ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] По настоящей заявке испрашивается приоритет по предварительной заявке U.S. № 62/788722, поданной 4 января 2019 г., и по предварительной заявке U.S. №62/863853, поданной 19 июня 2019 г., которые обе во всей своей полноте включены в настоящее изобретение в качестве ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0002] Настоящее изобретение в целом относится к терапевтическим средствам, которые можно использовать для модулирования белка активации фибробластов.
УРОВЕНЬ ТЕХНИКИ
[0003] Белок активации фибробластов (FAP), также называющийся, как FAPα, Seprase или α2-антиплазминконвертирующий фермент, представляет собой интегральную мембранную серинпротеазу типа II, которая относится к семейству пролилолигопептидазы S9, которое также включает ферменты DPPII, DPPIV, DPP8, DPP9 и PREP. Это семейство характеризуется, как обладающие экзодипептидилпептидазной активностью (DPP). FAP является единственным представителем, который также обладает эндопептидазной активностью (Aertgeerts, K., et al. J Biol Chem, 2005. 280(20): p. 19441-4). FAP обладает высокой степенью гомологии с DPPIV. Он в основном содержится в виде клеточного поверхностного гомодимера, но также сообщали, что он образует гетеродимеры с DPPIV in vivo (O'Brien, P., et al. Biochim Biophys Acta, 2008. 1784(9): p. 1130-45). Подразумевающиеся физиологические субстраты для эндопептидазной активности FAP включают α2-антиплазмин, коллаген типа I, желатин и фактор роста фибробластов 21 (FGF21) (Lee, K.N., et al., Biochemistry, 2009. 48(23): p. 5149-58), и для экзопептидазной активности включают нейропептид Y, натрийуретический пептид типа B, вещество P и пептид YY (Brokopp, C.E., et al., Eur Heart J, 2011. 32(21): p. 2713-22; Coppage, A.L., et al., PLoS One, 2016. 11(3): p. e0151269; Dunshee, D.R., et al., J Biol Chem, 2016. 291(11): p. 5986-96; Lee, K.N., et al., J Thromb Haemost, 2011. 9(5): p. 987-96).
[0004] FAP участвует в заболеваниях, включающих пролиферацию, ремоделирование ткани, хроническое воспаление и/или фиброз, включая, но не ограничиваясь только ими, фиброзное заболевание, заживление раны, келоидное образование, остеоартрит, ревматоидный артрит и родственные нарушения, включая разрушение хряща, атеросклеротическое заболевание и болезнь Крона.
[0005] Экспрессирование FAP связано с плохим прогнозом при некоторых типах рака, включая рак желудка, аденокарциному поджелудочной железы и печеночно-клеточную карциному (Wen, X., et al., Oncol Res, 2016; Cohen, S.J., et al., Pancreas, 2008. 37(2): p. 154-8; Ju, M.J., et al., Am J Clin Pathol, 2009. 131(4): p. 498-510) и при раке толстой кишки усиленное экспрессирование FAP было связано с более агрессивным заболеванием (Henry, L.R., et al., Clin Cancer Res, 2007. 13(6): p. 1736-41). Подразумевается, что FAPα при CAFs играет критические роли в регуляции противоопухолевого иммунного ответа путем индуцирования стимулирующего опухоль воспаления (Chen, L., et al., Biochem Biophys Res Commun, 2017; Wen, X., et al., Oncol Res, 2016; Hugo, W., et al., Cell, 2016. 165(1): p. 35-44).
[0006] Val-boroPro (талабостат, PT-100) является единственным ингибитором FAP, подвергающимся клиническим исследованиям. Это соединение впервые было разработано в качестве ингибитора DPPIV и затем исследовано в качестве ингибитора FAP несмотря на отсутствие селективности (Cunningham, C.C., Expert Opin Investig Drugs, 2007. 16(9): p. 1459-65). Это средство исследовали в фазе II для множества раковых заболеваний в комбинации со стандартной цитотоксичной химиотерапией, однако оно не соответствует концевым точкам для эффективности (Eager, R.M., et al., BMC Cancer, 2009. 9: p. 263; Narra, K., et al., Cancer Biol Ther, 2007. 6(11): p. 1691-9; Eager, R.M., et al., Clin Oncol R Coll Radiol, 2009. 21(6): p. 464-72). Два исследования в фазе III были закончены раньше, видимо, вследствие опасений, связанных с безопасностью и эффективностью (Jansen, K., et al., J Med Chem, 2014. 57(7): p. 3053-74). Поскольку Val-boroPro быстро теряет ингибирующую активность по отношению к протеазе вследствие циклизации после выдерживания при pH 7,8, эффективные концентрации трудно обеспечить у пациентов вследствие клинической токсичности, наблюдающейся при более высоких дозах (Narra, K., et al., Cancer Biol Ther, 2007. 6(11): p. 1691-9).
[0007] Задачей является улучшение селективности ингибитора FAP и характеристик ингибиторов для улучшения безопасности и эффективности in vivo.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0008] Настоящее изобретение относится к соединениям, их солям, фармацевтическим композициям из указанных выше и способам их получения и применения. Одним объектом является соединение формулы (I):
 (I)
(I)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где R, m, n, X, Y и L являются такими, как подробно описано в настоящем изобретении.
[0009] Одним объектом является соединение формулы (I):
(I)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где:
R означает водород, C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R независимо необязательно замещены посредством Rd;
m равно 0, 1, 2, 3 или 4;
n равно 0, 1, 2, 3 или 4,
где m+n равно 1, 2, 3 или 4;
X означает -C(=O)-, -O-, -CH(OH)-, -S-, -S(=O)- или -S(=O)2-;
L означает
(a)  , где
, где
* обозначает положение присоединения к фрагменту Y-X-,
** обозначает положение присоединения к остальной части молекулы,
Ra означает водород, C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве Ra независимо необязательно замещены посредством Re,
R1 и R2 независимо друг от друга и независимо в каждом случае означают водород, C1-C2 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R1 и R2 независимо необязательно замещены посредством Rf,
или R1 и R2 вместе с атомом или атомами углерода, к которым они присоединены, образуют 3- 8-членный циклоалкилен, необязательно замещенный посредством Rf,
q равно 1, 2 или 3,
R3 и R4 независимо друг от друга и независимо в каждом случае означают водород, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R3 и R4 независимо необязательно замещены посредством Rg,
или R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют 3- 8-членный циклоалкилен, необязательно замещенный посредством Rg, и
p равно 0, 1 или 2;
(b) 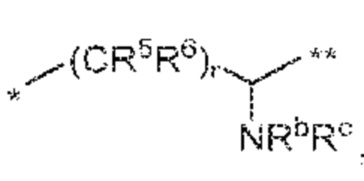 , где
, где
* обозначает положение присоединения к фрагменту Y-X-,
** обозначает положение присоединения к остальной части молекулы,
R5 и R6 независимо друг от друга и независимо в каждом случае означают H, C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R5 и R6 независимо необязательно замещены посредством Rh,
Rb и Rc независимо означают H, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил, C6-C14 арил, или -C(=O)OR17, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве Rb и Rc независимо необязательно замещены посредством Ri, и
r равно 1, 2 или 3; или
(c)  , где
, где
* обозначает положение присоединения к фрагменту Y-X-,
** обозначает положение присоединения к остальной части молекулы,
R7 и R8 независимо друг от друга и независимо в каждом случае означают водород, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R7 и R8 независимо необязательно замещены посредством Rj,
или R7 и R8 вместе с атомом углерода, к которому они присоединены, образуют 3- 8-членный циклоалкилен, необязательно замещенный посредством Rj,
R9 и R10 независимо друг от друга и независимо в каждом случае означают H, C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R9 и R10 независимо необязательно замещены посредством Rk,
s равно 1, 2 или 3,
t равно 1, 2 или 3,
где s+t равно 2, 3 или 4,
u равно 0 или 1, и
v равно 0 или 1;
Y означает C6-C9 арил, замещенный посредством R11, 6- 10-членный гетероарил, замещенный посредством R12 или 3-12-членный гетероциклил, замещенный посредством R13, где
каждый R11, R12 и R13 независимо означает C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C4-C8 циклоалкенил, 3-12-членный гетероциклил, 5-10-членный гетероарил, C6-C14 арил, -OR14, -NR15R16, -SR14, -NO2, -C=NH(OR14), -C(O)R14, -OC(O)R14, -C(O)OR14, -C(O)NR15R16, -NR14C(O)R15, -NR14C(O)OR15, -NR14C(O)NR15R16, -S(O)R14, -S(O)2R14, -NR14S(O)R15, -NR14S(O)2R15, -S(O)NR15R16, -S(O)2NR15R16 или -P(O)(OR15)(OR16), где C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C4-C8 циклоалкенил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R11, R12 и R13 замещены посредством RL;
R14, R15 и R16 независимо друг от друга и независимо в каждом случае означают водород, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил или 3-12-членный гетероциклил, где C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил и 3-12-членный гетероциклил в качестве R14, R15 и R16 независимо замещены C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой, C1-C6 пергалогеналкоксигруппой, C6-C14 арилом или C6-C14 арилоксигруппой где C6-C14 арил или C6-C14 арилоксигруппа необязательно дополнительно замещена галогеном, -OH, цианогруппой, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой или C1-C6 пергалогеналкоксигруппой; и где по меньшей мере один из R14, R15 и R16, если содержится, не означает водород;
RL означает C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил или 3-12-членный гетероциклил, где C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил или 3-12-членный гетероциклил в качестве RL замещен галогеном, -OH, цианогруппой, оксогруппой, -NH2, -NH-(3-12-членным гетероциклилом), -O-(3-12-членным гетероциклилом), C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой, C1-C6 пергалогеналкоксигруппой или C6-C14 арилом, где
C1-C6 алкил дополнительно необязательно замещен 3-12-членным гетероциклилом, где 3-12-членный гетероциклил дополнительно необязательно замещен C1-C6 алкилом,
3-12-членный гетероциклил -NH-(3-12-членного гетероциклила) и -O-(3-12-членного гетероциклила) дополнительно необязательно замещен C1-C6 алкилом, и
C6-C14 арил дополнительно необязательно замещен галогеном, -OH, цианогруппой, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой или C1-C6 пергалогеналкоксигруппой; и
Rd, Re, Rf, Rg, Rh, Ri, Rj и Rk независимо друг от друга и независимо в каждом случае означают галоген, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил, 3-12-членный гетероциклил, -OR14, -NR15R16, цианогруппу или нитрогруппу.
[0010] Одним объектом является соединение формулы (I):
(I)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где:
R означает водород, C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R независимо необязательно замещены посредством Rd;
m равно 0, 1, 2, 3 или 4;
n равно 0, 1, 2, 3 или 4,
где m+n равно 1, 2, 3 или 4;
X означает -C(=O)-, -O-, -CH(OH)-, -S-, -S(=O)- или -S(=O)2-;
L означает
(a) , где
* обозначает положение присоединения к фрагменту Y-X-,
** обозначает положение присоединения к остальной части молекулы,
Ra означает водород, C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве Ra независимо необязательно замещены посредством Re,
R1 и R2 независимо друг от друга и независимо в каждом случае означают водород, C1-C2 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R1 и R2 независимо необязательно замещены посредством Rf, или R1 и R2 вместе с атомом или атомами углерода, к которым они присоединены, образуют 3- 8-членный циклоалкилен, необязательно замещенный посредством Rf,
q равно 1, 2 или 3,
R3 и R4 независимо друг от друга и независимо в каждом случае означают водород, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R3 и R4 независимо необязательно замещены посредством Rg, или R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют 3- 8-членный циклоалкилен, необязательно замещенный посредством Rg, и
p равно 0, 1 или 2;
(b) , где
* обозначает положение присоединения к фрагменту Y-X-,
** обозначает положение присоединения к остальной части молекулы,
R5 и R6 независимо друг от друга и независимо в каждом случае означают H, C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R5 и R6 независимо необязательно замещены посредством Rh,
Rb и Rc независимо означают H, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил, C6-C14 арил, или -C(=O)OR17, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве Rb и Rc независимо необязательно замещены посредством Ri, и
r равно 1, 2 или 3; или
(c) , где
* обозначает положение присоединения к фрагменту Y-X-,
** обозначает положение присоединения к остальной части молекулы,
R7 и R8 независимо друг от друга и независимо в каждом случае означают водород, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R7 и R8 независимо необязательно замещены посредством Rj, или R7 и R8 вместе с атомом углерода, к которому они присоединены, образуют 3- 8-членный циклоалкилен, необязательно замещенный посредством Rj,
R9 и R10 независимо друг от друга и независимо в каждом случае означают H, C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R9 и R10 независимо необязательно замещены посредством Rk,
s равно 1, 2 или 3,
t равно 1, 2 или 3,
где s+t равно 2, 3 или 4,
u равно 0 или 1, и
v равно 0 или 1;
Y означает C6-C9 арил, замещенный посредством R11, 6- 10-членный гетероарил, замещенный посредством R12 или 3-12-членный гетероциклил, замещенный посредством R13, где
каждый R11, R12 и R13 независимо означает C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C4-C8 циклоалкенил, 3-12-членный гетероциклил, 5-10-членный гетероарил, C6-C14 арил, -OR14, -NR15R16, -SR14, -NO2, -C=NH(OR14), -C(O)R14, -OC(O)R14, -C(O)OR14, -C(O)NR15R16, -NR14C(O)R15, -NR14C(O)OR15, -NR14C(O)NR15R16, -S(O)R14, -S(O)2R14, -NR14S(O)R15, -NR14S(O)2R15, -S(O)NR15R16, -S(O)2NR15R16 или -P(O)(OR15)(OR16), где C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C4-C8 циклоалкенил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R11, R12 и R13 замещены посредством RL;
R14, R15 и R16 независимо друг от друга и независимо в каждом случае означают водород, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил или 3-12-членный гетероциклил, где C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил и 3-12-членный гетероциклил в качестве R14, R15 и R16 независимо замещены C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой, C1-C6 пергалогеналкоксигруппой, C6-C14 арилом или C6-C14 арилоксигруппой где C6-C14 арил или C6-C14 арилоксигруппа необязательно дополнительно замещена галогеном, -OH, цианогруппой, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой или C1-C6 пергалогеналкоксигруппой; и где по меньшей мере один из R14, R15 и R16, если содержится, не означает водород;
RL означает C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил или 3-12-членный гетероциклил, где C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил или 3-12-членный гетероциклил в качестве RL означает, замещенный галогеном, -OH, цианогруппой, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой, C1-C6 пергалогеналкоксигруппой или C6-C14 арилом, где C6-C14 арил дополнительно необязательно замещен галогеном, -OH, цианогруппой, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой или C1-C6 пергалогеналкоксигруппой; и
Rd, Re, Rf, Rg, Rh, Ri, Rj и Rk независимо друг от друга и независимо в каждом случае означают галоген, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил, 3-12-членный гетероциклил, -OR14, -NR15R16, цианогруппу или нитрогруппу.
[0011] Одним объектом является соединение формулы (I) или его фармацевтически приемлемая соль, где соединение обладает одной или большим количеством следующих характеристик:
(i) X означает -C(=O)-, -O- или -CH(OH)-;
(ii) L означает
(a) -NH-CR1R2-, такой как -NH-CH2- или -NH-CH(CH3)- или где R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют 3- 8-членный циклоалкилен, такой как циклопропилен;
(b) -CR5R6-CH(NRbRc)-, включая, но не ограничиваясь только ими, где R6, Rb и Rc все означают H и R5 означает H или C1-C6 алкил; или
(c)  , где * обозначает положение присоединения к фрагменту Y-X-, ** обозначает положение присоединения к остальной части молекулы, такой как
, где * обозначает положение присоединения к фрагменту Y-X-, ** обозначает положение присоединения к остальной части молекулы, такой как  ;
;
(iii) Y означает:
(a) C6-C9 арил, замещенный посредством R11, такой как 2,3-дигидро-1H-инден-2-ил, фенил и нафтил, которые замещены по меньшей мере одним R11;
(b) 6- 10-членный гетероарил, замещенный посредством R12, такой как пиридинил, пиримидинил, пиридин-2(1H)-онил и хинолин-6-ил, которые замещены по меньшей мере одним R12; или
(c) 3-12-членный гетероциклил, замещенный посредством R13, такой как 2H-пиран-2-ил, изоиндолинил, пиперидин-2-ил и пиперидинил, которые замещены по меньшей мере одним R13.
[0012] Другим объектом является соединение формулы (I), его фармацевтически приемлемая соль, стереоизомер или таутомер, где фрагмент -X-L- выбран из группы, состоящей из следующих:


и 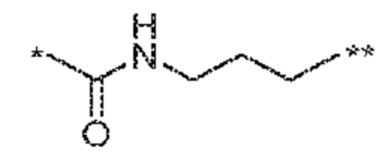 ; где * обозначает положение присоединения к фрагменту Y и ** обозначает положение присоединения к остальной части молекулы.
; где * обозначает положение присоединения к фрагменту Y и ** обозначает положение присоединения к остальной части молекулы.
[0013] Настоящее изобретение также относится к фармацевтической композиции, включающей соединение любой формулы, приведенной в настоящем изобретении, включая формулу (I), или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
[0014] Настоящее изобретение также относится к способу лечения заболевания или нарушения, опосредуемого белком активации фибробластов (FAP) у нуждающегося в нем индивидуума, включающему введение индивидууму терапевтически эффективного количества соединения, подробно описанного в настоящем изобретении, включая, но не ограничиваясь только ими, соединение формулы (I) или его фармацевтически приемлемую соль, или фармацевтическую композицию, содержащую такие соединение или соль. Такое заболевание или нарушение в одном объекте характеризуется пролиферацией, ремоделированием ткани, хроническим воспалением, ожирением, непереносимостью глюкозы или нечувствительностью к инсулину. В одном объекте заболеванием или нарушением является рак молочной железы, колоректальный рак, рак яичников, рак предстательной железы, рак поджелудочной железы, рак почки, рак легких, меланома, фибросаркома, саркома кости, саркома соединительной ткани, почечноклеточная карцинома, гигантоклеточная карцинома, плоскоклеточная карцинома, лейкоз, рак кожи, рак мягких тканей, рак печени, желудочно-кишечная карцинома или аденокарцинома. В предпочтительном объекте заболеванием или нарушением является метастатический рак почки, хронический лимфоцитарный лейкоз, аденокарцинома поджелудочной железы или немелкоклеточный рак легких. В другом объекте заболеванием или нарушением является фиброзное заболевание, заживление раны, келоидное образование, остеоартрит, ревматоидный артрит и родственные нарушения, включая разрушение хряща, атеросклеротическое заболевание, болезнь Крона или диабет типа II. Другим предпочтительным объектом является способ уменьшения роста опухоли, пролиферации опухоли или онкогенности у нуждающегося в нем индивидуума, включающий введение индивидууму соединения, подробно описанного в настоящем изобретении, такого как соединение формулы (I) или его фармацевтически приемлемая соль, или фармацевтической композиции из них.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0015] На фиг. 1A представлено разложение PRXS-AMC во времени при действии rhFAP. На фиг. 1B представлено разложение Z-Gly-Pro-AMC во времени при действии rhFAP.
[0016] На фиг. 2A представлено разложение PRXS-AMC во времени при действии rhPREP. На фиг. 2B представлено разложение Z-Gly-Pro-AMC во времени при действии rhPREP.
[0017] На фиг. 3A представлено разложение PRXS-AMC во времени при действии rhDPPIV. На фиг. 3B представлено разложение PRXS-AMC во времени при действии rhDPP9.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0018] В настоящем изобретении описаны соединения формулы (I):
 (I)
(I)
и их фармацевтически приемлемые соли, стереоизомеры или таутомеры. Соединения можно использовать для ингибирования белка активации фибробластов (FAPα). В некоторых вариантах осуществления соединение используют для лечения заболевания или нарушения, опосредуемого с помощью FAPα у индивидуума. Такие заболевания или нарушения могут включать или характеризоваться пролиферацией, ремоделированием ткани, хроническим воспалением, ожирение, непереносимостью глюкозы и/или нечувствительностью к инсулину. В некоторых вариантах осуществления соединение используют для лечения рака.
Определения
[0019] При использовании в настоящем изобретении, если явно не указано иное, термины в единственном числе также означают термины во множественном числе.
[0020] Указание "примерно" для значения или параметра в настоящем изобретении включает (и описывает) варианты осуществления, которые относятся к самому этому значению или параметру. Например, описание с указанием "примерно X" включает описание "X".
[0021] "Алкил" при использовании в настоящем изобретении означает и включает, если не указано иное, насыщенную линейную (т. е. неразветвленную) или разветвленную одновалентную углеводородную цепь или их комбинацию, содержащую указанное количество атомов углерода (т. е. C1-C10 означает от 1 до 10 атомов углерода). Конкретными алкильными группами являются такие, которые содержат от 1 до 20 атомов углерода ("C1-C20 алкил"), содержат от 1 до 10 атомов углерода ("C1-C10 алкил"), содержат от 6 до 10 атомов углерода ("C6-C10 алкил"), содержат от 1 до 6 атомов углерода ("C1-C6 алкил"), содержат от 2 до 6 атомов углерода ("C2-C6 алкил"), или содержат от 1 до 4 атомов углерода ("C1-C4 алкил"). Примеры алкильных групп включают, но не ограничиваются только ими, группы, такие как метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, н-пентил, н-гексил, н-гептил, н-октил, н-нонил, н-децил и т. п.
[0022] "Алкилен" при использовании в настоящем изобретении означает такие же остатки, как алкил, но двухвалентные. Конкретными алкиленовыми группами являются такие, которые содержат от 1 до 20 атомов углерода ("C1-C20 алкилен"), содержат от 1 до 10 атомов углерода ("C1-C10 алкилен"), содержат от 6 до 10 атомов углерода ("C6-C10 алкилен"), содержат от 1 до 6 атомов углерода ("C1-C6 алкилен"), от 1 до 5 атомов углерода ("C1-C5 алкилен"), от 1 до 4 атомов углерода ("C1-C4 алкилен") или от 1 до 3 атомов углерода ("C1-C3 алкилен"). Примеры алкилена включают, но не ограничиваются только ими, группы, такие как метилен (-CH2-), этилен (-CH2CH2-), пропилен (-CH2CH2CH2-), изопропилен (-CH2CH(CH3)-), бутилен (-CH2(CH2)2CH2-), изобутилен (-CH2CH(CH3)CH2-), пентилен (-CH2(CH2)3CH2-), гексилен (-CH2(CH2)4CH2-), гептилен (-CH2(CH2)5CH2-), октилен (-CH2(CH2)6CH2-) и т. п.
[0023] "Алкенил" при использовании в настоящем изобретении означает и включает, если не указано иное, ненасыщенную линейную (т. е. неразветвленную) или разветвленную одновалентную углеводородную цепь или их комбинацию, содержащую по меньшей мере одну двойную связь (т. е. содержащую по меньшей мере один фрагмент формулы C=C) и содержащую указанное количество атомов углерода (т. е. C2-C10 означает от 2 до 10 атомов углерода). Алкенильная группа может обладать "цис" или "транс" конфигурациями или, альтернативно обладать "E" или "Z" конфигурациями. Конкретными алкенильными группами являются такие, которые содержат от 2 до 20 атомов углерода ("C2-C20 алкенил"), содержат от 6 до 10 атомов углерода ("C6-C10 алкенил"), содержат от 2 до 8 атомов углерода ("C2-C8 алкенил"), содержат от 2 до 6 атомов углерода ("C2-C6 алкенил") или содержат от 2 до 4 атомов углерода ("C2-C4 алкенил"). Примеры алкенильной группы включают, но не ограничиваются только ими, группы, такие как этенил (или винил), проп-1-енил, проп-2-енил (или аллил), 2-метилпроп-1-енил, бут-1-енил, бут-2-енил, бут-3-енил, бута-1,3-диенил, 2-метилбута-1,3-диенил, пент-1-енил, пент-2-енил, гекс-1-енил, гекс-2-енил, гекс-3-енил и т. п.
[0024] "Алкинил" при использовании в настоящем изобретении означает и включает, если не указано иное, ненасыщенную линейную (т. е. неразветвленную) или разветвленную одновалентную углеводородную цепь или их комбинацию, содержащую по меньшей мере одну тройную связь (т. е. содержащий по меньшей мере один фрагмент формулы C≡C) и содержащую указанное количество атомов углерода (т. е. C2-C10 означает от 2 до 10 атомов углерода). Конкретными алкинильными группами являются такие, которые содержат от 2 до 20 атомов углерода ("C2-C20 алкинил"), содержат от 6 до 10 атомов углерода ("C6-C10 алкинил"), содержат от 2 до 8 атомов углерода ("C2-C8 алкинил"), содержат от 2 до 6 атомов углерода ("C2-C6 алкинил") или содержат от 2 до 4 атомов углерода ("C2-C4 алкинил"). Примеры алкинильной группы включают, но не ограничиваются только ими, группы, такие как этинил (или ацетиленил), проп-1-инил, проп-2-инил (или пропаргил), бут-1-инил, бут-2-инил, бут-3-инил и т. п.
[0025] "Алкоксигруппа" означает группу R-O-, где R означает алкил; и включает, например, метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу, н-бутоксигруппу, трет-бутоксигруппу, втор-бутоксигруппу, н-пентоксигруппу, н-гексилоксигруппу, 1,2-диметилбутоксигруппу и т. п. Аналогичным образом, "циклоалкоксигруппа" означает группу "циклоалкил-O-" и "арилоксигруппа" означает группу "арил-O-". "Замещенная алкоксигруппа" означает группу "замещенный алкил-O-". "Замещенная циклоалкоксигруппа" означает группу "замещенный циклоалкил-O-". "Замещенная арилоксигруппа" означает группу "замещенный арил-O-".
[0026] "Арил" или "Ar" при использовании в настоящем изобретении означает ненасыщенную ароматическую карбоциклическую группу, содержащую одно кольцо (например, фенил) или несколько конденсированных колец (например, нафтил или антрил) и эти конденсированные кольца могут быть или не быть ароматическими. Конкретными арильными группами являются такие, которые содержат от 6 до 14 кольцевых атомов углерода ("C6-C14 арил"). Арильная группа, содержащая более одного кольца, в которой по меньшей мере одно кольцо является неароматическим, может быть соединена с исходной структурой по положению ароматического кольца или по положению неароматического кольца. В одном варианте арильная группа, содержащая более одного кольца, в которой по меньшей мере одно кольцо является неароматическим, соединена с исходной структурой по положению ароматического кольца.
[0027] "Арилен" при использовании в настоящем изобретении означает такие же остатки, как арил, но двухвалентные. Конкретными ариленовыми группами являются такие, которые содержат от 6 до 14 кольцевых атомов углерода ("C6-C14 арилен").
[0028] "Циклоалкил" при использовании в настоящем изобретении означает и включает, если не указано иное, насыщенные циклические одновалентные углеводородные структуры, содержащие указанное количество атомов углерода (т. е. C3-C10 означает от 3 до 10 атомов углерода). Циклоалкил может состоять из одного кольца, такого как циклогексил, или нескольких колец, таких как адамантил. Циклоалкил, содержащий более одного кольца, может быть конденсированным, спироциклическим или мостиковым, или их комбинацией. Конкретными циклоалкильными группами являются такие, которые содержат от 3 до 12 кольцевых атомов углерода. Предпочтительным циклоалкилом является циклический углеводород, содержащий от 3 до 8 кольцевых атомов углерода ("C3-C8 циклоалкил"), содержащий от 3 до 6 атомов углерода ("C3-C6 циклоалкил") или содержащий от 3 до 4 кольцевых атомов углерода ("C3-C4 циклоалкил"). Примеры циклоалкила включают, но не ограничиваются только ими, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, норборнил и т. п.
[0029] "Циклоалкилен" при использовании в настоящем изобретении означает такие же остатки, как циклоалкил, но двухвалентные. Циклоалкилен может состоять из одного кольца или нескольких колец, которые могут быть конденсированными, спироциклическими или мостиковыми, или их комбинацией. Конкретными циклоалкиленовыми группами являются такие, которые содержат от 3 до 12 кольцевых атомов углерода. Предпочтительным циклоалкиленом является циклический углеводород, содержащий от 3 до 8 кольцевых атомов углерода ("C3-C8 циклоалкилен"), содержащий от 3 до 6 атомов углерода ("C3-C6 циклоалкилен") или содержащий от 3 до 4 кольцевых атомов углерода ("C3-C4 циклоалкилен"). Примеры циклоалкилена включают, но не ограничиваются только ими, циклопропилен, циклобутилен, циклопентилен, циклогексилен, циклогептилен, норборнилен и т. п. Циклоалкилен может соединяться с остальными структурами через тот же кольцевой атом углерода или разные атомы углерода. Если циклоалкилен соединяется с остальными структурами через два разных атома углерода, соединяющие связи могут находиться в цис- или транс-положении друг по отношению к другу. Например, циклопропилен может включать 1,1-циклопропилен и 1,2-циклопропилен (например, цис-1,2-циклопропилен или транс-1,2-циклопропилен), или их смесь.
[0030] "Циклоалкенил" означает и включает, если не указано иное, ненасыщенную циклическую неароматическую одновалентную углеводородную структуру, содержащую по меньшей мере одну двойную связь (т. е. содержащую по меньшей мере один фрагмент формулы C=C) и содержащую указанное количество атомов углерода (т. е. C3-C10 означает от 3 до 10 атомов углерода). Циклоалкенил может состоять из одного кольца, такого как циклогексенил, или нескольких колец, таких как норборненил. Предпочтительным циклоалкенилом является ненасыщенный циклический углеводород, содержащий от 3 до 8 кольцевых атомов углерода ("C3-C8 циклоалкенил"). Примеры циклоалкенильных групп включают, но не ограничиваются только ими, циклопропенил, циклобутенил, циклопентенил, циклогексенил, норборненил и т. п.
[0031] "Циклоалкенилен" при использовании в настоящем изобретении означает такие же остатки, как циклоалкенил, но двухвалентные.
[0032] "Гетероарил" при использовании в настоящем изобретении означает ненасыщенную ароматическую циклическую группу, содержащую от 1 до 14 кольцевых атомов углерода и по меньшей мере один кольцевой гетероатом, включая, но не ограничиваясь только ими, гетероатомы, такие как азот, кислород и серу. Гетероарильная группа может содержать одно кольцо (например, пиридил, фурил) или несколько конденсированных колец (например, индолизинил, бензотиенил), где конденсированные кольца могут быть или не быть ароматическими. Конкретными гетероарильными группами являются такие, которые содержат 5- 14-членные кольца, содержащие от 1 до 12 кольцевых атомов углерода и от 1 до 6 кольцевых гетероатомов, независимо выбранных из группы, включающей азот, кислород и серу, 5-10-членные кольца, содержащие от 1 до 8 кольцевых атомов углерода и от 1 до 4 кольцевых гетероатомов, независимо выбранных из группы, включающей азот, кислород и серу, или 5-, 6- или 7-членные кольца, содержащие от 1 до 5 кольцевых атомов углерода и от 1 до 4 кольцевых гетероатомов, независимо выбранных из группы, включающей азот, кислород и серу. В одном варианте конкретными гетероарильными группами являются моноциклические ароматические 5-, 6- или 7-членные кольца, содержащие от 1 до 6 кольцевых атомов углерода и от 1 до 4 кольцевых гетероатомов, независимо выбранных из группы, включающей азот, кислород и серу. В другом варианте конкретными гетероарил группами являются полициклические ароматические кольца, содержащие от 1 до 12 кольцевых атомов углерода и от 1 до 6 кольцевых гетероатомов, независимо выбранных из группы, включающей азот, кислород и серу. Гетероарильная группа, содержащая более одного кольца, в которой по меньшей мере одно кольцо является неароматическим, может быть соединена с исходной структурой по положению ароматического кольца или по положению неароматического кольца. В одном варианте гетероарильная группа, содержащая более одного кольца, в которой по меньшей мере одно кольцо является неароматическим, соединена с исходной структурой по положению ароматического кольца. Гетероарильная группа может быть соединена с исходной структурой по кольцевому атому углерода или по кольцевому гетероатому.
[0033] Когда это применимо, гетероарильную группу можно представить в таутомерной форме. Такие соединения следует считать гетероарилами, даже если некоторые таутомерные формы представляют собой, например, гетероциклил. Например, гетероарильную группу  можно представить в гетероциклической таутомерной форме
можно представить в гетероциклической таутомерной форме  . Независимо от того, какой таутомер приведен, группу считают гетероарилом.
. Независимо от того, какой таутомер приведен, группу считают гетероарилом.
[0034] "Гетероцикл", "гетероциклический" или "гетероциклил" при использовании в настоящем изобретении означает насыщенную или ненасыщенную неароматическую циклическую группу, содержащую одно кольцо или несколько конденсированных колец и содержащую от 1 до 14 кольцевых атомов углерода и от 1 до 6 кольцевых гетероатомов, таких как азот, сера или кислород и т. п. Гетероцикл, содержащий более одного кольца, может быть конденсированным, спироциклическим или мостиковым, или их комбинацией, но исключает гетероарильные группы. Таким образом, следует понимать, что в конденсированных "гетероциклах" один или большее количество из конденсированных колец может представлять собой циклоалкил, циклоалкенил или арил, но не гетероарил. Гетероциклильная группа может быть необязательно независимо замещена одним или большим количеством заместителей, описанных в настоящем изобретении. Конкретными гетероциклил группами являются такие, которые содержат 3- 14-членные кольца, содержащие от 1 до 13 кольцевых атомов углерода и от 1 до 6 кольцевых гетероатомов, независимо выбранных из группы, включающей азот, кислород и серу, 3-12-членные кольца, содержащие от 1 до 11 кольцевых атомов углерода и от 1 до 6 кольцевых гетероатомов, независимо выбранных из группы, включающей азот, кислород и серу, 3- 10-членные кольца, содержащие от 1 до 9 кольцевых атомов углерода и от 1 до 4 кольцевых гетероатомов, независимо выбранных из группы, включающей азот, кислород и серу, 3- 8-членные кольца, содержащие от 1 до 7 кольцевых атомов углерода и от 1 до 4 кольцевых гетероатомов, независимо выбранных из группы, включающей азот, кислород и серу, или 3- 6-членные кольца, содержащие от 1 до 5 кольцевых атомов углерода и от 1 до 4 кольцевых гетероатомов, независимо выбранных из группы, включающей азот, кислород и серу. В одном варианте гетероциклил включает моноциклические 3-, 4-, 5-, 6- или 7-членные кольца, содержащие от 1 до 2, от 1 до 3, от 1 до 4, от 1 до 5 или от 1 до 6 кольцевых атомов углерода и от 1 до 2, от 1 до 3 или от 1 до 4 кольцевых гетероатомов, независимо выбранных из группы, включающей азот, кислород и серу. В другом варианте гетероциклил включает полициклические неароматические кольца, содержащие от 1 до 12 кольцевых атомов углерода и от 1 до 6 кольцевых гетероатомов, независимо выбранных из группы, включающей азот, кислород и серу.
[0035] "Галоген" означает элементы группы 17, обладающие атомными номерами от 9 до 85. Предпочтительные галоген группы включают радикалы фтор, хлор, бром и йод. Если остаток замещен более, чем одним галогеном, его можно назвать с использованием префикса, соответствующего количеству присоединенных галогеновых фрагментов, например, дигалогенарил, дигалогеналкил, тригалогенарил и т. п. означают арил и алкил, замещенный двумя ("ди") или тремя ("три") галогеновыми группами, которыми может быть, но необязательно, один и тот же галоген; таким образом 4-хлор-3-фторфенил входит в объем дигалогенарила. Алкильная группа, в которой каждый водород заменен галогеновой группой, называется "пергалогеналкилом". Предпочтительной пергалогеналкильной группой является трифторметил (-CF3). Аналогичным образом, "пергалогеналкоксигруппа" означает алкоксигруппу, в которой галоген занимает место каждого H в углеводороде, образующем алкильный фрагмент алкоксигруппы. Примером пергалогеналкоксигруппы является трифторметоксигруппа (-OCF3).
[0036] "Карбонил" означает группу C=O.
[0037] "Оксогруппа" означает фрагмент =O.
[0038] "Необязательно замещенная" если не указано иное означает, что группа может быть незамещенной или замещенной одним или большим количеством (например, 1, 2, 3, 4 или 5) заместителей, указанных для этой группы, где заместители могут быть одинаковыми или разным. В одном варианте осуществления необязательно замещенная группа содержит один заместитель. В другом варианте осуществления необязательно замещенная группа содержит два заместителя. В другом варианте осуществления необязательно замещенная группа содержит три заместителя. В другом варианте осуществления необязательно замещенная группа содержит четыре заместителя. В некоторых вариантах осуществления необязательно замещенная группа содержит от 1 до 2, от 1 до 3, от 1 до 4, от 1 до 5, от 2 до 3, от 2 до 4, или от 2 до 5 заместителей. В одном варианте осуществления необязательно замещенная группа является незамещенной.
[0039] Если явно не указано иное, "индивидуум" при использовании в настоящем изобретении означает млекопитающее, включая, но не ограничиваясь только ими, примата, человека, крупный рогатый скот, лошадь, кошачьих, собачьих или грызунов. В одном варианте индивидуумом является человек.
[0040] При использовании в настоящем изобретении "лечение" или "лечить" означает подход для обеспечения благоприятных или желательных результатов, включая клинические результаты. Благоприятные или желательные результаты включают, но не ограничиваются только ими, один или большее количество из следующего: ослабление одного или большего количества симптомов, вызванных заболеванием, уменьшение тяжести заболевания, стабилизацию заболевания (например, предупреждение или замедление ухудшения заболевания), предупреждение или замедление распространения заболевания, замедление появления или рецидива заболевания, задержку или замедление прогрессирования заболевания, смягчение патологического состояния, обеспечение ремиссии (частичной или полной) заболевания, уменьшение дозы одного или большего количества других лекарственных средств, необходимых для лечения заболевания, усиление воздействия другого лекарственного средства, задержку прогрессирования заболевания, улучшение качества жизни и/или пролонгирование выживания. Способы, описанные в настоящем изобретении, включают любой один или большее количество этих аспектов лечения.
[0041] При использовании в настоящем изобретении термин "эффективное количество" означает такое количество соединения, предлагаемого в настоящем изобретении, которое должно быть эффективным для данной терапевтической формы. Как известно в данной области техники, эффективное количество может находиться в одной или большем количестве доз, т. е. для обеспечения желательного лечения может потребоваться одна доза или несколько доз. Эффективное количество можно рассматривать в контексте введения одного или большего количества терапевтических средств и можно считать, что одно средство дается в эффективном количестве, если вместе с одним или большим количеством других средств можно обеспечить или обеспечивается желательный или благоприятный результат. Подходящие дозы вводимых совместно соединений необязательно можно уменьшить вследствие объединенного воздействия (например, аддитивного или синергетического эффектов) соединений.
[0042] "Терапевтически эффективное количество" означает количество соединения или его соли, достаточное для обеспечения желательного терапевтического результата.
[0043] При использовании в настоящем изобретении "разовая дозированная форма" означает физически дискретные порции, пригодные для использования в качестве разовых доз, причем каждая порция содержит заданное количество активного ингредиента, согласно расчету обеспечивающее желательный терапевтический эффект вместе с необходимым фармацевтическим носителем. Разовые дозированные формы могут содержать одно или комбинированное лекарственно средство.
[0044] При использовании в настоящем изобретении "фармацевтически приемлемый" или "фармакологически приемлемый" означает материал, который не является нежелательным с биологической или иной точки зрения, например, материал можно включать в фармацевтическую композицию, вводимую пациенту без проявления сколько-нибудь значительных нежелательных биологических эффектов или взаимодействия вредным образом с любыми другими компонентами композиции, в которой он содержится. Фармацевтически приемлемые носители или инертные наполнители предпочтительно удовлетворяют требованиям необходимых стандартов и проверок при изготовлении и/или включены в Руководство по неактивным ингредиентам Управления по продовольствию и лекарствам США.
[0045] "Фармацевтически приемлемые соли" являются такими солями, которые сохраняют по меньшей мере часть биологической активности свободного (не являющегося солью) соединения и которые можно вводить индивидууму в качестве лекарственных средств или фармацевтических препаратов. Такие соли, например, включают: (1) соли присоединения с кислотами, образованные с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т. п.; или образованные с органическими кислотами, такими как уксусная кислота, щавелевая кислота, пропионовая кислота, янтарная кислота, малеиновая кислота, винная кислота и т. п.; (2) соли, образованные с кислым протоном, содержащимся в исходном соединении, замещаемым ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; или координирование с органическим основанием. Приемлемые органические основания включают этаноламин, диэтаноламин, триэтаноламин и т. п. Приемлемые неорганические основания включают гидроксид алюминия, гидроксид кальция, гидроксид калия, карбонат натрия, гидроксид натрия и т. п. Фармацевтически приемлемые соли можно получить in situ в процессе получения или с помощью отдельной реакции очищенного соединения, предлагаемого в настоящем изобретении, в форме свободной кислоты или основания с подходящим органическим или неорганическим основанием или кислотой соответственно и с выделением полученной таким образом соли при последующей очистке.
[0046] Термин "инертный наполнитель" при использовании в настоящем изобретении означает инертное или неактивное вещество, которое можно использовать при изготовлении лекарственного средства или фармацевтического средства, такого как таблетка, содержащего соединение, предлагаемое в настоящем изобретении, в качестве активного ингредиента. Термин инертный наполнитель может включать разные вещества, включая без наложения ограничений любое вещество, использующееся, как связующее, разрыхлитель, покрытие, средство для прессования/капсулирования, крем или примочка, смазывающее вещество, растворы для парентерального введения, материалы для жевательных таблеток, подсластитель или вкусовая добавка, суспендирующий/гелеобразующий агент или агент для мокрого гранулирования. Связующие включают, например, карбомеры, повидон, ксантановую камедь и т. п.; покрытия включают, например, ацетатфталат целлюлозы, этилцеллюлоза, геллановая камедь, мальтодекстрин, энтеросолюбильные покрытия и т. п.; средства для прессования/капсулирования включают, например, карбонат кальция, декстрозу, фруктозу dc (dc = "для прямого прессования"), мед dc, лактозу (ангидрат или моногидрат; необязательно в комбинации с аспартамом, целлюлозой или микрокристаллической целлюлозой), крахмал dc, сахарозу и т. п.; разрыхлители включают, например, натриевую соль кроскармеллозы, геллановую камедь, натриевую соль гликолята крахмала и т. п.; кремы или примочки включают, например, мальтодекстрин, каррагенаны и т. п.; смазывающие вещества включают, например, стеарат магния, стеариновую кислоту, стеарилфумарат натрия и т. п.; материалы для жевательных таблеток включают, например, декстрозу, фруктозу dc, лактозу (моногидрат, необязательно в комбинации с аспартамом или целлюлозой) и т. п.; суспендирующие/гелеобразующие агенты включают, например, каррагенан, натриевая соль гликолята крахмала, ксантановую камедь и т. п.; подсластители включают, например, аспартам, декстрозу, фруктозу dc, сорбит, сахарозу dc и т. п.; и агенты для мокрого гранулирования включают, например, карбонат кальция, мальтодекстрин, микрокристаллическую целлюлозу и т. п.
[0047] Следует понимать, что объекты и варианты осуществления, описанные в настоящем изобретении, как "включающие" включают "состоящие из" и "состоящие в основном из" варианты осуществления.
[0048] Если композиция описана, как "состоящая в основном из" перечисленных компонентов, композиция содержит явно указанные компоненты и может содержать другие компоненты, которые существенно не влияют на подвергающееся лечению заболевание или патологическое состояние, такие как микропримеси. Однако композиция или не содержит любые другие компоненты, которые существенно не влияют на подвергающееся лечению заболевание или патологическое состояние, кроме явно указанных компонентов; или, если композиция все же содержит дополнительные компоненты кроме указанных, которые существенно влияют на подвергающееся лечению заболевание или патологическое состояние, композиция не содержит находящиеся в достаточной концентрации или в достаточном количестве эти дополнительные компоненты, которые существенно влияют на подвергающееся лечению заболевание или патологическое состояние. Если способ описан, как "состоящий в основном из" перечисленных стадий, способ содержит перечисленные стадии и может содержать другие стадии, которые существенно не влияют на подвергающееся лечению заболевание или патологическое состояние, но способ не содержит какие-либо другие стадии, которые существенно влияют на подвергающееся лечению заболевание или патологическое состояние, кроме явно указанных стадий.
[0049] Если фрагмент указан, как замещенный "по меньшей мере одним" заместителем, это также включает раскрытие точно одного заместителя.
Соединения
[0050] Некоторые варианты осуществления относятся к соединению формулы (I):
(I)
его фармацевтически приемлемой соли, стереоизомеру или таутомеру, где:
R означает водород, C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R независимо необязательно замещены посредством Rd;
m равно 0, 1, 2, 3 или 4;
n равно 0, 1, 2, 3 или 4,
где m+n равно 1, 2, 3 или 4;
X означает -C(=O)-, -O-, -CH(OH)-, -S-, -S(=O)- или -S(=O)2-;
L означает
(a) , где
* обозначает положение присоединения к фрагменту Y-X-,
** обозначает положение присоединения к остальной части молекулы,
Ra означает водород, C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве Ra независимо необязательно замещены посредством Re,
R1 и R2 независимо друг от друга и независимо в каждом случае означают водород, C1-C2 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R1 и R2 независимо необязательно замещены посредством Rf,
или R1 и R2 вместе с атомом или атомами углерода, к которым они присоединены, образуют 3- 8-членный циклоалкилен, необязательно замещенный посредством Rf,
q равно 1, 2 или 3,
R3 и R4 независимо друг от друга и независимо в каждом случае означают водород, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R3 и R4 независимо необязательно замещены посредством Rg,
или R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют 3- 8-членный циклоалкилен, необязательно замещенный посредством Rg, и
p равно 0, 1 или 2;
(b) , где
* обозначает положение присоединения к фрагменту Y-X-,
** обозначает положение присоединения к остальной части молекулы,
R5 и R6 независимо друг от друга и независимо в каждом случае означают H, C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R5 и R6 независимо необязательно замещены посредством Rh,
Rb и Rc независимо означают H, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил, C6-C14 арил, или -C(=O)OR17, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве Rb и Rc независимо необязательно замещены посредством Ri, и
r равно 1, 2 или 3; или
(c) , где
* обозначает положение присоединения к фрагменту Y-X-,
** обозначает положение присоединения к остальной части молекулы,
R7 и R8 независимо друг от друга и независимо в каждом случае означают водород, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R7 и R8 независимо необязательно замещены посредством Rj,
или R7 и R8 вместе с атомом углерода, к которому они присоединены, образуют 3- 8-членный циклоалкилен, необязательно замещенный посредством Rj,
R9 и R10 независимо друг от друга и независимо в каждом случае означают H, C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R9 и R10 независимо необязательно замещены посредством Rk,
s равно 1, 2 или 3,
t равно 1, 2 или 3,
где s+t равно 2, 3 или 4,
u равно 0 или 1, и
v равно 0 или 1;
Y означает C6-C9 арил, замещенный посредством R11, 6- 10-членный гетероарил, замещенный посредством R12 или 3-12-членный гетероциклил, замещенный посредством R13, где
каждый R11, R12 и R13 независимо означает C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C4-C8 циклоалкенил, 3-12-членный гетероциклил, 5-10-членный гетероарил, C6-C14 арил, -OR14, -NR15R16, -SR14, -NO2, -C=NH(OR14), -C(O)R14, -OC(O)R14, -C(O)OR14, -C(O)NR15R16, -NR14C(O)R15, -NR14C(O)OR15, -NR14C(O)NR15R16, -S(O)R14, -S(O)2R14, -NR14S(O)R15, -NR14S(O)2R15, -S(O)NR15R16, -S(O)2NR15R16 или -P(O)(OR15)(OR16), где C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C4-C8 циклоалкенил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R11, R12 и R13 замещены посредством RL;
R14, R15 и R16 независимо друг от друга и независимо в каждом случае означают водород, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил или 3-12-членный гетероциклил, где C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил и 3-12-членный гетероциклил в качестве R14, R15 и R16 независимо замещены C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой, C1-C6 пергалогеналкоксигруппой, C6-C14 арилом или C6-C14 арилоксигруппой где C6-C14 арил или C6-C14 арилоксигруппа необязательно дополнительно замещена галогеном, -OH, цианогруппой, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой или C1-C6 пергалогеналкоксигруппой; и где по меньшей мере один из R14, R15 и R16, если содержится, не означает водород;
RL означает C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил или 3-12-членный гетероциклил, где C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил или 3-12-членный гетероциклил в качестве RL означает, замещенный галогеном, -OH, цианогруппой, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой, C1-C6 пергалогеналкоксигруппой или C6-C14 арилом, где C6-C14 арил дополнительно необязательно замещен галогеном, -OH, цианогруппой, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой или C1-C6 пергалогеналкоксигруппой; и
Rd, Re, Rf, Rg, Rh, Ri, Rj и Rk независимо друг от друга и независимо в каждом случае означают галоген, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил, 3-12-членный гетероциклил, -OR14, -NR15R16, цианогруппу или нитрогруппу.
[0051] Для описаний в настоящем изобретении, следует понимать, что каждое описание, изменение, вариант осуществления или аспект фрагмента можно объединить с каждым описанием, изменением, вариантом осуществления или аспектом других фрагментов так, как если бы каждое описание, изменение, вариант осуществления или аспект фрагмента было указано явно и по отдельности. Например, каждое описание, изменение, вариант осуществления или аспект, приведенный в настоящем изобретении применительно к R формулы (I) можно объединить с каждым описанием, изменением, вариантом осуществления или аспектом Y, X, L, m и/или n так, как если бы каждая и всякая комбинация была указана явно и по отдельности. Также следует понимать, что все описания, изменения, варианты осуществления или аспекты формулы (I), когда это применимо, в равной степени относятся к другим формулам, подробно описанным в настоящем изобретении, и одинаково описаны так, как если бы каждое и всякое описание, изменение, вариант осуществления или аспект были указаны явно и по отдельности для всех формул. Например, все описания, изменения, варианты осуществления или аспекты формулы (I), когда это применимо, в равной степени относятся к любым формулам, указанным в настоящем изобретении, и одинаково описаны так, как если бы каждое и всякое описание, изменение, вариант осуществления или аспект были указаны явно и по отдельности для всех формул. Это относится к любой другой формуле, приведенной в настоящем изобретении.
[0052] В некоторых вариантах осуществления соединение формулы (I) описывается формулой (Ia):
 (Ia)
(Ia)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X, L, R, m и n являются такими, как определено для формулы (I).
[0053] В некоторых вариантах осуществления соединение формулы (I) описывается формулой (Ib):
 (Ib)
(Ib)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X, L, R, m и n являются такими, как определено для формулы (I).
[0054] В некоторых вариантах осуществления соединения формулы (I), где m равно 1 и n равно 1, соединение описывается формулой (II):
 (II)
(II)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X, L и R являются такими, как определено для формулы (I).
[0055] В некоторых вариантах осуществления соединение формулы (II) описывается формулой (IIa):
 (IIa)
(IIa)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X, L и R являются такими, как определено для формулы (I).
[0056] В некоторых вариантах осуществления соединение формулы (II) описывается формулой (IIb):
 (IIb)
(IIb)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X, L и R являются такими, как определено для формулы (I).
[0057] В некоторых вариантах осуществления соединения формулы (II), где L означает -NH-CH2-, соединение описывается формулой (III):
 (III)
(III)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I).
[0058] В некоторых вариантах осуществления соединение формулы (III) описывается формулой (IIIa):
 (IIIa)
(IIIa)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I). В некоторых таких вариантах осуществления X означает -C(=O)-. В некоторых таких вариантах осуществления X означает -C(=O)- и R означает водород. В некоторых таких вариантах осуществления X означает -C(=O)- и Y означает  .
.
[0059] В некоторых вариантах осуществления соединение формулы (III) описывается формулой (IIIb):
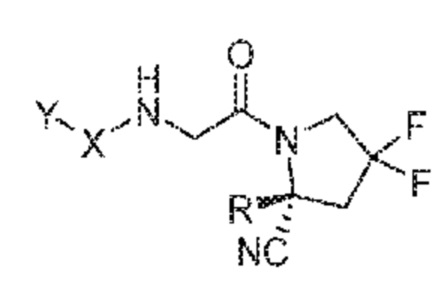 (IIIb)
(IIIb)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I). В некоторых таких вариантах осуществления X означает -C(=O)-. В некоторых таких вариантах осуществления X означает -C(=O)- и R означает водород. В некоторых таких вариантах осуществления X означает -C(=O)- и Y означает .
[0060] В некоторых вариантах осуществления соединение формулы (III) описывается формулой (III-1):
 (III-1)
(III-1)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y и X являются такими, как определено для формулы (I).
[0061] В некоторых вариантах осуществления соединения формулы (II), где L означает -NH-CH(CH3)-, соединение описывается формулой (IV):
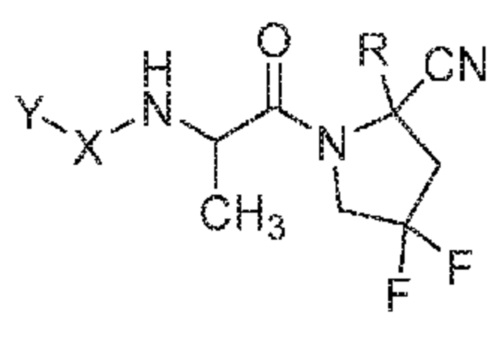 (IV)
(IV)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I). В одном варианте осуществления соединения формулы (IV) атом углерода, содержащий метильную группу от L, находится в конфигурации S. В одном варианте осуществления соединения формулы (IV) атом углерода, содержащий метильную группу от L, находится в конфигурации R.
[0062] В некоторых вариантах осуществления соединение формулы (IV) описывается формулой (IVa):
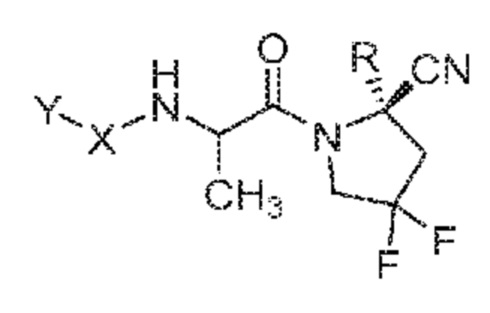 (IVa)
(IVa)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I). В одном варианте осуществления соединения формулы (IVa) атом углерода, содержащий метильную группу от L, находится в конфигурации S. В одном варианте осуществления соединения формулы (IVa) атом углерода, содержащий метильную группу от L, находится в конфигурации R.
[0063] В некоторых вариантах осуществления соединение формулы (IV) описывается формулой (IVb):
 (IVb)
(IVb)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I). В одном варианте осуществления соединения формулы (IVb) атом углерода, содержащий метильную группу от L, находится в конфигурации S. В одном варианте осуществления соединения формулы (IVb) атом углерода, содержащий метильную группу от L, находится в конфигурации R.
[0064] В некоторых вариантах осуществления соединения формулы (II), соединение описывается формулой (V):
 (V)
(V)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I).
[0065] В некоторых вариантах осуществления соединение формулы (V) описывается формулой (Va):
 (Va)
(Va)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I).
[0066] В некоторых вариантах осуществления соединение формулы (V) описывается формулой (Vb):
 (Vb)
(Vb)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I).
[0067] В некоторых вариантах осуществления соединения формулы (II), соединение описывается формулой (VI):
 (VI)
(VI)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I).
[0068] В некоторых вариантах осуществления соединение формулы (VI) описывается формулой (VIa):
 (VIa)
(VIa)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I).
[0069] В некоторых вариантах осуществления соединение формулы (VI) описывается формулой (VIb):
 (VIb)
(VIb)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I).
[0070] В некоторых вариантах осуществления соединения формулы (II), где L означает -CH2-CH(NH2)-, соединение описывается формулой (VII):
 (VII)
(VII)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I). В одном варианте осуществления соединения формулы (VII) атом углерода, содержащий группу -NH2 от L, находится в конфигурации S. В одном варианте осуществления соединения формулы (VII), атом углерода, содержащий группу -NH2 от L, находится в конфигурации R.
[0071] В некоторых вариантах осуществления соединение формулы (VII) описывается формулой (VIIa):
 (VIIa)
(VIIa)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I). В одном варианте осуществления соединения формулы (VIIa), атом углерода, содержащий группу -NH2 от L, находится в конфигурации S. В одном варианте осуществления соединения формулы (VIIa), атом углерода, содержащий группу -NH2 от L, находится в конфигурации R.
[0072] В некоторых вариантах осуществления соединение формулы (VII) описывается формулой (VIIb):
 (VIIb)
(VIIb)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I). В одном варианте осуществления соединения формулы (VIIb), атом углерода, содержащий группу -NH2 от L, находится в конфигурации S. В одном варианте осуществления соединения формулы (VIIb), атом углерода, содержащий группу -NH2 от L, находится в конфигурации R.
[0073] В некоторых вариантах осуществления соединения формулы (II), где L означает -CH(CH3)-CH(NH2)-, соединение описывается формулой (VIII):
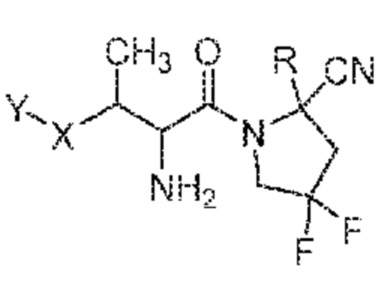 (VIII)
(VIII)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I). В одном варианте осуществления соединения формулы (VIII), атом углерода, содержащий группу -NH2 от L, находится в конфигурации S. В одном варианте осуществления соединения формулы (VIII), атом углерода, содержащий группу -NH2 от L, находится в конфигурации R. В одном варианте осуществления соединения формулы (VIII) атом углерода, содержащий метильную группу от L, находится в конфигурации S. В одном варианте осуществления соединения формулы (VIII) атом углерода, содержащий метильную группу от L, находится в конфигурации R.
[0074] В некоторых вариантах осуществления соединение формулы (VIII) описывается формулой (VIIIa):
 (VIIIa)
(VIIIa)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I). В одном варианте осуществления соединения формулы (VIIIa), атом углерода, содержащий группу -NH2 от L, находится в конфигурации S. В одном варианте осуществления соединения формулы (VIIIa), атом углерода, содержащий группу -NH2 от L, находится в конфигурации R. В одном варианте осуществления соединения формулы (VIIIa) атом углерода, содержащий метильную группу от L, находится в конфигурации S. В одном варианте осуществления соединения формулы (VIIIa) атом углерода, содержащий метильную группу от L, находится в конфигурации R.
[0075] В некоторых вариантах осуществления соединение формулы (VIII) описывается формулой (VIIIb):
 (VIIIb)
(VIIIb)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I). В одном варианте осуществления соединения формулы (VIIIb), атом углерода, содержащий группу -NH2 от L, находится в конфигурации S. В одном варианте осуществления соединения формулы (VIIIb), атом углерода, содержащий группу -NH2 от L, находится в конфигурации R. В одном варианте осуществления соединения формулы (VIIIb) атом углерода, содержащий метильную группу от L, находится в конфигурации S. В одном варианте осуществления соединения формулы (VIIIb) атом углерода, содержащий метильную группу от L, находится в конфигурации R. В некоторых вариантах осуществления соединения формулы (II), соединение описывается формулой (IX):
 (IX)
(IX)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I). В одном варианте осуществления соединения формулы (IX) 1,3-циклобутилен является цис-изомером. В одном варианте осуществления соединения формулы (IX) 1,3-циклобутилен является транс-изомером.
[0076] В некоторых вариантах осуществления соединение формулы (IX) описывается формулой (IXa):
 (IXa)
(IXa)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I). В одном варианте осуществления соединения формулы (IXa) 1,3-циклобутилен является цис-изомером. В одном варианте осуществления соединения формулы (IXa) 1,3-циклобутилен является транс-изомером.
[0077] В некоторых вариантах осуществления соединение формулы (IX) описывается формулой (IXb):
 (IXb)
(IXb)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I). В одном варианте осуществления соединения формулы (IXb) 1,3-циклобутилен является цис-изомером. В одном варианте осуществления соединения формулы (IXb) 1,3-циклобутилен является транс-изомером.
[0078] В некоторых вариантах осуществления соединения формулы (II), где L означает -NH-CH2-, соединение описывается формулой (X):
 (X)
(X)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I).
[0079] В некоторых вариантах осуществления соединение формулы (X) описывается формулой (Xa):
 (Xa)
(Xa)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I).
[0080] В некоторых вариантах осуществления соединение формулы (X) описывается формулой (Xb):
 (Xb)
(Xb)
его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y, X и R являются такими, как определено для формулы (I).
[0081] Один вариант относится к соединению формулы (I), его фармацевтически приемлемой соли, стереоизомеру или таутомеру, где X означает -C(=O)-, -O- или -CH(OH)-. Другой вариант относится к соединению формулы (I), его фармацевтически приемлемой соли, стереоизомеру или таутомеру, где X означает -S-, -S(=O)- или -S(=O)2-. В некоторых вариантах осуществления соединения формулы (I), его фармацевтически приемлемой соли, стереоизомера или таутомера X означает -C(=O)-. В других вариантах осуществления соединения формулы (I), его фармацевтически приемлемой соли, стереоизомера или таутомера X означает -O-. Все варианты X в равной степени применимы к любым применимым формулам, приведенным в настоящем изобретении, таким как формулы Ia, Ib, II, IIa, IIb, III, IIIa, IIIb, IV, IVa, IVb, V, Va, Vb, VI, VIa, VIb, VII, VIIa, VIIb, VIII, VIIIa и VIIIb.
[0082] В некоторых вариантах осуществления соединения формулы (I), его фармацевтически приемлемой соли, стереоизомера или таутомера L означает 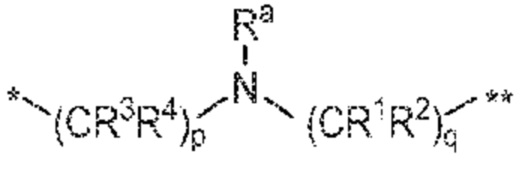 . В предпочтительном таком варианте осуществления Ra означает H и R1, R2, R3 и R4, если содержатся, все означают H. В одном варианте осуществления L означает и X означает -C=O. В другом варианте осуществления L означает , X означает -C=O и p равно 0.
. В предпочтительном таком варианте осуществления Ra означает H и R1, R2, R3 и R4, если содержатся, все означают H. В одном варианте осуществления L означает и X означает -C=O. В другом варианте осуществления L означает , X означает -C=O и p равно 0.
[0083] В некоторых вариантах осуществления L означает . В одном предпочтительном варианте осуществления R1 и R2 присоединены к одному атому углерода. В другом предпочтительном варианте, R1 и R2 присоединены к разным атомам углерода.
[0084] В некоторых вариантах осуществления L означает -N(Ra)-CR1R2- (т. е. p равно 0). В одном предпочтительном варианте осуществления L означает -NH-CR1R2-. В другом предпочтительном варианте L означает -NH-CH2-. В другом предпочтительном варианте L означает -NH-CH(CH3)-. В другом предпочтительном варианте L означает -NH-CR1R2-, где R1 и R2 вместе с атомом или атомами углерода, к которым они присоединены, образуют 3- 8-членный циклоалкилен (например, циклопропилен).
[0085] В некоторых вариантах осуществления L означает -N(Ra)-(CR1R2)3- (т. е. p равно 0). В одном предпочтительном варианте осуществления L означает -NH-(CR1R2)3-. В другом предпочтительном варианте L означает -NH-(CH2)3-. В другом предпочтительном варианте L означает -NH-(CR1R2)3-, где R1 и R2 двух несоседних атомов углерода вместе с атомами углерода, к которым они присоединены, и промежуточным атомом углерода образуют 3- 8-членный циклоалкилен (например, 1,3-циклобутилен).
[0086] В других вариантах осуществления соединения формулы (I), его фармацевтически приемлемой соли, стереоизомера или таутомера L означает  . В одном таком варианте осуществления X означает -C=O. В другом таком варианте осуществления X означает -O-. В другом таком варианте осуществления X означает -CH(OH)-. В еще одном таком варианте осуществления X означает -S-. В еще одном таком варианте осуществления X означает -S(=O)-. В еще одном таком варианте осуществления X означает -S(=O)2. В одном воплощении таких вариантов осуществления r равно 1. В другом воплощении таких вариантов осуществления r равно 2. В еще одном воплощении таких вариантов осуществления r равно 3. В любом варианте осуществления, где L означает в одном варианте Rb и Rc оба означают H. В любом варианте осуществления, где L означает в другом варианте Rb и Rc оба означают H, r равно 1, R5 означает H и R6 означает C1-C6 алкил, такой как метил.
. В одном таком варианте осуществления X означает -C=O. В другом таком варианте осуществления X означает -O-. В другом таком варианте осуществления X означает -CH(OH)-. В еще одном таком варианте осуществления X означает -S-. В еще одном таком варианте осуществления X означает -S(=O)-. В еще одном таком варианте осуществления X означает -S(=O)2. В одном воплощении таких вариантов осуществления r равно 1. В другом воплощении таких вариантов осуществления r равно 2. В еще одном воплощении таких вариантов осуществления r равно 3. В любом варианте осуществления, где L означает в одном варианте Rb и Rc оба означают H. В любом варианте осуществления, где L означает в другом варианте Rb и Rc оба означают H, r равно 1, R5 означает H и R6 означает C1-C6 алкил, такой как метил.
[0087] В некоторых из этих вариантов осуществления L означает -CR5R6-CH(NRbRc)- (т. е. r равно 1). В одном предпочтительном варианте осуществления L означает -CH(R5)-CH(NH2)-, включая, но не ограничиваясь только ими, варианты, где R5 означает водород или C1-C6 алкил. В одном предпочтительном варианте осуществления L означает -CH2-CH(NH2)-. В другом предпочтительном варианте L означает -CH(CH3)-CH(NH2)-.
[0088] В некоторых вариантах осуществления соединения формулы (I), его фармацевтически приемлемой соли, стереоизомера или таутомера L означает  . В одном таком варианте осуществления X означает -C=O. В другом таком варианте осуществления X означает -O-. В другом таком варианте осуществления X означает -S-. В еще одном таком варианте осуществления X означает -S(=O)-. В еще одном таком варианте осуществления X означает -S(=O)2. В предпочтительном варианте L означает и u равно 0. В другом варианте L означает u равно 0 и X выбран из группы, состоящей из следующих: -C=O, -O-, -S-, -S(=O)- и -S(=O)2. В любом варианте осуществления или варианте, где L означает , в одном объекте s равно 1 и t равно 1.
. В одном таком варианте осуществления X означает -C=O. В другом таком варианте осуществления X означает -O-. В другом таком варианте осуществления X означает -S-. В еще одном таком варианте осуществления X означает -S(=O)-. В еще одном таком варианте осуществления X означает -S(=O)2. В предпочтительном варианте L означает и u равно 0. В другом варианте L означает u равно 0 и X выбран из группы, состоящей из следующих: -C=O, -O-, -S-, -S(=O)- и -S(=O)2. В любом варианте осуществления или варианте, где L означает , в одном объекте s равно 1 и t равно 1.
[0089] В одном варианте L означает 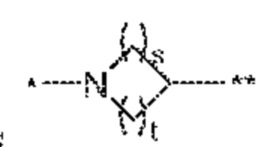 . В другом предпочтительном варианте L означает
. В другом предпочтительном варианте L означает  . В одном варианте L означает и X выбран из группы, состоящей из следующих: -C=O, -O-, -S-, -S(=O)- и -S(=O)2.
. В одном варианте L означает и X выбран из группы, состоящей из следующих: -C=O, -O-, -S-, -S(=O)- и -S(=O)2.
[0090] В одном варианте настоящее изобретение относится к соединению формулы (I), его фармацевтически приемлемой соли, стереоизомеру или таутомеру, где фрагмент -X-L- выбран из группы, состоящей из следующих:


 и
и  .
.
где * обозначает положение присоединения к фрагменту Y и ** обозначает положение присоединения к остальной части молекулы.
[0091] Другой вариант относится к соединению формулы (I), его фармацевтически приемлемой соли, стереоизомеру или таутомеру, где фрагмент -X-L- выбран из группы, состоящей из следующих:

 и
и  ,
,
где * обозначает положение присоединения к фрагменту Y и ** обозначает положение присоединения к остальной части молекулы.
[0092] Одним объектом является соединение формулы (I) или его фармацевтически приемлемая соль, где соединение обладает одной или большим количеством следующих характеристик:
(i) X означает -C(=O)-, -O- или -CH(OH)-;
(ii) L означает:
(a) -NH-(CR1R2)q-, где R1 и R2 независимо друг от друга и независимо в каждом случае означают водород или C1-C2 алкил или группы R1 и R2, присоединенные к одному атому углерода, вместе с атомом углерода, к которому они присоединены, образуют 3- 5-членный циклоалкилен или группы R1 и R2, присоединенные к двум разным атомам углерода, вместе с атомами углерода, к которым они присоединены, образуют 3- 5-членный циклоалкилен (примеры таких фрагментов -NH-(CR1R2)q- включают 
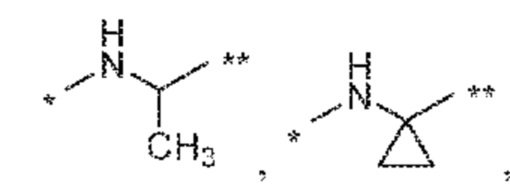 и
и  ;
;
(b) -CR5R6-CH(NH2)-, где R5 и R6 независимо друг от друга и независимо в каждом случае означают водород или C1-C2 алкил (примеры таких фрагментов -CR5R6-CH(NH2)- включают  и
и  ; или
; или
(c)  ,
,
где * обозначает положение присоединения к фрагменту Y-X- и ** обозначает положение присоединения к остальной части молекулы; и
(iii) Y означает:
(a) C6-C9 арил, замещенный посредством R11, такой как 2,3-дигидро-1H-инден-2-ил, фенил и нафтил, которые замещены по меньшей мере одним R11;
(b) 6- 10-членный гетероарил, замещенный посредством R12, такой как пиридинил, пиримидинил, пиридин-2(1H)-онил и хинолин-6-ил, которые замещены по меньшей мере одним R12; или
(c) 3-12-членный гетероциклил, замещенный посредством R13, такой как 2H-пиран-2-only, изоиндолинил, пиперидин-2-only и пиперидинил, которые замещены по меньшей мере одним R13.
[0093] В одном воплощении этого варианта (i) применяются (ii)(a) и (iii)(a). В другом варианте (i) применяются (ii)(a) и (iii)(b). В другом варианте (i) применяются (ii)(a) и (iii)(c). В другом варианте (i) применяются (ii)(b) и (iii)(a). В другом варианте (i) применяются (ii)(b) и (iii)(b). В другом варианте (i) применяются (ii)(b) и (iii)(c). В другом варианте (i) применяются (ii)(c) и (iii)(a). В другом варианте (i) применяются (ii)(c) и (iii)(b). В другом варианте (i) применяются (ii)(c) и (iii)(c).
[0094] Все варианты L или комбинации X и L в равной степени применяются в настоящем изобретении в формулах, таких как формулы Ia, Ib, II, IIa, IIb, III, IIIa, IIIb, IV, IVa, IVb, V, Va, Vb, VI, VIa, VIb, VII, VIIa, VIIb, VIII, VIIIa и VIIIb.
[0095] В некоторых вариантах осуществления Y означает C6-C9 арил, замещенный одним или большим количеством R11, 6- 10-членный гетероарил, замещенный одним или большим количеством R12 или 3-12-членный гетероциклил, замещенный одним или большим количеством R13. В одном варианте Y означает замещенный с помощью от 1 до 3 R11, R12 или R13 фрагментов, которые могут быть одинаковыми или разными.
[0096] В некоторых вариантах осуществления Y означает C6-C9 арил, замещенный посредством R11. В одном объекте Y означает фенил, замещенный посредством R11. В одном варианте Y означает замещенный с помощью от 1 до 3 R11 фрагментов, которые могут быть одинаковыми или разными.
[0097] В некоторых вариантах осуществления Y означает 6- 10-членный гетероарил, замещенный посредством R12. В одном варианте Y означает 6-членный гетероарил, замещенный посредством R12. В некоторых вариантах осуществления Y означает пиридин, замещенный посредством R12. В некоторых вариантах осуществления Y означает пиридин-4-ил, замещенный посредством R12. В некоторых вариантах осуществления Y означает пиридин-4-ил, замещенный посредством R12 в положении 3. В некоторых вариантах осуществления Y означает хинолинил, замещенный посредством R12. В некоторых вариантах осуществления Y означает хинолин-4-ил, замещенный посредством R12. В некоторых вариантах осуществления Y означает хинолин-4-ил, замещенный посредством R12 в положении 7. В некоторых вариантах осуществления Y означает 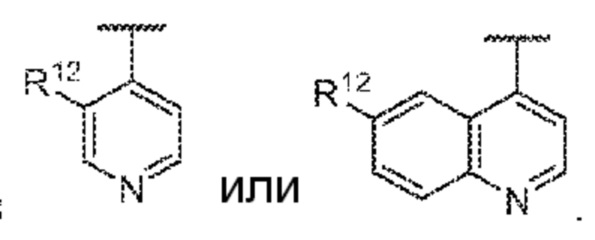 .
.
[0098] В одном варианте R12 означает C1-C6 алкил, замещенный посредством RL. В другом варианте R12 означает C2-C6 алкенил, замещенный посредством RL. В другом варианте R12 означает C2-C6 алкинил, замещенный посредством RL. В еще одном варианте R12 означает 3-12-членный гетероциклил, замещенный посредством RL.
[0099] В некоторых вариантах осуществления RL означает C6-C14 арил, замещенный галогеном, -OH, цианогруппой, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой, C1-C6 пергалогеналкоксигруппой или C6-C14 арилом. В некоторых вариантах осуществления RL означает 5-10-членный гетероарил, замещенный галогеном, -OH, цианогруппой, оксогруппой, -NH2, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой или C1-C6 пергалогеналкоксигруппой. В некоторых вариантах осуществления RL означает 3-12-членный гетероциклил, замещенный галогеном, -OH, цианогруппой, оксогруппой, -NH2, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой или C1-C6 пергалогеналкоксигруппой.
[0100] В одном варианте R12 означает -NR14C(O)R15. В некоторых вариантах осуществления по меньшей мере один из R14 и R15 означает C1-C6 алкил или C6-C14 арил, где C1-C6 алкил или C6-C14 арил в качестве R14 и R15 независимо замещены C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой, C1-C6 пергалогеналкоксигруппой, C6-C14 арилом или C6-C14 арилоксигруппой, где C6-C14 арил или C6-C14 арилоксигруппа необязательно дополнительно замещена галогеном, -OH, цианогруппой, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой или C1-C6 пергалогеналкоксигруппой. В некоторых вариантах осуществления по меньшей мере один из R14 и R15 означает C1-C6 алкил или C6-C14 арил, где C1-C6 алкил или C6-C14 арил в качестве R14 и R15 независимо замещены C1-C6 алкоксигруппой, C1-C6 пергалогеналкоксигруппой, C6-C14 арилом или C6-C14 арилоксигруппой, где C6-C14 арил или C6-C14 арилоксигруппа необязательно дополнительно замещена галогеном, -OH, цианогруппой, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой или C1-C6 пергалогеналкоксигруппой.
[0101] В одном варианте R12 означает -NR14R15. В некоторых вариантах осуществления один из R14 и R15 означает водород и второй означает C1-C6 алкил, где C1-C6 алкил замещен C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой, C1-C6 пергалогеналкоксигруппой, C6-C14 арилом или C6-C14 арилоксигруппой, где C6-C14 арил или C6-C14 арилоксигруппа необязательно дополнительно замещена галогеном, -OH, цианогруппой, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой или C1-C6 пергалогеналкоксигруппой.
[0102] В некоторых вариантах осуществления Y означает B’ или его таутомер. Таким образом, следует понимать, что в некоторых вариантах осуществления Y означает  , где z равно 0, 1, 2, 3, 4 или 5;
, где z равно 0, 1, 2, 3, 4 или 5;  указывает на таутомерию между A’ и B’; и R12 и R13 одинаковы для любой пары таутомеров. В некоторых вариантах осуществления Y означает D’ или его таутомер. Таким образом, следует понимать, что в некоторых вариантах осуществления Y означает
указывает на таутомерию между A’ и B’; и R12 и R13 одинаковы для любой пары таутомеров. В некоторых вариантах осуществления Y означает D’ или его таутомер. Таким образом, следует понимать, что в некоторых вариантах осуществления Y означает  , где z равно 0, 1, 2, 3, 4 или 5; указывает на таутомерию между C’ и D’; и R12 и R13 одинаковы для любой пары таутомеров.
, где z равно 0, 1, 2, 3, 4 или 5; указывает на таутомерию между C’ и D’; и R12 и R13 одинаковы для любой пары таутомеров.
[0103] В некоторых вариантах осуществления Y означает 3-12-членный гетероциклил, замещенный посредством R13. В некоторых вариантах осуществления Y означает замещенный изоиндолин-2-ил. В некоторых вариантах осуществления Y означает пиперидин-2-он-5-ил, замещенный посредством R13.
[0104] В настоящем изобретении также предложены соли соединений, указанных в настоящем изобретении, такие как фармацевтически приемлемые соли. Настоящее изобретение также включает любую или все стереохимические формы, включая любые энантиомерные или диастереоизомерные формы и любые таутомеры или другие формы описанных соединений.
[0105] Некоторые из соединений, описанных в настоящем изобретении, находятся в равновесии с таутомерной формой. Например, амид A является таутомерной формой B и имидовая кислота B является таутомерной формой A. Аналогичным образом, амид C является таутомерной формой D и имидовая кислота D является таутомерной формой C. Амид A находится в равновесии с таутомерной формой имидовой кислоты B и амид C находится в равновесии с таутомерной формой имидовой кислоты D. Независимо от того, какая таутомерная форма приведена, специалист с общей подготовкой в данной области техники понимает, что в соединения включены таутомеры и амид и имидовая кислота. 
[0106] Соединение, подробно описанное в настоящем изобретении, в одном объекте может быть в очищенной форме и композиции, содержащие соединение в очищенных формах, подробно описаны в настоящем изобретении. Предложены композиции, содержащие соединение, подробно описанное в настоящем изобретении, или его фармацевтически приемлемую соль, стереоизомер или таутомер, такие как композиции в основном чистых соединений. В некоторых вариантах осуществления композиция, содержащая соединение, подробно описанное в настоящем изобретении, или его фармацевтически приемлемую соль, стереоизомер или таутомер находится в основном в чистой форме. Если не указано иное, "в основном чистая" означает композицию, которая содержит не более 35% примеси, где примесь означает соединение, не являющееся соединением, образующим большую часть композиции, или его фармацевтически приемлемой солью, стереоизомером или таутомером. В некоторых вариантах осуществления предложена композиция в основном чистого соединения или его фармацевтически приемлемой соли, стереоизомера или таутомера, где композиция содержит не более 25%, 20%, 15%, 10% или 5% примеси. В некоторых вариантах осуществления предложена композиция в основном чистого соединения или его фармацевтически приемлемой соли, стереоизомера или таутомера, где композиция содержит не более 3%, 2%, 1% или 0,5% примеси.
[0107] Типичные соединения приведены в таблице 1.
Таблица 1













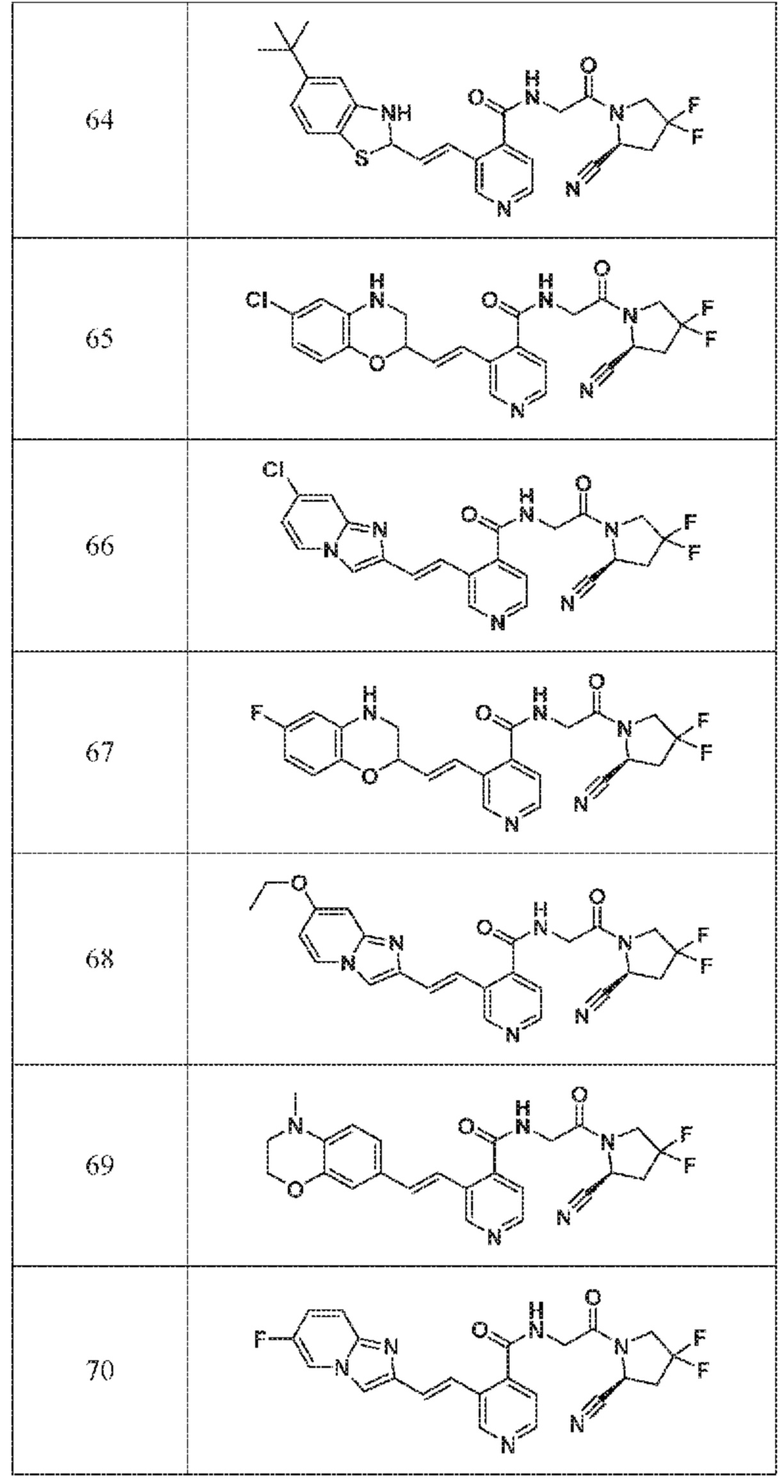

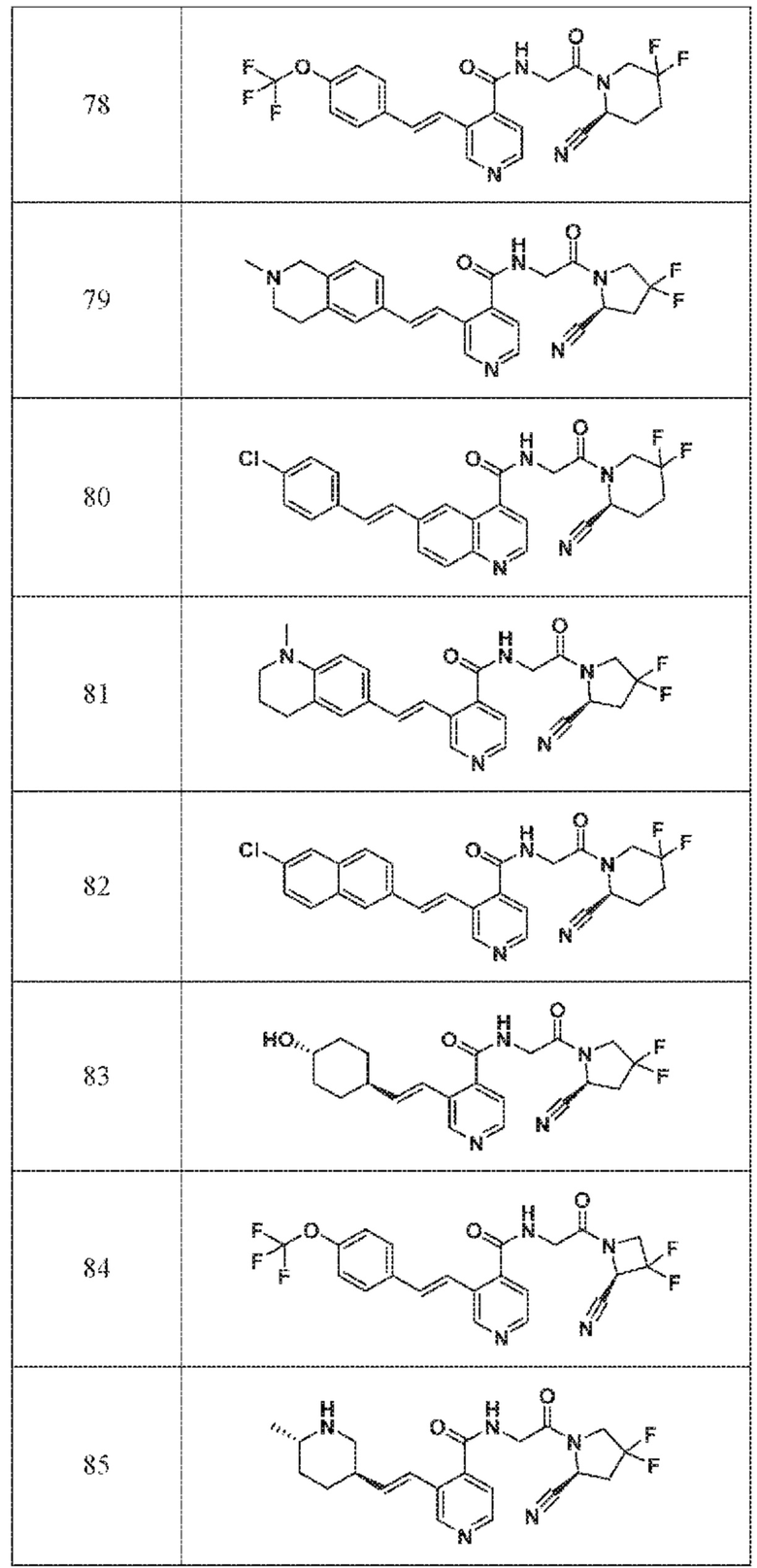
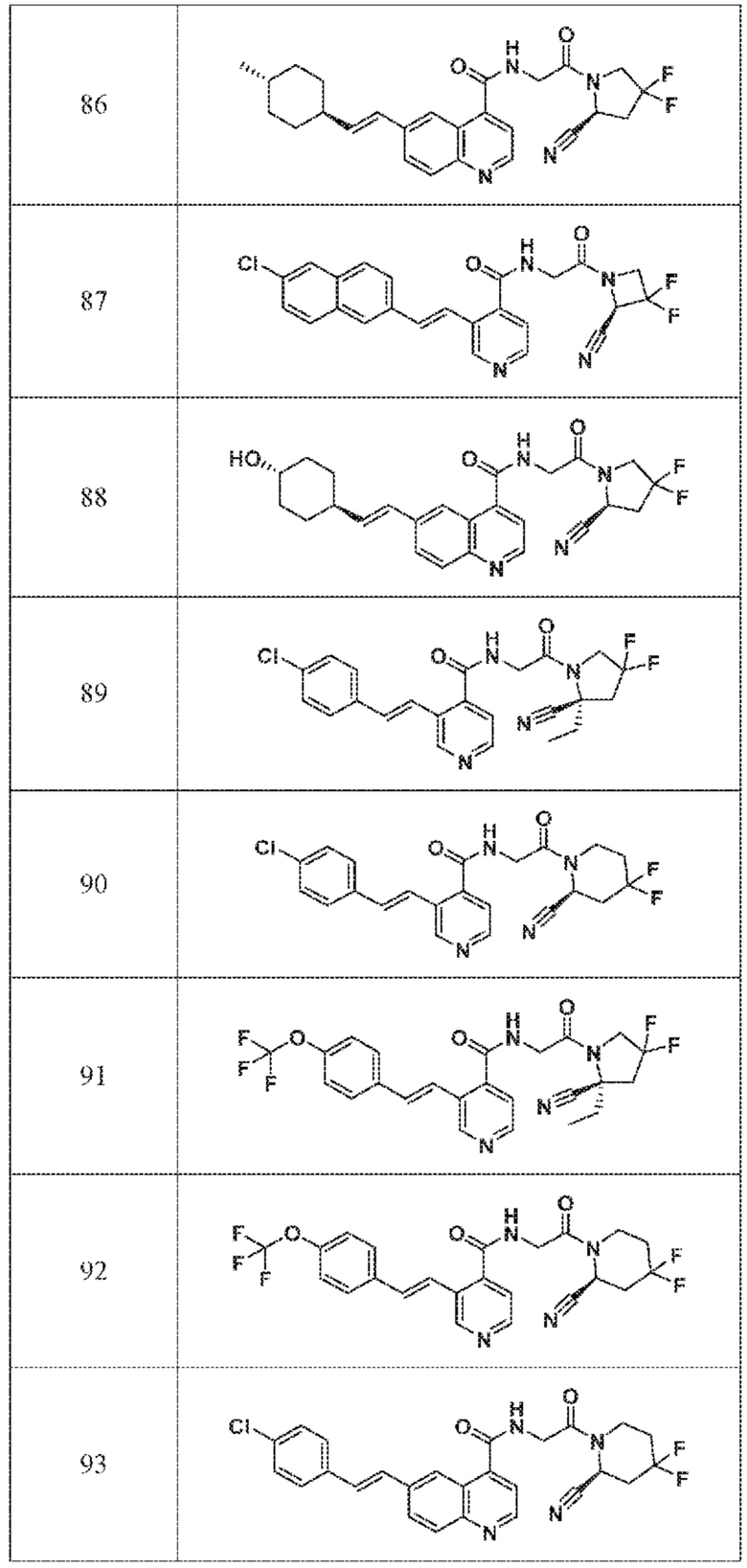
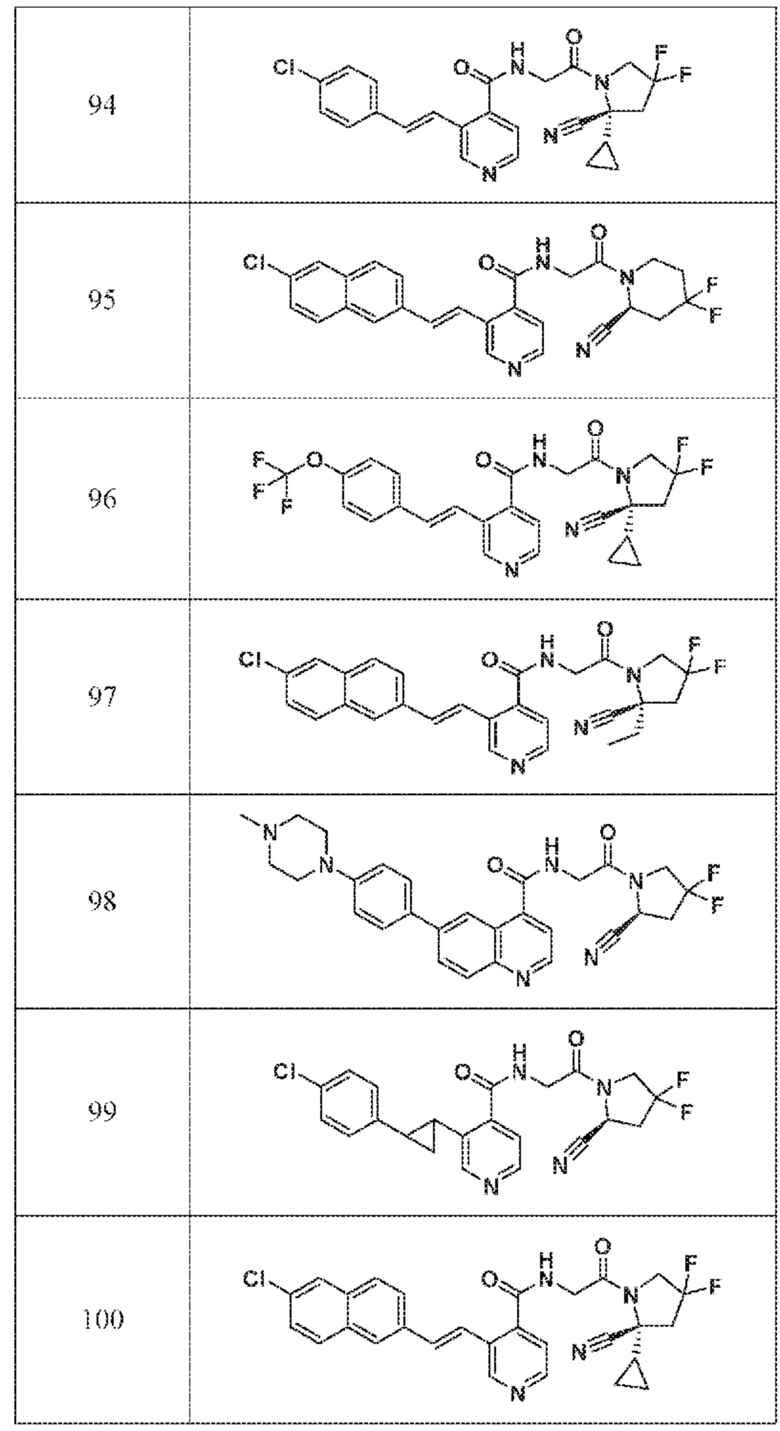



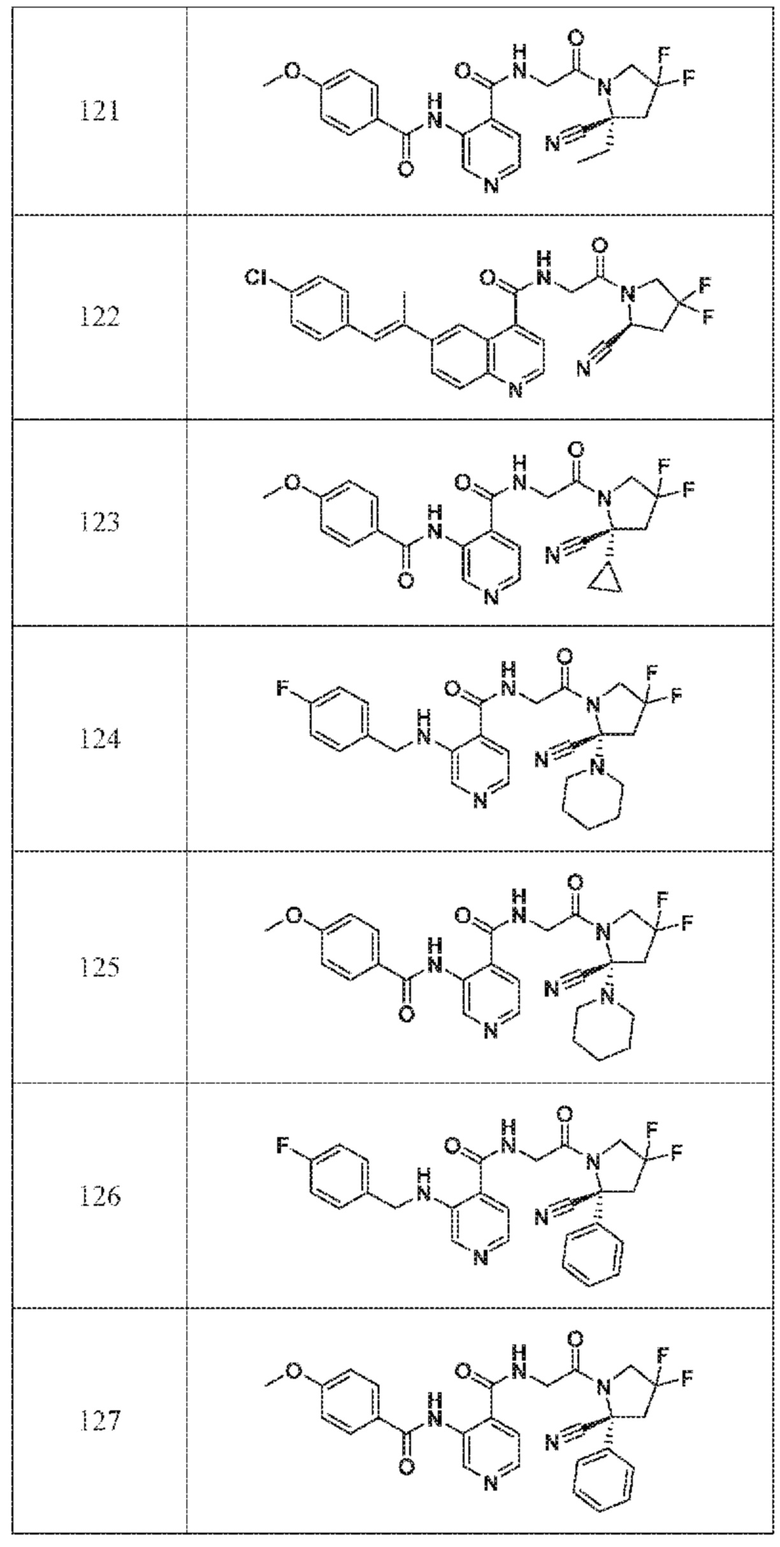




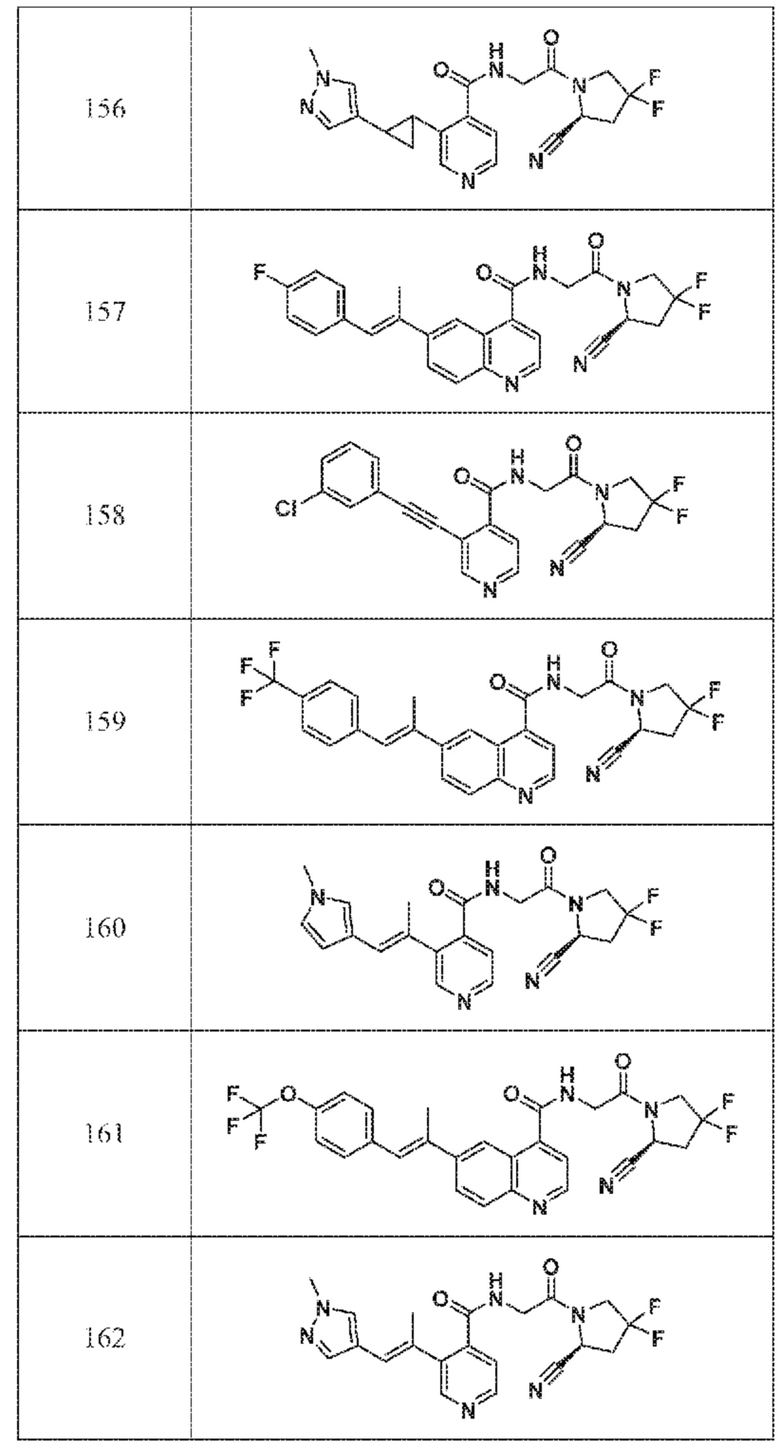



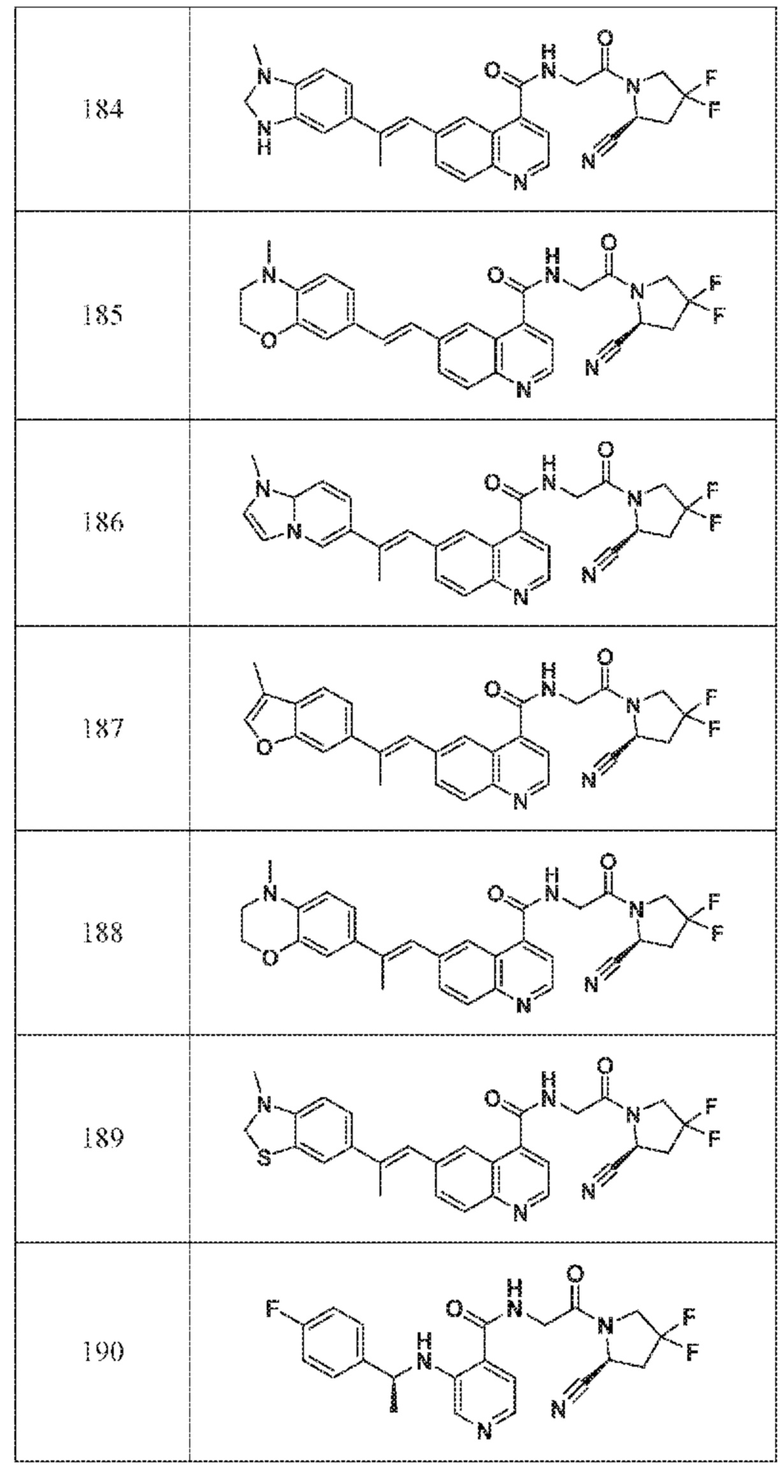



[0108] Некоторые соединения, приведенные в таблице 1, существуют в виде таутомеров. Независимо от того, какой таутомер указан, включены все таутомерные формы.
[0109] В некоторых вариантах осуществления настоящее изобретение относится к соединению, описанному в таблице 1, его таутомеру, или соли любого из указанных выше и к его применениям. В некоторых вариантах осуществления настоящее изобретение относится к соединению, описанному в таблице 1, или его фармацевтически приемлемой соли. В некоторых вариантах осуществления настоящее изобретение относится к соединению, описанному в таблице 1, или его таутомеру, или соли любого из указанных выше и к их применениям. В некоторых вариантах осуществления настоящее изобретение относится к соединению, описанному в таблице 1, или его фармацевтически приемлемой соли .
[0110] В некоторых вариантах осуществления предложено соединение, выбранное из числа соединений №№ 1-208 или их стереоизомера (включая смесь двух или большего количества их стереоизомеров), или его соли. В некоторых вариантах осуществления соединением является соль соединения, выбранного из числа соединений №№ 1-208, или его стереоизомер.
[0111] В одном варианте соединение, подробно описанное в настоящем изобретении, выбрано из группы, состоящей из следующих:
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-фенилацетамидо)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((4-фторбензил)амино)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-метоксибензамидо)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((1-(4-фторфенил)этил)амино)изоникотинамид;
3-(2-(4-хлор-3-фторфенокси)ацетамидо)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
2-(4-(бис(4-фторфенил)метил)пиперазин-1-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-фторбензил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-фторбензил)изоникотинамид;
3-(4-хлорстирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
3-(4-хлорфенетил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-фторфенетил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-фторстирил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-фторфенил)проп-1-ен-1-ил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-фторфенил)аллил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-метоксистирил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-(трифторметокси)фенетил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-(трифторметил)фенетил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-фторфенил)пропил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-(трифторметокси)фенил)проп-1-ен-1-ил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-(трифторметил)фенил)проп-1-ен-1-ил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-метоксифенил)аллил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-(трифторметокси)фенил)пропил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-(трифторметил)фенил)пропил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-(трифторметил)стирил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-метоксифенетил)изоникотинамид;
3-(2-(4-хлорфенил)проп-1-ен-1-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
3-(2-(4-хлорфенил)аллил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
3-(2-(4-хлорфенил)пропил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-метоксифенил)проп-1-ен-1-ил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-(трифторметокси)фенил)аллил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-(трифторметил)фенил)аллил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-метоксифенил)пропил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-(трифторметокси)стирил)изоникотинамид;
6-(4-хлорстирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(4-(трифторметил)фенил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((4-метоксифенил)этинил)изоникотинамид;
3-((4-хлорфенил)этинил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(6-(трифторметил)пиридин-3-ил)хинолин-4-карбоксамид;
6-(2-(4-хлорфенил)проп-1-ен-1-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид;
3-(2-(6-хлорнафталин-2-ил)винил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-((4-метоксифенил)этинил)хинолин-4-карбоксамид;
6-(2-(6-хлорнафталин-2-ил)винил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-метоксипиридин-3-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(4-фторстирил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-метилпиридин-3-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-(трифторметил)пиридин-3-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(4-метоксистирил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(4-(трифторметокси)стирил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(5-метилпиридин-2-ил)винил)хинолин-4-карбоксамид;
6-(4-(трет-бутил)стирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(4-(трифторметил)стирил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(6-(4-метилпиперазин-1-ил)пиридин-3-ил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(1-метил-1H-индол-3-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(5-фторбензофуран-2-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(1-метил-1H-индазол-3-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(6-фторбензофуран-2-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(1-метилпиперидин-4-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(5-метоксибензофуран-2-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-фторциклогексил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(5-метил-2,3-дигидробензо[d]тиазол-2-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4,4-дифторциклогексил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(6-метил-2,3-дигидробензо[d]тиазол-2-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-метил-3,4-дигидро-2H-бензо[b][1,4]оксазин-2-ил)винил)изоникотинамид;
3-(2-(5-(трет-бутил)-2,3-дигидробензо[d]тиазол-2-ил)винил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
3-(2-(6-хлор-3,4-дигидро-2H-бензо[b][1,4]оксазин-2-ил)винил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
3-(2-(7-хлоримидазо[1,2-a]пиридин-2-ил)винил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(6-фтор-3,4-дигидро-2H-бензо[b][1,4]оксазин-2-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(7-этоксиимидазо[1,2-a]пиридин-2-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-метил-3,4-дигидро-2H-бензо[b][1,4]оксазин-7-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(6-фторимидазо[1,2-a]пиридин-2-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(1-метил-1H-бензо[d]имидазол-5-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(1-метилпиперидин-4-ил)винил)хинолин-4-карбоксамид;
3-(2-(6-хлор-1H-бензо[d]имидазол-2-ил)винил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(4-метилпиперазин-1-ил)винил)хинолин-4-карбоксамид;
3-(2-(5-хлор-1H-бензо[d]имидазол-2-ил)винил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
3-(4-хлорстирил)-N-(2-(2-циано-5,5-дифторпиперидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(1-метил-1H-бензо[d]имидазол-2-ил)винил)изоникотинамид;
N-(2-(2-циано-5,5-дифторпиперидин-1-ил)-2-оксоэтил)-3-(4-(трифторметокси)стирил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)винил)изоникотинамид;
6-(4-хлорстирил)-N-(2-(2-циано-5,5-дифторпиперидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(1-метил-1,2,3,4-тетрагидрохинолин-6-ил)винил)изоникотинамид;
3-(2-(6-хлорнафталин-2-ил)винил)-N-(2-(2-циано-5,5-дифторпиперидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-гидроксициклогексил)винил)изоникотинамид;
N-(2-(2-циано-3,3-дифторазетидин-1-ил)-2-оксоэтил)-3-(4-(трифторметокси)стирил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(6-метилпиперидин-3-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(4-метилциклогексил)винил)хинолин-4-карбоксамид;
3-(2-(6-хлорнафталин-2-ил)винил)-N-(2-(2-циано-3,3-дифторазетидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(4-гидроксициклогексил)винил)хинолин-4-карбоксамид;
3-(4-хлорстирил)-N-(2-(2-циано-2-этил-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
3-(4-хлорстирил)-N-(2-(2-циано-4,4-дифторпиперидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-2-этил-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-(трифторметокси)стирил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпиперидин-1-ил)-2-оксоэтил)-3-(4-(трифторметокси)стирил)изоникотинамид;
6-(4-хлорстирил)-N-(2-(2-циано-4,4-дифторпиперидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид;
3-(4-хлорстирил)-N-(2-(2-циано-2-циклопропил-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
3-(2-(6-хлорнафталин-2-ил)винил)-N-(2-(2-циано-4,4-дифторпиперидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-2-циклопропил-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-(трифторметокси)стирил)изоникотинамид;
3-(2-(6-хлорнафталин-2-ил)винил)-N-(2-(2-циано-2-этил-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(4-(4-метилпиперазин-1-ил)фенил)хинолин-4-карбоксамид;
3-(2-(4-хлорфенил)циклопропил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
3-(2-(6-хлорнафталин-2-ил)винил)-N-(2-(2-циано-2-циклопропил-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
3-(2-(4-хлорфенил)циклобут-1-ен-1-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
3-(4-хлорстирил)-N-(2-(2-циано-4,4-дифтор-2-(пиперидин-1-ил)пирролидин-1-ил)-2-оксоэтил)изоникотинамид;
3-(2-(4-хлорфенил)циклопент-1-ен-1-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифтор-2-(пиперидин-1-ил)пирролидин-1-ил)-2-оксоэтил)-3-(4-(трифторметокси)стирил)изоникотинамид;
N-(4-((2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)карбамоил)пиридин-3-ил)-5-(трифторметокси)-2,3-дигидробензо[d]тиазол-2-карбоксамид;
3-(2-(6-хлорнафталин-2-ил)винил)-N-(2-(2-циано-4,4-дифтор-2-(пиперидин-1-ил)пирролидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(4-((2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)карбамоил)пиридин-3-ил)-7-этоксиимидазо[1,2-a]пиридин-2-карбоксамид;
3-(4-хлорстирил)-N-(2-(2-циано-4,4-дифтор-2-фенилпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
6-(2-(4-хлорфенил)циклопропил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифтор-2-фенилпирролидин-1-ил)-2-оксоэтил)-3-(4-(трифторметокси)стирил)изоникотинамид;
N-(2-(2-циано-5,5-дифторпиперидин-1-ил)-2-оксоэтил)-3-(4-метоксибензамидо)изоникотинамид;
N-(2-(2-циано-4,4-дифторпиперидин-1-ил)-2-оксоэтил)-3-((4-фторбензил)амино)изоникотинамид;
3-(2-(6-хлорнафталин-2-ил)винил)-N-(2-(2-циано-4,4-дифтор-2-фенилпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпиперидин-1-ил)-2-оксоэтил)-3-(4-метоксибензамидо)изоникотинамид;
3-((4-(трет-бутил)фенил)этинил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-3,3-дифторазетидин-1-ил)-2-оксоэтил)-3-((4-фторбензил)амино)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((1-метил-1H-пиррол-3-ил)этинил)изоникотинамид;
N-(2-(2-циано-3,3-дифторазетидин-1-ил)-2-оксоэтил)-3-(4-метоксибензамидо)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((1-метил-1H-пиразол-4-ил)этинил)изоникотинамид;
N-(2-(2-циано-2-этил-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((4-фторбензил)амино)изоникотинамид;
N-(2-(2-циано-2-этил-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-метоксибензамидо)изоникотинамид;
6-(1-(4-хлорфенил)проп-1-ен-2-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид;
N-(2-(2-циано-2-циклопропил-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-метоксибензамидо)изоникотинамид;
N-(2-(2-циано-4,4-дифтор-2-(пиперидин-1-ил)пирролидин-1-ил)-2-оксоэтил)-3-((4-фторбензил)амино)изоникотинамид;
N-(2-(2-циано-4,4-дифтор-2-(пиперидин-1-ил)пирролидин-1-ил)-2-оксоэтил)-3-(4-метоксибензамидо)изоникотинамид;
N-(2-(2-циано-4,4-дифтор-2-фенилпирролидин-1-ил)-2-оксоэтил)-3-((4-фторбензил)амино)изоникотинамид;
N-(2-(2-циано-4,4-дифтор-2-фенилпирролидин-1-ил)-2-оксоэтил)-3-(4-метоксибензамидо)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(4-фторфенил)проп-1-ен-1-ил)хинолин-4-карбоксамид;
3-((1-(4-хлор-3-фторфенил)этил)амино)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-(трифторметил)фенокси)ацетамидо)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-метоксифенокси)ацетамидо)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(4-(трифторметил)фенил)проп-1-ен-1-ил)хинолин-4-карбоксамид;
3-(1-(4-хлорфенил)проп-1-ен-2-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
3-(1-(4-хлор-2-метилфенил)проп-1-ен-2-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(4-(трифторметокси)фенил)проп-1-ен-1-ил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(1-метил-1H-пиразол-4-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(1-метил-1H-пиррол-3-ил)проп-1-ен-1-ил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((4-фторфенил)этинил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-гидроксипиридин-3-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(5-метоксибензофуран-2-карбоксамидо)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(4-метоксифенил)проп-1-ен-1-ил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(5-(трифторметокси)-2,3-дигидробензо[d]тиазол-2-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-метилциклогексил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-метоксипиридин-3-ил)проп-1-ен-1-ил)хинолин-4-карбоксамид;
6-(2-(4-(трет-бутил)фенил)проп-1-ен-1-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид;
N-(2-(2-циано-5,5-дифторпиперидин-1-ил)-2-оксоэтил)-3-((4-фторбензил)амино)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-метилпиридин-3-ил)проп-1-ен-1-ил)хинолин-4-карбоксамид;
3-(4-хлорстирил)-N-(2-(2-циано-3,3-дифторазетидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-(трифторметил)пиридин-3-ил)проп-1-ен-1-ил)хинолин-4-карбоксамид;
N-(2-(2-циано-2-циклопропил-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((4-фторбензил)амино)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-гидроксипиридин-3-ил)проп-1-ен-1-ил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(1-метил-1H-пиррол-3-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(5-метилпиридин-2-ил)проп-1-ен-1-ил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(1-метил-1H-пиразол-4-ил)проп-1-ен-1-ил)изоникотинамид;
6-(2-(6-хлорнафталин-2-ил)проп-1-ен-1-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(1-метил-1H-пиразол-4-ил)циклопропил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(1-(4-фторфенил)проп-1-ен-2-ил)хинолин-4-карбоксамид;
3-((3-хлорфенил)этинил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(1-(4-(трифторметил)фенил)проп-1-ен-2-ил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(1-(1-метил-1H-пиррол-3-ил)проп-1-ен-2-ил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(1-(4-(трифторметокси)фенил)проп-1-ен-2-ил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(1-(1-метил-1H-пиразол-4-ил)проп-1-ен-2-ил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(1-(6-метоксипиридин-3-ил)проп-1-ен-2-ил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(1-метил-1H-пиррол-3-ил)циклопропил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(1-(4-метоксифенил)проп-1-ен-2-ил)хинолин-4-карбоксамид;
3-((3-(трет-бутил)фенил)этинил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
6-(1-(4-(трет-бутил)фенил)проп-1-ен-2-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(3,5-диметилизоксазол-4-ил)винил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(1-(6-метилпиридин-3-ил)проп-1-ен-2-ил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(3,5-диметилизоксазол-4-ил)проп-1-ен-1-ил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(1-(6-(трифторметил)пиридин-3-ил)проп-1-ен-2-ил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(1-(3,5-диметилизоксазол-4-ил)проп-1-ен-2-ил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(1-(6-гидроксипиридин-3-ил)проп-1-ен-2-ил)хинолин-4-карбоксамид;
3-((4-хлор-2-метилфенил)этинил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(1-(5-метилпиридин-2-ил)проп-1-ен-2-ил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((4-(трифторметил)фенил)этинил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(3-метилбензофуран-6-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((2,4-диметоксифенил)этинил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(3-метил-2,3-дигидробензо[d]тиазол-6-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((6-метоксипиридин-3-ил)этинил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(1-метил-2,3-дигидро-1H-бензо[d]имидазол-5-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((4-(трифторметокси)фенил)этинил)изоникотинамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(1-метил-1,8a-дигидроимидазо[1,2-a]пиридин-6-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(1-метил-2,3-дигидро-1H-бензо[d]имидазол-5-ил)проп-1-ен-1-ил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(4-метил-3,4-дигидро-2H-бензо[b][1,4]оксазин-7-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(1-метил-1,8a-дигидроимидазо[1,2-a]пиридин-6-ил)проп-1-ен-1-ил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(3-метилбензофуран-6-ил)проп-1-ен-1-ил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(4-метил-3,4-дигидро-2H-бензо[b][1,4]оксазин-7-ил)проп-1-ен-1-ил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(3-метил-2,3-дигидробензо[d]тиазол-6-ил)проп-1-ен-1-ил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(2-метилпиридин-4-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(2-метоксипиридин-4-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(2-(трифторметил)пиридин-4-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(4-(2-гидроксипропан-2-ил)фенил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(1-оксо-1,2,3,4-тетрагидроизохинолин-6-ил)винил)изоникотинамид;
6-(2-(2-аминопиридин-4-ил)винил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид;
6-(2-(6-аминопиридин-3-ил)винил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-оксо-5,6-дигидропиридин-3-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-((4-метилпиперазин-1-ил)метил)пиридин-3-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-(пиперазин-1-илметил)пиридин-3-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-(морфолинометил)пиридин-3-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-((1-метилпиперидин-4-ил)метил)пиридин-3-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-(пиперидин-4-илметил)пиридин-3-ил)винил)хинолин-4-карбоксамид;
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-((1-метилпиперидин-4-ил)амино)пиридин-3-ил)винил)хинолин-4-карбоксамид; и
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-((1-метилпиперидин-4-ил)окси)пиридин-3-ил)винил)хинолин-4-карбоксамид,
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(2-гидроксипиридин-4-ил)винил)хинолин-4-карбоксамид,
N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(1-оксоизоиндолин-5-ил)винил)изоникотинамид,
или его фармацевтически приемлемая соль. Кроме того, настоящее изобретение относится, если это применимо, к любому и всем стереоизомерам соединений, приведенным в настоящем изобретении, включая геометрические изомеры (например, цис/транс изомеры или E/Z изомеры), энантиомеры, диастереоизомеры или их смеси в любом отношении, включая рацемические смеси.
[0112] Соединения, приведенные в настоящем изобретении, могут содержаться, как соли, даже если соли не указаны, и следует понимать, что настоящее изобретение включает все соли и сольваты соединений, приведенных в настоящем изобретении, а также несолевые и несольватированные формы соединения, как должен понимать специалист в данной области техники. В некоторых вариантах осуществления также предложены и описаны соли соединений настоящее изобретение относится к фармацевтически приемлемым солям. Если в соединении содержится один или большее количество фрагментов третичных аминов, также предложены и описаны N-оксиды.
[0113] Если для любых соединений, описанных в настоящем изобретении, могут содержаться таутомерные формы, то включены каждая и все таутомерные формы, даже если одна или некоторые таутомерные формы явно не указаны. Специально указанные таутомерные формы могут быть или не быть преобладающими формами в растворах или при использовании способами, описанными в настоящем изобретении.
[0114] Настоящее изобретение также включает любую или все стереохимические формы, включая любые энантиомерные или диастереоизомерные формы описанных соединений, таких как соединения таблицы 1. Указанная структура или название включает все возможные стереоизомеры приведенного соединения. Все формы соединений также включены в объем настоящего изобретения, такие как кристаллические или некристаллические формы соединений. В объем настоящего изобретения также включены композиции, содержащие соединение, предлагаемое в настоящем изобретении, такие как композиция в основном чистого соединения, включая его конкретную стереохимическую форму, или композиция, содержащая смеси соединений, предлагаемых в настоящем изобретении, в любом отношении, включая две или большее количество стереохимических форм, такие как рацемическая или нерацемическая смесь.
[0115] Настоящее изобретение также включает изотопно меченые и/или изотопно обогащенные формы соединений, описанных в настоящем изобретении. Соединения, описанные в настоящем изобретении, могут содержать находящиеся в не встречающемся в природе отношении изотопы одного или большего количества атомов, которые образуют такие соединения. В некоторых вариантах осуществления соединение является изотопно меченым, такое как изотопно меченое соединение формулы (I) или его варианты, описанные в настоящем изобретении, где часть одного или большего количества атомов заменена на изотоп того же элемента. Типичные изотопы, которые можно включить в соединения, предлагаемые в настоящем изобретении, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, хлора, такие как 2H, 3H, 11C, 13C, 14C 13N, 15O, 17O, 32P, 35S, 18F, 36Cl. Некоторые изотопно меченые соединения (например, с помощью 3H и 14C) применимы для исследования распределения соединения или субстрата в ткани. Включение более тяжелых изотопов, таких как дейтерий (2H), может обеспечить некоторые терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличенным временем полувыведения или возможностью применения меньших доз и, следовательно в некоторых случаях может быть предпочтительным.
[0116] Изотопно меченые соединения, предлагаемые в настоящем изобретении, обычно можно получить по стандартным методикам, известным специалистам в данной области техники, или по методикам, аналогичным описанным в прилагаемых примерах, с использованием подходящих изотопно меченых реагентов вместо соответствующих немеченых реагентов.
[0117] Предложены изделия, содержащие соединение, описанное в настоящем изобретении, или его соль или сольват в подходящем контейнере. Контейнером может быть сосуд, склянка, ампула, предварительно заполненный шприц, мешок для внутривенного введения и т. п.
[0118] Предпочтительно, если соединения, подробно описанные в настоящем изобретении, перорально биодоступны. Однако соединения также можно приготовить для парентерального (например, внутривенного) введения.
[0119] Одно или несколько соединений, описанных в настоящем изобретении, можно использовать для приготовления лекарственного средства путем объединения соединения или соединений в качестве активного ингредиента с фармакологически приемлемым носителем, который известен в данной области техники. В зависимости от терапевтической формы лекарственного средства носитель может находиться в разных формах. В одном варианте приготовление лекарственного средства предназначено для применения в любом из способов, раскрытых в настоящем изобретении, например, для лечения рака.
Общие методики синтеза
[0120] Соединения, предлагаемые в настоящем изобретении, можно получить по многим методикам, в целом описанным ниже, и, точнее, описанным в примерах ниже в настоящем изобретении (таким как схемы, приведенные ниже в примерах). В последующих описаниях процедур символы, использующиеся в приведенных формулах, описывают группы, указанные выше для формул в настоящем изобретении.
[0121] Если желательно получить конкретный энантиомер соединения, его можно получить из соответствующей смеси энантиомеров по любой обычной подходящей методике выделения или разделения энантиомеров. Таким образом, например, диастереоизомерные производные можно получить по реакции смеси энантиомеров, например, рацемата и подходящего хирального соединения. Затем диастереоизомеры можно разделить обычными средствами, например, путем кристаллизации и извлечения желательного энантиомера. В другой процедуре разделения рацемат можно разделить с использованием хиральной высокоэффективной жидкостной хроматографии. Альтернативно, при желании конкретный энантиомер можно получить путем использования соответствующего хирального промежуточного продукта в одной из описанных процедур.
[0122] Хроматографию, перекристаллизацию и другие обычные процедуры разделения при желании также можно использовать для промежуточных или конечных продуктов и получить конкретный изомер соединения или иным образом очистить продукт реакции.
[0123] Также включены сольваты соединения, предлагаемого в настоящем изобретении, его фармацевтически приемлемой соли, стереоизомера или таутомера. Сольваты содержат стехиометрическое или нестехиометрическое количество растворителя и часто образуются во время кристаллизации. Гидраты образуются, когда растворителем является вода, или алкоголяты когда растворителем является спирт.
[0124] Соединения формулы (I-1) можно получить в по схеме 1, где R, R1, R2, Y, m, n и q являются такими, как подробно описано в настоящем изобретении для формулы (I) или любого ее варианта, подробно описанного в настоящем изобретении; Z и Z1 означают отщепляющиеся группы; и PG1 означает защитную группу аминогруппы.
Схема 1
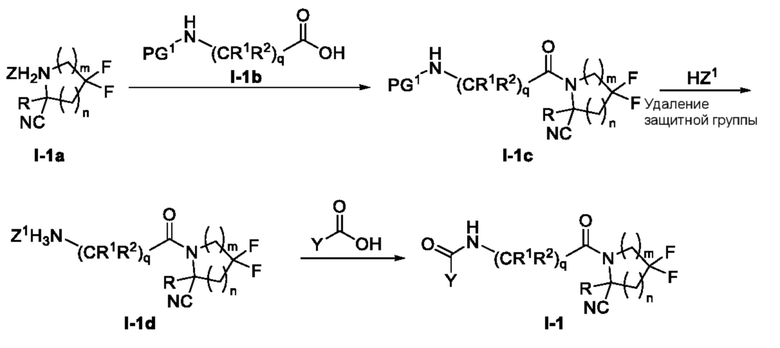
[0125] Сочетание соединения формулы (I-1a) с соединением формулы (I-1b) в присутствии реагента сочетания (например, HATU, HOBt, или PyBOP) дает соединение формулы (I-1c). Удаление защитной группы аминогруппы соединения формулы (I-1c) в кислой среде (например, HCl или pTsOH) дает соединение формулы (I-1d) в виде соли, которую вводят в сочетание с карбоновой кислотой в присутствии реагента сочетания (например, HATU, HOBt, или PyBOP) и получают соединение формулы (1-1).
[0126] Типичный вариант осуществления методики получения, приведенной на схеме 1, представлен на схеме 1a.
Схема 1a
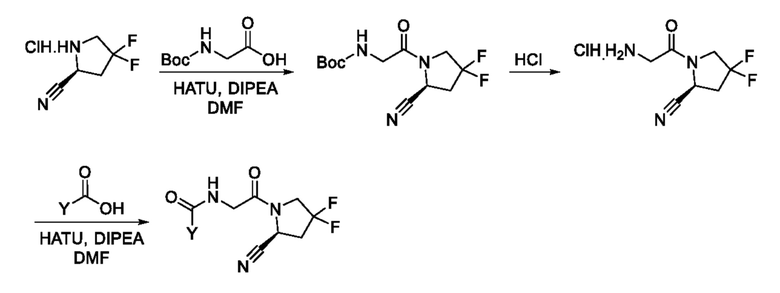
[0127] В некоторых вариантах осуществления Y означает 6- 10-членный гетероарил, замещенный посредством R12. В другом варианте осуществления Y означает пиридин-4-ил, замещенный посредством R12 в положении 3, где  представлен соединением формулы (II-1).
представлен соединением формулы (II-1).
Схема 2

[0128] Следует понимать, что приведенные выше схемы можно модифицировать и получить разные соединения, предлагаемые в настоящем изобретении, путем выбора подходящих реагентов и исходных веществ. Общее описание защитных групп и их применения см. P.G.M. Wuts and T.W. Greene, Greene's Protective Groups in Organic Synthesis 4th edition, Wiley-Interscience, New York, 2006.
Фармацевтические композиции и препараты
[0129] Фармацевтические композиции любых соединений, подробно описанных в настоящем изобретении, входят в объем настоящего изобретения. Таким образом, настоящее изобретение включает фармацевтические композиции, содержащие соединение, подробно описанное в настоящем изобретении, или его фармацевтически приемлемую соль, стереоизомер или таутомер и фармацевтически приемлемый носитель или инертный наполнитель. В одном объекте фармацевтически приемлемой солью является соль присоединения с кислотой, такая как соль, образованная с неорганической или органической кислотой. Фармацевтические композиции могут находиться в форме, применимой для перорального, буккального, парентерального, назального, местного или ректального введения, или в форме, применимой для введения с помощью ингаляции.
[0130] Соединение, подробно описанное в настоящем изобретении, в одном объекте может быть в очищенной форме и композиции, содержащие соединение в очищенных формах, подробно описаны в настоящем изобретении. Предложены композиции, содержащие соединение, подробно описанное в настоящем изобретении, или его фармацевтически приемлемую соль, стереоизомер или таутомер, такие как композиции в основном чистых соединений. В некоторых вариантах осуществления композиция, содержащая соединение, подробно описанное в настоящем изобретении, его фармацевтически приемлемую соль, стереоизомер или таутомер находится в основном в чистой форме.
[0131] В одном варианте соединения в настоящем изобретении являются синтетическими соединениями, приготовленными для введения индивидууму. В другом варианте предложены композиции, содержащие соединение в основном в чистой форме. В другом варианте настоящее изобретение включает фармацевтические композиции, содержащие соединение, подробно описанное в настоящем изобретении, и фармацевтически приемлемый носитель. В другом варианте предложены способы введения соединения. Очищенные формы, фармацевтические композиции и способы введения соединений являются подходящими для любых соединения или их формы, подробно описанной в настоящем изобретении.
[0132] Соединение, подробно описанное в настоящем изобретении, или его соль можно приготовить для любого доступного пути доставки, включая пероральную, мукозальную (например, назальную, сублингвальную, вагинальную, буккальнм или ректальную), парентеральную (например, внутримышечную, подкожную или внутривенную), местную или чрескожную форму доставки. Соединение или его соль можно приготовить с подходящими носителями и получить формы доставки, которые включают, но не ограничиваются только ими, таблетки, каплеты, капсулы (такие как капсулы из твердого желатина или капсулы из мягкого эластичного желатина), облатки, пастилки, лепешки, камеди, дисперсии, суппозитории, мази, припарки (примочки), пасты, порошки, повязки, кремы, растворы, пластыри, аэрозоли (например, назальный спрей или спрей для ингаляции), гели, суспензии (например, водные или неводные жидкие суспензии, жидкие эмульсии масло-в-воде или жидкие эмульсии вода-в-масле), растворы и эликсиры.
[0133] Одно или несколько соединений, описанных в настоящем изобретении их фармацевтически приемлемую соль, стереоизомер или таутомер можно использовать для приготовления препарата, такого как фармацевтический препарат, путем объединения соединения или соединений, их фармацевтически приемлемой соли, стереоизомера или таутомера в качестве активного ингредиента с фармацевтически приемлемым носителем, таким как указанные выше. В зависимости от терапевтической формы системы (например, чрескожный пластырь или пероральная таблетка) носитель может находиться в разных формах. Кроме того, фармацевтические препараты могут содержать консерванты, солюбилизаторы, стабилизаторы, повторно смачивающие агенты, эмульгаторы, подсластители, красители, регуляторы и соли для регулирования осмотического давления, буферы, агенты для нанесения покрытия или антиоксиданты. Препараты, содержащие соединение также могут содержать другие вещества, которые обладают ценными терапевтическими характеристиками. Фармацевтические препараты можно получить по известным фармацевтическим методикам. Подходящие препараты описаны, например, в публикации Remington’s Pharmaceutical Sciences, Mack Publishing Company, Philadelphia, PA, 20th ed. (2000), которая включена в настоящее изобретение в качестве ссылки.
[0134] Соединения, описанные в настоящем изобретении, можно вводить индивидуумам в виде обычно использующейся пероральной композиций, такой как таблетки, таблетки с покрытием и гелевые капсулы в твердой или мягкой оболочке, эмульсии или суспензии. Примерами носителей, которые можно использовать для приготовления таких композиций, являются лактоза, кукурузный крахмал или его производные, тальк, стеарат или их соли и т. п. Приемлемыми носителями для гелевых капсул с мягкой оболочкой являются, например, растительные масла, воск, жиры, полужидкие и жидкие полиолы и т. д. Кроме того, фармацевтические препараты могут содержать консерванты, солюбилизаторы, стабилизаторы, повторно смачивающие агенты, эмульгаторы, подсластители, красители, регуляторы и соли для регулирования осмотического давления, буферы, агенты для нанесения покрытия или антиоксиданты.
[0135] Также описаны композиции, содержащие соединение, предлагаемое в настоящем изобретении. В одном варианте композиция содержит соединение или его соль и фармацевтически приемлемый носитель или инертный наполнитель. В другом варианте предложена композиция в основном чистого соединения. В некоторых вариантах осуществления композиция предназначена для использования в качестве лекарственного средства в медицине или ветеринарии. В некоторых вариантах осуществления композиция предназначена для использования в способе, описанном в настоящем изобретении. В некоторых вариантах осуществления композиция предназначена для использования для лечения заболевания или нарушения, описанного в настоящем изобретении.
Способы применения и применения
[0136] Соединения и композиции, подробно описанные в настоящем изобретении, такие как фармацевтическая композиция, содержащая соединение любой формулы, приведенной в настоящем изобретении, его фармацевтически приемлемую соль, стереоизомер или таутомер и фармацевтически приемлемый носитель или инертный наполнитель можно использовать в способах введения и лечения, предлагаемых в настоящем изобретении. Соединения и композиции также можно использовать в способах in vitro, таких как in vitro способы введения соединения или композиции в клетки для скрининга и/или для проведения анализов по контролю качества.
[0137] Настоящее изобретение относится к способу лечения заболевания или нарушения у нуждающегося в нем индивидуума, включающему введение соединения, описанного в настоящем изобретении или любого его варианта осуществления, варианта или аспекта, или его фармацевтически приемлемой соли. В некоторых вариантах осуществления соединение, его фармацевтически приемлемую соль или композицию вводят индивидууму в дозе и/или способом введения, описанным в настоящем изобретении.
[0138] Соединения или их соли, описанные в настоящем изобретении, и композиции, описанные в настоящем изобретении, предположительно эффективны для лечения разных заболеваний и нарушений. В некоторых вариантах осуществления соединение или его соль, описанную в настоящем изобретении, или композицию, описанную в настоящем изобретении, можно использовать в способе лечения заболевания или нарушения, опосредуемого белком активации фибробластов (FAP). В некоторых вариантах осуществления заболевание или нарушение характеризуется пролиферацией, ремоделированием ткани, фиброзом, хроническим воспалением, избыточным потреблением алкоголя или аномальным метаболизмом.
[0139] В некоторых вариантах осуществления соединение или его соль, описанную в настоящем изобретении, или композицию, описанную в настоящем изобретении, можно использовать в способе лечения заболевания или нарушения, опосредуемого физиологическим субстратом пептидазной активности FAP. В некоторых вариантах осуществления пептидазная активность FAP является эндопептидазной активностью. В некоторых вариантах осуществления физиологическим субстратом эндопептидазной активности FAP является α2-антиплазмин, коллаген типа I, желатин и фактор роста фибробластов 21 (FGF21). В некоторых вариантах осуществления пептидазная активность FAP является экзопептидазной активностью. В некоторых вариантах осуществления физиологическим субстратом экзопептидазной активности FAP является нейропептид Y, натрийуретический пептид типа B, вещество P и пептид YY. В некоторых вариантах осуществления соединение или его соль, описанную в настоящем изобретении, или композицию, описанную в настоящем изобретении, можно использовать в способе лечения заболевания или нарушения, опосредуемого с помощью FGF21.
[0140] В некоторых вариантах осуществления соединение или его соль, описанную в настоящем изобретении, или композицию, описанную в настоящем изобретении, можно использовать в способе лечения связанного с FGF21 нарушения, такого как ожирение, диабет типа I и типа II, панкреатит, дислипидемия, гиперлипидемические патологические состояния, неалкогольная жировая инфильтрация печени (NAFLD), неалкогольный стеатогепатит (NASH), резистентность к инсулину, гиперинсулинемия, непереносимость глюкозы, гипергликемия, метаболический синдром, острый инфаркт миокарда, гипертензия, сердечно-сосудистые заболевания, атеросклероз, заболевание периферической артерии, апоплексия, сердечная недостаточность, заболевание коронарной артерии сердца, заболевание почек, осложнения при диабете, невропатия, гастропарез, нарушение, связанное с сильной инактивирующей мутацией инсулинового рецептора, и другие метаболические нарушения. В некоторых вариантах осуществления связанным с FGF21 нарушением является диабет, ожирение, дислипидемия, метаболический синдром, неалкогольная жировая инфильтрация печени, неалкогольный стеатогепатит или сердечно-сосудистые заболевания.
[0141] В некоторых вариантах осуществления соединение или его соль, описанную в настоящем изобретении, или композицию, описанную в настоящем изобретении, можно использовать в способе лечения заболевания или нарушения, характеризующегося пролиферацией, ремоделированием ткани, фиброзом, хроническим воспалением, избыточным потреблением алкоголя или аномальным метаболизмом.
[0142] В некоторых вариантах осуществления соединение или его соль, описанную в настоящем изобретении, или композицию, описанную в настоящем изобретении, можно использовать в способе лечения рака, такого как рак молочной железы, колоректальный рак, рак яичников, рак предстательной железы, рак поджелудочной железы, рак почки, рак легких, меланома, фибросаркома, саркома кости, саркома соединительной ткани, почечноклеточная карцинома, гигантоклеточная карцинома, плоскоклеточная карцинома, лейкоз, рак кожи, рак мягких тканей, рак печени, желудочно-кишечная карцинома или аденокарцинома. В некоторых вариантах осуществления соединение, соль, или композицию можно использовать в способе лечения метастатический рак почки, хронический лимфоцитарный лейкоз, аденокарцинома поджелудочной железы или немелкоклеточный рак легких.
[0143] В некоторых вариантах осуществления введение соединения, соли или композиции уменьшает рост опухоли, пролиферацию опухоли, или онкогенности у индивидуума. В некоторых вариантах осуществления соединение, соль, или композицию можно использовать в способе уменьшения роста опухоли, пролиферации опухоли или онкогенности у нуждающегося в нем индивидуума. В некоторых вариантах осуществления рост опухоли замедляется или останавливается. В некоторых вариантах осуществления рост опухоли уменьшается по меньшей мере примерно на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% или более. В некоторых вариантах осуществления уменьшается размер опухоли. В некоторых вариантах осуществления предупреждается или замедляется метастазирование опухоли. В некоторых вариантах осуществления рост опухоли, пролиферация опухоли или онкогенность сравнима с ростом опухоли, пролиферацией опухоли или онкогенностью у индивидуума до введения соединения, соли или композиции. В некоторых вариантах осуществления рост опухоли, пролиферация опухоли или онкогенность сравнима с ростом опухоли, пролиферацией опухоли или онкогенностью у аналогичного индивидуума или группы индивидуумов. Методики измерения роста опухоли, пролиферации опухоли и онкогенности известны в данной области техники, например, проводятся путем повторяющейся визуализации индивидуума.
[0144] В некоторых вариантах осуществления соединение или его соль, описанную в настоящем изобретении, или композицию, описанную в настоящем изобретении, можно использовать в способе лечения фиброзного заболевания, тромбоза, заживления раны, келоидного образования, остеоартрита, ревматоидного артрита и родственных нарушений, включая разрушение хряща, атеросклеротическое заболевание, болезнь Крона, цирроз печени, идиопатический фиброз легких, гипертрофию миокарда, диастолическую дисфункцию, ожирение, непереносимость глюкозы, нечувствительность к инсулину или сахарный диабет. В некоторых вариантах осуществления циррозом печени является вызванный вирусный гепатитом, вызванный алкоголем или билиарный цирроз. В некоторых вариантах осуществления сахарным диабетом является диабет типа II. В некоторых вариантах осуществления заболеванием или нарушением является фиброзная дегенерация печени.
[0145] В некоторых вариантах осуществления настоящее изобретение относится к способу ингибирования FAP. Соединения или их соли, описанные в настоящем изобретении, и композиции, описанные в настоящем изобретении, предположительно эффективны для ингибирования FAP.
[0146] В некоторых вариантах осуществления способ ингибирования FAP включает ингибирование FAP в клетке путем введения или доставки в клетку соединений, описанных в настоящем изобретении, или его фармацевтически приемлемой соли, или фармацевтической композиции, описанной в настоящем изобретении. В некоторых вариантах осуществления клеткой является фибробласт, такой как миофибробласт, келоидный фибробласт, связанный с раком фибробласт (CAF) или реакцинноспособный стромальный фибробласт, из числа других клеток, связанных с экспрессией FAP.
[0147] В некоторых вариантах осуществления способ ингибирования FAP включает ингибирование FAP в опухоли или плазме путем введения или доставки в опухоль или плазму соединений, описанных в настоящем изобретении, или его фармацевтически приемлемой соли, или фармацевтической композиции, описанной в настоящем изобретении.
[0148] В некоторых вариантах осуществления ингибирование FAP включает ингибирование эндопептидазной и/или экзопептидазной активности FAP. В некоторых вариантах осуществления FAP ингибируется по меньшей мере примерно на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 98% или более. Ингибирование FAP можно определить по методикам, известным в данной области техники.
[0149] В некоторых вариантах осуществления соединение, его соль или композиция ингибирует FAP с IC50, равной менее примерно 1 мкМ, например, менее примерно 750 нМ, 600 нМ, 500 нМ, 300 нМ, 200 нМ, 100 нМ, 80 нМ, 60 нМ, 40 нМ, 25 нМ или менее. В некоторых вариантах осуществления соединение, его соль или композиция ингибирует FAP с IC50, равной примерно от 7 нМ до 1 мкМ, например, примерно от 10 нМ до 600 нМ, от 15 нМ до 200 нМ или от 20 нМ до 180 нМ. В некоторых объектах половинная максимальная ингибирующая концентрация (IC50) является мерой эффективности вещества для ингибирования конкретной биологической или биохимической функции. В некоторых объектах IC50 является количественной мерой, которая показывает, сколько ингибитора необходимо для ингибирования в два раза данного биологического процесса или компонента процесса, такого как фермент, клетка, рецептор клетки или микроорганизма. Методики определения IC50 in vitro и in vivo известны в данной области техники.
[0150] В некоторых вариантах осуществления соединения или их соли описанные в настоящем изобретении, и композиции, описанные в настоящем изобретении, вводят в количестве, при котором активность DPPII, DPPIV, DPP8, DPP9 и/или PREP не ингибируется или ингибируется в меньшей степени. В некоторых вариантах осуществления ингибирование FAP по меньшей мере или по меньшей мере примерно в 2 раза сильнее, чем ингибирование активности DPPII, DPPIV, DPP8, DPP9 и/или PREP, например, сильнее по меньшей мере или по меньшей мере примерно в 3 раза, 4 раза, 5 раз, 8 раз, 10 раз, 15 раз, 30 раз, 50 м, 60 раз, 75 раз или 100 раз.
[0151] Например и если не ограничиваться теорией, то можно полагать, что DPP9 является цитоплазматической DPP и также относится к подсемейству S9B пролин-селективных растворимых протеаз. Ингибирование активности DPP9 в макрофагах активирует инфламмасому Nlrp1b. Активация этого пути приводит к пироптозу, провоспалительной форме гибели клеток (Okondo MC et al. 2017; Okondo MC et al. 2018), сопутствующей активации каспазы-1 и последующей активации pro-IL-1β и pro-IL-18. В сингенной модели на мышах MC38 показано, что Val-boroPro неселективный ингибитор DPP, ингибирует рост рака с сопутствующей повышающей регуляцией иммуностимулирующих цитокинов и инфильтрацией опухоли клетками противораковых типов, включая клетки CD8+ T, M1-макрофаги и клетки NK при объединении с иммунными контрольными точками анти-PD1. Цитоплазматический опухолевый антиген RU134-42 является природным субстратом для DPP9 и эндогенный DPP9 ограничивает представление пептида RU134-42. Эти данные свидетельствуют о роли DPP9 в представлении антигена (Geiss-Friedlander R et al, 2009). В миелоидных клетках человека CARD8 опосредует индуцируемый ингибитором DPP8/9 зависимый от прокаспазы-1β пироптоз. Ингибиторы DPP8/9 индуцируют пироптоз в большинстве линий клеток острого миелолейкоза (AML) человека и в образцах первичного AML, но не в клетках многих других линий и эти ингибиторы ингибируют прогрессирование AML человека в моделях на мышах. Val-boroPro приводил к составляющему 97% снижению опухолевой нагрузки по сравнению с контролем с введением разбавителя в модели диссеминированных лейкозных клеток MV4;11 на мышах NSG (Johnson et al., 2018).
[0152] Настоящее изобретение относится к способу усиления иммунного ответа у индивидуума, включающему введение индивидууму соединения, описанного в настоящем изобретении, или его фармацевтически приемлемой соли, или фармацевтической композиции, описанной в настоящем изобретении. В некоторых вариантах осуществления у индивидуума имеется рак. В некоторых вариантах осуществления усиленный иммунный ответ направлен на опухолевые или раковые клетки. Например и если не ограничиваться теорией, то можно полагать, что FAP подавляет иммунные ответы, в особенности в контексте рака, поэтому ингибирование FAP может усилить иммунный ответ индивидуума. Соответственно, настоящее изобретение относится к способам лечения рака у нуждающегося в нем индивидуума, включающим введение индивидууму соединения, описанного в настоящем изобретении, или его фармацевтически приемлемой соли, или фармацевтической композиции, описанной в настоящем изобретении, где иммунный ответ индивидуума усиливается.
[0153] Настоящее изобретение относится к способу усиления степени экспрессии FGF21 у индивидуума, включающему введение индивидууму соединения, описанного в настоящем изобретении, или его фармацевтически приемлемой соли, или фармацевтической композиции, описанной в настоящем изобретении. Настоящее изобретение также относится к способу увеличения содержания FGF21 или аналога FGF21 у индивидуума, включающему введение индивидууму соединения, описанного в настоящем изобретении, или его фармацевтически приемлемой соли, или фармацевтической композиции, описанной в настоящем изобретении. В некоторых вариантах осуществления способ дополнительно включает введение FGF21 или аналога FGF21, такого как мутировавший FGF21, пэгилированный FGF21, PF-05231023 или LY2405319.
[0154] FGF21 является пептидным эндокринным гормоном, секретируемым преимущественно печенью (Markan, K.R. et al. Semin Cell Dev Biol, 2016, 53: 85-93). При попадании в кровоток FGF21 действует путем передачи сигналов в специфические ткани, регулирующие метаболизм углеводов и липидов (Kharitonenkov, A., et al., J Clin Invest, 2005, 115(6): 1627-35). FGF21 стимулирует потребление глюкозы адипоцитами и предположительно защищает от ожирения и нечувствительности к инсулину. Фармакологическое введение FGF21 в моделях диабета и ожирения на животных значительно уменьшает ожирение, резистентность к инсулину, дислипидемию, жировую инфильтрацию печени и гипергликемию у грызунов (Markan, K.R. et al. Semin Cell Dev Biol, 2016, 53: 85-93). Аналогичные клинические исследования показали, что аналоги FGF21 эффективны для индуцирования потери массы и коррекции гиперинсулинемии, дислипидемии и гипоадипонектинемии у индивидуумов с ожирением с диабетом типа 2 (Gaich, G., et al., Cell Metab, 2013, 18(3): p. 333-40; Dong, J.Q., et al., Br J Clin Pharmacol, 2015, 80(5): 1051-63.
[0155] Например и если не ограничиваться теорией, то можно полагать, что FAP является ферментом, ответственным за расщепление и инактивацию FGF21; поэтому ингибирование FAP может увеличить степени экспрессии FGF21 и может усилить действие эндогенного и/или экзогенного FGF21. FGF21 взаимодействует с FGFR1 через его N-конец и с β-Klotho через его C-конец. Этот C-концевой участок FGF21 важен для активации рецепторного комплекса для инициирования передачи сигналов (Micanovic, R., et al., J Cell Physiol, 2009, 219(2): 227-34; Yie, J., et al., FEBS Lett, 2009, 583(1): 19-24). Недавно FAPα идентифицировали, как протеазу, ответственную за инактивацию циркуляции FGF21 путем расщепления C-конца по Pro171 (Dunshee, D.R., et al., J Biol Chem, 2016, 291(11): 5986-96; Coppage, A.L., et al., PLoS One, 2016, 11(3): e0151269; Zhen, E.Y., et al., Biochem J, 2016, 473(5): 605-14). У грызутов и приматов период полувыведения экзогенно введенного FGF21 человека является коротким (~ 0,5-2 ч) вследствие опосредуемого FAP разложения фермента и восприимчивости к почечному клиренсу (Hager, T., et al., Anal Chem, 2013, 85(5): 2731-8; Xu, J., et al., Am J Physiol Endocrinol Metab, 2009, 297(5): E1105-14; Kharitonenkov, A., et al., Endocrinology, 2007, 148(2): 774-81). Обычные стратегии увеличения периода полувыведения значительно улучшили PK характеристики этих аналогов FGF21 in vivo; однако в этих аналогах все же сохраняется протеолитическая обработка (Hecht, R., et al., PLoS One, 2012, 7(11): e49345; Mu, J., et al., Diabetes, 2012, 61(2): 505-12; Camacho, R.C., et al., Eur J Pharmacol, 2013, 715(1-3): 41-5).
[0156] Соответственно, настоящее изобретение относится к способам лечения сахарного диабета, нечувствительности к инсулину и/или ожирения у нуждающегося в нем индивидуума, включающим введение индивидууму соединения, описанного в настоящем изобретении, или его фармацевтически приемлемой соли, или фармацевтической композиции, описанной в настоящем изобретении. В некоторых вариантах осуществления способ дополнительно включает введение FGF21 или аналога FGF21. В некоторых вариантах осуществления аналогом FGF21 является пэгилированный FGF21, PF-05231023 или LY2405319. Кроме того, настоящее изобретение относится к способам лечения сахарного диабета, нечувствительности к инсулину и/или ожирения у нуждающегося в нем индивидуума, включающим введение индивидууму соединения, описанного в настоящем изобретении, или его фармацевтически приемлемой соли, или фармацевтической композиции, описанной в настоящем изобретении, где экспрессия FGF21 усиливается. В некоторых вариантах осуществления сахарным диабетом является диабет типа II.
[0157] В некоторых вариантах осуществления индивидуумом является млекопитающее. В некоторых вариантах осуществления индивидуумом является примат, крупный рогатый скот, овца, свинья, лошадь, собака, кошка, кролик или грызун. В некоторых вариантах осуществления индивидуумом является человек. В некоторых вариантах осуществления у индивидуума имеется любое из заболеваний или нарушений, раскрытых в настоящем изобретении. В некоторых вариантах осуществления для индивидуума имеется опасность развития любого из заболеваний или нарушений, раскрытых в настоящем изобретении.
[0158] В некоторых вариантах осуществления индивидуумом является человек. В некоторых вариантах осуществления человек находится в возрасте, равном по меньшей мере примерно или примерно 21, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80 или 85 лет. В некоторых вариантах осуществления человеком является ребенок. В некоторых вариантах осуществления человек находится в возрасте, равном менее примерно или примерно 21, 18, 15, 12, 10, 8, 6, 5, 4, 3, 2 или 1 год.
[0159] Кроме того, настоящее изобретение относится к применениям соединения, описанного в настоящем изобретении или его фармацевтически приемлемой соли, или фармацевтической композиции, описанной в настоящем изобретении, для приготовления лекарственного средства. В некоторых вариантах осуществления приготовление лекарственного средства предназначено для лечения нарушения или заболевания, описанного в настоящем изобретении. В некоторых вариантах осуществления приготовление лекарственного средства предназначено для предупреждения и/или лечения нарушения или заболевания, опосредуемого с помощью FAP.
Комбинированная терапия
[0160] В настоящем изобретении также предложены соединения или их соли описанные в настоящем изобретении, и композиции, описанные в настоящем изобретении, можно вводить вместе с дополнительным средством лечения любого из заболеваний и нарушений, раскрытых в настоящем изобретении.
[0161] В некоторых вариантах осуществления (a) соединение, описанное в настоящем изобретении, или его фармацевтически приемлемую соль, или фармацевтическую композицию, описанную в настоящем изобретении, и (b) дополнительное средство вводят последовательно, вводят совместно или вводят одновременно. В некоторых вариантах осуществления (a) соединение, описанное в настоящем изобретении, или его фармацевтически приемлемую соль, или фармацевтическую композицию, описанную в настоящем изобретении, и (b) дополнительное средство вводят с разделением по времени, равным примерно 15 мин или менее, например, примерно 10, 5 или 1 мин или менее. В некоторых вариантах осуществления (a) соединение, описанное в настоящем изобретении, или его фармацевтически приемлемую соль, или фармацевтическую композицию, описанную в настоящем изобретении, и (b) дополнительное средство вводят с разделением по времени, равным примерно 15 мин или более, например, примерно 20, 30, 40, 50, 60 или большего количества минут. (a) Любое из следующих, соединение, описанное в настоящем изобретении, или его фармацевтически приемлемую соль, или фармацевтическую композицию, описанную в настоящем изобретении, и (b) дополнительное средство можно вводить первым. В некоторых вариантах осуществления (a) соединение, описанное в настоящем изобретении, или его фармацевтически приемлемую соль, или фармацевтическую композицию, описанную в настоящем изобретении, и (b) дополнительное средство вводят одновременно.
[0162] В некоторых вариантах осуществления дополнительное средство направлено на иммунный контрольный белок. В некоторых вариантах осуществления дополнительное средство является антителами, которые направлены на иммунный контрольный белок. В некоторых вариантах осуществления дополнительное средство направлено на PD-1, PD-L1, PD-L2, CTLA4, TIM3, LAG3, CCR4, OX40, OX40L, IDO и A2AR. В некоторых вариантах осуществления дополнительное средство является антителами к PD-1, антителами к PD-L1 или антителами к CTLA-4.
[0163] В некоторых вариантах осуществления дополнительным средством является индуктор экспрессии FGF21, такой как агонист PPARα. В одном варианте осуществления агонистом PPARα является фибрат или фенофибрат. В некоторых вариантах осуществления дополнительным средством является FGF-21 или аналог FGF-21. В некоторых вариантах осуществления аналогом является мутировавший FGF21 и/или пэгилированный FGF21. В некоторых вариантах осуществления аналогом FGF-21 является PF-05231023 или LY2405319.
[0164] В некоторых вариантах осуществления дополнительным средством является комплексный агонист KLB/FGFR, антагонист DDPIV, агонист рецептора GLP-1 или агонист глюкагонового рецептора.
[0165] Настоящее изобретение относится к способу усиления иммунного ответа у индивидуума, включающему введение индивидууму (a) соединения, описанного в настоящем изобретении, или его фармацевтически приемлемой соли, или фармацевтической композиции, описанной в настоящем изобретении, и (b) средства, которое направлено на иммунный контрольный белок. В некоторых вариантах осуществления у индивидуума имеется рак. В некоторых вариантах осуществления усиленный иммунный ответ направлен на опухолевые или раковые клетки.
[0166] Кроме того, настоящее изобретение относится к способам лечения рака у нуждающегося в нем индивидуума, включающим введение индивидууму (a) соединения, описанного в настоящем изобретении, или его фармацевтически приемлемой соли, или фармацевтической композиции, описанной в настоящем изобретении, и (b) средства, которое направлено на иммунный контрольный белок, где иммунный ответ индивидуума усиливается.
[0167] Настоящее изобретение относится к способу усиления степени экспрессии FGF21 у индивидуума, включающему введение индивидууму (a) соединения, описанного в настоящем изобретении, или его фармацевтически приемлемой соли, или фармацевтической композиции, описанной в настоящем изобретении, и (b) средства, которое индуцирует экспрессию FGF21.
[0168] Кроме того, настоящее изобретение относится к способам лечения сахарного диабета, нечувствительности к инсулину и/или ожирения у нуждающегося в нем индивидуума, включающим введение индивидууму (a) соединения, описанного в настоящем изобретении, или его фармацевтически приемлемой соли, или фармацевтической композиции, описанной в настоящем изобретении, и (b) средства, которое индуцирует экспрессию FGF21, где экспрессия FGF21 усиливается. В некоторых вариантах осуществления сахарным диабетом является диабет типа II.
[0169] В настоящем изобретении также предложены соединения или их соли, описанные в настоящем изобретении, и композиции, описанные в настоящем изобретении, которые вводят в качестве части режима лечения, который включает режим тренировки, такой как тренировка для развития силы или сердечно-сосудистая тренировка. В некоторых вариантах осуществления соединения или их соли, описанные в настоящем изобретении, и композиции, описанные в настоящем изобретении, вводят с дополнительным средством и в качестве части режима лечения, который включает режим тренировки, такой как тренировка для развития силы или сердечно-сосудистая тренировка. В некоторых вариантах осуществления режим тренировки включает тренировку по меньшей мере один раз в неделю, например, два раза в неделю, 3 раза в неделю, 4 раза в неделю, 5 раз в неделю, 6 раз в неделю или 7 раз в неделю. В некоторых вариантах осуществления режим тренировки включает тренировку в течение по меньшей мере одного дня в неделю, например, двух дней в неделю, 3 дней в неделю, 4 дней в неделю, 5 дней в неделю, 6 дней в неделю или 7 дней в неделю. В некоторых вариантах осуществления режим тренировки включает тренировку один раз в день, два раза в день или 3 раза в день. В некоторых вариантах осуществления режим тренировки включает тренировку в течение не менее 10 мин за сессию, например, в течение не менее 15 мин, 20 мин, 25 мин, 30 мин, 35 мин, 40 мин, 45 мин, 50 мин, 55 мин, 1 ч, 1,25 ч или 1,5 ч.
Введение и способ введения
[0170] Доза соединения, вводимого индивидууму (такому как человек) может меняться в зависимости от конкретного соединения или его соли, методики введения и конкретного заболевания, подвергающегося лечению, такого как тип и стадия рака. В некоторых вариантах осуществления количество соединения или его соли представляет собой терапевтически эффективное количество.
[0171] Эффективное количество соединения в одном объекте может составлять дозу, равную от примерно 0,01 до примерно 100 мг/кг. Эффективные количества или дозы соединений, предлагаемых в настоящем изобретении, можно определить по стандартным методикам, таким как моделирование, повышение дозы или клинические исследования, принимая во внимание обычные факторы, например, режим или путь введения или доставки лекарственного средства, фармакокинетику средства, тяжесть и течение заболевания, подвергающегося лечению, состояние здоровья субъекта и массу. Типичная доза находится в диапазоне примерно от 0,7 мг до 7 г в сутки, или примерно от 7 мг до 350 мг в сутки, или примерно от 350 мг до 1,75 г в сутки или примерно от 1,75 до 7 г м.
[0172] Любой из способов, предлагаемых в настоящем изобретении, в одном объекте может включать введение индивидууму фармацевтической композиции, которая содержит эффективное количество соединения, предлагаемого в настоящем изобретении, его фармацевтически приемлемой соли, стереоизомера или таутомера и фармацевтически приемлемый инертный наполнитель.
[0173] Соединение или композицию, предлагаемую в настоящем изобретении, можно вводить индивидууму в соответствии с эффективным режимом введения в течение желательного периода времени или продолжительности, например, не менее примерно одного месяца, не менее примерно 2 месяцев, не менее примерно 3 месяцев, не менее примерно 6 месяцев или не менее примерно 12 месяцев или дольше, что с небольшими изменениями может быть продолжительностью жизни индивидуума. В одном варианте соединение вводят один раз в сутки или в периодическом режиме. Соединение можно вводить индивидууму непрерывно (например, не реже одного раза в сутки) в течение времени. Частота введения также может быть реже одного раза в сутки, например, введение примерно один раз в неделю. Частота введения также может быть чаще одного раза в сутки, например, два или три раза в сутки. Частота введения также может быть переменной, включая перерыв введения лекарственного средства (например, введение один раз в сутки в течение 7 суток, затем без введения в течение 7 суток с повторением в каждый раз в течение 14 суток, например, примерно 2 месяца, примерно 4 месяца, примерно 6 месяцев или более). Любую частоту введения можно использовать для любых соединений, описанных в настоящем изобретении, вместе с любыми другими введениями, описанными в настоящем изобретении.
Изделия и наборы
[0174] Настоящее изобретение дополнительно относится к изделиям, включающим соединение, описанное в настоящем изобретении его фармацевтически приемлемую соль, стереоизомер или таутомер, композицию, описанную в настоящем изобретении, или одну или большее количество разовых доз, описанных в настоящем изобретении, в подходящей упаковке. В некоторых вариантах осуществления изделие предназначено для применения в любом из способов, описанных в настоящем изобретении. Подходящая упаковка известна в данной области техники и включает, например, флаконы, сосуды, ампулы, бутылки, банки, гибкую упаковку и т. п. Изделие можно дополнительно стерилизовать и/или герметизировать.
[0175] Настоящее изобретение дополнительно относится к наборам для осуществления способов, предлагаемых в настоящем изобретении, которые включают одно или большее количество соединений, описанных в настоящем изобретении, или композицию, содержащую соединение, описанное в настоящем изобретении. В наборах можно использовать любое из соединений, раскрытых в настоящем изобретении. В одном варианте в наборе используется соединение, описанное в настоящем изобретении, его фармацевтически приемлемая соль, стереоизомер или таутомер. Наборы можно использовать для любого одного или большего количества применений, описанных в настоящем изобретении, и, соответственно, они могут содержать инструкции для лечения любого заболевания или нарушения, описанного в настоящем изобретении, например, для лечения рака.
[0176] Наборы обычно включают подходящую упаковку. Наборы могут включать один или большее количество контейнеров, содержащих любое соединение, описанное в настоящем изобретении. Каждый компонент (если имеется более одного компонента) можно упаковать в отдельные контейнеры или некоторые компоненты можно объединить в одном контейнере, если это позволяет перекрестная реакционная способность и срок хранения.
[0177] Наборы могут представлять собой разовые дозированные формы, объемные упаковки (например, многодозовые упаковки) или дозы, меньше разовых. Например, при условии, что они содержат достаточные дозы соединения, раскрытые в настоящем изобретении, и/или дополнительные фармацевтически активные соединения, применимые для заболевания, подробно описанного в настоящем изобретении, которые обеспечивают эффективное лечение индивидуума в течение длительного периода времени, например, любого из следующих: неделя, 2 недели, 3 недели, 4 недели, 6 недель, 8 недель, 3 месяца, 4 месяца, 5 месяцев, 7 месяцев, 8 месяцев, 9 месяцев или более. Наборы также могут включать множество разовых доз соединений и инструкции для применения и могут быть упакованы в количествах, достаточных для хранения и применения в аптеках (например, больничных аптеках и универсальных аптеках).
[0178] Наборы необязательно могут включать набор инструкций, обычно печатных инструкций, хотя применимы также электронные носители (например, магнитная дискета или оптический диск), содержащие инструкции, относящиеся к применению компонента (компонентов) способов, предлагаемых в настоящем изобретении. Инструкции, включенные в набор, обычно включают информацию о компонентах и их введении индивидууму.
[0179] Настоящее изобретение можно дополнительно понять с помощью следующих примеров, которые приведены для иллюстрации и не являются ограничивающими.
[0180] Вся цитированная литература, такая как публикации, патенты, заявки на патенты и опубликованные заявки на патенты, во всей своей полноте включена в настоящее изобретение в качестве ссылки.
ПРИМЕРЫ
Примеры синтеза
[0181] Химические реакции в описанных примерах синтеза можно легко адаптировать для получения целого ряда других соединений, предлагаемых в настоящем изобретении, и альтернативные методики получения соединений, предлагаемых в настоящем изобретении, представляются входящими в объем настоящего изобретения. Например, синтез не приведенных в качестве примеров соединений, предлагаемых в настоящем изобретении можно легко провести с помощью модификаций, очевидных для специалистов в данной области техники, например, путем соответствующей защиты мешающих групп, путем использования других подходящих реагентов, известных в данной области техники, кроме описанных, или путем внесения стандартных изменений в условия проведения реакций. Альтернативно, другие реакции, раскрытые в настоящем изобретении или известные в данной области техники, применимы для получения других соединений, предлагаемых в настоящем изобретении.
Пример S1
Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-фенилацетамидо)изоникотинамида

[0182] Соединение 1a. При перемешивании к раствору 2-фенилуксусной кислоты (0,500 г, 3,67 ммоля, 1,0 экв.) в DMF (10 мл) добавляли метил-3-аминоизоникотинат (0,558 г, 3,67 ммоля, 1,0 экв.) и HATU (2,8 г, 7,35 ммоля, 2,0 экв.) при КТ. Полученную реакционную смесь перемешивали в течение 10 мин при КТ и добавляли DIPEA (2,0 мл, 11,50 ммоля, 3,0 экв.). Реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS. Реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (20 мл×4), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-40% этилацетата в гексане в качестве элюента) и получали метил-3-(2-фенилацетамидо)изоникотинат (0,460 г, 46% выход ) в виде желтого полужидкого вещества.
[0183] LCMS 271,2 [0M+H]+
[0184] Соединение 1b. При перемешивании к раствору метил-3-(2-фенилацетамидо)изоникотината (0,290 г, 1,07 ммоля, 1 экв.) в THF и воде (6 мл: 3 мл) добавляли LiOH (0,051 г, 2,14 ммоля, 2,0 экв.). Смесь перемешивали при температуре окружающей среды в течение 16 ч. Образование продукта подтверждали с помощью LCMS. Реакционную смесь разбавляли водой (20 мл). Водный слой промывали этилацетатом (10 мл), водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали 3-(2-фенилацетамидо)изоникотиновую кислоту (0,420 г. количественный выход) в виде почти белого твердого вещества
[0185] LCMS 257,2 [0M+H]+
[0186] Соединение 1. При перемешивании к раствору 3-(2-фенилацетамидо)изоникотиновой кислоты (0,200 г, 0,713 ммоля, 1 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,161 г, 0,713 ммоля, 1,0 экв.), HOBt (0,115 г, 0,856 ммоля, 1,2 экв.) и EDC.HCl (0,164 г, 0,856 ммоля, 1,2 экв.). Смесь перемешивали при КТ в течение 10 мин. Триэтиламин (0,145 г, 1,427 ммоля, 2,0 экв.) добавляли и смесь перемешивали при температуре окружающей среды в течение 16 ч. Образование продукта подтверждали с помощью LCMS. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (40 мл×2). Объединенные органические экстракты промывали водой (25 мл×5). Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении и получали неочищенный продукт. Полученный неочищенный продукт очищали с помощью HPLC с обращенной фазой и получали (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-фенилацетамидо)изоникотинамид (0,070 г, 21% выход) в виде желтого твердого вещества.
[0187] LCMS 428,3 [0M+H]+
[0188] 1H NMR (400MHz, DMSO-d6) δ 10,44 (br. s., 1 H), 9,41 (s, 1 H), 9,21 (br. s., 1 H), 8,42 (d, J=4,8 Hz, 1 H), 7,56 (d, J=4,8 Hz, 1 H), 7,43-7,15 (m, 4 H), 5,16 (d, J=8,8 Hz, 1 H), 4,31 (d, J=12,7 Hz, 2 H), 4,22-4,08 (m, 3 H), 3,76 (s, 2 H), 2,93 (br. s., 1 H), 2,84 (d, J=15,8 Hz, 2 H).
Пример S2
Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((4-фторбензил)амино)изоникотинамида

[0189] Соединение 2a. При перемешивании к раствору метил-3-бромизоникотината (0,500 г, 2,3148 ммоля, 1 экв.) добавляли (4-фторфенил)метанамин (0,289 г, 2,3148 ммоля, 1 экв.) и CS2CO3(1,5 г, 4,6296 ммоля, 2 экв.) в диоксане (15,0 мл). Полученную смесь продували азотом в течение 10 мин, затем добавляли Pd2(dba)3 (0,106 г, 0,1157 ммоля, 0,05 экв.) и xantphos (0,134 г, 0,2314 ммоля, 0,1 экв.), повторно продували азотом в течение 10 мин. Реакционную смесь нагревали при 120°C в течение ночи. За протеканием реакции следили с помощью LCMS. Реакционную смесь разбавляли водой (30 мл), экстрагировали с помощью EtOAc (2×50 мл). Объединенные органические слои промывали водой (30 мл), рассолом (30 мл), сушили над Na2SO4, концентрировали и получали неочищенный продукт. Неочищенный продукт очищали с помощью колоночной хроматографии (0-50% этилацетата в гексане в качестве элюента) и получали искомый метил-3-((4-фторбензил)амино)изоникотинат (200 мг, 33,00%) в виде почти белого твердого вещества.
[0190] LCMS: 262,1 [0M+H]+
[0191] 1H NMR; (400MHz, DMSO-d6) δ8,18 (s, 1 H), 7,83 (s, 2 H), 7,43 (d, 2H), 7,34(d, 2H), 7,17 (s, 1 H), 4,57 (d, J=5,7 Hz, 2 H), 3,86 (s, 3 H)
[0192] Соединение 2b. При перемешивании к раствору соединения метил-3-((4-фторбензил)амино)изоникотината (0,200 г, 0,7692 ммоля, 1 экв.) в THF (8 мл) и воде (4 мл) добавляли LiOH (0,040 г, 1,5384 ммоля, 2 экв.). Смесь перемешивали при 80°C в течение ночи. Образование продукта подтверждали с помощью LCMS. Реакционную смесь концентрировали и разбавляли водой (10 мл) и промывали этилацетатом (2×10 мл). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали соединение 3-((4-фторбензил)амино)изоникотиновую кислоту (170 мг, 89,94%) в виде белого твердого вещества.
[0193] LCMS: 247,2 [0M+H]+
[0194] 1H NMR; (400MHz, DMSO-d6) δ11,70 (s, 1H), 8,18 (s, 1 H), 7,83 (s, 2 H), 7,43 (d, 2H), 7,34(d, 2H), 7,17 (s, 1 H), 4,57 (d, J=5,7 Hz, 2 H).
[0195] Соединение 2. При перемешивании к раствору 3-((4-фторбензил)амино)изоникотиновой кислоты (0,170 г, 0,8130 ммоля, 1 экв.), HATU(0,464 г, 1,2195 ммоля, 1,5 экв.) в DMF (10 мл) через 10 мин добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,219 г, 0,9756 ммоля, 1,2 экв.) и реакционную смесь перемешивали в течение 10 мин, затем по каплям добавляли DIPEA (0,3 мл, 1,2195 ммоля, 1,5 экв.) и реакционную смесь перемешивали в течение 16 ч при КТ. За протеканием реакции следили с помощью LCMS и TLC, обрабатывали путем добавления холодной воды (50 мл) к реакционной массе и экстрагировали этилацетатом (3×50 мл) и все собранные органические слои трижды промывали водой и один раз бикарбонатом натрия и один раз рассолом. Органический слой сушили над безводным Na2SO4, концентрировали при пониженном давлении, неочищенный продукт очищали с помощью хроматографии с обращенной фазой и получали искомый продукт (S)-N-(2-(2-циано-4, 4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((4-фторбензил)амино)изоникотинамид (30 мг, 11%) в виде белого твердого вещества.
[0196] LCMS: 418,2 [0M+H]+
[0197] 1H NMR (400MHz, DMSO-d6) δ 8,99 (br. s., 1 H), 8,10 (s, 1 H), 7,88 (d, J=4,8 Hz, 2 H), 7,49 (d, J=5,3 Hz, 1 H), 7,44-7,35 (m, 2 H), 7,17 (t, J=9,0 Hz, 2 H), 5,10 (d, J=7,5 Hz, 1 H), 4,47 (d, J=5,3 Hz, 4 H), 4,12 (d, J=6,6 Hz, 2 H), 2,82 (d, J=16,7 Hz, 2 H).
Пример S3
Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-метоксибензамидо)изоникотинамида
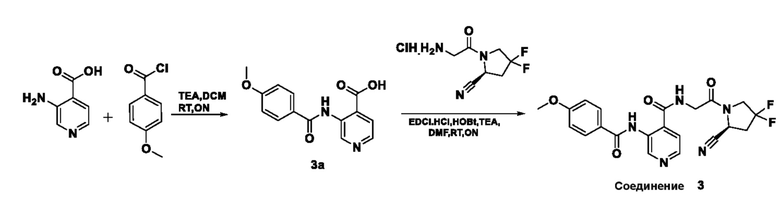
[0198] Соединение 3a. К раствору 3-аминоизоникотиновой кислоты (0,5 г, 3,628 ммоля, 1,0 экв.) в диоксане (10 мл) добавляли 4-метоксибензоилхлорид (1,0 мл, 7,246 ммоля, 2,0 экв.). Полученную реакционную смесь перемешивали при КТ в течение 10 мин. TEA (1,5 мл, 10,87 ммоля, 3,0 экв.) по каплям добавляли при КТ. Полученную реакционную смесь перемешивали в течение ночи при КТ. Образование продукта подтверждали с помощью LCMS. После завершения реакции реакционную смесь концентрировали досуха и разбавляли водой (50 мл). Водный слой промывали с помощью EtOAc (2×20 мл). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали 3-(4-метоксибензамидо)изоникотиновую кислоту (количественный выход) в виде желтого твердого вещества.
[0199] LCMS 273,1 [0M+H]+
[0200] Соединение 3. При перемешивании к раствору 3-(4-метоксибензамидо)изоникотиновой кислоты (0,10 г, 0,367 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,082 г, 0,367 ммоля, 1,0 экв.), EDCI.HCl (0,141 г, 0,734 ммоля, 2,0 экв.) и HOBt (0,099 г, 0,737 ммоля, 2,0 экв.). Смесь перемешивали при КТ в течение 10 мин. Добавляли TEA (0,4 мл) и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (50 мл×3). Объединенные органические экстракты промывали водой (50 мл×5), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (5% MeOH в DCM в качестве элюента) и получали (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-метоксибензамидо)изоникотинамид (0,015 г, 9,2% выход) в виде почти белого твердого вещества.
[0201] LCMS 444,2 [0M+H]+
[0202] 1H NMR (400MHz, DMSO-d6) δ 11,59 (s, 1 H), 9,75 (s, 1 H), 9,44 (br. s., 1 H), 8,49 (d, J=4,8 Hz, 1 H), 7,99-7,83 (m, J=8,8 Hz, 2 H), 7,77 (d, J=5,3 Hz, 1 H), 7,25-6,97 (m, J=8,8 Hz, 2 H), 5,12 (d, J=6,1 Hz, 1 H), 4,33 (br. s., 1 H), 4,29-4,07 (m, 2 H), 2,91 (br. s., 3 H).
Пример S4
Синтез N-(2-((S)-2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((1-(4-фторфенил)этил)амино)изоникотинамида

[0203] Соединение 4a. При перемешивании к раствору метил-3-бромизоникотината (0,500 г, 2,3148 ммоля, 1,0 экв.) добавляли 1-(4-фторфенил)этан-1-амин (0,350 г, 2,540 ммоля, 1,0 экв.) и CS2CO3(1,5 г, 4,6296 ммоля, 2 экв.) в диоксане (20 мл). Полученную смесь продували азотом в течение 10 мин, затем добавляли Pd2(dba)3 (0,110 г, 0,115 ммоля, 0,05 экв.) и xantphos (0,135 г, 0,231 ммоля, 0,1 экв.), повторно продували азотом в течение 10 мин. Реакционную смесь нагревали при 120°C в течение ночи. За протеканием реакции следили с помощью LCMS. Реакционную смесь разбавляли водой (30 мл), экстрагировали с помощью EtOAc (2×50 мл). Объединенный органический слой промывали водой (30 мл), рассолом (30 мл), сушили над Na2SO4, концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-50% этилацетата в гексане в качестве элюента) и получали метил-3-((1-(4-фторфенил)этил)амино)изоникотинат (200 мг, 31,0%) в виде почти белого твердого вещества.
[0204] LCMS: 275,1 [0M+H]+
[0205] 1H NMR (400MHz, хлороформ-d) δ 7,98 (s, 1 H), 7,87 (d, J=5,3 Hz, 1 H), 7,62 (d, J=5,3 Hz, 1 H), 7,38-7,29 (m, 1 H), 7,00 (t, J=8,6 Hz, 2 H), 4,67 (t, J=6,4 Hz, 1 H), 3,93 (s, 3 H), 1,63-1,49 (m, 3 H).
[0206] Соединение 4b. При перемешивании к раствору метил-3-((1-(4-фторфенил)этил)амино)изоникотината (0,200 г, 0,727 ммоля, 1,0 экв.) в THF (10 мл) и воде (4 мл) добавляли LiOH (0,035 г, 1,45 ммоля, 2 экв.). Смесь перемешивали при 80°C в течение ночи. Образование продукта подтверждали с помощью LCMS. Реакционную смесь концентрировали и разбавляли водой (10 мл) и промывали этилацетатом (2×10 мл). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали литиевую соль 3-((1-(4-фторфенил)этил)амино)изоникотиновой кислоты (0,250 г, количественный выход) в виде белого твердого вещества.
[0207] LCMS: 261,2 [0M+H]+
[0208] 1H NMR (400MHz, DMSO-d6) δ 9,56 (d, J=7,5 Hz, 1 H), 7,65-7,57 (m, 2 H), 7,52 (d, J=4,8 Hz, 1 H), 7,38 (dd, J=5,7, 8,8 Hz, 2 H), 7,11 (t, J=9,0 Hz, 2 H), 4,64 (t, J=6,8 Hz, 1 H), 1,42 (d, J=7,0 Hz, 3 H).
[0209] Соединение 4. При перемешивании к раствору литиевой соли 3-((1-(4-фторфенил)этил)амино)изоникотиновой кислоты (0,250 г, 0,936 ммоля, 1,0 экв.) в DMF (5 мл) добавляли EDC.HCl (0,269 г, 1,404 ммоля, 1,5 экв.), HOBt (0,152 г, 1,123, 1,2 экв.), (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,219 г, 0,9756 ммоля, 1,2 экв.), DMAP (0,002 г, 0,009 ммоля, 0,01 экв.), затем добавляли триэтиламин (0,4 мл, 2,808 ммоля, 3,0 экв.). Полученную реакционную смесь перемешивали при КТ в течение 16 ч. За протеканием реакции следили с помощью LCMS и TLC, реакционную смесь разбавляли водой и экстрагировали этилацетатом (2×50 мл). Объединенный органический слой промывали водой (20 мл×4). Органический слой сушили над безводным Na2SO4, концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии с обращенной фазой и получали N-(2-((S)-2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((1-(4-фторфенил)этил)амино)изоникотинамид (0,012 г, 3%) в виде белого твердого вещества.
[0210] LCMS: 432,2 [0M+H]+
[0211] 1H NMR (400MHz, DMSO-d6) δ 9,02 (br. s., 1 H), 7,96-7,89 (m, 1 H), 7,83 (d, J=4,8 Hz, 1 H), 7,52-7,33 (m, 2 H), 7,19-7,05 (m, 1 H), 5,12 (d, J=8,3 Hz, 1 H), 4,88-4,72 (m, 1 H), 4,31 (br. s., 1 H), 4,22-4,02 (m, 2 H), 2,92 (br. s., 1 H), 2,83 (d, J=18,9 Hz, 1 H), 1,45 (d, J=6,6 Hz, 3 H).
Пример S5
Синтез (S)-3-(2-(4-хлор-3-фторфенокси)ацетамидо)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамида

[0212] Соединение 5a. При перемешивании к раствору 3-аминоизоникотиновой кислоты (0,2 г, 1,45 ммоля, 1,0 экв.) в DMF (5 мл) добавляли 2-(4-хлор-3-фторфенокси)уксусную кислоту (0,59 г, 2,898 ммоля, 2,0 экв.), EDCI.HCl (0,556 г, 2,898 ммоля, 2,0 экв.) и HOBt (0,391 г, 2,898 ммоля, 2,0 экв.). Смесь перемешивали при КТ в течение 10 мин. Добавляли TEA (0,4 мл) и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (50 мл×3). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали 3-(2-(4-хлор-3-фторфенокси)ацетамидо)изоникотиновую кислоту (количественный выход) в виде почти белого твердого вещества.
[0213] LCMS 325,1 [0M+H]+
[0214] Соединение 5. При перемешивании к раствору 3-(2-(4-хлор-3-фторфенокси)ацетамидо)изоникотиновой кислоты (0,20 г, 0,617 ммоля, 1,0 экв.) в DMF (15 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,138 г, 0,617 ммоля, 1,0 экв.), EDCI.HCl (0,141 г, 1,234 ммоля, 2,0 экв.) и HOBt (0,166 г, 1,234 ммоля, 2,0 экв.). Смесь перемешивали при КТ в течение 10 мин. Добавляли TEA (2,0 мл) и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (50 мл×3). Объединенные органические экстракты промывали водой (50 мл×5), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (5% MeOH в DCM в качестве элюента) и получали (S)-3-(2-(4-хлор-3-фторфенокси)ацетамидо)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид (0,025 г, 8,1% выход) в виде почти белого твердого вещества.
[0215] LCMS 444,2 [0M+H]+
[0216] 1H NMR 1H NMR (400MHz, DMSO-d6) δ 11,37 (s, 1 H), 9,63 (s, 1 H), 9,35 (br. s., 1 H), 8,50 (d, J=4,8 Hz, 1 H), 7,73 (d, J=5,3 Hz, 1 H), 7,52 (t, J=8,8 Hz, 1 H), 7,19 (d, J=11,0 Hz, 1 H), 6,96 (d, J=6,1 Hz, 1 H), 5,11 (d, J=6,6 Hz, 1 H), 4,81 (s, 2 H), 4,32 (br. s., 1 H), 4,27-3,99 (m, 2 H), 2,92 (br. s., 2 H), 2,84 (d, J=11,4 Hz, 1 H)
Пример S6
Синтез (S)-2-(4-(бис(4-фторфенил)метил)пиперазин-1-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамида

[0217] Соединение 6a. При перемешивании к раствору метил-2-фторизоникотината (0,050 г, 0,32 ммоля, 1,0 экв.) в DMF (3 мл) добавляли 1-(бис(4-фторфенил)метил)пиперазин (0,093 г, 0,32 ммоля, 1,0 экв.). Реакционную смесь нагревали при 100°C в течение 16 ч. Образование продукта подтверждали с помощью TLC и LCMS. Реакционную смесь разбавляли водой (15 мл) и экстрагировали этилацетатом (20 мл×3). Объединенный органический слой промывали водой (12 мл×6). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенное соединение очищали с помощью хроматографии Combiflash с нормальной фазой и получали метил-2-(4-(бис(4-фторфенил)метил)пиперазин-1-ил)изоникотинат (0,015 г, 11% выход ) в виде почти белого твердого вещества.
[0218] LCMS 424,2 [0M+H]+
[0219] Соединение 6b. При перемешивании к раствору метил-2-(4-(бис(4-фторфенил)метил)пиперазин-1-ил)изоникотината (0,120 г, 0,28 ммоля, 1 экв.) в THF и воде (1:1)(6 мл) добавляли LiOH.H2O (0,018 г, 0,42 ммоля, 2,0 экв.). Смесь перемешивали при КТ в течение 16 ч. Образование продукта подтверждали с помощью LCMS. Реакционную смесь разбавляли водой (20 мл), промывали этилацетатом (15 мл×2). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали 2-(4-(бис(4-фторфенил)метил)пиперазин-1-ил)изоникотиновую кислоту (0,100 г) в виде почти белого твердого вещества.
[0220] LCMS 410,2 [0M+H]+
[0221] Соединение 6. При перемешивании к раствору 2-(4-(бис(4-фторфенил)метил)пиперазин-1-ил)изоникотиновой кислоты (0,100 г, 0,24 ммоля, 1,0 экв.) в DMF (3 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,068 г, 0,3 ммоля, 1,2 экв.), EDC.HCl (0,057 г, 0,3 ммоля, 1,2 экв.), HOBT (0,041 г, 0,3 ммоля, 1,2 экв.), DMAP (0,001 г) и реакционную смесь перемешивали при КТ в течение 10 мин, затем добавляли TEA (0,073 г, 0,72 ммоля, 3,0 экв.). Реакционную смесь перемешивали при КТ в течение 16 ч. Образование продукта подтверждали с помощью LCMS. После завершения реакции реакционную смесь разбавляли водой, что приводило к образованию осадка. Полученное твердое вещество отфильтровывали и промывали охлажденной льдом водой, сушили в вакууме. Неочищенный продукт очищали с помощью флэш-хроматографии и получали (S)-2-(4-(бис(4-фторфенил)метил)пиперазин-1-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид (0,006 г, 4,26% выход) в виде почти белого твердого вещества.
[0222] LCMS 581,4 [0M+H]+
[0223] 1H NMR (400 MHz, DMSO-d6) δ 8,91 (t, J=6,0 Hz, 1H), 8,21 (d, J=5,2 Hz, 1H), 7,48 (dd, J=8,5, 5,5 Hz, 4H), 7,19-7,10 (m, 5H), 7,01 (d, J=5,3 Hz, 1H), 5,08 (dd, J=9,4, 2,8 Hz, 1H), 4,43 (s, 1H), 4,28 (td, J=11,3, 10,4, 5,9 Hz, 1H), 4,11 (qd, J=14,2, 11,8, 4,4 Hz, 3H), 3,55 (t, J=4,8 Hz, 4H), 3,31 (s, 2H), 2,98-2,72 (m, 2H), 2,40 (t, J=4,9 Hz, 5H), 1,54 (s, 1H), 1,38-1,16 (m, 9H), 0,85 (t, J=6,5 Hz, 1H).
Пример S7
Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-фторбензил)изоникотинамида

[0224] Соединение 7a. При перемешивании к раствору метил-3-бромизоникотината (0,100 г, 0,46 ммоля, 1,0 экв.) в диоксане (4 мл) и воде (1 мл) добавляли K2CO3 (0,032 г, 0,23 ммоля, 0,5 экв.), 2-(2-фторбензил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,163 г, 0,69 ммоля, 1,5 экв.) и Pd(PPh3)Cl2 (0,016 г, 0,023 ммоля, 0,05 экв.). Реакционную смесь нагревали при 160°C в течение 1 ч в микроволновом реакторе. Образование продукта подтверждали с помощью TLC и LCMS. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (15 мл×3). Объединенный органический слой промывали рассолом (30 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии Combiflash с нормальной фазой и получали метил-3-(2-фторбензил)изоникотинат (0,025 г, 22% выход) в виде желтого полужидкого вещества.
[0225] LCMS 246,0 [0M+H]+
[0226] Соединение 7b. При перемешивании к раствору метил-3-(2-фторбензил)изоникотината (0,130 г, 0,53 ммоля, 1 экв.) в THF и воде (3:1)(4 мл) добавляли LiOH.H2O (0,045 г, 1,06 ммоля, 2,0 экв.). Смесь перемешивали при КТ в течение 16 ч. Образование продукта подтверждали с помощью LCMS. Реакционную смесь разбавляли водой (25 мл), промывали этилацетатом (20 мл). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали 3-(2-фторбензил)изоникотиновую кислоту (0,120 г, количественный выход).
[0227] LCMS 231,9 [0M+H]+
[0228] Соединение 7. При перемешивании к раствору 3-(2-фторбензил)изоникотиновой кислоты (0,130 г, 0,56 ммоля, 1,0 экв.) в DMF (4 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,153 г, 0,68 ммоля, 1,2 экв.), EDC.HCl (0,129 г, 0,68 ммоля, 1,2 экв.), HOBT (0,092 г, 0,68 ммоля, 1,2 экв.), DMAP (0,001 г) и реакционную смесь перемешивали при КТ в течение 10 мин, затем добавляли TEA (0,170 г, 1,68 ммоля, 3,0 экв.). Реакционную смесь перемешивали при КТ в течение 16 ч. Образование продукта подтверждали с помощью LCMS. Реакционную смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (30 мл×2). Объединенный органический слой промывали водой (20 мл×6) и рассолом (40 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью HPLC с обращенной фазой и получали (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-фторбензил)изоникотинамид (0,002 г) в виде почти белого твердого вещества.
[0229] LCMS 403,3 [0M+H] +
[0230] 1H NMR (400 MHz, DMSO-d6) δ 8,95 (t, J=5,8 Hz, 1H), 8,54 (d, J=5,0 Hz, 1H), 8,41 (s, 1H), 7,38 (d, J=4,9 Hz, 1H), 7,19 (ddt, J=39,1, 20,6, 7,2 Hz, 4H), 5,21-5,03 (m, 1H), 4,43-3,99 (m, 6H), 3,02-2,73 (m, 2H).
Пример S8
Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-фторбензил)изоникотинамида

[0231] Соединение 8a. При перемешивании к раствору метил-3-бромизоникотината (0,100 г, 0,46 ммоля, 1,0 экв.) в диоксане (4 мл) и воде (1 мл) добавляли K2CO3 (0,032 г, 0,23 ммоля, 0,5 экв.), 2-(4-фторбензил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,163 г, 0,69 ммоля, 1,5 экв.) и Pd(PPh3)Cl2 (0,016 г, 0,023 ммоля, 0,05 экв.). Реакционную смесь нагревали при 160°C в течение 1 ч в микроволновом реакторе. Образование продукта подтверждали с помощью TLC и LCMS. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (15 мл×3). Объединенный органический слой промывали рассолом (30 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии Combiflash с нормальной фазой и получали и получали метил-3-(4-фторбензил)изоникотинат (0,025 г, 22% выход) в виде желтого полужидкого вещества.
[0232] LCMS 246,0 [0M+H]+
[0233] Соединение 8b. При перемешивании к раствору метил-3-(4-фторбензил)изоникотинат (0,200 г, 0,82 ммоля, 1 экв.) в THF и воде (4:1)(5 мл) добавляли LiOH.H2O (0,069 г, 1,64 ммоля, 2,0 экв.). Смесь перемешивали при КТ в течение 16 ч. Образование продукта подтверждали с помощью LCMS. Реакционную смесь разбавляли водой (25 мл), промывали этилацетатом (20 мл). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали 3-(4-фторбензил)изоникотиновую кислоту (0,180 г) в виде почти белого твердого вещества.
[0234] LCMS 231,9 [0M+H]+
[0235] Соединение 8. При перемешивании к раствору 3-(4-фторбензил)изоникотиновой кислоты (0,250 г, 1,08 ммоля, 1,0 экв.) в DMF (4 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,293 г, 1,3 ммоля, 1,2 экв.), EDC.HCl (0,247 г, 1,3 ммоля, 1,2 экв.), HOBT (0,176 г, 1,3 ммоля, 1,2 экв.), DMAP (0,001 г) и реакционную смесь перемешивали при КТ в течение 10 мин, затем добавляли TEA (0,327 г, 3,24 ммоля, 3,0 экв.). Реакционную смесь перемешивали при КТ в течение 16 ч. Образование продукта подтверждали с помощью LCMS. Реакционную смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (30 мл×2). Объединенный органический слой промывали водой (20 мл×5) и рассолом (40 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью HPLC с обращенной фазой и получали чистый (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-фторбензил)изоникотинамид (0,005 г) в виде почти белого твердого вещества.
[0236] LCMS 403,3 [0M+H]+
[0237] 1H NMR (400 MHz, DMSO-d6) δ 8,96 (t, J=5,8 Hz, 1H), 8,54 (s, 1H), 8,51 (d, J=5,0 Hz, 1H), 7,39-7,27 (m, 3H), 7,07 (t, J=8,8 Hz, 2H), 5,13 (dd, J=9,4, 2,7 Hz, 1H), 4,28 (td, J=12,4, 6,4 Hz, 1H), 4,21- 4,03 (m, 5H), 2,99-2,74 (m, 2H).
Пример S9
Синтез (S, E)-3-(4-хлорстирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамида

[0238] Соединение 9a. К раствору этил-3-бромизоникотината (0,2 г, 0,925 ммоля, 1,0 экв.) в диоксане (8 мл) и воде (2 мл) добавляли (E)-2-(4-хлорстирил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,366 г, 1,39 ммоля, 1,5 экв.), K2CO3 (0,384 г, 2,78 ммоля, 3,0 экв.) и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,033 г, 0,046 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 120°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции смесь фильтровали через слой целита, промывали этилацетатом (100 мл). Фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (0-20% этилацетата в гексане в качестве элюента) и получали метил-(E)-3-(4-хлорстирил)изоникотинат (0,120 г, 47,43% выход) в виде желтого твердого вещества.
[0239] LCMS 274,5 [0M+H]+
[0240] 1H NMR (400 MHz, DMSO-d6) δ 9,11 (s, 1 H), 8,64 (d, J=5,3 Hz, 1 H), 7,78-7,67 (m, 2 H), 7,63 (d, J=8,3 Hz, 2 H), 7,48 (d, J=8,3 Hz, 2 H), 7,36 (d, J=16,7 Hz, 1 H), 3,91 (s, 3 H).
[0241] Соединение 9b. При перемешивании к раствору метил-(E)-3-(4-хлорстирил)изоникотината (0,1 г, 0,366 ммоля, 1,0 экв.) в THF (5 мл) и воде (5 мл) добавляли LiOH (0,023 г, 0,0,549 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и 1H NMR спектроскопии. Реакционную смесь концентрировали и разбавляли водой (10 мл) и промывали этилацетатом (10 мл×2). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали (E)-3-(4-хлорстирил)изоникотиновую кислоту (количественный выход) в виде почти белого твердого вещества.
[0242] LCMS 260,0 [0M+H]+
[0243] 1H NMR (400 MHz, DMSO-d6) δ 8,80 (s, 1 H), 8,30 (d, J=4,8 Hz, 1 H), 7,87 (d, J=16,7 Hz, 1 H), 7,63-7,49 (m, J=8,3 Hz, 2 H), 7,47-7,35 (m, J=8,8 Hz, 2 H), 7,28 (d, J=4,8 Hz, 1 H), 7,19 (d, J=16,7 Hz, 1 H).
[0244] Соединение 9. При перемешивании к раствору (E)-3-(4-хлорстирил)изоникотиновой кислоты (0,11 г, 0,424 ммоля, 1,0 экв.) в DMF (3 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,096 г, 0,424 ммоля, 1,0 экв.), EDCI.HCl (0,122 г, 0,636 ммоля, 1,5 экв.) и HOBt (0,086 г, 0,48 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 10 мин. Добавляли триэтиламин (0,25 мл) и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл×2). Объединенные органические экстракты промывали водой (20 мл×4), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт кристаллизовали из эфира и получали (S, E)-3-(4-хлорстирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид (0,18 г, 98,36% выход) в виде почти белого твердого вещества.
[0245] LCMS 431,2 [0M+H]+
[0246] 1H NMR (400 MHz, DMSO-d6) δ 9,14 (s, 1 H), 9,06-8,94 (m, 1 H), 8,54 (d, J=4,8 Hz, 1 H), 7,83-7,61 (m, 3 H), 7,60-7,41 (m, 3 H), 7,36 (d, J=4,8 Hz, 1 H), 5,25-5,10 (m, 1 H), 4,31 (d, J=11,8 Hz, 1 H), 4,24-3,96 (m, 3 H), 2,94 (br. s., 1 H), 2,92-2,78 (m, 1 H).
Пример S10
Синтез (S)-3-(4-хлорфенетил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамида

[0247] Соединение 10a. К раствору этил-3-бромизоникотината (0,2 г, 0,925 ммоля, 1,0 экв.) в диоксане (8 мл) и воде (2 мл) добавляли (E)-2-(4-хлорстирил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,366 г, 1,39 ммоля, 1,5 экв.), K2CO3 (0,384 г, 2,78 ммоля, 3,0 экв.) и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,033 г, 0,046 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 120°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции смесь фильтровали через слой целита, промывали этилацетатом (100 мл). Фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (0-20% этилацетата в гексане в качестве элюента) и получали метил-(E)-3-(4-хлорстирил)изоникотинат (0,120 г, 47,43% выход) в виде желтого твердого вещества.
[0248] LCMS 274,5 [0M+H]+
[0249] 1H NMR (400 MHz, DMSO-d6) δ 9,11 (s, 1 H), 8,64 (d, J=5,3 Hz, 1 H), 7,78-7,67 (m, 2 H), 7,63 (d, J=8,3 Hz, 2 H), 7,48 (d, J=8,3 Hz, 2 H), 7,36 (d, J=16,7 Hz, 1 H), 3,91 (s, 3 H).
[0250] Соединение 10b. Раствор метил-(E)-3-(4-хлорстирил)изоникотината (0,12 г, 0,44 ммоля, 1,0 экв.) в метаноле (15 мл) продували газообразным N2 в течение 10 мин, затем добавляли Pd/C (0,030 г). Затем полученную реакционную смесь продували газообразным H2 в течение 10 ч. Образование продукта подтверждали с помощью LCMS. После завершения реакции смесь фильтровали через слой целита, промывали метанолом (30 мл). Фильтрат концентрировали при пониженном давлении и получали метил-3-(4-хлорфенетил)изоникотинат (0,105 г, 86,77% выход) в виде белого твердого вещества.
[0251] LCMS 276,2 [0M+H]+
[0252] 1H NMR (400MHz, DMSO-d6) δ 8,63-8,51 (m, 2 H), 7,67 (d, J=4,9 Hz, 1 H), 7,33 (d, J=8,3 Hz, 2 H), 7,20 (d, J=8,3 Hz, 2 H), 3,96-3,78 (m, 3 H), 3,15 (dd, J=6,6, 9,0 Hz, 2 H), 2,90-2,79 (m, 2 H).
[0253] Соединение 10c. При перемешивании к раствору метил-3-(4-хлорфенетил)изоникотината (0,105 г, 0,38 ммоля, 1,0 экв.) в THF (5 мл) и воде (5 мл) добавляли LiOH.H2O (0,024 г, 0,0,57 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и 1H NMR спектроскопии. Реакционную смесь концентрировали и разбавляли водой (10 мл) и промывали этилацетатом (10 мл×2). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали (E)-3-(4-хлорстирил)изоникотиновую кислоту (количественный выход) в виде белого твердого вещества.
[0254] LCMS 262,1 [0M+H]+
[0255] Соединение 10. При перемешивании к раствору (E)-3-(4-хлорстирил)изоникотиновой кислоты (0,07 г, 0,268 ммоля, 1,0 экв.) в DMF (3 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,06 г, 0,268 ммоля, 1,0 экв.), HATU (0,152 г, 0,402 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 10 мин. Добавляли DIPEA (0,2 мл) и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл×2). Объединенные органические экстракты промывали водой (20 мл×4), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (5% MeOH в DCM в качестве элюента) и получали (S)-3-(4-хлорфенетил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид (0,015 г, 13% выход) в виде почти белого твердого вещества.
[0256] LCMS 433,2 [0M+H]+
[0257] 1H NM (DMSO-d6,400MHz): δ 9,01 (br. s., 1 H), 8,57 (d, J=4,9 Hz, 1 H), 8,53 (s, 1 H), 7,46 (d, J=4,9 Hz, 1 H), 7,30 (q, J=8,3 Hz, 4 H), 5,13 (d, J=9,8 Hz, 1 H), 4,32 (br. s., 1 H), 4,12-4,22 (m, 2 H), 4,10 (br. s., 1 H), 2,97-3,13 (m, 2 H), 2,84-2,95 (m, 2 H), 2,81 (br. s., 2 H).
Пример S11
Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-фторфенетил)изоникотинамида
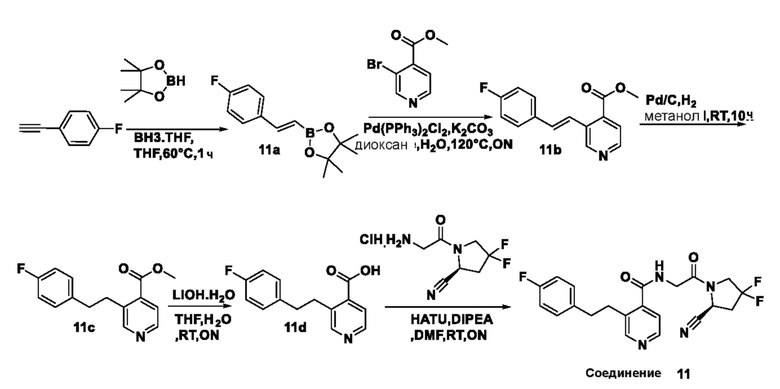
[0258] Соединение 11a. При перемешивании к раствору 1-этинил-4-фторбензола (500 мг, 4,16 ммоля, 1,0 экв.) в THF (10 мл) добавляли 4,4,5,5-тетраметил-1,3,2-диоксаборолан (800 мг, 6,25 ммоля, 1,5 экв.), BH3.THF (2 мл, 2,08 ммоля, 0,5 экв.) и продували азотом при КТ. Смесь нагревали в течение 1 ч при 60°. Образование продукта подтверждали с помощью TLC. После завершения реакции смесь разбавляли раствором NH4Cl (20 мл) и экстрагировали этилацетатом (20 мл×2). Объединенные органические экстракты промывали водой (20 мл×4), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (10% этилацетат) и получали (E)-2-(4-фторстирил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (350 мг, 13% выход) в виде желтого масла.
[0259] LCMS 249,2 [0M+H]+
[0260] 1H NMR (DMSO-d6, 400 MHz) δ 7,65 (dd, J=8,3, 5,7 Hz, 2 H), 7,29 (d, J=18,4 Hz, 1 H), 7,20 (t, J=8,8 Hz, 2 H), 6,09 (d, J=18,4 Hz, 1 H), 1,13-1,32 част./млн (m, 12 H).
[0261] Соединение 11b. К раствору метил-3-бромизоникотината (0,2 г, 0,921 ммоля, 1,0 экв.) в диоксане (4 мл) добавляли (E)-2-(4-фторстирил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,3 г, 1,38 ммоля, 1,5 экв.), K2CO3 (0,384 г, 2,78 ммоля, 3,0 экв.) и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,032 г, 0,046 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 120°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции смесь фильтровали через слой целита, промывали этилацетатом (100 мл). Фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (0-20% этилацетата в гексане в качестве элюента) и получали метил-(E)-3-(4-фторстирил)изоникотинат (0,05 г, 47,43% выход) в виде желтого твердого вещества.
[0262] LCMS 258,0 [0M+H]+
[0263] 1H NMR (DMSO-d6, 400 MHz) δ 9,07 (s, 1 H), 8,59 (d, J=4,9 Hz, 1 H), 7,48-7,76 (m, 4 H), 7,33 (d, J=16,6 Hz, 1 H), 7,22 (t, J=8,6 Hz, 2 H), 3,87 част./млн (s, 3 H).
[0264] Соединение 11c. К раствору метил-(E)-3-(4-фторстирил)изоникотинат (0,10 г, 0,389 ммоля, 1,0 экв.) в метаноле (15 мл) продували газообразным N2 в течение 10 мин, затем добавляли Pd/C (0,030 г). Затем полученную реакционную смесь продували газообразным H2 в течение 10 ч. Образование продукта подтверждали с помощью LCMS. После завершения реакции смесь фильтровали через слой целита, промывали метанолом (30 мл). Фильтрат концентрировали при пониженном давлении и получали метил-3-(4-фторфенетил)изоникотинат (0,1 г, 86,77% выход) в виде белого твердого вещества.
[0265] LCMS 260,2 [0M+H]+
[0266] 1H NMR (DMSO-d6, 400MHz) δ 8,57 (br. s., 2 H), 7,65 (d, J=4,9 Hz, 1 H), 7,16-7,29 (m, 2 H), 7,00-7,12 (m, 2 H), 3,88 (s, 3 H), 3,06-3,19 (m, 2 H), 2,77-2,88 част./млн (m, 2 H).
[0267] Соединение 11d. При перемешивании к раствору метил-3-(4-фторфенетил)изоникотинат (0,100 г, 0,38 ммоля, 1,0 экв.) в THF (5 мл) и воде (5 мл) добавляли LiOH.H2O (0,024 г, 0,583 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и 1H NMR спектроскопии. Реакционную смесь концентрировали и разбавляли водой (10 мл) и промывали этилацетатом (10 мл×2). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали 3-(4-фторфенетил)изоникотиновую кислоту (количественный выход) в виде белого твердого вещества.
[0268] LCMS 246,2 [0M+H]+
[0269] 1H NMR (DMSO-d6, 400MHz) δ 8,21 (s, 1 H), 8,13 (s, 1 H), 7,13-7,29 (m, 3 H), 7,06 (t, J=8,8 Hz, 2 H), 2,90-3,03 (m, 2 H), 2,72-2,87 част./млн (m, 2 H).
[0270] Соединение 11. При перемешивании к раствору (E)-3-(4-фторстирил)изоникотиновой кислоты (0,09 г, 0,367 ммоля, 1,0 экв.) в DMF (3 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,08 г, 0,367 ммоля, 1,0 экв.), HATU (0,209 г, 0,550 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 10 мин. DIPEA (0,2 мл) добавляли и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл×2). Объединенные органические экстракты промывали водой (20 мл×4), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (5% MeOH в DCM в качестве элюента) и получали (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-фторфенетил)изоникотинамид (0,060 г, 13% выход) в виде почти белого твердого вещества.
[0271] LCMS 417,3 [0M+H]+
[0272] 1H NMR (DMSO-d6, 400MHz) δ 8,94 (br. s., 1 H), 8,35-8,55 (m, 2 H), 7,34 (d, J=4,4 Hz, 1 H), 7,16-7,31 (m, 2 H), 7,07 (t, J=8,8 Hz, 2 H), 5,13 (d, J=7,8 Hz, 1 H), 4,31 (br. s., 1 H), 3,95-4,20 (m, 3 H), 3,01 (d, J=9,3 Hz, 2 H), 2,87 (d, J=8,8 Hz, 2 H), 2,80 част./млн (br. s., 2 H).
Пример S12
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-фторстирил)изоникотинамида

[0273] Соединение 12a. При перемешивании к раствору 1-этинил-4-фторбензола (500 мг, 4,16 ммоля, 1,0 экв.) в THF (10 мл) добавляли 4,4,5,5-тетраметил-1,3,2-диоксаборолан (800 мг, 6,25 ммоля, 1,5 экв.), BH3.THF (2 мл, 2,08 ммоля, 0,5 экв.) и продували азотом при КТ. Смесь нагревали в течение 1 ч при 60°. Образование продукта подтверждали с помощью TLC. После завершения реакции смесь разбавляли раствором NH4Cl (20 мл) и экстрагировали этилацетатом (20 мл×2). Объединенные органические экстракты промывали водой (20 мл×4), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (10% этилацетат) и получали искомый (E)-2-(4-фторстирил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (350 мг, 13% выход) в виде желтого масла.
[0274] LCMS 249,2 [0M+H]+
[0275] 1H NMR (DMSO-d6, 400MHz) δ 7,65 (dd, J=8,3, 5,7 Hz, 2 H), 7,29 (d, J=18,4 Hz, 1 H), 7,20 (t, J=8,8 Hz, 2 H), 6,09 (d, J=18,4 Hz, 1 H), 1,13-1,32 част./млн (m, 12 H).
[0276] Соединение 12b. К раствору метил-3-бромизоникотината (0,2 г, 0,921 ммоля, 1,0 экв.) в диоксане (4 мл) добавляли (E)-2-(4-фторстирил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,3 г, 1,38 ммоля, 1,5 экв.), K2CO3 (0,384 г, 2,78 ммоля, 3,0 экв.) и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,032 г, 0,046 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 120°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции смесь фильтровали через слой целита, промывали этилацетатом (100 мл). Фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (0-20% этилацетата в гексане в качестве элюента) и получали метил-(E)-3-(4-фторстирил)изоникотинат (0,05 г, 47,43% выход) в виде желтого твердого вещества.
[0277] LCMS 258,0 [0M+H]+
[0278] 1H NMR (DMSO-d6, 400MHz) δ 9,07 (s, 1 H), 8,59 (d, J=4,9 Hz, 1 H), 7,48-7,76 (m, 4 H), 7,33 (d, J=16,6 Hz, 1 H), 7,22 (t, J=8,6 Hz, 2 H), 3,87 част./млн (s, 3 H).
[0279] Соединение 12c. При перемешивании к раствору метил-(E)-3-(4-фторстирил)изоникотината (0,300 г, 1,167 ммоля, 1,0 экв.) в THF (4 мл) и воде (4 мл) добавляли LiOH.H2O (0,074 г, 1,751 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и 1H NMR спектроскопии. Реакционную смесь концентрировали и разбавляли водой (20 мл) и промывали этилацетатом (10 мл×2). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали (E)-3-(4-фторстирил)изоникотиновую кислоту (количественный выход) в виде белого твердого вещества.
[0280] LCMS 244,0 [0M+H]+
[0281] Соединение 12. При перемешивании к раствору (E)-3-(4-фторстирил)изоникотиновой кислоты (0,1 г, 0,412 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,093 г, 0,412 ммоля, 1,0 экв.), EDCI.HCl (0,119 г, 0,618 ммоля, 1,5 экв.) и HOBt (0,084 г, 0,618 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 10 мин. Добавляли TEA (0,3 мл) и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл×2). Объединенные органические экстракты промывали водой (20 мл×4), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (5% MeOH в DCM в качестве элюента) и получали (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-фторстирил)изоникотинамид (0,1 г, 58,8% выход) в виде белого твердого вещества.
[0282] LCMS 415,2 [0M+H]+
[0283] 1H NMR (DMSO-d6, 400MHz) δ 9,13 (s, 1 H), 8,99 (t, J=5,9 Hz, 1 H), 8,53 (d, J=4,8 Hz, 1 H), 7,76 (dd, J=8,6, 5,5 Hz, 2 H), 7,64 (d, J=16,7 Hz, 1 H), 7,48 (d, J=16,7 Hz, 1 H), 7,36 (d, J=5,3 Hz, 1 H), 7,11-7,26 (m, 2 H), 5,19 (dd, J=9,4, 2,9 Hz, 1 H), 4,25-4,36 (m, 1 H), 4,00-4,24 (m, 3 H), 3,92 (d, J=5,7 Hz, 1 H), 2,78-2,97 част./млн (m, 2 H).
Пример S13
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-фторфенил)проп-1-ен-1-ил)изоникотинамида и (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-фторфенил)аллил)изоникотинамида

[0284] Соединения 13a и 14a. К раствору метил-3-бромизоникотината (1,0 г, 4,629 ммоля, 1,0 экв.) в THF (20 мл) добавляли 1-фтор-4-(проп-1-ен-2-ил)бензол (1,0 г, 6,94 ммоля, 1,5 экв.), TEA (1,4 мл, 10,18 ммоля, 2,2 экв.), затем добавляли Pd(OAc)2 (0,52 г, 0,23 ммоля, 0,05 экв.) и трифенилфосфин (0,12 г, 0,4629 ммоля, 0,1 экв.) Полученную реакционную смесь нагревали при 120°C в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь фильтровали через слой целита, промывали этилацетатом (100 мл). Фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (0-20% этилацетата в гексане в качестве элюента) и получали смесь метил-(E)-3-(2-(4-фторфенил)проп-1-ен-1-ил)изоникотината и метил-3-(2-(4-фторфенил)аллил)изоникотината (0,05 г, 4%) в виде желтого масла.
[0285] LCMS 272,1 [0M+H]+
[0286] Соединения 13b и 14b. При перемешивании к раствору смеси метил-(E)-3-(2-(4-фторфенил)проп-1-ен-1-ил)изоникотината и метил-3-(2-(4-фторфенил)аллил)изоникотината (0,067 г, 0,24 ммоля, 1,0 экв.) в THF (5 мл) и воде (5 мл) добавляли LiOH.H2O (0,016 г, 0,371 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS. Реакционную смесь концентрировали и разбавляли водой (10 мл) и промывали этилацетатом (5 мл×2). Водный слой подкисляли с помощью 6 н. HCl (pH~5-6), твердый осадок отфильтровывали и сушили в вакууме и получали смесь (E)-3-(2-(4-фторфенил)проп-1-ен-1-ил)изоникотиновой кислоты и 3-(2-(4-фторфенил)аллил)изоникотиновой кислоты (45 мг,73%) в виде почти белого твердого вещества.
[0287] LCMS 258,0 [0M+H]+
[0288] Соединения 13 и 14. При перемешивании к раствору (E)-3-(2-(4-фторфенил)проп-1-ен-1-ил)изоникотиновой кислоты и 3-(2-(4-фторфенил)аллил)изоникотиновой кислоты (0,045 г, 0,175 ммоля, 1,0 экв.) в DMF (2 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,040 г, 0,175 ммоля, 1,0 экв.), EDCI.HCl (0,05 г, 0,263 ммоля, 1,5 экв.) и HOBt (0,036 г, 0,263 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 10 мин. Добавляли TEA (0,2 мл) и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (10 мл×2). Объединенные органические экстракты промывали водой (10 мл×4), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (5% MeOH в DCM в качестве элюента) и получали смесь соединений, которую дополнительно очищали с помощью HPLC с обращенной фазой и получали (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-фторфенил)проп-1-ен-1-ил)изоникотинамид (0,022 г) в виде желтого твердого вещества и (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-фторфенил)аллил)изоникотинамид (0,008 г) в виде белого твердого вещества.
[0289] LCMS 429,2 [0M+H]+
[0290] 1H NMR (соединение 13) (400MHz, DMSO-d6) δ 8,92 (br. s., 1 H), 8,73 (br. s., 1 H), 8,64 (br. s., 1 H), 7,74-7,57 (m, 2 H), 7,55 (d, J=4,8 Hz, 1 H), 7,23 (t, J=8,8 Hz, 2 H), 7,07-6,97 (m, 1 H), 5,12 (d, J=9,2 Hz, 1 H), 4,30 (d, J=11,0 Hz, 1 H), 4,15 (d, J=4,4 Hz, 1 H), 4,08 (d, J=9,6 Hz, 2 H), 3,01-2,73 (m, 3 H), 2,17 (s, 3 H).
[0291] 1H NMR (соединение 14) (400MHz, DMSO-d6) δ 8,89 (br. s., 1 H), 8,49 (d, J=4,8 Hz, 1 H), 8,43 (s, 1 H), 7,54 (dd, J=5,5, 8,6 Hz, 2 H), 7,35 (d, J=5,3 Hz, 1 H), 7,12 (t, J=8,8 Hz, 2 H), 5,47 (s, 1 H), 5,13 (d, J=6,1 Hz, 1 H), 4,99 (s, 1 H), 4,27 (d, J=11,8 Hz, 1 H), 4,20-3,95 (m, 3 H), 3,17 (d, J=5,7 Hz, 1 H), 2,90 (br. s., 1 H), 2,82 (d, J=17,5 Hz, 1 H), 2,67 (br. s., 1 H).
Пример S14
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-метоксистирил)изоникотинамида

[0292] Стадия 1: Синтез (E)-2-(4-метоксистирил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана. При перемешивании к раствору 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (1,0 г, 4,16 ммоля, 1,1 экв.) в сухом THF (20 мл) добавляли CuCl (3,7 мг, 0,038 ммоля, 0,01 экв.), Xanthphos (22 мг, 0,038 ммоля, 0,01 экв.) и NaOtBu (0,435 г, 4,536 ммоля, 1,2 экв.) при КТ. Смесь перемешивали при КТ в течение 30 мин. 1-Этинил-4-метоксибензол (0,5 г, 3,78 ммоля, 1,0 экв.), растворенный в THF (5 мл) добавляли к указанной выше реакционной смеси и перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью TLC. После завершения реакции смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл×2). Объединенные органические экстракты промывали водой (20 мл×2), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (5% EtOAc в гексане в качестве элюента) и получали (E)-2-(4-метоксистирил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,8 г, 86,5% выход) в виде желтого масла.
[0293] LCMS 261,2 [0M+H]+
[0294] 1H NMR (DMSO-d6, 400MHz) δ 7,46-7,60 (m, J=8,8 Hz, 2 H), 7,25 (d, J=18,4 Hz, 1 H), 6,84 -6,97 (m, J=8,8 Hz, 2 H), 5,96 (d, J=18,4 Hz, 1 H), 3,77 (s, 3 H), 1,23 (s, 12 H).
[0295] Стадия 2: Синтез метил-(E)-3-(4-метоксистирил)изоникотината. К раствору метил-3-бромизоникотината (0,2 г, 0,926 ммоля, 1,0 экв.) в диоксане (20 мл) и воде (1 мл) добавляли (E)-2-(4-метоксистирил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,361 г, 1,39 ммоля, 1,5 экв.), Na2CO3 (0,2 г, 1,85 ммоля, 2,0 экв.) и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,033 г, 0,0463 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции смесь фильтровали через целит®, промывали этилацетатом (100 мл). Фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью флэш-хроматографии (0-20% этилацетата в гексане в качестве элюента) и получали метил-(E)-3-(4-метоксистирил)изоникотинат (0,25 г, 99% выход) в виде желтого твердого вещества.
[0296] LCMS 270,0 [0M+H]+
[0297] 1H NMR (DMSO-d6,400MHz) δ 9,10 (s, 1 H), 8,59 (d, J=4,8 Hz, 1 H), 7,68 (d, J=4,8 Hz, 1 H), 7,45-7,60 (m, 3 H), 7,33 (d, J=16,2 Hz, 1 H), 6,99 (d, J=8,3 Hz, 2 H), 3,91 (s, 2 H), 3,67-3,83 (m, 3 H).
[0298] Стадия 3: Синтез (E)-3-(4-метоксистирил)изоникотиновой кислоты. При перемешивании к раствору метил-(E)-3-(4-метоксистирил)изоникотината (0,250 г, 0,929 ммоля, 1,0 экв.) в THF (10 мл) и воде (10 мл) добавляли LiOH.H2O (0,056 г, 1,39 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и 1H NMR спектроскопии. Реакционную смесь концентрировали и разбавляли водой (20 мл) и промывали этилацетатом (10 мл×2). Водный слой подкисляли с помощью 6 н. HCl (pH ~ 5-6), образовавшийся твердый осадок отфильтровывали и сушили в вакууме и получали (E)-3-(4-метоксистирил)изоникотиновую кислоту (0,15 г, 63,5%) в виде желтого твердого вещества.
[0299] LCMS 255,9 [0M+H]+
[0300] Стадия 4: Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-метоксистирил)изоникотинамида. При перемешивании к раствору (E)-3-(4-метоксистирил)изоникотиновой кислоты (0,12 г, 0,47 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,158 г, 0,705 ммоля, 1,5 экв.), HATU (0,268 г, 0,705 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 10 мин. DIPEA (0,4 мл) добавляли и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл×2). Объединенные органические экстракты промывали водой (20 мл×4), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (5% MeOH в DCM в качестве элюента), затем с помощью HPLC с обращенной фазой и получали (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-метоксистирил)изоникотинамид (0,06 г, 30% выход) в виде белого твердого вещества.
[0301] LCMS 427,3 [0M+H]+
[0302] 1H NMR (DMSO-d6, 400MHz) δ 9,11 (s, 1 H), 8,97 (t, J=5,9 Hz, 1 H), 8,49 (d, J=4,8 Hz, 1 H), 7,57-7,68 (m, J=8,8 Hz, 2 H), 7,51 (d, J=16,2 Hz, 1 H), 7,41 (d, J=16,7 Hz, 1 H), 7,33 (d, J=5,3 Hz, 1 H), 6,85-7,03 (m, J=8,8 Hz, 2 H), 5,18 (dd, J=9,2, 2,2 Hz, 1 H), 4,27-4,38 (m, 1 H), 3,99-4,21 (m, 3 H), 3,66-3,87 (m, 3 H), 2,75-2,98 (m, 2 H).
Пример S15
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-(трифторметокси)фенил)проп-1-ен-1-ил)изоникотинамида

[0303] Стадия 1: Синтез (E)-4,4,5,5-тетраметил-2-(2-(4-(трифторметокси)фенил)проп-1-ен-1-ил)-1,3,2-диоксаборолана. При перемешивании к раствору 1-этинил-4-(трифторметокси)бензола (1,0 г, 5,37 ммоля, 1,0 экв.) в THF (10 мл) добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (1,49 г, 5,90 ммоля, 1,1 экв.), CuCl (0,005 г, 0,050 ммоля, 0,01 экв.), Xanthphos ( 0,030 г, 0,050 ммоля, 0,01 экв.), KOtBu(0,71 г, 0,63 ммоля, 1,2 экв.), затем добавляли метилйодид (1,3 мл, 21,2 ммоля, 4,0 экв.). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью TLC. После завершения реакции реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (20 мл×4), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (0-10% этилацетата в гексане) и получали (E)-4,4,5,5-тетраметил-2-(2-(4-(трифторметокси)фенил)проп-1-ен-1-ил)-1,3,2-диоксаборолан (0,700 г, 39% выход) в виде желтого масла.
[0304] LCMS 328,1 [0M+H]+
[0305] 1H NMR (400 MHz, хлороформ-d) δ 7,40-7,58 (m, 2 H) 7,16 (m, J=8,33 Hz, 2 H) 5,73 (d, J=0,88 Hz, 1 H) 2,29-2,44 (m, 3 H) 1,14-1,42 (m, 12 H).
[0306] Стадия 2: Синтез метил-(E)-3-(2-(4-(трифторметокси)фенил)проп-1-ен-1-ил)изоникотината. К раствору метил-3-бромизоникотината (0,220 г, 1,018 ммоля, 1,0 экв.) в смеси диоксан:вода (4:2 мл) добавляли (E)-4,4,5,5-тетраметил-2-(2-(4-(трифторметокси)фенил)проп-1-ен-1-ил)-1,3,2-диоксаборолан (0,400 г, 1,22 ммоля, 1,2 экв.), K2CO3 (0,284 г, 2,037 ммоля, 2,0 экв.) и смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh2)Cl2 (0,035 г, 0,050 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 120°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции реакционную смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (50 мл×2). Объединенные органические экстракты промывали водой (10 мл×2), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-20% этилацетата в гексане в качестве элюента) и получали метил-(E)-3-(2-(4-(трифторметокси)фенил)проп-1-ен-1-ил)изоникотинат (0,110 г, 35% выход) в виде желтого твердого вещества.
[0307] LCMS 338,1 [0M+H]+
[0308] 1H NMR (400 MHz, DMSO-d6) δ 8,57-8,82 (m, 2 H) 7,64-7,78 (m, 2 H) 7,42 (d, J=7,89 Hz, 2 H) 7,16 (s, 1 H) 3,75-3,99 (m, 3 H) 2,09 (d, J=1,32 Hz, 3 H).
[0309] Стадия 3: Синтез (E)-3-(2-(4-(трифторметокси)фенил)проп-1-ен-1-ил)изоникотиновой кислоты. При перемешивании к раствору метил-(E)-3-(2-(4-(трифторметокси)фенил)проп-1-ен-1-ил)изоникотината (0,100 г, 0,296 ммоля, 1,0 экв.) в THF (4 мл) и воде (4 мл) добавляли LiOH.H2O (0,024 г, 0,593 ммоля, 2,0 экв.). Смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и 1H NMR спектроскопии. Реакционную смесь разбавляли водой (20 мл) и промывали этилацетатом (10 мл×2). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали (E)-3-(2-(4-(трифторметокси)фенил)проп-1-ен-1-ил)изоникотиновую кислоту (0,100 г, количественный выход) в виде белого твердого вещества.
[0310] LCMS 324,0 [0M+H]+
[0311] Стадия 4: Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-(трифторметокси)фенил)проп-1-ен-1-ил)изоникотинамида. При перемешивании к раствору (E)-3-(2-(4-(трифторметокси)фенил)проп-1-ен-1-ил)изоникотиновой кислоты (0,1 г, 0,308 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,069 г, 0,308 ммоля, 1,0 экв.), EDCI.HCl (0,070 г, 0,370 ммоля, 1,2 экв.) и HOBt (0,049 г, 0,370 ммоля, 1,2 экв.), затем добавляли TEA (0,2 мл). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (25 мл×2). Объединенные органические экстракты промывали водой (10 мл×4), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (5% MeOH в DCM в качестве элюента) и получали (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-(трифторметокси)фенил)проп-1-ен-1-ил)изоникотинамид (0,030 г, 20,0% выход) в виде белого твердого вещества.
[0312] LCMS 495,3 [0M+H]+
[0313] 1H NMR (400 MHz, DMSO-d6) δ 8,89 (t, J=5,92 Hz, 1 H) 8,69 (s, 1 H) 8,60 (d, J=4,82 Hz, 1 H) 7,74 (m, J=8,77 Hz, 2 H) 7,47 (d, J=4,82 Hz, 1 H) 7,39 (m, J=8,33 Hz, 2 H) 7,01-7,13 (m, 1 H) 5,13 (d, J=6,58 Hz, 1 H) 4,24-4,35 (m, 1 H) 3,92-4,20 (m, 3 H) 2,69-2,95 (m, 2 H) 2,18 (s, 3 H).
Пример S16
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-(трифторметил)фенил)проп-1-ен-1-ил)изоникотинамида

[0314] Стадия 1: Синтез (E)-4,4,5,5-тетраметил-2-(2-(4-(трифторметил)фенил)проп-1-ен-1-ил)-1,3,2-диоксаборолана. При перемешивании к раствору 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (0,818 г, 3,23 ммоля, 1,1 экв.) в сухом THF (20 мл) добавляли CuCl (3,0 мг, 0,0294 ммоля, 0,01 экв.), Xanthphos (0,017 мг, 0,0294 ммоля, 0,01 экв.) и NaOtBu (0,34 г, 3,53 ммоля, 1,2 экв.) при КТ. Смесь перемешивали при КТ в течение 30 мин. 1-Этинил-4-(трифторметил)бензол (0,5 г, 2,94 ммоля, 1,0 экв. ), растворенный в THF (5 мл), и метилйодид (0,8 мл, 11,764 ммоля, 4,0 экв.) добавляли к указанной выше реакционной смеси, полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью TLC. После завершения реакции смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл×2). Объединенные органические экстракты промывали водой (20 мл×2), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (5% EtOAc в гексане в качестве элюента) и получали (E)-4,4,5,5-тетраметил-2-(2-(4-(трифторметил)фенил)проп-1-ен-1-ил)-1,3,2-диоксаборолан (0,8 г, 87,2% выход) в виде желтого масла.
[0315] 1H NMR (DMSO-d6, 400 MHz) δ 7,81 (d, J=7,9 Hz, 2 H), 7,59-7,74 (m, 2 H), 5,75 (s, 1 H), 2,30-2,43 (m, 3 H), 1,18-1,31 (m, 12 H).
[0316] Стадия 2: Синтез метил-(E)-3-(2-(4-(трифторметил)фенил)проп-1-ен-1-ил)изоникотината. К раствору метил-3-бромизоникотината (0,1 г, 0,463 ммоля, 1,0 экв.) в диоксане (5 мл) и воде (1 мл) добавляли (E)-4,4,5,5-тетраметил-2-(2-(4-(трифторметил)фенил)проп-1-ен-1-ил)-1,3,2-диоксаборолан (0,216 г, 0,694 ммоля, 1,5 экв.), Na2CO3 (0,1 г, 0,936 ммоля, 2,0 экв.) и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,016 г, 0,0235 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции смесь фильтровали через слой целита, промывали этилацетатом (50 мл). Фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью флэш-хроматографии (0-20% этилацетата в гексане в качестве элюента) и получали метил-(E)-3-(2-(4-(трифторметил)фенил)проп-1-ен-1-ил)изоникотинат (0,06 г, 40,26% выход) в виде желтого масла.
[0317] LCMS 322,2 [0M+H]+
[0318] 1H NMR (DMSO-d6, 400MHz) δ 8,63-8,73 (m, 2 H), 7,71-7,84 (m, 5 H), 7,25 (s, 1 H), 3,79-3,95(m, 3 H), 2,06-2,20 (m, 3 H)
[0319] Стадия 3: Синтез (E)-3-(2-(4-(трифторметил)фенил)проп-1-ен-1-ил)изоникотиновой кислоты. При перемешивании к раствору метил-(E)-3-(2-(4-(трифторметил)фенил)проп-1-ен-1-ил)изоникотината (0,06 г, 0,186 ммоля, 1,0 экв.) в THF (2 мл) и воде (2 мл) добавляли LiOH.H2O (0,012 г, 0,28 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и 1H NMR спектроскопии. Реакционную смесь концентрировали и разбавляли водой (20 мл) и промывали этилацетатом (10 мл×2). Водный слой подкисляли с помощью 6 н. HCl (pH ~ 5-6), твердый осадок отфильтровывали и сушили в вакууме и получали (E)-3-(2-(4-(трифторметил)фенил)проп-1-ен-1-ил)изоникотиновую кислоту (0,035 г, 61,4%) в виде почти белого твердого вещества.
[0320] LCMS 308,1 [0M+H]+
[0321] Стадия 4: Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-(трифторметил)фенил)проп-1-ен-1-ил)изоникотинамида. При перемешивании к раствору (E)-3-(2-(4-(трифторметил)фенил)проп-1-ен-1-ил)изоникотиновой кислоты (0,035 г, 0,114 ммоля, 1,0 экв.) в DMF (3 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,026 г, 0,114 ммоля, 1,5 экв.), EDCI.HCl (0,033 г, 0,171 ммоля, 1,5 экв.) и HOBt (0,023 г, 0,171 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 10 мин. TEA (0,1 мл) добавляли и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (10 мл×2). Объединенные органические экстракты промывали водой (10 мл×4), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью HPLC с обращенной фазой и получали (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-(трифторметил)фенил)проп-1-ен-1-ил)изоникотинамид (0,007 г, 12,86% выход) в виде белого твердого вещества.
[0322] LCMS 479,2[0M+H]+
[0323] 1H NMR (DMSO-d6, 400MHz) δ 8,91 (d, J=5,7 Hz, 1 H), 8,72 (s, 1 H), 8,63 (d, J=4,8 Hz, 1 H), 7,84 (d, J=8,3 Hz, 2 H), 7,76 (d, J=8,3 Hz, 2 H), 7,50 (d, J=4,8 Hz, 1 H), 7,13-7,20 (m, 1 H), 5,14 (d, J=7,5 Hz, 1 H), 4,23-4,34 (m, 1 H), 4,03-4,23 (m, 2 H), 2,68-2,94 (m, 3 H), 2,21 (s, 3 H).
Пример S17
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-(трифторметил)стирил)изоникотинамида
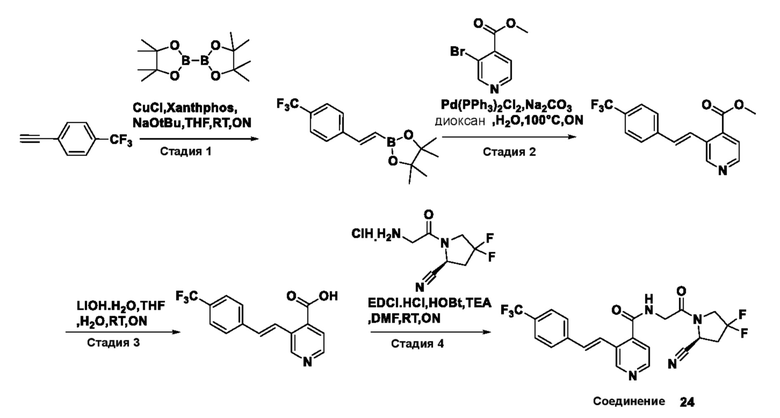
[0324] Стадия 1: Синтез (E)-4,4,5,5-тетраметил-2-(4-(трифторметил)стирил)-1,3,2-диоксаборолана. При перемешивании к раствору 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (0,818 г, 3,23 ммоля, 1,1 экв.) в сухом THF (20 мл) добавляли CuCl (3,0 мг, 0,0294 ммоля, 0,01 экв.), Xanthphos (0,017 г, 0,0294 ммоля, 0,01 экв.) и NaOtBu (0,34 г, 3,53 ммоля, 1,2 экв.) при КТ. Смесь перемешивали при КТ в течение 30 мин. 1-Этинил-4-(трифторметил)бензол (0,5 г, 2,94 ммоля, 1,0 экв. ) в THF (5 мл) добавляли к указанной выше реакционной смеси и полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью TLC. После завершения реакции смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл×2). Объединенные органические экстракты промывали водой (20 мл×2), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (5% EtOAc в гексане в качестве элюента) и получали (E)-4,4,5,5-тетраметил-2-(4-(трифторметил)стирил)-1,3,2-диоксаборолан (0,6 г, 68,5% выход) в виде коричневого твердого вещества.
[0325] 1H NMR (DMSO-d6, 400MHz) δ 7,81 (d, J=9,2 Hz, 2 H), 7,66-7,79 (m, 2 H), 7,35 (s, 1 H), 7,40 (s, 1 H), 6,32 (d, J=18,4 Hz, 1 H), 1,22-1,35 (m, 10 H), 1,13-1,22 (m, 2 H).
[0326] Стадия 2: Синтез метил-(E)-3-(4-(трифторметил)стирил)изоникотината. К раствору метил-3-бромизоникотината (0,3 г, 1,39 ммоля, 1,0 экв.) в диоксане (20 мл) и воде (2 мл) добавляли (E)-4,4,5,5-тетраметил-2-(4-(трифторметил)стирил)-1,3,2-диоксаборолан (0,496 г, 1,67 ммоля, 1,2 экв.), Na2CO3 (0,3 г, 2,78 ммоля, 2,0 экв.) и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,049 г, 0,0694 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции смесь фильтровали через слой целита, промывали этилацетатом (100 мл). Фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью флэш-хроматографии (0-20% этилацетата в гексане в качестве элюента) и получали метил-(E)-3-(4-(трифторметил)стирил)изоникотинат (0,4 г, 93,9% выход) в виде желтого масла.
[0327] LCMS 308,1 [0M+H]+
[0328] 1H NMR (DMSO-d6, 400MHz) δ 9,15 (s, 1 H), 8,68 (d, J=4,8 Hz, 1 H), 7,71-7,87 (m, 5 H), 7,43 (s, 1 H), 7,48 (s, 1 H), 3,83-3,96 (m, 3 H).
[0329] Стадия 3: Синтез (E)-3-(4-(трифторметил)стирил)изоникотиновой кислоты. При перемешивании к раствору метил-(E)-3-(4-(трифторметил)стирил)изоникотината (0,4 г, 1,303 ммоля, 1,0 экв.) в THF (5 мл) и воде (5 мл) добавляли LiOH.H2O (0,092 г, 2,21 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и 1H NMR спектроскопии. Реакционную смесь концентрировали и разбавляли водой (20 мл) и промывали этилацетатом (10 мл×2). Водный слой подкисляли с помощью 6 н. HCl (pH ~ 5-6), твердый осадок отфильтровывали и сушили в вакууме и получали (E)-3-(4-(трифторметил)стирил)изоникотиновую кислоту (0,355 г, 93%) в виде желтого твердого вещества.
[0330] LCMS 294,1 [0M+H]+
[0331] Стадия 4: Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-(трифторметил)стирил)изоникотинамида. При перемешивании к раствору (E)-3-(4-(трифторметил)стирил)изоникотиновой кислоты (0,07 г, 0,239 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,54 г, 0,239 ммоля, 1,5 экв.), HOBt (0,05 г, 0,359 ммоля, 1,5 экв.) и EDCI.HCl (0,07 г, 0,359 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 10 мин. TEA (0,2 мл) добавляли и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл×2). Объединенный органический слой промывали водой (20 мл×4), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью флэш-хроматографии (5% MeOH в DCM в качестве элюента) и получали (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-(трифторметил)стирил)изоникотинамид (0,05 г, 45% выход) в виде почти белого твердого вещества.
[0332] LCMS 465,2 [0M+H]+
[0333] 1H NMR (DMSO-d6, 400MHz) δ (s, 1 H), 8,92-9,05 (m, 1 H), 8,57 (d, J=4,8 Hz, 1 H), 7,82-7,97 (m, 3 H), 7,75 (d, J=8,3 Hz, 2 H), 7,59 (d, J=17,1 Hz, 1 H), 7,39 (d, J=5,3 Hz, 1 H), 5,21 (d, J=6,1 Hz, 1 H), 4,28-4,37 (m, 1 H), 4,03-4,27 (m, 2 H), 2,73-3,02 (m, 3 H).
Пример S18
Синтез (S, E)-3-(2-(4-хлорфенил)проп-1-ен-1-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамида

[0334] Стадия 1: Синтез (E)-2-(2-(4-хлорфенил)проп-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана. При перемешивании к раствору 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (1,4 г, 5,514 ммоля, 1,5 экв.) в сухом THF (10 мл) добавляли CuCl (18 мг, 0,183 ммоля, 0,05 экв.), Xanthphos (0,106 г, 0,183 ммоля, 0,05 экв.) и KOtBu (0,5 г, 4,4 ммоля, 1,2 экв.) при КТ. Реакционную смесь перемешивали при КТ в течение 30 мин. Добавляли 1-хлор-4-этинилбензол (0,5 г, 3,78 ммоля, 1,0 экв.) в THF (5 мл), затем добавляли метилйодид (1 мл, 15,12 ммоля, 4,0 экв.). Полученную реакционную смесь нагревали при 60°C в течение ночи. Образование продукта подтверждали с помощью TLC. После завершения реакции смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл×2). Объединенные органические экстракты промывали водой (20 мл×2), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (2% EtOAc в гексане в качестве элюента) и получали (E)-2-(2-(4-хлорфенил)проп-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,57 г, 33,81% выход) в виде белого твердого вещества.
[0335] LCMS 279,0 [0M+H]+
[0336] 1H NMR (DMSO-d6, 400MHz) δ 7,46-7,60 (m, J=8,8 Hz, 2 H), 7,35-7,45 (m, J=8,8 Hz, 2 H), 5,67 (s, 1 H), 2,32 (s, 3 H), 1,06-1,32 част./млн (m, 12 H).
[0337] Стадия 2: Синтез метил-(E)-3-(2-(4-хлорфенил)проп-1-ен-1-ил)изоникотината. К раствору метил-3-бромизоникотината (0,15 г, 0,694 ммоля, 1,0 экв.) в диоксане (10 мл) и воде (0,5 мл) добавляли (E)-2-(2-(4-хлорфенил)проп-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,29 г, 1,04 ммоля, 1,5 экв.), Na2CO3 (0,15 г, 1,38 ммоля, 2,0 экв.) и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,024 г, 0,035 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции смесь фильтровали через целит®, промывали этилацетатом (50 мл). Фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью флэш-хроматографии (0-20% этилацетата в гексане в качестве элюента) и получали метил-(E)-3-(2-(4-хлорфенил)проп-1-ен-1-ил)изоникотинат (0,050 г, 25% выход) в виде желтого масла.
[0338] LCMS 288,2 [0M+H]+
[0339] Стадия 3: Синтез (E)-3-(2-(4-хлорфенил)проп-1-ен-1-ил)изоникотиновой кислоты. При перемешивании к раствору метил-(E)-3-(2-(4-хлорфенил)проп-1-ен-1-ил)изоникотината (0,050 г, 0,174 ммоля, 1,0 экв.) в THF (2 мл) и воде (2 мл) добавляли LiOH.H2O (0,011 г, 0,26 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и 1H NMR спектроскопии. Реакционную смесь концентрировали и разбавляли водой (4 мл) и промывали этилацетатом (2 мл×2). Водный слой подкисляли с помощью 6 н. HCl (pH ~ 5-6), образовавшийся твердый осадок отфильтровывали и сушили в вакууме и получали (E)-3-(2-(4-хлорфенил)проп-1-ен-1-ил)изоникотиновую кислоту (0,04 г, 84,21%) в виде желтого твердого вещества.
[0340] LCMS 274,2 [0M+H]+
[0341] 1H NMR (DMSO-d6, 400MHz) δ 13,61 (br. s., 1 H), 8,57-8,70 (m, 2 H), 7,77 (d, J=5,3 Hz, 1 H), 7,55-7,67 (m, J=8,3 Hz, 2 H), 7,41-7,52 (m, J=8,8 Hz, 2 H), 7,18 (s, 1 H), 2,08 (s, 3 H).
[0342] Стадия 4: (S, E)-3-(2-(4-хлорфенил)проп-1-ен-1-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид. При перемешивании к раствору (E)-3-(2-(4-хлорфенил)проп-1-ен-1-ил)изоникотиновой кислоты (0,04 г, 0,146 ммоля, 1,0 экв.) в DMF (2 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,05 г, 0,219 ммоля, 1,5 экв.), HATU (0,09 г, 0,219 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 10 мин. Добавляли DIPEA (0,1 мл) и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (10 мл×2). Объединенные органические экстракты промывали водой (5 мл×4), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью HPLC с обращенной фазой и получали (S, E)-3-(2-(4-хлорфенил)проп-1-ен-1-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид (0,008 г, 13,84% выход) в виде белого твердого вещества.
[0343] LCMS 445,2 [0M+H]+
[0344] 1H NMR (DMSO-d6, 400MHz) δ 8,88 (t, J=5,9 Hz, 1 H), 8,68 (s, 1 H), 8,60 (d, J=4,8 Hz, 1 H), 7,64 (d, J=8,3 Hz, 2 H), 7,32-7,55 (m, 3 H), 7,10 (s, 1 H), 5,06-5,19 (m, 1 H), 4,28 (br. s., 1 H), 3,95-4,17 (m, 2 H), 2,91 (br. s., 2 H), 2,82 (d, J=15,8 Hz, 2 H), 2,17 (s, 3 H).
Пример S19
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-метоксифенил)проп-1-ен-1-ил)изоникотинамида

[0345] Стадия 1: Синтез (E)-2-(2-(4-метоксифенил)проп-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана. При перемешивании к раствору 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (1,4 г, 4,16 ммоля, 1,5 экв.) в сухом THF (10 мл) добавляли CuCl (0,019 г, 0,189 ммоля, 0,05 экв.), Xanthphos (0,109 г, 0,189 ммоля, 0,05 экв.) и KOtBu (0,509 г, 4,536 ммоля, 1,2 экв.) при КТ. Смесь перемешивали при КТ в течение 30 мин. 1 Добавляли -этинил-4-метоксибензол (0,5 г, 3,78 ммоля, 1,0 экв.) в THF (5 мл), затем метилйодид (1 мл, 15,12 ммоля, 4,0 экв.) добавляли к указанной выше реакционной смеси, полученную реакционную смесь нагревали при 60°C в течение ночи. Образование продукта подтверждали с помощью TLC. После завершения реакции смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл×2). Объединенные органические экстракты промывали водой (20 мл×2), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (2% EtOAc в гексане в качестве элюента) и получали (E)-2-(2-(4-метоксифенил)проп-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,35 г, 33,81% выход) в виде белого твердого вещества.
[0346] LCMS 261,2 [0M+H]+
[0347] 1H NMR (DMSO-d6, 400MHz) δ7,38-7,57 (m, J=8,8 Hz, 2 H), 6,76-6,94 (m, J=8,8 Hz, 2 H), 5,58 (s, 1 H), 3,67-3,80 (m, 3 H), 3,33 (s, 3 H), 1,17-1,32 (m, 12 H).
[0348] Стадия 2: Синтез метил-(E)-3-(2-(4-метоксифенил)проп-1-ен-1-ил)изоникотината. К раствору метил-3-бромизоникотината (0,15 г, 0,694 ммоля, 1,0 экв.) в диоксане (10 мл) и воде (0,5 мл) добавляли (E)-2-(2-(4-метоксифенил)проп-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,285 г, 1,04 ммоля, 1,5 экв.), K2CO3 (0,2 г, 1,85 ммоля, 2,0 экв.) и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,024 г, 0,035 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции смесь фильтровали через целит®, промывали этилацетатом (50 мл). Фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью флэш-хроматографии (0-20% этилацетата в гексане в качестве элюента) и получали метил-(E)-3-(2-(4-метоксифенил)проп-1-ен-1-ил)изоникотинат (0,150 г, 76,53% выход) в виде желтого масла.
[0349] LCMS 284,1 [0M+H]+
[0350] Стадия 3: Синтез (E)-3-(2-(4-метоксифенил)проп-1-ен-1-ил)изоникотиновой кислоты. При перемешивании к раствору метил-(E)-3-(2-(4-метоксифенил)проп-1-ен-1-ил)изоникотината (0,250 г, 0,929 ммоля, 1,0 экв.) в THF (10 мл) и воде (10 мл) добавляли LiOH.H2O (0,056 г, 1,39 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и 1H NMR спектроскопии. Реакционную смесь концентрировали и разбавляли водой (20 мл) и промывали этилацетатом (10 мл×2). Водный слой подкисляли с помощью 6 н. HCl (pH ~ 5-6), образовавшийся твердый осадок отфильтровывали и сушили в вакууме и получали (E)-3-(2-(4-метоксифенил)проп-1-ен-1-ил)изоникотиновую кислоту (0,08 г, 38,27%) в виде желтого твердого вещества.
[0351] LCMS 270,1 [0M+H]+
[0352] 1H NMR (DMSO-d6, 400MHz) δ13,59 (br. s., 1 H), 8,51-8,67 (m, 2 H), 7,73 (d, J=4,8 Hz, 1 H), 7,42-7,57 (m, J=8,8 Hz, 2 H), 7,08 (s, 1 H), 6,87-7,04 (m, J=8,3 Hz, 2 H), 3,78 (s, 3 H), 2,07 (s, 3 H).
[0353] Стадия 4: (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-метоксифенил)проп-1-ен-1-ил)изоникотинамид. При перемешивании к раствору (E)-3-(2-(4-метоксифенил)проп-1-ен-1-ил)изоникотиновой кислоты (0,08 г, 0,297 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,1 г, 0,446 ммоля, 1,5 экв.), EDCI.HCl (0,09 г, 0,446 ммоля, 1,5 экв.) и HOBt (0,06 г, 0,446 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 10 мин. Добавляли TEA (0,2 мл) и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл×2). Объединенные органические экстракты промывали водой (20 мл×4), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью HPLC с обращенной фазой и получали (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(4-метоксифенил)проп-1-ен-1-ил)изоникотинамид (0,042 г, 32,3% выход) в виде белого твердого вещества.
[0354] LCMS 441,3 [0M+H]+
[0355] 1H NMR (DMSO-d6, 400MHz) δ 8,84 (br. s., 1 H), 8,66 (s, 1 H), 8,57 (d, J=4,8 Hz, 1 H), 7,55 (d, J=8,8 Hz, 2 H), 7,44 (d, J=5,3 Hz, 1 H), 6,81-7,08 (m, 3 H), 5,11 (d, J=9,6 Hz, 1 H), 4,27 (br. s., 1 H), 4,13 (d, J=6,1 Hz, 2 H), 3,77 (s, 3 H), 2,83 (br. s., 2 H), 2,67 (d, J=1,8 Hz, 2 H), 2,15 (s, 3 H).
Пример S20
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-(трифторметокси)стирил)изоникотинамида

[0356] Стадия 1: Синтез (E)-4,4,5,5-тетраметил-2-(4-(трифторметокси)стирил)-1,3,2-диоксаборолана. При перемешивании к раствору 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (0,750 г, 2,92 ммоля, 1,1 экв.) в сухом THF (10 мл) добавляли CuCl (0,002 г, 0,020 ммоля, 0,01 экв.), Xanthphos (0,015 г, 0,020 ммоля, 0,01 экв.) и KOtBu (0,361 г, 3,22 ммоля, 1,2 экв.) при КТ. Смесь перемешивали при КТ в течение 30 мин. 1-Этинил-4-(трифторметокси)бензол (0,5 г, 2,368 ммоля, 1,0 экв.) в THF (5 мл) добавляли к указанной выше реакционной смеси, полученную реакционную смесь нагревали при 60°C в течение ночи. Образование продукта подтверждали с помощью TLC. После завершения реакции смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл×2). Объединенные органические экстракты промывали водой (20 мл×2), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (2% EtOAc в гексане в качестве элюента) и получали (E)-4,4,5,5-тетраметил-2-(4-(трифторметокси)стирил)-1,3,2-диоксаборолан (0,35 г, 41% выход) в виде почти белого твердого вещества.
[0357] LCMS 315,0 [0M+H]+
[0358] 1H NMR (400 MHz, DMSO-d6) δ 7,73 (d, J=8,77 Hz, 1 H) 7,27-7,43 (m, 2 H) 6,18 (d, J=18,86 Hz, 1 H) 1,24 (s, 9 H).
[0359] Стадия 2: Синтез (E)-3-(4-(трифторметокси)стирил)изоникотиновой кислоты. К раствору метил-3-бромизоникотината (0,200 г, 1,62 ммоля, 1,0 экв.) в диоксане (5 мл) и воде (2 мл) добавляли (E)-4,4,5,5-тетраметил-2-(4-(трифторметокси)стирил)-1,3,2-диоксаборолан (0,350 г, 1,94 ммоля, 1,2 экв.), K2CO3 (0,256 г, 3,24 ммоля, 2,0 экв.) и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,033 г, 0,081 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 120°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции реакционную смесь разбавляли водой (10 мл). Водный слой промывали этилацетатом (5 мл×2). Водный слой отделяли и сушили вымораживанием и получали (E)-3-(4-(трифторметокси)стирил)изоникотиновую кислоту (0,150 г, 50% выход) в виде почти белого твердого вещества.
[0360] LCMS 310,1 [0M+H]+
[0361] Стадия 3: (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-(трифторметокси)стирил)изоникотинамид. При перемешивании к раствору (E)-3-(4-(трифторметокси)стирил)изоникотиновой кислоты (0,150 г, 0,483 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,108 г, 0,483 ммоля, 1,1 экв.), EDCI.HCl (0,185 г, 0,966 ммоля, 2,0 экв.) и HOBt (0,136 г, 0,966 ммоля, 2,0 экв.).Полученную реакционную смесь перемешивали при КТ в течение 10 мин. TEA (0,3 мл) добавляли и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (75 мл×2). Объединенные органические экстракты промывали водой (25 мл×4), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью HPLC с обращенной фазой и получали (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(4-(трифторметокси)стирил)изоникотинамид (0,020 г, 8% выход) в виде белого твердого вещества.
[0362] LCMS 481,2 [0M+H]+
[0363] 1H NMR (400 MHz, DMSO-d6) δ 9,15 (s, 1 H) 9,02 (t, J=5,70 Hz, 1 H) 8,54 (d, J=5,26 Hz, 1 H) 7,84 (d, J=8,33 Hz, 2 H) 7,75 (d, J=16,66 Hz, 1 H) 7,53 (d, J=16,66 Hz, 1 H) 7,30-7,46 (m, 3 H) 5,20 (dd, J=9,21, 2,63 Hz, 1 H) 4,27-4,38 (m, 1 H) 3,99-4,27 (m, 3 H) 2,78-3,07 (m, 2 H).
Пример S21
Синтез (S, E)-6-(4-хлорстирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамида

[0364] Стадия 1: Синтез (E)-6-(4-хлорстирил)хинолин-4-карбоновой кислоты. К раствору 6-бромхинолин-4-карбоновой кислоты (0,05 г, 0,199 ммоля, 1,0 экв.) в диоксане (4 мл) и воде (2 мл) добавляли (E)-2-(4-хлорстирил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,079 г,298 ммоля, 1,5 экв.), K2CO3 (0,055 г, 0,397 ммоля, 2,0 экв.) и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh2)Cl2 (0,008 г, 0,01 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции реакцию останавливали водой (10 мл) и водный слой промывали этилацетатом (10 мл). Водный слой отделяли и подкисляли с помощью 6 н. HCl (pH ~ 5-6), образовавшийся твердый осадок отфильтровывали и сушили в вакууме и получали (E)-6-(4-хлорстирил)хинолин-4-карбоновую кислоту (0,06 г, 96% выход) в виде желтого твердого вещества.
[0365] LCMS 310,1 [0M+H]+
[0366] 1H NMR (400 MHz, DMSO-d6) δ 8,98 (d, J=4,38 Hz, 1H), 8,75 (s, 1H), 8,24 (d, J=8,33 Hz, 1H), 8,10 (d, J=8,77 Hz, 1H), 7,91 (d, J=4,38 Hz, 1H), 7,75 (d, J=8,33 Hz, 2H), 7,54-7,67 (m, 1H), 7,33-7,54 (m, 3H).
[0367] Стадия 2: Синтез (S, E)-6-(4-хлорстирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамида. При перемешивании к раствору (E)-6-(4-хлорстирил)хинолин-4-карбоновой кислоты (0,06 г, 0,194 ммоля, 1,0 экв.) в DMF (3 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,065 г, 0,291 ммоля, 1,5 экв.), EDCI.HCl (0,056 г, 0,291 ммоля, 1,5 экв.) и HOBt (0,040 г, 0,291 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 10 мин. Добавляли триэтиламин (0,1 мл) и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (20 мл×2). Объединенные органические экстракты промывали водой (10 мл×4), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт обогащали с помощью флэш-хроматографии (5% метанола в DCM в качестве элюента), затем очищали с помощью HPLC с обращенной фазой и получали (S, E)-6-(4-хлорстирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид (0,020 г, 21,5% выход) в виде желтого твердого вещества.
[0368] LCMS 481,2 [0M+H]+
[0369] 1H NMR (400 MHz, DMSO-d6) δ 9,18 (br. s., 1H), 8,94 (d, J=3,95 Hz, 1H), 8,60 (s, 1H), 7,96-8,15 (m, 2H), 7,73 (d, J=8,77 Hz, 2H), 7,51-7,61 (m, 2H), 7,30-7,49 (m, 3H), 5,23 (d, J=6,14 Hz, 1H), 4,11-4,40 (m, 3H), 2,87 (d, J=17,10 Hz, 3H).
Пример S22
Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(4-(трифторметил)фенил)хинолин-4-карбоксамида

[0370] Стадия 1: Синтез 6-(4-(трифторметил)фенил)хинолин-4-карбоновой кислоты. К раствору 6-бромхинолин-4-карбоновой кислоты (0,05 г, 0,199 ммоля, 1,0 экв.) в диоксане (4 мл) и воде (2 мл) добавляли (4-(трифторметил)фенил)бороновую кислоту (0,057 г, 298 ммоля, 1,5 экв.), K2CO3 (0,055 г, 0,397 ммоля, 2,0 экв.) и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh2)Cl2 (0,008 г, 0,009 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции реакцию останавливали водой (10 мл) и водный слой промывали этилацетатом (10 мл). Водный слой отделяли и подкисляли с помощью 6 н. HCl (pH ~ 5-6), образовавшийся твердый осадок отфильтровывали и сушили в вакууме и получали 6-(4-(трифторметил)фенил)хинолин-4-карбоновую кислоту (0,054 г, 82,53% выход) в виде почти белого твердого вещества.
[0371] LCMS 318 [0M+H]+
[0372] 1H NMR (DMSO-d6, 400MHz) δ 8,98-9,19 (m, 2 H), 8,12-8,35 (m, 2 H), 7,97-8,09 (m, 3 H), 7,92 (d, J=8,3 Hz, 2 H).
[0373] Стадия 2: Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(4-(трифторметил)фенил)хинолин-4-карбоксамида. При перемешивании к раствору 6-(4-(трифторметил)фенил)хинолин-4-карбоновой кислоты (0,05 г, 0,158 ммоля, 1,0 экв.) в DMF (3 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,053 г, 0,236 ммоля, 1,5 экв.), EDCI.HCl (0,045 г, 0,236 ммоля, 1,5 экв.) и HOBt (0,032 г, 0,236 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 10 мин. Добавляли триэтиламин (0,1 мл) и полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (20 мл×2). Объединенные органические экстракты промывали водой (10 мл×4), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт обогащали с помощью флэш-хроматографии (5% метанола в DCM в качестве элюента), затем очищали с помощью HPLC с обращенной фазой и получали (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(4-(трифторметил)фенил)хинолин-4-карбоксамид (0,016 г, 23,37% выход) в виде белого твердого вещества.
[0374] LCMS 489,3 [0M+H]+
[0375] 1H NMR (400 MHz, DMSO-d6) δ 9,18-9,31 (m, 1H), 9,02 (d, J=3,95 Hz, 1H), 8,97 (d, J=1,75 Hz, 1H), 8,27-8,36 (m, 2H), 8,11-8,27 (m, 3H), 7,90 (d, J=8,33 Hz, 1H), 7,62 (d, J=4,38 Hz, 1H), 5,15-5,30 (m, 1H), 4,24-4,40 (m, 2H), 4,05-4,24 (m, 1H), 2,95 (br. s., 1H), 2,87 (d, J=16,66 Hz, 2H).
Пример S23
Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((4-метоксифенил)этинил)изоникотинамида

[0376] Стадия 1: Синтез метил-3-((4-метоксифенил)этинил)изоникотината. К раствору метил-3-бромизоникотината (1000 мг, 4,6 ммоля, 1,0 экв.) в DMF (10 мл) добавляли 1-этинил-4-метоксибензол (611 мг, 4,6 ммоля, 1,0 экв.), CuI (87 мг, 0,46 ммоля, 0,1 экв.), DIPEA (1186 мг, 9,2 ммоля, 2,0 экв.), затем добавляли pd(pph3)2Cl2 (161 мг, 0,23 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 80°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×2), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (0-30% EtOAc в гексане в качестве элюента) и получали метил-3-((4-метоксифенил)этинил)изоникотинат (300 мг, 24% выход) в виде желтого твердого вещества.
[0377] LCMS 267,0 [0M+H]+
[0378] Стадия 2: Синтез 3-((4-метоксифенил)этинил)изоникотиновой кислоты. При перемешивании к раствору метил-3-((4-метоксифенил)этинил)изоникотината (300 мг, 1,11 ммоля, 1,0 экв.) в THF (10 мл) и воде (05 мл) добавляли LiOH (53 мг, 2,23 ммоля, 2,0 экв.). Смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и 1H NMR спектроскопии. Реакционную смесь разбавляли водой (15 мл) и промывали этилацетатом (15 мл). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали 3-((4-метоксифенил)этинил)изоникотиновую кислоту (400 мг, количественный выход ) в виде белого твердого вещества.
[0379] LCMS 253,0 [0M+H]+
[0380] Стадия 3: Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((4-метоксифенил)этинил)изоникотинамида. При перемешивании к раствору 3-((4-метоксифенил)этинил)изоникотиновой кислоты (100 мг, 0,39 ммоля, 1,0 экв.) в DMF (05 мл) добавляли HATU (296 мг, 0,78 ммоля, 2,0 экв.), (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (89 мг, 0,39 ммоля,1,0 экв.), затем добавляли DIPEA (151 мг, 1,17 ммоля, 3,0 экв.). Полученную реакционную смесь перемешивали в течение ночи при КТ. Образование продукта подтверждали с помощью TLC и LCMS. После завершения реакции реакционную смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (40 мл×2). Органический слой промывали водой (20 мл×4), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью HPLC с обращенной фазой и получали (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-((4-метоксифенил)этинил)изоникотинамид (05 мг, 04% выход) в виде желтого твердого вещества.
[0381] LCMS 425,2 [0M+H]+
[0382] 1H NMR (400 MHz, DMSO-d6) δ 8,92 (br. s., 1H), 8,82 (s, 1H), 8,64 (d, J=5,26 Hz, 1H), 7,45-7,65 (m, 3H), 6,99 (d, J=8,77 Hz, 2H), 5,13 (d, J=7,89 Hz, 1H), 4,29 (d, J=15,35 Hz, 2H), 4,21 (br. s., 2H), 3,80 (s, 3H), 2,79-2,98 (m, 2H).
Пример S24
Синтез (S)-3-((4-хлорфенил)этинил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамида

[0383] Стадия 1: Синтез метил-3-((4-хлорфенил)этинил)изоникотината. К раствору метил-3-бромизоникотината (1 г, 4,62 ммоля, 1,0 экв.) в DMF (10 мл) добавляли 1-хлор-4-этинилбензол (0,935 г, 6,88 ммоля, 1,5 экв.) и TEA (1,38 г, 2,28 ммоля, 3,0 экв.) при КТ. Полученную реакционную смесь продували газообразным N2 в течение 5 мин, затем добавляли Pd(PPh2)Cl2 (0,160 г, 0,229 ммоля, 0,05 экв.). Реакционную смесь нагревали при 80°C в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×4), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (0-30% этилацетата в гексане в качестве элюента) и получали метил-3-((4-хлорфенил)этинил)изоникотинат (0,408 г, 32% выход) в виде желтого твердого вещества.
[0384] LCMS 272,1 [0M+H]+
[0385] Стадия 2: Синтез 3-((4-хлорфенил)этинил)изоникотиновой кислоты. При перемешивании к раствору метил-3-((4-хлорфенил)этинил)изоникотината (0,05 г, 0,183 ммоля, 1,0 экв.) в THF (3 мл) добавляли LiOH.H2O (0,009 г, 0,220 ммоля, 1,2 экв.) и H2O (1 мл) при КТ. Смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции реакционную смесь разбавляли водой (10 мл) и промывали этилацетатом (10 мл). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали 3-((4-хлорфенил)этинил)изоникотиновую кислоту (0,050 г, количественный выход) в виде желтого твердого вещества.
[0386] LCMS 258,1 [0M+H]+
[0387] Стадия 3: Синтез (S)-3-((4-хлорфенил)этинил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамида. При перемешивании к раствору 3-((4-хлорфенил)этинил)изоникотиновой кислоты (0,150 г, 0,583 ммоля, 1,0 экв.) в DMF (7 мл) добавляли EDCI.HCl (0,167 г, 0,875 ммоля, 1,5 экв.),HOBt (0,118 г, 0,875 ммоля, 1,5 экв.) и (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,196 г, 0,875 ммоля, 1,5 экв.), затем добавляли TEA (0,176 г, 1,749 ммоля, 3,0 экв.). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции реакционную смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (50 мл×2). Объединенные органические экстракты промывали водой (20 мл×4), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью HPLC с обращенной фазой и получали (S)-3-((4-хлорфенил)этинил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид (0,024 г, 9,6% выход) в виде белого твердого вещества.
[0388] LCMS 429,3 [0M+H]+
[0389] 1H NMR (400 MHz, DMSO-d6) δ 8,97 (br. s., 1 H) 8,86 (s, 1 H) 8,68 (d, J=4,82 Hz, 1 H) 7,64 (d, J=8,77 Hz, 2 H) 7,43-7,60 (m, 3 H) 5,14 (d, J=7,02 Hz, 1 H) 4,06-4,36 (m, 4 H) 2,76-3,00 (m, 2 H).
Пример S25
Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(6-(трифторметил)пиридин-3-ил)хинолин-4-карбоксамида

[0390] Стадия 1: Синтез 6-(6-(трифторметил)пиридин-3-ил)хинолин-4-карбоновой кислоты. К раствору 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-(трифторметил)пиридина (100 мг, 0,36 ммоля, 1,0 экв.) в смеси диоксан:вода (05:02 мл) добавляли 6-бромхинолин-4-карбоновую кислоту (93 мг, 0,36 ммоля, 1,0 экв.), Na2CO3 (77 мг, 0,72 ммоля, 2,0 экв.), затем добавляли Pd(PPh3)2Cl2 (13 мг, 0,018 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции реакционную смесь разбавляли водой (30 мл). Водный слой экстрагировали этилацетатом (50 мл×2) разделяли и сушили вымораживанием в лиофилизаторе и получали 6-(6-(трифторметил)пиридин-3-ил)хинолин-4-карбоновую кислоту (100 мг,) в виде почти белого твердого вещества.
[0391] LCMS 319,0 [0M+H]+
[0392] Стадия 2: Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(6-(трифторметил)пиридин-3-ил)хинолин-4-карбоксамида. При перемешивании к раствору 6-(6-(трифторметил)пиридин-3-ил)хинолин-4-карбоновой кислоты (100 мг, 0,31 ммоля, 1,0 экв.) в DMF (05 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (71 мг, 0,31 ммоля, 1,0 экв.), HOBT (63 мг, 0,46 ммоля, 1,5 экв.) и EDC.HCl (89 мг, 0,46 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 10 мин. Добавляли триэтиламин (0,10 мл) и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл×2). Объединенные органические экстракты промывали водой (20 мл×4), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью HPLC с обращенной фазой и получали (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(6-(трифторметил)пиридин-3-ил)хинолин-4-карбоксамид (15 мг, 10% выход) в виде почти белого твердого вещества.
[0393] LCMS 490,3 [0M+H]+
[0394] 1H NMR (400 MHz, DMSO-d6) δ 9,30 (s, 1H), 9,23 (br. s., 1H), 9,05 (d, J=4,39 Hz, 1H), 8,99 (s, 1H), 8,65 (d, J=10,96 Hz, 1H), 8,37 (br. s., 1H), 8,26 (d, J=8,77 Hz, 1H), 8,06 (d, J=8,33 Hz, 2H), 7,65 (d, J=4,38 Hz, 1H), 5,21 (d, J=7,02 Hz, 1H), 4,27-4,39 (m, 2H), 4,12 (d, J=11,40 Hz, 2H), 2,91 (d, J=17,10 Hz, 2H).
Пример S26
Синтез (S, E)-6-(2-(4-хлорфенил)проп-1-ен-1-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамида

[0395] Стадия 1: Синтез (E)-6-(2-(4-хлорфенил)проп-1-ен-1-ил)хинолин-4-карбоновой кислоты. К раствору 6-бромхинолин-4-карбоновой кислоты (0,3 г, 1,14 ммоля, 1,0 экв.) в диоксане (4 мл) и воде (2 мл) добавляли (E)-2-(2-(4-хлорфенил)проп-1-ен-1-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,288 г, 0,91 ммоля, 0,8 экв.), K2CO3 (0,241 г, 2,28 ммоля, 2,0 экв.) и смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh2)Cl2 (0,042 г, 0,057 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции реакционную смесь разбавляли водой (30 мл) и промывали этилацетатом (50 мл×2). Водный слой отделяли и сушили в лиофилизаторе и получали (E)-6-(2-(4-хлорфенил)проп-1-ен-1-ил)хинолин-4-карбоновую кислоту (0,350 г, количественный выход) в виде почти белого твердого вещества.
[0396] LCMS 324,1 [0M+H]+
[0397] Стадия 2: Синтез (S, E)-6-(2-(4-хлорфенил)проп-1-ен-1-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамида. При перемешивании к раствору (E)-6-(4-хлорстирил)хинолин-4-карбоновой кислоты (0,35 г, 1,08 ммоля, 1,0 экв.) в DMF (5 мл) добавляли EDCI.HCl (0,311 г, 1,62 ммоля, 1,5 экв.),HOBt (0,218 г, 1,62 ммоля, 1,5 экв.) и (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,364 г, 1,62 ммоля, 1,5 экв.), затем добавляли триэтиламин (0,327 г, 3,24 ммоля, 3,0 экв.). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции реакционную смесь разбавляли водой (25 мл) и экстрагировали этилацетатом (50 мл×2). Объединенные органические экстракты промывали водой (20 мл×4), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью флэш-хроматографии (0-5% MeOH в DCM в качестве элюента), затем с помощью HPLC с обращенной фазой и получали (S, E)-6-(2-(4-хлорфенил)проп-1-ен-1-ил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид (0,02 г, 3,7% выход) в виде коричневого твердого вещества.
[0398] LCMS 495,4 [0M+H]+
[0399] 1H NMR (400 MHz, DMSO-d6) δ 9,16 (t, J=5,92 Hz, 1 H) 8,97 (d, J=4,38 Hz, 1 H) 8,41 (s, 1 H) 8,09 (d, J=8,77 Hz, 1 H) 7,87 (d, J=8,33 Hz, 1 H) 7,67 (d, J=8,77 Hz, 2 H) 7,58 (d, J=4,38 Hz, 1 H) 7,47 (d, J=8,77 Hz, 2 H) 7,11 (s, 1 H) 5,16 (d, J=7,02 Hz, 1 H) 4,08-4,38 (m, 4 H) 2,78-2,99 (m, 2 H) 2,32 (s, 3 H).
Пример S27
Синтез (S, E)-3-(2-(6-хлорнафталин-2-ил)винил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамида
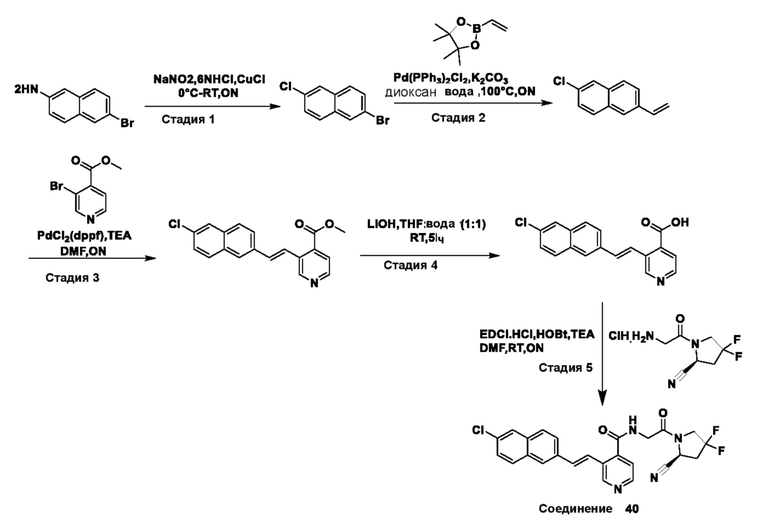
[0400] Стадия 1: Синтез 2-бром-6-хлорнафталина. При перемешивании к раствору 6-хлорнафталин-2-амина (1,0 г, 4,504 ммоля, 1,0 экв.) в 6 н. HCl (10 мл) добавляли NaNO2 (0,372 г, 5,405 ммоля, 1,2 экв.) в воде (10 мл) при 0°C и полученную реакционную смесь перемешивали при КТ в течение 1 ч. Раствор CuCl (2,2 г, 22,522 ммоля, 5,0 экв.) в 6 н. HCl (10 мл) повторно добавляли и перемешивали при КТ в течение ночи. После завершения реакции (следили с помощью TLC), реакционную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (150 мл×3). Объединенные органические экстракты промывали водой (50 мл×2) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (гексан в качестве элюента) и получали 2-бром-6-хлорнафталин (0,600 г, 55,5% выход) в виде белого твердого вещества.
[0401] 1H NMR (400 MHz, DMSO-d6) δ 8,27 (s, 1 H) 8,10 (s, 1 H) 7,97 (d, J=8,77 Hz, 1 H) 7,90 (d, J=8,77 Hz, 1 H) 7,70 (dd, J=8,77, 1,75 Hz, 1 H) 7,59 (dd, J=8,99, 1,97 Hz, 1 H).
[0402] Стадия 2: Синтез 2-хлор-6-винилнафталина. При перемешивании к раствору 2-бром-6-хлорнафталина (0,250 г, 1,061 ммоля, 1,0 экв.) в диоксане (9 мл) добавляли 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолан (0,395 г, 1,562 ммоля, 1,5 экв.) и раствор K2CO3 (0,287 г, 2,083 ммоля, 2,0 экв.) в воде (3 мл), смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,036 г, 0,0520 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью TLC. После завершения реакции реакционную смесь разбавляли водой (50 мл) экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×2) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (гексан в качестве элюента) и получали 2-хлор-6-винилнафталин (0,150 г, 76,92% выход) в виде белого твердого вещества.
[0403] 1H NMR (400 MHz, хлороформ-d) δ 7,52-7,82 (m, 4 H) 7,40 (dd, J=8,77, 2,19 Hz, 1 H) 7,26 (s, 1 H) 6,86 (dd, J=17,54, 10,96 Hz, 1 H) 5,88 (d, J=17,54 Hz, 1 H) 5,36 (d, J=10,96 Hz, 1 H).
[0404] Стадия 3: Синтез метил-(E)-3-(2-(6-хлорнафталин-2-ил)винил)изоникотината. При перемешивании к раствору 2-хлор-6-винилнафталина (0,250 г, 1,32 ммоля, 1,0 экв.) в DMF (10 мл) добавляли метил-3-бромизоникотинат (0,574 г, 2,65 ммоля, 2,0 экв.) и триэтиламин (0,575 мл, 3,983 ммоля, 3,0 экв.). Полученную реакционную смесь продували газообразным N2 в течение 5 мин, затем добавляли pd(dppf)Cl2 (0,097 г, 0,132 ммоля, 0,1 экв.). Реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью TLC. После завершения реакции реакционную смесь разбавляли водой (50 мл) экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×2) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-15% EA/гексан в качестве элюента) и получали метил-(E)-3-(2-(6-хлорнафталин-2-ил)винил)изоникотинат (0,120 г, 28% выход) в виде белого твердого вещества.
[0405] LCMS 324,2 [0M+H]+
[0406] 1H NMR (400 MHz, хлороформ-d) δ 9,06 (s, 1 H) 8,63 (d, J=5,26 Hz, 1 H) 8,00 (d, J=16,22 Hz, 1 H) 7,89 (s, 1 H) 7,66-7,84 (m, 4 H) 7,44 (dd, J=8,77, 2,19 Hz, 1 H) 7,19-7,28 (m, 2 H) 3,99 (s, 3 H).
[0407] Стадия 4: Синтез (E)-3-(2-(6-хлорнафталин-2-ил)винил)изоникотиновой кислоты. При перемешивании к раствору метил-(E)-3-(2-(6-хлорнафталин-2-ил)винил)изоникотината (0,120 г, 0,370 ммоля, 1,0 экв.) в THF (6 мл) добавляли раствор LiOH.H2O (0,031 г, 0,741 ммоля, 2,0 экв.) в воде (6 мл). Реакционную смесь перемешивали при КТ в течение 5 ч. Образование продукта подтверждали с помощью TLC. Реакционную смесь разбавляли водой (20 мл) и промывали этилацетатом (25 мл×3). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали соединение (E)-3-(2-(6-хлорнафталин-2-ил)винил)изоникотиновую кислоту (0,100 г, 87% выход) в виде белого твердого вещества.
[0408] LCMS 310,0 [0M+H]+
[0409] 1H NMR (400 MHz, DMSO-d6) δ 8,85 (s, 1 H) 8,31 (d, J=4,82 Hz, 1 H) 8,02 (s, 1 H) 7,98 (br. s., 1 H) 7,87-7,98 (m, 2 H) 7,79 (d, J=7,45 Hz, 1 H) 7,52 (dd, J=8,77, 1,75 Hz, 1 H) 7,35 (d, J=16,66 Hz, 1 H) 7,27 (d, J=4,82 Hz, 1 H).
[0410] Стадия 5: Синтез (S, E)-3-(2-(6-хлорнафталин-2-ил)винил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамида. При перемешивании к раствору (E)-3-(2-(6-хлорнафталин-2-ил)винил)изоникотиновой кислоты (0,100 г, 0,323 ммоля, 1,0 экв.) в DMF (3 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,109 г, 0,485 ммоля, 1,5 экв.), HOBt (0,065 г, 0,485 ммоля, 1,5 экв.) и EDCI.HCl (0,092 г, 0,485 ммоля, 1,5 экв.), затем добавляли TEA (0,09 мл, 0,647 ммоля, 2,0 экв.). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции реакционную смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (50 мл×2). Объединенные органические экстракты промывали водой (20 мл×2) и рассолом (20 мл). Органический слой отделяли и сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью флэш-хроматографии (0-5% MeOH в DCM в качестве элюента) и получали (S, E)-3-(2-(6-хлорнафталин-2-ил)винил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)изоникотинамид (0,030 г, 20% выход) в виде почти белого твердого вещества.
[0411] LCMS 481,1 [0M+H]+
[0412] 1H NMR (400 MHz, DMSO-d6) δ 9,20 (s, 1 H) 9,06 (t, J=5,70 Hz, 1 H) 8,55 (d, J=4,82 Hz, 1 H) 8,18 (s, 1 H) 7,97-8,07 (m, 2 H) 7,92 (d, J=8,77 Hz, 1 H) 7,82 (d, J=16,66 Hz, 1 H) 7,65 (d, J=16,66 Hz, 1 H) 7,48-7,61 (m, 1 H) 7,38 (d, J=4,82 Hz, 1 H) 5,23 (d, J=6,58 Hz, 1 H) 4,34 (t, J=11,62 Hz, 1 H) 4,23 (br. s., 1 H) 4,03-4,23 (m, 2 H) 2,76-3,05 (m, 3 H).
Пример S28
Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-((4-метоксифенил)этинил)хинолин-4-карбоксамида

[0413] Стадия 1: Синтез 6-((4-метоксифенил)этинил)хинолин-4-карбоновой кислоты. К раствору 6-бромхинолин-4-карбоновой кислоты (250 мг, 1,0 ммоля, 1,0 экв.) в DMF (07 мл) добавляли 1-этинил-4-метоксибензол (204 мг, 1,2 ммоля, 1,5 экв.), Cs2CO3 (487 мг, 1,5 ммоля, 1,5 экв.), Pd(dppf)cl2 (35 мг, 0,05 ммоля, 0,05 экв.). Полученную реакционную смесь перемешивали при 80°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции реакционную смесь разбавляли водой (30 мл) и промывали этилацетатом (10 мл×2). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали 6-((4-метоксифенил)этинил)хинолин-4-карбоновую кислоту (100 мг, 33% выход) в виде почти белого твердого вещества.
[0414] LCMS 304,0 [0M+H]+
[0415] Стадия 2: Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-((4-метоксифенил)этинил)хинолин-4-карбоксамида. При перемешивании к раствору 6-((4-метоксифенил)этинил)хинолин-4-карбоновой кислоты (100 мг, 0,33 ммоля, 1,0 экв.) в DMF (05 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (75 мг, 0,33 ммоля,1,0 экв.), EDC.HCl (95 мг, 0,49 ммоля, 1,5 экв.), HOBT (67 мг, 0,49 ммоля, 1,5 экв.) и TEA (0,2 мл, 0,99 ммоля, 3,0 экв.). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью TLC и LCMS. После завершения реакции реакционную смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (50 мл×2). Органический слой промывали водой (20 мл×4), рассолом (20 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью HPLC с обращенной фазой и получали (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-((4-метоксифенил)этинил)хинолин-4-карбоксамид (05 мг, 4%) в виде желтого твердого вещества.
[0416] LCMS 475,3 [0M+H]+
[0417] 1H NMR (400MHz, DMSO-d6) δ 9,20 (br. s., 1 H), 9,01 (d, J=4,4 Hz, 1 H), 8,55 (s, 1 H), 8,10 (d, J=8,8 Hz, 1 H), 7,89 (d, J=10,5 Hz, 1 H), 7,67-7,51 (m, 3 H), 7,01 (d, J=8,8 Hz, 2 H), 5,20 (d, J=9,2 Hz, 1 H), 4,36-4,19 (m, 3 H), 4,16 (br. s., 1 H), 3,81 (s, 3 H), 2,86 (d, J=17,1 Hz, 2 H).
Пример S29
Синтез (S, E)-6-(2-(6-хлорнафталин-2-ил)винил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамида

[0418] Стадия 1: Синтез (E)-6-(2-(6-хлорнафталин-2-ил)винил)хинолин-4-карбоновой кислоты. При перемешивании к раствору 2-хлор-6-винилнафталина (0,100 г, 0,531 ммоля, 1,0 экв.) в DMF (5 мл) добавляли 6-бромхинолин-4-карбоновую кислоту (0,134 г, 0,531 ммоля, 1,0 экв.) и триэтиламин (0,230 мл, 1,595 ммоля, 3,0 экв.). Полученную реакционную смесь продували газообразным N2 в течение 5 мин, затем добавляли Pd(dppf)Cl2 (0,038 г, 0,0531 ммоля, 0,1 экв.). Реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью TLC. Реакционную смесь охлаждали до RT, разбавляли водой (50 мл) и промывали этилацетатом (50 мл×3). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали (E)-6-(2-(6-хлорнафталин-2-ил)винил)хинолин-4-карбоновую кислоту (0,100 г, 52% выход) в виде желтого твердого вещества.
[0419] LCMS 360,0 [0M+H]+
[0420] 1H NMR (400 MHz, DMSO-d6) δ 8,94 (d, J=4,38 Hz, 1 H) 8,83 (br. s., 1 H) 8,24 (d, J=8,33 Hz, 1 H) 8,17 (br. s., 1 H) 8,09 (d, J=8,33 Hz, 1 H) 8,04 (d, J=7,89 Hz, 2 H) 7,76-8,00 (m, 2 H) 7,48-7,75 (m, 1 H) 6,90 (dd, J=17,32, 11,18 Hz, 1 H) 6,00 (d, J=17,54 Hz, 1 H) 5,39 (d, J=10,52 Hz, 1 H).
[0421] Стадия 2: Синтез (S, E)-6-(2-(6-хлорнафталин-2-ил)винил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамида. При перемешивании к раствору (E)-6-(2-(6-хлорнафталин-2-ил)винил)хинолин-4-карбоновой кислоты (0,100 г, 0,278 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,094 г, 0,417 ммоля, 1,5 экв.), HOBt (0,056 г, 0,417 ммоля, 1,5 экв.) и EDC.HCl (0,079 г, 0,417 ммоля, 1,5 экв.), затем добавляли TEA (0,1 мл). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×3) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-5% MeOH в DCM в качестве элюента), затем с помощью HPLC с обращенной фазой и получали (S, E)-6-(2-(6-хлорнафталин-2-ил)винил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид (0,015 г, 10% выход) в виде желтого твердого вещества.
[0422] LCMS 531,4 [0M+H]+
[0423] 1H NMR (400 MHz, DMSO-d6) δ 9,20 (t, J=5,70 Hz, 1 H) 8,93 (d, J=4,38 Hz, 1 H) 8,70 (s, 1 H) 8,14 (br. s., 2 H) 8,05-8,12 (m, 1 H) 7,87-8,05 (m, 4 H) 7,76 (d, J=16,66 Hz, 1 H) 7,43-7,67 (m, 3 H) 5,17-5,36 (m, 1 H) 4,12-4,41 (m, 4 H) 2,96 (br. s., 1 H) 2,89 (d, J=9,21 Hz, 1 H).
Пример S30
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-метоксипиридин-3-ил)винил)хинолин-4-карбоксамида
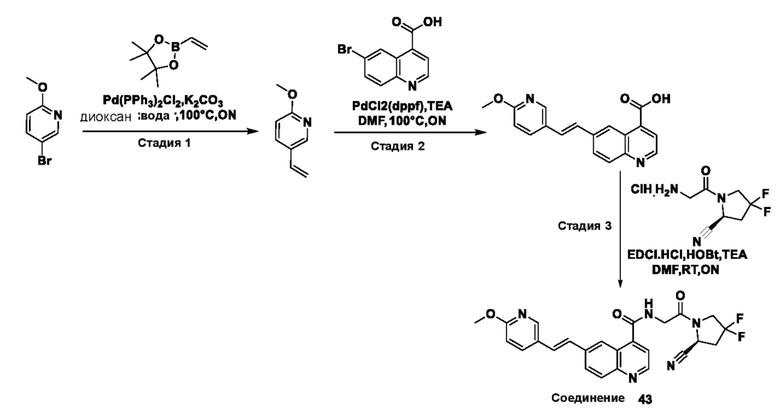
[0424] Стадия 1: Синтез 5-этенил-2-метоксипиридина. При перемешивании к раствору 5-бром-2-метоксипиридина (0,500 г, 2,659 ммоля, 1,0 экв.) в диоксане (8 мл) добавляли 2-винил-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,614 г, 3,989 ммоля, 1,5 экв.) и раствор K2CO3 (0,734 г, 5,319 ммоля, 2,0 экв.) в воде (4 мл), и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,093 г, 0,132 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью TLC. Реакционную смесь охлаждали до RT, разбавляли водой (50 мл) экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×2) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-5% EA в гексане в качестве элюента) и получали 5-этенил-2-метоксипиридин (0,250 г, 89% выход) в виде желтого полужидкого вещества.
[0425] 1H NMR (400 MHz, хлороформ-d) δ 8,12 (d, J=2,63 Hz, 1 H) 7,69 (dd, J=8,77, 2,63 Hz, 1 H) 6,72 (d, J=8,77 Hz, 1 H) 6,55-6,68 (m, 1 H) 5,64 (d, J=17,54 Hz, 1 H) 5,21 (d, J=10,96 Hz, 1 H) 3,87-4,04 (m, 3 H).
[0426] Стадия 2: Синтез (E)-6-(2-(6-метоксипиридин-3-ил)винил)хинолин-4-карбоновой кислоты. При перемешивании к раствору 6-бромхинолин-4-карбоновой кислоты (0,200 г, 0,793 ммоля, 1,0 экв.) в DMF (5 мл) добавляли 5-этенил-2-метоксипиридин (0,125 г, 1,190 ммоля, 1,5 экв.) и триэтиламин (0,34 мл, 2,380 ммоля, 3,0 экв.). Полученную реакционную смесь продували газообразным N2 в течение 5 мин, затем добавляли Pd(dppf)Cl2 (0,058 г, 0,079 ммоля, 0,1 экв.). Реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. Реакционную смесь охлаждали до RT, разбавляли водой (20 мл) промывали этилацетатом (10 мл×2). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали (E)-6-(2-(6-метоксипиридин-3-ил)винил)хинолин-4-карбоновую кислоту (0,200 г, 82% выход) в виде желтого твердого вещества.
[0427] LCMS 307,1 [0M+H]+
[0428] 1H NMR (400 MHz, DMSO-d6) δ 10,28 (s, 1 H) 8,89 (d, J=4,38 Hz, 1 H) 8,73 (s, 1 H) 8,40 (d, J=2,19 Hz, 1 H) 8,09-8,22 (m, 2 H) 8,03 (d, J=9,21 Hz, 1 H) 7,76 (d, J=3,95 Hz, 1 H) 7,28-7,54 (m, 2 H) 6,87 (d, J=8,77 Hz, 1 H) 3,88 (s, 3 H).
[0429] Стадия 3: Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-метоксипиридин-3-ил)винил)хинолин-4-карбоксамида. При перемешивании к раствору (E)-6-(2-(6-метоксипиридин-3-ил)винил)хинолин-4-карбоновой кислоты (0,200 г, 0,653 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,220 г, 0,980 ммоля, 1,5 экв.), HOBt (0,132 г, 0,980 ммоля, 1,5 экв.) и EDC.HCl (0,187 г, 0,980 ммоля, 1,5 экв.), затем добавляли TEA (0,18 мл). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×3) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-5% MeOH в DCM в качестве элюента) и получали (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-метоксипиридин-3-ил)винил)хинолин-4-карбоксамид (0,070 г, 22,43% выход) в виде желтого твердого вещества.
[0430] LCMS 478,4 [0M+H]+
[0431] 1H NMR (400 MHz, DMSO-d6) δ 9,17 (t, J=5,92 Hz, 1 H) 8,93 (d, J=4,38 Hz, 1 H) 8,52 (s, 1 H) 8,41 (d, J=2,19 Hz, 1 H) 8,03-8,20 (m, 3 H) 7,56 (d, J=4,38 Hz, 1 H) 7,53 (d, J=16,66 Hz, 1 H) 7,39 (d, J=16,66 Hz, 1 H) 6,88 (d, J=8,77 Hz, 1 H) 5,23 (dd, J=9,21, 3,07 Hz, 1 H) 4,11-4,40 (m, 4 H) 3,89 (s, 3 H) 2,97 (d, J=8,77 Hz, 1 H) 2,87 (d, J=14,03 Hz, 1 H).
Пример S31
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(4-фторстирил)хинолин-4-карбоксамида
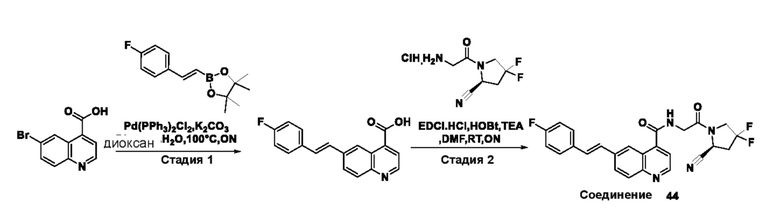
[0432] Стадия 1: Синтез (E)-6-(4-Фторстирил)хинолин-4-карбоновой кислоты. К раствору 6-бромхинолин-4-карбоновой кислоты (0,341 г, 1,36 ммоля, 0,8 экв.) и (E)-2-(4-фторстирил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,5 г, 1,70 ммоля, 1,0 экв.) в диоксане (10 мл) и воде (1 мл) добавляли K2CO3 (0,357 г, 3,4 ммоля, 2,0 экв.) и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh2)Cl2 (0,059 г, 0,085 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции реакцию останавливали водой (10 мл) и водный слой промывали этилацетатом (10 мл×2). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали (E)-6-(4-фторстирил)хинолин-4-карбоновую кислоту (0,250 г, количественный выход) в виде желтого твердого вещества.
[0433] LCMS 294,2 [0M+H]+
[0434] Стадия 2: Синтез (S, E)-6-(4-фторстирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамида. При перемешивании к раствору (E)-6-(4-хлорстирил)хинолин-4-карбоновой кислоты (0,1 г, 0,34 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,114 г, 0,51 ммоля, 1,5 экв.), EDC.HCl (0,097 г, 0,51 ммоля, 1,5 экв.) и HOBt (0,068 г, 0,51 ммоля, 1,5 экв.), затем добавляли TEA (0,1 мл). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×3) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-5% MeOH в DCM в качестве элюента), затем с помощью HPLC с обращенной фазой и получали (S, E)-6-(4-фторстирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид (0,015 г, 10% выход) в виде почти белого твердого вещества.
[0435] LCMS 465,4 [0M+H]+
[0436] 1H NMR (400 MHz, DMSO-d6) δ 9,18 (br. s., 1 H) 8,94 (d, J=4,39 Hz, 1 H) 8,58 (s, 1 H) 8,09 (t, J=9,43 Hz, 2 H) 7,67-7,82 (m, 2 H) 7,48-7,64 (m, 2 H) 7,42 (s, 1 H) 7,24 (t, J=8,77 Hz, 2 H) 5,22 (br. s., 1 H) 4,29 (t, J=5,92 Hz, 4 H) 3,17 (d, J=5,26 Hz, 1 H) 2,90 (br. s., 2 H).
Пример S32
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-метилпиридин-3-ил)винил)хинолин-4-карбоксамида

[0437] Стадия 1: Синтез 2-(метил)-5-винилпиридина. При перемешивании к раствору 5-бром-2-(метил)пиридина (0,500 г, 2,906 ммоля, 1,0 экв.) в диоксане (8 мл) добавляли 2-винил-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,671 г, 4,360 ммоля, 1,5 экв.) и раствор K2CO3 (0,802 г, 5,813 ммоля, 2,0 экв.) в воде (4 мл), и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,102 г, 0,145 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью TLC. Реакционную смесь охлаждали до RT, разбавляли водой (50 мл) экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×2) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-15% EA в гексане в качестве элюента) и получали 2-(метил)-5-винилпиридин (0,170 г, 49,27% выход) в виде желтого полужидкого вещества.
[0438] 1H NMR (400 MHz, DMSO-d6) δ 8,49 (d, J=2,19 Hz, 1 H) 7,82 (dd, J=7,89, 2,19 Hz, 1 H) 7,23 (d, J=8,33 Hz, 1 H) 6,73 (dd, J=17,76, 11,18 Hz, 1 H) 5,78-5,98 (m, 1 H) 5,32 (d, J=10,96 Hz, 1 H) 2,37-2,48 (m, 3 H).
[0439] Стадия 2: Синтез (E)-6-(2-(6-метилпиридин-3-ил)винил)хинолин-4-карбоновой кислоты. При перемешивании к раствору 6-бромхинолин-4-карбоновой кислоты (0,150 г, 0,595 ммоля, 1,0 экв.) в диоксане (5 мл) добавляли 2-(метил)-5-винилпиридин (0,106 г, 0,892 ммоля, 1,5 экв.) и триэтиламин (0,257 мл, 1,785 ммоля, 3,0 экв.). Полученную реакционную смесь продували газообразным N2 в течение 5 мин, затем добавляли Pd(dppf)Cl2 (0,043 г, 0,059 ммоля, 0,1 экв.). Реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. Реакционную смесь охлаждали до RT, разбавляли водой (50 мл) промывали этилацетатом (20 мл×2). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали (E)-6-(2-(6-метилпиридин-3-ил)винил)хинолин-4-карбоновую кислоту (0,150 г, 87% выход) в виде желтого твердого вещества.
[0440] LCMS 291,1 [0M+H]+
[0441] 1H NMR (400 MHz, DMSO-d6) δ 9,56 (br. s., 1 H) 8,91 (d, J=3,95 Hz, 1 H) 8,76 (s, 1 H) 8,70 (br. s., 1 H) 8,18 (d, J=9,65 Hz, 2 H) 8,06 (d, J=8,33 Hz, 1 H) 7,79 (d, J=3,95 Hz, 1 H) 7,57 (d, J=16,66 Hz, 1 H) 7,43 (d, J=16,22 Hz, 1 H) 7,29 (d, J=7,89 Hz, 1 H) 2,33 (br. s., 3 H).
[0442] Стадия 3: Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-метилпиридин-3-ил)винил)хинолин-4-карбоксамида. При перемешивании к раствору (E)-6-(2-(6-метилпиридин-3-ил)винил)хинолин-4-карбоновой кислоты (0,200 г, 0,689 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,232 г, 1,034 ммоля, 1,5 экв.), HOBt (0,139 г, 1,034 ммоля, 1,5 экв.) и EDC.HCl (0,197 г, 1,034 ммоля, 1,5 экв.), затем добавляли TEA (0,19 мл). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×3) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-5% MeOH в DCM в качестве элюента) и получали (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-метилпиридин-3-ил)винил)хинолин-4-карбоксамид (0,070 г, 22,08% выход) в виде почти белого твердого вещества.
[0443] LCMS 462,4 [0M+H]+
[0444] 1H NMR (400 MHz, DMSO-d6) δ 9,18 (t, J=5,92 Hz, 1 H) 8,94 (d, J=4,38 Hz, 1 H) 8,71 (d, J=1,75 Hz, 1 H) 8,58 (s, 1 H) 7,94-8,23 (m, 3 H) 7,40-7,65 (m, 3 H) 7,29 (d, J=7,89 Hz, 1 H) 5,23 (dd, J=9,21, 3,07 Hz, 1 H) 4,11-4,41 (m, 4 H) 2,96 (br. s., 1 H) 2,87 (d, J=15,35 Hz, 1 H) 2,49 (s, 3 H).
Пример S33
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-(трифторметил)пиридин-3-ил)винил)хинолин-4-карбоксамида

[0445] Стадия 1: Синтез 2-(трифторметил)-5-винилпиридина. При перемешивании к раствору 5-бром-2-(трифторметил)пиридина (0,500 г, 2,212 ммоля, 1,0 экв.) в диоксане (8 мл) добавляли 2-винил-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,511 г, 3,312 ммоля, 1,5 экв.) и раствор K2CO3 (0,616 г, 4,424 ммоля, 2,0 экв.) в воде (4 мл), и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,077 г, 0,110 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью TLC. Реакционную смесь охлаждали до RT, разбавляли водой (50 мл) экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×2) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-5% EA в гексане в качестве элюента) и получали 2-(трифторметил)-5-винилпиридин (0,200 г, 52,35% выход) в виде желтого полужидкого вещества.
[0446] 1H NMR (400 MHz, DMSO-d6) δ 8,77-8,97 (m, 1 H) 8,14-8,26 (m, 1 H) 7,88 (d, J=7,89 Hz, 1 H) 6,88 (dd, J=17,76, 11,18 Hz, 1 H) 6,17 (d, J=17,98 Hz, 1 H) 5,58 (d, J=11,40 Hz, 1 H).
[0447] Стадия 2: Синтез (E)-6-(2-(6-(трифторметил)пиридин-3-ил)винил)хинолин-4-карбоновой кислоты. При перемешивании к раствору 6-бромхинолин-4-карбоновой кислоты (0,100 г, 0,396 ммоля, 1,0 экв.) в диоксане (10 мл) добавляли 2-(трифторметил)-5-винилпиридин (0,102 г, 0,595 ммоля, 1,5 экв.) и триэтиламин (0,171 мл, 1,190 ммоля, 3,0 экв.). Полученную реакционную смесь продували газообразным N2 в течение 5 мин, затем добавляли Pd(dppf)Cl2 (0,029 г, 0,0396 ммоля, 0,1 экв.). Реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. Реакционную смесь охлаждали до RT, разбавляли водой (50 мл) промывали этилацетатом (20 мл×2), Водный слой подкисляли с помощью 1 н. HCl и экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (20 мл×2) и рассолом (20 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении и получали (E)-6-(2-(6-(трифторметил)пиридин-3-ил)винил)хинолин-4-карбоновую кислоту (0,100 г, 73,52% выход) в виде желтого твердого вещества.
[0448] LCMS 345,1 [0M+H]+
[0449] 1H NMR (400 MHz, DMSO-d6) δ 13,91 (br. s., 1 H) 9,06 (s, 1 H) 9,00 (d, J=4,38 Hz, 1 H) 8,82 (s, 1 H) 8,41 (d, J=8,77 Hz, 1 H) 8,26 (d, J=9,21 Hz, 1 H) 8,13 (d, J=8,77 Hz, 1 H) 7,81-7,99 (m, 3 H) 7,59 (d, J=16,66 Hz, 1 H).
[0450] Стадия 3: Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-(трифторметил)пиридин-3-ил)винил)хинолин-4-карбоксамида. При перемешивании к раствору (E)-6-(2-(6-(трифторметил)пиридин-3-ил)винил)хинолин-4-карбоновой кислоты (0,100 г, 0,290 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,098 г, 0,436 ммоля, 1,5 экв.), HOBt (0,058 г, 0,436 ммоля, 1,5 экв.) и EDC.HCl (0,083 г, 0,436 ммоля, 1,5 экв.), затем добавляли TEA (0,08 мл). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×3) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-5% MeOH в DCM в качестве элюента) и получали (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-(трифторметил)пиридин-3-ил)винил)хинолин-4-карбоксамид (0,095 г, 63% выход) в виде белого твердого вещества.
[0451] LCMS 516,4 [0M+H]+
[0452] 1H NMR (400 MHz, DMSO-d6) δ 9,20 (t, J=5,70 Hz, 1 H) 9,05 (s, 1 H) 8,98 (d, J=3,95 Hz, 1 H) 8,68 (s, 1 H) 8,40 (d, J=7,45 Hz, 1 H) 8,03-8,27 (m, 2 H) 7,93 (d, J=8,33 Hz, 1 H) 7,81 (s, 1 H) 7,66-7,74 (m, 1 H) 7,60 (d, J=4,39 Hz, 1 H) 5,24 (d, J=7,02 Hz, 1 H) 4,26-4,50 (m, 2 H) 4,00-4,26 (m, 2 H) 2,76-3,03 (m, 2 H).
Пример S34
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(4-метоксистирил)хинолин-4-карбоксамида
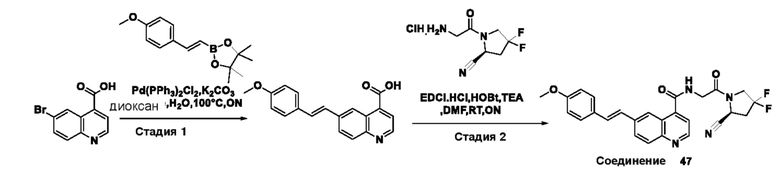
[0453] Стадия 1: Синтез (E)-6-(4-метоксистирил)хинолин-4-карбоновой кислоты. К раствору 6-бромхинолин-4-карбоновой кислоты (0,383 г, 1,52 ммоля, 0,8 экв.) и (E)-2-(4-метоксистирил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,5 г, 1,91 ммоля, 1,0 экв.) в диоксане (10 мл) и воде (1 мл) добавляли K2CO3 (0,404 г, 3,82 ммоля, 2,0 экв.) и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh2)Cl2 (0,067 г, 0,095 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции реакцию останавливали водой (20 мл) и водный слой промывали этилацетатом (10 мл×2). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали (E)-6-(4-метоксистирил)хинолин-4-карбоновую кислоту (0,200 г, 44% выход ) в виде желтого твердого вещества.
[0454] LCMS 306,0 [0M+H]+
[0455] Стадия 2: Синтез (S, E)-6-(4-метоксистирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамида. При перемешивании к раствору (E)-6-(4-хлорстирил)хинолин-4-карбоновой кислоты (0,2 г, 0,65 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,220 г, 0,98 ммоля, 1,5 экв.), EDC.HCl (0,188 г, 0,98 ммоля, 1,5 экв.) и HOBt (0,132 г, 0,98 ммоля, 1,5 экв.), затем добавляли TEA (0,2 мл). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×3) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-5% MeOH в DCM в качестве элюента), затем с помощью HPLC с обращенной фазой и получали (S, E)-6-(4-метоксистирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид (0,045 г, 14% выход) в виде почти белого твердого вещества.
[0456] LCMS 477,4 [0M+H]+
[0457] 1H NMR (400 MHz, DMSO-d6) δ 9,17 (t, J=5,92 Hz, 1 H) 8,91 (d, J=3,95 Hz, 1 H) 8,52 (s, 1 H) 8,10 (dd, J=8,99, 1,53 Hz, 1 H) 8,05 (d, J=8,77 Hz, 1 H) 7,65 (m, J=8,33 Hz, 2 H) 7,47-7,59 (m, 2 H) 7,28 (d, J=16,22 Hz, 1 H) 6,97 (m, J=8,77 Hz, 2 H) 5,23 (dd, J=9,21, 2,63 Hz, 1 H) 4,11-4,41 (m, 4 H) 3,79 (s, 3 H) 2,78-3,05 (m, 2 H).
Пример S35
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(4-(трифторметокси)стирил)хинолин-4-карбоксамида

[0458] Стадия 1: Синтез (E)-6-(4-трифторметоксистирил)хинолин-4-карбоновой кислоты. К раствору 6-бромхинолин-4-карбоновой кислоты (0,140 г, 0,56 ммоля, 0,8 экв.) и (E)-2-(4-трифторметоксистирил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,220 г, 0,70 ммоля, 1,0 экв.) в диоксане (10 мл) и воде (1 мл) добавляли K2CO3 (0,147 г, 1,4 ммоля, 2,0 экв.) и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh2)Cl2 (0,024 г, 0,035 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции реакцию останавливали водой (20 мл) и водный слой промывали этилацетатом (10 мл×2). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали (E)-6-(4-метоксистирил)хинолин-4-карбоновую кислоту (0,100 г, 50% выход) в виде желтого твердого вещества.
[0459] LCMS 360,1 [0M+H]+
[0460] Стадия 2: Синтез (S, E)-6-(4-метоксистирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамида. При перемешивании к раствору (E)-6-(4-хлорстирил)хинолин-4-карбоновой кислоты (0,1 г, 0,27 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,093 г, 0,41 ммоля, 1,5 экв.), EDCI.HCl (0,078 г, 0,41 ммоля, 1,5 экв.) и HOBt (0,055 г, 0,41 ммоля, 1,5 экв.), затем добавляли TEA (0,1 мл). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×3) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-5% MeOH в DCM в качестве элюента), затем с помощью HPLC с обращенной фазой и получали (S, E)-6-(4-метоксистирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид (0,045 г, 32% выход) в виде почти белого твердого вещества.
[0461] LCMS 531,4 [0M+H]+
[0462] 1H NMR (400 MHz, DMSO-d6) δ 9,19 (t, J=5,70 Hz, 1 H) 8,94 (d, J=4,38 Hz, 1 H) 8,62 (s, 1 H) 8,04-8,17 (m, 2 H) 7,83 (m, J=8,77 Hz, 2 H) 7,53-7,70 (m, 2 H) 7,45-7,53 (m, 1 H) 7,39 (m, J=8,33 Hz, 2 H) 5,23 (dd, J=9,21, 2,19 Hz, 1 H) 4,11-4,41 (m, 4 H) 2,96 (br. s., 2 H).
Пример S36
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(5-метилпиридин-2-ил)винил)хинолин-4-карбоксамида

[0463] Стадия 1: Синтез 5-(метил)-2-винилпиридина. При перемешивании к раствору 2-бром-5-метилпиридина (0,500 г, 2,906 ммоля, 1,0 экв.) в диоксане (8 мл) добавляли 2-винил-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,671 г, 4,360 ммоля, 1,5 экв.) и раствор K2CO3 (0,802 г, 5,813 ммоля, 2,0 экв.) в воде (4 мл), и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,102 г, 0,145 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью TLC. Реакционную смесь охлаждали до RT, разбавляли водой (50 мл) экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×2) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-15% EA в гексане в качестве элюента) и получали 5-(метил)-2-винилпиридин (0,300 г, 86,95% выход) в виде желтого полужидкого вещества.
[0464] 1H NMR (400 MHz, хлороформ-d) δ 8,40 (s, 1 H) 7,45 (d, J=7,02 Hz, 1 H) 7,17-7,34 (m, 1 H) 6,79 (dd, J=17,54, 10,96 Hz, 1 H) 6,12 (d, J=17,54 Hz, 1 H) 5,42 (d, J=10,96 Hz, 1 H) 2,20-2,45 (m, 3 H).
[0465] Стадия 2: Синтез (E)-6-(2-(5-метилпиридин-2-ил)винил)хинолин-4-карбоновой кислоты. При перемешивании к раствору 6-бромхинолин-4-карбоновой кислоты (0,100 г, 0,396 ммоля, 1,0 экв.) в DMF (5 мл) добавляли 5-(метил)-2-винилпиридин (0,070 г, 0,595 ммоля, 1,5 экв.) и триэтиламин (0,171 мл, 1,190 ммоля, 3,0 экв.). Полученную реакционную смесь продували газообразным N2 в течение 5 мин, затем добавляли Pd(dppf)Cl2 (0,014 г, 0,019 ммоля, 0,1 экв.). Реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. Реакционную смесь охлаждали до RT, разбавляли водой (60 мл) промывали этилацетатом (20 мл×2). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали (E)-6-(2-(5-метилпиридин-2-ил)винил)хинолин-4-карбоновую кислоту (0,100 г, 86% выход) в виде желтого твердого вещества.
[0466] LCMS 291,0 [0M+H]+
[0467] 1H NMR (400 MHz, DMSO-d6) δ 9,37 (br. s., 1 H) 8,90-9,03 (m, 1 H) 8,82 (s, 1 H) 8,45 (s, 1 H) 8,20 (d, J=7,89 Hz, 1 H) 8,01-8,13 (m, 1 H) 7,75-7,90 (m, 2 H) 7,54-7,75 (m, 2 H) 7,45 (d, J=16,22 Hz, 1 H) 2,33 (s, 3 H).
[0468] Стадия 3: Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(5-метилпиридин-2-ил)винил)хинолин-4-карбоксамида. При перемешивании к раствору (E)-6-(2-(5-метилпиридин-2-ил)винил)хинолин-4-карбоновой кислоты (0,100 г, 0,344 ммоля, 1,0 экв.) в DMF (2 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,116 г, 0,517 ммоля, 1,5 экв.), HOBt (0,069 г, 0,517 ммоля, 1,5 экв.) и EDC.HCl (0,098 г, 0,517 ммоля, 1,5 экв.), затем добавляли TEA (0,09 мл). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×3) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-5% MeOH в DCM в качестве элюента) и получали (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(5-метилпиридин-2-ил)винил)хинолин-4-карбоксамид (0,070 г, 44,30% выход) в виде почти белого твердого вещества.
[0469] LCMS 462,4 [0M+H]+
[0470] 1H NMR (400 MHz, DMSO-d6) δ 9,19 (t, J=5,92 Hz, 1 H) 8,95 (d, J=3,95 Hz, 1 H) 8,58 (s, 1 H) 8,47 (s, 1 H) 8,17 (d, J=10,09 Hz, 1 H) 8,08 (d, J=8,77 Hz, 1 H) 7,79 (d, J=16,22 Hz, 1 H) 7,44-7,65 (m, 3 H) 5,25 (d, J=7,45 Hz, 1 H) 4,11-4,40 (m, 4 H) 2,77-3,05 (m, 2 H) 2,33 (s, 3 H).
Пример S37
Синтез (S, E)-6-(4-(трет-бутил)стирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамида
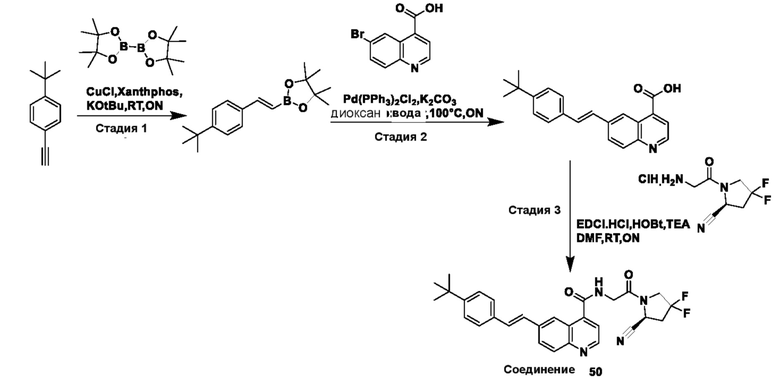
[0471] Стадия 1: Синтез 2-[02-(4-трет-бутилфенил)этенил]-4,4,5,5-тетраметил-1,3,2-диоксаборолана. При перемешивании к раствору 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан (0,200 г, 1,265 ммоля, 1,0 экв.) в THF (5 мл) добавляли CuCl(I) (0,001 г, 0,012 ммоля, 0,01 экв.) и Xanthphos (0,007 г, 0,012 ммоля, 0,01 экв.). Полученную реакционную смесь перемешивали при КТ в течение 15 мин. Через 15 мин добавляли трет-бутоксид калия (0,170 г, 1,518 ммоля, 1,2 экв.) и 1-трет-бутил-4-этинилбензол (0,200 г, 1,265 ммоля, 1,0 экв.). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью TLC. После завершения реакции реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (80 мл×3). Объединенные органические экстракты промывали водой (20 мл×3) и рассолом (20 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (5% EA-гексан в качестве элюента) и получали 2-[02-(4-трет-бутилфенил)этенил]-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,100 г, 27,62% выход) в виде желтого твердого вещества.
[0472] LCMS 287,0 [0M+H]+
[0473] 1H NMR (400 MHz, хлороформ-d) δ 7,41-7,48 (m, 2 H) 7,31-7,41 (m, 2 H) 7,26 (s, 1 H) 6,12 (d, J=18,42 Hz, 1 H) 1,31 (s, 12 H).
[0474] Стадия 2: Синтез (E)-6-(4-(трет-бутил)стирил)хинолин-4-карбоновой кислоты. При перемешивании к раствору 6-бромхинолин-4-карбоновой кислоты (0,100 г, 0,396 ммоля, 1,0 экв.) в диоксане (3 мл) добавляли 2-[02-(4-трет-бутилфенил)этенил]-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,136 г, 0,476 ммоля, 1,2 экв.) и раствор K2CO3 (0,109 г, 0,792 ммоля, 2,0 экв.) в воде (1 мл), и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,013 г, 0,019 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. Реакционную смесь охлаждали до RT, разбавляли водой (50 мл) промывали этилацетатом (20 мл×3). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали (E)-6-(4-(трет-бутил)стирил)хинолин-4-карбоновую кислоту (0,100 г, 76% выход) в виде желтого твердого вещества.
[0475] LCMS 332,1 [0M+H]+
[0476] 1H NMR (400 MHz, DMSO-d6) δ 8,64-8,78 (m, 2 H) 8,55 (s, 1 H) 8,01 (d, J=7,89 Hz, 1 H) 7,89 (d, J=8,33 Hz, 1 H) 7,59 (d, J=8,33 Hz, 2 H) 7,37-7,51 (m, 2 H) 7,33 (d, J=6,58 Hz, 2 H) 1,30 (s, 9 H).
[0477] Стадия 3: Синтез (S, E)-6-(4-(трет-бутил)стирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамида. При перемешивании к раствору (E)-6-(4-(трет-бутил)стирил)хинолин-4-карбоновой кислоты (0,100 г, 0,302 ммоля, 1,0 экв.) в DMF (2 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,101 г, 0,453 ммоля, 1,5 экв.), HOBt (0,061 г, 0,453 ммоля, 1,5 экв.) и EDC.HCl (0,086 г, 0,453 ммоля, 1,5 экв.), затем добавляли TEA (0,09 мл). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×3) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-5% MeOH в DCM в качестве элюента) и получали (S, E)-6-(4-(трет-бутил)стирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамид (0,060 г, 39,73% выход) в виде почти белого твердого вещества.
[0478] LCMS 503,4 [0M+H]+
[0479] 1H NMR (400 MHz, DMSO-d6) δ 9,18 (br. s., 1 H) 8,92 (d, J=3,95 Hz, 1 H) 8,57 (s, 1 H) 8,13 (d, J=8,77 Hz, 1 H) 8,06 (d, J=9,21 Hz, 1 H) 7,63 (d, J=8,33 Hz, 2 H) 7,49-7,60 (m, 2 H) 7,29-7,49 (m, 3 H) 5,23 (d, J=8,77 Hz, 1 H) 4,26-4,46 (m, 2 H) 4,05-4,26 (m, 2 H) 2,96 (br. s., 1 H) 2,88 (d, J=15,79 Hz, 1 H) 1,30 (s, 9 H).
Пример S38
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(4-(трифторметил)стирил)хинолин-4-карбоксамида
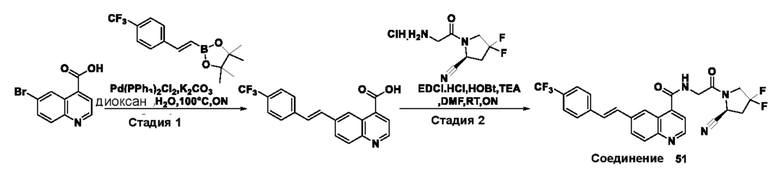
[0480] Стадия 1: Синтез (E)-6-(4-трифторметил)стирил)хинолин-4-карбоновой кислоты. К раствору 6-бромхинолин-4-карбоновой кислоты (0,140 г, 0,56 ммоля, 0,8 экв.) и (E)-2-(4-трифторметил)стирил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,220 г, 0,70 ммоля, 1,0 экв.) в диоксане (10 мл) и воде (1 мл) добавляли K2CO3 (0,147 г, 1,4 ммоля, 2,0 экв.) и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh2)Cl2 (0,024 г, 0,035 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции реакцию останавливали водой (20 мл) и водный слой промывали этилацетатом (10 мл×2). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали (E)-6-(4-трифторметилстирил)хинолин-4-карбоновую кислоту (0,250 г, количественный выход) в виде желтого твердого вещества.
[0481] LCMS 360,1 [0M+H]+
[0482] Стадия 2: Синтез (S, E)-6-(4-трифторметилстирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин-4-карбоксамида. При перемешивании к раствору (E)-6-(4-трифторметил)стирил)хинолин-4-карбоновой кислоты (0,250 г, 0,726 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,245 г, 1,09 ммоля, 1,5 экв.), EDC.HCl (0,209 г, 1,09 ммоля, 1,5 экв.) и HOBt (0,145 г, 1,09 ммоля, 1,5 экв.), затем добавляли TEA (0,2 мл). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×3) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-5% MeOH в DCM в качестве элюента), затем с помощью HPLC с обращенной фазой и получали (S, E)-6-(4-трифторметилстирил)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)хинолин- 4-карбоксамид (0,008 г, 32% выход) в виде почти белого твердого вещества.
[0483] LCMS 515,3[0M+H]+
[0484] 1H NMR (400 MHz, DMSO-d6) δ 9,21 (s, 1 H) 8,96 (d, J=4,38 Hz, 1 H) 8,69 (s, 1 H) 8,16 (d, J=8,77 Hz, 1 H) 8,10 (d, J=8,77 Hz, 1 H) 7,93 (m, J=8,33 Hz, 2 H) 7,72-7,80 (m, 2 H) 7,66 (d, J=14,47 Hz, 1 H) 7,53-7,61 (m, 1 H) 5,24 (d, J=8,77 Hz, 1 H) 4,18-4,36 (br. s., 4 H) 2,96 (br. s., 2 H).
Пример S39
Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(6-(4-метилпиперазин-1-ил)пиридин-3-ил)хинолин-4-карбоксамида

[0485] Стадия 1: Синтез 6-(6-(4-метилпиперазин-1-ил)пиридин-3-ил)хинолин-4-карбоновой кислоты. При перемешивании к раствору 6-бромхинолин-4-карбоновой кислоты (0,250 г, 0,992 ммоля, 1,0 экв.) в диоксане (5 мл) добавляли пинаколиновый эфир 2-(4-метилпиперазин-1-ил)пиридин-5-бороновой кислоты (0,360 г, 1,190 ммоля, 1,5 экв.), добавляли K2CO3 (0,273 г, 1,984 ммоля, 2,0 экв.) в воде (2 мл) и смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,034 г, 0,049 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. Реакционную смесь охлаждали до RT, разбавляли водой (60 мл) промывали этилацетатом (25 мл×3). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали 6-(6-(4-метилпиперазин-1-ил)пиридин-3-ил)хинолин-4-карбоновую кислоту (0,250 г, 72% выход) в виде желтого твердого вещества.
[0486] LCMS 349,0 [0M+H]+
[0487] 1H NMR (400 MHz, DMSO-d6) δ 8,94 (s, 1 H) 8,73 (d, J=4,38 Hz, 1 H) 8,53 (d, J=2,19 Hz, 1 H) 7,96 (s, 2 H) 7,88-7,93 (m, 1 H) 7,47 (d, J=4,38 Hz, 1 H) 6,98 (d, J=8,77 Hz, 1 H) 3,55 (d, J=5,26 Hz, 4 H) 2,39-2,44 (m, 4 H) 2,23 (s, 3 H).
[0488] Стадия 2: Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(6-(4-метилпиперазин-1-ил)пиридин-3-ил)хинолин-4-карбоксамида. При перемешивании к раствору 6-(6-(4-метилпиперазин-1-ил)пиридин-3-ил)хинолин-4-карбоновой кислоты (0,100 г, 0,287 ммоля, 1,0 экв.) в DMF (2 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,082 г, 0,431 ммоля, 1,5 экв.), HOBt (0,058 г, 0,431 ммоля, 1,5 экв.) и EDC.HCl (0,082 г, 0,431 ммоля, 1,5 экв.), затем добавляли TEA (0,1 мл). Полученную реакционную смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл×2). Объединенные органические экстракты промывали водой (50 мл×3) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-5% MeOH в DCM в качестве элюента), затем с помощью HPLC с обращенной фазой и получали (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(6-(4-метилпиперазин-1-ил)пиридин-3-ил)хинолин-4-карбоксамид (0,115 г, 77% выход) в виде почти белого твердого вещества.
[0489] LCMS 520,5 [0M+H]+
[0490] 1H NMR (400 MHz, DMSO-d6) δ 9,17 (t, J=5,92 Hz, 1 H) 8,94 (d, J=4,38 Hz, 1 H) 8,65 (dd, J=6,14, 2,19 Hz, 2 H) 8,16-8,23 (m, 1 H) 8,07-8,16 (m, 2 H) 7,56 (d, J=4,38 Hz, 1 H) 6,98 (d, J=9,21 Hz, 1 H) 5,19 (dd, J=9,21, 2,63 Hz, 1 H) 4,22-4,42 (m, 3 H) 4,08-4,22 (m, 1 H) 3,50-3,65 (m, 4 H) 2,78-3,01 (m, 2 H) 2,36-2,46 (m, 4 H) 2,23 (s, 3 H).
Пример S40
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-гидроксипиридин-3-ил)винил)хинолин-4-карбоксамида

[0491] Стадия 1: Синтез 2-хлор-5-винилпиридина. При перемешивании к раствору 5-бром-2-хлорпиридина (1,0 г, 5,19 ммоля, 1,0 экв.) в THF (16 мл) добавляли 2-винил-4,4,5,5-тетраметил-1,3,2-диоксаборолан (1,20 г, 7,79 ммоля, 1,5 экв.) и раствор Na2CO3 (2,75 г, 25,98 ммоля, 5,0 экв.) в воде (4 мл). Полученную смесь продували газообразным N2 в течение 10 мин., затем добавляли Pd(PPh3)4 (0,120 г, 0,103 ммоля, 0,2 экв.). Полученную реакционную смесь нагревали при 75°C в течение ночи. Образование продукта подтверждали с помощью TLC. Реакционную смесь охлаждали до RT, разбавляли водой (50 мл) экстрагировали этилацетатом (80 мл×3). Объединенные органические экстракты промывали водой (80 мл×2) и рассолом (80 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-5% EA в гексане в качестве элюента) и получали 2-хлор-5-винилпиридин (0,600 г, 83% выход) в виде масла.
[0492] 1H NMR (400 MHz, хлороформ-d) δ 8,37 (d, J=2,63 Hz, 1 H) 7,70 (dd, J=8,33, 2,63 Hz, 1 H) 7,23-7,32 (m, 1 H) 6,67 (dd, J=17,54, 10,96 Hz, 1 H) 5,81 (d, J=17,54 Hz, 1 H) 5,41 (d, J=11,40 Hz, 1 H).
[0493] Стадия 2: Синтез (E)-6-(2-(6-хлорпиридин-3-ил)винил)хинолин-4-карбоновой кислоты. При перемешивании к раствору 6-бромхинолин-4-карбоновой кислоты (0,300 г, 1,190 ммоля, 1,0 экв.) в смеси DMF (4 мл) и диоксана (4 мл) добавляли 2-хлор-5-винилпиридин (0,248 г, 1,785 ммоля, 1,5 экв.) и триэтиламин (0,5 мл, 3,57 ммоля, 3,0 экв.). Полученную реакционную смесь продували газообразным N2 в течение 5 мин, затем добавляли Pd(dppf)Cl2 (0,043 г, 0,059 ммоля, 0,05 экв.) и реакционную смесь нагревали при 120°C в течение ночи. Образование продукта подтверждали с помощью LCMS. Реакционную смесь охлаждали до RT, разбавляли этилацетатом (100 мл), фильтровали через целит® и фильтрат промывали водой (100 мл×2), водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали (E)-6-(2-(6-хлорпиридин-3-ил)винил)хинолин-4-карбоновую кислоту (0,250 г, 67% выход) в виде желтого твердого вещества.
[0494] LCMS 311,0 [0M+H]+
[0495] Стадия 3: Синтез (E)-6-(2-(6-гидроксипиридин-3-ил)винил)хинолин-4-карбоновой кислоты. При перемешивании к раствору (E)-6-(2-(6-хлорпиридин-3-ил)винил)хинолин-4-карбоновой кислоты (0,200 г, 0,643 ммоля, 1,0 экв.) в диоксане (2 мл) и воде (2 мл) добавляли концентрированную HCl (0,5 мл). Полученную реакционную смесь нагревали при 140°C в течение 48 ч. Образование продукта подтверждали с помощью LCMS. Реакционную смесь охлаждали до RT и концентрировали при пониженном давлении и подвергали сушке вымораживанием в лиофилизаторе и получали (E)-6-(2-(6-гидроксипиридин-3-ил)винил)хинолин-4-карбоновую кислоту (0,110 г, 58% выход) в виде желтого твердого вещества.
[0496] LCMS 293,0 [0M+H]+
[0497] Стадия 4: Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-гидроксипиридин-3-ил)винил)хинолин-4-карбоксамида. При перемешивании к раствору (E)-6-(2-(6-гидроксипиридин-3-ил)винил)хинолин-4-карбоновой кислоты (0,100 г, 0,342 ммоля, 1,0 экв.) в DMF (4 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,092 г, 0,410 ммоля, 1,2 экв.), HOBt (0,069 г, 0,513 ммоля, 1,5 экв.) и EDCI.HCl (0,098 г, 0,513 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 5 мин. Добавляли триэтиламин (0,09 мл) и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (40 мл) и экстрагировали этилацетатом (50 мл×3). Объединенные органические экстракты промывали водой (60 мл×3) и рассолом (60 мл), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью HPLC с обращенной фазой и получали (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-гидроксипиридин-3-ил)винил)хинолин-4-карбоксамид (0,040 г, 15% выход) в виде желтого твердого вещества.
[0498] LCMS 464,4 [0M+H]+
[0499] 1H NMR (400 MHz, DMSO-d6) δ 9,14 (t, J=5,92 Hz, 1 H) 8,90 (d, J=4,38 Hz, 1 H) 8,39 (s, 1 H) 8,04 (s, 1 H) 7,96 (d, J=7,02 Hz, 1 H) 7,62 (br. s., 1 H) 7,54 (d, J=3,95 Hz, 1 H) 7,33 (d, J=16,22 Hz, 1 H) 7,12 (d, J=16,22 Hz, 1 H) 6,41 (d, J=9,65 Hz, 1 H) 5,20 (d, J=6,58 Hz, 1 H) 4,23-4,40 (m, 3 H) 4,09-4,23 (m, 1 H) 2,95 (br. s., 1 H) 2,87 (d, J=17,10 Hz, 2 H).
Пример S41
Синтез N-(2-((S)-2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(((S)-1-(4-фторфенил)этил)амино)изоникотинамида

[0500] Стадия 1: Синтез метил (S)-3-((1-(4-фторфенил)этил)амино)изоникотината. При перемешивании к раствору метил-3-бромизоникотината (0,500 г, 2,31 ммоля, 1,0 экв.) и (S)-1-(4-фторфенил)этан-1-амин (0,350 г, 2,54 ммоля, 1,0 экв.) в диоксане (10 мл) добавляли CS2CO3(1,5 г, 4,62 ммоля, 2 экв.).Полученную смесь продували азотом в течение 10 мин, затем добавляли Pd2(dba)3 (0,110 г, 0,115 ммоля, 0,05 экв.) и xanthphos (0,135 г, 0,231 ммоля, 0,1 экв.). Реакционную смесь нагревали при 120°C в течение ночи. За протеканием реакции следили с помощью TLC и LCMS. Реакционную смесь разбавляли водой (50 мл), экстрагировали с помощью EtOAc (3×80 мл). Объединенный органический слой промывали водой (2×60 мл), рассолом (60 мл), сушили над Na2SO4, концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-50% этилацетата в гексане в качестве элюента) и получали метил (S)-3-((1-(4-фторфенил)этил)амино)изоникотинат (0,210 г, 34% выход) в виде масла.
[0501] LCMS: 275,0 [0M+H] +
[0502] 1H NMR: (400 MHz, хлороформ-d) δ 7,97 (s, 1 H) 7,78-7,90 (m, 2 H) 7,61 (d, J=5,26 Hz, 1 H) 7,22-7,36 (m, 2 H) 6,95-7,07 (m, 2 H) 4,60-4,76 (m, 1 H) 3,87-4,00 (s, 3 H) 1,59 (d, J=7,02 Hz, 3 H).
[0503] Стадия 2: Синтез литиевой соли (S)-3-((1-(4-фторфенил)этил)амино)изоникотиновой кислоты. При перемешивании к раствору метил (S)-3-((1-(4-фторфенил)этил)амино)изоникотинат (0,200 г, 0,727 ммоля, 1,0 экв.) в THF (10 мл) и воде (4 мл) добавляли LiOH (0,035 г, 1,45 ммоля, 2 экв.). Смесь перемешивали при 80°C в течение ночи. Образование продукта подтверждали с помощью LCMS. Реакционную смесь концентрировали и разбавляли водой (50 мл) и промывали этилацетатом (2×50 мл). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали литиевую соль (S)-3-((1-(4-фторфенил)этил)амино)изоникотиновой кислоты (0,200 г, количественный выход) в виде желтого твердого вещества.
[0504] LCMS: 261,0 [0M+H] +
[0505] Стадия 3: Синтез N-(2-((S)-2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(((S)-1-(4-фторфенил)этил)амино)изоникотинамида. При перемешивании к раствору литиевой соли (S)-3-((1-(4-фторфенил)этил)амино)изоникотиновой кислоты (0,200 г, 0,769 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,259 г, 1,153 ммоля, 1,5 экв.), HATU (0,438 г, 1,153 ммоля, 1,5 экв.) и DIPEA (0,198 г, 1,538 ммоля, 2,0 экв.). Полученную реакционную смесь перемешивали при КТ в течение ночи. За протеканием реакции следили с помощью LCMS и TLC, реакционную смесь разбавляли водой (30 мл) экстрагировали этилацетатом (3×40 мл). Объединенный органический слой промывали водой (40 мл×4), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии с обращенной фазой и получали N-(2-((S)-2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(((S)-1-(4-фторфенил)этил)амино)изоникотинамид (0,050 г, 15%) в виде почти белого твердого вещества.
[0506] LCMS: 432,4 [0M+H] +
[0507] 1H NMR: (400 MHz, DMSO-d6) δ 9,03 (t, J=5,70 Hz, 1 H) 7,89-7,95 (m, 2 H) 7,83 (d, J=5,26 Hz, 1 H) 7,48 (d, J=5,26 Hz, 1 H) 7,40 (dd, J=8,55, 5,48 Hz, 2 H) 7,15 (t, J=8,77 Hz, 2 H) 5,13 (dd, J=9,21, 2,63 Hz, 1 H) 4,81 (t, J=6,58 Hz, 1 H) 4,25-4,39 (m, 1 H) 3,99-4,24 (m, 3 H) 2,89-3,02 (m, 1 H) 2,83 (d, J=18,42 Hz, 1 H) 1,45 (d, J=6,58 Hz, 3 H).
Пример S42
Синтез N-(2-((S)-2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(((R)-1-(4-фторфенил)этил)амино)изоникотинамида

[0508] Стадия 1: Синтез метил-(R)-3-((1-(4-фторфенил)этил)амино)изоникотината. При перемешивании к раствору метил-3-бромизоникотината (0,500 г, 2,31 ммоля, 1,0 экв.) и (S)-1-(4-фторфенил)этан-1-амина (0,350 г, 2,54 ммоля, 1,0 экв.) в диоксане (10 мл) добавляли CS2CO3(1,5 г, 4,62 ммоля, 2 экв.).Полученную смесь продували азотом в течение 10 мин, затем добавляли Pd2(dba)3 (0,110 г, 0,115 ммоля, 0,05 экв.) и xanthphos (0,135 г, 0,231 ммоля, 0,1 экв.). Реакционную смесь нагревали при 120°C в течение ночи. За протеканием реакции следили с помощью TLC и LCMS. Реакционную смесь разбавляли водой (50 мл), экстрагировали с помощью EtOAc (3×80 мл). Объединенный органический слой промывали водой (2×60 мл), рассолом (60 мл), сушили над Na2SO4, концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-50% этилацетата в гексане в качестве элюента) и получали метил-(R)-3-((1-(4-фторфенил)этил)амино)изоникотинат (0,220 г, 35% выход) в виде масла.
[0509] LCMS: 275,0 [0M+H] +
[0510] 1H NMR (400 MHz, хлороформ-d) δ 7,97 (s, 1 H) 7,78-7,90 (m, 2 H) 7,61 (d, J=5,26 Hz, 1 H) 7,22-7,36 (m, 2 H) 6,95-7,07 (m, 2 H) 4,60-4,76 (m, 1 H) 3,87-4,00 (s, 3 H) 1,59 (d, J=7,02 Hz, 3 H).
[0511] Стадия 2: Синтез литиевой соли (R)-3-((1-(4-фторфенил)этил)амино)изоникотиновой кислоты. При перемешивании к раствору метил-(R)-3-((1-(4-фторфенил)этил)амино)изоникотината (0,200 г, 0,727 ммоля, 1,0 экв.) в THF (10 мл) и воде (4 мл) добавляли LiOH (0,035 г, 1,45 ммоля, 2 экв.). Смесь перемешивали при 80°C в течение ночи. Образование продукта подтверждали с помощью LCMS. Реакционную смесь концентрировали и разбавляли водой (50 мл) и промывали этилацетатом (2×50 мл). Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали литиевую соль (R)-3-((1-(4-фторфенил)этил)амино)изоникотиновой кислоты (0,200 г, количественный выход) в виде желтого твердого вещества.
[0512] LCMS: 261,0 [0M+H] +
[0513] Стадия 3: Синтез N-(2-((S)-2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(((R)-1-(4-фторфенил)этил)амино)изоникотинамида. При перемешивании к раствору литиевой соли (R)-3-((1-(4-фторфенил)этил)амино)изоникотиновой кислоты (0,200 г, 0,769 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,259 г, 1,15 ммоля, 1,5 экв.), HATU (0,438 г, 1,15 ммоля, 1,5 экв.) и DIPEA (0,198 г, 1,53 ммоля, 2,0 экв.). Полученную реакционную смесь перемешивали при КТ в течение ночи. За протеканием реакции следили с помощью LCMS и TLC, реакционную смесь разбавляли водой (30 мл) экстрагировали этилацетатом (3×40 мл). Объединенный органический слой промывали водой (40 мл×2). Органический слой сушили над безводным Na2SO4, концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии с обращенной фазой и получали N-(2-((S)-2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(((R)-1-(4-фторфенил)этил)амино)изоникотинамид (0,050 г, 15% выход) в виде почти белого твердого вещества.
[0514] LCMS: 432,4 [0M+H] +
[0515] 1H NMR (400 MHz, DMSO-d6) δ 9,03 (t, J=5,70 Hz, 1 H) 7,89-7,95 (m, 2 H) 7,83 (d, J=5,26 Hz, 1 H) 7,48 (d, J=5,26 Hz, 1 H) 7,40 (dd, J=8,55, 5,48 Hz, 2 H) 7,15 (t, J=8,77 Hz, 2 H) 5,13 (dd, J=9,21, 2,63 Hz, 1 H) 4,81 (t, J=6,58 Hz, 1 H) 4,25-4,39 (m, 1 H) 3,99-4,24 (m, 3 H) 2,89-3,02 (m, 1 H) 2,83 (d, J=18,42 Hz, 1 H) 1,45 (d, J=6,58 Hz, 3 H).
Пример S43
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(2-метилпиридин-4-ил)винил)хинолин-4-карбоксамида

[0516] Стадия 1: Синтез 2-(метил)-4-винилпиридина. При перемешивании к раствору 4-бром-2-(метил)пиридина (0,500 г, 2,90 ммоля, 1,0 экв.) в диоксане (8 мл) добавляли 2-винил-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,671 г, 4,36 ммоля, 1,5 экв.) и K2CO3 (0,802 г, 5,81 ммоля, 2,0 экв.) в воде (4 мл). Полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,102 г, 0,145 ммоля, 0,05 экв.). Реакционную смесь нагревали при 120°C в течение ночи. Образование продукта подтверждали с помощью TLC. Реакционную смесь охлаждали до RT, разбавляли водой (50 мл) экстрагировали этилацетатом (100 мл×3). Объединенные органические экстракты промывали водой (50 мл×3), рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (0-15% этилацетата в гексане в качестве элюента) и получали 2-(метил)-4-винилпиридин (0,170 г, 49% выход) в виде желтого полужидкого вещества.
[0517] 1H NMR (400 MHz, хлороформ-d) δ 8,44 (d, J=5,26 Hz, 1 H) 7,05-7,19 (m, 2 H) 6,63 (dd, J=17,76, 10,74 Hz, 1 H) 5,94 (d, J=17,54 Hz, 1 H) 5,45 (d, J=10,96 Hz, 1 H) 2,55 (s, 3 H).
[0518] Стадия 2: Синтез (E)-6-(2-(6-метилпиридин-3-ил)винил)хинолин-4-карбоновой кислоты. При перемешивании к раствору 6-бромхинолин-4-карбоновой кислоты (0,200 г, 0,79 ммоля, 1,0 экв.) в DMF (5 мл) добавляли 2-(метил)-4-винилпиридин (0,141 г, 1,19 ммоля, 1,5 экв.) и триэтиламин (0,34 мл, 2,38 ммоля, 3,0 экв.). Полученную реакционную смесь продували газообразным N2 в течение 5 мин, затем добавляли Pd(dppf)Cl2 (0,058 г, 0,079 ммоля, 0,1 экв.). Реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. Реакционную смесь охлаждали до RT, разбавляли водой (40 мл) промывали этилацетатом (40 мл×2), Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали (E)-6-(2-(6-метилпиридин-3-ил)винил)хинолин-4-карбоновую кислоту (0,200 г, 86% выход) в виде желтого твердого вещества.
[0519] LCMS 291,1 [0M+H]+
[0520] Стадия 3: Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-метилпиридин-3-ил)винил)хинолин-4-карбоксамида. При перемешивании к раствору (E)-6-(2-(6-метилпиридин-3-ил)винил)хинолин-4-карбоновой кислоты (0,200 г, 0,689 ммоля, 1,0 экв.) в DMF (4 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,232 г, 1,034 ммоля, 1,5 экв.), HOBt (0,139 г, 1,034 ммоля, 1,5 экв.) и EDCI.HCl (0,197 г, 1,034 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 10 мин. триэтиламин (0,19 мл,) добавляли и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (60 мл×3). Объединенные органические экстракты промывали водой (50 мл×3) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (5% MeOH в DCM в качестве элюента), затем с обращенной фазой и получали (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-метилпиридин-3-ил)винил)хинолин-4-карбоксамид (0,090 г, 28% выход) в виде почти белого твердого вещества.
[0521] LCMS 462,4 [0M+H]+
[0522] 1H NMR (400 MHz, DMSO-d6) δ 9,20 (t, J=6,14 Hz, 1 H) 8,96 (d, J=3,95 Hz, 1 H) 8,69 (s, 1 H) 8,44 (d, J=4,82 Hz, 1 H) 8,12 (q, J=8,62 Hz, 2 H) 7,71 (d, J=16,22 Hz, 1 H) 7,58 (d, J=4,39 Hz, 1 H) 7,51-7,56 (m, 2 H) 7,46 (d, J=4,82 Hz, 1 H) 5,25 (d, J=6,58 Hz, 1 H) 4,12-4,41 (m, 4 H) 2,81-3,03 (m, 2 H).
Пример S44
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(2-метоксипиридин-4-ил)винил)хинолин-4-карбоксамида
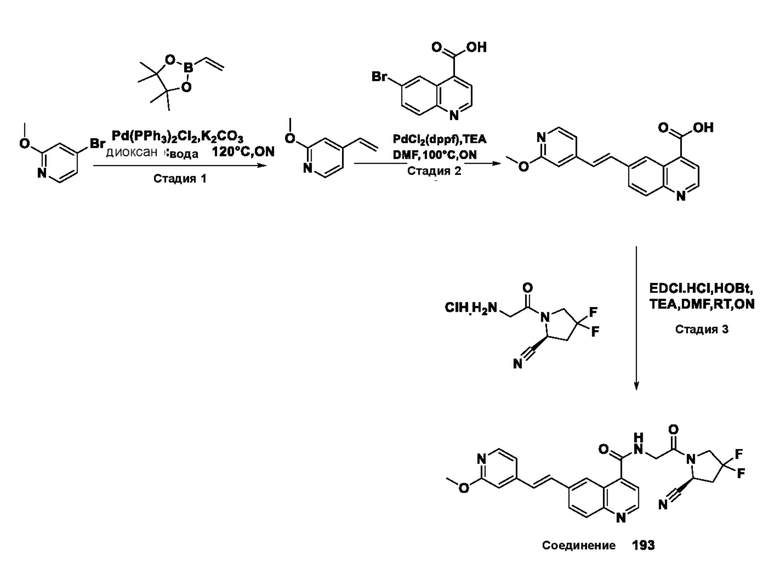
[0523] Стадия 1: Синтез 4-этенил-2-метоксипиридина. При перемешивании к раствору 4-бром-2-метоксипиридина (0,500 г, 2,66 ммоля, 1,0 экв.) в диоксане (8 мл) добавляли 2-винил-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,614 г, 3,99 ммоля, 1,5 экв.) и K2CO3 (0,734 г, 5,32 ммоля, 2,0 экв.) в воде (4 мл), и полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,093 г, 0,132 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 120°C в течение ночи. Образование продукта подтверждали с помощью TLC. Реакционную смесь охлаждали до RT, разбавляли водой (50 мл) экстрагировали этилацетатом (50 мл×3). Объединенные органические экстракты промывали водой (50 мл×2) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (0-10% этилацетата в гексане в качестве элюента) и получали 4-этенил-2-метоксипиридин (0,180 г, 50% выход) в виде масла.
[0524] 1H NMR (400 MHz, хлороформ-d) δ 8,11 (d, J=5,26 Hz, 1 H) 6,91 (dd, J=5,26, 1,32 Hz, 1 H) 6,69 (s, 1 H) 6,62 (dd, J=17,54, 10,96 Hz, 1 H) 5,91 (d, J=17,54 Hz, 1 H) 5,44 (d, J=10,52 Hz, 1 H) 3,94 (s, 3 H).
[0525] Стадия 2: Синтез (E)-6-(2-(6-метоксипиридин-3-ил)винил)хинолин-4-карбоновой кислоты. При перемешивании к раствору 6-бромхинолин-4-карбоновой кислоты (0,200 г, 0,793 ммоля, 1,0 экв.) в DMF (5 мл) добавляли 4-этенил-2-метоксипиридин (0,160 г, 1,190 ммоля, 1,5 экв.) и триэтиламин (0,34 мл, 2,380 ммоля, 3,0 экв.). Полученную реакционную смесь продували газообразным N2 в течение 5 мин, затем добавляли Pd(dppf)Cl2 (0,058 г, 0,079 ммоля, 0,1 экв.). Реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. Реакционную смесь охлаждали до RT, разбавляли водой (40 мл) промывали этилацетатом (40 мл×3), Водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали (E)-6-(2-(6-метоксипиридин-3-ил)винил)хинолин-4-карбоновую кислоту (0,200 г, 82% выход) в виде желтого твердого вещества.
[0526] LCMS 307,1 [0M+H]+
[0527] Стадия 3: Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-метоксипиридин-3-ил)винил)хинолин-4-карбоксамида. При перемешивании к раствору (E)-6-(2-(6-метоксипиридин-3-ил)винил)хинолин-4-карбоновой кислоты (0,200 г, 0,653 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,220 г, 0,980 ммоля, 1,5 экв.), HOBt (0,132 г, 0,980 ммоля, 1,5 экв.) и EDCI.HCl (0,187 г, 0,980 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 10 мин. Добавляли триэтиламин (0,18 мл) и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл×3). Объединенные органические экстракты промывали водой (100 мл×3) и рассолом (100 мл), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (5% MeOH в DCM в качестве элюента) и получали S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-метоксипиридин-3-ил)винил)хинолин-4-карбоксамид (0,110 г, 35% выход) в виде желтого твердого вещества.
[0528] LCMS 478,4 [0M+H]+
[0529] 1H NMR (400 MHz, DMSO-d6) δ 9,20 (br. s., 1 H) 8,97 (d, J=4,39 Hz, 1 H) 8,60 (s, 1 H) 8,16 (d, J=5,70 Hz, 2 H) 8,07-8,13 (m, 1 H) 7,70 (d, J=16,22 Hz, 1 H) 7,59 (d, J=4,39 Hz, 1 H) 7,49 (d, J=16,66 Hz, 1 H) 7,31 (d, J=5,70 Hz, 1 H) 7,04 (s, 1 H) 5,22 (d, J=6,58 Hz, 1 H) 4,24-4,41 (m, 2 H) 4,10-4,24 (m, 1 H) 3,87 (s, 3 H) 2,96 (br. s., 1 H) 2,88 (d, J=12,28 Hz, 1 H).
Пример S45
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(2-(трифторметил)пиридин-4-ил)винил)хинолин-4-карбоксамида

[0530] Стадия 1: Синтез 2-(трифторметил)-4-винилпиридина. При перемешивании к раствору 4-бром-2-(трифторметил)пиридина (0,500 г, 2,21 ммоля, 1,0 экв.) в диоксане (8 мл) добавляли 2-винил-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,511 г, 3,31 ммоля, 1,5 экв.) и K2CO3 (0,610 г, 4,42 ммоля, 2,0 экв.) в воде (4 мл). Полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(PPh3)Cl2 (0,077 г, 0,110 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 120°C в течение ночи. Образование продукта подтверждали с помощью TLC. Реакционную смесь охлаждали до RT, разбавляли водой (100 мл) экстрагировали этилацетатом (100 мл×3). Объединенные органические экстракты промывали водой (100 мл×2) и рассолом (100 мл), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (0-10% этилацетата в гексане в качестве элюента) и получали 2-(трифторметил)-4-винилпиридин (0,210 г, 55% выход) в виде желтого полужидкого вещества.
[0531] 1H NMR (400 MHz, хлороформ-d) δ8,68 (d, J=5,26 Hz, 1 H) 7,66 (s, 1 H) 7,45 (d, J=3,95 Hz, 1 H) 6,73 (dd, J=17,76, 10,74 Hz, 1 H) 6,07 (d, J=17,54 Hz, 1 H) 5,62 (d, J=10,96 Hz, 1 H).
[0532] Стадия 2: Синтез (E)-6-(2-(6-(трифторметил)пиридин-3-ил)винил)хинолин-4-карбоновой кислоты. При перемешивании к раствору 6-бромхинолин-4-карбоновой кислоты (0,200 г, 0,793 ммоля, 1,0 экв.) в диоксане (4 мл) добавляли 2-(трифторметил)-4-винилпиридин (0,205 г, 1,190 ммоля, 1,5 экв.) и триэтиламин (0,34 мл, 2,380 ммоля, 3,0 экв.). Полученную реакционную смесь продували газообразным N2 в течение 5 мин, затем добавляли Pd(dppf)Cl2 (0,058 г, 0,0793 ммоля, 0,1 экв.). Реакционную смесь нагревали при 120°C в течение ночи. Образование продукта подтверждали с помощью LCMS. Реакционную смесь охлаждали до RT, разбавляли водой (50 мл) промывали этилацетатом (50 мл×2), водный слой отделяли и сушили вымораживанием в лиофилизаторе и получали (E)-6-(2-(6-(трифторметил)пиридин-3-ил)винил)хинолин-4-карбоновую кислоту (0,200 г, 73% выход) в виде желтого твердого вещества.
[0533] LCMS 345,1 [0M+H]+
[0534] Стадия 3: Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-(трифторметил)пиридин-3-ил)винил)хинолин-4-карбоксамида. При перемешивании к раствору (E)-6-(2-(6-(трифторметил)пиридин-3-ил)винил)хинолин-4-карбоновой кислоты (0,200 г, 0,581 ммоля, 1,0 экв.) в DMF (4 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,196 г, 0,872 ммоля, 1,5 экв.), HOBt (0,117 г, 0,872 ммоля, 1,5 экв.) и EDCI.HCl (0,166 г, 0,872 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 10 мин. триэтиламин (0,2 мл) добавляли и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (40 мл) и экстрагировали этилацетатом (50 мл×2). Объединенные органические экстракты промывали водой (50 мл×2) и рассолом (50 мл), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (5% MeOH в DCM в качестве элюента), затем с обращенной фазой и получали (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(2-(6-(трифторметил)пиридин-3-ил)винил)хинолин-4-карбоксамид (0,09 г, 3% выход) в виде почти белого твердого вещества.
[0535] LCMS 516,4 [0M+H]+
[0536] 1H NMR (400 MHz, DMSO-d6) δ 9,22 (br. s., 1 H) 8,99 (d, J=4,38 Hz, 1 H) 8,75 (d, J=4,38 Hz, 1 H) 8,68 (s, 1 H) 8,10-8,24 (m, 2 H) 7,90-8,02 (m, 2 H) 7,69 (s, 1 H) 7,59-7,65 (m, 1 H) 5,23 (d, J=7,45 Hz, 1 H) 4,36 (br. s., 1 H) 4,30 (br. s., 3 H) 2,88 (d, J=12,28 Hz, 2 H).
Пример S46
Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(4-(2-гидроксипропан-2-ил)фенил)хинолин-4-карбоксамида
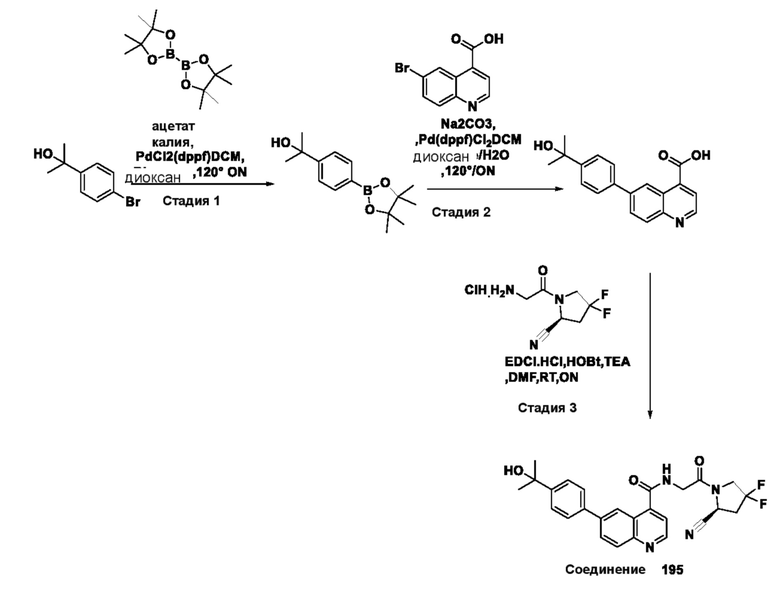
[0537] Стадия 1: Синтез 2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропан-2-ола. К раствору 2-(4-бромфенил)пропан-2-ола (0,3 г, 1,401 ммоля, 1,0 экв.) в диоксане (08 мл) добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (0,42 г, 1,681 ммоля, 1,0 экв.), ацетат калия (0,41 г, 4,203 ммоля, 3,0 экв.). Полученную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(dppf)Cl2DCM (0,057 г, 0,070 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 120°C в течение ночи. Образование продукта подтверждали с помощью TLC. После завершения реакции смесь разбавляли водой (20 мл×2) и экстрагировали этилацетатом (40 мл×2). Объединенные органические экстракты промывали водой (10 мл×2), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии и получали 2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропан-2-ол (0,230 г, 68% выход) в виде почти белого твердого вещества.
[0538] 1H NMR (400 MHz, DMSO-d6) δ 7,61 (m, J=8,33 Hz, 2 H) 7,47 (m, J=8,33 Hz, 2 H) 5,04 (s, 1 H) 1,40 (s, 6 H) 1,28 (s, 12 H).
[0539] Стадия 2: Синтез 6-(4-(2-гидроксипропан-2-ил)фенил)хинолин-4-карбоновой кислоты. К раствору 2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропан-2-ола (0,2 г, 0,763 ммоля, 1,0 экв.) в диоксане (5 мл) добавляли 6-бромхинолин-4-карбоновую кислоту (0,192 г, 0,763 ммоля, 1,0 экв.), Na2CO3(0,161 г, 1,526 ммоля, 1,0 экв.) в H2O (2 мл). Полученную реакционную смесь продували газообразным N2 в течение 10 мин, затем добавляли Pd(dppf)Cl2 DCM (0,031 г, 0,038 ммоля, 0,05 экв.) и нагревали при 120°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции смесь разбавляли водой (20 мл) и промывали этилацетатом (20 мл×2). Водный слой подвергали сушке вымораживанием в лиофилизаторе и получали 6-(4-(2-гидроксипропан-2-ил)фенил)хинолин-4-карбоновую кислоту (0,150 г, 64% выход) в виде почти белого твердого вещества.
[0540] LCMS 308,2 [0M+H]+
[0541] 1H NMR (400 MHz, DMSO-d6) δ 9,00 (s, 1 H) 8,75 (d, J=4,38 Hz, 1 H) 7,97 (d, J=3,51 Hz, 2 H) 7,66 (m, J=8,33 Hz, 2 H) 7,58 (m, J=8,33 Hz, 2 H) 7,48 (d, J=4,38 Hz, 1 H) 5,08 (br. s., 1 H) 1,46 (s, 6 H).
[0542] Стадия 3: Синтез (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(4-(2-гидроксипропан-2-ил)фенил)хинолин-4-карбоксамида. При перемешивании к раствору 6-(4-(2-гидроксипропан-2-ил)фенил)хинолин-4-карбоновой кислоты (0,150 г, 0,488 ммоля, 1,0 экв.) в DMF (4 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,109 г, 0,488 ммоля, 1,0 экв.), EDCI.HCl (0,111 г, 0,585 ммоля, 1,2 экв.) и HOBt (0,078 г, 0,585 ммоля, 1,2 экв.). Смесь перемешивали при КТ в течение 10 мин. Добавляли TEA (0,1 мл) и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (40 мл×2). Объединенные органические экстракты промывали водой (20 мл×4), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью HPLC с обращенной фазой и получали (S)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-6-(4-(2-гидроксипропан-2-ил)фенил)хинолин-4-карбоксамид (0,015 г, 6% выход) в виде почти белого твердого вещества.
[0543] LCMS 479,5 [0M+H]+
[0544] 1H NMR (400 MHz, DMSO-d6) δ 9,13-9,24 (m, 1 H) 8,97 (d, J=4,39 Hz, 1 H) 8,72 (s, 1 H) 8,06-8,25 (m, 2 H) 7,83 (d, J=8,33 Hz, 2 H) 7,52-7,68 (m, 3 H) 5,13-5,22 (m, 1 H) 5,08 (br. s., 1 H) 4,32-4,40 (m, 1 H) 4,06-4,32 (m, 3 H) 2,79-3,06 (m, 2 H) 1,47 (br. s., 6 H).
Пример S47
Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(1-оксо-1,2,3,4-тетрагидроизохинолин-6-ил)винил)изоникотинамида

[0545] Стадия 1: Синтез 6-винил-3,4-дигидроизохинолин-1(2H)-она. К раствору 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолана (0,500 г, 3,24 ммоля, 1,0 экв.) и 6-бром-3,4-дигидроизохинолин-1(2H)-она (0,587 г, 2,59 ммоля, 0,8 экв.) в диоксане (10 мл) и воде (2 мл) добавляли Na2CO3 (0,686 г, 6,48 ммоля, 2,0 экв.). Полученную реакционную смесь продували газообразным N2 в течение 10 мин., затем добавляли Pd(PPh3)Cl2 (0,114 г, 0,162 ммоля, 0,05 экв.). Полученную реакционную смесь нагревали при 80°C в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл×2). Объединенный органический слой промывали водой (50 мл×2), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-30% этилацетата в гексане в качестве элюента) и получали 6-винил-3,4-дигидроизохинолин-1(2H)-он (0,250 г, 64% выход) в виде желтого полужидкого вещества.
[0546] LCMS 174,1 [0M+H]+
[0547] Стадия 2: Синтез метил-(E)-3-(2-(1-оксо-1,2,3,4-тетрагидроизохинолин-6-ил)винил)изоникотината). К раствору 6-винил-3,4-дигидроизохинолин-1(2H)-она (0,150 г, 0,867 ммоля, 1,0 экв.) в диоксане (1 мл) и DMF (2 мл) добавляли метил-3-бромизоникотинат (0,280 г, 1,30 ммоля, 1,5 экв.), добавляли P(O-tol)3(0,052 г, 0,173 ммоля, 0,2 экв.) и DIPEA (0,335 г, 2,601 ммоля, 3,0 экв.).Полученную реакционную смесь продували газообразным N2 в течение 10 мин., затем добавляли Pd(OAc) (0,019 г, 0,086 ммоля, 0,1 экв.). Полученную реакционную смесь нагревали при 100°C в течение ночи. Образование продукта подтверждали с помощью LCMS. После завершения реакции смесь разбавляли водой (50 мл) экстрагировали этилацетатом (100 мл). Объединенные органические экстракты промывали водой (10 мл×4), сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (0-5% MeOH в DCM в качестве элюента) и получали метил-(E)-3-(2-(1-оксо-1,2,3,4-тетрагидроизохинолин-6-ил)винил)изоникотинат (0,1 г, 37% выход) в виде желтого твердого вещества.
[0548] LCMS 309,1 [0M+H]+
[0549] Стадия 3: Синтез (E)-3-(2-(1-оксо-1,2,3,4-тетрагидроизохинолин-6-ил)винил)изоникотиновой кислоты. При перемешивании к раствору метил-(E)-3-(2-(1-оксо-1,2,3,4-тетрагидроизохинолин-6-ил)винил)изоникотината (0,1 г, 0,323 ммоля, 1,0 экв.) в THF (3 мл) и воде (4 мл) добавляли LiOH.H2O (0,016 г, 0,388 ммоля, 1,2 экв.). Смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS. Реакционную смесь концентрировали при пониженном давлении и разбавляли водой (20 мл). Водный слой промывали этилацетатом (10 мл×2) и сушили вымораживанием в лиофилизаторе и получали (E)-3-(2-(1-оксо-1,2,3,4-тетрагидроизохинолин-6-ил)винил)изоникотиновую кислоту (0,094 г, количественный выход) в виде желтого твердого вещества.
[0550] LCMS 295,0 [0M+H]+
[0551] Стадия 4: Синтез (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(1-оксо-1,2,3,4-тетрагидроизохинолин-6-ил)винил)изоникотинамида. При перемешивании к раствору (E)-3-(2-(1-оксо-1,2,3,4-тетрагидроизохинолин-6-ил)винил)изоникотиновой кислоты (0,1 г, 0,340 ммоля, 1,0 экв.) в DMF (5 мл) добавляли (S)-4,4-дифтор-1-глицилпирролидин-2-карбонитрилгидрохлорид (0,114 г, 0,510 ммоля, 1,5 экв.), HOBt (0,068 г, 0,510 ммоля, 1,5 экв.) и EDCI.HCl (0,097 г, 0,510 ммоля, 1,5 экв.). Смесь перемешивали при КТ в течение 10 мин. Добавляли триэтиламин (0,103 г, 1,02 ммоля, 3,0 экв.) и смесь перемешивали при КТ в течение ночи. Образование продукта подтверждали с помощью LCMS и TLC. После завершения реакции смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл×2). Объединенные органические экстракты промывали водой (20 мл×4), сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии (0-5% MeOH в DCM в качестве элюента) и дополнительно очищали с помощью HPLC с обращенной фазой и получали (S, E)-N-(2-(2-циано-4,4-дифторпирролидин-1-ил)-2-оксоэтил)-3-(2-(1-оксо-1,2,3,4-тетрагидроизохинолин-6-ил)винил)изоникотинамид (0,007 г, 4% выход) в виде почти белого твердого вещества.
[0552] LCMS 466,5 [0M+H]+
[0553] 1H NMR (400 MHz, DMSO-d6) δ 9,17 (s, 1 H) 9,04 (br. s., 1 H) 8,55 (d, J=4,82 Hz, 1 H) 7,90 (br. s., 1 H) 7,86 (d, J=7,89 Hz, 2 H) 7,60-7,72 (m, 2 H) 7,52 (d, J=16,66 Hz, 2 H) 7,37 (d, J=5,26 Hz, 1 H) 5,19 (d, J=9,21 Hz, 1 H) 4,33 (br. s., 1 H) 4,20 (d, J=6,14 Hz, 1 H) 4,12 (d, J=11,40 Hz, 2 H) 3,38 (t, 2H) 2,96 (t, 2 H) 2,83 (m, 2 H).
Биологические примеры
Пример B1
Ингибирование FAPα исследуемыми соединениями оценивали с помощью анализов ферментативной активности in vitro
[0554] Исследование активности ферментной экзопептидазы FAPα (дипептидазы). Для исследования исходной активности ферментной экзопептидазы FAPα человека 40 нг FAPα рекомбинантной (rhFAPα, R&S system, #3715-SE) инкубировали со 100 мкМ пептида Z-Gly-Pro-AMC (BACHEM, #L-1145) в буфере для анализа FAPα (50 мМ Tris pH 7,4, 100 мМ NaCl, 0,1 мг/мл бычьего сывороточного альбумина) в течение 1 ч при 37°C в защищенных от действия света черных 96-луночных планшетах (Nunc, #237108). Для исследования активности ингибирования ферментной экзопептидазы FAPα исследуемыми соединениями все исследуемые соединения предварительно инкубировали с ферментом в течение 15 мин при 37°C, затем реакцию начинали путем добавления субстрата в черные 96-луночные планшеты (Nunc, #237108). высвобождение 7-амино-4-метилкумарина (AMC) детектировали путем измерения интенсивности флуоресценции при Ex/Em 380/460 нм с помощью многофункционального устройства для считывания микропланшетов (Synergy 4, Biotek). Все измерения проводили дважды. Val-boroPro, неспецифический ингибитор пролилпептидазы, использовали в качестве положительного контроля. Ингибирование rmFAPα или rhFAPα активности ферментной экзопептидазы при 1 мкМ в процентах определяли для некоторых соединений, приведенных в таблице 2. Для расчетов средние значения результатов измерений для реакций, содержащих только разбавитель и субстрат, без фермента, использовали в качестве холостых значений и их вычитали из остальных значений. Ингибирование в процентах рассчитывали с использованием средних значений результатов измерений для реакций, содержащих разбавитель, фермент и субстрат при максимальной ферментативной активности. Кроме того, IC50 для ферментной экзопептидазной активности rmFAPα или rhFAPα некоторых соединений также приведены в таблице 2. Измерения проводили однократно.
[0555] Исследование активности ферментной эндопептидазы FAPα (коллагеназы). Для исследования исходной активности ферментной экзопептидазы FAPα 50 нг рекомбинантной FAPα человека (rhFAPα) (R&S system, #3715-SE) разводили в буфере для анализа FAPα (50 мМ Tris pH 7,4, 100 мМ NaCl, 0,1 мг/мл бычьего сывороточного альбумина) инкубировали с 5 мкг раствора субстрата DQ коллагена (Molecular Probes #D-12060) в течение 5 ч при 37°C и в защищенных от действия света 384-луночных планшетах OptiPlate™ (Perkin Elmer, #384-F). Для исследования активности ингибирования ферментной экзопептидазы FAPα исследуемыми соединениями все исследуемые соединения предварительно инкубировали с ферментом в течение 30 мин при 37°C, затем реакцию начинали путем добавления субстрата в 384-луночные планшеты OptiPlate™ (Perkin Elmer, #384-F). Гидролиз коллагена исследовали путем измерения интенсивности флуоресценции при Ex/Em 495/515 нм с помощью многофункционального устройства для считывания микропланшетов (Synergy 4, Biotek). Все измерения проводили однократно. Val-boroPro, неспецифический ингибитор пролилпептидазы, использовали в качестве положительного контроля. IC50 для ферментной экзопептидазной активности rhFAPα (определенные по исследованию коллагеназы) некоторых соединений также приведены в таблице 2.
Таблица 2: Ингибирование экзопептидазы или эндопептидазы rmFAPα или rhFAPα исследуемыми соединениями
Эталонное соединение: Соединение 60, описанное в публикации Jansen, K., et al., J Med Chem, 2014. 57(7): p. 3053-74; для ингибирования, %: +++ означает >50% ингибирование при 1 мкМ исследуемого соединения; ++ означает 25% <% ингибирование < 50% при 1 мкМ исследуемого соединения; + означает <25% ингибирование при 1 мкМ; для IC50: +++ означает IC50<1 мкМ; ++ означает 1 мкМ < IC50<10 мкМ; + означает IC50 >10 мкМ; - означает, что соединение не исследовали; rhFAPα: рекомбинантный белок активации фибробластов альфа человека; эндо: эндопептидаза; экзо: экзопептидаза; inh: ингибирование.
Пример B2
Селективность ингибирования FAPα исследуемыми соединениями оценивали путем сопоставления с другими представителями семейства пролилолигопептидазы S9: DPPIV, PREP и DPP9
Исследование ферментативной активности DPPIV
[0556] Для исследования исходной активности дипептидилпептидазы-4 (DPPIV) 40 нг рекомбинантной DPPIV человека (rhDPPIV) (R&S system, #1180-SE) инкубировали со 400 мкМ H-Gly-Pro-pNA субстрата (BACHEM, #L-1880) в буфере для анализа DPPIV (25 мМ Tris, pH 8,3) в течение 30 мин при 37°C в защищенных от действия света черных 96-луночных планшетах (Nunc, #237108). Для исследования ингибирования DPPIV исследуемыми соединениями исследуемые соединения предварительно инкубировали с ферментом в течение 15 мин при 37°C, затем реакцию начинали путем добавления субстрата в черные 96-луночные планшеты (Nunc, #237108). Высвобождение пара-нитроанилина (pNA) детектировали путем измерения поглощения при 405 нм с помощью многофункционального устройства для считывания микропланшетов (Synergy 4, Biotek). Все измерения проводили трижды. Val-boroPro, неспецифический ингибитор пролилпептидазы, использовали в качестве положительного контроля.
Исследование ферментативной активности PREP
[0557] Для исследования исходной активности пролилэндопептидазы (PREP) 20 нг рекомбинантной PREP человека (rhPREP) (R&S system, #4308-SE) инкубировали со 100 мкМ пептида Z-Gly-Pro-AMC (BACHEM, #L-1145) в буфере для анализа PREP (25 мМ Tris, 250 мМ NaCl, 10 мМ DTT, pH 7,5) в течение 30 мин при 37°C в защищенных от действия света черных 96-луночных планшетах (Nunc, #237108). Для исследования активности ингибирования PREP исследуемыми соединениями исследуемые соединения предварительно инкубировали с ферментом в течение 15 мин при 37°C, затем реакцию начинали путем добавления субстрата в черные 96-луночные планшеты (Nunc, #237108). высвобождение 7-амино-4-метилкумарина (AMC) детектировали путем измерения интенсивности флуоресценции при Ex/Em 380/460 нм с помощью многофункционального устройства для считывания микропланшетов (Synergy 4, Biotek). Все измерения проводили трижды. Val-boroPro, неспецифический ингибитор пролилпептидазы, использовали в качестве положительного контроля.
Исследование ферментативной активности DPP9
[0558] Для исследования исходной активности дипептидилпептидазы 9 (DPP9) 40 нг рекомбинантной DPP9 (rhDPP9) человека (R&S system, #5419-SE) инкубировали со 100 мкМ пептида H-Gly-Pro-AMC (BACHEM, #L-1215) в буфере для анализа DDP9 (50 мМ HEPES, pH 8) в течение 30 мин при 37°C в черных 96-луночных планшетах (Nunc, #237108). Для исследования активности ингибирования rhDPP9 исследуемыми соединениями исследуемые соединения предварительно инкубировали с ферментом в течение 15 мин при 37°C, затем реакцию начинали путем добавления субстрата в черные 96-луночные планшеты (Nunc, #237108). Высвобождение 7-амино-4-метилкумарина (AMC) детектировали путем измерения интенсивности флуоресценции при Ex/Em 380/460 нм с помощью многофункционального устройства для считывания микропланшетов (Synergy 4, Biotek). Все измерения проводили трижды. Val-boroPro, неспецифический ингибитор пролилпептидазы, использовали в качестве положительного контроля.
[0559] Для установления того, являются ли новые ингибиторы FAPα селективными или ингибируют ли они также и другие пролилпептидазы, IC50 некоторых исследуемых соединений, эталонного соединения (соединение 60, описанное в публикации Jansen, K., et al., J Med Chem, 2014. 57(7): p. 3053-74), и Val-boroPro исследовали, как указано в таблице 3A.
Таблица 3A: Селективность ингибирования FAPα исследуемыми соединениями
Эталонное соединение: Соединение 60, описанное в публикации Jansen, K., et al., J Med Chem, 2014. 57(7): p. 3053-74; для IC50: +++ означает IC50<1 мкМ; ++ означает 1 мкМ < IC50<10 мкМ; + означает IC50 >10 мкМ; - означает соединение не исследовали; rhFAPα: рекомбинантный белок активации фибробластов альфа человека; rhDPPIV: рекомбинантная дипептидилпептидаза-4 человека; rhDPP9: рекомбинантная дипептидилпептидаза 9 человека; экзо: экзопептидаза.
[0560] Кроме того, для исследования активности ингибирования дипептидилпептидазы 9 (DPP9) аликвоты по 10 мкл разведенных типичных соединений, эталонных соединений или разбавителя смешивали в черном 96-луночном планшете с 40 мкл буфера для анализа DDP9 (25 мМ Tris-HCl pH 8,0 и 0,01% BSA), содержащего 10 нг рекомбинантной DPP9 человека (rhDPP9) (Cat. No. #5419-SE, R&D systems). Соединениям давали возможность взаимодействовать с ферментом в течение 15 мин при 37°C до инициирования реакции путем добавления 50 мкл синтетического дипептидного субстрата, 200 мкМ H-Gly-ProAMC (Cat. No. #L-1215, Bachem) в буфере для анализа DPPIV. Реакции проводили в течение 30 мин при 37°C при защите от воздействия света. Высвобождение AMC детектировали путем измерения интенсивности флуоресценции при Ex/Em 380/460 нм с помощью многофункционального устройства для считывания микропланшетов. Результаты для ингибирования других пролилпептидаз семейства S9 типичными соединениями приведены в таблице 3B.
Таблица 3B: Ингибирование других пролилпептидаз семейства S9 типичными соединениями
Эталонное соединение: Соединение 60, описанное в публикации Jansen, K., et al., J Med Chem, 2014. 57(7): p. 3053-74; для ингибирования, %: +++ означает >50% ингибирование при 1 мкМ исследуемого соединения; ++ означает 25% <% ингибирование < 50% при 1 мкМ исследуемого соединения; + означает <25% ингибирование при 1 мкМ; для IC50: +++ означает IC50<1 мкМ; ++ означает 1 мкМ < IC50<10 мкМ; + означает IC50 >10 мкМ; rhFAPα: рекомбинантный белок активации фибробластов альфа человека; экзо: экзопептидаза; inh: ингибирование; rhFAP: рекомбинантный белок активации фибробластов человека; rhDPPIV: рекомбинантная дипептидилпептидаза-4 человека; rhDPP9: рекомбинантная дипептидилпептидаза 9 человека; rhPREP: рекомбинантная пролилэндопептидаза человека.
Пример B3
Проверка селективности субстрата PRXS-AMC для исследований активности FAPα
[0561] Активность FAPα можно исследовать с помощью общего анализа интенсивности флуоресценции дипептидилпептидазы с использованием пептидного субстрата, присоединенного к подвергающемуся химическому тушению красителю, такому как Ala-Pro-7-амино-4-трифторметилкумарин (AFC), или субстрата, содержащего консенсусный Gly-Pro дипептид, такого как Z-Gly-Pro-AMC (Levy, M.T., et al., Hepatology, 1999, 29(6): 1768-78; Santos, A.M., et al., J Clin Invest, 2009, 119(12): 3613-25; Park, J.E., et al., J Biol Chem, 1999, 274(51): 36505-12; Niedermeyer, J., et al., Mol Cell Biol, 2000, 20(3): 1089-94; Narra, K., et al., Cancer Biol Ther, 2007, 6(11): 1691-9; Lee, K.N., et al., J Thromb Haemost, 2011, 9(5): 987-96; Li, J., et al., Bioconjug Chem, 2012, 23(8): 1704-11). На эти субстраты, вероятно, также направлены и другие находящиеся в кровотоке пролинспецифические эндопептидазы, такие как PREP, которая может содержаться при реакции. В отличие от этого, патентованный субстратный реагент под названием PRXS-AMC может обеспечить специфический мониторинг активности FAPα.
[0562] Для проверки высокой селективности этого патентованного субстрата проводили исследования ферментативной активности FAP, DPPIV, PREP и DPP9 с использованием Z-Gly-Pro-AMC или PRXS-AMC, описанных в примерах B1 и B2.
[0563] Для исследования ферментативной активности FAPα, DPPIV, DPP9 и PREP рекомбинантные ферменты человека использовали при конечных концентрациях, равных 5, 2,5, 2,5 и 5 нМ соответственно. Z-Gly-Pro-AMC или PRXS-AMC использовали при конечных концентрациях, равных 25, 50, 100 и 200 мкМ. Реакции проводили в течение 60 мин при 37°C и защищали от воздействия света. Высвобождение AMC детектировали путем измерения интенсивности флуоресценции при Ex/Em 380/460 нм с помощью многофункционального устройства для считывания микропланшетов в кинетическом режиме. Измерения проводили однократно. Полученная зависимость интенсивности флуоресценции от времени для PRXS-AMC и Z-gly-pro-AMC в присутствии rhFAPα приведена на фиг. 1A и фиг. 1B соответственно; полученная зависимость интенсивности флуоресценции от времени для PRXS-AMC и Z-gly-pro-AMC в присутствии rhPREP приведена на фиг. 2A и фиг. 2B соответственно; и полученная зависимость интенсивности флуоресценции от времени для PRXS-AMC в присутствии rhDPPIV или rhDPP9 приведена на фиг. 3A и фиг. 3B соответственно.
[0564] PRXS-AMC подвергается процессингу в меньшей степени, чем Z-Gly-Pro-AMC близкородственной пролилолигопертидазой PREP при сходных концентрациях (см. фиг. 2A-2B). PRXS-AMC не подвергается процессингу с помощью DPPIV или DPP9 (фиг. 3A-3B). Кроме того, PRXS-AMC обладает повышенной растворимостью в водных буферах.
Пример B4
Ферментативная активность в плазме
Ферментативная активность FAPα в плазме мышей
[0565] Примерно 500 мкл цельной крови от одной мыши C57BL/6 собирали в пробирки BD Microtainer® (K2) EDTA (#365974, Becton Dickinson и Co.) путем терминального прокола сердца. Образец крови сразу центрифугировали примерно при 9000 g при 4°C в течение 5 мин. Плазму отделяли и хранили при -80°C аликвотами по 300 мкл. Для исследования исходной ферментативной экзопептидазной активности FAPα 5 мкл размороженной плазмы разводили (1:5) буфером cFAP (100 мМ Tris-HCl, 400 мМ NaCl, 50 мМ салициловой кислоты, 1 мМ EDTA, pH 7,5) и смешивали с 35 мкл того же буфера и затем предварительно инкубировали с исследуемыми соединениями в разных концентрациях или разбавителем DMSO с использованием 10 мкл в течение 15 мин при 37°C в черных 96-луночных планшетах (Nunc, #237108). После предварительной инкубации к смеси добавляли 50 мкл 200 мкМ PRXS-AMC. Исследование проводили в течение 1 ч при 37°C при защите от действия света. Высвобождение 7-амино-4-метилкумарина (AMC) детектировали путем измерения интенсивности флуоресценции при длине волны возбуждения, равной 380 нм, и при длине волны испускания, равной 460 нм, с помощью многофункционального устройства для считывания микропланшетов (Synergy 4, Biotek). Все измерения проводили не менее одного раза. Результаты приведены в таблице 4.
Ферментативная активность DPPIV в плазме мышей
[0566] Примерно 500 мкл цельной крови от одной мыши C57BL/6 собирали в пробирки BD Microtainer® (K2) EDTA (#365974, Becton Dickinson и Co.) путем терминального прокола сердца. Образец крови сразу центрифугировали примерно при 9000 g при 4°C в течение 5 мин. Плазму отделяли и хранили при -80°C аликвотами по 300 мкл. Для исследования исходной ферментативной экзопептидазной активности DPPIV 5 мкл размороженной плазмы разводили (1:5) буфером (100 мМ Tris-HCl, 400 мМ NaCl, 50 мМ салициловой кислоты, 1 мМ EDTA, pH 7,5) и смешивали с 35 мкл того же буфера и затем предварительно инкубировали с исследуемыми соединениями в разных концентрациях или разбавителем DMSO с использованием 10 мкл в течение 15 мин при 37°C в черных 96-луночных планшетах (Nunc, #237108). После предварительной инкубации к смеси добавляли 50 мкл 200 мкМ дипептидного субстрата H-Gly-Pro-AMC (Bachem, #L-1225). Исследование проводили в течение 1 ч при 37°C. Высвобождение 7-амино-4-метилкумарина (AMC) детектировали путем измерения интенсивности флуоресценции при длине волны возбуждения, равной 360 нм, и при длине волны испускания, равной 460 нм, с помощью многофункционального устройства для считывания микропланшетов (Synergy 4, Biotek). Все измерения проводили не менее двух раз. Результаты приведены в таблице 4.
Таблица 4: Ингибирование и специфичность исследуемых соединений в биологических образцах
Эталонное соединение: Соединение 60, описанное в публикации Jansen, K., et al., J Med Chem, 2014. 57(7): p. 3053-74; IC50: +++ означает IC50<1 мкМ; ++ означает 1 мкМ < IC50<10 мкМ; + означает IC50 >10 мкМ; экзо: экзопептидаза.
Пример B5
Ex vivo ингибирование активности FAPα в кровотоке по данным для плазмы разных видов
Плазма человека
[0567] Кровь человека брали у здоровых молодых добровольцев. Пробы крови собирали в пробирки с покрытием из EDTA-K2 по методике венопункции, осторожно перемешивали, затем хранили на льду и центрифугировали при 2500×g в течение 15 мин при 4°C. После отделения плазмы пробы хранили при -80°C аликвотами по 300 мкл.
[0568] Для исследования ингибиторной активности типичных исследуемых соединений для FAPα в кровотоке по данным для плазмы человека 20 мкл размороженной плазмы смешивали с 20 мкл буфера cFAP (100 мМ Tris-HCl, 400 мМ NaCl, 50 мМ салициловой кислоты, 1 мМ EDTA, pH 7,5) и 10 мкл типичных исследуемых соединений в разных концентрациях или разбавителя (DMSO).
[0569] Типичным соединениям давали возможность взаимодействовать с ферментом в течение 15 мин при 37°C. После предварительной инкубации 50 мкл 200 мкМ PRXS-AMC субстрата добавляли во все смеси. Все реакции проводили в течение 1 ч при 37°C при защите от действия света. Высвобождение AMC детектировали путем измерения интенсивности флуоресценции при длине волны возбуждения/испускания, равной 380/460 нм, с помощью многофункционального устройства для считывания микропланшетов. Все измерения проводили однократно.
[0570] Значения IC50 для типичных исследуемых соединений для FAPα в кровотоке людей приведены в таблице 5.
Плазма хомяка
[0571] Самцов хомяков Golden Syrian предоставлял National Laboratory Animal Center (NLAC) in Taiwan. Животных содержали в гигиенической среде при регулируемых температуре (20-24°C) и влажности (50% - 80%) в циклах 12 ч освещение/затемнение. Предоставляли свободный доступ к стандартному лабораторному корму [0MFG (Oriental Yeast Co., Ltd. Japan)] и стерилизованной в автоклаве водопроводной воде. Все условия проведения этого исследования, включая содержание, эксперименты и удаление животных соответствовали общим положениям руководства "Guide for the Care and Use of Laboratory Animals: Eighth Edition" (National Academies Press, Washington, D.C., 2011). Кроме того, протокол содержания и использования животных был рассмотрен и утвержден IACUC at Pharmacology Discovery Services Taiwan, Ltd.
[0572] Сразу после умерщвления хомяков пробы крови собирали путем терминального прокола сердца собирали в пробирки с покрытием из EDTA-K2, осторожно перемешивали, затем хранили на льду и центрифугировали при 2500×g в течение 15 мин при 4°C. После отделения плазмы пробы хранили при -80°C аликвотами по 300 мкл.
[0573] Для исследования типичных соединений в плазме хомяков использовали протокол, аналогичный описанному для плазмы человека, размороженную плазму разводили в соотношении 1:2 в буфере cFAP. В черном 96-луночном планшете 5 мкл разведенной плазмы хомяков смешивали с 35 мкл того же буфера и 10 мкл типичных исследуемых соединений при разных концентрациях или разбавителя (DMSO).
[0574] Типичным исследуемым соединениям давали возможность взаимодействовать с ферментом в течение 15 мин при 37°C. После предварительной инкубации 50 мкл 200 мкМ PRXS-AMC субстрата добавляли во все смеси. Все реакции проводили в течение 1 ч при 37°C при защите от действия света. Высвобождение AMC детектировали путем измерения интенсивности флуоресценции при длине волны возбуждения/испускания, равной 380/460 нм, с помощью многофункционального устройства для считывания микропланшетов. Все измерения проводили однократно.
[0575] Значения IC50 для типичных исследуемых соединений для FAPα в кровотоке по данным для плазмы хомяков приведены в таблице 5.
Таблица 5: Ингибирование ex-vivo активности FAPα в кровотоке типичными соединениями по данным для плазмы человека и хомяков.
Для IC50: +++ означает IC50<1 мкМ; ++ означает 1 мкМ < IC50<10 мкМ; + означает IC50 >10мкМ.
Пример B6
Исследования цитотоксичности в линиях клеток лейкоза человека
[0576] Линии клеток острого миелолейкоза человека (AML) и не-AML приобретали у ATCC и выращивали в соответствии с их инструкциями. В день проведения эксперимента линии клеток AML и не-AML высевали в белый 96-луночный планшет в 100 мкл среды для выращивания, содержащей разбавитель (DMSO) или типичное соединение, предлагаемое в настоящем изобретении, при разных концентрациях. Через 48 ч после обработки основанную на люминесценции жизнеспособность клеток определяли с помощью анализа Cell-Titer Glo (CTG) в соответствии с инструкциями изготовителя (Cat.No.: G7573, Promega). Жизнеспособность клеток в процентах рассчитывали путем нормирования интенсивности флуоресценции на среднее значение для обработанных разбавителем лунок при допущении о максимальной жизнеспособности (100%). В каждом эксперименте все обработки проводили трижды и приводили в виде ингибирования в % жизнеспособности ± стандартное отклонение (SD).
[0577] Val-boroPro, неспецифический ингибитор пролилпептидазы, использовали в качестве положительного контроля, описанного в публикации Johnson DC et al, Nature Medicine, 2018. IC50 для некоторых соединений по данным исследования цитотоксичности с использованием линии клеток AML человека MV4-11 приведены в таблице 6.
Таблица 6: Ингибирование жизнеспособности линии клеток AML человека некоторыми соединениями.
Для IC50: +++ означает IC50<3 мкМ; ++ означает 3 мкМ < IC50<10 мкМ; + означает IC50 >10 мкМ.
ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ
[0578] Вариант осуществления 1. Соединение формулы (I):
(I)
или его фармацевтически приемлемая соль, где:
R означает водород, C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R независимо необязательно замещены посредством Rd;
m равно 0, 1, 2, 3 или 4;
n равно 0, 1, 2, 3 или 4,
где m+n равно 1, 2, 3 или 4;
X означает -C(=O)-, -O-, -CH(OH)-, -S-, -S(=O)- или -S(=O)2-;
L означает
(a) , где
* обозначает положение присоединения к фрагменту Y-X-,
** обозначает положение присоединения к остальной части молекулы,
Ra означает водород, C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве Ra независимо необязательно замещены посредством Re,
R1 и R2 независимо друг от друга и независимо в каждом случае означают водород, C1-C2 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R1 и R2 независимо необязательно замещены посредством Rf,
или R1 и R2 вместе с атомом или атомами углерода, к которым они присоединены, образуют 3- 8-членный циклоалкилен, необязательно замещенный посредством Rf,
q равно 1, 2 или 3,
R3 и R4 независимо друг от друга и независимо в каждом случае означают водород, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R3 и R4 независимо необязательно замещены посредством Rg,
или R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют 3- 8-членный циклоалкилен, необязательно замещенный посредством Rg, и
p равно 0, 1 или 2;
(b) , где
* обозначает положение присоединения к фрагменту Y-X-,
** обозначает положение присоединения к остальной части молекулы,
R5 и R6 независимо друг от друга и независимо в каждом случае означают H, C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R5 и R6 независимо необязательно замещены посредством Rh,
Rb и Rc независимо означают H, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил, C6-C14 арил, или -C(=O)OR17, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве Rb и Rc независимо необязательно замещены посредством Ri, и
r равно 1, 2 или 3; или
(c) , где
* обозначает положение присоединения к фрагменту Y-X-,
** обозначает положение присоединения к остальной части молекулы,
R7 и R8 независимо друг от друга и независимо в каждом случае означают водород, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R7 и R8 независимо необязательно замещены посредством Rj,
или R7 и R8 вместе с атомом углерода, к которому они присоединены, образуют 3- 8-членный циклоалкилен, необязательно замещенный посредством Rj,
R9 и R10 независимо друг от друга и независимо в каждом случае означают H, C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил или C6-C14 арил, где C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R9 и R10 независимо необязательно замещены посредством Rk,
s равно 1, 2 или 3,
t равно 1, 2 или 3,
где s+t равно 2, 3 или 4,
u равно 0 или 1, и
v равно 0 или 1;
Y означает C6-C9 арил, замещенный посредством R11, 6- 10-членный гетероарил, замещенный посредством R12 или 3-12-членный гетероциклил, замещенный посредством R13, где
каждый R11, R12 и R13 независимо означает C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C4-C8 циклоалкенил, 3-12-членный гетероциклил, 5-10-членный гетероарил, C6-C14 арил, -OR14, -NR15R16, -SR14, -NO2, -C=NH(OR14), -C(O)R14, -OC(O)R14, -C(O)OR14, -C(O)NR15R16, -NR14C(O)R15, -NR14C(O)OR15, -NR14C(O)NR15R16, -S(O)R14, -S(O)2R14, -NR14S(O)R15, -NR14S(O)2R15, -S(O)NR15R16, -S(O)2NR15R16 или -P(O)(OR15)(OR16), где C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C4-C8 циклоалкенил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R11, R12 и R13 замещены посредством RL;
R14, R15 и R16 независимо друг от друга и независимо в каждом случае означают водород, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил или 3-12-членный гетероциклил, где C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил и 3-12-членный гетероциклил в качестве R14, R15 и R16 независимо замещены C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой, C1-C6 пергалогеналкоксигруппой, C6-C14 арилом или C6-C14 арилоксигруппой где C6-C14 арил или C6-C14 арилоксигруппа необязательно дополнительно замещена галогеном, -OH, цианогруппой, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой или C1-C6 пергалогеналкоксигруппой; и где по меньшей мере один из R14, R15 и R16, если содержится, не означает водород;
RL означает C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил или 3-12-членный гетероциклил, где C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил или 3-12-членный гетероциклил в качестве RL означает, замещенный галогеном, -OH, цианогруппой, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой, C1-C6 пергалогеналкоксигруппой или C6-C14 арилом, где C6-C14 арил дополнительно необязательно замещен галогеном, -OH, цианогруппой, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой или C1-C6 пергалогеналкоксигруппой; и
Rd, Re, Rf, Rg, Rh, Ri, Rj и Rk независимо друг от друга и независимо в каждом случае означают галоген, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил, 3-12-членный гетероциклил, -OR14, -NR15R16, цианогруппу или нитрогруппу.
[0579] Вариант осуществления 2. Соединение варианта осуществления 1 или его соль, где X означает -C(=O)-.
[0580] Вариант осуществления 3. Соединение варианта осуществления 1 или его соль, где X означает -O-.
[0581] Вариант осуществления 4. Соединение варианта осуществления 1 или его соль, где X означает -CH(OH)-.
[0582] Вариант осуществления 5. Соединение любого из вариантов осуществления 1-4 или его соль, где L означает -NH-CR1R2-.
[0583] Вариант осуществления 6. Соединение варианта осуществления 5 или его соль, где L означает -NH-CH2-.
[0584] Вариант осуществления 7. Соединение варианта осуществления 5 или его соль, где L означает -NH-CH(CH3)-.
[0585] Вариант осуществления 8. Соединение варианта осуществления 5 или его соль, где L означает -NH-CR1R2-, где R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют 3- 8-членный циклоалкилен.
[0586] Вариант осуществления 9. Соединение варианта осуществления 8 или его соль, где R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют циклопропилен.
[0587] Вариант осуществления 10. Соединение любого из вариантов осуществления 1-4 или его соль, где L означает -CR5R6-CH(NRbRc)-.
[0588] Вариант осуществления 11. Соединение варианта осуществления 10 или его соль, где L означает -CR5R6-CH(NRbRc)-, где R6, Rb и Rc означают H и R5 означает H или C1-C6 алкил.
[0589] Вариант осуществления 12. Соединение любого из вариантов осуществления 1-4 или его соль, где L означает , где * обозначает положение присоединения к фрагменту Y-X-, ** обозначает положение присоединения к остальной части молекулы.
[0590] Вариант осуществления 13. Соединение варианта осуществления 12 или его соль, где L означает , где * обозначает положение присоединения к фрагменту Y-X- и ** обозначает положение присоединения к остальной части молекулы.
[0591] Вариант осуществления 14. Соединение варианта осуществления 1 или его соль, где фрагмент -X-L- выбран из группы, состоящей из следующих:
и ; где * обозначает положение присоединения к фрагменту Y и ** обозначает положение присоединения к остальной части молекулы.
[0592] Вариант осуществления 15. Соединение любого из вариантов осуществления 1-14 или его соль, где Y означает C6-C9 арил, замещенный посредством R11.
[0593] Вариант осуществления 16. Соединение любого из вариантов осуществления 1-14 или его соль, где Y означает 6- 10-членный гетероарил, замещенный посредством R12.
[0594] Вариант осуществления 17. Соединение варианта осуществления 16 или его соль, где Y означает пиридин-4-ил, замещенный посредством R12 в положении 3.
[0595] Вариант осуществления 18. Соединение варианта осуществления 16 или 17 или его соль, где R12 означает C1-C6 алкил, замещенный посредством RL.
[0596] Вариант осуществления 19. Соединение варианта осуществления 16 или 17 или его соль, где R12 означает C2-C6 алкенил, замещенный посредством RL.
[0597] Вариант осуществления 20. Соединение варианта осуществления 16 или 17 или его соль, где R12 означает 3-12-членный гетероциклил, замещенный посредством RL.
[0598] Вариант осуществления 21. Соединение любого из вариантов осуществления 18-20 или его соль, где RL означает C6-C14 арил, замещенный галогеном, -OH, цианогруппой, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой, C1-C6 пергалогеналкоксигруппой или C6-C14 арилом.
[0599] Вариант осуществления 22. Соединение варианта осуществления 16 или 17 или его соль, где R12 означает -NR14C(O)R15.
[0600] Вариант осуществления 23. Соединение варианта осуществления 22 или его соль, где по меньшей мере один из R14 и R15 означает C1-C6 алкил или C6-C14 арил, где C1-C6 алкил или C6-C14 арил в качестве R14 и R15 независимо замещены C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой, C1-C6 пергалогеналкоксигруппой, C6-C14 арилом или C6-C14 арилоксигруппой, где C6-C14 арил или C6-C14 арилоксигруппа необязательно дополнительно замещена галогеном, -OH, цианогруппой, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой или C1-C6 пергалогеналкоксигруппой.
[0601] Вариант осуществления 24. Соединение любого из вариантов осуществления 1-14 или его соль, где Y означает 3-12-членный гетероциклил, замещенный посредством R13.
[0602] Вариант осуществления 25. Соединение любого из вариантов осуществления 1-24 или его соль, где m=n=1.
[0603] Вариант осуществления 26. Соединение любого из вариантов осуществления 1-25 или его соль, где R означает водород.
[0604] Вариант осуществления 27. Соединение, приведенное в таблице 1, или его соль.
[0605] Вариант осуществления 28. Фармацевтическая композиция, включающая соединение любого из вариантов осуществления 1-27 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
[0606] Вариант осуществления 29. Способ лечения заболевания или нарушения, опосредуемого белком активации фибробластов (FAP) у нуждающегося в нем индивидуума, включающий введение индивидууму терапевтически эффективного количества соединения любого из вариантов осуществления 1-27 или его фармацевтически приемлемой соли, или фармацевтической композиции варианта осуществления 28.
[0607] Вариант осуществления 30. Способ лечения заболевания или нарушения, характеризующегося пролиферацией, ремоделированием ткани, хроническим воспалением, ожирением, непереносимостью глюкозы или нечувствительностью к инсулину у нуждающегося в нем индивидуума, включающий введение индивидууму терапевтически эффективного количества соединения любого из вариантов осуществления 1-27 или его фармацевтически приемлемой соли, или фармацевтической композиции варианта осуществления 28.
[0608] Вариант осуществления 31. Способ вариантов осуществления 29 или 30, где заболеванием или нарушением является рак молочной железы, колоректальный рак, рак яичников, рак предстательной железы, рак поджелудочной железы, рак почки, рак легких, меланома, фибросаркома, саркома кости, саркома соединительной ткани, почечноклеточная карцинома, гигантоклеточная карцинома, плоскоклеточная карцинома, лейкоз, рак кожи, рак мягких тканей, рак печени, желудочно-кишечная карцинома или аденокарцинома.
[0609] Вариант осуществления 32. Способ варианта осуществления 31, где заболеванием или нарушением является метастатический рак почки, хронический лимфоцитарный лейкоз, аденокарцинома поджелудочной железы или немелкоклеточный рак легких.
[0610] Вариант осуществления 33. Способ варианта осуществления 29 или 30, где заболеванием или нарушением является фиброзное заболевание, заживление раны, келоидное образование, остеоартрит, ревматоидный артрит и родственные нарушения, включая разрушение хряща, атеросклеротическое заболевание, болезнь Крона или диабет типа II
[0611] Вариант осуществления 34. Способ уменьшения роста опухоли, пролиферации опухоли или онкогенности у нуждающегося в нем индивидуума, включающий введение индивидууму соединения любого из вариантов осуществления 1-27 или его фармацевтически приемлемой соли, или фармацевтической композиции варианта осуществления 28.
[0612] Вариант осуществления 35. Способ ингибирования FAP у индивидуума, включающий введение индивидууму соединения любого из вариантов осуществления 1-27 или его фармацевтически приемлемой соли, или фармацевтической композиции варианта осуществления 28.
[0613] Вариант осуществления 36. Способ ингибирования FAP в клетке, включающий введение или доставку в клетку соединения любого из вариантов осуществления 1-27 или его фармацевтически приемлемой соли, или фармацевтической композиции варианта осуществления 28, или его метаболита.
[0614] Вариант осуществления 37. Способ варианта осуществления 36, где клеткой является фибробласт.
[0615] Вариант осуществления 38. Способ варианта осуществления 36 или 37, где клеткой является связанный с раком фибробласт (CAF) или реакционноспособный стромальный фибробласт.
[0616] Вариант осуществления 39. Способ ингибирования FAP в опухоли, включающий введение или доставку в опухоль соединения любого из вариантов осуществления 1-27 или его фармацевтически приемлемой соли, или фармацевтической композиции варианта осуществления 28, или его метаболита.
[0617] Вариант осуществления 40. Способ ингибирования FAP в плазме, включающий введение или доставку в плазму соединения любого из вариантов осуществления 1-27 или его фармацевтически приемлемой соли, или фармацевтической композиции варианта осуществления 28, или его метаболита.
[0618] Вариант осуществления 41. Способ любого из вариантов осуществления 35-40, где ингибирование FAP включает ингибирование эндопептидазной активности FAP.
[0619] Вариант осуществления 42. Способ любого из вариантов осуществления 35-40, где ингибирование FAP включает ингибирование экзопептидазной активности FAP.
[0620] Вариант осуществления 43. Способ усиления иммунного ответа у индивидуума, включающий введение (a) ингибитора иммунных контрольных точек и (b) соединения любого из вариантов осуществления 1-27 или его фармацевтически приемлемой соли, или фармацевтической композиции варианта осуществления 28.
[0621] Вариант осуществления 44. Способ усиления степени экспрессии FGF21 у индивидуума, включающий введение индивидууму соединения любого из вариантов осуществления 1-27 или его фармацевтически приемлемой соли, или фармацевтической композиции варианта осуществления 28.
[0622] Вариант осуществления 45. Способ варианта осуществления 44, дополнительно включающий введение индуктора экспрессии FGF21.
[0623] Вариант осуществления 46. Способ варианта осуществления 45, где индуктором экспрессии FGF21 является агонист PPARα.
[0624] Вариант осуществления 47. Способ варианта осуществления 46, где агонистом PPARα является фибрат или фенофибрат.
[0625] Вариант осуществления 48. Композиция варианта осуществления 28 для применения в качестве лекарственного средства в медицине или ветеринарии.
[0626] Вариант осуществления 49. Применение соединения любого из вариантов осуществления 1-27 или его фармацевтически приемлемой соли, или фармацевтической композиции варианта осуществления 28, для приготовления лекарственного средства для предупреждения и/или лечения нарушения или заболевания, опосредуемого с помощью FAP.
[0627] Вся цитированная литература, такая как публикации, патенты, заявки на патенты и опубликованные заявки на патенты, во всей своей полноте включена в настоящее изобретение в качестве ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| Соединения, активные по отношению к бромодоменам | 2015 |
|
RU2743074C2 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ ИНФЕКЦИИ | 2021 |
|
RU2780103C1 |
| ИНГИБИРУЮЩИЕ JAK СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИМИДИНА И СПОСОБЫ | 2010 |
|
RU2567238C2 |
| ИНГИБИРУЮЩИЕ JAK СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИМИДИНА И СПОСОБЫ | 2010 |
|
RU2675857C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКОГО ЦИАНОЕНОНА КАК МОДУЛЯТОРЫ KEAP1 | 2021 |
|
RU2822828C1 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ-ИНФЕКЦИИ | 2020 |
|
RU2780101C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗО[1,2-b]ПИРИДАЗИНЫ, ЗАМЕЩЕННЫЕ ИМИДАЗО[1,5-b]ПИРИДАЗИНЫ, РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЙ | 2017 |
|
RU2773333C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2800292C2 |
| НОВЫЕ БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ | 2020 |
|
RU2830113C2 |
| АНАЛОГИ ДИАЗОНАМИДА | 2012 |
|
RU2608308C2 |



Изобретение относится к соединению формулы (I), где R, L, X, Y, m определены в формуле изобретения, и содержащим его фармацевтическим композициям, предназначенным для модуляции белка активации фибробластов (FAP). Соединения и композиции можно использовать в качестве лекарственных средств для лечения заболеваний или нарушений, опосредуемых белком активации фибробластов (FAP), включая гиперпролиферативные заболевания. 10 н. и 22 з.п. ф-лы, 3 ил., 6 табл., 47 пр.
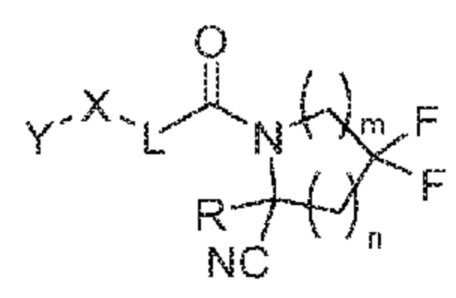
(I)
1. Соединение формулы (I):
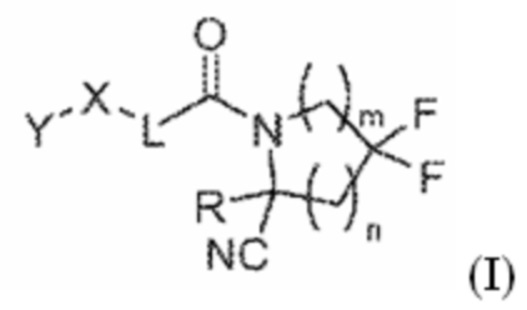
или его фармацевтически приемлемая соль, стереоизомер или таутомер, где:
R означает водород, C1-C6 алкил, C3-C8 циклоалкил, 3-12-членный гетероциклил или C6-C14 арил;
m равно 1 или 2;
n равно 1 или 2,
где m+n равно 2 или 3;
X означает -C(=O)-;
L означает *-NH-CH2-**,
где
* обозначает положение присоединения к фрагменту Y-X-, и
** обозначает положение присоединения к остальной части молекулы,
Y означает 6-10-членный гетероарил, замещенный посредством R12, где
каждый R12 означает C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C4-C8 циклоалкенил, 3-12-членный гетероциклил, 5-10-членный гетероарил, C6-C14 арил, -NR15R16 или -NR14C(O)R15, где C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C3-C8 циклоалкил, C4-C8 циклоалкенил, 3-12-членный гетероциклил, 5-10-членный гетероарил и C6-C14 арил в качестве R12 замещены посредством RL;
R14, R15 и R16 независимо друг от друга и независимо в каждом случае означают водород, C1-C6 алкил, C6-C14 арил или 5-10-членный гетероарил, где C1-C6 алкил, C6-C14 арил и 5-10-членный гетероарил в качестве R14, R15 и R16 независимо замещены C1-C6 алкоксигруппой, C1-C6 пергалогеналкоксигруппой, C6-C14 арилом или C6-C14 арилоксигруппой, где C6-C14 арил или C6-C14 арилоксигруппа необязательно дополнительно замещена галогеном, C1-C6 пергалогеналкилом или C1-C6 алкоксигруппой; и где по меньшей мере один из R14, R15 и R16, если содержится, не означает водород;
RL означает C1-C6 алкил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил или 3-12-членный гетероциклил, где C1-C6 алкил, C3-C8 циклоалкил, C6-C14 арил, 5-10-членный гетероарил или 3-12-членный гетероциклил в качестве RL замещен галогеном, -OH, оксогруппой, -NH2, -NH-(3-12-членным гетероциклилом), -O-(3-12-членным гетероциклилом), C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой, C1-C6 пергалогеналкоксигруппу или C6-C14 арилом, где
C1-C6 алкил дополнительно необязательно замещен 3-12-членным гетероциклилом, где 3-12-членный гетероциклил дополнительно необязательно замещен C1-C6 алкилом, и
C6-C14 арил дополнительно необязательно замещен галогеном;
где термин “гетероарил” означает ненасыщенную ароматическую циклическую группу, содержащую от 1 до 14 кольцевых атомов углерода и по меньшей мере один кольцевой гетероатом, включая, но не ограничиваясь только ими, гетероатомы, такие как азот, кислород и сера; и
где термин “гетероциклил” означает насыщенную или ненасыщенную неароматическую циклическую группу, содержащую одно кольцо или несколько конденсированных колец и содержащую от 1 до 14 кольцевых атомов углерода и от 1 до 6 кольцевых гетероатомов, таких как азот, сера или кислород.
2. Соединение по п. 1, его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y представляет собой пиридинил, замещенный R12.
3. Соединение по п. 1, его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y представляет собой пиридин-4-ил, замещенный R12.
4. Соединение по п. 1 или его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y означает пиридин-4-ил, замещенный посредством R12 в положении 3.
5. Соединение по п. 1, его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y представляет собой хинолинил, замещенный R12.
6. Соединение по п. 1, его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y представляет собой хинолин-4-ил, замещенный R12.
7. Соединение по п. 1, его фармацевтически приемлемая соль, стереоизомер или таутомер, где Y представляет собой хинолин-4-ил, замещенный R12 в положении 7.
8. Соединение по любому из пп. 1-7 или его фармацевтически приемлемая соль, стереоизомер или таутомер, где R12 означает C1-C6 алкил, замещенный посредством RL.
9. Соединение по любому из пп. 1-7 или его фармацевтически приемлемая соль, стереоизомер или таутомер, где R12 означает C2-C6 алкенил, замещенный посредством RL.
10. Соединение по любому из пп. 1-7, его фармацевтически приемлемая соль, стереоизомер или таутомер, где R12 означает 3-12-членный гетероциклил, замещенный посредством RL.
11. Соединение по любому из пп. 1-10 или его фармацевтически приемлемая соль, стереоизомер или таутомер, где RL означает C6-C14 арил, замещенный галогеном, -OH, C1-C6 алкилом, C1-C6 пергалогеналкилом, C1-C6 алкоксигруппой, C1-C6 пергалогеналкоксигруппой или C6-C14 арилом.
12. Соединение по любому из пп. 1-11 или его фармацевтически приемлемая соль, стереоизомер или таутомер, где m=n=1.
13. Соединение по любому из пп. 1-12 или его фармацевтически приемлемая соль, стереоизомер или таутомер, где R означает водород.
14. Соединение, выбранное из группы, состоящей из:
 ,
, ,
, ,
, ,
, ,
,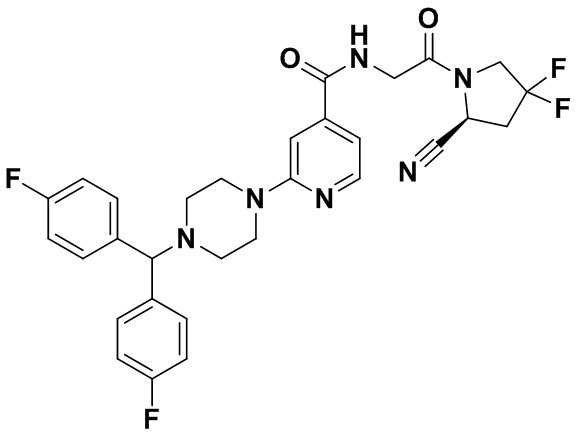 ,
, ,
,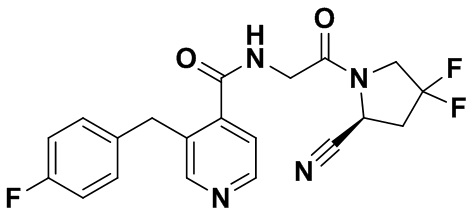 ,
, ,
,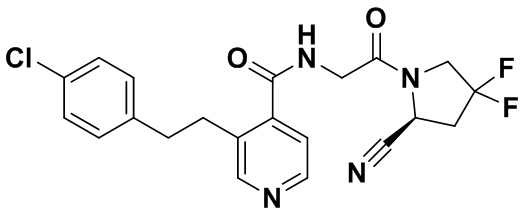 ,
, ,
, ,
,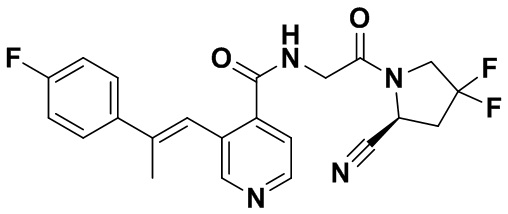 ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
,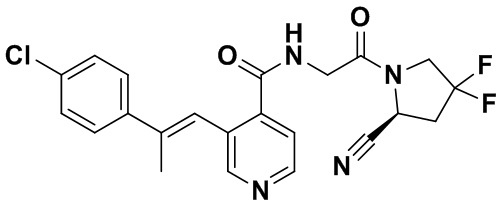 ,
, ,
, ,
,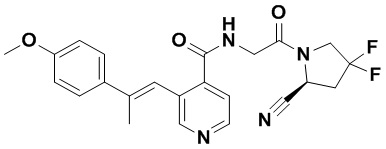 ,
, ,
, ,
, ,
, ,
,
 ,
,
 ,
,
 ,
,
 ,
,
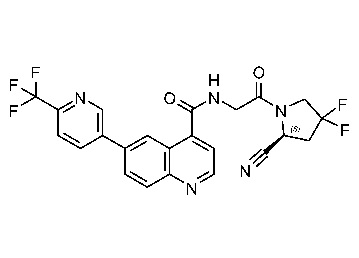 ,
,
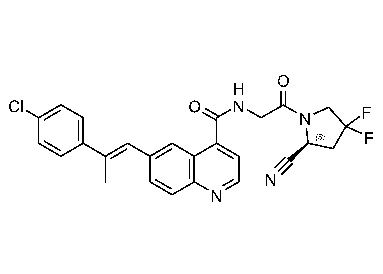 ,
,
 ,
,
 ,
,
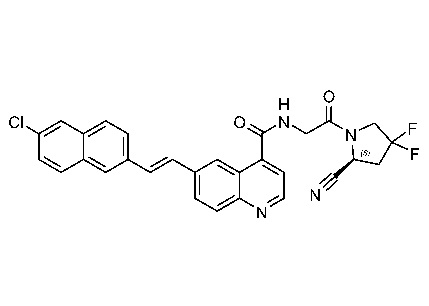 ,
,
 ,
,
 ,
, ,
,
 ,
,
 ,
,
 ,
,
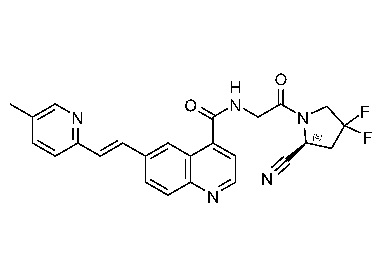 ,
,
 ,
,
 ,
,
 ,
,
 ,
, ,
,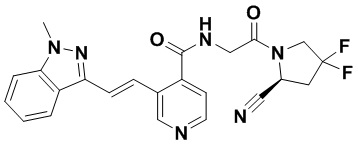 ,
, ,
,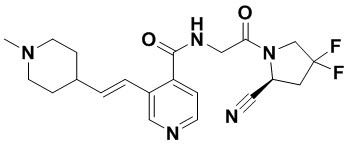 ,
, ,
, ,
, ,
,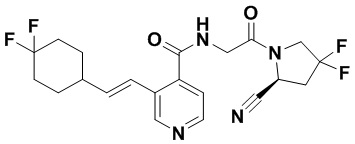 ,
, ,
, ,
,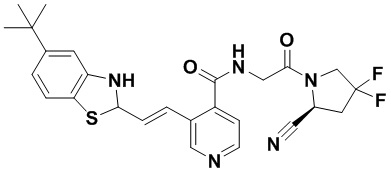 ,
, ,
, ,
,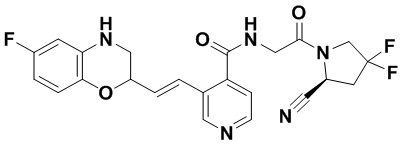 ,
,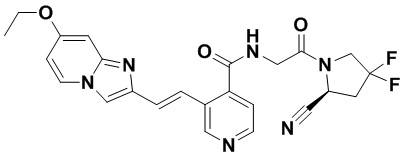 ,
, ,
,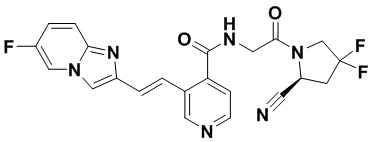 ,
, ,
,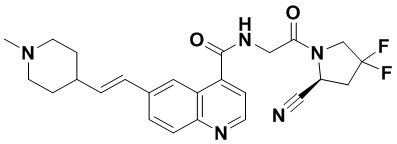 ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
,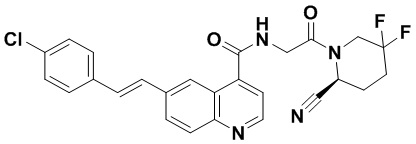 ,
, ,
, ,
,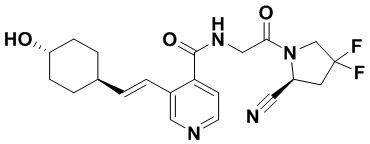 ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
,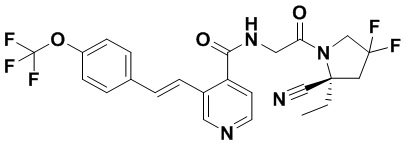 ,
, ,
,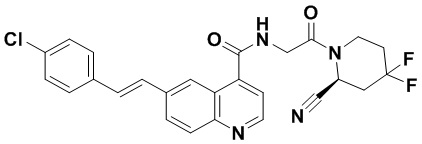 ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
,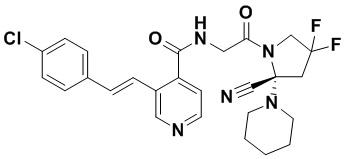 ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
,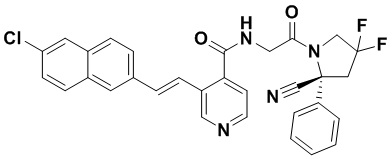 ,
,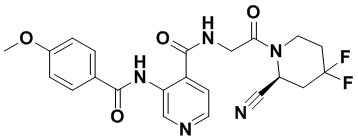 ,
, ,
,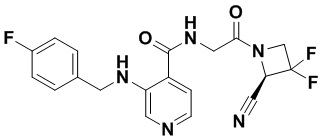 ,
, ,
, ,
, ,
, ,
, ,
,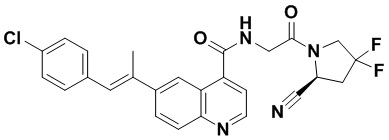 ,
, ,
,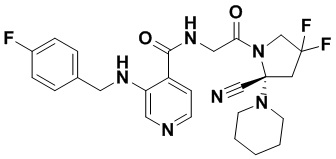 ,
, ,
, ,
, ,
, ,
, ,
, ,
,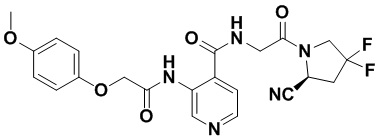 ,
,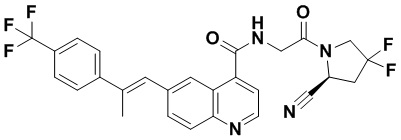 ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
,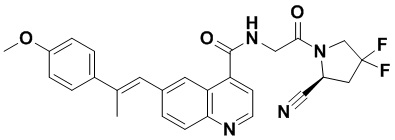 ,
, ,
,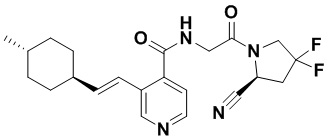 ,
, ,
,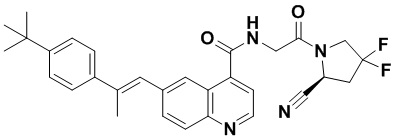 ,
,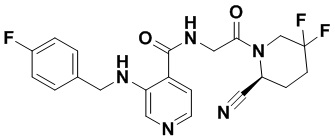 ,
, ,
,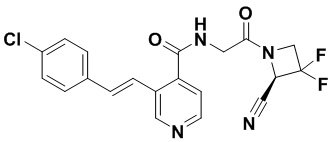 ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
,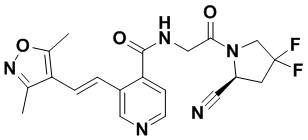 ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
,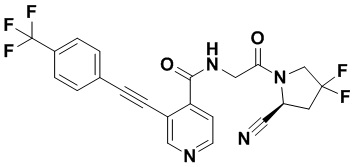 ,
,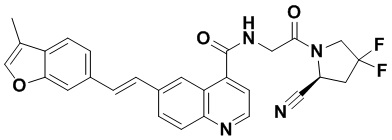 ,
, ,
,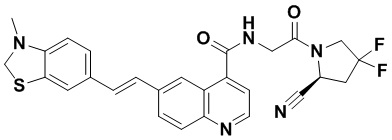 ,
,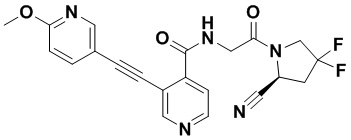 ,
,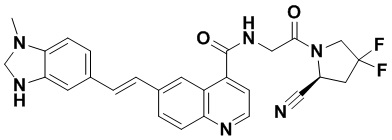 ,
, ,
, ,
, ,
, ,
, ,
, ,
, ,
,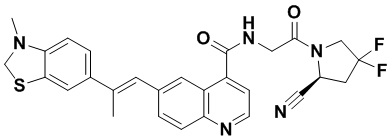 ,
,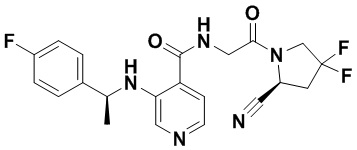 ,
, ,
,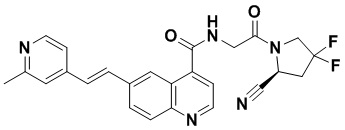 ,
, ,
, ,
, ,
, ,
, ,
, ,
,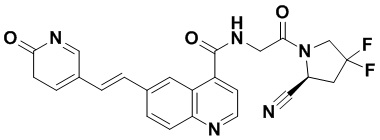 ,
,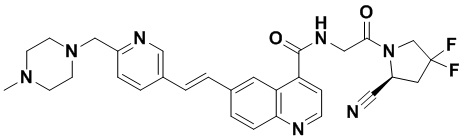 ,
,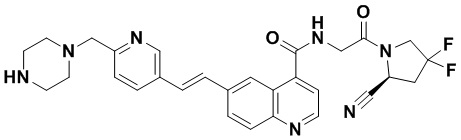 ,
, ,
,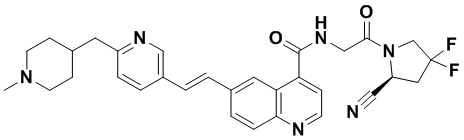 ,
, ,
, ,
, ,
, и
и
или его фармацевтически приемлемая соль, стереоизомер или таутомер.
15. Фармацевтическая композиция, содержащая эффективное количество соединения по любому из пп. 1-14 или его фармацевтически приемлемую соль, стереоизомер или таутомер и фармацевтически приемлемый носитель, для применения в лечении заболевания или нарушения, опосредуемого белком активации фибробластов (FAP).
16. Способ лечения заболевания или нарушения, опосредуемого белком активации фибробластов (FAP), у нуждающегося в нем индивидуума, включающий введение индивидууму терапевтически эффективного количества соединения по любому из пп. 1-14 или его фармацевтически приемлемой соли, стереоизомера или таутомера или фармацевтической композиции по п. 15.
17. Способ по п. 16, где заболеванием или нарушением является рак молочной железы, колоректальный рак, рак яичников, рак предстательной железы, рак поджелудочной железы, рак почки, рак легких, меланома, фибросаркома, саркома кости, саркома соединительной ткани, почечноклеточная карцинома, гигантоклеточная карцинома, плоскоклеточная карцинома, лейкоз, рак кожи, рак мягких тканей, рак печени, желудочно-кишечная карцинома или аденокарцинома.
18. Способ по п. 17, где заболеванием или нарушением является метастатический рак почки, хронический лимфоцитарный лейкоз, аденокарцинома поджелудочной железы или немелкоклеточный рак легких.
19. Способ по п. 16, где заболеванием или нарушением является фиброзное заболевание, заживление раны, келоидное образование, остеоартрит, ревматоидный артрит и родственные нарушения, включая разрушение хряща, атеросклеротическое заболевание, болезнь Крона или диабет типа II.
20. Способ ингибирования FAP у индивидуума, включающий введение индивидууму соединения по любому из пп. 1-14 или его фармацевтически приемлемой соли, стереоизомера или таутомера или фармацевтической композиции по п. 15.
21. Способ ингибирования FAP в клетке, включающий введение или доставку в клетку соединения по любому из пп. 1-14 или его фармацевтически приемлемой соли, стереоизомера или таутомера или фармацевтической композиции по п. 15.
22. Способ по п. 21, где клеткой является фибробласт.
23. Способ по п. 21 или 22, где клеткой является связанный с раком фибробласт (CAF) или реакционноспособный стромальный фибробласт.
24. Способ ингибирования FAP в опухоли, включающий введение или доставку в опухоль соединения по любому из пп. 1-14 или его фармацевтически приемлемой соли, стереоизомера или таутомера или фармацевтической композиции по п. 15.
25. Способ ингибирования FAP в плазме, включающий введение или доставку в плазму соединения по любому из пп. 1-14 или его фармацевтически приемлемой соли, стереоизомера или таутомера или фармацевтической композиции по п. 15.
26. Способ по любому из пп. 20-25, где ингибирование FAP включает ингибирование эндопептидазной активности FAP.
27. Способ по любому из пп. 20-25, где ингибирование FAP включает ингибирование экзопептидазной активности FAP.
28. Способ усиления степени экспрессии FGF21 у индивидуума, включающий введение индивидууму соединения по любому из пп. 1-14 или его фармацевтически приемлемой соли, стереоизомера или таутомера или фармацевтической композиции по п. 15.
29. Способ по п. 28, дополнительно включающий введение индуктора экспрессии FGF21.
30. Способ по п. 29, где индуктором экспрессии FGF21 является агонист PPARα.
31. Способ по п. 30, где агонистом PPARα является фибрат или фенофибрат.
32. Применение соединения по любому из пп. 1-14 или его фармацевтически приемлемой соли, стереоизомера или таутомера или фармацевтической композиции по п. 15 для приготовления лекарственного средства для предупреждения и/или лечения нарушения или заболевания, опосредуемого с помощью FAP.
| WO 2007085895, 02.08.2007 | |||
| WO 2009116067 A2, 24.09.2009 | |||
| УСТРОЙСТВО ДЛЯ УПРАВЛЕНИЯ ЗВУКАМИ КАТОДНЫХ МУЗЫКАЛЬНЫХ ПРИБОРОВ | 1922 |
|
SU7410A1 |
| WO 2013107820 A1, 25.07.2013 | |||
| WO 2017189569 A1, 02.11.2017 | |||
| WO 2003002553 A2, 09.01.2003 | |||
| WO 2018111989 A1, 21.06.2018 | |||
| Koen Jansen et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
