Ссылка на родственные заявки
По настоящей заявке испрашивается приоритет и преимущество в соответствии с заявкой на патент Китая № 201610435947.4, поданной в Китайское национальное управление интеллектуальной собственности 16 июня 2016 г., раскрытие которой полностью включено в настоящий документ посредством ссылки.
Область техники, к которой относится настоящее изобретение
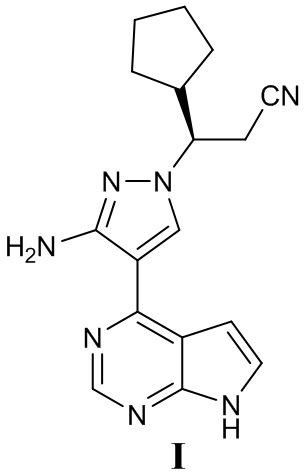
Настоящая заявка относится к области медицинской химии. В частности, настоящая заявка относится к кристаллу пирролопиримидинового соединения (3R)-3-[3-амино-4-(7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил]-3-циклопентилпропионитрила в качестве JAK-ингибитора, кристаллической композиции, фармацевтической композиции, способу их получения и применения.
Предшествующий уровень техники настоящего изобретения
Янус-киназа (JAK) представляет собой нерецепторный тип тирозинкиназ (PTK), который находится в клетках и передает сигнал стимуляции цитокинов через путь JAK-STAT. Посредством пути JAK-STAT химический сигнал вне клетки передается в промотор гена на эндонуклеарной ДНК через клеточную мембрану и, наконец, воздействует на ДНК в клетке, изменяя уровень ее транскрипции и активности. Путь JAK-STAT в основном состоит из трех компонентов: (1) рецептор; (2) янус-киназа (JAK) и (3) передатчик сигнала и активатор транскрипции (STAT) белка. Рецептор может быть активирован интерфероном, интерлейкином, фактором роста или другим химическим мессенджером, и такая активация приводит к фосфорилированию самой JAK. Затем белок STAT связывается с фосфорилированным рецептором, так что STAT фосфорилируется с помощью JAK. После этого фосфорилированный белок STAT выделяется из рецептора, затем димеризуется и транслоцируется в ядро клетки, тем самым связываясь со специфическим участком ДНК и изменяя транскрипцию (Scott, M. J., C. J. Godshall et al. (2002). “Jaks, STATs, Cytokines, and Sepsis” Clin Diagn Lab Immunol 9(6): 1153-9).
Семейство JAK играет роль в цитокинзависимой регуляции пролиферации и функции клеток, участвующих в иммунном ответе. В настоящее время известно четыре представителя семейства JAK млекопитающих: JAK1, JAK2, JAK3 и TYK2 (тирозинкиназа 2). Белки JAK характеризуются размером в диапазоне от 120 кДа до 140 кДа и содержат 7 консервативных доменов гомологии JAK (JH). Один из них представляет собой функциональный домен каталитической киназы, а другой представляет собой домен псевдокиназы, который эффективно выполняет регуляторную функцию и/или действует как сайт стыковки для STAT (Scott, Godshall et al. 2002, выше).
В настоящее время сообщается о различных ингибиторах янус-киназы. В заявке на патент Китая № 201410784461.2 с датой подачи 16 декабря 2014 г. раскрыто несколько JAK-ингибиторов (содержание которых полностью включено в настоящий документ посредством ссылки), включая в себя соединение (3R)-3-[3-амино-4-(7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил]-3-циклопентилпропионитрил, представленное формулой I:
В дополнение к терапевтической эффективности разработчики лекарственных средств пытаются предложить подходящую форму активной молекулы, обладающей свойствами лекарственного средства. С точки зрения получения коммерчески жизнеспособного способа получения или с точки зрения получения фармацевтической композиции, содержащей активное соединение, химическая стабильность, стабильность в твердом состоянии и срок годности активного ингредиента представляют собой очень важные факторы. Следовательно, для разработки лекарственного средства очень важно обеспечить подходящую форму лекарственного средства, характеризующегося желательными свойствами.
Краткая сущность настоящего изобретения
Согласно одному аспекту в настоящей заявке предусмотрен кристалл A соединения, представленного формулой I
где дифракционная рентгенограмма (XRD) кристалла A соединения, представленного формулой I, характеризуется дифракционными пиками при 2θ, равными 9,35°±0,2°, 11,93°±0,2°, 16,32°±0,2°, 21,23°±0,2° 23,13±0,2° и 25,58±0,2°.
Согласно другому аспекту в настоящей заявке предусмотрен способ получения кристалла A соединения, представленного формулой I, и способ предусматривает следующие стадии:
1) растворение соединения, представленного формулой I, в кристаллизующем растворителе, причем кристаллизующий растворитель выбирают из метанола, этанола, н-пропанола, изопропанола, н-бутанола, изобутанола, трет-бутанола, монометилового эфира этиленгликоля, диэтилового эфира, изопропилового эфира, метил-трет-бутилового эфира, диоксана, тетрагидрофурана, 2-метилтетрагидрофурана, ацетона, 1-бутанона, 2-бутанона, этилацетата, этилформиата, метилацетата, изопропилацетата, дихлорметана, хлороформа, воды или смешанного растворителя из любых двух или более из указанных выше растворителей; а также
2) кристаллизация соединения, представленного формулой I.
Согласно другому аспекту в настоящей заявке предусмотрена кристаллическая композиция, в которой кристалл A соединения, представленного формулой I, составляет 50% или более, предпочтительно 80% или более, более предпочтительно 90% или более и наиболее предпочтительно 95% или более от массы кристаллической композиции.
Согласно другому аспекту в настоящей заявке предусмотрена фармацевтическая композиция, причем фармацевтическая композиция содержит эффективное количество кристалла A соединения, представленного формулой I, или кристаллической композиции, содержащей кристалл A соединения, представленного формулой I.
Согласно другому аспекту в настоящей заявке предусмотрено применение кристалла A соединения, представленного формулой I, или описанной выше кристаллической композиции или фармацевтической композиции при получении лекарственного средства для лечения или профилактики опосредованного янус-киназой заболевания.
Согласно другому аспекту в настоящей заявке предусмотрен кристалл B соединения, представленного формулой I,
где дифракционная рентгенограмма (XRD) кристалла B соединения, представленного формулой I, характеризуется дифракционными пиками при 2θ, равными 8,97°±0,2°, 9,39°±0,2°, 12,90°±0,2°, 17,70°±0,2° 20,31°±0,2° и 23,63°±0,2°.
Согласно другому аспекту в настоящей заявке предусмотрен способ получения кристалла B соединения, представленного формулой I, и способ предусматривает следующие стадии:
1) растворение соединения, представленного формулой I, в ацетонитриле; а также
2) кристаллизация соединения, представленного формулой I.
Согласно другому аспекту в настоящей заявке предусмотрена кристаллическая композиция, в которой кристалл B соединения, представленного формулой I, составляет 50% или более, предпочтительно 80% или более, более предпочтительно 90% или более и наиболее предпочтительно 95% или более от массы кристаллической композиции.
Согласно другому аспекту в настоящей заявке предусмотрена фармацевтическая композиция, причем фармацевтическая композиция содержит эффективное количество кристалла B соединения, представленного формулой I, или кристаллической композиции, содержащей кристалл B соединения, представленного формулой I.
Согласно другому аспекту в настоящей заявке предусмотрено применение кристалла B соединения, представленного формулой I, или описанной выше кристаллической композиции или фармацевтической композиции при получении лекарственного средства для лечения или профилактики опосредованного янус-киназой заболевания.
Краткое описание графических материалов
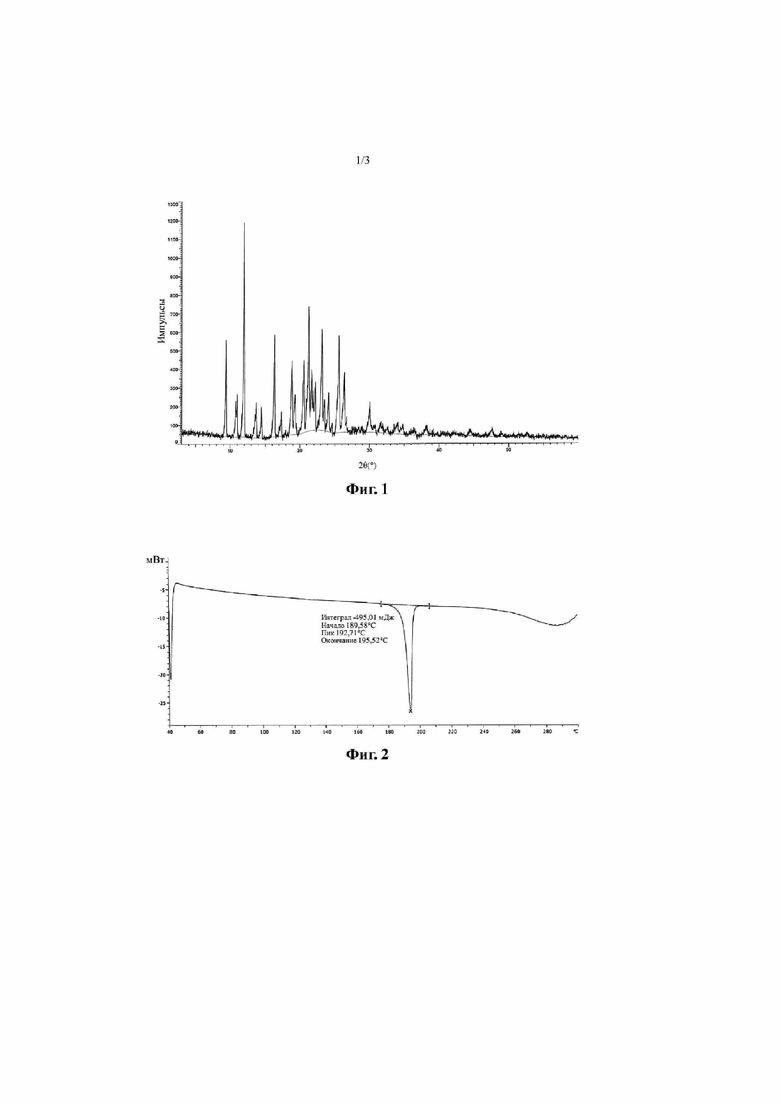
Фиг. 1 представляет собой дифракционную рентгенограмму кристалла A соединения, представленного формулой I (способ 1 в примере 2).
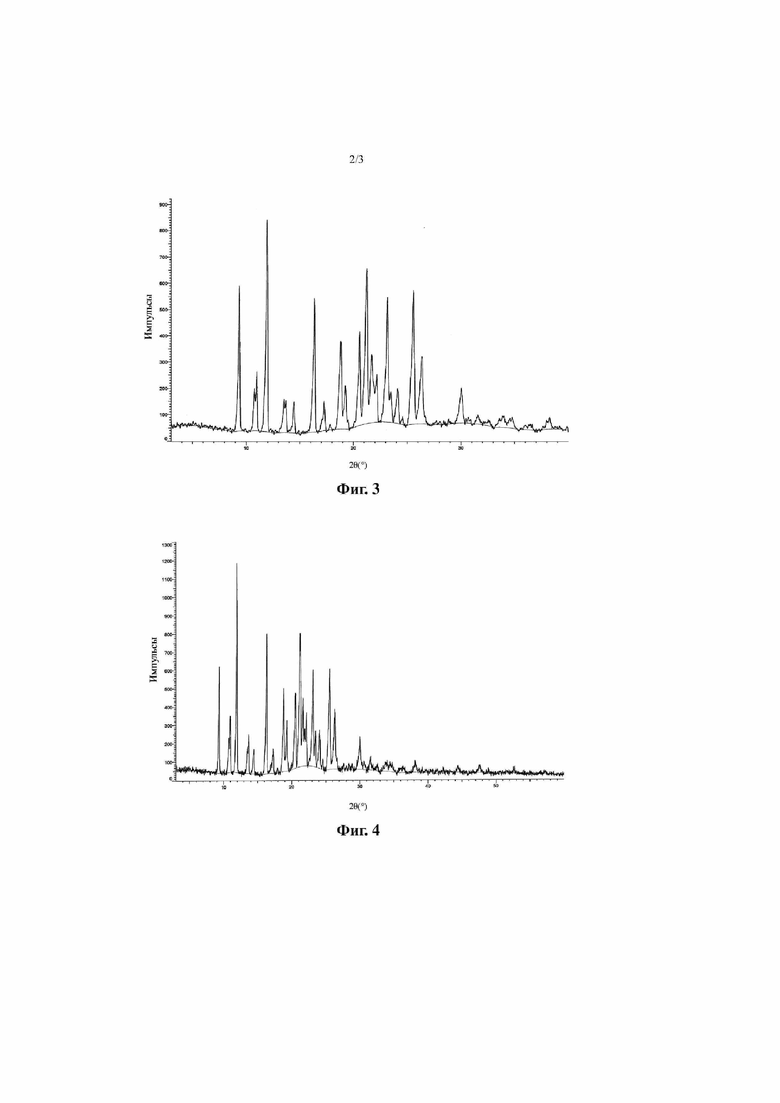
Фиг. 2 представляет собой спектр дифференциальной сканирующей калориметрии кристалла A соединения, представленного формулой I (способ 1 в примере 2).
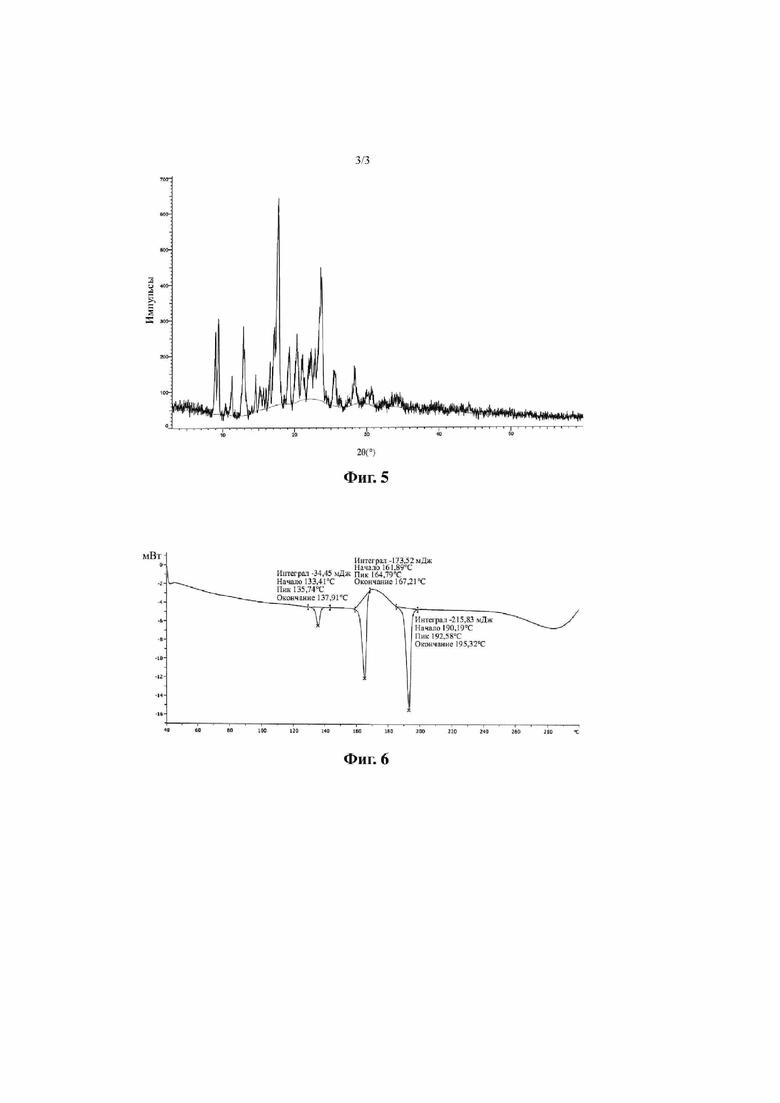
Фиг. 3 представляет собой дифракционную рентгенограмму кристалла A соединения, представленного формулой I (способ 2 в примере 2, этанол-этилацетат (4:1)).
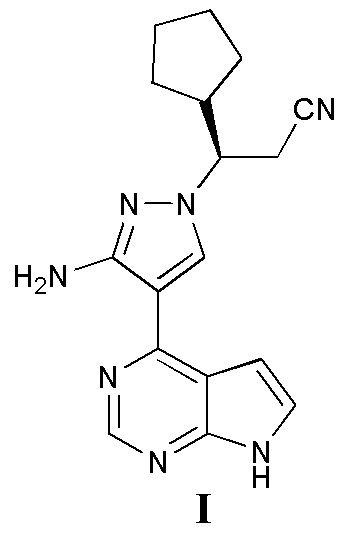
Фиг. 4 представляет собой дифракционную рентгенограмму кристалла A соединения, представленного формулой I (способ 3 в примере 2).
Фиг. 5 представляет собой дифракционную рентгенограмму кристалла B соединения, представленного формулой I (пример 3).
Фиг. 6 представляет собой спектр дифференциальной сканирующей калориметрии кристалла B соединения, представленного формулой I (пример 3).
Подробное описание настоящего изобретения
Согласно одному аспекту в настоящей заявке предусмотрен кристалл A соединения, представленного формулой I:
где дифракционная рентгенограмма (XRD) кристалла A соединения, представленного формулой I, характеризуется дифракционными пиками при 2θ, равными 9,35°, 11,93°, 16,32°, 21,23°, 23,13° и 25,58°±0,2°; как правило характеризуется дифракционными пиками при 2θ, равными 9,35°, 11,93°, 16,32°, 18,82°, 20,54°, 21,23°, 23,13° и 25,58°±0,2°; более типично характеризуется дифракционными пиками при 2θ, равными 9,35°, 10,93°, 11,93°, 14,46°, 16,32°, 18,82°, 20,54°, 21,23°, 21,66°, 23,13°, 25,58° и 26,34°±0,2°; и, кроме того, как правило, характеризуется дифракционными пиками при 2θ, равными 9,35°, 10,93°, 11,93°, 14,46°, 16,32°, 17,28°, 18,82°, 19,25°, 20,54°, 21,23°, 21,66°, 22,15°, 23,13°, 24,09°, 25,58° и 26,34°±0,2°.
Согласно некоторым вариантам осуществления настоящей заявки на дифракционной рентгенограмме (XRD) кристалла A соединения, представленного формулой I настоящей заявки, пик, имеющий самую высокую относительную интенсивность, появляется в положении дифракционного пика при 2θ, равном 11,93°, 16,32° или 21,23°±0,2°; и предпочтительно, пик, имеющий самую высокую относительную интенсивность, появляется в положении дифракционного пика при 2θ, равном 11,93°±0,2°.
Согласно некоторым вариантам осуществления настоящей заявки на дифракционной рентгенограмме (XRD) кристалла A соединения, представленного формулой I настоящей заявки, пики, имеющие три верхние относительные интенсивности, появляются в положениях дифракционных пиков при 2θ, равных 9,35°, 11,93°, 16,32°, 21,23°, 23,13° или 25,58°±0,2°.
Согласно некоторым вариантам осуществления настоящей заявки пики дифракционной рентгенограммы кристалла A соединения, представленного формулой I настоящей заявки, имеют следующие характеристики:
Согласно некоторым вариантам осуществления настоящей заявки дифракционная рентгенограмма кристалла A соединения, представленного формулой I, показана на фиг. 1.
Согласно некоторым вариантам осуществления настоящей заявки спектр дифференциальной сканирующей калориметрии кристалла A соединения, представленного формулой I, показан на фиг. 2.
Согласно некоторым вариантам осуществления настоящей заявки дифракционная рентгенограмма кристалла A соединения, представленного формулой I, показана на фиг. 3.
Согласно некоторым вариантам осуществления настоящей заявки дифракционная рентгенограмма кристалла A соединения, представленного формулой I, показана на фиг. 4.
Согласно другому аспекту в настоящей заявке предусмотрен способ получения кристалла A соединения, представленного формулой I, и способ предусматривает следующие стадии:
1) растворение соединения, представленного формулой I, в кристаллизующем растворителе, причем кристаллизующий растворитель выбирают из метанола, этанола, н-пропанола, изопропанола, н-бутанола, изобутанола, трет-бутанола, монометилового эфира этиленгликоля, диэтилового эфира, изопропилового эфира, метил-трет-бутилового эфира, диоксана, тетрагидрофурана, 2-метилтетрагидрофурана, ацетона, 1-бутанона, 2-бутанона, этилацетата, этилформиата, метилацетата, изопропилацетата, дихлорметана, хлороформа, воды или смешанного растворителя любых двух или более из указанных выше растворителей; а также
2) кристаллизация соединения, представленного формулой I, и, возможно, фильтрация, промывание и/или сушка полученного твердого вещества.
Согласно некоторым вариантам осуществления настоящей заявки кристаллизующий растворитель для получения кристалла A соединения, представленного формулой I, представляет собой этанол, изопропиловый эфир, этилацетат, ацетон, дихлорметан, воду или смешанный растворитель из любых двух или более из вышеуказанных растворителей; и предпочтительно этанол, смешанный растворитель из этанола и этилацетата, смешанный растворитель из этанола и воды, смешанный растворитель из этанола и изопропилового эфира, ацетона, этилацетата или дихлорметана.
Согласно некоторым вариантам осуществления настоящей заявки кристаллизующий растворитель для получения кристалла A соединения, представленного формулой I, предпочтительно представляет собой этанол или смешанный растворитель, содержащий этанол; и более предпочтительно другой растворитель в смешанном растворителе, содержащем этанол, выбирают из метанола, н-пропанола, изопропанола, н-бутанола, изобутанола, трет-бутанола, монометилового эфира этиленгликоля, диэтилового эфира, изопропилового эфира, метил-трет-бутилового эфира, диоксана, тетрагидрофурана, 2-метилтетрагидрофурана, ацетона, 1-бутанона, 2-бутанона, этилацетата, этилформиата, метилацетата, изопропилацетата, дихлорметана, хлороформа или воды.
Согласно некоторым вариантам осуществления настоящей заявки при получении кристалла A соединения, представленного формулой I, отношение количества соединения, представленного формулой I (по массе, в единицах г), к количеству кристаллизующего растворителя (по объему, в единицах мл) находится в диапазоне от 1:5 до 1:50, предпочтительно 1:7,5, 1:10, 1:12, 1:15, 1:18, 1:20, 1:25, 1:30, 1:35, 1:40, 1:45 или 1:50 и более предпочтительно от 1:7,5 до 1:30.
Согласно некоторым вариантам осуществления настоящей заявки, когда кристаллизующий растворитель для получения кристалла A соединения, представленного формулой I, представляет собой смешанный растворитель, содержащий этанол, содержание этанола (по объему) составляет от 10% до 90%; и предпочтительно 10%, 20%, 25%, 30%, 33%, 40%, 50%, 60%, 66%, 70%, 75%, 80% или 90%.
Согласно некоторым вариантам осуществления настоящей заявки, когда кристаллизующий растворитель для получения кристалла A соединения, представленного формулой I, представляет собой смешанный растворитель, содержащий этанол, отношение этанола к другому растворителю (по объему) находится в диапазоне от 9:1 до 1:9; и предпочтительно 9:1, 8:1, 7:1, 6:1, 5:1, 4:1, 3:1, 2:1, 1:1, 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8 или 1:9.
Согласно некоторым вариантам осуществления настоящей заявки кристаллизация может быть осуществлена путем охлаждения, например, охлаждения до температуры 0°C-5°C для кристаллизации. Согласно некоторым вариантам осуществления настоящей заявки кристаллизация может быть осуществлена путем концентрирования при пониженном давлении.
Согласно другому аспекту в настоящей заявке предусмотрена кристаллическая композиция, содержащая кристалл A соединения, представленного формулой I. Согласно некоторым вариантам осуществления настоящей заявки кристалл A соединения, представленного формулой I, составляет 50% или более, предпочтительно 80% или более, более предпочтительно 90% или более и наиболее предпочтительно 95% или более от массы кристаллической композиции.
Согласно другому аспекту в настоящей заявке предусмотрена фармацевтическая композиция, содержащая кристалл A соединения, представленного формулой I, причем фармацевтическая композиция содержит эффективное количество кристалла A соединения, представленного формулой I, или кристаллической композиции, содержащей кристалл А соединения, представленного формулой I. Кроме того, фармацевтическая композиция может дополнительно содержать фармацевтически приемлемый носитель, вспомогательное вещество и/или среду или может не содержать их.
Согласно другому аспекту в настоящей заявке предусмотрено применение кристалла A соединения, представленного формулой I, или описанной выше кристаллической композиции или фармацевтической композиции, при получении лекарственного средства для лечения или профилактики опосредованного янус-киназой заболевания.
Согласно другому аспекту в настоящей заявке предусмотрен способ лечения или профилактики опосредованного янус-киназой заболевания, предусматривающий введение нуждающемуся в этом млекопитающему терапевтически эффективного количества кристалла A соединения, представленного формулой I, или описанной выше кристаллической композиции или фармацевтической композиции.
Согласно другому аспекту в настоящей заявке предусмотрен кристалл A соединения, представленного формулой I, или описанная выше кристаллическая композиция или фармацевтическая композиция для применения в лечении или профилактике опосредованного янус-киназой заболевания.
Согласно другому аспекту в настоящей заявке предусмотрен кристалл B соединения, представленного формулой I:
где дифракционная рентгенограмма (XRD) кристалла B соединения, представленного формулой I, характеризуется дифракционными пиками при 2θ, равными 8,97°, 9,39°, 12,90°, 17,70°, 20,31° и 23,63°±0,2°; как правило, характеризуется дифракционными пиками при 2θ, равными 8,97°, 9,39°, 12,90°, 16,54°, 17,70°, 19,20°, 20,31°, 22,78° и 23,63°±0,2°; и более типично характеризуется дифракционными пиками при 2θ, равными 8,97°, 9,39°, 11,24°, 12,90°, 14,56°, 16,54°, 17,70°, 19,20°, 20,31°, 22,23°, 22,78°, 23,63° и 25,55°±0,2°.
Согласно некоторым вариантам осуществления настоящей заявки на дифракционной рентгенограмме (XRD) кристалла B соединения, представленного формулой I настоящей заявки, пик, имеющий самую высокую относительную интенсивность, появляется в положении дифракционного пика при 2θ, равном 9,39°, 17,70° или 23,63°±0,2°; и предпочтительно, пик, имеющий наивысшую относительную интенсивность, появляется в положении дифракционного пика при 2θ, равном 17,70°±0,2°.
Согласно некоторым вариантам осуществления настоящей заявки пики рентгеновской дифракции кристалла B соединения, представленного формулой I настоящей заявки, имеют следующие характеристики:
Согласно некоторым вариантам осуществления настоящей заявки дифракционная рентгенограмма кристалла B соединения, представленного формулой I, показана на фиг. 5.
Согласно некоторым вариантам осуществления настоящей заявки спектр дифференциальной сканирующей калориметрии кристалла B соединения, представленного формулой I, показан на фиг. 6.
Кристалл B соединения, представленного формулой I, в соответствии с настоящей заявкой представляет собой ацетонитрилат соединения, представленного формулой I, причем молярное отношение соединения, представленного формулой I, к ацетонитрилу находится в диапазоне от 1:0,5 до 1:2,0 и предпочтительно составляет 1:0,5, 1:1, 1:1,5 или 1:2,0.
Согласно другому аспекту в настоящей заявке предусмотрен способ получения кристалла B соединения, представленного формулой I, и способ предусматривает следующие стадии:
1) растворение соединения, представленного формулой I, в ацетонитриле; а также
2) кристаллизация соединения, представленного формулой I, и, возможно, фильтрация, промывание и/или сушка полученного твердого вещества.
Согласно некоторым вариантам осуществления настоящей заявки при получении кристалла B соединения, представленного формулой I, соотношение количества соединения, представленного формулой I (по массе, в единицах г), к количеству кристаллизующего растворителя ацетонитрила (по объему, в единицах мл) находится в диапазоне от 1:5 до 1:50, предпочтительно 1:7,5, 1:10, 1:12, 1:15, 1:18, 1:20, 1:25, 1:30, 1:35, 1:40, 1:45 или 1:50 и более предпочтительно в диапазоне от 1:10 до 1:25.
Согласно некоторым вариантам осуществления настоящей заявки кристаллизация может быть осуществлена путем охлаждения, например, путем охлаждения до температуры 0°С-5°С для кристаллизации.
Согласно другому аспекту в настоящей заявке предусмотрена кристаллическая композиция, содержащая кристалл B соединения, представленного формулой I. Согласно некоторым вариантам осуществления настоящей заявки кристалл B соединения, представленного формулой I, составляет 50% или более, предпочтительно 80% или более, более предпочтительно 90% или более и наиболее предпочтительно 95% или более от массы кристаллической композиции.
Согласно другому аспекту в настоящей заявке предусмотрена фармацевтическая композиция, содержащая кристалл B соединения, представленного формулой I, причем фармацевтическая композиция содержит эффективное количество кристалла B соединения, представленного формулой I, или кристаллической композиции, содержащей кристалл В соединения, представленного формулой I. Кроме того, фармацевтическая композиция может дополнительно содержать фармацевтически приемлемый носитель, вспомогательное вещество и/или среду или может не содержать их.
Согласно другому аспекту в настоящей заявке предусмотрено применение кристалла B соединения, представленного формулой I, или описанной выше кристаллической композиции или фармацевтической композиции при получении лекарственного средства для лечения или профилактики опосредованного янус-киназой заболевания.
Согласно другому аспекту в настоящей заявке предусмотрен способ лечения или профилактики опосредованного янус-киназой заболевания, предусматривающий введение нуждающемуся в этом млекопитающему терапевтически эффективного количества кристалла B соединения, представленного формулой I, или описанной выше кристаллической композиции или фармацевтической композиции.
Согласно другому аспекту в настоящей заявке предусмотрен кристалл B соединения, представленного формулой I, или описанная выше кристаллическая композиция или фармацевтическая композиция для применения в лечении или профилактике опосредованного янус-киназой заболевания.
В настоящей заявке дифракционные рентгенограммы измеряют следующим способом: прибор: рентгеновский дифрактометр Bruker D8 ADVANCE; способ: мишень: Cu: K-альфа; длина волны λ = 1,54179 Ǻ; напряжение трубки: 40 кВ; ток трубки: 40 мА; диапазон сканирования: 4-40o; скорость сканирования: 0,1 с/шаг, 0,02°/шаг.
В настоящей заявке используется следующий способ дифференциальной сканирующей калориметрии (ДСК): прибор: дифференциальный сканирующий калориметр DSC-1 Mettler; способ: образцы (-5 мг) испытывают на алюминиевой чашке для ДСК при температуре от 30°C до 300°C и при скорости нагрева 10°C/мин.
Следует отметить, что в рентгеновском дифракционном спектре дифракционная картина кристаллического соединения, как правило, характерна для конкретной кристаллической формы. Относительные интенсивности полос (особенно под малыми углами) могут варьировать в зависимости от преимущественных эффектов ориентации, обусловленных различиями состояний кристаллов, размеров частиц и другими условиями измерений. Следовательно, относительные интенсивности дифракционных пиков не характерны для конкретной кристаллической формы. Именно на относительные положения пиков, а не на их относительные интенсивности следует обращать больше внимания при оценке того, является ли кристаллическая форма такой же, как известная кристаллическая форма. Кроме того, что касается любой данной кристаллической формы, может быть небольшая ошибка в положении пиков, что также хорошо известно в области кристаллографии. Например, положение пика может сдвигаться из-за изменения температуры, движения образца или калибровки прибора и т.д. при анализе образца, и погрешность измерения значения 2θ иногда составляет приблизительно ±0,2°. Соответственно, эту ошибку следует учитывать при идентификации кристаллической структуры. Как правило, положение пика выражается в виде угла 2θ или периода решетки d в рентгенограмме, и простым соотношением преобразования между ними является d = λ/2sinθ, где d представляет собой период решетки, λ представляет собой длину волны падающего рентгеновского луча и θ представляет собой угол дифракции. Для одной и той же кристаллической формы того же самого соединения положение пиков в его рентгенограмме в целом имеет сходство, и ошибка относительных интенсивностей может быть больше. Кроме того, необходимо указать, что из-за некоторых факторов, таких как уменьшенное содержание, части дифракционных линий могут отсутствовать при идентификации смеси. В это время даже полоса может быть характерна для данной кристаллической формы без зависимости от всех полос образца высокой чистоты.
Следует отметить, что ДСК используется для измерения температуры теплового перехода при поглощении или выделении тепла вследствие изменения кристаллической структуры или плавления кристалла. При непрерывном анализе одной и той же кристаллической формы одного и того же соединения погрешность температуры теплового перехода и температуры плавления, как правило, находится в диапазоне приблизительно ±5°C. Когда говорят, что соединение имеет заданный пик или температуру плавления ДСК, это означает, что пик или температура плавления ДСК может варьировать в диапазоне ±5°С. ДСК предоставляет вспомогательный способ для установления различий между разными кристаллическими формами. Различные кристаллические формы могут быть идентифицированы по их характерно различным температурам перехода.
Заболевание, опосредованное янус-киназой, согласно настоящей заявке включает в себя, но без ограничения, опухоль (например, лимфому, лейкоз). Лимфома в соответствии с настоящей заявкой включает в себя, но без ограничения, болезнь Ходжкина или неходжкинскую лимфому, а неходжкинская лимфома включает в себя, но без ограничения, В-клеточную лимфому или Т-клеточную лимфому. Лейкоз в соответствии с настоящей заявкой включает в себя, но без ограничения, острый лимфобластный лейкоз, хронический лимфоцитарный лейкоз, острый миелоидный лейкоз и хронический миелоцитарный лейкоз.
В настоящей заявке термин «фармацевтическая композиция» относится к композиции из одного или нескольких соединений по настоящей заявке и носителя, вспомогательного вещества и/или среды, общепринятых в настоящей области техники для транспортировки биологически активного соединения в организм (например, человека). Задачей фармацевтической композиции является облегчение введения соединения по настоящему изобретению в организм.
Термин «носитель» определяется как соединение, которое облегчает введение соединения в клетку или ткань. Например, диметилсульфоксид (ДМСО), как правило, используется в качестве носителя, поскольку его легко использовать для введения некоторых органических соединений в клетки или ткани организмов.
Термин «фармацевтически приемлемый носитель» включает в себя, но без ограничения, любой адъювант, вспомогательное вещество, скользящее вещество, подсластитель, разбавитель, консервант, краситель/красящее вещество, ароматизатор, поверхностно-активное вещество, смачивающее средство, диспергатор, суспендирующее средство, стабилизатор, изотоническое средство, растворитель или эмульгатор, одобренный Национальным управлением по лекарственным средствам как приемлемый для применения у людей или домашнего скота.
Термин «терапевтически эффективное количество» относится к количеству соединения по настоящей заявке, и когда его вводят млекопитающему, предпочтительно человеку, его достаточно, чтобы реализовать лечение вирусной инфекции у млекопитающего, предпочтительно у человека, как определяется в дальнейшем. Количество соединения по настоящей заявке, образующего «терапевтически эффективное количество», изменяется в зависимости от соединения, патологического состояния и его тяжести, пути введения и возраста подлежащего лечению млекопитающего, но может быть условно определено специалистами обычной квалификации в настоящей области техники на основании собственных знаний и раскрытии настоящей заявки.
Используемый в настоящем документе термин «лечение» охватывает лечение вирусной инфекции у млекопитающего, предпочтительно вирусной инфекции у человека, и включает в себя:
(i) ингибирование вирусной инфекции, т.е. прекращение ее развития;
(ii) облегчение вирусной инфекции; т.е. способствование регрессу вирусной инфекции; или же
(iii) облегчение симптомов, вызванных вирусной инфекцией.
Все растворители, используемые в настоящей заявке, доступны на рынке и могут использоваться без дальнейшей очистки. Реакции, как правило, проводят в атмосфере инертного азота в безводном растворителе.
В настоящей заявке данные протонного ядерного магнитного резонанса записывают на спектрометре BRUKER AVANCE III HD 500M; химический сдвиг выражают в миллионных долях от тетраметилсилана; а масс-спектр измеряют с помощью Waters ACQUITY UPLC+XEVO G2 QTof. Масс-спектрометр оснащен источником электрораспыления ионов (ESI), работающим в положительном или отрицательном режиме.
Кристалл A и кристалл B соединения, представленного формулой I в соответствии с настоящей заявкой, обладают преимуществами высокой чистоты, высокой кристалличности и хорошей стабильности. Кроме того, способы получения кристалла A и кристалла B соединения, представленного формулой I, в соответствии с настоящей заявкой просты, используемые в них растворители недороги и легко доступны, а условия кристаллизации мягкие. Поэтому способы пригодны для промышленного производства.
Следующие примеры предоставлены для дополнительной иллюстрации технических решений настоящей заявки не ограничивающим образом. Они должны рассматриваться не как ограничивающие объем настоящего изобретения, а просто как иллюстративное описание и типичные представители настоящего изобретения. Растворители, реагенты и исходные материалы, используемые в настоящей заявке, представляют собой химически чистые или аналитически чистые продукты, доступные на рынке.
Пример 1: (3R)-3-{3-амино-4-{7Н-пирроло[2,3-d]пиримидин-4-ил}-1Н-пиразол-1-ил}-3-циклопентилпропаннитрил (I)
Стадия A: 3-циклопентилакриловая кислота
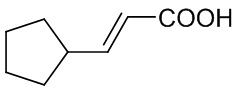
Циклопентилкарбальдегид (344,4 г, 3,51 моль, 1,17 экв.) добавляли по каплям к раствору пропандиовой кислоты в концентрации 5М (312 г, 3,0 моль, 1,0 экв.) в пиридине при комнатной температуре. После завершения добавления полученную смесь перемешивали в течение 10 минут. Затем по каплям медленно добавляли пиперидин (6,2 г, 0,075 моль, 0,025 экв.). После завершения добавления полученную смесь перемешивали при комнатной температуре в течение 1 часа. Полученную смесь нагревали до температуры 70-80°С, перемешивали в течение 8 часов и концентрировали при пониженном давлении, чтобы выпарить растворитель. Остаток доводили с помощью концентрированной соляной кислоты до рН 3,0 и трижды экстрагировали этилацетатом. Органические фазы объединяли и промывали раствором гидроксида натрия в концентрации 2,5 М пять раз. Водную фазу доводили с помощью концентрированной соляной кислоты до рН 3,0 и трижды экстрагировали этилацетатом. Органические слои объединяли, трижды промывали водой, промывали насыщенным раствором соли, сушили безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 3-циклопентилакриловой кислоты (391,2 г, выход: 93%). 1H ЯМР (500 МГц, CDCl3) δ: 7,08 (дд, J=15,6, 8,1 Гц, 1H), 5,81 (дд, J=15,6, 1,1 Гц, 1H), 11,25 (с, 1H), 2,64 (м, 1H), 1,63 (м, 2Н), 1,42 (м, 2Н), 1,86 (м, 2Н), 1,72 (м, 2Н); HRMS (ESI) рассчитано для C8H12O2 [M-H]- 139,0765; найдено: 139,0760.
Стадия B: 5-циклопентилпиразолидин-3-он
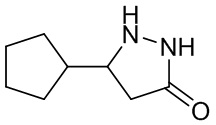
Гидразингидрат в концентрации 80% (253,5 г, 4,05 моль, 1,5 экв.) добавляли по каплям к циклопентилакриловой кислоте (378 г, 2,7 моль, 1,0 экв.) при перемешивании при комнатной температуре. Полученную смесь нагревали до температуры от 70 до 80°С, перемешивали в течение 6 часов, охлаждали до температуры от 0 до 10°С, перемешивали для кристаллизации и фильтровали. Осадок на фильтре дважды промывали водой и высушивали под давлением на воздухе при температуре 45°С в течение 12 часов, получая 5-циклопентилпиразолидин-3-он (292,5 г, выход 68%).
Стадия C: R-5-циклопентилпиразолидин-3-он-D-тартрат.
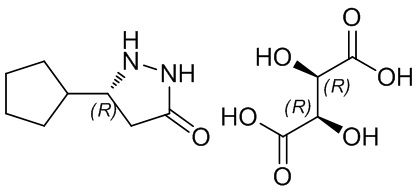
D-винную кислоту (135 г, 0,9 моль, 0,5 экв.) добавляли к раствору 5-циклопентилпиразолидин-3-она (278 г, 1,8 моль, 1,0 экв.) в ацетоне при перемешивании при комнатной температуре, перемешивали для реакции в течение 2 часов для кристаллизации и фильтровали. Осадок на фильтре 5 раз очищали ацетоном и сушили под давлением при температуре 50°С с получением R-5-циклопентилпиразолидин-3-он-D-тартрата (241 г, выход 88%, значение э.и. 99,5%).
Стадия D: R-5-циклопентилпиразолидин-3-он
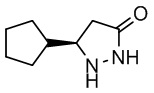
R-5-циклопентилпиразолидин-3-он-D-тартрат (228 г, 0,75 моль, 1,0 экв.) добавляли к раствору гидроксида натрия в концентрации 4М (52,2 г, 2,61 моль, 1,74 экв.) при перемешивании при комнатной температуре, и полученную смесь экстрагировали дихлорметаном. Органические слои объединяли, сушили безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением R-5-циклопентилпиразолидин-3-она (100,6 г, выход 85,2%, значение э.и. 99,5%). 1H-ЯМР (500 МГц, CDCl3) δ 8,93 (с, 1H), 5,15 (с, 1H), 1,89 (м, 1H), 1,67 (м, 2H), 1,55 (м, 2H), 1,47 (м, 2H), 1,26 (м, 1H), 1,14 (м, 1H); HRMS (ESI) рассчитано для C8H14N2O [М+Н]+ 155,1179; найдено: 155,1183.
Стадия E: 4-хлор-7-{[2-(триметилсилил)этокси]метил}-7H-пирроло[2,3-d]пиримидин
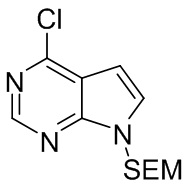
Раствор 4-хлорпирроло[2,3-d]пиримидина (200 г, 1,3 моль, 1,0 экв.) в N,N-диметилформамиде добавляли к NaH в концентрации 60% (62,4 г, 1,56 моль, 1,2 экв.) в ледяной ванне. После завершения добавления полученную смесь перемешивали для реакции при комнатной температуре в течение 1 часа. 2-(триметилсилил)этоксиметилхлорид (SEMCl, 260 г, 1,56 моль, 1,2 экв.) медленно добавляли по каплям при охлаждении на ледяной бане. После завершения добавления полученную смесь перемешивали для реакции на ледяной бане в течение 1 часа и реакцию гасили водой. Полученную смесь экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором соли, сушили безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением остатка, который очищали посредством колоночной хроматографии на силикагеле с получением 4-хлор-7-{[2-(триметилсилил)этокси]метил}-7Н-пирроло[2,3-d]пиримидина (312,2 г, выход 91,8%). 1H-ЯМР (500 МГц, CDCl3): δ 8,64 (с, 1H), 7,38 (д, J=3,6 Гц, 1H), 6,65 (д, J=3,6 Гц, 1H), 5,64 (с, 2H), 3,52 (т, J=8,2 Гц, 2H), 0,90 (т, J=8,2 Гц, 2H), -0,07 (с, 9H); HRMS (ESI) рассчитано для C12H18N3OSi [М+Н]+ 284,0980; найдено: 284,0995.
Стадия F: Этил-2-циано-2-{7-{[2-(триметилсилил)этокси]метил}-7H-пирроло[2,3-d]пиримидин-4-ил}ацетат
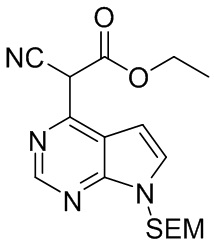
Карбонат калия (207 г, 1,5 моль, 3,0 экв.) добавляли к раствору 4-хлор-7-{[2-(триметилсилил)этокси]метил}-7Н-пирроло[2,3-d]пиримидина (142 г, 0,5 моль, 1,0 экв.) и этилцианоацетата (85 г, 0,75 моль, 1,5 экв.) в DMF при перемешивании при комнатной температуре. Полученную смесь нагревали до температуры 120ºC, перемешивали для реакции в течение 4 часов и затем охлаждали до комнатной температуры. Реакционную смесь гасили водой, перемешивали для кристаллизации и фильтровали. Осадок на фильтре промывали водой и высушивали под давлением при температуре 50ºC, получая этил-2-циано-2-{7-{[2-(триметилсилил)этокси]метил}-7H-пирроло[2,3-d]пиримидин-4-ил}ацетат (167 г, выход 92,6%). 1H-ЯМР (500 МГц, CDCl3): δ13,46 (с, 1H), 8,45 (с, 1H), 7,56 (д, J=3,6 Гц, 1H), 7,18 (д, J=3,6 Гц, 1H), 5,56 (с, 2H), 4,32 (кв, J=7,1 Гц, 2H), 3,52 (т, J=8,2 Гц, 2H), 1,27 (т, J=7,1 Гц, 3H), 0,83 (т, J=8,2 Гц, 2H), -0,08 (с, 9H); HRMS (ESI) рассчитано для C17H24N4O3Si [М+Н]+ 361,1690; найдено: 361,1699.
Стадия G: 2-{7-{[2-(триметилсилил)этокси]метил}-7H-пирроло[2,3-d]пиримидин-4-ил}ацетонитрил
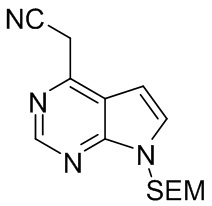
Хлорид натрия (263 г, 4,5 моль, 10 экв.) добавляли к смешанному раствору этил-2-циано-2-{7-{[2-(триметилсилил)этокси]метил}-7Н-пирроло[2,3-d]пиримидин-4-ил}ацетата (162,2 г, 0,45 моль, 1,0 экв.) в N-метилпирролидоне и воде при перемешивании при комнатной температуре. Полученную смесь нагревали до температуре 160-170°С и перемешивали в течение 30 часов. Реакцию гасили водой. Полученную смесь экстрагировали этилацетатом. Органическую фазу промывали насыщенным раствором соли, сушили безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии на силикагеле с получением 2-(7-{[2-(триметилсилил)этокси]метил}-7Н-пирроло[2,3-d]пиримидин-4-ил)ацетонитрила (98,6 г, выход 76%). 1H-ЯМР (500 МГц, CDCl3): δ 8,18 (с, 1H), 7,77 (д, J=3,4 Гц, 1H), 6,83 (д, J=3,4 Гц, 1H), 5,65 (с, 2H), 4,56 (с, 2H), 3,52 (т, J=7,6 Гц, 2H), 0,82 (т, J=7,6 Гц, 2H), -0,10 (с, 9H); HRMS (ESI) рассчитано для C14H20N4OSi [М+Н]+ 289,1479; найдено: 289,1498.
Стадия H: 3-(диметиламино)-2-{7-{[2-(триметилсилил)этокси]метил}-7H-пирроло[2,3-d]пиримидин-4-ил}акрилонитрил
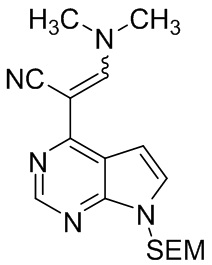
DMF-DMA (119 г, 1,0 моль, 3,0 экв.) добавляли к раствору 2-{7-{[2-(триметилсилил)этокси]метил}-7H-пирроло[2,3-d]пиримидин-4-ил}ацетонитрила (95 г, 0,33 моль, 1,0 экв.) в DMF. Полученную смесь нагревали до температуры кипения с обратным холодильником в течение 2 часов, а затем охлаждали до комнатной температуры. Добавляли воду, и полученную смесь перемешивали для кристаллизации и фильтровали. Осадок на фильтре промывали водой и высушивали под давлением при температуре 50ºC, получая 3-(диметиламино)-2-(7-{[2-(триметилсилил)этокси]метил}-7H-пирроло[2,3-d]пиримидин-4-ил)акрилонитрил (106,5 г, выход 94%).
1H-ЯМР (500 МГц, CDCl3): δ 8,50 (с, 1H), 8,38 (с, 1H), 7,26 (д, J=3,7 Гц, 1H), 7,18 (д, J=3,7 Гц, 1H), 5,56 (с, 2H), 3,49 (т, J=8,4 Гц, 2H), 3,43 (с, 3H), 3,23 (с, 3H), 0,87 (т, J=8,4 Гц, 2H), -0,10 (с, 9H); HRMS (ESI) рассчитано для C17H25N5OSi [М+Н]+ 344,1901; найдено: 344,1907.
Стадия I: (R)-3-{3-амино-4-{7-{[2-(триметилсилил)этокси]метил}-7H-пирроло[2,3-d]пиримидин-4-ил}-1H-пиразол-1-ил}-3-циклопентил-пропионовая кислота
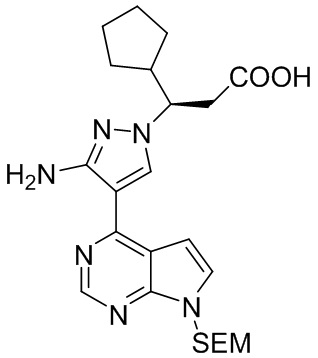
Ацетат калия (1,5 экв.) добавляли к раствору 3-(диметиламино)-2-{7-{[2-(триметилсилил)этокси]метил}-7Н-пирроло[2,3-d]пиримидин-4-ил}нитрила акриловой кислоты (68,7 г, 0,2 моль, 1,0 экв.) и R-5-циклопентилпиразолидин-3-она (37,0 г, 0,24 моль, 1,2 экв.) в N-метилпирролидоне при перемешивании при комнатной температуре. Полученную смесь нагревали до температуры 120-130°С и перемешивали в течение 12 часов. Реакцию гасили водой и полученную смесь экстрагировали этилацетатом. Органический слой трижды промывали водой, промывали насыщенным раствором соли и сушили безводным сульфатом натрия. После фильтрации остаток концентрировали при пониженном давлении и очищали посредством колоночной хроматографии на силикагеле с получением (R)-3-{3-амино-4-{7-{[2-(триметилсилил)этокси]метил}-7Н-пирроло[2,3-d]пиримидин-4-ил}-1Н-пиразол-1-ил}-3-циклопентил-пропионовой кислоты (37,6 г, выход 40,1%, значение э.и. 99,8%). 1H-ЯМР (500 МГц, CDCl3): δ 8,74 (с, 1H), 7,96 (с, 1H), 7,32 (д, J=3,4 Гц, 1H), 6,67 (д, J=3,4 Гц, 1H), 5,63 (м, 2H), 4,19 (т, J=8,2 Гц, 2H), 3,52 (м, 1H), 3,52 (т, J=8,4 Гц, 2H), 3,09 (дд, J=16,7, 8,2 Гц, 1H), 2,87 (д, J=16,7 Гц, 1H), 2,41 (м, 1H), 1,87 (м, 1H), 1,69 (м, 1H), 1,60 (м, 2H), 1,51 (м, 2H), 1,15. (м, 1H), 0,91 (т, J=8,4 Гц, 2H), -0,06 (с, 9H); HRMS (ESI) рассчитано для C17H25N5OSi [М+Н]+ 471,2534; найдено: 471,2538.
Стадия J: (R)-3-{3-(2,5-диоксопиррол-1-ил)-4-{7-{[2-(триметилсилил)этокси]метил}-7H-пирроло[2,3-d]пиримидин-4-ил}-1Н-пиразол-1-ил}-3-циклопентил-пропионовая кислота
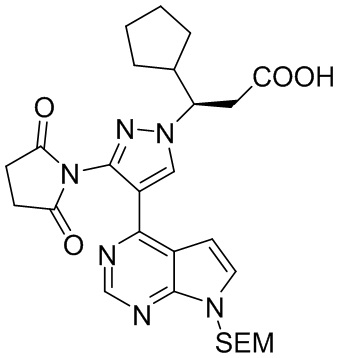
Янтарный ангидрид (10,4 г, 104 ммоль, 1,4 экв.) добавляли к раствору (R)-3-{3-амино-4-{7-{[2-(триметилсилил)этокси]метил}-7H-пирроло[2,3-d]пиримидин-4-ил}-1Н-пиразол-1-ил}-3-циклопентил-пропионовой кислоты в концентрации 0,2 М (35,0 г, 74,3 ммоль, 1,0 экв.) в метилбензоле при перемешивании при комнатной температуре. Под защитой газообразного азота полученную смесь нагревали с обратным холодильником для реакции (отвод воды) в течение 14 часов. Растворитель выпаривали концентрированием при пониженном давлении. Остаток растворяли в этилацетате и промывали водой, насыщенным раствором бикарбоната натрия и насыщенным раствором соли. Слой этилацетата сушили и обесцвечивали безводным сульфатом натрия и активированным углем при перемешивании, фильтровали и концентрировали при пониженном давлении, получая (R)-3-{3-(2,5-диоксопиррол-1-ил)-4-{7-{[2-(триметилсилил)этокси]метил}-7Н-пирроло[2,3-d]пиримидин-4-ил}-1Н-пиразол-1-ил}-3-циклопентил-пропионовую кислоту (39 г , 70,6 ммоль, выход 95%). 1H-ЯМР (500 МГц, CDCl3): δ 8,65 (с, 1H), 8,28 (с, 1H), 7,28 (д, J=3,7 Гц, 1H), 6,62 (д, J=3,7 Гц, 1H), 5,59 (д, J=11,1 Гц, 1H), 5,53 (д, J=11,1 Гц, 1H), 4,44 (тд, J=9,9, 3,2 Гц, 1H), 3,48 (м, 2H), 3,02 (дд, J=16,8, 10,0 Гц, 1H), 2,83 (м, 1H), 2,43 (м, 1H), 1,78 (м, 1H), 1,69 (м, 1H), 1,61 (м, 1H), 1,52 (м, 1H) 1,51 (м, 1Н), 1,50 (м, 2Н), 1,14 (м, 1Н), 0,88 (м, 2Н), -0,07 (с, 9Н); HRMS (ESI) рассчитано для C27H36N6O5Si [М+Н]+ 553,2589; найдено: 553,2603.
Стадия K: (R)-3-циклопентил-3-[3-(2,5-диоксопиррол-1-ил)-4-(7-{[2-(триметилсилил)этокси]метил}-7H-пирроло[2],3-d]пиримидин-4-ил)-1Н-пиразол-1-ил]пропанамид
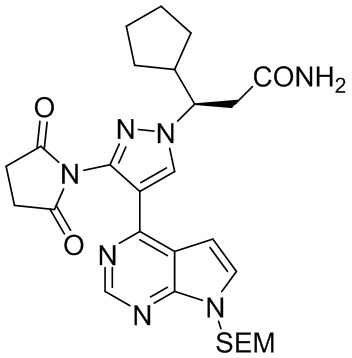
Оксалилхлорид (20,0 г, 158 ммоль, 2,5 экв.) добавляли по каплям к раствору (R)-3-{3-(2,5-диоксопиррол-1-ил)-4-{7-{[2-(триметилсилил)этокси]метил}-7Н-пирроло[2,3-d]пиримидин-4-ил}-1Н-пиразол-1-ил}-3-циклопентил-пропионовой кислоты в концентрации 0,18 М (35,0 г, 63,3 ммоль, 1,0 экв.) в дихлорметане при перемешивании на ледяной бане и под защитой газообразного азота. После завершения добавления по каплям добавляли DMF (0,1 г, 1,3 ммоль, 0,02 экв.) и полученную смесь перемешивали при комнатной температуре для реакции в течение 1 часа и концентрировали при пониженном давлении, чтобы выпарить растворитель. Полученную смесь растворяли в ТГФ, который сушили натриевыми палочками и снова выпаривали, и полученную смесь по каплям добавляли к раствору водного аммиака в концентрации 2М (20,0, 0,32 моль, 5,0 экв.) в ТГФ. Полученную смесь перемешивали на ледяной бане для реакции в течение 30 минут, концентрировали при пониженном давлении для выпаривания ТГФ, охлаждали на ледяной бане в течение 2 часов для кристаллизации и фильтровали. Осадок на фильтре промывали водой и высушивали под давлением при температуре 50°C для получения (R)-3-циклопентил-3-{3-(2,5-диоксопиррол-1-ил)-4-{7-{[2-(триметилсилил)этокси]метил}-7Н-пирроло[2,3-d]пиримидин-4-ил}-1Н-пиразол-1-ил}пропанамида (29,8 г, выход 85,5%).
1H-ЯМР (500 МГц, CDCl3): δ8,65 (с, 1H), 8,24 (с, 1H), 7,32 (д, J=3,7 Гц, 1H), 6,63 (д, J=3,7, 1H), 6,12 (с, 1H), 5,60 (д, J=11,1 Гц, 1H), 5,56 (д, J=11,1 Гц, 1H), 5,44 (с, 1H), 4,40 (тд, J=10,6, 3,2 Гц, 1H) 3,47 (дд, J=9,1, 7,5 Гц, 2H), 2,99 (дд, J=14,4, 11,0 Гц, 1H), 2,91 (с, 4H), 2,67 (дд, J=14,4, 3,3 Гц, 1H), 2,48 (м, 1Н), 1,84 (м, 1Н), 1,66 (м, 1Н), 1,58 (м, 2Н), 1,57 (м, 1Н), 1,50 (м, 1Н), 1,31 (м, 1Н), 1,21 (м, 1Н), 0,88 (дд, 9,1, 7,5, 2Н), -0,08 (с, 9Н); HRMS (ES) рассчитано для C27H37N7O4Si [М+Н]+ 552,2749; найдено: 552,2759.
Стадия L: (R)-3-циклопентил-3-[3-(2,5-диоксопиррол-1-ил)-4-(7-{[2-(триметилсилил)этокси]метил}-7H-пирроло[2],3-d]пиримидин-4-ил)-1Н-пиразол-1-ил]пропионитрил
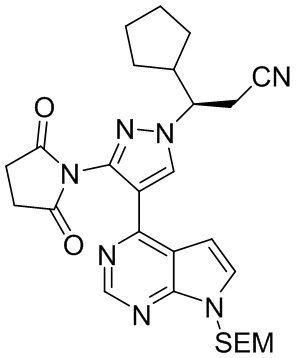
Оксихлорид фосфора (27,8 г, 181 ммоль, 4,0 экв.) добавляли по каплям к раствору (R)-3-циклопентил-3-{3-(2,5-диоксопиррол-1-ил)-4-{7-{[2-(триметилсилил)этокси]метил}-7Н-пирроло[2,3-d]пиримидин-4-ил}-1Н-пиразол-1-ил}пропанамида в концентрации 0,2 М (25 г, 45,3 ммоль, 1,0 экв.) в дихлорметане при перемешивании на ледяной бане. После завершения добавления полученную смесь перемешивали при комнатной температуре для взаимодействия в течение 2 часов. Реакцию гасили водой. Органический слой промывали водой, сушили и обесцвечивали безводным сульфатом магния и активированным углем при перемешивании. После фильтрации растворитель удаляли концентрированием при пониженном давлении для получения (R)-3-циклопентил-3-{3-(2,5-диоксопиррол-1-ил)-4-{7-{[2-(триметилсилил))этокси]метил}-7Н-пирроло[2,3-d]пиримидин-4-ил}-1Н-пиразол-1-ил}пропионитрила (22,2 г, 41,7 ммоль, выход 92%).
1H-ЯМР (500 МГц, CDCl3): δ8,70 (с, 1H), 8,35 (с, 1H), 7,35 (д, J=3,7 Гц, 1H), 6,66 (д, J=3,7 Гц, 1H), 5,62 (д, J=10,8 Гц, 1H), 5,58 (д, J=10,8 Гц, 1H), 4,30 (м, 1H), 3,50 (м, 2H), 3,09 (дд, J=16,8, 4,3 Гц, 1H), 3,01 (дд, J=16,8, 4,3 Гц, 1H), 2,94 (с, 4H), 2,62 (м, 1H), 1,96 (м, 1H), 1,69 (м, 2H), 1,60 (м, 1H) 1,58 (м, 2H), 1,27 (м, 2H), 0,90 (т, J=8,3 Гц, 2H), -0,06 (с, 9H); HRMS (ESI) рассчитано для C27H35N7O3Si [М+Н]+ 534,2643; найдено: 534,2657.
Стадия M: (R)-3-циклопентил-3-{3-(2,5-диоксопиррол-1-ил)-4-{(7-гидроксилметил)-7H-пирроло[2,3-d]пиримидин-4-ил}-1Н-пиразол-1-ил}пропионитрил
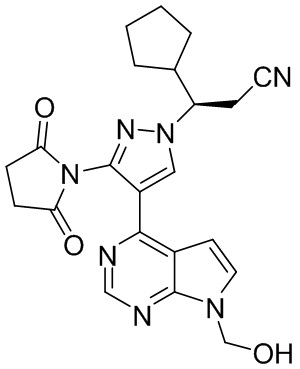
Раствор трифторида бора в концентрации 47% (34 г, 112,5 ммоль, 3,0 экв.) в диэтиловом эфире по каплям добавляли к раствору (R)-3-циклопентил-3-{3-(2,5-диоксопиррол)-1-ил)-4-{7-{[2-(триметилсилил)этокси]метил}-7Н-пирроло[2,3-d]пиримидин-4-ил}-1Н-пиразол-1-ил}пропионитрила в концентрации 0,2 М (20 г, 37,5 ммоль, 1,0 экв.) в дихлорметане при перемешивании на ледяной бане. После завершения добавления полученную смесь перемешивали при комнатной температуре, чтобы она прореагировала в течение 4 часов. Реакцию гасили водой. Полученную смесь доводили 10%-ным раствором NaOH до рН 6-7 и экстрагировали этилацетатом. Органический слой промывали водой, промывали насыщенным раствором соли и сушили безводным сульфатом магния при перемешивании. После фильтрации фильтрат концентрировали при пониженном давлении с получением (R)-3-циклопентил-3-[3-(2,5-диоксопиррол-1-ил)-4-(7-гидроксилметил)-7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил]пропионитрила (14,4 г, выход 88,5%).
1H-ЯМР (500 МГц, CDCl3): δ8,54 (с, 1H), 8,31 (с, 1H), 7,31 (д, J=3,7 Гц, 1H), 6,52 (д, J=3,7 Гц, 1H), 5,68 (д, J=10,9 Гц, 1H), 5,61 (д, J=10,9 Гц, 1H), 4,32 (м, 1H), 3,13 (дд, J=17,2, 7,9 Гц, 1H), 3,03 (дд, J=17,2, 4,3 Гц, 1H), 2,94 (с, 4H), 2,62 (м, 1H), 1,98 (м, 1H), 1,74 (м, 1H), 1,65 (м, 1H), 1,64 (м, 2H) 1,30 (м, 1Н); 1,29 (м, 2Н); HRMS (ESI) рассчитано для C22H23N7O3 [М+Н]+ 434,1935; найдено: 434,1944.
Стадия N: (R)-3-[3-амино-4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил]-3-циклопентилпропаннитрил (I)
Гидразингидрат в концентрации 80% (8,7 г, 138 ммоль, 5,0 экв.) добавляли по каплям к раствору (R)-3-{3-(2,5-диоксопиррол-1-ил)-4-{(7-гидроксиметил)-7Н-пирроло[2,3-d]пиримидин-4-ил}-1Н-пиразол-1-ил}-3-циклопентилпропаннитрила в концентрации 0,2 М (12 г, 27,7 ммоль, 1,0 экв.) в метаноле при перемешивании при комнатной температуре. После завершения добавления полученную смесь нагревали до температуры кипения с обратным холодильником в течение 8 часов и концентрировали при пониженном давлении, чтобы выпарить растворитель. Остаток растворяли в этилацетате, промывали водой, промывали насыщенным солевым раствором и сушили безводным сульфатом натрия в течение ночи. После фильтрации фильтрат концентрировали при пониженном давлении с получением (R)-3-[3-амино-4-(7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил]-3-циклопентилпропаннитрила (I) (7,7 г, выход 87%, значение э.и. 99,8%).
1H-ЯМР (500 МГц, CDCl3): δ11,73 (с, 1H), 8,79 (с, 1H), 8,06 (с, 1H), 7,32 (д, J=3,5 Гц, 1H), 6,62 (д, J=3,5 Гц, 1H), 5,03 (с, 2H), 4,05 (тд, J=9,5, 3,5 Гц, 1H), 3,12 (дд, J=17,1, 8,9 Гц, 1H), 2,91 (дд, J=17,1, 3,6 Гц, 1Н), 2,54 (м, 1Н), 1,74 (м, 1Н), 1,63 (м, 4Н), 1,27 (м, 1Н), 1,26 (м, 2Н); HRMS (ESI) рассчитано для C17H19N7 [M+H]+ 322,1775; найдено: 322,1783.
Пример 2: Кристалл A соединения, представленного формулой I
Способ 1
2,0 г соединения, представленного формулой I, полученного в примере 1, добавляли к 24 мл безводного этанола. Полученную смесь нагревали с обратным холодильником до получения прозрачного раствора, охлаждали до температуры 0-5°С, перемешивали в течение 4 часов для кристаллизации и фильтровали. Осадок на фильтре промывали 2 мл безводного этанола и сушили при пониженном давлении при температуре 50°С, получая 1,62 г продукта (выход 81%).
Способ 2
4 части 2,0 г соединения, представленного формулой I, полученного в примере 1, добавляли к 20 мл смешанного растворителя из безводного этанола и этилацетата (4:1, 2:1, 1:1, 1:4), соответственно. Полученные смеси нагревали с обратным холодильником до получения прозрачного раствора, охлаждали до температуры 0-5°С, перемешивали в течение 4 часов для кристаллизации и фильтровали. Осадок на фильтре промывали 2 мл этилацетата и сушили при пониженном давлении при температуре 50°С, получая 1,34 г, 1,06 г, 1,00 г и 1,60 г продуктов (выход: 67%, 53%, 50%, 80%).
Способ 3
2,0 г соединения, представленного формулой I, полученного в примере 1, добавляли к 20 мл смешанного раствора безводного этанола и воды (4:1). Полученную смесь нагревали с обратным холодильником до получения прозрачного раствора, охлаждали до температуры 0-5°С, перемешивали в течение 4 часов для кристаллизации и фильтровали. Осадок на фильтре промывали 2 мл безводного этанола и сушили при пониженном давлении при температуре 50°С, получая 1,6 г продукта (выход 80%).
Способ 4
2,0 г соединения, представленного формулой I, полученного в примере 1, добавляли к 15 мл ацетона. Полученную смесь нагревали с обратным холодильником до получения прозрачного раствора, охлаждали до температуры 0-5°С, перемешивали в течение 4 часов для кристаллизации и фильтровали. Осадок на фильтре промывали 2 мл ацетона и сушили при пониженном давлении при температуре 50°С, получая 1,22 г продукта (выход 61%).
Способ 5
2,0 г соединения, представленного формулой I, полученного в примере 1, добавляли к 50 мл этилацетата. Полученную смесь нагревали с обратным холодильником до получения прозрачного раствора. Растворитель выпаривали путем концентрирования при пониженном давлении, получая 1,98 г продукта (выход 99%).
Способ 6
2,0 г соединения, представленного формулой I, полученного в примере 1, добавляли к 60 мл дихлорметана. Полученную смесь нагревали с обратным холодильником до получения прозрачного раствора. Растворитель выпаривали путем концентрирования при пониженном давлении, получая 2,0 г продукта (выход 100%).
Способ 7
2,0 г соединения, представленного формулой I, полученного в примере 1, добавляли к 24 мл безводного этанола. Полученную смесь нагревали с обратным холодильником до получения прозрачного раствора. 120 мл изопропилового эфира добавляли по каплям. Полученную смесь охлаждали до температуры 0-5°С, перемешивали в течение 4 часов для кристаллизации и фильтровали. Осадок на фильтре промывали 2 мл изопропилового эфира и сушили при пониженном давлении при температуре 50°С, получая 1,56 г продукта (выход 78%).
Типичная рентгенограмма и типичный спектр ДСК кристалла A соединения, представленного формулой I, показаны на фиг. 1 и фиг. 2, соответственно (способ 1 в примере 2).
Другая типичная рентгенограмма кристалла A соединения, представленного формулой I, показана на фиг. 3 (способ 2 в примере 2, этанол-этилацетат (4:1)).
Еще одна типичная рентгенограмма кристалла A соединения, представленного формулой I, показана на фиг. 4 (способ 3 в примере 2).
Пример 3: Кристалл B соединения, представленного формулой I
2,0 г соединения, представленного формулой I, полученного в примере 1, добавляли к 25 мл ацетонитрила. Полученную смесь нагревали с обратным холодильником до получения прозрачного раствора, охлаждали до температуры 0-5°С, перемешивали в течение 4 часов для кристаллизации и фильтровали. Осадок на фильтре промывали 2 мл ацетонитрила и сушили при пониженном давлении при температуре 50°С, получая 1,82 г продукта (выход 91%).
Кристалл B соединения, представленного формулой I, представляет собой ацетонитрилат соединения, представленного формулой I. Типичная рентгенограмма и типичный спектр ДСК кристалла B соединения, представленного формулой I, показаны на фиг. 5 и фиг. 6, соответственно.
Пример 4: Испытание на стабильность
Кристалл А, полученный в способе 1 примера 2, и кристалл В, полученный в примере 3, помещали в открытый чистый контейнер при температуре 60°С и отбирали образцы для обнаружения в дни 5 и 10, соответственно. Результаты обнаружения сравнивали с первоначальным результатом обнаружения в 0-й день, результаты испытаний показаны в таблице ниже:
Пример 5. Анализы биологической активности.
1. Анализ на ферментативную активность (IC50) соединений
Платформу для испытания киназной активности JAK2 (дикого типа) создавали на основе анализа гомогенной флуоресценции с временным разрешением (HTRF), и с использованием платформы исследовали активности соединений. Соединения подвергали трехкратному градиентному разбавлению 100% ДМСО с начальной концентрацией 1 мМ (всего 11 разведений). 4 мкл каждого разведения добавляли к 96 мкл реакционного буфера (50 мМ HEPES, pH 7,4, 10 мМ MgCl2, 1 мМ EGTA, 0,01% Tween-20, 0,005% BAS, 2 мМ DTT) и гомогенно перемешивали. 2,5 мкл полученной жидкости затем добавляли в 384-луночный планшет (OptiPlate-384, доступный от PerkinElmer), а затем добавляли 5 мкл киназы JAK2 (доступной от Carna). Смесь гомогенно перемешивали центрифугированием. Затем добавляли 2,5 мкл смеси АТФ (конечная концентрация соответствует соответствующему значению Km) и TK-пептида (HTRF® KinEASE™-TK, доступный от Cisbio), чтобы инициировать реакцию (общий объем реакции составляет 10 мкл). 384-луночный планшет помещали в инкубатор и проводили реакцию в течение 120 минут при температуре 23°С. Затем реакцию прекращали добавлением 5 мкл меченого криптатом Eu3+ антитела к фосфотирозину (доступного от Cisbio) и 5 мкл стрептавидина-XL-665 (HTRF® KinEASE™-TK, доступного от Cisbio). Планшет инкубировали в инкубаторе в течение 1 часа, а затем считывали значения флуоресценции в Envision (доступном от PerkinElmer). Длина волны возбуждения составляла 320 нм, а длины волн излучения для обнаружения составляли 665 нм и 620 нм. Ферментативную активность представляли отношением двух считываний на двух длинах волн излучения. Ферментативную активность для каждого соединения исследовали при 11 концентрациях, и значения IC50 для соединений получали путем расчета данных с использованием программного обеспечения GraFit6.0 (Erithacus Software). Как видно из результатов, как значение IC50 соединения, представленного формулой I, так и значение IC50 контрольного руксолитиниба составляли менее 20 нМ.
2. Анализ эффективности на мышиной модели подкожной ксенотрансплантатной опухоли
Голые мыши Balb/c класса SPF представляли собой самок в возрасте 5-6 недель. 0,1 мл суспензии клеток Ba/F3-JAK2V617F в бессывороточной культуральной среде (содержащей 1×107 клеток, 50% MatriGel) вводили подкожно в правый бок каждой мыши. Когда средний объем опухоли достигал приблизительно 500 мм3, мышей с опухолями умерщвляли. Опухолевые ткани отбирали в асептических условиях и разрезали на маленькие кусочки, которые подкожно имплантировали в оба бока голых мышей Balb/c. Когда средний объем опухоли достигал приблизительно 100 мм3, каждую мышь маркировали в соответствии с серийными номерами и измеряли размеры опухолей и массу тела, соответственно. Этих мышей случайным образом распределяли от меньшего к большему с точки зрения объема опухоли, и каждую группу животных соответствующим образом корректировали, чтобы получить средние массы тела групп на одном уровне. Пять групп представляли собой отрицательную контрольную группу, положительную контрольную группу, группу с низкой дозой, группу с умеренной дозой и группу с высокой дозой, соответственно, и в каждой группе было пять мышей. Введение начинали в день распределения, дважды в день в течение 14 дней. Во время введения объемы опухоли и массу тела измеряли два раза в неделю. Мышей умерщвляли в конце эксперимента, а селезенку выделяли и взвешивали.
В ходе эксперимента измеряли максимальный продольный диаметр (L) и максимальный поперечный диаметр в вертикальном направлении (W) опухоли для расчета объема опухоли (V) в соответствии с V (мм3) = L×W2/2. Коэффициент ингибирования роста опухоли TGI (%) = 100%× (1-(Tt-T0)/(Vt-V0)), где Tt представляет собой средний объем опухоли, измеренный каждый раз в группе лечения; T0 представляет собой средний объем опухоли в группе лечения при распределении; Vt представляет собой средний объем опухоли, измеренный каждый раз в контрольной группе; и V0 представляет собой средний объем опухоли контрольной группы при распределении.
Результаты показаны в таблице ниже.
Из данных, представленных в таблице, можно видеть, что гидрохлорид соединения, представленного формулой I, исследовали на эффект ингибирования опухоли in vivo на модели мышей с опухолями Ba/F3-JAK2V617F, и было обнаружено, что он проявляет дозозависимый ингибирующий эффект на рост опухоли Ba/F3-JAK2V617F, и эффект подавления опухоли был очень значительным. После того, как гидрохлорид соединения, представленного формулой I, (100 мг/кг), вводили перорально дважды в день в течение 14 дней, коэффициент ингибирования роста опухоли (TGI) достигал 85,8%, тогда как для положительного контроля руксолитиниба (100 мг/кг) в эквивалентных условиях коэффициент ингибирования роста опухоли (TGI) составлял только 64,5%. Гидрохлорид соединения, представленного формулой I (50 мг/кг), также проявлял замечательный эффект подавления опухоли, и TGI достигал 68,4%, что было сопоставимо с эффектом подавления опухоли положительного контроля руксолитиниба (100 мг/кг).
3. Фармакокинетический анализ у взрослых самцов/самок крыс SD
Здоровых взрослых самок крыс SD получали от Beijing Vital River Laboratory Animal Technology Co., Ltd. Крыс делили на две группы по три крысы на группу и раздельно перорально вводили суспензию исследуемого образца (30 мг/кг) однократным внутрижелудочным введением. Перед экспериментом животные голодали в течение ночи, и время голодания составляло от 10 часов до введения и до 4 часов после введения. После введения забор крови проводили через 0,25 часа, 0,5 часа, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа. После того как животных наркотизированы изофлураном с использованием аппарата для анестезии для мелких животных, из венозного сплетения глазного дна брали 0,4 мл цельной крови и помещали в пробирку с антикоагулянтом гепарином. При температуре 4°С образец центрифугировали при 4200 об/мин в течение 5 минут, и плазму переносили в центрифужную пробирку и хранили при температуре -80°С до начала анализа. Образец в плазме экстрагировали способом осаждения белка, а жидкость экстракта анализировали способом ЖХ/МС/МС.
Примечание: а. Данные получены из обзора фармакологии, опубликованного FDA (Управление по контролю за продуктами и лекарственными средствами США).
Данные PK крыс (30 мг/кг РО) показали, что данные по соединению, представленному формулой I, превосходят данные по руксолитинибу.
4. Фармакокинетический анализ у взрослых гончих
В этом исследовании использовали четыре здоровых взрослых гончих, доступных от Beijing Marshall Biotechnology Co., Ltd. Исследование проводили два раза: в первый раз животным (двум самцам и двум самкам) вводили однократную внутривенную инъекцию в дозе 5 мг/кг; во второй раз той же группе животных (два самца и две самки) проводили через неделю однократное внутрижелудочное введение в дозе 10 мг/кг. Перед экспериментом животные, которых будут подвергать внутрижелудочному введению, голодали в течение ночи, и время голодания составляло от 10 часов до введения и до 4 часов после введения. Группа животных, которых подвергали внутривенному введению, получала пищу. После введения забор крови проводили через 0,083 часа, 0,25 часа, 0,5 часа, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа в группе внутривенного введения. После введения забор крови проводили через 0,25 часа, 0,5 часа, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа в группе внутрижелудочного введения. После того, как животных слегка наркотизировали изофлураном, брали 0,4 мл цельной крови из орбитального венозного сплетения с помощью стеклянной пробирки для сбора крови и помещали в пробирку с антикоагулянтом гепарином. При температуре 4°С образец центрифугировали при 4200 об/мин в течение 5 минут, и плазму переносили в центрифужную пробирку и хранили при температуре -80°С до начала анализа. Образец в плазме экстрагировали способом осаждения белка, а жидкость экстракта анализировали способом ЖХ/МС/МС.
Примечание: а. Данные получены из обзора фармакологии, опубликованного FDA (Управление по контролю за продуктами и лекарственными средствами США).
Данные по PK собак (10 мг/кг РО, 5 мг/кг IV) показали, что AUC соединения, представленного формулой I, при внутривенном введении была сравнима с таковой для положительного контроля руксолитиниба, но биодоступность соединения, представленного формулой I, при пероральном введении превосходила таковую у положительного контроля руксолитиниба (114% против 57%).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРРОЛОПИРИМИДИНОВОЕ СОЕДИНЕНИЕ | 2015 |
|
RU2701206C2 |
| СОЛЬ ИНГИБИТОРА SYK И ЕЕ КРИСТАЛЛИЧЕСКАЯ ФОРМА | 2019 |
|
RU2818103C2 |
| БОРАТНОЕ ПРОИЗВОДНОЕ АЗЕТИДИНА | 2019 |
|
RU2802287C2 |
| КРИСТАЛЛИЧЕСКАЯ ИЛИ АМОРФНАЯ ФОРМА АГОНИСТОВ FXR, ПРЕДСТАВЛЯЮЩИХ СОБОЙ ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2800751C2 |
| Соединения триазоло-пиримидина и их применение | 2019 |
|
RU2802866C2 |
| СОЕДИНЕНИЕ-ИНГИБИТОР JAK И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2820445C2 |
| АНАЛОГ ПИРИДО[1,2-A]ПИРИМИДОНА, ЕГО КРИСТАЛЛИЧЕСКАЯ ФОРМА, ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2016 |
|
RU2753696C2 |
| СТАБИЛЬНАЯ КРИСТАЛЛИЧЕСКАЯ ФОРМА ТИПИРАЦИЛА ГИДРОХЛОРИДА И СПОСОБ ЕЕ КРИСТАЛЛИЗАЦИИ | 2014 |
|
RU2640417C2 |
| ПРОИЗВОДНЫЕ 2,4-ДИЗАМЕЩЕННОГО ФЕНИЛЕН-1,5-ДИАМИНА И ИХ ПРИМЕНЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КОМПОЗИЦИИ, ПОЛУЧЕННЫЕ ИЗ НИХ | 2015 |
|
RU2649001C1 |
| КРИСТАЛЛЫ ПРОИЗВОДНЫХ 6,7-НЕНАСЫЩЕННОГО-7-КАРБАМОИЛМОРФИНАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2643807C1 |
Изобретение относится к кристаллу A и кристаллу B ацетонитрилата соединения, представленного формулой I, фармацевтическим композициям на их основе, их применению для получения лекарственного средства для лечения или профилактики опосредованного янус-киназой заболевания. Кристалл A и кристалл B соединения, представленного формулой I, обладают высокой чистотой и хорошей стабильностью и пригодны для промышленного производства. 8 н. и 5 з.п. ф-лы, 6 ил., 6 табл., 5 пр.
1. Кристалл А соединения, представленного формулой I:
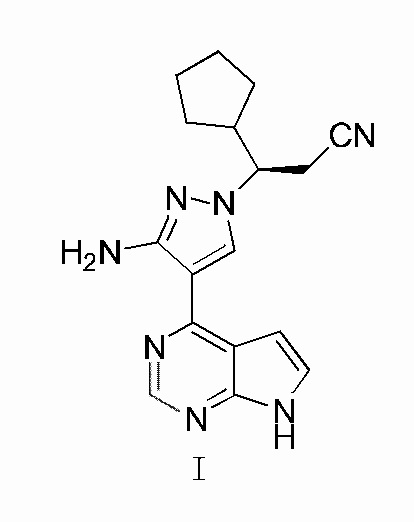
где рентгенограмма кристалла A соединения, представленного формулой I, характеризуется дифракционными пиками при 2θ, равными 9,35°±0,2°, 11,93°±0,2°, 16,32°±0,2°, 21,23°±0,2°, 23,13°±0,2° и 25,58°±0,2°.
2. Кристалл А соединения, представленного формулой I, по п. 1, где рентгенограмма кристалла A соединения, представленного формулой I, характеризуется дифракционными пиками при 2θ, равными 9,35°±0,2°, 11,93°±0,2°, 16,32°±0,2°, 18,82°±0,2°, 20,54°±0,2°, 21,23°±0,2°, 23,13°±0,2° и 25,58°±0,2°.
3. Кристалл А соединения, представленного формулой I, по п. 1, где рентгенограмма кристалла A соединения, представленного формулой I, характеризуется дифракционными пиками при 2θ, равными 9,35°±0,2°, 10,93°±0,2°, 11,93°±0,2°, 14,46°±0,2°, 16,32°±0,2°, 18,82°±0,2°, 20,54°±0,2°, 21,23°±0,2°, 21,66°±0,2°, 23,13°±0,2°, 25,58°±0,2° и 26,34°±0,2°.
4. Кристалл А соединения, представленного формулой I, по п. 1, где рентгенограмма кристалла A соединения, представленного формулой I, характеризуется дифракционными пиками при 2θ, равными 9,35°±0,2°, 10,93°±0,2°, 11,93°±0,2°, 14,46°±0,2°, 16,32°±0,2°, 17,28°±0,2°, 18,82°±0,2°, 19,25°±0,2°, 20,54°±0,2°, 21,23°±0,2°, 21,66°±0,2°, 22,15°±0,2°, 23,13°±0,2°, 24,09°±0,2°, 25,58°±0,2° и 26,34°±0,2°.
5. Фармацевтическая композиция для лечения или профилактики опосредованного янус-киназой заболевания, содержащая эффективное количество кристалла A соединения, представленного формулой I, по любому из пп. 1-4.
6. Применение кристалла А соединения, представленного формулой I, по любому из пп. 1-4 при получении лекарственного средства для лечения или профилактики опосредованного янус-киназой заболевания.
7. Применение фармацевтической композиции по п. 5 при получении лекарственного средства для лечения или профилактики опосредованного янус-киназой заболевания.
8. Кристалл B ацетонитрилата соединения, представленного формулой I:
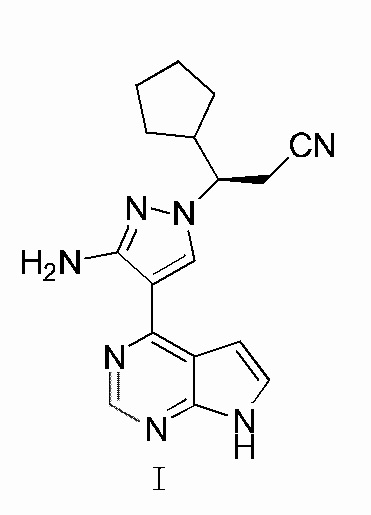
где молярное отношение соединения, представленного формулой I, к ацетонитрилу находится в диапазоне 1:1, а дифракционная рентгенограмма кристалла B ацетонитрилата соединения, представленного формулой I, характеризуется дифракционными пиками при 2θ, равными 8,97°±0,2°, 9,39°±0,2°, 12,90°±0,2°, 17,70°±0,2°, 20,31°±0,2° и 23,63°±0,2°.
9. Кристалл B ацетонитрилата соединения, представленного формулой I, по п. 8, где дифракционная рентгенограмма кристалла B ацетонитрилата соединения, представленного формулой I, характеризуется дифракционными пиками при 2θ, равными 8,97°±0,2°, 9,39°±0,2°, 12,90°±0,2°, 16,54°±0,2°, 17,70°±0,2°, 19,20°±0,2°, 20,31°±0,2°, 22,78°±0,2° и 23,63°±0,2°.
10. Кристалл B ацетонитрилата соединения, представленного формулой I, по п. 8, где дифракционная рентгенограмма кристалла B ацетонитрилата соединения, представленного формулой I, характеризуется дифракционными пиками при 2θ, равными 8,97°±0,2°, 9,39°±0,2°, 11,24°±0,2°, 12,90°±0,2°, 14,56°±0,2°, 16,54°±0,2°, 17,70°±0,2°, 19,20°±0,2°, 20,31°±0,2°, 22,23°±0,2°, 22,78°±0,2°, 23,63°±0,2° и 25,55°±0,2°.
11. Фармацевтическая композиция для лечения или профилактики опосредованного янус-киназой заболевания, содержащая эффективное количество кристалла B ацетонитрилата соединения, представленного формулой I, по любому из пп. 8-10.
12. Применение кристалла B ацетонитрилата соединения, представленного формулой I, по любому из пп. 8-10 при получении лекарственного средства для лечения или профилактики опосредованного янус-киназой заболевания.
13. Применение фармацевтической композиции по п. 11 при получении лекарственного средства для лечения или профилактики опосредованного янус-киназой заболевания.
| WO 2007070514 A1, 21.06.2007 | |||
| WO 2016026975 A1, 25.02.2016 | |||
| ТВЕРДЫЕ ФОРМЫ | 2007 |
|
RU2496780C2 |
| Машина для валяния войлока | 1929 |
|
SU19784A1 |

