Область техники, к которой относится изобретение
Настоящее изобретение в целом относится к области синтеза полипептидов и, конкретнее, к линейному жидкофазному подходу к гексапептиду WNT (от англ. wingless - бескрылый и integrated - интегрированный) Foxy-5. Настоящее изобретение, кроме того, относится к жидкофазному синтезу новых три-, тетра- и пентапептидных фрагментов Foxy-5.
Предшествующий уровень техники изобретения
Foxy-5 представляет собой формилированный, полученный из WNT5A гексапептид и миметик WNT-5A с потенциальной противометастатической активностью, который в настоящее время разрабатывается в качестве лекарственного средства-кандидата для предупреждения распространения опухоли при некоторых распространенных формах рака.
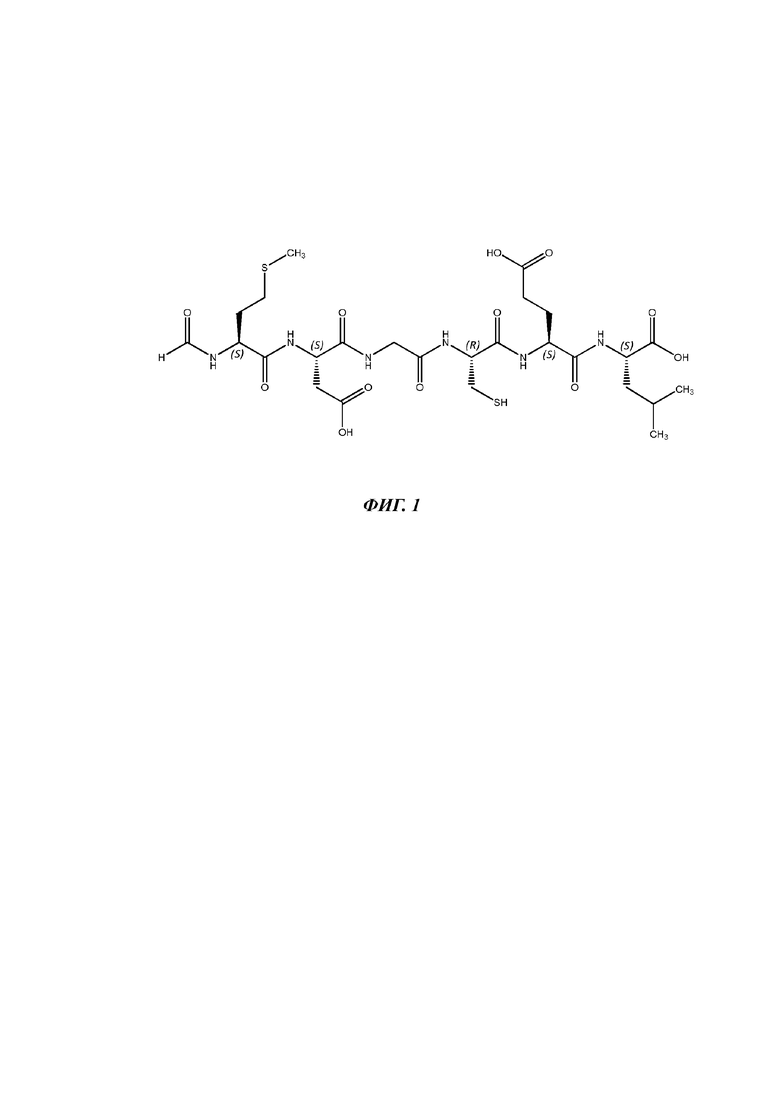
Foxy-5 характеризуется аминокислотной последовательностью For-Met-Asp-Gly-Cys-Glu-Leu-OH (SEQ ID NO 1, фигура 1).
При внутривенном введении Foxy-5 связывается с рецепторами WNT5A и активирует их, преимущественно с рецепторами из семейства Frizzled, которое активирует опосредованную WNT5A передачу сигнала.
Foxy-5 предназначен для компенсации дефицита белка WNT5A в опухолевой ткани, отмечаемого у пациентов с раком толстой кишки, с целью снижения риска метастазирования. Субанализ недавнего ретроспективного исследования пациентов с колоректальным раком на стадии III показывает, что доля пациентов с низкой экспрессией WNT5A значительно выше доли, наблюдаемой в предыдущих исследованиях у пациентов с колоректальным раком (KPP) стадии II. Пациенты с опухолями КРР стадии III отличаются от таковых со стадией II главным образом наличием опухолевых клеток в лимфатических узлах, соседних с первичной опухолью, благодаря чему эти опухоли более агрессивны и быстрее прогрессируют. Низкий уровень WNT5A наблюдался почти у 70 процентов пациентов на стадии III по сравнению с приблизительно 45 процентами пациентов с менее запущенными стадиями опухоли. Это подтверждает гипотезу о том, что уровень WNT5A существенно влияет на течение заболевания.
На основании завершенного исследования фазы 1b с Foxy-5, направленного на регистрирование профиля безопасности, фармакокинетических показателей и определения дозы лекарственного средства-кандидата для фазы 2, Foxy-5 в настоящее время представлен для исследования в фазе 2 клинического испытания, в котором лечение пациентов с раком толстой кишки будут начинать во время постановки диагноза до осуществления хирургического вмешательства. Лечение рассчитано максимум на 12 недель или до начала химиотерапии.
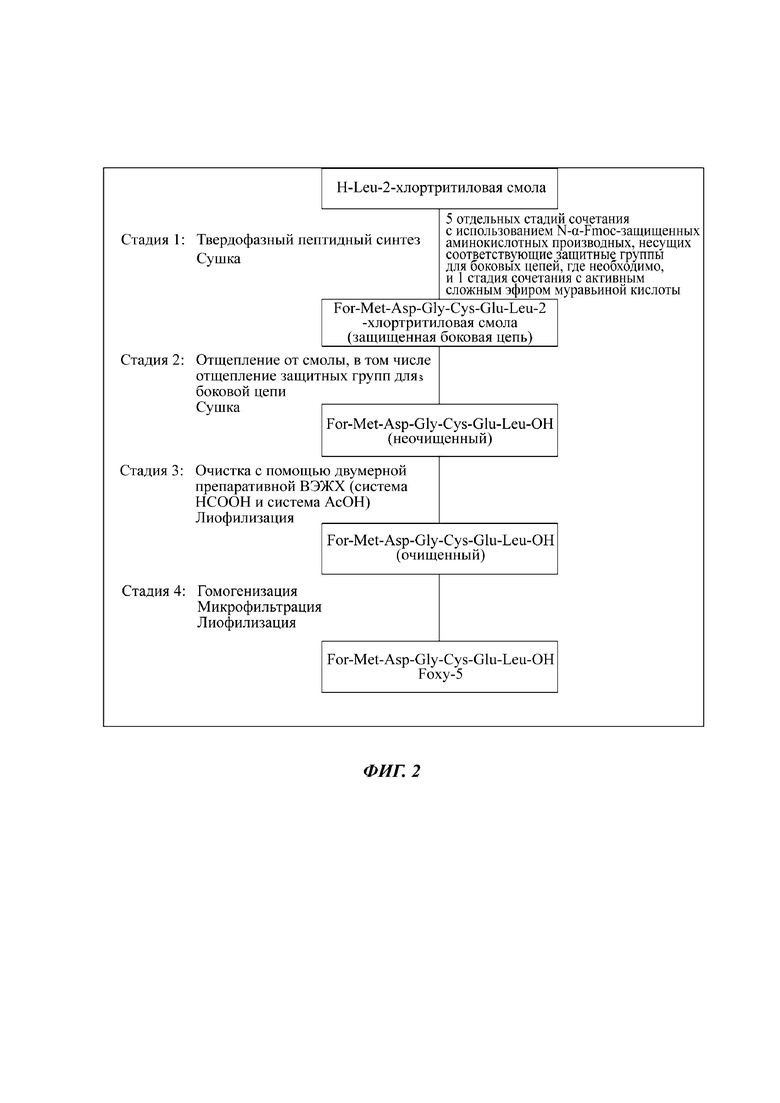
Foxy-5 и способ его получения описаны в публикации международной заявки на патент №WO 06130082 А1. Активный фармацевтический ингредиент АФИ для доклинических и клинических исследований, проводимых до настоящего времени, получали путем классического твердофазного пептидного синтеза (SPPS, от англ. solid phase peptide synthesis), при котором Foxy-5 получается линейным путем «1 плюс 1 плюс 1 плюс 1 плюс 1 плюс 1», см. фигуру 2.
Последовательность For-Met-Asp-Gly-Cys-Glu-Leu-OH, таким образом, собирается на 2-хлортритильной смоле, несущей С-концевую аминокислоту Leu, с использованием Fmoc-стратегии (от англ. Fluorenylmethoxycarbonyl флуоренилметилоксикарбонил). Синтез проводится в реакторе для SPPS и состоит из чередующихся процедур сочетания, ацетилирования и снятия защитной группы N-α. Сочетание проводят в N,N-диметилформамиде (ДМФ) или в ДМФ/дихлорметане (ДХМ) в качестве растворителя. Оно состоит из сочетания производного N-a-защищенной аминокислоты с предыдущей аминокислотой в присутствии активирующего реагента и основания, при необходимости. Муравьиную кислоту сочетают в виде активного сложного эфира без активирующих средств.
Если сочетание не завершено, его можно продолжить или повторить процедуру. Чтобы избежать образования последовательностей с делецией в качестве побочных продуктов, проводят систематическую процедуру ацетилирования (кэппинга) после стадии сочетания или, если проводят повторное сочетание, после стадий повторного сочетания, с использованием ДМФ, уксусного ангидрида и пиридина.
За ацетилированием следует процедура снятия защитной группы N-α, что предусматривает промывку смолы с помощью ДМФ, отщепление Fmoc-группы пиперидином в ДМФ и последующие промывки с помощью ДМФ. В случае неполного расщепления процедуру снятия защитной группы N-α, описанную выше, можно повторять. Для каждой отдельной стадии добавляют растворители и/или реагенты и реакционную смесь перемешивают, а затем фильтруют для удаления растворителей и/или реагентов из смолы.
Процедуры сочетания, ацетилирования и снятия защиты N-α повторяют до тех пор, пока смола не будет нести полную пептидную последовательность For-Met-Asp-Gly-Cys-Glu-Leu-OH. После окончательного сочетания активного сложного эфира муравьиной кислоты ацетилирование не проводят.SPPS завершают промывкой пептидной смолы с помощью ДМФ и изопропилового спирта (ИПС) и последующей сушкой при пониженном давлении.
Отщепление пептида от смолы и сопутствующее отщепление защитных групп боковой цепи осуществляют путем обработки пептидной смолы с помощью трифторуксусной кислоты (ТФУ) в присутствии подходящих акцепторов (например, воды и 1,2-этандитиола (EDT, от англ. - ethanedithiol)). Затем полученный неочищенный пептид очищают с помощью двумерной препаративной высокоэффективной жидкостной хроматографии (ВЭЖХ) на колонке с обращенной фазой при градиентном элюировании ацетонитрилом (АЦН) (системы муравьиной кислоты и уксусной кислоты).
Объединенные фракции надлежащей чистоты лиофилизируют. Лиофилизат анализируют с помощью ВЭЖХ и необязательно повторно очищают с помощью двумерной препаративной ВЭЖХ, как указано выше, в случае несоответствия установленным критериям чистоты.
С помощью описанного выше подхода SPPS было создано достаточно материала для доклинического и раннего клинического исследований, но для дальнейших клинических исследований и возможных коммерческих целей требуется синтез, лучше подходящий для крупномасштабного синтеза, с помощью которого можно снизить стоимость товаров и сделать доступными более крупные партии Foxy-5.
Таким образом, существует потребность в надежном способе синтеза, который может обеспечить Foxy-5 в масштабе нескольких килограммов, как для дальнейшего клинического испытания, так и для возможных коммерческих целей.
Краткое описание графических материалов
На фигуре 1 показана химическая структура Foxy-5. Foxy-5 представляет собой неразветвленный пептид, состоящий из шести аминокислот с формилированным N-концом. Все оптически активные аминокислотные остатки находятся в L-кон фигурации. Молекулярная формула Foxy-5 представляет собой C26H42N6O12S2, а молекулярная масса составляет 694,8 г/моль (средняя масса).
На фигуре 2 показана схема синтеза для способа SPPS для Foxy-5.
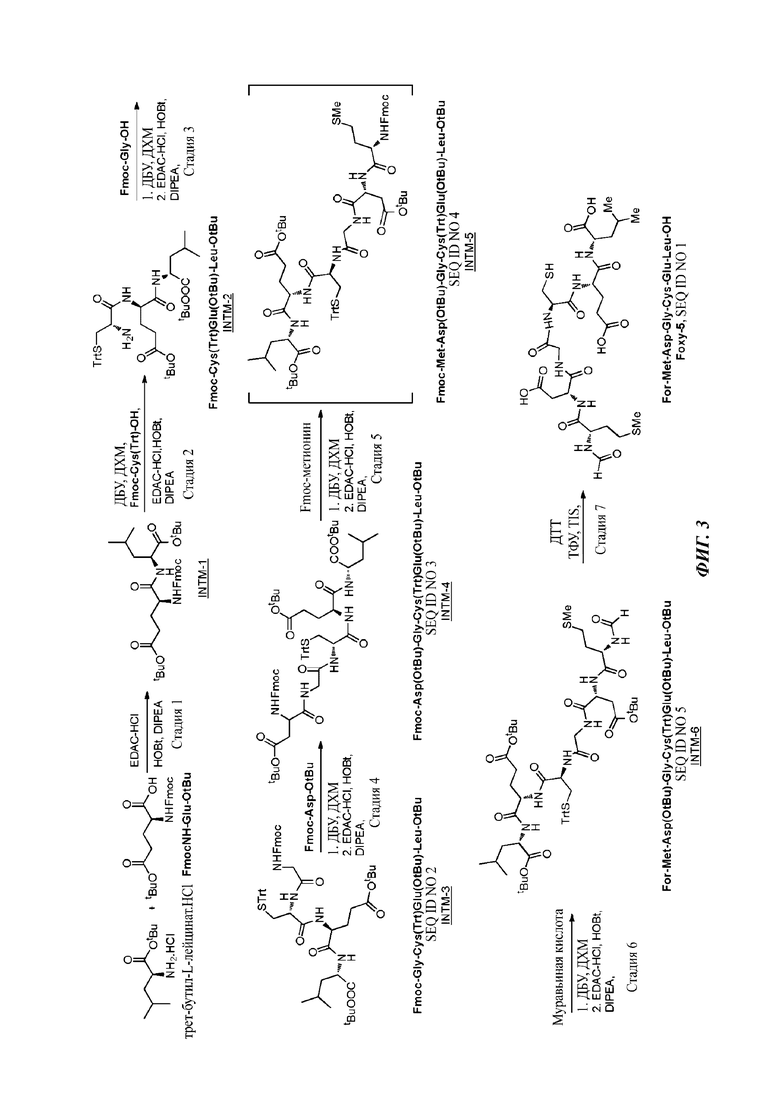
На фигуре 3 проиллюстрирована линейная стратегия «1 плюс 1 плюс 1 плюс 1 плюс 1 плюс 1» для образования Foxy-5.
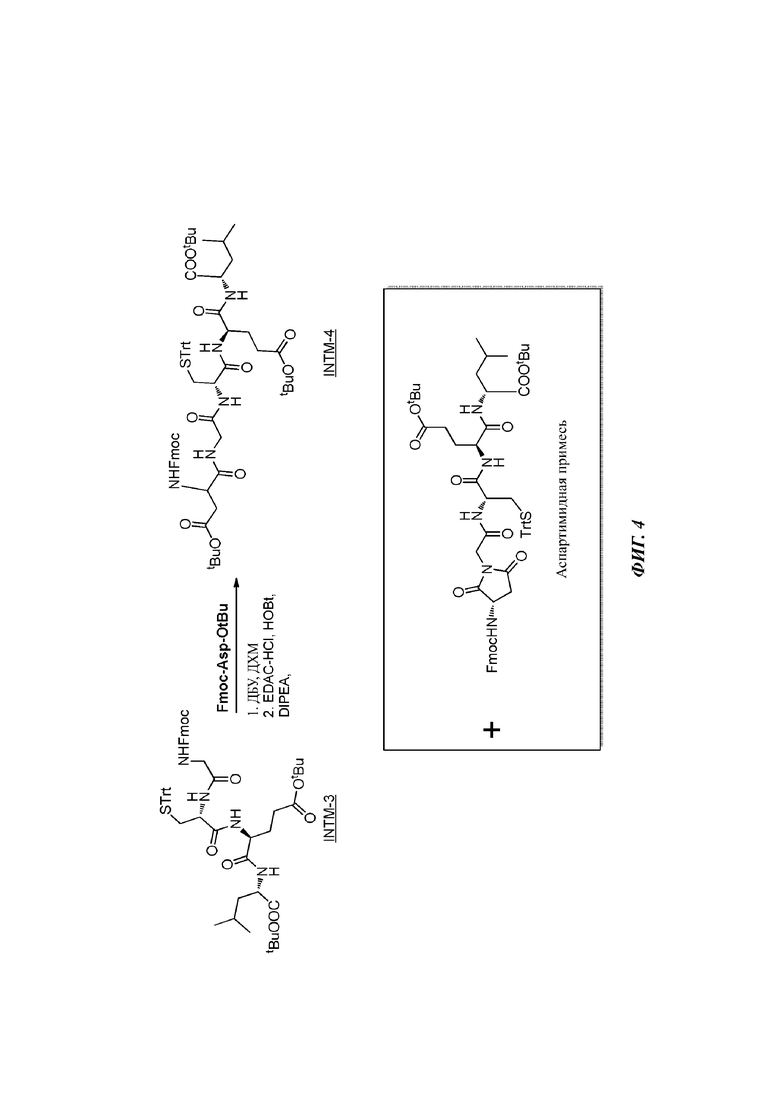
На фигуре 4 проиллюстрировано образование «аспартимидной примеси».
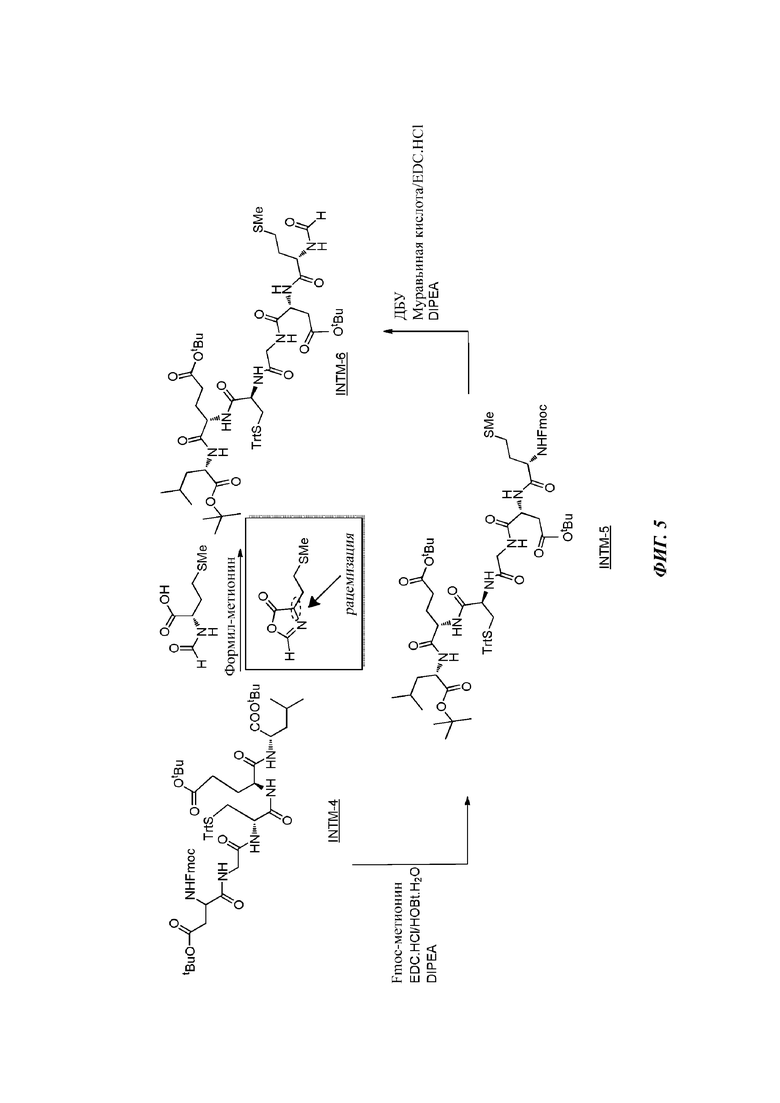
На фигуре 5 проиллюстрирована роль формильной группы в наблюдаемой реакции эпимеризации.
Аббревиатуры
Fmoc - флуо ре нил метокси карбон ил
Tfa (от англ. trifluoroacetyl) -трифторацетил
Tsoc (от англ. 4-toluenesulfonylethyloxycarbonyl) - 4-толуолсульфонилэтилоксикарбонил
Mesoc (от англ. methylsylfonylethyloxycarbonyl - метилсульфонилэтилоксикарбонил
Реос (от англ. 2-(triphenylphosphono)ethyloxycarbonyl) - 2-(трифенилфосфоно)этилоксикарбонил
Суос (от англ. 2-cyano-t-butyloxycarbonyl) - 2-циано-трет-бутилоксикарбонил и
Pht (от англ. phthalyl) - фталил
Nsc (от англ. 2-(4-nitrophenylsulfonyl)ethoxycarbonyl) - 2-(4-нитрофенилсульфонил)этоксикарбонил
Вос (от англ. tert-butyloxycarbonyl) - трет-бутилоксикарбонил For (от англ. formyl) - формил
Trt (от англ. triphenylmethyl (trityl)) - трифенилметил (тритил)
tBu (от англ. ferf-butyl) - трет-бутил
ТГФ - тетрагидрофуран
ДИПЭ = диизопропиловый эфир
ДМФ - N,N-диметилформамид
ТФУ - трифторуксусная кислота
TIS (от англ. triisopropylsilane) - триизопропилсилан
HOBt (от англ. 1-hydroxybenzotriazole) - 1-гидроксибензотриазол
ДХМ - дихлорметан
EDAC (от англ. 1 -ethyl-3-(3'-dimethylaminopropyl)carbodiimide).HCl) - 1-этил-3-(3'-диметиламинопропил)карбодиимид.HCl
DIPEA (от англ. diisopropylamine) - диизопропиламин ДБУ -1,8-диазабицикло[5,4,0]ундец-7-ен
Аббревиатуры аминокислот
Met - метионин
Asp - аспарагин
Gly - глицин
Cys - цистеин
Glu - глутаминовая кислота
Leu - лейцин
Foxy-5 - For-Met-Asp-Gly-Cys-Glu-Leu
Краткое раскрытие изобретения
Настоящее изобретение относится к линейному жидкофазному способу получения формилированного гексапептида, известного как «Foxy-5» (т.е. For-Met-Asp-Gly-Cys-Glu-Leu-OH, (SEQ ID NO 1)), а также к его различным три-, тетра-, пента- и гексапептидным фрагментам, в том числе его защищенным производным. Способ, представленный в настоящем документе, обладает многими преимуществами над традиционными твердофазными синтезами, включая без ограничения низкую стоимость сырьевых материалов, простоту очистки промежуточных соединений процесса, простоту сборки фрагментов, высокую хиральную чистоту и адаптируемость к коммерческому масштабу, что будет раскрыто более подробно ниже.
Таким образом, основная цель настоящего изобретения заключается в обеспечении масштабируемого пути синтеза Foxy-5. Следующая цель заключается в идентификации и характеристике подходящих ключевых промежуточных соединений для указанного масштабируемого пути синтеза с целью последующего изготовления лекарственного вещества согласно GMP (от англ. Good Manufacturing Practice -надлежащей производственной практике).
Ввиду стоимости продукции и трудоемкого масштабирования, обычно связанного с твердофазной химией, основное внимание уделялось разработке жидкофазных химических путей. Настоящее изобретение относится к линейному («1 плюс 1 плюс 1 плюс 1 плюс 1 плюс 1») жидкофазному подходу для получения Foxy-5 или его промежуточных соединений и предшественников.
В отличие от традиционных, конвергентных жидкофазных подходов для гексапептидов, при которых отдельно получают ди- или трипептиды с последующим сочетанием, авторы настоящего изобретения неожиданно обнаружили, что линейный путь, при котором пептидную последовательность Foxy-5 собирают с помощью последовательного жидкофазного сочетания защищенных производных аминокислот Met, Asp, Gly, Cys, Glu и Leu, может быть осуществлен очень эффективно.
Основное решение настоящего изобретения заключается во введении формильной (For) группы в N-концевой метионин. Как будет обсуждаться далее в настоящей заявке, эту конкретную химическую стадию было сложнее всего осуществить с хорошим выходом и высокой химической и оптической чистотой.
Авторы настоящего изобретения обнаружили, что гексапептидное производное формулы PG-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OR может отлично служить в качестве предшественника Foxy-5, и предпочтительно, если R выбран из С1-С6алкила, такого как метил или трет-бутил, и PG представляет собой чувствительную к основанию защитную группу для азота, т.е. защитную группу, которая является стабильной в кислотных условиях, но которая может быть отщеплена от пептида в щелочных/основных условиях. В контексте настоящего изобретения подходящими являются защитные группы, такие как Fmoc, Tfa, Tsoc, Mesoc, Peoc, Суос или Nsc, предпочтительно Fmoc.
Таким образом, согласно первому аспекту настоящее изобретение относится к гекса пептид ному производному формулы PG-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OR, в котором R выбран из С1-С6алкила, такого как метил или трет-бутил, и PG представляет собой чувствительную к основанию защитную группу, такую как Fmoc, Tfa, Tsoc, Mesoc, Peoc, Суос или Nsc.
Гексапептидное производное согласно первому аспекту можно превратить в желаемый гексапептид Foxy-5 путем снятия защиты с N с последующим сочетанием с муравьиной кислотой и, наконец, снятием общей защиты в виде остальных защитных групп.
Таким образом, согласно второму аспекту настоящее изобретение относится к способу получения гексапептида Foxy-5 (SEQ ID NO 1), при этом способ включает:
а. обеспечение гексапептидного производного согласно первому аспекту,
b. удаление защитной группы PG из указанного гексапептидного производного,
c. сочетание полученного продукта стадии b) с муравьиной кислотой или ее активным сложным эфиром с получением защищенного производного Foxy-5 For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(aBu)-Leu-OR,
d. снятие общей защиты с полученного защищенного производного Foxy-5 стадии с) с получением Foxy-5 в неочищенной форме,
e. необязательно выполнение дополнительных стадий очистки и
f. необязательно осаждение образованного гексапептида Foxy-5 в виде щелочной или кислой соли в твердой форме,
при этом R выбран из С1-С6алкила, такого как метил или трет-бутил, и PG представляет собой чувствительную к основанию защитную группу, такую как Fmoc, Tfa, Tsoc, Mesoc, Peoc, Суос или Nsc.
Гексапептидное производное согласно первому аспекту может быть получено любым способом превращения, таким как твердофазный синтез или жидкофазные способы, и, что наиболее удобно, разработанным авторами настоящего изобретения способом, раскрываемым в настоящем документе ниже.
Согласно третьему аспекту настоящее изобретение относится к способу получения гексапептидного производного согласно первому аспекту, включающему следующие стадии:
a. обеспечения защищенного L-лейцинового производного PG-Leu-OR,
b. удаления защитной группы PG из указанного защищенного L-лейцинового производного с последующим сочетанием с PG-Glu-OtBu с получением защищенного дипептида PG-Glu(OtBu)-Leu-OR,
c. удаления защитной группы PG из указанного защищенного дипептид с последующим сочетанием с PG-Cys(Trt)-OH с получением защищенного трипептида PG-Cys(Trt)-Glu(OtBu)-Leu-OR,
d. удаления защитной группы PG из указанного защищенного трипептида с последующим сочетанием с PG-Gly-OH с получением защищенного тетрапептида PG-Gly-Cys(Trt)-Glu(OtBu)-Leu-OR,
e. удаления защитной группы PG из указанного защищенного тетрапептида в основных условиях с последующим удалением избытка основания с получением разблокированного тетрапептида H-Gly-Cys(Trt)-Glu(OtBu)-Leu-OR,
f. сочетания указанного разблокированного тетрапептида с PG-Asp(OtBu) в основных условиях с последующим удалением избытка основания с получением защищенного пентапептида PG-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OR,
g. удаления защитной группы PG из указанного разблокированного пентапептида с последующим сочетанием с муравьиной кислотой или ее активным сложным эфиром с получением защищенного гексапептида PG-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(0tBu)-Leu-OR,
при этом R выбран из С1-С6алкила, такого как метил или трет-бутил, и PG представляет собой чувствительную к основанию защитную группу, такую как Fmoc, Tfa, Tsoc, Mesoc, Peoc, Суос или Nsc.
Далее будут дополнительно обсуждаться раскрываемые выше аспекты настоящего изобретения, в том числе его предпочтительные варианты осуществления.
Согласно четвертому аспекту настоящего изобретения раскрывается способ получения гексапептида Foxy-5 в защищенной форме на основе последовательного сочетания трипептидного промежуточного соединения INTM-2 Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu с защищенными производными аминокислот Gly, Asp и Met, с последующим снятием защиты с N и сочетанием с муравьиной кислотой.
Согласно пятому аспекту настоящего изобретения раскрывается способ получения гексапептида Foxy-5 в защищенной форме на основе последовательного сочетания тетрапептидного промежуточного соединения INTM-3 Fmoc-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu с защищенными производными аминокислот Asp и Met, с последующим снятием защиты с N и сочетанием с муравьиной кислотой.
Согласно следующим аспектам настоящего изобретения раскрываются следующие пептиды:
Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu,
Fmoc-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 2),
Fmoc-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 3),
Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 4),
For-Met-Asp(aBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 5),
Cys-Glu-Leu,
Gly-Cys-Glu-Leu (SEQ ID NO 6),
Asp-Gly-Cys-Glu-Leu (SEQ ID NO 7),
Met-Asp-Gly-Cys-Glu-Leu (SEQ ID NO 8),
Подробное раскрытие изобретения
Как упоминалось в приведенном выше кратком описании, разработали линейный жидкофазный подход для сборки гексапептидной последовательности Foxy-5, который далее будет обсуждаться более подробно.
Общий подход заключался в защите всех аминокислот в виде производных О-tBu, N-Fmoc. Более того, целью был синтез, который был максимально сокращен, что позволило избежать времязатратного и дорогостоящего выделения промежуточных соединений. Предпочтительно, чтобы каждую стадию в последовательности можно было выполнять без выделения продукта, допуская лишь незначительные стадии очистки, такие как водная обработка растворами органических растворителей и обработки с помощью пробки оксида кремния («флэш-хроматография»), например, для удаления избытка основания.
Линейный синтез начинают с получения в две сокращенных стадии (стадия 1 плюс 2) трипептида INTM-2 Fmoc-Cys (Trt)Glu(OtBu)-Leu-OtBu путем сочетания трет-бутил-Ьлейцината.HCl с Fmoc-Glu(OtBu) в дихлорметане (ДХМ) в качестве реакционного растворителя. Реакционную смесь обрабатывают водой и солевым раствором и раствор ДХМ используют непосредственно на стадии 2 без выделения промежуточного дипептида INTM-1 Fmoc-Glu(OtBu)Leu-OtBu.
На стадии 2 осуществляют реагирование раствора INTM-1 в ДХМ с Fmoc-Cys(Trt)-OH с получением желаемого трипептида INTM-2 Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu. Реакции протекают хорошо и повторно дают общий выход приблизительно 68% за две стадии. INTM-2 может быть очищен хроматографией на силикагеле (100-200) в виде твердого вещества белого цвета. Полученный раствор в ДХМ также можно оставлять как есть и использовать непосредственно на стадии 3 в сочетании с Fmoc-Gly-OH.
На стадии 3 INTM-2 подвергают снятию защиты с Fmoc с помощью ДБУ и сочетают с Fmoc-Gly-OH в ДХМ с получением тетрапептида INTM-3 Fmoc-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu. После завершения реакции слой ДХМ промывают водой и солевым раствором. Конечный органический слой затем концентрируют от 50 объемов до 10-15 объемов и оставляют как есть для следующей стадии (стадии 4) без выделения.
На стадии 4 раствор INTM-3 в ДХМ сначала обрабатывают с помощью ДБУ для осуществления снятия защиты с группы Fmoc. После разблокировки реакционную массу пропускают через колонку с пробкой из оксида кремния для удаления ДБУ перед сочетанием тетрапептида с Fmoc-Asp-OtBu в присутствии EDC.HCI, гидрата HOBt и DIPEA. После завершения реакции реакционную массу снова пропускают через колонку с пробкой из оксида кремния для удаления все еще присутствующего DIPEA. Удаление ДБУ и DIPEA, как описано, необходимо для подавления образования нежелательной примеси аспартимида, см. фигуру 4. При выполнении данного способа наблюдали выходы приюлизительно 60% INTM-4 Fmoc-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu за две стадии (стадия 3 плюс 4) в масштабе 100-160 г. В присутствии ДБУ наблюдаемый выход составлял всего приблизительно 25%.
На стадии 5 INTM-4 подвергают снятию защиты с Fmoc с помощью ДБУ в ДХМ в качестве растворителя и сочетают с Fmoc-метионином с получением промежуточного соединения INTM-5 Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 4) в неочищенной форме. Очистку проводят колоночной хроматографией с использованием ДХМ/ТГФ в качестве элюента. Очищенный продукт суспендируют в ДИПЭ с получением твердого вещества белого цвета.
Первоначально была предпринята попытка выполнить сочетание INTM-4 непосредственно с N-формилметионином (For-Met-OH) для получения гексапептида INTM-6, но обнаружили, что эта стратегия синтеза приводит к частичной эпимеризации в конечном продукте, поскольку For-Met-OH обратимо циклизируется в условиях реакции с образованием оксазолидона, который вызывает рацемизацию реагента For-Met-OH. См. фигуру 5. Напротив, использование Fmoc-метионина с последующим снятием защиты с помощью ДБУ с группы Fmoc и сочетанием с муравьиной кислотой или ее активным сложным эфиром приводит к желаемому промежуточному гексапептиду INTM-6. Общее снятие его защиты (групп Tit и O-tBu) дает Foxy-5 в неочищенной форме, которая может быть далее очищена, например, с помощью хроматографии, и/или осаждена в виде твердого вещества, такого как соль присоединения кислоты или щелочи.
Таким образом, согласно первому аспекту настоящее изобретение относится к гексапептидному производному формулы PG-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OR, в котором R выбран из С1-С6алкила, такого как метил или трет-бутил, и PG представляет собой чувствительную к основанию защитную группу, такую как Fmoc, Tfa, Tsoc, Mesoc, Peoc, Суос или Nsc.
Согласно варианту осуществления первого аспекта PG представляет собой Fmoc.
Согласно другому варианту осуществления в первом аспекте раскрывается гексапептидное производное формулы Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OR, в котором R выбран из С1-С6алкила, такого как метил или трет-бутил.
Согласно предпочтительному варианту осуществления в первом аспекте гексапептидное производное характеризуется формулой Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(0tBu)-Leu-OtBu (SEQ ID NO 4).
Согласно второму аспекту настоящее изобретение относится к способу получения гексапептида Foxy-5 (SEQ ID NO 1), при этом способ включает:
g. обеспечение гексапептидного производного согласно первому аспекту,
h. удаление защитной группы PG из указанного гексапептидного производного,
i. сочетание полученного продукта стадии b) с муравьиной кислотой или ее активным сложным эфиром с получением защищенного производного Foxy-5 For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(aBu)-Leu-OR,
j. снятие общей защиты с полученного защищенного производного Foxy-5 стадии с) с получением Foxy-5 в неочищенной форме,
k. необязательно выполнение дополнительных стадий очистки и
I. необязательно осаждение образованного гексапептида Foxy-5 в виде щелочной или кислой соли в твердой форме,
при этом R выбран из С1-С6алкила, такого как метил или трет-бутил, и PG представляет собой чувствительную к основанию защитную группу, такую как Fmoc, Tfa, Tsoc, Mesoc, Peoc, Суос или Nsc.
Согласно варианту осуществления второго аспекта PG представляет собой Fmoc.
Согласно другому варианту осуществления второго аспекта алкильная группа R представляет собой трет-бутил.
Согласно другому варианту осуществления второго аспекта сочетание с муравьиной кислотой или ее активным сложным эфиром осуществляют в растворе.
Согласно другому варианту осуществления второго аспекта полученный неочищенный Foxy-5 очищают с помощью хроматографии, такой как хроматография с обращенной фазой.
Согласно другому варианту осуществления второго аспекта полученный Foxy-5 осаждают в виде щелочной или кислой соли в твердой форме.
Согласно другому варианту осуществления второго аспекта полученный Foxy-5 выделяют в виде кристаллической щелочной или кислой соли.
Согласно третьему аспекту настоящее изобретение относится к способу получения гексапептидного производного согласно первому аспекту, включающему следующие стадии:
n. обеспечения защищенного L-лейцинового производного PG-Leu-OR,
i. удаления защитной группы PG из указанного защищенного L-лейцинового производного с последующим сочетанием с PG-Glu-OtBu с получением защищенного дипептида PG-Glu(OtBu)-Leu-OR,
j. удаления защитной группы PG из указанного защищенного дипептид с последующим сочетанием с PG-Cys(Trt)-OH с получением защищенного трипептида PG-Cys(Trt)-Glu(OtBu)-Leu-OR,
k. удаления защитной группы PG из указанного защищенного трипептида с последующим сочетанием с PG-Gly-OH с получением защищенного тетрапептида PG-Gly-Cys(Trt)-Glu(OtBu)-Leu-OR,
I. удаления защитной группы PG из указанного защищенного тетрапептида в основных условиях с последующим удалением избытка основания с получением разблокированного тетрапептида H-Gly-Cys(Trt)-Glu(OtBu)-Leu-OR,
m. сочетания указанного разблокированного тетрапептида с PG-Asp(OtBu) в основных условиях с последующим удалением избытка основания с получением защищенного пентапептида PG-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OR,
n. удаления защитной группы PG из указанного разблокированного пентапептида с последующим сочетанием с муравьиной кислотой или ее активным сложным эфиром с получением защищенного гексапептида PG-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(0tBu)-Leu-OR,
при этом R выбран из С1-С6алкила, такого как метил или трет-бутил, и PG представляет собой чувствительную к основанию защитную группу, такую как Fmoc, Tfa, Tsoc, Mesoc, Peoc, Суос или Nsc.
Согласно варианту осуществления третьего аспекта PG представляет собой Fmoc.
Согласно варианту осуществления третьего аспекта все стадии b)-g) выполняют в растворе.
Согласно следующему варианту осуществления гексапептидное производное первого аспекта получают способом согласно третьему аспекту.
Согласно предпочтительному варианту осуществления третьего аспекта чувствительная к основанию защитная группа PG представляет собой Fmoc, а алкильная группа R представляет собой трет-бутил.
Согласно другому варианту осуществления третьего аспекта по меньшей мере две последовательных стадии сочетания, например, две, три или четыре стадии, выполняют без выделения продукта.
Таким образом, настоящее изобретение согласно предпочтительному варианту осуществления относится к следующей последовательности стадий для получения гексапептида Foxy-5 в неочищенной форме:
1. сочетание Leu-OtBu с Fmoc-Glu-OtBu с получением дипептида INTM-1 Fmoc-Glu(OtBu)-Leu-OtBu, с последующим
2. сочетанием его с Fmoc-Cys(Trt)-OH с получением трипептида INTM-2 Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu, с последующим
3. сочетанием его с Fmoc-Gly-OH с получением защищенного тетрапептида INTM-3 Fmoc-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 2), с последующим
4. сочетанием его с Fmoc-Asp-OtBu с получением защищенного пентапептида INTM-4 Fmoc-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 3), с последующим
5. сочетанием с Fmoc-Met с получением защищенного гексапептида INTM-5 Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 4), с последующей
6. реакцией его с муравьиной кислотой с получением Foxy-5 в защищенной форме, т.е. INTM-6 For-Met-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 5), с последующим
7. снятием общей защиты с групп t-Bu и Tit с получением Foxy-5, т.е. For-Met-Asp-Gly-Cys-Glu-Leu-OH (SEQ ID NO 1), в неочищенной форме.
Согласно четвертому аспекту раскрывается способ получения гексапептида Foxy-5 в защищенной форме, т.е. INTM-6 For-Met-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 5), на основе последовательного сочетания нового трипептидного промежуточного соединения INTM-2 Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu с защищенными производными аминокислот Gly, Asp и Met с последующим снятием защиты с N и сочетанием с муравьиной кислотой.
Согласно варианту осуществления промежуточное соединение INTM-2 Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu получают с помощью твердофазного синтеза. Согласно предпочтительному варианту осуществления указанное промежуточное соединение INTM-2 получают с помощью жидкофазного синтеза.
Согласно пятому аспекту раскрывается способ получения гексапептида Foxy-5 в защищенной форме, т.е. INTM-6 For-Met-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 5), на основе последовательного сочетания нового трипептидного промежуточного соединения INTM-3 Fmoc-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu с защищенными производными аминокислот Asp и Met с последующим снятием защиты с N и сочетанием с муравьиной кислотой.
Согласно варианту осуществления промежуточное соединение INTM-3 Fmoc-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu получают с помощью твердофазного синтеза. Согласно предпочтительному варианту осуществления указанное промежуточное соединение INTM-3 получают с помощью жидкофазного синтеза.
Согласно следующим аспектам настоящего изобретения раскрываются следующие пептиды:
Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu,
Fmoc-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 2),
Fmoc-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 3),
Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-aBu (SEQ ID NO 4),
For-Met-Asp(aBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 5),
Cys-Glu-Leu,
Gly-Cys-Glu-Leu (SEQ ID NO 6),
Asp-Gly-Cys-Glu-Leu (SEQ ID NO 7),
Met-Asp-Gly-Cys-Glu-Leu (SEQ ID NO 8).
Экспериментальная часть
Пример 1. Трипептид INTM-2 Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu, стадия 1 плюс 2
Сочетали 50 г трет-бутиллейцината.HCl в N2 с 175 г (2,0 экв.) Fmoc-Glu-(OtBu) в смеси растворителей 2025 мл дихлорметана (27 об.) и 225 мл ТГФ (3 об.) в присутствии EDAC.HCl (2,0 экв.), HOBt (2,0 экв.) и DIPEA (5,0 экв.) сначала при 0-5°С в течение 1 часа, а затем при 15-20°С в течение 1 часа с получением дипептида INTM-1 Fmoc-Glu-(OtBu)-Leu-OtBu. Идентификацию выполняли с помощью протонного ядерного магнитного резонанса 1Н ЯМР и масс-спектрометрии. Для реакции с Fmoc-Cys(Trt)-OH на следующей стадии пропускали выделение продукта и использовали раствор дихлорметана непосредственно после обработки водой. Последующую реакцию полученного дипептида INTM-1 Fmoc-Glu-(OtBu)-Leu-OtBu, таким образом, выполняли с использованием раствора дихлорметана, упомянутого выше. Снятие защиты с Fmoc осуществляли с помощью ДБУ и сочетания с 204 г (1,1 экв.) Fmoc-Cys(Trt)-OH в присутствии EDAC. HCl (2,0 экв.), HOBt (2,0 экв.) и DIPEA (4 экв.) с получением неочищенного трипептида INTM-2, который очищали с помощью хроматографии на силикагеле (100-200) с использованием EtOAc-гексамина в качестве элюента с получением трипептида INTM-2 Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu в виде твердого вещества грязно-белого цвета (148 г, 68% общий выход за две реакции сочетания, чистота 93,8% с помощью ВЭЖХ).
Раскрываемые выше сокращенные реакции стадии 1 плюс 2 повторяли с 75 г /лрет-бутиллейцината.НС! с получением 208 г INTM-2.
Пример 2. Тетрапептид Fmoc-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu, стадия 3 Осуществляли реагирование INTM-2 (Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu), полученного как раскрывается в настоящем документе выше, с ДБУ в ДХМ (50 об.) с осуществлением снятия защиты с Fmoc, а затем осуществляли реагирование с 1,3 экв. Fmoc-Gly-OH в присутствии DIPEA (3 экв.), EDC.HCl (2,0 экв.) и HOBt (2,0 экв.) с получением защищенного тетрапептида INTM-3 Fmoc-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 2). С помощью тонкослойной хроматографии (ТСХ) наблюдали надлежащее превращение и слой ДХМ промывали водой и солевым раствором. Конечный органический слой концентрировали до 10-15 об. и использовали как есть на следующей стадии без выделения.
Пример 3. Пентапептид Fmoc-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu, стадия 4
Сначала осуществляли реагирование ключевого промежуточного соединения INTM-3 Fmoc-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu, полученного как раскрывается в настоящем документе выше, в виде концентрированного раствора в ДХМ с ДБУ с осуществлением снятия защиты с Fmoc. Перед переходом к следующей стадии сочетания реакционную массу пропускали через пробку из оксида кремния для удаления ДБУ, который, как выявили в предыдущих экспериментах, индуцирует образование нежелательного аспартимидного побочного продукта. Удаление ДБУ перед сочетанием с Fmoc-Asp-OtBu эффективно подавляет образованием аспартимида. После обработки с помощью пробки из оксида кремния осуществляли реагирование раствора ДХМ с Fmoc-Asp(OtBu) в присутствии DIPEA, EDAC.HCl (1,2 экв.) и HOBt (1,2 экв.) с получением защищенного пентапептида INTM-4 Fmoc-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 3). Идентификацию продукта выполняли с помощью 1Н ЯМР и масс-спектрометрии.
Сокращенные реакции (стадия 3 плюс стадия 4) повторяли два раза, начиная с 148 г и 190 г INTM-2, с получением 107 и 178 г INTM-4, соответственно (58,1% и 75,4% от теоретического выхода).
Пример 4. Гексапептид Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu, стадия 5
Осуществляли снятие защиты с Fmoc полученного пентапептида INTM-4 из примера 3 с помощью ДБУ и сочетание с Fmoc-Met в ДХМ-ТГФ (50 об. плюс 10 об.) в качестве растворителя в присутствии DIPEA (3,0 экв.), EDC.HCl (2,0 экв.) и HOBt.H2O (2,0 экв.) с получением защищенного производного Foxy-5 INTM-5 Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 4) в неочищенной форме. Очистку выполняли с помощью колоночной хроматографии с использованием ДХМ/ТГФ в качестве элюента. Очищенный продукт суспендировали в ДИПЭ с получением твердого вещества белого цвета.
Очистку выполняли несколько раз в разных условиях, таких как осаждение с помощью антирастворителей и хроматография. Выяснили, что наилучшим решением является колоночная хроматография с последующим суспендированием в ДИПЭ, что в масштабе 25 г обеспечивало выходы с химической чистотой 78% и 95,4%. Реакцию повторяли в масштабе 80 г с обеспечением 74 г продукта (выход 83%) с химической чистотой 94,1%.
Пример 5. Гексапептид INTM-6 For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu,стадия 6
Осуществляли снятие защиты с Fmoc полученного пентапептида INTM-5 из примера 4 с помощью ДБУ в ДХМ (50 об.) с последующим сочетанием с муравьиной кислотой (3,0 экв.) в присутствии EDC.HCl (4,0 экв.), HOBt.H2O (4,0 экв.) и DIPEA (4,0 экв.) с получением INTM-6 For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 5).
Реакцию (стадия 6) выполняли три раза с 4,18 и 18 г INTM-5, соответственно, с получением выходов 75-83% и химической чистоты от 67,5 до 77,2%. Реакцию повторяли с 10 экв. муравьиной кислоты в масштабе 30 г с получением 17 г продукта (выход 67%) с химической чистотой 88,8%.
Пример 6. Гексапептид For-Met-Asp-Gly-Cys-Glu-Leu-OH (Foxy-5), стадия 7 Снимали общую защиту с 17 г полученного гексапептида из примера 5 (с групп Trt и tBu) путем растворения под N2 и перемешивания в коктейле ТФУ (10 o6.)/(i-Pr)3SiH (TIS, 1,7 об.)/дитиотреитола (ДТТ) (1,7 экв.) в течение 15-30 минут при 10-15°С. Далее реакционную смесь нагревали до 25-30°С и перемешивали в течение 1-2 часов при этой температуре. Затем реакционную массу концентрировали при пониженном давлении до 2-3 объемов. После завершения реакции добавляли ТГФ (5 об.) и перемешивание продолжали еще 10-15 минут при 25-30°С. Затем медленно добавляли метил-трет-бутиловый эфир МТБЭ (30 об.) для осаждения неочищенного продукта, который получали в виде твердого вещества с количественным выходом (12,3 г).
Неочищенный продукт окончательно очищали с помощью хроматографии с обращенной фазой, которая давала желаемый гексапептид For-Met-Asp-Gly-Cys-Glu-Leu-OH (Foxy-5) с чистотой 98,3%, при использовании следующих условий способа.
Среда: Luna С18 (3) (изготовитель Phenomenex), размер пор: 10 мкм.
ID (от англ. - inner diameter - внутренний диаметр) колонки: ID 50 мм × 250 мм (Novasep), скорость потока: 50,0 мл/минута.
Приготовление образца: 100 г образца растворяли в 4 мл разбавителя, фильтровали через 0,45-мкм фильтр.
Приготовление бусрера
Буфер-А: готовили буфер на основе 0,10 плюс/минус 0,01% трифторуксусной кислоты в воде путем смешивания 10 мл трифторуксусной кислоты с 10 л очищенной воды.
Буфер-В: готовили буфер на основе 0,10 плюс/минус 0,01% трифторуксусной кислоты в ацетонитриле путем смешивания 5 мл трифторуксусной кислоты с 5 л ацетонитрила.
Операционная процедура
1. Уравновешивали колонку с помощью [буфер А:буфер В] в отношении (95:5) 3-5 объемами колонки со скоростью потока 5,0 мл/минута.
2. Загружали раствор образца в колонку.
3. Программировали хроматографическую систему, подключенную к колонке, для выполнения программы градиента, как показано ниже, чтобы начать элюирование продукта.
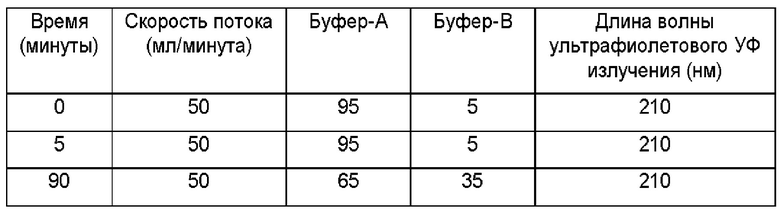
4. Собирали следующие фракции: переднюю, верхнюю и заднюю.
Примечание: Время элюирования может варьировать от цикла к циклу в зависимости от масштаба.
5. После элюирования пика сразу же промывали колонку водой и ацетонитрилом в отношении 20:80 (% об./об.) 2 объемами колонки.
6. Отправляли фракции на анализ чистоты.
7. Хранили фракции при -20°С плюс/минус 2°С.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> WntResearch AB
<120> ЛИНЕЙНЫЕ ЖИДКОФАЗНЫЕ ПУТИ ДЛЯ ГЕКСАПЕПТИДОВ WNT
<130> 138534
<160> 8
<170> BiSSAP 1.3.6
<210> 1
<211> 6
<212> Белок
<213> Искусственная последовательность
<220>
<221> Посттрансляционная модификация остатка
<222> 1
<223> Формилметионин
<220>
<223> Синтетический пептид
<400> 1
Met Asp Gly Cys Glu Leu
1 5
<210> 2
<211> 4
<212> Белок
<213> Искусственная последовательность
<220>
<221> Посттрансляционная модификация остатка
<222> 1
<223> (Флуоренилметоксикарбонил)-глицин
<220>
<223> Синтетический пептид
<220>
<221> Посттрансляционная модификация остатка
<222> 2
<223> Тритилцистеин
<220>
<221> Посттрансляционная модификация остатка
<222> 3
<223> Сложный трет-бутиловый эфир глютаминовой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 4
<223> Сложный трет-бутиловый эфир лейцина
<400> 2
Gly Cys Glu Leu
1
<210> 3
<211> 5
<212> Белок
<213> Искусственная последовательность
<220>
<221> Посттрансляционная модификация остатка
<222> 1
<223> Сложный трет-бутиловый эфир (флуоренилметоксикарбонил)-аспарагиновой
кислоты
<220>
<223> Синтетический пептид
<220>
<221> Посттрансляционная модификация остатка
<222> 3
<223> Тритилцистеин
<220>
<221> Посттрансляционная модификация остатка
<222> 4
<223> Сложный трет-бутиловый эфир глютаминовой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 5
<223> Сложный трет-бутиловый эфир лейцина
<400> 3
Asp Gly Cys Glu Leu
1 5
<210> 4
<211> 6
<212> Белок
<213> Искусственная последовательность
<220>
<221> Посттрансляционная модификация остатка
<222> 1
<223> (Флуоренилметоксикарбонил)-метионин
<220>
<223> Синтетический пептид
<220>
<221> Посттрансляционная модификация остатка
<222> 2
<223> Сложный трет-бутиловый эфир аспарагиновой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 4
<223> Тритилцистеин
<220>
<221> Посттрансляционная модификация остатка
<222> 5
<223> Сложный трет-бутиловый эфир глютаминовой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 6
<223> Сложный трет-бутиловый эфир лейцина
<400> 4
Met Asp Gly Cys Glu Leu
1 5
<210> 5
<211> 6
<212> Белок
<213> Искусственная последовательность
<220>
<221> Посттрансляционная модификация остатка
<222> 1
<223> Формилметионин
<220>
<223> Синтетический пептид
<220>
<221> Посттрансляционная модификация остатка
<222> 2
<223> Сложный трет-бутиловый эфир аспарагиновой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 4
<223> Тритилцистеин
<220>
<221> Посттрансляционная модификация остатка
<222> 5
<223> Сложный трет-бутиловый эфир глютаминовой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 6
<223> Сложный трет-бутиловый эфир лейцина
<400> 5
Met Asp Gly Cys Glu Leu
1 5
<210> 6
<211> 4
<212> Белок
<213> Искусственная последовательность
<220>
<223> Синтетический пептид
<400> 6
Gly Cys Glu Leu
1
<210> 7
<211> 5
<212> Белок
<213> Искусственная последовательность
<220>
<223> Синтетический пептид
<400> 7
Asp Gly Cys Glu Leu
1 5
<210> 8
<211> 6
<212> Белок
<213> Искусственная последовательность
<220>
<223> Синтетический пептид
<400> 8
Met Asp Gly Cys Glu Leu
1 5
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ЖИДКОФАЗНЫЕ ПУТИ ДЛЯ ГЕКСАПЕПТИДОВ WNT | 2019 |
|
RU2801268C2 |
| ГЛИКОЗИЛИРОВАННЫЙ ПЕПТИД GLP-1 | 2009 |
|
RU2543157C2 |
| ПЕПТИД GLP-1 С ПРИСОЕДИНЕННОЙ ОЛИГОСАХАРИДНОЙ ЦЕПЬЮ | 2008 |
|
RU2539829C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЛИКОПЕПТИДА, ИМЕЮЩЕГО СИАЛИРОВАННУЮ САХАРНУЮ ЦЕПЬ, И СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО СИАЛИЛГЛИКОАСПАРАГИНА | 2012 |
|
RU2586524C2 |
| ПЕПТИДЫ В КОМБИНАЦИИ С ИНГИБИТОРАМИ ИММУННЫХ КОНТРОЛЬНЫХ ТОЧЕК ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ РАКА | 2020 |
|
RU2826955C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДА | 2008 |
|
RU2478105C2 |
| АНАЛОГИ ЭФР(А) С ЗАМЕСТИТЕЛЯМИ - ЖИРНЫМИ КИСЛОТАМИ | 2017 |
|
RU2747877C2 |
| НЕОАНТИГЕНЫ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2813924C2 |
| ПРОИЗВОДНЫЕ FGF21 И ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2729011C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЛИКОПРОТЕИНА И СПОСОБ СКРИНИНГА | 2009 |
|
RU2520240C2 |
Изобретение относится к гексапептидному производному формулы PG-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OR, где R выбран из С1-С6алкила, такого как метил или трет-бутил, и PG представляет собой Fmoc, способу его получения и способу получения Foxy-5 (SEQ ID NO 1) с применением указанного гексапептидного производного линейным жидкофазным путем, обеспечивающим простоту очистки промежуточных соединений процесса, простоту сборки фрагментов, высокую хиральную чистоту и адаптируемость к коммерческому масштабу. 3 н. и 9 з.п. ф-лы, 5 ил., 1 табл., 6 пр.
1. Гексапептидное производное формулы PG-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OR, где R выбран из С1-С6алкила, такого как метил или трет-бутил, и PG представляет собой Fmoc.
2. Гексапептидное производное по п. 1, характеризующееся формулой Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 4).
3. Способ получения гексапептидного производного по п. 1 или 2, включающий следующие стадии:
a. обеспечения защищенного L-лейцинового производного PG-Leu-OR,
b. удаления защитной группы PG из указанного защищенного L-лейцинового производного с последующим сочетанием с PG-Glu-OtBu с получением защищенного дипептида PG-Glu(OtBu)-Leu-OR,
c. удаления защитной группы PG из указанного защищенного дипептида с последующим сочетанием с PG-Cys(Trt)-OH с получением защищенного трипептида PG-Cys(Trt)-Glu(OtBu)-Leu-OR,
d. удаления защитной группы PG из указанного защищенного трипептида с последующим сочетанием с PG-Gly-OH с получением защищенного тетрапептида PG-Gly-Cys(Trt)-Glu(OtBu)-Leu-OR,
e. удаления защитной группы PG из указанного защищенного тетрапептида в основных условиях с последующим удалением избытка основания с получением разблокированного тетрапептида H-Gly-Cys(Trt)-Glu(OtBu)-Leu-OR,
f. сочетания указанного разблокированного тетрапептида с PG-Asp(OtBu) в основных условиях с последующим удалением избытка основания с получением защищенного пентапептида PG-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OR,
g. удаления защитной группы PG из указанного пентапептида с последующим сочетанием с PG-Met с получением защищенного гексапептида PG-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OR,
где R выбран из С1-С6алкила, такого как метил или трет-бутил, и PG представляет собой Fmoc.
4. Способ по п. 3, где по меньшей мере две последовательные стадии сочетания, например, две, три или четыре стадии, выполняют без выделения продукта.
5. Способ по п. 3 или 4, где очистку промежуточного соединения, такую как с помощью флэш-хроматографии, выполняют по меньшей мере один раз между двумя последовательными стадиями сочетания, например, один, два, три или четыре раза, без выделения продукта.
6. Способ по любому из пп. 3-5, где все стадии b)-g) выполняют в растворе.
7. Способ получения Foxy-5 (SEQ ID NO 1), при этом способ включает:
a. обеспечение гексапептидного производного по п. 1 или 2 способом по п. 3,
b. удаление защитной группы с азота на Met-конце из указанного гексапептидного производного,
c. сочетание полученного гексапептида стадии b) с муравьиной кислотой или ее активным сложным эфиром с получением защищенного производного Foxy-5 For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OR,
d. снятие общей защиты полученного защищенного производного Foxy-5 стадии с) с получением Foxy-5 в неочищенной форме,
e. необязательно выполнение дополнительных стадий очистки и
f. необязательно получение Foxy-5 в твердой форме.
8. Способ по п. 7, где по меньшей мере две последовательные стадии реакции, например, две, три или четыре стадии, выполняют без выделения продукта.
9. Способ по п. 7 или 8, где очистку промежуточного соединения, такую как с помощью флэш-хроматографии, выполняют по меньшей мере один раз между двумя последовательными стадиями реакции, например, один, два или три раза, без выделения продукта.
10. Способ по любому из пп. 7-9, где Foxy-5 в неочищенной форме очищают с помощью хроматографии, такой как хроматография с обращенной фазой.
11. Способ по любому из пп. 7-10, где Foxy-5 выделяют в твердой форме, либо в виде гексапептида как такового, либо в виде его соли присоединения кислоты или щелочи.
12. Способ по любому из пп. 7-11, где твердая форма Foxy-5 представляет собой лиофилизат, аморфный порошок или кристаллическое соединение.
| WO 2016092378 A1, 16.06.2016 | |||
| ПРИМЕНЕНИЕ ПРОИЗВОДНЫХ ПЕПТИДА WNT5-A ДЛЯ ЛЕЧЕНИЯ МЕЛАНОМЫ И РАКА ЖЕЛУДКА | 2009 |
|
RU2517190C2 |
| WO 2006130082 A1, 07.12.2006 | |||
| JENEI VERONIKA ET AL, "A t-butyloxycarbonyl-modified Wnt5a-derived hexapeptide functions as a potent antagonist of Wnt5a-dependent melanoma cell invasion", PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES OF THE UNITED STATES OF AMERICA 2009, vol | |||
| Светоэлектрический измеритель длин и площадей | 1919 |
|
SU106A1 |
| Способ изготовления звездочек для французской бороны-катка | 1922 |
|
SU46A1 |
