ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка претендует на приоритет согласно пункту 35 U.S.С. § 119(e) предварительной патентной заявки US 62/153699, поданной 28 апреля 2015 г., номер патентного реестра ILMNP008P, предварительной патентной заявки US 62/193469, поданной 16 июля 2015 г., номер патентного реестра ILMNP008P2, и предварительной патентной заявки US 62/269485, поданной 18 декабря 2015 г., номер патентного реестра ILMNP008P3, содержания которых полностью включены в настоящее описание посредством ссылки для всех целей.
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
Настоящая заявка содержит Перечень последовательностей, который представлен в электронном виде в формате ASCII и, таким образом, полностью включен в настоящее описание посредством ссылки. Указанная копия ASCII, созданная 20 апреля 2016 г., озаглавлена ILMNP008WO_ST25.txt, и ее размер составляет 1164 байт.
ОБЛАСТЬ ТЕХНИКИ
Методика секвенирования следующего поколения обеспечивает постоянно увеличивающуюся скорость секвенирования, которая позволяет достигать большей глубины секвенирования. Однако, поскольку на точность и чувствительность секвенирования влияют ошибки и шумы, имеющие различное происхождение, например, обусловленные дефектами образца, проведением ПЦР (полимеразной цепной реакции) в процессе создания библиотеки, обогащением, объединением в кластеры (группировкой) и секвенированием, увеличение одной лишь глубины секвенирования не может обеспечить обнаружения последовательностей, имеющих очень низкую аллельную частоту, таких как последовательности, находящиеся во внеклеточной ДНК (сокращенно "вкДНК", англ. cfDNA, от cell-free DNA) плода, содержащейся в плазме крови матери, в циркулирующей опухолевой ДНК (сокращенно "цоДНК", англ. ctDNA, от circulating tumor DNA), в субклональных мутациях патогенов. Таким образом, желательно создать способы определения последовательностей молекул ДНК при наличии их в малых количествах и/или при низкой аллельной частоте при одновременном подавлении неточностей секвенирования, обусловленных ошибками различного происхождения.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Рассмотренные воплощения изобретения относятся к способам, установке, системам и компьютерным программным продуктам, применяемым для определения последовательностей фрагментов нуклеиновых кислот с помощью уникальных молекулярных индексов (англ. unique molecular indices, сокращенно "UMI"). В различных примерах осуществления применение способов секвенирования позволяет определять последовательности фрагментов нуклеиновых кислот, принадлежащих к обеим цепочкам (нитям) фрагментов нуклеиновых кислот.В некоторых примерах осуществления в способах применяют физические UMI, находящиеся на одной или на обеих цепочках адаптеров секвенирования. В некоторых примерах осуществления в способах также применяют виртуальные UMI, находящиеся на обеих цепочках фрагментов нуклеиновых кислот.
Один из аспектов изобретения относится к способу секвенирования молекул нуклеиновых кислот из образца с применением уникальных молекулярных индексов (UMI). Каждый из уникальных молекулярных индексов (UMI) представляет собой олигонуклеотидную последовательность, которая может быть использована для идентификации индивидуальной молекулы фрагмента двухцепочечной ДНК (также называемой двухнитевой ДНК), имеющегося в образце. Способ включает: (а) прикрепление адаптеров к обоим концам фрагментов двухцепочечной ДНК, находящейся в образце, где каждый адаптер включает двухцепочечную гибридизованную область, одноцепочечное 5'-плечо, одноцепочечное 3'-плечо и физический UMI на одной цепочке или на каждой цепочке адаптеров, в результате чего получают продукты присоединения адаптера к ДНК; (b) амплификацию обеих цепочек продуктов присоединения адаптера к ДНК с образованием множества амплифицированных полинуклеотидов; (с) секвенирование множества амплифицированных полинуклеотидов, в результате чего получают множество прочтений (также называемых "ридами", от англ. "read"), каждое из которых ассоциировано с физическим UMI; (d) идентификацию множества физических UMI, ассоциированных с множеством прочтений; (е) идентификацию множества виртуальных UMI, ассоциированных с множеством прочтений, где каждый виртуальный UMI представляет собой последовательность, находящуюся во фрагменте ДНК в образце; и (f) определение последовательностей фрагментов двухцепочечной ДНК, находящейся в образце, с использованием множества прочтений, полученных в этапе (с), множества физических UMI, идентифицированных при выполнении этапа (d), и множества виртуальных UMI, идентифицированных в этапе (е). В некоторых примерах осуществления способ включает операцию (f), включающую: (i) для каждого из одного или более фрагментов двухцепочечной ДНК, находящейся в образце, объединение (1) прочтений, имеющих первый физический UMI и по меньшей мере один виртуальный UMI в направлении 5'-3', и (2) прочтений, имеющих второй физический UMI и по меньшей мере один виртуальный UMI в направлении 5'-3', для определения консенсусной нуклеотидной последовательности; и (и) для каждого из одного или более фрагментов двухцепочечной ДНК, находящейся в образце, определение последовательности с помощью консенсусной нуклеотидной последовательности.
В некоторых примерах осуществления множество физических UMI включает случайные UMI. В некоторых примерах осуществления множество физических UMI включает неслучайные UMI. В некоторых примерах осуществления каждый из неслучайных UMI отличается от любого другого неслучайного UMI адаптеров по меньшей мере двумя нуклеотидами в соответствующих положениях последовательности неслучайных UMI. В некоторых примерах осуществления множество физических UMI включает не более чем приблизительно 10000, приблизительно 1000, приблизительно 500 или приблизительно 100 уникальных неслучайных UMI. В некоторых примерах осуществления множество физических UMI включает приблизительно 96 уникальных неслучайных UMI.
В некоторых примерах осуществления рассмотренных выше способов прикрепление адаптеров к обоим концам фрагментов двухцепочечной ДНК включает связывание адаптеров с обоими концами фрагментов двухцепочечной ДНК. В некоторых примерах осуществления операция (f) включает применение прочтений, имеющих общий физический UMI и общий виртуальный UMI, для определения последовательности фрагмента ДНК образца.
В некоторых примерах осуществления рассмотренных выше способов множество физических UMI включает менее 12 нуклеотидов. В некоторых примерах осуществления множество UMI включает не более 6 нуклеотидов. В некоторых примерах осуществления множество UMI включает не более 4 нуклеотидов.
В некоторых примерах осуществления каждый из адаптеров включает физический UMI на каждой цепочке адаптеров в двухцепочечной гибридизованной области. В некоторых примерах осуществления физический UMI находится на конце двухцепочечной гибридизованной области, причем этот конец двухцепочечной гибридизованной области противоположен 3'-плечу или 5'-плечу или расположен на расстоянии одного нуклеотида от этого конца двухцепочечной гибридизованной области. В некоторых примерах осуществления каждый из адаптеров включает тринуклеотид 5'-TGG-3' или тринуклеотид 3'-АСС-5', находящийся в двухцепочечной гибридизованной области по соседству с физическим UMI. В некоторых примерах осуществления каждый из адаптеров включает последовательность праймера прочтения («рида», от англ. "read") на каждой цепочке двухцепочечной гибридизованной области.
В некоторых примерах осуществления каждый из адаптеров включает физический UMI только на одной из цепочек адаптеров одноцепочечного 5'-плеча или одноцепочечного 3'-плеча. В некоторых таких примерах осуществления этап (f) включает: (i) объединение прочтений, имеющих один и тот же первый физический UMI, в первую группу с целью получения первой консенсусной нуклеотидной последовательности; (ii) объединение прочтений, имеющих один и тот же второй физический UMI, во вторую группу с целью получения второй консенсусной нуклеотидной последовательности; и (iii) определение из первой и второй консенсусных нуклеотидных последовательностей последовательности одного из фрагментов двухцепочечной ДНК, содержащихся в образце. В некоторых примерах осуществления этап (iii) включает: (1) получение третьей консенсусной нуклеотидной последовательности из информации о местоположении и информации о последовательности первой и второй консенсусных нуклеотидных последовательностей, и (2) определение последовательности одного из фрагментов двухцепочечной ДНК из третьей консенсусной нуклеотидной последовательности. В некоторых примерах осуществления операция (е) включает идентификацию множества виртуальных UMI, в то время как каждый из адаптеров включает физический UMI только на одной из цепочек адаптеров в области одноцепочечного 5'-плеча или области одноцепочечного 3'-плеча. В некоторых примерах осуществления этап (f) включает: (i) объединение прочтений, имеющих первый физический UMI и по меньшей мере один виртуальный UMI в направлении 5'-3', и прочтений, имеющих второй физический UMI и по меньшей мере один виртуальный UMI в направлении 5'-3', для определения консенсусной нуклеотидной последовательности; и (ii) определение последовательности одного из фрагментов двухцепочечной ДНК, содержащихся в образце, на основании данных консенсусной нуклеотидной последовательности.
В некоторых примерах осуществления каждый из адаптеров включает физический UMI на каждой цепочке адаптеров в двухцепочечной области адаптеров, причем физический UMI одной цепочки комплементарен физическому UMI другой цепочки. В некоторых примерах осуществления операция (f) включает: (i) объединение прочтений, имеющих первый физический UMI, по меньшей мере один виртуальный UMI и второй физический UMI в направлении 5'-3', и прочтений, имеющих второй физический UMI, по меньшей мере один виртуальный UMI и первый физический UMI в направлении 5'-3', для определения консенсусной нуклеотидной последовательности; и (ii) определение последовательности одного из фрагментов двухцепочечной ДНК, содержащихся в образце, на основании данных консенсусной нуклеотидной последовательности.
В некоторых примерах осуществления каждый из адаптеров включает первый физический UMI, находящийся на 3'-плече адаптера, и второй физический UMI, находящийся на 5'-плече адаптера, причем первый физический UMI и второй физический UMI не комплементарны друг другу. В некоторых таких примерах осуществления этап (f) включает: (i) объединение прочтений, имеющих первый физический UMI, по меньшей мере один виртуальный UMI и второй физический UMI в направлении 5'-3', и прочтений, имеющих третий физический UMI, по меньшей мере один виртуальный UMI и четвертый физический UMI в направлении 5'-3', для определения консенсусной нуклеотидной последовательности; и (ii) определение последовательности одного из фрагментов двухцепочечной ДНК, содержащихся в образце, на основании данных консенсусной нуклеотидной последовательности.
В некоторых примерах осуществления по меньшей мере некоторые из виртуальных UMI получены из подпоследовательностей, находящихся на концах или вблизи концов фрагментов двухцепочечной ДНК, содержащихся в образце.
В некоторых примерах осуществления один или более физических UMI и/или один или более виртуальных UMI уникальным образом связаны (ассоциированы) с фрагментом двухцепочечной ДНК, содержащимся в образце.
В некоторых примерах осуществления фрагменты двухцепочечной ДНК, содержащиеся в образце, включают более приблизительно 1000 фрагментов ДНК.
В некоторых примерах осуществления множество виртуальных UMI включают UMI, включающих от приблизительно 6 п.о. до приблизительно 24 п.о (где "п.о." означает пару оснований; англ. bp, сокращение от "base pair"). В некоторых примерах осуществления множество виртуальных UMI включают UMI, включающие от приблизительно 6 п. о. до приблизительно 10 п.о.
В некоторых примерах осуществления рассмотренных выше способов получение множества прочтений в результате осуществления операции (с) включает: получение двух прочтений парных концов от каждого из амплифицированных полинуклеотидов, где два прочтения парных концов включают длинное прочтение и короткое прочтение, причем длинное прочтение имеет большую длину, чем короткое прочтение. В некоторых таких примерах осуществления операция (f) включает: объединение пар прочтений, ассоциированных с первым физическим UMI, в первую группу и объединение пар прочтений, ассоциированных со вторым физическим UMI во вторую группу, где первый и второй физические UMI уникальным образом ассоциированы с двухцепочечным фрагментом, находящимся в образце; и определение последовательности двухцепочечного фрагмента, содержащегося в образце, на основании информации о последовательности длинных прочтений, содержащихся в первой группе, и информации о последовательности длинных прочтений, содержащихся во второй группе. В некоторых примерах осуществления длина длинного прочтения составляет приблизительно 500 п.о. или более. В некоторых примерах осуществления длина короткого прочтения составляет приблизительно 50 п.о. или менее.
В некоторых примерах осуществления способ включает подавление ошибок, возникающих при проведении одной или более из следующих операций: ПЦР, создания библиотеки, объединения в кластеры и секвенирования.
В некоторых примерах осуществления амплифицированные полинуклеотиды включают аллель, аллельная частота которой составляет менее приблизительно 1%.
В некоторых примерах осуществления амплифицированные полинуклеотиды включают молекулу внеклеточной ДНК, полученную из опухоли, и аллель, указывающую на наличие опухоли.
В некоторых примерах осуществления секвенирование множества амплифицированных полинуклеотидов включает получение прочтений, включающих по меньшей мере приблизительно 100 п.о.
Другой аспект настоящего изобретения относится к способу секвенирования молекул нуклеиновых кислот из образца, где способ включает: (а) присоединение адаптеров к обоим концам фрагментов двухцепочечной ДНК, находящейся в образце, где каждый адаптер включает двухцепочечную гибридизованную область, одноцепочечное 5'-плечо, одноцепочечное 3'-плечо и физический уникальный молекулярный индекс (UMI), находящийся на одноцепочечном 5'-плече или одноцепочечном 3'-плече; (b) амплификацию обеих цепочек продуктов лигирования (сшивания), полученных при выполнении этапа (а), в результате чего получают множество одноцепочечных амплифицированных полинуклеотидов; (с) секвенирование множества амплифицированных полинуклеотидов, в результате чего получают множество прочтений, каждое из которых ассоциировано с физическим UMI; (d) идентификацию множества физических UMI, ассоциированных с множеством прочтений; и (е) определение последовательностей фрагментов двухцепочечной ДНК, находящейся в образце, на основании данных множества последовательностей, полученных при осуществлении этапа (с), и множества физических UMI, идентифицированных при выполнении этапа (d).
Дополнительный аспект изобретения относится к способу секвенирования молекул нуклеиновых кислот из образца. Способ включает: (а) присоединение адаптеров к обоим концам фрагментов двухцепочечной ДНК, находящейся в образце, где каждый адаптер включает двухцепочечную гибридизованную область, одноцепочечное 5'-плечо, одноцепочечное 3'-плечо и физический уникальный молекулярный индекс (UMI), включающий менее 12 нуклеотидов, на одной цепочке или на каждой цепочке адаптеров; (b) амплификацию обеих цепочек продуктов лигирования, полученных при выполнении этапа (а), в результате чего получают множество одноцепочечных амплифицированных полинуклеотидов, каждый из которых включает физический UMI; (с) секвенирование множества амплифицированных полинуклеотидов, в результате чего получают множество прочтений, каждое из которых ассоциировано с физическим UMI; (d) идентификацию множества физических UMI, ассоциированных с множеством прочтений; и (е) определение последовательностей фрагментов двухцепочечной ДНК, находящейся в образце, с использованием множества прочтений, полученных в этапе (с), и множества физических UMI, идентифицированных при выполнении этапа (d).
Другой аспект настоящего изобретения относится к способу получения дуплексного (двухнитевого) адаптера секвенирования, имеющего физический UMI на каждой цепочке. Способ включает: предоставление предварительного адаптера секвенирования, включающего двухцепочечную гибридизованную область, два одноцепочечных плеча и "липкий" (выступающий) конец, включающий 5'-CCANNNNANNNNTGG-3' на конце двухцепочечной гибридизованной области, отстоящий еще дальше от двух одноцепочечных плеч; достройку (удлинение) одной цепочки двухцепочечной гибридизованной области с использованием в качестве матрицы (шаблона) липкого конца, что приводит к получению продукта достройки; и применение рестрикционного фермента Хсm1 для расщепления двухцепочечного конца продукта достройки и получения, таким образом, дуплексного адаптера секвенирования, имеющего физический UMI на каждой цепочке. В некоторых примерах осуществления предварительный адаптер секвенирования включает последовательность праймера прочтения на каждой цепочке.
Другой аспект настоящего изобретения относится к компьютерному программному продукту, включающему энергонезависимый машиночитаемый носитель, на котором хранится программный код, выполнение которого одним или более процессорами компьютерной системы приводит к осуществлению компьютерной системой способа получения информации о строении интересующей последовательности, находящейся в образце, причем в способе применяют уникальные молекулярные индексы (UMI). Программный код включает: (а) код для получения прочтений множества амплифицированных полинуклеотидов, где множество амплифицированных полинуклеотидов получено амплификацией фрагментов двухцепочечной ДНК, содержащейся в образце, включающем интересующую последовательность, и присоединение адаптеров к фрагментам двухцепочечной ДНК; (b) код для идентификации множества физических UMI в прочтениях множества амплифицированных полинуклеотидов, где каждый физический UMI находится в адаптере, присоединенном к одному из фрагментов двухцепочечной ДНК; (с) код для идентификации множества виртуальных UMI в полученных прочтениях множества амплифицированных полинуклеотидов, где каждый виртуальный UMI находится в индивидуальной молекуле одного из фрагментов двухцепочечной ДНК; и (d) код для определения последовательностей фрагментов двухцепочечной ДНК на основании данных прочтений множества амплифицированных полинуклеотидов, множества физических UMI и множества виртуальных UMI, что приводит к уменьшению погрешностей в прочитанных последовательностях фрагментов двухцепочечной ДНК. В некоторых примерах осуществления каждый из адаптеров включает двухцепочечную гибридизованную область, одноцепочечное 5'-плечо, одноцепочечное 3'-плечо и физический уникальный молекулярный индекс (UMI) на одной цепочке или на каждой цепочке адаптеров.
В некоторых примерах осуществления код для определения последовательностей фрагментов двухцепочечной ДНК включает: (i) код для объединения прочтений, имеющих один и тот же первый физический UMI, в первую группу с целью получения первой консенсусной нуклеотидной последовательности; (ii) код для объединения прочтений, имеющих один и тот же второй физический UMI, во вторую группу с целью получения второй консенсусной нуклеотидной последовательности; и (iii) код для определения последовательности одного из фрагментов двухцепочечной ДНК, содержащихся в образце, на основании данных первой и второй консенсусных нуклеотидных последовательностей.
В некоторых примерах осуществления код для определения последовательностей фрагментов двухцепочечной ДНК включает: (i) код для объединения прочтений последовательности, имеющих первый физический UMI, по меньшей мере один виртуальный UMI и второй физический UMI в направлении 5'-3', и прочтений последовательности, имеющих второй физический UMI, по меньшей мере один виртуальный UMI и первый физический UMI в направлении 5'-3', для определения консенсусной нуклеотидной последовательности; и (ii) код для определения последовательности одного из фрагментов двухцепочечной ДНК, содержащихся в образце, на основании данных консенсусной нуклеотидной последовательности.
Дополнительный аспект изобретения относится к компьютерной системе, включающей: один или более процессоров; системную память; и один или более машиночитаемых (пригодных для чтения компьютером) носителей для хранения информации. На носителе хранятся инструкции, доступные для компьютерного исполнения, которые позволяют компьютерной системе осуществлять способ получения информации о структуре интересующей последовательности, находящейся в образце, с применением уникальных молекулярных индексов (UMI), которые представляют собой олигонуклеотидные последовательности, которые могут быть использованы для идентификации индивидуальных молекул фрагментов двухцепочечной ДНК, содержащихся в образце. Инструкции включают: (а) получение прочтений множества амплифицированных полинуклеотидов, где множество амплифицированных полинуклеотидов получено амплификацией фрагментов двухцепочечной ДНК, содержащейся в образце, включающем интересующую последовательность, и присоединение адаптеров к фрагментам двухцепочечной ДНК; (b) идентификацию множества физических UMI в полученных прочтениях множества амплифицированных полинуклеотидов, причем каждый физический UMI находится в адаптере, присоединенном к одному из фрагментов двухцепочечной ДНК; (с) идентификацию множества виртуальных UMI в полученных прочтениях множества амплифицированных полинуклеотидов, где каждый виртуальный UMI находится в индивидуальной молекуле одного из фрагментов двухцепочечной ДНК; и (d) определение последовательностей фрагментов двухцепочечной ДНК с помощью последовательностей множества амплифицированных полинуклеотидов, множества физических UMI и множеств виртуальных UMI, что приводит к уменьшению погрешностей в прочитанных последовательностях фрагментов двухцепочечной ДНК.
В некоторых примерах осуществления определение последовательностей фрагментов двухцепочечной ДНК включает: (i) объединение прочтений, имеющих один и тот же первый физический UMI, в первую группу с целью получения первой консенсусной нуклеотидной последовательности; (ii) объединение прочтений, имеющих один и тот же второй физический UMI, во вторую группу с целью получения второй консенсусной нуклеотидной последовательности; и (iii) определение из первой и второй консенсусных нуклеотидных последовательностей последовательности одного из фрагментов двухцепочечной ДНК.
В некоторых примерах осуществления определение последовательностей фрагментов двухцепочечной ДНК включает: (i) объединение прочтений, имеющих первый физический UMI, по меньшей мере один виртуальный UMI и второй физический UMI в направлении 5'-3', и прочтений, имеющих второй физический UMI, по меньшей мере один виртуальный UMI и первый физический UMI в направлении 5'-3', для определения консенсусной нуклеотидной последовательности; и (ii) определение последовательности одного из фрагментов двухцепочечной ДНК из консенсусной нуклеотидной последовательности.
Один из аспектов изобретения относится к способам секвенирования молекул нуклеиновых кислот из образца с помощью неслучайных уникальных молекулярных индексов (UMI). Способы включают: (а) прикрепление адаптеров к обоим концам фрагментов ДНК, находящейся в образце, где каждый адаптер включает двухцепочечную гибридизованную область, одноцепочечное 5'-плечо, одноцепочечное 3'-плечо и неслучайный уникальный молекулярный индекс (UMI) на одной цепочке или на каждой цепочке адаптеров, в результате чего получают продукты присоединения адаптера к ДНК; (b) амплификацию продуктов присоединения адаптера к ДНК с образованием множества амплифицированных полинуклеотидов; (с) секвенирование множества амплифицированных полинуклеотидов, в результате чего получают множество прочтений, ассоциированных с множеством неслучайных UMI; (d) идентификацию прочтений, имеющих общий неслучайный UMI, из множества прочтений; и (е) определение последовательности по меньшей мере части фрагмента ДНК, находящегося в образце, имеющего прикрепленный адаптер с общим неслучайным UMI, из идентифицированных прочтений, имеющих общий неслучайный UMI.
В некоторых примерах осуществления способ дополнительно включает: выбор из прочтений, имеющих общий неслучайный UMI, прочтений, имеющих как общий неслучайный UMI, так и общее положение прочтения; при этом для определения последовательности фрагмента ДНК в этапе (е) применяют только прочтения, имеющие как общий неслучайный UMI, так и общее положение прочтения в эталонной последовательности (также называемой референсной последовательностью или последовательностью сравнения). В некоторых примерах осуществления каждый неслучайный UMI отличается от любого другого неслучайного UMI по меньшей мере двумя нуклеотидами, находящимися в соответствующих положениях последовательности неслучайных UMI.
Другой аспект изобретения относится к способам секвенирования молекул нуклеиновых кислот из образца с помощью неслучайных уникальных молекулярных индексов (UMI). В некоторых примерах осуществления способ включает: (а) прикрепление адаптеров к обоим концам фрагментов двухцепочечной ДНК, находящейся в образце, где каждый адаптер включает двухцепочечную гибридизованную область, одноцепочечное 5'-плечо, одноцепочечное 3'-плечо и неслучайный уникальный молекулярный индекс (UMI) на одной цепочке или на каждой цепочке адаптеров, в результате чего получают продукты присоединения адаптера к ДНК, причем неслучайный UMI может быть объединен с другой информацией для выполнения уникальной идентификации индивидуальной молекулы фрагментов двухцепочечной ДНК; (b) амплификацию обеих цепочек продуктов присоединения адаптера к ДНК с образованием множества амплифицированных полинуклеотидов; (с) секвенирование множества амплифицированных полинуклеотидов, в результате чего получают множество прочтений, каждое из которых ассоциировано с неслучайным UMI; (d) идентификацию множества неслучайных UMI, ассоциированных с множеством прочтений; и (е) применение множества прочтений и множества неслучайных UMI для определения последовательностей фрагментов двухцепочечной ДНК, содержащихся в образце.
В некоторых примерах осуществления применение множества прочтений и множества неслучайных UMI для определения последовательностей фрагментов двухцепочечной ДНК, содержащихся в образце, включает: идентификацию прочтений, имеющих общий неслучайный UMI, и применение идентифицированных прочтений для определения последовательности фрагмента ДНК, содержащегося в образце. В некоторых примерах осуществления применение множества прочтений и множества неслучайных UMI для определения последовательностей фрагментов двухцепочечной ДНК, содержащихся в образце, включает: идентификацию прочтений, имеющих общий неслучайный UMI и общее положение прочтения, и применение идентифицированных прочтений для определения последовательности фрагмента ДНК, находящегося в образце.
В некоторых примерах осуществления применение множества прочтений и множества неслучайных UMI для определения последовательностей фрагментов двухцепочечной ДНК, содержащихся в образце, включает: идентификацию прочтений, имеющих общий неслучайный UMI и общий виртуальный UMI, где общий виртуальный UMI находится во фрагменте ДНК, содержащемся в образце; и применение идентифицированных прочтений для определения последовательности фрагмента ДНК, содержащегося в образце.
В некоторых примерах осуществления применение множества прочтений и множества неслучайных UMI для определения последовательностей фрагментов двухцепочечной ДНК, содержащихся в образце, включает: идентификацию прочтений, имеющих общий неслучайный UMI, общее положение прочтения и общий виртуальный UMI, где общий виртуальный UMI находится во фрагменте ДНК, содержащемся в образце; и применение идентифицированных прочтений для определения последовательности фрагмента ДНК, содержащегося в образце.
В некоторых примерах осуществления каждый неслучайный UMI отличается от любого другого неслучайного UMI, находящихся в адаптерах, по меньшей мере двумя нуклеотидами, находящимися в соответствующих положениях последовательности неслучайных UMI. В некоторых примерах осуществления каждый из адаптеров включает физический UMI на каждой цепочке адаптеров в двухцепочечной гибридизованной области. В некоторых примерах осуществления множество неслучайных UMI включает не более чем приблизительно 10000, приблизительно 1000 или приблизительно 100 уникальных неслучайных UMI. В некоторых примерах осуществления множество неслучайных UMI включает приблизительно 96 уникальных неслучайных UMI.
В некоторых примерах осуществления каждое из множества прочтений включает неслучайный UMI. В некоторых примерах осуществления каждое из множества прочтений либо включает неслучайный UMI, либо ассоциировано с неслучайным UMI через прочтение парных концов. В некоторых примерах осуществления каждый из множества амплифицированных полинуклеотидов имеет неслучайный UMI на одном конце или имеет первый неслучайный UMI на первом конце и второй неслучайный UMI на втором конце.
Для определения последовательностей фрагментов ДНК с помощью рассмотренных способов также раскрыты система, установка и компьютерные программные продукты.
Один из аспектов изобретения относится к компьютерному программному продукту, включающему энергонезависимый машиночитаемый носитель, на котором хранится программный код, который при его исполнении одним или более процессорами компьютерной системы приводит к тому, что компьютерная система осуществляет способ получения информации о структуре интересующей последовательности, находящейся в образце, с применением уникальных молекулярных индексов (UMI). Программный код включает инструкции для выполнения рассмотренных выше способов.
Несмотря на то, что рассмотренные в настоящей работе примеры в основном касаются человека, и приведенное словесное разъяснение в основном нацелено на нужды человека, рассмотренные в описании концепции применимы к нуклеиновым кислотам, получаемым из любого вируса, растения, организма животного или другого организма и из популяций таких организмов (метагеномов, вирусных популяций и т.д.) Эти и другие признаки настоящего изобретения станут более очевидными после прочтения нижеследующего описания, сопровождаемого графическими материалами, и прилагаемой формулы изобретения, или могут быть освоены на практике при воплощении раскрытого изобретения.
ВКЛЮЧЕНИЕ ПОСРЕДСТВОМ ССЫЛКИ
Все патенты, патентные заявки и другие публикации, включающие все последовательности, рассмотренные в таких цитируемых документах, включены в настоящее описание посредством ссылки в том же объеме, как если бы в каждом отдельном случае было ясно указано, что каждая индивидуальная публикация, патент или патентная заявка включена в настоящее описание посредством ссылки. Все цитируемые документы в соответствующей части полностью включены в настоящее описание посредством ссылки для целей, указанных в контексте их цитирования в настоящем описании. Однако цитирование любого документа не должно рассматриваться как признание того, что он относится к предшествующему уровню техники по отношению к настоящему описанию.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На Фиг. 1А представлена блок-схема примера последовательности рабочих операций, в которых используют UMI для секвенирования фрагментов нуклеиновых кислот.
На Фиг. 1В представлен фрагмент/молекула ДНК и адаптеры, применяемые на начальных этапах последовательности рабочих операций, показанных на Фиг. 1А.
На Фиг. 2А схематично представлены пять различных структур адаптеров, которые могут быть использованы в различных примерах осуществления.
На Фиг. 2В представлен гипотетический процесс, при котором происходит "прыжок" UMI при осуществлении ПЦР с участием адаптеров, имеющих два физических UMI на двух плечах.
На Фиг. 2С представлен способ получения адаптеров, имеющих UMI на обеих цепочках адаптеров в двухцепочечной области; в качестве последовательности распознавания для рестрикционного фермента Хcm1 в способе применяют последовательность длиной 15-мер (SEQ ID NO:1).
На Фиг. 2D представлена схема адаптера, имеющего Р7 плечо верхней цепочки (SEQ ID NO:2) и Р5 плечо нижней цепочки (SEQ ID NO:3).
На Фиг. 2Е схематично представлена структура неслучайного UMI, который обеспечивает механизм обнаружения ошибок, возникающих в последовательности UMI при осуществлении способа секвенирования.
На Фиг. 3А и 3В представлены схемы, на которых показаны материалы и продукты реакции сшивки (лигирования) адаптеров с двухцепочечными фрагментами в соответствии с некоторыми из способов, рассмотренных в настоящей работе.
На Фиг. 4А-4Е представлены пути подавления различных источников ошибок при определении последовательности фрагмента двухцепочечной ДНК способами, рассмотренными в настоящей работе.
На Фиг. 5 схематично представлено применение физических UMI и виртуальных UMI для эффективного получения длинных прочтений парных концов.
На Фиг. 6 представлена блок-схема рассредоточенной системы для обработки испытуемого образца.
На Фиг. 7А и на Фиг. 7В представлены экспериментальные данные по эффективности подавления ошибок способами, рассмотренными в настоящей работе.
На Фиг. 8 представлены данные, показывающие, что использование только лишь информации о положении для объединения прочтений имеет тенденцию к объединению прочтений, которые на самом деле получены из различных исходных молекул.
На Фиг. 9 изображены эмпирические данные, которые показывают, что применение неслучайного UMI и информации о положении для объединения прочтений может привести к получению более точной оценки фрагментов, чем использование одной лишь информации о положении.
На Фиг. 10 в виде таблицы представлены различные виды ошибок, возникающие после обработки в трех образцах со случайными UMI.
На Фиг. 11А представлена чувствительность и селективность распознавания соматической мутации и вариации числа копий (англ. copy number variation, сокращенно CNV) в образце геномной ДНК с помощью двух способов объединения с использованием двух различных инструментов: VarScan и Denovo.
На Фиг. 11B-D представлена селективность (т.е. доля ложных распознаваний) распознавания соматической мутации и CNV в трех образцах вкДНК при повышении величин входных сигналов образцов, полученная двумя способами объединения с использованием двух различных инструментов: VarScan и Denovo.
СВЕДЕНИЯ, ПОДТВЕРЖДАЮЩИЕ ВОЗМОЖНОСТЬ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам, установке, системам и компьютерных программным продуктам для секвенирования нуклеиновых кислот, в частности, нуклеиновых кислот, присутствующих в ограниченном количестве или низкой концентрации, таких как внеклеточная ДНК (вкДНК) плода в плазме крови матери или циркулирующая опухолевая ДНК (цоДНК) в крови пациента, имеющего раковое заболевание.
Если не указано иное, осуществление способов и управление системами, рассмотренными в настоящей работе, включает применение традиционных методик и установки, обычно применяемых в молекулярной биологии, микробиологии, в очистке белка, конструировании белка, секвенировании белка и ДНК, а также в области рекомбинантных ДНК, которая также относится к данной области техники. Такие методики и установка известны специалистам в данной области техники и рассмотрены во множестве статей и справочных публикаций (см., например, Sambrook с соавт., "Molecular Cloning: A Laboratory Manual," третье издание (Cold Spring Harbor), [2001]).
Числовые диапазоны включают граничные значения диапазона. Согласно изобретению, каждое из приведенных в настоящей работе максимальных числовых граничных значений включает каждое из меньших числовых граничных значений, как если бы такие меньшие числовые граничные значения были ясно указаны в описании. Каждое из приведенных в настоящей работе минимальных числовых граничных значений включает каждое из больших числовых граничных значений, как если бы такие большие числовые граничные значения были ясно указаны в описании. Каждый из приведенных в настоящей работе числовых диапазонов включает более узкий числовой диапазон, который попадает в более широкий числовой диапазон, как если бы такие более узкие числовые диапазоны были ясно указаны в настоящей работе.
Приведенные в тексте настоящей заявки заголовки не имеют ограничивающего значения.
Если в тексте не указано иное, все технические и научные термины, используемые в настоящей работе, имеют значение, обычно приписываемое им специалистами в данной области техники. Различные научные словари, которые включают термины, упоминаемые в настоящей работе, хорошо известны и доступны специалистам в данной области техники. Несмотря на то, что любой из способов и материалов, аналогичных или эквивалентных способам и материалам, рассмотренным в настоящей работе, применяют при выполнении или исследовании примеров осуществления, рассмотренных в настоящей работе, некоторые способы и материалы рассмотрены ниже.
Термины, приведенные в следующем разделе, более полно раскрыты в тексте настоящей заявки. Следует понимать, что настоящее изобретение не ограничено конкретными рассмотренными здесь методологией, протоколами и реагентами, поскольку они могут различаться в зависимости от контекста, в котором их применяют специалисты в данной области техники.
ОПРЕДЕЛЕНИЯ
Упоминание в настоящей работе единственного числа включает множественное число, если из контекста не ясно иное.
Если не указано иное, порядок элементов в нуклеиновых кислотах указан слева направо в направлении 5'-3', и порядок элементов в аминокислотных последовательностях указан слева направо в направлении от аминогруппы к карбоксигруппе, соответственно.
Уникальные молекулярные индексы (UMI) представляют собой последовательности нуклеотидов, прикрепленные к молекулам ДНК или идентифицированные в молекулах ДНК, которые могут быть использованы для отличия индивидуальных молекул ДНК друг от друга. Поскольку UMI применяют для идентификации молекул ДНК, они также называются уникальными молекулярными идентификаторами. См., например, Kivioja, Nature Methods 9, 72-74 (2012). Для определения того, получены ли прочитанные последовательности из одной или из другой исходной молекулы ДНК, UMI могут быть секвенированы вместе с теми молекулами ДНК, с которыми они связаны. Используемый в настоящей работе термин "UMI" относится как к информации о последовательности полинуклеотида, так и к реальному полинуклеотиду как таковому.
Обычно проводят секвенирование множества экземпляров, получаемых из одной исходной молекулы. В случае секвенирования синтезом с применением методики секвенирования Illumina, перед подачей в проточную ячейку исходная молекула может быть амплифицирована с помощью ПЦР. В отсутствие ПЦР амплификации или после ее проведения, индивидуальные молекулы ДНК, находящиеся в проточной ячейке, подвергают мостиковой амплификации или амплификации ЕхАmр с образованием кластера. Каждая молекула в кластере получена из одной и той же исходной молекулы ДНК, но подвергнута отдельному секвенированию. Для коррекции ошибок и для других целей важно установить, являются все прочтения, получаемые из одного кластера, прочтениями, полученными из одной исходной молекулы. Применение UMI делает такую группировку возможной. Молекула ДНК, которую копируют посредством амплификации или другим способом с целью получения множества экземпляров молекулы ДНК, называется исходной молекулой ДНК.
UMI похожи на штрих-коды, которые обычно применяют для того, чтобы отличить прочтения, получаемые из одного образца, от прочтений, получаемых из других образцов, но отличие состоит в том, что UMI применяют для того, чтобы отличить одну исходную молекулу ДНК от другой в том случае, когда проводят совместное секвенирование множества молекул ДНК. Поскольку в одном образце может содержаться намного больше молекул ДНК, чем образцов в одной серии секвенирования, то в серии секвенирования количество различающихся UMI обычно намного превышает количество различающихся штрих-кодов.
Как было отмечено, UMI могут быть присоединены к индивидуальным молекулам ДНК или идентифицированы в индивидуальных молекулах ДНК. В некоторых примерах осуществления UMI могут быть присоединены к молекулам ДНК способами, которые позволяют произвести физическое связывание или присоединение UMI к молекулам ДНК, например, посредством лигирования или транспозиции с применением полимеразы, эндонуклеазы, транспозаз и т.д. Получаемые таким образом "прикрепленные" UMI также называются физическими UMI. В некоторых контекстах они также могут называться экзогенными UMI. UMI, идентифицированные в структуре исходных молекул ДНК, называются виртуальными UMI. В некоторых контекстах виртуальные UMI также могут называться эндогенными UMI.
Физические UMI могут быть определены множеством способов. Например, они могут представлять собой случайные, псевдослучайные или частично случайные или неслучайные нуклеотидные последовательности, которые вводят в адаптеры или иным образом включают в исходные молекулы ДНК, которые впоследствии подвергают секвенированию. В некоторых примерах осуществления физические UMI могут быть настолько уникальными, что каждый из них, как ожидается, будет уникальным образом идентифицировать любую заданную исходную молекулу ДНК, находящуюся в образце. Создают набор адаптеров, каждый из которых содержит физический UMI, и эти адаптеры прикрепляют к фрагментам или другим исходным молекулам ДНК, которые затем подвергают секвенированию, и каждая из индивидуальных секвенированных молекул содержит UMI, который позволяет отличить ее от всех других фрагментов. В таких примерах осуществления для уникальной идентификации фрагментов ДНК, содержащихся в образце, может быть использовано очень большое количество различных физических UMI (например, от нескольких тысяч до миллионов).
Разумеется, для обеспечения такой уникальности, физический UMI должен иметь достаточную длину для любой и каждой исходной молекулы ДНК. В некоторых примерах осуществления, для обеспечения уникальной идентификации каждой исходной молекулы ДНК, во время проведения секвенирования может быть использован менее уникальный молекулярный идентификатор в комбинации с другими методиками идентификации. В таких примерах осуществления множество фрагментов или адаптеров могут иметь один и тот же физический UMI. Для уникальной идентификации прочтений, полученных из одной исходной молекулы/фрагмента ДНК, физический UMI может быть использован в комбинации с другой информацией, такой как локализация выравнивания или виртуальные UMI. В некоторых примерах осуществления адаптеры включают физические UMI, ограниченные до относительно небольшого количества неслучайных последовательностей, например, до 96 неслучайных последовательностей. Такие физические UMI также называются неслучайными UMI. В некоторых примерах осуществления неслучайные UMI могут быть объединены с информацией о положении в последовательности и/или с виртуальными UMI для идентификации прочтений, отнесенных к одной и той же исходной молекуле ДНК. Идентифицированные прочтения могут быть объединены для получения консенсусной последовательности, которая отражает последовательность исходной молекулы ДНК, рассмотренной в настоящей работе.
"Виртуальный уникальный молекулярный индекс" или "виртуальный UMI" представляет собой уникальную подпоследовательность в исходной молекуле ДНК. В некоторых примерах осуществления виртуальные UMI находятся на концах или вблизи концов исходной молекулы ДНК. Одно или более из таких уникальных концевых положений может само по себе или в комбинации с другой информацией уникальным образом идентифицировать исходную молекулу ДНК. В зависимости от количества различных исходных молекул ДНК и количества нуклеотидов в виртуальном UMI, один или более виртуальных UMI могут уникальным образом идентифицировать исходные молекулы ДНК, находящиеся в образце. В некоторых случаях для идентификации исходной молекулы ДНК требуется комбинация из двух виртуальных уникальных молекулярных идентификаторов. Такие комбинации могут быть чрезвычайно редкими и, возможно, содержаться в образце только в единственном числе. В некоторых случаях один или более виртуальных UMI в комбинации с одним или более физическим UMI могут совместно уникальным образом идентифицировать исходную молекулу ДНК.
"Случайным UMI" может считаться физический UMI, выбранный в качестве случайного образца, с замещением или без замещения, из набора UMI, состоящего из всех возможных различных олигонуклеотидных последовательностей, имеющих заданную одну или более длину последовательности. Например, если каждый UMI в наборе UMI имеет n нуклеотидов, то набор включает 4∧n UMI, имеющих последовательности, которые различаются между собой. Случайный образец, выбранный из 4∧n UMI, представляет собой случайный UMI.
Напротив, используемый в настоящей работе термин "неслучайный UMI" означает физический UMI, который не является случайным UMI. В некоторых примерах осуществления доступные неслучайные UMI определяют заранее перед проведением соответствующего эксперимента или перед соответствующим применением. В некоторых примерах осуществления для создания последовательностей в наборе или для выбора образца из набора применяют правила получения неслучайного UMI. Например, последовательности в наборе могут быть созданы таким образом, чтобы последовательности содержали специальную структуру или структуры. В некоторых примерах осуществления каждая последовательность отличается от любой другой последовательности в наборе определенным количеством (например, 2, 3 или 4) нуклеотидов. Таким образом, ни одна неслучайная последовательность UMI не может быть превращена в любую другую доступную неслучайную последовательность UMI заменой такого количества нуклеотидов, которое составляет меньше заданного количества. В некоторых примерах осуществления неслучайный UMI выбран из набора UMI, включающего такое количество UMI, которое меньше количества всех возможных UMI при определенной длине последовательности. Например, неслучайный UMI, содержащий 6 нуклеотидов, может быть выбран из общего количества, составляющего 96 различных последовательностей (вместо общего количества возможных разных последовательностей, составляющего 4∧6=4096). В других примерах осуществления последовательности выбраны из набора неслучайным образом. Напротив, некоторые последовательности выбираются с более высокой вероятностью, чем другие последовательности.
В тех примерах осуществления, в которых неслучайные UMI выбраны из набора, включающего количество последовательностей, которое меньше количества всех возможных различных последовательностей, количество неслучайных UMI меньше, иногда значительно меньше количества исходных молекул ДНК. В таких примерах осуществления для идентификации прочтений последовательности, полученных от одной и той же исходной молекулы ДНК, информация из неслучайного UMI может быть скомбинирована с другой информацией, такой как виртуальный UMI и/или информация о последовательности.
Термин "прочтения парных концов" относится к прочтениям, полученным при секвенировании парных концов, в котором получают одно прочтение каждого конца фрагмента нуклеиновой кислоты. Секвенирование парных концов включает фрагментацию ДНК с образованием последовательностей, называемых вставками. Согласно некоторым протоколам, например, применяемым Illumina, прочтения из более коротких вставок (например, в диапазоне, составляющем приблизительно от десятков до сотен п.о.) называются прочтениями парных концов с короткими вставками или просто прочтениями парных концов. Напротив, прочтения более длинных вставок (например, порядка нескольких тысяч п.о.) называются прочтениями сопряженных пар. Согласно настоящему изобретению, возможно применение как прочтений парных концов с короткими вставками, так и прочтений сопряженных пар с длинными вставками, и оба эти вида прочтений одинаково применимы для осуществления способа определения последовательностей фрагментов ДНК. Таким образом, термин "прочтения парных концов" может относиться как к прочтениям парных концов с короткими вставками, так и к прочтениям сопряженных пар с длинными вставками, которые дополнительно рассмотрены в настоящей работе ниже. В некоторых примерах осуществления прочтения парных концов включают прочтения, включающие приблизительно от 20 п.о. до 1000 п.о. В некоторых примерах осуществления прочтения парных концов включают прочтения, включающие приблизительно от 50 п.о. до 500 п.о., приблизительно от 80 п.о. до 150 п.о. или приблизительно 100 п.о.
Используемый в настоящей работе термин "выравнивание" относится к способу сравнения прочтения с эталонной последовательностью, что позволяет определить: содержится ли последовательность прочтения в эталонной последовательности. В процессе выравнивания производятся попытки локализации (картирования) прочтения на эталонной последовательности, но это не всегда приводит к выравниванию прочтения по эталонной последовательности. Если эталонная последовательность содержит прочтение, то прочтение может быть локализовано в эталонной последовательности или в некоторых примерах осуществления в определенном участке эталонной последовательности. В некоторых случаях выравнивание просто указывает на то, является ли прочтение элементом конкретной эталонной последовательности (т.е. присутствует или отсутствует прочтение в эталонной последовательности). Например, выравнивание прочтения по эталонной последовательности 13-й хромосомы человека позволяет выяснить присутствует ли прочтение в эталонной последовательности 13-й хромосомы. Инструмент, помощью которого получают эту информацию, может быть назван инструментом для проверки принадлежности множеству. В некоторых случаях выравнивание дополнительно указывает на то местоположение в эталонной последовательности, где находится прочтение. Например, если эталонная последовательность представляет собой целую последовательность генома человека, то выравнивание может указывать на то, что прочтение находится на 13-й хромосоме, и может дополнительно указывать на то, что прочтение находится в определенной цепочке и/или сайте 13-й хромосомы. В некоторых сценариях инструменты для выравнивания недостаточно совершенны, поскольку а) обнаруживают не все достоверные выравнивания, и b) некоторые из полученных выравниваний недостоверны. Это происходит по различным причинам: например, прочтения могут содержать ошибки, и секвенированные прочтения могут отличаться от эталонного генома из-за гаплотипических различий. В некоторых примерах применения инструменты для проведения выравнивания включают встроенный допуск на несоответствие, который допускает некоторые степени несоответствия пар оснований, но при этом позволяет производить выравнивание прочтений по эталонной последовательности. Это может способствовать идентификации достоверного выравнивания прочтения, которое в противном случае было бы не найдено.
Выровненные прочтения представляют собой одну или более последовательностей, для которых показано, что они имеют порядок молекул нуклеиновых кислот, соответствующий известной эталонной последовательности, такой как эталонный геном. Выровненное прочтение и его положение, которое определено на эталонной последовательности, составляют маркер (метку) последовательности. Выравнивание может быть произведено вручную, хотя обычно для этого применяют компьютерный алгоритм, поскольку произвести выравнивание прочтений вручную для осуществления способов, рассмотренных в настоящей работе, в течение разумного периода времени было бы невозможно. Одним из примеров алгоритма выравнивания последовательностей является компьютерная программа эффективного локального выравнивания нуклеотидных данных Efficient Local Alignment of Nucleotide Data (сокращенно ELAND), распространяемая компанией Illumina в виде части программы Genomics Analysis (геномный анализ). В альтернативном варианте для выравнивания прочтений относительно эталонных геномов может быть использован фильтр Блума или аналогичный инструмент для проверки принадлежности множеству. См. патентную заявку US 14/354528, поданную 25 апреля 2014 г., содержание которой полностью включено в настоящее описание посредством ссылки. Соответствие прочтения последовательности при выравнивании может составлять 100% соответствия последовательности или менее 100% (т.е. неполное соответствие).
Используемый в настоящей работе термин "картирование" означает соотнесение последовательности прочтения при помощи выравнивания с более крупной последовательностью, например, эталонным геномом.
Термины "полинуклеотид", "нуклеиновая кислота" и "молекулы нуклеиновых кислот" используются взаимозаменяемо и относятся к последовательности нуклеотидов (т.е. рибонуклеотидов для РНК и дезоксирибонуклеотидов для ДНК), связанных ковалентными связями, в которой 3'-положение пентозы в одном нуклеотиде присоединено через группировку сложного фосфодиэфира к 5'-положению пентозы в следующем нуклеотиде. Нуклеотиды включают последовательности любых форм нуклеиновой кислоты, включающих, без ограничений, молекулы РНК и ДНК, такие как молекулы внеклеточной ДНК (вкДНК). Термин "полинуклеотид" включает, без ограничений, одно- и двухцепочечные полинуклеотиды.
Используемый в настоящей работе термин "испытуемый образец" относится к образцу, обычно полученному из биологической жидкости, клетки, ткани, органа или организма, который включает нуклеиновую кислоту или смесь нуклеиновых кислот, содержащую по меньшей мере одну последовательность нуклеиновой кислоты, которую необходимо исследовать на наличие вариации числа копий и других генетических изменений, таких как, без ограничений, однонуклеотидный полиморфизм, инсерции (вставки), делеции и структурные изменения. В некоторых примерах осуществления образец содержит по меньшей мере одну последовательность нуклеиновой кислоты, число копий которой, как полагают, изменилось. Такие образцы включают, без ограничений, слюну/ротовую жидкость, околоплодную жидкость, кровь, фракцию крови или образцы, полученные биопсией тонкой иглой, мочу, перитонеальную жидкость, плевральную жидкость и подобные вещества. Несмотря на то, что образец часто получают из организма человека (например, пациента), могут быть проведены исследования образцов, получаемых из организма любого млекопитающего, примеры которых включают, без ограничений, собак, кошек, лошадей, коз, овец, рогатый скот, свиней и т.д., а также смешанные популяции, такие как микробные популяции, получаемые из дикой природы, или вирусные популяции, получаемые из организмов пациентов. Образец может быть исследован непосредственно, в том виде, в котором он получен из биологического источника, или после предварительной обработки, проводимой для модификации характера образца. Например, такая предварительная обработка может включать получение плазмы крови из крови, разбавление вязких жидкостей и т.д. Способы предварительной обработки также могут включать, без ограничений, фильтрование, осаждение, разбавление, перегонку, смешивание, центрифугирование, замораживание, лиофилизацию, концентрацию, амплификацию, фрагментацию нуклеиновой кислоты, инактивацию мешающих компонентов, добавление реагентов, лизис и т.д. Если к образцу применяют такие способы предварительной обработки, то способы предварительной обработки обычно таковы, что интересующая нуклеиновая кислота (кислоты) остается в испытуемом образце, иногда в концентрации, пропорциональной концентрации в необработанном испытуемом образце (например, образце, который не подвергался какой-либо предварительной обработке (обработкам)). Такие "обработанные" образцы также считаются биологическими "испытуемыми" образцами для способов, рассмотренных в настоящей работе.
Используемый в настоящей работе термин "секвенирование нового поколения (СНП)" относится к способам секвенирования, которые позволяют проводить параллельные массовые секвенирования клонально амплифицированных молекул и единичных молекул нуклеиновых кислот.Неограничивающие примеры СНП включают секвенирование синтезом с применением обратимых окрашивающих терминаторов и секвенирование лигированием.
Термин "прочтение" относится к последовательности прочтения участков образца нуклеиновой кислоты. Обычно, но не обязательно, прочтение представляет собой короткую последовательность последовательно соединенных пар оснований, находящуюся в образце. Прочтение может быть представлено символически последовательностью пар оснований в виде А, Т, С и G в части образца вместе с вероятностной оценкой точности присутствия основания (показатель качества). Оно может храниться в запоминающем устройстве и может быть соответствующим образом обработано для определения его соответствия эталонной последовательности или другим критериям. Прочтение может быть передано непосредственно из установки секвенирования или получено опосредованно из хранящейся информации о последовательности, относящейся к образцу. В некоторых случаях прочтение представляет собой последовательность ДНК достаточной длины (например, по меньшей мере приблизительно 20 п.о.), которая может быть использована для идентификации более длинной последовательности или области, например, она может быть выровнена и локализована на хромосоме или в геномной области или гене.
Термины "сайт" и "участок выравнивания" используются взаимозаменяемо и относятся к уникальному положению (т.е. идентификатору (англ. ID) хромосомы, положению и ориентации хромосомы) на эталонном геноме. В некоторых примерах осуществления сайт может представлять собой положение остатка, маркера последовательности или сегмента на эталонной последовательности.
Используемый в настоящей работе термин "эталонный геном" или "эталонная последовательность" относится к любой конкретной известной последовательности генома, частичной или полной, любого организма или вируса, которая может быть использована в качестве эталонной для идентифицируемых последовательностей субъекта. Например, эталонный геном человека, а также множества других организмов, предоставлен Национальным центром биотехнологической информации (National Center for Biotechnology Information) на сайте ncbi.nlm.nih.gov. Термин "геном" относится к полной генетической информации организма или вируса, представленной в виде последовательностей нуклеиновых кислот.Однако следует понимать, что "полный" является относительным понятием, поскольку предполагается, что даже эталонный геном золотого стандарта включает пропуски («гэпы») и ошибки.
В различных примерах осуществления эталонная последовательность значительно длиннее выравниваемых по ней прочтений. Например, она может быть по меньшей мере приблизительно в 100 раз длиннее или по меньшей мере приблизительно в 1000 раз длиннее, или по меньшей мере приблизительно в 10000 раз длиннее, или по меньшей мере приблизительно в 105 раз длиннее, или по меньшей мере приблизительно в 106 раз длиннее, или по меньшей мере приблизительно в 107 раз длиннее.
В одном из примеров эталонная последовательность представляет собой последовательность, равную по длине геному человека. Такие последовательности могут быть названы геномными эталонными последовательностями. В другом примере эталонная последовательность ограничена определенной хромосомой человека, такой как 13-я хромосома. В некоторых примерах осуществления эталонная Y хромосома представляет собой последовательность Y хромосомы генома человека, версия hg19. Такие последовательности могут быть названы эталонными последовательностями хромосомы. Другие примеры эталонных последовательностей включают геномы других видов, а также хромосомы, субхромосомные области (такие как цепочки) и т.д. любых видов.
В некоторых примерах осуществления длина эталонной последовательности для выравнивания может составлять от приблизительно 1 до приблизительно 100 длин прочтения. В таких примерах осуществления выравнивание и секвенирование рассматриваются как целевые выравнивание или секвенирование, а не выравнивание или секвенирование целого генома. В этих примерах осуществления эталонная последовательность обычно включает последовательность гена и/или другую ограниченную интересующую последовательность.
В различных примерах осуществления эталонная последовательность представляет собой консенсусную последовательность или другую комбинацию, полученную от множества индивидуумов. Однако в некоторых примерах применения эталонная последовательность может быть получена из организма конкретного индивидуума.
Используемый в настоящей работе в отношении нуклеиновой кислоты или смеси нуклеиновых кислот термин "полученный" относится к процессу получения нуклеиновой кислоты (кислот) из источника из происхождения. Например, в одном из примеров осуществления смесь нуклеиновых кислот, полученная из двух различных геномов, означает, что нуклеиновые кислоты, например, вкДНК, были выделены из клеток в результате естественного процесса, такого как некроз или апоптоз. В другом примере осуществления смесь нуклеиновых кислот, полученная из двух различных геномов, означает, что нуклеиновые кислоты были извлечены из двух различных типов клеток субъекта.
Используемый в настоящей работе термин "биологическая жидкость" относится к жидкости, полученной из биологического источника, и включает, например, кровь, сыворотку крови, плазму крови, слюну, промывную жидкость, спинномозговую жидкость, мочу, сперму, пот, слезную жидкость, слюнные отделения и подобные жидкости. Используемые в настоящей работе термины "кровь", "плазма крови" и "сыворотка крови" включают исключительно их фракции или обработанные части. Аналогично, если образец получают при биопсии, взятии мазка, смыва и т.д., то термин "образец" включает исключительно обработанную фракцию или часть, полученную при биопсии, взятии мазка, смыва и т.д.
Используемый в настоящей работе термин "хромосома" относится к генному носителю наследственной информации живой клетки, который образован хроматиновыми цепочками, включающими ДНК и белковые компоненты (в частности, гистоны). В настоящей работе используется традиционная и признанная во всем мире система нумерации индивидуальных хромосом генома человека.
Используемый в настоящей работе термин "длина полинуклеотида" относится к абсолютному количеству молекул нуклеиновых кислот (нуклеотидов) в последовательности или в участке эталонного генома. Термин "длина хромосомы" относится к известной длине хромосомы, выраженной в парах оснований, например, представленной в библиотеке NCBI36/hg18 хромосом человека на Интернет-ресурсе |genome|.|ucsc|.|edu/cgi-bin/hgTracks?hgsid=167155613&chromlnfoPage=.
Используемый в настоящей работе термин "праймер" относится к выделенному олигонуклеотиду, который может быть начальной точкой синтеза в том случае, если его помещают в условия, подходящие для синтеза продукта достройки (продукта удлинения) (например, условия, которые включают присутствие нуклеотидов, индуцирующий агент, такой как полимераза ДНК, необходимые ионы и молекулы и подходящие температура и рН). Предпочтительно, для достижения максимальной эффективности амплификации, праймер может быть одноцепочечным, но в альтернативном варианте он может быть двухцепочечным. Если праймер двухцепочечный, то сначала, перед проведением синтеза продуктов достройки, его подвергают обработке для разделения цепочек. Праймер может представлять собой олигодезоксирибонуклеотид. Праймер имеет достаточную длину для инициирования синтеза продуктов достройки в присутствии индуцирующего агента. Точные значения длин праймеров зависят от множества факторов, примеры которых включают температуру, источник праймера, применяемый способ и параметры, используемые для конструирования праймера.
ВВЕДЕНИЕ И ОСНОВНАЯ ЧАСТЬ
Методика секвенирования нового поколения (СНП) быстро развивается, предоставляя новые инструменты для улучшения исследований и науки, а также здравоохранения и служб, использующих генетическую и связанную с ней биологическую информацию. Способы СНП включают параллельные исследования множества молекул, что позволяет постоянно повышать скорость определения информации о последовательности биомолекул. Однако, множество способов СНП и связанных с ними методик манипуляции с образцами имеют тенденцию к генерированию ошибок, в результате чего получаемые последовательности имеют относительно высокое содержание ошибок, которое составляет от одной ошибки в нескольких сотнях пар оснований до одной ошибки в нескольких тысячах пар оснований. Такое количество ошибок иногда бывает допустимым для определения наследственной генетической информации, такой как герминативные мутации, поскольку такая информация согласована в большинстве соматических клеток, из которых получают множество копий одного и того же генома испытуемого образца. Ошибка, получаемая в результате прочтения одной копии последовательности, оказывает небольшое или устраняемое влияние, если множество копий той же последовательности прочтены без ошибок. Например, если ошибочное прочтение одной копии последовательности не может быть полностью выровнено относительно эталонной последовательности, то оно может быть просто выброшено из анализа. Не содержащие ошибок прочтения других копий той же последовательности могут предоставлять достаточно информации для достоверного анализа. В альтернативном варианте вместо отбрасывания прочтения, в котором пара оснований отличается от других прочтений той же последовательности, можно не принимать во внимание отличающуюся пару оснований, принимая ее за результат ошибки известного или неизвестного происхождения.
Однако такие подходы к коррекции ошибок не дают полезного результата при обнаружении последовательностей с низкими аллельными частотами, таких как субклональные, соматические мутации, обнаруживаемые в нуклеиновых кислотах опухолевой ткани, циркулирующей опухолевой ДНК, вкДНК плода в низкой концентрации в плазме крови матери, устойчивые к воздействию медикаментов мутации патогенов и т.д. В этих примерах один фрагмент ДНК может содержать интересующую соматическую мутацию на сайте последовательности, в то время как многие другие фрагменты на том же сайте последовательности не имеют интересующей мутации. При таком сценарии, в традиционном секвенировании прочтения последовательности или пары оснований из мутировавшего фрагмента ДНК могут быть не учтены или неверно интерпретированы, что, таким образом, приводит к потере информации, необходимой для обнаружения интересующей мутации.
Из-за различного происхождения ошибок, повышение глубины секвенирования само по себе не может обеспечить обнаружения соматических вариаций с очень низкой аллельной частотой (например, <1%). В некоторых примерах осуществления, рассмотренных в настоящей работе, предоставлены дуплексные способы секвенирования, в которых происходит эффективное подавление ошибок в тех ситуациях, в которых получают низкие сигналы от достоверных интересующих последовательностей, таких как образцы с низкой аллельной частотой. В этих способах применяют виртуальные уникальные молекулярные индексы (UMI) в комбинации с короткими физическими уникальными молекулярными индексами, помещенными на одном плече или на обоих плечах адаптеров секвенирования, таких как адаптер Illumina TruSeq®. Эти примеры осуществления основаны на стратегии создания физических UMI на последовательностях адаптеров и виртуальных UMI на фрагментах последовательностей ДНК образца. В некоторых примерах осуществления для подавления ошибок также применяют выравнивание положений прочтений.
Например, если множество прочтений (или пар прочтений) имеют общий физический UMI и выравниваются в пределах одного и того же интервала (ограниченный диапазон положений) на эталонной последовательности, ожидается, что прочтения получены из одного фрагмента ДНК. Физические UMI, виртуальные UMI и выровненные положения, связанные с прочтениями, обеспечивают получение "индексов", которые, как таковые или в комбинации, уникальным образом ассоциированы с определенным фрагментом двухцепочечной ДНК, получаемой из образца. Использование таких индексов позволяет идентифицировать множество прочтений, полученных из одного фрагмента ДНК (одной молекулы), который может представлять собой один из множества фрагментов одного и того же геномного сайта. Использование множества прочтений, полученных из одной молекулы ДНК, позволяет эффективно корректировать ошибки. Например, методика секвенирования позволяет получать консенсусную нуклеотидную последовательность (далее называемую "консенсусной последовательностью") из множества прочтений, полученных из одного и того же фрагмента ДНК, причем при коррекции достоверная информация о последовательности этого фрагмента ДНК не отбрасывается.
Структура адаптеров может обеспечивать наличие физических UMI, которые позволяют определить, из какой цепочки фрагмента ДНК получены прочтения. В некоторых примерах осуществления этим пользуются для определения первой консенсусной последовательности для прочтений, полученных из одной цепочки фрагмента ДНК, и второй консенсусной последовательности для комплементарной цепочки. Во многих примерах осуществления консенсусная последовательность включает пары оснований, обнаруженные во всех или в большинстве прочтений, но из нее исключаются пары оснований, появляющиеся лишь в нескольких прочтениях. Могут быть выбраны различные критерии консенсуса. Способ объединения прочтений на основе UMI или участков выравнивания для получения консенсусной последовательности также называется "объединением" прочтений. Использование физических UMI, виртуальных UMI и/или участков выравнивания позволяет устанавливать то, что прочтения, составленные для первой и второй консенсусных последовательностей, получены из одного двухцепочечного фрагмента. Таким образом, в некоторых примерах осуществления третью консенсусную последовательность определяют, используя первую и вторую консенсусные последовательности, полученные для одной и той же молекулы/фрагмента ДНК; при этом третья консенсусная последовательность включает пары оснований, общие для первой и второй консенсусных последовательностей, но в нее не включены пары оснований, не согласующиеся между этими двумя последовательностями. В альтернативных примерах осуществления вместо сравнения двух консенсусных последовательностей, полученных из двух цепочек, непосредственно может быть получена только одна консенсусная последовательность, составляемая при объединении всех прочтений, полученных из обеих цепочек одного фрагмента. Наконец, последовательность фрагмента может быть определена из третьей или единственной консенсусной последовательности, которая включает пары оснований, согласующиеся во всех прочтениях, полученных из обеих цепочек фрагмента.
В различных примерах осуществления для подавления ошибок объединяют прочтения двух цепочек фрагмента ДНК. Однако в некоторых примерах осуществления в способе физические и виртуальные UMI создают на одноцепочечных фрагментах нуклеиновой кислоты (например, ДНК или РНК) и для подавления ошибок объединяют прочтения, имеющие одинаковые физические и виртуальные UMI. Для захвата одноцепочечных фрагментов нуклеиновых кислот в образце могут быть использованы различные способы.
В некоторых примерах осуществления для определения исходного полинуклеотида, по которому определяют прочтения, в способе комбинируют различные типы индексов. Например, в способе для идентификации прочтений, полученных из одной молекулы ДНК, могут использоваться как физические, так и виртуальные UMI. При использовании наряду с физическими UMI второй формы UMI, физические UMI могут быть более короткими, чем в случае применения только физических UMI для анализа исходного полинуклеотида. Такой подход имеет минимальное влияние на характеристики получения библиотеки и не требует увеличения длины прочтения при секвенировании.
Применение рассмотренных способов включает:
- Подавление ошибок при обнаружении соматической мутации. Например, обнаружение мутации с аллельной частотой менее 0,1% чрезвычайно важно при жидкой биопсии для обнаружения циркулирующей опухолевой ДНК.
- Коррекцию предварительного фазирования, фазирования и других ошибок секвенирования для получения высококачественных длинных прочтений (например, 1×1000 п.о.).
- Уменьшение продолжительности цикла для фиксированной длины прочтения и коррекция повышенного фазирования и предварительного фазирования этим способом.
- Применение UMI на обеих сторонах фрагмента для создания виртуальных длинных прочтений парных концов. Например, сшивка прочтения 2×500 проведением 500+50 на дупликатах.
Пример последовательности рабочих операций для секвенирования фрагментов нуклеиновых кислот с применением UMI
На Фиг. 1А представлена блок-схема примера последовательности 100 рабочих операций, в которой для секвенирования фрагментов нуклеиновых кислот используют UMI. В операции 102 получают фрагменты двухцепочечной ДНК. Фрагменты ДНК могут быть получены, например, фрагментацией геномной ДНК, сбором ДНК, фрагментированной в естественных условиях (например, вкДНК или цоДНК), или синтезом фрагментов ДНК из РНК. В некоторых примерах осуществления для синтеза фрагментов ДНК из РНК сначала производят очистку информационной (матричной) РНК с помощью выбранной poly(A) (полиаделиновой кислоты) или исчерпанием рибосомной РНК, затем выбранную мРНК подвергают химической фрагментации и превращают в одноцепочечную кДНК с помощью случайного примирования с использованием гексамера. Получают комплементарную цепочку кДНК для создания двухцепочечной кДНК, которая уже готова для создания библиотеки. Для получения фрагментов двухцепочечной ДНК из геномной ДНК (гДНК), вводимую гДНК подвергают фрагментации, например, посредством гидродинамического разрезания, распыления, ферментативной фрагментации и т.д., что приводит к созданию фрагментов подходящей длины, например, приблизительно 1000 п.о., 800 п.о., 500 или 200 п.о. Например, при распылении за короткий период времени могут быть получены куски ДНК, составляющие менее 800 п.о. В этом способе получают двухцепочечные фрагменты ДНК, содержащие 3'- и/или 5'-липкие концы.
На Фиг. 1В представлен фрагмент/молекула ДНК и адаптеры, применяемые на начальных этапах последовательности 100 рабочих операций, представленной на Фиг. 1А. Несмотря на то, что на Фиг. 1В представлен только один двухцепочечный фрагмент, одновременно в последовательности рабочих операций может быть получено от тысяч до миллионов фрагментов образца. При фрагментации ДНК физическими способами получают гетерогенные концы, включающие смесь 3'-липких концов, 5'-липких концов и тупых (ровных) концов. Липкие концы будут иметь различные длины; концы могут быть фосфорилированными или нефосфорилированными. Один из примеров фрагментов двухцепочечной ДНК, полученных при фрагментации геномной ДНК в операции 102, показан в виде фрагмента 123 на Фиг. 1В.
На левом конце фрагмента 123 имеется 3'-липкий конец, а на правом конце имеется 5'-липкий конец; фрагмент отмечен обозначениями ρ и ϕ, которые указывают на две находящиеся во фрагменте последовательности, которые могут быть использованы в качестве виртуальных UMI; виртуальные UMI при использовании их как таковых или в комбинации с физическими UMI адаптера, который сшивают с фрагментом, могут уникальным образом идентифицировать фрагмент.UMI уникальным образом связаны с единственным фрагментом ДНК образца, который включает исходный полинуклеотид и его комплементарную цепочку. Физический UMI представляет собой последовательность олигонуклеотида, связанную с исходным полинуклеотидом, его комплементарной цепочкой или с полинуклеотидом, полученным из исходного полинуклеотида. Виртуальный UMI представляет собой последовательность олигонуклеотида в исходном полинуклеотиде, его комплементарной цепочке или в полинуклеотиде, полученном из исходного полинуклеотида. Согласно этой схеме, физический UMI также может быть назван внешним UMI, а виртуальный UMI - внутренним UMI.
В действительности каждая из двух последовательностей риф означает две комплементарные последовательности на одном геномном сайте, но для простоты они указаны только на одной из цепочек в некоторых двухцепочечных фрагментах, показанных на изображении. Виртуальные UMI, такие как ρ и ϕ, могут быть применены на более позднем этапе последовательности рабочих операций для упрощения идентификации прочтений, получаемых из одной или обеих цепочек одного исходного фрагмента ДНК. После такой идентификации прочтений, они могут быть объединены с образованием консенсусной последовательности.
Если фрагменты ДНК получены физическими способами, то последовательность 100 рабочих операций включает выполнение операции 104 репарации концов, в которой получают фрагменты с тупыми концами, имеющие 5'-фосфорилированные концы. В некоторых примерах осуществления в этом этапе липкие концы, полученные при фрагментации, превращают в тупые концы под действием Т4 ДНК-полимеразы и фермента Кленова. Экзонуклеазная активность этих ферментов в направлении 3'-5' приводит к удалению 3'-липких концов, а полимеразная активность в направлении 5'-3' приводит к заполнению 5'-липких концов. Кроме того, в этой реакции под действием Т4-полинуклеотидкиназы протекает фосфорилирование 5'-концов во фрагментах ДНК. Фрагмент 125 на Фиг. 1В представляет собой один из примеров продукта репарации концов с образованными тупыми концами.
Следующей после репарации концов операцией последовательности 100 рабочих операций является операция 106 аденилирования 3'-концов фрагментов, которая также называется образованием А-хвоста или образованием dA-хвоста, поскольку она включает присоединение одного dATP (дезоксиаденозинтрифосфата) к 3'-концам тупых фрагментов для предотвращения их сшивания друг с другом при проведении реакции присоединения адаптера. Двухцепочечная молекула 127, представленная на Фиг. 1В, имеет А-хвостовой фрагмент, включающий тупые концы с 3'-dA липкими концами и 5'-фосфатными концами. Как показано позицией 129 на Фиг. 1В, единственный Т-нуклеотид на 3'-конце каждого из двух адаптеров секвенирования обеспечивает наличие липкого конца, комплементарного 3'-dA липкому концу, на каждом конце вставки для присоединения к вставке двух адаптеров.
После аденилирования 3'-концов последовательность 100 рабочих операций включает операцию 108 присоединения частично двухцепочечных адаптеров к обоим концам фрагментов. В некоторых примерах осуществления применяемые в реакции адаптеры включают различающиеся между собой олигонуклеотиды, и эти олигонуклеотиды образуют физические UMI для связывания прочтений последовательности с одним исходным полинуклеотидом, который может представлять собой одно- или двухцепочечный фрагмент ДНК. Поскольку все физические UMI олигонуклеотиды различны, два UMI олигонуклеотида, присоединенные к двум концам конкретного фрагмента, различаются между собой. Кроме того, два физических UMI конкретного фрагмента отличаются от физических UMI любого другого фрагмента. Таким образом, два физических UMI уникальным образом связаны с конкретным фрагментом.
Позицией 129 на Фиг. 1В показаны два адаптера, которые должны быть присоединены к двухцепочечному фрагменту, который включает два виртуальных UMI ρ и ϕ, расположенные вблизи концов фрагмента. Структуры представленных адаптеров аналогичны структурам адаптеров секвенирования, созданных Компанией Illumina, поскольку в различных примерах осуществления изобретения для получения прочтений и обнаружения интересующей последовательности могут быть использованы методики СНП, разработанные Компанией Illumina. Адаптер, показанный в левой части, включает на плече Р5 физический UMI α, а адаптер, показанный в правой части, включает на плече Р5 физический UMI β. На цепочке адаптеров, имеющей 5'-денатурированный конец, в направлении от 5' к 3' имеется последовательность Р5, физический UMI (α или β) и последовательность праймера прочтения 2. На цепочке адаптеров, имеющей 3'-денатурированный конец, в направлении от 3' к 5' имеется последовательность Р7', последовательность индекса и последовательность праймера прочтения 1. Олигонуклеотиды Р5 и Р7' комплементарны праймерам амплификации, связанным с поверхностями проточных ячеек, применяемых согласно концепции секвенирования Illumina. В некоторых примерах осуществления последовательность индекса представляет собой средства для отслеживания источника происхождения образца, что позволяет производить мультиплексирование множества образцов в программе секвенирования. В различных примерах осуществления могут быть использованы другие конструкции адаптеров и программ секвенирования. Адаптеры и методика секвенирования дополнительно рассмотрены в последующих разделах. В реакции, представленной на Фиг. 1В, происходит присоединение определенных последовательностей к 5' и 3'-концам каждой цепочки в геномном фрагменте. Продукт 131 лигирования, полученный из рассмотренного выше фрагмента, показан на Фиг. 1В. Этот продукт 131 лигирования включает физический UMI α, виртуальный UMI ρ и виртуальный UMI ϕ на верхней цепочке в направлении 5'-3'. Продукт лигирования также включает физический UMI β, виртуальный UMI ϕ и виртуальный UMI ρ на нижней цепочке в направлении 5'-3'. Продукт лигирования и содержащиеся в нем физические UMI и виртуальные UMI, показанные позицией 132, аналогичны соответствующим элементам, изображенным в верхней половине Фиг. 3А. Настоящее изобретение также относится к способам применения методик секвенирования и адаптеров, отличающимся от методик секвенирования и адаптеров, предоставляемых Illumina.
В некоторых примерах осуществления продукты таких реакций лигирования очищают и/или классифицируют по размерам с помощью электрофореза на агарозном геле или магнитных гранул. После классификации по размерам ДНК подвергают ПЦР амплификации для обогащения фрагментами, которые содержат адаптеры на обоих концах. См. блок 110. В нижней половине Фиг. 3А показано, что обе цепочки продукта лигирования подвергаются ПЦР амплификации, при которой получают две серии фрагментов, содержащих различные физические UMI (α и β). Каждая из двух серий имеет только один физический UMI. Обе серии имеют виртуальные UMI ρ и ϕ, но порядок виртуальных UMI относительно физических UMI в них различается: α-ρ-ϕ и β-ϕ-ρ. В некоторых примерах осуществления производят очистку продуктов ПЦР и выбор размерного диапазона матриц, подходящего для последующего создания кластеров.
Затем, согласно последовательности 100 рабочих операций, выполняют кластерную амплификацию продуктов ПЦР согласно концепции, разработанной Illumina. См. операцию 112. При объединении продуктов ПЦР в кластеры могут быть собраны библиотеки для мультиплексирования, содержащие, например, до 12 образцов на дорожке; при этом на адаптерах создают различные последовательности индексов для отслеживания различных образцов.
После проведения кластерной амплификации может быть проведено секвенирование синтезом согласно концепции Illumina с целью получения прочтений секвенирования. См. операцию 114. Несмотря на то, что рассмотренные в настоящей работе адаптеры и способы секвенирования основаны на концепции Illumina, вместо них или в добавление к ним могут быть использованы другие методики секвенирования, в частности, способы СНП.
Также ожидается, что прочтения секвенирования, полученные из сегмента, показанного на Фиг. 1В и 3А, включают UMI α-ρ-ϕ или β-ϕ-ρ. Эту особенности применяют в последовательности 100 рабочих операций для объединения прочтений, имеющих один и тот же физический UMI (или множество физических UMI) и/или один и тот же виртуальный UMI (или множество виртуальных UMI), в одну или более групп, в результате чего получают одну или более консенсусных последовательностей. См. операцию 116. Консенсусная последовательность включает нуклеотидные основания, которые согласуются или соответствуют критерию консенсуса во всех прочтениях в сводной группе. Как показано в операции 116, физические UMI, виртуальные UMI и информация о положении могут быть объединены различным способами для объединения прочтений с целью получения консенсусных последовательностей для определения последовательности фрагмента или по меньшей мере его части. В некоторых примерах осуществления для объединения прочтений физические UMI объединяют с виртуальными UMI. В других примерах осуществления для объединения прочтений объединяют физические UMI и локализацию прочтения. Информация о локализации прочтения может быть получена с помощью различных методик, в которых применяют различные способы определения локализации, например, геномные координаты прочтений, локализацию на эталонной последовательности или положение в хромосоме. В других примерах осуществления для объединения прочтений объединяют физические UMI, виртуальные UMI и локализацию прочтений.
Наконец, согласно последовательности 100 рабочих операций, одну или более консенсусных последовательностей используют для определения последовательности нуклеиновых кислот во фрагменте, содержащемся в образце. См. операцию 118. Эта операция может включать принятие последовательности нуклеиновых кислот во фрагменте в качестве третьей консенсусной последовательности или единственной консенсусной последовательности, как указано выше.
В одном из конкретных примеров осуществления, который включает операции, аналогичные операциям 108-119, способ секвенирования молекул нуклеиновых кислот из образца с помощью неслучайных UMI включает следующие этапы: (а) прикрепление адаптеров к обоим концам фрагментов ДНК, находящейся в образце, где каждый адаптер включает двухцепочечную гибридизованную область, одноцепочечное 5'-плечо, одноцепочечное 3'-плечо и неслучайный UMI, в результате чего получают продукты присоединения адаптера к ДНК; (b) амплификацию продуктов присоединения адаптера к ДНК с образованием множества амплифицированных полинуклеотидов; (с) секвенирование множества амплифицированных полинуклеотидов, в результате чего получают множество прочтений, ассоциированных с множеством неслучайных UMI; (d) идентификацию прочтений, имеющих общий неслучайный UMI и общее положение прочтения, из множества прочтений; и (е) определение последовательности по меньшей мере части фрагмента ДНК из идентифицированных прочтений.
В различных примерах осуществления полученные прочтения последовательности ассоциированы с физическими UMI (например, случайными или неслучайными UMI). В таких примерах осуществления UMI либо представляет собой часть последовательности прочтения, либо часть последовательности другого прочтения, причем то, что другое прочтение и требуемое прочтение происходят из одного фрагмента, известно, например, из прочтения парных концов или специальной информации о локализации, такой как виртуальные UMI.
В некоторых примерах осуществления прочтения последовательности представляют собой прочтения парных концов. Каждое прочтение либо включает неслучайный UMI, либо связано с неслучайным UMI через прочтение парных концов. В некоторых примерах осуществления длины прочтений составляют меньше длин фрагментов ДНК или меньше половины длины фрагментов. В таких случаях полная последовательность целого фрагмента иногда не определяется. Напротив, определяются два конца фрагмента. Например, длина фрагмента ДНК может составлять 500 п.о., из которых могут быть получены два прочтения парных концов длиной 100 п.о. В этом примере могут быть определены 100 оснований на каждом конце фрагмента, а 300 п.о. в середине фрагмента могут не определяться в отсутствии информации, получаемой из других прочтений. В некоторых примерах осуществления, если два прочтения парных концов имеют достаточную длину для того, чтобы они перекрывались, полная последовательность целого фрагмента может быть определена из двух прочтений. Для примера - см. пример, рассмотренный при описании Фиг. 5.
В некоторых примерах осуществления каждый неслучайный UMI отличается от любого другого неслучайного UMI по меньшей мере двумя нуклеотидами в соответствующих положениях последовательности неслучайных UMI. В различных примерах осуществления множество неслучайных UMI включает не более чем приблизительно 10000, 1000 или 100 уникальных неслучайных UMI. В некоторых примерах осуществления множество неслучайных UMI включает 96 уникальных неслучайных UMI.
В некоторых примерах осуществления в двухцепочечной области адаптера содержится дуплексный неслучайный UMI, и каждое прочтение включает первый неслучайный UMI на одном конце и второй неслучайный UMI на другом конце.
Адаптеры и UMI
Адаптеры
Кроме структуры адаптеров, рассмотренной выше в примере последовательности рабочих операций, в различных примерах осуществления способов и систем согласно изобретению, могут быть использованы другие конструкции адаптеров. На Фиг. 2А схематично представлены пять различных структур адаптеров, содержащих один или более UMI, которые могут быть использованы в различных примерах осуществления.
На Фиг. 2A(i) представлен стандартный двухиндексный адаптер TruSeq®, предоставляемый Illumina. Адаптер частично состоит из двух цепочек и получен сплавлением двух олигонуклеотидов, соответствующих этим двум цепочкам. Две цепочки содержат такое количество комплементарных пар оснований (например, 12-17 п.о.), которые позволяют прикреплять два олигонуклеотида к концу, который должен быть сшит с фрагментом дцДНК (т.е. двухцепочечной ДНК). Фрагмент дцДНК, предназначенный для прикрепления к обоим концам прочтений парных концов, также называется вставкой. Другие пары оснований некомплементарны по отношению к этим двум цепочкам, в результате чего вилкообразный адаптер имеет два свободных липких конца. В примере, представленном на Фиг. 2A(i), комплементарные пары оснований представляют собой часть последовательности праймера прочтения 2 и последовательности праймера прочтения 1. Далее по ходу транскрипции относительно последовательности праймера прочтения 2 находится однонуклеотидный 3'-Т липкий конец, который представляет собой липкий конец, комплементарный однонуклеотидному 3'-А липкому концу фрагмента дцДНК, подвергаемого секвенированию, который может способствовать гибридизации двух указанных липких концов. Последовательность праймера прочтения 1 находится на 5'-конце комплементарной цепочки, к которой присоединена фосфатная группа. Фосфатная группа способствует сшиванию 5'-конца последовательности праймера прочтения 1 с 3'-А липким концом фрагмента ДНК. На цепочке, содержащей свободный липкий 5'-конец (верхняя цепочка), в направлении 5'-3' адаптер включает последовательность Р5, последовательность индекса i5 и последовательность праймера прочтения 2. На цепочке, содержащей свободный липкий 3'-конец, в направлении 3'-5' адаптер включает последовательность Р7', последовательность индекса i7 и последовательность праймера прочтения 1. Олигонуклеотиды Р5 и Р7' комплементарны праймерам амплификации, закрепленным на поверхности проточных ячеек платформы секвенирования Illumina. В некоторых примерах осуществления последовательности индексов представляют собой средства для отслеживания источника происхождения образца, что позволяет проводить мультиплексирование множества образцов на платформе секвенирования.
На Фиг. 2A(ii) представлен адаптер, имеющий единственный физический UMI, заменяющий область индекса i7 в стандартном двухиндексном адаптере, показанном на Фиг. 2A(i). Эта структура адаптера совпадает со структурой, показанной в примере последовательности рабочих операций, рассмотренном выше при описании Фиг 1В. В некоторых примерах осуществления физические UMI α и β сконструированы таким образом, что они находятся только на 5'-плече двухцепочечных адаптеров; в результате этого получают продукты лигирования, имеющие только один физический UMI на каждой цепочке. Напротив, размещение физических UMI в обеих цепочках адаптеров приводит к образованию продуктов лигирования, имеющих два физических UMI на каждой цепочке, что приводит к удвоению времени и стоимости, затрачиваемых на секвенирование физических UMI. Однако, настоящее изобретение включает способы, в которых применяют физические UMI, находящиеся на обеих цепочках адаптеров, которые показаны на Фиг. 2A(iii)-2A(vi), которые обеспечивают доступ к дополнительной информации, которая может быть использована для объединения различных прочтений с целью получения консенсусных последовательностей.
В некоторых примерах осуществления физические UMI в адаптерах включают случайные UMI. В некоторых примерах осуществления физические UMI в адаптерах включают неслучайные UMI.
На Фиг. 2A(iii) представлен адаптер, содержащий два физических UMI, добавленные в стандартный двухиндексный адаптер. Показанные физические UMI могут представлять собой случайные UMI или неслучайные UMI. Первый физический UMI расположен против хода транскрипции относительно последовательности индекса i7, а второй физический UMI расположен против хода транскрипции относительно последовательности индекса i5. На Фиг. 2A(iv) представлен адаптер, также содержащий два физических UMI, добавленные в стандартный двухиндексный адаптер. Первый физический UMI расположен далее по ходу транскрипции относительно последовательности индекса i7, а второй физический UMI расположен далее по ходу транскрипции относительно последовательности индекса i5. Аналогично, эти два физических UMI могут представлять собой случайные UMI или неслучайные UMI.
Адаптер, содержащий два физических UMI на двух плечах одноцепочечной области, такой как адаптеры, показанные на Фиг. 2A(iii) и 2A(iv), может связывать две цепочки фрагмента двухцепочечной ДНК, если a priori или a posteriori известна информация, связывающая два некомплементарных физических UMI. Например, исследователь может знать последовательности UMI 1 и UMI 2 до их введения в адаптер, структура которого представлена на Фиг. 2A(iv). Такая информация о связях может быть использована для заключения о том, что прочтения, содержащие UMI 1 и UMI 2, получены из двух цепочек фрагмента ДНК, к которому был присоединен адаптер. Таким образом, могут быть объединены не только прочтения, имеющие одинаковый физический UMI, но и прочтения, имеющие любой из двух некомплементарных физических UMI. Интересно отметить, что рассмотренное ниже явление, называемое "прыжки UMI", может затруднять распознавание связи между физическими UMI в одноцепочечных областях адаптеров.
Два физических UMI, находящиеся на двух цепочках адаптеров, показанных на Фиг. 2A(iii) и Фиг 2A(iv), не располагаются на одном и том же сайте и не комплементарны друг другу. Однако настоящее изобретение относится к способам, в которых применяют физические UMI, расположенные на одном и том же сайте двух цепочек адаптера, и/или UMI, комплементарные друг другу. На Фиг. 2A(v) представлен дуплексный адаптер, в котором два физических UMI комплементарны в двухцепочечной области на конце или вблизи конца адаптера. В некоторых примерах осуществления физический UMI, расположенный вблизи конца адаптера, может включать 1 нуклеотид, 2 нуклеотида, 3 нуклеотида, 4 нуклеотида, 5 нуклеотидов или приблизительно 10 нуклеотидов от конца двухцепочечной области адаптера, где конец расположен в области, противоположной раздвоенной части адаптера. Два физических UMI могут представлять собой случайные UMI или неслучайные UMI. На Фиг. 2A(vi) представлен адаптер, аналогичный адаптеру, показанному на Фиг. 2A(v), но имеющий меньшую длину; при этом он не включает последовательностей индексов или последовательностей Р5 и Р7' комплементарных праймерам амплификации, находящимся на поверхности проточной ячейки. Аналогично, два физических UMI могут представлять собой случайные UMI или неслучайные UMI.
По сравнению с адаптерами, имеющими один или более одноцепочечных физических UMI на одноцепочечных плечах, адаптеры, содержащие двухцепочечный физический UMI в двухцепочечной области, могут обеспечивать непосредственную связь между двумя цепочками фрагмента двухцепочечной ДНК, к которому присоединен адаптер, что показано на Фиг. 2A(v) и Фиг. 2A(vi). Поскольку две цепочки двухцепочечного физического UMI комплементарны друг другу, связь между двумя цепочками двухцепочечного UMI по определению отражается комплементарными последовательностями и может быть установлена без привлечения информации, полученной a priori или a posteriori. Эта информация может быть использована для подтверждения того, что прочтения, содержащие две комплементарные последовательности двухцепочечного физического UMI, содержащегося в адаптере, получены из того же фрагмента ДНК, к которому был пришит адаптер; при этом, две комплементарные последовательности физического UMI присоединены к 3'-концу одной цепочки и 5'-концу другой цепочки фрагмента ДНК. Таким образом, можно объединить не только прочтения, имеющие на двух концах одинаковый порядок двух последовательностей физического UMI, но и прочтения, имеющие на двух концах обратный порядок двух комплементарных последовательностей.
В некоторых примерах осуществления может быть предпочтительным применение относительно коротких физических UMI, поскольку короткие физические UMI легче вводить в адаптеры. Кроме того, более короткие физические UMI быстрее и легче секвенировать в амплифицированных фрагментах. Однако, при чрезмерном укорочении физических UMI общее количество различающихся физических UMI может стать меньше количества молекул адаптеров, требуемых для обработки образца. Для создания достаточного количества адаптеров один и тот же UMI будет необходимо создавать в двух или более молекулах адаптеров. При таком сценарии адаптеры, имеющие одинаковый физический UMI, могут быть соединены с множеством исходных молекул ДНК. Однако такие короткие физические UMI могут обеспечивать достаточно информации, если их комбинируют с другими источниками информации, такими как виртуальные UMI и/или участки выравнивания прочтений, уникальным образом идентифицируя прочтения как прочтения, полученные из конкретного исходного полинуклеотида или фрагмента ДНК, содержащегося в образце. Это происходит потому, что даже если одинаковые физические UMI будут присоединены к двум разным фрагментам, то маловероятно, что два разных фрагмента также будут иметь одинаковые участки выравнивания или совпадающие подпоследовательности, которые служат виртуальными UMI. Таким образом, если два прочтения имеют одинаковые короткие физические UMI и одинаковый участок выравнивания (или одинаковый виртуальный UMI), то вероятно, что эти два прочтения получены из одного фрагмента ДНК.
Кроме того, в некоторых примерах осуществления объединение прочтений основано на двух физических UMI, находящихся на двух концах вставки. В таких примерах осуществления для определения источника фрагментов ДНК объединяют два очень коротких физических UMI (например, длиной 4 п.о.), и общая длина двух физических UMI обеспечивает информацией, достаточной для распознавания различных фрагментов.
В различных примерах осуществления физические UMI включают приблизительно 12 пар оснований или менее, приблизительно 11 пар оснований или менее, приблизительно 10 пар оснований или менее, приблизительно 9 пар оснований или менее, приблизительно 8 пар оснований или менее, приблизительно 7 пар оснований или менее, приблизительно 6 пар оснований или менее, приблизительно 5 пар оснований или менее, приблизительно 4 пар оснований или менее или приблизительно 3 пар оснований или менее. В некоторых примерах осуществления, в которых физические UMI представляют собой неслучайные UMI, UMI включают приблизительно 12 пар оснований или менее, приблизительно 11 пар оснований или менее, приблизительно 10 пар оснований или менее, приблизительно 9 пар оснований или менее, приблизительно 8 пар оснований или менее, приблизительно 7 пар оснований или менее или приблизительно 6 пар оснований.
Прыжки UMI могут влиять на выявление связей между физическими UMI на одном плече или обоих плечах адаптеров, таких как адаптеры, представленные на Фиг. 2A(ii)-(iv). Было показано, что при связывании этих адаптеров с фрагментами ДНК продукты амплификации могли включать большее количество фрагментов, имеющих уникальные физические UMI, превышающее реальное число фрагментов в образце.
Кроме того, при использовании адаптеров, имеющих физические UMI на обоих плечах, амплифицированные фрагменты, имеющие общий физический UMI на одном конце, предположительно имеют другой общий физический UMI на другом конце. Однако, иногда это не так. Например, в продукте реакции одной реакции амплификации некоторые фрагменты могут иметь первый физический UMI и второй физический UMI на обоих концах; другие фрагменты могут иметь второй физический UMI и третий физический UMI, а другие фрагменты могут иметь первый физический UMI и третий физический UMI; при этом еще одни фрагменты могут иметь третий физический UMI и четвертый физический UMI, и т.д. В этом примере определение происхождения таких амплифицированных фрагментов (фрагмента) может быть затруднительным. Очевидно, что при проведении амплификации один физический UMI может быть "заменен" другим физическим UMI.
В одном из возможных подходов к решению проблемы прыжков UMI только фрагменты, имеющие оба общих UMI, рассматриваются как фрагменты, происходящие из одной исходной молекулы, в то время как фрагменты, имеющие только один общий UMI, исключаются из анализа. Однако, некоторые из фрагментов, имеющих только один общий физический UMI, в действительности могут происходить из одной молекулы, как и фрагменты, имеющие оба общих физических UMI. При исключении из рассмотрения фрагментов, имеющих только один общий физический UMI, может быть потеряна полезная информация. В другом возможном подходе любые фрагменты, имеющие один общий физический UMI, рассматриваются как фрагменты, происходящие из одной и той же исходной молекулы. Но в этом подходе не разрешается объединять два физических UMI, находящиеся на двух концах фрагментов, для проведения анализа далее по ходу транскрипции. Кроме того, при любом подходе, например, одном из описанных выше, фрагменты, имеющие общие первый и второй физические UMI, не считаются полученными из той же исходной молекулы, что и фрагменты, имеющие общие третий и четвертый физические UMI. Это может соответствовать истине не во всех случаях. В третьем подходе проблема прыжков UMI может быть решена с помощью применения адаптеров в сочетании с физическими UMI на обеих цепочках одноцепочечной области, таких как адаптеры, представленные на Фиг. 2A(v)-(vi). Третий подход более подробно раскрыт ниже после описания предполагаемого механизма прыжка UMI.
На Фиг. 2В представлен предполагаемый процесс прыжка UMI, происходящий в реакции ПЦР, в котором участвуют адаптеры, несущие на двух плечах два физических UMI. Два физические UMI могут представлять собой случайные UMI или неслучайные UMI. Реальный механизм прыжка UMI и предполагаемый процесс, рассмотренный в настоящей работе, не влияют на применимость адаптеров и способов, рассмотренных в настоящей работе. Реакция ПЦР начинается с предоставления по меньшей мере одного двухцепочечного исходного фрагмента 202 ДНК и адаптеров 204 и 206. Адаптеры 204 и 206 аналогичны адаптерам, представленным на Фиг. 2A(iii)-(iv). Адаптер 204 имеет последовательность адаптера Р5 и физический UMI α1 на 5'-плече. Адаптер 204 также имеет последовательность адаптера Р7' и физический UMI α2 на 3'-плече. Адаптер 206 имеет последовательность адаптера Р5 и физический UMI β2 на 5'-плече и последовательность адаптера Р7' и физический UMI β1 на 3'-плече. Затем способ включает лигирование адаптера 204 и адаптера 206 к фрагменту 202 и получение продукта 208 лигирования. Затем способ включает денатурацию продукта 208 лигирования, которая приводит к образованию одноцепочечного денатурированного фрагмента 212. На этом этапе реакционная смесь часто включает остаточные адаптеры. Поскольку даже в том случае, когда согласно способу уже были удалены излишки адаптеров, например, с помощью гранул для твердофазной обратимой иммобилизации (англ. Solid Phase Reversible Immobilization, сокращенно SPRI), некоторые адаптеры все еще остаются в реакционной смеси. Такой оставшийся адаптер изображен в виде адаптера 210, аналогичного адаптеру 206, за исключением того, что на плечах 3' и 7' адаптера 210 имеются физические UMI γ1 и γ2, соответственно. В условиях денатурации, в которых образуется денатурированный фрагмент 212, также образуется олигонуклеотид 216 денатурированного адаптера, в котором физический UMI γ1 располагается вблизи последовательности адаптера Р7'.
Реакция ПЦР включает обработку (примирование, от англ. priming) денатурированного фрагмента 212 праймером 214 ПЦР и достройку праймера 214, что приводит к образованию двухцепочечного фрагмента, который затем подвергается денатурации с образованием одноцепочечного промежуточного фрагмента 220, комплементарного фрагменту 212. В процессе ПЦР также происходит примирование денатурированного олигонуклеотида 216 праймером 218 ПЦР и достройка праймера 218, что приводит к образованию двухцепочечного фрагмента, который затем подвергается денатурации с образованием одноцепочечного промежуточного олигонуклеотида 222 адаптера, комплементарного фрагменту 212. Перед проведением следующего цикла ПЦР амплификации промежуточные олигонуклеотиды 222 адаптера гибридизуются с образованием промежуточного фрагмента 220, расположенного вблизи конца Р7' и далее по ходу транскрипции относительно физического UMI β1. Гибридизованная область соответствует одноцепочечным областям адаптера 206 и адаптера 210, поскольку эти одноцепочечные области имеют одну и ту же общую последовательность.
Гибридизованный продукт, получаемый из промежуточного фрагмента 220 и промежуточного олигонуклеотида 222 адаптера, образует матрицу, в которую затем может быть введен Р7' ПЦР праймер 224, помещаемый на 5'-конец олигонуклеотида 222, и матрица может быть достроена. Во время достройки, после достижения конца промежуточного олигонуклеотида 222 адаптера, матрица достройки переключается на промежуточный фрагмент 220. Переключение матрицы представляет собой возможный механизм прыжка UMI. После достройки и денатурации образуется одноцепочечный фрагмент 226, который, в целом, комплементарен промежуточному фрагменту 220, за исключением того, что он имеет в промежуточном фрагменте 220 физический UMI γ1 вместо физического UMI β1. Аналогично, одноцепочечный фрагмент 226 идентичен фрагменту 212, за исключением того, что он включает физический UMI γ1 вместо физического UMI β1.
В некоторых примерах осуществления изобретения применение адаптеров, имеющих физические UMI на обеих цепочках двухцепочечной области адаптера, таких как адаптеры, представленные на Фиг. 2A(v)-(vi), может предотвратить или снизить частоту прыжков UMI. Это может объясняться тем фактом, что физические UMI в двухцепочечной области одного адаптера отличаются от физических UMI во всех других адаптерах. Это способствует уменьшению комплементарности между промежуточными олигонуклеотидами адаптеров и промежуточными фрагментам, что позволяет избежать гибридизации, такой как в случае, показанном для промежуточного олигонуклеотида 222 и промежуточного фрагмента 220, что способствует уменьшению частоты или предотвращению прыжка UMI.
Случайные физические UMI и неслучайные физические UMI
В некоторых примерах осуществления адаптеров, рассмотренных выше, физические UMI в адаптерах включают случайные UMI. В некоторых примерах осуществления каждый случайный UMI отличен от любого другого случайного UMI, имеющегося во фрагментах ДНК. Другими словами, случайные UMI выбраны случайным образом, без замещения, из набора UMI, включающего все возможные различные UMI заданной длины последовательности (последовательностей). В других примерах осуществления случайные UMI выбраны случайным образом с замещением. Существует вероятность, что в этих примерах осуществления два адаптера могут иметь одинаковый UMI.
В некоторых примерах осуществления физические UMI, находящиеся в адаптерах, включают неслучайные UMI. В некоторых примерах осуществления множество адаптеров включают одинаковые последовательности неслучайных UMI. Например, набор из 96 различных неслучайных UMI может быть распределен среди 100000 различающихся молекул/фрагментов, полученных из образца. В некоторых примерах осуществления каждый неслучайный UMI набора отличается от любого другого UMI набора двумя нуклеотидами. Другими словами, необходимо, чтобы в каждом неслучайном UMI по меньшей мере два из присутствующих в нем нуклеотида были заменены перед проведением сравнения с последовательностью любого другого неслучайного UMI, применяемого в секвенировании. В других примерах осуществления каждый неслучайный UMI из набора отличается от любого другого UMI набор тремя или более нуклеотидами.
На Фиг. 2С представлен способ получения адаптеров, имеющих случайные UMI на обеих цепочках адаптеров в двухцепочечной области, и при этом два адаптера на двух цепочках комплементарны друг другу. Для осуществления способа сначала подбирают адаптер 230 секвенирования, содержащий гибридизованную двухцепочечную область и два одноцепочечных плеча. Полученный адаптер аналогичен адаптеру, показанному на Фиг. 2A(v). В представленном здесь примере последовательность D7XX соответствует последовательности индекса i7, показанной на Фиг. 2A(v), последовательность SBS12' соответствует последовательности праймера прочтения 1, показанной на Фиг. 2A(v), D50X соответствует последовательности индекса i5, показанной на Фиг. 2A(v), и SBS3 соответствует последовательности праймера прочтения 2, показанной на Фиг. 2A(v). Адаптер секвенирования 232 включает 15-мерный липкий конец CCANNNNANNNNTGG (SEQ ID NO:1) на конце двухцепочечной гибридизованной области, расположенный против хода транскрипции относительно последовательности SBS12' праймера прочтения. Буквой N обозначены случайные нуклеотиды, четыре из которых, расположенные между А и TGG, будут применены для создания физического UMI на 5'-конце цепочки SBS12'. Липкий конец 15-мера может быть распознан рестрикционным ферментом Хсm1, поскольку Хсm1 распознает 15-мер, имеющий ССА на 5'-конце и TGG на 3'-конце. Затем способ 230 включает достройку 3'-конца цепочки SPS3 с использованием 15-мера в качестве матрицы достройки, в результате чего образуется продукт 234 достройки. Продукт 234 достройки содержит тирозин в середине 15-мера на цепочке SBS3, соответствующий аденозину на цепочке SBS12'. Остаток тирозина становится остатком на 3'-конце двухцепочечной области в адаптере, представляющем собой конечный продукт способа 230. Остаток тирозина может гибридизоваться с остатком аденозина на 3' А-хвосте вставки.
Далее способ 230 включает применение рестрикционного фермента Xcm1 для обработки только что достроенного конца продукта 234 достройки. Хст1 представляет собой рестрикционную эндонуклеазу, которая распознает 15-меры, содержащие ССА на 5'-конце и TGG на 3'-конце, и под ее действием как фосфодиэстеразы цепочка нуклеиновых кислот расщепляется за счет разрезания фосфодиэфирной связи между 8 и 9 нуклеотидами, если считать с 5'-конца САА. За счет этого механизма расщепления происходит расщепление двухцепочечного конца продукта 234 достройки непосредственно далее по ходу транскрипции относительно остатка аденозина, находящегося на цепочке SBS12', и далее по ходу транскрипции относительно остатка тирозина, находящегося на цепочке SBS3. Расщепление приводит к образованию адаптера 236, который содержит четыре случайных нуклеотида на 5'-конце в своей двухцепочечной области, расположенной против хода транскрипции относительно последовательности SBS12'. Адаптер 236 также включает тирозиновый липкий конец и четыре случайных нуклеотида на 3'-конце двухцепочечной области, расположенной далее по ходу транскрипции относительно последовательности SBS3. Расположенные на каждой цепочке четыре случайных нуклеотида образуют физический UMI, и два физических UMI, находящиеся на двух цепочках, комплементарны друг другу.
На Фиг. 2D представлена схема адаптера, имеющего цепочку SBS13 верхнего плеча (SEQ ID NO:2) и цепочку SBS3 нижнего плеча (SEQ ID NO:3), на которой показаны нуклеотиды, присутствующие в адаптере. Этот адаптер аналогичен адаптеру 236, показанному на Фиг. 2С, но при этом он содержит четыре пары оснований, находящиеся между сайтом распознавания Хсm1 и последовательностями прочтения адаптера. Кроме того, адаптер, показанный на Фиг. 2D, представляет собой укороченную версию адаптера 236, из которой удалены Р7/Р5 и последовательность индекса, что повышает стабильность адаптера. На верхней цепочке адаптера (SEQ ID NO:2), в двухцепочечной области, начиная от 5'-конца, содержатся четыре случайных нуклеотида, подходящих для физического UMI, за которыми следует TGG, представляющий собой сайт распознавания рестрикционного фермента Хсm1, и затем TCGC, расположенный против хода транскрипции относительно последовательности прочтения. Нуклеотиды TCGC включены для придания адаптеру стабильности. В некоторых примерах осуществления их присутствие необязательно.
Для обеспечения стабильности, во время получения адаптера, подготовки и обработки образца могут быть добавлены нуклеотиды. Было показано, что при введении дополнительных TCGC оснований даже при комнатной температуре повышается эффективность ренатурации олигонуклеотидов верхней и нижней цепочек, которую проводят для создания исходной матрицы адаптера. Поскольку достройку Кленова и расщепление под действием Хст1, выполняемые при получении адаптера, производят при более высоких температурах (30°С и 37°С, соответственно), добавление TCGC может повысить стабильность адаптера. Для повышения стабильности адаптера, кроме TCGC, могут быть использованы другие последовательности или нуклеотиды различной длины.
В некоторых примерах осуществления в адаптер для других целей могут быть введены дополнительные последовательности, не имеющие функции стабилизации, которые не влияют на способность адаптера обеспечивать присутствие уникальных индексов во фрагментах ДНК. Нижняя цепочка адаптера (SEQ ID NO:3) в двухцепочечной области комплементарна верхней цепочке, за исключением того, что она включает липкий конец Т на 3'-конце. Четыре случайных нуклеотида на нижней цепочке составляют второй физический UMI.
Случайные UMI, такие как UMI, представленные на Фиг. 2С и 2D, подходят для создания большего количества уникальных UMI, чем неслучайные UMI с той же длиной последовательности. Другими словами, случайные UMI с большей вероятностью оказываются уникальными, чем неслучайные UMI. Однако в некоторых примерах осуществления получить неслучайные UMI может быть легче, или они могут иметь более высокую эффективность конверсии. В том случае, когда неслучайные UMI скомбинированы с другой информацией, такой как положение в последовательности и виртуальные UMI, они могут служить эффективным механизмом для индексации молекул, из которых получены фрагменты ДНК.
В различных примерах осуществления неслучайные UMI идентифицируют, учитывая различные факторы, которые включают, без ограничений, средства для обнаружения ошибок в последовательностях UMI, эффективность конверсии, совместимость с данным видом анализа, содержание GC, гомополимеры и факторы, влияющие на способ получения.
Например, неслучайные UMI могут быть сконструированы таким образом, чтобы они способствовали обнаружению ошибок. На Фиг. 2Е схематично представлена структура неслучайного UMI, обеспечивающая создание механизма обнаружения ошибок, которые появляются в последовательности UMI при осуществлении способа секвенирования. Как видно из показанной структуры, каждый из неслучайных UMI содержит шесть нуклеотидов и отличается от любого другого UMI по меньшей мере двумя нуклеотидами. Как показано на Фиг. 2Е, неслучайный UMI 244 отличается от неслучайного UMI 242 первыми двумя нуклеотидами слева, что показано подчеркиванием нуклеотидов Т и G в UMI 244 в отличие от нуклеотидов А и С в UMI 242. UMI 246 представляет собой последовательность, идентифицированную как часть прочтения, и он отличается от всех других находящихся в адаптерах UMI, которые получены в способе. Поскольку последовательность UMI в прочтении, вероятно, получена из UMI, находящегося в адаптере, ошибка, по-видимому, произошла в процессе секвенирования, например, во время амплификации или секвенирования. UMI 242 и UMI 244 показаны, как два UMI, наиболее схожие с UMI 246 прочтения. Можно отметить, что UMI 246 отличается от UMI 242 одним нуклеотидом, т.е. первым нуклеотидом слева, который представляет собой Т вместо А. Кроме того, UMI 246 также отличается от UMI 244 одним нуклеотидом, который, в этом случае, представляет собой второй нуклеотид слева, т.е. С вместо G. Поскольку UMI 246 в прочтении отличается и от UMI 242, и от UMI 244 одним нуклеотидом, из представленной информации нельзя заключить, получен ли UMI 246 из UMI 242 или из UMI 244. Однако, во множестве других сценариев ошибки UMI в прочтениях не одинаково отличаются от двух наиболее схожих с UMI. Как показано в примере, среди UMI 248, UMI 242 и UMI 244 также имеются два UMI, наиболее схожих с UMI 248. Можно отметить, что UMI 248 отличается от UMI 242 одним нуклеотидом, третьим нуклеотидом слева, который представляет собой А вместо Т. Напротив, UMI 248 отличается от UMI 244 тремя нуклеотидами. Таким образом, невозможно определить, получен ли UMI 248 из UMI 242 или из UMI 244, и ошибка, по-видимому, проявилась в третьем нуклеотиде слева.
Виртуальные UMI
Рассматривая виртуальные UMI, следует отметить, что те виртуальные UMI, которые определяют на концах или относительно концевых положений исходных молекул ДНК, могут уникальным или почти уникальным образом определять индивидуальные исходные молекулы ДНК, если локализация концевых положений в целом случайна, как это бывает при проведении фрагментации и в природных вкДНК. Если образец содержит относительно мало исходных молекул ДНК, виртуальные UMI сами могут уникальным образом идентифицировать индивидуальные исходные молекулы ДНК. Применение комбинации из двух виртуальных UMI, каждый из которых ассоциирован с другим концом исходной молекулы ДНК, повышает вероятность того, что виртуальные UMI могут без дополнительной информации уникальным образом идентифицировать исходные молекулы ДНК. Разумеется, в тех случаях, когда один или два виртуальных UMI не могут без дополнительной информации уникальным образом идентифицировать исходные молекулы ДНК, может быть применена комбинация таких виртуальных UMI с одним или более физическими UMI.
Если два прочтения получены из одного фрагмента ДНК, то две подпоследовательности, содержащие одинаковые пары оснований, также будут иметь ту же самую относительную локализацию в прочтениях. Напротив, если два прочтения получены из двух разных фрагментов ДНК, то маловероятно, что две подпоследовательности, содержащие одинаковые пары оснований, будут иметь совершенно одинаковую относительную локализацию в прочтениях. Таким образом, если две или более подпоследовательности из двух или более прочтений имеют одинаковые пары оснований и одинаковую относительную локализацию в двух или более прочтениях, то можно заключить, что эти два или более прочтения получены из одного фрагмента.
В некоторых примерах осуществления подпоследовательности, находящиеся на концах или вблизи концов фрагмента ДНК, используют в качестве виртуальных UMI. Такой выбор конструкции имеет ряд практических преимуществ. Во-первых, относительные локализации этих подпоследовательностей в прочтениях легко определяются, поскольку они находятся в начале или вблизи начала прочтений, и системе не нужно использовать отступ для нахождения виртуального UMI. Кроме того, поскольку первыми подвергаются секвенированию пары оснований на концах фрагментов, эти пары оснований доступны, даже если прочтения оказываются относительно короткими. Кроме того, пары оснований, определенные ранее в длинном прочтении, с меньшей вероятностью содержат ошибки секвенирования, чем пары оснований, определенные позднее. Однако в других примерах осуществления подпоследовательности, локализованные вдали от концов прочтений, могут служить виртуальными UMI, но при этом, возможно, что их относительные положения в прочтении необходимо будет уточнить для того, чтобы определить, получены ли эти прочтения из одного и того же фрагмента.
В качестве виртуальных UMI может быть использована одна или более подпоследовательностей, содержащихся в прочтении. В некоторых примерах осуществления виртуальными UMI служат две подпоследовательности, каждая из которых определена на разных концах исходной молекулы ДНК. В различных примерах осуществления виртуальные UMI содержат приблизительно 24 пар оснований или менее, приблизительно 20 пар оснований или менее, приблизительно 15 пар оснований или менее, приблизительно 10 пар оснований или менее, приблизительно 9 пар оснований или менее, приблизительно 8 пар оснований или менее, приблизительно 7 пар оснований или менее, или приблизительно 6 пар оснований или менее. В некоторых примерах осуществления виртуальные UMI содержат приблизительно от 6 до 10 пар оснований. В других примерах осуществления виртуальные UMI содержат приблизительно от 6 до 24 пар оснований.
Объединение прочтений и получение консенсусных последовательностей
В различных примерах осуществления, в которых применяют UMI, множество последовательностей прочтений, имеющих одинаковый UMI (одинаковые UMI), объединяют, получая одну или более консенсусных последовательностей, которые затем используют для определения последовательности исходной молекулы ДНК. Множества различающихся прочтений могут быть получены из различных представителей одной и той же исходной молекулы ДНК, и эти прочтения могут быть сравнены, и, как показано в настоящей работе, получена консенсусная последовательность. Копии могут быть получены амплификацией исходной молекулы ДНК перед проведением секвенирования; при этом отдельные операции секвенирования выполняют на отдельных продуктах амплификации, каждый из которых имеет общую последовательность исходной молекулы ДНК. Разумеется, при амплификации могут появиться ошибки, вследствие которых последовательности разных продуктов амплификации будут иметь различия. В соответствии с некоторыми методиками секвенирования, такими как секвенирование синтезом согласно методике Illumina, исходная молекула ДНК или продукт ее амплификации образует кластер из ДНК молекул, связанных с участком проточной ячейки. Совокупность молекула кластера дает прочтение. Обычно для составления консенсусной последовательности требуется по меньшей мере два прочтения. Подходящие примеры глубины секвенирования, пригодной для создания консенсусных прочтений при низких аллельных частотах (например, приблизительно 1% или менее) в рассмотренных примерах осуществления, составляют 100, 1000 и 10000.
В некоторых примерах осуществления в консенсусную последовательность включаются нуклеотиды, согласующиеся для 100% прочтений, имеющих общий UMI или комбинацию UMI. В других примерах осуществления критерий консенсуса может составлять менее 100%. Например, может быть использован 90% критерий консенсуса, что означает, что пары оснований, присутствующие в 90% или более прочтений группы, включаются в консенсусную последовательность. В различных примерах осуществления может быть установлен критерий консенсуса, составляющий приблизительно 30%, приблизительно 40%, приблизительно 50%, приблизительно 60%, приблизительно 70%, приблизительно 80%, приблизительно 90%, приблизительно 95% или приблизительно 100%.
Объединение по физическим UMI и виртуальным UMI
Для объединения прочтений, включающих множество UMI, могут быть использованы различные методики. В некоторых примерах осуществления прочтения, имеющие общий физический UMI, могут быть объединены с образованием консенсусной последовательности. В некоторых примерах осуществления, если общий физический UMI представляет собой случайный UMI, то случайный UMI может быть достаточно уникальным для идентификации конкретной молекулы, из которой получен фрагмент ДНК, содержащийся в образце. В других примерах осуществления, если общий физический UMI представляет собой неслучайный UMI, то сам по себе UMI может быть недостаточно уникальным для идентификации конкретной исходной молекулы. В любом случае для создания индекса исходной молекулы физический UMI может быть скомбинирован с виртуальным UMI.
В рассмотренном выше примере последовательности рабочих операций, которая представлена на Фиг. 1В, 3А и 4, некоторые прочтения включают α-ρ-ϕ UMI, в то время как другие включают прочтения β-ϕ-ρ UMI. Физический UMI α служит для получения прочтений, имеющих α. Если все адаптеры, применяемые в последовательности рабочих операций, имеют различные физические UMI (например, различные случайные UMI), то все прочтения, имеющие α в области адаптера, с большой вероятностью получены из одной и той же цепочки фрагмента ДНК. Аналогично, физически UMI β служит для получения прочтений, имеющих β, каждое из которых получено из одной и той же комплементарной цепочки фрагмента ДНК. Таким образом, для получения одной консенсусной последовательности следует объединить все прочтения, включающие α, и для получения другой консенсусной последовательности следует объединить все прочтения, включающие β. Это показано на Фиг. 4В-4С как объединение первого уровня. Поскольку все прочтения в группе получены из одного исходного полинуклеотида, содержащегося в образце, то пары оснований, включаемые в консенсусную последовательность, с большой долей вероятности отражают истинную последовательность исходного полинуклеотида, в то время как пара оснований, исключенная из консенсусной последовательности, с большой долей вероятности отражает вариацию или ошибку, вкравшуюся в последовательность рабочих операций.
Кроме того, виртуальные UMI ρ и ϕ могут предоставлять информацию, подходящую для установления того, получены ли прочтения, включающие один или оба виртуальных UMI, из одного исходного фрагмента ДНК. Поскольку виртуальные UMI ρ и ϕ находятся внутри исходных фрагментов ДНК, применение виртуальных UMI на практике не создает непроизводительных расходов при подготовке или секвенировании. После получения последовательностей физических UMI из прочтений, одна или более подпоследовательностй, находящихся в прочтении, могут быть приняты за виртуальные UMI. Если виртуальные UMI включают достаточное количество пар оснований и имеют одну и ту же относительную локализацию в прочтениях, то они могут уникальным образом идентифицировать прочтения как прочтения, полученные из исходного фрагмента ДНК. Таким образом, прочтения, имеющие один или оба виртуальных UMI ρ и ϕ, могут быть объединены с образованием консенсусной последовательности. Комбинация виртуальных UMI и физических UMI может предоставить информацию для проведения объединения второго уровня, если только один физический UMI определен в консенсусной последовательности первого уровня в каждой цепочке, например, как показано на Фиг. 3А и Фиг. 4А-4С. Однако, в некоторых примерах осуществления объединение второго уровня с помощью виртуального UMI может быть затруднительным, если имеется слишком много исходных молекул ДНК или фрагментация не рандомизирована.
В альтернативных примерах осуществления прочтения, имеющие два физических UMI на обоих концах, такие как прочтения, показанные на Фиг. 3В и Фиг. 4D и 4Е, могут быть объединены в объединение второго уровня на основании комбинации физических UMI и виртуальных UMI. Это особенно полезно, если физические UMI слишком коротки для уникальной идентификации исходных фрагментов ДНК без использования виртуальных UMI. В этих примерах осуществления объединение второго уровня может быть выполнено с применением физических дуплексных UMI, показанных на Фиг. 3В, посредством объединения консенсусных прочтений α-ρ-ϕ-β и консенсусных прочтений β-ϕ-ρ-α из одной молекулы ДНК, в результате чего получают консенсусную последовательность, включающую нуклеотиды, согласующиеся во всех прочтениях.
Применение UMI и схемы объединения, рассмотренной в настоящей работе, позволяет, в различных примерах осуществления, подавлять различные источники ошибок, возникающих в определенной последовательности фрагмента, даже если фрагмент включает аллели с очень низкими аллельными частотами. Прочтения, имеющие общие UMI (физические и/или виртуальные) группируют вместе. Объединение сгруппированных прочтений позволяет устранить вариации (однонуклеотидные вариации (англ. single nucleotide variation, сокращенно SNV) и мелкие вставки), появляющиеся в результате ПЦР, в процессе создания библиотеки, объединения в кластеры, и устранить ошибки секвенирования. На Фиг. 4А-4Е представлено подавление различных источников ошибок при определении последовательности фрагмента двухцепочечной ДНК способом, раскрытым в примере последовательности рабочих операций. Представленные прочтения включают UMI α-ρ-ϕ или β-ϕ-ρ, показанные на Фиг. 3А и 4А-4С, и UMI α-ρ-ϕ-β или β-ϕ-ρ-α, показанные на Фиг. 3В, 4D и 4Е. На Фиг. 3А и 4А-4С UMI α и β представляют собой однонитевые физические UMI. На Фиг. 3В, 4D и 4Е UMI α и β представляют собой дуплексные UMI. Виртуальные UMI ρ и ϕ локализованы на концах фрагмента ДНК.
Способ применения однонитевых физических UMI, показанных на Фиг. 4А-4С, сначала включает объединение прочтений, имеющих один и тот же физический UMI α или β, то есть выполнение объединения первого уровня. В результате объединения первого уровня получают консенсусную последовательность α для прочтений, имеющих физический UMI α, и эти прочтения получены из одной цепочки двухцепочечного фрагмента. В результате объединения первого уровня также получают консенсусную последовательность β для прочтений, имеющих физический UMI β, и эти прочтения получены из другой цепочки двухцепочечного фрагмента. В результате объединения второго уровня, из консенсусной последовательности α и консенсусной последовательности β в способе получают третью консенсусную последовательность. Третья консенсусная последовательность содержит консенсусные пары оснований из прочтений, имеющих одинаковые дуплексные виртуальные UMI ρ и ϕ, и эти прочтения получены из двух комплементарных цепочек исходного фрагмента. Наконец, за последовательность фрагмента двухцепочечной ДНК принимают третью консенсусную последовательность.
Способ применения дуплексных физических UMI, показанный на Фиг. 4D-4E, сначала включает объединение прочтений, имеющих физические UMI α и β в порядке α β в направлении 5'-3', которое показано как объединение первого уровня. В результате объединения первого уровня получают консенсусную последовательность α-β для прочтений, имеющих физические UMI α и β, причем эти прочтения получены из первой цепочки двухцепочечного фрагмента. В результате объединения первого уровня также получают консенсусную последовательность β-α для прочтений, имеющих физические UMI β и α в порядке β α в направлении 5'-3', причем эти прочтения получены из второй цепочки, комплементарной первой цепочке двухцепочечного фрагмента. В результате объединения второго уровня в способе из консенсусной последовательности α-β и консенсусной последовательности β-α получают третью консенсусную последовательность. Третья консенсусная последовательность содержит консенсусные пары оснований из прочтений, имеющих одинаковые дуплексные виртуальные UMI ρ и ϕ, причем эти прочтения получены из двух цепочек фрагмента. Наконец, за последовательность фрагмента двухцепочечной ДНК принимают третью консенсусную последовательность.
На Фиг. 4А представлено подавление ошибок секвенирования при объединении первого уровня. Ошибки секвенирования возникают на платформе секвенирования после получения образца и создания библиотеки (например, при ПЦР амплификации). Ошибки секвенирования могут вводить различные ошибочные основания в различные прочтения. Истинные позитивные основания показаны сплошными буквами, в то время как ложные позитивные основания показаны штриховыми буквами. Ложные позитивные нуклеотиды в различных прочтениях в серии α-ρ-ϕ исключают из консенсусной последовательности α. Истинный позитивный нуклеотид "А", показанный на левых концах серии прочтений α-ρ-ϕ оставляют в консенсусной последовательности α. Аналогично, ложные позитивные нуклеотиды в различных прочтениях в серии β-ϕ-ρ исключают из консенсусной последовательности β, но при этом оставляют истинный позитивный нуклеотид "А". Как показано, при объединении первого уровня ошибки секвенирования могут быть эффективно удалены. На Фиг. 4А также представлено необязательное объединение второго уровня, основанное на виртуальных UMI ρ и ϕ. В результате объединения второго уровня ошибки могут быть дополнительно удалены способом, рассмотренным выше, но эти ошибки не показаны на Фиг. 4А.
Ошибки ПЦР появляются до кластерной амплификации. Таким образом, одна ошибочная пара оснований, попавшая в одноцепочечную ДНК при ПЦР, может быть амплифицирована в процессе кластерной амплификации, и, таким образом, может появиться во множестве кластеров и прочтений. Как показано на Фиг. 4В и Фиг. 4D, ложная позитивная пара оснований, введенная в результате ошибки ПЦР, может появиться во множестве прочтений. Основание "Т" в серии прочтений α-ρ-ϕ (Фиг. 4В) или α-β (Фиг. 4D) и основание "С" в серии прочтений β-ϕ-ρ (Фиг. 4В) или β-α (Фиг. 4D) являются такими ошибками ПЦР. Напротив, как показано на Фиг. 4А, ошибки секвенирования появляются в одном или нескольких прочтениях одной серии. Поскольку ошибки ПЦР секвенирования появляются во множестве прочтений серии, объединение прочтений первого уровня в цепочку не приводит к удалению ошибок ПЦР, несмотря на то, что в результате объединения первого уровня удаляются ошибки секвенирования (например, G и А были удалены из серии α-ρ-ϕ на Фиг. 4В и серии α-ρ на Фиг. 4D). Однако, поскольку ошибка ПЦР введена в одноцепочечную ДНК, комплементарная цепочка исходного фрагмента и полученные из нее прочтения обычно не содержат той же ошибки ПЦР. Таким образом, объединение второго уровня на основании прочтений двух цепочек исходного фрагмента позволяет эффективно удалять ошибки ПЦР, как показано в нижней части Фиг. 4В и 4D.
В некоторых платформах секвенирования появляются гомополимерные ошибки, которые приводят к введению небольших ошибочных вставок в гомополимеры из повторяющихся одиночных нуклеотидов. На Фиг. 4С и 4Е представлена коррекция гомополимерных ошибок способами, рассмотренными в настоящей работе. В серии прочтений α-ρ-ϕ (Фиг. 4С) или α-ρ-ϕ-β (Фиг. 4Е) два нуклеотида "Т" были удалены из второго сверху прочтения, и один нуклеотид "Т" был удален из третьего сверху прочтения. В сериях прочтений β-ϕ-ρ (Фиг. 4С) или β-ϕ-ρ-α (Фиг. 4Е) один нуклеотид "А" был вставлен в первое сверху прочтение. Аналогично ошибкам секвенирования, представленным на Фиг. 4А, гомополимерные ошибки появляются после ПЦР амплификации, в результате чего различные прочтения имеют разные гомополимерные ошибки. В результате объединения первого уровня ошибки вставки могут быть эффективно удалены.
Консенсусные последовательности могут быть получены объединением прочтений, имеющих один или более общих неслучайных UMI и один или более общих виртуальных UMI. Кроме того, как показано ниже, для получения консенсусных последовательностей также может быть использована информация о положении.
Объединение по положению
В некоторых примерах осуществления прочтения обрабатывают для выравнивания по эталонной последовательности с целью определения участков выравнивания прочтений на эталонной последовательности (локализации). Однако в некоторых примерах осуществления, не рассмотренных выше, локализацию устанавливают помощью анализа сходств k-меров и выравнивания прочтение-прочтение. Второй пример осуществления имеет два преимущества: во-первых, с помощью этого способа можно объединять (после исправления ошибок) прочтения, которые не соответствуют эталону из-за гаплотипических различий или транслокаций, и во-вторых, способ не зависит от алгоритма программы выравнивания, что исключает возможность появления обусловленных работой программы выравнивания артефактов (ошибок в программе выравнивания). В некоторых примерах осуществления прочтения, имеющие одинаковую информацию о локализации, могут быть объединены с образованием консенсусных последовательностей для определения последовательности исходных фрагментов ДНК. В некоторых случаях способ выравнивания также называется способом картирования. Последовательности прочтений выравнивают для нахождения их положения (локализации) на эталонной последовательности. Как показано в настоящей работе, для выравнивания прочтений по эталонной последовательности могут быть применены различные инструменты и алгоритмы для проведения выравнивания. Обычно в алгоритмах выравнивания некоторые прочтения успешно выравниваются по эталонной последовательности, в то время как другие прочтения не могут быть успешно выровнены или могут быть плохо выровнены по эталонной последовательности. Прочтения, успешно выровненные по эталонной последовательности, связывают с сайтами на эталонной последовательности. Выровненные прочтения и связанные с ними сайты также называются маркерами (метками) последовательностей. Некоторые последовательности прочтений, содержащие большое количество повторов, бывает труднее выровнять по эталонной последовательности. Если прочтение выровнено по эталонной последовательности так, что количество несоответствующих оснований превышает определенный критерий, прочтение считается плохо выровненным. В различных примерах осуществления прочтения считаются плохо выровненными, если они выровнены с по меньшей мере приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 несоответствиями. В других примерах осуществления прочтения считаются плохо выровненными, если при их выравнивании доля несоответствующих оснований составляет по меньшей мере приблизительно 5%. В других примерах осуществления прочтения считаются плохо выровненными, если при их выравнивании доля несоответствующих оснований составляет по меньшей мере приблизительно 10%, 15% или 20%.
В некоторых примерах осуществления в рассмотренных способах для индексации молекул, из которых получены фрагменты ДНК, комбинируют информацию о положении с информацией физических UMI. Прочтения последовательности, имеющие одинаковые положения прочтения и одинаковый неслучайный или случайный физический UMI, могут быть объединены с образованием консенсусной последовательности для определения последовательности фрагмента или его части. В некоторых примерах осуществления прочтения последовательности, имеющие одинаковые положения прочтения, одинаковый неслучайный физический UMI и случайный физический UMI, могут быть объединены с образованием консенсусной последовательности. В таких примерах осуществления адаптер может включать как неслучайный физический UMI, так и случайный физический UMI. В некоторых примерах осуществления прочтения последовательности, имеющие одинаковые положения прочтения и одинаковый виртуальный UMI, могут быть объединены с образованием консенсусной последовательности.
Информация о положении прочтения может быть получена различными способами. Например, в некоторых примерах осуществления для отыскания информации о положении прочтения могут быть применены геномные координаты. В некоторых примерах осуществления для получения информации о положении прочтения может быть использовано положение на эталонной последовательности, по которой выровнено прочтение. Например, для получения информации о положении прочтения могут быть использованы положения начала и окончания прочтения на хромосоме. В некоторых примерах осуществления положения прочтений считаются одинаковыми, если они имеют идентичную информацию о положении. В некоторых примерах осуществления положения прочтений считаются одинаковыми, если различия между информациями о положении меньше определенного критерия. Например, два прочтения, имеющие стартовые геномные положения, отличающиеся менее чем на 2, 3, 4 или 5 пар оснований, могут считаться прочтениями, имеющими одно и то же положение прочтения. В других примерах осуществления положения прочтений считаются одинаковыми, если информация об их положениях может быть превращена в и соответствовать конкретному пространственному положению. Эталонная последовательность может быть создана до секвенирования, например, она может представлять собой хорошо известную и широко используемую последовательность генома человека, или она может быть определена из прочтений, полученных в процессе секвенирования образца.
Независимо от типа платформы и протокола секвенирования, по меньшей мере часть нуклеиновых кислот, содержащихся в образце, подвергают секвенированию, в котором получают десятки тысяч, сотни тысяч или миллионы прочтений последовательности, например, прочтений 100 п.о. (пар оснований). В некоторых примерах осуществления прочтения последовательности включают приблизительно 20 п.о., приблизительно 25 п.о., приблизительно 30 п.о., приблизительно 35 п.о., приблизительно 36 п.о., приблизительно 40 п.о., приблизительно 45 п.о., приблизительно 50 п.о., приблизительно 55 п.о., приблизительно 60 п.о., приблизительно 65 п.о., приблизительно 70 п.о., приблизительно 75 п.о., приблизительно 80 п.о., приблизительно 85 п.о., приблизительно 90 п.о., приблизительно 95 п.о., приблизительно 100 п.о., приблизительно 110 п.о., приблизительно 120 п.о., приблизительно 130, приблизительно 140 п.о., приблизительно 150 п.о., приблизительно 200 п.о., приблизительно 250 п.о., приблизительно 300 п.о., приблизительно 350 п.о., приблизительно 400 п.о., приблизительно 450 п.о., приблизительно 500 п.о., приблизительно 800 п.о., приблизительно 1000 п.о. или приблизительно 2000 п.о.
В некоторых примерах осуществления прочтения выравнивают по эталонному геному, например, hg19. В других примерах осуществления прочтения выравнивают по части эталонного генома, например, хромосоме или сегменту хромосомы. Прочтения, уникальным образом локализованные на эталонном геноме, называются маркерами последовательности. В одном из примеров осуществления из прочтений получены по меньшей мере приблизительно 3×106 установленных маркеров последовательности, по меньшей мере приблизительно 5×106 установленных маркеров последовательности, по меньшей мере приблизительно 8×106 установленных маркеров последовательности, по меньшей мере приблизительно 10×106 установленных маркеров последовательности, по меньшей мере приблизительно 15×106 установленных маркеров последовательности, по меньшей мере приблизительно 20×106 установленных маркеров последовательности, по меньшей мере приблизительно 30×106 установленных маркеров последовательности, по меньшей мере приблизительно 40×106 установленных маркеров последовательности или по меньшей мере приблизительно 50×106 установленных маркеров последовательности, которые уникальным образом локализованы на эталонном геноме.
Применение
В различных вариантах применения стратегии коррекции ошибок, рассмотренные в настоящей работе, могут дать одно или более следующих преимуществ: (i) обнаружение соматических мутаций с очень низкой аллельной частотой, (ii) уменьшение продолжительности цикла за счет уменьшения количества ошибок фазирования / предварительного фазирования и/или (iii) увеличение длины прочтения за счет повышения качества распознаваний оснований в последней части прочтений и т.д. Применение и обоснование необходимости обнаружения соматических мутаций с низкой аллельной частотой обсуждены выше.
В некоторых примерах осуществления методики, рассмотренные в настоящей работе, позволяют производить надежное распознавание аллелей, частота которых составляет приблизительно 2% или менее или приблизительно 1% или менее, или приблизительно 0,5% или менее. Такие низкие частоты обычны для вкДНК, получаемых из опухолевых клеток пациентов, страдающих раковыми заболеваниями. В некоторых примерах осуществления методики, рассмотренные в настоящей работе, позволяют идентифицировать редкие штаммы в метагеномных образцах, а также обнаруживать редкие вариации в вирусных или других популяциях, в тех случаях, когда, например, пациент был инфицирован множеством вирусных штаммов и/или подвергался медицинскому лечению.
В некоторых примерах осуществления методики, рассмотренные в настоящей работе, позволяют сократить продолжительность цикла химического секвенирования. При укороченной продолжительности цикла повышается вероятность возникновения ошибок секвенирования, которые могут быть исправлены способами, рассмотренными выше.
В некоторых примерах осуществления, включающих применение UMI, длинные прочтения могут быть получены секвенированием парных концов с использованием прочтений асимметричной длины, полученных для пары прочтений парных концов (ПК) на двух концах сегмента. Например, пара прочтений, содержащая 50 п.о. в одном прочтении парных концов и 500 п.о. в другом прочтении парных концов, может быть "сшита" с другой парой прочтений с образованием длинного прочтения, содержащего 1000 п.о. В таких примерах осуществления может быть достигнута более высокая скорость секвенирования при определении длинных фрагментов с низкими аллельными частотами.
На Фиг. 5 схематично представлен пример эффективного получения длинных прочтений парных концов в областях применения указанного типа с помощью физических UMI и виртуальных UMI. Библиотеки, составленные из обеих цепочек одинаковых фрагментов ДНК, собирают в кластеры в проточной ячейке. Размер вставки в библиотеке составляет более 1 тысячи пар оснований (Kb). Секвенирование выполняют с использованием прочтений асимметричной длины (например, Прочтение 1=500 п.о., Прочтение 2=50 п.о.) для обеспечения нужного качества прочтений длиной 500 п.о. Сшивание двух цепочек позволяет создавать прочтения ПК длиной 1000 п.о. посредством секвенирования всего лишь 500+50 п.о.
Образцы
Образцы, используемые для определения последовательности фрагмента ДНК, могут включать образцы, взятые из любой клетки, жидкости, ткани или органа, которые включают нуклеиновые кислоты, интересующие последовательности которых нужно определить. В некоторых примерах осуществления, включающих диагностику раковых заболеваний, из физиологических жидкостей субъекта, например, крови или плазмы крови, может быть получена циркулирующая опухолевая ДНК. В некоторых примерах осуществления, включающих диагностику плода, из физиологической жидкости матери предпочтительно получают внеклеточные нуклеиновые кислоты, например, внеклеточную ДНК (вкДНК). Внеклеточные нуклеиновые кислоты, включающие внеклеточную ДНК, могут быть получены различными способами, известными в данной области техники, из биологических образцов, примеры которых включают, без ограничений, плазму крови, сыворотку крови и мочу (см., например, Fan с соавт., Proc. Natl. Acad. Sci. 105:16266-16271 [2008]; Koide с соавт., Prenatal Diagnosis 25:604-607 [2005]; Chen с соавт., Nature Med. 2: 1033-1035 [1996]; Lo с соавт., Lancet 350: 485-487 [1997]; Botezatu с соавт., Clin Chem. 46: 1078-1084, 2000; и Su с соавт., J. Mol. Diagn. 6: 101-107 [2004]).
В различных примерах осуществления, перед использованием образца (например, перед созданием библиотеки секвенирования), присутствующие в образце нуклеиновые кислоты (например, ДНК или РНК) могут быть специфичным или неспецифичным образом обогащены. Неспецифичное обогащение ДНК, содержащейся в образце, производят полногеномной амплификацией геномных фрагментов ДНК образца, которое может быть применено для повышения концентрации содержащейся в образце ДНК перед созданием библиотеки секвенирования вкДНК. Способы полногеномной амплификации известны в данной области техники. Примеры способов полногеномной амплификации включают ПЦР с вырожденным олигонуклеотидным праймером (англ. degenerate oligonucleotide-primed PCR, сокращенно DOP), ПЦР с удлинением праймера (англ. primer extension, сокращенно PEP) и амплификация с множественным вытеснением цепи (англ. multiple displacement amplification, сокращено MDA). В некоторых примерах осуществления обогащение ДНК в образце не производят.
Образцы, включающие нуклеиновые кислоты, обрабатываемые способами, рассмотренными в настоящей работе, обычно включают биологический образец ("испытуемый образец"), рассмотренный выше. В некоторых примерах осуществления нуклеиновые кислоты, подвергаемые секвенированию, очищают или выделяют любым из множества хорошо известных способов.
Соответственно, в некоторых примерах осуществления образец включает или по существу состоит из очищенного или выделенного полинуклеотида, или он может включать образцы, такие как образец ткани, образец биологической жидкости, образец клетки и подобные образцы. Подходящие образцы биологических жидкостей включают, без ограничений, кровь, плазму крови, сыворотку крови, пот, слезную жидкость, мокроту, мочу, ушные выделения, лимфу, слюну, спинномозговую жидкость, продукты деструкции, суспензию костного мозга, влагалищные выделения, трансцервикальные смывы, мозговую жидкость, асцит, молоко, секреторные выделения дыхательных путей, кишечника и мочеполовых путей, околоплодную жидкость, молоко и образцы лейкофореза. В некоторых примерах осуществления образец представляет собой образец, который легко получают при помощи неинвазивных процедур, например, кровь, плазму крови, сыворотку крови, пот, слезную жидкость, мокроту, мочу, стул, ушные выделения, слюну или кал. В некоторых примерах осуществления образец представляет собой образец периферической крови или фракции плазмы крови и/или сыворотки крови из образца периферической крови. В других примерах осуществления биологический образец представляет собой смыв или мазок, образец биопсии или клеточную культуру. В другом примере осуществления образец представляет собой смесь двух или более биологических образцов, например, биологический образец может включать два или более образца биологических жидкостей, образец ткани и образец клеточной культуры. Используемые в настоящей работе термины "кровь", "плазма крови" и "сыворотка крови" охватывают фракции или обработанные части крови. Аналогично, если образец отобран при проведении биопсии, смыва, мазка и т.д., то термин "образец" охватывает обработанные фракции или части, полученные проведении биопсии, смыва, мазка и т.д.
В некоторых примерах осуществления образцы могут быть получены из источников, примеры которых включают, без ограничений, образцы различных индивидуумов, образцы, полученные на различных этапах развития одного или разных индивидуумов, образцы, полученные от разных индивидуумов, имеющих заболевания (например, индивидуумов, которые предположительно имеют генетическое нарушение), нормальных индивидуумов, образцы, полученные на различных этапах заболевания индивидуума, образцы, полученные от индивидуума, подвергаемого различным видам лечения заболевания, образцы, полученные от индивидуумов, подвергаемых действию различных факторов окружающей среды, образцы, полученные от индивидуумов с предрасположенностью к патологии, образцы, полученные от индивидуумов, на которых действует возбудитель инфекционного заболевания, и подобные образцы.
В одном из иллюстративных и неограничивающих примеров осуществления образец представляет собой образец, полученный из организма матери, то есть полученный из организма беременной самки, например беременной женщины. В этом примере, для проведения пренатальной диагностики потенциальных хромосомных аномалий плода, образец может быть проанализирован способами, рассмотренными в настоящей работе. Образец, полученный из организма матери, может представлять собой образец ткани, образец биологической жидкости или клеточный образец. Неограничивающие примеры биологических жидкостей включают кровь, плазму крови, сыворотку крови, пот, слезную жидкость, мокроту, мочу, ушные выделения, лимфу, слюну, спинномозговую жидкость, продукты деструкции, суспензию костного мозга, влагалищные выделения, трансцервикальные смывы, мозговую жидкость, асцит, молоко, секреторные выделения дыхательных путей, кишечника и мочеполовых путей и образцы лейкофореза.
В некоторых примерах осуществления образцы также могут быть получены из культивируемых in vitro тканей, клеток или других источников, содержащих полинуклеотиды. Культивируемые образцы могут быть отобраны из источников, примеры которых включают, без ограничений, культуры (например, ткани или клетки), сохраняемые в различных средах и условиях (например, рН, давлении или температуре), культуры (например, ткани или клетки), сохраняемые в течение различных периодов времени, культуры (например, ткани или клетки), на которые воздействуют различные факторы или реагенты (например, потенциальный лекарственный препарат или модулятор), или культуры различных типов тканей и/или клеток.
Способы выделения нуклеиновых кислот из биологических источников хорошо известны, и выбор способа зависит от природы источника. Специалист в данной области техники может при необходимости с легкостью выделить нуклеиновые кислоты из источника для выполнения способа, рассмотренного в настоящей работе. В некоторых примерах может быть предпочтительной фрагментация молекул нуклеиновых кислот в образце нуклеиновой кислоты. Фрагментация может быть случайной или специфичной; в последнем случае ее проводят, например, посредством расщепления под действием рестрикционной эндонуклеазы. Способы случайной фрагментации хорошо известны в данной области техники, и включают, например, ограниченное расщепление ДНКзой, щелочную обработку и физическое расщепление.
Создание библиотеки секвенирования
В различных примерах осуществления секвенирование может быть выполнено на различных платформах секвенирования, для которых требуется создание библиотеки секвенирования. Создание обычно включает фрагментацию ДНК (разрушение ультразвуком, распылением или разрезанием), последующую репарацию ДНК и зачистку концов (тупого конца или А-липкого конца) и лигирование адаптера, специфичное для данной платформы. В одном из примеров осуществления в способах, рассмотренных в настоящей работе, могут быть использованы методики секвенирования нового поколения (СНП), которые позволяют индивидуально секвенировать множество образцов, как в виде геномных молекул (однонитевое секвенирование), так и объединенных образцов, включающих проиндексированные геномные молекулы (например, мультиплексное секвенирование), за один цикл секвенирования. В таких способах может быть получено до нескольких миллиардов прочтений последовательностей ДНК. В различных примерах осуществления последовательности геномных нуклеиновых кислот и/или проиндексированных геномных нуклеиновых кислот могут быть определены с помощью, например, методик секвенирования нового поколения (СНП), рассмотренных в настоящей работе. Как указано в настоящей работе, в различных примерах осуществления анализ огромного количества данных о последовательности, полученный способами СНП, может быть выполнен с помощью одного или более процессоров.
В различных примерах осуществления применение таких методик секвенирования не включает создание библиотек секвенирования.
Однако в некоторых примерах осуществления способы секвенирования, рассматриваемые в настоящей работе, включают создание библиотек секвенирования. Согласно одной иллюстративной методике, создание библиотеки секвенирования включает получение случайного набора модифицированных адаптером фрагментов ДНК (например, полинуклеотидов), готовых к секвенированию. Содержащие полинуклеотиды библиотеки секвенирования могут быть созданы из ДНК или РНК, включая эквиваленты и аналоги, как ДНК, так и кДНК, например, ДНК или кДНК, которые комплементарны или копируют ДНК, полученные из матрицы РНК, под действием обратной транскриптазы. Полинуклеотиды могут быть получены в двухцепочечной форме (например, дцДНК, такие как геномные фрагменты ДНК, кДНК, продукты ПЦР амплификации и подобные вещества) или в некоторых примерах осуществления полинуклеотиды могут быть получены в одноцепочечной форме (например, однонитевая ДНК, РНК и т.д.) и затем могут быть превращены в форму дцДНК. В качестве иллюстративного примера можно отметить, что в некоторых примерах осуществления молекулы одноцепочечной мРНК могут быть скопированы в молекулы двухцепочечной кДНК, подходящие для создания библиотеки секвенирования. Точная последовательность первичных полинуклеотидных молекул обычно не важна для способа создания библиотеки и может быть как известной, так и неизвестной. В одном из примеров осуществления полинуклеотидные молекулы представляют собой молекулы ДНК. В частности, в некоторых примерах осуществления полинуклеотидные молекулы составляют полный генетический комплект организма или по существу полный генетический комплект организма и представляют собой молекулы геномной ДНК (например, клеточной ДНК, внеклеточной ДНК (вкДНК) и т.д.), которые обычно включают и интронную последовательность, и экзонную последовательность (кодирующую последовательность), а также некодирующие регуляторные последовательности, такие как промоторные и энхансерные последовательности. В некоторых примерах осуществления первичные полинуклеотидные молекулы включают молекулы ДНК генома человека, например, молекулы вкДНК, находящиеся в периферической крови беременного субъекта.
Созданию библиотек секвенирования для некоторых платформ секвенирования СНП способствует использование полинуклеотидов, включающих определенный диапазон размеров фрагмента. Создание таких библиотек обычно включает фрагментацию крупных полинуклеотидов (например, клеточной геномной ДНК) с образованием полинуклеотидов, имеющих размер в целевом диапазоне.
В способах и системах секвенирования, рассмотренных в настоящей работе, могут быть использованы прочтения парных концов. Длина фрагмента или вставки больше длины прочтения и иногда больше суммы длин двух прочтений.
В некоторых иллюстративных примерах осуществления нуклеиновая кислота (кислоты) образца получена в виде геномной ДНК, которую подвергают фрагментации с образованием фрагментов, длина которых превышает приблизительно 50, 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 2000 или 5000 пар оснований, которые могут быть легко обработаны способами СНП. В некоторых примерах осуществления прочтения парных концов получены из вставок, содержащих приблизительно 100-5000 п.о. В некоторых примерах осуществления длины вставок составляют приблизительно 100-1000 п.о. Иногда их получают в виде обычных прочтений парных концов с короткими вставками. В некоторых примерах осуществления длины вставок составляют приблизительно 1000-5000 п.о. Иногда их получают в виде прочтений сопряженных пар с длинными вставками, рассмотренных выше.
В некоторых примерах осуществления длинные вставки конструируют для анализа очень длинных последовательностей. В некоторых примерах осуществления для получения прочтений, отделенных друг от друга тысячами пар оснований могут быть использованы прочтения сопряженных пар. В этих примерах осуществления вставки или фрагменты включают от сотен до тысяч пар оснований и имеют два адаптера с биотиновым соединением на двух концах вставки. Затем адаптерами с биотиновым соединением соединяют два конца вставки с образованием закольцованной молекулы, которую затем дополнительно подвергают фрагментации. Субфрагмент, включающий адаптер с биотиновым соединением, и два конца первоначальной вставки выбирают для секвенирования на платформе, которая предназначена для секвенирования более коротких фрагментов.
Фрагментация может быть выполнена любым из множества способов, известных специалистам в данной области техники. Например, фрагментация может быть выполнена механическими средствами, примеры которых включают, без ограничений, распыление, разрушение ультразвуком и гидроразрушение. Однако при механической фрагментации основная цепь ДНК обычно расщепляется по С-О, Р-O и С-С связям, что приводит к образованию гетерогенной смеси тупых и 3'- и 5'-липких концов с разрушенными С-О, Р-O и/или С-С связями (см., например, Alnemri, Liwack, J. Biol. Chem. 265:17323-17333 [1990]; Richards, Boyer, J. Mol. Biol. 11:327-240 [1965]), которые, возможно, будет необходимо восстановить, поскольку в них может отсутствовать 5'-фосфат, необходимый для протекания последующих ферментативных реакций, например, лигирования (пришивания) адаптеров секвенирования, которые нужны для получения ДНК для секвенирования.
Напротив, вкДНК, обычно существует в виде фрагментов, содержащих менее приблизительно 300 пар оснований, и, следовательно, для создания библиотеки секвенирования с использованием образцов вкДНК фрагментация обычно не требуется.
Обычно независимо от того, были ли полинуклеотиды фрагментированы искусственно (например, фрагментированы in vitro) или существовали в природе в виде фрагментов, их превращают в ДНК с тупыми концами, содержащую 5'-фосфаты и 3'-гидроксил. В стандартных протоколах, например, протоколах для секвенирования с применением, например, платформы Illumina, рассмотренной выше в примере последовательности рабочих операций, показанной на Фиг. 1А и 1В, для пользователей описаны этапы создания библиотеки, включающие репарацию концов ДНК, находящейся в образце, очистку продуктов репарации концов перед аденилированием или образованием dA-хвоста на 3'-концах и очистку продуктов образования dA-хвоста перед прикреплением адаптера.
Различные примеры осуществления способов создания библиотеки секвенирования, рассмотренные в настоящей работе, позволяют не выполнять один или более этапов, которые в стандартных протоколах обычно необходимо выполнять для получения модифицированного ДНК продукта, который может быть подвергнут секвенированию способами СНП. Примерами способов создания библиотеки секвенирования являются сокращенный способ (англ. abbreviated, сокращенно ABB), одноэтапный способ и двухэтапный способ, которые раскрыты в патентной заявке US 13/555037, поданной 20 июля 2012 г., содержание которой полностью включено в настоящее описание посредством ссылки.
Способы секвенирования
В рассмотренных в настоящей работе способах и установке может быть применена методика секвенирования нового поколения (СНП), которая позволяет производить параллельное массовое секвенирование. В некоторых примерах осуществления клонально амплифицированные ДНК матрицы или единичные молекулы ДНК подвергают параллельному массовому секвенированию внутри проточной ячейки (например, рассмотренной в публикации Volkerding с соавт.Clin Chem 55:641-658 [2009]; Metzker М. Nature Rev. 11:31-46 [2010]). Методики секвенирования СНП включают, без ограничений, пиросеквенирование, секвенирование синтезом с обратимыми окрашивающими терминаторами, секвенирование лигированием олигонуклеотидного зонда и ионное полупроводниковое секвенирование. ДНК из индивидуальных образцов могут быть подвергнуты секвенированию по отдельности (однонитевое секвенирование), или ДНК из множества образцов могут быть объединены и подвергнуты секвенированию в виде индексированных геномных молекул (мультиплексное секвенирование) за один цикл секвенирования с образованием до нескольких сотен миллионов прочтений последовательностей ДНК. Примеры методик секвенирования, которые могут быть применены для получения информации о последовательности способом согласно настоящему изобретению, более подробно рассмотрены ниже.
Некоторые методики секвенирования коммерчески доступны; их примеры включают платформу секвенирования гибридизацией, созданную Affymetrix Inc. (Sunnyvale, СА), и платформы секвенирования синтезом, созданные 454 Life Sciences (Bradford, СТ), Illumina/Solexa (Hayward, СА) и Helicos Biosciences (Cambridge, MA), и платформу секвенирования лигированием, созданную Applied Biosystems (Foster City, СА), рассмотренные ниже. Кроме секвенирования единичных молекул, выполняемого синтезом, которое было разработано Компанией Helicos Biosciences, другие методики секвенирования единичных молекул включают, без ограничений, методику SMRT™, созданную Pacific Biosciences, методику ION TORRENT™ и секвенирование в нанопорах, разработанное, например, Oxford Nanopore Technologies.
Несмотря на то, что автоматизированный способ Сенгер считается методикой «первого поколения», секвенирование по Сенгеру, включающее автоматизированное секвенирование по Сенгеру, также может быть использовано в способах, рассмотренных в настоящей работе. Дополнительные подходящие способы секвенирования включают, без ограничений, методики визуализации нуклеиновых кислот, например, атомно-силовую микроскопию (англ. atomic force microscopy, сокращенно AFM) или трансмиссионную электронную микроскопию (англ. transmission electron microscopy, сокращенно ТЕМ). Иллюстративные методики секвенирования более подробно рассмотрены ниже.
В некоторых примерах осуществления способы согласно изобретению включают получение информации о последовательности нуклеиновых кислот в испытуемом образце параллельным массовым секвенированием миллионов фрагментов ДНК, а именно секвенированием синтезом и секвенированием с использованием обратимого терминатора, разработанными Illumina (которые, например, рассмотрены в публикации Bentley с соавт., Nature 6:53-59 [2009]). Матричная ДНК может представлять собой геномную ДНК, например, клеточную ДНК или вкДНК. В некоторых примерах осуществления в качестве матрицы применяют геномную ДНК из выделенных клеток, и эту ДНК фрагментируют с образованием фрагментов длиной несколько сотен пар оснований. В других примерах осуществления в качестве матрицы применяют вкДНК или циркулирующую опухолевую ДНК (цоДНК), и фрагментация не требуется, поскольку вкДНК или цоДНК существуют в виде коротких фрагментов. Например, вкДНК плода циркулирует в кровеносной системе в виде фрагментов длиной приблизительно 170 пар оснований (п.о.) (Fan с соавт., Clin Chem 56:1279-1286 [2010]), и перед секвенированием фрагментация этой ДНК не требуется. Методика секвенирования, созданная Illumina, основана на присоединении фрагментированной геномной ДНК к плоской, оптически прозрачной поверхности, на которой закреплены олигонуклеотидные якоря. Концы матричной ДНК подвергают репарации с образованием 5'-фосфорилированных тупых концов, и для присоединения одного основания А к 3'-концу тупых фосфорилированных фрагментов ДНК используют полимеразное действие фрагмента Кленова. Это присоединение подготавливает фрагменты ДНК для сшивания с олигонуклеотидными адаптерами, которые имеют липкий конец из одного Т основания на 3'-конце, что повышает эффективность сшивания. Адаптерные олигонуклеотиды комплементарны якорным олигонуклеотидам, находящимся в проточной ячейке. В условиях ограниченного разбавления модифицированную адаптером одноцепочечную матричную ДНК помещают в проточную ячейку и иммобилизуют гибридизацией с якорными олигонуклеотидами. Присоединенные фрагменты ДНК подвергают достройке и мостиковой амплификации для достижения сверхвысокой плотности секвенирования в проточной ячейке, содержащей сотни миллионов кластеров, каждый из которых содержит приблизительно 1000 копий одной и той же матрицы. В одном из примеров осуществления фрагментированную случайным образом геномную ДНК подвергают ПЦР амплификации, а затем ее подвергают кластерной амплификации. В альтернативном варианте применяют безамплификационное создание геномной библиотеки, и фрагментированную случайным образом геномную ДНК обогащают, применяя только кластерную амплификацию (Kozarewa с соавт., Nature Methods 6:291-295 [2009]). В некоторых примерах применения матрицы секвенируют, применяя надежную методику секвенирования ДНК синтезом с использованием четырех меченых (окрашенных) нуклеотидов, в которой применяют обратимые терминаторы, меченые удаляемым флуоресцентным красителем. Высокочувствительное флуоресцентное обнаружение выполняют, применяя лазерную накачку и явление полного внутреннего отражения. Короткие прочтения последовательности, содержащие от приблизительно десятков до нескольких сотен пар оснований, выравнивают по эталонному геному, и с помощью специально разработанного программного конвейера для анализа данных идентифицируют уникальное расположение коротких прочтений последовательности на эталонном геноме. После завершения первого прочтения, матрицы могут быть восстановлены in situ для выполнения второго прочтения с противоположного конца фрагментов. Таким образом, может быть использовано либо секвенирование одного конца, либо секвенирование парных концов фрагментов ДНК.
В различных примерах осуществления изобретения может быть использовано секвенирование синтезом, которое позволяет производить секвенирование парных концов. В некоторых примерах осуществления платформа Illumina для секвенирования синтезом включает объединение фрагментов в кластеры. Объединение в кластеры представляет собой процесс, при котором каждая молекула фрагмента подвергается изотермической амплификации. В некоторых примерах осуществления, таких как рассматриваемый пример, фрагмент содержит два разных адаптера, присоединенные к двум концам фрагмента; эти адаптеры позволяют фрагменту гибридизироваться с двумя разными олигонуклеотидами, находящимися на поверхности дорожки в проточной ячейке. Фрагмент дополнительно включает или соединен с двумя последовательностями индексов, которые расположены на двух концах фрагмента, и эти последовательности индексов представляют собой метки для идентификации различных образцов при мультиплексном секвенировании. В некоторых платформах секвенирования подвергаемые секвенированию фрагменты, находящиеся на обоих концах, также называются вставками.
В одном из примеров осуществления проточная ячейка для объединения в кластеры платформы Illumina представляет собой стеклянный слайд с дорожками. Каждая дорожка представляет собой стеклянный канал, на который нанесено сплошное покрытие из двух типов олигонуклеотидов (например, олигонуклеотидов Р5 и Р7'). Первый из двух типов находящихся на поверхности олигонуклеотидов способен к гибридизации. Этот олигонуклеотид комплементарен первому адаптеру, находящемуся на одном конце фрагмента. Полимераза создает цепочку, комплементарную гибридизованному фрагменту. Двухцепочечную молекулу подвергают денатурации, и исходную матричную цепочку удаляют промывкой. Оставшуюся цепочку подвергают клональной амплификации с использованием мостика параллельно с множеством других оставшихся цепочек.
В процессе мостиковой амплификации и в других способах секвенирования, включающих объединение в кластеры, цепочка складывается, и вторая адаптерная область на втором конце цепочки гибридизуется с олигонуклеотидами второго типа, находящимися на поверхности проточной ячейки. Полимераза генерирует комплементарную цепочку, образуя двухцепочечную мостиковую молекулу. Эту двухцепочечную молекулу подвергают денатурации, получая две одноцепочечные молекулы, закрепленные в проточной ячейке с помощью двух разных олигонуклеотидов. Этот процесс повторяют снова и снова, и он протекает одновременно в миллионах кластеров, приводя к клональной амплификации всех фрагментов. По завершении мостиковой амплификации, обратные цепочки отщепляют и удаляют промывкой, оставляя только прямые цепочки. Для предотвращения нежелательного примирования, 3'-концы блокируют.
После объединения в кластеры начиняют секвенирование с достройкой первого праймера секвенирования для получения первого прочтения. В каждом цикле меченые флуоресцентной меткой нуклеотиды конкурируют за присоединение к растущей цепи. Присоединяется лишь один в соответствии с последовательностью матрицы. После присоединения каждого нуклеотида кластер возбуждают с помощью источника света, и происходит испускание характеристического флуоресцентного сигнала. Количество циклов определяет длину прочтения. Длина волны и интенсивность испускаемого сигнала позволяют распознавать основания. В заданном кластере все идентичные цепочки прочитываются одновременно. Сотни миллионов кластеров подвергаются параллельному массовому секвенированию. По завершении первого прочтения, продукт прочтения удаляют промывкой.
В следующем этапе протоколов, включающих использование двух индексированных праймеров, праймер с индексом 1 вводят в область индекса 1 матрицы и гибридизируют с этой областью. Области индексов обеспечивают идентификацию фрагментов, необходимую для демультиплексирования (разделения на потоки) образцов в способе мультиплексного секвенирования. Прочтение с индексом 1 получают аналогично первому прочтению. После завершения прочтения с индексом 1, продукт прочтения удаляют промывкой и снимают защиту с 3'-конца цепочки. Матричная цепочка затем складывается и связывается со вторым олигонуклеотидом, фиксированным в проточной ячейке. Последовательность индекса 2 прочитывается тем же образом, что и последовательность индекса 1. Затем, при выполнении заключительного этапа, продукт прочтения с индексом 2 удаляют промывкой.
После прочтения двух индексов, прочтение 2 инициирует достройку вторых олигонуклеотидов, находящихся в проточной ячейке, под действием полимераз, что приводит к образованию двухцепочечного мостика. Эта двухцепочечная ДНК денатурирована и имеет блокированный 3'-конец. Исходная прямая цепочка отщепляется, и ее удаляют промывкой; при этом обратная цепочка остается. Прочтение 2 начинается с введения праймера секвенирования прочтения 2. Как и при операциях с прочтением 1, этапы секвенирования повторяют до достижения требуемой длины. Продукт прочтения 2 удаляют промывкой. В целом, в способе образуются миллионы прочтений, которые представляют все фрагменты. Последовательности из объединенных библиотек образца разделяют на основании информации, содержащейся в уникальных индексах, которые были введены в процессе подготовки образца. Для каждого образца группируют прочтения со схожей протяженностью (хроматографических) пиков оснований. Прямые и обратные прочтения объединяют в пары, создавая непрерывные последовательности. Эти непрерывные последовательности выравнивают по эталонному геному для идентификации варианта.
Рассмотренный выше пример секвенирования синтезом включает прочтение парных концов, которое применяют во множестве примеров осуществления способов согласно изобретению. Секвенирование парных концов включает 2 прочтения, полученные от двух концов фрагмента. Прочтения парных концов применяют для атрибуции неоднозначных выравниваний. Секвенирование парных концов позволяет пользователю выбирать длину вставки (или фрагмента, подвергаемого секвенированию) и секвенировать любой из концов вставки, получая высококачественные, подходящие для выравнивания данные о последовательности. Поскольку расстояние между каждыми парными прочтениями известно, в алгоритмах выравнивания эта информация может быть использована для более точной локализации прочтений в повторяющихся областях. Это приводит к улучшенному выравниванию прочтений, в особенности, в повторяющихся областях генома, плохо поддающихся секвенированию. С помощью секвенирования парных концов можно обнаруживать перестановки, включая инсерции и делеции (инсерционно-делеционный полиморфизм) и инверсии.
В прочтениях парных концов могут быть использованы вставки различной длины (т.е. подвергаемые секвенированию фрагменты различного размера). Согласно стандартному значению, используемому в настоящем описании, прочтения парных концов применяют для соотнесения с прочтениями, получаемыми из вставок различной длины. В некоторых примерах для отличия прочтений парных концов с короткими вставками от прочтений парных концов с длинными вставками последние специально названы прочтениями сопряженных пар. В некоторых примерах осуществления, включающих прочтения сопряженных пар, два адаптера с биотиновым соединением сначала присоединяют к двум концам относительно длинной вставки (например, размером несколько тысяч пар оснований (Kb)). Затем адаптеры с биотиновым соединением соединяют два конца вставки с образованием кольцеобразной молекулы. Затем, при дальнейшей фрагментации кольцеобразной молекулы может быть получен субфрагмент, включающий адаптеры с биотиновым соединением. Субфрагмент, включающий два конца исходного фрагмента в противоположном последовательности порядке, может быть подвергнут секвенированию согласно процедуре, рассмотренной выше при описании секвенирования парных концов с короткими вставками. Более подробно секвенирование сопряженных пар с помощью платформы Illumina имеется в онлайн публикации, содержание которой полностью включено в настоящее описание посредством ссылки, по приведенному ниже адресу: res.illumina.com/documents/products/technotes/technote_nextera_matepair_data_proces sing.pdf
После секвенирования фрагментов ДНК, прочтения последовательности заранее заданной длины, например, 100 п.о., локализуют с помощью картирования (выравнивания) по известному эталонному геному. Картированные прочтения и соответствующие им локализации на эталонной последовательности также называются метками. В другом примере осуществления этой процедуры локализацию производят с помощью общих k-меров и выравнивания прочтение-прочтение. В анализах согласно многим примерам осуществления, рассмотренным в настоящей работе, используют прочтения, которые или плохо выравниваются, или не могут быть выровнены, а также выровненные прочтения (метки). В одном из примеров осуществления эталонная геномная последовательность представляет собой последовательность NCBI36/hg18, которая представлена в Интернете на сайте genome.ucsc.edu/cgi-bin/hgGateway?org=Human&db=hg18&hgsid=166260105). В альтернативном варианте эталонная геномная последовательность представляет собой последовательность GRCh37/hg19 или GRCh38, которые представлены в Интернете на сайте genome.ucsc.edu/cgi-bin/hgGateway. Другие источники общедоступной информации о последовательностях включают GenBank, dbEST, dbSTS, EMBL (сокращение от "European Molecular Biology Laboratory", т.е. Европейская лаборатория молекулярной биологии) и DDBJ (сокращение от "DNA Databank of Japan", т.е. Банк ДНК данных Японии). Доступен ряд компьютерных алгоритмов для выравнивания последовательностей, которые включают, без ограничений, BLAST (Altschul с соавт., 1990), BLITZ (MPsrch) (Sturrock & Collins, 1993), FASTA (Person & Lipman, 1988), BOWTIE (Langmead с соавт., Genome Biology 10:R25.1-R25.10 [2009]) или ELAND (Illumina, Inc., San Diego, CA, USA). В одном из примеров осуществления один из концов клонально удлиненных копий молекул вкДНК плазмы крови подвергали секвенированию и обрабатывали с помощью биоинформатического анализа выравнивания, предназначенного для анализатора генома Illumina, в котором применяют программное обеспечение для эффективного крупномасштабного выравнивания баз данных нуклеотидов (англ. Efficient Large-Scale Alignment of Nucleotide Databases, сокращенно ELAND).
В одном из иллюстративных неограничивающих примеров осуществления способы, рассмотренные в настоящей работе, включают получение информации о последовательностях нуклеиновых кислот, содержащихся в испытуемом образце, посредством использования методики секвенирования единичной молекулы, основанной на методике истинного секвенирования единичной молекулы Helicos (англ. Helicos True Single Molecule Sequencing, сокращенно tSMS) (например, рассмотренной в публикации Harris T.D. с соавт., Science 320:106-109 [2008]). Согласно tSMS методике, образец ДНК расщепляют на цепочки, содержащие приблизительно от 100 до 200 нуклеотидов, и к 3'-концу каждой цепочки ДНК присоединяют поли-А последовательность. Каждую цепочку метят присоединением аденозинового нуклеотида, меченого флуоресцентной меткой. Затем цепочки ДНК гибридизируются с нуклеотидами проточной ячейки, которая содержит миллионы сайтов захвата олиго-Т, иммобилизованных на поверхности проточной ячейки. В некоторых примерах осуществления плотность матриц может составлять приблизительно 100 миллионов матриц/см2. Затем проточную ячейку помещают в устройство, например, секвенатор HeliScope™, и освещают лазерным лучом поверхность проточной ячейки, обозначая положение каждой матрицы. Камера на приборе с зарядовой связью может картировать (локализовать) положения матриц на поверхности проточной ячейки. Затем флуоресцентную метку матрицы отщепляют и удаляют промывкой. Реакция секвенирования начинается с введения ДНК полимеразы и меченого флуоресцентной меткой нуклеотида. Праймером служит олиго-Т нуклеиновая кислота. Полимераза вводит меченые нуклеотиды в праймер матрично-управляемым образом. Полимеразу и оставшиеся свободными нуклеотиды удаляют. Матрицы, которые направляли введение меченого флуоресцентной меткой нуклеотида выявляют при помощи визуализации поверхности проточной ячейки. После визуализации выполняют этап отщепления для удаления флуоресцентной метки, и способ повторяют, используя другие меченые флуоресцентной меткой нуклеотиды, до достижения требуемой длины прочтения. Информацию о последовательности собирают после каждого этапа добавления нуклеотида. При полногеномном секвенировании для создания библиотек секвенирования с использованием методик секвенирования единичной молекулы не выполняют или обычно пропускают этап ПЦР амплификации, и такие способы позволяют производить прямые определения образца вместо определений копий этого образца.
В одном из иллюстративных неограничивающих примеров осуществления способы, рассмотренные в настоящей работе, включают получение информации о последовательности нуклеиновых кислот, содержащихся в испытуемом образце, посредством использования 454-секвенирования (Roche) (например, рассмотренного в публикации Margulies, М. с соавт.Nature 437:376-380 [2005]). 454-секвенирование обычно включает два этапа. При выполнении первого этапа ДНК нарезают на фрагменты, содержащие приблизительно 300-800 пар оснований, так, чтобы эти фрагменты имели тупые концы. Затем к концам фрагментов пришивают олигонуклеотидные адаптеры. Адаптеры служат праймерами для амплификации и секвенирования фрагментов. Фрагменты могут быть присоединены к гранулам для захвата ДНК, например, к гранулам с покрытием из стрептавидина, например, с помощью адаптера В, который содержит 5'-биотиновую метку. Фрагменты, присоединенные к гранулам, подвергают ПЦР амплификации внутри капель эмульсии "масло-вода". В результате на каждой грануле образуется множество копий клонально амплифицированных фрагментов ДНК. При выполнении второго этапа гранулы фиксируют в лунках (например, лунках объемом порядка пиколитра). Пиросеквенирование выполняют параллельно на каждом фрагменте ДНК. Присоединение одного или более нуклеотидов вызывает испускание светового сигнала, который записывается камерой на приборах с зарядовой связью, находящейся в секвенаторе. Интенсивность сигнала пропорциональна количеству включаемых в структуру нуклеотидов. В пиросеквенировании анализируют пирофосфат (PPi), который высвобождается при присоединении нуклеотида. PPi превращается в АТФ под действием АТФ-сульфурилазы в присутствии аденозин-5'-фосфосульфата. АТФ необходим люциферазе для превращения люциферина в оксилюциферин, и в результате этой реакции испускается свет, параметры которого определяют и анализируют.
В другом иллюстративном неограничивающем примере осуществления способы, рассмотренные в настоящей работе, включают получение информации о последовательности нуклеиновых кислот, содержащихся в испытуемом образце, посредством использования методики SOLiD™ (Applied Biosystems). При секвенировании лигированием SOLiD™, геномную ДНК разрезают на фрагменты, и для получения библиотеки фрагментов к 5' и 3'-концам фрагментов пришивают адаптеры. В альтернативном варианте внутренние адаптеры могут быть введены присоединением адаптеров к 5' и 3'-концам фрагментов, образованием кольцеобразных фрагментов, расщеплением кольцеобразного фрагмента с образованием внутреннего адаптера и присоединением адаптеров к 5' и 3'-концам получаемых фрагментов с образованием библиотеки прочтений сопряженных пар (длинных прочтений). Затем в микрореакторах, содержащих гранулы, праймеры, матрицу и ПЦР компоненты, подготавливают популяции клональных гранул. После проведения ПЦР, матрицы подвергают денатурации, и гранулы обогащают для отделения гранул с достроенными матрицами. 3'-концы матриц на выбранных гранулах подвергают модификации, которая позволяет закреплять их на стеклянном слайде. Последовательность может быть определена последовательной гибридизацией и лигированием частично случайных олигонуклеотидов с центральным определенным основанием (или парой оснований), которое было идентифицировано специфичным флуорофоров. После регистрации цвета прикрепленный олигонуклеотид отщепляют и удаляют, и затем повторяют способ.
В другом иллюстративном неограничивающем примере осуществления способы, рассмотренные в настоящей работе, включают получение информации о последовательности нуклеиновых кислот, содержащихся в испытуемом образце, посредством использования методики секвенирования единичной молекулы в режиме реального времени (англ. single molecule, real-time, сокращенно SMRT™), разработанной Pacific Biosciences. При секвенировании SMRT, во время синтеза ДНК производят визуализацию непрерывного введения меченых красителем нуклеотидов. Единичные молекулы ДНК-полимеразы присоединяют к нижней поверхности индивидуальных датчиков с нулевой модой длины волны (датчиков ZMW, от англ. zero-mode wavelength), в которых регистрируется информация о последовательности, по мере встраивания фосфосвязанных нуклеотидов в растущую цепочку праймера. Датчик ZMW включает изолирующую структуру, которая позволяет наблюдать встраивание единичного нуклеотида под действием ДНК-полимеразы на фоне флуоресцентных нуклеотидов, который быстро рассеивается за пределами ZMW датчика (например, в течение микросекунд). Для встраивания нуклеотида в растущую цепочку обычно требуется несколько миллисекунд. В течение этого времени флуоресцентная метка возбуждается, испускает флуоресцентный сигнал, и флуоресцентная метка отщепляется. Регистрация соответствующего флуоресцентного сигнала красителя указывает на то, какое именно основание было встроено. Способ повторяют до построения последовательности.
В другом иллюстративном неограничивающем примере осуществления способы, рассмотренные в настоящей работе, включают получение информации о последовательности нуклеиновых кислот, содержащихся в испытуемом образце, посредством использования секвенирования в нанопорах (например, рассмотренного в публикации Soni G.V., Meller A. Clin. Chem. 53: 1996-2001 [2007]). Методики анализа секвенирования ДНК в нанопорах были созданы несколькими компаниями, среди которых можно отметить, например, Oxford Nanopore Technologies (Oxford, United Kingdom), Sequenom, NABsys и подобные организации. Секвенирование в нанопорах представляет собой методику секвенирования единичной молекулы, в которой единичную молекулу ДНК секвенируют непосредственно по мере ее прохождения через нанопору. Нанопора представляет собой небольшое отверстие, обычно порядка 1 нанометра в диаметре. Погружение нанопоры в проводящую жидкость и приложение разности потенциалов (напряжения) приводит к протеканию через нанопору небольшого электрического тока в результате ионной проводимости. На величину протекающего электрического тока влияет размер и форма нанопоры. По мере прохождения молекулы ДНК через нанопору, каждый нуклеотид молекулы ДНК в разной степени блокирует нанопору, в разной степени изменяя величину электрического тока, проходящего через нанопору. Таким образом, изменение величины электрического тока по мере прохождения молекулы ДНК через нанопору позволяет прочитывать последовательность ДНК.
В другом иллюстративном неограничивающем примере осуществления способы, рассмотренные в настоящей работе, включают получение информации о последовательности нуклеиновых кислот, содержащихся в испытуемом образце, посредством использования чувствительной к химическим веществам матрицы полевых транзисторов (англ. chemical-sensitive field effect transistor, сокращенно chemFET) (например, рассмотренной в опубликованной патентной заявке US 2009/0026082). В одном из примеров этой методики молекулы ДНК могут быть помещены в реакционные камеры, и матричные молекулы могут быть гибридизованы с праймером секвенирования, связанным с полимеразой. Введение одного или более трифосфатов в новую цепочку нуклеиновой кислоты с закреплением на 3'-конце праймера секвенирования может быть распознано устройством chemFET по изменению величины электрического тока. Матрица транзисторов может включать множество датчиков chemFET. В другом примере единичные нуклеиновые кислоты могут быть присоединены к гранулам, нуклеиновые кислоты могут быть амплифицированы на гранулах, и индивидуальные гранулы могут быть помещены в индивидуальные реакционные камеры матрицы chemFET, в которой каждая камера имеет датчик chemFET, и может быть выполнено секвенирование нуклеиновых кислот.
В другом примере осуществления методикой секвенирования ДНК является ионное полупроводниковое секвенирование единичной молекулы, в котором полупроводниковая методика объединена с простым химическим секвенированием для непосредственного перевода химически кодированной информации (А, С, G, Т) в цифровую информацию (0, 1) с помощью полупроводникового чипа. В природных условиях, если нуклеотид встраивается под действием полимеразы в цепочку ДНК, в качестве побочного продукта высвобождается ион водорода. В методике ионного полупроводникового секвенирования Ion Torrent для выполнения множественного параллельного биохимического секвенирования используют матрицу (чип), содержащую обработанные с помощью микромеханической методики лунки, расположенные с высокой плотностью. Каждая лунка содержит молекулу ДНК, отличную от молекул в других лунках. Под лунками расположен слой, чувствительный к ионам, а под ним расположен датчик ионов. При присоединении нуклеотида, например, С, в матрицу ДНК и последующем его встраивании в цепочку ДНК высвобождается ион водорода. Заряд этого иона изменяет рН раствора, что может быть обнаружено датчиком ионов Ion Torrent. Секвенатор - по существу самый маленький в мире твердофазный рН-метр - обнаруживает соответствие оснований пикам (на хроматограмме), переходя непосредственно от химической информации к цифровой информации. Затем секвенатор Ion Personal Genome Machine (PGM™) последовательно заполняет матрицу (чип) сначала одним, а затем другим нуклеотидом. Если последующий нуклеотид, который подают в матрицу, не соответствует, то изменение напряжения не будет зарегистрировано, и соответствие основанию не будет найдено. Если в цепочке ДНК имеются два идентичных основания, то напряжение удвоится, и матрица запишет соответствие двух идентичных оснований. Прямое обнаружение позволяет регистрировать встраивание нуклеотида в течение нескольких секунд.
В другом примере осуществления способ согласно изобретению включает получение информации о последовательности нуклеиновых кислот, содержащихся в испытуемом образце, посредством использования секвенирования гибридизацией. Секвенирование гибридизацией включает контакт множества полинуклеотидных последовательностей с множеством полинуклеотидных зондов, где каждый из множества полинуклеотидных зондов необязательно может быть прикреплен к подложке (субстрату). Подложка может иметь плоскую поверхность, включающую массив известных нуклеотидных последовательностей. Для распознавания полинуклеотидных последовательностей, содержащихся в образце, может быть использована схема гибридизации. В других примерах осуществления каждый зонд закреплен на грануле, например, на магнитной грануле или подобном объекте. Для идентификации множества полинуклеотидных последовательностей, содержащихся в образце, может быть определена и использована гибридизация на гранулах.
В некоторых примерах осуществления способов, рассмотренных в настоящей работе, прочтения последовательностей содержат приблизительно 20 п.о. (пар оснований), приблизительно 25 п.о., приблизительно 30 п.о., приблизительно 35 п.о., приблизительно 40 п.о., приблизительно 45 п.о., приблизительно 50 п.о., приблизительно 55 п.о., приблизительно 60 п.о., приблизительно 65 п.о., приблизительно 70 п.о., приблизительно 75 п.о., приблизительно 80 п.о., приблизительно 85 п.о., приблизительной 90 п.о., приблизительно 95 п.о., приблизительно 100 п.о., приблизительно 110 п.о., приблизительно 120 п.о., приблизительно 130, приблизительно 140 п.о., приблизительно 150 п.о., приблизительно 200 п.о., приблизительно 250 п.о., приблизительно 300 п.о., приблизительно 350 п.о., приблизительно 400 п.о., приблизительно 450 п.о. или приблизительно 500 п.о. Ожидается, что технический прогресс вскоре позволит получать прочтения одного конца, длина которых превышает 500 п.о., и при генерации прочтений парных концов позволит получать прочтения, длина которых превышает приблизительно 1000 п.о. В некоторых примерах осуществления прочтения парных концов используют для определения интересующих последовательностей, которые включают прочтения последовательности, содержащие приблизительно от 20 п.о. до 1000 п.о., приблизительно от 50 п.о. до 500 п.о. или от 80 п.о. до 150 п.о. В различных примерах осуществления прочтения парных концов используют для анализа интересующей последовательности. Длина интересующей последовательности превышает длину прочтений. В некоторых примерах осуществления длина интересующей последовательности превышает приблизительно 100 п.о., 500 п.о., 1000 п.о. или 4000 п.о. Для определения хромосомного происхождения секвенированной молекулы нуклеиновой кислоты производят картирование прочтений последовательности, сравнивая последовательности прочтений с эталонной последовательностью, и при этом не требуется специальной генетической информации о последовательности. Для объяснения незначительного полиморфизма, который может вызвать различия между эталонным геномом и геномами смешанного образца, может быть допущено небольшое несоответствие (0-2 несоответствия на прочтение). В некоторых примерах осуществления прочтения, выровненные по эталонной последовательности, используют в качестве якорных прочтений, и прочтения, которые образуют пары с якорными прочтениями, но не могут быть выровнены или плохо выравниваются по эталонной последовательности, используют как заякоренные прочтения. В некоторых примерах осуществления плохо выровненные прочтения могут иметь относительно высокий процент несоответствий на одно прочтение, например, по меньшей мере приблизительно 5%, по меньшей мере приблизительно 10%, по меньшей мере приблизительно 15% или по меньшей мере приблизительно 20% несоответствий на одно прочтение.
Для одного образца обычно получают множество маркеров (меток) последовательности (т.е. прочтений, выровненных по эталонной последовательности). В некоторых примерах осуществления из локализации прочтений на эталонном геноме для одного образца получают по меньшей мере приблизительно 3×106 маркеров последовательности, по меньшей мере приблизительно 5×106 маркеров последовательности, по меньшей мере приблизительно 8×106 маркеров последовательности, по меньшей мере приблизительно 10×106 маркеров последовательности, по меньшей мере приблизительно 15×106 маркеров последовательности, по меньшей мере приблизительно 20×106 маркеров последовательности, по меньшей мере приблизительно 30×106 маркеров последовательности, по меньшей мере приблизительно 40×106 маркеров последовательности, или по меньшей мере приблизительно 50×106 маркеров последовательности, содержащих, например, 100 п.о. В некоторых примерах осуществления все прочтения последовательности локализованы во всех областях эталонного генома, что позволяет создать прочтения по всему геному. В других примерах осуществления прочтения локализованы на интересующей последовательности.
Установка и системы для секвенирования с применением UMI
Анализ данных секвенирования и получение из них диагноза обычно выполняют с помощью различных компьютерных алгоритмов и программ. Таким образом, в некоторых примерах осуществления применяют способы, в которых используют данные, хранящиеся в или передаваемые через одну или более компьютерных систем или других систем обработки. Примеры осуществления, рассмотренные в настоящей работе, также относятся к установке для выполнения таких операций. Установка может быть сконструирована специально для требуемых целей, или она может представлять собой неспециализированный компьютер (или группу компьютеров), селективно активизированных или перенастроенных с помощью компьютерной программы и/или структуры данных, хранящейся в компьютере. В некоторых примерах осуществления группа процессоров выполняет некоторые или все перечисленные аналитические операции совместно (например, через сеть или обработку данных в облаке) и/или параллельно. Процессор или группа процессоров, предназначенных для выполнения способов, рассмотренных в настоящей работе, может состоять из устройств различных типов, которые включают микроконтроллеры и микропроцессоры, такие как программируемые устройства (например, CPLD (от англ. Complex Programmable Logic Device, т.е. сложное устройство с программируемой логикой) и FPGA (от англ. Field Programmable Gate Array, т.е. матрица логических элементов с эксплуатационным программированием)) и непрограммируемые устройства, такие как вентильная матрица на специализированных интегральных схемах (ASIC, от англ. Application Specific Integrated Circuit) или неспециализированные микропроцессоры.
Один из примеров осуществления относится к системе, подходящей для определения последовательности с низкой аллельной частотой, находящейся в испытуемом образце, включающем нуклеиновые кислоты, где система включает секвенатор, предназначенный для помещения в него образца нуклеиновой кислоты и получения из образца информации о последовательности нуклеиновой кислоты; процессор; и машиночитаемый носитель данных, на котором хранятся инструкции, предназначенные для выполнения этим процессором с целью определения интересующей последовательности, находящейся в испытуемом образце, посредством: (а) приема последовательностей множества амплифицированных полинуклеотидов, где множество амплифицированных полинуклеотидов получено амплификацией фрагментов двухцепочечной ДНК, содержащейся в образце, содержащем интересующую последовательность, и присоединением адаптеров к фрагментам двухцепочечной ДНК; (b) идентификации множества физических UMI, каждый из которых находится в одном из множества амплифицированных полинуклеотидов, где каждый физический UMI получен из адаптера, присоединенного к одному из фрагментов двухцепочечной ДНК; (с) идентификации множества виртуальных UMI, каждый из которых находится в одном из множества амплифицированных полинуклеотидов, где каждый виртуальный UMI получен из индивидуальной молекулы одного из фрагментов двухцепочечной ДНК; и (а) определения последовательностей фрагментов двухцепочечной ДНК с использованием последовательностей множества амплифицированных полинуклеотидов, множества физических UMI и множества виртуальных UMI, что приводит к уменьшению погрешностей в прочитанных последовательностях фрагментов двухцепочечной ДНК.
Другой пример осуществления относится к системе, включающей секвенатор, предназначенный для помещения в него образца нуклеиновой кислоты и получения из образца информации о последовательности нуклеиновой кислоты; процессор; и машиночитаемый носитель данных, на котором хранятся инструкции, предназначенные для выполнения этим процессором с целью определения интересующей последовательности, находящейся в испытуемом образце. Инструкции включают: (а) прикрепление адаптеров к обоим концам фрагментов ДНК, находящейся в образце, где каждый адаптер включает двухцепочечную гибридизованную область, одноцепочечное 5'-плечо, одноцепочечное 3'-плечо и неслучайный уникальный молекулярный индекс (UMI) на одной цепочке или на каждой цепочке адаптеров, в результате чего получают продукты присоединения адаптера к ДНК; (b) амплификацию продуктов присоединения адаптера к ДНК с образованием множества амплифицированных полинуклеотидов; (с) секвенирование множества амплифицированных полинуклеотидов, в результате чего получают множество прочтений, ассоциированных с множеством неслучайных UMI; (d) идентификацию прочтений, имеющих общий неслучайный UMI, из множества прочтений; и (е) определение последовательности по меньшей мере части фрагмента ДНК, находящегося в образце, имеющего прикрепленный адаптер с общим неслучайным UMI, из идентифицированных прочтений, имеющих общий неслучайный UMI. В некоторых примерах осуществления инструкции дополнительно включают: выбор прочтений, имеющих как общий неслучайный UMI, так и общее положение прочтения, из прочтений, имеющих общий неслучайный UMI, причем для определения последовательности фрагмента ДНК в этапе (е) применяют только прочтения, имеющие как общий неслучайный UMI, так и общее положение прочтения в эталонной последовательности.
В другом примере осуществления инструкции включают: (а) прикрепление адаптеров к обоим концам фрагментов двухцепочечной ДНК, находящейся в образце, где каждый адаптер включает двухцепочечную гибридизованную область, одноцепочечное 5'-плечо, одноцепочечное 3'-плечо и неслучайный уникальный молекулярный индекс (UMI) на одной цепочке или на каждой цепочке адаптеров, в результате чего получают продукты присоединения адаптера к ДНК; при этом неслучайный UMI может быть объединен с другой информацией для выполнения уникальной идентификации индивидуальной молекулы фрагментов двухцепочечной ДНК; (b) амплификацию обеих цепочек продуктов присоединения адаптера к ДНК с образованием множества амплифицированных полинуклеотидов; (с) секвенирование множества амплифицированных полинуклеотидов, в результате чего получают множество прочтений, каждое из которых ассоциировано с неслучайным UMI; (d) идентификацию множества неслучайных UMI, ассоциированных с множеством прочтений; и (е) применение множества прочтений и множества неслучайных UMI для определения последовательностей фрагментов двухцепочечной ДНК, содержащихся в образце.
В некоторых примерах осуществления любой из рассмотренных в настоящей работе систем секвенатор предназначен для проведения секвенирования нового поколения (СНП). В некоторых примерах осуществления секвенатор предназначен для проведения массового параллельного секвенирования посредством секвенирования синтезом с обратимыми окрашивающими терминаторами. В других примерах осуществления секвенатор предназначен для проведения секвенирования лигированием. В других примерах осуществления секвенатор предназначен для проведения секвенирования единичной молекулы.
Кроме того, некоторые примеры осуществления относятся к материальным и/или энергонезависимым машиночитаемым носителям или компьютерным программным продуктам, которые включают программные инструкции и/или данные (включая структуры данных) для выполнения различных компьютерных операций. Примеры машиночитаемых носителей включают, без ограничений, полупроводниковые запоминающие устройства, магнитные носители, такие как дисковые накопители, магнитные ленты, оптические носители, такие как компакт-диски (CD), магнитооптические носители и аппаратные устройства, специально предназначенные для хранения и выполнения программных инструкций, такие как постоянные запоминающие устройства (англ. Read Only Memory, сокращенно ROM) и оперативные запоминающие устройства (англ. Random Access Memory, сокращенно RAM). Контроль машиночитаемого носителя может осуществляться непосредственно или опосредованно конечным пользователем. Примеры непосредственно контролируемых носителей включают носители, находящиеся на устройстве (предприятии) пользователя, и/или носители, к которым нет доступа с других устройств. Примеры опосредованно контролируемых носителей включают носители, предоставляющие пользователю опосредованный допуск через внешнюю сеть и/или через обслуживание с общими ресурсами, такими как "облако". Примеры программных инструкций включают машинный код, такой как код, создаваемый составителем программы, и файлы, содержащие более высокие уровни кода, которые могут быть выполнены компьютером при использовании преобразователя (интерпретатора).
В различных примерах осуществления данные или информация, используемые в рассматриваемых способах и установке, представлены в электронном формате. Такие данные или информация могут включать прочтения и метки, полученные из образца нуклеиновой кислоты, эталонные последовательности (включая эталонные последовательности, которые предоставляют данные исключительно или в основном по полиморфизму), маркеры, такие как диагностические маркеры раковых заболеваний, рекомендации по лечению, диагнозы и подобные данные. Согласно настоящему изобретению, данные или другая информация, представляемая в электронном формате, доступна для хранения в машине и для передачи между машинами. Традиционно данные, находящиеся в электронном формате, представляют собой цифровые данные и могут храниться в виде битов и/или байтов в различных структурах данных, списках, базах данных и т.д. Данные могут быть записаны электронным, оптическим и подобным образом.
Один из примеров осуществления относится к компьютерному программному продукту для генерации выходного сигнала, обозначающего последовательность фрагмента интересующей ДНК в испытуемом образце. Компьютерный продукт может содержать инструкции для выполнения одного или более из рассмотренных выше способов определения интересующей последовательности. Как указано, компьютерный продукт может включать энергонезависимый и/или материальный машиночитаемый носитель, на котором записаны логические операции, которые способен выполнять компьютер, или компилируемые логические операции (например, инструкции), которые позволяют процессору определять интересующую последовательность. В одном из примеров компьютерный продукт включает машиночитаемый носитель, на котором записаны логические операции, которые способен выполнять компьютер, или компилируемые логические операции (например, инструкции), которые позволяют процессору диагностировать состояние или определять последовательность интересующей нуклеиновой кислоты.
Следует понимать, что маловероятно или в большинстве случаев совершенно невозможно, чтобы неподготовленный человек мог самостоятельно производить вычислительные операции способов, рассмотренных в настоящей работе. Например, для картирования одного полученного из образца прочтения, содержащего 30 п.о., на любой из хромосом человека могут уйти годы усилий, если картирование производят без использования вычислительной установки. На самом деле проблема усложняется еще и тем, что для надежных соотнесений мутаций с низкой аллельной частотой обычно требуются произвести картирование (локализацию) нескольких тысяч (например, по меньшей мере приблизительно 10000) или даже миллионов прочтений на одной или более хромосомах.
Способы, рассмотренные в настоящей работе, могут быть осуществлены с помощью системы для определения интересующей последовательности в испытуемом образце. Система может включать: (а) секвенатор, в который помещают нуклеиновые кислоты из испытуемого образца для получения из образца информации о последовательности нуклеиновой кислоты; (b) процессор; и (с) один или более машиночитаемых носителей для хранения информации, на которых хранятся инструкции, предназначенные для выполнения процессором с целью определения интересующей последовательности, находящейся в испытуемом образце. В некоторых примерах осуществления способы выполняются согласно инструкциям, которые хранятся на машиночитаемом носителе, причем на носителе записаны машиночитаемые инструкции для выполнения способа определения интересующей последовательности. Таким образом, один из примеров осуществления относится к компьютерному программному продукту, включающему энергонезависимый машиночитаемый носитель, на котором хранится программный код, при выполнении которого одним или более процессорами компьютерной системы происходит активация компьютерной системы, приводящая к осуществлению способа определения последовательностей фрагментов нуклеиновых кислот в испытуемом образце. Программный код может включать: (а) код для определения последовательностей множества амплифицированных полинуклеотидов, где множество амплифицированных полинуклеотидов получено амплификацией фрагментов двухцепочечной ДНК, содержащейся в образце, содержащем интересующую последовательность, и присоединением адаптеров к фрагментам двухцепочечной ДНК; (b) код для идентификации множества физических UMI, каждый из которых находится в одном из множества амплифицированных полинуклеотидов, где каждый физический UMI получен из адаптера, присоединенного к одному из фрагментов двухцепочечной ДНК; (с) код для идентификации множества виртуальных UMI, каждый из которых находится в одном из множества амплифицированных полинуклеотидов, где каждый виртуальный UMI получен из индивидуальной молекулы одного из фрагментов двухцепочечной ДНК; и (d) код для определения последовательностей фрагментов двухцепочечной ДНК из последовательностей множества амплифицированных полинуклеотидов, множества физических UMI и множества виртуальных UMI, что приводит к уменьшению погрешностей в прочитанных последовательностях фрагментов двухцепочечной ДНК.
В некоторых примерах осуществления физические UMI включают неслучайные UMI. В других примерах осуществления физические UMI включают случайные UMI.
Другой пример осуществления относится к компьютерному программному продукту, включающему энергонезависимый машиночитаемый носитель, на котором хранится программный код, при выполнении которого одним или более процессорами компьютерной системы происходит активация компьютерной системы, приводящая к осуществлению способа определения последовательностей фрагментов нуклеиновых кислот в испытуемом образце. Программный код может включать: (а) код для прикрепления адаптеров к обоим концам фрагментов ДНК, находящейся в образце, где каждый адаптер включает двухцепочечную гибридизованную область, одноцепочечное 5'-плечо, одноцепочечное 3'-плечо и неслучайный уникальный молекулярный индекс (UMI) на одной цепочке или на каждой цепочке адаптеров, в результате чего получают продукты присоединения адаптера к ДНК; (b) код для амплификации продуктов присоединения адаптера к ДНК с образованием множества амплифицированных полинуклеотидов; (с) код для секвенирования множества амплифицированных полинуклеотидов, в результате чего получают множество прочтений, ассоциированных с множеством неслучайных UMI; (а) код для идентификации прочтений, имеющих общий неслучайный UMI, из множества прочтений; и (е) код для определения последовательности по меньшей мере части фрагмента ДНК, находящегося в образце, имеющего прикрепленный адаптер с общим неслучайным UMI, из идентифицированных прочтений, имеющих общий неслучайный UMI.
В другом примере осуществления программные коды включают: (а) код для прикрепления адаптеров к обоим концам фрагментов двухцепочечной ДНК, находящейся в образце, где каждый адаптер включает двухцепочечную гибридизованную область, одноцепочечное 5'-плечо, одноцепочечное 3'-плечо и неслучайный уникальный молекулярный индекс (UMI) на одной цепочке или на каждой цепочке адаптеров, в результате чего получают продукты присоединения адаптера к ДНК, причем неслучайный UMI может быть объединен с другой информацией для выполнения уникальной идентификации индивидуальной молекулы фрагментов двухцепочечной ДНК; (b) код для амплификации обеих цепочек продуктов присоединения адаптера к ДНК с образованием множества амплифицированных полинуклеотидов; (с) код для секвенирования множества амплифицированных полинуклеотидов, в результате чего получают множество прочтений, каждое из которых ассоциировано с неслучайным UMI; (d) идентификацию множества неслучайных UMI, ассоциированных с множеством прочтений; и (е) код для применения множества прочтений и множества неслучайных UMI для определения последовательностей фрагментов двухцепочечной ДНК, содержащихся в образце.
В некоторых примерах осуществления инструкции могут дополнительно включать автоматическую запись существенной для способа информации. Медицинские данные пациента могут храниться, например, в лаборатории, кабинете врача, госпитале, учреждении здравоохранения, страховой компании или на вебсайте для хранения персональных медицинских данных. Дополнительно, на основании результатов выполненного процессором анализа, способ может дополнительно включать назначение, начало и/или изменение терапии пациенту-человеку, из организма которого был взят испытуемый образец. Это может включать выполнение одного или более дополнительных испытаний (тестов) или анализов дополнительных образцов, взятых из организма пациента.
Рассмотренные способы также могут быть осуществлены с помощью компьютерной системы обработки, адаптированной или предназначенной для выполнения способа определения интересующей последовательности. Один из примеров осуществления относится к компьютерной системе обработки, адаптированной или предназначенной для выполнения способа, рассмотренного в настоящей работе. В одном из примеров осуществления установка включает устройство для секвенирования, адаптированное или предназначенное для секвенирования по меньшей мере части молекул нуклеиновых кислот, содержащихся в образце, с целью получений информации о последовательности, рассмотренной в настоящей работе. Установка также может включать компоненты для обработки образца. Такие компоненты рассмотрены в настоящей работе.
Данные о последовательности или другие данные могут быть загружены в компьютер, или они могут непосредственно или опосредованно храниться на машиночитаемом носителе. В одном из примеров осуществления компьютерная система непосредственно соединена с устройством для секвенирования, которое считывает и/или анализирует последовательности нуклеиновых кислот, имеющиеся в образцах. Информация о последовательности или другая информация, регистрируемая этими инструментами, передается через интерфейс в компьютерную систему. В альтернативном варианте обработанные системой последовательности получают из источника хранения последовательностей, такого как база данных или другое хранилище. После обеспечения доступа в обрабатывающую установку запоминающее устройство или запоминающее устройство большой емкости запоминает в буфере или хранит, по меньшей мере временно, последовательности нуклеиновых кислот. Кроме того, запоминающее устройство может хранить количество маркеров различных хромосом или геномов и т.д. В памяти также могут храниться различные подпрограммы и/или программы для анализа представления последовательности или картированных данных. Такие программы/подпрограммы могут включать программы для выполнения статистических анализов и т.д.
В одном из примеров пользователь помещает образец в установку секвенирования. Данные собираются и/или анализируются установкой секвенирования, которая соединена с компьютером. Установленное на компьютере программное обеспечение позволяет собирать и/или анализировать данные. Данные могут храниться, могут быть показаны (через монитор или другое аналогичное устройство) и/или могут быть направлены в другой участок. Компьютер может быть соединен с Интернетом, который применяют для передачи данных на мобильное устройство, используемое удаленным пользователем (например, врачом, научным работником или аналитиком). Следует понимать, что перед передачей данные могут храниться и/или быть проанализированы. В некоторых примерах осуществления необработанные данные собирают и направляют удаленному пользователю или в удаленную установку, которая будет анализировать и/или хранить данные. Передача может быть осуществлена через Интернет, но также может быть осуществлена через спутниковую или другую связь. В альтернативном варианте данные могут храниться на машиночитаемом носителе, и носитель может быть отправлен конечному пользователю (например, почтой). Удаленный пользователь может находиться в том же или ином географическом расположении, примеры которого включают, без ограничений, здание, город, штат, страну или континент.
В некоторых примерах осуществления способы также включают сбор данных о множестве полинуклеотидных последовательностей (например, прочтения, маркеры и/или эталонные хромосомные последовательности) и направление этих данных в компьютер или другую вычислительную систему. Например, компьютер может быть соединен с лабораторным оборудованием, например, установкой для отбора образцов, установкой амплификации нуклеотидов, установкой секвенирования нуклеотидов или установкой гибридизации. Затем компьютер может собрать применяемые данные, отобранные лабораторным устройством. Данные могут храниться на компьютере в любом этапе, например, во время сбора данных в режиме реального времени, перед отправкой, во время или в комбинации с отправкой или после отправки. Данные могут храниться на машиночитаемом носителе, который может быть извлечен из компьютера. Собранные или хранящиеся данные могут быть переданы из компьютера в удаленное место, например, через локальную сеть или широкоохватную сеть, такую как Интернет. На удаленном участке переданные данные могут быть обработаны с помощью различных операций, рассмотренных ниже.
Неограничивающие примеры данных, находящихся в электронных форматах, которые можно хранить, передавать, анализировать и/или обрабатывать в системах и установке способами, рассмотренными в настоящей работе, включают следующие:
- Прочтения, полученные секвенированием нуклеиновых кислот в испытуемом образце
- Метки, полученные при выравнивании прочтений по эталонному геному или другой эталонной последовательности или последовательностям
- Эталонный геном или последовательность
- Пороги для заключения о признании испытуемого образца как поврежденного, неповрежденного, или непризнании его таковым
- Реальные заключения о медицинских состояниях, связанных с интересующей последовательностью
- Диагнозы (клинические состояния, связанные с заключениями)
- Рекомендации для проведения дополнительных тестов, полученные из заключений и/или диагнозов
- Планы по терапии и/или мониторингу, полученные из заключений и/или диагнозов
Эти различные типы данных могут быть получены, могут храниться, передаваться, могут быть проанализированы и/или обработаны в одном или более местоположений с помощью определенной установки. Имеется множество разнообразных вариантов обработки. В одном из частных случаев вся или большая часть этой информации хранится и используется в том месте, где была проведена обработка испытуемого образца, например, в офисе врача или в другой клинической обстановке. В другом, противоположном случае образец получают в одном месте, обрабатывают и необязательно секвенируют в другом месте, выравнивание прочтений и распознавания производят в одном или более других местах, а диагнозы, рекомендации и/или планы подготавливают в ином месте (которое может быть местом, где был взят образец).
В различных примерах осуществления прочтения получают на установке секвенирования и затем передают на удаленный участок, где их обрабатывают для определения интересующей последовательности. На этом удаленном участке, например, прочтения выравнивают по эталонной последовательности, получая якорные и заякоренные прочтения. Неограничивающие примеры операций обработки, которые могут быть выполнены в различных участках, включают следующие:
- Отбор образца
- Предварительная обработка образца перед секвенированием
- Секвенирование
- Анализ данных о последовательности и медицинское заключение
- Диагноз
- Сообщение диагноза и/или заключения пациенту или поставщику медицинских услуг
- Разработка плана дополнительного лечения, тестирования и/или мониторинга
- Выполнение плана
- Рекомендации
Любая из этих операций или более одной операции могут быть автоматизированы, как указано в настоящей работе. Обычно секвенирование, анализ данных о последовательности и медицинские заключения выполняют на компьютере. Другие операции могут быть выполнены вручную или автоматически.
На Фиг. 6 представлен один из примеров осуществления рассредоточенной системы, предназначенной для получения заключения или диагноза из испытуемого образца. Отбор испытуемого образца из организма пациента производят на участке 01 отбора образца. Затем образцы направляют в участок 03 обработки и секвенирования, где может быть произведена рассмотренная выше обработка и секвенирование испытуемого образца. Участок 03 включает установку для обработки образца, а также установку для секвенирования обработанного образца. Как указано в настоящей работе, результатом секвенирования является набор прочтений, которые обычно предоставляют в электронном формате и выкладывают сеть, такую как Интернет, что показано на Фиг. 6 условным обозначением 05.
Данные о последовательности направляют в удаленный участок 07, где производят анализ и составление заключения. Этот участок может включать одно или более мощных вычислительных устройств, таких как компьютеры или процессоры. После выполнения анализа находящимися на участке 07 вычислительными ресурсами и составления заключения из полученной информации о последовательности, заключение передают обратно в сеть 05. В некоторых примерах осуществления на участке 07 составляют не только заключение, но и соответствующий диагноз. Затем, как показано на Фиг. 6, заключение или диагноз передают через сеть обратно в участок 01 отбора образца. Как уже было указано, это лишь один из множества вариантов разделения различных операций, связанных с составлением заключения или диагноза между различными участками. Один из общих вариантов включает отбор и обработку образца и секвенирования в одном участке. Другой вариант включает обработку и секвенирование в том же участке, где производят анализ и составление заключения.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Пример 1
Подавление ошибок посредством применения случайного физического UMI и виртуального UMI
На Фиг. 7А и Фиг. 7В представлены экспериментальные данные, показывающие эффективность подавления ошибок способами, рассмотренными в настоящей работе. Экспериментаторы использовали деградированную в результате гидродинамического сдвига геномную ДНК NA12878. Использовали создание библиотеки TruSeq и обогащение с настраиваемой панелью (~130 Kb). Секвенирование проводили при 2×150 п.о. в быстром режиме HiSeq2500, и среднее целевое покрытие составляло ~10000Х. На Фиг. 7А представлен профиль частоты появления ошибки (аллельная частота второго по высоте основания) оснований, имеющих высокое качество определения (>Q30), полученный согласно стандартному способу (средняя частота появления ошибки 0,04%). На Фиг. 7В представлен профиль частоты появления ошибки при объединении/конвейерной обработке UMI (средняя частота появления ошибки 0,007%). Следует отметить, что эти результаты получены при использовании прототипа кода, то есть при усовершенствовании способов может быть достигнуто дополнительное снижение частоты появления ошибки.
Пример 2
Подавление ошибок посредством применения неслучайного физического UMI и положения
На Фиг. 8 представлены данные, показывающие, что один лишь учет информации о положении для объединения прочтений может приводить к объединению прочтений, которые на самом деле получены из различных исходных молекул. Это явление также называется конфликтом прочтений. В результате определяемое в способе количество фрагментов в образце может оказаться заниженным. На Фиг. 8 по оси Y отложено наблюдаемое количество фрагментов, полученное при объединении прочтений на основании одной лишь информации о положении. На Фиг. 8 по оси X отложено вычисленное количество фрагментов, с учетом различных генотипов, например, различных однонуклеотидных полиморфизмов (англ. Single Nucleotide Polymorphism, сокращенно SNP) и других генотипических различий. Как показано на изображении, наблюдаемое количество фрагментов меньше, чем количество фрагментов, уточненных по генотипу, что указывает на заниженную оценку и конфликт прочтений при использовании одной лишь информации о положении для объединения прочтений и идентификации фрагментов.
На Фиг. 9 представлены эмпирические данные, показывающие, что использование неслучайного UMI и информации о положении для объединения прочтений может дать более точную оценку количества фрагментов, чем одна лишь информация о положении. Неслучайный UMI представляет собой дуплексный UMI, содержащий 6 п.о., расположенный на двухцепочечном конце адаптера, и этот неслучайный UMI был выбран из 96 различных UMI. По оси Y отложено среднее количество объединенных фрагментов; результат объединения по положению представлен левым столбцом в каждой паре, а результат объединения по UMI и положению представлен правым столбцом в каждой паре столбцов. Три левых пары столбцов показывают данные, полученные от образцов внеклеточной ДНК, в виде трех последовательно возрастающих входных сигналов. Три правых пары столбцов показывают данные, полученные из трех образцов деградированной в результате гидродинамического сдвига геномной ДНК. Попарные сравнения результатов двух способов объединения показывают, что объединение на основании UMI и положения приводят к более высокой оценке количества фрагментов, чем использование одной лишь информации о положении для объединения. Сравнение двух способов объединения обнаруживает более значительные различия для образцов внеклеточной ДНК, чем для четырех образцов геномной ДНК. Кроме того, различие между образцами внеклеточной ДНК повышается по мере повышения входного сигнала образца. Полученные данные показывают, что объединение на основании неслучайного UMI и информации о положении позволяет нивелировать конфликт прочтений и заниженную оценку количества фрагментов, в особенности, для внеклеточной ДНК.
На Фиг. 10 представлены в виде таблицы различные ошибки, возникающие в трех обработанных образцах, имеющих случайные UMI. В первых трех строчках данных указан процент ошибок различного типа в 43 образцах. В последней строчке представлены частоты появления ошибки, усредненные по образцам. Как видно из таблицы, 97,58% UMI не содержат ошибок, и 1,07% UMI содержат одну исправляемую ошибку. Более 98,65% всех UMI могут быть использованы для индексации индивидуальных фрагментов ДНК. Многие из оставшихся UMI могут быть использованы в комбинации с контекстной информацией.
На Фиг. 11А представлены чувствительность и селективность распознавания соматической мутации и вариации числа копий (англ. copy number variation, сокращенно CNV) в образце геномной ДНК с помощью двух способов объединения с использованием двух различных инструментов: VarScan и Denovo. Применение инструмента VarScan, объединение на основании UMI и информации о положении позволяет достигать несколько более высокой чувствительности и заметно более высокой селективности (снижение доли ложных распознаваний), что видно из сдвига ROC-кривой в верхнюю левую часть изображения при использовании UMI и информации о положении. Применение инструмента Denovo, объединение на основании UMI и информации о положении позволяет заметно повысить чувствительность.
На Фиг. 11 В-С показана селективность (т.е. доля ложных распознаваний) распознавания соматической мутации и CNV в трех образцах вкДНК с последовательно возрастающими входными сигналами образцов, полученная двумя способами объединения с помощью двух разных инструментов: VarScan и Denovo. Применение инструмента VarScan, объединение на основании UMI и информации о положении позволило достичь значительно более высокой селективности (снижение доли ложных распознаваний) результатов, полученных для всех трех образцов. Применение инструмента Denovo, объединение на основании UMI и информации о положении позволило достичь более высокой селективности (более низкая частота ложных сигналов тревоги) только для образца, имеющего самый высокий входной сигнал.
Настоящее изобретение может иметь другие конкретные примеры осуществления, не выходящие за пределы объема изобретения или не противоречащие его основным характеристикам. Рассмотренные примеры осуществления представлены для иллюстрации изобретения и не ограничивают его объем. Таким образом, объем изобретения ограничен прилагаемыми пунктами формулы изобретения, а не приведенным выше описанием. Все изменения, не противоречащие существу и объему пунктов формулы изобретения и их эквивалентов, включены в объем изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ И СИСТЕМЫ ДЛЯ ПОЛУЧЕНИЯ НАБОРОВ УНИКАЛЬНЫХ МОЛЕКУЛЯРНЫХ ИНДЕКСОВ С ГЕТЕРОГЕННОЙ ДЛИНОЙ МОЛЕКУЛ И КОРРЕКЦИИ В НИХ ОШИБОК | 2018 |
|
RU2766198C2 |
| БИБЛИОТЕКИ ДЛЯ СЕКВЕНИРОВАНИЯ НОВОГО ПОКОЛЕНИЯ | 2014 |
|
RU2698125C2 |
| СЕКВЕНИРОВАНИЕ ПОЛИНУКЛЕОТИДНЫХ БИБЛИОТЕК С ВЫСОКОЙ ПРОПУСКНОЙ СПОСОБНОСТЬЮ И АНАЛИЗ ТРАНСКРИПТОМОВ | 2018 |
|
RU2790291C2 |
| СПОСОБ И КОМПОЗИЦИЯ ДЛЯ АНАЛИЗА КЛЕТОЧНЫХ КОМПОНЕНТОВ | 2016 |
|
RU2761432C2 |
| СПОСОБ И КОМПОЗИЦИЯ ДЛЯ АНАЛИЗА КЛЕТОЧНЫХ КОМПОНЕНТОВ | 2016 |
|
RU2717491C2 |
| СИСТЕМА АНАЛИЗА ДЛЯ ОРТОГОНАЛЬНОГО ДОСТУПА К БИОМОЛЕКУЛАМ И ИХ МЕЧЕНИЯ В КЛЕТОЧНЫХ КОМПАРТМЕНТАХ | 2017 |
|
RU2771892C2 |
| НЕИНВАЗИВНАЯ ДИАГНОСТИКА ПУТЕМ СЕКВЕНИРОВАНИЯ 5-ГИДРОКСИМЕТИЛИРОВАННОЙ БЕСКЛЕТОЧНОЙ ДНК | 2017 |
|
RU2742355C2 |
| СПОСОБЫ ИНКАПСУЛИРОВАНИЯ ОДИНОЧНЫХ КЛЕТОК, ИНКАПСУЛИРОВАННЫЕ КЛЕТКИ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2793717C2 |
| СПОСОБЫ ИНКАПСУЛИРОВАНИЯ ОДИНОЧНЫХ КЛЕТОК, ИНКАПСУЛИРОВАННЫЕ КЛЕТКИ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2750567C2 |
| ТРАНСПОЗИЦИЯ С СОХРАНЕНИЕМ СЦЕПЛЕНИЯ ГЕНОВ | 2015 |
|
RU2709655C2 |



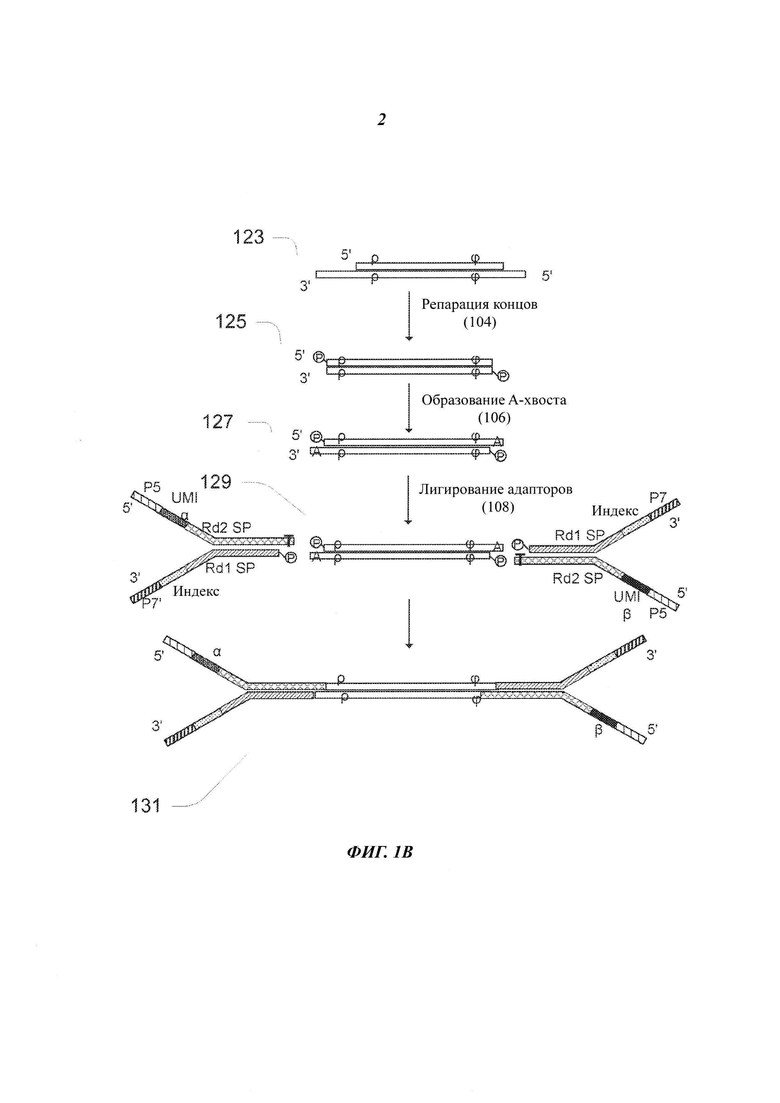

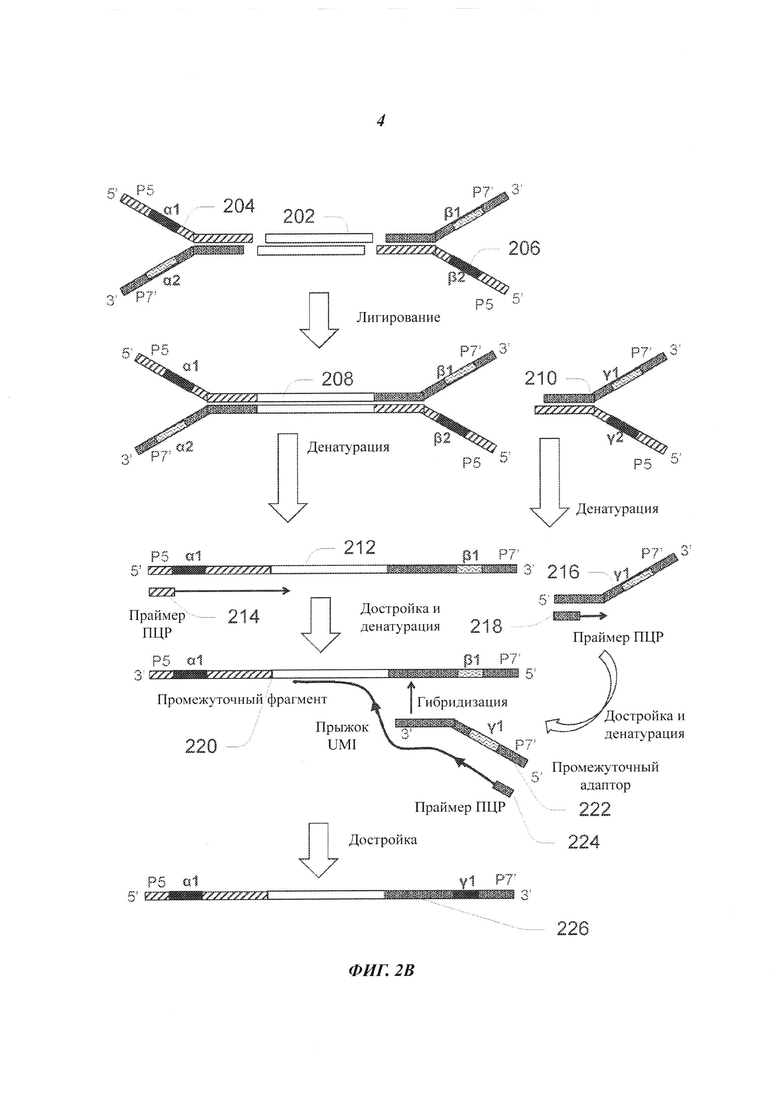
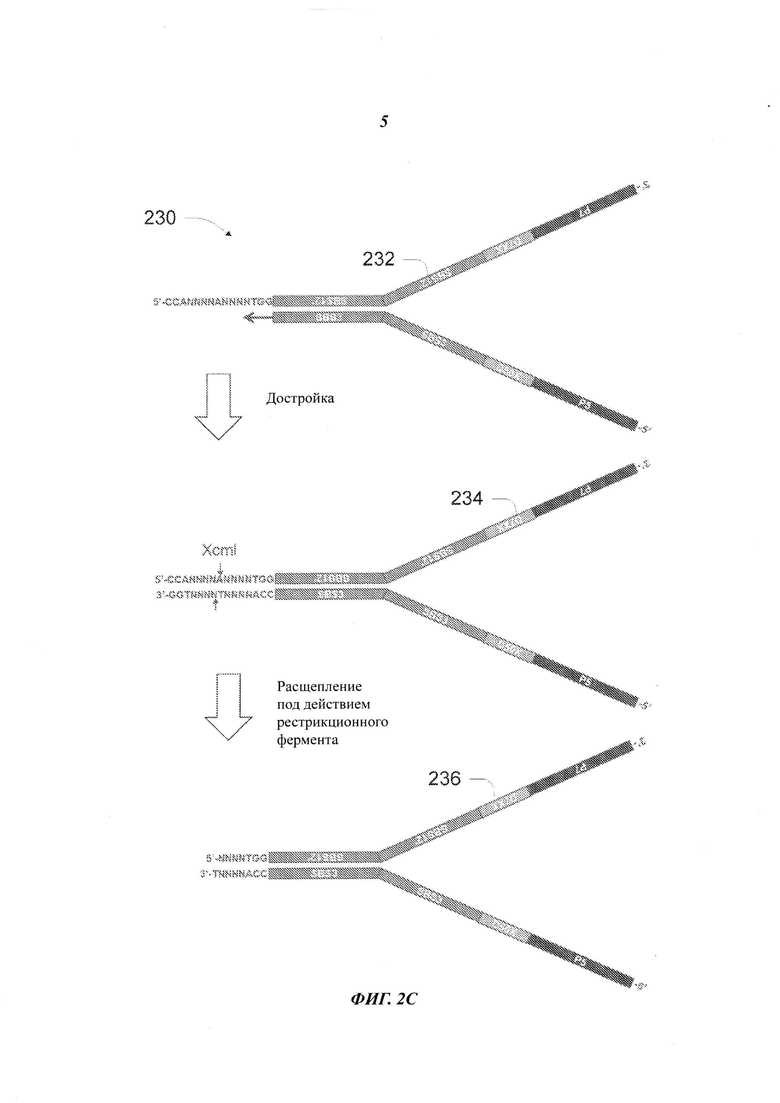

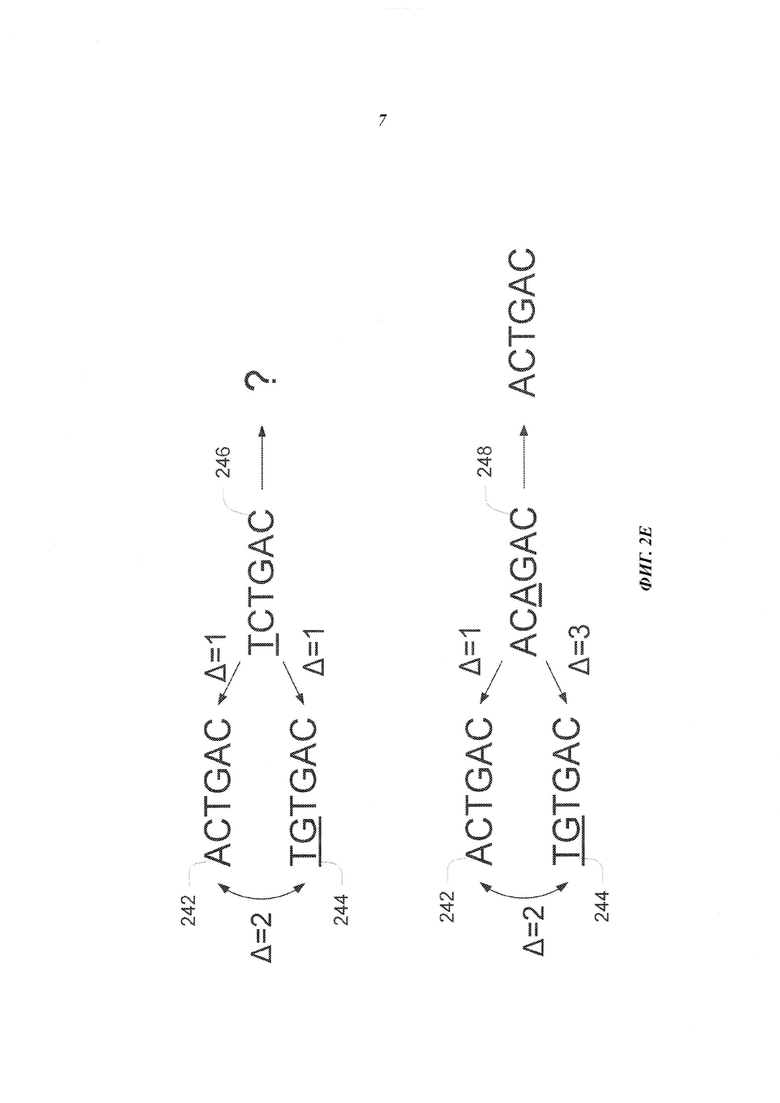




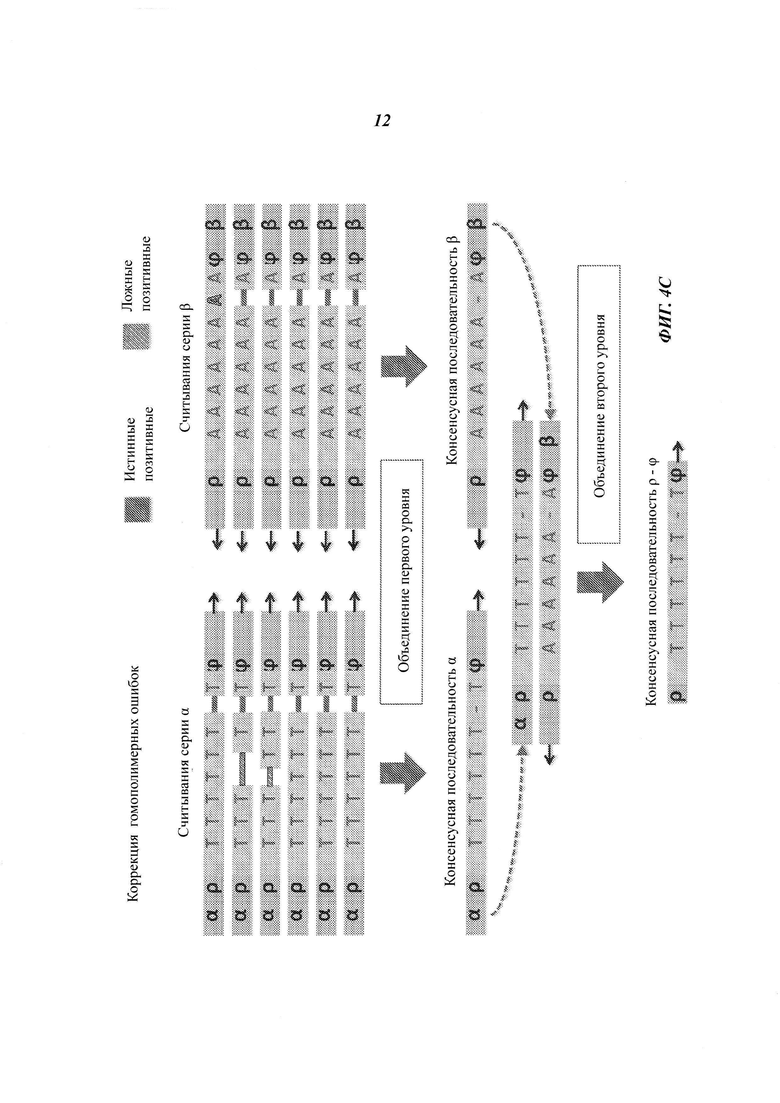

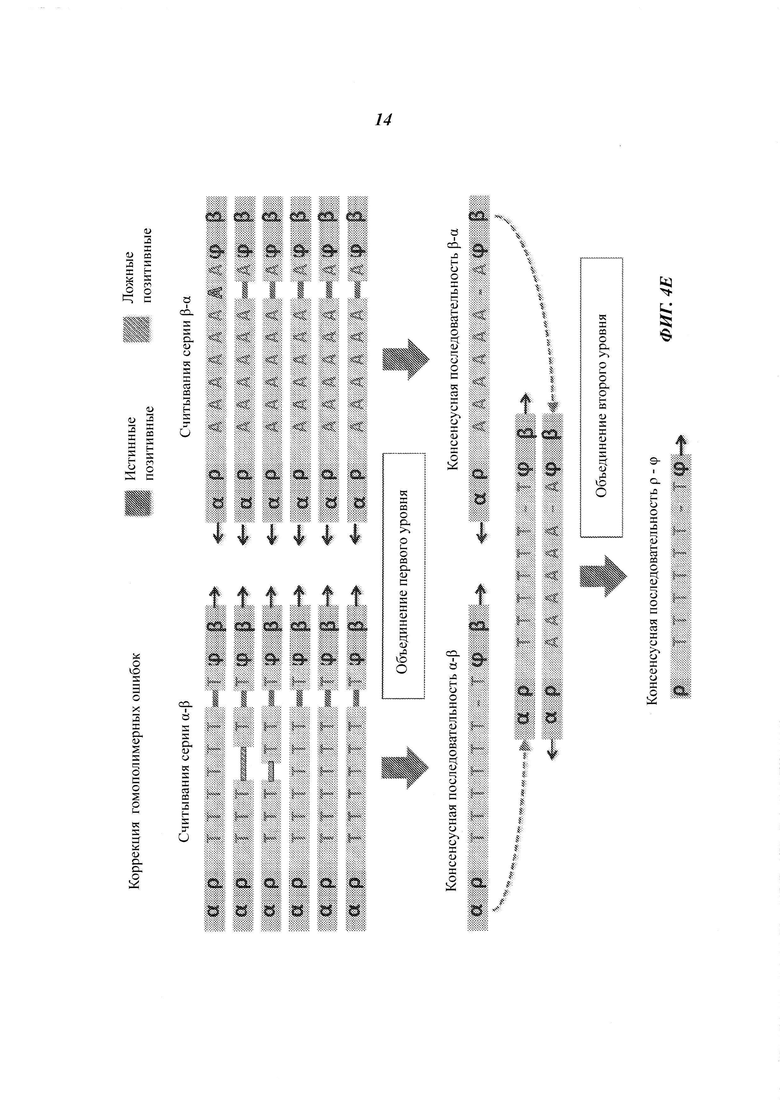
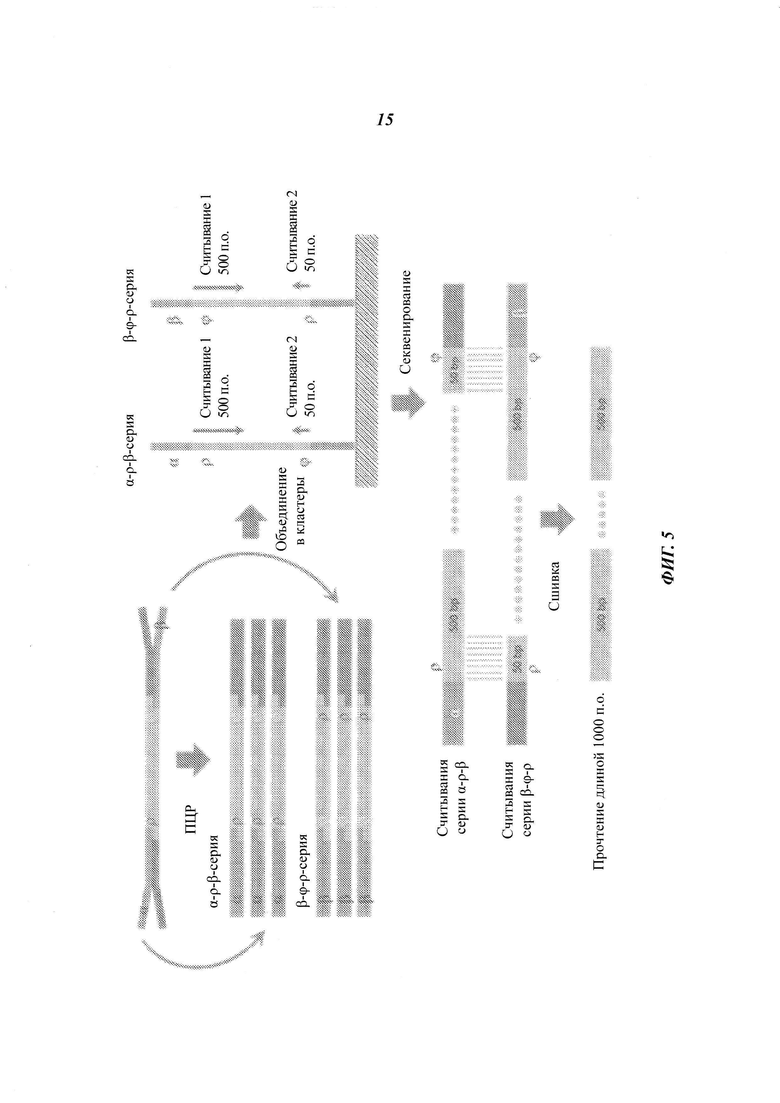
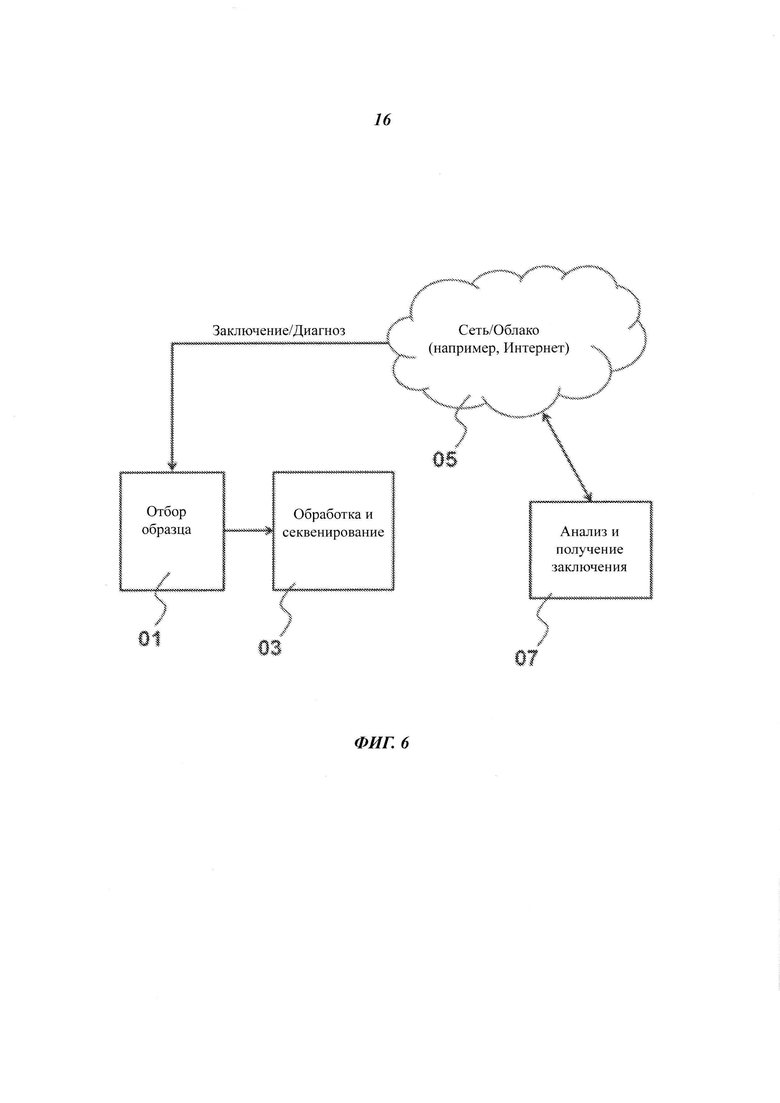


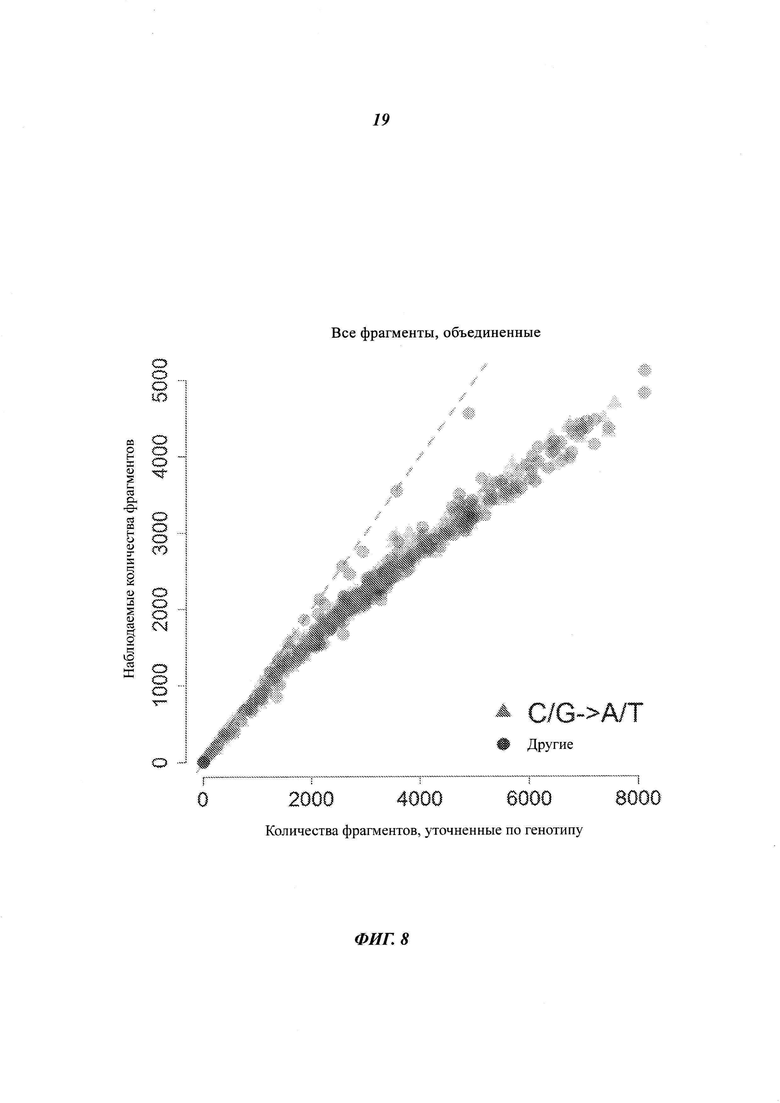
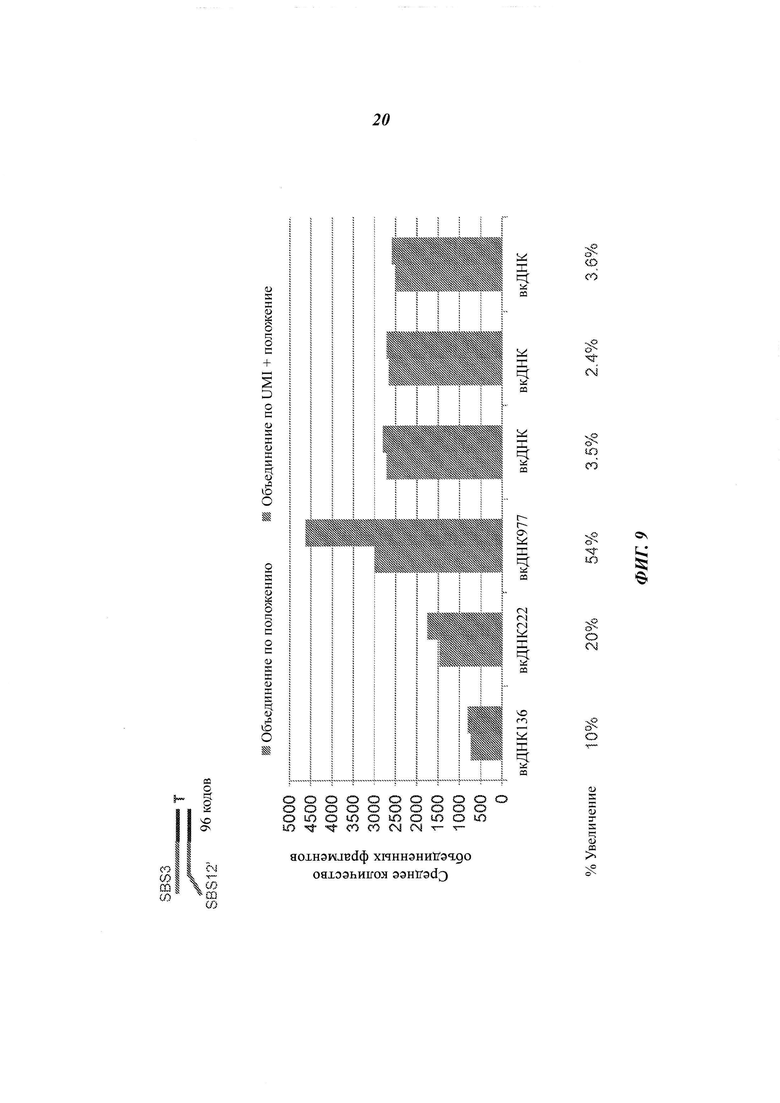
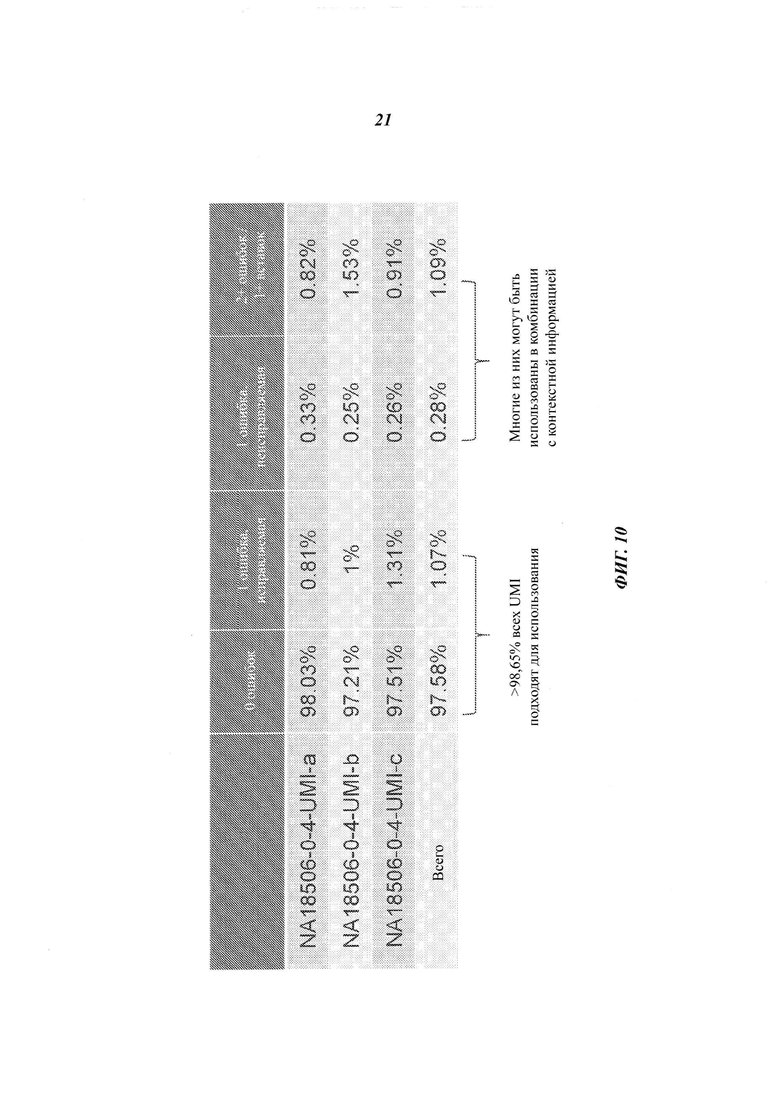
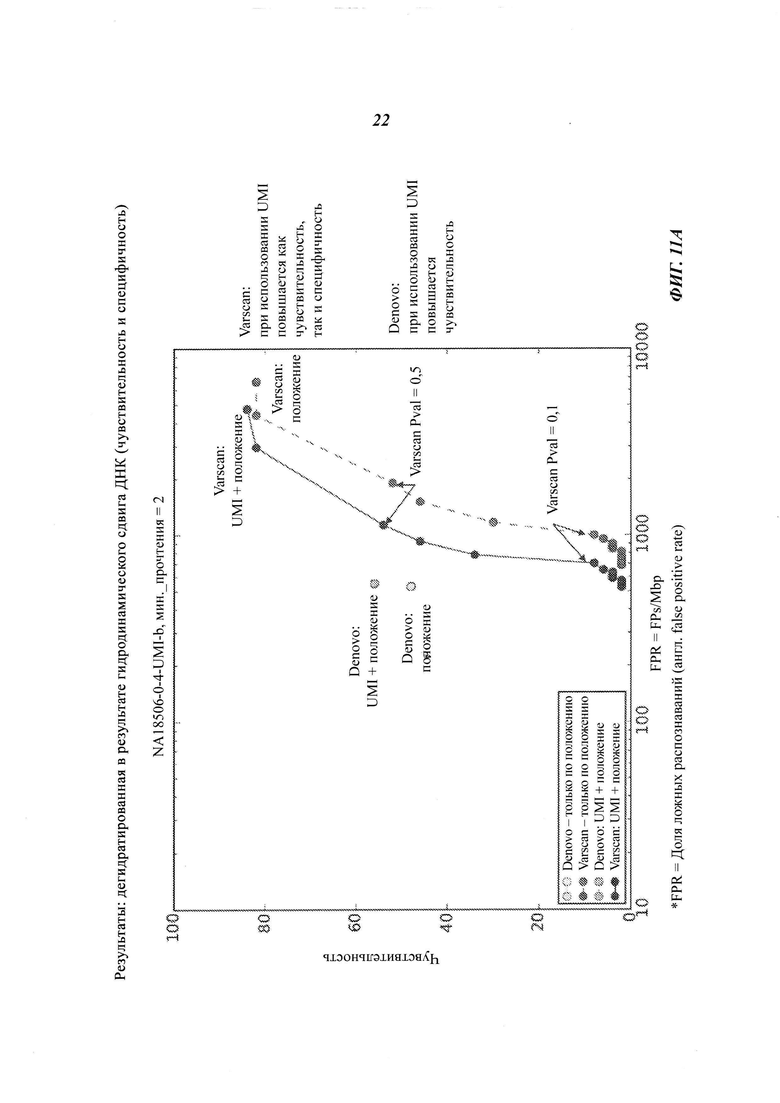
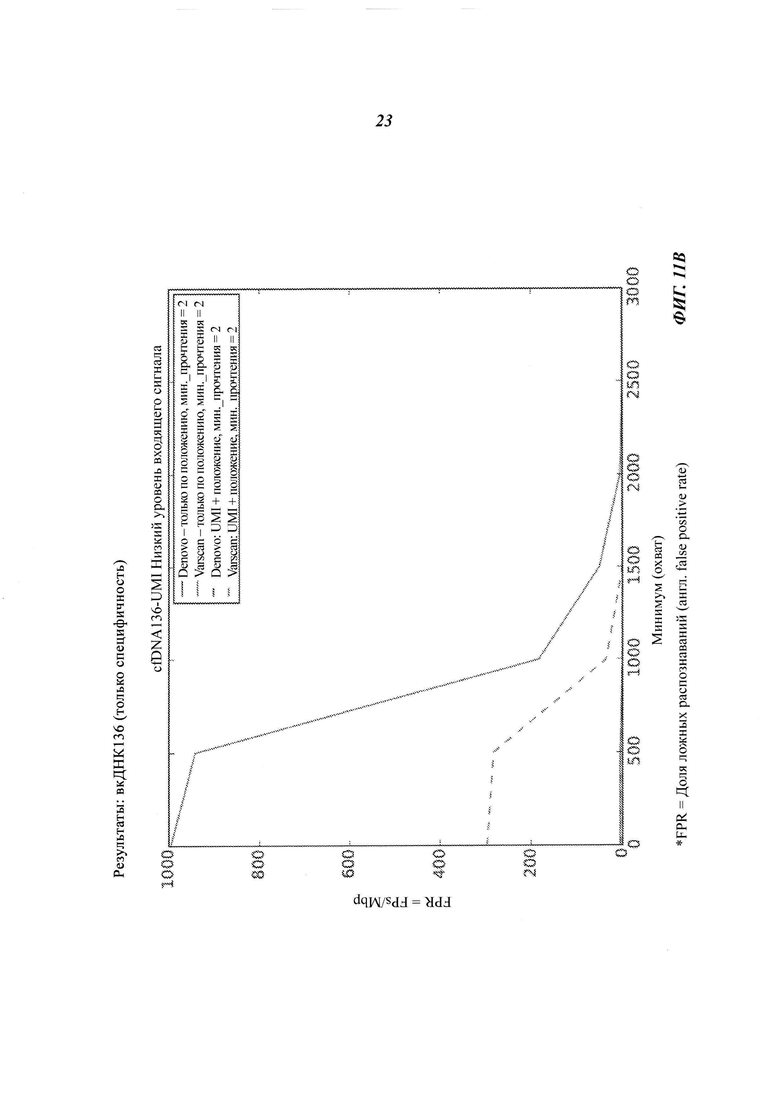
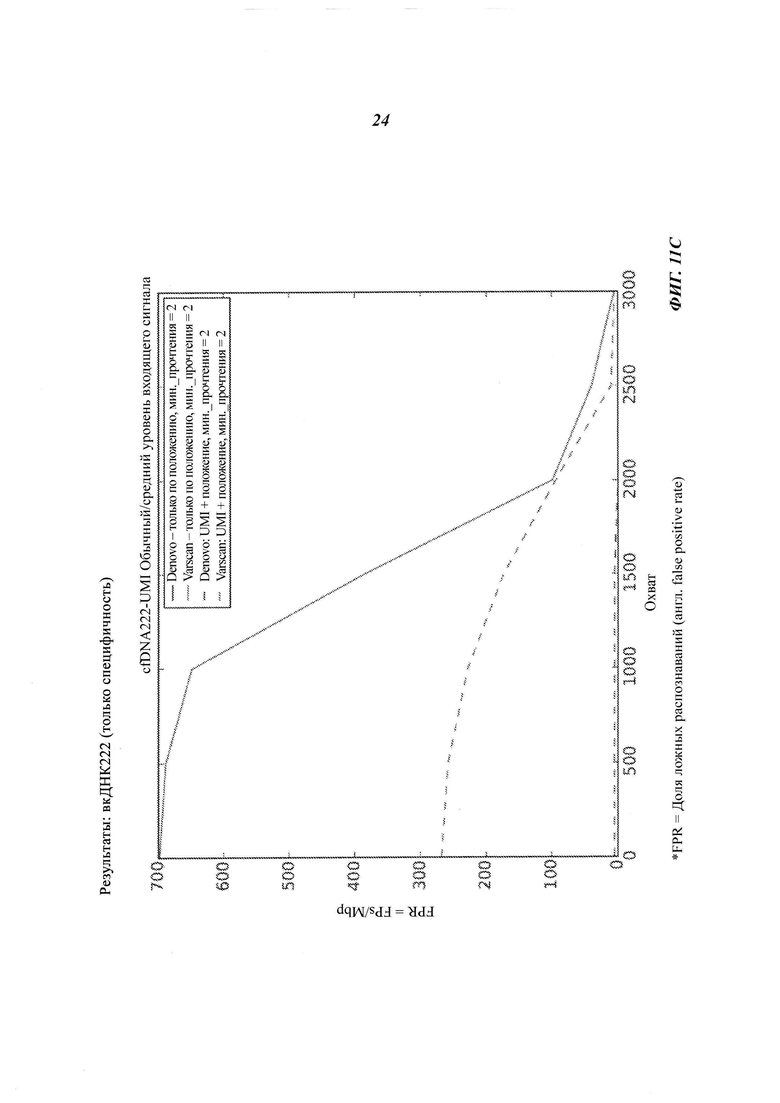
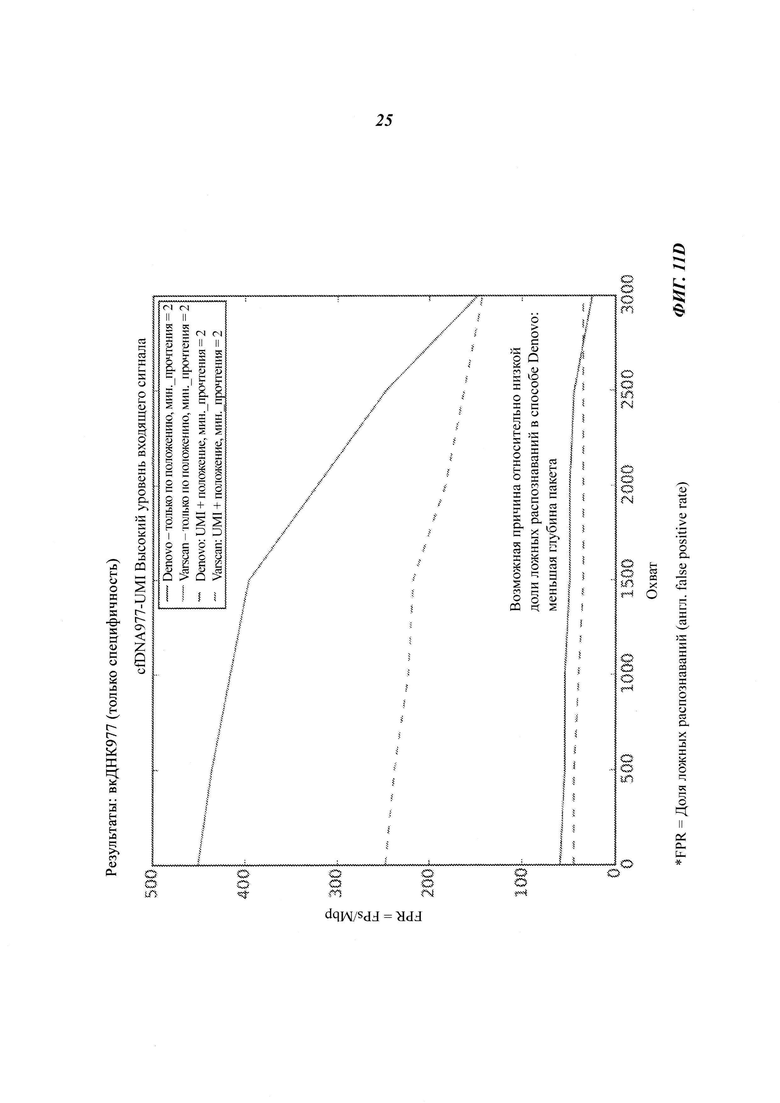
Настоящее изобретение относится к биотехнологии. Предложен способ секвенирования молекул нуклеиновых кислот с применением физических и виртуальных уникальных молекулярных индексов (UMI), где каждый UMI представляет собой олигонуклеотидную последовательность, которая может быть использована для идентификации индивидуальной молекулы фрагмента двухцепочечной ДНК, причем физический UMI представлен в составе адаптера, который присоединяется к фрагменту двухцепочечной ДНК, находящейся в образце, а виртуальный UMI представляет собой уникальную подпоследовательность в исходной молекуле ДНК. Данное изобретение может найти дальнейшее применение в способах анализа биологических образцов. 38 з.п. ф-лы, 11 ил., 2 пр.
1. Способ секвенирования молекул нуклеиновых кислот из образца с применением уникальных молекулярных индексов (UMI), где каждый уникальный молекулярный индекс (UMI) представляет собой олигонуклеотидную последовательность, которая может быть использована для идентификации индивидуальной молекулы фрагмента двухцепочечной ДНК, содержащейся в образце, включающий:
(a) прикрепление адаптеров к обоим концам фрагментов двухцепочечной ДНК, находящейся в образце, с получением продуктов присоединения адаптера к ДНК, где каждый адаптер включает двухцепочечную гибридизованную область, одноцепочечное 5’-плечо, одноцепочечное 3’-плечо и физический UMI на одной цепочке или на каждой цепочке адаптера, причем физический UMI выбран из множества физических UMI, и где каждый фрагмент двухцепочечной ДНК в образце содержит виртуальный UMI на одной цепочке или на каждой цепочке фрагмента двухцепочечной ДНК;
(b) амплификацию обеих цепочек продуктов присоединения адаптера к ДНК с образованием множества амплифицированных полинуклеотидов;
(c) секвенирование множества амплифицированных полинуклеотидов, в результате чего получают множество прочтений, каждое из которых содержит последовательность физического UMI, соответствующую физическому UMI на адаптированном фрагменте, и последовательность виртуального UMI, соответствующую виртуальному UMI на фрагменте двухцепочечной ДНК в образце;
(d) идентификацию множества последовательностей физических UMI для множества прочтений;
(e) идентификацию множества последовательностей виртуальных UMI для множества прочтений; и
(f) определение последовательностей фрагментов двухцепочечной ДНК, находящейся в образце, с использованием множества прочтений, полученных в этапе (c), множества последовательностей физических UMI, идентифицированных при выполнении этапа (d), и множества последовательностей виртуальных UMI, идентифицированных в этапе (e), и
где этап (f) включает:
(i) для каждого из фрагментов двухцепочечной ДНК, находящейся в образце, объединение первого множества прочтений, каждое из которых включает последовательность первого физического UMI из множества последовательностей физических UMI и последовательность первого виртуального UMI из множества последовательностей виртуальных UMI, для определения консенсусной нуклеотидной последовательности; и
(ii) определение последовательности фрагмента двухцепочечной ДНК, исходя из консенсусной нуклеотидной последовательности;
причем физический UMI физически присоединен к молекуле ДНК, а виртуальный UMI представляет собой уникальную подпоследовательность в исходной молекуле ДНК, и причем множество физических UMI включает неслучайные UMI.
2. Способ по п.1, в котором множество физических UMI включает случайные UMI.
3. Способ по п.2, в котором каждый неслучайный UMI отличается от любого другого неслучайного UMI, находящихся в адаптерах, по меньшей мере двумя нуклеотидами в соответствующих положениях последовательности неслучайных UMI.
4. Способ по п.3, в котором множество физических UMI включает не более чем приблизительно 10000 уникальных неслучайных UMI.
5. Способ по п.4, в котором множество физических UMI включает не более чем приблизительно 1000 уникальных неслучайных UMI.
6. Способ по п.5, в котором множество физических UMI включает не более чем приблизительно 500 уникальных неслучайных UMI.
7. Способ по п.6, в котором множество физических UMI включает не более чем приблизительно 100 уникальных неслучайных UMI.
8. Способ по п.7, в котором множество физических UMI включает приблизительно 96 уникальных неслучайных UMI.
9. Способ по п.1, в котором прикрепление адаптеров к обоим концам фрагментов двухцепочечной ДНК включает связывание адаптеров с обоими концами фрагментов двухцепочечной ДНК.
10. Способ по п.1, в котором множество физических UMI включает менее 12 нуклеотидов.
11. Способ по п.10, в котором множество UMI включает не более 6 нуклеотидов.
12. Способ по п.10, в котором множество UMI включает не более 4 нуклеотидов.
13. Способ по п.1, в котором каждый из адаптеров включает физический UMI на каждой цепочке адаптеров в двухцепочечной гибридизованной области.
14. Способ по п.13, в котором физический UMI находится на конце или вблизи конца двухцепочечной гибридизованной области, причем конец двухцепочечной гибридизованной области противоположен 3’-плечу или 5’-плечу.
15. Способ по п.14, в котором физический UMI находится на конце двухцепочечной гибридизованной области или на расстоянии одного нуклеотида от конца двухцепочечной гибридизованной области.
16. Способ по п.15, в котором каждый из адаптеров включает в двухцепочечной гибридизованной области тринуклеотид 5’-TGG-3’ или тринуклеотид 3’-ACC-5’, соседствующий с физическим UMI.
17. Способ по п.16, в котором каждый из адаптеров включает последовательность праймера прочтения на каждой цепочке двухцепочечной гибридизованной области.
18. Способ по п.1, в котором каждый из адаптеров включает физический UMI только на одной из цепочек адаптеров на одноцепочечном 5’-плече или одноцепочечном 3’-плече.
19. Способ по п.18, в котором этап (f) включает:
(i) объединение прочтений, имеющих один и тот же первый физический UMI, в первую группу с целью получения первой консенсусной нуклеотидной последовательности;
(ii) объединение прочтений, имеющих один и тот же второй физический UMI, во вторую группу с целью получения второй консенсусной нуклеотидной последовательности; и
(iii) определение из первой и второй консенсусных нуклеотидных последовательностей последовательности одного из фрагментов двухцепочечной ДНК, содержащихся в образце.
20. Способ по п.19, в котором этап (iii) включает: (1) получение третьей консенсусной нуклеотидной последовательности из информации о местоположении и информации о последовательности первой и второй консенсусных нуклеотидных последовательностей, и (2) определение последовательности одного из фрагментов двухцепочечной ДНК из третьей консенсусной нуклеотидной последовательности.
21. Способ по п.18, в котором этап (e) включает идентификацию множества виртуальных UMI, причем каждый из адаптеров включает физический UMI только на одноцепочечном 5’-плече или одноцепочечном 3’-плече.
22. Способ по п.21, в котором этап (f) включает:
(i) объединение прочтений, имеющих первый физический UMI и по меньшей мере один виртуальный UMI в направлении прочтения, и прочтений, имеющих второй физический UMI и по меньшей мере один виртуальный UMI в направлении прочтения, для определения консенсусной нуклеотидной последовательности; и
(ii) определение последовательности одного из фрагментов двухцепочечной ДНК, содержащихся в образце, на основании данных консенсусной нуклеотидной последовательности.
23. Способ по п.1, в котором каждый из адаптеров включает физический UMI на каждой цепочке адаптеров в двухцепочечной области адаптеров, причем физический UMI одной цепочки комплементарен физическому UMI другой цепочки.
24. Способ по п.22, в котором этап (f) включает:
(i) объединение прочтений, имеющих первый физический UMI, по меньшей мере один виртуальный UMI и второй физический UMI в направлении 5’-3’, и прочтений, имеющих второй физический UMI, по меньшей мере один виртуальный UMI и первый физический UMI в направлении 5’-3’, для определения консенсусной нуклеотидной последовательности; и
(ii) определение последовательности одного из фрагментов двухцепочечной ДНК, содержащихся в образце, на основании данных консенсусной нуклеотидной последовательности.
25. Способ по п.1, в котором каждый из адаптеров включает первый физический UMI на 3’-плече адаптера и второй физический UMI на 5’-плече адаптера, причем первый физический UMI и второй физический UMI не комплементарны друг другу.
26. Способ по п.24, в котором этап (f) включает:
(i) объединение прочтений, имеющих первый физический UMI, по меньшей мере один виртуальный UMI и второй физический UMI в направлении 5’-3’, и прочтений, имеющих третий физический UMI, по меньшей мере один виртуальный UMI и четвертый физический UMI в направлении 5’-3’, для определения консенсусной нуклеотидной последовательности; и
(ii) определение последовательности одного из фрагментов двухцепочечной ДНК, содержащихся в образце, на основании данных консенсусной нуклеотидной последовательности.
27. Способ по п.1, в котором по меньшей мере некоторые из виртуальных UMI получены из подпоследовательностей, находящихся на концах или вблизи концов фрагментов двухцепочечной ДНК, содержащихся в образце.
28. Способ по п.1, в котором один или более физических UMI и/или один или более виртуальных UMI уникальным образом ассоциированы с фрагментом двухцепочечной ДНК, содержащимся в образце.
29. Способ по п.1, в котором фрагменты двухцепочечной ДНК, содержащиеся в образце, включают более приблизительно 1000 фрагментов ДНК.
30. Способ по п.1, в котором множество виртуальных UMI включают UMI, содержащие от приблизительно 6 п.о. до приблизительно 24 п.о. (пар оснований).
31. Способ по п.30, в котором множество виртуальных UMI включают UMI, содержащие от приблизительно 6 п.о. до приблизительно 10 п.о.
32. Способ по п.1, в котором получение множества прочтений в операции (c) включает: получение двух прочтений парных концов от каждого из амплифицированных полинуклеотидов, где два прочтения парных концов включают длинное прочтение и короткое прочтение, причем длинное прочтение имеет большую длину, чем короткое прочтение.
33. Способ по п.32, в котором этап (f) включает:
объединение пар прочтений, ассоциированных с первым физическим UMI, в первую группу и объединение пар прочтений, ассоциированных со вторым физическим UMI, во вторую группу, где первый и второй физические UMI уникальным образом ассоциированы с двухцепочечным фрагментом, находящимся в образце; и
определение последовательности двухцепочечного фрагмента, содержащегося в образце, на основании информации о последовательности длинных прочтений, имеющихся в первой группе, и информации о последовательности длинных прочтений, имеющихся во второй группе.
34. Способ по п.32, в котором длина длинного прочтения составляет приблизительно 500 п.о. или более.
35. Способ по п.32, в котором длина короткого прочтения составляет приблизительно 50 п.о. или менее.
36. Способ по п.1, отличающийся тем, что способ включает подавление ошибок, возникающих при выполнении одной или более из следующих операций: ПЦР, создания библиотеки, объединения в кластеры и секвенирования.
37. Способ по п.1, в котором амплифицированные полинуклеотиды включают аллели с аллельными частотами, составляющими приблизительно менее 1%.
38. Способ по п.37, в котором амплифицированные полинуклеотиды включают молекулу внеклеточной ДНК, образованную в опухоли, и аллель является индикатором опухоли.
39. Способ по п.1, в котором секвенирование множества амплифицированных полинуклеотидов включает получение прочтений, содержащих по меньшей мере приблизительно 100 п.о.
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| PANG-CHUI S., YU-KEUNG M | |||
| "XcmI as a universal restriction enzyme for single-stranded DNA", Gene, 1993, 133(1):85-89 | |||
| KIVIOJA T | |||
| et al | |||
| "Counting absolute numbers of molecules using unique molecular identifiers." Nature, 2012, 9(1): 72-74 | |||
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| КРАСНОВ Я | |||
| М | |||
| и др | |||
| "Современные методы секвенирования ДНК (обзор)." Проблемы особо опасных инфекций, 2014, 2: 73-79. | |||

