Изобретение относится к способам получения наноразмерных и/или субмикронных порошков состава (Bi2O3)0.61(Fe2O3)0.22(WO3), представляющих собой смесь пирохлора состава (Bi2O3)0.52(Fe2O3)0.27(WO3) (далее - пирохлор) и фазы Ауривиллиуса Bi2WO6, и может быть использовано в технологиях получения материалов для фотокаталитических процессов окисления вредных органических веществ, в фотокаталитических процессах генерации водорода для нужд водородной энергетики, а также в качестве материалов проводящих слоев в солнечных элементах.
Впервые фаза со структурой пирохлора в системе Bi2O3-Fe2O3-WO3-(H2O) была получена и структурно охарактеризована в составе сложных композитов (см. Lomakin M.S., Proskurina O.V., Danilovich D.P., Panchuk V.V., Semenov V.G., Gusarov V.V. Hydrothermal Synthesis, Phase Formation and Crystal Chemistry of the pyrochlore/Bi2WO6 and pyrochlore/a-Fe2O3 Composites in the Bi2O3-Fe2O3-WO3 System, Journ. Solid State Chem., 2020, 282, 121064 DOI: 10.1016/j.jssc.2019.121064). Получение порошков проводили методом гидротермального синтеза, включавшим получение суспензии аморфного прекурсора, ее дальнейшую гидротермальную обработку, промывку полученного кристаллического осадка от посторонних растворимых солей и сушку кристаллического осадка. В работе (Lomakin M.S., Proskurina O.V., Gusarov V.V. Influence of hydrothermal synthesis conditions on the composition of the pyrochlore phase in the Bi2O3-Fe2O3-WO3 system, Nanosyst: Phys. Chem. Math., 2020, 11 (2), P. 246-251. DOI: 10.17586/2220-8054-2020-11-2-246-251) были впервые получены порошки фазы со структурой пирохлора без примеси других фаз аналогичным методом синтеза. Однако, хорошо известно (см., например, A. Fakharuddin, L. Schmidt-Mende, Hybrid organic/inorganic and perovskite solar cells, in: H. Tian, G. Boschloo, A. Hagfeldt (Eds.), Molecular Devices for Solar Energy Conversion and Storage, Green Chemistry and Sustainable Technology, Springer, Singapore, 2018, https://doi.org/10.1007/978-981-10-5924-7_5), что наиболее перспективным с точки зрения получаемых функциональных характеристик является использование в обозначенных выше процессах не однофазных, а многофазных материалов, поскольку ключевую роль во многом играет поверхность частиц и межфазные взаимодействия. Кроме того, получение нанокристаллических композиционных материалов, в которых размеры кристаллитов находятся в диапазоне от единиц до 20-30 нм, как правило, позволяет значительно улучшить их функциональные свойства. В связи с этим, наиболее перспективным является разработка метода синтеза, который позволил бы получать нанокристаллический композиционный материал и в то же время обладал бы рядом преимуществ (упрощение, удешевление, повышение экологичности технологии) над традиционными методами.
Известен способ синтеза оксидных наночастиц в гидротермальных условиях, в процессе которого в автоклавах протекает дегидратация предварительно осажденных гидроксидов [см. Альмяшева О.А., Гусаров В.В. Гидротермальный синтез наночастиц и нанокомпозитов в системе ZrO2 - Al2O3 - H2O // Альтернативная энергетика и экология. 2007. №1. С.113-115].
Недостатком известного способа синтеза, в котором кристаллизация целевого продукта происходит в гидротермальных условиях, является использование сосудов высокого давления - автоклавов, что не только увеличивает себестоимость этой технологии, но и требует строгого соблюдения правил безопасной эксплуатации такого оборудования. Кроме того, стадия гидротермальной обработки длится иногда часы или сутки, а после ее завершения необходимо производить отмывку кристаллического осадка от маточного раствора, содержащего посторонние соли, которая, как правило, проводится с использованием известного лабораторного приема - методом декантации (сливания маточного раствора). Эта стадия имеет значительную продолжительность, особенно, если речь идет о наноразмерных порошках, поскольку скорость их седиментации крайне мала, и нередко для ускорения этого процесса суспензию с кристаллическим осадком центрифугируют (что опять же, приводит к удорожанию технологии). Завершающей стадией, также имеющей значительную продолжительность, процесса синтеза в известном способе является сушка концентрированной суспензии промытого кристаллического осадка в керамических чашках в лабораторном сушильном шкафу.
Известен способ синтеза порошка ортогерманата висмута (см. RU2659268, МПК С30В 7/10, С30В 28/04, С30В 30/00, С30В 29/32, C09K 11/66, C09K 11/74, C01G 17/00, C01G 29/00, опубл. 29.06.2018), включающий смешивание реагентов - водного раствора нитрата висмута Bi(NO3)3⋅5H2O и оксида германия GeO2 - в стехиометрическом соотношении Bi/Ge - 4:3, добавление к полученной суспензии водного раствора аммиака (1,7-8 М) и последующую гидротермально-микроволновую обработку при температуре 140-220°С в течение 0,5-2,0 часов в тефлоновом автоклаве объемом, в 2-4 раза превышающим объем суспензии.
Недостатком известного способа синтеза оксидных соединений, в котором реализуется гидротермально-микроволновая обработка прекурсора, является необходимость использования и обслуживания дорогостоящего оборудования ограниченной производительности (микроволновых установок), что лимитирует переход от лабораторного уровня к промышленному и увеличивает стоимость единицы продукции. Кроме того, материалом сосудов (виал), работающих в условиях микроволнового нагрева реакционной смеси, является, как правило, боросиликатное стекло, которое может разрушаться при продолжительной работе в агрессивных средах при повышенных температурах и давлениях, что существенно сокращает спектр синтетических возможностей.
Известен способ получения порошков фаз кислородно-октаэдрического типа (см. RU 2448928, МПК С04В 35/626, С04В 35/46, В82В 3/00, опубл. 10.12.2011), реализующийся в несколько этапов: на первом этапе осуществляют синтез исходных нанокластеров, являющихся полимерными α-формами гидроксидов р- и d-элементов, которые осаждаются при температурах ниже 280 К из 0,1-0,3 М растворов нитратных комплексов этих элементов при рН 8±0,5 с помощью 5-10% раствора аммиака. На втором этапе к нанокластерам добавляют аммиачно-нитратный буферный раствор с насыщенными растворами нитратов различных элементов состава MeNO3, Me(NO3)2 и Me(NO3)3. Процесс синтеза фазы заданного состава проводят при температурах ниже 280 К и атмосферном давлении. Первичную и вторичную рекристаллизацию продуктов реакции осуществляют при температурах 600-700 К.
Недостатком известного способа синтеза порошков является необходимость использования осадителя (раствора аммиака) для изменения рН реакционной среды в щелочную сторону и перевода катионов металлов из раствора в осадок, что не только приводит к дополнительным финансовым затратам из-за использования дополнительного реактива, но и к необходимости проводить работы в вытяжном шкафу, поскольку раствор аммиака является очень летучей жидкостью. Кроме того, добавление осадителя в раствор солей, как правило, производится известным лабораторным приемом - по каплям, а при таком способе смешения на низком уровне остается и качество гомогенизации смеси. Это приводит к возникновению в растворе градиентов рН, поскольку концентрация осадителя распределяется по объему реакционного пространства неравномерно из-за неэффективного перемешивания раствора магнитной или лопастной мешалкой. Это существенно снижает производительность известного способа, поскольку для выравнивания концентрации осадителя следует производить перемешивание реакционной системы в течение длительного времени, что приводит к снижению выхода продукта в единицу времени с единицы массы исходных реагентов.
Известен способ синтеза оксидов со структурой перовскита (см. RU2440292, МПК С01В 13/18, опубл. 20.01.2012), заключающийся в механическом смешении исходных солей-предшественников в стехиометрическом отношении, микроволновой обработке смеси исходных солей в СВЧ-печи с последующей термической обработкой при температуре 500-900°С в течение 1-5 часов. В качестве исходных солей для синтеза используют кристаллогидраты нитратов редкоземельных, щелочно-земельных и переходных элементов, а также негидратированные нитраты или карбонаты тех же предшественников. Микроволновое излучение применяют при рабочей частоте 2,45 ГГц и мощности 600-1000 Вт в течение 3-10 минут.
Недостатками известного способа синтеза оксидов со структурой перовскита являются, во-первых, проведение на первой стадии синтеза ручного перемешивания исходных солей в агатовой ступке, что является малоэффективным способом гомогенизации сыпучих материалов, во-вторых, использование дорогостоящего оборудования (микроволновой установки) на промежуточной стадии синтеза, направленной на получение прекурсора кристаллического продукта, в-третьих, высокая температура термообработки (до 900°С) прекурсора, что приводит к заметному спеканию порошкообразного продукта и увеличению размерных параметров частиц и кристаллитов.
Известен способ получения наностержней двойного пирохлора Bi2Ti2O7 методом обратных мицелл и без использования подложки (см. US 8900537 B2, C01G 29/00, B01J 23/18, B82Y 30/00, B82Y 40/00, С01Р 2002/36, С01Р 2002/72, С01Р 2002/84, С01Р 2004/03, С01Р 2004/04, С01Р 2004/16, Y10T 428/2913, Y10T 428/298, Y10T 428/2982, опубл. 02.12.2014), заключающийся в отдельном смешении первого водного раствора, стабилизированного кислотой, включающего предшественник пирохлора А, и второго стабилизированного кислотой водного раствора, содержащего предшественник пирохлора В, с органическим раствором, включающим поверхностно-активное вещество, с образованием эмульсии масла в воде, после чего эквимолярные растворы первой и второй стабилизированных кислотой эмульсий масло в воде смешивают и затем в смесь первой и второй эмульсий масла в воде, стабилизированных кислотой, вводят осадитель, в результате чего образуется осадок, включающий предшественники пирохлора А и В. После этого осадок сушат для удаления летучих веществ. Затем осадок прокаливают в присутствии кислорода с образованием наноструктурированной фазы пирохлора (Bi2Ti2O7), имеющей стержнеобразную морфологию.
К недостаткам известного способа получения наностержней двойного пирохлора Bi2Ti2O7 следует отнести: использование органического поверхностно-активного вещества, что требует дополнительных затрат на приобретение этого реактива, а также затрат на его отмывку от лабораторной посуды (из-за его нерастворимости в воде); использование нелетучего осадителя на стадии образования осадка, включающего предшественники пирохлора А и В, может привести при его дальнейшем прокаливании к внедрению в структуру целевого продукта катионов натрия или калия, так как в известном способе отсутствует стадия промывки осадка перед его прокаливанием; известный способ обеспечивает формирование частиц целевого продукта только стержнеобразной морфологии.
Известен способ получения порошка титаната висмута (Bi4Ti3O12) со структурой фазы Ауривиллиуса, частицы которого представляют собой нанокубоид (см. CN 103274455, МПК C01G 23/00, B82Y 30/00, опубл. 10.12.2014), заключающийся в добавлении в раствор титаната висмута додецилбензолсульфоната натрия в концентрации 0,02-0,2 моль/л и перемешивании до полного растворения, добавлении затем водного раствора NaOH для получения осадка, термообработке полученного осадка в герметичном реакторе при температуре 200-230°С в течение 16-20 часов, промывке кристаллического осадка до нейтральной реакции среды и сушке кристаллического осадка в сушильной печи.
Основными недостатками известного способа являются: использование гидротермальной обработки для стабилизации кристаллической фазы титаната висмута, что требует проведения этой стадии в сосудах периодического действия, работающих под повышенным давлением, и влечет за собой все недостатки этого метода синтеза, описанные выше; наличие стадии промывки целевого продукта от посторонних примесей, что приводит к существенным дополнительным затратам времени на завершение процесса седиментации осадка, поскольку при декантации обогащенного ионами примесной соли раствора необходимо достичь пространственного разделения дисперсной фазы и дисперсионной среды водной-солевой суспензии целевого продукта; наличие стадии сушки концентрированной суспензии порошка целевого продукта в сушильном шкафу до постоянной массы, что, опять же, приводит к дополнительным затратам не только электроэнергии, но и времени.
Известен способ получения порошков сложного оксида висмута, железа и вольфрама со структурой фазы пирохлора (см. RU 2802703, МПК B01F 5/00, C01G 29/00, C01G 41/00, C01G 49/00, F26B 3/00, опубл. 31.08.2023), совпадающий с настоящим техническим решением по наибольшему числу существенных признаков и принятый за прототип. Способ-прототип включает приготовление раствора нитратов висмута и железа, раствора вольфрамата натрия и раствора щелочи (NaOH), взятых в стехиометрическом соотношении, обеспечивающем получение сложного оксида со структурой пирохлора состава (Bi2O3)0.42(Fe2O3)0.28(WO3), смешение исходных растворов в реакторе проточного смешения - микрореакторе с интенсивно закрученными потоками, с образованием суспензии аморфного прекурсора, имеющей рН=2-2,4, гидротермальную обработку суспензии аморфного прекурсора при температуре 190-210°С (при таких параметрах термообработки происходит полная кристаллизация аморфной фазы в целевой продукт - фазу со структурой пирохлора) в течение не менее 2 часов, промывку полученного порошка дистиллированной водой и сушку полученного порошка комбинацией распыления суспензии и псевдоожиженного слоя при температуре 80-90°С, при этом подачу сушильного агента под решетку псевдоожиженного слоя производят в импульсном режиме.
Можно выявить следующие недостатки известного способа-прототипа. Использование Na-содержащего реактива приводит к появлению в реакционной системе катиона Na+, который, во-первых, может внедряться в структуру целевого продукта, во-вторых, возникает необходимость проводить отмывку целевого кристаллического продукта от растворимой соли нитрата натрия (NaNO3). При применении гидротермальной обработки суспензии аморфного прекурсора для стабилизации кристаллической фазы целевого продукта имеет место активный массоперенос компонентов за счет наличия подвижной среды -гидротермального флюида, что приводит к быстрому росту кристаллитов и частиц целевого продукта. Для получения продукта заданной стехиометрии и фазового состава с использованием способа-прототипа необходимо, чтобы рН суспензии аморфного прекурсора составлял ~ 2-5, что достигается только за счет подщелачивания суспензии раствором NaOH, то есть необходимо использовать дополнительный реагент. При остывании гидротермального флюида, представляющего собой насыщенный раствор реагирующих компонентов при температуре гидротермальной обработки, происходит его пересыщение, что приводит к твердению из раствора аморфной фазы, являющейся примесью. Необходимость в способе-прототипе осуществлять сушку концентрированной суспензии порошка целевого продукта в сушилке комбинацией распыления суспензии и псевдоожиженного слоя (при 80-90°С), приводит к дополнительным затратам не только электроэнергии, но и времени.
Задачей настоящего технического решения является разработка способа получения порошка сложного оксида висмута, железа и вольфрама со структурой фазы пирохлора в составе двухфазного композита состава (Bi2O3)0.61(Fe2O3)0.22(WO3), представляющего собой смесь пирохлора состава (Bi2O3)0.52(Fe2O3)0.27(WO3) и фазы Ауривиллиуса Bi2WO6, который бы обеспечивал получение нанокристаллических частиц целевого продукта заданной стехиометрии и меньших размеров при упрощении, удешевлении и повышении экологичности технологии синтеза за счет, главным образом, сокращения количества, продолжительности и сложности стадий синтетического процесса, в том числе объемов потребляемой дистиллированной воды и сливаемых в окружающую среду жидких отходов.
Поставленная задача достигается тем, что способ получения порошка сложного оксида висмута, железа и вольфрама со структурой фазы пирохлора в составе двухфазного композита состава (Bi2O3)0.61(Fe2O3)0.22(WO3), представляющего собой смесь пирохлора состава (Bi2O3)0.52(Fe2O3)0.27(WO3) и фазы Ауривиллиуса Bi2WO6, включает приготовление раствора нитратов висмута и железа, раствора вольфрам-содержащего реактива, взятых в стехиометрическом соотношении, обеспечивающем получение указанного выше композиционного материала, смешение раствора нитратов висмута и железа с раствором вольфрам-содержащего реактива с образованием суспензии прекурсора, термообработку полученного порошка прекурсора на воздухе. Новым в способе является то, что в качестве вольфрам-содержащего реактива используют не содержащий натрия паравольфрамат аммония, для стабилизации кристаллических фаз целевого продукта полученную при смешении исходных растворов суспензию прекурсора подвергают не гидротермальной обработке, а выпаривают до полного испарения жидкости, полученный в результате выпаривания суспензии порошок прекурсора нагревают до температуры 280-300°С со скоростью 5-10°С/мин и выдерживают при этой температуре не менее 1 часа, затем нагревают до температуры 580-600°С со скоростью 10-15°С/мин и выдерживают при этой температуре в течение 4-5 часов, после чего остужают полученный порошок в естественном режиме.
Использование в качестве вольфрам-содержащего реактива паравольфрамата аммония позволяет избежать попадания в реакционную систему катионов натрия, что обеспечивает формирование кристаллического целевого продукта без примесей катионов натрия в структуре и позволяет исключить стадию промывки целевого продукта от нитрата натрия (NaNO3).
Отсутствие стадии подщелачивания суспензии прекурсора раствором NaOH позволяет сократить продолжительность синтеза и расходы на реактивы и избежать попадания примесного катиона натрия в реакционную систему.
Выпаривание суспензии прекурсора проводят с целью получения порошкообразного прекурсора (удаления жидкой фазы) и осуществляют в режиме кипения при нагревании суспензии на песчаной бане с помощью, например, бытовой электроплиты. Поскольку при любых значениях рН суспензии прекурсора, в том числе кислых, часть реагирующих компонентов находится в дисперсионной среде, декантация жидкости с осадка не проводится, иначе будет нарушена стехиометрия получаемого порошкообразного прекурсора и, как следствие, целевого продукта. При выпаривании суспензии прекурсора не происходит потерь реагирующих компонентов, поскольку из реакционной системы удаляют не раствор, а растворитель, что позволяет не только сохранить стехиометрию порошкообразного прекурсора, но и избежать сливов. Кроме того, отпадает необходимость осуществлять сушку концентрированной суспензии прекурсора как отдельную стадию, поскольку, выпаривание производится до полного удаления жидкости.
Термообработка порошка прекурсора в муфельной печи на воздухе является ключевой стадией синтеза, в результате которой происходит стабилизация кристаллических фаз целевого продукта. Эта стадия реализуется в два этапа, необходимость первого из которых обусловлена соображениями техники безопасности и направлена на безопасное удаление из реакционной системы побочного продукта реакции - нитрата аммония (NH4NO3). Первый этап термообработки заключается в нагревании порошка прекурсора от комнатной температуры до 280-300°С со скоростью 5-10°С/мин и выдержке при этой температуре не менее 1 часа. Такие параметры термообработки обусловлены поведением фазы нитрата аммония при нагревании, а именно, при температуре ~ 170°С происходит плавление, при нагреве выше этой температуры начинается постепенное разложение вещества, а при температуре 210°С происходит полное разложение по реакции:
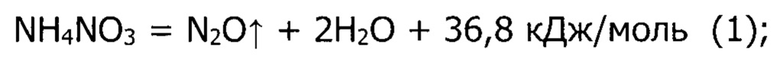
Температурный интервал (280-300°С), в котором производится первый этап термообработки порошка прекурсора, превышает температуру полного разложения (210°С) нитрата аммония по представленной выше реакции, с той целью, чтобы ускорить завершение процесса разложения. С термодинамической точки зрения, нитрат аммония выше температуры 210°С является неустойчивым, однако кинетика его разложения будет определяться температурой процесса: при 210°С скорость разложения будет минимальной, при нагревании до температур 280-300°С скорость разложения по реакции (1) повысится, но все еще не будет слишком высокой, чтобы привести к интенсивному выделению тепла, в то время как при еще большем повышении температуры (>300°С) разложение нитрата аммония будет уже происходить по другому механизму и с выделением большего количества теплоты и газообразных продуктов (детонация):
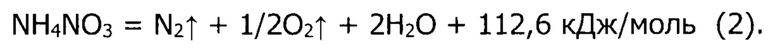
Верхняя граница диапазона температур, до которых производится нагревание порошка прекурсора на первом этапе термообработки, составляет 300°С, с той целью, чтобы не допустить слишком интенсивного выделения теплоты, при протекании разложения нитрата аммония по реакции (1), и в то же время не допустить протекания процесса разложения по реакции (2), которая по сути, является детонацией.
Такими же соображениями обусловлена скорость, с которой производится нагрев порошка прекурсора (5-10°С/мин). Такая скорость нагрева является оптимальной, поскольку не будет приводить к интенсивному разложению нитрата аммония, а скорости <5°С/мин будут сильно замедлять процесс синтеза, не приводя к положительному эффекту.
При протекании обоих процессов происходит образование сильных окислителей (газы N2O и О2), реакционная способность которых заметно возрастает при повышении температуры. Для полного удаления этих газообразных окислителей из реакционного пространства производят выдержку реакционной системы при температуре 280-300°С не менее 1 часа.
Второй этап термообработки производят непосредственно с целью стабилизации кристаллических фаз целевого продукта. Он заключается в нагревании реакционной системы от 280-300°С до 580-600°С со скоростью 10-15°С/мин и выдержке при этой температуре в течение 4-5 часов. Поскольку на первой стадии термообработки уже было обеспечено полное удаление взрывоопасного побочного продукта (NH4NO3), то дальнейший нагрев реакционной системы уже можно производить с более высокой скоростью (10-15°С/мин). На данном этапе скорость нагрева определяется лишь техническими возможностями используемой муфельной печи в рамках ее безопасной эксплуатации и не влияет на характеристики получаемого целевого продукта.
Верхняя граница диапазона температур изотермической выдержки (600°С) обусловлена следующими соображениями. Поскольку одна из целевых кристаллических фаз в исследуемой системе (фаза пирохлора) является фазой переменного состава, то положение границ области ее устойчивости будет зависеть от температуры, а именно, при повышении температуры выше 600°С (это показано авторами изобретения в их предыдущих работах) область устойчивости будет существенно уменьшаться, сходясь в точку при температуре ~ 700°С (выше этой температуры фаза пирохлора в исследуемой системе не существует). Другими словами, разнообразие химического состава фазы пирохлора, если ее синтез проводить при температурах >600°С, будет сильно сокращаться, а следовательно, будут сужаться возможности для варьирования функциональных свойств получаемых материалов. Кроме того, повышение температуры термообработки будет приводить к увеличению размерных параметров кристаллитов и частиц формирующихся фаз, а значит не обеспечит получения наноразмерного материала.
Нижняя граница диапазона температур изотермической выдержки (580°С) обусловлена, в первую очередь тем, что температура кристаллизации аморфной составляющей прекурсора находится в диапазоне ~500-550°С, следовательно, температура его термообработки должна быть выше этих значений, иначе в целевом продукте будет обнаруживаться аморфная фаза. Повышение температуры до 580°С произведено с той целью, чтобы ускорить процесс кристаллизации аморфной фазы, для завершения которого требуется тем меньше времени, чем выше будет температура.
В соответствии с одной из подзадач настоящего технического решения, как было отмечено выше, требуется обеспечить получение нанокристаллических частиц целевого продукта как можно меньших размеров, что, как правило, достигается определением оптимальных значений температуры и продолжительности изотермической выдержки. Другими словами, нужно подобрать такие величины этих параметров, при которых процессы кристаллизации аморфной фазы успевали бы завершиться до того момента, как отрастут кристаллиты и частицы целевого продукта. Таким оптимальным сочетанием является изотермическая выдержка при 580-600°С в течение 4-5 часов. Увеличение температуры и продолжительности изотермической выдержки приведет к возрастанию скорости роста кристаллитов и частиц, а также к сокращению разнообразия химического состава синтезируемых фаз пирохлора, в то время как уменьшение температуры и продолжительности изотермической выдержки приведет к обнаружению аморфной фазы в целевом продукте.
Настоящее техническое решение позволяет обеспечить получение порошкообразного продукта (композиционного материала) заданной стехиометрии (Bi2O3)0.61(Fe2O3)0.22(WO3), а именно, смеси сложных оксидов: пирохлора состава (Bi2O3)0.52(Fe2O3)0.27(WO3) и фазы Ауривиллиуса Bi2WO6, при этом размерные параметры частиц и кристаллитов пирохлора обнаруживаются в разы меньшими, чем в случае получения продукта по способу-прототипу, а среди других преимуществ настоящего технического решения перед способом-прототипом следует отметить упрощение, удешевление и повышение экологичности технологии синтеза.
Настоящее изобретение поясняется иллюстрациями, где:
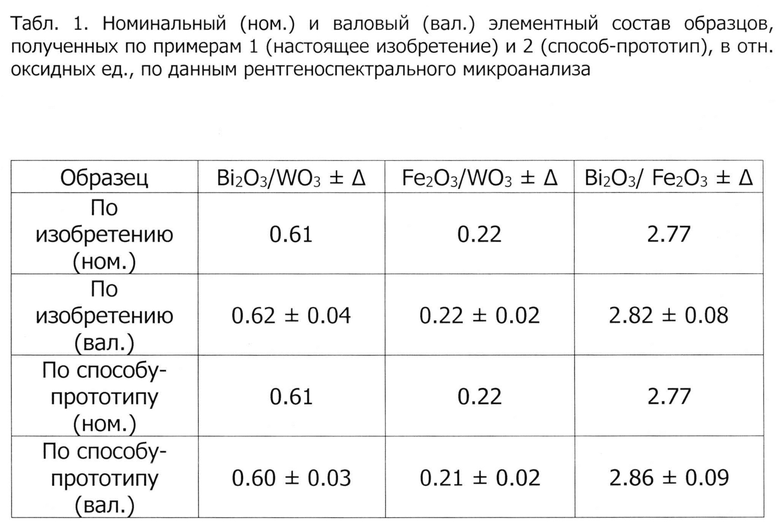
в табл.1 представлены номинальные и валовые (фактические) элементные составы образцов, полученных по примеру 1 (настоящее изобретение) и по примеру 2 (способ-прототип), в отн. оксидных ед., по данным рентгеноспектрального микроанализа;
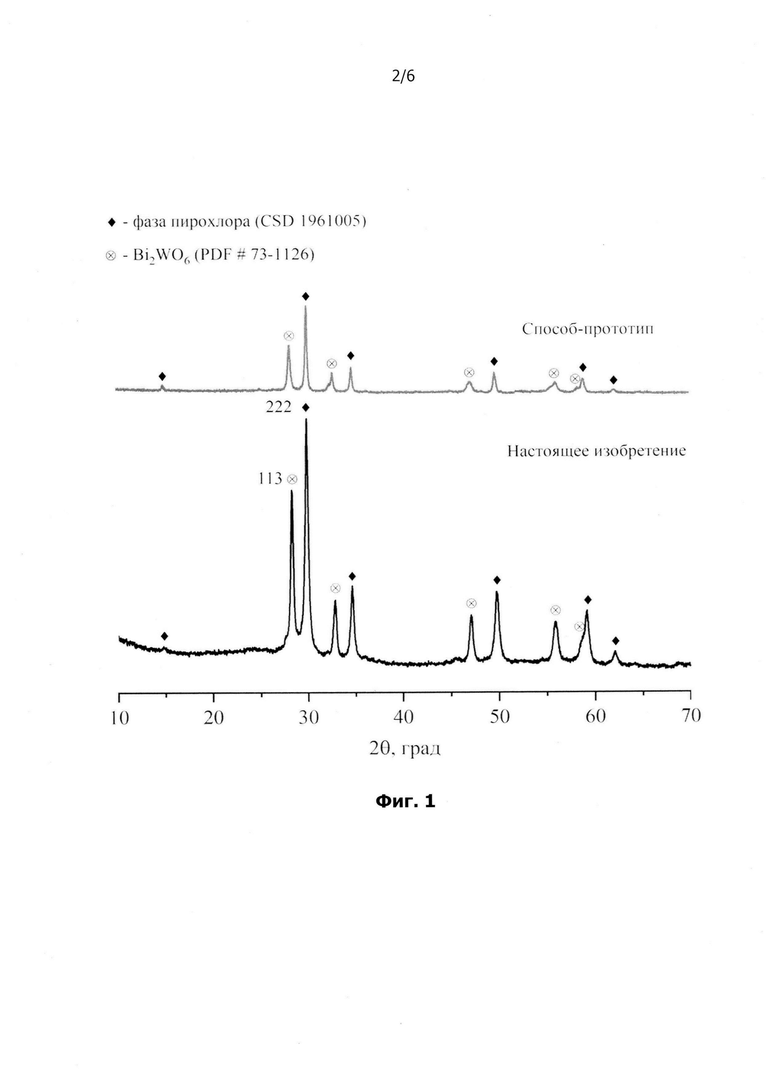
на фиг.1 представлены порошковые рентгеновские дифрактограммы (Cu-излучение) образцов, полученных по настоящему изобретению и по способу-прототипу;
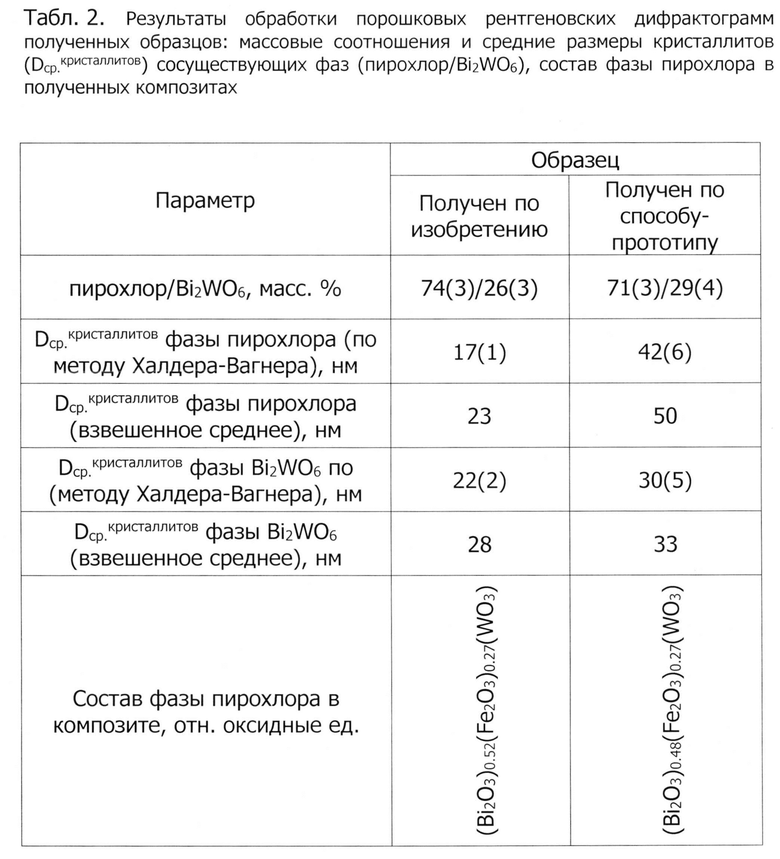
в табл.2 показаны результаты обработки порошковых рентгеновских дифрактограмм полученных образцов: массовые соотношения и средние размеры кристаллитов (Dcp.кристаллитов) сосуществующих фаз (пирохлор/Bi2WO6), состав фазы пирохлора в полученных композитах;
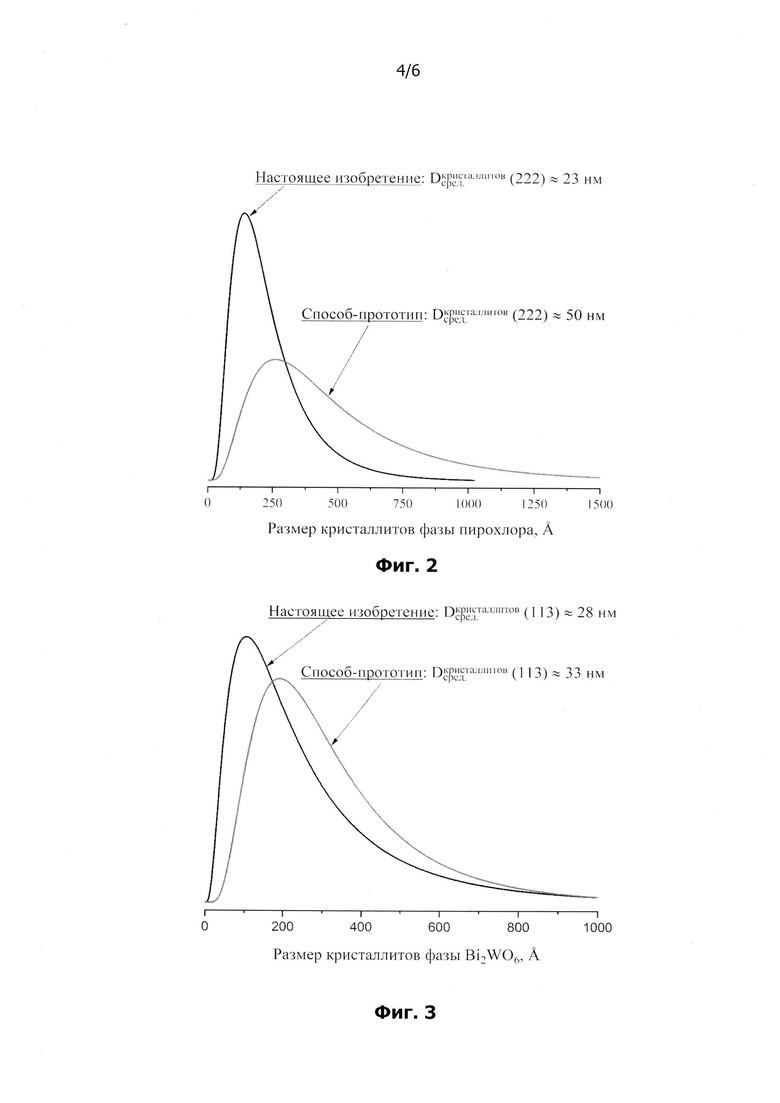
на фиг.2 представлены функции логнормального объемного распределения кристаллитов фазы пирохлора по размерам (по рефлексу 222) в образцах, полученных по настоящему изобретению и по способу-прототипу;
на фиг.3 представлены функции логнормального объемного распределения кристаллитов фазы Bi2WO6 по размерам (по рефлексу 113) в образцах, полученных по настоящему изобретению и по способу-прототипу;
на фиг.4 и на фиг.5 представлены микрофотографии разного увеличения образца, полученного по настоящему изобретению;
на фиг.6 представлены микрофотографии образца, полученного по способу-прототипу;
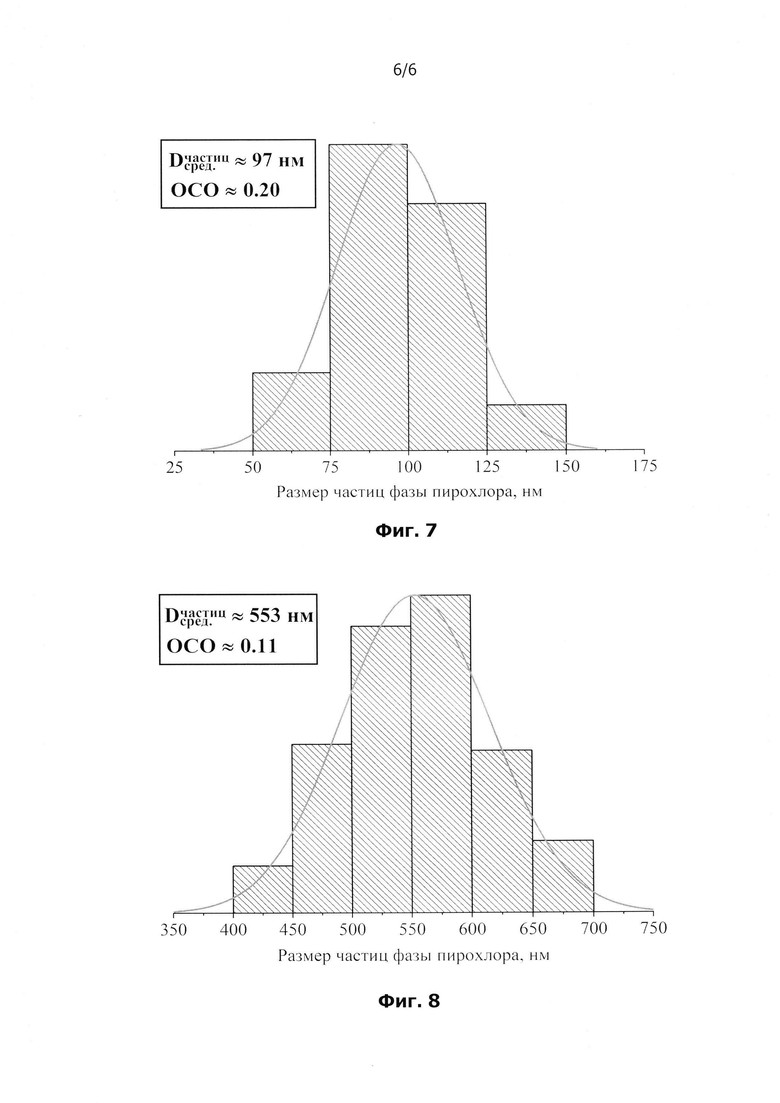
на фиг.7 и фиг.8 представлены гистограммы объемного распределения частиц фазы пирохлора по размерам и описывающие их функции нормального распределения в образцах, полученных по настоящему изобретению и по способу-прототипу (прим.: ОСО - относительное стандартное отклонение).
Настоящий способ получения наноразмерных и/или субмикронных порошков состава (Bi2O3)0.61(Fe2O3)0.22(WO3), представляющих собой смесь пирохлора состава (Bi2O3)0.52(Fe2O3)0.27(WO3) и фазы Ауривиллиуса Bi2WO6, осуществляют следующим образом.
Кристаллогидраты нитратов висмута и железа растворяют в азотной кислоте, а кристаллогидрат паравольфрамата аммония растворяют в дистиллированной воде. Водный раствор паравольфрамата аммония добавляют по каплям в кислый раствор нитратов висмута и железа при постоянном перемешивании последнего. Далее, полученная суспензия прекурсора переносится в стеклоуглеродный тигель и выпаривается на песчаной бане до полного удаления жидкости. Полученный сухой осадок (порошок прекурсора) переносят в керамический тигель и на первой стадии производят нагрев до температуры 280-300°С со скоростью 5-10°С/мин, затем выдерживают при этой температуре не менее 1 часа, а на второй стадии производят нагрев от 280-300°С до температуры 580-600°С со скоростью 10-15°С/мин, затем выдерживают при этой температуре в течение 4-5 часов, после чего происходит остывание порошкообразного продукта вместе с печью в естественном режиме.
Настоящее изобретение обеспечивает получение порошкообразного продукта (композиционного материала) заданной стехиометрии (Bi2O3)0.61(Fe2O3)0.22(WO3), а именно, смеси сложных оксидов: пирохлора состава (Bi2O3)0.52(Fe2O3)0.27(WO3) и фазы Ауривиллиуса Bi2WO6 с высокой степенью дисперсности, поскольку термообработка порошка прекурсора производится при достаточно низкой температуре (580-600°С), недостаточной для активации процессов спекания частиц целевого продукта с образованием крупных агрегатов. Это позволяет избежать стадии измельчения продукта термообработки, присущей всем высокотемпературным методам синтеза оксидных порошков.
Пример 1. Получение композиционного материала (пирохлор/Bi2WO6) в соответствии с настоящим изобретением осуществляли следующим образом. Соотношение исходных реагентов было подобрано таким образом, чтобы номинальный состав, заложенный по синтезу, соответствовал области двухфазного равновесия пирохлор/Bi2WO6 в системе Bi2O3-Fe2O3-WO3: 1.46 ммоль кристаллогидрата нитрата висмута (III) Bi(NO3)3⋅5H2O и 0.53 ммоль кристаллогидрата нитрата железа (III) Fe(NO3)3⋅9H2O было растворено в 20 мл 1М HNO3. Далее, 0.10 ммоль кристаллогидрата паравольфрамата (VI) аммония (NH4)10W12O41⋅5H2O (ос.ч.) растворяли в 60 мл дистиллированной воды (оставляли перемешиваться на магнитной мешалке в течение ~2 часов при нагреве до ~60°С до полного растворения соли) и полученный раствор по каплям добавляли (~1 капля/сек) в перемешиваемый магнитной мешалкой кислый раствор нитратов висмута и железа. После того, как весь раствор с вольфрамом был добавлен в раствор нитратов, образовавшаяся суспензия (~ 80 мл) дополнительно перемешивалась в течение ~1 часа. После перемешивания в течение этого времени суспензию переносили в стеклоуглеродные тигли (объемом 110 мл) и выпаривали на песчаной бане до сухого осадка (~3 часа). Следующим этапом синтеза являлась термообработка порошка прекурсора в керамическом тигле в муфельной печи на воздухе в две стадии. На первой стадии производили нагрев порошка прекурсора до температуры 280°С со скоростью 5°С/мин, затем образец выдерживали при этой температуре не менее 1 часа. По завершении изотермической выдержки на второй стадии производили нагрев от 280°С до температуры 580°С со скоростью 10°С/мин, затем образец выдерживали при этой температуре в течение 4 часов. После окончания изотермической выдержки программу нагрева печи завершают и производят остывание порошкообразного продукта вместе с печью в естественном режиме до комнатной температуры (~2 часа). Полученный таким образом продукт извлекается из керамического тигля и подвергается комплексному физико-химическому анализу. Результаты комплексного физико-химического анализа, представленные ниже, позволили определить химический и фазовый состав полученного композиционного материала, а также размерные параметры частиц и кристаллитов, которые оказались в разы меньшими, чем удается достичь при использовании известного способа.
Пример 2. Получение композиционного материала (пирохлор/Bi2WO6) в соответствии с настоящим изобретением осуществляли также, как в примере 1, но на первой стадии производили нагрев порошка прекурсора до температуры 300°С со скоростью 10°С/мин, а по завершении изотермической выдержки (1 час) на второй стадии производили нагрев от 300°С до температуры 600°С со скоростью 15°С/мин, затем образец выдерживали при этой температуре в течение 5 часов. Данные физико-химического анализа образца, полученного при таких условиях, показали, что его химический и фазовый состав, а также размерные параметры частиц и кристаллитов (данные не приводятся) в пределах погрешности не отличаются от таковых у образца, полученного по примеру 1. Таким образом, показано, что в пределах указанных диапазонов параметров термообработки обеспечена стабильность характеристик получаемого композиционного материала.
Пример 3. Получение порошка двухфазного композита (пирохлор/Bi2WO6) было также осуществлено с использованием известного способа-прототипа. Способ включал смешение раствора нитратов висмута и железа с раствором вольфрамата натрия с образованием суспензии, смешение суспензии с раствором щелочи для образования суспензии аморфного прекурсора, имеющей рН=2-2,4, гидротермальную обработку суспензии аморфного прекурсора, промывку полученного порошка и его сушку. Соотношение исходных реагентов было подобрано таким образом, чтобы номинальный состав, заложенный по синтезу, соответствовал области двухфазного равновесия пирохлор/Bi2WO6 в системе Bi2O3-Fe2O3-WO3, а приготовление растворов реагентов осуществляли следующим образом: 20.88 ммоль кристаллогидрата нитрата висмута (III), Bi(NO3)3⋅5H2O, и 7.54 ммоль кристаллогидрата нитрата железа (III), Fe(NO3)3⋅9H2O, было растворено в 300 мл 1М HNO3. Далее, 17.04 ммоль кристаллогидрата вольфрамата (VI) натрия, Na2WO4⋅2H2O, было растворено в 120 мл дистиллированной воды. Также был приготовлен раствор осадителя - 2М NaOH.
Смешение исходных растворов проводили в реакторе проточного смешения - микрореакторе с интенсивно закрученными потоками, гидротермальную обработку суспензии аморфного прекурсора проводили в проточном многоканальном автоклаве при температуре 210°С в течение 2 часов. Промывку полученного порошка и его отделение от маточного раствора осуществляли в электрофлотационной машине, а сушку полученного порошка вели комбинацией распыления суспензии и псевдоожиженного слоя при температуре 85°С. Подачу сушильного агента под решетку псевдоожиженного слоя производили в импульсном режиме. Полученный таким образом порошок был подвергнут анализу, результаты которого в сжатом виде излагаются ниже.
Сравнительный физико-химический анализ образцов, полученных по настоящему изобретению и по способу-прототипу показал следующее.
Результаты определения валового химического состава синтезированных образцов методом рентгеноспектрального микроанализа (далее - РСМД) представлены в табл.1. Полученные данные свидетельствуют о том, что валовые элементные составы образцов, полученных по изобретению и по способу-прототипу, в пределах погрешности соответствуют их номинальному составу, заложенному по синтезу. Однако, поскольку получение продукта по способу-прототипу осуществляется методом гидротермального синтеза, неизбежно наличие сливов, а именно, сначала сливается маточный раствор, а затем дистиллированной водой производится промывка кристаллического осадка от нитрата натрия, что способствует дополнительной нагрузке на окружающую среду, либо требует дополнительных энергетических и финансовых вложений на очистку этих сливов до приемлемого уровня экологической безопасности.
Таким образом, при получении композиционного материала по настоящему изобретению появляется возможность не только сохранить заданный по синтезу состав, но и сэкономить значительные объемы дистиллированной воды за счет отсутствия стадии промывки, а также избежать содержащих нитраты сливов.
Порошковые рентгеновские дифрактограммы образцов, полученных по настоящему изобретению и по способу-прототипу, представлены на фиг.1. Во всех образцах зафиксировано наличие двух кристаллических фаз: фазы со структурой пирохлора (CSD (Cambridge Structural Database) # 1961005) и фазы Ауривиллиуса (Bi2WO6, PDF # 73-1126). В результате обработки представленных дифрактограмм были получены массовые соотношения сосуществующих фаз, а также средние размеры и распределение по размерам кристаллитов сосуществующих фаз (табл.2, фиг.2-фиг.3). Установлено, что в образцах, полученных по настоящему изобретению и по способу-прототипу соотношение сосуществующих фаз (пирохлор/Bi2WO6) следующее: 74(3)/26(3) % и 71(3)/29(4) % соответственно по настоящему изобретению и по способу-прототипу. Отметим, что соотношение этих фаз в образцах определяется их валовым химическим составом, а также наличием/отсутствием аморфной фазы. Если говорить о валовом химическом составе, то, как было отмечено выше, в случае получения образцов по настоящему изобретению и по способу-прототипу он будет хорошо соответствовать заложенной по синтезу стехиометрии, то есть в обоих случаях имеется возможность заранее спрогнозировать, в каком массовом соотношении будут находиться сосуществующие в целевом продукте фазы, если известны данные о фазовых равновесиях в области синтезируемых составов и при условии, что система пришла в процессе синтеза к термодинамическому равновесию. Если говорить об аморфной фазе (это тот случай, когда система не пришла к состоянию термодинамического равновесия), то ранее было отмечено, что в продуктах гидротермального синтеза по способу-прототипу неизбежно наличие аморфной фазы, твердеющей при остывании гидротермального флюида, который содержит растворенные при повышенной температуре компоненты. Тем не менее, на представленных на фиг.1 рентгеновских дифрактограммах не удается надежно зафиксировать аморфное гало, которое бы свидетельствовало о наличии значительного количества аморфной фазы в каком-либо из полученных образцов. Отметим, что получать беспримесные двухфазные композиты (пирохлор/Bi2WO6) можно и с другими соотношением сосуществующих фаз, варьируя соотношение исходных реагентов в границах области составов, соответствующих такому двухфазному равновесию.
Средние размеры кристаллитов сосуществующих фаз были определены двумя методами (табл.2): методом Халдера-Вагнера и как взвешенное среднее функции логнормального объемного распределения кристаллитов по размерам по рефлексу 222 (для фазы пирохлора) и ИЗ (для фазы Bi2WO6). Средний размер кристаллитов фазы пирохлора в образце, полученном по настоящему изобретению, оказывается заметно меньше (~ 17(1) нм - метод Халдера-Вагнера и ~ 23 нм - взвешенное среднее), чем в образце, полученном по способу-прототипу (~ 42(6) нм - метод Халдера-Вагнера и ~ 50 нм - взвешенное среднее) в независимости от метода расчета. Аналогичная ситуация наблюдается в случае с фазой Bi2WO6, средний размер кристаллитов которой в образце, полученном по настоящему изобретению, составляет ~ 22(2) нм - метод Халдера-Вагнера и ~28 нм - взвешенное среднее, а в образце, полученном по способу-прототипу -~30(5) нм - метод Халдера-Вагнера и ~ 33 нм - взвешенное среднее. Функции логнормального объемного распределения кристаллитов фазы пирохлора по размерам по рефлексу 222 представлены на фиг.2, а фазы Bi2WO6 по рефлексу 113 на фиг.3. Средние размеры кристаллитов, определенные из представленных распределений (взвешенное среднее), обсуждались выше, поэтому обратим теперь внимание на уширение этих распределений. Если говорить о распределении по размерам кристаллитов фазы пирохлора, то в образце, полученном настоящему изобретению, наблюдается более узкое распределение (относительное стандартное отклонение меньше), чем в образце, полученном по способу-прототипу. Однако, в случае фазы Bi2WO6, для полученных образцов не наблюдается существенного различия в величинах относительных стандартных отклонений построенных распределений кристаллитов по размерам.
Таким образом, при получении композиционного материала по настоящему изобретению средние размеры кристаллитов сосуществующих фаз (как фазы пирохлора, так и фазы Bi2WO6) оказываются заметно меньшими, чем в образце, полученном по способу-прототипу.
На фиг.4-фиг.5 представлены микрофотографии образца, синтезированного по настоящему изобретению (с разным увеличением), полученные на сканирующем электронном микроскопе, а на фиг.6 представлена микрофотография образца, синтезированного по способу-прототипу. Во всех образцах наблюдается два морфологических мотива: условно сферические частицы фазы пирохлора и пластинчатые частицы фазы Bi2WO6, которые в случае образца, полученного по способу-прототипу, срастаются в более крупные агрегаты. Если говорить о частицах фазы пирохлора, то хорошо заметно, что их размер сильно зависит от способа получения образца. Для надежного определения в каждом полученном композите средних размеров частиц фазы пирохлора были построены гистограммы объемного распределения частиц по размерам, которые были описаны функциями нормального распределения (фиг.7-фиг.8). Средние размеры частиц фазы пирохлора, определенные из построенных распределений, составляют ~ 97 нм и 553 нм в образцах, полученных по изобретению (фиг.7) и по способу-прототипу (фиг.8) соответственно. Отметим, что для полученных образцов величины относительных стандартных отклонений, характерные для построенных распределений частиц по размерам, оказываются сопоставимы друг с другом. Если говорить о частицах фазы Bi2WO6, то в образце, полученном по изобретению, пластинчатые частицы этой фазы, толщиной ~250 нм, не срастаются в агрегаты, в то время как в образце, полученном по способу-прототипу, наблюдается заметное агрегирование пластинчатых частиц в цветок - подобные агрегаты, размер которых составляет ~5 мкм и более. Поскольку частицы фазы Bi2WO6 имеют заметную анизотропию формы, то определить их характерный размер и построить для них функции распределения по размерам затруднительно.
Таким образом, при получении композиционного материала настоящему изобретению средние размеры частиц фазы пирохлора оказываются в разы меньшими, чем в образце, полученном по способу-прототипу, причем то же самое можно сказать и про частицы фазы Bi2WO6, которые в случае получения композитов по настоящему изобретению, представляют собой отдельные пластинки, а в случае использования способа-прототипа, представляют собой крупные агрегаты размером ~5 мкм и более, составленные из сросшихся друг с другом пластинчатых частиц. Представленные результаты позволяют предполагать, что межфазное взаимодействие в композиционном материале, полученном в соответствии с настоящим изобретением, является более развитым, чем в случае его получения по способу-прототипу, за счет увеличения площади контакта фаз, что обусловлено уменьшением размеров частиц сосуществующих фаз.
Настоящее изобретение позволяет обеспечить получение нанокристаллического порошкообразного продукта (композиционного материала) заданной стехиометрии (Bi2O3)0.61(Fe2O3)0.22(WO3), а именно, смеси сложных оксидов: пирохлора состава (Bi2O3)0.52(Fe2O3)0.27(WO3) и фазы Ауривиллиуса Bi2WO6, при этом имеется возможность целенаправленно синтезировать смеси с определенным соотношением сосуществующих фаз, меняя заданную по синтезу стехиометрию, однозначно определяющую химический состав целевого продукта за счет того, что в процессе синтеза не происходит потерь реагирующих компонентов. Кроме того, настоящее изобретение позволяет обеспечить получение указанного композиционного материала со средними размерами кристаллитов сосуществующих фаз менее 30 нм, что превосходит аналогичные характеристики для случая использования способа-прототипа. Если же говорить о степени дисперсности композиционного материала, получаемого по способу, предлагаемому в настоящем изобретении, то она на порядок превышает этот показатель для случая использования способа-прототипа, что обеспечивает более развитое межфазное взаимодействие в таком композиционном материале и является существенным достоинством настоящего изобретения.
Таким образом, можно заключить, что способ, предложенный в настоящем изобретении, обеспечивает получение нанокристаллических частиц целевого продукта заданной стехиометрии и меньших размеров при упрощении, удешевлении и повышении экологичности технологии синтеза.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПОРОШКА СЛОЖНОГО ОКСИДА ВИСМУТА, ЖЕЛЕЗА И ВОЛЬФРАМА СО СТРУКТУРОЙ ФАЗЫ ПИРОХЛОРА С ИСПОЛЬЗОВАНИЕМ МИКРОРЕАКТОРА С ИНТЕНСИВНО ЗАКРУЧЕННЫМИ ПОТОКАМИ | 2022 |
|
RU2802703C1 |
| УСТРОЙСТВО ДЛЯ ГИДРОТЕРМАЛЬНОЙ ОБРАБОТКИ | 2023 |
|
RU2825758C1 |
| Микрореактор для синтеза наноразмерных частиц из растворов | 2021 |
|
RU2793562C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОРОШКОВ ФАЗ СЛОИСТЫХ ТИТАНАТОВ S- И P-ЭЛЕМЕНТОВ | 2011 |
|
RU2487849C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФОТОКАТАЛИЗАТОРА ДЛЯ РАЗЛОЖЕНИЯ ОРГАНИЧЕСКИХ ЗАГРЯЗНИТЕЛЕЙ | 2012 |
|
RU2478430C1 |
| Способ получения порошков фаз твёрдых растворов системы 0,75BiFeO-0,25Ba(ZrTi)O, легированных соединениями марганца | 2022 |
|
RU2787492C1 |
| Способ получения нанопорошков феррита висмута | 2019 |
|
RU2748446C2 |
| Получение керамики феррита висмута с высоким содержанием стехиометрического состава | 2023 |
|
RU2816609C1 |
| Способ получения нанопорошка феррита висмута | 2016 |
|
RU2641203C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОДНОФАЗНОГО НАНОПОРОШКА ФЕРРИТА ВИСМУТА | 2013 |
|
RU2556181C2 |

Изобретение относится к способам получения наноразмерных и/или субмикронных порошков сложного оксида висмута, железа и вольфрама, которые могут быть использованы в технологиях получения материалов для фотокаталитических процессов окисления вредных органических веществ, в фотокаталитических процессах генерации водорода для нужд водородной энергетики, а также в качестве материалов проводящих слоев в солнечных элементах. Композит состава (Bi2O3)0.61(Fe2O3)0.22(WO3), представляющий собой смесь пирохлора состава (Bi2O3)0.52(Fe2O3)0.27(WO3) и фазы Ауривиллиуса Bi2WO6, готовят из растворов нитрата висмута, нитрата железа и паравольфрамата аммония, взятых в стехиометрическом соотношении, путем их смешения с образованием суспензии. Полученную суспензию прекурсора выпаривают до полного испарения жидкости. Полученный порошок прекурсора нагревают до температуры 280-300°С со скоростью 5-10°С/мин и выдерживают при этой температуре не менее 1 часа, затем нагревают до температуры 580-600°С со скоростью 10-15°С/мин и выдерживают при этой температуре в течение 4-5 часов. Полученный порошок охлаждают. Изобретение позволяет упростить процесс получения нанокристаллических частиц заданной стехиометрии и меньших размеров порошкообразного продукта (Bi2O3)0.61(Fe2O3)0.22(WO3) и повысить экологичность технологии синтеза. 8 ил., 2 табл., 3 пр.
Способ получения порошка сложного оксида висмута, железа и вольфрама со структурой фазы пирохлора в составе двухфазного композита состава (Bi2O3)0.61(Fe2O3)0.22(WO3), представляющего собой смесь пирохлора состава (Bi2O3)0.52(Fe2O3)0.27(WO3) и фазы Ауривиллиуса Bi2WO6, включающий приготовление раствора нитратов висмута и железа, раствора вольфрамсодержащего реактива, взятых в стехиометрическом соотношении, обеспечивающем получение указанного выше композиционного материала, смешение раствора нитратов висмута и железа с раствором вольфрамсодержащего реактива с образованием суспензии прекурсора, термообработку полученного порошка прекурсора на воздухе, отличающийся тем, что в качестве вольфрамсодержащего реактива используют паравольфрамат аммония, полученную при смешении исходных растворов суспензию прекурсора выпаривают до полного испарения жидкости, полученный в результате выпаривания суспензии порошок прекурсора нагревают до температуры 280-300°С со скоростью 5-10°С/мин и выдерживают при этой температуре не менее 1 часа, затем нагревают до температуры 580-600°С со скоростью 10-15°С/мин и выдерживают при этой температуре в течение 4-5 часов, после чего остужают полученный порошок в естественном режиме.
| СПОСОБ ПОЛУЧЕНИЯ ПОРОШКА СЛОЖНОГО ОКСИДА ВИСМУТА, ЖЕЛЕЗА И ВОЛЬФРАМА СО СТРУКТУРОЙ ФАЗЫ ПИРОХЛОРА С ИСПОЛЬЗОВАНИЕМ МИКРОРЕАКТОРА С ИНТЕНСИВНО ЗАКРУЧЕННЫМИ ПОТОКАМИ | 2022 |
|
RU2802703C1 |
| CN 109529864 A, 29.03.2019 | |||
| JP S62230621 A, 09.10.1987 | |||
| CN 103086712 A, 08.05.2013 | |||
| LOMAKIN M.S | |||
| et al., Hydrothermal synthesis, phase formation and crystal chemistry of the pyrochlore/Bi2WO6 and pyrochlore/α-Fe2O3 composites in the Bi2O3-Fe2O3-WO3 system, Journal of Solid State Chemistry, 2020, vol | |||
| ПОРШНЕВОЙ ДВИГАТЕЛЬ | 1916 |
|
SU282A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| MA | |||

