Настоящее изобретение относится к аналитическим способам количественного определения полисорбатов в образцах биотерапевтических средств. Полисорбаты, такие как PS80, являются наиболее часто используемыми поверхностно-активными веществами в биотерапевтических препаратах.
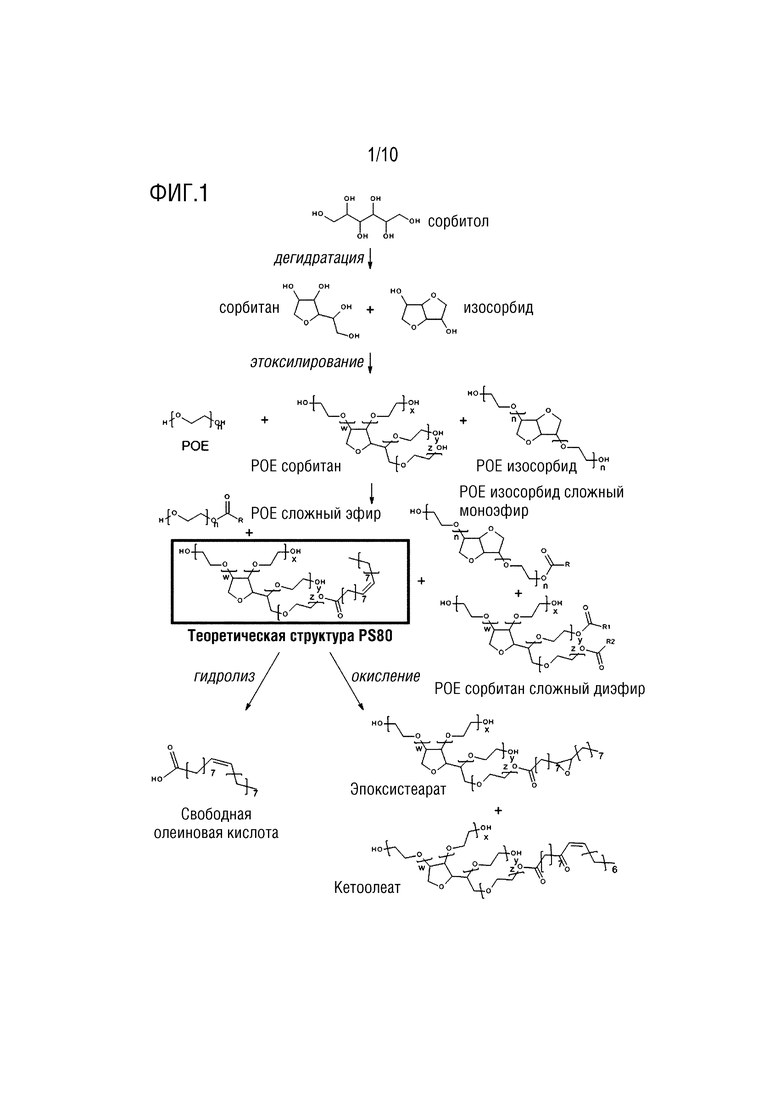
Полисорбаты представляют собой амфифильные неионные поверхностно-активные вещества, обычно используемые в составах биофармацевтических препаратов. Их основная роль заключается в защите белков и моноклональных антител (mAb) от агрегации, индуцированной на границе раздела фаз. Полисорбаты, такие как PS80, представляют собой гетерогенную смесь из более чем 1500 молекул, охватывающих широкий спектр физико-химических свойств и включающих множество жирных кислот. PS80 определяется как полиоксиэтилен сорбитан моноолеат, но вследствие способа его синтеза он представляет собой гетерогенную смесь, также известную своей подверженностью самоокислению и ферментативному гидролизу этерифицированных жирных кислот. Основной источник гетерогенности PS80 может быть отнесен на счет сложноэфирной связи и нескольких жирных кислот, участвующих в ней. Действительно, в нескольких работах сообщалось о присутствии больших количеств (до 34,1%) диэфиров, а иногда и сложных эфиров более высокого порядка (Borisov и соавт. Pharm. Biotechnol. 2015, 104, 1005-1018). Эта сложность представляет собой огромную проблему для количественной оценки PS80 и мониторинга деградации.
В недавнем обзоре (Martos и соавт. J.Pharm. Sci. 2017,106, 1722-1735) описаны различные способы мониторинга PS80. Представленные способы основаны на различных технологиях обнаружения, в основном с использованием жидкостной хроматографии (LC, ЖХ). Среди упомянутых способов те, которые позволяют количественно определять PS80, не являются чувствительными ко всем видам деградации PS80. Например, способы, основанные на жидкостной хроматографии в смешанном режиме с ELSD или CAD, подходят для количественной оценки PS80, но не являются наиболее чувствительными способами в случае деградации PS80. Способы, основанные на масс-спектрометрии (MS, МС), часто применяют для характеристики PS80, но не для количественного определения и мониторинга PS80 в среде контроля качества (QC). Последние достижения в области жидкостной хроматографии и масс-спектрометрии (LC-MS, ЖХ-МС) для определения характеристик PS80 основаны на характеристическом сигнале иона диоксоланилия (Borisov и соавт. Anal. Chem. 2011, 83, 3934-3942). В сообщениях об использовании этих характерных сигналов упоминается их полезность для интерпретации и идентификации видов PS80 и особенно состава жирных кислот. Его также использовали для выяснения структуры побочных продуктов окисления PS80 (Borisov и соавт. 2015, см. выше). До сих пор эти сигналы не использовались для количественной оценки PS80. Из полученных хроматограмм была получена только относительная или полуколичественная информация (Borisov и соавт. 2011, см. выше).
Следовательно, по-прежнему желательно предоставить эффективный и надежный способ, позволяющий осуществлять количественное определение полисорбатов в одном анализе.
Согласно одной из целей, настоящее изобретение предоставляет способ количественного определения по меньшей мере одного производного полисорбата в образце, при этом указанный способ включает в себя:
- этап проведения анализа LC-MS указанного образца на основе сигнала иона диоксоланилия;
- этап выполнения внутренней калибровки с использованием внутреннего стандарта указанного полисорбата.
Хотя использование внутреннего стандарта (IS) является хорошо известной практикой в хроматографии и масс-спектрометрии, об использовании IS для количественного определения полисорбатов никогда не сообщалось. Благодаря использованию внутреннего стандарта (IS) были улучшены линейность, повторяемость, точность и, следовательно, количественная оценка.
Одной из ключевых особенностей способа является использование внутреннего стандарта для внутренней калибровки, чтобы избежать проблем с матричными эффектами. Обычно этот внутренний стандарт имеет аналогичную химическую структуру и подвергается такой же фрагментации в источнике, как и заменитель интактного полисорбата. Согласно настоящему изобретению, способ основан на использовании характеристических ионов диоксоланилия сложного эфира жирных кислот, образующихся в результате диссоциации в источнике.
Посредством использования тщательно подобранного внутреннего стандарта, данный способ позволяет количественно определять полисорбаты в типичном составе с mAb.
Согласно одному из вариантов осуществления, полисорбаты представляют собой смесь соединений с формулой (I):
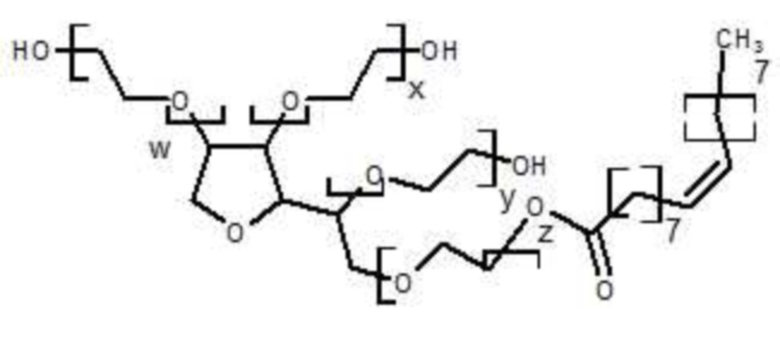
(I)
где w, x, y и z, одинаковые или разные, независимо представляют количество единиц полиэтиленгликоля в формуле (I) и отличаются от 0.
Согласно одному из вариантов осуществления, w+x+y+z=20, где полисорбат представляет собой, в частности, PS20 или PS80.
Согласно одному из вариантов осуществления, указанный полисорбат представляет собой PS20 или PS80. PS20 и PS80 являются коммерчески доступными (в частности, от компании Seppic-Puteaux, Франция).
Согласно одному из вариантов осуществления, в указанном анализе LC-MS используется одиночный квадрупольный масс-детектор, такой как QDa® (доступный от Waters). Посредством применения фрагментации в источнике в масс-детекторе QDa из полисорбата генерируются ионы диоксоланилия. QDa может выполнять несколько сигналов регистрации одиночных ионов (SIR) параллельно в режиме как положительной, так и отрицательной ионизации в одном аналитическом прогоне. Это позволяет количественно определять интактный полисорбат, искать характерные побочные продукты окисления полисорбата, а также количественно определять свободную олеиновую кислоту, побочный продукт гидролиза полисорбата. Как правило, в случае PS80 отслеживалось как минимум четыре сигнала SIR: один для иона диоксоланилия олеатного эфира, специфического иона интактного PS80, один для иона диоксоланилия IS, один для свободной олеиновой кислоты в режиме отрицательной ионизации (основные побочные продукты гидролиза PS80), один для иона диоксоланилия окисленного сложного олеатного эфира (основные побочные продукты окисленного PS80). Другие сигналы могут быть записаны в информационных целях.
Согласно одному из вариантов осуществления, способ позволяет отслеживать сигналы SIR относительно побочных продуктов деградации PS80: олеиновой кислоты (отрицательное отношение m/z 281,3) в случае гидролиза и окисленного сложного олеатного эфира (положительное отношение SIR m/z 325,3) в случае окисления.
Этот новый способ позволяет получить как минимум такой же уровень информации всего за один анализ, что сокращает время, затрачиваемое на анализ PS80. Информацию, собранную этим способом, намного легче понять, поскольку аналиты действительно являются специфичными либо для интактного PS80, либо для маркера деградированного PS80.
Следовательно, согласно одному из вариантов осуществления, способ также включает в себя этап обнаружения окисления и/или гидролиза указанного полисорбата. В отличие от существующих способов, настоящий способ позволяет количественно определять PS80 в составе независимо от того, произошла ли деградация (окисление или гидролиз) или адсорбция, которая может снизить общую концентрацию соединений PS80.
Фигура 2(b) иллюстрирует сигналы SIR при m/z 309,3 для количественного определения интактного PS80. Согласно одному из вариантов осуществления, способ позволяет количественно определять интактный PS80 с использованием подходящего суррогата, чувствительного как к окислению PS80, так и к гидролизу PS80. Сложные моноэфиры являются более чувствительными к деградации и могут использоваться в качестве суррогата для количественного определения полисорбатов. Согласно одному из вариантов осуществления, указанным конкретным суррогатом интактных полисорбатов могут быть моноэфиры. Для PS80 суррогатом является соединение, обозначенное пиком 2.1 на фигуре 2 (b). Обычно, в зависимости от условий эксперимента, этот пик имеет время удерживания от 6 до 7 мин; более конкретно, в экспериментальных условиях, описанных ниже в экспериментальной части, время удерживания пика составляло от 6,85 до 6,95, более конкретно, около 6,9 мин. При использовании данного суррогата применение данного способа является актуальным для количественного определения интактного PS80 в случае деградации в результате гидролиза или окисления. Ни один из ранее опубликованных способов не может этого сделать.
Используемый в настоящем описании термин «внутренний стандарт» относится к химическому соединению, которое добавляют в постоянном количестве к образцам, отрицательным контролям и калибровочным стандартам в химическом анализе. Затем это соединение можно использовать для калибровки, построив график отношения сигнала полисорбата к сигналу внутреннего стандарта в зависимости от концентрации полисорбата в стандартах. Внутренний стандарт представляет собой соединение, которое, как правило, очень похоже, но не идентично полисорбату в образцах, поскольку эффекты подготовки образца должны быть одинаковыми, в зависимости от количества каждого соединения, как для сигнала от внутреннего стандарта, так и для сигнала(-ов) от интересующего соединения в идеальном случае. Используемый внутренний стандарт, как правило, должен обеспечивать сигнал, который во многом похож на сигнал полисорбата, но отличается достаточно для того, чтобы прибор мог легко различить эти два сигнала.
Внутренний стандарт обычно должен быть аналогичен по структуре интересующему аналиту, иметь такое же время удерживания по сравнению с интересующим аналитом и должен подвергаться аналогичной фрагментации в источнике. Он также должен быть стабильным и не должен создавать помехи для компонентов образца.
Согласно одному из вариантов осуществления, внутренний стандарт (IS) может быть выбран из соединений, имеющих полиэтиленгликолевую цепь, этерифицированную одной или более карбоновыми кислотами, чтобы обладать такими же физико-химическими свойствами, что и полисорбат. Обычно часть карбоновой кислоты должна отличаться от жирных кислот, присутствующих в смеси полисорбатов.
Согласно одному из вариантов осуществления, внутренний стандарт представляет собой этоксилированную жирную кислоту с формулой (I):
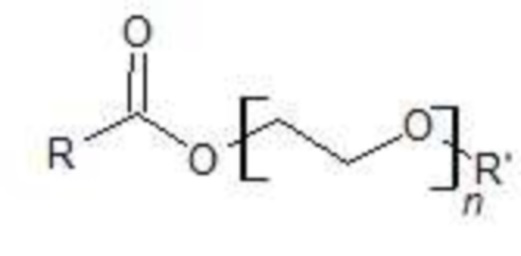 (I)
(I)
где:
 представляет собой остаток жирной кислоты,
представляет собой остаток жирной кислоты,
где R представляет собой C3-C24 линейный или разветвленный насыщенный алкил;
R’ представляет собой H или  , где R'' представляет собой C3-C24 линейный или разветвленный насыщенный алкил; и
, где R'' представляет собой C3-C24 линейный или разветвленный насыщенный алкил; и
n определяется как количество звеньев PEG (ПЭГ, полиэтиленгликоля) в общей формуле (I), при этом подразумевается, что соединение с формулой (I) находится в форме смеси одного или более соединений с формулой (I), где n, одинаковые или разные, находятся в диапазоне от 1 до 100;
и смеси указанного.
Согласно одному из вариантов осуществления, n представляет собой целое число от 1 до 100.
Согласно одному из вариантов осуществления, указанный внутренний стандарт выбран из:
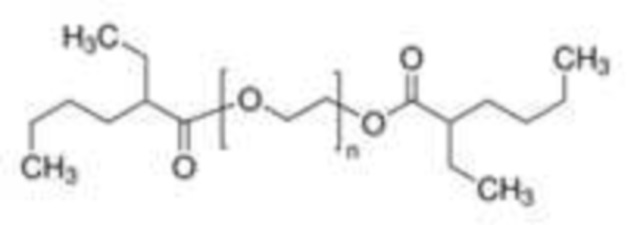
поли(этиленгликоль)бис(2-этилгексаноат)
где n, определенное, как указано выше, составляет от 1 до 100.
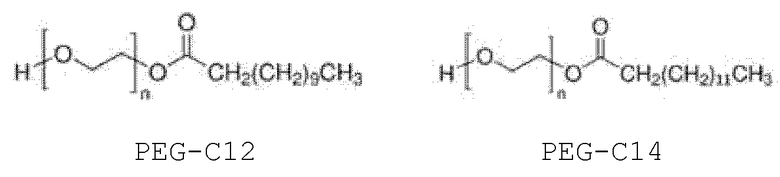
Более конкретно, внутренний стандарт представляет собой монолаурат полиэтиленгликоля (далее именуемый PEG-C12) или мономиристат полиэтиленгликоля (далее именуемый PEG-C14); еще более конкретно:
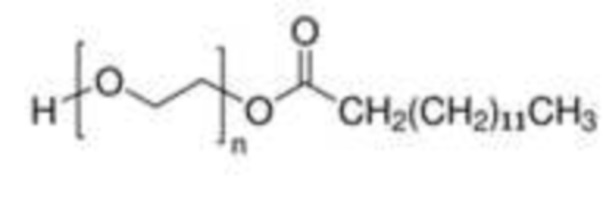 (миристат полиэтиленгликоля, PEG-C14).
(миристат полиэтиленгликоля, PEG-C14).
PEG-C12 и поли(этиленгликоль)бис(2-этилгексаноат) коммерчески доступны от Sigma-Aldrich (Saint Quentin Fallavier, Франция), и PEG-C14 используется в качестве компонента, присутствующего в PEG-C12.
В случае PEG-C12 n может быть таким, чтобы средняя молекулярная масса составляла от 300 до 600 г/моль, обычно около 400 г/моль.
В случае поли(этиленгликоля)бис(2-этилгексаноата) n может быть таким, чтобы средняя молекулярная масса составляла от 400 до 700 г/моль, обычно около 650 г/моль.
PEG-C14 может быть выделен из PEG-C12 или может использоваться через PEG-C12 в качестве компонента, присутствующего в PEG-C12.
Было обнаружено, что обнаружение полисорбата моноэфира и сложного эфира более высокого порядка и свободной олеиновой кислоты, а также чувствительность к полисорбату и свободной олеиновой кислоте можно регулировать посредством настройки подвижной фазы и/или напряжения на конусе.
Согласно одному из вариантов осуществления, подвижная фаза фазы LC-MS содержит градиент муравьиной кислоты. Обычно подвижная фаза представляет собой тройную подвижную фазу, содержащую воду, органический растворитель, такой как ацетонитрил или изопропанол, и кислоту, такую как муравьиная кислота или уксусная кислота. Обычно подвижная фаза состоит из воды, ацетонитрила и муравьиной кислоты с градиентом ацетонитрила и муравьиной кислоты, обычно с градиентом муравьиной кислоты в диапазоне от 0,01 до 0,1%. Обычно состав элюирующей фазы варьируется от начальной композиции, содержащей 80-90% воды, 10-20% ацетонитрила и 0,01-0,05% муравьиной кислоты, до конечной композиции, содержащей 0-10% воды, 90-99,9% ацетонитрила и 0,05%-0,1% муравьиной кислоты (по частям).
Напряжение на конусе может вызывать фрагментацию в источнике. Обычно напряжение на конусе ниже 100 В, предпочтительно, ниже 70 В. Обычно можно использовать разное напряжение на конусе (CV) для полисорбата и олеиновой кислоты. В качестве иллюстрации можно использовать CV 50 для PS80 и CV 15 для олеиновой кислоты для достижения более высокой чувствительности для PS80 и свободной олеиновой кислоты.
Согласно одному из вариантов осуществления, способ включает в себя начальный этап добавления внутреннего стандарта к образцу. Способ может также включать в себя этап обработки образца с целью осаждения белков, которые могут в нем содержаться. Указанная обработка может включать в себя добавление ацетонитрила к образцу и/или центрифугирование указанного образца.
Согласно одному из вариантов осуществления, указанный образец представляет собой биофармацевтический состав.
В данном контексте биофармацевтический состав представляет собой фармацевтическую композицию, содержащую по меньшей мере один биологический продукт, такой как белок. Соответственно, указанный образец может содержать по меньшей мере один терапевтический белок, биотерапевтический или биологический препарат, такой как моноклональное антитело (mAb) или его фрагмент, одноцепочечный вариабельный фрагмент (scFv), однодоменное антитело (VHH-антитело, т.е. нанотело), конъюгат антитело-лекарственное средство, биспецифическое антитело, триспецифическое антитело.
Согласно другой цели, настоящее изобретение также относится к способу мониторинга деградации по меньшей мере одного полисорбата в образце. Указанный способ включает в себя реализацию способа количественного определения указанных полисорбатов согласно изобретению.
Фигуры
На фигуре 1 представлена теоретическая структура и пути деградации PS80.
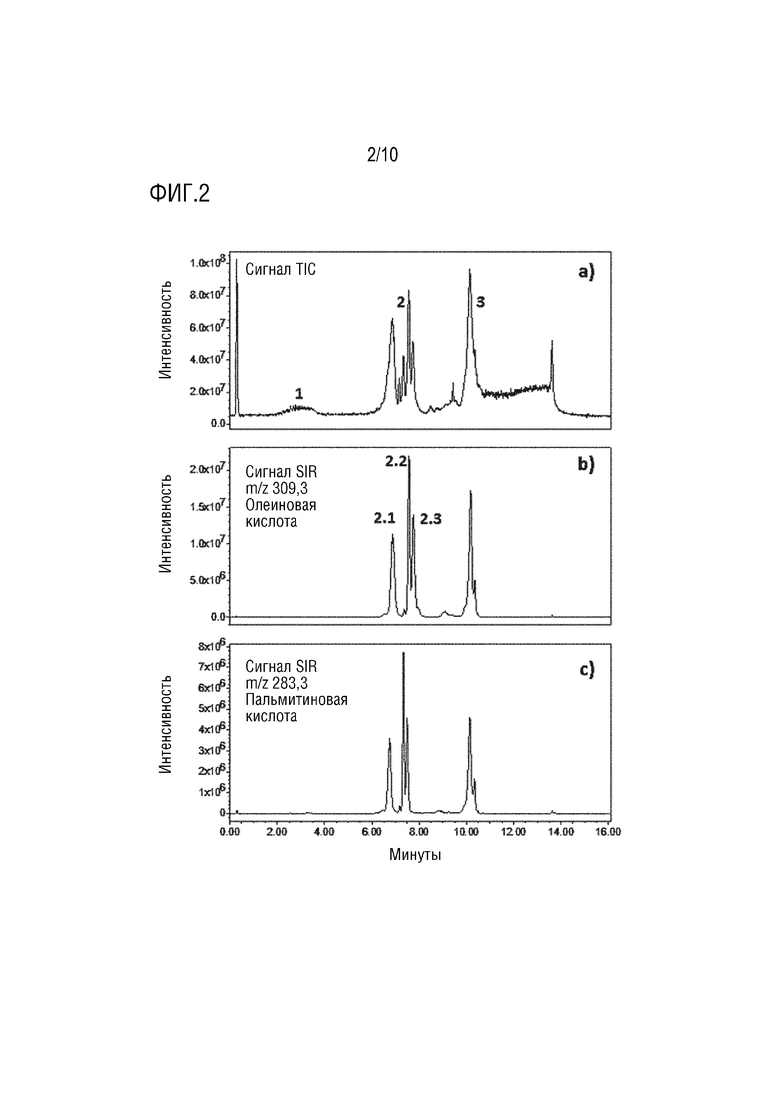
На фигуре 2 показана зависимость интенсивности (в условных единицах) от времени удерживания (в минутах). Проиллюстрирован характерный профиль полного ионного тока (TIC) PS80 с пиками, обозначенными как 1 - неэтерифицированные частицы, 2 - сложные моноэфиры и 3 - сложные эфиры более высокого порядка (a), запись одиночных ионов (SIR) для m/z 309,3, что указывает на элюирование олеатных соединений с пиками, обозначенными как 2.1 - POE сорбитана моноолеат, 2.2 - POE изосорбид моноолеат и 2.3 - POE моноолеат (b) и SIR для m/z 283.3, что указывает на элюирование разновидностей пальмитата (c).
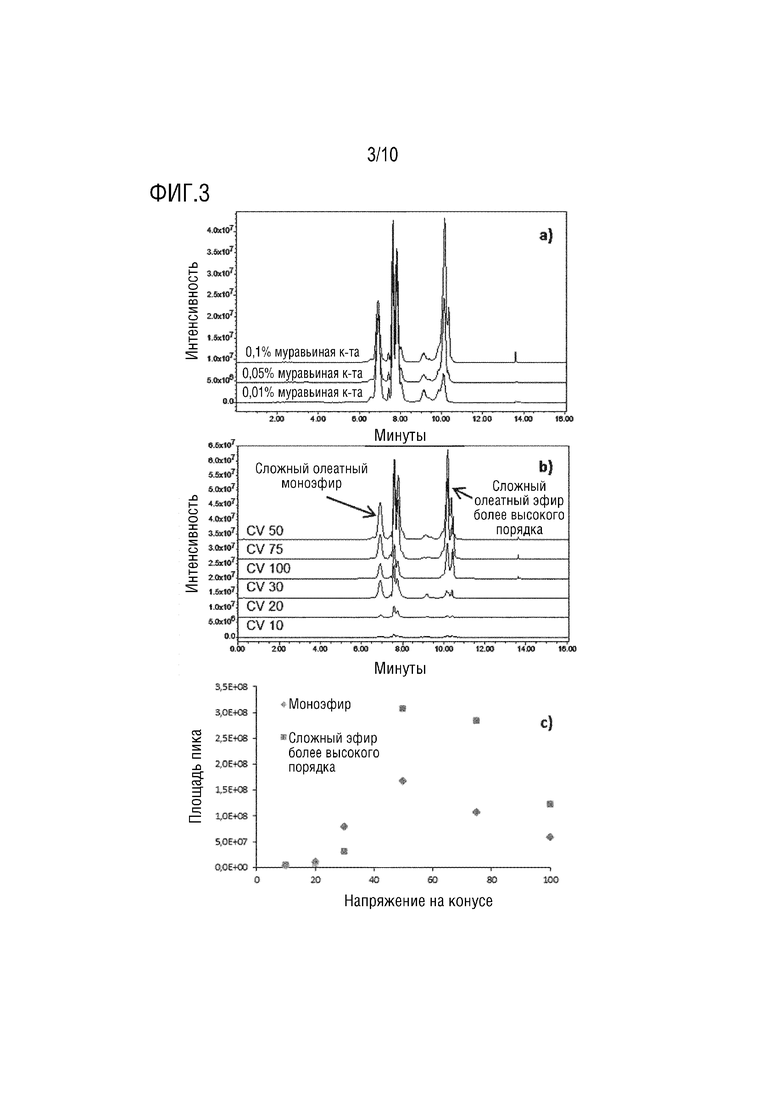
На фигуре 3 представлены хроматограммы ионов диоксоланилия олеатного эфира в режиме положительной ионизации с различным процентным содержанием муравьиной кислоты с CV 50 (a) и с разными CV при 0,1% муравьиной кислоты (b). Площадь пика, извлеченная из (b), нанесена на график зависимости напряжения на конусе для сложного моноэфира и сложного эфира более высокого порядка (c).
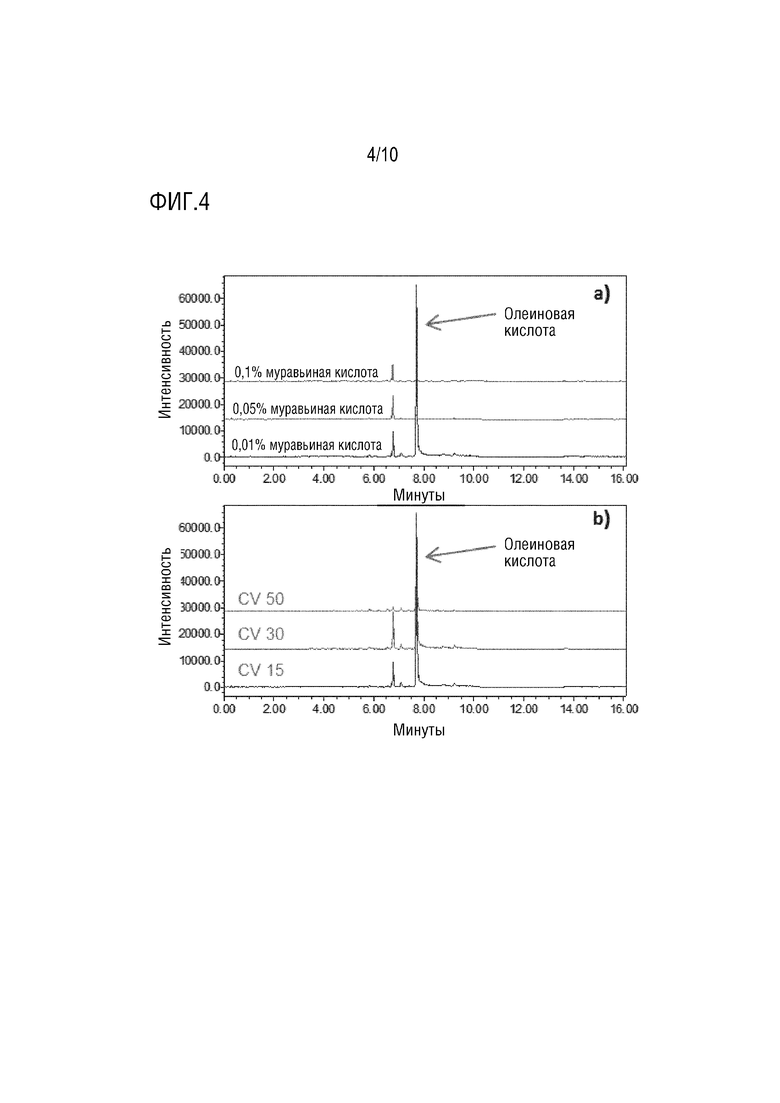
На фигуре 4 представлены хроматограммы олеиновой кислоты в режиме отрицательной ионизации с различным процентным содержанием муравьиной кислоты с CV 15 (a) и с разными CV при 0,01% муравьиной кислоте (b).
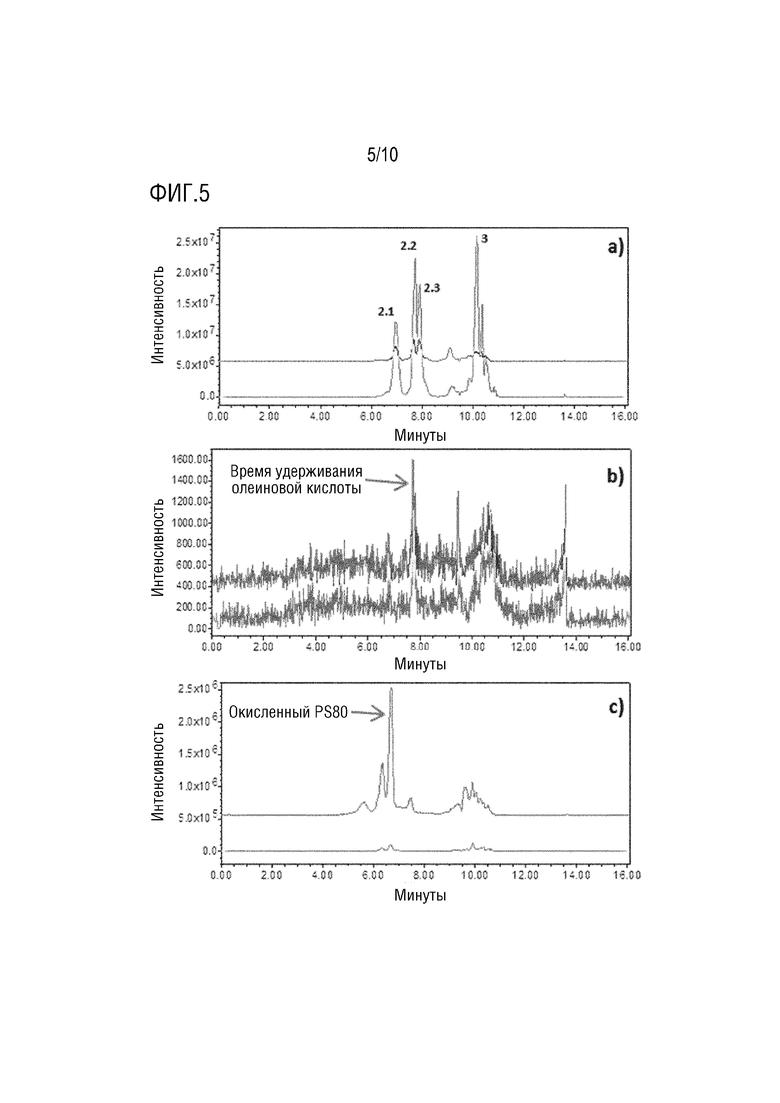
На фигуре 5 показаны типичные хроматограммы дифференциальной диагностики деградации PS80 в образцах, подвергнутых температурному стрессу (верхняя хроматограмма) и не подвергавшихся стрессу (нижняя хроматограмма). Подвиды олеата PS80 уменьшаются в подвергнутом температурному стрессу образце (a - положит. m/z 309,3). Не наблюдалось увеличения содержания олеиновой кислоты (b - отрицат. m/z 281,3), а количество окисленных побочных продуктов резко увеличилось (c - положит. m/z 325,3).
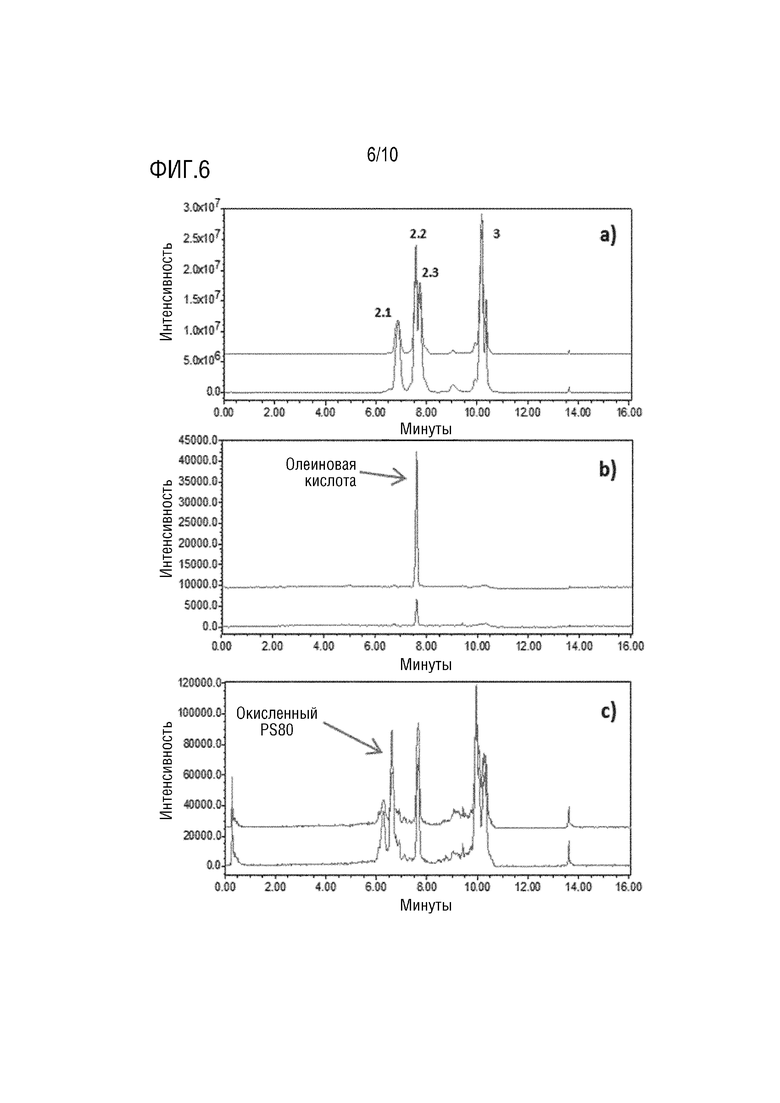
На фигуре 6 показаны типичные хроматограммы дифференциальной диагностики деградации PS80 в двух разных партиях A (верхняя хроматограмма) или B (нижняя хроматограмма). Подвиды олеата PS80 уменьшены в образце партии А (a - положит. m/z 309,3). Наблюдается значительное увеличение содержания олеиновой кислоты (b - отрицат. m/z 281,3), и окисленные побочные продукты не отличаются в двух партиях (c - положит. m/z 325,3).
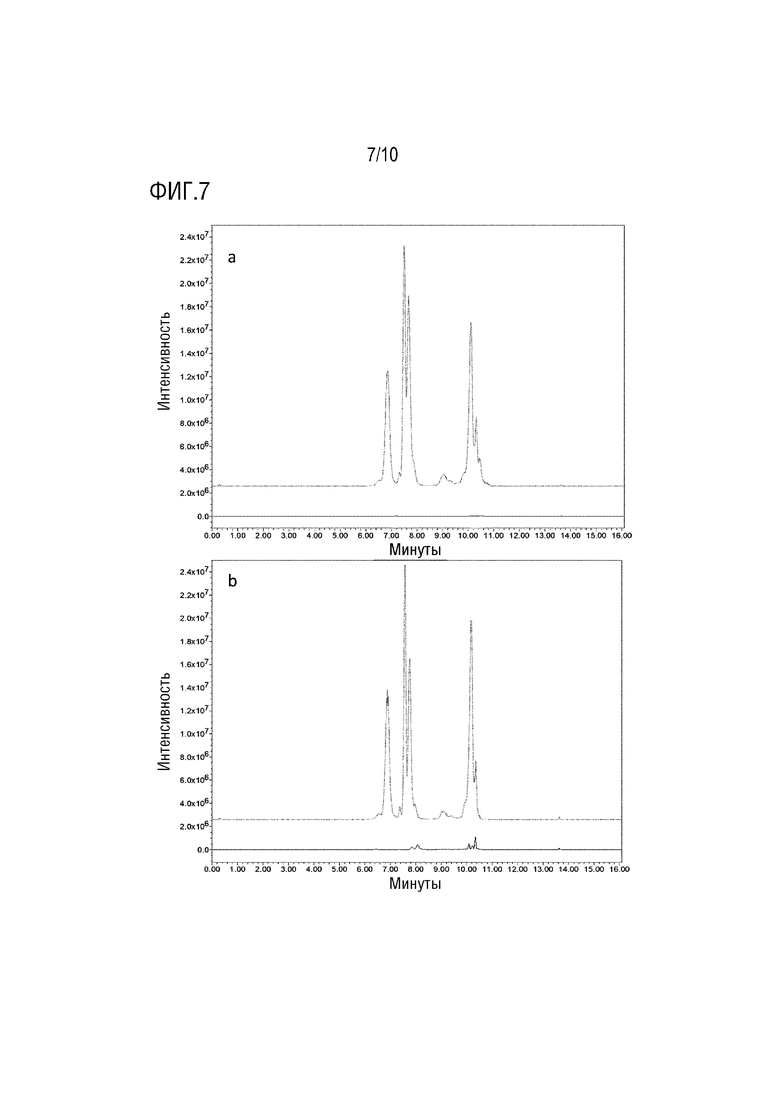
На фигуре 7 показаны сигналы SIR в режиме положительной ионизации при 309,3 m/z для образца PS80 при 50 мкг/мл (пунктирная линия) и внутреннего стандарта (сплошная линия) для PEG бис C8 (a) и PEG C12 и PEG C14 (b).
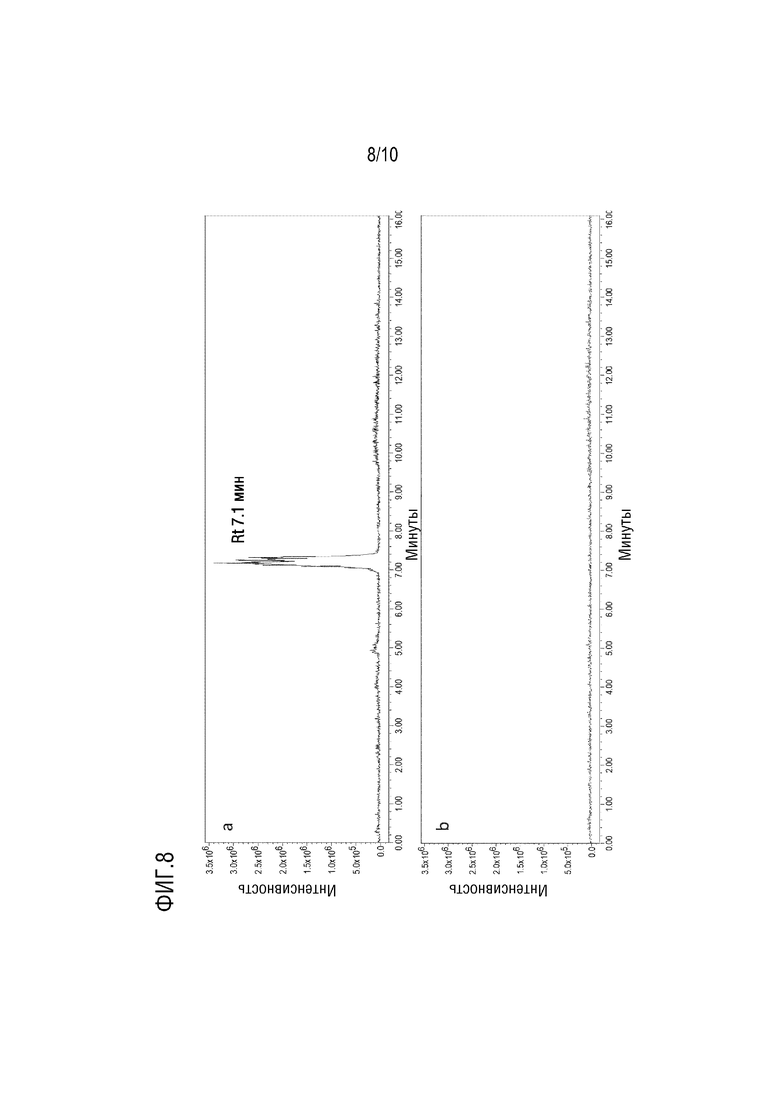
Фигура 8 представляет собой расширенную ионную хроматограмму в режиме положительной ионизации при 171 m/z специфическом сигнале PEG бис C8 для образца PS80 (пунктирная линия - панель b) и PEG бис C8 (сплошная линия - панель a). Приведено время удерживания (Rt) пика, используемого в качестве внутреннего стандарта.
Фигура 9 представляет собой хроматограмму извлеченных ионов в режиме положительной ионизации при 227,3 m/z специфическом сигнале PEG C12 для образца PS80 (пунктирная линия - панель b) и PEG бис C8 (сплошная линия - панель a). Приведено время удерживания (Rt) пика, используемого в качестве внутреннего стандарта.
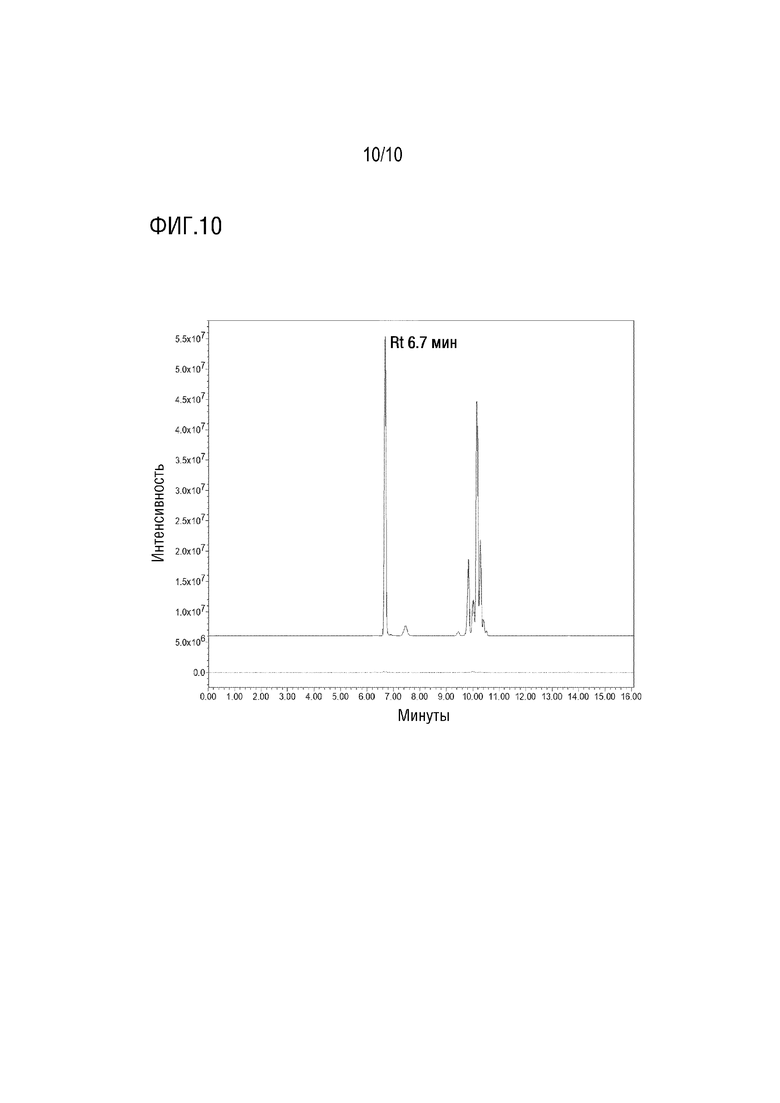
На Фигуре 10 показана запись одиночных ионов в режиме положительной ионизации при специфическом сигнале 255,3 m/z PEG C14 для образца PS80 (пунктирная линия) и PEG C14 (сплошная линия). Приведено время удерживания (Rt) пика, используемого в качестве внутреннего стандарта.
Примеры
Материалы. Полисорбат 80 был получен от Seppic (Puteaux, Франция). Для LC-MS 1 г полисорбата растворяли в 100 мл воды в мерной колбе с получением базового раствора 10 г/л, хранимого при 2-8°C в защищенном от света месте. Марка ацетонитрила для LC-MS была приобретена у Fisher Scientific (Illkirch, Франция). Использовалась очищенная вода из системы milliQ. Муравьиная кислота, монолаурат полиэтиленгликоля (PEG-C12, Mn=±400 г/моль), полиэтиленгликоль-бис-2-этилгексаноат (PEG-bis C8, Mn=±650 г/моль) и олеиновая кислота были приобретены у Sigma-Aldrich (Saint Quentin Fallavier, Франция). PEG-C12 использовался в качестве внутреннего стандарта. Базовый раствор PEG-C12 получали посредством растворения 500 мг в 100 мл ацетонитрила в мерной колбе с получением базового раствора 5 г/л. Рабочий раствор 100 мкг/мл готовили посредством разбавления в ацетонитриле. На протяжении всего исследования использовалась только одна партия PEG-C12. Исходный раствор олеиновой кислоты получали посредством растворения 100 мг в 100 мл ацетонитрила в мерной колбе с получением исходного раствора 1 г/л. Рабочий стандартный раствор для калибровки готовили в мерной колбе на 20 мл с конечной концентрацией PEG-C12 5 мкг/мл, с изменяющейся концентрацией PS80 от 5 до 75 мкг/мл и изменяющейся концентрацией олеиновой кислоты от 1 до 20 мкг/мл. Добавленный растворитель состоял из смеси вода/ацетонитрил (20%/80%).
LC-MS анализ. Обращенно-фазовое разделение проводили на системе Acquity UPLC, оснащенной масс-детектором QDa от Waters (Saint Quentin en Yvelines, Франция). Параметры QDa были установлены по умолчанию до оптимизации, подробно описанной ниже. Колонка Zorbax Sb-Aq (100 × 2,1 мм, 3,5 мкм) от Agilent (Les Ulis, Франция) работала при 50°С со скоростью потока 1 мл/мин в соответствии с Christiansen и соавт. (Pharmazie. 2011, 66, 666-671). Подвижные фазы А и В представляли собой, соответственно, воду + 0,1% муравьиной кислоты и ацетонитрил + 0,1% муравьиной кислоты. Аналитический градиент был следующим: 85% A с 15% B в течение 1 минуты с последующим линейным нарастанием до 60% B через 6 минут, выдерживание до 8 минут перед линейным нарастанием до 100% B через 10 минут, выдерживание еще 3 минуты с последующим возвращением к исходным условиям через 13,1 минуты еще на 3 минуты. Типичный градиент подвижной фазы описан в следующей таблице градиентов:
Таблица 1. График градиента подвижной фазы
Аналитические условия, такие как подвижная фаза и градиент, были настроены в соответствии с полученными результатами.
Подготовка образцов. Использовали образцы биотерапевтического препарата, содержащего mAb и различные вспомогательные вещества. Концентрация PS80 поддерживалась на уровне 200 мкг/мл. Подготовка образца состояла из этапа осаждения белка ацетонитрилом. К 80 мкл приготовленного mAb добавляли 20 мкл рабочего раствора внутреннего стандарта и 300 мкл ацетонитрила. После перемешивания смесь подвергали 10 мин центрифугированию на 1500 g при 10°C. Супернатант собирали и переносили во флаконы для ВЭЖХ (HPLC). Конечная концентрация составляла около 40 мкг/мл PS80 и 5 мкг/мл внутреннего стандарта.
Результаты и обсуждение
Разделение компонентов PS80 и идентификация сложных эфиров жирных кислот с диссоциацией «в источнике». Согласно большому количеству предыдущих публикаций, полисорбаты представляют собой гетерогенную смесь с большим разнообразием по химической структуре и концентрации этерифицированных соединений. Более того, природа жирных кислот, участвующих в сложноэфирной связи, может варьироваться. Согласно фармакопее ЕС олеиновая кислота и пальмитиновая кислота (соответственно, мононенасыщенные и насыщенные жирные кислоты) являются двумя основными жирными кислотами PS80. Серия ионов с низким соотношением m/z, характерных для эфира жирных кислот полиэтиленгликоля, называемых ионом диоксоланилия, была использована для идентификации этерифицированных видов PS80. Типичный профиль PS80 демонстрирует различные сложные эфиры жирных кислот от моно- до тетраэфиров (фигура 2a), в то время как регистрации одиночных ионов (SIR) m/z 309,3 и 283,3 специфична для видов сложного эфира олеата и сложного эфира пальмитата (фигура 2b и 2c). Хроматограммы, полученные с помощью SIR, было намного проще интерпретировать и интегрировать вследствие искусственного увеличения хроматографического разрешения. Идентификация различных видов сложных эфиров была основана на работе, опубликованной Borisov и соавт., 2015 (см. выше), о порядке элюирования полисорбатов в обращенно-фазовой хроматографии (фигуры 2а и 2b).
Разработка способа LC-MS. QDa может выполнять одновременный анализ в режиме отрицательной и положительной ионизации для источника ESI. Для того чтобы извлечь преимущество этой возможности, условия LC-MS были тщательно оптимизированы. Систематическая оценка процентного содержания муравьиной кислоты в подвижной фазе и напряжения на конусе (CV) источника ESI позволила получить одновременное разделение PS80 и продукта его деградации с высокой чувствительностью. Процентное содержание муравьиной кислоты от 0,01% до 0,1% мало влияло на обнаружение моноэфиров олеата, но было критическим для сложных эфиров более высокого порядка (фигура 3a). Повышенное напряжение на конусе приводило к увеличению сигнала, представленного площадью пика моноэфира и сложного эфира более высокого порядка (фигуры 3b и 3c), вплоть до CV50. Выше CV50 произошла резкая потеря сигнала. Это явление объяснялось фрагментацией в источнике. Повышенное напряжение на конусе, индуцированное увеличением фрагментации в источнике, приводило к образованию большего количества ионов диоксоланилия вплоть до CV50. Для более высокого CV интенсивная фрагментация в источнике привела к образованию вторичного фрагмента, объясняющего потерю сигнала.
Для обнаружения олеиновой кислоты в режиме отрицательной ионизации требовалось небольшое количество муравьиной кислоты в подвижной фазе - не более 0,01% (фигура 4а). Для обнаружения нефрагментированного молекулярного иона олеиновой кислоты требовалось низкое напряжение на конусе, чтобы свести к минимуму фрагментацию в источнике (фигура 4b).
Порядок элюирования между сложными моноэфирами, олеиновой кислотой и сложными эфирами более высокого порядка позволил установить трехкомпонентный градиент подвижной фазы, с тем чтобы независимо изменять процентное содержание ацетонитрила и муравьиной кислоты на протяжении всего анализа. Способ LC-MS был адаптирован из экспериментальной части следующим образом. Градиент подвижной фазы с 85% A (вода), 5% B (ацетонитрил) и 10% C (ацетонитрил+0,1% муравьиной кислоты) в течение 1 минуты с последующим линейным увеличением до 40% A, 50% B и 10% C с 6 минут удерживался 2 минуты. Был добавлен еще один подъем до 20% A, 70% B и 10% C за 9 минут. В 9,1 минуты было добавлено переключение на 18% A с 82% C, чтобы увеличить процентное содержание муравьиной кислоты до 0,1%, и линейное изменение продолжалось до 100% C в 10 минут и удерживалось в течение 3 минут, прежде чем вернуться к начальным условиям в 13,1 минуты. Переключение между подвижной фазой B и C через 9 минут было добавлено для изменения процентного содержания муравьиной кислоты в подвижной фазе с 0,01% до 0,1% при сохранении процентного содержания ацетонитрила в подвижной фазе на том же уровне градиента. Сигналы SIR в режиме положительной ионизации регистрировались с помощью CV50, тогда как сигналы SIR в режиме отрицательной ионизации регистрировались с помощью CV15.
Количественное определение полисорбата с помощью внешней калибровки. В ранее опубликованных способах PS80 или PS20 определяется количественно с использованием подходящего суррогата с внешней калибровочной кривой. Хорошо известными примерами являются способы, основанные на гидролизе и последующей этерификации в метаноле с образованием метилового эфира жирной кислоты, такого как метилолеат. Затем этот метилолеат считается суррогатом PS80 и подвергается количественному определению. В настоящем примере суррогатом для количественной оценки был пик 2.1 (фигура 2b), поскольку этот пик соответствует POE сорбитан моноолеату, который является структурой, близкой к теоретической структуре PS80. На основе этого пика была построена калибровочная кривая для семи уровней концентрации с концентрацией PS80 в водном растворе в диапазоне от 5 до 75 мкг/мл. Линейность оценивали с помощью невзвешенной линейной регрессии площади пика в зависимости от концентрации. Линейность была плохой с r²=0,977. Остатки показали квадратичное поведение, объясняющее плохие характеристики линейности. Воспроизводимость, оцененная путем введения 6 повторов каждого уровня калибровки, была плохой с RSD% площади пика выше 22%. Была выдвинута гипотеза, что эти плохие результаты имели место вследствие конкуренции во время фрагментации «в источнике», поскольку эта фрагментация плохо контролируется в источнике ESI QDa, одиночном квадруполе.
Сравнение внешней и внутренней калибровки. Для того чтобы решить эту проблему, был использован внутренний стандарт (IS) для выполнения внутренней калибровки вместо внешней калибровки. Наиболее часто используемым внутренним стандартом в LC-MS для количественной оценки являются дейтерированные соединения. Учитывая неоднородность смеси PS80, этот подход не рассматривался. Были выбраны соединения с аналогичной химической структурой и, следовательно, со сходными путями фрагментации в источниках: мономиристат полиэтиленгликоля (PEG-C14), монолаурат полиэтиленгликоля (PEG-C12) и бис-2-этилгексаноат полиэтиленгликоля (PEG-бис C8). PEG-С12 и PEG-бис-С8 коммерчески доступны (Sigma-Aldrich/Merck). PEG-C14 нельзя было купить у производителей классических химических соединений, но он был обнаружен как примесь PEG-C12.
Поскольку этот PEG-С14 состоит из PEG, этерифицированного насыщенными жирными кислотами С14, он подвергался такой же фрагментации, что привело к образованию характерных ионов диоксоланилия с m/z 255,3 в режиме положительной ионизации. Взаимодействие между PS80 и PEG-C14/PEG-C12/PEG бис C8 проверяли перед использованием его в качестве внутреннего стандарта. Ни один из сигналов от PS80 не создавал помех сигналу PEG-C14/PEG-C12/PEG бис C8 и наоборот. Время удерживания суррогатного пика PEG-C14/PEG-C12/PEG бис C8 и PS80 было близким, так что они подвергались одинаковой конкуренции во время фрагментации в источнике.
Отсутствие интерференции сигналов от PEG-C14/PEG-C12/PEG бис C8 с сигналами PS80 показано на фигуре 7.
Отсутствие интерференции сигналов от PS80 с сигналами PEG-C14/PEG-C12/PEG бис C8, а также времени удерживания интересующего пика от PEG-C14/PEG-C12/PEG бис C8 иллюстрируется фигурами 8-10.
Для сравнения, внутренняя калибровка была оценена с помощью того же набора экспериментов, что и внешняя калибровка. Линейность оценивали с помощью невзвешенной линейной регрессии отношения площадей пика суррогатного пика PS80 к IS в зависимости от концентрации. Линейность была хорошей с r2 более 0,999. Остатки распределялись более или менее случайным образом, а относительная систематическая ошибка не превышала 10%. Воспроизводимость, оцененная путем введения 6 повторов каждого уровня калибровки, также была хорошей с RSD% отношения площадей пиков ниже 6% для всех уровней калибровки. В таблице 2 сравниваются эти результаты воспроизводимости как для внешней, так и для внутренней калибровки с использованием мономиристата полиэтиленгликоля (PEG-C14), монолаурата полиэтиленгликоля (PEG-C12), полиэтиленгликоль бис 2 этилгексаноата (PEG-бис C8). Сравнение этих IS основано на линейности (r2) и повторяемости (RSD%).
Таблица 2. Значение RSD% площади пика (внешняя калибровка) и отношение площадей пиков (внутренняя калибровка) для 6 повторов (за исключением PEG бис C8, только 4 повтора)
На основе этих данных было показано, что внутренняя калибровка, такая как PEG-C12, PEG-C14 и PEG бис C8, демонстрирует более высокую повторяемость по сравнению с внешней калибровкой. Эти внутренние стандарты демонстрируют аналогичную повторяемость.
Количественное определение олеиновой кислоты. Олеиновая кислота является основным побочным продуктом гидролиза PS80. Несколько способов, описанных в Martos и соавт. (2017, см. выше), были разработаны для количественного определения свободных жирных кислот, включая олеиновую кислоту, высвобождаемых при гидролизе PS20 или PS80.
Благодаря особенностям QDa, сигнал SIR свободной олеиновой кислоты (m/z 281,3 в режиме отрицательной ионизации) был зарегистрирован в одном аналитическом прогоне вдоль сигналов SIR интактного и окисленного PS80. Олеиновая кислота была определена путем внешней калибровки с шестью калибровочными уровнями - от 1 мкг/мл до 20 мкг/мл - добавленными к калибровочному уровню PS80. Например, типичный уровень калибровки с 20 мкг/мл PS80 содержал также 5 мкг/мл внутреннего стандарта и 5 мкг/мл олеиновой кислоты. Линейность оценивали с помощью невзвешенной линейной регрессии площади пика в зависимости от концентрации. Линейность была хорошей с r2 более 0,997 и случайным распределением остатков. Воспроизводимость оценивалась аналогично PS80. Значения RSD% составляли от 6,0 до 13,1% с максимальным значением, наблюдаемым при концентрации 2 мкг/мл. Хотя значения RSD% были выше, чем ожидалось, анализ был признан удовлетворительным, поскольку эта информация будет использоваться только в случае проведения исследования. Было предположено, что эти высокие значения были вызваны высоким базовым шумом в режиме отрицательной ионизации вследствие состава подвижной фазы. Действительно, во время разработки был сделан компромисс в пользу обнаружения видов олеатного эфира PS80.
Применение к приготовленным образцам mAb. Способ применялся к различным случаям составов mAb с разными свойствами mAb и наполнителями, но каждый раз с 200 мкг/мл PS80.
mAb1 в концентрации 5 мг/мл использовали для оценки повторяемости подготовки образцов, а также точности способа. Были сделаны три препарата и проанализированы в течение трех дней подряд, в результате чего было определено девять концентраций PS80. Общее среднее значение составило 182 мкг/мл со значением RSD 3,1%. Эти результаты соответствовали предыдущим измерениям с классическим смешанным режимом анализа LC-CAD, где было найдено среднее значение 188 мкг/мл с 1,2% RSD по шести измерениям.
mAb2 в составе 20 мг/мл подвергали воздействию термического стресса в течение двух недель при 40°C. Результаты показали резкое снижение содержания PS80 с 198 до менее 25 мкг/мл. Больше информации было получено из других записанных сигналов. Хроматограмма видов сложного эфира пальмитата не показала различий между подвергнутыми и не подвергнутыми стрессу образцами, что указывает на то, что деградация влияет только на виды сложного эфира олеата, обычно наблюдаемые в случае окисления. Не было обнаружено следов свободной олеиновой кислоты, что исключает гидролиз как путь деградации. Сигналы SIR m/z 325,3 (в режиме положительной ионизации) показали большие различия между подвергнутыми и не подвергнутыми стрессу образцами (фигура 5). Ионы с m/z 325,3 были описаны как характерные побочные продукты окисления PS80 в виде полисорбата, модифицированного эпоксистеаратом или гидроксиолеатом. Здесь полная картина PS80 в этом деградированном образце была получена с помощью трех разных хроматограмм, записанным в одном анализе, что позволяет однозначно заключить, что снижение PS80 происходило вследствие окисления.
Другим примером является mAb3 в составе 50 мг/мл. Образцы из разных партий хранили при 5°C в течение почти месяца перед анализом. Результаты показали различия в содержании PS80 между партиями A и B соответственно 100 и 190 мкг/мл вместо 200 мкг/мл. Это проиллюстрировано на фигуре 6a при рассмотрении пика 2.1, суррогата для количественного определения PS80. Сигналы SIR окисленного PS80 не выявили разницы между партиями, что означало отсутствие увеличения побочных продуктов окисления PS80, особенно по сравнению с интенсивностью, наблюдаемой в случае mAb2. Свободная олеиновая кислота в растворе была обнаружена в проблемной партии А в концентрации около 25 мкг/мл. В партии B были обнаружены только небольшие следы олеиновой кислоты (фигура 6). Наличие свободной олеиновой кислоты свидетельствует о гидролизе PS80. Учитывая всю информацию, полученную в результате этого анализа, был сделан вывод, что деградация PS80 происходит вследствие гидролиза. Поскольку сложные эфиры более высокого порядка не подвергались влиянию, было предположено, что этот гидролиз имеет ферментативное происхождение.
Заключение. PS80 широко используется в биотерапевтических препаратах. В последнее время растет озабоченность по поводу стабильности полисорбатов в лекарственном препарате и их способности сохранять свою защитную роль против агрегации белков. Об эффективных способах измерения содержания PS80 и мониторинга его деградации сообщалось за последние несколько лет (Martos и соавт., см. выше). Среди них способы на основе LC-MS показали многообещающие результаты с точки зрения характеризации и полуколичественной информации. Характерные сигналы ионов диоксоланилия сложных эфиров жирных кислот использовали после CID «в источнике» для значительного упрощения хроматограмм и более легкой идентификации подклассов PS80. В упомянутой предыдущей работе способ не проверяли в количественном анализе. В настоящем исследовании была применена та же методология с целью использования ее для количественной оценки на уровне контроля качества с помощью одного квадрупольного масс-детектора. К сожалению, первые результаты количественной оценки подтвердили вывод Борисова (см. выше). Эта проблема была решена путем использования тщательно подобранного внутреннего стандарта с аналогичными химическими свойствами. Было показано, что градиент муравьиной кислоты позволяет с большей чувствительностью и одновременно обнаруживать как в режиме положительной, так и в отрицательной ионизации все представляющие интерес соединения от подклассов сложных эфиров PS80 до свободных жирных кислот.
Данный способ был успешно применен к различным случаям мониторинга PS80 и особенно к двум из них с отличительными особенностями деградации PS80. В этих двух случаях основная причина деградации PS80 была идентифицирована с использованием только одного 16-минутного анализа. Для достижения той же цели без этого способа потребовалось бы от трех до четырех различных способов: один для количественной оценки PS80 (смешанный режим LC-CAD) плюс один для профилирования PS80 (обращенно-фазовая CAD), а также другие способы для определения характеристических побочных продуктов (окисленный PS80 и свободная олеиновая кислота). Представленный в настоящем описании способ может эффективно заменить обширный набор аналитических инструментов, который требовался до сих пор.
Целью данного способа является обеспечение точного и стабильного измерения PS80, а также предоставление ценной информации для однозначного определения основной причины деградации PS80.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ИЗМЕРЕНИЯ КОЛИЧЕСТВА ПОЛИСОРБАТА-80 С ПРИМЕНЕНИЕМ ЩЕЛОЧНОГО ГИДРОЛИЗА ОБРАЗЦА С ПОСЛЕДУЮЩЕЙ ВЭЖХ | 2017 |
|
RU2670965C9 |
| Способ количественного определения леводопы в плазме крови | 2017 |
|
RU2665164C1 |
| Способ определения топирамата в плазме крови | 2016 |
|
RU2631613C1 |
| ГЛЮКОЗАМИНОВЫЕ ДИСАХАРИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2154068C2 |
| Способ количественного определения амантадина в плазме крови | 2017 |
|
RU2650968C1 |
| СОСТАВЫ ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ И РОДСТВЕННЫЕ СПОСОБЫ | 2008 |
|
RU2485975C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КАРНОЗИНА В БИОЛОГИЧЕСКИХ МАТЕРИАЛАХ | 2015 |
|
RU2585115C1 |
| ПРОИЗВОДНЫЕ 5Н-ФУРАН-2-ОНА ДЛЯ СТАБИЛИЗАЦИИ ОРГАНИЧЕСКОГО МАТЕРИАЛА | 2012 |
|
RU2605940C2 |
| КАПСУЛЫ С РАСТВОРИМЫМ ЭСТРАДИОЛОМ ДЛЯ ИНТРАВАГИНАЛЬНОГО ВВЕДЕНИЯ | 2013 |
|
RU2740059C2 |
| Способ контроля содержания противотуберкулёзных препаратов основного ряда и их токсичных метаболитов в плазме крови | 2018 |
|
RU2702998C1 |

Группа изобретений относится к области аналитической химии. Раскрыт способ количественного определения по меньшей мере одного полисорбата или продукта окисления и/или гидролиза полисорбата в образце, включающий в себя этап проведения анализа LC-MS указанного образца на основе сигнала иона диоксоланилия и этап выполнения внутренней калибровки с использованием внутреннего стандарта указанного полисорбата. Также раскрыт способ мониторинга деградации по меньшей мере одного полисорбата в образце. Группа изобретений обеспечивает эффективный и надежный способ, позволяющий осуществлять количественное определение полисорбатов в одном анализе. 2 н. и 11 з.п. ф-лы, 10 ил., 2 табл., 1 пр.
1. Способ количественного определения по меньшей мере одного полисорбата или продукта окисления и/или гидролиза полисорбата в образце, включающий в себя:
- этап проведения анализа LC-MS указанного образца на основе сигнала иона диоксоланилия;
- этап выполнения внутренней калибровки с использованием внутреннего стандарта указанного полисорбата,
где внутренний стандарт представляет собой этоксилированную жирную кислоту с формулой (I):
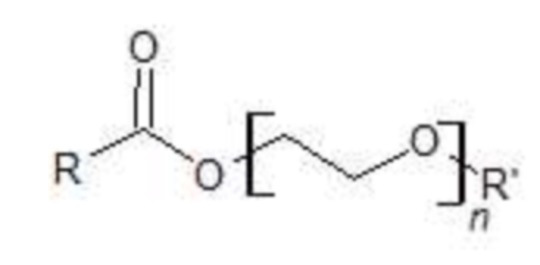 (I)
(I)
где:
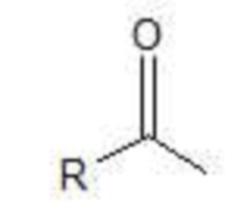 представляет остаток жирной кислоты,
представляет остаток жирной кислоты,
где R представляет собой C3-C24 линейный или разветвленный насыщенный алкил;
R’ представляет собой H или 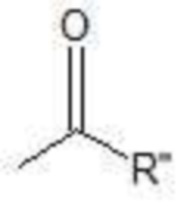 , где R’’ представляет собой C3-C24 линейный или разветвленный насыщенный алкил; и
, где R’’ представляет собой C3-C24 линейный или разветвленный насыщенный алкил; и
n составляет от 1 до 100;
или их смеси.
2. Способ по п. 1, в котором указанный полисорбат представляет собой PS20 или PS80.
3. Способ по любому из предшествующих пунктов, в котором указанный внутренний стандарт выбирают из:
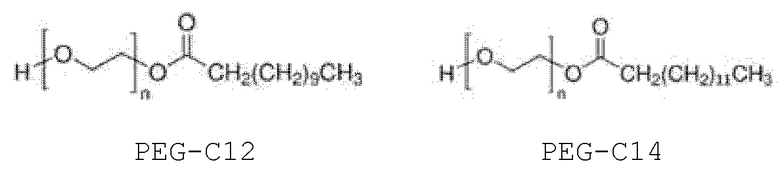
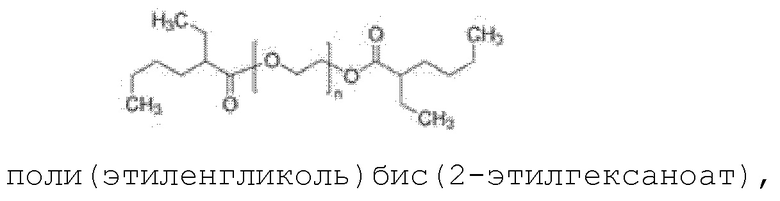
где n составляет от 1 до 100.
4. Способ по любому из предшествующих пунктов, в котором указанным внутренним стандартом является 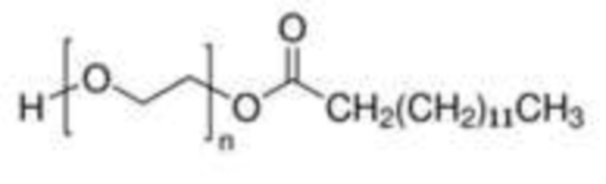 , и n составляет от 1 до 100.
, и n составляет от 1 до 100.
5. Способ по любому из предшествующих пунктов, в котором в указанном анализе LC-MS используется одиночный квадрупольный масс-детектор (QDa).
6. Способ по любому из предшествующих пунктов, в котором подвижная фаза LC-MS содержит градиент муравьиной кислоты.
7. Способ по любому из предшествующих пунктов, в котором подвижная фаза LC-MS представляет собой тройную подвижную фазу.
8. Способ по п. 7, в котором тройная подвижная фаза содержит воду, ацетонитрил и муравьиную кислоту.
9. Способ по любому из предшествующих пунктов, в котором указанный образец представляет собой биофармацевтический состав.
10. Способ по любому из предшествующих пунктов, в котором указанный образец содержит по меньшей мере один белок.
11. Способ по любому из предшествующих пунктов, в котором указанный образец содержит по меньшей мере одно моноклональное антитело.
12. Способ по любому из предшествующих пунктов, в котором указанный способ также включает в себя этап обнаружения окисления и/или гидролиза указанного полисорбата.
13. Способ мониторинга деградации по меньшей мере одного полисорбата в образце, включающий в себя реализацию способа по любому из предшествующих пунктов.
| BORISOV O.V | |||
| et al | |||
| Toward Understanding Molecular Heterogeneity of Polysorbates by Application of Liquid Chromatography-Mass Spectrometry with Computer-Aided Data Analysis // Anal | |||
| Chem., 2011, V.83, pp.3934-3942 | |||
| СПОСОБ ИЗМЕРЕНИЯ КОЛИЧЕСТВА ПОЛИСОРБАТА-80 С ПРИМЕНЕНИЕМ ЩЕЛОЧНОГО ГИДРОЛИЗА ОБРАЗЦА С ПОСЛЕДУЮЩЕЙ ВЭЖХ | 2017 |
|
RU2670965C9 |
| US 20150129766 A1, 14.05.2015 | |||
| ЛЕБЕДЕВ А.Т | |||
| Масс-спектрометрия в органической химии // Изд.: "Бином | |||
