[ОБЛАСТЬ ТЕХНИКИ]
Настоящее изобретение относится к соединениям производных пиримидина, эффективным для подавления роста раковых клеток, и вариантов их применения против рака.
[ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ]
Различные мутации в кина3Ном домене рецептора эпидермального фактора роста (РЭФР) обнаруживаются в виде онкогенных генов у пациентов с немелкоклеточным раком легких, и для того, чтобы их лечить, в качестве низкомолекулярного ингибитора киназ рецептора эпидермального фактора роста (РЭФР) используются гефитиниб, эрлотиниб, осимертиниб и т.п.
Среди них рак легких, обусловленный инсерционной мутацией в экзоне 20 РЭФР (мутацией со вставкой в экзоне 20 РЭФР), составляет 5% от всех мутаций, и для их лечения исследовались различные ингибиторы рецептора эпидермального фактора роста (ингибиторы РЭФР), однако до сих пор не существует препарата, одобренного в качестве стандартной терапии для лечения рака легких с инсерционной мутацией экзона 20 РЭФР, поэтому существует острая необходимость в разработке препарата, который может лечить это.
[ОПИСАНИЕ ИЗОБРЕТЕНИЯ]
[ТЕХНИЧЕСКАЯ ЗАДАЧА]
Соответственно, задачей, решаемой настоящим изобретением, является обеспечение соединений, пригодных для лечения рака легких, в частности, рака легких, проявляющего свойства мутации рецептора эпидермального фактора роста, и их медицинское применение.
[ТЕХНИЧЕСКОЕ РЕШЕНИЕ]
Для решения вышеупомянутой задачи настоящее изобретение предлагает соединение химической формулы 1, приведенной ниже, или его фармацевтически приемлемую соль:
[Химическая формула 1]
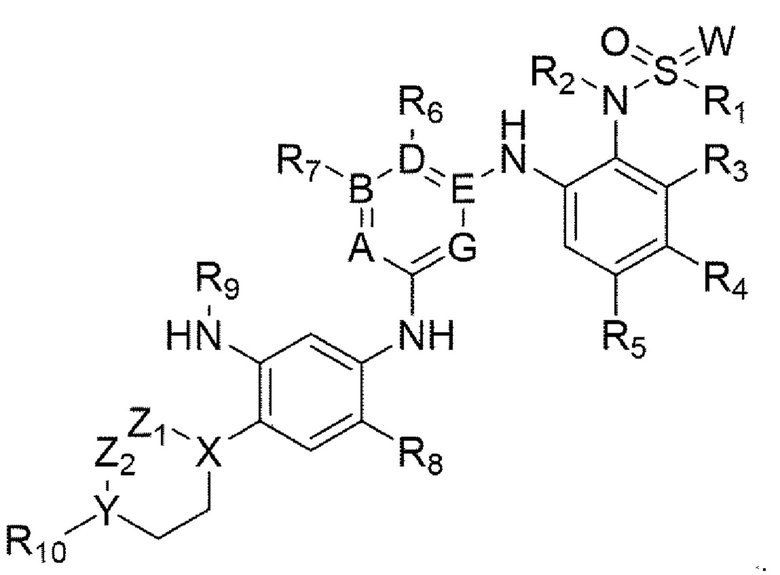
В химической формуле 1
А, В, D, Е и G представляют собой комбинации A=N, В=С, D=C, Е=С и G=N, или комбинации A=N, В=С, D=N, Е=С и G=C,
W представляет собой кислород или NH,
X и Y каждый независимо представляют собой СН, кислород или азот,
Z1 и Z2 каждый независимо отсутствуют, если X или Y представляет собой кислород, или каждый независимо представляют собой С1-С4-алкил, или каждый независимо состоят из атома углерода и связаны друг с другом с образованием 5-, 6- или 7-членного кольца вместе с X и Y, если X или Y не является кислородом,
R1 представляет собой C1-C4-алкил,
R2 представляет собой водород или С1-С4-алкил,
R3 и R4 каждый независимо представляют собой водород, галоген, ОН, ОМе, OEt, CN, CF3, C1-C4-алкил или связаны друг с другом с образованием 5-или 6-членного (конденсированного с фенилом) гетероарильного кольца, и
R5 представляет собой водород, галоген, ОН, ОМе, OEt, CN, CF3 или С1-С4-алкил,
R6 и R7 каждый независимо представляют собой водород, галоген, ОН, ОМе, OEt, CN, CF3, СООН, СОО-С1-С4-алкил, COO-C1-5-циклоалкил, С1-С4-алкил или соединены друг с другом с образованием 5- или 6-членного гетероарильного кольца,
R8 представляет собой водород, галоген, ОН, ОМе, OEt, CN, CF3 или С1-С4-алкил,
R9 представляет собой -С(O)-СН=СН2 или С1-С4-алкил, и
R10 отсутствует, представляет собой водород, галоген, С1-С4-алкил, ОН, ОМе, OEt, CN, CF3, NMe2, пиперазин, замещенный C1-С3-алкилом пиперазин, морфолин или замещенный C1-С3-алкилом морфолин.
Авторы настоящего изобретения подтвердили, что новые соединения согласно настоящему изобретению эффективны для лечения рака легких, в частности, рака легких, при котором проявляется мутация рецептора эпидермального фактора роста. В частности, соединения согласно настоящему изобретению пригодны для лечения рака легких с раковыми клетками, проявляющими свойства инсерционной мутации экзона 20 рецептора эпидермального фактора роста (РЭФР), среди рака легких.
В контексте данного документа «алкил» о3Начает насыщенный неразветвленный или разветвленный нециклический углеводород, содержащий от 1 до 10 атомов углерода (если число атомов углерода конкретно не ограничено). «Низший алкил» о3Начает неразветвленный или разветвленный алкил, имеющий от 1 до 4 атомов углерода. Типичный насыщенный неразветвленный алкил включает метил, этил, н-пропил, н-бутил, н-пентил, н-гексил, н-гептил, н-октил, н-нонил и н-децил, в то время как насыщенный разветвленный алкил включает изопропил, втор-бутил, изобутил, трет-бутил, изопентил, 2-метилгексил, 3-метилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилбутил, 2,3-диметилпентил, 2,4-диметилпентил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилпентил, 2,2-диметилгексил, 3,3-диметилпентил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилпентил, 3-этилпентил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, 2-метил-4-этилпентил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2-метил-4-этилгексил, 2,2-диэтилпентил, 3,3-диэтилгексил, 2,2-диэтилгексил.
В контексте данного документа «циклоалкил» о3Начает моноциклическое или полициклическое насыщенное кольцо, которое содержит атомы углерода и водорода и не содержит кратной связи углерод-углерод. Пример циклоалкильной группы включает (С3-С7)-циклоалкил (например, циклопропил, циклобутил, циклопентил, циклогексил и циклогептил), но не ограничен этим. В одном аспекте настоящего изобретения циклоалкил представляет собой (С3-С5)-циклоалкил. В другом аспекте настоящего изобретения циклоалкил представляет собой циклопропил. В одном примере циклоалкильная группа представляет собой моноциклическое или бициклическое кольцо.
В настоящем документе в тех случаях, когда охарактеризовано как «С1-6» или «от C1 до С6», это о3Начает, что число атомов углерода составляет от 1 до 6. Например, C1-6-алкил о3Начает алкил, имеющий от 1 до 6 атомов углерода.
В контексте настоящего документа термин «галоген» и «гало» о3Начает фтор, хлор, бром или йод.
В настоящем документе термин «арил» о3Начает карбоциклическую ароматическую группу, содержащую от 5 до 10 атомов в цикле. Типичный пример включает фенил, толил, ксилил, нафтил, тетрагидронафтил, антраценил, флуоренил, инденил, азуленил и т.п., но не ограничен этим. Карбоциклическая ароматическая группа может быть селективно замещена.
В контексте настоящего документа «гетероарил» представляет собой 5-10-членное гетероциклическое ароматическое кольцо, которое содержит по меньшей мере один гетероатом, выбранный из группы, состоящей из азота, кислорода и серы, и представляет собой 5-10-членное гетероциклическое ароматическое кольцо, содержащее по меньшей мере один атом углерода и имеющее моно- и бициклическую кольцевую систему. Типичный гетероарил представляет собой триазолил, тетразолил, оксадиазолил, пиридил, фурил, бензофуранил, тиофенил, бензотиофенил, хинолинил, пирролил, индолил, оксазолил, бензоксазолил, имидазолил, бензимидазолил, тиазолил, бензотиазолил, изоксазолил, пиразолил, изотиазолил, пиридазинил, пиримидинил, пиразинил, триазинил, циннолинил, фталазинил, хиназолинил, пиримидил, оксетанил, азепинил, пиперазинил, морфолинил, диоксанил, тиетанил и оксазолил.
В настоящем документе соединение, описываемое химической формулой 1, может быть использовано в форме соли, созданной неорганической или органической кислотой и, например, оно может быть использовано в форме соли, созданной одной или несколькими типами кислот, выбранных из группы, состоящей из соляной кислоты, бромоводородной кислоты, серной кислоты, фосфорной кислоты, азотной кислоты, уксусной кислоты, гликолевой кислоты, молочной кислоты, пировиноградной кислоты, малоновой кислоты, янтарной кислоты, глутаровой кислоты, фумаровой кислоты, яблочной кислоты, миндальной кислоты, винной кислоты, лимонной кислоты, аскорбиновой кислоты, пальмитиновой кислоты, малеиновой кислоты, бензойной кислоты, гидроксибензойной кислоты, фенилуксусной кислоты, коричной кислоты, салициловой кислоты, метансульфоновой кислоты, этансульфоновой кислоты, бензолсульфоновой кислоты, толуолсульфоновой кислоты и т.п.
В контексте настоящего документа термин «соединение согласно настоящему изобретению» о3Начает любое соединение химической формулы 1, а также клатраты, гидраты, сольваты или их полиморфные формы. Кроме того, термин «соединение согласно настоящему изобретению» имеет 3Начение, включающее фармацевтически приемлемые соли соединений согласно настоящему изобретению, если не упоминается его фармацевтически приемлемая соль. В одном примере соединение согласно настоящему изобретению может присутствовать в виде стереоизомерно чистых соединений (например, по существу не содержит других стереоизомеров (например, 85% э.и. или более, 90% э.и. или более, 95% э.и. или более, 97% э.и. или более, или 99% э.и. или более)). Другими словами, когда соединение химической формулы 1 согласно настоящему изобретению или его соль представляет собой таутомерный изомер и/или стереоизомер (например, геометрический изомер и конформационные изомеры), каждый из его выделенных изомеров и смесей также включается в объем соединений согласно настоящему изобретению. Если соединение согласно настоящему изобретению или его соль содержит асимметричный углерод в своей структуре, их оптически активные соединения и рацемические смеси также включаются в объем соединений согласно настоящему изобретению.
В контексте настоящего документа термин «полиморфная модификация» о3Начает твердую кристаллическую форму соединения согласно настоящему изобретению или его комплекс. Различные полиморфные модификации одного и того же соединения проявляют различные физические, химические и/или спектральные свойства. Различия в физических свойствах включают стабильность (например, термическую стабильность или светостабильность), сжимаемость и плотность (важные для разработки рецептуры и приготовления продукта), и скорость растворения (которая может влиять на биодоступность), но не ограничены этим. Различия в стабильности вызывают изменения в химической реакционной способности (например, ра3Ное окисление, в частности, ускоренное изменение окраски, когда в составе одна полиморфная модификация, по сравнению с теми случаями, когда в составе другая полиморфная модификация) или механических свойствах (например, очищенные частицы, сохраняемые в виде кинетически предпочтительных полиморфных модификаций, преобразуются в термодинамически более стабильные полиморфные модификации) или их обоих (очистка одной полиморфной модификации более подвержена ухудшению качества при высокой влажности). Другие физические свойства полиморфной модификации могут влиять на ее обработку. Например, одна полиморфная модификация может с большей вероятностью образовывать сольват, чем другая полиморфная модификация, например, из-за своей конфигурации или распределения частиц по размерам, или ее может быть сложнее отфильтровать или промыть.
В контексте настоящего документа термин «соединение растворителя» о3Начает соединение согласно настоящему изобретению или его фармацевтически приемлемую соль, включающие в себя стехиометрическое или нестехиометрическое количество растворителя, связанного нековалентными межмолекулярными силами. Предпочтительные растворители являются летучими, нетоксичными и могут вводиться людям в ничтожно малых количествах.
В контексте настоящего документа термин «гидрат» о3Начает соединение согласно настоящему изобретению или его фармацевтически приемлемую соль, включающую в себя стехиометрическое или нестехиометрическое количество воды, связанной нековалентными межмолекулярными силами.
В контексте настоящего документа термин «клатрат» о3Начает соединение согласно настоящему изобретению или его соль в форме кристаллической решетки, содержащей пространства (например, канал), которые удерживают гостевую молекулу (например, растворитель или воду).
В контексте настоящего документа термин «очищенный» о3Начает, что изолят обладает чистотой по меньшей мере 90% при выделении, причем в одном примере это о3Начает, что он обладает чистотой 95% или более, а в другом примере это о3Начает, что он обладает чистотой 99% или более, и в другом примере это о3Начает, что он обладает чистотой 99,9% или более.
В контексте настоящего документа «лечение» включает уничтожение, удаление, видоизменение или сдерживание первичной, локализованной или метастатической раковой ткани; а также минимизирует или замедляет распространение рака.
Неограничивающий пример соединения согласно настоящему изобретению включает, в частности, следующие соединения и их фармацевтически приемлемые соли.
N-(2-((5-хлор-2-((4-((2-(диметиламино)этил)(метил)амино)-5-(изопропиламино)-2-метоксифенил)амино)пиримидин-4-ил)амино)фенил)-N-метилметансульфонамид,
N-(2-((2-((4-((2-(диметиламино)этил)(метил)амино)-5-(изопропиламино)-2-метоксифенил)амино)тиено[3,2-d]пиримидин-4-ил)амино)фенил)-N-метилметансульфонамид,
N-(2-((2-((4-(4-(диметиламино)пиперидин-1-ил)-5-(изопропиламино)-2-метоксифенил)амино)тиено[3,2-d]пиримидин-4-ил)амино)фенил)-N-метилметансульфонамид,
N-(2-((2-((5-(изопропиламино)-2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)амино)фенил)-N-метилметансульфонамид,
N-(2-((5-хлор-2-((5-(изопропиламино)-2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)фенил)-N-метилметансульфонамид,
N-(2-((5-хлор-2-((4-(4-(диметиламино)пиперидин-1-ил)-5-(изопропиламино)-2-метоксифенил)амино)пиримидин-4-ил)амино)фенил)-N-метилметансульфонамид,
N-(4-метокси-5-((4-((2-(N-метилметилсульфонамидо)фенил)амино)тиено[3,2-d]пиримидин-2-ил)амино)-2-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)акриламид,
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-((2-(N-метилметилсульфонамидо)фенил)амино)тиено[3,2-d]пиримидин-2-ил)амино)фенил)акриламид,
N-(2-(4-(диметиламино)пиперидин-1-ил)-4-метокси-5-((4-((2-N-метилметилсульфонамидо)фенил)амино)тиено[3,2-d]пиримидин-2-ил)амино)фенил)акриламид,
N-(5-((5-хлор-4-((2-(метилсульфонамидо)фенил)амино)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид.
N-(5-((5-хлор-4-((5-N-метилметилсульфонамидо)хиноксалин-6-ил)амино)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид,
N-(5-((5-хлор-4-((5-(метилсульфонамидо)хиноксалин-6-ил)амино)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид,
N-(5-((5-хлор-4-((5-фтор-2-(N-метилметилсульфонамидо)фенил)амино)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метокси фенил)акриламид,
N-(5-((5-хлор-4-((5-фтор-2-(метилсульфонамидо)фенил)амино)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид,
изопропил
2-((5-акриламидо-4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)амино)-4-((2-(N-метилметилсульфонамидо)фенил)амино)пиримидин-5-карбоксилат,
циклопропил
2-((5-акриламидо-4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)амино)-4-((2-(N-метилметилсульфонамидо)фенил)амино)пиримидин-5-карбоксилат или
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((6-((2-(N-метилметилсульфонамидо)фенил)амино)пиримидин-4-ил)амино)фенил)акриламид.
В другом аспекте настоящее изобретение обеспечивает фармацевтическую композицию, содержащую терапевтически эффективную дозу соединения химической формулы 1 или его фармацевтически приемлемую соль согласно настоящему изобретению и фармацевтически приемлемый носитель.
В контексте настоящего документа «эффективная доза» о3Начает количество соединения согласно настоящему изобретению, достаточное для уничтожения, видоизменения, сдерживания или удаления первичных, локализованных или метастатических раковых клеток или раковой ткани; замедления или минимизации распространения рака; или обеспечения терапевтического эффекта при лечении или контроле течения рака, неопластического заболевания или опухоли. «Эффективная доза» также о3Начает количество соединения согласно настоящему изобретению, достаточное, чтобы вызвать гибель раковых или опухолевых клеток. «Эффективная доза» также о3Начает количество, достаточное для ингибирования или снижения активности клеток рака легких либо in vitro, либо in vivo.
В другом аспекте настоящее изобретение обеспечивает способ лечения заболевания или состояния, включающий введение терапевтически эффективной дозы соединения химической формулы 1 или его фармацевтически приемлемой соли нуждающемуся в этом субъекту, причем заболевание или состояние представляет собой рак легких. В другом аспекте субъектом является человек. В другом аспекте рак легких представляет собой рак легких, проявляющий свойства мутации рецептора эпидермального фактора роста (РЭФР). В другом аспекте рак легких представляет собой рак легких с раковыми клетками, проявляющими свойства инсерционной мутации экзона 20 рецептора эпидермального фактора роста (инсерционной мутации экзона 20 РЭФР).
Другими словами, настоящее изобретение обеспечивает медицинское применение, характеризующееся использованием соединения химической формулы 1 или его фармацевтически приемлемую соль согласно настоящему изобретению в качестве активного ингредиента. В одном аспекте медицинское применение настоящего изобретения представляет собой применение для лечения или профилактики заболевания или состояния, описанного в настоящем документе.
Соответственно, в другом аспекте, настоящее изобретение обеспечивает фармацевтическую композицию для лечения или профилактики рака легких, в частности, рака легких, экспрессирующего мутацию рецептора эпидермального фактора роста, содержащую соединение согласно настоящему изобретению или его фармацевтически приемлемую соль в качестве активного ингредиента.
Соединение согласно настоящему изобретению или его фармацевтически приемлемую соль обычно вводят в терапевтически эффективной дозе. Соединение согласно настоящему изобретению может быть введено любым подходящим путем в эффективной дозе для формы фармацевтической композиции, подходящей для этого пути введения, и надлежащего лечения. Эффективная доза может составлять, как правило, от примерно 0,0001 до примерно 200 мг/кг массы тела в день, предпочтительно от примерно 0,001 до примерно 100 мг/кг в день при однократном или дробном введении. В зависимости от возраста, биологического вида и заболевания или состояния, подлежащего лечению, может быть пригоден уровень дозы ниже нижнего предела этого диапазона. В других случаях тем не менее можно использовать большие дозы без вредных побочных эффектов. Большая доза может быть разделена на несколько меньших доз для введения в течение дня. Методы определения соответствующей дозы хорошо известны в данной области и, например, разрешается использовать документ Remington: The Science and Practice of Pharmacy, Mack Publishing Co., 20-е изд., 2000.
Для лечения рака легких соединение или его фармацевтически приемлемая соль, описанные в настоящем документе, могут быть введены различными методами следующим образом.
Пероральное введение
Соединение согласно настоящему изобретению может быть введено перорально, причем пероральный о3Начает понятие, содержащее проглатывание. При пероральном введении соединение согласно настоящему изобретению может поступать в желудочно-кишечный тракт или может всасываться непосредственно из полости рта в кровоток, например, при трансбуккальном или сублингвальном приеме.
Композиция, пригодная для перорального введения, может находиться в твердой, жидкой, гелевой или порошковой форме и может иметь такую лекарственную форму, как таблетки, пастилки, капсулы, гранулы, порошки и т.п.
Композиция для перорального введения может быть покрыта кишечнорастворимой оболочкой, по выбору, и может демонстрировать отсроченное или пролонгированное высвобождение благодаря кишечнорастворимой оболочке. Другими словами, композиция для перорального введения согласно настоящему изобретению может представлять собой лекарственную форму с немедленным или модифицированным типом высвобождения.
Жидкая лекарственная форма может содержать раствор, сироп и суспензию, и такая жидкая композиция может представлять собой форму, заключенную в мягкую или твердую капсулу. Этот состав может содержать фармацевтически приемлемый носитель, например, воду, этанол, полиэтиленгликоль, целлюлозу или масло. Лекарственная форма может также содержать один или несколько эмульгаторов и/или суспендирующих агентов.
В таблетированной лекарственной форме количество препарата в качестве активного ингредиента может присутствовать в составе в количестве примерно от 0,05% по массе до примерно 95% по массе от общей массы таблетки, в более общем случае примерно от 2% по массе до примерно 50% по массе относительно лекарственной формы. Дополнительно таблетка может содержать вещество для улучшения распадаемости таблеток, включенное в количестве примерно от 0,5% по массе до примерно 35% по массе, в более общем случае примерно от 2% по массе до примерно 25% по массе относительно рецептуры. В качестве примера вещества для улучшения распадаемости таблеток могут быть использованы лактоза, крахмал, натрия крахмал гликолят, кросповидон, кроскармеллоза натрия, мальтодекстрин или их смеси, но не ограничиваясь этим.
Подходящий глидант, содержащийся в части композиции в таблетке, может присутствовать в составе в количестве примерно от 0,1% по массе до примерно 5% по массе, и в качестве глиданта могут быть использованы тальк, диоксид кремния, стеариновая кислота, стеарат кальция, цинка или магния, стеарилфумарат натрия и т.п., но настоящее изобретение не ограничивается этими видами добавок.
В качестве связующего вещества в части композиции в таблетке могут быть использованы желатин, полиэтиленгликоль, сахар, камедь, крахмал, поливинилпирролидон, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза и т.п., а в качестве подходящего разбавителя в части композиции в таблетке могут быть использованы маннитол, ксилитол, лактоза, декстроза, сахароза, сорбитол, крахмал, микрокристаллическая целлюлоза и т.п., но настоящее изобретение не ограничивается этими видами добавок.
Солюбилизирующее средство, которое по выбору может быть включено в таблетку, может быть использовано в количестве примерно от 0,1% по массе до примерно 3% по массе от общей массы таблетки, и, например, для фармацевтической композиции согласно настоящему изобретению могут быть использованы, например, полисорбат, лаурилсульфат натрия, додецилсульфат натрия, пропиленкарбонат, простой моноэтиловый эфир диэтиленгликоля, диметилизосорбид, полиоксиэтиленгликолированное, натуральное или гидрогенизированное касторовое масло, HCOR™ (Nikkol), олеиловый сложный эфир, Gelucire™, моно/диглицерид каприловой/каприловой кислоты, сложный эфир сорбитана и жирной кислоты, Solutol HS™ и т.п., но настоящее изобретение не ограничивается этими конкретными видами солюбилизирующих средств.
Парентеральное введение
Соединение согласно настоящему изобретению может быть введено непосредственно в кровоток, мышцы или кишечник. Подходящий метод для парентерального введения включает внутривенное, внутримышечное, подкожное, внутриартериальное, внутрибрюшинное, интратекальное, внутричерепное введение и т.п. Подходящее средство для парентерального введения включает шприц (содержащий иглу и безыгольный шприц) и инфузионный метод.
Композиция для парентерального введения может представлять собой лекарственную форму с немедленным или модифицированным типом высвобождения, а модифицированный тип высвобождения может представлять собой отсроченный или пролонгированный тип высвобождения.
Большинство парентеральных лекарственных форм представляют собой жидкие композиции, и такие жидкие композиции представляют собой водный раствор, содержащий лекарственный ингредиент в соответствии с настоящим изобретением, соль, буфер, изотонический агент и т.п.
Парентеральные лекарственные формы могут быть также приготовлены в виде высушенной формы (например, лиофилизированной) или стерильного неводного раствора. Такие лекарственные формы могут быть использованы с подходящим носителем, таким как стерильная вода. При приготовлении парентерального раствора также могут использоваться усилители растворимости.
Местное применение
Соединение согласно настоящему изобретению может применяться местно, то есть дермально или трансдермально. Лекарственные формы для местного применения включают лосьон, раствор, крем, гель, гидрогель, мазь, пену, имплантат, пластырь и т.п. Фармацевтически приемлемый носитель для лекарственных форм местного применения может включать воду, спирт, минеральное масло, глицерин, полиэтиленгликоль и т.п. Местное применение может также осуществляться путем электропорации, ионофореза, фонофореза и т.п.
Композиция для местного применения может представлять собой лекарственную форму с немедленным или модифицированным типом высвобождения, а модифицированный тип высвобождения может представлять собой отсроченный или пролонгированный тип высвобождения.
[ПОЛЕ3НЫЕ ЭФФЕКТЫ]
Настоящее изобретение предлагает новые соединения производного пиримидина, обладающие активностью, ингибирующей раковые клетки, и их фармацевтически приемлемые соли. Производные пиримидина или их фармацевтически приемлемые соли в соответствии с настоящим изобретением могут эффективно ингибировать рост раковых клеток, экспрессирующих мутацию рецептора эпидермального фактора роста, в частности, раковых клеток, присутствующих в раке легких. Соответственно, соединения и их фармацевтически приемлемые соли в соответствии с настоящим изобретением пригодны для лечения рака легких.
[ПРИНЦИП ИЗОБРЕТЕНИЯ]
Здесь и далее, чтобы помочь пониманию настоящего изобретения, оно будет подробно описано ниже с помощью примеров и т.п. Тем не менее примеры согласно настоящему изобретению могут модифицироваться в различные другие формы, и объем настоящего изобретения не следует рассматривать как ограниченный следующими примерами. Примеры настоящего изобретения предоставлены для более полного описания настоящего изобретения для лиц со средними 3Наниями в области техники, к которой относится настоящее изобретение.
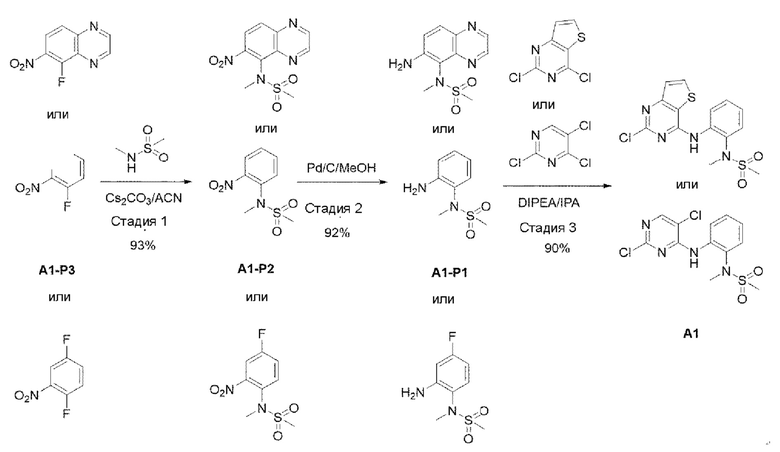
Соединения, представляемые химической формулой 1 выше согласно настоящему изобретению, могут быть легко синтезированы, например, обращаясь к методам, представленным ниже схемами реакций 1, 2, 3 и 4.
[Схема реакции 1]
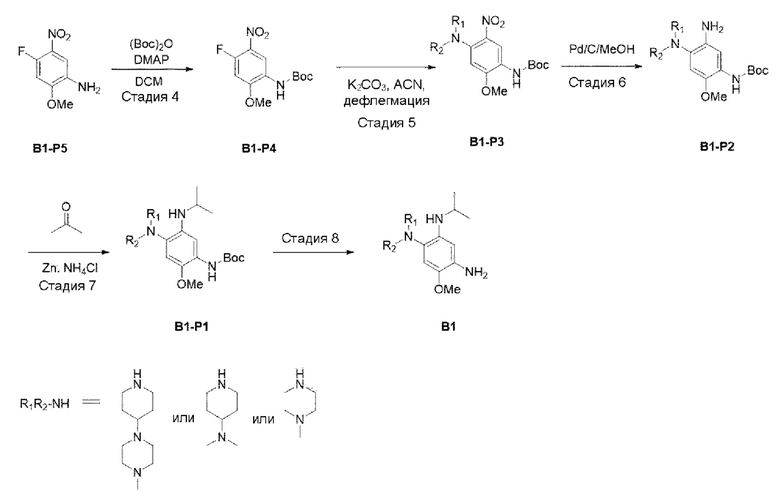
[Схема реакции 2]
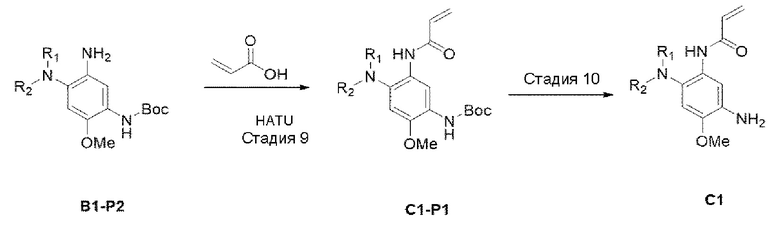
[Схема реакции 3]
[Схема реакции 4]
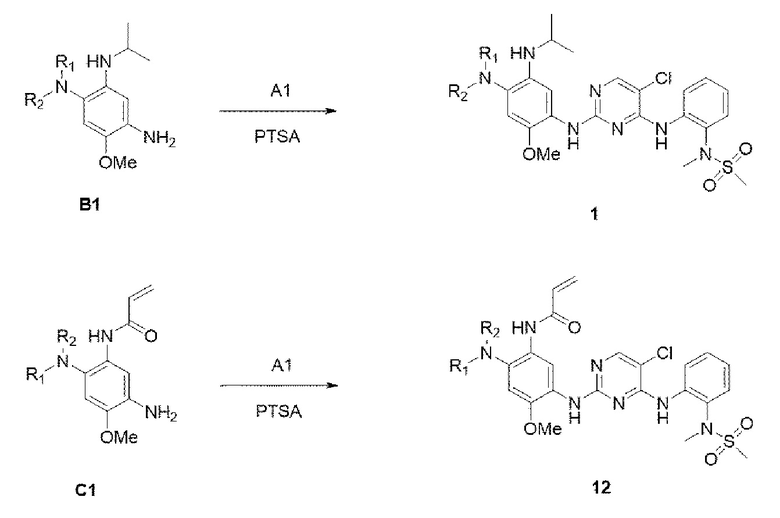
Пример синтеза: синтез
N-(2-((5-хлор-2-((4-((2-(диметиламино)этил)(метил)амино)-5-(изопропиламино)-2-метоксифенил)амино)пиримидин-4-ил)амино)фенил)-N-метилметансульфонамида и N-(5-((5-хлор-4-((2-(N-метилметилсульфонамидо)фенил)амино)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида
Стадия 1:
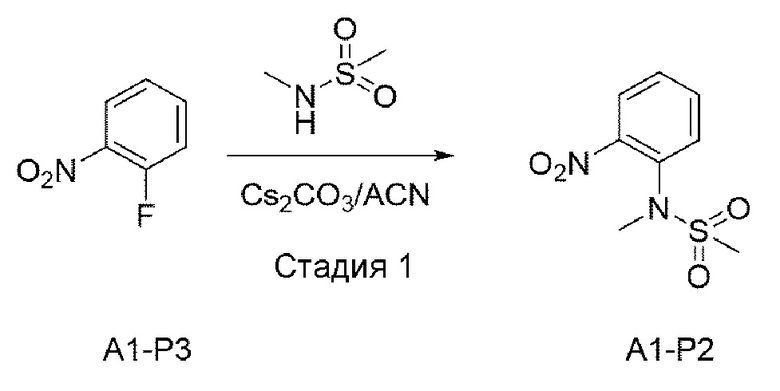
В вышеуказанной схеме реакции 1 структуру А1-Р3 (3 г, 21,262 ммоль) растворили ацетонитрилом (150 мл). При комнатной температуре добавили CS2CO3 (10,4 г, 31,892 ммоль) и N-метилметансульфонамид, и затем это перемешивали при 80°С в течение 12 часов. Осадок, оставшийся после реакции, отфильтровывали, и органический растворитель отгоняли при пониженном давлении для его удаления, и в результате получали А1-Р2. Его использовали для следующей реакции без специального процесса очистки.
Стадия 2:
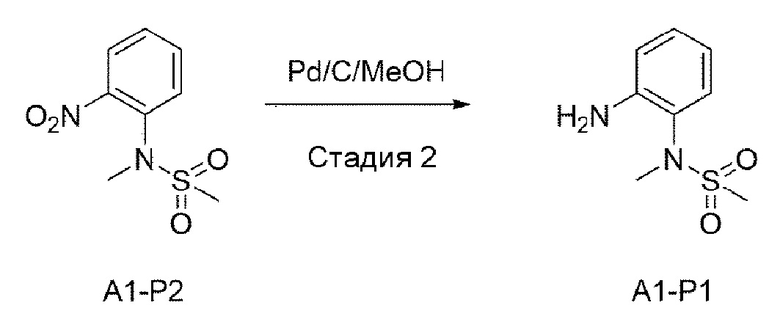
Структуру А1-Р2 (4,5 г, 19,545 ммоль), полученную на стадии 1, растворяли в смешанном растворителе из МеОН (100 мл) и DCM (дихлорметана) (50 мл), а затем добавляли 10% Pd/C (0,416 г, 3,909 ммоль). В среде водорода это перемешивали в течение 2 часов, а затем проводили фильтрацию с использованием целита и выпаривали при пониженном давлении для удаления растворителя с получением неочищенного А1-Р1. Этот сухой остаток промывали простым эфиром и н-пентаном и использовали для следующей реакции без специального процесса очистки.
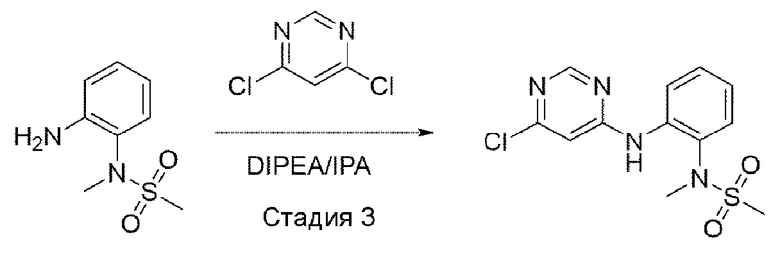
Стадия 3:
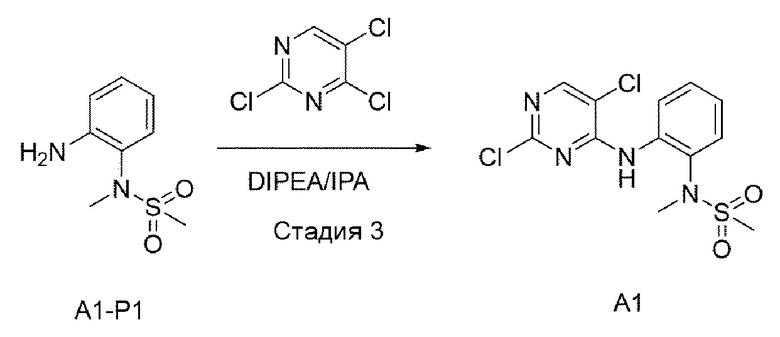
Структуру A1-P1 (8,3 r, 41,446 ммоль), полученную в предыдущей
реакции, растворяли в IPA (изопропиловом спирте) (200 мл), а затем при комнатной температуре добавляли 2,5,6-трихлорпиримидин (12,163 г, 66,314 ммоль) и DIPEA (диизопропилэтиламин) (21,428 г, 166 ммоль) и перемешивали при 90°С в течение 12 часов. После того, как реакция завершилась, выпаривали при пониженном давлении для удаления растворителя, а затем добавляли воду и экстрагировали с использованием DCM. Смешанный раствор снова промывали 2N HCl, органический слой отделяли и выпаривали при пониженном давлении для получения неочищенного А1. Его использовали для следующей реакции без специального процесса очистки.
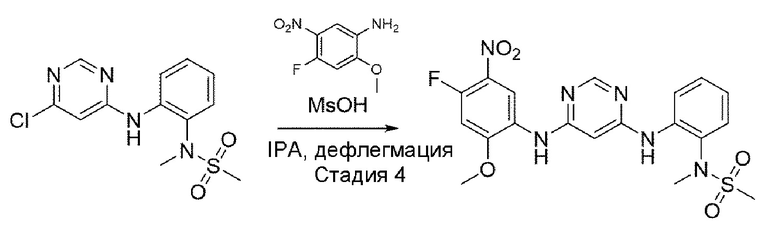
Стадия 4:
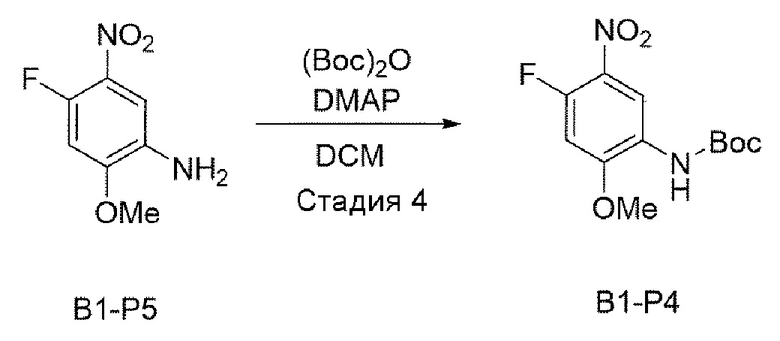
В приведенной выше схеме реакции 2 структуру В1-Р5 (4 г, 21,489 ммоль) растворили в DCM (100 мл), а затем при комнатной температуре добавили (Вос)2O (ди-трет-бутилдикарбонат) (4,690 г, 21,489 ммоль) и DMAP (4-диметиламинопиридин) (0,262 г, 2,149 ммоль) и перемешивали в течение 12 часов. После того, как реакция завершилась, реакционную смесь промывали 2N HCl, а затем высушивали с использованием Na2SO4 и отфильтровывали, и затем выпаривали при пониженном давлении с получением неочищенного В1-Р4. С помощью колоночной хроматографии получили выделенное чистое целевое соединение В1-Р4 (5,8 г, 97,5%).
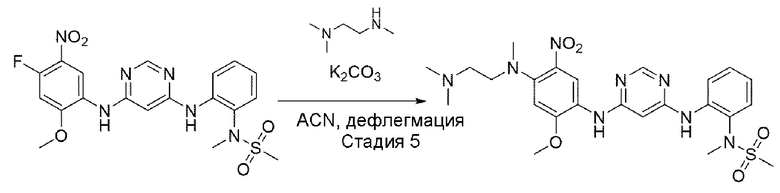
Стадия 5:
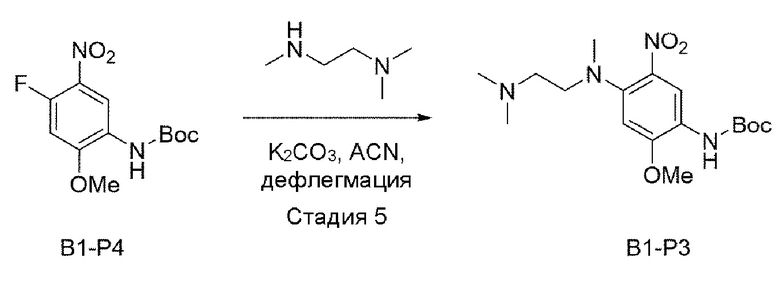
В приведенной выше схеме реакции 2 структуру В1-Р4 (2,2 г, 7,685 ммоль) растворяли в ацетонитриле (50 мл), а затем при комнатной температуре добавляли K2CO3 (2,124 г, 15,371 ммоль) и N,N,N'-триметилендиамин, а затем перемешивали при 80°С в течение 12 часов. После того, как реакция завершилась, продукты реакции отфильтровывали и выпаривали при пониженном давлении для получения неочищенного В1-Р3. Его использовали для следующей реакции без специального процесса очистки.
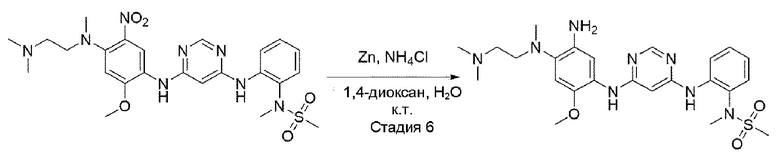
Стадия 6:
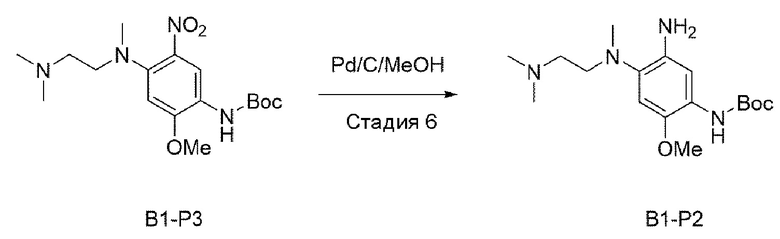
В приведенной выше схеме реакции 2 структуру В1-Р3 (2,8 г, 7,6 ммоль) растворяли в смешанном растворителе из МеОН (50 мл) и DCM (20 мл) и перемешивали в среде водорода в течение 4 часов. После того, как реакция завершилась, проводили фильтрацию с использованием целита, и растворитель удаляли отгонкой при пониженном давлении с получением неочищенного В1-Р2. Его использовали для следующей реакции без специального процесса очистки.
Стадия 7:
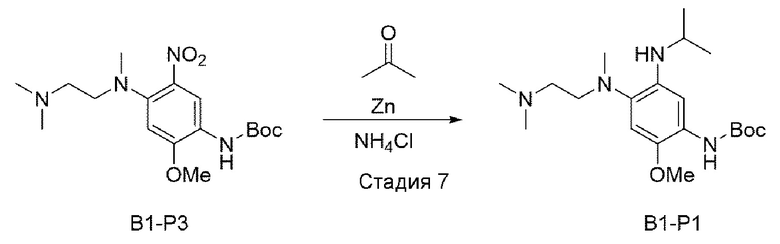
В вышеприведенной схеме реакции 2 добавляли 20 мл ацетона к В1-Р3 (580,8 мг, 1,00 ммоль) и порошку цинка (653,9 мг, 10,0 ммоль) и добавляли насыщенный водный раствор NH4Cl (10 мл), и реакцию доводили до конца при кипячении с обратным холодильником в течение 6 часов. После охлаждения при комнатной температуре порошок цинка удалили с использованием целита, и растворитель удалили при пониженном давлении, а затем полученный водный слой экстрагировали с использованием EtOAc (этилацетата). Из полученного органического слоя удалили растворитель при пониженном давлении, и очищали с использованием колоночной хроматографии.
Стадия 8:
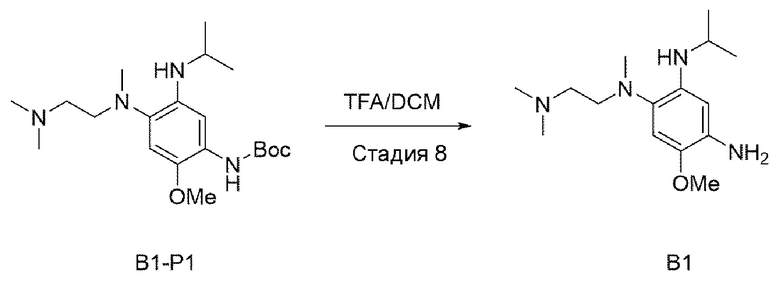
В приведенной выше схеме реакции 2 В1-Р1 (300 мг, 0,764 ммоль) растворяли в DCM (15 мл), а затем охлаждали до 0°С. Затем добавляли TFA (трифторуксусную кислоту) (0,6 мл, 7,643 ммоль), и повышали температуру до комнатной, и перемешивали в течение 3 часов. После того, как реакция завершилась, выпаривали при пониженном давлении для удаления растворителя, промывали DCM и насыщенным раствором NaHCO3, а затем органический слой отделяли и сушили, а потом выпаривали при пониженном давлении. Его использовали для следующей реакции без специального процесса очистки.
Стадия 9:
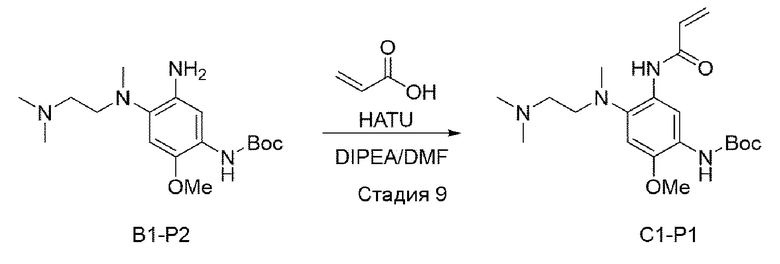
В приведенной выше схеме реакции 3 акриловую кислоту (75 мг, 1,034 ммоль) растворяли в DMF (диметилформамиде) (4 мл), а затем охлаждали до 0°С. Затем добавляли HATU (O-(7-Азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат) (393 мг, 1,034 ммоль) и DIPEA (400 мг, 3,103 ммоль), а затем добавляли В1-Р2 (350 мг, 1,034 ммоль), и реагенты перемешивали при комнатной температуре в течение 12 часов. После того, как реакция завершилась, экстрагировали с использованием Н2О и DCM. Органический слой отделяли, высушивали с использованием Na2SO4, а также отфильтровывали, а затем перегоняли при пониженном давлении. Используя колоночную хроматографию получали выделенное чистое целевое соединение В1-Р1 (220 мг, 55%).
Стадия 10:
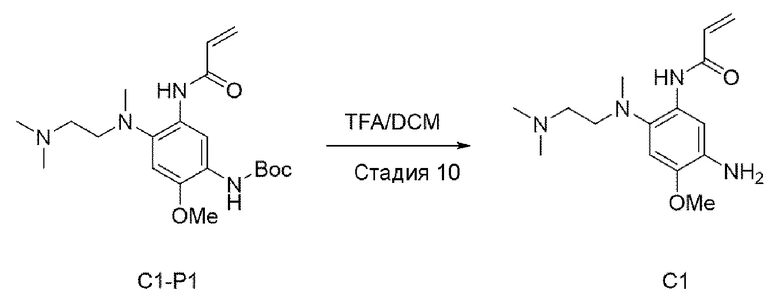
В приведенной выше схеме реакции 3 С1-Р1 (300 мг, 0,764 ммоль) растворяли в DCM (15 мл), а затем охлаждали до 0°С. Добавляли TFA (0,6 мл, 7,643 ммоль), и повышали температуру до комнатной температуры, а также перемешивали в течение 3 часов. После того, реакция была завершена, это выпаривали при пониженном давлении для удаления растворителя и промывали с использованием DCM и насыщенного раствора NaHCCb, и затем органический слой отделяли и высушивали, а потом выпаривали при пониженном давлении. Его использовали для следующей реакции без специального процесса очистки.
Стадия 11:
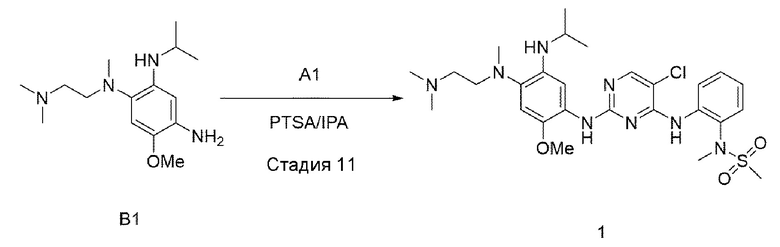
В приведенной выше схеме реакции 4 структуру В1 (50 мг, 0,178 ммоль) растворяли в изопропиловом спирте (3 мл), а затем добавляли А1 (71 мг, 0,205 ммоль) и PTSA (п-толуолсульфоновую кислоту) (48,8 мг, 0,257 ммоль) при комнатной температуре, и перемешивали при 90°С в течение 12 часов. После того, как реакция завершилась, выпаривали при пониженном давлении для удаления растворителя и экстрагировали водой и 10% MeOH/DCM. Органический слой отделяли и высушивали, и выпаривали при пониженном давлении с получением неочищенного соединения 1. Используя колоночную хроматографию, получили выделенное чистое соединение 1 (60 мг, 58%).
1H ЯМР (400 МГц, CDCl3) δ (ч/млн): 8,26-8,49 (2Н, м), 8,16 (1H, с) 8,09-8,08 (1H, д), 7,83 (1Н, с), 7,55-7,52 (1H, дд), 6,98 (1H, с), 6,97-6,77 (1H, дд), 5.72 (1Н, дд), 4,76 (1Н, м), 3,67 (3Н, с), 3,14 (3Н, с), 3,06 (3Н, с), 2,79 (2Н, м), 2,70 (3Н, с), 2,20 (8Н, уш.с), 1,11 (6Н, д). [М+Н]+: масса/заряд 591,17, найдено 592,2, ВЭЖХ чистота: 97,9%.
Стадия 12:
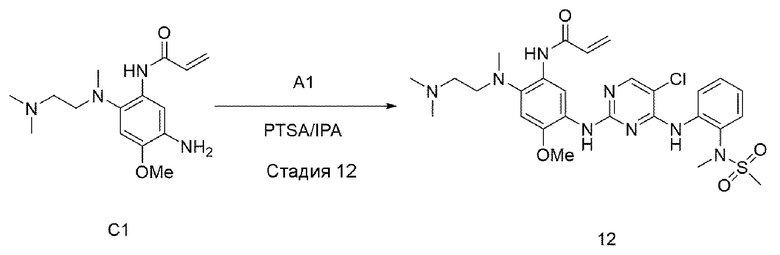
В приведенной выше схеме реакции 4 структуру С1 (50 мг, 0,171 ммоль) растворяли в изопропиловом спирте (3 мл), а затем добавляли А1 (71 мг, 0,205 ммоль) и PTSA (49,1 мг, 0,258 ммоль) при комнатной температуре, и перемешивали при 90°С в течение 12 часов. После того, как реакция завершилась, выпаривали при пониженном давлении для удаления растворителя и экстрагировали водой и 10% MeOH/DCM. Органический слой отделяли и высушивали, и выпаривали при пониженном давлении с получением неочищенного соединения 12. Используя колоночную хроматографию, получали выделенное чистое соединение 12 (65 мг, 63%).
1Н ЯМР (400 МГц, CDCl3) δ (ч/млн): 10,02 (1H, уш.с), 8,35-8,31 (2Н, м), 8,26 (1Н, с) 8,21-8,19 (1Н, д), 8,09 (1Н, с), 7,53-7,50 (1H, дд), 6,95 (1H, с), 6,18-6,14 (1H, дд), 5.72-5,68 (1H, дд), 4,31 (1H, м), 3,72 (3Н, с), 3,14 (3Н, с), 3,05 (3Н, с), 2,86 (2Н, м), 2,66 (3Н, с), 2,19 (8Н, уш.с), 1,12 (6Н, д). [М+Н]+: масса/заряд 591,17, найдено 592,2, ВЭЖХ чистота: 98,1%.
Пример 1. Получение N-(2-((5-хлор-2-((4-((2-(диметиламино)этил)(метил)амино)-5-(изопропиламино)-2-метоксифенил)амино)пиримидин-4-ил)амино)фенил)-N-метилметансульфонамида
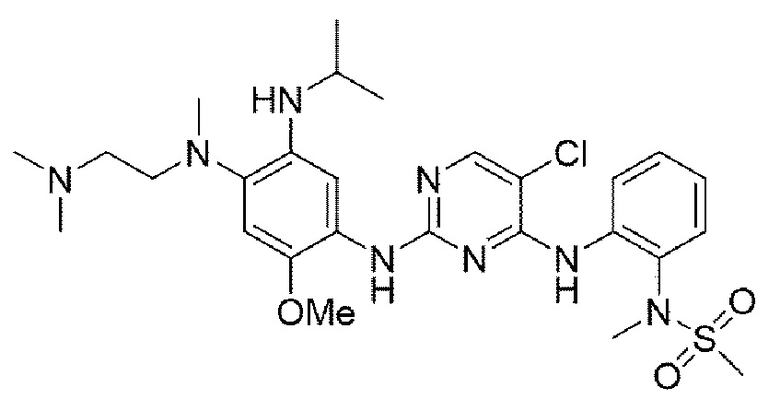
Конечное соединение синтезировали по вышеприведенной схеме реакции 4.
1Н ЯМР (400 МГц, CDCl3) δ (ч/млн): 8,26-8,49 (2Н, м), 8,16 (1H, с) 8,09-8,08 (1H, д), 7,83 (1H, с), 7,55-7,52 (1H, дд), 6,98 (1Н, с), 6,97-6,77 (1Н, дд), 5.72 (1H, дд), 4,76 (1H, м), 3,67 (3Н, с), 3,14 (3Н, с), 3,06 (3Н, с), 2,79 (2Н, м), 2,70 (3Н, с), 2,20 (8Н, уш.с), 1,11 (6Н, д). [М+Н]+: масса/заряд 591,17, найдено 592,2, ВЭЖХ чистота: 97,9%.
Пример 2. Получение N-(2-((2-((4-((2-(диметиламино)этил)(метил)амино)-5-(изопропиламино)-2-метоксифенил)амино)тиено[3,2-d]пиримидин-4-ил)амино)фенил)-N-метилметансульфонамида
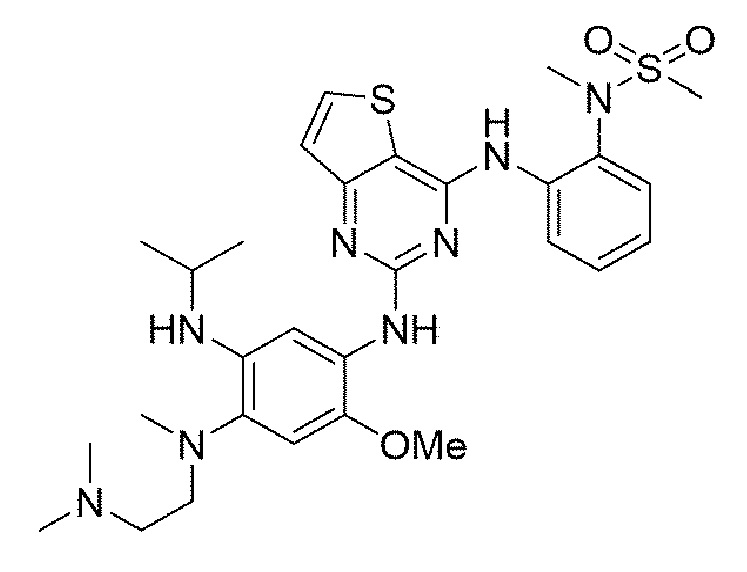
Конечное соединение синтезировали по вышеприведенной схеме реакции 4.
1Н ЯМР (400 МГц, DMSO-d6) δ 9,51 (с, 1Н), 9,20 (с, 1Н), 8,02 (с, 1Н), 7,63 (д, J=7,6 Гц, 1H), 7,45 (дд, J=7,5, 1,5 Гц, 1H), 7,39-7,32 (м, 2Н), 7,04 (тд, J=7,5, 1,5 Гц, 1Н), 6,76 (тд, J=7,5, 1,5 Гц, 1Н), 6,46 (с, 1Н), 5,51 (д, J=8,4 Гц, 1Н), 4,31 (м, 1Н), 3,85 (с, 3Н), 3,44-3,34 (уш.с, 2Н), 3,32 (с, 3Н), 3,26 (с, 3Н), 3,00 (с, 3Н), 2,57-2,50 (м, 8Н), 1,12 (д, J=6,9 Гц, 6Н). MS: ИЭР масса/заряд 613,1 [М+Н]+, ВЭЖХ чистота: 99,0%.
Пример 3. Получение N-(2-((2-((4-(4-(диметиламино)пиперидин-1-ил)-5-(изопропиламино)-2-метоксифенил)амино)тиено[3,2-а]пиримидин-4-ил)амино)фенил)-N-метилметансульфонамида
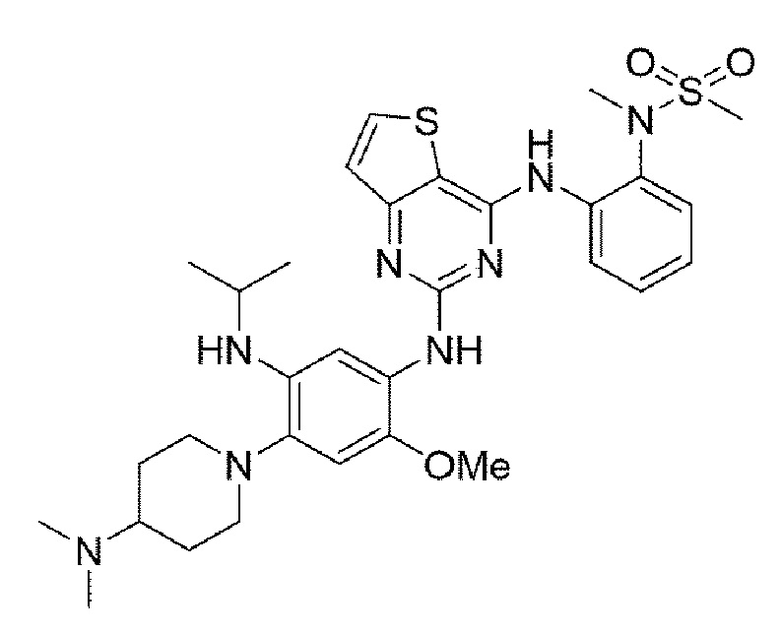
Конечное соединение синтезировали по вышеприведенной схеме реакции 4.
1Н ЯМР (400 МГц, DMS0-d6) δ 9,51 (с, 1Н), 9,20 (с, 1Н), 8,02 (с, 1Н), 7,63 (д, J=7,6 Гц, 1H), 7,45 (дд, J=7,5, 1,5 Гц, 1Н), 7,39-7,32 (м, 2Н), 7,04 (тд, J=7,5, 1,5 Гц, 1Н), 6,76 (тд, J=7,5, 1,5 Гц, 1H), 6,46 (с, 1Н), 5,51 (д, J=8,4 Гц, 1Н), 4,31 (м, 1H), 3,72 (с, 3Н), 3,14 (с, 3Н), 3,06 (с, 3Н), 3,00 (д, J=11,2 Гц, 2Н), 2,62 (дд, J=12,4, 10,2 Гц, 2Н), 2,19 (м, 8Н), 1,80 (д, J=11,4 Гц, 2Н), 1,65 (кв, J=11,3 Гц, 2Н), 1,12 (д, J=6,9 Гц, 6Н). MS: ИЭР масса/заряд 639,0 [М+Н]+, ВЭЖХ чистота: 98,5%.
Пример 4. Получение N-(2-((2-((5-(изопропиламино)-2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)амино)фенил)-N-метилметансульфонамида
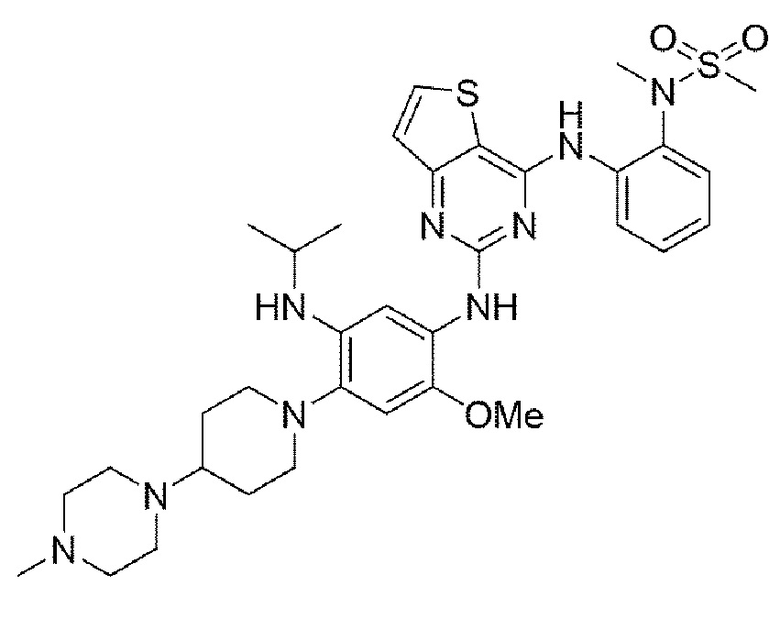
Конечное соединение синтезировали по вышеприведенной схеме реакции 4.
1Н ЯМР (400 МГц, DMSO-d6) δ 9,50 (с, 1Н), 9,22 (с, 1Н), 8,00 (с, 1Н), 7,61 (д, J=7,6 Гц, 1Н), 7,45 (дд, J=7,5, 1,5 Гц, 1Н), 7,37-7,33 (м, 2Н), 7,04 (тд, J=7,5, 1,5 Гц, 1Н), 6,76 (тд, J=7,5, 1,5 Гц, 1Н), 6,46 (с, 1Н), 5,51 (д, J=8,4 Гц, 1Н), 4,32 (м, 1Н), 3,70 (с, 3Н), 3,15 (с, 3Н), 3,04 (с, 3Н), 3,01 (д, J=11,2 Гц, 2Н),, 69-2,57 (м, 2Н), 2,55-2,48 (м, 4Н), 2,25 (м, 5Н), 2,12 (с, 3Н), 1,81 (д, J=11,9 Гц, 2Н), 1,67 (кв, J=11,4, 10,7 Гц, 2Н), 1.10 (д, J=6,9 Гц, 6Н). MS: ИЭР масса/заряд 694,1 [М+Н]+, ВЭЖХ чистота: 97,9%.
Пример 5. Получение N-(2-((5-хлор-2-((5-(изопропиламино)-2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)фенил)-N-метилметансульфонамида
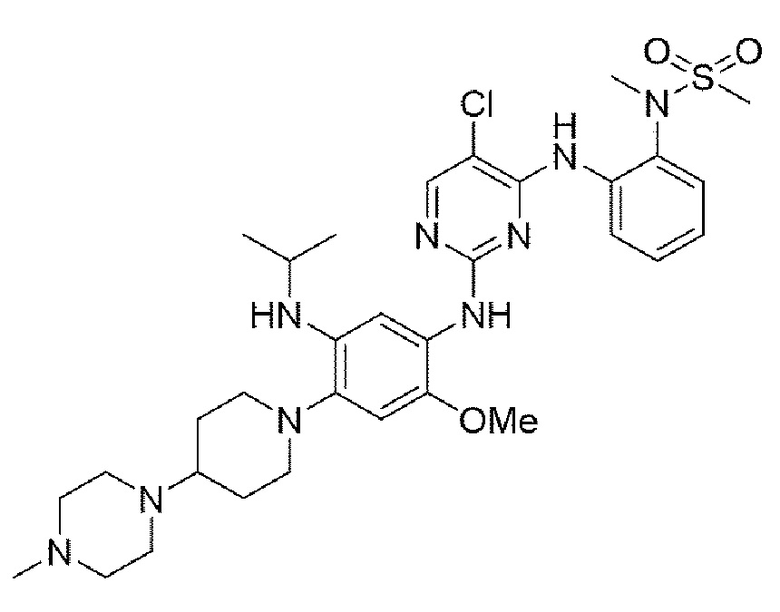
Конечное соединение синтезировали по вышеприведенной схеме реакции 4.
1Н ЯМР (400 МГц, CDCl3): δ (ч/млн): 10,02 (1Н, уш.с), 8,35-8,31 (2Н, м), 8,26 (1Н, с) 8,21-8,19 (1Н, д), 8,09 (1Н, с), 7,53-7,50 (1Н, дд), 6,95 (1Н, с), 6,18-6,14 (1Н, дд), 5,72-5,68 (1Н, дд), 4,31 (1Н, м), 3,72 (с, 3Н), 3,14 (с, 3Н), 3,06 (с, 3Н), 3,00 (2Н, д), 2,69-2,57 (2Н, м), 2,55-2,48 (4Н, м), 2,25 (5Н, м), 2,12 (3Н, с), 1,81 (2Н, д), 1,67 (2Н, кв), 1,12 (д, 6Н). MS: ИЭР масса/заряд 672,0 [М+Н]+, ВЭЖХ чистота: 98,2%.
Пример 6. Получение N-(2-((5-хлор-2-((4-(4-(диметиламино)пиперидин-1-ил)-5-(изопропиламино)-2-метоксифенил)амино)пиримидин-4-ил)амино)фенил)-N-метилметансульфонамида
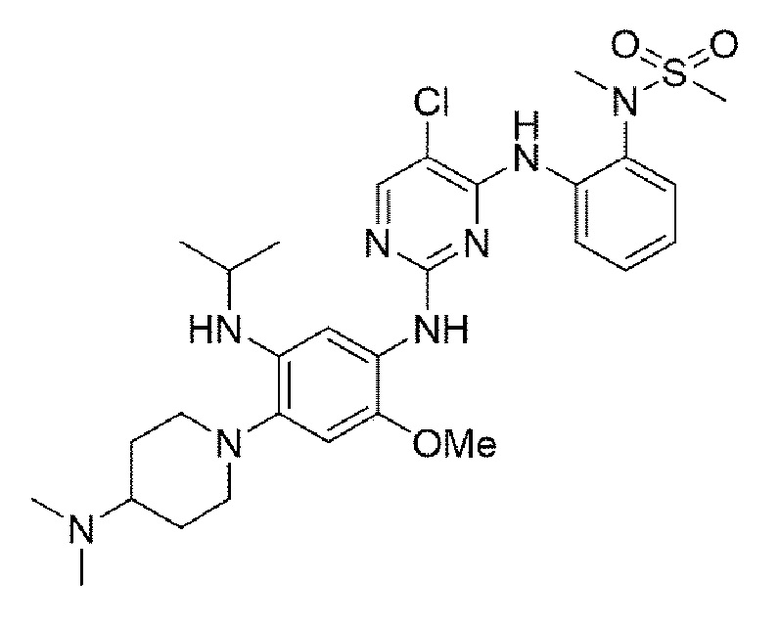
Конечное соединение синтезировали по вышеприведенной схеме реакции 4.
1H ЯМР (400 МГц, CDCl3): δ (ч/млн): 10,02 (1Н, уш.с), 8,37-8,33 (2Н, м), 8,23 (1Н, с) 8,20-8,17 (1Н, д), 8,09 (1H, с), 7,53-7,50 (1Н, дд), 6,95 (1H, с), 6,18-6,14 (1Н, дд), 5,72-5,68 (1Н, дд), 4,31 (1Н, м), 3,72 (с, 3Н), 3,14 (с, 3Н), 3,06 (с, 3Н), 3,00 (д, J=11,2 Гц, 2Н), 2,62 (2Н, дд), 2,19 (8Н, м), 1,80 (2Н, д), 1,65 (2Н, кв), 1,12 (6Н, д). MS: ИЭР масса/заряд 617,0 [М+Н]+, ВЭЖХ чистота: 97,6%.
Пример 7. Получение N-(4-метокси-5-((4-((2-(N-метилметилсульфонамидо)фенил)амино)тиено[3,2-d]пиримидин-2-ил)амино)-2-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)акриламида
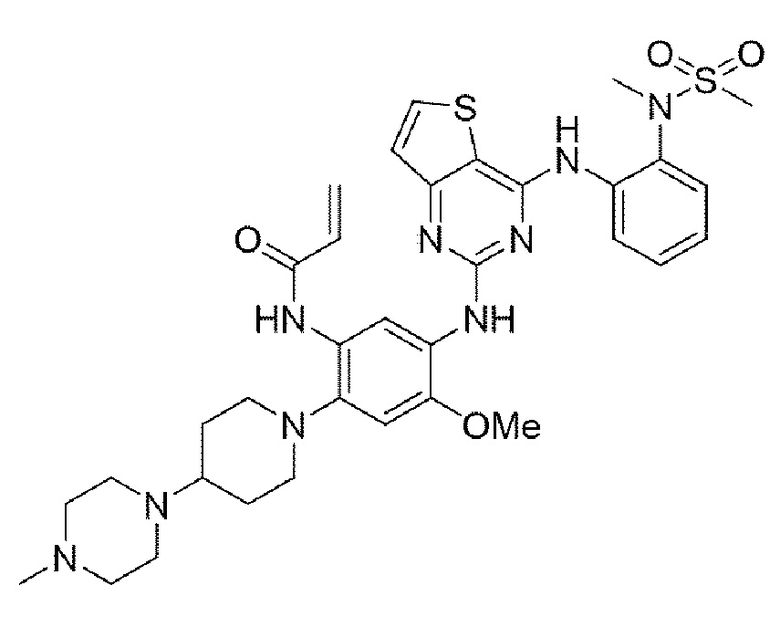
Конечное соединение синтезировали по вышеприведенной схеме реакции 4.
1H ЯМР (400 МГц, DMSO-d6) δ 10,05 (с, 1Н), 8,75 (с, 1H), 8,36 (с, 1H), 8,03 (д, J=5,3 Гц, 1Н), 8,00 (дд, J=8,1, 1,6 Гц, 1Н), 7,70 (с, 1Н), 7,52 (дд, J=7,9, 1,6 Гц, 1Н), 7,24 (тд, J=7,7, 1,6 Гц, 1Н), 7,20-7,11 (м, 2Н), 6,93 (с, 1H), 6,37 (м, 1Н), 6,18 (дд, J=16,9, 2,1 Гц, 1Н), 5,74-5,66 (м, 1H), 3,72 (с, 3Н), 3,14 (с, 3Н), 3,06 (с, 3Н), 3,00 (д, J=11,2 Гц, 2Н), 2,69-2,57 (м, 2Н), 2,55-2,48 (м, 4Н), 2,25 (м, 5Н), 2,12 (с, 3Н), 1,81 (д, J=11,9 Гц, 2Н), 1,67 (кв, J=11,4, 10,7 Гц, 2Н). MS: ИЭР масса/заряд 706,2 [М+Н]+, ВЭЖХ чистота: 98,5%.
Пример 8. Получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-((2-(N-метилметилсульфонамидо)фенил)амино)тиено[3,2-d]пиримидин-2-ил)амино)фенил)акриламида
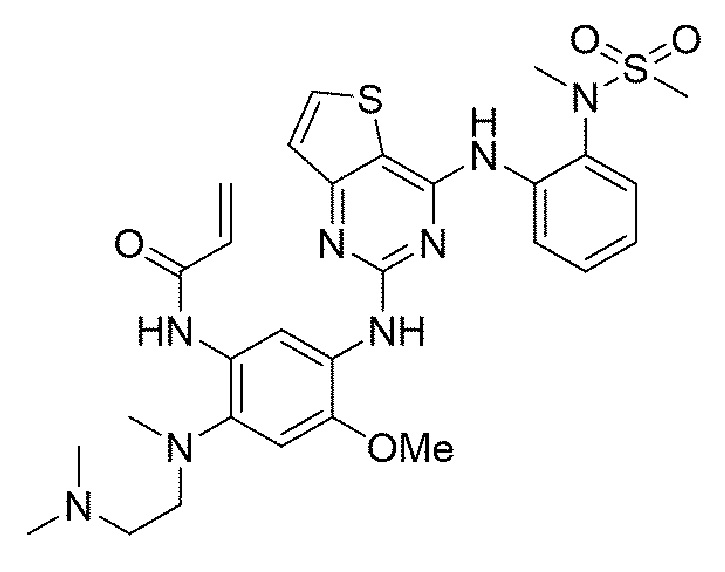
Конечное соединение синтезировали по вышеприведенной схеме реакции 4.
1Н ЯМР (400 МГц, DMSO-d6) δ 10,03 (с, 1Н), 8,74 (с, 1Н), 8,36 (с, 1Н), 8,03 (д, J=5,3 Гц, 1Н), 8,00 (дд, J=8,1, 1,6 Гц, 1Н), 7,71 (с, 1Н), 7,52 (дд, J=7,9, 1,6 Гц, 1Н), 7,24 (тд, J=7,7, 1,6 Гц, 1Н), 7,20-7,11 (м, 2Н), 6,93 (с, 1Н), 6,37 (м, 1Н), 6,18 (дд, J=16,9, 2,1 Гц, 1Н), 5,74-5,66 (м, 1Н), 3,75 (с, 3Н), 3,13 (с, 3Н), 3,01 (с, 3Н), 2,84 (уш.с, 2Н), 2,65 (с, 3Н), 2,37-2,07 (м, 8Н). MS: ИЭР масса/заряд 625,1 [М+Н]+, ВЭЖХ чистота: 97,9%.
Пример 9. Получение N-(2-(4-(диметиламино)пиперидин-1-ил)-4-метокси-5-((4-((2-(N-метилметилсульфонамидо)фенил)амино)тиено[3,2-а]пиримидин-2-ил)амино)фенил)акриламида
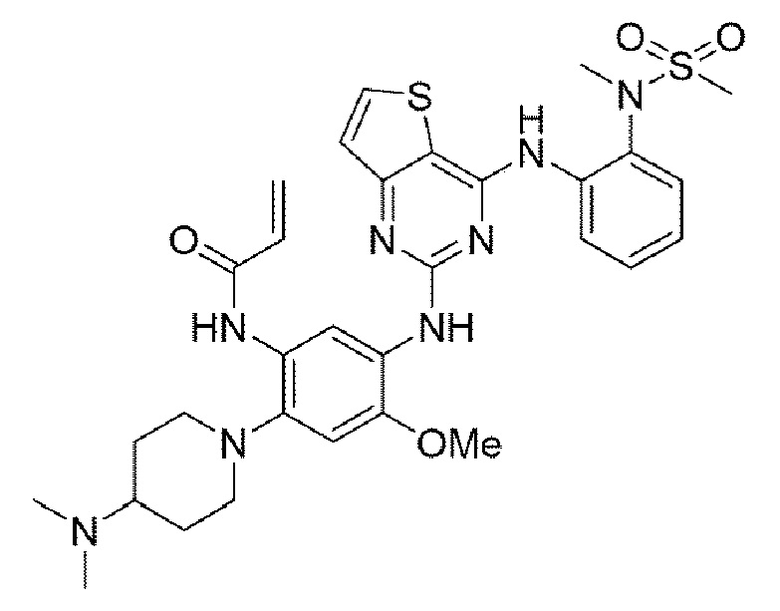
Конечное соединение получено по вышеприведенной схеме реакции 4.
1H ЯМР (400 МГц, DMSO-d6) δ 10,05 (с, 1H), 8,74 (с, 1Н), 8,35 (с, 1Н), 8,03 (д, J=5,3 Гц, 1Н), 8,00 (дд, J=8,1, 1,6 Гц, 1Н), 7,70 (с, 1Н), 7,52 (дд, J=7,9, 1,6 Гц, 1Н), 7,24 (тд, J=7,7, 1,6 Гц, 1H), 7,20-7,11 (м, 2Н), 6,93 (с, 1Н), 6,37 (м, 1H), 6,18 (дд, J=16,9, 2,1 Гц, 1H), 5,74-5,66 (м, 1Н), 3,72 (с, 3Н), 3,14 (с, 3Н), 3,06 (с, 3Н), 3,00 (д, J=11,2 Гц, 2Н), 2,62 (дд, J=12,4, 10,2 Гц, 2Н), 2,19 (с, 7Н), 1,80 (д, J=11,4 Гц, 2Н), 1,65 (кв, J=11,3 Гц, 2Н). MS: ИЭР масса/заряд 650,0 [М+Н]+, ВЭЖХ чистота: 98,1%.
Пример 10. Получение N-(5-((5-хлор-4-((2-(метилсульфонамидо)фенил)амино)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида
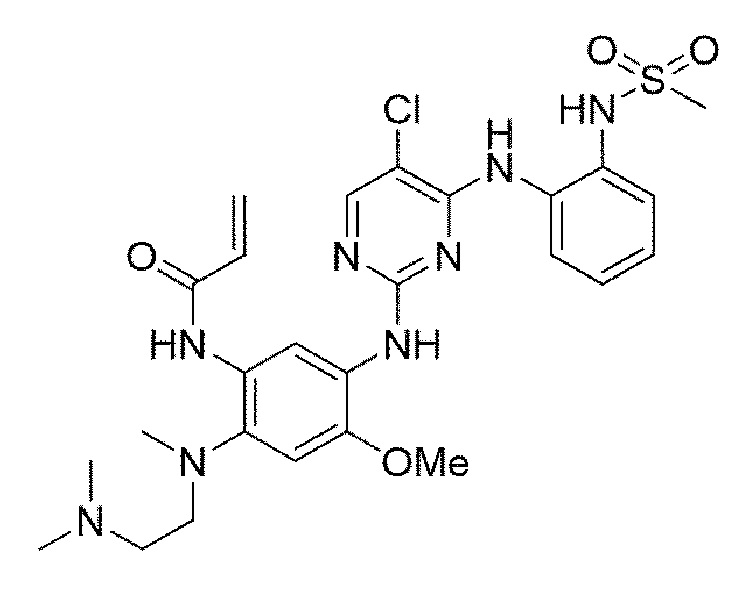
Конечное соединение синтезировано по вышеприведенной схеме реакции 4.
1Н ЯМР (400 МГц; DMSO-d6): δ (ч/млн): 10,02 (1Н, уш.с), 8,35-8,31 (2Н, м), 8,26 (1Н, с), 8,21-8,19 (1Н, д), 8,09 (1Н, с), 7,53-7,50 (1Н, дд), 7,12-7,04 (2Н, м), 6,95 (1Н, с), 6.37 (1Н, уш.с), 6,18-6,14 (1Н, дд), 5,72-5,68 (1Н, дд), 3,72 (3Н, с), 3,14 (3Н, с), 2,86 (2Н, м), 2,66 (3Н, с), 2,19 (8Н, уш.с). [М+Н]+,: масса/заряд 588,20, найдено 589,1, ВЭЖХ чистота: 98,2%.
Пример 11. Получение N-(5-((5-хлор-4-((5-(N-метилметилсульфонамидо)хиноксалин-6-ил)амино)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида
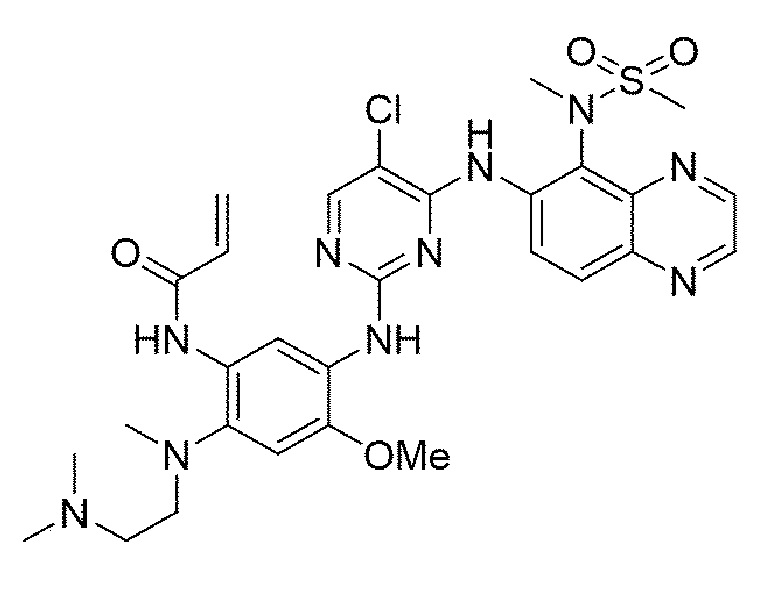
Конечное соединение синтезировано по вышеприведенной схеме реакции 4.
1Н ЯМР (400 МГц, DMSO-d6) δ 9,51 (д, J=7,3 Гц, 2Н), 9,43 (с, 1Н), 9,34 (д, J=7,5 Гц, 1Н), 8,58 (д, J=7,5 Гц, 1Н), 8,52 (с, 1Н), 7,58 (д, J=7,5 Гц, 1Н), 7,53 (д, J=7,3 Гц, 2Н), 6,41 (с, 1Н), 6,34 (дд, J=16,7, 10,0 Гц, 1H), 6,03 (дд, J=10,0, 3,1 Гц, 1Н), 5,93 (дд, J=16,7, 3,1 Гц, 1Н), 3,72 (с, 3Н), 3,14 (с, 3Н), 3,05 (с, 3Н), 2,86 (м, 2Н), 2,66 (с, 3Н), 2,19 (м, 8Н). MS: ИЭР масса/заряд 655,1 [М+Н]+, ВЭЖХ, чистота: 98,7%.
Пример 12. Получение N-(5-((5-хлор-4-((5-(метилсульфонамидо)хиноксалин-6-ил)амино)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида
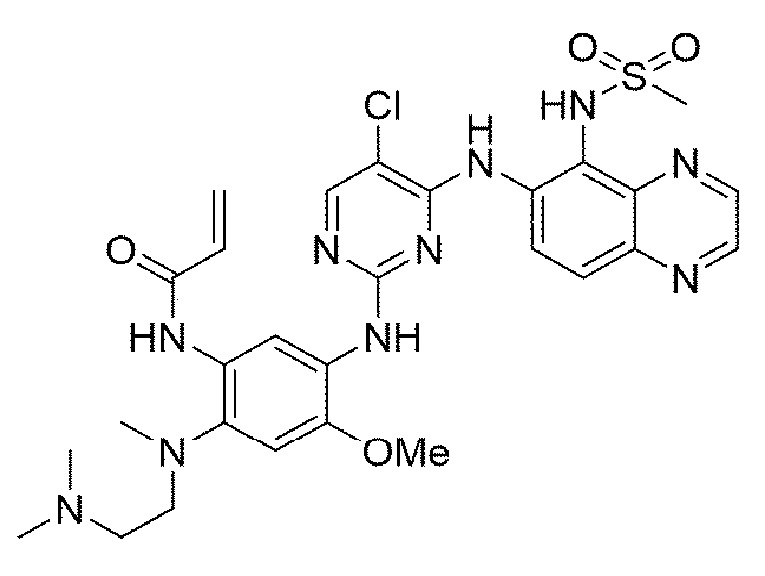
Конечное соединение синтезировали по вышеприведенной схеме реакции 4.
1Н ЯМР (400 МГц, DMSO-d6) δ 9,50 (д, J=7,3 Гц, 2Н), 9,43 (с, 1Н), 9,36 (д, J=7,5 Гц, 1Н), 8,58 (д, J=7,5 Гц, 1Н), 8,52 (с, 1Н), 7,56 (д, J=7,5 Гц, 1Н), 7,53 (д, J=7,3 Гц, 2Н), 6,41 (с, 1Н), 6,34 (дд, J=16,7, 10,0 Гц, 1H), 6,03 (дд, J=10,0, 3,1 Гц, 1H), 5,93 (дд, J=16,7, 3,1 Гц, 1H), 3,72 (с, 3Н), 3,12 (с, 3Н), 2,86 (м, 2Н), 2,66 (с, 3Н), 2,19 (м, 8Н). MS: ИЭР масса/заряд 641,0 [М+Н]+, ВЭЖХ чистота: 97,3%.
Пример 13. Получение N-(5-((5-хлор-4-((5-фтор-2-(N-метилметилсульфонамидо)фенил)амино)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида
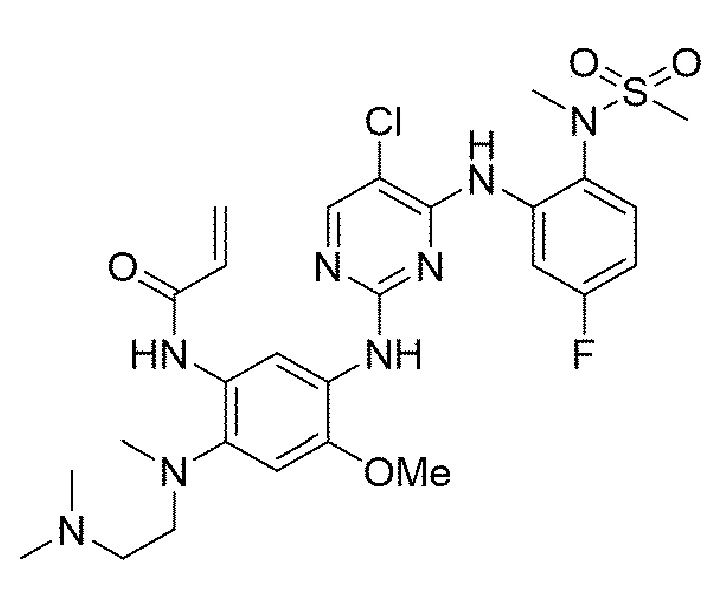
Конечное соединение синтезировано по вышеприведенной схеме реакции 4.
1H ЯМР (400 МГц, DMSO-d6) δ 9,49 (с, 1H), 9,42 (д, J=8,2 Гц, 2Н), 8,52 (с, 1H), 7,75 (дд, J=8,0, 1,5 Гц, 1Н), 7,54 (с, 1Н), 7,31 (дд, J=7,5, 5,0 Гц, 1Н), 6,93-6,84 (м, 1Н), 6,41 (с, 1Н), 6,34 (дд, J=16,7, 10,0 Гц, 1Н), 6,03 (дд, J=10,0, 3,1 Гц, 1H), 5,93 (дд, J=16,7, 3,1 Гц, 1Н), 3,72 (с, 3Н), 3,14 (с, 3Н), 3,05 (с, 3Н), 2,86 (уш.с, 2Н), 2,63 (с, 3Н), 2,20 (м, 8Н). MS: ИЭР масса/заряд 621,0 [М+Н]+, ВЭЖХ чистота: 98,9%.
Пример 14. Получение N-(5-((5-хлор-4-((5-фтор-2-(метилсульфонамидо)фенил)амино)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида
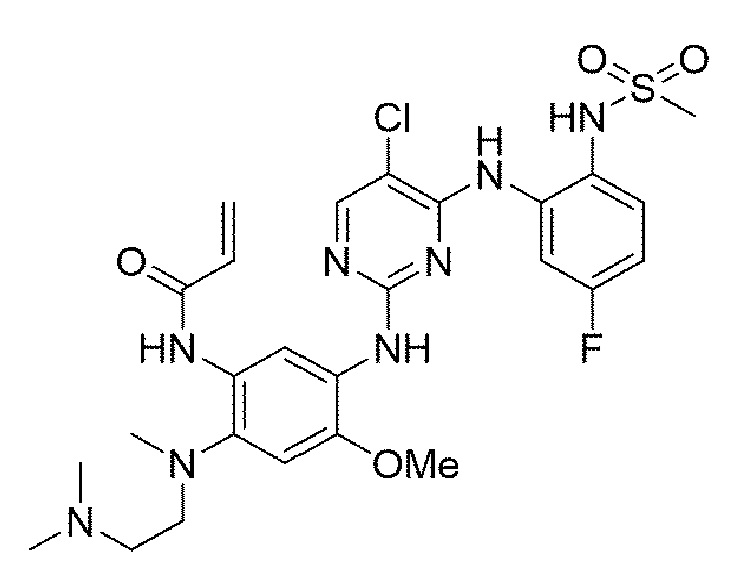
Конечное соединение синтезировали по вышеприведенной схеме реакции 4.
1Н ЯМР (400 МГц, DMSO-d6) δ 9,67 (с, 1Н), 9,54 (с, 1Н), 9,43 (д, J=2,5 Гц, 2Н), 8,52 (с, 1Н), 7,56-7,48 (м, 3Н), 6,84 (ддд, J=8,0, 7,5, 1,5 Гц, 1H), 6,41 (с, 1H), 6.34 (дд, J=16,7, 10,0 Гц, 1Н), 6,03 (дд, J=10,0, 3,1 Гц, 1H), 5,93 (дд, J=16,7, 3,1 Гц, 1H), 3,70 (с, 3Н), 3,16 (с, 3Н), 2,83 (уш.с, 2Н), 2,63 (с, 3Н), 2,18 (м, 8Н). MS: ИЭР масса/заряд 607,0 [М+Н]+, ВЭЖХ чистота: 98,7%.
Пример 15. Получение изопропил 2-((5-акриламидо-4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)амино)-4-((2-(N-метилметилсульфонамидо)фенил)амино)пиримидин-5-карбоксилата
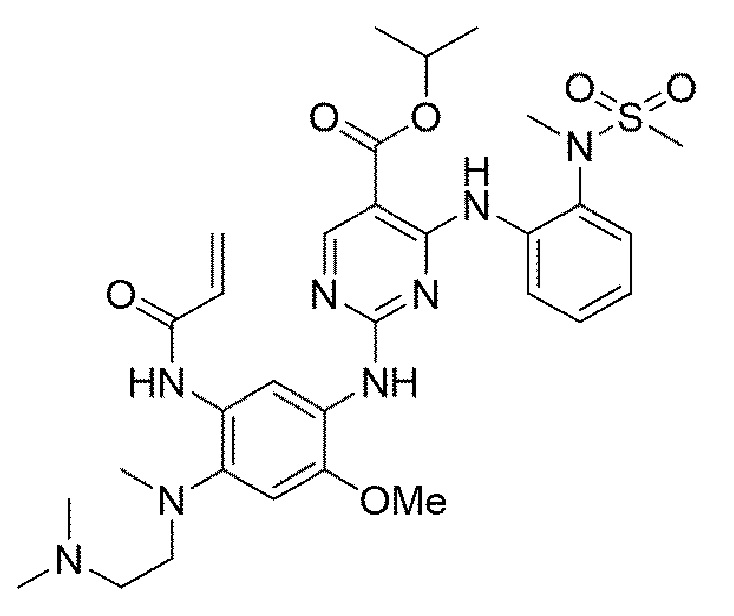
Конечное соединение синтезировано по вышеприведенной схеме реакции 4.
1H ЯМР (400 МГц, DMSO-d6) δ 10,22 (с, 1Н), 9,59-9,48 (м, 2Н), 8,84 (с, 1Н), 7,54 (с, 1Н), 7,18 (дд, J=7,4, 1,5 Гц, 1Н), 7,08-6,99 (м, 2Н), 6,95 (тд, J=7,1, 2,1 Гц, 1H), 6,41 (с, 1Н), 6,34 (дд, J=16,7, 10,1 Гц, 1H), 6,03 (дд, J=9,9, 3,1 Гц, 1Н), 5,93 (дд, J=16,7, 3,1 Гц, 1Н), 5,09 (г, J=6,2 Гц, 1Н), 3,81 (с, 3Н), 3,26 (т, J=6,8 Гц, 2Н), 3,12 (с, 3Н), 3,08 (с, 3Н), 2,83 (с, 3Н), 2,43 (м, 2Н), 2,11 (с, 6Н), 1,28 (д, J=6,2 Гц, 6Н), MS: ИЭР масса/заряд 655,0 [М+Н]+, ВЭЖХ чистота: 98,9%.
Пример 16. Получение циклопропил 2-((5-акриламидо-4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)амино)-4-((2-(N-метилметилсульфонамидо)фенил)амино)пиримидин-5-карбоксилата
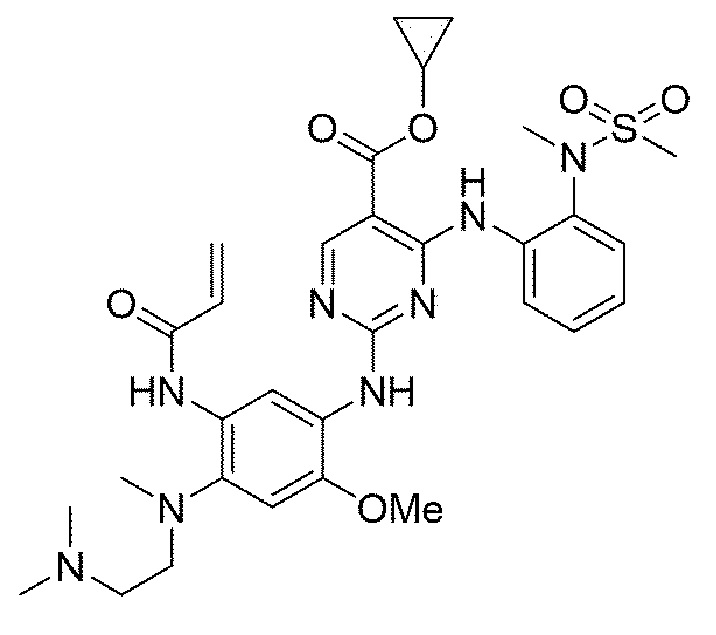
Конечное соединение синтезировано по вышеприведенной схеме реакции 4.
1H ЯМР (400 МГц, DMSO-d6) δ 10,20 (с, 1Н), 9,58-9,47 (м, 2Н), 8,82 (с, 1Н), 7,54 (с, 1Н), 7,18 (дд, J=7,4, 1,5 Гц, 1H), 7,08-6,99 (м, 2Н), 6,95 (тд, J=7,1, 2,1 Гц, 1Н), 6,41 (с, 1Н), 6,34 (дд, J=16,7, 10,1 Гц, 1Н), 6,03 (дд, J=9,9, 3,1 Гц, 1H), 5,93 (дд, J=16,7, 3,1 Гц, 1Н), 5,06 (м, 1Н), 3,79 (с, 3Н), 3,26 (т, J=6,8 Гц, 2Н), 3,15 (с, 3Н), 3,09 (с, 3Н), 2,80 (с, 3Н), 2,43 (м, 2Н), 2,11 (с, 6Н), 0,98-0,95 (м, 2Н), 0,88-0,85 (м, 2Н), MS: ИЭР масса/заряд 653,0 [М+Н]+, ВЭЖХ чистота: 98,6%.
Пример синтеза примера 17:
[Схема реакции 5]
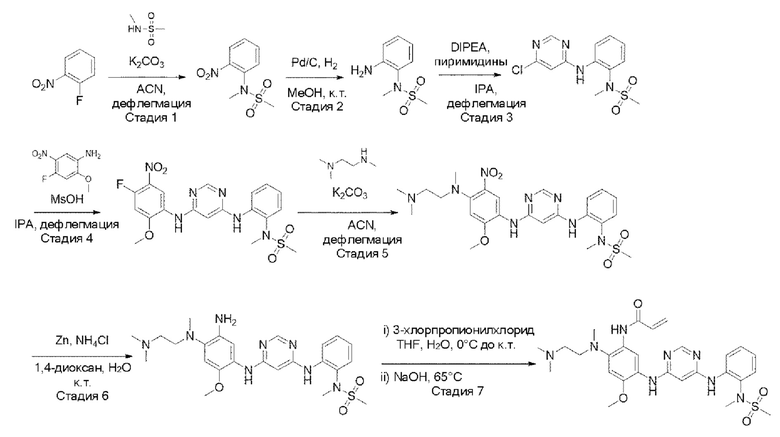
Стадия 1: синтез N-метил-N-(2-нитрофенил)метансульфонамида
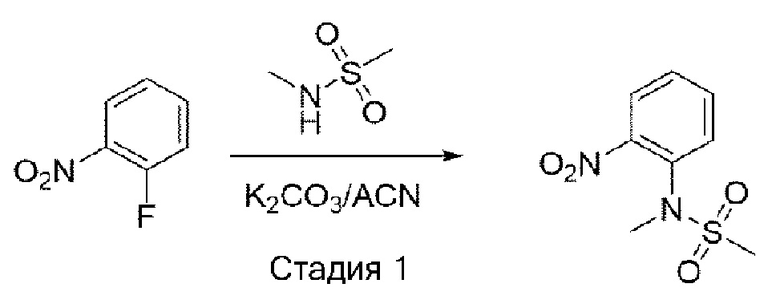
1-Фтор-2-нитробензол (1,0 экв.) растворили в ацетонитриле и добавили карбонат калия (2,0 экв.) и N-метилметансульфонамид (1,4 экв.) при комнатной температуре. Затем это перемешивали при 80°С всю ночь. После завершения реакции температуру понизили до комнатной температуры и провели фильтрацию. Фильтрат выпаривали при пониженном давлении с получением соединения. Его использовали для следующей реакции без процесса очистки.
Стадия 2: синтез N-(2-аминофенил)-N-метилметансульфонамида
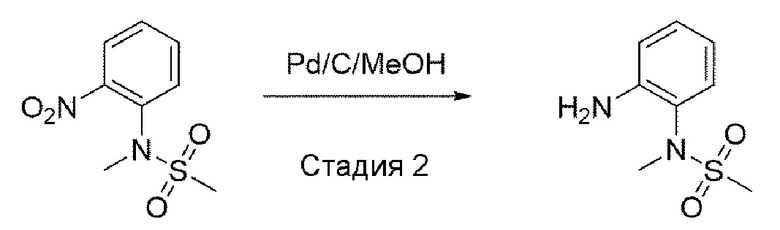
N-метил-N-(2-нитрофенил)метансульфонамид (1,0 экв.) растворили в метаноле и этилацетате (1:1) и добавили 10% палладированный уголь (0,2 экв). В атмосфере водорода это перемешивали в течение 2 часов. После завершения реакции провели фильтрацию с использованием целита, а затем фильтрат упарили при пониженном давлении. Используя простой этиловый эфир и пентан, кристаллизовали и провели фильтрацию с получением целевого соединения. Его использовали для следующей реакции без процесса очистки.
Стадия 3: синтез N-(2-((6-хлорпиримидин-4-ил)амино)фенил)-N-метилметансульфонамида
N-(2-аминофенил)-N-метилметансульфонамид (1,0 экв.) растворили в изопропиловом спирте, и добавили 2,4,5-трихлорпиридин (1,1 экв.) и N,N-диизопропилэтиламин (2,5 экв.) при комнатной температуре. Это перемешивали при 80°С всю ночь. После завершения реакции упаривали при пониженном давлении и экстрагировали, используя воду и дихлорметан. Органический слой промыли, используя 2N соляную кислоту. Органический слой упарили при пониженном давлении и провели колоночную хроматографию с получением целевого соединения. (50% гексан/этилацетат).
Стадия 4: синтез N-(2-((6-((4-фтор-2-метокси-5-нитрофенил)амино)пиримидин-4-ил)амино)фенил)-N-метилметансульфонамида
Производное пиримидина (1,0 экв.) растворяли в изопропиловом спирте и добавляли производное анилина (1,0 экв.) и метансульфоновую кислоту (1,3 экв.) при комнатной температуре. Это перемешивали при 80°С всю ночь. После завершения реакции упаривали при пониженном давлении и экстрагировали, используя смешанный раствор из воды и 10% метанола/дихлорметана. Органический слой упаривали при пониженном давлении и проводили колоночную хроматографию с получением целевого соединения. (50% гексан/этилацетат)
Стадия 5: синтез N-(2-((6-((4-((2-(диметиламино)этил)(метил)амино)-2-метокси-5-нитрофенил)амино)пиримидин-4-ил)амино)фенил)-N-метилметансульфонамида
N-(2-((6-((4-фтор-2-метокси-5-нитрофенил)амино)пиримидин-4-ил)ами но)фенил)-N-метилметансульфонамид (1,0 экв.) растворили в ацетонитриле и добавляли карбонат калия (3,0 экв.) и аминную цепь (1,2 экв.) при комнатной температуре. Это перемешивали всю ночь при кипячении с обратным холодильником. После завершения реакции температуру понизили до комнатной температуры и провели фильтрацию. Фильтрат выпаривали при пониженном давлении с получением целевого соединения. Его использовали для следующей реакции без процесса очистки.
Стадия 6: синтез N-(2-((6-((5-амино-4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)амино)пиримидин-4-ил)амино)фенил)-N-метилметансульфонамида
N-(2-((6-((4-((2-(диметиламино)этил)(метил)амино)-2-метокси-5-нитрофенил)амино)пиримидин-4-ил)амино)фенил)-N-метилметансульфонамид (1,0 экв.) растворили в 1,4-диоксане, и добавили цинк (10,0 экв.) и хлорид аммония (10,0 экв.) при комнатной температуре, и это перемешивали всю ночь. После завершения реакции температуру понизили до комнатной температуры, и после фильтрования на целите фильтрат упарили при пониженном давлении с получением соединения. Его использовали для следующей реакции без процесса очистки.
Стадия 7: синтез N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((6-((2-(N-метилметилсульфонамидо)фенил)амино)пиримидин-4-ил)амино)фенил)акриламида
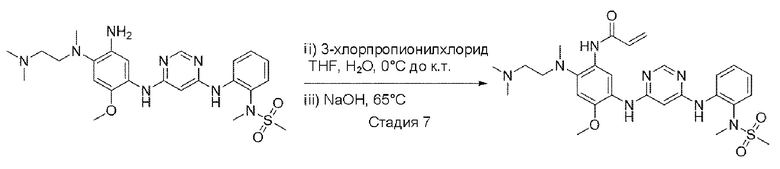
N-(2-((6-((5-амино-4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)амино)пиримидин-4-ил)амино)фенил)-N-метилметансульфонамид (1,0 экв.) растворили в тетрагидрофуране и воде и добавили 3-хлорпропионилхлорид (1,2 экв.) при 0±5°C. При той же температуре перемешивали в течение 15 минут. После завершения реакции при той же температуре добавили гидроксид натрия (4,0 экв). Повышая температуру реактора перемешивали при 65°С всю ночь. После завершения реакции растворитель удалили выпариванием при пониженном давлении и экстрагировали, используя смешанный раствор из воды и 10% метанола/дихлорметана. Органический слой упарили при пониженном давлении и провели колоночную хроматографию с получением целевого соединения. (10% метиловый спирт/дихлорметан).
Пример 17. N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((6-((2-(N-метилметилсульфонамидо)фенил)амино)пиримидин-4-ил)амино)фенил)акриламид
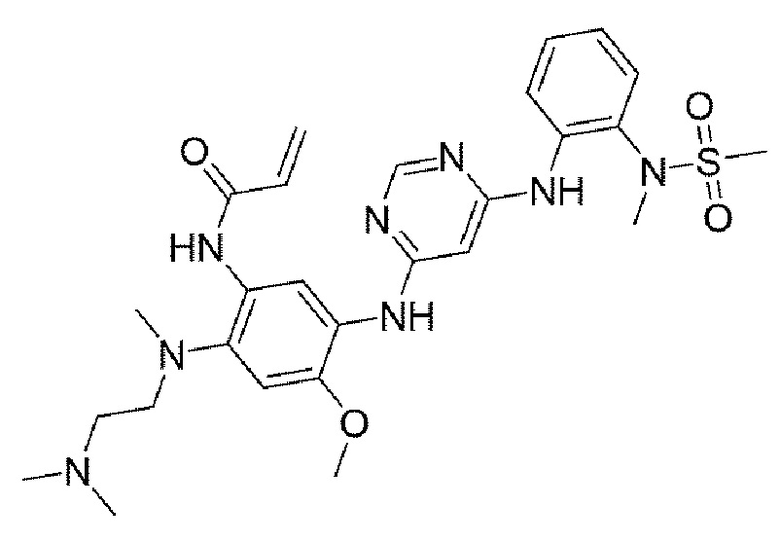
Выход: 23,0%, белое твердое вещество, 1Н ЯМР (400 МГц, DMSO-d6) 5 10,04 (с, 1Н), 8,42 (с, 1Н), 8,29 (с, 1Н), 8,09 (д, J=0,9 Гц, 1Н), 8,00 (с, 1Н), 7,79 (дд, J=8,2, 1,5 Гц, 1H), 7,46 (дд, J=8,0, 1,5 Гц, 1Н), 7,26 (ддд, J=8,4, 7,4, 1,6 Гц, 1H), 7,07 (тд, J=7,6, 1,5 Гц, 1Н), 6,93 (с, 1H), 6,34 (дд, J=16,9, 10,0 Гц, 1Н), 6,19 (дд, J=16,9, 2,2 Гц, 1H), 6,04 (д, J=1,1 Гц, 1Н), 5,71 (дд, J=10,0, 2,2 Гц, 1Н), 3,74 (с, 3Н), 3,10 (с, 3Н), 2,99 (с, 3Н), 2,80 (т, J=5,9 Гц, 2Н), 2,66 (с, 3Н), 2,26 (т, J=5,8 Гц, 2Н), 2,16 (с, 6Н). MS: ИЭР масса/заряд - 569,14 [М+Н]+.
Экспериментальный пример 1: Определение ингибирующего действия на рост раковых клеток
Поскольку клеточная линия рака легких с инсерционной мутацией РЭФР не поступает на рынок, предпринят метод для вставки 3-4 аминокислот в любой сайт в векторе экспрессии РЭФР дикого типа, используя сайт-направленный мутагенез. Сначала в настоящем изобретении использовали вектор, в котором NPG (неопентилглицины) в количестве 3 аминокислот были добавлены к сайту D770_N771 наиболее частых изменений, и использовали клеточную линию Ba/F3 для экспрессии этих генов. Клеточная линия Ba/F3 представляет собой мышиную IL3-зависимую про-В клеточную линию, показывающую рост клеток только тогда, когда добавляют IL-3 (интерлейкин-3), и имеет зависимость мутантного РЭФР от роста и гибели клеток в отсутствие IL-3, когда экспрессируется онкогенный мутантный РЭФР. Поскольку вещества, показывающие эффективное ингибирующее действие на мутантный РЭФР, подавляют рост клеток и индуцируют апоптоз, противораковое действие на клетки анализировали с помощью МТТ-теста.
Предварительно созданные стабильные клетки в количестве 1 X 104 клеток поместили в 96-луночный планшет и инкубировали всю ночь, а затем применяли соединения примеров дозозависимым образом. Через 72 часа добавили реагент МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий бромид), а через 3 часа добавили останавливающий буфер (10% SDS (натрия додецилсульфат)). После 24 часов инкубирования результат анализировали путем считывания при 595 им, и значение IC50 рассчитывали при концентрации, при которой каждое соединение подавляло рост клеток на 50%. Результат продемонстрирован в виде А, В, С и D в Таблице 1, приведенной ниже. При этом А означает IC50≤100 нМ, В означает IC50=100-300 нМ и С означает IC50=300-1000 нМ, и D означает IC50>1000 нМ. В качестве контрольного препарата использовали осимертиниб.
Измеренное значение ингибирующего действия на рост раковых клеток (GI50)
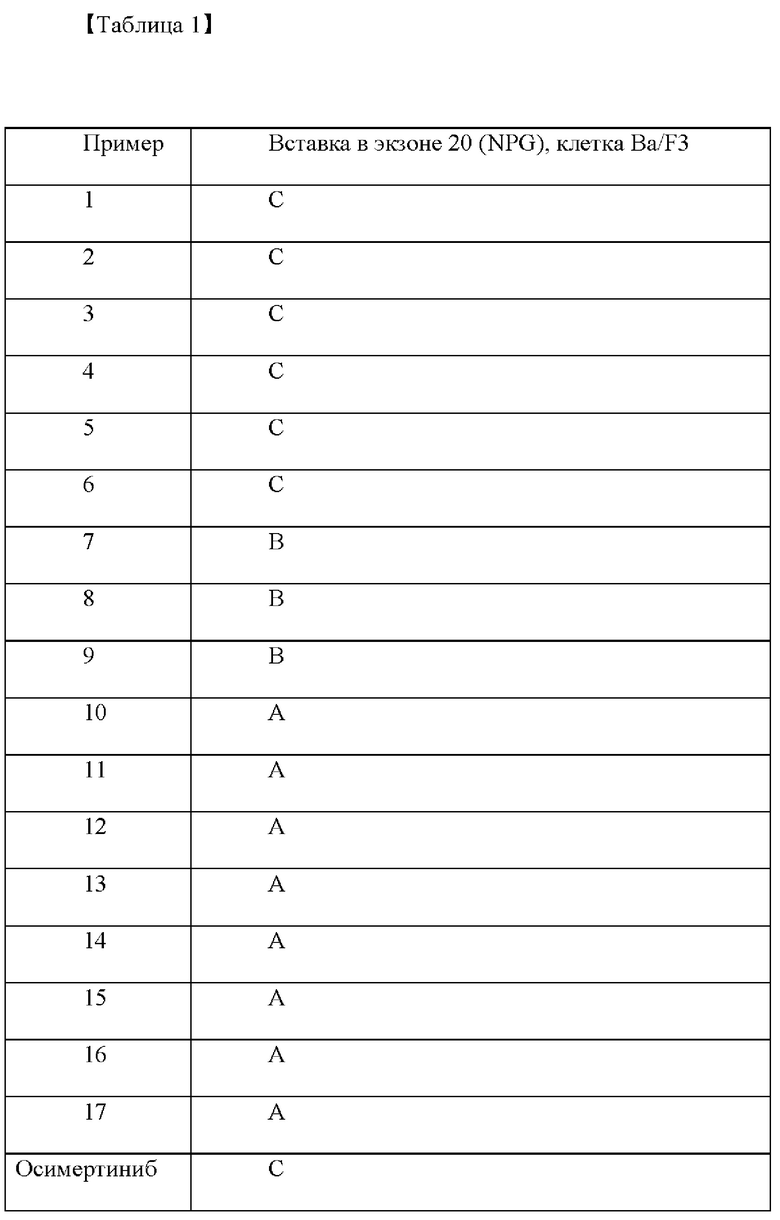
Как показано в таблице 1, соединения согласно настоящему изобретению проявляли очень хорошую активность по отношению к клеточной линии рака легких, экспрессирующей инсерционную мутацию экзона 20 рецептора эпидермального фактора роста. В частности, активность соединений примеров 10-17 была более высокого уровня. Напротив, активность контрольного препарата осимертиниба была относительно низкой.
Соответственно, настоящее изобретение предлагает новое производное пиримидина, которое может лечить рак легких, экспрессирующий инсерционную мутацию экзона 20 в рецепторе эпидермального фактора роста.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ, ОБЛАДАЮЩЕЕ ЭФФЕКТОМ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК, И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2744168C1 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ МОДУЛЯЦИИ КИНАЗНОЙ АКТИВНОСТИ МУТАНТОВ EGFR | 2024 |
|
RU2838180C1 |
| ПРОИЗВОДНОЕ БЕНЗАМИДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ РАКА, СОДЕРЖАЩАЯ ЕГО В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2022 |
|
RU2841260C2 |
| ПРОИЗВОДНЫЕ ПИРИДОПИРИМИДИНОНА И ИХ ИСПОЛЬЗОВАНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 2021 |
|
RU2818954C1 |
| СОЕДИНЕНИЕ 2-АМИНОПИРИМИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ ДАННОГО СОЕДИНЕНИЯ | 2015 |
|
RU2704129C2 |
| ИНГИБИТОР EGFR И ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2702631C2 |
| НОВОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ ПИРИМИДИНА, ПРОЯВЛЯЮЩЕЕ ИНГИБИРУЮЩЕЕ ДЕЙСТВИЕ НА РОСТ РАКОВЫХ КЛЕТОК, И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕГО | 2020 |
|
RU2834201C1 |
| НОВОЕ ПРОИЗВОДНОЕ ПИПЕРИДИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ АУТОТАКСИНА | 2022 |
|
RU2834850C2 |
| ПРОИЗВОДНЫЕ АНИЛИНПИРИМИДИНА И ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2734849C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ МОДУЛЯЦИИ КИНАЗНОЙ АКТИВНОСТИ МУТАНТОВ EGFR | 2015 |
|
RU2727700C2 |
Изобретение относится к соединению формулы 1 или его фармацевтически приемлемой соли, а также фармацевтической композиции для лечения или профилактики рака легких, экспрессирующего мутацию рецептора эпидермального фактора роста. Технический результат - обеспечение соединений, пригодных для лечения рака легких, экспрессирующего мутацию рецептора эпидермального фактора роста. В общей формуле 1 A, B, D, E и G представляют собой комбинации A=N, B=C, D=C, E=C и G=N или комбинации A=N, B=C, D=N, E=C и G=C, W представляет собой кислород или NH, X и Y каждый независимо представляют собой CH, кислород или азот, Z1 и Z2 каждый независимо представляют собой C1-C4-алкил или каждый независимо состоят из атома углерода и связаны друг с другом с образованием 6-членного гетероциклического кольца, содержащего 1 N, вместе с X и Y, при условии, что, когда Х представляет собой кислород, тогда Z1 отсутствует, и когда Y представляет собой кислород, тогда Z2 отсутствует, R1 представляет собой C1-C4-алкил, R2 представляет собой водород или C1-C4-алкил, R3 и R4 каждый независимо представляют собой водород, галоген, OH, OMe, OEt, CN, CF3, C1-C4-алкил или связаны друг с другом с образованием 5- или 6-членного гетероарильного кольца, которое содержит по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O и S, R5 представляет собой водород, галоген, OH, OMe, OEt, CN, CF3 или C1-C4-алкил, R6 и R7 каждый независимо представляют собой водород, галоген, OH, OMe, OEt, CN, CF3, COOH, COO-C1-C4-алкил, COO-C1-5-циклоалкил, C1-C4-алкил или соединены друг с другом с образованием 5- или 6-членного гетероарильного кольца, R8 представляет собой водород, галоген, OH, OMe, OEt, CN, CF3 или C1-C4-алкил, R9 представляет собой -C(O)-CH=CH2 или C1-C4-алкил, и R10 отсутствует, представляет собой водород, галоген, C1-C4-алкил, OH, OMe, OEt, CN, CF3, NMe2, пиперазин, замещенный C1-C3-алкилом пиперазин, морфолин или замещенный C1-C3-алкилом морфолин. 2 н. и 3 з.п. ф-лы, 1 табл., 18 пр.
 1
1
1. Соединение приведенной ниже химической формулы 1 или его фармацевтически приемлемая соль:
[Химическая формула 1]
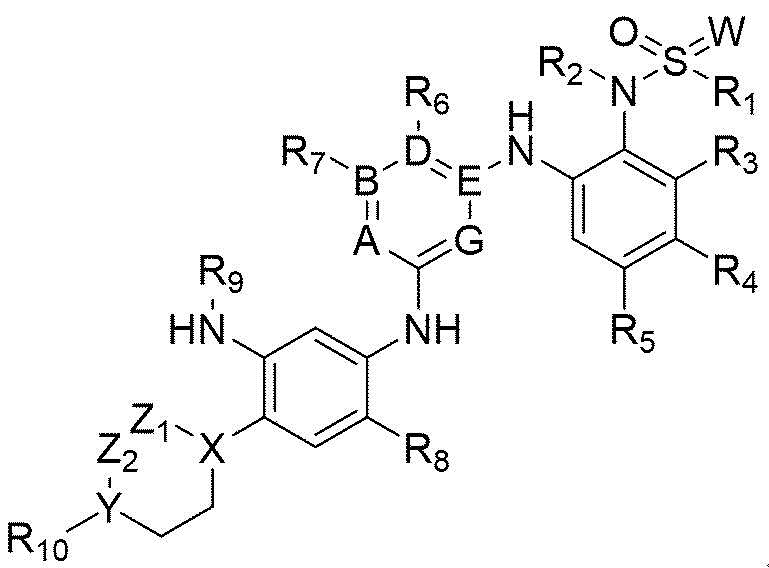 ,
,
в химической формуле 1
A, B, D, E и G представляют собой комбинации A=N, B=C, D=C, E=C и G=N, или комбинации A=N, B=C, D=N, E=C и G=C,
W представляет собой кислород или NH,
X и Y каждый независимо представляют собой CH, кислород или азот,
Z1 и Z2 каждый независимо представляют собой C1-C4-алкил или каждый независимо состоят из атома углерода и связаны друг с другом с образованием 6-членного гетероциклического кольца, содержащего 1 N, вместе с X и Y, при условии, что, когда Х представляет собой кислород, тогда Z1 отсутствует, и когда Y представляет собой кислород, тогда Z2 отсутствует,
R1 представляет собой C1-C4-алкил,
R2 представляет собой водород или C1-C4-алкил,
R3 и R4 каждый независимо представляют собой водород, галоген, OH, OMe, OEt, CN, CF3, C1-C4-алкил или связаны друг с другом с образованием 5- или 6-членного гетероарильного кольца, которое содержит по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O и S,
R5 представляет собой водород, галоген, OH, OMe, OEt, CN, CF3 или C1-C4-алкил,
R6 и R7 каждый независимо представляют собой водород, галоген, OH, OMe, OEt, CN, CF3, COOH, COO-C1-C4-алкил, COO-C1-5-циклоалкил, C1-C4-алкил или соединены друг с другом с образованием 5- или 6-членного гетероарильного кольца,
R8 представляет собой водород, галоген, OH, OMe, OEt, CN, CF3 или C1-C4-алкил,
R9 представляет собой -C(O)-CH=CH2 или C1-C4-алкил, и
R10 отсутствует, представляет собой водород, галоген, C1-C4-алкил, OH, OMe, OEt, CN, CF3, NMe2, пиперазин, замещенный C1-C3-алкилом пиперазин, морфолин или замещенный C1-C3-алкилом морфолин.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где соединение представляет собой
N-(2-((5-хлор-2-((4-((2-(диметиламино)этил)(метил)амино)-5-(изопропиламино)-2-метоксифенил)амино)пиримидин-4-ил)амино)фенил)-N-метилметансульфонамид, N-(2-((2-((4-((2-(диметиламино)этил)(метил)амино)-5-(изопропиламино)-2-метоксифенил)амино)тиено[3,2-d]пиримидин-4-ил)амино)фенил)-N-метилметансульфонамид,
N-(2-((2-((4-(4-(диметиламино)пиперидин-1-ил)-5-(изопропиламино)-2-метоксифенил)амино)тиено[3,2-d]пиримидин-4-ил)амино)фенил)-N-метилметансульфонамид,
N-(2-((2-((5-(изопропиламино)-2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)тиено[3,2-d]пиримидин-4-ил)амино)фенил)-N-метилметансульфонамид,
N-(2-((5-хлор-2-((5-(изопропиламино)-2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)фенил)-N-метилметансульфонамид,
N-(2-((5-хлор-2-((4-(4-(диметиламино)пиперидин-1-ил)-5-(изопропиламино)-2-метоксифенил)амино)пиримидин-4-ил)амино)фенил)-N-метилметансульфонамид,
N-(4-метокси-5-((4-((2-(N-метилметилсульфонамидо)фенил)амино)тиено[3,2-d]пиримидин-2-ил)амино)-2-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)акриламид,
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-((2-(N-метилметилсульфонамидо)фенил)амино)тиено[3,2-d]пиримидин-2-ил)амино)фенил)акриламид,
N-(2-(4-(диметиламино)пиперидин-1-ил)-4-метокси-5-((4-((2-(N-метилметилсульфонамидо)фенил)амино)тиено[3,2-d]пиримидин-2-ил)амино)фенил)акриламид, N-(5-((5-хлор-4-((2-(метилсульфонамидо)фенил)амино)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид,
N-(5-((5-хлор-4-((5-(N-метилметилсульфонамидо)хиноксалин-6-ил)амино)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид,
N-(5-((5-хлор-4-((5-(метилсульфонамидо)хиноксалин-6-ил)амино)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид,
N-(5-((5-хлор-4-((5-фтор-2-(N-метилметилсульфонамидо)фенил)амино)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид,
N-(5-((5-хлор-4-((5-фтор-2-(метилсульфонамидо)фенил)амино)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид,
изопропил 2-((5-акриламидо-4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)амино)-4-((2-(N-метилметилсульфонамидо)фенил)амино)пиримидин-5-карбоксилат,
циклопропил 2-((5-акриламидо-4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)амино)-4-((2-(N-метилметилсульфонамидо)фенил)амино)пиримидин-5-карбоксилат или
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((6-((2-(N-метилметилсульфонамидо)фенил)амино)пиримидин-4-ил)амино)фенил)акриламид.
3. Соединение или его фармацевтически приемлемая соль по п. 1, где R3 и R4 каждый независимо представляют собой водород, галоген, OH, OMe, OEt, CN, CF3, C1-C4-алкил или связаны друг с другом с образованием 6-членного гетероарильного кольца, содержащего 2N.
4. Фармацевтическая композиция для лечения или профилактики рака легких, экспрессирующего мутацию рецептора эпидермального фактора роста, отличающаяся тем, что содержит соединение или фармацевтически приемлемую соль по любому из пп. 1-3 в качестве активного ингредиента.
5. Фармацевтическая композиция для лечения или профилактики рака легких по п. 4, где мутация представляет собой инсерционную мутацию экзона 20.
| US 20170226065 A1, 10.08.2017 | |||
| KR 1020120047208 A, 11.05.2012 | |||
| WO 2014124230 A2, 14.08.2014 | |||
| KR 1020180135815 A, 21.12.2018 | |||
| Радиопередатчик с независимым возбуждением | 1930 |
|
SU26353A1 |

