ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к фармацевтическим составам, содержащим агонист опиоидных рецепторов в качестве активного ингредиента, к способам их получения и их терапевтическим применениям.
Более конкретно, изобретение относится к составу бупренорфина для буккального/сублингвального введения, обеспечивающему терапевтически эффективную дозировку для лечения абстинентного синдрома у новорожденных.
УРОВЕНЬ ТЕХНИКИ, К КОТОРОМУ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Абстинентный синдром новорожденных (NAS) представляет собой комплекс признаков и симптомов в постнатальном периоде, ассоциированных с внезапной отменой передаваемых от матери опиоидов.
Основные проявления включают повышение тонуса мышц, расстройство вегетативной нервной системы, раздражимость, низкий сосательный рефлекс и снижение набора массы тела.
В эпидемиологических исследованиях пристрастие матери к опиоидам является частым, с токсикологическими признаками употребления приблизительно у 1% рожденных детей (Vega WA et al. N Engl J Med 1993, 16:329(12), 850-854).
Кроме того, согласно Комитету по наркотикам Американской академии педиатрии, NAS возникает у 55%-94% детей, рожденных от матерей с опиоидной зависимостью.
Оптимальный способ лечения NAS еще не разработан.
Конкретные опиоидные средства, которые используют, включают морфина сульфат, морфин в растворе опиума для новорожденных, дезодорированную настойку опиума и метадон.
Описано, что введение композиций на основе морфина длится от 8 до 79 дней с общей продолжительностью приблизительно 30 дней.
Указанная длительность госпитализации является неоптимальной, вследствие препятствования материнской привязанности, возможной внутрибольничной инфекции и использования ресурсов.
Таким образом, существует потребность в усовершенствованных терапевтических средствах, которые могут безопасно снижать продолжительность госпитализации.
Бупренорфин является частичным агонистом опиоидных µ-рецепторов, который все в большей степени применяется для лечения опиоидной зависимости в высоких дозировках, для контроля умеренной острой боли у индивидуумов с непереносимостью опиоидов в низких дозировках и для контроля умеренной хронической боли в еще меньших дозах.
Это лекарственное средство имеет значительный метаболизм первого прохождения у взрослых, и, следовательно, его вводят сублингвальным и буккальным путем.
Бупренорфин имеет ряд характеристик, которые могут сделать его привлекательным средством для лечения NAS. Например, его длительное время полужизни и длительность действия препятствуют быстрому изменению занятости рецепторов, что может устранять симптомы отмены.
Однако опыт применения бупренорфина у новорожденных или в педиатрических популяциях является малым и существует ограниченная информация о применении сублингвального состава у новорожденных.
Более того, терапевтически эффективная дозировка у новорожденных зависит от их массы тела и тяжести злоупотребления матерью; таким образом, составы пленок с фиксированной дозой для сублингвального и буккального введения, такие как составы, коммерчески доступные или описанные в EP 2461795, не могут быть пригодными, поскольку невозможно точно скорректировать требуемое количество лекарственного средства.
Kraft W et al. описали результаты открытых клинических испытаний у новорожденных, касающихся лечения отмены опиатов (Pediatrics 2008, 122(3), e601-607; Addiction 2010, 106, 574-580; N Engl. J Med 2017, 376:2341-2348).
Авторы сделали заключение, что сублингвальный бупренорфин был безопасным с существенным преимуществом в отношении эффективности относительно стандартной терапии пероральным морфином.
Однако использовали состав раствора для приготовления непосредственно перед применением путем смешения бупренорфина гидрохлорида для инъекций в 30% этаноле и простого сиропа из сахарозы. Конечная концентрация составляла 0,075 мг/мл в виде свободного основания бупренорфина.
Вследствие присутствия этанола указанный состав может не быть пригодным для многократного введения новорожденным детям.
Более того, без надлежащей вязкости сублингвальные/буккальные дозированные формы в форме раствора могут доставлять варьирующие количества лекарственного вещества, поскольку они имеют низкое удержание в области применения.
В частности, учитывая объемы, которые, вероятно, будут вводиться, горизонтальное положение новорожденного после дозирования и использование пустышки, существует риск проглатывания лекарственного средства и всасывания в желудочно-кишечном тракте вместе сублингвального пути.
В других документах уровня техники, т.е. EP 0069600, US 2007/117828, US 2016/175295 и WO 03/080022, описаны составы на основе бупренорфина, однако характеристики, позволяющие быть им пригодными для введения новорожденных сублингвальным путем, отсутствуют.
Ввиду вышесказанного, все еще существует потребность в разработке более безопасного и эффективного состава для сублингального введения новорожденным, имеющего достаточный срок годности для хранения и применения.
Было бы преимущественным предоставление состава с достаточной вязкостью для удержания его под языком новорожденного или индивидуума, нуждающегося в лечении, и в тесном контакте со слизистой оболочкой, избегая проглатывания и всасывания через желудочно-кишечный тракт.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В первом аспекте настоящее изобретение относится к готовому для применения фармацевтическому составу в форме водного раствора для сублингвального и буккального введения, причем указанный состав содержит:
i) от 0,005 до 0,02% мас./об. бупренорфина или его фармацевтически приемлемой соли в качестве единственного активного ингредиента;
ii) от 0,5% до 10% мас./об. загустителя; и
iii) буферное вещество в количестве, обеспечивающем pH 5,0-7,0.
Его вязкость должна составлять от 500 до 2300 мПа с, предпочтительно от 700 до 2100 мПа с при 25±2°C.
В предпочтительном варианте осуществления изобретения загуститель представляет собой производное целлюлозы, более предпочтительно гидроксиэтилцеллюлозу (HEC) или натрий карбоксиметилцеллюлозу (NaCMC), еще более предпочтительно гидроксиэтилцеллюлозу.
Во втором аспекте изобретение относится к вышеупомянутому готовому для применения фармацевтическому составу для применения для лечения синдрома отмены опиоидов, предпочтительно синдрома отмены опиоидов у новорожденных.
В третьем аспекте изобретение относится к описанному выше готовому для применения фармацевтическому составу для изготовления лекарственного средства для лечения синдрома отмены опиоидов, предпочтительно синдрома отмены опиоидов у новорожденных.
Четвертый аспект относится к способу лечения синдрома отмены опиоидов у пациента, нуждающегося в этом, путем введения готового для применения состава по изобретению в течение периода, составляющего от 1 до 90 суток, предпочтительно от 3 до 70 суток.
Предпочтительно, пациент представляет собой новорожденного, страдающего синдромом отмены опиоидов у новорожденных.
Пятый аспект относится к способу получения готового для применения состава по изобретению.
Шестой аспект относится к шприцу, предварительно заполненному готовым для применения составом по изобретению.
Седьмой аспект относится к упаковке, содержащей фармацевтический состав по изобретению в форме либо готового для применения водного раствора, либо порошка для восстановления в подходящем водном носителе, в комбинации со шприцом и инструкциями по пероральному введению указанного фармацевтического состава пациенту, страдающему синдромом отмены опиоидов.
ОПРЕДЕЛЕНИЯ
В отношении бупренорфина, термины "лекарственное средство", "активный ингредиент" и "активное вещество" используются взаимозаменяемо.
Термины "новорожденные дети", "новорожденные" и "младенцы" используются взаимозаменяемо.
Термин "безопасный" означает фармацевтический состав, пригодный для сублингвального введения, хорошо переносимый новорожденными и лишенный эксципиентов, которые могут быть вредоносными, антигенными или токсичными для указанной выборки пациентов.
Термин "буккальное и сублингвальное введение" охватывает области слизистых оболочек, т.е. фармакологический путь введения, посредством которого вещества диффундируют в кровь через ткани под языком и через слизистую оболочку полости рта (ткани, которые выстилают полость рта), которые наиболее часто используют для системной доставки лекарственных средств. Буккальный/сублингвальный пути обходят ЖКТ и, таким образом, лекарственные средства, всасывающиеся таким образом, обходят печень и метаболизм первого прохождения и имеют прямой доступ к системному кровотоку.
Термин "pH микроокружения" относится к pH области полости рта пациента, непосредственно окружающей состав.
Для готового для применения состава выражение "физически стабильный" относится к составу, который в условиях длительного хранения (25°C±2°C, относительная влажность 60%±2%) по существу не демонстрирует оседания активного ингредиента и/или эксципиента при хранении в течение по меньшей мере одного месяца.
Вследствие аналитических затруднений, касающихся определения активного ингредиента в низкой концентрации, выражение "химически стабильный" относится к составу, который при хранении демонстрирует изменение содержания бупренорфина не более ±15% и отсутствие связанных с лекарственным средством продуктов деградации в количестве более 5% при хранении в течение по меньшей мере одного месяца.
Термин "биоэквивалентынй" означает достижение 80%-125% величин Cmax и AUC для данного активного ингредиента в другом продукте.
Термин "терапевтически эффективное количество" означает количество активного ингредиента, которое при доставке новорожденным обеспечивает желаемый биологический эффект.
Термин "лечение" относится к терапевтическому применению для паллиативного, излечивающего, облегчающего симптомы, уменьшающего симптомы, индуцирующего регрессию заболевания лечения.
Термин "по существу состоящий из" используют для указания на состав, содержащий только загуститель и буферное вещество в качестве основных эксципиентов. Например, он может содержать подсластитель и/или вкусовую добавку, но не эксципиенты, такие как усилители проницаемости.
ЧЕРТЕЖИ
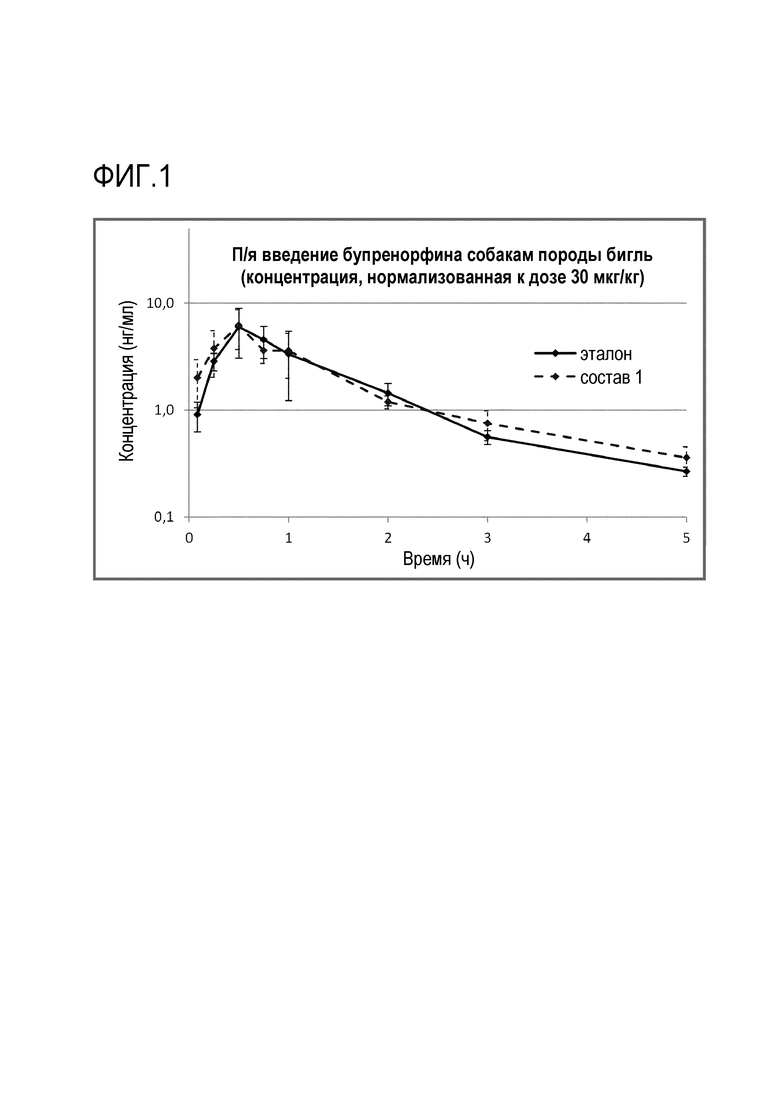
Фиг.1 - Профиль PK при введении состава 1 согласно примеру 1 против эталонного состава.
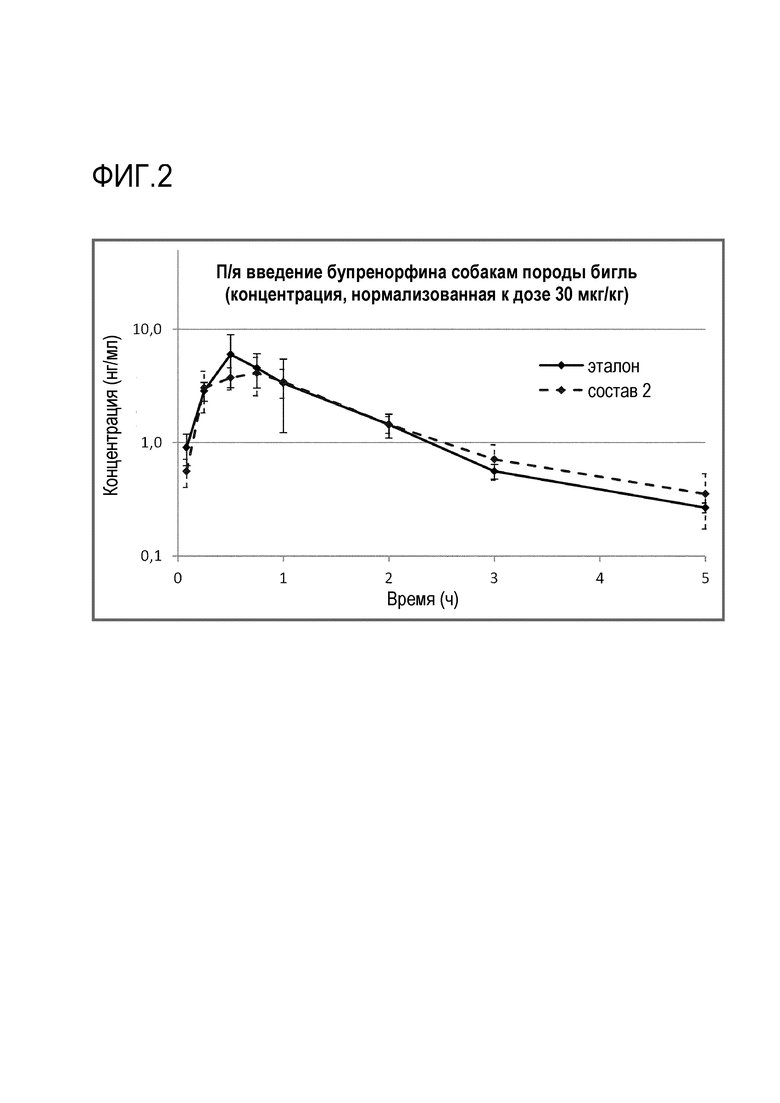
Фиг.2 - Профиль PK при введении состава 2 согласно примеру 1 против эталонного состава.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Благодаря своей активности в отношении опиоидных рецепторов бупренорфин можно успешно применять для лечения абстинентного синдрома, в частности, абстинентного синдрома новорожденных.
Таким образом, целью настоящего изобретения является предоставление безопасного фармацевтического состава для эффективного лечения посредством сублингвального введения пациентам, страдающим синдромом отмены опиатов (OWS), предпочтительно новорожденным, страдающим OWS новорожденных (далее NOWS).
Указанный безопасный состав содержит бупренорфин, растворенный в водном носителе.
Составы по изобретению можно вводить новорожденным, а также детям или подросткам.
Состав по изобретению может иметь форму сухого порошка для растворения непосредственно перед применением или форму готового для применения состава.
В случае выпуска в качестве сухого порошка для перерастворения, его можно получать известными способами, и он может быть предоставлен в качестве набора, содержащего a) порошковый фармацевтический состав; b) фармацевтически приемлемый водный носитель; c) шприц; d) контейнер для фармацевтического состава, водный носитель и шприц.
Предпочтительно используют готовый для применения состав.
Бупренорфин используют в форме основания или в форме фармацевтически приемлемой соли, образованной с неорганической или органической кислотой, такой как хлористоводородная кислота, бромистоводородная кислота, серная кислота, фосфорная кислота, азотная кислота, уксусная кислота, гликолевая кислота, молочная кислота, пировиноградная кислота, малоновая кислота, янтарная кислота, глутаровая кислота, фумаровая кислота, яблочная кислота, миндальная кислота, виннокаменная кислота, лимонная кислота, аскорбиновая кислота, пальмитиновая кислота, малеиновая кислота, гидроксималеиновая кислота, бензойная кислота, гидроксибензойная кислота, фенилуксусная кислота, коричная кислота, салициловая кислота, метансульфоновая кислота, бензолсульфоновая кислота и толуолсульфоновая кислота.
Предпочтительно бупренорфин присутствует в виде соли хлористводородной кислоты.
Преимущественно концентрация активного ингредиента составляет от 0,005 до 0,02% мас./об., предпочтительно от 0,006 до 0,01% мас./об. в расчете на свободное основание.
В конкретном варианте осуществления изобретения концентрация бупренорфина гидрохлорида составляет 0,0075% мас./об. в расчете на свободное основание.
Концентрация загустителя составляет от 0,5% до 10%, мас./об., предпочтительно от 0,6% до 8,0% мас./об. Тип и количество загустителя надлежащим образом выбирают для достижения достаточной вязкости для удержания состава насколько это возможно под языком пациента для минимизации всасывания через желудочно-кишечный тракт.
В то же время вязкость не должна быть слишком высокой, чтобы замедлять высвобождение активного ингредиента из матрикса и, таким образом, его локальное всасывание.
Более предпочтительно, концентрация загустителя может составлять от 1,0 до 6,0% мас./об.
В конкретных вариантах осуществления указанная концентрация составляет 1,0% мас./об., или 1,5% мас./об., или 2,0% мас./об., или 6,0% мас./об.
Преимущественно загуститель может быть выбран из растворимых в воде полисахаридов, таких как альгинаты, каррагенаны, пектин, растворимые в воде производные целлюлозы: алкилцеллюлозы гидроксиалкилцеллюлозы и гидроксиалкилалкилцеллюлозы, такие как метилцеллюлоза, гидроксиметилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилцеллюлоза, гидроксиэтилметилцеллюлоза, гидроксипропилметилцеллюлоза, гидроксибутилметилцеллюлоза, сложные эфиры целлюлозы и сложные эфиры гидроксиалкилцеллюлозы, такие как целлюлозы ацетат фталат (CAP), гидроксипропилметилцеллюлоза (HPMC); карбоксиалкилцеллюлозы, карбоксиалкилалкилцеллюлозы, сложные эфиры карбоксиалкилцеллюлозы, такие как карбоксиметилцеллюлоза и их соли щелочных металлов; растворимые в воде синтетические полимеры, такие как полиакриловые кислоты и сложные эфиры полиакриловых кислот, полиметакриловые кислоты и сложные эфиры полиметакриловых кислот, поливинилацетаты, поливинилспирты, поливинилацетатфталаты (PVAP), поливинилпирролидон (PVP) и поликротоновые кислоты; также пригодны фталированный желатин, желатина сукцинат, сшитый желатин, шеллак, растворимые в воде химические производные крахмала, катионно-модифицированные акрилаты и метакрилаты, обладающие, например, третичной или четвертичной аминогруппой, такой как диэтиламиноэтиловая группа, которая при желании может быть кватернизированной.
Предпочтительно загуститель представляет собой растворимое в воде производное целлюлозы, выбранное из группы, состоящей из гидроксиэтилцеллюлозы (HEC) или соли карбоксиметилцеллюлозы (CMC) с щелочным металлом, такой как соль натрия.
В действительности, загустители, принадлежащие указанным классам, могут обеспечить подходящую вязкость, в то время как в случае других средств класса камедей, таких как ксантановая смола, вязкость состава оказалась слишком высокой.
Преимущественно вязкость состава при 25±2°C составляет от 500 до 2300 мПа с (1 мПа с соответствует 1 сантипуазу), предпочтительно от 700 до 2100 мПа с. Вязкость можно определять любым способом, например, с использованием реометра.
Преимущественно pH состава по изобретению может составлять от 5,0 до 7,0, более преимущественно от 5,2 до 6,8, предпочтительно от 5,5 до 6,5.
В действительности, было обнаружено, что при pH выше 7,0 составы по изобретению могут демонстрировать более низкую химическую и физическую стабильность.
В противоположность тому, что описано в EP 2461795, было обнаружено, что указанный интервал pH, препятствующий преципитации активного ингредиента в слюне, способствует его всасыванию.
Без связи с теорией, это может быть следствием того, что не происходит сдвига в отношении pH микроокружения (поверхность контакта слюна/слизистая оболочка).
Результаты теста мукоагезии также продемонстрировали, что составы по изобретению наделены оптимальным свойством с точки зрения вязкости, чтобы позволить его удержание под языком пациента без замедления высвобождения активного ингредиента из матрикса, в частности, когда растворимое в воде производное целлюлозы используют в качестве загустителя.
Более того, в соответствии с экспериментами проницаемости in vitro, когда используют растворимое в воде производное целлюлозы, предпочтительные составы по изобретению имеют pH от 5,5 до 6,5 и количество загустителя, составляющее от 1,0% до 2,0% мас./об., еще более предпочтительно 1,5% мас./об. Предпочтительным загустителем этого класса может быть гидроксиэтилцеллюлоза. Указанный эксципиент является коммерчески доступным от Natrosol 250 HXTM.
Тесты проницаемости in vitro имитируют прохождение лекарственного средства через слизистую оболочку полости рта и позволяют определить скорость высвобождения.
Можно использовать любое буферное вещество, способное обеспечивать вышеупомянутое значение pH, например, фосфатные или цитратные буферы, такие как соли натрия или калия. Специалист в данной области способен определить надлежащее количество.
В предпочтительном варианте осуществления изобретения в качестве буферного вещества используют безводную лимонную кислоту и безводный цитрат натрия.
Состав по изобретению также может содержать другие эксципиенты, такие как вкусовые добавки и/или подсластители.
Вкусовые добавки могут быть выбраны из природных и синтетических ароматизирующих жидкостей. Иллюстративный перечень таких средств включает летучие масла, синтетические ароматизированные масла, вкусовые ароматизаторы, масла, жидкости, олеосмолы или экстракты из растений, листьев, цветков, плодов, стеблей и их комбинации. Репрезентативный перечень примеров включает мятные масла, какао и цитрусовые масла, такие как масла лимона, апельсина, винограда, лайма и грейпфрута, и фруктовые эссенции, включая вкусовые добавки яблока, груши, персика, винограда, клубники, малины, вишни, сливы, ананаса, абрикоса и других плодов.
Другие пригодные вкусовые добавки включают альдегиды и сложные эфиры, такие как бензальдегид (вишня, миндаль), цитраль, т.е. альфа-цитраль (лимон, лайм), нераль, т.е. бета-цитраль (лимон, лайм), деканаль (апельсин, лимон), альдегид C-8 (цитрусовые фрукты), альдегид C-9 (цитрусовые фрукты), альдегид C-12 (цитрусовые фрукты), толилальдегид (вишня, миндаль), 2,6-диметилоктанол (зеленые фрукты) и 2-додеканаль (цирусы, мандарин), их комбинации и т.п.
Подсластители могут быть выбраны из следующего неограничивающего перечня: глюкоза (кукурузный сироп), декстроза, инвертированный сахар, фруктоза и их комбинации; сахарин и его различные соли, такие как соль натрия; дипептидные подсластители, такие как аспартам; соединения дигидрохальконов, глицирризин; Stevia rebaudiana (стевиозид); производные сахарозы с хлором, такие как сукралоза; сахарные спирты, такие как сорбит, маннит, ксилит и т.п. Также предусматриваются гидролизаты гидрогенизированного крахмала и синтетический подсластитель 3,6-дигидро-6-метил-1-1-1,2,3-оксатиазин-4-он-2,2-диоксид, в частности, его соль калия (ацесульфам-K), и соли натрия и кальция. Также можно использовать другие подсластители.
Как правило, специалист в данной области способен выбрать подсластитель и/или вкусовую добавку среди тех, которые считаются безопасными для введения новорожденным.
Хотя и не предпочтительно, составы по изобретению также могут содержать усилители всасывания, такие как пропиленгликоль и производные полиоксил гидрогенизированного касторового масла, например, полиоксил 40 гидрогенизированного касторового масла (коммерчески доступного как Kolliphor RH 40TM).
В предпочтительном варианте осуществления изобретения состав имеет следующую композицию: 0,05-0,01% мас./об. бупренорфина гидрохлорида в расчете на свободное основание, 1,5% мас./об. гидроксиэтилцеллюлозы, 0,12% мас./об. безводной лимонной кислоты, 1,13% мас./об. безводного цитрата натрия, воды для инъекций, и его pH составляет 6,0±0,3.
В альтернативном предпочтительном варианте осуществления изобретения состав может иметь следующую композицию: 0,05-0,01% мас./об. бупренорфина гидрохлорида в расчете на свободное основание, 6,0% мас./об. натрий карбоксиметилцеллюлозы, 0,12% масс./об. безводной лимонной кислоты, 1,13% мас./об. безводного цитрата натрия, вода для инъекций, и ее pH составляет 6,0 ± 0,3.
Как более подробно описано в разделе "Примеры" ниже в модели на животных для сублингвального введения водные составы, имеющие вышеупомянутые композиции, оказались биоэквивалентными спиртовому составу, описанному в Kraft et al., Pediatrics 2008, 122(3), e601-607.
Не ограничиваясь теорией, это оказалось довольно неожиданным, поскольку этанол обычно способствует всасыванию.
Таким образом, в соответствии с предпочтительным вариантом осуществления изобретения, состав состоит только из фармацевтически приемлемой соли бупренорфина в качестве единственного активного ингредиента, загустителя, буферного вещества в надлежащем количестве для обеспечения pH от 5 до 7,0, и необязательно вкусовой добавки и/или подсластителя.
Состав по изобретению можно получать известными способами.
В конкретном варианте осуществления составы по изобретению получают посредством следующих стадий:
i) растворение подходящего количества бупренорфина или его фармацевтически приемлемой соли, предпочтительно его гидрохлорида, в воде в надлежащем контейнере для получения концентрированного прозрачного раствора;
ii) необязательно стерилизация раствора, полученного на стадии i), посредством фильтрации;
iii) параллельно растворение надлежащего количества буферного вещества в воде в подходящем мерном контейнере до получения прозрачного раствора;
iv) необязательно стерилизация раствора, полученного на стадии iii), путем нагревания;
v) добавление надлежащего объема концентрированного раствора бупренорфина к раствору буферного вещества для получения конечной желаемой концентрации бупренорфина, предпочтительно 0,05-0,01 мг/мл (в расчете на свободное основание), смешивая при непрерывном перемешивании;
vi) медленное добавление надлежащего количества загустителя к раствору согласно стадии iii) при постоянном перемешивании до тех пор, пока загуститель не растворится полностью и не будет получен прозрачный однородный раствор.
Предпочтительно, вода представляет собой воду для инъекций (WFI).
На стадии i) концентрация может варьироваться от 0,1 до 0,5 мг/мл, предпочтительно от 0,2 до 0,4 мг/мл, более предпочтительно она составляет 0,324 мг/мл.
Преимущественно контейнер на стадиях i) и iii) может быть изготовлен из любого подходящего материала, такого как пластмасса или стекло.
Процедуры стерилизации стадии ii) и iv) можно проводить способами, известными в данной области.
В частности, пористость фильтра на стадии ii) и температуру нагревания на стадии iv) могут быть надлежащим образом скорректированы специалистом в данной области.
Конечный состав можно распределять в асептических условиях в подходящие контейнеры.
Преимущественно состав по изобретению можно использовать для лечения абстинентного синдрома новорожденных любой тяжести.
Предпочтительно, состав по изобретению можно использовать для лечения пациентов, страдающих синдромом отмены опиоидов (OWS), с воздействием или без воздействия других лекарственных средств, более предпочтительно, для лечения новорожденных, страдающих NOWS.
Дозу и длительность лечения в любом случае будет корректировать лечащий врач в зависимости от массы тела новорожденного и тяжести абстинентного синдрома новорожденного.
Как правило, при использовании для лечения NOWS, состав наливают под язык шприцом, а затем в рот новорожденного вставляют пустышку для уменьшения глотания.
Можно использовать любой шприц, доступный в продаже, имеющий объем от 0,1 до 2,5 мл, более преимущественно от 0,5 до 2,0 мл.
Предпочтительно можно использовать шприцы объемом 0,5 мл или 1,0 мл.
Шприц может быть изготовлен из пластмассы, стекла или любого подходящего материала, предпочтительно из пластмассы, более предпочтительно из цикло-олефинового полимера (COP).
Например, могут быть пригодными шприцы от Becton Dickinson (New Jersey, США).
В предпочтительном варианте осуществления изобретения готовый для применения состав может быть предоставлен в виде предварительно заполненного шприца, предпочтительно в COP, и без наконечника Люэра.
Например, подходящие предварительно заполняемые шприцы коммерчески доступны от Gerresheimer AG (Dusseldorf, Германия).
При использовании для новорожденных указанные предварительно заполняемые шприцы должны быть без иглы, и они должны быть снабжены подходящей крышкой, предпочтительно достаточно большой, чтобы избежать обструкции у новорожденного в случае случайного проглатывания.
В альтернативном варианте осуществления может быть предоставлена упаковка, содержащая фармацевтический состав по изобретению в форме либо готового для применения водного раствора, либо порошка для восстановления в подходящем водном носителе, в комбинации с подходящим шприцом.
Изобретение иллюстрируется следующими примерами.
ПРИМЕРЫ
Пример 1
Целью первой группы экспериментов была оценка возможности получения составов водного раствора бупренорфина с достаточной вязкостью и pH, чтобы они оказались физически и химически стабильными при сохранении хорошего профиля всасывания.
Получали следующие составы:
*в расчете на основание.
Составы, аналогичные составу 1, получали с использованием 0,5%, 1,0% и 2,0% мас./об. гидроксиэтилцеллюлозы (состав 1’, состав 1", состав 1"’, соответственно).
Соответствующие составы получали в концентрации бупренорфина HCl 0,01 мг/мл в расчете на свободное основание.
Более того, получали эталонный состав в этаноле согласно идеям Kraft W et al. Pediatrics 2008, 122(3), e601-607.
Составы 1, 2, 3, 4 и 5 демонстрируют вязкость при 25±2°C в диапазоне 700-2100 мПа с (сП), в то время как состав 6 демонстрирует более высокую вязкость, т.е. 2500 мПа с (сП). В водном растворе последний состав образует что-то типа "мягкого геля", который сохраняется при условии, что систему не трогают.
Не было возможным определить вязкость эталонного состава в соответствии с Kraft, поскольку реометр, использованный для определения вязкости составов 1, 2, 3, 4, 5 и 6, не пригоден для жидкости, имеющей вязкость, сравнимую с ньютоновской жидкостью.
Пример 2
Составы 1, 2, 3 и 4 примера 1 при концентрации бупренорфина 0,01% мас./об. хранили в течение одного месяца в условиях длительного хранения, т.е. 25±2°C и относительная влажность 60%, и в ускоренных условиях, т.е. при 50±2°C, для оценки их физической и химической стабильности.
Стабильность также тестировали в условиях охлаждения.
Анализ бупренорфина и его примесей проводили посредством ВЭЖХ. Также тестировали следующие параметры: pH, вязкость и внешний вид.
Результаты в условиях длительного хранения представлены в таблице 1.
Таблица 1 - Стабильность составов 1, 2, 3 и 4 в соответствии с примером 1 в условиях длительного хранения.
(мПа с)
% против первоначального уровня
Результаты при 50±2°C представлены в таблице 2.
Таблица 2 - Стабильность составов 1, 2, 3 и 4 в соответствии с примером 1 при 50±2°C.
(мПа с)
Результаты, приведенные в таблицах 1 и 2, демонстрируют, что не наблюдается значительного повышения общего уровня продуктов деградации для составов 1 и 2 при pH=6,0, и их количество сохраняется значительно ниже предела 5,0%.
Примечательно, что уровень продуктов деградации возрастает после 4 недель при 50°C для состава 4, в то время как состав 3 оказался мутным, что указывает на то, что pH выше 7 может быть не оптимальным для химической и физической стабильности.
Пример 3
Составы 1 и 2 при концентрации бупренорфина 0,075 мг/мл в расчете на основание и эталонный состав вводили собакам посредством сублингвального введения.
Использовали трех самцов собак породы бигль, и им проводили все варианты введения с периодом вымывания, составляющим одну неделю. Составы вводили путем распределения дозы (максимум 2,5 мл) под язык в сублингвальную область, а затем удержания рта закрытым в течение приблизительно 45 с. Эту процедуру повторяли до тех пор, пока не вводили полный объем дозы (0,4 мл/кг).
Концентрации бупренорфина в плазме собак определяли с использованием валидированного биоаналитического способа ВЭЖХ-MS/MS, и их использовали для сравнения системной экспозиции для трех различных составов.
Как показано на фиг.1, состав 1 и эталонный состав продемонстрировали максимальную концентрацию в одно и то же время и после нормализации дозы имели практически равную максимальную концентрацию и AUC.
Состав 2 и эталонный состав сравнены на фиг.2: время максимальной концентрации в среднем наблюдалось несколько позднее для состава 2 (0,75 против 0,5 часов), и максимальная концентрация и AUC были несколько более низкими, чем для эталонного состава. Средняя относительная биодоступность составов 1 и 2 по сравнению с эталоном составляла 110% и 102%, соответственно.
Пример 4
Это исследование было предназначено для скрининга эффективности in vitro составов 1, 2, 3, 4 и 5 против эталонного состава согласно Kraft.
Методика эксперимента
Купрофан (мембраны на основе целлюлозы) содержит глицерин в качестве увлажнителя (для предупреждения высыхания и растрескивания). Его удаляли путем замачивания в дистиллированной воде. Мембраны (подготавливали предварительно вырезанные круги диаметром 22 мм) помещали в 500-мл дюрановую бутылку для замачивания и содержимое перемешивали путем вращения (20 об./мин). Для устранения глицерина проводили две дополнительных полных замены воды. Общее время замачивания/ополаскивания составляло ~46 часов перед испытанием высвобождения.
Мембраны купрофан устанавливали в качестве барьера между половинами индивидуально пронумерованных и слабо смазанных (смазка для высокого вакуума, Dow Corning, США) горизонтальных диффузионных ячеек Франца, с роговым слоем, обращенным к донорной камере. Площадь, доступная для диффузии, составляла приблизительно 1,2 см2, причем точная площадь и объем (в среднем ~3,4 мл) предварительно измерялись для каждой индивидуальной диффузионной ячейки.
Рецепторные камеры диффузионных ячеек заполняли известным объемом с уравновешенной температурой 1% (масс./об.) Brij O20 в pH 7,4 фосфатно-солевом буфере (BPBS) и закрывали. Диффузионные ячейки погружали в водяную баню постоянной температуры, поддерживая рецепторные камеры при 37,0±0,5°C в ходе эксперимента. Содержимое рецепторных камер непрерывно перемешивали посредством небольших покрытых PTFE магнитных элементов, управляемых погружными магнитными мешалками. Ячейкам позволяли уравновеситься перед дозированием (≥75 минут).
Тестируемые составы наносили на поверхность мембраны в дозе 500 мкл/см2 с использованием пипетки прямого вытеснения Microman M1000. Следили за тем, чтобы достигнуть полного покрытия поверхности без воздушных пузырьков). Донорные камеры накрывали с использованием Parafilm (Nesco) сразу после дозирования. Проводили три повторения для каждого активного вещества и было шесть контрольных ячеек, в каждую из которых дозировали отдельный состав плацебо. Отмечали точное время нанесения, и это время соответствовало нулевому моменту времени для этой ячейки. Мониторинг времени дозирования проводили очень тщательно вследствие частых моментов времени взятия образцов.
Дозы наносимых составов были равны 37,5 мкг/см2 бупренорфина HCl для всех ячеек активного вещества.
Проводили взятие 100 мкл образцов (с использованием цифровой пипетки) из каждой рецепторной камеры через 15 и 30 минут после дозирования. Удаленную жидкость заменяли только взятия образца через 30 минут. Образцы рецепторной фазы объемом 200 мкл получали через 60 и 120 минут после дозирования. Каждый образец помещали в предварительно маркированный 200-мкл конический стеклянный флакон (Agilent, Cheadle, Великобритания) и сразу накрывали крышкой (покрытая PTFE мембрана) перед замораживанием флакона (-20°C) до анализа.
После извлечения образцов рецепторной фазы объемом 200 мкл через 120 минут оставшуюся рецепторную фазу удаляли и помещали в 20-мл стеклянные флаконы (Chromacol, Великобритания), которые закрывали крышкой и замораживали (-20°C) для валидации анализа.
Анализ с использованием ультравысокоэффективной жидкостной хроматографии (УВЭЖХ) с УФ-детекцией использовали для анализа образцов в отношении содержания бупренорфина.
Образцы рецепторной фазы (в 200-мкл флаконах, хранившиеся при -20°C) подготавливали для анализа следующим образом. Флаконам позволяли разморозиться при комнатной температуре перед тщательным встряхиванием. Затем флаконы центрифугировали (3 минуты при 10000 об./мин.) и переносили в автоматический пробоотборник.
Выход бупренорфина в образцах рецепторной фазы через 2 ч оценивали посредством исследований с добавлением. С использованием совокупной рецепторной фазы (хранившейся в 20-мл флаконах), в образцы (одна ячейка из каждой группы активного вещества и контрольной группы) добавляли небольшую аликвоту калибровочного раствора (добавляемая концентрация 2,00 мкг/мл). После анализа образцов с добавлением и образцов без добавление вычисляли выход бупренорфина после вычитания вклада реципентной фазы. Средний выход при добавлении составлял 100,2% (0,79% RSD, n=17) и сходная величина была получена при добавлении только BPBS.
Результаты приведены в таблице 3.
Таблица 3 - Данные о проницаемости бупренорфина HCl (мкг/см2, среднее значение ± SE, n=3)
Как можно видеть, скорости высвобождения были лучшими для составов 1 и 3, чем для составов 2 и 4, имеющих pH выше 7,0.
Высвобождение из состава 1, содержавшего HEC, было более быстрым, чем как для состава 3, содержавшего NaCMC, так и для эталонного состава Kraft.
Включение усилителя всасывания значительно снижало высвобождение.
Пример 5 - Оценка мукоадгезивных свойств
Мукоадгезивные свойства составов по изобретению тестировали с использованием устройства "с наклонной плоскостью", как описано в Pescina, S., et al. Drug Dev Ind Pharm, 2017. 43(9): p. 1472-1479.
Для эксперимента использовали составы 1, 1’, 1" и 1"’ примера 1.
Приблизительно 30 мг, точно взвешенные, каждого состава наносили на эпителий пищевода (1,13 см2), предварительно приклеенный на чашку Петри (9 см2), наклоненную на 45°C. Состав промывали жидкостью, имитирующей слюнную жидкость (SSF, NaCl 8 г/л, Na2HPO4 2,38 г/л, KH2PO4 0,19 г/л, pH 6,8) со скоростью потока 1 мл/мин, с использованием шприцевого насоса.
Поддерживали температуру 25±2°C.
Раствор собирали в заданные моменты времени и инжектировали в ВЭЖХ для оценки смытого лекарственного средства.
Результаты представлены в таблице 4, где описан эффект вязкости (выраженный в качестве % загустителя) на % смытого бупренорфина со слизистой оболочки.
Вязкость состава, по-видимому, является критичной только при 0,5% (когда после 1 мин 60% состава смывалось со слизистой оболочки), в то время как составы, содержавшие 1%, 1,5% и 2,0% загустителя, продемонстрировали хорошие мукоадгезивные свойства.
Таблица 4
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ АТРОПИНА | 2018 |
|
RU2745603C1 |
| СОСТАВ ФЕНФЛУРАМИНА, СОВМЕСТИМЫЙ С КЕТОГЕННОЙ ДИЕТОЙ | 2018 |
|
RU2778848C2 |
| Фармацевтическая композиция на основе трамадола для офтальмологического применения | 2016 |
|
RU2744570C2 |
| СУБЛИНГВАЛЬНЫЕ ТАБЛЕТКИ С НИЗКИМИ ДОЗИРОВКАМИ ОПИОИДНЫХ АНАЛЬГЕТИКОВ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2007 |
|
RU2444360C2 |
| ЖИДКИЙ НАЗАЛЬНЫЙ СПРЕЙ, СОДЕРЖАЩИЙ НАЛТРЕКСОН В НИЗКОЙ ДОЗЕ | 2011 |
|
RU2552786C2 |
| СОСТАВЫ ДЛЯ ПАРЕНТЕРАЛЬНОЙ ДОСТАВКИ СОЕДИНЕНИЙ И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2539387C2 |
| СПОСОБ ПРЕДОТВРАЩЕНИЯ ЗЛОУПОТРЕБЛЕНИЯ СОДЕРЖАЩИМИ ОПИОИДЫ ЛЕКАРСТВЕННЫМИ ФОРМАМИ | 1998 |
|
RU2228180C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ПРИМЕНЕНИЯ ХОЛИНОВОГО ЭФИРА ЛИПОЕВОЙ КИСЛОТЫ | 2015 |
|
RU2735790C2 |
| СУБЛИНГВАЛЬНЫЕ КОМПОЗИЦИИ ДЕКСМЕДЕТОМИДИНА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2572692C2 |
| ПРИМЕНЕНИЕ МЕТИЛНАЛТРЕКСОНА ДЛЯ ЛЕЧЕНИЯ СИНДРОМА РАЗДРАЖЕННОГО КИШЕЧНИКА | 2004 |
|
RU2373936C2 |
Группа изобретений относится к области медицины и раскрывает готовый для применения фармацевтический состав в форме водного раствора для сублингвального и буккального введения, причем указанный состав содержит: 0,0075% мас./об. бупренорфина гидрохлорида в расчете на основание, 1,5% мас./об. гидроксиэтилцеллюлозы или 6,0% мас./об. натрий карбоксиметилцеллюлозы, 0,12% мас./об. безводной лимонной кислоты, 1,13% мас./об. безводного цитрата натрия, воды для инъекций. Также группа изобретений относится к способу получения готового для применения состава, к шприцу, предварительно заполненному составом и к упаковке, содержащей состав. Техническим результатом заявленной группы изобретений является безопасность и эффективность состава для сублингального введения новорожденным, имеющего достаточный срок годности для хранения и применения. При этом состав обладает достаточной вязкостью для удержания его под языком новорожденного или индивидуума, нуждающегося в лечении, и в тесном контакте со слизистой оболочкой, избегая проглатывания и всасывания через желудочно-кишечный тракт. 4 н. и 8 з.п. ф-лы, 5 табл., 2 ил., 5 пр.
1. Готовый для применения фармацевтический состав в форме водного раствора для сублингвального и буккального введения, причем указанный состав содержит:
0,0075% мас./об. бупренорфина гидрохлорида в расчете на основание, 1,5% мас./об. гидроксиэтилцеллюлозы или 6,0% мас./об. натрий карбоксиметилцеллюлозы, 0,12% мас./об. безводной лимонной кислоты, 1,13% мас./об. безводного цитрата натрия, воды для инъекций.
2. Состав по п.1 , где pH составляет от 5,2 до 6,8.
3. Состав по п.1 или 2, где pH составляет от 5,5 до 6,5.
4. Состав по любому из предшествующих пунктов, дополнительно содержащий подсластитель и/или вкусовую добавку.
5. Способ получения готового для применения состава по любому из пп.1-4, включающий стадии:
i) растворения подходящего количества бупренорфина или его гидрохлорида в воде в надлежащем контейнере для получения концентрированного прозрачного раствора;
iii) параллельно, растворения надлежащего количества буферного вещества в воде в подходящем мерном контейнере до тех пор, пока не будет получен прозрачный раствор;
iv) добавления надлежащего объема концентрированного раствора бупренорфина к раствору буферного вещества для достижения конечной желаемой концентрации бупренорфина при смешении посредством непрерывного перемешивания;
vi) медленного добавления надлежащего количества загустителя в раствор стадии iii) при постоянном перемешивании до тех пор, пока загуститель не растворится полностью и не будет получен однородный раствор.
6. Способ по п.5, дополнительно включающий стадию:
ii) стерилизации раствора, полученного на стадии i), фильтрованием.
7. Способ по п.5 или 6, дополнительно включающий стадии:
v) стерилизации раствора, полученного на стадии iii), путем нагревания.
8. Состав по любому из пп.1-4 для применения для лечения синдрома отмены опиоидов.
9. Состав по п.8, где синдром представляет собой синдром отмены опиоидов новорожденных.
10. Шприц, предварительно заполненный составом по любому из пп.1-4.
11. Упаковка, содержащая состав по любому из пп.1-4, в форме готового для применения водного раствора и инструкции по пероральному введению указанного фармацевтического состава пациенту, страдающему синдромом отмены опиоидов.
12. Упаковка по п.11, где пациентом является новорожденный, страдающий синдромом отмены опиоидов новорожденных.
| Способ получения расширяющегося цемента | 1946 |
|
SU69600A1 |
| WALTER K | |||
| KRAFT, et al., Sublingual Buprenorphine for Treatment of the Neonatal Abstinence Syndrome: A Randomized Trial, Pediatrics | |||
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| US 2007117828 A1, 24.05.2007 | |||
| US 2016175296 A1, 23.06.2016 | |||
| US 20050085440 A1, 21.04.2005 | |||
| WO 2012169934 A1, 13.12.2012. | |||

