Область техники
Настоящее изобретение относится к новому промежуточному соединению для получения длительно действующего конъюгата лекарственного средства, к содержащей его композиции и к способу получения длительно действующего конъюгата лекарственного средства с его использованием.
Предшествующий уровень техники
Физиологически активные полипептиды легко денатурируют вследствие низкой стабильности, расщепляются протеазами в крови и легко удаляются почками или печенью. Таким образом, чтобы поддержать в крови концентрацию и титр белкового лекарственного средства, содержащего физиологически активный полипептид в качестве фармакологического компонента, необходимо частое введение такого белкового лекарственного средства пациенту. Однако, поскольку большинство белковых лекарственных средств вводят пациентам в форме инъекции, частое введение посредством инъекции с целью поддержания концентрации в крови физиологически активного полипептида вызывает сильную боль у пациентов и увеличивает затраты на лечение. Чтобы решить эти проблемы, были предприняты усилия, направленные на максимальное увеличение эффективности белковых лекарственных средств путем повышения стабильности белковых лекарственных средств в крови и поддержания их концентрации в крови на высоком уровне в течение длительного периода времени. Однако такие длительно действующие композиции белковых лекарственных средств не должны вызывать иммунных ответов у пациентов при одновременном повышении стабильности белковых лекарственных средств и поддержании титра лекарственного средства на достаточно высоком уровне.
В качестве способа стабилизации белков, ингибирования контакта с протеазами и подавления почечного клиренса традиционно используют способ химического присоединения высокорастворимого полимера, такого как полиэтиленгликоль (далее обозначаемый как «ПЭГ») к поверхностям белковых лекарственных средств. Однако, хотя способа с использованием ПЭГ может увеличить продолжительность действия пептидного лекарственного средства in vivo в результате повышения молекулярной массы за счет присоединения ПЭГ, титр пептидного лекарственного средства значительно снижается при увеличении молекулярной массы, а выход может снижаться из-за низкой способности вступать во взаимодействие с пептидом.
Следовательно, в качестве метода увеличения периода полувыведения из сыворотки крови использовали конъюгат иммуноглобулинового фрагмента и физиологически активного полипептида и проводили различные исследования по улучшению способов его получения (выложенная для всеобщего ознакомления публикация корейской патентной заявки №10-2014-0109342).
Соответственно, неуклонно возрастает потребность в разработке эффективных способов получения длительно действующего конъюгата лекарственного средства путем упрощения существующего способа получения.
Описание
Техническая задача
Целью настоящего изобретения является предложение нового промежуточного соединения для получения длительно действующего конъюгата лекарственного средства.
Другой целью настоящего изобретения является предложение композиции для получения длительно действующего конъюгата лекарственного средства, включающей данное промежуточное соединение.
Другой целью настоящего изобретения является предложение способа получения длительно действующего конъюгата лекарственного средства с использованием данного промежуточного соединения.
Другой целью настоящего изобретения является предложение длительно действующего конъюгата лекарственного средства, полученного посредством данного способа получения.
Техническое решение
В одном аспекте настоящего изобретения предложено новое промежуточное соединение.
В одном воплощении настоящего изобретения предложено соединение, имеющее структуру приведенной ниже формулы 1, или его стереоизомер, сольват, или их фармацевтически приемлемая соль:
Измененное описание изобретения
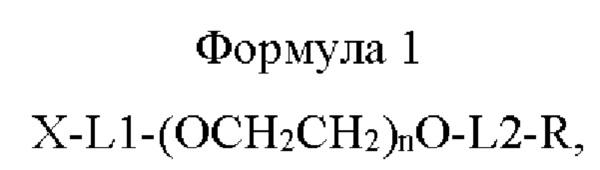
где в приведенной выше формуле 1
X представляет собой Fc-область иммуноглобулина;
L1 представляет собой C1-С6алкилен с прямой или разветвленной цепью;
L2 представляет собой -a1-CONH-, -a1-NHCO-, -a1-NHCO-a2-, -СОО-, -b1-СОО-, -СОО-b2- или -b1-СОО-b2-, и каждый из a1, а2, b1 и b2 независимо представляет собой C1-С6алкилен с прямой или разветвленной цепью;
n равно от 10 до 2400; и
R представляет собой радикал, выбранный из группы, состоящей из 2,5-диоксопирролидинила, 2,5-диоксопирролила, альдегида, малеимида, С6-С20 арилдисульфида, С5-С20 гетероарилдисульфида, винилсульфона, тиола, галогенированного ацетамида, сукцинимида, n-нитрофенилкарбоната, сложного тиоэфира и их производных.
В соединении или его стереоизомере, сольвате, либо их фармацевтически приемлемой соли согласно предшествующему воплощению в формуле 1 L1 представляет собой C1-С6алкилен с прямой или разветвленной цепью; L2 представляет собой -a1-NHCO- или -a1-NHCO-a2-; каждый из a1 и а2 независимо представляет собой C1-С6алкилен с прямой или разветвленной цепью; n равно от 200 до 250; и R представляет собой малеимид.
В соединении или его стереоизомере, сольвате, либо их фармацевтически приемлемой соли по любому из предшествующих воплощений X представляет собой Fc-область иммуноглобулина, содержащую последовательность шарнирной области на N-конце.
В соединении или его стереоизомере, сольвате, либо их фармацевтически приемлемой соли по любому из предшествующих воплощений X включает последовательность шарнирной области, модифицированную так, чтобы она включала только один остаток цистеина, посредством делетирования части аминокислотной последовательности из приведенных ниже аминокислот:
Glu-Ser-Lys-Tyr-Gly-Pro-Pro-Cys-Pro-Ser-Cys-Pro (SEQ ID NO: 7).
В соединении или его стереоизомере, сольвате, либо их фармацевтически приемлемой соли по любому из предшествующих воплощений последовательность шарнирной области включает аминокислотную последовательность с SEQ ID NO: 8 (Ser-Cys-Pro) или аминокислотную последовательность с SEQ ID NO: 9 (Pro-Ser-Cys-Pro).
В соединении или его стереоизомере, сольвате, либо их фармацевтически приемлемой соли по любому из предшествующих воплощений L1 соединен с реакционноспособной аминной или тиоловой группой, расположенной на одном конце X.
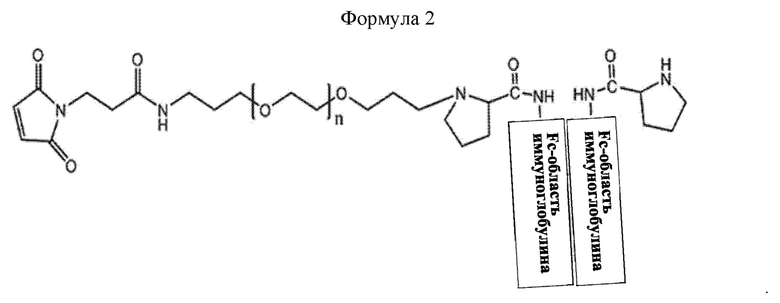
В соединении или его стереоизомере, сольвате, либо их фармацевтически приемлемой соли по любому из предшествующих воплощений соединение имеет структуру приведенной ниже формулы 2:
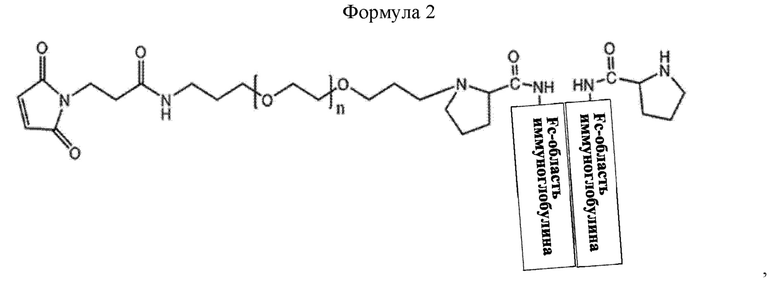
где в формуле 2 n равно от 200 до 250.
В соединении или его стереоизомере, сольвате, либо их фармацевтически приемлемой соли по любому из предшествующих воплощений X представляет собой Fc-область иммуноглобулина, происходящую из IgG, IgA, IgD, IgE или IgM.
В соединении или его стереоизомере, сольвате, либо их фармацевтически приемлемой соли по любому из предшествующих воплощений X представляет собой Fc-область иммуноглобулина, происходящую из IgG1, IgG2, IgG3 или IgG4.
В соединении или его стереоизомере, сольвате, либо их фармацевтически приемлемой соли по любому из предшествующих воплощений X представляет собой Fc-область иммуноглобулина в форме димера.
В соединении или его стереоизомере, сольвате, либо их фармацевтически приемлемой соли по любому из предшествующих воплощений X представляет собой Fc-область иммуноглобулина, включающую аминокислотную последовательность с SEQ ID NO: 10.
В соединении или его стереоизомере, сольвате, либо их фармацевтически приемлемой соли по любому из предшествующих воплощений соединение имеет массу от 40 кДа до 250 кДа.
Согласно другому аспекту настоящего изобретения предложена композиция для получения длительно действующего конъюгата лекарственного средства, включающая соединение или его стереоизомер, сольват, либо их фармацевтически приемлемую соль.
В одном воплощении композиция включает в себя соединение приведенной ниже формулы 1 или его стереоизомер, сольват, либо их фармацевтически приемлемую соль, где лекарственное средство представляет собой физиологически активный полипептид:
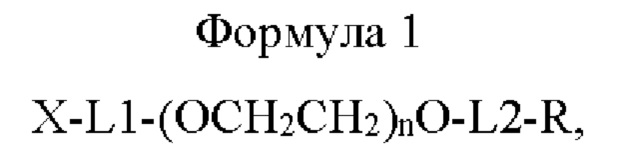
где в приведенной выше формуле 1
X представляет собой Fc-область иммуноглобулина;
L1 представляет собой C1-С6алкилен с прямой или разветвленной цепью;
L2 представляет собой -a1-CONH-, -a1-NHCO-, -a1-NHCO-a2-, -СОО-, -N-СОО-, -СОО-b2- или -b1-СОО-b2-, и каждый из a1, а2, b1 и b2 независимо представляет собой C1-С6алкилен с прямой или разветвленной цепью;
n равно от 10 до 2400; и
R представляет собой радикал, выбранный из группы, состоящей из 2,5-диоксопирролидинила, 2,5-диоксопирролила, альдегида, малеимида, С6-С20арилдисульфида, С5-С20 гетероарилдисульфида, винилсульфона, тиола, галогенированного ацетамида, сукцинимида, n-нитрофенилкарбоната, сложного тиоэфира и их производных.
В композиции по предшествующему воплощению
L1 представляет собой C1-С6алкилен с прямой или разветвленной цепью; L2 представляет собой -a1-NHCO- или -a1-NHCO-a2-;
каждый из a1 и а2 независимо представляет собой C1-С6алкилен с прямой или разветвленной цепью;
n равно от 200 до 250; и
R представляет собой малеимид.
В композиции по любому из предшествующих воплощений физиологически активный полипептид выбран из группы, состоящей из глюкагоноподобного пептида-1 (GLP-1), гранулоцитарного колониестимулирующего фактора (G-CSF), гормона роста человека (hGH), эритропоэтина (ЕРО), глюкагона, инсулина, рилизинг-фактора гормона роста; пептида, высвобождающего гормон роста; интерферонов, рецепторов интерферонов; рецепторов сопряженных с G-белком; интерлейкинов, рецепторов интерлейкинов, ферментов, интерлейкин-связывающего белка, цитокин-связывающего белка, фактора активации макрофагов, макрофагального пептида, фактора В-клеток, фактора Т-клеток, белка А, ингибитора аллергии; гликопротеина, ассоциированного с некрозом клеток; иммунотоксина, лимфотоксина, фактора некроза опухолей, онкосупрессора, фактора роста метастазов, α-1-антитрипсина, альбумина, α-лактальбумина, аполипопротеина Е, высокогликозилированного эритропоэтина, ангиопоэтинов, гемоглобина, тромбина; пептида, активирующего рецепторы тромбина; тромбомодулина, факторов крови VII, VIIa, VIII, IX и XIII, плазминоген-активирующего фактора, фибрин-связывающего пептида, урокиназы, стрептокиназы, гирудина, белка С, С-реактивного белка, ингибитора lenin, ингибитора коллагеназы, супероксиддисмутазы, лептина, фактора роста тромбоцитов, эпителиального фактора роста, эпидермального фактора роста, ангиостатина, ангиотензина, фактора роста костей, костного морфогенетического белка, кальцитонина, атриопептина; фактора, индуцирующего хрящеобразование; элкатонина; фактора, активирующего соединительную ткань; ингибитора пути тканевого фактора, фолликулостимулирующего гормона, лютеинизирующего гормона, рилизинг-фактора лютеинизирующего гормона, фактора роста нервов, гормона паращитовидной железы, релаксина, секретина, соматомедина, инсулиноподобного фактора роста, гормона коры надпочечников, холецистокинина, панкреатического полипептида, гастрин-высвобождающего пептида, кортикотропин-рилизинг-фактора, тиреотропного гормона, аутотаксина, лактоферрина, миостатина, инкретинов, желудочного ингибирующего полипептида (GIP), двойного агониста GLP-1/GIP, тройного агониста GLP-1/GIP/глюкагона, антигенов клеточной поверхности; вакцинных антигенов вирусного происхождения; моноклональных антител, поликлональных антител и фрагментов антител.
В композиции по любому из предшествующих воплощений, R в соединении, входящем в состав композиции, соединен с остатком цистеина лекарственного средства.
В композиции по любому из предшествующих воплощений, X представляет собой Fc-область иммуноглобулина, происходящую из IgG1, IgG2, IgG3 или IgG4.
В композиции по любому из предшествующих воплощений X представляет собой Fc-область иммуноглобулина, содержащую последовательность шарнирной области, модифицированную так, чтобы она включала только один остаток цистеина, посредством делетирования части аминокислотной последовательности из приведенных ниже аминокислот:
Glu-Ser-Lys-Tyr-Gly-Pro-Pro-Cys-Pro-Ser-Cys-Pro (SEQ ID NO: 7).
Согласно еще одному аспекту настоящего изобретения предложен способ получения длительно действующего конъюгата физиологически активного полипептида.
В одном воплощении способ получения включает получение конъюгата путем присоединения моно-ПЭГилированной Fc-области иммуноглобулина, полученной посредством соединения линкера приведенной ниже формулы 3 с N-концом Fc-области иммуноглобулина, содержащей последовательность шарнирной области, к физиологически активному полипептиду:
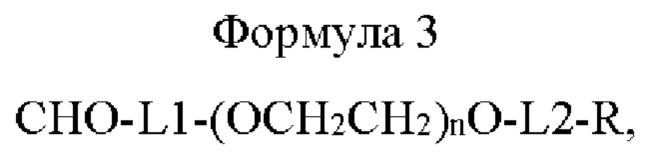
где в формуле 3
L1 представляет собой C1-С6алкилен с прямой или разветвленной цепью;
L2 представляет собой -a1-CONH-, -a1-NHCO-, -a1-NHCO-a2-, -СОО-, -N-СОО-, -СОО-b2- или -b1-СОО-b2-, и каждый из a1, а2, b1 и b2 независимо представляет собой C1-С6алкилен с прямой или разветвленной цепью;
n равно от 10 до 2400; и
R представляет собой радикал, выбранный из группы, состоящей из 2,5-диоксопирролидинила, 2,5-диоксопирролила, альдегида, малеимида, С6-С20 арилдисульфида, С5-С20 гетероарилдисульфида, винилсульфона, тиола, галогенированного ацетамида, сукцинимида, n-нитрофенилкарбоната, сложного тиоэфира и их производных.
В способе получения по предшествующему воплощению моно-ПЭГилированную Fc-область иммуноглобулина получают путем соединения линкера приведенной выше формулы 3 с N-концом Fc-области иммуноглобулина при рН 4,0-8,0 в присутствии восстанавливающего агента.
В способе получения по любому из предшествующих воплощений конъюгат получают путем присоединения линкера моно-ПЭГилированной Fc-области иммуноглобулина к физиологически активному полипептиду при рН 5,5-8,0.
В способе получения по любому из предшествующих воплощений получение конъюгата осуществляют путем приведения моно-ПЭГилированной Fc-области иммуноглобулина во взаимодействие с физиологически активным полипептидом в молярном соотношении от 1:1 до 1:3.
При получении согласно любому из предшествующих воплощений способ включает: получение моно-ПЭГилированной Fc-области иммуноглобулина путем соединения линкера формулы 3 с N-концом Fc-области иммуноглобулина; и получение конъюгата путем присоединения линкера моно-ПЭГилированной Fc-области иммуноглобулина, полученной на предыдущей стадии, к физиологически активному полипептиду.
При получении по любому из предшествующих воплощений, линкер моно-ПЭГилированной Fc-области иммуноглобулина соединяют с цистеином физиологически активного полипептида.
При получении по любому из предшествующих воплощений, способ включает: получение моно-ПЭГилированной Fc-области иммуноглобулина путем соединения линкера формулы 3 с N-концом Fc-области иммуноглобулина; очистку моно-ПЭГилированной Fc-области иммуноглобулина, полученной на предыдущей стадии, посредством анионообменной хроматографии в буферном растворе с рН 6,0-8,5; и получение конъюгата путем присоединения линкера моно-ПЭГилированной Fc-области иммуноглобулина, очищенной на предыдущей стадии, к физиологически активному полипептиду.
При получении по любому из предшествующих воплощений способ осуществляют без ультрафильтрации/диафильтрации после получения моно-ПЭГилированной Fc-области иммуноглобулина.
При получении по любому из предшествующих воплощений, способ дополнительно включает очистку конъюгата посредством гидрофобной хроматографии.
При получении по любому из предшествующих воплощений, в формуле 3 L1 представляет собой C1-С6алкилен с прямой или разветвленной цепью; L2 представляет собой -a1-NHCO- или -a1-NHCO-a2-; каждый из a1 и а2 независимо представляет собой C1-С6алкилен с прямой или разветвленной цепью; n равно от 200 до 250; и R представляет собой малеимид.
При получении по любому из предшествующих воплощений, линкер имеет структуру приведенной ниже формулы 4:
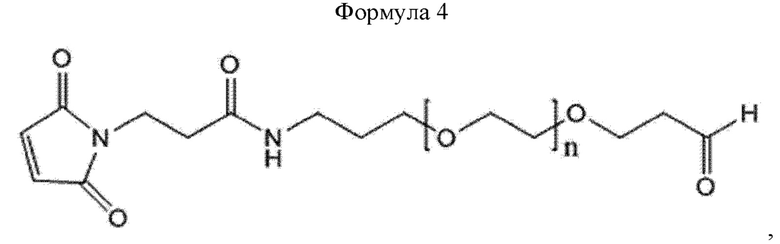
где в формуле 4 n равно от 200 до 250.
При получении по любому из предшествующих воплощений линкер имеет массу от 1 кДа до 100 кДа.
При получении по любому из предшествующих воплощений, физиологически активный полипептид выбран из группы, состоящей из глюкагоноподобного пептида-1 (GLP-1), гранулоцитарного колониестимулирующего фактора (G-CSF), гормона роста человека (hGH), эритропоэтина (ЕРО), глюкагона, инсулина, рилизинг-фактора гормона роста; пептида, высвобождающего гормон роста; интерферонов, рецепторов интерферонов; рецепторов, сопряженных с G-белком; интерлейкинов, рецепторов интерлейкинов, ферментов, интерлейкин-связывающего белка, цитокин-связывающего белка, фактора активации макрофагов, макрофагального пептида, фактора В-клеток, фактора Т-клеток, белка А, ингибитора аллергии; гликопротеина, ассоциированного с некрозом клеток; иммунотоксина, лимфотоксина, фактора некроза опухолей, онкосупрессора, фактора роста метастазов, α-1-антитрипсина, альбумина, α-лактальбумина, аполипопротеина Е, высокогликозилированного эритропоэтина, ангиопоэтинов, гемоглобина, тромбина; пептида, активирующего рецепторы тромбина; тромбомодулина, факторов крови VII, VIIa, VIII, IX и XIII, плазминоген-активирующего фактора, фибрин-связывающего пептида, урокиназы, стрептокиназы, гирудина, белка С, С-реактивного белка, ингибитора lenin, ингибитора коллагеназы, супероксиддисмутазы, лептина, фактора роста тромбоцитов, эпителиального фактора роста, эпидермального фактора роста, ангиостатина, ангиотензина, фактора роста костей, костного морфогенетического белка, кальцитонина, атриопептина; фактора, индуцирующего хрящеобразование; элкатонина; фактора, активирующего соединительную ткань; ингибитора пути тканевого фактора, фолликулостимулирующего гормона, лютеинизирующего гормона, рилизинг-фактора лютеинизирующего гормона, фактора роста нервов, гормона паращитовидной железы, релаксина, секретина, соматомедина, инсулиноподобного фактора роста, гормона коры надпочечников, холецистокинина, панкреатического полипептида, гастрин-высвобождающего пептида, кортикотропин-рилизинг-фактора, тиреотропного гормона, аутотаксина, лактоферрина, миостатина, инкретинов, желудочного ингибирующего полипептида (GIP), двойного агониста GLP-1/GIP, тройного агониста GLP-1/GIP/глюкагона, антигенов клеточной поверхности; вакцинных антигенов вирусного происхождения; моноклональных антител, поликлональных антител и фрагментов антител.
При получении по любому из предшествующих воплощений, физиологически активный полипептид представляет собой тройной агонист GLP-1/GIP/глюкагона или его аналог.
При получении по любому из предшествующих воплощений, физиологически активный полипептид включает одну из аминокислотных последовательностей с SEQ ID NO: 1-6.
При получении по любому из предшествующих воплощений, последовательность шарнирной области модифицируют так, чтобы она включала только один остаток цистеина, посредством делетирования части аминокислотной последовательности, имеющей приведенную ниже аминокислотную последовательность:
Glu-Ser-Lys-Tyr-Gly-Pro-Pro-Cys-Pro-Ser-Cys-Pro (SEQ ID NO: 7).
При получении по любому из предшествующих воплощений, последовательность шарнирной области включает аминокислотную последовательность с SEQ ID NO: 8 (Ser-Cys-Pro) или аминокислотную последовательность с SEQ ID NO: 9 (Pro-Ser-Cys-Pro).
При получении по любому из предшествующих воплощений, Fc-область иммуноглобулина происходит из IgG1, IgG2, IgG3 или IgG4.
Согласно еще одному аспекту настоящего изобретения, предложен длительно действующий конъюгат лекарственного средства, полученный с использованием данной композиции или данного способа.
Полезные эффекты
Согласно способу получения длительно действующего конъюгата лекарственного средства с использованием нового промежуточного соединения по настоящему изобретению, длительно действующий конъюгат лекарственного средства может быть получен с высоким выходом несмотря на то, что некоторые из традиционных процессов очистки исключены, и, в результате этого, эффективность получения длительно действующего конъюгата лекарственного средства может быть повышена.
Краткое описание графических материалов
На ФИГ. 1 показаны результаты анализа структуры моно-ПЭГилированной Fc-области иммуноглобулина посредством MALDI-TOF (время-пролетная масс-спектрометрии с матрично-активированной лазерной десорбцией/ионизацией).
Наилучший вариант осуществления изобретения
Далее будут подробно описаны воплощения настоящего изобретения.
При этом каждое описание и воплощение, раскрытое в настоящем изобретении, может быть использовано в данном описании изобретения для описания других описаний и воплощений. Другими словами, все комбинации различных компонентов, раскрытых в настоящем изобретении, включены в объем настоящего изобретения. Кроме того, объем настоящего изобретения не следует ограничивать приведенным ниже подробным описанием.
Кроме того, специалистам в данной области техники известны или они могут найти, используя только обычное экспериментирование, многие эквиваленты конкретных воплощений настоящего изобретения. Подразумевается, что такие эквиваленты включены в объем следующей ниже формулы изобретения.
По всему тексту описания использованы не только общепринятые однобуквенные коды и трехбуквенные обозначения природных аминокислот, но также и такие, обычно используемые для других аминокислот, 3-буквенные обозначения, как Aib (а-аминоизомасляная кислота), Sar (N-метилглицин) и а-метил-глутаминовая кислота. Кроме того, аминокислоты указаны в данном описании в виде аббревиатур в соответствии с правилами наименования IUPAC-IUB (Международный союз по теоретической и прикладной химии/Международный биохимический союз), следующим образом.
Согласно одному из аспектов настоящего изобретения предложено соединение, имеющее структуру приведенной ниже формулы 1 или его стереоизомер, сольват, либо их фармацевтически приемлемая соль:
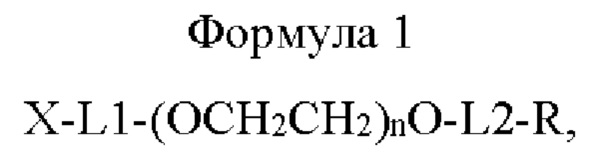
где в приведенной выше формуле 1
X представляет собой Fc-область иммуноглобулина;
L1 представляет собой C1-С6алкилен с прямой или разветвленной цепью;
L2 представляет собой -a1-CONH-, -a1-NHCO-, -a1-NHCO-a2-, -СОО-, -N-СОО-, -СОО-b2- или -b1-СОО-b2-, и каждый из a1, а2, b1 и b2 независимо представляет собой C1-С6алкилен с прямой или разветвленной цепью;
n равно от 10 до 2400; и
R представляет собой радикал, выбранный из группы, состоящей из 2,5-диоксопирролидинила, 2,5-диоксопирролила, альдегида, малеимида, С6-С20 арилдисульфида, С5-С20 гетероарилдисульфида, винилсульфона, тиола, галогенированного ацетамида, сукцинимида, n-нитрофенилкарбоната, сложного тиоэфира и их производных.
В настоящем изобретении соединение, имеющее структуру формулы 1 или его стереоизомер, сольват, либо их фармацевтически приемлемая соль представляет собой новое вещество, полученное с целью получения длительно действующего конъюгата, и также может упоминаться в настоящей заявке как «промежуточное соединение» или «промежуточное вещество».
В способе получения длительно действующего конъюгата лекарственного средства с использованием промежуточного соединения по настоящему изобретению стадии очистки посредством ультрафильтрации/диафильтрации и гидрофобной хроматографии могут быть исключены и, несмотря на отсутствие стадий очистки, можно в результате получить длительно действующий конъюгат лекарственного средства с высоким выходом.
Конкретно, промежуточное соединение находится в форме, в которой Fc-область иммуноглобулина соединена с линкером. В промежуточном соединении формулы 1, X может представлять собой Fc-область иммуноглобулина, и L1-(OCH2CH2)nO-L2-R может представлять собой линкер. Конкретно, в приведенной выше формуле 1, L1 представляет собой C1-С6алкилен с прямой или разветвленной цепью; L2 представляет собой -a1-NHCO- или -a1-NHCO-a2-; каждый из a1 и а2 независимо представляет собой C1-С6алкилен с прямой или разветвленной цепью; n равно от 200 до 250; и R представляет собой малеимид, не ограничиваясь этим.
L1 представляет собой место связывания с Fc-областью иммуноглобулина и может представлять собой C1-С6алкилен с прямой или разветвленной цепью, не ограничиваясь этим. L1 может быть соединен с реакционноспособной аминогруппой, расположенной на одном конце X, или остатком лизина в X, или реакционноспособной тиоловой группой, не ограничиваясь этим. R представляет собой место образования связи между промежуточным соединением и физиологически активным полипептидом и, конкретно, может включать реакционноспособную группу (например тиоловую, малеимидную, альдегидную и сукцинимидильную), способную связываться с цистеином, или с аминогруппой на N-конце, или с остатком лизина физиологически активного полипептида, не ограничиваясь этим.
В настоящем изобретении промежуточное соединение может иметь структуру приведенной ниже формулы 2, но не ограничивается этим:
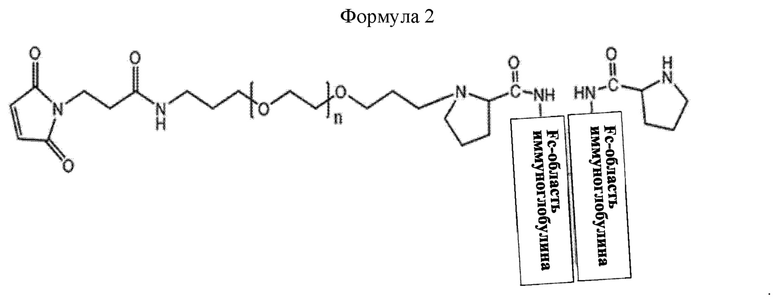
В приведенной выше формуле 2, n равно от 1 до 3000, от 10 до 2000, от 50 до 1000, от 100 до 700, от 150 до 300 или от 200 до 250.
Конкретно, промежуточное соединение может иметь массу от 40 кДа до 250 кДа, от 80 кДа до 200 кДа или от 100 кДа до 150 кДа, не ограничиваясь этим.
В настоящем изобретении X может представлять собой Fc-область иммуноглобулина, имеющую последовательность шарнирной области на N-конце и, в частности, последовательность шарнирной области может быть модифицирована так, чтобы она включала только один остаток цистеина, посредством делетирования части аминокислотной последовательности из приведенных ниже аминокислот, не ограничиваясь этим:
Glu-Ser-Lys-Tyr-Gly-Pro-Pro-Cys-Pro-Ser-Cys-Pro (SEQ ID NO: 7).
Используемый в данном описании термин «Fc-область иммуноглобулина» относится к области, содержащей константный домен 2 тяжелой цепи (СН2) и/или константный домен 3 тяжелой цепи (CH3), за исключением вариабельных доменов тяжелой цепи и легкой цепи иммуноглобулина. Fc-область иммуноглобулина может представлять собой компонент, составляющий группировку длительно действующего конъюгата лекарственного средства по настоящему изобретению.
Конкретно, X может представлять собой Fc-область иммуноглобулина, происходящую из IgG, IgA, IgD, IgE или IgM, более конкретно, Fc-область иммуноглобулина, происходящую из IgG1, IgG2, IgG3 или IgG4, не ограничиваясь этим.
В настоящем изобретении Fc-область иммуноглобулина может содержать конкретную последовательность шарнирной области на N-конце.
Использованный в данном описании термин «последовательность шарнирной области» относится к области, расположенной в тяжелой цепи и формирующей димер Fc-области иммуноглобулина посредством образования внутримолекулярной дисульфидной связи.
Использованный в данном описании термин «N-конец» относится к аминному концу белка или полипептида и может содержать аминокислотный остаток, расположенный на конце аминного конца или на расстоянии 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более аминокислот от окончания аминного конца. Fc-область иммуноглобулина по настоящему изобретению может содержать последовательность шарнирной области на N-конце, не ограничиваясь этим.
С учетом целей настоящего изобретения, последовательность шарнирной области может содержать только один остаток цистеина, поскольку остаток цистеина, расположенный в 8-ом или 11-ом положении последовательности шарнирной области с SEQ ID NO: 7, делетирован. Последовательность шарнирной области по настоящему изобретению может состоять из 3-12 аминокислот, включающих только один остаток цистеина, не ограничиваясь этим. Более конкретно, шарнирная область по настоящему изобретению может иметь следующую последовательность: Glu-Ser-Lys-Tyr-Gly-Pro-Pro-Pro-Ser-Cys-Pro, Glu-Ser-Lys-Tyr-Gly-Pro-Pro-Cys-Pro-Ser-Pro, Glu-Ser-Lys-Tyr-Gly-Pro-Pro-Cys-Pro-Ser, Glu-Ser-Lys-Tyr-Gly-Pro-Pro-Cys-Pro-Pro, Lys-Tyr-Gly-Pro-Pro-Cys-Pro-Ser, Glu-Ser-Lys-Tyr-Gly-Pro-Pro-Cys, Glu-Lys-Tyr-Gly-Pro-Pro-Cys, Glu-Ser-Pro-Ser-Cys-Pro, Glu-Pro-Ser-Cys-Pro, Pro-Ser-Cys-Pro, Glu-Ser-Lys-Tyr-Gly-Pro-Pro-Ser-Cys-Pro, Lys-Tyr-Gly-Pro-Pro-Pro-Ser-Cys-Pro, Glu-Ser-Lys-Tyr-Gly-Pro-Ser-Cys-Pro, Glu-Ser-Lys-Tyr-Gly-Pro-Pro-Cys, Lys-Tyr-Gly-Pro-Pro-Cys-Pro, Glu-Ser-Lys-Pro-Ser-Cys-Pro, Glu-Ser-Pro-Ser-Cys-Pro или Glu-Pro-Ser-Cys. Более конкретно, шарнирная область может включать аминокислотную последовательность с SEQ ID NO: 8 (Ser-Cys-Pro) или последовательность с SEQ ID NO: 9 (Pro-Ser-Cys-Pro), не ограничиваясь этим.
X может представлять собой димер, образованный двухцепочечными молекулами Fc-области иммуноглобулина в присутствии последовательности шарнирной области, и промежуточное соединение по настоящему изобретению может находиться в форме, где один конец линкера соединен с одной цепью Fc-области иммуноглобулина в димерной форме, не ограничиваясь этим. В настоящем изобретении X может представлять собой димер Fc-области иммуноглобулина, но этим не ограничивается.
Кроме того, X по настоящему изобретению может представлять собой Fc-область иммуноглобулина, включающую аминокислотную последовательность с SEQ ID NO: 10, не ограничиваясь этим.
При этом, Fc-область иммуноглобулина по настоящему изобретению может представлять собой расширенную Fc-область, содержащую часть константного домена 1 тяжелой цепи (СН1) или его целиком, и/или константный домен 1 легкой цепи (CL1), за исключением вариабельных доменов тяжелой цепи и легкой цепи иммуноглобулина, при условии, что Fc-область иммуноглобулина оказывает по существу такие же или улучшенные эффекты по сравнению с нативным типом. Кроме того, Fc-область иммуноглобулина может представлять собой область, где удалена довольно длинная часть аминокислотной последовательности, соответствующей СН2 и/или СН3.
Например, Fc-область иммуноглобулина по настоящему изобретению может включать 1) CH1-домен, СН2-домен, СН3-домен и СН4-домен, 2) СН1-домен и СН2-домен, 3) CH1-домен и СН3-домен, 4) СН2-домен и СН3-домен, 5) комбинацию из одного или более доменов, выбранного(ых) из CH1-домена, СН2-домена, СН3-домена и СН4-домена, и шарнирной области (или части шарнирной области) иммуноглобулина или 6) димер каждого из доменов, константного домена тяжелой цепи и константного домена легкой цепи. Однако настоящее изобретение этим не ограничивается.
Кроме того, Fc-область иммуноглобулина по настоящему изобретению содержит не только природную аминокислотную последовательность, но также производное этой последовательности. Термин «производное аминокислотной последовательности» относится к последовательности, которая отличается от природной аминокислотной последовательности наличием делеции, вставки, неконсервативной или консервативной замены, либо любой комбинации одной или более аминокислот в природной аминокислотной последовательности.
Например, в случае IgG Fc, аминокислотные остатки в положениях 214-238, 297-299, 318-322 или 327-331, которые, как известно, важны для связывания, могут быть использованы в качестве мишени, подходящей для модификации.
Также можно использовать другие различные производные, включая те из них, в которых делетирован сайт, способный к образованию дисульфидной связи, или удалены некоторые аминокислотные остатки с N-конца нативной формы Fc-области и к N-концу нативной формы Fc присоединен остаток метионина. Кроме того, для устранения эффекторных функций, может быть делетирован комплементсвязывающий сайт, такой как сайт связывания C1q, и может быть делетирован сайт, связанный с антителозависимой клеточно-опосредованной цитотоксичностью (ADCC). Методы получения таких производных последовательности Fc-области иммуноглобулина раскрыты в международных патентных публикациях WO 97/34631 и WO 96/32478.
В данной области техники известны аминокислотные замены в белках и пептидах, которые обычно не изменяют активности молекул (Н. Neurath, R. L. Hill, The Proteins, Academic Press, New York, 1979). Наиболее часто встречающимися заменами аминокислотных остатков являются замены Ala/Ser, Val/Ile, Asp/Glu, Thr/Ser, Ala/Gly, Ala/Thr, Ser/Asn, Ala/Val, Ser/Gly, Thy/Phe, Ala/Pro, Lys/Arg, Asp/Asn, Leu/Ile, Leu/Val, Ala/Glu и Asp/Gly. При необходимости, Fc-область может быть модифицирована посредством фосфорилирования, сульфатирования, акрилирования, гликозилирования, метилирования, фарнезилирования, ацетилирования и амидирования.
Описанные выше производные последовательности Fc-области представляют собой производные, которые обладают биологической активностью, эквивалентной активности Fc-области иммуноглобулина по настоящему изобретению, или улучшенной стабильностью структуры в отношении нагревания, рН или подобного.
Кроме того, такие Fc-области иммуноглобулинов могут быть получены из нативных форм, выделенных из людей и других животных, включая коров, коз, свиней, мышей, кроликов, хомячков, крыс и морских свинок, или могут представлять собой их рекомбинантные формы или производные, полученные из трансформированных клеток животных или микроорганизмов. С этой целью они могут быть получены из нативного иммуноглобулина путем выделения целых молекул иммуноглобулина из живого организма человека или животного и обработки этих молекул протеазой. Папаин расщепляет нативный иммуноглобулин на Fab- и Fc-области, а пепсин расщепляет нативный иммуноглобулин на фрагменты pF'c и F(ab)2. Эти фрагменты могут быть подвергнуты эксклюзионной хроматографии с выделением Fc или pF'c. В более конкретном воплощении, Fc-область человеческого происхождения представляет собой Fc-область рекомбинантного иммуноглобулина, полученного из микроорганизма.
Кроме того, Fc-область иммуноглобулина по настоящему изобретению может содержать природные гликаны, или содержать гликаны, увеличенные или уменьшенные по сравнению с природным типом, или находиться в дегликозилированной форме. Увеличение, уменьшение или удаление гликанов Fc-области иммуноглобулина может быть осуществлено любыми методами, обычно используемыми в данной области техники, такими как химический метод, ферментативный метод и метод генной инженерии с использованием микроорганизма. В связи с этим, Fc-область иммуноглобулина, полученная путем удаления гликанов, демонстрирует значительное уменьшение аффинности связывания с компонентом C1q и снижение или утрату антителозависимой цитотоксичности или комплеменезависимой цитотоксичности, и поэтому в живых организмах не индуцируются нежелательные иммунные ответы. На основании этого, дегликозилированная или агликозилированная Fc-область иммуноглобулина может быть более подходящей в качестве носителя лекарственного средства, исходя из целей настоящего изобретения.
Использованный в данном описании термин «дегликозилированная» относится к Fc-области, из которой ферментативно удален гликана, а термин «агликозилированная» Fc-области, которая негликозилирована и продуцируется в прокариотах, более конкретно, в Е. coli.
При этом, Fc-область иммуноглобулина может происходить из иммуноглобулина людей или животных, таких как коровы, козы, свиньи, мыши, кролики, хомячки, крысы или морские свинки. В более конкретном воплощении Fc-область иммуноглобулина может происходить из иммуноглобулина человека.
Кроме того, Fc-область иммуноглобулина может происходить из IgG, IgA, IgD, IgE или IgM, либо любой их комбинации или гибрида. В более конкретном воплощении Fc-область иммуноглобулина происходит из IgG или IgM, которые являются самыми распространенными белками в крови человека, и в еще более конкретном воплощении, она происходит из IgG, который, как известно, увеличивает периоды полувыведения лигандсвязывающих белков. В еще более конкретном воплощении Fc-область иммуноглобулина представляет собой Fc-область IgG4, и в наиболее конкретном воплощении Fc-область иммуноглобулина представляет собой агликозилированную Fc-область, происходящую из человеческого IgG4, но этим не ограничивается.
При этом, использованный в данном описании термин «комбинация», касающийся Fc-области иммуноглобулина, относится к образованию связи между полипептидом, в котором закодирована одноцепочечная Fc-область иммуноглобулина такого же происхождения, и одноцепочечным полипептидом другого происхождения с образованием димера или мультимера. То есть димер или мультимер может быть получен с использованием двух или более фрагментов Fc, выбранных из группы, состоящей из IgG Fc, IgA Fc, IgM Fc, IgD Fc и IgE Fc.
Использованный в данном описании термин «гибрид» означает, что последовательности, соответствующие двум или более Fc-областям иммуноглобулинов разного происхождения, присутствуют в одной цепи константного домена иммуноглобулина. В настоящем изобретении возможны различные гибридные формы. То есть, доменный гибрид может содержать от 1 до 4 доменов, выбранных из группы, состоящей из CH1, СН2, СН3 и СН4 из IgG Fc, IgM Fc, IgA Fc, IgE Fc и IgD Fc, и может дополнительно содержать шарнирную область.
При этом IgG также может быть подразделен на подклассы IgG1, IgG2, IgG3 и IgG4, которые могут быть объединены или гибридизированы в соответствии с настоящим изобретением. Предпочтительными являются подклассы IgG2 и IgG4 и наиболее предпочтительным является F-фрагмент из IgG4, редко обладающий эффекторными функциями, такими как комплементзависимая цитотоксичность (CDC).
Использованный в данном описании термин «линкер» относится к группировке, соединяющей лекарственное средство (например физиологически активный полипептид) с Fc-областью иммуноглобулина с образованием длительно действующего конъюгата лекарственного средства, и линкер может представлять собой пептидильный линкер или непептидильный линкер. Конкретно, линкер может быть представлен приведенной ниже формулой 3, не ограничиваясь этим:
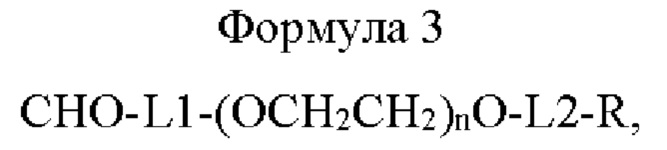
где в формуле 3
L1 представляет собой C1-С6алкилен с прямой или разветвленной цепью;
L2 представляет собой -a1-CONH-, -a1-NHCO-, -a1-NHCO-a2-, -СОО-, -N-СОО-, -СОО-b2- или -b1-СОО-b2-, и каждый из a1, а2, b1 и b2 независимо представляет собой C1-С6алкилен с прямой или разветвленной цепью;
n равно от 1 до 3000, от 10 до 2000, от 50 до 1000, от 100 до 700, от 150 до 300 или от 200 до 250; и
R представляет собой радикал, выбранный из группы, состоящей из 2,5-диоксопирролидинила, 2,5-диоксопирролила, альдегида, малеимида, С6-С20 арилдисульфида, С5-С20 гетероарилдисульфида, винилсульфона, тиола, галогенированного ацетамида, сукцинимида, n-нитрофенилкарбоната, сложного тиоэфира и их производных.
Линкер может содержать полиэтиленгликоль и имеет определенные химические структуры на обоих концах полиэтиленгликоля, не ограничиваясь этим.
Конкретно, в приведенной выше формуле 3, L1 может представлять собой C1-С6алкилен с прямой или разветвленной цепью; L2 может представлять собой -a1-NHCO- или -a1-NHCO-a2-; каждый из a1 и а2 может независимо представлять собой C1-С6алкилен с прямой или разветвленной цепью; n может составлять от 200 до 250; и R может представлять собой малеимид, и линкер может иметь массу от 1 кДа до 200 кДа, от 1 кДа до 150 кДа, от 1 кДа до 100 кДа, от 1 кДа до 50 кДа или от 1 кДа до 10 кДа, не ограничиваясь этим.
Кроме того, линкер может иметь структуру приведенной ниже формулы 4, не ограничиваясь этим:
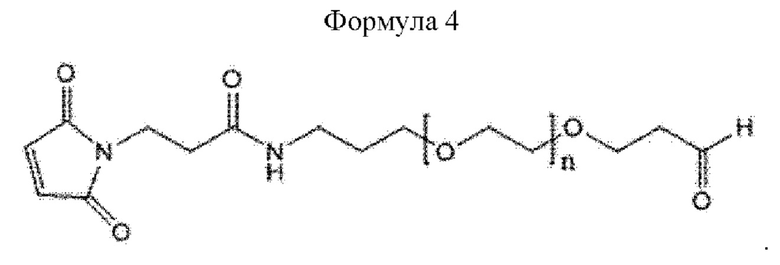
В приведенной выше формуле 4 n может составлять от 1 до 3000, от 10 до 2000, от 50 до 1000, от 100 до 700, от 150 до 300 или от 200 до 250.
Один конец линкера может быть соединен с Fc-областью иммуноглобулина, конкретно с N-концом Fc-области иммуноглобулина, более конкретно с последовательностью шарнирной области, расположенной на N-конце Fc-области иммуноглобулина, еще более конкретно с остатком пролина последовательности шарнирной области, с образованием промежуточного соединения, но этим не ограничивается.
В настоящем изобретении термин «фармацевтически приемлемый» относится к веществу, которое может быть эффективно использовано для предполагаемого применения в рамках фармако-медицинского заключения, не вызывая чрезмерных токсичности, раздражения или аллергических реакций.
Использованный в данном описании термин «фармацевтически приемлемая соль» относится к соли, происходящей из фармацевтически приемлемых неорганической кислоты, органической кислоты или основания. Примеры подходящей кислоты могут включать соляную кислоту, бромноватую кислоту, серную кислоту, азотную кислоту, перхлорную кислоту, фумаровую кислоту, малеиновую кислоту, фосфорную кислоту, гликолевую кислоту, молочную кислоту, салициловую кислоту, янтарную кислоту, толуол-пара-сульфоновую кислоту, винную кислоту, уксусную кислоту, лимонную кислоту, метансульфоновую кислоту, муравьиную кислоту, бензойную кислоту, малоновую кислоту, нафталин-2-сульфоновую кислоту и бензолсульфоновую кислоту. Примеры соли, происходящей из подходящего основания, могут включать соли щелочных металлов, таких как натрий и калий, щелочноземельных металлов, таких как магний, и аммония.
Настоящее изобретение включает не только соединение или его фармацевтически приемлемую соль, но также и полученный из них сольват.
Кроме того, соединение может быть представлено в форме энантиомера (R- или S-изомера), рацемата или диастереомера, либо любой их смеси в случае наличия асимметрического углеродного центра (отсутствующего углерода) в его заместителе. Кроме того, соединение может быть представлено в форме экзо- или эндо-изомера в случае наличия мостикового кольца, не ограничиваясь этим.
Согласно одному из аспектов настоящего изобретения предложена композиция, включающая соединение, имеющее структуру приведенной ниже формулы 1, или его стереоизомер, сольват, либо их фармацевтически приемлемую соль, где лекарственное средство представляет собой физиологически активный полипептид:

В приведенной выше формуле 1
X представляет собой Fc-область иммуноглобулина;
L1 представляет собой C1-С6алкилен с прямой или разветвленной цепью;
L2 представляет собой -a1-CONH-, -a1-NHCO-, -a1-NHCO-a2-, -СОО-, -b1-СОО-, -СОО-b2- или -b1-СОО-b2-, и каждый из a1, а2, b1 и b2 независимо представляет собой C1-С6алкилен с прямой или разветвленной цепью;
n равно от 1 до 3000, от 10 до 2000, от 50 до 1000, от 100 до 700, от 150 до 300 или от 200 до 250; и
R представляет собой радикал, выбранный из группы, состоящей из 2,5-диоксопирролидинила, 2,5-диоксопирролила, альдегида, малеимида, С6-С20 арилдисульфида, С5-С20 гетероарилдисульфида, винилсульфона, тиола, галогенированного ацетамида, сукцинимида, n-нитрофенилкарбоната, сложного тиоэфира и их производных.
Конкретно, в приведенной выше формуле 1, L1 представляет собой C1-С6алкилен с прямой или разветвленной цепью; L2 представляет собой -a1-NHCO- или -a1-NHCO-а2-; каждый из a1 и а2 независимо представляет собой C1-С6алкилен с прямой или разветвленной цепью; п равно от 200 до 250; и R представляет собой малеимид, не ограничиваясь этим.
Композиция по настоящему изобретению включает в себя промежуточное соединение и применяется для получения длительно действующего конъюгата лекарственного средства.
Конкретно, поскольку композиция по настоящему изобретению включает в себя промежуточное соединение, в котором линкер соединен с Fc-областью иммуноглобулина, то композиция по настоящему изобретению может взаимодействовать с физиологически активным полипептидом так, что линкер промежуточного соединения связывается с физиологически активным полипептидом с получением тем самым длительно действующего конъюгата лекарственного средства. Более конкретно, длительно действующий конъюгат лекарственного средства может быть получен в результате образования связи между радикалом R в формуле 1, соответствующим одному концу линкера, и остатком цистеина или аминогруппой, например, расположенной на N-конце, либо остатком лизина физиологически активного полипептида, не ограничиваясь этим.
Согласно настоящему изобретению любой физиологически активный полипептид в длительно действующем конъюгате лекарственного средства, который может быть получен с использованием данной композиции, может попадать в объем настоящего изобретения независимо от типа, размера, происхождения и тому подобного, при условии, что физиологически активный полипептид оказывает фармакологический эффект на заболевание. Примеры физиологически активного полипептида могут включать глюкагоноподобного пептида-1 (GLP-1), гранулоцитарного колониестимулирующего фактора (G-CSF), гормона роста человека (hGH), эритропоэтина (ЕРО), глюкагона, инсулина, рилизинг-фактора гормона роста; пептида, высвобождающего гормон роста; интерферонов, рецепторов интерферонов; рецепторов сопряженных с G-белком; интерлейкинов, рецепторов интерлейкинов, ферментов, интерлейкин-связывающего белка, цитокин-связывающего белка, фактора активации макрофагов, макрофагального пептида, фактора В-клеток, фактора Т-клеток, белка А, ингибитора аллергии; гликопротеина, ассоциированного с некрозом клеток; иммунотоксина, лимфотоксина, фактора некроза опухолей, онкосупрессора, фактора роста метастазов, α-1-антитрипсина, альбумина, α-лактальбумина, аполипо протеина Е, высокогликозилированного эритропоэтина, ангиопоэтинов, гемоглобина, тромбина; пептида, активирующего рецепторы тромбина; тромбомодулина, факторов крови VII, VIIa, VIII, IX и XIII, плазминоген-активирующего фактора, фибрин-связывающего пептида, урокиназы, стрептокиназы, гирудина, белка С, С-реактивного белка, ингибитора lenin, ингибитора коллагеназы, супероксиддисмутазы, лептина, фактора роста тромбоцитов, эпителиального фактора роста, эпидермального фактора роста, ангиостатина, ангиотензина, фактора роста костей, костного морфогенетического белка, кальцитонина, атриопептина; фактора, индуцирующего хрящеобразование; элкатонина; фактора, активирующего соединительную ткань; ингибитора пути тканевого фактора, фолликулостимулирующего гормона, лютеинизирующего гормона, рилизинг-фактора лютеинизирующего гормона, фактора роста нервов, гормона паращитовидной железы, релаксина, секретина, соматомедина, инсулиноподобного фактора роста, гормона коры надпочечников, холецистокинина, панкреатического полипептида, гастрин-высвобождающего пептида, кортикотропин-рилизинг-фактора, тиреотропного гормона, аутотаксина, лактоферрина, миостатина, инкретинов, желудочного ингибирующего полипептида (GIP), двойного агониста GLP-1/GIP, тройного агониста GLP-1/GIP/глюкагона, антигенов клеточной поверхности; вакцинных антигенов вирусного происхождения; моноклональных антител, поликлональных антител и фрагментов антител, но этим не ограничивается.
Более конкретно, физиологически активный полипептид может представлять собой глюкагоноподобный пептид-1 (GLP-1), глюкагон, инсулин, фермент, инкретин, желудочный ингибирующий полипептид (GIP), агонист GLР-1/GIP двойного действия или агонист GLP-1/GIP/глюкагона тройного действия, но этим не ограничивается.
Поскольку способ получения длительно действующего конъюгата лекарственного средства с использованием промежуточного соединения или композиции, содержащей его, согласно настоящему изобретению осуществляют посредством образования связи между промежуточным соединением, в котором линкер соединен с Fc-областью иммуноглобулина, и физиологически активным полипептидом, то для получения длительно действующего конъюгата лекарственного средства с применением промежуточного соединения или содержащей его композиции по настоящему изобретению можно использовать любой физиологически активный полипептид, содержащий аминокислотный остаток или реакционноспособную группу, которые способны связываться с промежуточным соединением, независимо от их типов.
В настоящем изобретении X может представлять собой Fc-область иммуноглобулина, происходящего из IgG1, IgG2, IgG3 или IgG4, не ограничиваясь этим.
Кроме того, X может представлять собой Fc-область иммуноглобулина, содержащую последовательность шарнирной области, модифицированную так, чтобы она включала только один остаток цистеина посредством делетирования части аминокислотной последовательности, составленной из приведенных ниже аминокислот, но этим не ограничивается этим:
Glu-Ser-Lys-Tyr-Gly-Pro-Pro-Cys-Pro-Ser-Cys-Pro (SEQ ID NO: 7).
В случае использования композиции по настоящему изобретению длительно действующий конъюгат лекарственного средства может быть получен без проведения ультрафильтрации/диафильтрации, и возможно, что может быть исключен один цикл гидрофобной хроматографии.
Конкретно, когда длительно действующий конъюгат лекарственного средства получают, приводя во взаимодействие промежуточное соединение или содержащую его композицию по настоящему изобретению с лекарственным средством, то чистота длительно действующего конъюгата лекарственного средства может составлять 70%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% или 99% либо больше, но этим не ограничиваться. Чистоту можно определить любым методом, хорошо известным в данной области техники, конкретно, посредством эксклюзионной высокоэффективной жидкостной хроматографии (SE-HPLC), обращенно-фазовой HPLC (RP-HPLC) и ионообменной HPLC (IE-HPLC), а также любым доступным в данной области техники методом. Более конкретно, чистота длительно действующего конъюгата лекарственного средства, полученного с использованием композиции по настоящему изобретению, может составлять 90% или более в случае SE-HPLC, 80% или более в случае RP-HPLC и 85% или более в случае IE-HPLC, но этим не ограничивается.
Кроме того, композиция по настоящему изобретению может дополнительно включать в себя буфер, стабилизатор, консервант, соль и тому подобное, необходимые для стабилизации промежуточного соединения и для получения длительно действующего конъюгата лекарственного средства, но этим не ограничивается.
Согласно другому аспекту настоящего изобретения предложен набор для получения длительно действующего конъюгата лекарственного средства, включающий в себя композицию. Набор может включать в себя реагент, руководство и тому подобное для получения длительно действующего конъюгата лекарственного средства, не ограничиваясь этим.
Согласно другому аспекту настоящего изобретения предложен способ получения длительно действующего конъюгата лекарственного средства.
Способ получения по настоящему изобретению представляет собой способ получения длительно действующего конъюгата лекарственного средства, при котором лекарственное средство присоединяют к Fc-области иммуноглобулина посредством линкера, конкретно, способ получения длительно действующего конъюгата лекарственного средства путем образования связи между промежуточным соединением и лекарственным средством, не ограничиваясь этим.
Конкретно, способ получения характеризуется последовательным осуществлением 1) присоединения линкера, содержащего полиэтиленгликоль (ПЭГ), к Fc-области иммуноглобулина и 2) присоединения линкера, который соединен с Fc-областью иммуноглобулина, к лекарственному средству (например, физиологически активному полипептиду или белку). Таким образом, способ получения характеризуется выполнением стадий в определенном порядке, т.е. выполнением первой стадии получения промежуточного соединения посредством присоединения линкера, содержащего ПЭГ, к Fc-области иммуноглобулина, и затем выполнением второй стадии присоединения лекарственного средства к промежуточному соединению. Альтернативно, способ получения по настоящему изобретению также может быть выполнен только путем проведения второй стадии получения длительно действующего конъюгата лекарственного средства путем осуществления взаимодействия между промежуточным соединением или данной композицией для получения длительно действующего конъюгата лекарственного средства, содержащего указанное выше, и лекарственным средством, без проведения первой стадии, но этим не ограничивается. Этот способ получения может быть назван в настоящей заявке как «способ получения в обратном порядке».
В настоящем изобретении в случае способа получения длительно действующего конъюгата лекарственного средства, осуществляемого путем получения сначала промежуточного соединения, а затем образования связи между этим промежуточным соединением и лекарственным средством, процессы очистки с использованием ультрафильтрации/диафильтрации и гидрофобной хроматографии могут быть исключены, и было подтверждено, что длительно действующий конъюгат лекарственного средства может быть получен с высоким выходом несмотря на исключение данных методов очистки.
Согласно общепринятому способу получения, при котором линкер сначала соединяют с физиологически активным полипептидом, а затем соединяют с Fc-областью иммуноглобулина без образования промежуточного соединения, когда соединенный с физиологически активным полипептидом линкер (например, линкер на основе полиэтиленгликоля) соединяют с Fc-областью иммуноглобулина, в качестве отдельного процесса после соединения линкера с физиологически активным полипептидом и перед связыванием данного объединенного продукта с Fc-областью иммуноглобулина требуется проведение ультрафильтрации/диафильтрации для снижения риска агрегации, которая может возникнуть вследствие условий низких значений рН (рН примерно 3,0) в буфере для уравновешивания и в элюирующем буфере, используемых для очистки линкера, соединенного с физиологически активным полипептидом, и для корректировки условий рН в реакции с использованием соответствующего буфера. Напротив, в способе получения по настоящему изобретению, при котором сначала получают промежуточное соединение посредством присоединения линкера к Fc-области иммуноглобулина, значение рН буфера, используемого для очистки линкера, соединенного с Fc-областью иммуноглобулина, является относительно высоким, и поэтому процесс ультрафильтрации/диафильтрации может быть исключен, и затем может быть осуществлен процесс образования связи между соединенным с Fc-областью иммуноглобулина линкером и физиологически активным полипептидом.
Таким образом, в способе получения по настоящему изобретению ультрафильтрацию/диафильтрацию можно не проводить после получения моно-ПЭГилированной Fc-области иммуноглобулина, но настоящее изобретение этим не ограничивается. В способе получения длительно действующего конъюгата лекарственного средства согласно настоящему изобретению рН раствора, используемого для очистки моно-ПЭГилированной Fc-области иммуноглобулина, существенно не отличается от рН раствора, используемого для последующей реакции, поэтому образование связи с лекарственным средством может быть осуществлено без проведения ультрафильтрации/диафильтрации. В результате исключения процесса ультрафильтрации/диафильтрации риск образования примесных агрегатов на стадии концентрирования может быть снижен, и способ получения может быть упрощен, в результате чего в случае коммерческого использования данной технологии можно ожидать эффекта снижения затрат.
Кроме того, способ получения по настоящему изобретению может дополнительно включать очистку конъюгата гидрофобной хроматографией, не ограничиваясь этим.
Конкретно, гидрофобная хроматография может быть проведена только один раз или более одного раза в зависимости от свойств лекарственного средства, входящего в состав длительно действующего конъюгата лекарственного средства, и типа и размера линкера.
Согласно способу получения по настоящему изобретению может быть уменьшено не только количество дорогого лекарственного средства, но также и количество не прореагировавших Fc-областей иммуноглобулина, поэтому процесс очистки гидрофобной хроматографией может быть исключен полностью или частично с получением эффекта уменьшения количества исходных веществ, необходимых для получения длительно действующего конъюгата лекарственного средства, и затрат на него по сравнению с традиционным способом.
При этом, несмотря на то, что в способе получения по настоящему изобретению исключены процессы ультрафильтрации/диафильтрации и гидрофобной хроматографии, которые выполнялись в случае традиционного способа получения длительно действующего конъюгата лекарственного средства, и осуществляется только процесс конечной очистки (например, один цикл гидрофобной хроматографии), его преимущество заключается в том, что сохраняется чистота конечного конъюгата, полученного согласно настоящему изобретению, по сравнению с традиционным способом получения. То есть, согласно способу получения по настоящему изобретению конечную чистоту можно поддерживать, хотя некоторые из процессов очистки исключены, в результате чего эффективность получения длительно действующего конъюгата лекарственного средства может быть улучшена.
Чистоту длительно действующего конъюгата лекарственного средства по настоящему изобретению можно определить любым методом, хорошо известным в данной области техники, и примерами таких методов могут быть SE-HPLC, RP-HPLC и IE-HPLC, не ограничиваясь этим.
Согласно способу получения по настоящему изобретению конечная чистота длительно действующего конъюгата лекарственного средства может составлять 90% или более, конкретно, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% или 99% или более, не ограничиваясь этим.
При этом, в способе получения по настоящему изобретению сначала получают моно-ПЭГилированную Fc-область иммуноглобулина, а затем ее соединяют с физиологически активным полипептидом, в результате чего может быть получен длительно действующий конъюгат лекарственного средства с более высоким выходом по сравнению с традиционным способом в отношении не только физиологически активного полипептида, но также и Fc-области иммуноглобулина.
В одном из воплощений настоящего изобретения было подтверждено, что выход длительно действующего конъюгата лекарственного средства, полученного способом получения по настоящему изобретению, был повышен в два раза или более по сравнению с выходом длительно действующего конъюгата лекарственного средства, полученного традиционным способом.
Конкретно, способ получения по настоящему изобретению относится к способу получения длительно действующего конъюгата лекарственного средства, включающему получение конъюгата путем присоединения моно-ПЭГилированной Fc-области иммуноглобулина, которую получают в результате соединения линкера приведенной ниже формулы 3 с N-концом Fc-области иммуноглобулина, содержащей последовательность шарнирной области, к лекарственному средству (физиологически активному полипептиду):
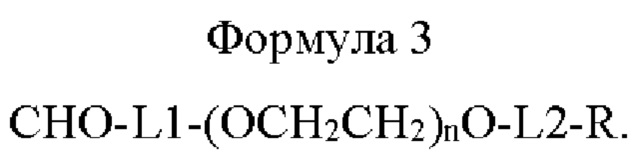
В приведенной выше формуле 3
L1 представляет собой C1-С6алкилен с прямой или разветвленной цепью;
L2 представляет собой -a1-CONH-, -a1-NHCO-, -a1-NHCO-a2-, -СОО-, -b1-СОО-, -СОО-b2- или -b1-СОО-b2-, и каждый из a1, а2, b1 и b2 независимо представляет собой C1-С6алкилен с прямой или разветвленной цепью;
n равно от 10 до 2400; и
R представляет собой радикал, выбранный из группы, состоящей из 2,5-диоксопирролидинила, 2,5-диоксопирролила, альдегида, малеимида, С6-С20арилдисульфида, С5-С20 гетероарилдисульфида, винилсульфона, тиола, галогенированного ацетамида, сукцинимида, n-нитрофенилкарбоната, сложного тиоэфира и их производных, не ограничиваясь этим.
Более конкретно,
способ получения может включать: получение моно-ПЭГилированной Fc-области иммуноглобулина путем соединения линкера приведенной выше формулы 3 с N-концом Fc-области иммуноглобулина; и получение конъюгата путем присоединения линкера в моно-ПЭГилированной Fc-области иммуноглобулина, полученной на описанной выше стадии, к физиологически активному полипептиду, или
способ получения может включать: получение моно-ПЭГилированной Fc-области иммуноглобулина путем соединения линкера формулы 3 с N-концом Fc-области иммуноглобулина; очистку моно-ПЭГилированной Fc-области иммуноглобулина, полученной на описанной выше стадии, анионообменной хроматографией в буферном растворе с рН 6,0-8,5; рН 6,0-8,0; рН 6,0-7,5; рН 6,0-7,0; рН 6,1-6,9; рН 6,2-6,8 или рН 6,3-6,7; и получение конъюгата путем присоединения линкера в моно-ПЭГилированной Fc-области иммуноглобулина, очищенной на описанной выше стадии, к GLP-1 и физиологически активному полипептиду, не ограничиваясь этим.
Кроме того, в способе получения по настоящему изобретению
(1) моно-ПЭГилированная Fc-область иммуноглобулина может быть получена путем соединения линкера формулы 3 с N-концом Fc-области иммуноглобулина в присутствии восстанавливающего агента при рН 4,0-8,0; рН 4,5-7,5; рН 5,5-7,5; рН 5,6-7,4; рН 5,7-7,3 или рН 5,8-7,2; и/или
(2) конъюгат может быть получен путем присоединения линкера в моно-ПЭГилированной Fc-области иммуноглобулина, к физиологически активному полипептиду при рН 5,5-8,0; рН 6,0-7,5 или рН 6,5-7,5, не ограничиваясь этим.
Кроме того, стадия получения конъюгата согласно способу получения по настоящему изобретению может быть проведена путем приведения во взаимодействие физиологически активного полипептида в количестве, эквивалентном количеству моно-ПЭГилированной Fc-области иммуноглобулина или превышающем его, и, конкретно, молярное соотношение между моно-ПЭГилированной Fc-областью иммуноглобулина и физиологически активным полипептидом может составлять от 1:1 до 1:10, от 1:1 до 1:7, от 1:1 до 1:5 или от 1:1 до 1:3, но этим не ограничиваться.
Использованный в данном описании термин «моно-ПЭГилированная Fc-область иммуноглобулина» относится к промежуточному веществу, которое получают в середине способа получения длительно действующего конъюгата лекарственного средства по настоящему изобретению, в котором одна молекула линкера, содержащего одну молекулу полиэтиленгликоля, соединена с Fc-областью иммуноглобулина. То есть, в настоящем изобретении термин «моно-ПЭГилированная Fc-область иммуноглобулина» может быть использован взаимозаменяемо с термином «промежуточное соединение» или «промежуточное вещество».
В настоящем изобретении Fc-область иммуноглобулина может представлять собой Fc-область иммуноглобулина, происходящего из IgG1, IgG2, IgG3 или IgG4, не ограничиваясь этим.
Кроме того, Fc-область иммуноглобулина может представлять собой Fc-область иммуноглобулина, содержащую последовательность шарнирной области, модифицированную так, чтобы она включала только один остаток цистеина посредством делетирования части приведенной ниже аминокислотной последовательности и, конкретно, последовательность шарнирной области может включать аминокислотную последовательность с SEQ ID NO: 8 (Ser-Cys-Pro) или с SEQ ID NO: 9 (Pro-Ser-Cys-Pro), не ограничиваясь этим, Glu-Ser-Lys-Tyr-Gly-Pro-Pro-Cys-Pro-Ser-Cys-Pro (SEQ ID NO: 7).
Термин «длительно действующий конъюгат лекарственного средства» по настоящему изобретению относится к конъюгату лекарственного средства, имеющему структуру, в которой лекарственное средство (физиологически активный полипептид), обладающее фармакологической активностью в организме, соединено с Fc-областью иммуноглобулина посредством линкера, и имеющему увеличенный период полувыведения. Исходя из целей настоящего изобретения длительно действующий конъюгат лекарственного средства может представлять собой конъюгат, в котором промежуточное соединение или моно-ПЭГилированная Fc-область иммуноглобулина соединено(а) с лекарственным средством, но этим не ограничивается.
Конкретно, лекарственное средство не ограничивается конкретными веществами при условии, что лекарственное средство оказывает профилактическое, терапевтическое или облегчающее действие на определенное заболевание и может представлять собой природные или неприродные белок, фермент, антитело, соединение или тому подобные вещества. Более конкретно, лекарственное средство может представлять собой физиологически активный полипептид или белок, еще более конкретно, физиологически активный полипептид может представлять собой глюкагоноподобный пептид-1 (GLP-1), гранулоцитарный колониестимулирующий фактор (G-CSF), гормон роста человека (hGH), эритропоэтин (ЕРО), глюкагон, инсулин, рилизинг-фактор гормона роста; пептид, высвобождающий гормон роста; интерфероны, рецепторы интерферонов; рецепторы, сопряженные с G-белком; интерлейкины, рецепторы интерлейкинов, ферменты, интерлейкин-связывающий белок, цитокин-связывающий белок, фактор активации макрофагов, макрофагальный пептид, фактор В-клеток, фактор Т-клеток, белок А, ингибитор аллергии; гликопротеин, ассоциированный с некрозом клеток; иммунотоксин, лимфотоксин, фактор некроза опухолей, онкосупрессор, фактор роста метастазов, α-1-антитрипсин, альбумин, α-лактальбумин, аполипопротеин Е, высокогликозилированный эритропоэтин, ангиопоэтин, гемоглобин, тромбин; пептид, активирующий рецепторы тромбина; тромбомодулин, факторы крови VII, VIIa, VIII, IX и XIII, плазминоген-активирующий фактор, фибрин-связывающий пептид, урокиназу, стрептокиназу, гирудин, белок С, С-реактивный белок, ингибитор lenin, ингибитор коллагеназы, супероксиддисмутазу, лептин, фактор роста тромбоцитов, эпителиальный фактор роста, эпидермальный фактор роста, ангиостатин, ангиотензин, фактор роста костей, костный морфогенетический белок, кальцитонин, атриопептин; фактор, индуцирующий хрящеобразование; элкатонин; фактор, активирующий соединительную ткань; ингибитор пути тканевого фактора, фолликулостимулирующий гормон, лютеинизирующий гормон, рилизинг-фактор лютеинизирующего гормона, фактор роста нервов, гормон паращитовидной железы, релаксин, секретин, соматомедин, инсулиноподобный фактор роста, гормон коры надпочечников, холецистокинин, панкреатический полипептид, гастрин-высвобождающий пептид, кортикотропин-рилизинг-фактор, тиреотропный гормон, аутотаксин, лактоферрин, миостатин, инкретины, желудочный ингибирующий полипептид (GIP), двойной агонист GLP-1/GIP, тройной агонист GLP-1/GIP/глюкагона, антигены клеточной поверхности; вакцинные антигены вирусного происхождения; моноклональные антитела, поликлональные антитела и фрагменты антител, но этим не ограничивается. Более конкретно, физиологически активный полипептид может представлять собой тройной агонист GLP-1/GIP/глюкагона, глюкагон или его аналог, но не ограничивается этим. Еще более конкретно, физиологически активный полипептид может включать одну из аминокислотных последовательностей, по существу состоять из, или состоять из одной из аминокислотных последовательностей с SEQ ID NO: 1-6, но этим не ограничивается.
Кроме того, в объем настоящего изобретения также входят любые варианты, производные и фрагменты физиологически активного полипептида.
Использованный в данном описании термин «вариант» относится к пептиду, имеющему аминокислотную последовательность, в которой одна или более аминокислот отличаются от аминокислот в нативном физиологически активном полипептиде, с сохранением при этом функций нативного физиологически активного полипептида, и такой вариант может быть получен путем замены, добавления, делеции, модификации или какого-либо комбинирования некоторых аминокислот в аминокислотной последовательности нативного физиологически активного полипептида.
Использованный в данном описании термин «производное» относится к пептиду, аналогу пептида или пептидомиметику, полученному посредством модификации одной или более аминокислот в нативном физиологически активном полипептиде посредством добавления, делеции или замены, для обеспечения такой же активности, что и у нативного физиологически активного полипептида.
Использованный в данном описании термин «фрагмент» относится к форме, полученной путем добавления к N-концу или С-концу либо делетирования с N-конца или С-конца одной или более аминокислот, и добавленная аминокислота может представлять собой любую аминокислоту, которая не существует в природе (например D-аминокислоту).
Способы получения такого варианта, производного и фрагмента физиологически активного полипептида могут быть использованы независимо или в комбиации. Например, в данное изобретение может быть включен любой физиологически активный полипептид, имеющий в аминокислотной последовательности одну или несколько других аминокислот и дезаминирование аминокислотного остатка на N-конце.
Производное физиологически активного полипептида включает биологически аналогичные и биологически улучшенные формы. Например, что касается биоаналогов, то биоаналогом может быть любой биоаналог фермента, входящий в состав длительно действующего конъюгата лекарственного средства по настоящему изобретению, хотя существует различие между известным ферментом и хозяином для его экспрессии, различие в природе и степени гликозилирования, и различие в степени замены конкретного аминокислотного остатка соответствующего фермента с учетом стандартной последовательности, где степень замены не является 100%-ной заменой. Физиологически активный полипептид и его вариант, производное и фрагмент могут быть получены из клеток животных, Е. coli, дрожжей, клеток насекомых, клеток растений, из живых животных и тому подобного посредством генетической рекомбинации, при этом способы получения этим не ограничиваются, и также можно использовать любые имеющиеся в продаже физиологически активные полипептиды и их варианты, производные и фрагменты.
Кроме того, физиологически активный полипептид и его вариант, производное и фрагмент могут включать аминокислотную последовательность, имеющую гомологию, составляющую по меньшей мере 80%, конкретно, по меньшей мере 90%, более конкретно, по меньшей мере 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% или 99%, и физиологически активный полипептид и его вариант, производное и фрагмент могут быть получены из микроорганизмов с использованием технологий генетической рекомбинации или иметься в продаже, не ограничиваясь этим.
Использованный в данном описании термин «гомология» относится к степени сходства между аминокислотными последовательностями белка дикого типа или кодирующими их нуклеотидными последовательностями и включает в себя последовательность, идентичную аминокислотной последовательности или нуклеотидной последовательности по настоящему изобретению на указанный выше или больший процент. Гомология может быть определена путем визуального сравнения последовательностей, но также может быть определена с использованием биоинформационного алгоритма, который обеспечивает получение результатов анализа степени гомологии путем выравнивания сравниваемых последовательностей. Гомология между двумя аминокислотными последовательностями может быть указана в процентах. Полезные автоматизированные алгоритмы могут быть использованы в модулях компьютерного программного обеспечения GAP, BESTFIT, FASTA и TFASTA из программного пакета Wisconsin Genetics (Genetics Computer Group, Madison, WI, USA). Алгоритмы автоматизированного выравнивания в модулях включают алгоритм Needleman и Wunsch, алгоритм Pearson и Lipman и алгоритм для работы с последовательностями Smith и Waterman. Другие полезные алгоритмы и методы определения гомологии при выравнивании являются автоматизированными в таком программном обеспечении, как FASTP, BLAST (средство поиска основного локального выравнивания), BLAST2, PSIBLAST и CLUSTAL W (кластерный анализ множественных выравниваний).
Информацию о последовательностях физиологически активного полипептида и его варианта, производного и фрагмента и кодирующих их нуклеотидных последовательностях можно получить из известной базы данных GenBank NCBI (Национальный центр биотехнологической информации) или подобной.
Замещенными или добавленными аминокислотами могут быть не только 20 аминокислот, обычно встречающихся в белках человека, но также нетипичные или неприродные аминокислоты. Коммерческие источники нетипичных аминокислот могут включать Sigma-Aldrich, ChemPep Inc. и Genzyme Pharmaceuticals. Пептиды, в состав которых входят эти аминокислоты, и типичные последовательности пептидов могут быть синтезированы и приобретены у коммерческих поставщиков, например у American Peptide Company, Bachem (США) или Anygen (Корея).
Кроме того, физиологически активный полипептид и его вариант, производное и фрагмент по настоящему изобретению могут находиться в различной форме, где N-конец и/или С-конец химически модифицированы или защищены группами органических молекул, или где к концам пептида могут быть присоединены аминокислоты, для защиты от протеаз in vivo с повышением его стабильности.
В частности, поскольку N- и С-концы химически синтезированных пептидов несут электрический заряд, то N-конец может быть ацетилирован и/или С-конец может быть амидирован для удаления зарядов, но данное воплощение не ограничивается этим.
Кроме того, пептид по настоящему изобретению включает все варианты, представленные в форме самого пептида, его соли (например фармацевтически приемлемой соли пептида) или его сольвата. Кроме того, пептид может находиться в любой фармацевтически приемлемой форме.
Типа соли конкретно не ограничен. Однако, соль предпочтительно находится в форме соли, безопасной и эффективной для субъекта, например млекопитающего, не ограничиваясь этим.
Использованный в данном описании термин «сольват» относится к комплексу пептида или его соли по настоящему изобретению и молекулы растворителя.
Хотя в настоящем изобретении пептид описан как «пептид, состоящий из определенной последовательности SEQ ID NO», это не исключает мутации, которая может возникать естественным образом или в результате присоединения бессмысловой последовательности вверх или вниз от аминокислотной последовательности с данным SEQ ID NO, или наличия в нем молчащей мутации, при условии, что пептид обладает активностью, идентичной или эквивалентной активности пептида, состоящего из данной аминокислотной последовательности, и даже когда имеется присоединение такой последовательности или такая мутация, это очевидным образом входит в объем настоящего изобретения.
Способ получения длительно действующего конъюгата лекарственного средства по настоящему изобретению может представлять собой способ получения конъюгата, в котором физиологически активный полипептид соединяется с Fc-областью иммуноглобулина посредством линкера, не ограничиваясь этим.
В настоящем изобретении соединение между линкером и Fc-областью иммуноглобулина может быть образовано посредством ковалентной связи или нековалентной связи между одним концом линкера и N-концом Fc-области иммуноглобулина, однако места связывания или способы связывания конкретно не ограничены. В частности, моно-ПЭГилированная Fc-область иммуноглобулина может быть получена путем соединения пролина на N-конце Fc-области иммуноглобулина с группой -СНО линкера, не ограничиваясь этим.
В настоящем изобретении линкер может иметь структуру приведенной ниже формулы 4, но этим не ограничивается:
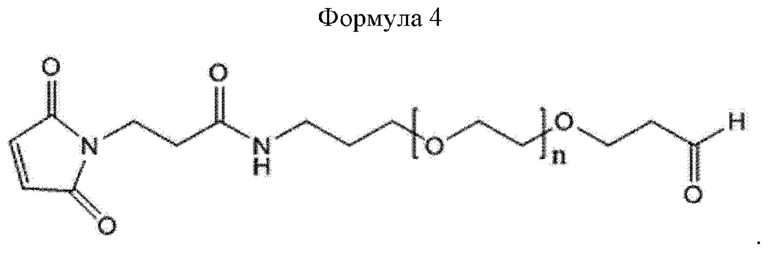
В приведенной выше формуле 4 п равно от 200 до 250.
Кроме того, масса линкера может составлять от 1 кДа до 200 кДа, от 1 кДа до 150 кДа, от 1 кДа до 100 кДа, от 1 кДа до 50 кДа или от 1 кДа до 10 кДа, не ограничиваясь этим.
Моно-ПЭГилированная Fc-область иммуноглобулина может иметь структуру приведенной ниже формулы 2, не ограничиваясь этим:
Способ получения по настоящему изобретению может быть осуществлен путем присоединения физиологически активного полипептида к одному концу моно-ПЭГилированной Fc-области иммуноглобулина, имеющей структуру формулы 2, не ограничиваясь этим.
Кроме того, другой конец линкера, который не присоединен к Fc-области иммуноглобулина, может быть присоединен к физиологически активному полипептиду, конкретно к группе -SH или к аминокислоте, содержащей группу -SH, или к цистеину физиологически активного полипептида, не ограничиваясь этим.
В настоящем изобретении длительно действующий конъюгат лекарственного средства может иметь структуру приведенной ниже формулы 5, не ограничиваясь этим:
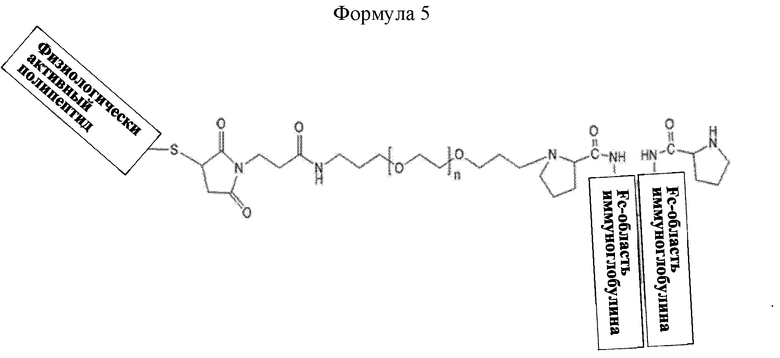
Формула 5 представляет структуру, в которой слева направо последовательно соединены физиологически активный полипептид, линкер и Fc-область иммуноглобулина, не ограничиваясь этим.
Когда длительно действующий конъюгат лекарственного средства получают в соответствии со способом получения по настоящему изобретению, в котором сначала получают моно-ПЭГилированную Fc-область иммуноглобулина и затем соединяют с физиологически активным полипептидом, была подтверждена возможность сохранения чистоты конечного конъюгата наряду с повышенным выходом по сравнению с традиционным способом получения, несмотря на исключение процессов ультрафильтрации/диафильтрации и гидрофобной хроматографии и проведение в способе получения по настоящему изобретению только заключительного процесса очистки (например одного цикла гидрофобной хроматографии).
Согласно другому аспекту настоящего изобретения предложен длительно действующий конъюгат лекарственного средства, полученный описанным выше способом.
Поскольку длительно действующий конъюгат лекарственного средства, полученный способом по настоящему изобретению, имеет увеличенный период полувыведения по сравнению с физиологически активным полипептидом, который не соединен с линкером или Fc-областью иммуноглобулина, то можно достичь благоприятных эффектов в приготовлении лекарственных средств.
Длительно действующий конъюгат лекарственного средства, полученный способом получения по настоящему изобретению, может быть использован в приготовления лекарственных средств или композиций для предупреждения, лечения и облегчения заболеваний.
Ниже настоящее изобретение будет описано более подробно со ссылкой на следующие ниже примеры. Однако следующие ниже примеры представлены только для иллюстрации настоящего изобретения, и объем настоящего изобретения ими не ограничивается.
Сравнительный пример: получение конъюгата путем присоединения ПЭГилированного физиологически активного полипептида к Fc-области иммуноглобулина ПЭГилированный физиологически активный полипептид присоединяли к Fc-области иммуноглобулина с получением длительно действующего конъюгата.
Сравнительный пример 1: получение конъюгата посредством присоединения ПЭГилированного аналога 1 тройного агониста GLP-1/GIP/глюкагона к Fc-области иммуноглобулина Чтобы осуществить ПЭГилирование физиологически активного полипептида (аналога 1 тройного агониста GLP-1/GIP/глюкагона, SEQ ID NO: 1) по остатку цистеина (группа -SH), проводили взаимодействие аналога 1 тройного агониста GLP-1/GIP/глюкагона с линкером, содержащим ПЭГ (малеимид-ПЭГ(10 кДа)-альдегид) (формула 4) в течение примерно 1 часа в молярном соотношении от 1:1,0 до 1:1,3 с концентрацией аналога 1 тройного агониста GLP-1/GIP/глюкагона примерно 3 г/л. Конкретно, данную реакцию проводили в 50 мМ трис-буфере, содержащем изопропанол (рН 7,5; 6°С±4°С). Чтобы получить моно-ПЭГилированный аналог 1 тройного агониста GLP-1/GIP/глюкагона, реакционный раствор разбавляли буфером для уравновешивания, содержащим цитрат натрия и этанол, до 20-кратного общего объема и очищали. Для этого моно-ПЭГилированный аналог 1 тройного агониста GLP-1/GIP/глюкагона очищали, используя колонку SP High Performance (колонка с сульфопропиловым носителем для высокоэффективной хроматографии (GE Healthcare, для катионообменной хроматографии), с использованием раствора, содержащего цитрат натрия, и этанол, и градиент концентрации хлорида калия. После разбавления водой очищенного раствора ПЭГилированного аналога 1 тройного агониста GLP-1/GIP/глюкагона буферный раствор заменяли на 0,1 М раствор фосфата калия посредством ультрафильтрации/диафильтрации (UF/DF), после чего концентрировали для извлечения результирующего продукта с конечной концентрацией примерно 3 г/л или более.
Моно-ПЭЕилированный аналог 1 тройного агониста GLP-1/GIP/глюкагона, полученный, как описано выше, присоединяли к Fc-области иммуноглобулина с получением длительно действующего конъюгата, как указано ниже.
Чтобы соединить альдегидную группу ПЭГ моно-ПЭГилированного аналога 1 тройного агониста GLP-1/GIP/глюкагона с амино-концом Fc-области иммуноглобулина, осуществляли взаимодействие моно-ПЭЕилированного аналога 1 тройного агониста GLP-1/GIP/глюкагона с Fc-областью иммуноглобулина в молярном соотношении 1:2 при температуре 6°С±4°С в течение примерно 12 часов с общей концентрацией белка (аналога 1 тройного агониста GLP-1/GIP/глюкагона и Fc-области иммуноглобулина) 30 г/л.
Для выделения и удаления непрореагировавших Fc-областей иммуноглобулина после реакции образования связи реакционный раствор очищали, используя колонку Butyl 4 Fast Flow (GE Healthcare, для гидрофобной хроматографии). В этом случае в реакционный раствор добавляли трис-буфер и хлорид натрия, и реакционный раствор очищали, используя раствор, содержащий бис-трис и градиент концентрации хлорида натрия.
После этого, используя колонку Source 15ISO (GE Healthcare), проводили гидрофобную хроматографию. Посредством этого процесса удаляли побочные продукты и получали конъюгат (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 1 тройного агониста GLP-1/GIP/глюкагона). В этом случае очистку проводили, используя буфер, содержащий цитрат натрия, и градиент концентрации сульфата аммония.
Сравнительный пример 2: получение конъюгата путем присоединения ПЭГилированного аналога 1 глюкагона к Fc-области иммуноглобулина
Чтобы осуществить ПЭЕилирование физиологически активного полипептида (аналог 1 глюкагона, SEQ ID NO: 4) по остатку цистеина (группа -SH), осуществляли взаимодействие аналога 1 глюкагона с линкером, содержащим ПЭГ (малеимид-ПЭГ 10 кДа-альдегид) (формула 1) в течение примерно 1 часа в молярном соотношении 1:1,3 с концентрацией аналога 1 глюкагона 3 г/л. Конкретно, данную реакцию проводили в 50 мМ трис-буфере, содержащем изопропанол (рН 7,3). Чтобы получить моно-ПЭГилированный аналог 1 глюкагона, реакционный раствор разбавляли буфером для уравновешивания, содержащим цитрат натрия и этанол, до 20-кратного общего объема и очищали. Для этого моно-ПЭГилированный аналог 1 глюкагона очищали, используя колонку SP High Performance (GE Healthcare, для катионообменной хроматографии), с использованием раствора, содержащего цитрат натрия, и этанол и градиент концентрации хлорида калия. После разбавления водой очищенного раствора ПЭГилированного аналога 1 глюкагона буферный раствор заменяли на 0,1 М раствор фосфата калия посредством ультрафильтрации/диафильтрации (UF/DF), после чего концентрировали для извлечения результирующего продукта с конечной концентрацией примерно 3 г/л или более.
Моно-ПЭГилированный аналог 1 глюкагона, полученный, как описано выше, присоединяли к Fc-области иммуноглобулина с получением длительно действующего конъюгата, как указано ниже.
Чтобы соединить альдегидную группу ПЭГ моно-ПЭГилированного аналога 1 глюкагона с амино-концом Fc-области иммуноглобулина, осуществляли взаимодействие моно-ПЭГилированного аналога 1 глюкагона с Fc-областью иммуноглобулина в молярном соотношении 1:5 при температуре 6°С±4°С в течение примерно 12 часов с общей концентрацией белка (аналога 1 глюкагона и Fc-области иммуноглобулина) 20 г/л.
Для выделения и удаления непрореагировавших Fc-областей иммуноглобулина после реакции образования связи, реакционный раствор очищали, используя колонку Butyl 4 Fast Flow (GE Healthcare, для гидрофобной хроматографии). В этом случае в реакционный раствор добавляли трис-буфер и хлорид натрия, и реакционный раствор очищали, используя раствор, содержащий бис-трис и градиент концентрации хлорида натрия.
После этого, используя колонку Source 15ISO (GE Healthcare), проводили гидрофобную хроматографию. Посредством данного процесса удаляли побочные продукты и получали конъюгат (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 1 глюкагона). В этом случае очистку проводили, используя буфер, содержащий цитрат натрия, и градиент концентрации сульфата аммония.
Авторы настоящего изобретения разработали способ, приведенный ниже, позволяющий эффективно получать конъюгат с высокой степенью чистоты, исключив процесс мембранной фильтрации и процесс очистки (гидрофобная хроматография на колонке Butyl 4 Fast Flow) из способа получения конъюгата в соответствии с описанными выше Сравнительными примерами 1 и 2.
Пример 1: получение моно-ПЭГилированной Fc-области иммуноглобулина Пример 1-1. Получение моно-ПЭГилированной Fc-области иммуноглобулина. Чтобы осуществить ПЭГилирование N-конца Fc-области иммуноглобулина (49,8 кДа), содержащей шарнирную область с последовательностью Pro-Ser-Cys-Pro на N-конце, Fc-область иммуноглобулина приводили во взаимодействие с линкером, содержащим ПЭГ (структура формулы 4; 10 кДа) в молярном соотношении (Fc-область иммуноглобулина:ПЭГ-содержащий линкер) 1:1 с концентрацией Fc-области иммуноглобулина 50 г/л при 6°С±4°С в течение примерно 4 часов.
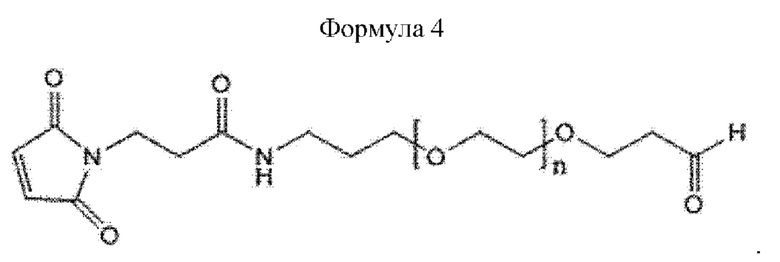
Конкретно, данную реакцию проводили в смеси, содержащей 5 мМ бис-трис-буфер (рН 6,5) и фосфат калия, и к ней добавляли 10 мМ NaCNBH3 (цианоборгидрид натрия) в качестве восстанавливающего агента. Чтобы получить моно-ПЭГилированную Fc-область иммуноглобулина, реакционный раствор разбавляли бис-трис-буфером и очищали.
В отличие от способа получения из описанных выше Сравнительных примеров, в котором моно-ПЭГилированный оксинтомодулин очищали катионообменной хроматографией, моно-ПЭГилированную Fc-область иммуноглобулина очищали с использованием колонки CaptoQ ImpRes (GE Healthcare, для анионообменной хроматографии), используя бис-трис-буфер и градиент концентрации хлорида натрия.
Пример 1-2. Анализ структуры моно-ПЭГилированной Fc-области иммуноглобулина
Структуру моно-ПЭГилированной Fc-области иммуноглобулина, полученной в примере 1-1, анализировали посредством MALDI-TOF и пептидного картирования. По данным метода MALDI-TOF молекулярная масса полученного продукта была идентична ожидаемой для моно-ПЭГилированной Fc-области иммуноглобулина (ФИГ. 1), а по данным пептидного картирования было подтверждено, что более 90% ПЭГ было бутилировано на N-конце Fc-области иммуноглобулина.
При этом в результате анализа моно-ПЭГилированной Fc-области иммуноглобулина (формулы 2), полученной в приведенном выше примере 1-1, с использованием методов SE-HPLC, RP-HPLC и IE-HPLC было подтверждено, что чистота составляла 90% или более в случае SE-HPLC, 90% или более в случае RP-HPLC и 80% или более в случае IE-HPLC.
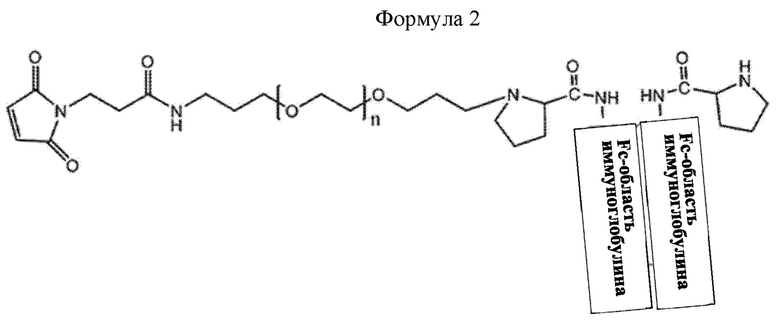
Пример 2: получение конъюгата путем присоединения ПЭЕилированной Fc-области иммуноглобулина к физиологически активному полипептиду
Длительно действующие конъюгаты получали, как указано ниже, путем присоединения моно-ПЭГилированной Fc-области иммуноглобулина, полученной в примере 1-1, к различным физиологически активным пептидам.
В отличие от способа получения из Сравнительных примеров, в котором моно-ПЭГилированный физиологически активный полипептид очищали катионообменной хроматографией и затем проводили замену буфера и концентрирование посредством ультрафильтрации/диафильтрации (UF/DF), моно-ПЭГилированную Fc-область иммуноглобулина приводили во взаимодействие с физиологически активным полипептидом посредством пептидного конъюгирования без проведения ультрафильтрации/диафильтрации. Длительно действующие конъюгаты, полученные, как описано выше, имели высокую степень чистоты, и поэтому один из двух циклов гидрофобной хроматографии можно было исключить в отличие от способа получения согласно Сравнительным примерам.
Пример 2-1. Получение конъюгата аналога 1 тройного агониста GLP-1/GIP/глюкагона
Длительно действующий конъюгат (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 1 тройного агониста GLP-1/GIP/глюкагона) получали посредством пептидного конъюгирования аналога 1 тройного агониста GLP-1/GIP/глюкагона (SEQ ID NO: 1) после анионообменной хроматографии Примера 1-1, без проведения ультрафильтрации/диафильтрации.

В связи с этим, для того, чтобы соединить малеимидную реакционноспособную группу на одном конце ПЭГ моно-ПЭГилированной Fc-области иммуноглобулина с аналогом 1 тройного агониста GLP-1/GIP/глюкагона, моно-ПЭГилированную Fc-область иммуноглобулина приводили во взаимодействие с аналогом 1 тройного агониста GLP-1/GIP/глюкагона в молярном соотношении 1:1 с концентрацией аналога 1 тройного агониста GLP-1/GIP/глюкагона 0,2 г/л при 6°С±4°С в течение примерно 2 часов.
Реакцию проводили в трис-С1 буфере (6°С±4°С), содержащем изопропанол. В результате анализа продукта, полученного после проведения взаимодействия, с использованием методов SE-HPLC, RP-HPLC и IE-HPLC было подтверждено, что чистота составляла 90% или более в случае SE-HPLC, 80% или более в случае RP-HPLC и 70% или более в случае IE-HPLC.
После этого полученный в результате взаимодействия продукт подвергали один раз гидрофобной хроматографии с использованием колонки Source 15ISO (GE Healthcare). Посредством этого процесса удаляли побочные продукты и получали конъюгат (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 1 тройного агониста GLP-1/GIP/глюкагона). В этом случае очистку проводили, используя буфер, содержащий цитрат натрия, и градиент концентрации сульфата аммония. Было подтверждено, что полученный при этом выход увеличивался примерно в два раза или более по сравнению с выходом в Сравнительном примере 1 при использовании того же количества аналога 1 тройного агониста GLP-1/GIP/глюкагона.
Элюированный конъюгат (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 1 тройного агониста GLP-1/GIP/глюкагона) анализировали с использованием методов SE-HPLC, RP-HPLC и IE-HPLC, и была подтверждена высокая степень чистоты, поскольку чистота составляла 90% или более в случае SE-HPLC, 90% или более в случае RP-HPLC и 90% или более в случае IE-HPLC.
Пример 2-2. Получение конъюгата аналога 2 тройного агониста GLP-1 / GIP/ глюкагона
Длительно действующий конъюгат (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 2 тройного агониста GLP-1/GIP/глюкагона) получали путем пептидного конъюгирования аналога 2 тройного агониста GLP-1/GIP/глюкагона (SEQ ID NO: 2) после анионообменной хроматографии Примера 1-1 без проведения ультрафильтрации/ диафильтрации.

При этом, чтобы соединить малеимидную реакционноспособную группу на одном конце ПЭГ моно-ПЭГилированной Fc-области иммуноглобулина с аналогом 2 тройного агониста GLP-1/GIP/глюкагона, моно-ПЭГилированную Fc-область иммуноглобулина приводили во взаимодействие с аналогом 2 тройного агониста GLP-1/GIP/глюкагона в молярном соотношении 1:1 с концентрацией аналога 2 тройного агониста GLP-1/GIP/глюкагона 0,2 г/л при 6°С±4°С в течение примерно 2 часов. Реакцию проводили в трис-С1 буфере (6°С±4°С), содержащем изопропанол. В результате анализа продукта, полученного после проведения взаимодействия, с использованием методов SE-HPLC, RP-HPLC и IE-HPLC было подтверждено, что чистота конъюгата длительного действия, содержащего аналог 2 тройного агониста GLP-1/GIP/глюкагона, составляла 90% или более в случае SE-HPLC, 80% или более в случае RP-HPLC и 70% или более в случае IE-HPLC.
После этого полученный в результате взаимодействия продукт подвергали один раз гидрофобной хроматографии с использованием колонки Source 15ISO (GE Healthcare). Посредством этого процесса удаляли побочные продукты и получали конъюгат (Fc-область иммуноглобулина)-(ПЭЕ-содержащий линкер)-(аналог 2 тройного агониста GLP-1/GIP/глюкагона). В этом случае очистку проводили, используя буфер, содержащий цитрат натрия, и градиент концентрации сульфата аммония. Было подтверждено, что полученный при этом выход увеличивался примерно в два раза или более по сравнению с выходом в Сравнительном примере 1 при использовании того же количества аналога 2 тройного агониста GLP-1/GIP/глюкагона.
Элюированный конъюгат (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 2 тройного агониста GLP-1/GIP/глюкагона) анализировали с использованием методов SE-HPLC, RP-HPLC и IE-HPLC, и была подтверждена высокая степень чистоты, поскольку чистота составляла 90% или более в случае SE-HPLC, 90% или более в случае RP-HPLC и 80% или более в случае IE-HPLC.
Пример 2-3. Получение конъюгата аналога 3 тройного агониста GLP-1/GIP/глюкагона
Длительно действующий конъюгат (Fc-область иммуноглобулина)-(ПЭЕ-содержащий линкер)-(аналог 3 тройного агониста GLP-1/GIP/глюкагона) получали посредством пептидного конъюгирования аналога 3 тройного агониста GLP-1/GIP/глюкагона (SEQ ID NO: 3) после анионообменной хроматографии Примера 1-1 без проведения ультрафильтрации/диафильтрации.

При этом, чтобы соединить малеимидную реакционноспособную группу на одном конце ПЭГ моно-ПЭГилированной Fc-области иммуноглобулина с цистеином аналога 3 тройного агониста GLP-1/GIP/глюкагона, моно-ПЭГилированную Fc-область иммуноглобулина приводили во взаимодействие с аналогом 3 тройного агониста GLP-1/GIP/глюкагона в молярном соотношении 1:1 с концентрацией аналога 3 тройного агониста GLP-1/GIP/глюкагона 0,2 г/л при 6°С±4°С в течение примерно 2 часов. Данную реакцию проводили в трис-С1 буфере (6°С±4°С), содержащем изопропанол. В результате анализа продукта, полученного после проведения взаимодействия, с использованием методов SE-HPLC, RP-HPLC и IE-HPLC было подтверждено, что чистота составляла 90% или более в случае SE-HPLC, 80% или более в случае RP-HPLC и 70% или более в случае IE-HPLC.
После этого полученный при взаимодействии продукт подвергали один раз гидрофобной хроматографии с использованием колонки Source 15ISO (GE Healthcare). Посредством этого процесса удаляли побочные продукты и получали конъюгат (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 3 тройного агониста GLP-1/GIP/глюкагона). В этом случае очистку проводили, используя буфер, содержащий цитрат натрия, и градиент концентрации сульфата аммония. Было подтверждено, что полученный при этом выход увеличивался примерно в два раза или более по сравнению с выходом в Сравнительном примере 1 при использовании того же количества аналога 3 тройного агониста GLP-1/GIP/глюкагона.
Элюированный конъюгат (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 3 тройного агониста GLP-1/GIP/глюкагона) анализировали с использованием методов SE-HPLC, RP-HPLC и IE-HPLC, и была подтверждена высокая степень чистоты, поскольку чистота составляла 90% или более в случае SE-HPLC, 90% или более в случае RP-HPLC и 90% или более в случае IE-HPLC.
Пример 2-4. Получение конъюгата аналога 1 глюкагона Длительно действующий конъюгат (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 1 глюкагона) получали посредством пептидного конъюгирования аналога 1 глюкагона (SEQ ID NO: 4) после анионообменной хроматографии Примера 1-1 без проведения ультрафильтрации/диафильтрации.

При этом, чтобы соединить малеимидную реакционноспособную группу на одном конце ПЭГ моно-ПЭГилированной Fc-области иммуноглобулина с аналогом 1 глюкагона, моно-ПЭГилированную Fc-область иммуноглобулина приводили во взаимодействие с аналогом 1 глюкагона в молярном соотношении 1:1 с концентрацией аналога 1 глюкагона 0,2 г/л при 6°С±4°С в течение примерно 2 часов. Данную реакцию проводили в трис-С1 буфере (6°С±4°С), содержащем изопропанол. В результате анализа продукта, полученного после проведения взаимодействия, с использованием методов SE-HPLC, RP-HPLC и IE-HPLC было подтверждено, что чистота конъюгата (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 1 глюкагона) составляла 90% или более в случае SE-HPLC, 70% или более в случае RP-HPLC и 70% или более в случае IE-HPLC.
После этого полученный при взаимодействии продукт подвергали один раз гидрофобной хроматографии с использованием колонки Source 15ISO (GE Healthcare). Посредством этого процесса удаляли побочные продукты и получали конъюгат (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 1 глюкагона). В этом случае очистку проводили, используя буфер, содержащий цитрат натрия, и градиент концентрации сульфата аммония. Было подтверждено, что полученный при этом выход увеличивался примерно в 1,5 раза или более по сравнению с выходом в Сравнительном примере 2 при использовании того же количества аналога 1 глюкагона.
Элюированный конъюгат (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 1 глюкагона) анализировали с использованием методов SE-HPLC, RP-HPLC и IE-HPLC, и была подтверждена высокая степень чистоты, поскольку чистота составляла 90% или более в случае SE-HPLC, 90% или более в случае RP-HPLC и 90% или более в случае IE-HPLC.
Пример 2-5. Получение конъюгата аналога 2 глюкагона
Длительно действующий конъюгат (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 2 глюкагона) получали посредством пептидного конъюгирования аналога 2 глюкагона (SEQ ID NO: 5) после анионообменной хроматографии Примера 1-1 без проведения ультрафильтрации/диафильтрации.

Glul6 (красный цвет) и Lys20 (красный цвет) соединены лактамным кольцом
При этом, чтобы соединить малеимидную реакционноспособную группу на одном конце ПЭГ моно-ПЭГилированной Fc-области иммуноглобулина с аналогом 2 глюкагона, моно-ПЭГилированную Fc-область иммуноглобулина приводили во взаимодействие с аналогом 2 глюкагона в молярном соотношении 1:1 с концентрацией аналога 2 глюкагона 0,2 г/л при 6°С±4°С в течение примерно 2 часов. Реакцию проводили в трис-С1 буфере (6°С±4°С), содержащем изопропанол. В результате анализа продукта, полученного после проведения взаимодействия, с использованием методов SE-HPLC, RP-HPLC и IE-HPLC было подтверждено, что чистота конъюгата (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 2 глюкагона) составляла 90% или более в случае SE-HPLC, 70% или более в случае RP-HPLC и 70% или более в случае IE-HPLC.
После этого полученный при взаимодействии продукт подвергали один раз гидрофобной хроматографии с использованием колонки Source 15ISO (GE Healthcare). Посредством этого процесса удаляли побочные продукты и получали конъюгат (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 2 глюкагона). В этом случае очистку проводили, используя буфер, содержащий цитрат натрия, и градиент концентрации сульфата аммония. Было подтверждено, что полученный при этом выход увеличивался примерно в 1,5 раза или более по сравнению с выходом в Сравнительном примере 2 при использовании того же количества аналога 2 глюкагона.
Элюированный конъюгат (Fc-область иммуноглобулина)-(ПЭЕ-содержащий линкер)-(аналог 2 глюкагона) анализировали с использованием методов SE-HPLC, RP-HPLC и IE-HPLC, и была подтверждена высокая степень чистоты, поскольку чистота составляла 90% или более в случае SE-HPLC, 90% или более в случае RP-HPLC и 90% или более в случае IE-HPLC.
Пример 2-6. Получение конъюгата аналога 3 глюкагона
Длительно действующий конъюгат (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 3 глюкагона) получали посредством пептидного конъюгирования аналога 3 глюкагона (SEQ ID NO: 6) после анионообменной хроматографии Примера 1-1 без проведения ультрафильтрации/диафильтрации.

При этом, чтобы соединить малеимидную реакционноспособную группу на одном конце ПЭГ моно-ПЭГилированной Fc-области иммуноглобулина с остатком цистеина аналога 3 глюкагона, моно-ПЭГилированную Fc-область иммуноглобулина приводили во взаимодействие с аналогом 3 глюкагона в молярном соотношении 1:1 с концентрацией аналога 3 глюкагона 0,2 г/л при 6°С±4°С в течение примерно 2 часов. Реакцию проводили в трис-С1 буфере (6°С±4°С), содержащем изопропанол. В результате анализа продукта, полученного после проведения взаимодействия, с использованием методов SE-HPLC, RP-HPLC и IE-HPLC было подтверждено, что чистота конъюгата (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 3 глюкагона) составляла 90% или более в случае SE-HPLC, 70% или более в случае RP-HPLC и 70% или более в случае IE-HPLC.
После этого полученный при взаимодействии продукт подвергали один раз гидрофобной хроматографии с использованием колонки Source 15ISO (GE Healthcare). Посредством этого процесса удаляли побочные продукты и получали конъюгат (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 3 глюкагона). В этом случае очистку проводили, используя буфер, содержащий цитрат натрия, и градиент концентрации сульфата аммония. Было подтверждено, что полученный при этом выход увеличивался примерно в 1,5 раза или более по сравнению с имеющимся выходом при использовании того же количества аналога 3 глюкагона.
Элюированный конъюгат (Fc-область иммуноглобулина)-(ПЭГ-содержащий линкер)-(аналог 3 глюкагона) анализировали с использованием методов SE-HPLC, RP-HPLC и IE-HPLC, и была подтверждена высокая степень чистоты, поскольку чистота составляла 90% или более в случае SE-HPLC, 90% или более в случае RP-HPLC и 90% или более в случае IE-HPLC.
В Таблицах 1 и 2 приведено сравнение результатов, полученных в способах из Сравнительных примеров и Примеров.

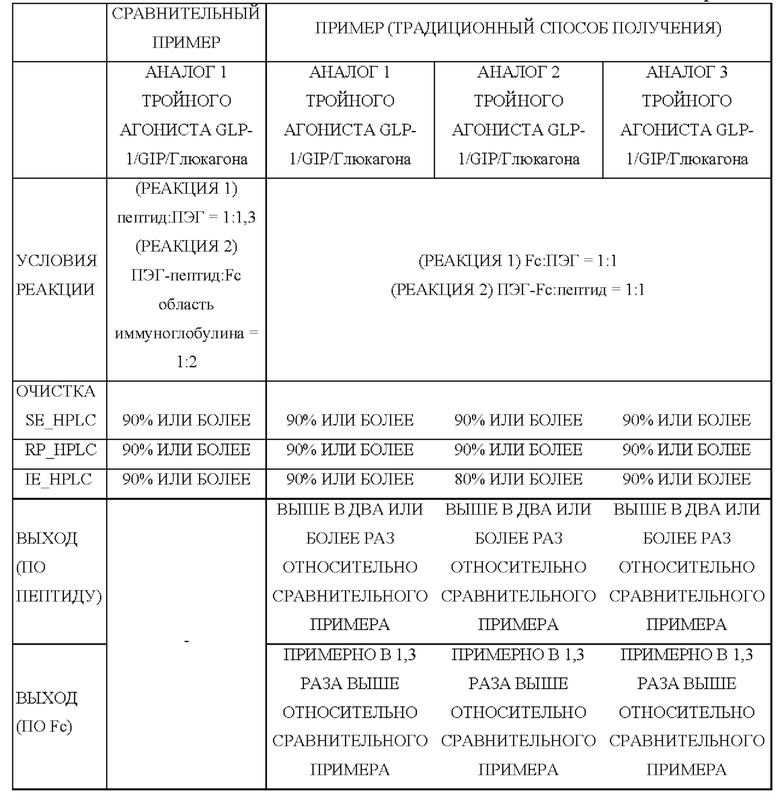
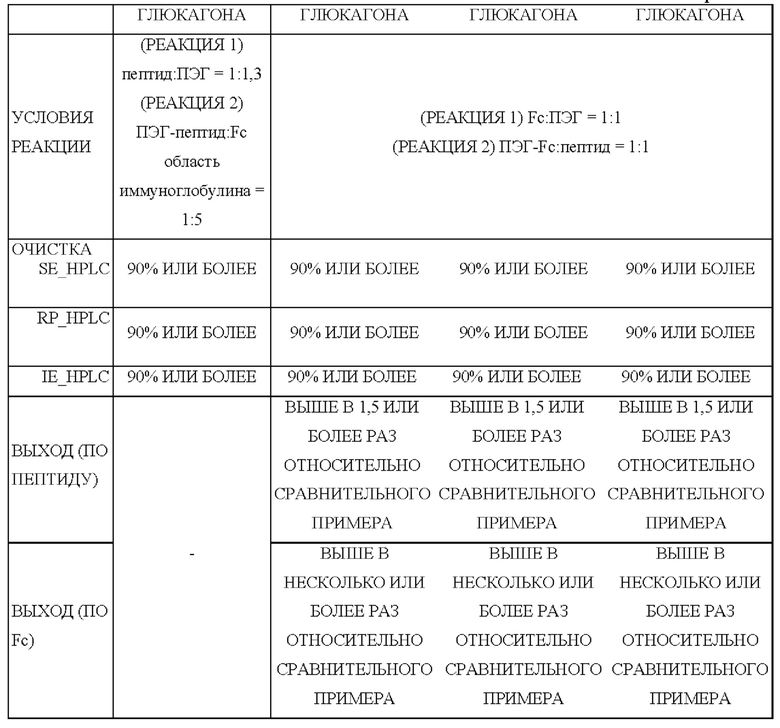
Приведенное выше описание настоящего изобретения представлено с целью иллюстрации, и специалистам в данной области техники должно быть понятно, что могут быть выполнены различные изменения и модификации без изменения технической концепции и существенных признаков настоящего изобретения. Таким образом, понятно, что описанные выше воплощения являются иллюстративными во всех аспектах и не ограничивают настоящее изобретение. Следовательно, объем раскрытия изобретения определяется не подробным описанием, а формулой изобретения и ее эквивалентами, и все варианты в пределах объема формулы изобретения и ее эквивалентов должны рассматриваться как включенные в раскрытие изобретения.
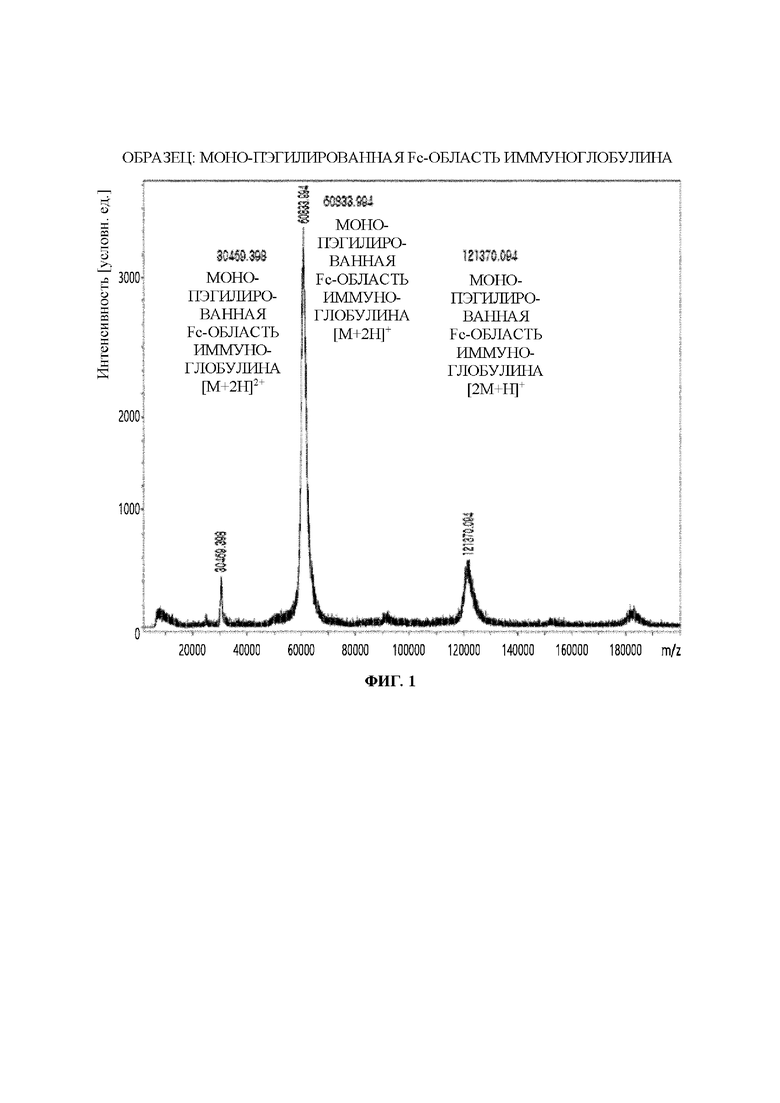
Изобретение относится к области биотехнологии. Описана группа изобретений, включающая промежуточное соединение для получения конъюгата лекарственного средства или его фармацевтически приемлемая соль, способ получения конъюгата физиологически активного полипептида и конъюгат лекарственного средства для приготовления лекарственных средств или композиций для предупреждения, лечения и облегчения заболеваний. В одном варианте реализации промежуточное соединение содержит Fc-область иммуноглобулина, содержащую на N-конце последовательность шарнирной области с аминокислотной последовательностью SEQ ID NO: 8 или 9. Изобретение расширяет арсенал средств для получения конъюгатов лекарственных средств. 3 н. и 20 з.п. ф-лы, 1 ил., 2 табл., 2 пр.
1. Промежуточное соединение для получения конъюгата лекарственного средства или его фармацевтически приемлемая соль, где соединение имеет структуру приведенной ниже формулы 2:
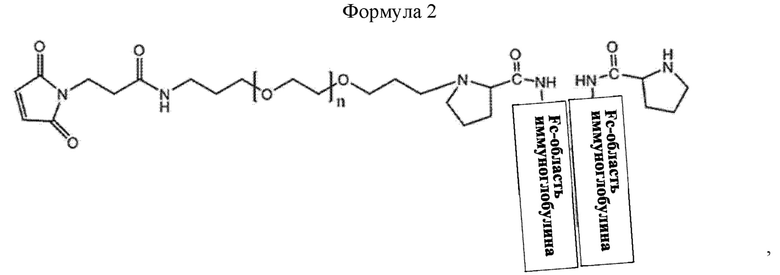
где в формуле 2 n равно от 200 до 250; и
где Fc-область иммуноглобулина содержит на N-конце последовательность шарнирной области, содержащую аминокислотную последовательность SEQ ID NO: 8 или SEQ ID NO: 9.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где Fc-область иммуноглобулина происходит из IgG, IgA, IgD, IgE или IgM.
3. Соединение или его фармацевтически приемлемая соль по п. 2, где Fc-область иммуноглобулина происходит из IgG1, IgG2, IgG3 или IgG4.
4. Соединение или его фармацевтически приемлемая соль по п. 1, где Fc-область иммуноглобулина находится в димерной форме.
5. Соединение или его фармацевтически приемлемая соль по п. 1, где Fc-область иммуноглобулина включает аминокислотную последовательность SEQ ID NO: 10.
6. Соединение или его фармацевтически приемлемая соль по п. 1, где соединение имеет массу от 40 кДа до 250 кДа.
7. Соединение или его фармацевтически приемлемая соль по п. 1, где Fc-область иммуноглобулина содержит последовательность шарнирной области, содержащую аминокислотную последовательность SEQ ID NO: 9.
8. Способ получения конъюгата физиологически активного полипептида, включающий:
получение конъюгата путем присоединения моно-ПЭГилированной Fc-области иммуноглобулина, полученной путем соединения линкера приведенной ниже формулы 4 с N-концом Fc-области иммуноглобулина, содержащей последовательность шарнирной области, содержащую аминокислотную последовательность SEQ ID NO: 8 или SEQ ID NO: 9, к физиологически активному полипептиду:
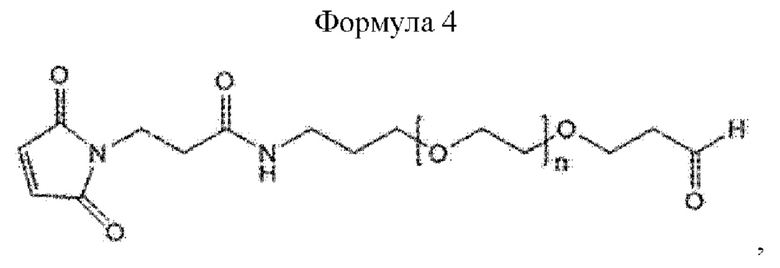
где в формуле 4 n равно от 200 до 250.
9. Способ по п. 8, где моно-ПЭГилированную Fc-область иммуноглобулина получают путем соединения линкера приведенной выше формулы 4 с N-концом Fc-области иммуноглобулина при рН 4,0-8,0 в присутствии восстанавливающего агента.
10. Способ по п. 8, где конъюгат получают путем соединения линкера моно-ПЭГилированной Fc-области иммуноглобулина с физиологически активным полипептидом при рН 5,5-8,0.
11. Способ по п. 8, где получение конъюгата осуществляют путем приведения моно-ПЭГилированной Fc-области иммуноглобулина во взаимодействие с физиологически активным полипептидом в молярном соотношении от 1:1 до 1:3.
12. Способ по п. 8, включающий:
получение моно-ПЭГилированной Fc-области иммуноглобулина путем соединения линкера формулы 4 с N-концом Fc-области иммуноглобулина; и
получение конъюгата путем присоединения линкера моно-ПЭГилированной Fc-области иммуноглобулина, полученной на предшествующей стадии, к физиологически активному полипептиду.
13. Способ по п. 12, где линкер моно-ПЭГилированной Fc-области иммуноглобулина присоединяют к цистеину физиологически активного полипептида.
14. Способ по п. 12, включающий:
получение моно-ПЭГилированной Fc-области иммуноглобулина путем
соединения линкера формулы 4 с N-концом Fc-области иммуноглобулина;
очистку моно-ПЭГилированной Fc-области иммуноглобулина, полученной на предшествующей стадии, посредством анионообменной хроматографией в буферном растворе с рН 6,0-8,5; и
получение конъюгата путем присоединения линкера моно-ПЭГилированной Fc-области иммуноглобулина, очищенной на предшествующей стадии, к физиологически активному полипептиду.
15. Способ по п. 12, который осуществляют без ультрафильтрации/диафильтрации после получения моно-ПЭГилированной Fc-области иммуноглобулина.
16. Способ по п. 8, дополнительно включающий очистку конъюгата посредством гидрофобной хроматографии.
17. Способ по п. 8, где линкер имеет массу от 1 кДа до 100 кДа.
18. Способ по п. 8, где физиологически активный полипептид выбран из группы, состоящей из глюкагоноподобного пептида-1 (GLP-1), гранулоцитарного колониестимулирующего фактора (G-CSF), гормона роста человека (hGH), эритропоэтина (ЕРО), глюкагона, инсулина, рилизинг-фактора гормона роста; пептида, высвобождающего гормон роста; интерферонов, рецепторов интерферонов; рецепторов сопряженных с G-белком; интерлейкинов, рецепторов интерлейкинов, ферментов, интерлейкин-связывающего белка, цитокин-связывающего белка, фактора активации макрофагов, макрофагального пептида, фактора В-клеток, фактора Т-клеток, белка А, ингибитора аллергии; гликопротеина, ассоциированного с некрозом клеток; иммунотоксина, лимфотоксина, фактора некроза опухолей, онкосупрессора, фактора роста метастазов, α-1-антитрипсина, альбумина, α-лактальбумина, аполипопротеина Е, высокогликозилированного эритропоэтина, ангиопоэтинов, гемоглобина, тромбина; пептида, активирующего рецепторы тромбина; тромбомодулина, факторов крови VII, VIIa, VIII, IX и XIII, плазминоген-активирующего фактора, фибрин-связывающего пептида, урокиназы, стрептокиназы, гирудина, белка С, С-реактивного белка, ингибитора lenin, ингибитора коллагеназы, супероксиддисмутазы, лептина, фактора роста тромбоцитов, эпителиального фактора роста, эпидермального фактора роста, ангиостатина, ангиотензина, фактора роста костей, костного морфогенетического белка, кальцитонина, атриопептина; фактора, индуцирующего хрящеобразование; элкатонина; фактора, активирующего соединительную ткань; ингибитора пути тканевого фактора, фолликулостимулирующего гормона, лютеинизирующего гормона, рилизинг-фактора лютеинизирующего гормона, фактора роста нервов, гормона паращитовидной железы, релаксина, секретина, соматомедина, инсулиноподобного фактора роста, гормона коры надпочечников, холецистокинина, панкреатического полипептида, гастрин-высвобождающего пептида, кортикотропин-рилизинг-фактора, тиреотропного гормона, аутотаксина, лактоферрина, миостатина, инкретинов, желудочного ингибирующего полипептида (GIP), двойного агониста GLP-1/GIP, тройного агониста GLP-1/GIP/глюкагона, антигенов клеточной поверхности; вакцинных антигенов вирусного происхождения; моноклональных антител, поликлональных антител и фрагментов антител.
19. Способ по п. 18, где физиологически активный полипептид представляет собой тройной агонист GLP-1/GIP/глюкагона или его аналог.
20. Способ по п. 19, где физиологически активный полипептид включает одну из аминокислотных последовательностей с SEQ ID NO: 1-6.
21. Способ по п. 8, где последовательность шарнирной области содержит аминокислотную последовательность SEQ ID NO: 9.
22. Способ по п. 8, где Fc-область иммуноглобулина происходит из IgG1, IgG2, IgG3 или IgG4.
23. Конъюгат лекарственного средства для приготовления лекарственных средств или композиций для предупреждения, лечения и облегчения заболеваний, содержащий структуру указанной ниже Формулы 5:
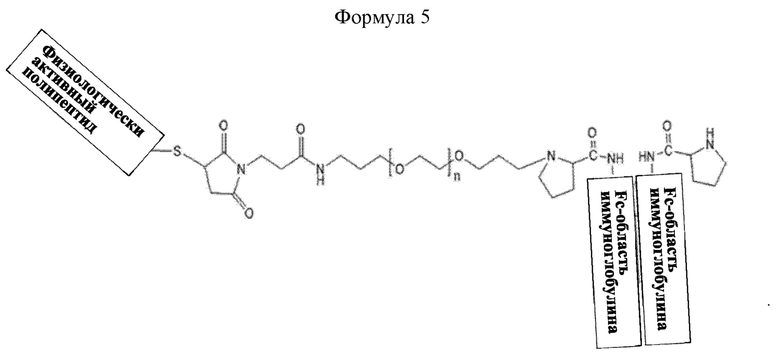
| WO 2019066609 A1 04.04.2019 | |||
| WO 2017155288 A1 14.09.2017 | |||
| WO 2018105988 A1 14.06.2018 | |||
| WO 2017052321 A1 30.03.2017 | |||
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ Fc-ОБЛАСТЬ ИММУНОГЛОБУЛИНА В КАЧЕСТВЕ НОСИТЕЛЯ | 2004 |
|
RU2352583C2 |
| БЕЛКОВЫЙ КОМПЛЕКС, ПОЛУЧЕННЫЙ С ИСПОЛЬЗОВАНИЕМ ФРАГМЕНТА ИММУНОГЛОБУЛИНА, И СПОСОБ ПОЛУЧЕНИЯ ТАКОГО КОМПЛЕКСА | 2004 |
|
RU2356909C2 |

