Область техники, к которой относится изобретение
Настоящее изобретение относится к способам лечения гнойного гидраденита с применением ингибиторов лейкотриен-A4-гидролазы (LTA4H).
Предпосылки изобретения
Гнойный гидраденит (HS) (также называемый инверсными угрями или болезнью Вернейля) представляет собой хроническое рецидивирующее воспалительное заболевание, для которого характерны глубоко расположенные узелки, свищевые ходы и абсцессы, которые приводят к фиброзу в подмышечных, паховых, складках груди и аногенитальных областях. (Revuz and Jemec (2016) Dermatol Clin 34:1-5; Jemec GB. (2012) N Engl J Med 366:158-64). Он связан с сильной болью и сопутствующими заболеваниями, включая метаболические, психиатрические и аутоиммунные нарушения, а также с повышенным риском развития рака кожи. (Revuz (2016); Shlyankevich et al. (2014) J Am Acad Dermatol 71:1144-50; Kohorst et al. (2015) J Am Acad Dermatol 73:S27-35; Wolkenstein et al. (2007) J Am Acad Dermatol 56:621-3).
Опубликованные коэффициенты распространенности HS варьируют от < 1% до 4% популяции. [Shlyankevich et al. (2014); Cosmatos et al. (2013) J Am Acad Dermatol 68:412-9; Davis et al. (2015) Skin Appendage Disord 1:65-73; Revuz et al. (2008) J Am Acad Dermatol 59:596-601; McMillan K. (2014) Am J Epidemiol 179:1477-83; Garg et al. (2017) J Am Acad Dermatol, 77(1):118-122; Jemec et al. (1996) J Am Acad Dermatol 35:191-4]. Однако истинную распространенность трудно установить, поскольку HS редко диагностируется, а оценки колеблются в зависимости от схемы исследования, популяции и географического местоположения. [Miller et al. (2016) Dermatol Clin 34:7-16]. Хотя Национальные институты здравоохранения (NIH) не классифицируют HS как редкое заболевание, эксперты обычно считают, что распространенность этого заболевания составляет < 1% от населения Соединенных Штатов Америки (США). [Cosmatos et al. (2013); Genetic and Rare Diseases Information Center. National Institutes of Health. Hidradenitis suppurativa. Доступно на: //rarediseases.info.nih.gov/diseases/6658/hidradenitis-suppurativa. Доступ 20 марта 2017 г.; Gulliver et al. (2016) Rev Endocr Metab Disord 17:343-51; Garg et al. 2007].
Современное лечение для HS заключается в использовании местных и/или системных антибиотиков, гормональных вмешательств, ретиноидов и, в некоторых случаях, иммунодепрессантов, биологических препаратов, таких как моноклональное антитело адалимумаб, представляющее собой ингибитор фактора некроза опухоли [TNF], являющееся единственным одобренным лекарственным средством для HS, и часто, в качестве крайней меры, обширное хирургическое удаление. [Gulliver et al. (2016); Zouboulis et al. (2015) J Eur Acad Dermatol Venereol 29:619-4414-16; Kimball et al. (2016) N Engl J Med 375:422-34]. Так как очаги поражения являются болезненными, пациенты часто нуждаются в анальгетиках и болеутолении.
Однако, контроль симптомов и устранение очагов поражения не являются устойчивыми среди средств лечения, и результаты рандомизированных контролируемых клинических испытаний, обеспечивающие доказательные данные, отсутствуют для большинства средств лечения, и только адалимумаб является одобренным. Хотя терапию антибиотиками применяют в качестве долгосрочного лечения в течение месяцев и даже лет, терапия антибиотиками может, таким образом, привести к развитию устойчивости к антибиотикам. Пероральное лечение с помощью ретиноидов вызывает опасения касательно тератогенности в популяции, ведущей активную половую жизнь и в большинстве представленной лицами женского пола. Более того, эффективность противовоспалительных лекарственных средств, таких как дапсон, фумараты и циклоспорин, основана на небольших диагностических исследованиях с варьирующими результатами, и эти молекулы не являются применяемыми систематически. Вследствие этих противоречивых результатов и тяжести заболевания HS пациенты с HS пользуются медицинским обслуживанием в дорогостоящих учреждениях (например, отделение неотложной помощи и стационарное лечение) с большей частотой, чем пациенты с другими хроническими воспалительными состояниями кожи. (Khalsa et al. (2016) J Am Acad Dermatol 73:609-14; Kirby et al. (2014) JAMA Dermatol 150:937-44). Поскольку не существует медицинского лечения для HS, а заболевание является изнурительным физически и психологически, то существует очевидная неудовлетворенная потребность в обеспечении безопасных и эффективных средств долгосрочного лечения пациентов с HS, в частности пероральных средств лечения.
КРАТКОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Хотя патогенез HS по-прежнему изучен не полностью, это хроническое кожное заболевание характеризуется большим количеством нейтрофилов и макрофагов в воспалительных очагах поражения при HS. Очевидно, что нейтрофилы и макрофаги являются ключевыми факторами патологического механизма гнойного гидраденита [Shah et al (2017), “The critical role of macrophages in the pathogenesis of hidradenitis suppurativa”. Inflamm. Res. 66(11): 931-945; [Lima et al (2016), “Keratinocytes and neutrophils are important sources of proinflammatory molecules in hidradenitis suppurativa”. Br. J. Dermatol. 174: 514-21; нейтрофилы и макрофаги являются основными продуцентами и клетками-мишенями липидного медиатора воспаления, представляющего собой лейкотриен B4 (LTB4), и противовоспалительного проразрешающего медиатора липоксина A4 (LXA4) (Serhan et al 2008, “Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators”. Nat. Rev. Immunol. p. 349-61). Провоспалительный липидный медиатор LTB4 представляет собой одну из наиболее эффективных хемотаксических молекул для нейтрофилов и моноцитов, и может стимулировать и активировать как нейтрофилы, так и макрофаги [Marc Peters-Golden and William R Henderson (2007) “Leukotrienes”, N Engl J Med 357:1841-1854]. Нанесение LTB4 на здоровую кожу вызывает миграцию IL-17+ нейтрофилов, вовлеченных в патологический механизм HS [Lima et al (2016), “Keratinocytes and neutrophils are important sources of proinflammatory molecules in hidradenitis suppurativa”. Br. J. Dermatol. 174: 514-21.], образование нейтрофильных абсцессов и гиперпролиферацию кератиноцитов. [Hendriks et al (2014) “Cutaneous application of leukotriene B4 as an in vivo model of psoriasis-like skin inflammation: an immunohistological study), Skin Pharmacol Physiol 27 (3):120-6. Значимость LTB4 была клинически продемонстрирована для родственной патологии кожи, представляющей собой воспалительное акне (Zouboulis et al 2003, “A new concept for acne therapy: a pilot study with zileuton, an oral 5-lipoxygenase inhibitor. Arch Dermatol” p. 668-70).
Самостоятельно проведенный липидомический анализ биоптатов HS показал сильное повышение уровня липидного медиатора LTB4 в пораженной коже пациентов с HS по сравнению со здоровой кожей. Кроме того, лейкоциты в очагах поражения являлись преобладающими, и они продемонстрировали сильно выраженную экспрессию 5-липоксигеназы и внутриклеточную локализацию данного фермента на перинуклеарной мембране, что является явным признаком активации 5-липоксигеназного пути, приводящего к продуцированию LTB4. (Christmas et al 1999, “Differential localization of 5- and 15- lipoxygenases to the nuclear envelope in RAW macrophages”. J. Biol. Chem. p. 25594-8). Транскриптомный анализ очагов поражения показал сверхэкспрессию генов 5-липоксигеназного пути ALOX5 и LTA4H, двух ферментов, ответственных за биосинтез LTB4 из арахидоновой кислоты [Marc Peters-Golden and William R Henderson (2007) “Leukotrienes”, N Engl J Med 357:1841-1854]. Ингибирование конечного и ограничивающего скорость фермента LTA4H 5-липоксигеназного пути может предотвращать биосинтез провоспалительного LTB4, но поддерживать или даже увеличивать биосинтез противовоспалительного LXA4. Кроме того, увеличение уровня LXA4 может влиять на фиброз в дополнение к противовоспалительному эффекту лекарственного средства ( et al. 2011, “Lipoxin A4 and benzo-lipoxin A4 attenuate experimental renal fibrosis”. FASEB J. p. 2967-79).
et al. 2011, “Lipoxin A4 and benzo-lipoxin A4 attenuate experimental renal fibrosis”. FASEB J. p. 2967-79).
Соединения, описанные в настоящем изобретении, ингибируют LTA4H и тем самым предотвращают биосинтез провоспалительного лейкотриена B4 (LTB4), но увеличивают образование противовоспалительного усиливающего разрешение воспаления липоксина A4 (LXA4). Оба липидных медиатора, как известно, играют важную роль в координации нейтрофильного воспаления. Ингибирование LTA4H представляет собой значимый механизм лечения опосредуемых нейтрофилами воспалительных состояний.
Следовательно, целью настоящего изобретения является предоставление нового способа лечения или предупреждения гнойного гидраденита (HS) у субъекта, нуждающегося в этом, включающего введение указанному субъекту эффективного количества ингибитора лейкотриен-A4-гидролазы (LTA4H).
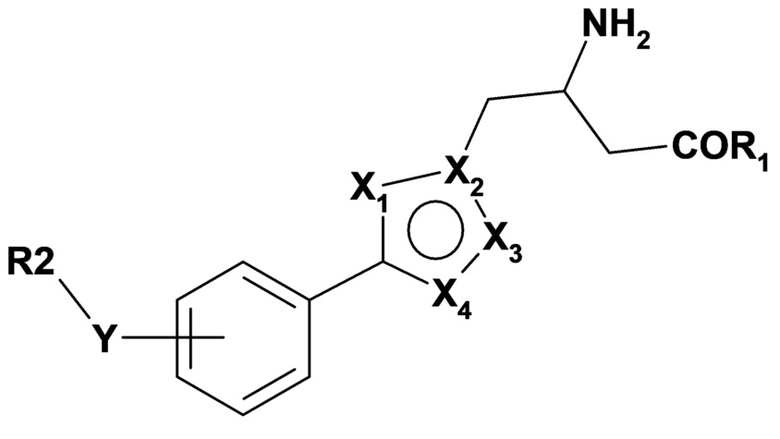
Более конкретно, настоящее изобретение относится к способу лечения или предупреждения гнойного гидраденита у субъекта, нуждающегося в этом, включающему введение указанному субъекту терапевтически эффективного количества соединения формулы (I):
 (I),
(I),
где
R1 представляет собой OH или NH2;
Y представляет собой O, S или CH2;
X1, X2, X3 и X4 представляют собой N; или
X1, X2, X3 и X4 выбраны из N, NH, C, CH и O при условии, что по меньшей мере два из X1, X2, X3 или X4 представляют собой N или NH;
R2 представляет собой C1-C6алкил, необязательно замещенный фенилом; C3-C6циклоалкил; фенил, необязательно являющийся замещенным галогеном, циано, C1-C6алкилом, необязательно замещенным галогеном, C1-C6алкокси или 5-6-членным гетероарильным кольцом, содержащим от 1 до 3 гетероатомов, выбранных из N, O и S; или 5-10-членный моно- или бициклический гетероарил, содержащий от 1 до 4 гетероатомов, выбранных из N, O и S, при этом указанный гетероарил необязательно является замещенным галогеном, циано или C1-C6алкилом, необязательно замещенным галогеном; или его фармацевтически приемлемой соли.
Кроме того, в настоящем изобретении дополнительно предусматривается способ лечения или предупреждения HS у субъекта, нуждающегося в этом, включающий введение указанному субъекту терапевтически эффективного количества ингибитора LTA4H с одним или несколькими терапевтическими средствами.
В другом аспекте настоящего изобретения в настоящем изобретении предусматривается ингибитор LTA4H для применения в лечении и/или предупреждении HS у пациента, нуждающегося в таком лечении и/или предупреждении. Более конкретно, настоящее изобретение относится к соединению формулы (I), как описано выше, для применения в лечении и/или предупреждении HS у пациента, нуждающегося в таком лечении и/или предупреждении.
Настоящее изобретение дополнительно относится к комбинациям ингибитора LTA4H с одним или несколькими дополнительными терапевтическими средствами для применения в лечении или предупреждении HS у пациента, нуждающегося в таком лечении и/или предупреждении.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фигуре 1 представлен иллюстративный спектр XRPD для кристаллической формы (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в ее свободной форме, обозначенной в данном документе как форма B, при этом по оси X отмечены градусы 2θ (2-тета), а по оси Y отмечена относительная интенсивность.
На фигуре 2 представлены иллюстративные результаты DSC для свободной формы (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты, обозначенной в данном документе как форма B.
На фигуре 3 представлены иллюстративные результаты TGA для свободной формы (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты, обозначенной в данном документе как форма В.
Более подробные перечни пиков XRPD для каждой из форм B представлены в таблице 1 ниже, в которой также представлены значения % относительной интенсивности (I/I0×100). Следует понимать, что на спектрах порошковой рентгеновской дифракции или на порошковых рентгеновских дифрактограммах присутствует естественная изменчивость в отношении значений, измеренных в градусах 2θ (°2θ), в результате, например, погрешности прибора (включая различия между приборами). В связи с этим следует понимать, что в измерениях пиков XRPD присутствует изменчивость 2θ составляющая до ±0,2°, и, тем не менее, такие значения пиков также будут считаться характерными для конкретной твердой формы кристаллических веществ, описанных в данном документе. Также следует понимать, что другие измеренные значения из экспериментов XRPD и экспериментов DSC/TGA, такие как относительная интенсивность и содержание воды, могут изменяться в результате, например, условий получения образца, и/или хранения, и/или окружающей среды, и измеренные значения также будут считаться характерными для конкретной твердой формы кристаллических веществ, описанных в данном документе.
На фигуре 4 представлен анализ очагов поражения у пациентов с HS путем иммуногистохимического исследования с применением окрашивания H&E.
На фигуре 5 представлен иммуногистохимический анализ биоптатов пораженной HS кожи.
На фигуре 6 представлена иллюстративная микрофотография, демонстрирующая 5-липоксигеназу, локализованную на ядерной мембране в многоядерной гигантской клетке CD68+.
На фигуре 7 представлен выполненный посредством LC-MS/MS анализ липидов из биоптатов пораженной HS кожи и кожи здоровых добровольцев в отношении содержания LTB4.
На фигуре 8 представлен выполненный посредством LC-MS/MS анализ липидов из биоптатов пораженной HS кожи и кожи здоровых добровольцев в отношении содержания 5-HETE.
На фигуре 9 представлен транскриптомный анализ для биоптатов пораженной HS кожи по сравнению с биоптатами здоровой кожи.
На фигуре 10 представлено подавление соединением из примера 1 биосинтеза провоспалительного LTB4 в биоптатах кожи пациентов с HS, как измерено с помощью LC-MS/MS.
На фигуре 11 представлено подавление соединением согласно варианту осуществления 3D биосинтеза провоспалительного LTB4 в биоптатах кожи пациентов с HS, как измерено с помощью LC-MS/MS.
На фигуре 12 представлено ингибирование высвобождения LTB4 из стимулированной ex-vivo крови у мышей, обработанных 3 мг/кг или 10 мг/кг (S)-3-амино-4-(5-(4-(4-хлорфенокси)-фенил)-2H-тетразол-2-ил)бутановой кислоты (соединением согласно варианту осуществления 3D).
На фигуре 13 представлены профили зависимости средней концентрации в плазме крови от времени после однократного перорального введения (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (соединения из примера 1) при разных дозах.
На фигуре 14 представлен профиль зависимости средней концентрации в плазме крови от времени после многократного перорального введения (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (соединения из примера 1) при разных дозах (фигура 14a: день 1; фигура 14b: день 12).
На фигуре 15 представлена концентрация LTB4 в стимулированной ex-vivo крови после перорального введения (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (соединения из примера 1) при измеренной концентрации соединения из примера 1 в плазме крови (взаимосвязь PK/PD).
На фигуре 16 представлено изменение LTB4 относительно исходного уровня (ингибирование в крови) после многократного перорального введения (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (соединения из примера 1) при разных дозах, измеренное в разное время (дни) после первой дозы.
На фигуре 17 представлено ингибирование LTB4 в коже и плазме крови после перорального введения (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (соединения из примера 1).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Используемый в данном документе термин “C1-C6алкил” относится к полностью насыщенному разветвленному или неразветвленному углеводородному фрагменту, содержащему до 6 атомов углерода. Если не предусмотрено иное, он относится к углеводородным фрагментам, содержащим от 1 до 6 атомов углерода, от 1 до 4 атомов углерода или от 1 до 2 атомов углерода. Иллюстративные примеры алкила включают без ограничения метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, изопентил, неопентил и н-гексил и т. п.
Используемый в данном документе термин “C1-C6алкокси” относится к алкил-O-, где алкил определен в данном документе выше. Иллюстративные примеры алкокси включают без ограничения метокси, этокси, пропокси, 2-пропокси, бутокси, трет-бутокси, пентилокси, гексилокси, циклопропилокси-, циклогексилокси- и т. п. Как правило, алкоксигруппы содержат от приблизительно 1 до 6 атомов углерода, от 1 до 4 атомов углерода или от 1 до 2 атомов углерода.
Используемый в данном документе термин “C1-C6алкил, необязательно замещенный галогеном” относится к C1-C6алкилу, как определено выше, который может быть замещен одним или несколькими атомами галогена. Примеры включают без ограничения трифторметил, дифторметил, фторметил, трихлорметил, 2,2,2-трифторэтил, 1-фторметил-2-фторэтил, 3-бром-2-фторпропил и 1-бромметил-2-бромэтил.
Используемый в данном документе термин "ди-C1-6алкиламино" относится к фрагменту формулы -N(Ra)-Ra, где каждый Ra представляет собой C1-6алкил, который может быть идентичным или отличающимся, как определено выше.
Используемый в данном документе термин "C3-C6циклоалкил" относится к насыщенным моноциклическим углеводородным группам из 3-6 атомов углерода. Циклоалкил может также называться карбоциклическим кольцом и наоборот с дополнительным указанием присутствующего числа атомов углерода. Если не указано иное, циклоалкил относится к циклическим углеводородным группам, содержащим от 3 до 6 атомов углерода в кольце или от 3 до 4 атомов углерода в кольце. Иллюстративные моноциклические углеводородные группы включают без ограничения циклопропил, циклобутил, циклопентил и циклогексил.
Используемый в данном документе термин “галоген” или “гало” относится к фтору, хлору, брому и йоду.
Используемый в данном документе термин "гетероарил" относится к 5-14-членной моноциклической, или бициклической, или трициклической системе ароматических колец, содержащей от 1 до 8 гетероатомов. Как правило, гетероарил представляет собой 5-10-членную кольцевую систему, содержащую от 1 до 4 гетероатомов, выбранных из N, S или O (например, 5-7-членный моноцикл или 8-10-членный бицикл) или 5-7-членную кольцевую систему. Предпочтительно термин “гетероарил” предусматривает 5-7-членный моноцикл. Типичные гетероарильные группы включают 2- или 3-тиенил, 2- или 3-фурил, 2- или 3-пирролил, 2-, 4- или 5-имидазолил, 3-, 4- или 5-пиразолил, 2-, 4- или 5-тиазолил, 3-, 4- или 5-изотиазолил, 2-, 4- или 5-оксазолил, 3-, 4- или 5-изоксазолил, 3- или 5-1,2,4-триазолил, 4- или 5-1,2,3-триазолил, тетразолил, 2-, 3- или 4-пиридил, 3- или 4-пиридазинил, 3-, 4- или 5-пиразинил, 2-пиразинил, и 2-, 4- или 5-пиримидинил.
Термин “гетероарил” также относится к группе, в которой гетероароматическое кольцо является сочлененным с одним или несколькими арильными, циклоалифатическими или гетероциклильными кольцами, где радикал или точка присоединения находятся на гетероароматическом кольце. Неограничивающие примеры включают 1-, 2-, 3-, 5-, 6-, 7- или 8-индолизинил, 1-, 3-, 4-, 5-, 6- или 7-изоиндолил, 2-, 3-, 4-, 5-, 6- или 7-индолил, 2-, 3-, 4-, 5-, 6- или 7-индазолил, 2-, 4-, 5-, 6-, 7- или 8-пуринил, 1-, 2-, 3-, 4-, 6-, 7-, 8- или 9-хинолизинил, 2-, 3-, 4-, 5-, 6-, 7- или 8-хинолил, 1-, 3-, 4-, 5-, 6-, 7- или 8-изохинолил, 1-, 4-, 5-, 6-, 7- или 8-фталазинил, 2-, 3-, 4-, 5- или 6-нафтиридинил, 2-, 3- , 5-, 6-, 7- или 8-хиназолинил, 3-, 4-, 5-, 6-, 7- или 8-циннолинил, 2-, 4-, 6- или 7-птеридинил, 1-, 2-, 3-, 4-, 5-, 6-, 7- или 8-4aH-карбазолил, 1-, 2-, 3-, 4-, 5-, 6-, 7- или 8-карбзаолил, 1-, 3-, 4-, 5-, 6-, 7-, 8- или 9-карболинил, 1-, 2-, 3-, 4-, 6-, 7-, 8-, 9- или 10-фенантридинил, 1- , 2-, 3-, 4-, 5-, 6-, 7-, 8- или 9-акридинил, 1-, 2-, 4-, 5-, 6-, 7-, 8- или 9-перимидинил, 2-, 3-, 4-, 5-, 6-, 8-, 9- или 10-фенатролинил, 1-, 2-, 3-, 4-, 6-, 7-, 8- или 9-феназинил, 1-, 2-, 3-, 4-, 6-, 7-, 8-, 9- или 10-фенотиазинил, 1-, 2-, 3-, 4-, 6-, 7-, 8-, 9- или 10-феноксазинил, 2-, 3-, 4-, 5-, 6- или l-, 3-, 4-, 5-, 6-, 7-, 8-, 9- или 10-бензизохинилинил, 2-, 3-, 4- или тиено[2,3-b]фуранил, 2-, 3-, 5-, 6-, 7-, 8-, 9-, 10 - или 11-7H-пиразинo[2,3-c]карбазолил, 2-, 3-, 5-, 6- или 7-2H-фуро[3,2-b]-пиранил, 2-, 3-, 4-, 5-, 7- или 8-5H-пиридо[2,3-d]-o-оксазинил, 1-, 3- или 5-1H-пиразолo[4,3-d]-оксазолил, 2-, 4- или 5-4H-имидазо[4,5-d]-тиазолил, 3-, 5- или 8-пиразинo[2,3-d]-пиридазинил, 2-, 3-, 5- или 6-имидазо[2,1-b]-тиазолил, 1-, 3-, 6-, 7-, 8- или 9-фуро[3,4-c]-циннолинил, 1-, 2-, 3-, 4-, 5-, 6-, 8-, 9-, 10- или 11-4H-пиридо[2,3-c]-карбазолил, 2-, 3-, 6-, или 7-имидазо[1,2-b][1,2,4]-триазинил, 7-бензо[b]тиенил, 2-, 4-, 5-, 6- или 7-бензоксазолил, 2-, 4-, 5-, 6- или 7-бензимидазолил, 2-, 4-, 4-, 5-, 6- или 7-бензотиазолил, 1-, 2-, 4-, 5-, 6-, 7-, 8- или 9-бензоксапинил, 2-, 4-, 5-, 6-, 7- или 8-бензоксазинил, 1-, 2-, 3-, 5-, 6-, 7-, 8-, 9-, 10- или 11-1H-пирроло[1,2-b][2]-бензaзапинил. Типичные сочлененные гетероарильные группы включают без ограничения 2-, 3-, 4-, 5-, 6-, 7- или 8-хинолинил, 1-, 3-, 4-, 5-, 6-, 7- или 8-изохинолинил, 2-, 3-, 4-, 5-, 6- или 7-индолил, 2-, 3-, 4-, 5-, 6- или 7-бензо[b]тиенил, 2-, 4-, 5-, 6- или 7-бензоксазолил, 2-, 4-, 5-, 6- или 7-бензимидазолил и 2-, 4-, 5-, 6- или 7-бензотиазолил.
Замещенный гетероарил представляет собой гетероарильную группу, содержащую один или несколько заместителей.
Используемый в данном документе термин “гетероциклил” относится к гетероциклической группе, которая является насыщенной или частично насыщенной, и предпочтительно является моноциклическим или полициклическим кольцом (в случае полициклического кольца, в частности, бициклическим, трициклическим или спироциклическим кольцом); и содержит от 3 до 24, более предпочтительно от 4 до 16, наиболее предпочтительно от 5 до 10 и наиболее предпочтительно 5 или 6 кольцевых атомов; где один или несколько, предпочтительно от одного до четырех, особенно один или два кольцевых атома являются гетероатомом (остальные атомы кольца, следовательно, представляют собой атомы углерода). Связывающее кольцо (т. е. кольцо, соединенное с молекулой) предпочтительно содержит от 4 до 12, особенно от 5 до 7 кольцевых атомов. Термин гетероциклил исключает гетероарил. Гетероциклическая группа может быть присоединена к гетероатому или атому углерода. Гетероциклил может включать конденсированные или мостиковые кольца, а также спироциклические кольца. Примеры гетероциклов включают тетрагидрофуран (THF), дигидрофуран, 1,4-диоксан, морфолин, 1,4-дитиан, пиперазин, пиперидин, 1,3-диоксолан, имидазолидин, имидазолин, пирролин, пирролидин, тетрагидропиран, дигидропиран, оксатиолaн, дитиолан, 1,3-диоксан, 1,3-дитиан, оксатиан, тиоморфолин и т. п.
Замещенный гетероциклил представляет собой гетероциклильную группу, независимо замещенную 1-4, как например одним, или двумя, или тремя, или четырьмя заместителями.
Используемый в данном документе термин "арил" относится к ароматической углеводородной группе, содержащей 6-20 атомов углерода в кольцевой части. Как правило, арил является моноциклическим, бициклическим или трициклическим арилом, содержащим 6-20 атомов углерода. Кроме того, термин "арил", применяемый в данном документе, относится к ароматическому заместителю, который может быть представлен одиночным ароматическим кольцом или несколькими ароматическими кольцами, сочлененными друг с другом. Неограничивающие примеры включают фенил, нафтил или тетрагидронафтил.
Замещенный арил представляет собой арильную группу, замещенную 1-5 (как например одним, или двумя, или тремя) заместителями, независимо выбранными из группы, состоящей из гидроксила, тиола, циано, нитро, C1-C4-алкила, C1-C4-алкенила, C1-C4-алкинила, C1-C4-алкокси, C1-C4-тиоалкила, C1-C4-алкенилокси, C1-C4-алкинилокси, галогена, C1-C4-алкилкарбонила, карбокси, C1-C4-алкоксикарбонила, амино, C1-C4-алкиламино, ди-C1-C4-алкиламино, C1-C4-алкиламинокарбонила, ди-C1-C4-алкиламинокарбонила, C1-C4-алкилкарбониламино, C1-C4-алкилкарбонил(C1-C4-алкил)амино, сульфонила, сульфамоила, алкилсульфамоила, C1-C4-алкиламиносульфонила, где каждая из вышеупомянутых углеводородных групп (например, алкиловые, алкениловые, алкиниловые, алкоксильные остатки) может быть дополнительно замещенной одним или несколькими остатками, независимо выбранными в каждом случае из галогеновых, гидроксильных или C1-C4-алкоксильных групп.
Используемые в данном документе термины “приблизительно” и “по сути” указывают в отношении таких характеристик, как эндотермы, эндотермический пик, экзотермы, смещения базовой линии и т. д., на то, что их значения могут изменяться. В отношении положений пиков рентгеновской дифракции термины “приблизительно” или “по сути” означают то, что учитываются типичная изменчивость положения и интенсивности пика. Например, специалист в данной области поймет, что положения пика (2θ) будут демонстрировать некоторую изменчивость между устройствами, обычно вплоть до 0,2°. Иногда изменчивость может составлять более 0,2° в зависимости от различий в калибровке устройств. Кроме того, специалист в данной области поймет, что значения относительной интенсивности пика будут демонстрировать изменчивость между устройствами, а также изменчивость, обусловленную степенью кристалличности, предпочтительной ориентацией, поверхностью полученного образца и другими факторами, известными специалистам в данной области, и должны рассматриваться исключительно как качественные показатели. В случае DSC изменение наблюдаемых значений температуры будет зависеть от скорости изменения температуры, а также методики получения образца и конкретного используемого прибора. Таким образом, приведенные в данном документе значения эндотермы/точки плавления, относящиеся к термограммам DSC/TGA, могут изменяться на ±5°C (и также считаются характерными для конкретной кристаллической формы, описанной в данном документе). При использовании в контексте других характеристик, таких как, например, процент по весу (% по весу), температуры реакций, термин “приблизительно” указывает на отклонение в ±5%.
Используемое в данном документе выражение “по сути чистая фаза”, в случае использования в отношении любой кристаллической формы соединения формулы I, означает соединение, характеризующееся чистотой фазы, составляющей более чем приблизительно 90% по весу, в том числе более чем приблизительно 90, 91, 92, 93, 94, 95, 96, 97, 98 и приблизительно 99% по весу, а также в том числе равной приблизительно 100% по весу соединения формулы I, в пересчете на вес соединения на безводной основе. Термин “чистая фаза” или “чистота фазы” в данном документе относится к однородности фазы в отношении конкретной твердой формы соединения формулы I и не подразумевает обязательно высокую степень химической чистоты при отсутствии прямого указания на это. Чистота фазы может быть определена в соответствии со способами, известными из уровня техники, например, с использованием XRPD, для выполнения количественного фазового анализа с использованием одного или нескольких подходов, известных из уровня техники, например, с помощью способа внешнего стандарта, прямых сопоставлений линейных (пиковых) характеристик, которые относятся к различным фазам в конкретных спектрах, или с помощью способа внутреннего стандарта. Однако количественное определение чистоты фазы с помощью XRPD может быть осложнено присутствием аморфного вещества. Соответственно, другие способы, которые можно применять для определения чистоты фазы, включают, например, ЯМР-спектроскопию твердого тела, рамановскую спектроскопию и/или инфракрасную спектроскопию. Специалист в данной области техники сможет легко разобраться в таких способах и в том, как применять такие дополнительные (или альтернативные) способы определения чистоты фазы.
Используемые в данном документе термины “соль” или “соли” относятся к соли присоединения кислоты или присоединения основания соединения для применения в способе согласно настоящему изобретению. “Соли” включают, в частности, “фармацевтически приемлемые соли”. Термин “фармацевтически приемлемые соли” относится к солям, которые сохраняют биологическую эффективность и свойства соединений по настоящему изобретению и которые, как правило, не являются нежелательными с биологической или иной точки зрения. Во многих случаях соединения для применения в способе согласно настоящему изобретению способны к образованию кислых и/или основных солей за счет присутствия амино- и/или карбоксильных групп или им подобных групп.
Фармацевтически приемлемые соли присоединения кислоты могут быть образованы неорганическими и органическими кислотами, например, ацетатные, аспартатные, бензоатные, безилатные, бромидные/гидробромидные, бикарбонатные/карбонатные, бисульфатные/сульфатные, камфорсульфонатные, хлоридные/гидрохлоридные, хлортеофиллонатные, цитратные, этандисульфонатные, фумаратные, глюцептатные, глюконатные, глюкуронатные, гиппуратные, гидройодидные/йодидные, изетионатные, лактатные, лактобионатные, лаурилсульфатные, малатные, малеатные, малонатные, манделатные, мезилатные, метилсульфатные, нафтоатные, напсилатные, никотинатные, нитратные, октадеканоатные, олеатные, оксалатные, пальмитатные, памоатные, фосфатные/гидрофосфатные/дигидрофосфатные, полигалактуронатные, пропионатные, стеаратные, сукцинатные, субсалицилатные, тартратные, тозилатные и трифторацетатные соли.
Неорганические кислоты, из которых могут быть получены соли, включают, например, хлористоводородную кислоту, бромистоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и т. п.
Органические кислоты, из которых могут быть получены соли, включают, например, уксусную кислоту, пропионовую кислоту, гликолевую кислоту, щавелевую кислоту, малеиновую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, лимонную кислоту, бензойную кислоту, миндальную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, толуолсульфоновую кислоту, сульфосалициловую кислоту и т. п. Фармацевтически приемлемые соли присоединения основания могут быть образованы с помощью неорганических и органических оснований.
Неорганические основания, из которых могут быть получены соли, включают, например, соли аммония и металлы из столбцов I - XII периодической таблицы элементов. В определенных вариантах осуществления соли получены из натрия, калия, аммония, кальция, магния, железа, серебра, цинка и меди; особенно подходящие соли включают соли аммония, калия, натрия, кальция и магния.
Органические основания, из которых могут быть получены соли, включают, например, первичные, вторичные и третичные амины, замещенные амины, включая встречающиеся в природе замещенные амины, циклические амины, основные ионообменные смолы и т. п. Определенные органические амины включают изопропиламин, бензатин, холинат, диэтаноламин, диэтиламин, лизин, меглюмин, пиперазин и трометамин.
Фармацевтически приемлемые соли для использования в настоящем изобретении можно синтезировать из основного или кислотного фрагмента с помощью традиционных химических способов. Обычно такие соли можно получать путем проведения реакции форм свободной кислоты таких соединений со стехиометрическим количеством подходящего основания (такого как гидроксид, карбонат, бикарбонат Na, Ca, Mg, или K и т. п.) или путем проведения реакции форм свободного основания таких соединений со стехиометрическим количеством подходящей кислоты. Такие реакции, как правило, проводят в воде или в органическом растворителе или в их смеси. Обычно желательно применять неводную среду, такую как простой эфир, этилацетат, этанол, изопропанол или ацетонитрил, при наличии соответствующей практической возможности. Перечни дополнительных подходящих солей можно найти, например, в “Remington's Pharmaceutical Sciences”, 20th ed., Mack Publishing Company, Easton, Pa., (1985); и в “Handbook of Pharmaceutical Salts: Properties, Selection, and Use” от Stahl and Wermuth (Wiley-VCH, Weinheim, Германия, 2002).
Любая формула, приведенная в данном документе, также подразумевает присутствие немеченых форм, а также меченых изотопом форм соединений. Меченые изотопом соединения характеризуются структурами, изображенными на формулах, приведенных в данном документе, за исключением того, что один или несколько атомов замещены атомом, характеризующимся выбранными атомной массой или массовым числом. Примеры изотопов, которые могут быть включены в соединения (т. е. ингибитор LTA4H, описанный в данном документе), включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2H, 3H, 11C, 13C, 14C, 15N, 18F, 31P, 32P, 35S, 36Cl, 125I соответственно. Настоящее изобретение включает различные меченые изотопом соединения, определенные в данном документе, например, соединения, в которых присутствуют радиоактивные изотопы, такие как 3H и 14C, или соединения, в которых присутствуют изотопы, не являющиеся радиоактивными, такие как 2H и 13C. Такие меченые изотопом соединения применимы в метаболических исследованиях (с применением 14C), исследованиях кинетических параметров реакций (например, с применением 2H или 3H), методиках выявления или визуализации, таких как позитронно-эмиссионная томография (PET) или однофотонная эмиссионная компьютерная томография (SPECT), включая анализы распределения лекарственного средства или субстрата в тканях, или при лучевой терапии пациентов. В частности, меченое 18F соединение может быть особенно востребованным для исследований с помощью PET или SPECT. Изотопно-меченные соединения формулы (I), как правило, можно получать с помощью традиционных методик, известных специалистам в данной области, или посредством способов, аналогичных описанным в сопутствующих примерах и способах получения, с использованием подходящих изотопно-меченых реагентов вместо немеченого реагента, используемого ранее.
Кроме того, замещение более тяжелыми изотопами, в частности, дейтерием (т. е. 2H или D), может обеспечивать определенные терапевтические преимущества, обусловленные более высокой метаболической стабильностью, например, увеличение периода полувыведения in vivo, или снижение требований в отношении дозировки, или улучшение в отношении терапевтического индекса. Следует понимать, что дейтерий в данном контексте рассматривается в качестве заместителя соединения формулы (I). Концентрация такого более тяжелого изотопа, конкретно дейтерия, может быть определена посредством коэффициента изотопного обогащения. Используемый в данном документе термин "коэффициент изотопного обогащения" означает отношение содержания изотопа к распространенности в природе указанного изотопа. В случае если заместитель в соединении по настоящему изобретению представляет собой указанный дейтерий, такое соединение характеризуется коэффициентом изотопного обогащения для каждого обозначенного атома дейтерия, составляющим по меньшей мере 3500 (введение 52,5% дейтерия при каждом обозначенном атоме дейтерия), по меньшей мере 4000 (введение 60% дейтерия), по меньшей мере 4500 (введение 67,5% дейтерия), по меньшей мере 5000 (введение 75% дейтерия), по меньшей мере 5500 (введение 82,5% дейтерия), по меньшей мере 6000 (введение 90% дейтерия), по меньшей мере 6333,3 (введение 95% дейтерия), по меньшей мере 6466,7 (введение 97% дейтерия), по меньшей мере 6600 (введение 99% дейтерия) или по меньшей мере 6633,3 (введение 99,5% дейтерия).
Фармацевтически приемлемые сольваты для применения в способе согласно настоящему изобретению включают таковые, где растворитель для кристаллизации может быть замещен изотопом, например, D2O, d6-ацетон, d6-DMSO.
Соединения для применения в способе согласно настоящему изобретению, т. е. соединения формулы (I), которые содержат группы, способные действовать в качестве доноров и/или акцепторов в отношении водородных связей, могут быть способны к образованию сокристаллов с подходящими средствами для образования сокристаллов. Такие сокристаллы могут быть получены из соединений формулы (I) посредством известных процедур получения сокристаллов. Такие процедуры включают измельчение, нагревание, совместную сублимацию, совместное плавление или приведение в контакт в растворе соединений формулы (I) со средством для образования сокристаллов в условиях кристаллизации и выделение сокристаллов, образованных таким образом. Подходящие средства для образования сокристаллов включают средства, описанные в WO 2004/078163. Следовательно, в настоящем изобретении дополнительно предусматриваются сокристаллы, содержащие соединение формулы (I), для применения в способе согласно настоящему изобретению.
Используемый в данном документе термин “введение” по отношению к соединению, например, ингибитору LTA4H (например, соединению формулы I или конкретному соединению, описанному в данном документе), или другому средству используется для обозначения доставки этого соединения пациенту любым путем.
Используемый в данном документе термин "фармацевтически приемлемый носитель" включает все возможные растворители, дисперсионные среды, покрытия, поверхностно-активные вещества, антиоксиданты, консерванты (например, антибактериальные средства, противогрибковые средства), средства для обеспечения изотоничности, замедляющие абсорбцию средства, соли, консерванты, стабилизаторы лекарственных средств, связующие вещества, вспомогательные вещества, разрыхляющие средства, смазывающие вещества, подслащивающие средства, ароматизирующие средства, красители и т. п. и их комбинации, которые будут известны специалистам в данной области техники (см., например, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289-1329). За исключением случаев, когда любой традиционный носитель является несовместимым с активным ингредиентом, предполагается его применение в терапевтических или фармацевтических композициях.
Термин "терапевтически эффективное количество" соединения для применения в способе согласно настоящему изобретению относится к количеству указанного соединения, которое будет вызывать биологический или медицинский ответ у субъекта, например, снижение или ингибирование активности фермента или белка, или уменьшать интенсивность проявлений симптомов HS, облегчать обусловленные HS состояния, замедлять или задерживать прогрессирование заболевания при HS или предупреждать HS. В одном неограничивающем варианте осуществления термин “терапевтически эффективное количество” относится к такому количеству соединения, которое при введении субъекту является эффективным для (1) по меньшей мере частичного облегчения, ингибирования, предупреждения и/или уменьшения интенсивности проявлений состояния HS. В другом неограничивающем варианте осуществления термин “терапевтически эффективное количество” относится к такому количеству соединения, которое при введении в клетку, или ткань, или неклеточный биологический материал, или среду является эффективным в отношении по меньшей мере частичного снижения или ингибирования активности LTA4H; или частичного или полного снижения или ингибирования экспрессии LTA4H.
Используемый в данном документе термин “субъект” относится к животному. Как правило, животное является млекопитающим. Субъект также относится, например, к приматам (например, людям, мужчинам или женщинам), коровам, овцам, козам, лошадям, собакам, кошкам, кроликам, крысам, мышам, рыбам, птицам и т. п. В определенных вариантах осуществления субъектом является примат. В еще других вариантах осуществления субъектом является человек. Термин “субъект” используется взаимозаменяемо с “пациентом”, когда он относится к человеку.
Используемая в данном документе фраза “популяция пациентов” означает группу пациентов. В некоторых вариантах осуществления раскрытых способов ингибитор LTA4H (например, соединение формулы I, или соединение, раскрытое в WO2015/092740, или любое соединение, описанное в данном документе) применяют для лечения популяции пациентов с HS.
Используемые в данном документе фразы “ранее не был подвергнут системному лечению HS” и “не получавший лечения” относятся к пациенту с HS, который ранее не был подвергнут лечению HS с помощью системного средства, например метотрексата, циклоспорина, или с помощью биологического средства (как например блокаторы IL-12 и IL-23, такие как устекинумаб и гуселькумаб, или с помощью ингибиторов TNF-альфа, такого как инфликсимаб, или с помощью блокатора IL-17, такого как секукинумаб, иксекизумаб и бродалумаб). Системные средства (т. е. средства, вводимые перорально, с помощью инъекции и т. д.) отличаются от локально действующих средств (например, местных средств и фототерапии) тем, что системные средства обладают системным (охватывающим весь организм) эффектом при доставке пациенту. В некоторых вариантах осуществления раскрытых способов, схем, вариантов применения, наборов и фармацевтических композиций пациенту ранее не вводили системное лечение для HS.
Используемая в данном документе фраза “ранее получал лечение с помощью системного средства для лечения HS” используется для обозначения пациента, который ранее подвергался лечению HS с использованием системного средства. Такие пациенты включают тех, которые ранее подвергались лечению с помощью биологических препаратов, таких как блокаторы IL-12 и IL-23, такие как устекинумаб и гуселькумаб, или с помощью ингибиторов TNF-альфа, как например инфликсимаб, или с помощью блокатора IL-17, такого как секукинумаб, иксекизумаб и бродалумаб, а также тех, которые ранее подвергались лечению с помощью небиологических препаратов, как например с помощью системного иммунодепрессанта или иммуномодуляторов (например циклоспорина, метотрексата и циклофосфамида), с помощью средств системного лечения, в том числе ретиноидов (как например изотретиноин), дапсона, метформина и перорального средства лечения на основе цинка. В некоторых вариантах осуществления настоящего изобретения пациенту ранее вводили системное средство для лечения HS. В некоторых вариантах осуществления пациенту ранее вводили системное средство для HS (например, метотрексат, циклоспорин), однако пациенту ранее не вводили системное биологическое лекарственное средство (т. е. лекарственное средство, продуцируемое живым организмом, например, антитела, рецепторы-ловушки и т. д.) для лечения HS (например, секукинумаб, устекинумаб, иксекизумаб, бродалумаб, ингибиторы TNF-альфа (этанерцепт, адалимумаб, ремикад и т. д.). В данном случае пациента называют “не получавшим лечение с помощью биологического средства”. В некоторых вариантах осуществления пациент является не получавшим лечение с помощью биологического средства.
Используемые в данном документе термины “осуществление отбора” и “отобранный”, используемые в отношении пациента, означают, что конкретный пациент специально выбран из большей группы пациентов на основании того (вследствие того), что конкретный пациент отвечает заранее определенным критериям. Подобным образом, “селективное лечение” относится к обеспечению лечением пациента, имеющего конкретное заболевание, причем этот пациент специально выбран из большей группы пациентов на основании того, что конкретный пациент отвечает заранее определенному критерию. Подобным образом, “селективное введение” относится к введению лекарственного средства пациенту, который специально выбран из большей группы пациентов на основании того (вследствие того), что конкретный пациент отвечает заранее определенному критерию. Под осуществлением отбора, селективным лечением и селективным введением подразумевается, что пациенту предоставляется персонализированная терапия, основанная на личной истории пациента (например, предыдущие терапевтические вмешательства, например, предыдущее лечение с помощью биологических препаратов), биологии (например, конкретных генетических маркерах) и/или проявлении (например, несоответствии определенным диагностическим критериям), вместо применения стандартной схемы лечения, основанной исключительно на принадлежности пациента к большей группе. Отбор, применительно к способу лечения в контексте данного документа, не относится к случайному лечению пациента, отвечающего конкретному критерию, но скорее относится ко взвешенному решению, принимаемому в отношении введения лекарственного средства пациенту на основании того, что пациент отвечает конкретному критерию. Таким образом, селективное лечение/введение отличается от стандартного лечения/введения, в ходе которого обеспечивается доставка конкретного лекарственного средства всем пациентам, имеющим конкретное заболевание, независимо от их личного анамнеза, проявлений заболевания и/или биологических особенностей. В некоторых вариантах осуществления пациент выбран для лечения на основании наличия HS.
Используемый в данном документе термин “подавлять”, "подавление" или “подавляющий” относятся к снижению выраженности или супрессии рассматриваемого состояния, симптома, или нарушения, или заболевания или значительному уменьшению исходного уровня активности в отношении биологической активности или процесса.
Используемые в данном документе выражения “лечить”, “осуществлять лечение” или "лечение" любого заболевания или нарушения относятся в одном варианте осуществления к уменьшению интенсивности проявлений заболевания или нарушения (т. е. к замедлению, или остановке, или снижению развития или прогрессирования заболевания или по меньшей мере одного из его клинических симптомов). В другом варианте осуществления выражения “лечить”, "осуществлять лечение" или "лечение" относятся к облегчению или уменьшению интенсивности проявлений по меньшей мере одного физического параметра, включая такие, которые могут не ощущаться пациентом. В еще одном варианте осуществления “лечить”, "осуществлять лечение" или "лечение" относится к модулированию заболевания или нарушения либо физически (например, путем стабилизации явного симптома), либо физиологически (например, путем стабилизации физического параметра), либо с помощью и того, и другого. Более конкретно, термин “осуществление лечения” заболевания HS относится к лечению очагов поражения у пациентов с HS (по количеству или качеству, или по уменьшению их объема и размера), и/или к лечению абсцессов и воспалительных узелков и/или дренирующих свищей у пациентов с HS, и/или к снижению количества рубцов, и/или к снижению функциональных ограничений, связанных с рубцеванием. Осуществление лечения заболевания HS также относится к частичному уменьшению болевого ощущения, усталости и/или зуда, связанных с HS, к уменьшению выделения гноя и уменьшению запаха, связанного с выделением гноя, и/или к улучшению качества жизни, и/или к снижению нарушенной трудоспособности пациентов с HS.
Используемый в данном документе термин "профилактика" означает задержку возникновения, или развития, или прогрессирования заболевания или нарушения. Более конкретно, термин “осуществление предупреждения” заболевания HS означает осуществление предупреждения обострений HS и/или появления новых очагов поражения; осуществление предупреждения рубцевания и осуществление предупреждения функциональных ограничений, связанных с рубцеванием, и/или, в частности, осуществление предупреждения хирургических вмешательств по причине HS.
Как используется в данном документе, субъект “нуждается в” лечении, если для такого субъекта будет обеспечиваться биологическая, медицинская польза или польза в отношении качества жизни в результате такого лечения.
Используемые в данном документе термины в форме единственного числа и подобные термины, используемые в контексте настоящего изобретения (в частности, в контексте формулы изобретения), следует истолковывать как охватывающие как формы единственного числа, так и формы множественного числа, если в данном документе не указано иное или это явно не противоречит контексту.
Любой асимметричный атом (например, углерод или подобный) соединения(соединений) для применения в способе согласно настоящему изобретению может находиться в рацемической или энантиомерно обогащенной форме, например, в (R)-, (S)- или (R, S)-конфигурации. В определенных вариантах осуществления каждый асимметрический атом характеризуется по меньшей мере 50% энантиомерным избытком, по меньшей мере 60% энантиомерным избытком, по меньшей мере 70% энантиомерным избытком, по меньшей мере 80% энантиомерным избытком, по меньшей мере 90% энантиомерным избытком, по меньшей мере 95% энантиомерным избытком или по меньшей мере 99% энантиомерным избытком в (R)- или (S)-конфигурации. Заместители при атомах с ненасыщенными двойными связями могут, если это возможно, присутствовать в цис- (Z)- или транс- (E)-форме.
Соответственно, соединение для использования в способе согласно настоящему изобретению может находиться в форме одного из возможных изомеров, ротамеров, атропоизомеров, таутомеров или их смесей, например, в виде по сути чистых геометрических (цис- или транс-) изомеров, диастереомеров, оптических изомеров (антиподов), рацематов или их смесей. Для большей ясности, термин “возможные изомеры” не будет включать позиционные изомеры.
Любые полученные в результате смеси изомеров могут быть разделены на основании физико-химических отличий составляющих компонентов на чистые или по сути чистые геометрические или оптические изомеры, диастереомеры, рацематы, например, посредством хроматографии и/или фракционной кристаллизации.
Любые полученные в результате рацематы конечных продуктов или промежуточных соединений могут быть разделены на оптические антиподы посредством известных способов, например, посредством разделения их диастереомерных солей, полученных с помощью оптически активных кислоты или основания, и выделения оптически активных кислотного или основного соединений. В частности, основный фрагмент таким образом может быть использован для разделения соединений по настоящему изобретению на их оптические антиподы, например путем фракционной кристаллизации соли, образованной с оптически активной кислотой, например винной кислотой, дибензоилвинной кислотой, диацетилвинной кислотой, ди-O, O'-p-толуоилвинной кислотой, миндальной кислотой, яблочной кислотой или камфор-10-сульфоновой кислотой. Рацемические продукты также могут быть разделены с помощью хиральной хроматографии, например, жидкостной хроматографии высокого давления (HPLC) с применением хиральной неподвижной фазы.
Кроме того, соединения для применения в способе согласно настоящему изобретению, включая их соли, также могут быть получены в форме их гидратов или включают другие растворители, применяемые для их кристаллизации. Соединения для применения в способе согласно настоящему изобретению могут по своему технологическому замыслу образовывать сольваты с фармацевтически приемлемыми растворителями (в том числе водой); следовательно, предполагается, что настоящее изобретение охватывает применение соединений, как указано в данном документе, как в сольватированной, так и в несольватированной форме. Термин "сольват" относится к молекулярному комплексу соединения для применения в способе согласно настоящему изобретению (в том числе его фармацевтически приемлемых солей) с одной или несколькими молекулами растворителя. Такие молекулы растворителя представляют собой таковые, широко применяемые в фармацевтической области, которые известны как нетоксичные для реципиента, например, вода, этанол и т. п. Термин "гидрат" относится к комплексу, где молекулой растворителя является вода.
Соединения для применения в способе согласно настоящему изобретению включают соли, гидраты, сольваты и их полиморфы.
Ингибиторы LTA4H
В одном варианте осуществления под номером один настоящее изобретение относится к способу лечения или предупреждения HS у субъекта, нуждающегося в этом, включающему введение указанному субъекту терапевтически эффективного количества ингибитора LTA4H.
В варианте осуществления 1A настоящее изобретение относится к способу лечения или предупреждения HS у субъекта, нуждающегося в этом, включающему введение указанному субъекту терапевтически эффективного количества ингибитора LTA4H, как описано в WO2014/164658.
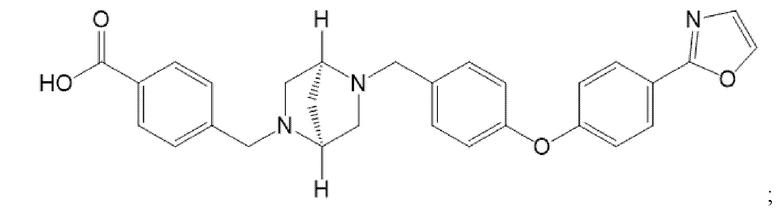
В варианте осуществления 1B настоящее изобретение относится к способу лечения или предупреждения HS у субъекта, нуждающегося в этом, включающему введение указанному субъекту терапевтически эффективного количества ингибитора LTA4H, который представляет собой:
или его фармацевтически приемлемой соли.
В варианте осуществления 1C настоящее изобретение относится к способу лечения или предупреждения HS у субъекта, нуждающегося в этом, включающему введение указанному субъекту терапевтически эффективного количества ингибитора LTA4H, который представляет собой 4-(((1S,4S)-5-(4-(4-(оксазол-2-ил)фенокси)бензил)-2,5-диазабицикло[2.2.1]гептан-2-ил)метил)бензойную кислоту; или его фармацевтически приемлемой соли.
В варианте осуществления 1D настоящее изобретение относится к способу лечения или предупреждения HS у субъекта, нуждающегося в этом, включающему введение указанному субъекту терапевтически эффективного количества ингибитора LTA4H, который представляет собой ацебилустат, также известный как CTX 4430.
В варианте осуществления два настоящее изобретение относится к способу лечения или предупреждения HS у субъекта, нуждающегося в этом, включающему введение указанному субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли:
(I),
где
R1 представляет собой OH или NH2;
Y представляет собой O, S или CH2;
X1, X2, X3 и X4 представляют собой N; или
X1, X2, X3 и X4 выбраны из N, NH, C, CH и O при условии, что по меньшей мере два из X1, X2, X3 или X4 представляют собой N или NH;
R2 представляет собой C1-C6алкил, необязательно замещенный фенилом; C3-C6циклоалкил; фенил, необязательно являющийся замещенным галогеном, циано, C1-C6алкилом, необязательно замещенным галогеном, C1-C6алкокси или 5-6-членным гетероарильным кольцом, содержащим от 1 до 3 гетероатомов, выбранных из N, O и S; или 5-10-членный моно- или бициклический гетероарил, содержащий от 1 до 4 гетероатомов, выбранных из N, O и S, при этом указанный гетероарил необязательно является замещенным галогеном, циано или C1-C6алкилом, необязательно замещенным галогеном; или его фармацевтически приемлемой соли.
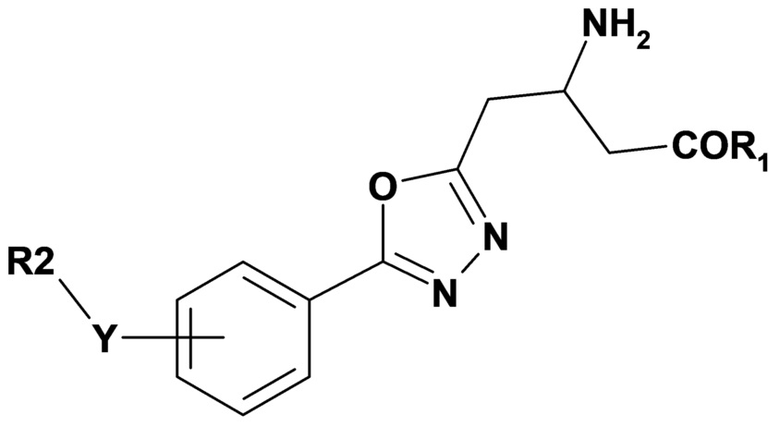
Вариант осуществления 2A относится к способу в соответствии с вариантом осуществления 2, включающему введение субъекту терапевтически эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли,
 (II),
(II),
где переменные R1, R2 и Y имеют значения, определенные в варианте осуществления 1.
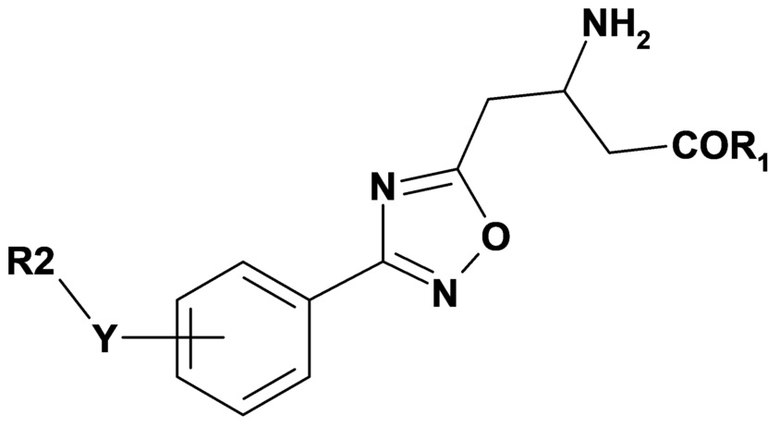
Вариант осуществления 2B относится к способу в соответствии с вариантом осуществления 2, включающему введение субъекту терапевтически эффективного количества соединения формулы (III) или его фармацевтически приемлемой соли,
 (III),
(III),
где переменные R1, R2 и Y имеют значения, определенные в варианте осуществления 1.
Вариант осуществления 2C относится к способу согласно варианту осуществления 2, включающему введение субъекту терапевтически эффективного количества соединения формулы (IV) или его фармацевтически приемлемой соли,
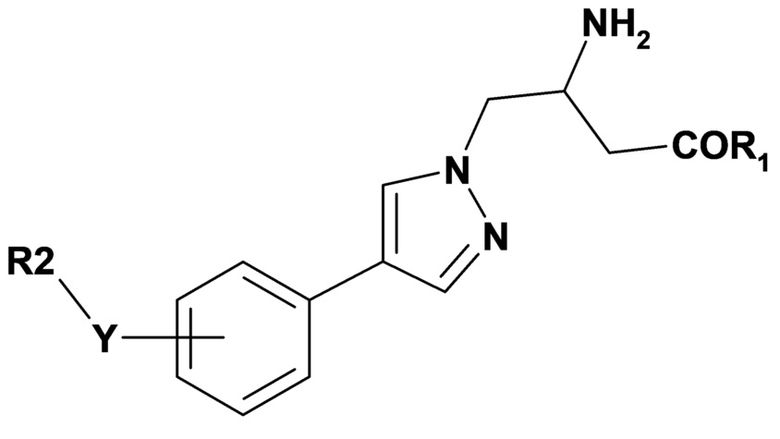 (IV),
(IV),
где переменные R1, R2 и Y имеют значения, определенные в варианте осуществления 1.
Вариант осуществления 2D относится к способу в соответствии с вариантом осуществления 2, включающему введение субъекту терапевтически эффективного количества соединения формулы (V) или его фармацевтически приемлемой соли;
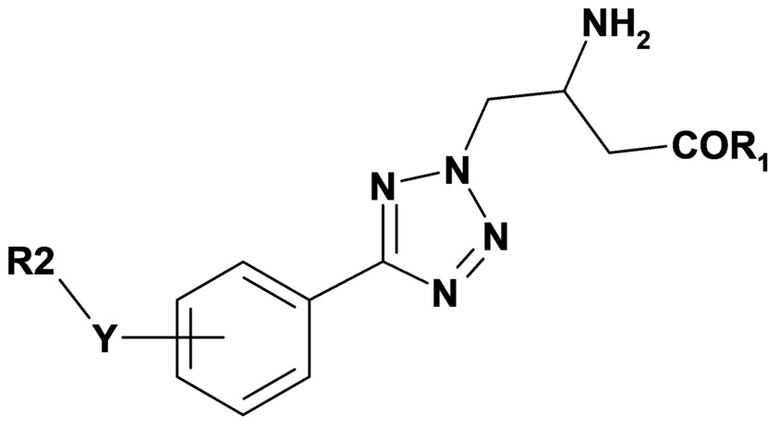 (V),
(V),
где переменные R1, R2 и Y имеют значения, определенные в варианте осуществления 1.
Вариант осуществления 2E настоящего изобретения относится к способу в соответствии с любым из вариантов осуществления 2 и 2A-2D, включающему введение субъекту терапевтически эффективного количества соединения формулы (I), (II), (III), (IV) или (V) или его фармацевтически приемлемой соли, где Y представляет собой O; и
R2 представляет собой фенил, необязательно являющийся замещенным галогеном, циано, C1-C6алкилом, необязательно замещенным галогеном, C1-C6алкокси или 5-6-членным гетероарильным кольцом, содержащим от 1 до 3 гетероатомов, выбранных из N, O и S; или
R2 представляет собой 5-10-членный моно- или бициклический гетероарил, содержащий от 1 до 4 гетероатомов, выбранных из N, O и S, при этом указанный гетероарил необязательно является замещенным галогеном, циано или C1-C6алкилом, необязательно замещенным галогеном.
Вариант осуществления 2F настоящего изобретения относится к способу в соответствии с любым из вариантов осуществления 2 и 2A-2D, включающему введение субъекту терапевтически эффективного количества соединения формулы (I), (II), (III), (IV) или (V) или его фармацевтически приемлемой соли, где Y представляет собой CH2; и
R2 представляет собой фенил, необязательно являющийся замещенным галогеном, циано, C1-C6алкилом, необязательно замещенным галогеном, C1-C6алкокси или 5-6-членным гетероарильным кольцом, содержащим от 1 до 3 гетероатомов, выбранных из N, O и S; или
R2 представляет собой 5-10-членный моно- или бициклический гетероарил, содержащий от 1 до 4 гетероатомов, выбранных из N, O и S, при этом указанный гетероарил необязательно является замещенным циано, галогеном или C1-C6алкилом, необязательно замещенным галогеном.
Вариант осуществления 2G настоящего изобретения относится к способу в соответствии с любым из вариантов осуществления 2 и 2A-2D, включающему введение субъекту терапевтически эффективного количества соединения формулы (I), (II), (III), (IV) или (V) или его фармацевтически приемлемой соли; где Y представляет собой O; и
R2 представляет собой C1-C6алкил, необязательно замещенный фенилом; или C3-C6циклоалкил.
Вариант осуществления 2H настоящего изобретения относится к способу в соответствии с любым из вариантов осуществления 2 и 2A-2D, включающему введение субъекту терапевтически эффективного количества соединения формулы (I), (II), (III), (IV) или (V) или его фармацевтически приемлемой соли; где Y представляет собой CH2; и
R2 представляет собой C1-C6алкил, необязательно замещенный фенилом; или C3-C6циклоалкил.
Вариант осуществления 2I относится к способу в соответствии с любым из вариантов осуществления 2 и 2A-2H, включающему введение субъекту терапевтически эффективного количества соединения формулы (I), (II), (III), (IV) или (V) или его фармацевтически приемлемой соли или его фармацевтически приемлемой соли, где Y присоединен к фенильному фрагменту в пара-положении.
Вариант осуществления 2J относится к способу в соответствии с любым из вариантов осуществления 2 и 2A-2H, включающему введение субъекту терапевтически эффективного количества соединения формулы (I), (II), (III), (IV) или (V) или его фармацевтически приемлемой соли, где Y присоединен к фенильному фрагменту в мета-положении.
Вариант осуществления 2K относится к способу в соответствии с любым из вариантов осуществления 2 и 2A-2J, включающему введение субъекту терапевтически эффективного количества соединения формулы (I), (II), (III), (IV) или (V) или его фармацевтически приемлемой соли, где R1 представляет собой OH.
Вариант осуществления 2L относится к способу в соответствии с любым из вариантов осуществления 2 и 2A-2K, включающему введение субъекту терапевтически эффективного количества соединения формулы (I), (II), (III), (IV) или (V) или его фармацевтически приемлемой соли; где аминогруппа характеризуется (R)-конфигурацией.
Вариант осуществления 2M относится к способу в соответствии с любым из вариантов осуществления 2 и 2A-2K, включающему введение субъекту терапевтически эффективного количества соединения формулы (I), (II), (III), (IV) или (V) или его фармацевтически приемлемой соли; где аминогруппа характеризуется (S)-конфигурацией.
Вариант осуществления 2N относится к способу в соответствии с вариантом осуществления 2, включающему введение субъекту терапевтически эффективного количества соединения формулы (I) и/или его фармацевтически приемлемой соли, где соединение раскрыто в WO2015/092740 [номер дела патентного поверенного PAT056044-WO-PCT]; т. е. соединение выбрано из:
(R)-3-амино-4-(5-(4-(бензо[d]тиазол-2-илокси)фенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(4-((5-хлорпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(4-(4-(оксазол-2-ил)-фенокси)фенил)-2H-тетразол-2-ил)-бутановой кислоты;
(R)-3-амино-4-(5-(3-(4-хлорфенокси)фенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(4-(4-хлорфенокси)-фенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(4-(4-фторфенокси)-фенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(4-(3-хлор-4-фторфенокси)фенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(4-(п-толилокси)фенил)-2H-тетразол-2-ил)бутановой кислоты;
(S)-3-амино-4-(5-(3-феноксифенил)-2H-тетразол-2-ил)бутановой кислоты;
(S)-3-амино-4-(5-(4-(бензо[d]тиазол-2-илокси)фенил)-2H-тетразол-2-ил)бутановой кислоты;
(S)-3-амино-4-(5-(4-(4-хлорфенокси)-фенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(3-фенэтоксифенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(4-фенэтоксифенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(4-(бензилокси)фенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(3-(бензилокси)фенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(4-бутоксифенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(4-(пентилокси)фенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(3-((5-(трифторметил)пиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(4-((5-(трифторметил)пиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(3-(бензо[d]тиазол-2-илокси)фенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(3-(3,5-дифторфенокси)фенил)-2H-тетразол-2-ил)бутановой кислоты;
(S)-3-амино-4-(5-(4-(п-толилокси)фенил)-2H-тетразол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(4-(4-фторфенокси)фенил)-1,3,4-оксадиазол-2-ил)бутановой кислоты;
(R)-3-амино-4-(5-(4-(4-хлорфенокси)фенил)-1,3,4-оксадиазол-2-ил)бутановой кислоты;
(R)-3-амино-4-(3-(4-(4-хлорфенокси)фенил)-1,2,4-оксадиазол-5-ил)бутановой кислоты;
(R)-3-амино-4-(3-(4-(4-хлорфенокси)фенил)-1,2,4-оксадиазол-5-ил)бутанамида;
(S)-3-амино-4-(4-(4-(4-хлорфенокси)фенил)-1H-пиразол-1-ил)бутановой кислоты; и
(S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты.
В варианте осуществления 3A настоящее изобретение относится к способу в соответствии с вариантом осуществления 2, включающему введение субъекту терапевтически эффективного количества соединения формулы (I), где соединение представляет собой (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановую кислоту; или его фармацевтически приемлемой соли.
В варианте осуществления 3B настоящее изобретение относится к способу в соответствии с вариантом осуществления 2, включающему введение субъекту терапевтически эффективного количества соединения формулы (I), где соединение представляет собой (R)-3-амино-4-(5-(4-фенэтоксифенил)-2H-тетразол-2-ил)бутановую кислоту; или его фармацевтически приемлемой соли.
В варианте осуществления 3C настоящее изобретение относится к способу в соответствии с вариантом осуществления 2, включающему введение субъекту терапевтически эффективного количества соединения формулы (I), где соединение представляет собой (R)-3-амино-4-(5-(4-(4-хлорфенокси)-фенил)-2H-тетразол-2-ил)бутановую кислоту; или его фармацевтически приемлемой соли.
В варианте осуществления 3D настоящее изобретение относится к способу в соответствии с вариантом осуществления 2, включающему введение субъекту терапевтически эффективного количества соединения формулы (I), где соединение представляет собой (S)-3-амино-4-(5-(4-(4-хлорфенокси)-фенил)-2H-тетразол-2-ил)бутановую кислоту; или его фармацевтически приемлемой соли.
Соединения любой из формул (I)-(V) и соединения в соответствии с любым из вариантов осуществления 2, 2A-2N, 3 и 3A-3C для применения в способе согласно настоящему изобретению раскрыты в WO2015/092740, который включен в данном документе посредством ссылки.
В варианте осуществления 3E настоящее изобретение относится к способу в соответствии с вариантом осуществления 2, включающему введение субъекту терапевтически эффективного количества соединения формулы (I), где соединение представляет собой кристаллическую форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в ее свободной форме (т. е. несолевой форме); или его фармацевтически приемлемой соли.
В варианте осуществления 3F настоящее изобретение относится к способу в соответствии с вариантом осуществления 21, где кристаллическая форма характеризуется по меньшей мере одной из следующих характеристик:
(i) порошковой рентгеновской дифрактограммой, содержащей характеристические пики, выраженные в 2θ при значении 2θ составляющем 22,6 ± 0,2°, значении 2θ составляющем 24,1 ± 0,2° и значении 2θ составляющем 26,3 ± 0,2°, измеренных при температуре, составляющей приблизительно 25°C, и при длине волны рентгеновского излучения, λ, составляющей 1,5418 Å;
(ii) порошковой рентгеновской дифрактограммой, содержащей четыре или более значений 2θ выбранных из группы, состоящей из значения 2θ составляющего 11,3 ± 0,2°, значения 2θ составляющего 12,8 ± 0,2°, значения 2θ составляющего 15,2 ± 0,2°, значения 2θ составляющего 19,7 ± 0,2°, значения 2θ составляющего 20,0 ± 0,2°, значения 2θ составляющего 20,3 ± 0,2°, значения 2θ составляющего 21,0 ± 0,2°, значения 2θ составляющего 22,6 ± 0,2°, значения 2θ составляющего 24,1 ± 0,2°, значения 2θ составляющего 24,4 ± 0,2°, значения 2θ составляющего 25,1 ± 0,2°, значения 2θ составляющего 26,3 ± 0,2°, значения 2θ составляющего 28,5 ± 0,2°, и значения 2θ составляющего 30,0 ± 0,2°, измеренных при температуре, составляющей приблизительно 25°C, и при длине волны рентгеновского излучения, λ, составляющей 1,5418 Å;
(iii) порошковой рентгеновской дифрактограммой, содержащей пять или более значений 2θ выбранных из группы, состоящей из значения 2θ составляющего 11,3 ± 0,2°, значения 2θ составляющего 12,8 ± 0,2°, значения 2θ составляющего 15,2 ± 0,2°, значения 2θ составляющего 19,7 ± 0,2°, значения 2θ составляющего 20,0 ± 0,2°, значения 2θ составляющего 20,3 ± 0,2°, значения 2θ составляющего 21,0 ± 0,2°, значения 2θ составляющего 22,6 ± 0,2°, значения 2θ составляющего 24,1 ± 0,2°, значения 2θ составляющего 24,4 ± 0,2°, значения 2θ составляющего 25,1 ± 0,2°, значения 2θ составляющего 26,3 ± 0,2°, значения 2θ составляющего 28,5 ± 0,2°, и значения 2θ составляющего 30,0 ± 0,2°, измеренных при температуре, составляющей приблизительно 25°C, и при длине волны рентгеновского излучения, λ, составляющей 1,5418 Å;
(iv) спектром рентгеновской дифракции, по сути таким же, как показанный на фиг. 1 спектр порошковой рентгеновской дифракции;
(v) термограммой дифференциальной сканирующей калориметрии (DSC), по сути такой же, как показанная на фигуре 2; и
(vi) диаграммой термогравиметрического анализа (TGA), по сути такой же, как показанная на фигуре 3.
Кристаллическая форма согласно вариантам осуществления 3D и 3E раскрыта в PAT058189-WO-PCT, который является PCT/CN2018/000278, поданной 31 июля 2018 г., которая включена в данный документ посредством ссылки.
Способы лечения и применения ингибиторов LTA4H при HS
Раскрытые ингибиторы LTA4H (т. е. соединения любой из формул от (I) до (V) и соединения согласно любому из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E, или их фармацевтически приемлемая соль) могут применяться in vitro, ex vivo или включаются в фармацевтические композиции и вводятся in vivo для лечения пациентов с HS (например пациентов-людей).
HS представляет собой хроническое воспалительное состояние с рубцеванием, затрагивающее главным образом интертригинозную кожу подмышечного, пахового, инфрамаммарного, генито-анального и перинеального участков тела. Также его называют инверсными угрями. Диагноз HS устанавливают на основании трех диагностических критериев: типичные очаги поражения (глубоко расположенные болезненные узелки, [невскрывшиеся] фурункулы в ранних первичных очагах поражения или абсцессы, дренирующие свищи, перекрывающиеся рубцы и открытые комедоны “черные точки” во вторичных очагах поражения); типичная топография (подмышечные впадины, пах, половые органы, перинеальные и перианальные участки, ягодицы, инфра- и интермаммарные участки; а также хронический и рецидивирующий характер (Margesson and Danby (2014) Best Practices and Res. Clin. Ob. And Gyn 28:1013-1027). Физическая степень HS может быть классифицирована с использованием клинического стадирования согласно Херли, показанного ниже в таблице 2.
Таблица 2. Стадии HS согласно Херли. В сущности, пациент, у которого имеется стадия III согласно Херли, может иметь стадию III с дистрофическими изменениями, но с активными очагами поражения стадии I или II.
HS заключается в закупорке фолликулов, разрыве протоков и вторичном воспалении. У пациентов сперва закупоривается фолликулярной проток, что со временем приводит к истечению из протока и горизонтальному разрыву в дерму. В случае, если не происходит восстановления сально-волосяного фолликула (FPSB), фрагменты фолликула стимулируют три реакции, с которых начинается течение заболевания HS. Первой является воспалительная реакция, запускаемая врожденной иммунной системой, вызывающая нагноение и разрушение тканей и приводящая к реакциям на инородное тело и обширному рубцеванию. Вторая реакция приводит к образованию эпителизированных свищей, которые могут развиваться из стволовых клеток, происходящих из одного FPSB, которые переживают разрушение, вызванное воспалительной реакцией. В ходе третьей реакции в большинстве случаев образуется инвазивная пролиферативная желатинозная масса, состоящая из геля, содержащего воспалительные клетки, и, как предполагается, предшественников эпителизированных элементов, описанных выше. (См. Margesson and Danby (2014). Используемая в данном документе фраза “замедление прогрессирования заболевания HS” означает замедление скорости развития любого из аспектов течения заболевания HS, описанного выше, в частности воспалительной реакции. В некоторых вариантах осуществления настоящего изобретения лечение ингибитором LTA4H замедляет прогрессирование заболевания HS.
Рецидив HS у пациента включает развитие папул, пустул или воспалительных узелков, возникновение боли и зуда, абсцессов, дренирования и любой их комбинации. Используемый в данном документе термин “обострение HS” (и т. п.) определяется как повышение количества абсцессов и воспалительных узелков (AN) на по меньшей мере 25% с минимальным повышением в два раза AN по сравнению с исходным уровнем.
В некоторых вариантах осуществления настоящего изобретения лечение в соответствии с раскрытыми способами с помощью ингибиторов LTA4H предупреждает обострения HS, снижает тяжесть обострений HS и/или снижает частоту обострений HS. В некоторых вариантах осуществления в случае, если популяцию пациентов с HS лечат в соответствии с раскрытыми способами, то менее чем 5%, менее чем 10%, менее чем 15% или менее чем 20% пациентов испытывают обострение в ходе лечения, например в течение первых 16 недель лечения.
Используемая в данном документе фраза “снижение тяжести обострений HS” и т. п. означает снижение интенсивности обострения HS, например снижение количества и/или размера абсцессов и/или воспалительных узелков, снижение интенсивности конкретного компонента обострения (например снижение количества, размера, толщины и т. д. абсцессов и/или воспалительных узелков, снижение степени раздражения кожи (зуда, боли) и т. д.) и/или уменьшение продолжительности времени сохранения обострения (или его компонента).
Используемая в данном документе фраза “снижение частоты обострений HS” и т. п. означает снижение числа случаев обострений HS, например снижение числа абсцессов и/или воспалительных узелков. Со снижением частоты обострений HS пациент будет испытывать меньше рецидивов HS. Частота обострений может быть оценена посредством мониторинга пациента с течением времени, чтобы определить, снижается ли частота случаев обострений.
Используемая в данном документе фраза “предупреждение обострений HS” означает устранение будущих обострений HS и/или компонентов обострения.
Эффективность лечения HS можно оценивать с использованием различных известных способов и средств, которые обеспечивают оценку состояния при заболевании HS и/или клинического ответа при HS. Некоторые примеры включают, например, стадирование согласно Херли, систему оценки степени тяжести (SAHS), балл по шкале Сарториуса, балл по модифицированный шкале Сарториуса, показатель общей оценки врача при HS (HS-PGA), визуальную аналоговую шкалу (VAS) или цифровую рейтинговую шкалу (NRS) для оценки боли, связанной с кожей, индекс качества жизни при заболеваниях кожи (DLQI), клинический ответ при HS, основанный на сумме абсцессов и воспалительных узелков (HiSCR), HiSCR по упрощенной шкале, EuroQuol-5D (EQ5D), госпитальную шкалу тревоги и депрессии, использование ресурсов здравоохранения, индекс тяжести гнойного гидраденита (HSSI), индекс производительности труда (WPI), площадь воспаленной поверхности тела (BSA), индекс тяжести инверсных угрей (AISI) и т. д. (см., например, Deckers and Prens (2016) Drugs 76:215-229; Sartorius et al. (2009) Br. J. Dermatol 161:831-39; Chiricozzi et al. (2015) Wounds 27(10):258-264). В некоторых вариантах осуществления эффективность способа по настоящему изобретению, раскрытого в данном документе, может оцениваться посредством показателя общей оценки врача при HS (HS-PGA), системы оценки степени тяжести (SAHS), цифровой рейтинговой шкалы (NRS), индекса качества жизни при заболеваниях кожи (DLQI), клинического ответа при HS, основанного на сумме абсцессов и воспалительных узелков (HiSCR), и/или HiSCR по упрощенной шкале. Предпочтительно эффективность лечения HS, раскрытого в данном документе, может оцениваться по клиническому ответу при HS, основанному на сумме абсцессов и воспалительных узелков (HiSCR), и/или HiSCR по упрощенной шкале.
В некоторых вариантах осуществления у пациента с HS достигается HiSCR в ответ на лечение HS. В некоторых вариантах осуществления при лечении популяции пациентов с HS в соответствии с раскрытыми способами у по меньшей мере 30%, по меньшей мере 40%, по меньшей мере 50%, по меньшей мере 60% или по меньшей мере 70% достигается HiSCR к неделе 16 лечения.
В других вариантах осуществления эффективность лечения HS, раскрытого в данном документе, может быть измерена по разнице между коэффициентом ответивших на лечение у пациентов, получавших лечение (то есть пациента, у которого достигается HiSCR-ответ на лечение HS соединением по настоящему изобретению), и коэффициентом ответивших на лечение пациентов, получавших плацебо, к неделе 16 лечения. В некоторых вариантах осуществления эта разница в коэффициенте ответивших на лечение, измеренная посредством HiSCR, составляет по меньшей мере 15%, по меньшей мере 25%, по меньшей мере 30% или по меньшей мере 35%.
Предпочтительными системами оценки ответа на лечение являются HiSCR, HiSCR по упрощенной шкале, NRS (особенно NRS30), система оценки степени тяжести (SAHS), HS-PGA, количество очагов воспаления (количество абсцессов, воспалительных узелков и/или дренирующих свищей) и DLQI.
Клинический ответ при гнойном гидрадените (HiSCR) является мерой клинического ответа на лечение HS. HiSCR-ответ на лечение (по сравнению с исходным уровнем) представляет собой следующее: 1) снижение на по меньшей мере 50% абсцессов и воспалительных узелков, при этом 2) количество абсцессов не увеличивается, и при этом 3) количество дренирующих свищей не увеличивается. Используемые в данном документе термины “HiSCR по упрощенной шкале” или “sHiSCR” относятся к модифицированному HiSCR, который не предусматривает сравнение количества абсцессов с количеством абсцессов на исходном уровне при оценке прогрессирования очагов поражения. В предпочтительных вариантах осуществления у пациента с HS достигается HiSCR по упрощенной шкале в ответ на лечение HS. В некоторых вариантах осуществления при лечении популяции пациентов с HS в соответствии с раскрытыми способами у по меньшей мере 40%, по меньшей мере 50%, по меньшей мере 60% или по меньшей мере 70% достигается HiSCR по упрощенной шкале к неделе 16 лечения.
Боль можно оценивать с использованием цифровой рейтинговой шкалы (NRS). В некоторых вариантах осуществления у пациента с HS достигается улучшение по NRS в ответ на лечение HS. NRS30 определяется как снижение интенсивности боли на по меньшей мере 30% и снижение на по меньшей мере 1 единицу от исходного уровня общей оценки пациента (PGA) интенсивности боли в коже от исходного уровня у пациентов с показателем 3 или выше на исходном уровне. В некоторых вариантах осуществления у пациента с HS достигается NRS30 в ответ на лечение HS. В некоторых вариантах осуществления при лечении популяции пациентов с HS в соответствии с раскрытыми способами у по меньшей мере 25%, по меньшей мере 30%, по меньшей мере 40%, по меньшей мере 50% или по меньшей мере 60% достигается NRS30 к неделе 16 лечения. В предпочтительном аспекте данного варианта осуществления при лечении популяции пациентов с HS в соответствии с раскрытыми способами у по меньшей мере 30% достигается NRS30 к неделе 16 лечения. В некоторых вариантах осуществления в ответ на лечение в соответствии с заявленными способами пациент испытывает быстрое снижение интенсивности боли, что измерено посредством VAS или NRS, уже через 1 неделю после первоначального введения дозы.
Система оценки степени тяжести (SAHS) описана в JAMA Dermatol 2018, 154(3): 330-335, Hassam et al. Тяжесть HS может быть оценена по балльной шкале SAHS, согласно которой исследуются следующие пункты: количество вовлеченных областей (левая подмышечная, правая подмышечная, левая субмаммарная, правая субмаммарная, межмаммарная или грудная, брюшная, лобковая, паховая левая, паховая правая, генитальная, перианальная или перинеальная, левая ягодичная, правая ягодичная и другие [например шея, ретроаурикулярная]), количество воспалительных и/или болезненных очагов поражения, кроме свищей (ILOF) и количество свищей. Эти оцениваемые врачом пункты были дополнены 2 пунктами, о которых сообщают пациенты: пациентов просили указать количество новых фурункулов или количество существующих фурункулов, которые воспалились в течение последних 4 недель, и оценить текущую тяжесть боли (NRS) наиболее симптоматически выраженного очага поражения в ходе повседневной деятельности (например сидя, двигаясь или работая) по числовой оценочной шкале. Балльная шкала SAHS представляет собой совокупную оценку всей собранной выше информации. Легкий случай HS определяется по балльной шкале SAHS как 4 или меньше. Средний случай HS определяется по шкале SASH как 5-8 баллов, а тяжелый случай HS определяется по шкале SASH как 9 баллов или выше.
В некоторых вариантах осуществления у пациента с HS достигается улучшение по балльной шкале SAHS в ответ на лечение HS. В некоторых вариантах осуществления у пациента с HS достигается снижение по меньшей мере на один балл по сравнению с исходным уровнем по балльной шкале SAHS в ответ на лечение HS. В других вариантах осуществления у пациента с HS достигается снижение на по меньшей мере два балла или по меньшей мере 3 балла по сравнению с исходным уровнем по балльной шкале SAHS в ответ на лечение HS. Предпочтительно, что показатель по балльной шкале SAHS составляет по меньшей мере 4 на исходном уровне до лечения ингибитором LTA4H.
DLQI является наиболее общепринятым средством для определения качества жизни при дерматологических заболеваниях. Он состоит из вопросов, касающихся влияния кожного заболевания на ощущения и различные аспекты повседневной жизни в течение последней недели. Каждый вопрос оценивается в баллах от 0 (вообще отсутствует) до 3 (очень много). Максимальный показатель составляет в общей сложности 30 баллов, при этом 0-1 считается отсутствием эффекта, 2-5 небольшим, 6-10 умеренным, 11-20 очень существенным и 21-30 исключительно существенным эффектом в отношении жизни пациента. (См. Finlay and Khan (1994) Clin Exp Dermatol 19:210-16). В некоторых вариантах осуществления у пациента с HS достигается улучшение по DLQI в ответ на лечение HS.
В некоторых вариантах осуществления в ответ на лечение в соответствии с заявленными способами пациент испытывает быстрое снижение уровня CRP, что измерено посредством стандартного анализа CRP или высокочувствительного анализа CRP (hsCRP), уже через 1 неделю после первоначального введения дозы. Используемые в данном документе термины “C-реактивный белок” и “CRP” относятся к C-реактивному белку в сыворотке крови, белку в плазме крови, часто используемому в качестве индикатора ответа острой фазы на воспаление. Уровень CRP в плазме крови может быть представлен в любой концентрации, например мг/дл, нмоль/л. Уровни CRP могут быть измерены посредством ряда стандартных анализов, например радиальной иммунодиффузии, электроиммуноанализа, иммунотурбидиметрии, ELISA, турбидиметрических способов, флуоресцентного поляризационного иммуноанализа и лазерной нефелометрии. Для тестирования уровня CRP можно использовать стандартный тест в отношении CRP или высокочувствительный тест в отношении CRP (hs-CRP) (т. е. высокочувствительный тест, посредством которого можно измерять низкие уровни CRP в образце с применением лазерной нефелометрии). Наборы для выявления уровней CRP могут быть приобретены у различных компаний, например у Calbiotech, Inc, Cayman Chemical, Roche Diagnostics Corporation, Abazyme, DADE Behring, Abnova Corporation, Aniara Corporation, Bio-Quant Inc., Siemens Healthcare Diagnostics и т. д.
Балл по шкале Сарториуса для HS (также называемый балльным показателем HS или HSS) получают посредством подсчета вовлеченных областей, узелков и свищевых ходов у пациента с HS. (Sartorius et al. (2003) Br J Dermatol 149:211-13). Балл по модифицированной шкале Сарториуса для HS представляет собой пересмотр исходной версии HSS посредством внесения незначительных упрощений, которые сделали его применение более практичным, например меньшее количество конкретных очагов поражения для включения в показатель, изменение количества пунктов, присвоенных каждому параметру, и т. д. (Sartorius et al. (2009) Br. J Dermatol. 161:831-839). В некоторых вариантах осуществления у пациента с HS достигается улучшенный модифицированный балл по шкале Сарториуса для HS в ответ на лечение HS.
Общая оценка врача при HS (HS-PGA) представляет собой 6-балльную оценочную шкалу (баллы в диапазоне от 0 до 5) на основании количества очагов поражения при HS (т. е. абсцессов, дренирующих свищей, воспалительных узелков и невоспалительных узелков). (Kimball AB, Kerdel F, Adams D et al Adalimumab for the treatment of moderate to severe hidradenitis suppurativa: a parallel randomized trial. Ann Intern Med 2012; 157: 846-855). В некоторых вариантах осуществления у пациента с HS достигается улучшенная HS-PGA в ответ на лечение HS. В некоторых вариантах осуществления у пациента с HS достигается снижение на по меньшей мере 2 балла по сравнению с исходным уровнем по шкале HS-PGA в ответ на лечение HS. Предпочтительно, что балльный показатель HS-PGA составляет по меньшей мере 3 на исходном уровне до лечения ингибитором LTA4H.
В некоторых вариантах осуществления, если популяцию пациентов с HS лечат в соответствии с раскрытыми способами, по меньшей мере 40%, по меньшей мере 50%, по меньшей мере 60%, по меньшей мере 70%, по меньшей мере 80% или по меньшей мере 90% пациентов, которые отвечали на лечение к неделе 16 (например пациенты, достигающие HiSCR или HiSCR по упрощенной шкале к неделе 16), характеризуются устойчивым ответом после завершения лечения (например через 3 месяца или через 6 месяцев после завершения лечения или через 12 месяцев после завершения лечения). В другом аспекте данного варианта осуществления по меньшей мере 40% пациентов или по меньшей мере 50% пациентов, которые отвечали на лечение (например пациенты, достигшие HiSCR или HiSCR по упрощенной шкале, например к неделе 16), характеризуются устойчивым ответом после завершения лечения (например через 3 месяца после завершения лечения). Предпочтительно по меньшей мере 70% пациентов, которые ответили на лечение (например пациенты, достигшие HiSCR или HiSCR по упрощенной шкале к неделе 16), характеризуются устойчивым ответом после завершения лечения (например через 3 месяца после завершения лечения). Используемый в данном документе термин “устойчивый” означает, что результат или цель (например снижение интенсивности боли, снижение интенсивности воспаления) в значительной степени сохраняются в течение указанного периода времени.
Зуд, связанный с очагом поражения, можно оценить посредством анкетирования пациента. Пациента просят оценить зуд, связанный с очагом поражения, от 0 (отсутствие зуда) до 10 (наихудший возможный зуд). В некоторых вариантах осуществления, если популяцию пациентов с HS лечат в соответствии с раскрытыми способами, балльный показатель зуда улучшается на по меньшей мере 2 балла, предпочтительно на по меньшей мере 3 балла. Более того, по сравнению с группой плацебо разница между группой лечения и группой плацебо составляет по меньшей мере 1 балл.
Запах, вызванный дренированием очага поражения, можно оценить посредством анкетирования пациента. Пациента просят оценить запах, вызванный дренированием очага поражения, от 1 (отсутствие запаха), 2 (слабый запах), 3 (умеренный запах) до 4 (сильный запах). В некоторых вариантах осуществления, если популяцию пациентов с HS лечат в соответствии с раскрытыми способами, балльный показатель зуда улучшается на по меньшей мере 1 балл, предпочтительно на по меньшей мере 2 балла. Более того, по сравнению с группой плацебо разница между группой лечения и группой плацебо составляет по меньшей мере 1 балл.
Влияние HS на способность выполнять работу можно оценить посредством анкетирования пациента. Пациента просят оценить, насколько HS влияет на способность выполнять работу, от 1 (вообще не влияет), 2 (незначительно), 3 (умеренно), 4 (значительно) до 5 (неспособен выполнять какую-либо работу). В некоторых вариантах осуществления, если популяцию пациентов с HS лечат в соответствии с раскрытыми способами, балльный показатель зуда улучшается на по меньшей мере 1 балл, предпочтительно на по меньшей мере 2 балла. Более того, по сравнению с группой плацебо разница между группой лечения и группой плацебо составляет по меньшей мере 1 балл.
Ингибиторы LTA4H, раскрытые в данном документе, могут применяться в качестве фармацевтической композиции и лекарственной формы при объединении с фармацевтически приемлемым носителем. Такая композиция может содержать, в дополнение к ингибитору LTA4H, носители, различные разбавители, наполнители, соли, буферы, стабилизаторы, солюбилизаторы и другие материалы, известные из уровня техники. Характеристики носителя будут зависеть от пути введения. Фармацевтическая композиция для применения в способе по настоящему изобретению может быть составлена для конкретных путей введения, таких как пероральное введение, парентеральное введение и ректальное введение и т. д. Кроме того, фармацевтические композиции для применения в способе по настоящему изобретению могут быть получены в твердой форме (включая без ограничения капсулы, таблетки, пилюли, гранулы, порошки или суппозитории) или в жидкой форме (включая без ограничения растворы, суспензии или эмульсии). Фармацевтические композиции для применения в способе по настоящему изобретению могут быть подвергнуты традиционным фармацевтическим операциям, таким как стерилизация, и/или могут содержать традиционные инертные разбавители, смазывающие средства или буферные средства, а также вспомогательные средства, такие как консерванты, стабилизаторы, смачивающие средства, эмульгаторы и буферы и т. д. Фармацевтические композиции и лекарственные формы для применения в настоящем изобретении могут содержать одно или несколько средств, которые снижают скорость, с которой соединение (т. е. ингибитор LTA4H, описанный в данном документе) в качестве активного ингредиента будет разлагаться. Такие средства, которые упоминаются в данном документе как “стабилизаторы”, включают без ограничения антиоксиданты, такие как аскорбиновая кислота, буферы для поддержания pH или солевые буферы и т. д.
Как правило, фармацевтические композиции для применения в настоящем изобретении представляют собой таблетки или капсулы, содержащие активный ингредиент вместе с
a) наполнителями, например лактозой, декстрозой, сахарозой, маннитом, сорбитом, целлюлозой и/или глицином;
b) смазывающими средствами, например двуокисью кремния, тальком, стеариновой кислотой, ее магниевой или кальциевой солью (например стеаратом магния) и/или полиэтиленгликолем;
c) связующими веществами, например алюмосиликатом магния, крахмальной пастой, желатином, трагакантом, метилцеллюлозой, например гидроксипропилметилцеллюлозой (HPMC или гипромеллозой), натрий-карбоксиметилцеллюлозой и/или поливинилпирролидоном; при необходимости
d) разрыхлителями, например кросповидоном, видами крахмала, агаром, альгиновой кислотой или ее натриевой солью или шипучими смесями;
e) веществом, способствующим скольжению, таким как двуокись кремния, коллоидный безводный/коллоидный диоксид кремния и/или
f) абсорбентами, красителями, ароматизаторами и подсластителями.
Таблетки могут быть покрыты либо пленочной оболочкой, либо энтеросолюбильной оболочкой в соответствии со способами, известными из уровня техники.
Подходящие для перорального введения композиции включают эффективное количество соединения формулы I (или соединения согласно любому из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E) в форме таблеток, пастилок, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул, или сиропов, или настоек. Композиции, предназначенные для перорального применения, получают в соответствии с любым способом, известным из уровня техники, предназначенным для изготовления фармацевтических композиций, и такие композиции могут содержать одно или несколько средств, выбранных из группы, состоящей из подсластителей, ароматизаторов, красителей и консервантов, с целью получения препаратов, которые являются фармацевтически эстетичными и приятными на вкус. Таблетки могут содержать активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми вспомогательными веществами, которые являются подходящими для изготовления таблеток. Такие вспомогательные вещества представляют собой, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и разрыхляющие средства, например кукурузный крахмал или альгиновая кислота; связующие средства, например крахмал, желатин или аравийская камедь; и смазывающие средства, например стеарат магния, стеариновая кислота или тальк. Таблетки являются непокрытыми или покрытыми посредством известных методик для задержки распада и всасывания в желудочно-кишечном тракте и обеспечения таким образом устойчивого действия в течение более длительного периода. Например, можно использовать материал для обеспечения замедленного действия, такой как глицерилмоностеарат или глицерилдистеарат. Составы для перорального применения могут быть представлены в виде твердых желатиновых капсул, где активный ингредиент смешан с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где активный ингредиент смешан с водой или масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом.
Подходящие композиции для трансдермального применения содержат эффективное количество соединения для применения в способе по настоящему изобретению с подходящим носителем. Носители, подходящие для трансдермальной доставки, включают абсорбируемые фармакологически приемлемые растворители для содействия прохождению через кожу пациента. Например, трансдермальные устройства представлены в форме перевязочного материала, содержащего поддерживающий элемент, резервуар, содержащий соединение, необязательно с носителями, необязательно барьер, контролирующий скорость, для доставки соединения через кожу пациента с контролируемой и предварительно определенной скоростью в течение длительного периода времени, а также средство для прикрепления устройства к коже.
В предпочтительном варианте осуществления подходящая композиция представляет собой композицию для перорального введения.
В предпочтительных вариантах осуществления раскрытых способов, путей применения и наборов ингибитор LTA4H (например соединение согласно любому из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E, или его фармацевтически приемлемая соль) помещен в фармацевтический состав, где указанный фармацевтический состав дополнительно содержит один или несколько наполнителей в количестве 70-95% по весу, разрыхлитель в количестве 3-5% по весу и связующее вещество в количестве 2-3% по весу. В конкретном аспекте данного варианта осуществления состав дополнительно предусматривает смазывающее средство в количестве 1% по весу и вещество, способствующее скольжению, в количестве 0,5% по весу.
В некоторых вариантах осуществления раскрытых способов, путей применения и наборов фармацевтический состав представлен в форме капсулы или таблетки. В некоторых вариантах осуществления раскрытых способов, путей применения и наборов фармацевтический состав представлен в форме таблетки.
В некоторых вариантах осуществления раскрытых способов, путей применения и наборов фармацевтический состав представлен в форме капсулы. В конкретном аспекте данного варианта осуществления содержимое капсулы содержит соединение согласно любому из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E, или его фармацевтически приемлемую соль, наполнитель, состоящий из микрокристаллической целлюлозы и маннита, где количество наполнителя составляет 70-95% по весу от всего содержимого капсулы, кросповидон в количестве 3-5% по весу от всего содержимого капсулы, гипромеллозу в количестве 2-3% по весу от всего содержимого капсулы, стеарат магния в количестве приблизительно 1% по весу от всего содержимого капсулы, двуокись кремния и коллоидный диоксид кремния в количестве приблизительно 0,5% по весу от всего содержимого капсулы.
Фармацевтические композиции для применения в раскрытых способах также могут содержать дополнительные терапевтические средства для лечения конкретного целевого нарушения. Например, фармацевтическая композиция может также содержать противовоспалительные средства. Такие дополнительные факторы и/или средства могут быть включены в фармацевтическую композицию для получения синергического эффекта с ингибитором LTA4H, описанным в данном документе, или для минимизации побочных эффектов, вызванных ингибитором LTA4H, описанным в данном документе.
Различные виды терапии можно с пользой комбинировать с раскрытыми ингибиторами LTA4H, такими как соединения любой из формул от (I) до (V) или соединение согласно любому из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E, или его фармацевтически приемлемая соль, в ходе лечения HS.
Следовательно, ингибиторы LTA4H по настоящему изобретению можно вводить либо одновременно с одним или несколькими другими терапевтическими средствами, либо перед или после их введения. Ингибиторы LTA4H для применения в способе по настоящему изобретению можно вводить отдельно от других средств посредством того же или другого пути введения или вместе с ними в одной фармацевтической композиции.
В одном варианте осуществления настоящее изобретение относится к способу лечения или предупреждения HS у субъекта, включающему введение субъекту продукта, содержащего соединение согласно любой из формул от (I) до (V) (или соединение согласно любому из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E) или его фармацевтически приемлемую соль и по меньшей мере одно другое терапевтическое средство в качестве объединенного препарата для одновременного, раздельного или последовательного применения в терапии. Необязательно фармацевтическая композиция для применения в способе по настоящему изобретению может содержать фармацевтически приемлемое вспомогательное вещество, как описано выше.
Продукты, представленные в виде объединенного препарата для применения в способе по настоящему изобретению, включают композицию, содержащую соединение согласно любой из формул от (I) до (V) (или соединение согласно любому из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E) или его фармацевтически приемлемую соль и другое терапевтическое средство(средства) совместно в одной и той же фармацевтической композиции, или соединение согласно любой из формул от (I) до (V) (или соединение согласно любому из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E) или его фармацевтически приемлемую соль и другое терапевтическое средство(средства) в отдельной форме, например в форме набора.
В одном варианте осуществления в настоящем изобретении предусмотрен набор для применения в способе по настоящему изобретению, содержащий две или более отдельные фармацевтические композиции, по меньшей мере одна из которых содержит соединение согласно любой из формул от (I) до (V) (или соединение согласно любому из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E) или его фармацевтически приемлемую соль. В одном варианте осуществления набор содержит средства для раздельного содержания указанных композиций, такие как контейнер, разделенная бутылка или разделенный пакет из фольги. Примером такого набора является блистерная упаковка, как правило, применяемая для упаковывания таблеток, капсул и т. п.
Набор по настоящему изобретению можно применять для введения различных лекарственных форм, например для перорального и парентерального применения, для введения отдельных композиций с различными интервалами между введениями доз или для титрования отдельных композиций относительно друг друга. В целях содействия соблюдению режима лечения набор по настоящему изобретению, как правило, содержит инструкции по введению.
В видах комбинированной терапии по настоящему изобретению ингибитор LTA4H, описанный в данном документе, и другое терапевтическое средство могут быть изготовлены и/или составлены одним и тем же или различными производителями. Более того, ингибиторы LTA4H, описанные в данном документе, и другое терапевтическое средство могут быть объединены в средство комбинированной терапии: (i) до передачи врачам комбинированного препарата (например в случае с набором, содержащим соединение для применения в способе по настоящему изобретению и другое терапевтическое средство); (ii) самими врачами (или под руководством врача) незадолго до введения; (iii) самими пациентами, например во время последовательного введения соединения (ингибитора LTA4H) и другого терапевтического средства.
Соответственно, в настоящем изобретении предусмотрено применение соединения согласно любой из формул от (I) до (V) (или соединения согласно любому из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E) или его фармацевтически приемлемой соли для лечения или предупреждения HS, где лекарственный препарат получен для введения с другим терапевтическим средством. В настоящем изобретении также предусмотрено применение другого терапевтического средства для лечения или предупреждения HS, где лекарственный препарат вводится с соединением согласно любой из формул от (I) до (V) (или соединением согласно любому из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E) или его фармацевтически приемлемой солью.
В настоящем изобретении также предусмотрено соединение согласно любой из формул от (I) до (V) (или соединение согласно любому из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E) или его фармацевтически приемлемая соль для применения в способе лечения или предупреждения HS, где указанное соединение получено для введения с другим терапевтическим средством. В настоящем изобретении также предусмотрено другое терапевтическое средство для применения в способе лечения или предупреждения HS, где другое терапевтическое средство получено для введения с соединением согласно любой из формул от (I) до (V) (или соединением согласно любому из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E) или его фармацевтически приемлемой солью.
В настоящем изобретении также предусмотрено соединение согласно любой из формул от (I) до (V) (или соединение согласно любому из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E) или его фармацевтически приемлемая соль для применения в способе лечения или предупреждения HS, где указанное соединение вводится с другим терапевтическим средством. В настоящем изобретении также предусмотрено другое терапевтическое средство для применения в способе лечения или предупреждения HS, где другое терапевтическое средство вводится с соединением согласно любой из формул от (I) до (V) (или соединением согласно любому из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E) или его фармацевтически приемлемой солью.
В настоящем изобретении также предусмотрено применение соединения согласно любой из формул от (I) до (V) (или соединения согласно любому из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E) или его фармацевтически приемлемой соли для лечения и/или предупреждения HS у пациента, нуждающегося в таком лечении и/или предупреждении, где пациент ранее (например, в период до 24 часов перед этим) подвергался лечению другим терапевтическим средством. В настоящем изобретении также предусмотрено применение другого терапевтического средства для лечения или предупреждения HS у пациента, нуждающегося в этом, где пациент ранее (например, в период до 24 часов перед этим) подвергался лечению соединением согласно любой из формул от (I) до (V) (или соединением согласно любому из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E) или его фармацевтически приемлемой солью.
Такие объединенные терапевтические средства включают средства лечения для местного применения (кремы [нестероидные или стероидные], жидкости для промывания, антисептики), системные средства лечения (например с биологическими препаратами, антибиотиками или химическими веществами), антисептики, фотодинамическую терапию и хирургическое вмешательство (лазер, дренирование или разрезание, иссечение).
Примерами антибиотиков для перорального применения являются тетрациклины и рифампицин.
Неограничивающие примеры средств для лечения HS для местного применения для применения с раскрытыми ингибиторами LTA4H включают бензоилпероксид, стероидные кремы для местного применения, антибиотики аминогликозидной группы для местного применения, такие как клиндамицин, гентамицин и эритромицин, крем на основе резорцина, отшелушивающие средства на основе йода и хлоргексидин.
Неограничивающие примеры средств для лечения HS, применяемых в системном лечении для применения с раскрытыми ингибиторами LTA4H, включают антагонисты IL-17 (иксекизумаб, бродалумаб, секукинумаб CJM112), а также антагонисты IL17A/F, такие как бимекизумаб, или антагонисты IL17C, такие как MOR106, блокаторы фактора некроза опухоли альфа (TNF-альфа) (такие как Enbrel® (этанерцепт), Humira® (адалимумаб), Remicade® (инфликсимаб) и Simponi® (голимумаб)), блокаторы интерлейкина 12/23 (такие как Stelara® (устекинумаб), тазоцитиниб и бриакинумаб), блокаторы IL-23 (такие как гуселкумаб, тилдракизумаб и рисанкизумаб) ингибиторы p19, ингибиторы PDE4, такие как апремиласт или Otezla®), ингибиторы пути комплемента, такие как ингибиторы фактора B (например соединения, раскрытые в WO2015/009616, или LNP023, который также известен как 4-((2S,4S)-4-этокси-1-((5-метокси-7-метил)-1H-индол-4-ил)метил)пиперидин-2-ил)бензойная кислота), ингибиторы C5a (IFX-001, CCX168, также известный как авакопан), антагонисты IL-1 (канакинумаб, гевокизумаб, рилонацепт, анакинра, MaBp1 (XBiotech)), ингибиторы инфламмасомы, такие как ингибиторы NLRP3 и NLRP5, ингибиторы CXCR1/2, антагонисты IL-18, антагонисты IL-6, антагонисты IL-36, антагонисты CD20, антагонисты CTLA4, антагонисты IL-8, деплеторы B-клеток (в частности антагонисты CD20, такие как ритуксимаб, а также BAFF-R и антагонисты CD40, такие как искалимаб (CFZ533)), антагонисты IL-21, антагонисты IL-22, антагонисты VEGF, антагонисты CXCL, антагонисты MMP и антагонисты дефензина (например рецепторы-ловушки, антагонистические антитела и т. д.), а также ингибиторы JAK широкого спектра действия для перорального применения или более специфические ингибиторы TYK2 или JAK 1, JAK2 или JAK 3.
Дополнительные средства для лечения HS для применения в комбинации с раскрытыми ингибиторами LTA4H в ходе лечения HS включают ретиноиды, такие как ацитретин (например Soriatane ®) и изотретиноин, средства, подавляющие иммунную систему (например рапамицин, блокаторы T-клеток [например Amevive® (алефацепт) и Raptiva® [эфализумаб]), циклоспорин, метотрексат, микофенолата мофетил, микофеноловую кислоту, лефлуномид, такролимус и т. д.), гидроксимочевину (например Hydrea®), сульфасалазин, 6-тиогуанин, фумараты (например диметилфумарат и сложные эфиры фумаровой кислоты), азатиоприн, колхицин, алитретиноин, стероиды, кортикостероиды, цертолизумаб, мометазон, розиглитазон, пиоглитазон, ботулотоксин, триамцинолон, IFX-1 (InflaRx), LY-3041658 (Eli Lilly), TE-2232 (Immunwork), NSAID, ингибиторы COX, рецептурные наркотические средства, кетопрофен, кодеин, габапентин, прегабалин, фентанил, антибиотики (для местного применения, для перорального применения, для внутривенного введения) (например клиндамицин, рифампин, тетрациклин, сарециклин, доксициклин, миноциклин, лимециклин, триметоприм-сульфаметоксазол, эритромицин, цефтриаксон, моксифлоксацин, метронидазол, отдельно или в виде комбинаций), кортикостероид (для введения путем инъекции или для перорального применения), антиандрогенную/гормональную терапию (контрацептивы для перорального применения, спиронолактон, финастерид, дутастерид, прогестерон IUD, ацетат ципротерона, этинилоэстрадиол, гестоден, норгестимат, дезогестрел, дроспиренон, спиронолактон), триамцинолона ацетонид, MEDI8968, гидроксихлорохин, дапсон, метформин, адапален, азелаиновую кислоту и цинк.
Предпочтительные комбинации для применения в раскрытых наборах, способах и путях применения включают PDE4i и JAKi, а также антибиотики (все для перорального применения).
Примеры ингибиторов JAK для применения в комбинации представляют собой BMS986165, INCB054707, руксолитиниб, аброцитиниб, тофацитиниб и барицитиниб. Другие примеры ингибиторов JAK представляют собой соединения, раскрытые в WO2017/089985, WO2018/055550 и WO2018/055551.
Специалист в данной области техники сможет определить подходящие дозы вышеуказанных средств для лечения HS для совместной доставки с раскрытыми ингибиторами LTA4H.
Ингибитор LTA4H, например соединение в соответствии с любым из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E или его фармацевтически приемлемая соль, в целях удобства вводится перорально. Продолжительность пероральной терапии с применением фармацевтической композиции по настоящему изобретению будет варьироваться в зависимости от тяжести заболевания, подлежащего лечению, а также состояния и индивидуального ответа каждого отдельного пациента. Медицинский работник примет решение о соответствующей продолжительности пероральной терапии и времени введения средства терапии с использованием фармацевтической композиции по настоящему изобретению. В некоторых вариантах осуществления пациент подвергается лечению HS в соответствии с заявленными способами в течение по меньшей мере 16 недель, по меньшей мере 24 недель, по меньшей мере 36 недель, по меньшей мере 48 недель или по меньшей мере 52 недель. В некоторых вариантах осуществления HS у пациента подвергают лечению в условиях длительного применения.
В одном варианте осуществления фармацевтическая композиция согласно настоящему изобретению для применения в предупреждении или лечении HS может быть представлена в однократной дозировке, составляющей приблизительно 1-500 мг активного(-ых) ингредиента(-ов) для субъекта весом приблизительно 50-70 кг, или приблизительно 1-250 мг, или приблизительно 1-150 мг, или приблизительно 1-100 мг, или приблизительно 1-50 мг активных ингредиентов. Терапевтически эффективная дозировка соединения, фармацевтической композиции зависит от видовой принадлежности субъекта, веса тела, возраста и индивидуального состояния, тяжести нарушения, представляющего собой контраст-индуцированную нефропатию. Врач, клиницист или ветеринар средней квалификации может легко определить эффективное количество каждого из активных ингредиентов, необходимое для предупреждения, лечения или подавления прогрессирования нарушения или заболевания.
Предпочтительный состав представляет собой композицию в форме капсулы или таблетки, содержащую от приблизительно 1 мг до приблизительно 160 мг ингибитора LTA4H или соединения в соответствии с любым из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E или его фармацевтически приемлемой соли и одно или несколько вспомогательных веществ, независимо выбранных из наполнителей, дезинтегрантов, связующих веществ и необязательно смазывающего вещества и вещества, способствующего скольжению. В предпочтительном варианте осуществления композиция в форме капсулы или таблетки содержит от приблизительно 5 мг до приблизительно 80 мг ингибитора LTA4H или соединения в соответствии с любым из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E или его фармацевтически приемлемой соли и одно или несколько вспомогательные веществ, независимо выбранных из наполнителей, дезинтегрантов, связующих веществ и необязательно смазывающего вещества и вещества, способствующего скольжению. В еще одном варианте осуществления композиция в форме капсулы или таблетки содержит приблизительно 1 мг, приблизительно 5 мг, приблизительно 10 мг, приблизительно 20 мг, приблизительно 30 мг, приблизительно 40 мг и приблизительно 50 мг ингибитора LTA4H или соединения в соответствии с любым из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E или его фармацевтически приемлемой соли и одно или несколько вспомогательных веществ, независимо выбранных из наполнителей, дезинтегрантов, связующих веществ и необязательно смазывающего вещества и вещества, способствующего скольжению.
В данном документе раскрыты способы лечения гнойного гидраденита (HS), включающие ежедневное пероральное введение пациенту, нуждающемуся в этом, дозы от приблизительно 1 мг до приблизительно 160 мг, или от приблизительно 10 мг до приблизительно 100 мг, или от приблизительно 20 мг до приблизительно 60 мг ингибитора LTA4H (т. е. соединения в соответствии с любым из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E или его фармацевтически приемлемой соли). Указанные дозы могут вводиться пациенту с использованием либо схемы введения доз один раз в день, либо схемы введения доз два раза в день. В другом варианте осуществления способ включает пероральное введение пациенту, нуждающемуся в этом, дозы от приблизительно 10 мг до приблизительно 30 мг ингибитора LTA4H (т. е. соединения в соответствии с любым из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E или его фармацевтически приемлемой соли), осуществляемое два раза в день (BID). В предпочтительном аспекте данного варианта осуществления способ включает пероральное введение дозы, составляющей приблизительно 20 мг ингибитора LTA4H (т. е. соединения в соответствии с любым из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E или его фармацевтически приемлемой соли), указанному пациенту, осуществляемое два раза в день (BID).
В данном документе раскрыты способы лечения гнойного гидраденита (HS), включающие пероральное введение пациенту, нуждающемуся в этом, суточной дозы от приблизительно 1 мг до приблизительно 160 мг, или от приблизительно 10 мг до приблизительно 100 мг, или от приблизительно 20 до приблизительно 60 мг ингибитора LTA4H, где ингибитор LTA4H представляет собой (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановую кислоту или ее фармацевтически приемлемую соль. В предпочтительном аспекте данного варианта осуществления способ включает пероральное введение дозы от приблизительно 10 мг до приблизительно 30 мг (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты или ее фармацевтически приемлемой соли указанному пациенту, осуществляемое два раза в день. В наиболее предпочтительном аспекте данного варианта осуществления способ включает пероральное введение дозы, составляющей приблизительно 20 мг (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты или ее фармацевтически приемлемой соли, указанному пациенту, осуществляемое два раза в день.
В данном документе раскрыты способы лечения гнойного гидраденита (HS), включающие пероральное введение пациенту, нуждающемуся в этом, суточной дозы от приблизительно 1 мг до приблизительно 160 мг, или от приблизительно 10 мг до приблизительно 100 мг, или от приблизительно 20 мг до приблизительно 60 мг ингибитора LTA4H, где ингибитор LTA4H представляет собой кристаллическую форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в ее свободной форме, как описано в данном документе. В предпочтительном аспекте данного варианта осуществления способ включает пероральное введение дозы от приблизительно 10 мг до приблизительно 30 мг кристаллической формы (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты, как описано в данном документе, указанному пациенту, осуществляемое два раза в день. В наиболее предпочтительном аспекте данного варианта осуществления способ включает пероральное введение дозы, составляющей приблизительно 20 мг кристаллической формы (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты, как описано в данном документе, указанному пациенту, осуществляемое два раза в день.
В данном документе раскрыт ингибитор LTA4H (т. е. соединение в соответствии с любым из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E или его фармацевтически приемлемая соль) для применения в лечении гнойного гидраденита (HS), где ингибитор LTA4H вводится перорально пациенту, нуждающемуся в этом, в суточной дозе от приблизительно 1 мг до приблизительно 160 мг, или от приблизительно 4 мг до приблизительно 100 мг, или от приблизительно 10 мг до приблизительно 100 мг, или от приблизительно 20 мг до приблизительно 60 мг, или от приблизительно 5 мг до приблизительно 80 мг (например, приблизительно 20 мг, приблизительно 30 мг, приблизительно 40 мг или приблизительно 80 мг). В предпочтительном аспекте данного варианта осуществления ингибитор LTA4H (например, соединение в соответствии с любым из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E или его фармацевтически приемлемая соль) вводится перорально пациенту, нуждающемуся в этом, в дозе от приблизительно 5 мг QD до приблизительно 40 мг BID. В еще одном предпочтительном аспекте данного варианта осуществления ингибитор LTA4H (например, соединение в соответствии с любым из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E или его фармацевтически приемлемая соль) вводится перорально пациенту, нуждающемуся в этом, в дозе от приблизительно 10 мг до приблизительно 30 мг два раза в день (например, приблизительно 10 мг BID, приблизительно 15 мг BID, предпочтительно приблизительно 20 мг BID). В наиболее предпочтительном аспекте данного варианта осуществления ингибитор LTA4H (например, соединение в соответствии с любым из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E или его фармацевтически приемлемая соль) вводится перорально пациенту, нуждающемуся в этом, в дозе, составляющей приблизительно 20 мг два раза в день.
В данном документе раскрыт ингибитор LTA4H, который представляет собой (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановую кислоту или ее фармацевтически приемлемую соль, для применения в лечении гнойного гидраденита (HS), где LTA4H вводят перорально пациенту, нуждающемуся в этом, в суточной дозе от приблизительно 1 мг до приблизительно 160 мг, или от приблизительно 4 мг до приблизительно 100 мг, или от приблизительно 10 мг до приблизительно 100 мг, или от приблизительно 20 мг до приблизительно 60 мг, или от приблизительно 5 мг до приблизительно 80 мг (например, приблизительно 20 мг, приблизительно 30 мг, приблизительно 40 мг или приблизительно 80 мг). В предпочтительном аспекте данного варианта осуществления (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановая кислота или ее фармацевтически приемлемая соль вводятся перорально пациенту, нуждающемуся в этом, в дозе от приблизительно 5 мг QD до приблизительно 40 мг BID. В еще одном предпочтительном аспекте данного варианта осуществления (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановая кислота или ее фармацевтически приемлемая соль вводятся перорально пациенту, нуждающемуся в этом, в дозе от приблизительно 10 мг до приблизительно 30 мг два раза в день (например, приблизительно 10 мг BID, приблизительно 15 мг BID, предпочтительно приблизительно 20 мг BID). В наиболее предпочтительном варианте осуществления (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановая кислота или ее фармацевтически приемлемая соль вводятся перорально пациенту, нуждающемуся в этом, в дозе, составляющей приблизительно 20 мг два раза в день.
В данном документе раскрыт ингибитор LTA4H, который представляет собой кристаллическую форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в ее свободной форме, как описано в данном документе, для применения в лечении гнойного гидраденита (HS), где LTA4H вводится перорально пациенту, нуждающемуся в этом, в суточной дозе от приблизительно 1 мг до приблизительно 160 мг, или от приблизительно 4 мг до приблизительно 100 мг, или от приблизительно 10 мг до приблизительно 100 мг, или от приблизительно 20 мг до приблизительно 60 мг, или от приблизительно 5 мг до приблизительно 80 мг (например, приблизительно 20 мг, приблизительно 30 мг, приблизительно 40 мг или приблизительно 80 мг). В предпочтительном аспекте данного варианта осуществления кристаллическая форма (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в ее свободной форме, как описано в данном документе, вводится перорально пациенту, нуждающемуся в этом, в дозе от приблизительно 10 мг QD до приблизительно 40 мг BID. В еще одном предпочтительном аспекте данного варианта осуществления кристаллическая форма (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в ее свободной форме, как описано в данном документе, вводится перорально пациенту, нуждающемуся в этом, в дозе от приблизительно 10 мг до приблизительно 30 мг два раза в день (например, приблизительно 10 мг BID, приблизительно 15 мг BID, предпочтительно приблизительно 20 мг BID). В наиболее предпочтительном варианте осуществления кристаллическая форма (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в ее свободной форме, как описано в данном документе, вводится перорально пациенту, нуждающемуся в этом, в дозе приблизительно 20 мг два раза в день.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов, доза ингибитора LTA4H составляет от приблизительно 10 мг до приблизительно 30 мг BID или приблизительно 20 мг BID.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов, у пациента достигается устойчивый ответ после одного года лечения, как измерено с помощью клинического ответа при гнойном гидрадените (HiSCR) (по упрощенной шкале), цифровой рейтинговой шкалы (NRS), общей оценки врача при гнойном гидрадените (HS-PGA) или индекса качества жизни при заболеваниях кожи (DLQI).
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов до лечения с помощью ингибитора LTA4H, как описано в данном документе, пациент ранее получал лечение с помощью системного средства для лечения HS. В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов системное средство выбрано из группы, состоящей из местного средства для лечения, антибиотика, средства, подавляющего иммунную систему, ингибитора TNF-альфа, антагониста IL-1 и их комбинаций.
В некоторых вариантах осуществления раскрытых способов, вариантов применения и наборов до лечения с помощью LTA4H, как описано в данном документе, пациент ранее не получал лечение с помощью системного средства или местного средства для лечения HS.
В одном варианте осуществления раскрытых способов, вариантов применения и наборов ингибитор LTA4H, как описано в данном документе (например, соединение в соответствии с любым из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E или его фармацевтически приемлемая соль), вводится в комбинации с по меньшей мере одним антибиотиком, ингибитором JAK, ингибитором TYK2, ингибитором PDE4 или иммунодепрессантом.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов доза ингибитора LTA4H, как описано в данном документе (например, соединения в соответствии с любым из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E или его фармацевтически приемлемой соли), составляет от приблизительно 10 мг до приблизительно 30 мг BID. В других предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов доза ингибитора LTA4H (например, соединения в соответствии с любым из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E или его фармацевтически приемлемой соли), составляет приблизительно 20 мг.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов у пациента имеется HS со степенью тяжести от средней до тяжелой.
Используемая в данном документе фраза “степень тяжести от средней до тяжелой” относится к заболеванию HS, при котором у пациентов имеется большее либо равное 3 количество активных воспалительных очагов поражения [т. е. глубоких воспалительных очагов поражения, таких как абсцессы и/или воспалительные узелки], не более 10 свищей, и при этом по меньшей мере две анатомические области должны быть вовлечены в очаги поражения HS.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов пациент является взрослым. В некоторых вариантах осуществления раскрытых способов, вариантов применения и наборов пациент с HS является взрослым с HS со степенью тяжести от средней до тяжелой.
В некоторых вариантах осуществления раскрытых способов, вариантов применения и наборов пациент является пациентом подросткового возраста (с возрастом 12 лет или старше). В некоторых вариантах осуществления пациент является пациентом подросткового возраста, у которого имеется HS со степенью тяжести от средней до тяжелой.
В некоторых вариантах осуществления раскрытых способов, вариантов применения и наборов у пациента было диагностировано наличие HS на протяжении по меньшей мере одного года.
В некоторых вариантах осуществления раскрытых способов, вариантов применения и наборов у пациента отсутствует обширное рубцевание в результате HS (т. е. менее 20 свищей, являющихся дренирующими или не являющихся дренирующими, или менее 10 свищей).
В некоторых вариантах осуществления раскрытых способов, вариантов применения и наборов у пациента ранее наблюдался недостаточный ответ на традиционную системную терапию в отношении HS.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов до лечения с помощью ингибитора LTA4H, как описано в данном документе (например, соединения в соответствии с любым из вариантов осуществления 1, 2, 2A-2N, 3 и 3A-3E или его фармацевтически приемлемой соли), пациент характеризуется баллом по шкале согласно HS-PGA, который больше или равняется 3.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов у пациента достигается HiSCR (по упрощенной шкале) к неделе 16 лечения.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов у пациента достигается NRS30 к неделе 16 лечения.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов у пациента наблюдается уменьшение обострений HS к неделе 16 лечения.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов у пациента достигается уменьшение показателя, которое составляет меньше или равняется 6, как измерено с помощью DLQI, к неделе 16 лечения.
В предпочтительных вариантах осуществления, в случае если раскрытые способы, варианты применения или наборы применяют для лечения популяции пациентов с HS со степенью тяжести от средней до тяжелой, у по меньшей мере 40% указанных пациентов достигается HiSCR по упрощенной шкале к неделе 16 лечения в ответ на указанную стадию введения.
В другом предпочтительном варианте осуществления, в случае если раскрытые способы, варианты применения или наборы применяют для лечения популяции пациентов с HS со степенью тяжести от средней до тяжелой, разница между коэффициентом ответивших на лечение пациентов (например, пациентов, у которых достигается HiSCR-ответ на лечение HS) и коэффициентом ответивших на лечение пациентов, получавших плацебо, к неделе 16 лечения составляет по меньшей мере 15%, по меньшей мере 25% или по меньшей мере 30%.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов у пациента наблюдается уменьшение балла по модифицированной шкале Сарториуса, например, после 16 недель лечения.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов у пациента наблюдается улучшение согласно DLQI, например, после 16 недель лечения.
В предпочтительных вариантах осуществления, в случае если раскрытые способы, варианты применения или наборы применяют для лечения популяции пациентов с HS со степенью тяжести от средней до тяжелой, у по меньшей мере 25% (и предпочтительно у по меньшей мере 30%) указанных пациентов достигается ответ, соответствующий NRS30, например, к неделе 16 лечения в ответ на указанную стадию введения.
В предпочтительных вариантах осуществления, в случае если раскрытые способы, варианты применения или наборы применяют для лечения популяции пациентов с HS со степенью тяжести от средней до тяжелой, у менее чем 15% указанных пациентов наблюдается обострение HS в течение лечения в ответ на указанную стадию введения (например, в течение 16 недель лечения).
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов пациента дополнительно лечат с помощью по меньшей мере одного препарата для местного применения и по меньшей мере одного антисептика в комбинации с ингибитором LTA4H, как описано в данном документе.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов пациент подвергается лечению с помощью ингибитора LTA4H, как описано в данном документе, в течение по меньшей мере 16 недель, по меньшей мере 24 недель, по меньшей мере 36 недель, по меньшей мере 48 недель или по меньшей мере 52 недель. Наиболее предпочтительно пациент подвергается лечению в течение по меньшей мере 16 недель.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов у пациента наблюдается быстрое уменьшение болевого ощущения, как измерено с помощью VAS или NRS, уже через одну неделю после введения ему первой дозы ингибитора LTA4H.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов у пациента наблюдается быстрое уменьшение уровня CRP, как измерено с использованием стандартного анализа CRP, уже через одну неделю после первой дозы антитела к IL-17 или его антигенсвязывающего фрагмента.
В предпочтительных вариантах осуществления раскрытых способов, вариантов осуществления и наборов ингибитор LTA4H представляет собой (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановую кислоту или ее фармацевтически приемлемую соль.
В предпочтительных вариантах осуществления настоящего изобретения ингибитор LTA4H представляет собой кристаллическую форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в ее свободной форме (в данном документе обозначенной как форма B).
В предпочтительных вариантах осуществления ингибитор LTA4H представляет собой форму B, как описано в данном документе, представленную в виде по сути чистой фазы.
Аспекты, преимущественные признаки и предпочтительные варианты осуществления настоящего изобретения, которые обобщены в следующих пунктах, соответственно отдельно или в комбинации, способствуют решению задачи настоящего изобретения.
Пункт 1. Способ лечения или предупреждения гнойного гидраденита (HS), включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества ингибитора LTA4H.
Пункт 2. Способ по п. 1, где ингибитор LTA4H представляет собой соединение формулы (I) или его фармацевтически приемлемую соль:
(I),
где
R1 представляет собой OH или NH2;
Y представляет собой O, S или CH2;
X1, X2, X3 и X4 представляют собой N; или
X1, X2, X3 и X4 выбраны из N, NH, C, CH и O при условии, что по меньшей мере два из X1, X2, X3 или X4 представляют собой N или NH;
R2 представляет собой C1-C6алкил, необязательно замещенный фенилом; C3-C6циклоалкил; фенил, необязательно являющийся замещенным галогеном, циано, C1-C6алкилом, необязательно замещенным галогеном, C1-C6алкокси или 5-6-членным гетероарильным кольцом, содержащим от 1 до 3 гетероатомов, выбранных из N, O и S; или 5-10-членный моно- или бициклический гетероарил, содержащий от 1 до 4 гетероатомов, выбранных из N, O и S, при этом указанный гетероарил необязательно является замещенным галогеном, циано или C1-C6алкилом, необязательно замещенным галогеном; или его фармацевтически приемлемую соль.
Пункт 3. Способ по пп. 1-2, где ингибитор LTA4H представляет собой (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановую кислоту или ее фармацевтически приемлемую соль.
Пункт 4. Способ по любому из вышеуказанных пунктов, где ингибитор LTA4H помещен в фармацевтический состав, где указанный фармацевтический состав содержит один или несколько фармацевтически приемлемых носителей, каждый из которых независимо выбран из наполнителя, смазывающего вещества, связующего вещества, дезинтегранта и вещества, способствующего скольжению.
Пункт 5. Способ по п. 4, где фармацевтический состав находится в форме таблетки или капсулы.
Пункт 6. Способ по любому из вышеуказанных пунктов, где ингибитор LTA4H вводят в суточной дозе от приблизительно 10 мг до приблизительно 100 мг.
Пункт 7. Способ по любому из вышеуказанных пунктов, где ингибитор LTA4H вводят в комбинации с одним или несколькими терапевтическими средствами.
Пункт 8. Способ по любому из пп. 1-6, где пациента дополнительно подвергают лечению с помощью по меньшей мере одного лекарственного препарата для местного применения и по меньшей мере одного антисептика в комбинации с ингибитором LTA4H.
Пункт 9. Способ по любому из пп. 1-8, где до лечения с помощью ингибитора LTA4H пациента ранее не подвергали лечению с помощью системного средства или местного средства для лечения HS.
Пункт 10. Способ по любому из вышеуказанных пунктов, где пациента выбирают в соответствии с одним из следующих критериев:
a) у пациента имеется HS со степенью тяжести от средней до тяжелой;
b) до лечения с помощью ингибитора LTA4H пациент характеризуется баллом по шкале согласно HS-PGA, который больше или равняется 3;
c) до лечения с помощью ингибитора LTA4H у пациента имеется по меньшей мере 3 воспалительных очага поражения; или
d) до лечения с помощью ингибитора LTA4H у пациента отсутствует обширное рубцевание (менее 10 свищей) вследствие HS.
Пункт 11. Способ по любому из вышеуказанных пунктов, где у указанного пациента достигается по меньшей мере одно из следующего (например к неделе 16 лечения):
a) HiSCR по упрощенной шкале;
b) уменьшение обострений HS;
c) NRS30;
d) уменьшение показателя, которое составляет меньше или равняется 6, что измерено посредством DLQI; и/или
e) улучшение согласно DLQI.
Пункт 12. Способ по любому из вышеуказанных пунктов, где в случае применения указанного способа для лечения популяции пациентов с HS со степенью тяжести от средней до тяжелой у по меньшей мере 40% указанных пациентов достигается HiSCR по упрощенной шкале, например, к неделе 16 лечения.
Пункт 13. Способ по любому из пп. 1-11, где в случае применения указанного способа для лечения популяции пациентов с HS со степенью тяжести от средней до тяжелой у по меньшей мере 25% указанных пациентов достигается ответ, соответствующий NRS30, например к неделе 16 лечения.
Пункт 14. Способ по любому из пп. 1-11, где в случае применения указанного способа для лечения популяции пациентов с HS со степенью тяжести от средней до тяжелой у менее чем 15% указанных пациентов наблюдается обострение HS в течение лечения, например в течение 16 недель лечения.
Пункт 15. Способ по любому из вышеуказанных пунктов, где пациента подвергаются лечению с помощью ингибитора LTA4H в течение по меньшей мере 16 недель.
Пункт 16. Способ по любому из пп. 1-11, где у пациента уже через одну неделю после первой дозы ингибитора LTA4H наблюдается по меньшей мере одно из следующего:
a) быстрое уменьшение болевого ощущения, что измерено посредством VAS или NRS,
b) быстрое уменьшение уровня CRP, что измерено посредством стандартного анализа CRP.
Пункт 17. Способ по любому из вышеуказанных пунктов, где у указанного пациента достигается устойчивый ответ после завершения лечения (например, через 3 месяца после завершения лечения), что измерено по количеству воспалительных очагов поражения, посредством клинического ответа при гнойном гидрадените (HiSCR), цифровой рейтинговой шкалы (NRS), балла по модифицированной шкале Сарториуса для HS, общей оценки врачом при гнойном гидрадените (HS-PGA) или индекса качества жизни при заболеваниях кожи (DLQI).
Пункт 18. Способ по п. 17, где у указанного пациента достигается устойчивый ответ после завершения лечения (например, через 3 месяца после завершения лечения), что измерено посредством HiSCR по упрощенной шкале (sHiSCR).
Пункт 19. Способ по любому из вышеуказанных пунктов, где ингибитор LTA4H вводят в дозе от приблизительно 10 мг до приблизительно 30 мг два раза в день.
Пункт 20. Ингибитор LTA4H для применения в лечении и/или предупреждении гнойного гидраденита (HS) у пациента, нуждающегося в таком лечении и/или предупреждении.
Пункт 21. Ингибитор LTA4H для применения по п. 20, где указанный ингибитор LTA4H представляет собой соединение формулы (I) или его фармацевтически приемлемую соль:
(I),
где
R1 представляет собой OH или NH2;
Y представляет собой O, S или CH2;
X1, X2, X3 и X4 представляют собой N; или
X1, X2, X3 и X4 выбраны из N, NH, C, CH и O при условии, что по меньшей мере два из X1, X2, X3 или X4 представляют собой N или NH;
R2 представляет собой C1-C6алкил, необязательно замещенный фенилом; C3-C6циклоалкил; фенил, необязательно являющийся замещенным галогеном, циано, C1-C6алкилом, необязательно замещенным галогеном, C1-C6алкокси или 5-6-членным гетероарильным кольцом, содержащим от 1 до 3 гетероатомов, выбранных из N, O и S; или 5-10-членный моно- или бициклический гетероарил, содержащий от 1 до 4 гетероатомов, выбранных из N, O и S, при этом указанный гетероарил необязательно является замещенным галогеном, циано или C1-C6алкилом, необязательно замещенным галогеном.
Пункт 22. Ингибитор LTA4H для применения по п. 20 или п. 21, где указанный ингибитор LTA4H представляет собой (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановую кислоту или ее фармацевтически приемлемую соль.
Пункт 23. Ингибитор LTA4H для применения по любому из пп. 20-22, где ингибитор LTA4H помещен в фармацевтический состав, где указанный фармацевтический состав содержит один или несколько фармацевтически приемлемых носителей, каждый из которых независимо выбран из наполнителя, смазывающего вещества, связующего вещества, дезинтегранта и вещества, способствующего скольжению.
Пункт 24. Ингибитор LTA4H для применения по п. 23, где фармацевтический состав находится в форме таблетки или капсулы.
Пункт 25. Фармацевтическая композиция, содержащая ингибитор LTA4H, который представляет собой соединение формулы I или его фармацевтически приемлемую соль, совместно с одним или несколькими фармацевтически приемлемыми носителями, для применения в лечении и/или предупреждении HS у пациента, нуждающегося в таком лечении и/или предупреждении.
Пункт 26. Ингибитор LTA4H для применения по любому из пп. 20-24 или фармацевтическая композиция для применения по п. 25, где ингибитор LTA4H вводится в суточной дозе от приблизительно 10 мг до приблизительно 100 мг.
Пункт 27. Ингибитор LTA4H для применения по любому из пп. 20-24 и п. 26 или фармацевтическая композиция для применения по п. 25 или п. 26, где ингибитор LTA4H или фармацевтическая композиция, содержащая его, вводятся в комбинации с одним или несколькими вторыми терапевтическими средствами.
Пункт 28. Комбинированный препарат, содержащий терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли и одно или несколько терапевтических средств, для применения в лечении и/или предупреждении HS у пациента, нуждающегося в таком лечении и/или предупреждении.
Пункт 29. Ингибитор LTA4H для применения по любому из пп. 20-24 и п. 26, фармацевтическая композиция для применения по любому из п. 25 и п. 26, где пациент дополнительно подвергается лечению с помощью по меньшей мере одного лекарственного препарата для местного применения и по меньшей мере одного антисептика в комбинации с ингибитором LTA4H.
Пункт 30. Ингибитор LTA4H для применения по любому из пп. 20-24, 26, 27 и п. 29, фармацевтическая композиция для применения по любому из пп. 25-27 и п. 29 или комбинация по п. 28 или п. 29, где до лечения с помощью ингибитора LTA4H пациент ранее не был подвергнут лечению с помощью системного средства или местного средства для лечения HS.
Пункт 31. Ингибитор LTA4H для применения по любому из пп. 20-24, 26, 27 и п. 29, фармацевтическая композиция для применения по любому из пп. 25-27 и п. 29 или комбинация по п. 28 или п. 29, где пациент выбран в соответствии с одним из следующих критериев:
a) у пациента имеется HS со степенью тяжести от средней до тяжелой;
b) до лечения с помощью ингибитора LTA4H пациент характеризуется баллом по шкале согласно HS-PGA, который больше или равняется 3;
c) до лечения с помощью ингибитора LTA4H у пациента имеется по меньшей мере 3 воспалительных очага поражения; или
d) до лечения с помощью ингибитора LTA4H у пациента отсутствует обширное рубцевание (менее 10 свищей) вследствие HS.
Пункт 32. Ингибитор LTA4H для применения по любому из пп. 20-24, 26, 27 и пп. 29-31, фармацевтическая композиция для применения по любому из пп. 25-27 и пп. 29-31 или комбинация по любому из пп. 27-31, где у указанного пациента к неделе 16 лечения достигается по меньшей мере одного из следующего:
a) HiSCR по упрощенной шкале;
b) уменьшение обострений HS;
c) NRS30;
d) уменьшение показателя, которое составляет меньше или равняется 6, что измерено посредством DLQI; и/или
e) улучшение согласно DLQI.
Пункт 33. Ингибитор LTA4H для применения по любому из пп. 20-24, 26, 27 и пп. 29-31, фармацевтическая композиция для применения по любому из пп. 25-27 и пп. 29-31 или комбинация по любому из пп. 27-31, где к неделе 16 лечения у по меньшей мере 40% указанных пациентов достигается HiSCR по упрощенной шкале; или у по меньшей мере 25% указанных пациентов достигается ответ, соответствующий NRS30; или у менее чем 15% указанных пациентов наблюдается обострение HS.
Пункт 34. Ингибитор LTA4H для применения по любому из пп. 20-24, 26, 27 и пп. 29-31, фармацевтическая композиция для применения по любому из пп. 25-27 и пп. 29-31 или комбинация по любому из пп. 27-31, где у пациента уже через одну неделю после первой дозы ингибитора LTA4H наблюдается по меньшей мере одно из следующего:
a) быстрое уменьшение болевого ощущения, что измерено посредством VAS или NRS, и
b) быстрое уменьшение уровня CRP, что измерено посредством стандартного анализа CRP.
Пункт 35. Ингибитор LTA4H для применения по любому из пп. 20-24, 26, 27 и пп. 29-34, фармацевтическая композиция для применения по любому из пп. 25-27 и пп. 29-34 или комбинация по любому из пп. 27-34, где у указанного пациента достигается устойчивый ответ после завершения лечения (например, через 3 месяца после завершения лечения), что измерено по количеству воспалительных очагов поражения, посредством клинического ответа при гнойном гидрадените (HiSCR), цифровой рейтинговой шкалы (NRS), балла по модифицированной шкале Сарториуса для HS, общей оценки врачом при гнойном гидрадените (HS-PGA) или индекса качества жизни при заболеваниях кожи (DLQI).
Пункт 36. Ингибитор LTA4H, фармацевтическая композиция или комбинация по п. 35, где у указанного пациента достигается устойчивый ответ после завершения лечения, например через 3 месяца после завершения лечения, что измерено посредством HiSCR по упрощенной шкале (sHiSCR).
Пункт 37. Ингибитор LTA4H для применения по любому из пп. 20-24, 26, 27 и пп. 29-34, фармацевтическая композиция для применения по любому из пп. 25-27 и пп. 29-34 или комбинация по любому из пп. 27-34, где ингибитор LTA4H вводится в дозе от приблизительно 10 мг до приблизительно 30 мг два раза в день.
Общая информация
Подробности одного или нескольких вариантов осуществления настоящего изобретения приведены в сопутствующем вышеизложенном описании. Хотя при осуществлении на практике или тестировании настоящего изобретения могут применяться любые способы и материалы, аналогичные или эквивалентные тем, которые описаны в настоящем изобретении, в данном документе описаны предпочтительные способы и материалы. Другие признаки, объекты и преимущества настоящего изобретения будут очевидны из описания и из формулы изобретения. В описании и прилагаемой формуле изобретения форма единственного числа включает ссылки на множественное число, если контекст явно не предписывает иное. Если не указано иное, то все технические и научные термины, используемые в данном документе, имеют то же значение, которое обычно понятно специалисту средней квалификации в данной области, к которой относится настоящее изобретение. Все патенты и публикации, процитированные в данном описании, включены в него посредством ссылки. Следующие примеры представлены для того, чтобы более полно проиллюстрировать предпочтительные варианты осуществления настоящего изобретения. Данные примеры никоим образом не следует истолковывать как ограничивающие объем раскрытого объекта изобретения, определяемого в прилагаемой формуле изобретения.
ПРИМЕРЫ
Сокращение
AE нежелательный эффект
AUC площадь под кривой
AUClast площадь под кривой зависимости концентрации в плазме крови (или сыворотки крови или крови) от времени
с момента времени ноль до момента времени, в котором концентрация поддается количественному определению (масса x время/объем)
AUCtau площадь под кривой зависимости концентрации в плазме крови (или сыворотки крови или крови) от времени
с момента времени ноль до окончания интервала введения доз тау (масса x время/объем)
b.i.d. или BID два раза в день
CI доверительный интервал
Cмакс. максимальная концентрация после введения лекарственного средства
Cмакс., ss максимальная концентрация в состоянии равновесия
ECG электрокардиограмма
EoS окончание исследования
PK фармакокинетика
PD фармакодинамика
q.d. или QD один раз в день
,ss в состоянии равновесия
Tмакс. предельная продолжительность периода времени до достижения максимальной концентрации после введения лекарственного средства
T1/2 конечный период полувыведения
Пример 1. (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановая кислота (кристаллическая форма B)
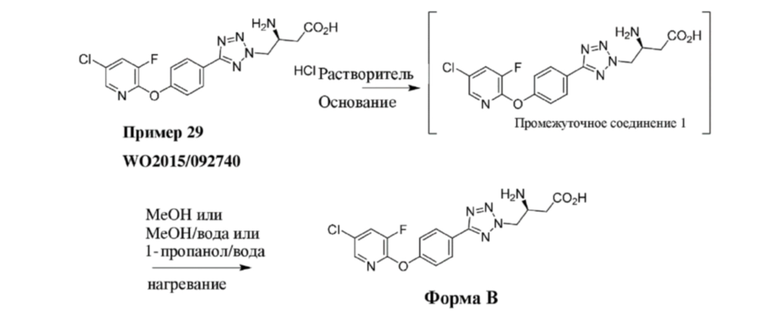
Соединение из примера 29, описанное в WO2015/092740 (28 г, 35 ммоль), и смесь растворителей, содержащую 360 г воды и 40 г THF, смешивали вместе и перемешивали в течение 20 минут. Смесь фильтровали и pH фильтрата доводили до pH=5 с помощью водного раствора NaHCO3. Перемешивание продолжали в течение 18 ч, после чего смесь фильтровали с получением свободной кислоты (Int-1) во влажном осадке весом 25,6 г, который применяли для получения формы B полиморфа без дополнительной очистки.
Взвешивали 505 мг свободной кислоты (Int-1) в стеклянном флаконе объемом 20 мл и добавляли 6 мл метанола. Взвесь нагревали до 50°C и перемешивали в течение 4 дней с применением магнитной мешалки. Суспензию охлаждали до комнатной температуры и фильтровали. Извлеченное твердое вещество высушивали при 40°C в течение 2,5 ч под вакуумом. Белое твердое вещество анализировали посредством XRPD, DSC и TGA (фигуры 1-3 соответственно).
Альтернативные способы получения кристаллической формы B описаны в заявке на патент № PCT/CN2018/000278, поданной 31-го июля 2018 года (номер дела патентного поверенного PAT058189-WO-PCT).
Порошковая рентгеновская дифракция
Порошковые рентгеновские дифрактограммы (XRPD) получали с применением Bruker Discovery D8 в геометрии на отражение (фигура 1). Порошки анализировали с применением держателя образца с Si-подложкой и нулевым фоном. Излучение представляло собой Cu Kα (l=1,5418 Å). Дифрактограммы получали при значениях 2-тета, составляющих от 2° до 40°.
Таблица 1
Данные порошковой рентгеновской дифракции для кристаллической формы B
DSC:
дифференциальную сканирующую калориметрию выполняли для каждой кристаллической формы с применением прибора от TA Instruments (DSC 2500) (фигура 2). Для каждого анализа 2-4 мг образца помещали в алюминиевый тигель T-zero, который закрывали крышкой с отверстием для иглы. Скорость нагревания составляла 10°C в минуту в диапазоне значений температуры от 30 до 300°C. Значения температуры приведены в градусах Цельсия (°C), и значения энтальпии приведены в Джоулях на грамм (Дж/г). На графиках эндотермические пики показаны направленными вниз. Эндотермический пик плавления (точку плавления) оценивали по экстраполированной температуре начала плавления. Точность измеренной температуры образца посредством данного способа составляет приблизительно ±1°C, и теплота плавления может быть измерена с относительной погрешностью, составляющей приблизительно ±5%.
Иллюстративные результаты DSC, полученные с применением кристаллических форм B, показаны на фигуре 2.
Форма B: эндотерма плавления: Tначала=197,4°C (плавление при разложении)
Термогравиметрический анализ (TGA):
кривые TGA получали с применением TA Instrument Q5000. 5-15 мг образца помещали в алюминиевый тигель и герметично закрывали. Непосредственно перед анализом автоматизированный пробоотборник пробивал отверстие в закрытом тигле. Измерения для кривой TGA проводили в диапазоне значений температуры 30-300°C с шагом 10°C/мин. Рассчитывали LoD (потеря веса при высушивании) при температуре от 40°C до 150°C. Строили график зависимости потери веса от измеренной температуры образца. Значения температуры приведены в градусах Цельсия (°C), а потеря веса - в %.
Иллюстративные результаты TGA, полученные с применением кристаллических форм B, показаны на фигуре 3.
Пример 2.
Состав на основе соединения из примера 1 в виде капсулы
Компоненты оболочки одной капсулы
Регламентирование (ЕС) 231/2012: регламент комиссии (ЕС), устанавливающий спецификацию для пищевых добавок. Капсулы приобретали у Lonza (теперь Capsugel)
Пример 3. Анализ образцов биопсии кожи пациентов с HS указывает на сильную активацию пути 5-липоксигеназы в лейкоцитах из очагов поражения
Нейтрофилы и макрофаги, основная клеточная мишень для ингибиторов LTA4H, часто встречаются в очагах поражения HS. Гистологический анализ очагов поражения HS посредством окрашивания с помощью H&E продемонстрировал большое количество миелоидных клеток, таких как нейтрофилы и макрофаги, в очагах поражения HS у пациентов. Парафиновые срезы толщиной три мкм нарезали и окрашивали гематоксилином и эозином. Автоматизированное иммуногистохимическое окрашивание для 5-липоксигеназы (клон: EP6072(2), Abcam, Соединенное Королевство) и CD68 (клон KP-1, Dako, Дания) проводили с помощью прибора для иммуноокрашивания Ventana Discovery XT (Roche Diagnostics, Швейцария). Специфические изотипические контроли применяли в качестве отрицательных контролей. Все образцы биопсии оцифровывали с применением сканера слайдов ScanScope XT (Aperio, Leica Biosystems, Швейцария) с объективом x40.
Макрофаги идентифицировали посредством окрашивания антителом к CD68. Совместное окрашивание антителом к 5-липоксигеназе демонстрировало сильную экспрессию в ядре фермента 5-липоксигеназы в миелоидных клетках, обнаруженных в пораженной коже пациентов с HS. Изображения с большим увеличением продемонстрировали экспрессию фермента 5-липоксигеназы на ядерной мембране многих макрофагов, что является признаком активации пути (Christmas et al., 1999 “Differential localization of 5- and 15- lipoxygenases to the nuclear envelope in RAW macrophages”. J. Biol. Chem. p. 25594-8). Транслокация 5-липоксигеназы к ядерной мембране активирует фермент и обеспечивает образование продукта его биосинтеза LTA4, который является субстратом LTA4H, который превращает его в LTB4. (Фигуры 4, 5, 6)
Фигура 4. Образец биопсии очага поражения, полученный от пациента с HS. Парафиновый срез, окрашенный гематоксилином и эозином, продемонстрировал смешанные воспалительные инфильтраты тяжелой степени, содержащие большие количества полиморфноядерных клеток и макрофагов (масштабный отрезок соответствует 4 мм).
Фигура 5. Образцы биопсии кожи с HS иммуногистохимически окрашивали в отношении 5-липоксигеназы. Окрашивание продемонстрировало сильную экспрессию 5-липоксигеназы в миелоидных клетках. Профиль экспрессии 5-липоксигеназы был либо диффузным в ядре (короткие стрелки), либо располагался на ядерной мембране (длинные стрелки, масштабный отрезок соответствует 50 мкм).
Фигура 6. Иллюстративная микрофотография, демонстрирующая 5-липоксигеназу (коричневый), локализованную на ядерной мембране в многоядерной гигантской клетке CD68+ (красный, масштабный отрезок соответствует 30 мкм).
Пример 4. Липидoмный анализ образцов биопсии кожи пациентов с HS
Замороженные образцы биопсии кожи человека из здоровой кожи или извлеченные очаги поражения HS (в среднем 500 мг) пульверизировали с применением механической силы (Covaris) и гомогенизировали в 4 мл меченых дейтерием внутренних стандартов, содержащих метанол (ISTD), с применением гомогенизатора (циклы по 2×20 сек при 5000 об/мин, Precellys), выдержанного при низкой температуре. Затем образцы хранили при -20°C/45 мин для обеспечения осаждения белка и центрифугировали при 13000 об/мин в течение 10 мин при 4°C. Супернатанты затем выпаривали (Genevac) до полного высушивания и восстанавливали в 70 мкл смеси метанол:вода (50%/50%; объем/объем) с последующим вводом в систему для LC-MS/MS. Калибровочные кривые составляли заново посредством серийных разбавлений чистых стандартов в смеси метанол:вода (50%/50%, объем/объем), содержащих ISTD, и количественное определение осуществляли посредством сравнения соотношений площади пика метаболитов с ISTD в образцах по сравнению с калибровочными растворами.
Две аналитические платформы использовали для получения данных в зависимости от доступности инструмента: 1) система для UPLC класса Н Waters, соединенная с трехквадрупольным масс-спектрометром (QQQ) Waters XevoTQS, и 2) Dionex Ultimate 3000RS UPLC, соединенная с Sciex6500+ QQQ. Разделения липидов достигали с применением колонок с обращенной фазой ACQUITY UPLC BEH C18, 1,7 мкм, 2,1×50 мм, и CORTECS UPLC C18, 1,6 мкм, 2,1 × 50 мм, нагретых до 40°C соответственно. Скорость потока устанавливали на 0,180 мл/мин, и подвижная фаза состояла из смеси вода-уксусная кислота (99,99/0,01; объем/объем) (A) и смеси метанол-ацетонитрил-уксусная кислота (B) (50/50/0,01; объем/объем/объем). Градиент элюента поддерживали на уровне 45% от B в течение 2 мин, затем на уровне 45-65% от B в течение 2-16 минут, 65-100% в течение 16-19 минут и 100% от B в течение 19-22 мин. С 23-30 мин колонку заново уравновешивали до исходных условий. Ионизацию проводили с применением источника ESI, который работал в режиме определения отрицательно заряженных ионов. Параметры фрагментации настраивали для каждого соединения по отдельности.
Для выбора пиков и количественного анализа применяли программное обеспечение от конкретного производителя (TargetLyx для Waters и Analyst/Multiquant для данных Sciex). Дальнейшую обработку данных, включая подстановку недостающих значений (замену на среднее значение для группы), логарифмическое преобразование (основание 2), коррекцию групповых эффектов (на основании масштабирования по среднему значению для каждого образца) и групповые сравнения (эмпирические t-критерии по Байесу) проводили в R (версия 3.4.2) с пакетами программного обеспечения Metabolomics и NormalizeMets (доступными от CRAN). (Anal. Chem., 2015, 87 (7), pp 3606-3615). (Фигуры 7 и 8)
Липиды экстрагировали из образцов биопсии кожи 14 здоровых пациентов и 16 пациентов с HS и анализировали посредством LC-MS/MS.
Фигура 7. Продукт LTA4H, LTB4, характеризовался повышенным уровнем в пораженной коже по сравнению с непораженной кожей (изображено в log2 средних масштабированных концентраций [фмоль/мг]).
Фигура 8. Уровень медиатора 5S,6R DiHETE пути 5-липоксигеназы был также повышен, что указывает на сильную активацию пути 5-липоксигеназы в очагах поражения HS (изображено в log2 средних масштабированных концентраций [фмоль/мг]). *** p<0,001 с применением двустороннего непарного t-критерия.
Пример 5. Транскриптомный анализ образцов биопсии кожи с HS по сравнению с образцами биопсии здоровой кожи
Транскриптомный анализ 18 образцов биопсии кожи с HS по сравнению с 8 образцами биопсии здоровой кожи демонстрирует повышенную транскриптомную регуляцию генов пути 5-липоксигеназы.
Из быстрозамороженной ткани кожи получали гомогенат с применением рекомендованных буферов из мини-набора Qiagen RNeasy. Общую РНК из клеток экстрагировали по протоколу производителя. кДНК из образцов получали из того же исходного количества РНК с применением высокопроизводительного набора для синтеза кДНК High capacity cDNA reverse Transcription Kit (Applied Biosystems). Образцы обрабатывали в CiToxLAB France на микрочипах Affymetrix HG_U133_Plus2. Нормализованные данные RMA анализировали с применением GeneSpring 11.5.1 (Agilent Technologies, Санта-Клара, Калифорния) и результаты интерпретировали с применением программного обеспечения Illumina BaseSpace Correlation Engine и Qiagen IPA. Сперва данные подвергали стандартному контролю QC с помощью CiToxLAB и в GeneSpring (PCA, контроли гибридизации). В дальнейшем их подвергали фильтрации по уровням экспрессии до наборов зондов выше 20-го процентиля в 100% образцов в любом из условий перед дальнейшим анализом.
Пораженная кожа пациентов с HS демонстрировала повышенные уровни генов пути 5-липоксигеназы, включая LTA4H. Экспрессия LTA4H была высокой как в здоровой, так и в пораженной коже (фигура 9).
На фигуре 9 показан нормализованный уровень экспрессии целевых генов. Можно определить значительное повышение уровня экспрессии генов 5-липоксигеназы ALOX5, ALOX5AP и LTA4H в образцах 18 пациентов с HS по сравнению с 8 контрольными образцами от здоровых пациентов, что свидетельствует о повышении уровня биосинтеза LTB4 в соответствии с данными липидомики. *** P<0,001; ** P<0,01; * P<0,05 с применением двустороннего непарного t-критерия.
Пример 6. Соединение из примера 1 и соединение согласно варианту осуществления 3D подавляют биосинтез провоспалительного LTB4 в образцах биопсии стимулированной кожи пациентов с HS
Образцы биопсии кожи пациентов с HS культивировали в среде для культивирования клеток в течение 2 ч в присутствии или в отсутствие 5 мкМ ингибиторов LTA4H, выбранных из соединения из примера 1 и соединения согласно варианту осуществления 3D. Образцы биопсии затем стимулировали в течение 2 ч с помощью 10 мкг/мл ионофора для индукции полной активации пути 5-липоксигеназы в воспалительных клетках из очагов поражения. Образцы биопсии кожи затем собирали, липиды экстрагировали и анализировали с получением LTB4 посредством LC-MS/MS, как описано выше. В стимулированных образцах биопсии кожи с очагами поражения HS образовывалось большое количество липидного медиатора LTB4. Образцы биопсии кожи, которые обрабатывали соединением из примера 1 или соединением согласно варианту осуществления 3D, практически не высвобождали LTB4. (Фигуры 10 и 11)
Образцы биопсии пораженной кожи пациентов с HS оставляли без обработки или полностью стимулировали ионофором с индукцией биосинтеза LTB4 в присутствии или в отсутствие ингибитора LTA4H
Фигура 10: соединение 1 представляет собой соединение из примера 1;
Фигура 11: соединение 2 представляет собой вариант осуществления 3D
Наличие ингибитора LTA4H полностью подавляло способность образцов биопсии пораженной кожи пациентов с HS к образованию LTB4. Количества LTB4 представлены в фмоль/мг ткани. Показаны иллюстративные результаты, полученные с помощью одного донора для одного соединения из нескольких протестированных.
Фигура 12: мышей обрабатывали либо средой-носителем в дозе 3 мг/кг, либо соединением 2 в дозе 10 мг/кг (соединение согласно варианту осуществления 3D). Через 22 часа после введения последней дозы получали кровь, разбавленную в соотношении 1:3 и стимулированную в течение 15 мин ex-vivo с помощью 10 мг/мл кальциевого ионофора с индукцией образования LTB4. Высвобождение LTB4 количественно определяли посредством EIA LTB4 и демонстрировали более чем 90% подавление LTB4 в обеих обработанных группах. Изображены средние значения для 4 мышей на группу +/- стандартная ошибка среднего.
Пример 7. Клинические данные из испытания, впервые проводившегося на человеке (FIH)
Соединение из примера 1 изучали в исследовании FIH, разработанном для характеристики его предварительной безопасности, переносимости и PK у взрослых здоровых субъектов. Исследование состояло из части 1, однократной нарастающей дозы (SAD), и части 2, многократной нарастающей дозы (MAD). Соединение из примера 1 вводили перорально в виде одной дозы QD или BID после приема пищи или после голодания в диапазоне дозы от 5 мг до 2 раз по 100 мг. Соединение из примера 1 также вводили перорально посредством введения многократных доз после голодания в течение 12 дней в диапазоне дозы от 5 мг QD до 80 мг BID.
Часть 1 (SAD)
Часть 1 представляла собой рандомизированное, с ослеплением субъекта и частичным ослеплением исследователя, плацебо-контролируемое исследование однократных нарастающих пероральных доз. Дозу вводили либо в один прием, либо в два приема с интервалом в 12 часов (N=69). Восемь субъектов рандомизировали в каждой когорте (за исключением когорты 7, где рандомизировали семь субъектов, и когорты 9, где рандомизировали шесть субъектов) для получения либо соединения из примера 1, либо соответствующего плацебо в соотношении 6:2 (активное соединение: плацебо) для испытания девяти уровней дозы.
Субъектов распределяли в одну из следующих когорт:
когорта 1: однократная пероральная доза 5 мг или соответствующее плацебо
когорта 2: однократная пероральная доза 10 мг или соответствующее плацебо
когорта 3: однократная пероральная доза 20 мг или соответствующее плацебо
когорта 4: однократная пероральная доза 30 мг или соответствующее плацебо
когорта 5: однократная пероральная доза 45 мг или соответствующее плацебо
когорта 6: однократная пероральная доза 70 мг или соответствующее плацебо
когорта 7: общая пероральная доза 140 мг или соответствующее плацебо, разделенные на две дозы по 70 мг, принимаемые с интервалом в 12 ч
когорта 8: общая пероральная доза 200 мг или соответствующее плацебо, разделенные на две дозы по 100 мг, принимаемые с интервалом в 12 ч
когорта 9: общая пероральная доза 80 мг или соответствующее плацебо, разделенные на две дозы по 40 мг, принимаемые с интервалом в 12 ч
часть 1: режим (однократная нарастающая доза) (qd) - обзор исследования
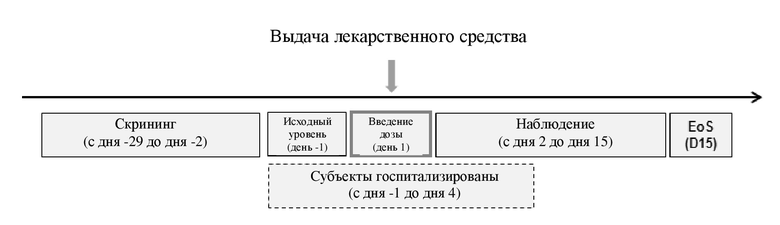
часть 1: режим (однократная нарастающая доза) (прием разделенной дозы 1 раз в день) - обзор исследования
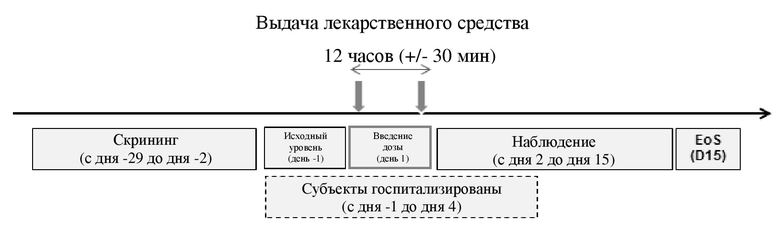
Часть 2 (MAD)
Часть 2 представляла собой рандомизированное, с ослеплением субъекта и частичным ослеплением исследователя, плацебо-контролируемое исследование MAD, в котором восемь субъектов рандомизировали по пяти когортам, где они получали либо соединение из примера 1, либо соответствующее плацебо в соотношении 6:2 (активное вещество: плацебо).
Субъектов распределяли в одну из следующих когорт:
когорта 1: многократная (qd) пероральная суточная доза 5 мг или соответствующее плацебо
когорта 2: многократная (qd) пероральная суточная доза 15 мг или соответствующее плацебо
когорта 3: многократная пероральная суточная доза 20 мг или соответствующее плацебо с введением два раза в день (bid)
когорта 4: многократная пероральная суточная доза 40 мг или соответствующее плацебо с введением два раза в день (bid)
когорта 5: многократная пероральная суточная доза 80 мг или соответствующее плацебо с введением два раза в день (bid)
часть 2: режим многократной нарастающей дозы (qd) - обзор исследования
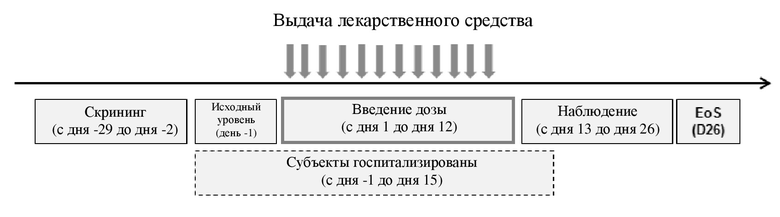
часть 2: режим многократной нарастающей дозы (bid) - обзор исследования
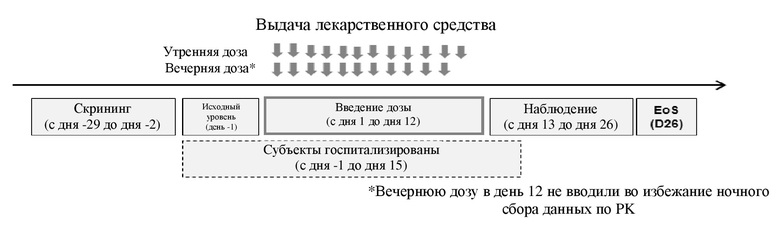
Безопасность для человека и переносимость
В части 1 (однократная нарастающая доза, SAD) вышеуказанного исследования здоровые субъекты получали однократную дозу соединения из примера 1 не более максимальной общей суточной дозы 200 мг (два раза по 100 мг с введением с интервалом 12 ч). В части 2 (многократная нарастающая доза, MAD) исследования субъекты получали дозы не более 80 мг BID в течение 12 дней. Никакие нежелательные явления не привели к прекращению исследования. Самые высокие дозы в обеих частях были безопасными, и максимально переносимая доза не была установлена.
У некоторых субъектов, получавших лечение соединением из примера 1 или плацебо в частях SAD или MAD данного исследования, наблюдалось асимптомное повышение уровня липазы. У троих из получавших лечение субъектов (одного из части SAD с самой низкой дозой 5 мг, двоих из части MAD с самой высокой дозой в 80 мг BID) и одного субъекта, получавшего лечение с помощью плацебо, наблюдалось повышение уровня липазы. У трех субъектов с повышенным уровнем липазы повышение уровня липазы сопровождалось умеренным повышением уровня амилазы (≤ 1,5 ULN). Все субъекты были асимптомными и события были временными, быстро возвращающимися к нормальным уровням, в то время как субъекты продолжали получать соединение из примера 1. Никакие из значительных данных, полученных при физикальном обследовании, показателей жизненно важных функций или ЭКГ не были связаны с соединением из примера 1.
Следовательно, соединение из примера 1 было хорошо переносимым у здоровых субъектов при дозах не более 80 мг bid (суточной дозе 160 мг) в течение 12 дней.
Данные по фармакокинетике у человека
Фармакокинетическое поведение соединения из примера 1 оценивали у здоровых субъектов после приема однократных и многократных пероральных доз. Рассчитанные параметры PK представляли собой стандартные параметры, применяемые для измерения времени действия лекарственного средства в системном кровотоке после получения однократных или многократных доз соединения из примера 1.
Часть 1 (SAD)
Профили зависимости средней концентрации соединения из примера 1 в плазме крови от времени показаны на фигуре 13.
После однократного перорального введения соединения в дозе 5, 10, 20, 30, 45, 70, два раза в дозе 70 (разделенный прием, 2 идентичные дозы вводили с интервалом 12 ч), два раза в дозе 100 (разделенный прием) и два раза в дозе 40 мг (разделенный прием) время действия в плазме крови соединения из примера 1 повышалось с дозой, и медианное значение Tмакс., находящееся в диапазоне от 1 до 1,5 часов после введения дозы, указывало на быструю абсорбцию. После достижения пиковой концентрации первоначально пиковые значения концентрации в плазме крови снижались очень быстро; в более поздних точках времени скорость снижения концентрации значительно замедлилась и средний кажущийся период полувыведения (T1/2) находился в диапазоне от 245 до 513 часов. С увеличением дозы от 5 до 100 мг (т. е. 20-кратным) Cмакс. повысилась в 21,7 раза; AUC0-24ч повысилась в 75 раз с 40-кратным повышением суточной дозы (с 5 мг до 100 мг bid). Коэффициент вариации в процентах (CV%) находился в диапазоне от 18,5 до 41,4 для Cмакс. и от 6,4 до 26,3 для AUC0-24ч.
Для диапазона дозы от 5 мг до 2 раз в день по 100 мг (40-кратной) для AUC и от 5 мг до 100 мг для Cмакс. (20-кратной) угловой коэффициент и соответствующий 90% CI для Cмакс. составлял 1,08 (1,01, 1,15), для AUClast угловой коэффициент составлял 1,00 с 90% CI (0,957; 1,04), и для AUC0-24 угловой коэффициент составлял 1,20 с 90% CI (1,15, 1,24). Пропорциональность дозе на протяжении введения всего диапазона доз была продемонстрирована для AUClast, но не для Cмакс. и AUC0-24ч, где данные указывают на незначительное сверхпропорциональное повышение времени действия с дозой. Однако для Cмакс. и AUC0-24ч PK может считаться пропорциональной дозе при не более чем 4,50-кратном и 2,57-кратном повышении дозы соответственно.
Часть 2 (MAD)
Профили зависимости средней концентрации соединения в плазме крови от времени показаны на фигуре 14а (день 1) и 14b (день 12).
После перорального введения первой дозы соединения из примера 1 в день 1 для всех групп доз (5 и 15 мг qd после голодания; 20, 40 и 80 мг bid после приема пищи) значения концентрации в плазме крови соединения из примера 1 повышались зависимым от дозы образом с медианным значением Tмакс., находящимся в диапазоне от 1 до 2,5 часов.
С повышением дозы соединения из примера 1 с 5 мг qd до 80 мг bid (т. е. 16-кратным для интервала введения дозы) значения Cмакс. и AUC в день 12 (AUC0-12ч,ss для когорт 20, 40 и 80 мг bid и AUC0-24ч,ss для когорт 5 и 15 мг qd) повышались 17,9-кратно и 20,9-кратно соответственно. Для диапазона доз от 5 до 80 мг (16-кратного) угловой коэффициент и соответствующий 90% CI для Cмакс,ss составлял 1,19 (1,08, 1,30), и для AUCtau, ss коэффициент составлял 1,07 с 90% CI (1,01; 1,12). Как для Cмакс,ss, так и для AUCtau, ss не была продемонстрирована пропорциональность дозе на протяжении введения всего диапазона доз. Однако PK для Cмакс,ss и AUCtau может считаться пропорциональной дозе при не более чем 2,12- и 6,29-кратном повышении дозы соответственно. Критерии пропорциональности дозе были соблюдены для AUCtau в диапазоне доз от 5 мг qd до 40 мг bid. % CV находился в диапазоне от 17,1 до 30,2% для Cмакс. и от 11,7 до 18,1 для AUC0-24ч.
Во всем изученном диапазоне доз значение Cмакс,ss было от 1,39 до 1,19 раз выше, чем после введения однократной дозы (среднее значение Cмакс. (день 12)/Cмакс. (день 1)). Для AUC (AUC0-12 и AUC0-24) средние соотношения составляли от 2,08 до 1,27. Это указывает на небольшое накопление соединения из примера 1 с дня 1 до достижения состояния равновесия. Для самой низкой дозы 5 мг qd наблюдали среднее 2,1-кратное накопление до достижения состояния равновесия, но для всех более высоких доз накопление было ≤ 1,4-кратным. Небольшое накопление в день 1 режима bid (среднее значение Rнакопл., день 1 от 1,09 до 1,29) позволяет предположить, что время до достижения состояния равновесия является коротким. В 4 из 5 групп доз утренние значения концентрации до введения доз выше в день 3 по сравнению с днем 2, но различия между значениями концентрации небольшие. С дня 3 до дня 4 постоянного или значительного повышения утренних значений концентрации до введения дозы не наблюдали. Это демонстрирует, что состояние равновесия достигается быстро, т. е. примерно в день 3.
Для перорального введения BID доз соединения из примера 1 после приема пищи равновесное время действия достигается в течение 3-4 дней после введения дозы. В состоянии равновесия критерии пропорциональности дозе были соблюдены для AUCtau от 5 мг QD до 40 мг BID.
Фармакодинамические данные для человека
Биомаркер PD в крови: лейкотриен B4 (LTB4) в крови
Определение лейкотриена B4 (LTB4) в плазме крови ex vivo из стимулированной цельной крови
На животных моделях степень подавления LTB4 в стимулированной крови ex-vivo хорошо коррелировала со степенью терапевтического эффекта (фигура 10), в частности со снижением количества нейтрофилов в воспаленных тканях.
Определение PD в периферической крови из стимулированной крови ex-vivo с LTB4 является прогностическим для терапевтической эффективности и, следовательно, подходит для мониторинга целевого подавления в испытании FIH.
Фигура 10: мышей обрабатывали либо средой-носителем в дозе 3 мг/кг, либо соединением 2 в дозе 10 мг/кг. Через 22 часа после введения последней дозы получали кровь, разбавленную в соотношении 1:3 и стимулированную в течение 15 мин ex-vivo с помощью 10 мг/мл кальциевого ионофора с индукцией образования LTB4. Высвобождение LTB4 количественно определяли посредством EIA LTB4 и демонстрировали более чем 90% подавление LTB4 в обеих обработанных группах. Изображены средние значения для 4 мышей на группу +/- стандартная ошибка среднего.
Следовательно, концентрации LTB4 определяли в плазме крови из стимулированной цельной крови ex-vivo посредством способа LC-MS/MS.
Биоаналитический способ для LTB4
Стимуляцию еx-vivo проводили в месте проведения клинического исследования в соответствии со следующей процедурой:
цельную кровь (500 мкл) стимулировали в течение 30 мин с применением кальциевого ионофора A23187. Затем собирали супернатант плазмы крови и замораживали при -80°C в течение по меньшей мере 24 часов до применения.
Количественное определение LTB4 посредством LC-MS/MS проводили следующим образом:
Плазму крови человека обрабатывали посредством органического осаждения с последующим разделением посредством обращенно-фазовой высокоэффективной жидкостной хроматографии с выявлением посредством тандемной масс-спектрометрии.
Хроматография:
обращенно-фазовое разделение на Agilent Technologies 1290 Infinity HPLC и Zorbax SB-C18, колонке Rapid Resolution HD 1,8 мкм (100×2,1 мм) при 60°C и скорости потока 400 мкл/мин.
Общее аналитическое время проведения анализа составляет 11,0 минут.
Выявление:
AB Sciex QTrap 5500 MS/MS Turbo Spray Ion Drive в режиме определения отрицательно заряженных ионов.
Образец от каждого субъекта и для каждого момента времени получали и анализировали в трех аликвотах. Аликвоты один и два содержали плазму крови из стимулированной цельной крови ex-vivo, в то время как аликвота три в качестве отрицательного контроля содержала плазму крови из нестимулированной цельной крови. Образцы с CV > 25% для первых двух аликвот и/или с выявленными значениями в аликвоте три были исключены из результатов.
Данные по концентрации LTB4 рассчитывали в виде средней концентрации в аликвотах один и два. Концентрацию в аликвоте три не включали в расчет. Изменение по сравнению с исходным уровнем и процент подавления (процент от исходного уровня) рассчитывали для каждого биомаркера. Исходный уровень представлял собой среднее значение исходного уровня и измерений через 1 день и через 1 час.
Чтобы избежать пропущенных значений при расчете изменений по сравнению с исходным уровнем и процента подавления, значения ниже, чем LLOQ, заменяли на 0,5 x LLOQ, и значения выше верхнего предела количественного определения (ULOQ) заменяли на 1,5 x ULOQ.
Результаты:
результаты для первых когорт SAD указывали на то, что высвобождение LTB4 стимулированными клетками крови оставалось на уровне 25-50% от исходного уровня через 24 часа при дозе 5 мг. Наблюдали повышение подавления через 24 часа при повышенной дозе. Примерно 50% подавление через 24 ч после введения дозы наблюдали при дозе 10 мг и временное максимальное подавление (> 90%) после достижения Cмакс. наблюдали при дозе 20 мг. (Фигура 15)
Дополнительные когорты в части SAD подтвердили зависимое от дозы подавление LTB4. Примерно 81%, 86% и 89% подавление через 24 ч после введения дозы наблюдали при дозе 30 мг, 45 мг и 70 мг соответственно.
Данные для состояния равновесия из когорт 1-3 в части MAD подтверждают зависимое от дозы подавление LTB4. Примерно 75%, 90% и 99% подавление наблюдали в образцах, собранных до введения дозы в день 9 (состояние равновесия) при дозе 5 мг (Q.D.), 15 мг (Q.D.) и 20 мг (B.I.D.) соответственно. (Фигура 16)
Целевое подавление, составляющее 90-99%, поддерживали при дозах 15 мг (Q.D.) и 20 мг (B.I.D.) в части MAD исследования.
В заключение, целевое подавление гидролазы лейкотриена A4 в состоянии равновесия, оцененное в образцах крови ex-vivo, составляло в среднем 75% при дозе 5 мг QD, 90% при дозе 15 мг QD и 99% при дозе 20 мг QD bid.
Биомаркер содержимого волдыря: лейкотриен B4 (LTB4) в крови в содержимом волдыря, индуцированного кантаридином.
Индуцированные кантаридином кожные волдыри
Для оценки PD-эффекта соединения из примера 1 на потенциальный целевой орган индуцировали появление кожных волдырей на руке субъектов в одной выбранной когорте части SAD и выбранных когортах части MAD.
В частях 1 и 2 клинического исследования (SAD и MAD) индуцировали появление кожных волдырей на руке
субъектов в выбранных когортах. Для этого диски из фильтровальной бумаги диаметром 10 мм, смоченные раствором кантаридина (таким как кантарон), прикладывали к руке субъекта, что индуцировало появление эпидермального волдыря, и содержимое волдыря собирали примерно через 24 часа (+/- 1 ч) после прикладывания.
Образцы содержимого волдырей собирали в день 1 до введения дозы и день 2 до введения дозы в когорте 4 SAD, а также в день 1 до введения дозы и день 9 до введения дозы в когортах 2-5 MAD.
Получение образцов для анализа лейкотриенов и липоксина А4 в содержимом волдырей человека
Образцы содержимого волдырей получали замороженными на сухом льду и хранили при -70°C или ниже до анализа.
В день анализа образцы доставали из морозильной камеры и немедленно извлекали после
размораживания.
Процедура для лейкотриенов и липоксина A4 в содержимом волдырей человека
90 мкл содержимого волдырей (образец или образец QC) обогащали с помощью 5 мкл раствора внутреннего стандарта IS-L-2 (дейтерированные лейкотриены и липоксин в концентрации 50 нг/мл) и встряхивали в микроцентрифужной пробирке. Добавляли 90 мкл метанола с 0,1% муравьиной кислоты. Затем в течение 5 минут перемешивали посредством встряхивания и в течение 10 минут обрабатывали ультразвуком на льду. Образцы инкубировали при -20°C в морозильной камере в течение 40 минут. Затем
центрифугировали при 16000 об/мин (27500xg) при 4°C в течение 20 минут. 160 мкл супернатанта переносили на фильтры Millipore 5kDa MWCO. Фильтры MWCO перед этим очищали посредством центрифугирования с пропусканием через них 500 мкл смеси метанол/вода 1/1. Загруженные фильтры центрифугировали при 16000 об/мин (27500xg) при 4°C в течение 40 минут (или до отфильтровывания всей жидкости). 140 мкл фильтрата
переносили в стеклянные флаконы для HPLC, объединяли с 108 мкл воды и кратковременно встряхивали. Если объем содержимого волдырей составлял менее 90 мкл, применяли воду для доведения объема до 90 мкл и фиксировали коэффициент разбавления.
Высокоэффективная жидкостная хроматография
HPLC проводили с применением системы для UHPLC Agilent, оснащенной 2 насосами, одним насосом для UHPLC (модель G4220B) для аналитического градиента и одним насосом для четырехкомпонентных смесей (модель G1311B) в качестве загрузочного насоса.
Результаты
В части 1 исследования содержимое волдырей оценивали в группе, получавшей лечение в дозе 30 мг, образец собирали через 24 часа и было обнаружено, что средняя концентрация соединения из примера 1 составляла 4,67 нг/мл. Значения концентрации содержимого волдырей были в среднем примерно в два раза выше, чем концентрации в плазме крови в это же время, и составляли только 14% от соответствующих значений концентрации в крови.
В части 2 исследования содержимое волдырей анализировали в группе, получавшей лечение в дозе 15 мг qd, а также в группах, получавших лечение в дозе 20, 40 и 80 мг, образцы собирали до введения дозы в день 9 (содержимое волдырей). Средние значения концентрации соединения из примера 1 повышались при дозе от 4,62 нг/мл при 15 мг qd до 233 нг/мл при 80 мг bid для содержимого волдырей. Значения концентрации в содержимом волдырей были схожими со значениями концентраций в плазме крови с соотношениями содержимое волдырей/плазма крови, составляющими около 2.
Подавление LTB4 в коже и плазме крови (% изменения от исходного уровня) показано на фигуре 17. Суточная доза 40 мг (20 мг BID) демонстрирует практически полное подавление LTB4 в крови в состоянии равновесия, и при такой же дозе достигали 90% подавления LTB4 образцах содержимого кожных волдырей человека, где появление кожного волдыря индуцировали посредством местного нанесения кантаридина.
Пример 8. Эффективность и безопасность (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты (форма B) у взрослых пациентов с HS со степенью тяжести от умеренной до тяжелой.
Ниже предусмотрены детали дизайна клинического исследования с демонстрацией эффективности (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты по сравнению с плацебо.
Дизайн клинического исследования
Ослепление субъектов и исследователей позволяет осуществлять объективную оценку субъективных данных, таких как результаты подсчета очагов поражения при HS или общие показатели HS-PGA, а также нежелательных явлений.
Рандомизированное, с ослеплением субъекта и исследователя, плацебо-контролируемое многоцентровое и параллельное исследование проводят для оценки эффективности, безопасности и переносимости нескольких активных соединений для лечения, таких как соединение из примера 1, у субъектов с гнойным гидраденитом (HS) со степенью тяжести от умеренной до тяжелой. После периода скрининга продолжительностью примерно 4 недели планируется период лечения длительностью 16 недель, после которого следует наблюдение для оценки безопасности продолжительностью примерно 12 недель. Субъекты получают 20 мг соединения из примера 1 BID p.o. или плацебо BID p.o.
Субъекты, включенные в данное исследование, являются взрослыми мужчинами и женщинами в возрасте от 18 до 65 лет с диагнозом HS со степенью тяжести от умеренной до тяжелой с рецидивирующими очагами воспаления в течение по меньшей мере 12 месяцев. Требования к субъекту для включения в исследование заключаются в том, что у него должно быть по меньшей мере 3 очага воспаления. Оценку на исходном уровне можно начинать с дня -7 с завершением оценки в день -1 до лечения в день 1. Все результаты оценивания безопасности на исходном уровне должны быть доступны до начала введения доз и соответствовать критериям отбора. Рандомизация осуществляется с помощью системы интерактивной технологии рандомизации (IRT).
Выбранная первичная клиническая конечная точка представляет собой HiSCR по упрощенной шкале (клинический ответ при гнойном гидрадените) после 16 недель лечения.
В день 113 (неделя 17) после проведения оценки безопасности и других оценок все субъекты вступают в период последующего наблюдения без введения какого-либо лекарственного средства. Если это обосновано медицинскими показаниями и если не было выявлено никаких потенциальных проблем, связанных с безопасностью (после обсуждения со спонсором), в течение этого периода последующего наблюдения субъекты могут получать запрещенные ранее лекарственные препараты.
Оценки безопасности и эффективности проводят при последующих визитах в день 141 (неделя 21) и день 197 (неделя 29). Собирают данные по фармакокинетике (PK) и фармакодинамике (PD). Ослепление сохраняют для исследователя и субъекта до завершения исследования. Завершающий визит проводится в день 197 (неделя 29), что включает оценку по завершению исследования с последующим завершением участия в исследовании.
Примерно 45 субъектов рандомизируют, из них 30 вводят активное вещество и 15 плацебо. В день 1 20 мг соединения из примера 1 или соответствующего ему плацебо предоставляют субъектам в месте проведения исследования. Персонал в месте проведения исследования инструктирует субъекта о том, как принимать лекарственное средство (BID), включая необходимость приема жидкости (предпочтительно воды) и приема лекарственного средства во время или вскоре после утреннего и вечернего приемов пищи. На момент начала исследования проводят введение лекарственного средства и клинические оценки, а также оценки PK, фармакодинамики (PD) и необходимые оценки безопасности. Субъектов освобождают от участия в исследовании в день завершения всех оценок при условии отсутствия опасений в отношении безопасности. Субъекты возвращаются в центр проведения исследования в день 8 (неделя 2), день 15 (неделя 3) и день 29 (неделя 5), и затем с интервалами в один месяц: день 57 (неделя 9), день 85 (неделя 13) и день 113 (неделя 17) соответственно. В ходе этих визитов проводят оценки безопасности и выбранных параметров эффективности и собирают образцы для анализа PK/PD.
Первичная цель заключается в том, чтобы показать предварительную эффективность лечения соединением из примера 1 у субъектов с HS через 16 недель лечения по сравнению с плацебо. После 16-недельного периода лечения начинается период последующего наблюдения в течение 12 недель для наблюдения за устойчивостью эффекта, который может сохраняться или возрастать после 16 недель лечения.
Планируется рандомизация 2:1, основанная на том факте, что предпочтение отдается воздействию активных веществ с ограничением при этом воздействия плацебо на субъектов.
Исследование включает несколько клинических конечных точек для более качественной оценки свойств данных выбранных конечных точек:
для данного исследования в качестве основной конечной точки был выбран HiSCR по упрощенной шкале. Упрощенный HiSCR определяется как 50% уменьшение общего количества абсцессов и воспалительных узелков без какого-либо увеличения количества дренирующих свищей.
Очаги воспаления при HS будут считаться как отдельные очаги (воспалительные узелки, абсцессы и дренирующие свищи) в типичных анатомических областях. В дополнение к подсчету будет использоваться шкала общей оценки (общая оценка врачом при гнойном гидрадените или HS-PGA), а также совокупный балльный показатель (оценка степени тяжести гнойного гидраденита или SAHS).
Будут использоваться несколько результатов лечения, основанных на опросе пациента, включая индекс качества жизни при заболеваниях кожи (DLQI). И наконец, поскольку с точки зрения субъекта боль, связанная с кожей, является наиболее важным симптомом, для оценки боли включена числовая оценочная шкала (NRS).
Дополнительная информация о клинических оценках:
HS-PGA (общая оценка врачом при гнойном гидрадените): Показатель будет применяться в качестве цели исследования для оценки HS, и он был применен и описан в Kimball AB, Kerdel F, Adams D, et al (2012) Adalimumab for the treatment of moderate to severe Hidradenitis suppurativa: a parallel randomized trial. Ann Intern Med;157:846-55.
Балльная шкала SAHS представляет собой комплексный балльный показатель (Hessam S, Scholl L, Sand M, et al (2018) A Novel Severity Assessment Scoring System for Hidradenitis Suppurativa. JAMA Dermatol;154(3):330-335), и он будет получен из собранной информации о количестве воспалительных очагов, количестве свищей и шкале оценки боли NRS. Кроме того, в обеих когортах будут собраны анатомические области и новые или обостренные существующие фурункулы.
Боль в области кожи - NRS (числовая оценочная шкала боли): NRS для боли, связанной с кожей, применяли в исследованиях адалимумаба (Kimball et al. (2016) N Engl J Med 375:422-34), и она будет применена в дальнейшем, поскольку боль, связанная с HS, является наиболее тяжелым бременем для пациента (Matusiak et al (2017) J Am Acad Dermatol;76:670-5). Боль, связанная с HS, будет регистрироваться в среднем за последние 24 часа и в худшем случае (за последние 24 часа).
Другие результаты лечения, основанные на опросе пациента (PRO), будут включать индекс качества жизни при заболеваниях кожи (DLQI) в качестве средства для оценки качества жизни (QoL), связанного с дерматологией, с подтвержденными балльными шкалами, доступными во многих странах и на многих языках. Также включена общая оценка пациентом.
Цели исследования и конечные точки
Частота изменений лабораторных показателей безопасности, показателей жизненно важных функций и параметров ЭКГ
Физикальное обследование и показатели жизненно важных функций на исходном уровне и периодически до визита по завершению исследования
Количество воспалительных узелков с течением времени
Количество абсцессов с течением времени
Количество дренирующих свищей с течением времени
Количество невоспаленных узелков с течением времени
Обострение определяется как повышение на по меньшей мере 25% общего количества абсцессов и воспалительных узелков (количество AN) с минимальным повышением на 2 AN относительно исходного уровня.
Общая оценка пациентом
Доля пациентов, у которых достигается NRS30 после 16 недель лечения среди пациентов с исходным уровнем боли в коже, соответствующим NRS, который больше или равняется 3
Количество новых фурункулов или существующих фурункулов, появившихся за последние четыре недели.
Ключевые критерии включения
Субъекты мужского и женского пола в возрасте от 18 до 65 лет с клинически диагностированным HS в течение по меньшей мере 12 месяцев до скрининга
Всего по меньшей мере 3 воспалительных очага поражения, т. е. абсцессов и/или воспалительных узелков, и
не более 10 свищей, и
по меньшей мере две анатомические области должны быть вовлечены в очаги поражения HS
Минимальный вес тела субъекта должен составлять 50 кг (включительно)
Ключевые критерии исключения
Применение экспериментальных лекарственных средств к моменту скрининга, или в пределах 30 дней после включения в исследование, или в течение 5 периодов полувыведения после включения в исследование, или до тех пор, пока ожидаемый фармакодинамический эффект не вернется к исходному уровню, но не ранее любого из указанных; или дольше, если этого требуют местные нормативные акты.
Применение биологических препаратов, блокирующих IL12 и IL23, таких как устекинумаб или гуселкумаб, в течение последних 6 месяцев до рандомизации/начала лечения.
Применение нацеливающихся на B-клетки или истощающих B-клетки биологических препаратов или подобных им, таких как ритуксимаб или белилумаб, в течение 12 месяцев; для пациента, который ранее получал эти лекарственные средства, количество B-клеток должно находиться в пределах нормального диапазона.
Применение отличных от вышеуказанных биологических иммуномодулирующих средств (например адалимумаба, секукинумаба, этанерцепта, инфликсимаба и т. д.) за 3 месяца до рандомизации/начала лечения или 5 периодов полувыведения (но не ранее любого из указанных).
Применение ритуксимаба или белилумаба в течение 12 месяцев.
Применение любого системного лечения HS в течение последних 4 недель до рандомизации (например ретиноидов или других иммуномодулирующих терапевтических средств, например метотрексата, циклоспорина А, кортикостероидов).
Применение циклофосфамида в течение последних 6 месяцев.
Применение системных антибиотиков для лечения HS в течение последней недели до рандомизации/начала лечения.
Если используется спиронолактон или другие антиандрогены (финастерид, ацетат ципротерона и т. д.) (для HS), то для участия подходят только те пациенты, которые получают фиксированную дозу в течении последних 3 месяцев и которые планируют продолжать ее прием в течение исследования.
Хирургическое лечение HS за последние 4 недели до рандомизации/начала лечения. Хирургическое лечение не включает нерегулярную эксцизионную биопсию.
Получение высокой болюсной дозы любого кортикостероида (> 1 мг/кг) в течение 3 месяцев.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА LTA4H | 2019 |
|
RU2808992C2 |
| НОВЫЕ ПРИМЕНЕНИЯ | 2019 |
|
RU2836384C2 |
| Фармацевтические комбинации | 2017 |
|
RU2759669C2 |
| КОМПОЗИЦИИ СОЕДИНЕНИЙ И ПУТИ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2659068C1 |
| ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ СПИРОПИПЕРИДИНИЛА И ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2021 |
|
RU2839891C1 |
| СОЕДИНЕНИЕ ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕЙРОГЕННОЙ ОРТОСТАТИЧЕСКОЙ ГИПОТЕНЗИИ | 2017 |
|
RU2723095C1 |
| АМИНОТЕТРАГИДРОПИРАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ-IV ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ ДИАБЕТА | 2010 |
|
RU2550508C2 |
| ЛЕЧЕНИЕ ГИПЕРТЕНЗИИ И/ИЛИ ПРЕДОТВРАЩЕНИЕ И ЛЕЧЕНИЕ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ У МЛЕКОПИТАЮЩЕГО, ПОЛУЧАЮЩЕГО ТЕРАПИЮ АНТИКОАГУЛЯНТАМИ | 2011 |
|
RU2564941C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ПЕРОРАЛЬНЫЕ ДОЗИРОВАННЫЕ ФОРМЫ ПРОЛЕКАРСТВА ЛЕВОДОПЫ И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2537137C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМБИНАЦИИ АНТАГОНИСТА РЕЦЕПТОРА АНГИОТЕНЗИНА И ИНГИБИТОРА NEP | 2006 |
|
RU2459809C2 |
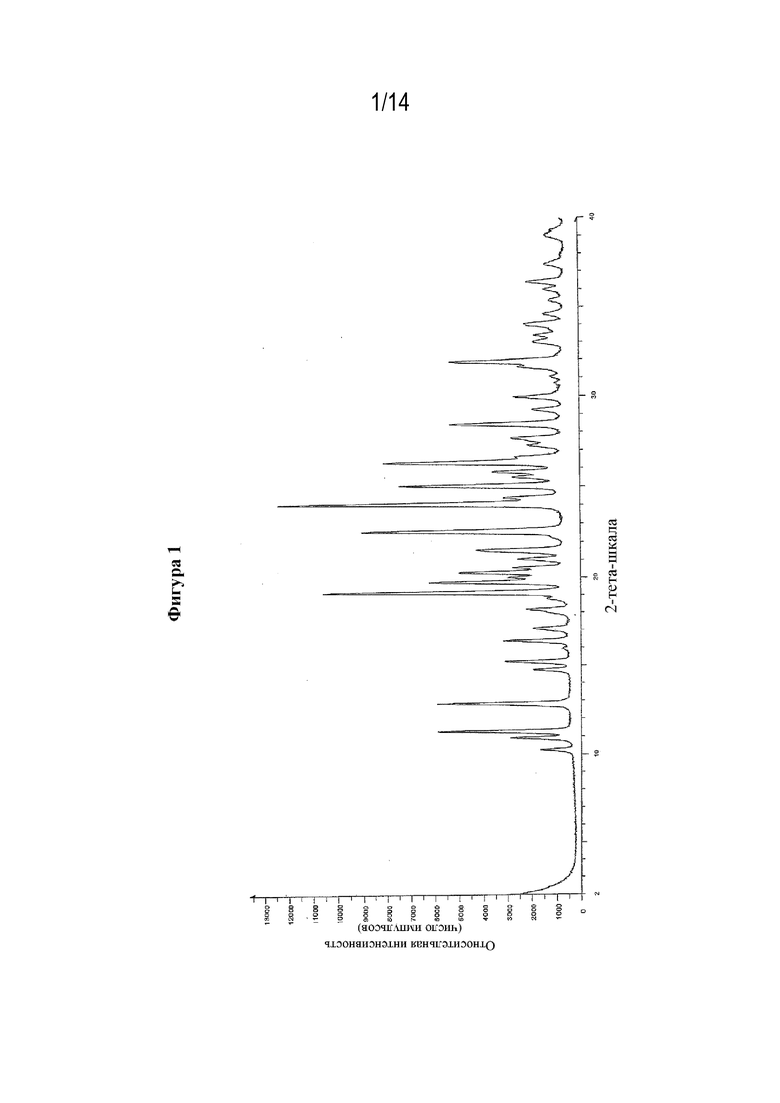
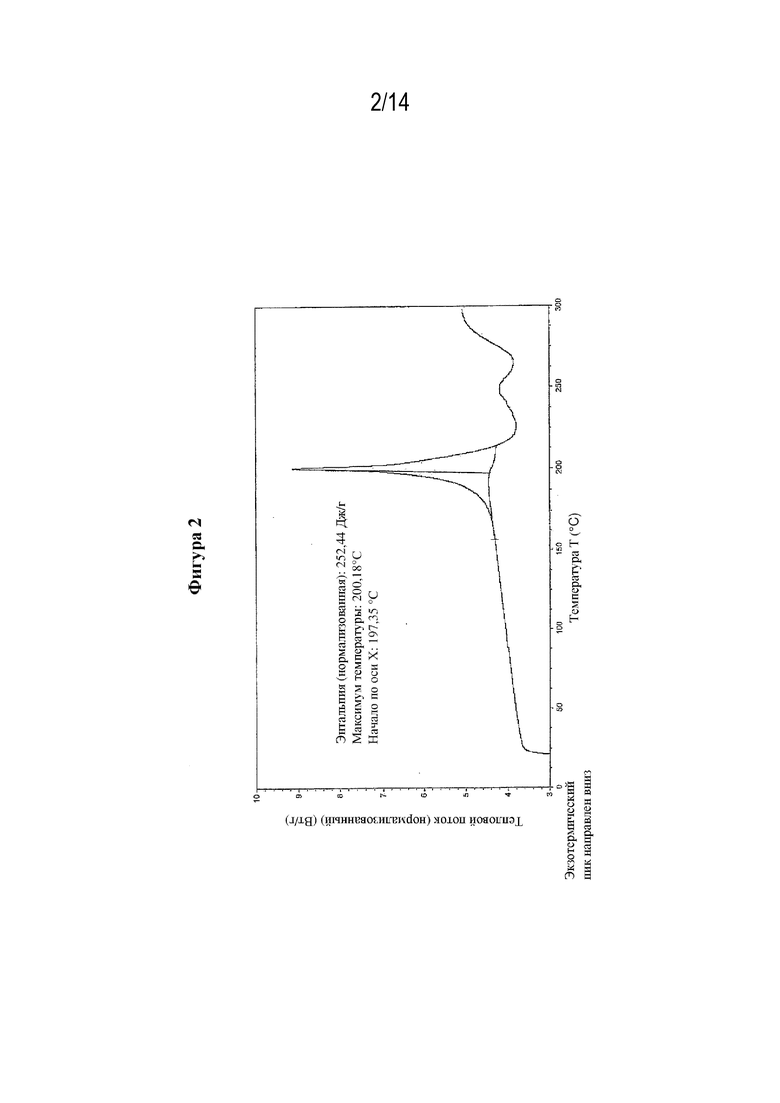
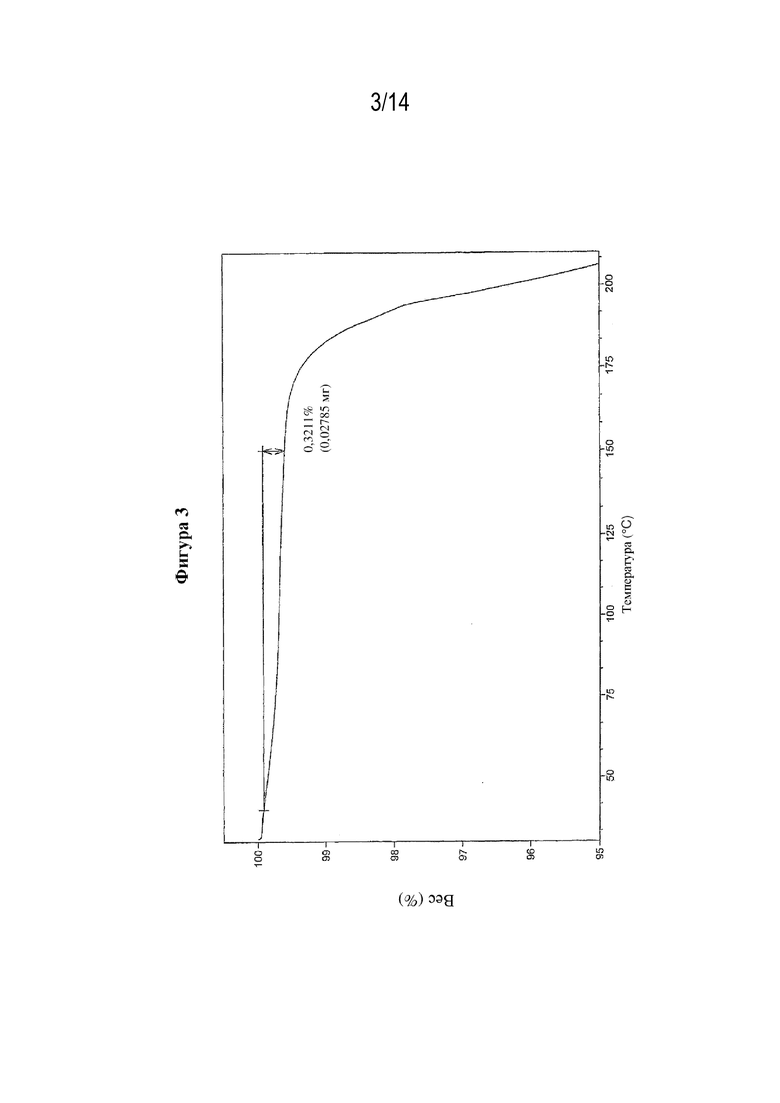


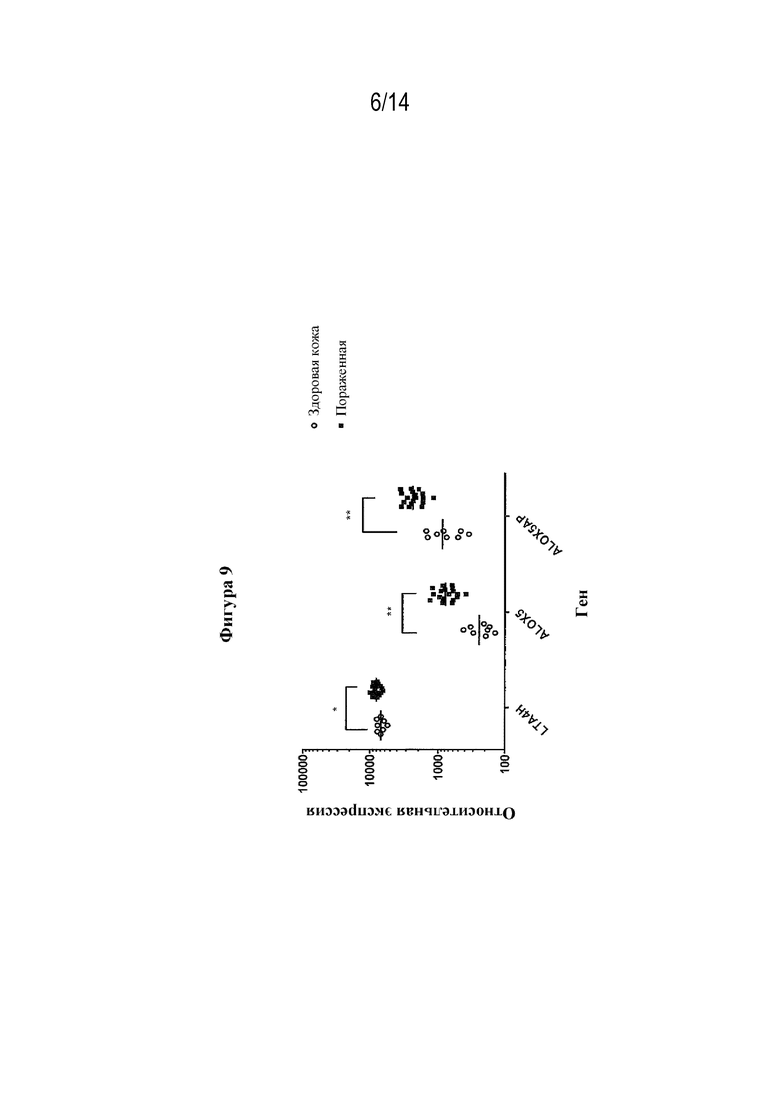
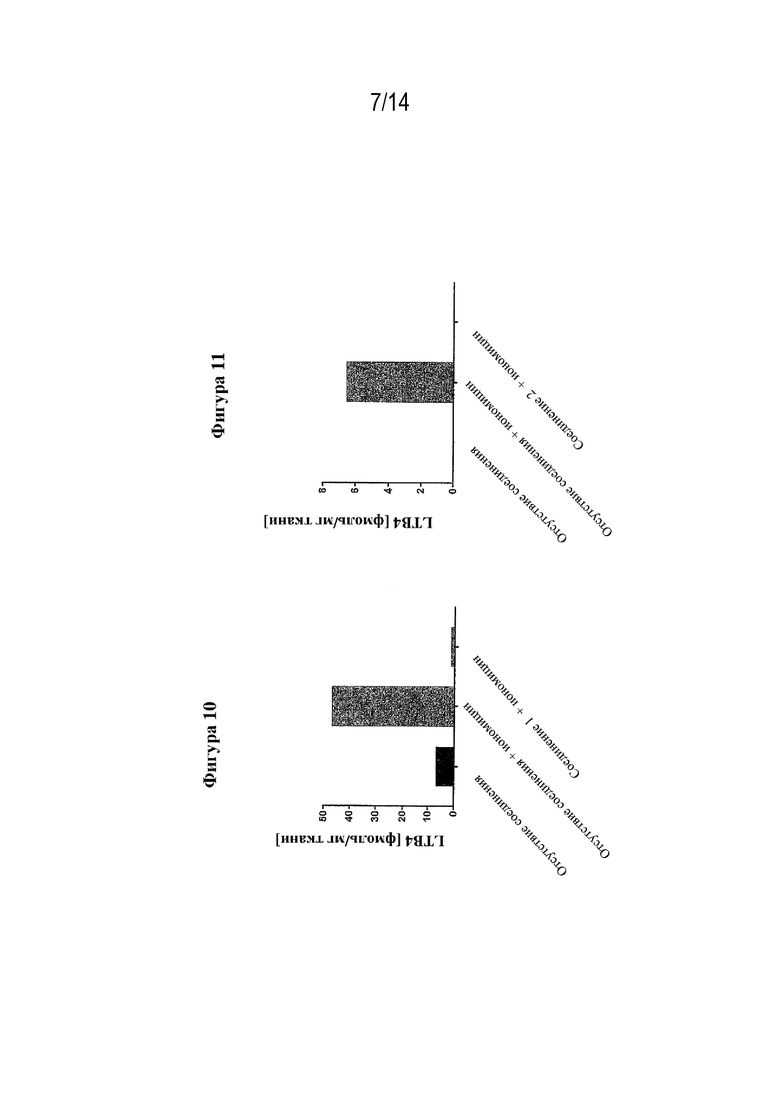
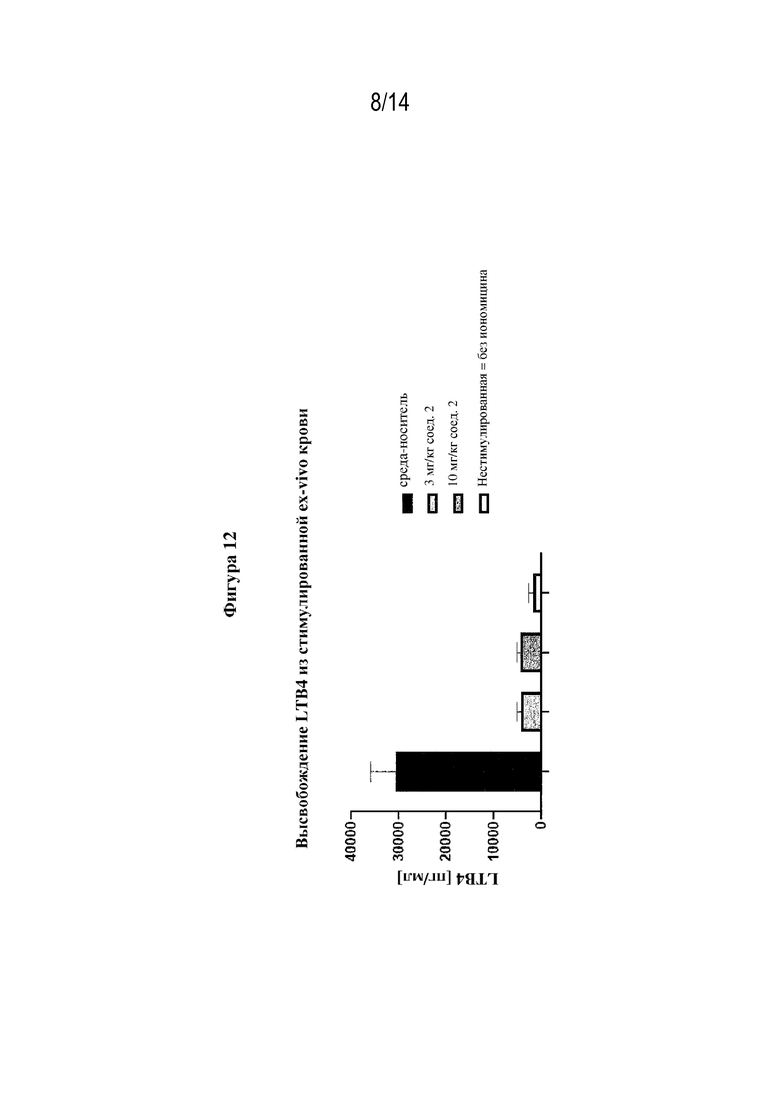
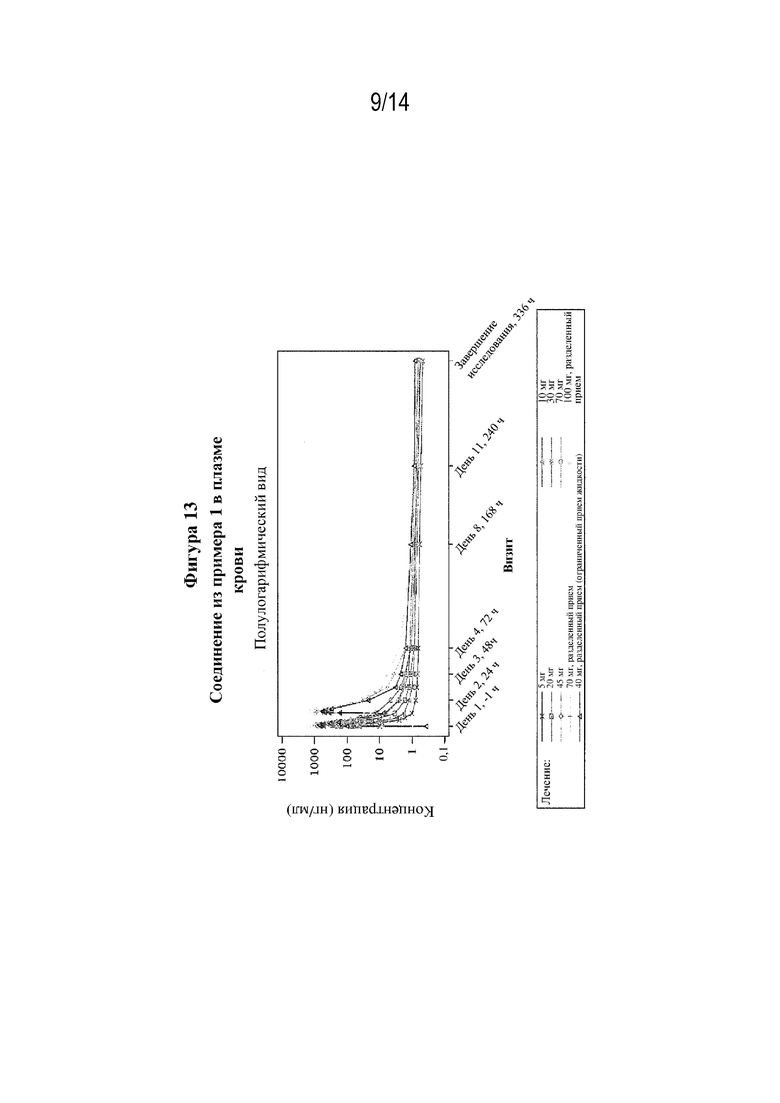
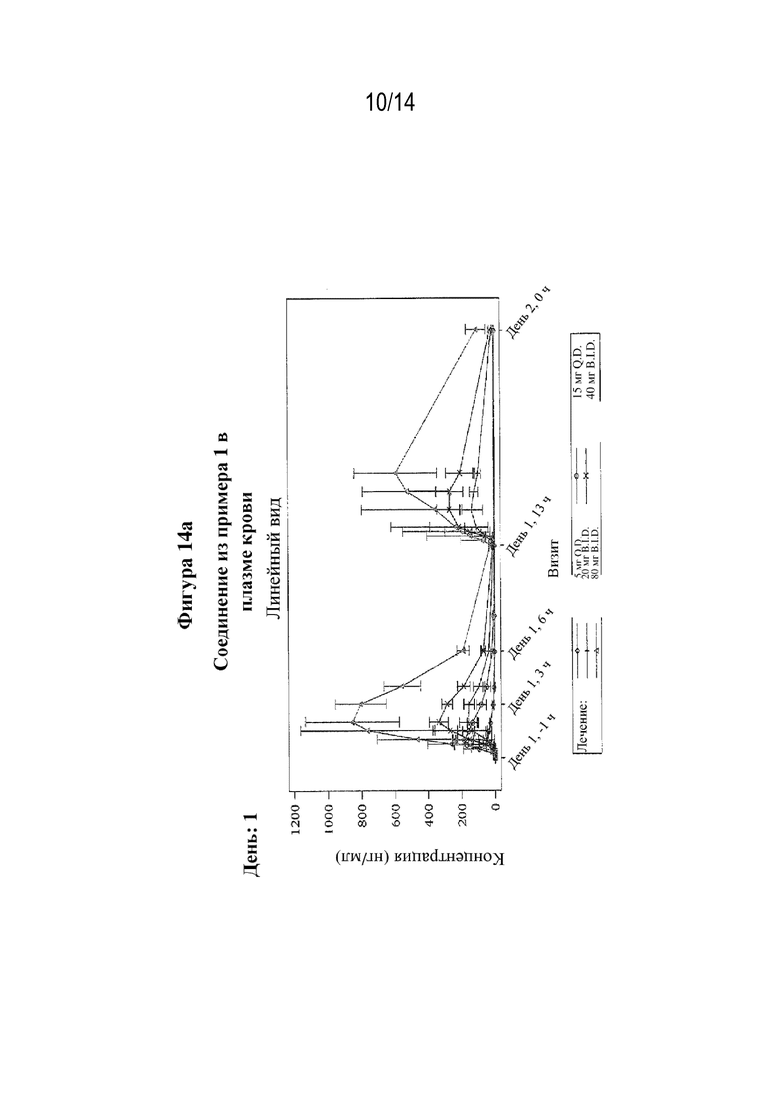
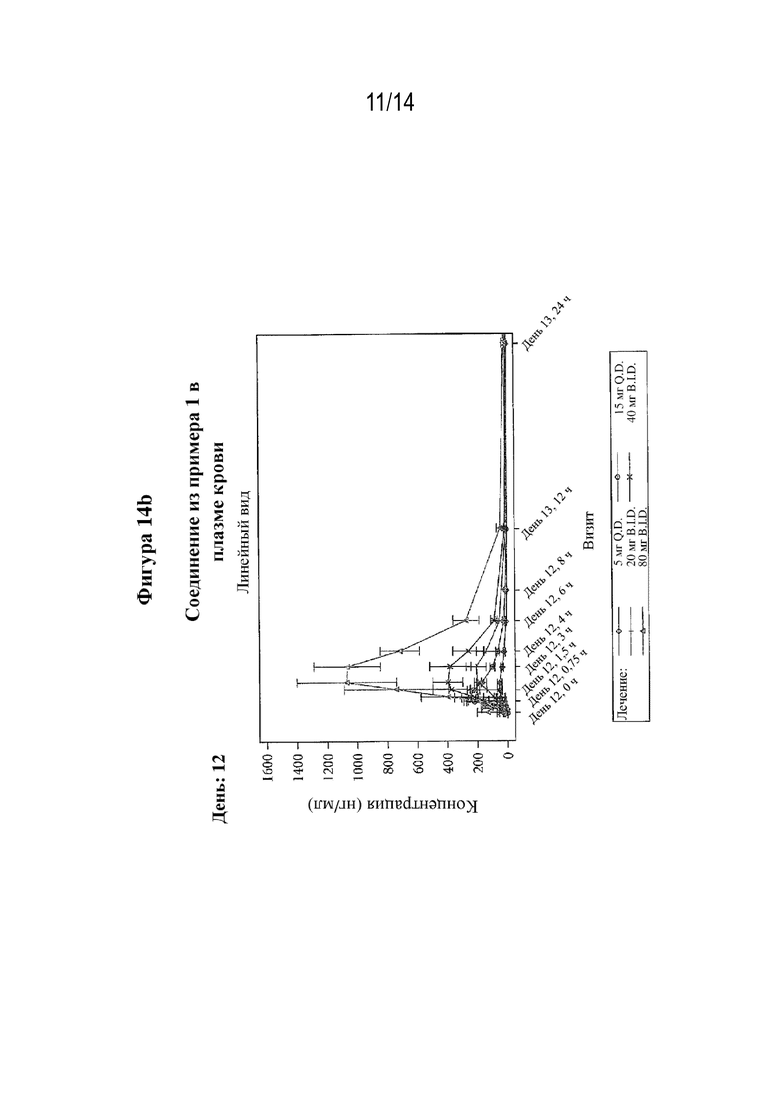
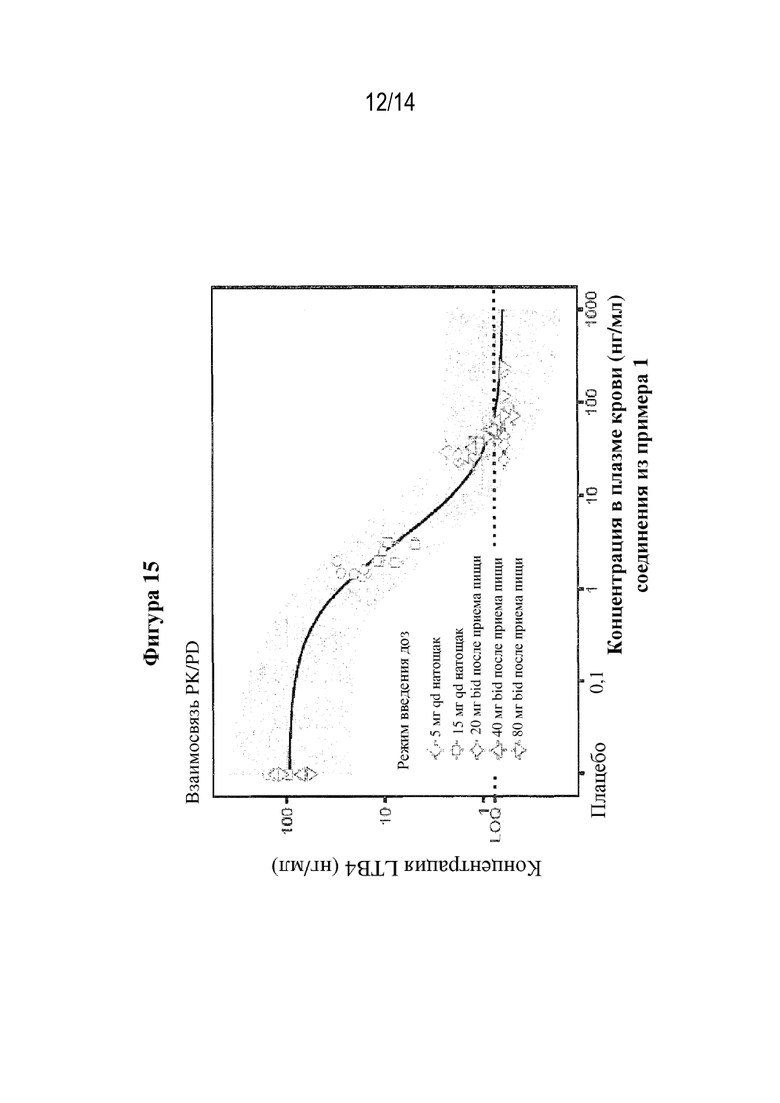
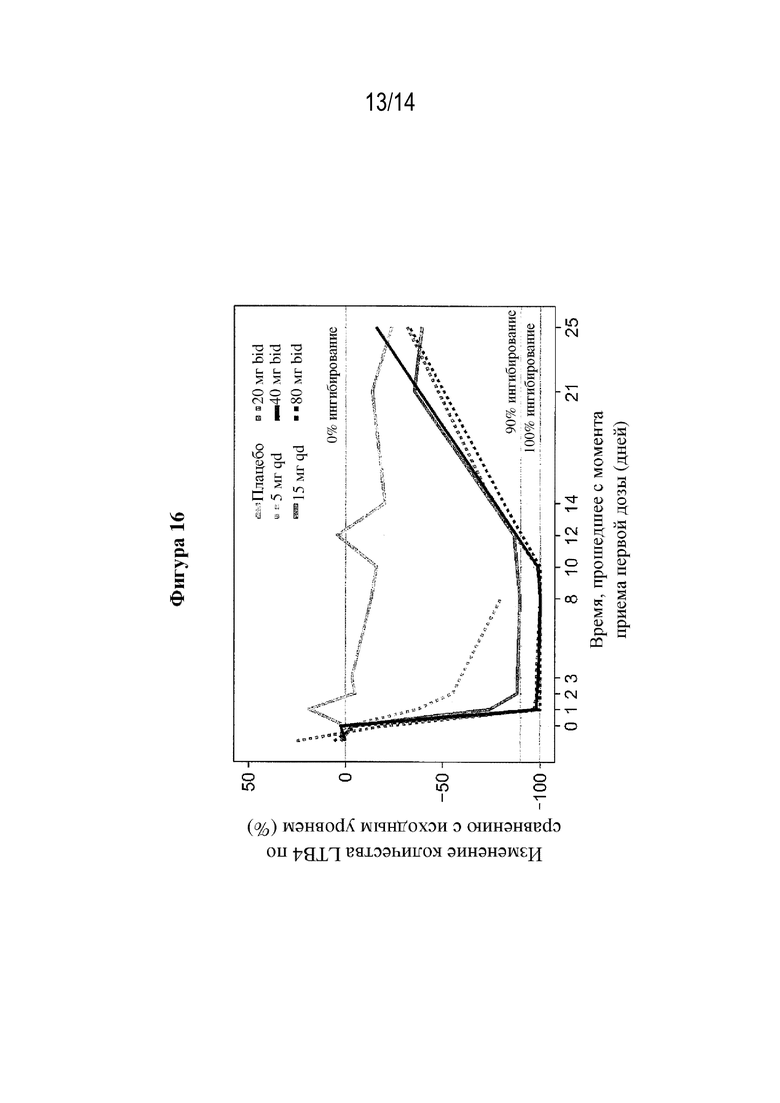
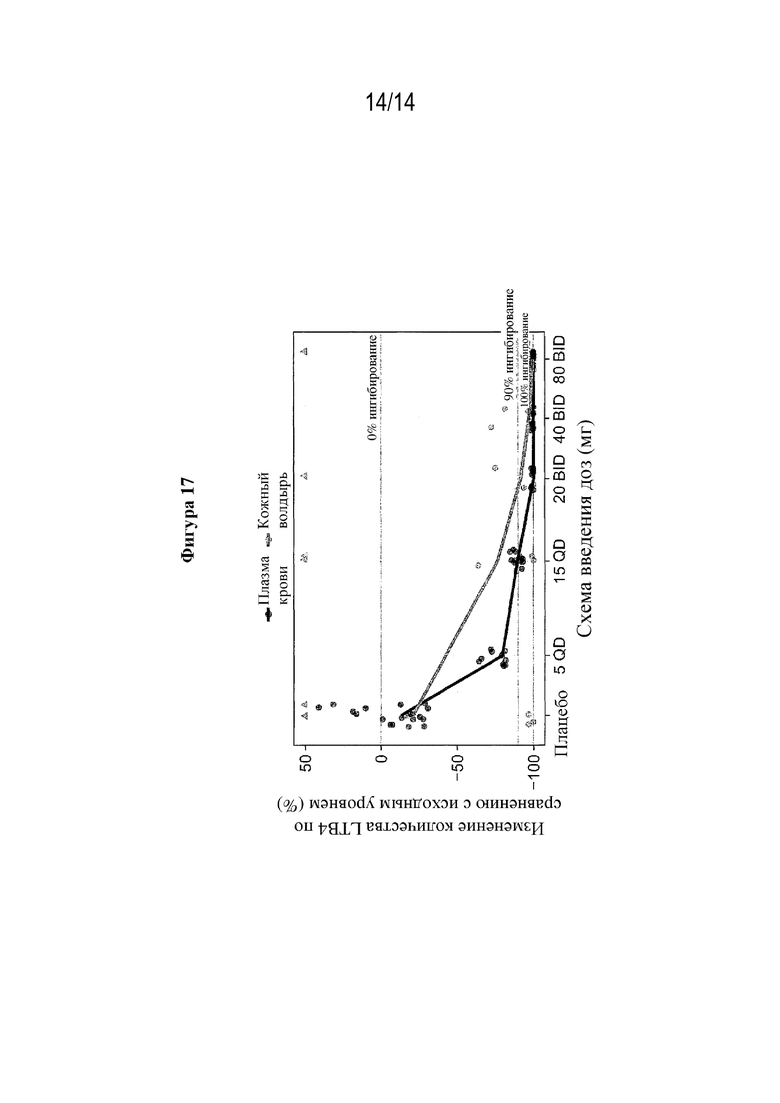
Изобретение относится к способам лечения гнойного гидраденита, а именно к применению ингибитора LTA4H для лечения или предупреждения гнойного гидраденита у пациента, нуждающегося в таком лечении. Указанный ингибитор LTA4H представляет собой (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановую кислоту или ее фармацевтически приемлемую соль; или указанный ингибитор LTA4H представляет собой кристаллическую форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в ее свободной форме, где указанная кристаллическая форма характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические пики при 2θ при 22,6±0,2 °2θ, 24,1±0,2 °2θ и 26,3±0,2 °2θ, измеренных при температуре приблизительно 25°C, и при длине волны рентгеновского излучения, λ, составляющей 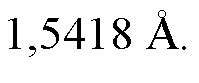 Изобретение также относится к применению фармацевтической композиции на основе указанного соединения или его кристаллической формы. Технический результат заключается в лечении указанным соединением или его кристаллической формой гнойного гидраденита. 2 н. и 8 з.п. ф-лы, 17 ил., 2 табл., 8 пр.
Изобретение также относится к применению фармацевтической композиции на основе указанного соединения или его кристаллической формы. Технический результат заключается в лечении указанным соединением или его кристаллической формой гнойного гидраденита. 2 н. и 8 з.п. ф-лы, 17 ил., 2 табл., 8 пр.
1. Применение ингибитора LTA4H для лечения или предупреждения гнойного гидраденита у пациента, нуждающегося в таком лечении или предупреждении,
где указанный ингибитор LTA4H представляет собой (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановую кислоту или ее фармацевтически приемлемую соль; или указанный ингибитор LTA4H представляет собой кристаллическую форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в ее свободной форме, где указанная кристаллическая форма характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические пики при 2θ при 22,6±0,2 °2θ, 24,1±0,2 °2θ и 26,3±0,2 °2θ, измеренных при температуре приблизительно 25°C, и при длине волны рентгеновского излучения, λ, составляющей 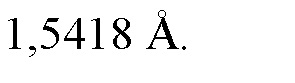
2. Применение ингибитора LTA4H по п. 1, где кристаллическая форма (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в ее свободной форме характеризуется следующими данными порошковой рентгеновской дифракции, XRPD:
3. Применение ингибитора LTA4H по п. 1 или 2, где ингибитор LTA4H помещен в фармацевтический состав, где указанный фармацевтический состав содержит один или несколько фармацевтически приемлемых носителей, каждый из которых независимо выбран из наполнителя, смазывающего вещества, связующего вещества, дезинтегранта и вещества, способствующего скольжению.
4. Применение ингибитора LTA4H по п. 3, где фармацевтический состав находится в форме таблетки или капсулы.
5. Применение фармацевтической композиции, содержащей ингибитор LTA4H, который представляет собой (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановую кислоту, или ее фармацевтически приемлемую соль, или кристаллическую форму (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в ее свободной форме, где указанная кристаллическая форма характеризуется порошковой рентгеновской дифрактограммой, содержащей характеристические пики при 2θ при 22,6±0,2 °2θ, 24,1±0,2 °2θ и 26,3±0,2 °2θ, измеренных при температуре приблизительно 25°C, и при длине волны рентгеновского излучения λ, составляющей  совместно с одним или несколькими фармацевтически приемлемыми носителями, для лечения или предупреждения гнойного гидраденита у пациента, нуждающегося в таком лечении или предупреждении.
совместно с одним или несколькими фармацевтически приемлемыми носителями, для лечения или предупреждения гнойного гидраденита у пациента, нуждающегося в таком лечении или предупреждении.
6. Применение по п. 5, где кристаллическая форма (S)-3-амино-4-(5-(4-((5-хлор-3-фторпиридин-2-ил)окси)фенил)-2H-тетразол-2-ил)бутановой кислоты в ее свободной форме, характеризуется следующими данными порошковой рентгеновской дифракции, XRPD:
7. Применение ингибитора LTA4H по любому из пп. 1-4 или применение фармацевтической композиции по любому из пп. 5, 6, где ингибитор LTA4H вводится в суточной дозе от приблизительно 10 мг до приблизительно 100 мг и где термин «приблизительно» указывает на отклонение ±5%.
8. Применение ингибитора LTA4H по любому из пп. 1-4, 7 и 8, или применение фармацевтической композиции по любому из пп. 5-8, где до лечения с помощью ингибитора LTA4H пациент ранее не был подвергнут лечению с помощью системного средства или местного средства для лечения гнойного гидраденита.
9. Применение ингибитора LTA4H по любому из пп. 1-4, 7 и 8, или применение фармацевтической композиции по любому из пп. 5-8, где пациент выбран в соответствии с одним из следующих критериев:
a) у пациента имеется гнойный гидраденит со степенью тяжести от средней до тяжелой;
b) до лечения с помощью ингибитора LTA4H пациент характеризуется баллом по шкале согласно HS-PGA, который больше или равняется 3;
c) до лечения с помощью ингибитора LTA4H у пациента имеется по меньшей мере 3 воспалительных очага поражения; или
d) до лечения с помощью ингибитора LTA4H у пациента отсутствует обширное рубцевание (менее 10 свищей) вследствие гнойного гидраденита.
10. Применение ингибитора LTA4H по любому из пп.1-4, 7 и 8, или применение фармацевтической композиции по любому из пп. 5-8, где ингибитор LTA4H вводится в дозе от приблизительно 10 мг до приблизительно 30 мг два раза в день и где термин «приблизительно» указывает на отклонение ±5%.
| WO 2018107158 A1, 14.06.2018 | |||
| WO 2018107158 A1, 14.06.2018 | |||
| WO 2018107167 A1, 14.06.2018 | |||
| WO 2018107167 A1, 14.06.2018 | |||
| WO 2015092740 A1, 25.06.2015 | |||
| WO 2014164658 A1, 09.10.2014 | |||
| СПОСОБ ЛЕЧЕНИЯ | 2012 |
|
RU2621148C2 |
| RU 2015137976 A, 20.04.2017 | |||
| Прибор для дезодорации воздуха в помещениях | 1928 |
|
SU17618A1 |

