Настоящее изобретение относится к фармации, химии, медицине, биологии, а именно, к определению фтивазида.
Уровень техники
Фтивазид является синтетическим химиотерапевтическим средством, используемом при лечении туберкулеза. Препараты на основе фтивазида были разработаны отечественными учеными в середине XX века, путем взаимодействия ванилина и изониазида. Высокая активность фтивазида в отношении микобактерий туберкулеза обусловлена ингибированием роста микобактерий, а также повреждением их мембраны.
Из уровня техники известен способ определения фтивазида, в котором пробу анализируемого вещества сначала растворяют в щелочи, затем последовательно обрабатывают перекисью водорода, сульфатом гидроксиламина, хлоридом железа и проводят фотометрирование полученного раствора [1]. Предложенный метод предполагает проведение многоэтапной пробоподготовки, что, в свою очередь, снижает точность, чувствительность, воспроизводимость и надежность способа.
В работе [2] изложен способ определения изониазида и его гидразонов основанный на многократной обработке анализируемой пробы различными реагентами и последующим проведением фотоэлектроколориметрии. Ввиду долгой и сложной подготовки испытуемого образца, а также невысокой точности полученных результатов данный метод считается устаревшим.
Известен другой способ количественного определения фтивазида с применением титрования 0,1 М раствором хлорной кислоты в смеси муравьиной кислоты и уксусного ангидрида [3]. Описанный способ является трудоемким и требует проведения длительных испытаний, что влечет за собой большой расход дорогостоящих реагентов.
Также, известен способ определения фтивазида, основанный на методе тонкослойной хроматографии с использованием смеси растворителей: хлороформ - этиловый спирт 95% - ледяная уксусная кислота - 25% раствор аммиака [4]. Основным недостатком описанного способа является его многостадийность, трудоемкость и длительность исполнения.
Известен способ количественного определения фтивазида, основанный на взаимодействии раствора фтивазида в серной кислоте с фотогенерированным йодом, в результате этого взаимодействия происходит уменьшение силы тока. По калибровочному графику изменения силы тока и времени генерации йода и рассчитывается содержание фтивазида [5]. Данный метод не подтверждает отсутствия влияния на полученные результаты сходных по строению примесей фтивазида, и, следовательно, является неселективным.
Другой известный способ количественного определения фтивазида основан на реакции фтивазида с 1,2-нафтохинон-4-сульфонатом натрия, приводящей к образованию окрашенных продуктов реакции, которые далее анализируются методом спектрофотометрии [6]. Длительная и сложная пробоподготовка представленного способа делает его непригодным для рутинного анализа.
Способ определения фтивазида, описанный в аттестованной методике, включенной в нормативные документы лекарственных средств, включает в себя применение метода спектрофотометрии при длине волны 275 нм с использованием внешнего стандарта. Однако известный способ является кумулятивным, неселективным методом, что может привести к завышенным результатам содержания фтивазида в многокомпонентных лекарственных препаратах.
В известном способе [7, 8, 9] определения родственного соединения фтивазида - изониазида применяется широко известный метод высокоэффективной жидкостной хроматографии (далее - ВЭЖХ). ВЭЖХ зарекомендовала себя как современный, эффективный, селективный и высокочувствительный метод. Однако в связи с рядом отличительных особенностей фтивазида, в частности его плохая растворимость в органических растворителях и быстрая деградация в кислой среде, способы, разработанные для количественного определения изониазида, не могут быть использованы для количественного определения фтивазида в лекарственных препаратах. Данный способ выбран в качестве прототипа заявленного способа количественного определения фтивазида.
В связи с этим технической задачей является разработка простого, селективного, высокочувствительного способа количественного определения фтивазида с применением ВЭЖХ для оптимизации временных затрат при проведения испытаний.
Раскрытие сущности изобретения
Технической задачей является разработка селективного, высокочувствительного, воспроизводимого способа количественного определения фтивазида с применением высокоэффективной жидкостной хроматографии (далее - ВЭЖХ) в изократическом режиме для оптимизации временных затрат при проведения испытаний.
Техническим результатом заявленного технического решения является высокая чувствительность, селективность, эффективность, сокращение времени, необходимого для количественного определения фтивазида.
Технический результат заявленного изобретения достигается за счет:
- применения высокоэффективной жидкостной хроматографии (далее - ВЭЖХ);
- использования ацетонитрила в качестве растворителя;
- использования подвижной фазы определенного качественного и количественного состава;
- применения ультрафиолетового детектора с длинной волны 330 нм;
- проведения хроматографического анализа в изократическом режиме элюирования.
Способ определения фтивазида, отличающийся тем, что раствор фтивазида с концентрацией 0,2 мг/мл в ацетонитриле, обрабатывают в ультразвуковой бане, фильтруют, затем в условиях высокоэффективной жидкостной хроматографии в изократическом режиме с использованием ультрафиолетового детектора с длинной волны 330 нм, неподвижной фазы при температуре 30°С и двухкомпонентной подвижной фазы, состоящей из 0,1М буферного раствора аммония ацетата, доведенного рН до 7,0 и ацетонитрила в соотношении 75% об./об. : 25% об./об., соответственно, где скорость потока компонентов подвижной фазы 1 мл/мин, проводят хроматографирование, затем проводят количественный расчет содержания фтивазида.
Способ, дополнительно характеризующийся тем, что фтивазид может быть в форме субстанции или таблеток.
Способ, дополнительно характеризующийся тем, что фильтрование проводят через мембранный фильтр диаметром 0,45 мкм.
Способ, дополнительно характеризующийся тем, что при определении фтивазида, его примеси определяют с использованием ультрафиолетового детектора с длинной волны 238 нм.
Краткое описание чертежей и иных материалов (Приложение 1-9)
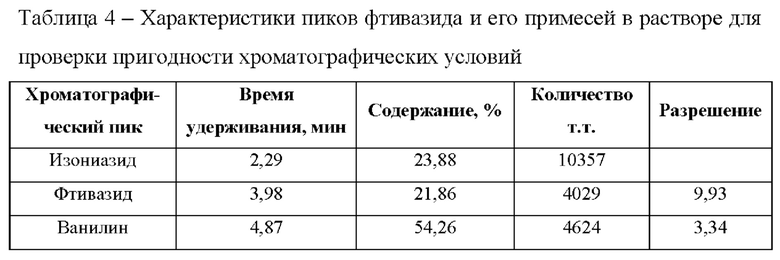
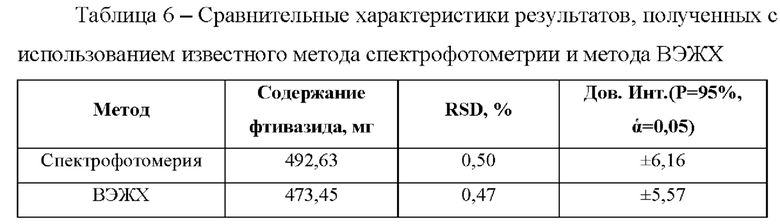
Фиг. 1 Хроматограмма стандартного раствора, полученная способом определения фтивазида на колонке Kinetex С18. 1 - фтивазид.
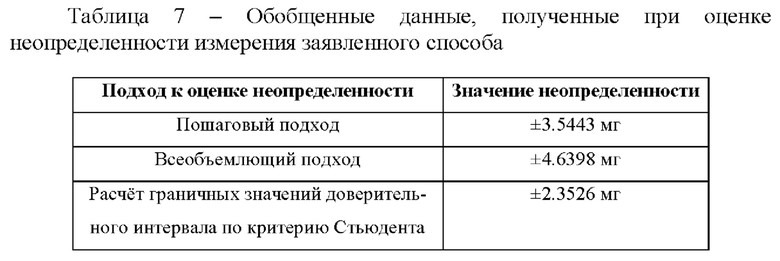
Фиг. 2 Хроматограмма испытуемого раствора, приготовленного с использованием таблеток фтивазида. 1 - фтивазид, 2 - ванилин.
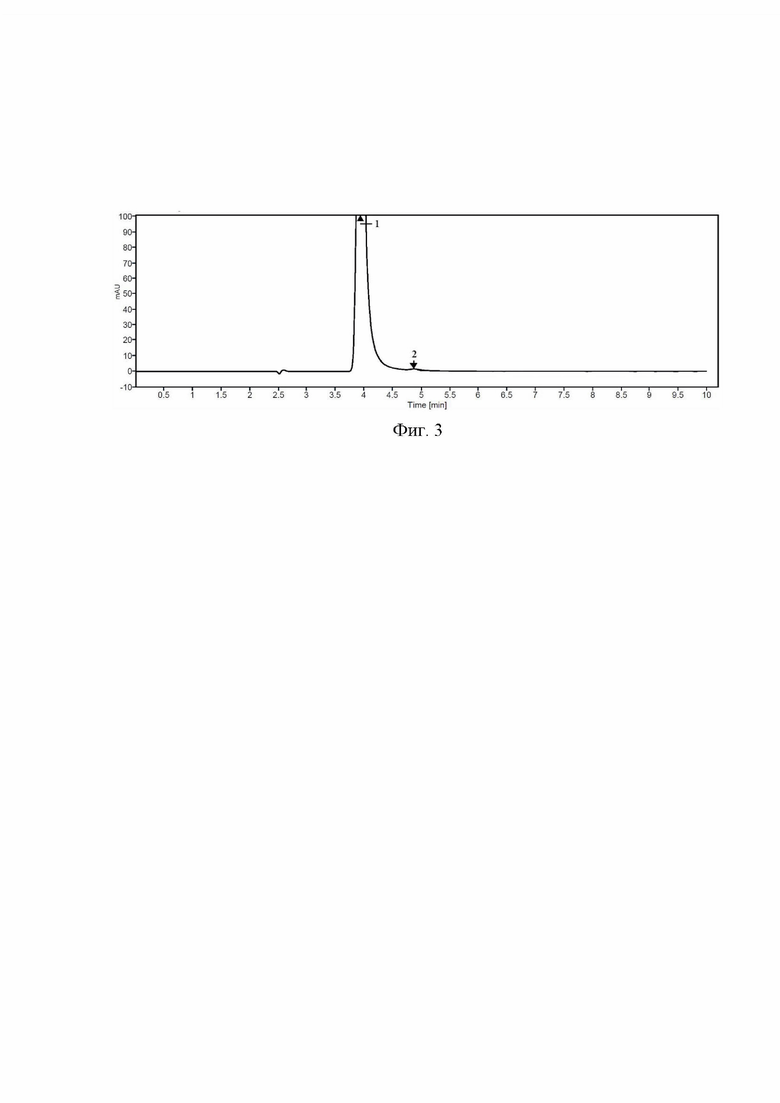
Фиг. 3 Хроматограмма испытуемого раствора, приготовленного с использованием субстанции-порошка фтивазида. 1 - фтивазид, 2 - ванилин.
Фиг. 4 Хроматограмма раствора для проверки пригодности хроматографической системы, полученная при кислотном разложении ОДМ раствором соляной кислоты. 1 - фтивазид, 2 - ванилин, 3 - изониазид.
Фиг. 5 Зависимость площади пика (S) фтивазида от его концентрации (С) в растворе, которая выражается в виде уравнения: у=13104х-10.871 и коэффициента корреляции: R2=0.9997.
Фиг. 6 Хроматограммы стандартного раствора, полученные при изменении состава подвижной фазы. 1 - фтивазид при 27% ацетонитрила, 2 - фтивазид при 25% ацетонитрила, 3 - фтивазид при 23% ацетонитрила.
Фиг. 7 Хроматограммы стандартного раствора, полученные при изменении температуры колонки. 1 - фтивазид при 35°С, 2 - фтивазид при 25°С, 3 - фтивазид 30°С.
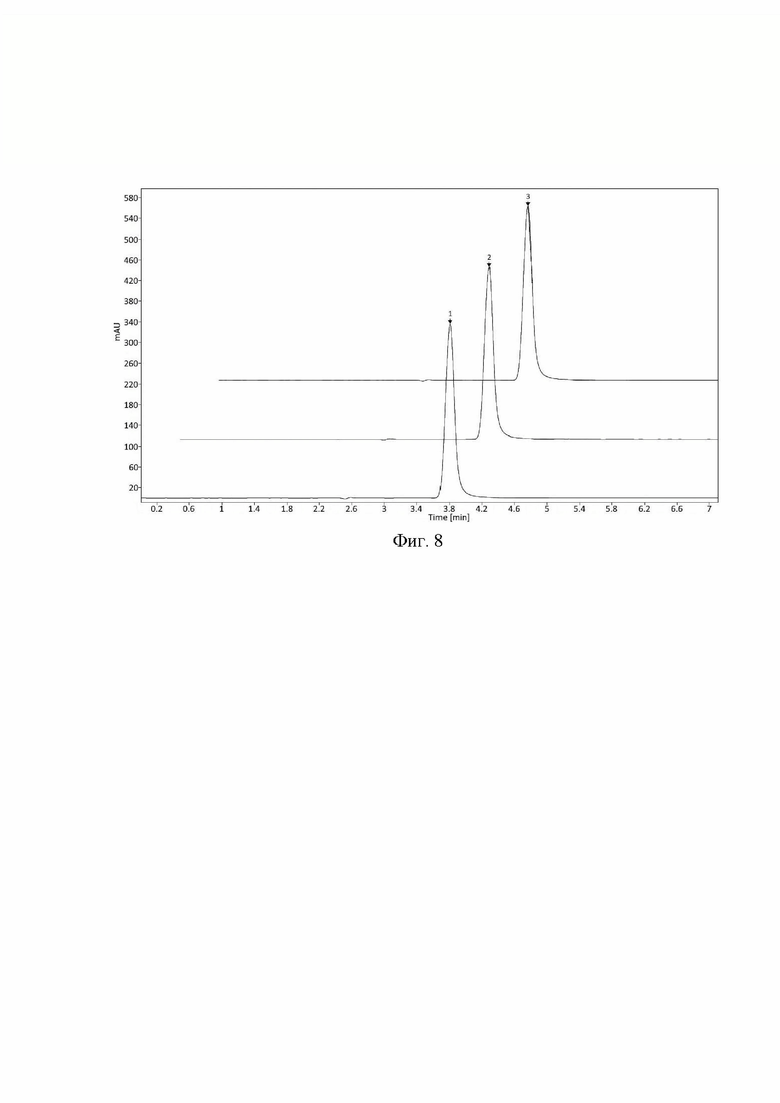
Фиг. 8 Хроматограммы стандартного раствора, полученные при длины волны детектора. 1 - фтивазид при 332 нм, 2 - фтивазид при 330 нм, 3 - фтивазид 328 нм.
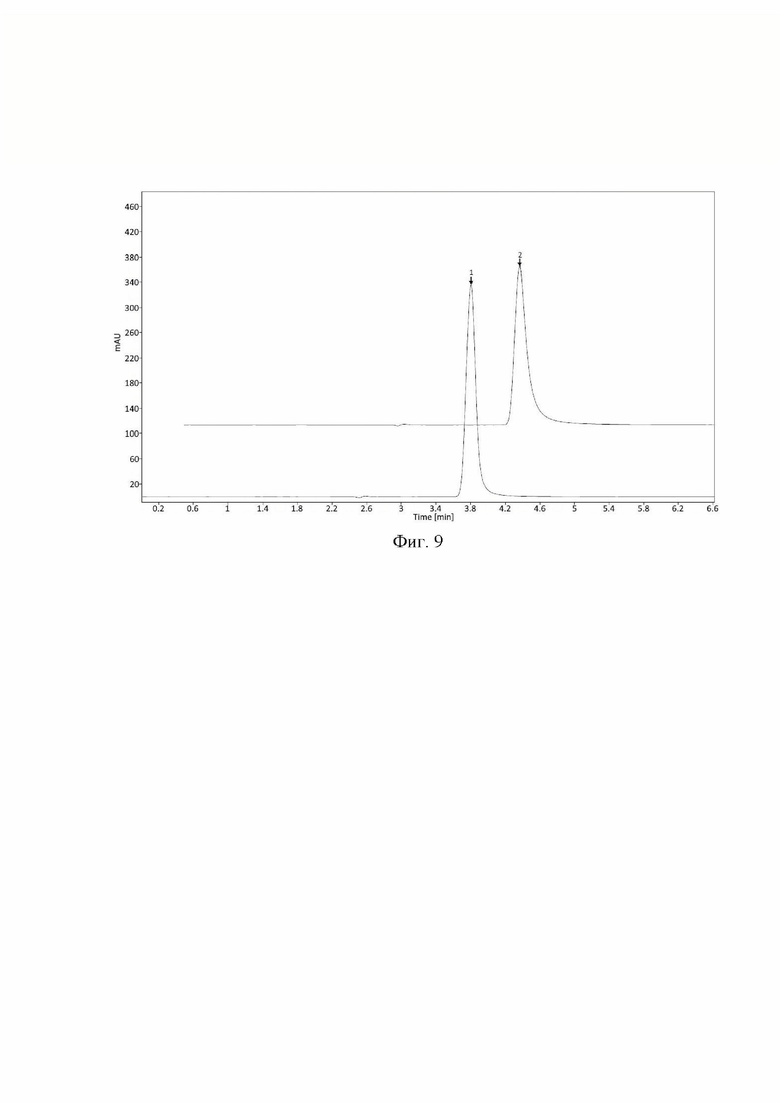
Фиг. 9 Хроматограммы стандартного раствора, полученные при изменении колонки. 1 - фтивазид при использовании колонки Kinetex ХВ-С18 (SN: 00G-4605-E0), 2 - фтивазид при использовании колонки Kinetex C18(SN: 00G-4601-E0).
Осуществление изобретения
Для разработки заявленного способа были выбраны: препарат фтивазида в форме таблеток, субстанция фтивазида, стандартные образцы изониазида и ванилина.
Фтивазид представляет собой соединение группы гидразона, который в кислой среде может гидролизоваться с образованием изониазида и ванилина.
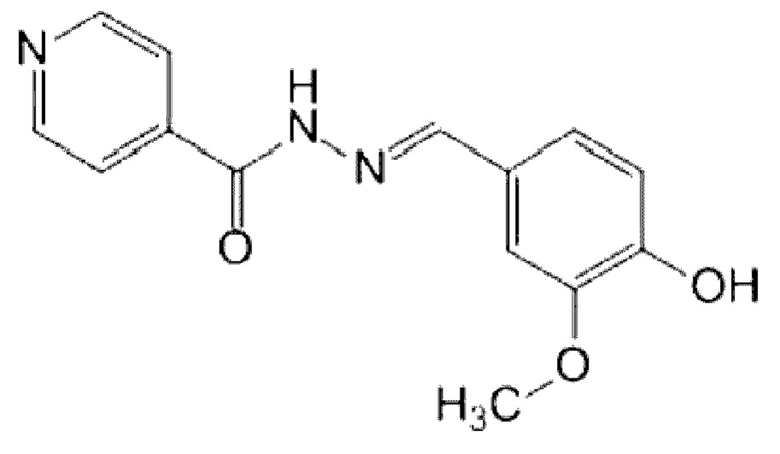
4-Пиридинкарбоновой кислоты [(4-гидрокси-3-метоксифенил)метилен] гидразид (C14H13N3O3).
Оценка количественного содержания действующего вещества в лекарственных средствах (ЛС) является важной задачей при их производстве и хранении. В настоящее время наиболее востребованы способы количественного определения с применением высокоэффективной жидкостной хроматографии (ВЭЖХ), характеризующейся высокой селективностью, эффективностью и чувствительностью.
Проведение пробоподготовки заключается в приготовлении испытуемого раствора фтивазида в ацетонитриле с концентрацией фтивазида 0,2 мг/мл с последующей обработкой в ультразвуковой бане в течение 30 минут, фильтрации полученного раствора через мембранный фильтр с диаметром пор 0,45 мкм и переносе раствора в хроматографические виалы с дальнейшим помещением в автоматический пробоотборник хроматографа.
Для проведения хроматографирования (ВЭЖХ) растворов была использована двухкомпонентная подвижная фаза, состоящая из ОДМ буферного раствора аммония ацетата, доведенного рН до 7,0 раствором аммиака или раствором ледяной уксусной кислоты, и ацетонитрила в объемном соотношении 75%: 25%. Для достижения наилучшей воспроизводимости заявленного способа, а также для сокращения времени анализа было выбрано изократическое элюирование без изменения состава подвижной фазы.
Возможная деградация фтивазида в кислой среде была экспериментально подтверждена обнаружением двух дополнительных пиков при хроматографировании раствора фтивазида, растворенного в 0,1 М растворе хлороводородной кислоте. Обнаруженные пики были идентифицированы по стандартным образцам изониазида и ванилина. Таким образом, можно прийти к выводу, что водные растворы кислот приводят к разложению фтивазида на его прекурсоры.
Известно, что фтивазид практически нерастворим в воде [3]. В связи с этим, выбор растворителя играет важную роль при анализе лекарственных средств на основе фтивазида. Поэтому в ходе разработки заявленного способа возникла первостепенная необходимость выбора растворителя для приготовления испытуемого раствора.
В качестве возможного использования растворителя в заявленном способе были исследованы следующие реагенты: метанол, диметилсульфоксид (ДМСО), диметилформамид (ДМФА), ацетонитрил, этанол 96%. Все растворы готовили с приблизительно одинаковой концентрацией фтивазида (0,2 мг/мл) и хроматографировали (3 инъекции) в одних и тех же условиях. Результаты исследования приведены в таблице 1.
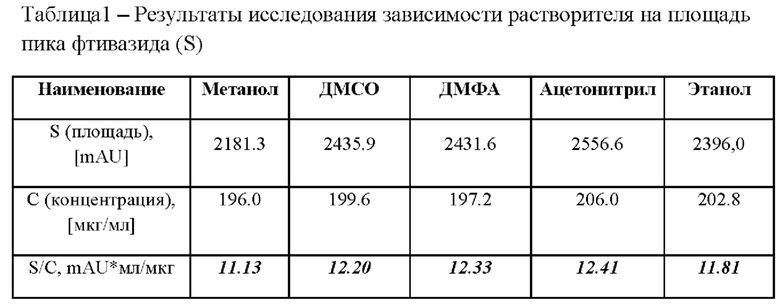
Для наглядности результаты представлены в виде отношения площади пика фтивазида к концентрации фтивазида в испытуемом растворе (табл. 1).
Исходя из представленных данных, можно прийти к выводу, что ацетонитрил зарекомендовал себя наилучшим образом из исследуемых органических растворителей. Ацетонитрил может обеспечить достаточную растворимость фтивазида для проведения испытаний на количественное определение без губительного эффекта на молекулу фтивазида. К тому же ацетонитрил имеет низкое УФ-поглощение и не так токсичен, как многие другие органические растворители. При этом ацетонитрил используемый в качестве добавки к подвижной фазе не будет оказывать существенного влияния на получаемые результаты.
Значение рН буферного раствора в нейтральном диапазоне около 7,0 обеспечивает оптимальные условия для стабильности испытуемых растворов, а выбор ацетонитрила в качестве компонента подвижной фазы позволяет добиться ускоренного анализа без потери эффективности.
Испытания проводили на жидкостном хроматографе, оборудованном диодно-матричным детектором с диапазоном длин волн 190-640 нм, позволяющим выбрать оптимальную длину волны. В ходе исследований было выявлено, что длина волны равная 330 нм отвечает за максимум на спектре поглощения фтивазида, что обеспечивает высокую чувствительность заявленного способа. Однако следует обратить внимание на то, что при выбранной длине волны показатель поглощения изониазида и ванилина стремиться к минимуму, что будет обозначать отсутствие данных примесей на хроматограмме испытуемого раствора даже при их значительном содержании в растворе. Таким образом, для выявления примесей фтивазида рекомендуется использовать другую длину волны. Так, для проверки пригодности хроматографической системы разрешение между пиками фтивазида и его примесями рассчитывали, используя длину волны УФ - детектора 238 нм.
Условия хроматографирования:
Объем вводимой пробы: 3 мкл.
Компонентный состав подвижной фазы: 0,1 М буферного раствора аммония ацетата - ацетонитрил в соотношении 75% об./об.: 25% об./об. (±2.0% ацетонитрила)
Скорость потока двух компонентной подвижной фазы: 1 мл/мин.
Температура колонки: 30°С±5°С
Длина волны детектора: 330 нм ±2 нм (для количественного определения фтивазида), 238 нм (для определения примесей фтивазида).
Возможность осуществления заявленного способа раскрыта в следующих примерах.
Пример 1. Для количественного определения фтивазида в таблетках готовят испытуемый раствор, содержащий препарат фтивазида в виде таблеток.
Для приготовления испытуемого раствора отбирают 20 таблеток, определяют среднюю массу и тщательно растирают в однородный порошок. Около 20 мг (точная навеска) полученного порошка помещают в мерную колбу вместимостью 100 мл, растворяют в ацетонитриле, обрабатывают ультразвуком в течение 30 мин при периодическом встряхивании и охлаждают до комнатной температуры. Затем доводят объем раствора до метки тем же растворителем, перемешивают и фильтруют через мембранный фильтр (политетрафторэтилен, PTFE) с диаметром пор 0,45 мкм. Полученный раствор переносят в хроматографические виалы, где концентрация раствора составляет 0,2 мг/мл, и помещают их в автоматический пробоотборник хроматографа.
Для приготовления стандартного раствора берут 20 мг стандартного образца фтивазида и помещают в мерную колбу вместимостью 100 мл, растворяют в ацетонитриле, обрабатывают ультразвуком до растворения при периодическом встряхивании и охлаждают до комнатной температуры. Затем доводят объем раствора до метки тем же растворителем, перемешивают и фильтруют через мембранный фильтр с диаметром пор 0,45 мкм.
Полученный раствор переносят в хроматографические впалы, где концентрация раствора составляет 0,2 мг/мл, и помещают их в автоматический пробоотборник хроматографа.
Приготовление буферного раствора с рН 7.0 осуществляют следующим образом: 7,71 г аммония ацетата помещают в мерную колбу вместимостью 1000 мл, прибавляют 800 мл воды, доводят рН раствора до значения 7.0 с помощью ледяной уксусной кислоты или раствора аммиака, доводят объем раствора до метки водой и перемешивают.
Для приготовления подвижной фазы смешивают буферный раствор с рН 7.0 и ацетонитрил в объемном соотношении 75%: 25% и дегазируют любым способом известным специалисту из данной области техники.
Хроматографические условия:
Колонка: Kinetex С18, 250 х 4.6 мм, 5 мкм
Объем вводимой пробы: 3 мкл;
Скорость потока подвижной фазы: 1,0 мл/мин;
Температура колонки: 30°С;
Детектор: Ультрафиолетовый детектор с длиной волны 330 нм.
Хроматографируют по 3 мкл растворы в следующей последовательности: растворитель в виде ацетонитрила, стандартный раствор, испытуемый раствор. На Фиг. 1 и 2 представлены типичные хроматограммы стандартного раствора и испытуемого растворов соответственно, в таблице 2 приведены основные характеристики пика фтивазида. На хроматограмме испытуемого раствора (фиг.2) также присутствует пик ванилина, разрешение которого с пиком фтивазида составило 4,34.

Содержание фтивазида, рассчитывают методом внешнего стандарта, который основан на сравнении сигнала (площади пика), полученного на хроматограммах испытуемого раствора (Si), и сигнала, полученного на хроматограммах раствора стандартного образца (S0). Таким образом, при расчете содержания фтивазида в таблетках (X, мг) используют следующую формулу:
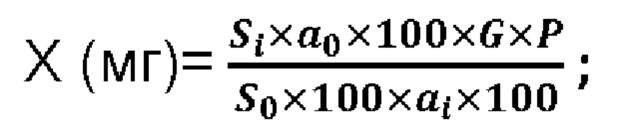
Si - площадь пика фтивазида на хроматограммах испытуемого раствора;
S0- площадь пика фтивазида на хроматограммах стандартного раствора;
а0 - навеска СО фтивазида, мг;
ai - навеска порошка растертых таблеток, мг;
Р - содержание фтивазида в СО фтивазида, %;
G - средняя масса одной таблетки, в мг;
Данным примером подтверждено, что заявленный технический результат в заявленном способе достигается. Рассчитанное методом внешнего стандарта содержание фтивазида составляет 473.46 мг (94.69%).
Пример 2. Количественное определение фтивазида в порошке субстанции проводят способом, описанным в примере 1.
Данным примером подтверждено, что заявленный технический результат в заявленном способе достигается. Рассчитанное методом внешнего стандарта содержание фтивазида составляет 988,11 мг (98.81%).
Относительное стандартное отклонение площадей пика фтивазида (RSD S), рассчитанное по хроматограммам испытуемого раствора в форме таблеток (Фиг. 2), а также по хроматограммам испытуемого раствора в форме субстанции-порошка (Фиг. 3) составляют не более 1,0% каждое, а времена удерживания пика фтивазида совпадают (табл. 3).
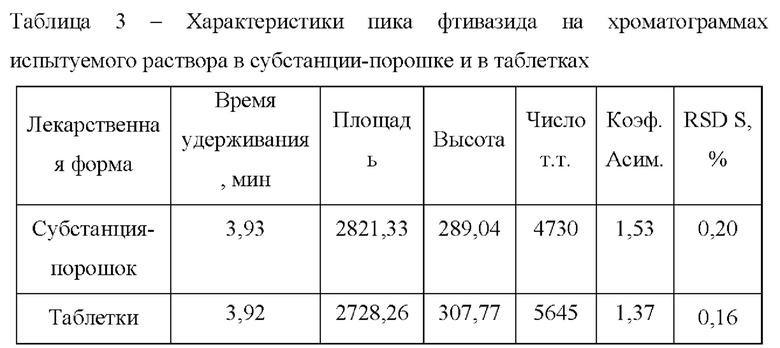
Приведенный пример демонстрирует сопоставимость результатов, что подтверждает возможность реализации заявленного способа для количественного определения фтивазида в различных лекарственных формах.
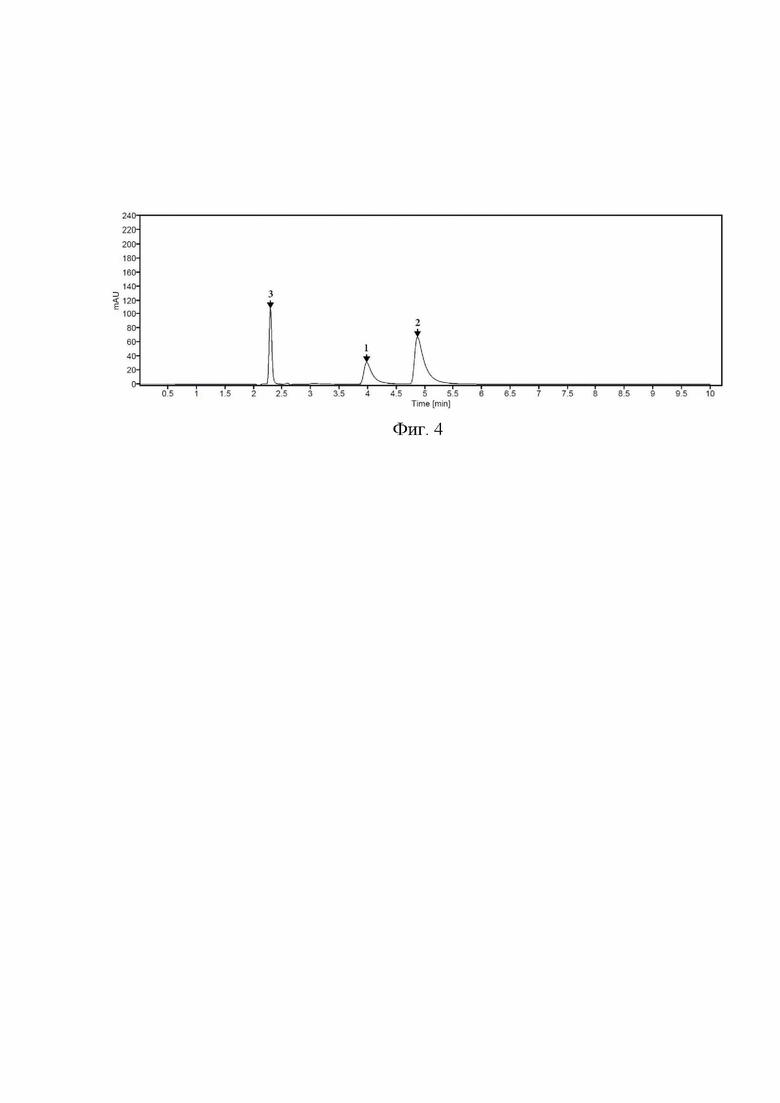
Пример 3. Для определения основных примесей, образующихся при деструкции молекулы фтивазида, субстанцию фтивазида растворяли в 0,1 М растворе хлороводородной кислоты для получения раствора с концентрацией 0,2 мг/мл. Приготовленный раствор хроматографировали с использованием длины волны детектора 238 нм. Полученная хроматограмма представлена на Фиг. 4. Идентификацию пиков примесей проводили с применением стандартных образцов изониазида и ванилина.
Было обнаружено, что при кислотном разложение фтивазида наблюдается значительный рост ванилина (до 54%), а также изониазида (до 24%). Значение разрешения между пиками фтивазида и его примесей составляет не менее 1,5, удовлетворяет критериям приемлемости по фармакопейной статье.
Пример 4. Линейность способа в аналитической области подтверждается линейной зависимостью площади пика исследуемого вещества от его концентрации в растворе (Фиг. 5). В разработанном способе, аналитическая область содержания фтивазида составляет 80-120%. Исследование проводилось на примере фтивазида в следующих концентрациях: 0.16, 0.18, 0.20, 0.22, 0.24 мг/мл, соответствующих 80, 90, 100, 110, 120%, а также раствор с пределом количественного определения (ПКО) (0.2 мкг/мл, что соответствует 0.1%).
Полученные данные обрабатывали методом наименьших квадратов, который заключается в нахождении коэффициентов а, b и коэффициента корреляции г линейной модели:
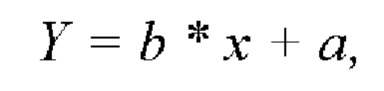
где коэффициент х - обозначает концентрацию фтивазида, Y - площадь пика фтивазида, b - угловой коэффициент модели, а - свободный член.
В результате расчетов коэффициент корреляции составил величину 0,9997, что доказывает линейность заявленного способа по фармакопейной статье.
Пример 5. Устойчивость заявленного способа количественного определения фтивазида к небольшим изменениям условий хроматографирования была подтверждена испытанием на возможное влияние состава подвижной фазы, температуры колоночного термостата, длины волны детектора и различных серий хроматографической колонки.
Хроматографирование стандартного раствора фтивазида с концентрацией 0,2 мг/мл проводили при изменении следующих параметров:
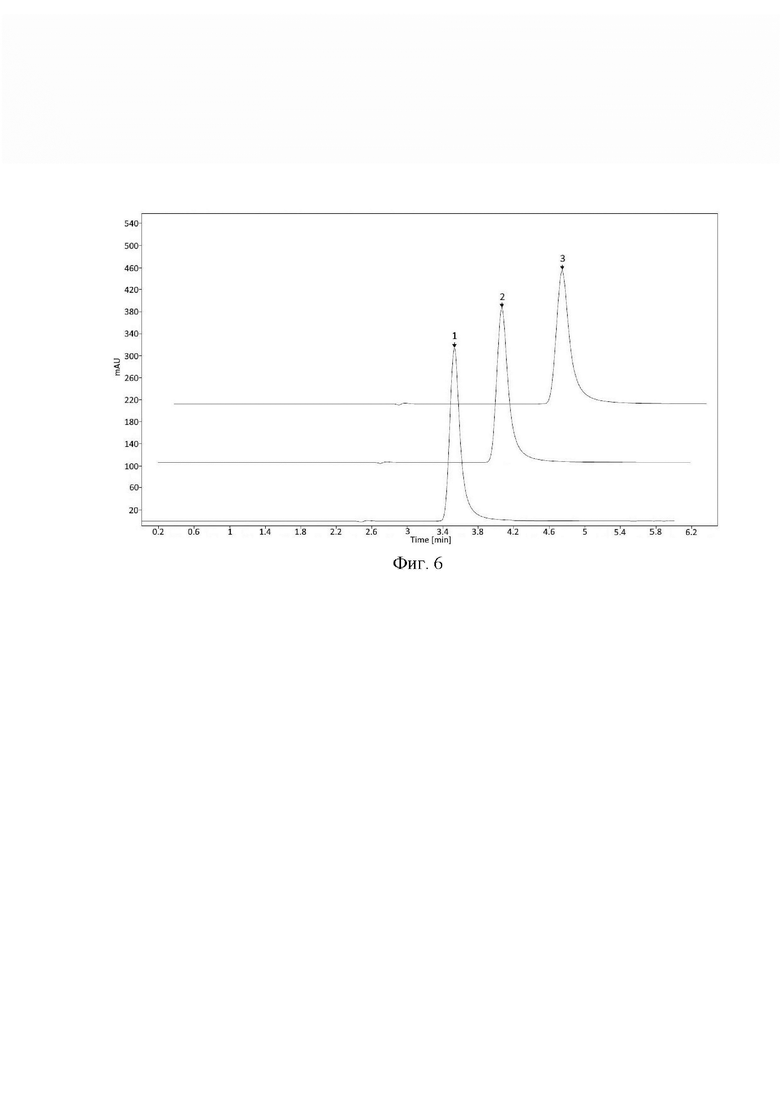
- изменение состава подвижной фазы в диапазоне ±2.0%) ацетонитрила (AcN) от заданного значения (25%) в подвижной фазе (Фиг. 6);
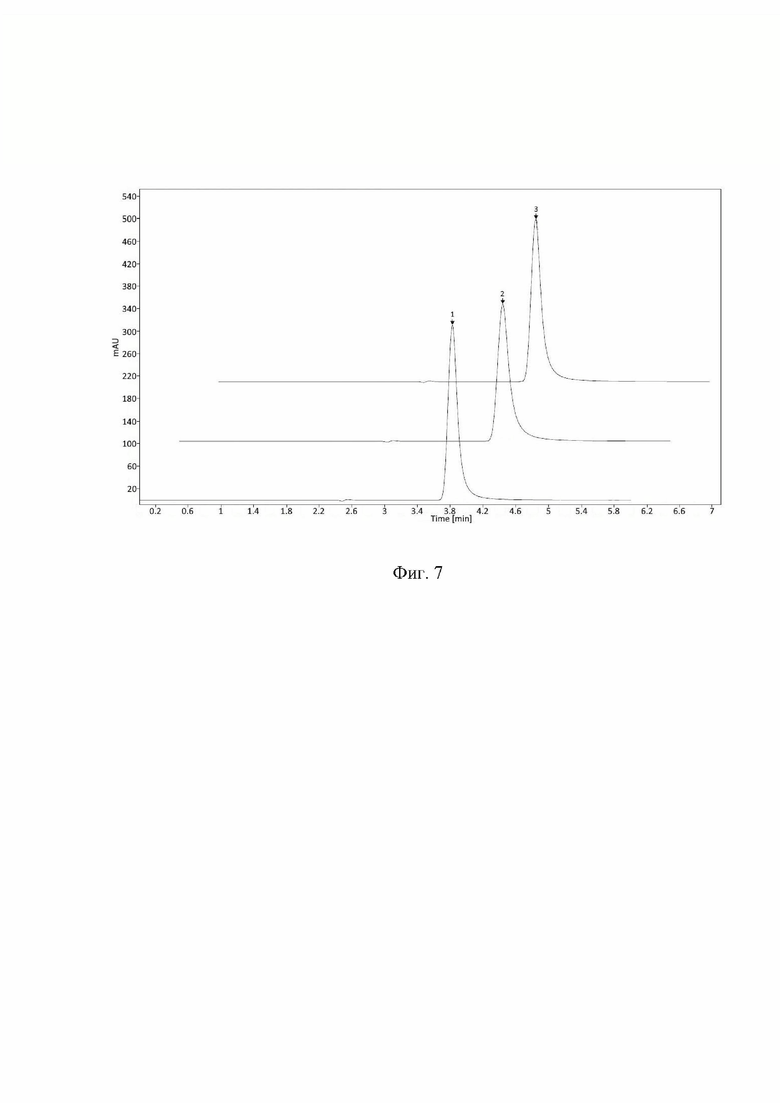
- изменение температуры колоночного термостата в диапазоне ±5°С от заданного значения (30°С) (Фиг. 7);
- изменение длины волны УФ-детектора в диапазоне ±2 нм от заданного значения (330 нм) (Фиг. 8);
- использование аналогичных хроматографических колонок (Фиг. 9). Критерием приемлемости является сохранение следующих параметров:
- относительно стандартное отклонение площади пика фтивазида (RSD S) составляет не более 1.0%;
- относительно стандартное отклонение времени удерживания пика фтивазида (RSD t) составляет не более 1.0%;
- фактор асимметрии пика фтивазида составляет не более 2.0; -число теоретических тарелок (т.т.) по пику фтивазида составляет не менее 2000;
- изменение площади пика фтивазида (AS) относительно стандартных условий методики находится в диапазоне ±5%;
Результаты проведенных тестов, а также их соответствие критериям приемлемости приведены в таблице 5:
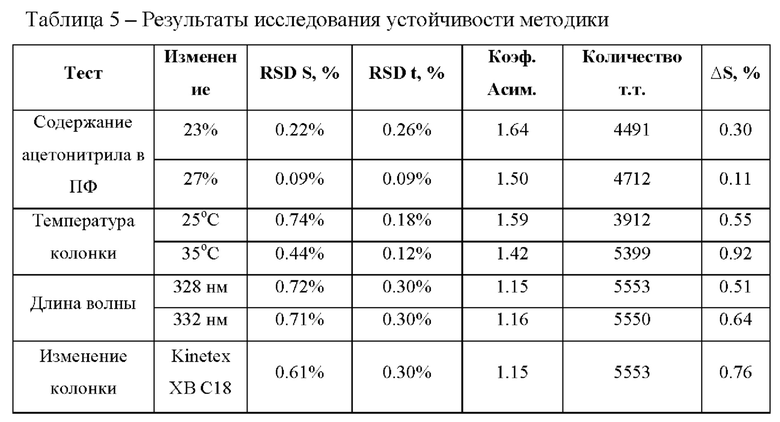
При внесении изменений в условия хроматографирования и приготовления подвижной фазы все критерии приемлемости выполняются, следовательно, методику можно считать устойчивой к влиянию различных изменений в параметрах метода.
Пример 6. Правильность предлагаемого изобретения была доказана при помощи сравнения полученных результатов разработанным способом с ранее валидированной методикой спектрофотометрии (табл. 6).
Для приготовления испытуемого раствора точную навеску порошка растертых таблеток, соответствующую около 100 мг фтивазида, помещают в мерную колбу вместимостью 100 мл, прибавляют 50 мл хлористоводородной кислоты раствора 0,1 М и встряхивают при нагревании на водяной бане в течение 5 мин. Охлаждают раствор до комнатной температуры, доводят объем раствора тем же растворителем до метки и фильтруют. В мерную колбу вместимостью 100 мл помещают 1,0 мл полученного фильтрата и доводят объем раствора хлористоводородной кислоты раствором 0,1 М до метки. Концентрация фтивазида в полученном растворе составляла 0,01 мг/мл.
Раствор стандартного образца фтивазида готовили аналогичным способом с использованием стандартного образца фтивазида. Концентрация фтивазида в стандартном растворе составляла 0,01 мг/мл.
Измерение оптической плотности испытуемого раствора и раствора стандартного образца фтивазида проводили на спектрофотометре в максимуме поглощения при длине волны 275 нм в кювете с толщиной слоя 1 см, используя в качестве раствора сравнения хлористоводородной кислоты раствор 0,1 М.
Содержание фтивазида в препарате (X) вычисляют по формуле:
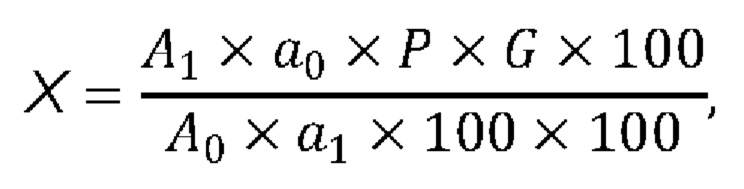
где А1 - оптическая плотность испытуемого раствора;
A0 - оптическая плотность раствора стандартного образца фтивазида;
а 1 - навеска порошка растертых таблеток, мг;
a 0 - навеска стандартного образца фтивазида, мг;
Р - содержание фтивазида в стандартном образце фтивазида, %;
G - средняя масса одной таблетки, мг;
В качестве критерия приемлемости было определено стандартное отклонение полученных результатов (RSD).
Для оценки сходимости результатов, полученных по разработанному способу и по известной методике, готовили и анализировали на количественное содержание фтивазида шесть растворов. По полученным результатам было рассчитано среднее арифметическое значение содержания фтивазида, относительное стандартное отклонение (RSD), а также доверительный интервал (табл. 5).
При использовании заявленного способа с применением ВЭЖХ обнаруженное содержание фтивазида составляет меньшее значение, чем по методу спектрофотометрии, что может указывать на небольшое содержание примесей в препарате, которые дают вклад в рассчитанное содержание фтивазида. Полученные результаты при использовании заявленного способа количественного определения фтивазида не только сопоставимы с результатами известного способа, но и демонстрируют то, что заявленный способ является более селективным и точным.
Пример 7. Для подтверждения возможности промышленного применения заявленного способа была оценена неопределенность измерения путем трех различных методов, основанных на пошаговом и всеобъемлющим подходах, а также на расчетах граничных значений доверительного интервала по критерию Стьюдента.
При пошаговом подходе оценки неопределенности измерения были определены ее основные источники, а также вклад каждого из них. При использовании всеобъемлющего подхода неопределенности были использованы данные, полученные при оценке внутрилабораторной прецизионности и правильности заявленного способа. По критерию Стьюдента был проведен расчет граничных значений доверительного интервала, который также может характеризовать степень неопределенности. Сводные данные о неопределенности измерений, полученной разными способами, обобщены и представлены в таблице 7.
Заявленное техническое решение раскрыто довольно подробно, с целью ясности понимания, и не должно рассматриваться, как ограничивающее объем притязаний.
Представленные примеры служат только для цели иллюстрации и раскрытия заявленного способа, и не должны рассматриваться как ограничивающие объем притязаний настоящего изобретения.
Промышленная применимость
Все представленные примеры подтверждают возможность применения заявленного способа в фармации, химии, медицине, а также в фармацевтической промышленности и смежных отраслях.
Таким образом, поставленная техническая задача, а именно, разработка способа количественного определения фтивазида выполнена.
Список литературы:
1. Калашников В.П., Мышка А.Ф., Способ количественного определения ларусана и/или фтивазида, заявка SU 1177733 А, C01N 21/78.
2. Калашников В.П., Мышка А.Ф. Способ определения изониазида и его гидразонов в фармакопейных препаратах, заявка SU 1236354 A1, G01N 21/78.
3. Фармакопейная статья ФС 2.1.0347. Фтивазид моногидрат.Фтивазид. Государственная фармакопея Российской Федерации. XV издание.
4. Илларионова Е.А., Абрамова Л.В., Илларионов А.И. Способ определения чистоты фтивазида и метазодина, патент RU 2225205 С2, МПК А61К 31/4409, А61К 31/455, А61Р 31/06.
5. Турусова Е.В., Насакин О.Е., Лыщиков А.Н. Способ определения производных нитрофурана, пиразола, изоникатиновой кислоты, теоаминокислот в лекарственных формах, патент RU 2479840 С2, МПК G01N 33/15.
6. Мехтиханов С.Д., Шапиев Б.И., Гебекова З.Г., Количественное определение изониазида и фтивазида в лекарственных формах, Известия ДГПУ, 2013; 4: 26.
7. Zhou Z, Chen L, Liu P, Shen M, Zou F. Simultaneous Determination of Isoniazid, Pyrazinamide, Rifampicin and Acetylisoniazid in Human Plasma by High-Performance Liquid Chromatography. Analytical Sciences, 2010, 26(11), 1133-1138. doi: 10.2116/analsci.26.1133.
8. Bhandari R, Kaur IP. A Sensitive HPLC Method for Determination of Isoniazid in Rat Plasma, Brain, Liver and Kidney. J Chromat Separation Techniq, 2012, 3(3), doi:10.4172/2157-7064.1000128.
9. Chellini PR, Eduardo BL, Franco PH, Nogueira FH, Cesar 1С, Pianetti GA, Development and Validation of an HPLC Method for Simultaneous Determination of Rifampicin, Isoniazid, Pyrazinamide, and Ethambutol Hydrochloride in Pharmaceutical Formulations. Journal of AOAC International, 2015, 98(5), 1234-1239. doi: 10.5740/jaoacint. 14-237.
| название | год | авторы | номер документа |
|---|---|---|---|
| Определение полисорбата 80 в биологических лекарственных препаратах | 2023 |
|
RU2812788C1 |
| Способ количественного определения аскорбиновой кислоты в лекарственных растительных препаратах | 2023 |
|
RU2801885C1 |
| Способ количественного определения сирингина в коре сирени обыкновенной | 2021 |
|
RU2782620C1 |
| Способ количественного определения алоэнина в свежих листьях алоэ древовидного | 2021 |
|
RU2780977C1 |
| СПОСОБ ИЗМЕРЕНИЯ КОЛИЧЕСТВА ПОЛИСОРБАТА-80 С ПРИМЕНЕНИЕМ ЩЕЛОЧНОГО ГИДРОЛИЗА ОБРАЗЦА С ПОСЛЕДУЮЩЕЙ ВЭЖХ | 2017 |
|
RU2670965C9 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ЭЛЕУТЕРОЗИДА В В КОРНЕВИЩАХ И КОРНЯХ ЭЛЕУТЕРОКОККА КОЛЮЧЕГО | 2022 |
|
RU2796764C1 |
| Определение стабилизаторов углеводной природы в биологически активных препаратах | 2023 |
|
RU2816030C1 |
| Способ количественного определения глицина в биологических лекарственных препаратах методом гидрофильной высокоэффективной жидкостной хроматографии | 2019 |
|
RU2700831C1 |
| Способ определения арбутина в листьях толокнянки | 2023 |
|
RU2802173C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ЭСТРОГЕНА И ДЕКСПАНТЕНОЛА В ДВУХКОМПОНЕНТНОМ ЛЕКАРСТВЕННОМ ПРЕПАРАТЕ МЕТОДОМ ВЭЖХ | 2011 |
|
RU2476873C1 |

Использование: для количественного определения фтивазида. Сущность изобретения заключается в том, что раствор фтивазида с концентрацией 0,2 мг/мл в ацетонитриле, обрабатывают в ультразвуковой бане, фильтруют, затем в условиях высокоэффективной жидкостной хроматографии в изократическом режиме с использованием ультрафиолетового детектора с длиной волны 330 нм, неподвижной фазы при температуре 30°С и двухкомпонентной подвижной фазы, состоящей из 0,1 М буферного раствора аммония ацетата, доведенного рН до 7,0, и ацетонитрила в соотношении 75 % об./об. : 25 % об./об. соответственно, где скорость потока компонентов подвижной фазы 1 мл/мин, проводят хроматографирование, затем проводят количественный расчет содержания фтивазида. Технический результат: обеспечение возможности количественного определения фтивазида с высокой чувствительностью и селективностью, а также обеспечение возможности сокращения времени, необходимого для количественного определения фтивазида. 3 з.п. ф-лы, 9 ил., 7 табл.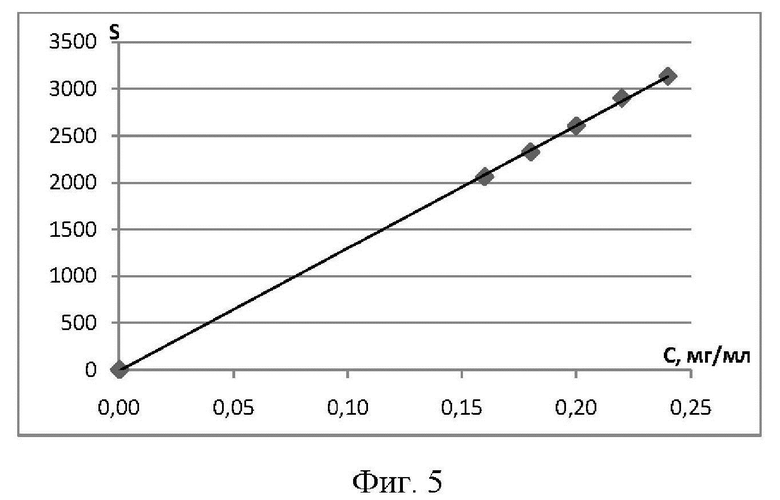
1. Способ определения фтивазида, отличающийся тем, что раствор фтивазида с концентрацией 0,2 мг/мл в ацетонитриле, обрабатывают в ультразвуковой бане, фильтруют, затем в условиях высокоэффективной жидкостной хроматографии в изократическом режиме с использованием ультрафиолетового детектора с длиной волны 330 нм, неподвижной фазы при температуре 30°С и двухкомпонентной подвижной фазы, состоящей из 0,1 М буферного раствора аммония ацетата, доведенного рН до 7,0, и ацетонитрила в соотношении 75 % об./об. : 25 % об./об., соответственно, где скорость потока компонентов подвижной фазы 1 мл/мин, проводят хроматографирование, затем проводят количественный расчет содержания фтивазида.
2. Способ по п.1, дополнительно характеризующийся тем, что фтивазид может быть в форме субстанции или таблеток.
3. Способ по п.1, дополнительно характеризующийся тем, что фильтрование проводят через мембранный фильтр диаметром 0,45 мкм.
4. Способ по п.1, дополнительно характеризующийся тем, что при определении фтивазида его примеси определяют с использованием ультрафиолетового детектора с длиной волны 238 нм.
| Способ количественного определения ларусана и/или фтивазида | 1984 |
|
SU1177733A1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ЧИСТОТЫ ФТИВАЗИДА И МЕТАЗИДА | 2001 |
|
RU2225205C2 |
| Способ определения изониазида и его гидразонов в фармакопейных препаратах | 1984 |
|
SU1236354A1 |
| CN 117665188 A, 08.03.2024 | |||
| EP 4021505 A1, 06.07.2022. | |||

