфичностью экспрессии терапевтических генов в раковых клетках и с недостаточно эффективным эффектом байстэндер при проведении экспериментов на пациентах в клинике.
Специфичность экспрессии в раковых клетках может задаваться либо путем его специфичной доставки в эти клетки, либо путем создания условий специфичности экспрессии трансгена в заданной ткани. Последний метод ввиду его простоты используется чаще под названием траскрипционного таргетинга. Для этого при конструировании векторов используют промоторы и энхансеры, специфично работающие в опухолях данной ткани (Robson THirst DG (2003) J Biomed Biotechnol 2003:110-13 7); (Saukkonen KHemminki A (2004) Expert Opin Biol Ther 4:683-96).
Правильный выбор промотора для транскрипционного таргетинга в ГНЭПТ представляет собой одну из самых важных проблем. Такие промоторы должны сочетать способность обеспечивать сильную и специфичную экспрессию трансгенов. Однако обычно используемые для этих целей природные раковоспецифичные промоторы значительно слабее по сравнению с сильными конститутивными промоторами, такими как CMV или SV40 (Lu В and Makhija SK et al. (2005) Gene Ther 12:330-8); (Rein DT and Breidenbach M et al. (2004) J Gene Med 6:1281-9); (Van Houdt WJ and Haviv YS et al. (2006) J Neurosurg 104:583-92). Кроме того, они обычно активны в ограниченном числе типов раковых клеток (Adachi Y and Reynolds PN et al. (2001) Cancer Res 61:7882-8); (Lee SE and Jin RJ et al. (2000) Anticancer Res 20:417-22); (Yamamoto M and Alemany R et al. (2001) Mol Ther 3:385-94). Даже относительно сильные широко используемые раковые промоторы, такие как промотор phSurv гена BIRC5, кодирующего игнгибитор апоптоза сурвивин, или промотор гена hTERT (phTERT), кодирующий каталитическую субъединицу теломеразы, существенно слабее упомянутых конститутивных промоторов CMV или SV40 (Hsu CP and Miaw J et al. (2003) Eur J Surg Oncol 29:594-9). Вдобавок, активность промоторов сильно варьирует в разных опухолях, затрудняя подбор доз пролекарства для получения терапевтического эффекта и меняя терапевтический индекс препарата от опухоли к опухоли. Например, активность phSurv промотора в разных опухолях варьирует от 0.3 до 16% относительно промотора CMV (Chen JS and Liu JC et al. (2004) Cancer Gene Ther 11:740-7); (Konopka К and Spain С et al. (2009) Cell Mol Biol Lett 14:70-89), активность phTERT промотора варьирует в 20 раз в различных клеточных линиях (Gu J and Fang В (2003) Cancer Biol Ther 2:S64-70).
Использовали два принципиальных подхода для увеличения эффективности раковоспецифической экспрессии терапевтических генов:
1. Бимодальные терапевтические системы. В одной из таких систем, общий принцип которой демонстрируется фиг.1Б, в клетку вводятся два вектора. В одном из них (активационном) находится ген, кодирующий трансактиватор транскрипции, например Tat белок вируса иммунодефицита под контролем раково-специфического промотора, тогда как в другом (терапевтическом), терапевтический трансген под контролем промотора, содержащего TAR элемент этого вируса. При совместной экспрессии Таt белок связывается с TAR элементом и активирует промотор, который содержит этот элемент. Таким образом, удается в десятки раз повысить уровень экспрессии терапевтического трансгена (Mingaleeva RN and Chernov IP et al. (2010) Mol Biol (Mosk) 44:507-14). В другой системе также используют два вектора (Фиг.14В). Первый содержит ген рекомбиназы Cre бактериофога P1 (Kuzmin DV and Vinogradova TV et al. (2010) The Open Gene Therapy Journal 3:31-39) под контролем раково-специфичного промотора. Второй - терапевтический ген, соединенный с конститутивным сильным промотором, который изолирован от гена транскрипционным терминатором, расположенным между сайтами рекомбиназы Cre. При помещении двух векторов в одну и ту же клетку рекомбиназа вырезает терминатор транскрипции, промотор второго вектора соединяется с трансгеном и начинается активная транскрипция последнего, уровень которой определяется силой конститутивного промотора, соединенного с трансгеном. Обе системы увеличивают эффективность транскрипции, но ее специфичность определяется раковым промотором, используемым в активационном векторе. И если этот промотор ограничен в своей активности узким спектром раковых опухолей, то активация происходит только в них. Кроме того, такая двойная система удорожает и делает менее удобным терапевтическое использование.
1. Использование комбинированных и двойных (химерных) промоторов.
Химерные промоторы могут включать в себя комбинации известных промоторов друг с другом или с отдельными гетерологичными регуляторными элементами с целью увеличить силу и специфичность экспрессии в раковых клетках (Wu С and Lin J et al. (2009) Mol Ther 17:2058-66). Примером химерного промотора может служить комбинация phTERT с минимальным промотором цитомегаловируса (phTERT-CMV) (Davis JJ and Wang L et al. (2006) Cancer Gene Ther 13:720-3) или с ТАТА боксом, который отсутствует в нативном phTERT. Как правило, авторы различных исследований, направленных на эффективное использование гибридных промоторов, идут по пути максимального увеличения эффективности и специфичности экспрессии в определенном виде рака. Примером такого подхода служит работа Poulsen et al. (Poulsen TT and Pedersen N et al. (2008) Cancer Gene Ther 15:563-75). В этой работе были идентифицированы два гена высокоэкспрессируемых в мелкоклеточном раке легких. Один из них кодирует фактор транскрипции hASH1, а другой, EZH2 относится к члену семейства Поликомб. Когда промоторы этих генов были соединены в одну конструкцию, то образующийся химерный промотор был способен вызывать сильную экспрессию трансгена специфично в клетках SCLC. Такой промотор способен инициировать экспрессию генов убийц в SCLC, но не в других видах рака.
Такая идеология максимальной спецификации активности промотора в определенном типе рака является распространенной (см., например (Farokhimanesh S and Rahbarizadeh F et al. (2010) Biotechnol Prog 26:505-11). Использование строго специфичных к данной опухоли промоторов и других регуляторных элементов имеет в качестве преимущества максимальное снижение побочных эффектов за счет понижения экспрессии трансгенов в нормальных тканях. Однако недостатком таких подходов является их неуниверсальный характер и связанное с этим неизбежное увеличение стоимости препаратов, основанных на таких промоторах. Компромиссным вариантом является использование более универсальных раковоспецифических промоторов, способных работать в широком спектре опухолей, но не в нормальных клетках. Несколько увеличивая риск поражения нормальных тканей, такой подход является более экономически оправданным: одни и те же конструкции могут использоваться в лечении широкого спектра опухолей. Существует и еще одно важное соображение в пользу использования промоторов более широкого спектра действия. Оно связано с плохо изученной специфичностью экспрессии генов в метастазах данной опухоли. Нет строгой гарантии, что узкоспецифический промотор, хорошо работающий в первичной опухоли, сохранит эту способность во всех ее метастазах. Использование универсальных промоторов снижает вероятность инактивации промотора в метастазах.
Авторы разработали новые системы транскрипционного контроля экспрессии генов-убийц, позволяющие создание новых технологий генной терапии рака. Авторы открыли, что комбинированные промоторы, состоящие из двух промоторов, в отличие от предыдущих работ, активных в широком спектре раковых опухолей, функционально не являются механической суммой двух составляющих промоторов, а образуют новый функциональный многопрофильный промотор, отличающийся от всех известных промоторов как по точке начала транскрипции, так и по повышенной активности в широком спектре опухолей. Авторы также обнаружили, что искусственный мутантный вариант BIRC5 промотора обладает расширенным спектром опухолей, в которых он активен, и также является многопрофильным промотором. Способы, основанные на транскрипционном контроле генов убийц открытыми авторами промоторами и их производными в отличие от технологий молекулярной таргетной терапии, не требуют специфического знания мишеней в раковых клетках и обеспечивают их убийство за счет образования внутри последних токсинов, убивающих раковую клетку. Токсины, также как хемиотерапевтические агенты, направлены на ингибирование систем репликации раковой клетки, но, в отличие от хемиотерапевтических средств, поставляются в раковую клетку не извне, а образуются внутри нее, таким образом, резко снижая токсичность лечения и повышая терапевтический индекс препаратов. Способы, использующие такие промоторы и их производные, в отличие от предыдущих предлагавшихся технологий, позволяют использовать одну и ту же генно-терапевтическую систему для лечения не одного типа опухолей, а широкого спектра опухолей, что делает такую систему более универсальной и экономически целесообразной. Широкая опухолевая специфичность, в отличие от предлагавшихся ранее узко специфичных терапий, значительно повышает вероятность убийства метастаз опухоли при системном введении препарата.
Раскрытие изобретения
Изобретение относится к многопрофильным промоторам, обеспечивающим внутри раковой клетки экспрессию гена фермента, способного превращать внутри раковой клетки нетоксическое соединение (пролекарство) в токсин в большинстве раковых, но не в нормальных клетках. Это приводит к превращению поставляемого извне пролекарства внутри раковой клетки в токсин, вызывающий убийство расширенного по сравнению с известными аналогами спектра опухолей, а синтезируемый внутри раковой клетки токсин диффундирует в окружающие раковые клетки.
Более конкретно изобретение относится к многопрофильным промоторам, содержащим расширенную по сравнению с известными промоторами совокупность участков узнавания белков-факторов транскрипции, необходимый набор которых существует в большинстве раковых, но не в нормальных клетках, и совокупность которых, связываясь с участками узнавания промотора, обеспечивает синергетическую активацию промотора в более широком спектре опухолевых клеток, чем известные промоторы. Также это относится к многопрофильным промоторам, содержащим измененную, по сравнению с известными промоторами, совокупность участков узнавания белков-факторов транскрипции, необходимый набор которых существует в большинстве раковых, но не в нормальных клетках, и совокупность которых, связываясь с участками узнавания промотора, обеспечивает синергетическую активацию промотора в более широком спектре опухолевых клеток, чем известные промоторы.
Еще более конкретно изобретение относится к многопрофильным промоторам, являющимся тандемной комбинацией BIRC5 и TERT промоторов или производным тандемной комбинации BIRC5 и TERT промоторов, образованной в результате делеции, замены, инсерции или иной мутации. Или к многопрофильным промоторам, являющимся искусственными мутантами промотора сурвивина мыши или искусственными мутантами промотора обратной транскриптазы теломеразы мыши или человека, или искусственными мутантами любого опухолеспецифичного промотора высших эукариот, в частности человека и мыши, или промоторами гена высших эукариот, участвующего в репликации ДНК, или искусственными мутантами BIRC5 промотора с пониженным ингибированием активности посредством фактора р53. И где в качестве активатора выступает ТАТ белок вируса иммунодефицита человека, и где в качестве сайта связывания активатора выступает TAR последовательность вируса иммунодефицита человека.
В другом аспекте изобретение относится к экспрессирующему вектору, содержащему описанный многофункциональный промотор. Конкретнее экспрессирующий вектор представляет собой вирусный вектор или экспрессирующий вектор представляет собой невирусный вектор.
В другом аспекте изобретение относится к способу избирательного убийства раковых, но не нормальных клеток, заключающемуся в синтезе токсинов внутри раковых клеток, включающему доставку в опухоль генной конструкции, состоящей из гена фермента, способного превращать внутри раковой клетки нетоксичное соединение - пролекарство - в токсин, и описанного многопрофильного промотора, при этом синтезируемый внутри раковой клетки токсин диффундирует в окружающие раковые клетки.
Конкретнее, где кодируемый фермент представляет собой тимидинкиназу вируса простого герпеса. Или где кодируемый фермент представляет собой мутантную или иначе измененную тимидинкиназу вируса простого герпеса. Или где кодируемый фермент представляет собой цитозиндезаминазу дрожжей. Или где кодируемый фермент представляет собой цитозиндезаминазу дрожжей, соединенную в один белок с урацилфосфорибозилтрансферазой дрожжей. Или где кодируемый фермент представляет собой мутантную или иначе измененную цитозиндезаминазу дрожжей.
Предложен способ избирательного убийства раковых, но не нормальных клеток, путем синтеза токсинов внутри раковых клеток и распространения их в окружающие раковые клетки, заключающийся в доставке в опухоль генной конструкции, состоящей из гена фермента, способного превращать внутри раковой клетки нетоксичное соединение - пролекарство в токсин, и многопрофильного промотора, обеспечивающего экспрессию этого гена в большинстве раковых, но не в нормальных клетках, за счет того, что многопрофильный промотор содержит расширенную, по сравнению с известными промоторами, совокупность участков узнавания белков-факторов транскрипции, необходимый набор которых существует в большинстве раковых, но не в нормальных клетках, и совокупность которых, связываясь с участками узнавания промотора, обеспечивает синергетическую активацию промотора в более широком спектре опухолевых клеток, чем известные природные промоторы, что вызывает направляемый активным промотором синтез фермента, превращение пролекарства в токсин и убийство расширенного, по сравнению с известными аналогами, спектра опухолей (Фиг.14А).
В соответствии с изобретением в опухоль доставляются две генные конструкции, одна из которых содержит ген фермента, способного превращать внутри раковой клетки нетоксичное соединение - пролекарство в токсин, и активируемого промотора, при активации обеспечивающего экспрессию этого гена, тогда как вторая является активирующей конструкцией, содержащей многопрофильный промотор, активный в большинстве раковых, но не в нормальных клетках, присоединенный к гену активатора, кодирующему белок активатор, способный активировать неактивный промотор. Многопрофильный промотор в раковых клетках вызывает транскрипцию гена активатора и синтез белка активатора, который связывается с соответствующей последовательностью в активируемом промоторе, активирует его и вызывает направляемый активным промотором синтез фермента, превращение пролекарства в токсин и убийство расширенного, по сравнению с известными аналогами, спектра опухолей (Фиг.14Б).
В соответствии с изобретением синтез токсинов осуществляется внутри раковых клеток и распространяется в окружающие раковые клетки. В опухоль доставляются две генные конструкции, одна из которых содержит ген фермента, способного превращать внутри раковой клетки нетоксичное соединение - пролекарство в токсин, и блокированного промотора, при деблокировании обеспечивающего экспрессию этого гена, тогда как вторая является деблокирующей конструкцией, содержащей многопрофильный промотор, активный в большинстве раковых, но не в нормальных клетках, присоединенный к гену деблокатора, кодирующему белок деблокатор, способный деблокировать блокированный промотор. Многопрофильный промотор в раковых клетках вызывает транскрипцию гена деблокатора, синтез белка деблокатора, который вырезает блокирующую последовательность из блокированного промотора, активирует его и вызывает направляемый деблокированным промотором синтез фермента, превращение пролекарства в токсин и убийство расширенного, по сравнению с известными аналогами, спектра опухолей (Фиг.14В).
Техническим результатом, обеспечиваемым настоящей группой изобретений, является то, что направляемый многопрофильным промотором синтез фермента приводит к превращению поставляемого извне пролекарства внутри раковой клетки в токсин, вызывающий убийство расширенного, по сравнению с известными аналогами, спектра опухолей.
Краткое описание чертежей
Фиг.1. Схематическое изображение экспрессионных конструкций. Справа приведено название конструкций. phSurv - промотор гена сурвивина человека; phTERT - промотор гена теломеразной обратной транскриптазы человека; LUC - ген люциферазы светлячка.
Фиг.2. Схематическое изображение экспрессионных конструкций с указанием идентифицированных стартов инициации транскрипции. phSurv - промотор гена сурвивина человека; phTERT - промотор гена теломеразной обратной транскриптазы человека; LUC - ген люциферазы светлячка. Стрелками указаны сайты инициации транскрипции. Цифры над стрелками обозначают количество клонов с данным сайтом инициации транскрипции. Для каждой конструкции были проанализированы 12 клонов.
Фиг.3. Активность двойных промоторов в различных клеточных линиях. По оси ординат приведена активность люциферазы относительно базальной активности в клетках, трансфицированных беспромоторным вектором BV-pGL3. По оси абсцисс обозначены типы клеточных линий, в которых проводилось измерение промоторной активности. В правом верхнем углу приведены названия конструкций, используемых в работе. Высота столбцов отражает среднее значение люциферазной активности в четырех независимых экспериментах, приведены стандартные ошибки среднего значения (SEM).
Фиг.4. Детекция белка р53 в трансфицированных клетках. Фракция, соответствующая по массе 53 кДа - белок р53, 36 кДа - GAPDH. М - набор белков с молекулярными массами 9-200 кДа, слева обозначены молекулярные массы маркерных белков в кДа. Справа указаны молекулярные массы белков р53 и GAPDH. Сверху обозначены номера образцов: 1 - нетрансфицированные клетки Calu-1; 2, 3, 4, 5 - клетки Calu-1, котрансфицированные плазмидами pSurv4-pGL3 и pCMV-p53 в количестве 0, 1, 10 и 50 нг, соответственно; 6, 7, 8, 9 - клетки Calu-1, котрансфицированные плазмидами 20G/T-pGL3 и pCMV-p53 в количестве 0, 1, 10 и 50 нг, соответственно; 10, 11, 12, 13 - клетки Calu-1, котрансфицированные Δ11-13-pGL3 и pCMV-p53 в количестве 0, 1, 10 и 50 нг, соответственно.
Фиг.5. Активность многопрофильных промоторов pSurv4, Δ11-13 и 20G/T в клетках Calu-1, трансфицированных плазмидой pCMV-p53. Клетки были котрансфицированы репортерными плазмидами phSurv-pGL3, Δ11-13-pGL3 и 20G/T-pGL3 совместно с разными количествами (0, 1, 10 и 50 нг) плазмиды pCMV-p53. За единицу принимали относительную люциферазную активность в клетках, трансфицированных соответствующей плазмидой в отсутствии pCMV-p53. Высота столбиков отражает среднее значение люциферазной активности как минимум трех трансфекций, барами указана величина стандартной ошибки среднего (SEM).
Фиг.6. Анализ экспрессии гена тимидинкиназы HSV в трансфицированных клетках НЕК293. Клетки линии НЕК293 трансфицировали конструкциями TK_pGL3 (беспромоторный вектор), pSV40_TK_pGL3 (промотор SV40), pCMV_TK_pGL3 (промотор CMV с энхансером) и pSurv4_TK_pGL3 (промотор гена сурвивина Psurv4) для экспрессии гена тимидинкиназы HSV. Слева приведен маркер молекулярных масс белков. Молекулярная масса тимидинкиназы HSV - 43 кДа.
Фиг.7. Микрофотографии клеток линии НТ1080, трансфицированных следующими конструкциями: А - pEGFP-N1 (отрицательный контроль), В - pCMV-HSVtk-pGL3, С - phTERT-HSVtk-pGL3, D - phSurv-HSVtk-pGL3 после культивирования в течение 72 часов в питательной среде, содержащей раствор ганцикловира в концентрации 0, 20, 200 мкМ. Над фотографиями приведены концентрации ганцикловира.
Фиг.8. Выживаемость клеток, трансфицированных векторами, экспрессирующими FCU1 в присутствии 5-FC. Клетки трансфицировали векторами, содержащими ген FCU1 без промотора (pGL3-(no promoter)-FCU1) под контролем промотора гена BIRC5 (pGL3-pBIRC5-1.5-FCU1) либо промотора CMV (pGL3-pCMV-FCU1). Спустя 24 часа после трансфекции к клеткам добавляли 5-фторцитозин (5-FC). После 120 часов инкубации в среде с 5-FC клетки окрашивали красителем MTS и измеряли оптическую плотность клеточных экстрактов. Высота столбцов отражает процент выживших клеток спустя 120 ч после начала эксперимента. Выживаемость рассчитывали как процентное отношение оптической плотности в экстрактах клеток, обработанных 5-FC, к оптической плотности в экстрактах клеток, не обработанных 5-FC. Приведены стандартные ошибки среднего (SEM).
Фиг.9. Вестерн-блот анализ белков HSV-tk и GAPDH в экстрактах клеток Calu-1 и НТ1080: 1 - нетрансфицированные клетки; 2 - клетки, трансфицированные контрольной плазмидой pEGFP-N1; 3, 4, 5, 6 - клетки, трансфицированные pCMV/E-tk, pSurv-tk, phTERT-CMV-tk, ΔLTRep-tk, соответственно; 7 - клетки, трансфицированные pSurv-tat и ΔLTRep-tk; 8 - phTERT-CMV-tat и ΔLTRep-tk.
Фиг.10. Влияние внутриопухолевой инъекции полиплекса с геном BSVtk на скорость роста опухолей, вызванных инокуляцией клеток LLC1 мышам C57black. Введение полиплекса проводили на 17 и 21 день после инокуляции клеток, внутрибрюшинные инъекции ганцикловира в дозе 75 мг/кг (дважды в день) проводили в течение 10 дней, начиная с 19 дня после инокуляции клеток за исключением дня повторного введения полиплексов. Данные представляют собой средние значения для группы из 8 животных, указанные на графике разбросы представляют собой стандартную ошибку среднего. По оси абсцисс указано время, прошедшее с момента инокуляции клеток.
Фиг.11. Влияние внутриопухолевой инъекции полиплекса с геном HSVtk на выживаемость мышей с опухолями, вызванных инокуляцией клеток LLC1 мышам C57black.
Фиг.12. Влияние внутриопухолевой инъекции полиплекса с геном HSVtk на скорость роста опухолей, вызванных инокуляцией клеток мышиной меланомы Клаудмана S91 мышам DBA/2y. Клетки были подкожно инокулированы мышам линии DBA/2y по 1 миллиону клеток на мышь, на 14 день, когда опухоли достигли размера 50 мм3, раствор полиплексов был введен внутрь опухолей в объеме 50 мкл. Начиная со следующего дня, мыши получали интраперитонеальные инъекции ганцикловира 9 мг/кг 2 раза в день с интервалом 11-13 часов. На третий день интратуморальную инъекцию повторяли и вечером этого дня ганцикловир не вводили (всего мыши получили инъекцию 5 доз полиплекса и ганцикловира в течение 15 дней). На графике представлены результаты (среднее ± стандартная ошибка среднего) для мышей, получавших HSVtk сурвивинового (•), 9 мышей, и теломеразного промоторов (▲), 10 мышей, а также контрольная группа (♦), 15 мышей.
Фиг.13. Влияние внутривенного введения полиплекса на основе блок-сополимера ПЭИ-ПЭГ-МК1С и HSVtk под контролем теломеразного промотора на продолжительность жизни мышей с экспериментальными опухолями меланомы Клаудмана. Выживаемость мышей группы, получавшей полиплекс с HSVtk (7 животных), достоверно отличаются по тесту Мантела-Кокса (p<0,03, определено при помощи программного пакета GraphPad Prism 5) от выживаемости мышей контрольной группы (9 животных), получавшей только ганцикловир по той же схеме, что и опытная группа.
Таблица 1. Относительная активность двойных phSurv-phTERT и phTERT-phSurv в различных клеточных линиях. Приведено среднее значение люциферазной активности как минимум трех трансфекций ± величина стандартной ошибки среднего (SD).
Таблица 2. Активность составных частей многопрофильного двойного промотора в различных клеточных линиях. За единицу принимали активность люциферазы в экстрактах клеток, несущих экспрессионную конструкцию phTERT-pGL3. Приведено среднее значение люциферазной активности как минимум трех трансфекций ± величина стандартной ошибки среднего (SD).
Таблица 3. Размеры опухолей у животных разных групп на момент начала лечения.
Таблица 4. Ингибирование роста опухолей у животных, получавших полиплекс с HSVtk под контролем различных промоторов и ганцикловир.
* процент ингибирования рассчитывали как отношение среднего размера опухоли опытной группы по отношению к среднему размеру контрольной группы, скорректированному согласно различиям в среднем размере опухоли на момент начала лечения (см. таблицу 3). Приведен средний процент ингибирования с 4 по 14 после начала лечения (от второго введения полиплекса до момента гибели первого животного в контрольной группе);
** достоверность отличия рассчитывали по критерию Вилкоксона; здесь и далее достоверность различия оценивали при помощи программы Statistica 7 (StatSoft).
Таблица 5. Средние размеры опухолей у животных на 14 день с момента первого введения полиплекса.
* достоверность отличия при сравнении каждой группы, получавшей полиплекс с HSVtk, с контрольной группой по критерию Манна-Уитни.
Примеры
Пример 1.
Конструирование экспрессионных векторов на основе двойных раковоспецифичных промоторов phTERT-phSurv и phSurv-phTERT
Промотор гена сурвивина человека (phSurv) длиной 1498 п.о. [Mityaev М. 2008. Functional significance of a putative sp1 transcription factor binding site in the survivin gene promoter. Biochemistry (Mosc). 73. 1183-91] клонировали в конструкцию phTERT-pGL3 на основе плазмиды pGL3-basic (Promega, США), несущую промотор обратной транскриптазы теломеразы человека (phTERT) и репортерный ген люциферазы светлячка. Промотор phSurv вставляли в конструкцию phTERT-pGL3 в прямой ориентации в двух положениях относительно промотора phTERT. Для этого получали кДНК гена BIRC5 длиной 1498 п.о. в результате гидролиза плазмиды phSurv-pGL3 [Mityaev M. 2008. Functional significance of a putative sp1 transcription factor binding site in the survivin gene promoter. Biochemistry (Mosc).73. 1183-91] ферментами рестрикции BglII и HindIII с последующей обработкой фрагментом Кленова. Затем полученный фрагмент кДНК гена сурвивина человека лигировали с вектором phTERT-pGL3, предварительно линеаризованной по сайту узнавания рестриктазой HindIII либо по сайту узнавания рестриктазой KpnI и обработанной большой субъединицей ДНК полимеразы I E.coli. Таким образом, в первом случае был получен вектор phTERT-phSurv-pGL3, несущий ген люциферазы под контролем двойного промотора phTERT-hSurv, где промотор phTERT находится выше промотора phSurv относительно старт-кодона гена люциферазы светлячка. Во втором случае получили вектор phSurv-phTERT-pGU, в котором промотор phSurv находится выше промотора phTERT относительно старт-кодона гена люциферазы. Структура полученных экспрессионных кассет проиллюстрирована на фиг.1.
Пример 2.
Определение активности и точек начала транскрипции полифункциональных промоторов phTERT-phSurv и phSurv-phTERT
В клетки линий Calu-1 и A375 были введены конструкции phSurv-hTERT-pGL3, phTERT-hSurv-pGL3 и контрольные конструкции phSurv-pGL3, phTERT-pGL3, через 48 часов после введения конструкций клетки собирали, выделяли суммарную РНК с использованием набора RNeasy Mini Kit ("Qiagen", США) и обрабатывали ДНКазойI ("Promega") согласно протоколам производителя. Для определения старта транскрипции использовали набор реагентов FirstChoice RLM-RACE Kit (Ambion, США) согласно рекомендациям производителя. Полученные в результате проведения полимеразной цепной реакции 5'RACE продукты (кДНК с присоединенным 5'RACE адаптером) анализировали с помощью электрофоретического разделения в агарозном геле. Далее 5'RACE продукты клонировали в вектор pAL-TA (Evrogene, Россия) и определяли их нуклеотидную последовательность. Для каждой конструкции было проанализировано не менее 12 клонов, содержащих 5'RACE продукт.
В результате проведенного анализа было показано наличие одной или двух явно выраженных точек инициации транскрипции для одиночных промоторов phSurv и phTert (фиг.2). Для двойных промоторов phTERT-phSurv, phSurv-phTERT было продемонстрировано рассеивание сайтов инициации транскрипции: промотор phTERT-phSurv содержал пять точек инициации транскрипции, phSurv-phTERT - шесть (фиг.2). В промоторе phTERT-phSurv по сравнению с промотором phSurv отсутствует «мажорная» точка инициации транскрипции и увеличивается общее число стартов транскрипции. В промоторе phSurv-phTERT также не наблюдается наличия доминирующего сайта инициации транскрипции, а количество точек инициации транскрипции увеличивается в три раза в клеточной линии Calu-1 относительно промотора phTERT.
Пример 3.
Создание полипрофильного модифицированного промотора гена сурвивина человека
1) Для получения мутантных форм промотора сурвивина, содержащих делеции и замены в сайте связывания р53, плазмиду phSurv-pGL3 [Mityaev MV, Functional significance of a putative sp1 transcription factor binding site in the survivin gene promoter. Biochemistry (Mosc) 2008; 73:1183-1191] гидролизовали по сайтам узнавания рестриктазой SacII, расположенным внутри последовательности промотора phSurv с последующим внутримолекулярным лигированием плазмиды. Полученную плазмиду получали в препаративных количествах, гидролизовали по сайту SacII и лигировали с фрагментами двуцепочечной ДНК из п.2) данного Примера.
2) Для внесения делеции в размере трех нуклеотидов (делеция Δ11-13) или замены двадцатого нуклеотида G на Т (замена 20G/T) в последовательность сайта связывания р53 промотора phSurv использовали химически синтезированные олигонуклеотиды. Для отжига олигонуклеотидов друг на друга их смешивали попарно в эквимолярных количествах и инкубировали при 100°C в течение 3 минут, после чего медленно охлаждали до 25°C. Полученные таким образом короткие фрагменты двухцепочечной ДНК содержали модифицированные сайты связывания р53 и участки узнавания рестриктазой SacII.
Пример 4.
Доставка генетических конструкций в эукариотические клетки
1) Генетические конструкции в молярном соотношении один из векторов из Примера 1 или из Примера 3 и вспомогательный нормировочный вектор pRL-TK («Promega», США) 10:1 доставляли внутрь клеток в клеточных культурах с использованием Липофектамина 2000 («Invitrogen», США) согласно рекомендациям производителя.
2) Генетические конструкции, несущие под контролем промотора сурвивина человека ген тимидинкиназы вируса простого герпеса или ген рекомбинантной цитозиндизаминазы дрожжей, доставляли в клетки, культивируемые в шестилуночном планшете с использованием Липофектамина 2000 («Invitrogen», США). Для трансфекции использовали 4 мкг плазмидной ДНК на лунку шестилуночного планшета.
3) Генетические конструкции ΔLTRep-HSVtk и phSurv-tat или phTERT в молярном соотношении ΔLTRep-HSVtk и phSurv-tat или phTERT 1:4 (количественное соотношение 2 мкг:8 мкг) доставляли в клетки, культивируемые в флаконе (25 см2) с использованием Липофектамина 2000 («Invitrogen», США).
Пример 5.
Определение активности многопрофильных промоторов в раковых клетках
Через 48 часов после введения в клетки одной из конструкций из Примера 1 или из Примера 3 и нормировочной плазмиды pRL-TK измеряли активности люцифераз светлячка и Renilla reniformis в клеточных экстрактах с помощью набора реактивов Dual-Luciferase Reporter Assay System («Promega», США). Значения активности люциферазы светлячка нормировали относительно активности люциферазы R.Reniformis плазмиды pRL-TK с целью снижения погрешности, связанной с различной эффективностью доставки экспрессионных конструкций в клетки в серии независимых экспериментов.
Среднюю люциферазную активность и стандартную ошибку среднего рассчитывали по формуле sx-bar=(s2/n)1/2, где s - совокупная дисперсия, n - число наблюдений.
В результате проведенного анализа было установлено, что активность двойных тандемных промоторов phTERT-phSurv, phSurv-phTERT часто превосходит активность обоих одиночных phTERT, phSurv промоторов. Относительная активность исследованных промоторов в разных клеточных линиях приведена на фиг.3 и в таблице 1. Как видно из таблицы 1, активности двойных промоторов phTERT-phSurv и phSurv-phTERT примерно соответствуют сумме активностей отдельных промоторов. Особенно это соблюдается для phSurv-phTERT тандема. Активность phTERT-phSurv тандема в двух случаях (клеточные линии НТ1080 и А375) заметно ниже и не превышает активности даже отдельных промоторов. Усредненная по клеточным линиям активность промотора phSurv-phTERT несколько ниже, чем усредненная активность суммы отдельных промоторов. Активность тандема phTERT-phSurv ниже активности тандема phSurv-phTERT.
Эти данные позволяют заключить, что использование тандемных промоторов для экспрессии генов предпочтительнее, чем использование каждого из одинарных промоторов, и phSurv-phTERT тандем предпочтительнее phTERT-phSurv тандема по активности экспрессии в разных клетках.
Пример 6.
Определение спектра активности составных частей многопрофильного двойного промотора в раковых клетках
В клеточные линии NCI-H1299, Calu-1, НТ1080, PANC-1, HCT116, AsPC1, MelCher, A431, T47D и MelKor вводили конструкции, содержащие репортерный ген люциферазы светлячка под контролем промотора phTERT, либо его модификации минимальным промотором цитомегаловируса (phTERT-CMV) или синтетическим ТАТА-боксом (phTERT-TATA), либо phSurv или его модификации phSurvD или промотора сурвивина мыши (pmSurv). Транскрипционную активность данных промоторов в вышеперечисленных клеточных линиях определяли, как описано в Примере 5. Результаты анализа приведены в таблице 2. По результатам анализа установлено, что базовый промотор phTERT является наименее активным среди промоторов на его основе, а наиболее оптимальной версией промотора phTERT является его модификация phTERT-CMV, показавшая максимальную активность в 8 из 10 клеточных линий (таблица 2). Аналогичный анализ для промотора сурвивина и его вариантов показал, что наиболее активным вариантом в подавляющем большинстве клеточных линий (в 8 из 10) является промотор сурвивина мыши pmSurv, в то время как базовый промотор сурвивина человека phSurv4 проявлял наименьшую активность.
Пример 7.
Детекция белка р53 методом Вестерн-блот анализа в трансфицированных раковых клетках
Наличие белка р53 в клетках, предварительно трансфицированных одной из конструкций из Примера 3 и плазмидой pCMV-p53, регистрировали с помощью Вестерн-блот анализа. Через 48 часов после введения конструкций клетки разрушали нагреванием в лизирующем буфере. Полученные лизаты клеток подвергали денатурирующему электрофорезу белков в полиакриламидном геле по Лэммли.
Далее проводили перенос фракционированных белков из геля на мембрану и анализировали методом Вестерн-блоттинга с моноклональными антителами, специфически узнающими белок р53. Для контроля количества белка в клеточных экстрактах использовали окраску с антителами к глицеральдегид-3-фосфат дегидрогеназе (GAPDH).
Белок р53 не обнаруживался в нетрансфицированных клетках и клетках линии Calu-1, трансфицированных только плазмидами, содержащими различные варианты промотора сурвивина, без одновременной трансфекции плазмидой pCMV-p53 (фиг.4). Такой результат объясняется отсутствием в клетках гена, кодирующего белок р53. При трансфекции клеток возрастающим количеством плазмиды pCMV-p53 происходило соответствующее значительное увеличение содержания р53 в клетках. Содержание GAPDH незначительно различалось в разных пробах, что свидетельствует о небольшой разнице в содержании тотального белка в исследуемых пробах.
Пример 8.
Анализ активности модифицированного промотора сурвивина человека в раковых клетках, экзогенно экспрессирующих белок р53
В раковые клетки, в норме не продуцирующие белок р53, доставляли одну из конструкций из Примера 3 и стандартную конструкцию pCMV-p53 (набор «р53 Dominant-Negative Vector Set»), несущую ген, кодирующий белок р53, под контролем сильного промотора цитомегаловируса. В каждом эксперименте конструкции доставляли в клетки совместно с нормировочной плазмидой pRL-TK. Через 48 часов после введения конструкций в клетки проводили определение активности мутантных форм промотора сурвивина человека, как описано в Примере 5.
Как видно на фиг.5, увеличение содержания белка р53 в клетках линии Calu-1 вызывает сильную репрессию активности промоторов phSurv4 и 20G/T (см. также результаты Вестерн-блот анализа на фиг.4). При этом динамика снижения активности данных промоторов сходна. Активность промоторного варианта Δ11-13 увеличивалась при котрансфекции 1 нг плазмиды pCMV-p53. При дальнейшем увеличении содержания белка р53 в клетках активность промотора снижалась, но медленней, чем в случае других промоторов.
Пример 9
Детекция белка тимидинкиназы вируса простого герпеса (HSVtk) с помощью Вестерн-блот анализа в трансфицированных раковых клетках
Наличие белка HSVtk в клетках, в которые предварительно были введены конструкции, содержащие ген HSVtk под контролем различных промоторов, в частности промотора phSurv, регистрировали с помощью Вестерн-блот анализа, как описано в Примере 7, за исключением использования моноклональных антител, специфически узнающих белок HSV-tk.
В результате анализа было установлено, что количество рекомбинантной тимидинкиназы вируса простого герпеса в клетках, трансфицированных конструкцией phSurv-HSVtk-pGL3 значительно выше, чем количество тимидинкиназы в клетках, трансфицированных контрольной плазмидой pSV40-HSVtk-pGL3, содержащей ген HSVtk под контролем конститутивного промотора вируса SV40 (фиг.6). При этом эффективность экспрессионной конструкции pSurv-HSVtk-pGL3 ниже, чем эффективность конструкции pGL3-pCMV-HSVtk, что объясняется разницей в активности промотора phSurv и промотора CMV.
Пример 10
Определение активности тимидинкиназы вируса простого герпеса
Способность клеточных экстрактов, содержащих тимидинкиназу вируса простого герпеса, фосфорилировать ганцикловир была проверена способом, описанным [Mercer К. 2002. Mutation of herpesvirus thymidine kinase to generate ganciclovir-specific kinases for use in cancer gene therapies. Protein Eng. 15. 903-11]. Инкубационная проба содержала: H3-GCV, АТФ, BSA, буфер В и клеточный экстракт. Реакцию запускали добавлением клеточного экстракта. Экстракты клеток инкубировали при 37°C в течение 1-24 часов в зависимости от типа клеток. Реакцию останавливали нанесением инкубационной смеси на мишени из анионообменной бумаги. Подсохшие мишени опускали в раствор формиата аммония, далее промывали 70% этанолом, сушили на воздухе и определяли их радиоактивность в сцинтилляторе. Вычисляли разницы между радиоактивностью образцов с экстрактами трансфицированных и нетрансфицированных эукариотических клеток. Полученные значения отражали в количестве фосфорилированного ганцикловира (GCV) в пробах в импульсах/мин. Их пересчет на количество образующегося фосфорилированного GCV проводили с помощью калибровочной кривой, отражающей зависимость радиоактивности (имп./мин) H3-GCV от его количества.
Пример 11.
Функциональный тест на способность HSV-tk подавлять рост опухолевых клеток
В раковые клетки вводили экспрессионные конструкции pEGFP-N1 (Clonetech, США), pCMV-HSVtk-pGL3, phTERT-HSVtk-pGL3, phSurv-HSVtk-pGL3, как описано в Примере 4 п.2). Спустя 48 часов после трансфекции к клеткам добавляли раствор ганцикловира различной концентрации («Cymeven Roche», Швейцария) в среде DMEM/F12 (1:1) с антибиотиками. Через 72 часа после добавления ганцикловира клетки фотографировали при помощи фазово-контрастного микроскопа ТЕ2000, «Nikon». Как видно на микрофотографиях (фиг.7), инкубация клеток, содержащих конструкции pCMV-HSVtk-pGL3, phTERT-HSVtk-pGL3 и phSurv-HSVtk-pGL3 с ганцикловиром, приводит к появлению большого количества округлившихся клеток, находящихся в состоянии апотоза, и их гибели, при этом эффект конструкции pCMV-HSVtk-pGL3, несущий ген HSVtk, под контролем сильного неспецифичного промотора цитомегаловируса в сочетании с ганцикловиром сравним с эффектом, оказываемым на клетки конструкциями phTERT-HSVtk-pGL3 или phSurv-HSVtk-pGL3 в комбинации с ганцикловиром (фиг.7, B, C, D).
Пример 12
Тест на цитотоксичность рекомбинантной цитозиндизаминазы дрожжей с последующим добавлением 5-фторцитозина
В раковые клетки доставляли плазмидную ДНК, несущую ген рекомбинантной цитозиндизаминазы дрожжей под контролем промотора phSurv, как описано в Примере 4, п.2. Спустя 48 часов после введения ДНК клетки пересевали на 96-луночный планшет в количестве 2000 клеток на лунку, далее к клеткам добавляли раствор 5-фторцитозина (Sigma Chemical Со.) в концентрации 0, 50, 200 и 500 мкМ. Через 72 часа после добавления 5-фторцитозина проводили окрашивание клеток красителем MTS (CellTiter 96 Non-Radioactive Cell Proliferation Assay (MTS), Promega, США) для определения количества выживших клеток. Введение экспрессионных конструкций pGL3-pCMV-FCU1, несущей ген рекомбинантной цитозиндезаминазы под контролем сильного неспецифичного промотора цитомегаловируса, и pG13-pBIRC5-1.5-FCU1, содержащей ген цитозиндезаминазы под контролем опухолеспецифичного промотора гена сурвивина человека, в сочетании с 5-фторцитозином приводит к гибели клеток линии НЕК293 и Calu-1 (фиг.8). Цитотоксический эффект на раковые клетки конструкции pGL3-pBIRC5-1.5-FCU1 в сочетании с 5-фторцитозином сравним с эффектом, оказываемым контрольной конструкцией pGL3-pCMV-FCU1 (в случае использования конструкции pGl3-pBIRC5-1.5-FCU1 погибает 70% раковых клеток конструкции pGL3-pCMV-FCU1 - 85%).
Пример 13.
Оценка способности Tat-TAR-системы ВИЧ-1 усиливать экспрессию гена HSV-tk в опухолевых клетках
Согласно литературным источникам 5'LTR ВИЧ-1 обладает свойствами индуцибельной промоторной системы, в которой вирусный белок tat связывается с участком мРНК, соответствующим TAR-последовательности LTR, что приводит к трансактивации промотора LTR и к усилению транскрипции подконтрольных генов. В отсутствии белка tat экспрессия генов минимальна. Для проверки способности tat-TAR-системы усиливать экспрессию гена HSV-tk были получены следующие конструкции: ΔLTRep-HSVtk, несущая ген HSVtk под контролем LTR ВИЧ-1, phSurv-tat и phTERT-CMV-tat, содержащие ген вирусного белка tat под опухолеспецифичными промоторами phSurv и phTERT-CMV, соответственно. В клетки линии Calu-1 и НТ1080 вводили либо бинарные системы ΔLTRep-HSVtk+phSurv-tat или ΔLTRep-HSVtk+phTERT-CMV-tat либо отдельные конструкции pSurv-HSVtk, phTERT-CMV-HSVtk, pCMV-HSVtk, как описано в Примере 4, п.3. Спустя 48 часов после введения конструкций в клетки готовили клеточные экстракты и анализировали уровень экспрессии трансгена методом иммуноблотинга с использованием антител к HSV-tk, как описано в Примере 9. В качестве отрицательных контролей использовали экстракты нетрансфицированных клеток, а также экстракты клеток, трансфицированных контрольной плазмидой pEGFP-N1 («Clontech», США). В обеих клеточных линиях уровень экспрессии гена HSVtk под контролем phSurv и phTERT-CMV был очень низким и слабо детектировался методом Вестерн-блот (фиг.8, д.4, 5), в отличие от сильной экспрессии под контролем конститутивного промотора pCMV (фиг.8, д.3). Однако, в случае, когда клетки трансфицировали бимодальными системами, происходила активная потенциация экспрессии HSV-tk с промотора LTR HIV-1, что приводило к повышению уровня экспрессии данного гена (фиг.8, д.7 и 8). В обеих клеточных линиях уровень экспрессии гена HSV-tk в бинарной системе с опухолеспецифичными промоторами phTERT-CMV и phSurv был сопоставим с уровнем экспрессии этого гена в конструкции pCMV-HSVtk (фиг.4.13, 8 и 3). Таким образом, бимодальная система tat-TAR позволяет получать высокий уровень экспрессии подконтрольного гена, сравнимый с уровнем одной из самых сильных промоторных систем pCMV.
Пример 14.
Влияние внутриопухолевого введения полиплексов с геном HSVtk под контролем опухолеспецифических промоторов на скорость роста опухолей при суицидной генотерапии у мышей с легочной карциномой Льюиса
Суицидную генотерапию проводили, используя терапевтический ген HSVtk, доставляемый в клетки экспериментальных опухолей мышей при помощи полиплексов, и пролекарство ганцикловир.
Для работы использовали мышей линии C57black/6J (Филиал «Столбовая» ГУ НЦБМТ РАМН), средний вес на начало эксперимента - 19,8±0,2 (здесь и далее среднее ± стандартная ошибка среднего). Животные содержались на стандартном гранулированном сухом корме.
Клетки линии карциномы легкого мыши линии LLC1 - карциномы Льюиса - культивировали на среде DMEM/F12 (Gibco) с 10% эмбриональной сыворотки крупного рогатого скота в CO2-инкубаторе в газовой смеси, состоящей из воздуха с 5% CO2. Для получения экспериментальных опухолей клетки LLC1 инокулировали подкожно в заднюю правую часть спины в количестве 1·106 клеток на животное в объеме 20 мкл, в среде DMEM/F12. Предварительно (за 1-2 дня до начала эксперимента) с участка, в который вводили клетки (1,5-2 см2), волосы были удалены под авертиновым наркозом. Начиная с 13 дня с момента инокуляции, после появления у животных пальпируемых опухолей, производили измерение размеров опухолей (длина, ширина, высота) при помощи электронного штангенциркуля. Объем опухоли рассчитывали по формуле эллипсоида. Критерием эвтаназии служило достижение опухоли 10% от массы животного.
Для приготовления полиплексов использовали следующие плазмиды: в качестве контроля - pGL3 basic vector (Promega, содержит ген люциферазы Photinus pyralis, не содержит промотора); pSurv4_TK_pGL3 (содержит ген HSVtk, под контролем промоторной области гена сурвивина человека); phTERT_TK (содержит ген HSVtk под контролем промотора теломеразы человека). Все плазмиды были выделены при помощи набора EndoFree Plasmid Maxi Kit (Qiagen). Все полиплексы были приготовлены с использованием блок-сополимера полиэтиленимин-полиэтиленгликоль-(ТАТ-пептид) (ПЭИ-ПЭГ-ТАТ), синтезирован в лаборатории молекулярной генетики внутриклеточного транспорта ИБГ РАН. Полиплексы готовили при помощи быстрого смешивания раствора ПЭИ-ПЭГ-ТАТ с раствором ДНК в изотоническом растворе глюкозы с 10 мМ 4-(2-гидроксиэтил)-1-пиперазинэтансульфоната, pH 7, в конечной концентрации полиплекса 80 мкг ДНК в миллилитре при соотношении азот:фосфат = 10. Препараты полиплекса готовили в день эксперимента в стерильных условиях. Перед введением животным в препараты добавляли гентамицин до конечной концентрации 50 мкг/мл.
Введение полиплекса проводили при помощи внутриопухолевой инъекции в объеме 50 мкл на животное при помощи инсулинового шприца (BD Micro-Fine Plus 0,5 мл, 12,7 мм × 0,33 мм) на 17 и 21 день после инокуляции клеток животным. Каждая опытная группа животных, получавших полиплекс с геном HSVtk под контролем различных промоторов, и ганцикловир, состояла из 8 животных, в качестве контрольной группы (8 животных) использовали группу, получавшую полиплекс с геном люциферазы pGL3 basic vector. Животные были равномерно распределены по группам согласно измеренным размерам опухолей. Средний размер опухолей на момент начала лечения был 38 мм3, средние размеры опухолей для каждой из групп приведены в таблице 3.
Ганцикловир (Hoffmann-La Roche, 4 мг/мл в стерильном растворе Хэнкса с 50 мкг/мл гентамицина) вводили интраперитонеально два раза в день в течение 10 дней в дозе 75 мг/кг на введение, начиная со следующего дня после введения полиплекса. В день повторного введения полиплекса ганцикловир не вводили.
Как показали результаты проведенных измерений, рост опухолей у животных, получавших внутриопухолевые инъекции полиплекса с геном HSVtk, был заметно медленнее, чем у животных контрольной группы (фиг.10). Выраженных отличий в размерах опухолей у животных групп, получавших полиплекс с терапевтическим геном под контролем сурвивинового и теломеразного промоторов, обнаружено не было.
Усредненный по времени лечения средний размер опухолей у групп, получавших полиплекс с HSVtk под контролем различных промоторов, достоверно отличался от среднего размера опухолей животных контрольной группы (таблица 4).
Средние размеры опухоли на момент гибели первого животного в контрольной группе (14 день с момента первого введения полиплекса) приведены в таблице 5. Средний размер опухоли у животных, получавших полиплекс с HSVtk под контролем теломеразного и цитомегаловирусного промоторов, достоверно отличался от среднего размера опухоли в контрольной группе.
Полученные данные указывают на наличие достоверного противоопухолевого эффекта тимидинкиназы под опухолеспецифическими промоторами в сочетании с введением ганцикловира при доставке гена в клетки опухоли карциномы Льюиса при внутриопухолевом введении полиплекса с терапевтическим геном.
Пример 15.
Влияние внутриопухолевого введения полиплексов с геном HSVtk под контролем опухолеспецифических промоторов на продолжительность жизни животных при суицидной генотерапии у мышей с легочной карциномой Льюиса
Двукратное внутриопухолевое введение полиплексов с плазмидами, несущими ген HSVtk под контролем опухолеспецифических промоторов, и 10-дневный курс ганцикловира (условия проведения эксперимента указаны в Примере 14) приводило к достоверному увеличению продолжительности жизни мышей с экспериментальными опухолями карциномы Льюиса. Средняя продолжительность жизни мышей, получавших лечение полиплексом с геном HSVtk и последующим введением ганцикловира, увеличивалась на 30-40 процентов по сравнению с контрольной группой и достоверно отличалась от нее (фиг.2, таблица 6).
Пример 16.
Влияние внутриопухолевого введения полиплексов с геном HSVtk под контролем опухолеспецифических промоторов на рост опухолей при суицидной генотерапии у мышей с меланомой Клаудмана
Для эксперимента использовали мышей линии DBA/2y (Филиал «Столбовая» ГУ НЦБМТ РАМН). Животные содержались на стандартном гранулированном сухом корме. Клетки меланомы Клаудмана S91 (клон М3) культивировали на среде DMEM/F12 (Gibco) с 10% эмбриональной коровьей сыворотки в CO2-инкубаторе в газовой смеси, состоящей из воздуха с 5% СО2. Для получения экспериментальных опухолей клетки инокулировали подкожно в заднюю правую часть спины в количестве 1·106 на животное в объеме 20 мкл, в среде DMEM/F12. Предварительно (за 1-2 дня до начала эксперимента) с участка, в который вводили клетки (1,5-2 см2), волосы были удалены под авертиновым наркозом. Начиная с 14 дня с момента инокуляции, после появления у животных пальпируемых опухолей производили измерение размеров опухолей (длина, ширина, высота) при помощи электронного штангенциркуля. Объем опухоли рассчитывали по формуле эллипсоида. Критерием эвтаназии служило достижение опухоли объема 1500 мм3. Полиплексы с соотношением азот:фосфат равном 10 были приготовлены с использованием блок-сополимера полиэтиленимин-полиэтиленгликоль-ТАТ-пептид (ПЭИ-ПЭГ-ТАТ), синтезирован в лаборатории молекулярной генетики внутриклеточного транспорта ИБГ РАН.
Для приготовления полиплексов использовали следующие плазмиды: pSurv4_TK_pGL3 (содержит ген HSVtk под контролем промоторной области гена сурвивина человека); phTERT_TK (содержит ген HSVtk под контролем промотора теломеразы человека). Плазмиды были выделены при помощи набора EndoFree Plasmid Maxi Kit (Qiagen). Полиплексы готовили при помощи быстрого смешивания раствора блок-сополимера с раствором ДНК в изотоническом растворе глюкозы с 10 мМ 4-(2-гидроксиэтил)-1-пиперазинэтансульфоната, pH 7, в конечной концентрации полиплекса 80 мкг ДНК в миллилитре. Препараты полиплекса готовили в день эксперимента в стерильных условиях. Перед введением животным в препараты добавляли гентамицин до конечной концентрации 50 мкг/мл.
Результаты проведенного эксперимента показали (Фиг.12), что внутриопухолевое введение HSVtk приводит к торможению роста опухоли по отношению к контролю на 46% для гена под контролем сурвивинового промотора и на 61% под контролем теломеразного промотора (p<0,05 и p<0,005 по t-критерию Стьюдента).
Полученные данные указывают на наличие достоверного противоопухолевого эффекта тимидинкиназы под опухолеспецифическими промоторами в сочетании с введением ганцикловира при доставке гена в клетки опухоли меланомы Клаудмана при внутриопухолевом введении полиплекса с терапевтическим геном.
Пример 17.
Влияние внутривенного введения полиплексов с геном HSVtk под контролем опухолеспецифического промотора на продолжительность жизни животных-опухоленосителей при суицидной генотерапии у мышей с меланомой Клаудмана
Особенностью меланом является ее высокая метастазирурующая способность, поэтому предпочтительным типом введения генотерапевтического средства для лечения меланом является внутривенное системное введение. Экспрессия рецептора меланоцит-стимулирующего гормона (□-МСГ) - одного из типов меланокортиновых рецепторов (MC1R, melanocortin 1 receptor), - как правило, заметно увеличена как в меланомных клеточных линиях, так и в меланомах человека по сравнению с нормальными меланоцитами. Для диагностики и возможного лечения важным является также то, что иммуноцитохимическое прокрашивание срезов различных тканей показало, что экспрессия этих рецепторов на меланомах значительно превышает их экспрессию на всех нормальных тканях. □-МСГ представляет собой пептид, состоящий из 13 аминокислот. Его аминокислотная последовательность одинакова у многих видов млекопитающих - человека, мыши, крупного рогатого скота и т.д. □-МСГ может связываться с четырьмя меланокортиновыми рецепторами человека (со всеми, кроме MC2R), однако обладает наиболее высоким сродством к MC1R и наиболее специфичен к нему, его сродство к MC3R ниже в 260 раз, к MC4R - в 5500 раз и к MC5R в 47500 раз. Среди других пептидов существуют лиганды с большей специфичностью, чем □-МСГ, которые взаимодействуют только с MC1R, их сродство к другим меланокортиновым рецепторам практически не измеряется. Последовательность одного из таких пептидов была использована для создания специфичного для меланом полиплекса. Блок-сополимер ПЭИ-ПЭГ - (специфический МК1с пептид) был синтезирован по схеме, использованной для создания полиплексов с ТАТ-пептидом.
Для эксперимента использовали мышей линии DBA/2y (Филиал «Столбовая» ГУ НЦБМТ РАМН). Животные содержались на стандартном гранулированном сухом корме. Клетки меланомы Клаудмана S91 (клон М3) культивировали на среде DMEM/F12 (Gibco) с 10% эмбриональной коровьей сыворотки в CO2-инкубаторе в газовой смеси, состоящей из воздуха с 5% CO2. Для получения экспериментальных опухолей клетки инокулировали подкожно в заднюю правую часть спины в количестве 1·106 на животное в объеме 20 мкл, в среде DMEM/F12. Предварительно (за 1-2 дня до начала эксперимента) с участка, в который вводили клетки (1,5-2 см2), волосы были удалены под авертиновым наркозом. Объем опухоли рассчитывали по формуле эллипсоида по данным измерения размеров опухолей (длина, ширина, высота) при помощи электронного штангенциркуля. Критерием эвтаназии служило достижение опухоли объема 1500 мм3. Полиплексы с соотношением азот:фосфат равном 20 были приготовлены с использованием блок-сополимера ПЭИ-ПЭГ-MK1c, синтезированного в лаборатории молекулярной генетики внутриклеточного транспорта ИБГ РАН
Для лечения опухоли меланомы Клаудмана S91 использовали внутривенное введение полиплекса на 3 и 8 день после подкожной инокуляции опухоли и внутрибрюшинное введение ганцикловира, 25 мг/кг, 2 раза в день, 7 инъекций после введения полиплекса, с однодневным перерывом между курсами. Введение специфического полиплекса (с блок-сополимером ПЭИ-ПЭГ-МК1С), несущего ген HSVtk под контролем теломеразного промотора, приводил к торможению роста опухолей по сравнению с контролем на 42%. Суицидная терапия с HSVtk под контролем опухолеспецифического промотора приводила также к достоверному увеличению продолжительности жизни мышей, несущих экспериментальные меланомные опухоли (Фиг.13). Медиана выживаемости у группы животных, получавших полиплекс с терапевтическим геном и ганцикловир, составила 39 дней по сравнению с 25 днями у контрольной группы, получавшей только ганцикловир (различие в 1,56 раза).
Полученные данные указывают на наличие достоверного противоопухолевого эффекта тимидинкиназы под опухолеспецифическим промотором в сочетании с введением ганцикловира при доставке гена в клетки опухоли меланомы Клаудмана при внутривенном введении специфического полиплекса с терапевтическим геном.
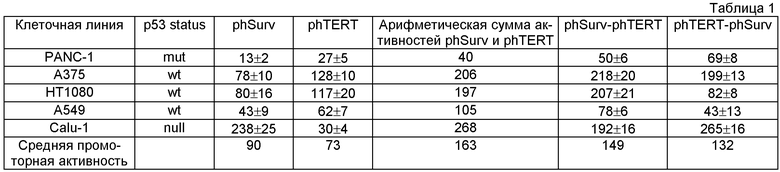
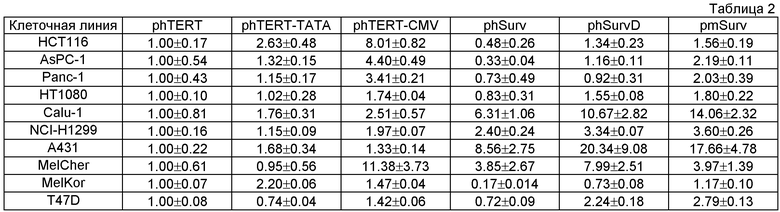
| название | год | авторы | номер документа |
|---|---|---|---|
| УНИВЕРСАЛЬНЫЕ РАКОВОСПЕЦИФИЧНЫЕ ПРОМОТОРЫ И ИХ ИСПОЛЬЗОВАНИЕ В ПРОТИВООПУХОЛЕВОЙ ТЕРАПИИ | 2013 |
|
RU2539764C2 |
| ФАРМАКОЛОГИЧЕСКАЯ КОМБИНАЦИЯ ПОЛИКАТИОННОГО НОСИТЕЛЯ ПЭГ-ПЭИ-ТАТ С ЗАКЛЮЧЕННОЙ В НЕМ ПЛАЗМИДОЙ, НЕСУЩЕЙ ТЕРАПЕВТИЧЕСКИЕ ГЕНЫ HSVtk И GM-CSF ДЛЯ ЦЕЛЕЙ ГЕНОТЕРАПИИ ОПУХОЛЕВЫХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2575077C2 |
| УНИВЕРСАЛЬНЫЙ ПРОМОТОР ДЛЯ ЭКСПРЕССИИ ТЕРАПЕВТИЧЕСКИХ ГЕНОВ В КЛЕТКАХ МЛЕКОПИТАЮЩИХ | 2013 |
|
RU2551784C1 |
| Лекарственная комбинация для ген-иммунной терапии | 2022 |
|
RU2792683C1 |
| Генотерапевтический препарат FCU1-BsFm/PP для лечения солидных опухолей | 2023 |
|
RU2824977C1 |
| СПОСОБЫ И КОМПОНЕНТЫ ИНДУКЦИИ ОПУХОЛЬ-СПЕЦИФИЧЕСКОЙ ЦИТОТОКСИЧНОСТИ | 1998 |
|
RU2214280C2 |
| Гистоны и биодеградируемые липиды как средство для доставки нуклеиновых кислот в клетки эукариот | 2015 |
|
RU2637371C2 |
| ОПРЕДЕЛЕНИЕ ТРАНСФОРМИРУЮЩЕЙ СПОСОБНОСТИ АГЕНТОВ | 1998 |
|
RU2214598C2 |
| ПРОМОТОР ДЛЯ ТКАНЕСПЕЦИФИЧЕСКОЙ ЭКСПРЕССИИ ГЕНОВ В ГЕРМИНАЛЬНЫХ ТКАНЯХ МЛЕКОПИТАЮЩИХ | 2010 |
|
RU2459870C2 |
| СРЕДСТВО ДЛЯ ГЕННОЙ ТЕРАПИИ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ | 2010 |
|
RU2447150C1 |
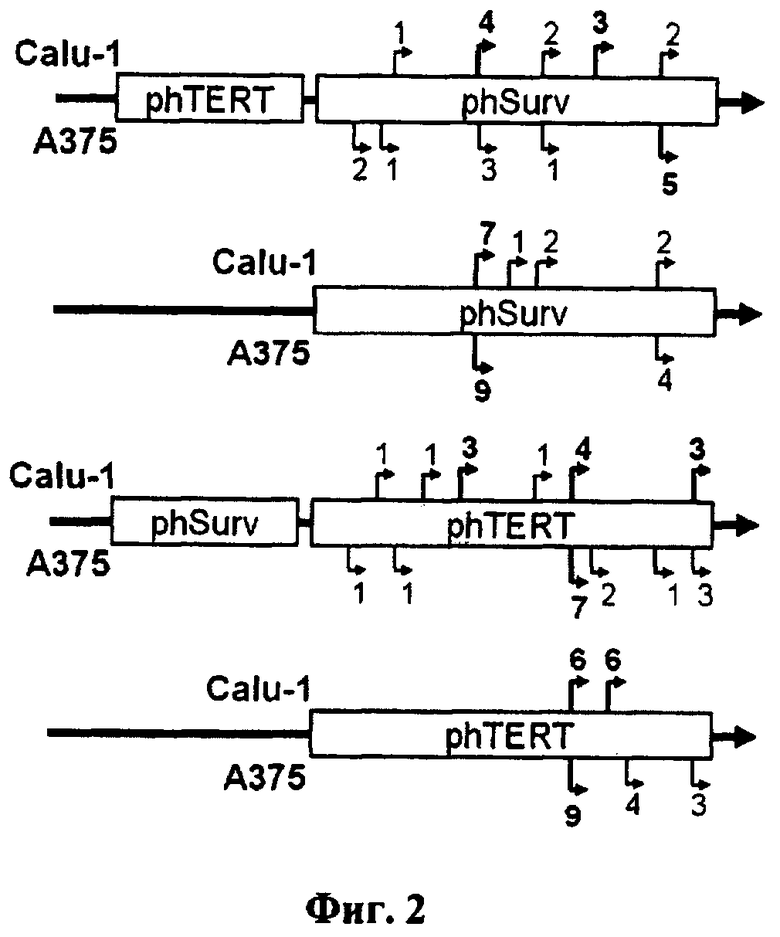
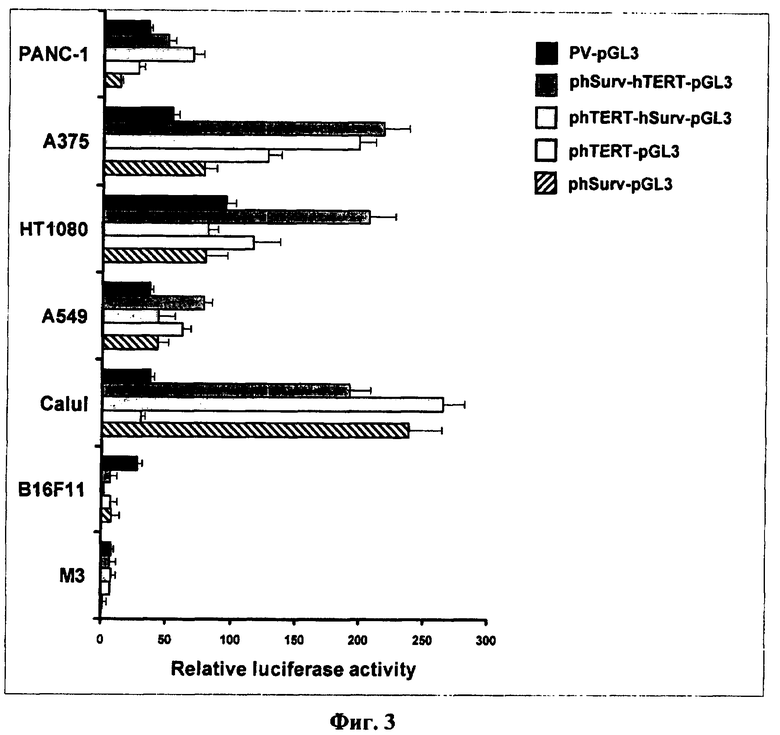

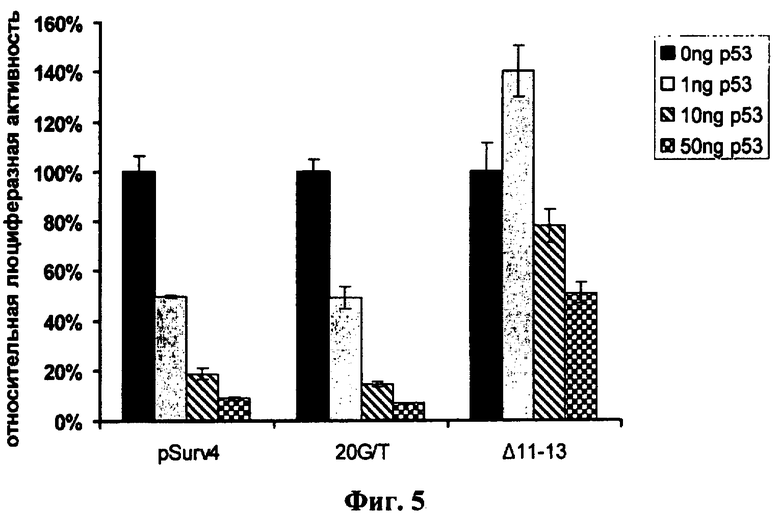
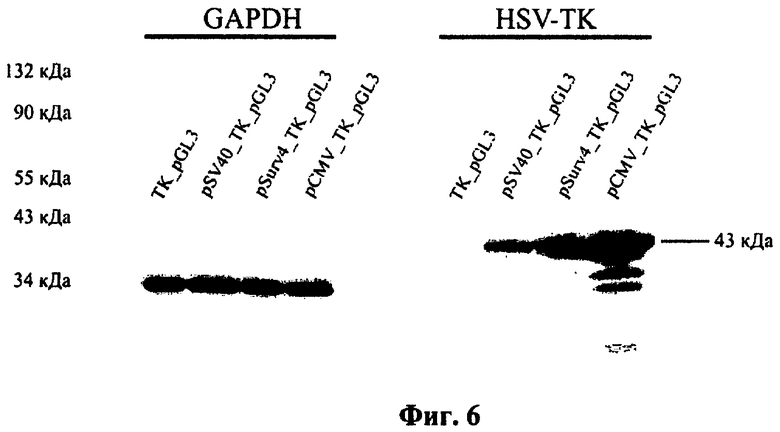
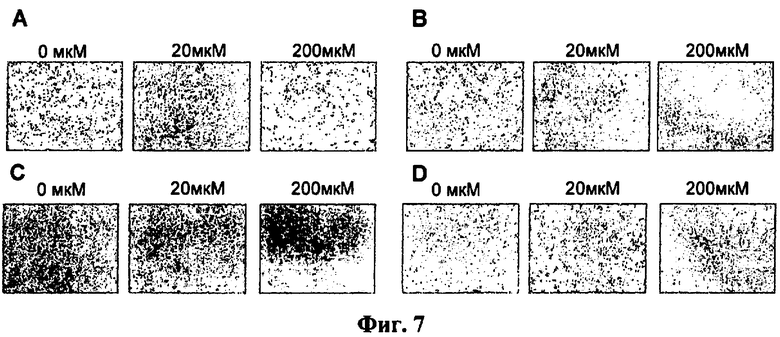
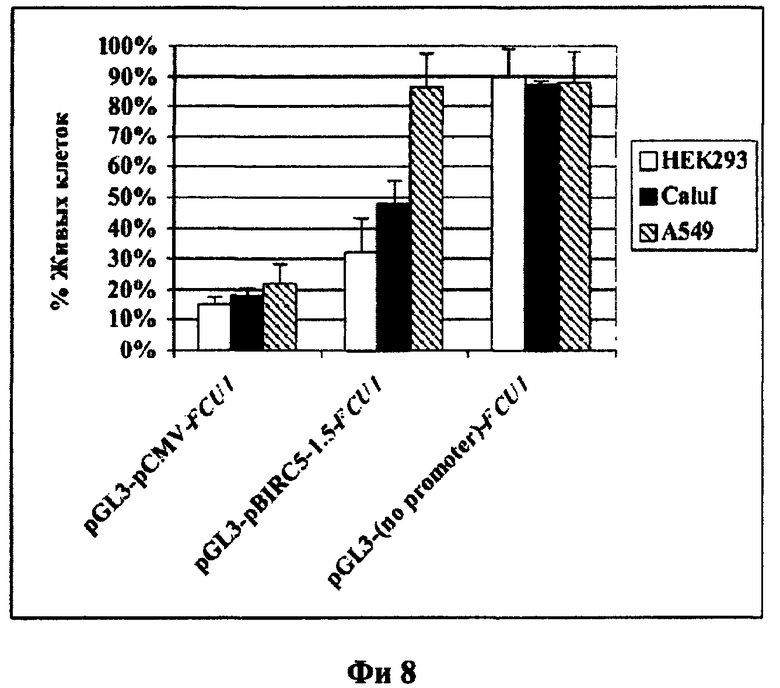
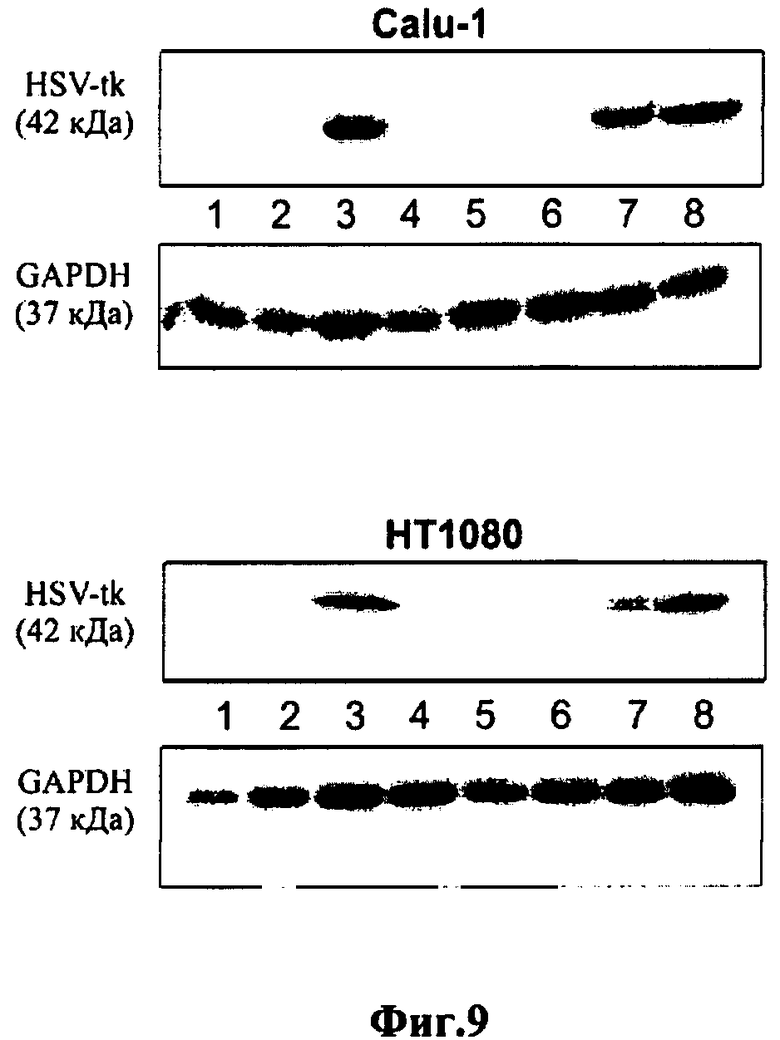
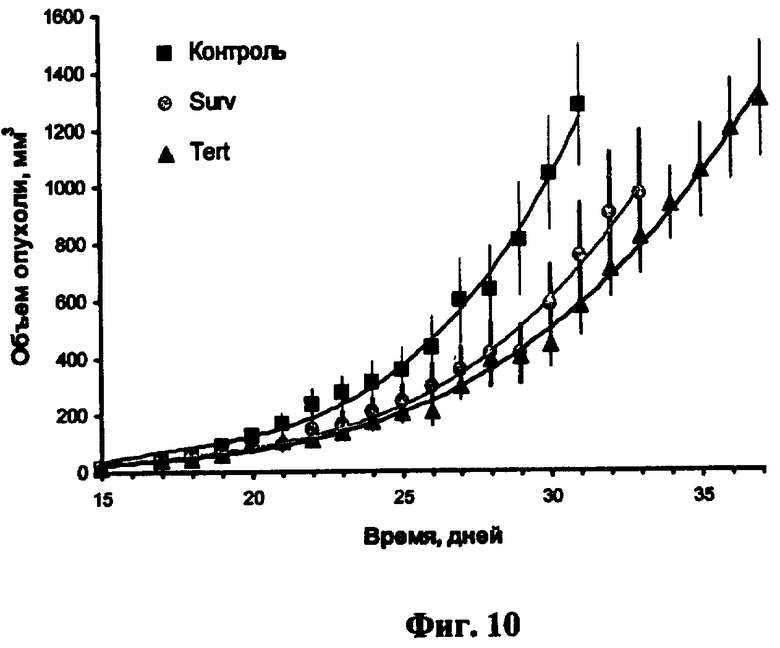
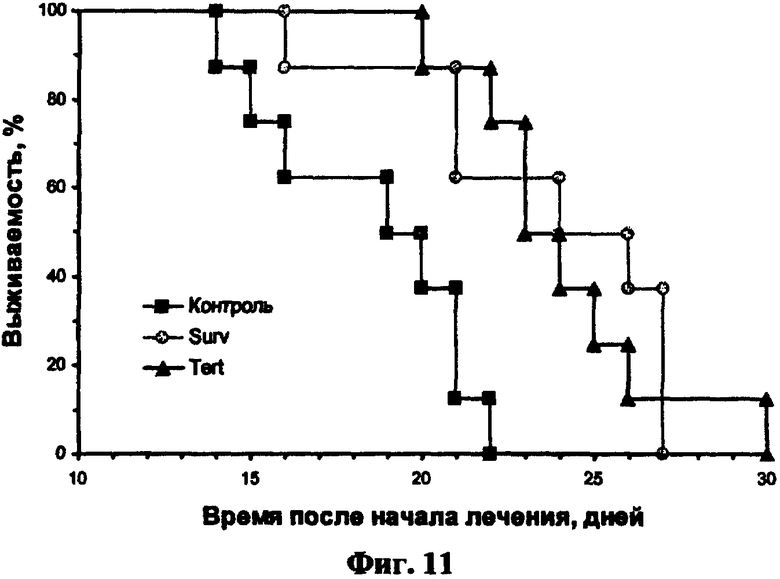
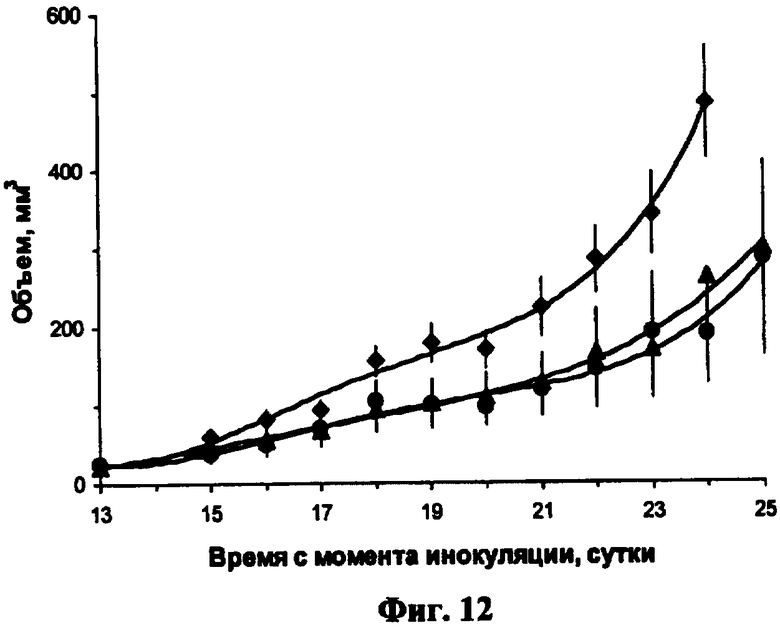
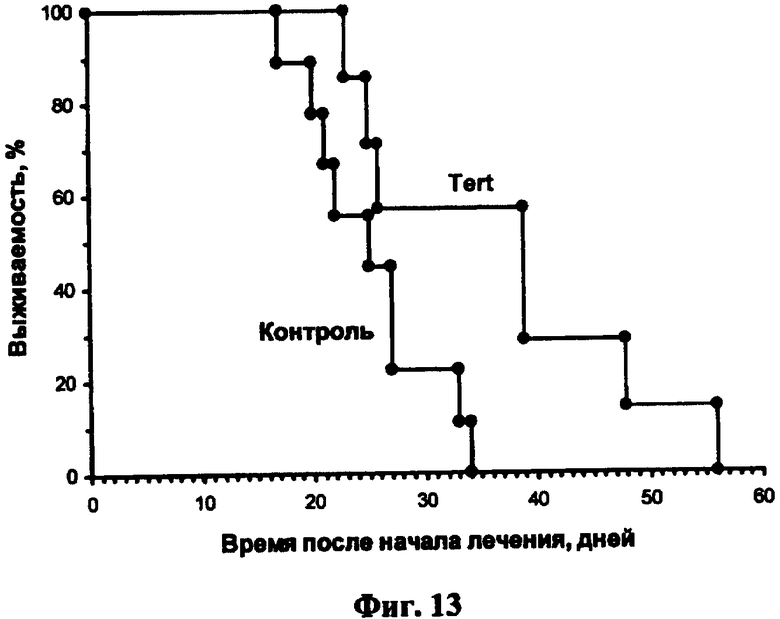
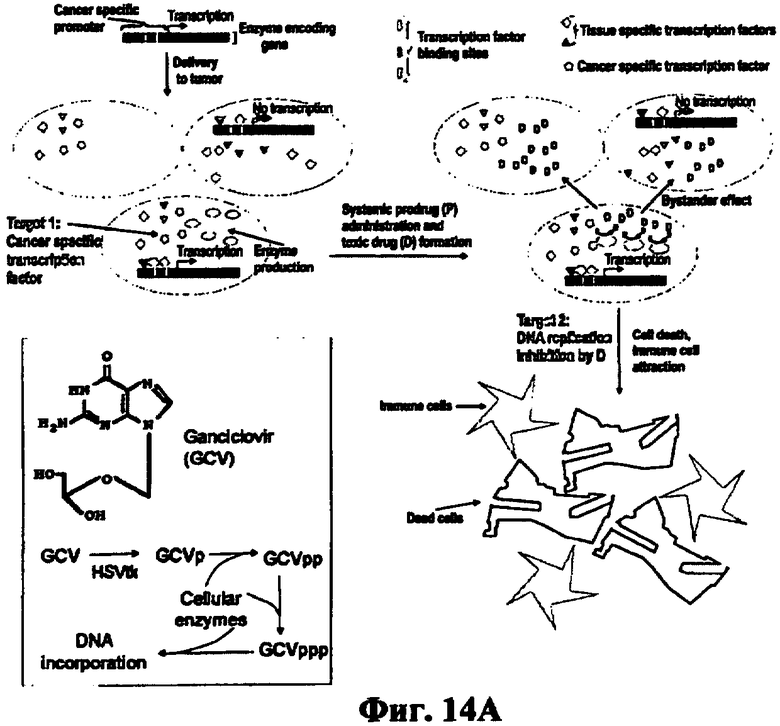
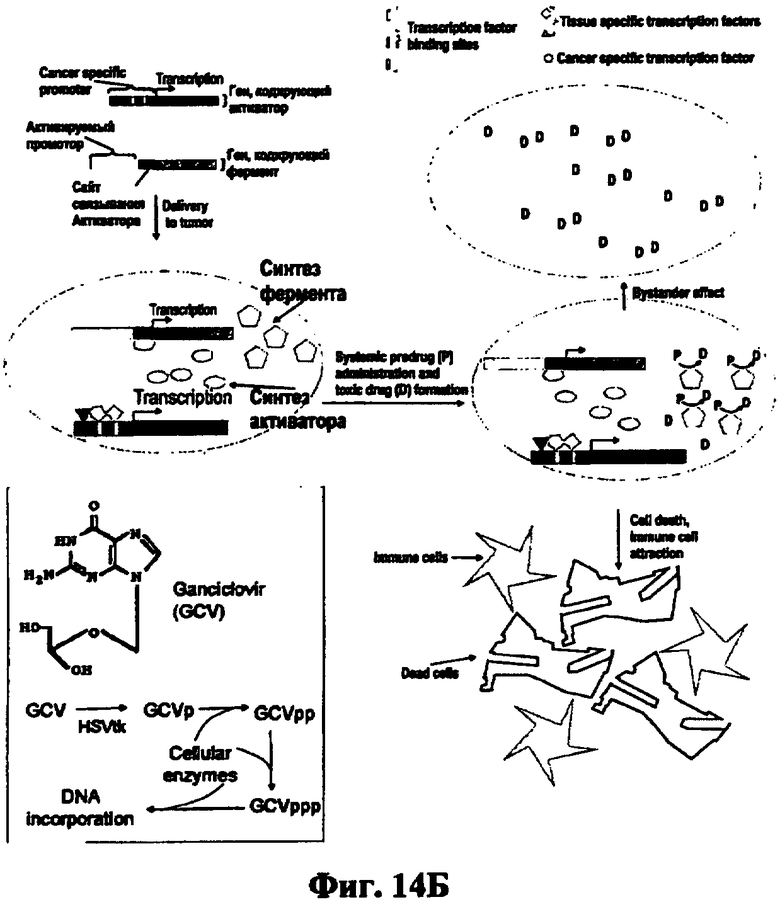
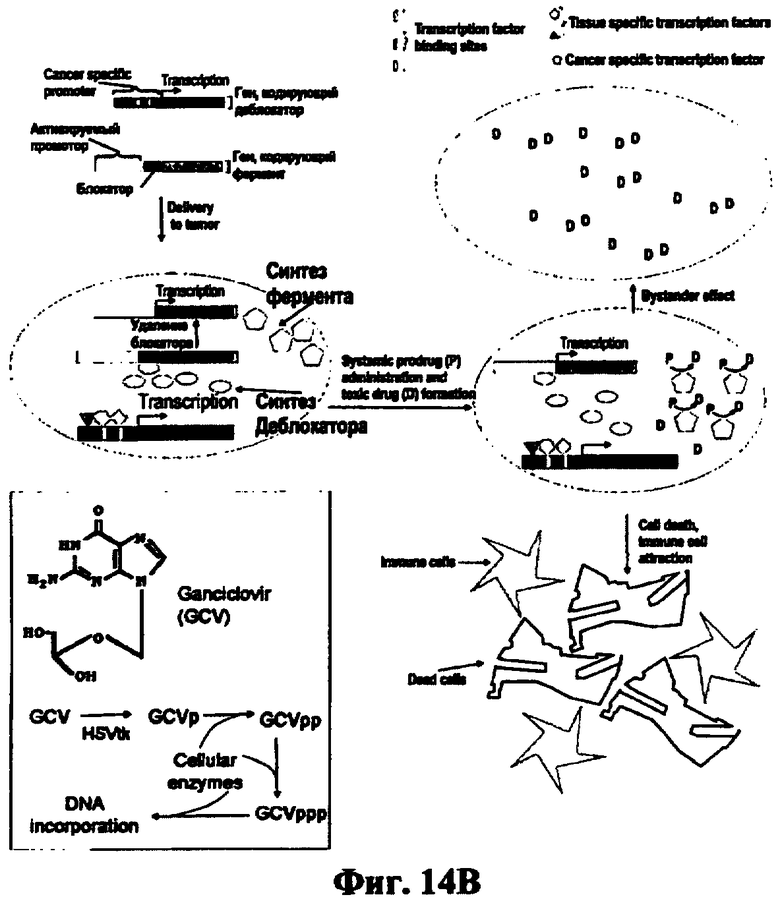
Изобретение относится к биотехнологии, в частности генной инженерии, и может быть использовано для лечения новообразований различной природы. Описан многопрофильный промотор, обеспечивающий внутри раковых клеток экспрессию гена фермента, способного превращать внутри раковых клеток, но не в нормальных клетках, нетоксичное соединение - пролекарство в токсин, содержащий измененную или расширенную по сравнению с известными промоторами совокупность участков узнавания белков-факторов транскрипции, необходимый набор которых существует в большинстве раковых, но не в нормальных клетках, и представляющий тандемную комбинацию BIRC5 и TERT промоторов. Также описан экспрессирующий вектор, содержащий многофункциональный промотор. Предложен способ убийства раковых клеток, приводящий к синтезу токсинов внутри раковых клеток. Изобретение обеспечивает новые системы транскрипционного контроля экспрессии генов-убийц, позволяющие создавать новые технологии генной терапии рака. 4 н. и 16 з.п. ф-лы, 5 табл., 17 пр., 16 ил.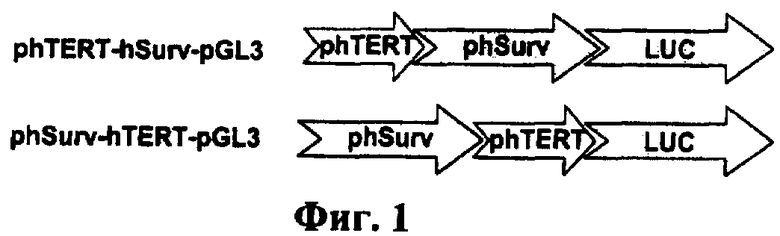
1. Многопрофильный промотор, обеспечивающий внутри раковых клеток экспрессию гена фермента, способного превращать внутри раковых клеток, но не в нормальных клетках, нетоксичное соединение - пролекарство в токсин, содержащий измененную или расширенную, по сравнению с известными промоторами, совокупность участков узнавания белков-факторов транскрипции, необходимый набор которых существует в большинстве раковых, но не в нормальных клетках, и представляющий тандемную комбинацию BIRC5 и TERT промоторов или производное тандемной комбинации BIRC5 и TERT промоторов, образованное в результате делеции, замены, инсерции или иной мутации промотора, не влияющих на активность промотора, но при этом меняющих его структуру.
2. Экспрессирующий вектор, содержащий многофункциональный промотор по п.1, соединенный с транскрибируемой последовательностью, которая при транскрипции продуцирует информационную РНК, кодирующую фермент, превращающий нетоксичное - пролекарство во внутриклеточный токсин.
3. Экспрессирующий вектор по п.2, где вектор представляет собой вирусный вектор.
4. Экспрессирующий вектор по п.2, где вектор представляет собой невирусный вектор.
5. Способ избирательного убийства раковых, но не нормальных клеток, приводящий к синтезу токсинов внутри раковых клеток, включающий доставку в опухоль генной конструкции, состоящей из гена фермента, способного превращать внутри раковой клетки нетоксическое соединение - пролекарство в токсин, и многопрофильного промотора по п.1, или доставку двух генных конструкций, одна из которых состоит из гена фермента, способного превращать внутри раковой клетки нетоксичное соединение - пролекарство в токсин, и блокированного промотора, при деблокировании обеспечивающего экспрессию этого гена, а вторая является деблокирующей конструкцией, содержащей многопрофильный промотор по п.1, присоединенный к гену деблокатора, кодирующего белок деблокатор, способный деблокировать неактивный блокированный промотор, при этом синтезируемый внутри раковой клетки токсин диффундирует в окружающие раковые клетки.
6. Способ по п.5, где кодируемый фермент представляет собой тимидинкиназу вируса простого герпеса, или цитозиндезаминазу дрожжей, или цитозиндезаминазу дрожжей, соединенную в один белок с урацилфосфорибозилтрансферазой дрожжей.
7. Способ по п.5, в котором генная конструкция вводится в количестве достаточном, чтобы разрушить раковые клетки и метастазы.
8. Способ по п.5, в котором генная конструкция вводится в экспрессирующем векторе в составе полиплексной конструкции.
9. Способ по п.8, в котором экспрессирующий вектор вводится внутрь раковых клеток.
10. Способ лечения рака млекопитающих, включающий введение животному, требующему такого лечения, терапевтически эффективного количества экспрессирующего вектора по п.2.
11. Способ по п.10, в которой экспрессирующий вектор вводится в составе полиплексной конструкции.
12. Способ по п.10, в котором экспрессирующий вектор вводится внутрь опухолевых клеток животного.
13. Способ по п.10, в котором большинство неопухолевых клеток не убиваются при обычном для неопухолевых клеток низком уровне факторов транскрипции, необходимых для активации многопрофильного промотора.
14. Способ по п.10, где кодируемый фермент является тимидинкиназой вируса простого герпеса, дополнительно включающий введение животному эффективных количеств пролекарства, в качестве которого используется ганцикловир.
15. Способ по п.10, где кодируемый фермент является цитозиндезаминазой дрожжей, дополнительно включающий введение животному эффективных количеств пролекарства, в качестве которого используется 5-фторцитозин.
16. Способ по п.10, где кодируемый фермент представляет собой цитозиндезаминазу дрожжей, соединенную в один белок с урацилфосфорибозилтрансферазой дрожжей.
17 Способ по п.10, где рак представляет собой метастатическую опухоль.
18. Способ по п.10, где рак представляет собой солидную опухоль.
19. Способ по п.10, где рак представляет собой метастатический рак, ассоциированный с одной из групп, выбранной из рака мочевого пузыря, рака молочной железы, рака шейки матки, рака толстой кишки, рака легких, рака простаты, рака головы и шеи.
20. Способ по п.10, в котором синтез фермента приводит к превращению поставляемого извне пролекарства внутри раковой клетки в токсин, при этом синтезируемый внутри раковой клетки токсин диффундирует в окружающие раковые клетки и убивает их.
| RU 2007137122 A, 20.04.2009 | |||
| US 20030157065 A1, 21.08.2003 | |||
| WO 2002010368 A1, 07.02.2002. |

