Изобретение относится к новому способу получения карбодиимидов, предпочтительно мономерных, стерически затрудненных ароматических карбодиимидов. Карбодиимиды зарекомендовали себя во многих сферах применения, например, в качестве средств защиты от гидролиза термопластичных полимеров, полиолов, полиуретанов, триглицеридов, смазочных масел и других материалов.
Синтез карбодиимидов согласно уровню техники осуществляют исходя из изоцианатов, которые подвергают карбодиимидизации посредством щелочного или гетероциклического катализа с отщеплением диоксида углерода. При этом монофункциональные или полифункциональные изоцианаты можно превращать в мономерные или полимерные карбодиимиды.
Обычно используемыми катализаторами являются соединения щелочных или щелочно-земельных металлов, а также фосфорсодержащие гетероциклические соединения (смотри Angew. Chem. 1962, 74, сс. 801-806, и Angew. Chem. 1981, 93, сс. 855-866).
Полное удаление, как правило, используемого фосфорсодержащего катализатора является затруднительным. Поскольку карбодиимиды предпочтительно используют при получении полиуретанов, присутствие фосфора даже в виде следов рассматривается как чрезвычайно негативное обстоятельство и подлежит исключению. Кроме того, получаемые мономерные карбодиимиды должны характеризоваться максимально низким индексом цвета и максимально низким содержанием изоцианатов, что, как правило, может быть технически достигнуто только либо посредством сопровождаемой потерями выхода дистилляции или многократной кристаллизации, либо посредством трудоемкого превращения в присутствии диоксида углерода (смотри европейские заявки на патент ЕР-А 0602477 и ЕР-А 2855573).
С учетом вышеизложенного в основу настоящего изобретения была положена задача предложить улучшенный способ получения определенных мономерных карбодиимидов, позволяющий получать мономерные карбодиимиды с низким индексом цвета, которые в идеальном случае не должны содержать органические фосфорные соединения и непревращенные исходные продукты, предпочтительно изоцианаты, а, следовательно, могут применяться при получении и/или стабилизации полиуретановых систем.
Неожиданно было обнаружено, что указанные выше задачи решаются благодаря предлагаемому в изобретении способу, который включает следующие стадии:
- карбодиимидизацию мономерных ароматических изоцианатов в присутствии катализатора,
- отделение низкокипящих соединений и катализатора от продукта реакции в пленочном выпарном аппарате и
- дистилляцию остатка в дополнительном пленочном выпарном аппарате.
Таким образом, объектом настоящего изобретения является способ получения мономерных ароматических карбодиимидов, который включает следующие стадии:
- карбодиимидизацию мономерных ароматических изоцианатов в присутствии катализатора,
- отделение низкокипящих соединений и катализатора от продукта реакции в пленочном выпарном аппарате и
- дистилляцию остатка в дополнительном пленочном выпарном аппарате.
При этом под мономерными ароматическими карбодиимидами согласно изобретению подразумеваются карбодиимиды, содержащие замещенные алкилом с 1-6 атомами углерода и/или алкокси с 1-6 атомами углерода фенильные остатки, которые присоединены к азоту карбодиимидной группы непосредственно через атом углерода фенильного остатка или через алкильную группу фенильного остатка.
При этом особенно предпочтительными являются мономерные карбодиимиды формулы (I):
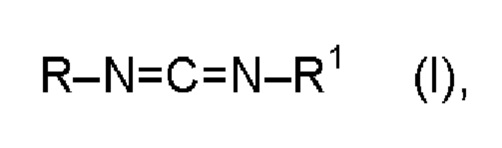
в которой
R и R1 означают 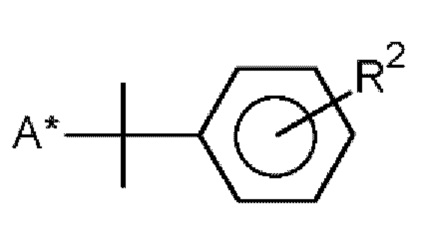 и/или
и/или 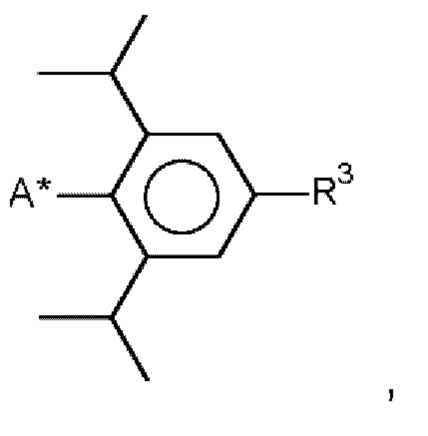
R2 означает изопропенил, трет-бутил или алкокси с 1-6 атомами углерода,
R3 означает водород, метил, этил, изопропил, н-пропил и/или трет-бутил, предпочтительно метил, этил, изопропил, н-пропил и/или трет-бутил, и
А* означает присоединение к азоту карбодиимидной функциональной группы в формуле (I).
В случае предпочтительных карбодиимидов формулы (I) предлагаемый в изобретении способ осуществляют посредством:
a) превращения (карбодиимидизации) изоцианатов, выбранных из группы, включающей R-N=C=O и/или R1-N=C=O, причем R и R1 такие, как указано выше, в присутствии катализатора,
b) отделения низкокипящих соединений и катализатора от полученного на стадии а) продукта реакции в пленочном выпарном аппарате и
c) дистилляцию образовавшегося на стадии b) остатка в дополнительном пленочном выпарном аппарате.
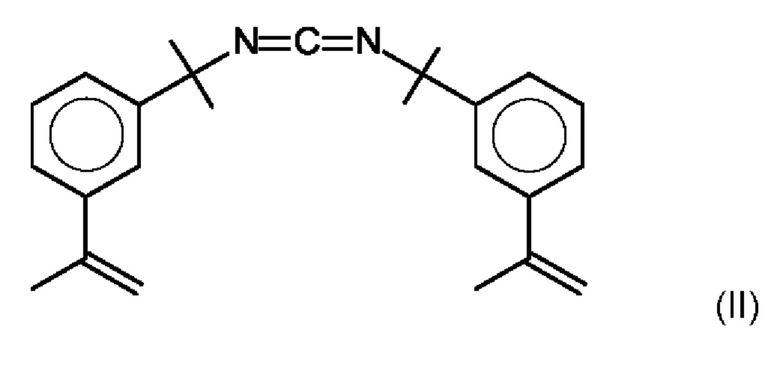
В предпочтительном варианте осуществления предлагаемого в изобретении способа карбодиимидом является соединение формулы (II):
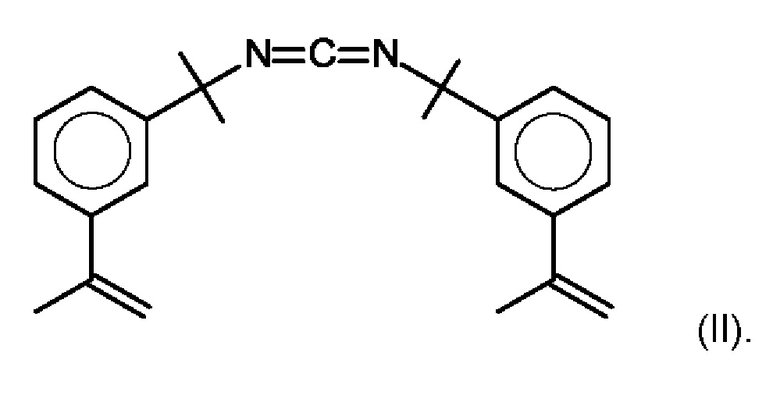
В другом предпочтительном варианте осуществления предлагаемого в изобретении способа карбодиимидом является соединение формулы (I) R-N=C=N-R1,
в которой R и R1 соответственно означают 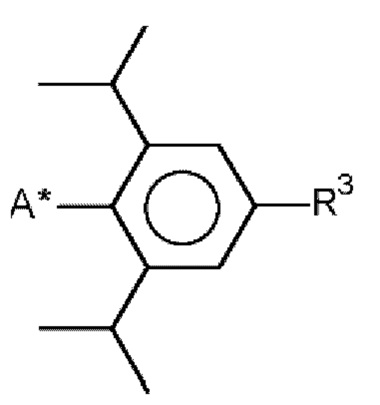 , R3 означает изопропильный остаток и А* означает присоединение к азоту карбодиимидной функциональной группы в формуле (I).
, R3 означает изопропильный остаток и А* означает присоединение к азоту карбодиимидной функциональной группы в формуле (I).
Карбодиимидизацию изоцианатов в присутствии катализатора на стадии а) предлагаемого в изобретении способа осуществляют по реакции конденсации с отщеплением диоксида углерода, например, как описано в Angew. Chem. 93, сс. 855-866 (1981), немецкой заявке на патент DE-A-1130594 или Tetrahedron Letters 48 (2007), сс. 6002-6004.
Карбодиимидизацию можно осуществлять как в массе, так и в растворителе. Кроме того, карбодиимидизацию можно начать в массе и завершить после добавления растворителя. В качестве растворителя предпочтительно используют бензины, бензол и/или алкилбензолы.
В качестве изоцианатов предпочтительно используют 3-изопропенил-α,α-диметилбензилизоцианат и/или 2,4,6 триизопропилфенилизоцианат.
В одном варианте осуществления изобретения в качестве катализаторов для получения соединений формулы (I) предпочтительно используют фосфорные соединения. Предпочтительно используют фосфоленоксиды, фосфолидины или фосфолиноксиды, а также соответствующие сульфиды. В качестве катализатора особенно предпочтительно используют алкил-фосфоленоксид с алкилом с 1-6 атомами углерода, предпочтительно метилфосфоленоксид.
Реакцию предпочтительно осуществляют в температурном интервале от 40 до 200°С.
В другом предпочтительном варианте осуществления предлагаемого в изобретении способа непосредственно после карбодиимидизации выполняют фильтрование полученного на стадии а) продукта реакции. В этом случае фильтрование предпочтительно осуществляют через патронный или мешочный фильтр, при необходимости добавляя вспомогательные средства фильтрования, в частности, предпочтительно кизельгур или активированный уголь.
Фильтрование предпочтительно осуществляют при температурах от 30 до 100°С.
Непосредственно после карбодиимидизации и при необходимости выполненного фильтрования из полученного на стадии а) продукта реакции в пленочном выпарном аппарате выделяют низкокипящие соединения и катализатор.
Согласно изобретению пленочным выпарным аппаратом является испаритель, жидкости в котором испаряются в тонкой пленке. Подобный аппарат предпочтительно приводится в действие в вакууме, а следовательно, он пригоден для разделения смесей в щадящем режиме. Согласно изобретению в качестве пленочного выпарного аппарата предпочтительно используют снабженный ротационными очистителями испаритель с падающей пленкой, например, поставляемый фирмой VTA GmbH & Co. KG.
Низкокипящими соединениями согласно изобретению предпочтительно являются образующиеся в процессе карбодиимидизации побочные продукты или непревращенные эдукты/изоцианаты с температурами кипения, которые по меньшей мере на 50°С, предпочтительно по меньшей мере на 60°С, особенно предпочтительно по меньшей мере на 70°С ниже температур кипения полученного, как указано выше, мономерного карбодиимида или соответствующей смеси карбодиимидов.
Разделение на стадии b) предпочтительно осуществляют при температуре от 150 до 200°С, предпочтительно от 160 до 190°С, особенно предпочтительно от 170 до 180°С и давлении от 0,1 до 5 мбар, предпочтительно от 0,2 до 1 мбар, особенно предпочтительно от 0,3 до 0,6 мбар.
В другом предпочтительном варианте осуществления изобретения разность температур между стадией b) и стадией с) составляет 5°С, предпочтительно 10°С.
В другом предпочтительном варианте осуществления изобретения температура на стадии с) на 5°С, предпочтительно на 10°С превышает температуру на стадии b).
В другом варианте осуществления изобретения при удалении низкокипящих соединений можно использовать так называемые разделяющие агенты. Разделяющими агентами предпочтительно являются бензолы, замещенные алкилом с 1-12 атомами углерода, и/или дибензолы и/или пирролидоны. Предпочтительным разделяющим агентом является N-метилпирролидон, N-этилпирролидон и/или ксилол.
На стадии с) осуществляют дистилляцию образующегося на стадии b) остатка в дополнительном пленочном выпарном аппарате. При этом дистилляцию предпочтительно осуществляют в дополнительном пленочном выпарном аппарате. В качестве дополнительного пленочного выпарного аппарата предпочтительно используют аппарат для испарения в глубоком вакууме.
Аппаратами для испарения в глубоком вакууме согласно изобретению являются модифицированные пленочные выпарные аппараты, конденсатор которых встроен внутрь выпарного цилиндра. Благодаря этому расстояние, которое должны преодолеть пары от пленки продукта в выпарном аппарате до конденсатора, является чрезвычайно коротким.
При этом образующийся на стадии b) остаток согласно изобретению прежде всего содержит полученные, как указано выше, мономерные карбодиимиды и образовавшиеся из них побочные продукты.
Дистилляцию образующегося на стадии b) остатка предпочтительно осуществляют на стадии с) при температуре от 160 до 220°С, предпочтительно от 165 до 210°С, особенно предпочтительно от 190 до 205°С и давлении от 0,05 до 5 мбар, предпочтительно от 0,1 до 1 мбар, особенно предпочтительно от 0,2 до 0,6 мбар.
В другом предпочтительном варианте осуществления изобретения стадии b) и с) осуществляют непрерывно без выделения образующегося на стадии b) остатка.
Полученные предлагаемым в изобретении способом мономерные карбодиимиды предпочтительно характеризуются значением b* индекса цвета менее 10 единиц (измерение осуществляют методом CIE l*a*b* согласно стандарту ISO 11664-4), а также низким содержанием мономерного изоцианата (менее 0,1%) и содержанием фосфора менее 1 ч.н.млн.
Другим объектом настоящего изобретения является применение полученных предлагаемым в изобретении способом мономерных карбодиимидов для формирования стойких к гидролизу систем на основе полиуретана, предпочтительно термопластичного полиуретана, мягкого пенополиуретана, жесткого пенополиуретана, а также термолитьевых полиуретановых эластомеров.
Согласно изобретению любые указанные выше и в дальнейшем описании общие или предпочтительные остатки, индексы, параметры и пояснения, а также соответствующие диапазоны и предпочтительные диапазоны можно любым образом комбинировать друг с другом.
Приведенные ниже примеры служат для более подробного пояснения настоящего изобретения и не ограничивают его объема.
Примеры осуществления изобретения
CDI 1 2,6-диизопропилфенилкарбодиимид
CDI 2 карбодиимид формулы (II):
CDI 3 карбодиимид формулы (I) R-N=C=N-R1, в которой R и R1 соответственно означают
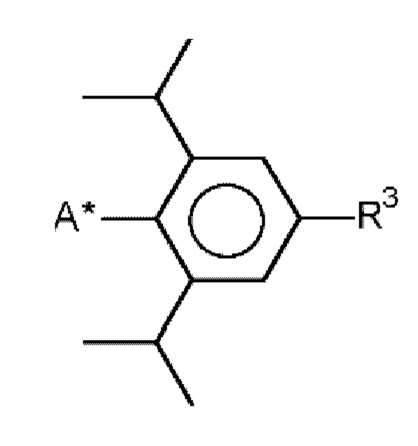 , причем R3 означает изопропил и А* означает присоединение к азоту карбодиимидной функциональной группы в формуле (I).
, причем R3 означает изопропил и А* означает присоединение к азоту карбодиимидной функциональной группы в формуле (I).
Карбодиимидизация
Карбодиимиды CDI 1, CDI 2 и CDI 3 получают в реакторе из специальной стали посредством превращения соответствующих изоцианатов: 2,6-диизопропилфенилизоцианата (для получения CDI 1), 3-изопропенил-α,α-диметилбензилизоцианата (для получения CDI 2) и 2,4,6-три-изопропилфенилизоцианата (для получения CDI 3), причем превращение осуществляют в присутствии от 200 до 500 ч.н.млн метилфосфоленоксида в качестве катализатора при температуре от 160 до 170°С и отщеплении диоксида углерода. Карбодиимидизацию осуществляет до тех пор, пока остаточное содержание изоцианата не составит менее 1%.
Полученные при этом карбодиимиды CDI 1, CDI 2 и CDI 3 затем направляют на описанную ниже дистилляцию.
a) Дистилляция и перегонка в пленочном выпарном аппарате согласно европейской заявке на патент ЕР-А-0602477 (сравнительный пример)
Сначала карбодиимиды CDI 1, CDI 2 и CDI 3 в периодическом режиме на колонке Вигре в примерном температурном интервале от 200 до 220°С и давлении от 0,3 до 0,4 мбар отделяют от низкокипящих продуктов, а затем непрерывно отгоняют в пленочном выпарном аппарате фирмы VTA GmbH & Co. KG при температуре от 190 до 200°С давлении 0,4 мбар.
b) Дистилляция в двух пленочных выпарных аппаратах (пример согласно изобретению)
Сначала карбодиимиды CDI 1, CDI 2 и CDI 3 в пленочном выпарном аппарате фирмы VTA GmbH & Co. KG освобождают от низкокипящих продуктов при температуре от 160 до 170°С и давлении 1,0 мбар и отгоняют остаток во втором пленочном выпарном аппарате, идентичном первому пленочному выпарному аппарату, при температуре от 200 до 205°С и давлении 0,4 мбар.
Цвет карбодиимидов определяют методом CIE l*a*b* согласно стандарту ISO 11664-4. Оценивают значение b*.
Остаточное содержание мономерного изоцианата определяют методом ВЭЖХ, содержание фосфора измеряют методом рентгенофлуоресцентного анализа.
Экспериментальные результаты приведены в таблице 1.
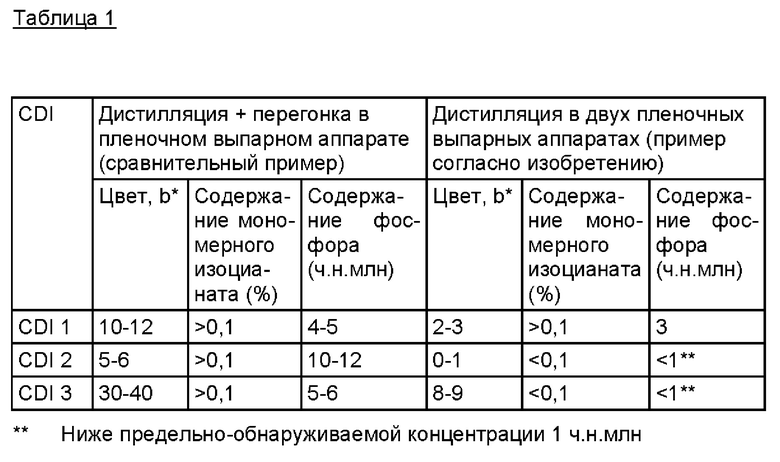
Полученные предлагаемым в изобретении способом карбодиимиды CDI 1, CDI 2 и CDI 3 по сравнению с карбодиимидами, полученными способом по уровню техники, обладают улучшенными свойствами.
Как следует из приведенных в таблице 1 данных, особенно высоким качеством (низким индексом цвета, низким содержанием токсичного мономерного изоцианата получают и отсутствием обнаруживаемого содержания органических фосфорных соединений) прежде всего характеризуются предпочтительные карбодиимиды CDI 2 и CDI 3.
Последние идеально пригодны для использования при получении и/или стабилизации полиуретановых систем.
Изобретение относится к способу получения мономерных ароматических карбодиимидов. Способ осуществляют посредством следующих стадий: a) карбодиимидизации мономерных ароматических изоцианатов в присутствии катализатора, b) отделения низкокипящих соединений и катализатора от продукта реакции со стадии а) в тонкопленочном выпарном аппарате, c) дистилляции остатка со стадии b) в дополнительном тонкопленочном выпарном аппарате. При этом остаток со стадии b) включает полученные мономерные ароматические карбодиимиды и образованные из них побочные продукты. Предлагаемый способ позволяет получать мономерные ароматические карбодиимиды с низким индексом цвета, которые не содержат органических фосфорных соединений и непревращенных исходных продуктов и могут применяться при получении и/или стабилизации полиуретановых систем. 10 з.п. ф-лы, 1 табл., 3 пр.
1. Способ получения мономерных ароматических карбодиимидов, отличающийся тем, что способ осуществляют посредством следующих стадий:
a) карбодиимидизации мономерных ароматических изоцианатов в присутствии катализатора,
b) отделения низкокипящих соединений и катализатора от продукта реакции со стадии а) в тонкопленочном выпарном аппарате,
c) дистилляции остатка со стадии b) в дополнительном тонкопленочном выпарном аппарате, причем остаток со стадии b) включает полученные мономерные ароматические карбодиимиды и образованные из них побочные продукты.
2. Способ по п. 1, отличающийся тем, что мономерными ароматическими карбодиимидами являются карбодиимиды формулы (I):
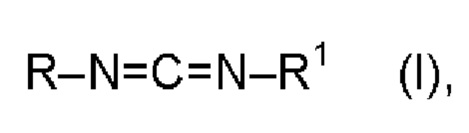
в которой
R и R1 означают 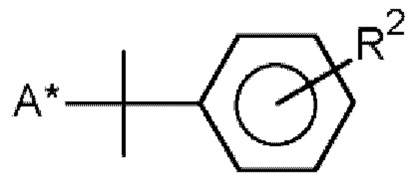 и/или
и/или 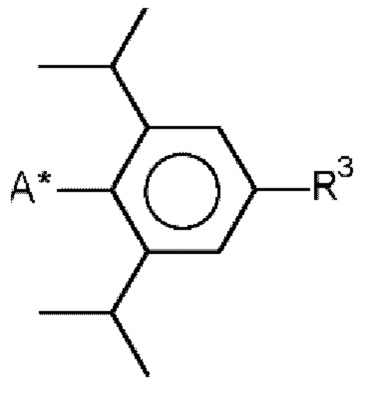 ,
,
R2 означает изопропенил, трет-бутил или алкокси с 1-6 атомами углерода,
R3 означает водород, метил, этил, изопропил, н-пропил и/или трет-бутил, предпочтительно метил, этил, изопропил, н-пропил и/или трет-бутил, и
А* означает присоединение к азоту карбодиимидной функциональной группы в формуле (I).
3. Способ по п. 2, отличающийся тем, что карбодиимид соответствует формуле (I):
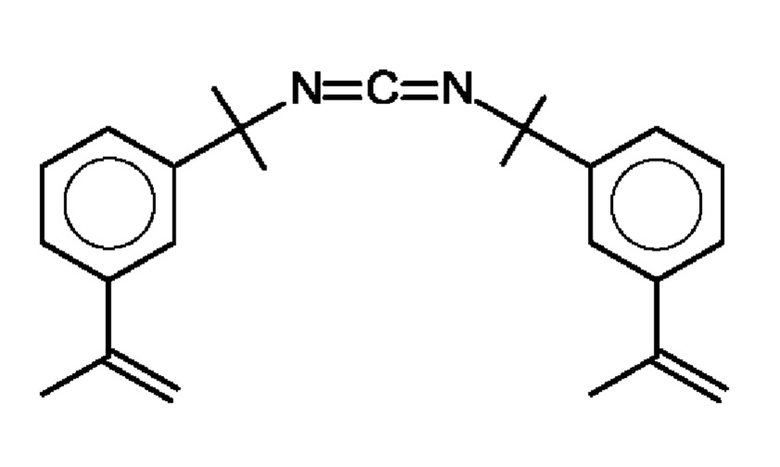
4. Способ по п. 1, отличающийся тем, что стадии дистилляции b) и с) осуществляют непрерывно.
5. Способ по п. 1, отличающийся тем, что дистилляцию на стадии b) осуществляют при температуре от 150 до 220°С, предпочтительно от 160 до 200°С, особенно предпочтительно от 170 до 190°С, и давлении от 0,1 до 5 мбар, предпочтительно от 0,2 до 1 мбар, особенно предпочтительно от 0,3 до 0,6 мбар.
6. Способ по п. 1, отличающийся тем, что при удалении низкокипящих соединений в качестве выводящего агента используют алкилзамещенные бензолы с 1-12 атомами углерода в алкиле и/или дибензолы, и/или пирролидоны.
7. Способ по п. 1, отличающийся тем, что температура на стадии с) на 5°С, предпочтительно на 10°С, выше температуры на стадии b).
8. Способ по п. 1, отличающийся тем, что дистилляцию на стадии с) осуществляют при температуре от 160 до 220°С, предпочтительно от 165 до 210°С, особенно предпочтительно от 180 до 205°С, и давлении от 0,05 до 5 мбар, предпочтительно от 0,1 до 1 мбар, особенно предпочтительно от 0,2 до 0,6 мбар.
9. Способ по п. 1, отличающийся тем, что в качестве изоцианатов используют 3-изопропенил-α,α-диметилбензилизоцианат (TMI) и/или 2,4,6-триизопропилфенилизоцианат (TRIPI).
10. Способ по п. 1, отличающийся тем, что в качестве тонкопленочного выпарного аппарата на стадии с) используют аппарат для испарения в глубоком вакууме.
11. Способ по одному из пп. 1-10, отличающийся тем, что получаемые таким образом карбодиимиды формулы (I) используют для получения стойких к гидролизу систем на основе полиуретана (PU), предпочтительно термопластичного полиуретана (TPU), мягкого и жесткого пенополиуретана, а также термолитьевых полиуретановых эластомеров.
| Устройство для обработки воды | 1976 |
|
SU602477A1 |
| RU 2014153664 A, 27.07.2016. | |||
