ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] Настоящая заявка испрашивает приоритет в соответствии с § 119(e) Раздела 35 Свода законов США по предварительной заявке на патент США №62/447,851, имеющей название: «Способы и системы для получения и коррекции ошибок наборов уникальных молекулярных индексов с гетерогенной длиной молекул» ("METHODS AND SYSTEMS FOR GENERATION AND ERROR-CORRECTION OF UNIQUE MOLECULAR INDEX SETS WITH HETEROGENEOUS MOLECULAR LENGTHS"), поданной 18 января 2017 г., которая включена в настоящую заявку посредством ссылки полностью для любых целей.
ОБЛАСТЬ ТЕХНИКИ
[0002] Технология секвенирования нового поколения обеспечивает все более высокую скорость секвенирования, позволяющую добиться большей глубины секвенирования. Однако, поскольку на точность и чувствительность секвенирования влияют ошибки и шум из различных источников, например, вследствие дефектов образцов, ПНР при получении, обогащении, кластеризации и секвенировании библиотек, увеличение глубины секвенирования само по себе не может гарантировать детекцию последовательностей с очень низкой частотой аллелей, например, во внеклеточной фетальной ДНК (вкДНК) в материнской плазме, в циркулирующей опухолевой ДНК (цоДНК) и субклональных мутаций патогенов. Соответственно, желательна разработка способов определения последовательностей молекул ДНК в незначительных количествах и/или при низкой частоте аллелей с одновременным подавлением неточности секвенирования из-за различных источников ошибок.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0003] Раскрытые варианты реализации относятся к способам, устройству, системам и компьютерным программным продуктам для определения последовательностей фрагментов нуклеиновых кислот с использованием уникальных молекулярных индексов (UMI). Согласно некоторым вариантам реализации указанные UMI включают неслучайные UMI (NRUMI) или неслучайные уникальные молекулярные индексы с вариабельной длиной (vNRUMI).
[0004] Согласно одному аспекту настоящего изобретения предложены способы секвенирования молекул нуклеиновой кислоты из образца. Указанный способ включает: (а) обработку адаптерами фрагментов ДНК в указанном образце с получением продуктов ДНК-адаптер, при этом каждый адаптер содержит неслучайный уникальный молекулярный индекс, при этом неслучайные уникальные молекулярные индексы указанных адаптеров характеризуются по меньшей мере двумя разными длинами молекул и образуют набор неслучайных уникальных молекулярных индексов с вариабельной длиной (vNRUMI); (b) амплификацию указанных продуктов ДНК-адаптер с получением множества амплифицированных полинуклеотидов; (с) секвенирование множества амплифицированных полинуклеотидов с получением таким образом множества ридов (прочтений), ассоциированных с набором vNRUMI; (d) идентификацию среди множества ридов тех ридов, которые ассоциированы с одним и тем же неслучайным уникальным молекулярным индексом с вариабельной длиной (vNRUMI); и (е) определение последовательности фрагмента ДНК в указанном образце с использованием ридов, ассоциированных с одним и тем же vNRUMI.
[0005] Согласно некоторым вариантам реализации идентификация ридов, ассоциированных с одним и тем же vNRUMI, включает получение для каждого рида из указанного множества ридов показателей выравнивания для указанного набора vNRUMI, при этом каждый из полученных показателей является индикатором сходства между частичной последовательностью рида и vNRUMI, где указанная частичная последовательность находится в той области указанного рида, где предположительно расположены нуклеотиды, происходящие из указанного vNRUMI.
[0006] Согласно некоторым вариантам реализации указанные показатели выравнивания основаны на совпадениях нуклеотидов и изменениях нуклеотидов между указанной частичной последовательностью рида и указанным vNRUMI. Согласно некоторым вариантам реализации указанные изменения (редактирования) нуклеотидов включают замены, добавления и делеции нуклеотидов. Согласно некоторым вариантам реализации каждый показатель выравнивания включает штраф за несовпадения в начале последовательности, однако не включает штраф за несовпадения в конце указанной последовательности.
[0007] Согласно некоторым вариантам реализации получение показателя выравнивания между ридом и vNRUMI включает: (а) вычисление показателя выравнивания между vNRUMI и каждой из всех возможных префиксных последовательностей частичной последовательности указанного рида; (b) вычисление показателя выравнивания между частичной последовательностью указанного рида и каждой из всех возможных префиксных последовательностей указанного vNRUMI; и (с) получение максимального показателя выравнивания из показателей выравнивания, вычисленных по (а) и (b), в качестве показателя выравнивания между указанным ридом и указанным vNRUMI.
[0008] Согласно некоторым вариантам реализации указанная частичная последовательность имеет длину, равную длине самого длинного vNRUMI в наборе vNRUMI. Согласно некоторым вариантам реализации идентификация ридов, ассоциированных с одним и тем же vNRUMI, по (d) дополнительно включает: выбор для каждого рида из указанного множества ридов по меньшей мере одного vNRUMI из набора vNRUMI на основании показателей выравнивания; и ассоциацию каждого рида из указанного множества ридов по меньшей мере с одним vNRUMI, выбранным для указанного рида.
[0009] Согласно некоторым вариантам реализации выбор по меньшей мере одного vNRUMI из набора vNRUMI включает выбор vNRUMI с самым высоким значением показателя выравнивания в наборе vNRUMI. Согласно некоторым вариантам реализации указанный по меньшей мере один vNRUMI включает два или более vNRUMI.
[0010] Согласно некоторым вариантам реализации указанный способ дополнительно включает выбор одного из двух или более vNRUMI, в качестве одного и того же vNRUMI по (d) и (е).
[0011] Согласно некоторым вариантам реализации адаптеры для применения по (а) получают путем: (i) обеспечения набора последовательностей олигонуклеотидов по меньшей мере с двумя разными длинами молекул; (ii) выбора поднабора последовательностей олигонуклеотидов из набора последовательностей олигонуклеотидов, при этом все редакционные расстояния между последовательностями олигонуклеотидов указанного поднабора последовательностей олигонуклеотидов соответствуют пороговому значению, причем указанный поднабор последовательностей олигонуклеотидов образует набор vNRUMI; и (iii) синтеза адаптеров, каждый из которых включает двуцепочечную гибридизованную область, одноцепочечное 5'-плечо, одноцепочечное 3'-плечо и по меньшей мере один vNRUMI из набора vNRUMI. Согласно некоторым вариантам реализации указанное пороговое значение равно 3. Согласно некоторым вариантам реализации указанный набор vNRUMI включает vNRUMI из 6 нуклеотидов и vNRUMI из 7 нуклеотидов.
[0012] Согласно некоторым вариантам реализации определение по (е) включает объединение ридов, ассоциированных с одним и тем же vNRUMI в группу с получением консенсусной последовательности нуклеотидов для последовательности фрагмента ДНК в указанном образце. Согласно некоторым вариантам реализации указанную консенсусную последовательность нуклеотидов получают частично на основе показателей качества ридов.
[0013] Согласно некоторым вариантам реализации определение по (е) включает: идентификацию среди ридов, ассоциированных с одним и тем же vNRUMI, ридов, которые характеризуются тем же положением или аналогичными положениями рида в референсной последовательности, и определение последовательности фрагмента ДНК с использованием ридов, которые (i) ассоциированы с одним и тем же vNRUMI и (ii) отличаются тем же положением рида или аналогичными положениями рида в референсной последовательности.
[0014] Согласно некоторым вариантам реализации указанный набор vNRUMI включает не более чем приблизительно 10000 разных vNRUMI. Согласно некоторым вариантам реализации указанный набор vNRUMI включает не более чем приблизительно 1000 разных vNRUMI. Согласно некоторым вариантам реализации указанный набор vNRUMI включает не более чем приблизительно 200 разных vNRUMI.
[0015] Согласно некоторым вариантам реализации обработка адаптерами фрагментов ДНК в указанном образце включает обработку адаптерами обоих концов фрагментов ДНК в указанном образце.
[0016] Согласно другому аспекту настоящее изобретение относится к способам получения адаптеров для секвенирования, включающим: (а) обеспечение набора последовательностей олигонуклеотидов по меньшей мере с двумя разными длинами молекул; (b) выбор поднабора последовательностей олигонуклеотидов из набора последовательностей олигонуклеотидов, где все редакционные расстояния между последовательностями олигонуклеотидов указанного поднабора последовательностей олигонуклеотидов соответствуют пороговому значению, и указанный поднабор последовательностей олигонуклеотидов образует набор неслучайных уникальных молекулярных индексов с вариабельной длиной (vNRUMI); и (с) синтез множества адаптеров для секвенирования, причем каждый адаптер для секвенирования включает двуцепочечную гибридизованную область, одноцепочечное 5'-плечо, одноцепочечное 3'-плечо и по меньшей мере один vNRUMI из набора vNRUMI.
[0017] Согласно некоторым вариантам реализации (b) включает: (i) выбор последовательности олигонуклеотидов из набора последовательностей олигонуклеотидов; (ii) добавление выбранного олигонуклеотида к расширяемому набору последовательностей олигонуклеотидов и удаление выбранного олигонуклеотида из набора последовательностей олигонуклеотидов для получения сокращенного набора последовательностей олигонуклеотидов; (iii) выбор текущей последовательности олигонуклеотидов из сокращенного набора, которая приводит к максимизации функции расстояния, где функция расстояния представляет собой минимальное редакционное расстояние между указанной текущей последовательностью олигонуклеотидов и любыми последовательностями олигонуклеотидов в расширяемом наборе, причем указанная функция расстояния соответствует пороговому значению; (iv) добавление указанного текущего олигонуклеотида к расширяемому набору и удаление указанного текущего олигонуклеотида из сокращенного набора; (v) повторение (iii) и (iv) один или более раз; и (vi) обеспечение расширяемого набора в качестве поднабора последовательностей олигонуклеотидов, образующих набор vNRUMI.
[0018] Согласно некоторым вариантам реализации (v) включает повторение (iii) и (iv) до тех пор, пока функция расстояния не перестанет соответствовать пороговому значению.
[0019] Согласно некоторым вариантам реализации (v) включает повторение (iii) и (iv) до тех пор, пока расширяемый набор не достигает заданного размера.
[0020] Согласно некоторым вариантам реализации указанная текущая последовательность олигонуклеотидов или последовательность олигонуклеотидов в расширяемом наборе короче самой длинной последовательности олигонуклеотидов в наборе последовательностей олигонуклеотидов, и указанный способ дополнительно включает, до осуществления (iii), (1) прибавление тиминового основания, или тиминового основания наряду с любым из четырех оснований к текущей последовательности олигонуклеотидов или последовательности олигонуклеотидов в расширяемом наборе, с формированием таким образом дополненной последовательности такой же длины, что и самая длинная последовательность олигонуклеотидов в наборе последовательностей олигонуклеотидов; и (2) использование дополненной последовательности для вычисления минимального редакционного расстояния. Согласно некоторым вариантам реализации указанные редакционные расстояния представляют собой расстояния Левенштейна. Согласно некоторым вариантам реализации указанное пороговое значение равно 3.
[0021] Согласно некоторым вариантам реализации указанный способ дополнительно включает, до осуществления (b), удаление определенных последовательностей олигонуклеотидов из набора последовательностей олигонуклеотидов с получением отфильтрованного набора последовательностей олигонуклеотидов; и обеспечение отфильтрованного набора последовательностей олигонуклеотидов в качестве набора последовательностей олигонуклеотидов, из которого выбирают поднабор.
[0022] Согласно некоторым вариантам реализации указанные определенные последовательности олигонуклеотидов включают последовательности олигонуклеотидов, включающие три или более последовательных идентичных оснований. Согласно некоторым вариантам реализации указанные определенные последовательности олигонуклеотидов включают последовательности олигонуклеотидов, общее число гуаниновых и цитозиновых оснований в которых меньше 2, и последовательности олигонуклеотидов, общее число гуаниновых и цитозиновых оснований в которых больше 4.
[0023] Согласно некоторым вариантам реализации указанные определенные последовательности олигонуклеотидов включают последовательности олигонуклеотидов, включающие одно и то же основание в последних двух положениях. Согласно некоторым вариантам реализации указанные определенные последовательности олигонуклеотидов включают последовательности олигонуклеотидов, включающие частичную последовательность, совпадающую со 3'-концом одного или более праймеров для секвенирования.
[0024] Согласно некоторым вариантам реализации указанные определенные последовательности олигонуклеотидов включают последовательности олигонуклеотидов, включающие тиминовое основание в последнем положении указанных последовательностей олигонуклеотидов.
[0025] Согласно некоторым вариантам реализации указанный набор vNRUMI включает vNRUMI из 6 нуклеотидов и vNRUMI из 7 нуклеотидов.
[0026] Согласно дополнительному аспекту настоящее изобретение относится к способу секвенирования молекул нуклеиновой кислоты из образца, в том числе (а) обработку адаптерами фрагментов ДНК в указанном образце с получением продуктов ДНК-адаптер, где каждый адаптер содержит неслучайный уникальный молекулярный индекс, при этом неслучайные уникальные молекулярные индексы указанных адаптеров характеризуются по меньшей мере двумя разными длинами молекул и образуют набор неслучайных уникальных молекулярных индексов с вариабельной длиной (vNRUMI); (b) амплификацию указанных продуктов ДНК-адаптер с получением множества амплифицированных полинуклеотидов; (с) секвенирование множества амплифицированных полинуклеотидов с получением таким образом множества ридов, ассоциированных с набором vNRUMI; и (d) идентификацию среди множества ридов тех ридов, которые ассоциированы с одним и тем же неслучайным уникальным молекулярным индексом с вариабельной длиной (vNRUMI).
[0027] Согласно некоторым вариантам реализации указанный способ дополнительно включает подсчет ридов, ассоциированных с одним и тем же vNRUMI.
[0028] Согласно другому аспекту настоящее изобретение относится к способу секвенирования молекул нуклеиновой кислоты из образца, в том числе (а) обработку адаптерами фрагментов ДНК в указанном образце с получением продуктов ДНК-адаптер, где каждый адаптер содержит уникальный молекулярный индекс (UMI), причем уникальные молекулярные индексы (UMI) указанных адаптеров характеризуются по меньшей мере двумя разными длинами молекул и образуют набор уникальных молекулярных индексов (vUMI) с вариабельной длиной; (b) амплификацию указанных продуктов ДНК-адаптер с получением множества амплифицированных полинуклеотидов; (с) секвенирование множества амплифицированных полинуклеотидов с получением таким образом множества ридов, ассоциированных с указанным набором vUMI; и (d) идентификацию среди множества ридов тех ридов, которые ассоциированы с одним и тем же уникальным молекулярным индексом с вариабельной длиной (vUMI).
[0029] Согласно некоторым вариантам реализации указанный способ дополнительно включает определение последовательности фрагмента ДНК в указанном образце с использованием ридов, ассоциированных с одним и тем же vUMI.
[0030] Согласно некоторым вариантам реализации указанный способ дополнительно включает подсчет ридов, ассоциированных с одними и теми же vUMI.
[0031] Согласно еще одному аспекту настоящее изобретение относится к способу секвенирования молекул нуклеиновой кислоты из образца, в том числе (а) обработку адаптерами фрагментов ДНК в указанном образце с получением продуктов ДНК-адаптер, при этом каждый адаптер содержит уникальный молекулярный индекс (UMI) в наборе уникальных молекулярных индексов (UMI); (b) амплификацию указанных продуктов ДНК-адаптер с получением множества амплифицированных полинуклеотидов; (с) секвенирование множества амплифицированных полинуклеотидов с получением таким образом множества ридов, ассоциированных с указанным набором UMI; (d) получение для каждого рида из указанного множества ридов показателей выравнивания для указанного набора UMI, при этом каждый из полученных показателей является индикатором сходства между частичной последовательностью рида и UMI; (е) идентификацию среди множества ридов тех ридов, которые ассоциированы с одним и тем же UMI, с использованием указанных показателей выравнивания; и (е) определение последовательности фрагмента ДНК в указанном образце с использованием ридов, ассоциированных с одним и тем же UMI.
[0032] Согласно некоторым вариантам реализации указанные показатели выравнивания основаны на совпадениях нуклеотидов и изменений нуклеотидов между частичной последовательностью указанного рида и указанного UMI. Согласно некоторым вариантам реализации каждый показатель выравнивания включает штрафы за несовпадения в начале последовательности, однако не включает штрафы за несовпадения в конце указанной последовательности. Согласно некоторым вариантам реализации указанный набор UMI включает UMI по меньшей мере с двумя разными длинами молекул.
[0033] Также предложены осуществляющие раскрытые способы система, устройство и компьютерные программные продукты для определения последовательностей фрагментов ДНК.
[0034] Согласно одному аспекту настоящего изобретения предложен компьютерный программный продукт, включающий энергонезависимый машиночитаемый носитель, где хранится программный код, который при исполнении одним или более процессорами компьютерной системы, приводит к осуществлению указанной компьютерной системой способа определения информации о представляющей интерес последовательности в образце с использованием уникальных молекулярных индексов (UMI). Указанный программный код включает инструкций для выполнения вышеописанных способов.
[0035] Хотя приведенные в настоящем документе примеры относятся к человеку и терминология в первую очередь касается вопросов, затрагивающих человека, принципы, описанные в настоящем документе, применимы к нуклеиновым кислотам из любого вируса, растения, животного или другого организма, и их популяций (метагеномов, вирусных популяций и т.п.) Указанные и другие признаки настоящего изобретения будут более понятны после изучения приведенного ниже описания, сопровождаемого чертежами, и прилагаемой формулы изобретения, или могут быть изучены в ходе практической реализации настоящего изобретения согласно описанию здесь и далее в настоящем документе.
ВКЛЮЧЕНИЕ ПОСРЕДСТВОМ ССЫЛОК
[0036] Все патенты, патентные заявки и другие публикации, в том числе все последовательности, раскрытые к указанных источниках, упоминаемые в настоящем документе, явным образом включены в настоящий документ посредством ссылок, в той же степени, как если бы каждая индивидуальная публикация, патент или патентная заявка была конкретным и индивидуальным образом включена посредством ссылки. Все цитируемые документы, в релевантной части, включены в настоящий документ полностью посредством ссылок и для целей, диктуемых контекстом цитирования в настоящем документе. Тем не менее цитирование какого-либо документа не должно быть истолковано как допущение того, что они представляют известный уровень техники применительно к настоящему изобретению.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0037] На Фиг. 1А приведена функциональная диаграмма, иллюстрирующая пример технологической схемы с применением UMI для секвенирования фрагментов нуклеиновых кислот.
[0038] На Фиг. 1В представлены фрагмент/молекула ДНК и адаптеры, используемые на начальных этапах выполнения технологической схемы, представленной на Фиг. 1A.
[0039] На Фиг. 1С приведена блок-схема, отражающая процесс секвенирования фрагментов ДНК с применением vNRUMI для подавления ошибок.
[0040] Фиг. 1D иллюстрирует процесс 140 для получения адаптеров для секвенирования, содержащих vNRUMI.
[0041] На Фиг. 1Е приведены примеры того, как частичная последовательность рида или запрашиваемая последовательность (Q) может быть сравнена с двумя референсными последовательностями (S1 и S2) в наборе vNRUMI.
[0042] Фиг. 1F иллюстрирует примеры того, как показатели глокального выравнивания могут обеспечивать лучшее подавление ошибок, чем показателя глобального выравнивания.
[0043] Фиг. 2А схематически иллюстрирует пять разных вариантов дизайна адаптеров, которые могут быть использованы согласно различным вариантам реализации.
[0044] Фиг. 2В иллюстрирует гипотетический процесс, в ходе которого происходит перепрыгивание UMI в ПЦР-реакции, включающей адаптеры, содержащие два физических UMI на двух плечах.
[0045] На Фиг. 2С представлены данные, отражающие различия показателей качества ридов последовательностей при применении NRUMI и в контрольных условиях.
[0046] На Фиг. 3А и 3В приведены диаграммы, представляющие материалы и продукты реакций лигирования адаптеров с двуцепочечными фрагментами в соответствии с некоторыми способами согласно описанию в настоящем документе.
[0047] Фиг. 4А-4Е иллюстрирует то, как способы согласно описанию в настоящем документе может подавлять разные источники ошибки при определении последовательности фрагмента двуцепочечной ДНК.
[0048] Фиг. 5 схематически иллюстрирует применение физических UMI и виртуальных UMI для эффективного получения длинных парно-концевых ридов.
[0049] На Фиг. 6 приведена блок-схема рассредоточенной системы для обработки тестового образца.
[0050] Фиг. 7 иллюстрирует компьютерную систему, которая может служить в качестве вычислительного устройства в соответствии с определенными вариантами реализации.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0051] Настоящее изобретение относится к способам, устройствам, системам и компьютерным программным продуктам для секвенирования нуклеиновых кислот, в частности, нуклеиновых кислот, присутствующих в ограниченном количестве или в низкой концентрации, например, фетальной вкДНК в материнской плазме или циркулирующей опухолевой ДНК (цоДНК) в крови пациента с раковым заболеванием.
[0052] Диапазоны численных значений включают все задающие указанный диапазон числа. Предполагается, что каждое максимальное числовое ограничение, приведенное в настоящем описании, включает каждое более низкое числовое ограничение, так, как если бы такие более низкие числовые ограничения были явным образом приведены в настоящем документе. Каждое минимальное числовое ограничение, приведенное в настоящем описании, включает каждое из более высоких числовых ограничений, так, как если бы такие более высокие числовые ограничения были явным образом приведены в настоящем документе. Каждый числовой диапазон, приведенный в настоящем описании, включает каждый из более узких числовых диапазонов, попадающих в такой более широкий числовой диапазон, как если бы все такие более узкие числовые диапазоны были явным образом указаны в настоящем документе.
[0053] Заголовки в данном документе не предназначены для ограничения раскрываемого изобретения.
[0054] Если в настоящем документе не указано иное, все технические и научные термины в настоящем документе имеют значения, соответствующие общеизвестным специалисту в данной области техники. Различные научные словари, которые включают термины, включенные в настоящий документ, хорошо известны и доступны специалистам в данной области техники. Хотя при практической реализации или тестировании вариантов реализации, раскрытых в настоящем документе, находят применение любые способы и материалы, аналогичные или эквивалентные описанным в настоящем документе, приведено описание некоторых способов и материалов.
[0055] Термины, определения которых приведены непосредственно далее в настоящем документе, более полно описаны в настоящем описании в целом. Следует понимать, что настоящее изобретение не ограничено конкретными описанными методологией, протоколами и реагентами, поскольку они могут варьировать в зависимости от контекста, в котором их используют специалисты в данной области техники.
Определения
[0056] В настоящем документе термины в единственном числе, в том числе сопровождаемые определением «указанный(-ая, -ое)», включают соответствующие термины во множественном числе, если из контекста явным образом не следует иное.
[0057] Если не указано иное, нуклеиновые кислоты записаны слева направо в направлении 5'→3', а последовательности аминокислот записаны слева направо в направлении от аминоконца к карбоксильному концу, соответственно.
[0058] Уникальные молекулярные индексы (UMI) представляют собой последовательности нуклеотидов, которыми обрабатывают молекулы ДНК или которые идентифицируют в молекулах ДНК, и которые могут быть использованы для различения индивидуальных молекул ДНК. Поскольку UMI используют для идентификации молекул ДНК, их также называют уникальными молекулярными идентификаторами. См., например, Kivioja, Nature Methods 9, 72-74 (2012). UMI могут быть секвенированы наряду с молекулами ДНК, с которыми они ассоциированы, для определения того, представляют ли собой последовательности ридов последовательности из той или другой исходной молекулы ДНК. Термин «UMI» используют в настоящем документе для обозначения как информации о последовательности полинуклеотида, так и физического полинуклеотида per se.
[0059] Обычно секвенируют несколько копий единственной исходной молекулы. В случае секвенирования путем синтеза с использованием технологии секвенирования Illumina исходная молекула может быть ПЦР-амплифицирована перед доставкой в проточную ячейку. И в случае ПЦР-амплификации, и без нее индивидуальные молекулы ДНК, внесенные в проточную ячейку, подвергают мостиковой амплификации или амплификации ExAmp для получения кластера. Каждая молекула в кластере происходит из одной и той же исходной молекулы ДНК, но секвенируется отдельно. Для коррекции ошибок и других целей может быть важно определить, что все риды из одного кластера идентифицированы как происходящие из одной и той же исходной молекулы. UMI позволяют осуществить указанную группировку. Молекула ДНК, копируемая путем амплификации или иным образом с получением нескольких копий указанной молекулы ДНК, называется исходной молекулой ДНК.
[0060] Наряду с ошибками, ассоциированными с исходными молекулами ДНК, ошибки могут также происходить в области, ассоциированной с UMI. Согласно некоторым вариантам реализации ошибка последнего типа может быть скорректирована путем картирования последовательности рида на наиболее вероятный UMI из пула UMI.
[0061] UMI аналогичны штрихкодам, которые широко используются для различения ридов из одного образца и ридов из других образцов, но UMI, в отличие от штрихкодов, используют для различения одной исходной молекулы ДНК и другой при совместном секвенировании множества молекул ДНК. Поскольку в образце может быть намного больше молекул ДНК, чем образцов при одном прогоне секвенирования, обычно в прогоне секвенирования значительно больше отдельных UMI, чем отдельных штрихкодов.
[0062] Как уже упоминалось, UMI могут применяться для индивидуальных молекул ДНК или идентифицированы в индивидуальных молекулах ДНК. Согласно некоторым вариантам реализации UMI могут применяться для молекул ДНК с применением способов, обеспечивающих физическое соединение или физическую связь указанных UMI с молекулами ДНК, например, путем лигирования или транспозиции с помощью полимеразы, эндонуклеазы, транспозаз и т.п. Указанные «применяемые» UMI, соответственно, также называют физическими UMI. В некоторых контекстах они могут также быть названы экзогенными UMI. UMI, идентифицированные в исходных молекулах ДНК, называют виртуальными UMI. В определенном контексте виртуальные UMI могут также быть названы эндогенными UMI.
[0063] Физические UMI могут быть определены многими способами. Например, они могут представлять собой случайные, псевдослучайные или частично случайные, или неслучайные последовательности нуклеотидов, которые инсертируют в адаптеры или иным способом встраивают в исходные молекулы ДНК, подлежащие секвенированию. Согласно некоторым вариантам реализации указанные физические UMI могут быть настолько уникальными, что каждый из них предположительно однозначно идентифицирует любую заданную исходную молекулу ДНК, присутствующую в образце. Получают коллекцию адаптеров, каждый из которых содержит физический UMI, и указанные адаптеры присоединяют к фрагментам или другим исходным молекулам ДНК, подлежащим секвенированию, и каждая из индивидуальных секвенированных молекул содержит UMI, который помогает отличить его от всех других фрагментов. Согласно таким вариантам реализации, очень значительное число разных физических UMI (например, от нескольких тысяч до миллионов) может быть использовано для однозначной идентификации фрагментов ДНК в образце.
[0064] Конечно, физические UMI должны иметь достаточную длину для обеспечения указанной уникальности для всех и каждой из исходных молекул ДНК. Согласно некоторым вариантам реализации может быть использован менее уникальный молекулярный идентификатор в сочетании с другими методиками идентификации, чтобы обеспечить однозначную идентификацию каждой исходной молекулы ДНК в ходе процесса секвенирования. Согласно таким вариантам реализации несколько фрагментов или адаптеров могут содержать один и тот же физический UMI. Другая информация, например, о местоположении выравнивания или виртуальных UMI, может быть скомбинирована с физическим UMI для однозначной идентификации ридов как происходящих из единственной исходной молекулы/единственного исходного фрагмента ДНК. Согласно некоторым вариантам реализации адаптеры включают физические UMI, ограниченные относительно малым числом неслучайных последовательностей, например, 120 неслучайных последовательностей. Такие физические UMI также называют неслучайными UMI. Согласно некоторым вариантам реализации неслучайные UMI могут быть скомбинированы с информацией о положении последовательности, положением последовательности и/или виртуальными UMI для идентификации ридов, которые могут происходить из одной и той же исходной молекулы ДНК. Идентифицированные риды могут быть скомбинированы с получением консенсусной последовательности, которая отражает последовательность исходной молекулы ДНК согласно описанию в настоящем документе. С использованием физических UMI, виртуальных UMI и/или местоположений выравнивания, можно идентифицировать риды, содержащие одни и те же или родственные UMI, или характеризуются одним и тем же местоположением, которые могут затем быть скомбинированы с получением одной или более консенсусных последовательностей. Процесс комбинирования ридов с получением консенсусной последовательности также называют «объединением» ридов, которое подробнее описано здесь и далее в настоящем документе.
[0065] «Виртуальный уникальный молекулярный индекс», или «виртуальный UMI» представляет собой уникальную частичную последовательность в исходной молекуле ДНК. Согласно некоторым вариантам реализации виртуальные UMI локализованы на концах или возле концов исходной молекулы ДНК. Одно или более таких уникальных концевых положений могут, по отдельности или в сочетании с другой информацией, позволять однозначно идентифицировать исходную молекулу ДНК. В зависимости от числа отдельных исходных молекул ДНК и числа нуклеотидов в виртуальных UMI, один или более виртуальных UMI позволяют однозначно идентифицировать исходные молекулы ДНК в образце. В некоторых случаях необходима комбинация двух виртуальных уникальных молекулярных идентификаторов для идентификации исходной молекулы ДНК. Такие комбинации могут быть крайне редкими, вероятно, встречаясь в образце единственный раз. В некоторых случаях один или более виртуальных UMI в комбинации с одним или более физическим UMI вместе могут однозначно идентифицировать исходную молекулу ДНК.
[0066] «Случайный UMI» может рассматриваться как физический UMI, выбранный как случайный образец, с заменой или без замены, из набора UMI, состоящего из всех возможных разных последовательностей олигонуклеотидов, при условии наличия одной или более длин последовательностей. Например, если каждый UMI в наборе UMI содержит n нуклеотидов, указанный набор включает 4^n UMI, содержащих последовательности, отличающиеся друг от друга. Случайный образец, выбранный из 4^n UMI, представляет собой случайный UMI.
[0067] И напротив, «неслучайный UMI» (NRUMI) в настоящем документе относится к физическому UMI, не являющемуся случайным UMI. Согласно некоторым вариантам реализации неслучайные UMI предварительно задают для конкретного эксперимента или применения. Согласно некоторым вариантам реализации используют правила получения последовательностей для набора или выбора образца из указанного набора для получения неслучайного UMI. Например, последовательности набора могут быть получены таким образом, чтобы содержать конкретный паттерн или паттерны. Согласно некоторым вариантам реализации каждая последовательность отличается от всех других последовательностей в наборе конкретным числом (например, 2, 3 или 4) нуклеотидов. Таким образом, ни одна последовательность неслучайного UMI не может быть преобразована в любую другую доступную последовательность неслучайного UMI путем замены числа нуклеотидов, меньшего, чем указанное конкретное число. Согласно некоторым вариантам реализации набор NRUMI, используемых в процессе секвенирования, включает не все возможные UMI для конкретной длины последовательности. Например, набор NRUMI, содержащих 6 нуклеотидов, может включать в общей сложности 96 разных последовательностей, вместо в общей сложности 4^6=4096 возможных разных последовательностей.
[0068] Согласно некоторым вариантам реализации, если неслучайные UMI выбирают из набора, содержащего не все возможные разные последовательностей, число неслучайных UMI меньше, иногда значимо, чем число исходных молекул ДНК. Согласно таким вариантам реализации информация о неслучайных UMI может быть скомбинирована с другой информацией, такой как информация о виртуальных UMI, местоположениях ридов на референсной последовательности и/или последовательности ридов, для идентификации ридов последовательностей, происходящих из одной и той же исходной молекулы ДНК.
[0069] Термин «неслучайный молекулярный индекс с вариабельной длиной» (vNRUMI) относится к UMI в наборе vNRUMI, выбранных из пула UMI с вариабельными длинами молекул (или гетерогенной длиной) с использованием неслучайного процесса выбора. Термин vNRUMI используют для обозначения как молекулы UMI, так и последовательности UMI. Согласно некоторым вариантам реализации определенные UMI могут быть удалены из пула UMI с получением отфильтрованного пула UMI, который затем используют для получения набора vNRUMI.
[0070] Согласно некоторым вариантам реализации каждый vNRUMI отличается от всех других vNRUMI в наборе, используемом в процессе, по меньшей мере заданным редакционным расстоянием. Согласно некоторым вариантам реализации набор vNRUMI, используемый в процессе секвенирования, включает не все возможные UMI, при условии использования релевантных длин молекул. Например, набор vNRUMI, содержащих 6 и 7 нуклеотидов, может включать в общей сложности 120 разных последовательностей (вместо в общей сложности 46+47=20480 возможных разных последовательностей). Согласно другим вариантам реализации последовательности не выбирают из набора случайным образом. Вместо этого некоторые последовательности выбирают с большей вероятностью, чем другие последовательности.
[0071] Термином «длина молекулы» также обозначают длину последовательности, которая может быть измерена в нуклеотидах. Также термин «длина молекулы» используют взаимозаменяемо с терминами «размер молекулы», «размер ДНК» и «длина последовательности».
[0072] Редакционное расстояние представляет собой метрику для количественного определения различия двух строк (например, слов) путем вычисления минимального числа операций, которые требуются для трансформации одной строки в другую. В биоинформатике оно может использоваться для количественного определения сходства последовательностей ДНК, которые могут рассматриваться как строки из букв А, С, G и Т.
[0073] Разные формы редакционного расстояния используют разные наборы строковых операций. Расстояние Левенштейна представляет собой распространенный тип редакционного расстояния. Строковые операции расстояния Левенштейна учитывают число делеций, инсерций и замен символов в строке. Согласно некоторым вариантам реализации могут быть использованы другие варианты редакционных расстояний. Например, другие варианты редакционного расстояния могут быть получены путем ограничения набора операций. Расстояние для самой длинной общей частичной последовательности (LCS) представляет собой редакционное расстояние с инсерциями и делециями в качестве всего двух допускаемых операций изменения, каждая из которых имеет удельную цену. Аналогичным образом, при допущении исключительно замен получают расстояние Хемминга, ограниченное строками равной длины. Расстояние Джаро-Винклера может быть получено на основании редакционного расстояния при допущении исключительно транспозиций.
[0074] Согласно некоторым вариантам реализации разным строковым операциям может быть присвоен разный вес для редакционного расстояния. Например, операции замены может быть присвоен вес, равный 3, тогда как инделу может быть присвоен вес, равный 2. Согласно некоторым вариантам реализации совпадениям разных видов может быть присвоен разный вес. Например совпадению А-А может быть присвоен вес, в два раза превышающий вес совпадения G-G.
[0075] Показатель выравнивания представляет собой показатель, отражающий сходство двух последовательностей, определенный с применением способа выравнивания. Согласно некоторым вариантам реализации показатель выравнивания учитывает число изменений (например, делеций, инсерций и замен символов в строке). Согласно некоторым вариантам реализации показатель выравнивания учитывает число совпадений. Согласно некоторым вариантам реализации показатель выравнивания учитывает как число совпадений, так и число изменений. Согласно некоторым вариантам реализации числу совпадений и изменений присваивают равное значение для показателя выравнивания. Например, показатель выравнивания может быть вычислен как: число совпадений - число инсерций - число делеций - число замен. Согласно другим вариантам реализации числам совпадений и изменений может быть присвоен разный вес. Например, показатель выравнивания может быть вычислен как:
число совпадений × 5 - число инсерций × 4 - число делеций × 4 - число замен × 6.
[0076] Термин «парно-концевые риды» относится к ридам, полученным путем парно-концевого секвенирования, когда получают один рид с каждого конца фрагмента нуклеиновой кислоты. Парно-концевое секвенирование включает фрагментацию ДНК на последовательности, называемые вставками. В некоторых протоколах, например, некоторых протоколах, используемых Illumina, риды с более коротких вставок (например, порядка десятков - сотен п.о.) называются парно-концевыми ридами с короткими вставками или просто парно-концевыми ридами. И напротив, риды с более длинных вставок (например, порядка нескольких тысяч п.о.) называются сцепленно-концевыми ридами. Согласно настоящему изобретению могут быть использованы как парно-концевые риды с короткими вставками, так и сцепленно-концевые риды с длинными вставками, и их не дифференцируют в контексте процесса определения последовательностей фрагментов ДНК. Соответственно, термин «парно-концевые риды» может относиться как к парно-концевым ридам с короткими вставками, так и сцепленно-концевым ридам с длинными вставками, согласно более подробному описанию далее в настоящем документе. Согласно некоторым вариантам реализации парно-концевые риды включают риды длиной от приблизительно 20 п.о. до 1000 п.о. Согласно некоторым вариантам реализации парно-концевые риды включают риды с длиной приблизительно 50 п.о. - 500 п.о., приблизительно 80 п.о. - 150 п.о. или приблизительно 100 п.о.
[0077] В настоящем документе термины «выравнивание» и «выравнивающий» относятся к процессу сравнения рида с референсной последовательностью и определения таким образом того, содержит ли указанная референсная последовательность последовательность указанного рида. Процесс выравнивания, согласно настоящему документу, направлен на определение того, может ли рид быть картирован на референсную последовательность, однако не всегда приводит к выравниванию рида на референсную последовательность. Если референсная последовательность содержит рид, указанный рид может быть картирован на указанную референсную последовательность или, согласно некоторым вариантам реализации, на конкретное местоположение в референсной последовательности. В некоторых случаях выравнивание просто указывает на то, является ли рид компонентом конкретной референсной последовательности (т.е. присутствует или отсутствует указанный рид в референсной последовательности). Например, выравнивание рида на референсную последовательность для хромосомы 13 человека указывает на то, присутствует ли указанный рид в референсной последовательности хромосомы 13.
[0078] Конечно, инструменты для выравнивания отличаются многими дополнительными аспектами и используются во многих других вариантах применения в биоинформатике, не описанных в настоящей заявке. Например, выравнивания могут также применяться для определения степени сходства двух последовательностей ДНК двух разных видов, с получением таким образом показателя для оценки их близости на эволюционном дереве.
[0079] Согласно некоторым вариантам реализации настоящего изобретения выравнивание осуществляют между частичной последовательностью рида и vNRUMI в качестве референсной последовательности для определения показателя выравнивания согласно более подробному описанию далее в настоящем документе. Показатели выравнивания для рида и нескольких vNRUMI могут затем быть использованы для определения одного из vNRUMI, с которым будет ассоциирован или на который будет картирован рид.
[0080] В некоторых случаях выравнивание также указывает на местоположение в референсной последовательности, на которое картирован рид. Например, если референсная последовательность представляет собой последовательность полного генома человека, выравнивание может показывать, что рид присутствует на хромосоме 13, и может дополнительно показывать, что указанный рид находится на конкретной цепи и/или сайте хромосомы 13. В некоторых сценариях инструменты для выравнивания являются несовершенными, то есть а) обнаруживаются не все валидные выравнивания и b) некоторые полученные выравнивания являются невалидными. Это происходит по различным причинам, например, риды могут содержать ошибки, и секвенированные риды могут отличаться от референсного генома из-за различий гаплотипов. В некоторых вариантах применения инструменты для выравнивания включают встроенный допуск несовпадений, допускающий определенную степень несовпадения пар оснований и при этом все же позволяющий выравнивать риды на референсную последовательность. Это может помочь идентифицировать валидное выравнивание ридов, которое в ином случае было бы пропущено.
[0081] Выравненные риды представляют собой одну или более последовательностей, которые идентифицируют как совпадающие, в отношении порядка в их молекулах нуклеиновой кислоты, с известной референсной последовательностью, такой как референсный геном. Выравненный рид и его определенное местоположение на референсной последовательности представляют собой метку последовательности. Выравнивание может быть выполнено вручную, хотя, как правило, его осуществляют при помощи компьютерного алгоритма, поскольку невозможно было бы провести выравнивание ридов в рамках периода времени целесообразной продолжительности для осуществления способов согласно описанию в настоящем документе. Одним из примеров алгоритма на основе выравнивания последовательностей является способ глобально-локального (глокального) гибридного выравнивания для сравнения префиксной последовательности рида в vNRUMI согласно более подробному описанию здесь и далее в настоящем документе. Другой пример способа выравнивания представлен компьютерной программой для эффективного локального выравнивания данных о нуклеотидах (Efficient Local Alignment of Nucleotide Data, ELAND), которая распространяется в качестве части системы анализа Illumina Genomics Analysis. Как вариант, для выравнивания ридов на референсные геномы может быть использован фильтр Блума или аналогичный тестировщик принадлежности множеству. См. заявку на патент США №14/354528, поданную 25 апреля 2014 г., которая включена в настоящий документ полностью посредством ссылки. Совпадение рида последовательности при выравнивании может составлять 100% или менее чем 100% (т.е. несовершенное совпадение). Дополнительные способы выравнивания раскрыты в заявке на патент США №.15/130668 (номер дела поверенного ILMNP008), поданной 15 апреля 2016 года, которая включена в настоящий документ полностью посредством ссылки.
[0082] Термин «картирование» в настоящем документе относится к отнесению последовательности рида к последовательности большего размера, например, референсному геному, путем выравнивания.
[0083] Термины «полинуклеотид», «нуклеиновая кислота» и «молекулы нуклеиновой кислоты» используются взаимозаменяемо и относятся к ковалентно связанной последовательности нуклеотидов (т.е. рибонуклеотидам в случае РНК и дезоксирибонуклеотидам в случае ДНК), в которой пентоза одного нуклеотида в 3'-положении присоединена фосфодиэфирной группой к пентозе следующего нуклеотида в 5'-положении. Нуклеотиды включают последовательности любой формы нуклеиновой кислоты, в том числе, но не ограничиваясь перечисленными, молекул ДНК и ДНК, таких как молекулы неклеточной ДНК (вкДНК). Термин «полинуклеотид» включает, без ограничения, одноцепочечные и двуцепочечные полинуклеотиды.
[0084] Термин «тестовый образец» в настоящем документе относится к образцу, как правило, происходящему из биологической жидкости, клетки, ткани, органа или организма, который включает нуклеиновую кислоту или смесь нуклеиновых кислот, содержащую по меньшей мере одну последовательность нуклеиновой кислоты, подлежащей скринингу на вариации числа копий и другие генетические изменения, такие как, не ограничиваясь перечисленным, однонуклеотидный полиморфизм, инсерций, делеций и структурные вариации. Согласно определенным вариантам реализации образец содержит по меньшей мере одну последовательность нуклеиновой кислоты, число копий которой предположительно претерпевало вариации. Такие образцы включают, не ограничиваясь перечисленными, образцы мокроты/жидкости полости рта, амниотической жидкости, крови, фракции крови или взятых тонкой иглой биоптатов, мочи, жидкости брюшной полости, плевральной жидкости; и т.п. Хотя часто образец берут у субъекта-человека (например, пациента), указанные анализы могут быть использованы для образцов от любого млекопитающего, в том числе, но не ограничиваясь перечисленными, собак, кошек, лошадей, коз, овец, крупного рогатого скота, свиней и т.п., также смешанных популяций, таких как микробные популяции из природной среды или вирусные популяции от пациентов. Образец может быть использован непосредственно в том виде, в котором он получен из биологического источника, либо после предварительной обработки для модификации характеристик образца. Например, такая предварительная обработка может включать получение плазмы из крови, разведение вязких жидкостей; и т.д. Способы предварительной обработки могут также включать, не ограничиваясь перечисленным, фильтрацию, осаждение, разведение, дистилляцию, смешивание, центрифугирование, замораживание, лиофилизацию, концентрацию, амплификацию, фрагментацию нуклеиновой кислоты, инактивацию вмешивающихся компонентов, добавление реагентов, лизис и т.п. Если такие способы предварительной обработки используют в отношении образца, такие способы предварительной обработки, как правило, таковы, что нуклеиновая кислота или нуклеиновые кислоты, представляющие интерес, остаются в тестовом образце, иногда в концентрации, пропорциональной концентрации в необработанном тестовом образце (т.е., например, образце, который не подвергался воздействию любого такого способа или способов предварительной обработки). Такие «обработанные», или «процессированные» образцы также считаются биологическими «тестовыми» образцами в контексте способов, описанных в настоящем документе.
[0085] Термин «секвенирование нового поколения (NGS)» в настоящем документе относится к способам секвенирования которые позволяют проводить массово-параллельное секвенирование клонально амплифицированных молекул и одиночных молекул нуклеиновой кислоты. Неограничивающие примеры NGS включают секвенирование путем синтеза с применением терминаторов с обратимым красителем, и секвенирование путем лигирования.
[0086] Термин «рид» относится к риду последовательности части образца нуклеиновой кислоты. Как правило, хотя не обязательно, рид представляет собой короткую последовательность из непрерывных пар оснований в указанном образце. Рид может быть символически представлен последовательностью пар оснований А, Т, С и G из части образца совместно с вероятностной оценкой корректности оснований (показатель качества). Рид может быть сохранен на запоминающем устройстве и обработан надлежащим образом для определения того, совпадает ли он с референсной последовательностью или отвечает ли другим критериям. Рид может быть получен прямо из устройства для секвенирования или непрямо из сохраненной информации о последовательности, относящейся к образцу. В некоторых случаях рид представляет собой последовательность ДНК достаточной длины (например, по меньшей мере приблизительно 20 п.о.), которая может быть использована для идентификации последовательности или области большего размера, например, может быть выравнен и картирован на хромосому, или геномную область, или ген.
[0087] Термины «сайт» и «местоположение выравнивания» используются взаимозаменяемо и относятся к уникальному положению (т.е. идентификатору хромосомы, положению и ориентации хромосомы) на референсном геноме. Согласно некоторым вариантам реализации сайт может представлять собой положение остатка, метки последовательности или сегмента в референсной последовательности.
[0088] В настоящем документе термин «референсный геном» или «референсная последовательность» относится к любой конкретной известной генетической последовательности, частичной или полной, из любого организма или вируса, которые могут применяться в качестве референсных для идентифицированных последовательностей субъекта. Например, референсный геном для использования у субъектов-людей, а также многих других организмов, можно найти на вебсайте: ncbi.nlm.nih.gov Национального центра биотехнологической информации. «Геном» относится к полной генетической информации организма или вируса, представленной в виде последовательностей нуклеиновых кислот. Однако следует понимать, что «полный» является относительным понятием, поскольку даже «золотой стандарт» референсного генома предположительно будет содержать пропуски и ошибки.
[0089] Согласно некоторым вариантам реализации последовательность vNRUMI может быть использована в качестве референсной последовательности, на которую выравнивают префиксную последовательность рида. Указанное выравнивание позволяет получить показатель выравнивания между префиксной последовательностью рида и vNRUMI, который может быть использован для определения того, должны ли указанный рид и указанный vNRUMI быть ассоциированы в ходе процесса объединения ридов, ассоциированных с одним и тем же vNRUMI.
[0090] Согласно различным вариантам реализации указанная референсная последовательность имеет значимо больший размер, чем риды, которые на нее выравнивают. Например, указанная последовательность может быть по меньшей мере приблизительно в 100 раз больше, или по меньшей мере приблизительно в 1000 раз больше, или по меньшей мере приблизительно в 10000 раз больше, или по меньшей мере приблизительно в 105 раз больше, или по меньшей мере приблизительно в 106 раз больше, или по меньшей мере приблизительно в 107 раз больше.
[0091] Согласно одному примеру указанная референсная последовательность представляет собой последовательность полноразмерного генома человека. Такие последовательности могут называться геномными референсными последовательностями. Согласно другому примеру указанная референсная последовательность ограничена специфической хромосомой человека, такой как хромосома 13. Согласно некоторым вариантам реализации референсная Y-хромосома представляет собой последовательность Y-хромосомы из генома человека, версия hg19. Такие последовательности могут называться хромосомными референсными последовательностями. Другие примеры референсных последовательностей включают геномы других видов, а также хромосомы, субхромосомные области (такие как цепи) и т.п., любых видов.
[0092] Согласно некоторым вариантам реализации референсная последовательность для выравнивания может иметь длину, приблизительно в 1 - приблизительно в 100 раз превышающую длину рида. Согласно таким вариантам реализации выравнивание и секвенирование считают целевым выравниванием или секвенированием, вместо полногеномного выравнивания или секвенирования полного генома. Согласно указанным вариантам реализации указанная референсная последовательность, как правило, включает генную последовательность и/или другую представляющую интерес ограниченную последовательность. В этом смысле выравнивание частичной последовательности рида на vNRUMI представляет собой форму целевого выравнивания.
[0093] Согласно различным вариантам реализации референсная последовательность представляет собой консенсусную последовательность или другую комбинацию, происходящую из нескольких индивидуумов. Тем не менее референсная последовательность для определенных вариантов применения может быть взята от конкретного индивидуума.
[0094] Термин «происходящий» в настоящем документе в контексте нуклеиновой кислоты или смеси нуклеиновых кислот относится к способу, посредством которого указанную нуклеиновую кислоту или нуклеиновые кислоты получают из источника их происхождения. Например, согласно одному варианту реализации смесь нуклеиновых кислот, происходящая из двух разных геномов, подразумевает, что указанные нуклеиновые кислоты, например, вкДНК, естественным образом высвобождены клетками в результате естественных процессов, таких как некроз или апоптоз. Согласно другому варианту реализации смесь нуклеиновых кислот, происходящая из двух разных геномов, подразумевает, что указанные нуклеиновые кислоты экстрагированы из клеток субъекта двух разных типов.
[0095] Термин «биологическая жидкость» в настоящем документе относится к жидкости, взятой из биологического источника, и включает, например, кровь, сыворотку, плазму, мокроту, жидкость лаважа, спинномозговую жидкость, мочу, сперму, пот, слезы, слюну и т.п. В настоящем документе термины «кровь», «плазма» и «сыворотка» явным образом включают фракции или обработанные части фракций. Аналогичным образом, если образец получают из биоптата, смыва, мазка и т.п., указанный «образец» явным образом включает обработанную фракцию или часть фракции, происходящую из указанного биоптата, смыва, мазка и т.п.
[0096] В настоящем документе термин «хромосома» относится к носителю генов наследственности в живой клетке, происходящих из цепей хроматина, содержащих ДНК и белковые компоненты (в частности, гистоны). В настоящем документе используется стандартная международно признанная система нумерации индивидуальных хромосом генома человека.
[0097] Термин «праймер» в настоящем документе относится к выделенному олигонуклеотиду, который способен действовать как точка инициации синтеза в условиях, индуцирующих синтез продукта достройки (например, указанные условия включают нуклеотиды, индуцирующий агент, такой как ДНК-полимераза, необходимые ионы и молекулы, и подходящие температуру и рН). Праймер может быть предпочтительно одноцепочечным для максимальной эффективности при амплификации, однако, как вариант, может быть двуцепочечным. Двуцепочечный праймер сначала обрабатывают для разделения цепей до использования для получения продуктов достройки. Праймер может представлять собой олигодезоксирибонуклеотид. Праймер имеет достаточную длину для примирования синтеза продуктов достройки в присутствии индуцирующего агента. Точные длины праймеров зависят от многих факторов, в том числе температуры, источника праймера, применения способа и параметров, используемых для дизайна праймеров.
Введение и контекст
[0098] Технология секвенирования нового поколения (NGS) быстро развивалась, обеспечивая новые инструменты для прогресса исследовательской и научной работы, а также а также здравоохранения и услуг, основанных на генетической и родственной биологической информации. Способы NGS осуществляют массово-параллельным образом, что позволяет обеспечивать все более высокую скорость определения информации о последовательности биомолекул. Однако многие из способов NGS и ассоциированных методик манипуляций с образцами вводят ошибки, так что итоговые последовательности характеризуются относительно высоким коэффициентом ошибки, в диапазоне от одной ошибки на несколько сотен пар оснований до одной ошибки на несколько тысяч пар оснований. Такие коэффициенты ошибки иногда приемлемы при определении наследуемой генетической информации, такой как мутации зародышевой линии, поскольку такая информация стабильно присутствует в большинстве соматических клеток, обеспечивающих множество копий одного и того же генома в тестовом образце. Ошибка, произошедшая при прочтении одной копии последовательности, оказывает незначительное или устранимое влияние, если множество копий той же самой последовательности прочитаны без ошибки. Например, если ошибочный рид с одной копии последовательности не может быть надлежащим образом выравнен на референсную последовательность, он может быть просто исключен из анализа. Риды без ошибок с других копий той же последовательности могут все еще обеспечивать достаточную информацию для валидного анализа. Как вариант, вместо исключения рида, содержащего отличающуюся пару оснований относительно других ридов из той же последовательности, можно пренебречь указанной отличающейся парой оснований, как обусловленной известным или неизвестным источником ошибки.
[0099] Однако такие способы коррекции ошибок не вполне подходят для для детекции последовательностей с низкими частотами аллелей, таких как субклональные соматические мутации, обнаруживаемые в нуклеиновых кислотах из опухолевой ткани, циркулирующей опухолевой ДНК, низкоконцентрированной фетальной вкДНК в материнской плазме, лекарственно-устойчивых мутаций патогенов и т.п. В указанных примерах один фрагмент ДНК может нести соматическую мутацию, представляющую интерес, в сайте последовательности, тогда как многие другие фрагменты в том же сайте последовательности не содержат мутацию, представляющую интерес. При таком сценарии риды последовательностей или пары оснований из мутированного фрагмента ДНК могут быть не использованы или неверно интерпретированы при стандартном секвенировании, с потерей таким образом информации для детекции представляющей интерес мутации.
[00100] Ввиду указанных различных источников ошибок увеличение глубины секвенирования по отдельности не может гарантировать детекцию соматических вариаций с очень низкой частотой аллелей (например, <1%). Согласно некоторым вариантам реализации, описанным в настоящем документе, предложены способы дуплексного секвенирования, которые эффективно подавляют ошибки в ситуациях, когда сигналы валидных представляющих интерес последовательностей незначительны, например, в образцах с низкими частотами аллелей.
[00101] Уникальные молекулярные индексы (UMI) позволяют использовать информацию для нескольких ридов, чтобы подавлять шум при секвенировании. UMI, наряду с контекстной информацией, такой как положение выравнивания, позволяют проследить происхождение каждого рида до специфической оригинальной молекулы ДНК. При наличии нескольких ридов, полученных с одной и той же молекулы ДНК, могут быть использованы вычислительные способы для отделения фактических вариантов (т.е. вариантов, биологически присутствующих в оригинальных молекулах ДНК) от вариантов, искусственно введенных в результате ошибки секвенирования. Варианты могут включать, не ограничиваясь перечисленным, инсерции, делеции, мультинуклеотидные варианты, однонуклеотидные варианты и структурные варианты. Используя указанную информацию, можно установить истинную последовательность молекул ДНК. Авторы настоящего изобретения называют указанный вычислительный метод объединением ридов. Указанная технология уменьшения ошибок используется для нескольких важных вариантов применения. В контексте анализа неклеточной ДНК часто возникают важные варианты с экстремально низкой частотой (т.е. <1%); соответственно, ошибки секвенирования могут заглушать их сигнал. Подавление шума на основе UMI позволяет максимально точно распознавать указанные низкочастотные варианты. UMI и объединение ридов могут также помочь идентифицировать дубликаты ПЦР в данных с высоким охватом, обеспечивая более точные измерения частоты вариантов.
[00102] Согласно некоторым вариантам реализации используют случайные UMI, полученные путем присоединения случайной последовательности к молекулам ДНК, и указанные случайные последовательности использовали в качестве штрихкодирующих UMI. Однако согласно некоторым вариантам реализации применение набора целенаправленно разработанных неслучайных UMI обеспечивало упрощение получения. Так как указанный способ является неслучайным, указанные UMI называются неслучайными UMI (NRUMI). Согласно некоторым вариантам реализации набор NRUMI состоит из последовательностей однородной длины (например, длина n=6 нуклеотидов). В результате процесса присоединения А-хвостов, с помощью которого указанные молекулы NRUMI лигируют с молекулами ДНК, 7-й (n+1) рид инвариантным образом представляет собой тимин (Т). Указанная однородность может обуславливать ухудшение качества ридов, распространяющееся в ходе циклов прочтения в 3'-направлении от указанного основания. Указанный эффект проиллюстрирован на Фиг. 2С.
[00103] Хотя указанная проблема может быть менее выраженной в непрофилированных проточных ячейках при секвенировании с применением 4 красителей, ее выраженность предположительно будет возрастать в профилированных проточных ячейках при секвенировании с применением 2 красителей, когда распознавание оснований заведомо становится более сложным. Согласно некоторым вариантам реализации новый процесс применяют для получения наборов NRUMI со смешанной длиной, однозначной идентификации таких NRUMI с вариабельной длиной (vNRUMI) и коррекции ошибок в пределах указанных vNRUMI. Он обеспечивает разнообразие при формировании и различении ДНК-штрихкодов гетерогенной длины. Экспериментальные результаты показывают, что способ с vNRUMI является более надежным (т.е. способен лучше корректировать ошибки секвенирования), чем стандартные решения.
[00104] Согласно некоторым вариантам реализации используют «жадный» алгоритм итерационного конструирования наборов vNRUMI. При каждой итерации он выбирает последовательность из пула кандидатных vNRUMI таким образом, чтобы выбранная последовательность максимизировала минимальное расстояние Левенштейна между ней и каждым из уже выбранных vNRUMI. Если несколько последовательностей характеризуются одинаковым максимальным значением указанной метрики, алгоритм выбирает одну такую последовательность случайным образом, отдавая предпочтение последовательностям меньшей длины. Значение указанной метрики расстояния должно составлять по меньшей мере 3, чтобы обеспечивать хорошую коррекцию ошибок в итоговом наборе vNRUMI; если указанное условие не может быть выполнено, процесс прекращает добавление новых vNRUMI к набору, и возвращает набор в существующем в текущий момент состоянии. Весь указанный процесс может быть повторен с получением других наборов vNRUMI с аналогичными характеристиками.
[00105] Адаптеры могут включать физические UMI, позволяющие определить, из какой цепи фрагмента ДНК происходят риды. Согласно некоторым вариантам реализации указанное преимущество используют для определения первой консенсусной последовательности для ридов, происходящих из одной цепи фрагмента ДНК, и второй консенсусной последовательности для комплементарной цепи. Согласно многим вариантам реализации консенсусная последовательность включает нуклеотиды, детектированные во всех или в большинстве ридов, и исключает нуклеотиды, встречающиеся в немногих ридах. Могут быть использованы другие критерии для консенсусных последовательностей. Процесс комбинирования ридов на основе UMI или местоположений выравнивания с получением консенсусной последовательности также называют «объединением» ридов. Используя физические UMI, виртуальные UMI и/или места выравнивания можно определить, какие из ридов для первой и второй консенсусных последовательностей происходят из одного и того же двуцепочечного фрагмента. Соответственно, согласно некоторым вариантам реализации третью консенсусную последовательность определяют с использованием первой и второй консенсусных последовательностей, полученных для той же молекулы/фрагмента ДНК, при этом указанная третья консенсусная последовательность включает нуклеотиды, общие для первой и второй консенсусных последовательностей, и исключает не присутствующие стабильно в обеих указанных последовательностях нуклеотиды. Согласно альтернативным вариантам реализации только одну консенсусную последовательность получают непосредственно путем объединения всех ридов, происходящих из обеих цепей одного и того же фрагмента, вместо сравнения двух консенсусных последовательностей, полученных из двух цепей. Наконец, последовательность фрагмента может быть определена по третьей или единственной консенсусной последовательности, которая включает пары оснований, стабильно присутствующие во всех ридах, происходящих из обеих цепей фрагмента.
[00106] Согласно некоторым вариантам реализации в указанном способе сочетают разные типы индексов для определения исходного полинуклеотида, из которого происходят риды. Например, в указанном способе могут быть использованы как физические, так и виртуальные UMI для идентификации ридов, происходящих из единственной молекулы ДНК. За счет применения второй формы UMI наряду с физическими UMI можно использовать более короткие физические UMI, чем при использовании только физических UMI для определения исходного полинуклеотида. Указанный подход оказывает минимальное влияние на эффективность приготовления библиотеки, и не требует дополнительной длины ридов секвенирования.
[00107] Варианты применения раскрытых способов включают:
• Подавление ошибок при детекции соматических мутаций. Например, детекция мутации с частотой аллеля менее 0,1% имеет крайне важное значение при жидкой биопсии циркулирующей опухолевой ДНК.
• Коррекция префазирования, фазирования и других ошибок секвенирования для получения длинных ридов высокого качества (например, 1×1000 п.о.)
• Уменьшение продолжительности цикла для фиксированной длины ридов и коррекция повышенного фазирования и префазирования указанным способом.
• Использование UMI с обеих сторон фрагмента для создания виртуальных длинных парно-концевых ридов. Например, сшивка рида размером 2×500 п.о. с использованием дубликатов ридов размером 500 п.о. + 50 п.о.
• Количественное определение или подсчет фрагментов нуклеиновых кислот, относящихся к представляющей интерес последовательности.
Технологическая схема для секвенирования фрагментов нуклеиновых кислот с применением UMI
[00108] На Фиг. 1А приведена блок-схема, иллюстрирующая пример технологической схемы 100 для применения UMI для секвенирования фрагментов нуклеиновых кислот. Технологическая схема 100 иллюстрирует только некоторые варианты реализации. Следует понимать, что некоторые варианты реализации задействуют технологические схемы, включающие дополнительные операции, не проиллюстрированные в настоящем документе, тогда как другие варианты реализации могут пропускать некоторые из операций, проиллюстрированные в настоящем документе. Например, в некоторых вариантах осуществления не требуется операция 102 и/или операция 104. Технологическую схему 100 также используют для секвенирования полного генома. Согласно некоторым вариантам реализации, включающим целевое секвенирование, могут быть использованы операционные этапы гибридизации и обогащения определенных областей между операциями 110 и 112.
[00109] Операция 102 обеспечивает получение фрагментов двуцепочечной ДНК. Указанные фрагменты ДНК могут быть получены путем фрагментирования геномной ДНК, сбора естественным образом фрагментированной ДНК (например, вкДНК или цоДНК) или синтеза фрагментов ДНК из РНК, например. Согласно некоторым вариантам реализации для синтеза фрагментов ДНК из РНК матричную РНК или некодирующую РНК сначала очищают с применением выбора по поли-А-фрагментам или истощения рибосомальной РНК, после чего выбранную мРНК химически фрагментируют и преобразуют в одноцепочечную кДНК с применением случайного гексамерного примирования. Получают комплементарную цепь кДНК для создания двуцепочечной кДНК, готовой для конструирования библиотеки. Для получения двуцепочечных фрагментов ДНК из геномной ДНК (гДНК) входящую гДНК фрагментируют, например, посредством гидродинамического сдвига, распыления, ферментативной фрагментации и т.п., с получением фрагментов подходящей длины, например, приблизительно 1000 п.о., 800 п.о., 500 или 200 п.о. Например, при распылении ДНК может разлагаться на участки длиной менее 800 п.о. за короткие периоды времени. В указанном процессе получают двуцепочечные фрагменты ДНК.
[00110] Согласно некоторым вариантам реализации фрагментированная или поврежденная ДНК может быть обработана без необходимости дополнительной фрагментации. Например, фиксированную в формалине и залитую в парафин (FFPE) ДНК или определенные вкДНК иногда фрагментированы в достаточной мере для того, чтобы не требовался этап дополнительной фрагментации.
[00111] На Фиг. 1В показаны фрагмент/молекула ДНК и адаптеры, используемые на начальных этапах технологической схемы 100 на Фиг. 1А. Хотя на Фиг. 1B проиллюстрирован только один двуцепочечный фрагмент, по указанной технологической схеме могут быть получены тысячи или миллионы фрагментов образца одновременно. При фрагментации ДНК физическими способами образуются гетерогенные концы, содержащие смесь липких 3'-концевых участков, липких 5'-концевых участков и тупых концов. Липкие концы имеют варьирующую длину, и концы могут быть или не быть фосфорилированными. Пример двуцепочечных фрагментов ДНК, получаемых путем фрагментирования геномной ДНК в ходе операции 102, представлен фрагментом 123 на Фиг. 1В.
[00112] Фрагмент 123 содержит как липкий 3'-концевой участок слева, так и липкий 5'-концевой участок, который виден справа, и маркирован символами ρ и ϕ, обозначающими две последовательности фрагмента, которые могут быть использованы в качестве виртуальных UMI согласно некоторым вариантам реализации, которые, при использовании по отдельности или в комбинации с физическими UMI адаптера для лигирования с указанным фрагментом, могут однозначно идентифицировать указанный фрагмент. UMI однозначно ассоциированы с единственным фрагментом ДНК в образце, включающем исходный полинуклеотид и комплементарную ему цепь. Физический UMI представляет собой последовательность олигонуклеотида, соединенную с исходным полинуклеотидом, комплементарной ему цепью или полинуклеотидом, происходящим из исходного полинуклеотида. Виртуальный UMI представляет собой последовательность олигонуклеотида в составе исходного полинуклеотида, комплементарной ему цепи или полинуклеотида, происходящего из исходного полинуклеотида. В соответствии с указанной схемой физический UMI может также быть назван внешним или экзогенным UMI, а виртуальный UMI - внутренним или эндогенным UMI.
[00113] Каждая из двух последовательностей ρ и ϕ фактически относится к двум комплементарным последовательностям в одном геномном сайте, однако для простоты они отмечены только на одной цепи в некоторых из двуцепочечных фрагментов, представленных в настоящем документе. Виртуальные UMI, такие как ρ и ϕ, могут применяться на более позднем этапе технологической схемы, помогая идентифицировать риды, происходящие из одной или обеих цепей единственного исходного фрагмента ДНК. Идентифицированные таким образом риды могут быть объединены с получением консенсусной последовательности.
[00114] Если фрагменты ДНК получают физическими способами, технологическая схема 100 переходит к выполнению операции репарации концов 104, продуцирующей фрагменты с тупыми концами, содержащие 5'-фосфорилированные концы. Согласно некоторым вариантам реализации указанный этап преобразует липкие концевые участки, полученные путем фрагментации, в тупые концы с использованием ДНК-полимеразы Т4 и фермента Кленова. Экзонуклеазная 3'→5' активность указанных ферментов обеспечивает удаление липких 3'-концевых участков, а полимеразная 5'→3' активность обеспечивает заполнение липких 5'-концевых участков. Кроме того, полинуклеотидкиназа Т4 в указанной реакции фосфорилирует 5'-концы фрагментов ДНК. Фрагмент 125 на Фиг. 1В представляет собой пример продукта с тупыми концами после репарации концов.
[00115] После репарации концов технологическая схема 100 переходит к операции 106 для аденилирования 3'-концов фрагментов, также называемого присоединением А-хвостов или присоединением dA-хвостов, поскольку к 3'-концам тупых фрагментов добавляют единственный дАТФ для предотвращения их лигирования друг с другом во время реакции лигирования адаптеров. На двуцепочечной молекуле 127 на Фиг. 1В виден фрагмент с А-хвостом, содержащий тупые концы с липкими 3'-концевыми dA-участками и 5'-концы с фосфатами. Единственный «Т»-нуклеотид на 3'-конце каждого из двух адаптеров для секвенирования, как видно на элементе 129 с Фиг. 1В, обеспечивает образование липкого концевого участка, комплементарного липкому 3'-концевому dA-участку на каждом из концов вставки для лигирования с указанной вставкой двух адаптеров.
[00116] После аденилирования 3'-концов технологическая схема 100 переходит к операции 108 для лигирования частично двуцепочечных адаптеров с обоими концами фрагментов. Согласно некоторым вариантам реализации адаптеры, используемые в реакции, включают разные физические UMI для ассоциации ридов последовательностей с одиночным исходным полинуклеотидом, который может представлять собой одноцепочечный или двуцепочечный фрагмент ДНК. Согласно некоторым вариантам реализации набор физических UMI, используемый в реакции, представлен случайными UMI. Согласно некоторым вариантам реализации набор физических UMI, используемых в реакции, представлен неслучайными UMI (NRUMI). Согласно некоторым вариантам реализации набор физических UMI, используемых в реакции, представлен неслучайными UMI с вариабельной длиной (vNRUMI).
[00117] Элемент 129 на Фиг. 1В иллюстрирует два адаптера для лигирования с двуцепочечным фрагментом, который включает два виртуальных UMI ρ и ϕ возле концов указанного фрагмента. Указанные изображенные адаптеры основаны на адаптерах для секвенирования платформы Ulumina, поскольку согласно различным вариантам реализации может быть использована платформа для NGS от Illumina для получения ридов и детекции представляющей интерес последовательности. Адаптер, представленный слева, включает физический UMI α в двуцепочечной области, тогда как адаптер справа включает физический UMI β в двуцепочечной области. На цепи, содержащей денатурированный 5'-конец, в направлении 5'→3' адаптеры содержат последовательность Р5, индексную последовательность, последовательность праймера рида 2 и физический UMI (α или β). На цепи, содержащей денатурированный 3'-конец, в направлении 3'→5' адаптеры содержат последовательность Р7', индексную последовательность, последовательность праймера рида 1 и физический UMI (α или β).
[00118] Олигонуклеотиды Р5 и Р7' комплементарны праймерам для амплификации, связанным с поверхностью проточных ячеек платформы для секвенирования Illumina. Согласно некоторым вариантам реализации индексная последовательность обеспечивает способ отслеживания источника образца, что позволяет проводить мультиплексирование нескольких образцов на платформе для секвенирования. Согласно различным вариантам реализации могут применяться другие варианты дизайна адаптеров и платформы для секвенирования. Адаптеры и технология секвенирования подробнее описаны в следующих разделах.
[00119] Реакция, изображенная на Фиг. 1В, добавляет отдельные последовательности к геномному фрагменту. Продукт лигирования 120 из того же фрагмента, описанного выше, проиллюстрирован на Фиг. 1B. Указанный продукт лигирования 120 содержит физический UMI α, виртуальный UMI ρ, виртуальный UMI ϕ и физический UMI β на верхней цепи, в направлении 5'→3'. Продукт лигирования также содержит физический UMI β, виртуальный UMI ϕ, виртуальный UMI ρ и физический UMI α на нижней цепи, в направлении 5'→3'. Настоящим изобретением предусмотрено и осуществление способов с применением технологий секвенирования и адаптеров, не предоставляемых Illumina.
[00120] Хотя приведенные в настоящем документе примеры адаптеров содержат физические UMI на двуцепочечных областях адаптеров, согласно некоторым вариантам реализации используют адаптеры, содержащие физические UMI на одноцепочечных областях, такие как адаптеры (i) и (iv) на Фиг. 2А.
[00121] Согласно некоторым вариантам реализации продукты указанной реакции лигирования очищают и/или отбирают по размеру путем гель электрофореза в агарозном геле или на магнитных гранулах. Отобранные по размеру ДНК затем ПЦР-амплифицируют для обогащения фрагментами, содержащими адаптеры на обоих концах. См. блок 110. Как упоминалось выше, согласно некоторым вариантам реализации могут применяться операции гибридизации и обогащения определенных областей фрагментов ДНК для нацеливания на области для секвенирования.
[00122] Затем технологическая схема 100 переходит к кластерной амплификации ПЦР-продуктов, например, на платформе Illumina. См. операцию 112. Путем кластеризации ПЦР-продуктов может быть получен общий пул библиотек для мультиплексирования, например, включающего до 12 образцов на дорожку, при использовании разных индексных последовательностей на адаптерах, чтобы отслеживать разные образцы.
[00123] После кластерной амплификации посредством секвенирования путем синтеза на платформе Illumina могут быть получены риды секвенирования. См. операцию 114. Хотя адаптеры и процесс секвенирования, описанные в настоящем документе, основаны на платформе Illumina, другие технологии секвенирования, в частности, методы NGS, могут применяться вместо платформы Illumina или наряду с платформой Illumina.
[00124] Технологическая схема 100 может объединять риды, содержащие один и тот же или одни и те же физические UMI, и/или один и тот же или одни и те же виртуальные UMI, в одну или более групп, с получением таким образом одной или более консенсусных последовательностей. См. операцию 116. Согласно некоторым вариантам реализации указанные физические UMI представляют собой случайные UMI. Согласно некоторым вариантам реализации указанные физические UMI представляют собой неслучайные UMI. Согласно некоторым вариантам реализации указанные физические UMI представляют собой случайные UMI с вариабельной длиной. Согласно некоторым вариантам реализации указанные физические UMI представляют собой неслучайные UMI с вариабельной длиной (vNRUMI). Консенсусная последовательность включает нуклеотидные основания, стабильно присутствующие или отвечающие критерию консенсусной последовательности все всех ридах в объединенной группе. Согласно некоторым вариантам реализации физические UMI по отдельности могут обеспечивать достаточную информацию для мечения фрагментов ДНК для объединения ридов. Такие варианты реализации требуют достаточно значительного числа физических UMI для однозначного мечения фрагментов ДНК. Согласно другим вариантам реализации информация о физических UMI, виртуальных UMI и информация о положении могут быть скомбинированы различным образом для объединения ридов с получением консенсусных последовательностей для определения последовательности фрагмента или по меньшей мере его части. Согласно некоторым вариантам реализации физические UMI комбинируют с виртуальными UMI для объединения ридов. Согласно другим вариантам реализации физические UMI и положения ридов комбинируют для объединения ридов. Информация о положениях ридов может быть получена с помощью различных методик, задействующих разные способы определения положения, например, геномные координаты ридов, положения на референсной последовательности или положения на хромосоме. Согласно дополнительным вариантам реализации физические UMI, виртуальные UMI и положения ридов комбинируют для объединения ридов.
[00125] Наконец, технологическая схема 100 использует одну или более консенсусных последовательностей для определения последовательности фрагмента нуклеиновой кислоты из образца. См. операцию 118. Указанное определение может включать определение последовательности фрагмента нуклеиновой кислоты как третьей консенсусной последовательности или единственной консенсусной последовательности, согласно описанию выше.
[00126] Согласно конкретному варианту реализации, который включает операции, аналогичные операциям 108-119, способ секвенирования молекул нуклеиновой кислоты из образца с использованием неслучайных UMI включает следующее: (а) обработку адаптерами фрагментов ДНК в указанном образце с получением продуктов ДНК-адаптер, при этом каждый адаптер содержит NRUMI, и NRUMI указанных адаптеров характеризуются по меньшей мере двумя разными длинами молекул, образуя набор vNRUMI; (b) амплификацию указанных продуктов ДНК-адаптер с получением множества амплифицированных полинуклеотидов; (с) секвенирование множества амплифицированных полинуклеотидов с получением таким образом множества ридов, ассоциированных с набором vNRUMI; (d) идентификацию среди множества ридов тех ридов, которые ассоциированы с одним и тем же vNRUMI; и (е) определение последовательности фрагмента ДНК в указанном образце с использованием ридов, ассоциированных с одним и тем же vNRUMI.
[00127] Согласно другому варианту реализации для секвенирования молекул нуклеиновой кислоты используют случайный UMI с вариабельной длиной. Указанный способ включает: (а) обработку адаптерами фрагментов ДНК в указанном образце с получением продуктов ДНК-адаптер, при этом каждый адаптер содержит уникальный молекулярный индекс (UMI), и уникальные молекулярные индексы (UMI) указанных адаптеров характеризуются по меньшей мере двумя разными длинами молекул и образуют набор уникальных молекулярных индексов (vUMI) с вариабельной длиной; (b) амплификацию указанных продуктов ДНК-адаптер с получением множества амплифицированных полинуклеотидов; (с) секвенирование множества амплифицированных полинуклеотидов с получением таким образом множества ридов, ассоциированных с указанным набором vUMI; и (d) идентификацию среди множества ридов тех ридов, которые ассоциированы с одним и тем же неслучайным уникальным молекулярным индексом с вариабельной длиной (vUMI). Некоторые варианты реализации дополнительно включают определение последовательности фрагмента ДНК в указанном образце с использованием ридов, ассоциированных с одним и тем же vUMI.
[00128] Согласно некоторым вариантам реализации UMI, используемые для секвенирования фрагментов нуклеиновых кислот, могут представлять собой случайные UMI с фиксированной длиной, неслучайные UMI с фиксированной длиной, случайные UMI с вариабельной длиной, неслучайные UMI с вариабельной длиной или любую комбинацию перечисленного. Согласно указанным вариантам реализации способ секвенирования фрагментов нуклеиновых кислот включает: (а) обработку адаптерами фрагментов ДНК в указанном образце с получением продуктов ДНК-адаптер, при этом каждый адаптер содержит уникальный молекулярный индекс (UMI) в наборе уникальных молекулярных индексов (UMI); (b) амплификацию указанных продуктов ДНК-адаптер с получением множества амплифицированных полинуклеотидов; (с) секвенирование множества амплифицированных полинуклеотидов с получением таким образом множества ридов, ассоциированных с указанным набором UMI; (d) получение для каждого рида из указанного множества ридов показателей выравнивания для указанного набора UMI, при этом каждый из полученных показателей является индикатором сходства между частичной последовательностью рида и UMI; (е) идентификацию среди множества ридов тех ридов, которые ассоциированы с одним и тем же UMI, с использованием показателей выравнивания; и (е) определение последовательности фрагмента ДНК в указанном образце с использованием ридов, ассоциированных с одним и тем же UMI. Согласно некоторым вариантам реализации указанные показатели выравнивания основаны на совпадениях нуклеотидов и изменений нуклеотидов между частичной последовательностью указанного рида и указанного UMI. Согласно некоторым вариантам реализации каждый показатель выравнивания включает штрафы за несовпадения в начале последовательности, однако не включает штрафы за несовпадения в конце указанной последовательности.
[00129] Согласно некоторым вариантам реализации риды последовательностей представляют собой парно-концевые риды. Каждый рид либо включает неслучайный UMI, либо ассоциирован с неслучайным UMI за счет парно-концевого рида. Согласно некоторым вариантам реализации длины ридов меньше длин фрагментов ДНК или меньше половины длины фрагментов. В таких случаях полную последовательность целого фрагмента иногда не определяют, место этого определяют два конца фрагмента. Например, длина фрагмента ДНК может составлять 500 п.о., и из него могут происходить два парно-концевых рида длиной 100 п.о. В указанном примере могут быть определены 100 оснований на каждом из концов фрагмента, а 300 п.о. в середине фрагмента не могут быть определены без использования информации о других ридах. Согласно некоторым вариантам реализации, если два парно-концевых рида имеют достаточную длину, чтобы перекрываться, полная последовательность целого фрагмента может быть определена по указанным двум ридам. Например, см. пример, описанный в контексте Фиг. 5.
[00130] Согласно некоторым вариантам реализации адаптер содержит дуплексные неслучайные UMI в двуцепочечной области адаптера, и каждый рид включает первый неслучайный UMI на одном конце и второй неслучайный UMI на другом конце.
Способ секвенирования фрагментов нуклеиновых кислот с применением vNRUMI
[00131] Согласно некоторым вариантам реализации vNRUMI включены в адаптеры для секвенирования фрагментов ДНК. Указанные vNRUMI обеспечивают механизм подавления разных типов ошибок, происходящих при выполнении технологической схемы, такой как описанная выше. Некоторые из ошибок могут происходить в фазе обработки образцов, например, делеции, добавления и замены при обработке образцов. Другие ошибки могут происходить в фазе секвенирования. Некоторые ошибки могут быть локализованы в основаниях, происходящих из фрагментов ДНК, другие ошибки могут быть локализованы в основаниях, соответствующих UMI в адаптерах.
[00132] Согласно некоторым вариантам реализации предложен новый способ детекции и коррекции ошибок в vNRUMI и в ридах последовательностей. На высоком уровне, при наличии рида, содержащего (потенциально неверно прочтенный) vNRUMI и основания в 3'-направлении, процесс использует стратегию глобально-локального (глокального) гибридного выравнивания для определения совпадений первых нескольких оснований рида с известным vNRUMI, с получением таким образом показателей выравнивания между префиксными последовательностями рида и указанным известным vNRUMI. vNRUMI с самым высоким значением показателя глокального выравнивания определяют как vNRUMI, ассоциированный с ридом, что обеспечивает механизм для объединения указанного рида с другими ридами, ассоциированными с тем же vNRUMI, с коррекцией таким образом ошибок. Согласно некоторым вариантам реализации предложен следующий псевдокод для получения показателей глокального выравнивания и совпадающих vNRUMI с применением указанных показателей глокального выравнивания:


[00133] Стоит отметить, что используется нестандартная метрика расстояния. В большинстве других сопоставимых методов для ДНК-штрихкодов используются эвристические методы количественного определения редакционного расстояния, а именно, расстояние Левенштейна, расстояние Хэмминга или их производные. Теоретически, оценка выравнивания обеспечивает аналогичную метрику сходства последовательностей, но с одним ключевым различием: она обеспечивает подсчет совпадений помимо изменений. Эвристические методы, учитывающие совпадения, лежат в основе некоторых преимуществ определенных вариантов реализации NRUMI с вариабельной длиной.
[00134] Согласно некоторым вариантам реализации используют не традиционное глобальное выравнивание Нидлмана-Вунша и не традиционный способ локального выравнивания Смита-Уотермана, а новый гибридный способ. Это означает, что для выравнивания используют способ Нидлмана-Вунша в начале выравнивания, применяя штрафы за изменения в этой области, однако в конце выравнивания благоприятным образом эксплуатируют принципы локального выравнивания Смита-Уотермана, не применяя штрафы за изменения на конце. В указанном смысле данный способ выравнивания включает как глобальный, так и локальный компонент, и, соответственно, называется способом глокального выравнивания. В случае инсерционной или делеционной ошибки при секвенировании выравнивание существенно сдвигается. Указанный глобальный способ не включает штрафы за такое одиночное событие, более высокие по сравнению со штрафами за единственную точечную мутацию. Разрешение подвижных пропусков позволяет осуществить указанное.
[00135] Способ глокального выравнивания позволяет работать со штрихкодирующими пулами гетерогенной длины, что является признаком, отличающим его от стандартных методов.
[00136] При идентификации совпадений некоторые варианты реализации могут возвращать несколько совпадений vNRUMI, как «наилучшие», при наличии совпадающих результатов. Хотя приведенный выше псевдокод отображает только наилучший и второй наилучший возвращаемые наборы, согласно некоторым вариантам реализации может быть возвращено более чем всего два набора vNRUMI, например, второй наилучший набор, третий наилучший набор, четвертый наилучший набор и т.п. Обеспечивая больший объем информации о хороших совпадениях, указанный процесс может позволить лучшую коррекцию ошибок за счет объединения ридов, ассоциированных с одним или более кандидатными совпадающими vNRUMI. На Фиг. 1С представлена блок-схема, отражающая процесс секвенирования фрагментов ДНК с применением vNRUMI для подавления ошибок, происходящих в указанных фрагментах ДНК, и ошибок в UMI, которые используют для мечения исходных молекул фрагментов ДНК. Процесс 130 начинается с обработки адаптерами фрагментов ДНК в образце для получения продуктов ДНК-адаптер. См. блок 131. Каждый из адаптеров содержит неслучайный уникальный молекулярный индекс. Неслучайные уникальные молекулярные индексы указанных адаптеров характеризуются по меньшей мере двумя разными длинами молекул и образуют набор неслучайных молекулярных индексов с вариабельной длиной (vNRUMI).
[00137] Согласно некоторым вариантам реализации адаптер присоединен, лигирован, инсертирован, встроен или иными образом соединен с каждым концом фрагментов ДНК. Согласно некоторым вариантам реализации образец, содержащий фрагменты ДНК, представляет собой образец крови. Согласно некоторым вариантам реализации указанные фрагменты ДНК содержат фрагменты неклеточной ДНК. Согласно некоторым вариантам реализации указанные фрагменты ДНК включают неклеточную ДНК, происходящую из опухоли, и последовательность фрагментов ДНК в указанном образце указывает на наличие опухоли.
[00138] Процесс 130 продолжается амплификацией указанных продуктов ДНК-адаптер с получением множества амплифицированных полинуклеотидов. См. блок 132. Процесс 130 дополнительно включает секвенирование множества амплифицированных полинуклеотидов с получением таким образом множества ридов, ассоциированных с набором vNRUMI. См. блок 133. Кроме того, процесс 130 включает идентификацию ридов, ассоциированных с одним и тем же vNRUMI, среди множества ридов. См. блок 134. Наконец, процесс 130 включает определение последовательности фрагмента ДНК в указанном образце с использованием ридов, ассоциированных с одним и тем же vNRUMI.
[00139] Как упоминалось выше, процесс 130, проиллюстрированный на Фиг. 1С, обеспечивает способ секвенирования фрагментов ДНК с применением vNRUMI. Процесс 130 начинается с обработки адаптерами фрагментов ДНК из образца для получения продуктов ДНК-адаптер (блок 131). Процесс 130 также включает амплификацию указанных продуктов ДНК-адаптер с получением множества амплифицированных полинуклеотидов (блок 132); секвенирование амплифицированных полинуклеотидов с получением таким образом множества ридов, ассоциированных с набором vNRUMI (блок 133); идентификацию ридов, ассоциированных с одним и тем же vNRUMI (блок 134); и определение последовательности фрагментов ДНК в указанном образце с использованием ридов, ассоциированных с одним и тем же vNRUMI (блок 135). Указанный образец может представлять собой образец крови, образец плазмы, образец ткани или один из образцов согласно описанию в тексте настоящего документа. Согласно некоторым вариантам реализации адаптеры для применения на этапе 131 могут быть получены в результате такого процесса, как процесс 140, проиллюстрированный на Фиг. 1D.
[00140] Согласно некоторым вариантам реализации vNRUMI указанных адаптеров характеризуются по меньшей мере двумя разными длинами молекул. Согласно некоторым вариантам реализации указанный набор vNRUMI характеризуется по меньшей мере двумя разными длинами молекул. Согласно некоторым вариантам реализации указанные vNRUMI содержат шесть или семь нуклеотидов. Согласно некоторым вариантам реализации указанные vNRUMI характеризуются более чем двумя разными длинами молекул, например, характеризуются тремя, четырьмя, пятью, шестью, семью, восемью, девятью, десятью, двадцатью или более разными длинами молекул. Согласно некоторым вариантам реализации указанные длины молекул выбирают из диапазона 4-100. Согласно некоторым вариантам реализации указанные длины молекул выбирают из диапазона 4-20. Согласно некоторым вариантам реализации указанные длины молекул выбирают из диапазона 5-15.
[00141] Согласно некоторым вариантам реализации указанный набор vNRUMI включает не более чем приблизительно 10000 разных vNRUMI. Согласно некоторым вариантам реализации указанный набор vNRUMI включает не более чем приблизительно 1000 разных vNRUMI. Согласно некоторым вариантам реализации указанный набор vNRUMI включает не более чем приблизительно 200 разных vNRUMI.
[00142] Согласно некоторым вариантам реализации этап 134 идентификации ридов, ассоциированных с одним и тем же vNRUMI, включает получение для каждого рида из указанного множества ридов показателей выравнивания применительно к vNRUMI. Каждый показатель выравнивания указывает на сходство между частичной последовательностью рида и vNRUMI. Указанная частичная последовательность находится в той области указанного рида, где предположительно расположены нуклеотиды, происходящие из указанного vNRUMI. Другими словами, согласно некоторым вариантам реализации указанная частичная последовательность включает первые нуклеотиды в области, где, как ожидается, локализован vNRUMI. Согласно некоторым вариантам реализации размер указанной частичной последовательности равен размеру максимального vNRUMI в наборе vNRUMI.
[00143] Согласно некоторым вариантам реализации указанные показатели выравнивания основаны на совпадениях и несовпадениях/изменениях нуклеотидов между указанной частичной последовательностью рида и указанным vNRUMI. Согласно некоторым вариантам реализации указанные изменения нуклеотидов включают замены, добавления и делеции нуклеотидов. Согласно некоторым вариантам реализации указанный показатель выравнивания включает штрафы за изменения в начале последовательности (например, частичной последовательности рида или референсной последовательности vNRUMI), но не включает штрафы за изменения в конце указанной последовательности. Показатель выравнивания отражает сходство между частичной последовательностью рида и референсной последовательностью vNRUMI.
[00144] Согласно некоторым вариантам реализации получение показателя выравнивания между ридом и vNRUMI включает: (а) вычисление показателя выравнивания между vNRUMI и каждой из всех возможных префиксных последовательностей частичной последовательности указанного рида; (b) вычисление показателя выравнивания между частичной последовательностью указанного рида и каждой из всех возможных префиксных последовательностей указанного vNRUMI; и (с) получение максимального показателя выравнивания из показателей выравнивания, вычисленных по (а) и (b), в качестве показателя выравнивания между указанным ридом и указанным vNRUMI.
[00145] Согласно некоторым вариантам реализации частичная последовательность рида имеет длину, равную длине самого длинного vNRUMI в наборе vNRUMI.
[00146] Согласно некоторым вариантам реализации идентификация ридов,
ассоциированных с одним и тем же vNRUMI, включает выбор для каждого рида из указанного множества ридов по меньшей мере одного vNRUMI из набора vNRUMI на основании показателей выравнивания; и ассоциацию каждого рида из указанного множества ридов по меньшей мере с одним vNRUMI, выбранным для указанного рида. Согласно некоторым вариантам реализации выбор указанного по меньшей мере одного vNRUMI из набора vNRUMI включает выбор vNRUMI с самым высоким значением показателя выравнивания из указанного набора vNRUMI.
[00147] Согласно некоторым вариантам реализации идентифицируют один vNRUMI с самым высоким значением показателя выравнивания. Согласно некоторым вариантам реализации идентифицируют два или более vNRUMI с самым высоким значением показателя выравнивания. В таком случае контекстная информация о ридах может быть использована для выбора одного из двух или более vNRUMI, которые должны быть ассоциированы с ридами, для определения последовательности во фрагментах ДНК. Например, общее число ридов, идентифицированных для одного vNRUMI, может сравниваться с общим числом ридов, идентифицированных для другого vNRUMI, и по большему общему числу определяют тот vNRUMI, который следует использовать как указывающий на источник фрагмента ДНК. Согласно другому примеру информация о последовательности ридов или местоположений ридов на референсной последовательности может быть использована для выбора одного из идентифицированных vNRUMI, ассоциированных с ридами, и выбранный vNRUMI используют для определения источника ридов последовательностей.
[00148] Согласно некоторым вариантам реализации два или более из самых высоких показателей выравнивания могут быть использованы для идентификации двух или более vNRUMI как указывающих на потенциальный источник любого фрагмента. Контекстная информация может быть использована согласно упоминанию выше для определения того, какой из указанных vNRUMI указывает на фактический источник фрагмента ДНК.
[00149] На Фиг. 1Е представлены примеры того, как частичная последовательность рида или запрашиваемая последовательность (Q) может быть сравнена с двумя референсными последовательностями в наборе vNRUMI γ={S1,S2}={AACTTC, CGCTTTCG}. Запрашиваемая последовательность Q включает семь первых нуклеотидов из последовательности рида, при этом риды предположительно происходят из vNRUMI.
[00150] Запрашиваемая последовательность Q включает семь нуклеотидов GTCTTCG. Q имеет такую же длину, что и самая длинная vNRUMI в наборе vNRUMI γ. В таблице показателей выравнивания 150 приведены показатели выравнивания для префиксных последовательностей Q и S1. Например, в ячейке 151 приведен показатель выравнивания для префиксной последовательности Q (GTCTTC) и полной последовательности S1 (ААСТТС). Показатель выравнивания учитывает число совпадений между двумя последовательностями, а также число изменений между двумя последовательностями. Каждый совпадающий нуклеотид увеличивает показатель на 1; каждая делеция, добавление или замена снижает показатель на 1. В отличие от этого, расстояние Левенштейна представляет собой редакционное расстояние, не учитывающее число совпадений между двумя последовательностями, а учитывающее только число добавлений, делеций и замен.
[00151] При сравнении, нуклеотид за нуклеотидом, префиксной последовательности Q (GTCTTC) и S1 (ААСТТС) имеется несовпадение между G и А, несовпадение между Т и А, совпадение между С и С, совпадение между Т и Т, совпадение между Т и Т; и совпадение между С и С. Соответственно, показатель выравнивания для двух префиксных последовательностей равен 2, как указано в ячейке 151. Показатель выравнивания не включает штраф за конец последовательности Q, содержащий нуклеотид G.
[00152] В таблице показателей выравнивания 150 в крайнем правом столбце с выделенными жирным начертанием показателями выравнивания приведены показатели выравнивания между всеми возможными частичными последовательностями запрашиваемой последовательности Q и всеми возможными префиксными последовательностями референсной последовательности vNRUMI S1. В нижней строке таблицы показателей выравнивания 150 приведены показатели выравнивания между полной последовательностью S1 и всеми возможными префиксными последовательностями Q. Согласно различным вариантам реализации самый высокий показатель выравнивания в крайнем правом столбце и нижней строке выбирают в качестве показателя глокального выравнивания между Q и S1. В указанном примере ячейка 151 содержит самое высокое значение, которое определяют как показатель глокального выравнивания между Q и S1, или g(Q,S1).
[00153] Самый высокий показатель выравнивания в нижней строке и крайнем правом столбце используют в качестве показателя глокального выравнивания между двумя последовательностями. Разным строковым операциям присваивают равный показатель в проиллюстрированных здесь показателях выравнивания. Показатель выравнивания вычисляют следующим образом: число совпадений - число инсерций - число делеций - число замен = число совпадений - расстояние Левенштейна. Однако, как упоминалось выше, согласно некоторым вариантам реализации разным строковым операциям может быть присвоен разный показатель при вычислении показателя выравнивания. Например, согласно некоторым вариантам реализации (не показанным на Фиг. 1E) показатель выравнивания может быть вычислен следующим образом:
число совпадений × 5 - число инсерций × 4 - число делеций × 4 - число замен × 6,
или с использованием других значений показателей.
[00154] Согласно описанным выше вариантам реализации эффекты совпадений и изменений скомбинированы в показателях выравнивания линейным образом, а именно, с помощью сложения и/или вычитания. Согласно другим вариантам реализации эффекты совпадений и изменений в показателях выравнивания могут быть скомбинированы нелинейным образом, например, с помощью умножения или логарифмических операций.
[00155] Показатели выравнивания в крайнем правом столбце и нижней строке указывают на сходство между префиксными последовательностями, с одной стороны, и полной последовательностью, с другой стороны. Если начало префиксной последовательности не совпадает с началом полной последовательности, в показатель выравнивания включают штраф. В указанном смысле показатель выравнивания содержит глобальный компонент. С другой стороны, если конец префиксной последовательности не совпадает с концом полной последовательности, в показатель выравнивания последовательности не включают штраф. В указанном смысле показатель выравнивания содержит локальный компонент. Соответственно, показатели выравнивания в крайнем правом столбце и нижней строке могут быть описаны как показатели «глокального» выравнивания. Показатель глокального выравнивания между Q и S1 представляет собой максимальный показатель выравнивания в крайнем правом столбце и нижней строке, равный 2 и расположенный в ячейке 151 для префиксной последовательности GTCTTC Q и S1 (ААСТТС).
[00156] Расстояние Левенштейна между префиксной последовательностью GTCTTC Q и S1 (ААСТТС) также составляет 2, ввиду несовпадения между G и А, несовпадения между Т и А, а также четырех совпадений для СТТС. Для указанных двух последовательностей расстояние Левенштейна и показатель выравнивания равны.
[00157] В отличие от показателя глокального выравнивания, для получения чистого показателя глобального выравнивания, который представлен показателем выравнивания в нижнем правом углу таблицы 150, необходима полная последовательность Q, с одной стороны, и полная последовательность S1, с другой стороны.
[00158] В таблице 152 на Фиг. 1Е приведены показатели выравнивания для запрашиваемой последовательности Q и референсной последовательности S2 (CGCTTCG). Самый высокий показатель выравнивания в крайнем правом столбце и нижней строке расположен в ячейке 153 и равен 4. Это показатель глокального выравнивания между Q и S2, или g(Q,S2). Расстояние Левенштейна между Q и S2 идентично расстоянию Левенштейна между Q и S1 ввиду двух несовпадений между двумя последовательностями в обоих сравнениях. Однако g(Q,S2) выше g(Q,S1), поскольку совпадающих нуклеотидов между Q и S2 больше, чем между Q и S1. Таким образом, показатели глокального выравнивания учитывают не только изменения нуклеотидов (как расстояние Левенштейна), однако также совпадения нуклеотидов между последовательностями.
[00159] На Фиг. 1Е показано, что показатель глокального выравнивания может обеспечивать лучшую коррекцию ошибок, чем расстояние Левенштейна или редакционное расстояние, поскольку расстояние Левенштейна учитывает только число изменений в последовательности, тогда как показатель глокального выравнивания учитывает как число изменений, так и число совпадений между последовательностями. На Фиг. 1F приведен пример, иллюстрирующий тот факт, что показатель глокального выравнивания может обеспечивать лучшее подавление ошибок, чем показатель глобального выравнивания, поскольку показатель глокального выравнивания не включает избыточных штрафов за несовпадения, обусловленные инсерцией, делецией или заменой в конце последовательности.
[00160] В примере на Фиг. 1F используют другой набор последовательностей vNRUMI, γ={S1,S2}={TTGTGAC,GGCCAT}. При обработке образца процесс S1 используют для мечения молекулы ДНК. Последовательность указанной молекулы: 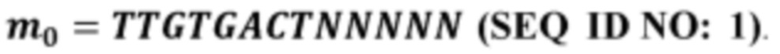 Во время секвенирования, происходит ошибка одиночной инсерции с инсерцией последовательности GCA в m0, образующей
Во время секвенирования, происходит ошибка одиночной инсерции с инсерцией последовательности GCA в m0, образующей  Для коррекции указанной ошибки и восстановления надлежащего UMI для указанной последовательности процесс берет первые 7 пар оснований в качестве запрашиваемой последовательности,
Для коррекции указанной ошибки и восстановления надлежащего UMI для указанной последовательности процесс берет первые 7 пар оснований в качестве запрашиваемой последовательности, 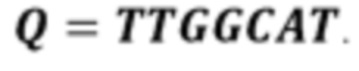 Процесс сравнивает Q с каждой последовательностью в γ.
Процесс сравнивает Q с каждой последовательностью в γ.
[00161] Получают таблицу показателей выравнивания 160 для g(Q, S1), показанную на Фиг. 1F. Аналогичным образом, получают таблицу показателей выравнивания 163 для g(Q, S2).
[00162] При использовании схемы глобального выравнивания вместо показателя глокального выравнивания используют показатель в нижнем правом углу в ячейках 161 и 164, значение которого в обоих случаях равно 2. Оптимальное выравнивание Q (TTGGCAT) и S1 (TTGTGAC) достигается путем выравнивания TTG-GCAT с TTGTG-AC, где черточками обозначены инсерции или пропуски. Указанное выравнивание включает 5 совпадений, 2 инсерций и 1 замену, обеспечивая показатель выравнивания 5-2-1=2. Оптимальное выравнивание Q (TTGGCAT) и S2 (GGCCAT) достигается путем выравнивания TTGGC-AT и --GGCCAT. Указанное выравнивание включает 5 совпадений и 3 инсерций, обеспечивая показатель выравнивания 5-3=2. При использовании показателя глобального выравнивания невозможно окончательно установить, какой из S1 и S2 с большей вероятностью представляет собой фактический vNRUMI.
[00163] Однако при применении схемы глокального выравнивания, использующей максимальное значение в последних строке и столбце, процесс получает показатель выравнивания, равный 3, для префиксной последовательности Q TTGGC и S1 (TTGTGAC), который становится глокальным показателем для S1 и превышает глокальный показатель для S2 (2). Соответственно, указанный процесс позволяет корректно ассоциировать Q с S1.
[00164] Возвращаясь к Фиг. 1С, этап 135 включает определение последовательности фрагмента ДНК в указанном образце с использованием ридов, ассоциированных с одним и тем же vNRUMI. Согласно некоторым вариантам реализации определение последовательности фрагмента ДНК включает объединение ридов, ассоциированных с одним и тем же vNRUMI, с получением консенсусной последовательности, что может быть выполнено согласно более подробному описанию здесь и далее в настоящем документе. Согласно некоторым вариантам реализации указанная консенсусная последовательность основана на показателях качества ридов, как и последовательность ридов. Согласно дополнительному или альтернативному варианту может быть использована другая контекстная информация, например, о положении ридов, для определения консенсусной последовательности.
[00165] Согласно некоторым вариантам реализации определение последовательности фрагмента ДНК также включает идентификацию ридов, занимающих одно и то же положение или аналогичные положения в референсной последовательности. Согласно указанному способу затем определяют последовательность фрагмента ДНК с использованием ридов, которые ассоциированы с одним и тем же vNRUMI и занимают одно и то же положение или аналогичные положения в референсной последовательности.
[00166] Согласно некоторым вариантам реализации определение последовательности фрагмента ДНК включает идентификацию среди ридов, ассоциированных с одним и тем же vNRUMI, ридов с общими виртуальными UMI или аналогичными виртуальными UMI, если общие виртуальные UMI обнаруживаются в указанном фрагменте ДНК. Указанный способ также включает определение последовательности фрагмента ДНК с применением только тех ридов, которые одновременно ассоциированы с одним и тем же vNRUMI и содержат одни и те же виртуальные UMI или клеточные виртуальные UMI.
[00167] Согласно некоторым вариантам реализации адаптеры для секвенирования, содержащие vNRUMI, могут быть получены с использованием процесса, изображенного на Фиг. 1D и дополнительно описанного здесь и далее в настоящем документе.
Дизайн UMI
Физические UMI
[00168] Согласно некоторым вариантам реализации описанных выше адаптеров физические UMI в указанных адаптерах включают случайные UMI. Согласно некоторым вариантам реализации каждый случайный UMI отличается от любого другого случайного UMI, которым обрабатывают фрагменты ДНК. Другими словами, случайные UMI выбирают случайным образом без замены из набора UMI, включающего все возможные разные UMI с учетом длин(ы) последовательности. Согласно другим вариантам реализации случайные UMI выбирают случайным образом с заменой. Согласно указанным вариантам реализации два адаптера могут содержать один и тот же UMI, что обусловлено случайным шансом.
[00169] Согласно некоторым вариантам реализации, используемые в процессе физические UMI представляют собой набор NRUMI, выбранных из пула кандидатных последовательностей с применением «жадного метода», который максимизирует различия между выбранными UMI, согласно более подробному описанию здесь и далее в настоящем документе. Согласно некоторым вариантам реализации указанные NRUMI имеют вариабельные или гетерогенные длины молекул, образуя набор vNRUMI. Согласно некоторым вариантам реализации пул кандидатных последовательностей фильтруют для удаления определенных последовательностей до перехода к выбору для набора UMI, используемого в реакции или в процессе.
[00170] Случайные UMI обеспечивают большее число уникальных UMI, чем неслучайные UMI при той же длине последовательности. Другими словами, случайные UMI с большей вероятностью будут уникальными, чем неслучайные UMI. Однако согласно некоторым вариантам реализации неслучайные UMI может быть легче получить, или они могут иметь более высокий коэффициент преобразования. В комбинации неслучайных UMI с другой информацией, например, положением последовательности и виртуальными UMI, они могут обеспечивать эффективный механизм для индексации исходных молекул фрагментов ДНК.
Конструирование vNRUMI
[00171] Согласно некоторым вариантам реализации адаптеры для секвенирования, содержащие vNRUMI, могут быть получены с использованием «жадного метода», представленного на Фиг. 1D. Указанный процесс включает (а) обеспечение набора последовательностей олигонуклеотидов с двумя разными длинами молекул; и (b) выбор поднабора последовательностей олигонуклеотидов из набора последовательностей олигонуклеотидов, отличающегося тем, что все редакционные расстояния между последовательностями олигонуклеотидов в указанном поднаборе соответствуют пороговому значению. Указанный поднабор последовательностей олигонуклеотидов образует набор vNRUMI. Указанный способ также включает (с) синтез множества адаптеров для секвенирования, где адаптер для секвенирования содержит двуцепочечную гибридизованную область, одноцепочечный 5'-конец, одноцепочечный 3'-конец согласно изображению на на Фиг. 2А, и по меньшей мере одного vNRUMI в наборе vNRUMI.
[00172] Фиг. 1D иллюстрирует процесс 140 для получения адаптеров для секвенирования, содержащих vNRUMI. Процесс 140 начинается с обеспечения набора последовательностей олигонуклеотидов (β) по меньшей мере с двумя разными длинами молекул. См. блок 141.
[00173] Согласно различным вариантам реализации неслучайные UMI получают с учетом различных факторов, в том числе, но не ограничиваясь перечисленным, способа детекции ошибки в последовательностях UMI, коэффициента преобразования, совместимости анализов, содержания GC, гомополимеров и ограничений производства.
[00174] Согласно некоторым вариантам реализации до операции 141 некоторые из указанных последовательностей олигонуклеотидов удаляют из полного набора всех возможных пермутаций нуклеотидов, при условии специфических длин молекул в наборе vNRUMI. Например, если указанные vNRUMI имеют длины молекул, равные шести и семи нуклеотидам, все возможные пермутаций последовательностей включают полный пул из 46+47=20480 последовательностей. Определенные последовательности олигонуклеотидов удаляют из пула для обеспечения набора последовательностей олигонуклеотидов β.
[00175] Согласно некоторым вариантам реализации последовательности олигонуклеотидов, содержащие три или более последовательных идентичных оснований, удаляют из пула для обеспечения набора β. Согласно некоторым вариантам реализации удаляют последовательности олигонуклеотидов, общее число гуаниновых и цитозиновых (G и С) оснований в которых меньше двух. Согласно некоторым вариантам реализации удаляют последовательности олигонуклеотидов, общее число гуаниновых и цитозиновых оснований в которых выше четырех. Согласно некоторым вариантам реализации удаляют последовательности олигонуклеотидов, содержащие одно и то же основание в последних двух положениях последовательности. Начало указанной последовательности располагается на конце, противоположном концу, присоединенному к фрагментам ДНК.
[00176] Согласно некоторым вариантам реализации удаляют последовательности олигонуклеотидов, содержащие частичную последовательность, совпадающую с 3'-концом любого из праймеров для секвенирования.
[00177] Согласно некоторым вариантам реализации удаляют последовательности олигонуклеотидов, содержащие тиминовое (Т) основание в последнем положении последовательностей нуклеотидов. Присоединенный к содержащему А-хвост концу обработанного фрагмента нуклеиновой кислоты vNRUMI обеспечивает получение частичной последовательности рида, содержащей последовательность vNRUMI и основание Т, отожженное с концом последовательности vNRUMI, поскольку основание Т комплементарно основанию А на А-хвосте. Путем отфильтровывания кандидатных последовательностей, содержащих основание Т в последнем положении, избегают смешивания таких кандидатных последовательностей и частичной последовательности ридов, происходящей из любых vNRUMI.
[00178] Процесс 140 переходит к выбору последовательности олигонуклеотидов (S0) из β. См. блок 142. Согласно некоторым вариантам реализации S0 может быть случайным образом выбран из набора последовательностей олигонуклеотидов.
[00179] Процесс 140 дополнительно включает добавление S0 к расширяемому набору γ последовательностей олигонуклеотидов и удаление S0 из набора β. См. блок 143.
[00180] Процесс 140 дополнительно включает выбор последовательности олигонуклеотидов Si из β, Si максимизирует функцию расстояния d(Si, γ), которое представляет собой минимальное редакционное расстояние между Si и любой последовательностью олигонуклеотидов в наборе γ. См. блок 144. Согласно некоторым вариантам реализации указанное редакционное расстояние представляет собой расстояние Левенштейна.
[00181] Согласно некоторым вариантам реализации, если последовательность короче максимальной длины vNRUMI, к концу указанной последовательности прибавляют одно или более оснований при вычислении расстояния Левенштейна или редакционного расстояния. Согласно некоторым вариантам реализации, если последовательность на одно основание короче максимальной длины vNRUMI, тиминовое (Т) основание добавляют к концу указанной последовательности. Указанное основание Т добавляют с учетом наличия липкого концевого участка с Т-основанием на конце адаптера, комплементарного А-основанию на конце фрагмента ДНК, подвергнутого обработке с присоединением dA-хвостов согласно описанию в тексте настоящего документа. Согласно некоторым вариантам реализации, если последовательность более чем на одно основание короче максимальной длины vNRUMI, к концу указанной последовательности добавляют Т-основание, а затем после Т-основания добавляют одно или более случайный оснований для создания последовательности с длиной молекулы, равной максимальной длине vNRUMI. Другими словами, может быть прибавлено несколько разных комбинаций случайных оснований после Т-основания для создания последовательностей, охватывающих все возможные наблюдаемые последовательности. Например, если vNRUMI имеют длины 6 и 8, могут быть получены четыре деривата 6-мера путем прибавления ТА, ТС, TG и ТТ.
[00182] Процесс 140 переходит к определению того, соответствует ли значение функции расстояния d(Si, γ) пороговому значению. Согласно некоторым вариантам реализации указанное пороговое значение может подразумевать, что функция расстояния (например, дополненное расстояние Левенштейна) принимала значение, равное по меньшей мере 3. Если значение функции расстояния d(Si, γ) соответствует порогу, процесс переходит к добавлению Si к расширяемому набору γ и удалению Si из набора β. См. ветвь решения типа «да» 145 и блок 146. Если функция расстояния не соответствует пороговому значению, процесс 140 не добавляет Si к расширяемому набору γ, а переходит к синтезу множества адаптеров для секвенирования, где каждый адаптер для секвенирования содержит по меньшей мере один vNRUMI в расширяемом наборе γ. См. ветвь решения типа «нет» 145 с указателем на блок 148.
[00183] После этапа 146 процесс 140 дополнительно включает операцию решения о том, должны ли быть рассмотрены дополнительные последовательности из набора β. В случае положительного решения процесс возвращается к блоку 144 для выбора дополнительных последовательностей олигонуклеотидов из набора β, максимизирующего функцию расстояния. При определении того, должны ли быть также рассмотрены дополнительные последовательности из набора β, могут быть учтены различные факторы. Например, согласно некоторым вариантам реализации после получения требуемого числа последовательностей уже нет необходимости рассмотрения процессом дополнительных последовательностей из данных набора последовательностей.
[00184] После решения об отсутствии необходимости рассмотрения дополнительных последовательностей процесс 140 переходит к синтезу множества адаптеров для секвенирования, где каждый адаптер содержит по меньшей мере один vNRUMI в наборе последовательностей γ. См. ветвь решения типа «нет» операции 147 с указателем на операция 148. Согласно некоторым вариантам реализации каждый адаптер для секвенирования содержит vNRUMI на одной цепи адаптеров для секвенирования. Согласно некоторым вариантам реализации адаптеры для секвенирования, принимающие любую из форм, проиллюстрированных на Фиг. 2А, синтезируют в ходе операции 148. Согласно некоторым вариантам реализации каждый адаптер для секвенирования содержит только один vNRUMI. Согласно некоторым вариантам реализации каждый адаптер содержит vNRUMI на каждой цепи адаптеров для секвенирования. Согласно некоторым вариантам реализации каждый адаптер для секвенирования содержит vNRUMI на каждой цепи адаптера для секвенирования в двуцепочечной гибридизованной области.
[00185] Согласно некоторым вариантам реализации указанный процесс может быть реализован приведенным ниже псевдокодом.


[00186] Далее приведен модельный пример для иллюстрации того, как vNRUMI могут быть получены в соответствии с процессом и алгоритмом, описанными выше. Указанный модельный пример показывает, как vNRUMI могут быть получены из пула, включающего пять кандидатных последовательностей, которые затем используют для картирования наблюдаемых ридов последовательностей. Отметим, что поскольку указанный модельный пример относится к значимо меньшему пространству последовательностей, чем те, которые были бы использованы/встречены на практике, не все аспекты характеристик vNRUMI могут быть рассмотрены.
[00187] В указанном модельном примере процесс направлен на конструирование набора из 3 последовательностей vNRUMI, начинающегося с набора 6-меров и 7-меров (но дает только 2 последовательности vNRUMI). Для простоты предположим, что все пространство возможных 6-меров и 7-меров состоит из следующих 5 последовательностей:
[00188] ААСТТС
[00189] ААСТТСА
[00190] AGCTTCG
[00191] CGCTTCG
[00192] CGCTTC
[00193] Отметим, что, как предполагается, все указанные 5 последовательностей прошли любые используемые биохимические фильтры. На очень высоком уровне указанный алгоритм получает поднабор из пула входящих последовательностей, максимизируя при этом редакционное расстояние (расстояние Левенштейна) между выбранными последовательностями. Это реализуется с использованием «жадного метода» - при каждой итерации алгоритм выбирает последовательность, которая максимизирует функцию расстояния. Функция расстояния, в указанном случае, представляет собой минимальное редакционное расстояние между последовательностью для добавления и любой последовательностью, уже вошедшей в набор. Указанная функция может быть математически выражена следующим образом:
[00194] d(s, γ) - min(levenshtein(s, x) ∀ х ∈ γ)
[00195] В примере ниже конструируемый набор vNRUMI (n=3) обозначен как у, набор входящих кандидатных последовательностей обозначен как β.
[00196] γ={ }, β={ААСТТС, AACTTCA, AGCTTCG, CGCTTCG, CGCTTC}
Поскольку в γ нет последовательностей, функция расстояния d не определена для каждой из 5 последовательностей. В случае совпадения результатов для наилучшего выбора всегда выбирают один из кандидатов с совпадающими результатами случайным образом, отдавая предпочтение более коротким последовательностям. В данном случае пример процесса выбирает 6-мерную последовательность ААСТТС. Он добавляет указанную последовательность в γ и удаляет ее из пула кандидатных последовательностей.
[00197] γ={ААСТТС}, β={AACTTCA, AGCTTCG, CGCTTCG, CGCTTC}
[00198] Вычисляют метрику расстояния d(s, γ) ∀ s ∈ β.
[00199] d(AACTTCA, γ)=1, поскольку требуется только одно изменение (добавление А) для перехода от единственного элемента в γ к ААСТТСА, и, соответственно, функция расстояния равна 1.
[00200] d(AGCTTCG, γ)=2, поскольку требуется два изменения для перехода от указанной последовательности к последовательности, уже вошедшей в γ.
[00201] d(CGCTTCG, γ)=3, поскольку требуется три изменения для перехода от указанной последовательности к последовательности, уже вошедшей в γ.
[00202] d{CGCTTC, γ)=2, поскольку сравниваемая последовательность представляет собой 6-мер, согласно некоторым вариантам реализации к ее концу добавляют основание «Т» для моделирования процесса отжига, в ходе которого основание Т, комплементарное «А»-хвосту, отжигают с последовательностью адаптера. Обоснованием является то, что позже при попытке специалистов идентифицировать NRUMI будет учитываться и первый 6-мер, и первый 7-мер. Добавление указанного основания Т гарантирует, что при рассмотрении 7-мера он все же не будет слишком близким любому другому NRUMI. По сравнению с ААСТТС, для 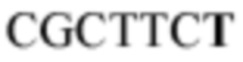 требуется два изменения.
требуется два изменения.
[00203] Поскольку максимальное значение функции расстояния, обеспечиваемое последовательностью CGCTTCG, равно 3, и указанное расстояние соответствует установленному нами минимальному порогу (равному 3), процесс добавляет CGCTTCG к γ и удаляет ее из β.
[00204] γ={ААСТТС, CGCTTCG}, β={AACTTCA, AGCTTCG, CGCTTC}
[00205] Затем процесс переходит к вычислению метрики расстояния d(s, γ) ∀ s ∈ β, поскольку число последовательностей в наборе vNRUMI меньше требуемого (3).
[00206] d(AACTTCA, γ)=1. Согласно произведенным на предыдущем этапе расчетам редакционное расстояние между указанной последовательностью и первой последовательностью vNRUMI, s1=ААСТТС, равно 1. Редакционное расстояние между указанной последовательностью и второй последовательностью vNRUMI, s2=CGCTTCG, равно 3. Функция расстояния принимает минимальное из всех редакционных расстояний между запрашиваемой последовательностью и любой существующей последовательностью, и min(3,1)=1, таким образом, значение функции расстояния равно 1.
[00207] d(AGCTTCG, γ)=1. Согласно произведенным на предыдущем этапе расчетам редакционное расстояние между указанной последовательностью и s1 равно 2. Редакционное расстояние между указанной последовательностью и s2 равно 1. Соответственно, значение функции расстояния равно меньшему значению из 2 и 1 (то есть 1).
[00208] d(CGCTTC, γ)=1. Как и ранее, процесс прибавляет Т к указанной последовательности с преобразованием ее в CGCTTCT. Расстояние между удлиненной запрашиваемой последовательностью и s1 равно 2, как было ранее определено. Расстояние между удлиненной запрашиваемой последовательностью и s2 равно 1, таким образом, значение функции расстояния равно 1.
[00209] После вычисления всех значений функции расстояния для всех кандидатных последовательностей видим, что ни одна из них не соответствует заданному нами инвариантному требованию: значению для редакционного расстояния не менее 3. Указанное требование обеспечивает очень малую вероятность изменения за счет случайных мутаций одной последовательности vNRUMI таким образом, чтобы она в какой-либо мере напоминала другую последовательность vNRUMI. Соответственно, мы возвращаем указанный набор из 2 последовательностей vNRUMI, γ={AACTTC, CGCTTCG}. Отметим, что указанные две последовательности vNRUMI соответствуют S1 и S2 на Фиг. 1Е, описанным выше, и они могут быть ассоциированы с ридами для определения исходного сегмента ридов согласно описанию применительно к Фиг. 1Е.
Виртуальные UMI
[00210] Переходя к виртуальным UMI, те виртуальные UMI, которые определены в концевых положениях или относительно концевых положений исходных молекул ДНК, могут однозначно или почти однозначно определять индивидуальные исходные молекулы ДНК, если местоположения концевых положений в общем случайны, как при некоторых процедурах фрагментации и для встречающихся в природе вкДНК. Если образец содержит относительно малое количество исходных молекул ДНК, виртуальные UMI могут сами по себе однозначно идентифицировать индивидуальные исходные молекулы ДНК. Использование комбинации двух виртуальных UMI, каждый из которых ассоциирован с другим концом исходной молекулы ДНК, увеличивает вероятность того, что виртуальные UMI сами по себе могут однозначно идентифицировать исходные молекулы ДНК. Разумеется, даже в ситуациях, когда один или два виртуальных UMI не могут сами по себе однозначно идентифицировать исходные молекулы ДНК, комбинация таких виртуальных UMI с одним или более физическими UMI может успешно это обеспечивать.
[00211] Если два рида происходят из одного и того же фрагмента ДНК, две частичные последовательности, содержащие одни и те же пары оснований, будут также характеризоваться одним и тем же относительным местоположением в ридах. И напротив, если два рида происходят из двух разных фрагментов ДНК, маловероятно, что две частичные последовательности, содержащие одни и те же пары оснований, будут характеризоваться одним и тем же относительным местоположением в ридах. Соответственно, если две или более частичных последовательностей из двух или более ридов содержат одни и те же пары оснований и одно и то же относительное местоположение на двух или более ридах, может быть сделан вывод, что указанные два или более ридов происходят из одного и того же фрагмента.
[00212] Согласно некоторым вариантам реализации частичные последовательности на концах или возле концов фрагмента ДНК используют в качестве виртуальных UMI. Выбор указанного дизайна обеспечивает некоторые практические преимущества. Во-первых, относительные местоположения указанных частичных последовательностей на ридах могут быть легко уточнены, поскольку они находятся в начале или возле начала ридов, и системе не нужно использовать смещение для обнаружения виртуальных UMI. Кроме того, поскольку пары оснований на концах фрагментов секвенируются первыми, указанные пары оснований доступны даже в том случае, если риды относительно короткие. Кроме того, пары оснований, определенные раньше в длинном риде, отличаются боле низким коэффициентом ошибок секвенирования, чем определенные позже. Тем не менее, согласно другим вариантам реализации, в качестве виртуальных UMI могут быть использованы частичные последовательности, локализованные на расстоянии от концов ридов, однако может быть необходимо уточнение их относительных положений на ридах, чтобы подтвердить, что риды получены из одного и того же фрагмента.
[00213] Одна или более частичных последовательностей в риде могут применяться в качестве виртуальных UMI. Согласно некоторым вариантам реализации две частичные последовательности, каждую из которых отслеживают с разных концов исходной молекулы ДНК, используют в качестве виртуальных UMI. Согласно различным вариантам реализации длина виртуальных UMI составляет приблизительно 24 пар оснований или менее, приблизительно 20 пар оснований или менее, приблизительно 15 пар оснований или менее, приблизительно 10 пар оснований или менее, приблизительно 9 пар оснований или менее, приблизительно 8 пар оснований или менее, приблизительно 7 пар оснований или менее, или приблизительно 6 пар оснований или менее. Согласно некоторым вариантам реализации длина виртуальных UMI составляет приблизительно 6-10 пар оснований. Согласно другим вариантам реализации длина виртуальных UMI составляет приблизительно 6-24 пары оснований.
Адаптеры
[00214] Наряду с дизайном адаптеров, описанным в примере технологической схемы 100 применительно к Фиг. 1А, выше, в различных вариантах реализации способов и систем согласно описанию в настоящем документе могут быть использованы другие варианты дизайна адаптеров. На Фиг. 2А схематически представлено пять разных вариантов дизайна адаптеров с UMI, которые могут быть задействованы в различных вариантах реализации.
[00215] На Фиг. 2A(i) показан стандартный адаптер с двойным индексом Illumina TruSeq®. Указанный адаптер является частично двуцепочечным и образован путем отжига двух олигонуклеотидов, соответствующих двум цепям. Указанные две цепи содержат ряд комплементарных пар оснований (например, 12-17 п.о.), что позволяет провести отжиг указанных двух олигонуклеотидов на конце для лигирования с фрагментом дцДНК. Фрагмент дцДНК для лигирования на обоих концах для парно-концевых ридов также называют вставкой. Другие пары оснований на указанных двух цепях не комплементарны, что приводит к образованию вилкообразного адаптера, содержащего два свободных липких концевых участка. В примере на Фиг. 2A(i) комплементарные пары оснований входят в состав последовательности праймера рида 2 и последовательности праймера рида 1. В 3'-направлении от последовательности праймера рида 2 находится однонуклеотидный липкий концевой участок 3'-Т, обеспечивающий образование липкого конца, комплементарного однонуклеотидному липкому концу 3'-А фрагмента дцДНК для секвенирования, что может облегчать гибридизацию двух липких концевых участков. Последовательность праймера рида 1 расположена на 5'-конце комплементарной цепи, к которой присоединена фосфатная группа. Фосфатная группа необходима для лигирования 5'-конца последовательности праймера рида 1 с липким концевым участком 3'-А фрагмента ДНК. На цепи, содержащей свободный липкий 5'-концевой участок (верхнюю цепь), адаптер содержит, в направлении 5'→3', последовательность Р5, индексную последовательность i5 и последовательность праймера рида 2. На цепи, содержащей свободный липкий 3'-концевой участок, адаптер содержит, в направлении 3'→5', последовательность Р7', индексную последовательность i7 и последовательность праймера рида 1. Олигонуклеотиды Р5 и Р7' комплементарны праймерам для амплификации, связанным с поверхностью проточных ячеек платформы для секвенирования Illumina. Согласно некоторым вариантам реализации индексные последовательности обеспечивают способ отслеживания источника образца, что позволяет проводить мультиплексирование нескольких образцов на платформе для секвенирования.
[00216] На Фиг. 2A(ii) показан адаптер, содержащий единственный физический UMI, заменяющий область индекса i7 стандартного адаптера с двойным индексом, показанного на Фиг. 2A(i). Указанный дизайн адаптера отражает представленный в примере технологической схемы, описанной выше применительно к Фиг. 1В. Согласно некоторым вариантам реализации дизайн физических UMI α и β подразумевает размещение только на 5'-плече двуцепочечных адаптеров, что приводит к получению продуктов лигирования, содержащих только один физический UMI на каждой цепи. В отличие от этого, включение физических UMI в обе цепи адаптеров приводит к получению продуктов лигирования с двумя физическими UMI на каждой цепи, что удваивает продолжительность и стоимость секвенирования физических UMI. Однако настоящее изобретение предусматривает реализацию способов, задействующих физические UMI на обеих цепях адаптеров, как показано на Фиг. 2A(iii) - 2A(vi), обеспечивающих дополнительную информацию, которую можно использовать для объединения разных ридов с получением консенсусных последовательностей.
[00217] Согласно некоторым вариантам реализации физические UMI в адаптерах включают случайные UMI. Согласно некоторым вариантам реализации физические UMI в адаптерах включают неслучайные UMI.
[00218] На Фиг. 2А(iii) показан адаптер, содержащий два физических UMI, добавленных к стандартному адаптеру с двойным индексом. Показанные физические UMI могут представлять собой случайные UMI или неслучайные UMI. Первый физический UMI расположен в 5'-направлении от индексной последовательности i7, а второй физический UMI расположен в 5'-направлении от индексной последовательности i5. На Фиг. 2A(iv) показан адаптер, также содержащий два физических UMI, добавленные к стандартному адаптеру с двойным индексом. Первый физический UMI расположен в 3'-направлении от индексной последовательности i7, а второй физический UMI расположен в 3'-направлении от индексной последовательности i5. Аналогичным образом, указанные два физических UMI могут представлять собой случайные UMI или неслучайные UMI.
[00219] Адаптер, содержащий два физических UMI на двух плечах одноцепочечной области, такие как показанные на 2А(iii) и 2A(iv), может соединять две цепи фрагмента двуцепочечной ДНК, если известна априорная или апостериорная информация об ассоциации двух некомплементарных физических UMI. Например, исследователь может знать последовательности UMI 1 и UMI 2 до интеграции их в один и тот же адаптер согласно дизайну, представленному на Фиг. 2A(iv). Указанная информация об ассоциации может быть использована для установления происхождения ридов, содержащих UMI 1 и UMI 2, из двух цепей фрагмента ДНК, с которым был лигирован указанный адаптер. Соответственно, могут быть объединены не только риды, содержащие одни и те же физические UMI, однако также риды, содержащие любой из двух некомплементарных физических UMI. Интересно, что, как дополнительно обсуждается ниже, явление, называемое «перепрыгиванием UMI», может осложнять установление ассоциаций между физическими UMI на одноцепочечных областях адаптеров.
[00220] Два физических UMI на двух цепях адаптеров на Фиг. 2А(iii) и Фиг. 2A(iv) не локализованы на одном и том же сайте и не комплементарны друг другу. Тем не менее настоящим изобретением предусмотрены способы, задействующие физические UMI, расположенные в одном и том же сайте на двух цепях адаптера и/или комплементарные друг другу. На Фиг. 2A(v) показан дуплексный адаптер, где два физических UMI комплементарны в двуцепочечной области на конце или возле конца адаптера. Указанные два физических UMI могут представлять собой случайные UMI или неслучайные UMI. На Фиг. 2A(vi) показан адаптер, аналогичный показанному на Фиг. 2A(v), но короче, однако он не содержит индексных последовательностей или последовательностей Р5 и Р7', комплементарных праймерам для амплификации на поверхности проточной ячейки. Аналогичным образом, указанные два физических UMI могут представлять собой случайные UMI или неслучайные UMI.
[00221] По сравнению с адаптерами, содержащими один или более одноцепочечных физических UMI на одноцепочечных плечах, адаптеры, содержащие двуцепочечный физический UMI в двуцепочечной области, могут обеспечивать прямую связь между двумя цепями фрагмента двуцепочечной ДНК, с которым лигируют указанный адаптер, как показано на Фиг. 2A(v) и Фиг. 2A(vi). Поскольку две цепи двуцепочечного физического UMI комплементарны друг другу, ассоциация между указанными двумя цепями двуцепочечного UMI по сути отражена в комплементарных последовательностях, и может быть установлена как без априорной, так и без апостериорной информации. Указанная информация может быть использована для установления того, что риды, содержащие две комплементарные последовательности двуцепочечного физического UMI адаптера, происходят из одного и того же фрагмента ДНК, с которым был лигирован указанный адаптер, но указанные две комплементарные последовательности физического UMI лигированы с 3'-концом на одной цепи и 5'-концом на другой цепи фрагмента ДНК. Соответственно, можно объединить не только риды, содержащие в одном и том же порядке две последовательности физических UMI на двух концах, но и риды, содержащие две комплементарные последовательности в обратном порядке на двух концах.
[00222] Согласно некоторым вариантам реализации может быть благоприятным использование относительно коротких физических UMI, поскольку короткие физические UMI проще ввести в состав адаптеров. Кроме того, более короткие физические UMI быстрее и проще секвенировать в амплифицированных фрагментах. Однако по мере значительного укорочения физических UMI общее число разных физических UMI может стать меньше, чем число адаптерных молекул, необходимое для обработки образцов. Для того, чтобы обеспечить достаточное число адаптеров, потребуется повторение одного и того же UMI в двух или более адаптерных молекулах. В таком сценарии адаптеры, содержащие одни и те же физические UMI, могут быть лигированы с несколькими исходными молекулами ДНК. Однако указанные короткие физические UMI могут обеспечивать достаточно информации в комбинации с другой информацией, такой как виртуальные UMI и/или места выравнивания ридов, для однозначной идентификации ридов как происходящих из конкретного исходного полинуклеотида или фрагмента ДНК в образце. Это обусловлено тем, что даже несмотря на то, что один и тот же физический UMI может быть лигирован с двумя разным фрагментами, маловероятно, что указанные два разных фрагмента будут также характеризоваться одними и теми же местами выравнивания, или совпадающими частичными последовательностями, которые служат в качестве виртуальных UMI. Соответственно, если два рида содержат один и тот же короткий физический UMI и характеризуются одним и тем же местом выравнивания (или содержат один и тот же виртуальный UMI), указанные два рида, вероятно, происходят из одного и того же фрагмента ДНК.
[00223] Кроме того, согласно некоторым вариантам реализации объединение ридов основано на двух физических UMI на двух концах вставки. Согласно таким вариантам реализации два очень коротких физических UMI (например, 4 п.о.) комбинируют для определения источника фрагментов ДНК, при этом комбинированная длина указанных двух физических UMI обеспечивает достаточную информацию для различения разных фрагментов.
[00224] Согласно различным вариантам реализации физические UMI содержат приблизительно 12 пар оснований или менее, приблизительно 11 пар оснований или менее, приблизительно 10 пар оснований или менее, приблизительно 9 пар оснований или менее, приблизительно 8 пар оснований или менее, приблизительно 7 пар оснований или менее, приблизительно 6 пар оснований или менее, приблизительно 5 пар оснований или менее, приблизительно 4 пар оснований или менее; или приблизительно 3 пар оснований или менее. Согласно некоторым вариантам реализации, отличающимся тем, что физические UMI представляют собой неслучайные UMI, указанные UMI содержат приблизительно 12 пар оснований или менее, приблизительно 11 пар оснований или менее, приблизительно 10 пар оснований или менее, приблизительно 9 пар оснований или менее, приблизительно 8 пар оснований или менее, приблизительно 7 пар оснований или менее; или приблизительно 6 пар оснований.
[00225] Перепрыгивание UMI может влиять на выводы об ассоциациях физических UMI на одном плече или обоих плечах адаптеров, например, в адаптерах с Фиг. 2A(ii)-(iv). Наблюдалось, что при обработке указанными адаптерами фрагментов ДНК продукты амплификации могут включать число фрагментов, содержащих уникальный физические UMI, превышающее фактическое число фрагментов в указанном образце.
[00226] Кроме того, при применении адаптеров, содержащих физические UMI на обоих плечах, предполагается, что амплифицированные фрагменты, содержащие общий физический UMI на одном конце, содержат другой общий физический UMI на другом конце. Однако иногда это не так. Например, в продукте реакции одной реакции амплификации некоторые фрагменты могут содержать первый физический UMI и второй физический UMI на двух концах; другие фрагменты могут содержать второй физический UMI и третий физический UMI; и другие фрагменты могут содержать первый физический UMI и третий физический UMI; дополнительные фрагменты могут содержать третий физические UMI и четвертый физический UMI, и так далее. В указанном примере исходный фрагмент или фрагменты для указанных амплифицированных фрагментов может быть трудно определить. По-видимому, в процессе амплификации физические UMI могут быть «подменены» другим физическим UMI.
[00227] Один из возможных способов решения указанной проблемы с перепрыгиванием UMI заключается в том, чтобы учитывать только фрагменты, в которых оба UMI общие, как происходящие из одной и той же исходной молекулы, тогда как фрагменты, содержащие только один общий UMI, исключают из анализа. Однако некоторые из указанных фрагментов, содержащих только один общий физический UMI, могут на самом деле происходить из той же молекулы, как и те, в которых оба физических UMI общие. При исключении из рассмотрения указанных фрагментов, содержащих всего один общий физический UMI, может быть утрачена полезная информация. Согласно другому возможному подходу рассматривают любые фрагменты, содержащие один общий физический UMI, как происходящие из одной и той же исходной молекулы. Однако указанный подход не позволяет комбинировать два физических UMI на двух концах фрагментов для последующего анализа. Кроме того, в рамках любого из указанных подходов, в примере выше фрагменты, содержащие общие первый и второй физические UMI, не будут рассматриваться как происходящие из той же исходной молекулы, что и фрагменты, содержащие общие третий и четвертый физические UMI. Это может быть истинным или нет. Третий подход может обеспечить решение проблемы перепрыгивания UMI за счет применения адаптеров с физическими UMI на обеих цепях одноцепочечной области, таких как адаптеры на Фиг. 2A(v)-(vi). Ниже представлено более подробное описание гипотетического механизма, лежащего в основе перепрыгивания UMI.
[00228] Фиг. 2В иллюстрирует гипотетический процесс, в ходе которого происходит перепрыгивание UMI при ПЦР-реакции, включающей адаптеры, содержащие физические UMI на обеих цепях в двуцепочечной области. Указанные два физических UMI могут представлять собой случайные UMI или неслучайные UMI. Фактический механизм, лежащий в основе перепрыгивания UMI, как и гипотетический процесс, описанный в настоящем документе, не влияет на полезность адаптеров и способов, предложенных в настоящем документе. ПЦР-реакция начинается с получения по меньшей мере одного двуцепочечного исходного фрагмента ДНК 202 и адаптеров 204 и 206. Адаптеры 204 и 206 аналогичны адаптерам, проиллюстрированным Фиг. 2А(iii)-(iv). Адаптер 204 содержит адаптерную последовательность Р5 и физический UMI α1 на 5'-плече. Адаптер 204 также содержит адаптерную последовательность Р7' и физический UMI α2 на 3'-плече. Адаптер 206 содержит адаптерную последовательность Р5 и физический UMI β2 на 5'-плече, и адаптерную последовательность Р7' и физический UMI β1 на 3'-плече. Процесс переходит к лигированию адаптера 204 и адаптера 206 с фрагментом 202, с получением продукта лигирования 208. Процесс переходит к денатурации продукта лигирования 208, что приводит к получению одноцепочечного денатурированного фрагмента 212. При этом реакционная смесь на указанной стадии часто включает остаточные адаптеры, поскольку некоторые адаптеры все же остаются в реакционной смеси, даже если процесс уже включал удаление избытка адаптеров, например, с применением гранул для твердофазной обратимой иммобилизации (SPRI-гранул). Такой остаточный адаптер показан как адаптер 210, аналогичный адаптеру 206, за исключением того, что адаптер 210 содержит физические UMI γ1 и γ2 на 3'- и 7'-плечах, соответственно. Денатурирующие условия, обеспечивающие получение денатурированного фрагмента 212, также обеспечивают получение денатурированного адаптерного олигонуклеотида 214, который содержит физический UMI γ2 возле адаптерной последовательности Р5.
[00229] Одноцепочечный адаптерный фрагмент 214 затем гибридизуют с одноцепочечным фрагментом ДНК 212, и в процессе ПЦР достраивают одноцепочечный адаптерный фрагмент 214 с получением промежуточной вставки 216, комплементарной фрагменту ДНК 212. В ходе различных циклов ПЦР-амплификации при ПЦР-достройке цепей Р7' адаптеров могут образовываться промежуточные адаптерные фрагменты 218, 220 и 222, в том числе разные физические UMI δ, ε, и ζ. Все промежуточные адаптерные фрагменты, 218, 220 и 222, содержат последовательность Р7' на 5'-конце, и, соответственно, содержат физические UMI δ, ε, и ζ. В последующих циклах ПНР промежуточные адаптерные фрагменты 218, 220 и 222 могут гибридизоваться с промежуточным фрагментом 216 или его ампликонами, поскольку 3'-концы промежуточных адаптерных фрагментов 218, 220 и 222 комплементарны области 217 промежуточной вставки 216. В результате ПЦР-достройки гибридизированных фрагментов образуются одноцепочечные фрагменты ДНК 224, 226 и 228. Фрагменты ДНК 224, 226 и 228 помечены тремя разными физическими UMI (δ, ε, и ζ) на 5'-конце, и физическими UMI γ2 на 3'-конце, указывающими на «перепрыгивание UMI», если разные UMI присоединяются к последовательностям нуклеотидов, происходящим из одного и того же фрагмента ДНК 202.
[00230] Согласно некоторым вариантам реализации настоящего изобретения применение адаптеров, содержащих физические UMI на обеих цепях двуцепочечной области адаптеров, таких как адаптеры на Фиг. 2A(v)-(vi), могут предотвращать или сокращать перепрыгивание UMI. Это может быть обусловлено тем фактом, что физические UMI на одном адаптере в двуцепочечной области отличаются от физических UMI на всех других адаптерах. Это помогает уменьшить комплементарность между промежуточными адаптерными олигонуклеотидами и промежуточными фрагментами, и избежать таким образом гибридизации, например, показанной для промежуточного олигонуклеотида 222 и промежуточного фрагмента 220, таким образом сокращая или предотвращая перепрыгивание UMI.
Объединение ридов и получение консенсусных последовательностей
[00231] Согласно различным вариантам реализации с применением UMI несколько ридов последовательностей, содержащих один и те же или одни и те же UMI, объединяют с получением одной или более консенсусных последовательностей, которые затем используют для определения последовательности исходной молекулы ДНК. Несколько отдельных ридов может быть получено с отдельных копий одной и той же исходной молекулы ДНК, и указанные риды могут сравниваться для получения консенсусной последовательности согласно описанию в настоящем документе. Копии могут быть получены путем амплификации исходной молекулы ДНК до секвенирования, таким образом отдельные операции секвенирования осуществляют на отдельных продуктах амплификации, имеющих общую последовательность исходной молекулы ДНК. Конечно, при амплификации могут быть введены ошибки, так что последовательности отдельных продуктов амплификации содержат различия. В контексте некоторых технологий секвенирования, таких как секвенирование путем синтеза от Illumina, исходная молекула ДНК или продукт ее амплификации образует кластер молекул ДНК, соединенную с областью проточной ячейки. Молекулы кластера в совокупности образуют рид. Как правило, по меньшей мере два рида необходимы для получения консенсусной последовательности. Примеры глубины секвенирования, подходящей для применения в предложенных вариантах реализации для создания консенсусных ридов при низкой частоте аллелей (например, приблизительно 1% или меньшей), представлены глубиной секвенирования, равной 100, 1000 и 10000.
[00232] Согласно некоторым вариантам реализации в консенсусную последовательность включают нуклеотиды, стабильно присутствующие в 100% ридов, содержащие общий UMI или общую комбинацию UMI. Согласно другим вариантам реализации консенсусный критерий может быть ниже 100%. Например, может быть использован консенсусный критерий 90%, что означает, что пары оснований, присутствующие в 90% или более ридов в группе, включают в консенсусную последовательность. Согласно различным вариантам реализации консенсусный критерий может быть установлен на уровне приблизительно 30%, приблизительно 40%, приблизительно 50%, приблизительно 60%, приблизительно 70%, приблизительно 80%, приблизительно 90%, приблизительно 95% или приблизительно 100%.
Объединение по физическим UMI и виртуальным UMI
[00233] Для объединения ридов, которые включают несколько UMI, может применяться несколько методик. Согласно некоторым вариантам реализации риды, содержащие общий физический UMI, могут быть объединены с получением консенсусной последовательности. Согласно некоторым вариантам реализации, если общий физический UMI представляет собой случайный UMI, указанный случайный UMI может быть достаточно уникальным для идентификации конкретной исходной молекулы фрагмента ДНК в образце. Согласно другим вариантам реализации, если общий физический UMI представляет собой неслучайный UMI, указанный UMI может не быть сам по себе достаточно уникальным для идентификации конкретной исходной молекулы. В любом случае физический UMI может быть скомбинирован с виртуальным UMI для получения индекса исходной молекулы.
[00234] В примере технологической схемы, описанной выше и изображенной на Фиг. 1 В, 3А и 4, некоторые риды включают UMI α-ρ-ϕ, тогда как другие включают UMI β-ϕ-ρ. Физический UMI α производит риды, содержащие α. Если все адаптеры, используемые в технологической схеме, содержат разные физические UMI (например, разные случайные UMI), все риды, содержащие α в адаптерной области, вероятно, происходят из одной и той же цепи фрагмента ДНК. Аналогичным образом, физический UMI β производит риды, содержащие β, все из которых происходят из одной и той же комплементарной цепи фрагмента ДНК. Соответственно, целесообразно объединение всех ридов, включающих α, для получения одной консенсусной последовательности, и объединение всех ридов, включающих β, для получения другой консенсусной последовательности. На Фиг. 4В-4С это показано как объединение первого уровня. Поскольку все риды в группе происходят из одного и того же исходного полинуклеотида в образце, пары оснований, включенные в консенсусная последовательность, вероятно, отражают истинную последовательность исходного полинуклеотида, тогда как пара оснований, исключенная из консенсусной последовательности, вероятно, отражает вариацию или ошибку, внесенную в технологическую схему.
[00235] Кроме того, виртуальные UMI ρ и ϕ могут обеспечивать информацию для определения того, какие риды, включающие один или оба виртуальных UMI, происходят из одного и того же исходного фрагмента ДНК. Поскольку виртуальные UMI ρ и ϕ являются внутренними для исходных фрагментов ДНК, использование виртуальных UMI не требует дополнительных ресурсов на практике при получении или секвенировании. После получения последовательностей физических UMI из ридов одна или более частичных последовательностей в ридах может быть определена в качестве виртуальных UMI. Если виртуальные UMI включают подходящие пары оснований и занимают одно и то же относительное местоположение на ридах, они могут однозначно идентифицировать указанные риды как происходящие из исходного фрагмента ДНК. Соответственно, риды, содержащие один из или оба виртуальных UMI ρ и ϕ, могут быть объединены с получением консенсусной последовательности. Комбинация виртуальных UMI и физических UMI может обеспечивать информацию для направления объединения второго уровня, когда задают только один физический UMI для консенсусной последовательности первого уровня каждой цепи, как показано на Фиг. 3А и Фиг. 4А-4С. Однако согласно некоторым вариантам реализации указанное объединение второго уровня с использованием виртуальных UMI может быть затруднительным при переизбытке входящих молекул ДНК или не рандомизированной фрагментации.
[00236] Согласно альтернативным вариантам реализации риды, содержащие два физических UMI на обоих концах, такие как приведенные на Фиг. 3В и Фиг. 4D и 4Е, могут быть объединены в объединение второго уровня на основании комбинации физических UMI и виртуальных UMI. Это может быть полезно, в частности, если физические UMI являются слишком короткими для однозначной идентификации исходных фрагментов ДНК без использования виртуальных UMI. Согласно указанным вариантам реализации может быть осуществлено объединение второго уровня, с использованием физических дуплексных UMI, представленных на Фиг. 3В, путем объединения консенсусных ридов α-ρ-ϕ-β и консенсусных ридов β-ϕ-ρ-α с одной и той же молекулы ДНК, с получением таким образом консенсусной последовательности, включающей нуклеотиды, стабильно присутствующие во всех ридах.
[00237] При применении UMI и схем объединения, описанных в настоящем документе, согласно различным вариантам реализации могут быть подавлены разные источники ошибки, влияющей на определенную последовательность фрагмента, даже если указанный фрагмент включает аллели с очень низкой частотой. Риды, содержащие одни и те же UMI (физические и/или виртуальные), группируют вместе. За счет объединения сгруппированных ридов могут быть элиминированы варианты (SNV и небольшие индели), обусловленные ошибками при ПЦР, получении библиотеки, кластеризации и секвенировании. Фиг. 4А-4Е иллюстрируют, каким образом способ, раскрытый на примере технологической схемы, может обеспечивать подавление различных источников ошибки при определении последовательности фрагмента двуцепочечной ДНК. Проиллюстрированные риды включают UMI α-ρ-ϕ- или β-ϕ-ρ на Фиг. 3А и 4А-4С; и UMI α-ρ-ϕ-β или β-ϕ-ρ-α на Фиг. 3В, 4D и 4Е. UMI α и β представляют собой одноплексные физические UMI на Фиг. 3А и 4А-4С. UMI α и β представляют собой дуплексные UMI на Фиг. 3В, 4D и 4Е. Виртуальные UMI ρ и ϕ локализованы на концах фрагмента ДНК.
[00238] Способ, задействующий одноплексные физические UMI, как показано на Фиг. 4А-4С, включает, во-первых, объединение ридов, содержащих один и тот же физический UMI α или β, показанное как объединение первого уровня. Объединение первого уровня дает консенсусную последовательность α для ридов, содержащих физический UMI α, которые происходят из одной цепи двуцепочечного фрагмента. Объединение первого уровня также дает консенсусную последовательность β для ридов, содержащих физический UMI β, которые происходят из другой цепи двуцепочечного фрагмента. При объединении второго уровня указанный способ дает третью консенсусную последовательность из консенсусной последовательности α и консенсусной последовательности β. Третья консенсусная последовательность соответствует консенсусным парам оснований из ридов, которые содержат одни и те же дуплексные виртуальные UMI ρ и ϕ, происходящих из двух комплементарных цепей исходного фрагмента. Наконец, определяют последовательность фрагмента двуцепочечной ДНК как третью консенсусную последовательность.
[00239] Способ, задействующий дуплексные физические UMI, как показано на Фиг. 4D-4E, включает, во-первых, объединение ридов, которые содержат физические UMI α и β в порядке α→β, в направлении 5'→3', показанные как объединение первого уровня. Объединение первого уровня дает консенсусную последовательность α-β для ридов, которые содержат физические UMI α и β, происходящих из первой цепи двуцепочечного фрагмента. Объединение первого уровня также дает консенсусную последовательность β-α для ридов, которые содержат физические UMI β и α в порядке β→α, в направлении 5'→3', происходящих из второй цепи, комплементарной первой цепи двуцепочечного фрагмента. При объединении второго уровня указанный способ дает третью консенсусную последовательность из консенсусной последовательности α-β и консенсусной последовательности β-α. Указанная третья консенсусная последовательность соответствует консенсусным парам оснований из ридов, которые содержат одни и те же дуплексные виртуальные UMI ρ и ϕ, происходящих из двух цепей фрагмента. Наконец, определяют последовательность фрагмента двуцепочечной ДНК как третью консенсусную последовательность.
[00240] На Фиг. 4А показано, каким образом объединение первого уровня может подавлять ошибки секвенирования. Ошибки секвенирования происходят на платформе для секвенирования после получения образцов и библиотек (например, при ПЦР-амплификации). Ошибки секвенирования могут вносить разные ошибочные основания в разные риды. Истинно-положительные основания обозначены буквами со сплошным контуром, тогда как ложноположительные основания обозначены буквами с пунктирным контуром. Ложноположительные нуклеотиды на разных ридах в семействе α-ρ-ϕ были исключены из консенсусной последовательности α. Истинно-положительный нуклеотид «А», изображенный слева на концах ридов семейства α-ρ-ϕ, сохраняют для консенсусной последовательности α. Аналогичным образом, ложноположительные нуклеотиды на разных ридах в семействе β-ϕ-ρ были исключены из консенсусной последовательности β, с сохранением истинно-положительного нуклеотида «А». Как показано на указанной фигуре, объединение первого уровня может эффективно устранять ошибки секвенирования. На Фиг. 4А также показано необязательное объединение второго уровня, основанное на виртуальных UMI ρ и ϕ. Указанное объединение второго уровня может дополнительно подавлять ошибки согласно объяснению выше, однако такие ошибки не проиллюстрированы на Фиг. 4А.
[00241] Ошибки ПНР происходят до амплификации с использованием кластеризации. Соответственно, одна ошибочная пара оснований, введенная в одноцепочечную ДНК в процессе ПЦР, может быть амплифицирована при амплификации с использованием кластеризации, появляясь таким образом в нескольких кластерах и ридах. Как показано на Фиг. 4В и Фиг. 4D, ложноположительная пара оснований, введенная в результате ошибки ПЦР, может появляться во многих ридах. Основание «Т» в семействе ридов α-ρ-ϕ (Фиг. 4 В) или α-β (Фиг. 4D), а также основание «С» в семействе ридов β-ϕ-ρ (Фиг. 4В) или β-α (Фиг. 4D) представляют собой такие ошибки ПЦР. Напротив, ошибки секвенирования, показанные на Фиг. 4А, появляются в одном риде или в небольшом числе ридов одного и того же семейства. Поскольку ошибки ПЦР-секвенирования появляются во многих ридах семейства, объединение ридов первого уровня в цепи не устраняет ошибки ПЦР, даже несмотря на то, что указанное объединение первого уровня устраняет ошибки секвенирования (например, G и А, устраненные из семейства α-ρ-ϕ на Фиг. 4В и семейства α-β на Фиг. 4D). Тем не менее, поскольку ПЦР вводит ошибку в одноцепочечную ДНК, комплементарная цепь исходного фрагмента и происходящие из нее риды обычно не содержат той же ошибки ПЦР. Соответственно, объединение второго уровня, основанное на ридах с двух цепей исходного фрагмента, может эффективно устранять ошибки ПЦР, как показано в нижней части Фиг. 4В и 4D.
[00242] В некоторых платформах для секвенирования возникают ошибки гомополимеров с внесением небольших инсерционно-делеционных ошибок (инделей) в гомополимеры из одного повторяющегося нуклеотида. Фиг. 4С и 4Е иллюстрируют коррекцию ошибок гомополимеров с применением способов, описанных в настоящем документе. В семействе ридов α-ρ-ϕ (Фиг. 4С) или α-ρ-ϕ-β (Фиг. 4Е) два «Т»-нуклеотида были удалены из второго рида сверху, и один «Т»-нуклеотид был удален из третьего рида сверху. В семействе ридов β-ϕ-ρ (Фиг. 4С) или β-ϕ-ρ-α (Фиг. 4Е) один из «Т»-нуклеотидов был инсертирован в первый рид сверху. Аналогично ошибке секвенирования, проиллюстрированной на Фиг. 4А, ошибки гомополимеров происходят после ПЦР-амплификации, соответственно, разные риды содержат разные ошибки гомополимеров. В результате объединение первого уровня может эффективно устранять инсерционно-делеционные ошибки.
[00243] Консенсусные последовательности могут быть получены путем объединения ридов, содержащих один или более общих неслучайных UMI и один или более общих виртуальных UMI. Кроме того, информация о положении может также быть использована для получения консенсусных последовательностей согласно приведенному ниже описанию.
Объединение по положению
[00244] Согласно некоторым вариантам реализации риды обрабатывают для выравнивания на референсную последовательность с определением местоположений выравнивания ридов на референсной последовательности (локализации). Однако согласно некоторым вариантам реализации, не проиллюстрированным выше, локализацию осуществляют с применением k-мерного анализа сходства и взаимного выравнивания ридов. Указанный второй вариант реализации обладает двумя преимуществами: во-первых, он способен объединять риды (корректировать ошибки), не совпадающие с референсной последовательностью ввиду различий гаплотипов или транслокаций; во-вторых, он не зависит от алгоритма выравнивателя, таким образом устраняя возможность возникновения индуцированных выравнивателем артефактов (ошибок выравнивателя). Согласно некоторым вариантам реализации риды, характеризующиеся одинаковой информацией о локализации, могут быть объединены с получением консенсусных последовательностей для определения последовательности исходных фрагментов ДНК. В некоторых контекстах процесс выравнивания называют также процессом картирования. Риды последовательностей подвергают процессу выравнивания для картирования на референсную последовательность. Для выравнивания ридов на референсную последовательность могут применяться различные инструменты и алгоритмы для выравнивания, описанные в тексте настоящего документа. Как обычно, алгоритмы выравнивания обеспечивают успешное выравнивание некоторых ридов на референсную последовательность, тогда как другие риды могут не быть успешно выравнены или могут быть выравнены на референсную последовательность неудовлетворительно. Риды, которые последовательно выравнивают на референсную последовательность, ассоциированы с сайтами на референсной последовательности. Выравненные риды и ассоциированные с ними сайты также называют метками последовательности. Для некоторых ридов последовательностей, которые содержат значительное число повторов, наблюдается тенденция к увеличению сложности выравнивания на референсную последовательность. При выравнивании рида на референсную последовательность с числом несовпадающих оснований, превышающим определенный критерий, рид считают выравненным неудовлетворительно. Согласно различным вариантам реализации считают, что риды выравнены неудовлетворительно, если при их выравнивании не совпадает по меньшей мере приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 оснований. Согласно другим вариантам реализации считают, что риды выравнены неудовлетворительно, если при их выравнивании не совпадает по меньшей мере приблизительно 5% оснований. Согласно другим вариантам реализации считают, что риды выравнены неудовлетворительно, если при их выравнивании не совпадает по меньшей мере приблизительно 10%, 15% или 20% оснований.
[00245] Согласно некоторым вариантам реализации раскрытые способы сочетают информацию о положении с информацией о физических UMI для индексации исходных молекул фрагментов ДНК. Риды последовательностей, характеризующиеся одним и тем же положением рида и одним и тем же неслучайным или случайным физическим UMI, могут быть объединены с получением консенсусной последовательности для определения последовательности фрагмента или его части. Согласно некоторым вариантам реализации риды последовательностей, характеризующиеся одним и тем же положением рида, одними и теми же неслучайными физическими UMI и случайными физическими UMI, могут быть объединены с получением консенсусной последовательности. Согласно таким вариантам реализации адаптер может включать как неслучайный физический UMI, так и случайный физический UMI. Согласно некоторым вариантам реализации риды последовательностей, характеризующиеся одним и тем же положением рида и одним и тем же виртуальным UMI, могут быть объединены с получением консенсусной последовательности.
[00246] Информация о положениях ридов может быть получена с применением различных методик. Например, согласно некоторым вариантам реализации для получения информации о положениях ридов могут быть использованы геномные координаты. Согласно некоторым вариантам реализации положение на референсной последовательности, на которое выравнивается рид, может быть использовано для получения информации о положении рида. Например, положения начала и окончания рида на хромосоме могут быть использованы для получения информации о положениях ридов. Согласно некоторым вариантам реализации положения ридов считают одинаковыми, если они характеризуются идентичной информацией о положении. Согласно некоторым вариантам реализации положения ридов считают одинаковыми, если различие информации о положении меньше определенного критерия. Например, два рида с положениями начала в геноме, которые различаются менее чем на 2, 3, 4 или 5 пар оснований, могут считаться ридами, характеризующимися одним и тем же положением. Согласно другим вариантам реализации положения ридов считают одинаковыми, если информация об их положении может быть приведена к конкретному пространству положений, где могут быть установлены совпадения. Референсная последовательность может быть обеспечена до секвенирования - например, она может представлять собой хорошо известную и широко используемую геномную последовательность человека - или она может быть определена на основании ридов, полученных при секвенировании образца.
[00247] Независимо от специфических платформы для секвенирования и протокола, по меньшей мере часть нуклеиновых кислот, содержащихся в указанном образце, секвенируют с получением десятков тысяч, сотен тысяч или миллионов ридов последовательностей, например, ридов размером 100 п.о. Согласно некоторым вариантам реализации риды последовательностей содержат приблизительно 20 п.о., приблизительно 25 п.о., приблизительно 30 п.о., приблизительно 35 п.о., приблизительно 36 п.о., приблизительно 40 п.о., приблизительно 45 п.о., приблизительно 50 п.о., приблизительно 55 п.о., приблизительно 60 п.о., приблизительно 65 п.о., приблизительно 70 п.о., приблизительно 75 п.о., приблизительно 80 п.о., приблизительно 85 п.о., приблизительно 90 п.о., приблизительно 95 п.о., приблизительно 100 п.о., приблизительно 110 п.о., приблизительно 120 п.о., приблизительно 130, приблизительно 140 п.о., приблизительно 150 п.о., приблизительно 200 п.о., приблизительно 250 п.о., приблизительно 300 п.о., приблизительно 350 п.о., приблизительно 400 п.о., приблизительно 450 п.о., приблизительно 500 п.о., приблизительно 800 п.о., приблизительно 1000 п.о. или приблизительно 2000 п.о.
[00248] Согласно некоторым вариантам реализации риды выравнивают на референсный геном, например, hg19. Согласно другим вариантам реализации риды выравнивают на часть референсного генома, например, хромосому или сегмент хромосомы. Риды, которые однозначно картируются на референсный геном, известны как метки последовательности. Согласно одному варианту реализации по меньшей мере приблизительно 3×106 соответствующих установленным требованиям меток последовательности, по меньшей мере приблизительно 5×106 соответствующих установленным требованиям меток последовательности, по меньшей мере приблизительно 8×106 соответствующих установленным требованиям меток последовательности, по меньшей мере приблизительно 10×106 соответствующих установленным требованиям меток последовательности, по меньшей мере приблизительно 15×106 соответствующих установленным требованиям меток последовательности, по меньшей мере приблизительно 20×106 соответствующих установленным требованиям меток последовательности, по меньшей мере приблизительно 30×106 соответствующих установленным требованиям меток последовательности, по меньшей мере приблизительно 40×106 соответствующих установленным требованиям меток последовательности, или по меньшей мере приблизительно 50×106 соответствующих установленным требованиям меток последовательности получают из ридов, которые однозначно картируются на референсный геном.
Варианты применения
[00249] Согласно различным вариантам применения стратегии коррекции ошибок, описанные в настоящем документе, могут обеспечивать одно или более из следующих преимуществ: (i) детекцию соматических мутаций с очень низкой частотой аллелей, (ii) уменьшение продолжительности цикла за счет подавления ошибок фазирования/префазирования; и/или (iii) увеличение длины ридов за счет повышения качества распознанных оснований в последующих частях ридов и т.п. Варианты применения и задачи, относящиеся к детекции соматических мутаций с низкой частотой аллелей обсуждаются выше.
[00250] Согласно некоторым вариантам реализации методики, описанные в настоящем документе, могут обеспечивать надежное распознавание аллелей с частотой приблизительно 2% или менее, или приблизительно 1% или менее, или приблизительно 0,5% или менее. Такие низкие частоты являются обычными для вкДНК, происходящей из опухолевых клеток пациента с раковым заболеванием. Согласно некоторым вариантам реализации методики, описанные в настоящем документе, могут обеспечивать идентификацию редких штаммов в метагеномных образцах, а также детекцию редких вариантов в вирусных или других популяциях, если, например, пациент был инфицирован несколькими вирусными штаммами и/или прошел медицинское лечение.
[00251] Согласно некоторым вариантам реализации методики, описанные в настоящем документе, могут обеспечивать меньшую продолжительность химического цикла секвенирования. Меньшая продолжительность цикла приумножает ошибки секвенирования, которые могут быть скорректированы с применением способа, описанного выше.
[00252] Согласно некоторым вариантам реализации, задействующим UMI, длинные риды могут быть получены путем парно-концевого секвенирования при использовании асимметричной длины для пары парно-концевых (ПК) ридов с двух концов сегмента. Например, пара ридов размером 50 п.о. в одном парно-концевом риде и 500 п.о. в другом парно-концевом риде может затем быть «сшита» с другой парой ридов для получения длинного рида размером 1000 п.о. Указанные варианты реализации могут обеспечивать более быстрое секвенирование для определения длинных фрагментов аллелей низкой частоты.
[00253] Фиг. 5 схематически иллюстрирует пример эффективного получения длинных парно-концевых ридов во вариантах применения указанного типа с использованием физических UMI и виртуальных UMI. Библиотеки с обеих цепей одних и тех же фрагментов ДНК кластеризуют на проточной ячейке. Размер вставки в библиотеке превышает 1 т.п.н. Секвенирование осуществляют с использованием асимметричных длин ридов (например, рид 1 = 500 п.о., рид 2 = 50 п.о.), чтобы гарантировать качество длинных ридом размером 500 п.о. Путем сшивания двух цепей могут быть получены длинные ПК-риды размером 1000 п.о. при секвенировании только 500+50 п.о.
Образцы
[00254] Образцы, которые используют для определения последовательности фрагмента ДНК, включают образцы, взятые из любой клетки, жидкости, ткани или органа, включающие нуклеиновые кислоты, в которых нужно определить представляющие интерес последовательности. Согласно некоторым вариантам реализации, включающим проведение диагностики рака, циркулирующая опухолевая ДНК может быть получена из биологической жидкости субъекта, например, крови или плазмы. Согласно некоторым вариантам реализации, включающим проведение диагностики у плода, благоприятно получение неклеточных нуклеиновых кислот, например, неклеточной ДНК (вкДНК) из жидкости организма матери. Неклеточные нуклеиновые кислоты, в том числе неклеточная ДНК, могут быть получены различными способами, известными в данной области техники, из биологических образцов, в том числе, но не ограничиваясь перечисленным, образцов плазмы, сыворотки и мочи (см., например, Fan et al., Proc Natl Acad Sci 105:16266-16271 [2008]; Koide et al., Prenatal Diagnosis 25:604-607 [2005]; Chen et al., Nature Med. 2: 1033-1035 [1996]; Lo et al., Lancet 350: 485-487 [1997]; Botezatu et al., Clin Chem. 46: 1078-1084, 2000; и Su et al., J Mol. Diagn. 6: 101-107 [2004]).
[00255] Согласно различным вариантам реализации нуклеиновые кислоты (например, ДНК или РНК), присутствующие в образце, могут быть обогащены специфическим или неспецифическим образом перед применением (например, до получения библиотеки для секвенирования). Неспецифическое обогащение образца ДНК относится к амплификации полного генома фрагментов геномной ДНК из образца, которая может быть использована для увеличения уровня ДНК в образце перед получением библиотеки вкДНК для секвенирования. Способы амплификации полного генома известны в данной области техники. ПЦР с примированием вырожденными олигонуклеотидами (DOP), методика ПЦР с достройкой праймеров (PEP) и амплификация с множественным вытеснением цепи (MDA) представляют собой примеры способов амплификации полного генома. Согласно некоторым вариантам реализации указанный образец не обогащен ДНК.
[00256] Образец, включающий нуклеиновые кислоты, к которым применяют способы, описанные в настоящем документе, как правило, включают биологический образец («тестовый образец») согласно описанию выше. Согласно некоторым вариантам реализации подлежащие секвенированию нуклеиновые кислоты очищают или выделяют любыми из ряда хорошо известных способов.
[00257] Соответственно, согласно определенным вариантам реализации образец включает или состоит по существу из очищенного или выделенного полинуклеотида, или он может включать образцы, такие как образец ткани, образец биологической жидкости, образец клеток и т.п. Подходящие образцы биологических жидкостей включают, не ограничиваясь перечисленными, кровь, плазму, сыворотку, пот, слезы, мокроту, мочу, мокроту, отделяемое из уха, лимфу, слюну, спинномозговую жидкость, лаважи, суспензию костного мозга, отделяемое влагалища, жидкость трансцервикального лаважа, жидкость головного мозга, асцит, молоко, выделения дыхательных, кишечных и мочеполовых путей, амниотическую жидкость, молоко и образцы лейкафереза. Согласно некоторым вариантам реализации указанный образец представляет собой образец, который может быть легко получен с помощью неинвазивных процедур, например, образец крови, плазмы, сыворотки, пота, слез, мокроты, мочи, стула, мокроты, отделяемого из ушей, слюны или кала. Согласно определенным вариантам реализации указанный образец представляет собой образец периферической крови, или фракции плазмы и/или сыворотки из образца периферической крови. Согласно другим вариантам реализации указанный биологический образец представляет собой смыв или мазок, биопсийный препарат или культуру клеток. Согласно другому варианту реализации указанный образец представляет собой смесь двух или более биологических образцов, например, биологический образец может включать два или более образцов биологической жидкости, образец ткани и образец клеточной культуры. В настоящем документе термины «кровь», «плазма» и «сыворотка» явным образом охватывают фракции или их обработанные части. Аналогичным образом, если образец получают из биоптата, смыва, мазка и т.п., указанный «образец» явным образом охватывает обработанную фракцию или часть, происходящую из указанного биоптата, смыва, мазка и т.п.
[00258] Согласно некоторым вариантам реализации образцы могут быть получены из источников, включающих, не ограничиваясь перечисленным, образцы от разных индивидуумов, образцы, взятые на разных стадиях развития одного и того же или разных индивидуумов, образцы от разных пораженных заболеванием индивидуумов (например, индивидуумов, предположительно страдающих генетическим расстройством), здоровых индивидуумов, образцы, полученные на разных стадиях заболевания у индивидуума, образцы, полученные от индивидуума, получающего разные виды лечения заболевания, образцы от индивидуумов, подвергающихся воздействию разных факторов окружающей среды, образцы от индивидуумов с предрасположенностью к патологии, образцы от индивидуумов, подвергающихся воздействию агента инфекционного заболевания, и т.п.
[00259] Согласно одному иллюстративному, однако неограничивающему варианту реализации указанный образец представляет собой материнский образец, который получают от беременной особи женского пола, например, от беременной женщины. В указанном случае образец может быть проанализирован с применением способов, описанных в настоящем документе, для обеспечения пренатальной диагностики потенциальных хромосомных аномалий плода. Материнский образец может представлять собой образец ткани, образец биологической жидкости или образец клеток. Биологическая жидкость включает, согласно неограничивающим примерам, кровь, плазму, сыворотку, пот, слезы, мокроту, мочу, мокроту, отделяемое из уха, лимфу, слюну, спинномозговую жидкость, лаважи, суспензию костного мозга, отделяемое влагалища, жидкость трансцервикального лаважа, жидкость головного мозга, асцит, молоко, выделения дыхательных, кишечных и мочеполовых путей; и образцы лейкафереза.
[00260] Согласно определенным вариантам реализации образцы могут также быть получены из культивированных in vitro тканей, клеток или других содержащих полинуклеотиды источников. Культивированные образцы могут быть взяты из источников, включающих, не ограничиваясь перечисленными, культуры (например, ткань или клетки), поддерживаемые на разных средах и в разных условиях (например, при разных значениях рН, давлении или температуре), культуры (например, ткань или клетки), поддерживаемые в течение периодов разной продолжительности, культуры (например, ткань или клетки), обработанные разными факторами или реагентами (например, кандидатным лекарственным средством или модулятором); или культуры разных типов ткани и/или клеток.
[00261] Способы выделения нуклеиновых кислот из биологических источников хорошо известны и различаются в зависимости от природы источника. Специалист в данной области техники может легко выделить нуклеиновые кислоты из источника так, как это требуется для способа, описанного в настоящем документе. В некоторых случаях может быть целесообразно фрагментировать молекулы нуклеиновой кислоты в образце нуклеиновой кислоты. Фрагментация может быть случайной или может быть специфической, что достигается, например, расщеплением рестрикционными эндонуклеазами. Способы случайной фрагментации хорошо известны в данной области техники и включают, например, ограниченное расщепление ДНКазой, обработку щелочами и физический сдвиг.
Получение библиотеки для секвенирования
[00262] Согласно различным вариантам реализации секвенирование может быть выполнено на различных платформах для секвенирования, требующих получения библиотеки для секвенирования. Получение, как правило, включает фрагментацию ДНК (обработку ультразвуком, распыление или сдвиг) с последующей репарацией ДНК и заключительной очисткой концов (тупых концов или липких А-концевых участков), и лигирование специфическими для платформы адаптерами. Согласно одному варианту реализации способы, описанные в настоящем документе, могут задействовать технологии секвенирования нового поколения (NGS), которые позволяют секвенировать несколько образцов индивидуально, как геномные молекулы (т.е. одноплексное секвенирование), или в виде общего пула образцов, содержащих индексированные геномные молекулы (например, мультиплексное секвенирование), за единственный прогон секвенирования. Указанные способы могут обеспечивать получение до нескольких миллиардов ридов последовательностей ДНК. Согласно различным вариантам реализации последовательности геномных нуклеиновых кислот и/или индексированных геномных нуклеиновых кислот могут быть определены с применением, например, технологий секвенирования нового поколения (NGS), описанных в настоящем документе. Согласно различным вариантам реализации анализ массивного объема данных последовательностей, полученных с применением NGS, может быть выполнен с применением одного или более процессоров согласно описанию в настоящем документе.
[00263] Согласно различным вариантам реализации применение таких технологий секвенирования не включает получение библиотек для секвенирования.
[00264] Однако согласно определенным вариантам реализации способы секвенирования, предусмотренные настоящим изобретением, включают получение библиотек для секвенирования. Согласно одному иллюстративному подходу получение библиотеки для секвенирования включает получение случайной коллекции модифицированных адаптерами фрагментов ДНК (например, полинуклеотидов), готовых для секвенирования. Библиотеки полинуклеотидов для секвенирования могут быть получены из ДНК или РНК, в том числе из эквивалентов и аналогов ДНК или кДНК, например, комплементарной ДНК или кДНК, или копии ДНК, полученной с РНК-матрицы под действием обратной транскриптазы. Указанные полинуклеотиды могут происходить из двуцепочечной формы (например, дцДНК, такой как фрагменты геномной ДНК, кДНК, продукты ПЦР-амплификации и т.п.), или, согласно некоторым вариантам реализации, указанные полинуклеотиды могут происходить из одноцепочечной формы (например, оцДНК, РНК и т.п.) и быть преобразованы в форму дцДНК. В иллюстративном примере, согласно некоторым вариантам реализации молекулы одноцепочечной мРНК могут быть копированы в двуцепочечные кДНК, подходящие для применения при получении библиотеки для секвенирования. Точная последовательность молекул первичных полинуклеотидов, как правило, несущественна для способа получения библиотеки, и может быть известна или неизвестна. Согласно одному варианту реализации молекулы полинуклеотидов представлены молекулами ДНК. Более конкретно, согласно некоторым вариантам реализации указанные молекулы полинуклеотидов представляют собой полный геномный набор организма или по существу полный геномный набор организма, и представляют собой молекулы геномной ДНК (например, клеточной ДНК, неклеточной ДНК (вкДНК) и т.п.), которые, как правило, включают и последовательность интрона и последовательность экзона (кодирующую последовательность), а также некодирующие регуляторные последовательности, такие как промоторные и энхансерные последовательности. Согласно некоторым вариантам реализации молекулы первичных полинуклеотидов содержат молекулы геномной ДНК человека, например, молекулы вкДНК, присутствующие в периферической крови беременного субъекта.
[00265] Получение библиотек для секвенирования для некоторых платформ для NGS-секвенирования облегчает применение полинуклеотидов, содержащих фрагменты с размерами в специфическом диапазоне. Получение таких библиотек, как правило, включает фрагментацию больших полинуклеотидов (например, клеточной геномной ДНК) для получения полинуклеотидов с размерами в требуемом диапазоне.
[00266] В способах и системах секвенирования согласно описанию в настоящем документе могут применяться парно-концевые риды. Длина фрагмента или вставки больше длины ридов, и иногда больше суммы длин двух ридов.
[00267] Согласно некоторым иллюстративным вариантам реализации образец нуклеиновой кислоты или нуклеиновых кислот получают в виде геномной ДНК, которую подвергают фрагментации на фрагменты длиннее приблизительно 50, 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 2000 или 5000 пар оснований, для которых могут быть легко применены способы NGS. Согласно некоторым вариантам реализации парно-концевые риды получают из вставок длиной приблизительно 100-5000 п.о. Согласно некоторым вариантам реализации длина указанных вставок составляет приблизительно 100-1000 п.о. Иногда их реализуют в виде стандартных парно-концевых ридов с короткими вставками. Согласно некоторым вариантам реализации длина указанных вставок составляет приблизительно 1000-5000 п.о. Иногда их реализуют в виде сцепленно-концевых ридов с длинными вставками согласно описанию выше.
[00268] Согласно некоторым вариантам реализации для оценки очень длинных последовательностей разрабатывают длинные вставки. Согласно некоторым вариантам реализации сцепленно-концевые риды могут применяться для получения ридов, разделенных тысячами пар оснований. Согласно указанным вариантам реализации длина вставок или фрагментов варьирует от сотен до тысяч пар оснований, при этом на двух концах вставки размещают два биотиновых соединительных адаптера. Впоследствии биотиновые соединительные адаптеры соединяют два конца вставки с образованием закольцованной молекулы, которую затем дополнительно фрагментируют. Субфрагмент, включающий биотиновые соединительные адаптеры и два конца исходной вставки, выбирают для секвенирования на платформе, дизайн которой предназначен для секвенирования более коротких фрагментов.
[00269] Фрагментация может осуществляться любым из ряда способов, известных специалистам в данной области техники. Например, фрагментация может осуществляться механическими способами, включающими, не ограничиваясь перечисленным, распыление, обработку ультразвуком и гидросдвиг. Однако механическая фрагментация, как правило, расщепляет ДНК-остов по связям С-О, Р-O и С-С, что приводит к образованию гетерогенной смеси тупых концов и липких 3' - и 5'-концов с разорванными связями С-О, Р-O и/ С-С (см., например, Alnemri andLiwack, J Biol. Chem 265:17323-17333 [1990]; Richards and Boyer, J Mol Biol 11:327-240 [1965]), которым может требоваться репарация, поскольку в них может отсутствовать 5'-фосфат, необходимый для последующих ферментативных реакций, например, лигирования адаптеров для секвенирования, необходимых для получения ДНК для секвенирования.
[00270] Напротив, вкДНК, как правило, представлена фрагментами с размером менее чем приблизительно 300 пар оснований и, следовательно, фрагментация обычно не является необходимой для формирования библиотеки для секвенирования с применением образцов вкДНК.
[00271] Как правило, и принудительно фрагментированные полинуклеотиды (например, фрагментированный in vitro), и существующие в виде фрагментов в естественных условиях полинуклеотиды преобразуют в ДНК с тупыми концами, содержащие 5'-фосфаты и 3'-гидроксил. Стандартные протоколы, например, протоколы для секвенирования с применением, например, платформы Ulumina согласно описанию в примере технологической схемы выше применительно к Фиг. 1А и 1В, предусматривают проведение пользователем репарации концов в образце ДНК, очищение продуктов после репарации концов до аденилирования или присоединения dA-хвостов по 3'-концам, и очищение продуктов присоединения dA-хвостов до этапов лигирования адаптеров при получении библиотеки.
[00272] Различные варианты реализации способов получения библиотек для секвенирования, описанных в настоящем документе, устраняют необходимость проведения одного или более из этапов, как правило, обязательных для стандартных протоколов получения модифицированного ДНК-продукта, который может быть секвенирован с применением NGS. Сокращенный способ (АВВ-способ), 1-этапный способ и 2-этапный способ представляют собой примеры способов получения библиотеки для секвенирования, с которыми можно ознакомиться в заявке на патент 13/555037, поданной 20 июля 2012 г., которая полностью включена в настоящий документ посредством ссылки.
Способы секвенирования
[00273] Способы и устройство, описанные в настоящем документе, могут задействовать технологию секвенирования нового поколения (NGS), которая позволяет проводить массово-параллельное секвенирование. Согласно некоторым вариантам реализации клонально амплифицированные ДНК-матрицы или одиночные молекулы ДНК секвенируют массово-параллельным образом в проточной ячейке (например, согласно описанию в Volkerding et al., Clin Chem 55:641-658 [2009]; Metzker M Nature Rev 11:31-46 [2010]). Технологии секвенирования NGS включают, не ограничиваясь перечисленными, пиросеквенирование, секвенирование путем синтеза с применением терминаторов с обратимыми красителями, секвенирование путем лигирования олигонуклеотидных зондов и ионное полупроводниковое секвенирование. ДНК из индивидуальных образцов могут быть секвенированы индивидуально (т.е. одноплексное секвенирование), или может быть получен общий пул ДНК из нескольких образцов и ДНК секвенирована как индексированные геномные молекулы (т.е. мультиплексное секвенирование) за единственный прогон секвенирования, с получением до нескольких сотен миллионов ридов последовательностей ДНК. Примеры технологий секвенирования, которые могут применяться для получения информации о последовательности в соответствии с предложенным способом, подробнее описаны в настоящем документе.
[00274] Некоторые технологии секвенирования коммерчески доступны, например, платформа для секвенирования путем гибридизации от Affymetrix Inc. (Саннивейл, Калифорния) и платформы для секвенирования путем синтеза от 454 Life Sciences (Брэдфорд, Коннектикут), Illumina/Solexa (Хейворд, Калифорния) и Helicos Biosciences (Кембридж, Массачусетс), а также платформа для секвенирования путем лигирования от Applied Biosystems (Фостер-Сити, Калифорния), согласно приведенному ниже описанию. Помимо одномолекулярного секвенирования, выполняемого с применением секвенирования путем синтеза от Helicos Biosciences, другие технологии одномолекулярного секвенирования включают, не ограничиваясь перечисленными, технологию SMRT™ от Pacific Biosciences, технологию ION TORRENT™ и секвенирование в нанопорах, разработанное, например, Oxford Nanopore Technologies.
[00275] Хотя автоматизированный способ Сэнгера считается технологией «первого поколения», секвенирование по Сэнгеру, в том числе автоматизированное секвенирование по Сэнгеру, может также быть использовано в способах, описанных в настоящем документе. Дополнительные подходящие способы секвенирования включают, не ограничиваясь перечисленными, технологии визуализации нуклеиновых кислот, например, атомно-силовую микроскопию (AFM) или трансмиссионную электронную микроскопию (ТЕМ). Иллюстративные технологии секвенирования подробнее описаны ниже.
[00276] Согласно некоторым вариантам реализации предложенные способы включают получение информации о последовательности для нуклеиновых кислот в тестовом образце путем массово-параллельного секвенирования миллионов фрагментов ДНК с применением секвенирования путем синтеза от Ulumina и химии секвенирования на основе обратимых терминаторов (например согласно описанию в источнике: Bentley et al., Nature 6:53-59 [2009]). ДНК-матрица может быть представлена геномной ДНК, например, клеточной ДНК или вкДНК. Согласно некоторым вариантам реализации геномную ДНК из выделенных клеток используют в качестве матрицы, и ее фрагментируют с получением фрагментов с длиной, равной нескольким сотням пар оснований. Согласно другим вариантам реализации вкДНК или циркулирующую опухолевую ДНК (цоДНК) используют в качестве матрицы, и фрагментация не требуется, поскольку вкДНК или цоДНК представлена короткими фрагментами. Например, фетальная вкДНК циркулирует в кровотоке в виде фрагментов длиной приблизительно 170 пар оснований (п.о.) (Fan et al., Clin Chem 56:1279-1286 [2010]), и фрагментация ДНК перед секвенированием не требуется. Технология секвенирования Ulumina основана на прикреплении фрагментированной геномной ДНК к плоской оптически прозрачной поверхности, с которой связаны якорные олигонуклеотиды. Проводят репарацию концов ДНК-матрицы с получением фосфорилированных тупых 5'-концов и используют полимеразную активность фрагмента Кленова для добавления единственного основания А к 3'-концу фосфорилированных фрагментов ДНК с тупыми концами. Указанное добавление подготавливает фрагменты ДНК для лигирования с олигонуклеотидными адаптерами, которые содержат липкий концевой участок из единственного основания Т на 3'-конце для увеличения эффективности лигирования. Указанные адаптерные олигонуклеотиды комплементарны якорным олигонуклеотидам в проточной ячейке. В условиях предельных разведений модифицированную адаптерами одноцепочечную ДНК-матрицу добавляют в проточную ячейку и иммобилизуют гибридизацией на якорных олигонуклеотидах. Присоединенные фрагменты ДНК достраивают и подвергают мостиковой амплификации для достижения в проточной ячейке секвенирования сверхвысокой плотности с сотнями миллионов кластеров, каждый из которых содержит приблизительно 1000 копий одной и той же матрицы. Согласно одному варианту реализации случайным образом фрагментированную геномную ДНК амплифицируют с помощью ПЦР до проведения кластерной амплификации. Как вариант, получают геномную библиотеку без секвенирования, и случайным образом фрагментированную геномную ДНК обогащают с применением только кластерной амплификации (Kozarewa et al., Nature Methods 6:291-295 [2009]). В некоторых вариантах применения матрицы секвенируют с использованием надежной технологии ДНК-секвенирования путем синтеза с 4 красителями, задействующей обратимые терминаторы с удаляемыми флуоресцентными красителями. Высокочувствительную детекцию флуоресценции обеспечивает использование лазерного возбуждения и оптики с полным внутренним отражением. Короткие риды последовательностей размером приблизительно от десятков до нескольких сотен пар оснований выравнивают на референсный геном, и идентифицируют уникальные картирования коротких ридов последовательностей на референсный геном с применением специально разработанного программного обеспечения для системы анализа данных. После завершения первого прочтения матрицы могут быть регенерированы in situ, что позволяет провести второе прочтение с противоположного конца фрагментов. Соответственно, могут применяться либо одностороннее, либо парно-концевое секвенирование фрагментов ДНК.
[00277] В различных вариантах реализации настоящего изобретения может быть использовано секвенирование путем синтеза, которое позволяет проводить парно-концевое секвенирование. Согласно некоторым вариантам реализации платформа секвенирования путем синтеза от Illumina включает кластеризацию фрагментов. Кластеризация представляет собой процесс, в ходе которого каждый фрагмент молекулы изотермически амплифицируют. Согласно некоторым вариантам реализации, как в примере, описанном в настоящем документе, указанный фрагмент содержит два разных адаптера, присоединенные к двум концам фрагмента, обеспечивающие гибридизацию указанного фрагмента с двумя разными олигонуклеотидами на поверхности дорожки проточной ячейки. Указанный фрагмент дополнительно включает две индексные последовательности или соединен с двумя индексными последовательностями на двух концах фрагмента, обеспечивающими метки для идентификации разных образцов при мультиплексном секвенировании. В некоторых платформах для секвенирования фрагмент, подлежащий секвенированию с обоих концов, также называют вставкой.
[00278] Согласно некоторому варианту реализации проточная ячейка для кластеризации в платформе Illumina представляет собой стеклянную пластину с дорожками. Каждая дорожка представляет собой стеклянный канал, покрытый «газоном» олигонуклеотидов двух типов (например, олигонуклеотидов Р5 и Р7'). Гибридизацию обеспечивает первый из двух типов олигонуклеотидов на поверхности. Указанный олигонуклеотид комплементарен первому адаптеру на конце фрагмента. Полимераза создает комплементарную цепь гибридизованного фрагмента. Двуцепочечную молекулу денатурируют, и отмывают оригинальную цепь матрицы. Оставшуюся цепь, параллельно с множеством других оставшихся цепей, клонально амплифицируют с использованием мостиков.
[00279] При мостиковой амплификации и в других способах секвенирования, задействующих кластеризацию, происходит наложение цепи, и вторая адаптерная область на втором конце цепи гибридизуется с олигонуклеотидами второго типа на поверхности проточной ячейки. Полимераза генерирует комплементарную цепь, образуя двуцепочечную мостиковую молекулу. Указанную двуцепочечную молекулу денатурируют, что приводит к получению двух одноцепочечных молекул, прикрепленных к проточной ячейке посредством двух разных олигонуклеотидов. Затем процесс многократно повторяется, и происходит одновременно для миллионов кластеров, что приводит к клональной амплификации всех фрагментов. После мостиковой амплификации обратные цепи отщепляют и отмывают, оставляя только прямые цепи. 3'-концы блокируют для предотвращения нежелательного примирования.
[00280] После кластеризации начинают секвенирование с достройки первого праймера для секвенирования для получения первого рида. В каждом цикле флуоресцентно меченые нуклеотиды конкурируют за добавление к растущей цепи. Происходит включение только одного нуклеотида на основании последовательности матрицы. После добавления каждого нуклеотида кластер возбуждают источником света, и он испускает характеристический флуоресцентный сигнал. Число циклов определяет длину рида. Длина волны излучения и интенсивность сигнала определяют распознанное основание. Для заданного кластера все идентичные цепи прочитываются одновременно. Сотни миллионов кластеров секвенируют массово-параллельным образом. После завершения первого прочтения продукт прочтения отмывают.
[00281] На следующем этапе протоколов, задействующих два индексных праймера, индексный праймер 1 вводят и гибридизуют с индексной областью 1 на матрице. Индексные области обеспечивают идентификацию фрагментов, подходящих для демультиплексирования образцов в процессе мультиплексного секвенирования. Индексный рид 1 получают аналогично первому риду. После завершения индексного прочтения 1 продукт прочтения отмывают и снимают защиту с 3'-конца цепи. Затем происходит наложение цепи матрицы и она связывается со вторым олигонуклеотидом на проточной ячейке. Прочтение индексной последовательности 2 проводят таким же образом, что и прочтение индексной последовательности 1. В завершение указанного этапа отмывают продукт индексного прочтения 2.
[00282] После прочтения двух индексов начинают 2 прочтение с применения полимераз для достройки вторых олигонуклеотидов в проточной ячейке с получением двуцепочечного мостика. Указанную двуцепочечную ДНК денатурируют и блокируют 3'-конец. Оригинальную прямую цепь отщепляют и отмывают, оставляя обратную цепь. Прочтение 2 начинают с введения праймера для секвенирования для 2 прочтения. Как при 1 прочтении, этапы секвенирования повторяют до достижения требуемой длины. Продукт 2 прочтения отмывают. Весь указанный процесс генерирует миллионы ридов, представляющих все фрагменты. Последовательности из общего пула библиотек образцов разделяют на основании уникальных индексов, введенных при получении образцов. В каждом образце риды с аналогичными отрезками распознанных оснований локально кластеризуются. Прямые и обратные риды спаривают с получением непрерывных последовательностей. Указанные непрерывные последовательности выравнивают на референсный геном для идентификации вариантов.
[00283] Описанный выше пример секвенирования путем синтеза включает парно-концевые риды, которые используют во многих вариантах реализации раскрытых способов. Парно-концевое секвенирование включает 2 рида из двух концов фрагмента. Парно-концевые риды используют для уточнения неоднозначных выравниваний. Парно-концевое секвенирование позволяет пользователям выбирать длину вставки (или фрагмента для секвенирования) и секвенировать любой конец вставки, с формированием высококачественных, позволяющих проводить выравнивание данных о последовательности. Поскольку известно расстояние между всеми парными ридами, алгоритмы выравнивание могут использовать указанную информацию для более прецизионного картирования ридов в повторяющихся областях. Это приводит к лучшему выравниванию ридов, в частности, в плохо поддающихся секвенированию повторяющихся областях генома. При парно-концевом секвенировании можно детектировать перестановки, в том числе инсерции и делеции (индели), а также инверсии.
[00284] Для парно-концевых ридов могут быть использованы вставки разной длины (т.е. для секвенирования фрагментов разного размера). По умолчанию в настоящем описании парно-концевые риды относятся к ридам, полученным с использованием вставок разной длины. В некоторых случаях для того, чтобы различать парно-концевые риды с короткими вставками и парно-концевые риды с длинными вставками, последние специально называют сцепленно-концевыми ридами. Согласно некоторым вариантам реализации, задействующим сцепленно-концевые риды, сначала два биотиновых соединительных адаптера присоединяют к двум концам относительно длинной вставки (например, длиной несколько т.п.н.). Затем биотиновыми соединительными адаптерами соединяют два конца указанной вставки с образованием закольцованной молекулы. Затем может быть получен субфрагмент, включающий указанные биотиновые соединительные адаптеры, путем последующего фрагментирования закольцованной молекулы. Указанный субфрагмент, включающий два конца исходного фрагмента в обратной последовательности, может затем быть секвенирован с помощью такой же процедуры, что и для парно-концевого секвенирования с короткими вставками, описанного выше. Более подробно сцепленно-концевое секвенирование с применением платформы Ulumina описано в онлайн-публикации, полностью включенной посредством ссылки в настоящий документ, по следующему адресу: res.illumina.com/documents/products/technotes/technote_nextera_matepair_data_pro cessing.pdf
[00285] После секвенирования фрагментов ДНК риды последовательностей, имеющие заранее заданную длину, например, 100 п.о., локализуют путем картирования (выравнивания) на известный референсный геном. Картированные риды и соответствующие им места на референсной последовательности также называют метки. Согласно другому варианту реализации указанной процедуры локализацию выполняют с использованием распределения k-меров и выравнивания ридов на риды. В анализах согласно многим вариантам реализации, описанных в настоящем документе, используются риды, которые либо неудовлетворительно выравниваются, либо не могут быть выравнены, наряду с выравниваемыми ридами (метки). Согласно одному варианту реализации последовательность референсного генома представляет собой последовательность NCBI36/hg18, доступную в сети Интернет по адресу genome.ucsc.edu/cgi-bin/hgGateway?org=Human&db=hg18&hgsid=166260105). Как вариант, в качестве последовательности референсного генома используют GRCh37/hg19 или GRCh38, доступную в сети Интернет по адресу: genome.ucsc.edu/cgi-bin/hgGateway. Другие источники общедоступной информации о последовательностях включают GenBank, dbEST, dbSTS, EMBL (Европейская молекулярно-биологическая лаборатория) и DDBJ (Японская база данных ДНК). Доступен ряд компьютерных алгоритмов для выравнивания последовательностей, в том числе, без ограничения, BLAST (Altschul et al., 1990), BLITZ (MPsrch) (Sturrock & Collins, 1993), FASTA (Person & Lipman, 1988), BOWTIE (Langmead et al., Genome Biology 10:R25.1-R25.10 [2009]) или ELAND (Ulumina, Inc., Сан-Диего, Калифорния, США). Согласно одному варианту реализации один конец клонально размноженных копий молекул вкДНК плазмы секвенируют и подвергают биоинформационному анализу выравнивания для анализатора Illumina Genome Analyzer, с использованием программного обеспечения для эффективного масштабного выравнивания баз данных нуклеотидов («Efficient Large-Scale Alignment of Nucleotide Databases)), ELAND).
[00286] Могут также применяться другие способы секвенирования для получения ридов последовательностей и их выравниваний. Дополнительные подходящие способы описаны во включенной полностью посредством ссылки заявке на патент США №15/130668, поданной 15 апреля 2016 г.
[00287] Согласно некоторым вариантам реализации способов, описанных в настоящем документе, длина ридов последовательностей составляет приблизительно 20 п.о., приблизительно 25 п.о., приблизительно 30 п.о., приблизительно 35 п.о., приблизительно 40 п.о., приблизительно 45 п.о., приблизительно 50 п.о., приблизительно 55 п.о., приблизительно 60 п.о., приблизительно 65 п.о., приблизительно 70 п.о., приблизительно 75 п.о., приблизительно 80 п.о., приблизительно 85 п.о., приблизительное п.о., приблизительно 95 п.о., приблизительно 100 п.о., приблизительно 110 п.о., приблизительно 120 п.о., приблизительно 130, приблизительно 140 п.о., приблизительно 150 п.о., приблизительно 200 п.о., приблизительно 250 п.о., приблизительно 300 п.о., приблизительно 350 п.о., приблизительно 400 п.о., приблизительно 450 п.о. или приблизительно 500 п.о. Ожидается, что технический прогресс позволит получать односторонние риды длиннее 500 п.о., что позволит получать риды длиннее чем приблизительно 1000 п.о. при создании парно-концевых ридов. Согласно некоторым вариантам реализации парно-концевые риды используют для определения представляющих интерес последовательностей, которые содержат ридов последовательностей длиной приблизительно от 20 п.о. до 1000 п.о., приблизительно от 50 п.о. до 500 п.о., или от 80 п.о. до 150 п.о. Согласно различным вариантам реализации парно-концевые риды используют для оценки представляющей интерес последовательности. Указанная представляющая интерес последовательность длиннее, чем риды. Согласно некоторым вариантам реализации представляющая интерес последовательность длиннее чем приблизительно 100 п.о., 500 п.о., 1000 п.о. или 4000 п.о. Картирование ридов последовательностей осуществляют путем сравнения последовательности ридов с референсной последовательностью для определения хромосомного происхождения секвенируемой молекулы нуклеиновой кислоты, и специфическая генетическая информация о последовательности не требуется. Может допускаться незначительная степень несовпадения (0-2 несовпадения на рид), обусловленная несущественными полиморфизмами, которые могут существовать в референсном геноме и геномах в смешанном образце. Согласно некоторым вариантам реализации риды, выравниваемые на референсную последовательность, используют в качестве якорных ридов, а риды, спаривающиеся с якорными ридами, но не выравнивающиеся на референсную последовательность или выравнивающиеся неудовлетворительно, используют в качестве заякоренных ридов. Согласно некоторым вариантам реализации неудовлетворительно выравненные риды могут содержать относительно большой процент несовпадений на рид, например, по меньшей мере приблизительно 5%, по меньшей мере приблизительно 10%, по меньшей мере приблизительно 15%, или по меньшей мере приблизительно 20% несовпадений на рид.
[00288] Как правило, для каждого образца получают множество меток последовательности (т.е. ридов, выравненных на референсную последовательность). Согласно некоторым вариантам реализации для каждого образца получают путем картирования ридов на референсный геном по меньшей мере приблизительно 3×106 меток последовательности, по меньшей мере приблизительно 5×106 меток последовательности, по меньшей мере приблизительно 8×106 меток последовательности, по меньшей мере приблизительно 10×106 меток последовательности, по меньшей мере приблизительно 15×106 меток последовательности, по меньшей мере приблизительно 20×106 меток последовательности, по меньшей мере приблизительно 30×106 меток последовательности, по меньшей мере приблизительно 40×106 меток последовательности, или по меньшей мере приблизительно 50×106 меток последовательности размером, например, 100 п.о. Согласно некоторым вариантам реализации все риды последовательностей картируют на все области референсного генома, что обеспечивает получение общегеномных ридов. Согласно другим вариантам реализации риды картируют на представляющую интерес последовательность.
Устройство и системы для секвенирования с применением UMI
[00289] Как очевидно, определенные варианты реализации настоящего изобретения задействуют процессы, происходящие под контролем инструкций и/или данных, хранящихся в одной или более компьютерных системах или передаваемых через них. Определенные варианты реализации также предусматривают устройство для выполнения указанных операций. Указанное устройство может быть специально разработано и/или сконструировано для требуемых целей, или может представлять собой компьютер общего назначения, избирательно сконфигурированный одной или более компьютерными программами и/или структурами данных, хранящимися на указанном компьютере или иным образом предоставляемыми указанному компьютеру. В частности, могут быть использованы различные машины общего назначения с программами, написанными в соответствии с принципами настоящего изобретения, или удобнее может быть сконструировать более специализированное устройство для выполнения требуемых этапов способа. Конкретная структура множества таких машин представлена и описана ниже.
[00290] Согласно определенным вариантам реализации также предложены функциональные средства (например, код и процессы) для хранения любых результатов (например, результатов запроса) или структур данных, генерируемых согласно описанию в настоящем документе. Такие результаты или структуры данных, как правило, хранят, по меньшей мере временно, на машиночитаемом носителе. Указанные результаты или структуры данных могут также быть выведены любым из множества способов, например, на экране, в печатном виде и т.п.
[00291] Примеры материальных машиночитаемых носителей, подходящих для применения компьютерных программных продуктов и вычислительного устройства согласно настоящему изобретению включают, не ограничиваясь перечисленными, магнитные носители, такие как жесткие диски, дискеты и магнитная лента; оптические носители, такие как ПЗУ на компакт-дисках; магнитооптические носители; полупроводниковые запоминающие устройства (например, флеш-память) и аппаратные устройства, специально выполненные с возможностью хранения и исполнения программных инструкций, такие как постоянные запоминающие (накопительные) устройства (ПЗУ) и оперативное запоминающее устройство (ОЗУ), и иногда специализированные интегральные схемы (ASIC), программируемые логические устройства (PLD) и среды передачи сигналов для доставки машиночитаемых инструкций, такие как локальные сети, сети широкого охвата и сеть Интернет. Данные и программные инструкции, предложенные согласно настоящему изобретению, могут также быть воплощены в несущей волне или другой транспортной среде (в том числе электропроводящих путях или световодах). Данные и программные инструкции согласно настоящему изобретению могут также быть воплощены в несущей волне или другой транспортной среде (например, оптических линиях, электрических линиях и/или воздушных волнах).
[00292] Примеры программных инструкций включают низкоуровневый код, такой как генерируемые компилятором, а также более высокоуровневый код, который может быть исполнен компьютером с использованием интерпретатора. Кроме того, программные инструкции могут представлять собой машинный код, исходный код и/или любой другой код, который прямо или непрямо контролирует работу вычислительной машины. Указанный код может точно определять ввод, вывод, вычисления, условные операторы, ветви, итерационные циклы и т.п.
[00293] Анализ данных секвенирования и основанную на нем диагностику, как правило, выполняют с применением различных машиноисполняемых алгоритмов и программ. Соответственно, определенные варианты реализации задействуют процессы с использованием данных, хранящихся в одной и более компьютерных системах или других системах обработки, или передаваемых через одну или более компьютерных систем или других систем обработки. Варианты реализации согласно описанию в настоящем документе также относятся к устройству для выполнения указанных операций. Указанное устройство может быть специально сконструировано для требуемых целей, или может представлять собой компьютер общего назначения (или группу компьютеров), избирательно активируемый или реконфигурируемый компьютерной программой и/или структурой данных, хранящейся на указанном компьютере. Согласно некоторым вариантам реализации группа процессоров выполняет некоторые или все из упомянутых аналитических операций совместно (например, через сеть или путем вычислений в облаке) и/или параллельно. Процессор или группа процессоров для выполнения способов, описанных в настоящем документе, могут относиться к разным типам, в том числе микроконтроллерам и микропроцессорам, таким как программируемые устройства (например, СПЛИС и ППВМ) и непрограммируемые устройства, такие как ASIC на основе вентильных матриц или микропроцессоры общего назначения.
[00294] Согласно одному варианту реализации предложена система для применения при определении последовательности с низкой частотой аллелей в тестовом образце, включающем нуклеиновые кислоты, включающая секвенатор для получения образца нуклеиновой кислоты и обеспечения информации о последовательности нуклеиновой кислоты из образца; процессор; и машиночитаемый носитель для хранения данных с хранящимися на них инструкциями для исполнения указанным процессором для определения представляющей интерес последовательности в тестовом образце путем: (а) обработки адаптерами фрагментов ДНК в указанном образце с получением продуктов ДНК-адаптер, где каждый адаптер содержит неслучайный уникальный молекулярный индекс, при этом неслучайные уникальные молекулярные индексы указанных адаптеров характеризуются по меньшей мере двумя разными длинами молекул и образуют набор неслучайных уникальных молекулярных индексов с вариабельной длиной (vNRUMI); (b) амплификации указанных продуктов ДНК-адаптер с получением множества амплифицированных полинуклеотидов; (с) секвенирования с применением секвенатора множества амплифицированных полинуклеотидов, с получением таким образом множества ридов, ассоциированных с набором vNRUMI; (d) идентификации процессором среди множества ридов тех ридов, которые ассоциированы с одним и тем же неслучайным уникальным молекулярным индексом с вариабельной длиной (vNRUMI); и (е) определения последовательности фрагмента ДНК в указанном образце с использованием ридов, ассоциированных с одним и тем же vNRUMI.
[00295] Согласно некоторым вариантам реализации любой из систем, предложенных в настоящем изобретении, секвенатор выполнен с возможностью осуществления секвенирования нового поколения (NGS). Согласно некоторым вариантам реализации указанный секвенатор выполнен с возможностью осуществления массово-параллельного секвенирования путем синтеза с применением терминаторов с обратимым красителем. Согласно другим вариантам реализации указанный секвенатор выполнен с возможностью осуществления секвенирования путем лигирования. Согласно другим дополнительным вариантам реализации указанный секвенатор выполнен с возможностью осуществления одномолекулярного секвенирования.
[00296] Согласно другому варианту реализации предложена система, включающая синтезатор нуклеиновых кислот, процессор и машиночитаемый носитель для хранения данных с хранящимися на нем инструкциями для исполнения указанным процессором для получения адаптеров для секвенирования. Указанные инструкции включают: (а) обеспечение указанным процессором набора последовательностей олигонуклеотидов по меньшей мере с двумя разными длинами молекул; (b) выбор указанным процессором поднабора последовательностей олигонуклеотидов из набора последовательностей олигонуклеотидов, отличающегося тем, что все редакционные расстояния между последовательностями олигонуклеотидов указанного поднабора последовательностей олигонуклеотидов соответствуют пороговому значению, и указанный поднабор последовательностей олигонуклеотидов образует набор неслучайных уникальных молекулярных индексов с вариабельной длиной (vNRUMI); и (с) синтез, с применением синтезатора нуклеиновых кислот, множества адаптеров для секвенирования, причем каждый адаптер для секвенирования содержит двуцепочечную гибридизованную область, одноцепочечное 5'-плечо, одноцепочечное 3'-плечо и по меньшей мере один vNRUMI из набора vNRUMI.
[00297] Кроме того, определенные варианты реализации относятся к материальным и/или энергонезависимым машиночитаемым носителям или компьютерным программным продуктам, которые включают программные инструкции и/или данные (в том числе структуры данных) для выполнения различных реализуемых компьютером операций. Примеры машиночитаемых носителей включают, не ограничиваясь перечисленными, полупроводниковые запоминающие устройства, магнитные носители, такие как дисковые накопители, магнитная лента, оптические носители, такие как компакт-диски, магнитооптические носители и аппаратные устройства, специально выполненные с возможностью хранения и выполнения программных инструкций, такие как постоянные запоминающие устройства (ПЗУ) и оперативные запоминающие устройства (ОЗУ). Конечный пользователь может прямо контролировать машиночитаемые носители, или конечный пользователь может непрямо контролировать указанные носители. Примеры прямо контролируемых носителей включают носители, локализованные на пользовательском оборудовании, и/или носители, которые не используются совместно с другими объектами. Примеры непрямо контролируемых носителей включают носители, к которым у пользователя имеется непрямой доступ через внешнюю сеть и/или сервис, обеспечивающий совместное использование ресурсов, такой как «облако». Примеры программных инструкций включают как машинный код, такой как генерируемый компилятором, и файлы, содержащие более высокоуровневый код, который может быть исполнен компьютером с использованием интерпретатора.
[00298] Согласно различным вариантам реализации данные или информация, используемые в предложенных способах и устройстве, предоставляются в электронном формате. Такие данные или информация могут включать риды и метки, происходящие из образца нуклеиновой кислоты, референсные последовательности (в том числе референсные последовательности, обеспечивающие исключительно или в первую очередь полиморфизмы), сигналы распознавания, такие как сигналы распознавания для диагностики рака, консультативные рекомендации, диагнозы и т.п. Согласно настоящему изобретению данные или другая информация, предоставляемые в электронном формате, доступны для хранения в машине и передачи между машинами. Обычно данные в электронном формате предоставляются в цифровом виде, и могут храниться в форме битов и/или байтов в различных структурах данных, перечнях, базах данных и т.п. Указанные данные могут быть представлены в электронном виде, оптическом виде и т.п.
[00299] Согласно одному варианту реализации предложен компьютерный программный продукт для формирования выводимых данных, указывающих на представляющую интерес последовательность фрагмента ДНК в тестовом образце. Указанный компьютерный продукт может содержать инструкции для реализации любого одного или более из вышеописанных способов определения представляющей интерес последовательности. Как было объяснено, указанный компьютерный продукт может включать энергонезависимый и/или материальный машиночитаемый носитель, содержащий машиноисполняемую или компилируемую логическую схему (например, инструкции), записанную на нем, чтобы обеспечивать определение процессором представляющей интерес последовательности. Согласно одному примеру указанный компьютерный продукт содержит машиночитаемый носитель, содержащий машиноисполняемую или компилируемую логическую схему (например, инструкции), записанную на нем, чтобы обеспечивать диагностику состояния или определение представляющей интерес последовательности нуклеиновой кислоты процессором.
[00300] Следует понимать, что в большинстве случаев выполнение вычислительных операций в способах согласно описанию в настоящем документе человеком без использования дополнительных средств нецелесообразно или даже невозможно. Например, картирование единственного рида размером 30 п.о. из образца на любую из хромосом человека без использования вычислительного устройства может потребовать нескольких лет работы. Конечно, указанная проблема усугубляется тем, что получение надежных сигналов распознавания мутаций с низкой частотой аллелей обычно требует картирования тысяч (например, по меньшей мере приблизительно 10000) или даже миллионов ридов на одну или более хромосом.
[00301] Способы согласно описанию в настоящем документе могут быть реализованы с применением системы для определения представляющей интерес последовательности в тестовом образце. Указанная система может включать: (а) секвенатор для загрузки нуклеиновых кислот из тестового образца, обеспечивающий информацию о последовательности нуклеиновой кислоты из указанного образца; (b) процессор; и (с) один или более машиночитаемых носителей для хранения данных с хранящимися на них инструкциями для исполнения указанным процессором для определения представляющей интерес последовательности в тестовом образце. Согласно некоторым вариантам реализации описание указанных способов обеспечивает машиночитаемый носитель с хранящимися на нем машиночитаемыми инструкциями для осуществления способа определения представляющей интерес последовательности. Соответственно, согласно одному варианту реализации предложен компьютерный программный продукт, включающий энергонезависимый машиночитаемый носитель, где хранится программный код, который при исполнении одним или более процессорами компьютерной системы обеспечивает осуществление компьютерной системой способа определения последовательностей фрагментов нуклеиновых кислот в тестовом образце. Указанный программный код может включать: (а) код для получения множества ридов с множества амплифицированных полинуклеотидов, отличающегося тем, что каждый полинуклеотид из указанного множества амплифицированных полинуклеотидов содержит адаптер, присоединенный к фрагменту ДНК, при этом указанный адаптер содержит неслучайный уникальный молекулярный индекс, причем неслучайные уникальные молекулярные индексы указанных адаптеров характеризуются по меньшей мере двумя разными длинами молекул, образуя набор неслучайных уникальных молекулярных индексов с вариабельной длиной (vNRUMI); (b) код для идентификации во множестве ридов тех ридов, которые ассоциированы с одними и теми же vNRUMI; и (с) код для определения с использованием ридов, ассоциированных с одним и тем же vNRUMI, последовательности фрагмента ДНК в указанном образце.
[00302] Согласно некоторым вариантам реализации указанные программные коды или инструкции могут дополнительно предусматривать автоматическую запись информации, относящейся к указанному способу. Медицинская документация пациента может вестись, например, в лаборатории, кабинете лечащего врача, больнице, организации здравоохранения, страховой компании или на вебсайте для личной медицинской документации. Кроме того, в зависимости от результатов осуществляемого процессорами анализа указанный способ может дополнительно включать назначение, инициацию и/или изменение лечения субъекта-человека, у которого был взят тестовый образец. Указанный способ может включать проведение одного или более дополнительных тестов или анализов на дополнительных образцах, взятых у указанного субъекта.
[00303] Раскрытые способы могут также быть реализованы с применением компьютерной системы обработки, сконфигурированной или выполненной с возможностью осуществления способа определения представляющей интерес последовательности. Согласно одному варианту реализации предложена компьютерная система обработки, сконфигурированная или выполненная с возможностью осуществления способа согласно описанию в настоящем документе. Согласно одному варианту реализации указанное устройство включает устройство для секвенирования, сконфигурированное или выполненное с возможностью секвенирования по меньшей мере части молекул нуклеиновой кислоты в образце для получения информации о последовательности описанного в различных разделах настоящего документа типа. Указанное устройство может также включать компоненты для обработки образца. Такие компоненты описаны в различных разделах настоящего документа.
[00304] Данные последовательности или другие данные могут быть введены в компьютер или сохранены на машиночитаемом носителе, прямо или непрямо. Согласно одному варианту реализации компьютерная система прямо сопряжена с устройством для секвенирования, которое считывает и/или анализирует последовательности нуклеиновых кислот из образцов. Информация о последовательностях или другая информация с таких инструментов поступает через интерфейс компьютерной системы. Как вариант, последовательности, обрабатываемые системой, поступают из источника хранения последовательностей, такого как база данных или другое хранилище. По мере поступления последовательностей в устройство для обработки, запоминающее устройство или накопительное устройство большой емкости буферизует или сохраняет, по меньшей мере временно, последовательности нуклеиновых кислот. Кроме того, указанное запоминающее устройство может сохранять количество меток для различных хромосом или геномов, и т.п. Указанное запоминающее устройство может также хранить различные подпрограммы и/или программы для анализа или представления данных последовательности или картированных данных. Такие программы/под программы могут включать программы для проведения статистических анализов и т.п.
[00305] Согласно одному примеру пользователь помещает образец в устройство для секвенирования. Данные собирают и/или анализируют на указанном устройстве для секвенирования, которое соединено с компьютером. Программное обеспечение на указанном компьютере позволяет производить сбор и/или анализ данных. Данные могут быть сохранены, выведены (с помощью монитора или другого аналогичного устройства) и/или отправлены в другое местоположение. Указанный компьютер может быть соединен с сетью Интернет, используемой для передачи данных на ручное устройство, используемое удаленным пользователем (например, лечащим врачом, научным работником или аналитиком). Следует понимать, что указанные данные могут быть сохранены и/или проанализированы до передачи. Согласно некоторым вариантам реализации собирают необработанные данные и отправляют удаленному пользователю или на устройство для анализа и/или хранения указанных данных. Передача может осуществляться через сеть Интернет, но может также осуществляться через спутниковое или другое соединение. Как вариант, данные могут быть сохранены на машиночитаемом носителе и указанный носитель может быть транспортирован к конечному пользователю (например, почтовым отправлением). Удаленный пользователь может находиться в том же или другом географическом местоположении, в том числе, но не ограничиваясь перечисленным, в том же или другом строении, городе, штате, стране или на том же или другом континенте.
[00306] Согласно некоторым вариантам реализации указанные способы также включают сбор данных о множестве последовательностей полинуклеотидов (например, ридов, меток и/или референсных хромосомных последовательностей) и отправку указанных данных на компьютер или в другую вычислительную систему. Например, компьютер может быть соединен с лабораторным оборудованием, например, с устройством для сбора образцов, устрйством для амплификации нуклеотидов, устройством для секвенирования нуклеотидов или устройством для гибридизации. Соответственно, указанный компьютер может собирать соответствующие данные, полученные лабораторным устройством. Данные могут быть сохранены на компьютере на любом этапе, например, при получении в реальном времени, до отправки, одновременно или совместно с отправкой, или после отправки. Указанные данные могут быть сохранены на машиночитаемом носителе, который может быть извлечен из компьютера. Собранные или хранимые данные могут быть переданы с компьютера в удаленное местоположение, например, по локальной сети или сети широкого доступа, такой как Интернет. В указанном удаленном местоположении над переданными данными могут быть выполнены различные операции согласно приведенному ниже описанию.
[00307] Типы форматированных электронным способом данных, которые могут быть сохранены, переданы, проанализированы и/или подвергнуты манипуляциям в системах, устройствах и способах согласно описанию в настоящем документе, включают, среди прочих, перечисленные ниже:
Риды, полученные путем секвенирования нуклеиновых кислот в тестовом образце
Метки, полученные путем выравнивания ридов на референсный геном или другую референсную последовательность или последовательности
Референсный геном или референсная последовательность
Пороговые значения для распознавания тестового образца либо как пораженного, либо как не пораженного, либо не дающего сигнала распознавания
Фактические сигналы распознавания медицинских состояний, связанных с представляющей интерес последовательностью
Диагнозы (клиническое состояние, ассоциированное с сигналами распознавания)
Рекомендации по проведению дополнительных тестов на основании сигналов распознавания и/или диагнозов
Планы лечения и/или мониторинга на основании сигналов распознавания и/или диагнозов.
[00308] Указанные различные типы данных могут быть получены, помещены на хранение, переданы, проанализированы и/или подвергнуты манипуляциям в одном или более местоположениях с использованием отдельного устройства. Возможности обработки представлены широким спектром вариантов. С одной стороны спектра указанную информацию полностью или по большей части хранят и используют в местоположении, где происходит обработка тестового образца, например, в кабинете врача или в других клинических условиях. В случае другой крайности образец получают в одном местоположении, обрабатывают и необязательно секвенируют в другом местоположении, выравнивают риды и получают сигналы распознавания в одном или более разных местоположениях, и диагнозы, рекомендации и/или планы получают также в другом местоположении (которое может представлять собой местоположение, где был получен указанный образец).
[00309] Согласно различным вариантам реализации риды получают на устройстве для секвенирования и затем передают в удаленный сайт, где их обрабатывают для определения представляющей интерес последовательности. В указанном удаленном местоположении, например, риды выравнивают на референсную последовательность с получением якорных и заякоренных ридов. В отдельных местоположениях могут применяться, в том числе, следующие операции обработки:
Взятие образцов
Обработка образцов, предваряющая секвенирование
Секвенирование
Анализ данных последовательности и получение медицинских сигналов распознавания
Диагностика
Оповещение о диагнозе и/или сигнале распознавания пациента или поставщика медицинских услуг
Разработка плана дальнейшего лечения, тестирования и/или мониторинга
Выполнение указанного плана
Консультирование
[00310] Любая одна или более из указанных операций может быть автоматизирована согласно описанию в тексте настоящего документа. Как правило, секвенирование и анализ данных последовательности, а также получение медицинских сигналов распознавания выполняют с применением вычислительных средств. Другие операции могут быть выполнены вручную или автоматически.
[00311] На Фиг. 6 показан один вариант реализации рассредоточенной системы для формирования сигнала распознавания или диагноза на основании тестового образца. Местоположение взятия образца 01 используют для получения тестового образца от пациента. Затем образцы передают в местоположение обработки и секвенирования 03, где тестовый образец может быть обработан и секвенирован согласно описанию выше. В местоположении 03 находится устройство для обработки образца, а также устройство для секвенирования обработанного образца. Результат секвенирования, согласно описанию в тексте настоящего документа, представляет собой коллекцию ридов, которые, как правило, предоставляются в электронном формате и передаются в сеть, такую как сеть Интернет, на что указывает ссылочный номер 05 на Фиг. 6.
[00312] Данные последовательности передают в удаленное местоположение 07, где осуществляют анализ и получают сигналы распознавания. Указанное местоположение может включать одно или более мощных вычислительных устройств, таких как компьютеры или процессоры. После завершения вычислительными средствами в местоположении 07 анализа и формирования сигнала распознавания из полученной информации о последовательности, указанный сигнал распознавания поступает обратно в сеть 05. Согласно некоторым вариантам реализации в местоположении 07 происходит формирование не только сигнала распознавания, но и ассоциированного с ним диагноза. Сигнал распознавания и/или диагноз затем передают по сети и они поступают назад в местоположение взятия образца 01, что проиллюстрировано Фиг. 6. Как было объяснено, представлен только один из многих возможных вариантов распределения по различным местоположениям различных операций, ассоциированных с формированием сигнала распознавания или диагноза. Один распространенный вариант предусматривает взятие образцов, обработку и секвенирование в одном местоположении. Другой вариант предусматривает обработку и секвенирование в том же местоположении, что и анализ и получение сигналов распознавания.
[00313] На Фиг. 7 проиллюстрирована, в формате простых блоков, типичная компьютерная система, которая, при надлежащих конфигурации или дизайне, может служить в качестве вычислительного устройства в соответствии с определенными вариантами реализации. Компьютерная система 2000 включает любое число процессоров 2002 (также называемых центральными процессорами, или ЦПУ), которые сопряжены с устройствами для хранения данных, в том числе первичным запоминающим устройством 2006 (как правило, оперативным запоминающим устройством, или ОЗУ), первичным запоминающим устройством 2004 (как правило, постоянным запоминающим устройством, или ROM). ЦПУ 2002 может быть представлен различными типами, в том числе микроконтроллерами и микропроцессорами, такими как программируемые устройства (например, СПЛИС и ППВМ) и непрограммируемые устройства, такие как ASIC на основе вентильных матриц или микропроцессоры общего назначения. Согласно представленному варианту реализации первичное запоминающее устройство 2004 однонаправленно переносит данные и инструкции в ЦПУ, а первичное запоминающее устройство 2006 используют, как правило, для двунаправленного переноса данных и инструкций. Оба указанных первичных запоминающих устройства могут включать любые подходящие машиночитаемые носители, такие как описанные выше. Накопительное устройство большой емкости 2008 также сопряжено двусторонним образом с первичным запоминающим устройством 2006, обеспечивает дополнительную емкость памяти и может включать любые из машиночитаемых носителей, описанных выше. Накопительное устройство большой емкости 2008 может быть использовано для хранения программ, данных и т.п., и, как правило, вторичный носитель для хранения данных, такой как жесткий диск. Часто такие программы, данные и т.п. временно копируют в первичную память 2006 для исполнения на ЦПУ 2002. Следует понимать, что информация, сохраняемая на накопительном устройстве большой емкости 2008, может, в подходящих случаях, быть передана стандартным образом на первичное запоминающее устройство 2004. Специальное накопительное устройство большой емкости, такое как ПЗУ на компакт-диске 2014, может также однонаправленно передавать данные на ЦПУ или на первичное запоминающее устройство.
[00314] ЦПУ 2002 также сопряжен с интерфейсом 2010, который соединяется с одним или более устройств ввода/вывода, таких как секвенатор нуклеиновых кислот (2020), синтезатор нуклеиновых кислот (2022), видеомониторы, шаровые манипуляторы, мыши, клавиатуры, микрофоны, дисплеи с сенсорным управлением, приемо-передаточные устройства для чтения карт, считыватели магнитных или бумажных лент, планшеты, стилусы, периферийные устройства распознавания голоса или рукописного ввода, USB-порты или другие хорошо известные устройства ввода, такие как, разумеется, другие компьютеры. Наконец, ЦПУ 2002 необязательно может быть сопряжен с внешним устройством, таким как база данных или компьютер, или телекоммуникационная сеть, с использованием внешнего соединения, схематично представленного в блоке 2012. Предполагается, что с таким соединением ЦПУ может получать информацию из сети, или выводить информацию в сеть в ходе выполнения этапов способа, описанного в настоящем документе. Согласно некоторым вариантам реализации секвенатор нуклеиновых кислот или синтезатор нуклеиновых кислот могут быть коммуникативно соединены с ЦПУ 2002 через сетевое соединение 2012 вместо интерфейса или наряду с интерфейсом 2010.
[00315] Согласно одному варианту реализации систему, такую как компьютерная система 2000, используют в качестве системы импорта данных, корреляции данных и системы для запросов, способной выполнять некоторые или все задачи, описанные в настоящем документе. Информация и программы, в том числе файлы данных, могут быть переданы через соединение 2012 для доступа или скачивания исследователем. Как вариант, такие информация, программы и файлы могут быть переданы исследователю на накопительном устройстве.
[00316] Согласно конкретному варианту реализации компьютерная система 2000 прямо сопряжена с системой регистрации данных, такой как микрочип, система высокопроизводительного скрининга или секвенатор нуклеиновых кислот (2020), которая захватывает данные из образцов. Данные из таких систем передаются через интерфейс 2010 для анализа системой 2000. Как вариант, данные для обработки системой 2000 передаются из источника, где хранятся данные, такого как база данных или другое хранилище релевантных данных. После попадания релевантных данных в устройство 2000 запоминающее устройство, такое как первичное запоминающее устройство 2006 или запоминающее устройство большой емкости 2008 буферизует или сохраняет, по меньшей мере временно, релевантные данные. Указанное запоминающее устройство может также сохранять различные подпрограммы и/или программы для импорта, анализа и представления данных, в том числе ридов последовательностей, UMI, кодов для определения ридов последовательностей, объединения ридов последовательностей и коррекции ошибок в ридах и т.п.
[00317] Согласно некоторым вариантам реализации компьютеры, используемые согласно настоящему изобретению, могут включать пользовательский терминал, который может быть представлен любым типом компьютера (например, стационарным компьютером, портативным компьютером, планшетом и т.п.), вычислительные платформы для передачи данных (например, кабель, спутниковые приставки, цифровые видеомагнитофоны и т.п.), карманные вычислительные устройства (например, КПК, почтовые клиенты и т.п.), мобильные телефоны; или вычислительные или коммуникационные платформы любого другого типа.
[00318] Согласно некоторым вариантам реализации компьютеры, используемые согласно настоящему изобретению, могут также включать серверную систему, коммуницирующую с пользовательским терминалом, которая может включать серверное устройство или децентрализированные серверные устройства, и может включать большие компьютеры, миникомпьютеры, суперкомпьютеры, персональные компьютеры или их комбинации. Может также применяться множество серверных систем без отступления от объема настоящего изобретения. Пользовательские терминалы и серверная система могут коммуницировать между собой через сеть. Указанная сеть может включать, например, проводные сети, такие как LAN (локальные сети), WAN (сети широкого охвата), MAN (городские вычислительные сети), ISDN (интегрированные сервисные цифровые сети) и т.п., а также беспроводные сети, такие как беспроводные LAN, CDMA, Bluetooth, сети спутниковой связи и т.п., без ограничения объема настоящего изобретения.
ЭКСПЕРИМЕНТЫ
Пример 1
Сравнение способа с vNRUMI и других способов со штрихкодами
[00319] В таблице 1 показана гетерогенность пар оснований NRUMI, сравниваемая с гетерогенностью пар оснований vNRUMI в соответствии с некоторыми вариантами реализации. Указанный набор из 120 vNRUMI состоит из 50 6-меров и 70 7-меров. Набор NRUMI полностью состоит из 218 6-меров, при этом минимальное редакционное расстояние между любыми двумя NRUMI превышает пороговое значение. В таблице 1 показано, что все из 218 или 128 штрихкодов присутствовали в равных количествах, например, по 1000 UMI каждого типа. Что касается 7-го основания, новый набор vNRUMI отличается значительно лучшей гетерогенностью по сравнению с оригинальным набором NRUMI, и значительно превышает рекомендованный минимум - 5% состава на каждое основание. Соответственно, понятно, что дизайн vNRUMI решает вышеописанную проблему отсутствия разнообразия пар оснований в определенных циклах. Другие наборы штрихкодов, состоявшие исключительно из 6-меров, характеризуются гетерогенностью для каждого основания, аналогичной гетерогенности оригинального набора NRUMI, представленного ниже.
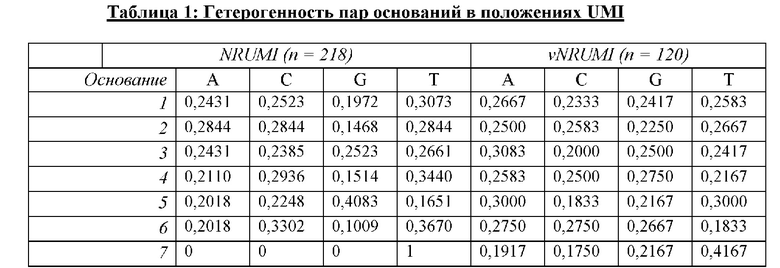
[00320] Используя представленные выше NRUMI и vNRUMI, проводили модельные исследования in silico для моделирования 10000 штрихкодов, вводили мутации в каждый отдельный штрихкод за счет независимых мутаций каждого основания и пытались восстановить оригинальную последовательность UMI. Для моделирования использовали частоту мутаций 2% для каждого основания (1% вероятность SNV, 1% вероятность индела с размером 1). Отметим, что указанная частота мутаций заметно выше типичных коэффициентов ошибок при секвенировании Illumina. Каждая из 10000 модельных последовательностей содержала по меньшей мере одну мутацию.
[00321] Для обеспечения дополнительного сравнения с другими способами, задействующими UMI, набор из 114 последовательностей NRUMI с длиной 6 нуклеотидов, сформированный в соответствии с существующим способом nxCode, также используют в указанном модельном исследовании. См. http://hannonlab.cshl.edu/nxCode/nxCode/main.html. Указанные последовательности подвергали такому же процессу мутирования, что и описанный выше. В способе nxCode используется вероятностная модель для определения мутаций, и полужадный подход для получения набора NRUMI с одинаковой длиной молекулы. Результаты сравнения наборов vNRUMI, NRUMI и nxCode представлены в таблице 2.

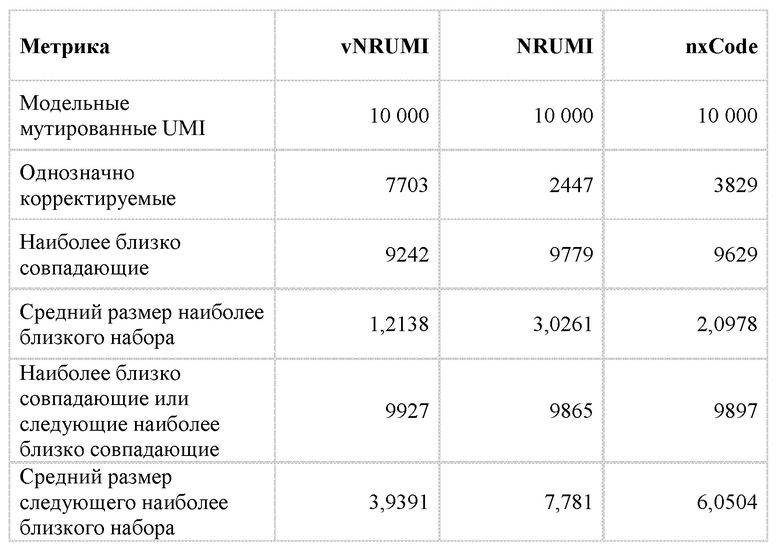
[00322] Набор vNRUMI содержит 120 UMI, из которых 50 UMI имеют длину 6 нуклеотидов и 70 UMI имеют длину 7 нуклеотидов. Набор NRUMI содержит 218 последовательностей с длиной 6. Стандартный способ nxCode задействует набор NRUMI из 114 последовательностей с длиной 6 нуклеотидов. Средний размер набора представляет собой средне число уникальных последовательностей, включенных в набор.
[00323] В таблице 2 однозначная коррекция определена как случай, когда набор наиболее близких соседей содержит только одну последовательность; другими словами, алгоритм поиска совпадающих UMI и коррекции, описанный выше, позволяет сделать недвусмысленное предположение относительно наиболее вероятного истинного vNRUMI. Отметим, что число таких однозначно корректируемых последовательностей значительно больше для способа с vNRUMI, чем для способа с NRUMI и nxCode. Также средний размер наиболее близкого/следующего наиболее близкого набора значительно меньше для способа с vNRUMI, чем при использовании других решений, а уровень содержания оригинального немутированного штрихкода в указанных наборах приблизительно одинаков. Это важно, поскольку при объединении ридов для выбора корректного UMI из указанных наиболее близкого/следующего наиболее близкого наборов используют контекстную информацию. Обеспечение уменьшения количества некорректных последовательностей на указанном этапе объединения ридов может снижать вероятность некорректного выбора на этом этапе, в конечном итоге повышая способность к подавлению шума и детекции вариантов.
[00324] Следует отметить, что способы с NRUMI и nxCode, как и другие существующие стратегии на основе штрихкодов, подразумевают, что все штрихкодирующие последовательности имеют одинаковую длину. При выполнении указанного моделирования, чтобы обеспечить прямое сравнение трех способов, оригинальные способы коррекции ошибок, описанные для способов с NRUMI и nxCode, не использовали, что могло ограничивать производительность способов NRUMI и nxCode. Тем не менее данные в таблице 2 дают представление о потенциальной возможности улучшения коррекции ошибок с помощью способа с vNRUMI, что дополнительно проиллюстрировано на следующем примере.
Пример 2
Восстановление фрагментов ДНК с использованием vNRUMI и NRUMI
[00325] В другой серии in silo исследований тестируют возможность восстановления ридов с использованием vNRUMI и NRUMI. В указанных исследованиях выбирают случайную мутацию COSMIC и генерируют единственный фрагмент ДНК, содержащий указанную мутацию. Средний размер фрагментов равен 166 при стандартном отклонении, равном 40. При моделировании случайный UMI добавляют к обоим концам указанного фрагмента. Она использует ART (см., например, https://www.niehs.nih.gov/research/resources/software/biostatistics/art/) для моделирования 10 парно-концевых ридов указанной молекулы UMI-фрагмент-UMI и выравнивания указанных ридов с использованием выравнивателя Барроуза-Уилера (BWA). См., например, http://bio-bwa.sourceforge.net/.
[00326] Затем процесс передает выравнивание на объединитель ридов собственной разработки, ReCo, для определения того, может ли он восстановить оригинальную последовательность фрагмента, и повторяют процесс для дополнительных ридов.
[00327] В таблице 3 приведены количества и проценты фрагментов, которые могут быть восстановлены.
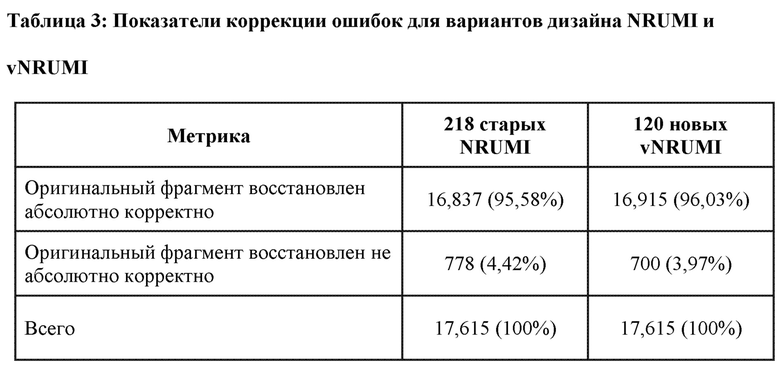
[00328] Способ с vNRUMI позволял восстановить больше фрагментов, чем способ с NRUMI фиксированной длины. Критерий хи-квадрат показывает, что различия являются значимыми. 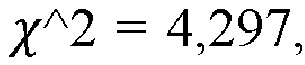 двустороннее Р-значение = 0,0382. При α=0,05 способ с vNRUMI обеспечивал статистически
двустороннее Р-значение = 0,0382. При α=0,05 способ с vNRUMI обеспечивал статистически 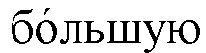 эффективность коррекции ошибок по сравнению со способом с NRUMI, наряду с устранением недостатков, свойственных способу с NRUMI.
эффективность коррекции ошибок по сравнению со способом с NRUMI, наряду с устранением недостатков, свойственных способу с NRUMI.
[00329] Стратегия NRUMI задействует наборы NRUMI гетерогенной длины. Это решает проблему различий пар оснований, которая приводила к снижению качества выравнивания.
[00330] Предложены новые способы формирования наборов UMI с вариабельной длиной, удовлетворяющих биохимическим ограничениям, и картирования неверно прочтенных UMI для коррекции UMI. Новый подход решает проблему снижения качества секвенирования, обусловленную штрихкодами постоянной длины. Использование схемы определения совпадений, учитывающей число совпадений и несовпадений, в отличие от простого отслеживания несовпадений, позволяет увеличить возможность коррекции ошибок. Указанные варианты реализации сопоставимы с существующими решениями или превосходят их, обеспечивая при этом дополнительную функциональность.
×[00331] Настоящее изобретение может быть реализовано в других специфических формах без отступления от его сути или существенных характеристик. Описанные варианты реализации следует рассматривать во всех отношениях исключительно иллюстративными и неограничивающими. Объем настоящего изобретения, соответственно, определен прилагаемой формулой изобретения, а не предшествующим описанием. Все изменения, которые соответствуют смыслу и диапазону эквивалентности формулы изобретения, включены в его объем.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> ILLUMINA, INC. / ИЛЛУМИНА, ИНК.
<120> METHODS AND SYSTEMS FOR GENERATION AND ERROR-CORRECTION OF
UNIQUE MOLECULAR INDEX SETS WITH HETEROGENEOUS MOLECULAR
LENGTHS /СПОСОБЫ И СИСТЕМЫ ДЛЯ ПОЛУЧЕНИЯ НАБОРОВ
УНИКАЛЬНЫХ МОЛЕКУЛЯРНЫХ ИНДЕКСОВ С ГЕТЕРОГЕННОЙ ДЛИНОЙ
МОЛЕКУЛ И КОРРЕКЦИИ В НИХ ОШИБОК
<130> ILMNP013WO
<140> PCT/US2018/012669
<141> 2018-01-05
<150> 62/447 851
<151> 2017-01-18
<160> 2
<170> PatentIn, версия 3.5
<210> 1
<211> 13
<212> DNA/ ДНК
<213> Описание искусственной последовательности: синтетический
полинуклеотид
<220>
<221> Другие признаки
<222> (9)..(13)
<223> n представляет собой a, c, g или t
<400> 1
ttgtgactnn nnn 13
<210> 2
<211> 16
<212> DNA/ ДНК
<213> Описание искусственной последовательности: синтетический
полинуклеотид
<220>
<221> Другие признаки
<222> (12)..(16)
<223> n представляет собой a, c, g или t
<400> 2
ttggcatgac tnnnnn 16
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОДАВЛЕНИЕ ОШИБОК В СЕКВЕНИРОВАННЫХ ФРАГМЕНТАХ ДНК ПОСРЕДСТВОМ ПРИМЕНЕНИЯ ИЗБЫТОЧНЫХ ПРОЧТЕНИЙ С УНИКАЛЬНЫМИ МОЛЕКУЛЯРНЫМИ ИНДЕКСАМИ (UMI) | 2016 |
|
RU2704286C2 |
| БИБЛИОТЕКИ ДЛЯ СЕКВЕНИРОВАНИЯ НОВОГО ПОКОЛЕНИЯ | 2014 |
|
RU2698125C2 |
| ТРАНСПОЗИЦИЯ С СОХРАНЕНИЕМ СЦЕПЛЕНИЯ ГЕНОВ | 2015 |
|
RU2709655C2 |
| ТАГМЕНТАЦИЯ С ПРИМЕНЕНИЕМ ИММОБИЛИЗОВАННЫХ ТРАНСПОСОМ С ЛИНКЕРАМИ | 2018 |
|
RU2783536C2 |
| ТРАНСПОЗИЦИЯ С СОХРАНЕНИЕМ СЦЕПЛЕНИЯ ГЕНОВ | 2015 |
|
RU2736728C2 |
| СПОСОБ ВЫЯВЛЕНИЯ МУТАЦИЙ В СЛОЖНЫХ СМЕСЯХ ДНК | 2014 |
|
RU2613489C2 |
| СПОСОБ АМПЛИФИКАЦИИ И ИДЕНТИФИКАЦИИ НУКЛЕИНОВЫХ КИСЛОТ | 2019 |
|
RU2811465C2 |
| КОНЪЮГАТЫ АФФИННАЯ МОЛЕКУЛА-ОЛИГОНУКЛЕОТИД И ИХ ПРИМЕНЕНИЯ | 2017 |
|
RU2763554C2 |
| КОНЪЮГАТЫ АФФИННАЯ ЧАСТЬ-ОЛИГОНУКЛЕОТИД И ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2778754C2 |
| КРУПНОМАСШТАБНЫЕ МОНОКЛЕТОЧНЫЕ БИБЛИОТЕКИ ТРАНСКРИПТОМОВ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2019 |
|
RU2773318C2 |
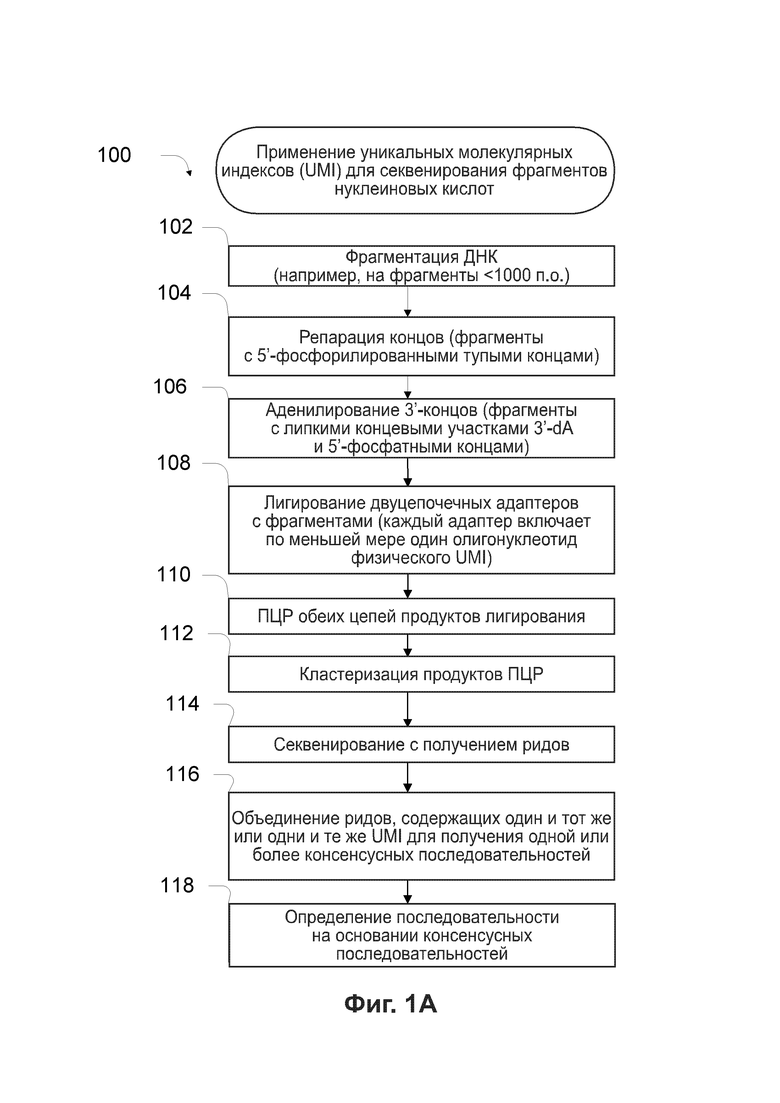
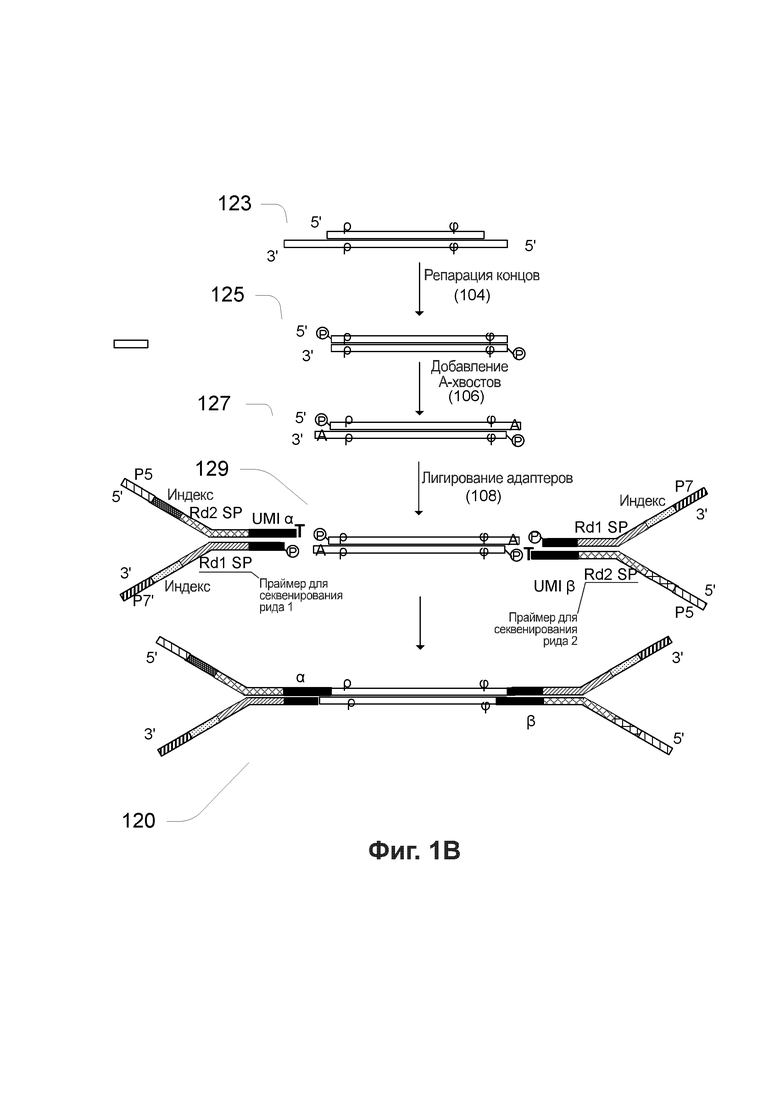
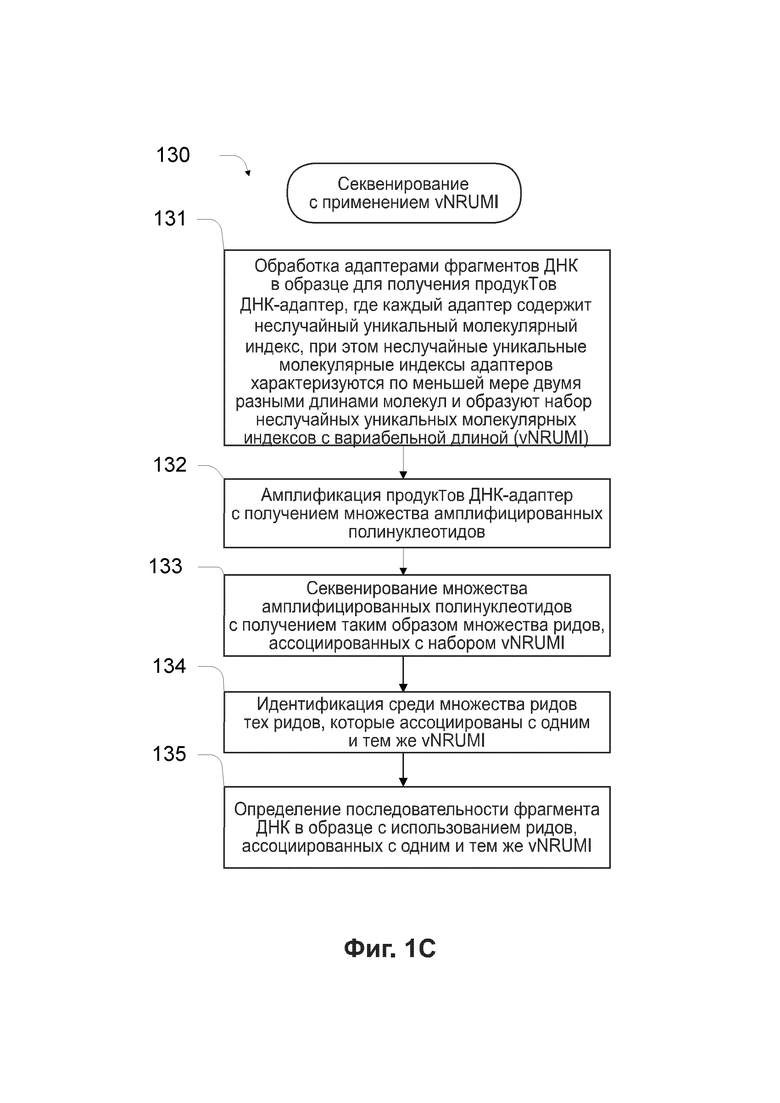
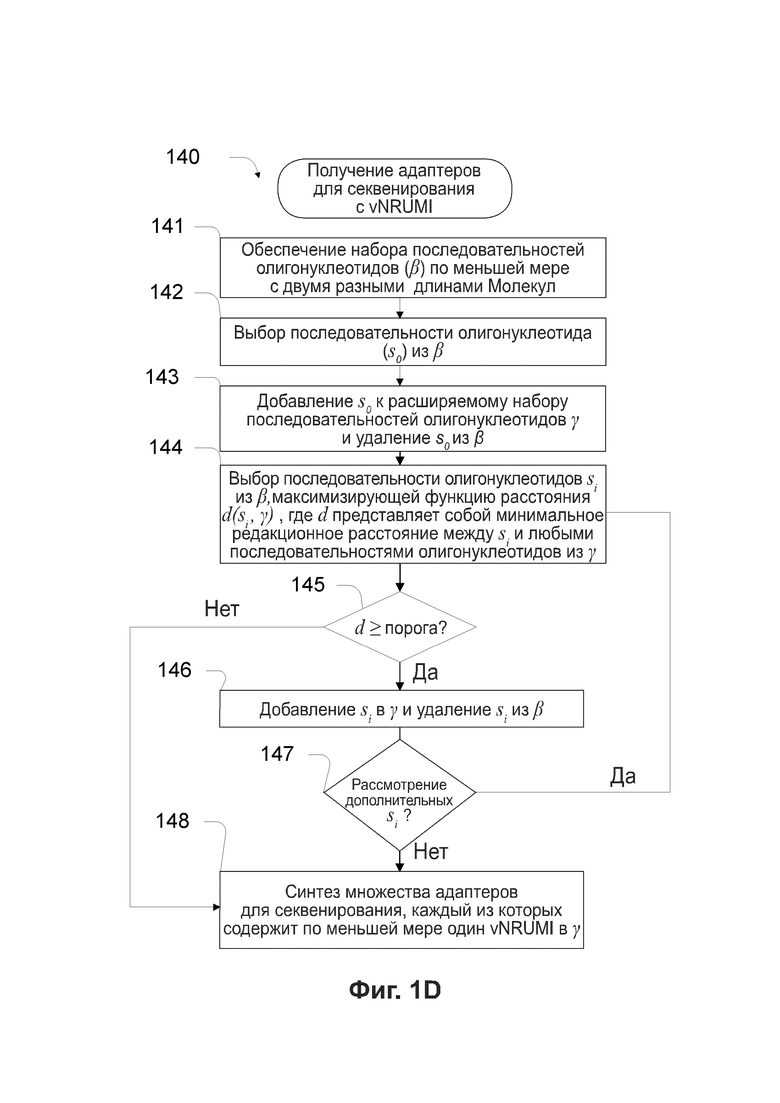


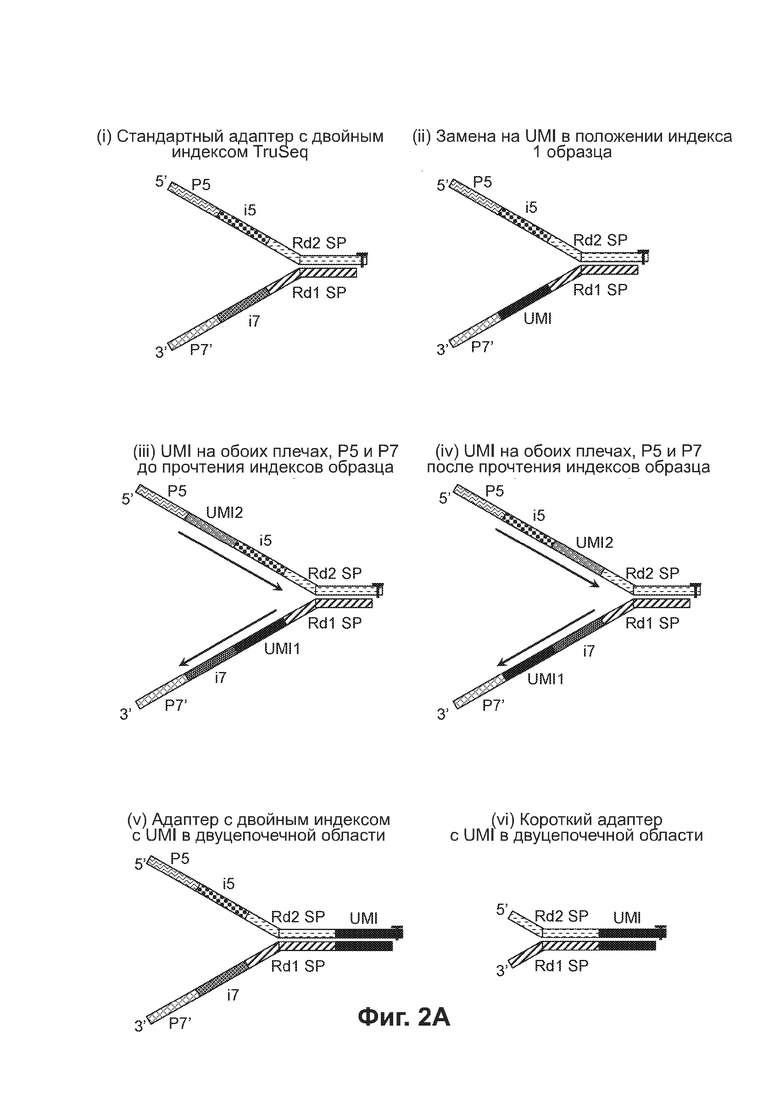
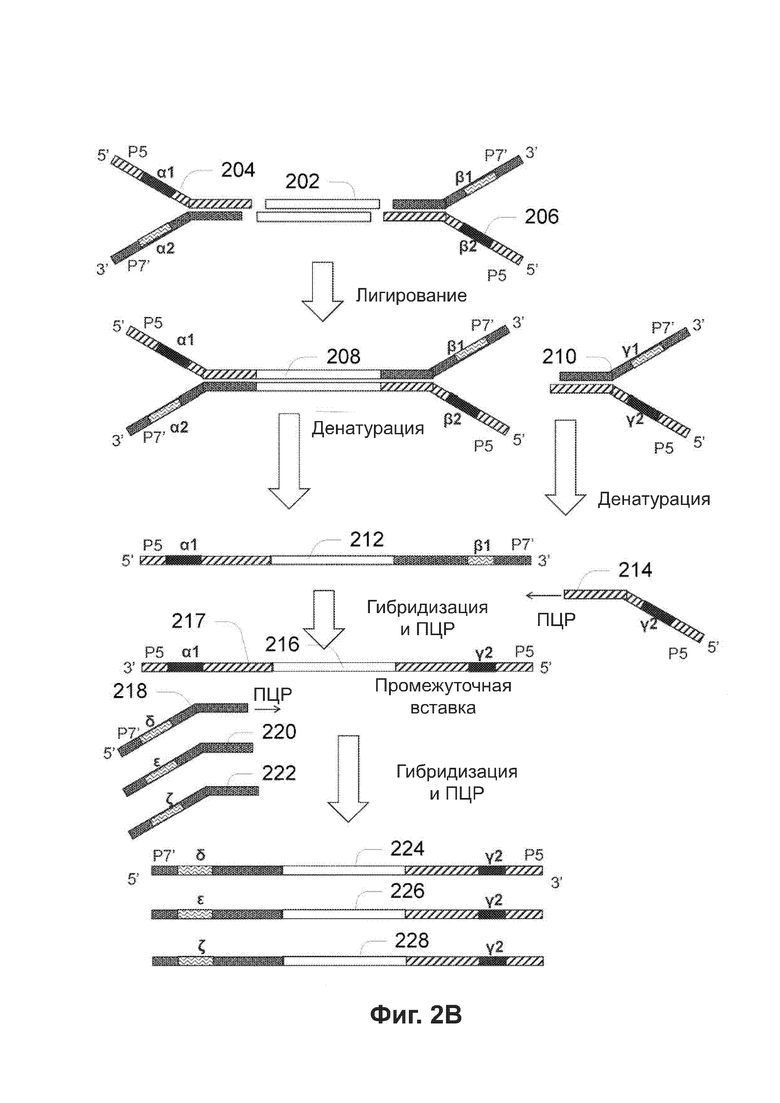
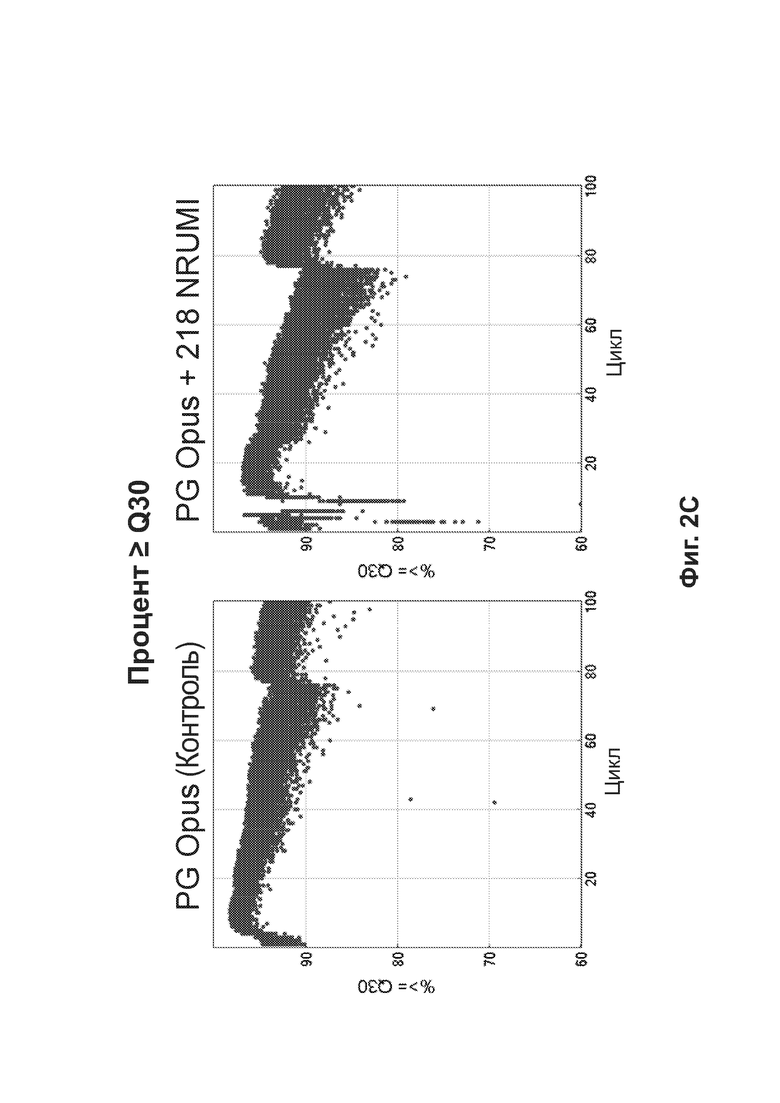
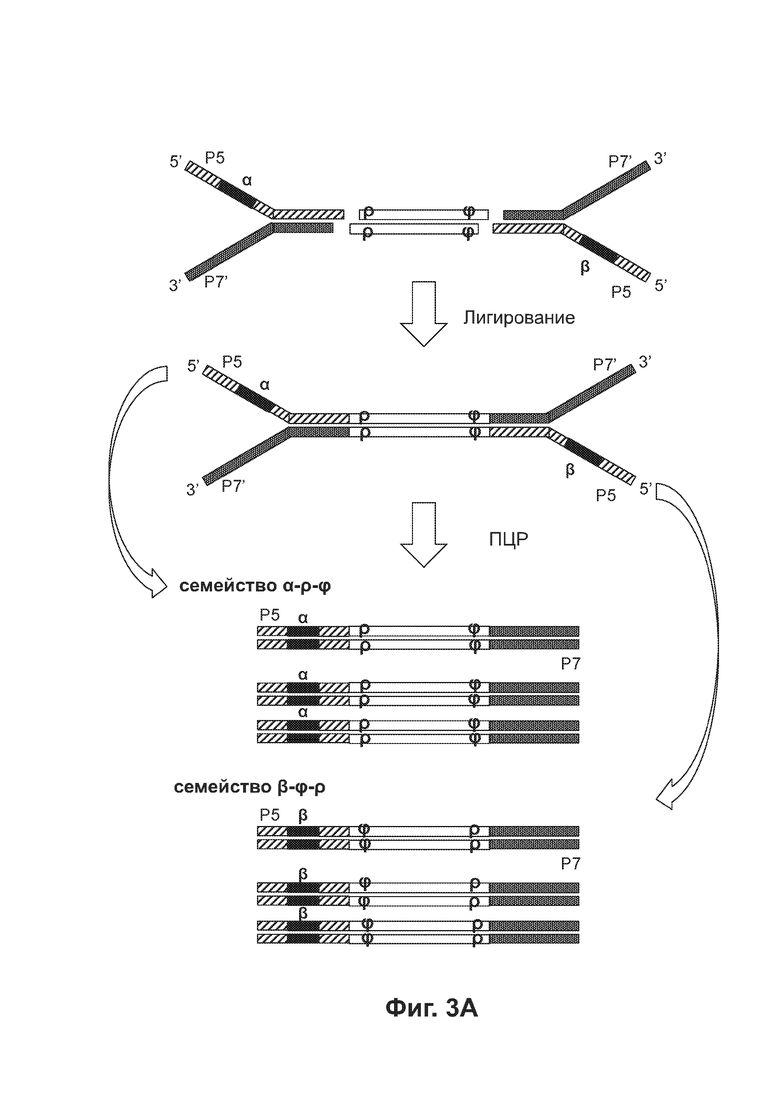
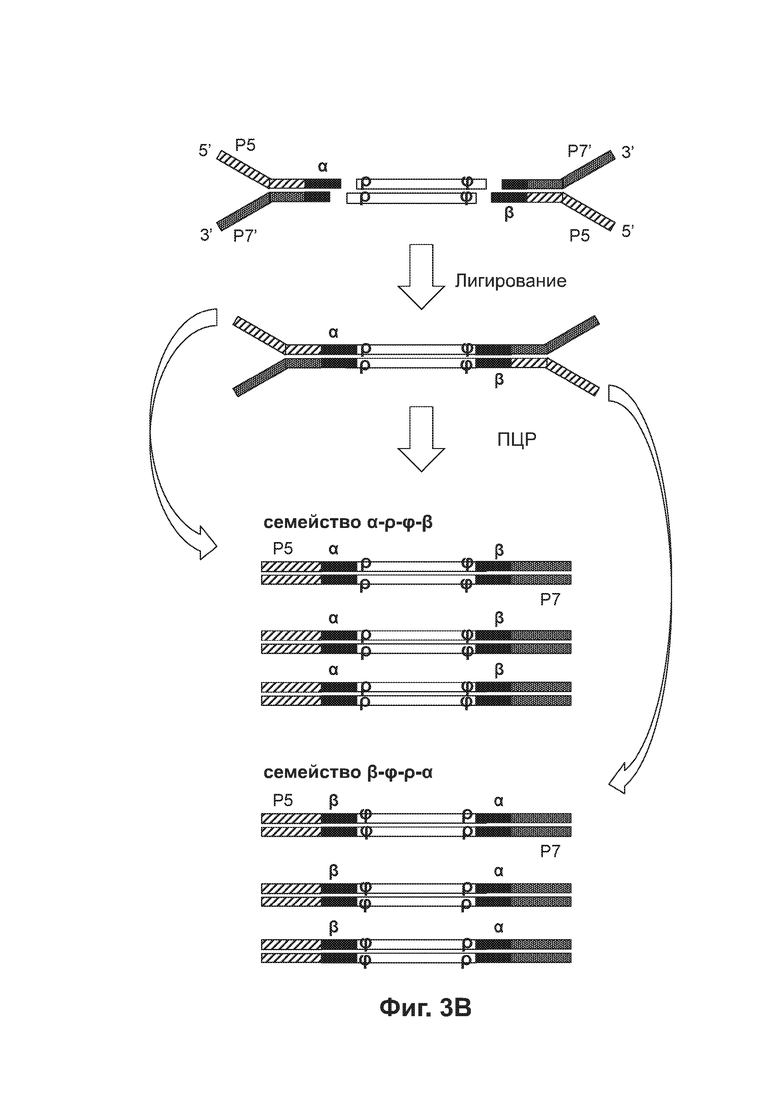
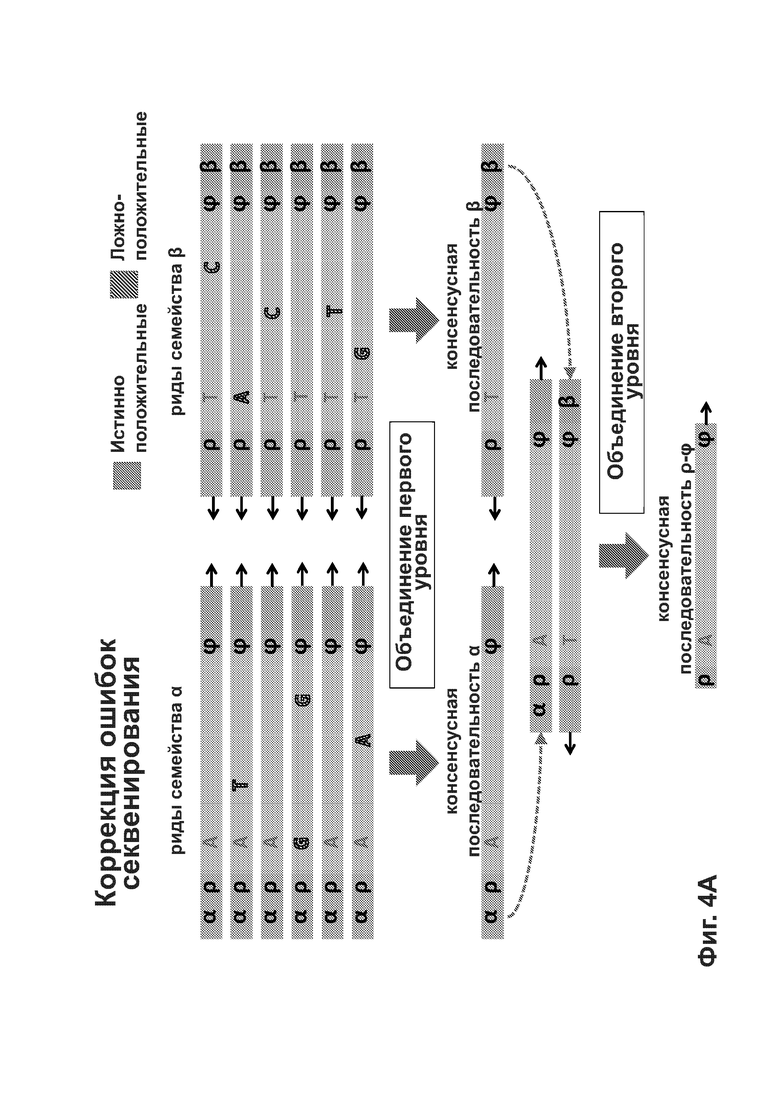
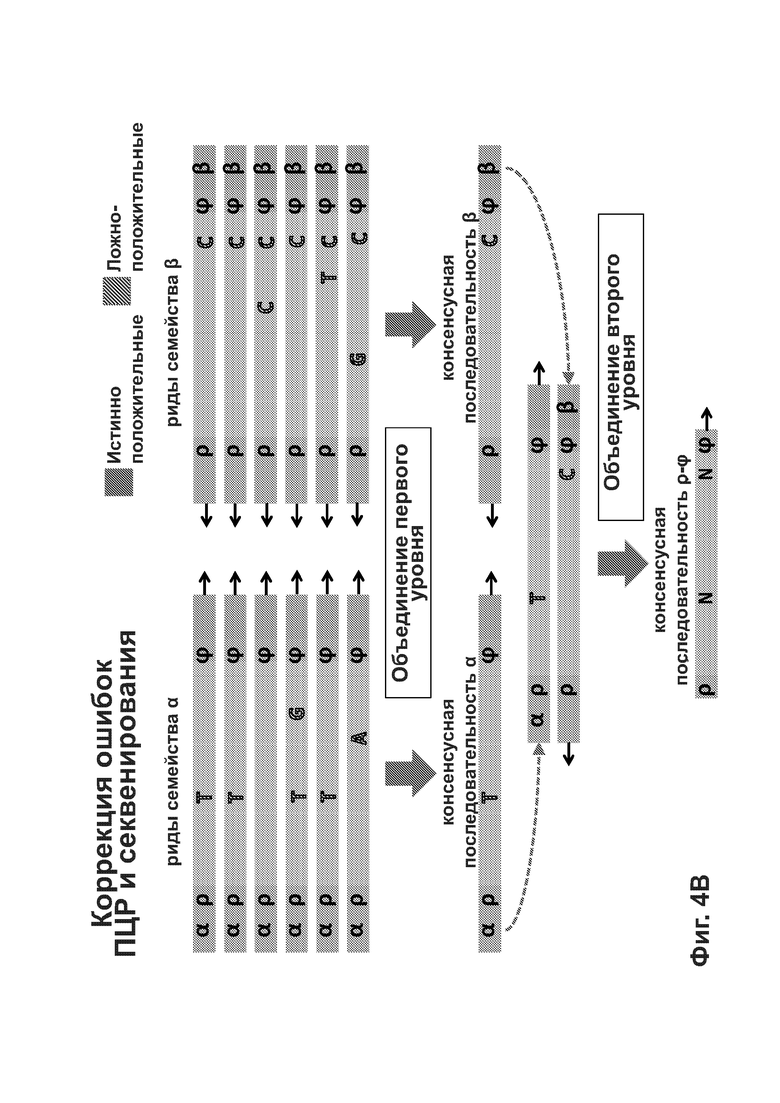
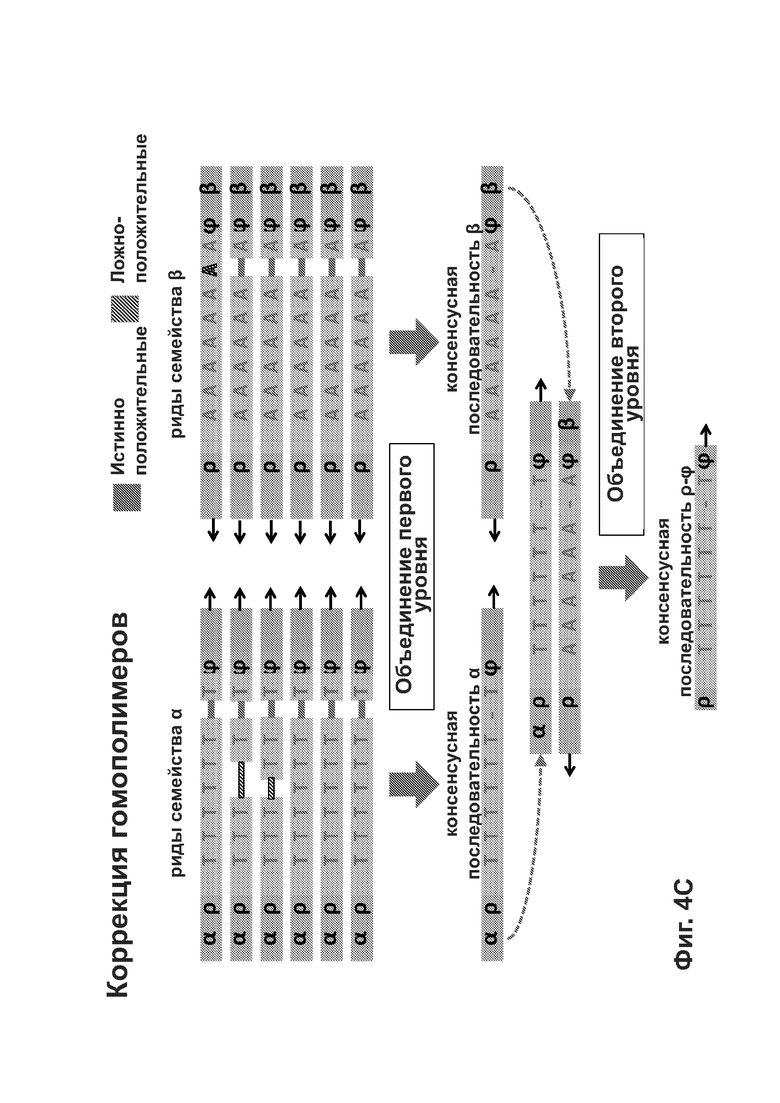
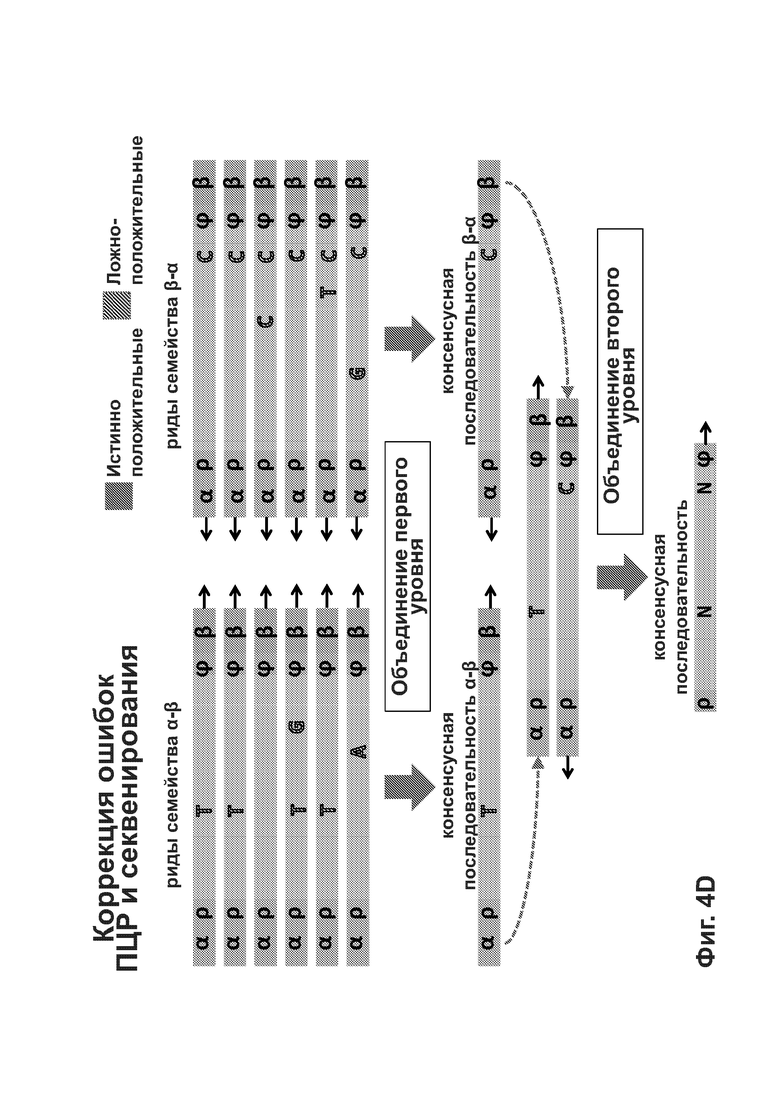
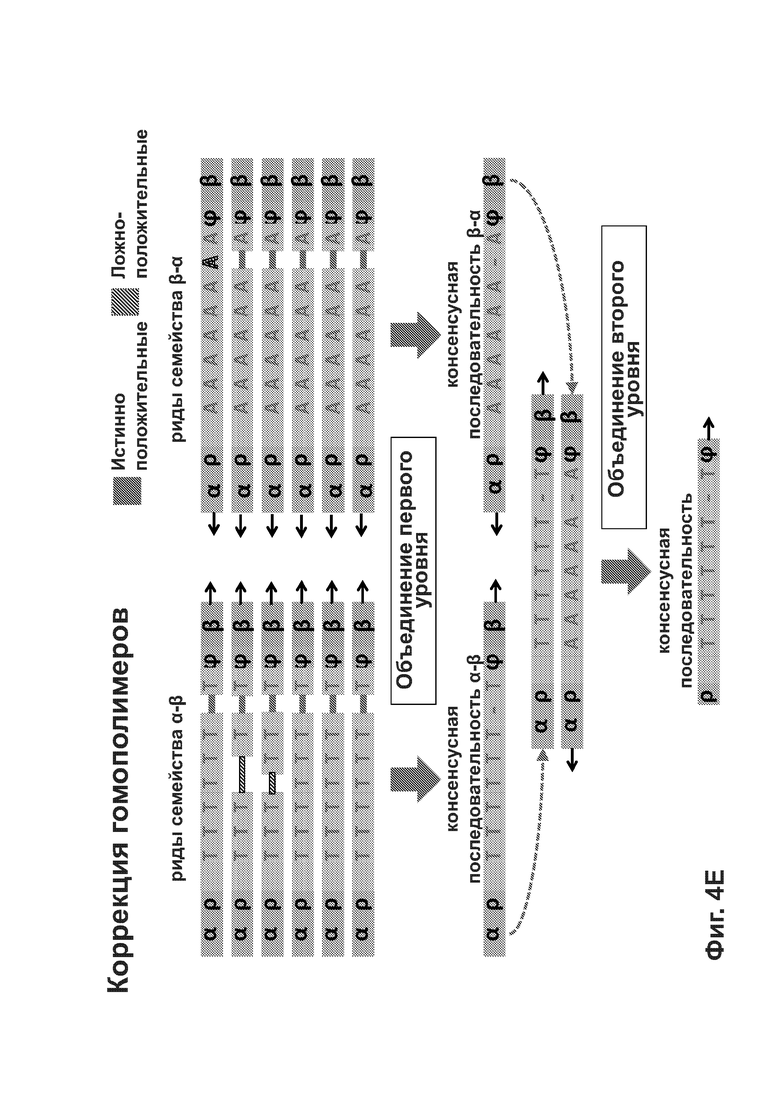
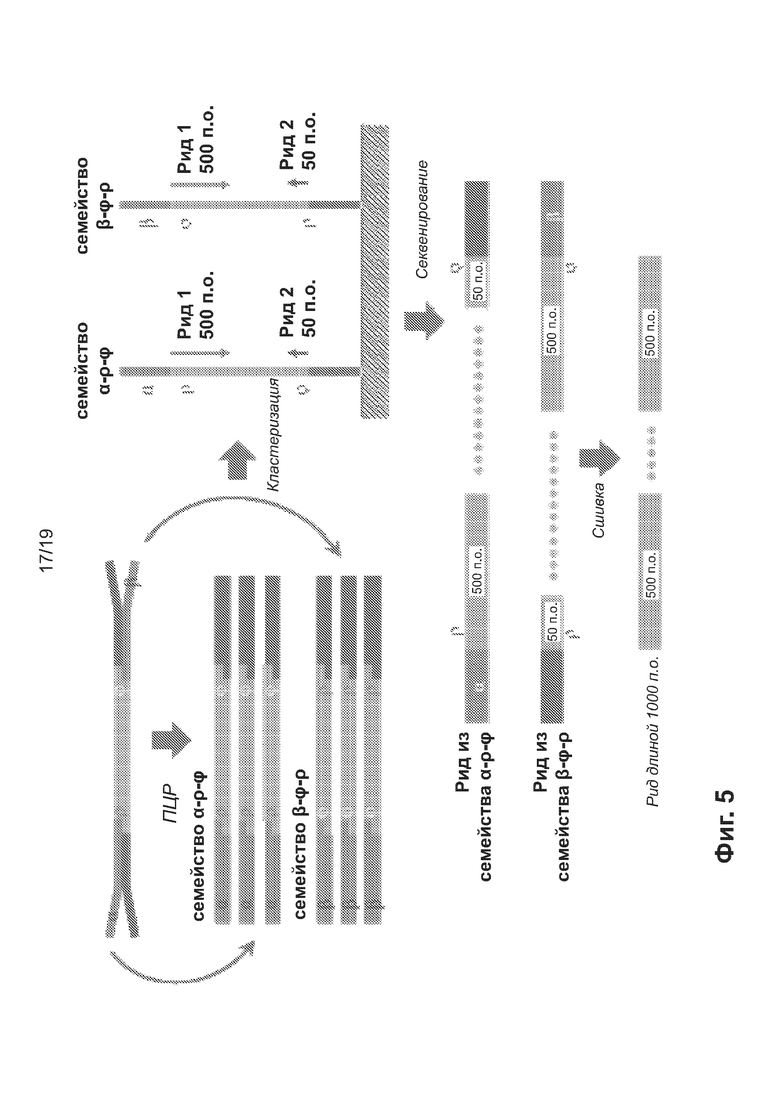
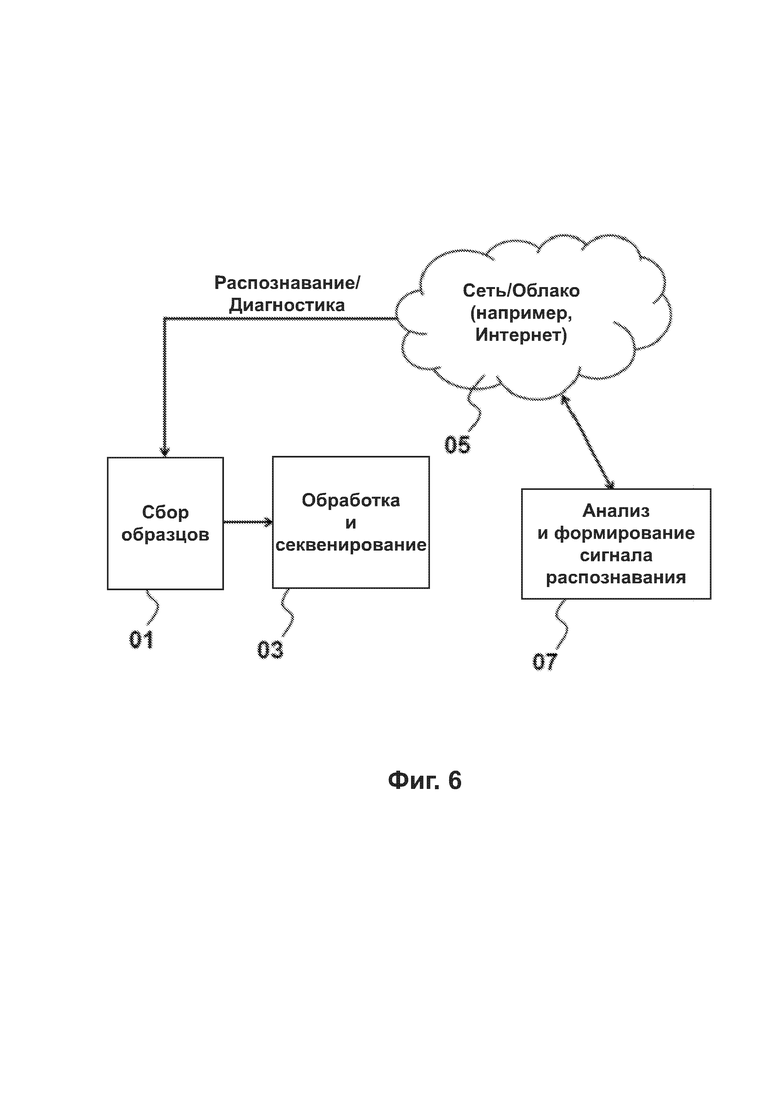
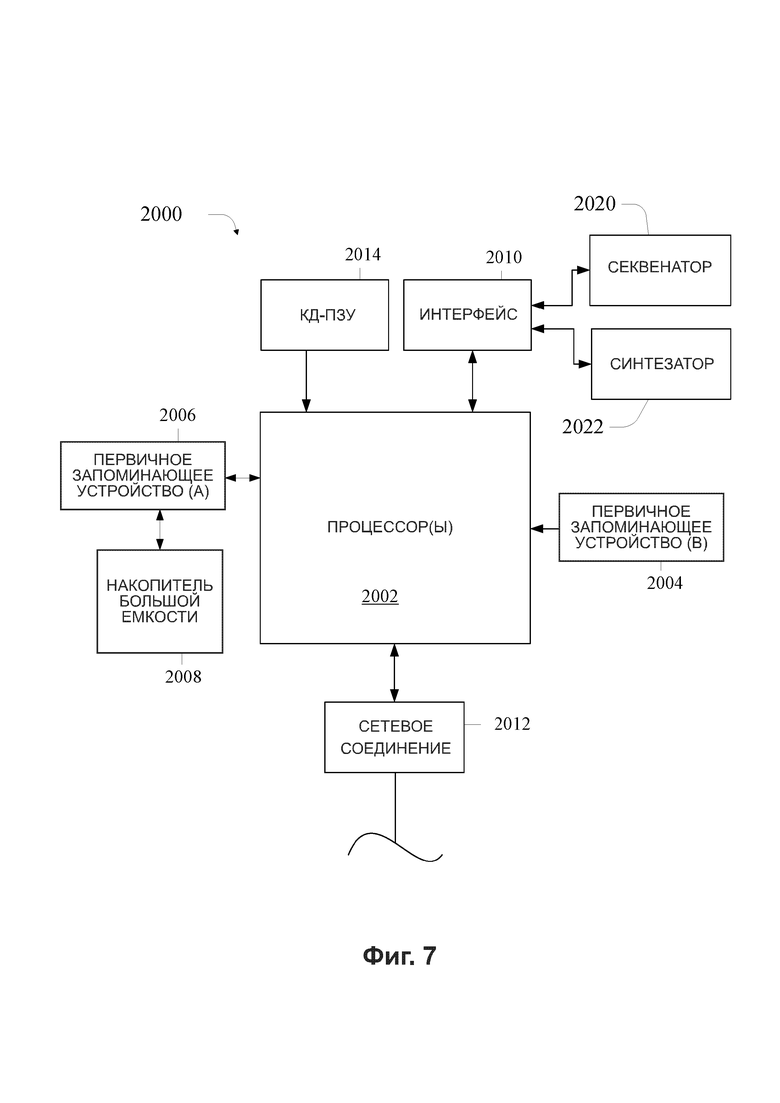
Группа изобретений относится к области биотехнологии. Предложен способ секвенирования молекул нуклеиновой кислоты (варианты), способ получения адаптеров для секвенирования, компьютерная система и компьютерный продукт для осуществления указанных способов. Способы секвенирования включают получение ДНК-адаптера, где каждый адаптер содержит неслучайный уникальный молекулярный индекс, при этом неслучайные уникальные молекулярные индексы адаптеров образуют набор неслучайных уникальных молекулярных индексов с вариабельной длиной, амплификацию ДНК-адаптера, секвенирование множества амплифицированных полинуклеотидов с получением множества ассоциированных с указанным набором ридов, идентификацию среди множества ридов, ассоциированных с одним и тем же набором ридов, получение для каждого рида показателей выравнивания для набора. Способ получения адаптеров включает обеспечение набора последовательностей олигонуклеотидов с разными длинами молекул, выбор поднабора последовательностей олигонуклеотидов из набора, где все редакционные расстояния между последовательностями олигонуклеотидов поднабора соответствуют пороговому значению, и поднабор последовательностей олигонуклеотидов образует набор неслучайных уникальных молекулярных индексов с вариабельной длиной, синтез множества адаптеров для секвенирования. Изобретения обеспечивают однозначную идентификацию каждой исходной молекулы ДНК в ходе процесса секвенирования. 7 н. и 36 з.п. ф-лы, 2 пр., 3 табл., 19 ил.
1. Способ секвенирования молекул нуклеиновой кислоты из образца, включающий
(a) обработку адаптерами фрагментов ДНК в указанном образце с получением продуктов, представляющих собой ДНК-адаптер,
где каждый адаптер содержит неслучайный уникальный молекулярный индекс, и
при этом неслучайные уникальные молекулярные индексы указанных адаптеров характеризуются по меньшей мере двумя разными длинами молекул и образуют набор неслучайных уникальных молекулярных индексов с вариабельной длиной (vNRUMI);
(b) амплификацию указанных продуктов ДНК-адаптер с получением множества амплифицированных полинуклеотидов;
(c) секвенирование указанного множества амплифицированных полинуклеотидов с получением таким образом множества ридов, ассоциированных с указанным набором vNRUMI;
(d) идентификацию среди указанного множества ридов тех ридов, которые ассоциированы с одним и тем же vNRUMI, включающую:
получение для каждого рида из указанного множества ридов показателей выравнивания для указанного набора vNRUMI, при этом каждый из показателей выравнивания является индикатором сходства между частичной последовательностью рида и vNRUMI, где указанная частичная последовательность находится в той области указанного рида, в которой предположительно расположены нуклеотиды, происходящие из указанного vNRUMI;
выбор для каждого рида из указанного множества ридов по меньшей мере одного vNRUMI из набора vNRUMI на основании показателей выравнивания; и
ассоциацию каждого рида из указанного множества ридов по меньшей мере с одним vNRUMI, выбранным для указанного рида;
(e) определение последовательности фрагмента ДНК в указанном образце с использованием ридов, ассоциированных с одним и тем же vNRUMI.
2. Способ по п. 1, отличающийся тем, что указанные показатели выравнивания основаны на совпадениях нуклеотидов и изменениях нуклеотидов между указанной частичной последовательностью рида и указанным vNRUMI.
3. Способ по п. 2, отличающийся тем, что указанные изменения нуклеотидов включают замены, добавления и делеции нуклеотидов.
4. Способ по п. 2, отличающийся тем, что каждый показатель выравнивания включает штраф за несовпадения в начале последовательности, но не включает штраф за несовпадения в конце указанной последовательности.
5. Способ по п. 4, отличающийся тем, что получение показателя выравнивания между ридом и vNRUMI включает:
(a) вычисление показателя выравнивания между vNRUMI и каждой из всех возможных префиксных последовательностей частичной последовательности указанного рида;
(b) вычисление показателя выравнивания между частичной последовательностью указанного рида и каждой из всех возможных префиксных последовательностей указанного vNRUMI; и
(c) получение максимального показателя выравнивания из показателей выравнивания, вычисленных согласно (a) и (b), в качестве показателя выравнивания между указанным ридом и указанным vNRUMI.
6. Способ по п. 1, отличающийся тем, что указанная частичная последовательность имеет длину, равную длине самого длинного vNRUMI в указанном наборе vNRUMI.
7. Способ по п. 6, отличающийся тем, что выбор указанного по меньшей мере одного vNRUMI из набора vNRUMI включает выбор vNRUMI с самым высоким значением показателя выравнивания в указанном наборе vNRUMI.
8. Способ по п. 6, отличающийся тем, что указанный по меньшей мере один vNRUMI включает два или более vNRUMI.
9. Способ по п. 8, дополнительно включающий выбор одного из двух или более vNRUMI в качестве одного и того же vNRUMI согласно (d) и (e).
10. Способ по п. 1, отличающийся тем, что адаптеры, которыми обрабатывают согласно (a), получают путем:
(i) обеспечения набора последовательностей олигонуклеотидов, обладающих по меньшей мере двумя разными длинами молекул;
(ii) выбора поднабора последовательностей олигонуклеотидов из набора последовательностей олигонуклеотидов, где все редакционные расстояния между последовательностями олигонуклеотидов указанного поднабора последовательностей олигонуклеотидов соответствуют пороговому значению, при этом указанный поднабор последовательностей олигонуклеотидов образует указанный набор vNRUMI; и
(iii) синтеза адаптеров, каждый из которых содержит двуцепочечную гибридизованную область, одноцепочечное 5’-плечо, одноцепочечное 3’-плечо и по меньшей мере один vNRUMI из указанного набора vNRUMI.
11. Способ по п. 10, отличающийся тем, что указанное пороговое значение равно 3.
12. Способ по п. 1, отличающийся тем, что указанный набор vNRUMI содержит vNRUMI из 6 нуклеотидов и vNRUMI из 7 нуклеотидов.
13. Способ по п. 1, отличающийся тем, что (e) включает объединение ридов, ассоциированных с одним и тем же vNRUMI, в группу с получением консенсусной последовательности нуклеотидов для последовательности указанного фрагмента ДНК в указанном образце.
14. Способ по п. 13, отличающийся тем, что указанную консенсусную последовательность нуклеотидов получают частично на основе показателей качества ридов.
15. Способ по п. 1, отличающийся тем, что (e) включает:
идентификацию среди ридов, ассоциированных с одним и тем же vNRUMI, ридов, которые характеризуются тем же положением или аналогичными положениями рида в референсной последовательности, и
определение последовательности указанного фрагмента ДНК с использованием ридов, которые (i) ассоциированы с одним и тем же vNRUMI и (ii) характеризуются тем же положением рида или аналогичными положениями рида в указанной референсной последовательности.
16. Способ по п. 1, отличающийся тем, что указанный набор vNRUMI включает не более чем 10 000 разных vNRUMI.
17. Способ по п. 16, отличающийся тем, что указанный набор vNRUMI включает не более чем 1000 разных vNRUMI.
18. Способ по п. 17, отличающийся тем, что указанный набор vNRUMI включает не более чем 200 разных vNRUMI.
19. Способ по п. 1, отличающийся тем, что обработка адаптерами указанных фрагментов ДНК в указанном образце включает обработку адаптерами обоих концов указанных фрагментов ДНК в указанном образце.
20. Способ получения адаптеров для секвенирования, включающий:
(a) обеспечение набора последовательностей олигонуклеотидов по меньшей мере с двумя разными длинами молекул;
(b) выбор поднабора последовательностей олигонуклеотидов из указанного набора последовательностей олигонуклеотидов, где все редакционные расстояния между последовательностями олигонуклеотидов указанного поднабора последовательностей олигонуклеотидов соответствуют пороговому значению, и указанный поднабор последовательностей олигонуклеотидов образует набор неслучайных уникальных молекулярных индексов с вариабельной длиной (vNRUMI), включающий:
(i) выбор последовательности олигонуклеотида из указанного набора последовательностей олигонуклеотидов;
(ii) добавление выбранного олигонуклеотида к расширяемому набору последовательностей олигонуклеотидов и удаление указанного выбранного олигонуклеотида из указанного набора последовательностей олигонуклеотидов для получения сокращенного набора последовательностей олигонуклеотидов;
(iii) выбор текущей последовательности олигонуклеотида из указанного сокращенного набора, которая максимизирует функцию расстояния, где функция расстояния представляет собой минимальное редакционное расстояние между указанной текущей последовательностью олигонуклеотида и любыми последовательностями олигонуклеотидов в указанном расширяемом наборе, причем указанная функция расстояния соответствует указанному пороговому значению;
(iv) добавление указанного текущего олигонуклеотида к указанному расширяемому набору и удаление указанного текущего олигонуклеотида из указанного сокращенного набора;
(v) повторение (iii) и (iv) по меньшей мере один раз; и
(vi) обеспечение указанного расширяемого набора в качестве указанного поднабора последовательностей олигонуклеотидов, образующих указанный набор vNRUMI; и
(c) синтез множества адаптеров для секвенирования, причем каждый адаптер для секвенирования содержит двуцепочечную гибридизованную область, одноцепочечное 5’-плечо, одноцепочечное 3’-плечо и по меньшей мере один vNRUMI из указанного набора vNRUMI.
21. Способ по п. 20, отличающийся тем, что (v) включает повторение (iii) и (iv) до тех пор, пока указанная функция расстояния не перестанет соответствовать указанному пороговому значению.
22. Способ по п. 20, отличающийся тем, что (v) включает повторение (iii) и (iv) до тех пор, пока указанный расширяемый набор не достигает заданного размера.
23. Способ по п. 20, отличающийся тем, что указанная текущая последовательность олигонуклеотида или последовательность олигонуклеотида в указанном расширяемом наборе короче самой длинной последовательности олигонуклеотидов в указанном наборе последовательностей олигонуклеотида, указанный способ дополнительно включает до осуществления (iii), (1) прибавление тиминового основания, или тиминового основания наряду с любым из четырех оснований к указанной текущей последовательности олигонуклеотида или к указанной последовательности олигонуклеотида в указанном расширяемом наборе, с формированием таким образом дополненной последовательности, имеющей такую же длину, что и указанная самая длинная последовательность олигонуклеотида в указанном наборе последовательностей олигонуклеотидов, и (2) использование указанной дополненной последовательности для вычисления минимального редакционного расстояния.
24. Способ по п. 20, отличающийся тем, что указанные редакционные расстояния представляют собой расстояния Левенштейна.
25. Способ по п. 20, отличающийся тем, что указанное пороговое значение равно трем.
26. Способ по п. 20, дополнительно включающий, до осуществления (b), удаление определенных последовательностей олигонуклеотидов из указанного набора последовательностей олигонуклеотидов с получением отфильтрованного набора последовательностей олигонуклеотидов; и обеспечение указанного отфильтрованного набора последовательностей олигонуклеотидов в качестве набора последовательностей олигонуклеотидов, из которого выбирают указанный поднабор.
27. Способ по п. 26, отличающийся тем, что указанные определенные последовательности олигонуклеотидов содержат последовательности олигонуклеотидов, включающие три или более последовательных идентичных оснований.
28. Способ по п. 26, отличающийся тем, что указанные определенные последовательности олигонуклеотидов содержат последовательности олигонуклеотидов, общее число гуаниновых и цитозиновых оснований в которых меньше 2, и последовательности олигонуклеотидов, общее число гуаниновых и цитозиновых оснований в которых больше 4.
29. Способ по п. 26, отличающийся тем, что указанные определенные последовательности олигонуклеотидов содержат последовательности олигонуклеотидов, включающие одно и то же основание в последних двух положениях.
30. Способ по п. 26, отличающийся тем, что указанные определенные последовательности олигонуклеотидов содержат последовательности олигонуклеотидов, включающие частичную последовательность, совпадающую со 3’-концом одного или более праймеров для секвенирования.
31. Способ по п. 26, отличающийся тем, что указанные определенные последовательности олигонуклеотидов содержат последовательности олигонуклеотидов, включающие тиминовое основание в последнем положении указанных последовательностей олигонуклеотидов.
32. Способ по п. 20, отличающийся тем, что указанный набор vNRUMI содержит vNRUMI из 6 нуклеотидов и vNRUMI из 7 нуклеотидов.
33. Способ секвенирования молекул нуклеиновой кислоты из образца, включающий
(a) обработку адаптерами фрагментов ДНК в указанном образце с получением продуктов, представляющих собой ДНК-адаптер,
где каждый адаптер содержит неслучайный уникальный молекулярный индекс, и
при этом неслучайные уникальные молекулярные индексы указанных адаптеров характеризуются по меньшей мере двумя разными длинами молекул и образуют набор неслучайных уникальных молекулярных индексов с вариабельной длиной (vNRUMI);
(b) амплификацию указанных продуктов ДНК-адаптер с получением множества амплифицированных полинуклеотидов;
(c) секвенирование указанного множества амплифицированных полинуклеотидов с получением таким образом множества ридов, ассоциированных с указанным набором vNRUMI; и
(d) идентификацию среди указанного множества ридов тех ридов, которые ассоциированы с одним и тем же vNRUMI, включающую:
получение для каждого рида из указанного множества ридов показателей выравнивания для указанного набора vNRUMI, при этом каждый из показателей выравнивания является индикатором сходства между частичной последовательностью рида и vNRUMI, где указанная частичная последовательность находится в той области указанного рида, в которой предположительно расположены нуклеотиды, происходящие из указанного vNRUMI, и при этом указанная частичная последовательность имеет длину, которая равняется длине самого длинного vNRUMI в указанном наборе vNRUMI.
34. Способ по п. 33, дополнительно включающий осуществление подсчета ридов, ассоциированных с одним и тем же vNRUMI.
35. Способ секвенирования молекул нуклеиновой кислоты из образца, включающий
(a) обработку адаптерами фрагментов ДНК в указанном образце с получением продуктов, представляющих собой ДНК-адаптер,
где каждый адаптер содержит уникальный молекулярный индекс (UMI), и
при этом уникальные молекулярные индексы (UMI) указанных адаптеров характеризуются по меньшей мере двумя разными длинами молекул и образуют набор уникальных молекулярных индексов (vUMI) с вариабельной длиной;
(b) амплификацию указанных продуктов ДНК-адаптер с получением множества амплифицированных полинуклеотидов;
(c) секвенирование указанного множества амплифицированных полинуклеотидов с получением таким образом множества ридов, ассоциированных с указанным набором vUMI; и
(d) идентификацию среди указанного множества ридов тех ридов, которые ассоциированы с одним и тем же уникальным молекулярным индексом с вариабельной длиной (vUMI), включающую:
получение для каждого рида из указанного множества ридов показателей выравнивания для указанного набора vUMI, при этом каждый из показателей выравнивания является индикатором сходства между частичной последовательностью рида и vUMI, где указанная частичная последовательность находится в той области указанного рида, в которой предположительно расположены нуклеотиды, происходящие из указанного vUMI;
выбор для каждого рида из указанного множества ридов по меньшей мере одного vUMI из набора vUMI на основании показателей выравнивания; и
ассоциацию каждого рида из указанного множества ридов по меньшей мере с одним vUMI, выбранным для указанного рида.
36. Способ по п. 35, дополнительно включающий определение последовательности фрагмента ДНК в указанном образце с использованием ридов, ассоциированных с одним и тем же vUMI.
37. Способ по п. 35, дополнительно включающий осуществление подсчета ридов, ассоциированных с одними и теми же vUMI.
38. Способ секвенирования молекул нуклеиновой кислоты из образца, включающий
(a) обработку адаптерами фрагментов ДНК в указанном образце с получением продуктов ДНК-адаптер, при этом каждый адаптер содержит уникальный молекулярный индекс (UMI) из набора уникальных молекулярных индексов (UMI);
(b) амплификацию указанных продуктов ДНК-адаптер с получением множества амплифицированных полинуклеотидов;
(c) секвенирование указанного множества амплифицированных полинуклеотидов с получением таким образом множества ридов, ассоциированных с указанным набором UMI;
(d) получение для каждого рида из указанного множества ридов показателей выравнивания для указанного набора UMI, при этом каждый из полученных показателей является индикатором сходства между частичной последовательностью рида и UMI;
(e) идентификацию среди указанного множества ридов тех ридов, которые ассоциированы с одним и тем же UMI, с использованием указанных показателей выравнивания, включающую:
получение для каждого рида из указанного множества ридов показателей выравнивания для указанного набора UMI, при этом каждый из показателей выравнивания является индикатором сходства между частичной последовательностью рида и UMI, где указанная частичная последовательность находится в той области указанного рида, в которой предположительно расположены нуклеотиды, происходящие из указанного UMI;
выбор для каждого рида из указанного множества ридов по меньшей мере одного UMI из набора UMI на основании показателей выравнивания; и
ассоциацию каждого рида из указанного множества ридов по меньшей мере с одним UMI, выбранным для указанного рида; и
(e) определение последовательности фрагмента ДНК в указанном образце с использованием ридов, ассоциированных с одним и тем же UMI.
39. Способ по п. 38, отличающийся тем, что указанные показатели выравнивания основаны на совпадениях нуклеотидов и изменениях нуклеотидов между частичной последовательностью указанного рида и указанным UMI.
40. Способ по п. 39, причем каждый показатель выравнивания включает штраф за несовпадения в начале последовательности, но не включает штраф за несовпадения в конце указанной последовательности.
41. Способ по п. 38, отличающийся тем, что указанный набор UMI содержит UMI по меньшей мере с двумя разными длинами молекул.
42. Компьютерный программный продукт для секвенирования молекул нуклеиновой кислоты из образца, при этом указанный компьютерный программный продукт содержит энергонезависимый машиночитаемый носитель, в котором хранится программный код, который при исполнении одним или более процессорами компьютерной системы приводит к осуществлению указанной компьютерной системой способа по любому из пп. 1-41.
43. Компьютерная система для секвенирования молекул нуклеиновой кислоты из образца, включающая:
один или более процессоров;
системную память; и
один или более машиночитаемых носителей для хранения данных с хранящимися на них машиноисполняемыми инструкциями, обеспечивающими осуществление указанной компьютерной системой способа по любому из пп. 1-41.
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Токарный резец | 1924 |
|
SU2016A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| ПРЯМОЙ ЗАХВАТ, АМПЛИФИКАЦИЯ И СЕКВЕНИРОВАНИЕ ДНК-МИШЕНИ С ИСПОЛЬЗОВАНИЕМ ИММОБИЛИЗИРОВАННЫХ ПРАЙМЕРОВ | 2011 |
|
RU2565550C2 |

