Ссылки
Содержания следующих документов включены в данную заявку посредством ссылки:
Патент США № 5 668 127;
Патент США № 6 087 358;
JP 2005330266;
WO 2004/033463;
WO 2007/075872;
WO 2008/140090;
WO 2008/008480;
WO 2009/120789;
WO 2011/151320;
WO 2011/093529;
WO 2011/087995;
WO 2011/014774;
WO 2011/014776;
WO 2013072903.
Область техники, к которой относится изобретение
[1] Настоящее изобретение относится к замещенному производному нитроимидазола, которое применяют главным образом для лечения соответствующих заболеваний, вызываемых микобактериальными инфекциями, такими как Mycobacterium tuberculosis, причем является особенно подходящим для лечения заболеваний, вызываемых устойчивыми Mycobacterium tuberculosis.
Предшествующий уровень техники
[2] Бактерии Mycobacterium tuberculosis являются возбудителем такого заболевания, как туберкулез. Это широко распространенное и смертельное инфекционное заболевание, поскольку согласно статистическим данным Всемирной организации здравоохранения, приблизительно более 800 миллионов человек инфицированы туберкулезом, при этом ежегодно от туберкулеза умирает 200 миллионов человек. За последнее десятилетие количество случаев заболевания туберкулезом возросло во всем мире на 20%, особенно в районах, страдающих от бедности. Если такая тенденция сохранится, то количество случаев заболевания туберкулезом, скорее всего, будет продолжать расти и в течение следующих двух десятилетий увеличится на 41%. Туберкулез всегда был основным инфекционным заболеванием, которое приводит к смерти среди взрослых, перенесших СПИД, в течение пятидесяти лет после первоначального применения химиотерапии. Осложнения туберкулеза приводят к появлению многих штаммов, устойчивых к лекарственным средствам и охваченных симбиотической связью со СПИДом. По сравнению с людьми с отрицательным результатом тестирования на ВИЧ, у людей с положительным результатом тестирования на ВИЧ и вместе с тем больных туберкулезом вероятность развития активной формы туберкулеза в 30 раз выше. В среднем от СПИДа умирают каждые три пациента, при этом смерть одного из них вызвана туберкулезом.
[3] В современном лечении туберкулеза применяют комбинацию ряда фармацевтических составов, рекомендованных Министерством здравоохранения США, что включает сперва применение комбинации из изониазида, рифампицина, пиразинамида и этамбутола в течение двух месяцев, а затем применение комбинации из изониазида и рифампицина отдельно в течение четырех месяцев. Для пациентов, инфицированных СПИД, применение такой комбинации лекарственных средств необходимо продлевать до семи месяцев. В случае пациентов, инфицированных туберкулезом с множественной лекарственной устойчивостью (MDR-TB), эту комбинацию лекарственных средств также необходимо дополнить другими фармацевтическими препаратами второй линии, такими как стрептомицин, канамицин, амикацин, капреомицин, этионамид, циклосерин, ципрофлоксацин и офлоксацин. Такая комбинация терапевтических лекарственных средств для пациентов, инфицированных MDR-TB (обычно при течении болезни свыше 2 лет), как правило, имеет более низкую активность и более тяжкие побочные действия по сравнению с лекарственными средствами первой линии, имеющимися на рынке в настоящее время.
[4] Следовательно, существует острая необходимость в таких новых производных нитроимидазооксазола, которые являлись бы высокоактивными противотуберкулезными лекарственными средствами как в аэробных (активная форма), так и анаэробных (латентная или тяжелая форма) условиях среды. Очевидно, что лекарственные средства, которые сокращают продолжительность лечения и уменьшают частоту обследований, могут привнести наибольшую пользу.
[5] В настоящее время продается препарат Дельтиба (деламанид), новый продукт компании Otsuka, как комбинированное лекарственное средство для лечения туберкулеза с множественной лекарственной устойчивостью, которое одобрено для лечения взрослых пациентов, принимая во внимание факторы устойчивости и толерантности к лекарственному средству. Подобным образом соединения нитроимидазооксазина PA-824 и TBA-354 (J. Org. Chem. 53; 8421-8439 (2010)) демонстрируют лучшую активность in vitro и in vivo при ингибировании Mycobacterium tuberculosis. Механизм действия PA-824 относится к высвобождению газа оксида азота (Singh et al., Science 322; 1392-1395 (2008)) и стадии восстановления включенных бактериальной глюкозо-6-фосфатдегидрогеназы (FGD1) и кофактора (F420) (Stover et al., Nature 405; 962-966 (2000)). В ходе исследований мутагенных штаммов дикого типа, FGD1 и F420, с помощью технологии генных чипов было обнаружено, что ведущую роль в данной серии стадий восстановления играет, судя по всему, неизвестный функциональный белок, состоящий из 151 аминокислоты (17,37 кДа), (Rv3547). Данная гипотеза также подтвердилась при исследовании восстановления FA-824. TBA-354 представляет собой производное нитроимидазооксазина, полученное из PA-824. Механизм действия деламанида заключается в ингибировании синтеза метокси- и кето-миколевой кислот, посредством чего происходит уничтожение бактерий, которые являются важными компонентами клеточной стенки Mycobacterium tuberculosis Производные нитроимидазола и способы лечения, направленные против Mycobacterium tuberculosis, были развернуто освещены ранее (патенты США №№ 5668127 и 6087358; Jiricek et al., WO2007075872A2; Tsubochi et al., WO2005042542A1 и WO2004033463Al; JP 2005330266A; THOMPSON et al., WO2011014776; MUSONDA et al., WO2013072903).
[6] В клинических исследованиях и испытаниях всех лекарственных средств против Mycobacterium tuberculosis наиболее совершенными и наиболее перспективными становятся производные нитроимидазола. Из вышеназванных патентных заявок можно видеть, что общие формулы основных соединений для лечения туберкулеза, в частности, для лечения туберкулеза с множественной лекарственной устойчивостью являются следующими (1 и 2):
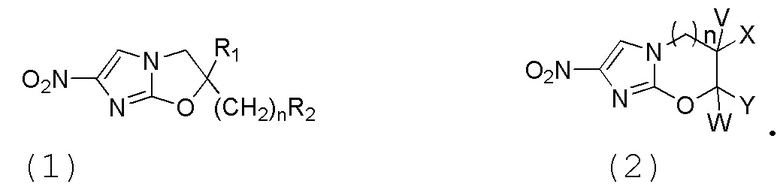 .
.
[7] Согласно исследованию были созданы две новые активные молекулы, OPC-67683 (деламанид) и TBA-354, для лечения туберкулеза, структуры которых являются следующими (3 и 4):
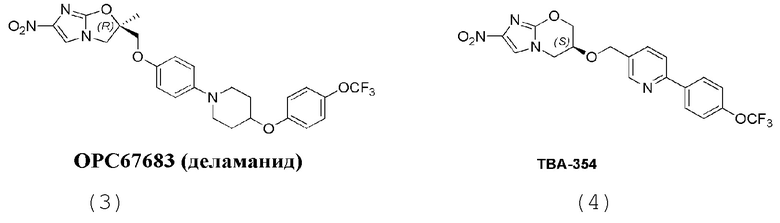
[8] Деламанид представляет собой производное нитро-2H-имидазооксазола, которое действует главным образом путем ингибирования биосинтеза миколевой кислоты и проявляет высокую активность против Mycobacterium tuberculosis как in vitro, так и in vivo. На 21 ноября 2013 г. дельтиба (деламанид) был условно одобрен Комитетом по использованию лекарственных препаратов для человека (Committee for Medicinal Products for Human Use, CHMP), и для лечения от Mycobacterium tuberculosis применяли 50 мг таблетки с энтеросолюбильным покрытием. В Европе он был официально выпущен 28 апреля 2014 г. 1 февраля 2008 г дельтиба признан лекарственным препаратом для лечения редких болезней. Настоящее изобретение направлено на разработку нового соединения нитроимидазола для лечения туберкулеза и туберкулеза с множественной лекарственной устойчивостью.
[9] Доказано, что хотя OPC-67683 и обладает клиническим эффектом при лечении туберкулеза с множественной лекарственной устойчивостью, он имеет дополнительно оптимизированные пространство в ходе проведения лечения и показатель эффективности лечения, при том что в этом тоже есть потребность. Доказано, что производные нитроимидазола по настоящему изобретению имеют лучшую водорастворимость и фармакокинетические свойства. Предполагается, что данное улучшение приведет к лучшему клиническому эффекту.
Описание настоящего изобретения
[10] В настоящем изобретении предлагается соединение, характеризующееся структурой формулы (I), его фармацевтически приемлемая соль или его оптический изомер,
 ,
,
[11] где
[12] кольцо A представляет собой 5-6-членный арил или гетероарил;
[13] X представляет собой N, C(R) или C;
[14] R представляет собой H, галоген, OH, CN, NO2 или выбран из группы, состоящей из амино, C1-6алкиламино, N,N-ди(C1-6алкил)амино, C1-6алкила, C1-6гетероалкила, C2-6алкенила, C2-6алкинила, C3-7циклоалкила, C3-7гетероциклоалкила, 5-7-членного арила и 5-7-членного гетероарила, каждый из которых необязательно замещен любым заместителем;
[15] каждый из V и W независимо выбран из группы, состоящей из метилена, -CH2CH2-, C(=O), -S(=O)- и -S(=O)2-, где метилен и -CH2CH2- необязательно замещены 1 или 2 R;
[16] Z представляет собой метилен, который необязательно замещен 1 или 2 R;
[17] L представляет собой одинарную связь, -O-, -S-, N(R), C(R)(R), -C(=O)-, -C(=S)-, -S(=O)- или -S(=O)2-;
[18] каждый из R1 и R2 независимо выбран из H, галогена, OH, CN, NO2 или каждый из R1 и R2 независимо выбран из группы, состоящей из амино, C1-6алкила, C1-6гетероалкила, C2-6алкенила, C2-6алкинила, C3-7циклоалкила, C3-7циклоалкил-C1-6алкила, C3-7гетероциклоалкила и 5-7-членного арил или гетероарил, каждый из которых необязательно замещен любым заместителем;
[19] необязательно заместитель R при Z и заместитель R при V присоединены к одному и тому же атому или атомной группе с образованием 5-7-членного кольца;
[20] необязательно фрагмент  может быть заменен
может быть заменен  ;
;
[21] также R2 может отсутствовать;
[22] m равняется 1, 2 или 3;
[23] n равняется 0, 1, 2 или 3;
[24] «гетеро» представляет собой гетероатом или гетероатомную группу, которые выбраны из группы, состоящей из -C(=O)NH-, -NH-, -C(=NH)-, -S(=O)2NH-, -S(=O)NH-, -O-, -S-, N, =O, =S, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- и -NHC(=O)NH-;
[25] количество гетероатомов или гетероатомных групп равняется независимо 0, 1, 2 или 3.
[26] В некоторых вариантах осуществления настоящего изобретения каждый из вышеупомянутых заместителей и R независимо выбран из группы, состоящей из H, F, Cl, Br, I, OH, CN, NH2, C1-4алкила и C1-4гетероалкила, где C1-4алкил или C1-4гетероалкил необязательно может быть дополнительно замещен 0-3 заместителями, которые выбраны из галогена, OH и/или NH2.
[27] В некоторых вариантах осуществления настоящего изобретения каждый из вышеупомянутых заместителей выбран из группы, состоящей из F, Cl, Br, I, CN, -CF3, -OCF3, -CH2CF3, OCH3 и (CH3)3COC(=O)-.
[28] В некоторых вариантах осуществления настоящего изобретения каждый из вышеупомянутых R1 и R2 независимо выбран из группы, состоящей из H, галогена, CN, или выбран из группы, состоящей из

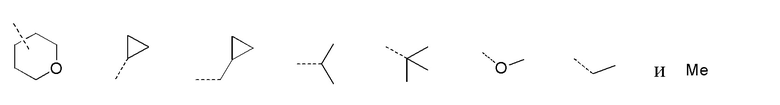 , каждый из которых является необязательно замещенным.
, каждый из которых является необязательно замещенным.
[29] В некоторых вариантах осуществления настоящего изобретения каждый из вышеупомянутых R1 и R2 независимо выбран из группы, состоящей из H, галогена, CN, или выбран из группы, состоящей из

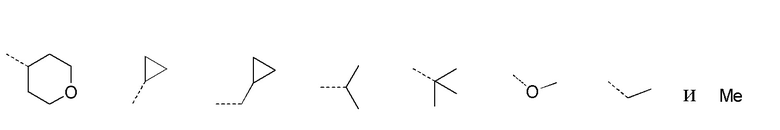 ,
,
каждый из которых является необязательно замещенным.
[30] В некоторых вариантах осуществления настоящего изобретения каждый из вышеупомянутых R1 и R2 независимо выбран из группы, состоящей из
 .
.
[31] В некоторых вариантах осуществления настоящего изобретения вышеупомянутый R выбран из группы, состоящей из H, Cl, Br, I, OH, CN, NH2, Me и Et.
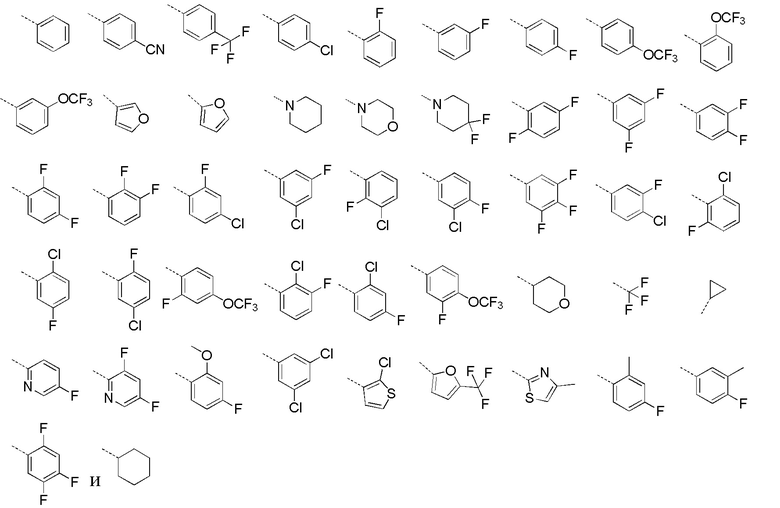
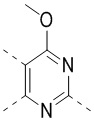
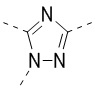
[32] В некоторых вариантах осуществления настоящего изобретения вышеупомянутое кольцо A выбрано из группы, состоящей из пиридила, тиазолила, оксазолила, имидазолила и пиримидинила.
[33] В некоторых вариантах осуществления настоящего изобретения вышеупомянутое кольцо A выбрано из группы, состоящей из
 ,
,  ,
, 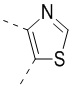 ,
,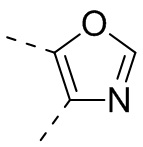 ,
,  ,
,  ,
, 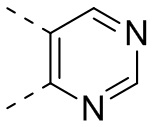 ,
, 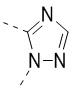 и
и 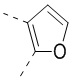 .
.
[34] В некоторых вариантах осуществления настоящего изобретения вышеупомянутый фрагмент 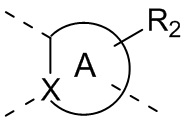 выбран из группы, состоящей из
выбран из группы, состоящей из 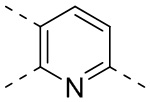 ,
,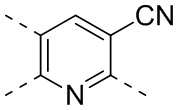 ,
, 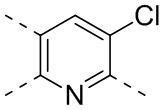 ,
, 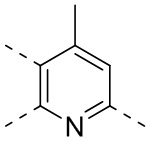 ,
, 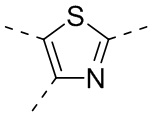 ,
, 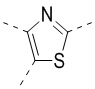 ,
, 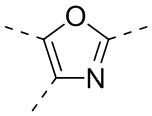 ,
, 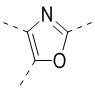 ,
, 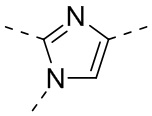 ,
, 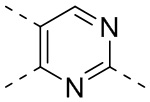 ,
,  ,
,  и
и 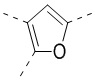 .
.
[35] В некоторых вариантах осуществления настоящего изобретения вышеупомянутый фрагмент 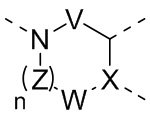 выбран из группы, состоящей из
выбран из группы, состоящей из 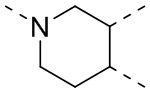 ,
, 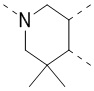 ,
, 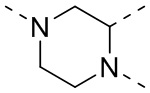 ,
, 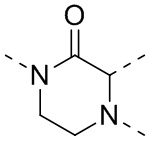 ,
, 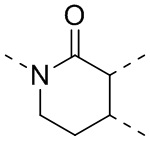 ,
, 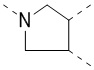 ,
, 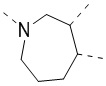 ,
, 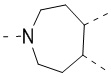 и
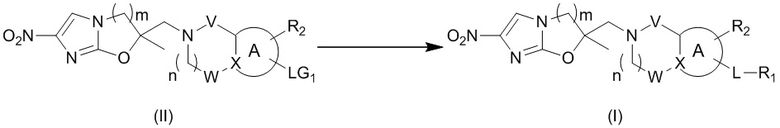
и  .
.
[36] В некоторых вариантах осуществления настоящего изобретения вышеупомянутый фрагмент  выбран из группы, состоящей из
выбран из группы, состоящей из  ,
,  ,
, 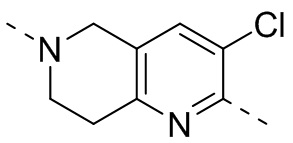 ,
, 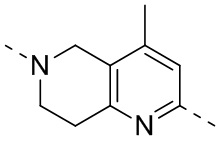 ,
, 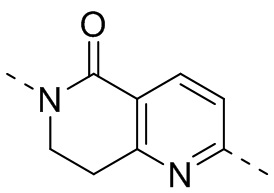 ,
, 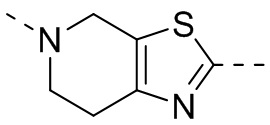 ,
, 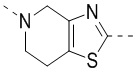 ,
, 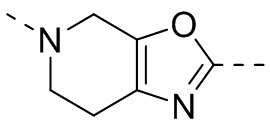 ,
, 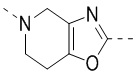 ,
,  ,
, 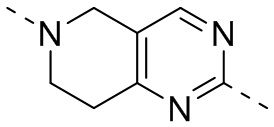 ,
, 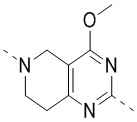 ,
, 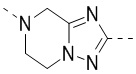 ,
, 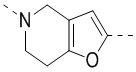 ,
, 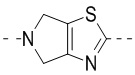 ,
, 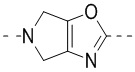 ,
, 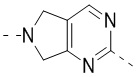 ,
, 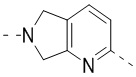 ,
, 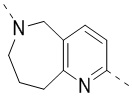 ,
, 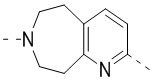 ,
, 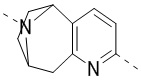 ,
, 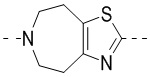 ,
, 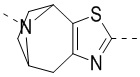 и
и  .
.
[37] В частности, соединения согласно настоящему изобретению выбраны из группы, состоящей из
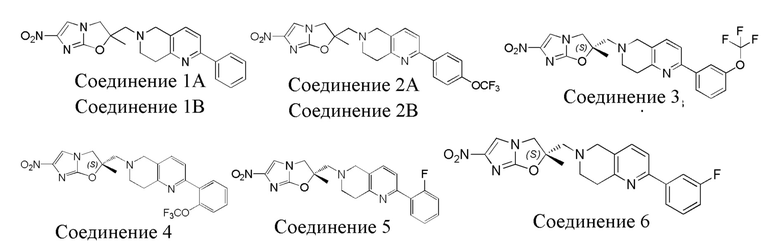
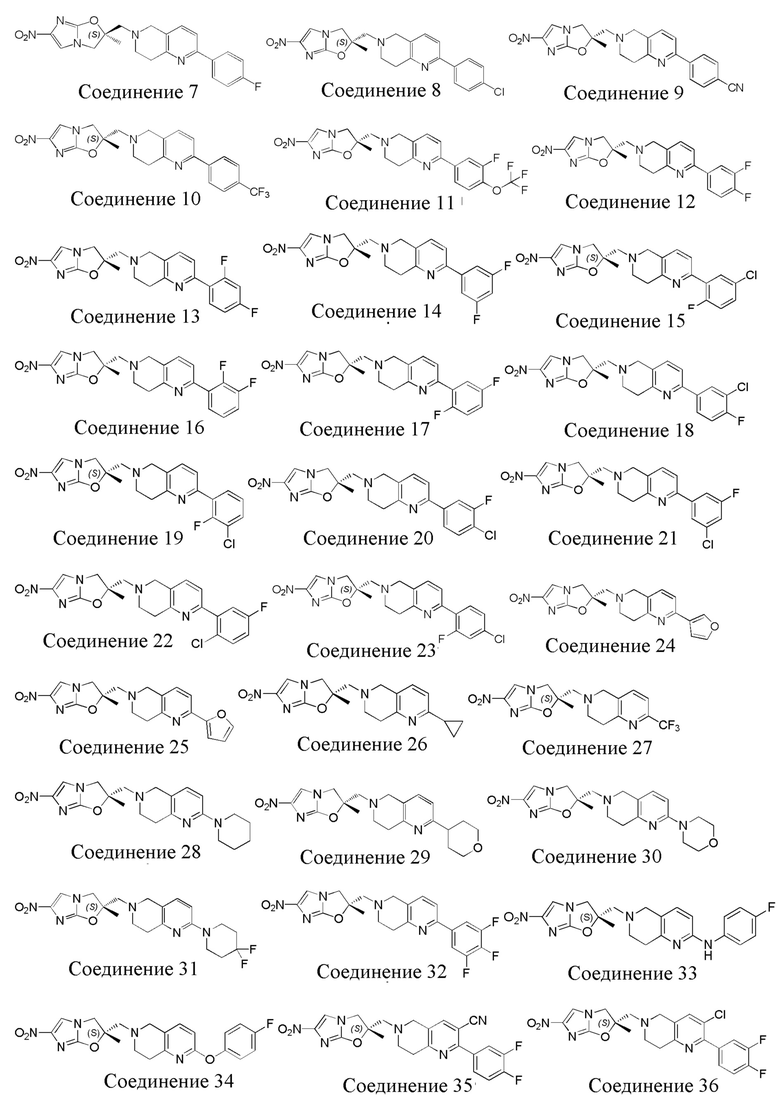
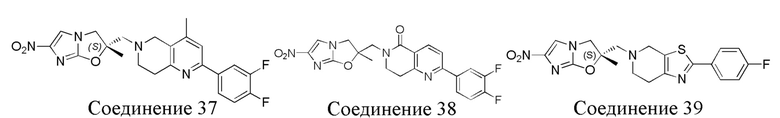
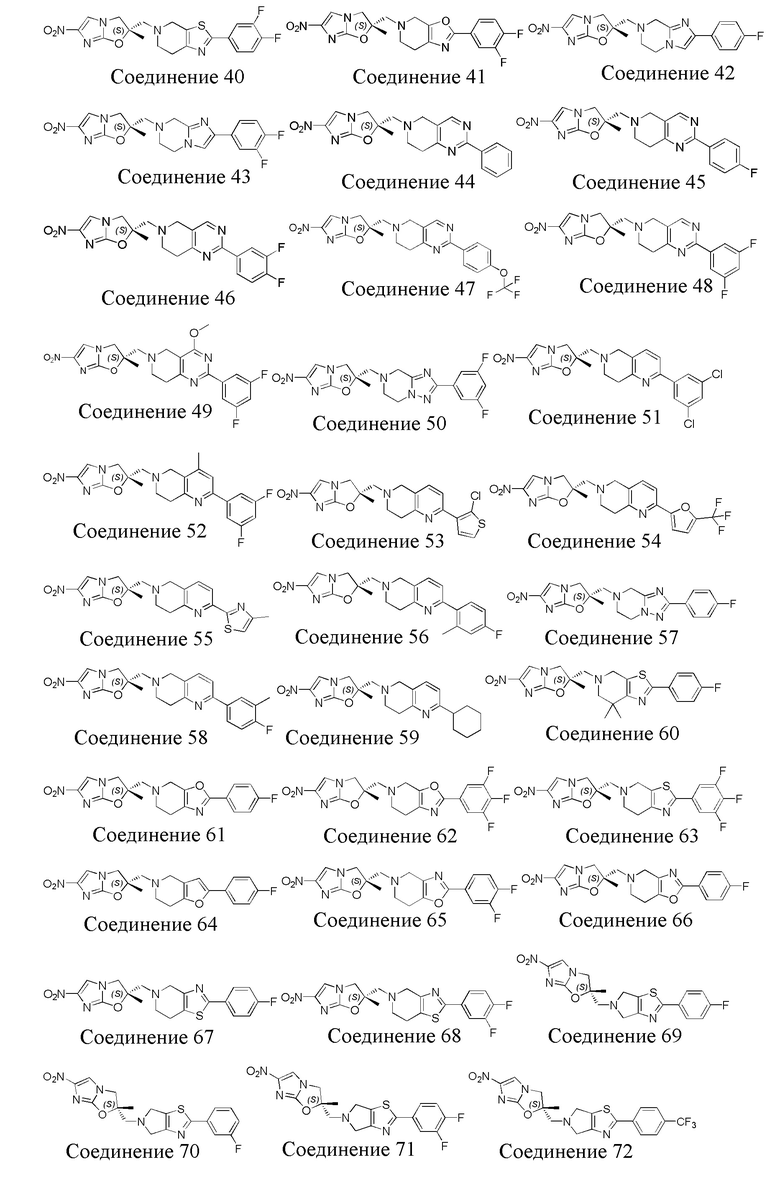
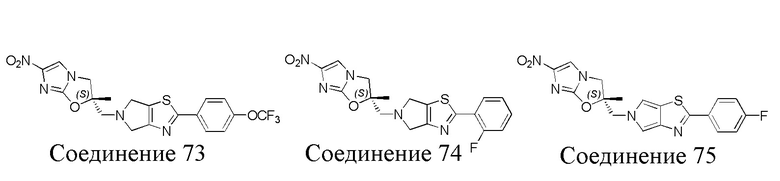
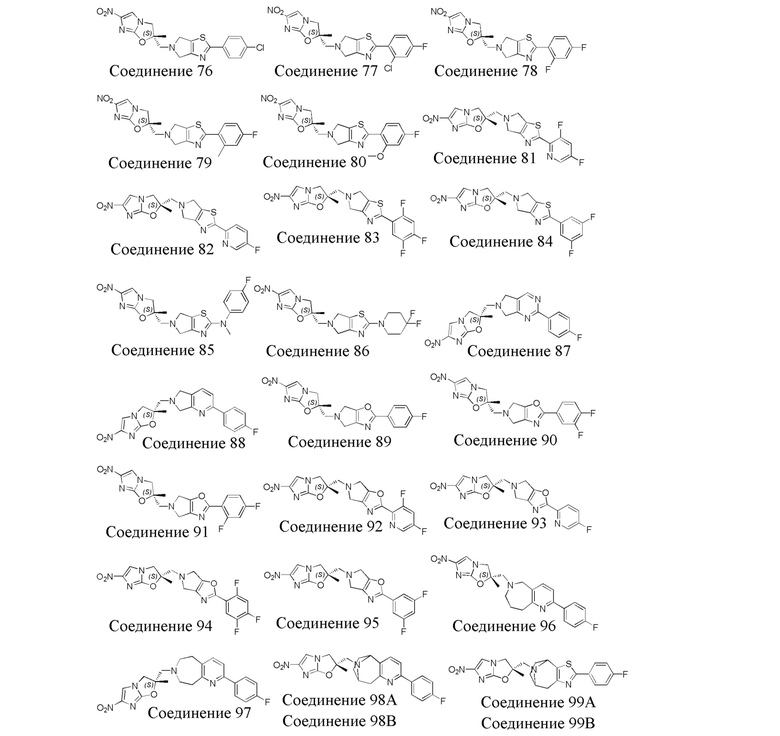 и
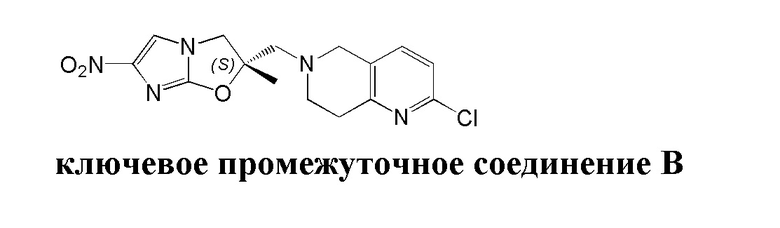
и 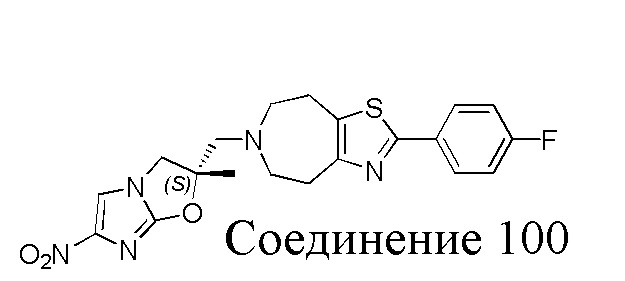 .
.
[38] В настоящем изобретении также предлагается фармацевтическая композиция, содержащая эффективное количество соединения формулы (I), его фармацевтически приемлемой соли, его оптического изомера, упомянутого выше, или фармацевтически приемлемый носитель.
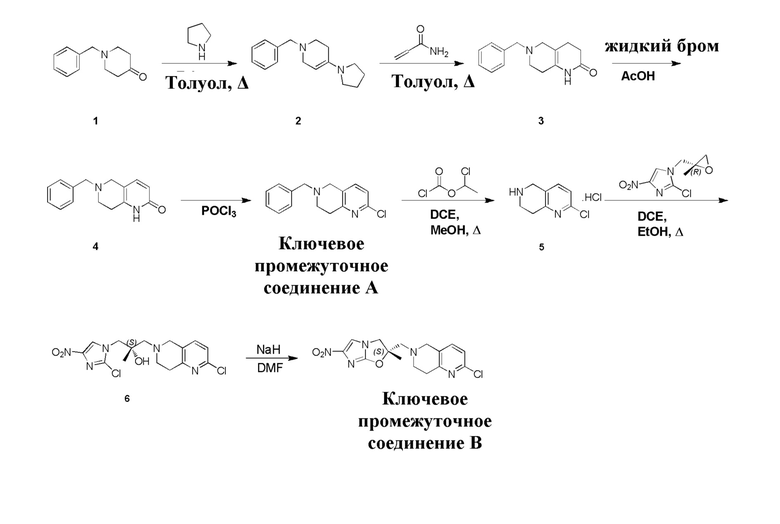
[39] В настоящем изобретении также предлагается применение соединения формулы (I), его фармацевтически приемлемой соли, его оптического изомера, упомянутого выше, или композиции, упомянутой выше, в изготовлении лекарственного препарата для лечения и профилактики инфекций, вызываемых Mycobacterium tuberculosis, или других микробных инфекций.
[40] В настоящем изобретении также предлагается способ получения соединения формулы (I), содержащего:
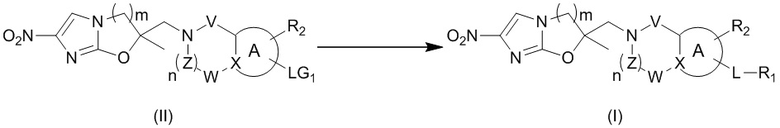
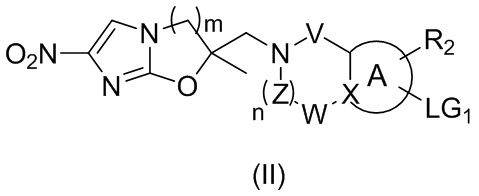
[41] или
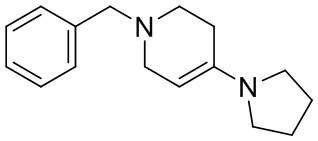
 ,
,
[42] где LG2 представляет собой подходящую уходящую группу, а другие переменные являются такими, как определено выше.
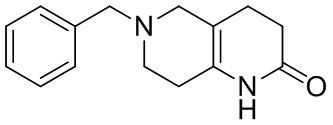
[43] В некоторых вариантах осуществления настоящего изобретения вышеупомянутый LG2 представляет собой галоген.
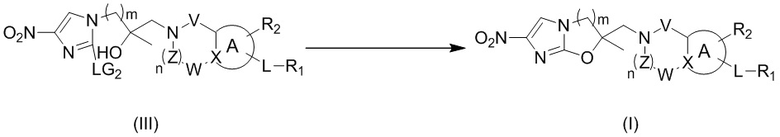
[44] В настоящем изобретении также предлагается промежуточное соединение для получения соединения формулы (I),
 ,
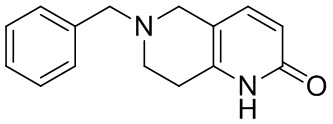
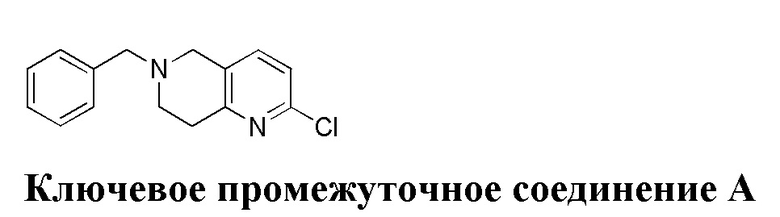
,
[45] где LG1 представляет собой подходящую уходящую группу, а другие переменные являются такими, как определено выше.
[46] В некоторых вариантах осуществления настоящего изобретения вышеупомянутый LG1 представляет собой галоген.
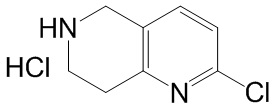
[47] В настоящем изобретении также предлагаются промежуточные соединения для получения соединения формулы (I) согласно п. 1 формулы настоящего изобретения, содержащие:
 .
.
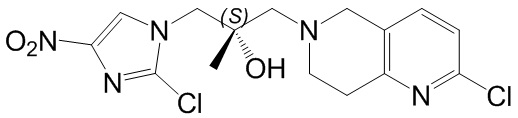
Соответствующие определения
[48] Если не указано иное, предполагается, что следующие термины и выражения, используемые в настоящем документе, имеют следующие значения. Конкретные термин или выражение при отсутствии точного определения не стоит считать неопределенным или неясным, а следует понимать в соответствии с обычным значением. Если в настоящем документе встречается торговое название, то оно относится к соответствующему продукту или к его активному ингредиенту.
[49] C1-12 выбран из группы, состоящей из C1, C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 и C12, C3-12 выбран из группы, состоящей из C3, C4, C5, C6, C7, C8, C9, C10, C11 и C12.
[50] C1-12алкил или гетероалкил, C3-12циклогидрокарбонил или гетероциклогидрокарбонил, C1-12алкил или гетероалкил, замещенные C3-12циклогидрокарбонилом или гетероциклогидрокарбонилом, включают без ограничения:
[51] C1-12алкил, C1-12алкиламино, N,N-ди(C1-12алкил)амино, C1-12алкокси, C1-12алкилацил, C1-12алкоксикарбонил, C1-12алкилсульфонил, C1-12алкилсульфинил, C3-12циклоалкил, C3-12циклоалкиламино, C3-12гетероциклоалкиламино, C3-12циклоалкокси, C3-12циклоалкилацил, C3-12циклоалкилоксикарбонил, C3-12циклоалкилсульфонил, C3-12циклоалкилсульфинил, 5-12-членный арил или гетероарил, 5-12-членный арилалкил или гетероарилалкил,
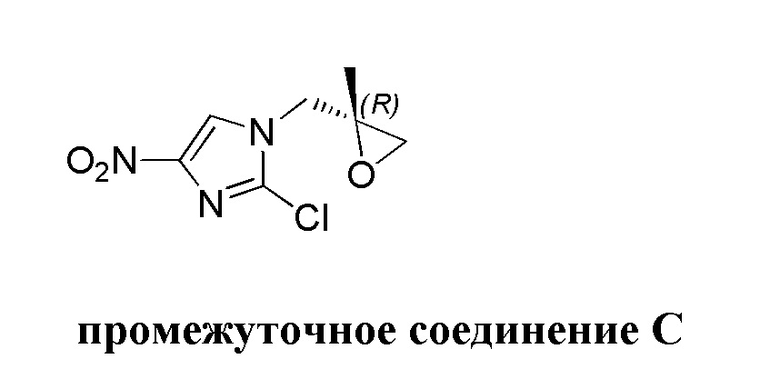
[52] метил, этил, н-пропил, изопропил, -CH2C(CH3)(CH3)(OH), циклопропил, циклобутил, пропилметилен, циклопропионил, бензилокси, трифторметил, аминометил, гидроксиметил, метоксил, формил, метоксикарбонил, метансульфонил, метилсульфинил, этокси, ацетил, этансульфонил, этоксикарбонил, диметиламино, диэтиламино, диметиламинокарбонил, диэтиламинокарбонил,
[53] N(CH3)2, NH(CH3), -CH2CF3, -CH2CH2CF3, -CH2CH2F, -CH2CH2S(=O)2CH3, -CH2CH2CN, -CH2CH(OH)(CH3)2, -CH2CH(F)(CH3)2, -CH2CH2F, -CH2CF3, -CH2CH2CF3, -CH2CH2NH2, -CH2CH2OH, -CH2CH2OCH3, -CH2CH2CH2OCH3, -CH2CH2N(CH3)2, -S(=O)2CH3, -CH2CH2S(=O)2CH3 и
[54] фенил, тиазолил, бифенил, нафтил, циклопентил, фурил, 3-пирролинил, пирролидинил, 1,3-диоксоланил, пиразолил, 2-пиразолинил, пиразолидинил, имидазолил, оксазолил, тиазолил, 1,2,3-оксазолил, 1,2,3-триазолил, 1,2,4-триазолил, 1,3,4-тиадиазолил, 4H-пиранил, пиридил, пиперидинил, 1,4-диоксанил, морфолинил, пиридазинил, пиримидинил, пиразинил, пиперазинил, 1,3,5-тритиил, 1,3,5-триазинил, бензoфуранил, бензoтиенил, индолил, бензимидазолил, бензoтиазолил, пуринил, хинолинил, изохинолинил, циннолинил или хиноксалинил.
[55] В настоящем документе термин «фармацевтически приемлемый» предназначен для тех соединений, материалов, композиций и/или составов, которые в рамках объективного врачебного мнения применимы для применения в контакте к тканям людей и животных, но без чрезмерной токсичности, раздражения, аллергических реакций или других проблем или осложнений, при этом также отвечают приемлемому соотношению пользы/риска.
[56] Термин «фармацевтически приемлемая соль» относится к соли соединения согласно настоящему изобретению, которую получают из данного соединения при помощи конкретного заместителя, раскрытого в настоящем изобретении, и относительно нетоксичной кислоты или щелочи. Если соединение согласно настоящему изобретению содержит относительно кислотную функциональную группу, то соль присоединения щелочи можно получить путем приведения соединения в нейтральной форме в контакт с достаточным количеством щелочи в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемая соль присоединения щелочи включает соль натрия, калия, кальция, аммония, органического аммония или магния или т. п. Если соединение согласно настоящему изобретению содержит относительно щелочную функциональную группу, то соль присоединения кислоты можно получить путем приведения соединения в нейтральной форме в контакт с достаточным количеством кислоты в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемой соли присоединения кислоты включают соль неорганической кислоты, причем неорганическая кислота включает такие кислоты, как хлористоводородная кислота, бромистоводородная кислота, азотная кислота, угольная кислота, бикарбонат, фосфорная кислота, гидрофосфат, дигидрофосфат, серная кислота, гидросульфат, йодистоводородная кислота, фосфористая кислота и т. д; и соль органической кислоты, причем органическая кислота включает такие кислоты, как уксусная кислота, пропионовая кислота, изомасляная кислота, малеиновая кислота, малоновая кислота, бензойная кислота, янтарная кислота, субериновая кислота, фумаровая кислота, молочная кислота, миндальная кислота, фталевая кислота, фенилсульфоновая кислота, п-толуолсульфоновая кислота, лимонная кислота, винная кислота, метилсульфоновая кислота и т. п; а также включают соль аминокислоты (например, аргинина и т. д.) и соль органической кислоты, такой как глюкуроновая кислота и т. п. (см. Berge et al., “Pharmaceutical Salts”, Journal of Pharmaceutical Science 66: 1-19 (1977)). Некоторое конкретное соединение согласно настоящему изобретению содержит как щелочную, так и кислотную функциональные группы с возможностью превращения в любую соль присоединения щелочи или присоединения кислоты.
[57] Предпочтительно нейтральную форму соединения восстанавливают путем приведения соли в контакт с основанием или кислотой традиционным способом, а затем выделения исходного соединения. Разница между исходной формой соединения и различными солевыми формами заключается в некоторых физических свойствах, как, например, отличающаяся растворимость в полярном растворителе.
[58] «Фармацевтически приемлемая соль» в настоящем изобретении представляет собой производные соединения согласно настоящему изобретению, где исходное соединение модифицировано путем образования соли при помощи кислоты или щелочи. Примеры фармацевтически приемлемой соли включают без ограничения соль неорганической кислоты или органической кислоты и щелочи, такой как амин, щелочного металла, или органическую соль с кислотным радикалом, таким как у карбоновой кислоты, и т. п. Фармацевтически приемлемая соль включает традиционные нетоксичные соли или соли четвертичного аммония исходного соединения, такие как соль, образованная при помощи нетоксичной неорганической кислоты или органической кислоты. Традиционная нетоксичная соль включает без ограничения такие соли, которые получены из неорганических кислот и органических кислот, причем неорганические кислоты или органические кислоты выбраны из 2-ацетоксибензойной кислоты, 2-изэтионовой кислоты, уксусной кислоты, аскорбиновой кислоты, фенилсульфоновой кислоты, бензойной кислоты, бикарбоната, угольной кислоты, лимонной кислоты, этилендиаминтетрауксусной кислоты, этандисульфоновой кислоты, этансульфоновой кислоты, фумаровой кислоты, глюкогептозы, глюконовой кислоты, глутаминовой кислоты, гликолевой кислоты, бромистоводородной кислоты, хлористоводородной кислоты, гидройодата, гидроксила, гидроксинафтойной кислоты, изэтионовой кислоты, молочной кислоты, лактозы, додекансульфоновой кислоты, малеиновой кислоты, яблочной кислоты, миндальной кислоты, метансульфоновой кислоты, азотной кислоты, щавелевой кислоты, памоевой кислоты, пантотеновой кислоты, фенилуксусной кислоты, фосфорной кислоты, полигалактуронана, пропионовой кислоты, салициловой кислоты, стеариновой кислоты, фолиновой кислоты, янтарной кислоты, аминосульфоновой кислоты, сульфаниловой кислоты, серной кислоты, дубильной кислоты, винной кислоты и п-толуолсульфоновой кислоты.
[59] Фармацевтически приемлемая соль согласно настоящему изобретению может быть получена посредством традиционного способа с помощью исходного соединения, содержащего кислотную или щелочную группу. Как правило, способ получения соли включает приведение данных соединений в формах свободных кислот или щелочей в реакцию со стехиометрическим количеством подходящих щелочей или кислот в воде, или органическом растворителе, или в смеси воды и органического растворителя. В общем, предпочтительно выбирать неводные среды, такие как простой эфир, этилацетат, этанол, изопропанол или ацетонитрил и т. п.
[60] За исключением формы соли в настоящем изобретении представлена форма пролекарства для соединения. Пролекарство соединения, описанного в настоящем изобретении, легко превращают в соединение согласно настоящему изобретению посредством химических изменений в физиологических условиях. Кроме того, пролекарство можно превращать в соединение согласно настоящему изобретению посредством химического или биохимического способа в среде in vivo.
[61] Некоторые соединения согласно настоящему изобретению могут существовать в виде несольватной или сольватной форм, в том числе гидратных форм. В общем, сольватная форма подобна несольватной форме, обе из которых включены в объем настоящего изобретения.
[62] Некоторые соединения согласно настоящему изобретению могут содержать асимметрические атомы углерода (оптический центр) или двойные связи. Рацемические изомеры, диастереомеры, геометрические изомеры и одинарные изомеры включены в объем настоящего изобретения.
[63] Графическое представление рацемического изомера, амбискалемического и скалемического или энантиомерно чистого соединения согласно настоящему изобретению взято из Maehr, J. Chem. Ed. 1985, 62: 114-120. Если не указано иное, абсолютная конфигурация стереоцентра представлена клиновидными и пунктирными линиями. Если соединение согласно настоящему изобретению содержит виниловую двойную связь или другой геометрический асимметрический центр и если не указано иное, то включены E-, Z-геометрические изомеры. Подобным образом, в объем настоящего изобретения включены все таутомерные формы.
[64] Соединение по настоящему изобретению может существовать в виде конкретного геометрического или стереоизомерного изомера. Настоящее изобретение предусматривает все соединения этого класса, включая цис- и транс-изомеры, (-)- и (+)-антимеры, (R)- и (S)-антимеры, диастереомеры, (D)-изомер, (L)-изомер, а также рацемические смеси и другие смеси, такие как энантиомерно или диастереомерно обогащенные смеси, при этом все такие смеси включены в объем настоящего изобретения. В таких заместителях, как алкил, могут находиться другие асимметричные атомы углерода. Все эти изомеры и их смеси включены в объем настоящего изобретения.
[65] Оптически активные (R)- и (S)-изомеры, (D)- и (L)-изомеры могут быть получены посредством асимметричного синтеза, или хиральных реагентов, или других обычных методик. Если желателен энантиомер соединения согласно настоящему изобретению, то для его получения можно применять асимметричный синтез или получение производных хиральных вспомогательных веществ, в случае которых выделяют получаемые в результате смеси диастереомеров, а вспомогательные группы расщепляют с получением чистого необходимого энантиомера. Либо же, если молекула содержит щелочную функциональную группу (такую как амино) или кислотные функциональные группы (такие как карбоксильная), то соль диастереомера образуется с помощью соответствующей оптически активной кислоты или щелочи, а затем чистый энантиомер можно повторно использовать после расщепления до соли диастереомера посредством способов, известных из уровня техники. Кроме того, разделение энантиомера и диастереомера, как правило, осуществляют посредством хроматографического способа, при этом в хроматографическом способе используют хиральную стационарную фазу и необязательно в комбинации с химическим способом получения производных (например, из амина образуется карбамат).
[66] Один или более атомов, из которых состоит соединение согласно настоящему изобретению, могут содержать неестественное соотношение атомных изотопов. Например, соединение может быть мечено радиоактивным изотопом, таким как тритий (3H), йод-125(125I) или C-14(14C). Все варианты в изотопной композиции соединения, раскрытого в настоящем изобретении, вне зависимости от радиоактивности, включены в объем настоящего изобретения.
[67] Термин «фармацевтически приемлемый носитель» относится к любому составу или несущей среде, способным доставлять эффективное количество активного вещества, раскрытого в настоящем изобретении, не оказывающим отрицательного воздействия на биологическую активность активного вещества и без каких-либо токсичных побочных эффектов на хозяина или пациента, при этом иллюстративный носитель включает воду, масло, растительного и минерального происхождения, основу для крема, основу для лосьона, основу для мази и т. п. Основа включает суспензию, загуститель, усилитель проникновения через кожу и т. п. Их составы хорошо известны специалисту в области косметических или местных лекарственных средств. В отношении других данных о носителе можно ссылаться на Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams & Wilkins (2005), содержание которого включено в данный раздел в качестве ссылки.
[68] Термин «вспомогательное средство» относится, как правило, к носителю, разбавителю и/или среде, необходимому для получения эффективной фармацевтической композиции.
[69] В отношении лекарственного средства или фармакологического активного средства термин «эффективное количество» или «терапевтически эффективное количество» относится к количеству лекарственного средства или состава, достаточному для достижения необходимых эффектов, но без токсичности. В отношении состава для перорального применения согласно настоящему изобретению «эффективное количество» одного активного вещества в композиции относится к количеству, необходимому для достижения необходимых эффектов в комбинации с другим активным веществом в композиции. Определение эффективного количества отличается для каждого субъекта, причем зависит от возраста и общего состояния пациента, а также от конкретного активного вещества. В одном из случаев соответствующее эффективное количество может быть определено специалистом в данной области техники согласно традиционным тестам.
[70] Термин «активный ингредиент», «терапевтическое средство», «активное вещество» или «активное средство» относится к химическому структурному элементу, с помощью которого можно эффективно лечить у целевого субъекта нарушение, болезнь или заболевание.
[71] Термин «замещенный» относится к одному или более атомам водорода на конкретном атоме, необязательно замещенном заместителем, в том числе дейтерием и вариантом водорода, при условии, что валентное состояние конкретного атома является нормальным, а соединение, полученное после замещения, является стабильным. Если заместитель представляет собой кетонную группу (т. е. =O), то это означает, что два атома водорода являются замещенными. Замещение кетонной группы не происходит в ариле. Термин «необязательно замещенный» означает, что названное может быть замещенным или быть незамещенным, если не указано иное, при этом тип и число заместителей может быть произвольным при соблюдении стабильности, доступной в химической структуре.
[72] Если любой параметр (например, R) демонстрирует появление в композиции или структуре соединения более одного раза, то определение каждого появления является независимым. Следовательно, например, если группа является замещенной 0-2 R, то группа может быть необязательно замещенной не более двух R, при этом R имеет независимый вариант в каждом случае. Кроме того, комбинация заместителей и/или их вариантов возможна, только если такая комбинация приведет к стабильному соединению.
[73] Если число групп соединения равняется 0, как например -(CRR)0-, то это означает, что группа соединения представляет собой одинарную связь. Если один из параметров выбран из одинарной связи, то это означает, что две группы, к которым он присоединен, являются непосредственно связанными, например, если L в A-L-Z представляет собой одинарную связь, то это означает, что структура фактически представляет собой собой A-Z.
[74] Если заместитель является незадействованным, то это означает, что заместитель отсутствует, например, если X в A-X незадействованный, то это означает, что структура фактически представляет собой A.
[75] Если связи заместителя могут быть поперечно соединены с двумя атомами кольца, то заместитель может быть связан с произвольными атомами в кольце. Если не указано, посредством какого атома приведенный заместитель соединен с общей структурной формулой, включая соединение, которое конкретно не упомянуто, заместитель может быть связан посредством любого из его атомов. Комбинация заместителей и/или их вариантов возможна только тогда, если такая комбинация приведет к стабильному соединению. Например, структурная единица или
или  означает, что соединение может произойти на любом атоме в циклогексиле или циклогексадиене.
означает, что соединение может произойти на любом атоме в циклогексиле или циклогексадиене.
[76] Заместитель алкильной и гетероалкильной группы, как правило, называется «алкильным заместителем», который может быть выбран без ограничения из группы, состоящей из -R’, -OR’, =O, =NR’, =N-OR’, -NR’R”, -SR’, галогена, -SiR’R”R”’, OC(O)R’, -C(O)R’, -CO2R’, -C(=O)NR’R”, -OC(O)NR’R”, -NR”C(O)R’, NR’ C(O)NR”R”’, -NR”C(O)2R’, -NR””’-C(NR’R”R’”)=NR””, NR”” C(NR’R”)=NR’”, -S(O)R’, -S(O)2R’, -S(O)2NR’R”, NR”SO2R’, -CN, –NO2, -N3, -CH(Ph)2 и фтор(C1-C4)алкила, при этом количество заместителей составляет от 0 до (2m’+1), где m’ представляет собой общее количество атомов углерода в группе. R’, R”, R”', R’’’’ и R’’’’’ независимо выбраны из H, замещенного или незамещенного гетероалкила, замещенного или незамещенного арила (например, арила, замещенного 1~3 атомами галогена), замещенного или незамещенного алкила, алкокси, тиоалкокси или аралкила. Если соединение согласно настоящему изобретению включает, например, больше одной группы R, то каждая из групп R является независимо выбранной, как и каждая из групп R’, R”, R”’, R”” и R””’, когда включено больше одной из них. Если R’ и R’’ присоединены к одному и тому же атому азота, то они могут образовывать 5-, 6- или 7-членное кольцо вместе с атомом азота. Например, -NR’R’’ включает без ограничения 1-пирролидинил и 4-морфолинил. Согласно вышеизложенному обсуждению касательно заместителя, специалист в данной области сможет понять, что термин «алкил» предназначен для включения группы, образованной за счет связывания атома углерода с неводородной группой, такой как галогенированный алкил (например, -CF3, -CH2CF3) и ацил (например, -C(O)CH3, -C(O)CF3, C(O)CH2OCH3 и т.п.).
[77] Подобно заместителю в алкильной группе, заместитель в арильной и гетероарильной группе, называемый, как правило, «арильным заместителем», может быть выбран, например, из -R’, -OR’, -NR’R”, -SR’, -галогена, -SiR’R”R”’, OC(O)R’, -C(O)R’, -CO2R’, -C(=O)NR’R”, -OC(O)NR’R”, -NR”C(O)R’, NR’ C(O)NR”R”’, -NR”C(O)2R’, -NR””’-C(NR’R”R’”)=NR””, NR”” C(NR’R”)=NR’”, -S(O)R’, -S(O)2R’, -S(O)2NR’R”, NR”SO2R’, -CN, –NO2, -N3, -CH(Ph)2, фтор(C1-C4)алкокси, фтор(C1-C4)алкила и т.п., при этом количество заместителей варьирует от 0 до суммарной открытой валентности ароматического кольца;где R’, R”, R”’, R”” и R””’ независимо и предпочтительно выбраны из H, замещенного или незамещенного алкила, замещенного или незамещенного гетероалкила, замещенного или незамещенного арила и замещенного или незамещенного гетероарила. Если соединение согласно настоящему изобретению включает, например, больше одной группы R, то каждая из групп R является независимо выбранной, как и каждая из групп R’, R”, R”’, R”” и R””’, когда включено больше одной из них.
[78] Два заместителя, присоединенные к смежным атомам в арильном или гетероарильном кольце, могут быть необязательно замещены заместителем с общей формулой -T-C(O)-(CRR’)q-U-, где T и U независимо выбраны из -NR-, -O-, CRR'- или одинарной связи, q представляет собой целое число от 0 до 3. В качестве альтернативы, два заместителя, присоединенные к смежным атомам в арильном или гетероарильном кольце, могут быть необязательно замещены заместителем с общей формулой -A (CH2)r B-, где A и B независимо выбраны из -CRR’-, -O-, -NR-, -S-, -S(O)-, S(O)2-, -S(O)2NR’- или одинарной связи, r представляет собой целое число от 1 до 4. Одинарная связь в новом кольце, образованная таким образом, необязательно может быть заменена двойной связью. В качестве альтернативы, два заместителя, присоединенные к смежным атомам в арильном или гетероарильном кольце, необязательно могут быть замещены заместителем с общей формулой -A(CH2)sX(CH2)dB-, где каждый из s и d независимо выбран из целых чисел от 0 до 3, X представляет собой -O-, -NR’, -S-, -S(O)-, -S(O)2- или -S(O)2NR’-. Заместители R, R’, R” и R”’ соответственно и предпочтительно выбраны из водорода и замещенного или незамещенного (C1-C6)алкила.
[79] Если не указано иное, термин «галогенированный» или «галоген» сам по себе или как часть другого заместителя относятся к атому фтора, хлора, брома или йода. Кроме того, термин «галогенированный алкил» предназначен для включения моногалогенированного алкила и полигалогенированного алкила. Например, термин «галогенированный (C1-C4)алкил» предназначен для включения, без ограничения, трифторметила, 2,2,2-трифторэтила, 4-хлорбутила, 3-бромпропила и т. п.
[80] Примеры галогенированного алкила включают без ограничения трифторметил, трихлорметил, пентафторэтил и пентахлорэтил. «Алкокси» означает, что алкильная группа с конкретным числом атомов углерода присоединена с помощью кислородного мостика. C1-6алкокси включает C1, C2, C3, C4, C5 и C6алкокси. Примеры алкокси включают без ограничения метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси и S-пентилокси. «Циклоалкил» включает насыщенную циклическую группу, такую как циклопропил, циклобутил или циклопентил. 3-7-членный циклоалкил включает C3, C4, C5, C6 и C7циклоалкил. «Алкенил» включает линейную или разветвленную углеводородную цепь, где любые стабильные участки на цепи характеризуются одной или более C-C двойными связями, как например, винил и пропенил.
[81] Термин «галоген» или «галогенид» относится к фтору, хлору, брому и йоду.
[82] Если не указано иное, термин «гетеро» относится к гетероатому или гетероатомной группе (т. е. группе, содержащей гетероатом), включая атомы за исключением углерода (C) и водорода (H) и группы, содержащие такие гетероатомы, например, включающие кислород (O), азот (N), серу (S), кремний (Si), германий (Ge), алюминий (Al), бор (B), -O-, -S-, =O, =S, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O), -S(=O)2- и необязательно замещенные -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O)2 N(H)- или -S(=O) N(H)-.
[83] Если не указано иное, «кольцо» относится к замещенному или незамещенному циклоалкилу, гетероциклоалкилу, циклоалкенилу, гетероциклоалкенилу, циклоалкинилу, гетероциклоалкинилу, арилу или гетероарилу. Кольцо включает одинарное кольцо, соединительное кольцо, спирокольцо, конденсированное кольцо или кольцо с внутренним мостиком. Число атомов в кольце обычно определяется как число членов кольца, например, «5-7-членное кольцо» представляет собой кольцо с замкнутой структурой из 5-7 атомов. Если не указано иное, кольцо необязательно содержит 1-3 гетероатома. Следовательно, «5-7–членное кольцо» включает, например, фенилпиридин и пиперидинил; с другой стороны, термин «5-7–членное гетероциклоалкильное кольцо» включает пиридил и пиперидинил, но не включает фенил. Термин «кольцо» также включает кольцевую систему, содержащую по меньшей мере одно кольцо, где каждое кольцо независимо отвечает вышеуказанному определению.
[84] Если не указано иное, термин «гетероцикл» или «гетероциклил» относится к стабильному моноциклическому, бициклическому или трициклическому кольцу, содержащему гетероатом или гетероатомную группу, при этом они могут быть насыщенными, частично ненасыщенными или ненасыщенными (ароматическими), они содержат атомы углерода и 1, 2, 3 или 4 гетероатома в кольце, которые независимо выбраны из группы, состоящей из N, O и S, где любой из гетероциклов может быть конденсирован до бензольного кольца с образованием бициклического кольца. Атомы азота и серы могут быть необязательно окислены (т. е. NO и S(O)p). Атом азота может быть замещенным или не замещенным (т. е. N или NR, где R представляет собой H или другой заместитель, определенный в настоящем документе). Гетероцикл может быть присоединен к боковой группе любого гетероатома или атома углерода с образованием стабильной структуры. Если полученное соединение является стабильным, то гетероцикл, описанный в настоящем документе, может быть замещен при атоме углерода или азота. Атом азота в гетероцикле необязательно является кватернизованным. В качестве предпочтительного варианта осуществления настоящего изобретения, если общее количество атомов S и O, содержащихся в гетероцикле, превышает 1, то эти гетероатомы не являются смежными друг по отношению к другу. В качестве другого предпочтительного варианта осуществления настоящего изобретения общее число атомов S и O в гетероцикле не превышает 1. Используемый в настоящем документе термин «ароматическая гетероциклическая группа» или «гетероарил» относится к стабильному 5-, 6-, 7-членному моноциклу или бициклу или 7-, 8-, 9- или 10-членному бициклическому гетероароматическому кольцу, которое содержит атомы углерода и 1, 2, 3 или 4 гетероатома в кольце, которые независимо выбраны из группы, состоящей из N, O и S. Атом азота может быть замещенным или незамещенным (т. е. N или NR, где R представляет собой H или другой заместитель, определенный в настоящем документе). Атомы азота и серы могут быть необязательно окислены (т. е. NO и S(O)p). Следует отметить, что общее число атомов S и O в гетероароматическом кольце не превышает 1. Кольца с внутренними мостиками также включены в определение гетероцикла. Если один или более атомов (т. е. C, O, N или S) присоединены к двум несмежным атомам углерода или атомам азота, то происходит образование кольца с внутренним мостиком. Предпочтительное кольцо с внутренним мостиком включает в себя без ограничения, один атом углерода, два атома углерода, один атом азота, два атома азота и одну группу углерод-азот. Следует отметить, что мостик всегда превращает моноциклическое кольцо в трициклическое кольцо. В кольце с внутренним мостиком заместитель в кольце также может находиться на мостике.
[85] Примеры гетероциклического соединения включают без ограничения акридинил, азоцинил, бензимидазолил, бензофуранил, бензомеркаптофуранил, бензомеркаптофенил, бензоксазолил, бензоксазолинил, бензотиазолил, бензотриазолил, бензотетразолил, бензоизоксазолил, бензоизотиазолил, бензоимидазолинил, карбазолил, 4aH-карбазолил, карболинил, хроманил, хромен, циннолинила декагидрохинолил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуранил, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1H-индазолил, индоалкенил, индолинил, индолизинил, индолил, 3H-индолил, изатиногруппу, изобензофуранил, пиранил, изоиндолил, изоиндолинил, изоиндолил, индолил, изохинолил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, изоксазолил, гидроксилиндил, пиримидил, фенантридинил, фенантролинил, феназин, фенотиазин, бензопуринил, феноксазинил, фталазинил, пиперазинил, пиперидил, оксопиперидинил, 4-оксопиперидинил, пиперонил, птеридил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, оксазолопиридин, пиридиноимидазол, пиридинотиазол, пиридил, пиримидинил, пирролидинил, пирролинил, 2H-пирролил, пирролил, пиразолил, хиназолинил, хинолил, 4H-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофурил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6H-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазил, изотиазолилтиенил, тиенил, тиофеноксазолил, тиофенотиазолил, тиофеноимидазолил, тиенил, триазинил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил и ксантенил. Также включено соединение с конденсированным кольцом и спирокольцом.
[86] Если не указано иное, термин «гидрокарбонил» или его конкретный представитель (такой как алкил, алкенил, алкинил, фенил и т. п.) сам по себе или как часть другого заместителя представляет собой линейный, разветвленный или циклический гидрокарбонил или их комбинацию, которые могут быть полностью насыщенными, моноциклическими или полициклическими ненасыщенными, могут быть монозамещенными, дизамещенными или полизамещенными, могут быть одновалентными (такими как метил), бивалентными (такими как метилен) или многовалентными (такими как метенил), могут включать бивалентные или многовалентные атомные группы с конкретным числом атомов углерода (как, например, C1-C10 относится к группе, имеющей 1~10 атомов углерода). Термин «алкил» включает без ограничения алифатический гидрокарбонил и ароматический гидрокарбонил, причем алифатический гидрокарбонил включает линейные и циклические структуры, в частности включает без ограничения алкил, алкенил и алкинил, а ароматический гидрокарбонил включает без ограничения 6-12–членный ароматический гидрокарбонил, такой как бензол, нафталин и т. п. В некоторых вариантах осуществления термин «гидрокарбонил» относится к линейной или разветвленной группам или к их комбинации, которые могут быть полностью насыщенными, моноциклическими или полициклическими ненасыщенными, могут включать дивалентные и поливалентные группы. Примеры насыщенного гидрокарбонила включают без ограничения гомологи или изомеры метила, этила, н-пропила, изопропила, н-бутила, трет-бутила, изо-бутила, втор-бутила, изо-бутила, циклогексила, (циклогексил)метила, циклопропилметила, н-амила, н-гексила, н-гептила, н-октила и т. п. Ненасыщенный алкил имеет одну или более двойную или тройную связь, примеры которого включают без ограничения винил, 2-пропенил, бутенил, кротил, 2-изопентенил, 2-бутадиенил, 2,4-(пентадиенил), 3-(1,4-пентадиенил), ацетенил, 1- и 3-пропинил, 3-бутинил и более сложные гомологи и изомеры.
[87] Если не указано иное, термин «гетерогидрокарбонил» или его конкретные представители (такие как гетероалкил, гетероалкенил, гетероалкинил, гетероарил и т. п.) сами по себе или же термин в сочетании с другим термином относятся к стабильному, линейному, разветвленному или циклическому гидрокарбонилу или к их комбинациям, которые состоят из конкретного числа атомов углерода и по меньшей мере одного гетероатома. В некоторых вариантах осуществления термин «гетерогидрокарбонил» сам по себе или же термин в сочетании с другим термином относится к стабильному, линейному, разветвленному гидрокарбонилу или к их комбинациям, которые состоят из конкретного числа атомов углерода и по меньшей мере одного гетероатома. В типичном варианте осуществления гетероатом выбран из группы, состоящей из B, O, N и S, в которой атомы азота и серы необязательно окислены, и атом азота необязательно кватернизован. Гетероатом или гетероатомная группа могут быть размещены в любом внутреннем положении гетерогидрокарбонила (включая положение, в котором гидрокарбонил присоединен к остальной части молекулы). Примеры включают без ограничения -CH2-CH2-O-CH3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -CH2-CH2, -S(O)-CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-CH3, -CH2-CH=N-OCH3 и -CH=CH-N(CH3)-CH3. Не более двух гетероатомов являются смежными, как например, -CH2-NH-OCH3.
[88] Термины «алкокси», «алкиламино» и «алкилтио» (или тиоалкокси) являются идиоматическими выражениями, которые относятся к алкильной группе, присоединенной к остальной части молекулы посредством атома кислорода, аминогруппы или атома серы, соответственно.
[89] Если не указано иное, термин «циклогидрокарбонил», «гетероциклогидрокарбонил» или их конкретный представитель (такой как арил, гетероарил, циклоалкил, гетероциклоалкил, циклоалкенил, гетероцикловинил, циклоалкинил, гетероциклоалкинил и т. п.) сам по себе или же термин в комбинации с другими терминами относится, соответственно, к циклическому «гидрокарбонилу», «гетерогидрокарбонилу». Кроме того, в отношении гетерогидрокарбонила или гетероциклогидрокарбонила (таких как гетероалкил, гетероциклоалкил), гетероатомы могут занимать положение, в котором гетероциклическое кольцо присоединено к остальной части молекулы. Примеры циклоалкила включают без ограничения циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил и т.п. Неограничивающие примеры гетероциклила включают 1-(1,2,5,6-тетрагидропиридинил), 1-пиперидил, 2-пиперидил, 3-пиперидил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофуранилиндол-3-ил, тетрагидротиофен-2-ил, тетрагидротиофен-3-ил, 1-пиперазинил и 2-пиперазинил.
[90] Если не указано иное, термин «арил» относится к полиненасыщенному ароматическому углеводородному заместителю, который может быть монозамещенным, дизамещенным или полизамещенным, может быть одновалентным, бивалентным или многовалентным. Он может быть моноциклическим или полициклическим (таким, что имеет от 1 до 3 колец, по меньшей мере одно из которых является ароматическим). Они конденсированы вместе или соединены посредством ковалентной связи. Термин «гетероарил» относится к арилу (или кольцу), содержащему от 1 до 4 гетероатомов. В иллюстративном варианте осуществления гетероатом выбран из группы, состоящей из B, N, O и S, в которой атомы азота и серы необязательно окислены, и атом азота необязательно кватернизован. Гетероарильная группа может быть присоединена к остальной части молекулы посредством гетероатома. Неограниченные примеры арила или гетероарила включают фенил, 1-нафтил, 2-нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 3-пиразолил, 2-имидазолил, 4-имидазолил, пиразинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-фуранил, 3-фуранил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-бензотиазолил, пуринил, 2-бензоимидазолил, 5-индолил, 1-изохинолил, 5-изохинолил, 2-хиноксалил, 5-хиноксалил, 3-хинолил и 6-хинолил. Любой из заместителей в арильной и гетероарильной кольцевой системе выбран из приемлемых заместителей, описанных ниже.
[91] В целях краткости, при использовании в сочетании с другими терминами (например, арилокси, арилтио, аралкил) арил включает определение арильного и гетероарильного кольца, определенного выше. Следовательно, термин «аралкил» предназначен для включения групп, в которых арил присоединен к алкилу (например, бензил, фенилэтил, пиридилметил), включая те алкилы, где атомы углерода (такие как метилен) замещены такими атомами, как атомы кислорода, к примеру, феноксиметил, 2-пиридилоксиметил-3-(1-нафтокси)пропил и т. п.
[92] Термин «уходящая группа» относится к функциональной группе или атому, которые могут быть замещены другими функциональной группой или атомом посредством реакции замещения (например, реакции нуклеофильного замещения). Например, иллюстративные уходящие группы включают трифлат; хлор, бром, йод; сульфонат, как например, мезилат, тозилат, п-бромбензолсульфонат, п-тозилат и т. п.; ацилокси, как например, ацетокси, трифторацетокси и т. д.
[93] Термин «защитная группа» включает без ограничения «защитную группу для аминогруппы», «защитную группу для гидоксигруппы» или «защитную группу для меркаптогруппы». Термин «защитная группа для аминогруппы» относится к защитной группе, которая является подходящей для предупреждения побочных реакций, возникающих при атоме азота аминогруппы. Иллюстративная защитная группа для аминогруппы включает без ограничения формил; ацил, такой как алканоил (как например, ацетил, трихлорацетил или трифторацетил); алкоксикарбонил, такой как трет-бутоксикарбонил (Boc); арилметоксикарбонил, такой как бензилоксикарбонил (Cbz) и 9-фторенилметоксикарбонил (Fmoc); арилметил, такой как бензил (Bn), трифенилметил (Tr), 1,1-бис-(4'-метоксифенил)метил; силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS), и т.п. Термин «защитная группа для гидроксигруппы» относится к защитной группе, которая является подходящей для предупреждения побочных реакций гидроксильной группы. Иллюстративная защитная группа для гидроксигруппы включает без ограничения алкил, такой как метил, этил и трет-бутил; ацил, такой как алканоил (как например, ацетил); арилметил, такой как бензил (Bn), п-метоксибензил (PMB), 9-фторенилметил (Fm) и дифенилметил (дифенилметил, DPM); силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS), и т. п.
[94] Соединение согласно настоящему изобретению можно получить посредством многих способов синтеза, которые хорошо известны специалисту в данной области техники, включая конкретные варианты осуществления, перечисленные далее, и их комбинацию с другими химическими способами синтеза и эквивалентными альтернативными способами, которые известны специалисту в данной области техники, при этом предпочтительные варианты осуществления включают без ограничения варианты осуществления настоящего изобретения.
[95] Используемые в настоящем изобретении растворители являются коммерчески доступными. В настоящем изобретении приняты следующие сокращения: водн. означает водный; экв. означает эквивалент, SEMCl означает (2-(хлорметокси)этил)триметилсилан, i-PrOH означает изопропанол; DCM означает дихлорметан; PE означает петролейный эфир; DIPEA означает N,N-диизопропилэтиламин; DMF означает N,N-диметилформамид, EtOAc означает этилацетат; EtOH означает этанол; MeOH означает метанол, THF означает тетрагидрофуран, DMSO означает диметилсульфоксид; AcOH означает уксусную кислоту; BOC означает трет-бутоксикарбонил, защитную группу для аминогруппы, Bn означает бензил; CuI означает йодид меди; AcOCu означает ацетат меди; Pd(OH)2 означает гидроксид палладия; RT означает комнатную температуру; POCl3 означает оксихлорид фосфора, Boc2O означает Boc-ангидрид, Bn2NH означает дибензиламин, (N-Bu)4Sn означает тетра-н-бутилолово, DMAP означает N,N-диметиламинопиридин; (NH4)2CO3 означает карбонат аммония; TFA означает трифторацетат; TFAA означает трифторуксусный ангидрид; TEA означает триэтиламин; DIBAl-H означает диизобутилалюмогидрид; NIS означает N-йодсукцинимид; Pd(PPh3P)2Cl2 означает бис(трифенилфосфин)палладия хлорид; DAST означает N,N-диэтилсульфид; N-BuSn означает н-бутилолово; Pd(PPh3)4 означает тетрафенилфосфин; LDA означает диизопропиламид лития; B(i-PrO)3 означает триизопропилборат; CsF означает фторид цезия; NaH означает гидрид натрия; TMSCF3 означает триметилтрифторметилсилан; MS означает молекулярное сито; Cbz означает бензилоксикарбонил; TBDMS означает трет-бутилдиметилсилил.
[96] Соединения названы самостоятельно или с помощью программного обеспечения ChemDraw®, или же названы в соответствии с каталогом поставщиков на нынешнем рынке.
Способы синтеза
[97] Соединение согласно настоящему изобретению может быть получено при помощи серии стадий синтеза в ходе различных способов синтеза, хорошо известных специалистам в данной области. Соединения согласно настоящему изобретению можно синтезировать, применяя описанные ниже способы синтеза, или их альтернативный вариант.
[98] Предпочтительные способы включают в себя, без ограничения изложенное ниже описание.
[99] В частности, соединение формулы (I) может быть получено путем приведения в реакцию промежуточного продукта реакции, характеризующегося формулой (II), с подходящими арилборной кислотой или боратом, если LG1 представляет собой подходящую уходящую группу в виде галогена (например, хлора, брома, йода) или подобного, или с ароматическим галогенидом, если LG1 представляет собой борную кислоту или борат. Реакцию осуществляют в подходящем растворителе (таком как диоксан/вода, толуол и т.п.), что требует использования подходящего основания (такого как фторид цезия, карбонат натрия, бикарбонат натрия) и подходящего катализатора (такого как Pd(dppf)Cl2, Pd(PPh3)4 и т.п.). Согласно схеме реакции 1 реакцию предпочтительно осуществляют при температуре от 80°C до 120°C:
Схема реакции 1
[100] Все переменные определены, как в формуле (I).
[101] Промежуточное соединение формулы (II) может быть получено согласно традиционным реакциям в ходе различных способов синтеза, хорошо известных специалистам в данной области. Например, промежуточное соединение формулы (II) может быть получено согласно схеме реакции 2:
Схема реакции 2
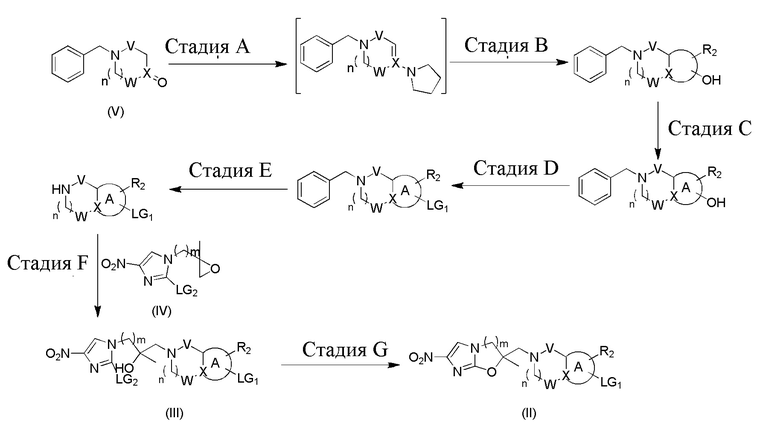
[102] Переменная LG1 представляет собой подходящую уходящую группу, такую как галоген, к примеру, хлор, бром, йод, борную кислоту или борат и т.п. Переменная LG2 представляет собой подходящую уходящую группу, такую как галоген (например, хлор, бром, йод и т.п.). Все другие переменные определены, как в формуле (I). На стадии A и стадии B схемы реакции 2 происходит удаление воды из защищенного бензилом кетона и пиррола в подходящем растворителе, таком как толуол, путем азеотропного способа при подходящей температуре, а затем циклизация при помощи акриламида или пропиоламида. На стадии C происходит ароматизация циклизированного продукта из предыдущей стадии под действием подходящего реактива, такого как жидкий бром, которую обычно необходимо осуществлять при условии нагревания. На следующей стадии D происходит реакция дегидратирующего реактива (например, оксихлорида фосфора, оксибромида фосфора и т.п.) с поступившим из предыдущей стадии продуктом при 110-130°C. На стадии E происходит удаление защитной группы для бензильной группы при атоме азота посредством гидрирования или химическими средствами, при этом растворителем обычно является метанол или этанол. На следующей стадии F обычно осуществляется реакции раскрытия цикла в полярном растворителе (например, метаноле, этаноле, изопропаноле) в присутствии основания, такого как DIPEA. На стадии G в присутствии подходящего основания (такого как гидрид натрия, ацетат натрия) и подходящего растворителя (такого как DMF или трет-бутилацетат) осуществляется реакция при 0-120°C с получением промежуточного соединения формулы (II).
[103] Очевидно, что продукт реакции можно выделить из реакционной среды в ходе реакции, упомянутой ранее и далее, и в случае необходимости дополнительно очистить способами очищения, известными специалистам в данной области, такими как экстракция и хроматография. Более конкретно, в случае продукта реакции, в котором присутствует больше одного енантиомера, соединение формулы (I) может быть выделено в виде изомеров способами разделения, известными специалистам в данной области, в частности, препаративной хроматографией, такой как препаративная HPLC, SFC или подобные.
[104] Соединение формулы (I) также можно получить путем циклизации соединения формулы (III) непосредственно под действием подходящего основания из схемы реакции 3:
Схема реакции 3
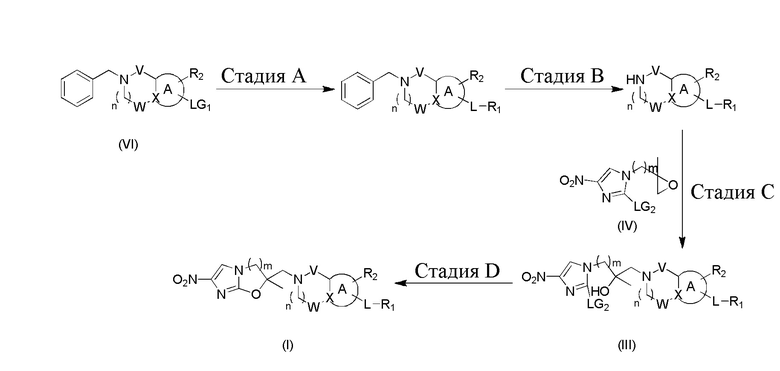
[105] Переменная LG1 представляет собой подходящую уходящую группу, такую как галоген, к примеру, хлор, бром, йод, борную кислоту или борат и т.п. Переменная LG2 представляет собой подходящую уходящую группу, такую как галоген, к примеру, хлор, бром, йод и т.п. Все другие переменные определены, как указано выше в формуле (I).
[106] На стадии A происходит реакция соединения формулы (VI) с подходящими арилборной кислотой или боратом, если LG1 представляет собой подходящую уходящую группу в виде галогена (например, хлора, брома, йода или подобного), или с ароматическим галогенидом, если LG1 представляет собой борную кислоту или борат. Для проведения реакции необходимо использовать подходящее основание (такое как фторид цезия, карбонат натрия, бикарбонат натрия), подходящий катализатор (такой как Pd(dppf)Cl2 и т.п.), при этом осуществлять ее в подходящем растворителе (таком как диоксан/вода, толуол и т.п.). Согласно схеме реакции 1 реакцию предпочтительно осуществлять при температуре от 80°C до 120°C. На стадии B происходит удаление защитной группы для бензильной группы при атоме азота посредством гидрирования или химическими средствами, такими как хлорэтилхлорформиат и метанол, при этом реакция, как правило, осуществляется при температуре от 60°C до 80°C. На стадии C для проведения реакции эпоксидированного промежуточного соединения с нуклеофилом обычно требуется подходящее основание (такое как DIPEA, ацетат натрия и т.п.), при этом она осуществляется в подходящем растворителе (таком как метанол, этанол, изопропанол, трет-бутанол) при температуре от 80 до 100°C. Следующая стадия D осуществляется в подходящем основании (таком как гидрид натрия, ацетат натрия) и подходящем растворителе (таком как DMF или трет-бутилацетат) при температуре от 0 до 120°C.
[107] Кроме того, соединение формулы (I) также можно получить по схеме реакции 4:
Схема реакции 4:
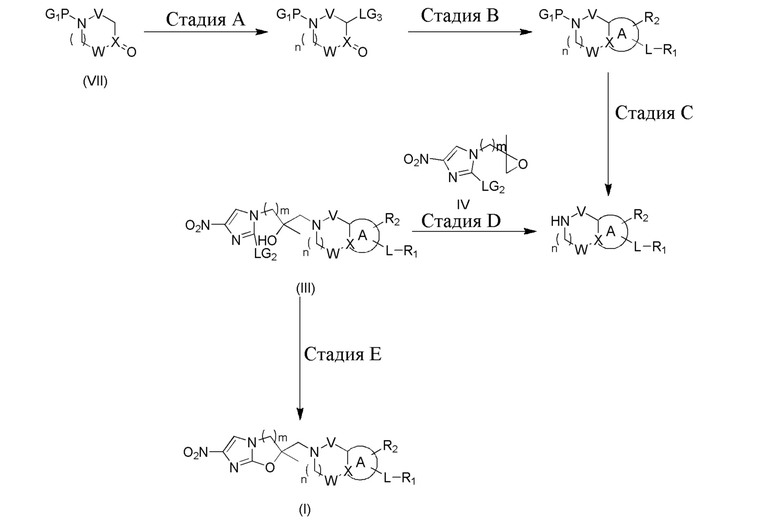
[108] X представляет собой собой C, переменная PG1 представляет собой подходящую уходящую группу, такую как трет-бутоксикарбонил, бензил, бензилоксикарбонил и подобное, LG2, LG3 представляет собой подходящую уходящую группу, такую как галоген, к примеру, хлор, бром, йод, N, N-диметиламинометилен и подобное. Все другие переменные определены, как в формуле (I).
[109] Схема реакции 4 включает в себя стадию A, которая заключается в приведении в реакцию пиперидона или пирролидона, которые имеют подходящую защитную группу при атоме азота, с N,N-диметилформамиддиметилацеталем или подходящим бромирующим реактивом (жидким бромом, бромсукцинимидом, бромидом меди, фенилтриметиламина трибромидом) в подходящем растворителе (таком как толуол, ксилол, DMF). Для проведения реакции обычно требуется более высокая температура от 50 до 140°C. На следующей стадии B осуществляется реакция полученного в результате аддукта с нуклеофилом в присутствии подходящего растворителя (такого как метанол, этанол, изопропанол, трет-бутанол, DMF) и подходящего основания (такого как триэтиламин, DIPEA или подобное). На стадии C выполняется удаление защитной группы PG1 посредством гидрирования или химическими средствами. На стадии D для проведения реакции эпоксидированного промежуточного соединения с нуклеофилом, как правило, требуется подходящее основание (такое как DIPEA, ацетат натрия и т.п.), при этом она осуществляется в подходящем растворителе (таком как метанол, этанол, изопропанол, трет-бутанол) при температуре от 80°C до 100°C. Следующая стадия E осуществляется в присутствии подходящего основания (такого как гидрид натрия, ацетат натрия) и подходящего растворителя (такого как DMF или трет-бутилацетат) при температуре от 0 до 120°C с получением соединения формулы (I).
[110] Промежуточные соединения из предыдущих схем могут быть коммерчески доступными или же их можно получить согласно общей схеме реакции, хорошо известной специалистам в данной области. Например, промежуточное соединение формулы (IV) можно получить согласно схеме реакции 5:
Схема реакции 5:
[111] Каждая из переменных LG2, LG3 в отдельности представляет собой подходящую уходящую группу, такую как галоген (например, хлор, бром, йод, метансульфонил и т.п.). Все другие переменные определены, как в формуле (I).
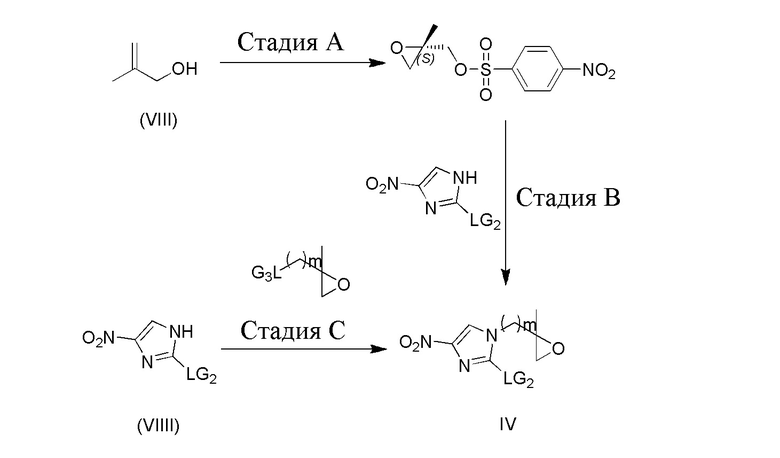
[112] Схема реакции 5 включает в себя стадию A, которая заключается в приведении аллилового спирта и пероксида кумола в реакцию окисления в присутствии соответствующего (+) диизопропилтартрата или (-) диизопропилтартрата и тетраизопропилтитаната. Реакция обычно осуществляется в подходящем растворителе, таком как дихлорметан, толуол. Для проведения реакции присоединения защитных групп к p-нитробензолсульфонилхлориду требуется подходящее основание (например, триэтиламин, диизопропилэтиламин, N,N-диметилпиридин), при этом реакция обычно осуществляется в температурном диапазоне от -20°C до 0°C. На следующей стадии B осуществляется перенос эпоксигруппы на нитроимидазол при нагревании ранее полученного промежуточного эпоксисоединения с неочищенным нитроимидазолом. Для проведения реакции требуется подходящий растворитель (такой как этанол, изопропанол, трет-бутанол, трет-бутилацетат и т.п.), подходящее основание (например, диизопропилэтиламин, карбонат калия и т.п.). Реакция обычно осуществляется в температурном диапазоне от 40°C до 100°C. На стадии C путем нагревания эпоксисоединения с уходящей группой и нитроимидазола можно также непосредственно получить оптически неактивное промежуточное соединение формулы (IV). Для проведения реакции требуется подходящий растворитель (такой как этанол, изопропанол, трет-бутиловый спирт, трет-бутилацетат и т.п.), подходящее основание (такое как диизопропилэтиламин, ацетат натрия и подобное). Кроме того, реакция обычно осуществляется в температурном диапазоне от 40°C до 100°C.
[113] Чтобы получить соединение согласно настоящему изобретению, специалистам в данной области иногда целесообразно модифицировать или выбрать стадию синтеза или схему реакции на основании существующих вариантов осуществления.
[114] Соединение, характеризующееся структурой формулы (I), также может быть получено собственно из соединения, характеризующегося структурой формулы (I), за счет превращения функциональных групп, хорошо известного в данной области.
[115] Химическую реакцию конкретного варианта осуществления согласно настоящему изобретению осуществляют в подходящем растворителе, при этом растворитель должен быть подходящим для химических превращений, а также реактивов и материалов, необходимых согласно настоящему изобретению. Чтобы получить соединение согласно настоящему изобретению, специалистам в данной области иногда целесообразно модифицировать или выбрать стадию синтеза или схему реакции на основании существующих вариантов осуществления.
[116] Важным аспектом в планировании любого пути синтеза в данной области является выбор подходящей защитной группы для реакционно-способной функциональной группы (такой как аминогруппа в настоящем изобретении). Для подготовленных практикующих специалистов «Protective Groups in Organic Synthesis, Wiley and Sons, 1991» под авторством Greene и Wuts является авторитетным источником в данной сфере. Все ссылочные материалы, цитируемые в настоящем документе, включены посредством ссылки во всей своей полноте.
[117] Соединения согласно настоящему изобретению могут быть получены различными способами синтеза, хорошо известными специалистам в данной области, включая конкретные варианты осуществления, перечисленные ниже, их варианты осуществления в сочетании с иными способами химического синтеза и равноценные заменяющие способы, известные специалистам в данной области. Предпочтительные варианты осуществления включают без ограничения варианты осуществления настоящего изобретения.
[118] Нижеследующие примеры дополнительно иллюстрируют настоящее изобретение, однако настоящее изобретение ими не ограничивается.
[119] Все растворители, используемые в настоящем изобретении, являются коммерчески доступными и могут применяться без дополнительного очищения. Реакцию, как правило, осуществляют в атмосфере инертного азота в безводном растворителе. Данные протонного ядерного магнитного резонанса регистрировали на спектрометре Bruker Avance III 400 (400 МГц) с химическим сдвигом, выраженным в единицах ppm, в слабом поле тетраметилсилана. Масс-спектр измеряли на Agilent 1200 Series plus 6110 (& 1956A). LC/MS или Shimadzu MS, включающем DAD: SPD-M20A (LC) и детектор Shimadzu Micromass 2020. Масс-спектрометр оснащен источником ионов с электрораспылением (ESI), функционирующим в положительном или отрицательном режиме.
[120] Соединения названы самостоятельно или с помощью программного обеспечения ChemDraw®, коммерчески доступные соединения названы в соответствии с каталогом поставщиков.
[121] Анализы высокоэффективной жидкостной хроматографией (HPLC) проводили с помощью системы Shimadzu LC20AB, оснащенной автоматическим пробоотборником Shimadzu SIL-20A и детектором Shimadzu DAD: SPD-M20A. Применяли колонку Xtimate C18 (заполнение насадочным материалом – 3 м, спецификация 2,1 × 300 мм). Способ 0-60AB_6 мин. включал: применение линейного градиента, начало элюирования 100% A (A представлял собой водный раствор 0,0675% TFA в воде) и окончание элюирования 60% B (B представлял собой раствор 0,0625% TFA в MeCN) в общей сложности в течение 4,2 мин., а затем элюирование 60% B в течение 1,0 мин. Затем колонку повторно уравновешивали в течение 0,8 мин. до 100:0 с общим временем прогона 6 мин. Способ 10-80AB_6 мин. включал: применение линейного градиента, начало элюирования 90% A (A представлял собой водный раствор 0,0675% TFA в воде) и окончание элюирования 80% B (B представлял собой раствор 0,0625% TFA в MeCN) в общей сложности в течение 4,2 мин., а затем элюирование 80% B в течение 1,0 мин. Затем колонку повторно уравновешивали в течение 0,8 мин. до 90:10 с общим временем прогона 6 мин. Температура колонки составляла 50°C, а скорость потока 0,8 мл/мин. Детектор на диодной матрице имел длину волны при сканировании от 200 до 400 нм.
[122] Тонкослойную хроматографию (TLC) проводили на силикагеле GF254 от Sanpont-group и пятна визуализировали посредством УФ-облучения. В некоторых случаях для визуализации пятен использовали также дополнительные способы. В таких случаях для визуализации соединения создавали пластины для TLC с йодом (получали путем добавления приблизительно 1 г I2 к 10 г силикагеля и тщательного смешивания), ванилином (получали путем растворения приблизительно 1 г ванилина в 100 мл 10% H2SO4), нингидрином (коммерчески доступна от Aldrich) или Magic Stain (получали путем тщательного смешивания (NH4)6Mo7O24•4H2O, 5 г (NH4)2Ce(IV)(NO3)6, 450 мл H2O и 50 мл концентрированной H2SO4). Флэш-хроматографию проводили с использованием 40-63 мкм (230-400 меш) силикагеля от Silicycle, применяя методики, аналогичные раскрытым в Still, W. C.; Kahn, M.; and Mitra, M. Journal of Organic Chemistry, 1978, 43, 2923-2925. Типичные растворители, используемые для флэш-хроматографии или тонкослойной хроматографии, представляли собой смесь дихлорметана/метанола, этилацетата/метанола и гексанов/этилацетата.
[123] Препаративную хроматографию проводили с помощью системы Gilson-281 Prep LC 322 с применением детектора Gilson UV/VIS-156. Применяемой колонкой была Agella Venusil ASB Prep C18, 5 мкм, 150 x 21,2 мм или Phenomenex Gemini C18, 5 мкм, 150 x 30 мм; Boston Symmetrix C18, 5 мкм, 150 x 30 мм или Phenomenex Synergi C18, 4 мкм, 150 x 30 мм. Узкие градиенты с ацетонитрилом/водой, где вода содержала 0,05% HCl, или 0,25% HCOOH, или 0,5% NH3•H2O, использовали для элюирования соединений со скоростью потока примерно 25 мл/мин. и общим временем прогона 8-15 мин.
[124] Анализы SFC проводили с помощью системы Agilent 1260 Infinity SFC с автоматическим пробоотборником Agilent 1260 и детектором Agilent DAD: 1260. Применяемой колонкой была Chiralcel OD-H 250 x 4,6 мм I.D., 5 мкм или Chiralpak AS-H 250 x 4,6 мм I.D., 5 мкм или Chiralpak AD-H 250 x 4,6 мм I.D., 5 мкм. Условия хроматографирования OD-H_5_40_2,35 мл: Chiralcel OD-H (спецификация 250 × 4,6 мм I.D., заполнение насадочным материалом 5 мкм); подвижная фаза представляла собой 40% этанол (0,05% DEA) в CO2; скорость потока составляла 2,35 мл/мин.; длина волны детектирования составляла 220 нм. Условия хроматографирования AS-H_3_40_2,35 мл: Chiralpak AS-H (спецификация 250 × 4,6 мм I.D., заполнение насадочным материалом 5 мкм); подвижная фаза представляла собой 40% метанол (0,05% DEA) в CO2; скорость потока составляла 2,35 мл/мин.; длина волны детектирования составляла 220 нм. Условия хроматографирования OD-H_3_40_2,35 мл: Chiralcel OD-H (спецификация 250 × 4,6 мм I.D., заполнение насадочным материалом 5 мкм); подвижная фаза представляла собой 40% метанол (0,05% DEA) в CO2; скорость потока составляла 2,35 мл/мин.; длина волны детектирования составляла 220 нм. Условия хроматографирования AD-H_2_50_2,35 мл: Chiralpak AS-H (спецификация 250 × 4,6 мм I.D., заполнение насадочным материалом 5 мкм); подвижная фаза представляла собой 50% метанол (0,1% MEA) в CO2; скорость потока составляла 2,35 мл/мин.; длина волны детектирования составляла 220 нм.
[125] Анализ посредством препаративной SFC проводили с помощью системы Waters Thar 80 Pre-SFC с применением УФ-детектора Gilson. Применяемой колонкой была Chiralcel OD-H 250 x 4,6 мм I.D., 5 мкм или Chiralpak AD-H 250 x 4,6 мм I.D., 5 мкм. Для элюирования соединения использовали узкие градиенты с этанолом или метанолом в CO2, где этанол или метанол содержали 0,05% NH3•H2O, или 0,05% DEA, или 0,1% MEA, при скорости потока от 40 до 80 мл/мин. и общем времени прогона 20-30 мин.
[126] Нижеследующие примеры дополнительно иллюстрируют настоящее изобретение, однако настоящее изобретение ими не ограничивается.
[127] Абсолютные пространственные конфигурации атомов углерода в хиральном центре конкретных соединений или промежуточных соединений или же конфигурации двойных связей экспериментально не исследовали. В данном случае изомеры, выделенные первыми посредством хиральной препаративной хроматографии, обозначены «A», а последующие обозначены «B». Любой средний специалист в данной области может отличить изомеры A и B при помощи конкретных методов, таких как ЯМР. Данный способ является наиболее подходящим способом определения пространственной конфигурации.
[128] Соединения нижеизложенных примеров получали, разделяли и описывали согласно предусмотренным в настоящем документе способам. Нижеперечисленные примеры являются всего лишь иллюстративными согласно объему настоящего изобретения и не предназначены быть исчерпывающими. В настоящем документе подробно описано настоящее изобретение, где также раскрыты его варианты осуществления, причем специалистам в данной области техники будут очевидны различные изменения и модификации в отношении конкретных вариантов осуществления согласно настоящему изобретению без отступления от сущности и объема настоящего изобретения.
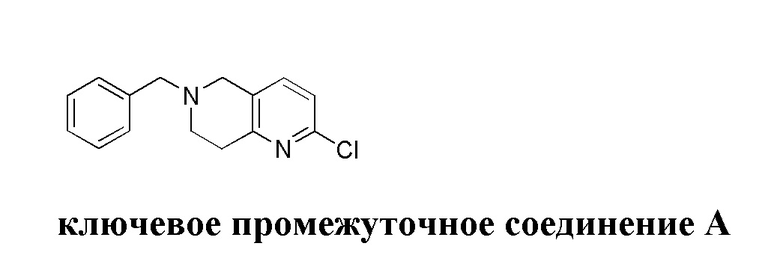
[129] Получение ключевых промежуточных соединений A, B и C.
[130] (S)-2-((2-Хлор-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
[131] Стадия 1.
[132] 1-Бензил-4-пирролидин-1-ил-3,6-дигидро-2H-пиридин
[133] Пирролидин (33,82 г, 475,56 ммоль, 1,00 экв.) добавляли к раствору 1-бензил-4-она (90,00 г, 475,56 ммоль, 1,00 экв.) в толуоле (70,00 мл) при 30°C в атмосфере газообразного азота. Смесь перемешивали при 130°C в течение 12 часов. Затем смесь концентрировали в вакууме при 45°C с получением 1-бензил-4-пирролидин-1-ил-3,6-дигидро-2H-пиридина (110,00 г, неочищенный) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительного очищения.
[134] Стадия 2.
[135] 6-Бензил-1,3,4,5,7,8-гексагидро-1,6-нафтиридин-2-он
[136] Пропан-2-енамид (52,35 г, 736,51 ммоль, 1,50 экв.) добавляли к раствору 1-бензил-4-пирролидин-1-ил-3,6-дигидро-2H-пиридина (119,00 г, 491,01 ммоль, 1,00 экв.) в толуоле (80,00 мл) при 30°C. Затем смесь перемешивали при 130°C в течение 12 часов, смесь охлаждали до 30°C и концентрировали при пониженном давлении при 45°C. Остаток промывали петролейным эфиром (100 мл), фильтровали и осадок концентрировали in vacuo с получением 6-бензил-1,3,4,5,7,8-гексагидро-1,6-нафтиридин-2-она (76,00 г, 313,63 ммоль, выход 63,88%) в виде желтого твердого вещества.
[137] Стадия 3.
[138] 6-Бензил-1,5,7,8-тетрагидро-1,6-нафтиридин-2-он
[139] 6-Бензил-1,3,4,5,7,8-гексагидро-1,6-нафтиридин-2-он (35,00 г, 144,44 ммоль, 1,00 экв.) растворяли в уксусной кислоте (200,00 мл). Смесь жидкого брома (23,08 г, 144,44 ммоль, 1,00 экв.) и уксусной кислоты (200,00 мл) добавляли к раствору при 0°C в атмосфере газообразного азота. Смесь перемешивали при 0°C в течение 30 минут, а затем нагревали до 110°C и перемешивали в течение 12 часов. Смесь охлаждали до 30°C и концентрировали при пониженном давлении при 45°C. Остаток выливали в водный раствор карбоната натрия (70 мл) и перемешивали в течение 10 минут. Твердое вещество отфильтровывали, промывали петролейным эфиром (30 мл) и сушили in vacuo с получением 6-бензил-1,5,7,8-тетрагидро-1,6-нафтиридин-2-она (35,00 г, неочищенный) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 241 (M + 1).
[140] Стадия 4.
[141] 6-Бензил-2-хлор-7,8-дигидро-5H-1,6-нафтиридин
[142] 6-Бензил-1,5,7,8-тетрагидро-1,6-нафтиридин-2-он (35,00 г, 145,65 ммоль, 1,00 экв.) порциями добавляли к оксихлориду фосфора (178,62 г , 1,16 моль, 8,00 экв.) при 30°C, после добавления смесь перемешивали в течение 10 минут, а затем смесь нагревали до 130°C в течение 12 часов. Смесь охлаждали и отгоняли оксихлорид фосфора при 50°C при пониженном давлении. Остаток разбавляли дихлорметаном и выливали в воду (500 мл), смесь подщелачивали насыщенным раствором карбоната натрия (500 мл). Водную фазу экстрагировали дихлорметаном (500 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (30 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 15/1, 7/1) с получением 6-бензил-2-хлор-7,8-дигидро-5H-1,6-нафтиридина (ключевого промежуточного соединения A) (17,00 г, 65,70 ммоль, выход 45,11%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 7,41-7,30 (m, 5H), 7,26 (d, J = 8,0 Гц, 1H), 7,10 (d, J = 8,0 Гц, 1H), 3,73 (s, 2H), 3,61 (s, 2H), 3,07-3,01 (m, 2H), 2,88-2,83 (m, 2H).
[143] Стадия 5.
[144] 2-Хлор-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорид
[145] 6-Бензил-2-хлор-7,8-дигидро-5H-1,6-нафтиридин (15,00 г, 57,97 ммоль, 1,00 экв.) растворяли в дихлорэтане (80,00 мл), добавляли 1-хлорэтилхлорформиат (12,43 г, 86,96 ммоль, 1,50 экв.) при 0°C в атмосфере газообразного азота. Смесь перемешивали при 0°C в течение 0,5 ч, а затем нагревали до 85°C и перемешивали в течение 12 часов. Смесь концентрировали, а затем растворяли в метаноле (30,00 мл), и далее смесь перемешивали при 80°C в течение дополнительных 2 часов. Смесь охлаждали и фильтровали с получением 2-хлор-5,6,7,8-тетрагидро-1,6-нафтиридина (6,30 г, неочищенный) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительного очищения.
[146] Стадия 6.
[147] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-3-(2-хлор-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-метилпропан-2-ол
[148] 2-Хлор-5,6,7,8-тетрагидро-1,6-нафтиридин (11,00 г, 65,24 ммоль, 1,00 экв.) и 2-хлор-1-[[(2R)-2-метилоксиран-2-ил] метил]-4-нитроимидазол (14,20 г, 65,24 ммоль, 1,00 экв.) растворяли в этаноле (150,00 мл), добавляли к раствору N, N-диизопропилэтиламина (21,08 г, 163,10 ммоль, 2,50 экв.) при 15°C в атмосфере газообразного азота. Смесь перемешивали при 85°C в течение 12 часов. Затем смесь охлаждали до 15°C и концентрировали при пониженном давлении при 60°C. К остатку добавляли воду (30 мл). Смесь экстрагировали этилацетатом (200 мл × 4) и объединенные органические фазы промывали насыщенным солевым раствором (100 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 20/1, 1/2) с получением (S)-1-(2-хлор-7,8-дигидро-5H-1,6-нафтиридин-6-ил)-3-(2-хлор-4-нитроимидазол-1-ил)-2-метилпропан-2-ола (20,00 г, 51,78 ммоль, выход 79,37%) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 386 (M + 1).
[149] Стадия 7.
[150] (S)-2-((2-Хлор-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол

[151] (S)-1-(2-Хлор-7,8-дигидро-5H-1,6-нафтиридин-6-ил)-3-(2-хлор-4-нитроимидазол-1-ил)-2-метилпропан-2-ол (9,00 г, 23,30 ммоль, 1,00 экв.) растворяли в DMF (80,00 мл) и NaH (1,12 г, 46,60 ммоль, 2,00 экв.) добавляли при -20°C в атмосфере газообразного азота. Смесь перемешивали при -20°C в течение 10 минут, а затем нагревали до -5°C и перемешивали в течение 10 минут. Затем смесь нагревали до 15°C и перемешивали в течение еще 10 минут. Смесь охлаждали до 0°C, а затем добавляли по каплям к водному раствору соляной кислоты (0,25 моль, 400 мл), и далее смесь подщелачивали водным раствором бикарбоната натрия до pH = 7-8. Осадок фильтровали и сушили с получением (S)-2-((2-хлор-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола (ключевого промежуточного соединения B) (6,70 г, 19,16 ммоль, выход 82,22%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ: 7,52 (s, 1H), 7,27 (s, 1H), 7,12 (d, J = 8,0 Гц, 1H), 4,39 (d, J = 9,6 Гц, 1H), 3,96 (d, J = 9,6 Гц, 1H), 3,82 (q, J = 15,3 Гц, 2H), 3,14-3,03 (m, 2H), 3,02-2,85 (m, 3H), 2,80 (d, J = 14,8 Гц, 1H), 1,68 (s, 3H). LCMS (ESI) масса/заряд: 350 (M+1).
[152] (R)-2-Хлор-1-((2-метилоксиран-2-ил)метил)-4-нитро-1H-имидазол

[153] Стадия 1.
[154] (S)-(2-Метилоксиран-2-ил)метил-4-нитробензолсульфонат
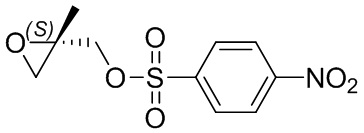
[155] 2-Метилпроп-2-ен-1-ол (15,00 г, 208,02 ммоль, 1,00 экв.) и (2S,3S)-диизопропилтартрат (2,92 г, 12,48 ммоль, 0,06 экв.)) добавляли к суспензии молекулярных сит 4A (15,00 г) в DCM (300,00 мл) при 10-30°C в атмосфере газообразного азота. Смесь охлаждали до температуры от -10 до 0°C и к реакционной смеси добавляли по каплям тетраизопропоксид титана (3,55 г, 12,48 ммоль, 0,06 экв.) с поддержанием температуры реакции на уровне от -15 до -5°C, и далее смесь перемешивали в течение 0,5 часа. В смесь добавляли по каплям (1-гидроперокси-1-метилэтил)бензол (63,32 г, 416,03 ммоль, 2,00 экв.), температуру поддерживали на уровне от -15 до -5°C. Через 3 часа к реакционной смеси добавляли триметилфосфит (25,83 г, 208,02 ммоль, 1,0 экв.) при температуре от -15 до -5°C. Через 20 минут к реакционной смеси добавляли 4-нитробензолсульфонилхлорид (46,10 г, 208,02 ммоль, 1,00 экв.), DMAP (1,27 г, 10,40 ммоль, 0,05 экв.). А затем добавляли по каплям триэтиламин (25,81 г, 208,02 ммоль, 1,0 экв.). После перемешивания смеси температуру постепенно повышали до 20°C. Через 1 час к реакционной смеси добавляли воду (100 мл), перемешивали в течение 20 минут и фильтровали. Фильтрат экстрагировали дихлорметаном (50 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (30 мл × 1), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (высота колонки: 250 мм, диаметр: 100 мм, силикагель 100-200 меш, петролейный эфир/этилацетат = 30/1, 5/1) с получением (S)-(2-метилоксиран-2-ил)метил-4-нитробензолсульфоната (24,00 г, 87,83 ммоль, выход 42,22%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 8,47-8,37 (m, 2H), 8,17-8,11 (m, 2H), 4,28 (d, J=10,9 Гц, 1H), 4,04 (d, J=10,9 Гц, 1H), 2,77-2,65 (m, 2H), 1,43-1,35 (m, 3H).
[156] Стадия 2.
[157] (R)-2-Хлор-1-((2-метилоксиран-2-ил)метил)-4-нитро-1H-имидазол

[158] Карбонат калия (56,21 г, 406,71 ммоль, 3,00 экв.) добавляли к смеси 2-хлор-4-нитро-1H-имидазола (20,00 г, 135,57 ммоль, 1,00 экв.) и (S)-(2-метилоксиран-2-ил)метил-4-нитробензолсульфоната (37,05 г, 135,57 ммоль, 1,00 экв.) в DMF (300 мл), реакционную смесь дегазировали и заменяли 3 раза азотом, затем смесь перемешивали при 60°C в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли, нейтрализовали насыщенным раствором карбоната натрия (200 мл). И полученную в результате смесь экстрагировали этилацетатом (50 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (50 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 5/1, 2/1) с получением (R)-2-хлор-1-((2-метилоксиран-2-ил)метил)-4-нитро-1H-имидазола (ключевого промежуточного соединения C) (17,86 г, 82,07 ммоль, выход 60,54%) в виде желтого масла. LCMS (ESI) масса/заряд: 218 (M + 1).
[159] Вариант осуществления 1
[160] 2-Метил-6-нитро-2-((2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
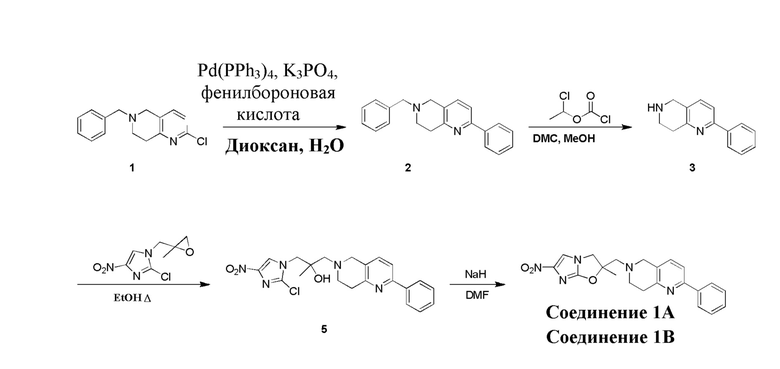
[161] Стадия 1.
[162] 6-Бензил-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин
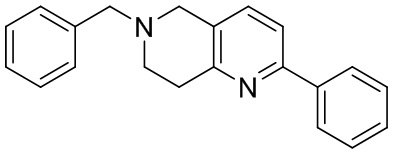
[163] Фосфат калия (2,23 г, 10,51 ммоль), фенилбороновую кислоту (1,03 г, 8,45 ммоль) и Pd(PPh3)4 (734,00 мг, 635,19 мкмоль) добавляли к смешанному раствору ключевого промежуточного соединения A (1,10 г, 4,25 ммоль) в диоксане/воде (11 мл, 10/1). Смесь перемешивали при 130°C в течение 3 часов. Смесь фильтровали и фильтрат концентрировали, а остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 10/0, 10/1) с получением 6-бензил-2-фенил-7,8-дигидро-5H-1,6-нафтиридина (750,00 мг, 58,75%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 7,96 (d, J = 7,2 Гц, 2H), 7,53-7,36 (m, 10H), 3,76 (s, 2H), 3,69 (s, 2H), 3,16 (t, J = 5,9 Гц, 2H), 2,93 (t, J = 5,9 Гц, 2H). LCMS (ESI) масса/заряд: 301 (M+1). LCMS (ESI) масса/заряд: 301 (M+1).
[164] Стадия 2.
[165] 2-Фенил-5,6,7,8-тетрагидро-1,6-нафтиридин
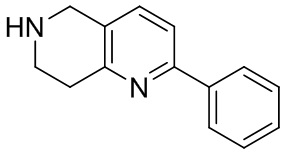
[166] 1-Хлорэтилхлорформиат (232,02 мг, 1,62 ммоль) добавляли к раствору 6-бензил-2-фенил-7,8-дигидро-5H-1,6-нафтиридина (375,00 мг, 1,25 ммоль) в дихлорэтане (15 мл) при 0°C. Смесь перемешивали при 0°C в течение 15 минут, а затем нагревали до 85°C и перемешивали в течение 12 часов. Смесь концентрировали при пониженном давлении и к остатку добавляли метанол (15 мл), а затем смесь перемешивали при 60°C в течение 1 часа. Смесь концентрировали с получением 2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорида (340,00 мг, неочищенный) в виде белого твердого вещества. LCMS (ESI) масса/заряд: 211 (M + 1).
[167] Стадия 3.
[168] 1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-2-метил-3-(2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)пропан-2-ол
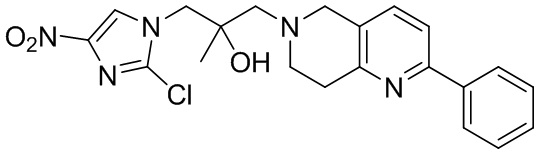
[169] 2-Хлор-1-((2-метилоксиран-2-ил)метил)-4-нитро-1H-имидазол (672,69 мг, 3,09 ммоль) добавляли к раствору 2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорида (500,00 мг, 2,38 ммоль) в этаноле (5 мл), смесь перемешивали при 70°C в течение 6 часов. Смесь концентрировали и остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 20/1, 2/1) с получением 1-(2-хлор-4-нитро-1H-имидазол-1-ил)-2-метил-3-(2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)пропан-2-ола (600,00 мг, неочищенный) в виде желтого масла. LCMS (ESI) масса/заряд: 428 (M + 1).
[170] Стадия 4.
[171] 2-Метил-6-нитро-2-((2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол

[172] NaH (67,20 мг, 2,80 ммоль) добавляли к раствору 1-(2-хлор-4-нитро-1H-имидазол-1-ил)-2-метил-3-(2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)пропан-2-ола (600,00 мг, 1,40 ммоль) в DMF (10 мл) при 0°C в атмосфере газообразного азота. Смесь перемешивали при 0°C в течение 1 часа. Смесь добавляли к перемешиваемой воде (40 мл) и осажденное твердое вещество отфильтровывали и перекристаллизовывали из этилацетата (50 мл) с получением белого твердого вещества, которое расщепляли посредством хиральной SFC (Chiralpak AD 250 × 30 мм I.D. 5 мкм, CO2 в сверхкритическом состоянии / EtOH (0,2% NH3H2O) =60/40, 80 мл/мин., 220 нм) с получением двух хиральных изомеров. Первое соединение 2-метил-6-нитро-2-((2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол назвали соединением 1A (40,60 мг, 7,19%), а полученное в результате второе соединение 2-метил-6-нитро-2-((2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол назвали соединением 1B (48,10 мг, 8,48%). Спектры ядерного магнитного резонанса были одинаковыми: 1H ЯМР (400 МГц, CDCl3): δ 7,94 (d, J = 7,2 Гц, 2H), 7,53-7,36 (m, 6H), 4,45 (d, J = 10,0 Гц, 1H), 3,97-3,86 (m, 3H), 3,16-2,99 (m, 5H), 2,81 (d, J = 14,8 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 392 (M+1).
[173] Вариант осуществления 2
[174] 2-Метил-6-нитро-2-((2-(4-(трифторметокси)фенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
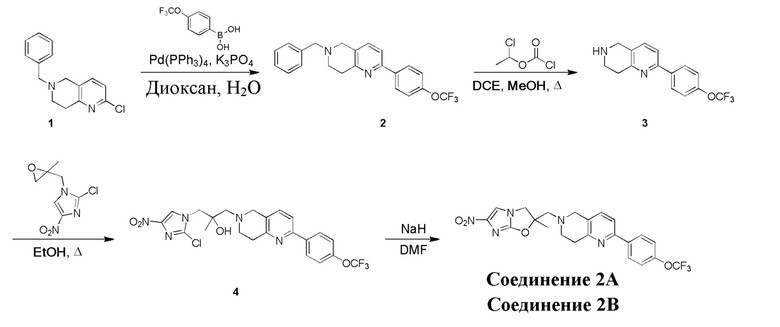
[175] Стадия 1.
[176] 6-Бензил-2-(4-(трифторметокси)фенил)-5,6,7,8-тетрагидро-1,6-нафтиридин
[177] Фосфат калия (2,46 г, 11,60 ммоль, 2,50 экв.) добавляли к смешанному раствору ключевого промежуточного соединения A (1,20 г, 4,64 ммоль, 1,00 экв.) в диоксане (5 мл) и воды (0,5 мл), добавляли Pd(PPh3)4 (536,18 мг, 464,00 мкмоль, 0,10 экв.), (4-(трифторметокси)фенил)бороновую кислоту (1,43 г, 6,96 ммоль, 1,50 экв.) при 0°C в атмосфере газообразного азота. Смесь перемешивали при 120°C в течение 3 часов. Смесь охлаждали и концентрировали при пониженном давлении при 50°C. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 50/1, 15/1) с получением 6-бензил-2-(4-(трифторметокси)фенил)-5,6,7,8-тетрагидро-1,6-нафтиридина (1,30 г, 3,38 ммоль, выход 72,89%) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 385 (M + 1).
[178] Стадия 2.
[179] 2-(4-(Трифторметокси)фенил)-5,6,7,8-тетрагидро-1,6-нафтиридин
[180] 1-Хлорэтилхлорформиат (725,28 мг, 5,07 ммоль, 1,50 экв.) добавляли к раствору 6-бензил-2-(4-(трифторметокси)фенил)-5,6,7,8-тетрагидро-1,6-нафтиридина (1,30 г, 3,38 ммоль) в дихлорэтане (20 мл) при 0°C в атмосфере газообразного азота. Смесь перемешивали при 15°C в течение 0,5 часа, а затем нагревали до 80°C в течение 12 часов. В смесь добавляли метанол (5 мл) и перемешивали ее в течение еще 4 часов, а затем охлаждали. Смесь концентрировали при пониженном давлении при 50°C. Остаток очищали посредством хроматографии на силикагеле (дихлорметан/метанол = 50/1, 1/10) с получением 2-(4-(трифторметокси)фенил)-5,6,7,8-тетрагидро-1,6-нафтиридина (500,00 мг, 1,70 ммоль, выход 50,27%) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 295 (M + 1).
[181] Стадия 3.
[182] 1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-2-метил-3-(2-(4-(трифторметокси)фенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)пропан-2-ол
[183] DIPEA (74,00 мг, 572,58 мкмоль, 0,42 экв.) добавляли к раствору 2-(4-(трифторметокси)фенил)-5,6,7,8-тетрагидро-1,6-нафтиридина (400,00 мг, 1,36 ммоль, 1,00 экв.) и 2-хлор-1-[(2-метилоксиран-2-ил)метил]-4-нитроимидазола (591,90 мг, 2,72 ммоль, 2,00 экв.) в этаноле (10 мл). Смесь перемешивали при 80°C в течение 12 часов. Смесь охлаждали и концентрировали при пониженном давлении при 50°C. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 15/1, 1/1) с получением 1-(2-хлор-4-нитро-1H-имидазол-1-ил)-2-метил-3-(2-(4-(трифторметокси)фенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)пропан-2-ола (500,00 мг, неочищенный) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 512 (M + 1).
[184] Стадия 4.
[185] 2-Метил-6-нитро-2-((2-(4-(трифторметокси)фенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
[186] NaH (28,13 мг, 1,17 ммоль, 2,00 экв.) добавляли к раствору 1-(2-хлор-4-нитро-1H-имидазол-1-ил)-2-метил-3-(2-(4-(трифторметокси)фенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)пропан-2-ола (300,00 мг, 586,07 мкмоль, 1,00 экв.) в DMF (5,00 мл). Смесь перемешивали при 0°C в течение 0,5 часа, и цвет раствора становился красным. Смесь добавляли к воде (10 мл) при 0°C и водную фазу экстрагировали этилацетатом (50 мл × 4). Объединенные органические фазы промывали насыщенным солевым раствором (20 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством препаративной хроматографии (GX-D; Boston Symmetrix C18 ODS-R 150 *30 мм*5 мкм; ацетонитрил 24% - 54%; вода (0,225% NH4OH); 25 мл/мин.) с получением смеси двух изомеров (90 мг), которые разделяли посредством хиральной SFC (Chiralpak AD 250×30 мм I.D. 5 мкм, CO2 в сверхкритическом состоянии / EtOH (0,2% NH3H2O) =60/40, 70 мл/мин., 220 нм) с получением двух хиральных изомеров; первое полученное соединение 2-метил-6-нитро-2-((2-(4-(трифторметокси)фенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол назвали соединением 2A (48,20 мг, 97,33 мкмоль, выход 16,61%, чистота 96%), а второе полученное соединение 2-метил-6-нитро-2-((2-(4-(трифторметокси)фенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол назвали соединением 2B (33,40 мг, 67,02 мкмоль, выход 11,44%, чистота 95,4%). Спектры ядерного магнитного резонанса были одинаковыми: 1H ЯМР (400 МГц, CDCl3): δ 7,98 (d, J = 8,8 Гц, 2H), 7,56-7,45 (m, 2H), 7,42-7,36 (m, 1H), 7,30 (d, J = 8,4 Гц, 2H), 4,43 (d, J = 9,6 Гц, 1H), 4,01-3,78 (m, 3H), 3,21-2,92 (m, 5H), 2,81 (d, J = 14,8 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 476 (M+1).
[187] Вариант осуществления 3
[188] (S)-2-Метил-6-нитро-2-((2-(3-(трифторметокси)фенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
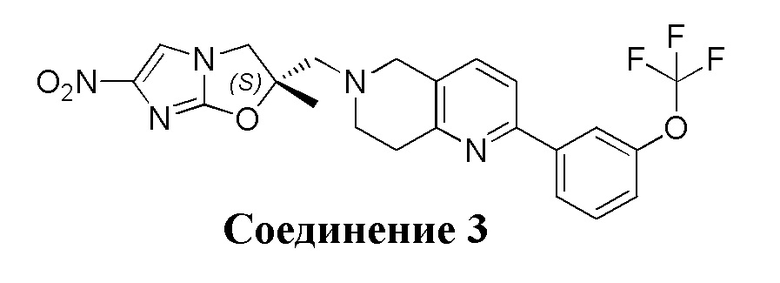
[189] Ключевое промежуточное соединение B (120,00 мг, 343,08 мкмоль, 1,00 экв.) и [3-(трифторметокси)фенил]бороновую кислоту (70,65 мг, 343,08 мкмоль, 1,00 экв.) растворяли в диоксане (5,00 мл) и добавляли к раствору воду (500,00 мкл), Pd(dppf)Cl2 (25,10 мг, 34,31 мкмоль, 0,10 экв.), карбонат натрия (90,91 мг, 857,70 мкмоль, 2,5 экв.) при 30°C в атмосфере газообразного азота. Смесь перемешивали при 110°C в течение 12 часов. После завершения реакции смесь охлаждали до 30°C и концентрировали при пониженном давлении при 45°C. Остаток выливали в воду (10 мл) и перемешивали в течение 5 минут. Водную фазу экстрагировали этилацетатом (30 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (10 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством препаративной тонкослойной хроматографии (петролейный эфир/этилацетат = 1/2,5) и препаративной хроматографии (GX-D; Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; ацетонитрил 24% - 54%; вода (0,225% муравьиная кислота) 25 мл/мин.) с получением (S)-2-метил-6-нитро-2-((2-(3-(трифторметокси)фенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазола – соединения 3 (12,00 мг, 24,62 мкмоль, выход 7,18%, чистота 97,54%). 1H ЯМР (400 МГц, CDCl3): δ 7,89-7,87 (d, J = 8,03 Гц, 1H), 7,84 (s, 1H), 7,52 (s, 1H), 7,51-7,46 (m, 2H), 7,41 (d, J = 8,16 Гц, 1H), 7,27 (d, J = 8,16 Гц, 1H), 4,44 (d, J = 9,66 Гц, 1H), 3,97 (d, J = 9,66 Гц, 1H), 3,95-3,84 (m, 2H), 3,18-3,01 (m, 5H), 2,82 (d, J = 14,81 Гц, 1H), 1,69 (s, 3H), LCMS (ESI) масса/заряд: 476 (M+1).
[190] Вариант осуществления 4
[191] (S)-2-Метил-6-нитро-2-((2-(2-(трифторметокси)фенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
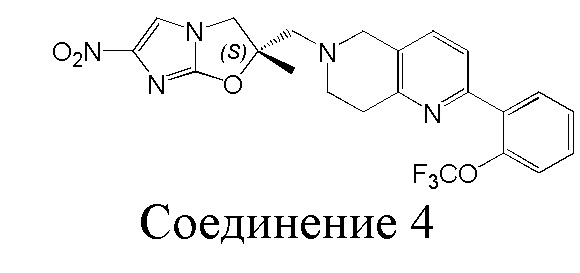
[192] Ключевое промежуточное соединение B (100,00 мг, 285,90 мкмоль, 1,00 экв.) и (2-(трифторметокси)фенил)бороновую кислоту (70,65 мг, 343,08 мкл, 1,20 экв.) растворяли в диоксане (5,00 мл) и воде (500,00 мкл). Добавляли к смеси карбонат натрия (60,61 мг, 571,80 мкмоль, 2,00 экв.), Pd(dppf)Cl2 (20,92 мг, 28,59 мкмоль, 0,10 экв.) при 15°C в атмосфере газообразного азота. Смешанный раствор перемешивали при 120°C в течение 12 часов. После завершения реакции смешанный раствор охлаждали до 15°C, фильтровали и фильтрат концентрировали in vacuo, Остаток очищали посредством препаративной хроматографии (GX-D; Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; ацетонитрил 24% - 54%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-метил-6-нитро-2-((2-(2-(трифторметокси)фенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазола – соединения 4 (15,00 мг, 29,97 мкмоль, выход 10,48%, чистота 95%). 1H ЯМР (400 МГц, CDCl3): δ 7,77 (d, J = 2,8 Гц, 1H), 7,55 (s, 1H), 7,49-7,31 (m, 5H), 4,45 (d, J = 9,6 Гц, 1H), 4,01-3,82 (m, 3H), 3,23-2,93 (m, 5H), 2,83 (d, J = 14,8 Гц, 1H), 1,70 (s, 3H). LCMS (ESI) масса/заряд: 476 (M+1).
[193] Вариант осуществления 5
[194] (S)-2-((2-(2-Фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
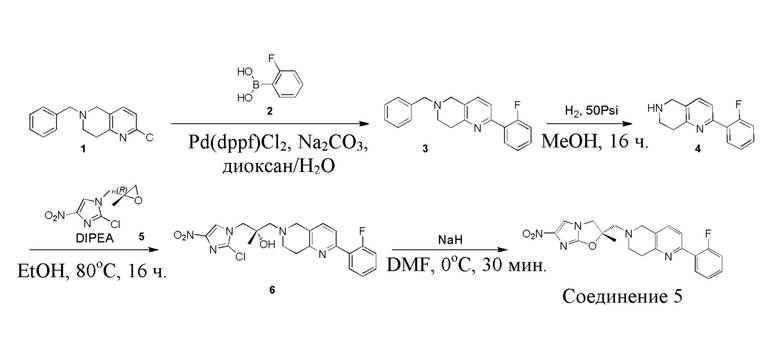
[195] Стадия 1.
[196] 6-Бензил-2-(2-фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридин
[197] Ключевое промежуточное соединение A (1,50 г, 5,80 ммоль, 1,00 экв.) и (2-фторфенил)бороновую кислоту (973,84 мг, 6,96 ммоль, 1,20 экв.) растворяли в диоксане (20,00 мл) и воде (5,00 мл), добавляли к смеси Pd(dppf)Cl2 (424,39 мг, 580,00 мкмоль, 0,10 экв.) и карбонат натрия (1,23 г, 11,60 ммоль, 2,00 экв.) в атмосфере газообразного азота. Смешанный раствор затем перемешивали при 100°C в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении для удаления растворителя, остаток разбавляли водой (20 мл) и экстрагировали этилацетатом (10 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (10 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 6-бензил-2-(2-фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридина (1,39 г, 4,37 ммоль, выход 75,27%) в виде коричневого твердого вещества.
[198] Стадия 2.
[199] 2-(2-Фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридин
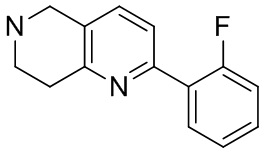
[200] 6-Бензил-2-(2-фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридин (1,39 г, 4,37 ммоль, 1,00 экв.) растворяли в метаноле (20,00 мл), добавляли Pd(OH)2/C (10%, 139 мг) в атмосфере газообразного азота. В смешанном растворе три раза производили замену на водород. Смешанный раствор нагревали до 50°C и перемешивали в течение 12 часов под H2 (50 фунтов/кв. дюйм). Реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного 2-(2-фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридина (1,05 г, неочищенный). При этом неочищенный продукт использовали на следующей стадии без дополнительного очищения.
[201] Стадия 3.
[202] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(2-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-метилпропан-2-ол
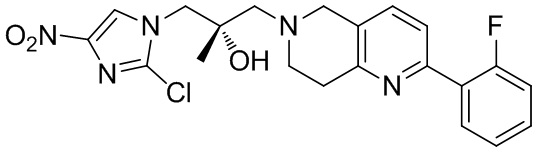
[203] 2-(2-Фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридин (300,00 мг, 1,31 ммоль, 1,00 экв.), (R)-2-хлор-1-((2-метилоксиран-2-ил)метил)-4-нитро-1H-имидазол (286,00 мг, 1,31 ммоль, 1,00 экв.) и DIPEA (507,91 мг, 3,93 ммоль, 3,00 экв.) растворяли в этаноле (20,00 мл). В смешанном растворе три раза производили замену на азот. Смешанный раствор затем перемешивали при 80°C в течение 12 часов в атмосфере газообразного азота. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток очищали посредством хроматографии на силикагеле (диоксид кремния, петролейный эфир/этилацетат = 5/1, 1/1) с получением (S)-1-(2-хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(2-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-метилпропан-2-ола (300,00 мг, 672,84 мкмоль, выход 51,36%) в виде желтого твердого вещества.
[204] Стадия 4.
[205] (S)-2-((2-(2-Фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
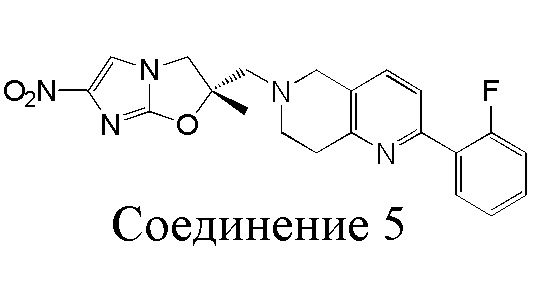
[206] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(2-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-метилпропан-2-ол (300,00 мг, 672,84 мкмоль, 1,00 экв.) растворяли в DMF (5,00 мл). NaH (32,30 мг, 807,41 мкмоль, 1,20 экв.) добавляли к смешанному раствору при 0°C и перемешивали при данной температуре в течение 30 минут. К реакционной смеси добавляли насыщенный раствор хлорида аммония (20 мл) при 0°C, а затем реакционную смесь разбавляли 20 мл воды и экстрагировали DCM (10 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (10 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством препаративной хроматографии (GX-E; Innoval C18 150*30 мм*5 мкм; ацетонитрил 12% - 42%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-((2-(2-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола –соединения 5 (101,80 мг, 248,65 мкмоль, выход 36,96%). 1H ЯМР (400 МГц, CDCl3): δ 7,94-7,87 (m, 1H), 7,57-7,54 (m, 1H), 7,53 (s, 1H), 7,42-7,33 (m, 2H), 7,28-7,23 (m, 1H), 7,19-7,11 (m, 1H), 4,44 (d, J = 8,0 Гц, 1H), 3,96 (d, J = 8,0 Гц, 1H), 3,92-3,83 (m, 2H), 3,22-3,08 (m, 2H), 3,08-2,94 (m, 3H), 2,82 (d, J = 12,0 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 409 (M+1).
[207] Вариант осуществления 6
[208] (S)-2-((2-(3-Фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
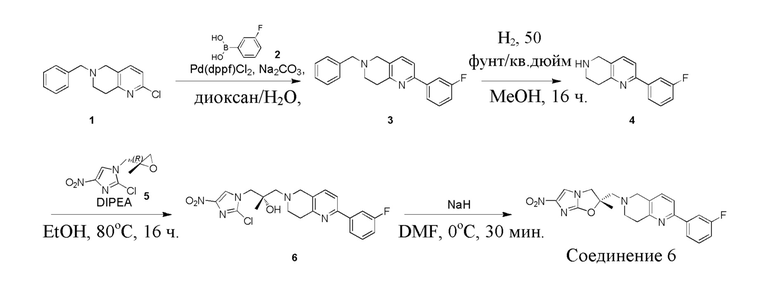
[209] Стадия 1.
[210] 6-Бензил-2-(3-фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридин
[211] Ключевое промежуточное соединение A (1,50 г, 5,80 ммоль, 1,00 экв.), (3-фторфенил)бороновую кислоту (973,36 мг, 6,96 моль, 1,20 экв.) растворяли в диоксане (20,00 мл) и воде (5,00 мл), добавляли к смешанному раствору Pd(dppf)Cl2 (424,17 мг, 579,71 мкмоль, 0,10 экв.) и карбонат натрия (1,23 г, 11,59 ммоль, 2,00 экв.) в атмосфере газообразного азота. Затем смесь перемешивали при 100°C в течение 12 часов в атмосфере газообразного азота. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли водой (20 мл) и экстрагировали этилацетатом (10 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (10 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 6-бензил-2-(3-фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридина (1,40 г, 4,40 ммоль, выход 75,81%) в виде коричневого твердого вещества.
[212] Стадия 2.
[213] 2-(3-Фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридин
[214] 6-Бензил-2-(3-фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридин (1,40 г, 4,40 ммоль, 1,00 экв.) растворяли в метаноле (20,00 мл). Добавляли Pd(OH)2/C (10%, 140 мг) в атмосфере газообразного азота. В смешанном растворе три раза производили замену на водород. Смесь нагревали до 50°C под H2 (50 фунтов/кв. дюйм) в течение 12 часов. Смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного 2-(3-фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридина (1,10 г, неочищенный). Неочищенный продукт использовали непосредственно на следующей стадии. При этом неочищенный продукт использовали на следующей стадии без дополнительного очищения.
[215] Стадия 3.
[216] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(3-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-метилпропан-2-ол
[217] 2-(3-Фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридин (300,00 мг, 1,31 ммоль, 1,00 экв.), (R)-2-хлор-1-((2-метилоксиран-2-ил)метил)-4-нитро-1H-имидазол (286,00 мг, 1,31 ммоль, 1,00 экв.), DIPEA (507,91 мг, 3,93 ммоль, 3,00 экв.) растворяли в этаноле (20,00 мл). Смесь перемешивали при 80°C в течение 12 часов в атмосфере газообразного азота. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (диоксид кремния, петролейный эфир/этилацетат = 5/1, 1/1) с получением (S)-1-(2-хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(3-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-метилпропан-2-ола (300,00 мг, 672,84 мкмоль, выход 51,36%) в виде желтого твердого вещества.
[218] Стадия 4.
[219] (S)-2-((2-(3-Фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
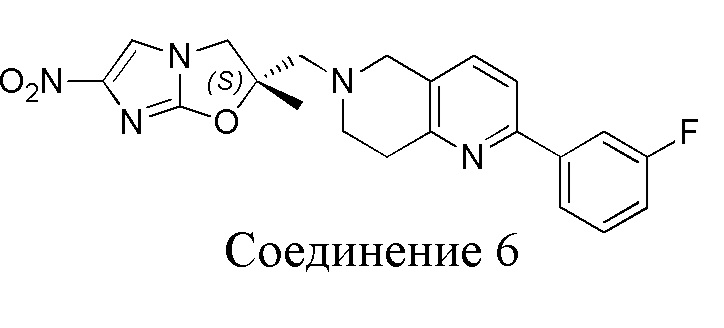
[220] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(3-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-метилпропан-2-ол (300,00 мг, 672,84 мкмоль, 1,00 экв.) растворяли в DMF (5,00 мл). Добавляли к смешанному раствору NaH (32,30 мг, 807,41 мкмоль, 1,20 экв.) при 0°C и перемешивали в течение 30 минут. Реакционную смесь гасили насыщенным раствором хлорида аммония (20 мл) при 0°C, затем разбавляли водой (20 мл) и экстрагировали DCM (10 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (10 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством препаративной хроматографии с получением (GX-E; Phenomenex Synergi C18 150*30мм*4мкм; ацетонитрил 15% - 45%; вода (0,13% HCl); 25 мл/мин.) (S)-2-((2-(3-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 6 (161,80 мг, 395,20 мкмоль, выход 58,74%). 1H ЯМР (400 МГц, CDCl3) δ: δ 7,76-7,66 (m, 2H), 7,53 (s, 1H), 7,50 (d, J = 8,0 Гц, 1H), 7,46-7,37 (m, 2H), 7,14-7,06 (m, 1H), 4,44 (d, J = 8,0 Гц, 1H), 3,97 (d, J = 12,0 Гц, 1H), 3,92-3,82 (m, 2H), 3,20-3,08 (m, 2H), 3,07-2,95 (m, 3H), 2,82 (d, J = 12,0 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 409 (M+1).
[221] Вариант осуществления 7
[222] (S)-2-((2-(4-Фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
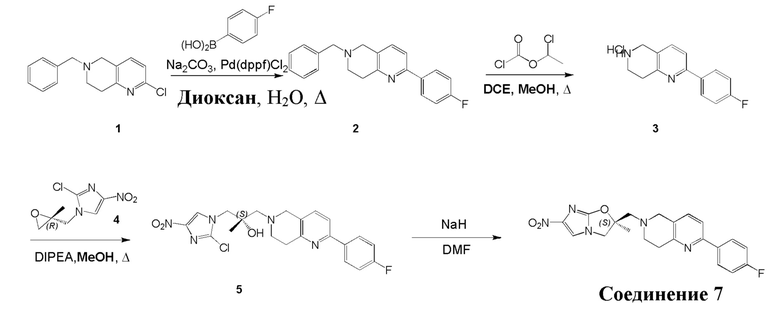
[223] Стадия 1.
[224] 6-Бензил-2-(4-фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридин
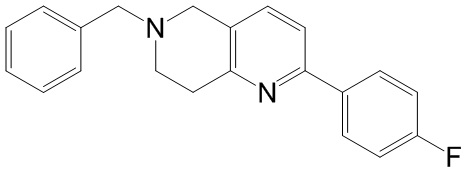
[225] Ключевое промежуточное соединение A (2,00 г, 7,73 ммоль, 1,00 экв.) и (4-фторфенил)бороновую кислоту (1,08 г, 7,73 ммоль, 1,00 экв.) растворяли в диоксане (20,00 мл) и воде (2,00 мл). Добавляли карбонат натрия (1,64 г, 15,46 ммоль, 2,00 экв.), Pd(dppf)Cl2 (565,60 мг, 773,00 мкмоль, 0,10 экв.) при 15°C в атмосфере газообразного азота. Смешанный раствор перемешивали при 110°C в течение 12 часов в атмосфере газообразного азота. После завершения реакции смешанный раствор охлаждали до 15°C и концентрировали при пониженном давлении при 50°C. Добавляли к остатку воду (20 мл) и водную фазу экстрагировали дихлорметаном (100 мл × 4). Объединенные органические фазы промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 20/1, 5/1) с получением 6-бензил-2-(4-фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридина (1,86 г, 5,84 ммоль, выход 75,57%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 8,00-7,89 (m, 2H), 7,47-7,30 (m, 7H), 7,15 (t, J = 8,8 Гц, 2H), 3,76 (s, 2H), 3,68 (s, 2H), 3,19-3,07 (m, 2H), 2,92 (t, J = 6,0 Гц, 2H).
[226] Стадия 2.
[227] 2-(4-Фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорид
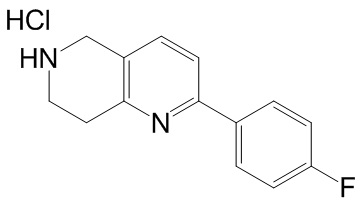
[228] 6-Бензил-2-(4-фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридин (1,86 г, 5,84 ммоль, 1,00 экв.) растворяли в дихлорэтане (20,00 мл). Добавляли 1-хлорэтилхлорформиат (1,25 г, 8,76 ммоль, 1,50 экв.) при 0°C в атмосфере газообразного азота. Смесь перемешивали при 0°C в течение 0,5 часа, а затем нагревали до 85°C и перемешивали в течение 12 часов. Затем смесь концентрировали для удаления растворителя, добавляли к остатку метанол (20,00 мл) и смесь перемешивали при 85°C в течение 2 часов. Смешанный раствор фильтровали и сушили с получением 2-(4-фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорида (1,54 г, неочищенный) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии.
[229] Стадия 3.
[230] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(4-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-метилпропан-2-ол
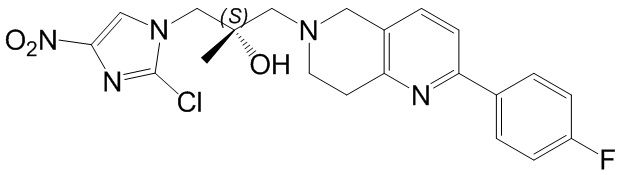
[231] 2-(4-Фторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорид (1,44 г, 6,75 ммоль, 1,00 экв.) и 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазол (1,47 г, 6,75 ммоль, 1,00 экв.) растворяли в этаноле (20,00 мл). Добавляли DIPEA (2,18 г, 16,88 ммоль, 2,50 экв.) при 15°C в атмосфере газообразного азота. Далее смесь перемешивали при 80°C в течение 12 часов, смесь охлаждали до 15°C и концентрировали при пониженном давлении при 60°C. Остаток очищали посредством хроматографии на силикагеле (диоксид кремния, петролейный эфир/этилацетат = 20/1, 1/3) с получением (S)-1-(2-хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(4-фторфенил)-7,8- дигидро-1,6-нафтиридин-6(5H)-ил)-2-метилпропан-2-ола (1,20 г, 2,69 ммоль, выход 39,87%) в виде желтого твердого вещества.
[232] Стадия 4.
[233] (S)-2-((2-(4-Фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
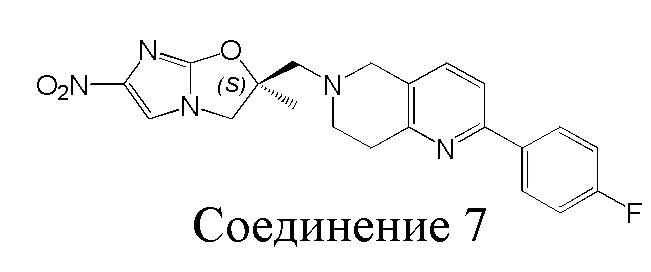
[234] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(4-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-метилпропан-2-ол (1,20 г, 2,69 ммоль, 1,00 экв.) растворяли в DMF (10,00 мл). Добавляли к смешанному раствору NaH (129,12 мг, 5,38 ммоль, 2,00 экв.) в атмосфере газообразного азота. Смешанный раствор затем перемешивали при -20°C в течение 10 минут, а потом при -5°C в течение 10 минут и при 15°C в течение 10 минут. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 20/1, 1/3) и полученный в результате продукт перемешивали в метаноле (20 мл) при 75°C в течение 0,5 часа, а затем охлаждали до 15°C, фильтровали и сушили с получением (S)-2-((2-(4-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 7 (711,10 мг, 1,72 ммоль, выход 63,99%, чистота 99,1%). 1H ЯМР (400 МГц, CDCl3): δ 7,93 (dd, J = 8,8, 5,5 Гц, 2H), 7,52 (s, 1H), 7,48-7,42 (m, 1H), 7,40-7,33 (m, 1H), 7,14 (t, J = 8,7 Гц, 2H), 4,43 (d, J = 9,6 Гц, 1H), 4,02-3,78 (m, 3H), 3,22-2,90 (m, 5H), 2,81 (d, J = 15,1 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 410 (M+1).
[235] Вариант осуществления 8
[236] (S)-2-((2-(4-Хлорфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
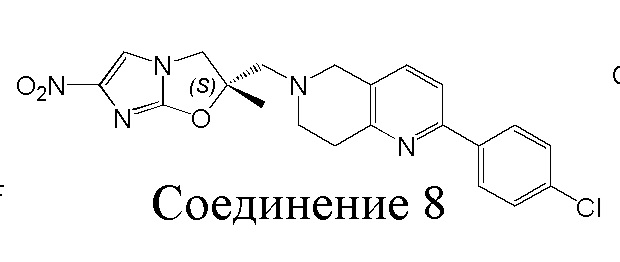
[237] Ключевое промежуточное соединение B (100,00 мг, 285,90 мкмоль, 1,00 экв.) и (4-хлорфенил)бороновую кислоту (53,65 мг, 343,08 мкмоль, 1,20 экв.) растворяли в диоксане (5,00 мл) и воде (500,00 мкл) при 15°C в атмосфере газообразного азота. Смесь перемешивали при 120°C в течение 12 часов, а затем охлаждали до 15°C, фильтровали. Фильтрат концентрировали при 50°C при пониженном давлении и остаток очищали посредством препаративной хроматографии (GX-D; Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; ацетонитрил 24% - 54%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-((2-(4-хлорфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 8 (10,80 мг, 24,26 мкмоль, выход 8,49%, чистота 95,67%). 1H ЯМР (400 МГц, CDCl3): δ 7,90 (d, J = 8,4 Гц, 2H), 7,55-7,34 (m, 5H), 4,43 (d, J = 9,5 Гц, 1H), 4,03-3,79 (m, 3H), 3,24 -2,92 (m, 5H), 2,81 (d, J = 14,8 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 426 (M+1).
[238] Вариант осуществления 9
[239] (S)-4-(6-((2-Метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)бензонитрил

[240] Ключевое промежуточное соединение B (120,00 мг, 343,08 мкмоль, 1,00 экв.) и (4-цианофенил)бороновую кислоту (60,49 мг, 411,70 мкмоль, 1,20 экв.) растворяли в диоксане (5,00 мл). Добавляли к раствору Pd(dppf)Cl2 (25,10 мг, 34,31 мкмоль, 0,10 экв.), карбонат натрия (90,91 мг, 857,70 мкмоль, 2,5 экв.) при 30°C в атмосфере газообразного азота. Смесь перемешивали при 110°C в течение 12 часов, затем охлаждали до 30°C и концентрировали при 45°C при пониженном давлении, и добавляли к остатку воду (10 мл). Водную фазу экстрагировали этилацетатом (20 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (10 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток подвергали предварительной обработке посредством тонкослойной хроматографии (петролейный эфир/этилацетат = 1/2,5), а затем его разделяли и очищали посредством препаративной хроматографии (GX-D; Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; ацетонитрил 24% - 54%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-4-(6-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)бензонитрила – соединения 9 (9,60 мг, 23,05 мкмоль, выход 6,72%, чистота 99,3%). 1H ЯМР (400 МГц, CDCl3): δ 8,09-8,07 (d, J = 8,28 Гц, 2H), 7,76-7,74 (d, J = 8,41 Гц, 2H), 7,55 (d, J = 8,03 Гц, 1H), 7,52 (s, 1H), 7,43-7,41 (d, J = 8,03 Гц, 1H), 4,43-4,41 (d, J = 9,66 Гц, 1H), 3,98-3,96 (d, J = 9,54 Гц, 1H), 3,92-3,84 (m, 2H), 3,17-3,00 (m, 5H), 2,82 (d, J = 14,81 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 417 (M+1).
[241] Вариант осуществления 10
[242] (S)-2-Метил-6-нитро-2-((2-(4-(трифторметил)фенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
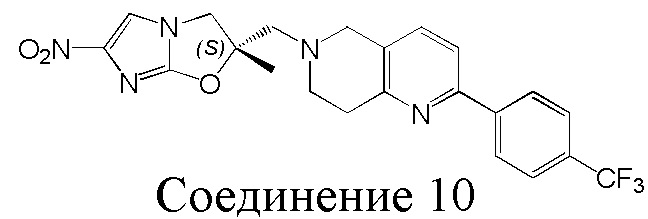
[243] Ключевое промежуточное соединение B (120,00 мг, 343,08 мкмоль, 1,00 экв.) и [4-(трифторметил) фенил]бороновую кислоту (65,16 мг, 343,08 мкмоль, 1,00 экв.) растворяли в диоксане (5,00 мл) и воде (500,00 мкл), добавляли карбонат натрия (72,73 мг, 686,17 мкмоль, 2,00 экв.), Pd(dppf)Cl2 (25,10 мг, 34,31 мкмоль, 0,10 экв.) при 15°C в атмосфере газообразного азота. Смешанный раствор перемешивали при 110°C в течение 12 часов и очищали посредством препаративной хроматографии (Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; ацетонитрил 24% - 54%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-метил-6-нитро-2-((2-(4-(трифторметил)фенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазола – соединения 10 (16,40 мг, 34,16 мкмоль, выход 9,96%, чистота 95,7%). 1H ЯМР (400 МГц, CDCl3): δ 8,07 (d, J = 8,0 Гц, 2H), 7,72 (d, J = 8,2 Гц, 2H), 7,59-7,48 (m, 2H), 7,42 (d, J = 8,0 Гц, 1H), 4,44 (d, J = 9,8 Гц, 1H), 4,04-3,81 (m, 3H), 3,23-2,93 (m, 5H), 2,82 (d, J = 14,8 Гц, 1H), 1,70 (s, 3H). LCMS (ESI) масса/заряд: 460 (M+1).
[244] Вариант осуществления 11
[245] (S)-2-((2-(3-Фтор-4-(трифторметокси)фенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
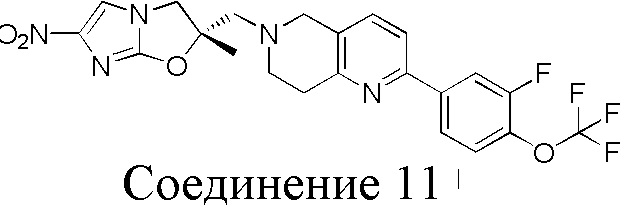
[246] Ключевое промежуточное соединение B (62,32 мг, 178,17 мкмоль, 1,00 экв.) и [3-фтор-4-(трифторметокси)фенил]бороновую кислоту (39,90 мг, 178,17 мкмоль, 1,00 экв.) растворяли в диоксане (5,00 мл), добавляли Pd(dppf)Cl2 (13,04 мг, 17,82 мкмоль, 0,10 экв.), карбонат натрия (47,21 мг, 445,42 мкмоль, 2,5 экв.) при 30°C в атмосфере газообразного азота. Смешанный раствор перемешивали при 110°C в течение 12 часов, а затем охлаждали и концентрировали при пониженном давлении при 45°C. Остаток выливали в воду (10 мл) и перемешивали в течение 5 минут. Водную фазу экстрагировали этилацетатом (30 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (10 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток подвергали первичной обработке посредством тонкослойной хроматографии (петролейный эфир/этилацетат = 1/2,5), а затем разделяли и очищали посредством препаративной хроматографии (GX-E; колонка: Innoval C18 150*30 мм*5 мкм; ацетонитрил 30% - 60%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-((2-(3-фтор-4-(трифторметокси)фенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 11 (10,70 мг, 20,82 мкмоль, выход 11,68%, чистота 96%). 1H ЯМР (400 МГц, CDCl3): δ 7,86 (dd, J = 11,36, 2,07 Гц, 1H), 7,85 (d, J = 8,53 Гц, 1H), 7,72 (s, 1H), 7,52-7,49 (m, 1H), 7,47-7,39 (m, 2H), 4,43 (d, J = 9,66 Гц, 1H), 3,96 (d, J = 9,66 Гц, 1H), 3,91-3,83 (m, 2H), 3,20- 2,99 (m, 5H), 2,82 (d, J = 14,93 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 494 (M+1).
[247] Вариант осуществления 12
[248] (S)-2-((2-(3,4-Дифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
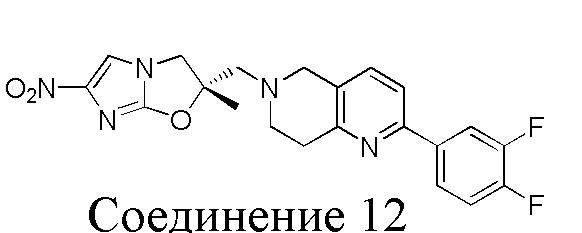
[249] Ключевое промежуточное соединение B (300,00 мг, 857,71 мкмоль, 1,00 экв,) и (3,4-дифторфенил)бороновую кислоту (162,53 мг, 1,03 ммоль, 1,20 экв,) растворяли в диоксане (5,00 мл) и воде (500,00 мкл). Добавляли карбонат натрия (113,64 мг, 1,07 ммоль, 2,50 экв,), Pd(dppf)Cl2 (62,76 мг, 85,77 мкмоль, 0,10 экв,) при 30°C в атмосфере газообразного азота. Смесь нагревали до 100°C, перемешивали в течение 12 часов и охлаждали, добавляли к смешанному раствору воду (10 мл) и водную фазу экстрагировали этилацетатом (30 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток подвергали первичной обработке посредством тонкослойной хроматографии (петролейный эфир/этилацетат = 1/1, 0/1), а затем разделяли и очищали посредством препаративной хроматографии (GX-D; Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; ацетонитрил 24% - 54%; вода (0,225% муравьиная кислота), 25 мл/мин.) с получением (S)-2-((2-(3,4-дифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 12 (5,70 мг, 13,34 мкмоль, выход 3,11%, чистота 95,0%). 1H ЯМР (400 МГц, CDCl3): δ 7,85 (ddd, J = 11,83, 7,83, 2,13 Гц, 1H), 7,83 (br, s,, 1H), 7,52 (s, 1H), 7,46-7,44 (m, 1H), 7,39-7,37 (m, 1H), 7,26-7,21 (m, 1H), 4,43 (d, J = 9,66 Гц, 1H), 3,96 (d, J = 9,54 Гц, 1H), 3,90-3,82 (m, 2H), 3,11 (d, J = 14,93 Гц, 2H), 3,01-2,99 (m, 3H), 2,83-2,79 (d, J = 14,81 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 428 (M+1).
[250] Вариант осуществления 13
[251] (S)-2-((2-(2,4-Дифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
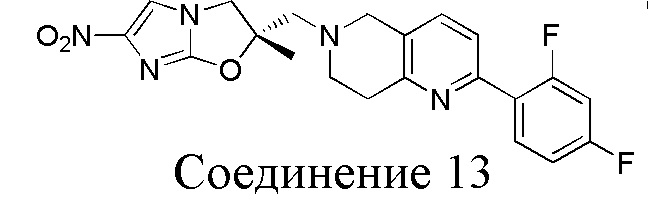
[252] Ключевое промежуточное соединение B (300,00 мг, 857,71 мкмоль, 1,00 экв,) и (2,4-дифторфенил)бороновую кислоту (162,53 мг, 1,03 ммоль, 1,20 экв,) растворяли в диоксане (5,00 мл) и воде (500,00 мкл). Затем добавляли карбонат натрия (113,64 мг, 1,07 ммоль, 2,50 экв.), Pd(dppf)Cl2 (62,76 мг, 85,77 мкмоль, 0,10 экв.) при 30°C в атмосфере газообразного азота. Далее смесь нагревали до 100°C и перемешивали в течение 12 часов. Смесь охлаждали до 30°C. Добавляли к смеси воду (10 мл) и экстрагировали этилацетатом (30 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 1/1, 0/1), а затем очищали посредством препаративной хроматографии (GX-D; Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; ацетонитрил 24% - 54%; вода (0,225% муравьиная кислота), 25 мл/мин.) с получением (S)-2-((2-(2,4-дифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 13 (35,70 мг, 83,53 мкмоль, выход 19,48%, чистота 97,0%). 1H ЯМР (400 МГц, CDCl3): δ 7,95-7,93 (td, J = 8,78, 6,65 Гц, 1H), 7,53-7,50 (m, 2H), 7,38-7,36 (d, J = 8,16 Гц, 1H), 7,00-6,99 (td, J = 8,03, 2,76 Гц, 1H), 6,93-6,88 (ddd, J = 11,11, 8,72, 2,51 Гц, 1H), 4,43 (d, J = 9,66 Гц, 1H), 3,96 (d, J = 9,66 Гц, 1H), 3,90-3,87 (m, 2H), 3,17-3,09 (m, 2H), 3,03-2,99 (m, 3H), 2,81 (d, J = 14,81 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 428 (M+1).
[253] Вариант осуществления 14
[254] (S)-2-((2-(3,5-Дифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
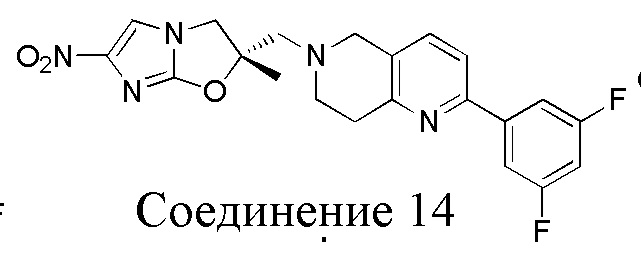
[255] Ключевое промежуточное соединение B (200 мг, 571,80 мкмоль, 1,00 экв.) и (3,5-дифторфенил)бороновую кислоту (628,98 мкмоль, 1,10 экв.) растворяли в диоксане (5,00 мл), добавляли Pd(dppf)Cl2 (42 мг, 57,18 мкмоль, 0,10 экв.) и фторид цезия (217 мг, 1,43 ммоль, 2,50 экв.) в атмосфере газообразного азота. Смесь нагревали до 110°C и перемешивали в течение 2 часов. Смесь охлаждали до 20°C и концентрировали при пониженном давлении. Остаток разбавляли водой и водную фазу экстрагировали этилацетатом (20 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (высота колонки: 250 мм, диаметр: 100 мм, силикагель 100-200 меш, петролейный эфир/этилацетат = 2/1, 1/1) с получением (S)-2-((2-(3,5-дифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 14 (26,00 мг, 59,62 мкмоль, выход 10,43%). 1H ЯМР (400 МГц, CDCl3): δ 7,52-7,46 (m, 4H), 7,41-7,39 (m, 1H), 6,84 (tt, J = 8,67, 2,31 Гц, 1H), 4,43 (d, J = 9,66 Гц, 1H), 4,98-3,87 (m, 3H), 3,17-2,84 (m, 5 H), 2,80 (d, J = 14,81 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 428 (M+1).
[256] Вариант осуществления 15
[257] (S)-2-((2-(5-Хлор-2-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
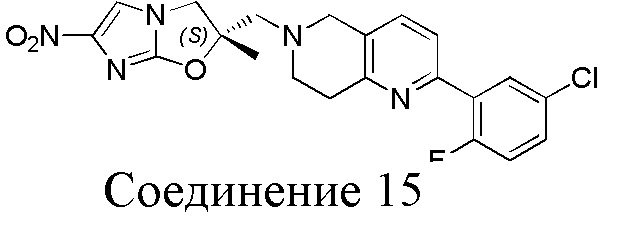
[258] Ключевое промежуточное соединение B (200,00 мг, 571,80 мкмоль, 1,00 экв.) и (5-хлор-2-фтор-фенил)бороновую кислоту (119,64 мг, 686,17 мкмоль, 1,20 экв.) растворяли в воде (500,00 мкл) и диоксане (3,00 мл), а затем добавляли Pd(dppf)Cl2 (41,84 мг, 57,18 мкмоль, 0,10 экв.) и фторид цезия (260,57 мг, 1,72 ммоль, 3,00 экв.). Смесь перемешивали при 110°C в течение 16 часов в атмосфере газообразного азота. Смесь концентрировали in vacuo и остаток очищали посредством препаративной хроматографии (GX-D; Boston Green ODS 150*30 5u; ацетонитрил 40% - 70%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-((2-(5-хлор-2-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 15 (40,09 мг, 88,24 мкмоль, выход 15,43%, чистота 97,7%). 1H ЯМР (400 МГц, CDCl3): δ 7,94 (dd, J = 6,78, 2,76 Гц, 1H), 7,53-7,56 (m, 2H), 7,39 (d, J =8,00 Гц, 1H), 7,32 (ddd, J = 8,75, 4,17, 2,76 Гц, 1H), 7,10 (dd, J = 10,42, 8,78 Гц, 1H), 4,44 (d, J = 9,66 Гц, 1H), 4,00-3,82 (m, 3H), 2,93-3,22 (m, 5H), 2,82 (d, J = 14,81 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 444,0 (M+1).
[259] Вариант осуществления 16
[260] (S)-2-((2-(2,3-Дифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
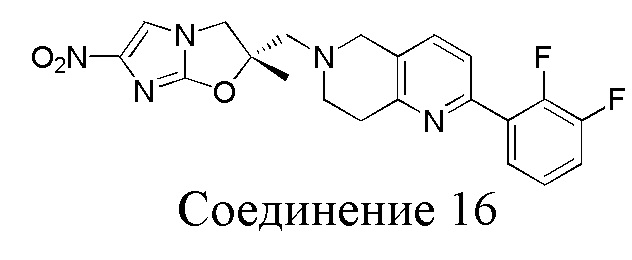
[261] Ключевое промежуточное соединение B (200,00 мг, 571,80 мкмоль, 1,00 экв.) и (2,3-дифторфенил)бороновую кислоту (135,44 мг, 857,70 мкмоль, 1,50 экв.), фторид цезия (173,71 мг, 1,14 ммоль, 2,00 экв.) растворяли в воде (300,00 мкл) и диоксане (3,00 мл). В смешанном растворе три раза производили замену на азот, добавляли Pd(dppf)Cl2 (41,84 мг, 57,18 мкмоль, 0,10 экв.) и перемешивали при 110°C в течение 12 часов. Остаток очищали посредством препаративной хроматографии (GX-A; Phenomenex Gemini C18 250*50 10u; ацетонитрил 38% - 68%; H2O (0,2% NH3.H2O); 25 мл/мин.) с получением (S)-2-((2-(2,3-дифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 16 (28,00 мг, 65,51 мкмоль, выход 11,46%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,11 (s, 1H), 7,73-7,43 (m, 4H), 7,37-7,22 (m, 1H), 4,29 (s, 1H), 4,10 (d, J = 10,54 Гц, 1H), 3,85 (d, J = 14,05 Гц, 2H), 2,98 (d, J =3,76 Гц, 6H), 1,60 (s, 3H). LCMS (ESI) масса/заряд: 428,0 (M+1).
[262] Вариант осуществления 17
[263] (S)-2-((2-(2,5-Дифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
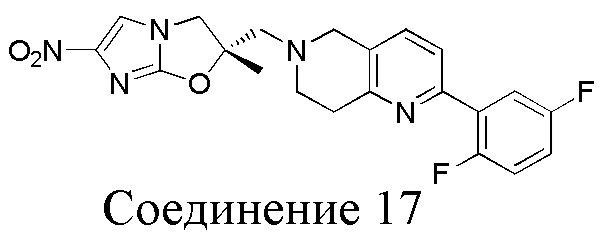
[264] Ключевое промежуточное соединение B (200,00 мг, 571,80 мкмоль, 1,00 экв.) и (2,5-дифторфенил)бороновую кислоту (108,3 мг, 686,16 мкмоль, 1,20 экв.) растворяли в толуоле (5,00 мл) и воде (500,00 мкл), добавляли к смешанному раствору фторид цезия (260,57 мг, 1,72 ммоль, 3,00 экв.) и Pd(dppf)Cl2 (41,84 мг, 57,18 мкмоль, 0,10 экв.) при 30°C в атмосфере газообразного азота, а затем нагревали до 100°C и перемешивали в течение 12 часов. Смесь охлаждали до 30°C и добавляли к смеси воду (10 мл), водную фазу экстрагировали этилацетатом (30 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток подвергали первичной обработке посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 1/1, 0/1), а затем разделяли и очищали посредством препаративной хроматографии (GX-D; Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; ацетонитрил 24% - 54%; вода (0,225% муравьиная кислота), 25 мл/мин.) с получением (S)-2-((2-(2,5-дифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 17 (51,50 мг, 118,69 мкмоль, выход 20,76%, чистота 98,5%). 1H ЯМР (400 МГц, CDCl3): δ 7,71-7,60 (ddd, J = 9,22, 6,02, 3,20 Гц, 1H), 7,59 (dd, J = 8,03, 2,26 Гц, 1H), 7,53 (s, 1H), 7,38 (d, J = 8,03 Гц, 1H), 7,12-7,05 (m, 2H), 4,44 (d, J = 9,66 Гц, 1H), 3,98 (d, J = 9,66 Гц, 1H), 3,91-3,84 (m, 2H), 3,16-3,09 (m, 2H), 3,04-3,00 (m, 3H), 2,83-2,80 (d, J = 14,81 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 428 (M+1).
[265] Вариант осуществления 18
[266] (S)-2-((2-(3-Хлор-4-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
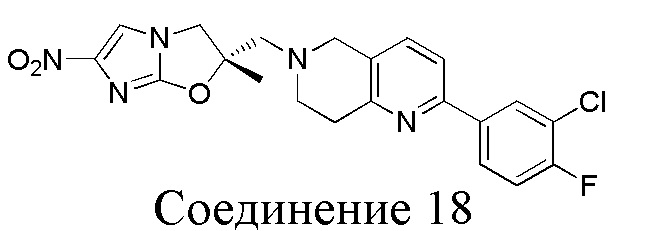
[267] Ключевое промежуточное соединение B (300,00 мг, 857,71 мкмоль, 1,00 экв.), (3-хлор-4-фтор-фенил)бороновую кислоту (224,32 мг, 1,29 ммоль, 1,50 экв.), фторид цезия (260,57 мг, ммоль, 2,00 экв.) растворяли в воде (500,00 мкл) и диоксане (5,00 мл), добавляли Pd(dppf)Cl2 (62,76 мг, 85,77 мкмоль, 0,10 экв.) в атмосфере газообразного азота и смесь перемешивали при 110°C в течение 12 часов. Смесь концентрировали и очищали посредством препаративной хроматографии (GX-G, Phenomenex Synergi C18 150*30 мм*4 мкм, ацетонитрил 45% - 75%; H2O (+0,0022 NH3.H2O); 25 мл/мин.) с получением (S)-2-((2-(3-хлор-4-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 18 (90,00 мг, 202,77 мкмоль, выход 23,64%). 1H ЯМР (400 МГц, DMSO-d6): δ 8,27-8,18 (m, 1H), 8,10 (s, 1H), 8,09-8,03 (m, 1H), 7,84-7,75 (m, 1H), 7,63-7,43 (m, 2H), 4,30 (d, J = 10,67 Гц, 1H), 4,16-4,06 (m, 1H), 3,82 (d, J = 12,67 Гц, 2H), 2,97 (d, J = 3,51 Гц, 6H), 1,60 (s, 3H). LCMS (ESI) масса/заряд: 444,1 (M+1).
[268] Вариант осуществления 19
[269] (S)-2-((2-(3-Хлор-2-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
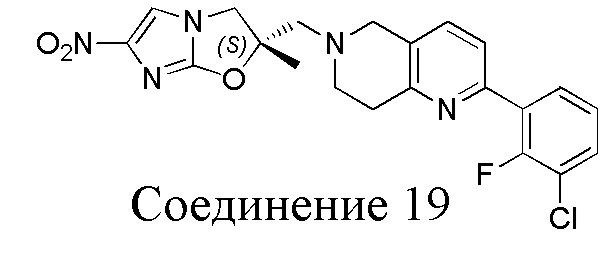
[270] Ключевое промежуточное соединение B (200,00 мг, 571,80 мкмоль, 1,00 экв.) и (3-хлор-2-фтор-фенил)бороновую кислоту (99,70 мг, 571,80 мкмоль, 1,00 экв.) растворяли в диоксане (3,00 мл) и воде (500,00 мкл), добавляли Pd(dppf)Cl2 (41,84 мг, 57,18 мкмоль, 0,10 экв.) и фторид цезия (260,57 мг, 1,72 ммоль, 3,00 экв.). Смесь перемешивали при 110°C в течение 16 часов в атмосфере газообразного азота, а затем концентрировали. Остаток очищали посредством препаративной хроматографии (GX-D; Boston Green ODS 150*30 5u; ацетонитрил 40% - 70%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-((2-(3-хлор-2-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 19 (47,80 мг, 101,12 мкмоль, выход 17,68%, чистота 93,9%). 1H ЯМР (400 МГц, CDCl3) δ 7,83-7,74 (m, 1H), 7,56-7,50 (m, 2H), 7,47-7,37 (m, 2H), 7,19 (td, J = 7,91, 0,88 Гц, 1H), 4,43 (d, J = 9,66 Гц, 1H), 3,99-3,86 (m, 3H), 3,24-2,91 (m, 5H), 2,82 (d, J = 14,81 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 444,1 (M+1).
[271] Вариант осуществления 20
[272] (S)-2-((2-(4-Хлор-3-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол

[273] Ключевое промежуточное соединение B (200 мг, 571,80 мкмоль, 1,00 экв.) и (4-хлор-3-фтор-фенил)бороновую кислоту (628,98 мкмоль, 1,10 экв.) растворяли в диоксане (5,00 мл), добавляли Pd(dppf)Cl2 (42 мг, 57,18 мкмоль, 0,10 экв.) и фторид цезия (217 мг, 1,43 ммоль, 2,50 экв.) в атмосфере газообразного азота. Смесь нагревали до 110°C и перемешивали в течение 2 часов. Смесь охлаждали до 20°C и концентрировали при пониженном давлении. Остаток разбавляли водой и водную фазу экстрагировали этилацетатом (20 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством препаративной хроматографии (прибор: GX-D; колонка: Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; подвижная фаза: MeCN: 25% - 55%; H2O (+0,0023 HCOOH), скорость: 25 мл/мин.; контролируемая длина волны: 220 нм/254 нм; длительность прогона: 10 мин./15 мин.; температура колонки: 20oC) с получением (S)-2-((2-(4-хлор-3-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола –соединения 20 (26,00 мг, 52,72 мкмоль, выход 9,22%). 1H ЯМР (400 МГц, CDCl3): δ 7,81 (d, J = 10,29 Гц, 1H), 7,68 (d, J = 8,78 Гц, 1H), 7,52-7,38 (m, 4H), 4,43 (d, J = 9,54 Гц, 1H), 3,98-3,66 (m, 3H), 3,16-2,79 (m, 6H), 1,69 (s, 1H). LCMS (ESI) масса/заряд: 444 (M+1).
[274] Вариант осуществления 21
[275] (S)-2-((2-(3-Хлор-5-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
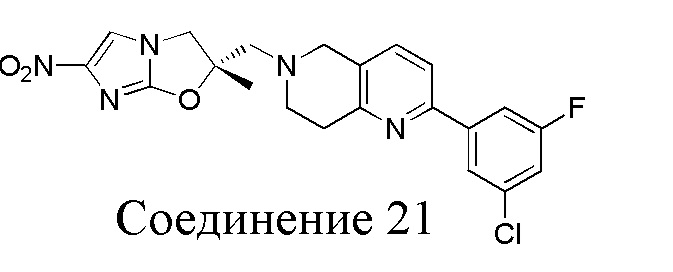
[276] Ключевое промежуточное соединение B (200 мг, 571,80 мкмоль, 1,00 экв.) и (3-хлор-5-фтор-фенил)бороновую кислоту (628,98 мкмоль, 1,10 экв.) растворяли в диоксане (5,00 мл), добавляли Pd(dppf)Cl2 (42 мг, 57,18 мкмоль, 0,10 экв.) и фторид цезия (217 мг, 1,43 ммоль, 2,50 экв.) в атмосфере газообразного азота. Смешанный раствор нагревали до 110°C и перемешивали в течение 2 часов, а затем охлаждали до 20°C. Смесь концентрировали при пониженном давлении. Остаток разбавляли водой и водную фазу экстрагировали этилацетатом (20 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством препаративной хроматографии (прибор: GX-D; колонка: Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; подвижная фаза: MeCN: 15% - 45%; H2O (+0,0023 HCOOH), скорость: 25 мл/мин.; контролируемая длина волны: 220 нм/254 нм; длительность прогона: 10 мин./15 мин.; температура колонки: 20oC) с получением (S)-2-((2-(3-хлор-5-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 21 (88,00 мг, 193,70 мкмоль, выход 33,88%). 1H ЯМР (400 МГц, CDCl3): δ 7,76 (s, 1H), 7,59 (dt, J = 9,47, 1,98 Гц, 1H), 7,53 (s, 1H), 7,50-7,43 (m, 1H), 7,43-7,35 (m, 1H), 7,12 (dt, J = 8,16, 2,07 Гц, 1H), 4,43 (d, J = 9,66 Гц, 1H), 3,98-3,68 (m, 3H), 3,34-2,88 (m, 5H), 2,82 (d, J = 14,81 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 444 (M+1).
[277] Вариант осуществления 22
[278] (S)-2-((2-(2-Хлор-5-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол

[279] Ключевое промежуточное соединение B (200 мг, 571,80 мкмоль, 1,00 экв.) и (2-хлор-5-фтор-фенил)бороновую кислоту (628,98 мкмоль, 1,10 экв.) растворяли в диоксане (5,00 мл), добавляли Pd(dppf)Cl2 (42 мг, 57,18 мкмоль, 0,10 экв.) и фторид цезия (217 мг, 1,43 ммоль, 2,50 экв.) в атмосфере газообразного азота. Смесь нагревали до 110°C и перемешивали в течение 2 часов. Смесь охлаждали до 20°C и концентрировали при пониженном давлении. Остаток разбавляли водой и водную фазу экстрагировали этилацетатом (20 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством препаративной хроматографии (прибор: GX-D; колонка: Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; подвижная фаза: MeCN: 35%-75%; H2O (+0,0023 HCOOH), скорость: 25 мл/мин.; контролируемая длина волны: 220 нм/254 нм; длительность прогона: 10 мин./15 мин.; температура колонки: 20oC) с получением (S)-2-((2-(2-хлор-5-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 22 (30,00 мг, 66,91 мкмоль, выход 11,70%). 1H ЯМР (400 МГц, CDCl3): δ 7,54 (s, 1H), 7,47-7,36 (m, 3H), 7,31 (dd, J = 8,97, 3,07 Гц, 1H), 7,04 (td, J = 8,22, 3,14 Гц, 1H), 4,44 (d, J = 9,66 Гц, 1H), 4,12-3,68 (m, 3H), 3,36-2,91 (m, 5H), 2,83 (d, J = 14,81 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 444 (M+1).
[280] Вариант осуществления 23
[281] (S)-2-((2-(4-Хлор-2-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
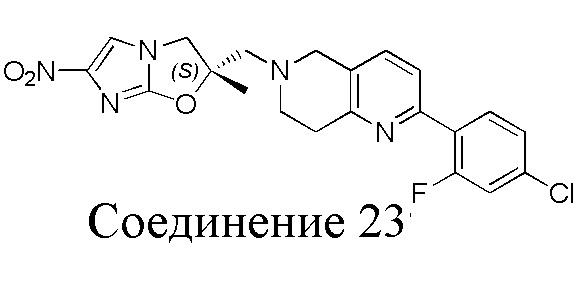
[282] Ключевое промежуточное соединение B (200,00 мг, 571,80 мкмоль, 1,00 экв.) и (4-хлор-2-фтор-фенил)бороновую кислоту (119,64 мг, 686,16 мкмоль, 1,20 экв.) растворяли в диоксане (3,00 мл) и воде (500,00 мкл). Добавляли Pd(dppf)Cl2 (41,84 мг, 57,18 мкмоль, 0,10 экв.) и фторид цезия (260,57 мг, 1,72 ммоль, 3,00 экв.) в атмосфере газообразного азота. Смесь перемешивали при 110°C в течение 16 часов и концентрировали. Остаток разделяли и очищали посредством препаративной хроматографии (GX-G; Phenomenex Synergi C18 150*30 мм*4 мкм; ацетонитрил 24% - 54%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-((2-(2-хлор-5-фторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 23 (56,26 мг, 125,10 мкмоль, выход 21,88%, чистота 98,7%). 1H ЯМР (400 МГц, CDCl3): δ 7,92 (t, J = 8,41 Гц, 1H), 7,57-7,50 (m, 2H), 7,38 (d, J = 8,03 Гц, 1H), 7,27-7,22 (m, 1H), 7,22-7,16 (m, 1H), 4,43 (d, J = 9,66 Гц, 1H), 4,0-3,81 (m, 3H), 3,23-2,90 (m, 5H), 2,81 (d, J = 14,81 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 444,0 (M+1).
[283] Вариант осуществления 24
[284] (S)-2-((2-(Фуран-3-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
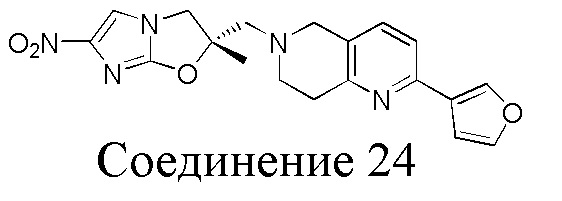
[285] Ключевое промежуточное соединение B (200,00 мг, 571,80 мкмоль, 1,00 экв.) и 3-фурилбороновую кислоту (76,77 мг, 686,16 мкмоль, 1,20 экв.) растворяли в диоксане (5,00 мл) и воде (500,00 мкл), добавляли карбонат натрия (121,21 мг, 1,14 ммоль, 2,00 экв.), Pd(PPh3)4 (66,07 мг, 57,18 мкмоль, 0,10 экв.) в атмосфере газообразного азота. Смесь перемешивали при 90°C в течение 12 часов. Смесь охлаждали до 30°C и концентрировали при пониженном давлении при 45°C. Остаток подвергали первичной обработке посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 10/1, 1/1), а затем разделяли и очищали посредством препаративной хроматографии (GX-D; Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; MeCN: 24% - 54%; H2O (+0,0025 FA); 25 мл/мин.) с получением (S)-2-((2-(фуран-3-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 24 (56,67 мг, 142,65 мкмоль, выход 24,95%, чистота 96%). 1H ЯМР (400 МГц, CDCl3): δ 7,99 (s, 1H), 7,51 (s, 1H), 7,48 (t, J = 1,69 Гц, 1H), 7,31-7,29 (m, 1H), 7,28-7,24 (m, 1H), 6,87 (d, J = 1,13 Гц, 1H), 4,43 (d, J = 9,66 Гц, 1H), 3,95 (d, J = 9,66 Гц, 1H), 3,81 (m, 2 H), 3,14-3,07 (m, 2H), 2,99-2,96 (m, 3H), 2,79 (d, J = 14,81 Гц, 1H), 1,68 (s, 3H). LCMS (ESI) масса/заряд: 382 (M+1).
[286] Вариант осуществления 25
[287] (S)-2-((2-(Фуран-2-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
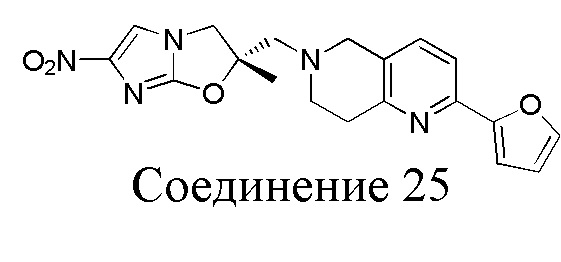
[288] Ключевое промежуточное соединение B (150,00 мг, 428,85 мкмоль, 1,00 экв.) и трибутил(2-фурил)станнан (229,73 мг, 643,28 мкмоль, 1,50 экв.) растворяли в дихлорэтане (10,00 мл), добавляли Pd(dppf)Cl2 (31,38 мг, 42,89 мкмоль, 0,10 экв.), хлорид лития (6,09 мг, 143,76 мкмоль, 1,00 экв.) при 30°C в атмосфере газообразного азота. Затем смесь нагревали до 120°C и перемешивали в течение 12 часов. Смесь охлаждали и концентрировали при пониженном давлении при 45°C. Добавляли к смеси воду (10 мл) и перемешивали в течение 5 минут. Водную фазу экстрагировали этилацетатом (30 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (10 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток подвергали первичной обработке посредством хроматографии на силикагеле (этилацетат), а затем разделяли и очищали посредством препаративной хроматографии (прибор: GX-D; Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; ацетонитрил 24% - 54%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-((2-(фуран-2-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 25 (31,70 мг, 83,12 мкмоль, выход 19,38%). 1H ЯМР (400 МГц, CDCl3): δ 7,53 (m, 1H), 7,52 (s, 1H), 7,47 (d, J = 8,16 Гц, 1H), 7,33 (d, J = 8,16 Гц, 1H), 6,99 (d, J = 3,39 Гц, 1H), 6,52 (dd, J = 3,39, 1,76 Гц, 1H), 4,43 (d, J = 9,54 Гц, 1H), 3,95 (d, J = 9,66 Гц, 1H), 3,85 (d, J = 14,18 Гц, 2H), 3,09 (d, J = 14,81 Гц, 2H), 3,01-2,97 (m, 3H), 2,79 (d, J = 14,81 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 382 (M+1).
[289] Вариант осуществления 26
[290] (S)-2-((2-Циклопропил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
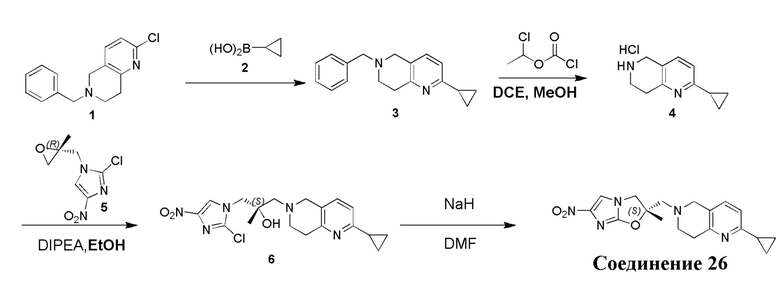
[291] Стадия 1.
[292] 6-Бензил-2-циклопропил-5,6,7,8-тетрагидро-1,6-нафтиридин

[293] Ключевое промежуточное соединение A (1,50 г, 5,80 ммоль, 1,00 экв.) и циклопропилбороновую кислоту (647,69 мг, 7,54 ммоль, 1,30 экв.) растворяли в толуоле (10,00 мл), добавляли ди(адамантан-1-ил)(бутил)фосфин (415,91 мг, 1,16 ммоль, 0,20 экв.), карбонат цезия (3,78 г, 11,60 ммоль, 2,00 экв.) и Pd(OAc)2 (130,22 мг, 580,00 мкмоль, 0,10 экв.) при 30°C в атмосфере газообразного азота. Затем смесь нагревали до 100°C и перемешивали в течение 12 часов. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 30/1) с получением 6-бензил-2-циклопропил-7,8-дигидро-5H-1,6-нафтиридина (1,30 г, 4,92 ммоль, выход 84,79%) в виде желтого твердого вещества. 1H ЯМР (300МГц, CDCl3): δ 7,35-7,29 (m, 2H), 7,27 (s, 1H), 7,25-7,21 (m, 1H), 7,21-7,17 (m, 1H), 7,05 (d, J =7,9 Гц, 1H), 6,71 (d, J = 7,9 Гц, 1H), 3,62 (s, 2H), 3,49 (s, 2H), 2,93-2,85 (m, 2H), 2,79-2,71 (m, 2H), 1,99-1,87 (m, 1H), 0,92-0,85 (m, 2H), 0,84-0,80 (m, 2H). LCMS (ESI) масса/заряд: 265 (M+1).
[294] Стадия 2.
[295] 2-Циклопропил-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорид
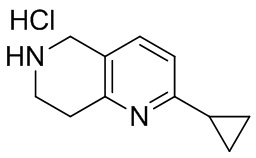
[296] 6-Бензил-2-циклопропил-7,8-дигидро-5H-1,6-нафтиридин (200,00 мг, 756,54 мкмоль, 1,00 экв.) растворяли в дихлорэтане (10,00 мл), добавляли 1-хлоркарбонилхлорид (162,24 мг, 1,13 ммоль, 1,50 экв.) при 0°C в атмосфере газообразного азота. Смесь перемешивали при 0°C в течение 20 минут, а затем нагревали до 80°C и перемешивали в течение 12 часов. Далее смесь концентрировали, добавляли к смеси метанол (10,00 мл) и перемешивали при 75°C в течение дополнительных 2 часов. Смесь охлаждали и концентрировали при пониженном давлении при 45°C. Остаток промывали дихлорметаном (20 мл × 3), фильтровали и сушили осадок с получением 2-циклопропил-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорида (170,00 мг, неочищенный) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительного очищения.
[297] Стадия 3.
[298] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-циклопропил-7,8-дигидро-5H-1,6-нафтиридин-6-ил)-2-метилпропан-2-ол
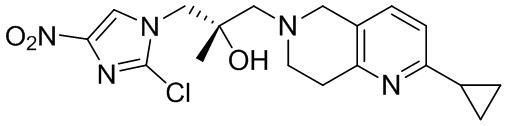
[299] 2-Циклопропил-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорид (550,00 мг, 3,16 ммоль, 1,00 экв.) и 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазол (893,94 мг, 4,11 ммоль, 1,30 экв.) растворяли в этаноле (20,00 мл), добавляли DIPEA (1,02 г, 7,90 ммоль, 2,50 экв.) при 30°C в атмосфере газообразного азота. Смесь перемешивали при данной температуре в течение 10 минут, а затем нагревали до 80°C и перемешивали в течение 12 часов. Смесь разбавляли водой (20 мл) и водную фазу экстрагировали этилацетатом (20 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (10 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 10/1, 2/1) с получением (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-циклопропил-7,8-дигидро-5H-1,6-нафтиридин-6-ил)-2-метилпропан-2-ола (400,00 мг, неочищенный) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 392 (M + 1).
[300] Стадия 4.
[301] (S)-2-((2-Циклопропил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
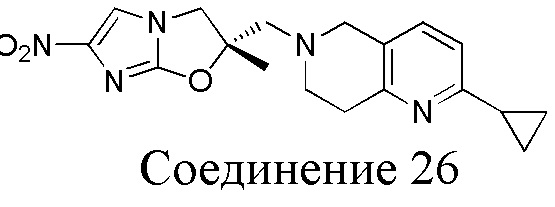
[302] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-циклопропил-7,8-дигидро-5H-1,6-нафтиридин-6-ил)-2-метилпропан-2-ол (440,00 мг, 1,12 ммоль, 1,0 экв.) растворяли в DMF (5,00 мл), добавляли NaH (67,37 мг, 1,68 ммоль, 1,50 экв.) при -45°C в атмосфере газообразного азота. Смесь перемешивали при температуре от -45 до -15°C в течение 2 часов. Остаток выливали в насыщенный водный раствор хлорида аммония (20 мл) и перемешивали при 0°C в течение 20 минут. Водную фазу экстрагировали этилацетатом (30 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (10 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток разделяли и очищали посредством препаративной хроматографии (GX-D; Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; ацетонитрил 20% - 54%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-((2-циклопропил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 26 (157,10 мг, 442,05 мкмоль, выход 39,47%). 1H ЯМР (400 МГц, CDCl3): δ 7,51 (s, 1H), 7,16 (d, J = 7,91 Гц, 1H), 6,82 (d, J = 8,03 Гц, 1H), 4,42 (d, J = 9,66 Гц, 1H), 3,93 (d, J = 9,54 Гц, 1H), 3,79 (q, J = 14,89 Гц, 2H), 3,07-3,04 (m, 2H), 2,94-2,85 (m, 3H), 2,76 (d, J = 14,81 Гц, 1H), 2,03-2,01 (m, 1H), 1,66 (s, 3H), 0,98-0,96 (m, 2H), 0,92-0,90 (m, 2H). LCMS (ESI) масса/заряд: 356 (M+1).
[303] Вариант осуществления 27
[304] (S)-2-Метил-6-нитро-2-((2-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
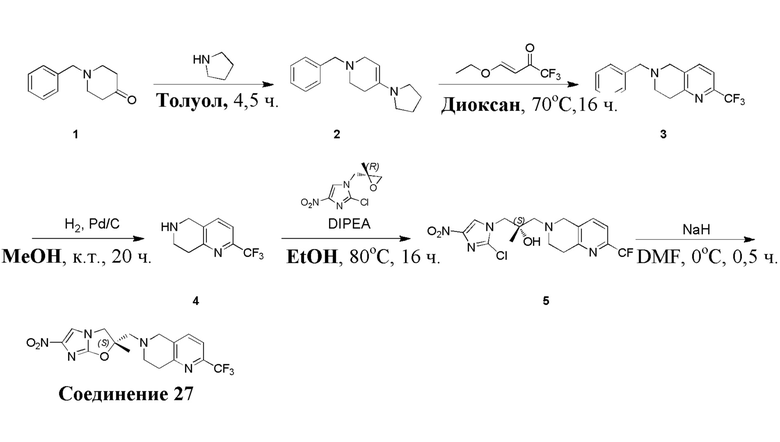
[305] Стадия 1.
[306] Бензил-4-пирролидин-1-ил-3,6-дигидро-2H-пиридин
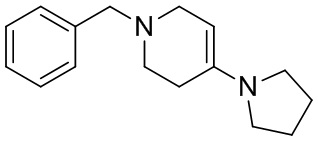
[307] 1-Бензил-4-он (10,00 г, 52,84 ммоль, 1,00 экв.) и пирролидин (4,51 г, 63,41 ммоль, 1,20 экв.) растворяли в толуоле (100 мл), удаляли воду при помощи водоотделителя при 110°C в течение 4,5 часа. Смесь охлаждали и концентрировали с получением 1-бензил-4-пирролидин-1-ил-3,6-дигидро-2H-пиридина (10,50 г, неочищенный) в виде желтого масла, которое использовали на следующей стадии без дополнительного очищения.
[308] Стадия 2.
[309] 6-Бензил-2-(трифторметил)-7,8-дигидро-5H-1,6-нафтиридин
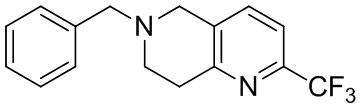
[310] 1-Бензил-4-пирролидин-1-ил-3,6-дигидро-2H-пиридин (2,50 г, 10,32 ммоль, 1,00 экв.) и (E)-4-этокси-1,1,1-трифтор-бут-3-ен-2-он (2,08 г, 12,38 ммоль, 1,20 экв.) растворяли в диоксане (30,00 мл). Смесь нагревали до 100°C и перемешивали в течение 4 часов. Затем добавляли ацетат аммония (2,39 г, 30,96 ммоль, 3,00 экв.) и перемешивали в течение дополнительных 16 часов. Смесь охлаждали и разбавляли водой. Водную фазу экстрагировали этилацетатом (100 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (высота колонки: 300 мм, диаметр 40 мм, силикагель 100-200 меш, петролейный эфир/этилацетат = 30/1) с получением 6-бензил-2-(трифторметил)-7,8-дигидро-5H-1,6-нафтиридина (1,50 г, 5,13 ммоль, выход 49,73%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 7,46-7,29 (m, 7H), 3,75 (s, 2H), 3,70 (s, 2 H), 3,15 (t, J = 5,90 Гц, 2H), 2,92 (t, J = 5,96 Гц, 2H).
[311] Стадия 3.
[312] 2-(Трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридин
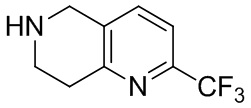
[313] 6-Бензил-2-(трифторметил)-7,8-дигидро-5H-1,6-нафтиридин (1,50 г, 5,13 ммоль, 1,00 экв.) растворяли в метаноле (20,00 мл) и добавляли к раствору Pd/C (100,00 мг). Смесь перемешивали при H2 (50 фунтов/кв. дюйм) в течение 16 часов. Смесь фильтровали и концентрировали с получением 2-(трифторметил)-5,6,7,8-тетрагидро-l,6-нафтиридина (850,00 мг, неочищенный) в виде грязно-белого твердого вещества, которое использовали на следующей стадии без дополнительного очищения.
[314] Стадия 4.
[315] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-2-метил-3-(2-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)пропан-2-ол

[316] 2-(Трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридин (200,00 мг, 989,22 мкмоль, 1,00 экв.) и (R)-2-хлор-1-((2-метилоксиран-2-ил)метил)-4-нитро-1H-имидазол (322,90 мг, 1,48 ммоль, 1,50 экв.) растворяли в этаноле (10,00 мл) и добавляли DIPEA (127,85 мг, 989,22 мкмоль, 1,00 экв.) в атмосфере газообразного азота. Смесь нагревали до 80°C и перемешивали в течение 16 часов. Смесь охлаждали и концентрировали при пониженном давлении. Остаток выливали в воду (50 мл) и экстрагировали этилацетатом (50 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo с получением (S)-1-(2-хлор-4-нитро-1H-имидазол-1-ил)-2-метил-3-(2-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)пропан-2-ола (310,00 мг, неочищенный) в виде коричневого масла. LCMS (ESI) масса/заряд: 420,1 (M+1).
[317] Стадия 5.
[318] (S)-2-Метил-6-нитро-2-((2-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол

[319] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-2-метил-3-(2-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)пропан-2-ол (100,00 мг, 238,21 мкмоль, 1,00 экв.) растворяли в DMF (2,00 мл), добавляли NaH (19,06 мг, 476,42 мкмоль, 2,00 экв.) при -20°C в атмосфере газообразного азота. Смесь перемешивали при -20°C в течение 1 часа. Смесь выливали в ледяную воду (вес/вес = 1/1) (10 мл) и перемешивали в течение 5 минут. Водную фазу экстрагировали этилацетатом (10 мл × 3). Объединенные органические фазы сушили над безводным Na2SO4, фильтровали и концентрировали in vacuo. Остаток разделяли и очищали посредством препаративной хроматографии (прибор: GX-G; колонка: Phenomenex Synergi C18 150*30 мм*4 мкм;подвижная фаза: MeCN: 20% - 60%; H2O (+0,0025 FA); скорость: 25 мл/мин.; контролируемая длина волны: 220 нм/254 нм; длительность прогона: 10 мин./15 мин.; температура колонки: 30oC) с получением (S)-2-метил-6-нитро-2-((2-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазола – соединения 27 (42,00 мг, 108,47 мкмоль, выход 45,54%, чистота 99%). 1H ЯМР (300 МГц, CDCl3) δ 7,52-7,47 (m, 3H), 4,39 (d, J = 9,8 Гц, 1H), 3,99-3,89 (m, 3H), 3,14-3,84 (m, 5H), 2,82 (d, J = 14,7 Гц, 1H), 1,68 (s, 3H). LCMS (ESI) масса/заряд: 384 (M+1).
[320] Вариант осуществления 28
[321] (S)-2-Метил-6-нитро-2-((2-(пиперидин-1-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
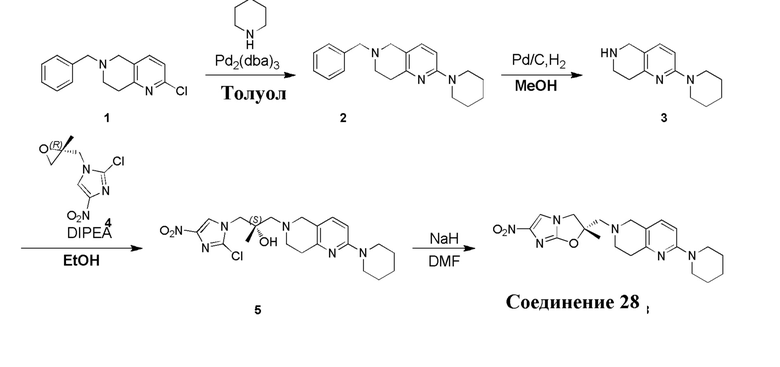
[322] Стадия 1.
[323] 6-Бензил-2-(1-пиперидинил)-7,8-дигидро-5H-1,6-нафтиридин
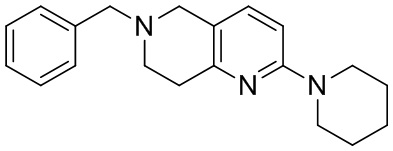
[324] Ключевое промежуточное соединение A (500,00 мг, 1,93 ммоль, 1,00 экв.) и пиперидин (328,68 мг, 3,86 ммоль, 2,00 экв.) растворяли в толуоле (5,00 мл), добавляли трет-бутоксид натрия (370,95 мг, 3,86 ммоль, 2,00 экв.), Pd2(dba)3 (88,37 мг, 96,5 мкмоль, 0,05 экв.), Xphos (92,01 мг, 193,00 мкмоль, 0,10 экв.) при 30°C в атмосфере газообразного азота. Смесь перемешивали при 100°C в течение 12 часов, затем охлаждали и концентрировали при пониженном давлении при 45°C. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 20/1, 5/1) с получением 6-бензил-2-(1-пиперидинил)-7,8-дигидро-5H-1,6-нафтиридина (550,00 мг, 1,79 ммоль, выход 92,70%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,43-7,39 (m, 2H), 7,39-7,33 (m, 2H), 7,33-7,29 (m, 1H), 7,09 (d, J = 8,5 Гц, 1H), 6,47 (d, J = 8,5 Гц, 1H), 3,71 (s, 2H), 3,52 (s, 2H), 3,51-3,46 (m, 4H), 2,91-2,87 (m, 2H), 2,84-2,80 (m, 2H), 1,67-1,61 (m, 6H). LCMS (ESI) масса/заряд: 308 (M+1).
[325] Стадия 2.
[326] 2-(1-Пиперидинил)-5,6,7,8-тетрагидро-1,6-нафтиридин
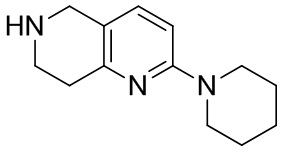
[327] 6-Бензил-2-(1-пиперидинил)-7,8-дигидро-5H-1,6-нафтиридин (500,00 мг, 1,63 ммоль, 1,00 экв.) растворяли в метаноле (20,00 мл), добавляли Pd/C (100,00 мг, 1,63 ммоль, 1,00 экв.) при 30°C в атмосфере газообразного азота. Смесь перемешивали при H2 (50 фунтов/кв. дюйм) при 30°C в течение 18 часов. Смесь фильтровали и концентрировали при пониженном давлении при 45°C с получением 2-(1-пиперидинил)-5,6,7,8-тетрагидро-1,6-нафтиридина (410,00 мг, неочищенный) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительного очищения.
[328] Стадия 3.
[329] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-2-метил-3-(2-(1-пиперидинил)-7,8-дигидро-5H-1,6-нафтиридин-6-ил)пропан-2-ол

[330] 2-(1-Пиперидинил)-5,6,7,8-тетрагидро-1,6-нафтиридин (200,00 мг, 920,34 мкмоль, 1,00 экв.) и (R)-2-хлор-1-((2-метилоксиран-2-ил)метил)-4-нитро-1H-имидазол (300,41 мг, 1,38 ммоль, 1,50 экв.) растворяли в этаноле (3,00 мл), добавляли DIPEA (59,47 мг, 460,17 мкмоль, 0,50 экв.) при 30°C в атмосфере газообразного азота. Смесь перемешивали при 80°C в течение 12 часов, охлаждали и концентрировали при пониженном давлении при 45°C. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 10/1, 1/1) с получением (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-2-метил-3-(2-(1-пиперидинил)-7,8-дигидро-5H-1,6-нафтиридин-6-ил)пропан-2-ола (200,00 мг, неочищенный) в виде желтого твердого вещества.
[331] Стадия 4.
[332] (S)-2-Метил-6-нитро-2-((2-(пиперидин-1-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
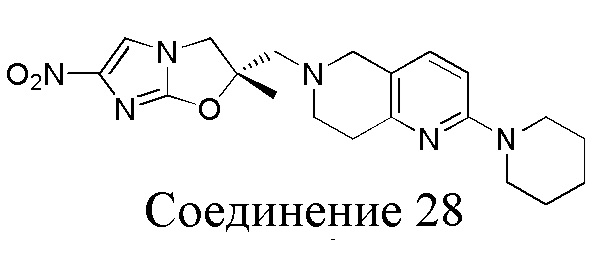
[333] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-2-метил-3-(2-(1-пиперидинил)-7,8-дигидро-5H-1,6-нафтиридин-6-ил)пропан-2-ол (139,00 мг, 319,60 мкмоль, 1,00 экв.) растворяли в DMF (5,00 мл) и добавляли NaH (25,57 мг, 639,20 мкмоль, 2,00 экв.) при -45°C в атмосфере газообразного азота. Далее смесь перемешивали при температуре от -45 до -15°C в течение 1 часа, добавляли воду (3 мл) и смесь перемешивали в течение 5 минут. Водную фазу экстрагировали дихлорметаном (20 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (10 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток обрабатывали посредством тонкослойной хроматографии (петролейный эфир/этилацетат = 1/2,5), а затем очищали посредством препаративной хроматографии (GX-D; Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; ацетонитрил 24% - 54%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-метил-6-нитро-2-((2-(пиперидин-1-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазола – соединения 28 (5,40 мг, 13,55 мкмоль, выход 4,24%). 1H ЯМР (400 МГц, CDCl3): δ 7,51 (s, 1H), 7,07 (d, J = 8,66 Гц, 1H), 6,46 (d, J = 8,53 Гц, 1H), 4,43 (d, J = 9,66 Гц, 1H), 3,91 (d, J = 9,54 Гц, 1H), 3,73-3,66 (m, 2H), 3,48 (br. S., 4H), 3,08-3,02 (m, 2H), 2,90-2,89 (m, 2H), 2,72 (d, J = 14,81 Гц, 2H), 1,65 (s, 3H), 1,63 (br. s., 6H). LCMS (ESI) масса/заряд: 399 (M+1).
[334] Вариант осуществления 29
[335] (S)-2-Метил-6-нитро-2-((2-(тетрагидро-2H-пиран-4-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
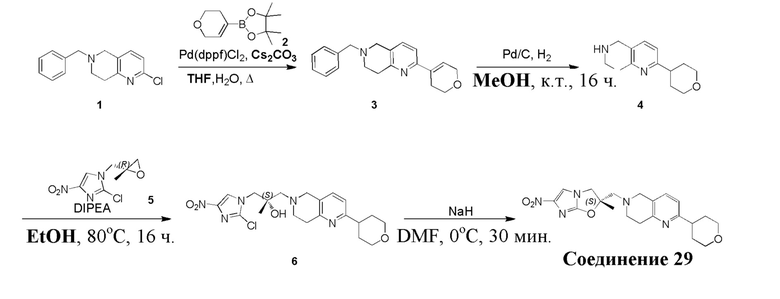
[336] Стадия 1.
[337] 6-Бензил-2-(3,6-дигидро-2H-пиран-4-ил)-5,6,7,8-тетрагидро-1,6-нафтиридин
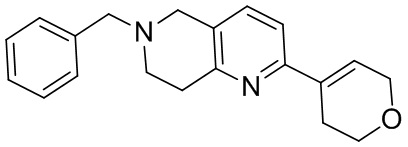
[338] Ключевое промежуточное соединение A (1,33 г, 5,14 ммоль, 1,20 экв.), 2-(3,6-дигидро-2H-пиран-4-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (900,00 мг, 4,28 ммоль, 1,00 экв.) растворяли в THF (20,00 мл) и воде (5,00 мл), добавляли к смешанному раствору Pd(dppf)Cl2 (313,17 мг, 428,00 мкмоль, 0,10 экв.) и карбонат цезия (2,79 г, 8,56 ммоль, 2,00 экв.) в атмосфере газообразного азота. Затем смесь перемешивали при 80°C в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, остаток разбавляли водой (20 мл) и экстрагировали этилацетатом (10 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (10 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали посредством хроматографии на силикагеле (диоксид кремния, PE: этилацетат = 5: 1) с получением 6-бензил-2-(3,6-дигидро-2H-пиран-4-ил)-5,6,7,8-тетрагидро-1,6-нафтиридина (960,00 мг, 3,13 ммоль, выход 73,20%) в виде белого твердого вещества. LCMS (ESI) масса/заряд: 308 (M + 1).
[339] Стадия 2.
[340] 2-(Тетрагидро-2H-пиран-4-ил)-5,6,7,8-тетрагидро-1,6-нафтиридин
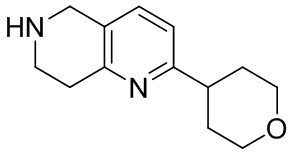
[341] 6-Бензил-2-(3,6-дигидро-2H-пиран-4-ил)-5,6,7,8-тетрагидро-1,6-нафтиридин (500,00 мг, 1,62 ммоль, 1,00) растворяли в метаноле (10,00 мл) и добавляли Pd/C (10%, 0,05 г) в атмосфере газообразного азота. В смешанном растворе три раза производили замену на водород и перемешивали при 28°C, H2 (50 фунтов/кв. дюйм) в течение 12 часов. Реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного 2-(тетрагидро-2H-пиран-4-ил)-5,6,7,8-тетрагидро-1,6-нафтиридина (400,00 мг, неочищенный) в виде бесцветного масла, которое использовали на следующей стадии без дополнительного очищения. LCMS (ESI) масса/заряд: 219 (M + 1).
[342] Стадия 3.
[343] (S)-2-Метил-2-((2-морфолино-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
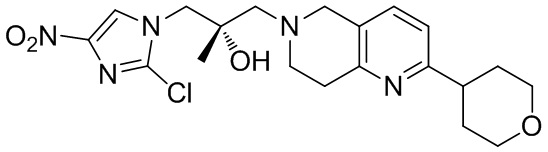
[344] 2-(Тетрагидро-2H-пиран-4-ил)-5,6,7,8-тетрагидро-1,6-нафтиридин (350,00 мг, 1,60 ммоль, 1,00 экв.), (R)-2-хлор-1-((2-метилоксиран-2-ил)метил)-4-нитро-1H-имидазол (417,81 мг, 1,92 ммоль, 1,20 экв.), DIPEA (620,35 мг, 4,80 ммоль, 3,00 экв.) добавляли к этанолу (10,00 мл), смесь перемешивали при 80°C в течение 12 часов в атмосфере газообразного азота. Реакционную смесь концентрировали при пониженном давлении и остаток очищали посредством хроматографии на силикагеле (диоксид кремния, дихлорметан/метанол = 20/1) с получением (S)-2-метил-2-((2-морфолино-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола (530,00 мг, 1,22 ммоль, выход 75,99%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,06 (s, 1H), 7,29 (d, J = 8,0 Гц, 1H), 7,01 (d, J = 8,0 Гц, 1H), 4,13-4,07 (m, 2H), 4,06 (s, 2H), 3,92-3,72 (m, 2H), 3,62-3,49 (m, 2H), 3,15-2,89 (m, 5H), 2,74-2,50 (m, 2H), 1,89-1,84 (m, 4H), 1,21 (s, 3H). LCMS (ESI) масса/заряд: 436/438 (M+1).
[345] Стадия 4.
[346] (S)-2-Метил-6-нитро-2-((2-(тетрагидро-2H-пиран-4-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
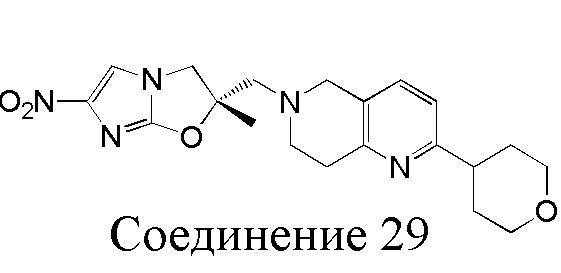
[347] (S)-2-Метил-2-((2-морфолино-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол (530,00 мг, 1,22 ммоль, 1,00 экв.) растворяли в DMF (5,00 мл), добавляли NaH (58,56 мг, 1,46 ммоль, 1,20 экв.) при 0°C. Смесь перемешивали при 0°C в течение 20 минут и гасили насыщенным раствором хлорида аммония (30 мл), а затем разбавляли водой (10 мл) и экстрагировали DCM (10 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (10 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток разделяли и очищали посредством препаративной хроматографии (GX-G; Phenomenex Synergi C18 150*30 мм*4 мкм; ацетонитрил 0% - 30%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-метил-6-нитро-2-((2-(тетрагидро-2H-пиран-4-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазола – соединения 29 (300,00 мг, 751,05 мкмоль, выход 61,56%). 1H ЯМР (400 МГц, CDCl3): δ 7,52 (s, 1H), 7,27 (d, J = 8,0 Гц, 1H), 6,98 (d, J = 8,0 Гц, 1H), 4,42 (d, J = 8,0 Гц, 1H), 4,13-4,06 (m, 2H), 3,95 (d, J = 12,0 Гц, 1H), 3,89-3,75 (m, 2H), 3,60-3,51 (m, 2H), 3,15-3,04 (m, 2H), 3,01-2,87 (m, 4H), 2,79 (d, J = 12,0 Гц, 1H), 1,90-1,81 (m, 4H), 1,68 (s, 3H). LCMS (ESI) масса/заряд: 399 (M+1).
[348] Вариант осуществления 30
[349] (S)-2-Метил-2-((2-морфолино-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
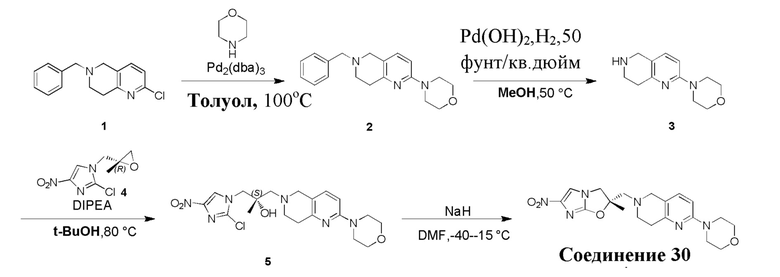
[350] Стадия 1.
[351] 4-(6-Бензил-7,8-дигидро-5H-1,6-нафтиридин-2-ил)морфолин
[352] Ключевое промежуточное соединение A (1,00 г, 3,86 ммоль, 1,00 экв.) и морфолин (672,57 мг, 7,72 ммоль, 2,00 экв.), Pd2(dba)3 (176,73 мг, 193,00 мкмоль, 0,05 экв.), трет-бутоксид натрия (741,92 мг, 7,72 ммоль, 2,00 экв.), Xphos (184,01 мг, 386,00 мкмоль, 0,10 экв.) растворяли в толуоле (20,00 мл) при 30°C в атмосфере газообразного азота. Затем смесь нагревали до 100°C и перемешивали в течение 12 часов. Смесь охлаждали и концентрировали при пониженном давлении при 45°C. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 20/1, 1/1) с получением 4-(6-бензил-7,8-дигидро-5H-1,6-нафтиридин-2-ил)морфолина (1,10 г, 3,56 ммоль, выход 92,11%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,43-7,39 (m, 2H), 7,36 (t, J = 7,3 Гц, 2H), 7,31 (d, J = 7,0 Гц, 1H), 7,15 (d, J = 8,4 Гц, 1H), 6,45 (d, J = 8,5 Гц, 1H), 3,86-3,81 (m, 4H), 3,72 (s, 2H), 3,54 (s, 2H), 3,49-3,44 (m, 4H), 2,92-2,87 (m, 2H), 2,85-2,81 (m, 2H). LCMS (ESI) масса/заряд: 310 (M+1).
[353] Стадия 2.
[354] 4-(6-Бензил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)морфолин
[355] 4-(6-Бензил-7,8-дигидро-5H-1,6-нафтиридин-2-ил)морфолин (700,00 мг, 2,26 ммоль, 1,00 экв.) растворяли в метаноле (20,00 мл), затем добавляли Pd(OH)2/C (10%, 20 мг). В смешанном растворе три раза производили замену на водород, а затем перемешивали при 50°C и H2 (50 фунтов/кв. дюйм) в течение 12 часов. Реакционную смесь фильтровали и фильтрат концентрировали с получением 4-(5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)морфолина (400,00 мг, неочищенный) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 220 (M + 1).
[356] Стадия 3.
[357] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-2-метил-3-(2-морфолино-7,8-дигидро-5H-1,6-нафтиридин-6-ил)пропан-2-ол
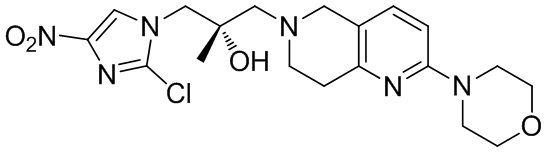
[358] 4-(5,6,7,8-Тетрагидро-1,6-нафтиридин-2-ил)морфолин (250,00 мг, 1,14 ммоль, 1,00 экв.) и (R)-2-хлор-1-((2-метилоксиран-2-ил)метил)-4-нитро-1H-имидазол (198,46 мг, 912,00 мкмоль, 0,80 экв.) растворяли в трет-бутаноле (3,00 мл) при 30°C в атмосфере газообразного азота. Затем добавляли DIPEA (147,34 мг, 1,14 ммоль, 1,00 экв.). Смесь перемешивали при 80°C в течение 12 часов, охлаждали и концентрировали при пониженном давлении при 45°C. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 10/1, 2/1) с получением (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-2-метил-3-(2-морфолино-7,8-дигидро-5H-1,6-нафтиридин-6-ил)пропан-2-ола (200,00 мг, 457,78 мкмоль, выход 40,16%) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 437 (M + 1).
[359] Стадия 4.
[360] (S)-2-Метил-2-((2-морфолино-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол

[361] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-2-метил-3-(2-морфолино-7,8-дигидро-5H-1,6-нафтиридин-6-ил)пропан-2-ол (200,00 мг, 457,78 мкмоль, 1,00 экв.) растворяли в DMF (5,00 мл), добавляли NaH (36,62 мг, 915,56 мкмоль, 2,00 экв.) при -45°C в атмосфере газообразного азота. Смесь перемешивали при температуре от -45 до 15°C в течение 2 часов, затем гасили хлоридом аммония (20 мл). Водную фазу экстрагировали этилацетатом (30 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством препаративной хроматографии (GX-A; Phenomenex Gemini C18 250*50 10u; (0,05% аммиак-ACN); 25 мл/мин.) с получением (S)-2-метил-2-((2-морфолино-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 30 (39,10 мг, 97,06 мкмоль, выход 21,2%, чистота 99,4%). 1H ЯМР (400 МГц, CDCl3): δ 7,51 (s, 1H), 7,13 (d, J = 8,53 Гц, 1H), 6,45 (d, J = 8,53 Гц, 1H), 4,43 (d, J = 9,66 Гц, 1H), 3,93 (d, J = 9,54 Гц, 1H), 3,83-3,81 (m, 4H), 3,72-3,68 (m, 2H), 3,47-3,44 (m, 4H), 3,07-3,04 (m, 2H), 2,76 (ddd, J = 12,02, 7,18, 4,77 Гц, 1H) 2,75-2,71 (m, 3H) 1,66 (s, 3H). LCMS (ESI) масса/заряд: 401 (M+1).
[362] Вариант осуществления 31
[363] (S)-2-((2-(4,4-Дифторпиперидин-1-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
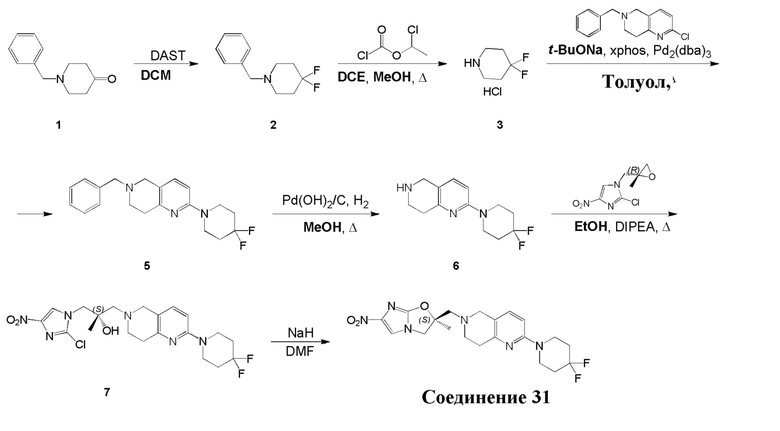
[364] Стадия 1.
[365] Бензил-4,4-дифторпиперидин
[366] 1-Бензил-4-он (1,00 г, 5,28 ммоль, 1,00 экв.) растворяли в DCM (10,00 мл) и добавляли DAST (2,56 г, 15,85 ммоль, 3,00 экв.) при 0°C в атмосфере газообразного азота. Смесь перемешивали при 0°C в течение 0,5 часа, затем нагревали до 15°C и перемешивали в течение 12 часов. Смесь добавляли к насыщенному раствору бикарбоната натрия (60 мл) при 0°C и экстрагировали смесь DCM (100 мл × 4). Объединенные органические фазы промывали насыщенным солевым раствором (40 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo с получением 1-бензил-4,4-дифтор-пиперидина (1,20 г, неочищенный) в виде черного твердого вещества, которое использовали на следующей стадии без дополнительного очищения. 1H ЯМР (400 МГц, CDCl3): δ 7,39-7,29 (m, 5H), 3,66-3,52 (m, 2H), 2,57 (t, J = 5,3 Гц, 4H), 2,02 (ddd, J = 19,7, 13,7, 5,8 Гц, 4H).
[367] Стадия 2.
[368] 4,4-Дифторпиперидина гидрохлорид
[369] 1-Бензил-4,4-дифторпиперидин (1,20 г, 5,68 ммоль, 1,00 экв.) растворяли в дихлорэтане (10,00 мл), добавляли 1-хлорэтилхлорформиат (1,22 г, 8,52 ммоль, 1,50 экв.) при 0°C в атмосфере газообразного азота. Смесь перемешивали при данной температуре в течение 0,5 ч, затем нагревали до 85°C и перемешивали в течение 12 часов. Смесь концентрировали, к остатку добавляли метанол (10,00 мл) и смесь перемешивали при 85°C в течение дополнительных 2 часов. Смесь охлаждали и концентрировали при пониженном давлении при 60°C с получением 4,4-дифторпиперидина гидрохлорида (580,00 мг, неочищенный) в виде черного твердого вещества, которое использовали на следующей стадии без дополнительного очищения.
[370] Стадия 3.
[371] 6-Бензил-2-(4,4-дифторпиперидин-1-ил)-5,6,7,8-тетрагидро-1,6-нафтиридин
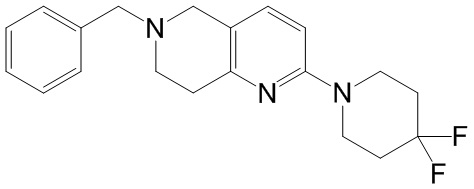
[372] 6-Бензил-2-хлор-7,8-дигидро-5H-1,6-нафтиридин (1,03 г, 3,98 ммоль, 1,00 экв.) и 4,4-дифторпиперидин (960,00 мг, 7,93 ммоль, 1,99 экв.) растворяли в толуоле (15,00 мл) при 15°C в атмосфере газообразного азота. А далее добавляли трет-бутоксид натрия (956,36 мг, 9,95 ммоль, 2,50 экв.), дициклогексил-[2-(2,4,6-триизопропил)фенил]фосфин (284,65 мг, 597,10 мкмоль, 0,15 экв.) и Pd2(dba)3 (291,61 мг, 318,45 мкмоль, 0,08 экв.). Смесь перемешивали при 110°C в течение 12 часов, охлаждали и концентрировали при пониженном давлении при 60°C. К остатку добавляли воду (20 мл) и водную фазу экстрагировали дихлорметаном (100 мл × 4). Объединенные органические фазы промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo с получением 6-бензил-2-(4,4-дифторпиперидин-1-ил)-5,6,7,8-тетрагидро-1,6-нафтиридина (700,00 мг, 1,67 ммоль, выход 41,92%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 7,49-7,30 (m, 5H), 7,21-7,06 (m, 1H), 6,58-6,47 (m, 1H), 3,78-3,66 (m, 5H), 3,64-3,46 (m, 2H), 2,86 (dd, J = 18,7, 5,4 Гц, 3H), 2,10-1,95 (m, 3H), 1,40-1,20 (m, 2H), 1,03-0,79 (m, 2H). LCMS (ESI) масса/заряд: 344 (M+1).
[373] Стадия 4.
[374] 2-(4,4-Дифторпиперидин-1-ил)-5,6,7,8-тетрагидро-1,6-нафтиридин
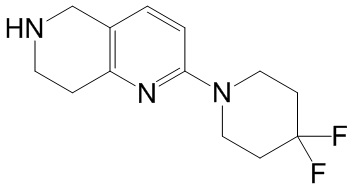
[375] 6-Бензил-2-(4,4-дифторпиперидин-1-ил)-5,6,7,8-тетрагидро-1,6-нафтиридин (600,00 мг, 1,75 ммоль, 1,00 экв.) растворяли в метаноле (30,00 мл) и добавляли Pd(OH)2/C (24,22 мг, 174,98 мкмоль, 0,10 экв.) при 15°C. Далее смесь перемешивали в течение 12 часов при 60°C, H2 (50 фунтов/кв. дюйм). Смесь фильтровали и фильтрат концентрировали, а остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 10/1, 1/10) с получением 2-(4,4-дифторпиперидин-1-ил)-5,6,7,8-тетрагидро-1,6-нафтиридина (250,00 мг, 987,01 мкмоль, выход 56,40%) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 254 (M + 1).
[376] Стадия 5.
[377] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(4,4-дифторпиперидин-1-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-метилпропан-2-ол
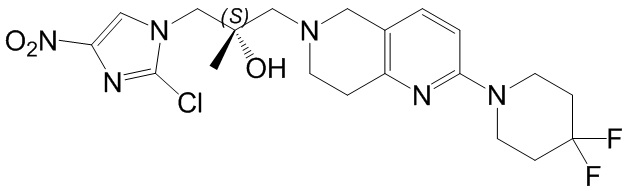
[378] 2-(4,4-Дифторпиперидин-1-ил)-5,6,7,8-тетрагидро-1,6-нафтиридин (100,00 мг, 394,80 мкмоль, 1,00 экв.) и 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазол (85,91 мг, 394,80 мкмоль, 1,00 экв.), DIPEA (127,56 мг, 987,00 мкмоль, 2,50 экв.) растворяли в трет-бутаноле (5,00 мл) при 15°C в атмосфере газообразного азота. Смесь перемешивали при 85°C в течение 12 часов, охлаждали и концентрировали при 60°C. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 20/1, 1/2) с получением (S)-1-(2-хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(4,4-дифторпиперидин-1-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-метилпропан-2-ола (200,00 мг, неочищенный) в виде желтого масла. LCMS (ESI) масса/заряд: 471 (M + 1).
[379] Стадия 6.
[380] (S)-2-((2-(4,4-Дифторпиперидин-1-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
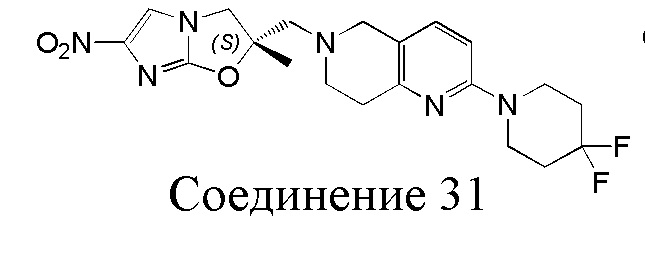
[381] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(4,4-дифторпиперидин-1-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-метилпропан-2-ол (200,00 мг, 424,72 мкмоль, 1,00 экв.) растворяли в DMF (5,00 мл) и добавляли NaH (20,39 мг, 849,44 мкмоль, 2,00 экв.) при -20°C в атмосфере газообразного азота. Смесь перемешивали при -20°C в течение 10 минут, а затем повышали температуру до 0°C и перемешивали в течение 10 минут. Затем смесь перемешивали при 15°C в течение дополнительных 10 минут и гасили насыщенным водным раствором хлорида аммония (50 мл). Смесь фильтровали и неочищенную сушили с получением неочищенного продукта, который очищали посредством препаративной хроматографии (GX-D; Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; ацетонитрил 24% - 54%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-((2-(4,4-дифторпиперидин-1-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 31 (15,00 мг, 33,88 мкмоль, выход 7,98%, чистота 98,12%). 1H ЯМР (400 МГц, метанол-d4): δ 7,81 (s, 1H), 7,21 (d, J = 8,4 Гц, 1H), 6,67 (d, J = 8,4 Гц, 1H), 4,40 (d, J = 10,3 Гц, 1H), 4,11 (d, J = 10,3 Гц, 1H), 3,75-3,61 (m, 6H), 3,11-2,98 (m, 2H), 2,95-2,85 (m, 2H), 2,77-2,67 (m, 1H), 2,66-2,55 (m, 1H), 2,06-1,91 (m, 4H), 1,66 (s, 3H). LCMS (ESI) масса/заряд: 435 (M+1).
[382] Вариант осуществления 32
[383] (S)-2-Метил-6-нитро-2-((2-(3,4,5-трифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
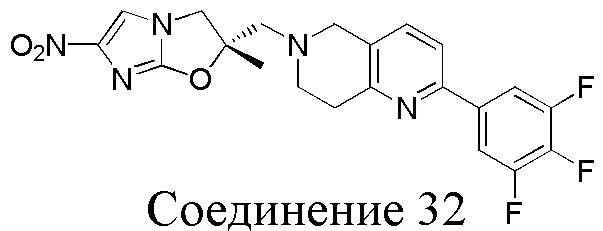
[384] Способ синтеза был таким, как в варианте осуществления 29.
[385] (S)-2-Метил-6-нитро-2-((2-(3,4,5-трифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол – соединение 32 (123,50 мг, 272,34 мкмоль, выход 52,49%, чистота 98,216%). 1H ЯМР (400 МГц, CDCl3) δ 7,68-7,59 (m, 2H), 7,52 (s, 1H), 7,46-7,37 (m, 2H), 4,43 (d, J = 12,0 Гц, 1H), 3,97 (d, J = 8,0 Гц, 1H), 3,95-3,81 (m, 2H), 3,24-2,92 (m, 5H), 2,82 (d, J = 12,0 Гц, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 446 (M+1).
[386] Вариант осуществления 33
[387] (S)-N-(4-Фторфенил)-6-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6,7,8-тетрагидро-1,6-нафтиридин-2-амин
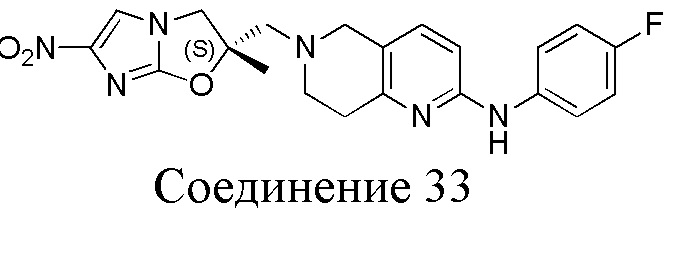
[388] Способ синтеза был таким, как в варианте осуществления 30.
[389] (S)-N-(4-Фторфенил)-6-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6,7,8-тетрагидро-1,6-нафтиридин-2-амин – соединение 33 (9,50 мг, 22,09 мкмоль, выход 6,79%, чистота 98,7%). 1H ЯМР (400 МГц, CDCl3): δ 8,47 (s, 1H), 7,97 (br. s., 1H), 7,53 (s, 1H), 7,25 (d, J = 4,6 Гц, 1H), 7,18 (d, J = 8,5 Гц, 1H), 7,05 (t, J = 8,5 Гц, 2H), 6,63 (d, J = 8,5 Гц, 1H), 4,41 (d, J = 9,5 Гц, 1H), 3,96 (d, J = 9,8 Гц, 1H), 3,79-3,65 (m, 2H), 3,12-3,04 (m, 2H), 2,94 (dd, J = 6,3, 11,4 Гц, 1H), 2,88-2,73 (m, 3H), 1,67 (s, 3H). LCMS (ESI) масса/заряд: 425 (M+1).
[390] Вариант осуществления 34
[391] (S)-2-((2-(4-Фторфенокси)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
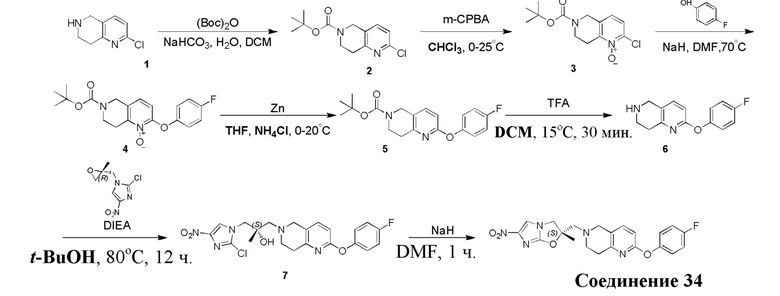
[392] Стадия 1.
[393] Трет-бутил-2-хлор-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилат
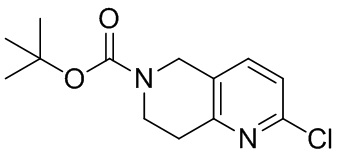
[394] 2-Хлор-5,6,7,8-тетрагидро-1,6-нафтиридин (850,00 мг, 4,14 ммоль, 1,00 экв.) и ди-трет-бутилдикарбонат (1,36 г, 6,22 ммоль, 1,50 экв.) растворяли в смешанном растворе дихлорметана (15,00 мл) и воды (15,00 мл), добавляли бикарбонат натрия (1,04 г, 12,43 ммоль, 3,00 экв.) при 15°C. Смесь перемешивали при 15°C в течение 2 часов. Смесь выливали в воду (30 мл) и водную фазу экстрагировали этилацетатом (50 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 50/1, 30/1) с получением трет-бутил-2-хлор-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилата (1,00 г, 3,72 ммоль, выход 89,75%) в виде белого твердого вещества.
[395] Стадия 2.
[396] Трет-бутил-2-хлор-1-оксо-7,8-дигидро-5H-1,6-нафтиридин-1-оний-6-карбоксилат
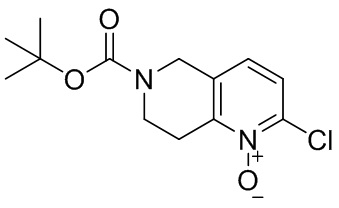
[397] Трет-бутил-2-хлор-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилат (1,20 г, 4,47 ммоль, 1,00 экв.) растворяли в хлороформе (20,00 мл) и добавляли к смеси м-хлорпербензойную кислоту (1,45 г, 6,71 ммоль, 1,50 экв.) при 0°C. Смесь перемешивали при 25°C в течение 12 часов. Затем смесь гасили насыщенным раствором сульфата натрия (20 мл) и экстрагировали дихлорметаном (40 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo с получением трет-бутил-2-хлор-1-оксо-7,8-дигидро-5H-1,6-нафтиридин-1-оний-6-карбоксилата (1,00 г, 3,51 ммоль, выход 78,52%) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии. LCMS (ESI) масса/заряд: 285 (M + 1).
[398] Стадия 3.
[399] 6-(Трет-бутоксикарбонил)-2-(4-фторфенокси)-5,6,7,8-тетрагидро-1,6-нафтиридин-1-оксид

[400] Трет-бутил-2-хлор-1-оксо-7,8-дигидро-5H-1,6-нафтиридин-1-оний-6-карбоксилат (600,00 мг, 2,11 ммоль, 1,00 экв.) и 4-фторфенол (283,46 мг, 2,53 ммоль, 1,20 экв.) растворяли в DMF (3,00 мл), добавляли NaH (168,57 мг, 4,21 ммоль, 2,00 экв.) при 0°C в атмосфере газообразного азота. Смесь перемешивали при 70°C в течение 12 часов, а затем охлаждали. Остаток выливали в воду (15 мл) и экстрагировали этилацетатом (30 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали, концентрировали in vacuo и остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 5/1, 1/3) с получением 6-(трет-бутоксикарбонил)-2-(4-фторфенокси)-5,6,7,8-тетрагидро-1,6-нафтиридин-1-оксида (510,00 мг, 1,42 ммоль, выход 67,07%) в виде желтого твердого вещества.
[401] Стадия 4.
[402] Трет-бутил-2-(4-фторфенокси)-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилат
[403] 6-(Трет-бутоксикарбонил)-2-(4-фторфенокси)-5,6,7,8-тетрагидро-1,6-нафтиридин-1-оксид (410,00 мг, 1,14 ммоль, 1,00 экв.) растворяли в хлориде аммония (3,00 мл) и THF (3,00 мл) и добавляли цинк (745,45 мг, 11,40 ммоль, 10,00 экв.) при 0°C в атмосфере газообразного азота. Смесь перемешивали при 15°C в течение 2 часов. Смесь фильтровали и фильтрат экстрагировали этилацетатом (30 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток затем очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 20/1, 10/1) с получением трет-бутил-2-(4-фторфенокси)-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилата (420,00 мг, неочищенный) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 7,41 (d, J = 8,4 Гц, 1H), 7,12-7,05 (m, 4H), 6,66 (d, J = 8,4 Гц, 1H), 4,55 (s, 2H), 3,72 (t, J = 5,8 Гц, 2H), 2,92-2,81 (m, 2H), 1,51 (s, 9H).
[404] Стадия 5.
[405] 2-(4-Фторфенокси)-5,6,7,8-тетрагидро-1,6-нафтиридин
[406] Трет-бутил-2-(4-фторфенокси)-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилат (200,00 мг, 580,75 мкмоль, 1,00 экв.) растворяли в дихлорметане (1,00 мл), добавляли TFA (66,24 мг, 580,75 мкмоль, 1,00 экв.) при 15°C и смесь перемешивали в течение 1 часа. Затем смесь концентрировали при пониженном давлении с получением 2-(4-фторфенокси)-5,6,7,8-тетрагидро-1,6-нафтиридина (177,00 мг, неочищенная соль TFA) в виде желтого масла.
[407] Стадия 6.
[408] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенокси)-7,8-дигидро-5H-1,6-нафтиридин-6-ил)-2-метилпропан-2-ол
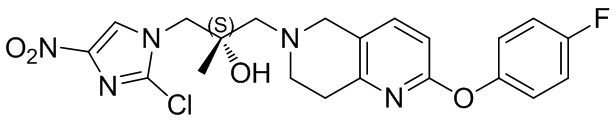
[409] 2-(4-Фторфенокси)-5,6,7,8-тетрагидро-1,6-нафтиридин (177,00 мг, 494,01 мкмоль, 1,00 экв., соль TFA) растворяли в трет-бутаноле (2,00 мл), добавляли DIPEA (191,54 мг, 1,48 ммоль, 3,00 экв.), 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазол (129,00 мг, 592,81 мкмоль, 1,20 экв.) при 15°C. Смесь перемешивали при 70°C в течение 12 часов, охлаждали и концентрировали при пониженном давлении при 45°C. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 10/1, 3/1) с получением (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенокси)-7,8-дигидро-5H-1,6-нафтиридин-6-ил)-2-метилпропан-2-ола (130,00 мг, 281,46 мкмоль, выход 56,97%) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 462 (M + 1).
[410] Стадия 7.
[411] (S)-2-((2-(4-Фторфенокси)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
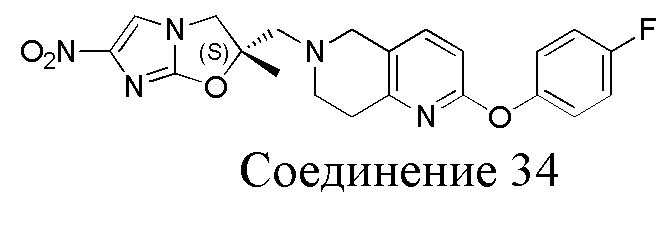
[412] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенокси)-7,8-дигидро-5H-1,6-нафтиридин-6-ил)-2-метилпропан-2-ол (160,00 мг, 346,42 мкмоль, 1,00 экв.) растворяли в DMF (2,00 мл), добавляли NaH (27,71 мг, 692,84 мкмоль, 2,00 экв.) при -45°C в атмосфере газообразного азота. Смесь перемешивали при температуре от -45 до 15°C в течение 1 часа. Далее смесь гасили насыщенным раствором хлорида аммония (20 мл) и перемешивали в течение 5 минут. Водную фазу экстрагировали этилацетатом (40 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток разделяли и очищали посредством препаративной хроматографии (GX-G; Phenomenex Synergi Max-RP 250*80 10u; ацетонитрил 30% - 60%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-((2-(4-фторфенокси)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 34 (56,00 мг, 129,00 мкмоль, выход 37,24%, чистота 98%). 1H ЯМР (400 МГц, CDCl3): δ 7,52 (s, 1H), 7,30 (br. s., 1H), 7,09 (s, 2H), 7,07 (d, J = 1,9 Гц, 2H), 6,60 (d, J = 8,3 Гц, 1H), 4,41 (d, J = 9,8 Гц, 1H), 3,95 (d, J = 9,7 Гц, 1H), 3,79 (q, J = 14,9 Гц, 2H), 3,11-3,03 (m, 2H), 2,93 (td, J = 6,0, 11,6 Гц, 1H), 2,84-2,74 (m, 3H), 1,68 (s, 3H). LCMS (ESI) масса/заряд: 426 (M+1).
[413] Вариант осуществления 35
[414] (S)-2-(3,4-Дифторфенил)-6-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбонитрил
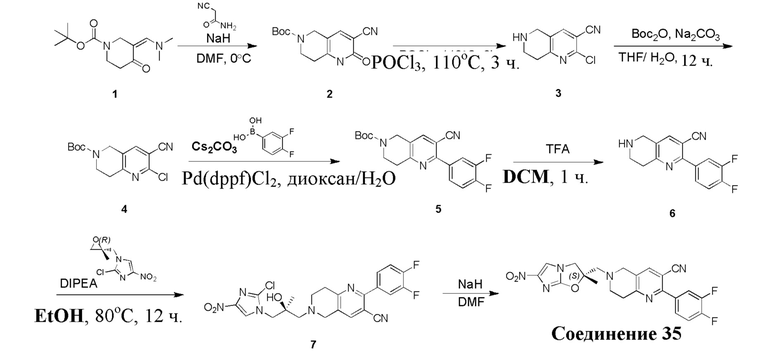
[415] Стадия 1.
[416] Трет-бутил-3-циано-2-оксо-1,2,7,8-тетрагидро-1,6-нафтиридин-6(5H)-карбоксилат
[417] (Z)-Трет-бутил-3-((диметиламино)метилен)-4-оксопиперидин-1-карбоксилат (16,00 г, 62,91 ммоль, 1,00 экв.) растворяли в DMF (120,00 мл), добавляли порциями NaH (5,03 г, 125,82 ммоль, 2,00 экв.) при 0°C и перемешивали в течение 1 часа. После добавления смесь перемешивали при данной температуре в течение дополнительных 30 минут, затем 2-цианоацетамид растворяли (5,55 г, 66,06 ммоль, 1,05 экв.) в DMF (80,00 мл), который добавляли по каплям к смешанному раствору, при этом температуру поддерживали постоянной. Полученную в результате смесь перемешивали при 28°C в течение 12 часов. Концентрировали полученную в результате смесь при пониженном давлении и остаток очищали посредством хроматографии на силикагеле (диоксид кремния, DCM / этилацетат = 1/1, 1: 5) с получением трет-бутил-3-циано-2-оксо-1,2,7,8-тетрагидро-1,6-нафтиридин-6(5H)-карбоксилата (1,00 г, 3,63 ммоль, выход 5,77%) в виде темно-коричневого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 8,04 (s, 1H), 4,24 (s, 2H), 3,55 (t, J = 4,0 Гц, 2H), 2,64 (t, J = 4,0 Гц, 2H), 1,42 (s, 9H).
[418] Стадия 2.
[419] 2-Хлор-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбонитрил
[420] Трет-бутил-3-циано-2-оксо-1,2,7,8-тетрагидро-1,6-нафтиридин-6(5H)-карбоксилат (2,00 г, 7,26 ммоль, 1,00 экв.) добавляли к оксихлориду фосфора (28,22 г, 184,05 ммоль, 25,35 экв.). Смесь перемешивали при 110°C в течение 3 часов. Реакционную смесь выливали в воду (1500 мл) с температурой 28°C, а затем доводили pH до 10 путем добавления карбоната натрия. Смесь экстрагировали DCM (500 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (500 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта 2-хлор-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбонитрила (2,00 г, неочищенный), который использовали непосредственно на следующей стадии.
[421] Стадия 3.
[422] Трет-бутил-2-хлор-3-циано-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилат
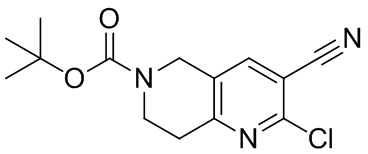
[423] 2-Хлор-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбонитрил (1,40 г, 7,23 ммоль, 1,00 экв.) растворяли в THF (20,00 мл) и воде (10,00 мл), добавляли ди-трет-бутилдикарбонат (3,16 г, 14,46 ммоль, 2,00 экв.) и карбонат натрия (2,30 г, 21,69 ммоль, 3,00 экв.). Смесь перемешивали при 25°C в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении, остаток разбавляли DCM (20 мл) и экстрагировали DCM (20 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (20 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (диоксид кремния, петролейный эфир/этилацетат = 10/1, 5/1) с получением трет-бутил-2-хлор-3-циано-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилата (700,00 мг, 2,38 ммоль, выход 32,96%) в виде желтого твердого вещества. 1H ЯМР (300МГц, CDCl3) δ 7,74 (s, 1H), 4,61 (s, 2H), 3,75 (t, J = 8,0 Гц, 2H), 3,03 (t, J = 8,0 Гц, 2H), 1,48 (s, 9H).
[424] Стадия 4.
[425] Трет-бутил-3-циано-2-(3,4-дифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилат
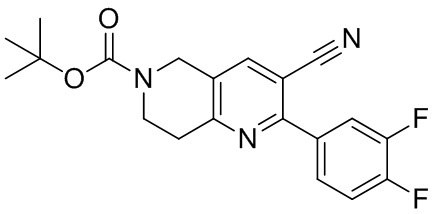
[426] Трет-бутил-2-хлор-3-циано-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилат (350,00 мг, 1,19 ммоль, 1,00 экв.), (3,4-дифторфенил)бороновую кислоту (225,50 мг, 1,43 ммоль, 1,20 экв.), карбонат цезия (775,45 мг, 2,38 ммоль, 2,00 экв.) растворяли в смешанном растворе диоксана (10,00 мл) и воды (4,00 мл), добавляли Pd(dppf)Cl2 (87,07 мг, 119,00 мкмоль, 0,10 экв.) в атмосфере газообразного азота. Затем смесь перемешивали при 110°C в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении, остаток разбавляли DCM (20 мл) и экстрагировали DCM (20 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (20 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (диоксид кремния, петролейный эфир/этилацетат =10/1, 5/1) с получением трет-бутил-3-циано-2-(3,4-дифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилата (420,00 мг, 1,13 ммоль, выход 95,04%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,53 (s, 1H), 8,32-8,25 (m, 1H), 8,24-8,19 (m, 1H), 7,31-7,22 (m, 1H), 4,65 (s, 2H), 3,81 (t, J = 6,0 Гц, 2H), 3,03 (t, J = 6,0 Гц, 2H), 1,53 (s, 9H).
[427] Стадия 5.
[428] 2-(3,4-Дифторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбонитрил
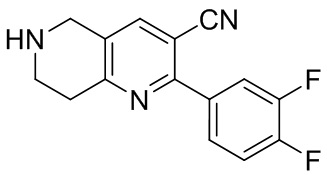
[429] Трет-бутил-3-циано-2-(3,4-дифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилат (246,00 мг, 662,39 мкмоль, 1,00 экв.) растворяли в DCM (2,00 мл), а затем добавляли TFA (75,52 мг, 662,39 мкмоль, 1,00 экв.) и смесь перемешивали при 25°C в течение 1 часа. Смесь концентрировали при пониженном давлении с получением продукта 2-(3,4-дифторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбонитрила (250,00 мг, неочищенный), который использовали непосредственно на следующей стадии без очищения.
[430] Стадия 6.
[431] (S)-6-(3-(2-Хлор-4-нитро-1H-имидазол-1-ил)-2-гидрокси-2-метилпропил)-2-(3,4-дифторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбонитрил
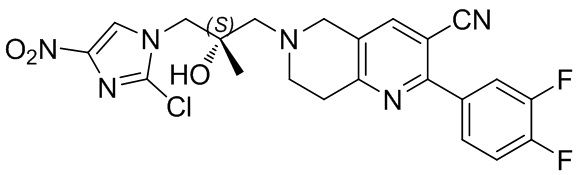
[432] 2-(3,4-Дифторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбонитрил (180,00 мг, 663,57 мкмоль, 1,00 экв.) и (R)-2-хлор-1-((2-метилоксиран-2-ил) метил)-4-нитро-1H-имидазол (173,28 мг, 796,28 мкмоль, 1,20 экв.) растворяли в трет-бутаноле (10,00 мл), затем добавляли DIPEA (257,28 мг, 1,99 ммоль, 3,00 экв.). Смесь перемешивали при 80°C в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали посредством хроматографии на силикагеле (диоксид кремния, петролейный эфир/этилацетат = 5/1, 1: 1) с получением (S)-6-(3-(2-хлор-4-нитро-1H-имидазол-1-ил)-2-гидрокси-2-метилпропил)-2-(3,4-дифторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбонитрила (230,00 мг, 470,47 мкмоль, выход 70,90%) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 489/491 (M + 1 / M + 3).
[433] Стадия 7.
[434] (S)-2-(3,4-Дифторфенил)-6-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбонитрил
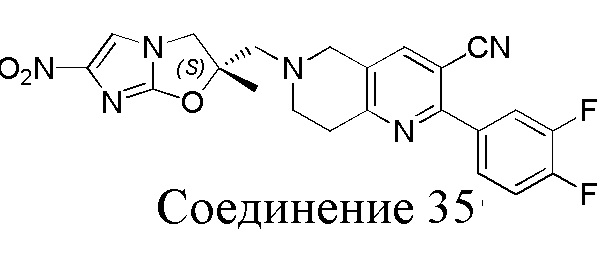
[435] (S)-6-(3-(2-Хлор-4-нитро-1H-имидазол-1-ил)-2-гидрокси-2-метилпропил)-2-(3,4-дифторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбонитрил (230,00 мг, 470,47 мкмоль, 1,00 экв.) растворяли в DMF (5,00 мл), добавляли NaH (22,58 мг, 564,56 мкмоль, 1,20 экв.) при 0°C. Смесь перемешивали при 0°C в течение 10 минут, гасили насыщенным раствором хлорида аммония (20 мл), затем разбавляли водой (20 мл) и экстрагировали DCM (20 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (20 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток разделяли и очищали посредством препаративной хроматографии (GX-F; Welch Ultimate AQ-C18 150*30 мм*5 мкм; ацетонитрил 43% - 73%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-(3,4-дифторфенил)-6-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбонитрила – соединения 35 (20,90 мг, 42,91 мкмоль, выход 9,12%, чистота 92,884%). 1H ЯМР (400 МГц, CDCl3) δ 7,80-7,66 (m, 3H), 7,53 (s, 1H), 7,36-7,29 (m, 1H), 4,37 (d, J = 12,0 Гц, 1H), 4,00 (d, J = 8,0 Гц, 1H), 3,93 (d, J=13,2 Гц, 2H), 3,25-2,98 (m, 5H), 2,84 (d, J = 16,0 Гц, 1H), 1,70 (s, 3H). LCMS (ESI) масса/заряд: 453 (M+1).
[436] Вариант осуществления 36
[437] (S)-2-((3-Хлор-2-(3,4-дифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
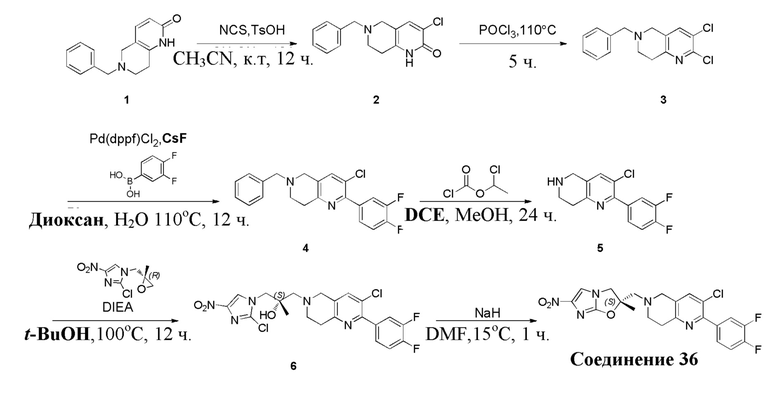
[438] Стадия 1.
[439] 6-Бензил-3-хлор-1,5,7,8-тетрагидро-1,6-нафтиридин-2-он

[440] 4-Метилбензолсульфоновую кислоту (4,30 г, 24,98 ммоль, 1,50 экв.) и 6-бензил-1,5,7,8-тетрагидро-1,6-нафтиридин-2-он (4,00 г, 16,65 ммоль, 1,00 экв.) растворяли в ацетонитриле (30,00 мл), затем добавляли NCS (3,33 г, 24,98 ммоль, 1,50 экв.). Смесь перемешивали при 25°C в течение 12 часов. Реакционную смесь гасили водой и экстрагировали реакционную смесь этилацетатом (250 мл × 2). Объединенные органические фазы концентрировали при пониженном давлении и остаток очищали посредством хроматографии на силикагеле (диоксид кремния, петролейный эфир/этилацетат = 3/1, дихлорметан/метанол = 10/1) с получением 6-бензил-3-хлор-5,6,7,8-тетрагидро-1,6-нафтиридин-2(1H)-она (220,00 мг, 750,37 мкмоль, выход 29,43%) в виде белого твердого вещества. LCMS (ESI) масса/заряд: 275,1 (M + 1).
[441] Стадия 2.
[442] 6-Бензил-2,3-дихлор-7,8-дигидро-5H-1,6-нафтиридин
[443] 6-Бензил-3-хлор-1,5,7,8-тетрагидро-1,6-нафтиридин-2-он (700,00 мг, 2,55 ммоль, 1,00 экв.) и оксихлорид фосфора (3,33 г, 21,73 ммоль, 8,53 экв.) растворяли в толуоле (5,00 мл), смесь нагревали при 100°C в течение 5 часов. Реакционную смесь выливали в воду (100 мл) при 25°C, доводили pH до приблизительно 9 за счет постепенного добавления раствора карбоната натрия и экстрагировали этилацетатом (200 мл × 2). Объединенные органические фазы концентрировали при пониженном давлении и остаток очищали посредством хроматографии на силикагеле (диоксид кремния, петролейный эфир/этилацетат = 80/1, 20/1) с получением 6-бензил-2,3-дихлор-7,8-дигидро-5H-1,6-нафтиридина (220,00 мг, 750,37 мкмоль, выход 29,43%) в виде белого твердого вещества. LCMS (ESI) масса/заряд: 293,0 (M + 1).
[444] Стадия 3.
[445] 6-Бензил-3-хлор-2-(3,4-дифторфенил)-7,8-дигидро-5H-1,6-нафтиридин
[446] 6-Бензил-2,3-дихлор-7,8-дигидро-5H-1,6-нафтиридин (200,00 мг, 682,15 мкмоль, 1,00 экв.), (3,4-дифторфенил)бороновую кислоту (0,295 мг, 613,94 мкмоль, 0,90 экв.), фторид цезия (207,24 мг, 1,36 ммоль, 2,00 экв.) растворяли в диоксане (3 мл) и воде (300,00 мкл), добавляли Pd(dppf)Cl2 (49,91 мг, 68,22 мкмоль, 0,10 экв.) в атмосфере газообразного азота и смесь перемешивали при 110°C в течение 12 часов. Смесь концентрировали in vacuo и остаток очищали посредством хроматографии на силикагеле (диоксид кремния, петролейный эфир/этилацетат = 50/1, 20: 1) с получением 6-бензил-3-хлор-2-(3,4-дифторфенил)-7,8-дигидро-5H-1,6-нафтиридина (170,00 мг, 458,44 мкмоль, выход 67,21%) в виде белого твердого вещества. LCMS (ESI) масса/заряд: 370,9 (M + 1).
[447] Стадия 4.
[448] 3-Хлор-2-(3,4-дифторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорид
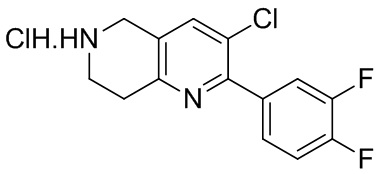
[449] 6-Бензил-3-хлор-2-(3,4-дифторфенил)-7,8-дигидро-5H-1,6-нафтиридин (70,00 г, 377,54 мкмоль, 1,00 экв.) и 1-хлорэтилхлорформиат (70,17 мг, 490,80 мкмоль, 1,30 экв.) растворяли в дихлорэтане (100,00 мл). Смесь перемешивали при 80°C в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении, добавляли к реакционной смеси метанол (100,00 мл) и смесь перемешивали при 80°C в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта 3-хлор-2-(3,4-дифторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорида (140,00 мг, неочищенный продукт, соляная кислота), который использовали на следующей стадии без дополнительного очищения. LCMS (ESI) масса/заряд: 280,9 (M + 1).
[450] Стадия 5.
[451] (2S)-1-(3-Хлор-2-(3,4-дифторфенил)-7,8-дигидро-5H-1,6-нафтиридин-6-ил)-3-(2-хлор-4-нитроимидазол-1-ил)-2-метилпропан-2-ол
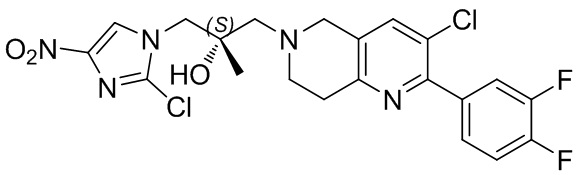
[452] 3-Хлор-2-(3,4-дифторфенил)-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорид (140,00 мг, 498,75 мкмоль, 1,00 экв.) растворяли в трет-бутаноле (5,00 мл), а затем добавляли DIPEA (161,15 мг, 1,25 ммоль, 2,50 экв.) и 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазол (130,24 мг, 598,50 мкмоль, 1,20 экв.). Смесь перемешивали при 100°C в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали посредством хроматографии на силикагеле (диоксид кремния, петролейный эфир/этилацетат = 20/1, 1/3) с получением (2S)-1-[3-хлор-2-(3,4-дифторфенил)-7,8-дигидро-5H-1,6-нафтиридин-6-ил]-3-(2-хлор-4-нитроимидазол-1-ил)-2-метилпропан-2-ола (80,00 мг, 160,54 мкмоль, выход 32,19%) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 499,8 (M + 1).
[453] Стадия 6.
[454] (S)-2-((3-Хлор-2-(3,4-дифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол

[455] (2S)-1-[3-Хлор-2-(3,4-дифторфенил)-7,8-дигидро-5H-1,6-нафтиридин-6-ил]-3-(2-хлор-4-нитроимидазол-1-ил)-2-метилпропан-2-ол (80,00 мг, 160,54 мкмоль, 1,00 экв.) растворяли в DMF (2,00 мл), добавляли NaH (3,85 мг, 160,54 мкмоль, 1,00 экв.) при -20°C и перемешивали в течение 30 минут, а затем смесь перемешивали при 15°C в течение 1 часа. Реакционную смесь гасили за счет добавления к воде (15 мл) при 0°C, а затем экстрагировали этилацетатом (100 мл × 2). Объединенные органические фазы концентрировали при пониженном давлении и остаток очищали посредством препаративной хроматографии (GX-A, Phenomenex Gemini C18 250*50 мм*10 мкм, ацетонитрил 50% - 80%; 0,05% аммиак-ACN; 25 мл/мин.) с получением (S)-2-((3-хлор-2-(3,4-дифторфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 36 (17,80 мг, 38,38 мкмоль, выход 23,91%, чистота 99,580%). 1H ЯМР (400 МГц, CDCl3) δ 7,53 (s, 1H), 7,45 (s, 1H), 7,28-7,20 (m, 1H), 4,40 (d, J =9,8 Гц, 1H), 4,03-3,79 (m, 3H), 3,22-3,07 (m, 2H), 3,05-2,75 (m, 4H), 1,69 (s, 3H) LCMS (ESI) масса/заряд: 462,1 (M+1).
[456] Вариант осуществления 37
[457] (S)-2-((2-(3,4-Дифторфенил)-4-метил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
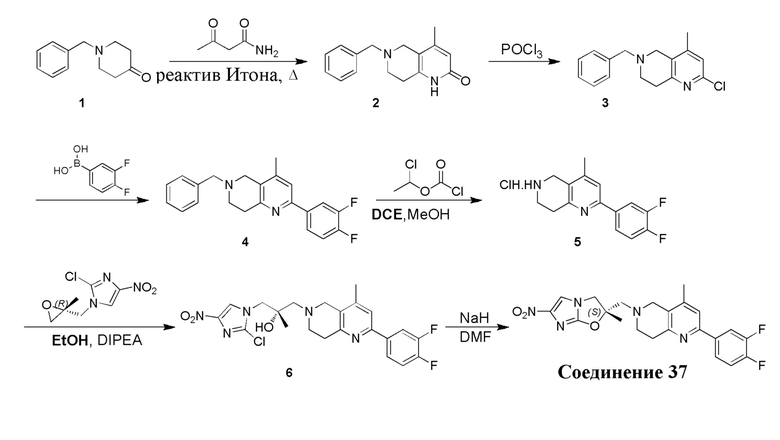
[458] Стадия 1.
[459] 6-Бензил-4-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-2(1H)-он
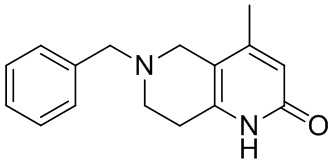
[460] Смесь 1-бензил-4-она (10,00 г, 52,84 ммоль, 1,00 экв.) и 3-оксобутанамида (5,88 г, 58,12 ммоль, 1,10 экв.) в реактиве Итона (20,00 мл) перемешивали при 110°C в течение 12 часов. Смесь добавляли к насыщенному водному раствору бикарбоната натрия (300 мл), чтобы довести pH до показателя > 7, и водную фазу экстрагировали этилацетатом (200 мл × 4). Объединенные органические фазы промывали насыщенным солевым раствором (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток выливали в ацетон (100 мл), а затем фильтровали и осадок сушили с получением 6-бензил-4-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-2(1H)-она (7,00 г, 27,52 ммоль, выход 52,09%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 7,46-7,25 (m, 5H), 6,25 (br, s, 1H), 3,76 (s, 2H), 3,42 (br, s, 2H), 2,84-2,64 (m, 4H), 2,10 (s, 3H).
[461] Стадия 2.
[462] 6-Бензил-2-хлор-4-метил-7,8-дигидро-5H-1,6-нафтиридин
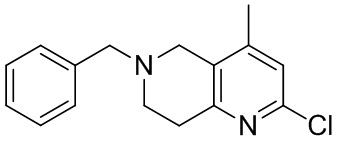
[463] 6-Бензил-4-метил-5,6,7,8-тетрагидро-1,6-нафтиридин-2(1H)-он (7,80 г, 30,67 ммоль, 1,00 экв.) добавляли к оксихлориду фосфора (97,44 г, 635,49 ммоль, 20,72 экв.). Смесь перемешивали при 110°C в течение 12 часов и охлаждали. Смесь гасили за счет добавления к воде (300 мл) и перемешивали в течение 30 минут. Водный слой затем подщелачивали до pH> 7 при помощи водного раствора бикарбоната натрия. Смесь экстрагировали DCM (200 мл × 4), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo с получением 6-бензил-2-хлор-4-метил-7,8-дигидро-5H-6-нафтиридина (5,00 г, неочищенный) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 273 (M + 1).
[464] Стадия 3.
[465] 6-Бензил-2-(3,4-дифторфенил)-4-метил-5,6,7,8-тетрагидро-1,6-нафтиридин
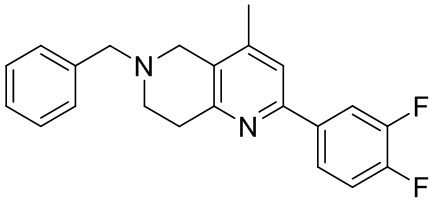
[466] 6-Бензил-2-хлор-4-метил-7,8-дигидро-5H-1,6-нафтиридин (1,87 г, 6,86 ммоль, 1,00 экв.) и (3,4-дифторфенил)бороновую кислоту (1,30 г, 8,23 ммоль, 1,20 экв.), фторид цезия (3,13 г, 20,58 ммоль, 3,00 экв.) растворяли в диоксане (25,00 мл) и воде (2,50 мл) при 15°C в атмосфере газообразного азота, а затем добавляли Pd(dppf)Cl2 (501,95 мг, 686,00 мкмоль, 0,10 экв.) и смесь перемешивали при 110°C в течение 12 часов. Смесь добавляли к воде (10 мл) и экстрагировали дихлорметаном (100 мл × 4). Объединенные органические фазы промывали насыщенным солевым раствором (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 20/1, 5/1) с получением 6-бензил-2-(3,4-дифторфенил)-4-метил-5,6,7,8-тетрагидро-1,6-нафтиридина (1,30 г, 3,71 ммоль, выход 54,08%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 7,83 (ddd, J = 2,1, 7,8, 11,7 Гц, 1H), 7,66 (ddd, J = 2,0, 4,2, 6,3 Гц, 1H), 7,46-7,30 (m, 6H), 7,27-7,18 (m, 1H), 3,83-3,78 (m, 2H), 3,64 (s, 2H), 3,11 (t, J = 5,8 Гц, 2H), 2,87 (t, J = 5,9 Гц, 2H), 2,23 (s, 3H).
[467] Стадия 4.
[468] 2-(3,4-Дифторфенил)-4-метил-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорид
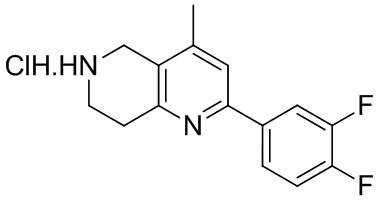
[469] 6-Бензил-2-(3,4-дифторфенил)-4-метил-5,6,7,8-тетрагидро-1,6-нафтиридин (1,30 г, 3,71 ммоль, 1,00 экв.) растворяли в дихлорэтане (20,00 мл), добавляли 1-хлоркарбонилхлорид (795,63 мг, 5,57 ммоль, 1,50 экв.) при 15°C в атмосфере газообразного азота и смесь перемешивали при 85°C в течение 12 часов. Затем смесь концентрировали, остаток добавляли в метанол (20, мл) и полученную в результате смесь перемешивали при 85°C в течение 2 часов. Смесь фильтровали, осадок собирали и сушили с получением 2-(3,4-дифторфенил)-4-метил-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорида (800,00 мг, 2,70 ммоль, выход 72,67%) в виде белого твердого вещества, которое использовали непосредственно на следующей стадии.
[470] Стадия 5.
[471] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(3,4-дифторфенил)-4-метил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-метилпропан-2-ол
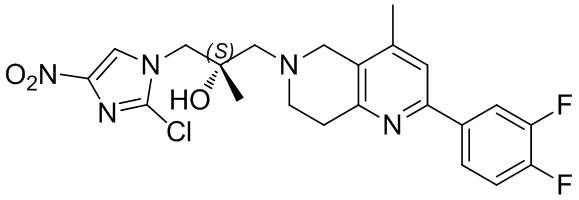
[472] 2-(3,4-Дифторфенил)-4-метил-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорид (800,00 мг, 2,70 ммоль, 1,00 экв.) и 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазол (705,06 мг, 3,24 ммоль, 1,20 экв.) растворяли в этаноле (20,00 мл), добавляли DIPEA (872,37 мг, 6,75 ммоль, 2,50 экв.) при 15°C в атмосфере газообразного азота. Смесь перемешивали при 80°C в течение 12 часов, охлаждали и концентрировали при 60°C при пониженном давлении и остаток добавляли к воде (10 мл). Водную фазу экстрагировали дихлорметаном (50 мл × 4), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (высота колонки: 250 мм, диаметр: 100 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 20/1, 1/1) с получением (S)-1-(2-хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(3,4-дифторфенил)-4-метил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-метилпропан-2-ола (800,00 мг, 1,67 ммоль, выход 62,00%) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 478 (M + l).
[473] Стадия 6.
[474] (S)-2-((2-(3,4-Дифторфенил)-4-метил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
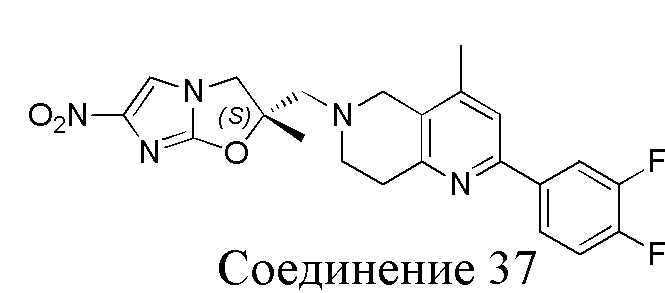
[475] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(3,4-дифторфенил)-4-метил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)-2-метилпропан-2-ол (400,00 мг, 837,01 мкмоль, 1,00 экв.) растворяли в DMF (5,00 мл) и добавляли NaH (40,18 мг, 1,67 ммоль, 2,00 экв.) при -20°C в атмосфере газообразного азота. Смесь перемешивали в течение 10 минут, а затем нагревали до -5°C в течение 10 минут. Затем смесь перемешивали при 15°C в течение дополнительных 10 минут. Смесь охлаждали до 0°C и гасили насыщенным водным раствором хлорида аммония (30 мл). Далее смесь фильтровали, собирали отфильтрованный осадок и сушили с получением неочищенного продукта, который очищали посредством препаративной хроматографии (GX-D; Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; ацетонитрил 24% - 54%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-((2-(3,4-дифторфенил)-4-метил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 37 (97,50 мг, 215,7 мкмоль, выход 25,78%, чистота 97,7%). 1H ЯМР (400 МГц, метанол-d4): δ 7,88-7,78 (m, 2H), 7,71 (d, J = 8,5 Гц, 1H), 7,46 (s, 1H), 7,38-7,30 (m, 1H), 4,45 (d, J = 10,5 Гц, 1H), 4,14 (d, J = 10,3 Гц, 1H), 3,84 (s, 2H), 3,18-3,08 (m, 2H), 3,05-2,90 (m, 3H), 2,88-2,78 (m, 1H), 2,30 (s, 3H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 442 (M+1).
[476] Вариант осуществления 38
[477] 2-(3,4-Дифторфенил)-6-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-7,8-дигидро-1,6-нафтиридин-5(6H)-он
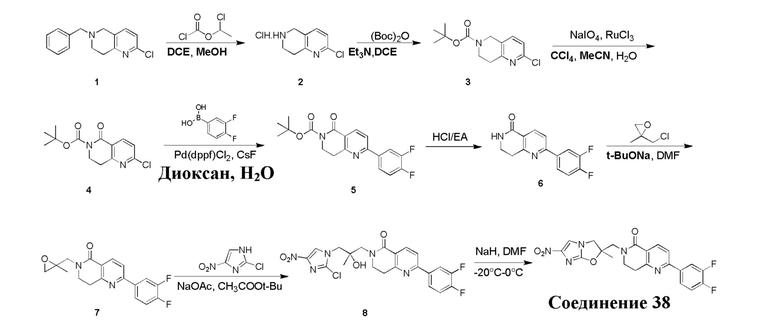
[478] Стадия 1.
[479] 2-Хлор-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорид
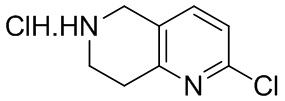
[480] 6-Бензил-2-хлор-7,8-дигидро-5H-1,6-нафтиридин (2,70 г, 10,43 ммоль, 1,00 экв.) растворяли в дихлорэтане (50,00 мл), добавляли 1-хлорэтилхлорформиат (2,24 г, 15,65 ммоль, 1,50 экв.) при 15°C в атмосфере газообразного азота. Смесь перемешивали при 85°C в течение 12 часов. Затем смесь концентрировали для удаления растворителя. Добавляли к остатку метанол (50,00 мл), а затем нагревали до 80°C и перемешивали в течение 2 часов. Смесь фильтровали, осадок собирали и сушили с получением 2-хлор-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорида (2,00 г, 9,75 ммоль, выход 93,50%) в виде белого твердого вещества, которое использовали непосредственно на следующей стадии.
[481] Стадия 2.
[482] Трет-бутил-2-хлор-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилат
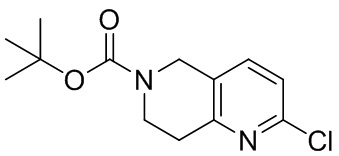
[483] 2-Хлор-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорид (2,00 г, 9,75 ммоль, 1,00 экв.) растворяли в DCM (20,00 мл) и добавляли триэтиламин (2,47 г, 24,38 ммоль, 2,50 экв.) и ди-трет-бутилдикарбонат (3,19 г, 14,63 ммоль, 1,50 экв.) при 15°C в атмосфере газообразного азота. Смесь перемешивали при 15°C в течение 12 часов. Смесь выливали в воду (30 мл) и водную фазу экстрагировали дихлорметаном (100 мл × 4). Объединенные органические фазы промывали насыщенным солевым раствором (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo с получением трет-бутил-2-хлор-7,8-дигидро-5H-1,6-нафтиридин-6-карбоксилата (2,50 г, 9,30 ммоль, выход 95,41%) в виде белого твердого вещества. LCMS (ESI) масса/заряд: 269 (M + 1).
[484] Стадия 3.
[485] Трет-бутил-2-хлор-5-оксо-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилат
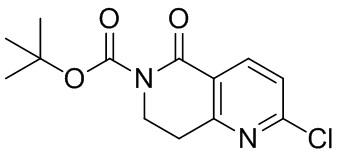
[486] Пентахлорат натрия (5,49 г, 25,68 ммоль, 3,00 экв.) и RuCl3 (532,58 мг, 2,57 ммоль, 0,30 экв.) добавляли к смешанному раствору трет-бутил-2-хлор-7,8-дигидро-5H-1,6-нафтиридин-6-карбоксилата (2,30 г, 8,56 ммоль, 1,00 экв.) в ацетонитриле (740,00 мкл), четыреххлористого углерода (37,00 мл) и воды (14,80 мл) при 15°C в атмосфере газообразного азота. Смесь перемешивали при 15°C в течение 12 часов. Смесь выливали в воду (20 мл), а затем экстрагировали дихлорметаном (100 мл × 4). Объединенные органические фазы промывали насыщенным солевым раствором (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (высота колонки: 250 мм, диаметр: 100 м, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 20/1 - 10/1) с получением трет-бутил-2-хлор-5-оксо-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилата (1,80 г, 6,37 ммоль, выход 74,38%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 8,38 (d, J = 8,3 Гц, 1H), 7,37 (d, J = 8,0 Гц, 1H), 4,13-4,04 (m, 2H), 3,24-3,12 (m, 2H), 1,60 (s, 9H). LCMS (ESI) масса/заряд: 283 (M+1).
[487] Стадия 4.
[488] Трет-бутил-2-(3,4-дифторфенил)-5-оксо-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилат
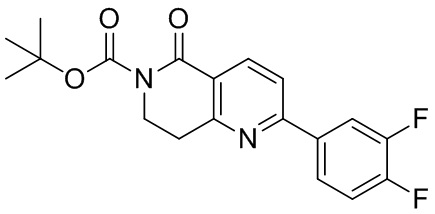
[489] Трет-бутил-2-хлор-5-оксо-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилат (1,80 г, 6,37 ммоль, 1,00 экв.) растворяли в диоксане (30,00 мл) и воде (3,00 мл), а затем добавляли (3,4-дифторфенил)бороновую кислоту (1,21 г, 7,64 ммоль, 1,20 экв.), фторид цезия (2,90 г, 19,11 ммоль, 3,00 экв.). Добавляли Pd(dppf)Cl2 (466,09 мг, 637,00 мкмоль, 0,10 экв.) при 15°C в атмосфере газообразного азота. Смесь перемешивали при 110°C в течение 12 часов. Смесь охлаждали до 15°C и концентрировали при пониженном давлении. Добавляли к остатку воду (20 мл) и водную фазу экстрагировали дихлорметаном (100 мл × 4). Объединенные органические фазы промывали насыщенным солевым раствором (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (высота колонки: 250 мм, диаметр: 100 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 20/1, 10/1) с получением трет-бутил-2-(3,4-дифторфенил)-5-оксо-7,8-дигидро-1,6-нафтиридин-6-карбоксилата (1,40 г, 3,89 ммоль, выход 60,99%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 8,49 (d, J = 8,3 Гц, 1H), 7,99 (ddd, J = 2,0, 7,8, 11,5 Гц, 1H), 7,82 (ddd, J = 2,0, 4,1, 6,4 Гц, 1H), 7,73 (d, J = 8,3 Гц, 1H), 7,35-7,29 (m, 1H), 4,18-4,08 (m, 2H), 3,28 (t, J = 6,4 Гц, 2H), 1,62 (s, 10H).
[490] Стадия 5.
[491] 2-(3,4-Дифторфенил)-7,8-дигидро-1,6-нафтиридин-5(6H)-он
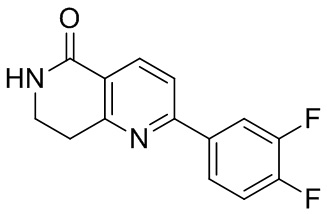
[492] Трет-бутил-2-(3,4-дифторфенил)-5-оксо-7,8-дигидро-1,6-нафтиридин-6-карбоксилат (500,00 мг, 1,39 ммоль, 1,00 экв.) растворяли в соляной кислоты/этилацетат (10,00 мл) и раствор перемешивали при 15°C в течение 1 часа. Смесь концентрировали до сухого состояния с получением 2-(3,4-дифторфенил)-7,8-дигидро-6H-1,6-нафтиридин-5-она (350,00 мг, 1,34 ммоль, выход 96,76%) в виде белого твердого вещества. LCMS (ESI) масса/заряд: 261 (M + 1).
[493] Стадия 6.
[494] 2-(3,4-Дифторфенил)-6-((2-метилоксиран-2-ил)метил)-7,8-дигидро-1,6-нафтиридин-5(6H)-он
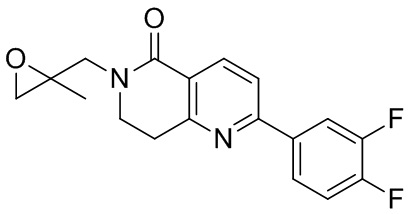
[495] Трет-бутоксид калия (215,59 мг, 1,92 ммоль, 2,00 экв.) добавляли к раствору 2-(3,4-дифторфенил)-7,8-дигидро-6H-1,6-нафтиридин-5-она (250,00 мг, 960,65 мкмоль, 1,00 экв.) и 2-(хлорметил)-2-метил-оксирана (153,54 мг, 1,44 ммоль, 1,50 экв.) в DMF (3,00 мл) при 15°C в атмосфере газообразного азота. Смесь перемешивали при 110°C в течение 3 часов. Добавляли к смеси воду (10 мл) и смесь экстрагировали этилацетатом (50 мл × 4). Объединенные органические фазы промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (высота колонки: 250 мм, диаметр 10 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 20/1, 1/1) с получением 2-(3,4-дифторфенил)-6-((2-метилоксиран-2-ил)метил)-7,8-дигидро-1,6-нафтиридин-5(6H)-она (240,00 мг, неочищенный) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 331 (M + 1).
[496] Стадия 7.
[497] 6-(3-(2-Хлор-4-нитро-1H-имидазол-1-ил)-2-гидрокси-2-метилпропил)-2-(3,4-дифторфенил)-7,8-дигидро-1,6-нафтиридин-5(6H)-он
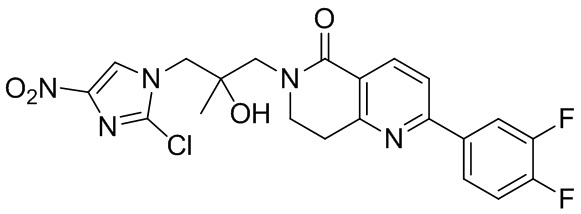
[498] 2-(3,4-Дифторфенил)-6-((2-метилоксиран-2-ил)метил)-7,8-дигидро-1,6-нафтиридин-5(6H)-он (150,00 мг, 454,09 мкмоль, 1,00 экв.) и 2-хлор-4-нитро-1H-имидазол (66,99 мг, 454,09 мкмоль, 1,00 экв.) растворяли в трет-бутилацетате (5,00 мл), добавляли ацетат натрия (37,25 мг, 454,09 мкмоль, 1,00 экв.) при 15°C в атмосфере газообразного азота. Смесь перемешивали при 110°C в течение 3 часов. Смесь охлаждали и концентрировали при пониженном давлении при 70°C. Остаток очищали посредством препаративной тонкослойной хроматографии (петролейный эфир/этилацетат = 1/1) с получением 6-(3-(2-хлор-4-нитро-1H-имидазол-1-ил)-2-гидрокси-2-метилпропил)-2-(3,4-дифторфенил)-7,8-дигидро-1,6-нафтиридин-5(6H)-она (40 мг, неочищенный) в виде белого твердого вещества. LCMS (ESI) масса/заряд: 478 (M + l).
[499] Стадия 8.
[500] 2-(3,4-Дифторфенил)-6-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-7,8-дигидро-1,6-нафтиридин-5(6H)-он

[501] 6-(3-(2-Хлор-4-нитро-1H-имидазол-1-ил)-2-гидрокси-2-метилпропил)-2-(3,4-дифторфенил)-7,8-дигидро-1,6-нафтиридин-5(6H)-он (30,00 мг, 83,71 мкмоль, 1,00 экв.) растворяли в DMF (2,00 мл), добавляли NaH (4,02 мг, 167,42 мкмоль, 2,00 экв.) при -20°C в атмосфере газообразного азота. Смесь перемешивали при -20°C в течение 10 минут, затем нагревали до 0°C и перемешивали в течение 10 минут, а далее перемешивали при 15°C в течение 10 минут. Смесь охлаждали до 0°C и гасили насыщенным водным раствором хлорида аммония (20 мл). Смесь экстрагировали этилацетатом (30 мл × 4). Объединенные органические фазы промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo, а остаток очищали посредством препаративной хроматографии (GX-D; Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; ацетонитрил 24% - 54%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением 2-(3,4-дифторфенил)-6-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-7,8-дигидро-1,6-нафтиридин-5(6H)-она – соединения 38 (8,00 мг, 18,12 мкмоль, выход 21,65%). 1H ЯМР (400 МГц, метанол-d4): δ 8,35 (d, J = 8,3 Гц, 1H), 8,13-8,02 (m, 1H), 7,91 (d, J = 8,3 Гц, 2H), 7,84 (s, 1H), 7,46-7,35 (m, 1H), 4,68-4,47 (m, 1H), 4,34-4,16 (m, 2H), 3,96-3,78 (m, 2H), 3,31-3,14 (m, 2H), 3,08-2,96 (m, 1H), 1,76 (s, 3H). LCMS (ESI) масса/заряд: 442 (M+1).
[502] Вариант осуществления 39
[503] (S)-2-((2-(4-Фторфенил)-6,7-дигидротиазоло[5,4-c]пиридин-5(4H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
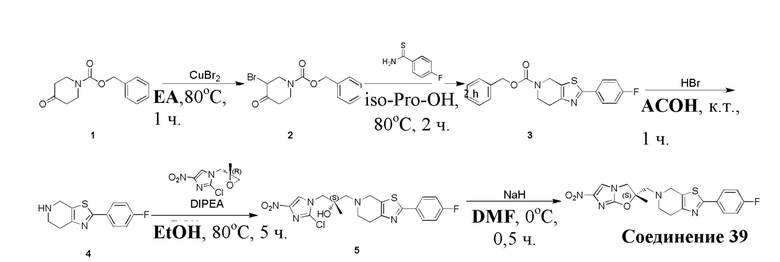
[504] Стадия 1.
[505] Бензил-3-бром-4-оксопиперидин-1-карбоксилат
[506] Бромид меди (3,83 г, 17,14 ммоль, 2,00 экв.) добавляли к раствору бензил-4-оксопиперидин-1-карбоксилата (2,00 г, 8,57 ммоль, 1,00 экв.) в этилацетате (10,00 мл) в атмосфере газообразного азота. Смесь перемешивали при 80°C в течение 1 часа. Смесь фильтровали и концентрировали при пониженном давлении с получением бензил-3-бром-4-оксопиперидин-1-карбоксилата (2,40 г, неочищенный), который использовали непосредственно на следующей стадии.
[507] Стадия 2.
[508] Бензил-2-(4-фторфенил)-6,7-дигидротиазоло[5,4-c]пиридин-5(4H)-карбоксилат
[509] Бензил-3-бром-4-оксопиперидин-1-карбоксилат (2,40 г, 7,69 ммоль, 1,00 экв.) и 4-фтортиобензамид (1,19 г, 7,69 ммоль, 1,00 экв.) растворяли в изопропаноле (20,00 мл), нагревали до 80°C и перемешивали в течение 2 часов. Смесь концентрировали при пониженном давлении и остаток очищали посредством хроматографии на силикагеле (высота колонки: 300 мм, диаметр: 50 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 30/1, 5/1) с получением бензил-2-(4-фторфенил)-6,7-дигидротиазоло[5,4-c]пиридин-5(4H)-карбоксилата (1,20 г, 3,26 ммоль, выход 42,36%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 8,12-7,73 (m, 2H), 7,49-7,31 (m, 5H), 7,19-7,06 (m, 2H), 5,22 (s, 2H), 4,78 (br. s., 2H), 3,89 (br. s., 2H), 2,98 (br. s., 2H).
[510] Стадия 3.
[511] 2-(4-Фторфенил)-4,5,6,7-тетрагидротиазоло[5,4-c]пиридин
[512] Бензил-2-(4-фторфенил)-6,7-дигидротиазоло[5,4-c]пиридин-5(4H)-карбоксилат (1,20 г, 3,26 ммоль, 1,00 экв.) растворяли в AcOH (5,00 мл). Добавляли раствор HBr в уксусной кислоте (48%, 5 мл) при 20°C и перемешивали в течение 1 часа. Смесь фильтровали, отфильтрованный осадок промывали этилацетатом и сушили in vacuo с получением 2-(4-фторфенил)-4,5,6,7-тетрагидротиазоло[5,4-c]пиридина (800,00 мг, 2,54 ммоль, выход 77,85%) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии.
[513] Стадия 4.
[514] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-6,7-дигидро-4H-тиазоло[5,4-c]пиридин-5-ил)-2-метилпропан-2-ол
[515] 2-Хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазол (150,00 мг, 689,31 мкмоль, 1,10 экв.) и 2-(4-фторфенил)-4,5,6,7-тетрагидротиазоло[5,4-c]пиридин (197,52 мг, 626,64 мкмоль, 1,00 экв.) растворяли в этаноле (5,00 мл), затем добавляли DIPEA (202,47 мг, 1,57 ммоль, 2,50 экв.). Смешанный раствор перемешивали при 80°C в течение 5 часов, охлаждали и концентрировали при пониженном давлении. Остаток разбавляли водой и экстрагировали этилацетатом (30 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo с получением (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-6,7-дигидро-4H-тиазоло[5,4-c]пиридин-5-ил)-2-метилпропан-2-ола (250,00 мг, неочищенный), который использовали непосредственно на следующей стадии.
[516] Стадия 5.
[517] (S)-2-((2-(4-Фторфенил)-6,7-дигидротиазоло[5,4-c]пиридин-5(4H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
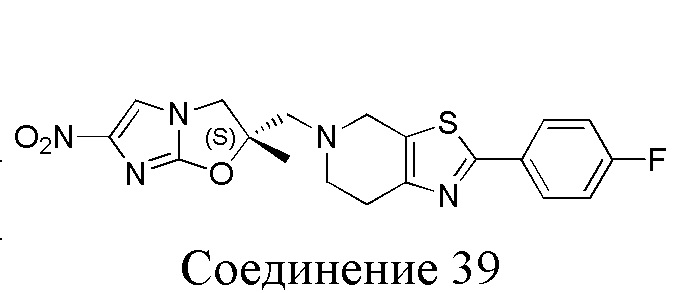
[518] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-6,7-дигидро-4H-тиазоло[5,4-c]пиридин-5-ил)-2-метилпропан-2-ол (150,00 мг, 331,93 мкмоль, 1,00 экв.) растворяли в DMF (3,00 мл), добавляли NaH (26,55 мг, 663,86 мкмоль, 2,00 экв.) при 0°C в атмосфере газообразного азота и перемешивали в течение 30 минут. Смесь выливали в ледяную воду (вес/вес = 1/1) (20 мл) и перемешивали в течение 10 минут. Водную фазу экстрагировали этилацетатом (20 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток разделяли и очищали посредством препаративной хроматографии (GX-A; Phenomenex Gemini C18 250*50 10u; 0,225% FA-ACN; начиная с 25 до 55; скорость потока (25 мл/мин.)) с получением (S)-2-((2-(4-фторфенил)-6,7-дигидротиазоло[5,4-c]пиридин-5(4H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 39 (20,00 мг, 47,18 мкмоль, выход 14,21%, чистота 98%). 1H ЯМР (400 МГц, CDCl3): δ 7,99-7,79 (m, 1H), 7,55 (s, 1H), 7,12 (t, J = 8,7 Гц, 2H), 4,42 (d, J = 9,7 Гц, 1H), 4,08-3,84 (m, 3H), 3,24-3,10 (m, 2H), 3,09-2,95 (m, 1H), 2,94-2,69 (m, 3H), 1,67 (s, 3H); LCMS (ESI) масса/заряд: 416 (M+1).
[519] Вариант осуществления 40
[520] (2S)-2-((2-(3,4-Дифторфенил)-6,7-дигидро-4H-тиазоло[5,4-c]пиридин-5-ил)метил)-2-метил-6-нитро-3H-имидазо[2,1-b]оксазол
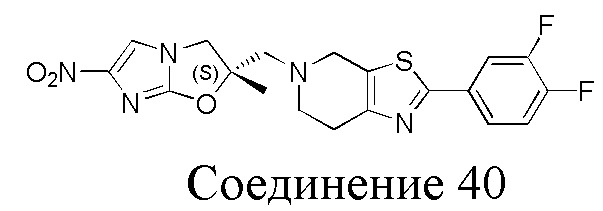
[521] Способ синтеза был таким, как в варианте осуществления 39.
[522] (2S)-2-((2-(3,4-Дифторфенил)-6,7-дигидро-4H-тиазоло[5,4-c]пиридин-5-ил)метил)-2-метил-6-нитро-3H-имидазо[2,1-b]оксазол – соединение 40 (30,00 мг, 92,29 мкмоль, 43,37%). 1H ЯМР (300МГц, CDCl3): δ 7,78-7,71 (m, 1H), 7,65-7,56 (m, 2H), 7,26-7,20 (m, 1H), 4,41 (d, J = 9,8 Гц, 1H), 4,06-3,85 (m, 3H), 3,23-2,95 (m, 3H), 2,93-2,68 (m, 3H), 1,67 (s, 3H); LCMS (ESI) масса/заряд: 434 (M+1).
[523] Вариант осуществления 41
[524] (S)-2-(3,4-Дифторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-4,5,6,7-тетрагидрооксазолo[5,4-c]пиридин
[525] Стадия 1.
[526] 1-Бензил-3,6-дигидро-2H-пиридин
[527] 1-Бензилпиридин-1-ия бромид (53,00 г, 211,89 ммоль, 1,00 экв.) растворяли в метаноле (500,00 мл), добавляли боргидрид натрия (12,02 г, 317,83 ммоль, 1,50 экв.). Смесь перемешивали при 25°C в течение 12 часов и концентрировали при пониженном давлении. Остаток разбавляли водой (250 мл) и экстрагировали этилацетатом (500 мл × 2). Объединенные органические слои концентрировали при пониженном давлении с получением l-бензил-3,6-дигидро-2H-пиридина (30,00 г, неочищенный), который использовали непосредственно на следующей стадии.
[528] Стадия 2.
[529] 4-Бензил-7-окса-4-азабицикло[4.1.0]гептан
[530] 1-Бензил-3,6-дигидро-2H-пиридин (10,00 г, 57,72 ммоль, 1,00 экв.) растворяли в воде (190,00 мл) и трифторуксусной кислоте (24,02 г, 210,68 ммоль, 3,65 экв.) при 25°C и перемешивали при данной температуре в течение 1 часа. Затем смесь нагревали до 35°C и добавляли бромсукцинимид (20,55 г, 115,44 ммоль, 2,00 экв.), далее смешанный раствор перемешивали в течение 5 часов, охлаждали до 25°C, добавляли к смешанному раствору гидроксид натрия (2,31 г, 57,72 ммоль, 1,00 экв.) и ацетонитрил (50,00 мл), а затем смешанный раствор перемешивали в течение 12 часов и концентрировали при пониженном давлении для удаления растворителя. Остаток экстрагировали этилацетатом (500 мл × 2). Объединенные органические слои концентрировали при пониженном давлении и остаток очищали посредством хроматографии на силикагеле (диоксид кремния, петролейный эфир/этилацетат = 10/1, 3: 1) с получением 4-бензил-7-окса-4-азабицикло[4.1.0]гептана (5,00 г, 26,42 ммоль, выход 45,77%) в виде желтого твердого вещества.
[531] Стадия 3.
[532] 4-Азидо-1-бензилпиперидин-3-ол
[533] 4-Бензил-7-окса-4-азабицикло[4.1.0]гептан (5,00 г, 26,42 ммоль, 1,00 экв.) и перхлорат лития (2,81 г, 26,42 ммоль, 1,00 экв.) растворяли в ацетонитриле (30,00 мл), добавляли азид натрия (2,23 г, 34,35 ммоль, 1,30 экв.) в атмосфере газообразного азота и смесь нагревали до 80°C, перемешивали в течение 16 часов. Смесь выливали в водный бикарбонат натрия (50 мл) и перемешивали в течение 10 минут. Водную фазу экстрагировали этилацетатом (50 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo с получением 4-азидо-1-бензил-пиперидин-3-ола (5,50 г, неочищенный), который использовали непосредственно на следующей стадии.
[534] Стадия 4.
[535] 4-Амино-1-бензилпиперидин-3-ол
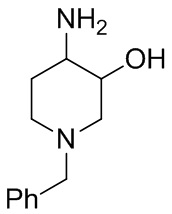
[536] 4-Азидо-1-бензил-пиперидин-3-ол (5,50 г, 23,68 ммоль, 1,00 экв.) растворяли в тетрагидрофуране (50,00 мл) и воде (2,00 мл), добавляли порциями трифенилфосфин (12,42 , 47,36 ммоль, 2,00 экв.) при 15°C и смесь перемешивали в течение 16 часов, а затем концентрировали in vacuo. Остаток очищали посредством разделения на силикагеле (высота колонки: 300 мм, диаметр: 50 мм, силикагель от 100 до 200 меш, дихлорметан/метанол = 50/1) с получением 4-амино-1-бензил-пиперидин-3-ола (3,40 г, 16,48 ммоль, выход 69,60%) в виде желтого твердого вещества.
[537] Стадия 5.
[538] N-(1-Бензил-3-гидрокси-4-пиперидинил)-3,4-дифторбензамид
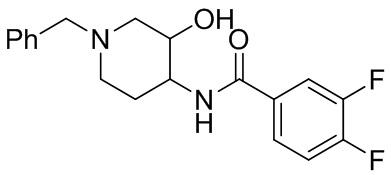
[539] 4-Амино-1-бензил-пиперидин-3-ол (2,00 г, 9,70 ммоль, 1,00 экв.) и 3,4-дифторбензойную кислоту (1,53 г, 9,70 ммоль, 1,00 экв.) растворяли в дихлорметане (30,00 мл), затем добавляли EDCI (3,72 г, 19,40 ммоль, 2,00 экв.), HOBT (2,62 г, 19,40 ммоль, 2,00 экв.), триэтиламин (3,93 г, 38,80 ммоль, 4,00 экв.) при 15°C и смесь перемешивали в течение 12 часов. Далее смесь разбавляли водой (20 мл) и водную фазу экстрагировали этилацетатом (40 мл × 3). Объединенные органические фазы промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток разделяли и очищали посредством тонкослойной хроматографии (петролейный эфир/этилацетат = 10/1, 1/3) с получением N-(1-бензил-3-гидрокси-4-пиперидинил)-3,4-дифторбензамида (1,70 г, 4,91 ммоль, выход 50,62%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 7,71-7,64 (m, 1H), 7,58-7,47 (m, 1H), 7,38-7,30 (m, 4H), 7,27-7,21 (m, 1H), 6,11 (d, J = 6,1 Гц, 1H), 3,92-3,82 (m, 1H), 3,66 (dt, J = 4,3, 9,3 Гц, 1H), 3,59 (s, 2H), 3,13 (dd, J = 3,2, 11,2 Гц, 1H), 2,87 (d, J = 11,8 Гц, 1H), 2,21-2,13 (m, 1H), 2,11-2,03 (m, 2H), 1,66 (dq, J = 4,3, 11,8 Гц, 1H). LCMS (ESI) масса/заряд: 347 (M+1).
[540] Стадия 6.
[541] 3,4-Дифтор-N-(3-гидроксипиперидин-4-ил)бензамид
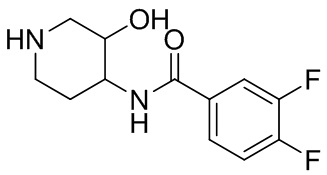
[542] N-(1-Бензил-3-гидрокси-4-пиперидинил)-3,4-дифторбензамид (1,70 г, 4,91 ммоль, 1,00 экв.) растворяли в метаноле (50,00 мл), добавляли Pd(OH)2/C (10%, 0,05 г) в атмосфере газообразного азота. Затем в смеси три раза производили замену на водород и смесь перемешивали при H2 (50 фунтов/кв. дюйм) при 50°C в течение 12 часов. Реакционную смесь фильтровали и фильтрат концентрировали с получением 3,4-дифтор-N-(3-гидрокси-4-пиперидинил)бензамида (1,20 г, неочищенный), который использовали на следующей стадии без дополнительного очищения. LCMS (ESI) масса/заряд: 257 (M + 1).
[543] Стадия 7.
[544] Бензил-4-(3,4-дифторбензамидо)-3-гидроксипиперидин-1-карбоксилат
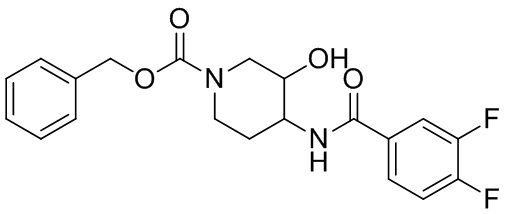
[545] Карбобензоксихлорид (878,75 мг, 5,15 ммоль, 1,10 экв.), 3,4-дифтор-N-(3-гидрокси-4-пиперидинил)бензамид (1,20 г, 4,68 ммоль, 1,00 экв.) растворяли в дихлорметане (30,00 мл), добавляли триэтиламин (1,42 г, 14,04 ммоль, 3,00 экв.) при 15°C и смесь перемешивали в течение 10 часов. Добавляли к смеси воду (20 мл) и водную фазу экстрагировали этилацетатом (40 мл × 3). Объединенные органические фазы промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток разделяли и очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 10/1, 1/1) с получением бензил-4-(3,4-дифторбензамидо)-3-гидроксипиперидин-1-карбоксилата (1,25 г, 3,20 ммоль, выход 68,42%) в виде белого твердого вещества. LCMS (ESI) масса/заряд: 391 (M + l).
[546] Стадия 8.
[547] Бензил-4-(3,4-дифторбензамидо)-3-оксопиперидин-1-карбоксилат

[548] Бензил-4-(3,4-дифторбензамидо)-3-гидроксипиперидин-1-карбоксилат (920,00 мг, 2,36 ммоль, 1,00 экв.) растворяли в дихлорметане (30,00 мл), добавляли DMP (3,00 г, 7,08 ммоль, 3,00 экв.) при 15°C и смесь перемешивали в течение 3 часов. Остаток затем выливали в водный раствор гидроксида натрия (0,5 н., 40 мл) и водную фазу экстрагировали дихлорметаном (60 мл × 3). Объединенные органические фазы промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 10/1, 1/3) с получением бензил-4-(3,4-дифторбензамидо)-3-оксопиперидин-1-карбоксилата (1,00 г, неочищенный) в виде желтого масла.
[549] Стадия 9.
[550] Бензил-2-(3,4-дифторфенил)-6,7-дигидро-4H-оксазолo[5,4-c]пиридин-5-карбоксилат
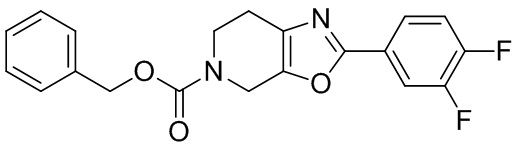
[551] Бензил-4-(3,4-дифторбензамидо)-3-оксопиперидин-1-карбоксилат (1,20 г, 3,09 ммоль, 1,00 экв.) добавляли к раствору оксихлорида фосфора (2,55 г, 16,63 ммоль, 5,38 экв.) в диоксане (20,00 мл) при 15°C в атмосфере газообразного азота. Далее смесь нагревали до 110°C и перемешивали в течение 3 часов. Остаток затем выливали в воду (50 мл) и перемешивали в течение 5 минут. Водную фазу экстрагировали этилацетатом (60 мл × 3) и объединенные органические фазы промывали солевым раствором (30 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (диоксид кремния, петролейный эфир/этилацетат = 1/0, 10/1) с получением бензил-2-(3,4-дифторфенил)-6,7-дигидро-4H-оксазолo[5,4-c]пиридин-5-карбоксилата (800,00 мг, 2,16 ммоль, выход 69,90%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3): δ 7,89-7,69 (m, 2H), 7,42-7,39 (m, 4H), 7,39-7,34 (m, 1H), 7,28-7,22 (m, 1H), 5,21 (s, 2H), 4,68 (br. s., 2H), 3,86 (br. s., 2H), 2,87-2,67 (m, 2H). LCMS (ESI) масса/заряд: 371 (M+1).
[552] Стадия 10.
[553] 2-(3,4-Дифторфенил)-4,5,6,7-тетрагидрооксазолo[5,4-c]пиридин
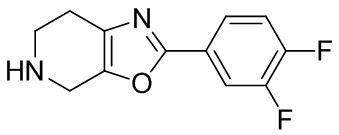
[554] Бензил-2-(3,4-дифторфенил)-6,7-дигидро-4H-оксазолo[5,4-c]пиридин-5-карбоксилат (800,00 мг, 2,16 ммоль, 1,00 экв.) растворяли в бромистоводородной кислоте/уксусной кислоте (20 мл) и перемешивали при 15°C в течение 3 часов. Смесь фильтровали и отфильтрованный осадок концентрировали in vacuo с получением 2-(3,4-дифторфенил)-4,5,6,7-тетрагидрооксазолo[5,4-c]пиридина (450,00 мг, 1,42 ммоль, выход 65,69%, гидробромид) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 237 (M + 1).
[555] Стадия 11.
[556] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-(3,4-дифторфенил)-6,7-дигидро-4H-оксазолo[5,4-c]пиридин-5-ил)-2-метилпропан-2-ол
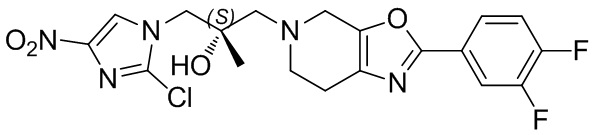
[557] 2-(3,4-Дифторфенил)-4,5,6,7-тетрагидрооксазолo[5,4-c]пиридин (200,00 мг, 630,66 мкмоль, 1,00 экв., гидробромид) и 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазол (137,24 мг, 630,66 мкмоль, 1,00 экв.) растворяли в трет-бутаноле (15,00 мл), затем добавляли DIPEA (244,52 мг, 1,89 ммоль, 3,00 экв.). Смесь перемешивали при 80°C в течение 12 часов, а затем охлаждали, концентрировали при 45°C и остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 10/1, 3/1) с получением (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(3,4-дифторфенил)-6,7-дигидро-4H-оксазолo[5,4-c]пиридин-5-ил)-2-метилпропан-2-ола (166,00 мг, 365,78 мкмоль, выход 58,00%) в виде желтого масла. LCMS (ESI) масса/заряд: 454 (M + 1).
[558] Стадия 12.
[559] (S)-2-(3,4-Дифторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-4,5,6,7-тетрагидрооксазолo[5,4-c]пиридин
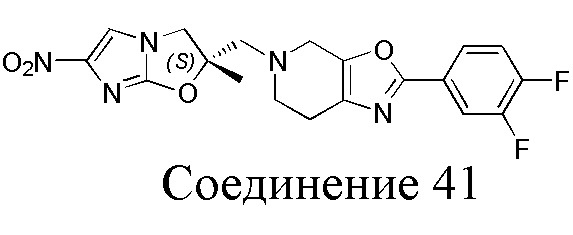
[560] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-(3,4-дифторфенил)-6,7-дигидро-4H-оксазолo[5,4-c]пиридин-5-ил)-2-метилпропан-2-ол (166,00 мг, 365,78 мкмоль, 1,00 экв.) растворяли в DMF (3,00 мл) и добавляли NaH (29,26 мг, 731,56 мкмоль, 2,00 экв.) при -45°C в атмосфере газообразного азота. Смесь перемешивали при температуре от -45 до -15°C в течение 2 часов. Затем смесь гасили насыщенным раствором хлорида аммония (20 мл). Водную фазу экстрагировали этилацетатом (50 мл × 2). Объединенные органические фазы промывали солевым раствором (20 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток затем очищали посредством препаративной хроматографии (GX-D; Boston Green ODS 150*30 5u; ацетонитрил 42% - 72%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-(3,4-дифторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-4,5,6,7-тетрагидрооксазолo[5,4-c]пиридина – соединения 41 (49,40 мг, 115,64 мкмоль, выход 31,61%, чистота 97,7%). 1H ЯМР (400 МГц, CDCl3): δ 7,85-7,78 (m, 1H), 7,74 (dd, J = 3,1, 7,7 Гц, 1H), 7,55 (s, 1H), 7,27-7,20 (m, 1H), 4,40 (d, J = 9,7 Гц, 1H), 3,97 (d, J = 9,7 Гц, 1H), 3,87 (s, 2H), 3,17-3,07 (m, 2H), 3,02-2,94 (m, 1H), 2,80 (d, J = 14,9 Гц, 1H), 2,65 (d, J = 1,9 Гц, 2H), 1,67 (s, 3H). LCMS (ESI) масса/заряд: 418 (M+1).
[561] Вариант осуществления 42
[562] (S)-2-((2-(4-Фторфенил)-5,6-дигидроимидазо[1,2-a]пиразин-7(8H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол

[563] Стадия 1.
[564] 2-Бром-1-(4-фторфенил)этанон

[565] 1-(4-Фторфенил)этанон (9,00 г, 65,15 ммоль, 1,00 экв.) растворяли в уксусной кислоте (100,00 мл), добавляли жидкий бром (10,41 г, 65,15 ммоль, 1,00 экв.) при 15°C и смесь перемешивали в течение 20 минут. Полученную в результате смесь затем перемешивали при 50°C в течение 12 часов, концентрировали при пониженном давлении и доводили pH смеси до 9 при помощи раствора карбоната натрия. Смесь экстрагировали этилацетатом (200 мл × 2). Объединенные органические слои концентрировали при пониженном давлении с получением остатка, который очищали посредством хроматографии на силикагеле (высота колонки: 250 мм, диаметр: 100 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 1/0) с получением 2-бром-1-(4-фторфенил)этанона (6,30 г, 29,03 ммоль, выход 44,56%) в виде белого твердого вещества. LCMS (ESI) масса/заряд: 218,8 (M + l).
[566] Стадия 2.
[567] 3-Хлорпиразин-2-амин
[568] 2,3-Дихлорпиразин (5,00 г, 33,56 ммоль, 1,00 экв.) смешивали с аммиаком (68,27 г, 1,95 моль, 58,04 экв.). Смесь перемешивали при 85°C в течение 12 часов, концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (высота колонки: 250 мм, диаметр: 100 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 50/1, 20/1) с получением 3-хлорпиразин-2-амина (700,00 мг, 5,40 ммоль, выход 16,09%) в виде белого твердого вещества.
[569] Стадия 3.
[570] 8-Хлор-2-(4-фторфенил)имидазо[1,2-a]пиразин
[571] 2-Бром-1-(4-фторфенил)этанон (3,02 г, 13,90 ммоль, 1,20 экв.) и 3-хлорпиразин-2-амин (1,50 г, 11,58 ммоль, 1,00 экв.) растворяли в диметиловом эфире этиленгликоля (20,00 мл), смесь нагревали до 35°C и перемешивали в течение 12 часов. Смесь концентрировали и очищали посредством хроматографии на силикагеле (высота колонки: 250 мм, диаметр: 100 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 20/1, 5/1) с получением 8-хлор-(4-фторфенил)имидазо[1,2-a]пиразина (900,00 мг, 3,63 ммоль, выход 31,38%) в виде белого твердого вещества. LCMS (ESI) масса/заряд: 247,9 (M + 1).
[572] Стадия 4.
[573] 2-(4-Фторфенил)-5,6,7,8-тетрагидроимидазо[1,2-a]пиразина гидрохлорид
[574] 8-Хлор-(4-фторфенил)имидазо[1,2-a]пиразин (900,00 мг, 3,63 ммоль, 1,00 экв.) растворяли в метаноле (20,00 мл) и добавляли Pd/C в атмосфере газообразного азота. Смешанный раствор затем перемешивали при 50°C, H2 (50 фунтов/кв. дюйм) в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении с получением 2-(4-фторфенил)-5,6,7,8-тетрагидроимидазо[1,2-a]пиразина гидрохлорида (850,00 мг, 2,93 ммоль, выход 80,70%), который использовали непосредственно на следующей стадии.
[575] Стадия 5.
[576] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-6,8-дигидро-5H-имидазо[1,2-a]пиразин-7-ил)-2-метилпропан-2-ол
[577] 2-(4-Фторфенил)-5,6,7,8-тетрагидроимидазо[1,2-a]пиразина гидрохлорид (400,00 мг, 1,58 ммоль, 1,00 экв.) растворяли в трет-бутаноле (10,00 мл), а затем добавляли DIPEA (509,42 мг, 3,94 ммоль, 2,50 экв.) и 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазол (514,65 мг, 2,36 ммоль, 1,50 экв.). Смесь нагревали до 100°C и перемешивали в течение 12 часов. Реакционный раствор выливали в воду (20 мл), а затем экстрагировали этилацетатом (50 мл × 2). Объединенные органические слои концентрировали досуха. Остаток очищали посредством хроматографии на силикагеле (высота колонки: 250 мм, диаметр: 100 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 5/1, 1/3) с получением (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-6,8-дигидро-5H-имидазо[1,2-a]пиразин-7-ил)-2-метилпропан-2-ола (200 мг, 459,93 мкмоль, выход 29,17%) в виде белого твердого вещества. LCMS (ESI) масса/заряд: 435,0 (M + 1).
[578] Стадия 6.
[579] (S)-2-((2-(4-Фторфенил)-5,6-дигидроимидазо[1,2-a]пиразин-7(8H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
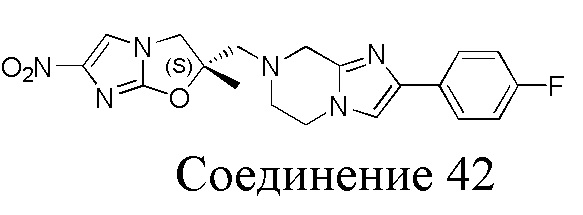
[580] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-6,8-дигидро-5H-имидазо[1,2-a]пиразин-7-ил)-2-метилпропан-2-ол (200,00 мг, 459,93 мкмоль, 1,00 экв.) растворяли в DMF (3,00 мл), добавляли NaH (11,04 мг, 459,93 мкмоль, 1,00 экв.) при -25°C и смесь перемешивали в течение 1 часа. Смесь выливали в воду (15 мл) при 0°C, а затем экстрагировали этилацетатом (30 мл × 2). Объединенные органические слои концентрировали до сухого состояния. Остаток очищали посредством препаративной хроматографии (GX-B, Phenomenex Synergi C18 150*30 мм*4 мкм, ацетонитрил 20% - 50%; 0,1% TFA-ACN; 25 мл/мин.) с получением (S)-2-((2-(4-фторфенил)-5,6-дигидроимидазо[1,2-a]пиразин-7(8H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 42 (70,60 мг, 176,28 мкмоль, выход 38,33%, чистота 99,476%). 1H ЯМР (400 МГц, метанол-d4): δ 7,86 (s, 1H), 7,78 (s, 1H), 7,71 (dd, J = 5,0, 8,7 Гц, 2H), 7,29 (t, J = 8,7 Гц, 2H), 4,47-4,38 (m, 1H), 4,31-4,05 (m, 5H), 3,30-3,09 (m, 3H), 1,70 (s, 3H). LCMS (ESI) масса/заряд: 399,0 (M+1).
[581] Вариант осуществления 43
[582] (S)-2-((2-(3,4-Дифторфенил)-5,6-дигидроимидазо[1,2-a]пиразин-7(8H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
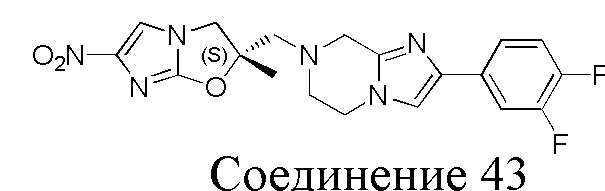
[583] Способ синтеза был таким, как в варианте осуществления 42.
[584] (S)-2-((2-(3,4-Дифторфенил)-5,6-дигидроимидазо[1,2-a]пиразин-7(8H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 43 (82,00 мг, 192,51 мкмоль, выход 21,79%, чистота 97,753%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, метанол-d4): δ 7,85 (d, J = 11,9 Гц, 2H), 7,69-7,62 (m, 1H), 7,54-7,40 (m, 2H), 4,42 (d, J = 10,7 Гц, 1H), 4,31-4,06 (m, 5H), 3,30-3,09 (m, 3H), 1,70 (s, 3H). LCMS (ESI) масса/заряд: 417,0 (M+1).
[585] Вариант осуществления 44
[586] (S)-2-Метил-6-нитро-2-((2-фенил-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
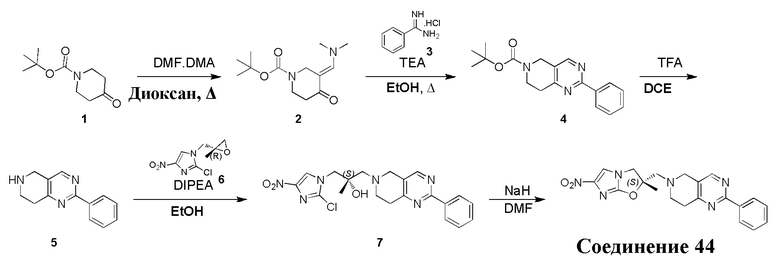
[587] Стадия 1.
[588] Трет-бутил-(3E)-3-((диметиламино)метилен)-4-оксопиперидин-1-карбоксилат
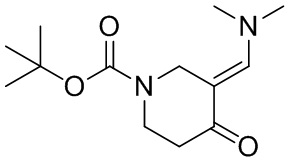
[589] Трет-бутил-4-оксопиперидин-1-карбоксилат (5,00 г, 25,09 ммоль, 1,00 экв.) растворяли в диоксане (30,00 мл), затем добавляли 1,1-диметокси-N,N-диметилметиламин (11,96 г, 100,36 ммоль, 4,00 экв.). Смесь перемешивали при 120°C в течение 16 часов, охлаждали и концентрировали при пониженном давлении при 50°C. Остаток разбавляли водой (50 мл) и перемешивали в течение 20 минут. Водную фазу экстрагировали этилацетатом (50 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (50 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (этилацетат/метанол = 20/1) с получением трет-бутил-(3E)-3-((диметиламино)метилен)-4-оксопиперидин-1-карбоксилата (3,00 г, 11,80 ммоль, выход 47,02%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3): δ 7,51 (s, 1H), 4,57 (s, 2H), 3,62 (t, J = 4,0 Гц, 1H), 3,13 (s, 7H), 2,46 (t, J = 4,0 Гц, 1H), 1,49 (s, 9H).
[590] Стадия 2.
[591] Трет-бутил-2-фенил-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-6-карбоксилат
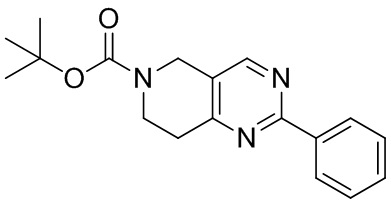
[592] Трет-бутил-(3E)-3-((диметиламино)метилен)-4-оксопиперидин-1-карбоксилат (3,00 г, 11,80 ммоль, 1,00 экв.) и бензамидин (1,85 г, 11,80 ммоль, 1,00 экв.) растворяли в этаноле (30,00 мл), затем добавляли триэтиламин (3,58 г, 35,40 ммоль, 3,00 экв.). Смесь перемешивали при 80°C в течение 2 часов, охлаждали и концентрировали при пониженном давлении. Остаток разбавляли водой (30 мл) и перемешивали в течение 20 минут. Водную фазу экстрагировали этилацетатом (30 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (30 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 3/1) с получением трет-бутил-2-фенил-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-6-карбоксилата (2,10 г, 6,74 ммоль, выход 57,15%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ: δ 8,54 (s, 1H), 8,46-8,36 (m, 2H), 7,54-7,44 (m, 3H), 4,64 (s, 2H), 3,80 (t, J = 4,0 Гц, 2H), 3,03 (t, J = 4,0 Гц, 2H), 1,53 (s, 9H). LCMS (ESI) масса/заряд: 312 (M+1).
[593] Стадия 3.
[594] 2-Фенил-5,6,7,8-тетрагидропиридо[4,3-d]пиримидин
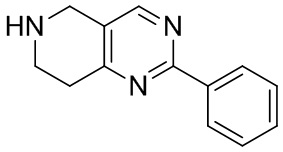
[595] Трет-бутил-2-фенил-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-6-карбоксилат (500,00 мг, 1,61 ммоль, 1,00 экв.) растворяли в дихлорметане (1,00 мл), добавляли трифторуксусную кислоту (183,09 мг, 1,61 ммоль, 1,00 экв.). Смесь перемешивали при 28°C в течение 2 часов и концентрировали при пониженном давлении при 50°C с получением соли 2-фенил-5,6,7,8-тетрагидропиридо[4,3-d]пиримидинтрифторуксусной кислоты (800,00 мг, неочищенная) в виде желтого масла. Продукт использовали на следующей стадии без дополнительного очищения.
[596] Стадия 4.
[597] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-2-метил-3-(2-фенил-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил)пропан-2-ол
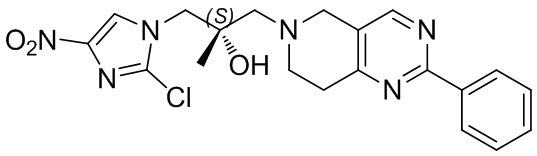
[598] 2-Фенил-5,6,7,8-тетрагидропиридо[4,3-d]пиримидин (340,00 мг, 1,61 ммоль, 1,00 экв.) и (R)-2-хлор-1-((2-метилоксиран-2-ил)метил)-4-нитро-1H-имидазол (420,42 мг, 1,93 ммоль, 1,20 экв.) растворяли в этаноле (10,00 мл), а затем добавляли DIPEA (623,99 мг, 4,83 ммоль, 3,00 экв.). Смесь перемешивали при 80°C в течение 12 часов, охлаждали и концентрировали при пониженном давлении при 50°C. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 2/1) с получением (S)-1-(2-хлор-4-нитро-1H-имидазол-1-ил)-2-метил-3-(2-фенил-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил)пропан-2-ола (650,00 мг, 1,52 ммоль, 94,14%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 8,46 (s, 1H), 8,43-8,36 (m, 2H), 8,06 (s, 1H), 7,52-7,45 (m, 3H), 4,10-4,06 (m, 2H), 3,96-3,78 (m, 2H), 3,19-2,99 (m, 4H), 2,77-2,56 (m, 2H), 1,22 (s, 3H). LCMS (ESI) масса/заряд: 429/431 (M+1).
[599] Стадия 5.
[600] (S)-2-Метил-6-нитро-2-((2-фенил-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
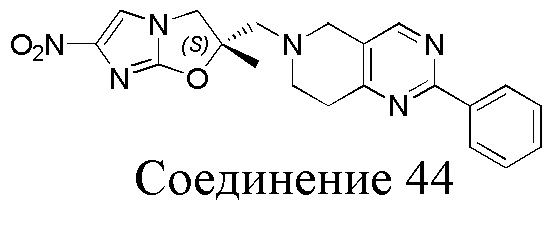
[601] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-2-метил-3-(2-фенил-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил)пропан-2-ол (350,00 мг, 816,10 мкмоль, 1,00 экв.) растворяли в DMF (5,00 мл), добавляли NaH (39,17 мг, 979,32 мкмоль, 1,20 экв.) при 0°C в атмосфере газообразного азота и смесь перемешивали в течение 30 мин. Реакционную смесь гасили насыщенным раствором хлорида аммония (20 мл), затем разбавляли водой (10 мл) и экстрагировали дихлорметаном (10 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (10 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной хроматографии (GX-E; Diamonsil 150*25 мм*5 мкм; ацетонитрил 20% - 50%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-метил-6-нитро-2-((2-фенил-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазола – соединения 44 (23,10 мг, 58,87 мкмоль, выход 7,21%). 1H ЯМР (400 МГц, CDCl3): δ 8,45 (s, 1H), 8,43-8,36 (m, 2H), 7,53 (s, 1H), 7,51-7,46 (m, 3H), 4,43-3,95 (m, 2H), 3,93-3,82 (m, 2H), 3,25-3,17 (m, 1H), 3,12 (d, J = 16,0 Гц, 1H), 3,05-2,89 (m, 3H), 2,83 (d, J = 12,0 Гц, 1H), 1,70 (s, 3H). LCMS (ESI) масса/заряд: 393 (M+1).
[602] Вариант осуществления 45
[603] (S)-2-((2-(4-Фторфенил)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
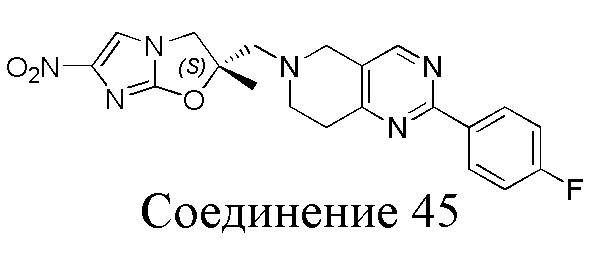
[604] Способ синтеза был таким, как в варианте осуществления 44.
[605] (S)-2-((2-(4-Фторфенил)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 45 (100,00 мг, 243,66 мкмоль, выход 27,22%). 1H ЯМР (400 МГц, CDCl3): δ 8,44-8,37 (m, 3H), 7,53 (s, 1H), 7,19-7,12 (m, 2H), 4,42-3,96 (m, 2H), 3,89-3,85 (m, 2H), 3,24-3,16 (m, 1H), 3,12 (d, J = 16,0 Гц, 1H), 3,03-2,87 (m, 3H), 2,83 (d, J = 16,0 Гц, 1H), 1,70 (s, 3H). LCMS (ESI) масса/заряд: 411 (M+1).
[606] Вариант осуществления 46
[607] (S)-2-((2-(3,4-Дифторфенил)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
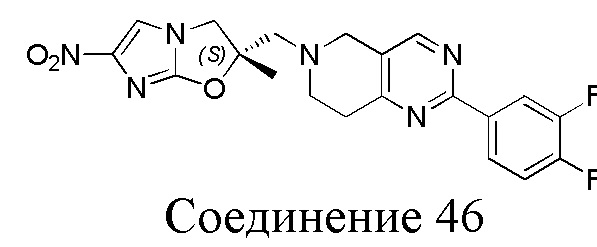
[608] Способ синтеза был таким, как в варианте осуществления 44.
[609] (S)-2-((2-(3,4-Дифторфенил)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 46 (20,80 мг, 47,05 мкмоль, выход 7,06%, чистота 96,9%). 1H ЯМР (400 МГц, CDCl3) δ 8,42 (s, 1H), 8,31-8,16 (m, 2H), 7,53 (s, 1H), 7,27-7,21 (m, 1H), 4,39 (d, J = 12,0 Гц, 1H), 3,99 (d, J = 12,0 Гц, 1H), 3,93-3,82 (m, 2H), 3,26-3,16 (m, 1H), 3,12 (d, J = 16,0 Гц, 1H), 3,04-2,88 (m, 3H), 2,83 (d, J = 16,0 Гц, 1H), 1,70 (s, 3H). LCMS (ESI) масса/заряд: 429 (M+1).
[610] Вариант осуществления 47
[611] (S)-2-Метил-6-нитро-2-((2-(4-(трифторметокси)фенил)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
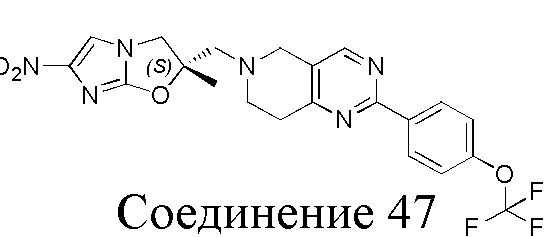
[612] Способ синтеза был таким, как в варианте осуществления 44.
[613] (S)-2-Метил-6-нитро-2-((2-(4-(трифторметокси)фенил)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол – соединение 47 (26,70 мг, 56,04 мкмоль, выход 5,75%). 1H ЯМР (400 МГц, CDCl3): δ 8,49-8,41 (m, 3H), 7,53 (s, 1H), 7,31 (d, J = 8,0 Гц, 2H), 4,43-3,95 (m, 2H), 3,94-3,82 (m, 2H), 3,25-3,16 (m, 1H), 3,12 (d, J = 16,0 Гц, 1H), 3,04-2,89 (m, 3H), 2,83 (d, J = 16,0 Гц, 1H), 1,70 (s, 3H). LCMS (ESI) масса/заряд: 477 (M+1).
[614] Вариант осуществления 48
[615] (S)-2-((2-(3,5-Дифторфенил)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
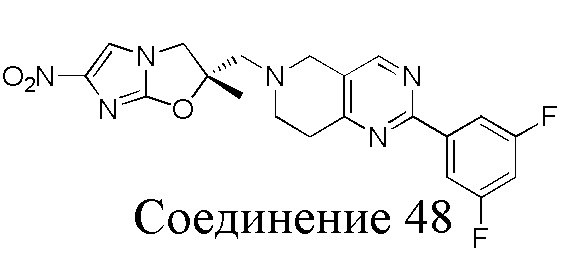
[616] Способ синтеза был таким, как в варианте осуществления 44.
[617] (S)-2-((2-(3,5-Дифторфенил)-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол –соединение 48 (25,20 мг, 57,67 мкмоль, выход 15,77%, чистота 98,030%). 1H ЯМР (400 МГц, CDCl3) δ 8,35 (s, 1H), 7,91-7,83 (m, 2H), 7,43 (s, 1H), 6,86-6,78 (m, 1H), 4,29 (d, J = 8,0 Гц, 1H), 3,89 (d, J = 8,0 Гц, 1H), 3,85-3,74 (m, 2H), 3,14-3,07 (m, 1H), 3,03 (d, J = 16,0 Гц, 1H), 2,96-2,79 (m, 3H), 2,75 (d, J = 16,0 Гц, 1H), 1,61 (s, 3H). LCMS (ESI) масса/заряд: 429 (M+1).
[618] Вариант осуществления 49
[619] (S)-2-((2-(3,5-Дифторфенил)-4-метокси-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
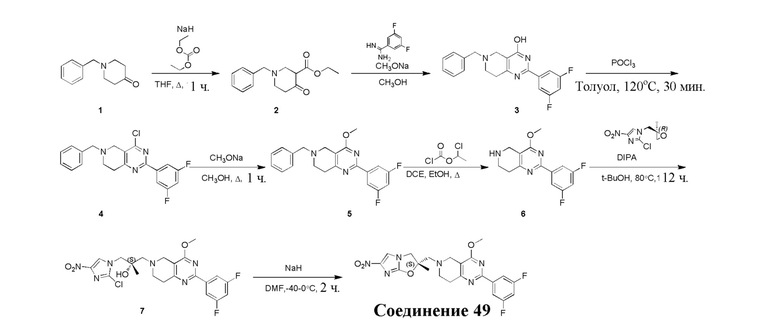
[620] Стадия 1.
[621] Этил-1-бензил-4-оксопиперидин-3-карбоксилат
[622] Гидрид натрия (2,11 г, 52,84 ммоль, 2,00 экв.) добавляли одной порцией к раствору 1-бензил-4-она (5,00 г, 26,42 ммоль, 1,00 экв.) в тетрагидрофуране (20,00 мл) при 15°C в атмосфере газообразного азота. Смесь перемешивали при 15°C в течение 30 минут. Затем добавляли к смеси диэтилкарбонат (6,24 г, 52,84 ммоль, 2,00 экв.) и перемешивали при 70°C в течение 1 часа. Смесь выливали в воду (50 мл) и перемешивали в течение 30 минут. Водную фазу экстрагировали этилацетатом (100 мл × 3). Объединенные органические фазы промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 30/1, 10/1) с получением этил-1-бензил-4-оксо-пиперидин-3-карбоксилата (4,50 г, 17,22 ммоль, выход 65,18%) в виде желтого масла.
[623] Стадия 2.
[624] 6-Бензил-2-(3,5-дифторфенил)-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-4-ол

[625] Метоксид натрия (496,13 мг, 9,18 ммоль, 2,00 экв.) добавляли к раствору этил-1-бензил-4-оксо-пиперидин-3-карбоксилата (1,20 г, 4,59 ммоль, 1,00 экв.) и 3,5-дифторбензамидина (931,63 мг, 5,97 ммоль, 1,30 экв.) в метаноле (15,00 мл). Смесь перемешивали при 80°C в течение 2 часов. Смесь концентрировали при пониженном давлении при 45°C, остаток выливали в воду (50 мл) и этилацетат (30 мл) и перемешивали в течение 5 минут. Фильтрат фильтровали и осадок сушили с получением 6-бензил-2-(3,5-дифторфенил)-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-4-ола (400,00 мг, 1,13 ммоль, выход 24,66%) в виде белого твердого вещества. LCMS (ESI) масса/заряд: 354,1 (M + 1).
[626] Стадия 3.
[627] 6-Бензил-4-хлор-2-(3,5-дифторфенил)-7,8-дигидро-5H-пиридо[4,3-d]пиримидин

[628] Оксихлорид фосфора (2,77 г, 18,07 ммоль, 8,86 экв.) добавляли к раствору 6-бензил-2-(3,5-дифторфенил)-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-4-ола (720,00 мг, 2,04 ммоль, 1,00 экв.) в толуоле (5,00 мл). Смесь перемешивали при 120°C в течение 2 часов и выливали смесь в смешанный раствор воды (50 мл) и этилацетата (50 мл). Смесь перемешивали в течение 5 минут, фильтровали и фильтрат сушили с получением 6-бензил-(3,5-дифторфенил)-7,8-дигидро-5H-пиридо[4,3-d]пиримидина (540,00 мг, 1,45 ммоль, выход 71,08%) в виде белого твердого вещества.
[629] Стадия 4.
[630] 6-Бензил-2-(3,5-дифторфенил)-4-метокси-7,8-дигидро-5H-пиридо[4,3-d]пиримидин
[631] Метоксид натрия (783,29 мг, 14,50 ммоль, 10,00 экв.) добавляли к раствору 6-бензил-(3,5-дифторфенил)-7,8-дигидро-5H-пиридо[4,3-d]пиримидина (540,00 мг, 1,45 ммоль, 1,00 экв.) в метаноле (10,00 мл). Смесь перемешивали при 70°C в течение 8 часов, добавляли к смешанному раствору воду (50 ) и перемешивали в течение 5 минут. Смесь фильтровали и отфильтрованный осадок сушили с получением 6-бензил-2-(3,5-дифторфенил)-4-метокси-7,8-дигидро-5H-пиридо[4,3-d]пиримидина (410,00 мг, 1,12 ммоль, выход 76,96%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,90-7,83 (m, 2H), 7,34-7,29 (m, 3H), 7,27-7,16 (m, 2H), 6,80 (tt, J = 2,4, 8,7 Гц, 1H), 3,98 (s, 3H), 3,68 (s, 2H), 3,48 (s, 2H), 2,92-2,85 (m, 2H), 2,78-2,70 (m, 2H).
[632] Стадия 5.
[633] 2-(3,5-Дифторфенил)-4-метокси-5,6,7,8-тетрагидропиридо[4,3-d]пиримидин
[634] 1-Хлорэтилхлорформиат (240,19 мг, 1,68 ммоль, 1,50 экв.) добавляли к раствору 6-бензил-2-(3,5-дифторфенил)-4-метокси-7,8-дигидро-5H-пиридо[4,3-d]пиримидина (410,00 мг, 1,12 ммоль, 1,00 экв.) в дихлорэтане (15,00 мл) при 0°C в атмосфере газообразного азота. Смесь перемешивали при 0°C в течение 30 минут и нагревали до 90°C. После перемешивания в течение 11,5 часа смесь концентрировали при 45°C при пониженном давлении, добавляли метанол (15 мл) и перемешивали при 90°C в течение 2 часов. Затем смесь концентрировали при пониженном давлении при 45°C, добавляли к остатку дихлорметан (30 мл) и перемешивали в течение 30 минут. Отфильтрованный осадок собирали с получением 2-(3,5-дифторфенил)-4-метокси-5,6,7,8-тетрагидропиридо[4,3-d]пиримидина (286,00 мг, 911,61 мкмоль, выход 81,39%, гидрохлорид) в виде белого твердого вещества.
[635] Стадия 6.
[636] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-(3,5-дифторфенил)-4-метокси-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-6-ил)-2-метилпропан-2-ол
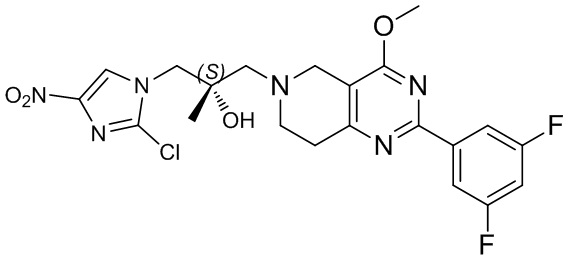
[637] 2-(3,5-Дифторфенил)-4-метокси-5,6,7,8-тетрагидропиридо[4,3-d]пиримидин (286,00 мг, 911,61 мкмоль, 1,00 экв., гидрохлорид) и 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазол (238,05 мг, 1,09 ммоль, 1,20 экв.) растворяли в трет-бутаноле (6,00 мл), добавляли диизопропиламин (235,63 мг, 1,82 ммоль, 2,00 экв.) в атмосфере газообразного азота. Смесь перемешивали при 80°C в течение 12 часов. Смесь охлаждали до 15°C и концентрировали при пониженном давлении при 45°C. Остаток очищали посредством хроматографии на силикагеле (диаметр: 250 мм, высота колонки: 100 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 10/1, 2/1) с получением (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(3,5-дифторфенил)-4-метокси-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-6-ил)-2-метилпропан-2-ола (400,00 мг, 808,28 мкмоль, выход 88,66%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,02 (s, 1H), 8,00-7,91 (m, 2H), 6,92 (tt, J = 2,3, 8,6 Гц, 1H), 4,11 (s, 3H), 4,07 (s, 2H), 3,82-3,67 (m, 2H), 3,63-3,51 (m, 2H), 3,02-2,98 (m, 2H), 2,77-2,57 (m, 2H), 1,35 (s, 3H).
[638] Стадия 7.
[639] (S)-2-((2-(3,5-Дифторфенил)-4-метокси-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
[640] Гидрид натрия (32,33 мг, 808,28 мкмоль, 2,00 экв.) добавляли к раствору (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(3,5-дифторфенил)-4-метокси-7,8-дигидро-5H-пиридо[4,3-d]пиримидин-6-ил)-2-метилпропан-2-ола (200,00 мг, 404,14 мкмоль, 1,00 экв.) в DMF (3,00 мл) при -45°C в атмосфере газообразного азота. Смесь перемешивали при температуре от -45°C до 0°C в течение 1 часа. Смесь выливали в насыщенный раствор хлорида аммония (50 мл) и перемешивали в течение 5 минут. Водную фазу экстрагировали этилацетатом (100 мл × 2). Объединенные органические фазы промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток промывали метанолом (30 мл × 2), фильтровали и собирали осадок с получением (S)-2-((2-(3,5-дифторфенил)-4-метокси-7,8-дигидропиридо[4,3-d]пиримидин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 49 (39,00 мг, 81,08 мкмоль, выход 20,06%, чистота 95,3%). 1H ЯМР (400 МГц, CDCl3) δ 7,85 (d, J = 7,7 Гц, 2H), 7,43 (s, 1H), 6,80 (t, J = 8,0 Гц, 1H), 4,29 (d, J = 9,5 Гц, 1H), 4,00 (s, 3H), 3,87 (d, J = 9,3 Гц, 1H), 3,72-3,56 (m, 2H), 3,11-2,98 (m, 2H), 2,89-2,67 (m, 4H), 1,60 (s, 3H). LCMS (ESI) масса/заряд: 459,1 (M+1).
[641] Вариант осуществления 50
[642] (S)-2-((2-(3,5-Дифторфенил)-5,6-дигидро-[1,2,4]триазоло[1,5-a]пиразин-7(8H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
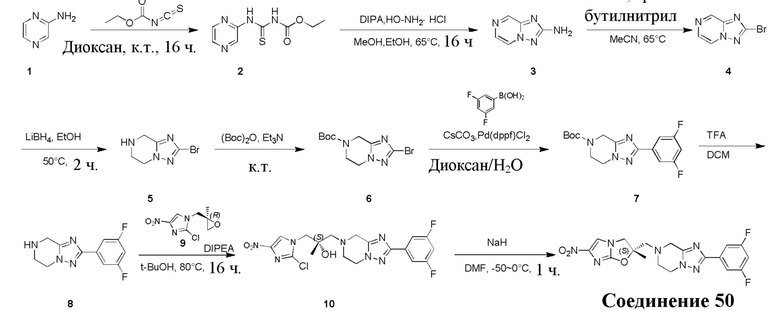
[643] Стадия 1.
[644] Этил-N-(пиразин-2-илтиокарбамоил)карбамат
[645] Этил-N-(тиометилен)карбаминовую кислоту (17,05 г, 130,00 ммоль, 1,24 экв.) добавляли по каплям к раствору пиразин-2-амина (10,00 г, 105,15 ммоль, 1,00 экв.) в диоксане (200,00 мл) при температуре от 0°C до 5°C в атмосфере газообразного азота. Смесь перемешивали при 15°C в течение 16 часов. Полученную в результате суспензию фильтровали и промывали дихлорметаном (200 мл) с получением этил-N-(пиразин-2-илтиокарбамоил)карбамата (13,60 г, 60,11 ммоль, выход 57,16%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 12,08 (s, 1H), 11,79 (s, 1H), 9,68 (s, 1H), 8,51 (s, 2H), 4,24 (m, 2H), 1,27 (m, 3H).
[646] Стадия 2.
[647] [1,2,4]Триазоло[1,5-a]пиразин-2-амин

[648] Гидрохлорид гидроксиламина (6,97 г, 100,24 ммоль, 1,80 экв.) и диизопропиламин (17,76 г, 137,55 ммоль, 2,47 экв.) добавляли к раствору этил-N-(пиразин-2-илтиокарбамоил)карбамата (12,60 г, 55,69 ммоль, 1,00 экв.) в метаноле (80,00 мл) и этаноле (80,00 мл). Смесь перемешивали при 65°C в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении до объема приблизительно 20 мл. Полученную в результате суспензию фильтровали, собирали твердое вещество и промывали раствором с соотношением дихлорметана к этанолу 60: 1 (90 мл) с получением [1,2,4]триазоло[1,5-a]пиразин-2-амина (5,30 г, 39,22 ммоль, выход 70,43%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 8,83 (s, 1H), 8,69-8,68 (d, J = 4,3 Гц, 1H), 7,97-7,96 (d, J = 4,3 Гц, 1H), 6,46 (s, 2H).
[649] Стадия 3.
[650] 2-Бром-[1,2,4]триазоло[1,5-a]пиразин
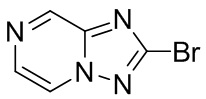
[651] [1,2,4]Триазоло[1,5-a]пиразин-2-амин (1,00 г, 7,40 ммоль, 1,00 экв.) добавляли к раствору бромида меди (1,98 г, 8,88 ммоль, 1,20 экв.) и трет-бутилнитрита (1,14 г, 11,10 ммоль, 1,50 экв.) в ацетонитриле (30 мл) при 65°C. После добавления смесь перемешивали при данной температуре в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении. Остаток разбавляли этилацетатом (150 мл) и промывали 1 н. соляной кислотой (100 мл) и насыщенным раствором хлорида аммония (100 мл). Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали посредством хроматографии на силикагеле (диоксид кремния, петролейный эфир/этилацетат = 1: 1) с получением 2-бром-[1,2,4]триазоло[1,5-a]пиразина (350,00 мг, 1,76 ммоль, выход 23,77%) в виде бледно-желтого твердого вещества. 1H ЯМР (300МГц, DMSO-d6) δ 9,38 (s, 1H), 9,10-9,09 (d, J = 4,3 Гц, 1H), 8,33-8,32 (d, J = 4,3 Гц, 1H).
[652] Стадия 4.
[653] 2-Бром-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразин
[654] Боргидрид лития (154,00 мг, 7,08 ммоль, 4,02 экв.) добавляли к раствору 2-бром-[1,2,4]триазоло[1,5-a]пиразина (350,00 мг, 1,76 ммоль, 1,00 экв.) в этаноле (10,00 мл). Смесь перемешивали при 50°C в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта в виде белого твердого вещества, которое использовали непосредственно на следующей стадии. LCMS (ESI) масса/заряд: 203/205 (M + 1) / (M + 2).
[655] Стадия 5.
[656] Трет-бутил-2-бром-5,6-дигидро-[1,2,4]триазоло[1,5-a]пиразин-7(8H)-карбоксилат
[657] Бикарбонат натрия (144,82 мг, 1,72 ммоль, 1,00 экв.) и Boc2O (379,98 мг, 1,74 ммоль, 1,01 экв.) добавляли к водному раствору (15 мл) 2-бром-5,6,7,8-тетрагидро[1,2,4]триазоло[1,5-a]пиразина (350,00 мг, 1,72 ммоль, 1,00 экв.). Смесь перемешивали при температуре от 10 oC до 20oC в течение 0,5 часов. Добавляли к реакционной смеси воду (50 мл) и экстрагировали этилацетатом (30 мл × 3). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением трет-бутил-2-бром-6,8-дигидро-5H-[1,2,4]триазоло[1,5-a]пиразин-7-карбоксилата (600,00 мг, неочищенный) в виде бледно-желтого твердого вещества. 1H ЯМР (300МГц, CDCl3) δ 4,72 (s, 2H), 4,23-4,13 (m, 2H), 3,99-3,89 (m, 2H), 1,50 (s, 9H).
[658] Стадия 6.
[659] Трет-бутил-2-(3,5-дифторфенил)-5,6-дигидро-[1,2,4]триазоло[1,5-a]пиразин-7(8H)-карбоксилат
[660] Карбонат цезия (1,08 г, 3,30 ммоль, 2,00 экв.) и Pd(dppf)Cl2 (60,00 мг, 82,00 мкмоль, 0,05 экв.) добавляли одной порцией к смешанному раствору трет-бутил-2-бром-6,8-дигидро-5H-[1,2,4]триазоло[1,5-a]пиразин-7-карбоксилата (500,00 мг, 1,65 ммоль, 1,00 экв.) и (3,5-дифторфенил)бороновой кислоты (260,00 мг, 1,65 ммоль, 1,00 экв.) в диоксане (10,00 мл) и воде (1,00 мл) в атмосфере газообразного азота. Смесь нагревали до 80°C и перемешивали в течение 16 часов. Смесь промывали водой (30 мл) и водную фазу экстрагировали этилацетатом (30 мл × 2). Объединенные органические фазы сушили над безводным Na2SO4, фильтровали и концентрировали in vacuo с получением остатка. Остаток очищали посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 20/1 – 5: 1) с получением трет-бутил-2-(3,5-дифторфенил)-6,8-дигидро-5H-[1,2,4]триазоло[1,5-a]пиразин-7-карбоксилата (310,00 мг, 921, мкмоль, выход 55,86%) в виде бледно-желтого твердого вещества. LCMS (ESI) масса/заряд: 337,1 (M + 1).
[661] Стадия 7.
[662] 2-(3,5-Дифторфенил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-a]пиразин
[663] Трифторуксусную кислоту (3,06 г, 26,84 ммоль, 29,12 экв.) добавляли к раствору трет-бутил-2-(3,5-дифторфенил)-6,8-дигидро-5H-[1,2,4]триазоло[1,5-a]пиразин-7-карбоксилата (310,00 мг, 921,69 мкмоль, 1,00 экв.) в дихлорметане (10,00 мл). Смесь перемешивали при 15°C в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении с получением остатка в виде темно-коричневого твердого вещества, которое использовали непосредственно на следующей стадии. LCMS (ESI) масса/заряд: 237,1 (M + 1).
[664] Стадия 8.
[665] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(3,5-дифторфенил)-5,6-дигидро-[1,2,4]триазоло[1,5-a]пиразин-7(8H)-ил)-2-метилпропан-2-ол
[666] 2-Хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазол (250,00 мг, 1,15 ммоль, 0,73 экв.) и диизопропиламин (1,01 г, 7,85 ммоль, 5,00 экв.) добавляли к раствору 2-(3,5-дифторфенил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-a]пиразина (550,00 мг, 1,57 ммоль, 1,00 экв., трифторацетат) в трет-бутаноле (10,00 мл). Смесь перемешивали при 80°C в течение 16 часов. Реакционную смесь промывали водой (50 мл) и экстрагировали этилацетатом (30 мл × 2). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 3/1 – 1/5) с получением (S)-1-(2-хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(3,5-дифторфенил)-5,6-дигидро-[1,2,4]триазоло[1,5-a]пиразин-7(8H)-ил)-2-метилпропан-2-ола (250,00 мг, 550,87 мкмоль, выход 35,09%) в виде бледно-желтого масла. LCMS (ESI) масса/заряд: 454,1 (M + l).
[667] Стадия 9.
[668] (S)-2-((2-(3,5-Дифторфенил)-5,6-дигидро-[1,2,4]триазоло[1,5-a]пиразин-7(8H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
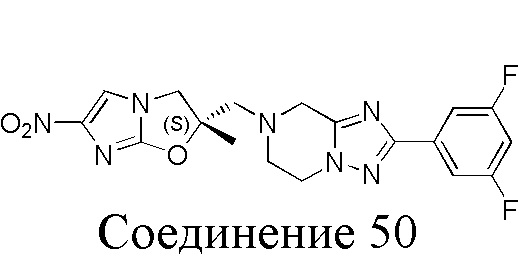
[669] Гидрид натрия (20,00 мг, 500,00 мкмоль, 2,27 экв.) добавляли к раствору (2S)-1-(2-хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(3,5-дифторфенил)-5,6-дигидро-[1,2,4]триазоло[1,5-a]пиразин-7(8H)-ил)-2-метилпропан-2-ола (100,00 мг, 220,35 мкмоль, 1,00 экв.) в DMF (5,00 мл) при -50°C в атмосфере газообразного азота. Смесь перемешивали при -50°C в течение 0,5 часа. Затем смесь нагревали до 0°C и перемешивали в течение дополнительных 0,5 часа. Реакционную смесь выливали в насыщенный раствор хлорида аммония (40 мл), а затем экстрагировали этилацетатом (30 мл × 2). Объединенные органические слои фильтровали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством препаративной разделительной хроматографии с получением (S)-2-((2-(3,5-дифторфенил)-5,6-дигидро-[1,2,4]триазоло[1,5-a]пиразин-7(8H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола (11,25 мг, 26,36 мкмоль, выход 11,96%, чистота 97,79%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,42 (br. s., 1H), 8,10 (s, 1H), 7,55 (d, J = 6,5 Гц, 2H), 7,31 (br. s., 1H), 4,31 (d, J = 10,8 Гц, 1H), 4,22-4,15 (m, 1H), 4,10 (d, J = 10,5 Гц, 1H), 4,02-3,90 (m, 3H), 3,19-3,15 (m, 2H), 3,08-3,04 (m, 2H), 1,59 (s, 3H). LCMS (ESI) масса/заряд: 418,2 (M+1).
[670] Вариант осуществления 51
[671] (S)-2-((2-(3,5-Дихлорфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
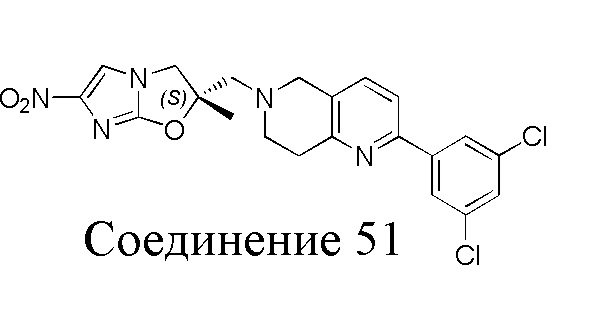
[672] Pd(dppf)Cl2 (20,92 мг, 28,59 мкмоль, 0,05 экв.), фторид цезия (260,57 мг, 1,72 ммоль, 3,00 экв.) добавляли одной порцией к смешанному раствору ключевого промежуточного соединения B (200,00 мг, 571,80 мкмоль, 1,00 экв.) и (3,5-дихлорфенил)бороновой кислоты (109,11 мг, 571,80 мкмоль, 1,00 экв.) в диоксане (5,00 мл) и воде (500,00 мкл) в атмосфере газообразного азота. Смесь перемешивали при 100°C в течение 6 часов. Смесь разбавляли этилацетатом, фильтровали и концентрировали. Остаток разделяли посредством препаративной разделительной хроматографии (GX-F; Phenomenex Synergi C18 150*25*10 мкм; 0,225% FA-ACN; начиная с 52 до 82; скорость потока (25 мл/мин.) с получением (S)-2-((2-(3,5-дихлорфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 51 (50,00 мг, 107,54 мкмоль, выход 18,81%, чистота 99%). 1H ЯМР (400 МГц, CDCl3) δ 7,86 (d, J = 1,9 Гц, 2H), 7,53 (s, 1H), 7,50-7,44 (m, 1H), 7,43-7,35 (m, 2H), 4,43 (d, J = 9,7 Гц, 1H), 4,07-3,66 (m, 3H), 3,30-2,88 (m, 5H), 2,82 (d, J = 14,9 Гц, 1H), 1,69 (s, 3H); LCMS (ESI) масса/заряд: 460 (M+1).
[673] Вариант осуществления 52
[674] (S)-2-((2-(3,5-Дифторфенил)-4-метил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
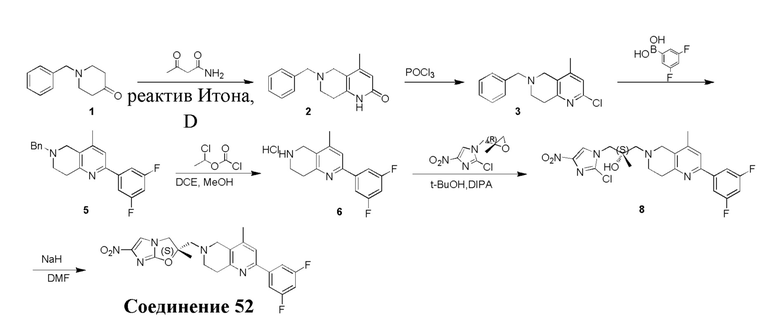
[675] Стадия 1.
[676] 6-Бензил-4-метил-1,5,7,8-тетрагидро-1,6-нафтиридин-2-он
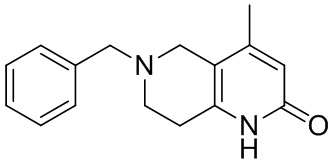
[677] 1-Бензил-4-он (4,00 г, 21,14 ммоль, 1,00 экв.) и 3-оксобутанамид (2,35 г, 23,25 ммоль, 1,10 экв.) растворяли в реактиве Итона (8,00 мл). Смесь перемешивали в течение 12 часов. Смесь добавляли к насыщенному водному раствору бикарбоната натрия (100 мл) для контроля pH> 7 и экстрагировали водный слой этилацетатом (100 мл × 4). Объединенные органические фазы промывали насыщенным солевым раствором (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток промывали ацетоном (60 мл), а затем фильтровали, чтобы собрать осадок, с получением 6-бензил-4-метил-1,5,7,8-тетрагидро-1,6-нафтиридин-2-она (2,80 г, 11,01 ммоль, выход 52,08%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6): δ 11,25 (br, s, 1H), 7,34 (d, J = 4,3 Гц, 4H), 7,27 (qd, J = 4,1, 8,6 Гц, 1H), 5,99 (s, 1H), 3,66 (s, 2H), 3,26 (s, 2H), 2,61-2,52 (m, 4H), 1,95 (s, 3H).
[678] Стадия 2.
[679] 6-Бензил-2-хлор-4-метил-7,8-дигидро-5H-1,6-нафтиридин
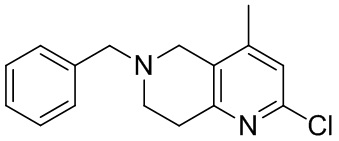
[680] 6-Бензил-4-метил-1,5,7,8-тетрагидро-1,6-нафтиридин-2-он (2,80 г, 11,01 ммоль, 1,00 экв.) и оксихлорид фосфора (9,00 мл) смешивали и перемешивали при 110°C в течение 12 часов. Смесь добавляли по каплям к ледяной воде (100 мл) и перемешивали смесь при 15°C в течение 0,5 часа. Добавляли к смеси насыщенный водный раствор бикарбоната натрия (100 мл), пока рН не станет > 7. Смесь экстрагировали дихлорметаном (100 мл × 4). Объединенные органические фазы промывали солевым раствором (100 мл) и органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo с получением 6-бензил-2-хлор-4-метил-7,8-дигидро-5H-1,6-нафтиридина (3,50 г, неочищенный) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 273 (M + 1).
[681] Стадия 3.
[682] 6-Бензил-2-(3,5-дифторфенил)-4-метил-7,8-дигидро-5H-1,6-нафтиридин
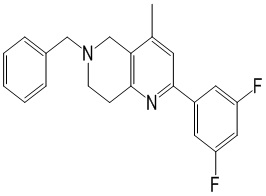
[683] Фторид цезия (694,94 мг, 4,58 ммоль, 2,50 экв.) и Pd(dppf)Cl2 (133,90 мг, 183,00 мкмоль, 0,10 экв.) добавляли одной порцией к смешанному раствору 6-бензил-2-хлор-4-метил-7,8-дигидро-5H-1,6-нафтиридина (500,00 мг, 1,83 ммоль, 1,00 экв.) и (3,5-дифторфенил)бороновой кислоты (346,77 мг, 2,20 ммоль, 1,20 экв.) в диоксане (10,00 мл) и воде (1,00 мл) в атмосфере газообразного азота. Смесь перемешивали при 110°C в течение 5 часов. Добавляли к смеси воду (10 мл), а затем экстрагировали дихлорметаном (100 мл × 3). Объединенные органические фазы промывали солевым раствором (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (высота колонки: 250 мм, диаметр: 100 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 40/1 – 15/1) с получением 6-бензил-2-(3,5-дифторфенил)-4-метил-7,8-дигидро-5H-1,6-нафтиридина (580,00 мг, 1,66 ммоль, выход 90,45%) в виде белого твердого вещества. 1H ЯМР (300МГц, CDCl3): δ 7,41 (d, J = 6,8 Гц, 2H), 7,32 (br, s, 3H), 7,27-7,16 (m, 3H), 6,78-6,65 (m, 1H), 3,70 (s, 2H), 3,55 (s, 2H), 3,08-2,95 (m, 2H), 2,83-2,71 (m, 2H), 2,14 (s, 3H).
[684] Стадия 4.
[685] 2-(3,5-Дифторфенил)-4-метил-5,6,7,8-тетрагидро-1,6-нафтиридина гидрохлорид
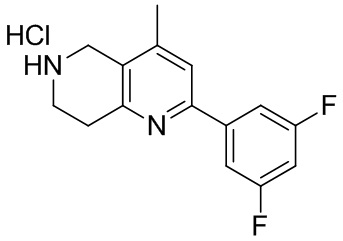
[686] 1-Хлоркарбонилхлорид (354,98 мг, 2,48 ммоль, 1,50 экв.) добавляли одной порцией к раствору 6-бензил-2-(3,5-дифторфенил)-4-метил-7,8-дигидро-5H-1,6-нафтиридина (580,00 мг, 1,66 ммоль, 1,00 экв.) в дихлорэтане (15,00 мл) при -15°C в атмосфере газообразного азота. Смесь перемешивали при 90°C в течение 12 часов. Затем смесь концентрировали до сухого состояния и добавляли к остатку метанол (10,00 мл). Далее полученную в результате смесь перемешивали при 90°C в течение дополнительного часа. Смесь концентрировали до сухого состояния с получением 2-(3,5-дифторфенил)-4-метил-5,6,7,8-тетрагидро-1,6-нафтиридина (400,00 мг, 1,35 ммоль, выход 81,56%, гидрохлорид) в виде белого твердого вещества.
[687] Стадия 5.
[688] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-(3,5-дифторфенил)-4-метил-7,8-дигидро-5H-1,6-нафтиридин-6-ил)-2-метилпропан-2-ол
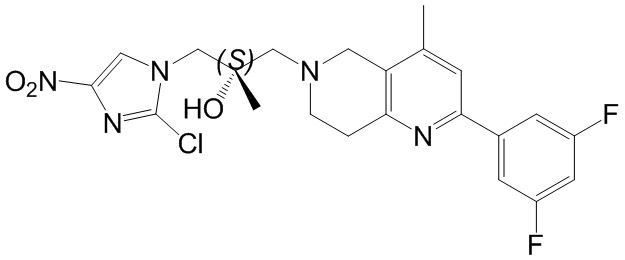
[689] 2-(3,5-Дифторфенил)-4-метил-5,6,7,8-тетрагидро-1,6-нафтиридин (400,00 мг, 1,35 ммоль, 1,00 экв., гидрохлорид) и 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазол (352,53 мг, 1,62 ммоль, 1,20 экв.) смешивали в трет-бутаноле (8,00 мл), добавляли одной порцией диизопропиламин (436,19 мг, 3,38 ммоль, 2,50 экв.) при 15°C в атмосфере газообразного азота. Смесь перемешивали при 100°C в течение 12 часов. Смесь концентрировали до сухого состояния. Остаток очищали посредством хроматографии на силикагеле (высота колонки: 250 мм, диаметр: 100 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 30/1 – 1/2) с получением (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(3,5-дифторфенил)-4-метил-7,8-дигидро-5H-1,6-нафтиридин-6-ил)-2-метилпропан-2-ола (260,00 мг, неочищенный) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 478 (M + l).
[690] Стадия 6.
[691] (2S)-2-((2-(3,5-Дифторфенил)-4-метил-7,8-дигидро-5H-1,6-нафтиридин-6-ил)метил)-2-метил-6-нитро-3H-имидазо[2,1-b]оксазол

[692] Гидрид натрия (43,60 мг, 1,09 ммоль, 2,00 экв.) добавляли одной порцией к раствору (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(3,5-дифторфенил)-4-метил-7,8-дигидро-5H-1,6-нафтиридин-6-ил)-2-метилпропан-2-ола (260,00 мг, 544,06 мкмоль, 1,00 экв.) в DMF (5,00 мл) при -20°C в атмосфере газообразного азота. Смесь перемешивали при -20°C в течение 10 минут, а затем нагревали до 0°C и перемешивали в течение 10 минут, далее нагревали до 15°C и перемешивали в течение еще 10 минут. Смесь добавляли по каплям к хлориду аммония (20 мл), затем смесь фильтровали и отфильтрованный осадок собирали с получением неочищенного продукта. Неочищенный продукт разделяли и очищали посредством препаративной разделительной хроматографии, проводимой в щелочной среде (GX-D; Boston Symmetrix C18 ODS-R 150*30 мм*5 мкм; ацетонитрил 24% - 54%; вода (0,225% NH4OH); 25 мл/мин.), с получением (2S)-2-((2-(3,5-дифторфенил)-4-метил-7,8-дигидро-5H-1,6-нафтиридин-6-ил)метил)-2-метил-6-нитро-3H-имидазо[2,1-b]оксазола – соединения 52 (60,00 мг, 135,51 мкмоль, выход 24,91%, чистота 99,7%). 1H ЯМР (400 МГц, CDCl3): δ 7,56-7,45 (m, 3H), 7,31 (s, 1H), 6,87-6,77 (m, 1H), 4,44 (d, J = 9,8 Гц, 1H), 3,97 (d, J = 9,5 Гц, 1H), 3,88-3,71 (m, 2H), 3,20-2,81 (m, 6H), 2,25 (s, 3H), 1,70 (s, 3H). LCMS (ESI) масса/заряд: 442 (M+1).
[693] Вариант осуществления 53
[694] (S)-2-((2-(2-Хлортиофен-3-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
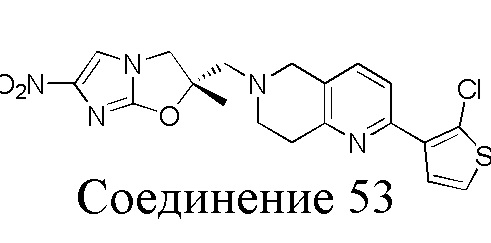
[695] Фторид цезия (173,71 мг, 1,14 ммоль, 2,00 экв.) и Pd(dppf)Cl2 (8,37 мг, 11,44 мкмоль, 0,02 экв.) добавляли к смешанному раствору (2S)-2-[(2-хлор-7,8-дигидро-5H-1,6-нафтиридин-6-ил)боксилфенетил]-2-метил-6-нитро-3H-имидазо[2,1-b]оксазола (200,00 мг, 571,80 мкмоль, 1,00 экв.) и (2-хлор-3-тиенил)бороновой кислоты (92,86 мг, 571,80 мкмоль, 1,00 экв.) в диоксане (3,00 мл) и воде (300,00 мкл). Смесь перемешивали при 70°C в течение 12 часов. Добавляли воду (50 мл) для гашения реакционной смеси и экстрагировали этилацетатом (50 мл × 3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток разделяли и очищали посредством препаративной разделительной хроматографии (GX-F;Phenomenex Synergi C18 150*25*10 мкм; ацетонитрил 30% - 60%; ACN (0,225% муравьиная кислота); 25 мл/мин.) с получением (2S)-2-((2-(2-хлортиофен-3-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 53 (26,30 мг, 59,55 мкмоль, выход 10,41%, чистота 97,8%). 1H ЯМР (400 МГц, метанол-d4): δ 7,82 (s, 1H), 7,65 (d, J = 6,15 Гц, 2H), 7,39 (d, J = 5,90 Гц, 1H), 7,31 (d, J = 5,77 Гц, 1H), 4,42 (d, J = 10,54 Гц, 1H), 4,15 (d, J = 10,54 Гц, 1H), 3,95 (s, 2H), 3,25-2,81 (m, 6H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 432,2 (M+1).
[696] Вариант осуществления 54
[697] (S)-2-Метил-6-нитро-2-((2-(5-(трифторметил)фуран-2-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
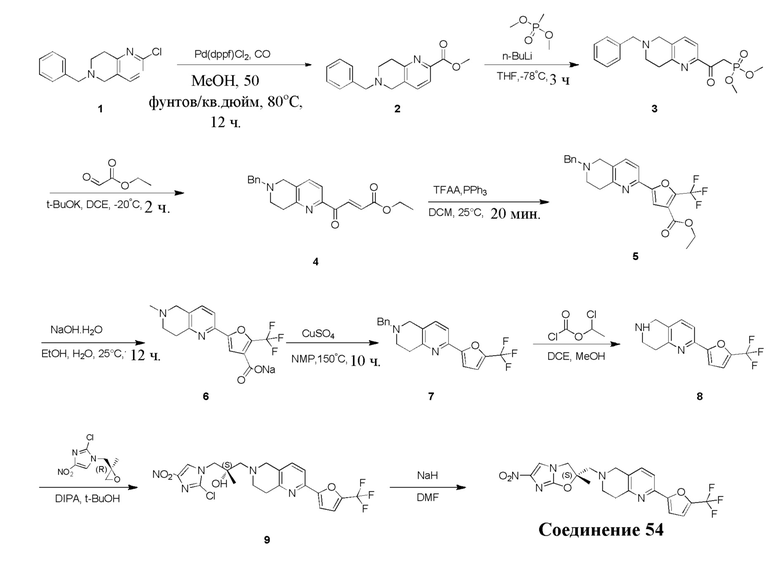
[698] Стадия 1.
[699] Метил-6-бензил-7,8-дигидро-5H-1,6-нафтиридин-2-карбоксилат
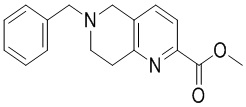
[700] Pd(dppf)Cl2 (706,96 мг, 966,00 мкмоль, 0,05 экв.), триэтиламин (3,65 г, 36,07 ммоль, 1,87 экв.) добавляли к раствору 6-бензил-2-хлор-7,8-дигидро-5H-1,6-нафтиридина (5,00 г, 19,32 ммоль, 1,00 экв.) в метаноле (50,00 мл) в атмосфере газообразного азота. Суспензию дегазировали при помощи монооксида углерода. Смесь перемешивали при 80°C в атмосфере монооксида углерода (50 фунтов/кв. дюйм) в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли, промывали карбонатом натрия (100 мл) и экстрагировали этилацетатом (500 мл × 2). Объединенные органические слои концентрировали при пониженном давлении с получением остатка. Остаток разделяли посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 50/1 – 5: 1) с получением метил-6-бензил-7,8-дигидро-5H-1,6-нафтиридин-2-карбоксилата (3,00 г, 10,63 ммоль, выход 55,00%) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 283,2 (M + 1).
[701] Стадия 2.
[702] 1-(6-Бензил-7,8-дигидро-5H-1,6-нафтиридин-2-ил)-2-диметоксифосфорил-этанон
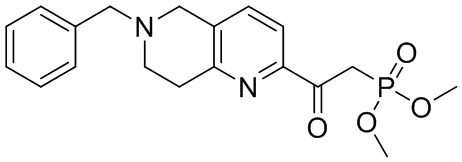
[703] N-бутиллитий (2,5 M, 10,62 мл, 3,00 экв.) добавляли по каплям к раствору метил-6-бензил-7,8-дигидро-5H-1,6-нафтиридин-2-карбоксилата (2,50 г, 8,85 ммоль, 1,00 экв.) в тетрагидрофуране (125,00 мл) при -78°C, через 30 минут после добавления смесь перемешивали при данной температуре в течение 30 минут, а затем добавляли раствор [метокси(метил)фосфорил]оксиметана (3,51 г, 28,32 ммоль, 3,20 экв.) в THF (125,00 мл) при -78°C. Полученную в результате смесь перемешивали при -78°C в течение 2 часов. Реакцию гасили за счет добавления хлорида аммония (30 мл) к реакционной смеси при -78°C, а затем экстрагировали этилацетатом (400 мл × 2). Объединенные органические слои концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 3/1 – 0: 1) с получением соединения 1-(6-бензил-7,8-дигидро-5H-1,6-нафтиридин-2-ил)-2-диметоксифосфорилэтанона (2,35 г, 6,28 ммоль, выход 70,93%) в виде бесцветного твердого вещества.
[704] Стадия 3.
[705] (E)-Этил-4-(6-бензил-7,8-дигидро-5H-1,6-нафтиридин-2-ил)-4-оксобут-2-еноат
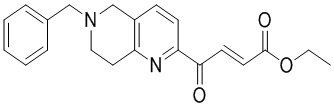
[706] Трет-бутоксид калия (845,24 мг, 7,53 ммоль, 1,20 экв.) и этил-2-оксоацетат (2,56 г, 12,55 ммоль, 2,00 экв.) добавляли к раствору 1-(6-бензил-7,8-дигидро-5H-1,6-нафтиридин-2-ил)-2-диметоксифосфорилэтанона (2,35 г, 6,28 ммоль, 1,00 экв.) в дихлорэтане (30,00 мл). Смесь перемешивали при -20°C в течение 2 часов. Остаток разделяли посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 1/0 – 20: 1) с получением соединения этил-(E)-этил-4-(6-бензил-7,8-дигидро-5H-1,6-нафтиридин-2-ил)-4-оксобут-2-еноата (1,50 г, 4,28 ммоль, выход 68,16%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 8,48 (d, J =15,9 Гц, 1H), 7,82 (d, J =7,9 Гц, 1H), 7,41-7,22 (m, 6H), 6,92 (d, J =15,9 Гц, 1H), 4,25-4,19 (m, 2H), 3,65 (d, J =16,9 Гц, 4H), 3,12-3,00 (m, 2H), 2,83 (t, J =6,0 Гц, 2H), 1,30-1,23 (m, 3H). LCMS (ESI) масса/заряд: 351,1 (M+1).
[707] Стадия 4.
[708] Этил-5-(6-бензил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)-2-(трифторметил)фуран-3-карбоксилат
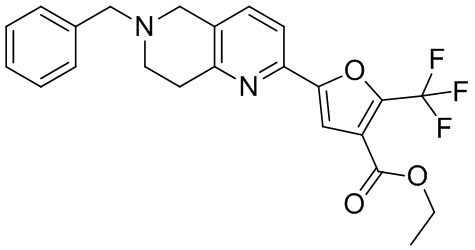
[709] (E)-этил-4-(6-бензил-7,8-дигидро-5H-1,6-нафтиридин-2-ил)-4-оксобут-2-еноат (1,50 г, 4,28 ммоль, 1,00 экв.) добавляли к раствору трифторуксусного ангидрида (1,35 г, 6,42 ммоль, 1,50 экв.) и трифенилфосфина (1,12 , 4,2 ммоль, 1,00 экв.) в дихлорметане (15,00 мл). Смесь перемешивали при 25°C в течение 0,5 часа. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 50/1 – 10: 1) с получением соединения этил-5-(6-бензил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)-2-(трифторметил)фуран-3-карбоксилата (1,30 г, 3,02 ммоль, выход 70,57%) в виде желтого твердого вещества.
[710] Стадия 5.
[711] Натрий-5-(6-бензил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)-2-(трифторметил)фуран-3-карбоксилат
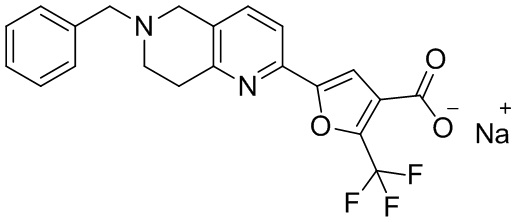
[712] Гидроксид натрия (260,80 мг, 6,52 ммоль, 4,00 экв.) добавляли к раствору этил-5-(6-бензил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)-2-(трифторметил)фуран-3-карбоксилата (700,00 мг, 1,63 ммоль, 1,00 экв.) в этаноле (7,00 мл) и воде (7,00 мл). Смесь перемешивали при 25°C в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении с удалением растворителя, с получением соединения 5-(6-бензил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)-2-(трифторметил)фуран-3-карбоксилата натрия (1,00 г, неочищенный) в виде желтого твердого вещества.
[713] Стадия 6.
[714] 6-Бензил-2-(5-(трифторметил)фуран-2-ил)-7,8-дигидро-5H-1,6-нафтиридин
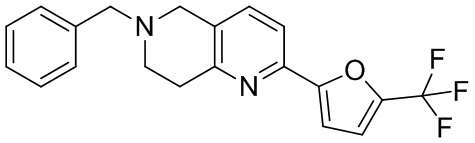
[715] Сульфат меди (37,67 мг, 236,00 мкмоль, 0,10 экв.) добавляли к раствору натрий-5-(6-бензил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)-2-(трифторметил)фуран-3-карбоксилата (1,00 г, 2,36 ммоль, 1,00 экв.) вN-метилпирролидоне (10,00 мл). Смесь перемешивали при 150°C в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток разделяли и очищали посредством препаративной пластины (диоксид кремния, петролейный эфир/этилацетат = 2: 1) с получением соединения 6-бензил-2-(5-(трифторметил)фуран-2-ил)-7,8-дигидро-5H-1,6-нафтиридина (500,00 мг, 1,40 ммоль, выход 59,12%) в виде желтого твердого вещества.
[716] Стадия 7.
[717] 2-[5-(Трифторметил)-2-фурил]-5,6,7,8-тетрагидро-1,6-нафтиридин
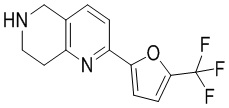
[718] 6-Бензил-2-(5-(трифторметил)фуран-2-ил)-7,8-дигидро-5H-1,6-нафтиридин (600,00 мг, 1,67 ммоль, 1,00 экв.) и 1-хлорэтилхлорформиат (358,14 мг, 2,51 ммоль, 1,50 экв.) растворяли в 1,2-дихлорэтане (6,00 мл). Смесь перемешивали при 80°C в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и добавляли метанол (6,00 мл). Смесь перемешивали при 80°C в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Доводили pH до приблизительно 9 за счет постепенного добавления раствора карбоната натрия и экстрагировали этилацетатом (100 мл × 2). Объединенные органические слои концентрировали при пониженном давлении с получением 2-[5-(трифторметил)-2-фурил]-5,6,7,8-тетрагидро-1,6-нафтиридина (200,00 мг, 745,63 мкмоль, выход 44,65%) в виде белого твердого вещества. LCMS (ESI) масса/заряд: 269,1 (M + 1).
[719] Стадия 8.
[720] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-2-метил-3-(2-(5-(трифторметил)фуран-2-ил)-7,8-дигидро-5H-1,6-нафтиридин-6-ил)пропан-2-ол
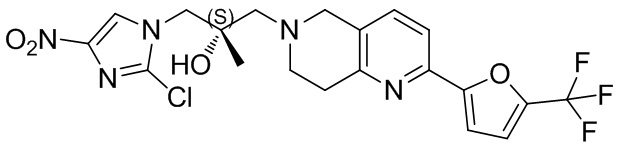
[721] Диизопропиламин (192,73 мг, 1,49 ммоль, 2,00 экв.) и 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазол (178,48 мг, 820,19 мкмоль, 1,10 экв.) добавляли к раствору 2-[5-(трифторметил)-2-фурил]-5,6,7,8-тетрагидро-1,6-нафтиридина (200,00 мг, 745,63 мкмоль, 1,00 экв.) в трет-бутаноле (5,00 мл). Смесь перемешивали при 100°C в течение 12 часов. Остаток очищали посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 10/1 – 1: 1) с получением (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-2-метил-3-(2-(5-(трифторметил)фуран-2-ил)-7,8-дигидро-5H-1,6-нафтиридин-6-ил)пропан-2-ола (260,00 мг, 535,16 мкмоль, выход 71,77%) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 486,2 (M + 1).
[722] Стадия 9.
[723] (S)-2-Метил-6-нитро-2-((2-(5-(трифторметил)фуран-2-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол

[724] Гидрид натрия (32,11 мг, 1,34 ммоль, 2,50 экв.) добавляли к раствору (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-2-метил-3-(2-(5-(трифторметил)фуран-2-ил)-7,8-дигидро-5H-1,6-нафтиридин-6-ил)пропан-2-ола (260,00 мг, 535,16 мкмоль, 1,00 экв.) в DMF (3,00 мл) при 0°C в течение 10 минут и смесь перемешивали при 15°C в течение 50 минут. Реакционную смесь гасили за счет добавления хлорида аммония (10 мл) при 0°C, а затем экстрагировали этилацетатом (100 мл × 2). Объединенные органические слои промывали хлоридом натрия (20 мл * 1) и концентрировали при пониженном давлении с получением остатка. Остаток разделяли и очищали посредством препаративной разделительной хроматографии (GX-I,YMC-Actus ODS-AQ 100*30 5u, ацетонитрил 24% - 54%; 0,1% TFA-ACN; 25 мл/мин.) с получением (S)-2-метил-6-нитро-2-((2-(5-(трифторметил)фуран-2-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазола – соединения 54 (48,00 мг, 105,28 мкмоль, выход 19,67%, чистота 98,561%). 1H ЯМР (400 МГц, CDCl3): δ 7,59-7,55 (m, 1H), 7,52 (s, 1H), 7,42-7,34 (m, 1H), 7,11-7,01 (m, 1H), 6,94-6,84 (m, 1H), 4,48-4,37 (m, 1H), 4,03-3,78 (m, 3H), 3,26-2,73 (m, 6H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 450,2 (M+1).
[725] Вариант осуществления 55
[726] (S)-2-Метил-2-((2-(4-метилтиазол-2-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
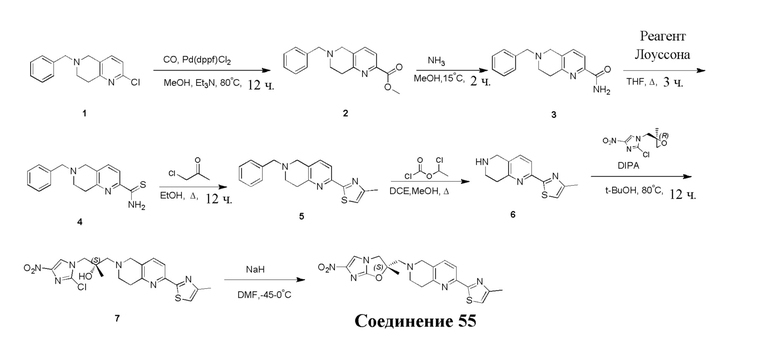
[727] Стадия 1.
[728] Метил-6-бензил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-карбоксилат

[729] Pd(dppf)Cl2 (1,07 г, 1,47 ммоль, 0,10 экв.) добавляли к раствору 6-бензил-2-хлор-7,8-дигидро-5H-1,6-нафтиридина (3,80 г, 14,69 ммоль, 1,00 экв.) в триэтиламине (2,00 мл) и метаноле (20,00 мл) в атмосфере газообразного азота. Суспензию дегазировали под вакуумом и продували монооксидом углерода. Смесь перемешивали при 80°C в течение 12 часов в атмосфере монооксида углерода (50 фунтов/кв. дюйм). Смесь фильтровали и концентрировали, а остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 10/1, 1/2) с получением метил-6-бензил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-карбоксилата (3,90 г, 13,81 ммоль, выход 94,04%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,93 (d, J = 7,9 Гц, 1H), 7,45 (d, J = 7,9 Гц, 1H), 7,42-7,30 (m, 5H), 4,01 (s, 3H), 3,75 (s, 2H), 3,71 (s, 2H), 3,18 (t, J = 6,0 Гц, 2H), 2,90 (t, J = 6,0 Гц, 2H).
[730] Стадия 2.
[731] 6-Бензил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-карбоксамид
[732] Через метанол (50,00 мл) пропускали аммиак (4,58 г, 269,20 ммоль, 20,00 экв.) при -50°C, через 10 минут добавляли к раствору метил-6-бензил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-карбоксилат (3,80 г, 13,46 ммоль, 1,00 экв.) при 15°C. Смесь перемешивали при 15°C в течение 120 минут. Реакционную смесь концентрировали с получением 6-бензил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-карбоксамида (3,30 г, 12,34 ммоль, выход 91,71%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,97 (d, J = 7,9 Гц, 1H), 7,46 (d, J = 7,9 Гц, 1H), 7,43-7,30 (m, 5H), 3,75 (s, 2H), 3,70 (s, 2H), 3,09-3,02 (m, 2H), 2,93-2,84 (m, 2H).
[733] Стадия 3.
[734] 6-Бензил-7,8-дигидро-5H-1,6-нафтиридин-2-карботиоамид
[735] Реагент Лоуссона (3,40 г, 8,42 ммоль, 1,50 экв.) добавляли к раствору 6-бензил-5,6,7,8-тетрагидро-1,6-нафтиридин-2-карбоксамида (1,50 г, 5,61 ммоль, 1,00 экв.) в тетрагидрофуране (30,00 мл) в атмосфере газообразного азота. Смесь перемешивали при 80°C в течение 4 часов. Смесь концентрировали при пониженном давлении при 45°C. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 10/1, 0/1) с получением 6-бензил-7,8-дигидро-5H-1,6-нафтиридин-2-карботиоамида (780,00 мг, 2,75 ммоль, выход 49,06%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,47 (d, J = 8,0 Гц, 1H), 7,45 (d, J = 7,8 Гц, 3H), 7,37 (d, J = 7,5 Гц, 3H), 3,96 (br. s., 2H), 3,84 (d, J = 7,7 Гц, 2H), 3,14 (br. s., 4H).
[736] Стадия 4.
[737] 2-(6-Бензил-7,8-дигидро-5H-1,6-нафтиридин-2-ил)-4-метилтиазол
[738] 1-Хлорпропан-2-он (1,48 г, 16,00 ммоль, 5,82 экв.) добавляли к раствору 6-бензил-7,8-дигидро-5H-1,6-нафтиридин-2-карботиоамида (780,00 мг, 2,75 ммоль, 1,00 экв.) в этаноле (15,00 мл) в атмосфере газообразного азота. Смесь перемешивали при 80°C в течение 12 часов. Смесь концентрировали при пониженном давлении при 45°C. Остаток промывали раствором с соотношением петролейного эфира к этилацетату 10: 1 (40 мл), фильтровали и отфильтрованный осадок сушили с получением 2-(6-бензил-7,8-дигидро-5H-1,6-нафтиридин-2-ил)-4-метилтиазола (719,00 мг, 2,24 ммоль, выход 81,34%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,91 (d, J = 7,9 Гц, 1H), 7,43-7,33 (m, 6H), 6,95 (s, 1H), 3,74 (s, 2H), 3,66 (s, 2H), 3,12-3,07 (m, 2H), 2,92-2,87 (m, 2H), 2,52 (s, 3H).
[739] Стадия 5.
[740] 4-Метил-2-(5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)тиазол
[741] 1-Хлорэтилхлорформиат (240,19 мг, 1,68 ммоль, 1,50 экв.) добавляли к раствору 2-(6-бензил-7,8-дигидро-5H-1,6-нафтиридин-2-ил)-4-метилтиазола (360,00 мг, 1,12 ммоль, 1,00 экв.) в дихлорэтане (10,00 мл) при 0°C в атмосфере газообразного азота. Смесь перемешивали при 100°C в течение 12 часов. Затем смесь охлаждали до 20°C и концентрировали при пониженном давлении при 45°C. Добавляли к остатку метанол (15,00 мл) и перемешивали при 80°C в течение 2 часов. Смесь концентрировали при пониженном давлении при 45°C, добавляли к остатку дихлорметан (20 мл) и перемешивали в течение 10 минут, фильтровали и собирали отфильтрованный осадок с получением 4-метил-2-(5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)тиазола (150,00 мг, 560,16 мкмоль, выход 50,01%, соляная кислота) в виде белого твердого вещества. 1H ЯМР (300МГц, DMSO-d6) δ 7,98 (d, J = 8,1 Гц, 1H), 7,81 (d, J = 8,1 Гц, 1H), 7,43 (s, 1H), 4,33 (br. s., 2H), 3,47 (br. s., 2H), 3,18-3,14 (m, 2H), 2,44 (s, 3H).
[742] Стадия 6.
[743] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-2-метил-3-(2-(4-метилтиазол-2-ил)-7,8-дигидро-5H-1,6-нафтиридин-6-ил)пропан-2-ол
[744] Диизопропиламин (144,79 мг, 1,12 ммоль, 2,00 экв.) добавляли к раствору 4-метил-2-(5,6,7,8-тетрагидро-1,6-нафтиридин-2-ил)тиазола (150,00 мг, 560,16 мкмоль, 1,00 экв., HCl) и 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазола (146,28 мг, 672,19 мкмоль, 1,20 экв.) в трет-бутаноле (10,00 мл) в атмосфере газообразного азота. Смесь перемешивали при 80°C в течение 12 часов. Смесь концентрировали при пониженном давлении при 45°C, добавляли к остатку воду (20 мл) и перемешивали в течение 5 минут. Водную фазу экстрагировали этилацетатом (30 мл × 3). Объединенные органические фазы промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток разделяли и очищали посредством препаративной тонкослойной хроматографии (петролейный эфир/этилацетат = 1/1) с получением (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-2-метил-3-(2-(4-метилтиазол-2-ил)-7,8-дигидро-5H-1,6-нафтиридин-6-ил)пропан-2-ола (73,00 мг, 162,61 мкмоль, выход 29,03%) в виде белого твердого вещества. 1H ЯМР (300МГц, CDCl3) δ 8,07 (s, 1H), 7,97 (d, J = 8,1 Гц, 1H), 7,41 (d, J = 8,1 Гц, 1H), 6,99 (s, 1H), 4,07 (s, 2H), 3,98-3,81 (m, 2H), 3,18-3,04 (m, 4H), 2,76-2,67 (m, 1H), 2,54 (s, 3H), 2,07 (s, 1H), 1,22 (s, 3H).
[745] Стадия 7.
[746] (S)-2-Метил-2-((2-(4-метилтиазол-2-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
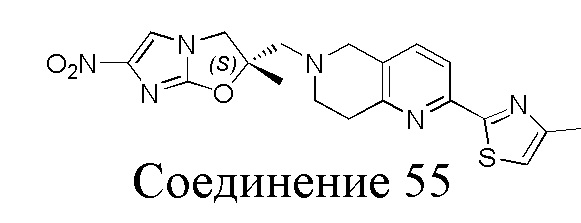
[747] Гидрид натрия (13,01 мг, 325,22 мкмоль, 2,00 экв.) добавляли одной порцией к раствору (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-2-метил-3-(2-(4-метилтиазол-2-ил)-7,8-дигидро-5H-1,6-нафтиридин-6-ил)пропан-2-ола (73,00 мг, 162,61 мкмоль, 1,00 экв.) в DMF (2,00 мл) при -45°C в атмосфере газообразного азота. Смесь перемешивали при температуре от -45 до 0°C в течение 10 минут. Остаток выливали в насыщенный раствор хлорида аммония (30 мл) и перемешивали в течение 5 минут. Водную фазу экстрагировали этилацетатом (50 мл × 3). Объединенные органические фазы промывали солевым раствором (20 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток разделяли и очищали посредством препаративной разделительной хроматографии (GX-F; Phenomenex Synergi C18 150*25*10 мкм; ацетонитрил 32% - 62%; вода (0,225%FA-ACN); 25 мл/мин.) с получением (S)-2-метил-2-((2-(4-метилтиазол-2-ил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 55 (27,96 мг, 65,35 мкмоль, выход 40,19%, чистота 96,4%). 1H ЯМР (400 МГц, CDCl3) δ 7,94 (d, J = 8,0 Гц, 1H), 7,52 (s, 1H), 7,39 (d, J = 8,0 Гц, 1H), 6,97 (s, 1H), 4,43 (d, J = 9,7 Гц, 1H), 3,96 (d, J = 9,5 Гц, 1H), 3,93-3,80 (m, 2H), 3,19-3,07 (m, 2H), 3,06-2,90 (m, 3H), 2,80 (d, J = 14,9 Гц, 1H), 2,53 (s, 3H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 413,1 (M+1).
[748] Вариант осуществления 56
[749] (S)-2-((2-(4-Фтор-2-метилфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
[750] Ключевое промежуточное соединение B (300,00 мг, 857,71 мкмоль, 1,00 экв.) и (4-фтор-2-метил-фенил)бороновую кислоту (198,07 мг, 1,29 ммоль, 1,50 экв.) растворяли в диоксане (5,00 мл) и воде (500,00 мкл), добавляли Pd(dppf)Cl2 (31,38 мг, 42,89 мкмоль, 0,05 экв.) и фторид цезия (260,57 мг, 1,72 ммоль, 2,00 экв.). Смесь перемешивали при 110°C в течение 10 часов. Реакционную смесь гасили за счет добавления воды (50 мл), а затем экстрагировали этилацетатом (50 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток разделяли и очищали посредством препаративной разделительной хроматографии (GX-F;Phenomenex Synergi C18 150*25*10 мкм; ацетонитрил 28% - 49%; ACN (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-((2-(4-фтор-2-метилфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 56 (104,70 мг, 239,60 мкмоль, выход 27,93%, чистота 96,9%). 1H ЯМР (400 МГц, CDCl3): δ 7,54 (s, 1H), 7,38 (d, J = 7,91 Гц, 1H), 7,33 (dd, J = 8,28, 6,02 Гц, 1H), 7,21-7,11 (m, 1H), 7,03-6,91 (m, 2H), 4,45 (d, J = 9,66 Гц, 1H), 4,03-3,80 (m, 3H), 3,22-2,92 (m, 5H), 2,83 (d, J = 14,81 Гц, 1H), 2,39-2,28 (s, 3H), 1,70 (s, 3H). LCMS (ESI) масса/заряд: 424,1 (M+1).
[751] Вариант осуществления 57
[752] (S)-2-((2-(4-Фторфенил)-5,6-дигидро-[1,2,4]триазоло[1,5-a]пиразин-7(8H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
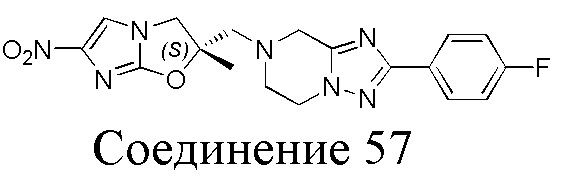
[753] Способ синтеза был таким, как в варианте осуществления 50.
[754] (S)-2-((2-(4-Фторфенил)-5,6-дигидро-[1,2,4]триазоло[1,5-a]пиразин-7(8H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 57 (6,85 мг, 16,20 мкмоль, выход 44,13%, чистота 94,46%). 1H ЯМР (400 МГц, метанол-d4) δ 8,02-7,96 (m, 2H), 7,86-7,82 (m, 1H), 7,20-7,14 (m, 2H), 4,67-4,60 (m, 1H), 4,45-4,38 (m, 1H), 4,15 (s, 2H), 4,07 (s, 1H), 4,00 (s, 2H), 3,24-3,14 (m, 2H), 3,09-3,02 (m, 1H), 1,69-1,65 (m, 3H). LCMS (ESI) масса/заряд: 400,1 (M+1).
[755] Вариант осуществления 58
[756] (S)-2-((2-(4-Фтор-3-метилфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
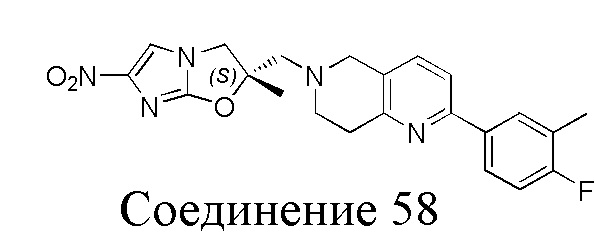
[757] Ключевое промежуточное соединение B (100,00 мг, 285,90 мкмоль, 1,00 экв.) и (4-фтор-3-метилфенил)бороновую кислоту (44,01 мг, 285,90 мкмоль, 1,00 экв.) растворяли в диоксане (1,00 мл) и воде (100,00 мкл), добавляли фторид цезия (86,86 мг, 571,80 мкмоль, 2,00 экв.) и Pd(dppf)Cl2 (20,92 мг, 28,59 мкмоль, 0,10 экв.). Смесь перемешивали при 110°C в течение 10 часов. Реакционную смесь гасили за счет добавления воды (50 мл), а затем экстрагировали этилацетатом (50 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток разделяли и очищали посредством препаративной разделительной хроматографии (GX-G;Phenomenex Synergi Max-RP 250*80 10u; ацетонитрил 20% - 50%; ACN (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-((2-(4-фтор-3-метилфенил)-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола (38,74 мг, 89,20 мкмоль, выход 31,20%, чистота 97,5%) – соединения 58. 1H ЯМР (400 МГц, CDCl3): δ 7,80 (d, J = 7,53 Гц, 1H), 7,74-7,68 (m, 1H), 7,53 (s, 1H), 7,47-7,43 (m, 1H), 7,38-7,34 (m, 1H), 7,08 (t, J = 8,97 Гц, 1H), 4,44 (d, J = 9,66 Гц, 1H), 4,02-3,79 (m, 3H), 3,22-2,91 (m, 5H), 2,81 (d, J = 14,81 Гц, 1H), 2,36 (s, 3H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 424,1 (M+1).
[758] Вариант осуществления 59
[759] (S)-2-((2-Циклогексил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол

[760] Стадия 1.
[761] 6-Бензил-2-(циклогексен-1-ил)-7,8-дигидро-5H-1,6-нафтиридин
[762] 6-Бензил-2-хлор-7,8-дигидро-5H-1,6-нафтиридин (200,00 мг, 772,95 мкмоль, 1,00 экв.), циклогексен-1-илбороновую кислоту (146,04 (1,65 ммоль, 1,50 экв.), Pd(dppf)Cl2 (56,56 мг, 77,30 мкмоль, 0,10 экв.) и фторид цезия (234,82 мг, 1,55 ммоль, 2,00 экв.) растворяли в диоксане (4,00 мл ) и воде (0,4 мл), а затем смесь перемешивали при 110°C в течение 12 часов в атмосфере газообразного азота. Реакционную смесь концентрировали при пониженном давлении, добавляли к остатку воду (10 мл) и экстрагировали этилацетатом (20 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток разделяли и очищали посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 20/1 – 10: 1) с получением 6-бензил-2-(циклогексен-1-ил)-7,8-дигидро-5H-1,6-нафтиридина (160,00 мг, 525,57 мкмоль, выход 68,00%) в виде желтого твердого вещества. 1H ЯМР (300МГц, CDCl3) δ 7,36-7,11 (m, 6H), 7,05-6,99 (m, 1H), 6,54 (s, 1H), 3,64 (s, 2H), 3,53 (s, 2H), 3,01-2,92 (m, 2H), 2,83-2,73 (m, 2H), 2,39 (br. s., 2H), 2,17 (d, J = 6,2 Гц, 2H), 1,77-1,66 (m, 2H), 1,65-1,55 (m, 2H).
[763] Стадия 2.
[764] 2-Циклогексил-5,6,7,8-тетрагидро-1,6-нафтиридин
[765] Гидроксид палладия/на угле (7,38 мг, 52,55 мкмоль, 0,10 экв.) добавляли к раствору 6-бензил-2-(циклогексен-1-ил)-7,8-дигидро-5H-1,6-нафтиридина (160,00 мг, 525,57 мкмоль, 1,00 экв.) в метаноле (5,00 мл) в атмосфере газообразного азота. Суспензию дегазировали и несколько раз производили замену на водород. Смесь перемешивали в атмосфере водорода (30 фунтов/кв. дюйм) при 15°C в течение 12 часов. Реакционную смесь фильтровали и концентрировали с получением 2-циклогексил-5,6,7,8-тетрагидро-1,6-нафтиридина (110,00 мг, 508,51 мкмоль, выход 96,75%, чистота 100%) в виде бесцветного масла.
[766] Стадия 3.
[767] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-циклогексил-7,8-дигидро-5H-1,6-нафтиридин-6-ил)-2-метилпропан-2-ол
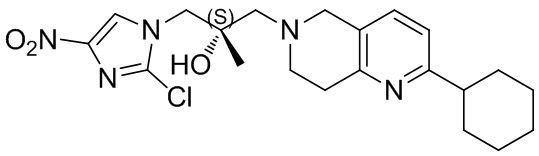
[768] Диизопропиламин (131,44 мг, 1,02 ммоль, 2,00 экв.) и 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазол (132,79 мг, 610,21 мкмоль, 1,20 экв.) добавляли к раствору 2-циклогексил-5,6,7,8-тетрагидро-1,6-нафтиридина (110,00 мг, 508,51 мкмоль, 1,00 экв.) в этаноле (5,00 мл). Смесь перемешивали при 80°C в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении, остаток разбавляли водой (20 мл) и экстрагировали этилацетатом (30 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток разделяли и очищали посредством препаративной тонкослойной хроматографии (диоксид кремния, этилацетат) с получением (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-циклогексил-7,8-дигидро-5H-1,6-нафтиридин-6-ил)-2-метилпропан-2-ола (80,00 мг, 135,87 мкмоль, выход 26,72%, чистота 73,7%) в виде желтого твердого вещества.
[769] Стадия 4.
[770] (S)-2-((2-Циклогексил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
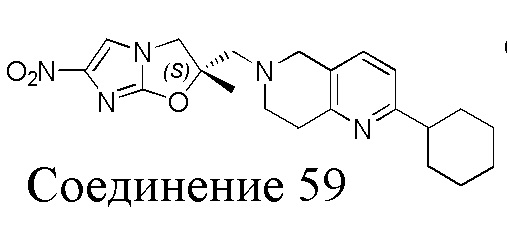
[771] Гидрид натрия (8,85 мг, 221,23 мкмоль, 1,20 экв.) добавляли к раствору (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-циклогексил-7,8-дигидро-5H-1,6-нафтиридин-6-ил)-2-метилпропан-2-ола (80,00 мг, 184,36 мкмоль, 1,00 экв.) в DMF (2,00 мл) при 0°C. Смесь перемешивали при 0°C в течение 1 часа. Реакционную смесь медленно добавляли к охлажденному насыщенному раствору хлорида аммония (10 мл), а затем экстрагировали этилацетатом (20 мл). Объединенные органические слои промывали солевым раствором (10 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной разделительной хроматографии (прибор: GX-D; колонка: Boston Green ODS 150*30 5u; подвижная фаза: 20% - 50%; H2O (+0,00225 FA); скорость: 25 мл/мин.; контролируемая длина волны: 220 нм/254 нм; длительность прогона: 10 мин./15 мин.; температура колонки: 30oC) с получением (S)-2-((2-циклогексил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 59 (70,00 мг, 174,71 мкмоль, выход 94,76%, чистота 99,2%). 1H ЯМР (400 МГц, CDCl3) δ 7,52 (s, 1H), 7,30 (s, 1H), 7,00 (d, J = 8,0 Гц, 1H), 4,42 (d, J = 9,7 Гц, 1H), 3,94 (d, J = 9,7 Гц, 1H), 3,87-3,74 (m, 2H), 3,15-2,90 (m, 5H), 2,84-2,67 (m, 2H), 2,02-1,72 (m, 5H), 1,67 (s, 3H), 1,52-1,23 (m, 5H). LCMS (ESI) масса/заряд: 398,2 (M+1).
[772] Вариант осуществления 60
[773] (S)-2-((2-(4-Фторфенил)-7,7-диметил-6,7-дигидротиазоло[5,4-c]пиридин-5(4H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
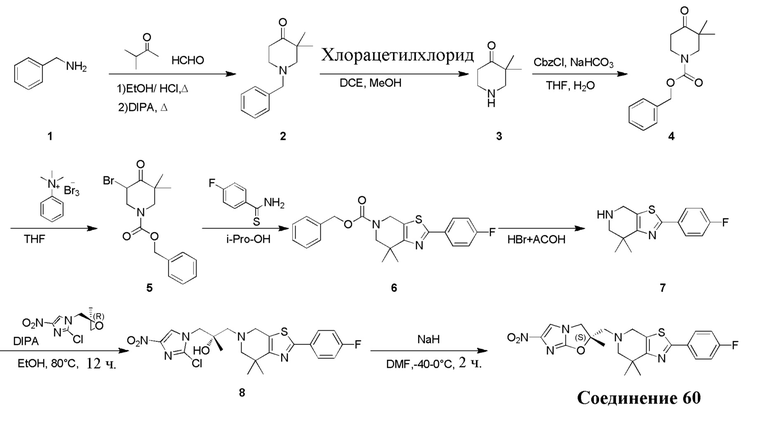
[774] Стадия 1.
[775] 1-Бензил-3,3-диметилпиперидин-4-он
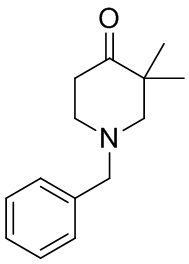
[776] Формальдегид (18,26 г, 225,00 ммоль, 2,25 экв.) растворяли в этаноле (100,00 мл) и к раствору медленно добавляли бензиламин (10,72 г, 100,00 ммоль, 1,00 экв.). Смесь перемешивали при 15°C в течение 1 часа. Затем смесь медленно добавляли к нагревающемуся с обратным холодильником раствору 3-метилбутан-2-она (8,61 г, 100,00 ммоль, 1,00 экв.) в этаноле (100,00 мл) и соляной кислоте (9,20 мл). Смесь перемешивали при 80°C в течение 12 часов. Затем смесь охлаждали до 15°C, а далее добавляли диизопропиламин (14,22 г, 110,00 ммоль, 1,10 экв.) и формальдегид (2,44 г, 30,00 ммоль, 0,30 экв.) и смесь перемешивали при 80°C в течение еще 7 часов. Доводили pH реакционной смеси до > 10 при помощи гидроксида калия, а затем экстрагировали этилацетатом (200 мл). Объединенные органические слои промывали солевым раствором (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 30/1 – 10: 1) с получением 1-бензил-3,3-диметилпиперидин-4-она (10,00 г, 46,02 ммоль, выход 46,02%) в виде бесцветного масла.
[777] Стадия 2.
[778] 3,3-Диметилпиперидин-4-он

[779] 1-Бензил-3,3-диметилпиперидин-4-он (5,00 г, 23,01 ммоль, 1,00 экв.) и 1-хлорэтилхлорформиат (6,58 г, 46,02 ммоль, 2,00 экв.) растворяли в дихлорэтане (50,00 мл). Смесь 3 раза дегазировали азотом, а затем смесь перемешивали при 80°C в течение 12 часов. Далее смесь концентрировали, добавляли метанол (50,00 мл) и смесь перемешивали при 80°C в течение дополнительных 4 часов. Реакционную смесь концентрировали для удаления MeOH, остаток промывали дихлорметаном (20 мл) и фильтровали. Собирали твердое вещество с получением 3,3-диметилпиперидин-4-она (3,00 г, 18,33 ммоль, выход 79,67%, гидрохлорид) в виде белого твердого вещества.
[780] 1H ЯМР (400 МГц, метанол-d4) δ 3,56 (t, J = 6,7 Гц, 1H), 3,37 (s, 1H), 3,25-3,07 (m, 1H), 3,01 (s, 1H), 2,76 (t, J = 6,7 Гц, 1H), 2,07-1,99 (m, 1H), 1,25 (s, 3H), 1,10 (d, J = 13,2 Гц, 3H).
[781] Стадия 3.
[782] Бензил-3,3-диметил-4-оксопиперидин-1-карбоксилат
[783] Бикарбонат натрия (1,28 г, 15,28 ммоль, 2,50 экв.) и CbzCl (1,25 г, 7,33 ммоль, 1,20 экв.) добавляли к смешанному раствору гидрохлорида 3,3-диметилпиперидин-4-она (1,00 г, 6,11 ммоль, 1,00 экв.) в тетрагидрофуране (5,00 мл) и воде (5,00 мл) при 0°C. Смесь перемешивали при 15°C в течение 12 часов. Реакционную смесь экстрагировали этилацетатом (20 мл). Объединенные органические слои промывали солевым раствором (10 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением бензил-3,3-диметил-4-оксопиперидин-1-карбоксилата (1,60 г, 4,81 ммоль, выход 78,66%, чистота 78,5%) в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 7,35-7,25 (m, 6H), 5,11 (s, 2H), 3,72 (t, J = 6,3 Гц, 2H), 3,42 (br. s., 2H), 2,44 (br. s., 2H), 1,02 (br. s., 6H).
[784] Стадия 4.
[785] Бензил-5-бром-3,3-диметил-4-оксопиперидин-1-карбоксилат
[786] Фенилтриметиламина трибромид (1,44 г, 3,83 ммоль, 1,00 экв.) добавляли к раствору бензил-3,3-диметил-4-оксопиперидин-1-карбоксилата (1,10 г, 4,21 ммоль, 1,10 экв.) в THF (10,00 мл). Смесь перемешивали при 15°C в течение 1 часа. Смесь гасили за счет добавления воды (10 мл), а затем экстрагировали этилацетатом (20 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного бензил-5-бром-3,3-диметил-4-оксопиперидин-1-карбоксилата (1,40 г, неочищенный) в виде желтого масла, которое использовали непосредственно на следующей стадии.
[787] Стадия 5.
[788] Бензил-2-(4-фторфенил)-7,7-диметил-4,6-дигидротиазоло[5,4-c]пиридин-5-карбоксилат
[789] Бензил-5-бром-3,3-диметил-4-оксопиперидин-1-карбоксилат (1,85 г, 5,44 ммоль, 1,00 экв.), 4-фтортиобензамид (843,89 мг, 5,44 ммоль, 1,00 экв.) растворяли в изопропаноле (20,00 мл), смесь дегазировали, три раза производили замену на азот и смеси позволяли прореагировать при 80°C в течение 12 часов. Реакционную смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (40 мл). Объединенные органические слои промывали солевым раствором 40 мл (20 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 20/1 – 10:1) с получением бензил-2-(4-фторфенил)-7,7-диметил-4,6-дигидротиазоло[5,4-c]пиридин-5-карбоксилата (800,00 мг, 2,02 ммоль, выход 37,09%) в виде бесцветного масла.
[790] Стадия 6.
[791] 2-(4-Фторфенил)-7,7-диметил-5,6-дигидро-4H-тиазоло[5,4-c]пиридин
[792] Бензил-2-(4-фторфенил)-7,7-диметил-4,6-дигидротиазоло[5,4-c]пиридин-5-карбоксилат (500,00 мг, 1,26 ммоль, 1,00 экв.) растворяли в растворе бромистого водорода/уксусной кислоты (5,00 мл), в смеси производили замену на азот, а затем ее перемешивали при 15°C в течение 2 часов. Реакционную смесь концентрировали для удаления части уксусной кислоты и отфильтровывали твердое вещество с получением 2-(4-фторфенил)-7,7-диметил-5,6-дигидро-4H-тиазоло[5,4-c]пиридина (400,00 мг, 1,17 ммоль, выход 92,86%, гидробромид) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,35-7,25 (m, 6H), 5,11 (s, 2H), 3,72 (t, J = 6,3 Гц, 2H), 3,42 (br. s., 2H), 2,44 (br. s., 2H), 1,02 (br. s., 6H).
[793] Стадия 6.
[794] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-7,7-диметил-4,6-дигидротиазоло[5,4-c]пиридин-5-ил)-2-метилпропан-2-ол
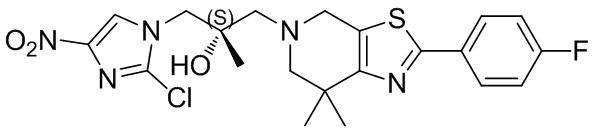
[795] 2-(4-Фторфенил)-7,7-диметил-5,6-дигидро-4H-тиазоло[5,4-c]пиридин (200,00 мг, 582,65 мкмоль, 1,00 экв., гидробромид), 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазол (152,15 мг, 699,18 мкмоль, 1,20 экв.) и диизопропиламин (225,91 мг, 1,75 ммоль, 3,00 экв.) растворяли в этаноле (5,00 мл), в смеси производили замену на азот, а затем ее перемешивали при 80°C в течение 12 часов. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (20 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 10/1 – 5/1) с получением (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-7,7-диметил-4,6-дигидротиазоло[5,4-c]пиридин-5-ил)-2-метилпропан-2-ола (200,00 мг, 416,71 мкмоль, выход 71,52%) в виде желтого масла. 1H ЯМР (300 МГц, CDCl3) δ 8,05 (s, 1H), 7,94-7,84 (m, 2H), 7,12 (t, J = 8,6 Гц, 2H), 4,09-3,78 (m, 4H), 2,90-2,57 (m, 4H), 1,47-1,35 (m, 6H), 1,31-1,26 (m, 3H).
[796] Стадия 7.
[797] (S)-2-((2-(4-Фторфенил)-7,7-диметил-6,7-дигидротиазоло[5,4-c]пиридин-5(4H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
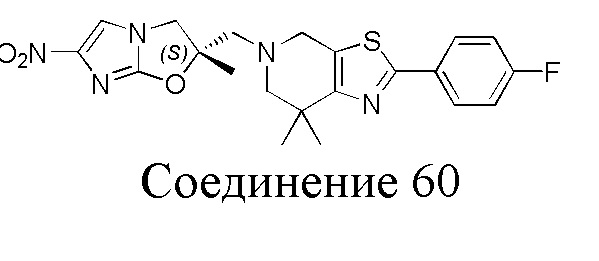
[798] Гидрид натрия (31,25 мг, 781,33 мкмоль, 1,50 экв.) добавляли к раствору (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-7,7-диметил-4,6-дигидротиазоло[5,4-c]пиридин-5-ил)-2-метилпропан-2-ола (250,00 мг, 520,89 мкмоль, 1,00 экв.) в DMF (5,00 мл) при 0°C. Смесь перемешивали при 0°C в течение 1 часа. Реакционную смесь добавляли к охлажденному насыщенному раствору хлорида аммония (20 мл) и гасили, а затем экстрагировали этилацетатом (20 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной разделительной хроматографии (прибор: GX-D; колонка: Boston Green ODS 150*30 5u; подвижная фаза: 58% - 88%; ACN (+0,00225 FA); скорость: 25 мл/мин.; контролируемая длина волны: 220 нм/254 нм; длительность прогона: 10 мин./15 мин.; температура колонки: 30oC) с получением (S)-2-((2-(4-фторфенил)-7,7-диметил-6,7-дигидротиазоло[5,4-c]пиридин-5(4H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 60 (72,40 мг, 163,25 мкмоль, выход 31,34%, чистота 100%). 1H ЯМР (400 МГц, CDCl3) δ 7,87 (dd, J = 5,3, 8,8 Гц, 1H), 7,52 (s, 1H), 7,11 (t, J = 8,6 Гц, 2H), 4,51 (d, J = 9,7 Гц, 1H), 3,96 (d, J = 9,7 Гц, 1H), 3,92-3,81 (m, 2H), 3,12 (d, J = 15,1 Гц, 1H), 2,89 (d, J = 11,5 Гц, 1H), 2,79 (d, J = 15,1 Гц, 1H), 2,66 (d, J = 11,5 Гц, 1H), 1,69 (s, 3H), 1,32 (s, 3H), 1,16 (s, 3H); LCMS (ESI) масса/заряд: 444,2 (M+1).
[799] Вариант осуществления 61
[800] (S)-2-(4-Фторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-4,5,6,7-тетрагидрооксазолo[5,4-c]пиридин
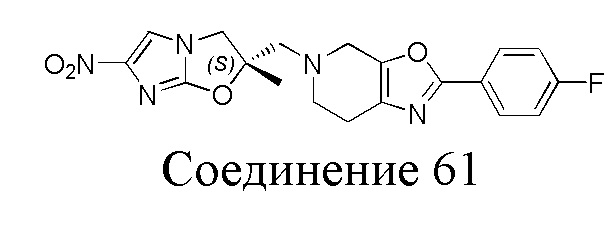
[801] Способ синтеза был таким, как в варианте осуществления 41.
[802] (S)-2-(4-Фторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-4,5,6,7-тетрагидрооксазолo[5,4-c]пиридин – соединение 61 (63,20 мг, 149,47 мкмоль, выход 8,12%, чистота 94,452%). 1H ЯМР (400 МГц, CDCl3) δ 7,94-7,86 (m, 2H), 7,46 (s, 1H), 7,06 (s, 2H), 4,35-4,29 (m, 1H), 3,91-3,85 (m, 1H), 3,77 (s, 2H), 3,09-2,97 (m, 2H), 2,93-2,84 (m, 1H), 2,75-2,67 (m, 1H), 2,56 (br. s., 2H), 1,58 (s, 3H). LCMS (ESI) масса/заряд: 400,2 (M+1).
[803] Вариант осуществления 62
[804] (S)-5-((2-Метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-2-(3,4,5-трифторфенил)-4,5,6,7-тетрагидрооксазолo[5,4-c]пиридин

[805] Способ синтеза был таким, как в варианте осуществления 41.
[806] (S)-5-((2-Метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-2-(3,4,5-трифторфенил)-4,5,6,7-тетрагидрооксазолo[5,4-c]пиридин – соединение 62 (222,70 мг, 484,42 мкмоль, выход 42,49%, чистота 94,7%). 1H ЯМР (400 МГц, CDCl3) δ 7,62 (t, J = 7,2 Гц, 2H), 7,55 (s, 1H), 4,45-4,35 (m, 1H), 3,98 (d, J = 9,8 Гц, 1H), 3,86 (s, 2H), 3,19-3,06 (m, 2H), 3,03-2,92 (m, 1H), 2,80 (d, J = 14,8 Гц, 1H), 2,70-2,58 (m, 2H), 1,68 (s, 3H). LCMS (ESI) масса/заряд: 436 (M+1).
[807] Вариант осуществления 63
[808] (S)-2-Метил-6-нитро-2-((2-(3,4,5-трифторфенил)-6,7-дигидротиазоло[5,4-c]пиридин-5(4H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
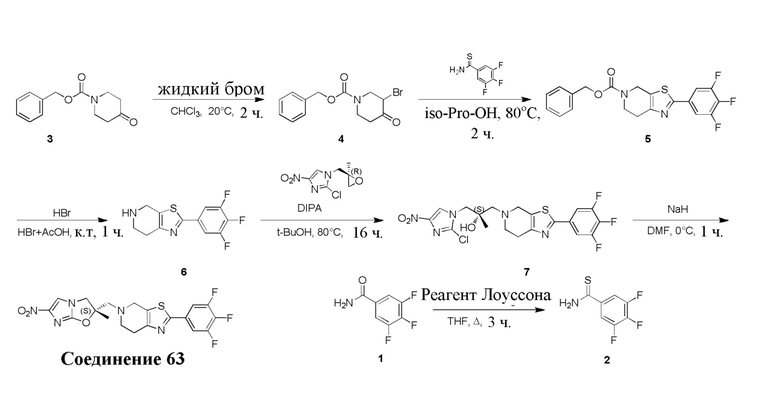
[809] Стадия 1.
[810] 3,4,5-Трифторбензотиоамид
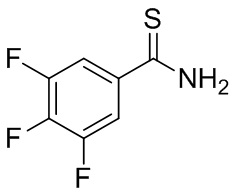
[811] Реагент Лоуссона (7,37 г, 18,22 ммоль, 1,10 экв.) добавляли к раствору 3,4,5-трифторбензамида (2,90 г, 16,56 ммоль, 1,00 экв.) в тетрагидрофуране (80,00 мл). Смесь перемешивали при 70°C в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя, остаток разбавляли водой (200 мл) и экстрагировали этилацетатом (30 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (30 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 5/1) с получением 3,4,5-трифторфенилтиоамида (4,35 г, неочищенный) в виде желтого твердого вещества.
[812] Стадия 2.
[813] Бензил-3-бром-4-оксопиперидин-1-карбоксилат
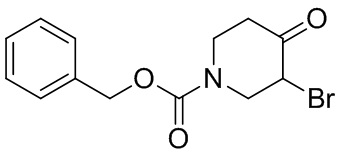
[814] Жидкий бром (3,43 г, 21,44 ммоль, 1,00 экв.) добавляли по каплям к раствору бензил-4-оксопиперидин-1-карбоксилата (5,00 г, 21,44 ммоль, 1,00 экв.) в хлороформе (50,00 мл) при 0°C в атмосфере газообразного азота. Смесь перемешивали при 25°C в течение 2 часов, реакционную смесь добавляли к охлажденному насыщенному раствору сульфита натрия (100 мл) и гасили, а затем экстрагировали дихлорметаном (50 мл × 3). Объединенные органические слои промывали насыщенным карбонатом натрия (100 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного бензил-3-бром-4-оксопиперидин-1-карбоксилата (7,10 г, неочищенный) в виде желтого масла, которое использовали непосредственно на следующей стадии.
[815] Стадия 3.
[816] Бензил-2-(3,4,5-трифторфенил)-6,7-дигидротиазоло[5,4-c]пиридин-5(4H)-карбоксилат
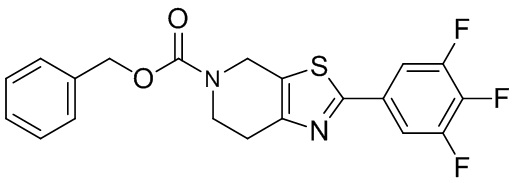
[817] 3,4,5-Трифторфенилтиоамид (4,35 г, 22,74 ммоль, 1,00 экв.) добавляли к раствору бензил-3-бром-4-оксопиперидин-1-карбоксилата (7,10 г, 22,74 ммоль, 1,00 экв.) в изопропаноле (50,00 мл). Смесь перемешивали при 80°C в течение 12 часов. Реакционную смесь концентрировали для удаления растворителя. Остаток очищали посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 5/1 – 1: 1, а затем дихлорметан : метанол = 50: 1) с получением бензил-2-(3,4,5-трифторфенил)-6,7-дигидротиазоло[5,4-c]пиридин-5(4H)-карбоксилата (1,00 г, неочищенный) в виде желтого масла. LCMS (ESI) масса/заряд: 405 (M + 1).
[818] Стадия 4.
[819] 2-(3,4,5-Трифторфенил)-4,5,6,7-тетрагидротиазоло[5,4-c]пиридин

[820] Бензил-2-(3,4,5-трифторфенил)-6,7-дигидротиазоло[5,4-c]пиридин-5(4H)-карбоксилат (1,00 г, 2,47 ммоль, 1,00 экв.) растворяли в растворе бромистого водорода/уксусной кислоты (3,00 мл). Смесь перемешивали при 25°C в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя с получением неочищенного 2-(3,4,5-трифторфенил)-4,5,6,7-тетрагидротиазоло[5,4-c]пиридина (1,00 г, неочищенный, гидробромид) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии. LCMS (ESI) масса/заряд: 271 (M + 1).
[821] Стадия 5.
[822] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-2-метил-3-(2-(3,4,5-трифторфенил)-6,7-дигидротиазоло[5,4-c]пиридин-5(4H)-ил)пропан-2-ол
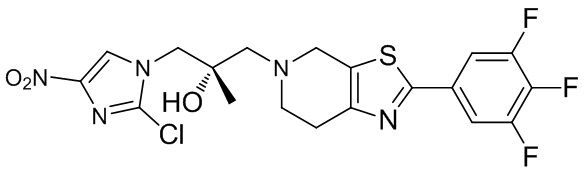
[823] Диизопропиламин (386,41 мг, 2,99 ммоль, 3,00 экв.) добавляли к раствору 2-(3,4,5-трифторфенил)-4,5,6,7-тетрагидротиазолo[5,4-c]пиридина (350,00 мг, 996,61 мкмоль, 1,00 экв., гидробромид) и (R)-2-хлор-1-((2-метилоксиран-2-ил)метил)-4-нитро-1H-имидазола (260,25 мг, 1,20 ммоль, 1,20 экв.) в этаноле (10,00 мл). Смесь перемешивали при 80°C в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и остаток очищали посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 5/1 – 1: 1) с получением (S)-1-(2-хлор-4-нитро-1H-имидазол-1-ил)-2-метил-3-(2-(3,4,5-трифторфенил)-6,7-дигидротиазоло[5,4-c]пиридин-5(4H)-ил)пропан-2-ола (200,00 мг, 409,94 мкмоль, выход 41,13%) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 488/490 (M + 1 / M + 3).
[824] Стадия 6.
[825] (S)-2-Метил-6-нитро-2-((2-(3,4,5-трифторфенил)-6,7-дигидротиазоло[5,4-c]пиридин-5(4H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
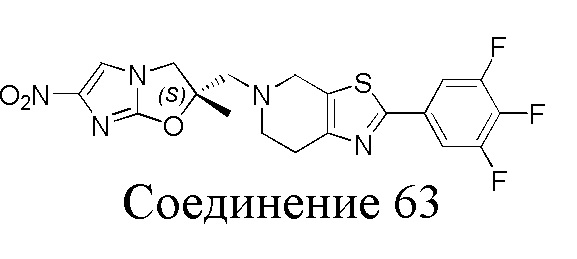
[826] Гидрид натрия (9,84 мг, 245,96 мкмоль, 1,20 экв.) добавляли к раствору (S)-1-(2-хлор-4-нитро-1H-имидазол-1-ил)-2-метил-3-(2-(3,4,5-трифторфенил)-6,7-дигидротиазоло[5,4-c]пиридин-5(4H)-ил)пропан-2-ола (100,00 мг, 204,97 мкмоль, 1,00 экв.) в DMF (2,00 мл) при 0°C. Смесь перемешивали при 0°C в течение 30 мин., гасили насыщенным раствором хлорида аммония (50 мл) и экстрагировали дихлорметаном (10 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (10 мл × 2), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной разделительной хроматографии (GX-F; Phenomenex Synergi C18 150*30 мм*4 мкм; ацетонитрил 50% - 80%; вода (0,225% муравьиная кислота); скорость: 25 мл/мин.) с получением (S)-2-метил-6-нитро-2-((2-(3,4,5-трифторфенил)-6,7-дигидротиазолo[5,4-c]пиридин-5(4H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазола – соединения 63 (23,00 мг, 50,67 мкмоль, выход 24,72%, чистота 99,448%). 1H ЯМР (400 МГц, CDCl3) δ 7,60-7,48 (m, 3H), 4,41 (d, J = 8,0 Гц, 2H), 4,03-3,91 (m, 3H), 3,22-3,10 (m, 2H), 3,09-2,99 (m, 1H), 2,91-2,83 (m, 2H), 2,79 (d, J = 16,0 Гц, 2H), 1,68 (s, 3H). LCMS (ESI) масса/заряд: 452 (M+1).
[827] Вариант осуществления 64
[828] (S)-2-(4-Фторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-4,5,6,7-тетрагидрофуро[3,2-c]пиридин
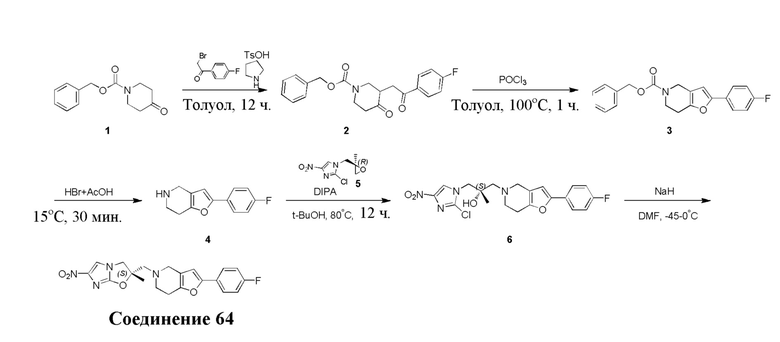
[829] Стадия 1.
[830] Бензил-3-(2-(4-фторфенил)-2-оксоэтил)-4-оксопиперидин-1-карбоксилат
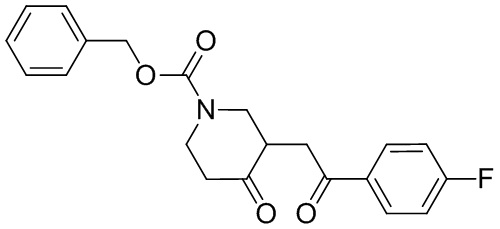
[831] Бензил-4-оксопиперидин-1-карбоксилат (5,00 г, 21,44 ммоль, 1,00 экв.) и пирролидин (6,10 г, 85,76 ммоль, 4,00 экв.), TsOH.H2O (407,83 мг, 2,14 ммоль, 0,10 экв.) растворяли в толуоле (20,00 мл). Смесь нагревали до 130°C и перемешивали, при этом отделяя воду при помощи отделителя воды. Через 12 часов, отделили приблизительно 0,5 мл воды и смесь концентрировали при 50°C. Неочищенный продукт бензил-4-пирролидин-1-ил-3,6-дигидро-2H-пиридин-1-карбоксилат (7,00 г, неочищенный) представлял собой коричневое масло. 2-Бром-1-(4-фторфенил)этанон (1,14 г, 5,24 ммоль, 1,00 экв.) добавляли одной порцией к раствору неочищенного продукта (1,50 г, 5,24 ммоль, 1,00 экв.) в толуоле (15,00 мл) при 10°C в атмосфере газообразного азота. Смесь перемешивали при 10°C в течение 12 часов, концентрировали, а остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 10/1, 2/1) с получением бензил-3-(2-(4-фторфенил)-2-оксоэтил)-4-оксопиперидин-1-карбоксилата (550,00 мг, 1,49 ммоль, выход 28,41%) в виде желтого масла.
[832] Стадия 2.
[833] Бензил-2-(4-фторфенил)-6,7-дигидрофуро[3,2-c]пиридин-5(4H)-карбоксилат
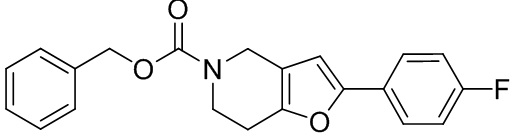
[834] Оксихлорид фосфора (1,81 г, 11,80 ммоль, 10,93 экв.) добавляли одной порцией к раствору бензил-3-(2-(4-фторфенил)-2-оксоэтил)-4-оксопиперидин-1-карбоксилата (400,00 мг, 1,08 ммоль, 1,00 экв.) в толуоле (6,00 мл) при 15°C в атмосфере газообразного азота. Смесь перемешивали при 100°C в течение 1 часа. Смесь выливали в воду (30 мл) и перемешивали в течение 5 минут. Водную фазу экстрагировали этилацетатом (50 мл × 3). Объединенные органические фазы промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 1/0, 10/1) с получением бензил-2-(4-фторфенил)-6,7-дигидрофуро[3,2-c]пиридин-5(4H)-карбоксилата (200,00 мг, 569,20 мкмоль, выход 52,70%) в виде желтого масла.
[835] Стадия 3.
[836] 2-(4-Фторфенил)-4,5,6,7-тетрагидрофуро[3,2-c]пиридин
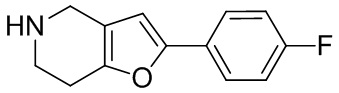
[837] Бензил-2-(4-фторфенил)-6,7-дигидрофуро[3,2-c]пиридин-5(4H)-карбоксилат (233,00 мг, 663,12 мкмоль, 1,00 экв.) растворяли в растворе бромистого водорода в уксусной кислоте (5 мл, 92,08 ммоль, 138,86 экв.). Смесь концентрировали при 45°C с получением твердого вещества 2-(4-фторфенил)-4,5,6,7-тетрагидрофуро[3,2-c]пиридина (200,00 мг, неочищенный, гидробромид). LCMS (ESI) масса/заряд: 218,1 (M + 1).
[838] Стадия 4.
[839] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил)-2-метилпропан-2-ол
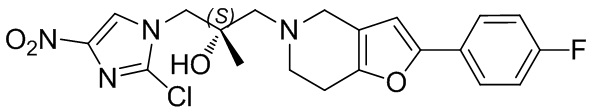
[840] Диизопропиламин (171,40 мг, 1,33 ммоль, 2,00 экв.) добавляли одной порцией к раствору 2-(4-фторфенил)-4,5,6,7-тетрагидрофуро[3,2-c]пиридина (197,71 мг, 663,12 мкмоль, 1,00 экв., гидробромид) и 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазола (158,73 мг, 729,43 мкмоль, 1,10 экв.) в трет-бутиловом спирте (10,00 мл) в атмосфере газообразного азота. Смесь перемешивали при 80°C в течение 12 часов и концентрировали ее при 45°C. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 1/0, 2/1) с получением (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил)-2-метилпропан-2-ола (190,00 мг, 436,93 мкмоль, выход 65,89%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,08 (s, 1H), 7,64-7,57 (m, 2H), 7,12-7,04 (m, 2H), 6,39 (s, 1H), 4,07-4,02 (m, 2H), 3,65 (d, J = 14,9 Гц, 2H), 3,12-2,95 (m, 2H), 2,85-2,78 (m, 2H), 2,70 (d, J = 14,1 Гц, 1H), 2,54 (d, J = 14,2 Гц, 1H), 1,20 (s, 3H).
[841] Стадия 5.
[842] (S)-2-(4-Фторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-4,5,6,7-тетрагидрофуро[3,2-c]пиридин
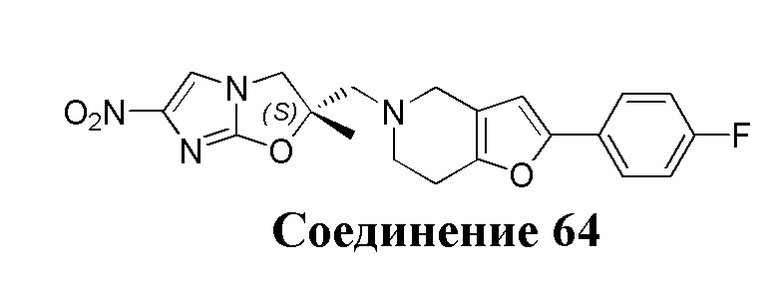
[843] Гидрид натрия (34,95 мг, 873,86 мкмоль, 2,00 экв.) добавляли одной порцией к раствору (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-6,7-дигидро-4H-фуро[3,2-c]пиридин-5-ил)-2-метилпропан-2-ола (190,00 мг, 436,93 мкмоль, 1,00 экв.) в DMF (5,00 мл) при -45°C в атмосфере газообразного азота. Смесь перемешивали при температуре от -45 до 0°C в течение 10 минут, выливали ее в насыщенный раствор хлорида аммония (30 мл) и перемешивали в течение 5 минут. Далее экстрагировали этилацетатом (50 мл × 3). Объединенные органические фазы промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством препаративной разделительной хроматографии (GX-D; Boston Green ODS 150*30 5u; ацетонитрил 40% - 70%; вода (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-(4-фторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-4,5,6,7-тетрагидрофуро[3,2-c]пиридина – соединения 64 (33,40 мг, 82,41 мкмоль, выход 18,86%, чистота 98,3%). 1H ЯМР (400 МГц, CDCl3) δ 7,58 (dd, J=5,3, 8,8 Гц, 2H), 7,54 (s, 1H), 7,06 (t, J=8,7 Гц, 2H), 6,36 (s, 1H), 4,42 (d, J=9,5 Гц, 1H), 3,94 (d, J=9,5 Гц, 1H), 3,63 (s, 2H), 3,19-3,06 (m, 2H), 2,98-2,88 (m, 1H), 2,74 (d, J=14,9 Гц, 1H), 2,71-2,56 (m, 2H), 1,67 (s, 3H). LCMS (ESI) масса/заряд: 399,1 (M+1).
[844] Вариант осуществления 65
[845] (S)-2-(3,4-Дифторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-4,5,6,7-тетрагидрооксазолo[4,5-c]пиридин
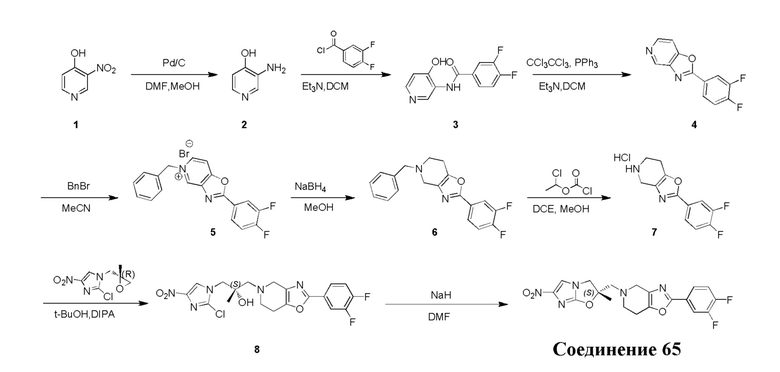
[846] Стадия 1.
[847] 3-Аминопиридин-4-ол
[848] Палладий на углероде (3,00 г, 214,13 ммоль, 1,00 экв.) добавляли одной порцией к смешанному раствору 3-нитропиридин-4-ола (30,00 г, 214,13 ммоль, 1,00 экв.) в метаноле (500,00 мл) и DMF (10,00 мл) при 15°C в атмосфере газообразного азота. Смесь перемешивали при 15°C в атмосфере водорода (25-40 фунтов/кв. дюйм) в течение 4 часов, фильтровали и концентрировали фильтрат до сухого состояния. Добавляли к остатку гидрохлорид/метанол (4 н., 100 мл) и смесь фильтровали, осадок промывали дихлорметаном (200 мл), фильтровали и собирали отфильтрованный осадок с получением 3-аминопиридин-4-ола (31,00 г, 211,50 ммоль, выход 98,77%, соляная кислота) в виде грязно-белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 14,44 (br, s, 1H), 7,96-7,84 (m, 2H), 7,27 (d, J = 6,3 Гц, 1H).
[849] Стадия 2.
[850] 3,4-Дифтор-N-(4-гидрокси-3-пиридил)бензамид
[851] Триэтиламин (41,42 г, 409,36 ммоль, 56,74 мл, 4,00 экв.) добавляли одной порцией к раствору 3-аминопиридин-4-ола (15,00 г, 102,34 ммоль, 1,00 экв, гидрохлорид) в дихлорметане (200,00 мл) при 0°C. Затем добавляли 3,4-дифторбензоилхлорид (25,30 г, 143,28 ммоль, 17,94 мл, 1,40 экв.) и полученную в результате смесь перемешивали при 15°C в течение 12 часов. Смесь фильтровали и отфильтрованный осадок промывали дихлорметаном (50 мл) с получением 3,4-дифтор-N-(4-гидрокси-3-пиридил)бензамида (30,00 г, неочищенный) в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 11,91 (br, s, 1H), 9,39 (s, 1H), 8,69 (br, s, 1H), 7,97 (ddd, J = 2,1, 7,8, 11,2 Гц, 1H), 7,84-7,77 (m, 1H), 7,73 (d, J = 5,1 Гц, 1H), 7,62 (td, J = 8,4, 10,3 Гц, 1H), 6,31 (d, J = 7,0 Гц, 1H). LCMS (ESI) масса/заряд: 251 (M+1).
[852] Стадия 3.
[853] 2-(3,4-Дифторфенил)оксазолo[4,5-c]пиридин

[854] Гексахлорэтан (35,48 г, 149,88 ммоль, 16,98 мл, 2,50 экв.), трифенилфосфин (47,17 г, 179,86 ммоль, 3,00 экв.) и триэтиламин (48,53 г, 479,62 ммоль, 66,48 мл, 8,00 экв.) растворяли в дихлорметане (200,00 мл), смесь перемешивали при 15°C в течение 0,5 часа, а затем добавляли порциями 3,4-дифтор-N-(4-гидрокси-3-пиридил)бензамид (15,00 г, 59,95 ммоль, 1,00 экв.). Полученную в результате смесь затем перемешивали при 15°C в течение 12 часов. Реакционную смесь добавляли по каплям к 1 н. соляной кислоте при 0°C и водный слой экстрагировали дихлорметаном (100 мл × 3). Затем доводили pH водного слоя до 8 с помощью насыщенного водного раствора бикарбоната натрия и экстрагировали водный слой дихлорметаном (100 мл × 3). Объединенные органические фазы промывали солевым раствором (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo с получением 2-(3,4-дифторфенил)оксазолo[4,5-c]пиридина (4,50 г, 19,38 ммоль, выход 32,33%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 9,13 (d, J = 0,8 Гц, 1H), 8,67-8,58 (m, 1H), 8,16-8,02 (m, 2H), 7,62-7,53 (m, 1H), 7,38 (td, J = 8,4, 9,3 Гц, 1H). LCMS (ESI) масса/заряд: 233 (M+1).
[855] Стадия 4.
[856] 5-Бензил-2-(3,4-дифторфенил)оксазолo[4,5-c]пиридин-5-ия бромид
[857] Бензилбромид добавляли (16,57 г, 96,90 ммоль, 11,51 мл, 5,00 экв.) к 2-(3,4-дифторфенил)оксазолo[4,5-c]пиридину (4,50 г, 19,38 ммоль, 1,00 экв.) в ацетонитриле (80,00 мл) при 0°C в атмосфере газообразного азота. Смесь перемешивали при 15°C в течение 12 часов. Смесь фильтровали и собирали отфильтрованный осадок с получением 5-бензил-2-(3,4-дифторфенил)оксазолo[4,5-c]пиридин-5-ия бромида (6,00 г, 14,88 ммоль, выход 76,78%) в виде белого твердого вещества. 1H ЯМР (400 МГц, метанол-d4) δ 9,78 (s, 1H), 9,12 (d, J = 7,0 Гц, 1H), 8,46 (d, J = 6,8 Гц, 1H), 8,37-8,21 (m, 2H), 7,69-7,55 (m, 3H), 7,54-7,42 (m, 3H), 6,04-5,95 (m, 2H). LCMS (ESI) масса/заряд: 323 (M+1).
[858] Стадия 5.
[859] 5-Бензил-2-(3,4-дифторфенил)-6,7-дигидро-4H-оксазолo[4,5-c]пиридин
[860] Боргидрид натрия (4,69 г, 124,00 ммоль, 10,00 экв.) добавляли к раствору 5-бензил-2-(3,4-дифторфенил)оксазолo[4,5-c]пиридин-5-ия бромида (5,00 г, 12,40 ммоль, 1,00 экв.) в метаноле (80,00 мл) при 0°C в атмосфере газообразного азота. Смесь перемешивали при 15°C в течение 12 часов. Добавляли к смеси воду (200 мл), а затем экстрагировали этилацетатом (200 мл × 4). Объединенные органические фазы промывали насыщенным солевым раствором (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали посредством хроматографии на силикагеле (высота колонки: 250 мм, диаметр: 100 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 20/1 – 5/1) с получением 5-бензил-2-(3,4-дифторфенил)-6,7-дигидро-4H-оксазолo[4,5-c]пиридина (3,80 г, неочищенный) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 7,81 (ddd, J = 2,1, 7,6, 11,0 Гц, 1H), 7,77-7,72 (m, 1H), 7,43-7,30 (m, 5H), 7,28-7,19 (m, 2H), 3,82-3,76 (m, 2H), 3,60-3,53 (m, 2H), 2,95-2,88 (m, 2H), 2,88-2,79 (m, 2H).
[861] Стадия 6.
[862] 2-(3,4-Дифторфенил)-4,5,6,7-тетрагидрооксазолo[4,5-c]пиридина гидрохлорид
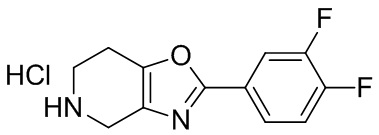
[863] 1-Хлорэтилхлорформиат (4,99 г, 34,92 ммоль, 3,00 экв.) добавляли одной порцией к раствору 5-бензил-2-(3,4-дифторфенил)-6,7-дигидро-4H-оксазолo[4,5-c]пиридина (3,80 г, 11,64 ммоль, 1,00 экв.) в дихлорэтане (60,00 мл) при 0°C в атмосфере газообразного азота. Смесь перемешивали при 100°C в течение 12 часов. Смесь концентрировали для удаления растворителя, добавляли к остатку метанол (80 мл) и перемешивали смесь при 80°C в течение 1 часа. Затем смесь концентрировали досуха и в остаток добавляли этилацетат (50 мл). Смесь фильтровали и собирали осадок с получением 2-(3,4-дифторфенил)-4,5,6,7-тетрагидрооксазолo[4,5-c]пиридина гидрохлорида (3,80 г, неочищенный, гидрохлорид) в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 9,85 (br, s, 2H), 8,01-7,93 (m, 1H), 7,87-7,79 (m, 1H), 7,69-7,59 (m, 1H), 4,20 (br, s, 2H), 3,52-3,49 (m, 2H), 3,12-3,03 (m, 2H).
[864] Стадия 7.
[865] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-(3,4-дифторфенил)-6,7-дигидро-4H-оксазолo[4,5-c]пиридин-5-ил)-2-метилпропан-2-ол
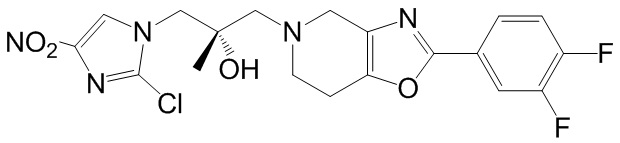
[866] Диизопропиламин (2,37 г, 18,34 ммоль, 3,20 мл, 2,50 экв.) добавляли одной порцией к раствору 2-(3,4-дифторфенил)-4,5,6,7-тетрагидрооксазолo[4,5-c]пиридина (2,00 г, 7,33 ммоль, 1,00 экв, гидрохлорид) и 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазола (1,60 г, 7,33 ммоль, 1,00 экв.) в трет-бутаноле (30,00 мл) при 15°C в атмосфере газообразного азота. Смесь перемешивали при 100°C в течение 12 часов. Смесь концентрировали досуха и остаток очищали посредством хроматографии на силикагеле (высота колонки: 250 мм, диаметр: 100 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 20/1 – 1/1) с получением (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(3,4-дифторфенил)-6,7-дигидро-4H-оксазолo[4,5-c]пиридин-5-ил)-2-метилпропан-2-ола (1,00 г, 2,20 ммоль, выход 30,06%) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 454 (M+1).
[867] Стадия 8.
[868] (S)-2-(3,4-Дифторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-4,5,6,7-тетрагидрооксазолo[4,5-c]пиридин

[869] Гидрид натрия (176,00 мг, 4,40 ммоль, чистота 60%, 2,00 экв.) добавляли одной порцией к раствору (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(3,4-дифторфенил)-6,7-дигидро-4H-оксазолo[4,5-c]пиридин-5-ил)-2-метилпропан-2-ола (1,00 г, 2,20 ммоль, 1,00 экв.) в DMF (10,00 мл) при -5°C в атмосфере газообразного азота. Смесь перемешивали при -5°C в течение 0,5 часа. Смесь добавляли по каплям к раствору хлорида аммония (100 мл), фильтровали и собирали отфильтрованный осадок с получением неочищенного продукта, который очищали посредством хроматографии на силикагеле (высота колонки: 250 мм, диаметр: 100 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 20/1 – 1/1) с получением (S)-2-(3,4-дифторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-4,5,6,7-тетрагидрооксазолo[4,5-c]пиридина (386,40 мг, 907,28 мкмоль, выход 41,24%, чистота 98,0%). 1H ЯМР (400 МГц, CDCl3) δ 7,80 (ddd, J = 2,0, 7,6, 10,9 Гц, 1H), 7,74 (ddd, J = 1,7, 4,0, 8,6 Гц, 1H), 7,55-7,51 (m, 1H), 7,24 (td, J = 8,3, 9,8 Гц, 1H), 4,37 (d, J = 9,7 Гц, 1H), 3,96 (d, J = 9,7 Гц, 1H), 3,77-3,65 (m, 2H), 3,23 (td, J = 4,9, 12,1 Гц, 1H), 3,16 (d, J = 15,1 Гц, 1H), 2,94 (ddd, J = 4,6, 7,9, 12,3 Гц, 1H), 2,78 (d, J = 15,1 Гц, 1H), 2,75-2,66 (m, 1H), 2,66-2,55 (m, 1H), 1,68 (s, 3H). LCMS (ESI) масса/заряд: 418 (M+1).
[870] Вариант осуществления 66
[871] (S)-2-(4-Фторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-4,5,6,7-тетрагидрооксазолo[4,5-c]пиридин
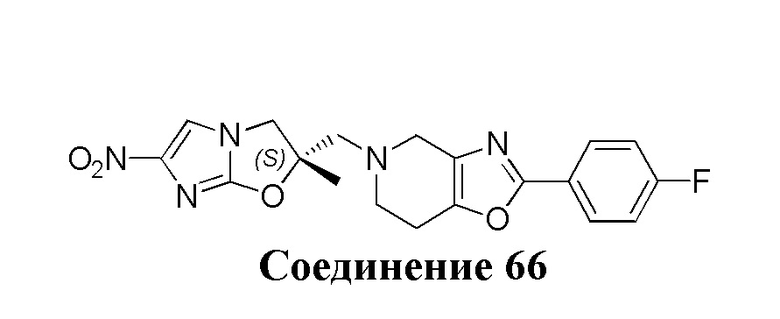
[872] Способ синтеза был таким, как в варианте осуществления 65.
[873] (S)-2-(4-Фторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-4,5,6,7-тетрагидрооксазолo[4,5-c]пиридин – соединение 66 (16,60 мг, 39,04 мкмоль, выход 21,27%, чистота 93,931%). 1H ЯМР (400 МГц, CDCl3) δ 7,94-7,84 (m, 2H), 7,45 (s, 1H), 7,09 (s, 2H), 4,32-4,22 (m, 1H), 3,96-3,70 (m, 3H), 3,31-3,17 (m, 2H), 3,04-2,70 (m, 4H), 1,63 (s, 3H). LCMS (ESI) масса/заряд: 400,2 (M+1).
[874] Вариант осуществления 67
[875] (S)-2-((2-(4-Фторфенил)-6,7-дигидротиазоло[4,5-c]пиридин-5(4H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
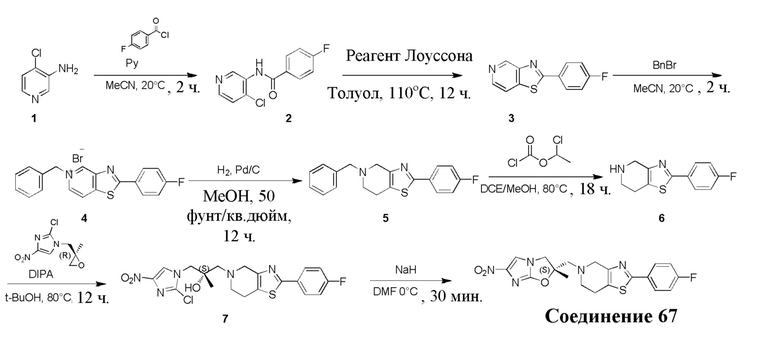
[876] Стадия 1.
[877] N-(4-Хлор-3-пиридинил)-4-фторбензамид
[878] Пиридин (2,46 г, 31,12 ммоль, 2,00 экв.) и 4-фторбензоилхлорид (2,71 г, 17,12 ммоль, 1,10 экв.) добавляли к раствору 4-хлорпиридин-3-амина (2,00 г, 15,56 ммоль, 1,00 экв.) в ацетонитриле (20,00 мл). Смесь перемешивали при 20°C в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении. Затем добавляли воду (100 мл), а далее смесь экстрагировали дихлорметаном (200 мл × 3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением N-(4-хлор-3-пиридинил)-4-фторбензамида (3,20 г, 12,77 ммоль, выход 82,07%) в виде белого твердого вещества. 1H ЯМР (300 МГц, CDCl3) δ 9,65 (s, 1H), 8,28 (d, J = 5,27 Гц, 1H), 8,12 (br. s., 1H), 7,93-7,83 (m, 2H), 7,33 (d, J = 5,09 Гц, 1H), 7,22-7,11 (m, 3H).
[879] Стадия 2.
[880] 2-(4-Фторфенил)тиазоло[4,5-c]пиридин
[881] Реагент Лоуссона (3,62 г, 8,94 ммоль, 0,70 экв.) добавляли к раствору N-(4-хлор-3-пиридинил)-4-фторбензамида (3,20 г, 12,77 ммоль, 1,00 экв.) в толуоле (50,00 мл). Смесь перемешивали при 110°C в течение 12 часов. Реакционную смесь концентрировали, добавляли к остатку насыщенный раствор бикарбоната натрия (100 мл), а затем экстрагировали дихлорметаном (150 мл × 3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (высота колонки: 300 мм, диаметр: 50 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 5/1, 3/1, 1/1) с получением 2-(4-фторфенил)тиазоло[4,5-c]пиридина (1,50 г, 6,51 ммоль, выход 51,01%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 9,28 (s, 1H), 8,46 (d, J = 5,52 Гц, 1H), 8,09-8,00 (m, 2H), 7,79 (d, J = 5,40 Гц, 1H), 7,21-7,09 (m, 2H).
[882] Стадия 3.
[883] 5-Бензил-2-(4-фторфенил)тиазоло[4,5-c]пиридин-5-ия бромид
[884] 2-(4-Фторфенил)тиазоло[4,5-c]пиридин (700,00 мг, 3,04 ммоль, 1,00 экв.) и бензилбромид (519,94 мг, 3,04 ммоль, 1,00 экв.) растворяли в ацетонитриле (5,00 мл). В смеси производили замену на азот, а затем перемешивали при 20°C в течение 2 часов. Смесь фильтровали и отфильтрованный осадок промывали дихлорметаном (20 мл). Собирали отфильтрованный осадок с получением 5-бензил-2-(4-фторфенил)тиазоло[4,5-c]пиридин-5-ия бромида (1,20 г, 2,99 ммоль, выход 98,36%) в виде белого твердого вещества.
[885] Стадия 4.
[886] 5-Бензил-2-(4-фторфенил)-4,5,6,7-тетрагидротиазоло[4,5-c]пиридин
[887] Гидроксид палладия/углерод (157,28 мг, 1,12 ммоль, 0,50 экв.) добавляли к раствору 5-бензил-2-(4-фторфенил)тиазоло[4,5-c]пиридин-5-ия бромида (900,00 мг, 2,24 ммоль, 1,00 экв.) в метаноле (200,00 мл) в атмосфере газообразного азота. Суспензию дегазировали и три раза производили замену на водород. Смесь перемешивали в водороде (50 фунтов/кв. дюйм) при 50°C в течение 12 часов. Реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении с получением 5-бензил-2-(4-фторфенил)-4,5,6,7-тетрагидротиазоло[4,5-c]пиридина (580,00 мг, 1,79 ммоль, выход 79,91%) в виде белого твердого вещества. 1H ЯМР (400 МГц, метанол-d4) δ 8,01-7,91 (m, 2H), 7,68-7,51 (m, 5H), 7,23 (t, J = 8,72 Гц, 2H), 4,59 (s, 2H), 4,49-4,37 (m, 2H), 3,74 (br. s., 2H), 3,38-3,33 (m, 2H).
[888] Стадия 5.
[889] 2-(4-фторфенил)-4,5,6,7-тетрагидро[4,5-c]пиридин

[890] 1-Хлорэтилхлорформиат (396,74 мг, 2,78 ммоль, 1,50 экв.) добавляли к раствору 5-бензил-2-(4-фторфенил)-4,5,6,7-тетрагидротиазолo[4,5-c]пиридина (600,00 мг, 1,85 ммоль, 1,00 экв.) в дихлорэтане (5,00 мл). Смесь перемешивали при 80°C в течение 16 часов. Дихлорэтан удаляли путем концентрирования, добавляли метанол (5,00 мл) и смесь перемешивали при 60°C в течение 2 часов. Смесь фильтровали и сушили осадок с получением 2-(4-фторфенил)-4,5,6,7-тетрагидро[4,5-c]пиридина (280,00 мг, 1,20 ммоль, выход 64,60%) в виде белого твердого вещества. LCMS (ESI) масса/заряд: 235 (M + 1).
[891] Стадия 6.
[892] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-6,7-дигидро-4H-тиазоло[4,5-c]пиридин-5-ил)-2-метилпропан-2-ол
[893] Диизопропиламин (220,65 мг, 1,71 ммоль, 2,00 экв.) добавляли к раствору 2-(4-фторфенил)-4,5,6,7-тетрагидро[4,5-c]пиридина (200,00 мг, 853,64 мкмоль, 1,00 экв.) и 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метилэтил]-4-нитроимидазола (222,91 мг, 1,02 ммоль, 1,20 экв.) в трет-бутаноле (2,00 мл). Смесь перемешивали при 80°C в течение 12 часов. Добавляли к реакционной смеси воду (50 мл), а затем экстрагировали этилацетатом (50 мл × 3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (высота колонки: 300 мм, диаметр: 50 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 5/1, 3/1) с получением (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-6,7-дигидро-4H-тиазоло[4,5-c]пиридин-5-ил)-2-метилпропан-2-ола (230,00 мг, 508,96 мкмоль, выход 59,62%) в виде желтого твердого вещества.
[894] Стадия 7.
[895] (S)-2-((2-(4-Фторфенил)-6,7-дигидротиазоло[4,5-c]пиридин-5(4H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
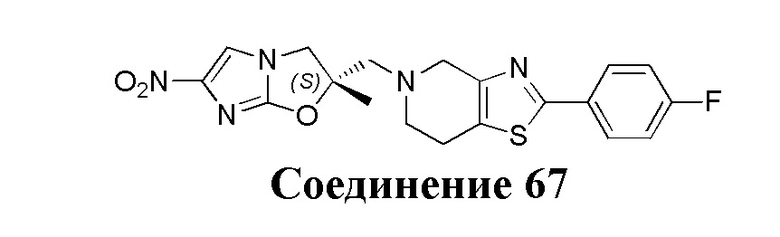
[896] Гидрид натрия (40,72 мг, 1,02 ммоль, 2,00 экв.) добавляли к раствору (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-6,7-дигидро-4H-тиазоло[4,5-c]пиридин-5-ил)-2-метилпропан-2-ола (230,00 мг, 508,96 мкмоль, 1,00 экв.) в DMF (3,00 мл). Смесь перемешивали при -45°C в течение 30 минут, а затем перемешивали при 0°C в течение 30 минут. Реакционную смесь добавляли к насыщенному раствору хлорида аммония (50 мл), а затем экстрагировали этилацетатом (50 мл × 3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством препаративной хроматографии (GX-I; Phenomenex Gemini 150*25 мм*10 мкм; ацетонитрил 36% - 66%; ACN (0,225% муравьиная кислота); 25 мл/мин.) с получением (S)-2-((2-(4-фторфенил)-6,7-дигидротиазоло[4,5-c]пиридин-5(4H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 67 (84,60 мг, 200,99 мкмоль, выход 39,49%, чистота 98,7%). 1H ЯМР (400 МГц, CDCl3-d) δ 7,83-7,72 (m, 2H), 7,44 (s, 1H), 7,09-6,98 (m, 2H), 4,31 (d, J = 9,66 Гц, 1H), 3,92-3,72 (m, 3H), 3,16-3,01 (m, 2H), 2,84-2,52 (m, 4H), 1,60 (s, 3H). LCMS (ESI) масса/заряд: 416 (M+1).
[897] Вариант осуществления 68
[898] (S)-2-((2-(3,4-Дифторфенил)-6,7-дигидротиазоло[4,5-c]пиридин-5(4H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
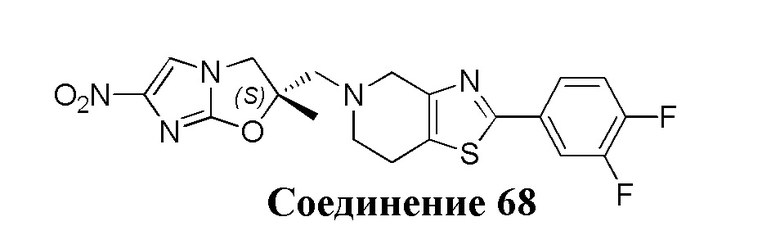
[899] Способ синтеза был таким, как в варианте осуществления 67.
[900] (S)-2-((2-(3,4-Дифторфенил)-6,7-дигидротиазоло[4,5-c]пиридин-5(4H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 68 (75,10 мг, 171,19 мкмоль, выход 32,18%, чистота 98,8%). 1H ЯМР (400 МГц, CDCl3) δ 7,73 (ddd, J = 2,2, 7,6, 11,0 Гц, 1H), 7,62-7,56 (m, 1H), 7,53 (s, 1H), 7,26-7,17 (m, 1H), 4,39 (d, J = 9,5 Гц, 1H), 3,99-3,82 (m, 3H), 3,28-3,11 (m, 2H), 2,92-2,74 (m, 3H), 2,73-2,61 (m, 1H), 1,69 (s, 3H). LCMS (ESI) масса/заряд: 434,2 (M+1).
[901] Вариант осуществления 69
[902] (S)-2-((2-(4-Фторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
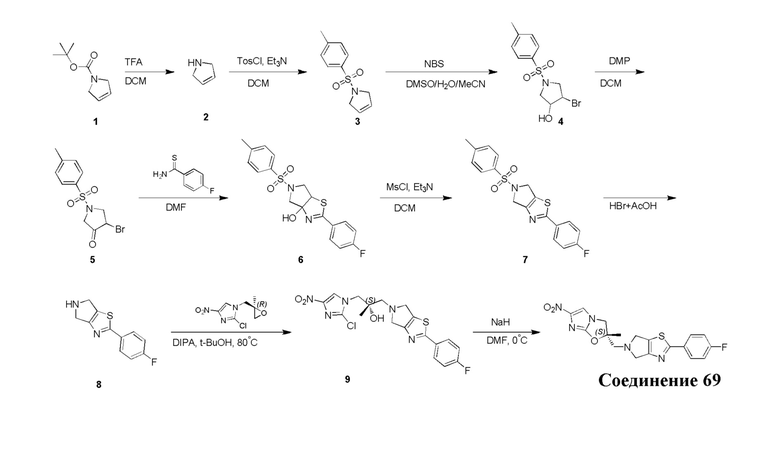
[903] Стадия 1.
[904] 2,5-Дигидро-lH-пиррол
[905] Трифторуксусную кислоту (9,18 г, 80,51 ммоль, 6,81 экв.) добавляли к раствору трет-бутил-2,5-дигидропиррол-1-карбоксилата (2,00 г, 11,82 ммоль, 1,00 экв.) в дихлорметане (6,00 мл), смесь перемешивали при 20°C в течение 1 часа. Смесь концентрировали при пониженном давлении с получением неочищенного продукта в виде темно-коричневого масла, которое использовали непосредственно на следующей стадии.
[906] Стадия 2.
[907] 1-Тозил-2,5-дигидро-1H-пиррол
[908] TosCl (2,70 г, 14,15 ммоль, 1,20 экв.) и триэтиламин (3,58 г, 35,37 ммоль, 3,00 экв.) добавляли к раствору 2,5-дигидро-1H-пиррола (2,16 г, 11,79 ммоль, 1,00 экв., TFA) в дихлорметане (10,00 мл). Смесь перемешивали при 20°C в течение 16 часов. Реакционную смесь промывали 1 M разбавленной соляной кислотой (30 мл), насыщенным раствором NaHCO3 (30 мл) и солевым раствором (30 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 1-(п-толуолсульфонил)-2,5-дигидропиррола (1,50 г, 6,72 ммоль, выход 56,98%) в виде коричневого твердого вещества.
[909] Стадия 3.
[910] 4-Бром-1-тозилпирролидин-3-ол
[911] NBS (1,79 г, 10,08 ммоль, 1,50 экв.) добавляли порциями к раствору 1-(п-толуолсульфонил)-2,5-дигидропиррола (1,50 г, 6,72 ммоль, 1,00 экв.) в DMSO (10,00 мл) и ацетонитриле (5,00 мл) при 0°C. Затем смесь перемешивали при 15°C в течение 16 часов. Добавляли к реакционной смеси воду (50 мл) и экстрагировали этилацетатом (30 мл × 2). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 10: 1 – 3: 1) с получением 4-бром-1-(п-толуолсульфонил)пирролидин-3-ола (1,58 г, 4,93 ммоль, выход 73,43%) в виде белого твердого вещества.
[912] Стадия 4.
[913] 4-Бром-1-тозилпирролидин-3-он

[914] Перйодинан Десс-Мартина (4,19 г, 9,87 ммоль, 2,00 экв.) добавляли к раствору 4-бром-1-(п-толуолсульфонил)пирролидин-3-ола (1,58 г, 4,93 ммоль, 1,00 экв.) в дихлорметане (15,00 мл). В смеси производили замену на азот и перемешивали при температуре от 10 до 15°C в течение 12 часов. Реакционную смесь промывали водным раствором бикарбоната натрия (50 мл × 2) и экстрагировали этилацетатом (30 мл × 2). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (диоксид кремния, петролейный эфир/этилацетат = 10: 1 – 1: 1) с получением 4-бром-1-(п-толуолсульфонил)пирролидин-3-она (980,00 мг, 3,08 ммоль, выход 62,47%) в виде желтого твердого вещества. 1H ЯМР (300МГц, CDCl3) δ 7,69-7,61 (d, J = 8,1 Гц, 2H), 7,35-7,28 (d, J = 7,8 Гц, 2H), 4,32-4,25 (t, J = 6,3 Гц, 1H), 4,00-3,91 (m, 1H), 3,61 (s, 2H), 3,55-3,47 (m, 1H), 2,39 (s, 3H).
[915] Стадия 5.
[916] 2-(4-Фторфенил)-5-тозил-4,5,6,6a-тетрагидро-3aH-пирроло[3,4-d]тиазол-3a-ол
[917] 4-Фтортиобензамид (430,00 мг, 2,77 ммоль, 1,00 экв.) добавляли к раствору 4-бром-1-(п-толуолсульфонил)пирролидин-3-она (880,00 мг, 2,77 ммоль, 1,00 экв.) в DMF (12,00 мл). Смесь перемешивали при 60°C в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении и остаток разбавляли этилацетатом (10 мл), фильтровали, собирали отфильтрованный осадок и очищали отфильтрованную жидкость посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 10:1, 3:1) с получением 2-(4-фторфенил)-5-тозил-4,5,6,6a-тетрагидро-3aH-пирроло[3,4-d]тиазол-3a-ола (900,00 мг, 2,49 ммоль) в виде грязно-белого твердого вещества.
[918] Стадия 6.
[919] 2-(4-Фторфенил)-5-тозил-5,6-дигидро-4H-пирроло[3,4-d]тиазол
[920] 2-(4-Фторфенил)-5-тозил-4,5,6,6a-тетрагидро-3aH-пирроло[3,4-d]тиазол-3a-ол (900,00 мг, 2,29 ммоль, 1,00 экв.), метансульфонилхлорид (444,00 мг, 3,88 ммоль, 1,69 экв.) и триэтиламин (730,00 мг, 7,21 ммоль, 3,15 экв.) растворяли в дихлорметане (20,00 мл), смесь дегазировали и перемешивали при 20°C в течение 16 часов в атмосфере газообразного азота. Добавляли воду (50 мл) и экстрагировали дихлорметаном (30 мл × 2). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 20: 1 – 1: 1) с получением 2-(4-фторфенил)-5-тозил-5,6-дигидро-4H-пирроло[3,4-d]тиазола (600,00 мг, 1,11 ммоль, выход 48,28%, чистота 69%) в виде светло-желтого твердого вещества.
[921] Стадия 7.
[922] 2-(4-Фторфенил)-5,6-дигидро-4H-пирроло[3,4-d]тиазол
[923] Раствор уксусной кислоты (81,72 мг, 1,01 ммоль, 1,00 экв.) добавляли к смешанному раствору 2-(4-фторфенил)-5-тозил-5,6-дигидро-4H-пирроло[3,4-d]тиазола (550,00 мг, 1,01 ммоль, 1,00 экв.) в воде (3,00 мл) и AcOH (15,00 мл). Смесь перемешивали при 20°C в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении, затем разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл). Водный слой подщелачивали NaOH, чтобы довести pH до 11, а затем экстрагировали этилацетатом (40 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 2-(4-фторфенил)-5,6-дигидро-4H-пирроло[3,4-d]тиазола (145,00 мг, 658,28 мкмоль, выход 65,18%) в виде бледно-желтого твердого вещества.
[924] Стадия 8.
[925] (S)-1-(2-Хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(4-фторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)-2-метилпропан-2-ол

[926] 2-Хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазол (158,00 мг, 726,07 мкмоль, 1,00 экв.) и диизопропиламин (481,00 мг, 3,72 ммоль, 5,12 экв.) добавляли к раствору 2-(4-фторфенил)-5,6-дигидро-4H-пирроло[3,4-d]тиазола (160,00 мг, 726,38 мкмоль, 1,00 экв.) в трет-бутаноле. Смесь перемешивали при 80°C в течение 16 часов. Добавляли воду (50 мл) и экстрагировали этилацетатом (30 мл × 2). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, остаток очищали посредством препаративной тонкослойной хроматографии (диоксид кремния, дихлорметан : метанол = 20: 1) с получением (S)-1-(2-хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(4-фторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)-2-метилпропан-2-ола (150,00 мг, 342,56 мкмоль, выход 47,16%) в виде бледно-желтого твердого вещества.
[927] Стадия 9.
[928] (S)-2-((2-(4-Фторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
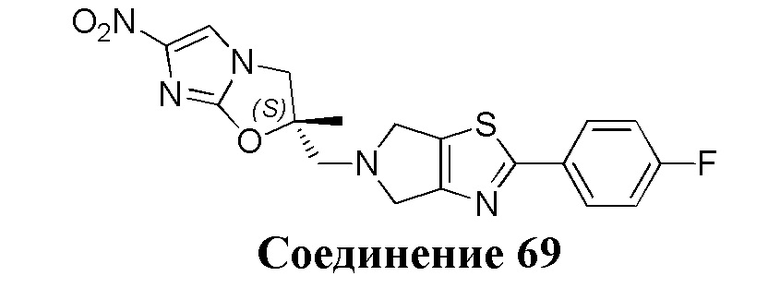
[929] Гидрид натрия (30,00 мг, 750,00 мкмоль, 2,19 экв.) добавляли к раствору (S)-1-(2-хлор-4-нитро-1H-имидазол-1-ил)-3-(2-(4-фторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)-2-метилпропан-2-ола (150,00 мг, 342,56 мкмоль, 1,00 экв.) в DMF (5,00 мл) при 0°C в атмосфере газообразного азота. Смесь перемешивали при 0°C в течение 1 часа. Реакционную смесь выливали в насыщенный раствор хлорида аммония (40 мл) при 0°C, а затем экстрагировали этилацетатом (30 мл × 2). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали этилацетатом с получением (S)-2-((2-(4-фторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола (17,71 мг, 40,90 мкмоль, выход 11,94%, чистота 92,71%). 1H ЯМР (400 МГц, CDCl3) δ 7,91-7,84 (m, 2H), 7,57 (s, 1H), 7,17-7,09 (t, J = 8,4 Гц, 2H), 4,53-4,48 (d, J = 9,6 Гц, 1H), 4,20 (d, J = 3,3 Гц, 3H), 4,13-4,07 (m, 1H), 4,03-3,97 (m, J = 9,6 Гц, 1H), 3,42 (d, J = 14,8 Гц, 1H), 3,12 (d, J = 14,8 Гц, 1H), 1,64-1,60 (m, 3H). LCMS (ESI) масса/заряд: 402,10 (M+1).
[930] Вариант осуществления 70
[931] (S)-2-((2-(3-Фторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол

[932] Способ синтеза был таким, как в варианте осуществления 69.
[933] (S)-2-((2-(3-Фторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 70 (67,10 мг, 165,99 мкмоль, выход 48,46%, чистота 99,3%). 1H ЯМР (400 МГц, CDCl3) δ 7,67-7,58 (m, 2H), 7,55 (s, 1H), 7,39 (dt, J = 5,8, 8,0 Гц, 1H), 7,11 (dt, J = 1,8, 8,3 Гц, 1H), 4,49 (d, J = 9,7 Гц, 1H), 4,26-4,17 (m, 3H), 4,14-4,06 (m, 1H), 3,98 (d, J = 9,7 Гц, 1H), 3,40 (d, J = 14,8 Гц, 1H), 3,11 (d, J = 14,7 Гц, 1H), 1,70 (s, 3H). LCMS (ESI) масса/заряд: 402,2 (M+1).
[934] Вариант осуществления 71
[935] (S)-2-((2-(3,4-Дифторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
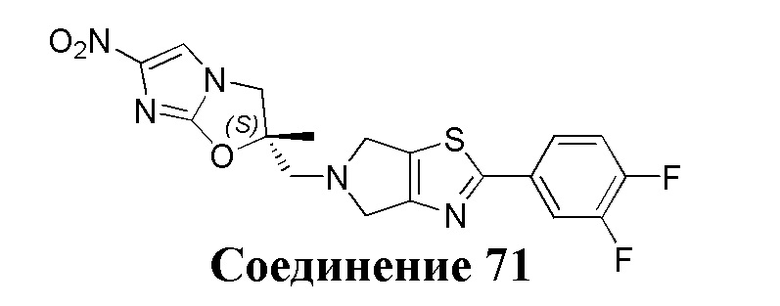
[936] Способ синтеза был таким, как в варианте осуществления 69.
[937] (S)-2-((2-(3,4-Дифторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 71 (49,80 мг, 115,42 мкмоль, выход 21,05%, чистота 97,2%). 1H ЯМР (400 МГц, CDCl3) δ 7,75 (ddd, J = 2,2, 7,5, 11,0 Гц, 1H), 7,65-7,58 (m, 1H), 7,56 (s, 1H), 7,25-7,19 (m, 1H), 4,49 (d, J = 9,7 Гц, 1H), 4,27-4,18 (m, 3H), 4,13-4,07 (m, 1H), 4,00 (d, J = 9,7 Гц, 1H), 3,42 (d, J = 14,8 Гц, 1H), 3,12 (d, J = 14,7 Гц, 1H), 1,72 (s, 3H). LCMS (ESI) масса/заряд: 420,2 (M+1).
[938] Вариант осуществления 72
[939] (S)-2-Метил-6-нитро-2-((2-(4-(трифторметил)фенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
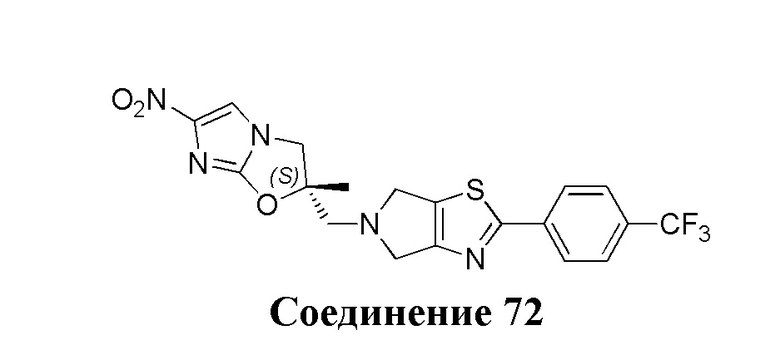
[940] Способ синтеза был таким, как в варианте осуществления 69.
[941] (S)-2-Метил-6-нитро-2-((2-(4-(трифторметил)фенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол – соединение 72 (24,30 мг, 52,53 мкмоль, выход 17,68%, чистота 97,59%). 1H ЯМР (400 МГц, CDCl3) δ 8,01 (d, J = 8,0 Гц, 2H), 7,70 (d, J = 8,3 Гц, 2H), 7,57 (s, 1H), 4,50 (d, J = 9,7 Гц, 1H), 4,29-4,20 (m, 3H), 4,16-4,09 (m, 1H), 4,00 (d, J = 9,7 Гц, 1H), 3,43 (d, J = 14,8 Гц, 1H), 3,13 (d, J = 14,7 Гц, 1H), 1,72 (s, 3H). LCMS (ESI) масса/заряд: 452,2 (M+1).
[942] Вариант осуществления 73
[943] (S)-2-Метил-6-нитро-2-((2-(4-(трифторметокси)фенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
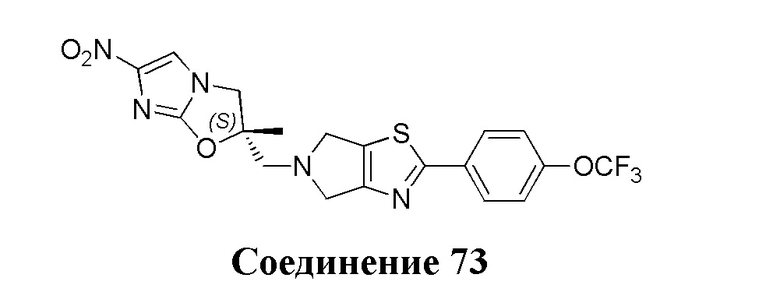
[944] Способ синтеза был таким, как в варианте осуществления 69.
[945] (S)-2-Метил-6-нитро-2-((2-(4-(трифторметокси)фенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол – соединение 73 (46,00 мг, 97,82 мкмоль, выход 16,43%, чистота 99,4%). 1H ЯМР (400 МГц, хлороформ-d)= 7,96-7,89 (m, 1H), 7,56 (s, 1H), 7,31-7,29 (m, 2H), 4,54-4,46 (m, 1H), 4,29-4,17 (m, 3H), 4,16-4,07 (m, 1H), 4,00 (d, J=9,7 Гц, 1H), 3,42 (d, J=14,7 Гц, 1H), 3,12 (d, J=14,7 Гц, 1H), 1,72 (s, 3H). LCMS (ESI) масса/заряд: 468,1 (M+1).
[946] Вариант осуществления 74
[947] (S)-2-((2-(2-Фторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
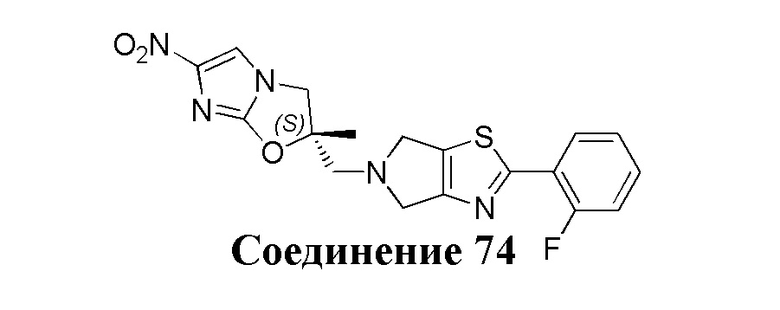
[948] Способ синтеза был таким, как в варианте осуществления 69.
[949] (S)-2-((2-(2-Фторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 74 (45,45 мг, 110,14 мкмоль, выход 34,45%, чистота 97,271%). 1H ЯМР (400 МГц, CDCl3) δ 8,23-8,16 (m, 1H), 7,56 (s, 1H), 7,44-7,36 (m, 1H), 7,28-7,17 (m, 2H), 4,51 (d, J = 8,0 Гц, 1H), 4,29-4,22 (m, 3H), 4,17-4,10 (m, 1H), 4,00 (d, J = 12,0 Гц, 1H), 3,43(d, J = 16,0 Гц, 1H), 3,13 (d, J = 16,0 Гц, 1H), 1,72 (s, 3H). LCMS (ESI) масса/заряд: 402 (M+1).
[950] Вариант осуществления 75
[951] (S)-2-((2-(4-Фторфенил)-5H-пирроло[3,4-d]тиазол-5-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
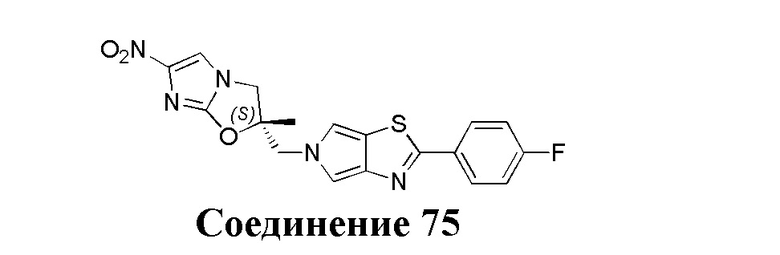
[952] Раствор (2S)-2-[[2-(4-фторфенил)-4,6-дигидропирроло[3,4-d]тиазол-5-ил]метил]-2-метил-6-нитро-3H-имидазо[2,1-b]оксазола (100,00 мг, 249,12 мкмоль, 1,00 экв.) в этилацетате (30,00 мл) перемешивали на воздухе при 80°C в течение 20 часов. Реакционную смесь концентрировали при пониженном давлении и остаток очищали посредством препаративной тонкослойной хроматографии (диоксид кремния, дихлорметан : метанол = 15: 1). Затем полученный в результате продукт промывали метанолом (5 мл) с получением (S)-2-((2-(4-фторфенил)-5H-пирроло[3,4-d]тиазол-5-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 75 (12,30 мг, 30,40 мкмоль, выход 12,20%, чистота 98,7%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,04 (s, 1H), 8,01-7,93 (m, 2H), 7,41-7,31 (m, 3H), 7,04 (d, J = 1,8 Гц, 1H), 4,69-4,54 (m, 2H), 4,29-4,16 (m, 2H), 1,56 (s, 3H). LCMS (ESI) масса/заряд: 400,2 (M+1).
[953] Вариант осуществления 76
[954] (S)-2-((2-(4-Хлорфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
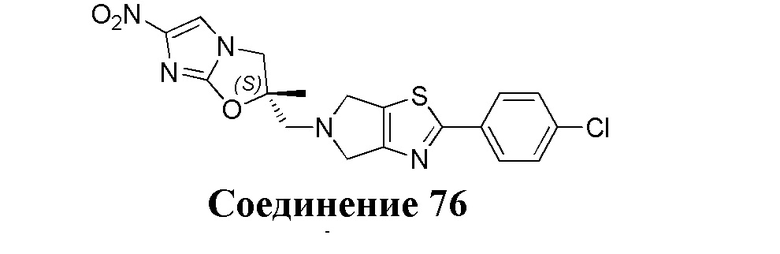
[955] Способ синтеза был таким, как в варианте осуществления 69.
[956] (S)-2-((2-(4-Хлорфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 76 (114,50 мг, 267,79 мкмоль, выход 60,83%, чистота 97,729%). 1H ЯМР (400 МГц, CDCl3) δ 7,83 (d, J =8,5Гц, 2H), 7,56 (s, 1H), 7,41 (d, J =8,5Гц, 2H), 4,50 (d, J =9,5Гц, 1H), 4,29-4,16 (m, 3H), 4,14-4,05 (m, 1H), 4,00 (d, J =9,8 Гц, 1H), 3,42 (d, J =14,8Гц, 1H), 3,11 (d, J =14,8Гц, 1H), 1,71 (s, 3H). LCMS (ESI) масса/заряд: 418,2 (M+1).
[957] Вариант осуществления 77
[958] (S)-2-((2-(2-Хлор-4-фторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
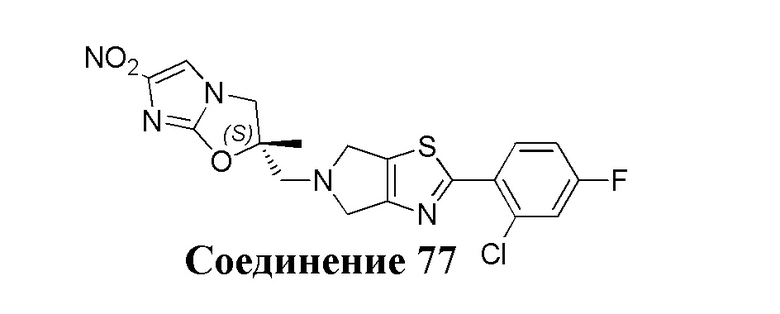
[959] Способ синтеза был таким, как в варианте осуществления 69.
[960] (S)-2-((2-(2-Хлор-4-фторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 77 (38,00 мг, 85,78 мкмоль, выход 27,01%, чистота 98,384%). 1H ЯМР (400 МГц, CDCl3) δ 8,13-8,06 (m, 1H), 7,56 (s, 1H), 7,27-7,23 (m, 1H), 7,13-7,07 (m, 1H), 4,50 (d, J = 8,0 Гц, 1H), 4,30-4,20 (m, 3H), 4,18-4,09 (m, 1H), 4,00 (d, J = 12,0 Гц, 1H), 3,42 (d, J = 12,0 Гц, 1H), 3,13 (d, J = 12,0 Гц, 1H), 1,72 (s, 3H). LCMS (ESI) масса/заряд: 436/438(M+1/M+3).
[961] Вариант осуществления 78
[962] (S)-2-((2-(2,4-Дифторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
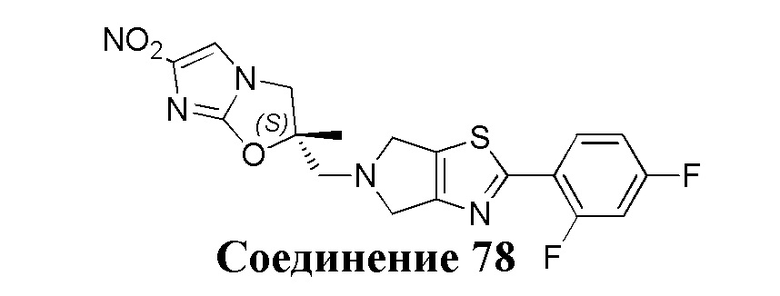
[963] Способ синтеза был таким, как в варианте осуществления 69.
[964] (S)-2-((2-(2,4-Дифторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 78 (117,40 мг, 271,83 мкмоль, выход 82,61%, чистота 97,107%). 1H ЯМР (400 МГц, CDCl3) δ 8,23-8,14 (m, 1H), 7,56 (s, 1H), 7,03-6,92 (m, 2H), 4,50 (d, J = 8,0 Гц, 1H), 4,28-4,21 (m, 3H), 4,16-4,08 (m, 1H), 4,00 (d, J = 12,0 Гц, 1H), 3,42 (d, J = 12,0 Гц, 1H), 3,12 (d, J = 12,0 Гц, 1H), 1,72 (s, 3H). LCMS (ESI) масса/заряд: 420 (M+1).
[965] Вариант осуществления 79
[966] (S)-2-((2-(4-Фтор-2-метилфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
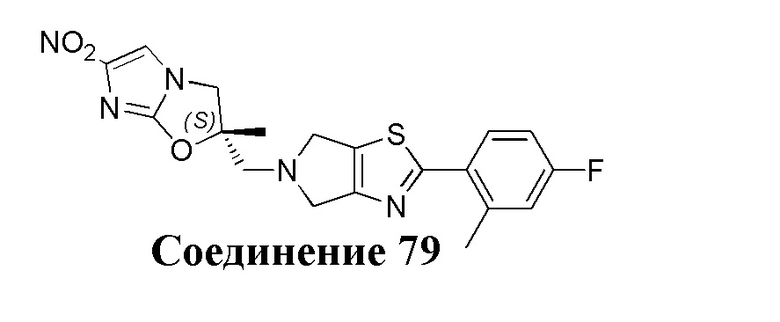
[967] Способ синтеза был таким, как в варианте осуществления 69.
[968] (S)-2-((2-(4-Фтор-2-метилфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 79 (332,10 мг, 786,28 мкмоль, выход 39,12%, чистота 98,36%). 1H ЯМР (400 МГц, CDCl3) δ 7,65-7,54 (m, 2H), 7,05-6,92 (m, 2H), 4,56-4,46 (m, 1H), 4,31-4,17 (m, 3H), 4,15-4,09 (m, 1H), 4,03-3,97 (m, 1H), 3,47-3,35 (m, 1H), 3,20-3,06 (m, 1H), 2,56 (s, 3H), 1,72 (s, 3H). LCMS (ESI) масса/заряд: 416,1 (M+1).
[969] Вариант осуществления 80
[970] (S)-2-((2-(4-Фтор-2-метоксифенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
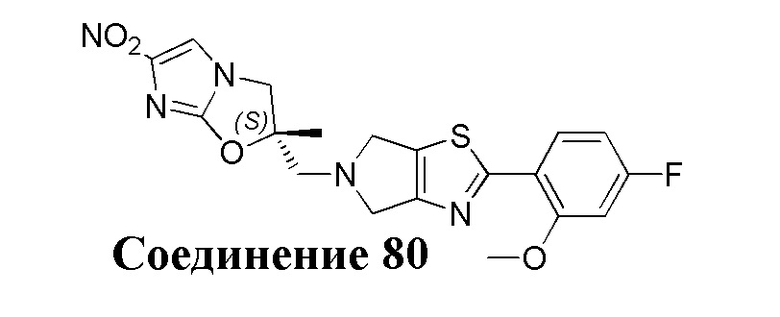
[971] Способ синтеза был таким, как в варианте осуществления 69.
[972] (S)-2-((2-(4-Фтор-2-метоксифенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 80 (47,90 мг, 105,87 мкмоль, выход 38,11%, чистота 95,36%). 1H ЯМР (400 МГц, хлороформ-d) δ = 8,21-8,12 (m, 1H), 7,47 (s, 1H), 6,73-6,61 (m, 2H), 4,43 (d, J = 9,7 Гц, 1H), 4,19-4,08 (m, 3H), 4,06-3,97 (m, 1H), 3,93 (s, 3H), 3,89 (d, J = 9,7 Гц, 1H), 3,32 (d, J = 14,7 Гц, 1H), 3,02 (d, J = 14,8 Гц, 1H), 1,62 (s, 3H). LCMS (ESI) масса/заряд: 432,1 (M+1).
[973] Вариант осуществления 81
[974] (S)-2-((2-(3,5-Дифторпиридин-2-ил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
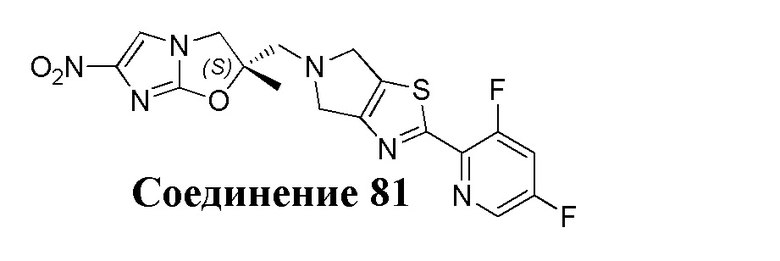
[975] Способ синтеза был таким, как в варианте осуществления 69.
[976] (S)-2-((2-(3,5-Дифторпиридин-2-ил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 81 (24,50 мг, 55,89 мкмоль, выход 36,48%, чистота 95,9%). 1H ЯМР (400 МГц, CDCl3) δ 8,30 (d, J = 2,1 Гц, 1H), 7,47 (s, 1H), 7,32-7,25 (m, 1H), 4,42 (d, J = 9,7 Гц, 1H), 4,25-4,13 (m, 3H), 4,10-4,03 (m, 1H), 3,90 (d, J = 9,8 Гц, 1H), 3,34 (d, J = 14,7 Гц, 1H), 3,05 (d, J = 14,7 Гц, 1H), 1,63 (s, 3H). LCMS (ESI) масса/заряд: 421,1 (M+1).
[977] Вариант осуществления 82
[978] (S)-2-((2-(5-Фторпиридин-2-ил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
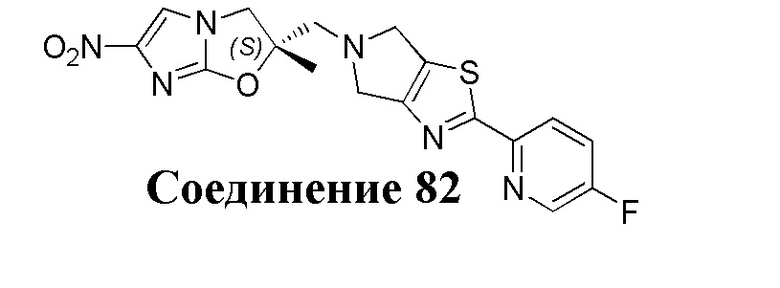
[979] Способ синтеза был таким, как в варианте осуществления 69.
[980] (S)-2-((2-(5-Фторпиридин-2-ил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол –соединение 82 (970,60 мг, 2,37 ммоль, выход 63,61%, чистота 98,1%). 1H ЯМР (400 МГц, CDCl3) δ 8,45 (d, J = 2,8 Гц, 1H), 8,11 (dd, J = 4,5, 8,7 Гц, 1H), 7,56 (s, 1H), 7,51 (dt, J = 2,8, 8,4 Гц, 1H), 4,51 (d, J = 9,5 Гц, 1H), 4,27-4,18 (m, 3H), 4,14-4,07 (m, 1H), 3,99 (d, J = 9,7 Гц, 1H), 3,42 (d, J = 14,7 Гц, 1H), 3,12 (d, J = 14,8 Гц, 1H), 1,72 (s, 3H). LCMS (ESI) масса/заряд: 403 (M+1).
[981] Вариант осуществления 83
[982] (S)-2-Метил-6-нитро-2-((2-(2,4,5-трифторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол
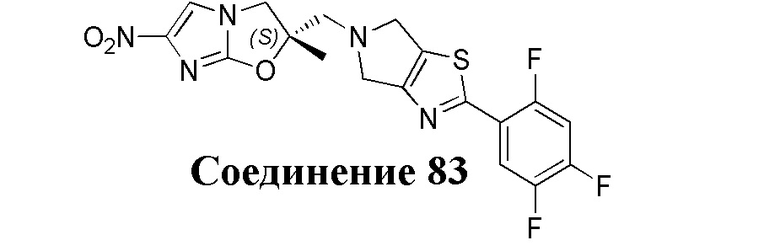
[983] Способ синтеза был таким, как в варианте осуществления 69.
[984] (S)-2-Метил-6-нитро-2-((2-(2,4,5-трифторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2,3-дигидроимидазо[2,1-b]оксазол – соединение 83 (50,80 мг, 114,98 мкмоль, выход 14,54%, чистота 99%). 1H ЯМР (400 МГц, CDCl3) δ 8,06 (ddd, J = 6,8, 8,8, 10,9 Гц, 1H), 7,61-7,49 (m, 1H), 7,12-7,01 (m, 1H), 4,50 (d, J = 9,7 Гц, 1H), 4,29-4,18 (m, 3H), 4,16-4,07 (m, 1H), 4,01 (d, J = 9,7 Гц, 1H), 3,42 (d, J = 14,7 Гц, 1H), 3,13 (d, J = 14,8 Гц, 1H), 1,72 (s, 3H). LCMS (ESI) масса/заряд: 438,0 (M+1).
[985] Вариант осуществления 84
[986] (S)-2-((2-(3,5-Дифторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
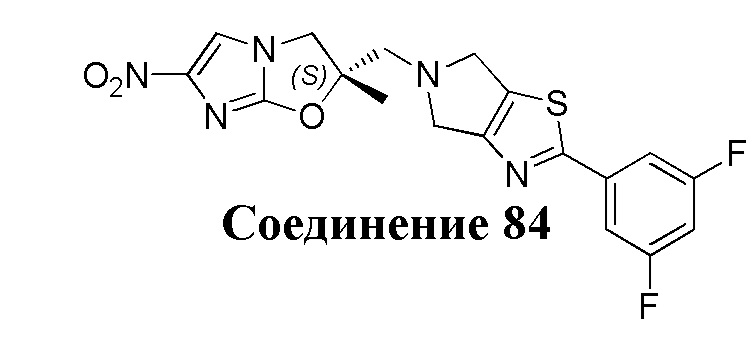
[987] Способ синтеза был таким, как в варианте осуществления 69.
[988] (S)-2-((2-(3,5-Дифторфенил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 84 (48,70 мг, 115,57 мкмоль, выход 27,67%, чистота 99,53%). 1H ЯМР (400 МГц, CDCl3) δ 7,56 (s, 1H), 7,47-7,39 (m, 2H), 6,87 (tt, J = 2,3, 8,7 Гц, 1H), 4,49 (d, J = 9,5 Гц, 1H), 4,28-4,19 (m, 3H), 4,15-4,08 (m, 1H), 4,00 (d, J = 9,7 Гц, 1H), 3,42 (d, J = 14,8 Гц, 1H), 3,12 (d, J = 14,7 Гц, 1H), 1,72 (s, 3H). LCMS (ESI) масса/заряд: 420,1 (M+1)
[989] Вариант осуществления 85
[990] (S)-N-(4-Фторфенил)-N-метил-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6-дигидро-4H-пирроло[3,4-d]тиазол-2-амин
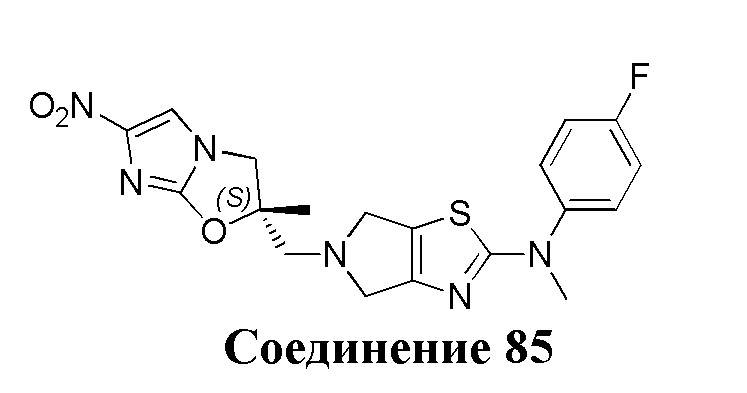
[991] Способ синтеза был таким, как в варианте осуществления 69.
[992] (S)-N-(4-Фторфенил)-N-метил-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6-дигидро-4H-пирроло[3,4-d]тиазол-2-амин – соединение 85 (27,10 мг, 62,20 мкмоль, выход 14,52%, чистота 98,8%). 1H ЯМР (400 МГц, CDCl3) δ 7,54 (s, 1H), 7,36-7,30 (m, 2H), 7,17-7,08 (m, 2H), 4,48 (d, J = 9,7 Гц, 1H), 4,09-3,87 (m, 5H), 3,46 (s, 3H), 3,35 (d, J = 14,7 Гц, 1H), 3,04 (d, J = 14,8 Гц, 1H), 1,67 (s, 3H). LCMS (ESI) масса/заряд: 431 (M+1).
[993] Вариант осуществления 86
[994] (S)-2-((2-(4,4-Дифторпиперидин-1-ил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол

[995] Способ синтеза был таким, как в варианте осуществления 69.
[996] (S)-2-((2-(4,4-Дифторпиперидин-1-ил)-4H-пирроло[3,4-d]тиазол-5(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 86 (9,30 мг, 20,98 мкмоль, выход 13,31%, чистота 96,2%). 1H ЯМР (400 МГц, CDCl3) δ 7,54 (s, 1H), 4,48 (d, J = 9,7 Гц, 1H), 4,08-4,00 (m, 3H), 3,98-3,85 (m, 2H), 3,66-3,58 (m, 4H), 3,35 (d, J = 14,7 Гц, 1H), 3,04 (d, J = 14,8 Гц, 1H), 2,15-2,02 (m, 4H), 1,68 (s, 3H). LCMS (ESI) масса/заряд: 427,0 (M+1).
[997] Вариант осуществления 87
[998] (S)-2-((2-(4-Фторфенил)-5H-пирроло[3,4-d]пиримидин-6(7H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
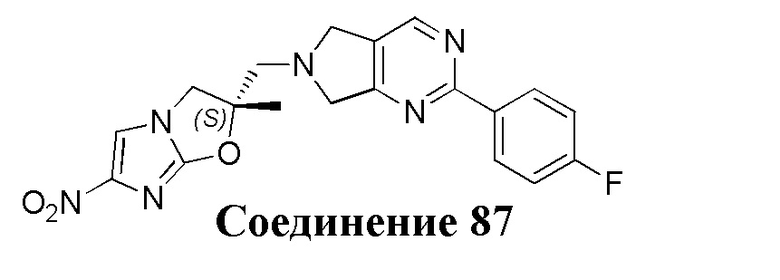
[999] Способ синтеза был таким, как в варианте осуществления 44.
[1000] (S)-2-((2-(4-Фторфенил)-5H-пирроло[3,4-d]пиримидин-6(7H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 87 (14,60 мг, 35,57 мкмоль, выход 13,21%, чистота 96,567%). 1H ЯМР (400 МГц, CDCl3) δ 8,58 (s, 1H), 8,47-8,36 (m, 2H), 7,55 (s, 1H), 7,19-7,11 (m, 2H), 4,46 (d, J = 8,0 Гц, 1H), 4,28-4,11 (m, 4H), 4,00 (d, J = 8,0 Гц, 1H), 3,33 (d, J = 12,0 Гц, 1H), 3,11 (d, J = 12,0 Гц, 1H), 1,73 (s, 3H). LCMS (ESI) масса/заряд: 397 (M+1).
[1001] Вариант осуществления 88
[1002] (S)-2-((2-(4-Фторфенил)-5H-пирроло[3,4-b]пиридин-6(7H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол

[1003] Стадия 1.
[1004] Диметилпиридин-2,3-дикарбоксилат
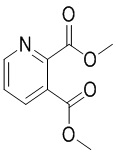
[1005] Концентрированную серную кислоту (17,61 г, 179,51 ммоль, 9,57 мл, 1,50 экв.) добавляли к раствору пиридин-2,3-дикарбоновой кислоты (20,00 г, 119,67 ммоль, 1,00 экв.) в метаноле (150,00 мл). Смесь перемешивали при 60°C в течение 10 часов. Добавляли раствор карбоната натрия, чтобы довести pH до приблизительно 9, и смесь экстрагировали этилацетатом (200 мл × 2). Объединенные органические слои концентрировали при пониженном давлении и остаток хроматографировали на силикагеле (SiO2, петролейный эфир/этилацетат = 20/1 – 3: 1) с получением диметилпиридин-2,3-дикарбоксилата (20,00 г, 102,47 ммоль, выход 85,63%) в виде белого твердого вещества.
[1006] Стадия 2.
[1007] Диметилпиридин-1-оксид-1-оний-2,3-дикарбоксилат
[1008] m-CPBA (28,74 г, 133,21 ммоль, чистота 80%, 1,30 экв.) добавляли к раствору диметилпиридин-2,3-дикарбоксилата (20,00 г, 102,47 ммоль, 1,00 экв.) в хлороформе (160,00 мл). Смесь перемешивали при 60°C в течение 4 часов. Водный раствор бикарбоната натрия (200 мл) добавляли к реакционной смеси и смесь экстрагировали дихлорметаном (500 мл × 4). Объединенные органические слои концентрировали при пониженном давлении, промывали этилацетатом (50 мл) и собирали отфильтрованный осадок путем фильтрования с получением диметилпиридин-1-оксид-1-оний-2,3-дикарбоксилата (17,50 г, неочищенный) в виде белого твердого вещества, которое использовали непосредственно на следующей стадии.
[1009] Стадия 3.
[1010] Диметил-6-хлорпиридин-2,3-дикарбоксилат
[1011] Оксихлорид фосфора (50,83 г, 331,50 ммоль, 30,81 мл, 5,00 экв.) добавляли к раствору диметилпиридин-1-оксид-1-оний-2,3-дикарбоксилата (14,00 г, 66,30 ммоль, 1,00 экв.) в диоксане (140,00 мл). Смесь перемешивали при 100°C в течение 2 часов. Реакционную смесь выливали в воду (100 мл), доводили pH до приблизительно 9 при помощи раствора карбоната натрия и экстрагировали этилацетатом (500 мл × 2). Объединенные органические слои концентрировали при пониженном давлении и остаток очищали посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 40/1 – 10: 1) с получением диметил-6-хлорпиридин-2,3-дикарбоксилата (9,50 г, 41,37 ммоль, выход 62,40%) в виде желтого масла.
[1012] Стадия 4.
[1013] Диметил-6-(4-фторфенил)пиридин-2,3-дикарбоксилат
[1014] Диметил-6-хлорпиридин-2,3-дикарбоксилат (5,00 г, 21,78 ммоль, 1,00 экв.), (4-фторфенил)бороновую кислоту (3,96 г, 28,31 ммоль, 1,30 экв.), Pd(dppf)Cl2 (796,65 мг, 1,09 ммоль, 0,05 экв.), карбонат натрия (4,62 г, 43,56 ммоль, 2,00 экв.) растворяли в диоксане (30,00 мл) и H2O (400,00 мкл), раствор дегазировали, производили замену на азот и перемешивали смесь при 80°C в течение 10 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и остаток разделяли посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 30/1 – 10: 1) с получением диметил-6-(4-фторфенил)пиридин-2,3-дикарбоксилата (5,00 г, 17,29 ммоль, выход 79,36%) в виде белого твердого вещества. LCMS (ESI) масса/заряд: 290,2 (M + 1).
[1015] Стадия 5.
[1016] [6-(4-Фторфенил)-2-(гидроксиметил)-3-пиридил]метанол
[1017] Хлорид кальция (2,30 г, 20,75 ммоль, 1,20 экв.) и боргидрид натрия (6,54 г, 172,90 ммоль, 10,00 экв.) добавляли к раствору диметил-6-(4-фторфенил)пиридин-2,3-дикарбоксилата (5,00 г, 17,29 ммоль, 1,00 экв.) в метаноле (100,00 мл). Смесь перемешивали при 15°C в течение 3 часов. Реакционную смесь экстрагировали этилацетатом (1 л × 2). Объединенные органические слои концентрировали при пониженном давлении и остаток разделяли и очищали посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 20/1 – 3: 1) с получением [6-(4-фторфенил)-2-(гидроксиметил)-3-пиридил]метанола (3,50 г, 15,01 ммоль, выход 86,79%) в виде белого твердого вещества.
[1018] Стадия 6.
[1019] 2,3-Бис(хлорметил)-6-(4-фторфенил)пиридин

[1020] Тионилхлорид (19,68 г, 165,42 ммоль, 12,00 мл, 25,73 экв.) добавляли к раствору [6-(4-фторфенил)-2-(гидроксиметил)-3-пиридил]метанола (1,50 г, 6,43 ммоль, 1,00 экв.) в дихлорметане (20,00 мл). Смесь перемешивали при 0°C в течение 0,5 часа. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя с получением 2,3-бис(хлорметил)-6-(4-фторфенил)пиридина (2,00 г, неочищенный) в виде желтого масла. LCMS (ESI) масса/заряд: 270,1 (M + 1).
[1021] Стадия 7.
[1022] 2-(4-Фторфенил)-6-тритил-5,7-дигидропирроло[3,4-b]пиридин
[1023] Диизопропиламин (2,87 г, 22,20 ммоль, 3,88 мл, 3,00 экв.) и тритиламин (2,88 г, 11,10 ммоль, 1,50 экв.) добавляли к раствору 2,3-бис(хлорметил)-6-(4-фторфенил)пиридина (2,00 г, 7,40 ммоль, 1,00 экв.) в DMF (60,00 мл). Смесь перемешивали при 80°C в течение 20 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя. Остаток экстрагировали этилацетатом (200 мл × 2). Объединенные органические слои концентрировали при пониженном давлении и остаток очищали посредством хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 1/0 – 50: 1) с получением 2-(4-фторфенил)-6-тритил-5,7-дигидропирроло[3,4-b]пиридина (1,50 г, неочищенный) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,80 (dd, J =5,4, 8,8 Гц, 2H), 7,53 (d, J =7,5 Гц, 6H), 7,33 (s, 2H), 7,26-7,18 (m, 8H), 7,11 (d, J =7,3 Гц, 4H), 7,03 (s, 2H), 4,04-3,90 (m, 4H).
[1024] Стадия 8.
[1025] 2-(4-Фторфенил)-6,7-дигидро-5H-пирроло[3,4-b]пиридин; 2,2,2-трифторуксусная кислота
[1026] Трифторуксусную кислоту (10,78 г, 94,54 ммоль, 7,00 мл, 28,74 экв.) добавляли к раствору 2-(4-фторфенил)-6-тритил-5,7-дигидропирроло[3,4-b]пиридина (1,50 г, 3,29 ммоль, 1,00 экв.) в метаноле (7,00 мл) и хлороформе (7,00 мл). Смесь перемешивали при 0°C в течение 0,5 часа и реакционную смесь промывали водой (150 мл × 2). Объединенные водные фазы концентрировали при пониженном давлении с получением 2-(4-фторфенил)-6,7-дигидро-5H-пирроло[3,4-b]пиридина, 2,2,2-трифторуксусной кислоты (1,00 г, неочищенный) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 215,2 (M + 1).
[1027] Стадия 9.
[1028] (2S)-1-(2-Бром-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-5,7-дигидропирроло[3,4-b]пиридин-6-ил)-2-метилпропан-2-ол
[1029] Диизопропиламин (1,18 г, 9,14 ммоль, 1,60 мл, 3,00 экв.) и 2-бром-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазол (1,20 г, 4,58 ммоль, 1,50 экв.) добавляли к раствору 2-(4-фторфенил)-6,7-дигидро-5H-пирроло[3,4-b]пиридина, 2,2,2-трифторуксусной кислоты (1,00 г, 3,05 ммоль, 1,00 экв.) в трет-бутаноле (50,00 мл). Смесь перемешивали при 40°C в течение 10 часов. Реакционную смесь концентрировали при пониженном давлении для удаления растворителя и очищали остаток посредством колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 10/1 – 3: 1) с получением (2S)-1-(2-бром-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-5,7-дигидропирроло[3,4-b]пиридин-6-ил)-2-метилпропан-2-ола (700,00 мг, неочищенный) в виде коричневого масла. LCMS (ESI) масса/заряд: 476,0 (M + 1).
[1030] Стадия 10.
[1031] (S)-2-((2-(4-Фторфенил)-5H-пирроло[3,4-b]пиридин-6(7H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
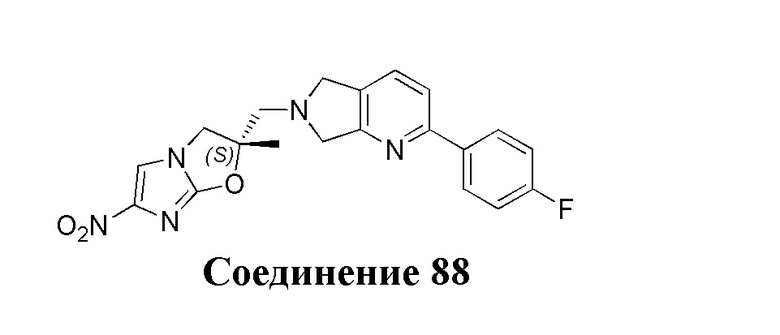
[1032] Гидрид натрия (70,54 мг, 2,94 ммоль, 2,00 экв.) добавляли к раствору (2S)-1-(2-бром-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-5,7-дигидропирроло[3,4-b]пиридин-6-ил)-2-метилпропан-2-ола (700,00 мг, 1,47 ммоль, 1,00 экв.) в DMF (4,00 мл). Смесь перемешивали при 0°C в течение 10 минут. Реакционную смесь добавляли к насыщенному водному раствору хлорида аммония (200 мл) при 0°C. Отфильтрованный осадок фильтровали и сушили. Отфильтрованный осадок очищали посредством препаративной тонкослойной хроматографии (диоксид кремния, DCM : метанол = 15: 1) с получением (S)-2-((2-(4-фторфенил)-5H-пирроло[3,4-b]пиридин-6(7H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазола – соединения 88 (243,60 мг, 600,70 мкмоль, выход 40,86%, чистота 97,5%). 1H ЯМР (400 МГц, CDCl3) δ 7,97-7,89 (m, 2H), 7,55 (s, 3H), 7,21-7,11 (m, 2H), 4,58-4,46 (m, 1H), 4,34-4,08 (m, 4H), 4,04-3,93 (m, 1H), 3,41-3,25 (m, 1H), 3,15-3,04 (m, 1H), 1,73 (s, 3H). LCMS (ESI) масса/заряд: 396,2 (M+1).
[1033] Вариант осуществления 89
[1034] (S)-2-(4-Фторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6-дигидро-4H-пирроло[3,4-d]оксазол
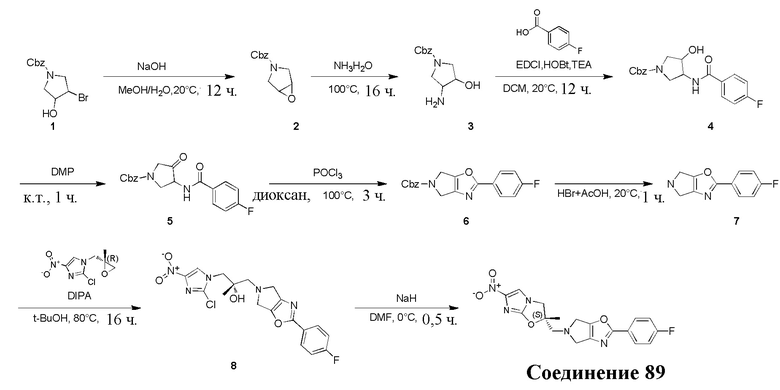
[1035] Стадия 1.
[1036] Бензил-6-окса-3-азабицикло[3,1,0]гексан-3-карбоксилат
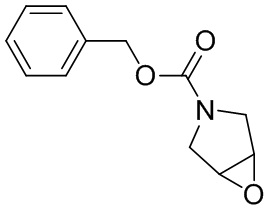
[1037] Гидроксид натрия (1,50 г, 37,49 ммоль, 1,50 экв.) добавляли к смешанному раствору бензил-3-бром-4-гидроксипирролидин-1-карбоксилата (7,50 г, 24,99 ммоль, 1,00 экв.) в метаноле (50,00 мл) и воде (50,00 мл). Смесь перемешивали при 20°C в течение 12 часов. Добавляли к смеси воду (200 мл), а затем экстрагировали этилацетатом (200 мл × 3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением бензил-6-окса-3-азабицикло[3,1,0]гексан-3-карбоксилата (350,00 мг, 1,60 ммоль, выход 95,81%) в виде слегка желтоватого масла. 1H ЯМР (300 МГц, CDCl3) δ 7,36-7,31 (m, 5H), 5,14-5,12 (m, 2H), 3,88 (t, J = 12,90 Гц, 1H), 3,68-3,63 (m, 2H), 3,43-3,37 (m, 3H).
[1038] Стадия 2.
[1039] Бензил-3-амино-4-гидроксипирролидин-1-карбоксилат
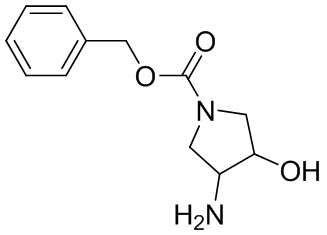
[1040] Смешивали бензил-6-окса-3-азабицикло[3,1,0]гексан-3-карбоксилат (5,40 г, 24,63 ммоль, 1,00 экв.) и аммиак (43,17 г, 1,23 моль, 47,43 мл, 50,00 экв.), затем смесь перемешивали при 100°C в течение 16 часов. Смесь концентрировали при пониженном давлении с получением бензил-3-амино-4-гидрокси-пирролидин-1-карбоксилата (5,00 г, 21,16 ммоль, выход 85,92%) в виде слегка желтоватого масла.
[1041] Стадия 3.
[1042] Бензил-3-(4-фторбензамидо)-4-гидроксипирролидин-1-карбоксилат
[1043] HOBt (2,57 г, 19,05 ммоль, 1,00 экв.), EDCI (7,30 г, 38,10 ммоль, 2,00 экв.) и триэтиламин (5,78 г, 57,15 ммоль, 7,92 мл, 3,00 экв.) добавляли к раствору бензил-3-амино-4-гидрокси-пирролидин-1-карбоксилата (4,50 г, 19,05 ммоль, 1,00 экв.) и 4-фторбензойной кислоты (2,67 г, 19,05 ммоль, 1,00 экв.) в дихлорметане (50,00 мл). Смесь перемешивали при 20°C в течение 12 часов. Добавляли к реакционной смеси воду (200 мл) и экстрагировали дихлорметаном (150 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (высота колонки: 300 мм, диаметр: 50 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 5/1, 2/1, 0/1) с получением бензил-3-(4-фторбензамидо)-4-гидроксипирролидин-1-карбоксилата (2,78 г, 7,76 ммоль, выход 40,73%) и бензил-3-[(4-фторбензоил)амино]-4-(4-фторбензоил)оксипирролидин-1-карбоксилата (2,20 г, 4,58 ммоль, выход 24,04%) в виде светло-желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 8,52 (d, J = 6,40 Гц, 1H), 7,92 (dd, J = 6,90, 5,77 Гц, 2H), 7,38-7,36 (m, 5H), 7,30 (d, J = 8,78 Гц, 2H), 5,08-5,06 (m, 2H), 4,23-4,11 (m, 2H), 3,65-3,57 (m, 2H), 3,29-3,16 (m, 2H).
[1044] Стадия 4.
[1045] Бензил-3-(4-фторбензамидо)-4-оксопирролидин-1-карбоксилат
[1046] Перйодинан Десс-Мартина (9,94 г, 23,44 ммоль, 7,26 мл, 2,00 экв.) добавляли к раствору 3-(4-фторбензамидо)-4-гидроксипирролидин-1-карбоксилата (4,20 г, 11,72 ммоль, 1,00 экв.) в DCM (3,00 мл) при 0°C. Смесь перемешивали при 20°C в течение 2 часов. Добавляли к реакционной смеси раствор сульфита натрия (100 мл) и раствор бикарбоната натрия (100 мл), полученную в результате смесь экстрагировали дихлорметаном (200 мл × 3) и объединенные органические слои промывали насыщенным солевым раствором (200 мл * 1), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением бензил-3-(4-фторбензамидо)-4-оксопирролидин-1-карбоксилата (4,00 г, 11,22 ммоль, выход 95,78%) в виде слегка желтоватого масла. 1H ЯМР (400 МГц, DMSO) δ 9,15 (d, J = 6,52 Гц, 1H), 7,92 (dd, J = 8,47, 5,58 Гц, 2H), 7,41-7,33 (m, 7H), 5,15 (s, 2H), 4,51 (d, J = 8,91 Гц, 1H), 4,09-3,77 (m, 3H), 3,40 (s, 1H).
[1047] Стадия 5.
[1048] Бензил-2-(4-фторфенил)-4,6-дигидропирроло[3,4-d]оксазол-5-карбоксилат
[1049] Оксихлорид фосфора (9,62 г, 62,74 ммоль, 5,83 мл, 5,59 экв.) добавляли к раствору бензил-3-(4-фторбензамидо)-4-оксопирролидин-1-карбоксилата (4,00 г, 11,22 ммоль, 1,00 экв.) в диоксане (5,00 мл). Смесь перемешивали при 100°C в течение 4 часов. После охлаждения смеси ее медленно добавляли к H2O (200 мл), фильтровали и отфильтрованный осадок промывали дихлорметаном (50 мл), собирали отфильтрованный осадок с получением бензил-2-(4-фторфенил)-4,6-дигидропирроло[3,4-d]оксазол-5-карбоксилата (1,50 г, 4,43 ммоль, выход 39,48%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 8,03 (dd, J = 7,40, 5,40 Гц, 2H), 7,44-7,36 (m, 7H), 5,18 (s, 2H), 4,68-4,60 (m, 2H), 4,50-4,41 (m, 2H).
[1050] Стадия 6.
[1051] 2-(4-Фторфенил)-5,6-дигидро-4H-пирроло[3,4-d]оксазол
[1052] Бензил-2-(4-фторфенил)-4,6-дигидропирроло[3,4-d]оксазол-5-карбоксилат (1,50 г, 4,43 ммоль, 1,00 экв.) растворяли в растворе бромистого водорода в уксусной кислоте (7,17 г, 88,60 ммоль, 4,81 мл, 20,00 экв.) и перемешивали при 20°C в атмосфере азота в течение 1 часа. Смесь концентрировали при пониженном давлении, добавляли дихлорметан (20 мл) и этилацетат (20 мл), фильтровали и отфильтрованный осадок сушили in vacuo с получением 2-(4-фторфенил)-5,6-дигидро-4H-пирроло[3,4-d]оксазола (1,00 г, 3,51 ммоль, выход 79,17%, гидробромид) в виде черного твердого вещества. 1H ЯМР (300 МГц, DMSO) δ 8,07-7,89 (m, 2H), 7,48-7,27 (m, 2H), 4,58-4,23 (m, 4H).
[1053] Стадия 7.
[1054] (2S)-1-(2-Хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-4,6-дигидропирроло[3,4-d]оксазол-5-ил)-2-метилпропан-2-ол
[1055] Диизопропиламин (906,60 мг, 7,01 ммоль, 1,23 мл, 2,00 экв.) добавляли к раствору 2-(4-фторфенил)-5,6-дигидро-4H-пирроло[3,4-d]оксазола (1,00 г, 3,51 ммоль, 1,00 экв., гидробромид) и 2-хлор-1-[[(2R)-2-метилоксиран-2-ил]метил]-4-нитроимидазола (916,57 мг, 4,21 ммоль, 1,20 экв.) в трет-бутаноле (20,00 мл). Смесь перемешивали при 80°C в течение 12 часов. Добавляли к реакционной смеси воду (50 мл), а затем экстрагировали этилацетатом (100 мл × 3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали посредством хроматографии на силикагеле (высота колонки: 300 мм, диаметр: 50 мм, силикагель от 100 до 200 меш, петролейный эфир/этилацетат = 5/1, 3/1, 1/1) с получением (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-4,6-дигидропирроло[3,4-d]оксазол-5-ил)-2-метилпропан-2-ола (700,00 мг, 1,66 ммоль, выход 47,28%) в виде желтого твердого вещества. LCMS (ESI) масса/заряд: 422,1 (M + 1).
[1056] Стадия 8.
[1057] (S)-2-(4-Фторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6-дигидро-4H-пирроло[3,4-d]оксазол
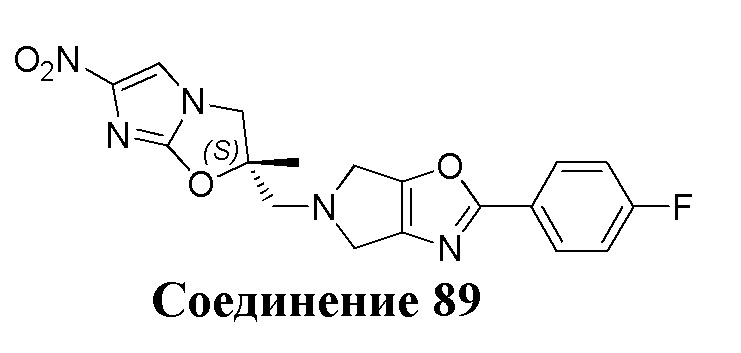
[1058] Гидрид натрия (132,76 мг, 3,32 ммоль, чистота 60%, 2,00 экв.) добавляли к раствору (2S)-1-(2-хлор-4-нитроимидазол-1-ил)-3-(2-(4-фторфенил)-4,6-дигидропирроло[3,4-d]оксазол-5-ил)-2-метилпропан-2-ола (700,00 мг, 1,66 ммоль, 1,00 экв.) в DMF (5,00 мл) при -45°C. Смесь перемешивали при -45°C в течение 30 минут, а затем перемешивали при 0°C в течение 30 минут. Реакционную смесь добавляли к насыщенному раствору хлорида аммония (150 мл), смесь фильтровали и отфильтрованный осадок очищали посредством препаративной разделительной хроматографии (GX-A; Phenomenex Gemini 150*25 мм*10 мкм; ацетонитрил 40% - 70%; вода (0,05% гидроксид аммония, об./об.); 25 мл/мин.) с получением (S)-2-(4-фторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6-дигидро-4H-пирроло[3,4-d]оксазола – соединения 89 (46,00 мг, 117,70 мкмоль, выход 7,09%, чистота 98,6%). 1H ЯМР (400 МГц, CDCl3) δ 7,98 (dd, J = 8,85, 5,33 Гц, 2H), 7,56 (s, 1H), 7,15 (t, J = 8,66 Гц, 2H), 4,46 (d, J = 9,66 Гц, 1H), 4,11-3,89 (m, 5H), 3,42 (d, J = 14,93 Гц, 1H), 3,10 (d, J = 14,81 Гц, 1H), 1,70 (s, 3H). LCMS (ESI) масса/заряд: 386,1 (M+1).
[1059] Вариант осуществления 90
[1060] (S)-2-(3,4-Дифторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6-дигидро-4H-пирроло[3,4-d]оксазол
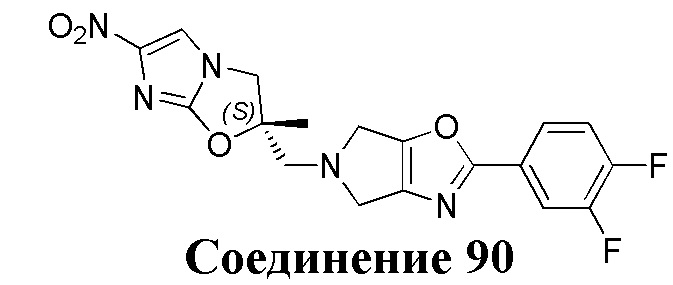
[1061] Способ синтеза был таким, как в варианте осуществления 89.
[1062] (S)-2-(3,4-Дифторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6-дигидро-4H-пирроло[3,4-d]оксазол – соединение 90 (24,80 мг, 59,27 мкмоль, выход 35,88%, чистота 96,4%). 1H ЯМР (400 МГц, CDCl3) δ 7,84-7,77 (m, 1H), 7,77-7,71 (m, 1H), 7,56 (s, 1H), 7,27-7,21 (m, 1H), 4,45 (d, J = 9,66 Гц, 1H), 4,10-3,89 (m, 5H), 3,42 (d, J = 14,93 Гц, 1H), 3,10 (d, J = 14,93 Гц, 1H), 1,70 (s, 3H).
[1063] Вариант осуществления 91
[1064] (S)-2-(2,4-Дифторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6-дигидро-4H-пирроло[3,4-d]оксазол
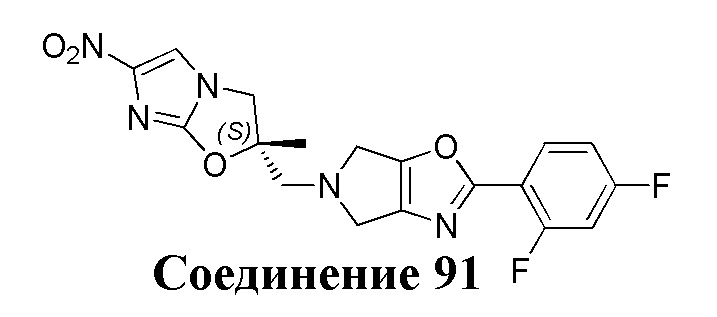
[1065] Способ синтеза был таким, как в варианте осуществления 89.
[1066] (S)-2-(2,4-Дифторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6-дигидро-4H-пирроло[3,4-d]оксазол – соединение 91 (31,30 мг, 75,82 мкмоль, выход 24,48%, чистота 97,7%). 1H ЯМР (400 МГц, CDCl3) δ 8,03-7,91 (m, 1H), 7,56 (s, 1H), 6,98 (d, J = 8,2 Гц, 2H), 4,49-4,42 (m, 1H), 4,16-3,89 (m, 5H), 3,48-3,37 (m, 1H), 3,17-3,03 (m, 1H), 1,70 (s, 3H). LCMS (ESI) масса/заряд: 404,1 (M+1).
[1067] Вариант осуществления 92
[1068] (S)-2-(3,5-Дифторпиридин-2-ил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6-дигидро-4H-пирроло[3,4-d]оксазол
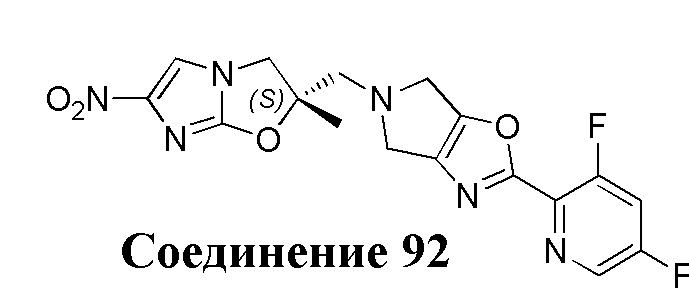
[1069] Способ синтеза был таким, как в варианте осуществления 89.
[1070] (S)-2-(3,5-Дифторпиридин-2-ил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6-дигидро-4H-пирроло[3,4-d]оксазол – соединение 92 (125,10 мг, 305,38 мкмоль, выход 51,10%, чистота 98,7%). 1H ЯМР (400 МГц, CDCl3) δ 8,48 (d, J = 2,1 Гц, 1H), 7,56 (s, 1H), 7,41 (ddd, J = 2,3, 7,8, 10,0 Гц, 1H), 4,43 (d, J = 9,7 Гц, 1H), 4,20-4,06 (m, 3H), 4,05-3,92 (m, 2H), 3,43 (d, J = 14,8 Гц, 1H), 3,13 (d, J = 14,8 Гц, 1H), 1,71 (s, 3H). LCMS (ESI) масса/заряд: 405,0 (M+1).
[1071] Вариант осуществления 93
[1072] (S)-2-(5-Фторпиридин-2-ил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6-дигидро-4H-пирроло[3,4-d]оксазол
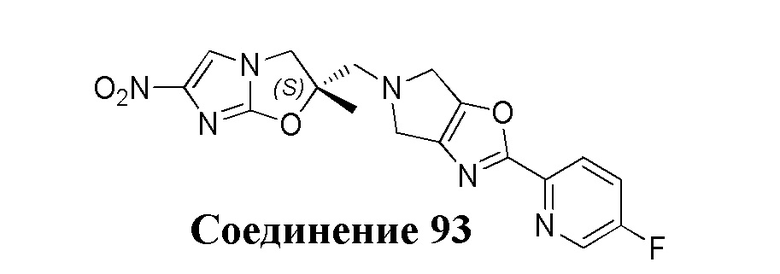
[1073] Способ синтеза был таким, как в варианте осуществления 89.
[1074] (S)-2-(5-Фторпиридин-2-ил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6-дигидро-4H-пирроло[3,4-d]оксазол – соединение 93 (175,46 мг, 445,96 мкмоль, выход 52,09%, чистота 98,194%). 1H ЯМР (400 МГц, CDCl3) δ 8,56 (d, J = 0,4 Гц, 1H), 8,13-8,07 (m, 1H), 7,57-7,51 (m, 2H), 4,43 (d, J = 12,0 Гц, 1H), 4,19-4,02 (m, 3H), 4,01-3,92 (m, 2H), 3,42 (d, J = 16,0 Гц, 1H), 3,11 (d, J = 16,0 Гц, 1H), 1,70 (s, 3H). LCMS (ESI) масса/заряд: 387 (M+1).
[1075] Вариант осуществления 94
[1076] (S)-5-((2-Метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-2-(2,4,5-трифторфенил)-5,6-дигидро-4H-пирроло[3,4-d]оксазол
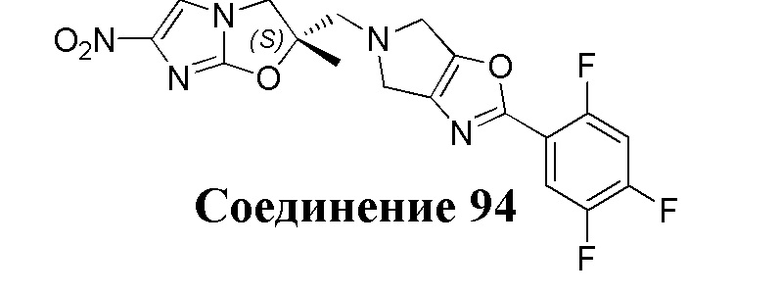
[1077] Способ синтеза был таким, как в варианте осуществления 89.
[1078] (S)-5-((2-Метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-2-(2,4,5-трифторфенил)-5,6-дигидро-4H-пирроло[3,4-d]оксазол – соединение 94 (491,75 мг, 1,14 ммоль, выход 57,13%, чистота 97,416%). 1H ЯМР (400 МГц, CDCl3) δ 7,89-7,74 (m, 1H), 7,57 (s, 1H), 7,15-7,02 (m, 1H), 4,50-4,39 (m, 1H), 4,14-3,90 (m, 5H), 3,48-3,38 (m, 1H), 3,16-3,07 (m, 1H), 1,71 (s, 3H). LCMS (ESI) масса/заряд: 422 (M+1).
[1079] Вариант осуществления 95
[1080] (S)-2-(3,5-Дифторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6-дигидро-4H-пирроло[3,4-d]оксазол
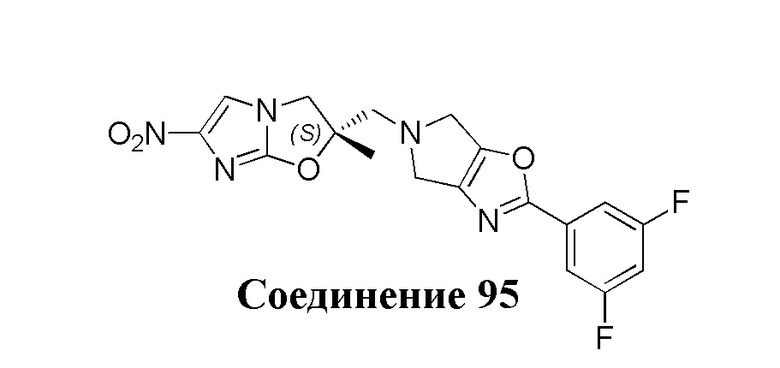
[1081] Способ синтеза был таким, как в варианте осуществления 89.
[1082] (S)-2-(3,5-Дифторфенил)-5-((2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол-2-ил)метил)-5,6-дигидро-4H-пирроло[3,4-d]оксазол –соединение 95 (21,60 мг, 53,29 мкмоль, выход 10,32%, чистота 99,5%). 1H ЯМР (400 МГц, CDCl3) δ 7,56 (s, 1H), 7,54-7,48 (m, 2H), 6,95-6,83 (m, 1H), 4,43 (s, 1H), 4,15-3,88 (m, 5H), 3,41 (s, 1H), 3,10 (d, J =14,8 Гц, 1H), 1,71 (s, 3H). LCMS (ESI) масса/заряд: 404,2 (M+1).
[1083] Вариант осуществления 96
[1084] (S)-2-((2-(4-Фторфенил)-8,9-дигидро-5H-пиридо[3,2-c]азепин-6(7H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
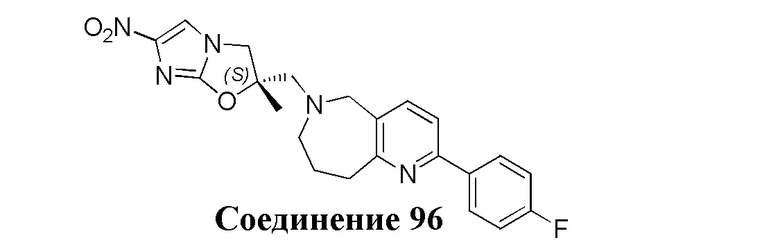
[1085] Способ синтеза был таким, как в варианте осуществления 7.
[1086] (S)-2-((2-(4-Фторфенил)-8,9-дигидро-5H-пиридо[3,2-c]азепин-6(7H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол –соединение 96 (85,50 мг, 196,26 мкмоль, выход 36,10%, чистота 97,2%). 1H ЯМР (400 МГц, CDCl3) δ 8,08-7,93 (m, 2H), 7,54-7,40 (m, 3H), 7,16 (t, J = 8,7 Гц, 2H), 4,36 (d, J = 9,5 Гц, 1H), 4,03-3,84 (m, 3H), 3,31-3,15 (m, 4H), 2,96 (d, J = 15,3 Гц, 1H), 2,54 (d, J = 15,3 Гц, 1H), 1,80 (br, s, 2H), 1,59 (s, 3H). LCMS (ESI) масса/заряд: 424 (M+1).
[1087] Вариант осуществления 97
[1088] (S)-2-((2-(4-Фторфенил)-8,9-дигидро-5H-пиридо[2,3-d]азепин-7(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
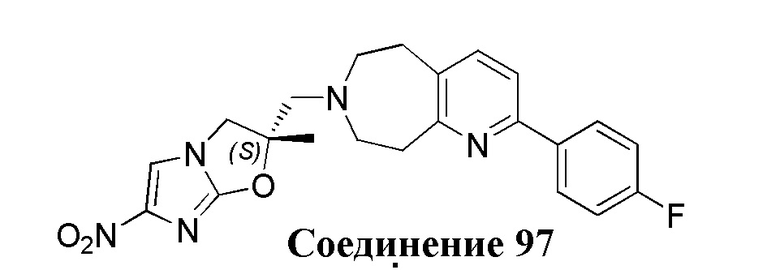
[1089] Способ синтеза был таким, как в варианте осуществления 7.
[1090] (S)-2-((2-(4-Фторфенил)-8,9-дигидро-5H-пиридо[2,3-d]азепин-7(6H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 97 (23,80 мг, 50,02 мкмоль, выход 9,20%, чистота 89%). 1H ЯМР (400 МГц, CDCl3) δ 8,11-7,89 (m, 2H), 7,56-7,46 (m, 1H), 7,45-7,34 (m, 2H), 7,15 (t, J = 8,7 Гц, 2H), 4,36 (d, J = 9,5 Гц, 1H), 4,00-3,89 (m, 1H), 3,19-2,62 (m, 10H), 1,72-1,57 (m, 3H). LCMS (ESI) масса/заряд: 424 (M+1).
[1091] Вариант осуществления 98
[1092] (2S)-2-((2-(4-Фторфенил)-6,7,8,9-тетрагидро-5H-5,8-эпиминоциклогепта[b]пиридин-10-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
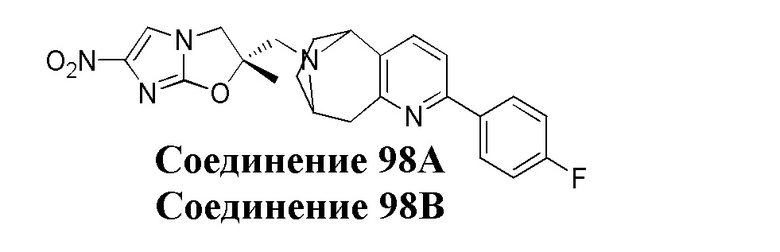
[1093] Способ синтеза был таким, как в варианте осуществления 7.
[1094] (2S)-2-((2-(4-Фторфенил)-6,7,8,9-тетрагидро-5H-5,8-эпиминоциклогепта[b]пиридин-10-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 98A (30,26 мг, 69,49 мкмоль, выход 32,79%, чистота 100%). 1H ЯМР (400 МГц, CDCl3) δ 7,94-7,87 (m, 2H), 7,45 (s, 1H), 7,42-7,38 (m, 1H), 7,36-7,31 (m, 1H), 7,19-7,10 (m, 2H), 4,39 (d, J = 9,7 Гц, 1H), 4,06 (d, J = 6,0 Гц, 1H), 3,93 (d, J = 9,7 Гц, 1H), 3,63 (t, J = 6,1 Гц, 1H), 3,39 (dd, J = 4,8, 17,9 Гц, 1H), 3,08 (d, J = 14,7 Гц, 1H), 2,75-2,64 (m, 2H), 2,29-2,08 (m, 2H), 1,83-1,78 (m, 1H), 1,73-1,63 (m, 4H). LCMS (ESI) масса/заряд: 436,3 (M+1).
[1095] (2S)-2-((2-(4-Фторфенил)-6,7,8,9-тетрагидро-5H-5,8-эпиминоциклогепта[b]пиридин-10-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 98B (41,29 мг, 94,82 мкмоль, выход 44,75%, чистота 100%). 1H ЯМР (400 МГц, CDCl3) δ 7,97-7,90 (m, 2H), 7,55 (s, 1H), 7,50-7,45 (m, 1H), 7,42-7,37 (m, 1H), 7,19-7,11 (m, 2H), 4,50 (d, J = 9,4 Гц, 1H), 4,03 (d, J = 6,0 Гц, 1H), 3,94 (d, J = 9,4 Гц, 1H), 3,71-3,62 (m, 1H), 3,22 (d, J = 14,6 Гц, 1H), 2,99 (d, J = 14,8 Гц, 1H), 2,80 (d, J = 14,8 Гц, 1H), 2,67 (d, J = 17,8 Гц, 1H), 2,15 (d, J = 11,7 Гц, 1H), 2,05-1,93 (m, 1H), 1,76 (d, J = 12,3 Гц, 2H), 1,60 (s, 3H). LCMS (ESI) масса/заряд: 436,3 (M+1).
[1096] Вариант осуществления 99
[1097] (2S)-2-((2-(4-Фторфенил)-5,6,7,8-тетрагидро-4H-5,8-эпиминоциклогепта[d]тиазол-9-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол
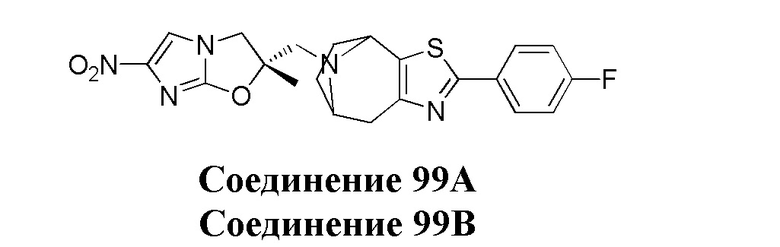
[1098] Способ синтеза был таким, как в варианте осуществления 69.
[1099] (2S)-2-((2-(4-Фторфенил)-5,6,7,8-тетрагидро-4H-5,8-эпиминоциклогепта[d]тиазол-9-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 99A (3,70 мг, 8,26 мкмоль, выход 2,47%, чистота 98,594%). 1H ЯМР (400 МГц, CDCl3) δ 7,91-7,84 (m, 2H), 7,53 (s, 1H), 7,16-7,09 (m, 2H), 4,35 (d, J = 8,0 Гц, 1H), 4,25 (d, J = 4,0 Гц, 1H), 3,95 (d, J = 8,0 Гц, 1H), 3,69-3,63 (m, 1H), 3,38-3,27 (m, 1H), 3,04 (d, J = 16,0 Гц, 1H), 2,80 (d, J = 16,0 Гц, 1H), 2,56 (d, J = 16,0 Гц, 1H), 2,19-2,10 (m, 2H), 1,99-1,91 (m, 1H), 1,67 (s, 3H), 1,69-1,61 (m, 1H). LCMS (ESI) масса/заряд: 442 (M+1).
[1100] (2S)-2-((2-(4-Фторфенил)-5,6,7,8-тетрагидро-4H-5,8-эпиминоциклогепта[d]тиазол-9-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 99B (3,70 мг, 8,33 мкмоль, выход 2,49%, чистота 99,431%). 1H ЯМР (400 МГц, CDCl3) δ 7,94-7,85 (m, 2H), 7,56 (s, 1H), 7,17-7,09 (m, 2H), 4,45 (d, J = 12,0 Гц, 1H), 4,31-4,26 (m, 1H), 3,95 (d, J = 8,0 Гц, 1H), 3,68-3,60 (m, 1H), 3,23-3,14 (m, 1H), 3,07 (d, J = 16,0 Гц, 1H), 2,78 (d, J = 12,0 Гц, 1H), 2,61-2,53 (m, 1H), 2,21-2,09 (m, 1H), 1,96 - 1,86 (m, 2H), 1,61 (s, 3H), 1,57-1,53 (m, 1H). LCMS (ESI) масса/заряд: 442 (M+1).
[1101] Вариант осуществления 100
[1102] (S)-2-((2-(4-Фторфенил)-7,8-дигидро-4H-тиазоло[4,5-d]азепин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол

[1103] Способ синтеза был таким, как в варианте осуществления 69.
[1104] (S)-2-((2-(4-Фторфенил)-7,8-дигидро-4H-тиазоло[4,5-d]азепин-6(5H)-ил)метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b]оксазол – соединение 100 (35,30 мг, 77,62 мкмоль, выход 10,33%, чистота 94,431%). 1H ЯМР (400 МГц, CDCl3) δ 7,87-7,78 (m, 2H), 7,56 (s, 1H), 7,13-7,07 (m, 2H), 4,40 (d, J = 8,0 Гц, 2H), 3,97 (d, J = 8,0 Гц, 2H), 3,24 (d, J = 16,0 Гц, 2H), 3,17-3,09 (m, 1H), 3,07-2,86 (m, 6H), 2,82 (d, J = 16,0 Гц, 2H), 2,76-2,66 (m, 1H), 1,67 (s, 3H). LCMS (ESI) масса/заряд: 430 (M+1).
[1105] Раздел фармакологии.
[1106] Часть I. Эффективность соединений против Mycobacterium tuberculosis in vitro исследовали с использованием штамма H37Rv.
[1107] В день проведения исследования соединение растворяли в чистом DMSO (Sigma 276855-2L) до концентрации 10 мг/мл, который использовали в качестве исходного раствора данного соединения. В v-образные лунки с 2 по 11 столбец 96-луночных планшетов вносили по 30 мкл DMSO (Axygen-wipp02280). В лунку столбца 2 добавляли 30 мкл исходного раствора соединения, а после смешивания из лунки столбца 2 отбирали 30 мкл и добавляли в лунку столбца 3, затем пипетировали и перемешивали. Такую процедуру выполняли до столбца 10. В столбце 11 медикамент отсутствовал, а содержалось лишь 30 мкл DMSO. Это был «исходный планшет» соединения. От столбца 2 по столбец 11 соответствующая концентрация соединения составляла 5, 2,5, 1,25, 0,625, 0,3125, 0,156, 0,078, 0,039, 0,02, 0 мг/мл. Для соединений с хорошей эффективностью исследуемую концентрацию соответствующим образом понижали. В качестве «дочернего планшета» использовали плоскодонный 96-луночный планшет (Greiner 655090). В лунки всех дочерних планшетов вносили по 98 мкл среды 7H9 (Sigma M0178). Из исходной панели отбирали 2 мкл соединения и вносили в соответствующий дочерний планшет. Линии A и H столбцов 1 и 12 из дочернего планшета содержали только среду 7H9.
[1108] Среду 7H9, содержащую 0,05% Твина 80, засевали штаммом H37Rv из криопробирки с глицерином и культивировали в течение 4 недель при 37°C на шейкере при 200 об/мин. Бактериальную жидкость дважды промывали средой 7H9, содержащей 0,05% Твин 80, и ресуспендировали в ту самую среду. Оптическую плотность бактериальной жидкости доводили до OD550 = 0,4-0,5 с использованием той самой среды. Бактериальной жидкостью заполняли микроцентрифужные пробирки и хранили при -80°C. Время хранения составляло не более 1 месяца. В день проведения исследования помещенную в пробирки бактериальную жидкость необходимо заморозить. Бактериальную жидкость разбавляли в 20 раз, а затем разбавляли в 50 раз средой 7H9, и в целом разбавление было 1000-кратным. Бактериальную жидкость использовали для засева дочернего планшета. Каждую лунку дочерней панели засевали 100 мкл бактериальной жидкости, причем в лунки столбца 12 вносили 100 мкл среды 7H9 без бактериальной жидкости.
[1109] Испытательный дочерний планшет помещали в термостат при 37°C, где поддерживали влажность >80%. Через неделю в лунку столбца 1, содержащую бактерии, и лунку столбца 12 без бактерий каждый день добавляли по 12,5 мкл среды 7H9, содержащей 20% Твин 80, и 20 мкл аламара синего (Invitrogen DAL1100), а затем продолжали культивировать в течение 24 часов и вести наблюдение. Если добавленный аламар синий становился розовым в течение 24 часов за счет бактериальной жидкости в лунке столбца 1, то во все лунки на испытательном планшете добавляли среду 7H9, содержащую 20% Твин 80, и аламар синий, причем после культивирования в течение 24 часов при 37°C измеряли значения флюоресценции.
[1110] Минимальную ингибирующую концентрацию (MIC) определяли как минимальную концентрацию лекарственного средства, вызывающую полное заметное невооруженным глазом прекращение изменения цвета аламара синего или же минимальную концентрацию лекарственного средства, вызывающую более 90% изменение цвета аламара синего, которое можно определить путем измерения флуоресценции. Результаты некоторых соединений представлены в таблице 1.
[1111] Часть II. Способы исследования эффективности соединений против Mycobacterium tuberculosis in vitro с использованием штамма Mycobacterium bovis BCG TMC1019 (ATCC35737).
[1112] В день проведения исследования соединение растворяли в чистом DMSO (Sigma 276855-2L) до концентрации 12,8 мг/мл, что использовали в качестве исходного раствора данного соединения. В v-образные лунки с 1 по 12 столбец 96-луночных планшетов вносили по 30 мкл DMSO (Axygen-wipp02280). В лунку столбца 1 вносили 30 мкл исходного раствора соединения. Из лунки столбца 1 отбирали 30 мкл и добавляли в лунку столбца 2, а затем пипетировали и смешивали. С помощью такой процедуры выполняли двойные градиентные разбавления до столбца 11. В столбце 12 соединение отсутствовало, и содержалось лишь 30 мкл DMSO. Все лунки в линии A и H содержали только по 30 мкл DMSO. Это был «исходный планшет» соединения. От столбца 1 по столбец 12 соответствующая концентрация соединения составляла 6,4, 3,2, 1,6, 0,8, 0,4, 0,2, 0,1, 0,05, 0,025, 0,0125, 0,00625 и 0 мг/мл. Для соединений с хорошей эффективностью исследуемую концентрацию соответствующим образом понижали. В качестве «дочернего планшета» использовали плоскодонный 96-луночный планшет (Greiner 655090). Из исходной панели отбирали 2 мкл соединения и вносили в соответствующий дочерний планшет. Линии A и H столбца 12 из дочернего планшета содержали только среду 7H9.
[1113] Среду 7H9, содержащую 0,05% Твина 80, засевали штаммом BCG из криопробирки с глицерином и культивировали в течение 4 недель при 37°C на шейкере при 200 об/мин. Бактериальную жидкость дважды промывали средой 7H9, содержащей 0,05% Твин 80, и ресуспендировали в ту самую среду. Оптическую плотность бактериальной жидкости доводили до OD550 = 0,4-0,5 с использованием той самой среды. Бактериальной жидкостью заполняли микроцентрифужные пробирки и хранили при -80°C. Время хранения составляло не более 1 месяца. В день проведения исследования помещенную в пробирки бактериальную жидкость необходимо заморозить. Бактериальную жидкость разбавляли в 20 раз, а затем разбавляли в 50 раз средой 7H9, и в целом разбавление было 1000-кратным. Бактериальную жидкость использовали для посева. Каждую лунку дочерней панели, за исключением линии А, засевали 100 мкл бактериальной жидкости, а в лунки линии А вносили 100 мкл среды 7H9 и без бактериальной жидкости. Конечные концентрации исследуемого соединения составляли 64, 32, 16, 8, 4, 1, 0,5, 0,25, 0,125, 0,0625 и 0 мкг/мл. Испытательный дочерний планшет культивировали в термостате при 37°C, где поддерживали влажность >80%.
[1114] Через неделю в лунку линии А без бактерий и лунку линии Н, содержащую бактерии, каждый день добавляли по 12,5 мкл среды 7H9, содержащей 20% Твин 80, и 20 мкл аламара синего (Invitrogen DAL1100), а затем продолжали культивировать в течение 24 часов и вести наблюдение. Если добавленный аламар синий становился розовым в течение 24 часов за счет бактериальной жидкости в лунке линии Н, то во все лунки на испытательном планшете добавляли аламар синий, причем после культивирования в течение 24 часов при 37°C отмечали минимальную ингибирующую концентрацию.
[1115] Минимальную ингибирующую концентрацию (MIC) определяли как минимальную концентрацию лекарственного средства, вызывающую полное заметное невооруженным глазом прекращение изменения цвета аламара синего или же минимальную концентрацию лекарственного средства, вызывающую более 90% изменение цвета аламара синего, которое можно определить путем измерения флуоресценции. Результаты некоторых соединений представлены в таблице 1.
[1116] Таблица 1. Активность некоторых молекул согласно настоящему изобретению против штамма Mycobacterium Bovis BCG и штамма Mycobacterium tuberculosis H37Rv in vitro
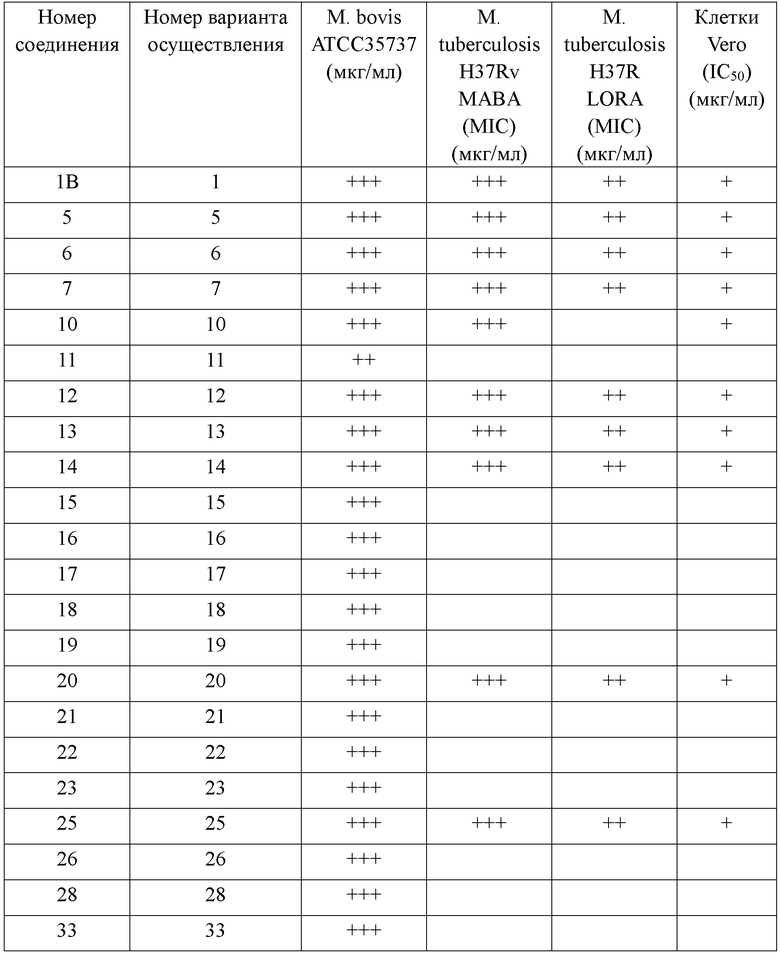
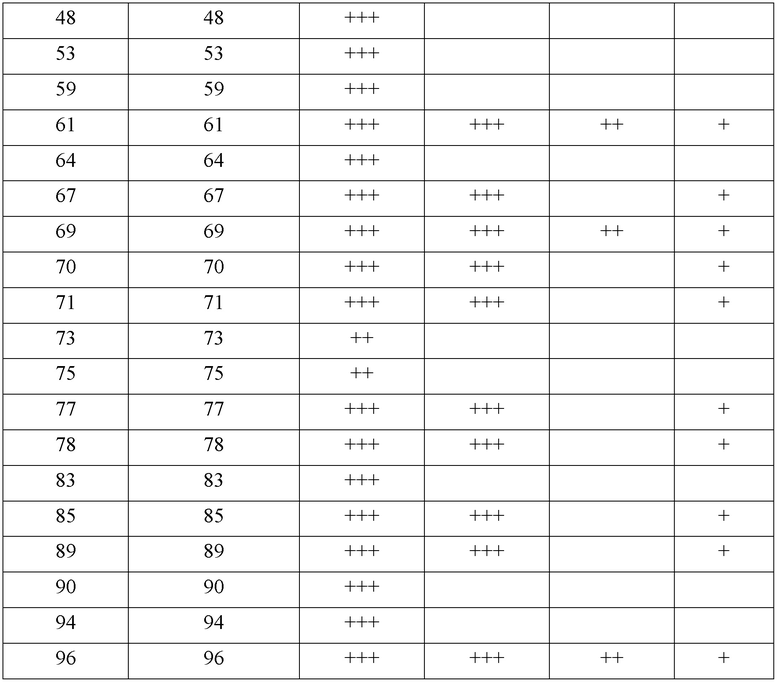
[1117] +++: <1; ++: 1~32; +: >32
[1118] Результаты показали, что соединения, рассматриваемые в настоящем изобретении, обладают хорошей ингибирующей активностью против обоих штаммов, Mycobacterium bovis BCG и Mycobacterium tuberculosis H37Rv, при этом минимальная ингибирующая концентрация всех молекул составляла <1 мкг/мл, и соединения согласно настоящему изобретению не были цитотоксичными.
[1119] Часть III. Эксперименты для оценки кинетической растворимости и двунаправленной проницаемости клеток MDR1-MDCK. Результаты исследования представлены в таблице 2.
[1120] 1. Исследование кинетической растворимости. Образец соединения взвешивали количественно и растворяли в чистом DMSO, чтобы конечная концентрация составляла 10 мМ. Исследуемое соединение и контрольное соединение (10 мМ исходный раствор DMSO, 10 мкл на лунку) добавляли в 96-луночный планшет, содержащий 490 мкл буфера на лунку. После перемешивания на вортексе в течение 2 минут образцы в планшете культивировали на шейкере при комнатной температуре (22 ± 2°C) в течение 24 часов. Затем 200 мкл образца переносили на пластину фильтрпресса MultiScreen (поликарбонатную пленку), фильтровали через вакуумный коллектор с миллипорами и собирали фильтрат. Концентрацию соединения в фильтрате определяли посредством HPLC-UV. Последовательно в хроматограф вводили образцы УФ-стандартных растворов с тремя разными концентрациями и образцы для проведения исследования на растворимость. Каждый образец вводили в хроматограф 2 раза, с помощью градуировочной кривой получали концентрацию и рассчитывали среднее значение.
[1121] 2. Эксперимент по определению двунаправленной проницаемости клеток MDR1-MDCK Клетки MDR1-MDCK, которые непрерывно экспрессируют P-гликопротеин человека (P-гликопротеин), высевали на 96-луночные планшеты со вставкой для культуры клеток и культивировали в течение 4–7 дней с получением монослоя агрегированных клеток. Для проверки качества монослоя клеток применяли оценку однонаправленной (A → B) проницаемости фенотерола (маркера низкой проницаемости) и пропранолола (маркера высокой проницаемости) и двунаправленной проницаемости дигоксина (P-гликопротеинового субстрата). Из трех контрольных соединений сделали две комплексные лунки.
[1122] Стандартные условия для экспериментов над исследуемым соединением, касающихся переноса, были следующими:
- исследуемая концентрация: 2 мкМ (DMSO ≤ 1%);
- повторение: n = 3;
- направление: двунаправленный перенос, состоящий из двух направлений: A → B и B → A;
- время культивирования: один момент времени, 2,5 часа;
- буфер для переноса: HBSS, pH 7,4;
- условия культивирования: 37°C, 5% CO2, относительная влажность 95%.
[1123] После культивирования растворы образцов, взятые из донорных лунок и принимающих лунок, сразу же смешивали с холодным раствором ацетонитрила, содержащим внутренний стандарт. И измеряли количество внутриклеточно накопленных соединений путем лизиса клеток с помощью холодного раствора ацетонитрила, содержащего внутренний стандарт. Анализ концентраций исследуемого соединения во всех образцах (включая первоначально вводимый раствор, надосадочную жидкость донорных лунок, принимающий раствор, клеточный лизат) проводили посредством LC/MS/MS-способа. Концентрацию исследуемого соединения выражали как соотношение площади его пика к площади пика внутреннего стандарта. Кинетическая растворимость (KS) соединений-кандидатов и данные проницаемости в монослое клеток MDR1-MDCK перечислены в таблице 2.
[1124] Таблица 2. Кинетическая растворимость и результаты двунаправленной проницаемости клеток MDR1-MDCK по отношению к некоторым молекулам согласно настоящему изобретению
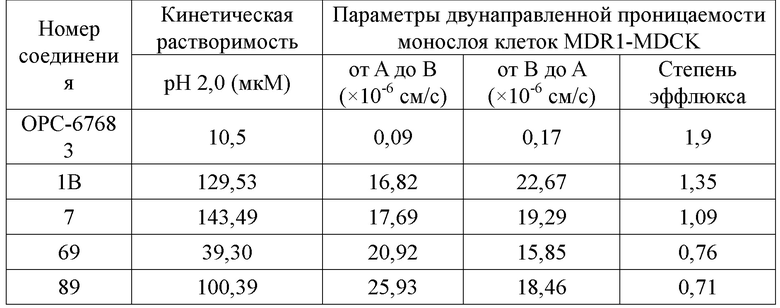
[1125] Разумеется, что вышеуказанные четыре соединения-кандидата превосходили OPC-67683 по кинетической растворимости. Хорошая растворимость благоприятна для изучения абсорбции и получения лекарственных средств in vivo. Не сложно понять, что предпочтительные молекулы являются высокопроницаемыми молекулами благодаря параметрам проницаемости. По сравнению с контрольным соединением (OPC-67683) преимущества являются очевидными. Хорошая проницаемость может дополнительно способствовать абсорбции лекарственных средств и достижению надлежащего действия против Mycobacterium tuberculosis.
[1126] Часть IV. Фармакокинетика in vivo
[1127] Фармакокинетические характеристики грызунов после внутривенного и перорального введения соединения исследовали согласно стандартному протоколу. В частности, соединения-кандидаты данного эксперимента внутривенно инъецировали и перорально вводили самцам мышей CD-1 в возрасте 7-10 недель. Препарат для перорального введения представлял собой 0,5% суспензию метилцеллюлозы, а препарат для внутривенной инъекции представлял собой прозрачный раствор из этанола/DMSO/полиэтиленгликоля 400/чистой воды (10: 10: 50: 30). Производили забор образцов плазмы и легкого, анализировали посредством LC-MS/MS-способа и рассчитывали фармакокинетические параметры. Фармакокинетические параметры соединения-кандидата 7 представлены в таблице 3.
[1128] Таблица 3. Фармакокинетические параметры in vivo
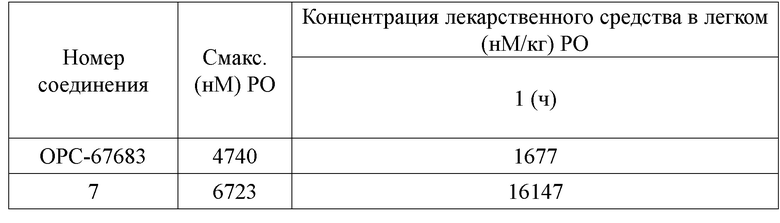
[1129] Было ясно, что фармакокинетические параметры новообнаруженного соединения 7 превосходят фармакокинетические параметры контрольного соединения (OPC-67683), причем следует отметить, что через 1 час после введения лекарственного средства 7 его концентрация в легком была намного выше концентрации контрольного соединения (OPC-67683), достигая более чем 9-кратного превышения по сравнению с контрольным соединением. Для пациентов с легочной инфекцией, вызываемой Mycobacterium tuberculosis, чем больше величина экспозиции лекарственного средства, тем лучше эффективность, что является очень важным.
[1130] Благодаря исследованию активности в отношении штамма H37Rv было обнаружено, что все молекулы, рассматриваемые в настоящем изобретении, обладают хорошей активностью против Mycobacterium tuberculosis in vitro. Из структуры видно, что все молекулы имеют атомы щелочного азота и способны образовывать соли, что благоприятно для улучшения растворимости молекул, поскольку тогда легче проводить изучение препарата, при этом экспериментальные данные касательно растворимости также подтверждают эту гипотезу. Исследования проницаемости показали, что большинство обнаруженных молекул являются высокопроницаемыми молекулами, что способствует распределению и абсорбции в организме, при этом ожидается, что будет достигнута лучшая эффективность. Фармакокинетические данные in vivo дополнительно подтверждают, что соединение 7 с превосходной растворимостью и высокой проницаемостью оказывает на легкое превосходное действие. Принимая во внимание собранные данные, нет оснований сомневаться в том, что данные молекулы будут проявлять лучшую эффективность, нежели контрольное соединение (OPC-67683), тем самым принося пользу большинству пациентов.
| название | год | авторы | номер документа |
|---|---|---|---|
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| БИЦИКЛИЧЕСКИЕ НИТРОИМИДАЗОЛЫ, КОВАЛЕНТНО СОЕДИНЕННЫЕ С ЗАМЕЩЕННЫМИ ФЕНИЛОКСАЗОЛИДИНОНАМИ | 2009 |
|
RU2504547C2 |
| ПРОИЗВОДНОЕ РЕЗОРЦИНА В КАЧЕСТВЕ ИНГИБИТОРА HSP90 | 2016 |
|
RU2697703C2 |
| НИТРОИМИДАЗООКСАЗИНОВЫЕ И НИТРОИМИДАЗООКСАЗОЛЬНЫЕ АНАЛОГИ И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2540860C2 |
| СОЕДИНЕНИЕ НА ОСНОВЕ ДИГИДРОНАФТИРИДИНОНА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ, И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2809869C1 |
| ИНГИБИТОРЫ TrkA КИНАЗЫ, ОСНОВАННЫЕ НА НИХ КОМПОЗИЦИИ И СПОСОБЫ | 2015 |
|
RU2672583C2 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ RORc | 2017 |
|
RU2757571C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ | 2019 |
|
RU2794894C2 |
| ИМИДАЗОПИРАЗИНОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE1 | 2016 |
|
RU2712219C2 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
Изобретение относится к новым производным нитроимидазола формулы (I), а также к фармацевтической композиции на их основе и применению для изготовления лекарственного препарата для лечения заболеваний, вызываемых Mycobacterium tuberculosis. Технический результат: получены новые соединения, обладающие ингибиторной активностью в отношении Mycobacterium tuberculosis, которые, кроме того, обладают высокой растворимостью и проницаемостью. 4 н. и 7 з.п. ф-лы, 1 табл.
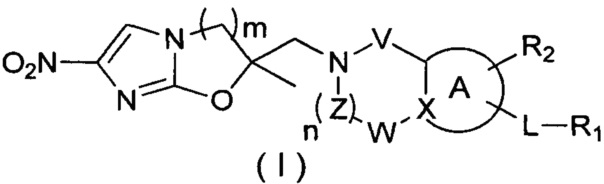
1. Соединение, характеризующееся структурой формулы (I), его фармацевтически приемлемая соль, его (R)-изомер или (S)-изомер,
где
кольцо А представляет собой 5-6-членный гетероарил;
X представляет собой N или С;
каждый из V и W независимо выбран из группы, состоящей из метилена, -СН2СН2- и С(=O), где метилен необязательно замещен 1 или 2 R;
Z представляет собой метилен, который необязательно замещен 1 или 2 R;
L представляет собой одинарную связь, -О- или N(R);
R представляет собой Н или С1-6алкил;
каждый из R1 и R2 независимо выбран из Н, галогена, CN или каждый из R1 и R2 независимо выбран из группы, состоящей из С1-6алкила, С1-6гетероалкила, С3-7циклоалкила, С3-7гетероциклоалкила, 6-членного арила, 5-7-членного гетероарила, каждый из которых необязательно замещен 1-3 заместителями, выбранными из группы, состоящей из F, Cl, Br, I, CN, С1-4алкила и С1-4гетероалкила, где С1-4алкил или С1-4гетероалкил необязательно может быть дополнительно замещен 1-3 заместителями, которые выбраны из галогена;
необязательно заместитель R при Z и заместитель R при V присоединены к одному и тому же атому или атомной группе с образованием 5-7-членного кольца;
необязательно фрагмент 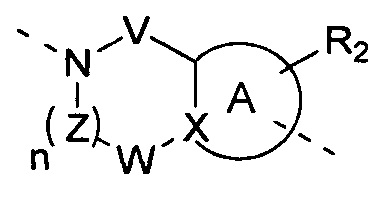 может быть заменен
может быть заменен 
также R2 может отсутствовать;
m равняется 1;
n равняется 0, 1 или 2;
«гетеро» представляет собой гетероатом, который выбран из группы, состоящей из -О-, -S- и N;
количество гетероатомов выбрано из 1, 2 или 3.
2. Соединение, его фармацевтически приемлемая соль, его (R)-изомер или (S)-изомер по п. 1, где R представляет собой Н или С1-4алкил;
и каждый из R1 и R2 необязательно замещен заместителем, выбранным из группы, состоящей из F, Cl, Br, I, CN, -CF3, -СН3, -OCF3, -CH2CF3 и ОСН3.
3. Соединение, его фармацевтически приемлемая соль, его (R)-изомер или (S)-изомер по п. 1 или 2, где каждый из R1 и R2 независимо выбран из группы, состоящей из Н, галогена, CN, или выбран из группы, состоящей из

 ,
,
каждый из которых является необязательно замещенным 1-3 заместителями, выбранными из группы, состоящей из F, Cl, Br, I, CN, С1-4алкила и С1-4гетероалкила, где С1-4алкил или С1-4гетероалкил необязательно может быть дополнительно замещен 1-3 заместителями, которые выбраны из галогена;
или каждый из R1 и R2 независимо выбран из группы, состоящей из Н, галогена, CN, или выбран из группы, состоящей из

каждый из которых является необязательно замещенным 1-3 заместителями, выбранными из группы, состоящей из F, Cl, Br, I, CN, С1-4алкила и С1-4гетероалкила, где С1-4алкил или С1-4гетероалкил необязательно может быть дополнительно замещен 1-3 заместителями, которые выбраны из галогена,
или каждый из R1 и R2 независимо выбран из группы, состоящей из
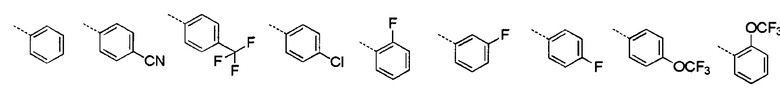


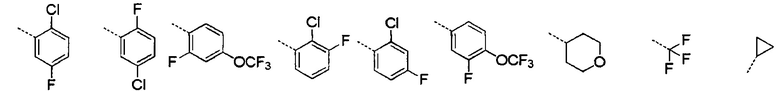

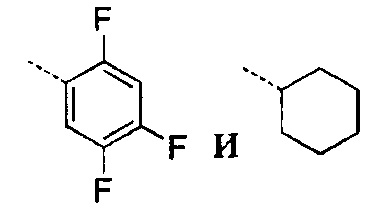 .
.
4. Соединение, его фармацевтически приемлемая соль, его (R)-изомер или (S)-изомер по п. 1 или 2, где R выбран из группы, состоящей из Н, Me и Et.
5. Соединение, его фармацевтически приемлемая соль, его (R)-изомер или (S)-изомер по п. 1 или 2, где кольцо А выбрано из группы, состоящей из пиридила, тиазолила, оксазолила, имидазолила и пиримидинила;
или кольцо А выбрано из группы, состоящей из

или фрагмент 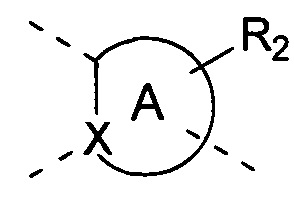 выбран из группы, состоящей из
выбран из группы, состоящей из 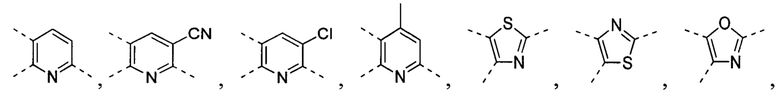
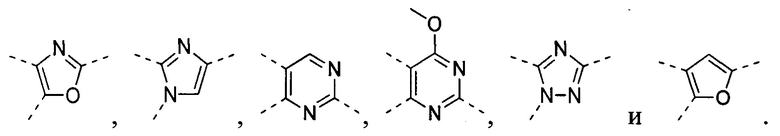
6. Соединение, его фармацевтически приемлемая соль, его (R)-изомер или (S)-изомер по п. 1 или 2, где фрагмент 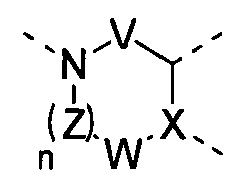 выбран из группы, состоящей из
выбран из группы, состоящей из 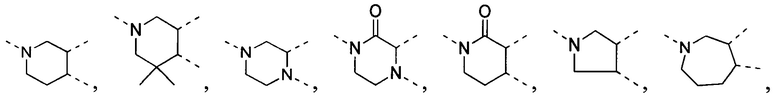

7. Соединение, его фармацевтически приемлемая соль, его (R)-изомер или (S)-изомер по п. 1 или 2, где фрагмент 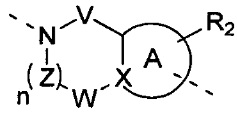 выбран из группы, состоящей из
выбран из группы, состоящей из
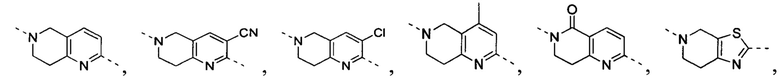
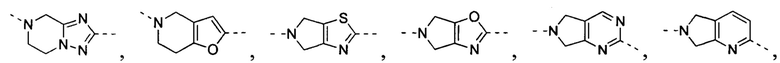
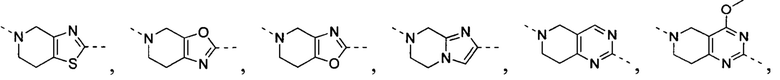

8. Соединение, его фармацевтически приемлемая соль, его (R)-изомер или (S)-изомер по п. 1 или 2, которые выбраны из группы, состоящей из

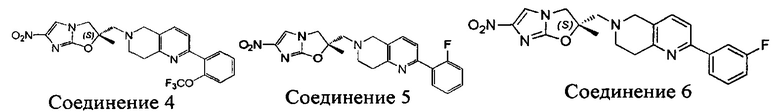
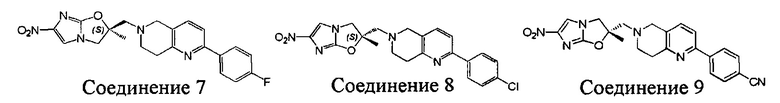
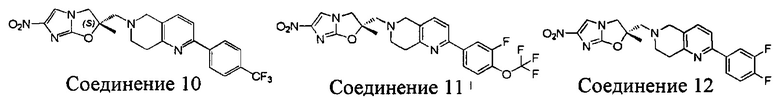
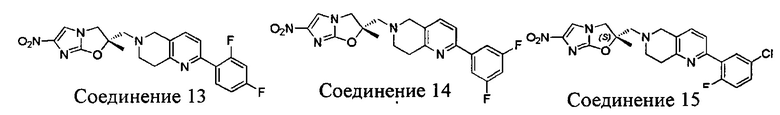
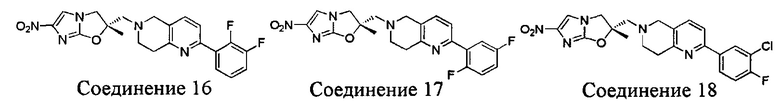
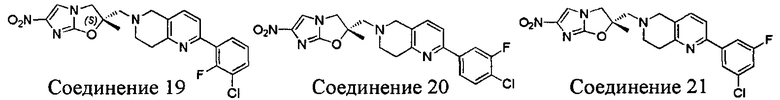
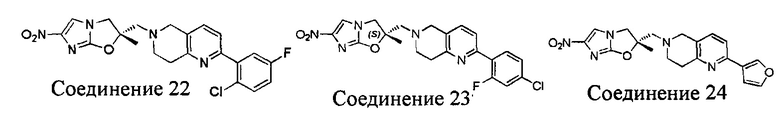
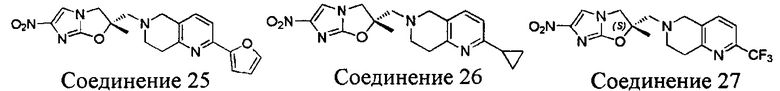
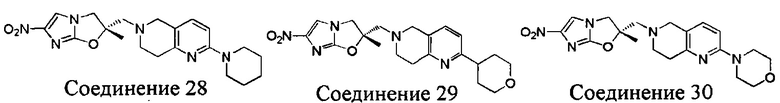

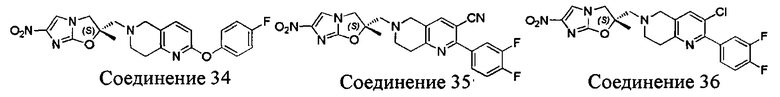
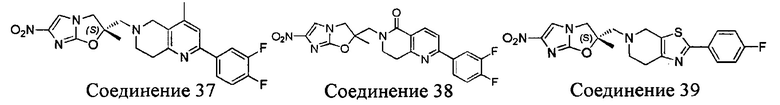
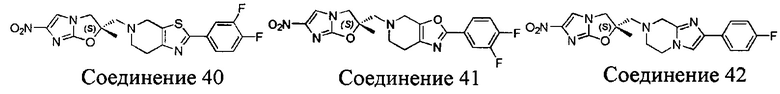

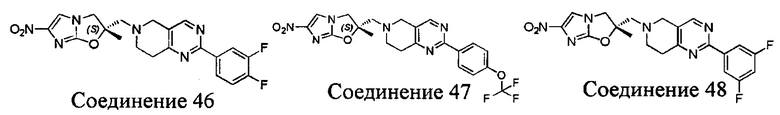
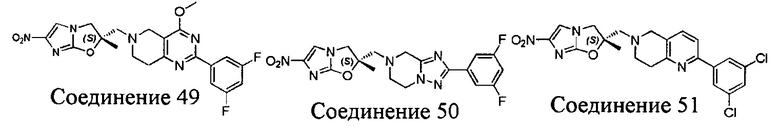

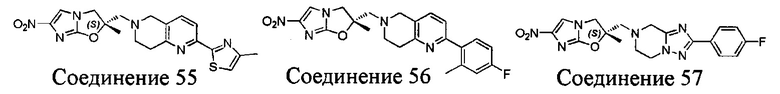

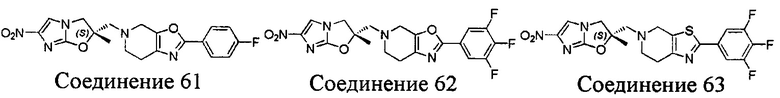
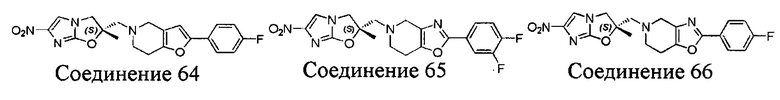
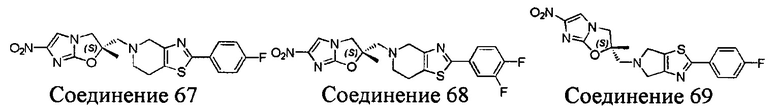
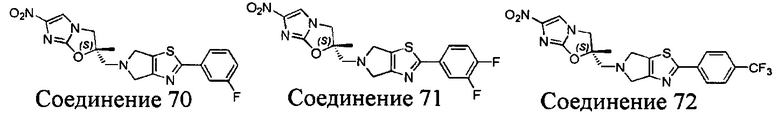
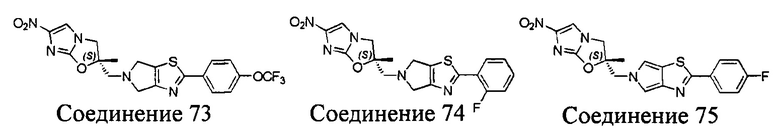
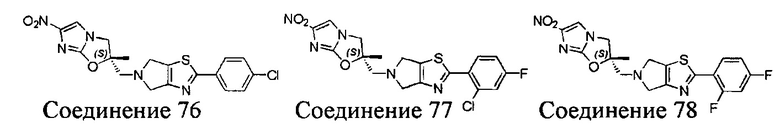
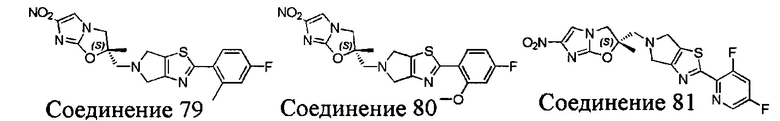
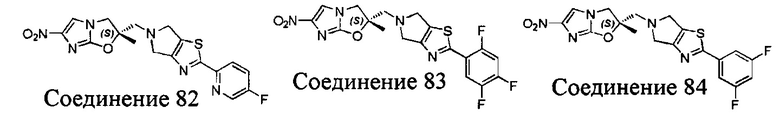
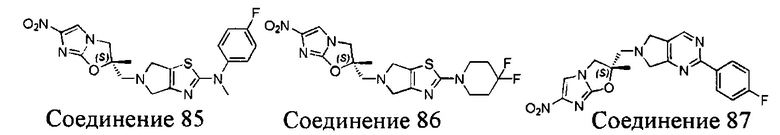
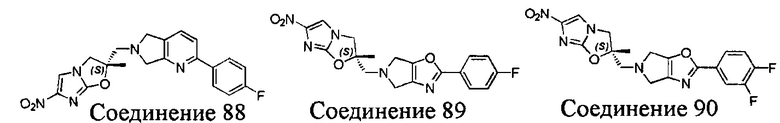


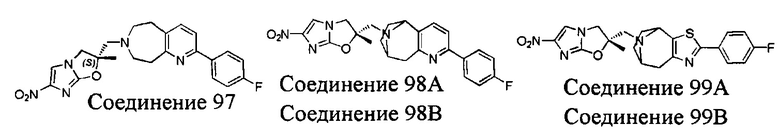

9. Фармацевтическая композиция для лечения или предупреждения инфекций, вызываемых Mycobacterium tuberculosis, содержащая эффективное количество соединения формулы (I), его фармацевтически приемлемой соли, его (R)-изомера или (S)-изомера по любому из пп. 1-8 или фармацевтически приемлемый носитель.
10. Применение соединения формулы (I), его фармацевтически приемлемой соли, его (R)-изомера или (S)-изомера по любому из пп. 1-8 в изготовлении лекарственного препарата для лечения и предупреждения инфекций, вызываемых Mycobacterium tuberculosis.
11. Применение композиции по п. 9 в изготовлении лекарственного препарата для лечения и предупреждения инфекций, вызываемых Mycobacterium tuberculosis.
| Навесное грузозахватное приспособление к погрузчику | 1987 |
|
SU1555267A1 |
| ПРОИЗВОДНЫЕ 2,3-ДИГИДРО-6-НИТРОИМИДАЗО [2,1-b] ОКСАЗОЛА ДЛЯ ЛЕЧЕНИЯ ТУБЕРКУЛЕЗА | 2004 |
|
RU2365593C2 |
| JP4787529 B2, 05.10.2011. | |||

