Изобретение относится к способам получения наноразмерных и/или субмикронных порошков сложного оксида со структурой фазы пирохлора состава (Bi2O3)0.42(Fe2O3)0.28(WO3) (далее - BFWO), которые за счет своих полупроводниковых свойств могут быть использованы в «зеленой» энергетике для создания фотоэлектрических преобразователей энергии, а также для фотокаталитической очистки и обеззараживания воздуха и воды. Уникальное электронное строение оксида BFWO, за счет которого в нем реализуются все четыре типа электронных переходов, а также характерное для структурного типа пирохлора явление сегнетоэлектричества, определяют возможности создания на основе BFWO новых композиционных функциональных материалов для электроники, катализа, водородной энергетики и других областей науки и технологии.
Впервые фаза со структурой пирохлора в системе Bi2O3-Fe2O3-WO3-(H2O) была получена и структурно охарактеризована в составе сложных композитов (см. Lomakin M.S., Proskurina O.V., Danilovich D.P., Panchuk V.V., Semenov V.G., Gusarov V.V. Hydrothermal Synthesis, Phase Formation and Crystal Chemistry of the pyrochlore/Bi2WO6 and pyrochlore/α-Fe2O3 Composites in the Bi2O3-Fe2O3-WO3 System, Journ. Solid State Chem., 2020, 282, 121064 DOI: 10.1016/j.jssc.2019.121064). Получение порошков проводили методом гидротермального синтеза, включавшим получение суспензии аморфного прекурсора, ее дальнейшую гидротермальную обработку, промывку полученного кристаллического осадка от посторонних растворимых солей и сушку кристаллического осадка.
Известен способ синтеза оксида титана (см. RU 2709093, МПК C01G 23/053, опубл. 13.12.2019), включающий приготовление водного раствора хлорида титана, гидролиз указанного раствора при добавлении аммиака с образованием осадка, значение рН в ходе гидролиза остается постоянным в диапазоне от 4,7 до 5,3. После стадии гидролиза проводят дополнительную стадию промывки осадка водой, выполняют отделение осадка от маточного раствора, термическую обработку осадка, включающую последовательные сушку и обжиг, проводимые в атмосфере воздуха. Сушку осадка проводят до постоянной массы при температуре (20-140)°С, а обжиг при температуре не выше 800°С в течение 0,5-24 часов.
Недостатком известного способа синтеза оксида титана является малая производительность, неприемлемая для промышленной реализации способа. Получение осадка гидроксида титана производится путем добавления по каплям раствора аммиака к раствору хлорида титана, т.е. для смешения растворов используется широко известный лабораторный прием, имеющий крайне малую производительность. Кроме того, при таком способе смешения на низком уровне остается и качество гомогенизации смеси, что осложняет поддержание постоянного значения рН во всем объеме реакционного пространства, т.е. в растворе возникают значительные градиенты рН, что приводит к неполному протеканию реакций, в результате чего снижается выход продукта с единицы вводимых в реактор реагентов.
Известен способ синтеза ориентированных кристаллов слоистых гидроксинитратов гадолиния (см. RU 2700509, МПК С30В 29/22, С30В 29/68, С30В 29/60 C01F 1/00, C01F 17/00, С01В 21/48, C09K 11/78, B01J 23/10, B01J 35/08, опубл. 17.09. 2019), включающий в себя стадии получения раствора нитрата гадолиния, приведения в контакт указанного выше раствора нитрата гадолиния и раствора аммиака, отделения сформировавшейся твердой фазы от маточного раствора, сушку, отличающийся тем, что на стадии приведения в контакт растворов значение рН образующейся суспензии поддерживают постоянным в интервале 7-9 единиц рН при концентрации гадолиния в растворе нитрата в диапазоне от 0,05 до 1 моль/л.
Недостатком известного способа синтеза ориентированных кристаллов слоистых гидроксинитратов гадолиния, также является низкая производительность, неприемлемая для промышленной реализации способа. Поскольку смешение растворов реагентов происходит при использовании известного лабораторного приема (добавления осадителя по каплям), то этой стадии синтеза присуще все те же недостатки, что были указаны выше. Следующая стадия, включающая отделение сформировавшейся твердой фазы от маточного раствора, также проводится с использованием известного лабораторного приема - методом декантации (сливания маточного раствора). Продолжительность этой стадии большая - необходимо либо ждать, пока осадок осядет на дно емкости, либо центрифугировать суспензию.
В том случае, когда необходимо провести осаждение двух и более катионов металлов, необходимо подобрать такие условия (обычно рН реакционной среды), при которых все катионы металлов были бы в составе нерастворимой фазы (осадка). Известен способ получения наноразмерных порошков твердого раствора железо-кобальт (см. RU 2432232, МПК B22F 9/24, В82В 3/00, опубл. 27.10.2011), включающий приготовление раствора солей гептагидрата сульфата железа и гексагидрата хлорида кобальта, его нагрев и осаждение щелочью катионов металлов в виде гидроксидов железа и кобальта при непрерывном перемешивании. В ходе перемешивания для осаждения гидроксидов железа и кобальта в раствор вводят 20-25 г твердой щелочи (сухого гидроксида натрия), после чего в раствор добавляют 20-40 мл раствора 65 масс. % гидразин гидрата и выдерживают в течение 5-20 мин.
Недостатком известного способа получения наноразмерных порошков твердого раствора железо-кобальт является плохое качество перемешивания реакционной среды и плохая гомогенизация реагирующих компонентов. При добавлении в раствор солей металлов сухой щелочи происходит резкое повышение значения рН среды вблизи твердых частиц растворяющейся щелочи, и, чтобы равномерно распределить образующиеся гидроксид-анионы по всему объему реакционного пространства недостаточно использовать «традиционное» перемешивание магнитной или лопастной мешалкой. Концентрация щелочи в реакционном пространстве в результате такого перемешивания будет перераспределяться очень медленно, то есть для полного осаждения катионов металлов необходимо производить непрерывное перемешивание в течение длительного времени, пока не будет выровнена концентрация щелочи по всему объему реактора. Это существенно снижает производительность известного способа, а также приводит к снижению выхода продукта с единицы массы исходных реагентов.
Известен способ получения пирохлора, включающий получение раствора, содержащего растворитель и предшественник металла или его соль, способные образовывать пирохлор, при этом предшественник металла или его соль растворяют в растворителе, подвергая затем раствор стадии сушки для получения не гелеобразного или не полимеризованного прекурсора пирохлора в порошкообразной форме, после чего подвергают прекурсор пирохлора стадии прокаливания для получения пирохлора (CN 105026316, МПК B01J 21/16; B01J 23/42; B01J 23/46; B01J 23/63; B01J 23/755; B01J 23/83; B01J 37/02; B01J 37/08; С01В 3/26; С01В 3/38; C01G 25/00; C01G 51/00; C01G 53/00; C01G 55/00, опубл. 04.11.2015). Известный способ получения пирохлора, включает:
(a) получение раствора, содержащего растворитель и прекурсор металла, способный образовывать пирохлор, при этом прекурсор металла растворяют в растворителе;
(b) подвергание раствора стадии сушки для получения не гелеобразного или не полимерного прекурсора пирохлора в форме порошка; и
(c) подвергание прекурсора пирохлора стадии прокаливания для получения пирохлора.
Кроме того, в известном способе растворителем является вода, метанол, этанол, пропанол, изопропанол, бутанол, ацетон или их смесь, вода или метанол, а прекурсором металла является соль металла или оксид металла, или их комбинация. Помимо этого, предшественник металла содержит трехвалентные ионы La, Се, Nd, Bi, Sc или Y и/или элементы Zr, Pt, Pd, Ni, Mo, Rh, Ru или Ir четырехвалентные ионы и/или Ba, двухвалентные, трехвалентные или четырехвалентные ионы Са, Cu, Mg, Ru, Rh, Pt, Pd, Ni, Co, Ir или Mo. При этом пирохлор имеет следующую структуру: AxBy-zCzO7, в А - трехвалентный ион La, Се, Nd, Bi, Sc или Y, где 0≤х≤2, В представляет собой четырехвалентный ион элементов Zr, Pt, Pd, Mo, Rh, Ru или Ir, где 0≤y-z≤2, и С представляет собой двухвалентный, трехвалентный или четырехвалентный ион Ba, Са, Cu, Mg, Ru, Rh, Pt, Pd, Ni, Co, Ir или Mo, где 0≤z≤2.
Недостатком известного способа синтеза пирохлоров является не только низкая производительность стадии образования промежуточных продуктов в растворе, но и малая производительность стадии сушки, неприемлемая для промышленной реализации способа. Сушка производится в керамических чашках в лабораторном сушильном шкафу, однако, при таком способе сушки площадь поверхности, с которой испаряется жидкость, ограничена площадью керамической чашки. Кроме того, затруднен отвод испаряющейся жидкости от поверхности суспензии, поскольку вентиляция в таких лабораторных сушильных шкафах не очень интенсивная, поэтому в сушильной камере в процессе сушки всегда наблюдается повышенное давление насыщенного водяного пара.
Известен способ (JP 2012049075, МПК C01G 55/00; Н01М 4/88; Н01М 4/90; Н01М 8/10, опубл. 08.03.2012) получения оксида пирохлорного типа, представленного формулой: А, включающий стадии: образования осадка оксида пирохлорного типа путем реакции между галогенидом или нитратом А и солью щелочного металла В, и обжига осадка оксида пирохлорного типа при температуре 250°С или выше и 900°С или ниже. Кроме того, способ получения оксида пирохлорного типа включает стадию приготовления 1-го водного раствора, который представляет собой водный раствор галогенида или нитрата А, и 2-го водного раствора, который представляет собой водный раствор соли щелочного металла В, стадию добавления по каплям 1-го водного раствора и или 2-го водного раствора в другой для проведения реакции с образованием осадка оксида пирохлорного типа; и этап обжига осадка оксида пирохлорного типа при температуре 250°С или выше и 900°С или ниже. Способ получения оксида пирохлорного типа дополнительно включает стадию предварительного диспергирования проводящего материала в 1-м водном растворе, либо во 2-м водном растворе перед стадией образования осадка оксида пирохлорного типа методом осаждения, В способе получения оксида пирохлорного типа температура реакции на стадии образования осадка составляет 0°С или выше и 60°С или ниже, а стадию обжига осадка оксида пирохлорного типа осуществляют в атмосфере инертного газа. В известном способе получения оксида пирохлорного типа А содержит металл А2, отличный от А1, и/или В содержит металл B2, отличный от В1, и каждый из А2 и В2 независимо и по меньшей мере один из них выбран из группы, состоящей из Be, Mg, Са, Sr, Ba, Sc, Y, Ti, Zr, Hf, V, Nb, Та, Cr, Mo, W, Mn, Re, Fe, Os, Co, Rh, Ni, Pd, Cu, Al, Ga, In, Ge, As, Sb, Bi, La, Ce, Pr, Nd, Pm, Sm, Eu, Gd, Dy, Ho, Er, Tm, Yb и Lu.
Недостатками известного способа получения пирохлоров являются невысокая производительность стадии получения аморфного осадка, а также плохое качество перемешивания реакционной среды и плохая гомогенизация реагирующих компонентов при его образовании. Другими словами, здесь обнаруживаются те же перечисленные выше недостатки, которые присущи смешению при использовании известного лабораторного приема - добавления по каплям растворов.
Известен способ получения порошков сложного оксида со структурой фазы пирохлора в системе Bi2O3-Fe2O3-WO3-(H2O), совпадающий с настоящим техническим решением по наибольшему числу существенных признаков и принятый за прототип (см. Lomakin M.S., Proskurina O.V., Sergeev A.A., Buryanenko I.V., Semenov V.G., Voznesenskiy S.S., Gusarov V.V. Crystal structure and optical properties of the Bi-Fe-W-O pyrochlore phase synthesized via a hydrothermal method, J. Alloys Compd., v. 889, 2021, 161598). Способ-прототип включает приготовление раствора нитратов висмута и железа, раствора вольфрамата натрия и раствора щелочи, взятых в стехиометрическом соотношении, обеспечивающем получение сложного оксида со структурой пирохлора состава (Bi2O3)0.25(Fe2O3)0.18(WO3), смешение раствора нитратов висмута и железа с раствором вольфрамата натрия с образованием суспензии, смешение суспензии с раствором щелочи для образования суспензии аморфного прекурсора, имеющей рН=2, 3, 7, гидротермальную обработку суспензии аморфного прекурсора в течение 24 часов при температуре 200°С, промывку полученного порошка дистиллированной водой и его сушку в течение 20 часов при температуре 80°С.
В способе-прототипе процесс гидротермальной обработки чрезвычайно длительный, и проводится в автоклавах (их объем составляет 50 мл), работающих в периодическом режиме, то есть за один цикл обрабатывается не более 40 мл суспензии в одном автоклаве (коэффициент заполнения объема равен 80%), в результате чего производительность по обработанной суспензии составляет не более 0,0017 л/час (при расчете на один автоклав). При расчетной концентрации твердой фазы в суспензии 28 г/л производительность по твердой фазе одной только стадии гидротермальной обработки составляет всего 0,048 г/ч. Такая производительность неприемлемо низка для производственных условий. Недостатками способа-прототипа является использование периодического процесса гидротермальной обработки, один цикл которой длится 24 часа; применение трудоемкой и продолжительной промывки кристаллического продукта методом декантации обогащенного посторонними ионами раствора (как правило, несколько циклов); использование низко производительных методов сушки, требующих до 20 часов обработки для получения нескольких граммов продукта.
Задачей настоящего технического решения является разработка способа получения порошка сложного оксида состава (Bi2O3)0.42(Fe2O3)0.28(WO3) со структурой фазы пирохлора, который бы обеспечивал высокую равномерность распределения (на уровне микросмешения, то есть молекулярном и ионном) исходных реагентов и осадителя в объеме реактора, приводящую к формированию продукта однородного состава и структуры; высокую производительность, обусловленную существенным снижением продолжительности стадий получения суспензии аморфного прекурсора соосаждением при высокоскоростном смешении растворов, гидротермальной обработки (в том числе охлаждения), промывки и сушки.
Поставленная задача достигается тем, что способ получения порошка сложного оксида состава (Bi2O3)0.42(Fe2O3)0.28(WO3) со структурой фазы пирохлора включает приготовление раствора нитратов висмута и железа, раствора вольфрамата натрия и раствора щелочи, взятых в стехиометрическом соотношении, обеспечивающем получение указанного выше сложного оксида, смешение раствора нитратов висмута и железа с раствором вольфрамата натрия с образованием суспензии, смешение суспензии с раствором щелочи для образования суспензии аморфного прекурсора, имеющей рН=2,0-2,4, гидротермальную обработку суспензии аморфного прекурсора, промывку полученного порошка (кристаллического осадка) и его сушку. Новым в способе является то, что смешение исходных растворов проводят в реакторе проточного смешения - микрореакторе с интенсивно закрученными потоками, при этом раствор нитратов висмута и железа подают в один тангенциальный патрубок, раствор вольфрамата натрия подают во второй тангенциальный патрубок, обеспечивая смешение этих растворов с образованием суспензии, а раствор щелочи подают в осевой патрубок, смешивая его с образовавшейся суспензией с получением суспензии аморфного прекурсора, гидротермальную обработку суспензии аморфного прекурсора проводят в проточном многоканальном автоклаве при температуре (190-210)°С в течение не менее 2 часов, причем промывку полученного порошка и его отделение от маточного раствора осуществляют в электрофлотационной машине, а сушку полученного порошка ведут комбинацией распыления суспензии и псевдоожиженного слоя при температуре (80-90)°С, при этом подачу сушильного агента под решетку псевдоожиженного слоя производят в импульсном режиме.
Гидротермальная обработка суспензии аморфного прекурсора в проточном многоканальном автоклаве позволяет распараллелить потоки суспензии и обеспечить равномерный подвод тепловой энергии к нагреваемой суспензии, и тем, самым, обеспечить минимальные градиенты температуры в суспензии. Это, в свою очередь, способствует образованию кристаллического продукта заданного фазового состава.
Гидротермальная обработка суспензии аморфного прекурсора, проводимая при продолжительности изотермической выдержки менее 2 часов, не приводит к формированию полностью закристаллизованного продукта, и наряду с фазой пирохлора присутствует заметное количество аморфной фазы. При продолжительности гидротермальной обработки более 2 часов не происходит заметных изменений ни в фазовом составе образцов, ни в размерных параметрах кристаллитов и частиц. В то же время, значительное увеличение продолжительности гидротермальной обработки нежелательно, поскольку приводит к снижению производительности устройства. В этой связи, оптимальной продолжительностью гидротермальной обработки суспензии аморфного прекурсора является 2 часа.
Гидротермальная обработка суспензии аморфного прекурсора, проводимая при температуре менее 190°С, должна осуществляться более длительное время, (более 2 часов), чтобы на выходе получить полностью закристаллизованный продукт, поскольку скорость кристаллизации будет тем ниже, чем ниже температура обработки. Это приведет к дополнительным затратам как по времени, так и по материальным ресурсам, поскольку необходимо будет увеличивать протяженность каналов проточного автоклава, чтобы увеличить продолжительность пребывания в нем суспензии аморфного прекурсора.
Гидротермальная обработка суспензии аморфного прекурсора при температуре более 210°С приведет, во-первых, к неоправданным дополнительным энергетическим затратам. Во-вторых, давление в системе при более высокой температуре гидротермальной обработки будет возрастать, что потребует использования более металлоемкого и дорогостоящего оборудования, а также приведет к необходимости дополнительной электроэнергии на нагрев и дополнительного времени на охлаждение. В-третьих, с точки зрения фазообразования, не будет наблюдаться никакого выигрыша в параметрах целевого продукта (составе, структуре, свойствах).
Сочетание сушки путем комбинации распыления суспензии и сушки в псевдоожиженном слое при температуре (80-90)°С обусловлено тем, что при температуре менее 80°С процесс испарения влаги будет недостаточно интенсивным, а при температуре более 90°С процесс сушки становится чрезмерно интенсивным, что может привести к ускоренному парообразованию в порах агломератов и их частичному разрушению, вследствие чего дисперсный состав высушенного порошка становится чрезмерно широким. При этом сушка распылением (с использованием различного вида форсунок или других устройств для диспергирования суспензии) позволяет осуществить быстрое первичное обезвоживание частиц, а доведение до окончательной влажности достигается при их пребывании в псевдоожиженном слое. Подача сушильного агента под решетку псевдоожиженного слоя в импульсном режиме позволяет подать необходимое количество сушильного агента (например, воздуха) в течение той части импульса, когда сушильный агент подается, а прекращение подачи сушильного агента позволяет предотвратить унос частиц из аппарата с сушильным агентом.
Настоящее техническое решение позволяет обеспечить получение суспензии аморфного прекурсора при комнатной температуре - в диапазоне от 20°С до 30°С и атмосферном давлении (в микрореакторе с интенсивно закрученными потоками), а в целом обеспечивает получение BFWO с высокой производительностью, поскольку каждая из стадий процесса имеет производительность, во много раз превышающую таковую для известных методов синтеза порошков пирохлоров.
Настоящее изобретение поясняется чертежом, где:
на фиг. 1 схематически изображен продольный разрез микрореактора с интенсивно закрученными потоками (раствор А - кислый раствор нитратов висмута и железа; раствор Б - водный раствор вольфрамата натрия; раствор В - 2М водный раствор щелочи NaOH).
в табл. 1 приведены условия проведения синтеза в микрореакторе с интенсивно закрученными потоками по настоящему изобретению;
в табл. 2 приведен элементный состав образцов, синтезированных при различной продолжительности изотермической (200°С) выдержки по настоящему изобретению.
в табл. 3 приведен элементный состав образцов, синтезированных при различной продолжительности изотермической (200°С) выдержки по способу-прототипу;
на фиг. 2 приведены рентгеновские дифрактограммы образцов порошка сложного оксида, синтезированных при различной продолжительности изотермической (200°С) выдержки по настоящему изобретению;
на фиг. 3 приведены рентгеновские дифрактограммы образцов порошка сложного оксида, синтезированных при различной продолжительности изотермической (200°С) выдержки по способу-прототипу;
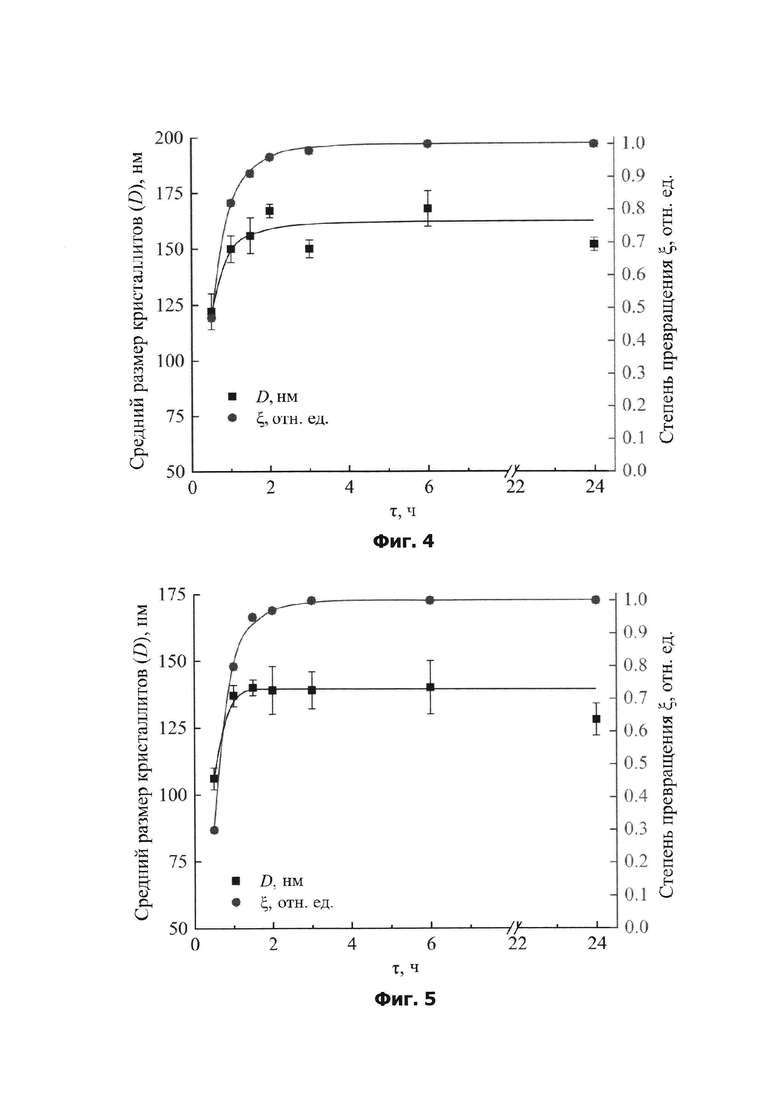
на фиг. 4 представлены зависимости степени превращения аморфной фазы в кристаллическую BFWO и среднего размера кристаллитов фазы BFWO от продолжительности изотермической (200°С) выдержки для образцов, синтезированных по настоящему изобретению;
на фиг. 5 представлены зависимости степени превращения аморфной фазы в кристаллическую BFWO и среднего размера кристаллитов фазы BFWO от продолжительности изотермической (200°С) выдержки для образцов, синтезированных по способу-прототипу;
на фиг. 6 представлены микрофотографии образцов, синтезированных при различной продолжительности изотермической (200°С) выдержки (а - 0,5 ч, в - 2 ч, д - 24 ч - с использованием настоящего изобретения; 6 - 0,5 ч, г - 2 ч, е - 24 ч - с использованием способа-прототипа).
Под первым этапом смешения (фиг. 1) подразумевается смешение раствора А (кислый раствор нитратов висмута и железа) с раствором Б (водный раствор вольфрамата натрия) с образованием суспензии MI, под вторым - смешение полученной суспензии MI с раствором В (раствором осадителя - NaOH) с образованием суспензии MII - суспензии аморфного прекурсора.
Микрореактор (фиг. 1) снабжен корпусом в форме конфузора 1, двумя или более тангенциальными патрубками 2, осевым патрубком 3 с соплом 4, на выходе из конфузора 1 установлена горловина 5, переходящая в диффузор 6, на выходе из которого образуется суспензия с аморфным прекурсором. Микрореактор называется так по той причине, что размер поперечного сечения его горловины 5 составляет, как правило, не более 3-4 мм, а объем горловины 5 - не более 150-250 мкл.
Настоящий способ получения порошка сложного оксида состава (Bi2O3)0.42(Fe2O3)0.28(WO3) со структурой фазы пирохлора осуществляют следующим образом.
Исходные растворы солей из емкостей (на фиг. 1 не показаны) подают насосами высокого давления в патрубки 2 микрореактора с заданными расходами. В первой зоне смешения в микрореакторе происходит смешение растворов А и Б умеренной интенсивности с удельной скоростью диссипации энергии порядка 100 Вт/кг (существенно превышающей интенсивность смешения в известных устройствах, включая перемешивание мешалками известных типов и тем более капельный метод, характеризующихся удельной скоростью диссипации энергии не выше 1 Вт/кг), после чего образовавшаяся суспензия MI попадает во вторую зону смешения, находящуюся ниже среза сопла 4, являющегося продолжением патрубка 3 и включающую объем горловины 5. Именно во второй зоне, имеющей объем менее 0,5 мл, трансформируется наибольшее количество вводимой в микрореактор механической энергии, и удельная скорость диссипации энергии достигает 10000 Вт/кг. По этой причине раствор осадителя (раствор В) и суспензия Ml, образовавшаяся после смешения растворов А и Б на первом этапе смешения, на втором этапе смешения подвергаются чрезвычайно интенсивному смешению, что создает условия гомогенизации, близкие к идеальным (индекс сегрегации достигает от 0,01 до 0,002, тогда как при использовании магнитной мешалки он составляет 0,5 и не зависит от скорости вращения мешалки). Образовавшаяся на выходе микрореактора суспензия аморфного прекурсора поступает на гидротермальную обработку в проточный многоканальный автоклав при температуре (190-210)°С в течение не менее 2 часов. При прохождении суспензии по каналам многоканального автоклава, обогреваемым при помощи высокотемпературных нагревательных элементов, в обрабатываемой суспензии вдоль длины каналов быстро возрастает температура и давление (в результате парообразования, а также расширения жидкости). Благодаря наличию большого количества параллельных каналов обеспечивается высокая производительность автоклава, а достаточно большая длина его каналов позволяет, с одной стороны, гарантировать необходимое время пребывания суспензии, поступающей из микрореактора на гидротермальную обработку, а с другой стороны обеспечивает поддержание высокого давления за счет гидравлического сопротивления. Из проточного автоклава продукт, полученный в результате гидротермальной обработки суспензии аморфного прекурсора и представляющий собой суспензию кристаллических частиц в маточном растворе, поступает на промывку в электрофлотационную машину. В электрофлотационной машине на катодах машины выделяются микропузырьки водорода, а на анодах - микропузырьки кислорода. Микроскопические пузырьки водорода и кислорода, поднимаясь сквозь слой обрабатываемой суспензии, содержащей наноразмерные и субмикронные частицы продукта, захватывают указанные частицы за счет адсорбционных сил, в результате чего частицы всплывают со слоем пены в верхнюю часть аппарата и выводятся из него. Очищенный от частиц (осветленный) раствор выводится из электрофлотационной машины и направляется на анализ и повторное использование, например, для извлечения соли NaNO3, или в дренаж. Концентрированная суспензия с частицами кристаллического продукта из электрофлотационной машины поступает далее в сушилку на сушку, которую ведут комбинацией распыления суспензии и псевдоожиженного слоя при температуре (80-90)°С, при этом подачу сушильного агента под решетку псевдоожиженного слоя производят в импульсном режиме, чтобы обеспечить, с одной стороны, разрушение межчастичных связей в слое и облегчить их выгрузку, с другой стороны, импульсный режим подачи сушильного агента препятствует уносу мелких частиц из слоя с потоком газа. В качестве дополнительного источника энергии при необходимости могут использоваться инфракрасные излучатели, установленные внутри сушилки на ее боковых или потолочных стенках соответственно. Комбинированный способ сушки позволяет существенно повысить производительность процесса сушки, обеспечив промышленный уровень производства.
Настоящее изобретение обеспечивает высокую равномерность распределения (на уровне микросмешения, т.е. молекулярном и ионном) исходных реагентов и осадителя в объеме реактора, приводящую к формированию продукта однородного состава и структуры, что достигается использованием микрореактора с интенсивно закрученными потоками, в котором интенсивность микросмешения определяется высокими значениями удельной скорости диссипации энергии (порядка 10000 Вт/кг) и значениями индекса сегрегации, близкими к идеальному микросмешению; и достижение высокой производительности способа получения порошка сложного оксида состава (Bi2O3)0.42(Fe2O3)0.28(WO3) со структурой фазы пирохлора.
Пример 1. Приготовление растворов в соответствии с настоящим изобретением осуществляли следующим образом: 28,00 ммоль кристаллогидрата нитрата висмута (III), Bi(NO3)3⋅5H2O (ос. ч.), и 18,67 ммоль кристаллогидрата нитрата железа (III) Fe(NO3)3⋅9H2O (ч.), было растворено в 66,7 мл 6 M HNO3 (ос. ч.), после чего в полученный раствор было добавлено 333,3 мл дистиллированной воды (раствор A: CBi≈0,070 M, CFe≈0,047 M). Далее 33,33 ммоль кристаллогидрата вольфрамата (VI) натрия, Na2WO4⋅2H2O (ос. ч.), растворяли в 238 мл дистиллированной воды (раствор Б: CW≈0,140 M). Растворы синхронно при помощи насосов объемного типа подавали в микрореактор (фиг. 1) - раствор А нитратов Bi+Fe - в один тангенциальный патрубок 2, раствор Б вольфрамата натрия - во второй тангенциальный патрубок 2, а раствор В (раствор щелочи) - в осевой патрубок 3. При этом благодаря конструкции микрореактора сначала происходит смешение растворов А и Б умеренной интенсивности в первой зоне смешения (в широкой части конфузора 1, примыкающей к тангенциальным патрубкам 2) с образованием суспензии MI, а затем, во второй зоне, на выходе из центрального сопла 4, суспензия MI интенсивно смешивается с раствором щелочи с образованием суспензии MII (далее - суспензия аморфного прекурсора). Объем зоны смешения растворов А и Б в используемом микрореакторе составлял 3,4 мл, а объем зоны смешения суспензии MI со щелочью составлял всего 0,35 мл. Расходы растворов контролировали при помощи электромагнитных расходомеров М2000 Bager Meter (погрешность измерений не превышала 2%). Расходы растворов были рассчитаны с учетом задаваемой стехиометрии продукта (Bi2O3)0,42(Fe2O3)0,28(WO3) и исходных концентраций реагентов. Расчетное значение рН суспензии составляло 2,0-2,4. Расходомеры были подключены к АЦП, который был соединен с ноутбуком с предустановленной на нем программой для сбора, обработки и хранения данных PowerGraph. Значения рН измеряли при помощи рН-метра Ohaus (электрод для суспензий и эмульсий - ST 230). Условия проведения синтеза в микрореакторе представлены в таблице 1. При условиях, представленных в таблице 1, расчетные значения скоростей составили: в тангенциальных патрубках wA=4,27 м/с, wB=2,52 м/с, на выходе из сопла wC=6,18 м/с. Скорости в горловине составили: тангенциальная (без учета потерь на трение) 33,2 м/с, осевая 7,97 м/с, полная 34,1 м/с.При таких высоких скоростях потоков жидкости уровень скорости диссипации энергии ε достигает порядка 10000 Вт/кг и выше, что обеспечивает высокое качество микросмешения. Согласно работе (Falk L, Commenge J.-M., 2010. Performance comparison of micromixers // Chem. Eng. Sci., 65, 405-411. DOI: 10.1016/j.ces.2009.05.045) именно удельная скорость диссипации энергии, практически независимо от геометрии аппарата, является ключевым параметром, от которого зависит эффективность микрореактора. В указанной работе по результатам исследований 8 типов микрореакторов получена обобщающая зависимость для времени микросмешения τm.

При уровне ε≥10000 Вт/кг расчет по формуле (1) дает значения τm≤2,4 мс. Значение 2,4 мс сопоставимо с временем смешения суспензии MI с раствором щелочи, которое происходит в объеме второй зоны (суммарный объем второй зоны составляет V2=0,35 мл) и составляет τ2=V2/(QA+QБ+QB)=3,0 мс, то есть микросмешение со щелочью осуществляется быстро и в малом объеме (0,35 мл). Смешение растворов А и Б в первой зоне не столь интенсивное, поскольку осуществляется с существенно меньшей скоростью диссипации энергии и в большем объеме (V1=3,4 мл). Время пребывания в первой зоне смешения растворов А и Б составляет 40 мс. Таким образом, в микрореакторе обеспечено время микросмешения не более 2,4 мс (вторая зона смешения), что является индикатором быстрого микросмешения. Другим индикатором высокого качества микросмешения являются низкие значения индекса сегрегации (от 0,002 до 0,01, а при использовании магнитной мешалки он составляет 0,5 и не зависит от скорости вращения мешалки; при идеальном микросмешении индекс сегрегации равен нулю, при полной сегрегации, то есть полном разделении ионов по объему, он равен единице). Эти количественные характеристики качества микросмешения обусловили высокие характеристики полученного продукта. Благодаря сочетанию высокой производительности и малого объема зон смешения достигается кратковременное время пребывания реагентов в зоне реакции, достаточное для нуклеации и роста частиц аморфного прекурсора, но недостаточное для чрезмерного роста агрегатов и агломератов. Получаемую в микрореакторе суспензию аморфного прекурсора непрерывно подавали в проточный многоканальный автоклав с параллельными каналами. С целью определения влияния продолжительности гидротермальной обработки на характеристики получаемого продукта, расчетное время пребывания в автоклаве (в секции изотермической выдержки) суспензии аморфного прекурсора составляло 0,5, 1, 1,5, 2, 3, 6 и 24 часа. При прохождении через зону нагрева суспензия быстро нагревалась до 200°С и далее (до зоны охлаждения) ее температура поддерживалась на этом уровне с погрешностью ±10°С в течение заданного промежутка времени. За зоной нагрева автоклава суспензия попадала в зону охлаждения, оснащенную охлаждающими элементами, где происходило быстрое охлаждение нагретой суспензии, так что на выходе из автоклава температура достигала 40-50°С. В каналах автоклава поддерживалось повышенное давление (порядка 16 атм) для предотвращения перехода суспензии из жидкого состояния в парообразное. Далее суспензия подавалась на промывку кристаллических частиц от маточного раствора в аппарат для промывки осадков - в электрофлотационную машину. Объем рабочей камеры электрофлотационной машины составил 210 л, ее длина составляла 3 м, ее ширина 120 мм и высота 800 мм (уровень жидкости около 585 мм). В рабочей камере электрофлотационной машины были равномерно установлены электроды - аноды и катоды. Концентрированная суспензия из нижней части электрофлотационной машины подавалась в распылительную головку сушилки. Сушилка представляла собой комбинированное устройство, в котором совмещены функции распылительной сушки и сушилки псевдоожиженного слоя, при этом подача сушильного агента (горячего воздуха) под решетку псевдоожиженного слоя производилась в импульсном режиме, с частотой 0,5 Гц (период импульсов составлял 2 с), при этом продолжительность стадии подачи воздуха под решетку составляла 0,2 с, а продолжительность стадии отвода воздуха из-под решетки составляла 1,8 с. При такой подаче воздуха происходило, с одной стороны, импульсное псевдоожижение слоя на стадии подачи, с другой, воздух с парами влаги успевал отводиться на стадии отвода. Результаты комплексного физико-химического анализа полученных образцов порошков, приведенные ниже, показали, что они практически не отличаются от таковых, полученных по известному способу-прототипу, однако, при этом достигается производительность, на несколько порядков превышающая производительность известного способа.
Пример 2. Получение порошка сложного оксида состава (Bi2O3)0.42(Fe2O3)0.28(WO3) со структурой фазы пирохлора было также осуществлено с использованием известного способа-прототипа. Способ заключается в последовательном использовании серии известных лабораторных приемов, а именно, в применении капельного метода подачи растворов реагентов на стадии получения суспензии аморфного прекурсора, в применении автоклавов периодического действия для гидротермальной обработки, в промывке декантацией, в сушке в полочном сушильном шкафу. Получение суспензии аморфного прекурсора было осуществлено капельной подачей (прямое соосаждение) растворов реагентов (при соотношении компонентов и концентрациях растворов аналогичных указанным в Примере 1). Методика получения образцов оксида BFWO капельной подачей растворов заключалась в следующем: 11,060 ммоль кристаллогидрата нитрата висмута (III), Bi(NO3)3⋅5H2O (ос. ч.), и 7,375 ммоль кристаллогидрата нитрата железа (III), Fe(NO3)3⋅9H2O (ч.), было растворено в 26,3 мл 6 M HNO3 (ос. ч.), после чего в полученный раствор было добавлено 131,7 мл дистиллированной воды (раствор A: CBi≈0,070 M, CFe≈0,047 M). Далее 13,165 ммоль кристаллогидрата вольфрамата (VI) натрия, Na2WO4⋅2H2O (ос. ч.), растворяли в 94 мл дистиллированной воды (раствор Б: CW≈0,140 M) и полученный раствор по каплям добавляли (QБ≈5,8 мл/мин) в перемешиваемый в лабораторном стакане на магнитной мешалке (800 об/мин) кислый раствор нитратов висмута и железа. Через 1 ч непрерывного перемешивания образовавшейся суспензии (КI) к ней по каплям добавляли раствор 2М NaOH (QB≈3,3 мл/мин) до рН ~ 2,2. В результате получали суспензию КII (далее - суспензия аморфного прекурсора). Полученную таким образом суспензию аморфного прекурсора после дополнительного перемешивания (800 об/мин) в течение ~ 1 часа переносили равными объемами в тефлоновые вкладыши (общий объем вкладыша - 50 мл; степень заполнения объема ~ 80%) и разогретые до 200°С стальные автоклавы, помещавшиеся затем в разогретую до такой же температуры печь. Через определенные промежутки времени (0.5, 1, 1.5, 2, 3, 6 и 24 ч) автоклавы извлекали из печи, охлаждали на воздухе, а полученные осадки несколько раз промывали декантацией и сушили при 80°С в течение 24 ч. Полученные таким образом порошки были подвергнуты анализу, результаты которого в сжатом виде излагаются ниже. Продолжительность процесса составляла не менее 3 суток, при этом получали ~1 г целевого кристаллического продукта (при расчете на один автоклав), то есть производительность составляла ~0,33 г/сут. При использовании настоящего изобретения процесс организован непрерывно, и при суммарной подаче растворов ~ 7 л/мин производительность по кристаллическому продукту составляет 145,8 г/мин, или 210 кг/сут (в 63000 раз больше, чем по способу-прототипу).
Был проведен сравнительный анализ образцов оксидов BFWO, полученных в примерах 1 и 2. Химический состав полученных образцов был определен методом рентгеноспектрального микроанализа. Полученные данные свидетельствуют о том, что валовые элементные составы всех образцов, независимо от условий синтеза (способа получения аморфного прекурсора и продолжительности его изотермической выдержки при гидротермальной обработке), хорошо соответствуют номинальному составу (табл. 2 - табл. 3). Рентгенофазовый анализ полученных порошков (фиг. 2 - фиг. 3) показал, что единственной кристаллической фазой во всех образцах является фаза со структурой кубического пирохлора в системе Bi2O3-Fe2O3-WO3.
Из представленных на фиг. 4 - фиг. 5 (правая OY) зависимостей следует, что с увеличением продолжительности изотермической выдержки от 0,5 до 2 ч степень превращения аморфной фазы в кристаллическую (ξ) увеличивается от ~45% до ~100% в случае образцов, полученных при использовании настоящего изобретения (фиг. 4), и от ~30% до ~100% в случае образцов, полученных известным способом-прототипом (фиг. 5).
Из представленных на фиг. 4 - фиг. 5 (левая OY) зависимостей следует, что с увеличением продолжительности изотермической выдержки от 0,5 до 1 ч средний размер кристаллитов (D) фазы пирохлора увеличивается от ~120 нм до ~155 нм в случае образцов, полученных при использовании настоящего изобретения (фиг. 4), и от ~100 нм до ~140 нм в случае образцов, полученных известным способом-прототипом (фиг. 5). При дальнейшем увеличении продолжительности изотермической выдержки от 1 до 24 ч средний размер кристаллитов в пределах погрешности не изменяется.
Средний размер частиц в образцах, полученных при использовании настоящего изобретения (фиг. 6 а, в, д), составляет ~400 нм (кроме образца 0,5 ч - ~340 нм), то есть, при увеличении продолжительности изотермической выдержки от 1 ч до 24 ч, средний размер частиц практически не изменяется. Схожим образом изменяется средний размер сферических частиц в случае образцов, полученных известным способом-прототипом (фиг. 6 6, г, е), - увеличивается от ~240 нм до ~300 нм при увеличении продолжительности изотермической выдержки от 0,5 ч до 1 ч, а при дальнейшем увеличении ее продолжительности уже практически не меняется.
Значения ширины запрещенной зоны Eg полученных порошков, рассчитанные по методу Тауца, равны 2,40±0,03 эВ и 2,35±0,03 эВ при использовании настоящего изобретения и известного способа-прототипа соответственно, что характеризует полученные порошки как широкозонные полупроводники.
Помимо представленных выше результатов было обнаружено, что на внутренних стенках тефлоновых вкладышей после извлечения из них продукта гидротермального синтеза (например, при т=6 ч), в случае образцов, полученных известным способом-прототипом, присутствует темно-красный налет, отсутствующий в случае образцов, полученных по настоящему изобретению. Данные по локальному рентгеноспектральному микроанализу, полученные с некоторой области такого налета, свидетельствуют о присутствии в нем фазы на основе оксида железа (III), поскольку количество других элементов (Bi и W), по сравнению с Fe, незначительно. В случае капельной подачи раствора NaOH в суспензию КI при использовании известного способа-прототипа имеет место неравномерное распределение гидроксид-анионов по объему реакционного пространства, поэтому в месте соприкосновения капель раствора NaOH с поверхностью суспензии КI образуется сильно щелочная среда. Катионы железа (III), находящиеся в дисперсионной среде, переходят в гидроксидную форму, устойчивую при рН, близких к 14. В это же время, в других объемах реакционного пространства повышение значения рН происходит гораздо медленнее из-за недостаточно интенсивного перемешивания реакционной среды при использовании магнитной мешалки. Как итог, недостаточное перемешивание приводит к тому, что среднее значение рН в реакционной системе постепенно растет при добавлении щелочи, но вместе с тем, в поверхностной области имеются объемы, в которых на протяжении всего процесса добавления щелочи рН близко к 14. По этой причине, в процессе гидротермальной обработки полученной таким образом суспензии аморфного прекурсора (КII) образуется примесное количество оксида железа (III), которое в основном сконцентрировано на стенках тефлонового вкладыша. Следует добавить, что указанная проблема усугубляется при переходе к промышленному масштабу оборудования, где качество микросмешения существенно хуже, чем в лабораторном масштабе.
При получении суспензии аморфного прекурсора по настоящему изобретению, за счет подачи растворов реагентов в микрореактор с закручивающимися потоками, в каждый момент времени с определенным объемом суспензии MI (во второй зоне) соприкасается одно и то же количество раствора NaOH, причем в этой зоне микрореактора смешение является высокоинтенсивным. Это приводит к скачкообразному повышению значения рН от ~1 до ~2,2 без дальнейшего его роста в какой-либо области реакционного пространства. В результате гидротермальной обработки полученной таким образом суспензии аморфного прекурсора (MII) не происходит пространственного разделения реагирующих компонентов и из кристаллических фаз образуется только фаза пирохлора заданного по синтезу состава.
Формирование примесной фазы оксида железа (III) в процессе гидротермальной обработки суспензии аморфного прекурсора (что наблюдается в случае использования известного способа-прототипа) должно сказываться на составе образующейся кристаллической фазы пирохлора, который должен в таком случае обедняться по оксиду железа. Состав кристаллической фазы (известно, что фаза пирохлора является фазой переменного состава), в свою очередь, будет влиять на функциональные свойства полученного порошкообразного материала, что наглядно иллюстрируется на примере исследованных оптических свойств.
Разброс значений Eg образцов, полученных при использовании настоящего изобретения, практически не наблюдается, в то время как присутствует в случае использования известного способа-прототипа. Наблюдаемый в последнем случае разброс значений объясняется некоторым различием в химическом составе фазы пирохлора в образцах, полученных известным способом-прототипом, что обусловлено плохим качеством микросмешения, приводящем к пространственному разделению реагирующих компонентов и формированию примесной фазы оксида железа (III).
В настоящем изобретении на стадии получения суспензии аморфного прекурсора за счет смешения растворов реагентов в микрореакторе с интенсивно закрученными потоками удалось избежать пространственного обособления компонентов при образовании аморфного прекурсора. Это обеспечило формирование кристаллов только фазы пирохлора с заданной стехиометрией при гидротермальной обработке суспензии аморфного прекурсора. Производительность микрореактора (при исследованных режимах работы) составила ~ 420 л/ч суспензии аморфного прекурсора. Это на порядки превышает производительность капельной подачи растворов реагентов.
Настоящее изобретение позволяет обеспечить высокую равномерность распределения (на уровне микросмешения) исходных реагентов и осадителя в объеме реактора; достичь высокой производительности на всех стадиях процесса.
Таким образом, настоящее изобретение обеспечивает непрерывность проведения процесса получения оксида BFWO на всех стадиях - от синтеза из растворов до сушки, обеспечивая при этом производительность 7 л/мин по суспензии аморфного прекурсора.
Табл. 1. Условия проведения синтеза в микрореакторе с интенсивно закрученными потоками (по настоящему изобретению)
QА, л/мин
QБ, л/мин
QB=QNaOH,
л/мин
суспензии аморфного прекурсора
Табл. 2. Валовый элементный состав образцов, синтезированных при различной продолжительности изотермической (200°С) выдержки, в отн. оксидных. ед., по данным рентгеноспектрального микроанализа (по настоящему изобретению)
Табл. 3. Валовый элементный состав образцов, синтезированных при различной продолжительности изотермической (200°С) выдержки, в отн. оксидных. ед., по данным рентгеноспектрального микроанализа (известный способ-прототип)
*отн. оксидные ед., соответствующие номинальному составу, заложенному по синтезу, равны. 0,42, 0,28 и 1,50, соответственно.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПОРОШКА СЛОЖНОГО ОКСИДА ВИСМУТА, ЖЕЛЕЗА И ВОЛЬФРАМА СО СТРУКТУРОЙ ФАЗЫ ПИРОХЛОРА | 2023 |
|
RU2825757C1 |
| УСТРОЙСТВО ДЛЯ ГИДРОТЕРМАЛЬНОЙ ОБРАБОТКИ | 2023 |
|
RU2825758C1 |
| Микрореактор для синтеза наноразмерных частиц из растворов | 2021 |
|
RU2793562C2 |
| Способ получения нанопорошка иттрий-алюминиевого граната | 2021 |
|
RU2761324C1 |
| Способ получения нанокристаллического порошка на основе диоксида циркония | 2022 |
|
RU2793893C1 |
| Способ получения порошков фаз твёрдых растворов системы 0,75BiFeO-0,25Ba(ZrTi)O, легированных соединениями марганца | 2022 |
|
RU2787492C1 |
| Способ получения нанопорошков феррита висмута | 2019 |
|
RU2748446C2 |
| Способ получения нанопорошка иттрий-алюминиевого граната | 2020 |
|
RU2741733C1 |
| Микрореактор-смеситель во встречными закрученными потоками | 2020 |
|
RU2741735C1 |
| ТЕРМОИНДИКАТОР ДЛЯ КУХОННОГО ИЗДЕЛИЯ | 2018 |
|
RU2780015C2 |



Изобретение относится к получению наноразмерных и/или субмикронных порошков сложного оксида со структурой пирохлора состава (Bi2O3)0.42(Fe2O3)0.28(WO3), которые могут быть использованы для создания фотоэлектрических преобразователей энергии, для фотокалитической очистки и обеззараживания воздуха и воды. Осуществляют смешивание раствора нитратов висмута и железа с раствором вольфрамата натрия с образованием суспензии, смешение суспензии с раствором щелочи для образования суспензии аморфного прекурсора, имеющей рН=2-2,4, гидротермальную обработку суспензии аморфного прекурсора, промывку полученного порошка и его сушку. Смешение исходных растворов проводят в реакторе проточного смешения - микрореакторе с интенсивно закрученными потоками. Гидротермальную обработку суспензии аморфного прекурсора проводят в проточном многоканальном автоклаве при температуре 190-210°С в течение не менее 2 часов. Промывку полученного порошка и его отделение от маточного раствора осуществляют в электрофлотационной машине, а сушку полученного порошка ведут комбинацией распыления суспензии и псевдоожиженного слоя при температуре 80-90°С. Подачу сушильного агента под решетку псевдоожиженного слоя производят в импульсном режиме. Обеспечивается получение порошка однородного состава и структуры и высокая производительность. 6 ил., 3 табл., 2 пр.
Способ получения порошка сложного оксида состава (Bi2O3)0.42(Fe2O3)0.28(WO3) со структурой фазы пирохлора, включающий приготовление раствора нитратов висмута и железа, раствора вольфрамата натрия и раствора щелочи, взятых в стехиометрическом соотношении, обеспечивающем получение указанного выше сложного оксида, смешение раствора нитратов висмута и железа с раствором вольфрамата натрия с образованием суспензии, смешение суспензии с раствором щелочи для образования суспензии аморфного прекурсора, имеющей рН=2-2,4, гидротермальную обработку суспензии аморфного прекурсора, промывку полученного порошка и его сушку, отличающийся тем, что смешение исходных растворов проводят в реакторе проточного смешения - микрореакторе с интенсивно закрученными потоками, при этом раствор нитратов висмута и железа подают в один тангенциальный патрубок, раствор вольфрамата натрия подают во второй тангенциальный патрубок, обеспечивая смешение этих растворов с образованием суспензии, а раствор щелочи подают в осевой патрубок, смешивая его с образовавшейся суспензией с получением суспензии аморфного прекурсора, гидротермальную обработку суспензии аморфного прекурсора проводят в проточном многоканальном автоклаве при температуре 190-210°С в течение не менее 2 часов, причем промывку полученного порошка и его отделение от маточного раствора осуществляют в электрофлотационной машине, а сушку полученного порошка ведут комбинацией распыления суспензии и псевдоожиженного слоя при температуре 80-90°С, при этом подачу сушильного агента под решетку псевдоожиженного слоя производят в импульсном режиме.
| LOMAKIN M.S | |||
| et.al | |||
| Crystal structure and optical properties of the Bi-Fe-W-O pyrochlore phase synthesized via a hydrothermal method | |||
| Journal of alloys and compounds, 31.12.2021, V.889, p.161598 | |||
| LOMAKIN M.S | |||
| et.al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
