Область техники, к которой относится изобретение
Настоящее изобретение в целом относится к области синтеза полипептидов и, конкретнее, к жидкофазному синтезу гексапептида WNT (от англ. wingless - бескрылый и integrated - интегрированный) Foxy-5. Настоящее изобретение, кроме того, относится к жидкофазному синтезу новых три-, тетра- и пентапептидов.
Предшествующий уровень техники изобретения
Foxy-5 представляет собой формилированный, полученный из Wnt5a гексапептид и миметик Wnt-5a с потенциальной противометастатической активностью, который в настоящее время разрабатывается в качестве лекарственного средства-кандидата для предупреждения распространения опухоли при некоторых распространенных формах рака.
Foxy-5 характеризуется аминокислотной последовательностью For-Met-Asp-Gly-Cys-Glu-Leu-OH (SEQ ID NO 1, фигура 1). При внутривенном введении Foxy-5 связывается с рецепторами wnt-5a, Frizzled-2 и -5 и активирует их, что активирует опосредованную wnt-5a передачу сигналов. Повышенная передача сигналов wnt-5a может ингибировать миграцию и инвазию эндотелиальных опухолевых клеток.
Foxy-5 предназначен для компенсации дефицита белка Wnt-5a в опухолевой ткани, отмечаемого у пациентов с раком толстой кишки, с целью снижения риска метастазирования. Субанализ недавнего ретроспективного исследования пациентов с коло ректальным раком на стадии III показывает, что доля пациентов с низкой экспрессией Wnt 5а значительно выше, чем наблюдалось в предыдущих исследованиях у пациентов с колоректальным раком (КРР) стадии II. Пациенты с опухолями КРР стадии III отличаются от таковых со стадией II главным образом наличием опухолевых клеток в лимфатических узлах, соседних с первичной опухолью, благодаря чему эти опухоли более агрессивны и быстрее прогрессируют. Низкий уровень Wnt-5a наблюдался почти у 70 процентов пациентов на стадии III по сравнению с приблизительно 45 процентами пациентов с менее запущенными стадиями опухоли. Это подтверждает гипотезу о том, что уровень Wnt-5a существенно влияет на течение заболевания.
На основании завершенного исследования фазы 1b с Foxy-5, направленного на регистрирование профиля безопасности, фармакокинетических показателей и определения дозы лекарственного средства-кандидата для фазы 2, Foxy-5 в настоящее время представлен для исследования в фазе 2 клинического испытания, в котором лечение пациентов с раком толстой кишки будут начинать во время постановки диагноза до осуществления хирургического вмешательства. Лечение рассчитано максимум на 12 недель или до начала химиотерапии.
Foxy-5 и способ его получения описаны в публикации международной заявки на патент №WO06130082 А1. Активный фармацевтический ингредиент АФИ для доклинических и клинических исследований, проводимых до настоящего времени, получали путем классического твердофазного пептидного синтеза (SPPS, от англ. solid phase peptide synthesis), при котором Foxy-5 получается линейным путем «1 плюс 1 плюс 1 плюс 1 плюс 1 плюс 1», см. фигуру 2.
Последовательность For-Met-Asp-Gly-Cys-Glu-Leu-OH, таким образом, собирается на 2-хлортритильной смоле, несущей С-концевую аминокислоту Leu, с использованием Fmoc-стратегии (от англ. Fluorenylmethoxycarbonyl флуоренилметилоксикарбонил). Синтез проводится в реакторе для SPPS и состоит из чередующихся процедур сочетания, ацетилирования и снятия защитной группы N-α. Сочетание проводят в N,N-диметилформамиде (ДМФ) или в ДМФ/дихлорметане (ДХМ) в качестве растворителя. Оно состоит из сочетания производного N-α-защищенной аминокислоты с предыдущей аминокислотой в присутствии активирующего реагента и основания, при необходимости. Муравьиную кислоту сочетают в виде активного сложного эфира без активирующих средств.
Если сочетание не завершено, его можно продолжить или повторить процедуру. Чтобы избежать образования последовательностей с делецией в качестве побочных продуктов, проводят систематическую процедуру ацетилирования (кэппинга) после стадии сочетания или, если проводят повторное сочетание, после стадий повторного сочетания, с использованием ДМФ, уксусного ангидрида и пиридина.
За ацетилированием следует процедура снятия защитной группы N-α, что предусматривает промывку смолы с помощью ДМФ, отщепление Fmoc-группы пиперидином в ДМФ и последующие промывки с помощью ДМФ. В случае неполного расщепления процедуру снятия защитной группы N-α, описанную выше, можно повторять. Для каждой отдельной стадии добавляют растворители и/или реагенты и реакционную смесь перемешивают, а затем фильтруют для удаления растворителей и/или реагентов из смолы.
Процедуры сочетания, ацетилирования и снятия защиты N-α повторяют до тех пор, пока смола не будет нести полную пептидную последовательность For-Met-Asp-Gly-Cys-Glu-Leu-OH. После окончательного сочетания активного сложного эфира муравьиной кислоты ацетилирование не проводят.SPPS завершают промывкой пептидной смолы с помощью ДМФ и изопропилового спирта (ИПС) и последующей сушкой при пониженном давлении.
Отщепление пептида от смолы и сопутствующее отщепление защитных групп боковой цепи осуществляют путем обработки пептидной смолы с помощью трифторуксусной кислоты (TFA, от англ. Trifluoroacetic acid) в присутствии подходящих акцепторов (например, воды и 1,2-этандитиола (EDT, от англ. - ethanedithiol)). Затем полученный неочищенный пептид очищают с помощью двумерной препаративной высокоэффективной жидкостной хроматографии (ВЭЖХ) на колонке с обращенной фазой при градиентном элюировании ацетонитрилом (АЦН) (системы муравьиной кислоты и уксусной кислоты).
Объединенные фракции надлежащей чистоты лиофилизируют. Лиофилизат анализируют с помощью ВЭЖХ и необязательно повторно очищают с помощью двумерной препаративной ВЭЖХ, как указано выше, в случае несоответствия установленным критериям чистоты.
С помощью описанного выше подхода SPPS было создано достаточно материала для доклинического и раннего клинического исследований, но для дальнейших клинических исследований и возможных коммерческих целей требуется синтез, лучше подходящий для крупномасштабного синтеза, с помощью которого можно снизить стоимость товаров и сделать доступными более крупные партии Foxy-5.
Таким образом, существует потребность в надежном способе синтеза, который может обеспечить Foxy-5 в масштабе нескольких килограммов, как для дальнейшего клинического испытания, так и для возможных коммерческих целей.
Краткое описание графических материалов
На фигуре 1 показана химическая структура Foxy-5. Foxy-5 представляет собой неразветвленный пептид, состоящий из шести аминокислот с формилированным N-концом. Все оптически активные аминокислотные остатки находятся в L-конфигурации. Молекулярная формула Foxy-5 представляет собой C26H42N6O12S2, а молекулярная масса составляет 694,8 г/моль (средняя масса).
На фигуре 2 показана схема синтеза для способа SPPS для Foxy-5.
На фигуре 3 показана химическая структура ключевого трипептида 1 (KT-1), т.е. For-Met-Asp(OtBu)-Gly (верхняя часть фигуры), и ключевого трипептида 1* (KT-1*), т.е. Fmoc-Met-Asp(OtBu)-Gly. Все оптически активные аминокислотные остатки находятся в L-конфигурации. Молекулярная формула KT-1 представляет собой C16H27N3O7S, а молекулярная масса составляет 405 г/моль (средняя масса).
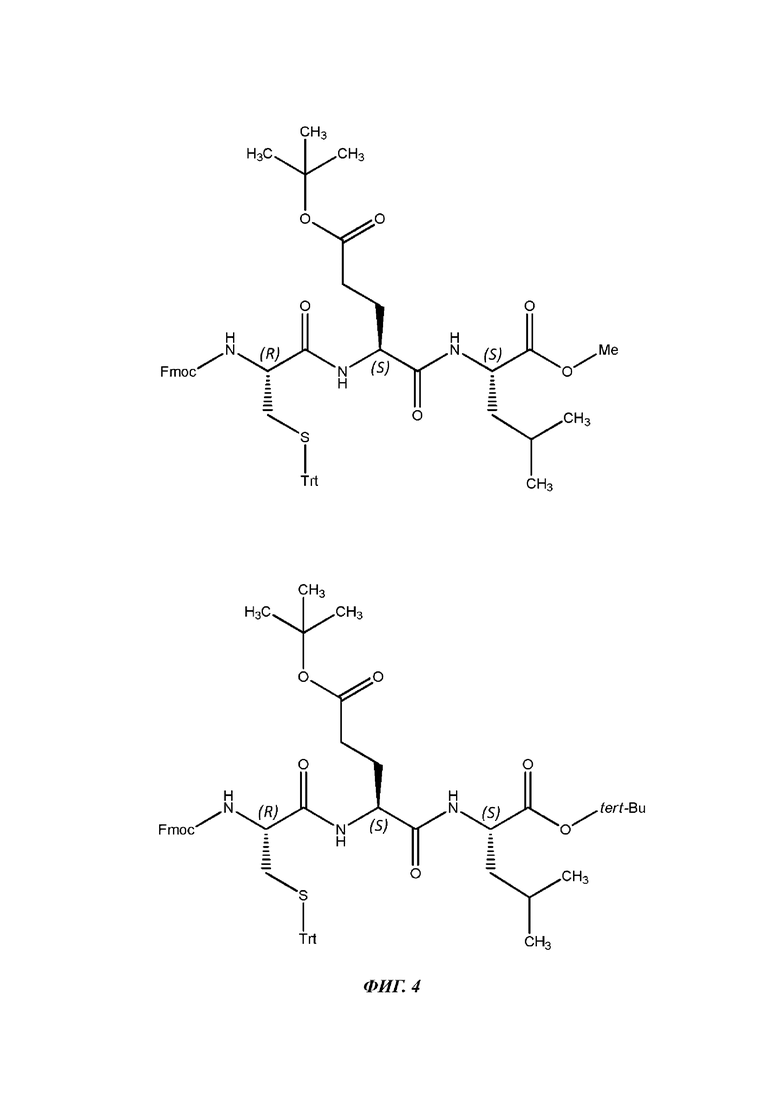
На фигуре 4 показана структура ключевого трипептида 2 (KT-2) Fmoc-Cys(Trt)Glu(OtBu)-Leu-OMe (верхняя часть) и ключевого трипептида 2* (KT-2*) Fmoc-Cys(Trt)Glu(OtBu)-Leu-O-Tpeт-Bu (нижняя часть).
На фигуре 5 иллюстрирована стратегия «2 плюс 2 плюс 2» для образования Foxy-5 на основе стратегии кислотно-лабильной защитной группы.
На фигуре 6 иллюстрирована стратегия «2 плюс 2 плюс 2» для образования Foxy-5 на основе гидрогенолитической стратегии защитной группы.
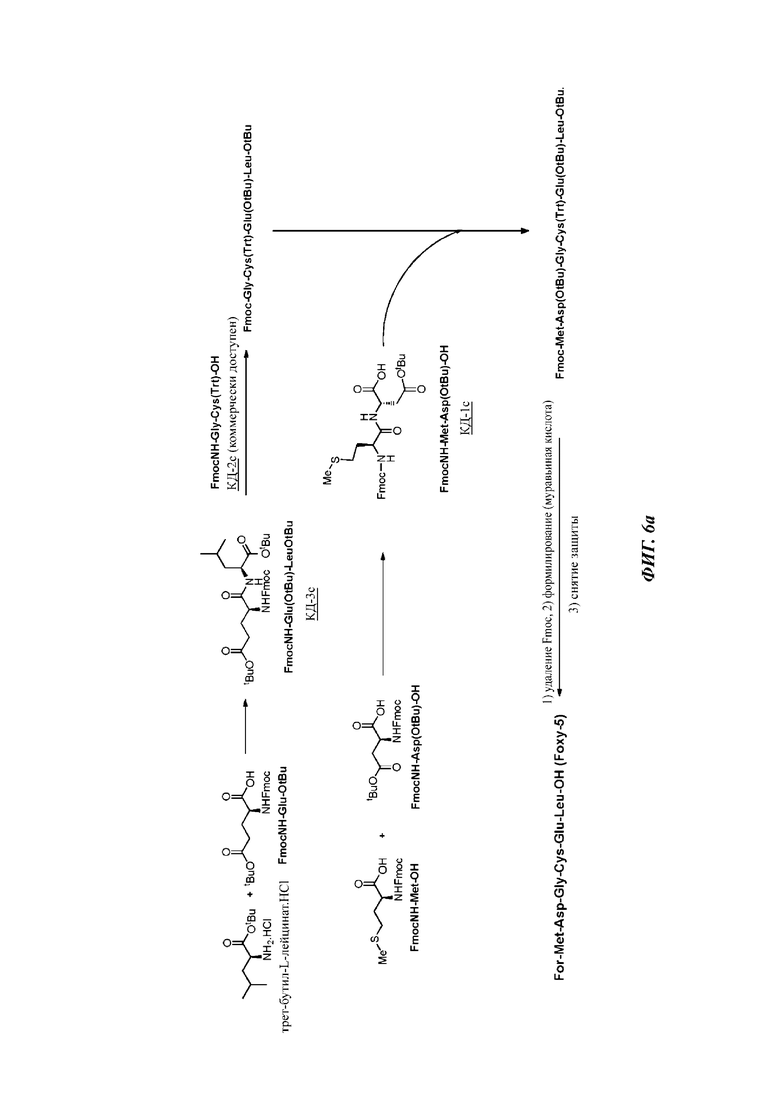
На фигуре 6а иллюстрирована стратегия «2 плюс 2 плюс 2» для образования Foxy-5 на основе стратегии основно-лабильной защитной группы.
На фигуре 7 иллюстрирована стратегия «3 плюс 3» для образования Foxy-5 на основе сочетания ключевого трипептида 1 (KT-1) с ключевым трипептидом 2 (KT-2) с последующим 1) омылением сложного метилового эфира лейцина и 2) снятием защиты трет-бутильной и тритильной защитных групп.
На фигуре 7а иллюстрирована стратегия «3 плюс 3» для образования Foxy-5 на основе сочетания ключевого трипептида 1* (KT-1*) с ключевым трипептидом 2* (KT-2*) с последующим снятием защитной группы Fmoc с помощью 1,8-диазабицикло[5,4,0]ундец-7-ена (ДБУ), сочетанием с муравьиной кислотой и снятием защиты трет-бутильной и тритильной защитных групп с получением желаемого Foxy-5.
На фигуре 8 иллюстрирована стратегия «3 плюс 1 плюс 1 плюс 1» для образования Foxy-5 на основе последовательного удлинения ключевого трипептида 2 (KT-2) с помощью аминокислот Gly, Asp и формил-Met.
На фигуре 8а иллюстрирована стратегия «3 плюс 1 плюс 1 плюс 1» для образования Foxy-5 на основе последовательного удлинения ключевого трипептида 2* (KT-2*) с помощью аминокислот Fmoc-Gly, Fmoc-Asp-OtBu и Fmoc-Met. Полученный в результате гексапептид подвергают снятию защиты Fmoc с помощью ДБУ, сочетанию с муравьиной кислотой и снятию защиты трет-бутильной и тритильной защитных групп с получением желаемого Foxy-5.
Аббревиатуры
Fmoc - флуоренилметоксикарбонил
Вое (от англ. tert - Butyloxycarbonyl) - трет-бутилоксикарбонил For (от англ. Formyl) - формил
Trt (от англ. Triphenyl methyl (Trityl)) - трифенилметил (тритил)
Cbz (от англ. carboxybenzyl) - карбоксибензил
ДЦГА - дициклогексиламин
tBu (от англ. tert - Butyl) - трет-бутил
ТГФ - тетрагидрофуран
ДМФ - N,N-диметилформамид
ТЭА - триэтиламин
Bn (от англ. Benzyl) - бензил
TFA - трифторуксусная кислота
TIS (от англ. Triisopropylsilane) - триизопропилсилан
HOBt (от англ. 1-Hydroxybenzotriazole)-1-гидроксибензотриазол
HOSu (от англ. N-Hydroxysuccinimide)-N-гидроксисукцинимид
ДХМ - дихлорметан
HOAt (от англ. 1-Hydroxy-7-azabenzotriazole)-1-гидрокси-7-азабензотриазол
EDAC (от англ. 1-Ethyl-3-(3'-dimethylaminopropyl)carbodiimide), HCl - 1-этил-3-(3'-диметиламинопропил)карбодиимид, HCl
DIPEA (от англ. Diisopropylamine) - диизопропиламин
DIPE (от англ. Diisopropylether) - диизопропиловый эфир
ДБУ - 1,8-диазабицикло[5,4,0]ундец-7-ен
Аббревиатуры аминокислот
Met - метионин
Asp - аспарагин
Gly - глицин
Cys- цистеин
Glu - глутаминовая кислота
Leu - лейцин
Foxy-5 - For-Met-Asp-Gly-Cys-Glu-Leu
Краткое раскрытие изобретения
Настоящее изобретение относится к некоторым жидкофазным способам получения формилированного гексапептида, известного как «Foxy-5» (т.е. For-Met-Asp-Gly-Cys-Glu-Leu-OH), а также к его различным три-, тетра-, пента- и гексапептидным фрагментам, в том числе его защищенным производным. Способы, представленные в настоящем документе, обладают рядом преимуществ над традиционными твердофазными синтезами, включая без ограничения низкую стоимость сырьевых материалов, простоту очистки промежуточных соединений процесса, простоту сборки фрагментов, высокую хиральную чистоту и адаптируемость к коммерческому масштабу, среди прочего, что будет раскрыто более подробно ниже.
Таким образом, основная цель настоящего изобретения заключается в обеспечении масштабируемого пути синтеза Foxy-5. Следующая цель заключается в идентификации и характеристике подходящих ключевых промежуточных соединений для указанного масштабируемого пути синтеза с целью последующего изготовления лекарственного вещества согласно GMP (от англ. Good Manufacturing Practice - надлежащей производственной практике).
Ввиду себестоимости продукции и трудоемкого масштабирования, обычно связанного с твердофазной химией, основное внимание уделялось разработке жидкофазных химических путей. Таким образом, настоящее изобретение относится к жидкофазным способам получения Foxy-5 или его промежуточных соединений и предшественников с использованием стратегий сочетания фрагментов.
Тестировали три разных подхода:
стратегию сочетания «2 плюс 2 плюс 2», при которой сочетают три дипептида с образованием конечного гексапептида,
стратегию сочетания «3 плюс 3», при которой сочетают два трипептида,
стратегию сочетания «4 плюс 1 плюс 1», при которой тетрапептид последовательно удлиняют, и
стратегию сочетания «3 плюс 1 плюс 1 плюс 1», при которой трипептид последовательно удлиняют.
В подходе «2 плюс 2 плюс 2» основную цепь Foxy-5 собирают путем сочетания трех независимых ключевых дипептидных фрагментов КД-1, КД-2 и КД-3. Разработали три разных подхода a, b и с, см. фигуру 5, фигуру 6 и фигуру 6а. Жидкофазный синтез фрагментов этих трех дипептидов сопровождался образованием гексапептида конвергентным образом. Стратегия защитной группы в подходе «2 плюс 2 плюс 2» была основана либо на гидрогенолизе N-концевых защитных групп (Cbz), либо на катализируемом основанием снятии защиты N-концевых защитных групп (Fmoc) с фрагментов и предусматривала применение кислотно-лабильной защитной группы на боковых цепях. Кроме того, все промежуточные соединения могут быть доступны с помощью жидкофазной химии. Для всех трех подходов «2 плюс 2 плюс 2» N-формильную группу Met вводят на конечной стадии синтеза после того, как произошло сочетание все трех дипептидных фрагментов.
В подходе «3 плюс 3» основную цепь Foxy-5 собирают путем жидкофазного сочетания фрагментов двух трипептидных фрагментов. В одной версии подхода «3 плюс 3» с использованием ключевых трипептидов KT-1 и KT-2 (фигура 7) формильную группу вводят на ранней стадии, т.е. при синтезе трипептидного фрагмента KT-1. Во второй версии подхода «3 плюс 3» формильную группу вводят вначале после того, как был образован гексапептидный каркас посредством сочетания двух разных трипептидных фрагментов KT-1* и KT-2* (фигура 7а).
Согласно первому аспекту настоящего изобретения представлен новый трипептид Met-Asp-Gly и его защищенные производные For-Met-Asp(OtBu)-Gly (ключевой трипептид-1, KT-1) и Fmoc-Met-Asp(OtBu)-Gly (ключевой трипептид-1*, KT-1*).
Согласно второму аспекту настоящего изобретения представлен новый трипептид Cys-Glu-Leu и его защищенные производные Fmoc-Cys(Trt)Glu(OtBu)-Leu-O-Me (ключевой трипептид-2, KT-2) и Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu (ключевой трипептид-2*, KT-2*).
Согласно третьему аспекту настоящего изобретения представлен способ получения гексапептида Foxy-5 в защищенной форме, основанный на сочетании новых трипептидных производных KT-1 и KT-2, в качестве альтернативы, основанный на сочетании новых трипептидных производных KT-1* и KT-2*, при этом сочетание может сопровождаться (в зависимости от фактического пути) либо снятием общей защиты, либо снятием защиты N с последующим N-формилированием и окончательным снятием защиты боковой цепи с получением желаемого гексапептида Foxy-5.
В подходе «3 плюс 1 плюс 1 плюс 1» основную цепь Foxy-5 собирают путем последовательного жидкофазного сочетания трипептидного фрагмента KT-2 или KT-2* с защищенными производными аминокислот Gly, Asp и Met с образованием либо Foxy-5, либо гексапептида Met-Asp-Gly-Cys-Glu-Leu-OH в защищенной форме. В зависимости от фактического пути снятие общей защиты или снятие защиты N с последующим N-формилированием и окончательным снятием защиты боковой цепи дает желаемый гексапептид Foxy-5.
Согласно четвертому аспекту настоящего изобретения представлен способ получения гексапептида Foxy-5 в защищенной форме, основанный на последовательном сочетании новых трипептидных производных KT-2 или KT-2* с защищенными производными аминокислот Gly, Asp и Met, при котором последовательность сочетаний может сопровождаться (в зависимости от определенного исходного трипептида) снятием общей защиты или снятием защиты N с последующим N-формилированием и окончательным снятием защиты боковой цепи, что дает желаемый гексапептид Foxy-5. Подробное раскрытие изобретения
Как упоминалось в приведенном выше кратком описании, разработали четыре разных жидкофазных подхода для сборки гексапептидной последовательности Foxy-5: путь «2 плюс 2 плюс 2» (три версии), путь «3 плюс 3» (две версии), путь «4 плюс 1 плюс 1» и путь «3 плюс 1 плюс 1 плюс 1» (2 версии). Далее стратегии обсуждаются более подробно.
Стратегия сочетания «2 плюс 2 плюс 2»
Путь А «2 плюс 2 плюс 2» (фигура 5)
Синтез первого ключевого дипептида КД-1а (Boc-Met-Asp(OtBu)-OH) начинается с сочетания коммерчески доступного Boc-Met-OH и HAsp(OtBu)-OMe. Полученный в результате дипептид Boc-Met-Asp(OtBu)-OMe затем подвергают селективному снятию защиты на С-конце.
Второй ключевой дипептид КД-2а (Cbz-Gly-Cys(ψMe,MePro)-OH) затем получают путем образования цистеинацетонида H-Cys(ψMe,MePro)-OH из H-Cys-ОН и ацетона с последующим сочетанием с сукцинимидным сложным эфиром Cbz-Gly-OSu.
Синтез третьего дипептида КД-3а (H-Glu(OtBu)-Leu-OtBu) предусматривает сочетание коммерчески доступных исходных материалов Cbz-Glu(OtBu)-OH и H-Leu-OtBu. Последующее снятие защитной группы Cbz путем гидрогенизации на Pd/C дает промежуточное соединение дипептид H-Glu(OtBu)-Leu-OtBu.
Синтез Foxy-5 с помощью пути А из трех ключевых дипептидов КД-1а, КД-2а и КД-3а окончательно инициируют путем сочетания дипептидов КД-2а Cbz-Gly-Cys(ψMe,MePro)-OH и КД-3а H-Glu(OtBu)-Leu-OtBu, что дает тетрапептид Gly-Cys(ψMe,MePro)-Glu(OtBu)-l_eu-OtBu (SEQ ID NO 5) после снятия защиты. Дипептид КД-1а Boc-Met-Asp(OtBu)-OH затем сочетают с тетрапептидом Gly-Cys(ψMe,MePro)-Glu(OtBu)-Leu-OtBu в присутствии стандартных реагентов сочетания. Снятие общей защиты осуществляют путем обработки полученного в результате защищенного гексапептида сильной кислотой. Затем последующее формилирование дает целевое соединение Foxy-5 в неочищенной форме.
Путь В «2 плюс 2 плюс 2» (фигура 6)
Синтез первого ключевого дипептида КД-1b (Cbz-Met-Asp(OBn)-OSu) предусматривает образование сукцинимидного сложного эфира Cbz-Met-OSu из соответствующего метионинового производного. Сочетание активированного сложного эфира с коммерчески доступным H-Asp(OBn)-OH при основных условиях дает дипептид Cbz-Met-Asp(OBn)-OH, который затем также активируют с помощью HOSu, что дает первый дипептид КД-1 Cbz-Met-Asp(OBn)-OSu.
Для второго ключевого дипептида КД-2b (H-Gly-Cys(Bn)-OH) цистеин сначала защищают бензилом на боковой цепи, что дает H-Cys(Bn)-OH. Затем коммерчески доступное активированное сукцинимидом глициновое структурное звено Boc-Gly-OSu сочетают с защищенным бензилом цистеиновым структурным звеном с получением желаемого дипептида КД-2b H-Gly-Cys(Bn)-OH после снятия защитной группы Boc.
Третий ключевой дипептид КД-3b (H-Glu(OBn)-Leu-OBn) образуют путем сочетания активированного сложного эфира Boc-Glu(OBn)-OSu и коммерчески доступного H-Leu-OBn.
Синтез Foxy-5 с помощью пути В из трех дипептидных фрагментов КД-1b, КД-2b и КД-3b окончательно инициируют путем сочетания защищенных ключевых дипептидов КД-1b (Cbz-Met-Asp(OBn)-Osu) и КД-2b (H-Gly-Cys(Bn)-OH) с получением защищенного тетрапептида Cbz-Met-Asp(OBn)-Gly-Cys(Bn)-OH (SEQ ID NO 12), который затем сочетают с КД-3b (H-Glu(OBn)-Leu-OBn) с получением Cbz-Met-Asp(OBn)-Gly-Cys(Bn)-Glu(OBn)-Leu-OBn (SEQ ID NO 13). Этот защищенный гексапептид затем подвергают снятию общей защиты путем гидрогенизации на Pd/C, что дает гексапептид Met-Asp-Gly-Cys-Glu-Leu-OH (SEQ ID NO 14), который на последней химической стадии формилируют с получением желаемого гексапептида Foxy-5 в неочищенной форме.
Следует отметить, что тетрапептиды Gly-Cys-Glu-Leu-OH (SEQ ID NO 8) и Met-Asp-Gly-Cys-OH (SEQ ID NO 11), а также два их защищенных производных Gly-Cys(ψMe,MePro)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 5) и Cbz-Met-Asp(OBn)-Gly-Cys(Bn)-OH (SEQ ID NO 12), раскрываемые в настоящем документе, представляют собой новые соединения. Подобным образом, защищенный гексапептид Cbz-Met-Asp(OBn)-Gly-Cys(Bn)-Glu(OBn)-Leu-OBn (SEQ ID NO 13) представляет собой новое соединение.
Путь С «2 плюс_2 плюс 2» (фигура 6а)
Синтез первого ключевого дипептида КД-1с Fmoc-Met-Asp(OtBu)-OH осуществляли путем сочетания коммерчески доступного Fmoc-Met-OH и Н-Asp(OtBu)-OH с дициклогексилкарбодиимидом (DCC, от англ. Dicyclohexylcarbodiimide).
Синтез второго ключевого дипептида КД-2с Fmoc-Gly-Cys(Trt)-OH не требовался, поскольку защищенный дипептид был коммерчески доступен.
Синтез третьего ключевого дипептида КД-3с Fmoc-Glu-(OtBu)-Leu-OtBu осуществляли путем сочетания коммерчески доступного трет-бутиллейцината.HCl с Fmoc-Glu-(OtBu) в ДХМ при большом разбавлении в присутствии EDAC.HCl, HOBt и DIPEA.
Синтез Foxy-5 с помощью пути С из трех дипептидных фрагментов КД-1 с, КД-2 с и КД-Зс окончательно инициируют путем сочетания защищенных ключевых дипептидов КД-3с Fmoc-Glu-(OtBu)-Leu-OtBu с КД-2 с Fmoc-Gly-Cys(Trt)-OH с получением тетрапептида Fmoc-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 6), который затем подвергают реагированию с КД-1 с Fmoc-Met-Asp(OtBu)-OH с получением защищенного гексапептида Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 4), который затем подвергают снятию защитной группы Fmoc с помощью ДБУ в ДХМ с последующим сочетанием с муравьиной кислотой в присутствии EDC.HCI (EDC, от англ. ethylene dichloride - этилендихлорид), HOBt.H2O и DIPEA с получением For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 15).
Этот защищенный гексапептид (SEQ ID NO 15) подвергали снятию общей защиты (групп Trt и tBu) путем растворения и перемешивания в коктейле TFA/(i-Pr)3SiH/дитиотреитол (ДТТ). После завершения реакции неочищенный продукт получали в виде твердого вещества с выходом 97% (9,5 г) путем осаждения с помощью ТГФ/метил-трет-бутиловый эфир (МТБЭ). Хроматографическая очистка давала желаемый гексапептид For-Met-Asp-Gly-Cys-Glu-Leu-OH (Foxy-5) с чистотой 97,6%.
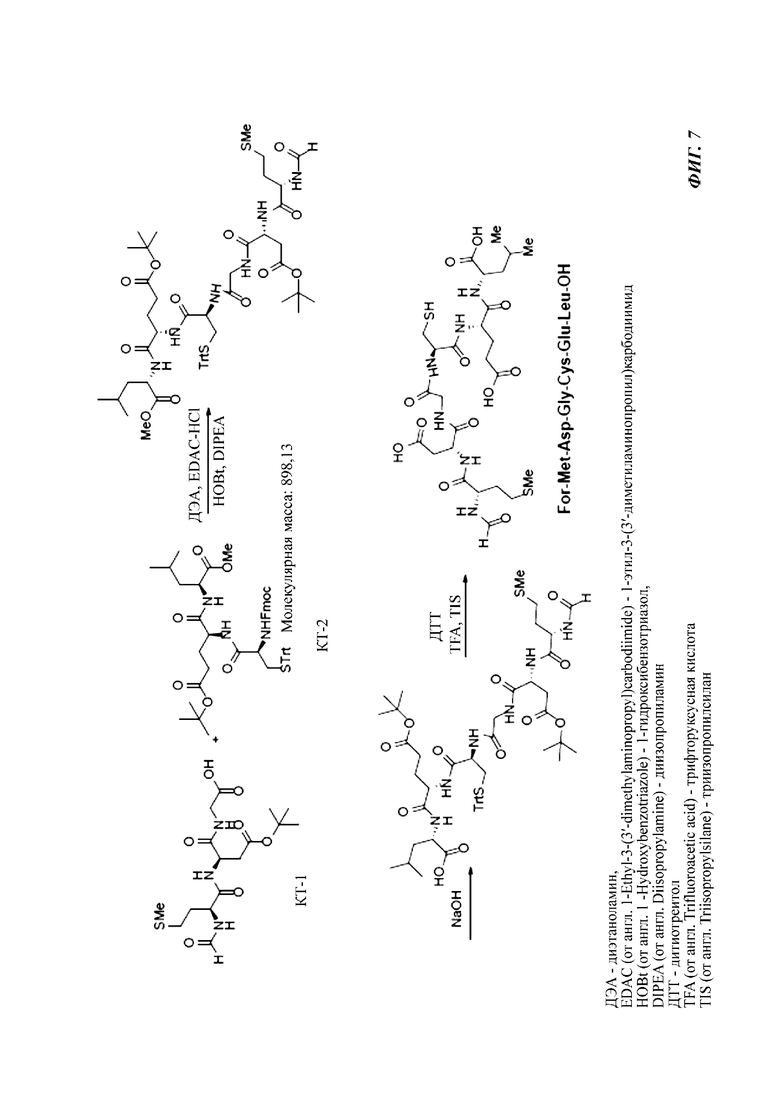
Стратегия сочетания «3 плюс 3» (фигуры 7 и 7а)
Синтез фрагмента KT-1 (For-Met-Asp(OtBu)-Gly-OH) инициируют путем превращения глицина в сложный метиловый эфир глицина. Сочетание его с коммерчески доступным Fmoc-Asp-(O-tBu) дает Fmoc-Asp(OtBu)-Gly-OMe. Снятие защитной группы Fmoc с последующим сочетанием с коммерчески доступным For-Met-OH дает For-Met-Asp(OtBu)-Gly-OMe. Последний при гидролизе сложного метилового эфира дает желаемый фрагмент KT-1.
Чтобы обеспечить необязательное позднее введение формильной группы (или другой ацильной группы), также получают модифицированную версию фрагмента KT-1 - KT-1* (Fmoc-Met-Asp(OtBu)-Gly-OH), при этом сначала подвергают дипептид Fmoc-Asp(OtBu)-Gly-OMe снятию защитной группы Fmoc с последующим сочетанием с коммерчески доступным Fmoc-Met-OH.
Синтез фрагмента KT-2 (Fmoc-Cys(Trt)Glu(OtBu)-Leu-OMe) инициируют путем превращения L-лейцина в его сложный метиловый эфир. Образование амидной связи его с коммерчески доступным Fmoc-Glu-(OtBu) дает Fmoc-Glu(OtBu)-Leu-OMe. Снятие защиты и дальнейшее сочетание с коммерчески доступным Fmoc-Cys(Trt)-OH дает желаемый фрагмент KT-2.
Синтез альтернативного фрагмента KT-2* (Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu) осуществляют путем сочетания трет-бутиллейцината. HCl с Fmoc-Glu-(OtBu) с получением Fmoc-Glu-(OtBu)-Leu-OtBu (т.е. КД-3с). Дипептид не выделяют, а сочетают непосредственно (после снятия защитной группы Fmoc с помощью ДБУ) с Fmoc-Cys(Trt)-OH с получением желаемого фрагмент KT-2*.
Синтез Foxy-5 из трипептидных фрагментов KT-1 и KT-2 осуществляют, начиная со снятия защитной группы Fmoc с KT-2. Последующее сочетание с KT-1 дает For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OMe. Омыление его при основных условиях дает For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OH, а последний при снятии трет-бутильной и тритильной защитных групп дает желаемый гексапептид Foxy-5 в неочищенной форме.
В качестве альтернативы (фигура 7а), синтез Foxy-5 из трипептидных фрагментов KT-1* и KT-2* осуществляют, начиная со снятия защитной группы Fmoc с KT-2*. Последующее сочетание с KT-1* дает Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 4), который затем подвергают снятию защитной группы Fmoc с помощью ДБУ с последующим сочетанием с муравьиной кислотой с получением For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu For-Met-Asp-Gly-Cys-Glu-Leu-OH. Этот защищенный гексапептид подвергают снятию общей защиты (групп Trt и tBu) с получением Foxy-5 в неочищенной форме. Хроматографическая очистка дает желаемый гексапептид For-Met-Asp-Gly-Cys-Glu-Leu-OH (Foxy-5) с чистотой 97,6%.
Стратегия сочетания «4 плюс 1 плюс 1»
Синтез Foxy-5 с помощью стратегии сочетания «4 плюс 1 плюс 1» инициируют путем последовательного сочетания тетрапептида Fmoc-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 6) с Fmoc-Asp-OtBu и Fmoc-Met с последующим удалением Fmoc, формилированием и снятием защиты. Сам тетрапептид обеспечивают путем сочетания дипептидных фрагментов КД-3с Fmoc-Glu-(OtBu)-Leu-OtBu и КД-2 с Fmoc-Gly-Cys(Trt)-OH, раскрываемых выше.
Затем тетрапептид Fmoc-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 6) сочетают с Fmoc-Asp-OtBu с получением защищенного пентапептида Fmoc-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 10) с последующим сочетанием с Fmoc-метионином с получением защищенного гексапептида Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 4). Затем его подвергают снятию защитной группы Fmoc с ДБУ с последующим сочетанием с муравьиной кислотой с получением For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 15). Этот защищенный гексапептид подвергают снятию общей защиты (групп Trt и tBu) с получением Foxy-5 в неочищенной форме. Хроматографическая очистка дает желаемый гексапептид For-Met-Asp-Gly-Cys-Glu-Leu-OH (Foxy-5) с чистотой 97,6%.
Стратегия сочетания «3 плюс 1 плюс 1 плюс 1» (фигуры 8 и 8а) Синтез Foxy-5 с помощью стратегии сочетания «3 плюс 1 плюс 1 плюс 1» инициируют путем последовательного сочетания фрагмента KT-2 (Fmoc-Cys(Trt)Glu(OtBu)-Leu-OMe) с Fmoc-Gly-OH с получением защищенного тетрапептида Fmoc-Gly-Cys(Trt)Glu(OtBu)-Leu-OMe (SEQ ID NOSEQ ID NO 7) с последующим сочетанием с Fmoc-Asp-OtBu с получением защищенного пентапептида Fmoc-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OMe (SEQ ID NO 9) с последующим сочетанием с формил-метионином с получением Foxy-5 в защищенной форме, т.е. For-Met-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OMe (SEQ ID NO 3).
Синтез самого Foxy-5 окончательно достигают с помощью той же стратегии, что и приведенная выше стратегия сочетания «3 плюс 3», т.е. путем омыления сложного метилового эфира лейцина с получением For-Met-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OH (SEQ ID NO 4) и окончательного удаления из него тритильной и трет-бутильной защитных групп с получением Foxy-5 в неочищенной форме.
В качестве альтернативы, синтез Foxy-5 с помощью стратегии сочетания «3 плюс 1 плюс 1 плюс 1» инициируют путем последовательного сочетания фрагмента KT-2* (Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu) с Fmoc-Gly-OH с получением защищенного тетрапептида Fmoc-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 6) с последующим сочетанием с Fmoc-Asp-OtBu с получением защищенного пентапептида Fmoc-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 10), который затем подвергают сочетанию с Fmoc-метионином с получением защищенного гексапептида Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 4). Его затем подвергают снятию защитной группы Fmoc с помощью ДБУ с последующим сочетанием с муравьиной кислотой с получением For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 15). Этот защищенный гексапептид подвергают снятию общей защиты (групп Trt и tBu) с получением Foxy-5 в неочищенной форме. Хроматографическая очистка дает желаемый гексапептид For-Met-Asp-Gly-Cys-Glu-Leu-OH (Foxy-5) с чистотой 97,6%.
Следует отметить, что трипептид For-Met-Asp-Gly-OH и защищенные производные, раскрываемые в настоящем документе, For-Met-Asp(OtBu)-Gly-OMe и For-Met-Asp(OtBu)-Gly-OH представляют собой новые соединения. Подобным образом, трипептид Met-Asp-Gly и раскрываемые в настоящем документе защищенные производные Fmoc-Met-Asp(OtBu)-Gly-OMe, Fmoc-Met-Asp(OtBu)-Gly-OtBu и Fmoc-Met-Asp(OtBu)-Gly-OH (KT-1*) представляют собой новые соединения. Подобным образом, трипептид Cys-Glu-Leu-OH и раскрываемые в настоящем документе защищенные производные Fmoc-Cys(Trt)Glu(OtBu)-Leu-ОМе, (Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu) и Cys-Glu-Leu-OMe представляют собой новые соединения.
Кроме того, следует отметить, что все тетрапептиды Fmoc-Gly-Cys(Trt)Glu(OtBu)-Leu-OMe (SEQ ID NO 7) и Fmoc-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 6), защищенные пентапептиды Fmoc-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OMe (SEQ ID NO 9) и Fmoc-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 10), а также защищенные производные Foxy-5 For-Met-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OMe (SEQ ID NO 3) и For-Met-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 15) представляют собой новые соединения.
Согласно другому аспекту настоящего изобретения, таким образом, представлен защищенный трипептидный фрагмент Foxy-5 (For-Met-Asp-Gly-Cys-Glu-Leu-OH), выбранный из:
a) PG1-Cys(PG2)Glu(OPG3)-Leu-OR1,
b) PG5-Met-Asp(OPG4)-Gly-OR2,
c) PG1-Gly-Cys(PG2)Glu(OPG3)-Leu-OR1,
при этом PG1 выбран из Н и защитных групп, таких как флуоренилметилоксикарбонил (Fmoc) или Boc, PG2 выбран из Н и защитных групп, выбранных из ацетонида (псевдопролина, ψMe,MePro), тритила (Trt), PG3 выбран из Н и защитных групп, выбранных из терт-бутила (tBu), PG5 представляет собой формил или чувствительную к основанию защитную группу, такую как флуоренилметилоксикарбонил (Fmoc), PG4 выбран из Н и защитных групп, выбранных из трет-бутила (tBu), a R1 и R2 независимо выбраны из Н и С1-С6алкила, такого как метил, этил или трет-бутил (tBu).
Согласно предпочтительному варианту осуществления представлен защищенный трипептидный фрагмент Foxy-5, который представляет собой PG1-Cys(PG2)Glu(OPG3)-Leu-OR1, при этом PG1,PG2, PG3 и определены выше. Согласно особенно предпочтительному варианту осуществления указанный трипептидный фрагмент представляет собой Fmoc-Cys(Trt)Glu(OtBu)-Leu-OMe.
Согласно другому предпочтительному варианту осуществления представлен защищенный трипептидный фрагмент Foxy-5, который представляет собой PG5-Met-Asp(OPG4)-Gly-OR2, при этом PG5, PG4 и R2 определены выше. Согласно особенно предпочтительному варианту осуществления указанный трипептидный фрагмент представляет собой Fmoc-Met-Asp(OtBu)-Gly-OtBu.
Согласно следующему аспекту настоящего изобретения представлен жидкофазный способ получения защищенного производного Foxy-5 PG5-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu, при этом способ предусматривает:
a. сочетание дипептидов Fmoc-Glu-(OtBu)-Leu-OtBu (КД-3с) с Fmoc-Gly-Cys(Trt)-OH (КД-2 с) с получением тетрапептида Fmoc-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 6) с последующим сочетанием с PG5-Met-Asp(OtBu)-OH, или
b. сочетание трипептида PG5-Met-Asp(OtBu)-Gly-OH с Fmoc-Cys(Trt)-Glu(OtBu)-Leu-OtBu (KT-2*), или
c. последовательное сочетание трипептида Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu (KT-2*) с Fmoc-Gly-OH, Fmoc-Asp-OtBu и PG5-Met, или
d. последовательное сочетание тетрапептида Fmoc-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 6) с Fmoc-Asp-OtBu и PG5-Met,
с последующей необязательной очисткой неочищенного гексапептида с помощью колоночной хроматографии с последующим осаждением в виде твердого вещества из органического растворителя, и при этом PG5 представляет собой формил или чувствительную к основанию защитную группу, такую как Fmoc.
Настоящее изобретение согласно следующему аспекту относится к жидкофазному способу превращения PG5-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu в Foxy-5 (For-Met-Asp-Gly-Cys-Glu-Leu-OH), при этом способ предусматривает либо для случая, когда PG5 представляет собой формил,
i. снятие защиты с For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 15) с получением Foxy-5 в неочищенной форме,
либо для случая, когда PG5 представляет собой чувствительную к основанию защитную группу, такую как Fmoc,
ii. удаление PG5 из гексапептида PG6-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu с последующим сочетанием с муравьиной кислотой с получением For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 15),
iii. снятие защиты с For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 15) с получением Foxy-5 в неочищенной форме,
а затем:
a. необязательную очистку неочищенного продукта с помощью колоночной хроматографии, необязательно с последующим осаждением в виде твердого вещества из органического растворителя,
b. необязательное осаждение образованного гексапептида Foxy-5 в виде щелочной соли или кислой соли в твердой форме, например, в кристаллической форме.
Согласно предпочтительному варианту осуществления PG5 представляет собой Fmoc.
Экспериментальная часть Пути «2 плюс 2 плюс 2»
Пример 1. Получение ключевого дипептида КД-1а Boc-Met-Asp(OtBu)-OH Синтез сложного метилового эфира Boc-Met-Asp(OtBu)-OMe целевого дипептида осуществляли следующим образом: Boc-Met-OH (2,6 г, 10,4 ммоль, 1,0 экв.) растворяли в ДМФ (сухом) (25 мл), добавляли DCC (2,2 г, 10,4 ммоль, 1,0 экв.) и HOAt (1,4 г, 10,4 ммоль, 1,0 экв.). В сухой 100-мл круглодонной колбе Н-Asp(OtBu)-OMe HCl (2,5 г, 10,4 ммоль, 1,0 экв.) объединяли с DIPEA (1,8 мл, 10,4 ммоль, 1,0 экв.) в ДМФ (сухом) (50 мл). Оба раствора охлаждали до О С.Через 10 минут раствор, содержащий аспартатное производное, каплями переносили в раствор метионина. Обеспечивали нагревание реакционной смеси до комнатной температуры в течение 72 часов. После обработки водным раствором кислоты (0,5 М лимонной кислоты) и основанием (насыщ. NaHCO3) полученный материал анализировали с помощью жидкостной хроматографии с тандемной масс-спектрометрией (ЖХ/МС). Хотя наблюдали некоторые сигналы растворителя (EtOAc) и примесей, эти сигналы соответствовали структуре соединения Boc-Met-Asp(OtBu)-OMe.
На следующей стадии промежуточное соединение сложный метиловый эфир Boc-Met-Asp(OtBu)-OMe (3,5 г, 8,1 ммоль, 1,0 экв.) растворяли в 1,4-диоксане/МеОН (14:1) (75 мл) и добавляли свежеприготовленный 3,6 М раствор NaOH (водн.), (2,3 мл, 8,5 ммоль, 1,0 экв.). Обеспечивали перемешивание реакционной смеси при комнатной температуре в течение 18 часов и отбирали образцы реакционной смеси. Анализ ЖХ/МС показал, что исходный материал был полностью израсходован. Основной сигнал через 2,031 минуты (70% площади пика) показывал массу целевой молекулы Boc-Met-Asp(OtBu)-OH (М составляла 420 г/моль, найдено: масса/заряд+(m/z+) составляющее 421 [М+Н]+, 443 [M+Na]+). Наблюдали побочный продукт, который можно было идентифицировать как Boc-Met-Asp-OH, и поэтому рекомендуется более короткое время реакции (менее 18 часов) для реакции омыления. Целевое соединение ключевой дипептид КД-1a (Boc-Met-Asp(OtBu)-OH) получали с выходом 63%, чистотой 70% по площади пика (ЖХ/МС).
Пример 2. Получение ключевого дипептида КД-2а Cbz-Gly-Cys(ψMe'MePro)-ОН
В 250-мл круглодонную колбу добавляли моногидрат L-цистеина гидрохлорида (5,0 г, 28,5 ммоль) и ацетон (80 мл, 1088 ммоль) (чистый для анализа). Затем реакционную смесь (0,36 молярн.) нагревали до появления конденсата в течение 1,5 часов. Через 30 минут белая суспензия превращалась в густую взвесь. Фильтрование и последующее удаление избытка ацетона давали белое аморфное твердое вещество 4,7 г. Анализ протонный ядерный магнитный резонанс (1Н ЯМР) показал, что осталось приблизительно 50% непрореагировавшего цистеина. Для улучшения выхода и качества пролинацетонида белое аморфное твердое вещество (4,72 г) обрабатывали второй раз ацетоном (200 мл) в более разбавленной реакционной смеси (0,14 М). Суспензию нагревали с обратным холодильником в течение 2 часов. Затем суспензию медленно охлаждали до комнатной температуры, а потом 30 минут при 0°С. Охлажденную льдом суспензию фильтровали через стеклянный фильтр р3 и промывали ацетоном (2 × 10 мл). Остаточный ацетон удаляли in vacuo. Желаемый ацетонид Н-Cys(ψMe'MePro)-ОН (4,4 г, 27 ммоль, выход 95%) получали с превосходными выходом и чистотой в виде белого порошка. Спектр 1Н ЯМР (диметилсульфоксид (ДМСО)) продукта соответствовал структуре целевой молекулы и не показывал сигналов исходного материала H-Cys-OH.
На следующей стадии ацетонидное промежуточное соединение Н-Cys(ψMe'MePro)-OH (1,0 г, 6,2 ммоль, 1,0 экв.) растворяли в ДМФ (50 мл) и добавляли триэтиламин (2,6 мл, 19 ммоль, 3,0 экв.). После перемешивания в течение 10 минут при комнатной температуре добавляли коммерчески доступный Cbz-Gly-OSu (2,4 г, 6,2 ммоль, 1,0 экв.) и обеспечивали перемешивание реакционной смеси в течение 18 часов, на протяжении которых смесь оставалась суспензией.
Реакционную смесь подкисляли путем добавления 4 М хлористоводородной кислоты в 1,4-диоксане (10,11 мл, 10,11 ммоль) до рН равного 4. В этот момент осаждалось твердое вещество, которое собирали путем фильтрования. Остаток в основном состоял из соли Et3N⋅HCl. К фильтрату добавляли 250 мл воды и экстрагировали его с помощью EtOAc (3 × 50 мл). Затем объединенную органическую фазу промывали солевым раствором (2 * 50 мл), сушили над Na2SO4, фильтровали и концентрировали с получением грязно-белого твердого вещества (1,58 г). Анализ ЖХ/МС объединенной органической фазы показывал основной сигнал через 2,003 минуты (77% по площади пика), что представляет дипептид Cbz-Gly-Cys(ψMe,MePro)-OH (М составляет 352 г/моль, найдено: m/z- составляющее 351 [М-Н]-, 703 [2М-Н]-). Через 1,779 минуты (9% по площади пика) наблюдали Cbz-Gly-OH (сложный сукцинимидный эфир Cbz-Gly-OSu через 1,887 минуты не наблюдали, что указывает на гидролиз исходного материала).
Затем водную фазу дополнительно подкисляли до рН равного 1 и снова экстрагировали с помощью EtOAc (3 × 50 мл). Объединенную органическую фазу промывали солевым раствором (3 × 50 мл), сушили над Na2SO4, фильтровали и концентрировали с получением желтоватого масла (2,60 г). Анализ ЖХ/МС показывал целевую молекулу через 2,006 минуты (66% по площади пика). Основной сигнал побочного продукта наблюдали через 1,778 минуты (20% по площади пика) и относили к Cbz-Gly-OH. Объединенный выход, без дополнительной очистки, дипептида Cbz-Gly-Cys(ψMe,MePro)-OH составлял 4,2 г (83%, на основании чистоты 70% по данным ЖХ/МС). Основную примесь идентифицировали как Cbz-Gly-OH.
Пример 2а. Получение альтернативного дипептида КД-1 с Fmoc-Met-Asp(OtBu)-OH
Fmoc-Met-Asp(OtBu)-OH Fmoc-Met-OSu получали согласно WO 1990008773 A1, in situ, с помощью реакции Fmoc-Met-OH (14,87 г), N-гидроксисукцинимида (HOSu, 5,52 г) и дициклогексилкарбодиимида (DCC, 8,26 г) в тетрагидрофуране (ТГФ, 200 мл) при 0°С в течение 3,5 часа. Осажденную дициклогексил мочевину (DCU, от англ. dicyclohexylurea) удаляли путем фильтрования и ТГФ фильтрат добавляли в холодный раствор H-Asp(OtBu)-OH в 220 мл воды/ТГФ 10:1, к которому было добавлено 40 мл N гидроксида натрия. После перемешивания реакционной смеси при комнатной температуре на протяжении ночи добавляли твердую лимонную кислоту (20 г) вместе с EtOAc (600 мл). Слой EtOAc отделяли, промывали 10% лимонной кислотой, солевым раствором и сушили (сульфатом магния). Выпаривание раствора EtOAc давало остаток, который растворяли в 200 мл EtOAc и обрабатывали дициклогексиламином (ДЦГА, 7,84 мл) с осаждением 17,93 г соли ДЦГА желаемого продукта, точка плавления 159-162°С.
Пример 3. Получение ключевого дипептида КД-3а H-Glu(OBn)-Leu-OBn В 250-мл круглодонной колбе охлажденный раствор коммерчески доступного Cbz-Glu(OtBu)-OH (5,0 г, 14,8 ммоль, 1,0 экв.) объединяли с HOAt (2,4 г, 17,8 ммоль, 1,2 экв.) и DCC (3,7 г, 17,8 ммоль, 1,2 экв.) в безводном MeCN (25 мл). В этот раствор добавляли смесь H-Leu-OtBu HCl (3,3 г, 14,8 ммоль, 1,0 экв.) и DIPEA (2,58 мл, 14,82 ммоль) в безводном MeCN (50 мл). Полученную в результате реакционную смесь (0,2 молярн.) перемешивали при 0°С с постепенным повышением температуры до комнатной температуры за 18 часов. Затем реакционную смесь концентрировали in vacuo с получением 9,1 г (макс, выход 83% на основании чистоты 70% по площади пика ВЭЖХ) густого желтого масла, которое медленно затвердевало при отстаивании в течение нескольких часов. Часть его (5,2 г) суспендировали в 50 мл EtOAc и добавляли 30 мл H2O. Органическую фазу промывали и выпаривали, что давало 2,9 г белого твердого вещества. Анализ ВЭЖХ показывал продукт через 4,105 минуты (76% по площади пика). Анализ 1Н ЯМР соответствовал целевой молекуле, и Cbz-Glu(OtBu)-Leu-OtBu получали с расчетным выходом 52%, средней чистотой 76% (на основании анализа ВЭЖХ) без хроматографии с выходом 63% и чистотой 95% на основании анализа 1Н ЯМР. Материал использовали в последующих реакциях без дополнительной очистки.
Наконец, после тщательного отбора катализатора далее использовали катализатор Pd/C Noblyst типа Р1141 для осуществления снятия защитной группы Cbz с получением целевого дипептида КД-3а H-Glu(OBn)-Leu-OBn. Для этой реакции Cbz-Glu(OtBu)-Leu-OtBu (1,5 г, 3,0 ммоль, 1,0 экв.) растворяли в МеОН (20 мл) и добавляли Pd (10% на активированном угле типа Noblyst Р1141) (500 мг, 470 мкмоль, 0,2 экв.). Через раствор барботировали Н2 в течение 1 часа. Затем обеспечивали перемешивание реакционной смеси в атмосфере водорода (1 атм.) в течение 18 часов. Анализ ВЭЖХ (проведенный через 18 часов) показывал полное превращение исходного материала и показывал в основном сигнал толуола через 3,391 минуты и сигнал целевой молекулы через 2,843 минуты. Реакционную смесь фильтровали через целит на стеклянном фильтре р3. Остаток промывали с помощью МеОН (3 × 1 мл) и фильтрат концентрировали in vacuo. Получали целевое соединение HGlu(OtBu)-Leu-OtBu в виде желтоватого масла, 1,02 г (выход 92%) с чистотой 95% по площади пика на основании анализа ВЭЖХ. Этот материал использовали для последующих реакций по мере его получения.
Пример 3а. Получение альтернативного ключевого дипептида КД-3с Fmoc-Glu-(OtBu)-Leu-OtBu
50 г трет-бутиллейцината.HCl сочетали с 1,3 экв. Fmoc-Glu-(OtBu) в дихлорметане при большом разбавлении (50 объем.) в присутствии EDAC, HCl (2,0 экв.), HOBt (2,0 экв.) и DIPEA (5,0 экв.) при начальной температуре 0-5°С в течение 1 часа, а затем при 15-20°С в течение 1 часа, что давало дипептид Fmoc-Glu-(OtBu)-Leu-OtBu (КД-3с). Идентичность подтверждали с помощью 1Н ЯМР и масс-спектрометрии. Для реакции с Fmoc-Gly-Cys(Trt)-OH выделение продукта не проводили и дихлорметановый раствор использовали непосредственно в примере 4а после водной обработки.
Пример 4. Получение тетрапептида Gly-Cys(ψMe,MPro)-Glu(OtBu)-l_eu-OtBu (SEQ ID NO 5)
Следующая реакция предусматривала сочетание дипептидов H-Glu(OtBu)-Leu-OtBu и Cbz-Gly-Cys(ψMe,MePro)-OH в присутствии DCC и HOAt.
Реакцию сочетания осуществляли следующим образом: H-Glu(OtBu)-Leu-OtBu (898 мг, 2,4 ммоль, 1,0 экв.) растворяли в сухом ДМФ (20 мл) и раствор охлаждали до 0°С. Затем добавляли DCC (498 мг, 2,4 ммоль, 1,0 экв.) и HOAt (328 мг, 2,4 ммоль, 1,0 экв.). К смеси добавляли Cbz-Gly-Cys(ψMe,MePro)-OH (850 мг, 2,4 ммоль, 1,0 экв.) и реакционную смесь перемешивали, обеспечивая ее постепенное нагревание до комнатной температуры в течение 18 часов.
Реакционную смесь фильтровали через целит в стеклянном фильтре р3. Остаток промывали с помощью ДМФ (2×5 мл). В фильтрат добавляли 100 мл Н20, при этом образовывался молочный коллоидный раствор. Добавляли EtOAc (50 мл). Разделение фаз было очень медленным, и было решено добавить приблизительно 20 г NaCl в смесь, после чего смесь слегка осветлялась и появлялось четкое разделение фаз. Затем водную фазу дважды экстрагировали с помощью EtOAc (50 мл). Полученная в результате водная фаза подвергалась отбору образцов для анализа ЖХ/МС и не содержала массы продукта. Объединенные органические фазы обрабатывали в соответствии со стандартной процедурой, в результате чего получали 1,41 г желтоватого масла, которое затвердевало при совместном выпаривании с Et20 до грязно-белого объемистого твердого вещества (1,41 г, выход 58% на основании чистоты 70%). Анализ ЖХ/МС показал, что присутствует приблизительно 6% по площади пика DCC, что соответствует сигналу через 2,034 минуты (М составляла 224 г/моль, найдено: m/z+ составляющее 225 [М+Н]+, 449 [M+Na]+). Через 2,297 минуты наблюдали неизвестное соединение с m/z+равным 452, 579 (16% по площади пика). Целевую молекулу Cbz-Gly-Cys(ψMe,MePro)-Glu(OtBu)-Leu-OtBu наблюдали через 2,404 минуты (76% по площади пика) (М составляла 706 г/моль, найдено: m/z+составляющее707 [М+Н]+, 729 [M+Na]+).
Затем осуществляли последующее снятие защитной группы Cbz с Cbz-Gly-Cys(ψMe,MePro)-Glu(OtBu)-Leu-OtBu с помощью катализатора Noblyst Р1141, который ранее упоминался в реакции снятия защитной группы с дипептида Cbz-Glu(OtBu)-Leu-OtBu. В этом конкретном случае выбирали данный катализатор, поскольку он демонстрировал хорошую реакционную способность в скрининговом эксперименте, и исходный материал был полностью превращен за время реакции 18 часов. Поэтому, данные условия также применяли для селективного снятия защитной группы Cbz с тетрапептида Cbz-Gly-Cys(ψMe,MePro)-Glu(OtBu)-Leu-OtBu. Первоначальная тестовая реакция показала, что для удовлетворительной скорости реакции гидрогенизация тетрапептида (Cbz-Gly-Cys(ψMe,MePro)-Glu(OtBu)-Leu-OtBu) требовала больше Pd/C, чем использовали для дипептида Cbz-Glu(OtBu)-Leu-OtBu.
Для этой реакции Cbz-Gly-Cys(ψMe,MePro)-Glu(OtBu)-Leu-OtBu (700 мг, 1,0 ммоль, 1,0 экв.) растворяли в МеОН (10 мл) и добавляли Pd/C (Noblyst Р1141, 10 мас./мас. % Pd, 60% H2O) (700 мг, 40 мкмоль, 4 мол. %). Обеспечивали перемешивание реакционной смеси в атмосфере водорода (1 атм.). Реакцию сопровождали анализом ВЭЖХ.
По прошествии 5 часов реакции все еще наблюдали 23% по площади пика исходного материала. Через 24 часа анализ ВЭЖХ показывал, что все еще оставалось 12% по площади пика исходного материала. Затем добавляли дополнительное количество катализатора Pd/C (200 мг). Через 72 часа общего времени реакции анализ ВЭЖХ показывал, что исходный материал был почти полностью израсходован (оставалось 0,84% по площади пика). Реакционную смесь фильтровали через гифло на стеклянном фильтре р3 и промывали с помощью МеОН (3×5 мл). Фильтрат концентрировали in vacuo с получением 550 мг желтоватого масла. Анализ ВЭЖХ показывал основной сигнал через 1,929 минуты (81% по площади пика), что отображало массу целевой молекулы (М составляла 572 г/моль, найдено: m/z+573 [М+Н]*).
Тетрапептид H-Gly-Cys(ψMe,MePro)-Glu(OtBu)-l_eu-OtBu (550 мг, 0,78 ммоль, выход 79%) получали с чистотой 80% (на основании ЖХ/МС).
Пример 4а. Получение альтернативного тетрапептида Fmoc-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 6)
Последующую реакцию полученного дипептида КД-3с Fmoc-Glu-(OtBu)-Leu-OtBu осуществляли с использованием дихлорметанового раствора, упоминаемого выше (пример 3а). Снятие защитной группы Fmoc достигали с помощью ДБУ и сочетание с 1,3 экв. Fmoc-Gly-Cys(Trt)-OH (коммерчески доступного) осуществляли в присутствии DIPEA (3 экв.), EDC.HCI (2,0 экв.) и HOBt (2,0 экв.) с получением защищенного тетрапептида Fmoc-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 6). Слой ДХМ промывали водой и солевым раствором, концентрировали до 10-15 об. и использовали как есть на следующей стадии (пример 5а) без выделения.
Пример 5. Получение гексапептида Boc-Met-AsprotBuJ-Gly-Cys(ψMe,MePro)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 2)
Затем осуществляли сочетание пептидов Boc-Met-Asp(OtBu)-OH и H-Gly-Cys(ψMe,MePro)-Glu(OtBu)-Leu-OtBu с получением гексапептида Boc-Met-Asp(OtBu)-Gly-Cys(ψMe,MePro)-Glu(OtBu)-Leu-OtBu в присутствии реагента сочетания DCC и с добавлением HOAt в ДМФ при комнатной температуре в течение 18 часов.
Для этой реакции Boc-Met-Asp(OtBu)-OH (200 мг, 0,48 ммоль, 1,0 экв.) растворяли в ДМФ (2,0 мл) и охлаждали в ледяной бане. В прозрачный раствор добавляли HOAt (65 мг, 0,48 ммоль, 1,0 экв.) и DCC (98 мг, 0,48 ммоль, 1,0 экв.). Через 15 минут добавляли HCys(ψMe,MePro)-Glu(OtBu)-Leu-OtBu (272 мг, 0,48 ммоль, 1,0 экв.) и реакционную смесь перемешивали в течение 18 часов при медленном нагревании до комнатной температуры. Затем реакционную смесь обрабатывали в соответствии со стандартной процедурой. Анализ ЖХ/МС обработанной партии показывал целевую молекулу Boc-Met-Asp(OtBu)-Gly-Cys(ψMe,MePro)-Glu(OtBu)-Leu-OtBu после времени удерживания 2,760 минуты (М составляла 974 г/моль, найдено: m/z+ составляющее975 [М+Н]+, 997 [M+Na]4). Целевую молекулу Boc-Met-Asp(OtBu)-Gly-Cys(ψMe,MePro)-Glu(OtBu)-Leu-OtBu получали с выходом 76% (420 мг, 0,36 ммоль) на основании чистоты 84% (ЖХ/МС) в виде объемистого белого твердого вещества.
Пример 5а. Получение альтернативного гексапептида Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 4) и его превращение в Foxy-5
Fmoc-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 6), полученный, как раскрывается выше в примере 4а, в виде концентрированного раствора ДХМ сначала подвергали реагированию с ДБУ для достижения снятия защитной группы Fmoc. Перед тем, как приступить к следующей стадии сочетания, реакционную массу пропускали через пробку оксида кремния для удаления ДБУ. После обработки пробкой оксида кремния раствор ДХМ подвергали реагированию (после высвобождения) с Fmoc-Met-Asp(OtBu)-OH (КД-1с), полученным в виде соли ДЦГА в приведенном выше примере 2а, в присутствии DIPEA (3,0 экв.), EDC.HCl (2,0 экв.) и HOBt.H2O (2,0 экв.) с получением защищенного производного Foxy-5 Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 4) в неочищенной форме. Затем его подвергали снятию защитной группы Fmoc с помощью ДБУ в ДХМ с последующим сочетанием с муравьиной кислотой в присутствии EDC.HCl, HOBt.H2O и DIPEA с получением For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 15). Этот защищенный гексапептид подвергали снятию общей защиты (групп Trt и tBu) путем растворения и перемешивания в коктейле TFA/(i-Pr)3SiH/ДТТ. После завершения реакции неочищенный продукт получали в виде твердого вещества путем осаждения с помощью ТГФ/МТБЭ. Хроматографическая очистка давала желаемый гексапептид For-Met-Asp-Gly-Cys-Glu-Leu-OH (Foxy-5) с выходом более 90% и чистотой приблизительно 97%.
Путь «3 плюс 3»
Пример 6. Получение фрагмента KT-1 и KT-1* (For-Met-Asp(OtBu)-Gly-OH и Fmoc-Met-Asp(OtBu)-Gly-OH, соответственно)
Пример 6а. Пример с использованием Fmoc-метионина (1 Для обеспечения возможности последующего необязательного введения формильной группы.) вместо формил-метионина при получении трипептида KT-1* (Fmoc-Met-Asp(OtBu)-Gly-OH)
Сложный метиловый эфир глицина (25 г) подвергали реагированию в ДМФ с Fmoc-Asp(OtBu)-OH в присутствии DIPEA (1,0 экв.), EDAC, HCl (2,0 экв.) и HOBt (2,0 экв.). После стандартной обработки неочищенный продукт очищали на силикагеле с использованием 15% EtOAc/н-гексана. Сочетали 1 г его с Fmoc-Met с получением защищенного трипептида Fmoc-Met-Asp(OtBu)-Gly-OMe (2,0 г, 72,7%). Идентичность подтверждали с помощью 1Н ЯМР и масс-спектрометрии.
Пример 6b. Получение фрагмента KT-1 (For-Met-Asp(OtBu)-Gly-OH) Сложный метиловый эфир глицина сочетали с Fmoc-Asp(OtBu)-OH в соответствии с процедурой, раскрываемой в примере 6а, с получением защищенного дипептида Fmoc-Asp(OtBu)-Gly-OMe. Снятие его защитной группы Fmoc достигали с помощью ДБУ, и сочетание с коммерчески доступным For-Met-ОН в ДХМ в присутствии DIPEA (1,0 экв.), EDAC.HCl (2,0 экв.) и HOBt (2,0 экв.) давало желаемый фрагмент KT-1.
Пример 7. Получение фрагмента KT-2 (Fmoc-Cys(Trt)Glu(OtBu)-Leu-OMe) Fmoc-лейцин (250 г) растворяли в метаноле и превращали в Fmoc-метиллейцинат с количественным выходом путем обработки тионилхлоридом. Снятие защитной группы Fmoc с ДБУ в дихлорметане давало желаемый сложный метиловый эфир лейцина (76 г, общий выход 74%). Идентичность подтверждали с помощью 1Н ЯМР и масс-спектрометрии. Далее сочетание 10 г сложного метилового эфира лейцина с Fmoc-Glu-(OtBu) в дихлорметане при большом разбавлении в присутствии EDAC, HCl (2,0 экв.) и HOBt (2,0 экв.) сначала при 0-5 С в течение 1 часа, а затем при 15-20 С в течение 1 часа давало дипептид Fmoc-Glu-(OtBu)-Leu-OMe (7,2 г, общий выход 55%). Идентичность подтверждали с помощью 1Н ЯМР и масс-спектрометрии. Для реакции с Fmoc-цистеином на следующей стадии выделение продукта не проводили и раствор дихлорметана использовали непосредственно после водной обработки. Последующую реакцию полученного дипептида Fmoc-Glu-(OtBu)-Leu-OMe осуществляли, таким образом, с использованием раствора дихлорметана, упоминаемого выше. Снятия защитной группы Fmoc достигали с помощью ДБУ, и сочетание с Fmoc-Cys(Trt)-ОН в присутствии EDAC, HCl (1,2 экв.) и HOBt (1,2 экв.) давало фрагмент KT-2 Fmoc-Cys(Trt)-Glu-(OtBu)-Leu-OMe (6,6 г, общий выход 88% за две реакции сочетания). Идентичность подтверждали с помощью 1Н ЯМР и масс-спектрометрии.
Пример 8. Получение альтернативного трипептидного фрагмента Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu (KT-2*).
50 г трет-бутиллейцината.НС1 сочетали с 1,3 экв. Fmoc-Glu-(OtBu) в дихлорметане при большом разбавлении (50 об.) в присутствии EDAC, HCl (2,0 экв.), HOBt (2,0 экв.) и DIPEA (5,0 экв.) сначала при 0-5 С в течение 1 часа, а затем при 15-20 С в течение 1 часа, что давало дипептид Fmoc-Glu-(OtBu)-Leu-OtBu (КД-3с). Идентичность подтверждали с помощью 1Н ЯМР и масс-спектрометрии. Для реакции с Fmoc-Cys(Trt)-OH на следующей стадии выделение продукта не проводили и раствор дихлорметана использовали непосредственно после водной обработки. Последующую реакцию полученного дипептида КД-3с осуществляли, таким образом, с использованием раствора дихлорметана, упоминаемого выше. Снятия защитной группы Fmoc достигали с помощью ДБУ, и сочетание с 1,3 экв. Fmoc-Cys(Trt)-OH в присутствии EDAC, HCl (1,2 экв.), HOBt (1,2 экв.) и DIPEA (5 экв.) давало неочищенный трипептид KT-2*, который очищали путем хроматографии на силикагеле (100-200) с использованием EtOAc-гексана в качестве элюента с получением трипептида KT-2* Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu в виде белого твердого вещества (148 г, общий выход 68% за две реакции сочетания). Идентичность подтверждали с помощью 1Н ЯМР и масс-спектрометрии.
Упомянутые выше многостадийные реакции повторяли с 75 г трет-бутиллейцината.HCl с получением 208 г KT-2*.
Пример 9а. Получение Foxy-5 путем сочетания трипептидного фрагмента KT-1 и KT-2 с последующим снятием защиты
Синтез Foxy-5 из двух трипептидных фрагментов KT-1 и KT-2 осуществляли, начиная со снятия защитной группы Fmoc с KT-2. Последующее сочетание с KT-1 давало For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OMe. Затем омыление при основных условиях давало For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OH, и последний при снятии O-tBu и тритильных защитных групп давал желаемый гексапептид Foxy-5 в неочищенной форме.
Пример 9b. Получение Foxy-5 путем сочетания трипептидного фрагмента KT-1* и KT-2* с последующим удалением Fmoc, формилированием и снятием защиты
Синтез Foxy-5 из двух трипептидных фрагментов KT-1* и KT-2* осуществляли, начиная со снятия защитной группы Fmoc с KT-2*. Последующее сочетание с KT-1* давало Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 4), который затем подвергали снятию защитной группы Fmoc с помощью ДБУ в ДХМ с последующим сочетанием с муравьиной кислотой в присутствии EDC.HCI, HOBt.H2Щ и DIPEA с получением For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 15). Этот защищенный гексапептид подвергали снятию общей защиты (групп Trt и tBu) путем растворения и перемешивания в коктейле TFA/(i-Pr)3SiH/ДТТ. После завершения реакции неочищенный продукт получали в виде твердого вещества путем осаждения с помощью ТГФ/МТБЭ. Хроматографическая очистка давала желаемый гексапептид For-Met-Asp-Gly-Cys-Glu-Leu-OH (Foxy-5) с выходом более 90% чистотой приблизительно 97%.
Путь «4 плюс 1 плюс 1»
Пример 10. Получение Foxy-5 путем последовательного сочетания тетрапептида Fmoc-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 6) с Fmoc-Asp-OtBu и Fmoc-Met с последующим удалением Fmoc, формилированием и снятием защиты
Fmoc-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 6), полученный, как раскрывается выше в примере 4а, сначала подвергали реагированию с ДБУ для достижения снятия защитной группы Fmoc. Прежде чем перейти к следующей стадии сочетания, реакционную массу пропускали через пробку оксида кремния для удаления ДБУ, который, как было обнаружено в предыдущих экспериментах, вызывает образование нежелательного побочного продукта аспартимида. Удаление ДБУ перед сочетанием с Fmoc-Asp-OtBu эффективно подавляет образование аспартимида. После обработки с помощью пробки из оксида кремния раствор ДХМ подвергали реагированию с Fmoc-Asp(OtBu) в присутствии DIPEA, EDAC, HCl (1,2 экв.) и HOBt (1,2 экв.) с получением защищенного пентапептида Fmoc-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 3). Идентичность продукта подтверждали с помощью 1Н ЯМР и масс-спектрометрии.
Снятия защитной группы Fmoc с полученного пентапептида Fmoc-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 3) достигали с помощью ДБУ, а сочетание с Fmoc-Met в ДХМ-ТГФ (50 об. плюс 10 об.) в качестве растворителя в присутствии DIPEA (3,0 экв.), EDC.HCl (2,0 экв.) и HOBt.H2O (2,0 экв.) давало защищенный гексапептид Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 4) в неочищенной форме.
Снятия защитной группы Fmoc с полученного гексапептида Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 4) из примера 4 достигали с помощью ДБУ в ДХМ (50 об.) с последующим сочетанием с муравьиной кислотой (3,0 экв.) в присутствии EDC.HCI (4,0 экв.), HOBt.H2O (4,0 экв.) и DIPEA (4,0 экв.) с получением For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 15), который подвергали снятию общей защиты (групп Trt и tBu) путем растворения и перемешивания в коктейле TFA (10 o6.)/(i-Pr)3SiH (TIS, 1,7 об.)/ДТТ (1,7 экв.) в течение 1,5 часа. После завершения реакции неочищенный продукт получали в виде твердого вещества с выходом 97% (9,5 г) путем осаждения с помощью ТГФ/МТБЭ. Хроматографическая очистка давала желаемый гексапептид For-Met-Asp-Gly-Cys-Glu-Leu-OH (Foxy-5) с чистотой 97,6%.
Путь «3 плюс 1 плюс 1 плюс 1»
Пример 11. Применимость линейного жидкофазного подхода к Foxy-5 KT-2 (Fmoc-Cys(Trt)Glu(OtBu)-Leu-OMe), полученный, как раскрывается выше, подвергали реагированию с ДБУ в ДХМ для достижения снятия защитной группы Fmoc, а затем с Fmoc-Gly-OH в присутствии DIPEA, EDAC, HCl (1,2 экв.) и HOBt (1,2 экв.) с получением защищенного тетрапептида Fmoc-Gly-Cys(Trt)-Glu(OtBu)-Leu-OMe (SEQ ID NO 7). Идентичность продукта подтверждали с помощью 1Н ЯМР и масс-спектрометрии. Неочищенный продукт далее подвергали реагированию аналогичным образом с ДБУ в ДХМ для достижения снятия защитной группы Fmoc, а затем с Fmoc-Asp(OtBu) в присутствии DIPEA, EDAC, HCl (1,2 экв.) и HOBt (1,2 экв.) с получением защищенного пентапептида Fmoc-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OMe (SEQ ID NO 9), который получали в виде белого твердого вещества после очистки на силикагеле и суспендирования в метаноле. Идентичность продукта подтверждали с помощью 1Н ЯМР и масс-спектрометрии. Снятие защитной группы Fmoc с полученного пентапептида достигали с помощью ДБУ, и сочетание с коммерчески доступным For-Met-OH в ДХМ в присутствии DIPEA (1,0 экв.), EDAC, HCl (2,0 экв.) и HOBt (2,0 экв.) давало защищенное производное Foxy-5 For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OMe (SEQ ID NO 3), которое при омылении при основных условиях давало For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OH (SEQ ID NO 4), a последний при снятии O-tBu и тритильной защитных групп давал желаемый гексапептид Foxy-5 в неочищенной форме.
Пример 12. Fmoc-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 6) KT-2* (Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu), полученный, как раскрывается выше в примере 8, подвергали реагированию с ДБУ в ДХМ (50 об.) для достижения снятия защитной группы Fmoc, а затем подвергали реагированию с 1,3 экв. Fmoc-Gly-OH в присутствии DIPEA (3 экв.), EDC.HCl (2,0 экв.) и HOBt (2,0 экв.) с получением защищенного тетрапептида Fmoc-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 6). Надлежащее превращение наблюдали с помощью тонкослойной хроматографии (ТСХ) и слой ДХМ промывали водой и солевым раствором. Конечный органический слой концентрировали до 10-15 об. и использовали как есть на следующей стадии без выделения.
Пример 13. Fmoc-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 10)
Fmoc-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 6), полученный, как раскрывается выше, в виде концентрированного раствора ДХМ, сначала подвергали реагированию с ДБУ для достижения снятия защитной группы Fmoc. Прежде чем перейти к следующей стадии сочетания, реакционную массу пропускали через пробку из оксида кремния для удаления ДБУ, который, как было обнаружено в предыдущих экспериментах, вызывает образование нежелательного побочного продукта аспартимида. Удаление ДБУ перед сочетанием с Fmoc-Asp-OtBu эффективно подавляет образование аспартимида. После обработки с помощью пробки из оксида кремния раствор ДХМ подвергали реагированию с Fmoc-Asp(OtBu) в присутствии DIPEA, EDAC, HCl (1,2 экв.) и HOBt (1,2 экв.) с получением защищенного пентапептида Fmoc-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 3). Идентичность продукта подтверждали с помощью 1Н ЯМР и масс-спектрометрии.
Многостадийные реакции повторяли дважды, начиная с 148 г и 220 г исходного материала, с получением 107 г и 163 г продукта, соответственно (58,1% и 59,2% от теоретического).
Пример 14. Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 4)
Снятия защитной группы Fmoc с полученного пентапептида Fmoc-Asp(OtBu)-Gly-Cys(Trt)Glu(OtBu)-Leu-OtBu (SEQ ID NO 10) из примера 13 достигали с помощью ДБУ, и сочетание с Fmoc-Met в ДХМ-ТГФ (50 об. плюс 10 об.) в качестве растворителя в присутствии DIPEA (3,0 экв.), EDC.HCl (2,0 экв.) и HOBt.H2O (2,0 экв.) давало защищенное производное Foxy-5 Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 4) в неочищенной форме. Очистку осуществляли с помощью колоночной хроматографии с использованием ДХМ/ТГФ в качестве элюента. Очищенный продукт суспендировали в DIPE с получением белого твердого вещества.
Очистку осуществляли несколько раз при разных условиях, таких как осаждение с антирастворителями и хроматография. Выяснили, что наилучшим решением является колоночная хроматография с последующим суспендированием в DIPE, что в масштабе 25 г давало выходы с химической чистотой 78% и 95,4%.
Пример 15. For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu, (SEQ ID NO 15)
Снятия защитной группы Fmoc с полученного гексапептида Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 4) из примера 14 достигали с помощью ДБУ в ДХМ (50 об.) с последующим сочетанием с муравьиной кислотой (3,0 экв.) в присутствии EDC.HCI (4,0 экв.), HOBt.H20 (4,0 экв.) и DIPEA (4,0 экв.) с получением For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ_ NO 15).
Реакцию осуществляли три раза с 4, 18 и 18 г исходного материала, соответственно, с получением выхода 75-83% и химической чистоты от 67,5 до 77,2%.
Пример 16. For-Met-Asp-Gly-Cys-Glu-Leu-OH (Foxy-5), (SEQ ID NO 1) 14 г полученного гексапептида из примера 15 подвергали снятию общей защиты (групп Trt и tBu) путем растворения и перемешивания в коктейле TFA (10 o6.)/(i-Pr)3SiH (TIS, 1,7 об.)/ДТТ (1,7 экв.) в течение 1,5 часа. После завершения реакции неочищенный продукт получали в виде твердого вещества с выходом 97% (9,5 г) путем осаждения с помощью ТГФ/МТБЭ. Хроматографическая очистка давала желаемый гексапептид For-Met-Asp-Gly-Cys-Glu-Leu-OH (Foxy-5) с чистотой 97,6%.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> WntResearch AB
<120> ЖИДКОФАЗНЫЕ ПУТИ ДЛЯ ГЕКСАПЕПТИДОВ WNT
<130> 138735
<160> 15
<170> BiSSAP 1.3.6
<210> 1
<211> 6
<212> Белок
<213> Искусственная последовательность
<220>
<221> Посттрансляционная модификация остатка
<222> 1
<223> Формил-метионин
<220>
<223> Пептид
<400> 1
Met Asp Gly Cys Glu Leu
1 5
<210> 2
<211> 6
<212> Белок
<213> Искусственная последовательность
<220>
<221> Посттрансляционная модификация остатка
<222> 1
<223> (Трет-бутоксикарбонил)-метионин
<220>
<223> Cинтетический пептид
<220>
<221> Посттрансляционная модификация остатка
<222> 2
<223> Сложный трет-бутиловый эфир аспарагиновой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 4
<223> Тритилцистеин
<220>
<221> Посттрансляционная модификация остатка
<222> 5
<223> Сложный трет-бутиловый эфир глютаминовой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 6
<223> Сложный трет-бутиловый эфир лейцина
<400> 2
Met Asp Gly Cys Glu Leu
1 5
<210> 3
<211> 5
<212> Белок
<213> Искусственная последовательность
<220>
<221> Посттрансляционная модификация остатка
<222> 1
<223> (Флуоренилметоксикарбонил)-сложный трет-бутиловый эфир
аспарагиновой кислоты
<220>
<223> Cинтетический пептид
<220>
<221> Посттрансляционная модификация остатка
<222> 3
<223> Тритилцистеин
<220>
<221> Посттрансляционная модификация остатка
<222> 4
<223> Сложный трет-бутиловый эфир глютаминовой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 5
<223> Сложный трет-бутиловый эфир лейцина
<400> 3
Asp Gly Cys Glu Leu
1 5
<210> 4
<211> 6
<212> Белок
<213> Искусственная последовательность
<220>
<221> Посттрансляционная модификация остатка
<222> 1
<223> Формил-метионин
<220>
<223> Cинтетический пептид
<220>
<221> Посттрансляционная модификация остатка
<222> 2
<223> Сложный трет-бутиловый эфир аспарагиновой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 4
<223> Тритилцистеин
<220>
<221> Посттрансляционная модификация остатка
<222> 5
<223> Сложный трет-бутиловый эфир глютаминовой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 6
<223> Сложный трет-бутиловый эфир лейцина
<400> 4
Met Asp Gly Cys Glu Leu
1 5
<210> 5
<211> 4
<212> Белок
<213> Искусственная последовательность
<220>
<223> Cинтетический пептид
<220>
<221> Посттрансляционная модификация остатка
<222> 2
<223> Цистеина ацетонид
<220>
<221> Посттрансляционная модификация остатка
<222> 3
<223> Сложный трет-бутиловый эфир глютаминовой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 4
<223> Сложный трет-бутиловый эфир лейцина
<400> 5
Gly Cys Glu Leu
1
<210> 6
<211> 4
<212> Белок
<213> Искусственная последовательность
<220>
<221> Посттрансляционная модификация остатка
<222> 1
<223> (Флуоренилметоксикарбонил)-глицин
<220>
<223> Синтетический пептид
<220>
<221> Посттрансляционная модификация остатка
<222> 2
<223> Тритилцистеин
<220>
<221> Посттрансляционная модификация остатка
<222> 3
<223> Сложный трет-бутиловый эфир глютаминовой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 4
<223> Сложный трет-бутиловый эфир лейцина
<400> 6
Gly Cys Glu Leu
1
<210> 7
<211> 4
<212> Белок
<213> Искусственная последовательность
<220>
<221> Посттрансляционная модификация остатка
<222> 1
<223> (Флуоренилметоксикарбонил)-глицин
<220>
<223> Cинтетический пептид
<220>
<221> Посттрансляционная модификация остатка
<222> 2
<223> Тритилцистеин
<220>
<221> Посттрансляционная модификация остатка
<222> 3
<223> Сложный трет-бутиловый эфир глютаминовой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 4
<223> Сложный трет-бутиловый эфир лейцина
<400> 7
Gly Cys Glu Leu
1
<210> 8
<211> 4
<212> Белок
<213> Искусственная последовательность
<220>
<223> Cинтетический пептид
<400> 8
Gly Cys Glu Leu
1
<210> 9
<211> 5
<212> Белок
<213> Искусственная последовательность
<220>
<221> Посттрансляционная модификация остатка
<222> 1
<223> (Флуоренилметоксикарбонил)-сложный трет-бутиловый эфир
аспарагиновой кислоты
<220>
<223> Синтетическая последовательность
<220>
<221> Посттрансляционная модификация остатка
<222> 3
<223> Тритилцистеин
<220>
<221> Посттрансляционная модификация остатка
<222> 4
<223> Сложный трет-бутиловый эфир глютаминовой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 5
<223> Сложный метиловый эфир лейцина
<400> 9
Asp Gly Cys Glu Leu
1 5
<210> 10
<211> 5
<212> Белок
<213> Искусственная последовательность
<220>
<221> Посттрансляционная модификация остатка
<222> 1
<223> (Флуоренилметоксикарбонил)-сложный трет-бутиловый эфир
аспарагиновой кислоты
<220>
<223> Синтетический пептид
<220>
<221> Посттрансляционная модификация остатка
<222> 3
<223> Тритилцистеин
<220>
<221> Посттрансляционная модификация остатка
<222> 4
<223> Сложный трет-бутиловый эфир глютаминовой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 5
<223> Сложный метиловый эфир лейцина
<400> 10
Asp Gly Cys Glu Leu
1 5
<210> 11
<211> 5
<212> Белок
<213> Искусственная последовательность
<220>
<223> Cинтетический пептид
<400> 11
Met Asp Gly Cys Cys
1 5
<210> 12
<211> 4
<212> Белок
<213> Искусственная последовательность
<220>
<221> Посттрансляционная модификация остатка
<222> 1
<223> Карбоксибензил-метионин
<220>
<223> Cинтетический пептид
<220>
<221> Посттрансляционная модификация остатка
<222> 2
<223> Сложный бензиловый эфир аспарагиновой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 4
<223> Бензилцистеин
<400> 12
Met Asp Gly Cys
1
<210> 13
<211> 6
<212> Белок
<213> Искусственная последовательность
<220>
<221> Посттрансляционная модификация остатка
<222> 1
<223> Карбоксибензил-метионин
<220>
<223> Cинтетический пептид
<220>
<221> Посттрансляционная модификация остатка
<222> 2
<223> Сложный бензиловый эфир аспарагиновой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 4
<223> Бензилцистеин
<220>
<221> Посттрансляционная модификация остатка
<222> 5
<223> Сложный бензиловый эфир глутаминовой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 6
<223> Сложный бензиловый эфир лейцина
<400> 13
Met Asp Gly Cys Glu Leu
1 5
<210> 14
<211> 6
<212> Белок
<213> Искусственная последовательность
<220>
<223> Cинтетический пептид
<400> 14
Met Asp Gly Cys Glu Leu
1 5
<210> 15
<211> 6
<212> Белок
<213> Искусственная последовательность
<220>
<221> Посттрансляционная модификация остатка
<222> 1
<223> (Трет-бутоксикарбонил)-метионин
<220>
<223> Cинтетический пептид
<220>
<221> Посттрансляционная модификация остатка
<222> 2
<223> Сложный трет-бутиловый эфир аспарагиновой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 4
<223> Тритилцистеин
<220>
<221> Посттрансляционная модификация остатка
<222> 5
<223> Сложный трет-бутиловый эфир глютаминовой кислоты
<220>
<221> Посттрансляционная модификация остатка
<222> 6
<223> Сложный трет-бутиловый эфир лейцина
<400> 15
Met Asp Gly Cys Glu Leu
1 5
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛИНЕЙНЫЕ ЖИДКОФАЗНЫЕ ПУТИ ДЛЯ ГЕКСАПЕПТИДОВ WNT | 2019 |
|
RU2799031C2 |
| ПЕПТИДЫ В КОМБИНАЦИИ С ИНГИБИТОРАМИ ИММУННЫХ КОНТРОЛЬНЫХ ТОЧЕК ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ РАКА | 2020 |
|
RU2826955C2 |
| ГЛИКОЗИЛИРОВАННЫЙ ПЕПТИД GLP-1 | 2009 |
|
RU2543157C2 |
| ПЕПТИД GLP-1 С ПРИСОЕДИНЕННОЙ ОЛИГОСАХАРИДНОЙ ЦЕПЬЮ | 2008 |
|
RU2539829C2 |
| АНАЛОГИ ИНСУЛИНОПОДОБНОГО ФАКТОРА РОСТА-1 (IGF-1), СОДЕРЖАЩИЕ АМИНОКИСЛОТНУЮ ЗАМЕНУ В ПОЛОЖЕНИИ 59 | 2010 |
|
RU2511577C2 |
| НЕОАНТИГЕНЫ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2813924C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЛИКОПЕПТИДА, ИМЕЮЩЕГО СИАЛИРОВАННУЮ САХАРНУЮ ЦЕПЬ, И СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО СИАЛИЛГЛИКОАСПАРАГИНА | 2012 |
|
RU2586524C2 |
| АНАЛОГИ ЭФР(А) С ЗАМЕСТИТЕЛЯМИ - ЖИРНЫМИ КИСЛОТАМИ | 2017 |
|
RU2747877C2 |
| ПРОИЗВОДНЫЕ FGF21 И ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2729011C2 |
| СИНТЕЗ ЛИКСИСЕНАТИДА С КЭППИРОВАНИЕМ | 2019 |
|
RU2782772C2 |








Настоящее изобретение относится к области биотехнологии, конкретно к области синтеза полипептидов. Изобретение раскрывает новый подход к жидкофазному синтезу гексапептида WNT Foxy-5, а также к его защищенным производным и пептидным фрагментам, позволяя получить более высокие выходы целевого продукта. 3 н. и 3 з.п. ф-лы, 16 пр., 8 ил.
1. Защищенный пептидный фрагмент для получения гексапептида Foxy-5 (For-Met-Asp-Gly-Cys-Glu-Leu-OH), выбранный из:
a) PG1-Gly-Cys(PG2)-Glu(OPG3)-Leu-OR1,
b) PG1-Asp(OPG3)-Gly-Cys(PG3)-Glu(OPG3)-Leu-OR1,
c) PG1-Met-Asp(OPG3)-Gly-Cys(PG2)-Glu(OPG3)-Leu-OR1,
d) For-Met-Asp(OPG3)-Gly-Cys(PG2)-Glu(OPG3)-Leu-OR1,
где For представляет собой формил, PG1 выбран из Н и чувствительных к основанию защитных групп, таких как флуоренилметилоксикарбонил (Fmoc), PG2 выбран из Н и тритила (Trt), PG3 выбран из Н и трет-бутила (tBu), a R1 выбран из Н и С1-С6 алкила, такого как метил, этил или трет-бутил (tBu), при условии, что PG1, PG2, PG3 и R1 одновременно не представляют собой водород.
2. Защищенный пептидный фрагмент для получения гексапептида Foxy-5 (For-Met-Asp-Gly-Cys-Glu-Leu-OH) по п. 1, выбранный из:
a) Fmoc-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 6),
b) Fmoc-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 3),
c) Fmoc-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu(SEQ ID NO 4),
d) For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 15).
3. Защищенный тетрапептидный фрагмент для получения гексапептида Foxy-5 по п. 1 или 2 в твердой форме.
4. Защищенный тетрапептидный фрагмент для получения гексапептида Foxy-5 по любому из пп. 1-3 в кристаллической форме.
5. Жидкофазный способ получения защищенного производного Foxy-5 PG1-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu, включающий:
a) последовательное сочетание трипептида Fmoc-Cys(Trt)Glu(OtBu)-Leu-OtBu (КТ-2*) с Fmoc-Gly-OH, Fmoc-Asp-OtBu и PG1-Met или
b) последовательное сочетание тетрапептида Fmoc-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 6) с Fmoc-Asp-OtBu и PG1-Met,
где:
i) снятия защитной группы Fmoc достигают с помощью 1,8-диазабицикло[5,4,0]ундец-7-ена (ДБУ), причем
ii) ДБУ удаляют из реакционной массы перед сочетанием с Fmoc-Asp-OtBu и причем
iii) PG1 представляет собой чувствительную к основанию защитную группу, такую как Fmoc.
6. Жидкофазный способ превращения PG1-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu в Foxy-5 (For-Met-Asp-Gly-Cys-Glu-Leu-OH), включающий:
i) удаление PG1 из гексапептида PG1-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu с последующим сочетанием с муравьиной кислотой с образованием For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 15),
ii) снятие защиты с For-Met-Asp(OtBu)-Gly-Cys(Trt)-Glu(OtBu)-Leu-OtBu (SEQ ID NO 15) с получением Foxy-5 в неочищенной форме, а затем:
a) необязательную очистку неочищенного продукта с помощью колоночной хроматографии, необязательно с последующим осаждением в виде твердого вещества из органического растворителя,
b) необязательное осаждение образованного гексапептида Foxy-5 в виде щелочной соли или кислой соли в твердой форме, например в кристаллической форме.
| HIDAKA Yu | |||
| et al.: " Disulfide Linkages in a Heat-Stable Enterotoxin (STp) Produced by a Porcine Strain of Enterotoxigenic Escherichia coli", BCSJ, 1988, v | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
| WO 2016092378 A1, 16.06.2016 | |||
| US 5516891 A, 14.05.1996 | |||
| BRUCKDORFER T | |||
| et al.: "From production of peptides in milligram amounts for research to multi-tons quantities for | |||
