Настоящее изобретение относится к фармации, химии, медицине, биологии, а именно, к определению компонентов капреомицина и его примесей. Может быть использовано в фармацевтической, химической или смежных отраслях.
Уровень техники
Лекарственные препараты капреомицина используются для лечения туберкулеза с множественной лекарственной устойчивостью. Капреомицин считается препаратом второй линии из-за его серьезных нежелательных эффектов, таких как гепатотоксичность и нефротоксичность, потеря слуха и аллергическая реакция [1]. Согласно литературным данным токсичность препаратов капреомицина в значительной степени зависит от содержащихся родственных примесей. При сравнении острой токсичности и фармакокинетических профилей двух препаратов капреомицина с различным содержанием примесей показано, что лекарственное средство с меньшим количеством примесей демонстрирует гораздо более низкую токсичность. [2]
Хроматографические методы определения продуктов деструкции антибиотиков широко внедрены в практику оценки качества лекарственных средств. В настоящее время наиболее распространена высокоэффективная жидкостной хроматографии (ВЭЖХ).
Наиболее сложными соединениями при разработке методик определения родственных примесей являются природные многокомпонентные антибиотики, как например, капреомицин.
Монографии на капреомицина сульфат представлены в Фармакопее США (USP) [3] и Международной фармакопее (IP) [4]. Методика из монографии USP предназначена для определения компонентного состава капреомицина и не предполагает определения содержания примесей. Методика монографии IP предназначена для определения, как компонентного состава, так и родственных примесей, однако, как было обнаружено, не является достаточно эффективной для определения ряда примесей. Также является очень ресурсозатратной в связи с большим расходом реагентов при проведении анализа.
Из уровня техники известен способ, разработанный Mallampati с соавторами [5], на основе ион-парной высокоэффективной жидкостной хроматографии, в которой в качестве ион-парного реагента использовали натрия 1-гексансульфонат, в данной работе было обнаружено только одиннадцать примесей, что не позволяет оценить содержание примесей. Кроме того, данная методика также является очень ресурсозатратной в связи с большим расходом реагентов при проведении анализа
В работе Liu и сотр. [6] предложена методика определения четырех основных примесей капреомицина на основе методики S. Mallampati, а также методика полупрепаративного выделения примесей на основе технологии хроматографии гидрофобных взаимодействий (HILIC). В данной работе были выделены и проанализированы основные примеси, полученные целенаправленной деструкцией отдельновыделенных компонентов капреомицина, но эффективность выделения других примесей была невысока, кроме того в данной работе было показано, что одна из основных примесей плохо отделяется в аналитической методике от одного из основных пиков, как и в работе S. Mallampati.
В статье Chopra и сотр. [7] был произведен анализ с помощью двухступенчатой хроматографии, совмещенной с масс-спектрометрией, который позволил определить структуру 14 (четырнадцати) примесей, однако данная методика слишком сложна, требовательна к оборудованию и, вследствие этого, не может быть использована для рутинных анализов экспертизы качества субстанции и препаратов капреомицина.
Аналогом заявленного способа выбран известный из уровня техники способ определения ванкомицина и его родственных примесей, где используют ультраэффективную жидкостную хроматографию (далее - УВЭЖХ) [8], Недостатками известного способа является то, что из-за полярности капреомицина, использовать данный способ невозможно в связи со слишком высокой элюирующей силой подвижной фазы.
В этой связи представляется актуальной разработка методики одновременного определения компонентов капреомицина и его родственных примесейей с использованием колонок, предназначенных для УВЭЖХ.
Технической задачей является разработка способа одновременного определения содержания основных компонентов капреомицина (IA, IB, IIA, IIB) и его родственных примесей с применением ион-парной УВЭЖХ хроматографии при использовании хроматографической колонки с частицами размером менее 2 мкм для повышения эффективности и разделительной способности хроматографической системы
Раскрытие сущности изобретения
Технической задачей является разработка точного и чувствительного способа одновременного определения компонентов капреомицина и его родственных примесей.
Техническим результатом является то, что был разработан способ определения компонентов капреомицина и его родственных примесей, характеризующийся чувствительностью и точностью.
Технический результат заявленного технического решения достигается благодаря тому, что используют:
- ультра эффективную жидкостную хроматографию (далее - УЭ2УХ) с использованием колонки с размером частиц 1,7 мкм:
- подвижную фазу определенного качественного и количественного состава при низкой скорости потока:
- температуру термостатирования колонки 70°С:
Способ определения капреомицина и его примесей с использованием ультраэффективной жидкостной хроматографии (УВЭЖХ) и характеризующийся тем, что используют раствор 1 с рН 9,8 и концентрацией 50 мМ и раствор 2 с рН 1,4 и концентрацией 50 мМ, далее раствор 1 и раствор 2 объединяют в соотношении 1:1 и разводят пятикратно, фильтруют, далее хроматографируют при двухфазном градиентом элюировании с использованием двухкомпонентной подвижной фазы со скоростью ее потока 0,1 мл/мин до температуры 70°С, а далее скорость потока двухкомпонентной подвижной фазы увеличивают до 0,2 мл/мин при температуре 70°С, где длина волны детектора 268 нм.
Способ, отличающийся тем, что раствор 1, представляет собой смесь раствора капреомицина 2 мг/мл и водного раствора аммиака 32% в соотношении 1000:3,23 выдержанный при 80°С в течении 24 часов.
Способ, отличающийся тем, что раствор 2, представляет собой смесь раствора капреомицина 2 мг/мл и трифторуксусной кислоты в соотношении 1000:3,83 выдержанный при 80°С в течении 24 часов.
Способ, отличающийся тем, что двухкомпонентной подвижной фаза состоит из фазы А и фазы Б.
Способ, отличающийся тем, что фаза А представляет собой композицию буферного раствора с рН 2,3 и ацетонитрила в объемном соотношении 95:5.
Способ, отличающийся тем, что фаза Б представляет собой композицию буферного раствора с рН 2,3 и ацетонитрила в объемном соотношении 90:10.
Способ, отличающийся тем, что буферный раствор представляет собой раствор, стоящий из раствора натрия дигидрофосфата и раствор натрия гексансульфонат и доведенные концентрированной фосфорной кислотой.
Полипептидный антибиотик капреомицин, продуцируемый Streptomyces capreolus, выделен в 1960 году [9] и используется как противотуберкулезное средство.
Химическое название капреомицина - (3S)-3,6-Диамино-N-[[(2S,5S,8E,11S,15S)-15-амино-11-[(4R)-2-амино-3,4,5,6-тетрагидропиримидин-4-ил]-8-[(карбамоиламино)метилиден]-2-(гидроксиметил)-3,6,9,12,16-пентаоксо-1,4,7,10,13-пентаазациклогексадец-5-ил]метил]гексанамид. Капреомицин представляет собой смесь четырех изоформ: IA, IB, IIA и IIB в соотношении примерно 25%, 67%, 3% и 6% соответственно [10]. Молекула капреомицина состоит из циклического пентапептида, содержащего два остатка 2,3-диаминопропионовой кислоты, один остаток уреидодегидроаланина, один остаток L-капреомицидина, один остаток серина (для IA и IIA) или аланина (для IB и IIB), а также остаток лизина в боковой цепи (для IA и IB), которые проявляют сильные основные свойства и обладают высокой полярностью. Например, циклический гуанидин, расположенный на боковой цепи L-капреомицидина, является одним из самых сильных известных органических оснований (pKa (14). [11]
Капреомицин - белое вещество. Растворим в воде с образованием бесцветного раствора. Практически нерастворим в большинстве органических растворителей. Применяется в форме капреомицина сульфата, характеризующегося следующей химической структурой:
Брутто формула капреомицина IA: C25H44N14O8 [11]. Молекулярная масса компонента IA: 668,7.
В связи с сильными основными свойствами и высокой полярностью капреомицина определение примесей в нем в значительной степени затруднено. Основные примеси капреомицина: стерео- и цис-транс- изомеры его изоформ [6]. Кроме того, примеси капреомицина могут быть результатами перестановки структурных элементов, заменой аминокислотных остатков и добавлением дополнительных групп [7].
В качестве объекта исследований использовали капреомицин сульфат в форме порошка, из которого были приготовлены растворы, используемые для анализа.
Пробоподготовку проводили в следующих условиях. Изготовили исходный водный раствор капреомицина с концентрацией 2 мг/мл. Для анализа примесей, образующихся в ходе деградации в щелочной или кислотной среде, приготовили растворы (см. таблицу 1):
Раствор 1 (смесь) готовили из исходного раствора капреомицина с концентрацией 2 мг/мл и добавления к нему водного раствора аммиака 32% (32% NH3 вод.р-р) в соотношении 1000:3,23, выдержанный при 80 °С в течении 24 часов, где концентрация в полученном растворе 50 мМ и pH 9,8.
Раствор 2 (смесь) готовили из исходного раствора капреомицина с концентрацией 2 мг/мл и добавления к нему трифторуксусной кислоты (CF3COOH) в соотношении 1000:3,83, выдержанный при 80 °С в течении 24 часов, где концентрация в полученном растворе 50 мМ и pH 1,4.
Для проверки разрешающей способности, готовили раствор 3, для чего смешивали раствор 1 и раствор 2 в равном соотношении 1:1 и в дальнейшем смесь растворов разбавляли водой в 5 раз. После приготовления все растворы пропускали через фильтр с диаметром пор 0,45 мкм, переносили в хроматографические виалы и помещали в автоматический пробоотборник хроматографа.
Испытания проводили на жидкостном хроматографе, предназначенном для работы в режиме обычной высокоэффективной жидкостной хроматографии (ВЭЖХ) и оборудованном следующими модулями: градиентным четырехканальным насосом, обладающим возможностью производить смешение четырех различных элюентов с максимальной скоростью потока 5 мл/мин с максимальным давление 400 бар или 10 мл/мин с максимальным давлением 200 бар; диодно-матричным детектором с диапазоном длин волн 190-800 нм; термостатируемым колоночным отделением.
Разделение проводили с использованием двухфазного градиентного режима элюирования, со следующим составом подвижной фазы:
Компонент подвижной фазы А, представляет собой композицию буферного раствора с рН 2,3 и ацетонитрила в объемном соотношении 95:5.
Компонент подвижной фазы Б, представляет собой композицию буферного раствора с рН 2,3 и ацетонитрила в объемном соотношении 90:10.
Буферный раствор для подвижных фаз А и Б представляет собой раствор 28 г/л (200 мМ) натрия дигидрофосфата и 4,7 г/л (25 мМ) натрия гексансульфоната в очищенной воде, со значением рН 2,3, доведенным с помощью концентрированной фосфорной кислоты.
Для проведения ультраэффективной жидкостной хроматографии на хроматографе, при давлении не более 400 бар, температуру термостатирования хроматографической колонки увеличили до 70 °C. Прогрев колонки до указанной температуры проводили при скорости потока элюента (~ 0,1 мл/мин). После прогрева колонки, что подтверждается установлением постоянного давления, скорость потока увеличивали до 0,2 мл/мин с сохранением температуры 70 °C и далее колонка уравновешивалась начальным составом подвижной фазы А (100%) до установления постоянного давления и выравнивания базовой линии.
Для реализации заявленного способа была использована колонка BEH C18 2,1 x 150 мм с размером частиц сорбента 1,7 мкм.
Условия хроматографирования:
- Объем вводимой пробы: 5 мкл.
- Скорость потока двухкомпонентной подвижной фазы: 0,2 мл/мин
- Температура колонки: 70°С
- Длина волны детектора: 268 нм
- Программа градиента:
На первой стадии с 0 по 80 минуту происходит градиентное элюирование всех компонентов и примесей капреомицина. Вторая и третья стадия с 80 по 95 минуту предназначены для промывки колонки и сохранения эффективности колонки при последовательных вколах.
Относительный компонентный состав рассчитывали по пропорциональному соотношению площадей четырех пиков капреомицина. Расчет содержания примесей проводили методом нормализации от суммы площадей всех пиков на хроматограмме.
Краткое описание чертежей и иных материалов (Приложения 1-10)
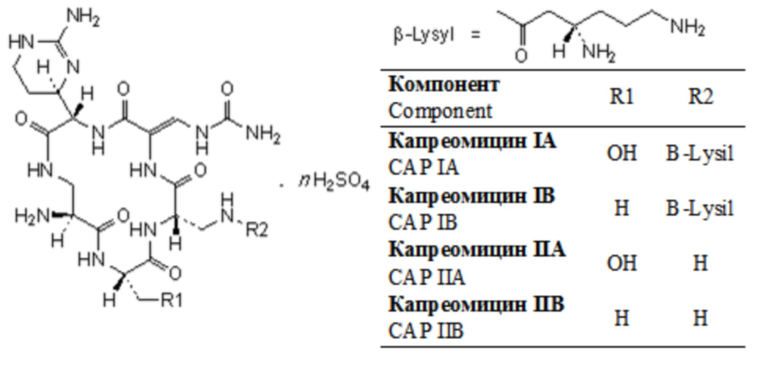
Фиг. 1. Хроматограмма исходного раствора субстанции капреомицина, полученная способом определения компонентов и родственных примесей капреомицина.
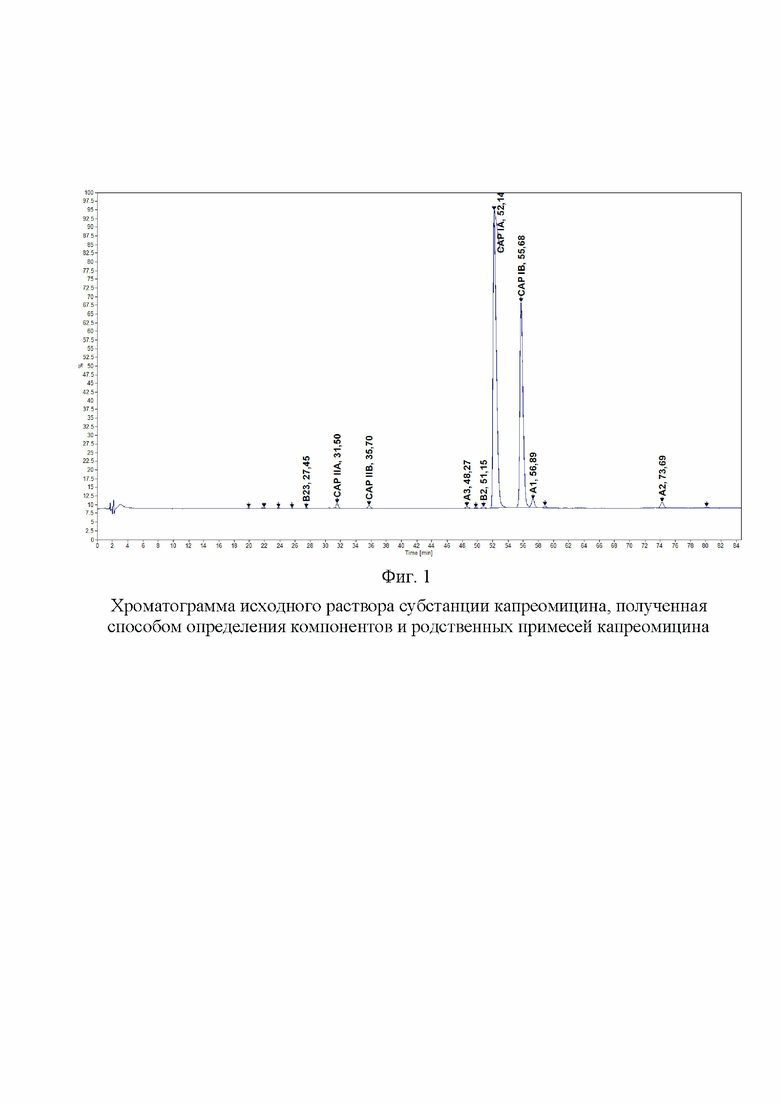
Фиг. 2. Хроматограмма раствора 1 (щелочное разложение), полученная способом определения компонентов и родственных примесей капреомицина
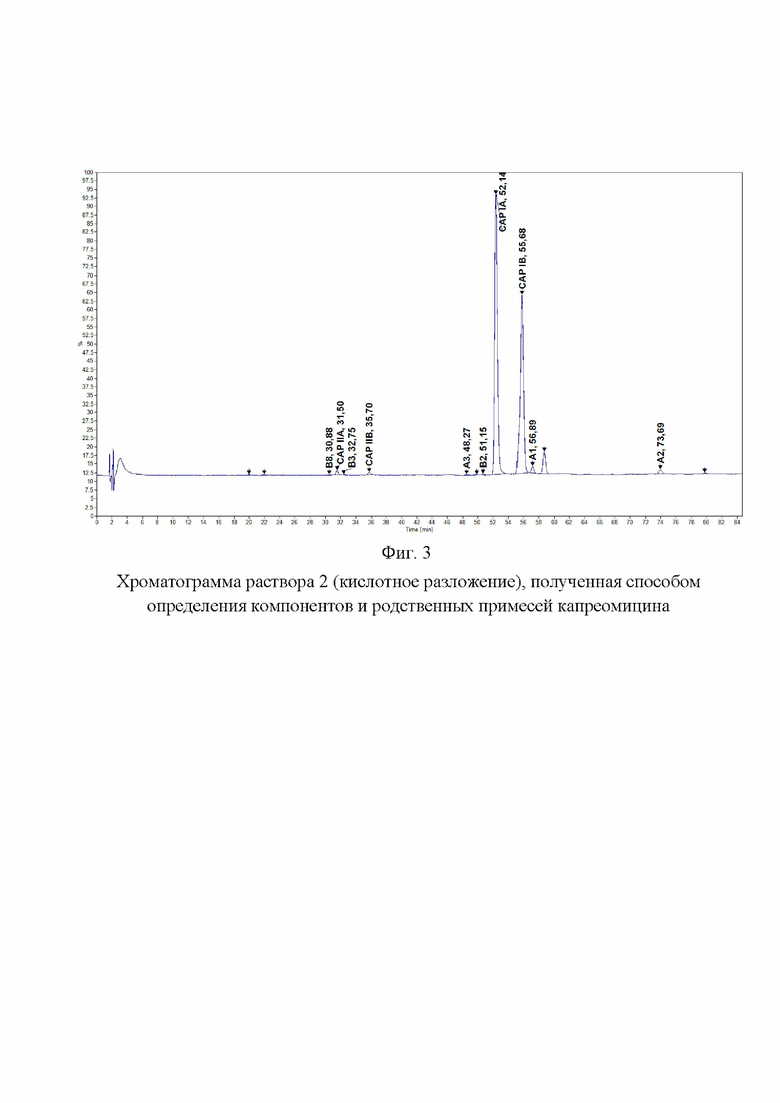
Фиг. 3. Хроматограмма раствора 2 (кислотное разложение), полученная способом определения компонентов и родственных примесей капреомицина.
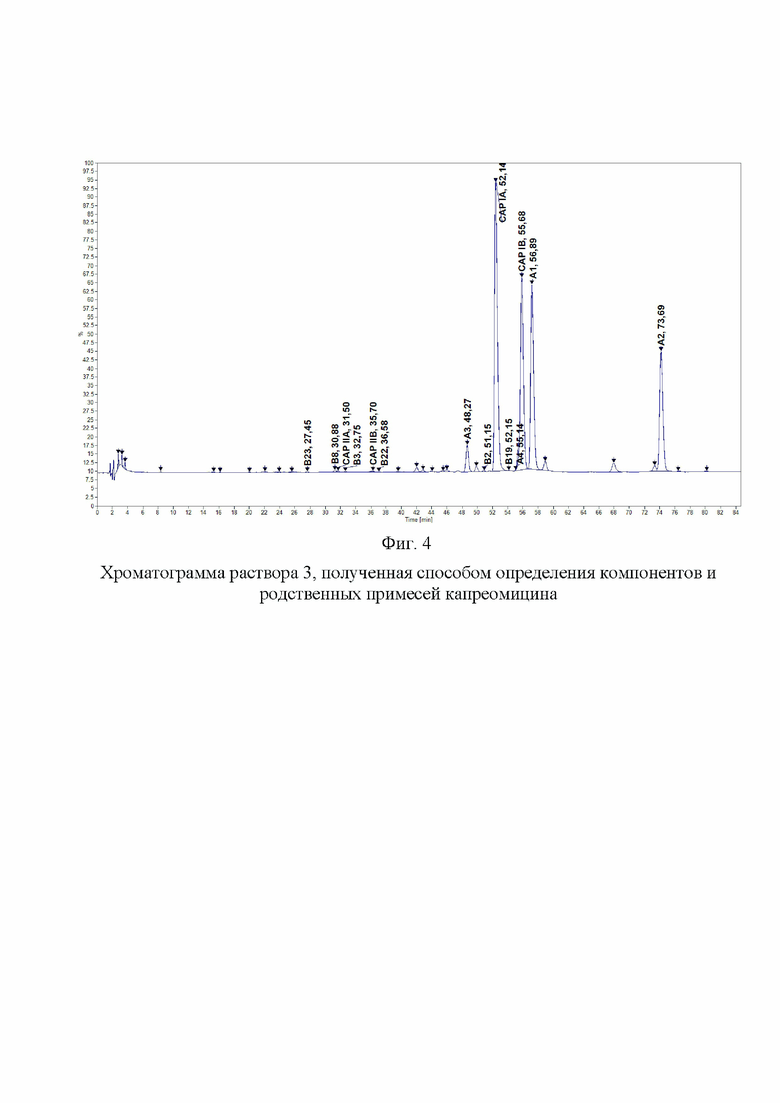
Фиг. 4. Хроматограмма раствора 3, полученная способом определения компонентов и родственных примесей капреомицина.
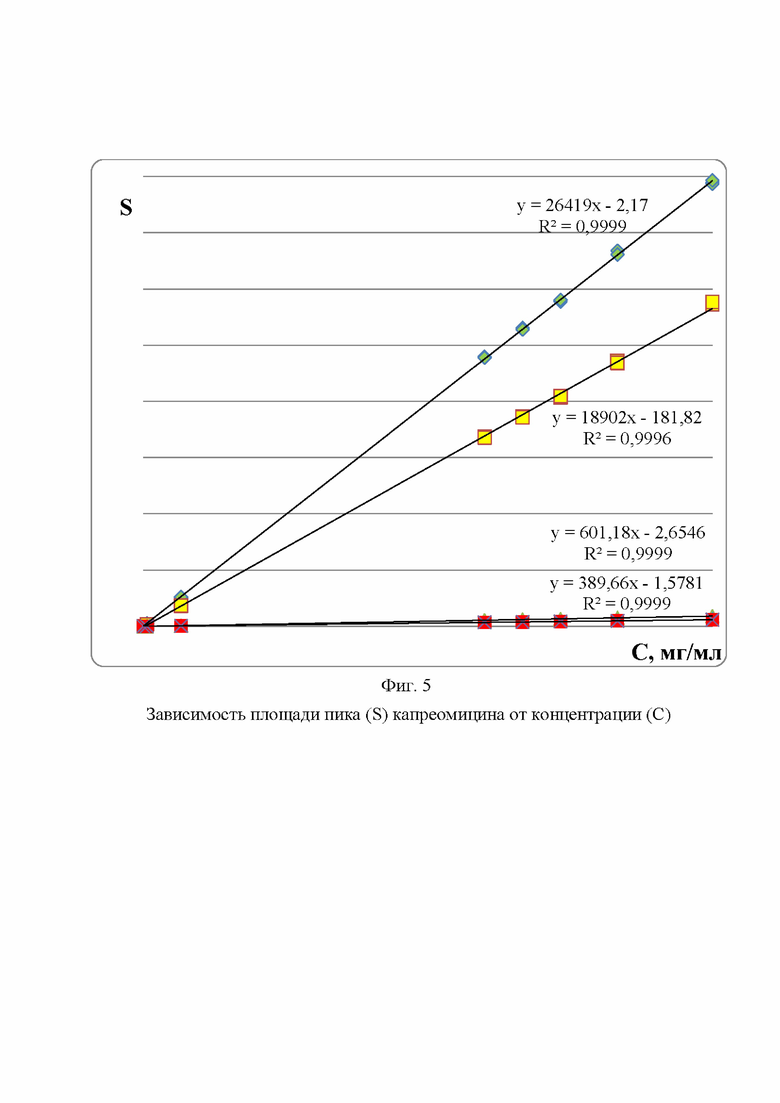
Фиг. 5. Зависимость площади пика (S) капреомицина от концентрации (С)
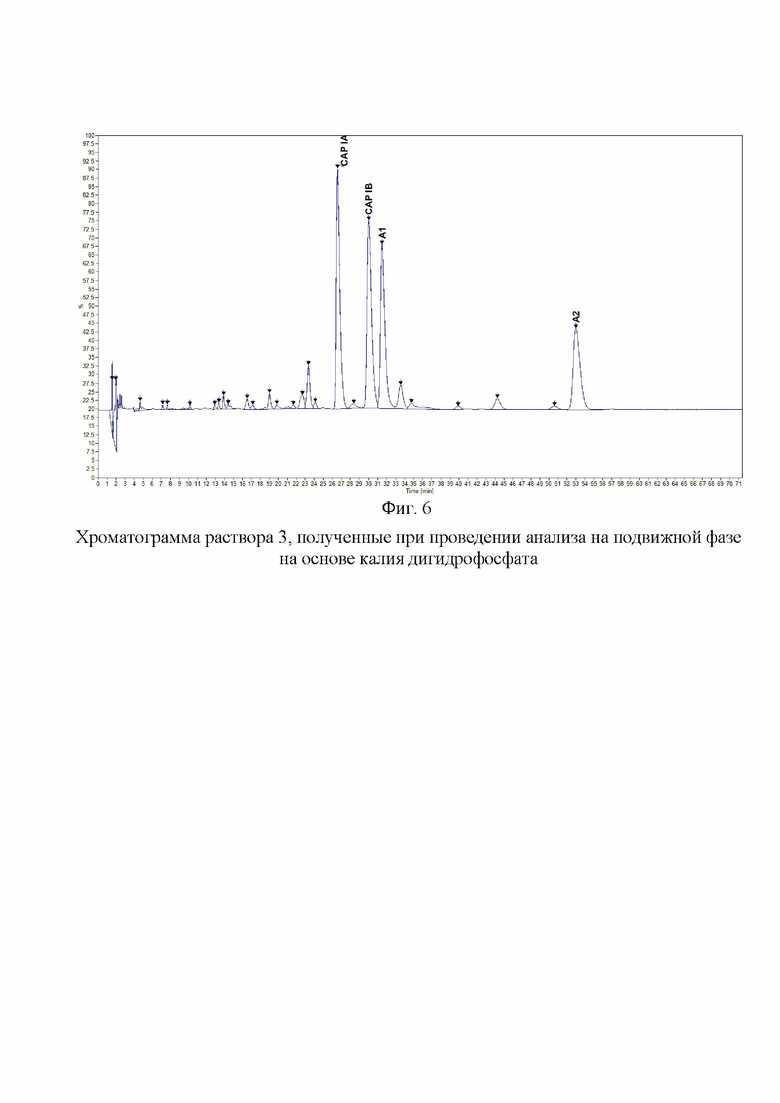
Фиг. 6. Хроматограмма раствора 3, полученные при проведении анализа на подвижной фазе на основе калия дигидрофосфата.
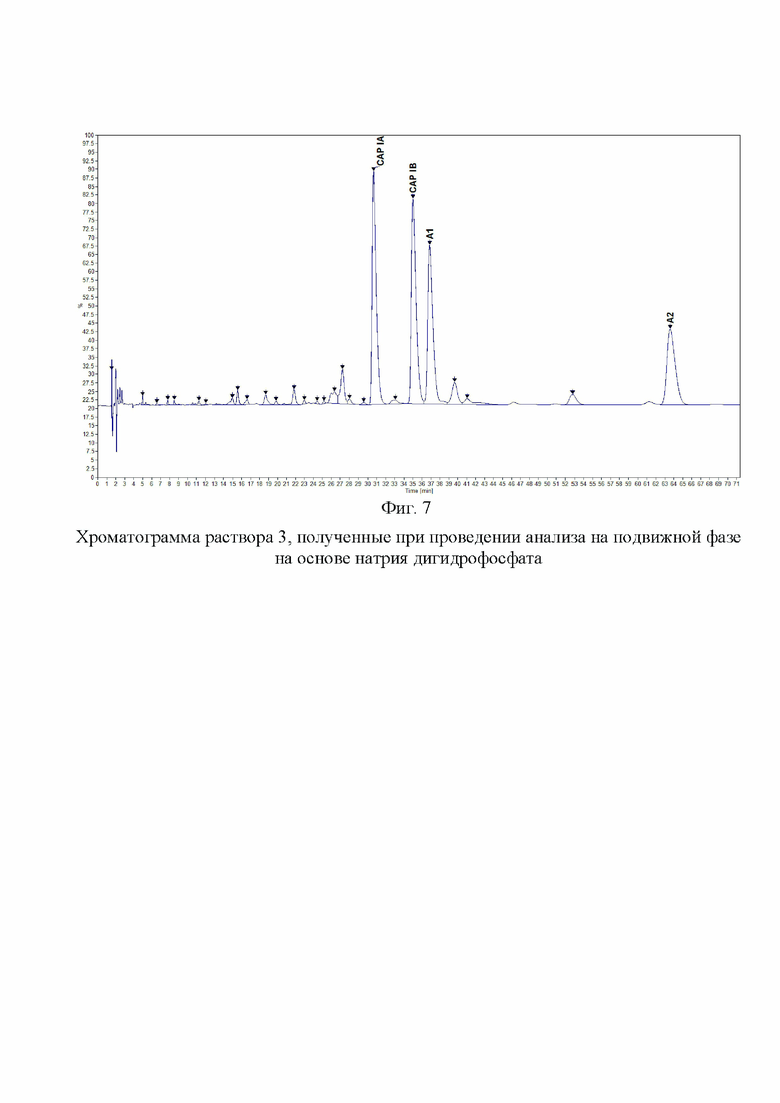
Фиг. 7. Хроматограмма раствора 3, полученные при проведении анализа на подвижной фазе на основе натрия дигидрофосфата.
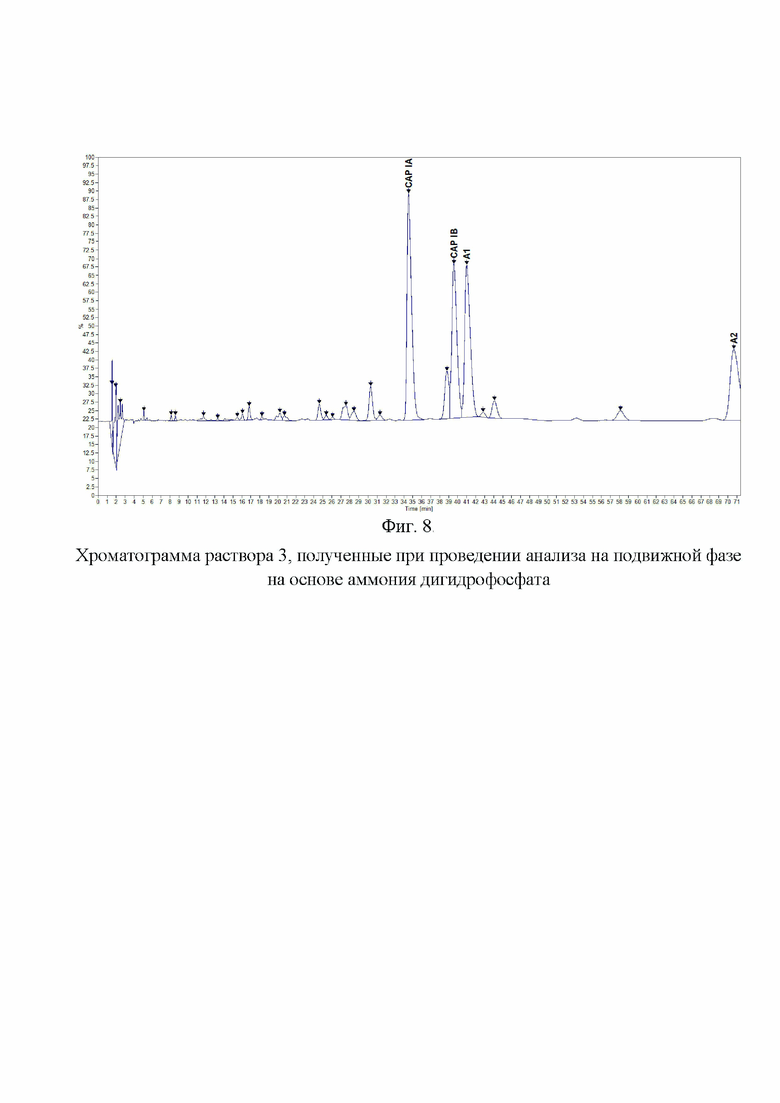
Фиг. 8. Хроматограмма раствора 3, полученные при проведении анализа на подвижной фазе на основе аммония дигидрофосфата.
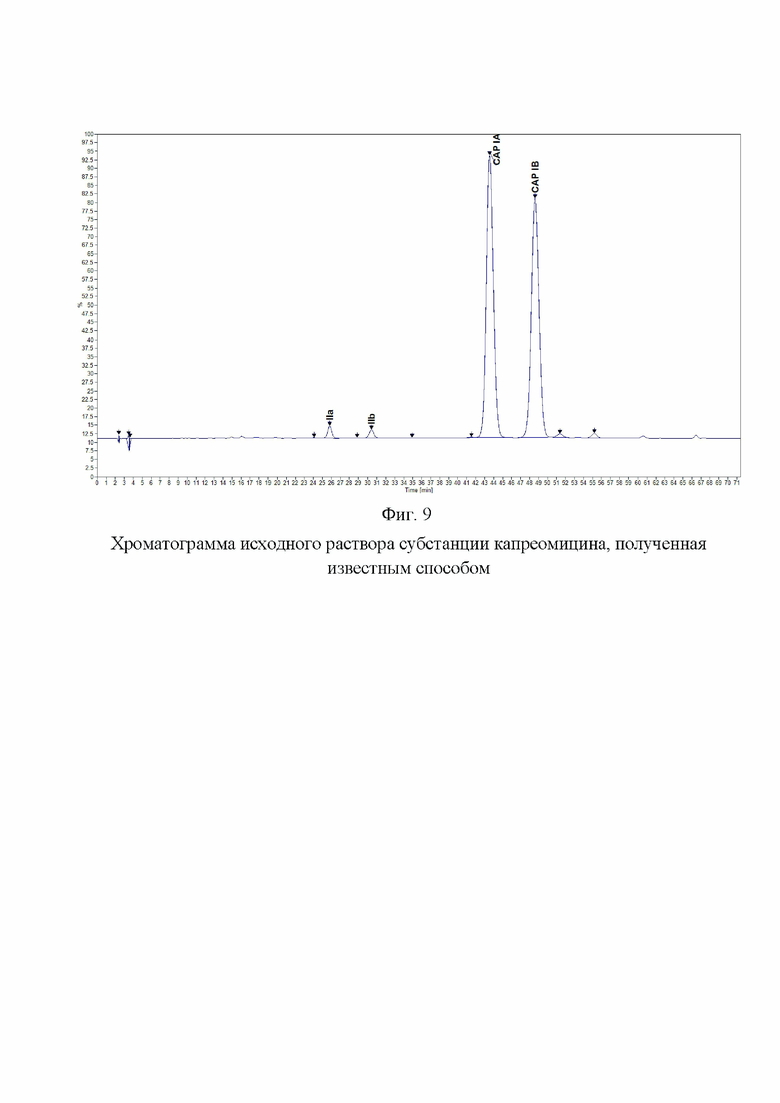
Фиг. 9. Хроматограмма исходного раствора субстанции капреомицина, полученная известным способом.
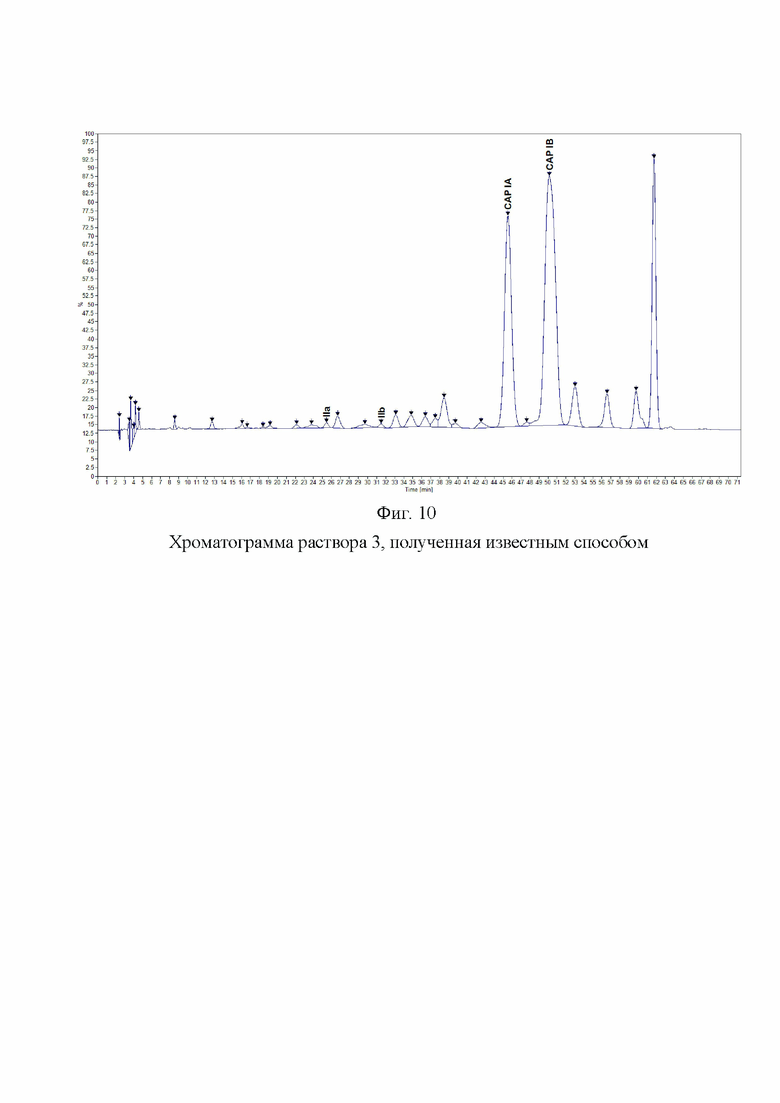
Фиг. 10. Хроматограмма раствора 3, полученная известным способом
Возможность осуществления заявленного способа раскрыта в следующих примерах.
Пример 1. Для определения компонентов капреомицина и его родственных примесей в порошке субстанции готовили испытуемый раствор, содержащий субстанцию-порошок капреомицина.
Испытуемый водный раствор капреомицина сульфата с исходной 0,2% и растворы 1, 2 и 3. переносили в хроматографические виалы и помещали их в автоматический пробоотборник хроматографа.
Состав буферного раствора рН 2,3: водный раствор 200 мМ натрия дигидрофосфат и 50 мМ натрия гексансульфоната, доведенный до рН 2,3 фосфорной кислотой.
Состав компонента подвижной фазы А: буферный раствор с рН 2,3 и ацетонитрил в объемном соотношении 95:5.
Состав компонента подвижной фазы Б: буферный раствор с рН 2,3 и ацетонитрил в объемном соотношении 90:10.
Хроматографические условия:
Колонка: BEH С18, 150 x 2,1 мм, 1,7 мкм
Объем вводимой пробы: 5 мкл;
Скорость потока подвижной фазы: 0,2 мл/мин;
Температура колонки: 70 °С;
Температура автосамплера: 5 °С;
Детектор: спектрофотометрический детектор, УФ с длиной волны 268 нм.
Программа градиента:
Хроматографировали по 5 мкл растворы в следующей последовательности: растворитель в виде очищенной воды, раствор подвижной фазы A, испытуемый раствор. На Фиг. 1, 2, 3 и 4 представлены типичные хроматограммы исходного раствора и растворов 1, 2 и 3.
Содержание капреомицина по данным фармакопейных статей должно составлять для суммы компонентов IA и IB не менее 90,0% от суммы всех пиков, а суммарное количество примесей - не более 7,0%.
Содержание компонентов капреомицина, единичных примесей, а также суммы всех примесей рассчитывали методом внутренней нормализации, который характеризуется тем, что процентное содержание капреомицина, либо содержание единичной примеси (Xi) в испытуемом образце рассчитывается путем определения площади пика компонента капреомицина, либо площади единичной примеси (Si) как часть от общей площади всех присутствующих на хроматограмме пиков (∑Si) за исключением пиков, соответствующих растворителю и подвижной фазе. Таким образом, при расчете процентного содержания компонентов капреомицина используют следующую формулу:
Данным примером подтвердили, что технический результат в заявленном способе достигается. Рассчитанное методом внутренней нормализации процентное содержание суммы компонентов капреомицина IA и IB и суммарное содержание его примесей составляет 95,99% и 4,01% соответственно, что удовлетворяет требованиям фармакопейных документов.
Пример 2. Линейность способа в аналитической области подтверждали линейной зависимостью площади пика исследуемого вещества от его концентрации в растворе (Фиг. 5). Исследование проводили по компонентам капреомицина в концентрациях, характерных для примесей в испытуемом растворе 0,00002 - 0,3%.
Полученные данные обрабатывали методом наименьших квадратов, который заключается в нахождении коэффициентов a, b и коэффициента корреляции R линейной модели: y=b*x+a,
где коэффициент х - обозначает концентрацию капреомицина, y - площадь пика капреомицина, b - угловой коэффициент модели, a - свободный член.
В результате расчетов коэффициент корреляции составил величину от 0,9996 до 0,9999 для разных пиков компонентов капреомицина, что доказывает линейность заявленной методики по фармакопейной статье.
Пример 3. Для подбора оптимальной хроматографической колонки выбрали несколько колонок. Выбранные колонки отличались друг от друга привитой фазой (октадецилсилильная, октилсилильная и фенилгексилсилильная), размером частиц (1,7, 1,9 и 5 мкм), степенью покрытия (доли углерода, от 10 до 20%), Характеристики колонок приведены в таблице 2.
Относительный компонентный состав рассчитывали в виде доли площади каждого из четырех пиков капреомицина от суммы площадей этих четырех пиков. Отмечали, что на хроматограммах, полученных на разных колонках, основная примесь, обозначенная как A1, имеет различную эффективность разделения с пиком IB.
Основным критерием выбора колонки было разделение данных двух пиков.
Относительный компонентный состав и критерии разделения пиков IB и A1, полученные при хроматографировании на разных колонках приведены в таблице 3.
Отсутствие пика примеси А1 на хроматограммах, полученных на некоторых колонках, а также отклонения в компонентном составе принимали как критерий непригодности использования колонки для данной технической задачи. Среди остальных колонок выбирали ту, на которой разрешение между требуемыми пиками было наибольшим. Исходя из полученных результатов, выбрали колонку BEH C18 (150×2,1 мм, 1,7 мкм), показавшую большее соответствие требованиям технической задачи.
Пример 4. Для анализа влияния на эффективность разделения катиона дигидрофосфата в составе подвижной фазы, которая составляет основу буферного раствора. Для этой цели были приготовлены отдельные подвижные фазы на основе дигидрофосфата калия, натрия и аммония. Концентрации исходных буферных растворов были одинаковы (200 мМ по катиону, при доведении pH до 2,3 ортофосфорной кислотой). К растворам солей добавляли 1-гексансульфоната натрия в количестве, чтобы его конечная концентрация составляла 25 мМ. Затем буферный раствор смешивали с ацетонитрилом в соотношении 94:6. Таким образом, все три подвижные фазы имели одинаковые молярные концентрации по катиону дигидрофосфата, значению pH, содержанию 1-гексансульфоната натрия и доле ацетонитрила. На Фиг. 6. представлена хроматограмма раствора 3, полученная на колонке BEH C18 (150×2,1 мм, 1,7 мкм) для подвижной фазы с использованием дигидрофосфата калия и на Фиг. 7. - для дигидрофосфата натрия. На данных хроматограммах были получены схожие профили пиков компонентов и примесей капреомицина. В случае применения дигидрофосфата натрия в сравнении с дигидрофосфатом калия было обнаружено незначительное увеличение времени удерживания при увеличении эффективности разделения основных пиков. При применении дигидрофосфата аммония (Фиг. 8) было обнаружено значительное изменение профиля хроматограммы с изменением порядка выхода пиков, а также меньшая разрешающая способность для основных пиков и меньшее общее количество детектируемых пиков примесей и увеличение времени удерживания всех пиков. После анализа полученных данных было принято решение использовать в составе подвижной фазы дигидрофосфат натрия.
Пример 5. Пример сравнения. Для сравнения эффективности заявленного способа провели его сравнение с известным способом.
Хроматографические условия известного способа:
Состав буферного раствора рН 2,3: 0,2 M калия дигидрофосфат, 50 мМ натрия гексансульфонат и фосфорной кислоты до значения рН 2,3 в воде.
Состав компонента подвижной фазы А: буферный раствор с рН 2,3 и ацетонитрил в объемном соотношении 95:5.
Состав компонента подвижной фазы Б: буферный раствор с рН 2,3 и ацетонитрил в объемном соотношении 85:15.
- Колонка: С18 (L1) 250 мм (4,6 мм, 5 мкм (ODS Hypersil, согласно IP)
- Скорость потока: 1 мл/мин
- УФ-детектирование при длине волны 268 нм.
- Объем вводимой пробы: 20 мкл (при концентрации 2 мг/мл, растворение в подвижной фазе A)
- Программа градиента:
Хроматограмма исходного раствора и раствора 3 представлена на Фиг. 9 и 10.
Было проведено сравнение хроматограмм, полученных заявленным способом, и хроматограмм, полученных известным способом. Заявленный способ позволил обнаружить разделения пиков капреомицина IB и примеси А1, что являлось основным критерием пригодности разрабатываемого способа. Относительный состав компонентов капреомицина определяется с большей точностью, поскольку правильно определяется содержание компонента IB. Заявленный способ позволяет обнаружить и оценить содержание значительно большего количества примесей, что показывает, что заявленный способ значительно более чувствителен и селективен, чем известный (фиг. 4, 10).
Представленные примеры не ограничивают объем притязаний настоящего изобретения и служат только для цели иллюстрации и раскрытия заявленного способа.
Промышленная применимость
Все приведенные примеры, подтверждают определение компонентов капреомицина и его родственных примесей.
Таким образом, поставленная техническая задача, а именно, разработка точного и чувствительного способа для определения компонентов капреомицина и его родственных примесей достигнута, что подтверждается приведенными примерами.
Применение заявленного способа возможно в фармации, химии, медицине, биологии.
Список литературы.
1. R.H. Browning, R.L. Donnerberg, Capreomycin/experiences in patient acceptance and toxicity, Ann. N. Y. Acad. Sci. 135 (1966) 1057-1064. https://doi.org/10.1111/j.1749-6632.1966.tb45546.x;
2. S.H. Lee, J. Shin, J.M. Choi, E.Y. Lee, D.H. Kim, J.W. Suh, J.H. Chang, The impurities of capreomycin make a difference in the safety and pharmacokinetic profiles, Int. J. Antimicrob. Agents 22 (2003) 81-83. https://doi.org/10.1016/S0924-8579(03)00124-9;
3. Capreomycin sulfate. United States Pharmacopoeia. USP 43-NF 38. Rockville, MD; 2020;
4. The International Pharmacopoeia - Tenth Edition, 2020 Capreomycin sulfates;
5. S. Mallampati, S. Huang, D. Ashenafi, E. VanHemelrijck, J. Hoogmartens, E. Adams, Development and validation of a liquid chromatographic method for the analysis of capreomycin sulfate and its related substances, J. Chromatogr. A 1216 (2009) 2449-2455. https://doi.org/10.1016/j.chroma.2009.01.031;
6. Liu, G.; Luan, B.; Liang, G.; Xing, L.; Huang, L.; Wang, C.; Xu, Y. Isolation and identification of four major impurities in capreomycin sulfate. J. Chromatogr. A 2018, 1571, 155-164. https://doi.org/10.1016/j.chroma.2018.08.015;
7. Chopra S, Pendela M, Hoogmartens J, Van Schepdael A, Adams E. Impurity profiling of capreomycin using dual liquid chromatography coupled to mass spectrometry. Talanta. 2012 Oct 15;100:113-22. https://doi.org/10.1016/j.talanta.2012.07.090;
8. Заявка на патент РФ №2021133328, 16.11.2021. Высочанская О.Н., Кулешова С.И., Симонова Е.П. "Способ определения ванкомицина В и его родственных примесей"//Патент России №2775300 С1. 2022. Бюл. №19;
9. E.B. Herr, M.E. Haney, G.E. Pittenger, C.E. Higgens. The chemical structure of capreomycin. Proc. Indiana Acad. Sci. 69 (1960) 134. https://doi.org/10.1007/BF01927571;
10. The Merck Index, 14th ed., Merck Research Laboratories, USA, 2006, pp.284;
11. A. Gobbi, G. Frenking, Y-conjugated compounds: the equilibrium geometries and electronic structures of guanidine, guanidinium cation, urea, and 1,1-diaminoethylene, J. Am. Chem. Soc. 115 (1993) 2362-2372. https://doi.org/10.1021/ja00059a035;
12. https://www.rlsnet.ru/active-substance/kapreomicin-1969 (дата обращения 25.11.2022).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОПРЕДЕЛЕНИЯ ПРОКАИНА В ПЛАЗМЕ КРОВИ | 2013 |
|
RU2537179C1 |
| Способ количественного определения фтивазида | 2024 |
|
RU2828350C1 |
| СПОСОБ ОДНОВРЕМЕННОГО ОПРЕДЕЛЕНИЯ ПРИМЕСЕЙ ЭТИЛЕНДИАМИНТЕТРАУКСУСНОЙ КИСЛОТЫ, ДИМЕТИЛСУЛЬФОКСИДА И N-ЭТИЛМАЛЕИМИДА В ФАРМАЦЕВТИЧЕСКИХ СУБСТАНЦИЯХ МЕТОДОМ ОБРАЩЕННО-ФАЗОВОЙ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2016 |
|
RU2621645C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ 2,4-ДИХЛОРФЕНОКСИУКСУСНОЙ КИСЛОТЫ В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 2011 |
|
RU2453848C1 |
| Способ количественного определения папаверина гидрохлорида и его родственных примесей в лекарственных средствах | 2021 |
|
RU2772608C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРЕГНЕНОВОГО ГЛИКОЗИДА "ХУДИКОЛ", ПОДАВЛЯЮЩЕГО АППЕТИТ | 2009 |
|
RU2414233C1 |
| СПОСОБ ОЧИСТКИ ЦИКЛИЧЕСКОГО ИЛИ НЕЦИКЛИЧЕСКОГО ПЕПТИДА | 2008 |
|
RU2461564C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТИОФОСФАТНЫХ ОЛИГОНУКЛЕОТИДОВ ДЛЯ ТЕРАПЕВТИЧЕСКОГО ПРИМЕНЕНИЯ | 2024 |
|
RU2836132C1 |
| Способ хроматографического разделения гидрокортизона ацетата, кортизона ацетата, метилпарагидроксибензоата, пропилпарагидроксибензоата методом обращенно-фазовой высокоэффективной жидкостной хроматографии | 2017 |
|
RU2653191C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КОЛИЧЕСТВЕННОГО СОСТАВА МНОГОКОМПОНЕНТНОГО ЛЕКАРСТВЕННОГО ПРЕПАРАТА ЖАРОПОНИЖАЮЩЕГО, АНТИАЛЛЕРГИЧЕСКОГО ДЕЙСТВИЯ | 2007 |
|
RU2342655C1 |
Изобретение относится к области к фармации, химии и медицины, а именно к способу определения капреомицина и его примесей. Способ определения капреомицина и его примесей с использованием ультраэффективной жидкостной хроматографии (УЭЖХ), характеризующийся тем, что используют раствор 1 с рН 9,8 с концентрацией 50 мМ, представляющий собой смесь раствора капреомицина 2 мг/мл и водного раствора аммиака 32% в соотношении 1000:3,23, и раствор 2 с рН 1,4 и концентрацией 50 мМ, представляющий собой смесь раствора капреомицина 2 мг/мл и трифторуксусной кислоты в соотношении 1000:3,83, раствор 1 и раствор 2 объединяют в соотношении 1:1 и разводят пятикратно, фильтруют, хроматографируют при двухфазном градиентном элюировании со скоростью ее потока 0,1 мл/мин до температуры 70°С, а далее скорость потока двухкомпонентной подвижной фазы увеличивают до 0,2 мл/мин при температуре 70°С с использованием двухкомпонентной подвижной фазы, состоящей из фазы А, представляющей собой композицию буферного раствора с рН 2,3 и ацетонитрила в объемом соотношении 95:5, и фазы Б, представляющей собой композицию буферного раствора с рН 2,3 и ацетонитрила в объемом соотношении 90:10, где буферный раствор для фазы А и Б представляет собой раствор, состоящий из раствора натрия дигидрофосфата и раствора натрия гексансульфоната, доведенных концентрированной фосфорной кислотой, длина волны детектора 268 нм, раствор 1 и раствор 2 выдерживывают при 80°С в течение 24 часов. Вышеописанный способ характеризуется чувствительностью и точностью и позволяет определять капреомицин и его примеси. 10 ил., 3 табл., 4 пр.
Способ определения капреомицина и его примесей с использованием ультраэффективной жидкостной хроматографии (УЭЖХ), характеризующийся тем, что используют раствор 1 с рН 9,8 с концентрацией 50 мМ, представляющий собой смесь раствора капреомицина 2 мг/мл и водного раствора аммиака 32% в соотношении 1000:3,23, и раствор 2 с рН 1,4 и концентрацией 50 мМ, представляющий собой смесь раствора капреомицина 2 мг/мл и трифторуксусной кислоты в соотношении 1000:3,83, раствор 1 и раствор 2 объединяют в соотношении 1:1 и разводят пятикратно, фильтруют, хроматографируют при двухфазном градиентном элюировании со скоростью ее потока 0,1 мл/мин до температуры 70°С, а далее скорость потока двухкомпонентной подвижной фазы увеличивают до 0,2 мл/мин при температуре 70°С с использованием двухкомпонентной подвижной фазы, состоящей из фазы А, представляющей собой композицию буферного раствора с рН 2,3 и ацетонитрила в объемом соотношении 95:5, и фазы Б, представляющей собой композицию буферного раствора с рН 2,3 и ацетонитрила в объемом соотношении 90:10, где буферный раствор для фазы А и Б представляет собой раствор, состоящий из раствора натрия дигидрофосфата и раствора натрия гексансульфоната, доведенных концентрированной фосфорной кислотой, длина волны детектора 268 нм, раствор 1 и раствор 2 выдерживают при 80°С в течение 24 часов.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| LI, Qian et al, HPLC determination for the contents/components and related substances of capreomycin sulfate and its preparations // Chinese Journal of Pharmaceutical Analysis, Vol | |||
| Нивелир для отсчетов без перемещения наблюдателя при нивелировании из средины | 1921 |
|
SU34A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| УСТРОЙСТВО ДЛЯ ИСПОЛЬЗОВАНИЯ ЭНЕРГИИ МОРСКИХ ВОЛН | 1923 |
|
SU1011A1 |
| G | |||
| Liu, et al., Isolation and identification | |||
