ВКЛЮЧЕНИЕ ПУТЕМ ССЫЛКИ
[0001] Форма запроса PCT подается одновременно с данной спецификацией в рамках настоящей заявки. Каждая заявка, в отношении которой в настоящей заявке испрашивается преимущество или приоритет, как указано в одновременно поданной форме запроса РСТ, полностью и для всех целей включена в настоящий документ путем ссылки.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
[0002] Экспансии повторов представляют собой особый класс микросателлитных и минисателлитных вариантов, включающих полиморфизмы коротких тандемных повторов (КТП). Экспансии повторов также называются динамическими мутациями вследствие их нестабильности в случаях, когда короткие тандемные повторы расширяются сверх определенных размеров. Генетические заболевания, вызванные нестабильными экспансиями повторов, включают в себя, помимо прочего, синдром ломкой X-хромосомы (FXS), болезнь Хантингтона и боковой амиотрофический склероз (АБС).
[0003] Выявление экспансий повторов важно для диагностирования и лечения определенных генетических заболеваний. Однако сложно определить последовательности повторов с помощью коротких прочтений, которые не полностью охватывают последовательность повторов. Таким образом, желательно разработать способы, в которых используются короткие прочтения, с целью выявления значимых с медицинской точки зрения экспансий повторов.
ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
[0004] Описанные варианты реализации относятся к способам, устройствам, системам и компьютерным программным продуктам, предназначенным для секвенирования геномных локусов, включая последовательности повторов, в том числе последовательности коротких тандемных повторов, которые могут быт связаны с генетическими расстройствами. В число способов входит выравнивание прочтений с графами последовательностей, каждый из которых представляет собой геномный локус, а также использование выровненных прочтений для генотипирования одной или более последовательностей повторов в геномном локусе. Графы последовательностей представляют собой направленные графы, каждый из которых включает по меньшей мере один собственный простой цикл, представляющий последовательность повторов.
[0005] В первом аспекте описания предложены способы генотипирования геномных локусов с помощью компьютера, включая последовательности повторов. Способы реализуют с применением компьютера, включающего в себя один или более процессоров и системную память. Данные способы могут применяться для генотипирования одной или более последовательностей повторов, каждая из которых содержит одну или более подпоследовательностей повторов. Способы включают: (a) сбор прочтений последовательности исследуемого образца из базы данных с применением одного или более процессоров; (b) выравнивание с помощью одного или более процессоров, прочтение одной или более последовательностей повторов, каждая из которых представлена графом последовательности, причем граф последовательности имеет структуру данных направленного графа, где вершины представляют нуклеотидные последовательности, а направленные ребра соединяют вершины, и при этом граф последовательности содержит один или более собственных простых циклов, причем каждый собственный простой цикл представляет собой подпоследовательность повторов, при этом каждая подпоследовательность повторов содержит повторы повторяющегося звена одного или более нуклеотидов; и (c) определение одним или более процессорами одного или более генотипов одной или более последовательностей повторов с применением прочтений последовательности, выравненных с одной или более последовательностями повторов.
[0006] В некоторых вариантах реализации последовательность повторов из одной или более последовательностей повторов содержит конкретное повторяющееся звено, содержащее по меньшей мере один частично определенный нуклеотид. В некоторых вариантах реализации конкретное повторяющееся звено содержит вырожденные кодоны.
[0007] В некоторых вариантах реализации один или более собственных простых циклов содержат два или более собственных простых циклов, представляющих две или более подпоследовательностей повторов.
[0008] В некоторых вариантах реализации граф последовательности дополнительно содержит два или более альтернативных путей для двух или более аллелей. В некоторых вариантах реализации два или более аллеля содержат делецию или замену. В некоторых вариантах реализации замена включает в себя однонуклеотидный вариант (ОНВ) или однонуклеотидный полиморфизм (ОНП). В некоторых вариантах реализации способ дополнительно включает в себя генотипирование двух или более аллелей с применением прочтений последовательностей, выровненных с двумя или более альтернативными путями. В некоторых вариантах реализации генотипирование двух или более аллелей включает в себя охват двух или более альтернативных путей к вероятностной модели для определения вероятностей двух или более аллелей. В некоторых вариантах реализации вероятностная модель моделирует вероятность аллеля в зависимости от охвата аллеля, причем функция выбрана из распределения Пуассона, отрицательного биномиального распределения, биномиального распределения или бета-биномиального распределения. В некоторых вариантах реализации параметр скорости распределения Пуассона оценивают по длине прочтения и средней глубине, наблюдаемой в геномном локусе.
[0009] В некоторых вариантах реализации способ дополнительно включает выравнивание, до (b), прочтения последовательности по эталонному геному для определения геномных координат прочтения последовательности, а также выбор подмножества прочтения последовательности по мере выравнивания прочтения последовательности с одной или более последовательностями повторов, каждая из которых представлена графом последовательности. В некоторых вариантах реализации подмножество прочтений последовательностей включает в себя прочтения, выровненные с областью, представленной графом последовательности, или с пространством вблизи нее. В некоторых вариантах реализации подмножество прочтений последовательностей включает в себя невыровненные прочтения, сопряженные прочтения которых сопоставляются с областью, представленной графом последовательности, или с пространством вблизи нее. В некоторых вариантах реализации подмножество прочтений последовательностей содержит прочтение последовательностей, выровненное с одной или более нецелевыми областями, которые являются известными горячими точками для прочтения неправильного выравнивания.
[0010] В некоторых вариантах реализации выравнивание прочтения последовательности с графом последовательности включает в себя: поиск соответствия кмер между прочтением последовательности и путем графа последовательности; и расширение соответствия кмер до полного выравнивания узлов и ребер графа последовательностей, включая один или более собственных простых циклов.
[0011] В некоторых вариантах реализации выравнивание прочтения последовательности с графом последовательности включает в себя сокращение графа путем удаления концов выравниваний с низким уровнем достоверности.
[0012] В некоторых вариантах реализации выравнивание прочтения последовательности с графом последовательности включает объединение выравниваний путем: выравнивания подпоследовательностей прочтения с графом последовательности; и объединения выравниваний подпоследовательностей для полного выравнивания прочтения последовательности.
[0013] В некоторых вариантах реализации способ дополнительно включает в себя генерирование графа последовательности на основании спецификации локуса, включающей в себя структуру геномного локуса.
[0014] В некоторых вариантах реализации прочтения последовательностей включают в себя парные концевые прочтения, а операция (c) включает в себя следующие составляющие: (i) определение базовых и закрепленных прочтений в парных концевых прочтениях, причем базовые прочтения являются выровненными с одной или более последовательностями повторов или с пространством рядом с ними, а закрепленные прочтения представляют собой невыровненные прочтения, которые сопряжены с закрепленными прочтениями; и (ii) определение одного или более генотипов для одной или более последовательностей повторов с применением по меньшей мере закрепленных прочтений.
[0015] В некоторых вариантах реализации операция (ii) включает в себя определение одного или более генотипов для одной или более последовательностей повторов с использованием базовых прочтений, а также закрепленных прочтений. В некоторых вариантах реализации базовые прочтения выровнены с точностью до около 5 т. п. н. последовательности повторов. В некоторых вариантах реализации невыровненные прочтения включают в себя прочтения, которые не могут быть выровнены или являются плохо выровненными с графом последовательности.
[0016] В некоторых вариантах реализации одна последовательность повторов или более включают в себя последовательность коротких тандемных повторов (КТП). В некоторых вариантах реализации экспансия КТП связана с синдромом ломкой X-хромосомы, боковым амиотрофическим склерозом (АБС), болезнью Хантингтона, атаксией Фридрейха, спиномозжечковой атаксией, спинобульбарной мышечной атрофией, миотонической дистрофией, болезнью Мачадо-Джозефа или дентато-рубро-паллидо-льюисовой атрофией.
[0017] В некоторых вариантах реализации способ дополнительно включает в себя использование секвенатора для создания парных концевых прочтений исследуемого образца.
[0018] В некоторых вариантах реализации способ дополнительно включает в себя извлечение исследуемого образца из организма субъекта.
[0019] В некоторых вариантах реализации исследуемый образец представляет собой образец крови, мочи, слюны или ткани.
[0020] В некоторых вариантах реализации повторяющееся звено включает в себя от 1 до 50 нуклеотидов.
[0021] В некоторых вариантах реализации прочтение является более коротким, чем по меньшей мере одна из одной или более последовательностей повторов.
[0022] В другом аспекте описания предложены системы для генотипирования геномных локусов, включая последовательности повторов. В некоторых вариантах реализации система включает в себя: системное запоминающее устройство и один или более процессоров, настроенных для выполнения следующих действий: (a) сбор с применением одного или более процессоров, прочтений последовательности исследуемого образца из базы данных; (b) выравнивание с помощью одного или более процессоров, прочтение одной или более последовательностей повторов, каждая из которых представлена графом последовательности, причем граф последовательности имеет структуру данных направленного графа, где вершины представляют нуклеотидные последовательности, а направленные ребра соединяют вершины, и при этом граф последовательности содержит один или более собственных простых циклов, причем каждый собственный простой цикл представляет собой подпоследовательность повторов, при этом каждая подпоследовательность повторов содержит повторы повторяющегося звена одного или более нуклеотидов; и (c) определение одним или более процессорами одного или более генотипов одной или более последовательностей повторов с использованием прочтений последовательности, выровненных с одной или более последовательностями повторов.
[0023] В некоторых вариантах реализации система также включает в себя секвенатор для секвенирования нуклеиновых кислот исследуемого образца.
[0024] В некоторых вариантах реализации один или более процессоров настроены для работы в рамках различных способов, описанных в настоящем документе.
[0025] В другом аспекте описания предлагается компьютерный программный продукт, включающий в себя машиночитаемый носитель, предназначенный для долговременного хранения информации, содержащий программный код, исполнение которого одним или более процессорами компьютерной системы приводит к реализации компьютерной системой описанных выше способов генотипирования геномных локусов, включая последовательности повторов. Программный код включает в себя (a) код для сбора прочтений последовательности исследуемого образца из базы данных; (b) код для выравнивания прочтений последовательности с одной или более последовательностями повторов, каждая из которых представлена на графе последовательности, причем граф последовательности имеет структуру данных направленного графа, где вершины представляют нуклеотидные последовательности и направленные ребра, соединяющие вершины, и при этом граф последовательности содержит один или более собственных простых циклов, причем каждый собственный простой цикл представляет собой подпоследовательность повторов, причем каждая подпоследовательность повторов содержит повторы повторяющегося звена одного или более нуклеотидов; и (c) код для определения одного или более генотипов одной или более последовательностей повторов с применением прочтений последовательности, выровненных с одной или более последовательностями повторов.
[0026] В некоторых вариантах реализации программный код включает в себя код для выполнения операций в рамках способов, описанных в настоящем документе.
[0027] Несмотря на то, что примеры и терминология, используемые в настоящем документе, относятся к организму человека, концепции, описанные в настоящем документе, применимы к геномам любых растений или животных. Пониманию этих и других целей и признаков настоящего описания способствует представленное ниже описание и прилагаемая формула изобретения; кроме того, необходимые данные могут быть получены при практической реализации содержания описания, представленного ниже.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0028] На Фиг. 1A представлена принципиальная схема, иллюстрирующая трудности выравнивания последовательности с последовательностью повторов на эталонной последовательности.
[0029] На Фиг. 1B представлена принципиальная схема, иллюстрирующая выравнивание прочтений последовательностей с применением парных концевых прочтений в соответствии с определенными вариантами реализации для преодоления трудностей, проиллюстрированных на Фиг. 1A.
[0030] На Фиг. 1C представлена блок-схема, иллюстрирующая процесс генотипирования геномного локуса, включающего в себя последовательность повторов, в соответствии с некоторыми вариантами реализации.
[0031] На Фиг. 1D проиллюстрирован первый граф последовательности, представляющий первый геномный локус.
[0032] На Фиг. 1E проиллюстрирован второй граф последовательности, представляющий второй геномный локус.
[0033] На Фиг. 1F проиллюстрирован третий граф последовательности, представляющий третий геномный локус
[0034] На Фиг. 1G представлена принципиальная схема процесса определения генотипов вариантов локуса HTT, включающего в себя две последовательности КТП, в соответствии с некоторыми вариантами реализации.
[0035] На Фиг. 1H представлена принципиальная схема процесса определения генотипов вариантов в локусе Lynch I, включающем ОНВ и КТП, в соответствии с некоторыми вариантами реализации. На левой панели Фиг. 1H представлена принципиальная схема общего процесса выполнения целевого генотипирования; на правой панели проиллюстрировано применение данного процесса в рамках вариантов генотипирования в локусе, связанном с синдромом Lynch I.
[0036] На Фиг. 2 представлена блок-схема, на которой проиллюстрировано изображение высокого уровня примера способа определения наличия или отсутствия экспансии последовательности повторов в образце.
[0037] На Фиг. 3 и 4 представлены блок-схемы, иллюстрирующие примеры способов обнаружения экспансии повторов с применением парных концевых прочтений.
[0038] На Фиг. 5 представлена блок-схема способа, в котором для определения экспансии повторов применяют невыровненные прочтения, не связанные с какой-либо исследуемой последовательностью повторов.
[0039] На Фиг. 6 представлена блок-схема дисперсной системы для обработки исследуемого образца.
[0040] На Фиг. 7 проиллюстрирована точность генотипирования КТП CAG и CCG в локусе HTT на основании смоделированных данных с применением различных способов.
ПОДРОБНОЕ ОПИСАНИЕ
[0041] Описание относится к способам, устройствам, системам и компьютерным программным продуктам, предназначенным для определения целевых экспансий повторов, в том числе экспансий последовательностей повторов, которые являются значимыми с медицинской точки зрения. Примеры экспансий повторов включают, без ограничений, экспансии, связанные с генетическими расстройствами, такими как синдром ломкой X-хромосомы, АБС, болезнь Хантингтона, атаксия Фридрейха, спиномозжечковая атаксия, спинобульбарная мышечная атрофия, миотоническая дистрофия, болезнь Мачадо-Джозефа и дентато-рубро-паллидо-льюисова атрофия.
[0042] Если не указано иное, практическая реализация способов и систем, описанных в настоящем документе, включает стандартные методики и устройства, широко применяемые в молекулярной биологии, микробиологии, при очистке белков, белковой инженерии, сфере секвенирования белков и ДНК и областях работы с рекомбинантными ДНК, которые находятся в рамках компетенции специалистов в данной области. Такие методики и устройство известны специалистам в данной области и описаны в многочисленных справочных публикациях (см. например, Sambrook et al., “Molecular Cloning: A Laboratory Manual,” Third Edition (Cold Spring Harbor), [2001]); и Ausubel et al., “Current Protocols in Molecular Biology” [1987]).
[0043] Числовые диапазоны включают в себя числа, определяющие диапазон. Предполагается, что каждое максимальное числовое ограничение, встречающееся в настоящем описании, включает в себя каждое меньшее числовое ограничение таким образом, как если бы такие меньшие числовые ограничения были явным образом указаны в настоящем документе. Каждое минимальное числовое ограничение, встречающееся в настоящем описании, будет включать каждое большее числовое ограничение таким образом, как если бы такие большие числовые ограничения были явным образом указаны в настоящем документе. Каждый числовой диапазон, встречающийся в настоящем описании, будет включать каждый более узкий числовой диапазон, который находится в пределах такого более широкого числового диапазона таким образом, как если бы все такие более узкие числовые диапазоны были явным образом указаны в настоящем документе.
[0044] Заголовки, представленные в настоящем документе, не имеют ограничительного характера относительно настоящего описания.
[0045] Если не указано иное, все технические и научные термины, используемые в настоящем документе, имеют общепринятое значение, понятное любому обычному специалисту в данной области. Специалистам в данной области хорошо известна и доступна разнообразная научная терминология, в том числе терминология, использующаяся в настоящем документе. Хотя любые методы и материалы, подобные или эквивалентные описанным в настоящем документе, находят применение при практическом применении или тестировании вариантов реализации, описанных в данном документе, были описаны некоторые методы и материалы.
[0046] Термины, определения которых приведены ниже, более полно описаны со ссылкой на полную версию спецификации. Следует понимать, что настоящее описание не ограничено исключительно конкретной методологией, протоколами и реагентами, поскольку они могут изменяться в зависимости от контекста, в рамках которого они используются специалистами в данной области.
Определения
[0047] Используемые в настоящем документе термины в единственном числе подразумевают такие же термины в множественном числе, если контекст не требует иного.
[0048] Если не указано иное, нуклеиновые кислоты записывают слева направо в ориентации от 5’ к 3’, а аминокислотные последовательности записывают слева направо в ориентации от амино к карбокси, соответственно.
[0049] Термин «множество» относится к более чем одному элементу. Например, термин используется в настоящем документе в отношении ряда молекул нуклеиновых кислот или прочтений последовательностей, достаточных для определения существенных различий в экспансиях повторов в исследуемых образцах и контрольных образцах с применением способов, описанных в настоящем документе.
[0050] Термин «последовательность повторов» относится к нуклеотидной последовательности, включающей повторяющиеся включения более короткой последовательности. В настоящем документе более короткая последовательность называется «повторяющимся звеном». Повторяющиеся случаи включения повторяющихся звеньев называются «повторами» или «копиями» повторяющегося звена. Во многих контекстах местоположение последовательности повторов связано с геном, кодирующим белок. В других ситуациях последовательность повторов может находиться в некодирующей области. Повторяющиеся звенья могут образовываться в последовательности повторов с разрывами или без разрывов между повторяющимися звеньями. Например, в нормальных образцах ген FMR1 обычно включает в себя разрыв AGG в повторах CGG, например, (CGG)10 + (AGG) + (CGG)9. Образцы с отсутствием разрыва, а также последовательности длинных повторов с малым количеством разрывов, склонны к экспансии повторов связанного гена, что может приводить к генетическим заболеваниям, поскольку повторы расширяются выше определенного числа. В различных вариантах реализации настоящего описания число повторов подсчитывается так же, как и количество повторов внутри рамки, независимо от наличия разрывов. Способы оценки повторов внутри рамки дополнительно описаны ниже.
[0051] В разнообразных вариантах реализации повторяющиеся звенья содержат от 1 до 100 нуклеотидов. Многими достаточно хорошо изученными повторяющимися звеньями являются тринуклеотидные или гексануклеотидные звенья. Некоторые другие повторяющиеся звенья, которые были хорошо изучены и являются применимыми к вариантам реализации, описанным в настоящем документе, содержат, без ограничений, звенья из 4, 5, 6, 8, 12, 33 или 42 нуклеотидов. См. например, Richards (2001) Human Molecular Genetics, Vol. 10, No. 20, 2187-2194. Области применения настоящего изобретения не ограничены конкретным количеством нуклеотидных оснований, описанных выше, при условии, что они являются относительно короткими по сравнению с последовательностью повторов, имеющей множество повторов или копий повторяющихся звеньев. Например, повторяющееся звено может содержать по меньшей мере 3, 6, 8, 10, 15, 20, 30, 40, 50 нуклеотидов. В качестве альтернативы или дополнения, повторяющееся звено может содержать не более около 100, 90, 80, 70, 60, 50, 40, 30, 20, 10, 6 или 3 нуклеотидов.
[0052] Последовательность повторов может расширяться в условиях развития и мутагенеза с образованием большего числа копий одного и того же повторяющегося звена. В данной области это называется «экспансией повторов». Данный процесс также называется «динамической мутацией» вследствие нестабильной природы экспансии повторяющегося звена. Было показано, что некоторые экспансии повторов связаны с генетическими заболеваниями и патологическими симптомами. Другие экспансии повторов являются недостаточно изученными. Описанные в настоящем документе способы могут применяться для определения как известных, так и новых экспансий повторов. В некоторых вариантах реализации последовательность повторов, имеющая экспансию повторов, длиннее, чем около 100, 150, 300 или 500 пар нуклеотидных оснований (п.н.о.). В некоторых вариантах реализации последовательность повторов, имеющая экспансию повторов, составляет более чем около 1 000 п.н.о., 2 000 п.н.о., 3 000 п.н.о., 4 000 п.н.о., 5 000 п.н.о. или 10 000 п.н.о. и т.д.
[0053] В теории графов вершина и ребро являются двумя базовыми единицами, из которых построены графы. Вершина или узел представляет собой одну из точек графа, которая может быть соединена ребрами. На диаграмме графа вершина может быть представлена в виде формы с меткой, а ребро может быть представлено линией (ненаправленным ребром) или стрелкой (направленным ребром), проходящей от одной вершины к другой.
[0054] Две вершины, соединенные ребром, считают конечными точками ребра. Вершина × считается смежной с другой вершиной y в случае, если граф содержит ребро (x, y).
[0055] Ненаправленный граф состоит из набора вершин и набора ненаправленных ребер (соединяющих неупорядоченные пары вершин), а направленный граф состоит из набора вершин и набора направленных ребер (соединяющих упорядоченные пары вершин).
[0056] В теории графов каждое ребро имеет две вершины (на гиперграфах их может быть больше), к которым оно прикреплено и которые называются его конечными точками. Ребра могут быть направленными или ненаправленными; Ненаправленные ребра также называют линиями, а направленные ребра также называют дугами или стрелками.
[0057] Направленное ребро представляет собой ребро, которое соединяет верхнюю и нижнюю вершины, причем верхняя вершина находится перед направленным ребром, а нижняя вершина находится после направленного ребра.
[0058] Ненаправленное ребро представляет собой ребро, соединяющее две вершины, причем любая из вершин может находиться перед другой на пути в графе.
[0059] В настоящем документе термины «цикл», «собственный простой цикл» и «одноузловой цикл» являются взаимозаменяемыми. Цикл имеет один узел и ребро, оба конца которых соединены с одним узлом.
[0060] Цикл представляет собой путь, содержащий две или более вершин, причем путь цикла начинается и заканчивается одной и той же вершиной. Простой цикл представляет собой цикл, который не имеет повторяющихся вершин или ребер, кроме от начальной и конечной вершин.
[0061] Циклический граф представляет собой граф, который содержит по меньшей мере один цикл.
[0062] Ациклический граф представляет собой граф, не содержащих циклов или собственных простых циклов.
[0063] Направленный ациклический граф (DAG) представляет собой направленный граф без каких-либо циклов или собственных простых циклов.
[0064] Путь в графе представляет собой последовательность вершин и ребер, в которой обе конечные точки ребра находятся смежно с ребром в последовательности. Путь в направленном графе имеет верхнюю вершину, которая находится перед направленным ребром (или дугой/стрелкой), и нижнюю вершину, которая находится после направленного ребра.
[0065] Распределение Пуассона представляет собой дискретное распределение вероятности, которое выражает вероятность заданного числа событий, происходящих в рамках фиксированного интервала времени или пространства, если эти события происходят с известной постоянной скоростью и независимо от времени с момента последнего события.
[0066] Полностью определенные символы основания включают G, A, T, C для обозначения гуанина, аденина, тимина и цитозина, соответственно.
[0067] Перечень частично определенных нуклеиновых кислот включает в себя, среди прочего, следующие составляющие:
[0068] Пурин (аденин или гуанин): R
[0069] Пиримидин (тимин или цитозин): Y
[0070] Аденин или тимин. W
[0071] Гуанин или цитозин: S
[0072] Аденин или цитозин: M
[0073] Гуанин или тимин: K
[0074] Аденин, тимин или цитозин: H
[0075] Гуанин, цитозин или тимин: B
[0076] Гуанин, аденин или цитозин: V
[0077] Гуанин, аденин или тимин. D
[0078] Гуанин, аденин, тимин или цитозин: N
[0079] Термин «парные концевые прочтения» относится к прочтениям, полученным путем парного концевого секвенирования, в результате которого получают по одному прочтению с каждого конца фрагмента нуклеиновой кислоты. Парное концевое секвенирование включает фрагментирование ДНК для образования последовательностей, называемых вставками. В некоторых протоколах, в том числе протоколах, используемых Illumina, прочтения с более коротких вставок (например, от десятков до сотен п.н.о.), называют парными концевыми прочтениями с короткими вставками или парными концевыми прочтениями. В противоположность этому, прочтения из более длинных вставок (например, порядка нескольких тысяч п.н.о.) называют прочтениями сопряженных пар. В настоящем описании могут применять парные концевые прочтения и короткими вставками и прочтения сопряженных пар с длинными вставками; при этом они не дифференцируются в отношении процесса анализа экспансий повторов. Следовательно, термин «парные концевые прочтения» может относиться как к парным концевым прочтениям с короткими вставками, так и к прочтениям сопряженных пар с длинными вставками, которые дополнительно описаны ниже в настоящем документе. В некоторых вариантах реализации парные концевые прочтения включают в себя прочтения от около 20 п.н.о. до 1 000 п.н.о. В некоторых вариантах реализации парные концевые прочтения включают в себя прочтения от около 50 п.н.о. до 500 п.н.о., от около 80 п.н.о. до 150 п.н.о. или около 100 п.н.о. Следует понимать, что два прочтения на парном конце не обязательно должны располагаться на крайнем конце секвенируемого фрагмента. Вместо этого одно или оба прочтения могут находиться вблизи конца фрагмента. Более того, способы, примеры которых приведены в настоящем документе в контексте парных концевых прочтений, можно осуществлять с любым из множества парных прочтений независимо от того, получены ли прочтения с конца или другой части фрагмента.
[0080] Используемый в настоящем документе термин «выравнивание» относится к процессу сравнения прочтения с эталонной последовательностью и определения того, содержит ли эталонная последовательность считываемую последовательность. В процессе выравнивания предпринимают попытки определения того, может ли прочтение быть сопоставлено с эталонной последовательностью, но не всегда приводить к выполнению прочтения, выровненного с эталонной последовательностью. Если эталонная последовательность содержит прочтение, то прочтение может сопоставляться с эталонной последовательностью или, в некоторых вариантах реализации, с конкретным местоположением в эталонной последовательности. В некоторых случаях выравнивание просто указывает, является ли прочтение членом конкретной эталонной последовательности (т.е. присутствует ли прочтение в эталонной последовательности или отсутствует). Например, выравнивание прочтения относительно эталонной последовательности для человеческой хромосомы 13 будет указывать, присутствует ли прочтение в эталонной последовательности для хромосомы 13. Инструмент, предоставляющий эту информацию, можно назвать модулем тестирования принадлежности множеству. В некоторых случаях выравнивание дополнительно указывает местоположение в эталонной последовательности, с которой сопоставляется прочтение. Например, если эталонная последовательность представляет собой человеческую последовательность целого генома, то выравнивание может указывать на наличие прочтения на хромосоме 13 и может дополнительно указывать на то, что прочтение находится на конкретной цепи и/или сайте хромосомы 13.
[0081] Выровненные прочтения представляют собой одну или более последовательностей, которые определены как совпадающие в соответствии с порядком их молекул нуклеиновой кислоты с известной эталонной последовательностью, такой как эталонный геном. Выровненное прочтение и его определенное местоположение на эталонной последовательности составляют метку последовательности. Выравнивание можно выполнять вручную, хотя обычно оно реализуется с помощью компьютерного алгоритма, поскольку невозможно выровнять прочтения за приемлемый период времени для реализации способов, описанных в настоящем документе. Одним примером алгоритма выравнивания последовательностей является компьютерная программа Efficient Local Alignment of Nucleotide Data (ELAND), распространяемая в качестве части технологического процесса геномного анализа Illumina. В альтернативном варианте реализации для выравнивания прочтений с эталонными геномами можно применять фильтр Блума или аналогичный модуль тестирования принадлежности множеству. См. Патентную заявку США № 14/354 528, поданную 25 апреля 2014 г., которая полностью включена в настоящий документ посредством ссылки. Согласование прочтения последовательности в процессе выравнивания может представлять собой 100%-ное или менее чем 100%-ное совпадение последовательности (т.е. неидеальное совпадение).
[0082] Используемый в настоящем документе термин «сопоставление» означает присвоение последовательности прочтений большей последовательности, например эталонному геному, путем выравнивания.
[0083] В некоторых случаях прочтение одного конца двух парных концевых прочтений выровнено с последовательностью повторов эталонной последовательности, а прочтение другого конца двух парных концевых прочтений не выровнено. В таких случаях спаренное прочтение, которое выровнено с последовательностью повторов эталонной последовательности, называется «базовым прочтением». Парное концевое прочтение, не выровненное с последовательностью повторов, но соединенное с базовым прочтением, называется закрепленным прочтением. Таким образом, невыровненное прочтение может закрепляться и связываться с последовательностью повторов. В некоторых вариантах реализации невыровненные прочтения включают в себя как прочтения, которые не могут быть выровнены с эталонной последовательностью, так и прочтения, которые являются плохо выровненными с эталонной последовательностью. Если прочтение выровнено с эталонной последовательностью, и при этом уровень ошибочно спаренных оснований находится выше определенного критерия, такое прочтение считается плохо выровненным. Например, в различных вариантах реализации прочтение считают плохо выровненным, если при его выравнивании были отмечены по меньшей мере около 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 несовпадений. В некоторых случаях оба прочтения пары выровнены с эталонной последовательностью. В таких случаях оба прочтения могут быть проанализированы в качестве «базовых прочтений» в различных вариантах реализации.
[0084] Термины «полинуклеотид», «нуклеиновая кислота» и «молекулы нуклеиновой кислоты» используются взаимозаменяемо и относятся к ковалентно связанной последовательности нуклеотидов (например, рибонуклеотидов для РНК и дезоксирибонуклеотидов для ДНК), в которой положение 3’ пентозы одного нуклеотида соединено фосфодиэфирной группой с положением 5’ пентозы следующего нуклеотида. нуклеотиды включают последовательности любой формы нуклеиновой кислоты, включая, без ограничений, молекулы РНК и ДНК, такие как свободно-клеточные ДНК (скДНК). Термин «полинуклеотид» включает, без ограничений, одно- и двухцепочечные полинуклеотиды.
[0085] Термин «исследуемый образец» в данном документе относится к образцу, как правило, полученному из биологической текучей среды, клетки, ткани, органа или организма, содержащего нуклеиновую кислоту или смесь нуклеиновых кислот, содержащие по меньшей мере одну нуклеотидную последовательность, которая должна быть проверена на предмет вариации числа копий. В определенных вариантах реализации образец имеет по меньшей мере одну нуклеотидную последовательность, число копий которой предположительно было подвержено изменениям. Такие образцы включают в себя, без ограничений, мокроту/жидкость ротовой полости, амниотическую жидкость, кровь, фракцию крови или образцы, полученные с применением тонкоигольной пункционной биопсии, а также мочу, перитонеальную жидкость, плевральную жидкость и т.п. Хотя образец часто отбирают от субъекта-человека (например, пациента), анализы можно использовать для вариации числа копий (CNV) в образцах, отобранных от любого млекопитающего, включая, без ограничений, собак, кошек, лошадей, коз, овец, крупный рогатый скот, свиней и т.д. Образец может быть использован непосредственно после получения из биологического источника или после предварительной обработки с целью изменения природы образца. Например, такая предварительная обработка может включать получение плазмы из крови, разбавление вязких жидкостей и т.д. Способы предварительной обработки могут также включать, без ограничений, фильтрацию, преципитацию, разбавление, дистилляцию, смешивание, центрифугирование, замораживание, лиофилизацию, концентрирование, амплификацию, фрагментацию нуклеиновых кислот, инактивацию интерферирующих компонентов, добавление реагентов, лизирование и т.п. В случае, если такие способы предварительной обработки используются относительно образца, такие способы предварительной обработки, как правило, подразумевают, что нуклеиновая кислота (кислоты) остается (остаются) в исследуемом образце, иногда в концентрации, являющейся пропорциональной их концентрации в необработанном исследуемом образце (например, в образце, который не подвергают такому виду (видам) предварительной обработки). Такие «обработанные» образцы по-прежнему считаются биологическими «тестовыми» образцами относительно способов, описанных в настоящем документе.
[0086] Контрольный образец может быть как отрицательным, так и положительным. Термин «отрицательный контрольный образец» или «незатронутый образец» относится к образцу, содержащему нуклеиновые кислоты, которые, как известно или ожидается, имеют последовательность повторов с числом повторов в диапазоне, который не является патогенным. Известно, что «положительный контрольный образец» или «затронутый образец» имеет последовательность повторов с количеством повторов в диапазоне, который является патогенным. Повторы, находящиеся в последовательности повторов в отрицательном контрольном образце, обычно не расширялись за пределы нормального диапазона, тогда как повторы, находящиеся в последовательности повторов в положительном контрольном образце, обычно расширялись за пределы нормального диапазона. Таким образом, нуклеиновые кислоты в исследуемом образце можно сравнить с одним или более контрольными образцами.
[0087] Термин «исследуемая последовательность» в настоящем документе относится к нуклеотидной последовательности, связанной с различиями в представлениях последовательности у здоровых и больных пациентов. Исследуемая последовательность может представлять собой последовательность повторов на хромосоме, которая расширяется при заболевании, в том числе, при наследственном заболевании. Исследуемая последовательность может представлять собой часть хромосомы, ген, кодирующую или некодирующую последовательность.
[0088] В настоящем документе термин «секвенирование следующего поколения (NGS)» относится к способам секвенирования, которые позволяют осуществлять массовое параллельное секвенирование клонально амплифицированных молекул и отдельных молекул нуклеиновых кислот. Не имеющие ограничительного характера примеры NGS включают в себя секвенирование путем синтеза с использованием терминаторов обратимых красителей, а также секвенирование путем лигирования.
[0089] Термин «параметр», используемый в настоящем документе, относится к числовому значению, характеризующему физическое свойство. Часто параметр используют для числовой характеризации набора количественных данных и/или численной зависимости между наборами количественных данных. Например, параметром является соотношение (или функция соотношения) между количеством меток последовательности, сопоставленных с хромосомой, и длиной хромосомы, с которой сопоставлены метки.
[0090] Термин «критерий распознавания» в настоящем документе относится к любому числу или количеству, которое используют в качестве предельного уровня для характеристики образца, такого как исследуемый образец, содержащий нуклеиновую кислоту и отобранный из организма субъекта, предположительно имеющего медицинское состояние. Пороговое значение можно сравнивать со значением параметра для определения того, указывает ли образец, выдающий такое значение параметра, на наличие медицинского состояния у субъекта. В определенных вариантах реализации пороговое значение рассчитывают с использованием набора контрольных данных и используют в качестве предела при выполнении диагностики экспансии повторов в организме. В некоторых вариантах реализации в случаях, если пороговое значение было превышено результатами, полученными способами, описанными в данном документе, у субъекта можно диагностировать экспансию повторов. В рамках способов, описанных в настоящем документе, соответствующие пороговые значения могут быть определены путем анализа значений, рассчитанных относительно обучающего набора образцов или контрольных образцов. Пороговые значения также можно рассчитать по эмпирическим параметрам, таким как глубина секвенирования, длина прочтения, длина последовательности повторов и т.д. В качестве альтернативы, затронутые образцы, определенно имеющие экспансию повторов, также могут использоваться для подтверждения того, что выбранные пороговые значения можно использовать для различения незатронутых образцов в рамках тестовой последовательности. Выбор порогового значения зависит от уровня достоверности, которую пользователь желает сделать для классификации. В некоторых вариантах реализации обучающий набор, используемый для определения соответствующих пороговых значений, содержит по меньшей мере 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 300, 400, 500, 600, 700, 800, 900, 1 000, 2 000, 3 000, 4 000 или более квалифицированных образцов. Применение больших наборов квалифицированных образцов для повышения диагностического значения пороговых значений может являться предпочтительным.
[0091] Термин «прочтение» относится к прочтению последовательности части образца нуклеиновой кислоты. Как правило, прочтение представляет собой короткую последовательность связных пар нуклеотидных оснований в образце. Прочтение может символически представлять собой последовательность пар нуклеотидных оснований (в виде ATCG) фрагмента образца. Он может храниться в запоминающем устройстве и обрабатываться при необходимости для определения того, соответствует ли он эталонной последовательности или другим критериям. Прочтение можно получать непосредственно с помощью аппарата для секвенирования или опосредованно из сохраненной информации о последовательности, относящейся к образцу. В некоторых случаях прочтение представляет собой последовательность ДНК достаточной длины (например, по меньшей мере около 25 п.н.о.), которую можно использовать для определения большей последовательности или области, например которая может быть выровнена и сопоставлена с хромосомой, геномной областью или геном.
[0092] Термин «прочтение генома» используют для обозначения прочтения любых сегментов всего генома субъекта.
[0093] Термин «сайт» относится к уникальному положению (т.е. идентификатор хромосомы, хромосомное положение и ориентация) в эталонном геноме. В некоторых вариантах реализации сайт может представлять собой остаток, метку последовательности или положение сегмента на последовательности.
[0094] Используемый здесь термин «эталонный геном» или «эталонная последовательность» относится к любой конкретной известной геномной последовательности, частичной или полной, любого организма или вируса, которая может использоваться для обозначения определенных последовательностей, полученных от субъекта. Например, эталонный геном, используемый для субъектов-людей, а также для многих других организмов, можно найти в Национальном центре биотехнологической информации по адресу ncbi.nlm.nih.gov. Термин «геном» относится к полной генетической информации об организме или вирусе, экспрессируемой в нуклеотидных последовательностях.
[0095] В разнообразных вариантах реализации эталонная последовательность значительно больше прочтений, которые выравнивают с ней. Например, он может быть по меньшей мере около в 100 раз больше, или по меньшей мере около в 1 000 раз больше, или по меньшей мере около в 10 000 раз больше, или по меньшей мере около в 105 раз больше, или по меньшей мере около в 106 раз больше, или по меньшей мере около в 107 раз больше.
[0096] В одном примере базовая последовательность представляет собой последовательность полноразмерного генома человека. Такие последовательности могут называться эталонными геномными последовательностями. В другом примере базовая последовательность ограничена конкретной хромосомой человека, такой как хромосома 13. В некоторых вариантах реализации эталонная хромосома Y представляет собой хромосомную последовательность Y из версии hg19 генома человека. Такие последовательности могут называться эталонными хромосомными последовательностями. Другие примеры эталонных последовательностей включают геномы других видов, а также хромосомы, субхромосомные области (такие как хроматиды) любых видов.
[0097] В некоторых вариантах реализации эталонная последовательность, необходимая для выполнения выравнивания, может иметь длину, превышающую длину прочтения в от около 1 до около 100 раз. В таких вариантах реализации выравнивание и секвенирование считают целевым выравниванием или секвенированием, а не выравниванием или секвенированием всего генома. В данных вариантах реализации эталонная последовательность, как правило, включает в себя исследуемый ген и/или последовательность повторов.
[0098] В разнообразных вариантах реализации эталонная последовательность представляет собой консенсусную последовательность или другую комбинацию, полученную от множества индивидуумов. Однако в некоторых приложениях эталонную последовательность можно отбирать у конкретного индивидуума.
[0099] Термин «клинически значимая последовательность» в настоящем документе обозначает нуклеотидную последовательность, точно или предположительно связанную с заболеванием, в том числе наследственным. Определение отсутствия или наличия клинически значимой последовательности может быть полезным при определении или подтверждении диагноза медицинского состояния, либо при прогнозировании развития заболевания.
[00100] Термин «полученный», используемый в настоящем документе, в контексте нуклеиновой кислоты или смеси нуклеиновых кислот означает, что нуклеиновая кислота (кислоты) получена (получены) из источника, из которого (которых) они происходят. Например, в одном варианте реализации смесь нуклеиновых кислот, полученных из двух разных геномов, означает, что нуклеиновые кислоты, например, скДНК, естественным образом высвобождались клетками посредством естественных процессов, таких как некроз или апоптоз. В другом варианте реализации термин «смесь нуклеиновых кислот, полученных из двух разных геномов» означает, что нуклеиновые кислоты экстрагировали из двух разных типов клеток пациента.
[00101] Термин «основанный» при использовании в контексте получения конкретного количественного значения в настоящем документе относится к использованию другого количества в качестве входных данных для расчета конкретного количественного значения в качестве выходных данных.
[00102] В настоящем документе термин «образец пациента» обозначает биологический образец, отобранный у пациента, т.е. лица, получающего медицинский уход или лечение. Образец пациента может представлять собой любой из образцов, описанных в настоящем документе. В некоторых вариантах реализации образец пациента отбирают с применением неинвазивных процедур, таким образцом может являться, например, образец периферической крови или фекалий. Способы, описанные в настоящем документе, могут применяться не только к людям. Таким образом, рассматривают разнообразные применения в сфере ветеринарии, и в этом случае образец пациента может представлять собой образец, отобранный у млекопитающего, не относящегося к человеку (например, представителя семейства кошачьих, свинообразных, лошадиных, бычьих и т.п.).
[00103] Термин «биологическая жидкость» в настоящем документе относится к жидкости, взятой из биологического источника, и включает, например, кровь, серозный экссудат, плазму, мокроту, лаваж, спинномозговую жидкость, мочу, семя, пот, слезную жидкость, слюну и т.п. Используемые в настоящем документе термины «кровь», «плазма» и «серозный экссудат» относятся к фракциям или их обработанным частям. Аналогичным образом, в случае, если образец получают путем выполнения биопсии, взятия смыва, мазка и т.д., термин «образец» обозначает обработанную фракцию или часть, полученную путем выполнения биопсии, взятия смыва, мазка и т.д.
[00104] Термин «соответствующий», используемый в настоящем документе, иногда относится к нуклеотидной последовательности, например, гену или хромосоме, которая присутствует в геноме разных субъектов и которая не обязательно имеет одинаковую последовательность во всех геномах, однако служит для определения, а не для предоставления генетической информации об исследуемой последовательности, например гене или хромосоме.
[00105] Термин «хромосома», используемый в настоящем документе, относится к несущему наследственность генному носителю живой клетки, который состоит из хроматиновых цепей, содержащих ДНК и белковые компоненты (в частности, гистоны). В настоящем документе используется стандартная международно признанная система нумерации отдельных хромосом генома человека.
[00106] Термин «длина полинуклеотида», используемый в настоящем документе, относится к абсолютному числу мономерных субъединиц (нуклеотидов) нуклеиновой кислоты в последовательности или в области эталонного генома. Термин «длина хромосомы» относится к известной длине хромосомы, заданной парами нуклеотидных оснований, например приведенная в сборке NCBI36/hg18 человеческой хромосомы, данные которой можно найти в сети Интернет по следующему адресу: |genome|.|ucsc|.|edu/cgi- bin/hgTracks?hgsid=167155613&chromInfoPage=
[00107] Термины «субъект» и «пациент», используемые в настоящем документе, обозначают субъекта-человека, а также субъекта, не являющегося человеком, такого как млекопитающее, беспозвоночное, позвоночное, грибок, дрожжевые грибы, бактерия и вирус. Несмотря на то, что примеры и терминология, используемые в настоящем документе, относятся к организму человека, концепции, описанные в настоящем документе, применимы к геномам любых растений или животных и могут применяться в области ветеринарной медицины, зоотехники, лабораторных исследований и т.п.
[00108] Используемый в настоящем документе термин «праймер» относится к выделенному олигонуклеотиду, который способен играть роль точки инициации синтеза при помещении в условия, являющиеся индуктивными по отношению к синтезу продукта достройки (например, такие условия подразумевают присутствие нуклеотидов, средства для индукции, такого как ДНК-полимераза, подходящую температуру и pH). Праймер может быть предпочтительно одноцепочечным для обеспечения максимальной эффективности амплификации, но также может быть двухцепочечным. В случае, если праймер является двухцепочечным, его сначала обрабатывают для разделения спиралей перед его использованием для получения продуктов достройки. Праймер может представлять собой олигодезоксирибонуклеотид. Праймер является достаточно длинным для инициации процесса синтеза продуктов достройки при наличии средства для индукции. Точные длины праймеров зависят от многих факторов, включая температуру, источники праймеров, а также способы и параметры, используемые при создании праймеров.
5.2. Введение
[00109] Короткие тандемные повторы (КТП) встречаются повсеместно в геноме человека. Несмотря на то, что биологическая составляющая КТП еще не полностью изучена, новые доказательства свидетельствуют о том, что КТП играют важную роль в основных клеточных процессах.
[00110] Экспансии повторов представляют собой это особый класс микросателлитных и минисателлитных вариантов, включающих полиморфизмы КТП. Экспансии повторов также называются динамическими мутациями вследствие их нестабильности в случаях, когда короткие тандемные повторы расширяются сверх определенных размеров. Экспансии КТП являются основной причиной более 20 тяжелых неврологических расстройств, включая боковой амиотрофический склероз, атаксию Фридрейха (FRDA), болезнь Хантингтона (БХ) и синдром ломкой X-хромосомы. В Таблице 1 представлено небольшое число патогенных экспансий повторов, отличных от последовательностей повторов в нормальных образцах. В столбцах проиллюстрированы гены, ассоциированные с последовательностями повторов, нуклеотидные последовательности повторяющихся звеньев, числа повторов повторяющихся звеньев в нормальных и патогенных последовательностях и заболевания, связанные с экспансиями повторов.
Таблица 1. Примеры патогенных экспансий повторов
[00111] Генетические заболевания, связанные с экспансиями повторов, во многих отношениях являются гетерогенными. размер повторяющегося звена, степень экспансии, местоположение относительно затронутого гена и патогенный механизм могут различаться в зависимости от заболевания. Например, АБС включает в себя экспансию повторов гексануклеотидов нуклеотидов GGGGCC в гене C9orf72, расположенном на коротком плече открытой рамки считывания 72 хромосомы 9. Напротив, синдром ломкой X-хромосомы связан с экспансией тринуклеотидного повтора CGG (триплетного повтора), влияющей на ген 1 (FMR1) на X-хромосоме, являющийся причиной задержки умственного развития при синдроме ломкой X-хромосомы. Экспансия повторов CGG может привести к неспособности экспрессировать белок FMRP, отсутствие которого приводит к задержке умственного развития при синдроме ломкой X-хромосомы и наличие которого является обязательным для нормального развития нервной системы. В зависимости от длины повтора CGG аллель можно классифицировать как нормальный (не подверженный влиянию синдрома), премутационный (подверженный риску развития расстройств, связанных с ломкостью X-хромосомы) или полностью мутировавший (обычно подверженный влиянию синдрома). В соответствии с различными оценками, существует от 230 до 4 000 повторов CGG в мутированных генах FMR1, вызывающих синдром ломкой X-хромосомы у больных, по сравнению с 60 до 230 повторов у носителей, склонных к атаксии, и от 5 до 54 повторов у здоровых субъектов. Экспансия повторов гена FMR1 является причиной проявлений аутизма, так как было обнаружено, что у около 5% пациентов, страдающих аутизмом, экспансия повторов гена FMR1 является причиной аутизма. McLennan, et al. (2011), Fragile × Syndrome, Current Genomics 12 (3): 216-224. Окончательная диагностика синдрома ломкой X-хромосомы включает в себя выполнения генетического тестирования для определения числа повторов CGG.
[00112] Во многих исследованиях были выявлены различные общие свойства заболеваний, связанных с экспансией повторов. Экспансия повторов или динамическая мутация обычно проявляется в виде увеличения числа повторов, причем скорость такой мутации связана с числом повторов. Редкие явления, такие как потеря разрыва повторов, могут привести к увеличению вероятности экспансии аллелей; такие события называются неблагоприятными событиями. Может существовать взаимосвязь между количеством повторов в последовательности повторов и тяжестью заболевания, вызванного экспансией повторов, и/или его развитием.
[00113] Таким образом, выявление экспансий повторов играет важную роль в диагностике и лечении различных заболеваний. Однако определение последовательностей повторов, особенно с использованием прочтений, которые не полностью охватывают последовательность повторов, сопряжена с рядом проблем. Во-первых, сложно выровнять повторы с эталонной последовательностью, поскольку отсутствует четкое взаимно-однозначное сопоставление между прочтением и эталонным геномом. Кроме того, даже если прочтение выровнено с эталонной последовательностью, прочтения часто являются слишком короткими для полного покрытия значимой с медицинской точки зрения последовательности повторов. Например, прочтения могут иметь длину около 100 п.н.о. Для сравнения, экспансия повторов может охватывать от сотен до тысяч пар нуклеотидных оснований. Например, при синдроме ломкой X-хромосомы ген FMR1 может иметь более 1 000 повторов, охватывающих более 3 000 п.н.о. Таким образом, прочтение длиной 100 п.н.о. не может сопоставлять полную длину экспансии повторов. Кроме того, сборка коротких прочтений в более длинную последовательность не позволяет преодолеть проблему коротких прочтений и длинных повторов, поскольку сборка коротких прочтений в более длинную последовательность является труднореализуемой из-за неоднозначности выравнивания повторов в рамках одного прочтения с повторами в другом прочтении.
[00114] Выравнивание является первичной причиной потери информации либо из-за неполноты эталонной последовательности, неуникального соответствия между прочтением и сайтами на эталонной последовательности, либо значительных отклонений от эталонной последовательности. Систематические ошибки секвенирования и другие проблемы, влияющие на точность прочтения, являются вторичной причиной неудач при обнаружении последовательностей повторов. В некоторых протоколах эксперимента около 7% прочтений не являются выровненными или по шкале MAPQ имеют балл, равный 0. Даже если исследователи работают над улучшением технологии секвенирования и средств анализа, всегда будут наблюдать значительное количество невыровненных и плохо выровненных прочтений. Варианты реализации способов выявления экспансий повторов, описанных в настоящем документе, основаны на определении невыровненных или плохо выровненных прочтений.
[00115] Способы, в которых используются длинные прочтения для обнаружения экспансии повторов, имеют определенные недостатки. В контексте секвенирования следующего поколения доступные в настоящее время технологии, в которых используются более длинные прочтения, являются более медленными и склонными к появлению ошибок, чем технологии, в которых используются более короткие прочтения. Более того, в некоторых областях применения невозможно выполнять длинные прочтения, например, при секвенировании свободно-клеточных ДНК. Свободно-клеточная ДНК, полученная из материнской крови, может быть использована для пренатальной генетической диагностики. Свободно-клеточная ДНК существует в виде фрагментов, которые, как правило, короче 200, при использовании свободно-клеточной ДНК. В вариантах реализации способов, описанных в настоящем документе, используются короткие прочтения для определения экспансий повторов, значимых с медицинской точки зрения.
[00116] Более того, традиционные способы не предназначены для обработки сложных локусов, содержащих множество повторов. Важными примерами таких локусов являются повтор CAG, вызывающий БХ, фланкированный повтором CCG, повтор GAA, вызывающий FRDA, фланкированный гомополимером аденозина, и повтор CAG, вызывающий спиномозжечковую атаксию 8 типа (SCA8), фланкированную повтором ACT. Еще более ярким примером является повтор CCTG в гене CNBP, экспансии которого вызывают миотоническую дистрофию 2 типа (DM2). Данный повтор является смежным с полиморфными повторами TG и TCTG (J. E. Lee and Cooper 2009), что особенно затрудняет точное выравнивание прочтений с этим локусом. Другим типом комплексного повтора является полиаланиновый повтор, на настоящий момент связанный с по меньшей мере девятью расстройствами (Shoubridge and Gecz 2012). Полиаланиновые повторы состоят из повторов кодонов a-аминокислот GCA, GCC, GCG или GCT.
[00117] Кластеры вариантов могут влиять на выравнивание и точность генотипирования (Lincoln et al. 2019). Варианты, смежные с полиморфными последовательностями низкой сложности, могут быть сопряжены с дополнительными проблемами, поскольку способы обнаружения вариантов могут приводить к выведению кластеров неверно представленных или ложных распознаваний вариантов в таких геномных областях. Это, в частности, связано с повышенной частотой возникновения ошибок в таких областях данных секвенирования (Benjamini and Speed 2012; Dolzhenko et al., 2017). Одним из примеров является однонуклеотидный вариант (ОНВ), смежный с гомополимером аденозина в гене MSH2, который вызывает синдром Линча I (Frogatt et al. 1999).
[00118] В рамках вариантов реализации, описанных в настоящем документе, могут выполнять обработку сложных локусов, как описано выше. В них используют граф последовательности в качестве общей гибкой модели каждого целевого локуса.
[00119] В некоторых вариантах реализации в рамках описанных способов решают вышеупомянутые проблемы при определении и распознавании экспансии повторов путем использования парного концевого секвенирования. Парное концевое секвенирование включает фрагментирование ДНК для образования последовательностей, называемых вставками. В некоторых протоколах, в том числе протоколах, используемых Illumina, прочтения с более коротких вставок (например, от десятков до сотен п.н.о.), называют парными концевыми прочтениями с короткими вставками или парными концевыми прочтениями. В противоположность этому, прочтения из более длинных вставок (например, порядка нескольких тысяч п.н.о.) называют прочтениями сопряженных пар. Как отмечалось выше, в различных вариантах реализации способов, описанных в настоящем документе, могут использовать как парные концевые прочтения с короткими вставками, так и прочтения сопряженных пар с длинными вставками.
[00120] Фиг. 1A представляет собой схематическую иллюстрацию, демонстрирующую определенные сложности при выравнивании прочтений последовательности с последовательностью повторов на эталонной последовательности, особенно при выравнивании прочтений последовательности, полученных из образца последовательности длинных повторов, имеющей экспансию повторов. В нижней части Фиг. 1A проиллюстрирована эталонная последовательность 101 с относительно короткой последовательностью повторов 103, изображенной вертикальными штриховыми линиями. В середине фигуры проиллюстрирована гипотетическая последовательность 105 образца, отобранного у пациента, имеющего последовательность длинных повторов 107 с экспансией повторов, которая такжеизображена вертикальными штриховыми линиями. В верхней части рисунка представлены прочтения последовательностей 109 и 111, проиллюстрированные в местоположениях соответствующих сайтов последовательности образца 105. В некоторых из данных прочтений последовательностей, например, прочтений 111, некоторые пары нуклеотидных оснований берут начало из последовательности длинных повторов 107, изображенной вертикальными штриховыми линиями и обведенной кругом. Прочтения 111, имеющие эти повторы, потенциально трудно поддаются выравниванию с эталонной последовательностью 101, поскольку такие повторы не имеют четких местоположений на эталонной последовательности 101. Поскольку эти потенциально невыровненные прочтения не могут быть четко связаны с последовательностью повторов 103 в эталонной последовательности 101, получение информации о последовательности повторов и экспансии последовательности повторов из этих потенциально невыровненных прочтений 111 является труднореализуемым. Кроме того, поскольку эти прочтения, как правило, являются более короткими, чем последовательность длинных повторов 107, имеющая экспансию повторов, и они не могут предоставлять исчерпывающую информацию о типе или местонахождении последовательности повторов 107. Кроме того, повторы в прочтениях 111 затрудняют их сборку из-за неоднозначности их местоположений на эталонной последовательности 101 и неоднозначности отношений между прочтениями 111. Прочтения, частично принадлежащие к последовательности длинных повторов 107 в образце, наполовину заштрихованные и наполовину закрашенные черным цветом в иллюстрации, могут быть выровнены основаниями, не принадлежащими к последовательности повторов 107. Если прочтения содержат слишком мало пар нуклеотидных оснований за пределами последовательности повторов 107, прочтения могут плохо выравниваться или вовсе не выравниваться. Таким образом, некоторые из этих прочтений с частичными повторами можно анализировать в качестве базовых прочтений, а другие анализировать в качестве закрепленных прочтений, как дополнительно описано ниже.
[00121] На Фиг. 1B представлена принципиальная схема, иллюстрирующая то, как парные концевые прочтения можно использовать в некоторых описанных вариантах реализации для преодоления трудностей, проиллюстрированных на Фиг. 1A. При парном концевом секвенировании секвенирование происходит с обоих концов фрагментов нуклеиновых кислот в исследуемом образце. В нижней части Фиг. 1B представлена эталонная последовательность 101 и последовательность образца 105, а также прочтения 109 и 111, эквивалентные проиллюстрированным на Фиг. 1A. Сверху на Фиг. 1B проиллюстрирован фрагмент 125, полученный из последовательности исследуемого образца 105, и область праймеров 131 прочтения 1, а также область праймеров 133 прочтения 2 для получения двух прочтений 135 и 137 парных концевых прочтений. Фрагмент 125 также упоминается в качестве вставки для парных концевых прочтений. В некоторых вариантах реализации вставки можно амплифицировать с применением или без применения ПЦР. Некоторые последовательности повторов, такие как последовательности, включающие большое количество повторов GC или GCC, не могут быть достаточно качественно секвенированы традиционными способами, включающими ПЦР-амплификацию. Амплификация таких последовательностей могут проводить без применения ПЦР. Амплификация других последовательностей могут проводить с применением ПЦР
[00122] Вставка 125, изображенная на Фиг. 1B, получена из участка или соответствует участку последовательности образца 105, сбоку от которой расположены две вертикальные стрелки, изображенные в нижней половине рисунка. В частности, вставка 125 содержит повторяющуюся секцию 127, соответствующую части длинного повтора 107 в последовательности образца 105. Длину вставок можно регулировать в зависимости от области применения. В некоторых вариантах реализации вставки могут быть несколько короче, чем исследуемая последовательность повторов или последовательность повторов, имеющая экспансию повторов. В других вариантах реализации вставки могут иметь длину, аналогичную последовательности повторов или последовательности повторов с экспансией повторов. В других вариантах реализации вставки могут быть даже несколько длиннее последовательности повторов или последовательности повторов с экспансией повторов. В некоторых вариантах реализации такие вставки могут представлять собой длинные вставки для выполнения секвенирования сопряженных пар в некоторых вариантах реализации, дополнительно описанных ниже. Как правило, прочтения, полученные из вставок, являются более короткими, чем последовательность повторов. Поскольку вставки длиннее прочтений, парные концевые прочтения могут лучше захватывать сигналы при более длительном отрезке последовательности повторов в образце, чем одиночные концевые прочтения.
[00123] Показанная вставка 125 имеет две области праймеров прочтения 131 и 133 на двух концах вставки. В некоторых вариантах реализации области праймеров прочтения являются свойственными вставке. В других вариантах реализации области праймеров вводят во вставку путем лигирования или достройки. На левом конце вставки изображена область праймеров 131 прочтения 1, которая позволяет гибридизировать праймер 132 прочтения 1 со вставкой 125. В результате достройки праймера 132 прочтения 1 генерируют первое прочтение или прочтение 1, обозначенное как 135. На правом конце вставки 125 изображена область праймеров 133 прочтения 2, которая позволяет гибридизировать праймер 134 прочтения 2 со вставкой 125, инициируя второе прочтение или прочтение 2, помеченное числом 137. В некоторых вариантах реализации вставка 125 может также включать в себя индексные области, снабженные штрихкодом (не проиллюстрированы на фигуре), что позволяет выполнять определениекацию различных образцов в процессе многоканального секвенирования. В некоторых вариантах реализации парные концевые прочтения 135 и 137 могут быть получены путем секвенирования Illumina с помощью платформ для синтеза. Пример процесса секвенирования, реализованного на такой платформе, дополнительно описан ниже в разделе «Способы секвенирования», в ходе которого создают два парных концевых прочтения и два индексных прочтения.
[00124] Затем парные концевые прочтения, полученные способом, изображенным на Фиг. 1B, могут быть выровнены с эталонной последовательностью 101, имеющей относительно короткую последовательность повторов 103. Таким образом, известно относительное местоположение и направление пары прочтений. Это позволяет косвенно связать невыровненное или плохо выровненное прочтение, например указанное в круге 111, с последовательностью относительно длинных повторов 107 в последовательности образца 105 посредством соответствующего парного прочтения 109, как указано в нижней части Фиг. 1B. В иллюстративном примере прочтения, полученные в результате парного концевого секвенирования, составляют около 100 п.н.о., а вставки составляют около 500 п.н.о. В данной иллюстративной конфигурации относительные местоположения двух парных концевых прочтений находятся на расстоянии около 300 пар нуклеотидных оснований от их 3’ концов и имеют противоположные направления. Соотношение между парами прочтений позволяет лучше связывать прочтения с областями повторов. В некоторых случаях первое прочтение в паре выравнивается с последовательностью, не имеющей повторов, фланкирующей область повторов на эталонной последовательности, а второе прочтение в паре не выравнивается с эталоном должным образом. Cм., например, информацию о паре прочтений 109а и 111а, представленных в нижней половине Фиг. 1B, где первым прочтением является левое прочтение 109а, а вторым прочтением является правое прочтение 111a. Учитывая спаривание двух прочтений 109а и 111а, второе прочтение 111а может быть связано с областью повторов 107 в последовательности образца 105, несмотря на то, что второе прочтение 111а не может быть выровнено с эталонной последовательностью 101. Зная расстояние и направление второго прочтения 111а относительно первого прочтения 109а, можно дополнительно определить местоположение второго прочтения 111а в пределах длинной области повторов 107. В случае, если между повторами во втором прочтении 111а существует разрыв, также можно определить местоположение разрыва относительно эталонной последовательности 101. В настоящем описании прочтение, такое как левое прочтение 109a, которое выровнено с эталоном, называется базовым прочтением. Прочтение, такое как правое прочтение 111а, которое не выровнено с эталонной последовательностью, но сопряжено с базовым прочтением, называют закрепленным прочтением. Таким образом, невыровненная последовательность может быть связана с экспансией повторов. Таким образом, можно использовать короткие прочтения для обнаружения длинных экспансий повторов. Хотя проблема обнаружения экспансий повторов обычно возрастает с увеличением длины экспансии из-за сложности выполнения секвенирования; способы, описанные в настоящем документе, позволяют обнаруживать более высокий сигнал от более длинных последовательностей экспансии повторов, чем от более коротких последовательностей экспансии повторов. Это связано с тем, что по мере увеличения длины последовательности повторов или экспансии повторов, в области экспансии будет закреплено больше прочтений, и большее количество прочтений будет иметь вероятность попадания в область повторов, а также при каждом прочтении может происходить больше повторов.
[00125] В некоторых вариантах реализации описанные способы включают анализ распределения частот количества повторов, обнаруженных в рамках базовых и закрепленных прочтений. В некоторых вариантах реализации анализируют исключительно закрепленные прочтения. В других вариантах реализации анализируют как базовые, так и закрепленные прочтения. Распределение исследуемого образца можно сравнить с критерием, полученным эмпирическим или теоретическим способом и использующимся для отделения незатронутых образцов от затронутых образцов. Таким образом, можно определять, имеется ли в исследуемом образце рассматриваемая экспансия повторов, и, соответственно, выполнять распознавание.
[00126] В способах и устройстве, описанных в настоящем документе, может быть использована технология секвенирования следующего поколения (NGS), которая позволяет осуществлять массовое параллельное секвенирование. В определенных вариантах реализации клонально амплифицированные матрицы ДНК или одиночные молекулы ДНК секвенируют внутри проточной кюветы путем массового параллельного секвенирования (например, как описано в публикации Volkerding et al. Clin Chem 55:641-658 [2009]; Metzker M Nature Rev 11:31-46 [2010]). Технологии секвенирования NGS включают в себя, без ограничений, пиросеквенирование, секвенирование путем синтеза с использованием терминаторов обратимых красителей, секвенирование путем лигирования олигонуклеотидных зондов и ионное полупроводниковое секвенирование. ДНК из отдельных образцов можно секвенировать по отдельности (т.е. выполнять одноканальное секвенирование) или ДНК из нескольких образцов можно объединять и секвенировать в виде индексированных геномных молекул (т.е. выполнять многоканальное секвенирование) в рамках одного сеанса секвенирования для создания до нескольких сотен миллионов прочтений последовательностей ДНК. Примеры технологий секвенирования, которые можно использовать для получения информации о последовательности в рамках настоящего способа, дополнительно описаны ниже.
[00127] Различные анализы экспансии повторов с применением образцов ДНК включают выравнивание или сопоставление прочтений последовательностей секвенатора с эталонной последовательностью. Эталонная последовательность может представлять собой последовательность целого генома, последовательность хромосомы, последовательность субхромосомной области и т.п. С точки зрения вычислительного процесса повторы создают неоднозначность при выравнивании, что, в свою очередь, может привести к ошибкам на уровне подсчета всей хромосомы. В разнообразных вариантах реализации парные концевые прочтения в сочетании с регулируемой длиной вставки могут помочь устранить неоднозначность при выравнивании последовательностей повторов и обнаруживать экспансию повторов.
Варианты генотипирования в локусе последовательности повторов с применением графа последовательности
[00128] На Фиг. 1C представлена блок-схема, иллюстрирующая процесс 140 генотипирования геномного локуса, содержащего последовательность повторов, в соответствии с некоторыми вариантами реализации. В некоторых вариантах реализации генетический локус заранее определен в каталоге вариантов, содержащем геномные положения и структуру локусов в геномных положениях. На Фиг. 1D, 1E и 1F проиллюстрированы три разных графа последовательности в соответствии с некоторыми вариантами реализации.
[00129] На Фиг. 1G проиллюстрирована принципиальная схема процесса определения генотипов вариантов локуса HTT, содержащего две последовательности КТП, в соответствии с некоторыми вариантами реализации. На панели (a) Фиг. 1G изображена часть каталога вариантов, содержащего геномные локусы и спецификации локусов, в частности, их структуру. Например, без учета повторов, последовательность в локусе HTT представляет собой CAGCAACAGCGG (Посл. №: 2); последовательность в локусе CNBP представляет собой CAGGCAGACA (Посл. №: 3).
[00130] На Фиг. 1H проиллюстрирована принципиальная схема процесса определения генотипов вариантов в локусе Lynch I, содержащем ОНВ и КТП, в соответствии с некоторыми вариантами реализации. На Фиг. 1H в рамке 162 проиллюстрирована общая структура спецификаций локуса, в рамке 163 отображен конкретный пример спецификации локуса Lynch I (MSH2).
[00131] В каталоге вариантов структуру локуса указывают с использованием ограниченного подмножества регулярного синтаксиса выражения. Например, область повторов, связанная с БХ, может определяться выражением (CAG)* CAACAG(CGG)* или Посл. №: 2 (без учета повторов), что означает, что она содержит переменные числа повторов CAG и CCG, разделенных разрывом CAACAG; область, связанная с областью FRDA, соответствует выражению (A)*(GAA)*; область, связанная с SCA8, соответствует выражению (CTA)*(CTG)*; область повторов DM2, состоящая из трех смежных повторов, определяется выражением (CAGG)*(CAGA)*(CA)* или Посл. №: 3 (без учета повторов); ОНВ MSH2 является смежным с гомополимером, вызывающим синдром Линча I, и соответствует выражению (A|T)(A)*.
[00132] Кроме того, регулярные выражения могут содержать многоаллельные или «вырожденные» основные символы, которые могут быть указаны с помощью обозначения «Международного союза теоретической и прикладной химии» (ИЮПАК) («Перечень частично определенных оснований в нуклеотидных последовательностях. Рекомендации», 1984. Номенклатурный комитет Международного союза биохимиков (НК МСБ) »1986).
[00133] Частично определенные основания, соответствующие основаниям в вырожденных кодонах, в настоящем документе называются вырожденными основаниями. Вырожденные основания позволяют представлять определенные классы несовершенных повторов ДНК, в рамках которых, например, различные основания могут встречаться в одном и том же положении. Используя это обозначение, полиаланиновые повторы могут кодироваться выражением (GCN)*, а полиглутаминовые повторы могут кодироваться выражением (CAR)*.
[00134] В некоторых вариантах реализации последовательность повторов, включенная в геномный локус, включает в себя последовательность коротких тандемных повторов (КТП). В некоторых вариантах реализации расширение FTR связано с синдромом ломкой X-хромосомы, боковым амиотрофическим склерозом (АБС), болезнью Хантингтона, атаксией Фридрейха, спиномозжечковой атаксией, спинобульбарной мышечной атрофией, миотонической дистрофией, болезнью Мачадо-Джозефа или дентато-рубро-паллидо-льюисовой атрофией.
[00135] Процесс 140 включает в себя сбор прочтений последовательностей нуклеиновых кислот исследуемого образца из базы данных. См. блок 142. В некоторых вариантах реализации прочтения нуклеотидных последовательностей изначально выровнены с эталонным геномом, но в данном случае в рамках процесса выполняется повторное выравнивание прочтений последовательностей с исследуемым геномным локусом, как описано ниже. В альтернативных вариантах реализации прочтения можно непосредственно выравнивать с графом последовательности без первоначального выравнивания с эталонным геномом.
[00136] В рамках процесса 140 выполняется выравнивание прочтений последовательности с последовательностью геномного локуса, включающей в себя одну или более последовательностей повторов. См. блок 144. Последовательность геномного локуса представлена данными, хранящимися в системной памяти, где также хранится структура данных графа последовательности. граф последовательности включает в себя направленный граф, где вершины представляют собой нуклеотидные последовательности, а направленные ребра соединяют вершины. нуклеотидная последовательность, представленная вершиной, включает в себя одно или более нуклеотидных оснований. Граф последовательности включает в себя один или более собственных простых циклов. Каждый собственный простой цикл представляет собой последовательность повторов одной или более последовательностей повторов. Каждая последовательность повторов включает в себя повторы повторяющегося звена одного или более нуклеотидов.
[00137] В некоторых вариантах реализации прочтения последовательностей изначально выравнивают с эталонным геномом для определения геномных координат прочтений до того, как подмножество первоначально выровненных прочтений будет выровнено с одним или более графами последовательностей, представляющими одну или более исследуемых последовательностей. В некоторых вариантах реализации первоначально выровненные прочтения выровнены с графами последовательностей для определения экспансий повторов в диапазоне от нескольких десятков до нескольких тысяч областей (каждая область соответствует графу последовательности). Общее число первоначально выровненных прочтений, которые повторно выравнивают с графами последовательностей во время каждого применения вариантов реализации, может находиться в диапазоне от тысяч до множества миллионов прочтений.
[00138] В некоторых вариантах реализации прочтения, изначально выровненные с исследуемой последовательностью или локусом, выбирают в качестве подмножества прочтений, затем подмножество выравнивают с последовательностями повторов, каждая из которых представлена в графе последовательности, причем граф последовательности имеет один или более собственных простых циклов, и эти собственные простые циклы представляют одну или более последовательностей повторов. В разнообразных вариантах реализации прочтение, находящееся в пределах около 10, 50, 100, 500, 1 000, 2 000, 3 000, 4 000, 5 000, 6 000, 7 000, 8 000, 9 000, 10 000, 50 000, 100 000 оснований исследуемой последовательности или локуса, считается находящимся вблизи исследуемой последовательности или локуса. В некоторых вариантах реализации прочтение, находящееся в пределах около 1 000, 2 000, 3 000, 4 000, 5 000, 6 000, 7 000, 8 000, 9 000 или 10 000 оснований исследуемого локуса находится рядом с исследуемым локусом. Некоторые из необработанных прочтений могут изначально являться плохо выравненными, поскольку они, в том числе, включают последовательности повторов, выравнивание которых сложно выполнить в однозначной манере. В некоторых вариантах реализации прочтения, изначально являющиеся плохо выравненными (например, по результатам измерения с применением оценки выравнивания), но при этом спаренные с прочтением, выровненным с исследуемым локусом или пространством вблизи него (в паре прочтений с парными концами), выровнены с графом последовательности. В некоторых вариантах реализации прочтения, изначально выровненные с нецелевыми областями, которые представляют собой известные горячие точки для прочтения неправильного выравнивания, выровнены с графом последовательности.
[00139] На Фиг. 1D, 1E и 1F проиллюстрированы три разных графа последовательности в соответствии с некоторыми вариантами реализации. На Фиг. 1D проиллюстрирован первый граф последовательности 1100, представляющий первый геномный локус, содержащий последовательность повторов, имеющую тринуклеотидное повторяющееся звено CAG. Первый граф последовательности 1100 включает в себя вершины 1102 и 1112, соответственно представляющие две фланкирующие последовательности. Первый граф последовательности также включает вершину 1106, представляющую собой последовательность повторов, содержащий тринуклеотидное повторяющееся звено CAG. Первый граф последовательности включает в себя направленное ребро 1104, соединяющее вершину 1102 (фланкирующая последовательность) и вершину 1106 (последовательность повторов CAG) в направлении от вершины 1102 к вершине 1106. Направление ребра указывает на относительное положение двух нуклеотидных последовательностей. Первый граф последовательности также включает в себя направленное ребро 1104, соединяющее вершину 1102 (фланкирующая последовательность) и вершину 1106 (последовательность повторов CAG) в направлении от вершины 1102 к вершине 1106. Первый граф последовательности также включает в себя направленное ребро 1110, соединяющее вершину 1106 (последовательность повторов CAG) и вершину 1112 (фланкирующая последовательность) в направлении от вершины 1106 к вершине 1112. Первый граф последовательности также включает в себя собственный простой цикл 1108, который представляет собой последовательность повторов, содержащую повторяющееся звено CAG (проиллюстрированное вершиной 1106), которое повторяется один или более раз. Путь, проходящий от начальной вершины к конечной вершине графа последовательности, представляет собой последовательность геномного локуса, которая может содержать нуклеотиды, находящиеся вблизи последовательности повторов, такой как фланкирующие последовательности.
[00140] На Фиг. 1E проиллюстрирован второй граф последовательности 1200, представляющий второй геномный локус. Второй граф последовательности 1200 включает в себя вершины 1202 и 1224, соответственно представляющие две фланкирующие последовательности. Второй граф последовательности также включает в себя вершину 1206 и вершину 1216, представляющие последовательность повторов, содержащую. тринуклеотидное повторяющееся звено CAG, и последовательность повторов, содержащую тринуклеотидное повторяющееся звено CCG соответственно. Второй граф последовательности также включает в себя вершину 1212, представляющую последовательность CAACAG, не имеющую повторов. Второй граф последовательности включает в себя направленные ребра 1204, 1210, 1214 и 1220. Данные направленные ребра направленно соединяют вершины 1202, 1206, 1212, 1216 и 1224, как изображено на иллюстрации. Второй граф последовательности также включает в себя собственный простой цикл 1208, который представляет собой последовательность повторов, содержащую повторяющееся звено CAG (проиллюстрированное вершиной 1206), которое повторяется один или более раз. Второй граф последовательности также включает в себя собственный простой цикл 1218, который представляет собой последовательность повторов, содержащую повторяющееся звено CCG (проиллюстрированное вершиной 1216), которое повторяется один или более раз.
[00141] На Фиг. 1F проиллюстрирован третий граф последовательности 1300, представляющий третий геномный локус. Третий граф последовательности 1300 аналогичен второму графу последовательности 1200, но включает в себя два альтернативных пути, представляющие два аллеля CAC и CAT. Два аллеля могут представлять собой аллели ОНВ или ОНП. Направленное ребро 1310, вершина 1312 и направленное ребро 1314 представляют собой первый аллель CAC. Направленное ребро 1316, вершина 1318 и направленное ребро 1320 представляют собой второй аллель САТ. Третий граф последовательности включает в себя элементы, иным образом аналогичные элементам второго графа последовательности, включая вершины 1302, 1306, 1322 и 1328. Он также включает собственные простые циклы 1308 и 1324, указывающие на последовательности повторов CAG и CCG. Он дополнительно включает в себя направленные ребра 1304 и 1326.
[00142] В некоторых вариантах реализации прочтения последовательностей выравнивают с графом последовательности с применением методик, описанных ниже.
[00143] 1. Индекс кмер строят на основании всего графа таким образом, что при наличии значения кмер из последовательности можно пересчитать все узлы графа, в которых начинается или заканчивается такой кмер. В некоторых случаях кмер может начинаться на одном узле и заканчиваться на другом узле.
[00144] 2. Относительно каждой точки графа регистрируют два подграфа: один в прямом направлении кмер, а другой в обратном направлении. На подграфах выполняют «разворачивание» экспансии повторов до оставшейся длины прочтения, при этом на них отсутствуют какие-либо узлы, расположенные дальше от места попадания кмер по сравнению с остальной длиной прочтения, и предполагается, что экспансия повторов не происходит. Процедура представляет собой метод поиска «в ширину» и используется для генерации структуры данных, содержащей следующие элементы:
[00145] - Последовательность всех последовательностей узлов (включая расширенные повторы) на подграфе
[00146] - Индекс узлов, который позволяет легко получать идентификатор узла из смещения в последовательности при выполнении поиска с возвращением по алгоритму Смита-Уотермана.
[00147] - Последовательность смещений концов узлов с входящими ребрами для каждого начального смещения узла
[00148] - Индекс для каждого узла, позволяющий легко определять, находится ли основание в начале узла или не в начале узла, а также подсчитывать все концевые смещения предшествующих узлов.
[00149] 3. Выравнивание служит для:
[00150] - Поддержания аффинных пробелов.
[00151] - Поиска наилучшего выравнивания (выравниваний) последовательности с учетом приведенной выше информации и матрицы штрафов.
[00152] Доступны два разных интерфейса:
[00153] - Приведены данные о наилучшем выравнивании и втором наилучшем результате оценки выравнивания.
[00154] - Весь массив наилучших выравниваний, а также второй наилучший результат оценки выравнивания.
[00155] Выравнивания представляют собой общие выравнивания, определяющие штраф за пробел между потенциальным кмер и началом выровненной последовательности. В некоторых вариантах реализации настраивают параметры времени компиляции.
[00156] Использующийся алгоритм заполнения матрицы доступен в двух вариантах реализации:
[00157] - Последовательные циклы со сложностью N*M.
[00158] - Последовательные циклы циклов фиксированного размера с параметром времени компиляции фиксированной длины, по умолчанию равным 16, которые gcc автоматически распознает и преобразует в векторные команды SSE или AVX на ЦП.
[00159] В некоторых вариантах реализации повторяющееся звено одной или более последовательностей повторов содержит по меньшей мере один частично определенный нуклеотид. В некоторых вариантах реализации конкретное повторяющееся звено содержит вырожденные кодоны.
[00160] В некоторых вариантах реализации один или более собственных простых циклов содержат два или более собственных простых циклов, представляющих две или более последовательностей повторов. См., например, Фиг. 1E, Фиг. 1F и Фиг. 1G, панель (b).
[00161] В некоторых вариантах реализации граф последовательности дополнительно содержит два или более альтернативных путей для двух или более аллелей. См., например, Фиг. 1F, ссылочные номера 1312 и 1318. См. также Фиг. 1H, ссылочные номера 165 и 167а, указывающие локус Lynch I (MSH2), где верхний путь включает в себя вершину для основания нуклеиновой кислоты A, а нижний путь включает в себя вершину для основания нуклеиновой кислоты T.
[00162] В некоторых вариантах реализации два или более аллеля содержат делецию или замену. В некоторых вариантах реализации замена включает в себя однонуклеотидный вариант (ОНВ) или однонуклеотидный полиморфизм (ОНП). См., например, Фиг. 1F, ссылочные номера 1312 и 1318.
[00163] В некоторых вариантах реализации выравнивание прочтения последовательности с графом последовательности включает в себя: поиск соответствия кмер между прочтением последовательности и путем графа последовательности и последующее расширение данного пути до полного выравнивания. В некоторых вариантах реализации выравнивание включает в себя выделение подграфа по пути; «разворачивание» циклов на подграфе для получения направленного ациклического графа; и выполнение выравнивания Смита-Уотермана относительно прочтения последовательности по направленному ациклическому графу.
[00164] В некоторых вариантах реализации выравнивание прочтения последовательности с графом последовательности включает в себя сокращение графа путем удаления концов выравниваний с низким уровнем достоверности. После выравнивания прочтения с графом в рамках способа выполняют поиск других аналогичных альтернативных выравниваний. Это осуществляется путем повторного выравнивания первоначального прочтения с путями графа, который перекрывает путь первоначального выравнивания. Это позволяет определить наличие низкого уровня достоверности одного или обоих концов изначального выравнивания; такой низкий уровень достоверности указывает на то, что они могли бы быть выровнены другим способом. Возможность обнаружения частей выравнивания с высоким и низким уровнем достоверности позволяет точно определить, какие генетические варианты поддержаны в рамках прочтения.
[00165] В некоторых вариантах реализации выравнивание прочтения последовательности с графом последовательности включает в себя объединение выравниваний путем: выравнивания подпоследовательностей прочтения с графом последовательности; и объединения выравниваний подпоследовательностей для полного выравнивания прочтения последовательности.
[00166] В некоторых вариантах реализации в рамках процесса также генерируют граф последовательности на основании спецификации локуса, включающей в себя структуру геномного локуса. В некоторых вариантах реализации спецификация локуса определяется в каталоге вариантов, как объяснено выше.
[00167] См. также на панелях (b)-(d) Фиг. 1G для схематических иллюстраций выравнивания прочтений с графом последовательности локуса HTT. На Фиг. 1H схематично изображены анализаторы локусов 164 для выполнения выравнивания прочтений с графом последовательности, в том числе отн. локуса Lynch I (165).
[00168] Способ 140 дополнительно включает в себя определение одного или более генотипов одной или более последовательностей повторов с применением прочтений последовательностей, выровненных с графом последовательности. См. блок 140. См. также на панели (e) Фиг. 1G изображено определение двух КТП (CAG и CCG) в локусе HTT. Последовательность слева, включая повторы CAG, представляет собой CAGCAGCAGCAGCAG (Посл. №: 4). Последовательность слева, включая повторы CCG, представляет собой CCGCCGCCGCCGCCG (Посл. №: 5).
[00169] На Фиг. 1H изображен модуль ПО Variant Genotyper (168) для определения вариантов в локусе Lynch I, включая ОНВ с аллелями A/T (169а) и повторяющимся мономером A (169b). На Фиг. 1H также представлены модули ПО Variant Analyzer (166) для управления данными о выравнивании последовательностей и их передачи в ПО Variant Genotyper (168), а также пути реализации ПО Variant Analyzer для работы с ОНВ с аллелями A/T (167а) и повторяющимся мономером A (167b). Результаты отн. локуса, полученные с помощью ПО Genotyper, проиллюстрированы на Фиг. 1H, рамка 170, там же представлен генотип ОНВ с аллелями A/T (171а) и повторяющимся мономером A (171b).
[00170] В некоторых вариантах реализации граф последовательности включает в себя два альтернативных пути для двух аллелей, а способ дополнительно включает в себя генотипирование двух или более аллелей с применением прочтений последовательностей, выровненных с двумя или более альтернативными путями. В некоторых вариантах реализации генотипирование двух или более аллелей включает в себя охват двух или более альтернативных путей к вероятностной модели для определения вероятностей двух или более аллелей. В некоторых вариантах реализации вероятностная модель моделирует вероятность аллеля в зависимости от охвата аллеля, причем функция выбрана из распределения Пуассона, отрицательного биномиального распределения, биномиального распределения или бета-биномиального распределения.
[00171] В некоторых вариантах реализации функция вероятности представляет собой распределение Пуассона, а ее параметр скорости оценивают по длине прочтения и средней глубине, наблюдаемой в геномном локусе.
[00172] В модели Пуассона вероятность аллеля выражена следующим образом:
[00173] P(Y=y) = (Cy × e-C)/y!
[00174] • y представляет собой охват прочтений основания
[00175] • C представляет собой среднюю глубину, наблюдаемую в геномном локусе
[00176] В некоторых вариантах реализации среднюю глубину C определяют следующим образом.
[00177] C=LN/G
[00178] • G представляет собой длину геномного локуса
[00179] • L представляет собой длину прочтения
[00180] • N представляет собой общее количество прочтений
Библиотека GraphTools
[00181] В некоторых вариантах реализации при работе с графами последовательностей применяют библиотеку GraphTools. В библиотеке присутствуют ключевые схематические изображения графов (сами графы, пути графов и выравнивания графов), операции с ними и алгоритмы для выравнивания линейных последовательностей с графами.
[00182] В некоторых вариантах реализации граф последовательности состоит из узлов и направленных ребер. Графы могут включать собственные простые циклы (ребра, соединяющие узел с самим собой), но не включать другие циклы. Узлы содержат последовательности, состоящие из базовых оснований и кодов вырожденных оснований, определенных ИЮПАК.
[00183] Путь в графе определяется последовательностью узлов, через которые проходит путь, а также начальной точкой пути на первом узле и конечной точкой на последнем узле. Положения указывают с использованием «полуоткрытой» системы координат с отсчетом от нуля. В библиотеке представлены обозначения множества операций на путях, включая расширение и сокращение путей, проверки перекрытия и объединение путей.
[00184] Выравнивания графов содержат кодовые обозначения того, как линейные запрашиваемые последовательности (обычно это последовательности прочтений) выравнивают с графами. В некоторых вариантах реализации выравнивание графа содержит путь графа и последовательность линейных выравниваний, определяющих выравнивание запрашиваемой последовательности с узлами пути графа. С применением соответствующих операций с путями, выравнивания графов могут быть сокращены или объединены с другими выравниваниями графов. Сокращение пути обеспечивает механизм устранения концов выравниваний с низким уровнем достоверности, в то время как объединение выравниваний используется алгоритмами выравнивания графа для объединения полного выравнивания запрашиваемой последовательности с выравниваниями подпоследовательностей (например, кмер). В некоторых вариантах реализации алгоритм выравнивания работает путем поиска соответствия кмер между запрашиваемой последовательностью и графом и последующего расширения этого соответствия до полного выравнивания. В некоторых вариантах реализации выравнивание включает в себя выделение подграфа по пути, совпадающему с соответствием кмер (с «разворачиванием» циклов в процессе). Затем выполняют выравнивание Смита-Уотермана относительно полученного направленного ациклического графа. В некоторых вариантах реализации алгоритм поддерживает определение штрафов за аффинные пробелы и записывает с применением циклов постоянной длины для генерации кода SIMD компиляторами.
[00185] В некоторых вариантах реализации путь в графе может быть получен с помощью алгоритма поиска, который включает расширение или сокращение пути путем увеличения или уменьшения количества повторов повторяющегося звена, представленного собственным простым циклом, до тех пор, пока выравнивание не достигнет критерия поиска или конвергенции (например, в случае, когда оценка выравнивания достигает максимального значения).
[00186] В некоторых вариантах реализации на основе графа последовательности генерируют множество путей в графе, причем каждый путь в графе представляет собой конкретное число повторов повторяющегося звена, представленного собственным простым циклом. Запрашиваемую последовательность выравнивают с множеством путей в графе, а затем путь, соответствующий критерию выравнивания, выбирают для выполнения графического выравнивания.
Структура способа применения
[00187] Некоторые варианты реализации разработаны в качестве общего способа выполнения целевого генотипирования вариантов (Фиг. 1H). В ходе каждого цикла программа предпринимает попытки генотипирования набора вариантов,
[00188] описанных в файле каталога вариантов. Варианты, расположенные в непосредственной близости друг от друга, сгруппированы в один и тот же локус. Структуру локуса указывают с применением ограниченного подмножества регулярного синтаксиса выражения (RE). RE содержат последовательности, расположенные в алфавитном порядке, состоящие из символов базовых оснований и кодов вырожденных оснований, определенных ИЮПАК; они должны содержать одно или более из следующих выражений (<последовательность>)?, (<последовательность a>|<последовательность b>), (<последовательность>)*, (<последовательность>)+, возможно, разделенных разрывами последовательностей. Данные выражения соответствуют вставкам/делециям, заменам, повторению последовательности 0 или более раз, а также повторению последовательности по меньшей мере один раз, соответственно. Кроме того, описание каждого локуса содержит набор основных областей для данного локуса, а также основные координаты каждого составляющего варианта.
[00189] Основная часть работы упорядочена по объектам класса LocusAnalyzer, который синтезирует граф последовательности, представляющий локус соответствующего RE в процессе инициализации. После инициализации анализатор локусов обрабатывает соответствующие прочтения путем их выравнивания с графом и дальнейшей передачи полученных выравниваний в ПО VariantAnalyzer, предназначенное для работы со всеми вариантами, содержащимся в локусе. ПО Variant Analyzer собирает информацию, важную с точки зрения процесса генотипирования связанного варианта, и передает ее ПО Genotyper, отвечающему за выполнение процесса генотипирования. Результаты, полученные каждой единицей ПО Genotyper, затем используются для создания выходного файла в формате VCF.
[00190] Например, в анализаторе LocusAnalyzer, отвечающем за обработку локуса патогенным вариантом, связанным с синдромом Lynch I, используют анализатор ОНВ и анализатор КТП (Фиг. S1, правая панель).
5.8. Применение ПО Genotyper для работы с делециями
[00191] Некоторые КТП могут иметь рядом небольшую вставку или делецию. Такие делеции моделируются в виде дополнительных подграфов фланкирующих последовательностей КТП. Количество прочтений, сопоставляемых с каждым аллелем (или путем в графе), моделируют с помощью распределения Пуассона, параметр скорости которого оценивают по средней глубине и длине прочтения, наблюдаемой в локусе. Вероятность генотипа рассчитывают по байесовской схеме.
5.9. Идентификация экспансий повторов
[00192] Используя описанные в настоящем документе варианты реализации, можно определить различные наследственные заболевания, относящиеся к экспансии повторов с высокой эффективностью, чувствительностью и/или селективностью по сравнению с традиционными способами. В некоторых вариантах реализации настоящего изобретения предложены способы определения и распознавания значимых с медицинской точки зрения экспансий повторов, таких как экспансия повторов CGG, вызывающая задержку умственного развития при синдроме ломкой X-хромосомы, с помощью прочтений последовательностей, которые не полностью охватывают последовательность повторов. Короткие прочтения, такие как прочтения 100 п.н.о., являются недостаточно длинными для выполнения секвенирования множества экспансий повторов. Однако при выполнении анализа описанными способами образцы с экспансией повторов демонстрируют статистически значимый избыток прочтений, содержащих большое число последовательности повторов. Кроме того, крайне большие экспансии повторов содержат невыровненные пары прочтений, где оба прочтения полностью или почти полностью состоят из последовательности повторов. Нормальные образцы используют для определения фоновых ожиданий.
[00193] Общепринятое убеждение заключается в том, что экспансию повторов невозможно обнаружить без применения прочтения, охватывающего весь повтор. В существующих подходах к обнаружению экспансий повторов используют целевое секвенирование с длинными прочтениями, и в некоторых случаях такие прочтения не являются успешными из-за недостаточной длины, вследствие чего с их помощью невозможно охватить последовательность повторов. Результаты некоторых описанных вариантов реализации были неожиданными, частично потому, что в них использованы нормальные (нецелевые) данные последовательности и длина прочтения около в 100 п.н.о., однако они обеспечивают очень высокую чувствительность при обнаружении экспансий повторов. Способы, изложенные в настоящем документе, позволяют обнаруживать число повторяющихся звеньев при экспансии повторов с помощью парных прочтений, имеющих длину вставки (т.е. два прочтения последовательностей и промежуточную последовательность), которая является более короткой, чем длина всей последовательности повторов.
[00194] Что касается деталей способов определения наличия экспансии повторов в соответствии с некоторыми вариантами реализации, на Фиг. 2 представлена блок-схема с изображением высокого уровня вариантов реализации для определения наличия или отсутствия экспансии повторов в последовательности повторов в образце. Последовательность повторов представляет собой нуклеотидную последовательность, содержащую повторяющиеся короткие последовательности, называемые повторяющимися звеньями. В приведенной выше Таблице 1 изложены примеры повторяющихся звеньев, количество повторов повторяющихся звеньев в повторяющихся последовательностях отн. нормальных и патогенных последовательностей, гены, связанные с последовательностями повторов, и заболевания, связанные с экспансией повторов. Процесс 200, проиллюстрированный на Фиг. 2, начинают с получения парных концевых прочтений исследуемого образца. См. блок 202. Парные концевые прочтения обрабатывают для выравнивания с эталонной последовательностью, содержащей исследуемую последовательность повторов. В некоторых контекстах процесс выравнивания также называют процессом сопоставления. Исследуемый образец содержит нуклеиновую кислоту и может иметь форму биологической жидкости, ткани и т.д., как дополнительно описано в разделе «Образец» ниже. Прочтения последовательностей подвергали процессу выравнивания для сопоставления с эталонной последовательностью. Для выполнения выравнивания прочтений с эталонной последовательностью могут использовать различные инструменты и алгоритмы выравнивания, как описано в других разделах настоящего описания. Обычно при выполнении алгоритмов выравнивания некоторые прочтения успешно выравнивают с эталонной последовательностью, тогда как другие прочтения могут выравнивать не настолько успешно, либо могут плохо выравниваться с эталонной последовательностью. Прочтения, которые последовательно выровнены с эталонной последовательностью, связаны с сайтами на эталонной последовательности. Выровненные прочтения и связанные с ними сайты также называются метками последовательностей. Как объяснено выше, выравнивание некоторых прочтений последовательностей, содержащих большое число повторов, с эталонной последовательностью, является более сложным. Если прочтение выровнено с эталонной последовательностью, и при этом уровень ошибочно спаренных оснований находится выше определенного критерия, такое прочтение считается плохо выровненным. В разнообразных вариантах реализации прочтения считают плохо выровненными, если они выровнены с по меньшей мере около 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 несовпадениями. В других вариантах реализации прочтения считают плохо выровненными, если они выровнены с по меньшей мере около 5% несовпадений. В других вариантах реализации прочтение считают плохо выровненным, если оно выровнено с по меньшей мере около 10%, 15% или 20% ошибочно спаренных оснований.
[00195] Как изображен на Фиг. 2, в процессе 200 описана определениекация базовых и закрепленных прочтений в парных концевых прочтениях. См. блок 204. Базовые прочтения считывают между парными концевыми прочтениями, которые выровнены с исследуемой последовательностью повторов или пространством рядом с ней. Например, базовое прочтение может быть выровнено с местоположением на эталонной последовательности, которая отделена от последовательности повторов длиной последовательности, которая меньше длины последовательности вставки. Длина разделения может быть меньше. Например, базовое прочтение может быть выровнено с местоположением на эталонной последовательности, которая отделена от последовательности повторов длиной последовательности, которая меньше длины последовательности базового прочтения или меньше общей длины последовательности базового прочтения и последовательности, которая соединяет базовое прочтение с закрепленным прочтением (длина вставки минус длина закрепленного прочтения). В некоторых вариантах реализации исследуемой последовательностью повторов может являться последовательность повторов в гене FMR1, включая повторы повторяющегося звена CGG. В нормальной эталонной последовательности последовательность повторов в гене FMR1 включает в себя около от 6 до 32 повторов повторяющегося звена CGG. Поскольку повторы расширяются до свыше 200 копий, существует вероятность патогенизации экспансии повторов, что вызывает синдром ломкой X-хромосомы. В некоторых вариантах реализации прочтение считают выровненным вблизи исследуемой последовательности в случаях, когда оно выровнено в пределах 1 000 п.н.о. от исследуемой последовательности повторов. В других вариантах реализации данный параметр может быть скорректирован, например, в пределах около 100 п.н.о., 200 п.н.о., 300 п.н.о., 400 п.н.о., 500 п.н.о., 600 п.н.о., 700 п.н.о., 800 п.н.о., 900 п.н.о., 1 500 п.н.о., 2 000 п.н.о., 3 000 п.н.о., 5 000 п.н.о. и т. д. Кроме того, в рамках процесса также определяют закрепленные прочтения, которые представляют собой прочтения, спаренные с базовыми прочтениями, но при этом они являются плохо выровненными или не могут быть выровнены с их эталонной последовательностью. Дополнительная информация о плохо выровненных прочтениях приведена выше.
[00196] Процесс 200 дополнительно включает в себя определение вероятности присутствия экспансии повторов последовательности повторов в исследуемом образце, на основании определенных закрепленных прочтений, по меньшей мере, частично. См. блок 206. Данный этап процесса определения может включать в себя разнообразные подходящие методы анализа и вычисления, как дополнительно описано ниже. В некоторых вариантах реализации в рамках процесса для определения вероятности наличия экспансии повторов применяют определенные базовые прочтения, а также закрепленные прочтения. В некоторых вариантах реализации количество повторов в определенных базовых и закрепленных прочтениях анализируют и сравнивают с одним или более критериями, полученными теоретическим путем, либо полученными из эмпирических данных затронутых контрольных образцов.
[00197] В разнообразных вариантах реализации, описанных в настоящем документе, повторы получают в качестве повторов внутри рамки, причем два повтора одного и того же повторяющегося звена попадают в одну и ту же рамку считывания. Рамка считывания представляет собой способ деления последовательности нуклеотидов в молекуле нуклеиновой кислоты (ДНК или РНК) на набор последовательных неперекрывающихся триплетов. Во время преобразования триплеты кодируют аминокислоты, и такие триплеты называются кодонами. Таким образом, любая последовательность имеет три возможных рамки считывания. В некоторых вариантах реализации повторы подсчитывают в соответствии с тремя различными рамками считывания, а один подсчет из трех, показавший наибольшее значение, считается количеством соответствующих повторов в прочтении.
[00198] Пример процесса, включающего в себя выполнение дополнительной операции и анализов, проиллюстрирован на Фиг. 3. На Фиг. 3 представлена блок-схема, иллюстрирующая процесс 300, необходимый для обнаружения экспансии повторов с использованием парных концевых прочтений, имеющих большое количество повторов. Способ 300 включает в себя дополнительные действия для обработки исследуемого образца. Процесс начинают с секвенирования исследуемого образца, содержащего нуклеиновые кислоты, для получения парных концевых прочтений. См. блок 302. В некоторых вариантах реализации исследуемый образец может быть получен и подготовлен разнообразным способами, как дополнительно описано ниже в разделе «Образцы». Например, исследуемый образец может представлять собой биологическую текучую среду, например, плазму или любой подходящий образец, как описано ниже. Образец может быть получен с применением неинвазивной процедуры, такой как обычный забор крови. В некоторых вариантах реализации исследуемый образец содержит смесь молекул нуклеиновых кислот, например, молекул скДНК. В некоторых вариантах реализации исследуемый образец представляет собой образец материнской плазмы, содержащий смесь молекул эмбриональной и материнской ДНК.
[00199] Перед секвенированием нуклеиновые кислоты экстрагируют из образца. Подходящие процессы экстракции и устройство описаны в других разделах настоящего документа. В некоторых вариантах реализации устройством выполняют совместную обработку ДНК из множества образцов для мультиплексирования библиотек и данных последовательности. В некоторых вариантах реализации устройством 20 выполняют обработку ДНК восьми или более исследуемых образцов параллельно. Как описано ниже, системой секвенирования могут обрабатывать извлекаемую ДНК для получения библиотеки кодированных (например, путем штрих-кодирования) фрагментов ДНК.
[00200] В некоторых вариантах реализации нуклеиновые кислоты в исследуемом образце можно дополнительно обрабатывать для подготовки библиотек для одноканального или многоканального секвенирования, как дополнительно описано ниже в разделе «Подготовка библиотек для секвенирования». После обработки и подготовки образцов секвенирование нуклеиновой кислоты могут выполнять разнообразными способами. В некоторых вариантах реализации можно применять разнообразные платформы и протоколы секвенирования следующего поколения, которые дополнительно описаны в разделе «Способы секвенирования» ниже.
[00201] Независимо от конкретной платформы и протокола секвенирования в блоке 302 по меньшей мере часть содержащихся в образце нуклеиновых кислот секвенируют для генерирования десятков тысяч, сотен тысяч или миллионов прочтений последовательностей, например, прочтений 100 п.н.о. В некоторых вариантах реализации прочтения включают в себя парные концевые прочтения. В других вариантах реализации, в том числе описанных ниже со ссылкой на Фиг. 5, в дополнение к парным концевым прочтениям для определения последовательности повторов можно применять одиночные концевые длинные прочтения, охватывающие более сотен тысяч или десятков тысяч оснований. В некоторых вариантах реализации прочтения последовательностей содержат около 20 п.н.о., около 25 п.н.о., около 30 п.н.о., около 35 п.н.о., около 36 п.н.о., около 40 п.н.о., около 45 п.н.о., около 50 п.н.о., около 55 п.н.о., около 60 п.н.о., около 65 п.н.о., около 70 п.н.о., около 75 п.н.о., около 80 п.н.о., около 85 п.н.о., около 90 п.н.о., около 95 п.н.о., около 100 п.н.о., около 110 п.н.о., около 120 п.н.о., около 130, около 140 п.н.о., около 150 п.н.о., около 200 п.н.о., около 250 п.н.о., около 300 п.н.о., около 350 п.н.о., около 400 п.н.о., около 450 п.н.о. или около 500 п.н.о. Ожидается, что при генерировании парных концевых прочтений технологические достижения обеспечат возможность одиночных концевых прочтений более 500 п.н.о. и прочтений более 1 000 п.н.о.
[00202] В процессе 300 описано выравнивание результатов парных концевых прочтений, полученных из блока 302, с эталонной последовательностью, включающей в себя последовательность повторов. См. блок 304. В некоторых вариантах реализации последовательность повторов имеет склонность к экспансии. Известно, что в некоторых вариантах реализации экспансия повторов связана с генетическим заболеванием. В других вариантах реализации экспансия повторов последовательности повторов ранее не была изучена для установления связи с генетическим заболеванием. Способы, описанные в настоящем документе, позволяют обнаруживать последовательность повторов и экспансию повторов независимо от любой связанной с ней патологии. В некоторых вариантах реализации прочтения выравнивают с эталонным геномом; пример: hg18. В других вариантах реализации прочтения выравнивают с участком эталонного генома, например, с хромосомой или сегментом хромосомы. Прочтения, однозначно сопоставленные с эталонным геномом, называются метками последовательностей. В одном варианте реализации по меньшей мере около 3 × 106 квалифицированных меток последовательности, по меньшей мере около 5 × 106 квалифицированных меток последовательности, по меньшей мере около 8 × 106 квалифицированных меток последовательности, по меньшей мере около 10 × 106 квалифицированных меток последовательности, по меньшей мере около 15 × 106 квалифицированных меток последовательности, по меньшей мере около 20 × 106 квалифицированных меток последовательностей, по меньшей мере около 30 × 106 квалифицированных меток последовательностей, по меньшей мере около 40 × 106 квалифицированных меток последовательностей или по меньшей мере около 50 × 106 квалифицированных меток последовательностей получают на основе прочтений, однозначно сопоставляемых с эталонным геномом.
[00203] В некоторых вариантах реализации в рамках процесса могут выполнять фильтрацию прочтений последовательности перед выравниванием. В некоторых вариантах реализации фильтрация прочтений представляет собой процесс качественной фильтрации, выполняемый с применением программного обеспечения секвенатора и направленный на отсечение ошибочных и низкокачественных прочтений. Например, программное обеспечение Illumina под названием Sequencing Control Software (SCS), а также Consensus Assessment of Sequence and Variation, применяют для отсечения ошибочных и низкокачественных прочтений путем преобразования исходных изображений-данных, генерированных в результате реакций секвенирования, в значения интенсивности, процессы распознавания оснований, оценки качества и дополнительные форматы для сбора биологически релевантной информации для этапа дальнейшего анализа.
[00204] В определенных вариантах реализации прочтения, полученные с помощью аппарата для секвенирования, представлены в электронном формате. Процесс выравнивания осуществляют с помощью вычислительного устройства, как описано ниже. Отдельные прочтения сравнивают с эталонным геномом, который часто является обширным (миллионы пар нуклеотидных оснований), чтобы определить точки, где прочтения однозначно соответствуют эталонному геному. В некоторых вариантах реализации в рамках выравниваний допускают ограниченное несоответствие между прочтениями и эталонным геномом. В некоторых случаях допускается несовпадение 1, 2, 3 или более соответствующих пар нуклеотидных оснований в эталонном геноме, и при этом по-прежнему выполняют сопоставление. В некоторых вариантах реализации прочтения считают выровненными прочтениями в случаях, когда прочтения выровнены с эталонной последовательностью, содержащей не более чем 1, 2, 3 или 4 пары нуклеотидных оснований. Соответственно, невыровненные прочтения представляют собой прочтения, которые не могут быть выровнены или являются плохо выровненными. Плохо выровненные прочтения представляют собой прочтения с большим количеством несовпадений, чем у выровненных прочтений. В некоторых вариантах реализации прочтения считают выровненными прочтениями в случаях, когда прочтения выровнены с эталонной последовательностью, содержащей не более 1%, 2%, 3%, 4%, 5% или 10% пар нуклеотидных оснований.
[00205] После выравнивания парных концевых прочтений с эталонной последовательностью, содержащей исследуемую последовательность повторов, в рамках процесса 300 выполняют определение базовых и закрепленных прочтений между парными концевыми прочтениями. См. блок 306. Как упоминалось выше, базовые прочтения представляют собой парные концевые прочтения, выровненные с последовательностью повторов или с пространством вблизи нее. В некоторых вариантах реализации базовые прочтения представляют собой парные концевые прочтения, которые выровнены в пределах 1 т. п. н. Последовательности повторов. Закрепленные прочтения являются спаренными с базовыми прочтениями, но при этом они не могут быть выровнены или являются плохо выровненными с эталонной последовательностью, как описано выше.
[00206] В рамках процесса 300 анализируют количество повторов повторяющихся звеньев в определенных базовых и/или закрепленных прочтениях для определения наличия или отсутствия экспансии последовательности повторов. Более конкретно, процесс 300 включает в себя применение числа повторов в прочтениях для получения чисел прочтений большого объема в базовых и/или закрепленных прочтениях. Прочтения большого объема представляют собой прочтения с большим количеством повторов, чем пороговое значение. В некоторых вариантах реализации прочтения большого объема получают только из закрепленных прочтений. В других вариантах реализации прочтения большого объема получают как с базовых, так и с закрепленных прочтений. Если в некоторых вариантах реализации число повторов приближается к максимально возможному числу повторов для прочтения, прочтение считают прочтением большого объема. Например, если прочтение составляет 100 п.н.о., а рассматриваемое повторяющееся звено составляет 3 п.н.о., максимальное число повторов составит 33. Другими словами, максимальное значение рассчитывают на основе длины парных концевых прочтений и длины повторяющегося звена. В частности, максимальное число повторов можно получить путем деления длины прочтения на длину повторяющегося звена и округления полученного значения в меньшую сторону. В данном примере в рамках различных вариантов реализации могут определять прочтения 100 п.н.о., имеющие по меньшей мере около 28, 29, 30, 31, 32 или 33 повторов в прочтениях большого объема. Количество повторов в прочтениях большого объема может регулироваться в большую или меньшую сторону на основании эмпирических факторов. В разнообразных вариантах реализации пороговое значение для прочтений большого объема составляет по меньшей мере около 80%, 85%, 90% или 95% от максимального числа повторов.
[00207] Затем в рамках процесса 300 определяют возможное присутствие экспансии повторов в последовательности повторов на основании количества прочтений большого объема. См. блок 310. В некоторых вариантах реализации в рамках анализа полученные прочтения большого объема сравнивают с критерием распознавания и выполняют определение вероятного присутствия экспансии повторов при превышении значений критерия. В некоторых вариантах реализации критерий распознавания получают на основе распределения прочтений контрольных образцов большого объема. Например, анализируют множество контрольных образцов, имеющих или предположительно имеющих нормальную последовательность повторов, и для контрольных образцов получают прочтения большого объема таким же образом, как описано выше. Можно выполнить распределение прочтений большого объема отн. контрольных образцов и оценить вероятность появления незатронутого образца с прочтениями большого объема, превышающими конкретное значение. Данная вероятность позволяет определять чувствительность и селективность с учетом критерия распознавания, установленного относительно данного конкретного значения. В некоторых вариантах реализации критерий распознавания устанавливают на пороговое значение, и таким образом вероятность появления незатронутого образца с прочтениями большого объема, превышающими пороговое значение, составляет менее 5%. Другими словами, p-значение меньше чем 0,05. В данных вариантах реализации по мере экспансии повторов последовательность повторов становится длиннее, и появляется возможность появления большего количества прочтений исключительно в рамках в последовательности повторов, а также появляется возможность получения большего количества прочтений образца. В разнообразных альтернативных вариантах реализации можно выбрать более заниженный критерий распознавания таким образом, чтобы вероятность появления незатронутого образца с прочтениями большого объема, превышающими пороговое значение, составляла менее, чем около 1%, 0,1%, 0,01%, 0,001%, 0,0001% и т.д. Следует понимать, что критерий распознавания можно отрегулировать в сторону повышения или понижения в зависимости от разнообразных факторов и необходимости повышения чувствительности или селективности испытания.
[00208] В некоторых вариантах реализации вместо применения эмпирического варианта получения критерия распознавания по количеству прочтений большого объема от контрольных образцов или в дополнение к нему теоретически может быть получен критерий распознавания для определения экспансии повторов. Можно рассчитать ожидаемое количество прочтений, которые полностью находятся в пределах диапазона повторения, с учетом количества параметров, включая длину парных концевых прочтений, длину последовательности, имеющей экспансию повторов, и глубину секвенирования. Например, можно применять глубину секвенирования для вычисления среднего расстояния между прочтениями в выровненном геноме. В случае, если отдельный образец секвенировали при 30-кратной глубине, общее количество секвенированных оснований равняется результату произведения размера генома на глубину. В случае применения настоящего способа к человеку, данное количество будет составлять около 3 × 109 30=9 × 1010. Если каждое прочтение имеет длину в 100 п.н.о., то для достижения данной глубины требуется всего 9 × 108 прочтений. Поскольку геном является диплоидным, половина данных прочтений представляет собой результаты секвенирования одной хромосомы/гаплотипа, а остальные прочтения представляют собой результаты секвенирования другой хромосомы/гаплотипа. На каждый гаплотип проводят по 4,5 × 108 прочтений, а при делении общего размера генома на это число получают среднее расстояние между начальными положениями каждого прочтения, т.е. в среднем 3 × 109/4,5 × 108=1 прочтение на каждые 6,7 п.н.о. Можно использовать данное число для оценки числа прочтений, которые будут полностью находиться в пределах последовательности повторов, на основе размера данной последовательности повторов у конкретного субъекта. В случае, если общий размер последовательности повторов составляет 300 п.н.о., любое прочтение, начинающееся в пределах первых 200 п.н.о. данной последовательности повторов, будет полностью находиться в пределах последовательности повторов (прочтения, начинающиеся в пределах последних 100 п.н.о., будут по меньшей мере частично находиться за пределами последовательности повторов на основании длин прочтений в 100 п.н.о.). Поскольку ожидается, что прочтение будет выравниваться на каждых 6,7 п.н.о., ожидается, что 200 п.н.о. / (6,7 п.н.о. на каждое прочтение) = 30 прочтений будут полностью выравниваться в пределах последовательности повторов. Несмотря на то, что данное число может изменяться, это позволяет оценить общее число прочтений, которые будут полностью находиться в пределах последовательности повторов при любом размере экспансии.
[00209] В некоторых вариантах реализации критерий распознавания рассчитывают на основании расстояния между первым и последним наблюдениями последовательности повторов в пределах прочтений, таким образом допуская мутации в последовательности повторов и ошибки секвенирования.
[00210] В некоторых вариантах реализации способ может дополнительно включать в себя диагностирование подверженности субъекта, от которого получен исследуемый образец, повышенному риску развития генетических заболеваний, таких как синдром ломкой X-хромосомы, АБС, болезнь Хантингтона, атаксия Фридрейха, спиномозжечковая атаксия, спинобульбарная мышечная атрофия, миотоническая дистрофия, болезнь Мачадо-Джозефа, дентато-рубро-паллидо-льюисова атрофия и т.д. Такой диагноз может основываться на определении вероятности присутствия экспансии повторов в исследуемом образце, а также на последовательности повторов и генов, связанных с экспансией повторов. В других вариантах реализации в случаях, если генетическое заболевание неизвестно, некоторые варианты реализации могут использоваться для обнаружения аномально большого количества повторов для выявления генетических предпосылок заболевания.
[00211] На Фиг. 4 представлена блок-схема, иллюстрирующая другой способ обнаружения экспансии повторов в соответствии с некоторыми вариантами реализации. Для определения наличия экспансии повторов в процессе 400 вместо прочтений большого объема применяют число повторов в парных концевых прочтениях исследуемого образца. Процесс 400 начинают с секвенирования исследуемого образца, содержащего нуклеиновую кислоту, для получения парных концевых прочтений. См. блок 402, эквивалентный блоку 302 процесса 300. Процесс 400 продолжают, путемвыравнивания парных концевых прочтений с эталонной последовательностью, содержащей последовательность повторов. См. блок 404, эквивалентный блоку 304 процесса 300. Процесс продолжают путем определения базовых значений и базовых прочтений в парных концевых прочтениях, причем базовые прочтения осуществляются с выравниванием с последовательностью повторов или пространством вблизи нее, а закрепленные прочтения представляют собой невыровненные прочтения, которые сопряжены с базовыми прочтениями. В некоторых вариантах реализации невыровненные прочтения включают в себя как прочтения, которые не могут быть выровнены, так и прочтения, плохо выровненные с эталонной последовательностью.
[00212] После определения базовых и закрепленных прочтений в рамках процесса 400 получают количество повторов в базовых и/или закрепленных прочтениях исследуемого образца. См. блок 408. Затем в рамках процесса получают распределение количества повторов в базовых и/или закрепленных прочтениях исследуемого образца. В некоторых вариантах реализации анализируют только количество повторов, полученное в рамках закрепленных прочтений. В других вариантах реализации анализируют повторы как закрепленных прочтений, так и базовых прочтений. Затем распределение числа повторов исследуемого образца сравнивают с распределением числа повторов одного или более контрольных образцов. См. блок 410. В некоторых вариантах реализации в рамках процесса выполняют определение присутствия экспансии повторов последовательности повторов в исследуемом образце в случае, если распределение исследуемого образца имеет статистически значимые отличия от распределения контрольных образцов. См. блок 412. В процессе 400 анализируют количество повторов относительно прочтений, включая прочтения большого объема и прочтения малого объема, которое отличается от процесса, в рамках которого анализируются только прочтения большого объема, как описано выше относительно процесса 300.
[00213] В некоторых вариантах реализации сравнение распределения исследуемого образца с распределением контрольных образцов включает в себя использование рангового критерия Манна-Уитни для определения значительности различий этих двух распределений. В некоторых вариантах реализации в рамках анализа выполняют определение вероятного присутствия экспансии повторов в исследуемом образце в случае, если распределение исследуемого образца в большей степени смещено к большему количеству повторов по сравнению с контрольными образцами, а p-значение рангового критерия Манна-Уитни составляет менее чем около 0,0001 или 0,00001. При необходимости можно выполнять корректировку р-значения для улучшения селективности или чувствительности теста.
[00214] В рамках процессов обнаружения экспансии повторов, описанной выше относительно Фиг. 2-4, применяют закрепленные прочтения, которые представляют собой невыровненные прочтения, спаренные с прочтениями, выровненными относительно исследуемой последовательности повторов. Вариации в данных процессах могут включать в себя поиск среди невыровненных прочтений таких пар прочтений, которые практически полностью состоят из последовательности повторов какого-либо типа для обнаружения новых ранее неопределенных экспансий повторов, которые могут являться значимыми с медицинской точки зрения. Данный способ не позволяет выполнить количественную оценку точного числа повторов, но является эффективным для определения крайних экспансий повторов или артефактов, которые должны быть помечены для дальнейшего выполнения количественного определения. В сочетании с более длинными прочтениями, в рамках данного способа можно выполнять как определение, так и количественную оценку повторов до 200 п.н.о. или более в пределах общей длины.
[00215] На Фиг. 5 проиллюстрирована блок-схема процесса 500, в котором для определения экспансии повторов применяют невыровненные прочтения, не связанные с какой-либо исследуемой последовательностью повторов. В рамках процесса 500 для обнаружения экспансии повторов можно применять невыровненные прочтения целого генома. Процесс начинают с секвенирования исследуемого образца, содержащего нуклеиновые кислоты, для получения парных концевых прочтений. См. блок 502. Процесс 500 продолжают путем выравнивания парных концевых прочтений с эталонным геномом. См. блок 504. Затем в рамках процесса выполняют определение невыровненных прочтений целого генома. Невыровненные прочтения включают в себя парные концевые прочтения, которые не могут быть выровнены или являются плохо выровненными с эталонной последовательностью. См. блок 506. Затем в рамках процесса анализируют число повторов повторяющегося звена в невыровненных прочтениях для определения вероятности наличия экспансии повторов в исследуемом образце. Такой анализ может не зависеть от какой-либо конкретной последовательности повторов. Такой анализ можно применять к разнообразным повторяющимся звеньям, и число повторов в различных повторяющихся звеньях исследуемого образца можно сравнить с числом повторяющихся звеньев в нескольких контрольных образцах. В данном анализе можно применять методы сравнения исследуемого образца с контрольными образцами, описанными выше. В случае, если в результате сравнения было обнаружено, что исследуемый образец имеет аномально большое число повторений повторяющегося звена, можно провести дополнительный анализ для определения наличия экспансии повторов конкретной исследуемой последовательности повторов в исследуемом образце. См. блок 510.
[00216] В некоторых вариантах реализации дополнительный анализ включает прочтение очень длинных последовательностей, которые потенциально могут охватывать длинные последовательности повторов, имеющие экспансии повторов, являющиеся значимыми с медицинской точки зрения. Прочтения, выполненные в рамках данного дополнительного анализа, длиннее парных концевых прочтений. В некоторых вариантах реализации для получения длинных прочтений используют одномолекулярное секвенирование или синтетическое секвенирование с применением длинных прочтений. В некоторых вариантах реализации связь между экспансией повторов и генетическим заболеванием является известной в данной области. Однако в других вариантах реализации связь между экспансией повторов и генетическим заболеванием может не являться известной в данной области.
[00217] В некоторых вариантах реализации анализ числа повторов повторяющегося звена в невыровненных прочтениях операции 510 включает в себя выполнение анализа большого объема, сравнимого с анализом, выполняемым в рамках операции 308 (см. Фиг. 3). Анализ включает в себя получение количества прочтений большого объема, причем прочтения большого объема представляют собой невыровненные прочтения с большим количеством повторений, чем пороговое значение; и сравнение числа прочтений большого объема прочтений в исследуемом образце с критерием распознавания. В некоторых вариантах реализации пороговое значение для прочтений большого объема составляет по меньшей мере около 80% от максимального числа повторов, причем максимальное значение рассчитывают как отношение длины парных концевых прочтений к длине повторяющегося звена. В некоторых вариантах реализации прочтения большого объема также включают в себя прочтения, сопряженные с невыровненными прочтениями, и имеющие больше повторений, чем пороговое значение.
[00218] В некоторых вариантах реализации перед дополнительным анализом операции 510 процесс дополнительно включает в себя (a) определение парных концевых прочтений, сопряженных с невыровненными прочтениями и выровненных с последовательностью повторов на эталонном геноме или с пространством вблизи нее; и (b) предоставление последовательности повторов в качестве конкретной исследуемой последовательности повторов для выполнения операции 510. Затем в рамках дополнительного анализа исследуемой последовательности повторов может использоваться любой из способов в соответствии с Фиг. 2-4, описанных выше.
Образцы
[00219] Образцы, применяемые для определения экспансии повторов, могут включать образцы, взятые из любой клетки, текучей среды, ткани или органа, включая нуклеиновые кислоты, в которых необходимо определить экспансию повторов в одной исследуемой последовательности повторов или более. В некоторых вариантах реализации, связанных с диагностикой плода, предпочтительно получать свободно-клеточные нуклеиновые кислоты, например, свободно-клеточную ДНК (скДНК), из жидкости материнского организма. Свободно-клеточные нуклеиновые кислоты, включая свободно-клеточную ДНК, можно получать разнообразными способами, известными в данной области, из биологических образцов, включая, без ограничений, плазму, серозный экссудат и мочу (см., например, Fan et al., Proc Natl Acad Sci 105:16266-16271 [2008]; Koide et al., Prenatal Diagnosis 25:604-607 [2005]; Chen et al., Nature Med. 2: 1033-1035 [1996]; Lo et al., Lancet 350: 485-487 [1997]; Botezatu et al., Clin Chem. 46: 1078-1084, 2000; and Su et al., J Mol. Diagn. 6: 101-107 [2004]).
[00220] В разнообразных вариантах реализации нуклеиновые кислоты (например, ДНК или РНК), присутствующие в образце, могут быть обогащены специфическим или неспецифическим образом перед применением (например, перед подготовкой библиотеки для секвенирования). В иллюстративных примерах, изображенных ниже, ДНК используются в качестве примера нуклеиновых кислот. Неспецифическое обогащение образца ДНК означает амплификацию всего генома фрагментов геномной ДНК образца, которые можно использовать для повышения уровня образца ДНК до момента подготовки библиотеки для секвенирования скДНК. Способы амплификации целого генома являются известными специалистам в данной области. Примерами способов амплификации целого генома является ПЦР, примированная вырожденным олигонуклеотидом (DOP), метод ПЦР с применением достройки праймера (PEP) и амплификация с множественным замещением (MDA). В некоторых вариантах реализации образец может представлять собой необогащенную ДНК.
[00221] Образец, содержащий нуклеиновые кислоты, к которым применяют способы, описанные в настоящем документе, обычно включает в себя биологический образец («исследуемый образец»), как описано выше. В некоторых вариантах реализации нуклеиновые кислоты, подлежащие проверке на предмет экспансии повторов, очищают или выделяют любым из хорошо известных способов.
[00222] Соответственно, в определенных вариантах реализации образец содержит или по существу состоит из очищенного или выделенного полинуклеотида, либо может содержать образцы, в том числе образец ткани, образец биологической текучей среды, клеточный образец и т. п. Подходящие образцы биологической текучей среды включают, без ограничений, образцы крови, плазмы, серозного экссудата, пота, слезной жидкости, мокроты, мочи, ушной жидкости, лимфы, слюны, спинномозговой жидкости, лаважа, суспензии костного мозга, влагалищной жидкости, трансцервикального лаважа, жидкости головного мозга, асцитической жидкости, секретов дыхательных, кишечных и мочеполовых путей, амниотической жидкости, молока и образцов лейкофореза. В некоторых вариантах реализации образец представляет собой образец, легко получаемый неинвазивными процедурами, например, кровь, плазму, серозный экссудат, пот, слезную жидкость, мокроту, мочу, ушную жидкость, слюну или фекалии. В некоторых вариантах реализации образец представляет собой образец периферической крови или фракции плазмы и/или серозного экссудата образца периферической крови. В других вариантах реализации биологический образец представляет собой материал, полученный путем выполнения биопсии, взятия смыва, мазка, либо клеточную культуру. В другом варианте реализации образец представляет собой смесь двух или более биологических образцов; например, биологический образец может включать в себя два или более образца биологической текучей среды, ткани или клеточной культуры. Используемые в настоящем документе термины «кровь», «плазма» и «серозный экссудат» относятся к фракциям или их обработанным частям. Аналогичным образом, в случае, если образец получают путем выполнения биопсии, взятия смыва, мазка и т.д., термин «образец» обозначает обработанную фракцию или часть, полученную путем выполнения биопсии, взятия смыва, мазка и т.д.
[00223] В некоторых вариантах реализации образцы могут быть получены из источников, включая, без ограничений, образцы, полученные от разных субъектов, образцы от одних и тех же или разных субъектов с разными стадиями развития, образцы от разных заболевших субъектов (например, субъекты с подозрением на наличие генетического заболевания), здоровых субъектов, образцы, полученные на разных стадиях заболевания пациента, образцы, полученные от пациента, подвергнутого различным способам лечения заболевания, образцы, полученные от пациентов, подвергнутых воздействию различных факторов окружающей среды, образцы, полученные от пациентов, предрасположенных к проявлению патологий, образцы, полученные от пациентов, подверженных воздействию инфекционного возбудителя заболевания и т.п.
[00224] В одном иллюстративном варианте реализации, не имеющем ограничительного характера, образец представляет собой образец, полученный из организма беременной женщины. В данном случае образец можно анализировать с использованием способов, описанных в настоящем документе, для выполнения пренатальной диагностики потенциальных хромосомных аномалий плода. Образец, полученный из организма матери, может представлять собой образец ткани, образец биологической текучей среды или клеточный образец. Термин «биологическая текучая среда» подразумевает, без ограничений, кровь, плазму, серозный экссудат, пот, слезную жидкость, мокроту, мочу, ушную жидкость, лимфу, слюну, спинномозговую жидкость, лаваж, суспензию костного мозга, влагалищную жидкость, трансцервикальный лаваж, жидкость головного мозга, асцитическую жидкость, секреты дыхательных, кишечных и мочеполовых путей, амниотическую жидкость, молоко и лейкоциты крови.
[00225] В некоторых вариантах реализации образцы также могут быть получены из тканей, клеток или других полинуклеотидсодержащих источников, культивированных в искусственных условиях. Культивируемые образцы могут получать из источников, включая, без ограничений, различные культуры (например, ткани или клетки), содержащиеся в различных средах и условиях (например, pH, давление и температура), культуры (например, ткани или клетки), хранящиеся в течение различных периодов времени, культуры (например, ткани или клетки), обработанные с применением различных факторов или реагентов (например, потенциальных лекарственных препаратов или модуляторов), либо культуры, принадлежащие к различным типам тканей и/или клеток.
[00226] Способы выделения нуклеиновых кислот из биологических источников являются хорошо изученными и могут различаться в зависимости от свойств конкретного источника. Специалисты в данной области могут выполнять выделение нуклеиновых кислот из источника по мере необходимости в рамках способа, описанного в настоящем документе. В некоторых случаях фрагментация молекул нуклеиновой кислоты в образце нуклеиновой кислоты может являться предпочтительной. Фрагментацию могут выполнять в случайном порядке, а также в специфическом порядке, например, при расщеплении рестрикционной эндонуклеазой. Способы случайной фрагментации, выполняемой в случайном порядке, хорошо известны в рамках данной области и включают в себя, например, ограниченное расщепление дезоксирибонуклеазой, обработку щелочью и физическое гидродинамическое фрагментирование.
Подготовка библиотек для секвенирования
[00227] В разнообразных вариантах реализации секвенирование можно проводить на разнообразных платформах для секвенирования, требующих подготовки библиотеки для секвенирования. Подготовка, как правило, включает в себя фрагментирование ДНК (обработку ультразвуком, пульверизацию или гидродинамическое фрагментирование) с последующей репарацией ДНК и обработкой концов (тупого конца или нависающего конца А), а также лигирование адаптера, характерное для используемой платформы. В одном варианте реализации в рамках способов, описанных в настоящем документе, можно использовать технологии секвенирования следующего поколения (NGS), которые позволяют проводить секвенирование множества образцов по отдельности в виде геномных молекул (т. е. одноканальное секвенирование) или в виде объединенных образцов, содержащих индексированные геномные молекулы (например, многоканальное секвенирование) в рамках одного цикла секвенирования. В рамках данных способов могут генерировать до нескольких сотен миллионов прочтений последовательностей ДНК. В разнообразных вариантах реализации последовательности геномных нуклеиновых кислот и/или индексированных геномных нуклеиновых кислот могут определять, например, с использованием технологий секвенирования следующего поколения (NGS), описанных в настоящем документе. В разнообразных вариантах реализации анализ большого объема данных последовательности, полученных с использованием NGS, могут выполнять с применением одного или более процессоров, как описано в настоящем документе.
[00228] В разнообразных вариантах реализации применение таких технологий секвенирования не включает в себя подготовку библиотек для секвенирования.
[00229] Однако в определенных вариантах реализации предусмотренные в настоящем документе способы секвенирования включают в себя подготовку библиотек для секвенирования. В одном иллюстративном подходе подготовка библиотек для секвенирования включает в себя получение случайного набора модифицированных адаптером фрагментов ДНК (например, полинуклеотидов), готовых к секвенированию. Библиотеки для секвенирования полинуклеотидов можно получить из ДНК или РНК, включая эквиваленты, аналоги ДНК или кДНК, например ДНК или кДНК, которые являются комплементарными, или из копийной ДНК, полученной из матрицы РНК, под действием обратной транскриптазы. Полинуклеотиды могут образовываться, имея двухцепочечную форму (например, дцДНК, такая как фрагменты геномной ДНК, кДНК, продукты ПЦР-амплификации и т. п.) или в определенных вариантах реализации полинуклеотиды могут быть образованы, имея одноцепочечную форму (например, оцДНК, РНК и т.п.), и преобразованы в форму дцДНК. В качестве примера в определенных вариантах реализации одноцепочечные молекулы мРНК могут быть скопированы в двухцепочечные кДНК, пригодные для применения при подготовке библиотеки для секвенирования. Точная последовательность первичных полинуклеотидных молекул, в целом, не является материалом для применения в рамках способа подготовки библиотеки, и может быть как известной, так и неизвестной. В одном варианте реализации полинуклеотидные молекулы представляют собой молекулы ДНК. Более конкретно, в определенных вариантах реализации молекулы полинуклеотида представляют собой весь генетический комплемент организма или по существу весь генетический комплемент организма и представляют собой молекулы геномной ДНК (например, клеточной ДНК, свободно-клеточной ДНК (скДНК) и т.д.), которые, как правило, включают как интронную последовательность, так и экзонную последовательность (кодирующую последовательность), а также некодирующие регуляторные последовательности, такие как промоторные и энхансерные последовательности. В некоторых вариантах реализации первичные полинуклеотидные молекулы содержат молекулы геномной ДНК человека, например, молекулы скДНК, присутствующие в периферической крови беременного пациента.
[00230] Получение библиотек для секвенирования для некоторых платформ секвенирования NGS облегчается применением полинуклеотидов, содержащих определенный диапазон размеров фрагментов. Получение таких библиотек обычно включает фрагментацию больших полинуклеотидов (например, клеточной геномной ДНК) для получения полинуклеотидов в желаемом диапазоне размеров для определения экспансии повторов.
[00231] Длина фрагмента или вставки больше длины прочтения и, как правило, больше суммы длин двух прочтений.
[00232] В некоторых примерах вариантов реализации образец нуклеиновой кислоты (нуклеиновых кислот) получают в виде геномной ДНК, которую (которые) подвергают фрагментации на фрагменты, содержащие около 100, 200, 300, 400, 500 или более пар нуклеотидных оснований и к которым можно применять способы NGS. В некоторых вариантах реализации парные концевые прочтения получают из вставок длиной около 100-5 000 п.н.о. В некоторых вариантах реализации вставки имеют длину около 100-1 000 п.н.о. Иногда их реализуют как обычные парные концевые прочтения с короткими вставками. В некоторых вариантах реализации вставки имеют длину около 1 000-5 000 п.н.о. Иногда их реализуют в виде прочтений сопряженных пар с длинными вставками, как описано выше.
[00233] В некоторых вариантах реализации длинные вставки могут использоваться для оценки очень длинных расширенных последовательностей повторов. В некоторых вариантах реализации для получения прочтений, разделенных тысячами пар нуклеотидных оснований, могут применяться прочтения сопряженных пар. В данных вариантах реализации вставки или фрагменты находятся в диапазоне от сотен до тысяч пар нуклеотидных оснований с двумя адаптерами биотинового соединения на двух концах вставки. Затем адаптеры биотинового соединения соединяют два конца вставки с образованием круглой молекулы, которая затем дополнительно фрагментируется. Для секвенирования на платформе, выполненной с возможностью секвенирования более коротких фрагментов, выбрана субфрагмент, включающий в себя адаптеры биотинового соединения и два конца исходной вставки.
[00234] Фрагментация могут осуществлять любым из ряда способов, известных специалистам в данной области. Например, фрагментацию можно выполнять механическими средствами, включая, без ограничений, пульверизацию, обработку ультразвуком и гидродинамическое фрагментирование. Однако механическая фрагментация, как правило, расщепляет каркас ДНК по связям C-O, P-O и C-C с образованием гетерогенной смеси тупых и 3'- и 5'-нависающих концов с разорванными связями C-O, P-O и C-C (см., например, Alnemri and Liwack, J Biol. Chem 265:17323-17333 [1990]; Richards and Boyer, J Mol Biol 11:327-240 [1965]), которым может потребоваться восстановление, поскольку в них может отсутствовать необходимый 5’-фосфат для проведения последующих ферментативных реакций, например, для лигирования адаптеров секвенирования, необходимых для получения ДНК для секвенирования.
[00235] Напротив, скДНК, как правило, существует в виде фрагментов менее около 300 пар нуклеотидных оснований, и, следовательно, фрагментация, как правило, не требуется для генерирования библиотеки для секвенирования с использованием образцов скДНК.
[00236] Как правило, независимо от того, подвергались ли полинуклеотиды искусственной фрагментации (например, фрагментации в искусственных условиях), либо они существуют в виде фрагментов в естественных условиях, они конвертируются в ДНК с тупыми концами, содержащие 5’-фосфаты и 3’-гидроксил. В стандартных протоколах, например, протоколах секвенирования с использованием, в том числе, платформы Illumina, как описано в других разделах настоящего документа, приведены инструкции по восстановлению конца образца ДНК, очищению продуктов с восстановленными концами перед присоединением к концу dA и очищению продуктов, присоединенных к концу dA перед этапами подготовки библиотеки с применением лигирования адаптера.
[00237] Разнообразные варианты реализации способов подготовки библиотек для секвенирования, описанные в настоящем документе, устраняют необходимость в выполнении одной или более стадий, выполнение которых обычно требуется в рамках стандартных протоколов для получения модифицированного продукта ДНК, который может быть секвенирован NGS. Сокращенный способ (сокр. способ), 1-стадийный способ и 2-стадийный способ представляют собой примеры способов подготовки библиотеки для секвенирования, которые можно найти в патентной заявке 13/555 037, поданной 20 июля 2012 г., содержание которой полностью включено в настоящий документ путем ссылки.
5.12. Способы секвенирования
[00238] Как указано выше, полученные образцы (например, библиотеки для секвенирования) секвенируют в рамках методики определения вариации (вариаций) числа копий. Могутт применять любую из множества технологий секвенирования.
[00239] На рынке представлены устройства и технологии секвенирования, такие как платформа для секвенирования путем гибридизации, производимая компанией Affymetrix Inc. (г. Саннивейл, штат Калифорния, США), а также платформы для секвенирования путем синтеза производства компании 454 Life Sciences (г. Брэдфорд, штат Коннектикут, США), Illumina/Solexa (г. Сан-Диего, штат Калифорния, США) и Helicos Biosciences (г. Кембридж, штат Массачусетс, США), и платформа для секвенирования путем лигирования производства компании Applied Biosystems (г. Фостер-Сити, штат Калифорния, США), как описано ниже. В дополнение к одномолекулярному секвенированию, которое выполняют с использованием секвенирования путем синтеза методом Helicos Biosciences, другие технологии одномолекулярного секвенирования включают, без ограничений, технологию SMRT™ компании Pacific Biosciences, технологию ION TORRENTTM и метод секвенирования через нанопоры, разработанный, например, компанией Oxford Nanopore Technologies.
[00240] Хотя автоматический метод Сэнгера считается технологией «первого поколения», в рамках применения способов, описанных в настоящем документе, также можно использовать секвенирование Сэнгера, включая автоматическое секвенирование Сэнгера. Дополнительные способы секвенирования включают, без ограничений, технологии визуализации нуклеиновых кислот, например, атомно-силовую микроскопию (АСМ) или просвечивающую электронную микроскопию (ТЭМ). Иллюстративные примеры технологий секвенирования более подробно описаны ниже.
[00241] В некоторых вариантах реализации описанные способы включают получение информации о последовательности нуклеиновых кислот в исследуемом образце путем массового параллельного секвенирования миллионов фрагментов ДНК с использованием секвенирования путем синтеза по методу Illumina и обратимого химического анализа способа секвенирования на основе терминатора (например, как описано в публикации Bentley et al., Nature 6:53-59 [2009]). Матричная ДНК может представлять собой геномную ДНК, например, клеточную ДНК или скДНК. В некоторых вариантах реализации в качестве матрицы используют геномную ДНК из выделенных клеток, затем ее фрагментируют для образования длин нескольких сотен пар нуклеотидных оснований. В других вариантах реализации в качестве матрицы используется скДНК, и выполнение фрагментации не требуется, поскольку скДНК существует в виде коротких фрагментов. Например, в кровотоке циркулирует скДНК плода в виде фрагментов длиной около в 170 пар нуклеотидных оснований (п.н.о.) (Fan et al., Clin Chem 56:1279-1286 [2010]), и фрагментация ДНК перед выполнением секвенирования не требуется. Технология секвенирования Illumina основана на закреплении фрагментированной геномной ДНК на прозрачной плоской поверхности, на которой находятся олигонуклеотидные якори. Матричную ДНК восстанавливают на конце для получения 5’-фосфорилированных тупых концов, а полимеразное действие фрагмента Кленова используют для добавления одного основания к 3’ концу тупых фосфорилированных фрагментов ДНК. В рамках данного добавления происходит подготовка фрагментов ДНК к лигированию с применением олигонуклеотидных адаптеров, которые имеют нависающий конец одного основания Т на 3’ конце для повышения эффективности лигирования. Олигонуклеотиды адаптера являются комплементарными по отношению к базовым олигонуклеотидам проточной кюветы (не следует путать с базовыми/закрепленными прочтениями в анализе экспансии повторов). В условиях лимитирования и разбавления модифицированную адаптером одноцепочечную матричную ДНК добавляли в проточную кювету и иммобилизовали путем гибридизации с базовыми олигонуклеотидами. Выполняют удлинение и мостиковую амплификацию присоединенных фрагментов ДНК для создания проточной кюветы со сверхвысокой плотностью секвенирования и сотнями миллионов кластеров, каждый из которых содержит около 1 000 копий одной и той же матрицы. В одном варианте реализации выполняется амплификация случайным образом фрагментированной геномной ДНК с использованием ПЦР до того, как ее подвергают кластерной амплификации. В качестве альтернативы используют способ подготовки неамплифицированной геномной библиотеки, а случайным образом фрагментированную геномную ДНК обогащают только с применением кластерной амплификации (Kozarewa et al., Nature Methods 6:291-295 [2009]). Матрицы секвенируют с использованием технологии глубокого четырехцветного секвенирования ДНК путем синтеза, в рамках которой используются обратимые терминаторы с удаляемыми флуоресцентными красителями. Высокочувствительное флуоресцентное детектирование выполняют при помощи возбуждения лазера и использования полного набора внутренних оптических отражающих элементов. Прочтение коротких последовательностей от около десятков до нескольких сотен пар нуклеотидных оснований выравнивают с эталонным геномом, и уникальное сопоставление прочтений коротких последовательностей с эталонным геномом определяют с помощью специально разработанного программного обеспечения для анализа данных. После завершения первого прочтения матрицы можно восстановить непосредственно на месте для обеспечения выполнения второго прочтения с противоположного конца фрагментов. Таким образом, можно использовать одиночное, либо парное концевое секвенирование фрагментов ДНК.
[00242] В разнообразных вариантах реализации настоящего описания можно использовать секвенирование путем синтеза, обеспечивающего парное концевое секвенирование. В некоторых вариантах реализации платформа для секвенирования путем синтеза производства компании Illumina имеет функцию кластеризации фрагментов. Кластеризация представляет собой процесс, в рамках которого каждая молекула фрагмента изотермически амплифицирована. В некоторых вариантах реализации в качестве примера, описанного в настоящем документе, фрагмент имеет два разных адаптера, присоединенных к двум концам фрагмента; такие адаптеры позволяют фрагменту гибридизироваться с двумя разными олигонуклеотидами на поверхности полосы проточной кюветы. Фрагмент дополнительно включает в себя две индексные последовательности на двух концах фрагмента или имеет связь с ними, причем индексные последовательности позволяют использовать метки для определения различных образцов при многоканальном секвенировании. В рамках работы с некоторыми платформами для секвенирования фрагмент, подлежащий секвенированию, также называют вставкой.
[00243] В некоторых вариантах реализации проточная кювета для кластеризации, использующаяся на платформе Illumina, представляет собой предметное стекло с полосами. Каждая полоса представляет собой стеклянный канал, покрытый олигонуклеотидами двух типов. Гибридизацию выполняют с применением первого из двух типов олигонуклеотидов, находящихся на поверхности. Данный олигонуклеотид является комплементарным по отношению к первому адаптеру на одном конце фрагмента. Полимераза создает комплементарную цепь гибридизированного фрагмента. Двухцепочечную молекулу денатурируют, а исходную матричную цепь вымывают. Оставшаяся цепь клонально амплифицируется посредством применения мостиков параллельно с остальными цепями.
[00244] При выполнении мостиковой амплификации вторая область адаптера на втором конце цепи гибридизируется с олигонуклеотидами второго типа на поверхности проточной кюветы. Полимераза создает комплементарную цепь, образуя двухцепочечную мостиковую молекулу. Данную двухцепочечную молекулу денатурируют, в результате чего образуются две одноцепочечные молекулы, присоединенные к проточной кювете двумя разными олигонуклеотидами. Затем процесс повторяет некоторое количество раз; данный процесс проводят одновременно с участием миллионов кластеров, что приводит к клональной амплификации всех фрагментов. После выполнения мостиковой амплификации обратные цепи отщепляют и вымывают, таким образом остаются только прямые цепи. 3’ концы блокируют для предотвращения нежелательного примирования.
[00245] После кластеризации процесс секвенирования начинают с удлинения первого праймера секвенирования, в результате чего получают первое прочтение. В рамках каждого цикла флуоресцентно-меченые нуклеотиды стремятся к добавлению в растущую цепь. На основе последовательности матрицы встраивается только один из них. После добавления каждого нуклеотида кластер возбуждают источником света, и излучается характерный флуоресцентный сигнал. Число циклов определяет длину прочтения. Длина волны излучения и интенсивность сигнала определяют процесс распознавания оснований. Для данного кластера считывание всех идентичных цепей выполняется одновременно. Сотни миллионов кластеров секвенируют путем массового параллельного секвенирования. После завершения первого прочтения продукт прочтения вымывают.
[00246] На следующей стадии работы в соответствии с протоколами с применением двух индексных праймеров, индексный праймер 1 вводят в индексную область 1 матрицы и гибридизируют с ней. Индексные области позволяют выполнять определение фрагментов, которые можно использовать для демультиплексирования образцов в рамках процесса многоканального секвенирования. Прочтение индекса 1 генерируют аналогично первому прочтению. После завершения прочтения индекса 1 продукт прочтения вымывают, а также снимают защиту 3’ конца цепи. Затем матричная цепь складывается и связывается со вторым олигонуклеотидом на проточной кювете. Последовательность индекса 2 считывают таким же образом, как и индекс 1. Затем по завершении стадии продукт прочтения индекса 2 вымывают.
[00247] После прочтения двух показателей прочтение 2 начинается с использования полимераз для выполнения экспансии олигонуклеотидов второй проточной кюветы, в результате чего образуется двухцепочечный мостик. Данную двухцепочечную ДНК денатурируют, а 3’ конец блокируют. Исходную прямая цепь отщепляют и вымывают, таким образом остается только обратная цепь. Прочтение 2 начинают с введения праймера секвенирования прочтения 2. Как и в случае с прочтением 1, выполнение стадий секвенирования повторяют до момента достижения нужной длины. Продукт прочтения 2 вымывают. В рамках данного процесса можно получить миллионы прочтений, представляющих все фрагменты. Последовательности из объединенных библиотек образцов разделяют на основании уникальных индексов, присвоенных во время подготовки образцов. С участием каждого образца проводят локальную кластеризацию прочтений аналогичных отрезков результатов распознавания оснований. Прочтения в прямом и обратном направлениях спаривают, в результате чего создают связные последовательности. Данные связные последовательности выравнивают с эталонным геномом для определения вариантов.
[00248] Пример секвенирования путем синтеза включает в себя использование парных концевых прочтений, которые используют во многих вариантах реализации описанных способов. При парном концевом секвенировании производят 2 прочтения с двух концов фрагмента. Для устранения неоднозначности при выравниваниях используют парные концевые прочтения. При выполнении парного концевого секвенирования пользователи могут выбирать длину вставки (или фрагмента, подлежащего секвенированию) и секвенировать оба конца вставки, что приводит к генерации высококачественных выравниваемых данных последовательности. Поскольку известно расстояние между всеми парными прочтениями, алгоритмы выравнивания могут использовать данную информацию для более точного сопоставления повторяющихся областей. Это позволяет добиться лучшего выравнивания прочтений, особенно в трудно поддающихся секвенированию повторяющихся областях генома. С применением парного концевого секвенирования можно выполнять обнаружение перестроек, в том числе вставок, делеций и инверсий.
[00249] Для парных концевых прочтений можно использовать вставку разной длины (т.е. разный размер фрагмента для секвенирования). В качестве исходного значения в настоящем описании для обозначения прочтений, полученных с различных длин вставок, используются парные концевые прочтения. В некоторых случаях для отличия парных концевых прочтений с короткими вставками от парных концевых прочтений с длинными вставками, последние, в частности, называются прочтениями сопряженных пар. В некоторых вариантах реализации, включающих прочтения сопряженных пар, два адаптера биотинового соединения сначала прикрепляют к двум концам относительно длинной вставки (например, длиной в несколько т.п.н.). Затем адаптеры биотинового соединения связывают два конца вставки с образованием круглой молекулы. Субфрагмент, объединяющий в себе адаптеры биотинового соединения, можно получить путем дополнительного фрагментирования круглой молекулы. Затем субфрагмент, включающий в себя два конца исходного фрагмента в противоположном порядке последовательности, можно секвенировать с применением той же процедуры, что и отн. описанного выше парного концевого секвенирования с короткой вставкой. Дополнительная информация о выполнении секвенирования сопряженных пар с использованием платформы Illumina представлена в онлайн-публикации по следующему адресу: res.illumina.com/documents/products/technotes/technote_nextera_matepair_data_processing.pdf. Данная публикация также полностью включена в настоящий документ путем ссылки.
[00250] После секвенирования фрагментов ДНК прочтения последовательностей заданной длины, например, 100 п.н.о., сопоставляют или выравнивают с известным эталонным геномом. Сопоставленные или выровненные прочтения и их соответствующие местоположения на эталонной последовательности также называются тегами. В анализах многих вариантов реализации, описанных в настоящем документе, для определения экспансии повторов используются прочтения, которые являются либо плохо выровненными, либо не могут быть выровнены в принципе, а также используются выровненные прочтения (метки). В одном варианте реализации эталонная геномная последовательность представляет собой последовательность NCBI36/hg18, данные которые можно найти в сети Интернет по следующему адресу: genome.ucsc.edu/cgi- bin/hgGateway?org=Human&db=hg18&hgsid=166260105). В альтернативном варианте реализации эталонная геномная последовательность представляет собой последовательность GRCh37/hg19, данные которые можно найти в сети Интернет по следующему адресу: genome.ucsc.edu/cgi-bin/hgGateway. К другим источникам общедоступной информации о последовательностях относятся база генетических данных, dbEST, dbSTS, EMBL (Европейская лаборатория по молекулярной биологии) и DDBJ (Банк данных ДНК Японии). Выравнивание последовательностей можно выполнять с применением ряда компьютерных алгоритмов, включая, без ограничений, BLAST (Altschul et al., 1990), BLITZ (MPsrch) (Sturrock & Collins, 1993), FASTA (Person & Lipman, 1988), BOWTIE (Langmead et al., Genome Biology 10:R25.1- R25.10 [2009]) или ELAND (Illumina, Inc., г. Сан-Диего, штат Калифорния, США). В одном варианте реализации один конец клонально расширенных копий молекул скДНК плазмы секвенируют и обрабатывают путем выполнения биоинформационного анализа выравниваний для анализатора генома Illumina, в котором применяется ПО Efficient Large-Scale Alignment of Nucleotide Databases (ELAND).
[00251] В одном иллюстративном варианте реализации, не имеющем ограничительного характера, способы, описанные в настоящем документе, включают получение информации о последовательности нуклеиновых кислот в исследуемом образце с использованием технологии одномолекулярного секвенирования Helicos True Single Molecule Sequencing (tSMS) (например, описанной в публикации Harris T.D. et al., Science 320:106-109 [2008]). Согласно методике tSMS образец ДНК расщепляют на цепи длиной около от 100 до 200 нуклеотидов, и добавляют последовательность polyA к 3’ концу каждой цепи ДНК. Каждую цепь помечают путем добавления флуоресцентно-меченого аденозина. Затем цепи ДНК гибридизируют с проточной кюветой, содержащей миллионы сайтов захвата T-нуклеотидов, иммобилизованных на поверхности проточной кюветы. В определенных вариантах реализации матрицы могут иметь плотность, равную около 100 миллионам матриц/см2. Проточную кювету загружают в прибор, например, секвенатор HeliScopeTM, а лазером освещают поверхность проточной кюветы, выявляя положение каждой матрицы. ПЗС-камеру могут использовать для сопоставления положения матриц на поверхности проточной кюветы. Затем флуоресцентную метку матрицы отщепляют и вымывают. Реакцию секвенирования начинают с введения ДНК-полимеразы и флуоресцентно-меченого нуклеотида. Т-олигонуклеотидная кислота служит в качестве праймера. Полимераза выполняет встраивание меченых нуклеотидов в праймер под управлением матрицы. Полимераза и невстроенные нуклеотиды удаляются. Матрицы, в которые направленно встроен флуоресцентно-меченый нуклеотид, распознают путем визуализации поверхности проточной кюветы. После выполнения визуализации флуоресцентную метку удаляют в рамках стадии расщепления, и процесс повторяют с участием других флуоресцентно-меченых нуклеотидов до момента достижения желаемой длины прочтения. На каждом этапе добавления нуклеотидов собирают информацию о последовательности. Секвенирование целого генома с помощью технологий одномолекулярного секвенирования устраняет необходимость ПЦР-амплификации при подготовке библиотек для секвенирования, также способы позволяют проводить прямое измерение образца вместо измерения копий этого образца.
[00252] В другом иллюстративном варианте реализации, не имеющем ограничительного характера, способы, описанные в настоящем документе, включают в себя получение информации о последовательности нуклеиновых кислот в исследуемом образце с помощью секвенирования 454 (Roche) (например, как описано в публикации Margulies, M. et al. Nature 437:376-380 [2005]). Секвенирование 454, как правило, включает в себя две стадии. На первой стадии ДНК подвергают гидродинамическому фрагментированию для разделения ее на фрагменты, имеющие около по 300-800 пар нуклеотидных оснований в каждом и тупые концы. Затем олигонуклеотидные адаптеры лигируют с концами фрагментов. Адаптеры служат в качестве праймеров для выполнения амплификации и секвенирования фрагментов. Фрагменты могут присоединять к микросферам для захвата ДНК, например, микросферам, покрытым стрептавидином, с использованием, например, адаптера B, который содержит 5‘-биотиновую метку. Фрагменты, присоединенные к микросферам, амплифицируют с помощью ПЦР в каплях масляно-водной эмульсии. В результате получают множество копий клонально амплифицированных фрагментов ДНК на каждой микросфере. На втором этапе микросферы захватывают лунками (например, пиколитровыми). Пиросеквенирование каждого фрагмента ДНК проводят параллельным способом. Добавлением одного или более нуклеотидов генерируют световой сигнал, который регистрируют ПЗС-камерой в аппарате для секвенирования. Сила сигнала является пропорциональной числу встроенных нуклеотидов. При выполнении пиросеквенирования используют пирофосфат (PPi), который высвобождается при добавлении нуклеотидов. PPi превращается в АТФ при помощи АТФ-сульфурилазы в присутствии аденозина 5’-фосфосульфата. Люцифераза взаимодействует с АТФ для преобразования люциферина в оксилюциферин, и в результате данной реакции генерируется световое излучение, которое затем измеряется и анализируется.
[00253] В другом иллюстративном варианте реализации, не имеющем ограничительного характера, способы, описанные в настоящем документе, включают в себя получение информации о последовательности нуклеиновых кислот в исследуемом образце с использованием технологии SOLiD™ (Applied Biosystems). При выполнении секвенирования путем лигирования в соответствии с технологией SOLiD™ геномную ДНК подвергают гидродинамическому фрагментированию для разделения ее на фрагменты, а адаптеры присоединяются к 5’ и 3’ концам фрагментов для генерирования библиотеки фрагментов. В альтернативном варианте реализации внутренние адаптеры могут вводить путем лигирования адаптеров с 5’ и 3’ концами фрагментов, округления фрагментов, расщепления круглого фрагмента для получения внутреннего адаптера и присоединения адаптеров к 5’ и 3’ концам полученных фрагментов для генерирования библиотеки сопряженных пар. Затем выполняется подготовка популяций клональных микросфер в микрореакторах, содержащих микросферы, праймеры, матрица и компоненты ПЦР. После ПЦР матрицы денатурируют и микросферы обогащают для разделения микросфер с удлиненными матрицами. Матрицы на выбранных микросферах подвергают модификации 3’, в результате которой устанавливают связи с предметным стеклом. Определение последовательности выполняют путем последовательной гибридизации и лигирования частично случайных олигонуклеотидов с центральным определенным основанием (или парой оснований), определяемым конкретным флуорофором. После регистрации цвета лигированный олигонуклеотид расщепляют и удаляют, затем процесс повторяют.
[00254] В другом иллюстративном варианте реализации, не имеющем ограничительного характера, способы, описанные в настоящем документе, включают в себя получение информации о последовательности нуклеиновых кислот в исследуемом образце с использованием технологии секвенирования в реальном времени (SMRT™) компании Pacific Biosciences. При секвенировании SMRT во время синтеза ДНК визуализируют непрерывное встраивание меченых красителем нуклеотидов. Одиночные молекулы ДНК-полимеразы прикрепляют к нижней поверхности отдельных детекторов длины волны с нулевой модой (детекторов ZMW), которые получают информацию о последовательности во время встраивания фосфосвязанных нуклеотидов в растущую праймерную цепь. Детектор ZMW содержит ограничивающую структуру, которая позволяет наблюдать встраивание одного нуклеотида с помощью ДНК-полимеразы на фоне флуоресцентных нуклеотидов, которые быстро диффундируют внутрь и за пределы ZMW (в течение микросекунд). Включение нуклеотида в растущую цепь, как правило, занимает несколько миллисекунд. В течение данного периода времени флуоресцентная метка возбуждается и продуцирует флуоресцентный сигнал, после чего флуоресцентная метка расщепляется. Измерение соответствующей флуоресценции красителя указывает на то, какое основание было встроено. Процесс выполняется повторно для получения последовательности.
[00255] В другом иллюстративном варианте реализации, не имеющем ограничительного характера, способы, описанные в настоящем документе, включают в себя получение информации о последовательности нуклеиновых кислот в исследуемом образце с помощью секвенирования через нанопоры (например, как описано в публикации Soni GV and Meller A. Clin Chem 53: 1996-2001 [2007]). Методики анализа ДНК путем секвенирования через нанопоры разработаны рядом компаний, включая, например, Oxford Nanopore Technologies (г. Оксфорд, Великобритания), Sequenom, NABsys и т.п. Секвенирование через нанопоры представляет собой технологию одномолекулярного секвенирования, при котором одиночную молекулу ДНК секвенируют непосредственно при прохождении через нанопору. Нанопора представляет собой небольшое отверстие диаметром, как правило, порядка 1 нанометра. Погружение нанопоры в электропроводящую текучую среду и подключение к ней источника потенциала (напряжения) приводит к появлению электрического тока небольшой силы вследствие проведения ионов через нанопоры. Сила протекающего тока зависит от размера и формы нанопоры. При прохождении молекулы ДНК через нанопору каждый нуклеотид молекулы ДНК перекрывает нанопору в разной степени, изменяя величину тока, проходящего через нанопору в разной степени. Таким образом, такое изменение тока при прохождении молекулы ДНК через нанопору позволяет получать прочтение последовательности ДНК.
[00256] В другом иллюстративном варианте реализации, не имеющем ограничительного характера, способы, описанные в настоящем документе, включают в себя получение информации о последовательности нуклеиновых кислот в исследуемом образце с использованием химически чувствительного транзистора с управляемым полем (chemFET) (например, как описано в опубликованной патентной заявке США № 2009/0026082). В одном примере реализации данной методики молекулы ДНК можно помещать в реакционные камеры, а матричные молекулы можно гибридизовать с праймером секвенирования, связанным с полимеразой. Встраивание одного или более трифосфатов в новую нуклеотидную цепочку на 3’ конце праймера секвенирования можно распознать с помощью chemFET по изменению тока. Массив может включать в себя множество датчиков chemFET. В другом примере отдельные нуклеиновые кислоты могут прикреплять к микросферам, также могут выполнять амплифицикацию нуклеиновых кислот на микросфере, и отдельные микросферы могут переносить в отдельные реакционные камеры на массиве chemFET, причем каждая из камер снабжена датчиком chemFET; кроме того, может проводиться секвенирование нуклеиновых кислот.
[00257] В другом варианте реализации технология секвенирования ДНК представляет собой одномолекулярное секвенирование с применением технологии Ion Torrent, которая объединяет полупроводниковую технологию с химическим анализом обычного способа секвенирования для прямого преобразования химически кодированной информации (A, C, G, T) в цифровую информацию (0, 1) с применением полупроводникового чипа. В природе, при встраивании полимеразой нуклеотида в цепь ДНК, побочный продукт в виде иона водорода высвобождается. В рамках технологии Ion Torrent используют массив микрообработанных лунок высокой плотности для выполнения данного биохимического процесса путем массового параллельного секвенирования. Каждая лунка содержит в себе отдельную молекулу ДНК. Под лунками находится чувствительный к ионам слой, а под ним находится ионный датчик. При добавлении нуклеотида, например C, в матрицу ДНК, и последующем встраивании в цепь ДНК высвобождается ион водорода. Заряд данного иона приведет к изменению pH-значения раствора, что, в свою очередь, регистрируют ионным датчиком Ion Torrent. Секвенатор, представляющий собой мельчайший в мире твердотельный измеритель значения pH, применяют для распознавания основания, конвертируя данные химического анализа в цифровые данные. Затем секвенатор Ion Personal Genome Machine (PGM™) последовательно заполняет чип нуклеотидами одним за другим. В случае несовпадения следующего нуклеотида, заполняющего чип. Изменения напряжения не будут регистрировать, и основания не будет распознавать. В случае, если на цепи ДНК имеются два идентичных основания, напряжение удваивается, и чип регистрирует два идентичных основания. Метод прямого обнаружения позволяет регистрировать включение нуклеотидов в течение нескольких секунд.
[00258] В другом варианте реализации настоящий способ включают в себя получение информации о последовательности нуклеиновых кислот в исследуемом образце с использованием секвенирования путем гибридизации. Секвенирование путем гибридизации содержит приведение множества полинуклеотидных последовательностей в контакт с множеством полинуклеотидных зондов, причем каждый из множества полинуклеотидных зондов может быть необязательно присоединен к субстрату. Субстрат может представлять собой плоскую поверхность, содержащую массив известных нуклеотидных последовательностей. Для определения полинуклеотидных последовательностей, присутствующих в образце, могут использовать паттерн для выполнения гибридизации с массивом. В других вариантах реализации каждый зонд прикрепляют к микросфере, например, к магнитной микросфере и т.п. Гибридизацию микросфер можно использовать для определения множества полинуклеотидных последовательностей в образце.
[00259] В некоторых вариантах реализации способов, описанных в настоящем документе, прочтения последовательностей имеют длину около в 20 п.н.о., около 25 п.н.о., около 30 bp, около 35 bp, около 40 bp, около 45 bp, около 50 п.н.о., около 55 п.н.о., около 60 п.н.о., около 65 п.н.о., около 70 п.н.о., около 75 п.н.о., около 80 п.н.о., около 85 п.н.о., около 90 п.н.о., около 95 п.н.о., около 100 п.н.о., около 110 п.н.о., около 120 п.н.о., около 130, около 140 п.н.о., около 150 п.н.о., около 200 п.н.о., около 250 п.н.о., около 300 п.н.о., около 350 п.н.о., около 400 п.н.о., около 450 п.н.о. или около 500 п.н.о. Ожидается, что при создании парных концевых прочтений технологические достижения обеспечат возможность одиночных концевых прочтений более 500 п.н.о. и прочтений более 1 000 п.н.о. В некоторых вариантах реализации для определения экспансии повторов используют парные концевые прочтения, которые содержат прочтения последовательностей длиной от около 20 п.н.о. до 1 000 п.н.о., от около 50 п.н.о. до 500 п.н.о. или от 80 п.н.о. до 150 п.н.о. В разнообразных вариантах реализации для оценки последовательности, имеющей экспансию повторов, применяют парные концевые прочтения. Последовательность, имеющая экспансию повторов, имеет большую длину, чем прочтения. В некоторых вариантах реализации последовательность, имеющая экспансию повторов, имеет большую длину, чем около 100 п.н.о., 500 п.н.о., 1 000 п.н.о. или 4 000 п.н.о. Сопоставление прочтений последовательностей осуществляется путем сравнения последовательности прочтений с последовательностью эталонного образца для определения хромосомного происхождения секвенированной молекулы нуклеиновой кислоты, и в таком случае специфическая информация о генетической последовательности не требуется. Допускается небольшая степень несоответствия (0-2 несовпадений на каждое прочтение) с учетом незначительных полиморфизмов, которые могут существовать между эталонным геномом и геномами в смешанном образце. В некоторых вариантах реализации прочтения, которые выровнены с эталонной последовательностью, используют в качестве базовых прочтений, а те прочтения, которые являются спаренными с базовыми прочтениями, но при этом не могут быть выровнены или являются плохо выровненными с эталонной последовательностью, используется в качестве закрепленных прочтений. В некоторых вариантах реализации плохо выровненные прочтения могут иметь относительно большое число несовпадений на каждое прочтение, например, по меньшей мере около 5%, 10%, 15% или 20% несовпадений на каждое прочтение.
[00260] Как правило, для каждого образца получают множество меток последовательности (т. е. Прочтений, выравненных с эталонной последовательностью). В некоторых вариантах реализации по меньшей мере около 3 × 106 меток последовательности, по меньшей мере около 5 × 106 меток последовательности, по меньшей мере около 8 × 106 меток последовательности, по меньшей мере около 10 × 106 меток последовательности, по меньшей мере около 15 × 106 меток последовательности, по меньшей мере около 20 × 106 меток последовательности, по меньшей мере около 30 × 106 меток последовательности, по меньшей мере около 40 × 106 меток последовательности или по меньшей мере около 50 × 106 меток последовательности, включающих, например, 100 п.н.о. Получают путем сопоставления прочтений с эталонным геномом относительно каждого образца. В некоторых вариантах реализации все прочтения последовательностей сопоставлены со всеми областями эталонного генома, обеспечивая возможность выполнения прочтений по всему геному. В других вариантах реализации прочтения сопоставляют с исследуемой последовательностью, например, хромосомой, сегментом хромосомы или исследуемой последовательностью повторов.
5.13. Устройство и системы для определения экспансии повторов
[00261] Анализ данных секвенирования и диагностики, полученных из них, как правило, выполняют с использованием различных компьютерных алгоритмов и программ. Таким образом, в некоторых вариантах реализации применяют процессы, включающие в себя использование данных, хранящихся или передаваемых посредством одной или более компьютерных систем или иных систем обработки. Варианты реализации, описанные в настоящем документе, также относятся к устройству, использующемуся для выполнения данных операций. Данное устройство может быть специально сконструировано для достижения требуемых целей, также таким устройством может быть компьютер общего назначения (или группа компьютеров), специально настраиваемый компьютерной программой и/или структурой данных, хранящейся в компьютере. В некоторых вариантах реализации группа процессоров отвечает за выполнение некоторых или всех указанных аналитических операций в совместном (например посредством сетевых или облачных вычислений) и/или параллельном порядке. Процессор или группа процессоров, использующиеся для реализации способов, описанных в настоящем документе, могут быть различных типов, включая микроконтроллеры и микропроцессоры, такие как программируемые устройства (например, СПЛИС и ППВМ), и непрограммируемые устройства, такие как специализированные ИС вентильной матрицы или микропроцессоры общего назначения.
[00262] В одном варианте реализации предлагается система определения генотипов вариантов в геномных локусах, включая последовательности повторов, также система включает в себя секвенатор для приема образцов нуклеиновой кислоты и предоставления информации о нуклеотидной последовательности таких образцов; процессор; и машиночитаемый носитель данных, содержащий инструкции, исполняемые на указанном процессоре и применяемые для генотипирования вариантов с применением следующих методов: (a) сбор прочтений последовательностей нуклеиновых кислот исследуемого образца из базы данных;(b) выравнивание прочтений последовательности с одной или более последовательностями повторов, каждая из которых представлена на графе последовательности, причем граф последовательности имеет структуру данных направленного графа, где вершины представляют нуклеотидные последовательности и направленные ребра, соединяющие вершины, и при этом граф последовательности содержит один или более собственных простых циклов, причем каждый собственный простой цикл представляет собой подпоследовательность повторов, причем каждая подпоследовательность повторов содержит повторы повторяющегося звена одного или более нуклеотидов; и (c) определение одного или более генотипов одной или более последовательностей повторов с использованием прочтений последовательности, выровненных с одной или более последовательностями повторов.
[00263] В некоторых вариантах реализации любой из систем, предложенных в настоящем документе, секвенатор настраивают для выполнения секвенирования следующего поколения (NGS). В некоторых вариантах реализации секвенатор настраивается для выполнения массового параллельного секвенирования с применением секвенирования путем синтеза с использованием терминаторов обратимых красителей. В других вариантах реализации секвенатор настраивается для выполнения секвенирования путем лигирования. В других вариантах реализации секвенатор настраивается для выполнения одномолекулярного секвенирования.
[00264] Кроме того, определенные варианты реализации относятся к материальным носителям и/или машиночитаемым носителям, предназначенным для долговременного хранения информации, или компьютерным программным продуктам, которые включают в себя программные команды и/или данные (включая структуры данных) для выполнения различных операций с помощью компьютера. Примеры машиночитаемых носителей включают в себя, без ограничений, полупроводниковые запоминающие устройства, магнитные носители, такие как дисковые накопители, магнитная лента, оптические носители, такие как диски, магнитооптические носители и аппаратные устройства, специально предусмотренные для хранения и выполнения программных команд, такие как постоянные запоминающие устройства (ПЗУ) и оперативные запоминающие устройства (ОЗУ). Машиночитаемые носители могут непосредственно или опосредованно управляться конечным пользователем. Примеры носителей с непосредственным управлением включают в себя носители, расположенные на объекте пользователя, и/или носители, данные с которых не являются общедоступными. Примеры носителей с опосредованным управлением включают в себя носители, опосредованно доступные пользователю через внешнюю сеть и/или сервис общего пользования, например, «облачный сервис». Примеры программных команд включают в себя как машинный код, в том числе создаваемый компилятором, так и файлы, содержащие код более высокого уровня, который может выполняться компьютером с использованием интерпретатора.
[00265] В разнообразных вариантах реализации данные, используемые в рамках описанных способов, а также используемые в устройстве, представлены в электронном формате. Такие данные могут включать в себя прочтения и метки, полученные из образца нуклеиновой кислоты, эталонные последовательности (включая эталонные последовательности, исключительно или преимущественно приводящие к образованию полиморфизмов), распознавания, такие как распознавания экспансии повторов, рекомендации, противопоказания, диагнозы и т.п. В соответствии с содержанием настоящего документа, данные в электронном формате могут хранить в устройстве и передавать между устройствами. Обычно данные в электронном формате предоставляют в цифровом виде и могут хранить в виде набора битов и/или байтов в различных структурах данных, списках, базах данных и т.д. Данные могут быть реализованы в электронном, оптическом и ином виде.
[00266] В одном варианте реализации предлагается компьютерный программный продукт для генерации выходных данных, указывающих на наличие или отсутствие экспансии повторов в исследуемом образце. Компьютерный продукт может содержать в себе инструкции по реализации любого одного или более из описанных выше способов определения экспансии повторов. Как объяснялось выше, компьютерный продукт может включать в себя материальный машиночитаемый носитель и/или носитель, предназначенный для долговременного хранения информации, содержащий записанный исполняемый или компилируемый программный код (например, инструкции), позволяющий процессору выполнять определение закрепленных прочтений и повторов в закрепленных прочтениях, а также наличие или отсутствие экспансии повторов. В одном примере компьютерный продукт содержит машиночитаемый носитель, содержащий записанный исполняемый или компилируемый программный код (например, инструкции), позволяющий процессору выполнять определение экспансии повторов и содержащий: процедуру получения данных секвенирования от по меньшей мере части молекул нуклеиновой кислоты, подвергнутых выравниванию с последовательностью повторов; машинный алгоритм для выполнения анализа экспансии повторов на основе указанных получаемых данных; и процедуру генерации выходных данных, указывающих на наличие, отсутствие и тип указанной экспансии повторов.
[00267] Информация о последовательности в рассматриваемом образце может быть сопоставлена с эталонными хромосомными последовательностями для определения парных концевых прочтений, выровненных с исследуемой последовательностью повторов или закрепленных на ней, а также для определения экспансии повторов последовательности повторов. В разнообразных вариантах реализации эталонные последовательности хранят в базе данных, такой как реляционная или объектно-ориентированная база данных.
[00268] Следует понимать, что в большинстве случаев выполнение вычислительных операций в рамках способов, описанных в настоящем документе, без посторонней помощи является непрактичным или даже невозможным. Например, для сопоставления одного прочтения из образца длиной в 30 п.н.о. с любой из хромосом человека может потребоваться несколько лет в случае, если не используется вычислительное устройство. Конечно, проблема усугубляется тем, что качественное распознавание экспансии повторов требует сопоставления тысяч (например, по меньшей мере около 10 000) или даже миллионов прочтений с одной хромосомой или более.
[00269] В разнообразных вариантах реализации необработанные прочтения последовательностей выравнивают с одним или более графами последовательностей, представляющими одну или более исследуемых последовательностей. В разнообразных вариантах реализации по меньшей мере 10 000, 100 000, 500 000, 1 000 000, 5 000 000 или 10 000 000 прочтений выровнены с одним или более графами последовательностей. В разнообразных вариантах реализации один или более графов последовательностей включают в себя по меньшей мере 1, 2, 5, 10, 50, 100, 500, 1 000, 5 000, 10 000 или 50 000 графов последовательностей.
[00270] В некоторых вариантах реализации необработанные прочтения последовательностей изначально выравнивают с эталонным геномом для определения геномных координат прочтений до того, как подмножество первоначально выровненных прочтений будет выровнено с одним или более графами последовательностей, представляющими одну или более исследуемых последовательностей. В разнообразных вариантах реализации по меньшей мере 10 000, 100 000, 500 000, 1 000 000, 5 000 000, 10 000 000 или 100 000 000 прочтений являются изначально выровненными с эталонным геномом. В некоторых вариантах реализации первоначально выровненные прочтения повторно выравнивают с графами последовательностей для определения экспансий повторов во множестве областей (каждая область соответствует графу последовательности). Общее число прочтений, которые повторно выравнивают с графами последовательностей во время каждого применения вариантов реализации, может находиться в диапазоне от тысяч до множества миллионов прочтений. В разнообразных вариантах реализации 10 000 000 прочтений повторно выравнивают с каждым графом последовательности. В разнообразных вариантах реализации один или более графов последовательностей включают в себя по меньшей мере 1, 2, 5, 10, 50, 100, 500, 1 000, 5 000, 10 000 или 50 000 графов последовательностей.
[00271] Способы, описанные в настоящем документе, могут реализовываться с применением системы для определения генотипов вариантов в геномном локусе, включающем в себя последовательность повторов. Система может включать в себя: (a) секвенатор для приема нуклеиновых кислот из исследуемого образца, предоставляющий информацию о нуклеотидной последовательности образца; (b) процессор; и (c) один или более машиночитаемых носителей данных, содержащий инструкции, исполняемые на указанном процессоре и применяемые для генотипирования вариантов в геномных локусах, включая последовательности повторов. В некоторых вариантах реализации инструкции по реализации способов могут получать с машиночитаемого носителя, на котором хранят машиночитаемые инструкции по выполнению определения экспансий повторов. Таким образом, в одном варианте реализации предлагается компьютерный программный продукт, содержащий машиночитаемый носитель, предназначенный для долговременного хранения информации, на котором хранится программный код, который при исполнении одним или более процессорами компьютерной системы приводит к реализации метода определенияикации экспансии повторов последовательности повторов в исследуемом образце, включающем нуклеиновые кислоты, где последовательность повторов включает повторы повторяющегося звена нуклеотидов. Программный код может включать в себя: (a) код для сбора прочтений последовательности исследуемого образца из базы данных; (b) код для выравнивания прочтений последовательности с одной или более последовательностями повторов, каждая из которых представлена на графе последовательности, причем граф последовательности имеет структуру данных направленного графа, где вершины представляют нуклеотидные последовательности и направленные ребра, соединяющие вершины, и при этом граф последовательности содержит один или более собственных простых циклов, причем каждый собственный простой цикл представляет собой подпоследовательность повторов, причем каждая подпоследовательность повторов содержит повторы повторяющегося звена одного или более нуклеотидов; и (c) код для определения одного или более генотипов одной или более последовательностей повторов с применением прочтений последовательности, выровненных с одной или более последовательностями повторов.
[00272] В некоторых вариантах реализации инструкции могут дополнительно включать в себя автоматическую регистрацию информации, относящейся к способу, например относящемуся к повторам и закрепленным прочтениям, а также к наличию или отсутствию указаний на наличие экспансии повторов в медицинской документации субъекта-человека, у которого отбирают исследуемый образец. Медицинскую документацию пациента могут вести и хранить, например, в лаборатории, кабинете врача, больнице, учреждении здравоохранения, страховой компании, либо метод использования личной медицинской карты может дополнительно включать в себя назначение, начало и/или изменение лечения пациента, у которого отбирается исследуемый образец. Данный процесс может включать в себя выполнение одного или более дополнительных тестов или анализов дополнительных образцов, отобранных у субъекта.
[00273] Описанные способы также могут выполнять с применением компьютерной системы обработки, специально адаптированной для выполнения определения экспансий повторов. В одном варианте реализации предлагается реализация компьютерной системы обработки, специально настроенной для осуществления последовательности действий в рамках способа, описанного в настоящем документе. В одном варианте реализации аппарат включает в себя устройство для секвенирования, специально настроенное с возможностью секвенирования по меньшей мере части молекул нуклеиновой кислоты в образце для получения информации о типе последовательности, описанной в других разделах настоящего документа. Аппарат может также включать в себя компоненты для обработки образца. Такие компоненты описаны в других разделах настоящего документа.
[00274] Последовательность или другие данные могут вводить в компьютер или сохранять на машиночитаемом носителе в прямой или косвенной манере. В одном варианте реализации компьютерная система непосредственно связана с устройством для секвенирования, которое выполняет считывание и/или анализ последовательностей нуклеиновых кислот, полученных из образцов. Получение последовательностей или других данных с применением таких устройств выполняется путем взаимодействия с интерфейсом компьютерной системы. В альтернативном варианте реализации последовательности, обрабатываемые системой, получают из места хранения последовательностей, такого как база данных или другое хранилище. После получения доступа к устройству обработки запоминающее устройство или запоминающее устройство большой емкости используется для буферизации или хранения (в т.ч. временного) последовательностей нуклеиновых кислот. Кроме того, в запоминающем устройстве может храниться число меток для различных хромосом или геномов и т.п. В запоминающем устройстве также могут храниться различные подпрограммы и/или программы для анализа представления последовательности или сопоставленных данных. Такие программы/подпрограммы могут включать в себя программы для выполнения статистического анализа и т.д.
[00275] В одном примере пользователь помещает образец в аппарат для секвенирования. Сбор и/или анализ данных осуществляется с помощью аппарата для секвенирования, соединенного с компьютером. Программное обеспечение, установленное на компьютере, позволяет собирать и/или анализировать данные. Данные могут хранить, отображать (с помощью монитора или иного аналогичного устройства) и/или отправлять в другие места. Компьютер может быть соединен с сетью Интернет, используемой для передачи данных на мобильное устройство, используемое удаленным пользователем (например, врачом, ученым или лаборантом). Следует понимать, что данные могут хранить и/или анализировать перед осуществлением такой передачи. В некоторых вариантах реализации необработанные данные собирают и удаленно отправляют пользователю или на устройство анализа и/или хранения данных. Передачу данных могут выполнять посредством их хранения на машиночитаемом носителе, причем такой носитель может быть отправлен конечному пользователю (например, по почте). Удаленный пользователь может находиться в том же или ином географическом местоположении, включая, без ограничений, здание, город, штат, страну или континент.
[00276] В некоторых вариантах реализации в рамках реализации способов также выполняют сбор данных множества полинуклеотидных последовательностей (например, прочтений, меток и/или эталонных хромосомных последовательностей) и отправку данных на компьютер или в другую вычислительную систему. Например, компьютер может быть подключен к лабораторному оборудованию, например, аппарату для сбора образцов, аппарату для амплификации нуклеотидов, аппарату для секвенирования нуклеотидов или аппарату для гибридизации. Затем компьютер может осуществлять сбор соответствующих данных, собранных с применением лабораторного оборудования. Данные могут хранить на компьютере на любом этапе, например, во время их сбора в режиме реального времени, перед их отправкой, во время отправки или после нее. Данные могут хранить на машиночитаемом носителе, который также может быть извлечен из компьютера. Собранные или сохраненные данные могут передавать с компьютера в удаленное местоположение, например через локальную сеть или глобальную сеть, такую как сеть Интернет. В удаленном местоположении с передаваемыми данными могут выполнять различные операции, как описано ниже.
[00277] Ниже описаны типы данных, отформатированных с применением электронных устройств, которые можно хранить, передавать, анализировать и/или обрабатывать в системах, устройстве и в рамках реализации способов, описанных в настоящем документе.
Прочтения, полученные путем секвенирования нуклеиновых кислот в исследуемом образце
Метки, полученные путем выравнивания прочтений с эталонным геномом или другой эталонной последовательностью или последовательностями
Эталонный геном или последовательность
Спецификация локуса, где указывается тип локуса, его местоположение и структура
Охват прочтений
Генотип вариантов
Граф последовательности
Пути графа
Информация о выравнивании графа
Фактические распознавания экспансии повторов
Диагнозы (клиническое состояние, связанное с соответствующими распознаваниями)
Рекомендации по выполнению дополнительных тестов, полученные на основе распознаваний и/или диагностики
Планы лечения и/или мониторинга, полученные на основе распознаваний и/или диагностики
[00278] Данные типы данных могут получать, сохранять, анализировать и/или обрабатывать в одном или более местах с помощью специализированных устройств. Насчитывается множество вариантов их обработки. В одном случае, вся или большую часть данной информации сохраняют и используют в месте обработки исследуемого образца, например, в кабинете врача или в иных клинических условиях. В ином случае, образец получают в одном местоположении, его обрабатывают и необязательно секвенируют в другом местоположении, прочтения выравнивают и выполняют распознавание в одном или более других местоположениях и подготавливают диагнозы, рекомендации и/или планы в еще одном местоположении (которое может представлять собой место отбора образца).
[00279] В разнообразных вариантах реализации прочтения генерируются с помощью аппарата для секвенирования, а затем передают на удаленный сайт, где обрабатывают для распознавания наличия экспансии повторов. Например, в данном удаленном местоположении прочтения выровнены с эталонной последовательностью для создания базовых и закрепленных прочтений. К операциям обработки, которые могут применять в различных местах, относятся следующие:
Сбор образцов
Обработка образцов перед секвенированием
Секвенирование
Анализ данных последовательности и распознавание наличия экспансии повторов
Диагностика
Передача информации о диагнозе и/или результате распознавания пациенту или медицинскому работнику разработка плана дальнейшего лечения, тестирования и/или мониторинга Реализация плана
Консультирование
[00280] Данные операции могут быть автоматизированы в соответствии с содержанием других разделов настоящего документа. Как правило, секвенирование, анализ данных последовательности и распознавание наличия экспансии повторов выполняются путем вычислений. Другие операции могут выполняться вручную или автоматически.
[00281] На Фиг. 6 проиллюстрирована одна реализация дисперсной системы для выполнения распознавания или диагностики на основании исследуемого образца. Место сбора образцов 01 используют для получения исследуемого образца у пациента. Затем образцы передают в место обработки и секвенирования 03, где исследуемые образцы обрабатывают и секвенируют, как описано выше. В местоположении 03 находится устройство для обработки образца, а также устройство для секвенирования обработанного образца. Результатом секвенирования, как описано в других разделах настоящего документа, является набор прочтений, которые, как правило, представлены в электронном формате и передают в сеть, такую как сеть Интернет; такой набор обозначен ссылочным номером 05 на Фиг. 6.
[00282] Данные последовательности передают в удаленное местоположение 07, в котором выполняют анализ и распознавание. В данном месте может находиться одно или более мощных вычислительных устройств, например, компьютеры или процессоры. После завершения анализа и выполнения распознавания на основе полученной информации о последовательности в местоположении 07, результаты данного распознавания возвращают в сеть 05. В некоторых вариантах реализации в местоположении 07 также генерируют не только результаты распознавания, но и информация о диагнозе. Затем результаты распознавания и/или диагностики передают по сети и возвращают в место сбора образцов 01, как изображено на Фиг. 6. Как объясняется выше, это лишь один из множества вариантов того, как различные операции, связанные с созданием результатов распознаваний или диагностики, могут разделяться между различными местоположениями. Один распространенный вариант реализации подразумевает выполнение сбора, обработки и секвенирования образцов в одном месте. Другой вариант включает в себя выполнение обработки и секвенирования в том же местоположении, где был выполнен анализ и создание результатов распознавания.
ЭКСПЕРИМЕНТАЛЬНЫЕ
Примеры
[00283] Программа была применена к смоделированному набору данных, содержащему широкий диапазон размеров повторов CAG и CCG в локусе HTT. HTT или хантингтин представляет собой ген заболевания, связанный с болезнью Хантингтона (БХ), нейродегенеративным расстройством, характеризующимся потерей нейронов полосатого тела. Считается, что это вызвано увеличенным нестабильным тринуклеотидным повтором в гене под названием хантингтин, который транслируется как полиглутаминовый повтор в продуцируемом белке. В нормальных контрольных образцах был определеницирован достаточно широкий диапазон тринуклеотидных повторов (9-35), а количества повторов, превышающие 40, были названы патологическими.
[00284] Как и ожидалось, точность, достигнутая с применением способов, соответствующих некоторым вариантам осуществления, была существенно выше в случаях, когда прочтения выравнивались с графом последовательности, содержащим оба повтора, по сравнению со случаями, когда оба повтора подвергались анализу независимо. Наблюдали аналогичное улучшение по сравнению с другим процессом генотипирования КТП (Фиг. 7).
[00285] Для демонстрации возможности вырождения генотипов повторов ДНК в рамках методов реализации мы проанализировали полиаланиновый повтор в гене PHOX2B в 150 контрольных образцах, отобранных у здоровых субъектов, и одного образца, в котором содержалась известная экспансия патогенного типа. PHOX2B содержит полиаланиновый повтор 20 кодонов, который может расширяться, что вызывает проявление врожденного центрального гиповентиляционного синдрома. В соответствии с известным описанием повтора (Amiel et al. 2003), все контрольные образцы, кроме нескольких, генотипировали в соотношении 20/20. В рамках вариантов реализации выполняли точное генотипирование единственного образца с экспансией в соотношении 20/27; Правильность данного генотипа была подтверждена в рамках секвенирования Сэнгера. Также в рамках вариантов реализации был правильно определен патогенный ОНВ, смежный с гомополимером аденозина в гене MSH2, в трех репликатах, полученных в результате полногеномного секвенирования, образца, полученного от компании SeraCare Life Sciences (см. раздел «Дополнительные материалы»).
[00286] Таким образом, мы разработали новый способ, удовлетворяющий потребность в более точном генотипировании сложных локусов. Данный способ позволяет выполнять генотипирование полиаланиновых повторов и разделение сложных областей, содержащих повторы в непосредственной близости от малых вариантов и других повторов. Ожидается, что гибкость используемой в настоящем документе структуры графа последовательности позволит реализовать множество новых способов распознавания вариантов.
Анализ КТП CAG и CCG в локусе HTT
[00287] Для каждой комбинации генотипов повторов CAG и CCG был смоделирован образец короткого прочтения с использованием WGSIM (Li, н/о). Мы установили значение длины прочтения на 150, значение расстояния между концами сопряжения - на 350, значение стандартного отклонения расстояния между концами сопряжения - на 50, частоту мутаций и частоту базовых ошибок - на 0,0010 и долю делеций - на 0. Число пар устанавливали таким образом, чтобы получить 40-кратный охват локуса. Прочтения были выровнены с эталоном GRCh37 с BWA-MEM 0.7.17-r1194-грязн. (Li 2013).
[00288] Мы проанализировали эти данные двумя способами с применением некоторых вариантов реализации. Во-первых, мы указали структуру локуса HTT с использованием экспрессии (CAG)*CAACAG(CCG)*, обеспечивая выравнивание прочтений с графом последовательности, содержащим оба повтора. Затем мы использовали некоторые варианты реализации для выполнения независимого анализа каждого повтора. В данном режиме прочтения были выровнены с графом, представляющим КТП CAG, также они были по отдельности выровнены с графом, представляющим КТП CCG. Наконец, мы проанализировали оба повтора независимо с использованием последних версий GangSTR и TredParse, в которых не используются графы последовательностей для выравнивания прочтений с областью повторов (Фиг. 7).
[00289] На Фиг. 7 проиллюстрирована точность генотипирования КТП CAG и CCG в локусе HTT на основе смоделированных данных. (a) Характеристики варианта реализации, в котором прочтения выровнены с графом последовательности, содержащим оба повтора; (b) характеристики варианта реализации, в котором повторы анализируют независимо друг от друга; (c) характеристики GangSTR; (d) характеристики TredParse. Мы измерили максимальное процентное отклонение прогнозируемых длин КТП от ожидаемых длин КТП каждого смоделированного образца. Пунктирными синими линиями указан генотип в эталонном геноме, а сплошными красными линиями - пороговое значение патогенных экспансий. В рамках данного варианта реализации точно предсказывают верные генотипы всех образцов при совместном генотипировании обоих образцов. Совместное генотипирование имеет большое преимущество перед раздельным генотипированием обоих повторов при помощи любого из трех инструментов, для которого характерна высокая относительная частота ошибок, особенно в случаях, когда один из повторов значительно длиннее другого.
Наборы данных
[00290] Данные полногеномного секвенирования без ПЦР, относящиеся к 150 неродственным контрольным образцам, использованные для анализа полиаланинового повтора PHOX2B, представляют собой группу Polaris Diversity. Данная группа состоит из образцов, выбранных из Международного ресурса по образцам генома (1000 Genomes Project Consortium et al. 2015) (www|.|internationalgenome|.|org/). Данные полногеномного секвенирования можно получить из Европейского архива генома-фенома (EGA; www|.|ebi|.|ac|.|uk/ega/home; PRJEB20654) и из архива прочтений последовательностей (АПП) Национального центра биотехнологической информации (АПП; www|.|ncbi|.|nlm|.|nih|.|gov/sra; bioproject:387148). Описание образцов (github|.|com/Illumina/Polaris/wiki/HiSeqX-Diversity-Cohort).
[00291] Образец, имеющий экспансию PHOX2B в соотношении 20/27, был предоставлен компанией Genetics Laboratories Molecular Genetics, медицинским исследовательским центром г. Адденбрук, штат Коннектикут, и Кембриджским университетом.
[00292] Образец под названием Seraseq Inherited Cancer DNA Mix v1, предоставленный компанией SeraCare Life Sciences, содержит определенно патогенные варианты, трудно поддающиеся распознаванию. Данные варианты были добавлены синтетическим методом в хорошо изученную клеточную линию GM24385; ожидаемые частоты таких вариантов составляют 50%. Одна из данный искусственных мутаций соответствует ОНВ в гене MSH2, который непосредственно примыкает к области длинного гомополимера А.
[00293] Подготовку трех репликатов образца SeraCare выполняли с использованием набора Illumina TruSeq PCR Free. Работу с репликатами проводили на одной полосе NovaSeq6000 с применением рабочего процесса XP, после чего проводилось секвенирование с длиной прочтения в 2 × 151. Репликаты анализировали с применением ПО Sentieon DNASeq FASTQ to VCF и ПО Whole Genome Resequencing v8.0.0 на облачной платформе Basespace (https://basespace.illumina.com). Sentieon можно рассматривать в качестве показателя эффективности распознавания вариантов с помощью программного пакета BWA-GATK производства Broad, в рамках которого реализуются такие же алгоритмы. Ни одна из единиц программного обеспечения не позволяла выполнить правильное определение ОНВ MSH2. Файлы Fastq и результаты анализа представлены в следующем разделе облачной платформы Basespace: https://basespace.illumina.com/s/HAQNxJyEtJLP
[00294] Содержание настоящего описания может быть реализовано в иных формах при условии сохранения его существенных характеристик. Описанные варианты реализации следует рассматривать только как иллюстративные варианты реализации, не имеющие ограничительного характера. Таким образом, область применения настоящего документа определяется соответствующими приложенными пунктами, а не приведенным выше описанием. Все изменения, соответствующие значению и диапазону эквивалентности пунктов, должны быть включены в область их применения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНСТРУМЕНТ НА ОСНОВЕ ГРАФОВ ПОСЛЕДОВАТЕЛЬНОСТЕЙ ДЛЯ ОПРЕДЕЛЕНИЯ ВАРИАЦИЙ В ОБЛАСТЯХ КОРОТКИХ ТАНДЕМНЫХ ПОВТОРОВ | 2020 |
|
RU2825664C2 |
| НАБОР ЗОНДОВ ДЛЯ АНАЛИЗА ОБРАЗЦОВ ДНК И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2016 |
|
RU2753883C2 |
| ПОДАВЛЕНИЕ ОШИБОК В СЕКВЕНИРОВАННЫХ ФРАГМЕНТАХ ДНК ПОСРЕДСТВОМ ПРИМЕНЕНИЯ ИЗБЫТОЧНЫХ ПРОЧТЕНИЙ С УНИКАЛЬНЫМИ МОЛЕКУЛЯРНЫМИ ИНДЕКСАМИ (UMI) | 2016 |
|
RU2704286C2 |
| ЖИВОТНЫЕ, ОТЛИЧНЫЕ ОТ ЧЕЛОВЕКА, ХАРАКТЕРИЗУЮЩИЕСЯ ЭКСПАНСИЕЙ ГЕКСАНУКЛЕОТИДНЫХ ПОВТОРОВ В ЛОКУСЕ C9ORF72 | 2017 |
|
RU2760877C2 |
| ФРЕЙМВОРК НА ОСНОВЕ ГЛУБОКОГО ОБУЧЕНИЯ ДЛЯ ИДЕНТИФИКАЦИИ ПАТТЕРНОВ ПОСЛЕДОВАТЕЛЬНОСТИ, КОТОРЫЕ ВЫЗЫВАЮТ ПОСЛЕДОВАТЕЛЬНОСТЬ-СПЕЦИФИЧНЫЕ ОШИБКИ (SSE) | 2019 |
|
RU2745733C1 |
| СПОСОБЫ И СИСТЕМЫ ДЛЯ ПОЛУЧЕНИЯ НАБОРОВ УНИКАЛЬНЫХ МОЛЕКУЛЯРНЫХ ИНДЕКСОВ С ГЕТЕРОГЕННОЙ ДЛИНОЙ МОЛЕКУЛ И КОРРЕКЦИИ В НИХ ОШИБОК | 2018 |
|
RU2766198C2 |
| БИОИНФОРМАЦИОННЫЕ СИСТЕМЫ,УСТРОЙСТВА И СПОСОБЫ ВЫПОЛНЕНИЯ ВТОРИЧНОЙ И/ИЛИ ТРЕТИЧНОЙ ОБРАБОТКИ | 2017 |
|
RU2750706C2 |
| БИОИНФОРМАЦИОННЫЕ СИСТЕМЫ, УСТРОЙСТВА И СПОСОБЫ ДЛЯ ВЫПОЛНЕНИЯ ВТОРИЧНОЙ И/ИЛИ ТРЕТИЧНОЙ ОБРАБОТКИ | 2017 |
|
RU2799750C2 |
| Способ обработки данных полногеномного секвенирования | 2023 |
|
RU2806429C1 |
| ОБНАРУЖЕНИЕ МУТАЦИЙ И ПЛОИДНОСТИ В ХРОМОСОМНЫХ СЕГМЕНТАХ | 2015 |
|
RU2717641C2 |
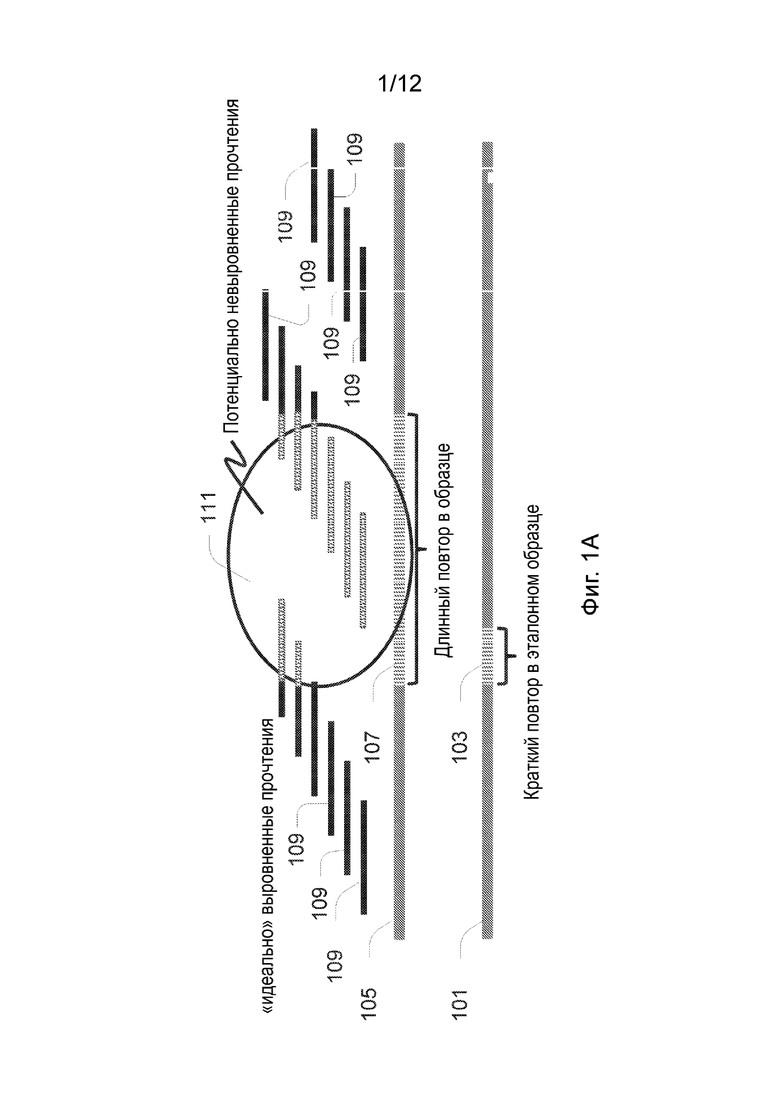
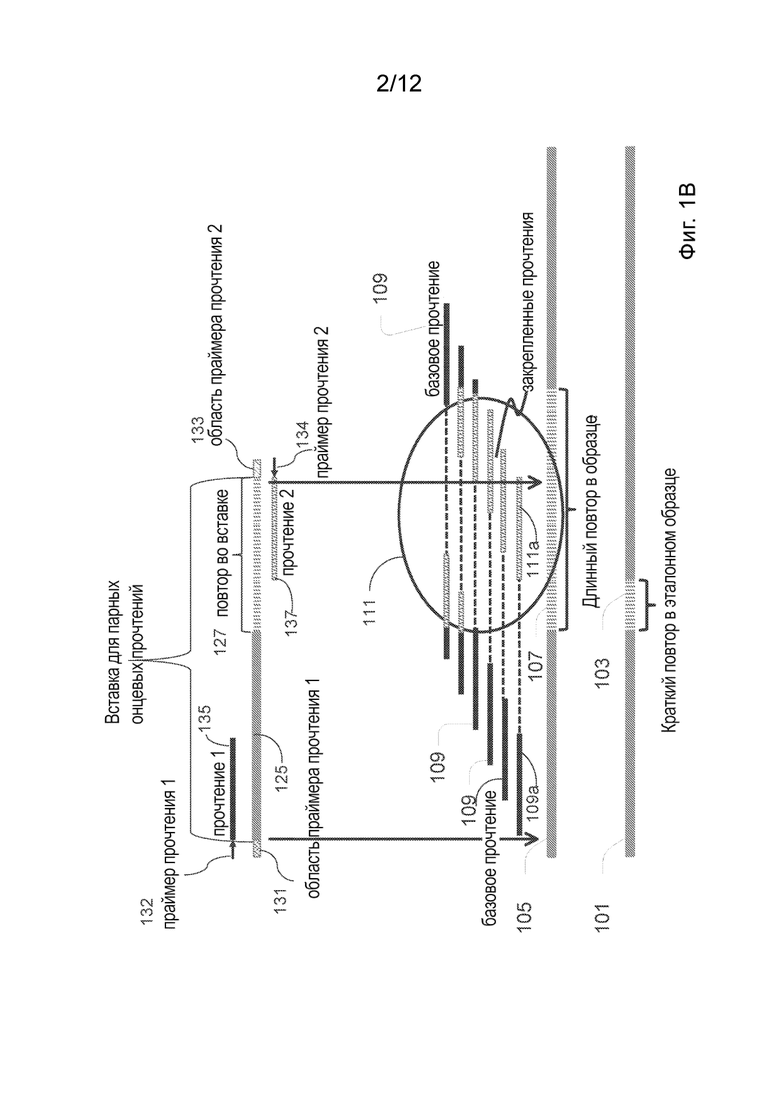
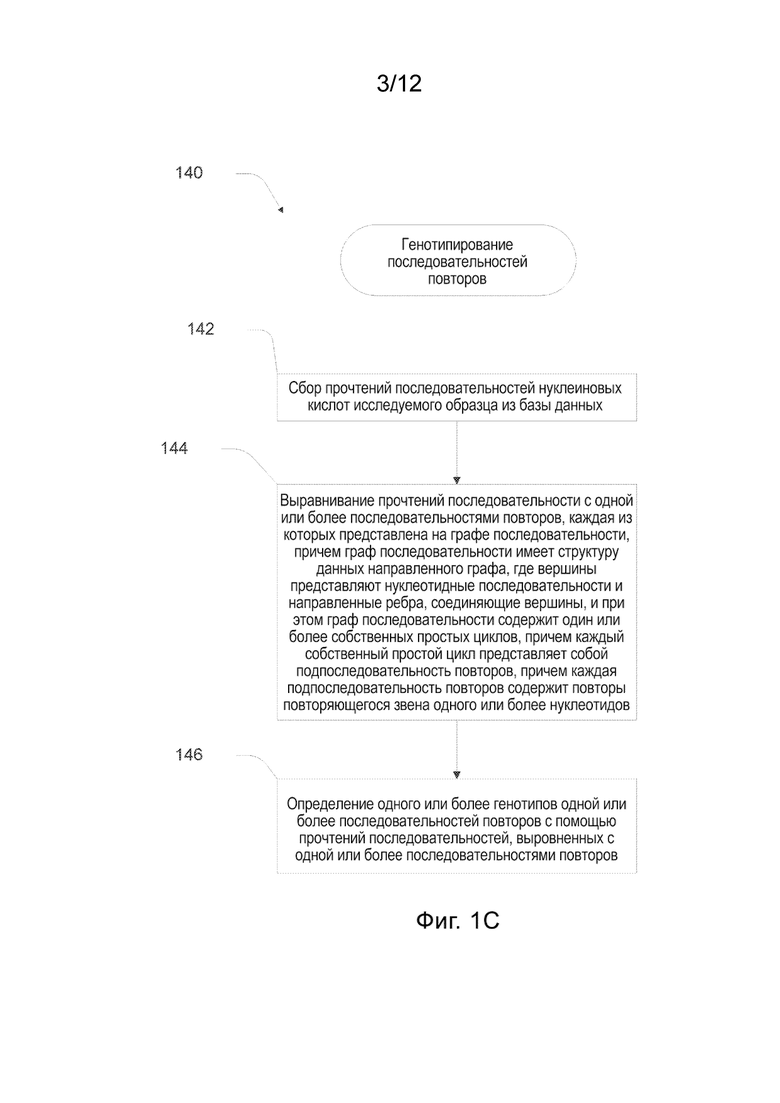
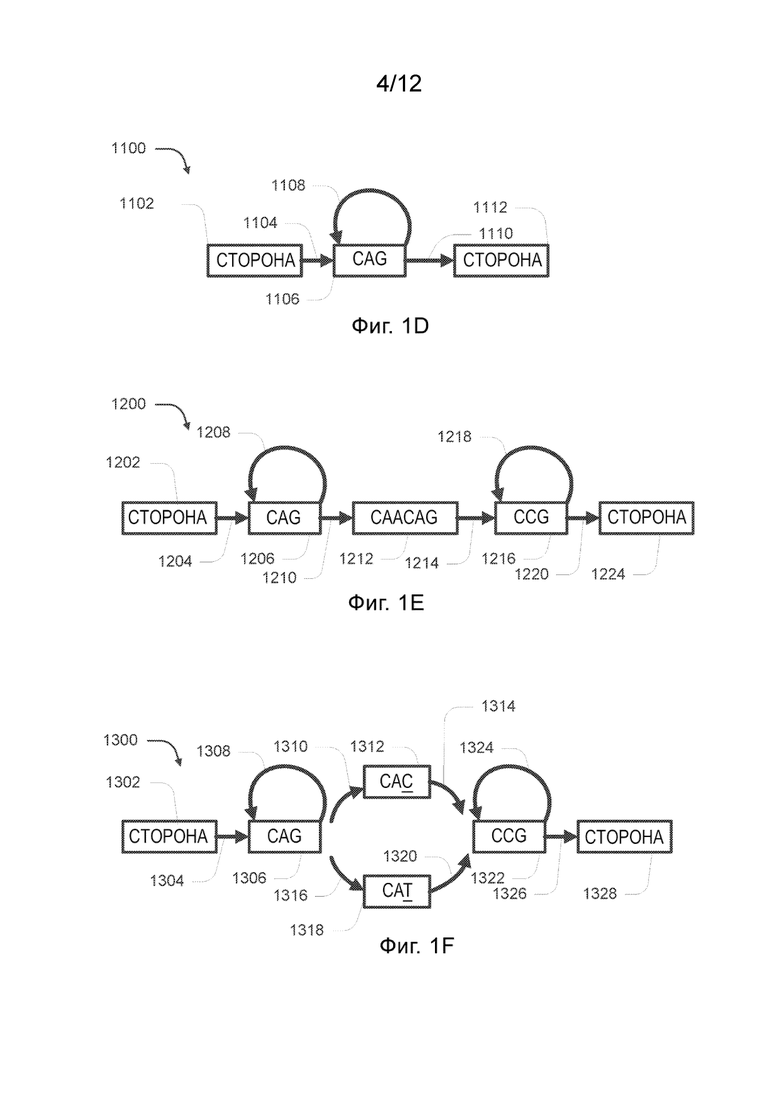
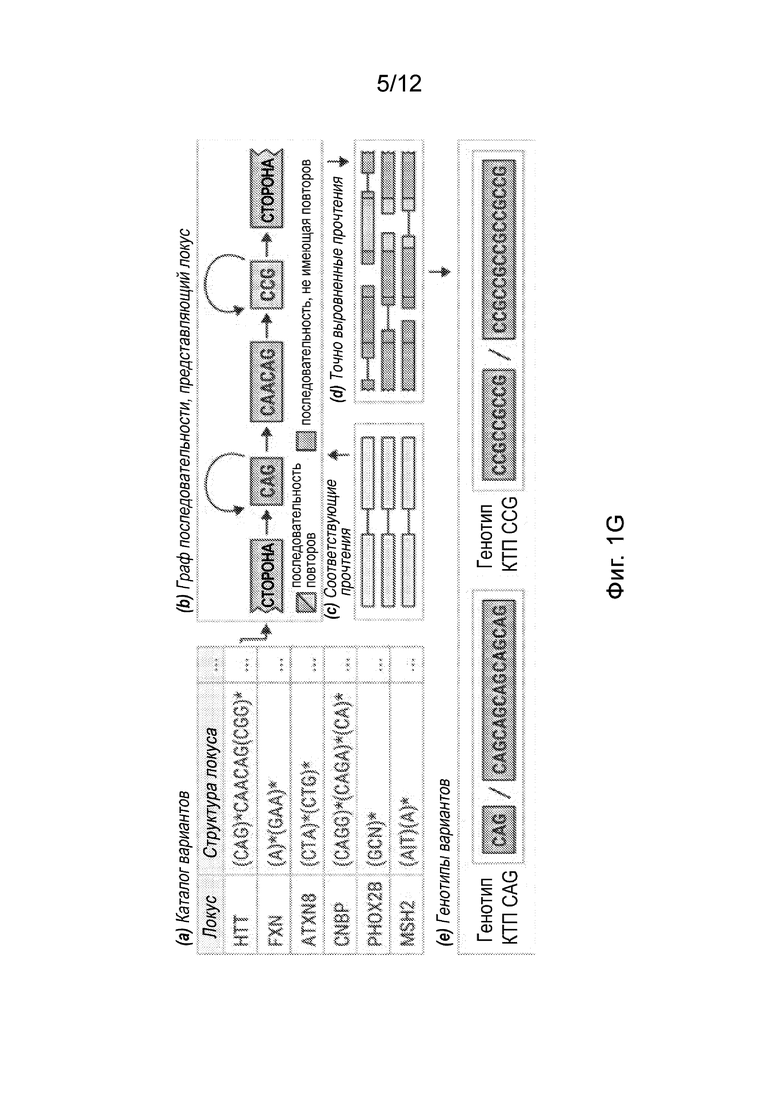


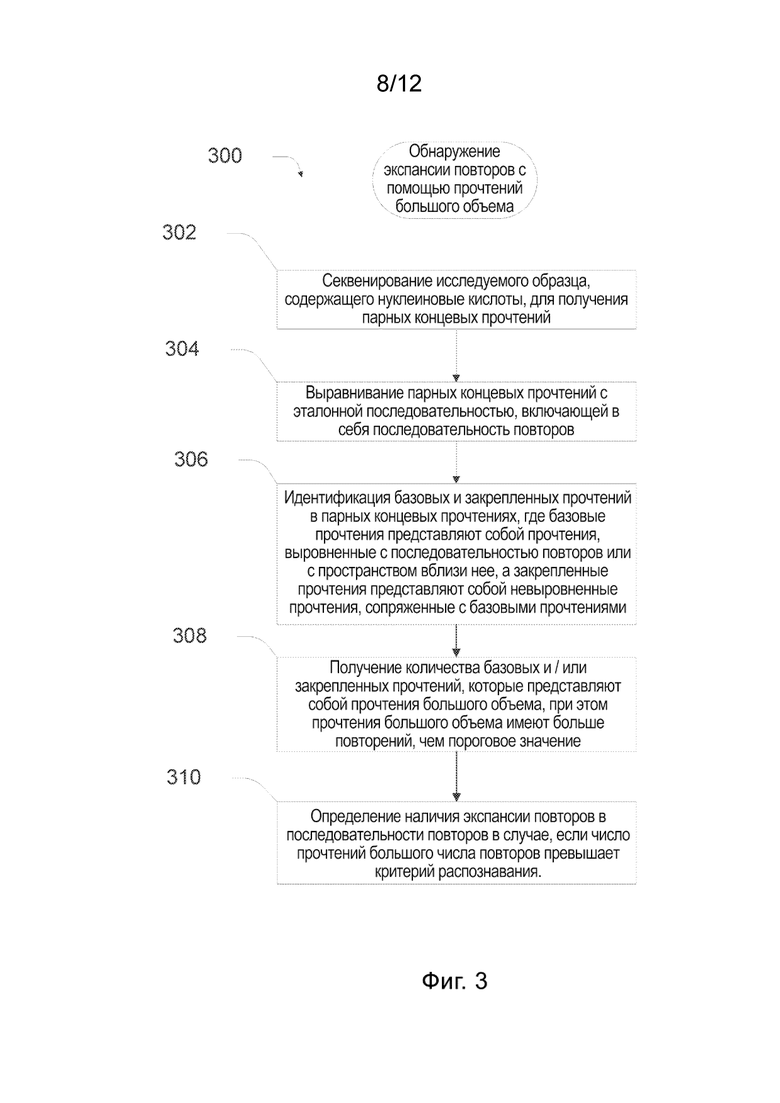
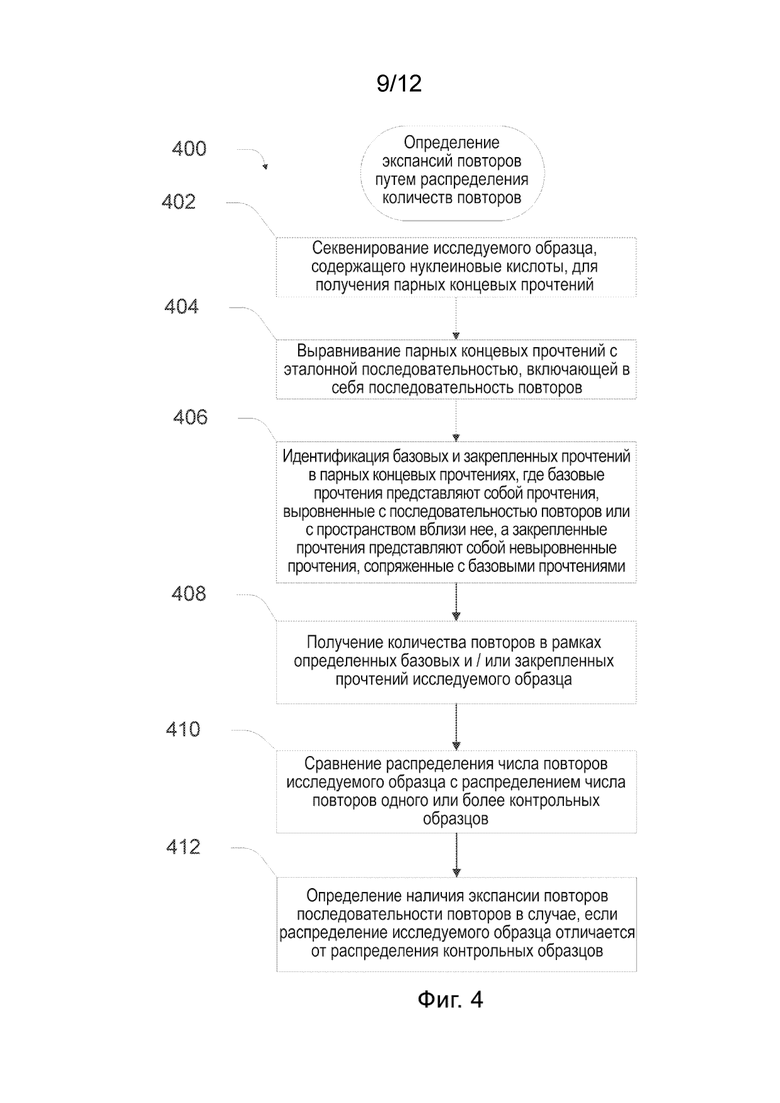
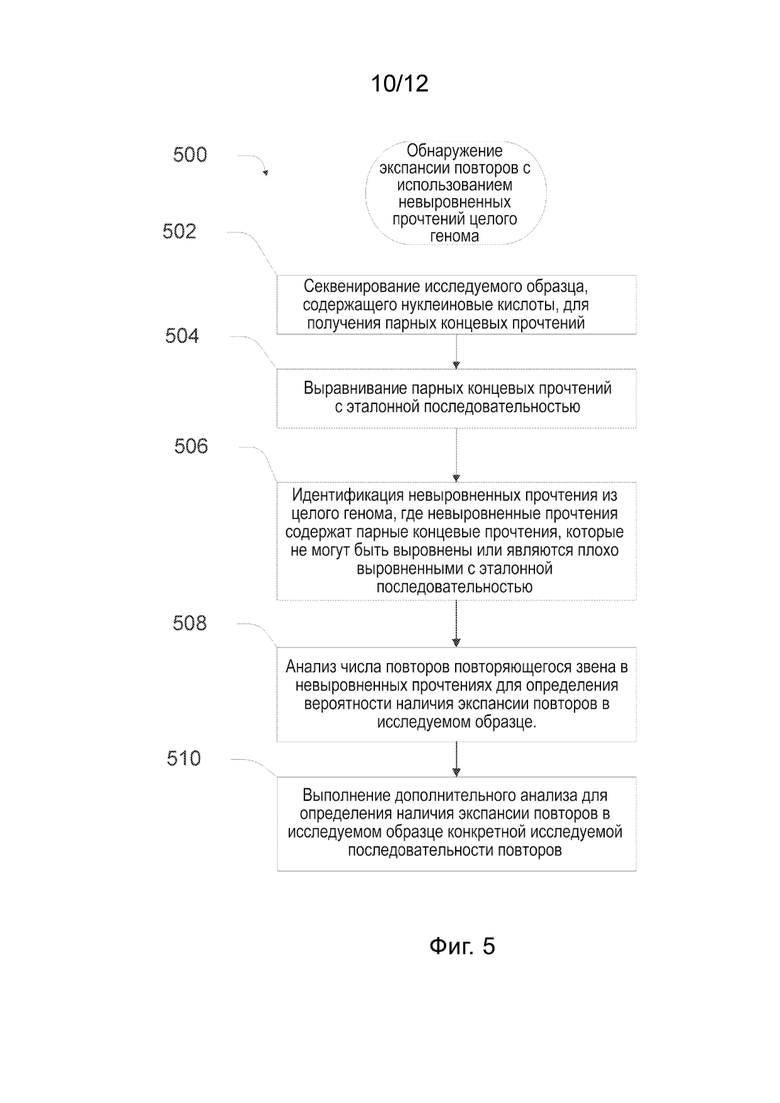
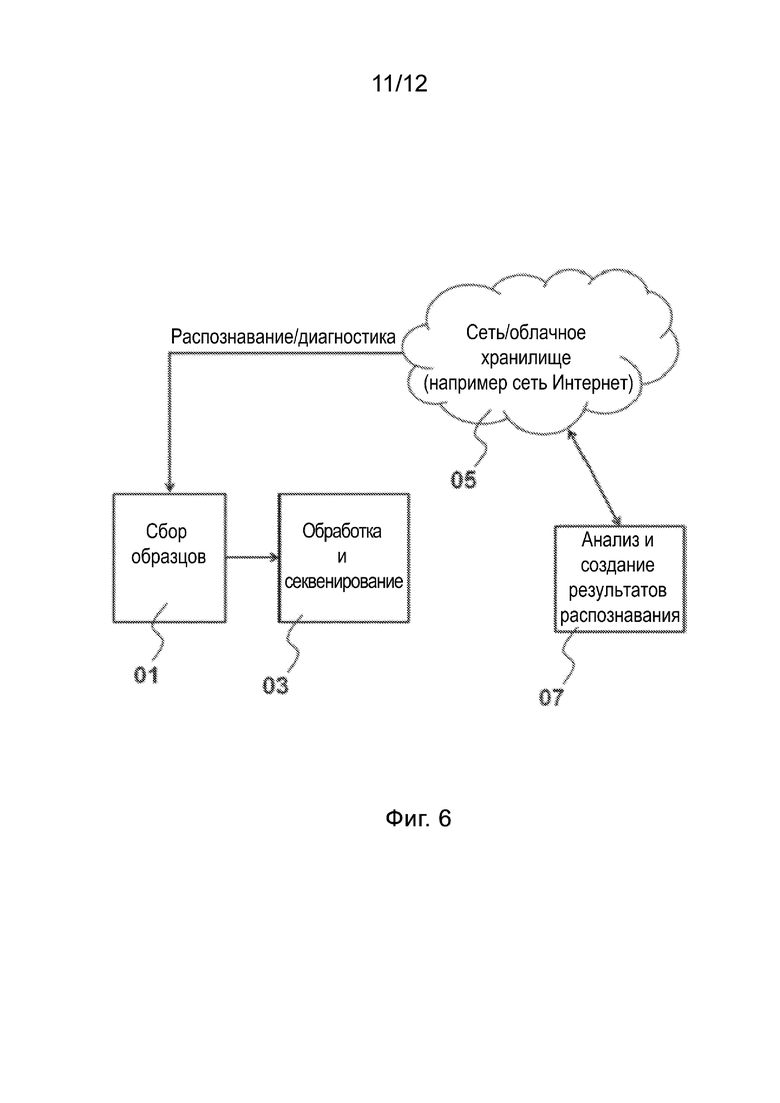

Изобретение относится к биоинформатике. Описанные варианты реализации относятся к способам, устройствам, системам, предназначенным для генотипирования последовательностей повторов, в том числе коротких тандемных повторов (CTR), которые являются значимыми с медицинской точки зрения. Способы включают выравнивание прочтений с последовательностью повторов, представленной графом последовательности, и использование выравненных прочтений для генотипирования последовательности повторов. Граф последовательности представляет собой направленный граф, который включает в себя по меньшей мере один собственный простой цикл, представляющий подпоследовательность повторов. В некоторых вариантах реализации прочтения представляют собой парные концевые прочтения, и для генотипирования последовательностей повторов можно использовать оба сопряженных прочтения каждой пары прочтений. Некоторые варианты реализации можно использовать для определения повторов вырожденных кодонов. Некоторые варианты реализации могут использоваться для генотипирования последовательностей повторов, каждая из которых включает в себя две или более подпоследовательностей повторов. Некоторые варианты реализации могут использоваться для генотипирования нуклеотидных последовательностей, каждая из которых включает по меньшей мере одну повторяющуюся подпоследовательность и другой генетический вариант, такой как вставка, делеция или замена. Изобретение расширяет арсенал средств для генотипирования. 2 н. и 29 з.п. ф-лы, 7 ил., 1 табл., 1 пр.
1. Способ, реализованный с применением компьютера, снабженного одним или более процессорами и системной памятью, для генотипирования одной или более последовательностей повторов, каждая из которых содержит одну или более подпоследовательностей повторов, включающий:
(a) генерирование и сбор прочтений последовательности исследуемого образца из базы данных с применением одного или более процессоров и секвенатора;
(b) выравнивание с помощью одного или более процессоров, прочтение одной или более последовательностей повторов, каждая из которых представлена графом последовательности, причем граф последовательности имеет структуру данных направленного графа, где вершины представляют нуклеотидные последовательности, а направленные ребра соединяют вершины, и при этом граф последовательности содержит один или более собственных простых циклов, причем каждый собственный простой цикл представляет собой подпоследовательность повторов, при этом каждая подпоследовательность повторов содержит повторы повторяющегося звена одного или более нуклеотидов;
(c) определение одним или более процессорами одного или более генотипов одной или более последовательностей повторов с использованием прочтений последовательности, выровненных с одной или более последовательностями повторов.
2. Способ по п. 1, в котором последовательность повторов из одной или более последовательностей повторов содержит конкретное повторяющееся звено, содержащее по меньшей мере один частично определенный нуклеотид.
3. Способ по п. 2, в котором конкретное повторяющееся звено содержит вырожденные кодоны.
4. Способ по любому из предшествующих пунктов, в котором один или более собственных простых циклов содержат два или более собственных простых циклов, представляющих две или более повторяющихся подпоследовательностей.
5. Способ по любому из предшествующих пунктов, в котором граф последовательности дополнительно содержит два или более альтернативных путей двух или более аллелей.
6. Способ по п. 5, в котором два или более аллеля содержат делецию или замену.
7. Способ по п. 5, в котором замена содержит однонуклеотидный вариант (ОНВ) или однонуклеотидный полиморфизм (ОНП).
8. Способ по п. 5, дополнительно включающий в себя генотипирование двух или более аллелей с применением прочтений последовательностей, выровненных с двумя или более альтернативными путями.
9. Способ по п. 8, в котором генотипирование двух или более аллелей включает в себя охват двух или более альтернативных путей к вероятностной модели для определения вероятностей двух или более аллелей.
10. Способ по п. 9, в котором вероятностная модель моделирует вероятность аллеля в зависимости от охвата аллеля, причем функция выбрана из распределения Пуассона, отрицательного биномиального распределения, биномиального распределения или бета-биномиального распределения.
11. Способ по п. 10, в котором параметр скорости распределения Пуассона оценивают по длине прочтения и средней глубине, наблюдаемой в геномном локусе.
12. Способ по любому из предшествующих пунктов, дополнительно включающий выравнивание до (b), прочтения последовательности по эталонному геному для определения геномных координат прочтения последовательности, а также выбор подмножества прочтения последовательности по мере выравнивания прочтения последовательности с одной или более последовательностями повторов, каждая из которых представлена графом последовательности.
13. Способ по п. 12, в котором подмножество прочтений последовательностей содержит прочтения, выровненные с областью, представленной графом последовательности, или с пространством вблизи нее.
14. Способ по п. 12, в котором подмножество прочтений последовательностей содержит невыровненные прочтения, сопряженные прочтения которых сопоставляются с областью, представленной графом последовательности, или с пространством вблизи нее.
15. Способ по п. 12, в котором подмножество прочтений последовательностей содержит прочтение последовательностей, выровненное с одной или более нецелевыми областями, которые являются известными горячими точками для прочтения неправильного выравнивания.
16. Способ по любому из предшествующих пунктов, в котором выравнивание прочтения последовательности с графом последовательности включает:
поиск соответствия кмер между прочтением последовательности и путем графа последовательности; и
расширение соответствия кмер до полного выравнивания узлов и ребер графа последовательности, включая один или более собственных простых циклов.
17. Способ по любому из предшествующих пунктов, в котором выравнивание прочтения последовательности с графом последовательности включает в себя сокращение графа путем удаления концов выравниваний с низким уровнем достоверности.
18. Способ по любому из предшествующих пунктов, в котором выравнивание прочтения последовательности с графом последовательности включает объединение выравниваний путем:
выравнивания подпоследовательностей прочтения с графом последовательности; и
объединения выравниваний подпоследовательностей для получения полного выравнивания прочтения последовательности.
19. Способ по любому из предшествующих пунктов, дополнительно включающий генерирование графа последовательности на основе спецификации локуса, содержащей структуру геномного локуса.
20. Способ по любому из предшествующих пунктов, в котором прочтения последовательностей содержат парные концевые прочтения, а операция (c) включает в себя:
(i) определение базовых и закрепленных прочтений в парных концевых прочтениях, причем базовые прочтения являются выровненными с одной или более последовательностями повторов или с пространством рядом с ними, а закрепленные прочтения представляют собой невыровненные прочтения, которые сопряжены с закрепленными прочтениями; и
(ii) определение одного или более генотипов для одной или более последовательностей повторов с применением по меньшей мере закрепленных прочтений.
21. Способ по п. 20, в котором операция (ii) включает в себя определение одного или более генотипов одной или более последовательностей повторов с применением базовых прочтений, а также закрепленных прочтений.
22. Способ по п. 20 или 21, в котором базовые прочтения выровнены с точностью до около 5 т. п. н. последовательности повторов.
23. Способ по любому из пп. 20-22, в котором невыровненные прочтения содержат прочтения, которые не могут быть выровнены или являются плохо выровненными с графом последовательности.
24. Способ по любому из предшествующих пунктов, в котором одна или более последовательности повторов содержат последовательность коротких тандемных повторов (КТП).
25. Способ по п. 24, в котором экспансия КТП связана с синдромом ломкой X-хромосомы, боковым амиотрофическим склерозом (АБС), болезнью Хантингтона, атаксией Фридрейха, спиномозжечковой атаксией, спинобульбарной мышечной атрофией, миотонической дистрофией, болезнью Мачадо-Джозефа или дентато-рубро-паллидо-льюисовой атрофией.
26. Способ по любому из предшествующих пунктов, дополнительно включающий применение секвенатора для генерирования парных концевых прочтений исследуемого образца.
27. Способ по любому из предшествующих пунктов, дополнительно включающий извлечение исследуемого образца из организма субъекта.
28. Способ по любому из предшествующих пунктов, в котором исследуемый образец представляет собой образец крови, образец мочи, образец слюны или образец ткани.
29. Способ по любому из предшествующих пунктов, в котором повторяющееся звено содержит от 1 до 50 нуклеотидов.
30. Способ по любому из предшествующих пунктов, в котором прочтения являются более короткими, чем по меньшей мере одна из последовательностей повторов.
31. Система для генотипирования одной или более последовательностей повторов, каждая из которых содержит одну или более подпоследовательностей повторов, содержащая:
системную память; и
один или более процессоров, выполненных с возможностью:
(a) генерирования с применением секвенатора и сбора прочтений последовательности исследуемого образца из базы данных;
(b) выравнивания прочтений последовательности с одной или более последовательностями повторов, каждая из которых представлена на графе последовательности, причем граф последовательности имеет структуру данных направленного графа, где вершины представляют нуклеотидные последовательности и направленные ребра, соединяющие вершины, и при этом граф последовательности содержит один или более собственных простых циклов, причем каждый собственный простой цикл представляет собой подпоследовательность повторов, причем каждая подпоследовательность повторов содержит повторы повторяющегося звена одного или более нуклеотидов;
(c) определения одного или более генотипов одной или более последовательностей повторов с помощью прочтений последовательностей, выровненных с одной или более последовательностями повторов.
| RU 2019100495 A, 31.01.2019 | |||
| WO 2017070096 A1, 27.04.2017 | |||
| RU 2016139287 A, 03.05.2018. |

