ОБЛАСТЬ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к соединениям, композициям и способам лечения инфекции вирусом иммунодефицита человека (ВИЧ). Более конкретно, настоящее изобретение обеспечивает новые ингибиторы ВИЧ, фармацевтические композиции, содержащие такие соединения, и способы использования этих соединений для лечения ВИЧ-инфекции. Настоящее изобретение также относится к способам получения соединений, описанных ниже.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Синдром приобретенного иммунодефицита (СПИД) является результатом заражения ВИЧ. ВИЧ продолжает оставаться одной из основных проблем общественного здравоохранения в мире. В 2015 году, по оценкам, 36,7 миллиона человек жили с ВИЧ (включая 1,8 миллиона детей), а глобальный показатель распространенности ВИЧ составил 0,8%. Подавляющее большинство из этого числа проживает в странах с низким и средним уровнем доходов. В том же году 1,1 миллиона человек умерли от болезней, связанных со СПИДом.
Современная терапия для ВИЧ-инфицированных состоит из комбинации одобренных антиретровирусных средств. В настоящее время одобрено около четырех десятков лекарственных средств для лечения ВИЧ-инфекции в виде схем приема отдельных средств, комбинаций с фиксированными дозами или одной таблетки; последние два содержат 2-4 одобренных средства. Эти лекарственные средства принадлежат к ряду различных классов, нацеленных либо на вирусный фермент, либо на функцию вирусного белка во время цикла репликации вируса. Таким образом, лекарственные средства классифицируются как нуклеотидные ингибиторы обратной транскриптазы (NRTI), ненуклеотидные ингибиторы обратной транскриптазы (NNRTI), ингибиторы протеазы (PI), ингибиторы переноса цепи интегразы (INSTI) или ингибиторы проникновения в клетку (один, маравирок, нацелен на белок CCR5 хозяина, а другой, энфувиртид, представляет собой пептид, нацеленный на область gp41 вирусного белка gp160). Кроме того, фармакокинетический усилитель (кобицистат или ритонавир) можно использовать в комбинациях с антиретровирусными средствами (ARV), которые требуют усиления.
Несмотря на арсенал средств и комбинаций лекарственных средств, по-прежнему существует медицинская потребность в новых антиретровирусных средствах. Высокая вирусная гетерогенность, лекарственная токсичность, проблемы с переносимостью и плохая комплаентность - все это может привести к неэффективности лечения и может привести к селекции вирусов с мутациями, которые придают резистентность к одному или нескольким антиретровирусным средствам или даже нескольким лекарственным средствам из всего класса (Beyrer, C., Pozniak A. HIV drug resistance - an emerging threat to epidemic control. N. Engl. J. Med. 2017, 377, 1605-1607; Gupta, R. K., Gregson J., et al. HIV-1 drug resistance before initiation or re-initiation of first-line antiretroviral therapy in low-income and middle-income countries: a systematic review and meta-regression analysis. Lancet Infect. Dis. 2017, 18, 346-355; Zazzi, M., Hu, H., Prosperi, M. The global burden of HIV-1 drug resistance in the past 20 years. PeerJ. 2018, DOI 10,7717/peerj,4848). Как результат, необходимы новые лекарственные средства, которые легче принимать, которые имеют высокие генетические барьеры для развития резистентности и обладают большей безопасностью по сравнению с существующими препаратами. В этом наборе вариантов новые механизмы действия (MOA), которые могут использоваться как часть предпочтительной антиретровирусной терапии (ART) могут по-прежнему играть важную роль, поскольку они должны быть эффективными против вирусов, резистентных к существующим средствам. Усовершенствования, которые могут облегчить прием лекарственных средств в течение длительных периодов времени или даже в течение всей жизни, могут включать все или некоторые из следующего: уменьшение побочных эффектов, уменьшение взаимодействий между лекарственными средствами, увеличение продолжительности между вводимыми дозами или альтернативные пути введения, соответствующие индивидуальным предпочтениям пациента. Цели повышения безопасности, безусловно, должны включать высокие терапевтические индексы в отношении любых токсичностей, которые могут вызвать прекращение приема, а также могут включать уменьшение побочных эффектов или уменьшение межлекарственных взаимодействий. Возможность использования меньшего общего количества лекарственных средств в комбинированном режиме также, вероятно, приведет к улучшению комплаентности и безопасности. Повышенная эффективность против мишени противовирусного средства, особенно если ее поддерживать в присутствии человеческой плазмы и сывороточного альбумина, также может привести к снижению дозы и может прямо и положительно повлиять на продолжительность введения и терапевтический индекс по сравнению с побочными эффектами и токсичностями. Подводя итог, можно сказать, что можно было бы достичь максимальных преимуществ для ВИЧ-инфицированных пациентов, если бы были найдены лекарственные средства против ВИЧ с новыми механизмами действия, которые также обладают другими преимуществами, описанными выше, способствующими долговременной комплаентности и безопасности.
Некоторые потенциально терапевтические соединения были описаны в данной области техники и изложены в Blair, Wade S. et.al. Antimicrobial Agents and Chemotherapy (2009), 53(12), 5080-5087, Blair, Wade S. et al. PLoS Pathogens (2010), 6(12), e1001220, Thenin-Houssier, Suzie; Valente, Susana T. Current HIV Research, 2016, 14, 270-282, а также в патентных заявках РСТ со следующими номерами: WO 2012065062, WO 2013006738, WO 2013006792, WO 2014110296, WO 2014110297, WO 2014110298, WO 2014134566, WO 2015130964, WO2015130966, WO 2016033243, WO2018035359 и WO2018203235.
В настоящее время в данной области необходимы дополнительные соединения, которые являются новыми и полезными для лечения ВИЧ. Кроме того, эти соединения должны обеспечивать преимущества для фармацевтического применения, например, что касается одного или нескольких механизмов их действия, связывания, эффективности ингибирования, селективности в отношении мишени, растворимости, профилей безопасности, биодоступности и/или уменьшения частоты дозирования. Также необходимы новые композиции и способы лечения, в которых используются эти соединения.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Вкратце, в одном аспекте настоящее изобретение раскрывает соединения, описанные ниже
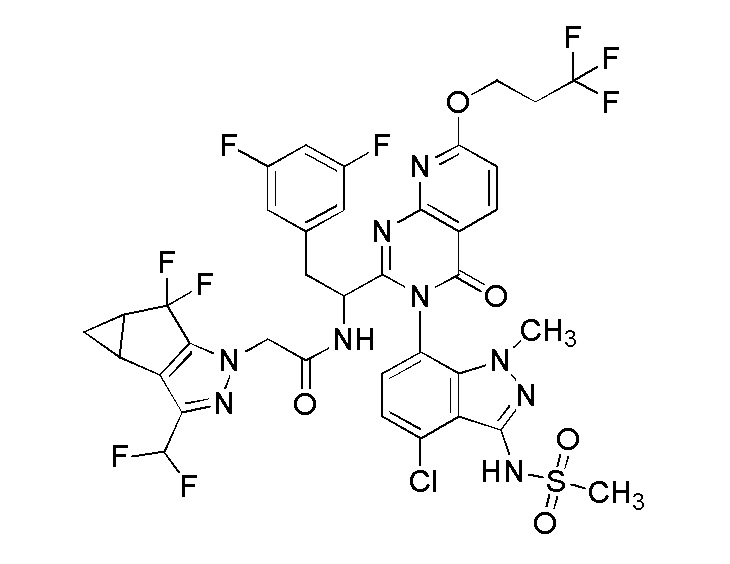
и их фармацевтически приемлемые соли (в дальнейшем “соединения и соли по изобретению”).
В другом аспекте настоящее изобретение раскрывает фармацевтическую композицию, включающую соединение или соль по изобретению.
В другом аспекте настоящее изобретение раскрывает способ лечения ВИЧ-инфекции у человека, включающий введение соединения или соли по изобретению.
В другом аспекте настоящее изобретение раскрывает соединение или соль по изобретению для применения в терапии.
В другом аспекте настоящее изобретение раскрывает соединение или соль по изобретению для применения в лечении ВИЧ-инфекции у человека.
В другом аспекте настоящее изобретение раскрывает применение соединения или соли по изобретению для получения лекарственного средства для лечения ВИЧ-инфекции у человека.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1 представляет профили средняя концентрация в плазме - время у крыс в исследовании, описанном ниже.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Предпочтительно соединения и соли по изобретению имеют стереохимию, показанную ниже
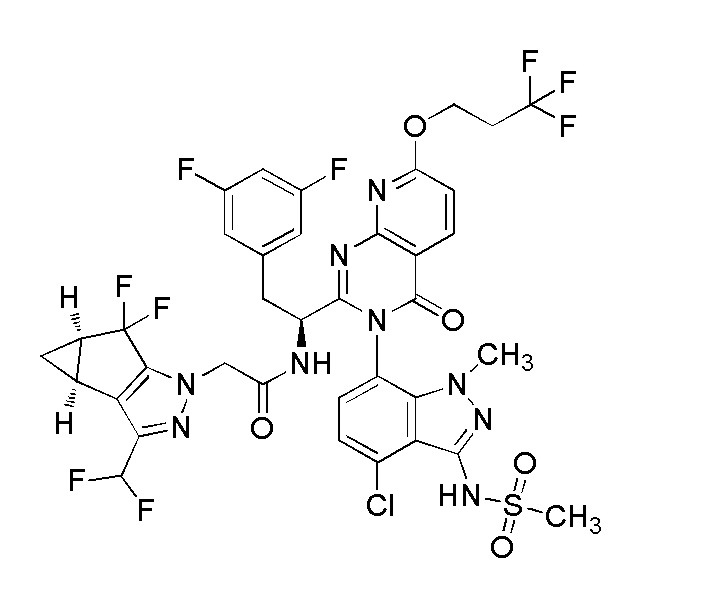 .
.
В другом аспекте соединения и соли по изобретению имеют стереохимию, показанную ниже
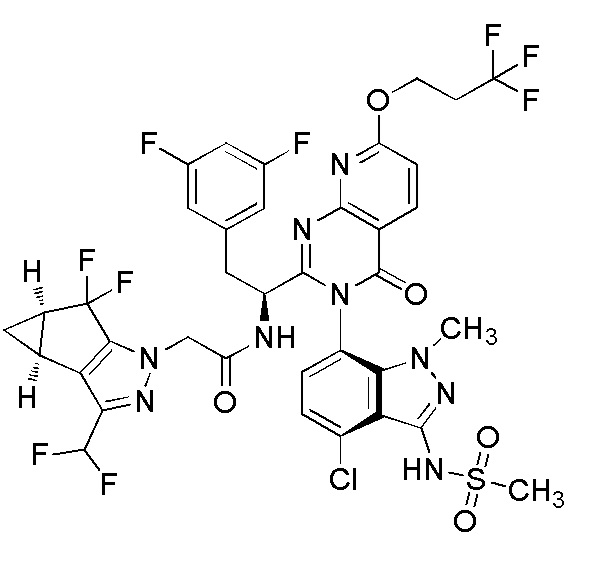 .
.
Соли по изобретению являются фармацевтически приемлемыми. Такие соли могут быть кислотно-аддитивными солями или основно-аддитивными солями. Обзор подходящих фармацевтически приемлемых солей см., например, в Berge et al, J. Pharm, Sci., 66, 1-19, 1977.
Типичные фармацевтически приемлемые кислотно-аддитивные соли включают, но не ограничиваются этим, 4-ацетамидобензоат, ацетат, адипат, альгинат, аскорбат, аспартат, бензолсульфонат (безилат), бензоат, бисульфат, битартрат, бутират, эдетат кальция, камфорат, камфорсульфонат (камзилат), капрат (деканоат), капроат (гексаноат), каприлат (октаноат), циннамат, цитрат, цикламат, диглюконат, 2,5-дигидроксибензоат, дисукцинат, додецилсульфат (эстолат), эдетат (этилендиаминтетраацетат), эстолат (лаурилсульфат), этан-1,2-дисульфонат (эдизилат), этансульфонат (эзилат), формиат, фумарат, галактарат (муцинат), гентизат (2,5-дигидроксибензоат), глюкогептонат (глюцептат), глюконат, глюкуронат, глутамат, глутарат, глицерофосфорат, гликолят, гексилрезорцинат, гиппурат, гидрабамин (N,N'-ди(дегидроабиетил)этилендиамин), гидробромид, гидрохлорид, гидроиодид, гидроксинафтоат, изобутират, лактат, лактобионат, лаурат, малат, малеат, малонат, манделат, метансульфонат (мезилат), метилсульфат, муцинат, нафталин-1,5-дисульфонат (нападисилат), нафталин-2-сульфонат (напсилат), никотинат, нитрат, олеат, пальмитат, п-аминобензолсульфонат, п-аминосалициклат, памоат (эмбонат), пантотенат, пектинат, персульфат, фенилацетат, фенилэтилбарбитурат, фосфат, полигалактуронат, пропионат, п-толуолсульфонат (тозилат), пироглутамат, пируват, салицилат, себацинат, стеарат, субацетат, сукцинат, сульфамат, сульфат, таннат, тартрат, теоклат (8-хлортеофиллинат), тиоцианат, триэтиодид, ундеканоат, ундециленат и валерат.
Типичные фармацевтически приемлемые основно-аддитивные соли включают, но не ограничиваются этим, соли алюминия, 2-амино-2- (гидроксиметил)-1,3-пропандиола (TRIS, трометамин), аргинина, бенетамина (N-бензилфенэтиламин), бензатина (N,N'-дибензилэтилендиамин), бис-(2-гидроксиэтил)амина, висмута, кальция, хлорпрокаина, холина, клемизола (1-п-хлорбензил-2-пирролидин-1'-илметилбензимидазол), циклогексиламина, дибензилэтилендиамина, диэтиламина, диэтилтриамина, диметиламина, диметилэтаноламина, дофамина, этаноламина, этилендиамина, L-гистидина, железа, изохинолина, лепидина, лития, лизина, магния, меглумина (N-метилглюкамин), пиперазина, пиперидина, калия, прокаина, хинина, хинолина, натрия, стронция, трет-бутиламина и цинка.
В одном варианте осуществления кислотно-аддитивные соли выбраны из гидрохлорида, гидробромида, гидроиодида, сульфата, бисульфата, нитрата, фосфата, гидрофосфата, ацетата, бензоата, сукцината, сахарата, фумарата, малеата, лактата, цитрата, тартрата, глюконата, камсилата, метансульфоната, этансульфоната, бензолсульфоната, п-толуолсульфоната и памоата. В одном варианте осуществления основно-аддитивные соли включают соли металлов (таких как натрий, калий, алюминий, кальций, магний и цинк) и соли аммония (такие как соли изопропиламина, диэтиламина, диэтаноламина). Другие соли (такие как трифторацетаты и оксалаты) можно использовать при изготовлении соединений и солей по настоящему изобретению, и они включены в объем изобретения.
Все возможные стехиометрические и нестехиометрические формы солей соединения по изобретению включены в объем изобретения. Кислотно- и основно-аддитивные соли могут быть получены квалифицированным химиком путем обработки соединения по изобретению соответствующей кислотой или основанием в подходящем растворителе с последующей кристаллизацией и фильтрацией.
Фармацевтический композиции по изобретению также включают фармацевтически приемлемый носитель, эксципиент и/или разбавитель. В одном варианте осуществления фармацевтические композиции по настоящему изобретению также включают фармацевтически приемлемый эксципиент.
В способе по настоящему изобретению предпочтительными путями введения являются пероральный и инъекционный для подкожной или внутримышечной доставки. Поэтому предпочтительные фармацевтические композиции включают композиции, подходящие для перорального введения (например, таблетки), и композиции, подходящие для инъекции, например, подкожной или внутримышечной инъекции.
В другом аспекте настоящее изобретение раскрывает способы предотвращения ВИЧ-инфекции у человека или снижения риска инфицирования, включающие введение соединения или соли по изобретению. Доконтактная профилактика (или PrEP) для людей из группы риска заражения ВИЧ предполагает ежедневный прием лекарственного средства, чтобы снизить шансы заразиться ВИЧ. Доказано, что PrEP снижает риск заражения. В контексте настоящей заявки “ВИЧ” или “вирус иммунодефицита человека” относится к ВИЧ-1 и/или к ВИЧ-2.
Считается, что соединения и соли по настоящему изобретению имеют своей биологической мишенью капсид ВИЧ, и, таким образом, их механизм действия заключается в изменении одним или несколькими способами функции капсида ВИЧ. Например, соединения и соли по настоящему изобретению могут действовать как ингибиторы капсида.
Соединения и соли по изобретению можно использовать отдельно или в комбинации с другими терапевтическими средствами. Комбинированная терапия в соответствии с настоящим изобретением, таким образом, включает введение по меньшей мере одного соединения или соли по изобретению и введение по меньшей мере одного другого средства, которое может быть полезным при лечении ВИЧ-инфекции. Соединения и соли по изобретению и любое другое фармацевтически активное средство(средства) можно вводить вместе или по отдельности, и если вводят по отдельности, введение может происходить одновременно или последовательно, в любом порядке. Например, соединение или соль по изобретению и другое средство можно сформулировать и вводить вместе в одной фармацевтической композиции, или их можно сформулировать и вводить по отдельности.
Количества соединения и солей по настоящему изобретению и другого фармацевтически активного средства(средств) и относительные интервалы между введениями будут выбраны для достижения желаемого комбинированного терапевтического эффекта. Введение в комбинации соединения по настоящему изобретению и его солей, сольватов или других фармацевтически приемлемых производных с другими лечебными средствами можно осуществлять в комбинации путем одновременного введения в: (1) объединенной фармацевтической композиции, включающей несколько соединений; или (2) отдельных фармацевтических композициях, каждая из которых включает одно из соединений. Альтернативно, комбинацию можно вводить раздельно последовательным образом, при этом одно лечебное средство вводят первым, а другое - вторым, или наоборот, и при необходимости разные средства можно вводить по разным схемам введения. Такое последовательное введение можно осуществлять через короткие промежутки времени или длительные промежутки времени. Количества соединения по изобретению или его солей и другого фармацевтически активного средства(средств) и относительные интервалы между введениями будут выбраны для достижения желаемого комбинированного терапевтического эффекта.
Таким образом, соединения и соли по настоящему изобретению можно использовать в комбинации с одним или несколькими средствами, полезными для профилактики или лечения ВИЧ. Такие средства включают, например, нуклеозидные ингибиторы обратной транскриптазы ВИЧ, ненуклеозидные ингибиторы обратной транскриптазы ВИЧ, ингибиторы протеазы ВИЧ, ингибиторы слияния ВИЧ, ингибиторы прикрепления ВИЧ, ингибиторы CCR5, ингибиторы CXCR4, ингибиторы почкования или созревания ВИЧ и ингибиторы интегразы ВИЧ. Подходящие другие средства включают, например, абакавир, атазанавир, биктегравир, каботегравир, дарунавир, делавирдин, диданозин, дидезоксиинозин, долутегравир, доравирин, эфавиренц, элвитегравир, эмтрицитабин, этавирин, фосампренавир, фостемсавир, GSK3640254, индинавир, слатравир, ламивудин, лопинавир, маравирок, нелфинавир, невирапин, ралтегравир, рилпиверин, ритонавир, саквинавир, слатравир, ставудин, типранавир, тенофовир, тенофовир алафенамид, тенофовир дизопроксил фумарат, залцитабин, зидовудин, антитела N6LS, GSK3739937/VH3739937 и S-648414. Дополнительные подходящие другие средства включают Долутегравир, ламивудин, Фостемсавир, Каботегравир, маравирок, рилпиверин, Реятаз, Тенофовир, Афенамид, EfDA, Доравирин и Презиата. Другие подходящие средства включают Долутегравир, ламивудин, Фостемсавир и Каботегравир. Предпочтительные средства включают, например, биктегравир, каботегравир, долутегравир, фостемсавир, ислатравир и ламивудин. Особенно предпочтительные средства включают, например, биктегравир, каботегравир, долутегравир, фостемсавир и ламивудин.
ПРИМЕРЫ
Получение бицикло[3.1.0]гексан-3-ола
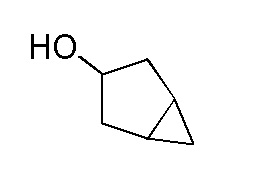
К перемешиваемому раствору циклопент-3-енола (130 г, 1545 ммоль) в DCM (1200 мл) в атмосфере N2 при 0-5°C добавляли по каплям раствор диэтилцинка в гексане (1,0 M, 3091 мл, 3091 ммоль) в течение 3 часов. К раствору при 0°C добавляли по каплям раствор дийодметана (249 мл, 3091 ммоль) в DCM (300 мл) в течение 1 часа. Реакционной смеси давали нагреться до 27°C, после чего наблюдали образование белого осадка. Смесь перемешивали в течение 16 часов. За ходом реакции наблюдали при помощи ТСХ (SiO2, 20% EtOAc/петрол. (петролейный эфир), Rf=0,3, УФ-неактивный, PMA-активный). Реакционную смесь гасили осторожным добавлением водн. насыщенного раствора NH4Cl (1,5 л). Смесь фильтровали через слой целита. Водный слой экстрагировали при помощи DCM (2×1л). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и затем концентрировали при пониженном давлении с получением неочищенного бицикло[3.1.0]гексан-3-ола в виде красной жидкости, 180 г. 1H ЯМР (400 МГц, CDCl3) δ=4,41-4,35 (м, 1H), 2,18-2,05 (м, 2H), 1,73 (д, J=13,9 Гц, 2H), 1,35-1,25 (м, 2H), 1,21-1,14 (м, 1H), 0,57-0,43 (м, 2H). ГХМС: m/z=98,1).
Получение бицикло[3.1.0]гексан-3-она
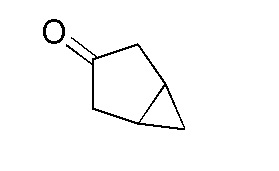
К перемешиваемому раствору бицикло[3.1.0]гексан-3-ола (210 г, 2054 ммоль) в DCM (5000 мл) в атмосфере N2 при 0°C добавляли порциями периодинан Десса-Мартина (954 г, 225 ммоль). Смеси давали нагреться до 27°C и затем перемешивали в течение 16 часов. За ходом реакции наблюдали при помощи ТСХ (SiO2, 20% Ацетон/Гекс., Rf=0,3, УФ-неактивный, PMA-активный). Реакционную смесь фильтровали через слой целита и фильтрат промывали водн. NaOH (1N, 8× 1 л). Объединенные водные фазы экстрагировали при помощи DCM (5×1 л). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и затем концентрировали при пониженном давлении (температура бани: 20°C) с получением неочищенного бицикло[3.1.0]гексан-3-она в виде коричневой жидкости. Жидкость дополнительно очищали нисходящей дистилляцией при 70°C с получением бицикло[3.1.0]гексан-3-она в виде бледно-желтой вязкой жидкости, 125 г (62%). 1H ЯМР (400 МГц, CDCl3) δ=2,61-2,54 (м, 2H), 2,17-2,12 (м, 2H), 1,54-1,46 (м, 2H), 0,92-0,86 (м, 1H), -0,01 -0,08 (м, 1H); ГХМС: M/Z=96,1.
Получение 2-(2,2-дифторацетил)бицикло[3.1.0]гексан-3-она
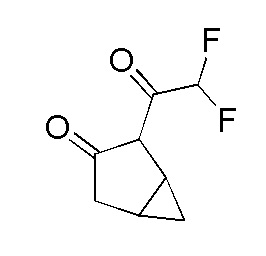
К перемешиваемому раствору бицикло[3.1.0]гексан-3-она (125 г, 1274 ммоль) в THF (1500 мл) в атмосфере N2 при -78°C добавляли LDA (2,0 M в THF, 0,701 л, 1402 ммоль). Раствор перемешивали в течение 1 часа при -78°C. К раствору добавляли медленно в течение 30 минут раствор этилдифторацетата (174 г, 1402 ммоль) в THF (300 мл), поддерживая температуру -78°C. Реакционной смеси давали нагреться до 27°C и затем перемешивали в течение 1 часа. За ходом реакции наблюдали при помощи ТСХ (SiO2, 20% Ацетон/Гексан, Rf=0,3, УФ-активный). Реакционную смесь гасили добавлением водн. HCl (1N, 2000 мл). Смесь перемешивали в течение 30 мин и затем экстрагировали при помощи EtOAc (3×1000 мл). Объединенные органические слои промывали насыщенным солевым раствором (1000 мл), сушили над безводным Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении с получением 2-(2,2-дифторацетил)бицикло[3.1.0]гексан-3-она в виде бледно-желтой вязкой жидкости, 180 г (71%). 1H ЯМР (400 МГц, CDCl3) δ=6,18 (т, J=54,8 Гц, 1H), 2,70-2,62 (м, 1H), 2,35 (д, J=19,4 Гц, 1H), 2,14 (шир. с, 1H), 1,26-1,21 (м, 1H), 1,04-1,03 (м, 1H), 0,22-0,21 (м, 1H), ЖХМС: M/Z=173,17).
Получение этил 2-(3-(дифторметил)-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетата.
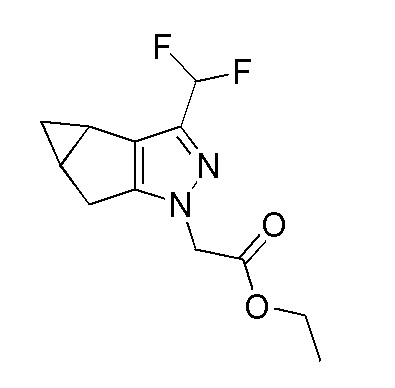
К перемешиваемому раствору 2-(2,2-дифторацетил)бицикло[3.1.0]гексан-3-она (180 г, 910 ммоль) в этаноле (2 л) в атмосфере N2 при 27°C добавляли этил 2-гидразинилацетат гидрохлорид (422 г, 2729 ммоль), затем серную кислоту (20 мл, 375 ммоль). Смесь перемешивали в течение 30 мин и затем нагревали до 100°C и перемешивали в течение 16 часов. За ходом реакции наблюдали при помощи ТСХ (SiO2, 20% Ацетон/Гексан, Rf=0,3, УФ-активный). Реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в EtOAc (2000 мл) и промывали водой (2×1 л), насыщенным солевым раствором (1,0 л), сушили над безводным Na2SO4, фильтровали и затем концентрировали при пониженном давлении. Полученный остаток подвергали колоночной хроматографии на силикагеле (петрол.:ацетон 100:0→98:2) с получением этил 2-(3-(дифторметил)-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетата в виде не совсем белого твердого вещества, 110 г (46%). 1H ЯМР (400 МГц, DMSO-d6) δ=6,86 (т, J=54,8 Гц, 1H), 4,93 (с, 2H), 4,14 (кв., J=7,2 Гц, 2H), 2,88-2,79 (м, 1H), 2,76-2,68 (м, 1H), 2,14-2,04 (м, 2H), 1,19 (т, J=7,2 Гц, 3H), 1,10-1,03 (м, 1H), 0,14 (кв., J=4,3 Гц, 1H).
Получение этил 2-(3-(дифторметил)-5-оксо-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетата.

К перемешиваемому раствору этил 2-(3-(дифторметил)-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетата (110 г, 422 ммоль) и Целита (395 г) в циклогексане (3,5 л) при 0°C порциями добавляли дихромат пиридиния (794 г, 2110 ммоль). К смеси в атмосфере азота добавляли по каплям трет-бутил гидропероксид (355 мл, 2130 ммоль) в течение 10 мин. Реакционную смесь нагревали до 27°C и затем перемешивали при этой температуре в течение 48 часов. За ходом реакции наблюдали при помощи ТСХ (SiO2, 30% Ацетон/петрол., Rf=0,4, УФ-активный). Реакционную смесь фильтровали и фильтровальную лепешку экстрагировали при помощи EtOAc (1000 мл). Фильтрат промывали насыщенным водн. Na2S2O3 (2×500 мл); насыщенным водн. FeSO4 (300 мл); и затем насыщенным солевым раствором (500 мл). Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (150 г).
Получение этил 2-(3-(дифторметил)-4,4a-дигидроспиро[циклопропа[3,4]циклопента[1,2-c]пиразол-5,2'-[1,3]дитиолан]-1(3bH)-ил)ацетата.
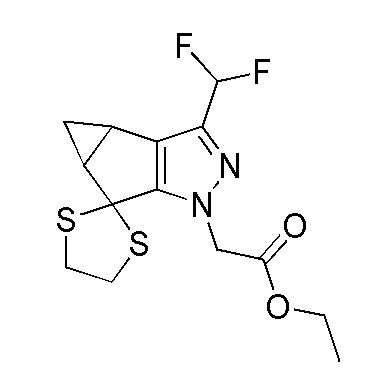
К перемешиваемому раствору этил 2-(3-(дифторметил)-5-оксо-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетата (75 г, 269 ммоль) в DCM (1500 мл) при 27°C в атмосфере азота добавляли этан-1,2-дитиол (43,0 мл, 511 ммоль) с последующим добавлением трифторида бора-уксусной кислоты (72,6 мл, 511 ммоль). Раствор перемешивали в течение 16 часов. За ходом реакции наблюдали при помощи ТСХ (SiO2, 20% Ацетон/Петрол., Rf=0,35, УФ-активный). После завершения реакционную смесь охлаждали до 0°C и гасили добавлением водн. насыщенного раствора NaHCO3 (500 мл). Смесь экстрагировали при помощи DCM (2×1000 мл). Объединенные органические слои промывали насыщенным солевым раствором (1000 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением коричневой жидкости. Это вещество подвергали колоночной хроматографии на силикагеле (Петрол.:EtOAc 95:5→90:10) с получением этил 2-(3-(дифторметил)-4,4a-дигидроспиро[циклопропа[3,4]циклопента [1,2-c]пиразол-5,2'-[1,3]дитиолан]-1(3bH)-ил)ацетата в виде не совсем белого твердого вещества, 80 г (74%). 1H-ЯМР (400 МГц, CDCl3) δ=6,61 (т, J=55,2 Гц, 1H), 5,00-4,85 (м, 2H), 4,29-4,19 (м, 2H), 3,55-3,46 (м, 4H), 2,63-2,53 (м, 1H), 2,49-2,38 (м, 1H), 1,30-1,24 (м, 4H), 0,65-0,60 (м, 1H). ЖХМС M+H=346,9.
Получение этил 2-(3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетата
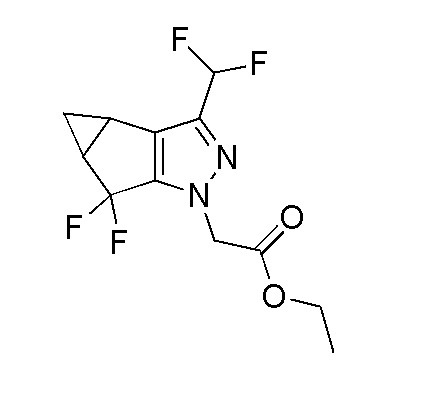
К перемешиваемому раствору 1,3-дибром-5,5-диметилимидазолидин-2,4-диона (26,3 г, 92 ммоль) в DCM (20 мл) при -70°C в атмосфере N2 добавляли HF-пиридин (2,460 г, 24,83 ммоль). Раствор перемешивали в течение 30 мин. К раствору добавляли раствор этил 2-(3-(дифторметил)-4,4a-дигидроспиро[циклопропа[3,4]циклопента[1,2-c]пиразол-5,2'-1,3]дитиолан]-1(3bH)-ил)ацетата (10 г, 25 ммоль) в DCM (20 мл). Реакционной смеси давали нагреться до -40°C и затем перемешивали при этой температуре в течение 1 часа. За ходом реакции наблюдали при помощи ТСХ (SiO2, 30% EtOAc/Петрол., Rf=0,3, УФ-неактивный). Реакционную смесь гасили добавлением водн. насыщ. NaHCO3 (200 мл). Смесь нагревали до комнатной температуры и затем экстрагировали при помощи EtOAc (2×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (50 мл); сушили над безводным Na2SO4; фильтровали; и концентрировали при пониженном давлении с получением коричневого твердого вещества. Это вещество подвергали колоночной хроматографии на силикагеле (Петрол.:EtOAc 100:0→75-25) с получением этил 2-(3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетата в виде бледно-желтого твердого вещества, 8,5 г (91%). 1H ЯМР (400 МГц, CDCl3) δ=6,62 (т, J=55,2 Гц, 1H), 4,82 (с, 2H), 4,30-4,18 (м, 2H), 2,51-2,37 (м, 2H), 1,42-1,35 (м, 1H), 1,31-1,23 (м, 3H), 1,14-1,08 (м, 1H). ЖХМС M+H=293,07.
Получение 2-(3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)уксусной кислоты
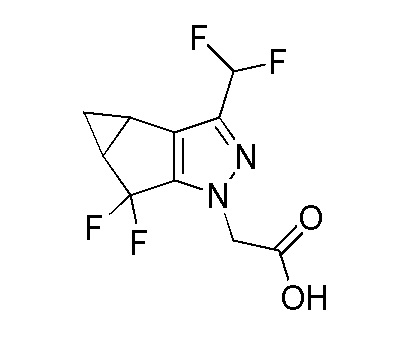
К перемешиваемому раствору этил 2-(3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетата (15 г, 50 ммоль) в THF (17 мл) и MeOH (66 мл) при 0°C в атмосфере N2 добавляли раствор LiOH (1,788 г, 74,7 ммоль) в воде (66 мл). Реакционной смеси давали нагреться до 27°C и затем перемешивали в течение 3 часов при этой температуре. За ходом реакции наблюдали при помощи ТСХ (SiO2, 5% MeOH/DCM, Rf=0,2, УФ Активный). После завершения реакционную смесь концентрировали при пониженном давлении; разбавляли водой (50 мл); и промывали при помощи EtOAc (2×250 мл) для удаления примесей. Водный слой доводили до pH 2-3 с использованием водн. HCl (1M), затем экстрагировали при помощи EtOAc (3×1000 мл). Объединенные органические слои сушили над безводным Na2SO4; фильтровали; и концентрировали при пониженном давлении с получением 2-(3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)уксусной кислоты в виде не совсем белого твердого вещества, 14 г (98%). ЖХМС M+H=265,15.
Разделение с получением 2-((3bS,4aR)-3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)уксусной кислоты и 2-((3bR,4aS)-3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)уксусной кислоты
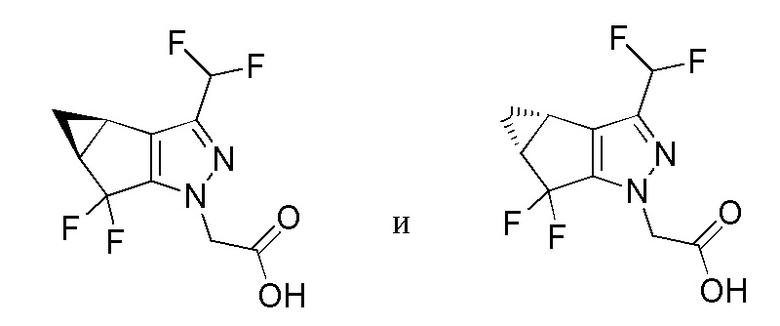
2-(3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)уксусную кислоту (5,5 г) растворяли в изопропаноле (20 мл). Раствор порциями подвергали хиральному разделению СФХ следующим образом: устройство=Thar 80; колонка=Chiralpak IC 30×250мм, 5 микрон; растворитель A=сверхкритический CO2; растворитель B=изопропанол с 0,5% изопропиламина (об/об); композиция элюента=70%A:30%B; скорость потока=65 г/мин; обратное давление=100 бар; температура=30°C; объем вводимой пробы=2,5 мл; детекция=220 нм. 2-((3bS,4aR)-3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)уксусную кислоту собирали в виде пика, элюирующего от 7,5 мин до 14 мин; 2-((3bR,4aS)-3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)уксусную кислоту собирали в виде пика, элюирующего от 2,7 мин до 5,8 мин. Для каждого энантиомера полученный раствор концентрировали при пониженном давлении и полученные твердые вещества растворяли в EtOAc, затем дважды промывали водн. раствором лимонной кислоты (1M), затем водой, затем насыщенным солевым раствором. Органический раствор сушили над Na2SO4; фильтровали; затем концентрировали в вакууме с получением разделенного энантиомера с извлечением 80-90%.
Получение 3-бром-6-хлор-2-фторбензонитрила
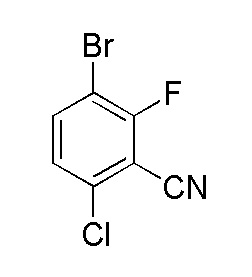
К перемешиваемому раствору 3-бром-6-хлор-2-фторбензальдегида (210,0 г, 0,89 моль, 1,0 экв.) в воде (2,1 л) при комнатной температуре добавляли гидроксиламин-O-сульфоновую кислоту (175,15 г, 1,55 моль, 1,75 экв.). Реакционную смесь нагревали до 50°C и перемешивали в течение 18 часов). Смесь охлаждали до комнатной температуры и перемешивали в течение 1-1,5 часов. Твердые вещества выделяли фильтрованием и затем промывали водой. Влажное твердое вещество сушили под вакуумом при 50°C в течение 12-15 часов с получением 3-бром-6-хлор-2-фторбензальдегида, 190,0 г (91%).
Получение 7-бром-4-хлор-1-метил-1H-индазол-3-амина
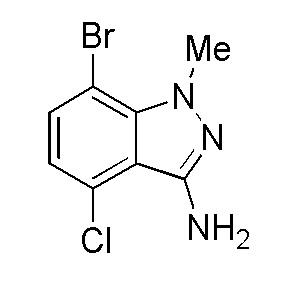
К раствору 3-бром-6-хлор-2-фторбензонитрила (360,0 г, 1,55 моль, 1,0 экв.) в этаноле (1,08 л) добавляли метилгидразинсульфат (1,11 кг, 7,73 моль, 5,0 экв.) с последующим добавлением триэтиламина (1,3 л, 9,3 моль, 6,0 экв.) при 25-35°C. Реакционную смесь нагревали до 110°C и выдерживали 15 часов (за ходом реакции наблюдали при помощи ТСХ). После завершения реакции смесь охлаждали до комнатной температуры. Добавляли воду (3,0 л) и смесь перемешивали в течение 1 часа при комнатной температуре. Твердые вещества выделяли фильтрованием и промывали водой. Влажное твердое вещество сушили под вакуумом при 50°C в течение 12-15 часов. Неочищенное твердое вещество очищали колоночной хроматографией (10% EA/гексан - 40% EA/Гексан) с получением продукта в виде бледно-желтого твердого вещества. Выход: 185,0 г (46,0%).
Получение N-(7-бром-4-хлор-1-метил-1H-индазол-3-ил)метансульфонамида
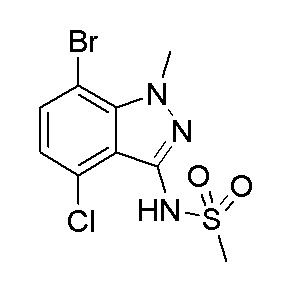
К раствору 7-бром-4-хлор-1-метил-1H-индазол-3-амина (1,40 г, 5,37 ммоль) в DCM (30 мл) добавляли основание Хунига (3,75 мл, 21,5 ммоль) и затем реакционную смесь охлаждали на ледяной бане и добавляли метансульфонилхлорид (1,26 мл, 16,1 ммоль). Реакционную смесь перемешивали при этой температуре в течение 1 часа (образование осадка). Смесь затем разбавляли дихлорметаном (100 мл) и промывали водой, 1 M HCl и насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток поглощали в EtOH (30 мл) и 10 мл 20% водн. NaOH. Полученную смесь нагревали с помощью теплового пистолета до тех пор, пока она не превратилась в гомогенный раствор, и перемешивали при комнатной температуре в течение 30 минут. Смесь разбавляли водой (80 мл) и подкисляли 1 N раствором HCl (60 мл). Осадок фильтровали, промывали водой и сушили в вакууме с получением указанного в заголовке продукта (1,5 г) в виде не совсем белого твердого вещества. 1H ЯМР (500 МГц, CDCl3) δ 7,48 (д, J=7,9 Гц, 1H), 7,24 (шир. с, 1H), 6,95 (д, J=7,9 Гц, 1H), 4,38 (с, 3H), 3,42 (с, 3H). ЖХ/МС (M+H)+=337,80.
Получение N-(7-бром-4-хлор-1-метил-1H-индазол-3-ил)-N-(4-метоксибензил)метансульфонамида
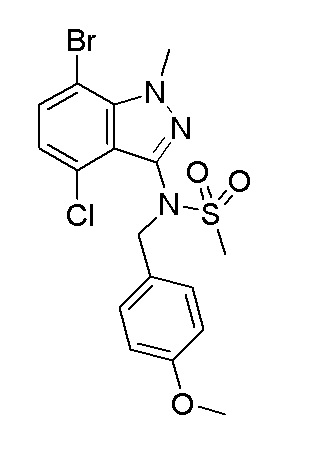
К смеси N-(7-бром-4-хлор-1-метил-1H-индазол-3-ил)метансульфонамида (1,3 г, 3,84 ммоль) и 1-(хлорметил)-4-метоксибензола (0,625 мл, 4,61 ммоль) в DMF (30 мл) добавляли карбонат цезия (1,626 г, 4,99 ммоль) и смесь нагревали при 80°C в течение 2 часов. Смесь выливали в воду (100 мл) и экстрагировали при помощи EtOAc (50 мл, 2×). Объединенный органический слой промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали при помощи Bioateg (0~35% EtOAc-гексан) с получением указанного в заголовке продукта (1,5 г) в виде белого пенистого вещества. 1H ЯМР (500 МГц, CDCl3) δ 7,44 (д, J=7,9 Гц, 1H), 7,31 (д, J=8,5 Гц, 2H), 6,99 (д, J=7,9 Гц, 1H), 6,84 (д, J=8,5 Гц, 2H), 4,99 (шир. с, 1H), 4,76 (шир. с, 1H), 4,40 (с, 3H), 3,80 (с, 3H), 3,01 (с, 3H).
Получение N-(7-амино-4-хлор-1-метил-1H-индазол-3-ил)-N-(4-метоксибензил)метансульфонамида
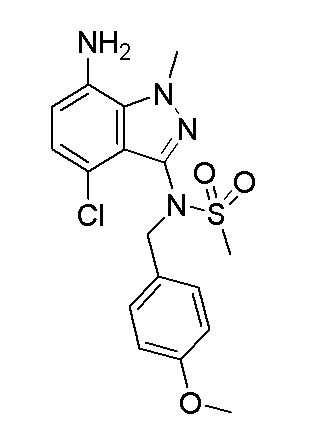
В соответствии с: Andersen, Jacob et al, Synlett 2005 (14), 2209-2213. К смеси N-(7-бром-4-хлор-1-метил-1H-индазол-3-ил)-N-(4-метоксибензил)метансульфонамида (600,0 мг, 1,308 ммоль), иодида меди (I) (49,8 мг, 0,262 ммоль), аскорбата натрия (518 мг, 2,62 ммоль) и (1R,2R)-N1,N2-диметилциклогексан-1,2-диамин (46,5 мг, 0,327 ммоль) в NMP (10 мл) добавляли раствор азида натрия (255 мг, 3,92 ммоль) в воде (2,0 мл). Затем смесь герметично закрывали и нагревали в микроволновой системе при 120°C в течение 2,5 часов. Смесь затем фильтровали через слой целита и слой промывали при помощи EtOAc. Фильтрат выливали в воду (100 мл) и экстрагировали при помощи EtOAc (50 мл, 2×). Объединенный органический слой промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и упаривали в вакууме. Остаток очищали при помощи Bioatge (5-100% EtOAc/гексан) с получением указанного в заголовке продукта (400 мг) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,33-7,29 (м, 2H), 6,89 (д, J=7,8 Гц, 1H), 6,85-6,79 (м, 2H), 6,48 (д, J=7,8 Гц, 1H), 5,11 (шир.с, 1H), 4,81 (шир.с, 1H), 4,30 (с, 3H), 3,80 (шир. с, 2H), 3,79 (с, 3H), 2,99 (с, 3H). ЖХ/МС (M+H)+=395,00.
Получение 2-амино-6-(бензилокси)никотиновой кислоты
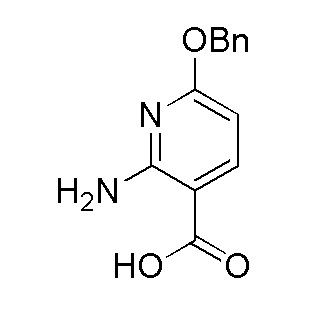
Раствор 2-амино-6-хлорникотиновой кислоты (5 г, 29 ммоль) и трет-бутоксида калия (9,75 г, 87 ммоль) в бензиловом спирте (97 мл) нагревали до 120°C в течение 3 часов. После охлаждения до температуры окружающей среды очень темную реакционную смесь добавляли к воде и промывали эфиром (×3). Водный слой затем подкисляли 0,5 М лимонной кислотой. Желто-коричневый осадок фильтровали с получением продукта (4,4 г, 62%), который использовали в следующей реакции без дополнительной очистки. 1H ЯМР (500 МГц, DMSO-d6) δ 12,40 (шир. с, 1H), 7,94 (д, J=8,55 Гц, 1H), 7,06-7,52 (м, 5H), 6,04 (д, J=8,24 Гц, 1H), 5,33 (с, 2H). ЖХ/МС: m/z=245,15 [M+1]+.
Получение N-[(6P)-7-{2-[(1S)-1-амино-2-(3,5-дифторфенил)этил]-7-гидрокси-4-оксо-3H,4H-пиридо[2,3-d]пиримидин-3-ил}-4-хлор-1-метил-1H-индазол-3-ил]-N-[(4-метоксифенил)метил]метансульфонамида
Схема:
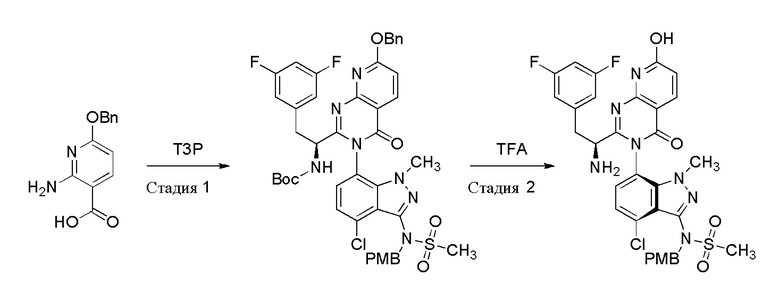
Стадия 1:
К суспензии (S)-2-((трет-бутоксикарбонил)амино)-3-(3,5-дифторфенил)пропановой кислоты (5,49 г, 18,23 ммоль) и 2-амино-6-(бензилокси)никотиновой кислоты (4,45 г, 18,23 ммоль) в ацетонитриле (92 мл) (желтый раствор) при -25°C добавляли пиридин (9,83 мл, 122 ммоль), затем 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (“T3P”, 45,2 мл, 76 ммоль). Реакционную смесь (ставшую прозрачным раствором после добавления T3P) перемешивали при температуре от -25°C до 10°C в течение 4,5 часов, затем добавляли N-(7-амино-4-хлор-1-метил-1H-индазол-3-ил)-N-(4-метоксибензил)метансульфонамид (6 г, 15,19 ммоль) и смесь перемешивали в течение 18 часов при нагревании до комнатной температуры. Реакционную смесь разбавляли этилацетатом, промывали 1N раствором NaOH, затем водой, затем 0,5 M лимонной кислотой, затем водой, затем сушили над Na2SO4 и концентрировали в вакууме. Полученный остаток очищали на силикагеле (330 г колонка RediSep Gold) с использованием 0-60% этилацетата в гексане более 15 CV (колоночных объемов), затем поддерживая при 60% EtOAc при 10 CV. Желаемые фракции объединяли и концентрировали с получением бледно-желтого твердого вещества (8,1 г, 9,14 ммоль, 60,1% выход), смеси трет-бутил N-[(1S)-1-[(3P,3P)-7-(бензилокси)-3-(4-хлор-3-{N-[(4-метоксифенил)метил]метансульфонамидо}-1-метил-1H-индазол-7-ил)-4-оксо-3H,4H-пиридо[2,3-d]пиримидин-2-ил]-2-(3,5-дифторфенил)этил]карбамата (основной) и трет-бутил N-[(1S)-1-[(3M,3M)-7-(бензилокси)-3-(4-хлор-3-{N-[(4-метоксифенил)метил]метансульфонамидо}-1-метил-1H-индазол-7-ил)-4-оксо-3H,4H-пиридо[2,3-d]пиримидин-2-ил]-2-(3,5-дифторфенил)этил]карбамата (минорный). ЖХ/МС: m/z=886,25 [M+1]+.
Стадия 2:
TFA (21,1 мл, 274 ммоль) добавляли к раствору трет-бутил (S)-(1-(7-(бензилокси)-3-(4-хлор-3-(N-(4-метоксибензил)метилсульфонамидо)-1-метил-1H-индазол-7-ил)-4-оксо-3,4-дигидропиридо[2,3-d]пиримидин-2-ил)-2-(3,5-дифторфенил)этил)карбамата (Продукт со стадии 1, 8,1 г, 9,14 ммоль) в дихлорметане (45,7 мл). Смесь перемешивали при комн. темп. в течение 2 часов. Полученный бледно-желтый раствор концентрировали. Остаток растворяли в этилацетате, затем промывали три раза 1 N раствором NaOH, затем сушили над Na2SO4 и затем концентрировали в вакууме с получением маслянистого остатка. Остаток очищали на силикагеле (330 г колонка RediSep Gold) градиентным методом Растворитель A:Растворитель B 65:35→0:100 (2 CV), затем 0:100 (9 CV); Растворитель A=гексан; Растворитель B=9:9:2 гексан:этилацетат:MeOH. Первый элюируемый изомер (основной) собирали и концентрировали в вакууме с получением N-[(6P)-7-{2-[(1S)-1-амино-2-(3,5-дифторфенил)этил]-7-гидрокси-4-оксо-3H,4H-пиридо[2,3-d]пиримидин-3-ил}-4-хлор-1-метил-1H-индазол-3-ил]-N-[(4-метоксифенил)метил]метансульфонамида (4,1 г, 5,89 ммоль, 64,5% выход). 1H ЯМР (500 МГц, DMSO-d6) δ 7,86-7,98 (м, 1 H) 7,15-7,37 (м, 4 H) 6,97-7,06 (м, 1 H) 6,70-6,89 (м, 4 H) 6,40-6,48 (м, 1 H) 4,70-4,88 (м, 2 H) 3,41-3,81 (м, 7 H) 3,20-3,28 (м, 1 H) 3,08-3,12 (м, 3 H) 2,71-2,79 (м, 1 H) 1,69-2,00 (м, 2 H). ЖХ/МС: m/z=696,20 [M+1]+.
Получение N-((S)-1-((3P)-3-(4-хлор-3-(N-(4-метоксибензил)метилсульфонамидо)-1-метил-1H-индазол-7-ил)-7-гидрокси-4-оксо-3,4-дигидропиридо[2,3-d]пиримидин-2-ил)-2-(3,5-дифторфенил)этил)-2-((3bS,4aR)-3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетамида
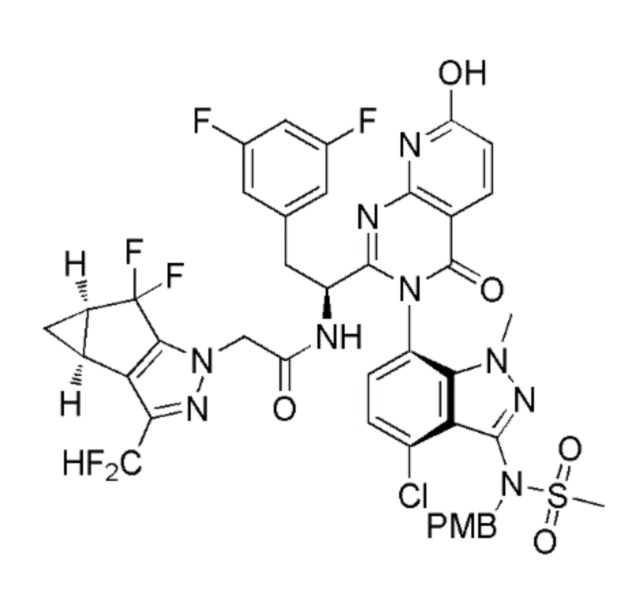
К перемешиваемому раствору N-[(6P)-7-{2-[(1S)-1-амино-2-(3,5-дифторфенил)этил]-7-гидрокси-4-оксо-3H,4H-пиридо[2,3-d]пиримидин-3-ил}-4-хлор-1-метил-1H-индазол-3-ил]-N-[(4-метоксифенил)метил]метансульфонамида (0,926 г, 1,330 ммоль)<autotext key="1AE0F6DD" name="[Reactants]" index="2" field="Reactants" type="field" length="192" /> в DMF (13 мл)<autotext key="1AE0F6DE" name="[Solvents]" index="1" field="Solvents" type="field" length="38" /> добавляли 2-((3bS,4aR)-3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)уксусную кислоту (0,351 г, 1,330 ммоль)<autotext key="1AE0F6DF" name="[Reactants]" index="1" field="Reactants" type="field" length="147" />, 2-(3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-1,1,3,3-тетраметилизоурония гексафторфосфат (V) (“HATU”, 0,531 г, 1,397 ммоль)<autotext key="1AE0F6E0" name="[Reactants]" index="3" field="Reactants" type="field" length="116" /> и DIPEA (0,581 мл, 3,33 ммоль)<autotext key="1AE0F6E1" name="[Reactants]" index="4" field="Reactants" type="field" length="27" />. Реакционную смесь перемешивали в течение 2 часов, затем реакционную смесь разбавляли водой и экстрагировали этилацетатом. Объединенные EtOAc экстракты промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Неочищенный продукт очищали флэш-хроматографией на силикагеле с использованием 10-100% этилацетата в гексане с получением N-((S)-1-((3P)-3-(4-хлор-3-(N-(4-метоксибензил)метилсульфонамидо)-1-метил-1H-индазол-7-ил)-7-гидрокси-4-оксо-3,4-дигидропиридо[2,3-d]пиримидин-2-ил)-2-(3,5-дифторфенил)этил)-2-((3bS,4aR)-3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетамида (1,1 г, 88%) в виде не совсем белого пенистого твердого вещества. ЖХ/МС: m/z=942,25 [M+1]+.
Получение Примера 1: N-((S)-1-((3P)-3-(4-хлор-1-метил-3-(метилсульфонамидо)-1H-индазол-7-ил)-4-оксо-7-(3,3,3-трифторпропокси)-3,4-дигидропиридо[2,3-d]пиримидин-2-ил)-2-(3,5-дифторфенил)этил)-2-((3bS,4aR)-3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетамид
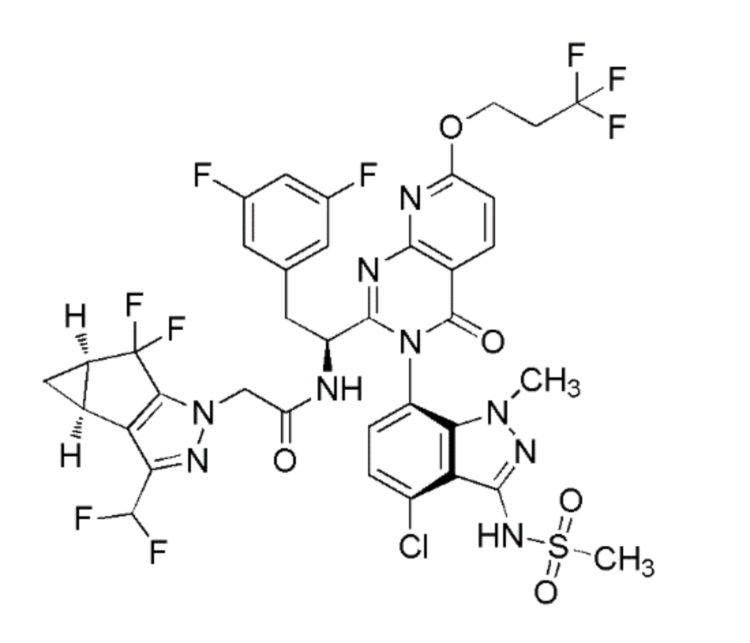
Раствор диизопропил (E)-диазен-1,2-дикарбоксилата (“DIAD”, 0,125 мл, 0,637 ммоль) в THF (0,2 мл) добавляли по каплям к смеси N-(1-((3P)-3-(4-хлор-3-(N-(4-метоксибензил)метилсульфонамидо)-1-метил-1H-индазол-7-ил)-7-гидрокси-4-оксо-3,4-дигидропиридо[2,3-d]пиримидин-2-ил)-2-(3,5-дифторфенил)этил)-2-((3bS,4aR)-3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетамида (0,2 г, 0,212 ммоль)), 3,3,3-трифторпропан-1-ола (0,073 г, 0,637 ммоль) и трифенилфосфина (0,178 г, 0,679 ммоль) в тетрагидрофуране (2,1 мл) при комн. темп. Реакционную смесь перемешивали в течение 18 часов при комн. темп. и затем концентрировали в вакууме. Остаток очищали на силикагеле (колонка 24 г RediSep Gold) с использованием градиента 0-60% этилацетата в гексане при более 15 CV, а затем поддерживая при 60% этилацетата в гексане при 5 CV. Фракции, содержащие чистый продукт, объединяли и затем концентрировали с получением желтого твердого вещества. Это твердое вещество помещали в смесь DCM (1 мл):TFA (0,5 мл); раствор охлаждали до 0°C; и к раствору добавляли трифторметансульфокислоту (0,057 мл, 0,637 ммоль). Смесь перемешивали в течение 1 часа и затем концентрировали в вакууме. Остаток растворяли в этилацетате; промывали 1 N раствором NaOH; промывали при помощи 0,5M лимонной кислоты; сушили над Na2SO4; фильтровали; и затем концентрировали в вакууме. Остаток подвергали хроматографии на силикагеле (24 г колонка RediSep Gold) с использованием 0-60% этилацетата в гексане более 20 CV, затем поддерживая при 60% этилацетата при 10 CV. Фракции, содержащие чистый продукт, объединяли и затем концентрировали в вакууме с получением N-(1-((6P)-3-(4-хлор-1-метил-3-(метилсульфонамидо)-1H-индазол-7-ил)-4-оксо-7-(3,3,3-трифторпропокси)-3,4-дигидропиридо[2,3-d]пиримидин-2-ил)-2-(3,5-дифторфенил)этил)-2-((3bS,4aR)-3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетамида (0,078 г, 0,081 ммоль, 38,0% выход) в виде коричневого твердого вещества. 1H ЯМР (500 МГц, Метанол-d4) δ ppm 8,46-8,53 (м, 1 H) 7,28-7,34 (м, 1 H) 7,19-7,24 (м, 1 H) 7,03-7,09 (м, 1 H) 6,53-6,81 (м, 4 H) 4,80 (дд, J=5,96, 2,98 Гц, 3 H) 4,49-4,62 (м, 2 H) 3,58-3,62 (м, 3 H) 3,40-3,49 (м, 1 H) 3,22-3,24 (м, 3 H) 3,06-3,14 (м, 1 H) 2,80-2,89 (м, 2 H) 2,37-2,44 (м, 2 H) 1,32-1,37 (м, 1 H) 0,96-1,01 (м, 1 H). Метод анализа ЖХМС: Колонка=Acquity UPLC BEH C18, 2,1×100 мм, частицы 1,7 мкм; Объем вводимой пробы=5,00 мкл; Скорость потока=0,80 мл/мин; Растворитель A=95:5 Вода:MeCN масс./ 0,1% об/об муравьиной кислоты; Растворитель B=5:95 Вода:MeCN масс./ 0,1% об/об муравьиной кислоты; Профиль элюирования=начальный %B: 0, конечный %B: 100, Градиент Время: 3,5 мин, затем поддержание при 100% B в течение 1 мин; Длина волны детекции 1=220 нм, длина волны 2=254 нм. Время удерживания ЖХМС=3,097 мин; m/z=918,05 [M+1]+.
Альтернативное получение N-(7-амино-4-хлор-1-метил-1H-индазол-3-ил)-N-(4-метоксибензил)метансульфонамида
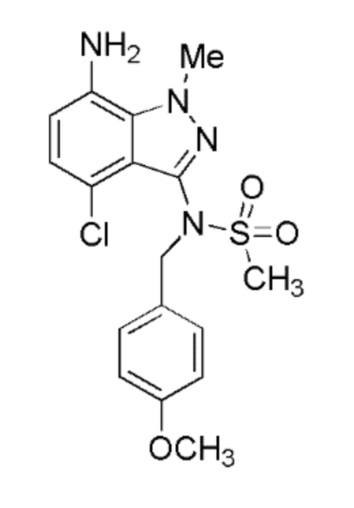
Схема синтеза:
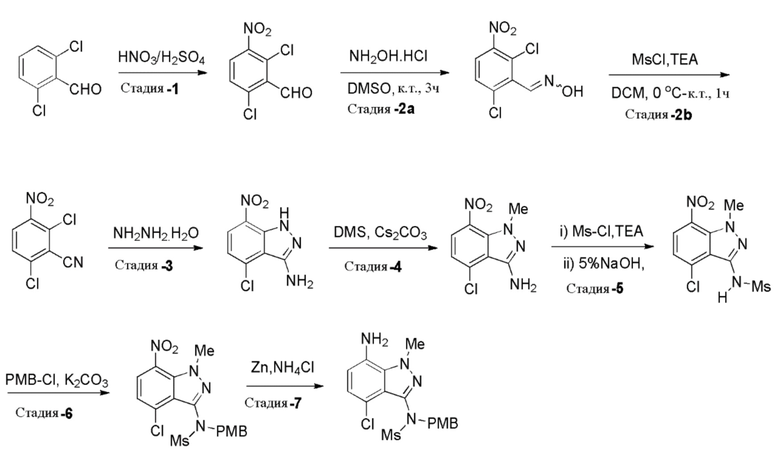
Стадия 1: Получение 2,6-дихлор-3-нитробензальдегида
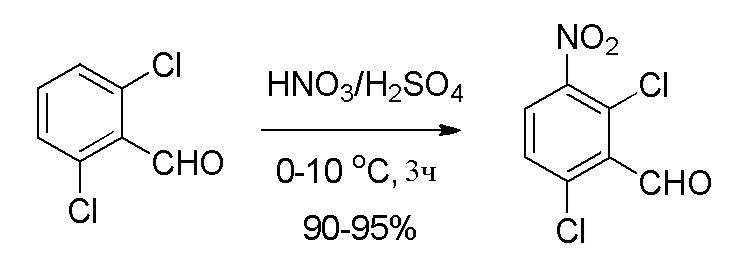
К раствору серной кислоты (H2SO4) (5,63 л, 4,5 об.) в круглодонной колбе при 0-5°C порциями добавляли 2,6-дихлорбензальдегид (1,25 кг, 7,10 моль, 1,0 экв.) при температуре ниже 15°C. Реакционную массу перемешивали при 0-5°C в течение 30 мин. Раствор свежеприготовленной смеси для нитрования [получали из конц. H2SO4 (0,425 л, 0,34 об.) и 70% HNO3 (0,85 кг, 13,49 моль, 1,30 экв.) при 0°C] добавляли к указанной выше реакционной смеси при температуре ниже 10°C [Примечание: реакция является слегка экзотермической (3-6°C); так что добавление предпочтительно при более низкой температуре]. Реакционную смесь перемешивали при 5-10°C в течение 2-3 часов. После завершения реакции (отслеживали при помощи ТСХ) ее гасили ледяной водой (18,75 л, 15 об.) при температуре ниже 25°C. Затем реакционной массе давали нагреться до комнатной температуры и перемешивали в течение 2 часов. Твердые вещества выделяли фильтрованием и затем промывали водой (2,5 л, 2,0 об.). Остаточную массу воды удаляли из твердых веществ, поддерживая вакуумную фильтрацию в течение 60-90 мин. Неочищенное влажное твердое вещество сначала сушили в атмосфере воздуха; затем в сушильном шкафу с горячим воздухом при 50-55°C в течение 10-12 часов (до содержания влаги не больше 5,0%) с получением высушенного указанного в заголовке продукта, 2,6-дихлор-3-нитробензальдегида (1,44 кг, 92% выход), в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 10. 44 (с, 1H), 7,88 (д, J=8,4 Гц, 1H), 7,56 (д, J=8,8 Гц, 1H).
Стадия 2: Получение 2,6-дихлор-3-нитробензонитрила
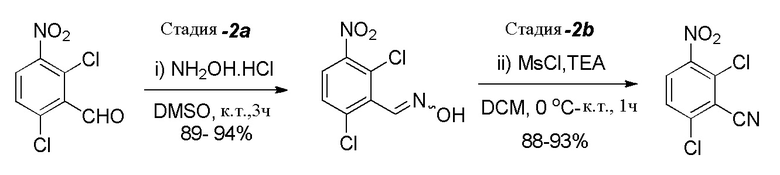
(Стадия-2a) К раствору DMSO (5,9 л, 5,0 об.)) в круглодонной колбе добавляли 2,6-дихлор-3-нитробензальдегид (1,17 кг, 5,31 моль, 1,0 экв.) при комнатной температуре. После перемешивания в течение 30 мин при комнатной температуре добавляли гидроксиламингидрохлорид (0,63 кг, 9,04 моль, 1,70 экв.) и реакционную массу перемешивали при комнатной температуре в течение 3 часов. После завершения реакции (отслеживали при помощи ТСХ) реакционную массу гасили добавлением ледяной воды (18,0 л, 15,0 об.) со скоростью, достаточной для поддержания температуры ниже 30°C (Наблюдение: твердые вещества образовывались при добавлении воды). Реакционную массу перемешивали при комнатной температуре в течение 60-90 мин. Твердые вещества выделяли фильтрованием; промывали водой (2,5 л, 2,0 об.); с последующей промывкой смесью ацетона и гексана (6,0 л, соотношение 1:1). Остаточную массу воды удаляли из твердых веществ, поддерживая вакуумную фильтрацию в течение 60-90 мин. Влажное твердое вещество сначала сушили на воздухе, а затем сушили в сушильном шкафу с горячим воздухом при 50-55°C в течение 10-12 часов (до содержания влаги не больше 1,0%) с получением высушенного целевого продукта, 2,6-дихлор-3-нитробензальдегидоксима (1,22 кг, 92% выход), в виде не совсем белого твердого вещества. Неочищенный продукт (который содержит 10-20% 2,6-дихлор-3-нитробензонитрила) использовали непосредственно на следующей стадии без дополнительной очистки.
(Стадия-2b) К перемешиваемому раствору неочищенного оксима (описанное выше получение, 1,13 кг, 4,80 моль, 1,0 экв.) в DCM (9,04 л, 8,0 об.) при 0-5°C добавляли триэтиламин (“TEA”, 1,02 кг, 10,09 моль, 2,1 экв.). После перемешивания в течение 5 мин медленно при 15°C добавляли метансульфонилхлорид (0,60 кг, 5,29 моль, 1,1 экв.) (Наблюдение: во время добавления отмечали экзотермический эффект). Затем реакционную массу перемешивали при комнатной температуре в течение 30-45 мин. После завершения реакции (прогресс реакции отслеживали при помощи ТСХ; подвижная фаза: 20% этилацетата в гексане) реакционную массу разбавляли водой (6,78 л, 6,0 об.); органический слой отделяли; и водный слой экстрагировали при помощи DCM (3,4 л, 3,0 об.). Объединенные органические слои промывали насыщенным солевым раствором (5,65 л, 5,0 об.); сушили над Na2SO4; и концентрировали под вакуумом. Полученные неочищенные твердые вещества растирали в порошок с гексаном (4,50 л, 4,0 об.) при комнатной температуре. Влажное вещество сушили в сушильном шкафу с горячим воздухом при 50-55°C в течение 5-6 часов с получением высушенного продукта, 2,6-дихлор-3-нитробензонитрила (0,95 кг, 91% выход), в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 8,07 (д, J=8,8 Гц, 1H), 7,63 (д, J=8,8 Гц, 1H).
Стадия 3: Получение 4-хлор-7-нитро-1H-индазол-3-амина
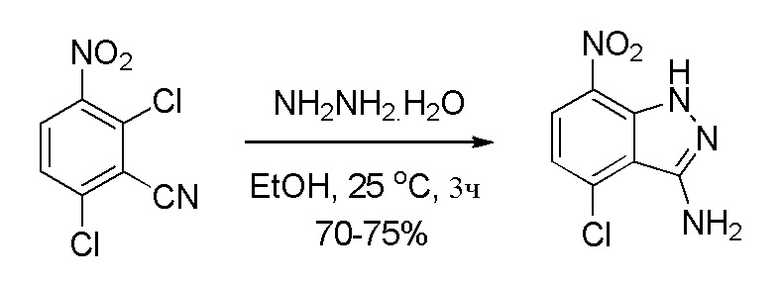
К перемешиваемому раствору 2,6-дихлор-3-нитробензонитрила (750,0 г, 3,45 моль, 1,0 экв.) в этаноле (7,5 л, 10,0 об.) при 15-20°C медленно добавляли гидразингидрат (519,0 г, 10,36 моль, 3,0 экв.), поддерживая температуру реакционной массы ниже 25°C (Наблюдение: добавление является слегка экзотермическим, и после добавления начнется образование твердого вещества). Температуру реакционной смеси медленно повышали до комнатной, а затем смесь перемешивали в течение 3 часов (наблюдение: количество твердых веществ за это время возрастало). После завершения реакции (отслеживали при помощи ТСХ) смесь разбавляли водой (7,5 л, 10,0 об.) и дополнительно перемешивали в течение 1 часа при комнатной температуре. Твердые вещества выделяли фильтрованием и затем промывали водой (2,25 л, 3,0 об.). Влажное твердое вещество промывали смесью ацетона (1,875 л, 2,5 об.) и гексана (1,875 л, 2,5 об.) в соотношении 1:1. Остаточную массу воды удаляли из твердых веществ, поддерживая вакуумную фильтрацию в течение 60-90 мин. Влажное твердое вещество в завершение сушили в сушильном шкафу с горячим воздухом в течение 7-8 часов при 50°C (до содержания влаги ниже 1,5%) с получением высушенного продукта, 4-хлор-7-нитро-1H-индазол-3-амина (549,0 г, 75% выход), в виде твердого вещества кирпично-красного цвета. 1H ЯМР (400 МГц, CDCl3): δ 10,36 (шир. с, 1H), 8,20 (д, J=8,4 Гц, 1H), 7,07 (д, J=8,40 Гц, 1H), 4,73 (шир. с, 2H).
Стадия 4: Получение 4-хлор-1-метил-7-нитро-1H-индазол-3-амина
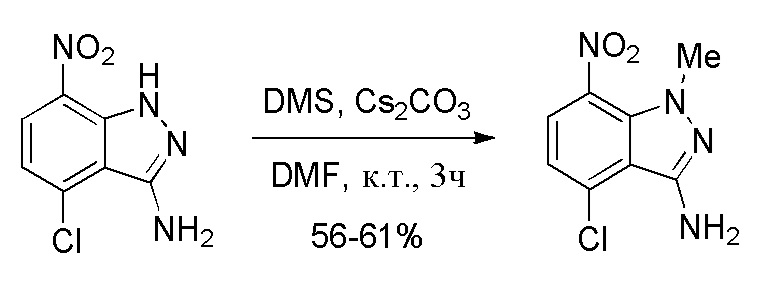
К перемешиваемому раствору 4-хлор-7-нитро-1H-индазол-3-амина (500 г, 0,42 моль, 1,0 экв.) в DMF (5,0 л, 10,0 об.) при 5-10°C медленно добавляли карбонат цезия (Cs2CO3) (1,91 кг, 5,88 моль, 2,5 экв.), поддерживая температуру реакционной массы ниже 10°C. После перемешивания в течение 5-10 мин добавляли диметилсульфат (326,3 г, 2,59 моль, 1,1 экв.), поддерживая температуру реакционной массы ниже 10°C (Примечание: медленное добавление предпочтительно для получения более благоприятной региоселективности). Затем температуру реакции медленно повышали до комнатной и перемешивание продолжали еще 2 часа при той же температуре. После завершения реакции (отслеживали при помощи ТСХ) реакционную массу гасили добавлением ледяной воды (15,0 л, 30,0 об.) и полученную смесь затем перемешивали в течение 6-8 часов при комнатной температуре. Твердые вещества выделяли фильтрованием и затем промывали водой (1,5 л, 3,0 об.). Влажное твердое вещество промывали IPA (1,5 л, 3,0 об.), затем гексаном (1,0 л, 2,0 об.). Остаточную массу воды удаляли из твердых веществ, поддерживая вакуумную фильтрацию в течение 60-90 мин. Влажное твердое вещество сушили в сушильном шкафу с горячим воздухом в течение 7-8 часов при 50°C (до содержания влаги ниже 1,0%). Выделенное вещество, 4-хлор-1-метил-7-нитро-1H-индазол-3-амин (319,0 г, 60% выход), использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3): δ 7,97 (д, J=8,32 Гц, 1H), 6,97 (д, J=8,24 Гц, 1H), 4,63 (шир. с, 2H), 3,96 (с, 3H).
Стадия 5: Получение N-(4-хлор-1-метил-7-нитро-1H-индазол-3-ил)метансульфонамида
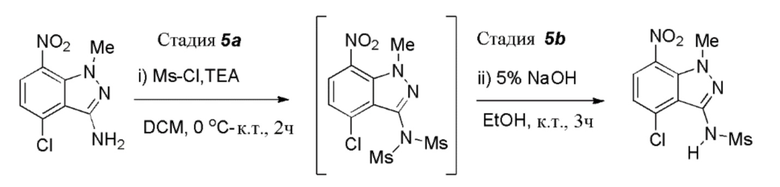
(Стадия 5a) К раствору 4-хлор-1-метил-7-нитро-1H-индазол-3-амина (625,0 г, 2,76 моль, 1,0 экв.) в DCM (6,25 л, 10,0 об.) при 0-5°C. добавляли триэтиламин (TEA) (837,0 г, 8,27 моль, 3,0 экв.); с последующим добавлением 4-диметиламинопиридина (DMAP) (20,60 г, 0,165 моль, 0,06 экв.). Реакционную массу перемешивали в течение 5-10 мин, затем медленно добавляли метансульфонилхлорид (MsCl) (790,0 г, 6,89 моль, 2,5 экв.), поддерживая температуру реакционной массы ниже 10°C. Реакционной смеси давали нагреться до комнатной температуры и затем перемешивали в течение 1,5-2,0 часов. После завершения реакции (отслеживали при помощи ТСХ) смесь разбавляли водой (6,25 л, 10,0 об.) и затем перемешивали при комнатной температуре в течение 15 мин. Органический слой отделяли и водный слой экстрагировали при помощи DCM (6,25 л, 10,0 об.). Объединенные органические слои промывали насыщенным солевым раствором (1,25 л, 2,0 об.), сушили над Na2SO4 и концентрировали с получением неочищенных твердых веществ. Твердые вещества растирали в порошок с гексаном (1,25 л, 2,0 об.) при комнатной температуре с получением промежуточного соединения, N-(4-хлор-1-метил-7-нитро-1H-индазол-3-ил)-N-(метилсульфонил)метансульфонамида, которое использовали непосредственно на следующей стадии.
(ii) К перемешиваемому раствору N-(4-хлор-1-метил-7-нитро-1H-индазол-3-ил)-N-(метилсульфонил)метансульфонамида (полученного выше) в этаноле (10,5 л, 20,0 об.) при комнатной температуре медленно добавляли водн. 5% раствор NaOH (4,38 л, 7,0 об.) [Примечание: медленное добавление предпочтительно через капельную воронку]. Реакционную массу перемешивали при той же температуре в течение 3 часов. После завершения реакции (отслеживали при помощи ТСХ) [приготовление образца для ТСХ анализа: ~1,0 мл образца, подкисленного водн. 2,0 N раствором HCl до достижения pH: 2-3, экстрагировали этилацетатом и анализировали органический слой при помощи ТСХ], реакционную массу охлаждали до 0-5°C и pH доводили до 2-3 добавлением водн. 2,0 N раствора HCl (3,13 л, 5,0 об.) при поддержании температуры реакции ниже 10°C [Примечание: осаждение происходило при добавлении HCl и увеличивалось при перемешивании]. Реакционную смесь нагревали до комнатной температуры и затем перемешивали в течение 1,5-2,0 часов. Полученные твердые вещества выделяли фильтрованием и затем промывали водой (1,25 л, 2,0 об.); с последующей промывкой гексаном (1,25 л, 2,0 об.). Остаточную массу воды удаляли из твердых веществ, поддерживая вакуумную фильтрацию в течение 60-90 мин. Влажное вещество сушили в сушильном шкафу с горячим воздухом при 50°C в течение 6-7 часов (до содержания влаги ниже 1,0%) с получением высушенного продукта, N-(4-хлор-1-метил-7-нитро-1H-индазол-3-ил)метансульфонамида (640,0 г, 76%), в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 8,05 (д, J=8,32 Гц, 1H), 7,32 (шир. с, 1H), 7,17 (д, J=8,28 Гц, 1H), 4,15 (с, 3H), 3,45 (с, 3H).
Стадия 6: Получение N-(4-хлор-1-метил-7-нитро-1H-индазол-3-ил)-N-(4-метоксибензил)метансульфонамида
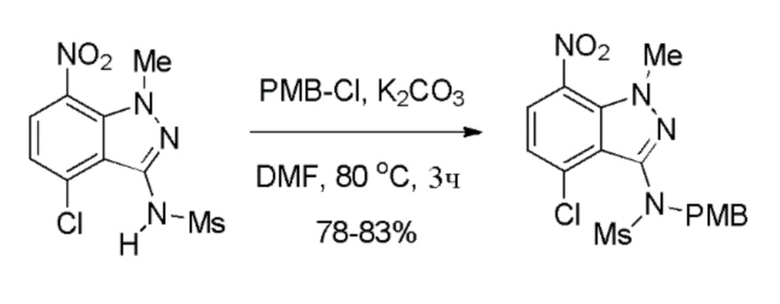
К смеси N-(4-хлор-1-метил-7-нитро-1H-индазол-3-ил)метансульфонамида (635,0 г, 2,08 моль, 1,0 экв.) и 1-(хлорметил)-4-метоксибензола (359,0 г, 2,30 моль, 1,1 экв.) в DMF (6,35 л, 10,0 об.) при комнатной температуре добавляли карбонат калия (374,7 г, 2,70 моль, 1,3 экв.). Реакционную смесь нагревали до 80-90°C и поддерживали при этой температуре в течение 3 часов. После завершения реакции (отслеживали при помощи ТСХ) смесь выливали в ледяную воду (19,05 л, 30,0 об.) [Примечание: медленное гашение при интенсивном перемешивании предпочтительно во избежание образования комков при осаждении продукта]. Полученные твердые вещества выделяли фильтрованием и промывали водой (1,90 л, 3,0 об.); затем твердые вещества промывали гексаном (1,27 л, 2,0 об.). Остаточную массу воды удаляли из твердых веществ, поддерживая вакуумную фильтрацию в течение 60-90 мин. Выделенное твердое вещество растворяли в этилацетате (12,7 л, 20,0 об.) и добавляли активированный уголь (63,5 г). Смесь нагревали до 60-70°C и затем перемешивали в течение 30-45 мин при этой температуре. Смесь фильтровали горячей (40-50°C) через слой целита, и слой целита затем экстрагировали этилацетатом (3,17 л, 5,0 об.). Объединенные фильтраты концентрировали досуха при пониженном давлении при температуре ниже 50°C. К твердым веществам добавляли этилацетат (0,635 л, 1,0 об.) при комнатной температуре. Полученную твердую суспензию перемешивали в течение 30 мин. Твердые вещества выделяли фильтрованием и затем промывали гексаном (1,27 л, 2,0 об.). Остаточную воду удаляли из твердых веществ, поддерживая вакуумную фильтрацию в течение 45-60 мин, с получением продукта N-(4-хлор-1-метил-7-нитро-1H-индазол-3-ил)-N-(4-метоксибензил) метансульфонамида (705,0 г, 80% выход) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 7,99 (д, J=8,24 Гц, 1H), 7,27 (д, J=8,68 Гц, 2H), 7,19 (д, J=8,24 Гц, 1H), 6,80 (д, J=8,44 Гц, 2H), 4,95-4,76 (м, 2H), 4,17 (с, 3H), 3,76 (с, 3H), 3,01 (с, 3H).
Стадия 7: Получение N-(7-Амино-4-хлор-1-метил-1H-индазол-3-ил)-N-(4-метоксибензил)метансульфонамида
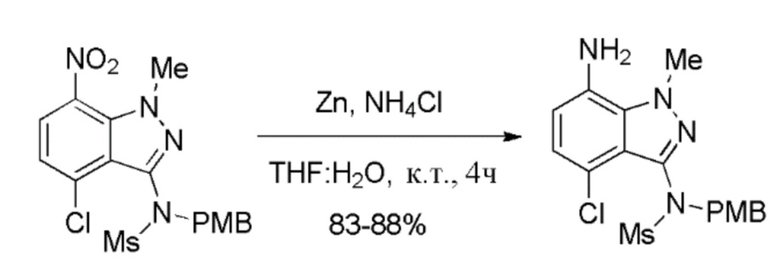
К перемешиваемой суспензии порошка цинка (540,0 г, 8,23 моль, 10,0 экв.) в смеси THF (3,50 л, 10,0 об.) и воды (7,0 л, 20,0 об.) при комнатной температуре добавляли хлорид аммония (NH4Cl) (449,0 г, 8,23 моль, 10,0 экв.). К смеси добавляли N-(4-хлор-1-метил-7-нитро-1H-индазол-3-ил)-N-(4-метоксибензил)метансульфонамид (350 г, 0,823 моль, 1,0 экв.) в THF (7,0 л, 20,0 об.). Реакционную смесь перемешивали при комнатной температуре в течение 3-4 часов. После завершения реакции (отслеживали при помощи ТСХ/ВЭЖХ в ходе процесса) смесь разбавляли этилацетатом (3,5 л, 10,0 об.) и водой (1,12 л, 2,5 об.). Смесь перемешивали в течение 15 мин. Реакционную массу фильтровали через слой целита, промывая этилацетатом (1,75 л, 5,0 об.). Двухфазный фильтрат собирали и фазы разделяли. Водный слой экстрагировали этилацетатом (3,50 л, 10,0 об.). Объединенные органические слои промывали насыщенным солевым раствором (3,50 л, 10 об.), сушили над Na2SO4 и затем концентрировали в вакууме с получением неочищенного твердого вещества. К неочищенному продукту добавляли MTBE (3,25 л, 10 об.) и суспензию перемешивали в течение 30 мин при комнатной температуре. Твердые вещества выделяли фильтрованием. Остаточную массу воды удаляли из твердых веществ, поддерживая вакуумную фильтрацию в течение 30-45 мин. Влажный продукт сушили в сушильном шкафу с горячим воздухом (50°C) в течение 2 часов с получением указанного в заголовке продукта, N-(7-амино-4-хлор-1-метил-1H-индазол-3-ил)-N-(4-метоксибензил)метансульфонамида (276,0 г, 85% выход), в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 7,29-7,26 (м, 2H), 6,86-6,79 (м, 2H), 6,42 (д, J=7,80 Гц, 1H), 4,99-4,70 (м, 2H), 4,25 (с, 3H), 3,77 (с, 5H), 2,98 (с, 3H).
Получение 2-амино-6-(3,3,3-трифторпропокси)никотиновой кислоты
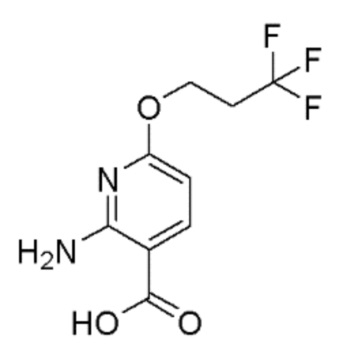
Схема синтеза:
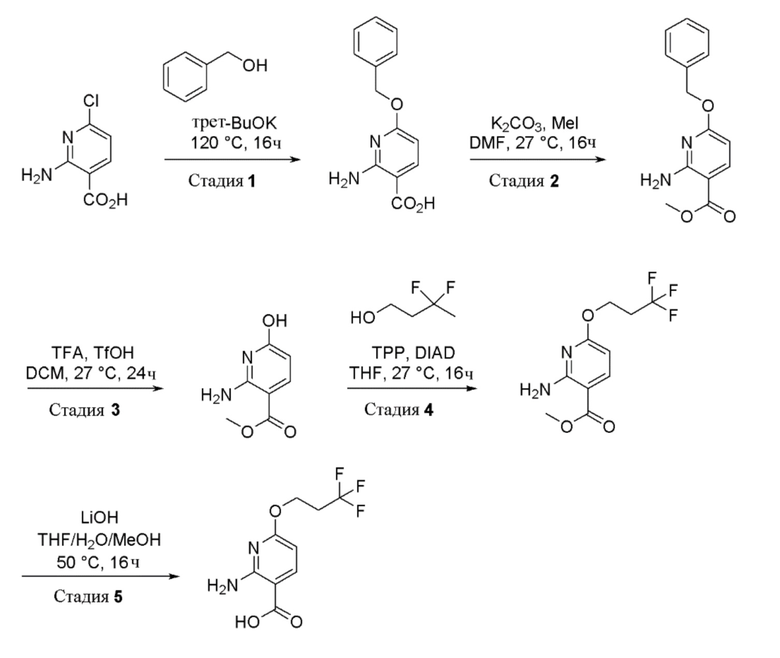
Стадия 1: Получение 2-амино-6-(бензилокси)никотиновой кислоты

К перемешиваемому раствору 2-амино-6-хлорникотиновой кислоты (200 г, 1159 ммоль) в бензиловом спирте (1400 мл, 13464 ммоль) при 26°C в атмосфере N2 добавляли трет-бутоксид калия (390 г, 3477 ммоль). Реакционную смесь нагревали до 120°C и перемешивали при этой температуре в течение 16 часов. Прогресс реакции отслеживали при помощи ТСХ (SiO2, 10% MeOH в DCM, Rf=0,5). По завершении реакционную смесь разбавляли водой (3 л) и экстрагировали диэтиловым эфиром (2×1000 мл). Органический слой отделяли и водный слой подкисляли до pH 4 с использованием водн. раствора лимонной кислоты (0,5 M). Осажденное твердое вещество собирали фильтрацией и затем сушили при пониженном давлении с получением 2-амино-6-(бензилокси)никотиновой кислоты в виде не совсем белого твердого вещества (220 г, выход=72%). 1H ЯМР (400 МГц, DMSO-d6) δ=12,56-12,32 (м, 1H), 7,97-7,91 (м, 1H), 7,52-7,41 (м, 2H), 7,38-7,11 (м, 5H), 6,03 (д, J=8,5 Гц, 1H), 5,39-5,31 (м, 2H). Чистота ЖХМС=93%; m/z=245,29 (M+H).
Стадия 2: Получение метил 2-амино-6-(бензилокси)никотината
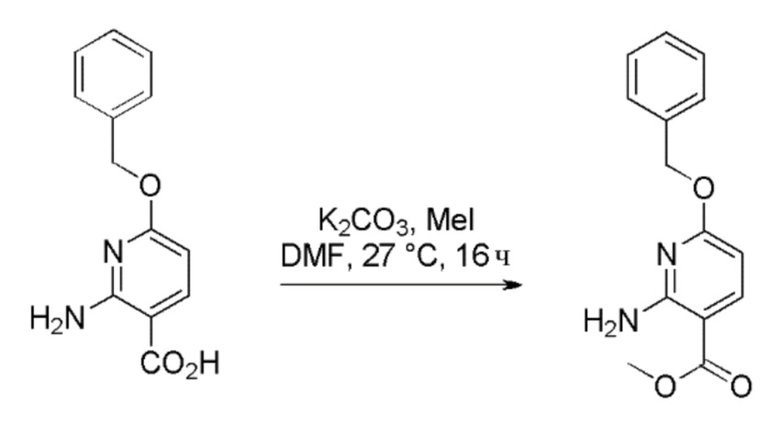
К перемешиваемому раствору 2-амино-6-(бензилокси)никотиновой кислоты (220 г, 901 ммоль) в DMF (2,5 л) при 26°C в атмосфере N2 медленно добавляли карбонат калия (373 г, 2702 ммоль) и йодметан (0,282 л, 4504 ммоль). Реакционную смесь перемешивали при 27°C в течение 16 часов. Прогресс реакции отслеживали при помощи ТСХ (SiO2, 40% EtOAc/Петрол., Rf=0,6). По завершении реакционную смесь разбавляли водой (5 л). Осажденное твердое вещество выделяли фильтрованием и затем сушили под вакуумом с получением метил 2-амино-6-(бензилокси)никотината в виде не совсем белого твердого вещества (220 г, выход=92%). 1H ЯМР (400 МГц, CDCl3) δ=8,00 (д, J=8,4 Гц, 1H), 7,42-7,40 (м, 2H), 7,39-7,35 (м, 2H), 7,34-7,31 (м, 1H), 6,01 (д, J=8,4 Гц, 1H), 5,33 (с, 2H), 3,84 (с, 3H). Чистота ЖХМС=97%, m/z=259,30 (M+H).
Стадия 3: Получение метил 2-амино-6-гидроксиникотината
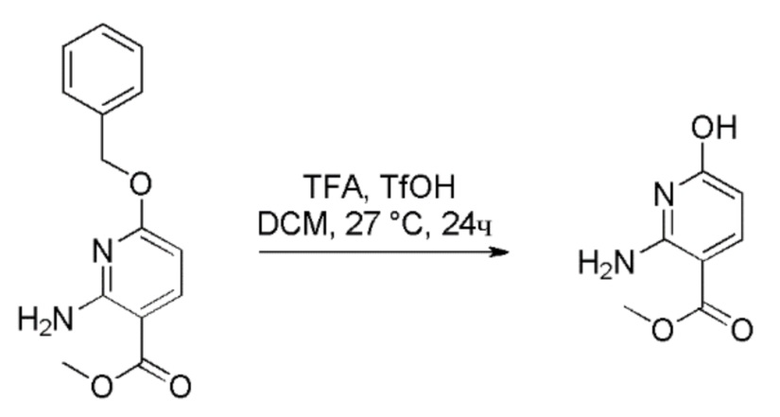
К перемешиваемому раствору метил 2-амино-6-(бензилокси)никотината (50 г, 190 ммоль) в DCM (500 мл) при 26°C в атмосфере N2 медленно добавляли TFA (800 мл) и трифторметансульфокислоту (25 мл, 282 ммоль). Реакционную смесь перемешивали при 26°C в течение 16 часов. Прогресс реакции отслеживали при помощи ТСХ (SiO2, EtOAc, Rf=0,2). По завершении летучие вещества удаляли под вакуумом с получением неочищенного продукта. Это вещество растирали в порошок с диэтиловым эфиром (3×1000 мл) и осажденное твердое вещество затем выделяли фильтрованием. К твердому веществу добавляли воду (2 л) и смесь затем оставляли на 5 часов. Твердое вещество собирали фильтрацией и промывали водой. Твердое вещество сушили под вакуумом с получением метил 2-амино-6-гидроксиникотината в виде не совсем белого твердого вещества (25 г, выход=78%). 1H ЯМР (300 МГц, DMSO-d6) δ=10,92-10,76 (м, 1H), 7,65 (д, J=9,5 Гц, 1H), 7,43-6,87 (м, 2H), 5,51 (д, J=9,5 Гц, 1H), 3,69 (с, 3H). Чистота ЖХМС=99,32%; m/z=169,32 (M+H). Отсутствие TFA и трифторметансульфокислоты в продукте подтверждали при помощи 19F-ЯМР. Продукт использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 4: Получение метил 2-амино-6-(3,3,3-трифторпропокси)никотината
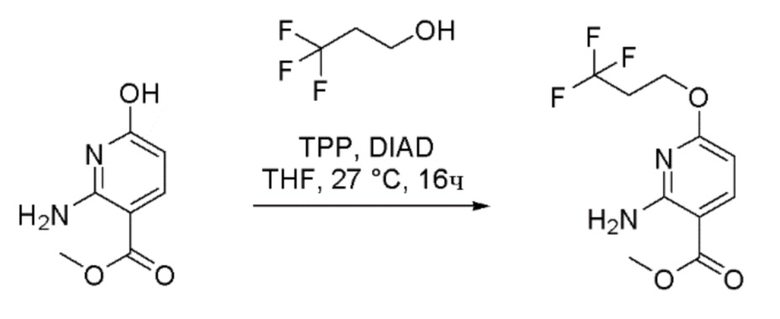
К перемешиваемому раствору метил 2-амино-6-гидроксиникотината (50 г, 297 ммоль) в THF (1000 мл) при 27°C в атмосфере азота добавляли трифенилфосфин (156 г, 595 ммоль). Реакционную массу охлаждали до 0°C и к реакционной массе добавляли по каплям диизопропилазодикарбоксилат (“DIAD” 116 мл, 595 ммоль). Раствор перемешивали в течение 30 мин. К раствору при 0°C добавляли раствор 3,3,3-трифторпропан-1-ола (52,4 мл, 595 ммоль) в THF (200 мл). Затем реакционной массе давали медленно нагреться до 27°C и затем перемешивали при этой температуре в течение 16 часов. За ходом реакции наблюдали при помощи ТСХ (SiO2, 10% EtOAc/Петрол. Rf=0,5). По завершении реакционную смесь разбавляли водой (500 мл) и экстрагировали при помощи EtOAc (2×500 мл). Объединенные органические слои промывали водой (500 мл) и затем насыщенным солевым раствором (500 мл). Органический слой сушили над безводным Na2SO4, фильтровали и затем концентрировали при пониженном давлении с получением неочищенного продукта в виде желтого полутвердого вещества (100 г). Это вещество очищали хроматографией на силикагеле, элюируя 5-10% EtOAc в петрол. Фракции, содержащие желаемый продукт, объединяли и концентрировали при пониженном давлении с получением метил 2-амино-6-(3,3,3-трифторпропокси)никотината в виде жидкости желтого цвета (50 г, 60% выход). 1H-ЯМР (400 МГц, CDCl3) δ: 8,01 (д, J=8,8 Гц, 1H), 7,21-6,85 (шир.с, 1H), 6,04 (д, J=8,4 Гц, 1H), 4,50 (т, J=6,6 Гц, 2H), 3,84 (с, 3H), 2,63-2,55 (м, 2H). Метод анализа ЖХМС: Колонка=Acquity BEH C18 (50 мм × 2,1 мм, 1,7 мкм); Подвижная фаза A=0,05% Муравьиной кислоты в воде; Подвижная фаза B=0,05% Муравьиной кислоты в CAN; Градиент=Время (мин) / %B: 0/3, 0,4/3, 2,5/98, 3,4/98, 3,5/3, 4/3; Темп. колонки.=35°C; Скорость потока=0,6 мл/мин. Результат ЖХМС: время удерживания=2,03 мин; наблюдаемый ион=265,15 (M+H); Чистота ЖХМС=93%.
Стадия 5: Получение 2-амино-6-(3,3,3-трифторпропокси)никотиновой кислоты
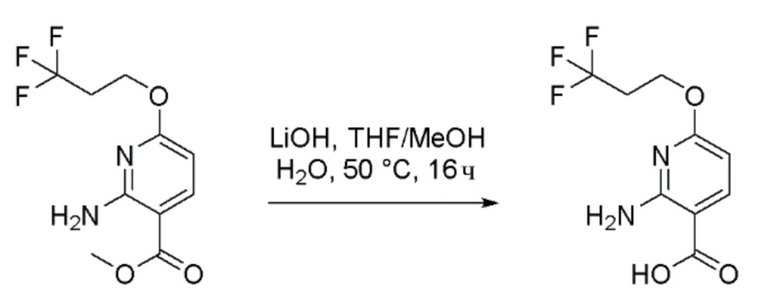
К перемешиваемому раствору метил 2-амино-6-(3,3,3-трифторпропокси)никотината (50 г, 189 ммоль) в тетрагидрофуране (THF) (500 мл), метаноле (150 мл) и воде (80 мл) при 0°C в атмосфере азота добавляли моногидрат гидроксида лития (22,66 г, 946 ммоль). Реакционную смесь перемешивали при 50°C в течение 16 часов. За ходом реакции наблюдали при помощи ТСХ (SiO2, 50% EtOAc/Петрол. Rf=0,3). По завершении реакционную смесь концентрировали при пониженном давлении с получением водного остатка. Остаток затем подкисляли до pH 4 добавлением 1N HCl. Полученный осадок собирали фильтрацией и промывали водой (500 мл), затем н-гексаном (400 мл) и затем сушили с получением 2-амино-6-(3,3,3-трифторпропокси)никотиновой кислоты в виде не совсем белого твердого вещества (45 г, 90% выход). 1H-ЯМР (400 МГц, DMSO-d6) δ: 12,47 (шир.с, 1H), 7,93 (д, J=8,8 Гц, 1H), 7,35 (шир.с, 2H), 5,98 (д, J=8,8 Гц, 1H), 4,44 (т, J=6,1 Гц, 2H), 2,84-2,73 (м, 2H). Продукт использовали непосредственно на следующей стадии без дополнительной очистки.
Альтернативное получение 2-амино-6-(3,3,3-трифторпропокси)никотиновой кислоты
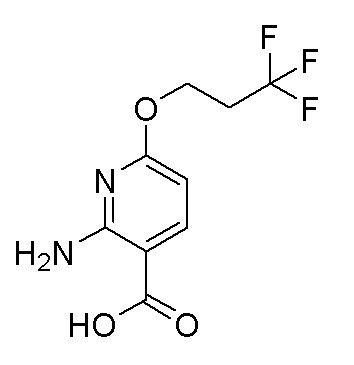
Схема синтеза:
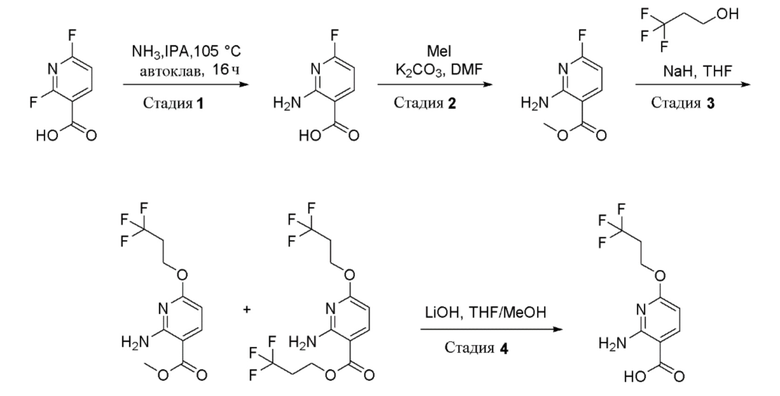
Стадия 1: Получение 2-амино-6-фторникотиновой кислоты
Смесь NH3 в воде (“25% NH3 в H2O”, 1 л, 4об.) и изопропаноле (6,5 л, 26 об.) при 0°C барботировали газообразным аммиаком в течение 1 часа. К смеси в автоклаве (25 л) добавляли 2,6-дифторникотиновую кислоту (250 г, 1571 ммоль). Затем реакционную смесь перемешивали при 105°C в течение 20 часов. За ходом реакции наблюдали при помощи ТСХ (SiO2, 80% EtOAc/Петрол. Rf=0,3). По завершении реакционной смеси давали остыть до 20°C и затем концентрировали при пониженном давлении при температуре ниже 20°C до объема 4-6 об. (1,5 л). Остаток растворяли в воде (5 л) и подкисляли до pH 2-3 добавлением 2N HCl (700 мл) и затем перемешивали в течение 2 часов. Полученный осадок собирали фильтрацией и промывали водой (4000 мл), затем н-гексаном (5000 мл) и затем сушили в вакуумной печи при 50°C с получением 2-амино-6-фторникотиновой кислоты в виде не совсем белого твердого вещества (250 г, 92% выход). Этот продукт смешивали с продуктом из других партий, полученным тем же способом, с получением 2000 г объединенного продукта. Остаточный растворитель удаляли из твердых веществ путем суспендирования твердых веществ в толуоле (10 л), а затем удаляя толуол дистилляцией. Полученные твердые вещества сушили в вакуумной печи при 60°C в течение 7 дней с получением 2-амино-6-фторникотиновой кислоты в виде не совсем белого твердого вещества (1,6 кг, 77% выход). 1H-ЯМР (400 МГц, DMSO-d6) δ: 12,94 (шир.с, 1H), 8,17 (т, J=8,8 Гц, 1H), 7,56 (шир.с, 2H), 6,25 (дд, J=8,2, 2,8 Гц, 1H). Метод ЖХМС: Колонка=Acquity BEH C18 (50 мм × 2,1 мм, 1,7 мкм); Подвижная фаза A=0,05% Муравьиной кислоты в воде; Подвижная фаза B=0,05% Муравьиной кислоты в ацетонитриле; Градиент=Время (мин) /%B: 0/3, 0,4/3, 2,5/98, 3,4/98, 3,5/3, 4/3; Температура Колонки=35°C; Скорость потока=0,6 мл/мин. Результат ЖХМС: время удерживания=1,24 мин; наблюдаемый ион=157,04 (M+H); Чистота ЖХМС=96%.
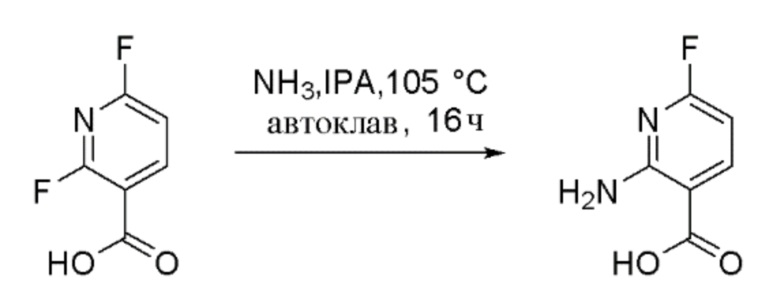
Стадия 2: Получение метил 2-амино-6-фторникотината

К перемешиваемому раствору 2-амино-6-фторникотиновой кислоты (150 г, 961 ммоль) и карбоната калия (398 г, 2882 ммоль) в DMF (1500 мл) добавляли йодметан (300 мл, 4804 ммоль). Реакционную смесь перемешивали при 27°C в течение 16 час в атмосфере азота. За ходом реакции наблюдали при помощи ТСХ (SiO2, 30% EtOAc/Петрол. Rf=0,7). По завершении реакционную смесь гасили добавлением ледяной воды (5000 мл). Полученный осадок собирали фильтрацией и промывали водой (2000 мл), затем н-гексаном (1000 мл) и затем сушили с получением метил 2-амино-6-фторникотината в виде коричневого твердого вещества (120 г, 70% выход). 1H-ЯМР (400 МГц, DMSO-d6) δ: 8,20 (т, J=8,8 Гц, 1H), 7,54 (шир.с, 2H), 6,29 (дд, J=8,4, 2,8 Гц, 1H), 3,82 (м, 3H). Метод ЖХМС: Колонка=Acquity BEH C18 (50 мм × 2,1 мм, 1,7 мкм); Подвижная фаза A=0,05% Муравьиной кислоты в воде; Подвижная фаза B=0,05% Муравьиной кислоты в ацетонитриле; Градиент=Время (мин) /%B: 0/3, 0,4/3, 2,5/98, 3,4/98, 3,5/3, 4/3; Темп. колонки.=35°C; Скорость потока=0,6 мл/мин. Результат ЖХМС: время удерживания=1,55 мин; наблюдаемый ион=171,07 (M+H); Чистота ЖХМС=96%. Продукт использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 3: Получение метил 2-амино-6-(3,3,3-трифторпропокси)никотината и 3,3,3-трифторпропил 2-амино-6-(3,3,3-трифторпропокси)никотината
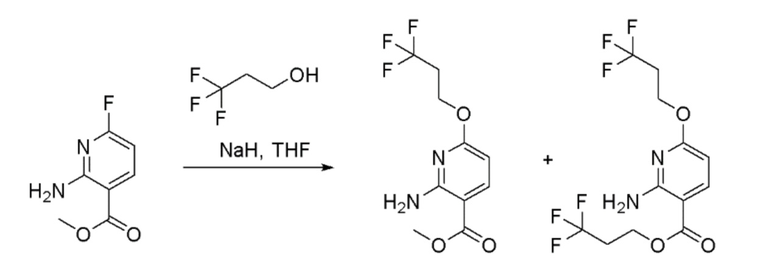
К перемешиваемому раствору метил 2-амино-6-фторникотината (25 г, 147 ммоль) и 3,3,3-трифторпропан-1-ола (15,54 мл, 176 ммоль) в THF (500 мл) при 0°C в атмосфере азота добавляли порциями гидрид натрия (60% дисперсия в масле, 8,82 г, 220 ммоль). Реакционную смесь перемешивали при 0°C в течение 30 мин и затем давали медленно нагреться до 27°C и перемешивали при этой температуре в течение 16 часов. За ходом реакции наблюдали при помощи ТСХ (SiO2, 10% EtOAc/Петрол. Rf=0,5). По завершении реакционную смесь охлаждали до 0°C и гасили насыщенным водным раствором NH4Cl (300 мл). Смесь экстрагировали при помощи EtOAc (2 × 500 мл). Объединенные органические слои промывали насыщенным солевым раствором (200 мл), сушили над безводным Na2SO4, фильтровали и затем концентрировали при пониженном давлении с получением метил 2-амино-6-(3,3,3-трифторпропокси)никотината в виде желтой жидкости (40 г). В реакции также наблюдали образование побочного продукта переэтерификации 3,3,3-трифторпропил 2-амино-6-(3,3,3-трифторпропокси)никотината. Метод ЖХМС: Колонка=Acquity BEH C18 (50 мм × 2,1 мм, 1,7 мкм); Подвижная фаза A=0,05% Муравьиной кислоты в воде; Подвижная фаза B=0,05% Муравьиной кислоты в ацетонитриле; Градиент=Время (мин)/%B: 0/97, 0,4/97, 2,5/2, 3,4/2, 3,5/97, 4,0/97; Темп. колонки.=35°C; Скорость потока=0,6 мл/мин. Результат ЖХМС: время удерживания=2,04 & 2,22 мин; наблюдаемый ион=265,18 & 347,29 (M+H); Чистота ЖХМС=57% метил 2-амино-6-(3,3,3-трифторпропокси)никотината и 15% 3,3,3-трифторпропил 2-амино-6-(3,3,3-трифторпропокси)никотината. Эту смесь неочищенных продуктов смешивали с двумя другими партиями неочищенных продуктов, полученных тем же способом (40 г и 50 г). Объединенное вещество (130 г) очищали хроматографией на силикагеле, элюируя 10-20% EtOAc в петрол. Фракции, содержащие желаемый продукт, объединяли и концентрировали при пониженном давлении с получением смеси 6:1 метил 2-амино-6-(3,3,3-трифторпропокси)никотината и 3,3,3-трифторпропил 2-амино-6-(3,3,3-трифторпропокси)никотината в виде бледно-желтой жидкости (100 г, 90% выход). 1H-ЯМР (400 МГц, CDCl3) δ: 8,02-7,97 (м, 1H), 7,04-6,48 (м, 1H), 6,08-6,03 (м, 1H), 4,52-4,47 (м, 2H), 3,83 (с, 3H), 2,64-2,54 (м, 2H). Метод ЖХМС: Колонка=Acquity BEH C18 (50 мм × 2,1 мм, 1,7 мкм); Подвижная фаза A=0,05% Муравьиной кислоты в воде; Подвижная фаза B=0,05% Муравьиной кислоты в ацетонитриле; Градиент=Время (мин)/%B: 0/97, 0,4/97, 2,5/2, 3,4/2, 3,5/97, 4,0/97; Темп. колонки.=35°C; Скорость потока=0,6 мл/мин. Результат ЖХМС: время удерживания=2,02 & 2,21 мин; наблюдаемый ион=264,97 & 346,97 (M+H); Чистота ЖХМС=66% метил 2-амино-6-(3,3,3-трифторпропокси)никотината и 11% 3,3,3-трифторпропил 2-амино-6-(3,3,3-трифторпропокси)никотината.
Стадия 4: Получение 2-Амино-6-(3,3,3-трифторпропокси)никотиновой кислоты
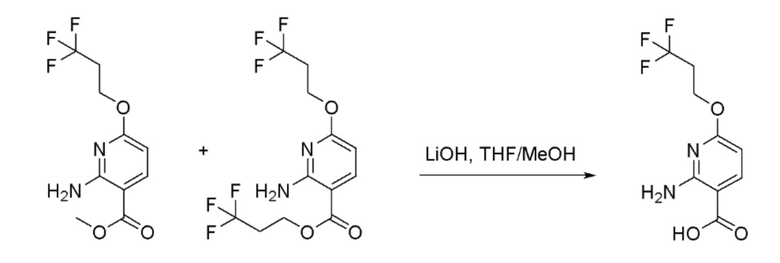
К перемешиваемому раствору метил 2-амино-6-(3,3,3-трифторпропокси)никотината и 3,3,3-трифторпропил 2-амино-6-(3,3,3-трифторпропокси)никотината (6:1, 100 г, 310 ммоль) в тетрагидрофуране (THF) (800 мл), метаноле (300 мл) и воде (200 мл) при 27°C в атмосфере азота добавляли моногидрат гидроксида лития (37,2 г, 1552 ммоль). Реакционную смесь перемешивали при 50°C в течение 8 часов. За ходом реакции наблюдали при помощи ТСХ (SiO2, 50% EtOAc/Петрол. Rf=0,3). По завершении реакционную смесь концентрировали при пониженном давлении и полученный водный остаток затем подкисляли до pH 4 добавлением 1N HCl. Полученный осадок собирали фильтрацией и промывали водой (2000 мл), затем н-гексаном (1000 мл) и затем сушили с получением 2-амино-6-(3,3,3-трифторпропокси)никотиновой кислоты в виде не совсем белого твердого вещества (80 г, 97% выход). 1H-ЯМР (400 МГц, DMSO-d6) δ: 12,47 (шир.с, 1H), 7,93 (д, J=8,4 Гц, 1H), 7,33 (шир.с, 2H), 5,99 (д, J=8,4 Гц, 1H), 4,45 (т, J=6,2 Гц, 2H), 2,83-2,74 (м, 2H). Метод ЖХМС: Колонка=Acquity BEH C18 (50 мм × 2,1 мм, 1,7 мкм); Подвижная фаза A=0,05% Муравьиной кислоты в воде; Подвижная фаза B=0,05% Муравьиной кислоты в ацетонитриле; Градиент=Время (мин)/%B: 0/3, 0,4/3, 2,5/98, 3,4/98, 3,5/3, 4/3; Темп. колонки.= 35°C; Скорость потока=0,6 мл/мин. Результат ЖХМС: время удерживания=1,73 мин; наблюдаемый ион=251,17 (M+H); Чистота ЖХМС=94%. Продукт использовали непосредственно на следующей стадии без дополнительной очистки.
Альтернативное получение соединения Примера 1: N-((S)-1-((3P)-3-(4-хлор-1-метил-3-(метилсульфонамидо)-1H-индазол-7-ил)-4-оксо-7-(3,3,3-трифторпропокси)-3,4-дигидропиридо[2,3-d]пиримидин-2-ил)-2-(3,5-дифторфенил)этил)-2-((3bS,4aR)-3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетамида
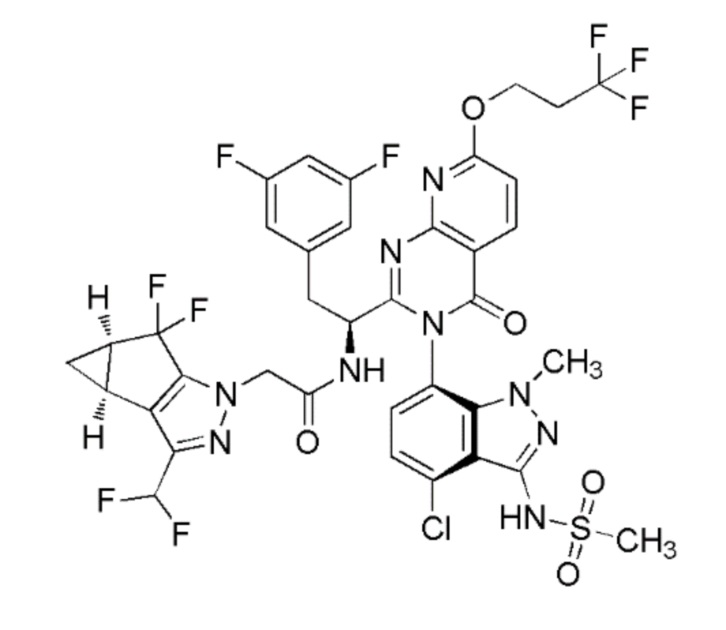
Схема синтеза:
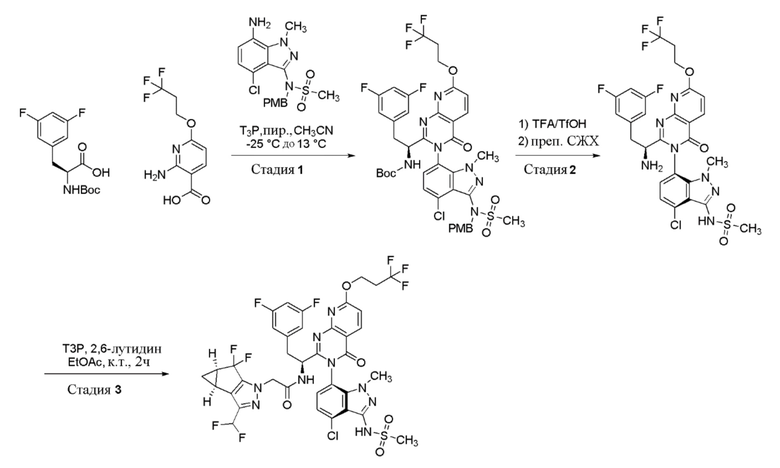
Стадия 1: Получение трет-бутил (S)-(1-(3-(4-хлор-3-(N-(4-метоксибензил)метилсульфонамидо)-1-метил-1H-индазол-7-ил)-4-оксо-7-(3,3,3-трифторпропокси)-3,4-дигидропиридо[2,3-d]пиримидин-2-ил)-2-(3,5-дифторфенил)этил)карбамата
К перемешиваемому раствору (S)-2-((трет-бутоксикарбонил)амино)-3-(3,5-дифторфенил)пропановой кислоты (62,3 г, 207 ммоль) и 2-амино-6-(3,3,3-трифторпропокси)никотиновой кислоты (55 г, 207 ммоль) в ацетонитриле (600 мл) в атмосфере азота при -25°C добавляли пиридин (41,8 мл, 517 ммоль). К полученной смеси добавляли по каплям в течение 15 мин 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (“T3P”, 50% масс. в EtOAc, 609 мл, 1033 ммоль). Раствор перемешивали при -25°C в течение 1 часа, затем давали медленно нагреться до 13°C и перемешивали в течение 5 часов. К раствору при 13°C добавляли N-(7-амино-4-хлор-1-метил-1H-индазол-3-ил)-N-(4-метоксибензил)метансульфонамид (82 г, 207 ммоль). Реакционной массе затем давали медленно нагреться до 27°C и затем перемешивали при этой температуре в течение 16 часов. За ходом реакции наблюдали при помощи ТСХ (SiO2, 40% EtOAc/Петрол. Rf=0,4). По завершении реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в EtOAc (500 мл) и затем промывали водн. раствором лимонной кислоты (0,5M, 2 × 500 мл), далее водн. раствором NaOH (1N, 3 × 500 мл). Органический слой сушили над Na2SO4, фильтровали и затем концентрировали при пониженном давлении с получением неочищенного продукта (180 г), который очищали хроматографией на силикагеле, элюируя 40-50% EtOAc в петрол. Фракции, содержащие желаемый продукт, объединяли и концентрировали при пониженном давлении с получением трет-бутил (S)-(1-(3-(4-хлор-3-(N-(4-метоксибензил)метилсульфонамидо)-1-метил-1H-индазол-7-ил)-4-оксо-7-(3,3,3-трифторпропокси)-3,4-дигидропиридо[2,3-d]пиримидин-2-ил)-2-(3,5-дифторфенил)этил)карбамата в виде не совсем белого твердого вещества (85 г, 39% выход). Продукт представлял собой смесь гомохиральных атропоизомеров (диастереомеров). Метод ЖХМС: Колонка=Acquity BEH C18 (50 мм × 2,1 мм, 1,7 мкм); Подвижная фаза A=0,05% Муравьиной кислоты в воде; Подвижная фаза B=0,05% Муравьиной кислоты в ацетонитриле; Градиент=Время (мин)/%B: 0/3, 0,4/3, 2,5/98, 3,4/98, 3,5/3, 4/3; Темп. колонки.=35°C; Скорость потока=0,6 мл/мин. Результат ЖХМС: время удерживания=2,46 мин; наблюдаемый ион=892,53 (M+H); Чистота ЖХМС=85%.
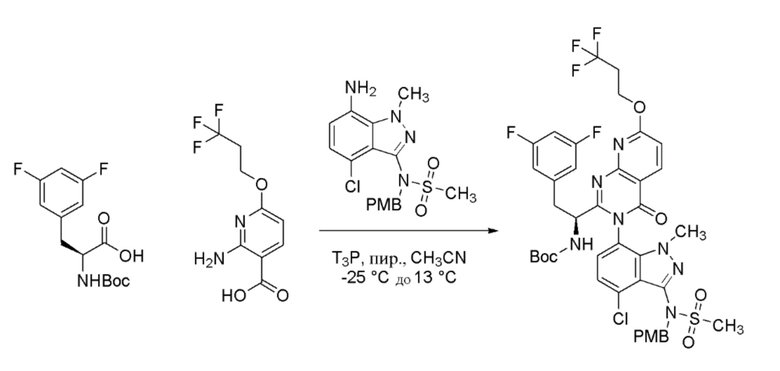
Стадия 2: Получение (S)-N-((6P)-7-((3P)-2-(1-амино-2-(3,5-дифторфенил)этил)-4-оксо-7-(3,3,3-трифторпропокси)пиридо[2,3-d]пиримидин-3(4H)-ил)-4-хлор-1-метил-1H-индазол-3-ил)метансульфонамида
К перемешиваемому раствору трет-бутил (S)-(1-(3-(4-хлор-3-(N-(4-метоксибензил)метилсульфонамидо)-1-метил-1H-индазол-7-ил)-4-оксо-7-(3,3,3-трифторпропокси)-3,4-дигидропиридо[2,3-d]пиримидин-2-ил)-2-(3,5-дифторфенил)этил)карбамата (85 г, 95 ммоль) в DCM (300 мл) при 0°C добавляли трифторуксусную кислоту (TFA, 294 мл, 3810 ммоль) с последующим добавлением трифторметансульфокислоты (25,4 мл, 286 ммоль). Раствору давали нагреться до 27°C и перемешивали в течение 1 часа в атмосфере азота. За ходом реакции наблюдали при помощи ТСХ (SiO2, 80% EtOAc/Петрол. Rf=0,3). По завершении летучие вещества удаляли под слабым потоком газообразного азота. Остаток растворяли в EtOAc (1000 мл) и промывали 2N раствором NaOH (2 × 500 мл) и затем насыщенным солевым раствором (500 мл). Органический слой сушили над Na2SO4, фильтровали и затем концентрировали при пониженном давлении с получением неочищенного продукта, который очищали хроматографией на силикагеле, элюируя 50-99% EtOAc в петрол. Фракции, содержащие желаемый продукт, объединяли и концентрировали при пониженном давлении с получением (S)-N-(7-(2-(1-амино-2-(3,5-дифторфенил)этил)-4-оксо-7-(3,3,3-трифторпропокси)пиридо[2,3-d]пиримидин-3(4H)-ил)-4-хлор-1-метил-1H-индазол-3-ил)метансульфонамида в виде бледно-желтого твердого вещества (63 г). Вещество представляло собой смесь гомохиральных атропоизомеров (диастереомеров) в соотношении 64:26, как определяли при помощи ЖХМС. Этот продукт смешивали с тремя дополнительными партиями продуктов, полученных с использованием той же процедуры. Объединенный продукт (195 г) растворяли в смеси метанол:ацетонитрил (80:20, 1300 мл) и этот раствор очищали препаративной СФХ с использованием следующего метода: Колонка=(R, R) Whelk-01 (250 × 30 × 5мкм); элюент=CO2:MeOH (65:35); Скорость потока=90 г/мин; Обратное давление=120 бар; Детекция=214 нм (УФ); Время накопления=14 мин; Нагрузка на инжекцию=430 мг. Чистый главный пик собирали и концентрировали при пониженном давлении с получением единственного стереоизомера (S)-N-((6P)-7-((3P)-2-(1-амино-2-(3,5-дифторфенил)этил)-4-оксо-7-(3,3,3-трифторпропокси)пиридо[2,3-d]пиримидин-3(4H)-ил)-4-хлор-1-метил-1H-индазол-3-ил)метансульфонамида в виде не совсем белого твердого вещества (113 г, 63% выход). 1H ЯМР (400 МГц, DMSO-d6) δ: 8,42 (д, J=8,8 Гц, 1H), 7,43 (д, J=8,1 Гц, 1H), 7,37 (д, J=8,1 Гц, 1H), 7,06 (д, J=8,8 Гц, 1H) 7,03-6,97 (м, 1H), 6,75-6,70 (м, 2H), 4,73-4,69 (м, 2H), 3,68 (с, 3H), 3,58-3,52 (м, 1H), 3,28-3,24 (м, 1H), 3,22 (с, 3H), 2,97-2,83 (м, 3H). Метод ЖХМС: Колонка=XBridge C18 (75 мм × 4,6 мм, 3,5 мкм); Подвижная фаза A=5 мМ бикарбоната аммония в воде; Подвижная фаза B= ацетонитрил; Градиент=Время (мин)/%B: 0/5, 0,5/5, 1,0/15, 4,0/98, 7,0/98, 7,5/5, 8,0/5; Темп. колонки=35°C; Скорость потока=1,3 мл/мин. Результат ЖХМС: время удерживания=4,03 мин; наблюдаемый ион=672,07 (M+H); Чистота ЖХМС=98%; ВЭЖХ Чистота=98%; Чистота по данным хиральной ВЭЖХ=98%.
Стадия 3: Получение N-((S)-1-((3P)-3-(4-хлор-1-метил-3-(метилсульфонамидо)-1H-индазол-7-ил)-4-оксо-7-(3,3,3-трифторпропокси)-3,4-дигидропиридо[2,3-d]пиримидин-2-ил)-2-(3,5-дифторфенил)этил)-2-((3bS,4aR)-3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетамида
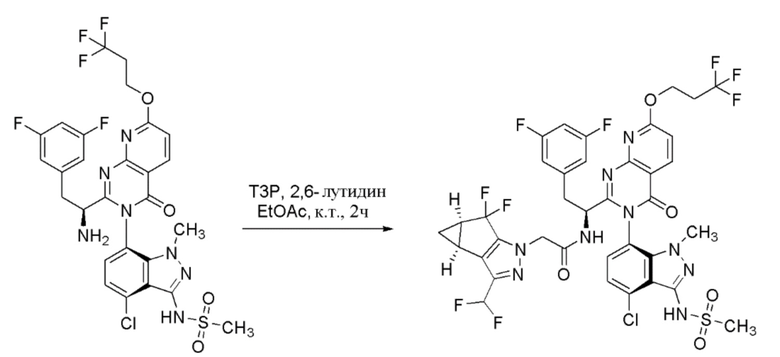
К перемешиваемому раствору (S)-N-((6P)-7-((3P)-2-(1-амино-2-(3,5-дифторфенил)этил)-4-оксо-7-(3,3,3-трифторпропокси)пиридо[2,3-d]пиримидин-3(4H)-ил)-4-хлор-1-метил-1H-индазол-3-ил)метансульфонамида (55 г, 82 ммоль) и 2-((3bS,4aR)-3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)уксусной кислоты (22,70 г, 86 ммоль) в этилацетате (818 мл) добавляли 2,6-лутидин (23,83 мл, 205 ммоль). К смеси добавляли по каплям 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинан 2,4,6-триоксид (“T3P”, 50% масс. в этилацетате) (97 мл, 164 ммоль), после чего внутренняя температура повышалась с 17°C до 24°C. Смесь перемешивали в течение 2 часов при комнатной температуре. Реакцию гасили добавлением воды (500 мл). Фазы разделяли и органическую фазу промывали водой (500 мл), затем сушили над Na2SO4. Смесь фильтровали и фильтрат концентрировали до 1/4 начального объема с получением неочищенного продукта в виде раствора в этилацетате.
Вторую партию продукта получали, следуя той же процедуре, модифицированной следующим образом: реакцию осуществляли с использованием (S)-N-((6P)-7-((3P)-2-(1-амино-2-(3,5-дифторфенил)этил)-4-оксо-7-(3,3,3-трифторпропокси)пиридо[2,3-d]пиримидин-3(4H)-ил)-4-хлор-1-метил-1H-индазол-3-ил)метансульфонамида (53,4 г, 79 ммоль), и количества всех других реагентов были соответственно скорректированы. Обработка завершалась промывкой насыщенным солевым раствором (300 мл) перед сушкой над MgSO4.
Неочищенные продукты объединяли и затем адсорбировали на целите. Полученный порошок подвергали хроматографии на силикагеле (3 кг колонка RediSep Gold), элюируя раствором 30-85% этилацетата в гексане. Фракции, содержащие желаемый продукт, объединяли и концентрировали при пониженном давлении с получением желтой пены. Вещество помещали в глубокий вакуум на 18 часов. Вещество превращали в тонкодисперсный порошок при помощи ступки и пестика и твердые вещества помещали в вакуумную печь при 50°C в течение 48 часов с получением N-((S)-1-((3P)-3-(4-хлор-1-метил-3-(метилсульфонамидо)-1H-индазол-7-ил)-4-оксо-7-(3,3,3-трифторпропокси)-3,4-дигидропиридо[2,3-d]пиримидин-2-ил)-2-(3,5-дифторфенил)этил)-2-((3bS,4aR)-3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетамида в виде желтого порошка (134,1 г, 90% выход). 1H ЯМР (500 МГц, DMSO-d6) δ ppm 9,84-9,91 (1 H, м) 9,49 (1 H, д, J=8,34 Гц) 8,47 (1 H, д, J=8,35 Гц) 7,79 (1 H, д, J=7,75 Гц) 7,49 (1 H, д, J=8,05 Гц) 7,11 (1 H, д, J=8,64 Гц) 6,80-7,09 (2 H, м) 6,66 (2 H, дд, J=8,20, 2,24 Гц) 4,69-4,75 (3 H, м) 4,57 (1 H, д, J=16,39 Гц) 4,48 (1 H, ддд, J=11,03, 8,35, 2,68 Гц) 3,51 (3 H, с) 3,42 (1 H, дд, J=14,01, 2,38 Гц) 3,20 (3 H, с) 3,05 (1 H, дд, J=14,01, 11,03 Гц) 2,89-2,99 (2 H, м) 2,42-2,48 (2 H, м) 1,32-1,40 (1 H, м) 0,81-0,86 (1 H, м). Метод ЖХМС: Колонка=Acquity UPLC BEH C18 (2,1×100 мм, частицы 1,7 мкм); Растворитель A=Вода:MeCN (95:5) с 0,1% об/об Муравьиной кислоты; Растворитель B=MeCN:Вода (95:5) с 0,1% об/об Муравьиной кислоты; Градиент=Время (мин)/%B: 0/0, 3,5/100, 4,5/100; Скорость потока=0,8 мл/мин. Результат ЖХМС: время удерживания=3,173 мин; наблюдаемая масса=917,95 (M+H). Чистота УВЭЖХ=99,8%.
Название Примера 1:
Соединение Примера 1, полученное выше, представляет собой гомохиральное вещество, обладающее осевой хиральностью. Осевая хиральность может быть описана с использованием номенклатуры P/M, как подробно описано в Золотой книге IUPAC (doi:10.1351/goldbook.A00547). Однако в настоящее время доступно лишь ограниченное количество программных инструментов, которые способны генерировать химические названия, содержащие номенклатуру P/M, и еще меньше вариантов доступно для преобразования названий химических веществ с использованием этой номенклатуры в структурные представления молекул. Поэтому для ясности и удобства ниже приведены несколько названий Примера 1:
Название соединения Примера 1, сгенерированное ChemDraw Ultra 12 (без номенклатуры P/M), представляет собой:
N-((S)-1-(3-(4-хлор-1-метил-3-(метилсульфонамидо)-1H-индазол-7-ил)-4-оксо-7-(3,3,3-трифторпропокси)-3,4-дигидропиридо[2,3-d]пиримидин-2-ил)-2-(3,5-дифторфенил)этил)-2-((3bS,4aR)-3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетамид
Химическое название соединения Примера 1, сгенерированное JChem для Excel (включая номенклатуру P/M), представляет собой:
N-[(1S)-1-[(3P,3P)-3-(4-хлор-3-метансульфонамидо-1-метил-1H-индазол-7-ил)-4-оксо-7-(3,3,3-трифторпропокси)-3H,4H-пиридо[2,3-d]пиримидин-2-ил]-2-(3,5-дифторфенил)этил]-2-[(2S,4R)-9-(дифторметил)-5,5-дифтор-7,8-диазатрицикло[4,3,0,0²,4]нона-1(6),8-диен-7-ил]ацетамид
Химическое название соединения, сгенерированное ChemDraw Ultra 12 с добавленной вручную номенклатурой P/M, представляет собой:
N-((S)-1-((3P)-3-(4-хлор-1-метил-3-(метилсульфонамидо)-1H-индазол-7-ил)-4-оксо-7-(3,3,3-трифторпропокси)-3,4-дигидропиридо[2,3-d]пиримидин-2-ил)-2-(3,5-дифторфенил)этил)-2-((3bS,4aR)-3-(дифторметил)-5,5-дифтор-3b,4,4a,5-тетрагидро-1H-циклопропа[3,4]циклопента[1,2-c]пиразол-1-ил)ацетамид.
Сравнительные испытания:
Соединение Примера 1 сравнивали с соединением Примера 60.2, описанным в WO2018203235 (Схема 1), в ряде испытаний. Для целей этих сравнений авторы изобретения решили использовать гомохиральное вещество каждого соединения, так как этот уровень чистоты является наиболее репрезентативным для того, что будет использоваться в клинических испытаниях на людях. В частности, ограниченное вращение вокруг указанной C-N связи индазола дает атропоизомеры (диастереомеры) как в Примере 1, так и в Примере 60.2, которые можно разделить хроматографией и которые не являются взаимопревращающимися при комнатной температуре. Таким образом, при помощи хроматографии авторы выделили в чистом виде стереоизомеры, изображенные на Схеме 2.
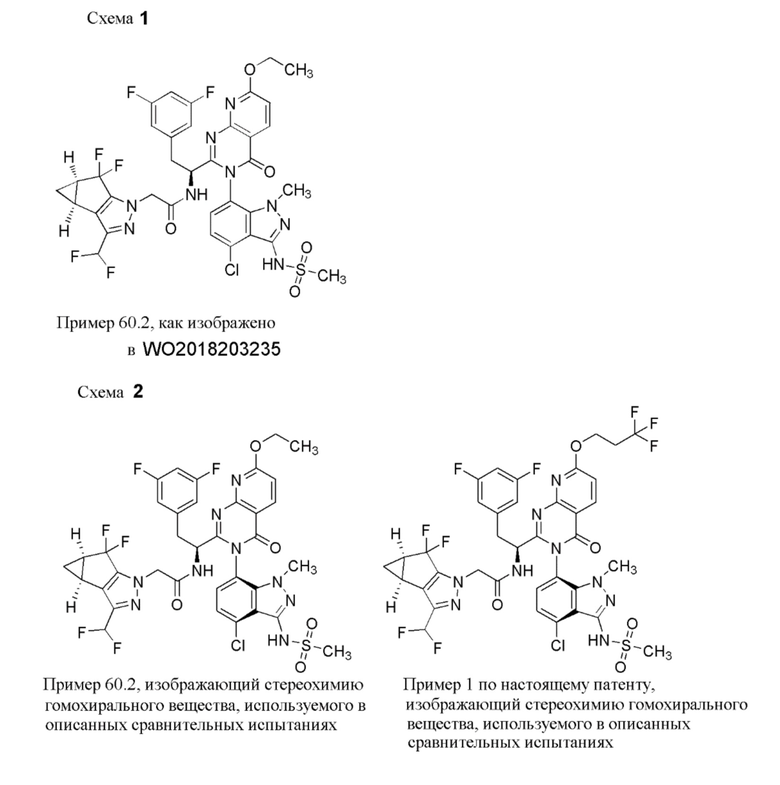
Общая процедура количественного определения соединения при помощи ЖХ-МС/МС:
Все образцы in vitro вводили в ЖХ-МС/МС систему Exion LC 4500 Triple QuadTM. В качестве аналитической колонки использовали Phenomenex C18 (C18, 4,6 мм × 50 мм, 5 мкм), поддерживаемую при комнатной температуре. Подвижная фаза А состояла из 0,1% (об/об) муравьиной кислоты в воде MilliQ. Подвижная фаза B состояла из 100% метанола. Скорость потока составляла 1 мл/мин. Градиент был следующим: подвижную фазу B линейно увеличивали с 5% до 90% за 0,7 мин, поддерживали при 90% в течение 1,4 мин и поддерживали при 5% в течение 0,7 мин.
Все образцы in vivo вводили в ЖХ-МС/МС систему Triple QuadTM 6500 с колонками, поддерживаемыми при 60°C. Подвижная фаза A состояла из H2O, 1 мМ NH4OAc, 0,025% муравьиной кислоты. Подвижная фаза B состояла из MeOH, 5 мМ NH4OAc. Скорость потока составляла 0,6 мл/мин. Колонка и градиент элюирования были выбраны из одного из общих методов анализа, описанных ниже.
Общий метод анализа A:
Колонка=Waters X-Bridge BEH C18 (2,1×50 мм, частицы 1,7 мкм); Градиент: Время(мин)/%B=0,0/10, 0,2/10, 0,8/90, 1,3/90, 1,31/10, 2,0/10.
Общий метод анализа B:
Колонка=Waters BEH C18 (2,1×50 мм, частицы 2,5 мкм); Градиент: Время(мин)/%B=0,0/10, 0,2/10, 0,8/90, 1,3/90, 1,31/10, 2,0/10.
Общий метод анализа C:
Колонка=Waters BEH C18 (2,1×50 мм, частицы 1,7 мкм); Градиент: Время(мин)/%B=0,0/2, 0,40/2, 0,7/65, 1,3/90, 1,9/90, 1,91/2, 2,5/2.
Процедура измерения активности и цитотоксичности:
Клетки МТ-2, клетки 293Т и клон провирусной ДНК вируса NL4-3 получали от NIH AIDS Research and Reference Reagent Program. Клетки МТ-2 размножали в среде RPMI 1640 с добавлением 10% термоинактивированной фетальной бычьей сыворотки (FBS), 100 мг/мл пенициллина G и до 100 единиц/мл стрептомицина. Клетки 293T размножали в среде DMEM, дополненной 10% термоинактивированной FBS, 100 мг/мл пенициллина G и 100 мг/мл стрептомицина. Рекомбинантный провирусный клон NL4-3, в котором часть гена nef была заменена геном люциферазы Renilla, использовали для создания эталонного вируса, используемого в этих исследованиях. Рекомбинантный вирус получали путем трансфекции рекомбинантного провирусного клона NL4-3 в клетки 293T с использованием реагента для трансфекции Transit-293 от Mirus Bio LLC (Madison, WI). Супернатант собирали через 2-3 дня, и определляли титры вируса, присутствующего в клетках МТ-2, используя активность фермента люциферазы в качестве маркера путем измерения активности фермента люциферазы. Люциферазу определяли количественно с использованием живого субстрата клеток EnduRen от Promega (Madison, WI). Осуществляли количественное определение противовирусной активности соединений в отношении рекомбинантного вируса путем измерения активности люциферазы в клетках МТ-2, инфицированных в течение 4-5 дней рекомбинантным вирусом, в присутствии серийных разведений соединения.
50% Эффективную концентрацию (EC50) рассчитывали с использованием экспоненциальной формы уравнения медианного эффекта, где (Fa)=1/[1+ (ED50/конц. лекарственного средства)m] (Johnson VA, Byington RT. Infectivity Assay. In Techniques in HIV Research. ed. Aldovini A, Walker BD. 71-76. New York: Stockton Press.1990). 50% Ингибирующую концентрацию (EC50) рассчитывали с использованием экспоненциальной формы уравнения медианного эффекта, где процент ингибирования=1/[1 + (EC50/концентрация лекарственного средства)m], где m представляет собой параметр, отражающий наклон кривой концентрация-ответ.
Цитотоксичность соединения и соответствующие значения CC50 определяли с использованием того же протокола, что и для противовирусного анализа, за исключением того, что использовали неинфицированные клетки. Цитотоксичность оценивали в день 4 в неинфицированных клетках MT2 с использованием колориметрического анализа на основе XTT (2,3-бис[2-Метокси-4-нитро-5-сульфофенил]-2H-тетразолий-5-карбоксианилидная внутренняя соль) (Sigma-Aldrich, St Louis, Mo).
Результаты:
Активности соединений Примера 1 и Примера 60.2 находятся в пределах погрешности первоначального анти-ВИЧ вирусологического анализа (EC50 Примера 1=25 ±8 пМ, EC50 Примера 60.2= 18 ±13 пМ). Следует отметить, что EC50 Примера 1 составляла 0,034 нМ при первоначальном испытании, однако, дальнейшие испытания привели к пересмотренному среднему значению 25 ±8 пМ. Измеренная цитотоксичность CC50 составляла >0,5 мкМ и >10 мкМ для Примера 1 и Примера 60.2, соответственно.
Процедура измерения метаболизма в микросомах печени:
Микросомы печени человека, крысы, собаки и обезьяны размораживали и разбавляли до конечной концентрации 1 мг/мл в 100 мМ калий-фосфатном буфере (pH 7,4). Тестируемые соединения и контроли получали при 100× конечной концентрации 1 мкМ в смеси ацетонитрил:вода 1:1 (об/об) и вносили аликвоты в микросомальную смесь. Смесь предварительно инкубировали при 37°C на водяной бане со встряхиванием в течение 10 минут. Инкубации осуществляли в двух повторах. В инкубации включали три контроля; варфарин, фенацетин и верапамил. После предварительной инкубации реакцию инициировали при помощи NADPH в конечной концентрации 1 мМ. Через 0, 5, 15, 30, 45 и 60 минут извлекали 25 мкл образца и гасили 300 мкл ацетонитрила, содержащего внутренний стандарт (телмисартан). Образцы встряхивали в течение 5 минут при 1200 об/мин и затем центрифугировали при 4000 об/мин в течение 10 мин. 100 мкл аликвоту супернатанта разбавляли трехкратно водой и вводили в систему ЖХ-МС/МС Exion LC 4500 Triple Quad. Результаты представляли как процент оставшегося исходного соединения и рассчитывали из соотношений площадей пиков тестируемого соединения, остающегося после в каждый момент времени, и сравнивали с инкубацией в момент времени 0.
Результаты:
Соединение Примера 1 в шесть раз более стабильно в микросомах печени собаки по сравнению с соединением Примера 60.2, и соединение Примера 1 по меньшей мере в два раза более стабильно в микросомах печени обезьяны по сравнению с соединением Примера 60.2. Эти данные предполагают, что соединение Примера 1 должно быть значительно более стабильным, чем соединение Примера 60.2 к метаболизму in vivo у собак и обезьян.
Процедура измерения метаболизма в гепатоцитах человека:
Криоконсервированные гепатоциты в суспензии от человека, обезьяны, собаки, крысы и мыши размораживали и разводили в предварительно нагретой средой Вильямса E (pH 7,4). Аликвоты суспензии гепатоцитов добавляли к рабочим растворам тестируемых соединений, полученных в предварительно нагретой среде Вильямса E (pH 7,4), для достижения конечных концентраций 0,5 мкМ в 0,5 × 106 клеток на миллилитр и ≤0,25% DMSO. Эти образцы инкубировали при 37°C в присутствии 5% диоксида углерода и встряхивали при 200 об/мин. Инкубации осуществляли однократно. В моменты времени 0, 10, 30, 60, 120 и 240 минут извлекали 50 мкл аликвоту инкубационных смесей и добавляли к 100 мкл раствора ацетонитрила, содержащего внутренний стандарт, и смесь встряхивали и затем центрифугировали при 4°C и 3500 об/мин в течение 15 минут. После завершения эксперимента образцы анализировали при помощи ЖХ-МС/МС. Результаты метаболической стабильности были представлены как процент оставшегося исходного тестируемого соединения. Этот процент рассчитывали путем деления отношения площадей пиков тестируемых соединений после инкубации (tx) на отношение площадей пиков тестируемых соединений в момент времени 0 (t0) непосредственно перед инкубацией.
Константу скорости элиминации (k, мин-1) рассчитывали методом нелинейной регрессии с использованием следующего уравнения:
Ct=C0 × e (−k × t)
где:
C0 - начальная концентрация, представленная как отношение площадей пиков (площадь пика тестируемого соединения/площадь пика внутреннего стандарта);
Ct - концентрация в момент времени t, представленная как отношение площадей (площадь пика тестируемого соединения/площадь пика внутреннего стандарта);
e - основание натурального логарифма
t - время (мин);
k - константа скорости элиминации (мин−1).
Период полувыведения (t1/2, мин) рассчитывали с использованием следующего уравнения:
где:
k - константа скорости элиминации (мин−1).
Внутренний клиренс in vitro (CLint, мл/мин/миллион клеток) рассчитывали с использованием следующего уравнения:
CLint=0,693/t1/2/n
где:
t1/2 - период полувыведения;
n - количество клеток на мл.
Результаты:
Период полувыведения соединения Примера 1 в гепатоцитах человека по расчетам составлял >480 минут, тогда как период полувыведения соединения Примера 60.2 в гепатоцитах человека по расчетам составлял 350 минут. Внутренний клиренс соединения Примера 60.2 в гепатоцитах человека составлял 0,465 мл/мин/г печени, что в 1,5 раза быстрее, чем клиренс 0,312 мл/мин/г печени, определенный для Примера 1.
Процедура измерения фармакокинетических (PK) параметров in vivo:
PK исследовали на самцах мышей CD1, крысах Wistar Han, обезьянах Cynomolgus и собаках породы бигль. Две группы животных (N=3 на группу) получали тестируемое соединение либо в виде внутривенной (в/в) дозы (1 мг/кг), либо через желудочный зонд (5 мг/кг раствора и суспензии). Лекарственное средство формулировали в 90% PEG 400, 10% этанола для в/в введения и в 90% PEG400, 5% этанола для перорального (п/о) введения. Образцы крови собирали через 0,167, 0,25, 0,5, 0,75, 1, 2, 3, 5, 7, 24, 48, 72 и 96 ч после в/в введения; 0,25, 0,5, 0,75, 1, 2, 3, 5, 7, 24, 48, 72 и 96 часов после перорального введения. Образцы крови собирали в пробирки с K3EDTA и центрифугировали при 1500-2000 × g для получения плазмы. Образцы плазмы хранили при -20°C до ЖХ-МС/МС анализа.
Все образцы in vivo вводили в систему ЖХ-МС/МС Exion LC 4500 Triple QuadTM, где колонку поддерживали при 60°C, и скорость потока составляла 0,6 мл/мин. Все параметры анализа ЖХ-МС/МС фиксировали в электронном виде в файлах исходных данных. РК образцы Крысы в/в, Собаки в/в, Обезьяны в/в и Обезьяны п/о анализировали общим методом анализа A. РК образцы Мыши в/в, Мыши п/о и Собаки п/о анализировали общим методом анализа B. Образцы Крысы п/о анализировали общим методом анализа C.
PK параметры получены с использованием некомпартментального анализа данных концентрация vs время (Phoenix WinNonlin v8.1). Пиковая концентрация (Cmax) и время для Cmax (Tmax) регистрировали непосредственно из экспериментальных наблюдений. Площадь под кривой от нулевого времени до последнего времени отбора проб [AUC0-T] и площадь под кривой от нулевого времени до бесконечности [AUCINF] рассчитывали с использованием линейно-логарифмического правила трапеций. Общий плазматический клиренс (CLTp), стационарный объем распределения (Vss), кажущийся период полувыведения (T-HALF) и среднее время удержания (MRT) оценивали после в/в введения. Оценки AUC и T-HALF осуществляли с использованием как минимум трех временных точек с поддающимися количественному определению концентрациями. Абсолютную пероральную биодоступность (F) оценивали как отношение значений AUC, нормализованных по дозе, после пероральной и внутривенной доз.
Результаты:
Фармакокинетические (PK) параметры в/в введения соединения Примера 1 и соединения Примера 60.2 измеряли у четырех доклинических видов: мыши, крысы, собаки и обезьяны. Пример 1 показал улучшенный клиренс у всех четырех видов по сравнению с Примером 60.2. Согласно результатам анализа в микросомах печени, описанного выше, различия в клиренсе были наиболее значительными для собак и обезьян, где клиренс был улучшен в 4,9 и 2,6 раза, соответственно. Аналогичным образом, период полужизни соединения Примера 1 в кровотоке у собак и обезьян был в 3,4 раза и в два раза выше, чем у соединения Примера 60.2, соответственно.
Прежде чем предполагаемое лекарственное средство может быть представлено для клинических испытаний на людях, безопасность соединения, как правило, должна быть оценена на двух доклинических видах: на одном грызуне и одном не-грызуне. Обычно это крысы, собаки или обезьяны. Одной из целей исследования безопасности in vivo является достижение концентраций лекарственного средства в кровотоке, которые во много раз превышают те, которые можно было бы ожидать, если бы человек получил эффективную дозу лекарственного средства. Кратная разница между концентрациями лекарственного средства, достигнутыми в исследовании безопасности, и концентрациями лекарственного средства, которые можно было бы ожидать у человека, принимающего эффективную дозу лекарственного средства, называют “резервом” безопасности. Достижение высоких резервов в исследовании безопасности важно, потому что с увеличением резервов растет и уверенность в том, что, если бы нежелательное событие, связанное с приемом лекарственных средств, было возможным, оно бы наблюдалось во время доклинической оценки безопасности.
Улучшенные PK параметры у крысы, собаки или обезьяны означают, что для достижения высокой концентрации лекарственного средства в кровотоке для этих доклинических видов потребуется более низкая доза соединения. Следовательно, обезьяна или собака, получившая дозу соединения Примера 1, сможет достичь более высоких резервов, чем при введении такой же дозы соединения Примера 60.2. Из-за практических ограничений размера дозы резерв (и, следовательно, достоверность), который может быть достигнут с использованием соединения Примера 1 в исследовании для оценки безопасности у не-грызунов, выше, чем резерв, который может быть достигнут с использованием соединения Примера 60.2.
Процедура аллометрического масштабирования доклинических PK параметров для прогнозирования дозы для человека:
Прогнозирование дозы для человека осуществляли с использованием программного обеспечения Phoenix WinNonlin (v 8.0) и Microsoft Excel с использованием расширения ModelRisk для моделирования популяции. Оценки CLTp для каждого из соединений для человека получали на основе среднего аллометрического масштабирования в/в данных мыши, крысы, обезьяны и собаки (коэффициент масштабирования массы тела 0,75) и среднего значения (коэффициент масштабирования массы тела 1,0) от всех видов для Vss. В/в параметры у человека (Vc, Ka, K12, K21, Kel) определяли с использованием масштабирования среднего времени удержания (MRT) для доклинических видов (мышь, крыса, собака и обезьяна) с использованием Vss и CLTp оценок у человека. Абсорбцию (Ka) определяли путем деконволюции (п/о) или из периода полувыведения (п/к) у доклинических видов (Ka=LN(2)/t1/2). Прогнозируемую п/о и п/к дозу для человека рассчитывали с учетом вариабельности между людьми и вычисляли для охвата 95% популяции людей.
Результаты:
PK параметры у доклинических видов обычно используют для прогнозирования PK параметров у человека до клинических испытаний на людях. Способы, используемые для этого прогнозирования, называют “аллометрическим масштабированием”, и они обычно обсуждаются и используются в литературе. Используя аллометрическое масштабирование, прогнозируемая пероральная доза один раз в сутки, необходимая для поддержания эффективной концентрации лекарственного средства в плазме у человека, в 7 раз ниже для Примера 1, чем для Примера 60.2. В частности, прогнозируемая для человека QD п/о доза Примера 1 составляет менее 10 мг, в то время как прогнозируемая для человека QD п/о доза Примера 60.2 составляет более 30 мг.
В то время как идиосинкратические реакции на лекарственные средства (т.е. аллергические реакции) непредсказуемы и серьезны по своей природе и, таким образом, представляют собой значительную клиническую проблему, было заявлено, что лекарственные средства, назначаемые в суточной дозе 10 мг или меньше, редко связаны, если вообще связаны, с высокой частотой идиосинкратических реакций на лекарственные средства (Uetrecht, J. P. New Concepts in Immunology Relevant to Idiosyncratic Drug Reactions: The “Danger Hypothesis” and Innate Immune System. Chem. Res. Toxicol. 1999, 12(5), 387-395, DOI:10.1021/tx980249i).
Процедура измерения фармакокинетических параметров в эксперименте in vivo с подкожным введением:
Лекарственное средство формулировали в смеси 1% Kolliphor P188/1% PEG3350/3,5% маннит/94,5% вода и затем вводили крысам Wistar Han в виде подкожной инъекции в дозе 20 мг/кг. Образцы крови собирали через 0,167, 0,25, 0,5, 0,75, 1, 2, 3, 5, 7, 24, 48, 72, 96 часов и затем каждые 3 дня в течение до 122 дней. Образцы крови собирали в пробирки с K3EDTA и центрифугировали при 1500-2000 × g для получения плазмы. Образцы плазмы хранили при -20°C до ЖХ-МС/МС анализа.
Результаты:
Приемлемость каждого соединения для подкожного (п/к) введения оценивали в п/к PK эксперименте на крысах. Как определено в этом эксперименте, кажущийся период полужизни соединения в плазме составлял 13 дней для Примера 1 и 11,5 дней для Примера 60.2. Значение AUC0-бесконечность для соединения Примера 1 составляло 4,941 дней*нг/мл (2,89% экстраполированного значения AUC). Значение AUC0-бесконечность для соединения Примера 60.2 составляло 609 дней*нг/мл (12,8% экстраполированного значения AUC). Биодоступность составила 93% для Примера 1 и 25% для Примера 60.2. Концентрации лекарственных средств поддерживали выше 7 нг/мл для всех животных в течение 73 дней для Примера 1 и в течение 24 дней для Примера 60.2 (Фиг. 1). Прогнозируемые подкожные дозы один раз в месяц (Q1M п/к) для человека рассчитывали с использованием кажущихся периодов полужизни и биодоступности, полученных из PK крысы п/к в сочетании с прогнозируемыми значениями клиренса для человека, полученными методом аллометрического масштабирования. Прогнозируемая доза Q1M п/к, необходимая для поддержания эффективной концентрации лекарственного средства в плазме у человека, в 15 раз ниже для Примера 1, чем для Примера 60.2.
Процедура измерения индукции цитохрома P450 в криоконсервированных гепатоцитах человека:
В соответствии с руководством FDA (“In Vitro Metabolism- and Transporter- Mediated Drug-Drug Interaction Studies Guidance for Industry”), способность соединения Примера 1 и соединения Примера 60.2 индуцировать экспрессию CYP2B6 испытывали с использованием гепатоцитов от тех же трех отдельных доноров (а не объединенных доноров), и изменения уровней мРНК фермента оценивали с использованием “метода кратных изменений”. В этом испытании кратность изменения в уровнях мРНК менее чем в 2 раза считается отрицательным результатом, а изменение в ≥2 раза считается положительным результатом.
Соединения испытывали в анализе индукции CYP с использованием индуцибельных криоконсервированных первичных гепатоцитов человека от трех доноров для определения способности вызывать индукцию (измеряемую по увеличению транскрипции мРНК) CYP2B6. Тестируемые соединения (конечная концентрация от 0,12 до 30 мкМ) инкубировали в течение 48 часов с первичными гепатоцитами человека, естественным образом экспрессирующими все ядерные рецепторы, участвующие в регуляции уровней экспрессии различных ферментов CYP. Свежие растворы тестируемых соединений и контролей разбавляли в среде для количественного определения и добавляли каждые 24 часа в течение двух последовательных дней до конечной концентрации DMSO 0,1%. По окончании инкубации оценивали целостность клеточных монослоев, плотность и жизнеспособность клеток для оценки эффектов цитотоксичности. Затем клетки солюбилизировали в буфере для лизиса клеток и из каждого образца очищали общую РНК. Затем образцы использовали в полимеразной цепной реакции с обратной транскрипцией (RT-PCR) для количественного определения специфических видов мРНК, кодирующих ген CYP2B6 человека.
Индукционный потенциал тестируемых соединений и контролей сравнивали с известным индуктором CYP2B6 фенобарбиталом (1000 мкМ). Результаты этого анализа выражены как кратность индукции. Кратность индукции рассчитывали как отношение уровня мРНК в клетках, обработанных тестируемым соединением, к уровню мРНК в клетках, обработанных только DMSO (контроль растворителем), базальному уровню мРНК, и, таким образом, представляет собой индукционный потенциал тестируемого соединения. Значения кратности индукции использовали для расчета значений процента активности от контроля, которые затем использовали в 4-параметрической модели логистической регрессии для определения значений EC50 и Emax (если наблюдалась индукция). Также параллельно оценивали цитотоксичность, чтобы избежать ложноположительных результатов индукции CYP из-за цитотоксичности. Следует избегать оценки и интерпретации потенциала индукции CYP при цитотоксических концентрациях.
Результаты:
Никакой цитотоксичности не наблюдали ни для одного соединения при любой из протестированных концентраций (до 30 мкМ). Для Примера 1 отрицательный результат (без индукции) был обнаружен у всех трех доноров. Для Примера 60.2 положительный результат (индукция) был обнаружен у 2 из 3 доноров (значения EC50 1,5 мкм и 1,8 мкм).
Индукция экспрессии фермента CYP признана первопричиной межлекарственных взаимодействий, приводящей к повышению клиренса лекарственного средства, являющегося объектом взаимодействия, метаболизм которого регулируется индуцированной изоформой CYP. Среди изоформ CYP CYP2B6 имеет особое значение в контексте лечения ВИЧ, поскольку эфавиренц (EFV), лекарственное средство, широко используемое для лечения ВИЧ (включено в список основных лекарственных средств Всемирной организации здравоохранения 2019 г.), в основном метаболизируется CYP2B6 (Ward, B. A., Gorski, J. C., Jones, D. R., Hall, S. D., Flockhard, D. A., Desta, Z. The Cytochrome P450 2B6 (CYP2B6) Is the Main Catalyst of Efavirenz Primary and Secondary Metabolism: Implication for HIV/AIDS Therapy and Utility of Efavirenz as a Substrate Marker of CYP2B6 Catalytic Activity, J. Pharmacol. Exp. Ther., 2003, 306, 287-300, DOI: 10.1124/jpet.103.049601.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ТРАНСГЛУТАМИНАЗЫ 2 (TG2) | 2019 |
|
RU2781370C2 |
| ИНГИБИТОРЫ РЕПЛИКАЦИИ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА | 2019 |
|
RU2812128C2 |
| СОЕДИНЕНИЯ | 2016 |
|
RU2725616C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛОПИРИДИНОНА В КАЧЕСТВЕ TTX-S БЛОКАТОРОВ | 2013 |
|
RU2646754C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ КАРБОНУКЛЕОЗИДА, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2017 |
|
RU2712944C1 |
| ТЕТРАЗАМЕЩЕННЫЕ АЛКЕНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2733741C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| ПИРРОЛОПИРАЗИНОВЫЕ ИНГИБИТОРЫ КИНАЗЫ | 2012 |
|
RU2673064C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
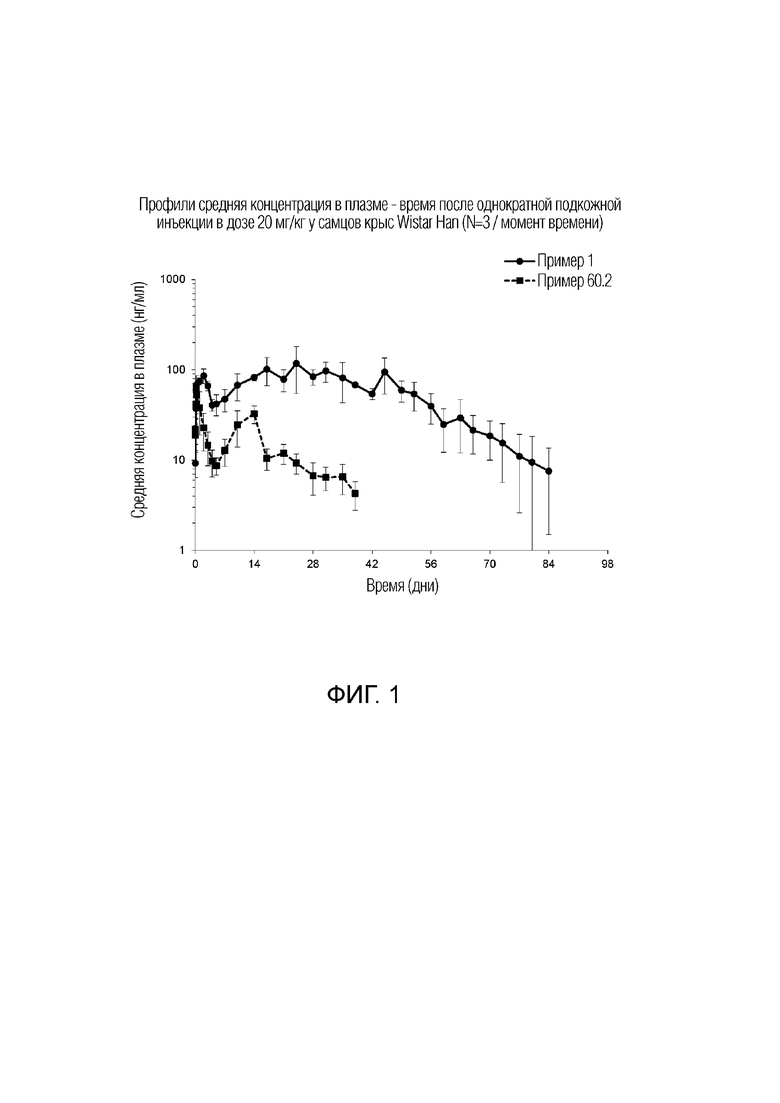
Группа изобретений относится к фармацевтической химии и включает соединение

а также фармацевтическую композицию на его основе, применение и способ лечения ВИЧ-инфекции. Технический результат - производное 3,4-дигидропиридо[2,3-d]пиримидинила, применяемое для лечения ВИЧ-инфекции, обладающее улучшенными фармакокинетическими параметрами. 5 н. и 3 з.п. ф-лы, 1 ил., 3 табл., 1 пр.
1. Соединение, которое представляет собой
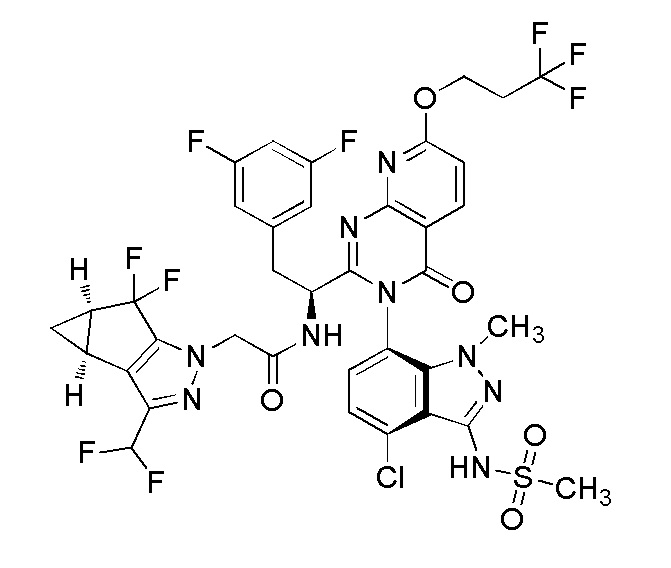 .
.
2. Фармацевтическая композиция для лечения ВИЧ инфекции, включающая эффективное количество соединения по п. 1, и дополнительно включающая фармацевтически приемлемый эксципиент.
3. Композиция по п. 2, подходящая для перорального введения, для внутримышечной инъекции или для подкожной инъекции.
4. Способ лечения ВИЧ-инфекции у человека, включающий введение соединения по п. 1.
5. Способ по п. 4, где указанное введение является пероральным.
6. Способ по п. 4, где указанное введение представляет собой внутримышечную инъекцию или подкожную инъекцию.
7. Применение соединения по п. 1 для лечения ВИЧ-инфекции.
8. Применение соединения по п. 1 в изготовлении лекарственного средства для лечения ВИЧ-инфекции.
| WO 2018203235 A1, 08.11.2018 | |||
| S | |||
| Swallow, Fluorine in medicinal chemistry | |||
| Progress in medicinal chemistry, 2015, Elsevier, pp.97-165 | |||
| WO 2018035359 A1, 22.02.2018 | |||
| КОМПОЗИЦИЯ И ПРОИЗВОДНЫЕ ЗАМЕЩЕННОГО АЗАИНДОЛОКСОАЦЕТАПИПЕРАЗИНА, ОБЛАДАЮЩИЕ ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ | 2003 |
|
RU2325389C2 |
