Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к способу получения тиазольного производного, пригодного в качестве антагониста аденозинового A2A рецептора, кристаллам тиазольного производного или его моногидрата, и подобным.
Уровень техники
Известно, что тиазольное производное, представленное следующей формулой (I), или его фармацевтически приемлемая соль обладает антагонистическим действием относительно аденозинового A2A рецептора и является пригодным в качестве терапевтического средства, например, для болезни Паркинсона (смотри патентные документы 1 и 2). Кроме того, известно тиазольное производное, пригодное в качестве терапевтического агента для расстройства сна, устойчивости к опиоидам в качестве анальгетиков, мигрени, двигательного расстройства, депрессии, тревожного расстройства и подобных (смотри патентные документы 3, 4, 5, 6, 7 и 8). В качестве данных тиазольных производных, являются в частности известными соединения, представленные следующими формулами (IA), (IB), (IC), (ID) и подобные, и подобные (смотри патентные документы 1, 3, 4, 5, 6, 7 и 8).
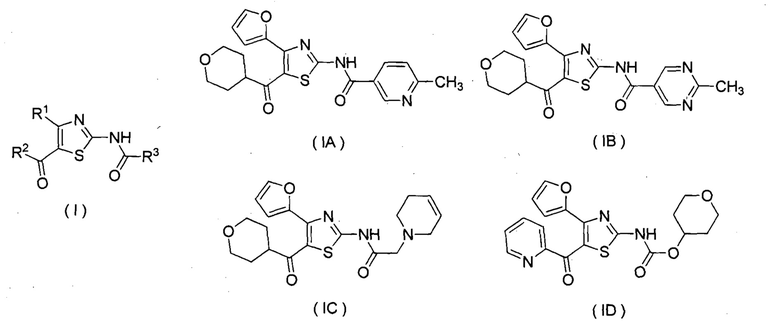
(где R1 представляет собой фурил, R2 представляет собой пиридил или тетрагидропиранил, R3 представляет собой арил, аралкил, ароматическую гетероциклическую группу, ароматический гетероциклилалкил, алифатический гетероциклилалкил или тетрагидропиранилокси, или данные группы замещены 1-3 заместителями, выбранными из группы, состоящей из галогена; низшего алкила, необязательно замещенного низшим алкокси или морфолино; низшего алкокси; низшего алканоила; и винила)
В качестве способов получения данных тиазольных производных, известны следующие три способа получения (схемы 1-3) (смотри патентный документ 1).
Схема 1

(где R10 представляет собой фурил или подобный, Hal представляет собой галоген, Ph представляет собой фенил, R12 представляет собой, как определено выше для R2, или подобный, и R11 представляет собой, как определено выше для R3, или подобный)
Схема 2

(где каждый R10, R11 и R12 представляет собой, как определено выше)
Схема 3

(где каждый R10, R11 и R12 представляет собой, как определено выше)
Помимо перечисленных выше, например, также является известным способ, включающий реакцию α-галогенметилкетона и производного N-(аминометилен)тиомочевины (смотри непатентные документы 1-4), способ, включающий реакцию α-галогенметилкетона и производного N-ацилтиомочевины (смотри непатентные документы 3-6) и подобные.
Более конкретно, соединения, представленные приведенными выше формулами (IA), (IB), (IC) и (ID), описывают в патентном документе 1 в качестве примеров 504, 508, 557 и 253.
[Список документов]
[патентные документы]
патентный документ 1: WO 2005/063743
патентный документ 2: WO 2006/137527
патентный документ 3: WO 2007/015528
патентный документ 4: WO 2009/145289
патентный документ 5: WO 2010/010908
патентный документ 6: WO 2010/126082
патентный документ 7: WO 2011/027805
патентный документ 8: WO 2011/027806
[непатентные документы]
непатентный документ 1: Indian Journal of Chemistry, 1970, vol. 8, p.1145
непатентный документ 2: Indian Journal of Chemistry, 1978, vol. 16B, p.749
непатентный документ 3: Journal of Chemical Society, Perkin Transactions I, 1979, p.1762
непатентный документ 4: Journal of Chemical Society, Perkin Transactions I, 1987, p.1153
непатентный документ 5: Zeitschrift für Chemie, 1974, vol. 14, p.470
непатентный документ 6: Indian Journal of Chemistry, 1986, vol. 25B, p.446.
СУЩНОСТЬ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Проблемы, которые решает настоящее изобретение
Цель настоящего изобретения заключается в обеспечении промышленного способа получения соединения, представленного формулой (I), которое обладает антагонистическим действием относительно аденозинового A2A рецептора и является пригодным в качестве терапевтического агента, например, для болезни Паркинсона, расстройства сна, устойчивости к опиоидам в качестве анальгетиков, мигрени, двигательного расстройства, депрессии, тревожного расстройства и подобных, и подобных. Кроме того, цель включает обеспечение кристаллов соединения, представленного формулой (IA), или его моногидрата, и способа их получения, и подобные.
Способы решения данных проблем
Настоящее изобретение относится к следующему (1)-(31).
(1) Способ получения соединения, представленного формулой (C), включающий (i) стадию реакции соединения, представленного формулой (A), и соединения, представленного формулой (B):

(где R1 представляет собой фурил, R4, R5 и R6 являются одинаковыми или отличными, и каждый представляет собой низший алкил или арил, R2 представляет собой пиридил или тетрагидропиранил, и X1 представляет собой галоген).
(2) Способ получения по (1), где X1 представляет собой атом хлора, атом брома или атом йода.
(3) Способ получения по (1), где X1 представляет собой атом брома.
(4) Способ получения по любому из (1)-(3), где каждый R4, R5 и R6 представляет собой метил.
(5) Способ получения по любому из (1)-(4), где R1 представляет собой 2-фурил.
(6) Способ получения соединения, представленного формулой (I), включающий стадию, описанную в любом из (1)-(5):

(где каждый R1 и R2 представляет собой, как определено выше, R3 представляет собой арил, аралкил, ароматическую гетероциклическую группу, ароматический гетероциклилалкил, алифатический гетероциклилалкил или тетрагидропиранилокси, или данные группы замещены 1-3 заместителями, выбранными из группы, состоящей из галогена; низшего алкила, необязательно замещенного низшим алкокси или морфолино; низший алкокси; низший алканоил; и винил).
(7) Способ получения по (6), дополнительно включающий (ii) стадию получения соединения, представленного формулой (D), обработкой соединения, представленного формулой (C), кислотой

(где каждый R1, R2, R4, R5 и R6 представляет собой, как определено выше), и
(iii) стадию получения соединения, представленного формулой (I), реакцией соединения, представленного формулой (D), и соединения, представленного формулой (E):
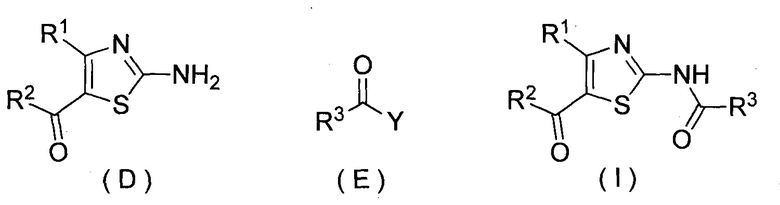
(где Y представляет собой галоген или гидрокси, и каждый R1, R2 и R3 представляет собой, как определено выше).
(8) Способ получения по (7), где кислота на стадии (ii) представляет собой хлористоводородную кислоту или трифторуксусную кислоту.
(9) Способ получения по (7) или (8), где Y представляет собой гидрокси.
(10) Способ получения по (9), где реакцию на стадии (iii) проводят в присутствии 1,3-дициклогексилкарбодиимида (DCC), 1-гидрохлорида этил-3-(3-диметиламинопропил)карбодиимида (EDC), 1,1ʹ-карбонилдиимидазола (CDI) или пропилфосфонового ангидрида (T3P).
(11) Способ получения по (9), где реакцию на стадии (iii) проводят в присутствии CDI.
(12) Способ получения по любому из (1)-(11), где R2 представляет собой 4-тетрагидропиранил.
(13) Способ получения по любому из (6)- (12), где R3 представляет собой 2-метилпиридин-5-ил, 2-метилпиримидин-5-ил, 5,6-дигидро-2H-пиридилметил или 4-тетрагидропиранилокси.
(14) Способ получения по любому из (6)-(12), где R3 представляет собой 2-метилпиридин-5-ил.
(15) Способ получения по любому из (6)-(11), где R1 представляет собой 2-фурил, R2 представляет собой 4-тетрагидропиранил, и R3 представляет собой 2-метилпиридин-5-ил.
(16) Способ получения соединения, представленного формулой (A), включающий стадию реакции соединения, представленного формулой (P), соединения, представленного формулой (Q), и тиоцианатной соли:

(где X2 представляет собой галоген, и каждый R1, R4, R5 и R6 представляет собой, как определено выше).
(17) Способ получения по (16), где тиоцианатная соль представляет собой тиоцианат натрия или тиоцианат калия.
(18) Способ получения по (16) или (17), где R1 представляет собой 2-фурил, и каждый R4, R5 и R6 представляет собой метил.
(19) Способ получения по любому из (16)-(18), где реакцию проводят в тетрагидрофуране (THF).
(20) Кристаллы соединения, представленного формулой (IA), где соединение представляет собой моногидрат:
 .
.
(21) Кристаллы по (20), которые имеют пики при 8,1° и 12,0° для углов дифракции (2θ±0,2°), как измерено порошковой дифракцией рентгеновских лучей.
(22) Кристаллы по (20) или (21), которые имеют пики при 16,3°, 21,8° и 23,0° для углов дифракции (2θ±0,2°), как измерено порошковой дифракцией рентгеновских лучей.
(23) Кристаллы по любому из (20)-(22), которые имеют пики при 14,7°, 21,1°, 24,4°, 24,7° и 28,3° для углов дифракции (2θ±0,2°), как измерено порошковой дифракцией рентгеновских лучей.
(24) Кристаллы соединения, представленного формулой (IA), где соединение представляет собой ангидрид:
 .
.
(25) Кристаллы по (24), которые имеют пики при 8,3° и 19,1° для углов дифракции (2θ±0,2°), как измерено порошковой дифракцией рентгеновских лучей.
(26) Кристаллы по (24) или (25), которые имеют пики при 21,2°, 23,8° и 27,0° для углов дифракции (2θ±0,2°), как измерено порошковой дифракцией рентгеновских лучей.
(27) Кристаллы по любому из (24)-(26), которые имеют пики при 12,6°, 16,5°, 19,5°, 20,8° и 22,4° для углов дифракции (2θ±0,2°), как измерено порошковой дифракцией рентгеновских лучей.
(28) Способ получения кристаллов, описанных по любому из (20)-(23), включающий стадию кристаллизации соединения, представленного формулой (IA), из ацетона-воды.
(29) Способ получения кристаллов, описанных по любому из (24)-(27), включающий стадию кристаллизации соединения, представленного формулой (IA), из изобутилового спирта.
(30) Способ получения по (28) или (29), где соединение, представленное формулой (IA), которое будут применять в качестве исходного соединения, представляет собой моногидрат.
(31) Способ получения по любому из (28)-(30), где соединение, представленное формулой (IA), которое будут применять в качестве исходного соединения, представляет собой соединение, полученное способом получения, описанным в любом из (6)-(15).
Эффекты настоящего изобретения
Согласно настоящему изобретению, обеспечивают способ получения соединения, представленного формулой (I), которое обладает антагонистическим действием относительно аденозинового A2A рецептора и является пригодным в качестве терапевтического агента, например, для болезни Паркинсона, расстройства сна, устойчивости к опиоидам в качестве анальгетиков, мигрени, двигательного расстройства, депрессии, тревожного расстройства и подобных, способ получения соединения, представленного формулой (C), которое является пригодным в качестве промежуточного соединения для получения соединения, представленного формулой (I), кристаллы соединения, представленного формулой (IA), или его моногидрата и способ их получения и подобные. Способы получения настоящего изобретения являются пригодными в качестве промышленных способов получения лекарственного вещества фармацевтического продукта. Кроме того, кристаллы соединения, представленного формулой (IA), или его моногидрата настоящего изобретения являются пригодными в качестве лекарственного вещества фармацевтического продукта.
Краткое описание чертежей
Фигура 1 показывает порошковую рентгеновскую дифрактограмму кристаллов моногидрата соединения IA (HA кристалл), где вертикальная ось показывает интенсивность дифракции (имп/сек), и горизонтальная ось показывает дифракционный угол (2θ. °).
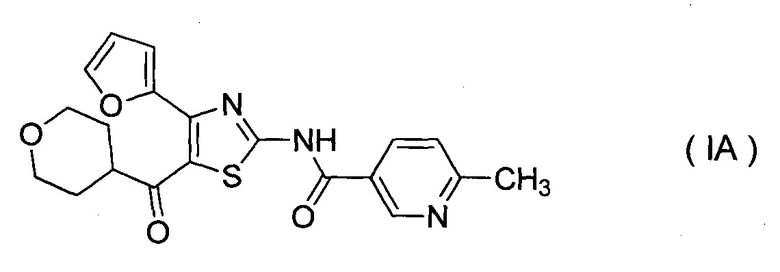
Фигура 2 показывает порошковую рентгеновскую дифрактограмму кристаллов ангидрида соединения IA (A кристалл), где вертикальная ось показывает интенсивность дифракции (имп/сек), и горизонтальная ось показывает дифракционный угол (2θ, °).
Фигура 3 показывает порошковую рентгеновскую дифрактограмму кристаллов 0,5 этанолата соединения IA (EA кристалл), где вертикальная ось показывает интенсивность дифракции (имп/сек), и горизонтальная ось показывает дифракционный угол (2θ, °).
Подробное описание настоящего изобретения
Далее, соединение, представленное формулой (I), называют соединением I. То же самое применяют к соединениям других номеров формул.
В определении каждой группы в формулах (I), (A), (B), (C), (E), (P) и (Q):
Примеры низшего алкила и низшей алкильной группы низшего алкокси и низшего алканоила включают линейный или разветвленный алкил, содержащий 1-10 атомов углерода, и его более конкретные примеры включают метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, неопентил, гексил, гептил, октил, нонил, децил и подобные.
Примеры аралкила включают аралкил, содержащий 7-16 атомов углерода, и его более конкретные примеры включают бензил, фенэтил, фенилпропил, фенилбутил, фенилпентил, фенилгексил, фенилгептил, фенилоктил, фенилнонил, фенилдецил, нафтилметил, нафтилэтил, нафтилпропил, нафтилбутил, нафтилпентил, нафтилгексил, антрилметил, антрилэтил и подобные.
Примеры арила включают арил, содержащий 6-14 атомов углерода, и его более конкретные примеры включают фенил, нафтил, азуленил, антрил и подобные.
Примеры ароматической гетероциклической группы включают 5-членную или 6-членную моноциклическую ароматическую гетероциклическую группу, содержащую, по меньшей мере, один атом, выбранный из атома азота, атома кислорода и атома серы, бициклическую или трициклическую конденсированную ароматическую гетероциклическую группу, где 3-8-членные кольца являются конденсированными, которая содержит, по меньшей мере, один атом, выбранный из атома азота, атома кислорода и атома серы, и подобных, и ее более конкретные примеры включают фурил, тиенил, пирролил, имидазолил, пиразолил, оксазолил, изоксазолил, оксадиазолил, тиазолил, изотиазолил, тиадиазолил, триазолил, тетразолил, пиридил, пиридазинил, пиримидин, пиразинил, триазинил, бензофуранил, бензотиофенил, бензоксазолил, бензотиазолил, изоиндолил, индолил, индазолил, бензимидазолил, бензотриазолил, оксазолипиримидин, Тиазолипиримидин, пирролопиридинил, пирролопиримидин, имидазопиридин, пуринил, хинолинил, изохинолинил, цинолинил, фталазинил, хиназолинил, хиноксалинил, нафтиридинил, фуро[2,3-b]пиридил, 6,7-дигидро-5Н-циклопента[b]пиридил, 7,8-дигидро-5Н-пирано[4,3-b]пиридил, 7,8-дигидро-5Н-тиопирано[4,3-b]пиридил и подобные.
Примеры ароматического гетероциклилалкила включают группу, в которой ароматическая гетероциклическая группа соединена с алкиленом. Примеры ароматической гетероциклической группы включают группы, перечисленные в качестве примеров приведенной выше ароматической гетероциклической группы, примеры алкилена включают алкилен, содержащий 1-10 атомов углерода, и его более конкретные примеры включают метилен, этилен, триметилен, пропилен, тетраметилен, пентаметилен, гексаметилен, гептаметилен, октаметилен, нонаметилен, декаметилен и подобные. Более конкретные примеры ароматического гетероциклилалкила включают пирролилметил, пирролилэтил, тиазолилметил, пиридилметил, пиридилэтил, пиримидинилметил, пиримидинилэтил, индолилметил, бензоимидазолилметил и подобные.
Примеры алифатического гетероциклилалкила включают группу, где алифатическая гетероциклическая группа соединена с алкиленом. Примеры алифатической гетероциклической группы включают 5-членную или 6-членную моноциклическую алифатическую гетероциклическую группу, содержащую, по меньшей мере, один атом, выбранный из атома азота, атома кислорода и атома серы, бициклическую или трициклическую конденсированную алифатическую гетероциклическую группу, где 3-8-членные кольца являются конденсированными, которая содержит, по меньшей мере, один атом, выбранный из атома азота, атома кислорода и атома серы, и подобные, и его более конкретные примеры включают азиридинил, азетидинил, пирролидинил, пиперидино, пиперидинил, азепанил, 1,2,5,6-тетрагидропиридил, имидазолидинил, пиразолидинил, пиперазинил, гомопиперазинил, пиразолинил, оксиранил, тетрагидрофуранил, тетрагидро-2Н-пиранил, 5,6-дигидро-2Н-пиранил, 5,6-дигидро-2Н-пиридил, оксазолидинил, морфолино, морфолинил, тиоксазолидинил, тиоморфолинил, 2Н-оксазолил, 2Н-тиоксазолил, дигидроиндолил, дигидроизоиндолил, дигидробензофуранил, бензимидазолидинил, дигидробензооксазолил, дигидробензотиоксазолил, бензодиоксолинил, тетрагидрохинолил, тетрагидроизохинолил, дигидро-2Н-хроманил, дигидро-1Н-хроманил, дигидро-2Н-тиохроманил, дигидро-1Н-тиохроманил, тетрагидрохиноксалинил, тетрагидрохиназолинил, дигидробензодиоксанил и подобные. Примеры алкилена включают алкилен, содержащий 1-10 атомов углерода, и его более конкретные примеры включают метилен, этилен, триметилен, пропилен, тетраметилен, пентаметилен, гексаметилен, гептаметилен, октаметилен, нонаметилен, декаметилен и подобные. Более конкретные примеры алифатического гетероциклилалкила включают 5,6-дигидро-2H-пиридилметил, 5,6-дигидро-2H-пиридилэтил, тетрагидро-2H-пиранилметил, 5,6-дигидро-2H-пиранилметил, 5,6-дигидро-2H-пиранилэтил, морфолинометил, морфолиноэтил, пиперазинилметил, оксазолидинилметил и подобные.
Каждый галоген обозначает атом фтора, хлора, брома, йода.
Способ получения соединения I (схема, показанная ниже) конкретно показан ниже.

(где M представляет собой атом металла, такой как натрий, калий и подобный, и каждый R1, R2, R3, R4, R5, R6, X1, X2 и Y представляет собой, как определено выше)
Стадия 1
Соединение A можно получить реакцией соединения P, соединения Q и тиоцианатной соли в подходящем растворителе. Данные три реагента (соединение P, соединение Q и тиоцианатная соль) могут реагировать одновременно, или соответствующие реагенты могут также реагировать последовательно в подходящем порядке. Предпочтительно, соединение A можно получить реакцией тиоцианатной соли и соединения P в подходящем растворителе, и затем добавлением соединения Q к полученной реакционной смеси, обеспечивая реакцию, согласно способу, описанному, например, в J.C.S. Perkin I, 1153 (1987) и подобным.
Тогда как количество каждого реагента, который будут применять, конкретно не ограничено, оно составляет, например, предпочтительно 0,8-1,5 эквивалента, более предпочтительно 0,9-1,2 эквивалента, соединения Q, и предпочтительно 0,8-1,5 эквивалента, более предпочтительно 0,9-1,2 эквивалента, тиоцианатной соли, оба относительно соединения P.
Примеры тиоцианатной соли включают тиоцианат натрия, тиоцианат калия и подобные, предпочтительно тиоцианат натрия.
Тогда как растворитель конкретно не ограничен, можно применять, например, алифатический углеводород, такой как пентан, гексан, гептан, циклогексан и подобный; ароматический углеводород, такой как толуол, ксилол и подобные; галогенированный углеводород, такой как дихлорметан, хлороформ, дихлорэтан и подобные; полярные растворители, такие как ацетонитрил, диметилсульфоксид (DMSO), N,N-диметилформамид (DMF), N,N-диметилацетамид (DMA), N-метил-2-пирролидон (NMP), 1,3-диметил-2-имидазолидинон (DMI) и подобные; эфиры, такие как диоксан, THF, диэтиловый эфир, циклопентилметиловый эфир, 1,2-диметоксиэтан (DME), диметиловый эфир этиленгликоля и подобные; сложные эфиры, такие как метилацетат, этилацетат, изопропилацетат и подобные, и подобные. Их можно применять отдельно или в виде их смеси. Предпочтительными являются THF и подобные. Количество растворителя, который будут применять, конкретно не ограничен, и, например, 1-50 объем/вес (об./вес) применяют относительно соединения P.
Реакцию проводят при температуре предпочтительно от -10°C до 150°C, более предпочтительно 0°C-100°C, обычно в течение 5 минут-72 часов.
Соединение P и соединение Q можно получить в качестве имеющихся в продаже продуктов.
Стадия 2
Соединение C можно получить реакцией соединения A и соединения B.
Соединение B применяют в количестве предпочтительно 0,1-5 эквивалента, более предпочтительно 0,5-2 эквивалента, даже более предпочтительно 0,9-1,3 эквивалента, относительно соединения A.
Тогда как реакцию можно проводить без растворителя, ее предпочтительно проводить в растворителе. Примеры растворителя включают алифатический углеводород, такой как пентан, гексан, гептан, циклогексан и подобные; ароматический углеводород, такой как толуол, ксилол и подобные; галогенированный углеводород, такой как дихлорметан, хлороформ, дихлорэтан и подобные; полярные растворители, такие как ацетонитрил, DMSO, DMF, DMA, NMP, DMI и подобные; эфиры, такие как диоксан, THF, диэтиловый эфир, циклопентилметиловый эфир, DME, диметиловый эфир этиленгликоля и подобные; сложные эфиры, такие как метилацетат, этилацетат, изопропилацетат и подобные; спирты, такие как метанол, этанол, пропанол, 2-пропанол и подобные; воду и подобные. Их можно применять отдельно или в виде их смеси. Предпочтительными являются полярные растворители, такие как MF, DMA, NMP, DMI и подобные. Тогда как количество растворителя, который будут применять, конкретно не ограничен, например, его применяют в количестве 0,5-20 об./вес относительно соединения A.
Реакцию проводят при температуре предпочтительно от -50°C до температуры кипения растворителя, который будут применять, более предпочтительно при температуре 10°C-70°C, даже более предпочтительно 30°C-50°C, в течение обычно 5 минут-100 часов.
На данной стадии, когда стерически затрудненная группа, например, трет-бутил, 1,1-диметилпропил, 1-метил-1-фенилэтил, тритил и подобная, присутствует в качестве заместителя -CR4R5R6 при одном атоме азота соединение A, требуемое триазольное кольцо можно селективно получить (соединение C получает селективно, а не соединение, представленное следующей формулой C1):

(где каждый R1, R2, R4, R5 и R6 представляет собой, как определено выше)
Кроме того, когда применяют подходящий полярный растворитель, такой как DMF, DMA, NMP, DMI и подобный, полученное соединение C можно осадить в твердом виде добавлением воды к реакционной смеси после завершения реакции, посредством этого соединение C можно получить общепринятым способом.
Кроме того, соединение C иногда получают в виде соли, содержащей X. В частности, например, когда X представляет собой атом хлора, можно получить гидрохлорид, и когда X представляет собой атом брома, можно получить гидробромид и подобный.
Соединение B можно получить в виде имеющего в продаже продукта или согласно известному способу (например, Organic Synthesis, IV, 193 (1988) и подобным). Когда применяют имеющийся в продаже продукт соединения B, его применяют после очистки, при необходимости.
Стадия 3
Соединение D можно получить обработкой соединения C кислотой без растворителя или в растворителе. Обработку проводят предпочтительно при температуре от -80°C до температуры кипения кислоты или растворителя, который будут применять, более предпочтительно при температуре 10°C-100°C, в течение обычно 1 мин-100 часов, предпочтительно 5 мин-24 часов. При необходимости, добавляют катионный скавенджер, такой как анизол и подобный. Катионный скавенджер применяют в количестве предпочтительно от 0,5 эквивалента до большого избытка, более предпочтительно 1-50 эквивалента, относительно соединения C.
Примеры кислоты включают галогеноводороды, такие как хлористоводородная кислота, бромистоводородная кислота, йодистовдородная кислота и подобные; сульфокислоты, такие как метансульфокислота, трифторметансульфокислота, бензолсульфокислота, пара-толуолсульфокислота и подобные; карбоновые кислоты, такие как уксусная кислота, пропионовая кислота, трихлоруксусная кислота, трифторуксусную кислоту, бензойная кислота, метилбензойная кислота, трихлорбензойная кислота, трифторбензойная кислота, пентафторбензойная кислота и подобные; серную кислоту; азотную кислоту и подобные, предпочтительно хлористоводородную кислоту, трифторуксусную кислоту и подобные, и ее применяют предпочтительно в количестве 0,5-200 эквивалента, более предпочтительно 1-50 эквивалента, относительно соединения C.
Примеры растворителя включают алифатические углеводороды, такие как пентан, гексан, гептан, циклогексан и подобные; ароматические углеводороды, такие как толуол, ксилол и подобные; галогенированные углеводороды, такие как дихлорметан, хлороформ, дихлорэтан и подобные; полярные растворители, такие как ацетонитрил, DMSO, DMF, DMA, NMP, DMI и подобные; эфиры, такие как диоксан, THF, диэтиловый эфир, циклопентилметиловый эфир, DME, диметиловый эфир этиленгликоля и подобные; сложные эфиры, такие как метилацетат, этилацетат, изопропилацетат и подобные; спирты, такие как метанол, этанол, пропанол, 2-пропанол и подобные; воду и подобные, предпочтительно толуол, диоксан, воду и подобные. Их применяют отдельно или в виде их смеси. Тогда как количество растворителя, который будут применять, конкретно не ограничено, например, его применяют в количестве 0,5-20 об./вес относительно соединения A.
Более предпочтительно, например, соединение D можно общепринято получить с высоким выходом обработкой соединения C 3-12 моль/л хлористоводородной кислотой при температуре 50°C-100°C.
После приведенной выше обработки, соединение D можно легко получить из реакционной смеси, например, нейтрализацией реакционной смеси подходящим основанием, таким как гидроксид лития, гидроксид натрия, гидроксид калия, карбонат калия, карбонат натрия и подобным. Соединение D можно также легко получить проведением обработки при упаривании получаемого побочно соединения с низкой температурой кипения, кислоты и подобного, применяя ловушку Дина-Старка и подобные. Для упаривания получаемого побочно соединения с низкой температурой кипения, кислоты и подобного, также является эффективным проведение реакции при пониженном давлении.
Стадия 4
Соединение I можно получить реакцией соединения D и соединения E.
(1) когда Y в соединении E представляет собой гидрокси, соединение I можно получить реакцией соединения D и соединения E в растворителе, в присутствии конденсирующего агента и, при необходимости, в присутствии добавки.
Соединение E предпочтительно применяют в количестве 0,8-5 эквивалента, более предпочтительно 1-2 эквивалента, относительно соединения D.
Примеры конденсирующего агента включают 1,3-дициклогексилкарбодиимид (DCC), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (EDC), 1,1ʹ-карбонилдиимидазол (CDI), пропилфосфоновый ангидрид (T3P) и подобные, и конденсирующего агента предпочтительно применяют в количестве 0,1-10 эквивалента, более предпочтительно 1-2 эквивалента, относительно соединения D.
Примеры добавки включают моногидрат 1-гидроксибензотриазола (HOBt H2O), N-гидроксисукцинимид (HOSu) и подобные, и добавку предпочтительно применяют в количестве 0,1-10 эквивалента, более предпочтительно 1-2 эквивалента, относительно соединения D.
Примеры растворителя включают спирты, такие как метанол, этанол и подобные; галогенированные углеводороды, такие как дихлорметан, хлороформ, 1,2-дихлорэтан и подобные; ароматические углеводороды, такие как толуол, ксилол и подобные; сложные эфиры, такие как метилацетат, этилацетат, изопропилацетат и подобные; эфиры, такие как диоксан, THF, диэтиловый эфир, циклопентилметиловый эфир, DME, диметиловый эфир этиленгликоля и подобные; полярные растворители, такие как ацетонитрил, DMSO, DMF, DMA, NMP, DMI и подобные; пиридин; воду и подобные, предпочтительно полярные растворители, такие как DMF, DMA, NMP, DMI и подобные. Их можно применять отдельно или в виде их смеси. Поскольку количество растворителя конкретно не ограничено, например, его применяют в количестве 0,5-20 об./вес относительно соединения D.
Реакцию проводят при температуре предпочтительно от 0°C до температуры кипения растворителя, который будут применять, более предпочтительно 20°C-100°C, обычно в течение 5 мин-100 часов.
Соединение E можно получить в виде имеющегося в продаже продукта.
(2) Когда Y в соединении E представляет собой галоген, соединение I можно получить реакцией соединения D и соединения E без растворителя или в растворителе, в присутствии подходящего основания, при необходимости.
Соединение E предпочтительно применяют в количестве 1-10 эквивалента, более предпочтительно 1-2 эквивалента, относительно соединения D.
Примеры основания включают карбонат калия, гидроксид калия, гидроксид натрия, трет-бутоксид калия, триэтиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 1,8-диазабицикло[5.4.0]-7-ундецен (DBU), 4-диметиламинопиридин (DMAP) и подобные, и основание предпочтительно применяют в количестве 1-10 эквивалента, более предпочтительно 1-2 эквивалента, относительно соединения D.
Примеры растворителя включают спирты, такие как метанол, этанол и подобные; галогенированные углеводороды, такие как дихлорметан, хлороформ, 1,2-дихлорэтан и подобные; ароматические углеводороды, такие как толуол, ксилол и подобные; сложные эфиры, такие как метилацетат, этилацетат, изопропилацетат и подобные; эфиры, такие как диоксан, THF, диэтиловый эфир, циклопентилметиловый эфир, DME, диметиловый эфир этиленгликоля и подобные; полярные растворители, такие как ацетонитрил, DMSO, DMF, DMA, NMP, DMI и подобные; пиридин; воду и подобные, предпочтительно метанол, этанол, дихлорметан, хлороформ, 1,2-дихлорэтан, толуол, этилацетат, ацетонитрил, диэтиловый эфир, THF, DME, диоксан, DMF, DMA, NMP, DMI, пиридин, воду и подобные. Их можно применять отдельно или в виде их смеси. Поскольку количество растворителя конкретно не ограничено, например, его применяют в количестве 0,5-20 об./вес относительно соединения D.
Реакцию проводят при температуре предпочтительно от -30°C до 150°C, более предпочтительно от комнатной температуры до 100°C, обычно в течение 5 мин-100 часов.
Соединение E можно получить в виде имеющегося в продаже продукта, или согласно известному способу, относящемуся к получению хлорангидрида кислоты, обычно применяемому в области химии органического синтеза.
В приведенных выше (1) и (2), когда применяют полярный растворитель, такой как DMF, DMA, NMP, DMI и подобный, соединение I можно высадить из реакционного раствора добавлением воды к реакционной смеси, посредством этого соединение I можно получить в твердом виде общепринятым способом.
Полученные в результате продукты и промежуточные соединения на каждой из приведенных выше стадий можно выделить и очистить, применяя способы выделения и очистки, обычно применяемые в области химии органического синтеза, например, фильтрование, экстракцию, промывку, сушку, концентрирование, перекристаллизацию, различные хроматографии и подобные. Полученные в результате продукты и промежуточные соединения на каждой стадии можно также подвергать следующей реакции без конкретной очистки.
Некоторые из промежуточных соединений и получаемых в результате продуктов, полученные на каждой стадии, могут содержать стереоизомеры, такие как геометрический изомер, оптический изомер и подобные, таутомер и подобные. Промежуточные соединения и получаемые в результате продукты в настоящем изобретении включают все возможные изомеры и их смеси, включая те, что приведены выше.
Кроме того, исходные соединения, которые будут применять на каждой стадии, и полученные промежуточные соединения и получаемые в результате продукты могут быть в виде соли или сольвата.
Когда требуется соль промежуточного соединения или полученного в результате продукта, полученных на каждой стадии, и промежуточное соединение или получаемый в результате продукт, получаемые на каждой стадии, находится в форме соли, его можно непосредственно очистить. Когда его получают в свободной форме, промежуточное соединение или получаемый в результате продукт, получаемый на каждой стадии, растворяют или суспендируют в подходящем растворителе, добавляют кислоту или основание, получая соль, и соль выделяют и очищают.
Соль исходного соединения, которую будут применять на каждой стадии, промежуточное соединение или получаемый в результате продукт, полученные на каждой стадии, включают, например, соль присоединения кислоты, соль металла, соль аммония, соль присоединения органического амина, соль присоединения аминокислоты и подобные. Примеры соли присоединения кислоты включают соли неорганических кислот, такие как гидрохлорид, гидробромид, нитрат, сульфат, фосфат и подобные, соли органических кислот, такие как ацетат, оксалат, малеат, фумарат, цитрат, бензоат, метансульфонат и подобные, и подобные; примеры соли металла включают соли щелочных металлов, такие как натриевая соль, калиевая и подобные, соли щелочноземельных металлов, такие как магниевая соль, кальциевая и подобные, алюминиевая соль, цинковая соль и подобные; примеры аммониевых солей включают соли аммония, тетраметиламмония и подобные; примеры соли присоединения органического амина включают соли морфолина, пиперидина и подобные; и примеры соли присоединения аминокислоты включают соли присоединения лизина, глицина, фенилаланина, аспарагиновой кислоты, глютаминовой кислоты и подобные.
Когда требуются сольваты исходного соединения соединение, которое будут применять на каждой стадии, полученное промежуточное соединение и получаемый в результате продукт, их можно непосредственно получить приведенным выше способом получения и подобными. Их можно также получить смешением исходного соединения соединение, которое будут применять на каждой стадии, полученного промежуточного соединения или получаемого в результате продукта с каждый растворителем, получая сольват, и их выделением и очисткой.
Согласно приведенному выше способу получения, соединение I можно общепринятым способом получить более короткой стадией, чем известными способами (например, WO2005/063743). Кроме того, способ получения может эффективно давать соединение I с определенной степенью чистоты и с хорошей воспроизводимостью, и является предпочтительным в качестве промышленного способа получения.
Как приводится выше, соединение I может присутствовать в виде его соли или его сольвата в добавление к свободной форме, и они могут присутствовать в виде кристаллов. Кристаллы соединения I или его соли или его сольвата могут обладать полиморфизмом и структурой кристалла. Например, соединение I включает кристаллы соединения I, кристаллы соли соединения I, кристаллы сольвата соединения I, кристаллы сольвата соли соединения I и их кристаллические полиморфы, их различные кристаллические структуры и подобные. Их более конкретные примеры включают кристаллы (A кристалл) безводного соединения IA, кристаллы (HA кристалл) моногидрата соединения IA, кристаллы 0,5 этанолата соединения IA (EA кристалл) и подобные, которые показаны в приведенных ниже примерах и сравнительных примерах. Данные кристаллические формы можно идентифицировать, например, измерением порошковой дифракции рентгеновских лучей, измеренные величины порошковой дифракции рентгеновских лучей, описанные в настоящем описании, получали проникающим способом. Измеренные величины (2θ) порошковой дифракции рентгеновских лучей иногда могут изменяться в пределах диапазона ±0,2°.
Кристаллы приведенного выше соединения IA или его моногидрата иногда получают непосредственно приведенным выше способом (стадия 4), например, их можно получить следующим способом.
В случае кристаллов (A кристалл) безводного соединения IA, соединение IA растворяют в изобутиловом спирте при температуре 50°C-108°C (температура кипения изобутилового спирта), предпочтительно 70°C-100°C, и смесь охлаждают при перемешивании до температуры от -5°C до комнатной температуры, посредством этого кристаллы можно получить с высоким выходом и с хорошей воспроизводимостью.
Изобутилового спирта применяют, например, в количестве 10-50 об./вес, предпочтительно 20-40 об./вес, более предпочтительно 20-30 об./вес, относительно соединения IA. Соединение IA в качестве исходного соединения может представлять собой соединение, полученное на приведенной выше стадии 4 или, например, соединение, полученное в WO 2005/063743, и оно конкретно не ограничено. Однако для сохранения качества фармацевтического продукта, предпочтительно применяют соединение, имеющее высокую чистоту. Более предпочтительно, можно применять, например, моногидрат соединения IA, полученный способом приведенного ниже примера 12 и подобным. Кроме того, затравку кристаллов (A кристалл), полученную отдельно, можно добавлять в процессе охлаждения, при необходимости.
Приведенную выше затравку кристаллов (A кристалл) можно получить растворением соединения IA, полученного согласно приведенной выше стадии 4 или WO 2005/063743 и подобным, в изобутиловом спирте при температуре 50°C-108°C, предпочтительно 70°C-100°C, охлаждением смеси при перемешивании, при необходимости, до температуры от -5°C до комнатной температуры. Более предпочтительно, кристаллы, полученные измельчением полученного кристалла струйной мельницей и подобным, можно применять в качестве затравки кристаллов.
В случае кристаллов (HA кристалл) моногидрата соединения IA, кроме того, соединение IA растворяют, например, в растворителе, содержащем существенное количество воды (например, DMF-вода, этанол-вода, ацетон-вода и подобные) предпочтительно при температуре от комнатной температуры до температуры кипения растворителя, который будут применять, и смесь охлаждают при перемешивании до от -5°C до комнатной температуры, посредством этого HA кристаллы можно получить с высоким выходом и хорошей воспроизводимостью. Для получения кристаллов с высокой чистотой, кристаллизацию более предпочтительно осуществлять из смешанного растворителя ацетон-вода.
Растворитель, содержащий существенное количество воды, применяют в количестве, например, 10-50 об./вес, предпочтительно 20-40 об./вес, более предпочтительно 20-30 об./вес, относительно соединения IA, которое изменяется в зависимости от типа растворителя, который будут применять. При необходимости, затравку кристаллов (HA кристалл), полученную отдельно, можно также добавлять при охлаждении.
Приведенную выше затравку кристаллов (HA кристалл) можно получить растворением соединения IA, полученного согласно приведенной выше стадии 4 или WO 2005/063743 и подобному, в смеси этанол-вода при температуре 50°C-100°C, предпочтительно 70°C-90°C, при необходимости, охлаждением смеси при перемешивании до температуры от -5°C до комнатной температуры. Более предпочтительно, кристалл, полученный измельчением полученного кристалла струйной мельницей и подобным, можно применять в качестве затравки кристаллов.
Приведенным выше способом, кристаллы соединения IA или его моногидрата можно эффективно получить с определенной степенью качества и с хорошей воспроизводимостью.
Как приводится выше, соединение IA включает кристаллические формы ангидрида, моногидрата, этанолата и подобных. Кристаллы, которые не превращаются в другую форму даже в жестких условиях, например, высокой температуре, высокой влажности и подобных, является особенно превосходным с точки зрения получения фармацевтического продукта, который требует обеспечения стабильности. Кроме того, кристаллы, обладающие превосходной пероральной абсорбционной способностью, требуются в качестве лекарственного вещества перорального фармацевтического препарата.
Например, в случае соединения IA, соединение IA, полученное согласно примеру 504 WO 2005/063743, представляет собой 0,5 этанолат (EA кристалл) (сравнительный пример 3), и кристаллы моногидрата, ангидрида и подобных можно получить с хорошей воспроизводимостью контролированием условий кристаллизации соединения IA. Из них, A кристалл и HA кристалл можно в частности получить способом, описанным в примерах 13-14, с хорошей воспроизводимостью. Из них, A кристалл обладает превосходной стабильностью (смотри экспериментальный пример 1), и его можно хранить с сохранением высокого качества в течение длительного периода времени.
A кристалл и HA кристалл обладают превосходной пероральной абсорбционной способностью (смотри экспериментальный пример 2), и являются предпочтительными в качестве лекарственного вещества фармацевтического продукта.
Кристаллы соединения IA или его моногидрата гранулируют измельчением и подобным, при необходимости, и их можно применять в качестве активного ингредиента фармацевтического препарата, например, агента для лечения и/или профилактики заболеваний, таких как болезнь Паркинсона, расстройство сна, устойчивость к опиоидам в качестве анальгетиков, мигрень, двигательное расстройство, депрессия, тревожное расстройство и подобные.
В то время как кристаллы соединения IA или его моногидрата можно вводить как есть, их обычно желательно обеспечивать в виде различных фармацевтических препаратов. Данные фармацевтические препараты применяют на животных или людях.
Фармацевтические препараты могут содержать, в качестве активного ингредиента, кристаллы соединения IA или его моногидрата отдельно или в виде смеси с любым другим активным ингредиентом для лечения. Данные фармацевтические препараты получают смешением активного ингредиента с одним или более видами фармацевтически приемлемых носителей (например, разбавителем, растворителем, вспомогательным веществом и подобными), и согласно любому способу, хорошо известному в технической области формулирования лекарственных средств.
В качестве пути введения, желательно применять путь, самый эффективный для лечения, который может быть пероральным или, например, парентеральным, таким как внутривенный и подобный.
Примеры формы введения включают таблетку, препарат местного применения, инъекцию и подобные.
Например, таблетку и подобную, пригодную для перорального введения, можно получить, применяя вспомогательные вещества, такие как лактоза и подобные, разрыхлители, такие как крахмал и подобные, смазывающие агенты, такие как стеарат магния и подобные, связующие, такие как гидроксипропилцеллюлоза и подобные, и подобные.
Примеры препарата для местного применения и подобные, подходящие для парентерального применения, включают мазь, крем, линимент, лосьон, припарку, пластырь, ленту и подобные. Например, мазь, крем и подобным можно получить растворением или смешением диспергированием активного ингредиента в основе, такой как белый вазелин и подобный. Кроме того, например, инъекцию и подобное можно получить, применяя разбавитель, растворитель и подобный, такой как соляной раствор, раствор глюкозы или смесь соляной воды и раствора глюкозы, и подобные.
Доза и частота введения кристаллов соединения IA или его моногидрата изменяются в зависимости от формы введения, возраста, веса тела пациентов, свойств или тяжести симптомов, которые будут лечить, и подобных. Для перорального введения, 0,01-1000 мг, предпочтительно 0,05-100 мг, обычно вводят взрослому от одной до нескольких порций в день. Для парентерального введения, такого как внутривенное введение и подобное, 0,001-1000 мг, предпочтительно 0,01-100 мг, обычно вводят взрослому от одного до нескольких раз в день. Однако данные дозы и частоты введения изменяются в зависимости от приведенных выше различных условий.
Настоящее изобретение более конкретно объясняется далее со ссылкой на примеры и сравнительные примеры, которые не считают ограничивающими.
Спектры ядерного магнитного резонанса (1H ЯМР), применяемые в примерах и сравнительных примерах, измеряли при 270 МГц или 300 МГц, и обменивающийся протон может явно не наблюдаться, в зависимости от соединения и условий измерения. Для указания мультиплетности сигнала, применяют обычно применяемые обозначения, где уш обозначает явно уширенный сигнал.
Порошковую дифракцию рентгеновских лучей (проникающий способ) измеряли измельчением образца в агатовой ступке, помещением его подходящего количества на плоскость для образца и измерением дифракционных пиков изменением дифракционного угла 2θ от 5° до 40°. Облучающие рентгеновские лучи представляли собой Kα лучи меди (CuKα лучи), и напряжение на лампе устанавливали на 5 кВ, электрический ток лампы устанавливали на 40 мА, шаг сканирования устанавливали на 0,017°, и время счета устанавливали на 0,56°/сек. Термический анализ проводили помещением приблизительно 2 мг образца в алюминиевую кювету, и измерением дифференциальной сканирующей калориметрии (DSC) при скорости повышения температуры 10°C/мин.
[Пример 1] Получение 4-(фуран-2-ил)-2-трет-бутиламинотиазол-5-илпиридин-2-илкетона (соединение C-1)
Соединение A-1 (228 мг, 1,0 ммоль), полученное в сравнительном примере 1, и гидробромид 2-(бромацетил)пиридина (281 мг, 1,0 ммоль) растворяли в DMF (2,8 мл), и смесь перемешивали при 40°C в течение 9 часов. К смеси добавляли воду (4,0 мл), и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным соляным раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат= 9:1), получая соединение C-1 (271 мг, 82%).
1H-ЯМР (CDCl3) δ 1,50 (с, 9H), 6,03 (уш с, 1H), 6,51 (дд, J=3,5 Гц, 1,8 Гц, 1H), 7,42 (ддд, J=7,9 Гц, 4,9 Гц, 1,0 Гц, 1H), 7,44 (дд, J=1,8 Гц, 0,7 Гц, 1H), 7,79 (дд, J=3,5 Гц, 0,7 Гц, 1H), 7,86 (тд, J=7,9 Гц, 1,8 Гц, 1H), 8,13 (ддд, J=7,9 Гц, 1,0 Гц, 0,9 Гц, 1H), 8,63 (ддд, J=4,9 Гц, 1,8 Гц, 0,9 Гц, 1H). LC/MS ESI(+) m/z 328 [M+H]+.
[Пример 2] Получение 4-(фуран-2-ил)-2-трет-бутиламинотиазол-5-илпиридин-3-илкетона (соединение C-2)
Соединение A-1 (218 мг, 0,96 ммоль), полученное в сравнительном примере 1, и гидробромид 3-(бромацетил)пиридина (278 мг, 1,0 ммоль) растворяли в DMF (2,0 мл), и смесь перемешивали при 40°C в течение 10 часов. К смеси добавляли воду (4,0 мл), и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным соляным раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат= 7:3), получая соединение C-2 (207 мг, 66%).
1H-ЯМР (CDCl3) δ 1,48 (с, 9H), 6,19 (уш с, 1H), 6,31 (дд, J=3,5 Гц, 1,8 Гц, 1H), 6,87 (дд, J=3,5 Гц, 0,6 Гц, 1H), 7,07 (дд, J=1,8 Гц, 0,6 Гц, 1H), 7,26 (дд, J=8,1 Гц, 5,0 Гц, 1H), 7,92 (ддд, J=8,1 Гц, 2,2 Гц, 1,7 Гц, 1H), 8,62 (дд, J=5,0 Гц, 1,7 Гц, 1H), 8,82 (дд, J=2,2 Гц, 0,7 Гц, 1H). LC/MS ESI(+) m/z 328 [M+H]+.
[Пример 3] Получение 4-(фуран-2-ил)-2-трет-бутиламинотиазол-5-илтетрагидропиран-4-илкетона (соединение C-3)
Соединение A-1 (30 мг, 0,13 ммоль), полученное в сравнительном примере 1, и 4-бромацетилтетрагидропиран (25 мг, 0,12 ммоль) растворяли в DMF (0,5 мл), и смесь перемешивали при 40°C в течение 8,5 часов. К смеси добавляли воду (0,5 мл), и выпавший осадок собирали фильтрованием. Полученный твердый остаток промывали водой (0,5 мл) и сушили при пониженном давлении, получая соединение C-3 (37 мг, 92%).
1H-ЯМР (CDCl3) δ 1,46 (с, 9H), 1,60-1,95 (м, 4H), 3,06 (тт, J=11,1 Гц, 3,9 Гц, 1H), 3,40 (дт, J=11,6 Гц, 2,2 Гц, 2H), 4,02 (ддд, J=11,6 Гц, 4,2 Гц, 2,2 Гц, 2H), 5,81 (уш с, 1H), 6,55 (дд, J=3,5 Гц, 1,8 Гц, 1H), 7,48 (дд, J=3,5 Гц, 0,7 Гц, 1H), 7,54 (дд, J=1,8 Гц, 0,7 Гц, 1H). LC/MS ESI(+) m/z 335 [M+H]+.
[Пример 4] Получение 4-(фуран-2-ил)-2-(1-метил-1-фенилэтил)аминотиазол-5-илтетрагидропиран-4-илкетона (соединение C-4)
Соединение A-2 (580 мг, 2,0 ммоль), полученное в сравнительном примере 2, и 4-бромацетилтетрагидропиран (416 мг, 2,0 ммоль) растворяли в DMF (2,0 мл), и смесь перемешивали при 40°C в течение 8 часов. При охлаждении на льду смесь нейтрализовали, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным соляным раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=4:1-1:1), получая соединение C-4 (690 мг, 87%).
1H-ЯМР (CDCl3) δ 1,53-1,83 (м, 4H), 1,76 (с, 6H), 2,79 (тт, J=12,9 Гц, 3,7 Гц, 1H), 3,29 (дт, J=11,6 Гц, 2,3 Гц, 2H), 3,94 (ддд, J=11,6 Гц, 3,6 Гц, 2,3 Гц, 2H), 6,54 (дд, J=3,5 Гц, 1,7 Гц, 1H), 6,76 (уш с, 1H), 7,28-7,42 (м, 3H), 7,44-7,51 (м, 3H), 7,54 (дд, J=1,7 Гц, 0,8 Гц, 1H).
[Пример 5] Получение 2-амино-4-(фуран-2-ил)тиазол-5-илпиридин-2-илкетона (соединение D-1)
Соединение C-1 (263 мг, 0,80 ммоль), полученное в примере 1, растворяли в концентрированной хлористоводородной кислоте (2,6 мл), и смесь перемешивали при 80°C в течение 3,5 часов. При охлаждении на льду, смесь нейтрализовали насыщенным водным раствором бикарбоната натрия, и выпавший осадок собирали фильтрованием. Полученный твердый остаток промывали водой (6,0 мл), и сушили при пониженном давлении, получая соединение D-1 (140 мг, 64%).
1H-ЯМР (DMSO-d6) δ 6,56 (дд, J=3,5 Гц, 1,7 Гц, 1H), 7,44 (дд, J=3,5 Гц, 0,7 Гц, 1H), 7,53-7,61 (м, 2H), 7,94-8,08 (м, 2H), 8,05 (уш с, 2H), 8,57 (ддд, J=5,0 Гц, 1,7 Гц, 0,9 Гц, 1H). LC/MS ESI(+) m/z 272 [M+H]+.
[Пример 6] Получение 2-амино-4-(фуран-2-ил)тиазол-5-илпиридин-3-илкетона (соединение D-2)
Соединение C-2 (207 мг, 0,63 ммоль), полученное в примере 2, растворяли в концентрированной хлористоводородной кислоте (2,0 мл), и смесь перемешивали при 80°C в течение 3,5 часов. При охлаждении на льду, смесь нейтрализовали насыщенным водным раствором бикарбоната натрия, и выпавший осадок собирали фильтрованием. Полученный твердый остаток промывали водой (3,0 мл), и сушили при пониженном давлении, получая соединение D-2 (127 мг, 74%).
1H-ЯМР (DMSO-d6) δ 6,40 (дд, J=3,5 Гц, 1,8 Гц, 1H), 6,80 (дд, J=3,5 Гц, 0,4 Гц, 1H), 7,28 (дд, J=1,8 Гц, 0,4 Гц, 1H), 7,35 (ддд, J=8,0 Гц, 4,9 Гц, 0,6 Гц, 1H), 7,85 (ддд, J=8,0 Гц, 2,2 Гц, 1,8 Гц, 1H), 8,15 (уш с, 2H), 8,60 (дд, J=4,9 Гц, 1,8 Гц, 1H), 8,63 (дд, J=2,2 Гц, 0,6 Гц, 1H). LC/MS ESI(+) m/z 272 [M+H]+.
[Пример 7] Получение 2-амино-4-(фуран-2-ил)тиазол-5-илтетрагидропиран-4-илкетона (соединение D-3)-(1)
Соединение C-3 (30 мг, 0,09 ммоль), полученное в примере 3, растворяли в концентрированной хлористоводородной кислоте (1,0 мл), и смесь перемешивали при 80°C в течение 1,5 часов. При охлаждении на льду, смесь нейтрализовали насыщенным водным раствором бикарбоната натрия, и выпавший осадок собирали фильтрованием. Полученный твердый остаток промывали водой (0,5 мл), и сушили при пониженном давлении, получая соединение D-3 (22 мг, 87%).
1H-ЯМР (CDCl3) δ 1,68-1,95 (м, 4H), 2,99 (тт, J=11,0 Гц, 3,9 Гц, 1H), 3,40 (дт, J=11,6 Гц, 2,4 Гц, 2H), 4,02 (ддд, J=11,6 Гц, 4,0 Гц, 2,4 Гц, 2H), 5,58 (уш с, 2H), 6,56 (дд, J=3,5 Гц, 1,8 Гц, 1H), 7,55 (д, J=3,5 Гц, 1H), 7,56 (д, J=1,8 Гц, 1H). LC/MS ESI(+) m/z 279 [M+H]+.
[Пример 8] Получение соединения D-3-(2)
Смесь соединения C-3 (80 г, 0,24 моль), полученного в примере 3, концентрированной хлористоводородной кислоты (200 мл) и воды (200 мл) перемешивали при 95-108°C в течение 4 часов при упаривании низкокипящих веществ. К смеси добавляли по каплям теплую воду 95°C (80 мл) при 90°C, и смесь перемешивали при той же температуре в течение 30 минут, и затем при 85°C в течение 30 минут, и охлаждали до 50°C. Добавляли по каплям метанол (240 мл). Смесь доводили до pH 9,5 водным раствором гидроксида натрия (8 моль/л), охлажденным до 5°C, и выпавший осадок собирали фильтрованием. Полученный твердый остаток промывали холодным 10% водным раствором метанола, и сушили при пониженном давлении, получая соединение D-3 (61 г, 92%).
[Пример 9] Получение соединения D-3-(3)
Соединение C-4 (50 мг, 0,13 ммоль), полученное в примере 4, растворяли в трифторуксусной кислоте (0,5 мл), и смесь перемешивали при 40°C в течение 3 часов. При охлаждении на льду, смесь нейтрализовали насыщенным водным раствором бикарбоната натрия, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным соляным раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали тонкослойной колоночной хроматографией на силикагеле (н-гексан:этилацетат= 1:1), получая соединение D-3 (21 мг, 59%).
[Пример 10] Получение моногидрата N-[4-(фуран-2-ил)-5-(тетрагидрофуран-4-карбонил)тиазол-2-ил]-6-метилпиридин-3-карбоксамида (соединение IA)
Смесь соединения D-3 (500 г, 1,80 моль), полученного в примере 7, активированного угля (C2, 25 г) и DMF (1,3 л) перемешивали при 60°C в течение 1 часа. Активированный уголь отфильтровывали при данной температуре, к фильтрату добавляли 6-метилникотиновую кислоту (419 г, 3,06 моль), CDI (495 г, 3,06 моль) и DMF (875 мл) при комнатной температуре, и смесь перемешивали при 90°C в течение 3 часов. Смесь охлаждали до 60°C, добавляли активированный уголь (C2, 50 г) и DMF (250 мл), и смесь перемешивали при данной температуре в течение 1 часа. Активированный уголь отфильтровывали при данной температуре, и промывали DMF (500 мл) при данной температуре. Фильтрат охлаждали до 40°C, добавляли по каплям воду (1,5 л) в течение 30 минут при перемешивании при данной температуре, и смесь перемешивали при данной температуре в течение 30 минут. Дополнительно добавляли по каплям воду (1,5 л). Смесь охлаждали до 5°C, перемешивали в течение 6 часов, и выпавший осадок собирали фильтрованием. Полученный твердый остаток промывали холодным 50% водным раствором метанола (1,0 л), и сушили при пониженном давлении, получая моногидрат соединения IA (619 г, выход 84%).
[Пример 11] получение кристалла моногидрата соединения IA (HA кристалл)
Моногидрат соединения IA (80 г, 0,19 моль), полученный в примере 10, растворяли смеси воды (0,56 л) и ацетона (2,24 л) при 55°C, добавляли активированный уголь (C2, 8 г), и смесь перемешивали в течение 30 минут. Активированный уголь отфильтровывали при данной температуре и промывали теплым 80% водным раствором ацетона (0,16 л). HA кристалл (0,40 г), полученный в примере 13, добавляли в качестве затравки кристаллов при перемешивании фильтрата при 40°C, и смесь охлаждали до 0°C в течение 3 часов и перемешивали при данной температуре в течение 15 часов. Выпавший осадок собирали фильтрованием, полученный твердый остаток промывали холодным 50% водным раствором ацетона (0,10 л), и сушили при пониженном давлении при 45°C в течение 48 часов, получая HA кристалл соединения IA (72 г, выход 90%).
Результаты измерения порошковой дифракции рентгеновских лучей: пики при дифракционных углах (2θ)=8,1, 12,0, 14,7, 16,3, 21,1, 21,8, 23,0, 24,4, 24,7, 28,3° (смотри фигуру 1).
Результаты измерения термического анализа (ДСК): эндотермические пики при приблизительно 133°C и приблизительно 209°C.
[Пример 12] Получение кристаллов ангидрида соединения IA (A кристалл)
Моногидрат соединения IA (300 г, 0,72 моль), полученный в примере 10, растворяли в изобутиловом спирте (8,1 л) при 80°C, добавляли активированный уголь (C2, 30 г), и смесь перемешивали в течение 30 минут. Активированный уголь отфильтровывали при данной температуре и промывали теплым изобутиловым спиртом (0,6 л). A кристалл (1,5 г), полученный в примере 14, добавляли в качестве затравки кристаллов к фильтрату при перемешивании фильтрата при 70°C. Смесь дополнительно перемешивали при данной температуре в течение 1 часа, охлаждали до 5°C в течение 6 часов и дополнительно перемешивали при данной температуре в течение 18 часов. Выпавший осадок собирали фильтрованием, полученный твердый остаток промывали холодным изобутиловым спиртом (0,60 л) и сушили при пониженном давлении при 50°C, получая A кристаллы (262 г, выход 93%).
Результаты измерения порошковой дифракции рентгеновских лучей: пики при дифракционных углах (2θ)=8,3, 12,6, 16,5, 19,1, 19,5, 20,8, 21,2, 22,4, 23,8, 27,0° (смотри фигуру 2).
Результаты измерения термического анализа (ДСК): эндотермический пик при приблизительно 210°C.
[Пример 13] Получение затравки кристаллов HA кристаллов
Моногидрат соединения IA (550 г, 1,38 моль), полученный в примере 10, растворяли в смеси воды (1,1 л) и этанола (9,9 л) при 80°C. Раствор добавляли по каплям к воде (1,1 л) в течение 20 минут, и смесь перемешивали при 20°C в течение 1,5 часов и дополнительно при 0°C в течение 2,5 часов. Выпавший осадок собирали фильтрованием, полученный твердый остаток промывали смесью (400 мл) холодной воды (320 мл) и холодного этанола (80 мл), и сушили при пониженном давлении при 55°C, получая HA кристаллы (525 г, выход 95%).
HA кристаллы (515 г) измельчали струйной мельницей, и измельченные кристаллы применяли в качестве затравки кристаллов.
Результаты измерения порошковой дифракции рентгеновских лучей: пики при дифракционных углах (2θ)=8,1, 12,0, 14,7, 16,3, 21,1, 21,8, 23,0, 24,4, 24,7, 28,3°.
[Пример 14] Получение затравки кристаллов A кристаллов
Моногидрат соединения IA (40 г, 96 ммоль), полученный в примере 10, растворяли в изобутиловом спирте (1,2 л) при 80°C, и раствор охлаждали до 5°C в течение 8 часов. Раствор дополнительно перемешивали при данной температуре в течение 16 часов, выпавший осадок собирали фильтрованием, полученный твердый остаток промывали холодным изобутиловым спиртом (80 мл) и сушили при пониженном давлении при 50°C, получая A кристаллы (37 г, выход 95%). A кристаллы (34 г) измельчали струйной мельницей. Измельченные кристаллы применяли в качестве затравки кристаллов.
Результаты измерения порошковой дифракции рентгеновских лучей: пики при дифракционных углах (2θ)= 8,3, 12,6, 16,5, 19,1, 19,5, 20,8, 21,2, 22,4, 23,8, 27,0°.
(Сравнительный пример 1) Получение 1-трет-бутил-3-(фуран-2-ил)карбонилтиомочевины (соединение A-1)
Тиоцианат натрия (0,91 г, 11 ммоль) суспендировали в THF (5,0 мл), 2-фуроилхлорид (1,0 мл, 10 ммоль) добавляли при 40°C, и смесь перемешивали в течение 10 минут. После охлаждения на льду, добавляли к смеси трет-бутиламин (1,1 мл, 11 ммоль), и смесь перемешивали при 40°C в течение 30 минут. Добавляли к смеси воду (10,0 мл), и смесь перемешивали при комнатной температуре в течение 30 минут. Выпавший осадок собирали фильтрованием, промывали водой (5,0 мл) и сушили при пониженном давлении, получая соединение A-1 (1,86 г, 81%).
1H-ЯМР (CDCl3) δ 1,59 (с, 9H), 6,59 (дд, J=3,6 Гц, 1,7 Гц, 1H), 7,29 (дд, J=3,6 Гц, 0,8 Гц, 1H), 7,57 (дд, J=1,7 Гц, 0,8 Гц, 1H), 8,86 (уш с, 1H), 10,62 (уш с, 1H). LC/MS (ESI(+)) m/z 227 [M+H]+.
(Сравнительный пример 2) Получение 1-(1-метил-1-фенилэтил)-3-(фуран-2-ил)карбонилтиомочевины (соединение A-2)
Тиоцианат натрия (922 мг, 11 ммоль) суспендировали в THF (5,0 мл), 2-фуроилхлорид (1,0 мл, 10 ммоль) добавляли при 40°C, и смесь перемешивали в течение 10 минут. После охлаждения на льду, добавляли к смеси 1-метил-1-фенилэтиламин (1,5 мл, 11 ммоль), и смесь перемешивали при комнатной температуре в течение 1,5 часов. Добавляли к смеси воду (10,0 мл), и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным соляным раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Этанол (3,0 мл) добавляли к полученному остатку, и смесь перемешивали, и выпавший осадок собирали фильтрованием и сушили при пониженном давлении, получая соединение A-2 (1,85 г, 63%).
1H-ЯМР (CDCl3) δ 1,90 (с, 6H), 6,60 (дд, J=3,6 Гц, 1,7 Гц, 1H), 7,20-7,42 (м, 5H), 7,33 (дд, J=3,6 Гц, 0,8 Гц, 1H), 7,57 (дд, J=1,7 Гц, 0,8 Гц, 1H), 8,91 (уш с, 1H), 11,05 (уш с, 1H).
(Сравнительный пример 3) Получение соединения IA способом, описанным в WO2005/063743 (получение 0,5 этанолата соединения IA (EA кристалл))
Тем же способом, как в примере 504 WO 2005/063743, соединение IA получали в виде бледно-коричевого твердого остатка. Подтверждали, что полученный твердый остаток представляет собой кристаллы 0,5 этанолата соединения IA (EA кристаллы) по различным спектральным данным (1H ЯМР спектр, порошковая рентгеновская дифракция, термический анализ, элементный анализ и подобные).
Результаты измерения порошковой дифракции рентгеновских лучей: пики при дифракционных углах (2θ)= 7,4, 9,0, 10,0, 12,7, 16,3, 17,7, 19,3, 19,8, 23,9, 27,2° (смотри фигуру 3).
Результаты измерения термического анализа (ДСК): эндотермические пики при приблизительно 154°C, приблизительно 198°C и приблизительно 208°C.
Экспериментальный пример 1: испытание на стабильность
Кристаллы, полученные в примере 12, хранили в условиях 40°C/75% RH (относительная влажность) и 40°C/90% RH (относительная влажность) в течение 6 месяцев каждый, и измеряли порошковую рентгеновскую дифракцию. При сравнении с рентгенограммой в момент начала хранения, рентгенограмма не показала любых изменений при обоих условиях. Подтверждали, что кристаллы являются стабильными даже после хранения в течение длительного периода времени в увлажненных условиях 40°C/75% RH (относительная влажность) и 40°C/90% RH (относительная влажность).
HA кристаллы, полученные в примере 11, хранили в течение 2 недель при 60°C, и затем измеряли порошковую дифракцию рентгеновских лучей. При сравнении с рентгенограммой в момент начала хранения, подтверждали, что рентгенограмма после хранения показывала характерные дифракционные пики A кристалла. Иными словами, считали, что часть HA кристаллов подверглась превращению в A кристаллы при 60°C.
Экспериментальный пример 2: испытание на абсорбционную способность
HA кристаллы и A кристаллы, полученные в примерах 11 и 12, вводили перорально мужским особям крыс, соответственно, и оценивали кинетику в плазме соединения IA. Результаты показаны в таблице 1 и таблице 2.
[Таблица 1]
Таблица 1 кинетика в плазме HA кристаллов
[Таблица 2]
Таблица 2 кинетика в плазме A кристаллов
Как результат, и HA кристаллы и A кристаллы показывали хорошую абсорбционную способность, и подтверждали, что они обладают превосходными свойствами в качестве терапевтических продуктов.
Концентрация в плазме увеличивалась более быстро для A кристаллов, чем для HA кристаллов.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Согласно настоящему изобретению, можно обеспечивать способ получения соединения, представленного формулой (I), который обладает антагонистическим действием относительно аденозинового A2A рецептора и является пригодным в качестве терапевтического агента, например, для болезни Паркинсона, расстройства сна, устойчивости к опиоидам в качестве анальгетиков, мигрени, двигательного расстройства, депрессии, тревожного расстройства и подобных, кристалл соединения, представленного формулой (IA), или его моногидрат и способ его получения и подобные. Способы получения настоящего изобретения являются пригодными в качестве промышленных способов получения лекарственного вещества фармацевтического продукта. Кроме того, кристаллическая форма соединения, представленного формулой (IA), или его моногидрата настоящего изобретения является пригодной в качестве лекарственного вещества фармацевтического продукта.
Данная заявка основана на патентной заявке No. 2015-2964 поданной в Японии, содержание которой включено полностью в настоящее изобретение.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ИНДОЛОПИРРОЛОКАРБАЗОЛА | 2003 |
|
RU2337105C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИАЗОЛОПИРИДИНОВОГО СОЕДИНЕНИЯ | 2017 |
|
RU2752384C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ОПТИЧЕСКИ АКТИВНОЕ СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ АКТИВНОСТЬЮ АГОНИСТА РЕЦЕПТОРА ТРОМБОПОЭТИНА, И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ ДЛЯ ЭТОГО | 2008 |
|
RU2476429C2 |
| ЦИКЛОАЛКАН-1,3-ДИАМИНОВОЕ ПРОИЗВОДНОЕ | 2019 |
|
RU2793247C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ФЕНИЛАЛАНИНА С ХИНАЗОЛИНДИОНОВЫМ СКЕЛЕТОМ И ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ, ПРИМЕНЯЕМЫХ ПРИ ПОЛУЧЕНИИ ТАКИХ ПРОИЗВОДНЫХ | 2007 |
|
RU2469028C2 |
| КРИСТАЛЛ ПРОИЗВОДНОГО 1,3,5-ТРИАЗИНА ИЛИ ЕГО СОЛЬВАТА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2020 |
|
RU2837449C1 |
| КРИСТАЛЛЫ ПРОИЗВОДНЫХ 6,7-НЕНАСЫЩЕННОГО-7-КАРБАМОИЛМОРФИНАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2607084C2 |
| МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2347784C2 |
| КРИСТАЛЛЫ ПРОИЗВОДНЫХ 6,7-НЕНАСЫЩЕННОГО-7-КАРБАМОИЛМОРФИНАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2643807C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 7Н-ПИРРОЛО[2,3-d]ПИРИМИДИНА И ИХ СОКРИСТАЛЛОВ | 2017 |
|
RU2779212C2 |

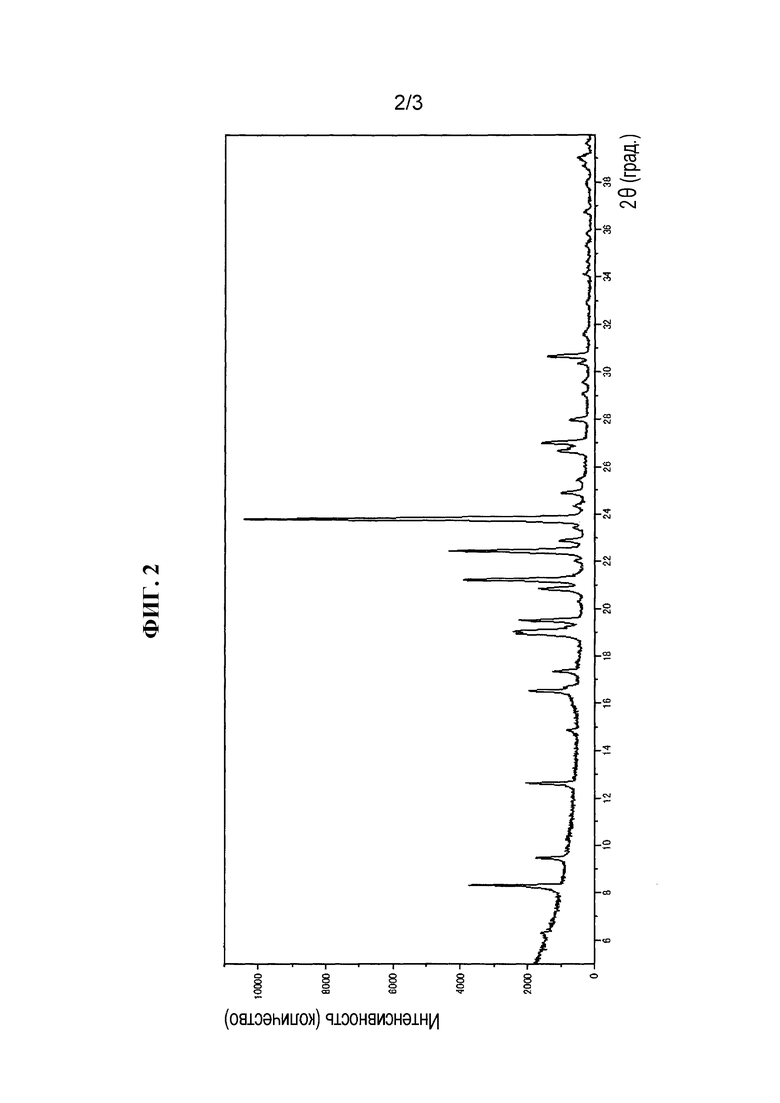

Изобретение относится к способу получения тиазольного производного, представленного формулой (I), включающему:
(i) способ получения соединения, представленного формулой (C), включающий (i) стадию реакции соединения, представленного формулой (A), и соединения, представленного формулой (B) при температуре между 10°C и 70°C в растворителе, выбранном из группы, состоящей из ароматических углеводородов, галогенированных углеводородов, полярных растворителей, простых эфиров, сложных эфиров, спиртов и воды, используемых отдельно или в виде их смеси:
 ;
;
(ii) стадию получения соединения, представленного формулой (D), обработкой соединения, представленного формулой (C), кислотой:
 , и
, и
(iii) стадию получения соединения, представленного формулой (I), реакцией соединения, представленного формулой (D), и соединения, представленного формулой (E):
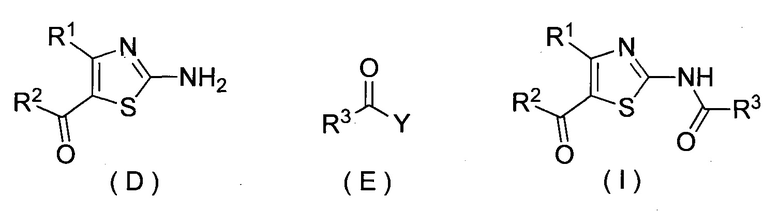
Технический результат: разработан новый промышленный способ получения соединения, представленного формулой (I), которое обладает антагонистическим действием относительно аденозинового A2A рецептора и является пригодным в качестве терапевтического агента, например, для болезни Паркинсона, расстройства сна, устойчивости к опиоидам в качестве анальгетиков, мигрени, двигательного расстройства, депрессии, тревожного расстройства и подобных. 2 н. и 12 з.п. ф-лы, 2 табл., 14 пр., 3 ил.
1. Способ получения соединения, представленного формулой (C), включающий (i) стадию реакции соединения, представленного формулой (A), и соединения, представленного формулой (B) при температуре между 10°C и 70°C в растворителе, выбранном из группы, состоящей из ароматических углеводородов, галогенированных углеводородов, полярных растворителей, простых эфиров, сложных эфиров, спиртов и воды, используемых отдельно или в виде их смеси:

где R1 представляет собой фурил, R4, R5 и R6 являются одинаковыми или отличными, и каждый представляет собой низший алкил или арил, R2 представляет собой пиридил или тетрагидропиранил, и X1 представляет собой галоген.
2. Способ получения по п.1, где X1 представляет собой атом хлора, атом брома или атом йода.
3. Способ получения по п.1, где X1 представляет собой атом брома.
4. Способ получения по любому из пп.1-3, где каждый R4, R5 и R6 представляет собой метил.
5. Способ получения по любому из пп.1-4, где R1 представляет собой 2-фурил.
6. Способ получения соединения, представленного формулой (I), включающий:
(i) стадию, описанную в любом из пп.1-5;
(ii) стадию получения соединения, представленного формулой (D), обработкой соединения, представленного формулой (C), кислотой:
 , и
, и
(iii) стадию получения соединения, представленного формулой (I), реакцией соединения, представленного формулой (D), и соединения, представленного формулой (E):

где каждый R1, R2, R4, R5 и R6 представляет собой, как определено в любом из пп.1-5, R3 представляет собой арил, аралкил, ароматическую гетероциклическую группу, ароматический гетероциклилалкил, алифатический гетероциклилалкил или тетрагидропиранилокси, или данные группы замещены 1-3 заместителями, выбранными из группы, состоящей из галогена; низшего алкила, необязательно замещенного низшим алкокси или морфолино; низшего алкокси; низшего алканоила; и винила,
Y представляет собой галоген или гидрокси.
7. Способ получения по п.6, где кислота на стадии (ii) представляет собой хлористоводородную кислоту или трифторуксусную кислоту.
8. Способ получения по п.6 или 7, где Y представляет собой гидрокси.
9. Способ получения по п.8, где реакцию на стадии (iii) проводят в присутствии 1,3-дициклогексилкарбодиимида, гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида, 1,1'-карбонилдиимидазола (CDI) или пропилфосфонового ангидрида.
10. Способ получения по п.8, где реакцию на стадии (iii) проводят в присутствии CDI.
11. Способ получения по любому из пп.1-10, где R2 представляет собой 4-тетрагидропиранил.
12. Способ получения по любому из пп.6-11, где R3 представляет собой 2-метилпиридин-5-ил, 2-метилпиримидин-5-ил, 5,6-дигидро-2H-пиридилметил или 4-тетрагидропиранилокси.
13. Способ получения по любому из пп.6-11, где R3 представляет собой 2-метилпиридин-5-ил.
14. Способ получения по любому из пп.6-10, где R1 представляет собой 2-фурил, R2 представляет собой 4-тетрагидропиранил, и R3 представляет собой 2-метилпиридин-5-ил.
| C.Friot, A.Reliquet, F.Reliquet & J.C.Meslin, 2,4-DIAMINO-1-THIA-3-AZABUTADIENES, INTERMEDIATES IN HETEROCYCLIC SYNTHESIS, Phosphorus, Sulfur, and Silicon and the Related Elements, 156, стр.135-149, 2000 | |||
| WO2006011631 A2, 02.02.2006 | |||
| US2007105919 A1, 10.05.2007 | |||
| RU 2008112184 A, 30.08.2006 | |||
| US4420478 A, 13.12.1983. |

