Настоящее изобретение относится к новой твердой дозированной форме с немедленным высвобождением, которая обеспечивает быстрое растворение и быструю абсорбцию напроксена в организме человека.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Напроксен представляет собой производное пропионовой кислоты. Это нестероидное противовоспалительное лекарственное средство и мощный ингибитор циклооксигеназы, ответственной за биосинтез простагландинов. Напроксен оказывает противовоспалительное, анальгетическое и жаропонижающее воздействие на организм человека. Напроксен и соли напроксена показаны для снижения температуры и обезболивания, например боли при артрите, боли при воспалении, мышечной боли, боли в спине, головной боли, мигрени, боли при менструальных спазмах, зубной боли и боли, связанной с ОРВИ.
В работе Davies and Anderson, Clinical Pharmacokinetics, 32(4):268-93 (1997) отмечается, что после перорального введения напроксен быстро и полностью абсорбируется, и уровень абсорбции обеспечивает аналогичное воздействие по сравнению с внутривенным введением, что определяется по площади под кривой зависимости концентрации от времени. Наличие пищи в желудке влияет на скорость, но не на уровень абсорбции. Максимальная концентрация в плазме, как правило, достигается в течение 1-2 часов после введения напроксена натрия.
Скорость до начала снижения боли является важной неудовлетворенной потребностью в области обезболивания. Повышение скорости и уровня абсорбции пероральных составов композиций было и остается предметом активных исследований. После того как пероральная твердая композиция с немедленным высвобождением попадает в желудок, она распадается и/или растворяется и проходит в тонкий кишечник, где активный ингредиент абсорбируется через стенки кишечника, попадая в систему кровообращения через воротную вену и печень, прежде чем достичь места действия. Для лекарственных средств без ограничения скорости абсорбции, таких как напроксен, быстрый распад и быстрое растворение активного ингредиента будет способствовать быстрой абсорбции in vivo. В патенте США № 9,757,455 (Roberts et. al) описаны составы, полученные в виде твердых дозированных форм с немедленным высвобождением, предназначенные для перорального введения целиком, которые обеспечивают быстрое растворение и быструю абсорбцию активного ингредиента, в том числе напроксена.
Вместе с тем напроксен отличается рН-зависимой растворимостью. При pH выше 5,4 напроксен остается в растворе. В условиях более низкого кислого рН напроксен натрия растворяется, но немедленно осаждается в виде мелкодисперсных коллоидных частиц напроксена. При осаждении в желудке напроксен должен попасть в тонкий кишечник до солюбилизации и повторного растворения. В результате может возникать задержка абсорбции.
В публикации заявки на патент US20070134317 описано данное явление и нешипучая форма напроксена натрия, содержащая гидрокарбонат натрия. В публикации описано образование агломератов осажденного напроксена с формированием более больших, плохо растворимых агломератов кристаллов напроксена и предложены составы, призванные свести к минимуму потенциальную плохую растворимость и биодоступность.
На текущий момент заявители обнаружили улучшенную твердую дозированную форму с немедленным высвобождением с определенным распределением частиц по размерам во внутригранулярной части и определенным распределением частиц по размерам в карбонатной части, что позволяет напроксену оставаться в растворе и обеспечивает более быстрое растворение и более быструю абсорбцию напроксена в организме человека. В частности, заявители обнаружили дозированную форму напроксена, которая при введении в организм человека натощак в течение 10 минут или менее обеспечивает среднюю концентрацию напроксена в плазме крови по меньшей мере 15-20 мкг/мл. Заявители также обнаружили дозированную форму напроксена, которая при введении в организм человека композиций, содержащих от 300 мг до 500 мг карбонатного соединения, после еды в течение 50 минут или менее обеспечивает среднюю концентрацию напроксена в плазме крови по меньшей мере 15-20 мкг/мл. Заявители дополнительно обнаружили дозированную форму напроксена, которая при введении в организм человека композиций, содержащих 500 мг карбонатного соединения, после еды в течение 25 минут или менее обеспечивает среднюю концентрацию напроксена в плазме крови по меньшей мере 15-20 мкг/мл.
ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложена улучшенная твердая дозированная форма напроксена натрия с немедленным высвобождением, которая обеспечивает быстрое растворение в желудке, позволяет напроксену оставаться в растворе и обеспечивает быструю абсорбцию напроксена. В частности, в настоящем изобретении предложена дозированная форма напроксена натрия, которая при введении в организм человека натощак в течение 10 минут или менее обеспечивает среднюю концентрацию напроксена в плазме крови по меньшей мере 15-20 мкг/мл. В настоящем изобретении также предложена дозированная форма напроксена, которая при введении в организм человека композиций, содержащих от 300 мг до 500 мг карбонатного соединения, после еды в течение 50 минут или менее обеспечивает среднюю концентрацию напроксена в плазме крови по меньшей мере 15-20 мкг/мл. В настоящем изобретении также предложена дозированная форма напроксена, которая при введении в организм человека композиций, содержащих 500 мг карбонатного соединения, после еды в течение 25 минут или менее обеспечивает среднюю концентрацию напроксена в плазме крови по меньшей мере 15-20 мкг/мл.
В одном варианте осуществления твердая дозированная форма напроксена натрия с немедленным высвобождением имеет внутригранулярное распределение частиц по размерам в диапазоне около 200-400 мкм. В другом варианте осуществления твердая дозированная форма напроксена натрия с немедленным высвобождением имеет распределение частиц карбоната по размерам в диапазоне около 50-200 мкм.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Описанные в настоящем документе чертежи приводятся исключительно в целях иллюстрации выбранных вариантов осуществления и не подразумевают ограничение объема настоящего описания.
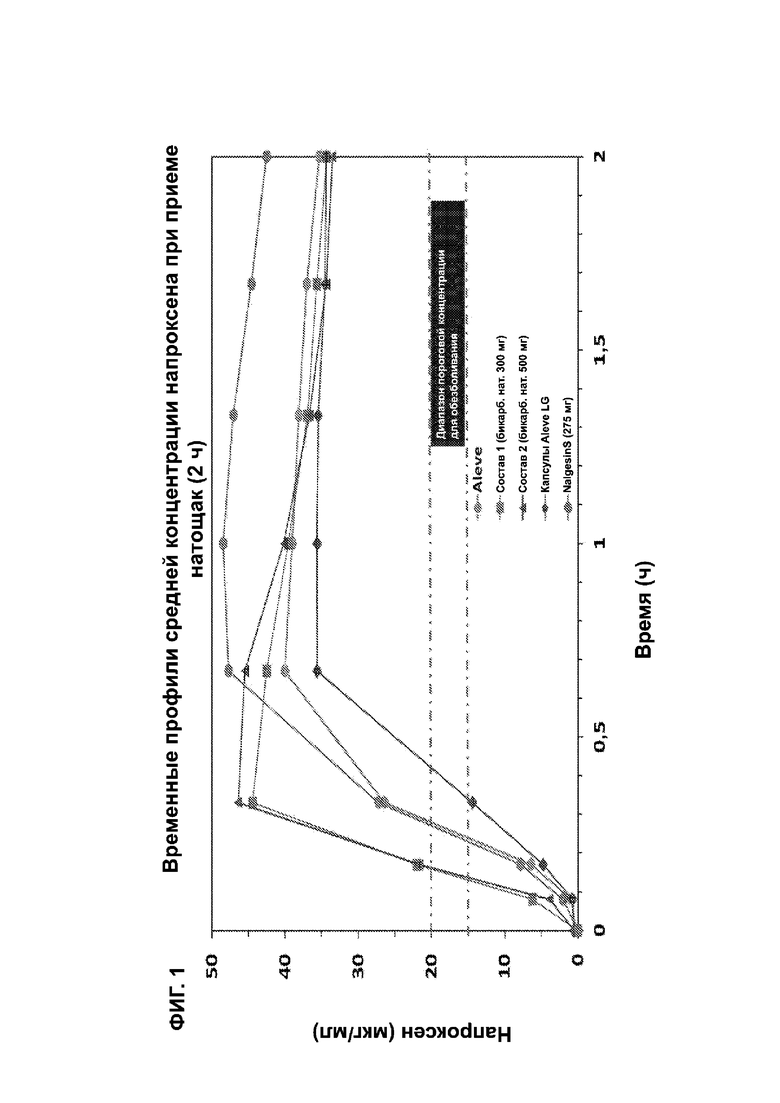
На ФИГ. 1 показаны временные профили средней концентрации напроксена при приеме натощак для исследования, описанного в примерах 1 и 2.
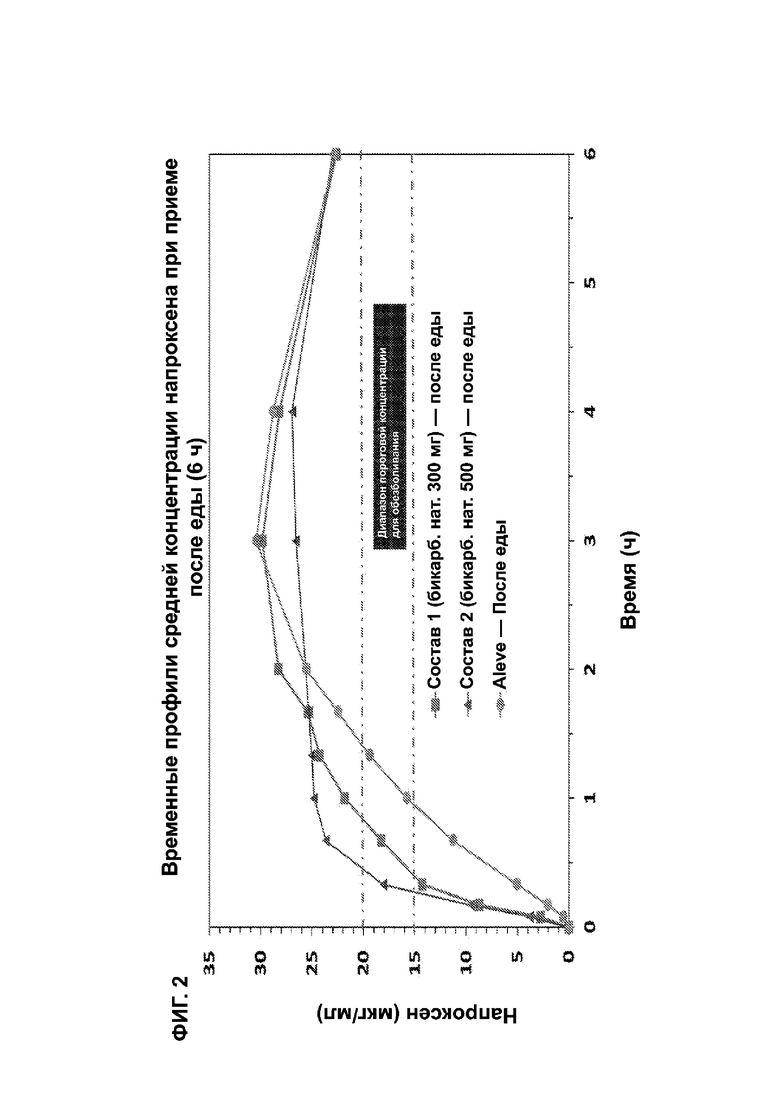
На ФИГ. 2 показаны временные профили средней концентрации напроксена при приеме после еды для исследования, описанного в примерах 1 и 2.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к применению композиции, которая обеспечивает определенное время достижения заданной средней (терапевтической) концентрации в плазме крови у пациентов-млекопитающих, которая коррелирует со снижением боли. Такая минимальная эффективная терапевтическая концентрация в плазме (МЭК) для напроксена в настоящем документе определяется равной от 15 до 20 мкг/мл. В одном варианте осуществления это рассматривается при введении натощак с конечным временем до МЭК в диапазоне от 7 минут до 9 минут. В одном варианте осуществления время до МЭК натощак составляет менее 20 минут, или менее 15 минут, или менее 10 минут. В другом варианте осуществления время до минимальной эффективной терапевтической концентрации после еды находится в диапазоне от 15 минут до 25 минут. В другом варианте осуществления время до МЭК после еды составляет менее 50 минут, или менее 40 минут, или менее 35 минут, или менее 30 минут, или менее 25 минут, или менее 20 минут.
Дополнительным предметом настоящего изобретения может быть время до максимальной концентрации в плазме. В одном варианте осуществления время до максимальной концентрации в плазме составляет менее 35 минут, или менее 30 минут, или менее 25 минут.
Композиция настоящего изобретения содержит внутригранулярную часть и внегранулярную часть. Внутригранулярная часть может содержать напроксен натрия, наполнители для прессования, связующие вещества и разрыхлители. Наполнители для прессования включают, без ограничений, микрокристаллическую целлюлозу, микрокристаллическую целлюлозу для прямого прессования, целлюлозы, нерастворимые в воде целлюлозы, крахмал, кукурузный крахмал и модифицированные крахмалы. Подходящие наполнители включают, без ограничений, крахмал и модифицированные крахмалы. Наполнитель можно добавлять в количестве от около 5 процентов до около 50 процентов или от около 10 процентов до около 40 процентов от массы таблетки. Приемлемые разрыхлители включают, без ограничений, натриевую соль гликолята крахмала, поперечно сшитый поливинилпирролидон, поперечно сшитую карбоксиметилцеллюлозу, крахмалы, микрокристаллическую целлюлозу и их смеси. Разрыхлители можно добавлять в количестве от около 0,5 процента до около 15 процентов или от около 1 процента до около 10 процентов от массы таблетки. В одном из вариантов осуществления разрыхлитель добавляют к внутригранулярной части в количестве от около 1 процента до около 3 процентов, а к внегранулярной части - от около 5 процентов до около 9 процентов от общей массы таблетки.
Материалы внегранулярной части включают карбонаты, наполнители для прессования, смазывающие вещества, агенты для повышения текучести и разрыхлители. Подходящие карбонаты включают бикарбонат калия и бикарбонат натрия. Подходящие смазывающие вещества включают стеарат магния и стеариновую кислоту. Смазывающее вещество и агент для повышения текучести можно добавлять в количестве от около 0,1 процента до около 5,0 процентов или от около 0,1 процента до около 2,0 процентов от массы таблетки. Подходящие агенты для повышения текучести включают диоксид кремния. В некоторых вариантах осуществления таблетка содержит менее 0,75 процента стеарата магния или менее 0,5 процента стеарата магния. В одном варианте осуществления наполнитель для прессования во внегранулярной части представляет собой микрокристаллическую целлюлозу. В этом варианте осуществления средний размер частиц микрокристаллической целлюлозы составляет менее 50 мкм или менее 30 мкм.
В одном варианте осуществления внегранулярная часть содержит «модулирующий pH агент» и включает в себя один или более из модулирующих pH агентов, которые изменяют рН водного раствора. Такие модулирующие агенты могут включать кислоты, основания или комбинацию одной или более кислот и/или оснований.
Карбонат может представлять собой любой фармацевтически приемлемый растворимый карбонат или его смесь и включает бикарбонат. Ссылка на термин «бикарбонат» или «карбонат» включает один агент или множество (то есть два или более) агентов. Предпочтительные карбонаты включают, без ограничений, бикарбонат натрия, карбонат натрия, бикарбонат калия, карбонат калия, бикарбонат кальция, карбонат кальция, бикарбонат магния, карбонат магния, бикарбонат аммония, карбонат аммония, глицина-карбонат натрия, глицин-карбонат динатрия, аргинин-карбонат, лизин-карбонат и/или другие фармацевтически приемлемые карбонаты или гомологи или их функциональные эквиваленты и их комбинации. Карбонат можно добавлять в количестве от около 20 процентов до около 50 процентов или от около 25 процентов до около 45 процентов от массы таблетки. В одном варианте осуществления внутригранулярная часть по существу не содержит карбоната. Используемый в настоящем документе термин «по существу не содержит» означает, что внутригранулярная часть содержит менее 0,1 процента карбоната от массы таблетки.
Карбонаты настоящего изобретения имеют средний размер частиц в диапазоне от около 50 мкм до около 200 мкм или от около 75 мкм до около 100 мкм. Размер частиц карбонатной части настоящего изобретения способствует растворению композиции. Было обнаружено, что чем больше размер частиц карбоната, тем медленнее профиль растворения композиции.
Внутригранулярная часть настоящего изобретения также отличается физическими характеристиками, которые способствуют растворению композиции. В одном варианте осуществления средний размер частиц внутригранулярной части составляет от около 200 мкм до 400 мкм или от около 200 мкм до 300 мкм.
Напроксен натрия отличается рН-зависимой растворимостью. При pH выше 5,4 напроксен натрия остается в растворе. При более низком кислом рН напроксен натрия растворяется, но немедленно осаждается в виде мелкодисперсных коллоидных частиц. При осаждении в желудке напроксен должен попасть в тонкий кишечник до повторного растворения, которое приводит к задержке абсорбции. Размер частиц внутригранулярной и карбонатной частей изобретения помогает удерживать активный ингредиент напроксен в растворе по мере его растворения в условиях кислого pH желудка. Карбонатная часть дозированной формы растворяется со скоростью, обеспечивающей увеличение pH среды в микро- и макросреде желудка, что облегчает растворение напроксена и позволяет напроксену оставаться в растворе, а также создает условия для начала абсорбции напроксена в желудке.
В некоторых вариантах осуществления объемная плотность внутригранулярной части составляет от около 0,5 до около 0,9 г/см3 или от около 0,5 до около 0,7 г/см3. В некоторых вариантах осуществления таблетку настоящего изобретения прессуют в определенных диапазонах силы сжатия, в том числе от около 18 килоньютонов до около 26 килоньютонов до твердости от около 10 килограмм-силы до около 17 килограмм-силы. В вариантах осуществления, в которых в таблетированную смесь вносят 300 мг бикарбоната, твердость таблетки составляет от около 10 килограмм-силы до около 16 килограмм-силы. В вариантах осуществления, в которых используют 500 мг карбоната, твердость таблетки составляет от около 11 килограмм-силы до около 17 килограмм-силы.
В данной области термин «твердость» обозначает разрушающую нагрузку в диаметральном направлении, измеряемую с помощью стандартного фармацевтического оборудования для определения твердости, такого как устройство для определения твердости Schleuniger. Для сравнения значений в случае таблеток различных размеров необходимо нормализовать разрушающую нагрузку к площади излома. Данное нормализованное значение, выраженное в тыс. ф/см2, в области техники иногда называют пределом твердости таблетки. Общее описание процесса оценки твердости таблетки можно найти в Leiberman et al., Pharmaceutical Dosage Forms--Tablets, Volume 2, 2.sup.nd ed., Marcel Dekker Inc., 1990, pp. 213-217, 327-329.
В других вариантах осуществления таблетки настоящего изобретения распадаются в воде. В этом случае время распада измеряют в ходе испытания на распадаемость и с помощью устройства, описанного в нормативном документе Фармакопеи США (USP) 28, раздел 701, с использованием деионизированной воды при температуре 37°C. Время распадаемости таблеток настоящего изобретения составляет менее 3 минут или менее 2 минут и 30 секунд. В других вариантах осуществления гранулы (внутригранулярная часть) перед сушкой проходят гранулирование и в виде влажного материала пропускаются через мельницу. В других вариантах осуществления дозированная форма может представлять собой таблетку, капсулу, порошок или иную единицу представления. Такие дозированные формы также могут включать в себя внутригранулярную и внегранулярную часть.
Карбонат предпочтительно присутствует в количестве от около 1% масс. до около 75% масс. перорального состава и в количестве, которое будет нейтрализовать от около 0,01 до 10 миллимолей соляной кислоты. Карбонат более предпочтительно присутствует в количестве от около 10% масс. до около 70% масс. перорального состава и в количестве, которое будет нейтрализовать от около 0,02 до 8 миллимолей соляной кислоты. Карбонатный компонент модулирующего рН агента присутствует в пероральном составе в количестве от около 1 мг до около 500 мг или от около 300 мг до 500 мг. Примеры других конкретных количеств карбоната включают от 8 до 850 мг в пероральном составе. Карбонат более предпочтительно присутствует в количестве от около 15 мг до 700 мг.
В одном варианте осуществления перорального состава карбонат представляет собой бикарбонат натрия и/или бикарбонат калия и присутствует в количестве от около 5% масс. до 75% масс. перорального состава.
Водопоглощающий агент может присутствовать в количестве от 5% масс. до 95% масс., или от 10% масс. до 90% масс., или более предпочтительно от 20% масс. до 60% масс. перорального состава и более предпочтительно от 30% масс. до 50% масс. перорального состава.
Соотношение водопоглощающего агента и модулирующего рН агента предпочтительно составляет от 0,1 : 1 до 20 : 1. Соотношение водопоглощающего агента и модулирующего рН агента более предпочтительно составляет от 0,3 : 1 до 15 : 1 или даже более предпочтительно от 0,5 : 1 до 8 : 1 по массе.
Как правило, по меньшей мере 50% терапевтического соединения растворяется из перорального состава в течение 300 секунд в устройстве 2 для растворения согласно USP в присутствии 900 мл 0,0033 н соляной кислоты при 30 об/мин и 37°C. В предпочтительном варианте осуществления по меньшей мере 55% терапевтического соединения растворяется из перорального состава в течение 300 секунд в устройстве 2 для растворения согласно USP в присутствии 900 мл 0,0033 н соляной кислоты при 30 об/мин и 37°C. В другом варианте осуществления по меньшей мере 50% растворяется в течение 240 секунд. В другом варианте осуществления по меньшей мере 75% растворяется из перорального состава в течение 180 секунд в устройстве 2 для растворения согласно USP в присутствии 900 мл фосфатного буфера с рН 7,4 при 50 об/мин и 37°C. В предпочтительном варианте осуществления по меньшей мере 95% высвобождается в течение 300 секунд в устройстве для растворения 2 согласно USP в присутствии 900 мл фосфатного буфера с рН 7,4 при 50 об/мин и 37°C.
Пример 1. Протокол оценки фармакокинетики (ФК) напроксена
Методология
Проводившееся исследование представляло собой открытое рандомизированное перекрестное исследование с однократной дозой, которое включало три части и пять отдельных периодов лечения. Участвовало тридцать здоровых пациент в возрасте от 18 до 55 лет. В исследуемой популяции было представлено не менее приблизительно 40% каждого пола.
Часть 1 исследования представляла собой проводимое натощак трехстороннее перекрестное исследование, в котором все пациенты были рандомизированы в шесть последовательностей курсов приема A, B и C в течение последовательных периодов, в сочетании с одним из курсов приема D и E в части 2 и одним из курсов приема F, G и H в части 3.
В части 1 курсы приема состояли из однократной дозы напроксена натрия в виде исследуемого состава 1 таблетки 220 мг (курс приема А), исследуемого состава 2 таблетки 220 мг (курс приема В) и таблетки Aleve® 220 мг (курс приема С), которые вводили вместе с приблизительно 240 мл воды после ночного голодания в течение по меньшей мере 10 часов.
В части 2 курсы приема представляли собой однократную дозу напроксена натрия в виде таблетки Nalgesin S® 275 мг (курс приема D) или Aleve 220 мг Liquid Gels® (курс приема E). В части 3 курсы приема представляли собой однократную дозу напроксена натрия в виде исследуемого состава 1 таблетки 220 мг (курс приема F), исследуемого состава 2 таблетки 220 мг (курс приема G) или таблетки Aleve® 220 мг (курс приема H) приблизительно через 30 минут после начала завтрака с высоким содержанием жиров. Дозу принимали перорально с приблизительно 240 мл воды.
Введение препарата разделял период вымывания, составляющий по меньшей мере 6 дней. В каждый период исследования для анализа фармакокинетики отбирали 17 проб крови в течение 1 часа до и через 5, 10, 20, 40, 60, 80, 100 минут, а также через 2, 3, 4, 6, 8, 12 и 24, 36 и 48 часов после введения лекарственного средства. Плазму отделяли и количественно определяли напроксен с помощью сертифицированного аналитического метода. Пациенты наблюдались для сообщения о любых нежелательных явлениях, которые могли возникнуть.
Цели
Часть 1. Сравнение биодоступности напроксена натрия при приеме следующих однократных доз:
исследуемый состав 1 таблетки 220 мг по сравнению с таблеткой Aleve® 220 мг при приеме натощак;
исследуемый состав 1 таблетки 220 мг по сравнению с исследуемым составом 2 таблетки 220 мг при приеме натощак; и
исследуемый состав 2 таблетки 220 мг по сравнению с таблеткой Aleve® 220 мг при приеме натощак.
Часть 2. Сравнение биодоступности напроксена натрия при приеме следующей однократной дозы:
исследуемый состав 1 таблетки 220 мг по сравнению с препаратами сравнения (таблетка Nalgesin S® 275 мг и Aleve 220 мг Liquid Gels®) натощак; и
исследуемый состав 2 таблетки 220 мг по сравнению с препаратами сравнения (таблетка Nalgesin S® 275 мг и Aleve 220 мг Liquid Gels®) натощак.
Часть 3. Оценка возможных эффектов приема пищи посредством сравнения биодоступности напроксена натрия при приеме следующих однократных доз:
исследуемый состав 1 таблетки 220 мг после еды по сравнению с исследуемым составом 2 таблетки 220 мг после еды;
исследуемый состав 1 таблетки 220 мг после еды по сравнению с таблеткой Aleve® 220 мг после еды; и
исследуемый состав 2 таблетки 220 мг после еды по сравнению с таблеткой Aleve® 220 мг после еды.
Исследуемые препараты, дозировка и способ введения:
один исследуемый состав 1 таблетки напроксена натрия 220 мг вводили перорально с приблизительно 240 мл воды натощак (курс приема А)
один исследуемый состав 2 таблетки напроксена натрия 220 мг вводили перорально с приблизительно 240 мл воды натощак (курс приема В)
один исследуемый состав 1 таблетки напроксена натрия 220 мг вводили перорально с приблизительно 240 мл воды после еды (курс приема F)
один исследуемый состав 2 таблетки напроксена натрия 220 мг вводили перорально с приблизительно 240 мл воды после еды (курс приема G)
Препараты сравнения, дозировка и способ введения:
одну таблетку Aleve® 220 мг вводили перорально с приблизительно 240 мл воды натощак (курс приема С)
одну таблетку Nalgesin S® 275 мг вводили перорально с приблизительно 240 мл воды натощак (курс приема D)
одну капсулу Aleve 220 мг Liquid Gel® вводили перорально с приблизительно 240 мл воды натощак (курс приема Е)
одну таблетку Aleve® 220 мг вводили перорально с приблизительно 240 мл воды после еды (курс приема Н)
Продолжительность исследования. Продолжительность исследования составила около девяти недель, которые включают продолжительность скрининга на пригодность (от одного до 28 дней до введения первой дозы) и пять отдельных периодов лечения. На протяжении каждого периода лечения пациенты оставались на месте проведения исследования.
Оценка данных. Фармакокинетика.
Для каждого пациента и препарата определяли следующие фармакокинетические (ФК) параметры с помощью некомпартментного анализа: Кмакс (максимальная концентрация в плазме), Вмакс (время до достижения максимальной концентрации) и концентрация напроксена в плазме через 5, 10, 20, 40, 60 и 80 минут (Кп5МИН Кп10МИН, Кп20МИН, Кп40МИН, Кп60МИН и Кп80МИН), λZ (константа скорости) и В1/2 (полувыведение).
Статистические способы
Предлагаемые вычисления размера выборки относятся к части 1 исследования. Если предположить, что интрасубъектный коэффициент вариации (КВ) составляет 13% для Кмакс, то размер выборки из 30 пациентов обеспечивал бы по меньшей мере 90% статистической мощности, чтобы гарантировать, что двусторонний 90% доверительный интервал для соотношения составит 80-125% препарата сравнения, при условии что истинное среднее отношение исследуемого препарата к препарату сравнения составит от 0,89 до 1,12. Оценку 13% интрасубъектного КВ наблюдали в проводившемся ранее исследовании биоэквивалентности напроксена. См. Product monograph ALEVE Liquid Gels Naproxen Sodium Tablets USP 220 mg Non-steroidal anti-inflammatory drug Analgesic, Antipyretic, Bayer Inc. Consumer Care, April 10, 2013. Control No. 162299; и Setiawati et al., Bioequivalence Study with Two Naproxen Sodium Tablet Formulations in Health Subjects, Journal of Bioequivalence & Bioavailability, 1(1): 28-33 (2009).
Анализ для Кмакс, а также концентраций напроксена в плазме через 40, 60 и 80 минут (Кп40 мин, Кп60 мин и Кп80 мин) проводили следующим образом.
Статистические сопоставления пар курсов приемов (A и B, A и C, B и C) опирались на логарифмически преобразованные (натуральный логарифм) данные фармакокинетических параметров. Для оценки средних показателей по методу наименьших квадратов и интрасубъектной дисперсии использовали анализ со смешанными эффектами модели дисперсии, которая в качестве фиксированных эффектов учитывала курс приема, период и последовательность лечения, а пациент в рамках последовательности рассматривался как случайный эффект. Использовали основанные на модели доверительные интервалы 90% для геометрического среднего соотношения Кмакс, соответствующего препарату сравнения.
Анализ проб плазмы
В течение каждого периода исследования в соответствующим образом маркированные пробирки для отбора крови K2EDTA vacutainer® собирали пробы крови (4 мл) для фармакокинетического анализа. Этикетки пробирок содержали следующую информацию (как минимум): номер протокола, идентификационный номер пациента, время отбора проб, период исследования и любой необходимый идентификационный код пробы для конкретного места исследования.
Пробы крови собирали перед введением дозы (до введения дозы) и в определенные моменты времени после введения каждой назначенной дозы. Фармакокинетические пробы собирали в точно заданное время относительно дозирования. Пробы, взятые вне 1-минутного интервала для моментов времени до 60 минут после введения дозы или вне 2-минутного интервала для моментов времени после 60 минут, будут отражены как отклонения от протокола. Точное время отбора проб отражено в исходном документе и средстве сбора данных (например, индивидуальной регистрационной карте).
После забора крови пробирку осторожно переворачивали приблизительно восемь раз после сбора и немедленно помещали в ледяную баню для переноса в центрифугу. Пробы хранили на льду и отделяли плазму в течение 90 минут. Любые отклонения, связанные с пробой крови для фармакокинетики и процессом обработки, регистрировали в соответствующем журнале.
Пробы центрифугировали на высокой скорости (~ 1500 g оборотов в минуту) в течение приблизительно 10 минут при номинальной температуре приблизительно 4 °C. После остановки центрифуги пробы возвращали в ледяную баню. Плазму отбирали двумя разделенными поровну аликвотами в соответствующим образом маркированную полипропиленовую пробирку для транспортировки (полипропиленовая пробирка с нажимной крышкой) (в которую может вмещать приблизительно 5 мл плазмы). На пробирки наносили устойчивые к замораживанию этикетки и/или маркировали их несмываемым маркером, при этом этикетки заполняли и закрепляли их на пробирке до переноса плазмы в пробирку.
Пробы помещали в морозильную камеру (номинальная температура приблизительно
-20 °C) в течение 90 минут с момента отбора и хранили до транспортировки. Время переноса проб в морозильную камеру регистрировали в системе учета проб. Пробы анализировали с использованием сертифицированного аналитического метода в соответствии со стандартными рабочими процедурами биоаналитической лаборатории. Диапазон измерений сертифицированного метода составлял от 0,5 мкг/мл до 100,000 мкг/мл.
Модель анализа результатов
С использованием некомпартментных методов определяли следующие фармакокинетические параметры однократной дозы для напроксена натрия в плазме:
Концентрации в плазме, измеренные через 5, 10, 20, 40, 60 и 80 минут (Кп5МИН, Кп10МИН, Кп20МИН, Кп40МИН, Кп60МИН и Кп80МИН) после введения дозы;
Максимальная концентрация в плазме (Кмакс);
Время до максимальной концентрации (Вмакс);
Полувыведение (В1/2);
Константа скорости выведения (λZ)
Параметры Кмакс для таблетки Nalgesin S® 275 мг (E) были представлены для данных с нормализованной дозой и без нормализованной дозы.
Модель анализа состояний натощак и после еды
Оценивали сравнение возможных эффектов приема пищи, сопоставляя биодоступность напроксена натрия при приеме следующих однократных доз:
исследуемый состав 1 таблетки (А) 220 мг натощак по сравнению с исследуемым составом 1 таблетки (F) 220 мг после еды
исследуемый состав 2 таблетки (В) 220 мг натощак по сравнению с исследуемым составом 2 таблетки (G) 220 мг после еды
таблетка Aleve® 220 мг (C) натощак по сравнению с таблеткой Aleve® 220 мг (H) после еды
исследуемый состав 1 таблетки (F) 220 мг после еды по сравнению с исследуемым составом 2 таблетки (G) 220 мг натощак
исследуемый состав 2 таблетки (G) 220 мг после еды по сравнению с таблеткой Aleve® 220 мг (Н) после еды
исследуемый состав 1 таблетки (F) 220 мг после еды по сравнению с таблеткой Aleve® 220 мг (Н) после еды
Статистические сравнения пар курсов приемов после еды и натощак (F по сравнению с A, G по сравнению с В, H по сравнению с C) для 1 набора параметров ФК анализировали с использованием парного t-критерия для логарифмически преобразованного (натуральный логарифм) фармакокинетического параметра. В каждом случае будут рассчитаны доверительные интервалы 90% для геометрического среднего отношения Кмакс, соответствующего эталонному курсу приема (F по сравнению с A, G по сравнению с B, H по сравнению с C).
Пример 2. Результаты для ФК и биоэквивалентности
Таблица 1. Средние параметры ФК для напроксена при приеме натощак
(300 мг бикарб. нат.) (n=29)
(135,9)
(81)
(39,5)
(0,17-2,00)
(500 мг бикарб. нат.) (n=27)
(16,5)
(138,4)
(76)
(36,2)
(0,17-1,00)
(115,7)
(78,2)
(0,67-3,00)
(177,4)
(335,6)
(0,20-12,00)
(163,1)
(91,8)
(0,67-1,67)
* относ. состава 1 (p < 0,05). Все параметры начальной абсорбции различались статистически значимым образом (p < 0,05) как для состава 1, так и для состава 2 по сравнению с Aleve, Aleve LG и Nalgesin при приеме натощак.
Таблица 2. Средние параметры ФК для напроксена при приеме после еды
(300 мг бикарб. нат.) (n=9)
(11,6)
(60,5)
(54,2)
(46,8)
(0,67-4)
(500 мг бикарб. нат.) (n=8)
(17,5)
(51)
(36,6)
(30,4)
(1,00-6,00)
(16,3)
(127,8)
(118)
(85,6)
(0,67-4,00)
* относ. натощак, † относ. Aleve после еды p < 0,05
Таблица 3. Средние параметры ФК для напроксена при приеме натощак
мл)
ч/мл)
ч/мл)
ч/мл)
ч/мл)
ч/мл)
ч/мл)
(0,17-1,00)
(0,67-3,00)
Таблица 4. Сводные данные о времени достижения эффективной концентрации в плазме крови
Бикарб. нат.: Бикарбонат натрия
* 15-20 мкг/мл считается минимальной эффективной концентрацией напроксена в плазме крови
** ФИГ. 1
*** ФИГ. 2
Пример 3. Композиции для применения в исследовании ФК
Для применения в исследовании ФК были получены следующие составы.
Таблица 5. Состав, включающий 220 мг напроксена натрия, 300 мг бикарбоната натрия (состав 1)
1: производства FMC Corporation под торговым названием Avicel®
2: производства Colorcon Corporation под торговым названием Starch 1500
3: производства Ashland Corporation под торговым названием PVP K29/32
4: производства BASF Corporation под торговым названием Kollidon® CL
5: вода, удаленная при высушивании гранулята
6: производства Evonik Corporation под торговым названием Aerosil®
7: производства Colorcon Corporation под торговым названием Opadry® II
Процедура гранулирования и таблетирования (для состава 1 (таблица 5) и состава 2 (таблица 6))
1. Материал для внутригранулярной части добавляли в гранулятор с высоким усилием сдвига; добавляли очищенную воду.
2. Гранулят выгружали из гранулятора и пропускали через Co-Mil для разделения по размерам, после чего переносили в сушилку с псевдоожиженным слоем.
3. Гранулят высушивали и повторно пропускали через Co-Mil; после чего смешивали с материалами внегранулярной части с образованием готовой смеси.
4. Смесь спрессовывали в таблетки с силой сжатия 18-26 килоньютонов, получая твердость 10-15 килограмм-силы.
5. Таблетки переносили в барабан для нанесения покрытия.
6. Раствор для нанесения пленочного покрытия распыляли на таблетки и высушивали.
Физические параметры внутригранулярной части и бикарбоната натрия
Объемная плотность внутригранулярной (гранулированной) части: 0,6 г/см3 ± 0,05
Плотность утряски внутригранулярной (гранулированной) части: 0,7 г/см3 ± 0,15
Распределение частиц по размерам во внутригранулярной (гранулированной) части по результатам ситового анализа: 222-372 мкм
Размер частиц бикарбоната натрия, USP: 90-95 мкм
Таблица 6. Состав, включающий 220 мг напроксена натрия, 500 мг бикарбоната натрия (состав 1)
1: производства FMC Corporation под торговым названием Avicel®
2: производства Colorcon Corporation под торговым названием Starch 1500
3: производства Ashland Corporation под торговым названием PVP K29/32
4: производства BASF Corporation под торговым названием Kollidon® CL
5: вода, удаленная при высушивании гранулята и нанесении оболочки
6: производства Evonik Corporation под торговым названием Aerosil®
7: производства Colorcon Corporation под торговым названием Opadry® II
Пример 3. Результаты растворения (in vitro)
Составы из таблицы 8 испытывали на растворение с использованием устройства для растворения 2 согласно USP в 900 мл 0,0033 н соляной кислоты при 30 об/мин и 37°C. Пробы отбирали в соответствующие моменты времени и анализировали с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ) на колонке Phenomenex Kinetex С18 (50 мм X 4,6 мм); с подвижной фазой 60 : 40 вода : метанол с 0,1% трифторуксусной кислоты; скорость потока 1,0 мл/мин; вводимый объем 10 мкл; УФ-детектор, установленный на 332 нм; и при температуре колонки 30 °C.
Таблица 7. Растворение в 0,0033 M HCL
Среда: 0,0033 M HCl, 30 об/мин, устройство 2 USP (лопастная мешалка)
Мин=минуты
LG=Liquigel
Составы из таблицы 8 также испытывали на растворение с использованием устройства для растворения 2 согласно USP в 900 мл фосфатного буфера с рН 7,4 при 50 об/мин и 37°C. Данные приведены в таблице 12. Способ растворения (среда, устройство, скорость, температура) был таким же, как и для таблеток напроксена натрия в соответствии с нормативным документом USP 41-NF 36. Способ анализа отобранных проб растворения был таким же, как и для таблицы 11.
Таблица 8. Растворение в фосфатном буфере с pH 7,4
Среда: Буфер с рН 7,4, 30 об/мин, устройство 2 USP (лопастная мешалка)
Мин=минуты
LG=Liquigel
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТИСМЫСЛОВЫЕ КОМПОЗИЦИИ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2009 |
|
RU2739302C2 |
| АНТИСМЫСЛОВЫЕ КОМПОЗИЦИИ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2009 |
|
RU2593939C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ АКОТИАМИДА И ИНГИБИТОРА ПРОТОННОЙ ПОМПЫ | 2019 |
|
RU2820820C2 |
| СПОСОБ ЛЕЧЕНИЯ ДЕФИЦИТА ВИТАМИНА B | 2008 |
|
RU2469728C2 |
| ФАРМАЦЕВТИЧЕСКАЯ ЛЕКАРСТВЕННАЯ ФОРМА ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ ДЛЯ УМЕНЬШЕНИЯ МЕЖИНДИВИДУАЛЬНОЙ ВАРИАБЕЛЬНОСТИ, В ПАРАЦЕТАМОЛ-СОДЕРЖАЩИХ СОСТАВАХ У ПАЦИЕНТА | 2009 |
|
RU2517139C2 |
| ПЕРОРАЛЬНАЯ ДОЗИРОВАННАЯ ФОРМА ГАБАПЕНТИНА ЗАМЕДЛЕННОГО ВЫСВОБОЖДЕНИЯ | 2005 |
|
RU2440112C2 |
| ДОЗИРОВАННАЯ ФОРМА ПАСТИЛКИ | 2018 |
|
RU2771806C2 |
| Аморфная твердая дисперсия, содержащая таксан, таблетка, содержащая такую дисперсию, и способ их получения | 2015 |
|
RU2684632C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ РЕИНА ИЛИ ДИАЦЕРЕИНА | 2008 |
|
RU2484816C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КЕТОРОЛАКА | 2017 |
|
RU2677663C1 |
Улучшенная твердая дозированная форма напроксена с немедленным высвобождением с определенным распределением частиц по размерам во внутригранулярной части и определенным распределением частиц по размерам в карбонатной части, что позволяет напроксену оставаться в растворе и обеспечивает быстрое растворение и быструю абсорбцию напроксена. В настоящем изобретении предложена дозированная форма напроксена, которая при введении в организм человека натощак в течение 10 минут или менее обеспечивает среднюю концентрацию напроксена в плазме крови по меньшей мере 15-20 мкг/мл. В настоящем изобретении предложены способы введения напроксена, направленные на лечение боли. 5 н. и 11 з.п. ф-лы, 2 ил., 8 табл., 3 пр.
1. Способ лечения боли, включающий введение твердой дозированной формы напроксена с немедленным высвобождением человеку натощак, указанная дозированная форма обеспечивает концентрацию напроксена в плазме крови по меньшей мере 15-20 мкг/мл в течение 10 минут или менее, в которой указанная дозированная форма включает внутригранулярную часть, включающую напроксен натрия и внегранулярную часть, включающую карбонатное соединение в количестве от 15 мг до 700 мг.
2. Способ лечения боли, включающий введение твердой дозированной формы напроксена с немедленным высвобождением человеку после еды, указанная дозированная форма обеспечивает концентрацию напроксена в плазме крови по меньшей мере 15-20 мкг/мл в течение 50 минут или менее, в которой указанная дозированная форма включает внутригранулярную часть, включающую напроксен натрия и внегранулярную часть, включающую карбонатное соединение в количестве от 300 мг до 500 мг.
3. Способ лечения боли, включающий введение состава напроксена натрия с немедленным высвобождением пациенту, нуждающемуся в этом, натощак, так чтобы уровень концентрации напроксена в плазме крови включал по меньшей мере 15-20 мкг/мл в течение 10 минут или менее, в котором указанный состав включает внутригранулярную часть, включающую напроксен натрия и внегранулярную часть, включающую карбонатное соединение в количестве от 15 мг до 700 мг.
4. Способ лечения боли, включающий введение состава напроксена натрия с немедленным высвобождением пациенту, нуждающемуся в этом, после еды, так чтобы уровень концентрации напроксена в плазме крови включал по меньшей мере 15-20 мкг/мл в течение 50 минут или менее, в котором указанный состав включает внутригранулярную часть, включающую напроксен натрия и внегранулярную часть, включающую карбонатное соединение в количестве от 300 мг до 500 мг.
5. Способ лечения боли, включающий введение твердой дозированной формы с немедленным высвобождением человеку, включающей внутригранулярную часть и внегранулярную часть, в которой внутригранулярная часть включает напроксен натрия, и внегранулярная часть включает растворимый карбонат, причем средний размер частиц растворимого карбоната составляет от 50 мкм до 200 мкм и причем средний размер частиц внутригранулярной части составляет от 200 мкм до 400 мкм.
6. Способ по п. 1 или 3, в котором дозированная форма или состав обеспечивают концентрацию напроксена в плазме крови по меньшей мере 15-20 мкг/мл в течение 7-9 минут.
7. Способ по п. 2 или 4, в котором дозированная форма или состав обеспечивают концентрацию напроксена в плазме крови по меньшей мере 15-20 мкг/мл в течение 25 минут или менее.
8. Способ по любому из пп. 1-4, в котором средний размер частиц растворимого карбоната составляет от 50 мкм до 200 мкм.
9. Способ по п. 5 или 8, в котором средний размер частиц растворимого карбоната составляет от 75 мкм до 100 мкм.
10. Способ по п. 1, или 3, или 5, в котором количество карбоната в дозированной форме составляет от 300 до 500 мг.
11. Способ по любому из пп. 1-4, в котором внутригранулярная часть включает наполнитель для прессования, связующее вещество и разрыхлитель, и причем средний размер частиц внутригранулярной части составляет от 200 мкм до 400 мкм.
12. Способ по п. 5 или 11, в котором средний размер частиц внутригранулярной части составляет от 200 мкм до 300 мкм.
13. Способ по п. 5, в котором растворимый карбонат выбран из группы, состоящей из карбоната натрия, бикарбоната натрия, карбоната кальция, карбоната магния, карбоната аммония, бикарбоната аммония, бикарбоната калия, глицин-карбоната натрия, глицин-карбоната динатрия, аргинин-карбоната и лизин-карбоната.
14. Способ по п. 5, в котором
(i) по меньшей мере 50% напроксена растворяется из твердой дозированной формы с немедленным высвобождением в течение 300 секунд в устройстве для растворения 2 согласно Фармакопее США (USP) в 900 мл 0,0033 н соляной кислоты при 30 об/мин и 37°С;
(ii) по меньшей мере 50% напроксена растворяется из твердой дозированной формы с немедленным высвобождением в течение 300 секунд в устройстве для растворения 2 согласно USP в 900 мл фосфатного буфера с рН 7,4 при 50 об/мин и 37°С;
(iii) по меньшей мере 75% напроксена растворяется из твердой дозированной формы с немедленным высвобождением в течение 600 секунд в устройстве для растворения 2 согласно USP в 900 мл фосфатного буфера с рН 7,4 при 50 об/мин и 37°С.
15. Способ по п. 14, в котором в подпункте (ii) объемная плотность внутригранулярной части составляет от 0,5 г/см3 до 0,9 г / см3.
16. Способ по п. 5, в котором дозированная форма отличается твердостью от 10 килограмм-силы до 17 килограмм-силы, определенной устройством для определения твердости Schleuniger, или в которой внутригранулярная часть дополнительно включает наполнитель для прессования, связующее вещество и разрыхлитель.
| WO 2000015195 A1, 23.03.2000 | |||
| US 2007134317 A1, 2007.06.14 | |||
| US 2009311327 A1, 2009.12.17 | |||
| WO 2013128858 A1, 2013.09.06 | |||
| US 2012148634 A1, 2012.06.14. |

