ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к области KDR-нацеленных пептид-фосфолипидных конъюгатов, которые используются в терапевтических и диагностических композициях, и, в частности, к способам их получения.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Ангиогенез представляет собой образование новых кровеносных сосудов из уже имеющейся сосудистой сети и играет важную роль не только в нормальных физиологических процессах, но также в патогенезе таких заболеваний, как злокачественная опухоль, ревматоидный артрит и диабетическая микроангиопатия. Например, ангиогенез участвует в переходе опухоли от гиперпластического к неопластическому росту. Таким образом, подавление связанных патологических процессов очень важно в терапевтических и диагностических исследованиях злокачественных опухолей.
В случай, когда ангиогенные факторы роста продуцируются, превосходя ингибиторы ангиогенеза, эндотелиальные клетки стимулируются к пролиферации. В числе известных и лучше всего охарактеризованных проангиогенных агентов или факторов роста семейство факторов роста эндотелия сосудов (VEGF) и, в частности, KDR (рецептор с доменом, содержащим киназную вставку, также известный как VEGFR-2 или Flk-1), представляют собой проангиогенные агенты или факторы роста, которые представляют больший интерес, поскольку демонстрируют более устойчивую экспрессию эндотелиальных клеток и доминирующий ангиогенный ответ1. Экспрессия KDR значительно активируется в ангиогенных сосудах, в частности в опухолях, вызывая сильный ангиогенный ответ2.
VEGF связывающая активность KDR in vivo имеет большое значение для ангиогенеза, поэтому возможность обнаруживать его активацию на эндотелиальных клетках или обнаруживать VEGF/KDR связывающие комплексы была бы чрезвычайно полезной при обнаружении или мониторинге ангиогенеза.
Известно, что для диагностических и терапевтических целей, например, для визуализации сосудов и внутренних органов, было бы особенно эффективно включать в газонаполненные эхоконтрастные препараты любую композицию нацеленного вектора, которая демонстрирует высокое сродство к связыванию с желаемой мишенью, такой как KDR.
Например, KDR нацеленные пептид-фосфолипидные конъюгаты можно использовать для получения газонаправленных эхоконтрастные препаратов.
Хорошо известно, что газонаполненные эхоконтрастные препараты являются высокоэффективными ультразвуковыми рефлекторами для эхографии. Например, введение в кровоток живых организмов суспензий газонаполненных микропузырьков в жидкости-носителе будет значительно усиливать визуализацию эхографии, тем самым способствуя визуализации внутренних анатомических структур, таких как кровеносные сосуды.
Одна из композиций нацеливающего вектора, которая демонстрирует высокую аффинность связывания с целевым KDR, или комплексом VEGF/KDR, представлена, например, следующим соединением (I), конъюгатом “нацеливающий пептид-фосфолипид” (липопептид), который впервые был описан в патентной заявке WO2007/067979 A2 и продемонстрировал высокую способность связываться с KDR-экспрессирующими тканями.
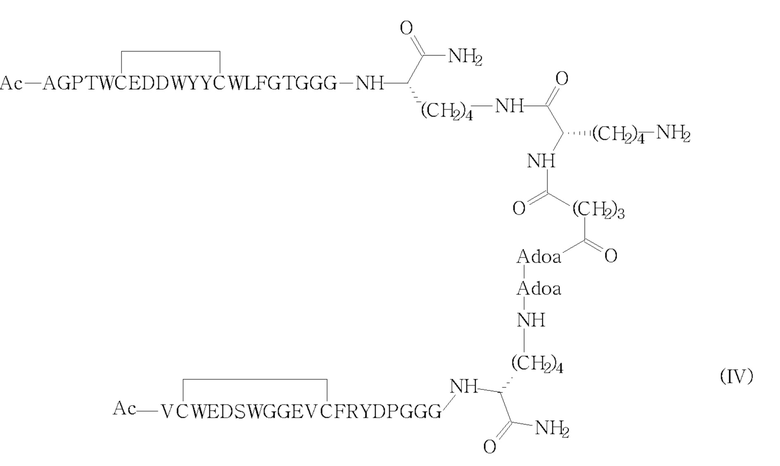
Указанное соединение (I), описанное в настоящем документе ниже и более подробно на фигуре 1, структурно состоит из гетеродимерного пептида, образованного двумя различными мономерными пептидными цепями (обе состоят из 23 аминокислот), связанного глутарильным линкером и конъюгированного с полиэтиленгликолевым фрагментом (PEG), таким как DSPE-PEG2000-NH2, через второй глутарильный линкер.

Способ получения KDR-связывающего пептид-фосфолипидного конъюгата (I) был описан в WO2007/067979 A2 (подробности смотри в приведенном здесь примере 5, страницы 52-54, и на фигуре 4).
Конъюгат получают, начиная с автоматизированного твердофазного синтеза пептидных мономеров, с последующим их сочетанием и активацией с использованием сукцинимидилглутарата (DSG) и последующим конъюгированием полученного производного с DSPE-PEG2000-NH2 посредством глутарильной связи.
Последняя стадия конъюгации также проиллюстрирована на следующей схеме 1.
Схема 1

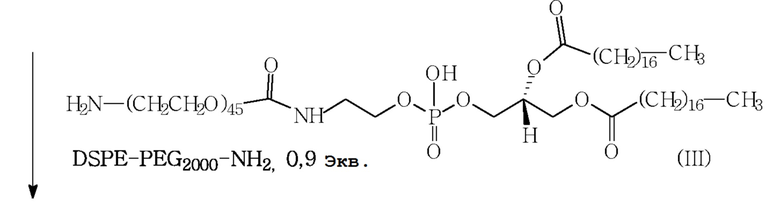

Из приведенного выше описания следует, что стадия конъюгации может иметь некоторые недостатки с точки зрения выхода и чистоты конечного продукта (см. параграф [0063]: “любой свободный фосфолипид может усложнить очистку и выделение конечного продукта”). Фактически, для предотвращения образования примесей и необходимости трудоемких очисток, фосфолипидный реагент DSPE-PEG2000-NH2 (III), который трудно отделить от конечного продукта (I) известными методами очистки, добавляется по умолчанию к сукцинимидил-дипептиду (II) (соотношение DSPE-PEG2000-NH2 и сукцинимидил-дипептид: от 0,9 до 1 эквивалента).
Однако этот подход, хотя и ограничивает примеси в конечном продукте, вызывает потерю ценного гетеродимера (II) и обеспечивает выход на стадии конъюгации не выше 60% или даже не выше 30%. Кроме того, конечное соединение после очистки методом препаративной ВЭЖХ, как описано в примере 5 в WO2007/067979, все еще содержит почти 2% примесей с профилем чистоты, который не соответствует требованиям, установленным государственными органами для фармацевтического продукта.
Другой недостаток известного способа связан с высоким содержанием трифторуксусной кислоты (TFA), которую добавляют в процессе синтеза для солюбилизации и в подвижных фазах для очистки методом препаративной ВЭЖХ. Кроме того, помимо того факта, что TFA считается фармацевтически неприемлемой солью, в случае, когда продукт хранится как соль TFA в форме лиофилизата при температуре 5°C или в растворе, наблюдается разложение, вероятно, определяемое TFA-активируемым кислотным гидролизом одного из сложных эфиров фосфолипидов и жирных кислот в димерном конъюгате с образованием лизосоединения в качестве нежелательной примеси, как описано в пунктах [0065] - [0066] в WO2007/067979. Таким образом, необходимо провести дополнительные обременительные и трудоемкие процедуры для “преобразования солей TFA конъюгата димерный пептид-фосфолипид” в другую более стабильную соль.
Вкратце, основные проблемы известного способа представлены неблагоприятной потерей дорогостоящего гетеродимера (II) во время стадии конъюгации с пегилированным фосфолипидом (III); сложной стадией очистки, снижающей чистоту и выход конечного продукта; и нестабильностью конечного продукта, полученного и хранимого в виде соли TFA.
Следовательно, описанный подход, хотя и является высоко эффективным, может быть в значительной степени обременительным и до сих пор не представляет собой действительный и промышленно применимый метод. До сих пор не соблюдаются показатели чистоты и эффективности производства.
В тоже время, для использования такого нацеленного пептид-фосфолипидного конъюгата in vivo для визуализации сосудов и внутренних органов было бы особенно полезно иметь эффективный способ крупномасштабного производства высокоочищенных форм продукта.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение обеспечивает новый способ получения пептид-фосфолипидного соединения (I), как определено выше, характеризующийся оптимизированными условиями стадий конъюгации и очистки. Этот процесс особенно эффективен для производства газонаполненных эхоконтрастных препаратов.
В этом контексте была обнаружена эффективная аналитическая процедура, значительно улучшающая отделение конечного соединения от побочных продуктов, тем самым обеспечивая увеличение количества исходного вещества (III) во время конъюгации и получить соединение (I) с более высокими выходами, с лучшим профилем чистоты и подходящим профилем стабильности, используемое для масштабирования всего процесса.
Соответственно, первым аспектом настоящего изобретения является способ получения соединения (I) или его фармацевтически приемлемых солей,

включающий стадию
(i) сочетания соответствующего промежуточного соединения сложного сукцинимидилового эфира (II)
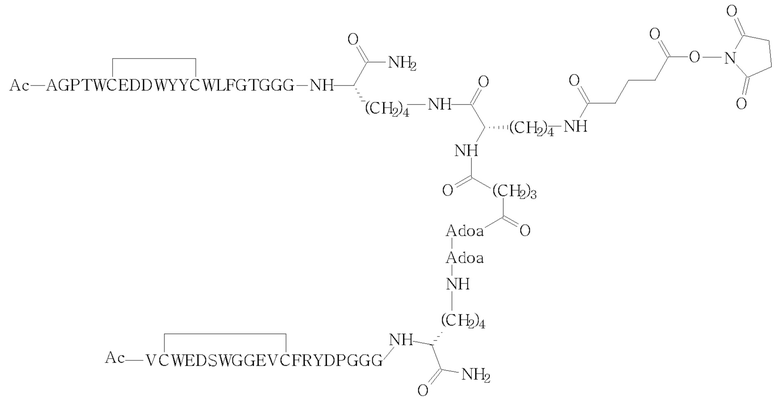 (II)
(II)
с фосфолипидом DSPE-PEG2000-NH2 (III)

в присутствии DIEA,
причем указанный фосфолипид (III) присутствует в избыточном количестве относительно соединения (II).
В предпочтительном варианте осуществления, сочетание проводят с 1,1 или более эквивалентами фосфолипида (III) на один эквивалент соединения (II).
В более предпочтительном варианте осуществления, сочетание проводят с двумя эквивалентами фосфолипида (III) на один эквивалент соединения (II).
В другом аспекте настоящее изобретение обеспечивает указанный способ, дополнительно включающий стадии:
(ii) выделение сырого продукта (I), извлеченного из реакционной смеси на стадии (i);
(iii) необязательное разбавление водой сырого продукта, полученного на стадии (ii), и добавление основания для достижения значения pH в диапазоне от 6 до 8; а также
(iv) очистка сырого продукта от раствора стадии (iii).
Добавление основания на стадии (iii) может способствовать полной солюбилизации сырого продукта в воде, поддерживая значение pH раствора в диапазоне от 6 до 8. Предпочтительно добавляется соответствующее количество 0,1н NaOH для достижения значения pH в диапазоне от 6,5 до 7,5, более предпочтительно для достижения значения pH, составляющего 7,3.
В соответствии с изобретением, очистка на стадии (iv) может быть осуществлена посредством обращенно-фазовой высокоэффективной жидкостной хроматографии (ОФ-ВЭЖХ) или посредством ионообменной хроматографии, или посредством одновременно ОФ-ВЭЖХ и ионообменной хроматографии.
В предпочтительном варианте осуществления, очистку проводят только методом ОФ-ВЭЖХ. В соответствии с изобретением, хроматографическое разделение посредством ОФ-ВЭЖХ достигается предпочтительно с использованием элюентов, имеющих значение pH от 6 до 8 и содержащих легко удаляемый углекислый аммоний. Оптимальная подвижная фаза может быть представлена, например, сочетанием элюента A, состоящего из 10 мМ AcONH4 в воде, и элюента B, состоящего из 10 мМ AcONH4 в смеси вода/ацетонитрил в соотношении 1/9.
В другом предпочтительном варианте осуществления, очистку проводят посредством ионообменной хроматографии. Предпочтительно такую хроматографию проводят с анионообменной смолой (например, смолой ANX Sepharose) и буферным раствором, выбранным из буферных растворов, которые обычно используются для ионной хроматографии, при pH предпочтительно в диапазоне от 7 до 8, необязательно с добавлением смешивающихся с водой растворителей, улучшающих солюбилизацию фосфолипидного фрагмента.
В соответствии с предпочтительным вариантом осуществления изобретения, очистку успешно проводят с использованием буфера Трис HCl/NaCl в качестве элюента. Подходящее хроматографическое разделение достигается, например, с использованием 0,05 М Трис HCl+0,10 М NaCl (pH 7,5) + 35% iPrOH в качестве буфера для фиксации и 0,05 М Трис HCl+1,00 М NaCl (pH 7,5) + 35% iPrOH в качестве элюирующего буфера.
В другом аспекте изобретения обеспечен вышеуказанный способ, в котором соединение (II) получают путем активации концевого алкиламинового фрагмента соответствующего промежуточного соединения формулы (IV) с ди(N-сукцинимидил)глутаратом (V), как показано на следующей схеме 2 (стадия i')).
Схема 2


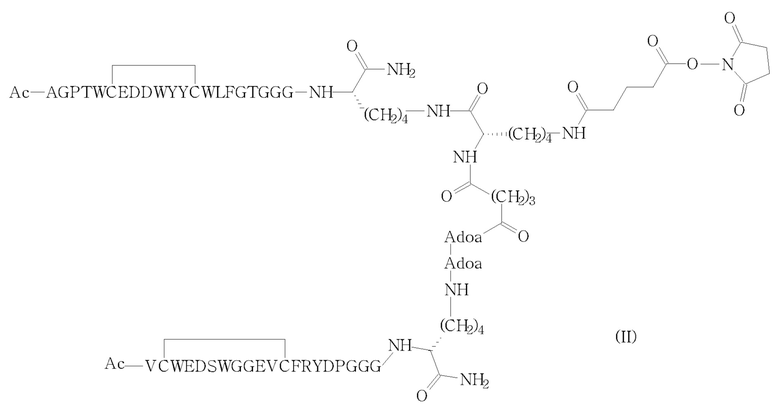
Таким образом, способ по изобретению характеризуется использованием избыточного количества фосфолипидного реагента DSPE-PEG2000-NH2 (III), который взаимодействует со всем присутствующим количеством активированного гетеродимера (II), например, для устранения любых потерь последнего дорогостоящего промежуточного соединения. Выходы стадий сочетания (i’) и, в частности, (i) теперь преимущественно выше по сравнению с выходами, полученными посредством ранее известной процедуры.
Фактически, предыдущий метод обеспечивает только выход ниже 60% димерного пептид-фосфолипидного конъюгата, который необходимо преобразовать из соли TFA в более стабильную соль, что дополнительно снижает эффективное извлечение конечного продукта.
В тоже время, настоящий способ обеспечивает получение соединения (I) с улучшенными выходами, по меньшей мере, 69%, и, что особенно важно, он обеспечивает более эффективные способы аналитического разделения и препаративной очистки конечного продукта от нежелательных примесей и удаления избыточных реагентов; таким образом, оптимизированные условия очистки позволяют получить конечный продукт с чистотой выше 99% после проведения ОФ-ВЭЖХ.
Следовательно, в соответствии с несколькими преимуществами, обеспечиваемыми настоящим способом, новая процедура синтеза соединения (I) устраняет недостатки ранее описанной процедуры и может быть особенно эффективной для увеличения масштаба и промышленного производства.
Описание изобретения
Краткое описание чертежей
На фиг.1 показана структура соединения (I), полученного в соответствии с изобретением.
На фиг.2 показана хроматограмма СВЭЖХ-ELSD(детекция по светорассеянию образца) соединения (I) после окончательной очистки методом ВЭЖХ.
Определения
В настоящем описании, если не указано иное, следующие термины имеют следующие значения.
Термин “гетеродимер” относится к молекуле, состоящей из двух полипептидных цепей, различающихся по составу, то есть по порядку, количеству или виду их аминокислотных остатков. В частности, упоминается в настоящем документе со ссылкой на соединение формулы (I) или его предшественников (II) и (IV), как определено выше.
Термин “пегилированный” относится к молекуле, которая ковалентно присоединена к полимерной цепи полиэтиленгликоля (PEG). Пегилированное соединение может быть получено путем инкубации реакционноспособного производного PEG, предпочтительно после функционализации на одном или обоих концах с реакционноспособным фрагментом, с молекулой-мишенью.
Термин “анионообменная твердая фаза” означает твердую подложку, способную осуществлять обмен анионов с раствором или суспензией, контактирующими с ней. Указанный контакт может быть осуществлен путем элюирования посредством колонки, заполненной соответствующей твердой фазой.
Подробное описание вариантов осуществления
Описанный в настоящем документе способ относится к получению соединения формулы (I), как определено выше, и имеет преимущества, заключающиеся в экономии количества дорогостоящего исходного материала, обеспечивая при этом конечный продукт с высокими выходами и оптимальной степенью чистоты.
Эти результаты могут быть достигнуты, в числе прочих, путем обнаружения двух эффективных методов очистки, которые можно применять по отдельности или в сочетании, обеспечивая эффективное удаление непрореагировавшего фосфолипида (III), добавляемого в избыточном количестве на стадии сочетания (i), вместе с любым другим нежелательным побочным продуктом.
Получение соединения (I), в соответствии со способом синтеза, описанным в WO2007/067979, обеспечивает активацию гетеродимера (IV), как определено выше, полученного, например, известными методами твердофазного синтеза, с ди(N-сукцинимидил)глутаратом (V) и последующее конъюгирование активированного гетеродимера (II) с DSPE-PEG2000-NH2 (III). Последняя стадия, как описано выше, осуществляется при недостатке фосфолипида, поэтому сочетание осуществляется только с неудовлетворительными результатами выхода.
С другой стороны, оптимизированный процесс, представленный настоящим изобретением, обеспечивает значительное улучшение стадии конъюгации, как подробно описано ниже.
Гетеродимер (IV) можно активировать, например, в соответствии с аналогичной процедурой, описанной в примере 5 WO2007/067979 (параграф [00124]), то есть взаимодействием предшественника гетеродимера с избыточным количеством ди(N-сукцинимидил)глутарата и основанием, таким как DIEA, например, с 5-кратным избытком обоих реагентов, для устранения сочетания гетеродимера. После завершения взаимодействия, смесь может быть разбавлена подходящим растворителем, таким как безводный этилацетат, для осаждения моно-NHS эфира гетеродимерного моноамида глутаровой кислоты (II), который затем выделяют и промывают с удалением оставшихся следовых количеств реагентов. Альтернативно, смесь может быть концентрирована для удаления растворителя, и сухой неочищенный продукт может быть промыт, например, EtOAc, и центрифугирован с извлечением твердого вещества из сосуда.
Сочетание с фосфолипидом
В соответствии с изобретением, и как более подробно описано в экспериментальной части, соединение формулы (II) инкубируют с избыточным количеством DSPE-PEG2000-NH2 (III), растворенным в ДМФ, и в присутствии основания, такого как DIEA. Соотношение между эквивалентами предшественника гетеродимера (II) и эквивалентами фосфолипида (III) составляет, по меньшей мере, 1:1,1, но более предпочтительно составляет от 1:1,1 до 1:5. Предпочтительно 1:2.
Завершение реакции сочетания можно контролировать посредством аналитической ВЭЖХ.
Выделение продукта
Неочищенный продукт может быть собран после концентрирования реакционной смеси. Например, часть избыточных реагентов может быть удалена посредством промывки сухой сырой нефти подходящим растворителем и центрифугирования смеси. Альтернативно, для ускорения осаждения конечного продукта, который затем может быть выделен фильтрованием и высушен, может добавляться растворитель, такой как этилацетат.
Предпочтительно, смесь очищают посредством хроматографии, так как описано ниже. Перед стадией хроматографии, реакционная смесь может быть концентрирована при пониженном давлении для извлечения неочищенного продукта, который затем растворяют в водной среде, такой как вода, необязательно путем добавления основания, такого как, например, 0,1н NH4OH, для ускорения полной солюбилизации при значении pH от 6 до 8, предпочтительно при значении pH около 7,3; прозрачный раствор предпочтительно фильтруют на фильтре с диаметром ячейки 0,2 мкм.
Хроматографическая очистка
В соответствии с изобретением, неочищенный продукт очищают посредством ОФ-ВЭЖХ, ионообменной хроматографии или обоими способами.
Обычно предпочтительным является разделение методом ОФ-ВЭЖХ, поскольку он является более эффективным для отделения чистого продукта от избыточного количества реагентов. Однако в случаях, когда остаточные следовые количества фосфолипида (III) остаются в конечном продукте (обычно более 1%), можно также добавить или провести быструю и надежную стадию ионообменной очистки.
Очистку препаративной ВЭЖХ в соответствии с изобретением предпочтительно проводят на препаративной колонке C4 для обращенного-фазной хроматографии, элюируя подвижной фазой, содержащей соль AcONH4. В одном из вариантов осуществления, подвижные фазы представлены водными растворами 10 мМ AcONH4 и 10 мМ AcONH4/ацетонитрил 1/9, смешанными в градиентной композиции, способной эффективно отделить продукт от фосфолипида.
Ионообменную очистку по изобретению эффективно проводить на анионообменной смоле, предпочтительно на слабой анионообменной смоле с группами третичного амина, присоединенными к основной матрице. Отделение конечного продукта от избыточного количества реагентов и других примесей осуществляется путем выбора подходящих буферов для фазы фиксации и элюирования. В соответствии с изобретением, оптимальные результаты были получены при использовании буферного раствора Трис-HCl/NaCl при различных концентрациях соли и при значении pH от около 7 до около 8, с добавлением относительного количества растворителя, такого как, например, iPrOH.
Изменение концентрации соли в буфере обеспечивает фиксирование продукта к твердой фазе при элюировании всех побочных продуктов, а затем элюировать и собирать чистый продукт.
Этот эффективный метод удаления всех следовых количеств фосфолипида (III) позволяет использовать этот реагент даже в большом избыточном количестве в процессе реакции конъюгации с гетеродимером (II).
Соответственно, как в широком смысле описано выше, настоящее изобретение обеспечивает более удобный и надежный способ получения соединения (I) с высокими выходами и степенью чистоты, который также можно применять в промышленном масштабе.
Фактически, продукт, полученный данным способом, по существу не содержит побочных продуктов и соответствует спецификациям чистоты, необходимым для его использования при производстве газонаполненных эхоконтрастных препаратов.
Далее настоящее изобретение будет проиллюстрировано примерами, которые не предназначены для ограничения его объема.
Экспериментальная часть
Материалы и оборудование
Растворители, такие как ДМФА и этилацетат, всегда использовались в чистом и высушенном виде, чтобы минимизировать время воздействия окружающего воздуха.
Гетеродимерный ацетат (IV) был предоставлен компанией Bachem (Bubendorf, Switzerland). Аммониевая соль DSPE-PEG2000-NH2 была приобретена у компании Avanti Polar Lipids Inc. (США).
Аналитическую ВЭЖХ с обращенной фазой выполняли на системе UFLC SHIMADZU, состоящей из бинарного насоса UFLC, контроллера UFLC (CBM-20A) и детектора ультрафиолетового и видимого диапазонов для ВЭЖХ (SPD-20A). Анализы выполняли с использованием линейного градиента фазы A (10 мМ AcONH4 в H2O) и фазы B (10 мМ AcONH4 в ACN/H2O 9/1) при 1,5 мл/мин с УФ-детектированием при длине волны 214 нм. Вводили 40 мкл, температура колонки составляла 25°C.
Препаративную ОФ-ВЭЖХ осуществляли на препаративной системе Shimadzu, состоящей из бинарного насоса для ВЭЖХ, коллектора фракций для ВЭЖХ (FRC-10A), ВЭЖХ контроллера (SCL-10A) и детектора ультрафиолетового и видимого диапазонов для ВЭЖХ (SPD-10AV). Система была оборудована колонкой Kromasil C4 300Å (10×250 мм). Очистку проводили элюированием с линейным градиентом фазы A (10 мМ AcONH4 в H2O) и фазы B (10 мМ AcONH4 в ACN/H2O 9/1) при 5 мл/мин с УФ-детектированием при длине волны 214 нм. Вводили 3 мл, и температура колонки составляла 25°C.
Чистоту конечного продукта определяли посредством системы ВЭЖХ AcquityTM Ultra Performance (Waters), оснащенной TUV детектором и колонкой Acquity BEH Phenyl 1,7 мкм (2,1×150 мм), или посредством ВЭЖХ системы Agilent 1100, оснащенной УФ-детектором и испарительным детектором по светорассеянию (ELSD Sedex 85) и колонкой Zorbax 300SB 3,5 мкм (3×150 мм).
Аббревиатуры для отдельных аминокислотных остатков являются общепринятыми: например, Asp или D соответствуют аспарагиновой кислоте, Gly или G соответствуют глицину, Arg или R соответствуют аргинину. Следует понимать, что указанные в настоящем документе аминокислоты имеют конфигурацию L-изомера, если не указано иное.
Перечень аббревиатур
Пример 1: Получение промежуточного соединения (II)
Перед конъюгацией с пегилированным фосфолипидом гетеродимер (IV) был активирован путем сочетания с ди(N-сукцинимидил)глутаратным фрагментом в качестве связывающего агента.
Раствор гетеродимера ацетата (49,08 мг) в 500 мкл ДМФА порциями (7 × 70 мкл) добавляли каждые 2 минуты к раствору дисукцинимидилглутарата с DIEA (130 мкл). Для предотвращения сочетания димеров, использовали избыточное количество DSG (5 экв.) и DIEA (5 экв.). После перемешивания при комнатной температуре в течение 30 минут после последнего добавления, активированный гетеродимер выделяли и анализировали методом ВЭЖХ для подтверждения завершения взаимодействия. Для этих анализов применялись следующие хроматографические условия:
Колонка: Phenomenex Luna 5 мкм C18 (250 × 4,6 мм)
Элюент A: 10 мМ AcONH4 в H2O
Элюент B: 10 мМ AcONH4 в H2O/ACN (1/9)
Скорость потока: 1,5 мл/мин
Детектор: УФ 214 нм
Градиент: от 25% до 52% подвижной фазы А
Время удерживания: 12,69 мин
Пример 2: Выделение соединения (II)
Соединение (II), полученное в примере 1, выделяли для удаления избыточного количества DSG из реакционной смеси. Суспензию концентрировали при пониженном давлении для удаления ДМФА. Сухой неочищенный продукт промывали 10 мл EtOAc и затем центрифугировали 3 мин при 2500 g. Супернатант сливали в круглодонную колбу объемом 100 мл, твердое вещество дважды промывали 15 мл EtOAc и сушили при пониженном давлении с получением 47,02 мг порошка белого цвета.
Пример 3: Синтез соединения (I)
Синтез соединения (I) выполняли в соответствии со стадией, представленной на схеме 1. Ход реакции отслеживали, используя аналитическую ВЭЖХ с обращенной фазой или ВЭЖХ с УФ-детектором при длине волны 220 нм или ELSD детектором.
Стадии i)-iii) Конъюгация и выделение продукта (I)
Образец аммониевой соли DSPE-PEG2000-NH2 (18 мкмоль, 50,23 мг, 2 экв.) растворяли в 300 мкл безводного ДМФА, а затем добавляли DIEA (2 экв.) для достижения общего объема, составляющего 315 мкл.
Соединение (II) солюбилизировали в 400 мкл ДМФА, затем добавляли пятью порциями к смеси DSPE-PEG2000-NH2 и DIEA и оставляли на ночь при перемешивании.
Аликвоту собирали для аналитического ВЭЖХ контроля, и профиль показал основной пик элюирования при времени удерживания около 12,5 мин. Затем смесь концентрировали при пониженном давлении с получением 105,5 мг неочищенного продукта.
Сначала добавляли 5 мл воды для достижения значения pH 4,8; однако для достижения полной солюбилизации и получения прозрачного раствора дополнительно добавляли около 20 капель 0,1н NH4OH с достижением pH 7,3. Затем раствор фильтровали на фильтре с диаметром ячейки 0,2 мкм и промывали с получением конечного объема, составляющего около 9 мл, готового для очистки методом препаративной ВЭЖХ.
Пример 4: Очистка соединения (I) методом ОФ-ВЭЖХ
Очистку конечного неочищеного продукта (I) посредством препаративной ВЭЖХ проводили с той же неподвижной фазой, что и для аналитического контроля реакции сочетания. Соответственно, колонку Kromasil 10 мкм 300Å C4 (250 × 10 мм) уравновешивали 10 мМ AcONH4 в смеси вода:ацетонитрил (1:9) перед загрузкой образца, разделенного на 3 аликвоты (3 × 3 мл). Жидкие фазы и условия элюирования, применяемые для очистки, подробно описаны ниже:
Колонка: Kromasil 10 мкм 300Å C4 (250 × 10 мм)
Элюент A: 10 мМ AcONH4 в H2O
Элюент B: 10 мМ AcONH4 в H2O/ACN (1/9)
Скорость потока: 5 мл/мин.
Объем введения: 3,0 мл
Температура колонки: 25°C
Детектор: УФ 214 нм
Градиент:
Сбор проводили фракциями по 10 мл, что обеспечивало получение 30 мл очищенного продукта для каждого цикла. Затем 90 мл, полученные в результате 3 циклов, концентрировали при пониженном давлении, удаляя большую часть ацетонитрила перед лиофилизацией. Конечный продукт выделяли в виде твердого вещества белого цвета (51,6 мг) с выходом 69% от исходного гетеродимера (IV).
Анализ очищенного продукта (I), проведенный методом СВЭЖХ-УФ, подтвердил чистоту, составляющую 99%. Не было обнаружено следовых количеств DSPE-PEG2000-NH2 (см. фиг. 2).
Пример 5: Очистка соединения (I) ионообменной хроматографией
Метод ионообменной хроматографии оптимизировали для дальнейшей очистки неочищенного продукта (I) в случае присутствия следовых количеств DSPE-PEG2000-NH2.
С этой целью использовали колонку, заполненную высокотекучей смолой ANX Sepharose 4 (GE Healthcare), со следующими буферами:
- для стадии фиксации: 0,05 М Трис HCl - 0,10 М NaCl - pH 7,5+35% iPrOH
- для стадии элюирования: 0,05 М Трис HCl - 1,00 М NaCl - pH 7,5+35% iPrOH
- для стадии обессоливания: 0,02 М Трис HCl pH 7,5
Два образца соединения (I) (растворы A и B, содержащие соответственно 0,5 мг и 1,5 мг соединения (I)) загружали в ANX колонку. Кроме того, аналогичный эксперимент проводили с DSPE-PEG2000-NH2 (растворы C и D, соответственно, содержащие 0,5 мг и 1,5 мг DSPE-PEG2000-NH2).
Анализ фракций показал, что DSPE-PEG2000-NH2 полностью элюируется первыми двумя объемами колонки (CV) с фиксирующим буфером. В тоже время соединение (I) не было обнаружено в первых двух фракциях, и это означает, что разделение могло быть эффективным, поскольку одно из них проходит напрямую, а другое соединение остается фиксированным на смоле.
Действительно, после прохождения в сумме 4 CV фиксирующего буфера, вводили элюирующий буфер, обеспечивая сбор соединения (I) по фракциям в двух CV (см. таблицу 1a, растворы A и B). Кроме того, во втором эксперименте было подтверждено, что DSPE-PEG2000-NH2 не элюируется после второй фракции даже с элюирующим буфером (таблица 1b, растворы C и D).
Пул собранных фракций пропускали через колонку cо смолой для обессоливания Sephadex G-25M перед лиофилизацией.
Таблица 1a - Удерживание и элюирование соединения (I) (растворы A и B) на колонке ANX
Таблица 1b - Удерживание и элюирование DSPE2000-NH2 (растворы C и D) на колонке ANX
Все фракции анализировали с использованием системы 1100 LC/MSD (Agilent) и количественно определяли по калибровочному стандарту с использованием УФ-детектора для соединения (I) или ELSD детектора для DSPE-PEG2000-NH2 (III).
Ссылки
1. Ferrara N. et al., “Vascular Endothelial Growth Factor: Basic Science and Clinical Progress”, Endocrine Reviews, 2004, 25(4), 581-611.
2. Veikkola T. et al., “Regulation of Angiogenesis via Vascular Endothelial Growth Factor Receptors”, Cancer Res., 2000, 60, 203-212.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТЫ ФОСФОЛИПИДОВ И НАПРАВЛЯЮЩИХ ВЕКТОРНЫХ МОЛЕКУЛ | 2006 |
|
RU2433137C2 |
| СПОСОБ ПОЛУЧЕНИЯ СУПРАМОЛЕКУЛЯРНЫХ ПЛАТИНОВЫХ СОЕДИНЕНИЙ | 2016 |
|
RU2795256C2 |
| Фармацевтическая композиция, включающая доксорубицин в составе фосфолипидных наночастиц, с использованием селективных молекул ДНК-аптамера для направленного транспорта в опухолевые клетки | 2021 |
|
RU2794798C1 |
| СОСТАВ НАЦЕЛЕННЫХ МИКРОВЕЗИКУЛ, НАПОЛНЕННЫХ ГАЗОМ | 2015 |
|
RU2725808C2 |
| Фармацевтическая композиция, включающая хлорин е6 в составе фосфолипидных наночастиц, с использованием специфического и проникающего пептидов как адресных молекул для направленного транспорта | 2020 |
|
RU2747485C1 |
| АНТАГОНИСТ ЛГ-РИЛИЗИНГ-ФАКТОРА (LHRH), КОМПЛЕКС, СПОСОБ ХИМИЧЕСКОЙ КАСТРАЦИИ И/ИЛИ ЛЕЧЕНИЯ ГОНАДОТРОПИНЗАВИСИМЫХ РАССТРОЙСТВ | 1994 |
|
RU2130464C1 |
| ПРОМЫШЛЕННЫЙ СПОСОБ ПОЛУЧЕНИЯ И ОЧИСТКИ РЕКОМБИНАНТНОГО ГОРМОНА РОСТА ЧЕЛОВЕКА ИЗ ТЕЛЕЦ ВКЛЮЧЕНИЯ | 2011 |
|
RU2473556C1 |
| СПОСОБ ПОЛИАЛКОКСИЛИРОВАНИЯ НУКЛЕИНОВЫХ КИСЛОТ, КОТОРЫЙ ПОЗВОЛЯЕТ ИЗВЛЕКАТЬ И ПОВТОРНО ИСПОЛЬЗОВАТЬ ИЗБЫТОК ПОЛИАЛКОКСИЛИРУЮЩЕГО РЕАГЕНТА | 2017 |
|
RU2765027C2 |
| МОДУЛЬНЫЙ МОЛЕКУЛЯРНЫЙ КОНЪЮГАТ ДЛЯ НАПРАВЛЕННОЙ ДОСТАВКИ ГЕНЕТИЧЕСКИХ КОНСТРУКЦИЙ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2012 |
|
RU2529034C2 |
| ДИАГНОСТИЧЕСКИЕ СОЕДИНЕНИЯ | 2005 |
|
RU2396272C9 |


Изобретение относится к новому эффективному способу получения KDR-нацеленного пептид-фосфолипидного конъюгата, который может быть использован в терапевтических и диагностических композициях и, в частности, при получении эхоконтрастных препаратов. 9 з.п. ф-лы, 2 ил., 1 табл., 5 пр.
1. Способ получения соединения (I) или его фармацевтически приемлемых солей,

включающий стадии
(i) сочетания соответствующего промежуточного соединения сложного сукцинимидилового эфира (II)
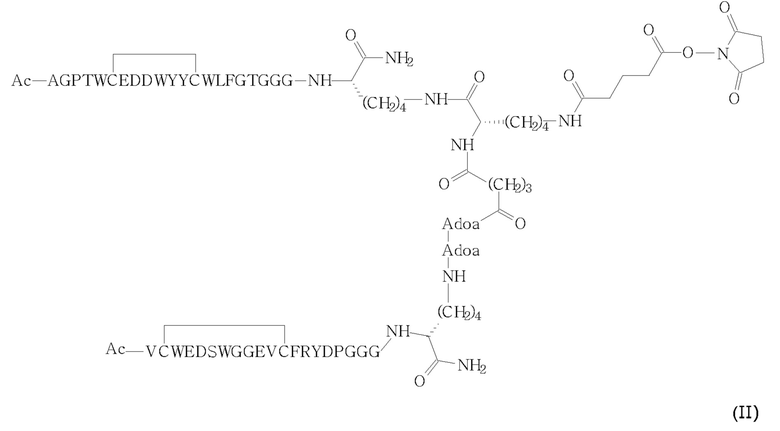
с фосфолипидом DSPE-PEG2000-NH2 (III)

в присутствии DIEA,
причем указанный фосфолипид (III) присутствует в избыточном количестве 1,1 или более эквивалентов относительно соединения (II);
(ii) выделение неочищенного продукта (I), извлеченного из реакционной смеси стадии (i);
(iii) необязательное разбавление водой сырого продукта, полученного на стадии (ii), и добавление основания для достижения значения pH от 6 до 8; и
(iv) очистка неочищенного продукта от раствора стадии (iii) методом ВЭЖХ, методом ионообменной хроматографии или обоими методами.
2. Способ по п. 1, отличающийся тем, что сочетание осуществляют с 2 эквивалентами фосфолипида (III) на один эквивалент соединения (II).
3. Способ по п. 1, отличающийся тем, что pH раствора, полученного на стадии (iii), доводят до значения 7,0-7,5.
4. Способ по п. 1, отличающийся тем, что очистку на стадии (iv) проводят методом высокоэффективной жидкостной хроматографии с обращенной фазой (ОФ-ВЭЖХ).
5. Способ по п. 1, отличающийся тем, что очистку стадии (iv) проводят методом ионообменной хроматографии.
6. Способ по п. 1, отличающийся тем, что очистку на стадии (iv) проводят как методом высокоэффективной жидкостной хроматографии с обращенной фазой (ОФ-ВЭЖХ), так и методом ионообменной хроматографии.
7. Способ по п. 4 или 6, отличающийся тем, что очистку методом ОФ-ВЭЖХ проводят с использованием подвижной фазы, содержащей соль AcONH4.
8. Способ по п. 5 или 6, отличающийся тем, что ионообменную хроматографию проводят с использованием слабой анионообменной смолы и буферного раствора при значении pH от 7 до 8.
9. Способ по п. 8, отличающийся тем, что элюирующий раствор представляет собой буфер 0,05 М Трис HCl+1,00 М NaCl+35% iPrOH.
10. Способ по п. 1, отличающийся тем, что соединение (II) получают путем активации ди(N-сукцинимидил)глутаратом соответствующего промежуточного соединения формулы (IV)
| КОНЪЮГАТЫ ФОСФОЛИПИДОВ И НАПРАВЛЯЮЩИХ ВЕКТОРНЫХ МОЛЕКУЛ | 2006 |
|
RU2433137C2 |
| FERRARA N | |||
| et al | |||
| "Vascular Endothelial Growth Factor: Basic Science and Clinical Progress", Endocrine Reviews, 2004, vol | |||
| Видоизменение пишущей машины для тюркско-арабского шрифта | 1923 |
|
SU25A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |

