Область техники, к которой относится изобретение
Настоящее изобретение относится к области медицины и, в частности, относится к аминонорборнановому производному в качестве ингибитора тирозинкиназы Брутона с высокой селективностью в отношении мутанта C481S, к содержащей его фармацевтической композиции, к способу его получения и к их применению при производстве лекарственного препарата.
Уровень техники
Путь передачи сигналов с участием B-клеточного рецептора (BCR) играет ключевую роль в созревании, дифференцировке и развитии B-клеток. Аберрантная BCR-опосредованная передача сигнала может привести к нарушению регуляции активации B-клеток и/или к выработке патогенных аутоантител, что приводит в результате к развитию множества заболеваний у человека, в том числе рака, аутоиммунных заболеваний, в том числе красной волчанки, хронической лимфоцитарной лимфомы, диффузной крупноклеточной лимфомы, фолликулярной лимфомы или хронического лимфоцитарного лейкоза, гетероиммунных заболеваний, в том числе воспалительных заболеваний, астмы и др.
Тирозинкиназа Брутона (BTK) является представителем семейства TEC — нерецепторных тирозинкиназ. Она играет ключевую роль в активации сигнального пути с участием BCR и является ключевым регулятором ранних стадий формирования B-клеток и активации и выживания зрелых B-клеток (Khan et al., Immunity 1995 3:283; Ellmeier et al., J Exp Med 2000 192:1611). BTK играет важную роль в регуляции пролиферации и апоптоза B-клеток (Islam and Smith, Immunol Rev 2000 178:49; Davis et al., Nature 2010 463:88-94), и поэтому ингибирование BTK можно использовать для лечения некоторых форм В-клеточной лимфомы и лейкоза (Feldhahn et al., J Exp Med 2005 201:1837).
Роль BTK в аутоиммунных и воспалительных заболеваниях была подтверждена на модельных мышах с дефицитом BTK. В доклинической мышиной модели системной красной волчанки (SLE) у мышей с дефицитом BTK наблюдается значительное уменьшение прогрессирования заболевания. Кроме того, мыши с дефицитом BTK устойчивы к развитию индуцируемого коллагеном артрита (Jansson and Holmdahl, Clin Exp Immunol 1993 94:459). Селективные ингибиторы BTK на мышиных моделях артрита характеризуются четкой взаимосвязью между дозой и эффектом (Pan et al., Chem.Med.Chem. 2007 2:58-61). В настоящее время проводятся клинические исследования по лечению артрита ингибиторами BTK.
Ибрутиниб (торговое название «имбрувика»), первый вышедший на рынок ингибитор BTK, имеет большой успех, при этом его годовые продажи в 2017 году достигали 2,6 миллиарда долларов США. Однако, как и в случае со многими другими противораковыми лекарственными средствами, у некоторых пациентов имеет место устойчивость к данному лекарственному средству. Было обнаружено, что основной причиной устойчивости к лекарственному средству является мутация C481S в киназе BTK. Ибрутиниб в фармакодинамическом контексте действует путем необратимого ковалентного связывания с остатком триптофана C481 в киназе BTK; однако, поскольку мутация C481S заменяет триптофан на серин, способность ибрутиниба связываться с остатком триптофана утрачивается.
Согласно клинической статистике, мутация BTK(C481S) связана с 87% пациентов с рецидивирующей хронической лимфомой (CLL) (Woyach et al., J Clin Oncol 2017 35:1437-1443) и приблизительно 80% пациентов с рецидивом лимфомы из клеток мантийной зоны (MCL) (Chiron et al., Cancer Discovery 2014 4(9): 1-14). Разработка ингибитора BTK, который эффективен против мутанта BTK(C481S), позволит преодолеть устойчивость к ибрутинибу, возникающую в результате мутации C481S.
Краткое описание изобретения
Техническая задача, которую необходимо решить с помощью настоящего изобретения, заключается в получении нового и нераскрытого соединения, являющегося ингибитором тирозинкиназы Брутона с высокой селективностью в отношении мутанта BTK(C481S), его фармацевтически приемлемой соли, сольвата, активного метаболита, полиморфа, сложного эфира, оптического изомера или пролекарства, в применении такого соединения в фармации и в создании способа предупреждения развития или лечения заболеваний, связанных с чрезмерной активностью BTK, у человека или млекопитающих путем применения раскрываемого в настоящем документе соединения.
Для решения данной технической задачи в настоящем изобретении взята на вооружение представленная далее техническая схема.
Предложено соединение с формулой (I):
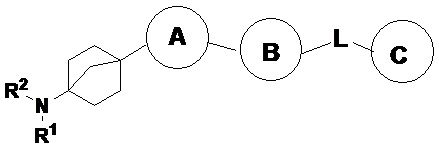
(I),
или его фармацевтически приемлемая соль, сольват, активный метаболит, полиморф, сложный эфир, оптический изомер или пролекарство, где
кольцо A выбрано из одной из следующих структур:
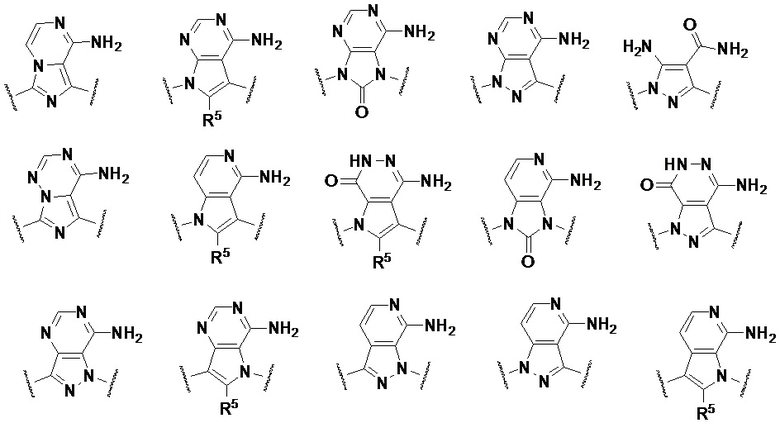
при этом R5 выбран из водорода, галогена, циано, гидроксила, алкинила, амино, C1-3-алкила, C1-3-алкокси, C1-3-галогеналкила, C1-3-галогеналкокси, C1-3-галогеналкиламино, C3-7-циклоалкила, C3-7-циклоалкокси и C3-7-циклоалкиламино;
кольцо B представляет собой замещенное или незамещенное ароматическое кольцо или гетероароматическое кольцо; кольцо C представляет собой замещенное или незамещенное ароматическое кольцо или гетероароматическое кольцо;
L представляет собой одинарную связь или одну из следующих структур:
R1 выбран из R3 и одной из следующих структур:
при этом R3 выбран из водорода, замещенного или незамещенного C1-6-алкила, замещенного или незамещенного C1-6-алкинила, замещенного или незамещенного C1-6-алкенила, замещенного или незамещенного C6-10-арила, замещенного или незамещенного C1-9-гетероарила, замещенного или незамещенного C3-7-циклоалкила и замещенного или незамещенного C2-7-гетероциклоалкиламино; и
при этом R4 выбран из водорода, замещенного или незамещенного C1-6-алкила, замещенного или незамещенного C6-10-арила, замещенного или незамещенного C1-9-гетероарила, замещенного или незамещенного C3-7-циклоалкила и замещенного или незамещенного C3-7-гетероциклоалкила; и
R2 выбран из H, замещенного или незамещенного C1-3-алкила, замещенного или незамещенного C3-7-циклоалкила, замещенного или незамещенного C2-7-гетероциклоалкила, замещенного или незамещенного C6-10-арила и замещенного или незамещенного C1-9-гетероарила.
R1 и R2 вместе с присоединенным к ним N образуют замещенное или незамещенное C2-7-гетероциклическое кольцо, а R3 и R4 вместе с присоединенным к ним N образуют или не образуют C3-7-гетероциклиламино или C3-9-гетероариламино.
В случае R3, предпочтительно, заместитель у замещенного C1-6-алкила, замещенного C1-6-алкинила, замещенного C1-6-алкенила, замещенного C6-10-арила, замещенного C1-9-гетероарила, замещенного C3-7-циклоалкила или замещенного C2-7-гетероциклоалкила выбран из одного или нескольких из галогена, циано, гидроксила, амино, замещенного или незамещенного ациламино, замещенного или незамещенного аминоацила, замещенного или незамещенного C1-4-алкила, замещенного или незамещенного C3-7-циклоалкила, замещенного или незамещенного C3-7-циклоалкокси, замещенного или незамещенного C1-4-алкиламино, ди[замещенного или незамещенного C1-4-алкил]амино, замещенного или незамещенного C3-7-циклоалкиламино, замещенного или незамещенного C3-7-гетероциклоалкиламино, замещенного или незамещенного C1-3-алкокси, замещенного или незамещенного C3-7-циклоалкокси, замещенного или незамещенного C6-10-арила и замещенного или незамещенного C3-7-гетероциклоалкила.
В случае R4, предпочтительно, заместитель у замещенного C1-6-алкила, замещенного C6-10-арила, замещенного C1-9-гетероарила, замещенного C3-7-циклоалкила или замещенного C3-7-гетероциклоалкила выбран из одного или нескольких из галогена, гидроксила, циано, амино, замещенного или незамещенного C1-4-алкенила, замещенного или незамещенного C3-7-циклоалкила, замещенного или незамещенного C3-7-циклоалкокси, замещенного или незамещенного C1-4-алкиламино, ди[замещенного или незамещенного C1-4-алкил]амино, замещенного или незамещенного C3-7-циклоалкиламино, замещенного или незамещенного C3-7-гетероциклоалкиламино, замещенного или незамещенного C1-3-алкокси, замещенного или незамещенного C3-7-циклоалкокси, замещенного или незамещенного C6-10-арила и замещенного или незамещенного C3-7-гетероциклоалкила.
Предпочтительно, кольцо A представляет собой одну из следующих структур:
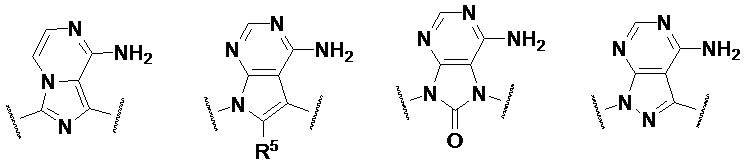
при этом R5 выбран из водорода, галогена, циано, гидроксила, алкинила, амино, C1-3-алкила, C1-3-алкокси, C1-3-галогеналкила, C1-3-галогеналкокси, C1-3-галогеналкиламино, C3-7-циклоалкила, C3-7-циклоалкокси и C3-7-циклоалкиламино;
в случае кольца B, предпочтительно, заместитель у замещенного ароматического кольца или гетероароматического кольца выбран из одного или нескольких из галогена, гидроксила, циано, амино, C1-3-алкила, C1-3-алкокси, C1-3-галогеналкила и C1-3-галогеналкокси.
Предпочтительно, кольцо B представляет собой одну из следующих структур:
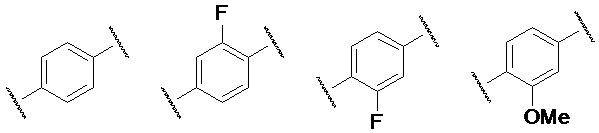
В случае кольца С, предпочтительно, заместитель у замещенного ароматического кольца или гетероароматического кольца выбран из одного или нескольких из галогена, гидроксила, циано, амино, C1-3-алкила, C1-3-алкокси, C1-3-галогеналкила и C1-3-галогеналкокси.
Предпочтительно, кольцо C представляет собой одну из следующих структур:
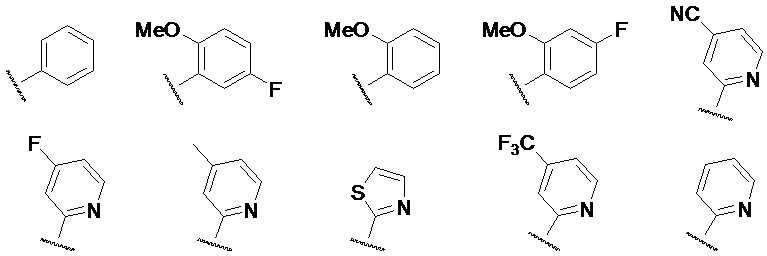
Предпочтительно, R2 выбран из H, а R1 выбран из 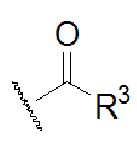 , где R3 выбран из водорода, замещенного или незамещенного C1-6-алкила, замещенного или незамещенного C1-6-алкинила, замещенного или незамещенного C1-6-алкенила, замещенного или незамещенного C6-10-арила, замещенного или незамещенного C1-9-гетероарила, замещенного или незамещенного C3-7-циклоалкила и замещенного или незамещенного C2-7-гетероциклоалкиламино.
, где R3 выбран из водорода, замещенного или незамещенного C1-6-алкила, замещенного или незамещенного C1-6-алкинила, замещенного или незамещенного C1-6-алкенила, замещенного или незамещенного C6-10-арила, замещенного или незамещенного C1-9-гетероарила, замещенного или незамещенного C3-7-циклоалкила и замещенного или незамещенного C2-7-гетероциклоалкиламино.
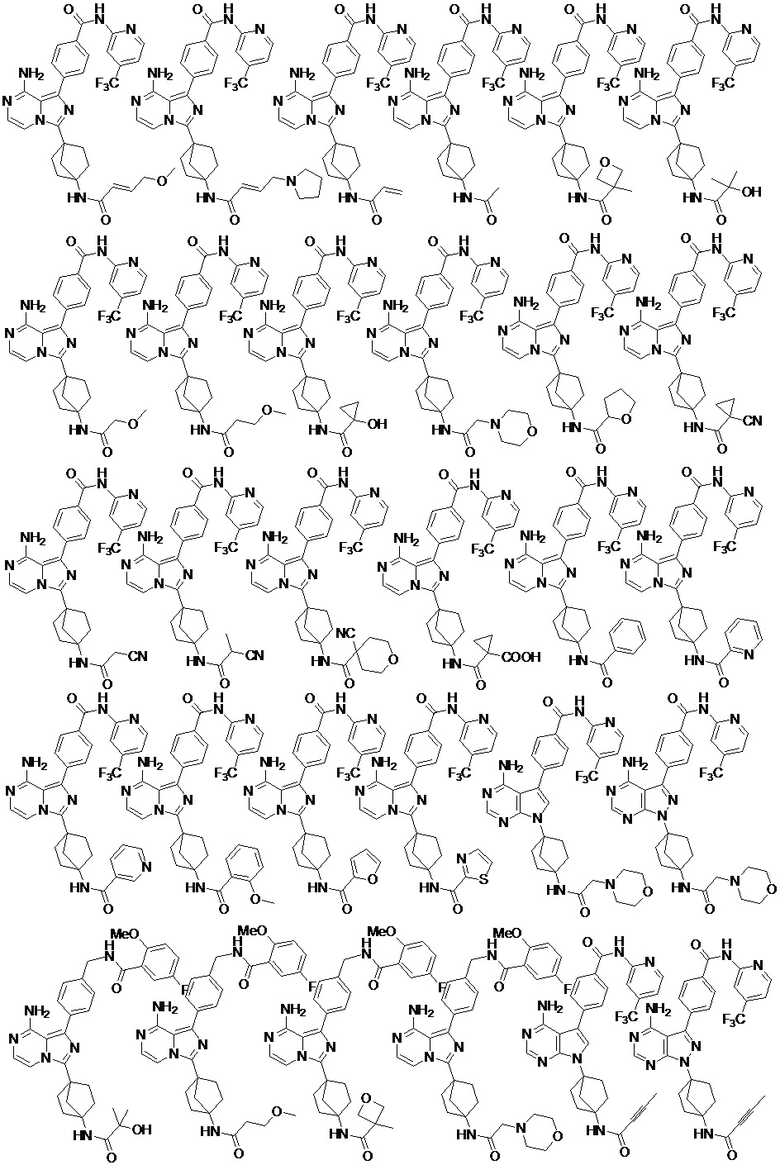
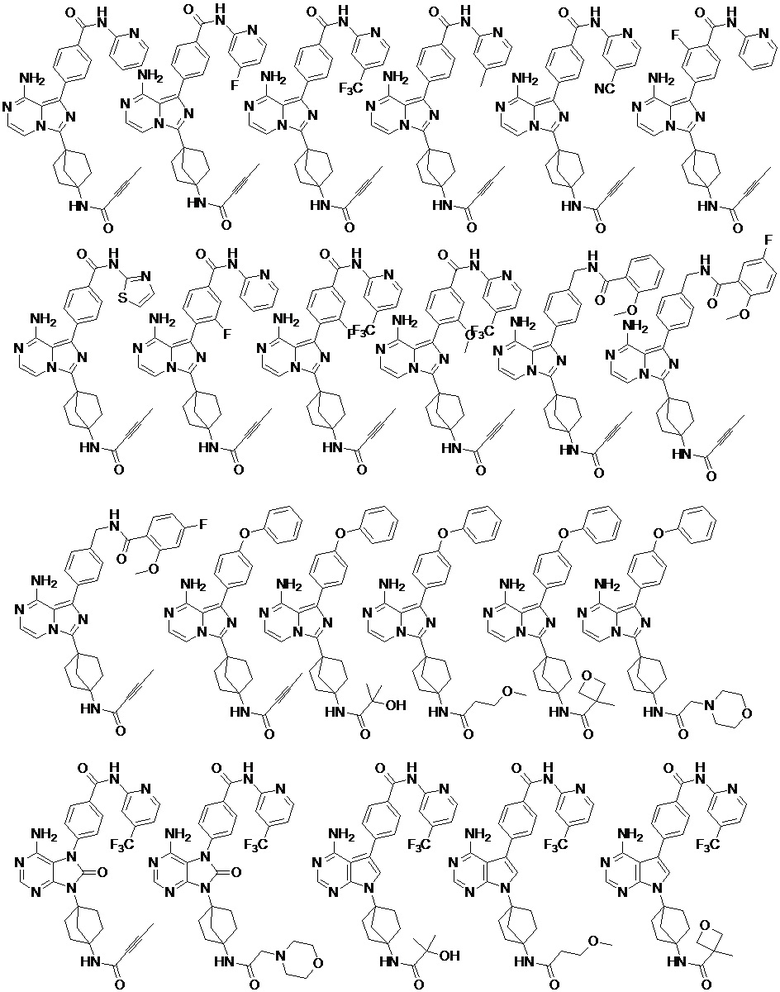
Наиболее предпочтительно, соединение выбрано из любой из следующих структур:
Предложено применение соединения по любому из приведенных выше аспектов при производстве лекарственного препарата для предупреждения развития или лечения гетероиммунного заболевания, аутоиммунного заболевания или рака,
где гетероиммунное заболевание, аутоиммунное заболевание или рак связаны с чрезмерной активностью тирозинкиназы Брутона, или
гетероиммунное заболевание, аутоиммунное заболевание или рак связаны с аберрантной пролиферацией B-клеток.
Кроме того, гетероиммунное заболевание представляет собой воспалительное заболевание или астму.
Кроме того, аутоиммунное заболевание представляет собой красную волчанку, хроническую лимфоцитарную лимфому, диффузную крупноклеточную лимфому, фолликулярную лимфому или хронический лимфолейкоз.
Предложена фармацевтическая композиция, содержащая одно или несколько соединений согласно любому из приведенных выше аспектов.
Предложен фармацевтический состав, содержащий терапевтически эффективное количество соединения согласно любому из приведенных выше аспектов и фармацевтически приемлемое вспомогательное вещество.
Фармацевтический состав составлен для перорального введения, парентерального введения, трансбуккального введения, назального введения, местного применения или ректального введения.
Фармацевтический состав предназначен для применения при лечении заболевания или патологического состояния, связанного с чрезмерной активностью тирозинкиназы Брутона, предусматривающего введение фармацевтического состава нуждающемуся в том человеку или млекопитающему; заболевание, связанное с чрезмерной активностью тирозинкиназы Брутона, представляет собой гетероиммунное заболевание, аутоиммунное заболевание или рак; гетероиммунное заболевание представляет собой воспалительное заболевание или астму; аутоиммунное заболевание представляет собой красную волчанку, хроническую лимфоцитарную лимфому, диффузную крупноклеточную лимфому, фолликулярную лимфому или хронический лимфолейкоз.
Настоящим изобретением предусмотрена стадия приведения фармацевтического состава в контакт с BTK, которая предусматривает анализ in vitro или in vivo.
Способ 1 получения указанного выше соединения I предусматривает: (S1) осуществление реакции сочетания Сузуки соединения IIIA с бороновой кислотой или боратом II с получением соединения IV; (S2) превращение соединения IV в гидрохлорид соединения V путем удаления бензилоксикарбонила трифторуксусной кислотой; и (S3) сочетание соединения V с органической кислотой с получением описанного выше соединения I;
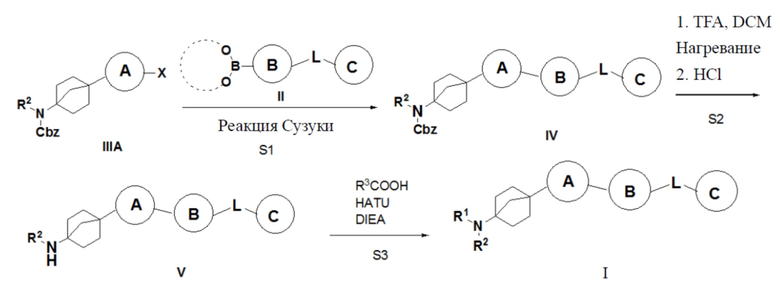
где X = галоген, а R2, R3, L, кольцо A, кольцо B и кольцо C являются такими, как описано выше.
Способ 2 получения указанного выше соединения I предусматривает: (A1) превращение соединения IIIA в гидрохлорид соединения VI путем удаления бензилоксикарбонила трифторуксусной кислотой; (A2) сочетание соединения VI с органической кислотой с получением соединения VII; и (A3) осуществление реакции сочетания Сузуки соединения VII с бороновой кислотой или боратом II с получением описанного выше соединения I;
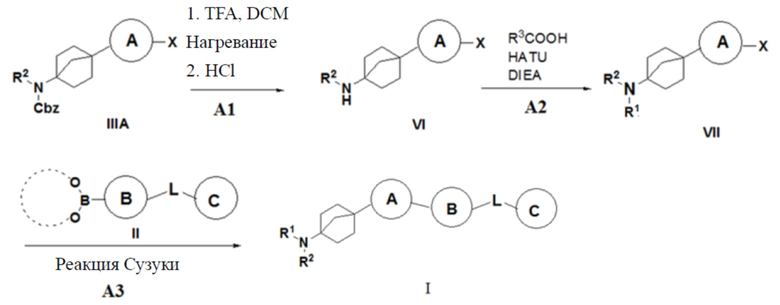
где X = галоген, а R2, R3, L, кольцо A, кольцо B и кольцо C являются такими, как описано выше.
Способ 3 получения указанного выше соединения I предусматривает: (B1) осуществление реакции сочетания Чана-Лама-Эванса соединения IIIB с бороновой кислотой II в присутствии катализа ацетатом меди с получением соединения VIII; (B2) превращение соединения VIII в гидрохлорид соединения IX путем удаления бензилоксикарбонила трифторуксусной кислотой; и (B3) сочетание соединения IX с органической кислотой с получением описанного выше соединения I;
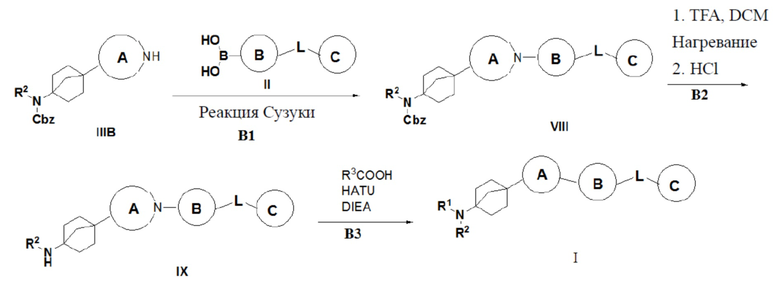
где R2, R3, L, кольцо A, кольцо B и кольцо C являются такими, как описано выше.
Каждый из продуктов, образующихся в результате реакций в способах 1, 2 и 3, можно получить с помощью традиционных методик разделения, в том числе без ограничения путем фильтрации, дистилляции, кристаллизации, хроматографического разделения и тому подобного. Исходные материалы, необходимые для синтеза, можно синтезировать самостоятельно или приобрести в коммерческих учреждениях, таких как Adrich или Sigma. Эти материалы можно охарактеризовать с помощью таких традиционных средств, как физические константы и спектральные данные. Описываемые в настоящем документе соединения можно синтезировать с получением одного оптического изомера или смеси оптических изомеров.
Верхними индексами букв в настоящем изобретении указан порядковый номер группы, а нижними индексами указано количество атомов. Например: R1, R2 и R3 обозначают с 1-ой по 3-ю группы R, а C1-4-алкил обозначает алкил, содержащий 1-4 атома C. Количество атомов C в заместителе не учитывается в основной цепи.
Краткое описание чертежей
На фиг. 1 представлена ксенотрансплантатная модель опухоли OCI-LY10.
На фиг. 2 представлена ксенотрансплантатная модель опухоли TMD-8.
Подробное описание изобретения
Настоящее изобретение можно лучше понять из приведенных далее примеров. Тем не менее, специалисты в настоящей области техники без труда поймут, что описанное в примерах используется лишь для иллюстрации настоящего изобретения, и оно не должно и не будет ограничивать настоящее изобретение, которое подробно описано в формуле изобретения.
Синтез промежуточного соединения I-5
Путь синтеза промежуточного соединения I-5
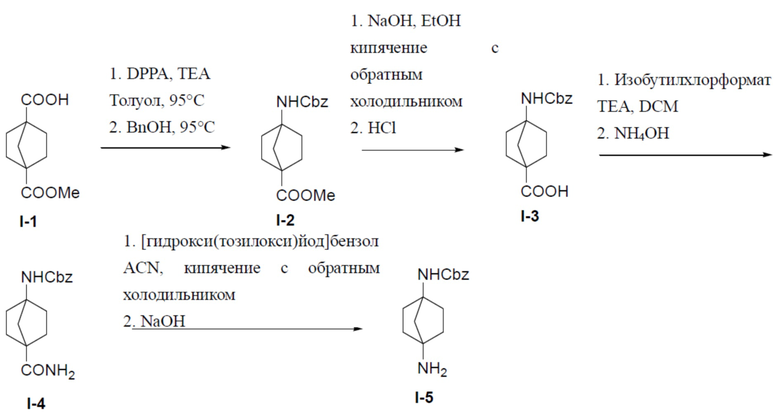
I-2: метил-4-(((бензилокси)карбонил)амино)бицикло[2.2.1]гептан-1-карбоксилат
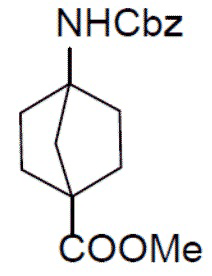
К раствору 4-(метоксикарбонил)бицикло[2.2.1]гептан-1-карбоновой кислоты (I-1) (3,5 г, 17,7 ммоль) и TEA (1,78 г, 17,7 ммоль) в толуоле (30 мл) добавляли DPPA (5,34 г, 19,5 ммоль). Смесь нагревали до 90°C и выдерживали при этой температуре в течение 2 часов. После охлаждения до комнатной температуры к смеси добавляли BnOH (1,9 г, 17,7 ммоль). Полученную в результате смесь перемешивали при температуре 90°С в течение 4 дней. После охлаждения до комнатной температуры смесь разводили этилацетатом и промывали водным раствором NaHCO3. Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали. Остаток очищали с помощью колоночной хроматографии (этилацетат/простой петролейный эфир = 1: 4) с получением искомого соединения I-2 (3 г, выход: 56%).
I-3: 4-(((бензилокси)карбонил)амино)бицикло[2.2.1]гептан-1-карбоновая кислота
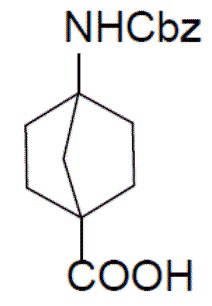
К раствору метил-4-(((бензилокси)карбонил)амино)бицикло[2.2.1]гептан-1-карбоксилата (I-2) (3 г, 9,9 ммоль) добавляли NaOH (792 мг, 19,8 ммоль) и смесь нагревали до 60°C и выдерживали при этой температуре в течение 10 часов. Смесь концентрировали, добавляли к ней воду (50 мл), а затем добавляли 1 н. водный раствор HCl для доведения pH до 4. Для экстракции добавляли этилацетат (20 мл × 3), а органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали с получением искомого соединения I-3 (2,2 г, выход: 77%), который непосредственно использовали на следующей стадии без очистки.
I-4: бензил-(4-карбамоилбицикло[2.2.1]гептан-1-ил)карбамат
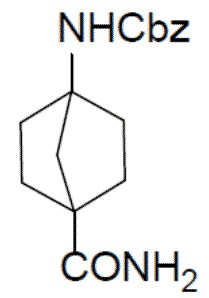
К раствору соединения I-3 (2 г, 6,9 ммоль) и Et3N (1 г, 10 ммоль) в DCM (20 мл) по каплям при 0°C добавляли изобутилхлорформиат (1,36 г, 10 ммоль). Смесь перемешивали при 0°C в течение 20 минут, по каплям добавляли NH4OH (10 мл) и затем перемешивали при комнатной температуре в течение 10 минут. Полученную смесь выливали в воду (30 мл) и отделяли органическую фазу. Водный раствор подвергали экстракции посредством DCM (15 мл ×2) и объединенные органические фазы сушили над Na2SO4, подвергали фильтрации и концентрировали. Остаток очищали колоночной хроматографией (элюент: EA/PE (1:1)–EA/MeOH (10:1)) с получением искомого соединения I-4 (1,7 г, выход: 85%).
I-5: бензил-(4-аминобицикло[2.2.1]гептан-1-ил)карбамат
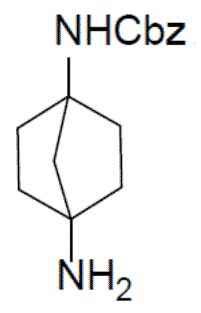
Раствор соединения I-4 (1,6 г, 5,55 ммоль) и [гидрокси(тозилокси)иод]бензола (2,17 г, 5,55 ммоль) в ACN (20 мл) кипятили в колбе с обратным холодильником в течение 1 часа. Выпаривали растворитель и добавляли 1 М NaOH (12 мл). Добавляли EA (15 мл × 2) для экстракции и органическую фазу сушили над Na2SO4, подвергали фильтрации и концентрировали. Остаток очищали колоночной хроматографией (элюент: DCM/MeOH = 10:1) с получением искомого соединения I-5 (920 мг, выход: 64%). LC-MS m/z = 261,1 [M+1]+.
Синтез промежуточной бороновой кислоты II
Путь синтеза промежуточного соединения II-1
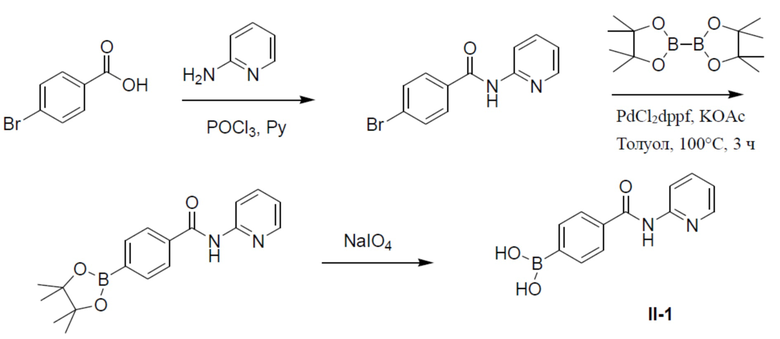
4-бром-N-(пиридин-2-ил)бензамид
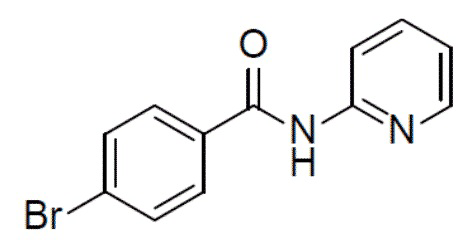
К смеси 4-бромбензойной кислоты (5 г, 24,8 ммоль) и пиридин-2-амина (4,68 г, 49 ммоль) в пиридине (30 мл) на ледяной бане по каплям добавляли POCl3 (11,4 г, 74 ммоль). Суспензию перемешивали при комнатной температуре в течение 20 мин. Реакционную смесь выливали в воду (100 мл) и подвергали экстракции этилацетатом (40 мл ×3). Органическую фазу промывали насыщенным водным раствором NaCl (50 мл ×2), сушили над безводным Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:9-1:1) с получением продукта 4-бром-N-(пиридин-2-ил)бензамид (3,28 г, 48%). LC-MS m/z = 277,0 [M+1]+.
N-(пиридин-2-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензамид
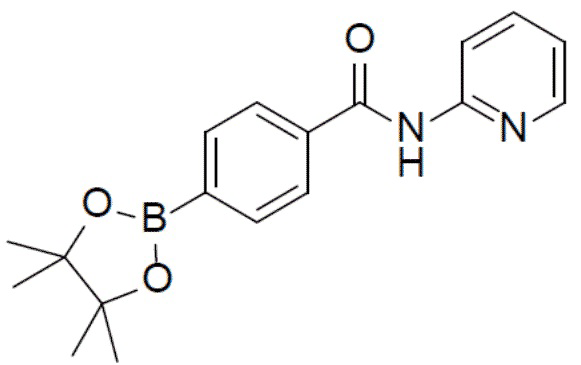
Смесь 4-бром-N-(пиридин-2-ил)бензамида (2 г, 7,22 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (2,75 г, 10,83 ммоль), PdCl2(dppf) (527 мг, 0,72 ммоль) и KOAc (235 мг, 2,4 ммоль) в толуоле (30 мл) нагревали до 110°C и выдерживали ее при данной температуре в течение 6 ч. Реакционную смесь упаривали и добавляли к ней воду (100 мл). Для экстракции добавляли этилацетат (40 мл × 2). Органическую фазу отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:4-1:1) с получением продукта N-(пиридин-2-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензамид (2 г, 85%).
II-1: (4-(пиридин-2-илкарбамоил)фенил)бориновая кислота
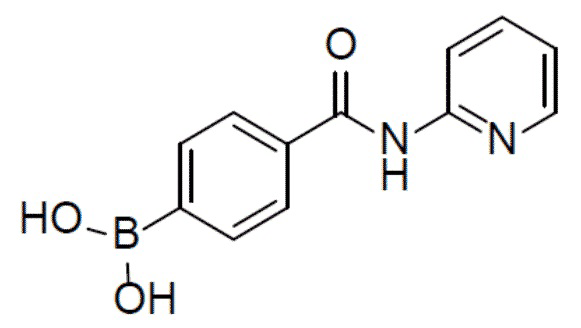
К раствору N-(пиридин-2-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензамида (2 г, 6,2 ммоль) в смешанном растворителе THF:H2O (24 мл:6 мл) добавляли NaIO4 (3,27 г, 18,6 ммоль) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли 2 н. водный раствор HCl (1,65 мл) и полученную в результате смесь перемешивали при комнатной температуре в течение 3 ч. Смесь разводили этилацетатом и промывали солевым раствором. Органическую фазу отделяли, сушили над безводным Na2SO4, подвергали фильтрации и концентрировали. Остаток очищали колоночной хроматографией (MeOH/DCM = 1:10) с получением продукта (4-(пиридин-2-илкарбамоил)фенил)бориновая кислота (II-1) (1,4 г, 93%). LC-MS m/z = 243,1 [M+1]+.
Путь синтеза промежуточного соединения II-2
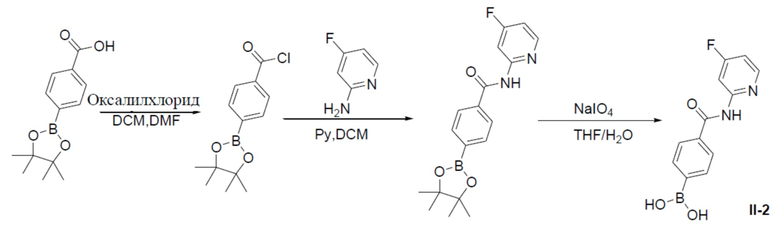
4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоилхлорид
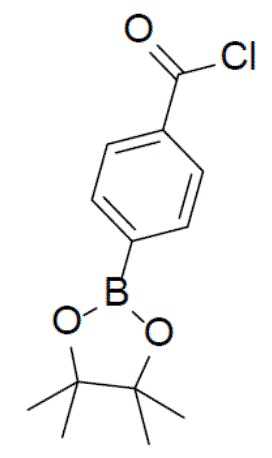
К раствору 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензойной кислоты (10 г, 40 ммоль) и 1 капли DMF в DCM (100 мл) на ледяной бане по каплям добавляли оксалилхлорид (10,2 г, 80 ммоль). Смесь перемешивали при 0°C в течение 30 минут, а затем нагревали до комнатной температуры и выдерживали при данной температуре в течение 3 ч. Затем смесь концентрировали с получением продукта, который непосредственно использовали на следующей стадии без очистки.
N-(4-фторпиридин-2-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензамид
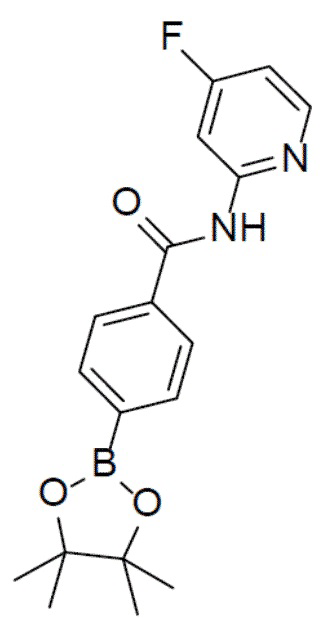
К раствору 4-фторпиридин-2-амина (421 мг, 3,76 ммоль) в пиридинe (3 мл) добавляли раствор 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)бензоилхлорида (1 г, 3,76 ммоль) в DCM (6 мл) и данную суспензию перемешивали при 0°C в течение 30 минут. Смесь выливали в воду и подвергали экстракции посредством DCM (20 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4 и упаривали, а остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:9) с получением продукта (1,04 г, 81%). LC-MS m/z = 343,2 [M+1]+.
II-2: (4-((4-фторпиридин-2-ил)карбамоил)фенил)бориновая кислота
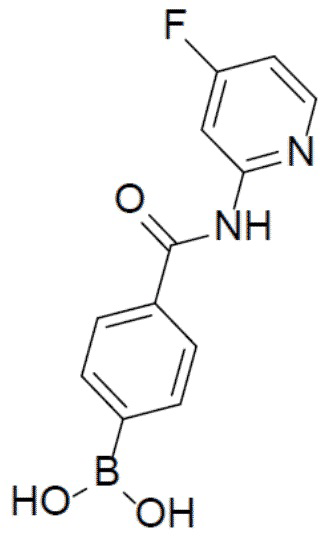
К раствору N-(4-фторпиридин-2-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензамида (1,04 г, 3,04 ммоль) в смешанном растворителе THF:H2O (24 мл:6 мл) добавляли NaIO4 (1,9 г, 9,12 ммоль) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли водный раствор HCl (1,65 мл) и полученную в результате смесь перемешивали при комнатной температуре в течение 3 ч. Смесь разводили этилацетатом и промывали солевым раствором. Органическую фазу отделяли, сушили над безводным Na2SO4, подвергали фильтрации и концентрировали. Остаток очищали колоночной хроматографией (MeOH/DCM = 1:10) с получением продукта II-2 (648 мг, 82%). LC-MS m/z = 261,1 [M+1]+.
II-3: (4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)бориновая кислота
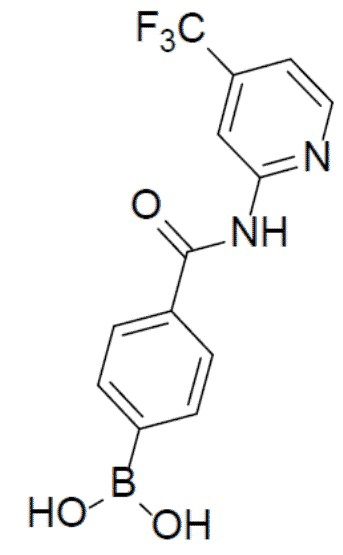
С 4-(трифторметил)пиридин-2-амином (609 мг, 3,76 ммоль) и 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)бензоилхлоридом (1 г, 3,76 ммоль) в качестве исходных материалов использовали тот же способ синтеза, что и для II-2, с получением искомого соединения (607 мг). LC-MS m/z = 311,1 [M+1]+.
II-4: (4-((4-метилпиридин-2-ил)карбамоил)фенил)бориновая кислота
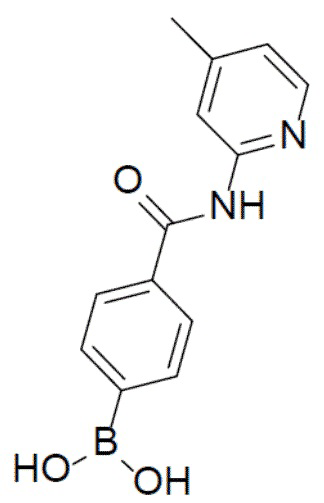
С 4-метил-пиридин-2-амином (406 мг, 3,76 ммоль) и 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)бензоилхлоридом (1 г, 3,76 ммоль) в качестве исходных материалов использовали тот же способ синтеза, что и для II-2, с получением искомого соединения (589 мг). LC-MS m/z = 257,1 [M+1]+.
II-5: (4-((4-цианопиридин-2-ил)карбамоил)фенил)бориновая кислота
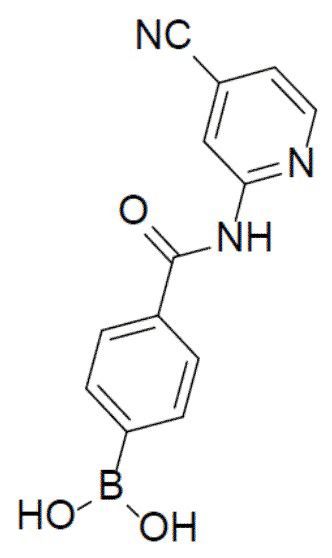
С 4-цианопиридин-2-амином (447 мг, 3,76 ммоль) и 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)бензоилхлоридом (1 г, 3,76 ммоль) в качестве исходных материалов использовали тот же способ синтеза, что и для II-2, с получением искомого соединения (465 мг). LC-MS m/z = 268,0 [M+1]+.
Путь синтеза промежуточного соединения II-6
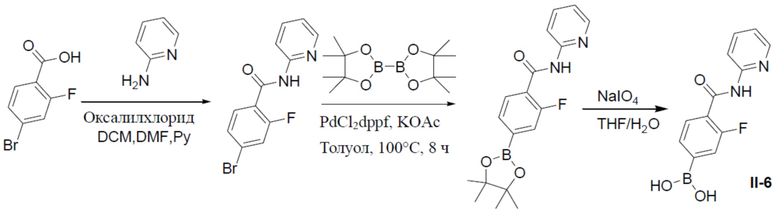
4-бром-2-фтор-N-(пиридин-2-ил)бензамид
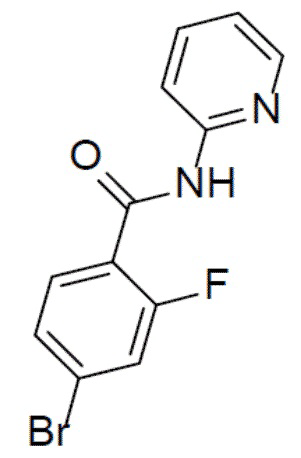
К раствору 4-бром-2-фторбензойной кислоты (1 г, 4,56 ммоль) в DCM (30 мл) на ледяной бане по каплям добавляли оксалилхлорид (1,16 г, 9,13 ммоль), а затем добавляли 1 каплю DMF. После перемешивания при комнатной температуре в течение 3 ч смесь концентрировали и растворяли в DCM (6 мл). Полученный в результате раствор добавляли к раствору пиридин-2-амина (428 мг, 4,56 ммоль) в пиридинe (3 мл) при 0°C и данную суспензию перемешивали при 0°C в течение 30 минут. Смесь выливали в воду и подвергали экстракции посредством DCM (20 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4 и упаривали, а остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:9) с получением продукта (1,05 г, 78%). LC-MS m/z = 295,0 [M+1]+.
2-фтор-N-(пиридин-2-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензамид
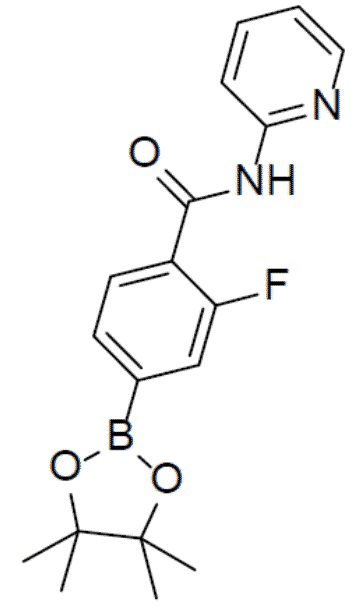
Раствор 4-бром-2-фтор-N-(пиридин-2-ил)бензамида (1,05 г, 3,55 ммоль), (4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,3,2-диоксаборолана (1,36 г, 5,3 ммоль), PdCl2(dppf) (260 мг, 0,36 ммоль) и KOAc (1,04 г, 10,65 ммоль) в толуоле (30 мл) нагревали до 110°C и выдерживали при данной температуре в течение 6 ч. Реакционную смесь упаривали, а затем добавляли к ней воду (100 мл). Для экстракции добавляли этилацетат (40 мл × 2). Органическую фазу отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:4-1:1) с получением продукта (971 мг, 80%).
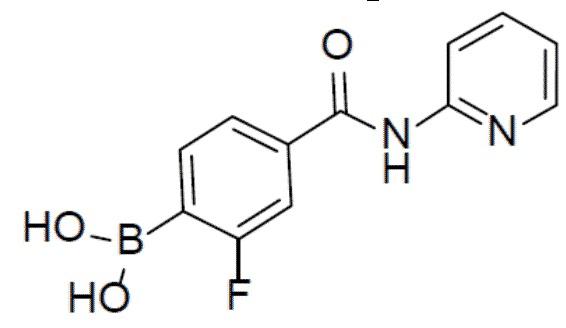
II-6: (3-фтор-4-(пиридин-2-илкарбамоил)фенил)бориновая кислота
К раствору 2-фтор-N-(пиридин-2-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензамида (970 мг, 2,84 ммоль) в смешанном растворителе THF:H2O (24 мл:6 мл) добавляли NaIO4 (1,8 г, 8,52 ммоль) и смесь перемешивали при комнатной температуре в течение 30 мин. Затем добавляли водный раствор HCl (1,65 мл). После перемешивания при комнатной температуре в течение 3 ч смесь разводили этилацетатом и промывали солевым раствором. Органическую фазу отделяли, сушили над безводным Na2SO4, подвергали фильтрации и концентрировали. Остаток очищали колоночной хроматографией (MeOH/DCM = 1:10) с получением продукта II-6 (605 мг, 82%). LC-MS m/z = 261,1 [M+1]+.
II-7: (4-(тиазол-2-илкарбамоил)фенил)бориновая кислота
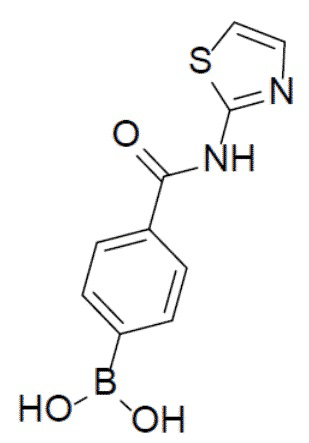
С тиазол-2-амином (376 мг, 3,76 ммоль) и 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)бензоилхлоридом (1 г, 3,76 ммоль) в качестве исходных материалов использовали тот же способ синтеза, что и для II-2, с получением искомого соединения II-7 (580 мг). LC-MS m/z = 249,1 [M+1]+.
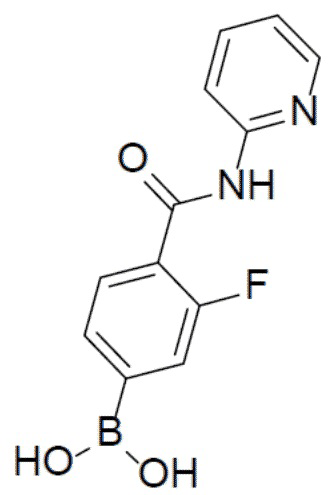
II-8: (2-фтор-4-(пиридин-2-илкарбамоил)фенил)бориновая кислота
С 4-бром-3-фторбензойной кислотой и пиридин-2-амином в качестве исходных материалов использовали тот же способ синтеза, что и для II-1, с получением искомого соединения II-8 (138 мг). LC-MS m/z = 261,0 [M+1]+.
II-9: (2-фтор-4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)бориновая кислота
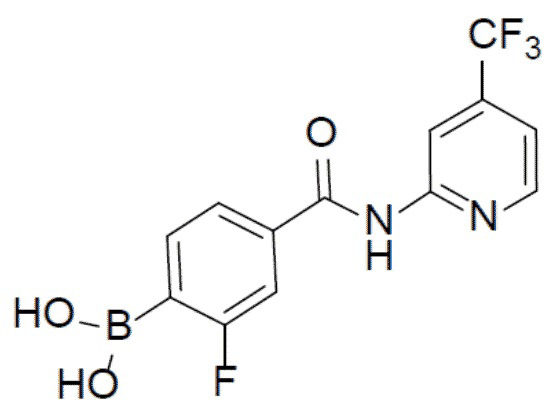
С 4-бром-3-фтор-бензойной кислотой и 4-(трифторметил)-пиридин-2-амином в качестве исходных материалов использовали тот же способ синтеза, что и для II-1, с получением искомого соединения II-9 (130 мг). LC-MS m/z = 329,0 [M+1]+.
II-10: (2-метокси-4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)бориновая кислота
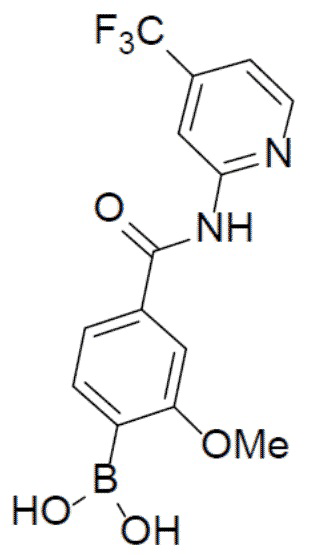
С 4-бром-3-метоксибензойной кислотой (1 г, 4,36 ммоль) и 4-(трифторметил)-пиридин-2-амином (706 мг, 4,36 ммоль) в качестве исходных материалов использовали тот же способ синтеза, что и для II-6, с получением искомого соединения II-10 (440 мг). LC-MS m/z = 341,0 [M+1]+.
Путь синтеза промежуточного бората II-11
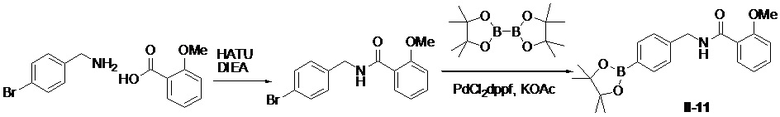
N-(4-бромбензил)-2-метоксибензамид
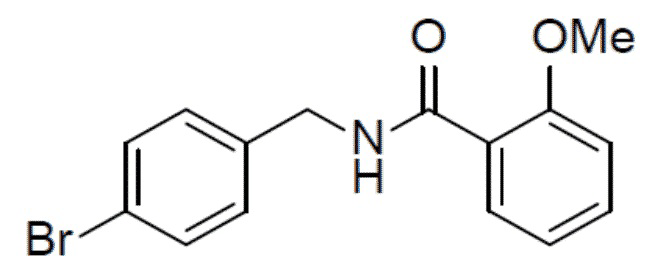
Раствор 4-бромбензиламина (1 г, 5,38 ммоль), 2-метоксибензойной кислоты (818 мг, 5,38 ммоль), HATU (2,45 г, 6,46 ммоль) и DIEA (1,39 г, 10,76 ммоль) в DMF (20 мл) перемешивали при комнатной температуре в течение 2 ч. Смесь выливали в воду (50 мл), подвергали фильтрации, промывали водой (30 мл × 2) и сушили с получением искомого соединения (1,6 г, выход: 93%), который непосредственно использовали на следующей стадии без очистки.
II-11: 2-метокси-N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)бензамид
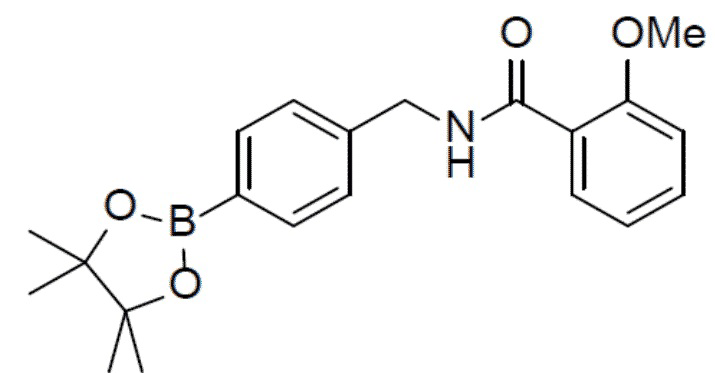
Раствор N-(4-бромбензил)-2-метоксибензамида (1,6 г, 5 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (1,9 г, 7,5 ммоль), PdCl2(dppf) (365 мг, 0,5 ммоль) и KOAc (1,47 г, 15 ммоль) в диоксане (30 мл) нагревали до 100°C и выдерживали его при данной температуре в течение 6 ч. Смесь концентрировали, добавляли воду (100 мл) и затем подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и концентрировали, а остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:9) с получением искомого соединения II-11 (1,5 г, выход: 82%).
II-12: 5-фтор-2-метокси-N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)бензамид
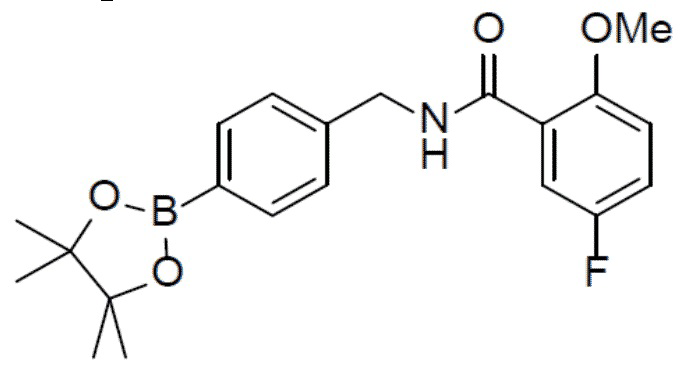
С 5-фтор-2-метоксибензойной кислотой (915 мг, 5,38 ммоль) и 4-бромбензиламином (1 г, 5,38 ммоль) в качестве исходных материалов использовали тот же способ синтеза, что и для II-11, с получением искомого соединения II-12 (900 мг).
II-13: 4-фтор-2-метокси-N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)бензамид
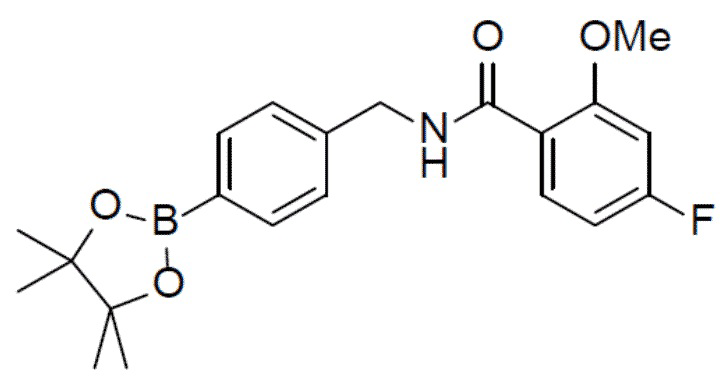
С 4-фтор-2-метоксибензойной кислотой (915 мг, 5,38 ммоль) и 4-бромбензиламином (1 г, 5,38 ммоль) в качестве исходных материалов использовали тот же способ синтеза, что и для II-11, с получением искомого соединения II-13 (1 г).
Путь синтеза промежуточного соединения A-4
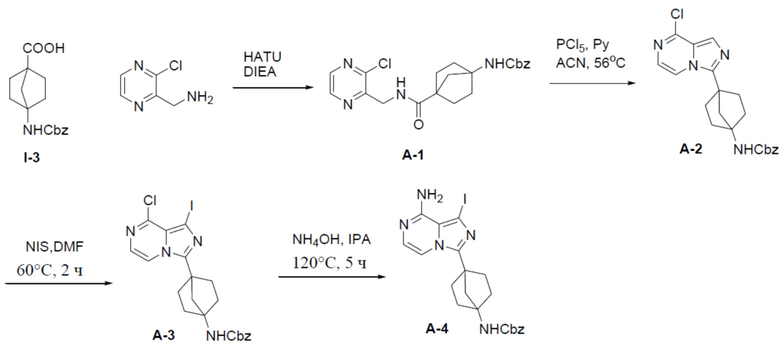
A-1: бензил(4-(((3-хлорпиразин-2-ил)метил)карбамоил)бицикло[2.2.1]гептан-1-ил)карбамат
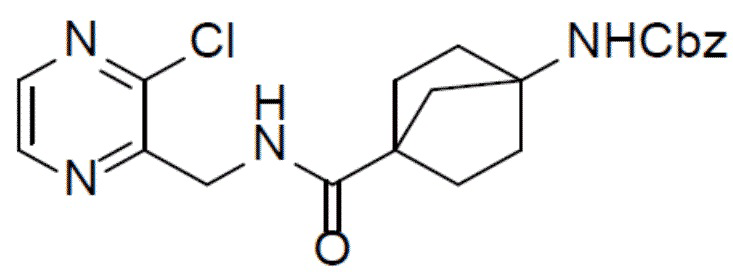
Раствор 4-(((бензилокси)карбонил)амино)бицикло[2.2.1]гептан-1-карбоновой кислоты (2 г, 6,9 ммоль), HATU (2,89 г, 7,6 ммоль), DIEA (3,56 г, 27,6 ммоль) и гидрохлорида (3-хлорпиразин-2-ил)метиламина (1,3 г, 7,24 ммоль) в DMF (20 мл) перемешивали при комнатной температуре в течение 6 ч. Смесь выливали в воду (100 мл) и подвергали экстракции этилацетатом (30 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:1-1:0) с получением искомого соединения A-1 (2 г, выход: 70%). LC-MS m/z = 414,9 [M+1]+.
A-2: бензил(4-(8-хлоримидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)карбамат
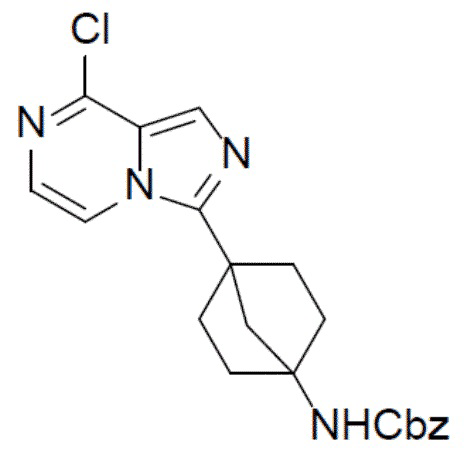
К раствору бензил(4N-(((3-хлорпиразин-2-ил)метил)карбамоил)бицикло[2.2.1]гептан-1-ил)карбамата (A-1) (2 г, 4,83 ммоль) в ACN (30 мл) добавляли пиридин (381 мг, 4,83 ммоль) и PCl5 (4 г, 19,32 ммоль) и смесь нагревали до 56°C и выдерживали ее при данной температуре в течение 1 ч. После охлаждения до комнатной температуры смесь медленно выливали в ледяной насыщенный водный раствор NaHCO3 (100 мл). При поддержании pH на уровне 9 смесь подвергали экстракции этилацетатом (30 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали. Остаток очищали с помощью колоночной хроматографии (этилацетат/простой петролейный эфир = 1: 1) с получением искомого соединения A-2 (1,5 г, выход: 78%).
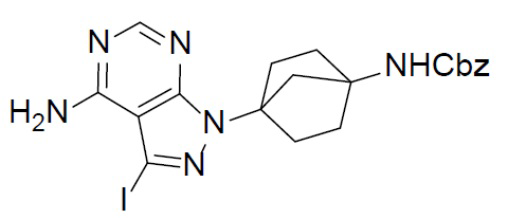
A-3: бензил(4-(8-хлор-1-йодимидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)карбамат
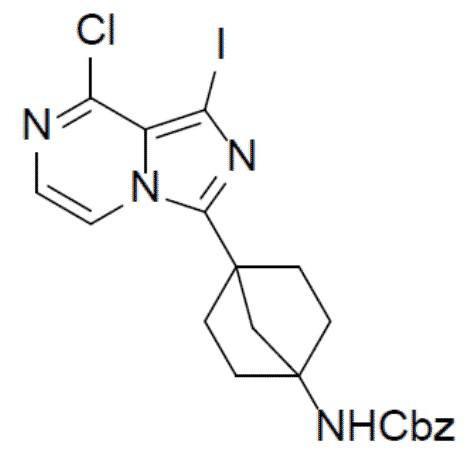
Смешанный раствор бензил(4-(8-хлоримидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)карбамата (A-2) (1,5 г, 3,79 ммоль) и NIS (1,13 г, 5,04 ммоль) в DMF (10 мл) нагревали до 60°C в условиях атмосферы N2 и перемешивали в течение 10 ч. После охлаждения до комнатной температуры смесь выливали в воду (100 мл) и подвергали экстракции этилацетатом (30 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 2:3) с получением искомого соединения A-3 (1,52 г, выход: 77%).
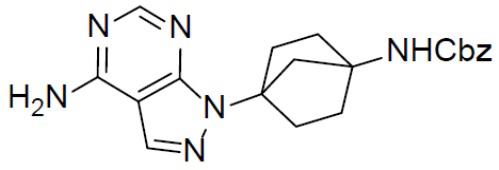
A-4: бензил(4-(8-амино-1-йодимидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)карбамат
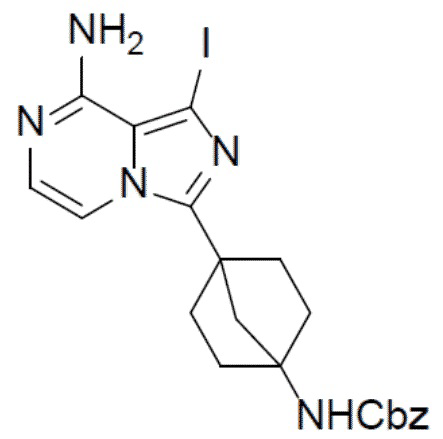
К суспензии соединения A-3 (1,5 г, 2,87 ммоль) в IPA (15 мл) добавляли NH4OH (3 мл) и смесь нагревали до 110°C и выдерживали при данной температуре в течение 6 ч. Затем смесь концентрировали и добавляли к ней насыщенный водный раствор NaHCO3 (20 мл). Для экстракции добавляли этилацетат (30 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:1) с получением искомого соединения A-4 (1,1 г, выход: 77%).
Путь синтеза промежуточного соединения B-3
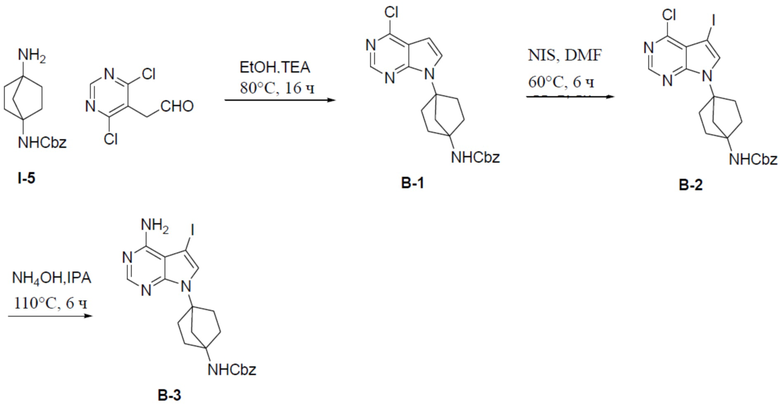
B-1: бензил(4-(4-хлор-7H-пирроло[2,3-d]пиримидин-7-ил)бицикло[2.2.1]гептан-1-ил)карбамат
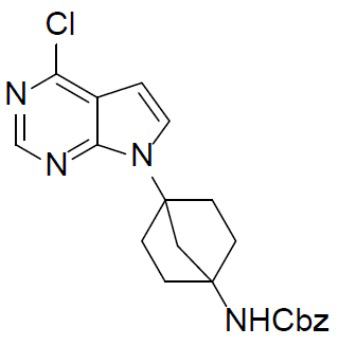
Раствор 2-(4,6-дихлорпиримидин-5-ил)ацетальдегида (735 мг, 3,8 ммоль), бензил(4-аминобицикло[2.2.1]гептан-1-ил)карбамата (1 г, 3,8 ммоль) и Et3N (389 мг, 3,8 ммоль) в EtOH (20 мл) нагревали до 80°C и выдерживали его при данной температуре в течение 16 ч. Смесь концентрировали, добавляли в нее воду (20 мл) и подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали, а остаток очищали колоночной хроматографией (EA/PE = 1:4) с получением искомого соединения B-1 (1,25 г, выход: 83%). LC-MS m/z = 397,1 [M+1]+.
B-2: бензил(4-(4-хлор-5-йод-7H-пирроло[2,3-d]пиримидин-7-ил)бицикло[2.2.1]гептан-1-ил) карбамат
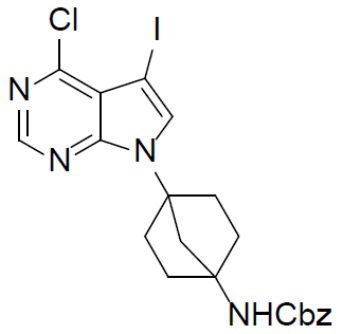
К раствору соединения B-1 (1,25 г, 3,16 ммоль) в DMF (10 мл) добавляли NIS (950 мг, 4,2 ммоль) и смесь нагревали до 60°C и выдерживали при данной температуре в течение 6 ч. Смесь выливали в воду (20 мл) и подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали, а остаток очищали колоночной хроматографией (EA/PE = 1:4) с получением искомого соединения (1,09 г, выход: 66%). LC-MS m/z = 523,1 [M+1]+.
B-3: бензил(4-(4-амино-5-йод-7H-пирроло[2,3-d]пиримидин-7-ил)бицикло[2.2.1]гептан-1-ил)карбамат
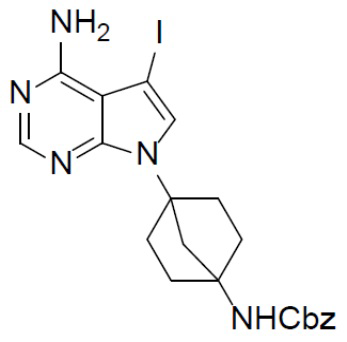
К раствору бензил(4-(4-хлор-5-йод-7H-пирроло[2,3-d]пиримидин-7-ил)бицикло[2.2.1]гептан-1-ил)карбамата (B-2) (1,09 г, 2,08 ммоль) в IPA (10 мл) добавляли NH4OH (2 мл) и смесь нагревали до 110°C и выдерживали при данной температуре в течение 6 ч. Смесь затем концентрировали, выливали в водный раствор NaHCO3 и подвергали экстракции посредством DCM (20 мл × 2). Органическую фазу отделяли, сушили, подвергали фильтрации и концентрировали, а остаток очищали колоночной хроматографией (MeOH/DCM = 1:20) с получением искомого соединения B-3 (900 мг, выход: 86%).
Путь синтеза промежуточного соединения C-4
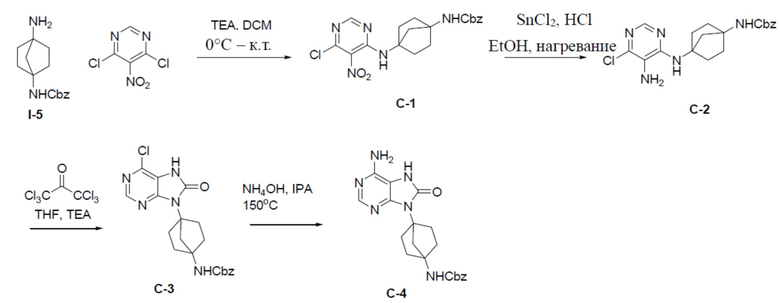
C-1: бензил(4-((6-хлор-5-нитропиримидин-4-ил)амино)бицикло[2.2.1]гептан-1-ил)карбамат
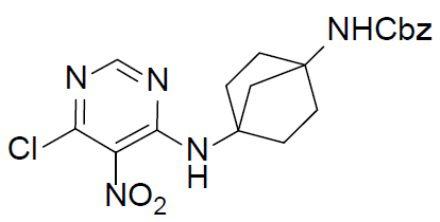
Раствор 4,6-дихлор-5-нитропиримидина (518 мг, 2,68 ммоль), бензил(4-аминобицикло[2.2.1]гептан-1-ил)карбамата (I-5) (698 мг, 2,68 ммоль) и Et3N (1,1 г, 10,72 ммоль) в DCM (20 мл) перемешивали при комнатной температуре в течение 4 ч. Растворитель удаляли, а остаток обрабатывали этилацетатом (50 мл) и промывали насыщенным водным раствором NaCl. Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и концентрировали, а остаток очищали колоночной хроматографией (EA/PE = 1:4) с получением искомого соединения C-1 (633 мг, выход: 57%).
C-2: бензил(4-((5-амино-6-хлорпиримидин-4-ил)амино)бицикло[2.2.1]гептан-1-ил)карбамат
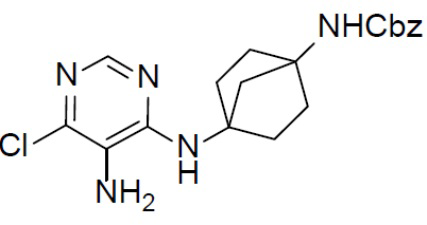
К раствору соединения C-1 (400 мг, 0,96 ммоль) в смешанном растворителе (EtOH/H2O = 20 мл/4 мл) добавляли порошок Fe (268 мг, 4,8 ммоль) и NH4Cl (254 мг, 4,8 ммоль). Смесь нагревали до закипания в течение 1 ч. После охлаждения до комнатной температуры смесь подвергали фильтрации и промывали посредством MeOH (10 мл). Фильтрат концентрировали и неочищенный продукт очищали колоночной хроматографией (EA/PE = 1:4) с получением искомого соединения C-2 (292 мг, выход: 79%).
C-3: бензил(4-(6-хлор-8-оксо-7,8-дигидропурин-9-ил)бицикло[2.2.1]гептан-1-ил)карбамат
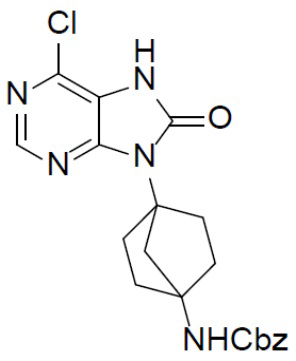
К раствору соединения C-2 (290 мг, 0,75 ммоль) в DCM (10 мл) при 0°C добавляли Et3N (166 мг, 1,5 ммоль) и трифосген (291 мг, 0,9 ммоль) и смесь перемешивали при 0°C в течение 2 ч. Затем смесь выливали в воду (20 мл) и отделяли органическую фазу, а затем водный раствор подвергали экстракции посредством DCM (10 мл × 2) для отделения органической фазы. Объединенные органические фазы сушили над Na2SO4 подвергали фильтрации и концентрировали с получением неочищенного C-3 (316 мг), которое непосредственно использовали на следующей стадии без очистки.
C-4: бензил(4-(6-амино-8-оксо-7,8-дигидропурин-9-ил)бицикло[2.2.1]гептан-1-ил)карбамат
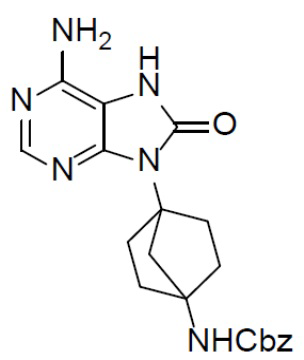
К раствору соединения C-3 (310 мг, 0,75 ммоль) в IPA (10 мл) добавляли NH4OH (2 мл) и смесь нагревали до 150°C и выдерживали при данной температуре в течение 24 ч. Затем смесь концентрировали, выливали в водный раствор NaHCO3 и подвергали экстракции посредством DCM (20 мл × 2). Органическую фазу отделяли, сушили, подвергали фильтрации и концентрировали, а остаток очищали колоночной хроматографией (MeOH/DCM = 1:20) с получением искомого соединения C-4 (100 мг, выход: 34%). LC-MS m/z = 395,1 [M+1]+.
Путь синтеза промежуточного соединения D-5
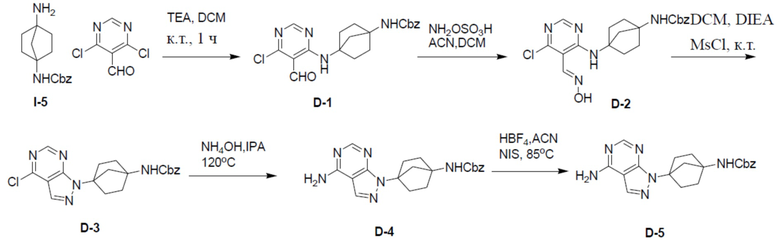
D-1: бензил(4-((6-хлор-5-формилпиримидин-4-ил)амино)бицикло[2.2.1]гептан-1-ил)карбамат
Смешанный раствор I-5 (1,47 г, 5,65 ммоль), 4,6-дихлорпиримидин-5-карбальдегида (1 г, 5,65 ммоль) и триэтиламина (1,15 г, 11,4 ммоль) DCM (20 мл) перемешивали на протяжении ночи при комнатной температуре. Реакционную смесь разводили посредством EA (50 мл) и промывали водой (30 мл × 2). Органическую фазу сушили над безводным Na2SO4, подвергали фильтрации и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: простой петролейный эфир/этилацетат = 1:1) с получением искомого продукта D-1 (1,24 г, 55%). LC-MS m/z = 401,0 [M+1]+.
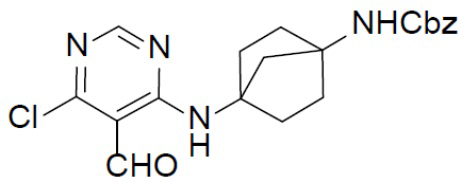
D-2: бензил-(E)-(4-((6-хлор-5-((гидроксиимино)метил)пиримидин-4-ил)амино)бицикло[2.2.1]гептан-1-ил)карбамат
Смешанный раствор из D-1 (1,2 г, 3,0 ммоль), гидроксиламин-O-сульфоновой кислоты (0,41 г, 3,6 ммоль) в DCM/ACN (50 мл/50 мл) перемешивали при комнатной температуре в течение 16 ч, а затем перемешивали при 50°C в течение 6 ч. После охлаждения смесь концентрировали до 10 мл и концентрат подвергали фильтрации и промывали посредством ACN (2 мл) с получением искомого продукта D-2 (1,0 г, 81%). LC-MS m/z = 416,0 [M+1]+.
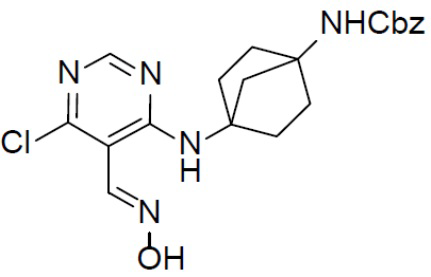
D-3: бензил(4-(4-хлор-1H-пиразоло[3,4-d]пиримидин-1-ил)бицикло[2.2.1]гептан-1-ил)карбамат
К раствору D-2 (1,0 г, 2,41 ммоль) в DCM (100 мл) добавляли DIEA (4 мл), а затем по каплям добавляли TsCl (0,23 мл, 2,9 ммоль). После перемешивания при комнатной температуре в течение 3 ч реакционную смесь обрабатывали водой (100 мл). Органическую фазу промывали солевым раствором (30 мл × 2), сушили над безводным Na2SO4, подвергали фильтрации и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: этилацетат/простой петролейный эфир = 1:6) с получением искомого продукта D-3 (400 мг, 42%). LC-MS m/z = 398,1 [M+1]+.
D-4: бензил(4-(4-амино-1H-пиразоло[3,4-d]пиримидин-1-ил)бицикло[2.2.1]гептан-1-ил)карбамат
Смесь из D-3 (400 мг, 1,0 ммоль) и гидроксида аммония (30%, 5 мл) в изопропаноле (20 мл) перемешивали в герметично закрытой пробирке при 120°C в течение 6 ч. Растворитель выпаривали, а остаток очищали колоночной хроматографией на силикагеле (элюент: PE/EA = 2:1) с получением искомого продукта D-4 (280 мг, 73,4%).
D-5: бензил(4-(4-амино-3-йод-1H-пиразоло[3,4-d]пиримидин-1-ил)бицикло[2.2.1]гептан-1-ил)карбамат
Смесь D-4 (190 мг, 0,5 ммоль), NIS (400 мг, 1,78 ммоль) и HBF4 (50%, 19,6 ммоль, 4 мл) в ACN (2,5 мл) нагревали до 85°C в герметично закрытой пробирке и перемешивали в течение 6 ч. После охлаждения смеси реакцию гасили с помощью насыщенного NaHCO3 и для экстракции добавляли EA (50 мл × 2). Органическую фазу сушили над безводным Na2SO4, подвергали фильтрации и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: PE/EA = 1:1-1:0) с получением искомого продукта D-5 (96 мг, 38,1%). LC-MS m/z = 505,0 [M+1]+.
Пример 1
Путь синтеза соединения A-7-n
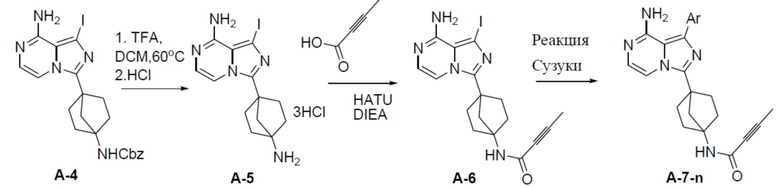
A-5: 3-(4-аминобицикло[2.2.1]гептан-1-ил)-1-йодимидазо[1,5-a]пиразин-8-амин
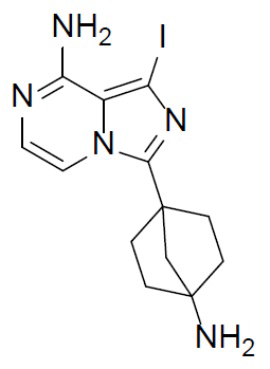
Раствор соединения A-4 (1 г, 1,99 ммоль) в смешанном растворителе TFA/DCM (10 мл/10 мл) нагревали до 60°C и выдерживали его при данной температуре в течение 6 ч. Смесь концентрировали и к ней добавляли DCM (20 мл × 2). Полученную в результате смесь концентрировали, растворяли в DCM (20 мл), добавляли к ней раствор HCl в диоксане (5 мл) и перемешивали при комнатной температуре в течение 10 мин. Смесь упаривали и добавляли к ней DCM (20 мл × 2), а полученную в результате смесь упаривали и добавляли к ней DME (20 мл). Смесь перемешивали при комнатной температуре в течение 30 минут, подвергали фильтрации, а затем промывали посредством DME (10 мл × 2). Полученное соединение A-5 непосредственно использовали на следующей стадии без очистки. LC-MS m/z = 370,1 [M+1]+.
A-6: N-(4-(8-амино-1-йодимидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)бут-2-инамид
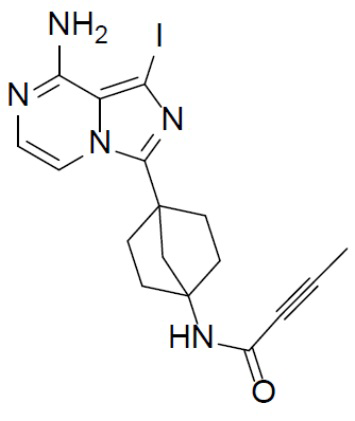
Раствор соединения A-5 (1,35 г, 2,8 ммоль), DIEA (3,28 г, 25,2 ммоль), бут-2-иновой кислоты (235 мг, 2,8 ммоль) и HATU (1,06 г, 2,8 ммоль) в DMF (20 мл) перемешивали при комнатной температуре в течение 30 минут. Смесь выливали в воду (30 мл) и подвергали экстракции этилацетатом (30 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 2:3) с получением искомого соединения A-6 (800 мг, выход: 65%).
A-7-1: 4-(8-амино-3-(4-(буд-2-инамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(пиридин-2-ил)бензамид
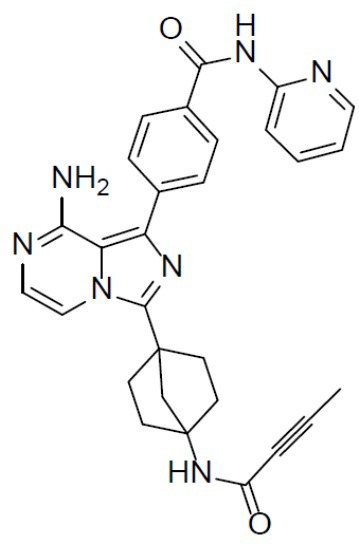
Раствор соединения A-6 (30 мг, 0,069 ммоль), (4-(пиридин-2-илкарбамоил)фенил)бориновой кислоты (II-1) (20 мг, 0,085 ммоль), Pd[PPh3]4 (8 мг, 0,0069 ммоль) и Cs2CO3 (45 мг, 0,138 ммоль) в смешанном растворителе DME/H2O (1,5 мл/0,3 мл) нагревали до 80°C и выдерживали его при данной температуре в течение 3 ч. Смесь концентрировали, добавляли к ней воду (20 мл) и подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:1) с получением искомого соединения A-7-1 (10 мг, выход: 30%). LC-MS m/z = 506,2 [M+1]+.
A-7-2: 4-(8-амино-3-(4-(бут-2-инамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(4-фторпиридин-2-ил)бензамид
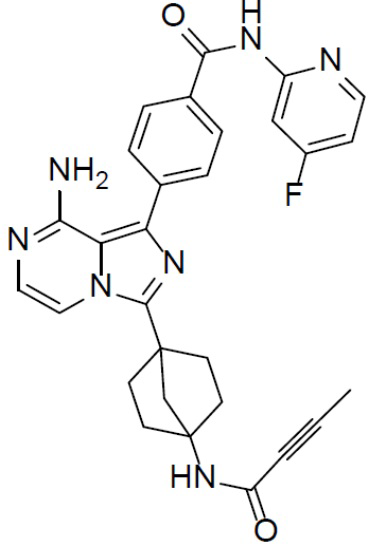
Раствор соединения A-6 (30 мг, 0,069 ммоль), (4-((4-фторпиридин-2-ил)карбамоил)фенил)бориновой кислоты (II-2) (22 мг, 0,085 ммоль), Pd[PPh3]4 (8 мг, 0,0069 ммоль) и Cs2CO3 (45 мг, 0,138 ммоль) в смешанном растворителе DME/H2O (1,5 мл/0,3 мл) нагревали до 80°C и выдерживали его при данной температуре в течение 3 ч. Смесь концентрировали, добавляли к ней воду (20 мл) и подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:1) с получением искомого соединения A-7-2 (15 мг, выход: 34%). LC-MS m/z = 524,0 [M+1]+.
A-7-3: 4-(8-амино-3-(4-(бут-2-инамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
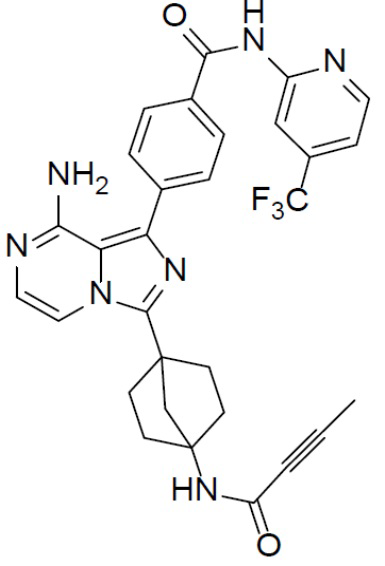
Раствор соединения A-6 (30 мг, 0,069 ммоль), (4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)бориновой кислоты (II-3) (26 мг, 0,085 ммоль), Pd[PPh3]4 (8 мг, 0,0069 ммоль) и Cs2CO3 (45 мг, 0,138 ммоль) в смешанном растворителе DME/H2O (1,5 мл/0,3 мл) нагревали до 80°C и выдерживали его при данной температуре в течение 3 ч. Смесь концентрировали, добавляли к ней воду (20 мл) и подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:1) с получением искомого соединения A-7-3 (12 мг, выход: 31%). LC-MS m/z = 574,2 [M+1]+.
A-7-4: 4-(8-амино-3-(4-(бут-2-инамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(4-метилпиридин-2-ил)бензамид
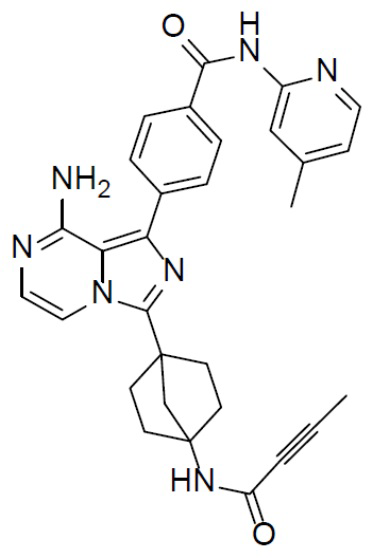
Раствор соединения A-6 (30 мг, 0,069 ммоль), (4-((4-метилпиридин-2-ил)карбамоил)фенил)бориновой кислоты (II-4) (22 мг, 0,085 ммоль), Pd[PPh3]4 (8 мг, 0,0069 ммоль) и Cs2CO3 (45 мг, 0,138 ммоль) в смешанном растворителе DME/H2O (1,5 мл/0,3 мл) нагревали до 80°C и выдерживали его при данной температуре в течение 3 ч. Смесь концентрировали, добавляли к ней воду (20 мл) и подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:1) с получением искомого соединения A-7-4 (14 мг, выход: 39%). LC-MS m/z = 520,2 [M+1]+.
A-7-5: 4-(8-амино-3-(4-(бут-2-инамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(4-цианопиридин-2-ил)бензамид
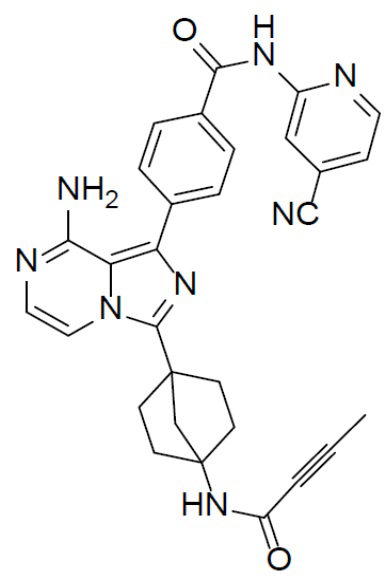
Раствор соединения A-6 (30 мг, 0,069 ммоль), (4-((4-цианопиридин-2-ил)карбамоил)фенил)бориновой кислоты (II-5) (23 мг, 0,085 ммоль), Pd[PPh3]4 (8 мг, 0,0069 ммоль) и Cs2CO3 (45 мг, 0,138 ммоль) в смешанном растворителе DME/H2O (1,5 мл/0,3 мл) нагревали до 80°C и выдерживали его при данной температуре в течение 3 ч. Смесь концентрировали, добавляли в нее воду (20 мл) и подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:1) с получением искомого соединения A-7-5 (12 мг, выход: 33%). LC-MS m/z = 531,0 [M+1]+.
A-7-6: 4-(8-амино-3-(4-(бут-2-инамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-2-фтор-N-(пиридин-2-ил)бензамид
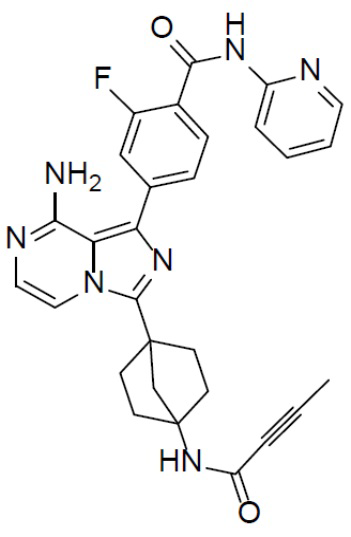
Раствор соединения A-6 (30 мг, 0,069 ммоль), (3-фтор-4-(пиридин-2-илкарбамоил)фенил)бориновой кислоты (II-6) (22 мг, 0,085 ммоль), Pd[PPh3]4 (8 мг, 0,0069 ммоль) и Cs2CO3 (45 мг, 0,138 ммоль) в смешанном растворителе DME/H2O (1,5 мл/0,3 мл) нагревали до 80°C и выдерживали его при данной температуре в течение 3 ч. Смесь концентрировали, добавляли в нее воду (20 мл) и подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:1) с получением искомого соединения A-7-6 (15 мг, выход: 42%). LC-MS m/z = 524,2 [M+1]+.
A-7-7: 4-(8-амино-3-(4-(бут-2-инамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(тиазол-2-ил)бензамид
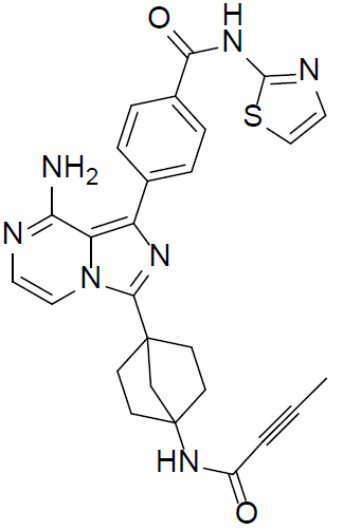
Раствор соединения A-6 (30 мг, 0,069 ммоль), (4-(тиазол-2-илкарбамоил)фенил)бориновой кислоты (II-7) (28 мг, 0,085 ммоль), Pd[PPh3]4 (8 мг, 0,0069 ммоль) и Cs2CO3 (45 мг, 0,138 ммоль) в смешанном растворителе DME/H2O (1,5 мл/0,3 мл) нагревали до 80°C и выдерживали его при данной температуре в течение 3 ч. Смесь концентрировали, добавляли в нее воду (20 мл) и подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:1) с получением искомого соединения A-7-7 (13 мг, выход: 37%). LC-MS m/z = 512,0 [M+1]+.
A-7-8: 4-(8-амино-3-(4-(бут-2-инамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-3-фтор-N-(пиридин-2-ил)бензамид
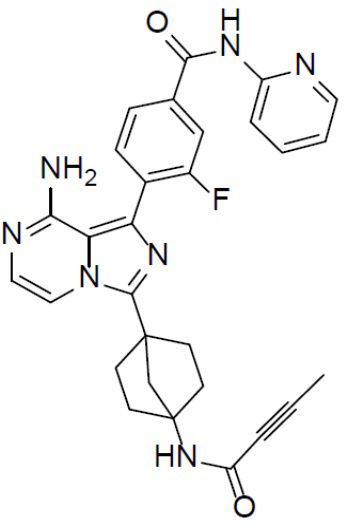
Раствор соединения A-6 (30 мг, 0,069 ммоль), (2-фтор-4-(пиридин-2-илкарбамоил)фенил)бориновой кислоты (II-8) (22 мг, 0,085 ммоль), Pd[PPh3]4 (8 мг, 0,0069 ммоль) и Cs2CO3 (45 мг, 0,138 ммоль) в смешанном растворителе DME/H2O (1,5 мл/0,3 мл) нагревали до 80°C и выдерживали его при данной температуре в течение 3 ч. Смесь концентрировали, добавляли в нее воду (20 мл) и подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:1) с получением искомого соединения A-7-8 (14 мг, выход: 39%). LC-MS m/z = 524,0 [M+1]+.
A-7-9: 4-(8-амино-3-(4-(бут-2-инамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-3-фтор-N-(4-(трифторметил)пиридин-2-ил)бензамид
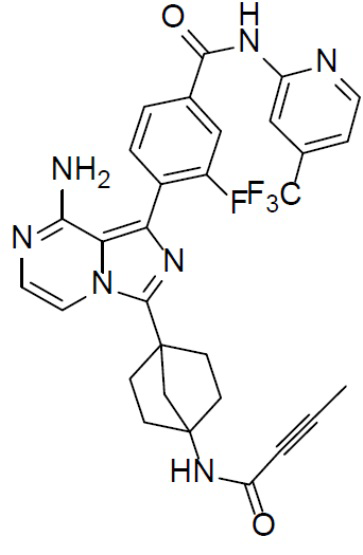
Раствор соединения A-6 (30 мг, 0,069 ммоль), (2-фтор-4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)бориновой кислоты (II-9) (28 мг, 0,085 ммоль), Pd[PPh3]4 (8 мг, 0,0069 ммоль) и Cs2CO3 (45 мг, 0,138 ммоль) в смешанном растворителе DME/H2O (1,5 мл/0,3 мл) нагревали до 80°C и выдерживали его при данной температуре в течение 3 ч. Смесь концентрировали, добавляли в нее воду (20 мл) и подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:1) с получением искомого соединения A-7-9 (19 мг, выход: 47%). LC-MS m/z = 592,0 [M+1]+.
A-7-10: 4-(8-амино-3-(4-(бут-2-инамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-3-метокси-N-(4-(трифторметил)пиридин-2-ил)бензамид
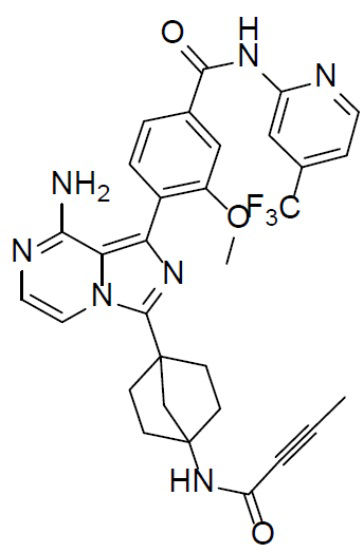
Раствор соединения A-6 (30 мг, 0,069 ммоль), (2-метокси-4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)бориновой кислоты (II-10) (29 мг, 0,085 ммоль), Pd[PPh3]4 (8 мг, 0,0069 ммоль) и Cs2CO3 (45 мг, 0,138 ммоль) в смешанном растворителе DME/H2O (1,5 мл/0,3 мл) нагревали до 80°C и выдерживали его при данной температуре в течение 3 ч. Смесь концентрировали, добавляли в нее воду (20 мл) и подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:1) с получением искомого соединения A-7-10 (16 мг, выход: 38%). LC-MS m/z = 604,0 [M+1]+.
A-7-11: N-(4-(8-амино-3-(4-(бут-2-инамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)бензил)-2-метоксибензамид
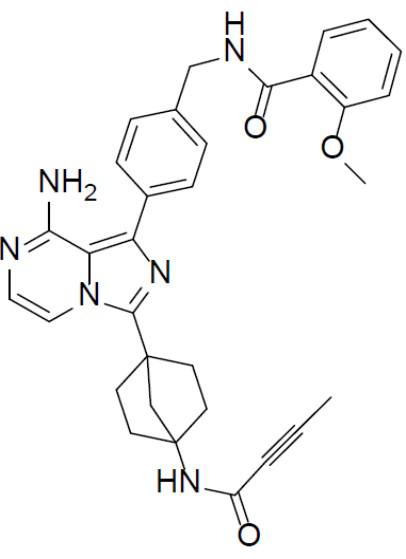
Раствор соединения A-6 (30 мг, 0,069 ммоль), 2-метокси-N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)бензамида (II-11) (31 мг, 0,085 ммоль), Pd[PPh3]4 (8 мг, 0,0069 ммоль) и Cs2CO3 (45 мг, 0,138 ммоль) в смешанном растворителе DME/H2O (1,5 мл/0,3 мл) нагревали до 80°C и выдерживали его при данной температуре в течение 3 ч. Смесь концентрировали, добавляли в нее воду (20 мл) и подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:1) с получением искомого соединения A-7-11 (21 мг, выход: 50%). LC-MS m/z = 549,3 [M+1]+.
A-7-12: N-(4-(8-амино-3-(4-(бут-2-инамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)бензил)-5-фтор-2-метоксибензамид
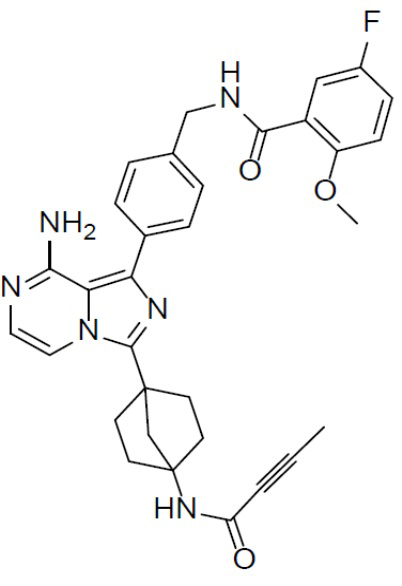
Раствор соединения A-6 (30 мг, 0,069 ммоль), 5-фтор-2-метокси-N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)бензамида (II-12) (33 мг, 0,085 ммоль), Pd[PPh3]4 (8 мг, 0,0069 ммоль) и Cs2CO3 (45 мг, 0,138 ммоль) в смешанном растворителе DME/H2O (1,5 мл/0,3 мл) нагревали до 80°C и выдерживали его при данной температуре в течение 3 ч. Смесь концентрировали, добавляли в нее воду (20 мл) и подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:1) с получением искомого соединения A-7-12 (24 мг, выход: 56%). LC-MS m/z = 567,0 [M+1]+.
A-7-13: N-(4-(8-амино-3-(4-(бут-2-инамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)бензил)-4-фтор-2-метоксибензамид
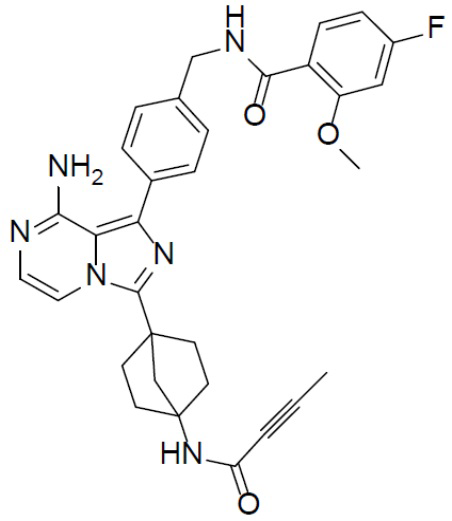
Раствор соединения A-6 (30 мг, 0,069 ммоль), 4-фтор-2-метокси-N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)бензамида (II-13) (33 мг, 0,085 ммоль), Pd[PPh3]4 (8 мг, 0,0069 ммоль) и Cs2CO3 (45 мг, 0,138 ммоль) в смешанном растворителе DME/H2O (1,5 мл/0,3 мл) нагревали до 80°C и выдерживали его при данной температуре в течение 3 ч. Смесь концентрировали, добавляли в нее воду (20 мл) и подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:1) с получением искомого соединения A-7-13 (26 мг, выход: 60%). LC-MS m/z = 567,1 [M+1]+.
A-7-14: N-(4-(8-амино-1-(4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)бут-2-инамид
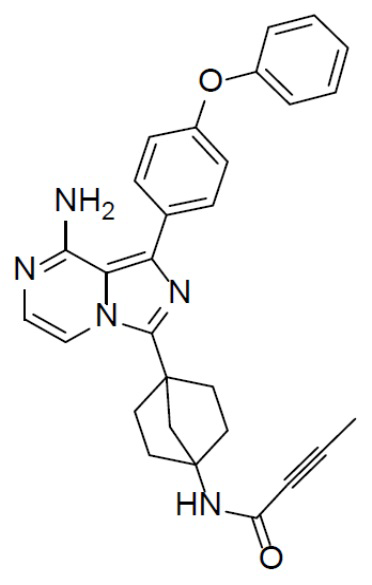
Раствор соединения A-6 (30 мг, 0,069 ммоль), 4-феноксифенилбориновой кислоты (18 мг, 0,085 ммоль), Pd[PPh3]4 (8 мг, 0,0069 ммоль) и Cs2CO3 (45 мг, 0,138 ммоль) в смешанном растворителе DME/H2O (1,5 мл/0,3 мл) нагревали до 80°C и выдерживали его при данной температуре в течение 3 ч. Смесь концентрировали, добавляли в нее воду (20 мл) и подвергали экстракции этилацетатом (20 мл × 2). Органическую фазу отделяли, сушили над Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (этилацетат/простой петролейный эфир = 1:1) с получением искомого соединения A-7-14 (19 мг, выход: 49%). LC-MS m/z = 478,0 [M+1]+.
Путь синтеза соединения A-10-n
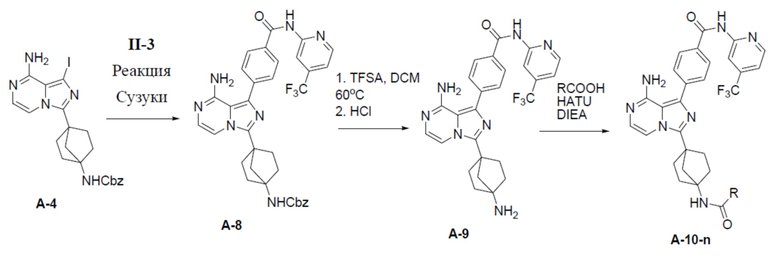
A-8: бензил(4-(8-амино-1-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)карбамат
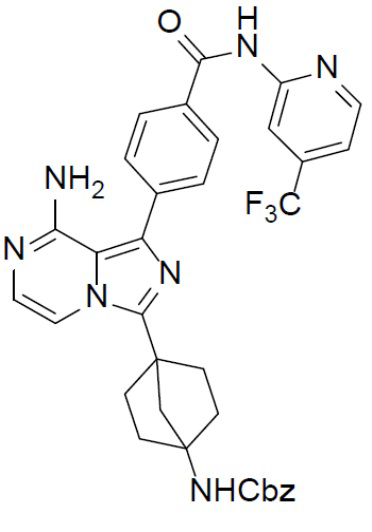
Раствор бензил(4-(8-амино-1-йодимидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)карбамата (A-4) (300 мг, 0,6 ммоль), (4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)бориновой кислоты (II-3) (229 мг, 0,738 ммоль), Pd[PPh3]4 (69 мг, 0,06 ммоль) и Cs2O3 (239 мг, 0,738 ммоль) в смешанном растворителе DME:H2O (2,5 мл:0,5 мл) нагревали до 80°C и перемешивали на протяжении ночи. Смесь концентрировали, а остаток очищали колоночной хроматографией (метанол/DCM = 1:30) с получением искомого соединения A-8 (265 мг, выход: 69%).
A-9: 4-(8-амино-3-(4-аминобицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
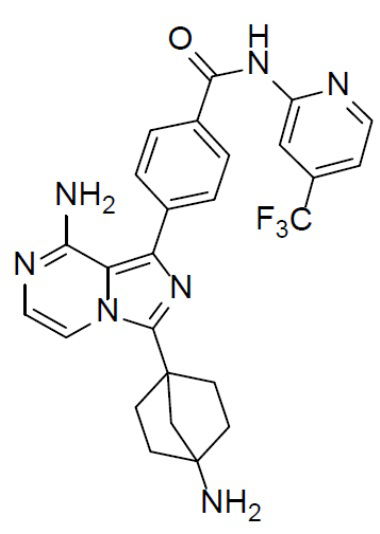
Раствор соединения A-8 (265 мг, 0,42 ммоль) в смешанном растворителе DCM/TFA (10 мл:10 мл) нагревали до 60°C и перемешивали в течение 18 ч. После этого смесь упаривали до сухого состояния, добавляли DCM (20 мл × 2) и полученную в результате смесь концентрировали. Остаток растворяли в DCM (30 мл), а затем данный раствор добавляли к раствору HCl в диоксане. После этого смесь упаривали до сухого состояния, затем к нему добавляли DCM (20 мл × 2) и полученную в результате смесь концентрировали с последующим добавлением простого изопропилового эфира (30 мл). После перемешивания в течение 2 ч смесь подвергали фильтрации и промывали простым изопропиловым эфиром (10 мл × 2) с получением гидрохлорида искомого продукта A-9 (196 мг), который непосредственно использовали на следующей стадии без очистки.
A-10-1: (E)-4-(8-амино-3-(4-(4-метоксибут-2-енамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
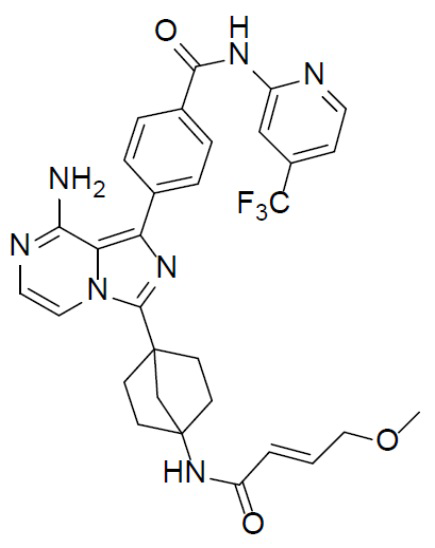
Раствор соединения A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и (E)-4-метоксибут-2-еновой кислоты (2,8 мг, 0,024 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (5 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (элюент: DCM/MeOH = 20:1) с получением продукта A-10-1 (5 мг, выход: 35%). LC-MS m/z = 606,1 [M+1]+.
A-10-2: (E)-4-(8-амино-3-(4-(4-(тетрагидропиррол-1-ил)бут-2-енамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
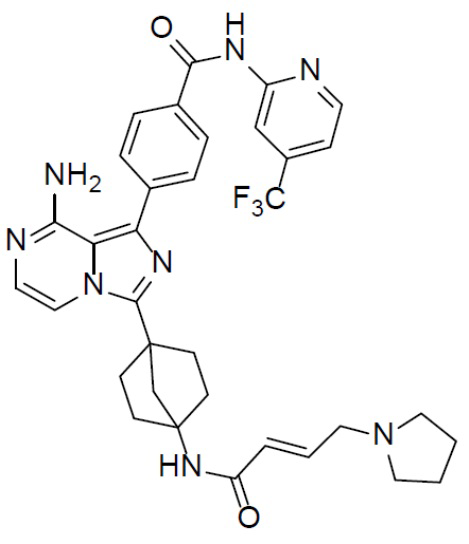
Раствор соединения A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и (E)-4-(тетрагидропиррол-1-ил)-бут-2-еновой кислоты (4 мг, 0,024 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (5 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-2 (6 мг, выход: 38%). LC-MS m/z = 645,0 [M+1]+.
A-10-3: 4-(3-(4-акриламидбицикло[2.2.1]гептан-1-ил)-8-аминоимидазо[1,5-a]пиразин-1-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
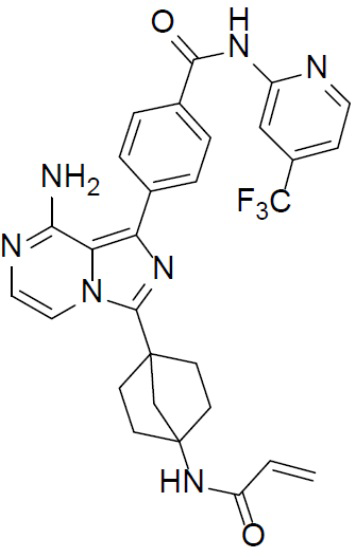
Соединение A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и акриловую кислоту (2 мг, 0,024 ммоль) перемешивали при комнатной температуре в течение 1 ч, а затем данную смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-3 (5 мг, выход: 38%). LC-MS m/z = 562,0 [M+1]+.
A-10-4: 4-(3-(4-ацетамидбицикло[2.2.1]гептан-1-ил)-8-аминоимидазо[1,5-a]пиразин-1-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
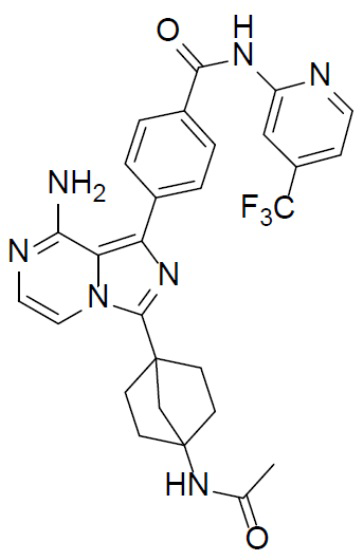
Соединение A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и уксусную кислоту (1,5 мг, 0,024 ммоль) перемешивали при комнатной температуре в течение 1 ч, а затем данную смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-4 (4 мг, выход: 31%). LC-MS m/z = 550,0 [M+1]+.
A-10-5: N-(4-(8-амино-1-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)-3-метилоксетан-3-карбоксамид
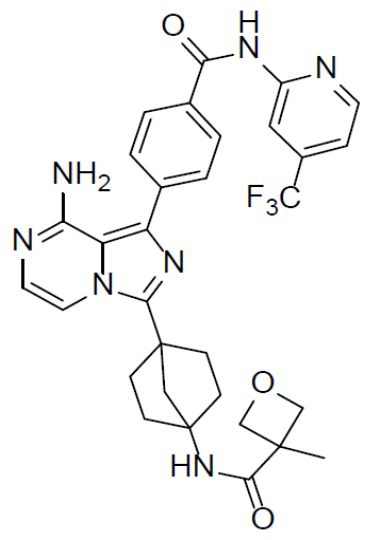
Раствор соединения A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и 3-метилоксетан-3-карбоновой кислоты (3 мг, 0,024 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (5 мл) и промывали этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-5 (6 мг, выход: 40%). LC-MS m/z = 606,1 [M+1]+.
A-10-6: 4-(8-амино-3-(4-(2-гидрокси-2-метилпропанамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
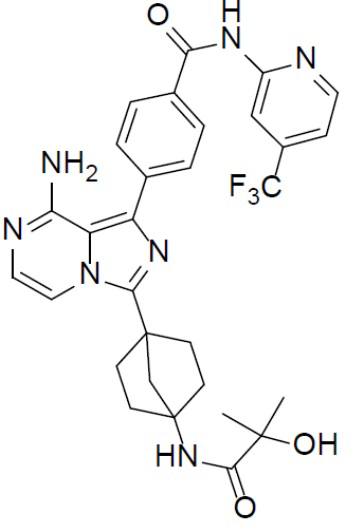
Соединение A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и 2-гидрокси-2-метилпропионовую кислоту (2,5 мг, 0,024 ммоль) перемешивали при комнатной температуре в течение 1 ч, а затем данную смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-6 (6 мг, выход: 41%). LC-MS m/z = 594,1 [M+1]+.
A-10-7: 4-(8-амино-3-(4-(2-метоксиацетамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
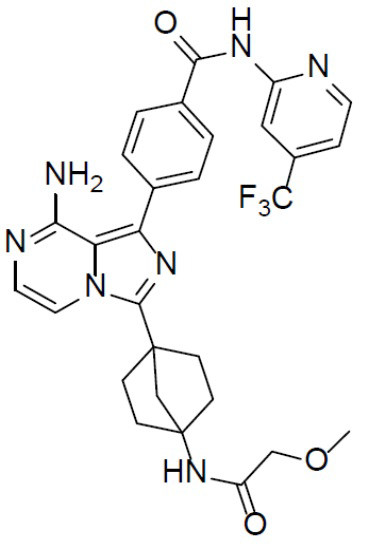
Соединение A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и 2-метоксиуксусную кислоту (2 мг, 0,024 ммоль) перемешивали при комнатной температуре в течение 1 ч, а затем данную смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-7 (5 мг, выход: 36%). LC-MS m/z = 580,1 [M+1]+.
A-10-8: 4-(8-амино-3-(4-(3-метоксипропанамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
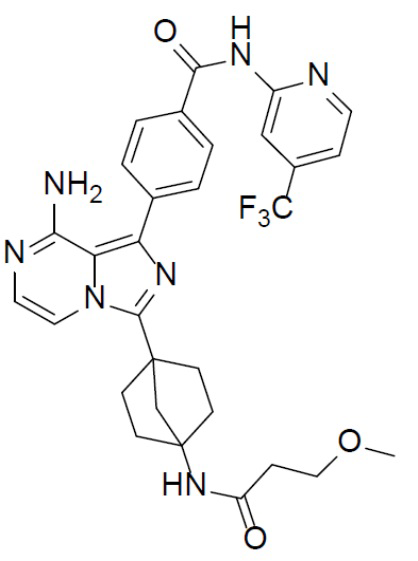
Соединение A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и 3-метоксипропионовую кислоту (2,5 мг, 0,024 ммоль) перемешивали при комнатной температуре в течение 1 ч, а затем данную смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-8 (5 мг, выход: 35%). LC-MS m/z = 594,1 [M+1]+.
A-10-9: 4-(8-амино-3-(4-(1-гидроксициклопропанкарбоксамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
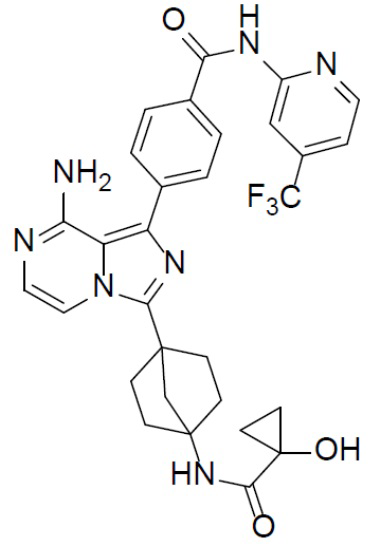
Раствор соединения A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и 1-гидроксициклопропанкарбоновой кислоты (2,5 мг, 0,024 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (5 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-9 (5 мг, выход: 36%). LC-MS m/z = 592,0 [M+1]+.
A-10-10: 4-(8-амино-3-(4-(2-морфолиноацетамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
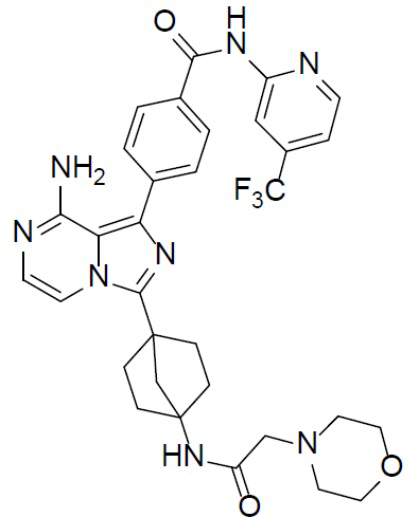
Соединение A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и 2-морфолинуксусную кислоту (3,5 мг, 0,024 ммоль) перемешивали при комнатной температуре в течение 1 ч, а затем данную смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-10 (7 мг, выход: 47%). LC-MS m/z = 635,0 [M+1]+.
A-10-11: N-(4-(8-амино-1-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)тетрагидрофуран-2-карбоксамид
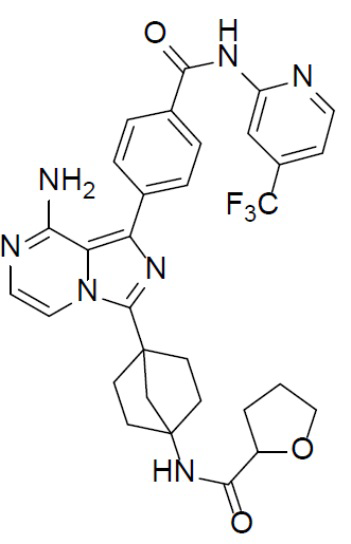
Раствор соединения A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и тетрагидрофуран-2-карбоновой кислоты (2,8 мг, 0,024 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (5 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-11 (5 мг, выход: 35%). LC-MS m/z = 606,0 [M+1]+.
A-10-12: 4-(8-амино-3-(4-(1-цианоциклопропанкарбоксамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
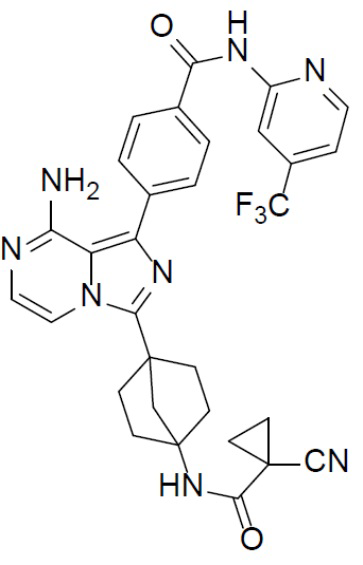
Соединение A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и 1-цианоциклопропанкарбоновую кислоту (2,7 мг, 0,024 ммоль) перемешивали при комнатной температуре в течение 1 ч, а затем данную смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-12 (6 мг, выход: 42%). LC-MS m/z = 601,3 [M+1]+.
A-10-13: 4-(8-амино-3-(4-(2-цианоацетамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
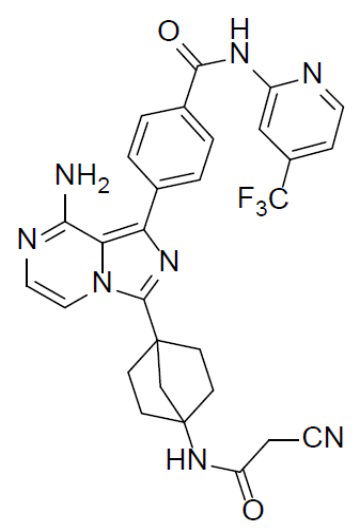
Соединение A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и 2-цианоуксусную кислоту (2 мг, 0,024 ммоль) перемешивали при комнатной температуре в течение 1 ч, а затем данную смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-13 (6 мг, выход: 44%). LC-MS m/z = 575,2 [M+1]+.
A-10-14: 4-(8-амино-3-(4-(2-цианопропанамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
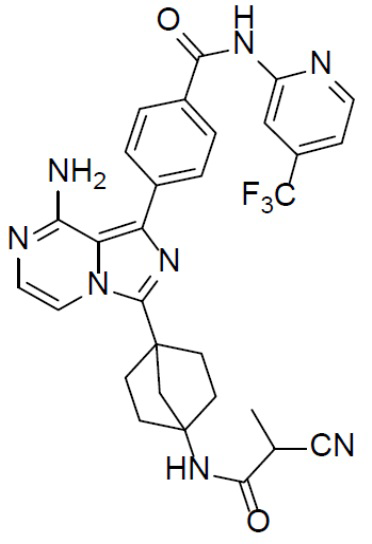
Соединение A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и 2-цианопропионовую кислоту (2,4 мг, 0,024 ммоль) перемешивали при комнатной температуре в течение 1 ч, а затем данную смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-14 (5 мг, выход: 36%). LC-MS m/z = 589,3 [M+1]+.
A-10-15: N-(4-(8-амино-1-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)-4-цианотетрагидро-2H-пиран-4-карбоксамид
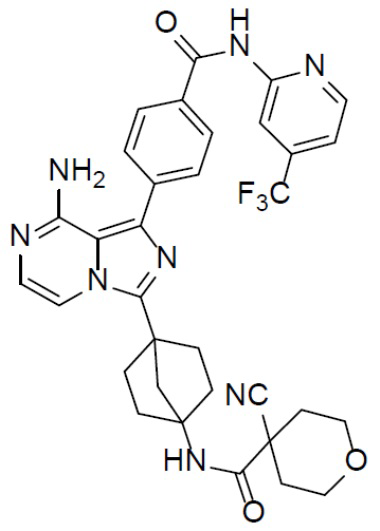
Соединение A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и 4-цианотетрагидро-2H-пиран-4-карбоновую кислоту (3,7 мг, 0,024 ммоль) перемешивали при комнатной температуре в течение 1 ч, а затем данную смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-15 (6 мг, выход: 39%). LC-MS m/z = 645,0 [M+1]+.
A-10-16: 1-((4-(8-амино-1-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)карбамоил)циклопропан-1-карбоновая кислота
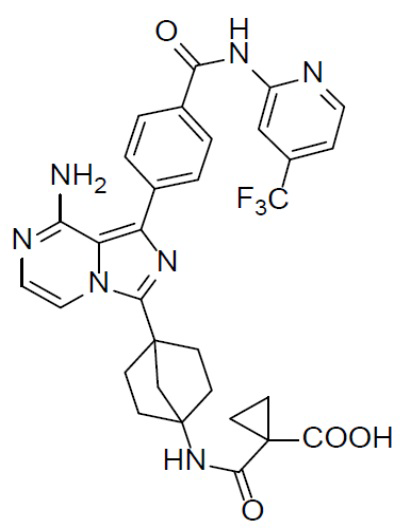
Раствор соединения A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и циклопропан-1,1-дикарбоновой кислоты (3 мг, 0,024 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (5 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-16 (6 мг, выход: 40%). LC-MS m/z = 555,3 [M+1]+.
A-10-17: 4-(8-амино-3-(4-бензамидобицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
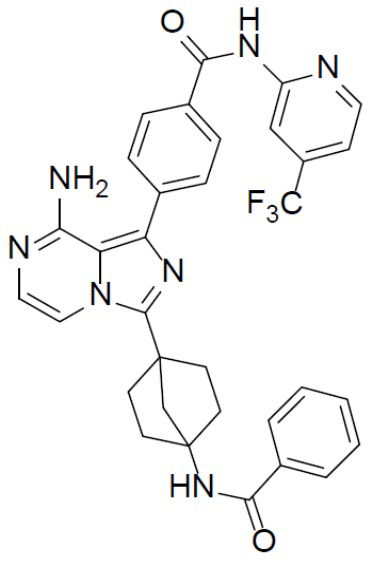
Соединение A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и бензойную кислоту (3 мг, 0,024 ммоль) перемешивали при комнатной температуре в течение 1 ч, а затем данную смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-17 (8 мг, выход: 53%). LC-MS m/z = 612,2 [M+1]+.
A-10-18: N-(4-(8-амино-1-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)пиколинамид
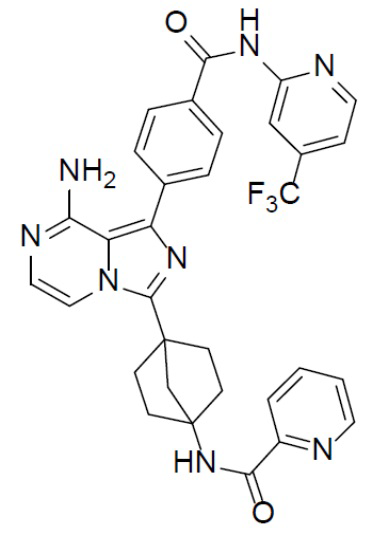
Соединение A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и 2-пиколиновую кислоту (3 мг, 0,024 ммоль) перемешивали при комнатной температуре в течение 1 ч, а затем данную смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-18 (9 мг, выход: 60%). LC-MS m/z = 613,2 [M+1]+.
A-10-19: N-(4-(8-амино-1-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)никотинамид
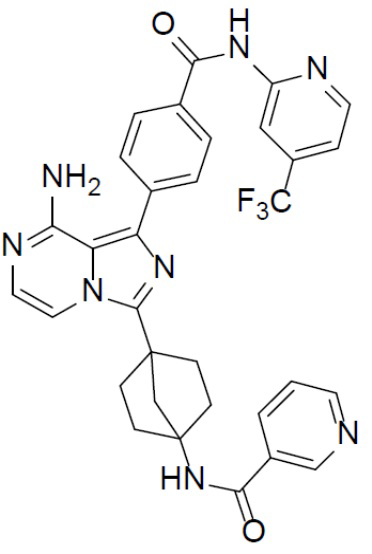
Соединение A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и никотиновую кислоту (3 мг, 0,024 ммоль) перемешивали при комнатной температуре в течение 1 ч, а затем данную смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-19 (8 мг, выход: 54%). LC-MS m/z = 613,2 [M+1]+.
A-10-20: N-(4-(8-амино-1-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)-2-метоксибензамид
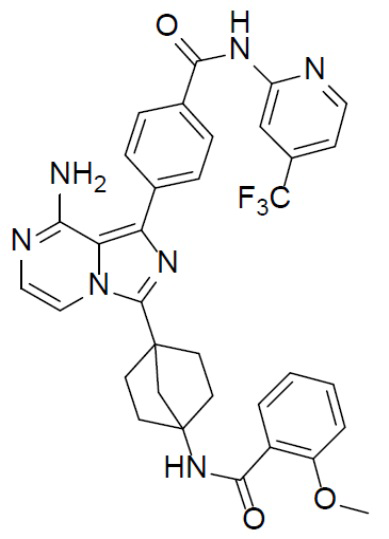
Соединение A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и 2-метоксибензойную кислоту (3,6 мг, 0,024 ммоль) перемешивали при комнатной температуре в течение 1 ч, а затем данную смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-20 (8 мг, выход: 52%). LC-MS m/z = 642,3 [M+1]+.
A-10-21: N-(4-(8-амино-1-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)фуран-2-карбоксамид
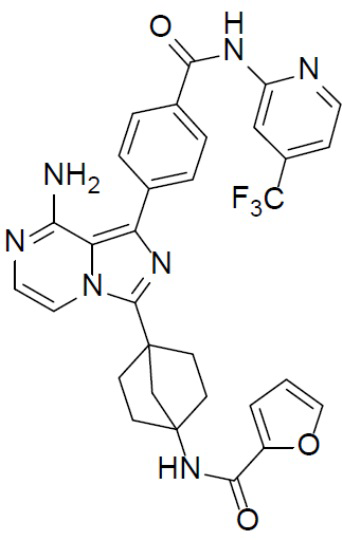
Раствор соединения A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и фуран-2-карбоновой кислоты (2,7 мг, 0,024 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (5 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-21 (6 мг, выход: 43%). LC-MS m/z = 602,2 [M+1]+.
A-10-22: N-(4-(8-амино-1-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)тиазол-2-карбоксамид
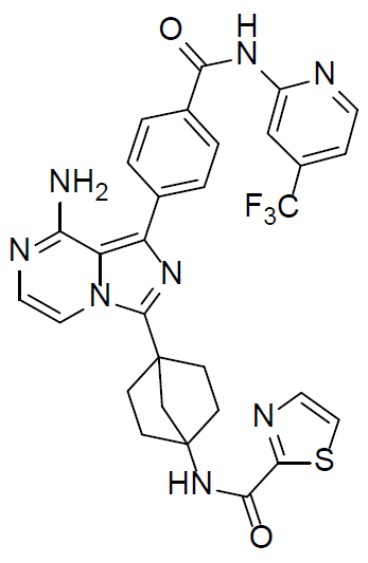
Раствор соединения A-9 (15 мг, 0,024 ммоль), HATU (9,12 мг, 0,024 ммоль), DIEA (16 мг, 0,12 ммоль) и тиазол-2-карбоновой кислоты (3 мг, 0,024 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (5 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-10-22 (6 мг, выход: 40%). LC-MS m/z = 619,2 [M+1]+.
Путь синтеза соединения A-13-n
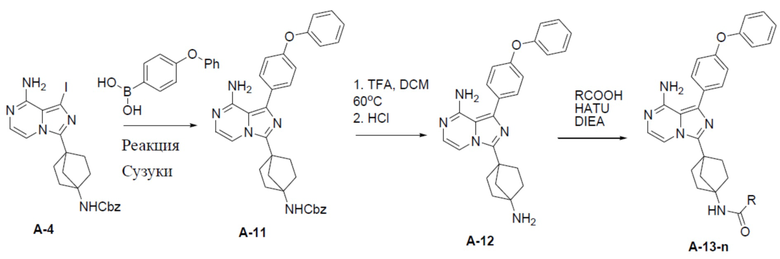
A-11: бензил(4-(8-амино-1-(4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)карбамат
Раствор бензил(4-(8-амино-1-йодимидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)карбамата (A-4) (300 мг, 0,6 ммоль), 4-феноксибензойной кислоты (158 мг, 0,738 ммоль), Pd[PPh3]4 (69 мг, 0,06 ммоль) и Cs2O3 (239 мг, 0,738 ммоль) в смешанном растворителе DME:H2O (2,5 мл:0,5 мл) нагревали до 80°C и перемешивали на протяжении ночи. Затем смесь концентрировали, а остаток очищали колоночной хроматографией с получением искомого соединения A-11 (268 мг, выход: 82%).
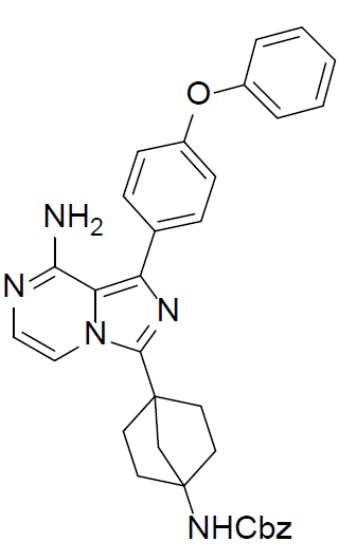
A-12: 3-(4-аминобицикло[2.2.1]гептан-1-ил)-1-(4-феноксифенил)имидазо[1,5-a]пиразин-8-амин
Раствор соединения A-11 (260 мг, 0,48 ммоль) в смешанном растворителе DCM/TFA (10 мл:10 мл) нагревали до 60°C и перемешивали в течение 18 ч. После этого смесь упаривали до сухого состояния, добавляли DCM (20 мл × 2) и полученную в результате смесь концентрировали. Остаток растворяли в DCM (30 мл), а затем данный раствор добавляли к раствору HCl в диоксане. После этого смесь упаривали до сухого состояния, затем к нему добавляли DCM (20 мл × 2) и полученную в результате смесь концентрировали с последующим добавлением простого изопропилового эфира (30 мл). После перемешивания в течение 2 ч смесь подвергали фильтрации и промывали простым изопропиловым эфиром (10 мл × 2) с получением гидрохлорида искомого продукта A-12 (200 мг), который непосредственно использовали на следующей стадии без очистки.
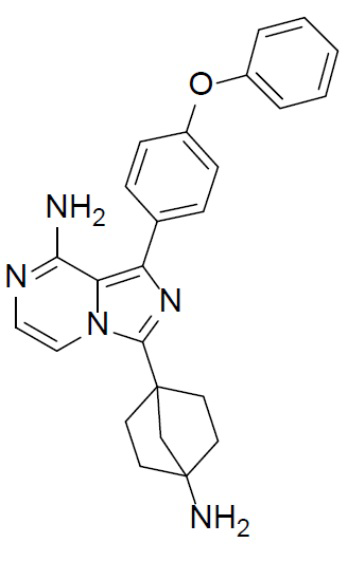
A-13-1: N-(4-(8-амино-1-(4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)-2-гидрокси-2-метилпропанамид
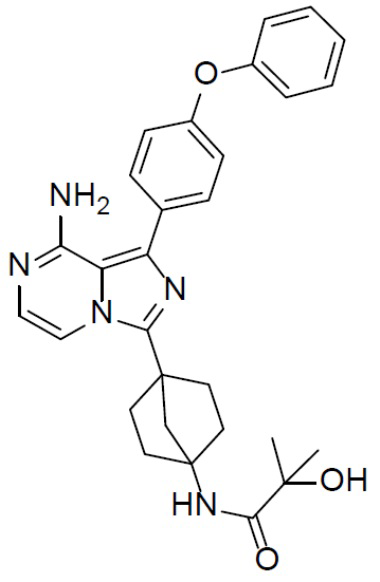
Соединение A-12 (15 мг, 0,025 ммоль), HATU (9,3 мг, 0,025 ммоль), DIEA (19 мг, 0,15 ммоль) и 2-гидрокси-2-пропионовой кислоты (3 мг, 0,029 ммоль) перемешивали при комнатной температуре в течение 1 ч, а затем данную смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-13-1 (8 мг, выход: 57%). LC-MS m/z = 498,4 [M+1]+.
A-13-2: N-(4-(8-амино-1-(4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)-3-метоксипропанамид
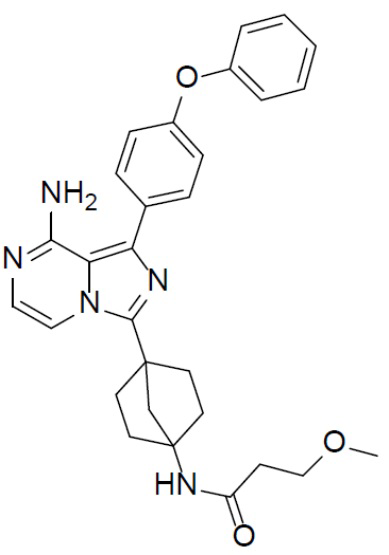
Соединение A-12 (15 мг, 0,025 ммоль), HATU (9,3 мг, 0,025 ммоль), DIEA (19 мг, 0,15 ммоль) и 3-метоксипропионовую кислоту (3 мг, 0,029 ммоль) перемешивали при комнатной температуре в течение 1 ч, а затем данную смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-13-2 (6 мг, выход: 40%). LC-MS m/z = 498,7 [M+1]+.
A-13-3: N-(4-(8-амино-1-(4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)-3-метилоксетан-3-карбоксамид
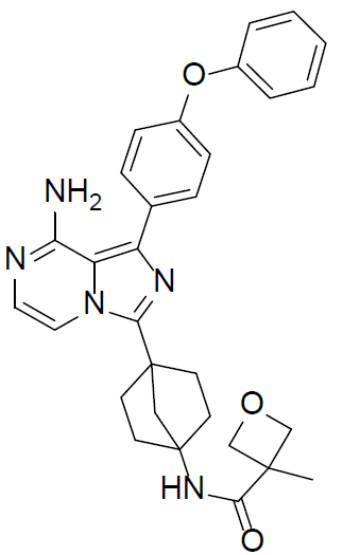
Раствор соединения A-12 (15 мг, 0,025 ммоль), HATU (9,3 мг, 0,025 ммоль), DIEA (19 мг, 0,15 ммоль) и 3-метилоксетан-3-карбоновой кислоты (3,4 мг, 0,029 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (5 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-13-3 (8 мг, выход: 53%). LC-MS m/z = 510,2 [M+1]+.
A-13-4: N-(4-(8-амино-1-(4-феноксифенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)-2-морфолиноацетамид
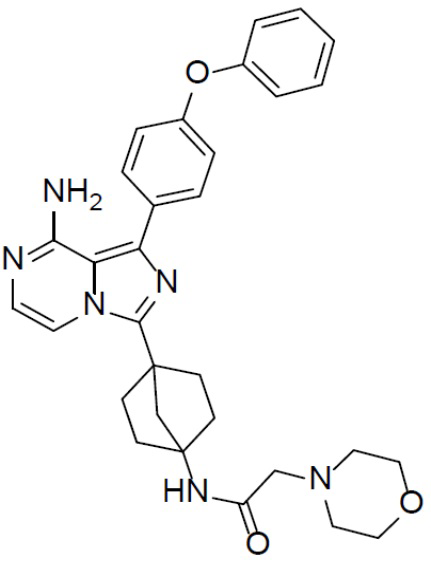
Раствор соединения A-12 (15 мг, 0,025 ммоль), HATU (9,3 мг, 0,025 ммоль), DIEA (19 мг, 0,15 ммоль) и 2-морфолиноуксусной кислоты (4,2 мг, 0,029 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-13-4 (9 мг, выход: 58%).
Путь синтеза соединения A-16-n
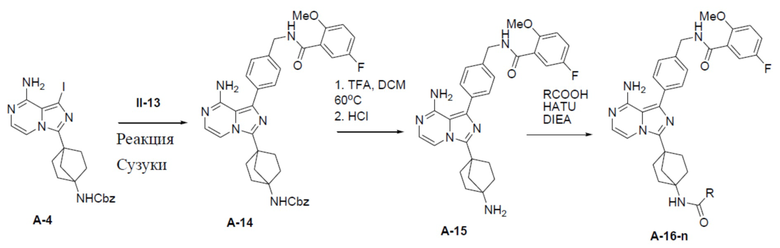
A-14: бензил (4-(8-амино-1-(4-((5-фтор-2-метоксибензамидо)метил)фенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)карбамат
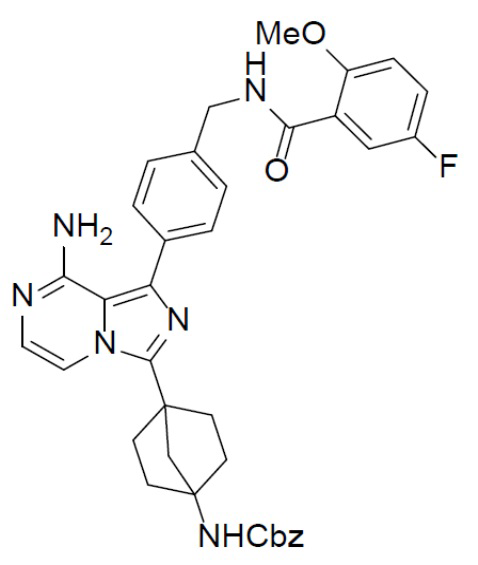
Раствор бензил(4-(8-амино-1-йодимидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)карбамата (A-4) (300 мг, 0,6 ммоль), 5-фтор-2-метокси-N-(4-(4,4,5,5- тетраметил-1,3,2-диоксаборолан-2-ил)бензил)бензамида (II-12) (284 мг, 0,738 ммоль), Pd[PPh3]4 (69 мг, 0,06 ммоль) и Cs2O3 (239 мг, 0,738 ммоль) в смешанном растворителе DME:H2O (2,5 мл:0,5 мл) нагревали до 80°C и перемешивали на протяжении ночи. Затем смесь концентрировали, а остаток очищали колоночной хроматографией с получением искомого соединения A-14 (285 мг, выход: 75%).
A-15: N-(4-(8-амино-3-(4-аминобицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)бензил)-5-фтор-2-метоксибензамид
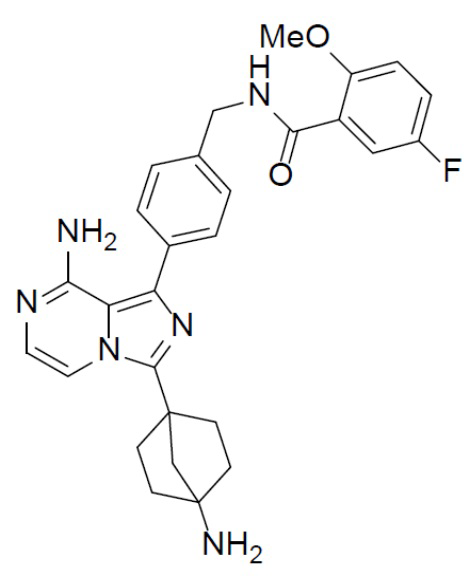
Раствор соединения A-14 (280 мг, 0,44 ммоль) в смешанном растворителе DCM/TFA (10 мл:10 мл) нагревали до 60°C и перемешивали в течение 18 ч. После этого смесь упаривали до сухого состояния, добавляли DCM (20 мл × 2) и полученную в результате смесь концентрировали. Остаток растворяли в DCM (30 мл), а затем данный раствор добавляли к раствору HCl в диоксане (10 мл). После этого смесь упаривали до сухого состояния, затем к нему добавляли DCM (20 мл × 2) и полученную в результате смесь концентрировали с последующим добавлением простого изопропилового эфира (30 мл). После перемешивания в течение 2 ч смесь подвергали фильтрации и промывали простым изопропиловым эфиром (10 мл × 2) с получением гидрохлорида искомого продукта A-15 (180 мг), который непосредственно использовали на следующей стадии без очистки.
A-16-1: N-(4-(8-амино-3-(4-(2-гидрокси-2-метилпропанамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)бензил)-5-фтор-2-метоксибензамид
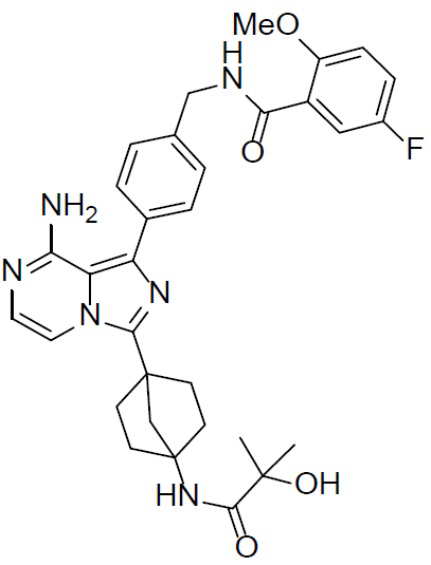
Раствор соединения A-15 (15 мг, 0,025 ммоль), HATU (9,3 мг, 0,025 ммоль), DIEA (19 мг, 0,15 ммоль) и 2-гидрокси-2-пропионовой кислоты (2,6 мг, 0,025 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-16-1 (6 мг, выход: 40%). LC-MS m/z = 587,3 [M+1]+.
A-16-2: N-(4-(8-амино-3-(4-(3-метоксипропанамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)бензил)-5-фтор-2-метоксибензамид
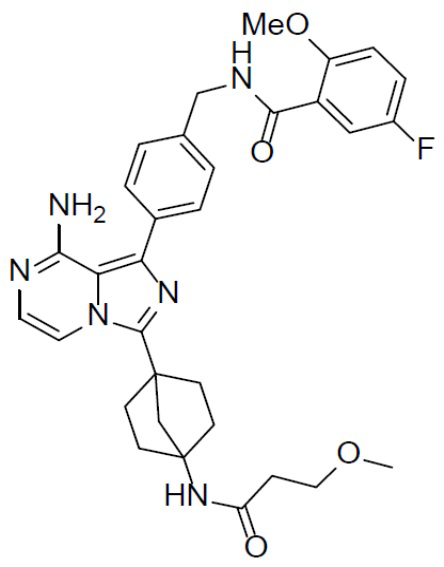
Раствор соединения A-15 (15 мг, 0,025 ммоль), HATU (9,3 мг, 0,025 ммоль), DIEA (19 мг, 0,15 ммоль) и 3-метоксипропионовой кислоты (2,6 мг, 0,025 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-16-2 (8 мг, выход: 40%). LC-MS m/z = 587,3 [M+1]+.
A-16-3: N-(4-(8-амино-1-(4-((5-фтор-2-метоксибензамидо)метил)фенил)имидазо[1,5-a]пиразин-3-ил)бицикло[2.2.1]гептан-1-ил)-3-метилоксетан-3-карбоксамид
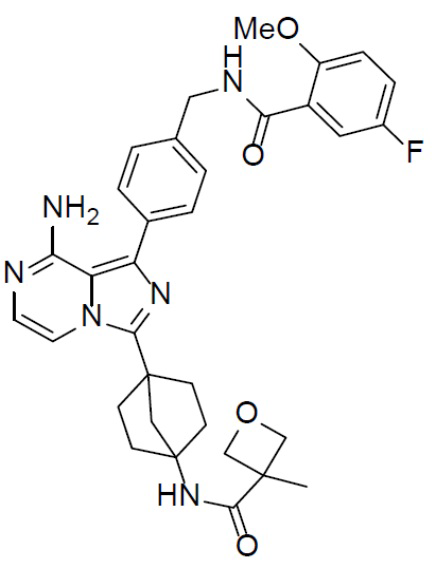
Раствор соединения A-15 (15 мг, 0,025 ммоль), HATU (9,3 мг, 0,025 ммоль), DIEA (19 мг, 0,15 ммоль) и 3-метилоксетан-3-карбоновой кислоты (2,9 мг, 0,025 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (5 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-16-3 (10 мг, выход: 66%). LC-MS m/z = 599,3 [M+1]+.
A-16-4: N-(4-(8-амино-3-(4-(2-морфолиноацетамидо)бицикло[2.2.1]гептан-1-ил)имидазо[1,5-a]пиразин-1-ил)бензил)-5-фтор-2-метоксибензамид
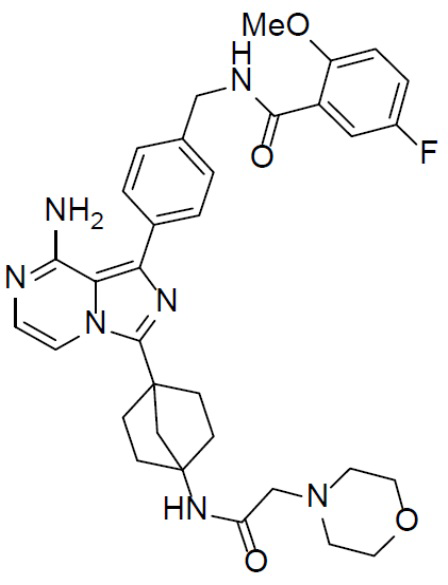
Раствор соединения A-15 (15 мг, 0,025 ммоль), HATU (9,3 мг, 0,025 ммоль), DIEA (19 мг, 0,15 ммоль) и 2-морфолиноуксусной кислоты (3,6 мг, 0,025 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта A-16-4 (9 мг, выход: 57%). LC-MS m/z = 628,3 [M+1]+.
Путь синтеза соединения B-6-n
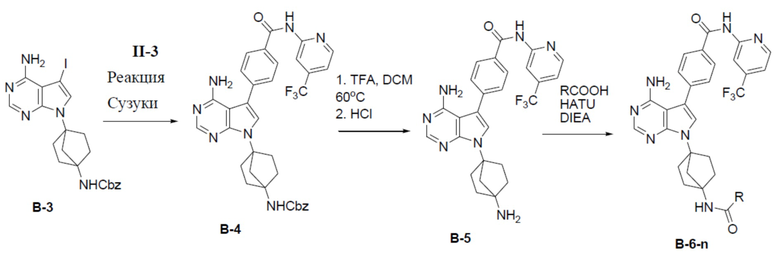
B-4: бензил(4-(4-амино-5-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)-7H-пирроло[2,3-d]пиримидин-7-ил)бицикло[2.2.1]гептан-1-ил)карбамат
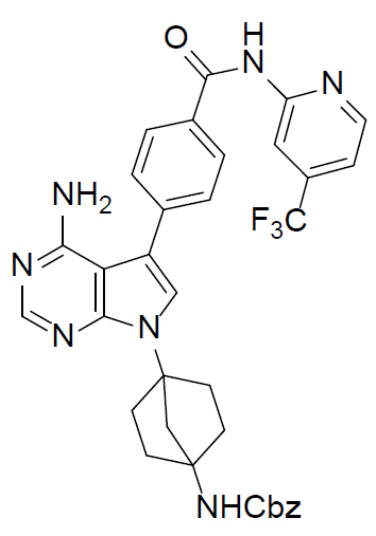
Раствор соединения B-3 (200 мг, 0,4 ммоль), (4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)бориновой кислоты (II-3) (152 мг, 0,49 ммоль), Pd[PPh3]4 (46 мг, 0,04 ммоль) и Cs2CO3 (159 мг, 0,49 ммоль) в смешанном растворителе DME:H2O (2,5 мл:0,5 мл) нагревали до 80°C и перемешивали на протяжении ночи. Смесь концентрировали, а остаток очищали колоночной хроматографией (метанол/DCM = 1:40) с получением искомого соединения B-4 (187 мг, выход: 73%).
B-5: 4-(4-амино-7-(4-аминобицикло[2.2.1]гептан-1-ил)-7H-пирроло[2,3-d]пиримидин-5-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
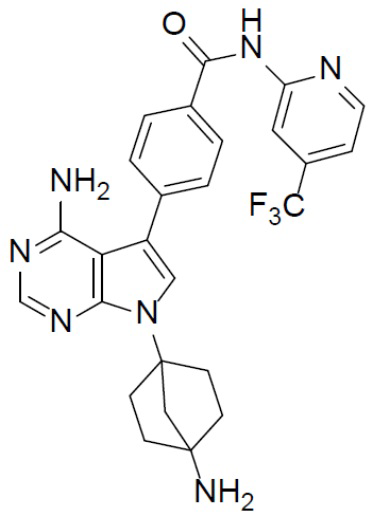
Раствор соединения B-4 (187 мг, 0,29 ммоль) в смешанном растворителе DCM/TFA (10 мл:10 мл) нагревали до 60°C и перемешивали в течение 18 ч. После этого смесь упаривали до сухого состояния, добавляли DCM (20 мл × 2) и полученную в результате смесь концентрировали. К остатку добавляли DCM (30 мл), а затем данную смесь добавляли к раствору HCl в диоксане (10 мл). После этого смесь упаривали до сухого состояния, затем снова добавляли DCM (20 мл × 2) и полученную в результате смесь упаривали до сухого состояния с последующим добавлением простого изопропилового эфира (30 мл). После перемешивания в течение 2 ч смесь подвергали фильтрации и промывали простым изопропиловым эфиром (10 мл × 2) с получением гидрохлорида искомого продукта B-5, который непосредственно использовали на следующей стадии без очистки.
B-6-1: 4-(4-амино-7-(4-(бут-2-инамидо)бицикло[2.2.1]гептан-1-ил)-7H-пирроло[2,3-d]пиримидин-5-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
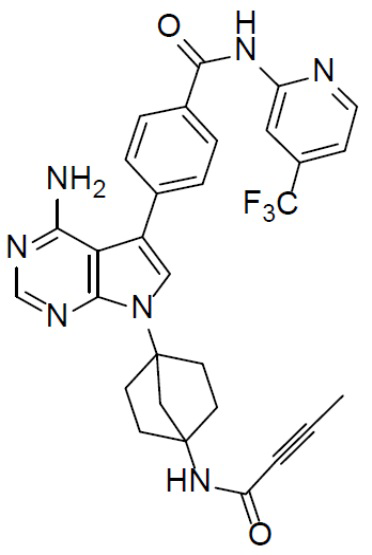
Раствор соединения B-5 (19 мг, 0,031 ммоль), HATU (12 мг, 0,031 ммоль), DIEA (24 мг, 0,19 ммоль) и бут-2-иновой кислоты (2,6 мг, 0,031 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта B-6-1 (11 мг, выход: 59%). LC-MS m/z = 574,3 [M+1]+.
B-6-2: 4-(4-амино-7-(4-(2-гидрокси-2-метилпропанамидо)бицикло[2.2.1]гептан-1-ил)-7H-пирроло[2,3-d]пиримидин-5-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
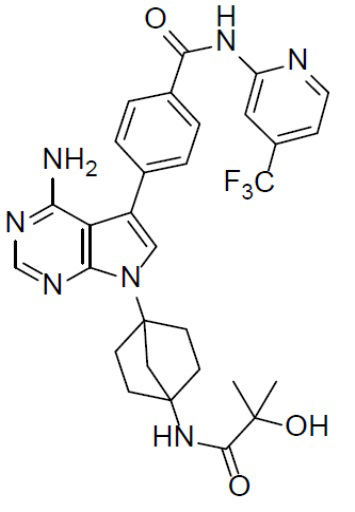
Раствор соединения B-5 (19 мг, 0,031 ммоль), HATU (12 мг, 0,031 ммоль), DIEA (24 мг, 0,19 ммоль) и 2-гидрокси-2-метилпропионовой кислоты (3,2 мг, 0,031 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта B-6-2 (9 мг, выход: 49%). LC-MS m/z = 594,2 [M+1]+.
B-6-3: 4-(4-амино-7-(4-(3-метоксипропанамидо)бицикло[2.2.1]гептан-1-ил)-7H-пирроло[2,3-d]пиримидин-5-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
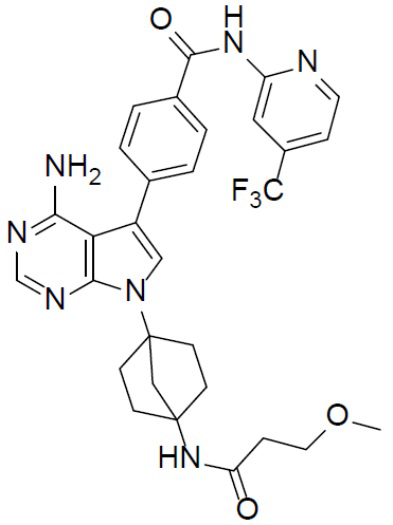
Раствор соединения B-5 (19 мг, 0,031 ммоль), HATU (12 мг, 0,031 ммоль), DIEA (24 мг, 0,19 ммоль) и 3-метоксипропионовой кислоты (3,2 мг, 0,031 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта B-6-3 (12 мг, выход: 66%). LC-MS m/z = 594,2 [M+1]+.
B-6-4: N-(4-(4-амино-5-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)-7H-пирроло[2,3-d]пиримидин-7-ил)бицикло[2.2.1]гептан-1-ил)-3-метилоксетан-3-карбоксамид
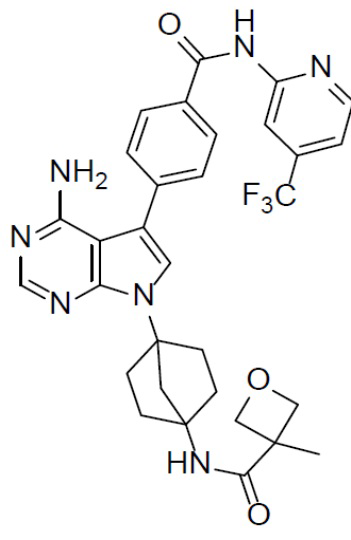
Раствор соединения B-5 (19 мг, 0,031 ммоль), HATU (12 мг, 0,031 ммоль), DIEA (24 мг, 0,19 ммоль) и 3-метилоксетан-3-карбоновой кислоты (3,6 мг, 0,031 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта B-6-4 (12 мг, выход: 64%). LC-MS m/z = 606,3 [M+1]+.
B-6-5: 4-(4-амино-7-(4-(2-морфолиноацетамидо)бицикло[2.2.1]гептан-1-ил)-7H-пирроло[2,3-d]пиримидин-5-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
Раствор соединения B-5 (19 мг, 0,031 ммоль), HATU (12 мг, 0,031 ммоль), DIEA (24 мг, 0,19 ммоль) и 2-морфолиноуксусной кислоты (4,5 мг, 0,031 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта B-6-5 (11 мг, выход: 58%). LC-MS m/z = 635,2 [M+1]+.
Путь синтеза соединения C-7-n
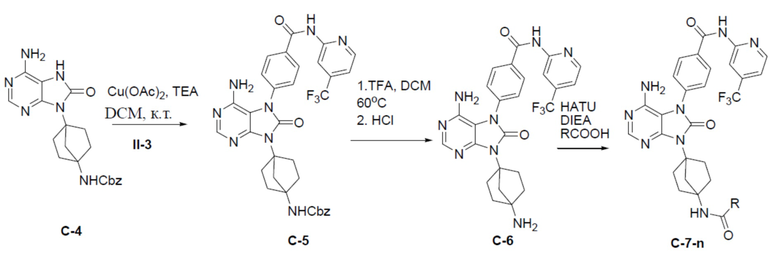
C-5: бензил(4-(6-амино-8-оксо-7-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)-7,8-дигидро-9H-пурин-9-ил)бицикло[2.2.1]гептан-1-ил)карбамат
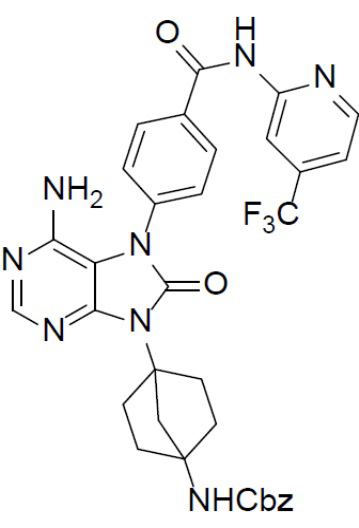
Раствор соединения C-4 (100 мг, 0,25 ммоль), (4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)бориновой кислоты (II-3) (160 мг, 0,5 ммоль), ацетaта меди (60 мг, 0,3 ммоль) и Et3N (30 мг, 0,3 ммоль) в DCM (10 мл) перемешивали при комнатной температуре в течение 24 ч, а затем смесь выливали в воду (20 мл) и подвергали экстракции этилацетатом (10 мл × 2). Органическую фазу отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (EA/PE = 1:1 – EA) с получением искомого соединения C-5 (47 мг, выход: 29%). LC-MS m/z = 657,3 [M-1]-.
C-6: 4-(6-амино-9-(4-аминобицикло[2.2.1]гептан-1-ил)-8-оксо-8,9-дигидро-7H-пурин-7-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
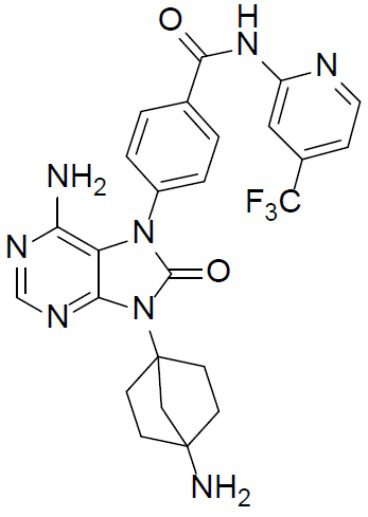
Раствор соединения C-5 (45 мг, 0,09 ммоль) в смешанном растворителе DCM/TFA (10 мл:10 мл) нагревали до 60°C и перемешивали в течение 18 ч. После этого смесь упаривали до сухого состояния, добавляли DCM (20 мл × 2) и полученную в результате смесь концентрировали. Остаток растворяли в DCM (30 мл), а затем данный раствор добавляли к раствору HCl в диоксане (10 мл). После этого смесь упаривали до сухого состояния, затем к нему добавляли DCM (20 мл × 2) и полученную в результате смесь концентрировали с последующим добавлением простого изопропилового эфира (30 мл). После перемешивания в течение 2 ч смесь подвергали фильтрации и промывали простым изопропиловым эфиром (10 мл × 2) с получением гидрохлорида искомого продукта C-6, который непосредственно использовали на следующей стадии без очистки.
C-7-1: 4-(6-амино-9-(4-(бут-2-инамидо)бицикло[2.2.1]гептан-1-ил)-8-оксо-8,9-дигидро-7H-пурин-7-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
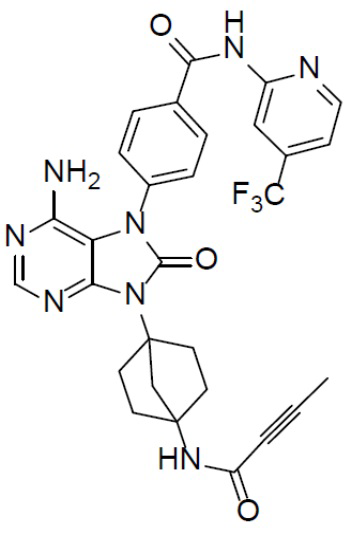
Раствор соединения C-6 (13 мг, 0,022 ммоль), HATU (9 мг, 0,022 ммоль), DIEA (16,8 мг, 0,13 ммоль) и бут-2-иновой кислоты (1,8 мг, 0,022 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта C-7-1 (5 мг, выход: 38%). LC-MS m/z = 591,2 [M+1]+.
C-7-2: 4-(6-амино-9-(4-(2-морфолиноацетамидо)бицикло[2.2.1]гептан-1-ил)-8-оксо-8,9-дигидро-7H-пурин-7-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
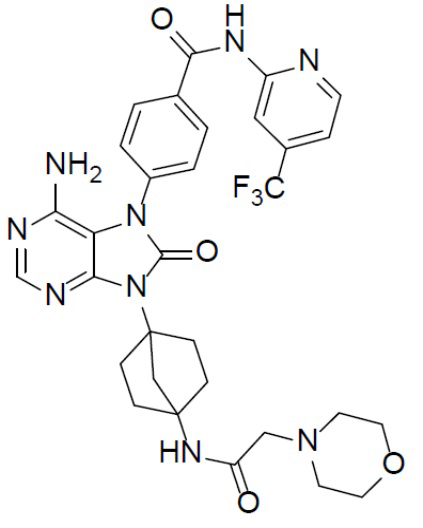
Раствор соединения C-6 (13 мг, 0,022 ммоль), HATU (9 мг, 0,022 ммоль), DIEA (16,8 мг, 0,13 ммоль) и 2-морфолиноуксусной кислоты (3,2 мг, 0,022 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 1 ч, а затем смесь выливали в воду (1 мл) и подвергали экстракции этилацетатом (5 мл × 2). Органическую фазу промывали насыщенным водным раствором NaCl, отделяли, сушили над безводным Na2SO4, подвергали фильтрации и упаривали до сухого состояния. Остаток очищали колоночной хроматографией (DCM/MeOH = 20:1) с получением продукта C-7-2 (6 мг, выход: 42%). LC-MS m/z = 652,2 [M+1]+.
Путь синтеза соединения D-8-n
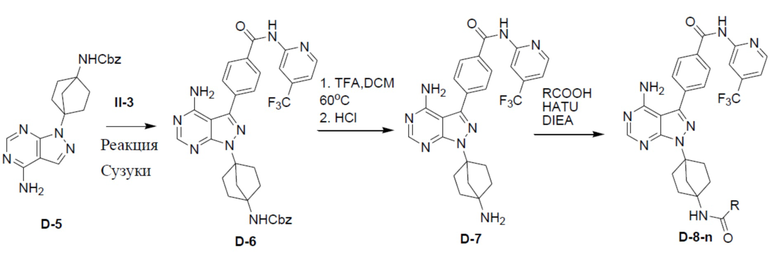
D-6: бензил(4-(4-амино-3-(4-((4-(трифторметил)пиридин-2-ил)карбамоил)фенил)-1H-пиразоло[3,4-d]пиримидин-1-ил)бицикло[2.2.1]гептан-1-ил)карбамат
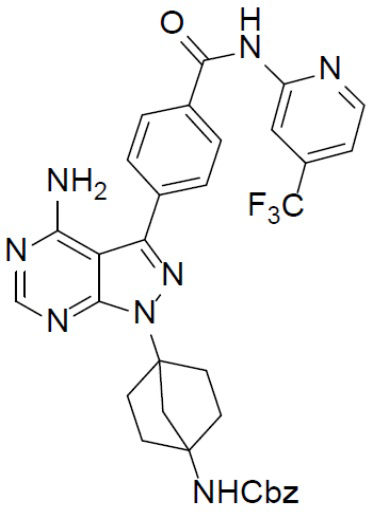
Смешанный раствор D-5 (96 мг, 0,19 ммоль), II-3 (73 мг, 0,234 ммоль), Pd[PPh3]4 (22 мг, 0,019 ммоль) и Cs2CO3 (76,4 мг, 0,234 ммоль) в DME/H2O (9 мл/1 мл) продували с помощью N2 в течение 1 минуты для удаления кислорода, а затем нагревали до 80°C в герметично закрытой пробирке и перемешивали в течение 12 ч. После этого смесь охлаждали, реакцию гасили солевым раствором (20 мл) и для экстракции добавляли EA (50 мл × 2). Органическую фазу сушили над безводным Na2SO4, подвергали фильтрации и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: PE/EA = 1:1) с получением искомого продукта D-6 (42 мг, 34,4%). LC-MS m/z = 643,2 [M+1]+.
D-7: 4-(4-амино-1-(4-аминобицикло[2.2.1]гептан-1-ил)-1H-пиразоло[3,4-d]пиримидин-3-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
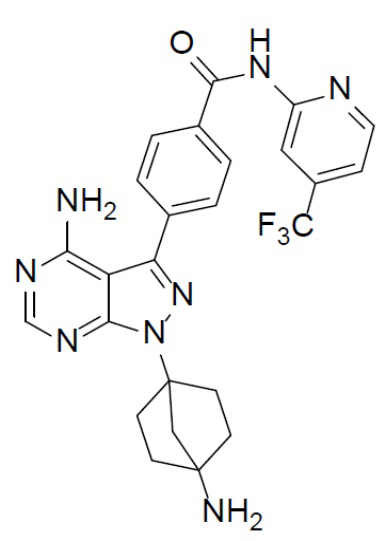
Смесь D-6 (42 мг, 0,065 ммоль) в TFA/DCM (1 мл/1 мл) перемешивали при 60°C в течение 12 ч. После этого смесь охлаждали, удаляли растворитель путем упаривания. Затем добавляли раствор HCl в диоксане (2 мл) и полученную в результате смесь перемешивали в течение 10 минут и концентрировали. К остатку добавляли простой изопропиловый эфир (10 мл), перемешивали и концентрировали. Описанную выше процедуру повторяли два раза. Твердое вещество подвергали фильтрации, промывали простым изопропиловым эфиром и сушили с получением гидрохлорида искомого продукта D-7 (30 мг, 75%).
D-8-1: 4-(4-амино-1-(4-(бут-2-инамидо)бицикло[2.2.1]гептан-1-ил)-1H-пиразоло[3,4-d]пиримидин-3-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
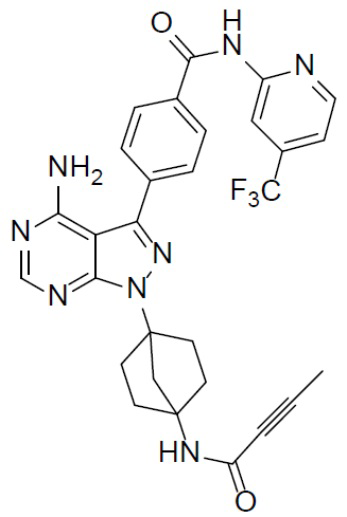
Смешанный раствор D-7 (15 мг, 0,025 ммоль), бут-2-иновой кислоты (2 мг, 0,024 ммоль), HATU (9,1 мг, 0,024 ммоль) и DIEA (0,041 мл, 0,241 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 4 ч. Реакционный раствор разводили с помощью EA (30 мл) и промывали солевым раствором, а органическую фазу сушили над безводным Na2SO4, подвергали фильтрации и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: PE/EA = 1:1) с получением искомого продукта D-8-1 (12 мг, 86%). LC-MS m/z = 575,1 [M+1]+.
D-8-2: 4-(4-амино-1-(4-(2-морфолиноацетамидо)бицикло[2.2.1]гептан-1-ил)-1H-пиразоло[3,4-d]пиримидин-3-ил)-N-(4-(трифторметил)пиридин-2-ил)бензамид
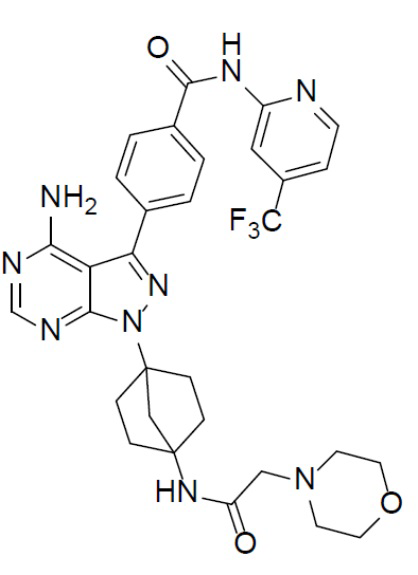
Смешанный раствор D-7 (15 мг, 0,025 ммоль), 2-морфолиноуксусной кислоты (3,5 мг, 0,024 ммоль), HATU (9,1 мг, 0,024 ммоль) и DIEA (0,041 мл, 0,241 ммоль) в DMF (1 мл) перемешивали при комнатной температуре в течение 4 ч. Реакционный раствор разводили с помощью EA (30 мл) и промывали солевым раствором, а органическую фазу сушили над безводным Na2SO4, подвергали фильтрации и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: PE/EA = 1:1) с получением искомого продукта D-8-2 (8,5 мг, 56%). LC-MS m/z = 636,3 [M+1]+.
1. Ингибирующая активность in vitro в отношении BTK(wt)/BTK(C481S) (определение значения IC50)
(1) Способ
Раствор субстрата готовили путем добавления натриевой соли субстрата поли(Glu, Tyr) (Sigma Aldrich, Сент-Луис, Миссури) к реакционному буферу для субстрата (20 мМ Hepes (pH 7,5), 10 мМ MgCl2, 1 мМ EGTA, 0,02% Brij35, 0,02 мг/мл BSA, 0,1 мМ Na3VO4, 2 мМ DTT и 1% DMSO) (конечная концентрация субстрата в реакционном растворе составляла 0,2 мкМ). Тестируемое соединение составляли в виде маточных растворов с концентрацией 10 мМ со 100% DMSO и для 10 доз производили 3-кратное серийное разведение в 384-луночном микропланшете с LDV (нижним недействующим объемом) из циклоолефинового сополимера. Киназу BTK(wt) или BTK(C481S) (рекомбинантный человеческий полноразмерный белок с гистидиновой меткой, экспрессированный в клетках насекомых, Invitrogen, Карлсбад, Калифорния) добавляли к раствору субстрата и осторожно перемешивали (конечная концентрация BTK в реакционном растворе составляла 8 нМ). Затем к реакционной смеси с киназой добавляли тестируемое соединение в 100% DMSO с помощью технологии акустического переноса жидкости (Echo 550; нанолитровый диапазон) (Labcyte Inc, Саннивейл, Калифорния) и инкубировали в течение 20 минут при комнатной температуре. Для инициации реакции к реакционной смеси добавляли 33P-ATP (удельная активность 10 мкКи/мкл) с последующей инкубацией при комнатной температуре в течение 2 часов. Небольшую часть реакционной смеси капали на ионообменную фильтровальную бумагу Р-81 (Whatman). После отмывания несвязанного фосфата от фильтровальной бумаги 0,75% фосфатным буфером (три раза) и сушки измеряли оставшуюся радиоактивность на фильтровальной бумаге. Киназную активность выражали как процент оставшейся киназной активности в тестируемом образце к киназной активности в холостом контроле в виде наполнителя (диметилсульфоксида). Значения IC50 рассчитывали путем аппроксимации кривой из полученных данных с использованием Prism (программного обеспечения GraphPad).
(2) Результаты
По сравнению с ингибитором BTK второго поколения акалабрутинибом, большинство раскрываемых в настоящем документе соединений обладали более сильной ингибирующей способностью в отношении активности фермента BTK(wt) дикого типа. Что еще более важно, большинство соединений характеризовались более высокой ингибирующей активностью в отношении мутантов BTK(C481S), а для некоторых даже наблюдали ингибирующую активность менее 0,1 нМ.
Таблица 1. Ингибирование активности фермента BTK(wt)/BTK(C481S) (A≤0,1 нМ; 0,1 нМ<B≤1 нМ; 1 нМ<C≤10 нМ; 10 нМ<D≤50 нМ)
BTK(wt)
BTK(C481S)
BTK(wt)
BTK(C481S)
2. Эксперимент по ингибированию пролиферации опухолевых клеток in vitro
(1) Способ
• Линию опухолевых клеток (TMD-8/OCY-LY10) суспендировали в RPMI1640 + FBS10% и культивировали в термостате (37°C, 5% CO2). Клетки периодически пересевали, а клетки для посева собирали в фазе логарифмического роста.
• Проводили окрашивание клеток трипановым синим и подсчитывали жизнеспособные клетки.
• Концентрацию клеток доводили до 7000 клеток/лунка с помощью среды.
• 90 мкл клеточной суспензии вносили в 96-луночный планшет, а в холостые контрольные лунки вносили культуральную среду без клеток.
• Клетки в 96-луночном планшете инкубировали на протяжении ночи в термостате (37°C, 5% CO2, 100% относительная влажность).
• Подготовка планшетов для хранения с 400× концентрацией соединений: тестируемые соединения и эталонное лекарственное средство растворяли в DMSO и 3× разводили от наивысшей концентрации (400 мкМ) до наиболее низкой концентрации (0,61 мкМ).
• Приготовление рабочих растворов с 10× концентрацией соединения: 78 мкл среды для культивирования клеток вносили в 96-луночный планшет с V-образным дном и при помощи пипетки переносили 2 мкл соединения из планшета для хранения с 400× концентрацией соединений в 96-луночный планшет. В контрольные лунки с наполнителем и в холостые контрольные лунки добавляли 2 мкл DMSO. После добавления соединения или DMSO смесь тщательно перемешивали с помощью пипетки.
• Добавление соединения: 10 мкл рабочего раствора с 10× концентрацией соединения (как показано в таблице 1) вносили в планшет для культивирования клеток. 10 мкл смеси DMSO и раствора культуры клеток вносили в контрольные лунки с наполнителем и в холостые контрольные лунки. Конечная концентрация DMSO составляла 0,25%.
• 96-луночный планшет с клетками снова помещали в термостат на 72 часа культивирования.
• Размораживали буфер CellTiter-Glo и оставляли его отстаиваться до комнатной температуры.
• Субстрат CellTiter-Glo оставляли отстаиваться до комнатной температуры.
• С целью приготовления рабочего раствора CellTiter-Glo в колбу с субстратом CellTiter-Glo добавляли буфер CellTiter-Glo для растворения.
• Колбу подвергали медленному встряхиванию на вортексе до полного растворения субстрата.
• Вынимали планшет для культивирования клеток и оставляли на 30 минут для уравновешивания до комнатной температуры.
• В каждую лунку вносили 50 мкл (что равно половине объема среды для культивирования клеток в каждой лунке) рабочего раствора CellTiter-Glo. Планшет с клетками заворачивали в алюминиевую фольгу для защиты от света.
• Планшет встряхивали на орбитальном шейкере в течение 2 минут для индукции лизиса клеток.
• Планшет оставляли на 10 минут отстояться в условиях комнатной температуры для стабилизации люминесцентных сигналов.
• Люминесцентные сигналы детектировали на планшет-ридере 2104 EnVision.
• Анализ данных: степень ингибирования (IR) тестируемого соединения рассчитывали по следующей формуле: IR (%) = (1 – (RLU соединения – RLU холостого контроля) / (RLU контрольного наполнителя – RLU холостого контроля)) × 100%. Значения степени ингибирования соединениями в различных концентрациях рассчитывали в Excel, после чего строили кривые ингибирования и рассчитывали значения IC50 с помощью GraphPad Prism.
(2) Результаты
В эксперименте по ингибированию пролиферации клеток В-клеточной лимфомы TMD-8 и OCI-LY10 соединение A-10-10 и ибрутиниб характеризовались сильной ингибирующей активностью в отношении пролиферации опухолевых клеток.
Таблица 2. Ингибирующая активность соединения A-10-10/ибрутиниба в отношении пролиферации опухолевых клеток
3. Фармакокинетическое исследование
(1) Способ
Самцов крыс SD не кормили на протяжении ночи перед тестом. Каждое из тестируемых лекарственных средств суспендировали в растворе 0,5% метилцеллюлозы (MC) и 0,1% SDS в деионизированной воде (вес/вес/объем) и внутрижелудочно вводили суспензию в количестве 5 мл/кг, в каждом случае с концентрацией 1 мг/мл. Через 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа после введения из орбитального венозного сплетения животных брали примерно 0,4 мл цельной крови, а затем помещали ее в пробирки с гепариновым антикоагулянтом. В каждый момент времени брали пробы у трех животных, а затем их умерщвляли и за счет этого сбор проб производили у разных особей. Образец цельной крови центрифугировали в течение 15 минут и центрифугировали в условиях 4°C/4200 об./мин в течение 5 минут. Все образцы плазмы до анализа хранили в холодильнике при температуре –80±15°C. Перед анализом образца проводили LC-MS/MS-анализ (тандемную масс-спектрометрию UPLC-Xevo TQD Waters I) с целью выявления соединений. Собранную плазму анализировали количественно, данные о концентрации в плазме крови в зависимости от времени у животных анализировали с помощью программного обеспечения WinNonlin (профессиональное издание, версия 5.2), для анализа концентрации использовали некомпартментную модель и рассчитывали фармакокинетические параметры тестируемых соединений.
Трех взрослых самцов биглей не кормили на протяжении ночи перед тестом. Каждое из тестируемых лекарственных средств суспендировали в растворе 0,5% метилцеллюлозы (MC) и 0,1% SDS в деионизированной воде (вес/вес/объем) и внутрижелудочно вводили суспензию в количестве 5 мл/кг, в каждом случае с концентрацией 1 мг/мл. Кровь брали через 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа после введения. Животных подвергали легкой анестезии хлоральгидратом, и из вены передних конечностей брали примерно 0,5 мл цельной крови, используя стеклянную пробирку для забора крови, и помещали в пробирку с гепариновым антикоагулянтом. Образец центрифугировали в условиях 4°C/4200 об./мин в течение 5 минут. Все образцы плазмы до анализа хранили в холодильнике при температуре –80±15°C. Перед анализом образца проводили LC-MS/MS-анализ (тандемную масс-спектрометрию UPLC-Xevo TQD Waters I) с целью выявления соединений. Собранную плазму анализировали количественно, данные о концентрации в плазме крови в зависимости от времени у животных анализировали с помощью программного обеспечения WinNonlin (профессиональное издание, версия 5.2), для анализа концентрации использовали некомпартментную модель и рассчитывали фармакокинетические параметры тестируемых соединений.
(1) Результаты
У соединения A-10-10 (вводимого внутрижелудочно, 5 мг/кг) наблюдали хорошее всасывание с Cmax 8323 нг/мл и 641 нг/мл у крыс и биглей соответственно, и оно имело довольно высокую концентрацию в крови (AUC0-INF = 73318 ч*нг/мл и 14867 ч*нг/мл).
Таблица 3. Фармакокинетические данные для крыс/биглей (самцов), подвергнутых внутрижелудочному введению (5 мг/кг) (t1/2: период полувыведения; Tmax: время пиковой концентрации; Cmax: максимальная концентрация в плазме; AUC0-INF: область под кривой зависимости времени 0-INF от концентрации)
4. Эксперимент по ингибированию pBTK на клетках HEK293, трансфицированных посредством BTK(wt) и BTK(C481S)
Основная причина лекарственной устойчивости к ибрутинибу заключается в мутации C481S у киназы BTK, и для преодоления лекарственной устойчивости к ибрутинибу большое значение имеет разработка ингибитора BTK, который может эффективно ингибировать клетки с модификацией BTK(C481S).
(1) Способ
• Эмбриональные клетки почки человека HEK293 временно трансфицировали вектором с человеческой полноразмерной BTK или BTK(C481S).
• Клетки распределяли в 96-луночном планшете с заранее определенной плотностью клеток.
• Каждое тестируемое соединение подвергали 3-кратному серийному разведению восемь раз с помощью 100% DMSO.
• Затем соединение разводили в среде для культивирования тканей до 10-кратной конечной аналитической концентрации и 5% DMSO.
• К клеткам в 96-луночном планшете (10-кратное разведение в культуральной среде) добавляли соединение, и его конечной концентрацией была 1× концентрация соединения и 0,5% DMSO. В случае положительных (с высоким уровнем сигнала) контролей клетки обрабатывали только 0,5% DMSO. В случае отрицательных (с низким уровнем сигнала) контролей клетки обрабатывали 20 мкМ ибрутинибом, а конечная концентрация составляла 0,5% DMSO.
• Клетки инкубировали с соединениями в течение 2 ч при 37°C.
• Клетки лизировали, а лизат переносили в планшет для ИФА с предварительно нанесенным покрытием антитела, захватывающего субстрат (человеческую BTK или BTK(C481S)).
• Планшет промывали, а затем инкубировали с антителом, связанным с HRP, для детекции общего фосфорилирования тирозина.
• Промывали планшет, а затем вносили субстрат для HRP. Поглощение считывали на 450 нм.
• Исходя из полученных значений оптической плотности положительных и отрицательных контролей, рассчитывали %-значение ингибирования по следующей формуле: (%INH = ((положительный контроль - образец) / (положительный контроль - отрицательный контроль)) × 100.
• Значение Z' рассчитывали по следующей формуле: 1 – ((3 × стандартное отклонение положительного + 3 × стандартное отклонение отрицательного) / (среднее положительного – среднее отрицательного)).
• С помощью GraphPad Prism строили график зависимости логарифма %-значения ингибирования от значения концентрации соединения.
• Значения IC50 определяли после экстраполяции для построения сигмоидальной кривой зависимости ответа от дозы.
(2) Результаты
Мутация C481S снижала ингибирование ибрутинибом фосфорилирования BTK у клеток HEK293 с 0,021 мкМ до 1,58 мкМ, тогда как раскрываемое в настоящем документе соединение A-10-10 оказывало сильное ингибирование (0,077 мкМ) в отношении клеток HEK293, трансфицированных посредством BTK(wt), и еще более сильное ингибирование (0,066 мкМ) в отношении клеток HEK293, трансфицированных посредством BTK(C481S).
Таблица 4. Значения ингибирования pBTK (IC50) в отношении клеток HEK293, трансфицированных посредством BTK(wt) и BTK(C481S)
1. Фармакодинамический эксперимент 1
(1) Способ
Самок мышей CB17/SCID с серьезным иммунодефицитом приобретали у компании Beijing Vital River Laboratory Animal Technology Co., Ltd. и разводили в помещениях для SPF-животных (без специфической патогенной микрофлоры). Клетки OCI-LY10 человека (Shanghai Junrui-UFBN0102) культивировали в культуральной среде RPMI 1640, содержащей 10% фетальной бычьей сыворотки, 100 ед./мл пенициллина и 100 мкг/мл стрептомицина, методом монослойной культуры in vitro в термостате (37°C, 5% CO2). Для пересева два раза в неделю производили стандартную обработку. При насыщении клеток до 80-90% и необходимом их количестве клетки собирали и подсчитывали. В правую часть спины каждой мыши подкожно инокулировали 0,2 мл (1×107) клеток OCI-LY10 (вместе с матригелем в объемном соотношении 1:1), и размер опухоли можно было измерять приблизительно через одну неделю после инокуляции. Размер опухоли измеряли с помощью штангенциркуля, а объем опухоли рассчитывали по следующей формуле: объем опухоли = (длина × ширина2) / 2. При достижении среднего объема опухоли 109 мм3 мышей разделяли на четыре группы (8 мышей в группе) для введения, т. е. на контрольную группу для введения наполнителя (5% DMSO + 20% HP-β-CD), группу внутрижелудочного введения ибрутиниба (25 мг/кг, один раз в сутки) и группу внутрижелудочного введения соединения A-10-10 (25 мг/кг, 50 мг/кг, два раза в сутки). Ибрутиниб или соединение A-10-10 растворяли в 5% DMSO + 20% HP-β-CD и вводили внутрижелудочно в количестве 10 мл/кг в течение 28 последовательных суток.
(2) Результаты
В случае соединения A-10-10 и ибрутиниба наблюдали чрезвычайно сильную противоопухолевую активность на ксенотрансплантатной модели опухоли OCI-LY10 (фиг. 1). Соединение A-10-10, вводимое внутрижелудочно (доза составляла 25 мг/кг, два раза в сутки; 50 мг/кг, два раза в сутки), могло значительно ингибировать рост диффузного штамма OCI-LY10 клеток крупноклеточной В-клеточной лимфомы (TGI = 110%, 110%; p<0,001, p<0,001). Через 21 сутки после введения все ксенотрансплантатные опухоли OCI-LY10 полностью исчезали в группе, получавшей 50 мг/кг. Через 28 суток после введения все ксенотрансплантатные опухоли OCI-LY10 также полностью исчезали в группе, получавшей 25 мг/кг. На 28-е сутки значение TGI у группы, получавшей контрольное соединение ибрутиниб в количестве 25 мг/кг, составляло 94% (p<0,001).
Влияние тестируемого вещества на массу тела у мышей с опухолью показано на фиг. 1. Мыши с опухолями демонстрировали хорошую переносимость тестируемого лекарственного средства А-10-10 при всех дозировках, и во всех группах обработки не наблюдали значимой потери массы тела.
2. Фармакодинамический эксперимент 2
(1) Способ
Самок мышей CB17/SCID с серьезным иммунодефицитом приобретали у компании Beijing Vital River Laboratory Animal Technology Co., Ltd. и разводили в помещениях для SPF-животных (без специфической патогенной микрофлоры). Клетки TMD-8 человека (Shanghai Junrui-UFBN1682) культивировали в культуральной среде RPMI 1640, содержащей 10% фетальной бычьей сыворотки, 100 ед./мл пенициллина и 100 мкг/мл стрептомицина, методом монослойной культуры in vitro в термостате (37°C, 5% CO2). Для пересева два раза в неделю производили стандартную обработку. При насыщении клеток до 80-90% и необходимом их количестве клетки собирали и подсчитывали. В правую часть спины каждой мыши подкожно инокулировали 0,2 мл (1×107) клеток TMD-8 (вместе с матригелем в объемном соотношении 1:1), и размер опухоли можно было измерять приблизительно через одну неделю после инокуляции. Размер опухоли измеряли с помощью штангенциркуля, а объем опухоли рассчитывали по следующей формуле: объем опухоли = (длина × ширина2) / 2. При достижении среднего объема опухоли 107 мм3 мышей разделяли на четыре группы (8 мышей в группе) для введения, т. е. на контрольную группу для введения наполнителя (5% DMSO + 20% HP-β-CD), группу внутрижелудочного введения ибрутиниба (25 мг/кг, один раз в сутки) и группу внутрижелудочного введения соединения A-10-10 (25 мг/кг, 50 мг/кг, два раза в сутки). Ибрутиниб или соединение A-10-10 растворяли в 5% DMSO + 20% HP-β-CD и вводили внутрижелудочно в количестве 10 мл/кг в течение 27 последовательных суток.
(2) Результаты
В случае соединения A-10-10 и ибрутиниба наблюдали чрезвычайно сильную противоопухолевую активность на ксенотрансплантатной модели опухоли TMD-8 (фиг. 2). Соединение A-10-10, вводимое внутрижелудочно (доза составляла 25 мг/кг, два раза в сутки; 50 мг/кг, два раза в сутки), могло значительно ингибировать рост диффузного штамма TMD-8 клеток крупноклеточной В-клеточной лимфомы (TGI = 94%, 104%; p<0,001, p<0,001). Через 27 суток после введения 5/8 ксенотрансплантатных опухолей TMD-8 исчезали в группе, получавшей 50 мг/кг. Значение TGI у группы, получавшей контрольное соединение ибрутиниб в количестве 25 мг/кг, составляло 90% (p<0,001).
Влияние тестируемого вещества на массу тела у мышей с опухолью показано на фиг. 2. Мыши с опухолями демонстрировали хорошую переносимость тестируемого лекарственного средства А-10-10 при всех дозировках, и во всех группах обработки не наблюдали значимой потери массы тела.
| название | год | авторы | номер документа |
|---|---|---|---|
| АМИДНОЕ ПРОИЗВОДНОЕ ПИРАЗОЛА | 2015 |
|
RU2658827C2 |
| НЕКОТОРЫЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ | 2015 |
|
RU2671494C2 |
| СШИТАЯ ИСКУССТВЕННАЯ НУКЛЕИНОВАЯ КИСЛОТА ALNA | 2019 |
|
RU2820226C2 |
| ИМИДАЗОПИРАЗИНОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE1 | 2016 |
|
RU2712219C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ВЕЩЕСТВА - АГОНИСТЫ GPR119 | 2017 |
|
RU2734500C2 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ ТРАНСПОРТЕРОВ | 2010 |
|
RU2552353C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2629118C2 |
| 2,5,6,7-ТЕТРАГИДРО-[1,4]ОКСАЗЕПИН-3-ИЛАМИНЫ ИЛИ 2,3,6,7-ТЕТРАГИДРО-[1,4]ОКСАЗЕПИН-5-ИЛАМИНЫ | 2011 |
|
RU2570796C2 |
| Производные 1-фенил-2-пиридинилалкиловых спиртов в качестве ингибиторов фосфодиэстеразы | 2013 |
|
RU2655170C2 |
| МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ КИНАЗ TRK | 2019 |
|
RU2778294C2 |
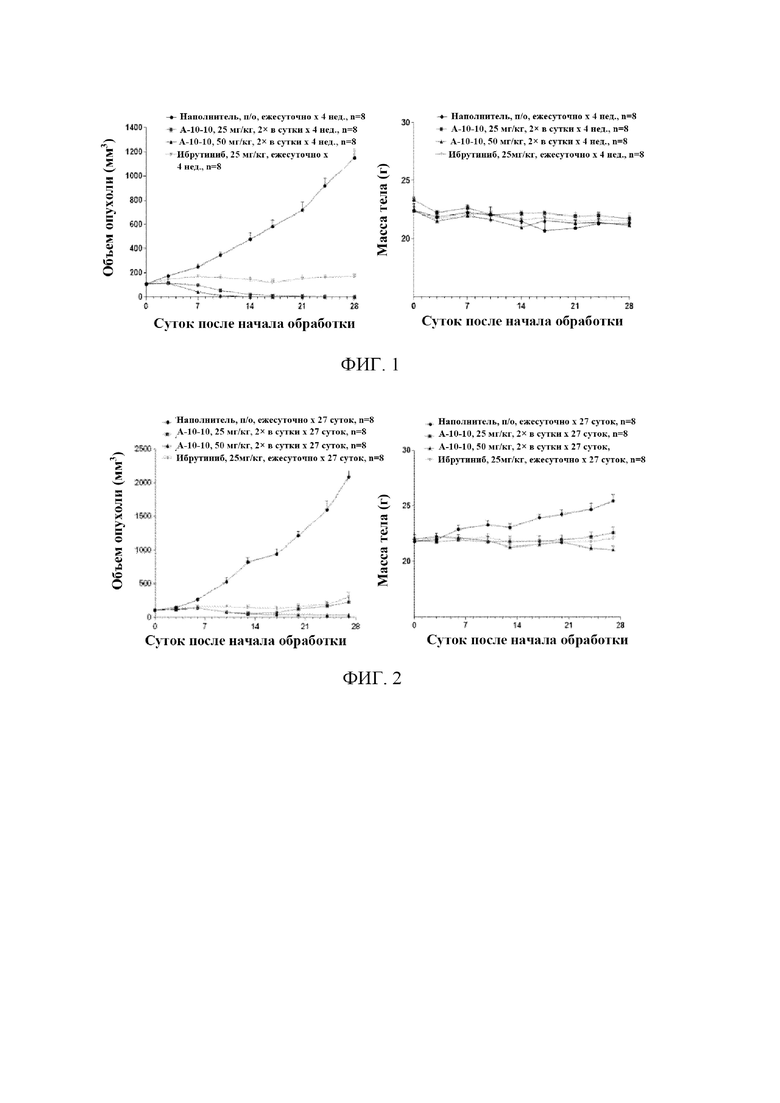
Группа изобретений относится к области органической химии и фармацевтики, а именно к аминонорборнановому производному в качестве ингибитора тирозинкиназы Брутона с высокой селективностью в отношении мутанта BTK(C481S). Раскрывается соединение с формулой I, или его фармацевтически приемлемая соль, где кольцо А, кольцо В, кольцо С и где значения L , R1, R2 и R5 такие, как представлено в формуле изобретения. Кроме того, раскрываются конкретное соединение, выбранное из группы, применение указанных выше соединений при производстве лекарственного препарата, фармацевтические композиции и составы для предупреждения развития или лечения гетероиммунного заболевания на основе данных соединений. Также раскрыт способ получения представленных соединений. Группа изобретений обеспечивает эффективное ингибирование тирозинкиназы Брутона с высокой селективностью в отношении мутанта BTK(C481S), что обеспечивает лечение гетероиммунного заболевания, аутоиммунного заболевания или рака. 14 н. и 8 з.п. ф-лы, 2 ил., 4 табл., 6 пр.
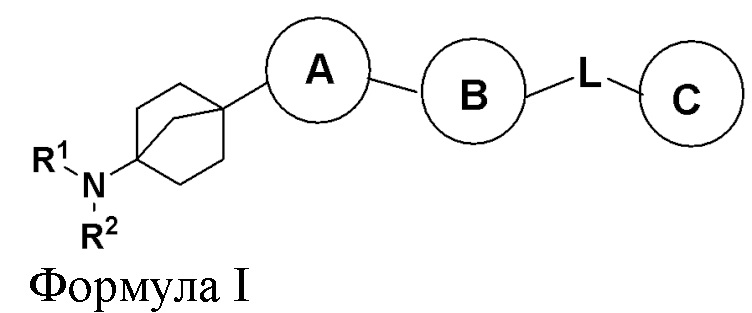
1. Соединение с формулой I
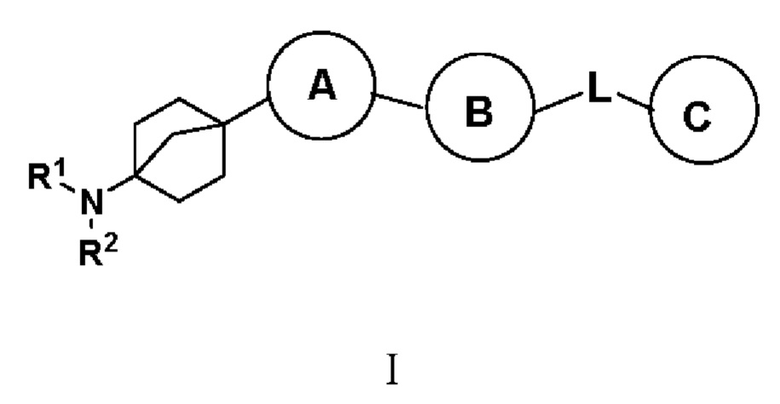
или его фармацевтически приемлемая соль, где кольцо А выбрано из одной из следующих структур:
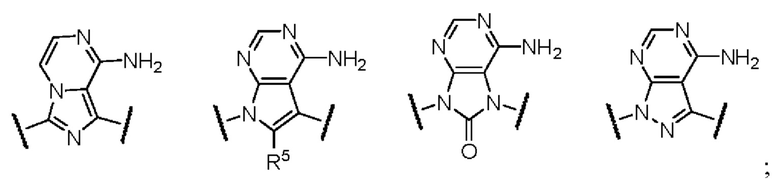
при этом R5 представляет собой водород;
кольцо В представляет собой
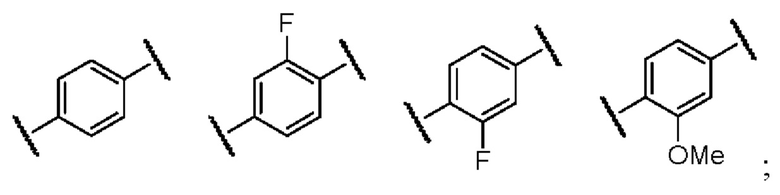
кольцо С представляет собой одну из следующих структур:
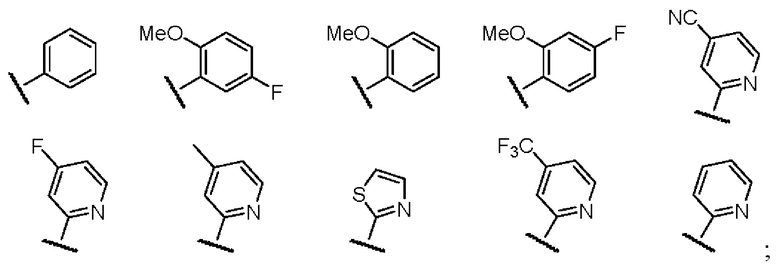
L представляет собой одну из следующих структур:
R1 представляет собой
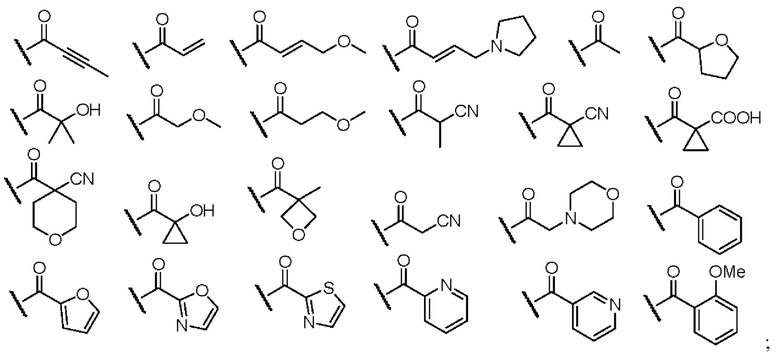
R2 выбран из Н.
2. Соединение, выбранное из одной из следующих структур:
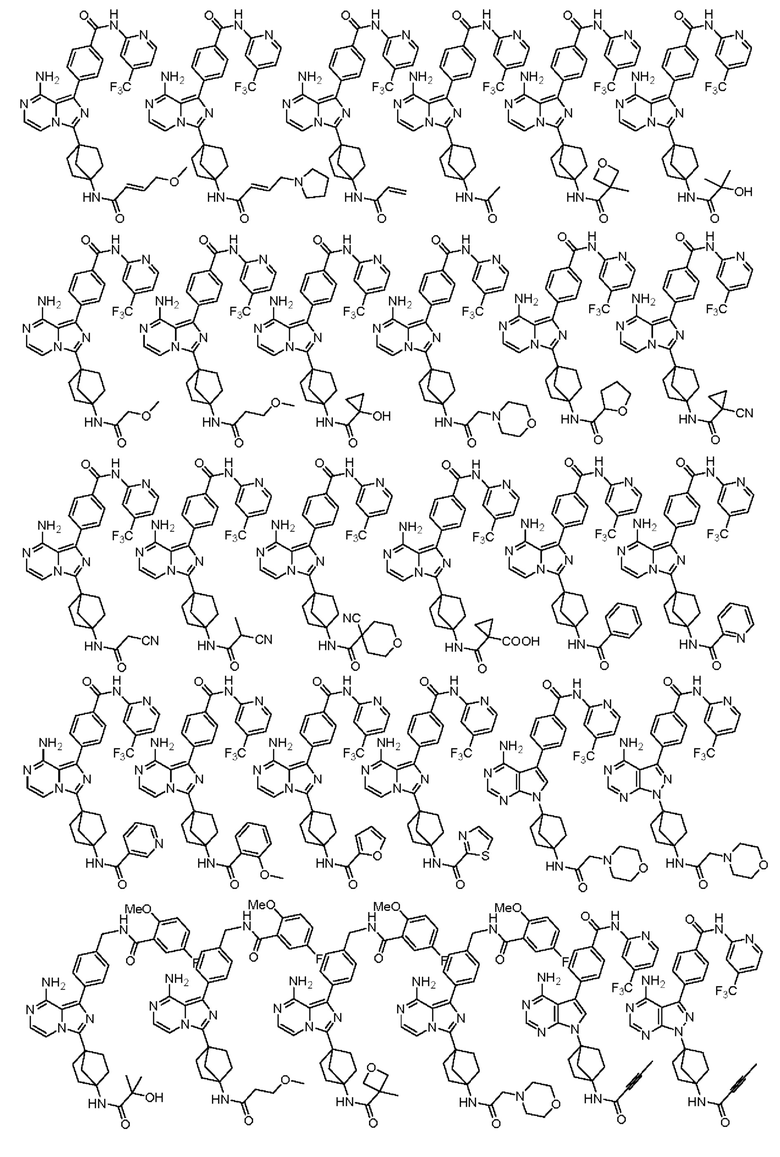
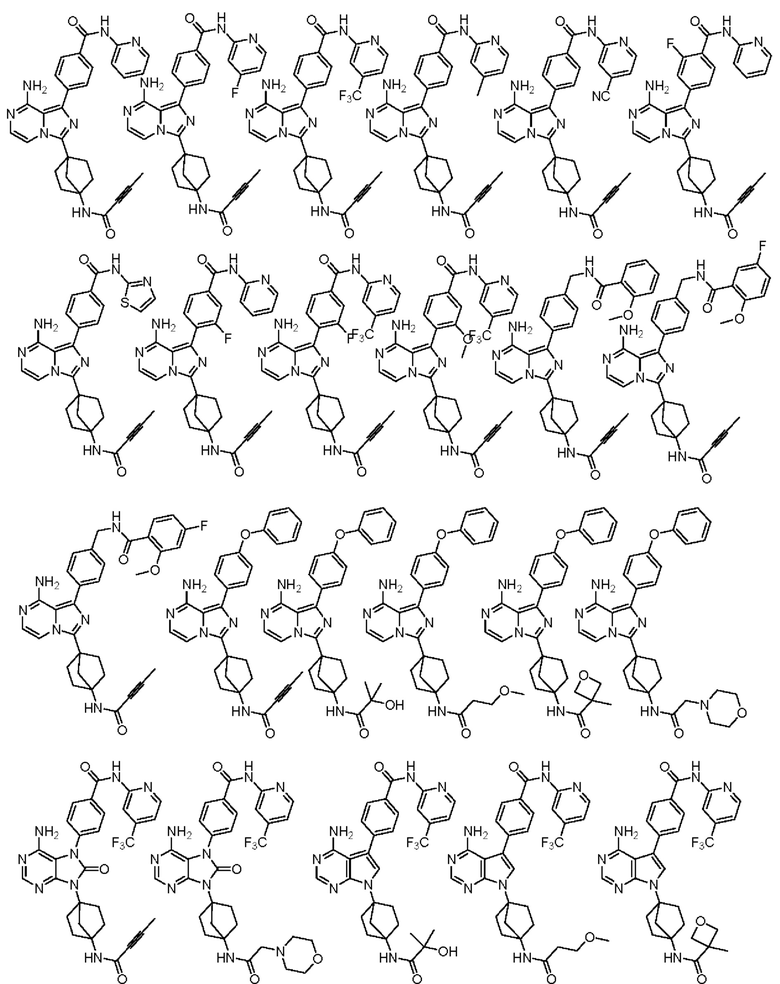
3. Применение соединения по п. 1 или 2 при производстве лекарственного препарата для предупреждения развития или лечения гетероиммунного заболевания.
4. Применение по п. 3, где гетероиммунное заболевание связано с чрезмерной активностью тирозинкиназы Брутона.
5. Применение по п. 3, где гетероиммунное заболевание связано с аберрантной пролиферацией В-клеток.
6. Применение по п. 3, где гетероиммунное заболевание представляет собой воспалительное заболевание или астму.
7. Применение соединения по п. 1 или 2 при производстве лекарственного препарата для предупреждения развития или лечения аутоиммунного заболевания.
8. Применение по п. 7, где аутоиммунное заболевание связано с чрезмерной активностью тирозинкиназы Брутона.
9. Применение по п. 7, где аутоиммунное заболевание связано с аберрантной пролиферацией В-клеток.
10. Применение по п. 7, где аутоиммунное заболевание представляет собой красную волчанку, хроническую лимфоцитарную лимфому, диффузную крупноклеточную лимфому, фолликулярную лимфому или хронический лимфолейкоз.
11. Применение соединения по п. 1 или 2 при производстве лекарственного препарата для предупреждения развития или лечения рака.
12. Применение по п. 11, где рак связан с чрезмерной активностью тирозинкиназы Брутона.
13. Применение по п. 11, где рак связан с аберрантной пролиферацией В-клеток.
14. Фармацевтическая композиция для предупреждения развития или лечения гетероиммунного заболевания, содержащая эффективное количество соединения по п. 1 или 2.
15. Фармацевтическая композиция для предупреждения развития или лечения аутоиммунного заболевания, содержащая эффективное количество соединения по п. 1 или 2.
16. Фармацевтическая композиция для предупреждения развития или лечения рака, содержащая эффективное количество соединения по п. 1 или 2.
17. Фармацевтический состав для предупреждения развития или лечения гетероиммунного заболевания, содержащий терапевтически эффективное количество соединения по п. 1 или 2 и фармацевтически приемлемое вспомогательное вещество.
18. Фармацевтический состав для предупреждения развития или лечения аутоиммунного заболевания, содержащий терапевтически эффективное количество соединения по п. 1 или 2 и фармацевтически приемлемое вспомогательное вещество.
19. Фармацевтический состав для предупреждения развития или лечения рака, содержащий терапевтически эффективное количество соединения по п. 1 или 2 и фармацевтически приемлемое вспомогательное вещество.
20. Способ получения соединения по п. 1 или 2, предусматривающий: (S1) осуществление реакции сочетания Сузуки соединения IIIA с бороновой кислотой или боратом II с получением соединения IV; (S2) превращение соединения IV в гидрохлорид соединения V путем удаления бензилоксикарбонила трифторуксусной кислотой; и (S3) сочетание соединения V с органической кислотой с получением I по п. 1:
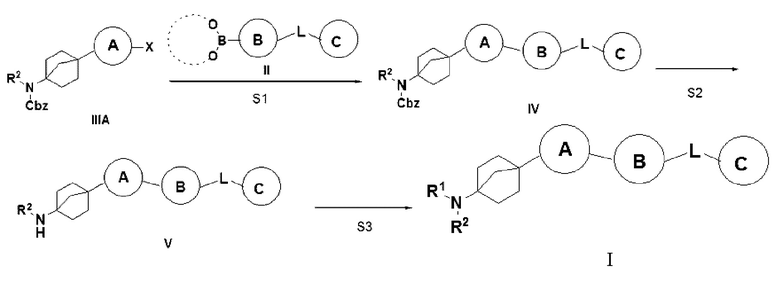
где X=галоген, a R2, R1, L, кольцо А, кольцо В и кольцо С являются такими, как описано в п. 1 или 2.
21. Способ получения соединения по п. 1 или 2, предусматривающий: (А1) превращение соединения IIIA в гидрохлорид соединения VI путем удаления бензилоксикарбонила трифторуксусной кислотой; (А2) сочетание соединения VI с органической кислотой с получением соединения VII; и (A3) осуществление реакции сочетания Сузуки соединения VII с бороновой кислотой или боратом II с получением соединения I по п. 1:
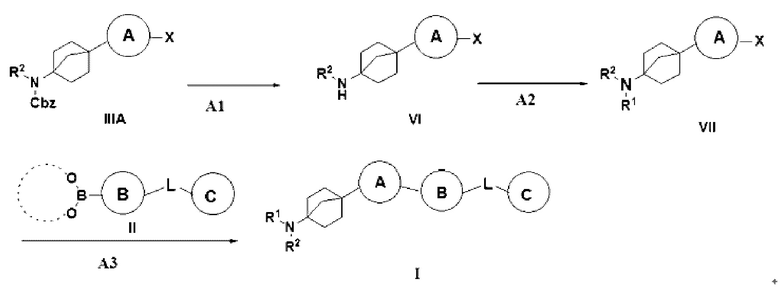
где X=галоген, a R2, R1, L, кольцо А, кольцо В и кольцо С являются такими, как описано в п. 1 или 2.
22. Способ получения соединения по п. 1 или 2, предусматривающий: (В1) осуществление реакции сочетания Чана-Лама-Эванса соединения IIIB с бороновой кислотой II в присутствии катализа ацетатом меди с получением соединения VIII; (В2) превращение соединения VIII в гидрохлорид соединения IX путем удаления бензилоксикарбонила трифторуксусной кислотой; и (В3) сочетание соединения IX с органической кислотой с получением соединения I по п. 1;
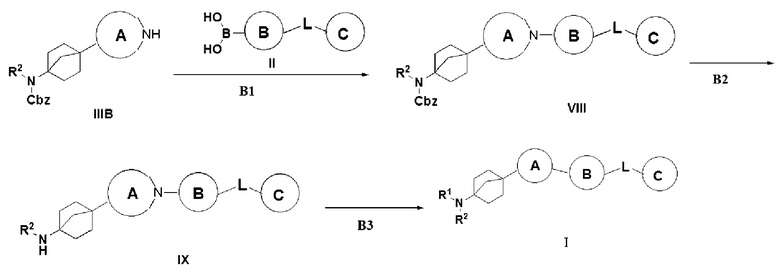
где R2, R1, L, кольцо А, кольцо В и кольцо С являются такими, как описано в п. 1 или 2.
| CN 107602564 A, 19.01.2018 | |||
| WO 2016109223 A1, 07.07.2016 | |||
| WO 2016109222 A1, 07.07.2016 | |||
| CN 106831789 A, 13.06.2017 | |||
| RU 2016115803 A, 09.11.2017 | |||
| EA 201692176 A1, 31.03.2017. |
