Область изобретения
Настоящее изобретение относится к способу очистки LNnT (от англ. lacto-N-neotetraose-лакто-N-неотетраоза, Gal(β1-4)GlcNAc(β1-3)Gal(β1-4)Glc). Более конкретно, настоящее изобретение относится к отделению побочных продуктов, примесей и/или загрязняющих веществ от LNnT, полученной в результате процесса ферментации, посредством способа очистки.
Предшествующий уровень техники
Человеческое грудное молоко считается наилучшим питанием для развития младенца. Оно состоит из жиров, белков, витаминов, минеральных веществ, микроэлементов и сложных олигосахаридов. Помимо лактозы человеческое грудное молоко, а также молоко других млекопитающих, содержит разные структурно различные олигосахариды, которые также известны, как олигосахариды грудного молока (ОГМ) (Usashima Т. et al., (2011) Milk 20 Oligosaccharides, Nova Biomedical Books, New York ISBN 978-1-61122-831-1). В настоящее время считают, что существует более 150 разных по структуре олигосахаридов, обнаруженных в человеческом грудном молоке. За очень редким исключением, с одной стороны, ОГМ характеризуются остатком дисахарида - лактозы на их восстанавливающих концах. С другой стороны, многие ОГМ содержат остаток фукозы, остаток галактозы или остаток N-ацетилнейраминовой кислоты на их невосстанавливающем конце. Кроме того, существуют линейные, а также разветвленные представители. Обычно, остатки моносахаридов ОГМ представляют собой D-глюкозу, D-галактозу, N-ацетилглюкозамин, L-фукозу и N-ацетилнейраминовую кислоту (последняя больше известна как сиаловая кислота или лактаминовая кислота). Важность ОГМ для вскармливания грудных детей непосредственно связана с их биологическими активностями, включая защиту новорожденных от патогенов, поддержание развития иммунной системы и когнитивных способностей грудного ребенка. Кроме того, ОГМ служат в качестве субстрата для полезных бактерий, подобно бифидобактериям или лактобациллам.
Из-за проблем, сопровождающих химических синтез олигосахаридов грудного молока, было разработано несколько ферментативных способов и ферментативных подходов. В частности, ферментативный поход требует очистки желательного олигосахарида от весьма сложного ферментационного бульона, содержащего несколько сотен разных отдельных соединений. Отдельно взятая углеводная фракция ферментационного бульона состоит из сложной смеси моно- и олигосахаридов, а также их производных, включая субстраты (например, лактозу, фруктозу, глюкозу, сахарозу и другие сахара, используемые в качестве источника углерода), биосинтетические промежуточные вещества, отдельные моносахариды (такие как глюкоза, галактоза, N-ацетил глюкоза мин, фукоза и N-ацетилнейраминовая кислота), побочные продукты метаболизма и другие олиго- и полисахариды, синтезированные микробом. Кроме того, структуры многих олигосахаридов, встречающихся в ферментационном бульоне, трудно идентифицировать (например, олигосахариды, продуцируемые в результате синтеза структур гликозилирования клеточной поверхности, подобных встречающимся в природе у хозяина, или олигосахариды, продуцируемые организмом в результате стресса). Таким образом, во многих случаях очистка биотехнологического продукта может быть гораздо более дорогой и времязатратной, чем его получение посредством ферментации.
В частности, ферментация лакто-N-неотетраозы часто ассоциирована с избыточным синтезом лакто-N-триозы II (LNT II, GlcNAc(β1-3)Gal(β1-4)Glc), биосинтетическим промежуточным веществом в биосинтезе лакто-N-неотетраозы, которое часто экспортируется из клетки в среду перед дальнейшим превращением в целевую LNnT. Кроме того, пара-лакто-N-неогексаоза (pLNnH, Gal(β1-3)GlcNAc(β1-3)Gal(β1-4)GlcNAc(β1-3)Gal(β1-4)Glc) часто встречается в качестве побочного продукта, когда LNnT в дальнейшем превращается в N-ацетилглюкозамин (GalNAc) и глюкозу вместо экспорта, и даже пара-лакто-N-неооктаозу (pLNnO, Gal(β1-4)GlcNAc(β1-3)Gal(β1-4)GlcNAc(β1-3)Gal(β1-4)Glc) была выявлена в виде дополнительно удлиненного производного. Последние побочные продукты представлены через глюкозиллактозу (GlcLac), галактозиллактозу (CalLac), глюкозилированную LNnT и галактозилированную LNnT, которые образуются во время ферментации. Однако, олигосахариды, которые несут GalNAc на невосстанавливающем конце, такие как лакто-N-триоза II или промежуточные вещества для pLNnH или pLNnO, могут быть эффективно удалены посредством обработки гликозидазой. Кроме того, автоклавирование (тепловая обработка) углеводов (например, сахарозы или лактозы) может приводить к образованию нежелательных побочных продуктов, таких как продукты альдольной реакции или реакции Майяра. Реакции изомеризации (например, превращение лактозы в лактулозу) могут, кроме того, в общем, приводить к большим загрязнениям и создавать олигосахариды-изомеры. Для того, чтобы избежать образования побочных продуктов из-за тепловой обработки, субстраты и источники С часто подвергают стерильной фильтрации, что, однако, вызывает риск загрязнения чужеродными агентами среды роста.
Для получения олигосахаридов грудного молока посредством микробной ферментации используют рекомбинантные микроорганизмы (рекомбинантные штаммы бактерий или дрожжей). Таким образом, данные способы ферментации основываются на генетически модифицированных организмах (ГМО), которые считаются крайне важными в секторе пищевой промышленности. Таким образом, требуемые продукты должны быть вследствие этого очищены от технологических остатков ГМО, таких как клетки, фрагменты клеток, эндотоксины и избыточные соли, для получения одобрения от регулирующих органов и потребителей. Лакто-N-неотетраоза, таким образом, должна быть очищена от ферментационного бульона, содержащего сложную смесь, состоящую из рекомбинантных нуклеиновых кислот, таких как ДНК и РНК, а также из рекомбинантных белков. Кроме того, микробная ферментация, в частности, содержит значимые количества эндотоксинов, когда используют E.coli. Однако, загрязнение продукта для потребления человеком рекомбинантной ДНК, эндотоксинами или белками, не считается приемлемой ни регулирующими органами, ни потребителями. Таким образом, любые нуклеиновые кислоты и белки, полученные из рекомбинантного микроорганизма, должны быть удалены из желательного олигосахарида грудного молока.
Известные способы очистки отдельных олигосахаридов являются технически сложными и часто неэкономичными, особенно, когда данные исходные соединения получают в виде смеси нескольких конструкций, построенных похожим образом, из ферментационного бульона, в частности, кода указанный олигосахарид предназначен для применений в пищу. Для промышленной очистки пригодных для применения в пищевой промышленности дисахаридов - лактозы или сахарозы из сложных смесей, таких как молочная сыворотка или меласса, разработаны промышленные способы в многотонном масштабе, которые включают множественные стадии кристаллизации. Однако, оказалось, что вплоть до сегодняшнего времени ОГМ, в общем и более конкретно лакто-N-неотетраозу, трудно очистить, особенно при получении из ферментационного бульона. В случае LNnT разработан способ синтетического получения и впоследствии кристаллизации полученного продукта, делающий возможным его применение в качестве пищевого ингредиента. Но в данном способе не содержится никаких побочных продуктов от ферментации, а способ дает почти чистый продукт, который затем выделяют по экономическим причинам в виде кристаллического вещества для дополнительной обработки впоследствии.
Ранняя работа по выделению, характеристики и кристаллизации разных ОГМ выполнена Richard Kuhn и соавторами в 1950-ых годах, где данные соединения были выделены из фракции углеводов молока матери посредством хроматографической очистки на колонках с активированным углем/целитом. Первый ОГМ, подлежащий кристаллизации, из Kuhn et. al., представлял собой лакто-N-биозу I (Kuhn et al.,Chem, Ber. 1954, 87(10), 1553-1560), за которым в последующие годы следовало огромное количество других углеводов: лактозо-N-тетраоза (Kuhn et al., Chem. Ber. 1953, 86(6), 827-830; Chem. Ber. 1954, 87(3), 289-300; Chem. Ber. 1956, 89(2), 504-511), лакто-N-фукопентаоза I (Kuhn et al., Chem. Ber. 1956, 89(11), 2514-523), 2'-фукозиллактоза (Kuhn et al., Chem. Ber. 1955, 88(8), 1135-1146; 1956, 89(11), 2513), лакто-N-триоза I и II (Kuhn et al., Chem. Ber. 1956, 89(4), 1027-1033) и лакто-N-неотетраоза (Kuhnetal., Chem. Ber. 1962, 95(11), 518-522).
Для очистки лакто-N-неотетраозы и близкородственного олигосахарида грудного молока лакто-N-тетраозы (Gal(β1-3)GlcNAc(β1-3)Gal(β1-4)Glc) использовали хроматографические способы, в частности, гель-фильтрационную хроматографию (Dumon et al., 2001 Glycoconj. J. 18(6), 465-474; Priem et al., Glycobiology 2002, 12(4), 235-240; Baumgartner et al., Chem. Bio. Chem. 2014 15(13), 1896-1900, Sprenger et al., 2017, J. Biotechnol. 258, 79-91). В данном отношении, очистка посредством гель-фильтрационой хроматографии не подходит для пищевых продуктов в промышленном масштабе, однако, хроматография с псевдодвижущимся слоем может считаться подходящим способом очистки LNnT для пищевой продукции, в сочетании с другими стадиями очистки, для получения очищенной LNnT в качестве конечного продукта.
При микробной ферментации лакто-N-неотетраозы могут быть образованы многие другие углеводы, такие как триозы (LNT II, GlcLac, GalLac), а также более длинноцепочечные олигосахариды, такие как гексаозы (pLNnH). Кроме того, продукты гидролиза полученных олигосахаридов образуются во время ферментации и процесса обработки. Используя способ очистки, который включает несколько стадий очистки, LNnT можно получать в больших количествах, более высокой чистоты и с более высоким выходом, что делает ее подходящей в качестве ингредиента в пищевой формуле или для применений в косметологии или медицине.
Способ по настоящему изобретению представляет рентабельную альтернативу способам предшествующего уровня техники, приводящим к получению продукта в виде твердого порошка, и является особенно релевантным для очистки лакто-N-неотетраозы для применений в питании и в частности для получения питательных продуктов для грудных детей и детей, начинающих ходить, лечебных пищевых продуктов, пищевых добавок или обычных пищевых продуктов.
Для преодоления всех данных недостатков известных способов согласно настоящему изобретению предложен новый способ очистки, простой, рентабельный и масштабируемый, для очистки лакто-N-неотетраозы, полученной в результате процесса ферментации.
Краткое изложение сущности изобретения
Настоящее изобретение относится к простому и экономичному способу очистки лакто-N-неотетраозы, полученной в результате микробной ферментации. Посредством микробной ферментации лакто-N-неотетраозы также могут быть образовано несколько других углеводов, таких как триозы, и более длинноцепочечные олигосахариды, такие как гексаозы. Кроме того, во время ферментации и процесса обработки образуются продукты гидролиза продуцируемых олигосахаридов.
Цель настоящего изобретения заключается в предложении простого, экономически эффективного и масштабируемого способа получения очищенной лакто-N-неотетраозы, полученной в результате микробной ферментации в качестве основного продукта, тогда как побочные продукты, примеси и/или другие загрязняющие вещества, как например, лакто-N-триозу II и/или пара-лакто-N-неогексаозу и/или пара-лакто-N-неооктаозу и/или глюкозиллактозу и/или галактозиллактозу, отделяют от такого основного продукта.
Еще одной целью настоящего изобретения является предоставление лакто-N-неотетраозы с чистотой 65% или больше, 70% или больше, 75% или больше, 80% или больше, 85% или больше, 90% или больше и 95% или больше.
Краткое описание графических материалов
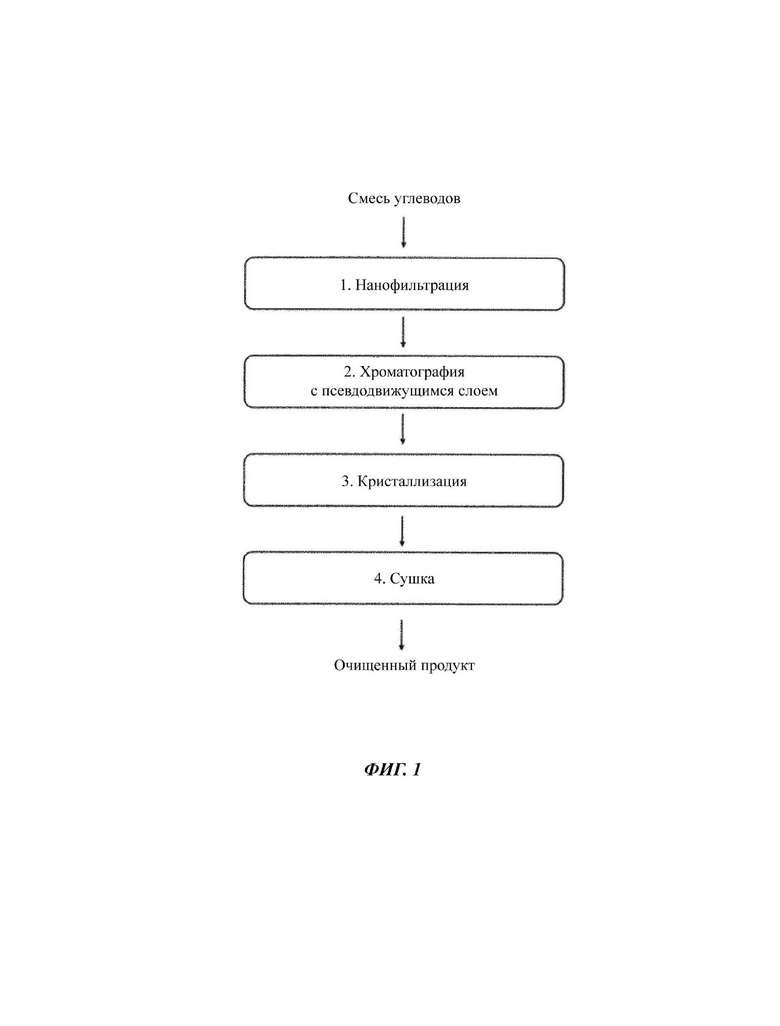
На Фиг. 1 показана общая схема стадий способа очистки согласно настоящему изобретению.
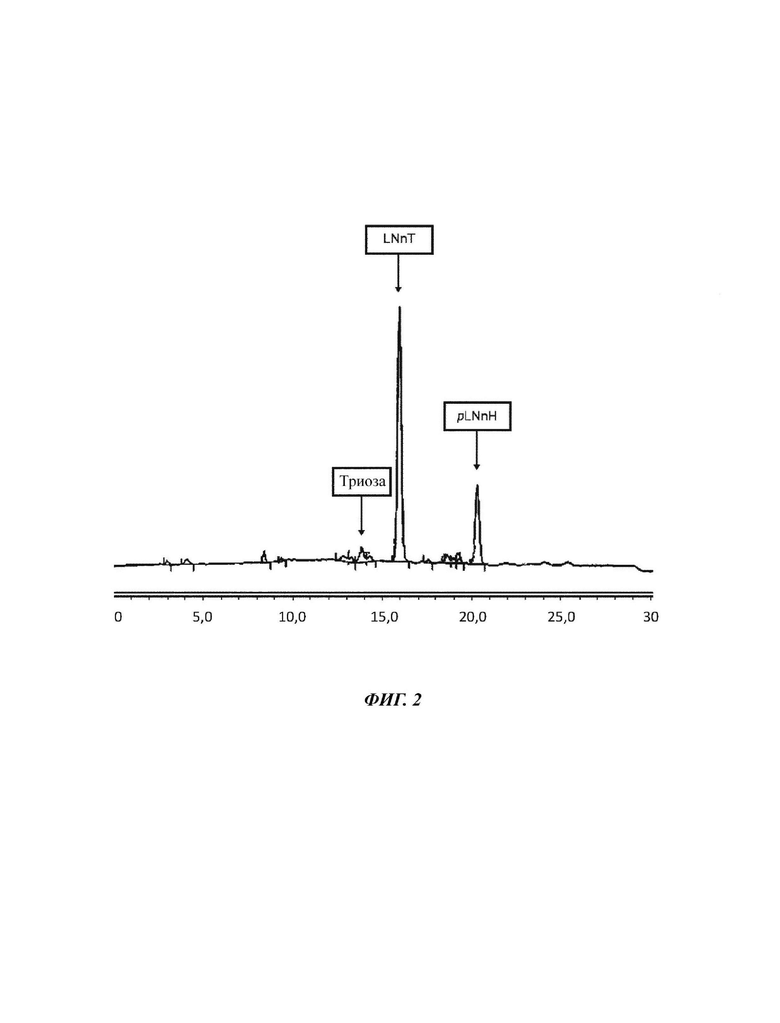
На Фиг. 2 показан анализ ретентата Примера 1 для объема ретентата 80 л.
На Фиг. 3 показан анализ ретентата Примера 1 для объема ретентата 60 л.
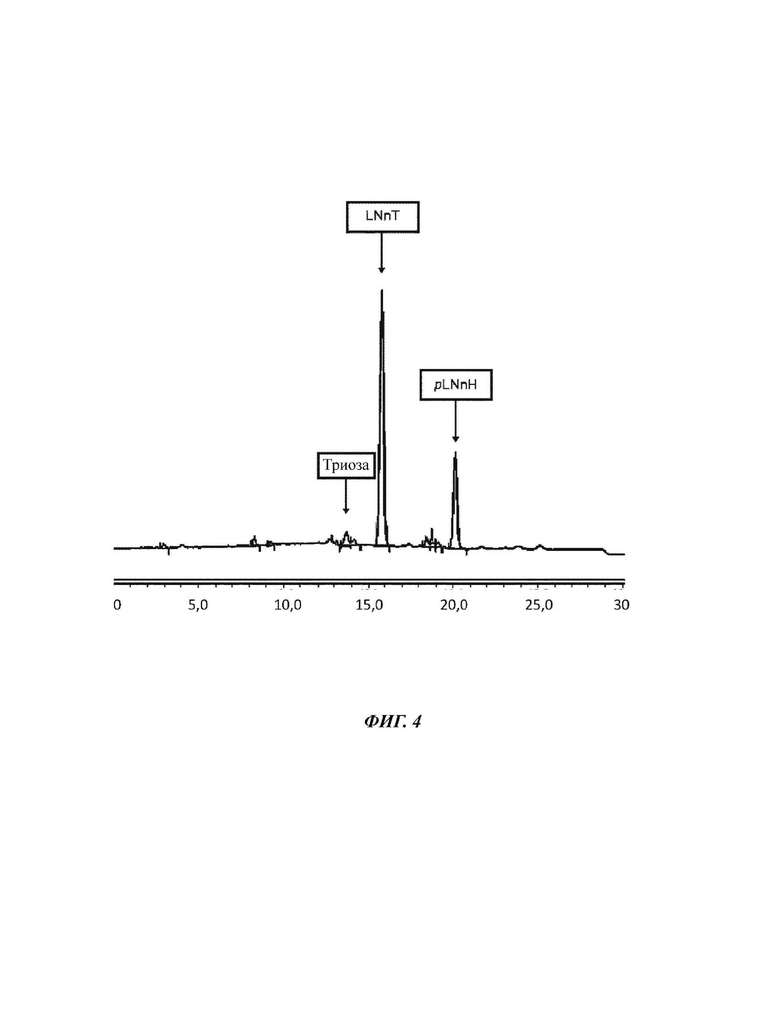
На Фиг. 4 показан анализ ретентата Примера 1 для объема ретентата 40 л.
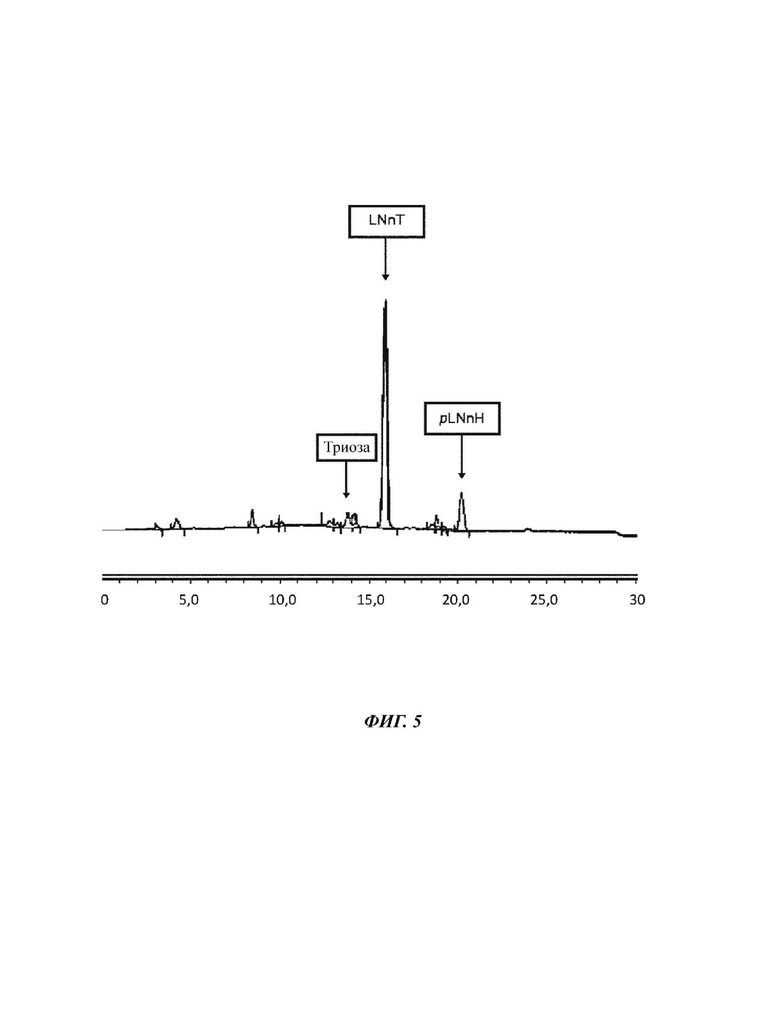
На Фиг. 5 показан анализ пермеата Примера 1 для объема ретентата пермеата 1-20 л.
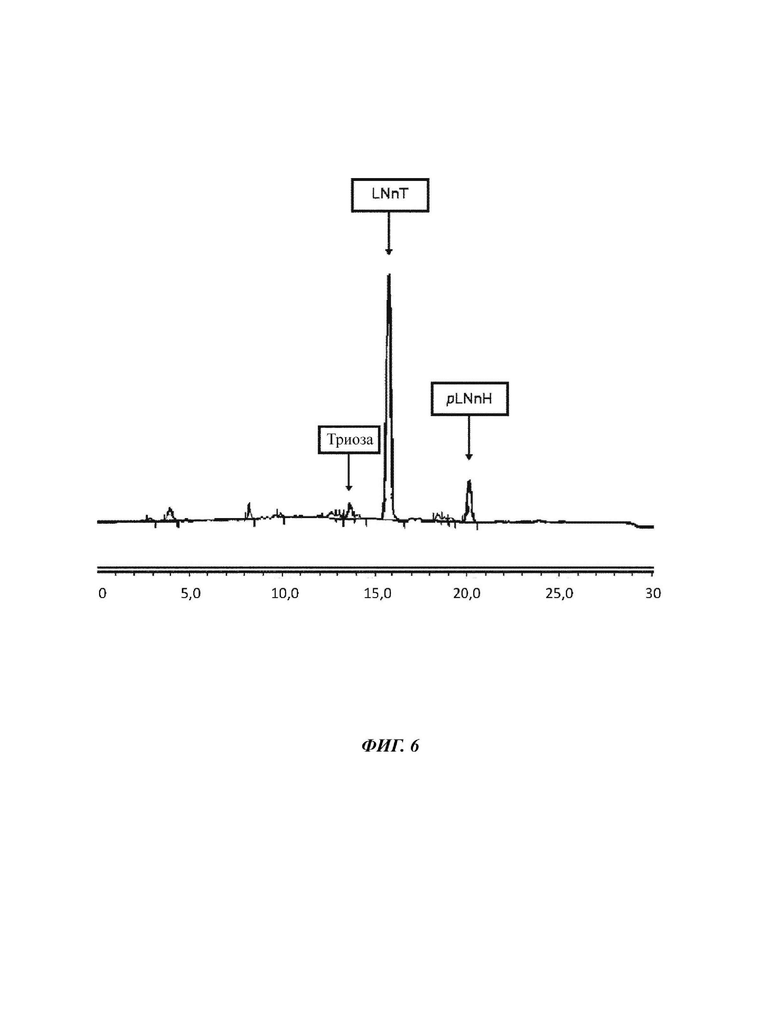
На Фиг. 6 показан анализ пермеата Примера 1 для объема ретентата пермеата 2-20 л.
На Фиг. 7 показан спектр HILIC-CAD (от англ. Hydrophilic interaction liquid chromatography coupled to a charged aerosol detector - жидкостная хроматография гидрофильного взаимодействия, соединенная с детектором заряженного аэрозоля) смеси углеводов, используемой в примере 4.
На Фиг. 8 показан спектр HILIC-CAD (жидкостная хроматография гидрофильного взаимодействия, соединенная с детектором заряженного аэрозоля) смеси углеводов, используемой в примере 4.
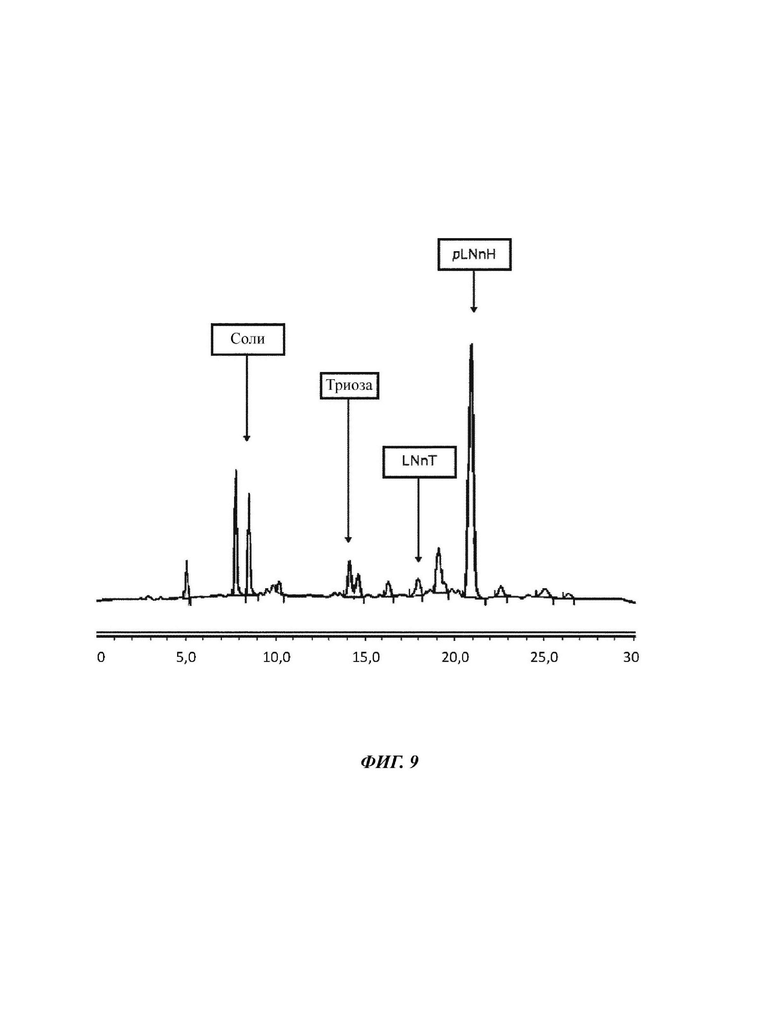
На Фиг. 9 показан спектр HILIC-CAD (жидкостная хроматография гидрофильного взаимодействия, соединенная с детектором заряженного аэрозоля) смеси углеводов, используемой в примере 4.
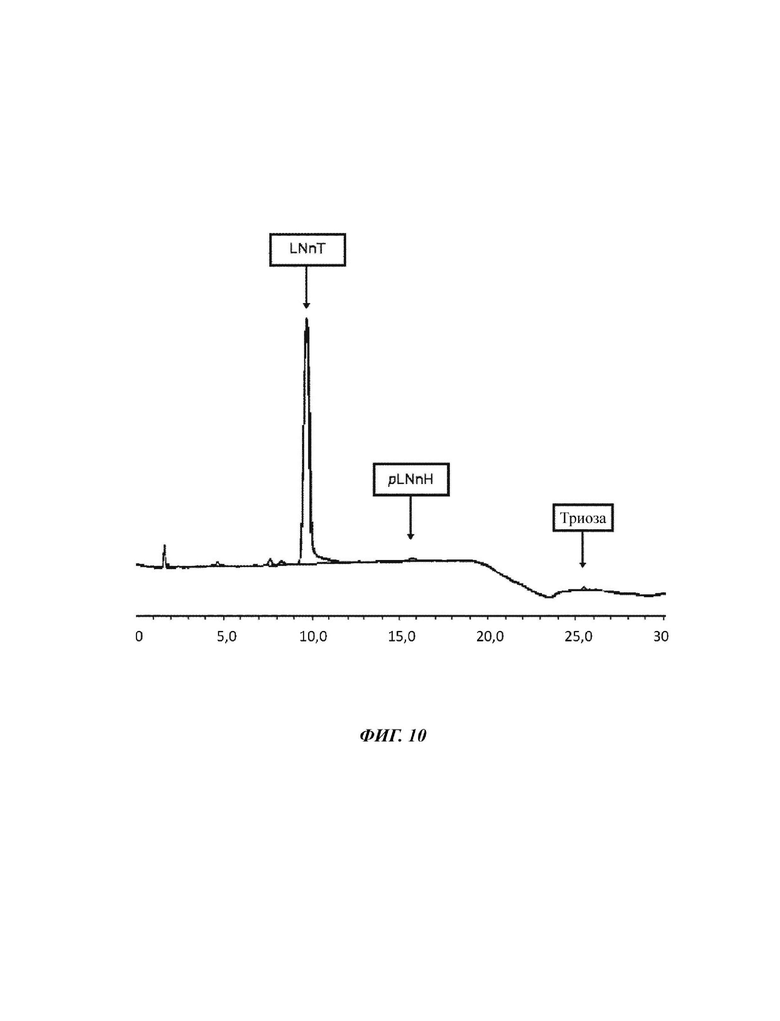
На Фиг. 10 показан спектр HPAEC-PAD (от англ. High performance anion exchange chromatography with pulsed amperometric detection - высокоэффективная анионообменная хроматография с импульсным амперометрическим детектированием) смеси углеводов, полученной после кристаллизации в примере 5.
На Фиг. 11 показан спектр HPAEC-PAD (высокоэффективная анионообменная хроматография с импульсным амперометрическим детектированием) смеси углеводов, полученной после первой кристаллизации в примере 6.
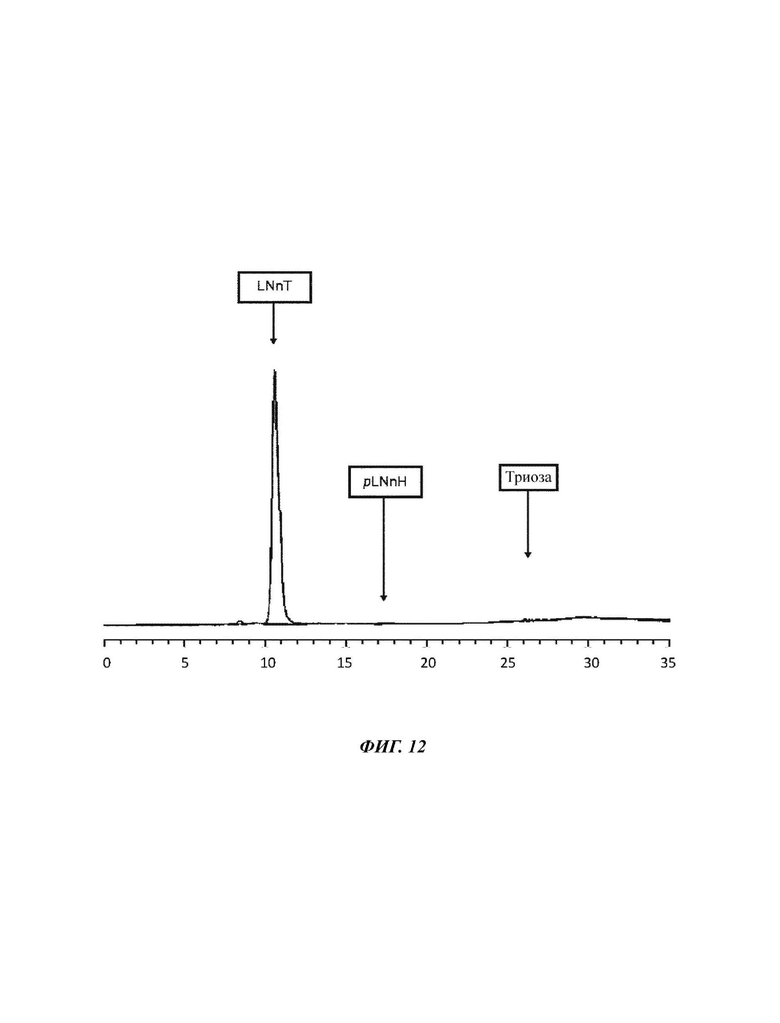
На Фиг. 12 показан спектр HPAEC-PAD (высокоэффективная анионообменная хроматография с импульсным амперометрическим детектированием) смеси углеводов, полученной после второй кристаллизации в примере 6.
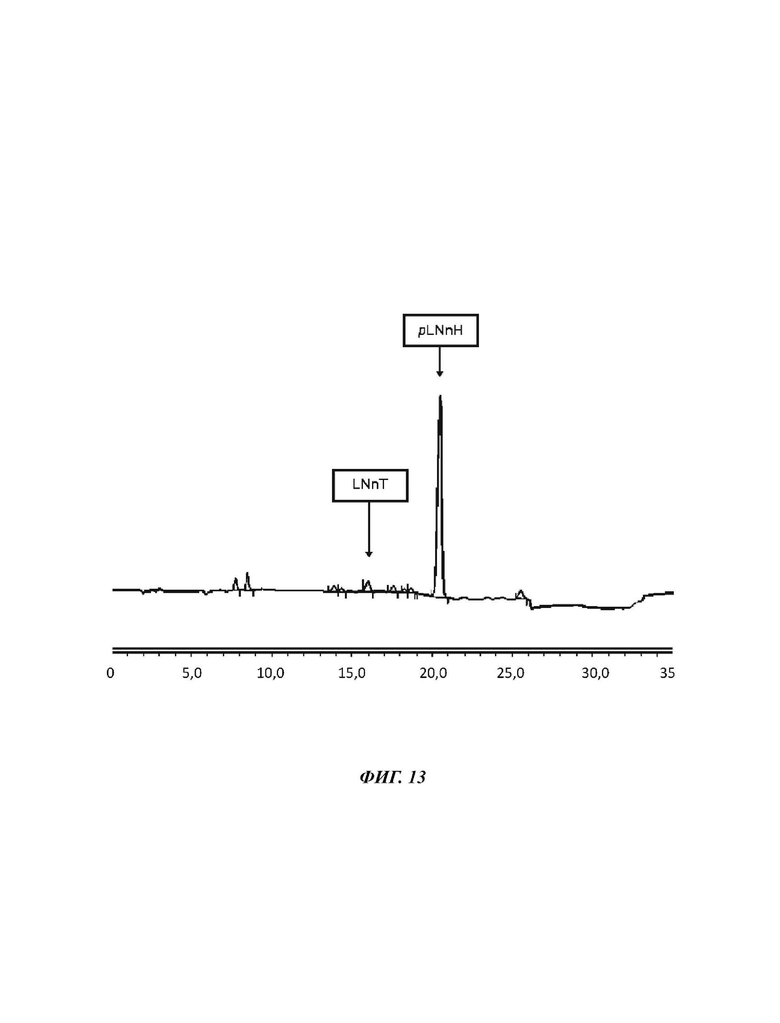
На Фиг. 13 показан спектр HILIC-CAD (жидкостная хроматография гидрофильного взаимодействия, соединенная с детектором заряженного аэрозоля) смеси углеводов, полученной после кристаллизации в примере 7.
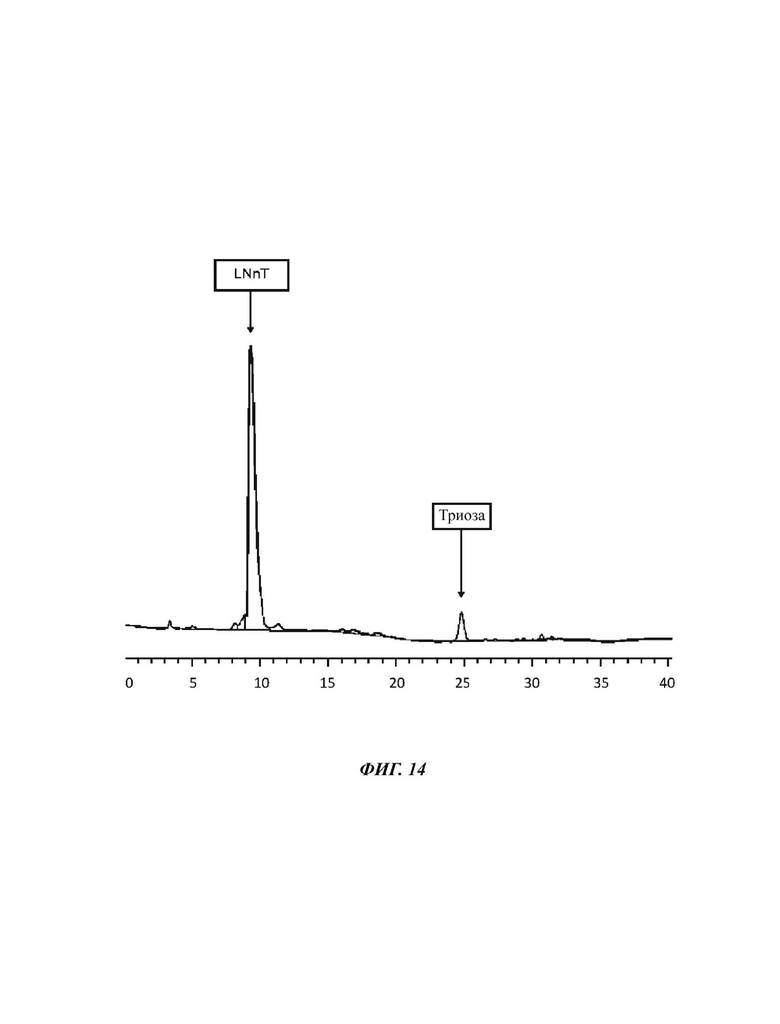
На Фиг. 14 показан спектр HPAEC-PAD (высокоэффективная анионообменная хроматография с импульсным амперометрическим детектированием) затравочных кристаллов, полученных посредством гель-фильтрации в примере 8.
На Фиг. 15 показан спектр HPAEC-PAD (высокоэффективная анионообменная хроматография с импульсным амперометрическим детектированием) исходного вещества LNnT, используемого в примере 9.
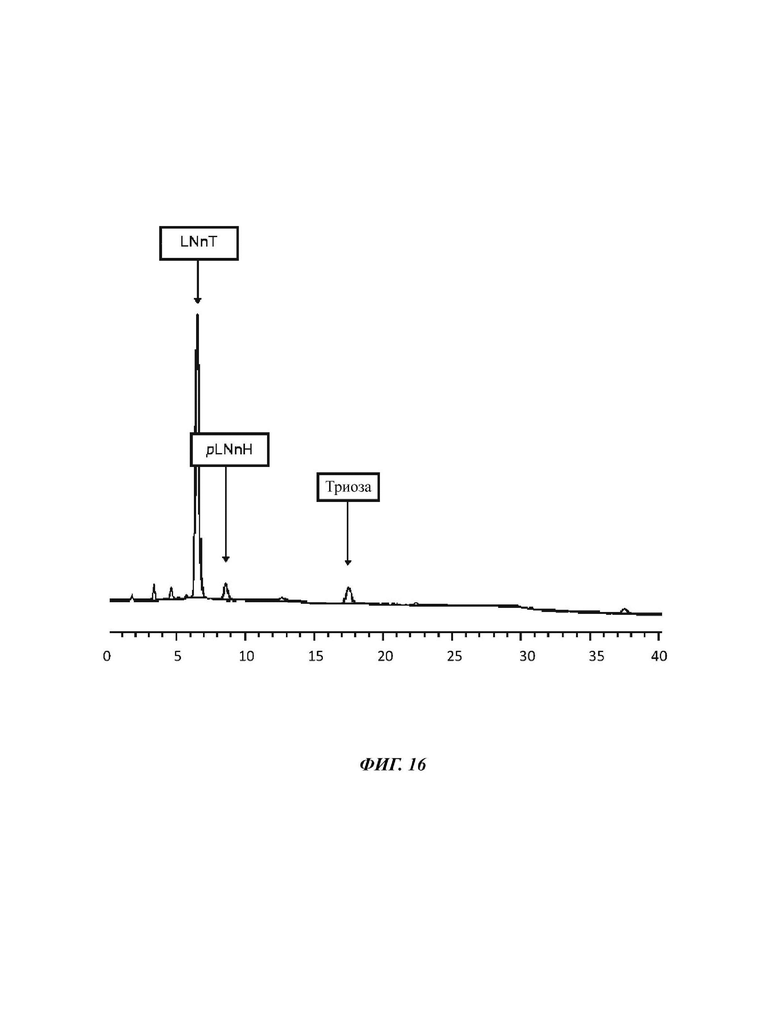
На Фиг. 16 показан спектр HPAEC-PAD (высокоэффективная анионообменная хроматография с импульсным амперометрическим детектированием) продукта LNnT, полученного в примере 9.
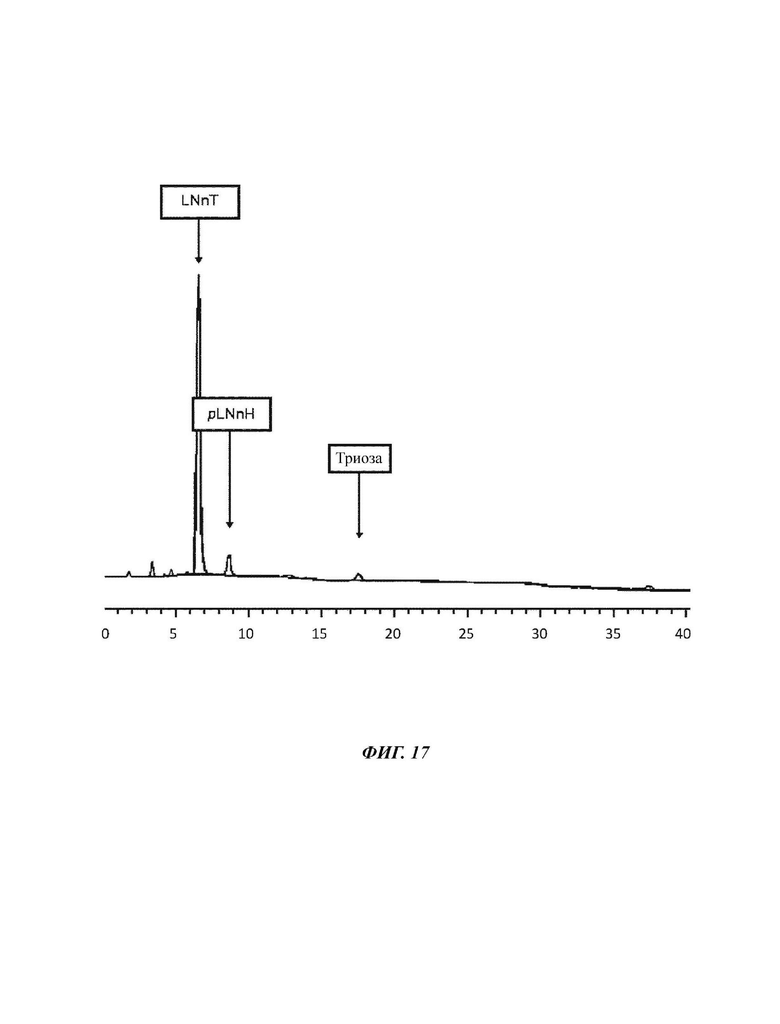
На Фиг. 17 показан спектр HPAEC-PAD (высокоэффективная анионообменная хроматография с импульсным амперометрическим детектированием) продукта LNnT, полученного в примере 10.
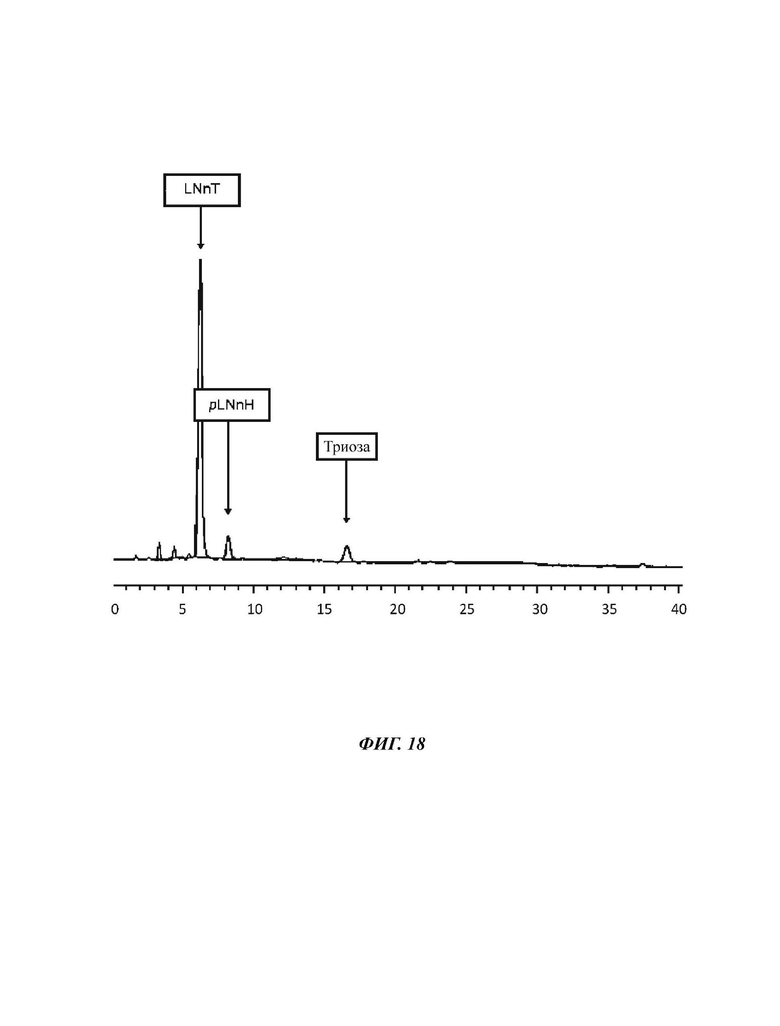
На Фиг. 18 показан спектр HPAEC-PAD (высокоэффективная анионообменная хроматография с импульсным амперометрическим детектированием) продукта LNnT, полученного в примере 11.
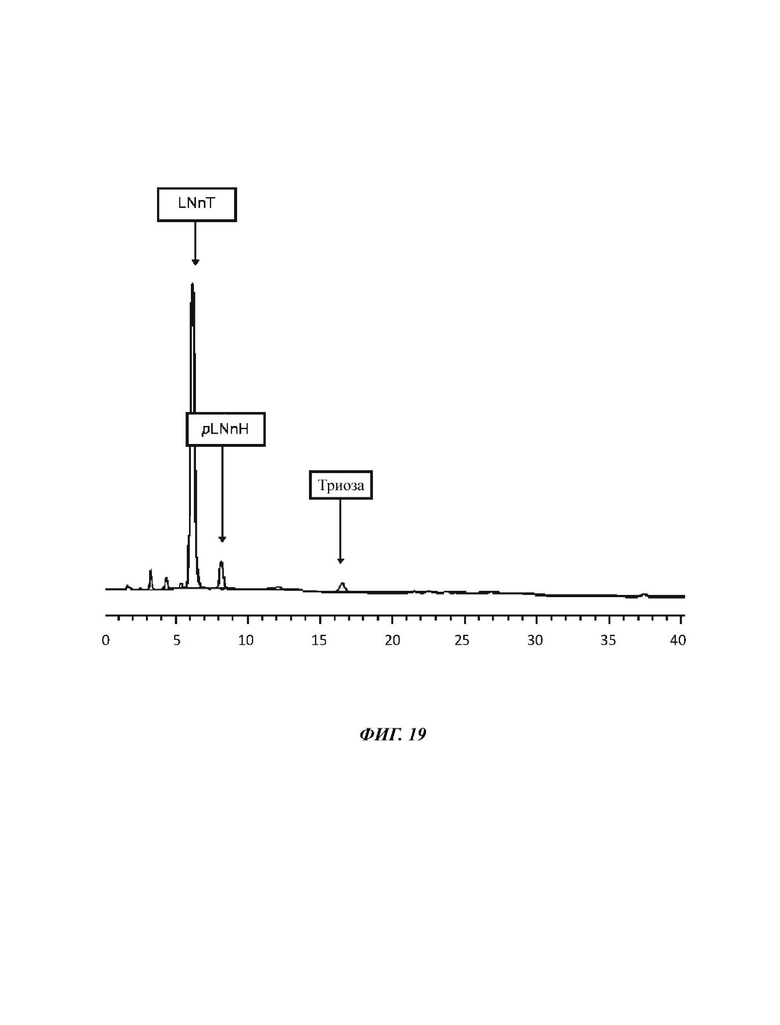
На Фиг. 19 показан спектр HPAEC-PAD (высокоэффективная анионообменная хроматография с импульсным амперометрическим детектированием) продукта LNnT, полученного в примере 12.
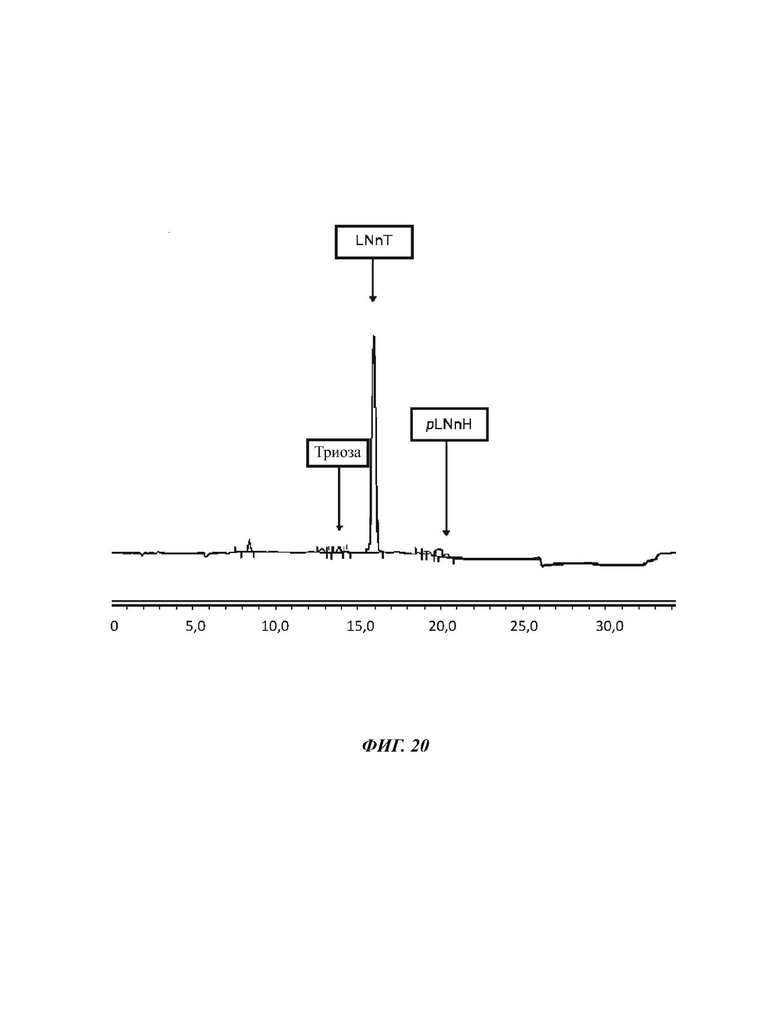
На Фиг. 20 показан спектр HILIC-CAD (жидкостная хроматография гидрофильного взаимодействия, соединенная с детектором заряженного аэрозоля) смеси углеводов, полученной после гомогенизации в примере 13.
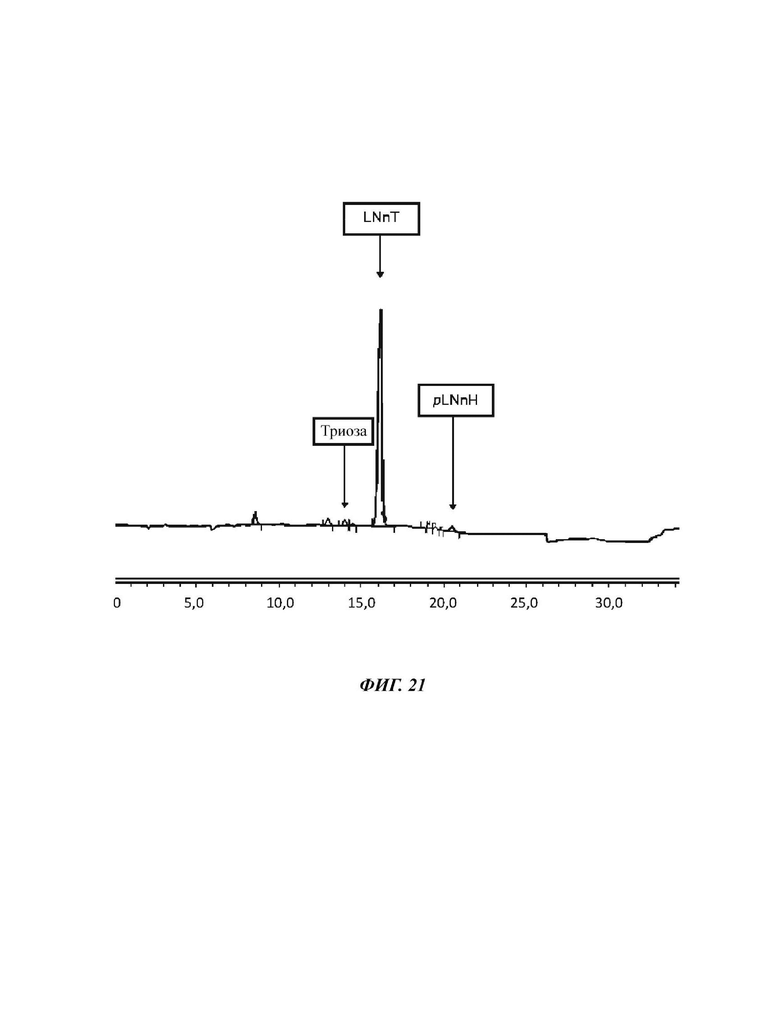
На Фиг. 21 показан спектр HILIC-CAD (жидкостная хроматография гидрофильного взаимодействия, соединенная с детектором заряженного аэрозоля) смеси углеводов, полученной после гомогенизации в примере 14.
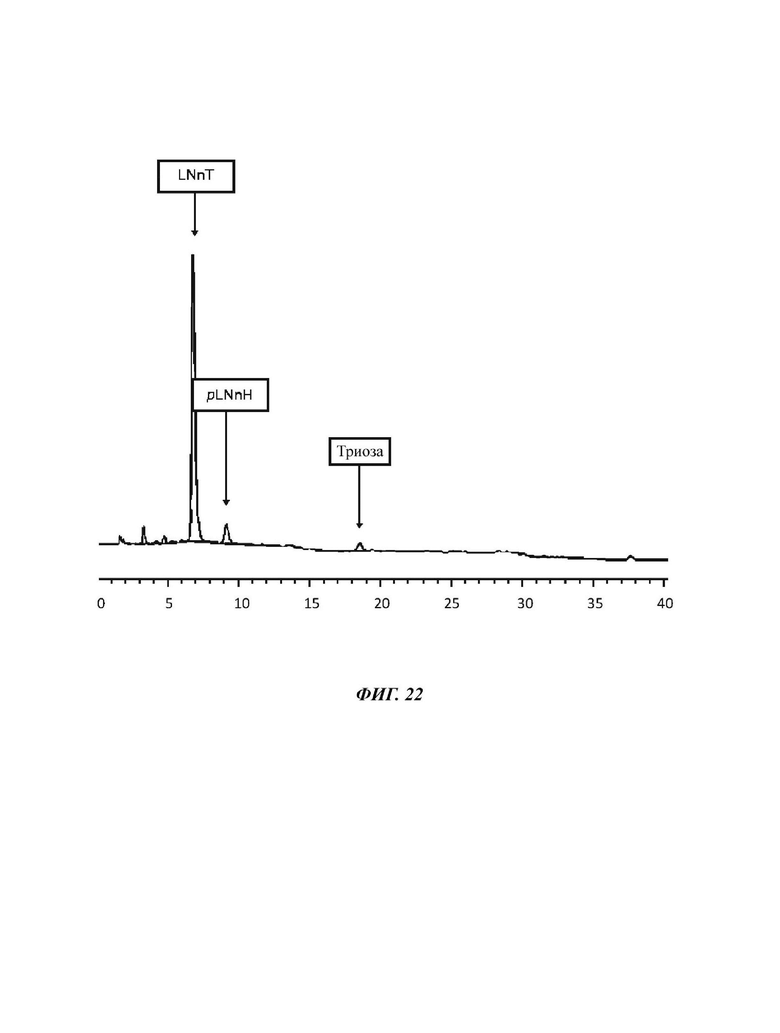
На Фиг. 22 показан спектр HPAEC-PAD (высокоэффективная анионообменная хроматография с импульсным амперометрическим детектированием) продукта LNnT, полученного в примере 15.
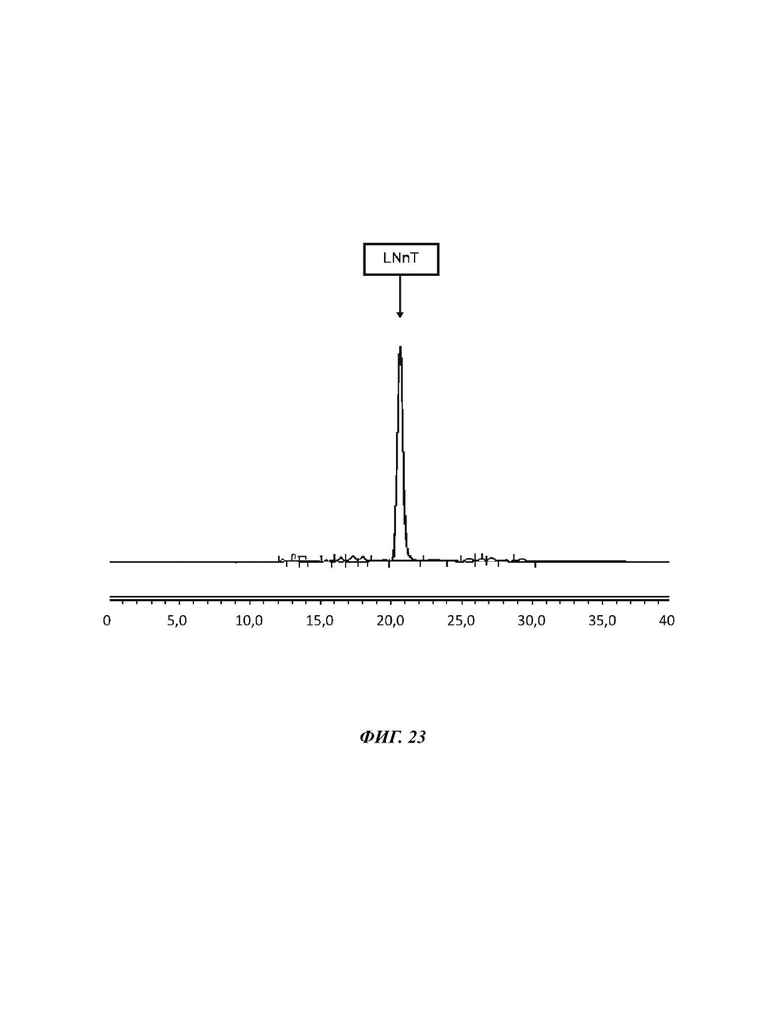
На Фиг. 23 показан спектр HILIC-CAD (жидкостная хроматография гидрофильного взаимодействия, соединенная с детектором заряженного аэрозоля) кристаллического исходного вещества LNnT, используемого в примерах 16-19.
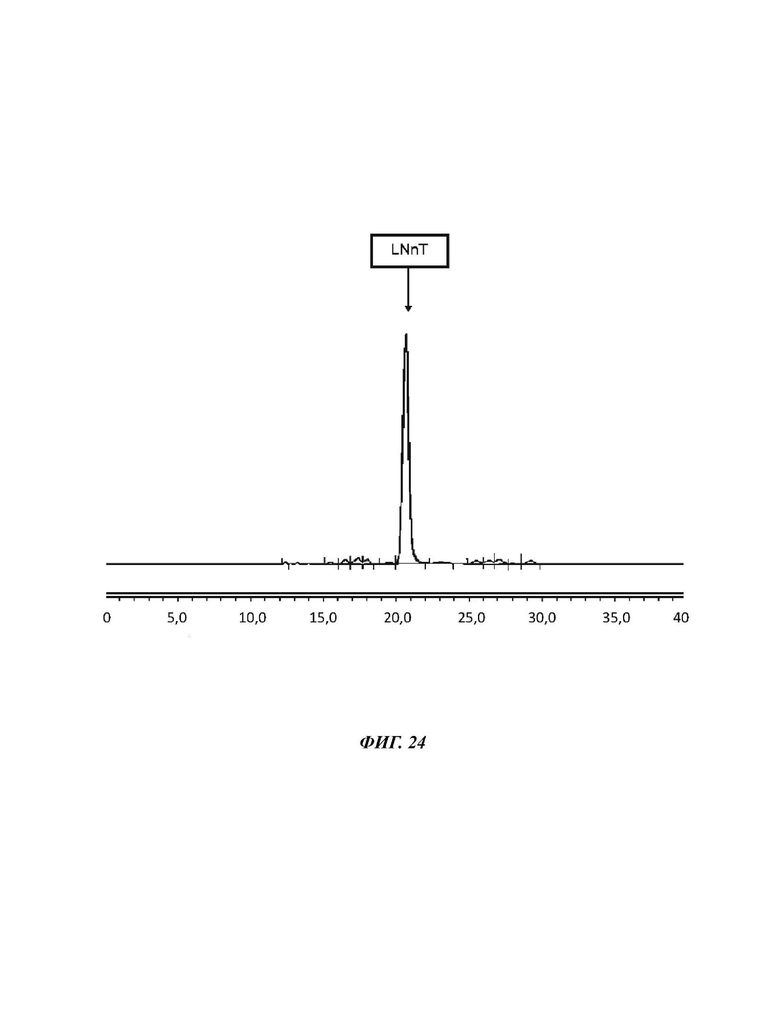
На Фиг. 24 показан спектр HILIC-CAD (жидкостная хроматография гидрофильного взаимодействия, соединенная с детектором заряженного аэрозоля) гомогенизированного продукта LNnT, полученного в примере 17.
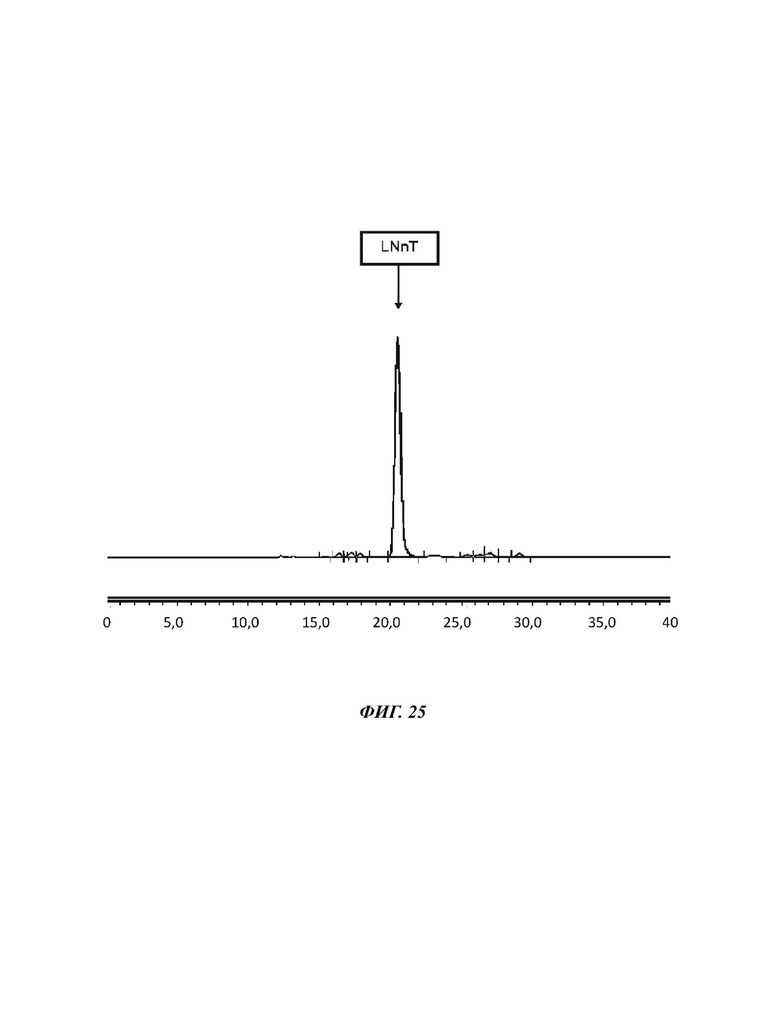
На Фиг. 25 показан спектр HILIC-CAD (жидкостная хроматография гидрофильного взаимодействия, соединенная с детектором заряженного аэрозоля) гомогенизированного продукта LNnT, полученного в примере 18.
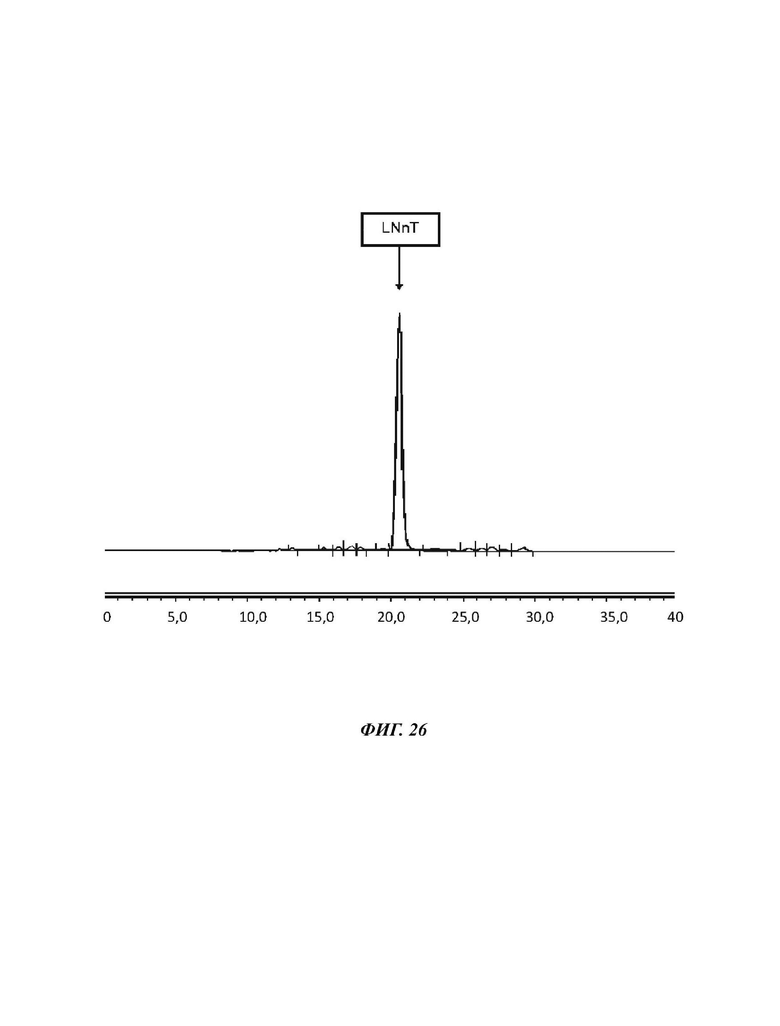
На Фиг. 26 показан спектр HILIC-CAD (жидкостная хроматография гидрофильного взаимодействия, соединенная с детектором заряженного аэрозоля) гомогенизированного продукта LNnT, полученного в примере 19.
На Фиг. 27 показан спектр HILIC-CAD (жидкостная хроматография гидрофильного взаимодействия, соединенная с детектором заряженного аэрозоля) гомогенизированного продукта LNnT, полученного в примере 20.
Подробное описание
Предложен простой, экономически эффективный и масштабируемый способ получения очищенной лакто-N-неотетраозы, полученной в результате процесса микробной ферментации, в качестве главного продукта, тогда как побочные продукты, примеси и/или другие загрязняющие вещества, как например, лакто-N-триозу II и/или глюкозиллактозу и/или галактозиллактозу и/или пара-лакто-N-неогексаозу и/или пара-лакто-N-неооктаозу, отделяют от такого основного продукта.
Согласно настоящему изобретению предложен способ очистки лакто-N-неотетраозы (LNnT), партиями или непрерывно, от ферментационного бульона, полученного способом микробной ферментации, где предложена очищенная LNnT с чистотой более 80%, более 85%, более 90% и/или более 95%. Ферментационный бульон содержит нейтральный ОГМ, биомассу, компоненты среды, загрязняющие вещества и углеводы, отличные от LNnT. Чистота LNnT в ферментационном бульоне составляет 60% или меньше.
В способе по изобретению настоящей заявки используется ферментационный бульон, содержащий LNnT и другие загрязняющие вещества, такие как триозы, гексаоза и/или тетраоза среди прочих, в качестве исходного материала, такой ферментационный бульон получен в результате процесса микробной ферментации. Чистота LNnT в ферментационном бульоне составляет 60% или меньше. Ферментационный бульон подвергают следующим стадиям очистки:
1) по меньшей мере одна стадия мембранной фильтрации раствора, причем раствор представляет собой ферментационный бульон, содержащий смесь углеводов, полученную в результате процесса микробной ферментации, и после использования на последующих стадиях стандартного протокола его подвергают нанофильтрации для значимого снижения содержания высших сахаридов, таких как пентаозы и/или гексаозы, до менее 10%, и чистота LNnT в фильтрованном растворе составляет более 60%, более 65% или более 70%;
2) по меньшей мере одна стадия хроматографии с SMB (от англ. simulated moving bed - псевдодвижущийся слой), установка оставшегося количества гексаозы ниже 5% для того, чтобы сделать подходящей кристаллизацию на следующей стадии, чистота LNnT в полученном очищенном растворе составляет более 75% или более 80%;
3) по меньшей мере одна стадия кристаллизации из воды с получением кристаллической массы, которую обрабатывают и промывают спиртом, смесью спирт/вода или растворителем или смесью растворитель/вода для того, чтобы смыть оставшиеся меньшие сахариды посредством стекания, и с установкой их концентрации ниже 3% с получением чистоты LNnT в полученных кристаллах более 85%, более 90% или более 95%;
4) по меньшей мере одна стадия гомогенизации полученной, высушенной кристаллической массы посредством лиофильной сушки, сушки распылением, сушки в барабанной сушилке/вальцовой сушки, сушки в вакуумном барабане/вальцовой сушки, сушки в ленточной сушилке или вакуумной сушки в ленточной сушилке;
где предложен продукт, содержащий очищенную LNnT с чистотой 90% или больше.
Мембранную фильтрацию в качестве первой стадии способа по настоящему изобретению используют для снижения содержания олигосахаридов с большей молекулярной массой, чем у требуемой LNnT меньшей молекулярной массы. Существует много разных типов мембран, которые могут быть использованы для мембранной фильтрации, но все они отличаются по своей пригодности в отношении углеводов. Наиболее распространенными материалами, используемыми для мембран, являются полимерные материалы и керамические мембранные модули. В то время, как мембраны-полимеры могут быть обработаны в виде полых волокон или в виде блоков за счет присущей им гибкости, керамические мембраны ограничиваются использованием в виде половолоконных блоков. Обе методики могут быть объединены в мембраны из комбинированных материалов или обработаны последовательно, однако в промышленности главным образом сосредоточены на получении гомогенных материалов по причине затрат и/или эффективности.
В случае желательного снижения колисества олигосахаридов, имеющих молекулярную, большую, чем молекулярная масса лакто-N-неотетраозы, используют стадию мембранной фильтрации и более конкретно стадию нанофильтрации. Нанофильтрация представляет собой мембранный процесс разделения под давлением, в основе которого лежит удержание растворенных молекул, ионов металлов и других частиц выше порога отсечения по молекулярной массе (MWCO - от англ. Molecular-weight cutoff). Мембраны, используемые в нанофильтрации, имеют размер пор 2 нм или меньше, который отличает их от более грубых мембран, используемых в других способах мембранной фильтрации, таких как ультрафильтрация и микрофильтрация.
По сравнению с другими стадиями мембранной фильтрации, например, обратным осмосом, при нанофильтрации используются соответственно более грубые мембраны и более низкое рабочее давление. Однако, мембраны, используемые для фильтрации, обычно ограничиваются и/или сильно зависят от своих свойств удерживания и/или устойчивости к температурному воздействию или воздействию химических веществ, таким образом, что применение данного способа существенно ограничивается применением воды и водных смесей. Лакто-N-неотетраоза имеет молекулярную массу 707,6 Да и сахариды большего размера, увеличенные на один или два моносахарида, соответственно, имеют молекулярную массу 869,3 Да (LNnT+Glc/Gal), 910,3 Да (LNnT+GlcNAc) и 1072,4 Да (LNnT+LacNAc). Таким образом, нанофильтрационные мембраны, предлагающие самый маленький возможный MWCO, представляют собой мембраны, используемые в настоящем изобретении. Мембраны, используемые в настоящем изобретении, имеют MWCO от 0,2 до 3,5 кДа, более предпочтительно от 0,2 до 2,0 кДа и даже более предпочтительно от 0,2 до 1,0 кДа.
Для достижения уменьшения количества сахаридов с большей молекулярной массой, чем у LNnT, или обычно сахаридов с большей молекулярной массой, чем у триозы, раствор смеси Сахаров готовят к процессу фильтрации посредством приложения давления к мембране. Для разделения используют три сахара: LNnT, гексаозу и триозу, которая также присутствует в качестве побочного продукта, происходящего из процесса микробной ферментации в смеси ферментационного бульона.
Целью данной первой стадии мембранной фильтрации в первом подходе является истощение гексаозы до тех пор, пока смесь Сахаров соответственно не будет содержать относительно меньше гексаозы для проведения последующих стадий SMB-хроматографии и кристаллизации как можно эффективнее. В качестве альтернативы, данная первая стадия мембранной фильтрации может быть использована во втором подходе для относительного истощения содержания тетраозы и гексаозы, по сравнению с триозой, в конечном итоге, с обогащением LNnT и pLNnH в ретентате для того, чтобы мочь получать очищенную LNnT после последующей стадии SMB-хроматографии и последующей стадии кристаллизации, таким образом, сначала удаляя низшие сахариды и затем удаляя высшие сахариды).
Водный раствор по меньшей мере двух, предпочтительно трех или более чем трех олигосахаридов, включая LNnT, из которых по меньше два отличаются по своей массе и все из которых происходят из процесса бактериальной ферментации и которые уже готовы к процессу очистки вплоть до сахарного компонента, используя микрофильтрацию, ультрафильтрацию, анионный и катионный обмен, обработку активированным углем и его фильтрацию, и/или диафильтрацию и электродиализ (ED - от англ. electrodialysis), подвергают нанофильтрации посредством приложения давления 100-5000 кПа, более предпочтительно 200-3000 кПа, более предпочтительно 300-1000 кПа и более предпочтительно 400-500 кПа. Порог MWCO мембраны составляет от 0,2 до 3,5 кДа, более предпочтительно от 0,2 до 2,0 кДа и более предпочтительно от 0,2 до 1,0 кДа. Предварительно установленная концентрация сахара раствора составляет от 0,01 до 70% содержания твердых веществ (DSC - от англ. dry solid content), более предпочтительно от 0,1 до 60% DSC, более предпочтительно от 1 до 50% DSC и более предпочтительно от 10 до 40% DSC. Коэффициент разведения равен 0,01-1. Соответственно, ретентат должен быть промыт дополнительными количествами воды, в зависимости от коэффициента разведения. DSC элюата, таким образом, изменяется на коэффициент 0,01-1. Со стадией мембранной фильтрации содержание гексаозы уменьшается ниже 20% в ретентате, более предпочтительно ниже 15% и более предпочтительно ниже 10%.
Чистота LNnT в фильтрованном растворе, полученном на стадии мембранной фильтрации, составляет более 60%, более 65% или более 70%.
В качестве дополнительного воплощения отделение триоз от смеси, также содержащей триозы, а также тетраозы, а также гексаозы, где данные три типа сахаридов могут представлять собой основные компоненты раствора Сахаров, делает возможной дальнейшую очистку посредством второй стадии мембранной фильтрации, в данном случае, нанофильтрации, с уменьшением гексаоз посредством выбора мембраны подходящего материала и размера пор и в конечном итоге с получением очищенного раствора LNnT.
В качестве дополнительного воплощения отделение гексаоз от смеси, также содержащей триозы, а также тетраозы, а также гексаозы, где данные три типа сахаридов могут быть основными компонентами раствора Сахаров, делает возможным дальнейшую очистку посредством второй стадии мембранной фильтрации, в данном случае, нанофильтрации, с уменьшением оставшихся триоз посредством выбора мембраны подходящего материала и размера пор и в конечном итоге с получением очищенного раствора LNnT.
Теперь фильтрованный раствор подвергают второй стадии хроматографии с псевдодвижущимся слоем (SMB-хроматографии), которая используется для регулирования содержания фракции, содержащей определенный сахар, такой как фракция гексаозы, в сравнении с фракциями, которые содержат тетраозу, посредством специально адаптированных параметров. Фильтрованный раствор, который представляет собой раствор, полученный с 1 стадии мембранной фильтрации, имеет содержание гексаозы ниже 20%, более предпочтительно ниже 15% и более предпочтительно ниже 10%.
Стадия SMB-хроматографии обеспечивает вторую стадию настоящего изобретения для отделения смеси ОГМ, чей экстракт или рафинат (или экстракт или рафинат низших или экстракт или рафинат высших сахаридов) содержит тетрасахарид лакто-N-неотетраозу в непрерывной хроматографии (или непрерывным образом), от фильтрованного раствора, содержащего нейтральный ОГМ-тетрасахарид лакто-N-неотетраозу и другие загрязняющие вещества - побочные продукты, где фильтрованный раствор содержит смесь лакто-N-неотетраозы и загрязняющих веществ, содержит или состоит из раствора, который получен на стадии 1, мембранной фильтрации, и где чистота раствора LNnT менее 90%.
Фильтрованный раствор применяют на по меньшей мере одной стадии очистки с использованием хроматографии с псевдодвижущимся слоем. Таким образом, должны быть получены два раствора, из которых один содержит требуемую лакто-N-неотетраозу. После применения данной стадии 2 SMB-хроматографии LNnT может главным образом находиться или в экстракте или в рафинате, или в результате отделения низших олигосахаридов, таких как триозы, или в результате отделения высшего сахарида, такого как гексаозы.
Используя указанную вторую стадию хроматографии с псевдодвижущимся слоем, получают LNnT с более высокой чистотой и непрерывным путем. Таким образом, большие количества ОГМ высокого качества могут быть получены очень удобным и экономичным путем. Стадия хроматографии с псевдодвижущимся слоем также является высоко стабильной даже без стадии регенерации материала колонки (например, катионного материала колонки), используемого на данной стадии. В предпочтительном воплощении чистота LNnT в фильтрованном растворе составляет более 60%, более 65% или более 70%. Термин «фильтрованный раствор» относится к раствору, содержащему LNnT перед стадией очистки хроматографии с одним псевдодвижущимся слоем и полученному на стадии 1, мембранной фильтрации, тогда как «очищенный раствор» относится к раствору после стадии хроматографии с псевдодвижущимся слоем.
Указанная меньшей мере одна стадия 2 хроматографии с одним псевдодвижущимся слоем имеет следующее:
i) по меньшей мере 4 колонки, предпочтительно по меньшей мере 8 колонок, более предпочтительно по меньшей мере 12 колонок, где по меньшей мере одна колонка содержит слабую или сильную катионообменную смолу, предпочтительно катионообменную смолу в Н+-форме, Ма+-форме, K+-форме или Са2+-форме; и/или
ii) четыре зоны I, II, III и IV с разными скоростями потока; и/или
iii) элюент, содержащий или состоящий из воды, предпочтительно этанола и воды, более предпочтительно 5-15 об. % этанола и 85-95 об. % воды, наиболее предпочтительно 9-11 об. % этанола и 89-91 об. % воды,
iv) рабочая температура 15°-60°С, предпочтительно 20°-55°С, более предпочтительно 25°-50°С.
Поскольку ОГМ, подлежащий очистке, представляет собой лакто-N-неотетраозу, по меньшей мере на одной стадии хроматографии с псевдодвижущимся слоем имеется следующее:
i) четыре зоны I, II, III и IV с разными скоростями потока, где скорости потока предпочтительно составляют: 22-32 мл/мин в зоне 1, 17-23 мл/мин в зоне II, 18-25 мл/мин в зоне III и/или 14-20 мл/мин в зоне IV; и/или
ii) скорость подачи 0,5-4 мл/мин, предпочтительно 2 мл/мин; и/или
iii) скорость потока элюента 6-12 мл/мин, предпочтительно 8 мл/мин; и/или
iv) время переключения 14-20 мин, предпочтительно 16-18 мин, более предпочтительно 17 мин.
Предпочтительно, по меньшей мере одна из колонок содержит 0,1-5000 кг катионообменной смолы, предпочтительно 0,2-500 кг катионообменной смолы, более предпочтительно 0,5-50 кг катионообменной смолы, наиболее предпочтительно 1,0-20 кг катионообменной смолы.
Важно, возможно пропорциональное увеличение количества катионообменного материала, скорости потока в разных зонах, скорости подачи, скорости потока элюента и/или времени переключения. Пропорциональное увеличение может представлять собой увеличение на коэффициент 2, 3, 4, 5, 6, 7, 8, 9, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 1000 или все возможные поправочные коэффициенты между указанными значениями.
В колонках сильная катионообменная смола может быть использована в качестве неподвижной фазы. Предпочтительно, катионообменная смола представляет собой смолу на основе сульфоновой кислоты, более предпочтительно смолу Purolite®PCR833H (Purolite, Ratingen, Германия), Lewatit MDS 2368 и/или Lewatit MDS 1368. Если в колонках используется катионообменная смола, она может быть регенерирована посредством серной кислоты. Серная кислота может быть использована в элюенте, предпочтительно при концентрации 10 мМ серной кислоты или меньше. (Сильная) катионообменная смола может находиться в Н+-форме или в Са2+-форме.
Рабочие температуры выше 60°С не являются предпочтительными при проведении хроматографии с псевдодвижущимся слоем. Обнаружено, что особенно в присутствии сильной катионообменной смолы (в Н+-форме или Са2+-форме) в качестве неподвижной фазы, применяемые нейтральные олигосахариды были значительно дестабилизированы, а именно деполимеризованы, что было неблагоприятным в отношении конечного выхода LNnT.
В одном предпочтительном воплощении изобретения очищенный раствор может быть применен на по меньшей мере одной дополнительной стадии очистки с использованием хроматографии с псевдодвижущимся слоем, где предложен очищенный раствор, содержащий нейтральный олигосахарид грудного молока с чистотой больше 85%, предпочтительно больше 90%; более предпочтительно больше 93%. В частности, согласно изобретению предложен продукт ОГМ, не содержащий рекомбинантную ДНК, и не содержащий белков штамма-хозяина.
На еще одной стадии хроматографии с псевдодвижущимся слоем имеется следующее:
i) по меньшей мере 4 колонки, предпочтительно по меньшей мере 8 колонок, более предпочтительно по меньшей мере 12 колонок, где по меньшей мере одна колонка содержит слабую или сильную катионообменную смолу, предпочтительно катионообменную смолу в Н+-форме, Na+-форме, K+-форме или Са2+-форме; и/или
ii) четыре зоны I, II, III и IV с разными скоростями потока, и/или
iii) элюент, содержащий или состоящий из воды, предпочтительно этанола и воды, более предпочтительно 5-15 об. % этанола и 85-95 об. % воды, наиболее предпочтительно 9-11 об. % этанола и 89-91 об. % воды, где элюент возможно дополнительно содержит серную кислоту, предпочтительно 10 мМ или меньше серной кислоты; более предпочтительно 2-5 мМ или меньше серной кислоты и/или
iv) рабочая температура 15°-60°С, предпочтительно 20°-55°С, более предпочтительно 25°-50°С.
Если ОГМ, подлежащий очистке, представляет собой лакто-N-неотетраозу, на еще одной стадии хроматографии с псевдодвижущимся слоем имеется следующее:
i) четыре зоны I, II, III и IV с разными скоростями потока, где скорости потока предпочтительно составляют: 22-32 мл/мин в зоне 1, 18-23 мл/мин в зоне II, 19-25 мл/мин в зоне III и/или 15-20 мл/мин в зоне IV; и/или
ii) скорость подачи 1-4 мл/мин, предпочтительно 2 мл/мин; и/или
iii) скорость потока элюента 6-12 мл/мин, предпочтительно 9 мл/мин; и/или
iv) время переключения 16-22 мин, предпочтительно 18-20 мин, более предпочтительно 19 мин.
В частности, по меньшей мере одна из колонок содержит 0,1-5000 кг катионообменной смолы, предпочтительно 0,2-500 кг катионообменной смолы, более предпочтительно 0,5-50 кг катионообменной смолы, наиболее предпочтительно 1,0-20 кг катионообменной смолы.
Важно, возможно пропорциональное увеличение количества катионообменного материала, скорости потока в разных зонах, скорости подачи, скорости потока элюента и/или времени переключения. Пропорциональное увеличение может представлять собой увеличение на коэффициент 2, 3, 4, 5, 6, 7, 8, 9, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 1000 или все возможные поправочные коэффициенты между указанными значениями. После стадии очистки с использованием хроматографии с псевдодвижущимся слоем, рН очищенного раствора может быть подведен до рН 7, предпочтительно посредством добавления основания, более предпочтительно посредством добавления NaOH (например, 0,2 М NaOH).
Чистота LNnT в очищенном растворе, полученном в результате стадии хроматографии с псевдодвижущимся слоем, составляет более 75% или более 80%.
Теперь данный очищенный раствор подвергают третьей стадии кристаллизации, которую используют для получения высоко очищенной LNnT. Очищенный раствор, полученный с 2 стадии SMB-хроматографии, имеет содержание гексаозы ниже 20%, более предпочтительно ниже 15% и более предпочтительно ниже 10%.
Стадия кристаллизации включает:
а) получение смеси по меньшей мере двух, более предпочтительно по меньшей мере трех или более олигосахаридов, из которых один представляет собой трисахарид или сахарид, менее случайная тетраоза, еще один представляет собой лакто-N-неотетраозу и еще один представляет собой лара-лакто-N-неогексаозу или олигосахарид, более случайная тетраоза упомянутых олигосахаридов, после стадий 1 или 2 получают очищенный раствор олигосахаридов, из которых одним является лакто-N-неотетраоза предпочтительно в концентрации по меньшей мере примерно 10% DSC, особенно по меньшей мере примерно 30% DSC, в частности, по меньшей мере примерно 50% DSC и с чистотой LNnT данной смеси по меньшей мере 15%, предпочтительно по меньшей мере примерно 25%, особенно по меньшей мере примерно 40%, в частности, по меньшей мере примерно 60%,
b) кристаллизация смеси указанных олигосахаридов при начальной температуре по меньшей мере 20-30°C, предпочтительно по меньшей мере примерно 30-40°C, предпочтительно по меньшей мере примерно 40-50°C и более предпочтительно по меньшей мере примерно 50-60°C и обеспечение кристаллизации насыщенной массы посредством охлаждения до по меньшей мере 40-50°C, предпочтительно по меньшей мере примерно 30-40°C, предпочтительно по меньшей мере примерно 20-30°C, предпочтительно по меньшей мере примерно 10-20°C и более предпочтительно по меньшей мере примерно 0-10°C для получения гомогенной кристаллической массы,
c) обработка полученной кристаллической массы строго смесью спиртового растворителя, такого как метанол, этанол или изопропанол или гликоль или глицерин или любой другой смешиваемый с водой спирт, или растворителя и воды, где содержание спирта или растворителя данного раствора составляет от 10 до 90 об. %, предпочтительно от 30 до 80 об. % и более предпочтительно от 50 до 70 об. %, с получением смеси раствора, содержащего спирт или растворитель, и кристаллической массы,
d) отфильтровывание фракции, содержащей спирт или растворитель и теперь содержащей сахар, кристаллической массы для снижения содержания низших сахаридов в кристаллической массе, предпочтительно способ промывки можно повторять по меньшей мере два раза для снижения содержания низших сахаридов ниже определенного порога, которое менее 10%, предпочтительно менее 5% и более предпочтительно менее 3%, перед подверганием указанной кристаллической массы четвертой стадии гомогенизации.
Количество спирта или растворителя, используемое для промывки кристаллической массы, не является критичным, но по меньшей мере примерно 40-100 л, предпочтительно примерно 30-70 л спирта или растворителя используют на 10 килограмм кристаллической массы. Предпочтительно, раствор спирта или растворителя затем отфильтровывают для удаления побочных продуктов- олигосахаридов с меньшей молекулярной массой. Раствор спирта или растворителя, после фильтрации и теперь содержащий меньше лакто-N- неотетраозы, можно использовать в еще одном цикле кристаллизации, при необходимости, чтобы получить больше лакто-N-неотетраозы. Однако, не только лакто-N-неотетраоза может быть очищена данным путем, но также трисахариды, происходящие из указанного ферментационного бульона или химического синтеза или биокатализа, а также высшие сахариды, такие как пентаозы, гексаозы, а также октаозы, могут быть очищены посредством вымывания низших олигосахаридов водными смесями, содержащими спирт или растворитель, из их масс кристаллизации.
Чистота LNnT в кристаллах, полученных на стадии кристаллизации, составляет более 80%, более 90% и более 95%.
Кристаллы подвергают четвертой стадии гомогенизации, которую используют для получения высоко гомогенного и очищенного продукта LNnT с содержанием остаточной воды не более 20%.
При гомогенизации кристаллов получают сухую лакто-N-неотетраозу из исходной кристаллической массы. Обычно посредством использования сушки осуществляется процесс массопереноса, состоящий из удаления воды или другого растворителя посредством выпаривания из твердой, полутвердой или жидкой фазы. Данный способ часто используют в качестве конечной стадии получения. Часто задействован источник тепла и агент для удаления пара, образующегося в результате данного процесса. В биологических продуктах, таких корм, зерно, и лекарственных средствах, таких как вакцины, растворитель, подлежащий удалению, почти неизменно представляет собой воду. Высушивание может являться синонимом сушки или считаться крайней формой сушки.
В наиболее общем случае в потоке газа, например, воздуха, используется тепло с помощью конвекции и уносится пар в виде влажности. Другие варианты представляют собой вакуумную сушку, где тепло поставляется с помощью проводимости или излучения (или микроволн), в то время как пар, образованный таким образом, удаляют посредством вакуумной системы. Еще одной опосредованной методикой является сушка в барабанной сушилке, при которой нагретая поверхность используется для обеспечения энергии, и аспираторы втягивают пар за пределами помещения.
В настоящем случае кристаллизованной лакто-N-неотетраозы несколько способов могут быть использованы для обеспечения гомогенизированного высушенного вещества с подходящими характеристиками продукта. Одним из наиболее распространенных используемых способов является сушка в печи или вакуумная сушка в печи. Однако, в случае неочищенной кристаллической массы сушка в печи или вакуумная сушка в печи может только обеспечить гомогенное вещество посредством объединения сушки и смешивания. Высушенная кристаллическая масса, полученная на стадии очистки кристаллизации, должна быть, таким образом, аккуратно распространена по поверхности с получением максимальной поверхности, с которой затем растворитель может испаряться во время процесса. При использовании стадии сушки в печи температура установлена на уровне 20-200°С, предпочтительно 25-100°С или даже более предпочтительно 30-50°С для того, чтобы привести к испарению растворителей. Растворители удаляют посредством потока воздуха. При применении вакуумной сушки в печи температура установлена на уровне 20-200°С, предпочтительно 25-100°С или даже более предпочтительно 30-50°С для того, чтобы привести к испарению растворителей. Растворители удаляют посредством насоса, установленного на вакуум в интервале от 0,001 до 100 кПа, предпочтительно от 0,01 до 10 кПа и более предпочтительно от 0,1 до 1 кПа. Сушку запускают в двух случаях от 1 ч до 7 суток. Перед или после сушки материал перемешивают.
Во втором воплощении конечная гомогенизация не является обязательной, поскольку сушку материала проводят посредством использования сушки в печи или вакуумной сушки в печи после второго цикла кристаллизации, которую запускают после первой кристаллизации и последующего процесса сушки. Как в данном случае, имеет место второй и третий цикл кристаллизации, не нужен процесс промывки, не образуется негомогенного вещества. Обычно, во втором и третьем цикле кристаллизации, кристаллы растут лучше благодаря полученной более высокой чистоте, и, таким образом, указанные кристаллы легче высушить после дополнительных стадий кристаллизации.
В третьем воплощении способ гомогенизации представляет собой лиофильную сушку. В данном способе кристаллы, полученные после сушки массы кристаллизации, растворяют в воде перед подверганием процессу лиофильной сушки. Таким образом, кристаллы LNnT растворяют в воде в концентрации, находящейся в интервале 0,1-70% DCS, предпочтительно 1-50% DSC и более предпочтительно 5-25% DSC перед заморозкой и перед подверганием действию прибора для лиофильной сушки, который выделяет воду и высвобождает пенистый - сухой и гомогенный сахарный продукт типа геля.
В идеале, применение лиофильной сушки, в отличие от большинства других способов сушки, приводит к получению полностью безводного или по существу не содержащего воду продукта, который опосредованно обеспечивает определение содержания воды образца. В частности, с углеводами, такими как ОГМ, из которых одним является используемая LNnT, определение остаточной воды является сложным, поскольку, во-первых, они плохо растворимы в безводных растворителях и, во-вторых, спиртовые функциональные группы (ОН-группы) обладают похожими -такими же спектроскопическими свойствами, как и вода.
В качестве четвертого воплощения способ гомогенизации представляет собой сушку распылением. Распылительная сушилка забирает поток жидкости и разделяет раствор или суспензию на твердую фазу и растворитель, превращающийся в пар. Твердая фаза обычно собирается в барабане или циклонном сепараторе. Входящий поток жидкости распыляют через сопло в горячий поток пара и выпаривают. Сопло обычно используют для создания капелек как можно более маленьких, максимизируя перенос тепла и скорость испарения воды. Распылительные сушилки могут сушить продукт очень быстро, по сравнению с другими способами сушки. Они также превращают раствор или суспензию в высушенный и гомогенизированный порошок за одну стадию.
Сушку распылением предпочтительно используют посредством установки концентрации раствора Сахаров на уровне от 1 до 70% DSC, предпочтительно от 10 до 60% DSC, предпочтительно от 20 до 50% DSC и более предпочтительно от 30 до 40% DSC. Раствор, содержащий сахара, затем пропускают под давлением через сопла распылительной сушилки с температурой на входе от 100 до 160°С, предпочтительно от 110 до 150°С и более предпочтительно от 120 до 140°С. Поток регулируют для поддержания температуры на выходе 50-80°С, предпочтительно 60-70°С, более предпочтительно от 66°С до 67°С. Используя данные установки, может быть получен гомогенный порошок, высушенный распылением.
В качестве пятого воплощения способ гомогенизации для получения лакто-N-неотетраозы представляет собой вальцовую сушку или сушку в барабанной сушилке или вакуумную вальцовую сушку. Вальцовая сушка или сушка в барабанной сушилке представляет собой способ, используемый для высушивания жидкостей из неочищенного негомогенного материала. В процессе сушки исходные ингредиенты сушат при относительно низких температурах над вращающимися валками большой мощности, которые производят листы высушенного и гомогенного продукта. Методики сушки в барабанной сушилке приводят к получению высушенного вещества, которое сразу же восстанавливается и сохраняет большую часть своего исходного вкуса, запаха, цвета и питательной ценности. Некоторые преимущества вальцовой сушки включают способность сушить вязкие негомогенные растворы, которые нельзя легко высушить другими способами, и барабанные сушилки могут быть легко очищены и просты в обращении и в обслуживании.
При использовании вальцовой сушки, температуру валков устанавливают на уровне 20-200°С, предпочтительно 50-150°С и боле предпочтительно 75-125°С для выпаривания растворителей. Растворители удаляют посредством потока воздуха. При использовании вакуумной вальцовой сушки температуру устанавливают на уровне 20-200°С, более конкретно 50-150°С и даже боле конкретно 75-125°С для выпаривания растворителей. Растворители удаляют посредством насоса, установленного на вакуум в интервале 0,01-100 кПА, предпочтительно 0,1-50 кПа и более предпочтительно 1-10 кПа. В обоих случаях валки вращаются при 0,1-100 оборотов/мин, предпочтительно от 1 до 10 оборотов/мин.
В качестве шестого воплощения способ гомогенизации для получения лакто-N-неотетраозы представляет собой сушку в ленточной сушилке или вакуумную сушку в ленточной сушилке. Сушка в ленточной сушилке или вакуумная сушка в ленточной сушилке представляет собой способ, используемый для сушки жидкостей из сырья. В процессе сушки исходные ингредиенты сушат при относительно низких температурах над движущимися лентами высокой производительности, которые производят высушенный гомогенный продукт.
Конечный порошковый продукт имеет чистоту LNnT в очищенном препарате 80% или больше, предпочтительно 85% или больше, более предпочтительно 90% или больше относительно сухого вещества очищенного препарата.
Настоящее изобретение описано относительно конкретных воплощений и со ссылкой на графические материалы, но данное изобретение не ограничивается ими, а только формулой изобретения. Кроме того, термины первый, второй и тому подобное в описании и в формуле изобретения используются для проведения различия между похожими элементами и не обязательно для описания последовательности, во времени, в пространстве, по рангу или любым другим образом. Следует понимать, что термины, используемые таким образом, являются взаимозаменяемыми в соответствующих обстоятельствах, и что воплощения изобретения, описанные в данном документе, способны работать в последовательностях, отличных от описанных или проиллюстрированных в данном документе.
Следует отметить, что термин «содержащий», используемый в формуле изобретения, не следует считать ограничивающимся средствами, перечисленными в дальнейшем; он не исключает других элементов или стадий. Таким образом, его следует считать определяющим наличие заявленных признаков, целых чисел, стадий или компонентов, на которые ссылаются, но он не исключает наличие или добавление одного или более других признаков, целых чисел, стадий или компонентов или их групп. Таким образом, объем выражения «устройство, содержащее средства А и В» не следует ограничивать устройствами, состоящими только из компонентов А и В. Оно означает, что в отношении настоящего изобретения, единственными релевантными компонентами устройства являются А и В.
Ссылка на всем протяжении данного описания изобретения на «одно воплощение» или «воплощение» означает, что конкретный признак, структура или характеристика, описанные в связи с данным воплощением, включены в по меньшей мере одно воплощение настоящего изобретения. Таким образом, появления фраз «в одном воплощении» или «в воплощении» в разных местах по всему объему данного описания изобретения не обязательно все относятся к одному и тому же воплощению, но могут. Кроме того, конкретные признаки, структуры или характеристики могут быть объединены любым подходящим образом, как будет очевидно среднему специалисту в данной области из данного раскрытия, в одном или более воплощениях.
Аналогично, следует понимать, что в описании иллюстративных воплощений изобретения разные признаки изобретения иногда сгруппированы вместе в одном единственном воплощении, фигуре или его описании в целях упрощения раскрытия и помощи в понимании одного или более из разных аспектов изобретения. Данный способ раскрытия, однако, не нужно считать отражающим мысль, что заявленное изобретение требует больше признаков, чем явным образом перечислены в каждом пункте. Скорее, как отражено в приведенной ниже формуле изобретения, аспекты изобретения заключаются менее во всех признаках одного вышеизложенного раскрытого воплощения. Таким образом, формула изобретения после подробного описания явным образом включена тем самым в данное подробное описание, причем каждый пункт отдельно стоит в виде отдельного воплощения данного изобретения.
Кроме того, в то время как некоторые воплощения, описанные в данном документе, включают некоторые, но не все признаки, включенные в другие воплощения, подразумевается, что комбинации признаков разных воплощений находятся в объеме изобретения и образуют разные воплощения, как будет понятно специалистам в данной области. Например, в приведенной ниже формуле изобретения любое из заявленных воплощений можно использовать в любой комбинации.
Кроме того, некоторые из воплощений описаны в данном документе как способ или комбинация элементов способа, которые могут быть реализованы посредством процессора компьютерной системы или с помощью других средств выполнения функции. Таким образом, процессор с необходимыми инструкциями для осуществления такого способа или элемента способа образует средство осуществления способа или элемента способа. Кроме того, описанный в данном документе элемент воплощения аппарата представляет собой пример средства осуществления функции, выполняемой элементом, с целью осуществления изобретения.
В описании и графических материалах, предоставленных в данном документе, изложены многочисленные конкретные подробности. Однако, понятно, что воплощения изобретения можно осуществлять на практике без данных конкретных подробностей. В других примерах хорошо известные способы, структуры и методики не были показаны подробно для того, чтобы не затруднять понимание данного описания.
Изобретение описано с помощью подробного описания нескольких воплощений изобретения. Ясно, что другие воплощения изобретения могут быть скомпонованы в соответствии со знаниями специалистов в данной области, не отклоняясь от истинной сущности или технической идеи изобретения, причем изобретение ограничено только условиями прилагаемой формулы изобретения.
Пример 1: Нанофильтрация LNnT-содержащей смеси углеводов объемом 80 л, содержащей два основных побочных продукта для отделения, с достижением, таким образом, истощения высших сахаридов, таких как pLNnH, и накопления меньших сахаридов, таких как триозы и LNnT.
80 л водного раствора смеси сахаров подвергают нанофильтрации 200 Да для истощения pLNnH. Содержание твердых веществ (DSC) раствора сахаров составляет 5,51%, и приведено соотношение 3-х рассматриваемых сахаров: 4,0% триозы, 62,0% LNnT и 20,8% pLNnH. Это соответствует эквиваленту общего количества сахаров 4,41 кг и количества LNnT 2,73 кг. Используя насос подачи, смесь сахаров подают при давлении примерно 400 кПа в циркуляцию фильтровальной установки. С циркуляционным насосом раствор сахаров подается насосом в круг при давлении 500 кПа вокруг керамической мембраны 200 Да для фильтрации через нее. В отношении пермеата фильтрованный раствор сахаров покидает мембрану при давлении сто кПа. После последующей фильтрации 20 литров каждого ретентат и пермеат анализируют в отношении состава сахаров и концентрации сахаров.
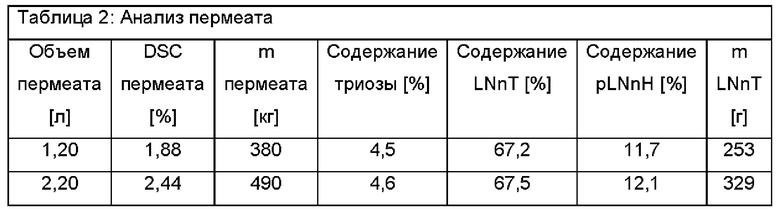
После 20 литров ретентат меняется следующим образом. 60 л ретентата теперь характеризуются 6,85% DSC, состоящими из 3,9% триозы, 61,7% LNnT и 22,3% pLNnH. Это соответствует эквиваленту общего количества Сахаров 4,11 кг и количества LNnT 2,54 кг. Пермеат имеет следующий состав: 1,88% DSC при составе 4,5% триозы, 67,2% LNnT и 11,7% pLNnH. Это соответствует в общей сложности 380 г сахара и количеству LNnT 253 г.
После еще 20 литров ретентат меняется следующим образом. Оставшиеся 40 л ретентата теперь характеризуются сухой массой 9,95% DSC с составом 3,9% триозы, 62,5% LNnT и 24,6% pLNnH. Это соответствует общему количеству Сахаров 3,98 кг и количеству LNnT 2,49 кг. 20 л пермеата имеют следующий состав: 2,44% DSC с составом 4,6% триозы, 67,5% LNnT и 12,1% pLNnH. Эквивалент общего количества сахара 490 г и количества LNnT 329 г.
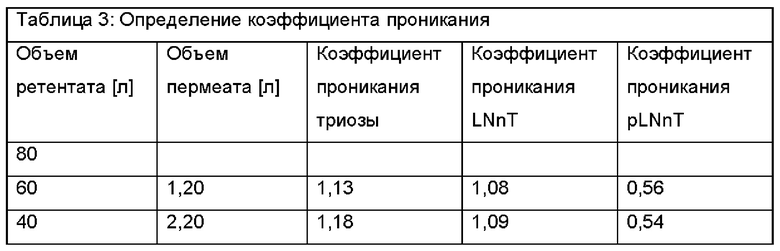
Данные результаты позволяют рассчитать коэффициенты проникания для каждого компонента - сахара. Триоза проникает на коэффициент примерно 1,15, LNnT - на коэффициент 1,09 и pLNnH - на коэффициент 0,55.
Результаты показаны в Таблице 1, Таблице 2 и Таблице 3.

Пример 2: Нанофильтрация LNnT-содержащей смеси углеводов объемом 35 л, содержащей два основных побочных продукта для разделения, и промежуточное добавление еще 10 л воды к подаваемому раствору с достижением истощения высших сахаридов, таких как pLNnH, и накопления меньших сахаридов, таких как триозы и LNnT
35 л водного раствора смеси Сахаров подвергают нанофильтрации 200 Да для истощения pLNnH. Содержание твердых веществ (DSC) раствора сахаров составляет 7,88%, и приведено соотношение 3-х рассматриваемых сахаров: 2,8% триозы, 58,0% LNnT и 26,1% pLNnH. Это соответствует эквиваленту общего количества сахаров 2,76 кг и количества LNnT 1,60 кг. Используя насос подачи, смесь сахаров подают при давлении примерно 400 кПа в систему циркуляции фильтровальной установки. Раствор сахаров подается насосом в круг при давлении 500 кПа вокруг керамической мембраны 200 Да, через которую осуществляют фильтрацию. В отношении пермеата фильтрованный раствор сахаров покидает мембрану при давлении сто кПа. Раствор сахаров закачивается через мембрану частями по 5 л, и оставшийся ретентат и полученный пермеат анализируют в отношении состава сахаров и концентрации сахаров. После первых 10 л к подаче два раза добавляют 5 л свежей воды.
После первых 5 литров ретентат меняется следующим образом. Теперь 30 л ретентата характеризуются 8,34% DSC, состоя из 2,2% триозы, 57,5% LNnT и 26,1% pLNnH. Это соответствует эквиваленту общего количества Сахаров 2,50 кг и количества LNnT 1,44 кг. Пермеат имеет следующий состав: 2,37% DSC при составе 3,5% триозы, 59,6% LNnT и 16,5% pLNnH. Это соответствует в общей сложности 120 г сахара и количеству LNnT 71 г.
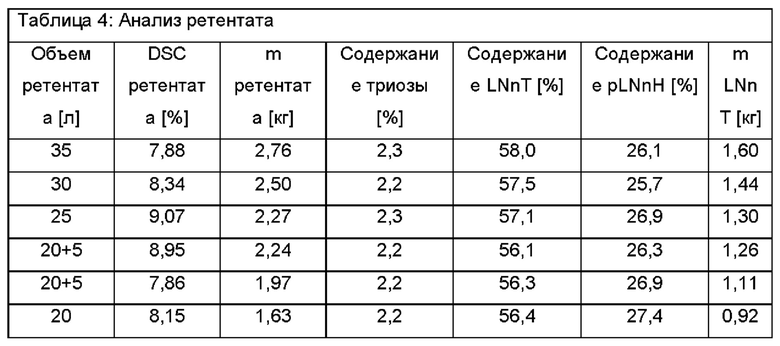
После еще 5 л ретентат изменяется следующим образом. Теперь оставшиеся 25 л ретентата характеризуется сухой массой 9,07% DSC с составом 2,3% триозы, 57,1% LNnT и 26,9% pLNnH. Это соответствует общему количеству Сахаров 2,27 кг в ретентате и количеству LNnT 1,30 кг. Объединенный пермеат 10 л имеет следующий состав: 2,21% DSC с составом 3,5% триозы, 61,1% LNnT и 16,3% pLNnH. Эквивалент общего количества сахара 220 г и количества LNnT 135 г.
Теперь 5 л воды добавляют к подаче перед дополнительной фильтрацией. После еще 5 л ретентат меняется следующим образом. Теперь оставшиеся 25 л ретентата характеризуются сухой масса 8,95% DSC с составом 2,2% триозы, 56,1% LNnT и 26,3% pLNnH. Это соответствует общему количеству сахара 2,24 кг в ретентате и количеству LNnT 1,26 кг. Объединенные 15 л пермеата имеют следующий состав: 2,24% DSC с составом 3,6% триозы, 60,4% LNnT и 16,6% pLNnH. Эквивалент общего количества сахара 340 г и количества LNnT 203 г.
Теперь еще 5 л воды добавляют к подаче перед дополнительной фильтрацией. После еще 5 л ретентат меняется следующим образом. Теперь оставшиеся 25 л ретентата характеризуются сухой массой 7,86% DSC с составом 2,2% триозы, 56,3% LNnT и 26,9% pLNnH. Это соответствует общему количеству сахара 1,97 кг в ретентате и количеству LNnT 1,11 кг. Объединенные 20 л пермеата имеют следующий состав: 1,94% DSC с составом 3,4% триозы, 61,3% LNnT и 16,5% pLNnH. Эквивалент общего количества сахара 390 г и количества LNnT 238 г.
После еще 5 л ретентат меняется следующим образом. Теперь оставшиеся 20 л ретентата характеризуются сухой массой 8,15% DSC с составом 2,2% триозы, 56,4% LNnT и 27,4% pLNnH. Это соответствует общему количеству Сахаров 1,63 кг в ретентате и количеству LNnT 919 г. Объединенные 25 л пермеата имеют следующий состав: 1,94% DSC с составом 3,5% триозы, 61,9% LNnT и 17,5% pLNnH. Эквивалент общего количества сахара 490 г и количества LNnT 300 г.
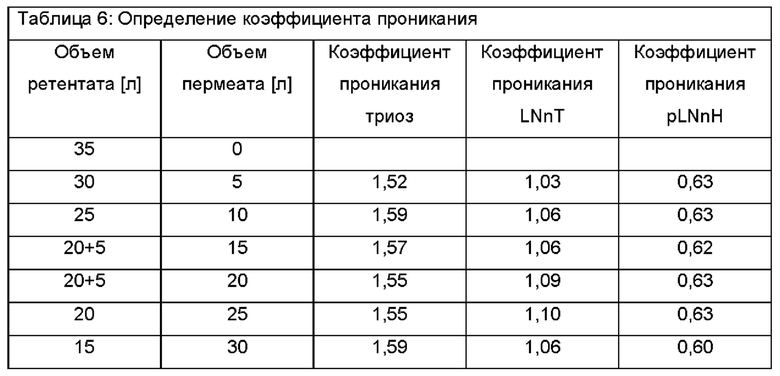
После последних 5 л объединенные 30 л пермеата имеют следующий состав: 2,51% DSC с составом 3,5% триозы, 59,7% LNnT и 16,5% pLNnH. Эквивалент общего количества сахара 750 г и количества LNnT 450 г. Данные результаты приводят к коэффициентам проникания для каждого компонента - сахара. Триоза проникает с коэффициентом примерно 1,55, LNnT - с коэффициентом 1,05 и pLNnH - с коэффициентом 0,62.
Результаты показаны в Таблице 4, Таблице 5 и Таблице 6.


Пример 3: Хроматография с псевдодвижущимся слоем LNnT-содержащей смеси углеводов, которая содержит два основных побочных продукта для отделения, с достижением истощения высших сахаридов, таких как pLNnH, ниже 3% и накопления меньших сахаридов, таких как триозы и LNnT
Для SMB-очистки прозрачный раствор, не содержащий частиц, содержащий указанные 3 олигосахарида, концентрировали до примерно 300 г/л с использованием вакуумного концентратора при 45°С. Для SMB-хроматографии использовали многокомпонентную систему SMB с закрытым контуром, оборудованную 12 колонками (колонки Prosep® с размерами: 40 мм × 740 мм (Latek, Eppelheim, Германия)), расположенными в зонах 2×4. Каждая колонка содержала 760 г сильной катионообменной смолы Purolite® PCR833H+(Purolite, Ratingen, Германия).
Система работала при 25°С со следующими установленными параметрами потока: скорость потока зоны I составляла 30,00 мл/мин, скорость потока зоны II устанавливали на уровне 21,00 мл/мин, скорость потока зоны III устанавливали на уровне 21,48 мл/мин, скорость потока зоны IV устанавливали на уровне 18,44 мл/мин, подачу устанавливали на уровне 3,00 мл/мин, скорость элюента устанавливали на уровне 11,56 мл/мин и время переключения устанавливали на уровне 17,92 мин. В качестве элюента использовали воду с 10% (об./об.) этанолом, пригодным для применения в пищевой промышленности. Сахариды, менее пентаозы, такие как триозы и LNnT, как например, меньшие продукты гидролиза, такие как лактоза, лакто-N-биоза, глюкоза, галактоза и N-ацетилгалактозамин, главным образом фракционировали в экстракт. Высшие олигосахариды, более точно сахариды, большие чем тетраозы, такие как пентаозы, гексаозы, такие как pLNnH, а также большие чем олигосахариды, такие как гептаозы или октаозы, а также остаточные загрязнения солями, происходящие из элюента или в результате загрузки смолы, фракционировали в рафинат. При описанных установках SMB-система могла непрерывно функционировать в течение по меньшей мере 3 месяцев.
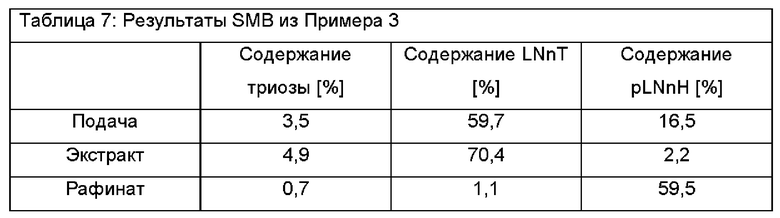
Используя данный протокол, чистота LNnT могла быть значимо увеличена. В данном первом примере использовали LNnT со следующими характеристиками: 12,7% DSC с составом 3,5% триозы, 59,7% LNnT и 16,5% pLNnH. После SMB-хроматографии экстракт имел следующий состав: 4,9% триозы, 70,4% LNnT и 2,2% pLNnT. Рафинат, в свою очередь, имел следующий состав: 0,7% триозы, 1,1% LNnT и 59,5% pLNnT. Выход очистки составлял приблизительно 80%.
Результаты показаны в Таблице 7.
Пример 4: Хроматография с псевдодвижущимся слоем LNnT-содержащей смеси углеводов, которая содержит два основных побочных продукта для отделения, с достижением истощения высших сахаридов, таких как pLNnH, ниже 3% и накопления меньших сахаридов, таких как триозы и LNnT
Для SMB-очистки прозрачный раствор, не содержащий частиц, содержащий указанные 3 олигосахарида, концентрировали до примерно 300 г/л с использованием вакуумного концентратора при 45°С. Для SMB-хроматографии использовали многокомпонентную систему SMB с закрытым контуром, оборудованную 12 колонками (колонки Prosep® с размерами: 40 мм × 740 мм (Latek, Eppelheim, Германия)), расположенными в зонах 2×4. Каждая колонка содержала 760 г сильной катионообменной смолы Purolite® PCR833H+ (Purolite, Ratingen, Германия).
Система работала при 25°С со следующими установленными параметрами потока: скорость потока зоны I составляла 30,00 мл/мин, скорость потока зоны II устанавливали на уровне 21,00 мл/мин, скорость потока зоны III устанавливали на уровне 21,48 мл/мин, скорость потока зоны IV устанавливали на уровне 18,44 мл/мин, подачу устанавливали на уровне 3,00 мл/мин, скорость элюента устанавливали на уровне 11,56 мл/мин и время переключения устанавливали на уровне 17,92 мин. В качестве элюента использовали воду с 10% (об./об.) этанолом, пригодным для применения в пищевой промышленности. Сахариды, менее пентаозы, такие как триозы и LNnT, как например, меньшие продукты гидролиза, такие как лактоза, лакто-N-биоза, глюкоза, галактоза и N-ацетилгалактозамин, главным образом фракционировали в экстракт. Высшие олигосахариды, более точно сахариды, большие, чем тетраоза, такие как пентаозы, гексаозы, такие как pLNnH, а также большие олигосахариды, такие как гептаозы или октаозы, а также остаточные загрязнения солями, происходящие из элюента или в результате загрузки смолы, фракционировали в рафинат. При описанных установках SMB-системы могли непрерывно функционировать в течение по меньшей мере 3 месяцев.
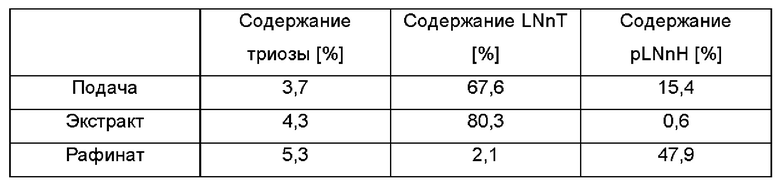
Используя данный протокол, чистота LNnT могла быть значимо увеличена. В данном втором примере использовали LNnT со следующими характеристиками: 3,7% триозы, 67,6% LNnT и 15,4% pLNnH. После SMB-хроматографии экстракт имел следующий состав: 4,3% триозы, 80,3% LNnT и 0,6% pLNnH. Рафинат, в свою очередь, имел следующий состав: 5,3% триозы, 2,1% LNnT и 47,9% pLNnH. Выход очистки составлял приблизительно 80%.
Результаты показаны в Таблице 8.

Пример 5: Кристаллизация LNnT-содержащей смеси углеводов, которая содержит один основной побочный продукт для отделения, с достижением истощения низших Сахаров, таких как триозы, ниже 3%
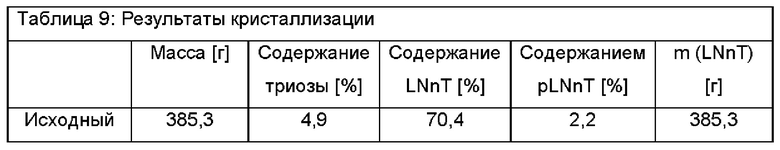
385,3 г смеси углеводов со следующим составом подвергали процессу кристаллизации. Триоза: 4,9%, LNnT: 70,4%, pLNnH: 2,2. Вещество растворяли в карусельной установке в 0,4 л деионизированной воды перед тем, как его DSC было установлено на уровне 70% посредством использования роторного испарителя при пониженном давлении. Указанную смесь углеводов удаляли из роторного испарителя перед ее хранением в условиях окружающей среды, которые обеспечивают медленную скорость охлаждения карусельной установки 10°/ч. Таким образом, слой твердого вещества начинал образовываться на поверхности смеси, приводя в конечном итоге к получению полностью кристаллической массы после 3 суток. После полной кристаллизации указанную кристаллическую массу активно перемешивали с двумя частями 70 об. % раствора этанола (70 об.% EtOH/30 об.% H2O), в конечном итоге удаляя полученный промывочный раствор при пониженном давлении посредством использования воронкообразного фриттового фильтра. Указанный способ промывки повторяли два раза, приводя к получению высушенной и очищенной кристаллической массы LNnT, которую сушили в вакуумной печи при 0,3 кПа и 35°С. Выход кристаллизации определяли как 228,0 г (59,2%); состав соединений определяли следующим образом: содержание триозы: 0,5%, содержание LNnT: 94,4%, содержание pLNnH: 1,2%. Однако, при рассмотрении чистоты LNnT исходного материала, а также кристаллического продукта, выход увеличивается до 79,3% ((94,4%*228 г)/(70,4%*385,3 г)).
Результаты показаны в Таблице 9:

Пример 6: Кристаллизация смеси углеводов, содержащих LNnT, которая содержит один основной побочный продукт для отделения, с достижением истощения низших сахаридов, таких как триозы, ниже 3%
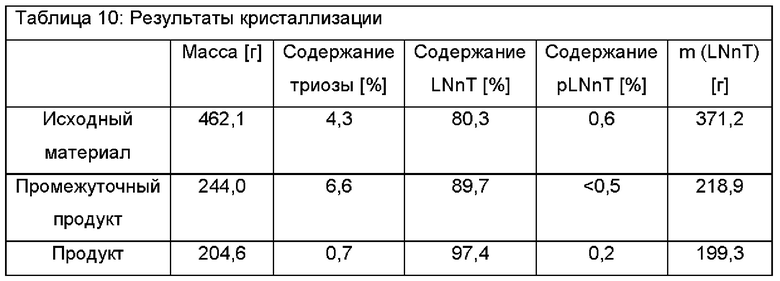
462,3 г смеси углеводов со следующим составом подвергали процессу кристаллизации. Триоза: 4,3%, LNnT: 80,3%, pLNnH: 0,6%. Ее количество LNnT определяли как 371,1 г. Вещество растворяли в карусельной установке в 0,5 л деионизированной воды перед тем, как его DSC было установлено на уровне 69% посредством использования роторного испарителя при пониженном давлении. Указанную смесь углеводов удаляли из роторного испарителя перед ее хранением в условиях окружающей среды, которые обеспечивают медленную скорость охлаждения карусельной установки 107 ч. Таким образом, слой твердого вещества начинал расти на поверхности смеси, приводя в конечном итоге к получению полностью кристаллической массы после 2 суток. После полной кристаллизации указанную кристаллическую массу активно перемешивали с двумя частями 70 об. % раствора этанола (70 об. % EtOH/30 об. % H2O), в конечном итоге удаляя полученный промывочный раствор при пониженном давлении посредством использования воронкообразного фриттового фильтра. Указанный способ промывки повторяли два раза, получая высушенную и очищенную кристаллическую массу LNnT, которую сушили в вакуумной печи при 0,3 кПа и 35°С. Выход кристаллизации определяли как 244,0 г (52,8%); состав соединений определяли следующим образом: содержание триозы: 6,6%, содержание LNnT: 89,7%, содержание pLNnH: меньше 0,5%. В данном случае очистка посредством кристаллизации не была успешной. Однако, указанный продукт использовали в еще одной кристаллизации.
244,0 г смеси углеводов направляли в процесс кристаллизации. Количество LNnT определяли как 218,9 г. Вещество растворяли в карусельной установке в 0,25 л деионизированной воды перед тем, как его DSC было установлено на уровне 70% посредством использования роторного испарителя при пониженном давлении. Указанную смесь углеводов удаляли из роторного испарителя перед ее хранением в условиях окружающей среды, которые обеспечивают медленную скорость охлаждения карусельной установки 107 ч. Таким образом, слой твердого вещества начинал расти на поверхности смеси, приводя в конечном итоге к получению полностью кристаллической массы после 5 суток. После полной кристаллизации указанную кристаллическую массу активно перемешивали с двумя частями 70 об. % раствора этанола, в конечном итоге удаляя полученный промывочный раствор при пониженном давлении посредством использования воронкообразного фриттового фильтра. Указанный способ промывки повторяли два раза, получая высушенную и очищенную кристаллическую массу LNnT, которую сушили в вакуумной печи при 0,3 кПа и 35°С. Выход кристаллизации определяли как 204,6 г (83,9%); состав соединений определяли следующим образом: содержание триозы: 0,7%, содержание LNnT: 97,4%, содержание pLNnH: 0,2%. При рассмотрении чистоты LNnT исходного вещества, а также кристаллического продукта, выход увеличивается до 91,1% ((97,4%*204,6 г)/(89,7%*244,0 г)). Результаты показаны в Таблице 10:
Пример 7: Кристаллизация смеси углеводов, содержащих pLNnH, которая содержит один основной побочный продукт для отделения, с достижением истощения низших сахаридов, таких как LNnT, ниже 4%
В карусельной установке 250 г смеси углеводов со следующим составом подвергали процессу кристаллизации: Триоза: меньше 0,5%, LNnT: 2,1%, pLNnH: 47,9%. Оставшиеся твердые вещества по существу состоят из остаточных солей. Вещество растворяли в карусельной установке в 0,5 л деионизированной воды перед тем, как его DSC было установлено на уровне 55% посредством использования роторного испарителя при пониженном давлении. Указанную смесь углеводов удаляли из роторного испарителя перед ее хранением в условиях окружающей среды, которые обеспечивают медленную скорость охлаждения карусельной установки 107 ч. Таким образом, слой твердого вещества начинал образовываться на поверхности смеси, приводя в конечном итоге к получению полностью кристаллической массы после 5 суток. После полной кристаллизации 50 г неочищенной указанной кристаллической массы активно смешивали с двумя частями 70 об. % раствора этанола (70 об. % EtOH/30 об. % H2O), в конечном итоге удаляя полученный промывочный раствор при пониженном давлении посредством использования воронкообразного фриттового фильтра. Указанный способ промывки повторяли два раза, получая высушенную и очищенную кристаллическую массу pLNnT, которую сушили в вакуумной печи при 0,3 кПа и 35°С. Выход кристаллизации определяли как 13,0 г (26,0%); состав соединений определяли следующим образом: содержание триозы: 0%, содержание LNnT: 3,75%, содержание pLNnH: 80,6%. Однако, при рассмотрении чистоты pLNnT исходного материала, а также кристаллического продукта, выход увеличивается до 58,3% ((80,6%*13,0 г)/(0,75*47,9%*50 г)) с учетом влажности высушенной кристаллической массы 25%.
Пример 8: Получение затравочных кристаллов LNnT из деионизированной воды
56%-ную смесь LNnT, полученную в результате ферментативного процесса, очищали посредством повторных стадий гель-фильтрации с помощью чистой воды в качестве растворителя (Biorad Bio-Gel® Р-2, тонкая очистка), в конечном итоге получая затравочные кристаллы LNnT после уменьшения объема посредством использования роторного испарителя. 10,0 г исходного материала, подверженного распылительной сушке, разводили до 50% DSC чистой водой и три раза фильтровали. На каждой стадии фильтрации преимущественно LNnT-содержащие фракции объединяли, повторно концентрировали в вакууме и еще раз разводили до 50% DSC и посылали на еще один раунд гель-фильтрации. После концентрирования при пониженном давлении получали 2,8 г гелеобразного вещества, которое кристаллизовалось при отстаивании. Продукт анализировали посредством HPAEC-PAD-хроматографии; чистота непромытых кристаллов составляла 83,1% LNnT с 6,3% неотделенной триозы в качестве основного побочного продукта.
Пример 9: Кристаллизация 200 г LNnT посредством добавления NaCl
Для предотвращения образования геля во время кристаллизации LNnT один массовый процент соли с пищевой совместимостью, такой как NaCl, добавляют к раствору. 200 г LNnT (15,0% триозы, 65,9% LNnT, 4,4% pLNnH; это эквивалентно 131,8 г чистой LNnT) и 2,0 г NaCl растворяют в 200 мл воды. Раствор затем устанавливают на значение DSC 70%. Некоторые затравочные кристаллы добавляют к высоковязкой суспензии, и LNnT дают кристаллизоваться при комнатной температуре в течение одних суток. После полной кристаллизации кристаллическую массу смешивают с двумя частями 80 об. % раствора этанола (80 об. % EtOH/20 об. % H2O) и центрифугируют в течение 15 мин при 6000 об./мин. Осажденную смесь углеводов суспендируют с помощью еще одной части 80 об. % раствора этанола и еще раз центрифугируют. Данный процесс повторяют. После трех циклов центрифугирования осадок подвергают лиофильной сушке. После первой кристаллизации получают 163,6 г LNnT. Чистота LNnT могла быть повышена на примерно 11% с 65,9% до 76,5% (6,5% триозы, 5,2 pLNnH) с получением 125,1 г чистой LNnT (выход 94,9%).
Пример 10: 2-ая Кристаллизация 200 г LNnT
158,6 г LNnT, взятые из Примера 9, кристаллизуют.Таким образом, исходный материал растворяют в 159 мл деионизированной воды и концентрируют до 70% DSC при пониженном давлении. Затравочные кристаллы добавляют к высоковязкой суспензии, и LNnT дают кристаллизоваться при комнатной температуре в течение 1 суток. После полной кристаллизации кристаллическую массу смешивают с двумя частями 80 об. % раствора этанола (80 об. % EtOH/20 об. % H2O) и центрифугируют в течение 15 мин при 6000 об./мин. Осажденную смесь углеводов суспендируют с еще одной частью 80 об. % раствора этанола и еще раз центрифугируют.Данный процесс повторяют. После трех циклов центрифугирования осадок повергают лиофильной сушке. 115,2 г LNnT получают после второй кристаллизации. Чистота LNnT могла быть повышена на примерно 6% с 76,5% до 82,4% (2,4% триозы, 5,7% pLNnH) с получением 94,9 г чистой LNnT (выход 78,2%).
Пример 11: Кристаллизация 100 г LNnT посредством добавления NaCl 100 г LNnT (15,0% триозы, 65,9% LNnT, 4,4% pLNnH; эквивалентно 131,8 г чистой LNnT) и 1,0 г NaCl растворяют в 100 мл воды и затем концентрируют при пониженном давлении до 70% DSC. Затравочные кристаллы добавляли к высоковязкой суспензии, и LNnT дают кристаллизоваться при комнатной температуре в течение 1 суток. После полной кристаллизации кристаллическую массу смешивают с двумя частями 80 об. % раствора этанола (80 об. % EtOH/20 об. % H2O) и центрифугируют в течение 15 мин при 6000 об./мин. Осажденный продукт суспендируют с еще одним объемным мл 80% EtOH и еще раз центрифугируют. Данный процесс повторяют. Затем, осадок повергают лиофильной сушке. 67,0 г LNnT получают после первой кристаллизации. Чистота вещества могла быть повышена на примерно 15% с 65,9% до 81,7% (6,2% триозы, 6,0% гексаозы) с получением 54,7 г чистой LNnT (выход 83,1%).
Пример 12: 2-ая Кристаллизация 100 г LNnT
50,0 г LNnT, взятые из Примера 11, кристаллизуют. Таким образом, исходный материал растворяют в 100 мл воды и концентрируют до 70% DSC при 60°С. Затравочные кристаллы добавляют к высоковязкой суспензии, и LNnT дают кристаллизоваться при комнатной температуре в течение 1 суток. После полной кристаллизации кристаллическую массу смешивают с двумя объемами 80 об. % раствора этанола (80 об. % EtOH/20 об. % H2O) и центрифугируют в течение 15 мин при 6000 об./мин. Осажденную смесь углеводов два раза суспендируют с еще одним объемом 80% EtOH и еще раз центрифугируют. Затем данный осадок повергают лиофильной сушке. 36,0 г LNnT получают после второй кристаллизации. Чистота вещества могла быть повышена на примерно 3% с 81,7% до 84,3% (2,9% триозы, 6,4% гексаозы) с получением 30,3 г чистой LNnT (выход 74,3%).
Пример 13: Гомогенизация LNnT посредством использования сушки распылением
228,0 г LNnT со следующим составом гомогенизировали посредством сушки распылением указанного продукта на проборе Buchi В-290: (0,5% триозы, 94,4% LNnT, 1,2% pLNnH). Таким образом, вещество растворяли в 1,5 л (15,2% DSC) воды, затем подвергали стерильной фильтрации (размер пор 45 мм) и подвергали сушке распылением при температуре на входе 130°С и на выходе 66°С. 178,0 г (78,1%) белого гомогенного порошка получали с остаточным содержанием воды 8,6%, определенным посредством титрования по Карлу Фишеру. Чистоту вещества определяли как 0,4% триозы, 94,4% LNnT и 1,1% pLNnH с получением 168,0 г чистой гомогенно высушенной распылением LNnT.
Пример 14. Гомогенизация LNnT посредством использования сушки распылением
204,0 г LNnT со следующим составом гомогенизировали посредством сушки распылением указанного продукта на проборе Buchi В-290: (0,7% триозы, 97,4% LNnT, 0,2% pLNnH). Таким образом, вещество растворяли в 1,2 л (14,6% DSC) воды, затем подвергали стерильной фильтрации (размер пор 45 мм) и подвергали сушке распылением при температуре на входе 130°С и на выходе 66°С. Получали 129,0 г (63,2%) белого гомогенного порошка с остаточным содержанием воды 7,1%, определенным посредством титрования по Карлу Фишеру. Чистоту вещества определяли как 0,3% триозы, 96,43% LNnT и меньше 0,25% pLNnH с получением 124,2 г чистой, гомогенно высушенной распылением LNnT.
Пример 15: Гомогенизация LNnT посредством использования сушки распылением
110,7 г LNnT со следующим составом гомогенизировали посредством сушки распылением указанного продукта на проборе Buchi В-290: (2,4% триозы, 82,4% LNnT, 5,7% pLNnH). Таким образом, вещество растворяли в 300 мл (27% DSC) воды, затем подвергали стерильной фильтрации (размер пор 45 мм) и подвергали сушке распылением при температуре на входе 130°С и температуре на выходе 66°С. Получали 71,9 г (65,0%) белого гомогенного порошка, который демонстрировал приблизительно такой же спектр, как и исходное вещество из Примера 10, как доказательство совместимости сушки распылением полученного вещества.
Пример 16: Сушка LNnT посредством использования сушки в печи
9,629 г кристаллической LNnT сушили в печи. После 7 суток при 35°С ее масса уменьшалась до 9,612 г, что соответствует 99,8%. Чистоту LNnT определяли как 88,7% перед и 88,2% после сушки, доказывая пригодность сушки в печи при 35°С.
Пример 17: Сушка LNnT посредством использования вакуумной сушки в печи
10,307 г кристаллической LNnT сушили в печи. После 7 суток при 35°С ее масса уменьшалась до 10,219 г, что соответствует 99,8%. Чистоту LNnT определяли как 88,7% перед и 90,3% после сушки, доказывая пригодность сушки в печи при 35°С.
Пример 18: Гомогенизация LNnT посредством использования лиофильной сушки
10,265 г кристаллической LNnT подвергали лиофильной сушке на проборе Christ BETA 2-8 LD плюс. Таким образом, указанный ОГМ растворяли в карусельной установке в 90 мл деионизированной воды и замораживали при -80°С перед подверганием процессу лиофильной сушки, устанавливали на уровне 0,2*10-3 кПа и охлаждали до -85°С. После 7 суток масса уменьшилась до 9,668 г, что соответствует 94,2%. Чистоту LNnT определяли как 88,7% перед и 90,2% после сушки, делая лиофильную сушку подходящим способом гомогенизации.
Пример 19: Гомогенизация LNnT посредством использования лиофильной сушки
15,404 г кристаллической LNnT подвергали лиофильной сушке на проборе Christ BETA 2-8 LD плюс. Таким образом, указанный ОГМ растворяли в карусельной установке в 60 мл деионизированной воды и замораживали при -80°С перед подверганием процессу лиофильной сушки, устанавливали на уровне 0,2*10-3 кПа и охлаждали до -85°С. После 7 суток масса уменьшилась до 14,566 г, что соответствует 94,2%. Чистоту LNnT определяли как 88,7% перед и 89,5% после сушки, делая лиофильную сушку подходящим способом гомогенизации.






Изобретение относится к области биотехнологии, а именно к очистке лакто-N-неотетраозы для получения питательных продуктов для грудных детей. Раскрывается способ очистки лакто-N-неотетраозы (LNnT) от ферментационного бульона, который включает стадии, на которых: берут ферментационный бульон, который содержит LNnT, биомассу, компоненты среды, загрязняющие вещества и углеводы, отличные от LNnT; подвергают ферментационный бульон по меньшей мере одной стадии мембранной фильтрации с использованием нанофильтрационных мембран, получая таким образом фильтрованный раствор, содержащий LNnT; подвергают фильтрованный раствор по меньшей мере одной стадии хроматографии с псевдодвижущимся слоем, получая таким образом очищенный раствор, который содержит LNnT; подвергают очищенный раствор по меньшей мере одной стадии кристаллизации из воды, получая таким образом кристаллическую массу; и подвергают такую кристаллическую массу по меньшей мере одной стадии гомогенизации, получая таким образом гомогенизированный и высушенный очищенный препарат LNnT; при этом очищенный препарат содержит LNnT с чистотой 80% или более. Изобретение обеспечивает эффективную очистку лакто-N-неотетраозы (LNnT) от ферментационного бульона, получая таким образом высокоочищенный порошок LNnT. 13 з.п. ф-лы, 27 ил., 10 табл., 19 пр.
1. Способ очистки лакто-N-неотетраозы (LNnT) от ферментационного бульона, который включает стадии, на которых:
- берут ферментационный бульон, который содержит LNnT, биомассу, компоненты среды, загрязняющие вещества и углеводы, отличные от LNnT;
- подвергают ферментационный бульон по меньшей мере одной стадии мембранной фильтрации с использованием нанофильтрационных мембран, получая таким образом фильтрованный раствор, содержащий LNnT;
- подвергают фильтрованный раствор по меньшей мере одной стадии хроматографии с псевдодвижущимся слоем, получая таким образом очищенный раствор, который содержит LNnT;
- подвергают очищенный раствор по меньшей мере одной стадии кристаллизации из воды, получая таким образом кристаллическую массу; и
- подвергают такую кристаллическую массу по меньшей мере одной стадии гомогенизации, получая таким образом гомогенизированный и высушенный очищенный препарат LNnT;
при этом очищенный препарат содержит LNnT с чистотой 80% или более.
2. Способ по п. 1, в котором стадия мембранной фильтрации представляет собой нанофильтрацию с использованием мембраны, которая имеет порог отсечения по молекулярной массе от 0,2 до 3,5 кДа.
3. Способ по любому из пп. 1, 2, в котором мембрана имеет порог отсечения по молекулярной массе от 0,2 до 2,0 кДа.
4. Способ по любому из пп. 1-3, в котором мембрана имеет порог отсечения по молекулярной массе от 0,2 до 1,0 кДа.
5. Способ по любому из пп. 1-4, в котором фильтрованный раствор, полученный на стадии мембранной фильтрации, содержит LNnT с чистотой более 60%, более 65% или более 70%.
6. Способ по п. 1, в котором стадия хроматографии с псевдодвижущимся слоем включает следующее:
i) по меньшей мере 4 колонки, предпочтительно по меньшей мере 8 колонок, более предпочтительно по меньшей мере 12 колонок, где по меньшей мере одна колонка содержит слабую или сильную катионообменную смолу, предпочтительно катионообменную смолу в Н+-форме, Na+-форме, K+-форме или Са2+-форме; и/или
ii) четыре зоны I, II, III и IV с разными скоростями потока; и/или
iii) элюент, содержащий или состоящий из воды, предпочтительно этанола и воды, более предпочтительно от 5 до 15 об. % этанола и от 85 до 95 об. % воды, наиболее предпочтительно от 9 до 11 об. % этанола и от 89 до 91 об. % воды,
iv) рабочую температуру от 15 до 60°С, предпочтительно от 20 до 55°С, более предпочтительно от 25 до 50°С.
7. Способ по п. 1 или 6, в котором стадия хроматографии с псевдодвижущимся слоем включает следующее:
i) четыре зоны I, II, III и IV с разными скоростями потока, где скорости потока предпочтительно составляют от 22 до 32 мл/мин в зоне 1, от 17 до 23 мл/мин в зоне II, от 18 до 25 мл/мин в зоне III и/или от 14 до 20 мл/мин в зоне IV; и/или
ii) скорость подачи от 0,5 до 4 мл/мин, предпочтительно 2 мл/мин; и/или
iii) скорость потока элюента от 6 до 12 мл/мин, предпочтительно 8 мл/мин;
и/или
iv) время переключения от 14 до 20 мин, предпочтительно от 16 до 18 мин, более предпочтительно 17 мин.
8. Способ по любому из пп. 1-7, в котором очищенный раствор, полученный в результате хроматографии с псевдодвижущимся слоем, содержит LNnT с чистотой более 75% или более 80%.
9. Способ по любому из пп. 1, 6-8, в котором очищенный раствор, полученный в результате по меньшей мере одной стадии хроматографии с псевдодвижущимся слоем, может подвергаться по меньшей мере еще одной дополнительной стадии очистки с использованием хроматографии с псевдодвижущимся слоем, при этом получают очищенный раствор, содержащий LNnT с чистотой более 85%, предпочтительно более 90%; более предпочтительно более 93%.
10. Способ по п. 1, в котором стадия кристаллизации включает:
- получение смеси по меньшей мере двух олигосахаридов, содержащей по меньшей LNnT в концентрации по меньшей мере примерно 1% DSC, предпочтительно по меньшей мере 10% DSC, предпочтительно по меньшей мере 30% DSC и более предпочтительно по меньшей мере примерно 50% DSC, и чистота такой LNnT в данной смеси составляет по меньшей мере примерно 15%, предпочтительно по меньшей мере примерно 25%, предпочтительно примерно 40% и более предпочтительно по меньшей мере примерно 60%;
- кристаллизацию смеси по меньшей мере двух олигосахаридов, полученной на стадии хроматографии с псевдодвижущимся слоем, при начальной температуре по меньшей мере примерно от 20 до 30°С, предпочтительно по меньшей мере примерно от 30 до 40°С, предпочтительно по меньшей мере примерно от 40 до 50°С и более предпочтительно по меньшей примерно от 50 до 60°С;
- предоставление возможности кристаллизации насыщенной массы посредством охлаждения до по меньшей мере 40-50°С, предпочтительно по меньшей мере примерно 30-40°С, предпочтительно по меньшей мере примерно 20-30°С, предпочтительно по меньшей мере примерно 10-20°С и более предпочтительно по меньшей мере примерно 0-10°С с получением гомогенной кристаллической массы,
- обработку кристаллической массы смесью спиртового растворителя или смешиваемого с водой спирта или растворителя и воды, где содержание спирта или растворителя в растворе составляет от 10 до 90 об. %, предпочтительно от 30 до 80 об. % и более предпочтительно от 50 до 70 об. % с получением смеси раствора, содержащего спирт или растворитель, и кристаллической массы; и
- отфильтровывание фракции, содержащей спирт или растворитель и теперь содержащей сахар, кристаллической массы для снижения содержания низших сахаридов в кристаллической массе.
11. Способ по п. 1 или 10, в котором содержание низших сахаридов составляет менее 10%, предпочтительно менее 5% и более предпочтительно менее 3%.
12. Способ по любому из пп. 1-11, в котором кристаллы, полученные на стадии кристаллизации, содержат LNnT с чистотой более 85%, более 90% и более 95%.
13. Способ по п. 1, в котором стадия гомогенизации может быть выбрана из лиофильной сушки, сушки распылением, вальцовой сушки или сушки в барабанной сушилке и/или сушки в ленточной сушилке или вакуумной сушки в ленточной сушилке.
14. Способ по любому из пп. 1-13, в котором чистота LNnT в очищенном препарате составляет 85% или больше, более предпочтительно 90% или больше, относительно сухого вещества очищенного препарата.
| WO 2015049331 A1, 09.04.2015 | |||
| EP 3494805 A1, 12.06.2019 | |||
| WO 2014086373 A1, 12.06.2014 | |||
| WO 2019003133 A1, 03.01.2019 | |||
| WO 2011100980 A1, 25.08.2011 | |||
| СПОСОБ ЭФФЕКТИВНОЙ ОЧИСТКИ НЕЙТРАЛЬНЫХ ОЛИГОСАХАРИДОВ ЧЕЛОВЕЧЕСКОГО МОЛОКА (ОЧМ), ПОЛУЧАЕМЫХ ПРИ МИКРОБИОЛОГИЧЕСКОЙ ФЕРМЕНТАЦИИ, КОНЦЕНТРАТ ОЧМ И ЕГО ПРИМЕНЕНИЕ | 2014 |
|
RU2682445C2 |

