Настоящее изобретение касается усовершенствованного непрерывного способа получения алкил(мет)акрилатов (С) путем непрерывной переэтерификации метил(мет)акрилата (А) со спиртами (В) с выделением свободного метанола (D) по реакции, описываемой следующим уравнением:
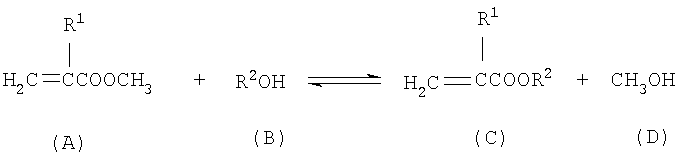
где R1 означает атом водорода или метил, R2 означает линейный, разветвленный или циклический алкильный или арильный остаток с 2-12 атомами углерода. Под группой R2 подразумевается, например, этил, н-пропил, изопропил, аллил, н-бутил, 1-метилпропил, 2-метилпропил, трет-бутил, н-пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 2,2-диметилпропил, н-гексил, 1-метилпентил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 1,1-диметилбутил, 2,2-диметилбутил, 3,3-диметидбутил, 1,2-диметилбутил, н-гептил, 1-метилгексил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 1,2-диметилпентил, 1,3-диметилпентил, 1,1-диметилпентил, 1,1,2,2-тетраметилпропил, бензил, н-октил, 2-этилгексил, н-нонил, 1-метилоктил, 2-метилоктил, н-децил, н-ундецил, 1-метилдецил, 2-метилдецил, н-додецил, 2,4-диэтилоктил, циклопентил, циклогексил, 4-трет-бутилциклогексил, циклогептил, циклододецил, 2-(диметиламино)этил, 3-(диметиламино)пропил, 4-(диметиламино)-бутил, 5-(диметиламино)пентил, 6-(диметиламино)гексил, 8-(диметиламино)октил, 10-(диметиламино)децил, 12-(диметиламино)додецил, 2-(диэтиламино)этил, 3-(диэтиламино)пропил, 4-(диэтиламино)бутил, 5-(диэтиламино)пентил, 6-(диэтиламино)гексил, 8-(диэтиламино)октил, 10-(диэтиламино)децил, 12-(диэтиламино)додецил, 2-(диизопропиламино)этил, 3-(диизопропиламино)пропил, 4-(диизопропиламино)бутил, 5-(диизопропиламино)пентил, 6-(диизопропиламино)гексил, 8-(диизопропиламино)октил, 10-(диизопропиламино)децил, 12-(диизопропиламино)додецил, 2-(дибутиламино)этил, 3-(дибутиламино)пропил, 4-(дибутиламино)бутил, 5-(дибутиламино)пентил, 6-(дибутиламино)гексил, 8-(дибутиламино)октил, 10-(дибутиламино)децил, 12-(дибутиламино)додецил, 2-(дигексиламино)этил, 3-(дигексиламино)пропил, 4-(дигексиламино)бутил, 5-(дигексиламино)пентил, 6-(дигексиламино)гексил, 8-(дигексиламино)октил, 10-(дигексиламино)децил, 12-(дигексиламино)додецил, 2-(метилэтиламино)этил, 2-(метилпропиламино)этил, 2-(метилизопропиламино)этил, 2-(метилбутиламино)этил, 2-(метилгексиламино)этил, 2-(метилоктиламино)этил, 2-(этилпропиламино)этил, 2-(этилизопропиламино)этил, 2-(этилбутиламино)этил, 2-(этилгексиламино)этил, 2-(этилоктиламино)этил, 3-(метилэтиламино)пропил, 3-(метилпропиламино)пропил, 3-(метилизопропиламино)пропил, 3-(метилбутиламино)пропил, 3-(метилгексиламино)пропил, 3-(метилоктиламино)пропил, 3-(этилпропиламино)пропил, 3-(этилизопропиламино)пропил, 3-(этилбутиламино)пропил, 3-(этилгексиламино)пропил, 3-(этилоктиламино)пропил, 4-(метилэтиламино)бутил, 4-(метилпропиламино)бутил, 4-(метилизопропиламино)бутил, 4-(метилбутиламино)бутил, 4-(метилгексиламино)бутил, 4-(метилоктиламино)бутил, 4-(этилпропиламино)бутил, 4-(этилизопропиламино)бутил, 4-(этилбутиламино)бутил, 4-(этилгексиламино)бутил, 4-(этилоктиламино)бутил, 2-(N-пиперидинил)этил, 3-(N-пиперидинил)пропил, 4-(N-пиперидинил)бутил, 5-(N-пиперидинил)пентил, 6-(N-пиперидинил)гексил, 8-(N-пиперидинил)октил, 10-(N-пиперидинил)децил, 12-(N-пиперидинил)додецил, 2-(N-пирролидинил)этил, 3-(N-пирролидинил)пропил, 4-(N-пирролидинил)бутил, 5-(N-пирролидинил)пентил, 6-(N-пирролидинил)гексил, 8-(N-пирролидинил)октил, 10-(N-пирролидинил)децил, 12-(N-пирролидинил)додецил, 2-(N-морфолино)этил, 3-(N-морфолино)пропил, 4-(N-морфолино)бутил, 5-(N-морфолино)пентил, 6-(N-морфолино)гексил, 8-(N-морфолино)октил, 10-(N-морфолино)децил, 12-(N-морфолино)додецил, 2-(N'-метил-N-пиперазинил)этил, 3-(N'-метил-N-пиперазинил)пропил, 4-(N'-метил-N-пиперазинил)бутил, 5-(N'-метил-N-пиперазинил)пентил, 6-(N'-метил-N-пиперазинил)гексил, 8-(N'-метил-N-пиперазинил)октил, 10-(N'-метил-N-пиперазинил)децил, 12-(N'-метил-N-пиперазинил)додецил, 2-(N'-этил-N-пиперазинил)этил, 3-(N'-этил-N-пиперазинил)пропил, 4-(N'-этил-N-пиперазинил)бутил, 5-(N'-этил-N-пиперазинил)пентил, 6-(N'-этил-N-пиперазинил)гексил, 8-(N'-этил-N-пиперазинил)октил, 10-(N'-этил-N-пиперазинил)децил, 12-(N'-этил-N-пиперазинил)додецил, 2-(N'-изопропил-N-пиперазинил)этил, 3-(N'-изопропил-N-пиперазинил)пропил, 4-(N'-изопропил-N-пиперазинил)бутил, 5-(N'-изопропил-N-пиперазинил)пентил, 6-(N'-изопропил-N-пиперазинил)гексил, 8-(N'-изопропил-N-пиперазинил)октил, 10-(N'-изопропил-N-пиперазинил)децил, 12-(N'-изопропил-N-пиперазинил)додецил, 3-оксабутил, 3-оксапентил, 2,2-диметил-4-оксапентил, 3,6-диоксагептил, 3,6-диоксаоктил, 3,6,9-триоксадецил, 3,6,9-триоксаундецил, 4-оксапентил, 4-оксагексил, 4-оксагептил, 4,8-диоксанонил, 4,8-диоксадецил, 4,8-диоксаундецил, 5-оксагексил или 5,10-диоксаундецил.
Кроме того, под R2OH могут подразумеваться этоксилированные и/или пропоксилированные спирты, а также смешанные этоксилированные/пропоксилированные спирты, например
R5-(O-CH2-CH2)x-OH или
R5-(O-CH(CH3)-CH2)x-OH или R5-(O-CH2-CH(CH3))x-OH,
в которых R5 означает алкил с 1-20 атомами углерода и
Х означает целое число от 10 до 20,
или этоксилированные и/или пропоксилированные аминоспирты
R3 2N(-CH2-CH2-O)y-H или
R3 2N(-CH(CH3)-CH2-O)y-H или R3 2N(-СН2СН(СН3)-О)y-N,
в которых y означает целое число от 1 до 4 и
R3 означает алкил с 1-6 атомами углерода, причем азот может также образовать с заместителями R3 пяти- - семи-членное кольцо, которое, при необходимости, может быть замещено одним или несколькими короткоцепочечными алкильными группами, например метилом, этилом или пропилом.
В способе согласно изобретению предпочтительно используют н-бутанол, изобутанол и 2-этилгексанол.
Усовершенствование по сравнению с уровнем техники состоит в том, что неожиданно оказалось возможным многократное использование тетраалкилтитаната (тетраалкоксититана) в качестве предпочтительного гомогенного катализатора путем рециркуляции. Благодаря этому значительно сокращается расход катализатора, а следовательно, существенно снижаются затраты на вспомогательные материалы, что приводит к резкому повышению рентабельности способа.
Алкил(мет)акрилаты могут быть получены путем осуществляемой различными непрерывными способами переэтерификации метил(мет)акрилата в присутствии катализаторов.
В европейской заявке на патент ЕР 0960877 (Elf Atochem S.А.) описан непрерывный способ получения сложных эфиров из метакрилатов и диалкиламиноспиртов. Диалкиламиноспирты подвергают взаимодействию с используемым в общем случае метилметакрилатом и получают диалкиламиноалкил(мет)акрилат следующим способом. Смесь используемых в качестве исходных продуктов метил(мет)акрилата и диалкиламиноспирта, содержащая в качестве гомогенного катализатора переэтерификации тетраалкилтитанат (например, тетрабутилтитанат, тетраэтилтитанат или тетра(2-этилгексил)титанат), а также, по меньшей мере, один ингибитор полимеризации (например, фенотиазин, трет-бутилкатехол, простой монометиловый эфир гидрохинона или гидрохинон), непрерывно вводят в трубчатый реактор, в котором при температуре 90-120°С происходит образование диалкиламино(мет)акрилата при одновременной непрерывной отгонке азеотропной смеси метил(мет)акрилата с метанолом. Сырую реакционную смесь (сырой эфир) направляют на первую перегонную колонну, из верхней части которой при пониженном давлении отбирают в основном освобожденный от катализатора продукт, а из куба выводят катализатор и небольшое количество диалкиламиноалкил(мет)акрилата. Отбираемый из верхней части первой перегонной колонны продукт направляют на вторую перегонную колонну, из верхней части которой при пониженном давлении отбирают низкокипящие продукты с низким содержанием диалкиламиноалкил(мет)акрилата, а из куба выводят остаток, содержащий основное количество диалкиламиноалкил(мет)акрилата и ингибитор (ингибиторы) полимеризации, который направляют на третью перегонную колонну. Третья перегонная колонна предназначена для ректификации при пониженном давлении, причем из ее верхней части отбирают чистый целевой эфир (диалкиламиноалкил(мет)акрилат), а в кубе остается в основном ингибитор (или ингибиторы) полимеризации. После дополнительной очистки на пленочном выпарном аппарате кубовый остаток первой перегонной колонны аналогично продукту, отбираемому из верхней части второй перегонной колонны, возвращают в реактор.
В указанном патенте отсутствуют подробности, касающиеся рециклизации гомогенного катализатора. Из приведенного в нем описания очистки кубового остатка первой перегонной колонны на пленочном выпарном аппарате можно сделать единственный вывод, что остаточный диалкиламиноалкил(мет)акрилат отделяют от катализатора и высококипящих побочных продуктов и возвращают в реактор.
В европейской заявке на патент ЕР 0968995 (Mitsubishi Gas Chemical Comp.) описан непрерывный способ получения сложных эфиров алкил(мет)акриловой кислоты с использованием реакционной колонны. При этом переэтерификацию осуществляют непосредственно в перегонной колонне (то есть реактор и колонна для отгонки азеотропной смеси метил(мет)акрилата и метанола совмещены, образуя единый аппарат), в которую непрерывно вводят исходные метил(мет)акрилат и спирт. Необходимый катализатор, в данном случае предпочтительно также соединение титана, находится внутри перегонной колонны. В случае использования гомогенного катализатора его непрерывно дозируют в перегонную колонну. Однако происходящее внутри перегонной колонны вымывание гомогенных катализаторов жидкой флегмой приводит к их повышенному расходу и загрязнению внутренних элементов колонны твердым осадком катализатора. В случае использования гетерогенного катализатора он находится внутри реакционной колонны. Однако недостатком позиционирования катализатора в перегонной колонне является повышенная потеря давления внутри нее, а регулярная очистка колонны связана с чрезвычайно высокими издержками. Кроме того, гетерогенные катализаторы могут утратить каталитическую активность, например, вследствие нежелательного протекания полимеризации.
В указанном патенте отсутствуют какие-либо сведения, касающиеся порядка рециклизации предварительно отделенного от сырого эфира гомогенного катализатора в реакционную колонну.
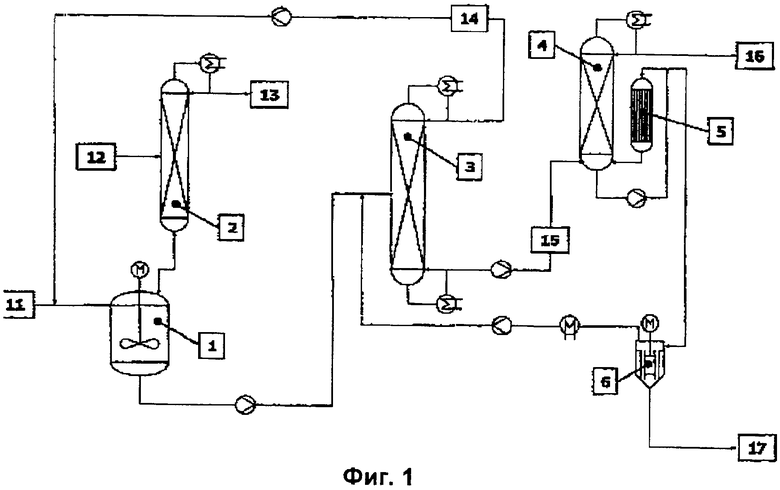
В немецкой заявке на патент DE 10200171.5 описан непрерывный способ получения алкил(мет)акрилатов путем переэтерификации метил(мет)акрилата более высококипящими по сравнению с метанолом спиртами, гораздо лучший нежели рассмотренные выше способы (смотри фиг.1).
Используемые в качестве исходных компонентов метил(мет)акрилат (ММА, 11) и спирт (12) непрерывно подают в пригодный реакционный аппарат (1), причем можно использовать как отдельную реакционную емкость, так и каскад, образованный несколькими последовательно соединенными реакционными емкостями. Для улавливания выделяющегося при переэтерификации свободного метанола все реакционные емкости целесообразно оборудовать системой отбора выпара в колонну азеотропной перегонки (2). Предпочтительно используемый в качестве гомогенного катализатора тетраалкилтитанат (количество тетраалоксититана по отношению к исходному ММА предпочтительно составляет 0,2-0,5 мас.%) аналогично ингибитору/ингибиторам полимеризации непрерывно дозируют в реакционный аппарат (1). Однако в качестве катализатора могут использоваться и любые другие известные из уровня техники, пригодные для переэтерификации катализаторы. Под ингибитором полимеризации подразумевается, например, простой монометиловый эфир гидрохинона в сочетании с кислородом. Поскольку исходный спирт может содержать воду (содержание воды в нем составляет от 0,05 до 0,005 мас.% в пересчете на н-бутанол), перед подачей в реакционный аппарат его предпочтительно подвергают обезвоживанию путем осуществляемой в колонне (2) азеотропной перегонки. При этом содержащуюся в спирте воду отбирают в верхней части перегонной колонны (2). Во избежание загрязнения азеотропной смеси метанола с ММА (13) исходным спиртом последний предпочтительно вводят в нижнюю часть перегонной колонны (2). Однако исходный спирт может быть обезвожен и на предварительно присоединенной обезвоживающей перегонной колонне, а также путем обработки с помощью агента обезвоживания, например молекулярного сита или путем мембранного разделения, например испарения через полунепроницаемую перегородку. Обезвоживание исходного спирта имеет большое значение, поскольку содержащаяся в нем вода может привести к необратимому повреждению катализатора (например, тетраалкилтитаната) в реакторе. Благодаря осуществлению стадии обезвоживания предотвращают гидролиз катализатора, а следовательно, исключают издержки, обусловленные повышенным расходом катализатора и проблемами, возникающими вследствие осаждения твердых веществ. Переэтерификацию в реакционном аппарате (1) осуществляют при температуре от 80 до 160°С. Предпочтительным является температурный интервал от 110 до 135°С. Чтобы оказать положительное воздействие на равновесие реакции, выделяющийся свободный метанол отгоняют из реакционной смеси на перегонной колонне (2) в виде азеотропной смеси с ММА (13). Реакционную смесь, в основном состоящую из алкил(мет)акрилата (целевого продукта) и непревращенных ММА и спирта, содержащую небольшие количества метанола, катализатора и ингибиторов полимеризации, а также чрезвычайно малое количество побочных продуктов, по истечении 0,5-3 часов пребывания в реакторе (предпочтительное время пребывания составляет 0,75-1,5 часа) направляют в непрерывно функционирующую перегонную колонну (3). В этой колонне при пониженном давлении, предпочтительно составляющем 20-200 мбар, выделяют компоненты, кипящие при более низкой температуре по сравнению с целевым эфиром, которыми преимущественно являются метанол, ММА и непревращенный исходный спирт. Указанные компоненты отбирают в верхней части перегонной колонны (3) и возвращают (14) в реактор. Благодаря такой рециркуляции в соответствии с общим материальным балансом технологического процесса достигают практически полного превращения исходных продуктов (ММА и спирта). Остающийся в кубе перегонной колонны (3) неочищенный сырой эфир (15), содержащий катализатор, ингибитор полимеризации, высококипящие побочные продукты и предпочтительно более 98 мас.% целевого эфира, непрерывно направляют на стадию дополнительной переработки (4, 5), осуществляемой путем вакуумной перегонки при предпочтительном давлении от 20 до 200 мбар. На данной стадии из верхней части колонны (4) непрерывно отбирают целевой эфир высокой степени чистоты (16). Если для отделения катализатора и ингибиторов полимеризации, а также высококипящих побочных продуктов от сырого эфира (15) используют обычную, предназначенную для вакуумной перегонки колонну согласно уровню техники, то в ее кубе вследствие недопустимо сильного температурного воздействия происходит разложение катализатора, а следовательно, выделение свободного исходного спирта с частичным образованием простых эфиров исходного спирта. По сравнению с целевым эфиром эти соединения (исходный спирт и простые эфиры исходного спирта) имеют более низкую температуру кипения, а следовательно, попадают в целевой продукт в качестве примесей, вызывая значительное ухудшение его качества. Данная проблема может быть решена благодаря использованию для отделения целевого эфира от катализатора и ингибиторов полимеризации, а также высококипящих побочных продуктов пленочного выпарного аппарата (5) со щадящим режимом испарения. Такими известными пригодными аппаратами являются выпарные аппараты с падающей пленкой, пленочные испарители и аппараты для перегонки в глубоком вакууме. Для обеспечения максимальной степени чистоты конечного продукта (содержания целевого эфира более 99,9 мас.%, спирта менее 120 частей на млн., ММА менее 10 частей на млн., простых эфиров менее 5 частей на млн. и показателя прозрачности (альфа) менее 1) используют дополнительную, последовательно присоединенную ступень отгонки (4) целевого эфира от высококипящих продуктов. Недостаток использования для этой цели единственного аппарата пленочного испарения состоит в невысокой эффективности очистки целевого эфира, вследствие чего высококипящие побочные продукты попадают в чистый целевой эфир (16). Данную проблему решают благодаря тому, что для отделения высококипящих побочных продуктов от чистого целевого эфира используют вакуумную ректификационную колонну (4), которую монтируют над пленочным выпарным аппаратом. После отделения катализатора и ингибиторов полимеризации, а также высококипящих побочных продуктов от сырого эфира в кубовом остатке остается определенное количество целевого эфира, в связи с чем кубовый остаток сохраняет высокую текучесть и способность к транспортированию. Чтобы при выведении катализатора, ингибиторов полимеризации и высококипящих побочных продуктов через шлюзовый затвор (17) свести к минимуму потери целевого эфира, необходимо дополнительно присоединить вакуумный испаритель (6) с предпочтительным интервалом рабочих давлений от 20 до 200 мбар. Для этой цели также оказался пригоден пленочный выпарной аппарат. Пригодными выпарными аппаратами в данном случае также являются известные выпарные аппараты с падающей пленкой, пленочные испарители и аппараты для перегонки в глубоком вакууме. Отбираемый из верхней части испарителя (6) целевой эфир в связи с недопустимо высоким содержанием в нем высококипящих компонентов не соответствует нормативам спецификации на чистый целевой эфир. Кроме того, он содержит исходный спирт и частично простые эфиры исходного спирта, образующиеся вследствие термического разрушения катализатора. По этим причинам отбираемый из верхней части испарителя (6) продукт не может быть направлен непосредственно в колонну (4), предназначенную для отделения целевого эфира от высококипящих продуктов, а напротив, его необходимо вернуть в реакционный аппарат (1) или предпочтительно в предназначенную для отгонки низкокипящих продуктов колонну (3), чтобы отделить эти продукты перед подачей в первый испаритель (5).
Указанный способ имеет недостаток, заключающийся в том, что подвергнутый многократному температурному воздействию, а следовательно, по всей вероятности поврежденный и частично дезактивированный гомогенный катализатор после отделения вместе с ингибиторами полимеризации и высококипящими побочными продуктами от целевого эфира полностью выводят через шлюзовый затвор (17) и не используют повторно путем рециклизации. В связи с довольно высокой стоимостью катализатора это приводит к повышенным расходам на вспомогательные материалы.
В немецкой заявке на патент DE 10127939 (BASF) описан непрерывный способ переэтерификации. Одной из задач этого изобретения является необходимость реализации не связанного с какими-либо проблемами повторного использования катализатора. Таким образом, в основу изобретения заявки DE 10127939 была положена задача разработки способа, не имеющего указанных выше недостатков и удовлетворяющего следующим критериям.
1. Исходные вещества (катализатор, стабилизатор, низкомолекулярный сложный эфир (мет)акриловой кислоты) должны иметь подходящую цену, пригодность для переработки, не сопровождаемую какими-либо проблемами, и доступность в объемах, приемлемых с технической точки зрения.
2. Катализатор должен быть стабилен при повышенной температуре и в присутствии небольших количеств влаги.
3. Потеря активности катализатора должна быть незначительной, и он должен быть пригоден для беспроблемного повторного использования.
4. В реакцию переэтерификации вместе с возвращаемым со стадии шлюзования катализатором переэтерификации не должен попадать какой-либо чужеродный спирт.
5. Пробег установки должен быть длительным, то есть должны быть сведены к минимуму проблемы, обусловленные полимеризацией, и должно использоваться оборудование, требующее как можно более редкого ремонта.
6. Должна существовать возможность непосредственного повторного использования или утилизации смеси побочных продуктов или азеотропа, состоящего из низкомолекулярного алканола и соответствующего сложного эфира.
7. Должна существовать возможность максимально полной регенерации остаточных ценных веществ, содержащихся в отработанных материальных потоках и побочных продуктах.
8. Переэтерификация предпочтительно должна осуществляться непрерывным способом.
9. Целевой эфир должен иметь высокую степенью чистоты (по меньшей мере, 99,9 мас.%), а в случае получения диалкиаминометил(мет)акрилатов количество образующихся этиленгликольди(мет)акрилата и винилоксиэтил(мет)акрилата должно быть незначительным, по возможности составляя не более 100 частей на млн.
10. Количество отработанных веществ по возможности должно быть минимальным, и они должны быть хорошо пригодны для переработки.
11. Степень превращения и выход должны быть высокими (более 95%).
12. Время пребывания реакционных компонентов в реакторе должно быть минимальным.
13. Способ в целом должен быть технически простым и рентабельным.
Хотя задача рециркуляции катализатора и была сформулирована в указанном изобретении заявки DE 10127939, в его описании отсутствуют какие-либо сведения, касающиеся целенаправленного возврата активного или сохранившего частичную активность катализатора. Кроме того, отсутствуют данные, касающиеся количественных характеристик рециркуляции катализатора и вероятного изменения качества целевого продукта, образующегося в результате использования полностью или частично возвращаемого катализатора переэтерификации.
Задача настоящего изобретения состоит в том, чтобы предоставить в распоряжение непрерывный способ взаимодействия сложных метиловых эфиров (мет)акриловой кислоты (А) со спиртами (В), имеющими более высокую температуру кипения по сравнению с метанолом (D), согласно которому используемый гомогенный катализатор можно многократно использовать путем частичной рециклизации. Под сложными эфирами (мет)акриловой кислоты (алкил(мет)акрилатами) в дальнейшем подразумеваются сложные эфиры и производные акриловой и метакриловой кислот. Кроме того, благодаря новому способу в распоряжение должен быть предоставлен продукт, имеющий более высокое качество по сравнению с аналогичной продукцией, поставляемой ныне на рынок сбыта. Вместе с тем новый способ получения алкил(мет)акрилатов должен быть связан с максимально низкими издержками, должен быть более энергетически выгоден и связан с меньшим расходом вспомогательных материалов (то есть должен быть менее дорогостоящим). Указанные задачи, а также другие, вытекающие из вступительного рассмотрения уровня техники задачи, не подвергнутые более детальному анализу, решаются следующим образом.
На основании анализа заданного Рëмом в немецкой заявке на патент DE 10200171.5 уровня техники неожиданно было обнаружено, что предпочтительно используемый в качестве гомогенного катализатора тетраалкилтитанат (тетраалкоксититан), несмотря на оказываемое на него в разных точках технологического процесса (кубе перегонных колонн и пленочных выпарных аппаратов) воздействие высоких тепловых нагрузок, все же постоянно сохраняет высокую каталитическую активность. Таким образом, предоставляется возможность многократного использования катализатора, которое выходит за рамки его однократного пропускания. Благодаря этому можно существенным образом сократить расходы на вспомогательные материалы и одновременно сохранить неизменными качественные показатели целевого продукта и его суммарный выход и выход на единицу объема реактора. Для достижения указанной цели дополнительно требуется лишь один разделитель потока и один насос. Регулирование расхода возвращаемого в реакционный аппарат (1) катализатора осуществляется на основании достигаемой в нем степени превращения спирта (В) или ММА (А) в качестве меры фактически сохраняющейся активности катализатора. В качестве дополнительного показателя фактически сохраняющейся активности катализатора можно использовать расход и состав рециркулируемой низкокипящей фракции (14).
В принципе возможны следующие варианты технического решения рециркуляции катализатора.
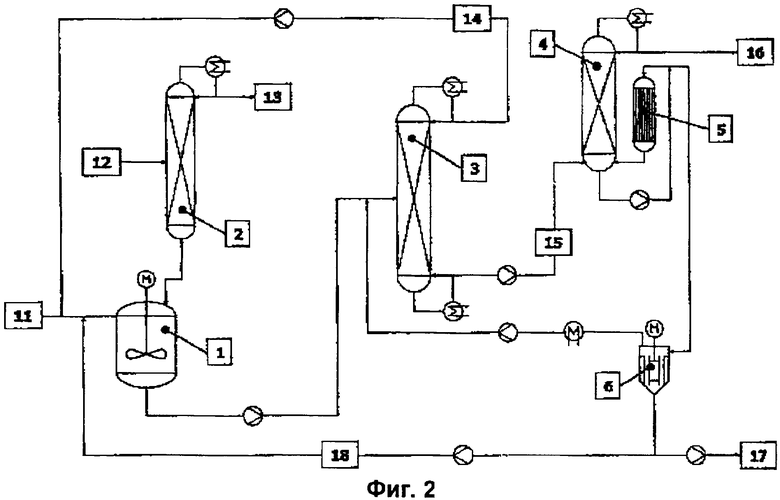
Вариант решения 1 (фиг.2).
Кубовый остаток вакуумного испарителя (6), содержащий катализатор, ингибиторы полимеризации, а также высококипящие побочные продукты и остаточное количество целевого эфира, разделяют на части и часть (18) возвращают в реакционный аппарат (1). Другую часть кубового остатка выводят через шлюзовый затвор (17). Выводимая через шлюзовый затвор часть кубового остатка (17) соответствует образующемуся в стационарном режиме количеству высококипящих побочных компонентов и сохраняющейся остаточной активности катализатора, содержащегося в кубовом остатке вакуумного испарителя (6). В отличие от технологического режима без рециклизации катализатора количество свежего катализатора в потоке (11) сокращается в соответствии с количеством возвращаемого катализатора.
Возвратный поток (18) образован 1-95 мас.%, предпочтительно 40-90 мас.% и еще более предпочтительно 60-85 мас.% кубового остатка вакуумного испарителя (6).
Вариант решения 2 (фиг.3).
Остающийся после отделения высокочистого целевого эфира в вакуумном выпарном аппарате (5) со щадящим режимом испарения кубовый остаток, содержащий катализатор, ингибиторы полимеризации, а также высококипящие побочные продукты и остаточное количество целевого эфира, разделяют на части и часть (18) возвращают в реакционный аппарат (1). Другую часть кубового остатка направляют в соответствии с уровнем техники в вакуумный испаритель (6), в котором выделяют бòльшую часть конечного эфира и возвращают его в реакционный аппарат (1) или предпочтительно в предназначенную для отгонки низкокипящих продуктов перегонную колонну (3). Кубовый остаток вакуумного испарителя (6), содержащий катализатор, ингибиторы полимеризации, а также высококипящие побочные продукты и остаточное количество конечного эфира, выводят через шлюзовый затвор (17). Выводимая через шлюзовый затвор (17) часть кубового остатка соответствует образующемуся в стационарном режиме количеству высококипящих побочных компонентов и сохраняющейся остаточной активности катализатора, содержащегося в кубовом остатке стадии вакуумной перегонки (4, 5). В отличие от технологического режима без рециклизации катализатора количество свежего катализатора в потоке (11) сокращается в соответствии с количеством возвращаемого катализатора.
Обратный поток (18) образован 1-95 мас.%, предпочтительно 40-90 мас.% и еще более предпочтительно 60-85 мас.% кубового остатка пленочного выпарного аппарата (5).
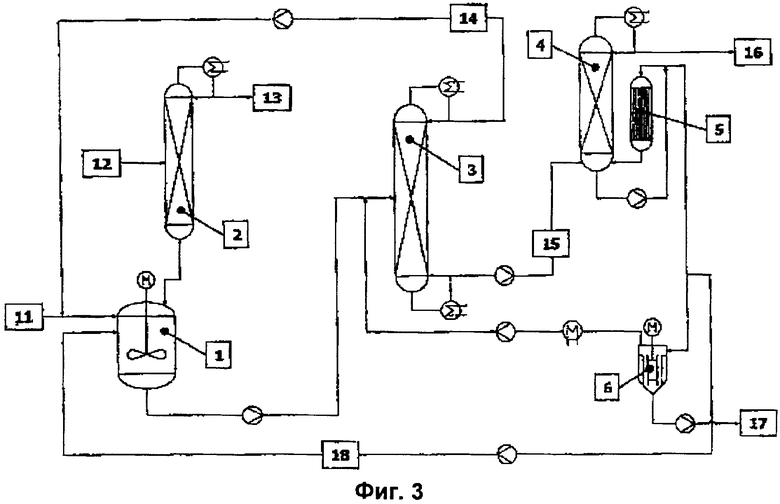
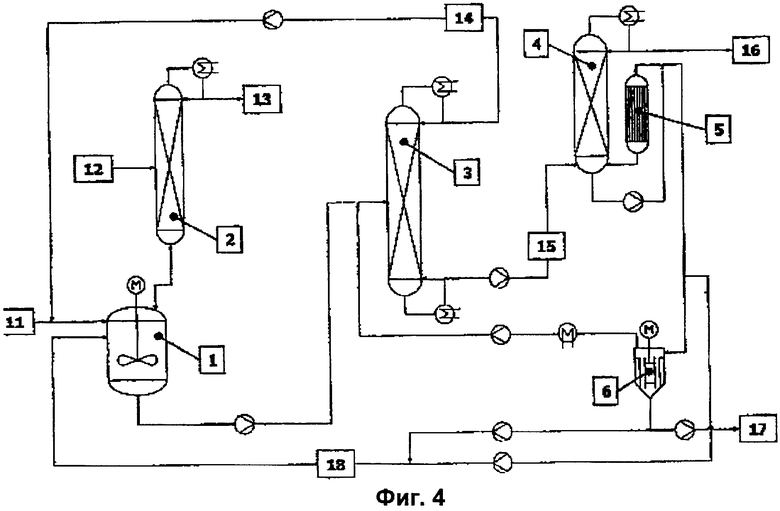
Вариант решения 3 (фиг.4).
Остающийся после отделения высокочистого конечного эфира кубовый остаток вакуумного выпарного аппарата (5) со щадящим режимом испарения, содержащий катализатор, ингибиторы полимеризации, а также высококипящие побочные продукты и остаточное количество конечного эфира, разделяют на части и часть (18) возвращают в реакционный аппарат (1). Другую часть кубового остатка в соответствии с уровнем техники направляют в вакуумный испаритель (6), в котором выделяют бòльшую часть целевого эфира и возвращают его в реакционный аппарат (1) или предпочтительно в предназначенную для отгонки низкокипящих продуктов перегонную колонну (3). Кубовый остаток вакуумного испарителя (6), содержащий катализатор, ингибиторы полимеризации, а также высококипящие побочные продукты и остаточное количество целевого эфира, разделяют на части и часть также возвращают в реакционный аппарат (1) в виде потока (18). Другую часть кубового остатка выводят через шлюзовый затвор (17). Выводимая через шлюзовый затвор (17) часть кубового остатка соответствует образующемуся в стационарном режиме количеству высококипящих побочных компонентов и сохраняющейся остаточной активности катализатора, содержащегося в рециклизуемом потоке (18). В отличие от рабочего режима без рециклизации катализатора количество свежего катализатора в потоке (11) сокращается в соответствии с количеством возвращаемого катализатора.
Обратный поток (18) образован 1-95 мас.%, предпочтительно 40-90 мас.% и еще более предпочтительно 60-85 мас.% от суммы кубового остатка пленочного выпарного аппарата (5) и кубового остатка вакуумного испарителя (6).
Примеры
Приведенные ниже примеры служат для пояснения способа согласно изобретению без его ограничения.
Примеры выполнены на экспериментальной установке в пилотном масштабе: расход исходных материалов (ММА и спирта) составлял 6-8 кг/ч, что соответствовало выходу целевого эфира 5-6 кг/ч. Состав опытной установки соответствовал представленной на фиг.3 технологической схеме или несколько отличался от нее.
Пример 1 (сравнительный). Получение н-бутилметакрилата/изобутилметакрилата без рециркуляции катализатора.
В качестве реакционного аппарата (1) использовали обогреваемую водяным паром емкость из нержавеющей стали с максимальным заполняемым объемом 15 литров. Реактор посредством линии отбора выпара был соединен со смонтированной над ним перегонной колонной (2). В качестве колонны для азеотропной отгонки (абсолютное давление в верхней части 1 атмосфера) использовали экспериментальную стеклянную колонну диаметром 0,1 м, на высоту 2 м заполненную насадкой из металлической ткани Sulzer CY. В средней части колонны (на уровне 1 м) располагался впускной штуцер, предназначенный для подачи исходного спирта. Выходящий из реактора поток реакционных продуктов непрерывно поступал в предназначенную для отгонки низкокипящих веществ перегонную колонну (3). В качестве перегонной колонны (3) использовали экспериментальную стеклянную колонну для вакуумной перегонки (абсолютное давление в верхней части 120 мбар) диаметром 0,1 м, на высоту 3,8 м заполненную насадкой из металлической ткани Sulzer CY. Впускной штуцер располагался на уровне 2 м. Куб колонны обогревали водяным паром. Конденсированный в верхней части колонны продукт (рециклизуемый поток 14) непрерывно возвращали в реактор. Вместо изображенного на фиг.3 пленочного выпарного аппарата с падающей пленкой (5) для непрерывной переработки кубового остатка (15) перегонной колонны (3) использовали обогреваемый горячим маслом пленочный испаритель с площадью рабочей поверхности А, равной 0,15 м2. Выпар из этого пленочного испарителя непрерывно подавали в верхнюю часть смонтированной над ним колонны (4), предназначенной для отгонки эфира от высококипящих продуктов. Речь при этом идет об экспериментальной стеклянной колонне для вакуумной перегонки (абсолютное давление в верхней части 120 мбар) диаметром 0,05 м, на высоту 0,5 м заполненной насадкой из металлической ткани Sulzer EX. Кубовый остаток непрерывно направляли во второй пленочный испаритель (6) с абсолютным давлением в верхней части 120 мбар, имеющий меньшие размеры (рабочая поверхность А равна 0,02 м2), также обогреваемый горячим маслом. Выпар этого второго пленочного испарителя конденсировали и, объединив с выходящими из реактора реакционными продуктами, непрерывно подавали в колонну для отгонки низкокипящих продуктов. Кубовый остаток непрерывно выводили из технологического процесса через шлюзовый затвор (17). Исходные продукты (ММА и спирт) непрерывно подавали посредством поршневых дозирующих насосов, причем катализатор (тетраалкилтитанат) дозировали в виде раствора в исходном ММА (согласно спецификации ММА не содержал влагу). Раствор катализатора в исходном ММА вводили непосредственно в реактор, а исходный спирт в среднюю часть колонны для азеотропной перегонки, предварительно нагрев его до внутренней температуры этой колонны. Непрерывную подачу раствора стабилизатора (0,2 мас.% простого монометилового эфира гидрохинона в ММА или целевом эфире) в возвратный поток перегонных колонн осуществляли со скоростью 50-100 г/ч посредством шланговых насосов. Непрерывное перемещение материальных потоков между отдельными узлами установки осуществляли посредством поршневых дозирующих насосов или благодаря всасывающему действию разрежения. По возможности избегали использования промежуточных емкостей (буферных объемов). Состав материальных потоков, то есть содержание ММА, спирта, метанола (МеОН) и целевого эфира, определяли с помощью газовых хроматографов.
Для непрерывного получения н-бутилметакрилата (н-БМА) в реакционный сосуд дозировали исходные реагенты (11): ММА (4 кг/ч), тетра-н-бутилтитанат (18 г/ч) и н-бутанол (2,7 кг/ч). Кроме того, в реактор направляли рециркулируемый из верхней части колонны для отгонки низкомолекулярных продуктов поток (2,8 кг/ч) следующего состава: 1,0 мас.% н-БМА, 38,3 мас.% н-бутанола, 57,3 мас.% ММА и 3,4 мас.% MeOH. Молярное соотношение ММА: н-бутанол в подаваемом в реактор потоке составляло 1,1:1. При времени пребывания реагентов в реакторе 1 час и азеотропной отгонке смеси ММА / МеОН (1,5 кг/ч) устанавливалась реакционная температура 115°С. Азеотропная смесь ММА / МеОН состояла из 82 мас.% МеОН и 18 мас.% ММА и содержала менее 5 частей на млн. н-бутанола. Выводимый из реактора поток реакционных продуктов (8 кг/ч) имел следующий состав: 64,6 мас.% н-БМА, 13,5 мас.% н-бутанола, 20,3 мас.% ММА, 1,3 мас.% МеОН и 0,3 мас.% побочных продуктов. Таким образом, выход н-БМА в расчете на единицу объема реактора составлял 570 кг/ч/м3. В связи с практически полным отделением более низкокипящих по сравнению с н-БМА компонентов в кубе колонны для отгонки низкокипящих продуктов получали сырой эфир (5,8 кг/ч), уже содержащий более 99,5 мас.% н-БМА, а также весь катализатор и стабилизатор. Таким образом, выход по н-бутанолу для процесса в целом составлял почти 100%. Выход по ММА для процесса в целом за вычетом заранее рассчитанных потерь ММА с азеотропной смесью ММА/МеОН также составлял почти 100%. При коэффициенте испарения (то есть отношении выпара к исходному потоку) в первом, более крупном пленочном испарителе, равном около 90%, в верхней части колонны для отгонки от высококипящих продуктов в конечном итоге получали 5,1 кг/ч чистого н-БМА следующего состава: свыше 99,92 мас.% н-БМА, менее 120 частей на млн. н-бутанола, менее 10 частей на млн. ММА, менее 5 частей на млн. ди-н-бутилового эфира, который имел показатель прозрачности (альфа) менее 0,2. При коэффициенте испарения во втором, имеющем меньшие размеры пленочном испарителе около 90%, общее количество выводимых через шлюзовый затвор продуктов (катализатора, стабилизатора, высококипящих побочных продуктов, н-БМА) составляло 0,1 кг/ч, а потери выхода н-БМА в расчете на чистый полученный н-БМА составляли менее 0,5 мас.%.
Расход катализатора (тетра-н-бутилтитаната) составлял 3,5 г/кг чистого н-БМА.
Для непрерывного получения изобутилметакрилата (изо-БМА) в реакционный сосуд дозировали исходные реагенты: ММА (3,4 кг/ч), тетраизобутилтитанат (19 г/ч) и изобутанол (2,36 кг/ч). Кроме того, в реактор направляли рециркулируемый из верхней части колонны для отгонки низкомолекулярных продуктов поток (2,4 кг/ч) следующего состава: 6,2 мас.% изо-БМА, 35,3 мас.% изобутанола, 56,3 мас.% ММА и 2,2 мас.% МеОН. Молярное соотношение ММА : изобутанол в направляемом в реактор потоке составляло 1,1:1. При времени пребывания реагентов в реакторе 1,2 часа и азеотропной отгонке смеси ММА / МеОН (1,26 кг/ч) устанавливалась реакционная температура 115°С. Азеотропная смесь ММА/МеОН состояла из 82 мас.% МеОН и 18 мас.% ММА и содержала менее 5 частей на млн. изобутанола. Выводимый из реактора поток реакционных продуктов (6,9 кг/ч) имел следующий состав: 67,3 мас.% изо-БМА, 12,0 мас.% изобутанола, 19,4 мас.% ММА, 0,8 мас.% МеОН и 0,5 мас.% побочных продуктов. Таким образом, выход изо-БМА в расчете на единицу объема реактора составлял 516 кг/ч/м3. В результате практически полного отделения более низкокипящих по сравнению с изо-БМА компонентов в кубе колонны для отгонки низкокипящих продуктов получали сырой эфир (5,0 кг/ч), уже содержащий более 99,5 мас.% изо-БМА, а также весь катализатор и стабилизатор. Следовательно, выход по изобутанолу для процесса в целом составлял почти 100%. Выход по ММА для процесса в целом за вычетом заранее рассчитанных потерь ММА с азеотропной смесью ММА0/МеОН также составлял почти 100%. При коэффициенте испарения (то есть отношении выпара к исходному потоку) в первом, более крупном пленочном испарителе, равном около 90%, в верхней части колонны для высококипящих продуктов в конечном итоге получали 4,5 кг/ч чистого изо-БМА следующего состава: свыше 99.9 мас.% изо-БМА, менее 150 частей на млн. изобутанола, менее 10 частей на млн. ММА, 0 частей на млн. диизобутилового эфира, который имел показатель прозрачности (альфа) менее 0,2. При коэффициенте испарения во втором, имеющем меньшие размеры пленочном испарителе около 90%, общее количество выводимых через шлюзовый затвор продуктов (катализатора, стабилизатора, высококипящих побочных продуктов, изо-БМА) составляло 0,05 кг/ч, а потери выхода изо-БМА в расчете на чистый полученный изо-БМА составляли менее 0,5 мас.%.
Расход катализатора (тетраизобутилтитаната) составлял 4,2 г/кг чистого изо-БМА.
Пример 2. Получение н-бутилметакрилата с полной рециркуляцией катализатора.
В качестве реакционного аппарата (1) использовали обогреваемый водяным паром сосуд из нержавеющей стали с максимальным загружаемым объемом 15 литров. Реактор посредством линии отбора выпара был соединен со смонтированной над ним перегонной колонной (2). В качестве колонны для азеотропной отгонки (абсолютное давление в верхней части 1 атмосфера) использовали экспериментальную стеклянную колонну диаметром 0,1 м, на высоту 2 м заполненную насадкой из металлической ткани Sulzer CY. В средней части колонны (на уровне 1 м) располагался впускной штуцер, предназначенный для подачи исходного спирта. Выходящий из реактора поток реакционных продуктов непрерывно поступал в предназначенную для отгонки низкокипящих веществ перегонную колонну (3). В качестве перегонной колонны (3) использовали экспериментальную стеклянную колонну для вакуумной перегонки (абсолютное давление в верхней части 120 мбар) диаметром 0,1 м, на высоту 3,8 м заполненную насадкой из металлической ткани Sulzer CY. Впускной штуцер располагался на уровне 2 м. Куб колонны обогревали водяным паром. Конденсированную верхнюю фракцию (рециклизованный поток 14) непрерывно возвращали в реактор. Вместо изображенного на фиг.3 пленочного выпарного аппарата с падающей пленкой (5) для непрерывной переработки кубового остатка (15) перегонной колонны (3) использовали обогреваемый горячим маслом пленочный испаритель с площадью рабочей поверхности А, равной 0,1 м2. Выпар из этого пленочного испарителя непрерывно подавали в верхнюю часть смонтированной над ним колонны (4), предназначенной для отгонки эфира от высококипящих продуктов. Речь при этом идет об экспериментальной стеклянной колонне для вакуумной перегонки (абсолютное давление в верхней части 150 мбар) диаметром 0,05 м, на высоту 0,5 м заполненной насадкой из металлической ткани Sulzer EX. Кубовый остаток непрерывно возвращали (18) в реакционный сосуд, а другую его часть непрерывно выводили из процесса через шлюзовый затвор (17). К моменту времени t, равному 0 час, кубовый остаток полностью непрерывно возвращали (18) в реакционный сосуд, то есть вообще не выводили через шлюзовый затвор (17) и в реактор не дозировали свежий катализатор. Исходные продукты (ММА и спирт) непрерывно подавали посредством поршневых дозирующих насосов, причем катализатор (тетраалкилтитанат) дозировали в виде раствора в исходном ММА (согласно спецификации ММА не содержал влагу). Раствор катализатора в исходном ММА вводили непосредственно в реактор, а исходный спирт в среднюю часть колонны для азеотропной перегонки, предварительно нагрев его до внутренней температуры этой колонны. Непрерывную подачу раствора стабилизатора (0,2 мас.% простого монометилового эфира гидрохинона в ММА или целевом эфире) в возвратный поток перегонных колонн осуществляли со скоростью 50-100 г/ч посредством шланговых насосов. Непрерывное перемещение материальных потоков между отдельными узлами установки осуществляли посредством поршневых дозирующих насосов или благодаря всасывающему действию разрежения. По возможности избегали использования промежуточных емкостей (буферных объемов). Состав материальных потоков, то есть содержание исходных ММА и спирта, МеОН и целевого эфира, определяли с помощью газовых хроматографов.
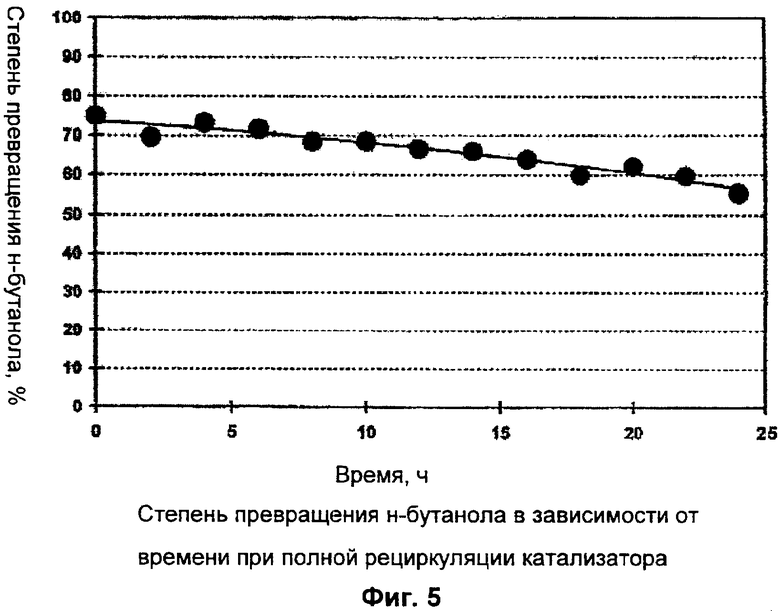
Для непрерывного получения н-БМА сначала в реакционный сосуд дозировали исходные реагенты: ММА (3,8 кг/ч), тетра-н-бутилтитанат (18 г/ч) и н-бутанол (2,7 кг/ч). Кроме того, в реактор направляли рециркулируемый поток из верхней части колонны для отгонки низкомолекулярных продуктов (3,0 кг/ч) следующего состава: 0,2 мас.% н-БМА, 31,1 мас.% н-бутанола, 66,4 мас.% ММА и 1,9 мас.% МеОН. Молярное соотношение ММА : н-бутанол в подаваемом в реактор потоке составляло 1,2:1. При времени пребывания реагентов в реакторе 1 час и азеотропной отгонке смеси ММА / МеОН (1,4 кг/ч) устанавливалась реакционная температура 115°С. Азеотропная смесь ММА / МеОН состояла из 80,4 мас.% МеОН и 19,6 мас.% ММА и содержала менее 5 частей на млн. н-бутанола. Выводимый из реактора поток реакционных продуктов (8,1 кг/ч) имел следующий состав: 63,8 мас.% н-БМА, 11,3 мас.% н-бутанола, 24,0 мас.% ММА, 0,7 мас.% МеОН и 0,2 мас.% побочных продуктов. Таким образом, степень превращения н-бутанола в реакторе составляла 74%. Начиная с момента времени t, равному 0 час, которому соответствовало полное прекращение выведения высококипящих продуктов через шлюзовый затвор (17) и добавления свежего катализатора, в течение последующих 24 часов наблюдалось превращение н-бутанола, служившее индикатором сохранения активности катализатора. Как следует из представленного на фиг.5 графика, неожиданно оказалось, что в течение этих 24 часов степень превращения н-бутанола снижалась всего на 18% (до 56%), хотя и было полностью прекращено как выведение высококипящих побочных продуктов через шлюзовый затвор, так и добавдение свежего катализатора, причем за указанный период катализатор почти 8 раз проходил через всю технологическую схему.
Пример 3. Получение изобутилметакрилата с частичной рециркуляцией катализатора.
В качестве реакционного аппарата (1) использовали обогреваемый водяным паром резервуар из нержавеющей стали с максимальным заполняемым объемом 15 литров. Реактор посредством линии отбора выпара был соединен со смонтированной над ним перегонной колонной (2). В качестве колонны для азеотропной отгонки (абсолютное давление в верхней части 1 атмосфера) использовали экспериментальную стеклянную колонну диаметром 0,1 м, на высоту 2 м заполненную насадкой из металлической ткани Sulzer CY. В средней части колонны (на уровне 1 м) располагался впускной штуцер, предназначенный для подачи исходного спирта. Выходящий из реактора поток реакционных продуктов непрерывно поступал в предназначенную для отгонки низкокипящих веществ перегонную колонну (3). В качестве перегонной колонны (3) использовали экспериментальную стеклянную колонну для вакуумной перегонки (абсолютное давление в верхней части 120 мбар) диаметром 0,1 м, на высоту 3,8 м заполненную насадкой из металлической ткани Sulzer CY. Впускной штуцер располагался на уровне 2 м. Куб колонны обогревали водяным паром. Конденсированную верхнюю фракцию (рециклизованный поток 14) непрерывно возвращали в реактор. Вместо изображенного на фиг.3 пленочного выпарного аппарата с падающей пленкой (5) для непрерывной переработки кубового остатка (15) перегонной колонны (3) использовали обогреваемый горячим маслом пленочный испаритель с площадью рабочей поверхности А, равной 0,1 м2. Выпар из этого пленочного испарителя непрерывно подавали в верхнюю часть смонтированной над ним колонны (4), предназначенной для отгонки эфира от высококипящих продуктов. Речь при этом идет об экспериментальной стеклянной колонне для вакуумной перегонки (абсолютное давление в верхней части 150 мбар) диаметром 0,05 м, на высоту 0,5 м заполненной насадкой из металлической ткани Sulzer EX. Кубовый остаток этой колонны делили на части, одну из которых (18) непрерывно возвращали в реакционный сосуд, а другую часть непрерывно выводили из процесса через шлюзовый затвор (17). Исходные продукты (ММА и спирт) непрерывно подавали посредством поршневых дозирующих насосов, причем катализатор (тетраалкилтитанат) дозировали в виде раствора в исходном ММА (согласно спецификации акрилат не содержал влагу). Раствор катализатора в исходном ММА вводили непосредственно в реактор, а исходный спирт в среднюю часть колонны для азеотропной перегонки, предварительно нагрев его до внутренней температуры этой колонны. Непрерывную подачу раствора стабилизатора (0,2% мас.% простого монометилового эфира гидрохинона в ММА или целевом эфире) в возвратный поток перегонных колонн осуществляли со скоростью 50-100 г/ч посредством шланговых насосов. Непрерывное перемещение материальных потоков между отдельными узлами установки осуществляли посредством поршневых дозирующих насосов или благодаря всасывающему действию разрежения. По возможности избегали использования промежуточных емкостей (буферных объемов). Состав материальных потоков, то есть содержание исходных ММА и спирта, МеОН и целевого эфира, определяли с помощью газовых хроматографов.
Для непрерывного получения изо-БМА сначала в реакционный сосуд дозировали исходные реагенты: ММА (2,78 кг/ч), тетраизобутилтитанат (3 г/ч) и изобутанол (1,9 кг/ч). Кроме того, в реактор направляли рециркулируемый поток из верхней части колонны для отгонки низкомолекулярных продуктов (2,2 кг/ч) следующего состава: 2,8 мас.% изо-БМА, 39,3 мас.% изобутанола, 56,2 мас.% ММА и 1,7 мас.% МеОН. Молярное соотношение ММА : изобутанол в подаваемом в реактор потоке составляло 1,1:1. При времени пребывания реагентов в реакторе 1,2 час и азеотропной отгонке смеси ММА/МеОН (1,04 кг/ч) устанавливалась реакционная температура 116°С. Азеотропная смесь ММА/МеОН состояла из 79 мас.% МеОН и 21 мас.% ММА и содержала менее 5 частей на млн. изобутанола. Выводимый из реактора поток реакционных продуктов (6,5 кг/ч) имел следующий состав: 66,8 мас.% изо-БМА, 13,1 мас.% изобутанола, 19,0 мас.% ММА, 0,5 мас.% МеОН и 0,6 мас.% побочных продуктов. Таким образом, выход изо-БМА в расчете на единицу объема реактора составлял 417 кг/ч/м3. В результате практически полного отделения более низкокипящих по сравнению с изо-БМА компонентов в кубе колонны для отгонки низкокипящих продуктов получали сырой эфир (4,25 кг/ч), уже содержащий более 99,5 мас.% изо-БМА, а также весь катализатор и стабилизатор. Следовательно выход по изобутанолу для процесса в целом составлял почти 100%. Выход по ММА для процесса в целом за вычетом заранее рассчитанных потерь ММА с азеотропной смесью ММА/МеОН также составлял почти 100%. При коэффициенте испарения (то есть отношении выпара к исходному потоку) в пленочном испарителе, равном около 90%, в верхней части колонны для высококипящих продуктов в конечном итоге получали 3,81 кг/ч чистого изо-БМА следующего состава: свыше 99,9 мас.% изо-БМА, менее 150 частей на млн. изобутанола, менее 10 частей на млн. ММА, 0 частей на млн. диизобутилового эфира, который имел показатель прозрачности (альфа) менее 0,2. При выведении через шлюзовый затвор 10% кубового остатка из пленочного испарителя (0,05 кг/ч), то есть при рециркуляции, составляющей 90% (0,45 кг/ч), расход катализатора (тетраизотитаната) составлял 0,8 г/кг чистого изо-БМА.
Таким образом, экономия катализатора по сравнению с примером 1 составляла около 80%.
Обозначения
1 - реакционный аппарат
2 - перегонная колонна для азеотропной отгонки
3 - перегонная колонна для отгонки низкокипящих компонентов
4 - перегонная колонна для отделения высококипящих компонентов
5 - пленочный выпарной аппарат
6 - пленочный выпарной аппарат
11 - исходные метил(мет)акрилат и катализатор
12 - исходный спирт
13 - азеотропная смесь метанола и метил(мет)акрилата
14 - рециркулируемый поток низкокипящих компонентов
15 - сырой эфир
16 - чистый эфир
17 - высококипящие продукты и катализатор
18 - рециклизация катализатора
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ АЛКИЛАМИНО(МЕТ)АКРИЛАМИДОВ | 2004 |
|
RU2374221C2 |
| ПОЛУЧЕНИЕ N,N-(ДИ)АЛКИЛАМИНОАЛКИЛ(МЕТ)АКРИЛАМИДА ИЛИ N,N-(ДИ)АЛКИЛАМИНОАЛКИЛ(МЕТ)АКРИЛАТА И ИХ ЧЕТВЕРТИЧНЫХ АММОНИЕВЫХ СОЛЕЙ В КАЧЕСТВЕ ФЛОКУЛИРУЮЩИХ ВСПОМОГАТЕЛЬНЫХ ВЕЩЕСТВ И ГЕЛЕОБРАЗУЮЩИХ АГЕНТОВ | 2017 |
|
RU2757390C2 |
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ АЛКИЛАМИНО(МЕТ)АКРИЛАМИДОВ | 2010 |
|
RU2546670C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИАЛКИЛМЕТАКРИЛАТНЫХ ПРИСАДОК И УСТАНОВКА ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2011 |
|
RU2466146C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОСОРБИДЭТОКСИЛАТДИ(МЕТ)АКРИЛАТА | 2016 |
|
RU2702013C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИ(МЕТ)АКРИЛАТА ИЗОСОРБИДА | 2016 |
|
RU2703461C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭФИРОВ (МЕТ)АКРИЛОВОЙ КИСЛОТЫ | 2009 |
|
RU2515985C2 |
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ БЕНЗИЛОВОГО СПИРТА | 1996 |
|
RU2176237C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭТИЛЕНГЛИКОЛЬДИМЕТАКРИЛАТА | 2008 |
|
RU2476420C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛ(МЕТ)АКРИЛАТОВ | 2006 |
|
RU2409552C2 |
Изобретение относится к усовершенствованному способу непрерывного получения алкил(мет)акрилатов путем переэтерификации метил(мет)акрилата со спиртами, имеющими более высокую температуру кипения по сравнению с метанолом, а именно к способу непрерывного получения высших сложных эфиров (мет)акриловой кислоты формулы (С)
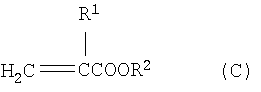
где R1 означает атом водорода или метил и R2 означает линейный, разветвленный или циклический алкильный или арильный остаток с 2-12 атомами углерода, путем переэтерификации сложных метиловых эфиров (мет)акриловой кислоты формулы (А)
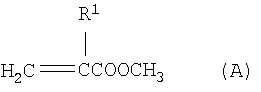
где R1 имеет вышеуказанное значение, высшими спиртами формулы (В)

где R2 имеет вышеуказанное значение, в присутствии катализатора или смеси катализаторов, в котором используют вакуумный испаритель и/или пленочный выпарной аппарат, предназначенный для обработки кубового остатка дистилляционной колонны для отделения высококипящих компонентов, в которой проводят очистку перегонкой целевого продукта, направляемого в нее из перегонной колонны выделения низкокипящих компонентов, с отделением из упомянутой дистилляционной колонны очищенного сложного эфира формулы (С) в качестве головного продукта, а кубовый остаток вакуумного испарителя и/или пленочного выпарного аппарата делят на части и часть кубового остатка подают в реакционный аппарат. Благодаря особой технологии переработки получают продукт, который имеет до последнего времени не достигнутые показатели качества. Кроме того, может быть обеспечен чрезвычайно высокий выход продукта в расчете на единицу объема реактора и его суммарный выход. Способ отличается, в частности, возможностью многократного использования используемого гомогенного катализатора, а следовательно, снижения расходов на вспомогательные материалы. 3 н. и 13 з.п. ф-лы, 5 ил.
1. Способ непрерывного получения высших сложных эфиров (мет)акриловой кислоты формулы (С)
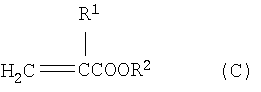
где R1 означает атом водорода или метил, и
R2 означает линейный, разветвленный или циклический алкильный или арильный остаток с 2-12 атомами углерода,
путем переэтерификации сложных метиловых эфиров (мет)акриловой кислоты формулы (А)
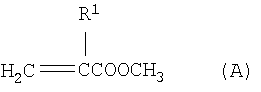
где R1 имеет вышеуказанное значение,
высшими спиртами формулы (В)

где R2 имеет вышеуказанное значение,
в присутствии катализатора или смеси катализаторов, в котором используют вакуумный испаритель, предназначенный для обработки кубового остатка дистилляционной колонны для отделения высококипящих компонентов, в которой проводят очистку перегонкой целевого продукта, направляемого в нее из перегонной колонны выделения низкокипящих компонентов, с отделением из упомянутой дистилляционной колонны очищенного сложного эфира формулы (С) в качестве головного продукта, а кубовый остаток вакуумного испарителя делят на части и часть кубового остатка подают в реакционный аппарат.
2. Способ по п.1, отличающийся тем, что в качестве спиртов используют н-бутанол, изобутанол или 2-этилгексанол.
3. Способ по п.1, отличающийся тем, что в качестве катализатора используют гомогенные катализаторы.
4. Способ по п.3, отличающийся тем, что в качестве катализатора используют титанат спирта формулы (В).
5. Способ по п.1, отличающийся тем, что в реакционный аппарат подают 1-95 мас.% кубового остатка вакуумного испарителя.
6. Способ по п.5, отличающийся тем, что в реакционный аппарат подают 40-90 мас.% кубового остатка вакуумного испарителя.
7. Способ по п.6, отличающийся тем, что в реакционный аппарат подают 60-85 мас.% кубового остатка вакуумного испарителя.
8. Способ непрерывного получения высших сложных эфиров (мет)акриловой кислоты формулы (С)
где R1 означает атом водорода или метил, и
R2 означает линейный, разветвленный или циклический алкильный или арильный остаток с 2-12 атомами углерода,
путем переэтерификации сложных метиловых эфиров (мет)акриловой кислоты формулы (А)
где R1 имеет вышеуказанное значение,
высшими спиртами формулы (В)
где R2 имеет вышеуказанное значение,
в присутствии катализатора или смеси катализаторов, в котором используют пленочный выпарной аппарат, предназначенный для обработки кубового остатка дистилляционной колонны для отделения высококипящих компонентов, в которой проводят очистку перегонкой целевого продукта, направляемого в нее из перегонной колонны выделения низкокипящих компонентов, с отделением из упомянутой дистилляционной колонны очищенного сложного эфира формулы (С) в качестве головного продукта, а кубовый остаток пленочного выпарного аппарата делят на части и часть кубового остатка подают в реакционный аппарат.
9. Способ по п.8, отличающийся тем, что в качестве спиртов используют н-бутанол, изобутанол или 2-этилгексанол.
10. Способ по п.8, отличающийся тем, что в качестве катализатора используют гомогенные катализаторы.
11. Способ по п.10, отличающийся тем, что в качестве катализатора используют титанат спирта формулы (В).
12. Способ по п.8, отличающийся тем, что в реакционный аппарат подают 1-95 мас.% кубового остатка пленочного выпарного аппарата.
13. Способ по п.12, отличающийся тем, что в реакционный аппарат подают 40-90 мас.% кубового остатка пленочного выпарного аппарата.
14. Способ по п.13, отличающийся тем, что в реакционный аппарат подают 60-85 мас.% кубового остатка пленочного выпарного аппарата.
15. Способ непрерывного получения высших сложных эфиров (мет)акриловой кислоты формулы (С)
где R1 означает атом водорода или метил, и
R2 означает линейный, разветвленный или циклический алкильный или арильный остаток с 2-12 атомами углерода,
путем переэтерификации сложных метиловых эфиров (мет)акриловой кислоты формулы (А)
где R1 имеет вышеуказанное значение,
высшими спиртами формулы (В)
где R2 имеет вышеуказанное значение,
в присутствии катализатора или смеси катализаторов, в котором используют пленочный выпарной аппарат и вакуумный испаритель, предназначенные для обработки кубового остатка дистилляционной колонны для отделения высококипящих компонентов, в которой проводят очистку перегонкой целевого продукта, направляемого в нее из перегонной колонны выделения низкокипящих компонентов, с отделением из упомянутой дистилляционной колонны очищенного сложного эфира формулы (С) в качестве головного продукта, а кубовый остаток пленочного выпарного аппарата делят на части и часть кубового остатка подают в реакционный аппарат и кубовый остаток вакуумного испарителя делят на части и подают в реакционный аппарат.
16. Способ по п.15, отличающийся тем, что в реакционный аппарат подают 1-95 мас.% от суммы кубового остатка пленочного выпарного аппарата и кубового остатка вакуумного испарителя.
| DE 10127939 A1, 29.05.2002 | |||
| US 4927954 A, 22.05.1990 | |||
| US 4876368 A, 24.10.1989 | |||
| Способ обнаружения возгораний | 1981 |
|
SU960877A1 |
| EP 0968995 A1, 05.01.2000 | |||
| СПОСОБ ПОЛУЧЕНИЯ ВЫСШИХ АЛКИЛОВЫХ ЭФИРОВ МЕТАКРИЛОВОЙ КИСЛОТЫ | 0 |
|
SU330160A1 |

