В данной заявке заявлен приоритет по предварительной заявке на патент Китая № 201910643061.2, поданной 16 июля 2019 года, полное содержание которой включено в данный документ посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Данное изобретение относится к области фармацевтических препаратов, медицинской химии и фармакологии и, более конкретно, относится к определенному типу ингибиторов аврора-киназы, способам их получения и их применению.
УРОВЕНЬ ТЕХНИКИ
Аврора-киназы представляют собой серин/треонинкиназы, которые играют важнейшие роли в дупликации центросомы, образовании веретена деления, хромосомном расщеплении и контрольной точке сборки веретена деления во время митоза [Cancer Metastasis Rev., 2003, 22, 451]. Существует три структурно и функционально родственные аврора-киназы, аврора-A, аврора-B и аврора-C. Аврора-A локализуется после центросомы на ранней стадии митоза и ассоциируется с микротрубочками веретена и митотическими полюсами во время метафазы и телофазы. Она необходима для дупликации, созревания и отделения центросомы, а также образования биполярных веретен и для управления входом и выходом из митоза [Nat. Rev. Cancer, 2005, 5, 42]. Аврора-B локализуется у центросомы вокруг хроматина на ранней стадии митоза, а также у митотического веретена в анафазе. Она играет ключевую роль в функционировании центросом, выравнивании и разделении хромосом, контрольной точке веретена деления и цитокинезе [Mol. Cancer Ther., 2009, 8, 2046-2056]. Роль аврора-C в митозе определена недостаточно. Она в высокой степени экспрессируется в семенниках и может играть специализированную роль у животных мужского пола [Proc Natl Acad Sci USA, 2002, 99 (24): 15440-15445].
Ген, кодирующий аврора-A, соответствует области 20q13.2, которая часто амплифицируется при раковых заболеваниях, включая рак молочной железы, рак толстой кишки, рак яичника и рак щитовидной железы. Эктопическая экспрессия аврора-A в нормальных клетках приводит к амплификации центросом, анеуплоидии, хромосомной нестабильности и удлинению теломер - характеристикам, ассоциирующимся с трансформированными клетками [J. Cell Sci., 2007, 120, 2987]. Сверхэкспрессия аврора-A или ее активирующего партнера TPX-2 предположительно способствует хромосомной нестабильности при раке человека. Кроме того, аврора-A регулирует функцию супрессоров опухоли и проапоптозных белков, таких как p53. Например, фосфорилирование p53 у Ser215 и Ser351 под действием аврора-A усиливает его функциональную инактивацию и деградацию, соответственно.
Ген, кодирующий аврора-B, соответствует 17p13.1, хромосомной области, которая иногда удалена или амплифицирована при некоторых видах рака [J. Clin. Pathol., 2007, 60 (2): 218-221]. Повышенная экспрессия мРНК и белка аврора-B наблюдается при раке толстой кишки, раке полости рта и немелкоклеточном раке легких. Аврора-B представляет собой субъединицу хромосомного пассажирского комплекса (CPC). Аврора-B регулирует митоз посредством фосфорилирования INCENP, CENP-A и сурвивина. Кроме того, показано также, что сверхэкспрессия аврора-B усиливает передачу сигналов ras.
Аврора-киназы стали терапевтическими мишенями для противораковых лекарственных средств благодаря их онкогенным свойствам. Разработаны ингибиторы аврора-киназы в качестве новых противораковых агентов, среди которых LY-3295668 с пиридиновой центральной структурой является специфическим ингибитором аврора-A на I фазе разработки [WO2016077161]. Структура LY-3295668 представлена ниже.
Однако LY-3295668 и другие ингибиторы аврора-A неизменно ассоциируются со слабой активностью, низкой пероральной биодоступностью или умеренной эффективностью in vivo. Таким образом, существует потребность в разработке новых ингибиторов аврора с улучшенной активностью in vitro и in vivo.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
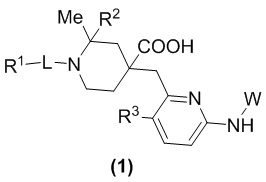
В данном изобретении предложен новый ингибитор киназы общей формулы (1), его оптический изомер, кристаллическая форма, фармацевтически приемлемая соль:
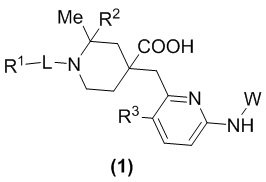
В формуле (1):
R1 представляет собой арил, гетероарил,  или
или  , где арил и гетероарил необязательно замещены 1-3 группами, выбранными из галогена, C1-C3 алкила, C1-C3 алкоксила, галогензамещенного C1-C3 алкила или галогензамещенного C1-C3 алкоксила;
, где арил и гетероарил необязательно замещены 1-3 группами, выбранными из галогена, C1-C3 алкила, C1-C3 алкоксила, галогензамещенного C1-C3 алкила или галогензамещенного C1-C3 алкоксила;
R2 представляет собой H или метил;
R3 представляет собой H или F;
W представляет собой 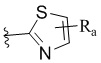 или
или 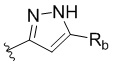 , где Ra представляет собой H, C1-C3 алкил или C3-C6 циклоалкил, Rb представляет собой H, C1-C3 алкил или C3-C6 циклоалкил; и
, где Ra представляет собой H, C1-C3 алкил или C3-C6 циклоалкил, Rb представляет собой H, C1-C3 алкил или C3-C6 циклоалкил; и
L представляет собой CH2, CO, CD2, CH(Me), C(Me)2, 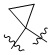 или
или 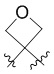 , когда W представляет собой или , и Rb представляет собой H, C2-C3 алкил или C3-C6 циклоалкил; или L представляет собой CO, CD2, CHMe, C(Me)2, или , когда W представляет собой , и Rb представляет собой метил.
, когда W представляет собой или , и Rb представляет собой H, C2-C3 алкил или C3-C6 циклоалкил; или L представляет собой CO, CD2, CHMe, C(Me)2, или , когда W представляет собой , и Rb представляет собой метил.
В другом предпочтительном варианте реализации в формуле (1) R1 представляет собой 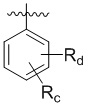 ,
, 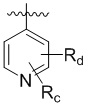 ,
, 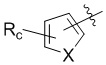 ,
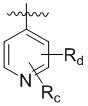
, 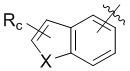 , или , где X представляет собой NH, O или S, Rc и Rd независимо представляют собой H, галоген, C1-C3 алкил, C1-C3 алкоксил, галогензамещенный C1-C3 алкил или галогензамещенный C1-C3 алкоксил.
, или , где X представляет собой NH, O или S, Rc и Rd независимо представляют собой H, галоген, C1-C3 алкил, C1-C3 алкоксил, галогензамещенный C1-C3 алкил или галогензамещенный C1-C3 алкоксил.
В другом предпочтительном варианте реализации в формуле (1) R1 представляет собой 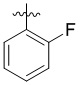 ,
,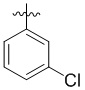 ,
, 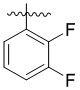 ,
, 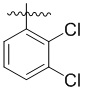 ,
, 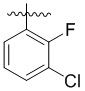 ,
, 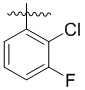 ,
, 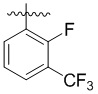 ,
, 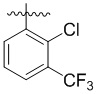 ,
, 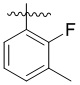 ,
, 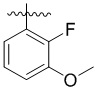 ,
, 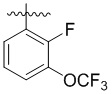 ,
, 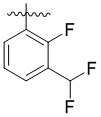 ,
, 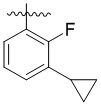 ,
, 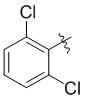 ,
, 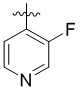 ,
, 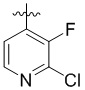 ,
,  ,
,  ,
,  , или .
, или .
В другом предпочтительном варианте реализации в формуле (1) W выбран из: ,
, 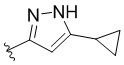 ,
, 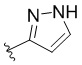 ,
, 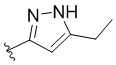 ,
, 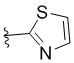 ,
, 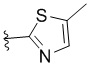 ,
, 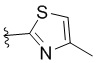 ,
, 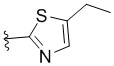 ,
, 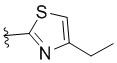 ,
,  или
или  ; и
; и
L представляет собой CH2, CO, CD2, CH(Me), C(Me)2, или , когда W представляет собой ,, , , , , , , или ; или L представляет собой CO, CD2, CHMe, C(Me)2, или , когда W представляет собой .
Посредством синтеза и тщательного изучения множества новых соединений, включая ингибирование аврора-киназы, авторами данного изобретения обнаружено, что для соединений общей формулы (1), если группа -L- изменена с CH2 на группу подходящего размера, такую как CD2, и/или если W представляет собой , то указанные соединения обладают чрезвычайно высокой ингибирующей активностью в отношении киназы аврора-A, при этом также существенно улучшена активность в отношении аврора-B и in vivo противоопухолевая активность.
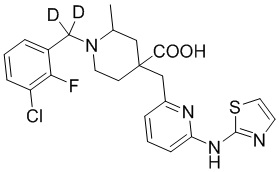
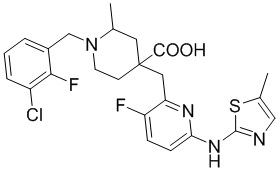
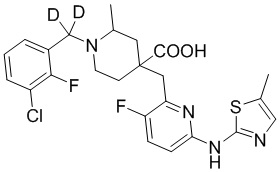
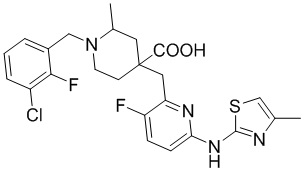
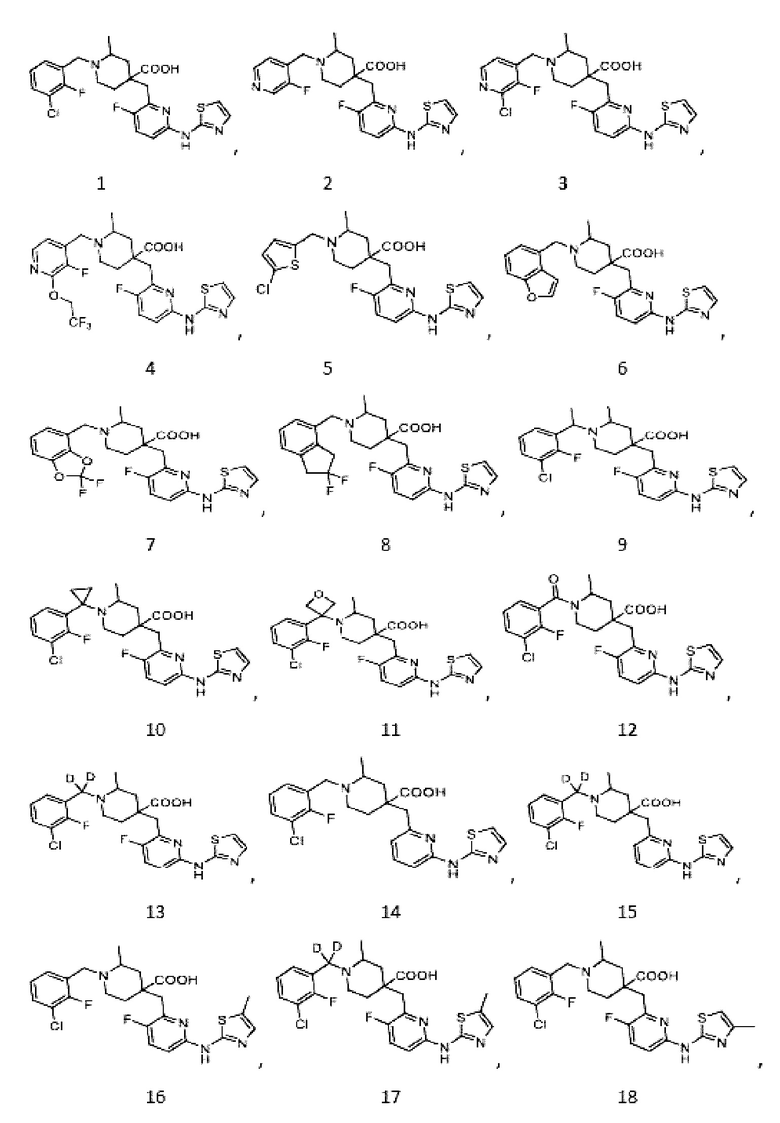
В другом предпочтительном варианте реализации соединение общей формулы (1) выбрано из соединений, указанных в таблице 1.
Таблица 1: Соединения по данному изобретению
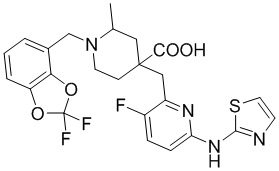
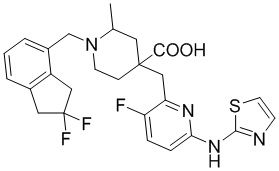
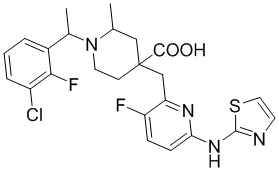
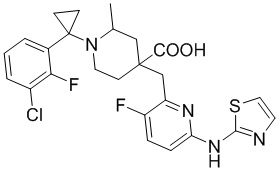
В другом варианте реализации данного изобретения предложена комбинированная фармацевтическая композиция, содержащая фармакологически приемлемое вспомогательное вещество или носитель и соединение формулы (1) по данному изобретению, его оптический изомер или его фармацевтически приемлемую соль в качестве активного ингредиента.
В другом варианте реализации данного изобретения предложено применение указанного соединения, его оптического изомера или его фармацевтически приемлемой соли в производстве препаратов для лечения заболеваний, связанных с аврора-киназой, в частности, для применения в противоопухолевых препаратах.
ПОДРОБНОЕ ОПИСАНИЕ ИЛЛЮСТРАТИВНЫХ ВАРИАНТОВ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ
В следующем разделе подробно описаны способы получения соединений общей формулы (1), но представленные конкретные способы не являются каким-либо ограничением данного изобретения.
Соединения формулы (1), описанные выше, могут быть синтезированы стандартными технологиями синтеза, общеизвестными технологиями или комбинацией способов, описанных в данном документе. Кроме того, растворители, температура и другие условия реакций, упомянутые в данном документе, могут варьироваться. Исходные материалы для синтеза соединений формулы (1) могут быть синтезированы или приобретены у коммерческих поставщиков, таких как, но не ограничиваясь ими, Aldrich Chemical Co. (Милуоки, штат Висконсин) или Sigma Chemical Co. (Сент-Луис, штат Миссури). Соединения, описанные в данном документе, и другие родственные соединения, содержащие другие заместители, могут быть синтезированы с использованием общеизвестных технологий и исходных материалов, включая те, которые описаны в публикациях March, ADVANCED ORGANIC CHEMISTRY, 4е изд. (Wiley 1992); Carey and Sundberg, ADVANCED ORGANIC CHEMISTRY, 4е изд., тома A и B (Plenum 2000, 2001), Green and Wuts, Protective Groups in Organic Synthesis, 3е изд., (Wiley 1999). …Общие способы получения предложенных соединений можно изменять, используя соответствующие реагенты и условия для внедрения различных групп в молекулярные формулы, представленные в данном документе.
В одном аспекте соединения, описанные в данном документе, получают общеизвестными способами. Однако условия процесса, такие как реагенты, растворители, основания, количество используемых соединений, температура реакций, время проведения реакций и т.п., не ограничены следующим пояснительным описанием. Соединения по данному изобретению также могут быть успешно получены с использованием необязательно комбинации различных синтетических способов, описанных в данном документе, или общеизвестных способов, такие комбинации могут быть без труда осуществлены специалистом в данной области техники. В другом аспекте данного изобретения также предложены способы получения соединений, представленных общей формулой (1), которые получены способом A или способом B:
Способ A включает следующие стадии: во-первых, исходные материалы A и B приводят во взаимодействие по реакции конденсации с получением соединения C в основных условиях с палладиевым катализатором и лигандом; во-вторых, соединение C подвергают снятию защитной Boc-группы в кислотных условиях с получением соединения D, затем соединение D подвергают взаимодействию с R1-L-X с получением соединения E, и, наконец, гидролизуют соединение E в кислотных или основных условиях с получением соединения формулы (1a).
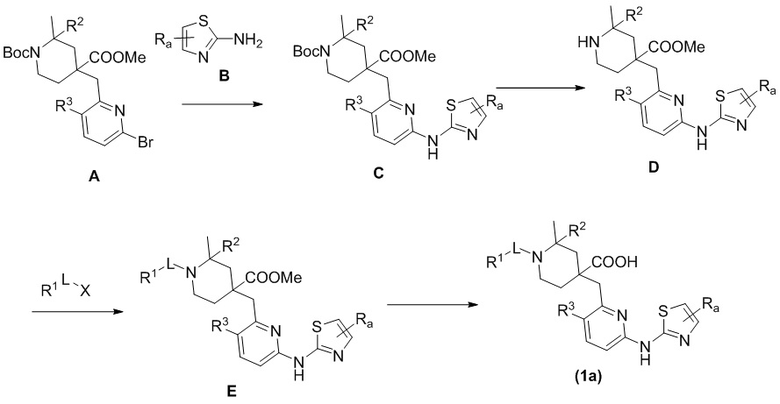
В вышеуказанной реакции R1, R2, R3, Ra и L имеют такие же значения, как указаны выше, X выбран из Br, Cl, OTf или OH.
Способ B включает следующие стадии: во-первых, приводят во взаимодействие исходные материалы F с B с получением соединения G, во-вторых, соединение G и H приводят во взаимодействие по реакции конденсации с получением соединения I в основных условиях с палладиевым катализатором и лигандом, и, наконец, гидролизуют соединение I в сильнокислотных условиях с получением соединения формулы (1b).
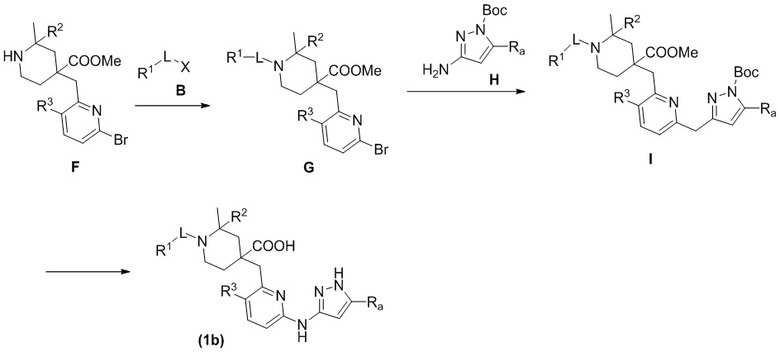
В вышеуказанной реакции R1, R2, R3, Ra и L имеют такие же значения, как указаны выше, X выбран из Br, Cl, OTf или OH.
ДОПОЛНИТЕЛЬНЫЕ ФОРМЫ СОЕДИНЕНИЙ
Термин «фармацевтически приемлемая соль» относится к такой форме соединения, которая не вызывает существенное раздражение у субъекта, которому она введена, и не подавляет биологическую активность и свойства указанного соединения. Соль соединения по данному изобретению относится к соли, обычно используемой в области органической химии, и может представлять собой, например, соль присоединения основания, если соединение содержит карбоксильную группу, или соль присоединения кислоты, если соединение содержит аминогруппу или основную гетероциклическую группу.
Пример соли присоединения основания включает соль щелочного металла, такую как соль натрия и соль калия, соль щелочноземельного металла, такую как соли кальция и магния; соль аммония, такую как соль триметиламина, соль триэтиламина, соль дициклогексиламина, соль этаноламина, соль диэтаноламина, соль триэтаноламина, соль прокаина, соль N,N'-дибензилэтилендиамина и т.п.
Пример соли присоединения кислоты включает соль неорганической кислоты, такую как гидрохлорид, сульфат, соль азотной кислоты и фосфат; соль органической кислоты, такую как ацетат, формиат, малеат, фумарат, цитрат, оксалат, аскорбат и другие соли органических кислот; сульфонат, такой как метансульфонат, бензолсульфонат, п-толуолсульфонат и т.п.
Следует понимать, что фармацевтически приемлемые соли включают формы присоединения растворителя или кристаллические формы, особенно сольваты или полиморфы. Сольваты содержат стехиометрическое или нестехиометрическое количество растворителей и селективно образуются во время кристаллизации с фармацевтически приемлемыми растворителями, такими как вода и этанол. Образуются гидраты, если растворителем является вода, или образуются алкоголяты, если растворителем является этанол. Сольваты соединения формулы (1) могут быть успешно получены или образованы в соответствии со способом, описанным в данном документе. Например, гидрат соединения формулы (1) без труда получают перекристаллизацией из смешанного растворителя, состоящего из воды/органического растворителя, и используемый органический растворитель включает, но не ограничивается ими, диоксан, тетрагидрофуран, этанол или метанол. Кроме того, соединения, упомянутые в данном документе, могут существовать в несольватированных или сольватированных формах. Таким образом, для описания соединений и способов, представленных в данном документе, сольватированные формы считаются эквивалентами несольватированных форм.
В других конкретных вариантах реализации соединения формулы (1) получают в различных формах, включая, но не ограничиваясь ими, аморфные, измельченные формы и формы наноразмерных частиц. Кроме того, соединения формулы (1) включают кристаллические формы и полиморфные формы. Полиморфные формы включают различные варианты кристаллической решетки соединений одного и того же элементного состава. Полиморфы обычно имеют разные профили рентгеновской дифракции, инфракрасные спектры, температуры плавления, плотность, твердость, кристаллическую форму, оптические и электрические свойства, стабильность и растворимость. Различные факторы, такие как растворитель, используемый для перекристаллизации, скорость кристаллизации и температура хранения, могут обусловливать преимущественное образование определенной кристаллической формы.
В другом аспекте соединения формулы (1) имеют один или более стереоцентров и, следовательно, существуют в форме рацемата, рацемической смеси, одного энантиомера, диастереомерного соединения и одного диастереомера. Асимметричные центры, которые могут существовать, зависят от свойств различных заместителей в молекуле. Каждый такой асимметричный центр в отдельности обусловливает образование двух оптических изомеров, и в объем данного изобретения входят все возможные оптические изомеры и диастереомерные смеси, а также чистые или частично чистые соединения. Данное изобретение включает все такие изомерные формы указанных соединений.
ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ
Соединения или композиции, описанные в данном документе, могут быть, в целом, использованы для ингибирования аврора-киназы, и, следовательно, могут быть использованы для лечения одного или более заболеваний, связанных с аврора-киназой. Таким образом, в некоторых вариантах реализации данного изобретения предложен способ лечения заболевания, опосредованного аврора-киназой, включающий стадию введения пациенту, нуждающемуся в этом, соединения по данному изобретению или его фармацевтически приемлемой композиции.
Раковые заболевания, которые можно лечить соединениями по данному изобретению, включают, но не ограничиваются ими, гематологические злокачественные заболевания (лейкоз, лимфома, миелома, включая множественную миелому, или миелодиспластический синдром) и солидные опухоли (такие раковые заболевания как рак простаты, молочной железы, легких, толстой кишки, поджелудочной железы, почек, яичников, мягких тканей, а также остеосаркома или стромальные опухоли).
СПОСОБ ВВЕДЕНИЯ
Соединение по данному изобретению и его фармацевтически приемлемые соли могут быть составлены в различные препараты, которые содержат соединение по данному изобретению или его фармацевтически приемлемые соли и фармацевтически приемлемые вспомогательные вещества или носители в безопасном и эффективном диапазоне количества. Среди них «безопасное и эффективное количество» означает, что указанное количество соединения может очевидно улучшать состояние, не вызывая серьезных побочных эффектов. Безопасную и эффективную дозу соединения определяют в соответствии с возрастом, заболеванием, курсом лечения и другими специфическими параметрами субъекта.
«Фармацевтически приемлемое вспомогательное вещество или носитель» относится к одному или более совместимых твердых или жидких наполнителей, или гелеобразных веществ, которые подходят для применения человеком и должны иметь достаточную чистоту и низкую токсичность. «Совместимость» в данном контексте означает, что каждый компонент в композиции может быть смешан с соединениями по данному изобретению и между собой, без существенного снижения эффективности указанного соединения. Примеры фармацевтически приемлемых вспомогательных веществ или носителей включают целлюлозу и ее производные (такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза натрия, ацетат целлюлозы и т.д.), желатин, тальк, твердые смазывающие вещества (такие как стеариновая кислота и стеарат магния), сульфат кальция, растительные масла (такие как соевое масло, кунжутное масло, арахисовое масло, оливковое масло и т.д.), полиолы (такие как пропиленгликоль, глицерин, маннит, сорбит и т.д.), эмульгаторы (такие как Tween), смачивающий агент (такой как додецилсульфат натрия), окрашивающий агент, вкусовой агент, стабилизатор, антиоксидант, консервант, апирогенная вода и т.д.
Соединения по данному изобретению можно вводить перорально, ректально, парентерально (внутривенно, внутримышечно или подкожно) или местно.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение смешано с по меньшей мере одним стандартным инертным вспомогательным веществом (или носителем), таким как цитрат натрия или фосфат дикальция, или с: …(a) наполнителем или компатибилизатором, таким как крахмал, лактоза, сахароза, глюкоза, маннит и кремниевая кислота; (b) связующими веществами, такими как гидроксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и гуммиарабик; (c) увлажнителем, таким как глицерин; (d) разрыхлителем, таким как агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые сложные силикаты и карбонат натрия; (e) агентами для замедления растворения, такими как парафин; (f) ускорителями абсорбции, такими как четвертичное аммониевое соединение; (g) смачивающими агентами, такими как цетиловый спирт и глицерилмоностеарат; (h) абсорбентом, таким как каолин; и (i) смазывающим веществом, таким как тальк, стеарат кальция, стеарат магния, твердый полиэтиленгликоль, лаурилсульфат натрия, или с их смесями. Лекарственная форма капсулы, таблетки и пилюли также может содержать буфер.
Твердые лекарственные формы, такие как таблетки, сахарные пилюли, капсулы, пилюли и гранулы, могут быть получены с использованием покрытий и оболочечных материалов, таких как оболочка, а также других материалов, известных в данной области техники. Они могут содержать замутнитель, и высвобождение активного соединения или соединение в такой композиции может высвобождаться замедленным образом в определенной части пищеварительного тракта. Примеры компонентов для заливки, которые могут быть использованы, представляют собой полимерные вещества и воски. При необходимости активное соединение также может образовывать микрокапсулы с одним или более из вышеуказанных вспомогательных веществ.
Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы или настойки. Помимо активного соединения, жидкая лекарственная форма может содержать инертные разбавители, обычно используемые в данной области техники, такие как вода или другие растворители, солюбилизаторы и эмульгаторы, например, этанол, изопропанол, этилкарбонат, этилацетат, пропиленгликоль, 1,3-бутандиол, диметилформамид и масла, в частности, хлопковое масло, арахисовое масло, масло зерновых зародышей, оливковое масло, касторовое масло, кунжутное масло или их смеси, и т.п.
Помимо указанных инертных разбавителей, композиция также может содержать адъюванты, такие как смачивающие агенты, эмульгаторы и суспендирующие агенты, подсластители, ароматизаторы и вкусовые добавки.
Помимо активных соединений, суспензия может содержать суспендирующие агенты, такие как этоксилированный изостеариловый спирт, полиоксиэтиленсорбит и сложные эфиры сорбита, микрокристаллическая целлюлоза, метоксид алюминия, агар или смеси указанных соединений, и т.п.
Композиция для парентерального введения может содержать физиологически приемлемый стерильный водный или неводный раствор, дисперсию, суспензию или эмульсию, а также стерильный порошок для разведения в стерильном растворе или дисперсии для инъекций. Пригодные водные и неводные носители, разбавители, растворители или вспомогательные вещества включают воду, этанол, полиолы и их соответствующие смеси.
Лекарственные формы соединений по данному изобретению для местного введения включают мази, порошки, пластыри, спреи и средства для ингаляции. Активный ингредиент смешивают в стерильных условиях с физиологически приемлемым носителем и любыми консервантами, буферами или газами-вытеснителями, которые могут быть необходимы.
Соединение по данному изобретению можно вводить отдельно или в комбинации с другими фармацевтически приемлемыми соединениями.
При использовании фармацевтической композиции используют безопасное и эффективное количество соединения по данному изобретению для введения млекопитающему (например, человеку), нуждающемуся в лечении, причем доза в момент введения является фармацевтически приемлемой эффективной дозой, и для человека с массой тела 60 кг суточная доза обычно составляет от 1 до 1000 мг, предпочтительно от 10 до 500 мг. Конечно, конкретная доза должна учитывать такие факторы как способ введения, состояние здоровья пациента и т.д., и такие факторы известны опытным врачам.
Вышеупомянутые признаки, упомянутые в данном изобретении, или признаки, упомянутые в вариантах реализации, можно комбинировать случайным образом. Все признаки, описанные в данном документе, можно использовать в любой форме композиции, и различные признаки, описанные в данном описании, можно менять на любой альтернативный признак, который служит для той же, эквивалентной или подобной цели. Таким образом, если не указано иное, описанные признаки являются лишь обобщенными примерами эквивалентных или подобных признаков.
Различные конкретные аспекты, признаки и преимущества описанных выше соединений, способов и фармацевтических композиций, подробно изложены в следующем описании. Следует понимать, что следующее подробное описание и примеры описывают конкретные варианты реализации лишь для справки. Специалисты в данной области техники после прочтения описания данного изобретения могут сделать различные изменения или модификации, и такие эквиваленты входят в объем данной заявки.
Во всех примерах 1H-ЯМР записывали на ЯМР спектрометре Varian Mercury 400, и химические сдвиги выражены как д (м.д.); силикагель для разделения имеет размер частиц 200-300 меш при отсутствии специального указания, а соотношение элюентов представляет собой объемное соотношение.
Ниже представлены сокращения, использованные в данном изобретении: ACN означает ацетонитрил; Ar означает аргон; CBr4 означает тетрабромид углерода; CDCl3 означает дейтерированный хлороформ; CD3OD означает дейтерированный метанол; ДХМ означает дихлорметан; DIPEA означает диизопропилэтиламин; Diox означает 1,4-диоксан; ДМФА означает диметилформамид; ДМСО означает диметилсульфоксид; EA означает этилацетат; EDCl означает гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида; ч. означает час; HOBt означает 1-гидроксибензотриазол; K2CO3 означает карбонат калия; KI означает йодид калия; K3PO4 означает фосфат калия; ЖХ-МС означает жидкостный масс-спектр; LiAlD4 означает дейтерид лития-алюминия; LiOH означает гидроксид лития; мл означает миллилитр; MeOH означает метанол; мин означает минуты; МС означает масс-спектр; ЯМР означает ядерный магнитный резонанс; Pd2(dba)3 означает трис(дибензилиденацетон)дипалладий; PE означает петролейный эфир; PPh3 означает трифенилфосфин; Tf2O означает трифторметансульфоновый ангидрид; Xantphos означает 9,9-диметил-4,5-бис(дифенилфосфино)ксантен.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
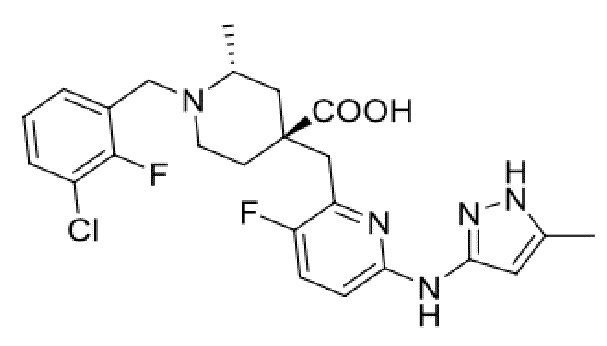
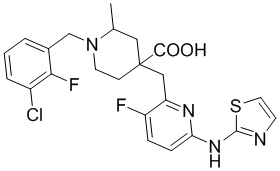
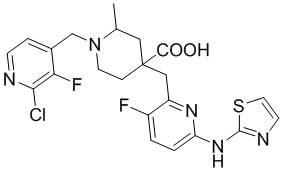
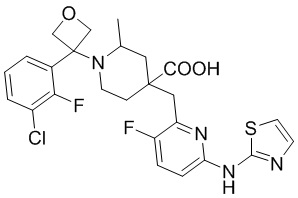
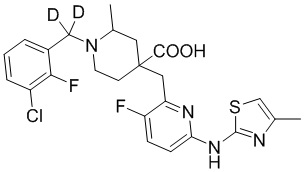
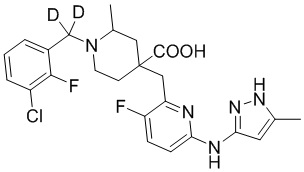
Пример 1. Синтез 1-(3-хлор-2-фторбензил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 1)
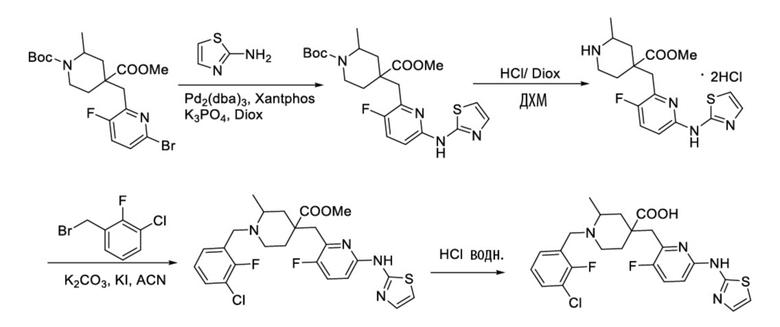
1-(трет-Бутил)-4-метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-1,4-дикарбоксилат
1-(трет-Бутил)-4-метил-4-((6-бром-3-фторпиридин-2-ил)метил)-2-метилпиперидин-1,4-дикарбоксилат (5 г, 11,23 ммоль, его синтезировали со ссылкой на способ, описанный в патенте WO2016077161), тиазол-2-амин (956 мг, 9,55 ммоль), безводный фосфат калия (6 г, 28,08 ммоль), Xantphos (650 мг, 1,123 ммоль) и диоксан (100 мл) добавляли в колбу объемом 250 мл, после замены воздуха на Ar добавляли Pd2(dba)3 (514 мг, 0,562 ммоль), нагревали до температуры кипения и проводили реакцию еще 5 часов под защитным слоем Ar. После завершения реакции, установленного по ЖХ-МС, концентрировали смесь при пониженном давлении и дополнительно очищали колоночной хроматографией (ДХМ/MeOH = от 50/0 до 50/1) с получением требуемого продукта в виде желтого твердого вещества (4,0 г, выход 89%), ИЭР-МС m/z: 465,2 [M+H]+.
Метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилат
1-(трет-Бутил)-4-метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-1,4-дикарбоксилат (4 г, 8,61 ммоль) добавляли в колбу объемом 100 мл, добавляли также ДХМ (20 мл) и HCl/диоксан (22 мл, 4 M, 88 ммоль), затем перемешивали при комнатной температуре в течение 20 часов. После завершения реакции, установленного по ЖХ-МС, концентрировали смесь, к остатку добавляли EA (30 мл) и перемешивали еще 30 минут, фильтровали и сушили с получением требуемого соединения в виде желтого твердого вещества (4,1 г, выход 100%), ИЭР-МС m/z: 365,2 [M+H]+.
Метил-1-(3-хлор-2-фторбензил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилат
Метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилат (800 мг, 1,83 ммоль), 1-(бромметил)-3-хлор-2-фторбензол (500 мг, 2,19 ммоль), K2CO3 (1,264 г, 9,15 ммоль), KI (20 мг) и ACN (20 мл) добавляли в колбу объемом 100 мл, затем оставляли смесь взаимодействовать при комнатной температуре на 2 часа. После завершения реакции, установленного по ЖХ-МС, к выпавшему в осадок твердому веществу добавляли воду (100 мл), затем фильтровали. Осадок на фильтре дважды промывали водой (20 мл*2) и затем суспендировали с PE (50 мл). После фильтрования дважды промывали осадок на фильтре PE (20 мл*2) и сушили с получением неочищенного продукта, метил-1-(3-хлор-2-фторбензил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилата (935 мг, выход 100% ), который использовали на следующей стадии без дополнительной очистки. ИЭР-МС m/z: 510,2 [M+H]+.
1-(3-Хлор-2-фторбензил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновая кислота
Метил-1-(3-хлор-2-фторбензил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилат (935 мг, 1,83 ммоль) добавляли в колбу объемом 100 мл, затем добавляли воду (15 мл) и конц. HCl (15 мл), нагревали до температуры кипения и проводили реакцию еще 5 часов. После завершения реакции, установленного по ЖХ-МС, концентрировали смесь и суспендировали остаток с ACN (30 мл) при комнатной температуре. После фильтрования промывали осадок на фильтре ACN (5 мл*2) и сушили с получением требуемого желтого порошка (818 мг, выход 90%).
1H ЯМР (400 МГц, ДМСО-d6) δ: 11,68 (с, 1H), 10,59 (с, 1H), 7,76 (т, J = 7,2 Гц, 1H), 7,73-7,68 (м, 1H), 7,61 (т, J = 9,2 Гц, 1H), 7,43 (д, J = 3,7 Гц, 1H), 7,34 (т, J = 7,9 Гц, 1H), 7,01 (к, J = 3,9 Гц, 2H), 4,72 (д, J = 13,3 Гц, 1H), 4,36 (дд, J = 13,6, 8,3 Гц, 1H), 3,88 (с, 2H), 3,26-3,21 (м, 2H), 3,09 (д, J = 12,9 Гц, 1H), 2,16-1,95 (м, 4H), 1,50 (д, J = 6,0 Гц, 3H); ИЭР-МС m/z: 493,1 [M+H]+.
Посредством синтеза из различных хиральных исходных материалов или посредством разделения хиральной СЖХ могут быть получены четыре различных оптических изомера соединения 1, и их структуры представлены ниже:
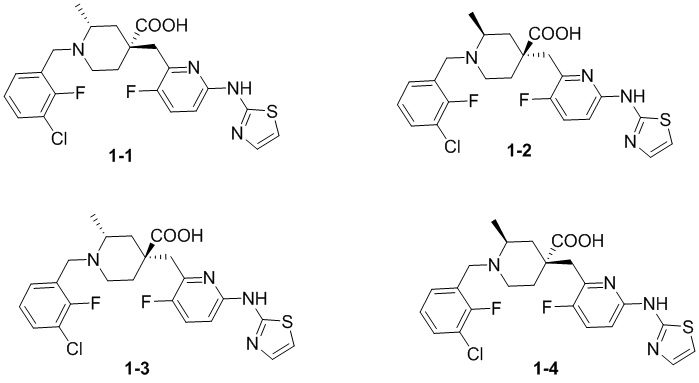
Соединения 1-1, 1-2, 1-3 и 1-4 обозначали следующим образом:
1-1: (2R,4R)-1-(3-хлор-2-фторбензил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновая кислота;
1-2: (2S,4S)-1-(3-хлор-2-фторбензил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновая кислота;
1-3: (2R,4S)-1-(3-хлор-2-фторбензил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновая кислота;
1-4: (2S,4R)-1-(3-хлор-2-фторбензил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновая кислота.
Другие соединения, описанные в данной заявке, также могут быть использованы для разделения соответствующих оптических изомеров таким же способом.
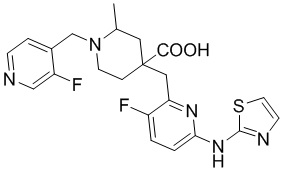
Пример 2. Синтез 4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-1-((3-фторпиридин-4-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 2)
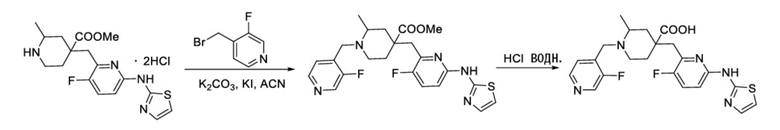
Используя гидрохлорид метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилата и 4-(бромметил)-3-фторпиридин в качестве исходных материалов, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, ДМСО-d6) δ: 12,45 (с, 1H), 11,45 (с, 1H), 8,74 (д, J = 1,4 Гц, 1H), 8,54 (д, J = 4,9 Гц, 1H), 8,03 (т, J = 5,7 Гц, 1H), 7,68 (т, J = 9,1 Гц, 1H), 7,50 (д, J = 4,0 Гц, 1H), 7,13 (к, J = 3,7, 3,3 Гц, 2H), 4,71 (д, J = 13,3 Гц, 1H), 4,43 (дд, J = 13,5, 7,9 Гц, 1H), 3,93 (с, 1H), 3,39 (дт, J = 14,8, 9,1 Гц, 1H), 3,30-3,23 (м, 2H), 3,09 (д, J = 13,1 Гц, 1H), 2,08 (т, J = 15,2 Гц, 3H), 1,92 (д, J = 15,2 Гц, 1H), 1,51 (д, J = 6,2 Гц, 3H); ИЭР-МС m/z: 460,2 [M+H]+.
Пример 3. Синтез 1-((2-хлор-3-фторпиридин-4-ил)метил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 3)
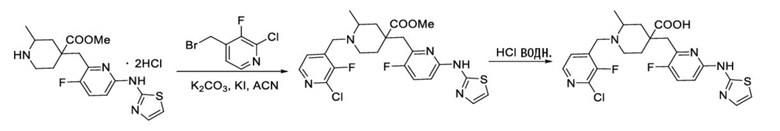
Используя гидрохлорид метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилата и 4-(бромметил)-2-хлор-3-фторпиридин в качестве исходных материалов, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, ДМСО-d6) δ: 11,22 (с, 1H), 9,82 (с, 1H), 8,39 (с, 1H), 7,68 (с, 1H), 7,57 (т, J = 9,1 Гц, 1H), 7,37 (д, J = 3,6 Гц, 1H), 6,98-6,89 (м, 2H), 4,76 (м, 1H), 4,41 (м, 1H), 3,89 (с, 2H), 3,23 (м, 3H), 2,13 (м, 2H), 1,82 (м, 2H), 1,36 (д, J = 6,4 Гц, 3H); ИЭР-МС m/z: 494,1 [M+H]+.
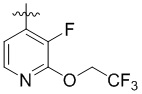
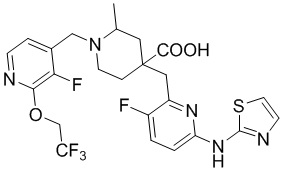
Пример 4. Синтез 1-((3-фтор-2-(2,2,2-трифторэтокси)пиридин-4-ил)метил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 4)

Используя гидрохлорид метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилата и 4-(бромметил)-3-фтор-2-(2,2,2-трифторэтокси)пиридин в качестве исходных материалов, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, метанол-d4) δ: 7,64 (дд, J = 8,3, 7,4 Гц, 1H), 7,32-7,27 (м, 2H), 7,23 (т, J = 7,9 Гц, 1H), 7,17 (дд, J = 7,8, 1,5 Гц, 1H), 7,02 (д, J = 7,2 Гц, 1H), 6,90- 6,86 (м, 2H), 3,84-3,77 (м, 2H), 3,73 (с, 2H), 3,44 (т, J = 5,1 Гц, 2H), 2,69 (т, J = 5,1 Гц, 2H), 2,64-2,58 (м, 3H), 1,53 (д, J = 6,0 Гц, 3H); ИЭР-МС m/z: 558,2 [M+H]+.
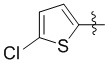
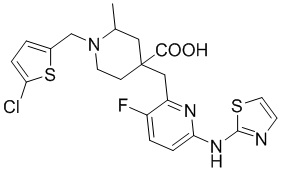
Пример 5. Синтез 1-((5-хлортиофен-2-ил)метил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 5)
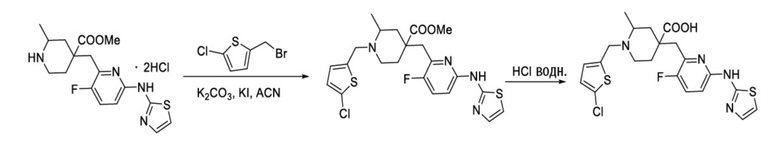
Используя гидрохлорид метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилата и 2-(бромметил)-5-хлортиофен в качестве исходных материалов, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, метанол-d4) δ: 7,69 (т, J = 8,7 Гц, 1H), 7,58 (с, 1H), 7,25 (к, J = 9,5, 7,5 Гц, 2H), 7,15 (дд, J = 9,0, 3,0 Гц, 1H), 7,00 (дд, J = 30,0, 3,5 Гц, 1H), 4,62-4,37 (м, 2H), 3,80 (д, J = 26,4 Гц, 2H), 3,39 (д, J = 6,4 Гц, 2H), 3,21-3,11 (м, 1H), 2,33-1,98 (м, 4H), 1,45 (д, J = 6,0 Гц, 3H); ИЭР-МС m/z: 481,1 [M+H]+.
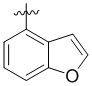
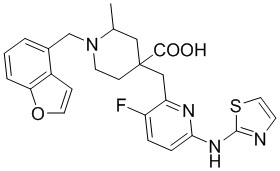
Пример 6. Синтез 1-(бензофуран-4-илметил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 6)
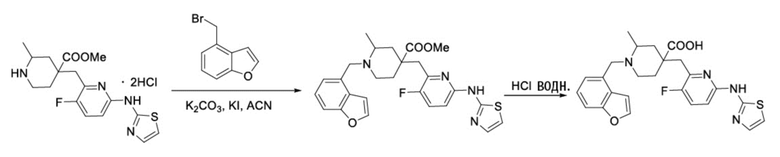
Используя гидрохлорид метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилата и 4-(бромметил)бензофуран в качестве исходных материалов, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, ДМСО-d6) δ: 7,62 (д, J = 7,2 Гц, 1H), 7,50 (т, J = 9,1 Гц, 1H), 7,32 (д, J = 3,4 Гц, 1H), 7,24 (дд, J = 7,1, 2,1 Гц, 1H), 7,19-7,11 (м, 2H), 6,89 (д, J = 5,1 Гц, 2H), 6,65 (д, J = 7,2 Гц, 1), 3,84 (д, J = 14,1 Гц, 1H), 3,69 (д, J = 14,2 Гц, 1H), 3,05 (с, 2H), 2,77-2,73 (м, 1H), 2,65-2,57 (м, 1H), 2,49- 2,42 (м, 1H), 1,84-1,75 (м, 1H), 1,72 - 1,63 (м, 2H), 1,57 (т, J = 11,9 Гц, 1H), 1,16 (д, J = 6,0 Гц, 3H); ИЭР-МС m/z: 481,2 [M+H]+.
Пример 7. Синтез 1-((2,2-дифторбензо[d][1,3]диоксол-4-ил)метил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 7)
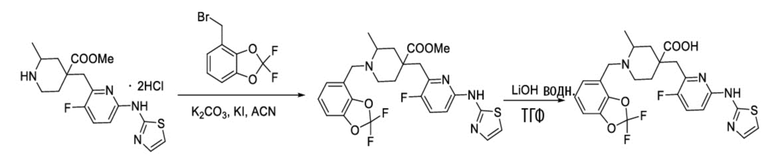
Используя гидрохлорид метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилата и 4-(бромметил)-2,2-дифторбензо[d][1,3]диоксол в качестве исходных материалов, получали промежуточный метил-1-((2,2-дифторбензо[d][1,3]диоксол-4-ил)метил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилат.
Вышеуказанное промежуточное соединение добавляли в колбу объемом 100 мл, затем добавляли ТГФ (10 мл), H2O (5 мл) и LiOH⋅H2O (79 мг, 1,88 ммоль), затем нагревали до 60°С и перемешивали в течение 5 часов под защитным слоем Ar. После завершения реакции, установленного по ЖХ-МС, концентрировали смесь до объема около 7,5 мл, остаток очищали обращенно-фазовой флэш-хроматографией с получением требуемого соединения (60 мг, выход 62%).
1H ЯМР (400 МГц, ДМСО-d6) δ: 7,50 (т, J = 9,1 Гц, 1H), 7,32 (д, J = 3,4 Гц, 1H), 7,24 (дд, J = 7,1, 2,1 Гц, 1H), 7,19-7,11 (м, 2H), 6,89 (д, J = 5,1 Гц, 2H), 3,94 (д, J = 14,1 Гц, 1H), 3,39 (д, J = 14,2 Гц, 1H), 3,05 (с, 2H), 2,75-2,71 (м, 1H), 2,61-2,52 (м, 1H), 2,45-2,40 (м, 1H), 1,83-1,79 (м, 1H), 1,75-1,62 (м, 2H), 1,54 (т, J = 11,9 Гц, 1H), 1,06 (д, J = 6,0 Гц, 3H); ИЭР-МС m/z: 521,2 [M+H]+.
Пример 8. Синтез 1-((2,2-дифтор-2,3-дигидро-1H-инден-4-ил)метил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 8)
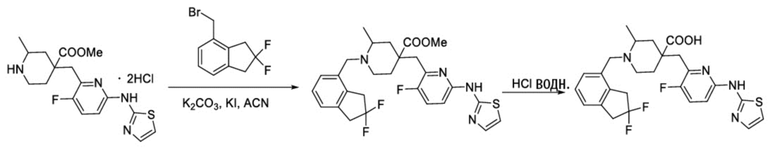
Используя гидрохлорид метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилата и 4-(бромметил)-2,2-дифтор-2,3-дигидро-1H-инден в качестве исходных материалов, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, ДМСО-d6) δ: 7,52 (т, J = 9,1 Гц, 1H), 7,30 (д, J = 3,4 Гц, 1H), 7,23 (дд, J = 7,1, 2,1 Гц, 1H), 7,19-7,11 (м, 2H), 6,89 (д, J = 5,0 Гц, 2H), 5,34-5,21 (м, 4H), 3,94 (д, J = 14,1 Гц, 1H), 3,39 (д, J = 14,2 Гц, 1H), 3,05 (с, 2H), 2,75-2,71 (м, 1H), 2,61-2,50 (м, 1H), 2,45-2,40 (м, 1H), 1,83-1,79 (м, 1H), 1,75-1,65 (м, 2H), 1,54 (т, J = 11,9 Гц, 1H), 1,07 (д, J = 6,0 Гц, 3H); ИЭР-МС m/z: 517,2 [M+H]+.
Пример 9. Синтез 1-(1-(3-хлор-2-фторфенил)этил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 9)
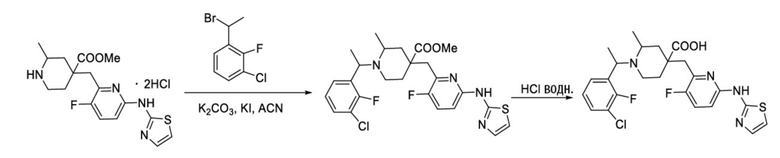
Используя гидрохлорид метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилата и 1-(1-бромэтил)-3-хлор-2-фторбензол в качестве исходных материалов, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, ДМСО-d6) δ: 12,27 (с, 1H), 11,15 (с, 1H), 7,55 (д, J = 8,1 Гц, 1H), 7,49-7,34 (м, 2H), 7,18 (т, J = 7,8 Гц, 1H), 7,02 (с, 1H), 6,86 (д, J = 8,3 Гц, 1H), 6,67 (д, J = 7,3 Гц, 1H), 4,41 (м, 1H), 3,11 (д, J = 16,6 Гц, 2H), 3,02 (м, 2H), 2,85 (м, 1H), 1,76-1,52 (м, 4H), 1,30 (д, J = 6,7 Гц, 3H), 1,06 (к, J = 7,1, 6,4 Гц, 3H); ИЭР-МС m/z: 507,2 [M+H]+.
Пример 10. Синтез 1-(1-(3-хлор-2-фторфенил)циклопропил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 10)
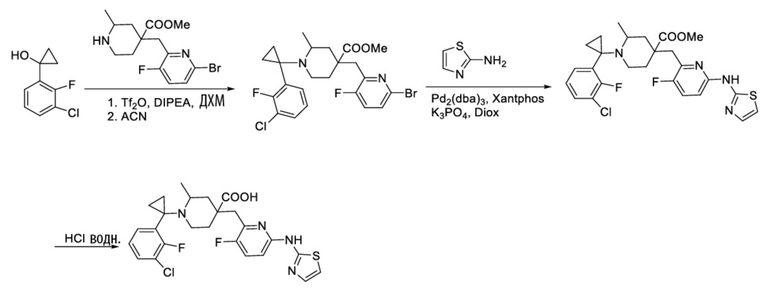
Метил-4-((6-бром-3-фторпиридин-2-ил)метил)-1-(1-(3-хлор-2-фторфенил)-
циклопропил)-2-метилпиперидин-4-карбоксилат
1-(3-Хлор-2-фторфенил)циклопропан-1-ол (400 мг, 2,145 ммоль) добавляли в колбу объемом 100 мл, затем добавляли сухой ДХМ (10 мл) и DIPEA (692 мг, 5,362 ммоль), охлаждали до -45°С под защитным слоем Ar, затем добавляли Tf2O (726 мг, 2,574 ммоль, в 10 мл ДХМ), перемешивали смесь в течение 2 часов при температуре от -50 до -40°С. Добавляли метил-4-((6-бром-3-фторпиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилат (444 мг, 1,287 ммоль, в 10 мл CH3CN), повышали температуру до комнатной температуры и перемешивали в течение 2 часов. После завершения реакции, установленного по ЖХ-МС, смесь гасили водой (20 мл), отделяли органическую фазу, экстрагировали ДХМ (20 мл), объединяли органическую фазу и концентрировали, остаток очищали колоночной хроматографией с получением требуемого соединения (284 мг, выход 43%), ИЭР-МС m/z: 533,2 [M+H]+.
1-(1-(3-Хлор-2-фторфенил)циклопропил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновая кислота
Используя метил-4-((6-бром-3-фторпиридин-2-ил)метил)-1-(1-(3-хлор-2-фторфенил)циклопропил)-2-метилпиперидин-4-карбоксилат в качестве исходного материала, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, ДМСО-d6) δ: 12,11 (с, 1H), 11,02 (с, 1H), 7,57 (д, J = 8,1 Гц, 1H), 7,43-7,33 (м, 2H), 7,15 (т, J = 7,8 Гц, 1H), 7,02 (с, 1H), 6,84 (д, J = 8,3 Гц, 1H), 6,65 (д, J = 7,3 Гц, 1H), 3,41 (д, J = 16,6 Гц, 2H), 3,02 (м, 3H), 2,23-1,89 (м, 4H), 1,30 (д, J = 6,7 Гц, 3H), 0,75-0,62 (м, 4H); ИЭР-МС m/z: 519,2 [M+H]+.
Пример 11. Синтез 1-(3-(3-хлор-2-фторфенил)оксетан-3-ил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 11)
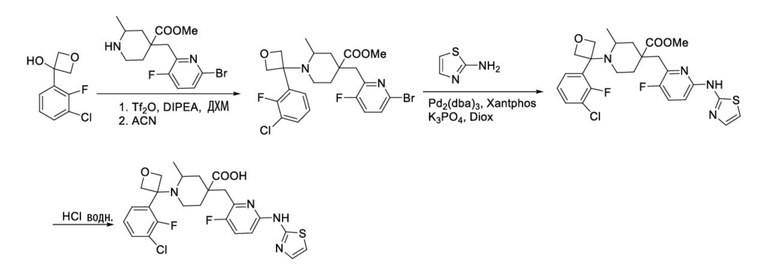
Используя метил-4-((6-бром-3-фторпиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилат и 3-(3-хлор-2-фторфенил)оксетан-3-ол в качестве промежуточных соединений, получали промежуточный метил-4-((6-бром-3-фторпиридин-2-ил)метил)-1-(3-(3-хлор-2-фторфенил)оксетан-3-ил)-2-метилпиперидин-4-карбоксилат таким же способом синтеза, как описан в примере 10.
Используя вышеуказанное промежуточное соединение и тиазол-2-амин в качестве исходных материалов, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, CD3OD) δ: 7,46 (м, 2H), 7,35-7,16 (м, 3H), 6,93 (дт, J = 8,9, 3,3 Гц, 1H), 6,87 (т, J = 4,1 Гц, 1H), 4,77 (с, 1H), 4,70-4,47 (м, 2H), 4,31 (д, J = 12,2 Гц, 1H), 4,14 (д, J = 8,0 Гц, 1H), 3,98 (ab, J = 29,9, 12,4 Гц, 1H), 3,67 (т, J = 11,3 Гц, 1H), 3,17-2,96 (м, 2H), 2,50-2,13 (м, 3H), 2,08-1,95 (м, 1H), 1,89 (д, J = 12,4 Гц, 1H), 1,41 (д, J = 6,9 Гц, 3H); ИЭР-МС m/z: 535,2 [M+H]+.
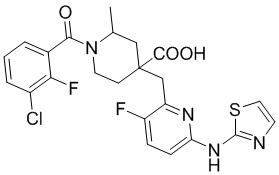
Пример 12. Синтез 1-(3-хлор-2-фторбензоил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 12)
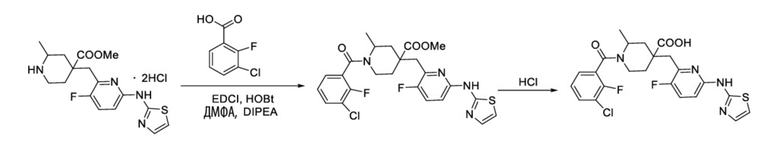
3-Хлор-2-фторбензойную кислоту (262 мг, 1,50 ммоль), ДМФА (20 мл), EDCI (431 мг, 2,25 ммоль), HOBt (304 мг, 2,25 ммоль) и DIPEA (970 мг, 7,52 ммоль) добавляли в колбу объемом 100 мл, перемешивали смесь при комнатной температуре в течение 30 минут под защитным слоем Ar, добавляли гидрохлорид метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилата (437 мг, 1,0 ммоль), затем перемешивали при комнатной температуре в течение 20 часов. После завершения реакции, установленного по ЖХ-МС, гасили смесь водой (40 мл), экстрагировали EA (50 мл* 2), объединяли с органической фазой и промывали насыщенным раствором NaCl, концентрировали, остаток очищали колоночной хроматографией с получением требуемого промежуточного соединения (365 мг, выход 70%).
Используя вышеуказанное промежуточное соединение в качестве исходного материала, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, CD3OD) δ: 7,68 (т, J = 9,0 Гц, 1H), 7,62-7,52 (м, 2H), 7,43 (дт, J = 12,0, 7,9 Гц, 1H), 7,27 (д, J = 7,5 Гц, 1H), 7,21 (д, J = 4,3 Гц, 1H), 7,12 (дд, J = 8,9, 3,0 Гц, 1H), 3,73 (с, 2H), 3,22-3,12 (м, 3H), 2,13-1,88 (м, 4H), 1,45 (д, J = 6,0 Гц, 3H); ИЭР-МС m/z: 475,2 [M+H]+.
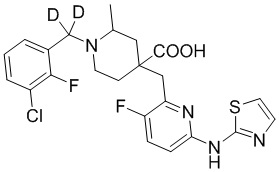
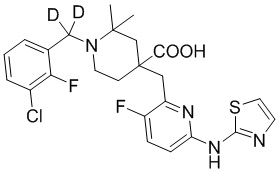
Пример 13. Синтез 1-((3-хлор-2-фторфенил)метил-d2)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 13)
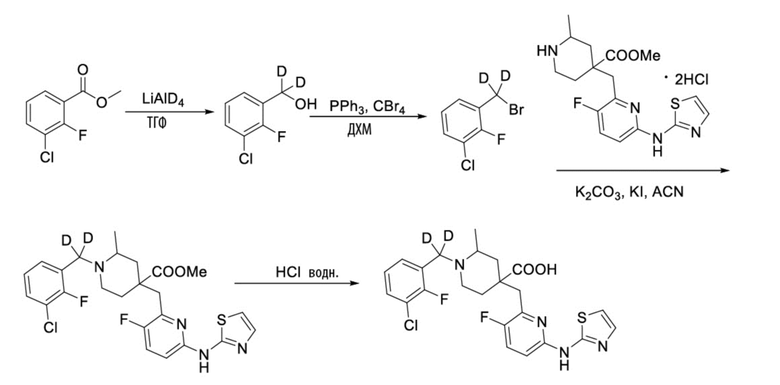
1-(Бромметил-d2)-3-хлор-2-фторбензол
Метил-3-хлор-2-фторбензоат (658 мг, 3,49 ммоль) и ТГФ (сухой, 10 мл) добавляли в колбу объемом 100 мл, по каплям добавляли LiAlD4 (146 мг, 3,49 ммоль) при охлаждении на ледяной бане, дополнительно перемешивали около 0,5 часа. После завершения реакции, по данным ТСХ (PE/EA = 10/1), смесь гасили водой (20 мл) при охлаждении на ледяной бане, добавляли насыщенный раствор NaCl (20 мл), экстрагировали EA (20 мл*2), объединяли органическую фазу и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного промежуточного (3-хлор-2-фторфенил)метан-d2-ола, который использовали на следующей стадии без дополнительной очистки.
(3-Хлор-2-фторфенил)метан-d2-ол, ДХМ (20 мл) и CBr4 (2 г, 6,03 ммоль) добавляли в колбу объемом 100 мл, по каплям добавляли PPh3 (1,371 г, 5,23 ммоль), перемешивали при комнатной температуре около 1 часа. После завершения реакции, установленного по ТСХ (PE/EA = 10/1), концентрировали смесь и очищали остаток колоночной хроматографией (PE, 800 мл) с получением 1-(бромметил-d2)-3-хлор-2-фторбензола в виде бесцветной жидкости (921 мг, выход 100%).
1H ЯМР (400 МГц, CDCl3) δ 7,34 (ддд, J = 8,3, 6,9, 1,7 Гц, 1H), 7,27 (ддд, J = 7,9, 6,5, 1,7 Гц, 1H), 7,05 (тд, J = 7,9, 1,2 Гц, 1H).
Метил-1-((3-хлор-2-фторфенил)метил-d2)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилат
Гидрохлорид метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилата (800 мг, 1,83 ммоль), 1-(бромметил-d2)-3-хлор-2-фторбензол (495 мг, 2,19 ммоль), K2CO3 (1,264 г, 9,15 ммоль), KI (20 мг) и ACN (20 мл) добавляли в колбу объемом 100 мл, перемешивали при комнатной температуре в течение около 2 часов. После завершения реакции, установленного по ТСХ, добавляли воду (100 мл) для осаждения твердого вещества, с последующим фильтрованием. Осадок на фильтре дважды промывали водой (20 мл*2) и затем суспендировали с PE (50 мл). После фильтрования промывали осадок на фильтре PE (20 мл*2) и сушили с получением неочищенного продукта, метил-1-((3-хлор-2-фторфенил)метил-d2)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилата (931 мг, выход 100%), который использовали на следующей стадии без дополнительной очистки. ИЭР-МС m/z: 509,2 [M+H]+.
1-((3-Хлор-2-фторфенил)метил-d2)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновая кислота
Метил-1-((3-хлор-2-фторфенил)метил-d2)-4-((3-фтор-6-(тиазол-2-иламино)-пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилат (931 мг, 1,83 ммоль) добавляли в колбу объемом 100 мл, затем добавляли воду (15 мл) и конц. HCl (15 мл), нагревали до 105°С при температуре кипения и дополнительно проводили реакцию в течение 5 часов. После завершения реакции, установленного по ЖХ-МС, концентрировали смесь и суспендировали остаток с ACN (30 мл) при комнатной температуре. После фильтрования промывали осадок на фильтре ACN (5 мл*2) и сушили с получением требуемого соединения в виде желтого порошка (815 мг, выход 90%).
1H ЯМР (400 МГц, CD3OD) δ: 7,73 (т, J = 8,7 Гц, 1H), 7,70-7,55 (м, 3H), 7,36-7,24 (м, 2H), 7,20 (дд, J = 8,8, 2,9 Гц, 1H), 3,96 (с, 2H), 3,57-3,44 (м, 2H), 3,36 (с, 1H), 2,41-1,96 (м, 4H), 1,53 (м, 3H); ИЭР-МС m/z: 495,1 [M+H]+.
Посредством синтеза из различных хиральных исходных материалов или посредством разделения хиральной СЖХ могут быть получены четыре различных оптических изомера соединения 13, и их структуры представлены ниже:
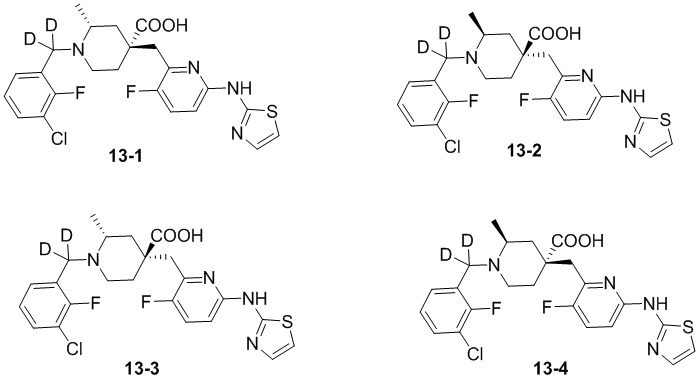
Соединения 13-1, 13-2, 13-3 и 13-4 обозначали следующим образом:
13-1: (2R,4R)-1-((3-хлор-2-фторфенил)метил-d2)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновая кислота;
13-2: (2S,4S)-1-((3-хлор-2-фторфенил)метил-d2)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновая кислота;
13-3: (2R,4S)-1-((3-хлор-2-фторфенил)метил-d2)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновая кислота;
13-4: (2S,4R)-1-((3-хлор-2-фторфенил)метил-d2)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновая кислота.
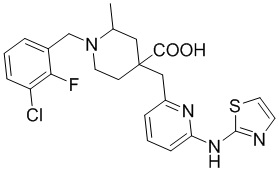
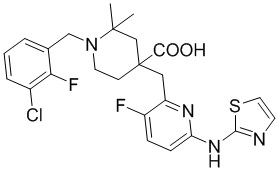
Пример 14. Синтез 1-(3-хлор-2-фторбензил)-2-метил-4-((6-(тиазол-2-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновой кислоты (соединение 14)
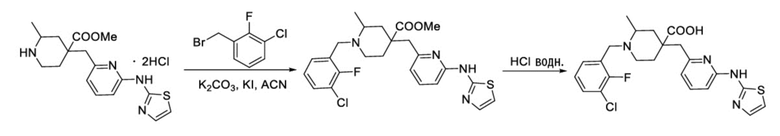
Используя гидрохлорид метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилата и 1-(бромметил)-3-хлор-2-фторбензол в качестве исходных материалов, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, ДМСО-d6) δ: 11,88 (с, 1H), 10,79 (с, 1H), 7,74 (т, J = 7,2 Гц, 1H), 7,72-7,63 (м, 2H), 7,55 (т, J = 9,2 Гц, 1H), 7,41 (д, J = 3,7 Гц, 1H), 7,32 (т, J = 7,9 Гц, 1H), 7, 06 (к, J = 3,9 Гц, 2H), 4,73 (д, J = 13,3 Гц, 1H), 4,38 (дд, J = 13,6, 8,3 Гц, 1H), 3,68 (с, 2H), 3,26-3,24 (м, 2H), 3,02 (д, J = 12,9 Гц, 1H), 2,15-1,91 (м, 4H), 1,52 (д, J = 6,0 Гц, 3H); ИЭР-МС m/z: 507,1 [M+H]+.
Пример 15. Синтез 1-((3-хлор-2-фторфенил)метил-d2)-2-метил-4-((6-(тиазол-2-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновой кислоты (соединение 15)
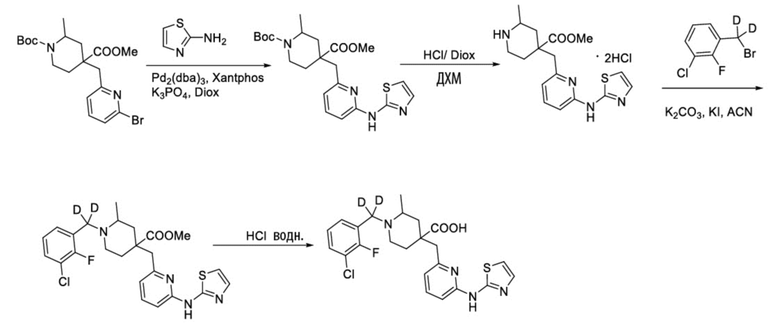
Используя метил-1-(трет-бутил)-4-метил-4-((6-бром-3-фторпиридин-2-ил)метил)-2-метилпиперидин-1,4-дикарбоксилат и тиазол-2-амин в качестве исходных материалов, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, CD3OD) δ: 7,78 (т, J = 8,7 Гц, 1H), 7,70-7,55 (м, 3H), 7,36-7,24 (м, 3H), 7,23 (дд, J = 8,8, 2,9 Гц, 1H), 3,92 (с, 2H), 3,57-3,44 (м, 2H), 3,36 (м, 1H), 2,41-1,96 (м, 4H), 1,55 (м, 3H); ИЭР-МС m/z: 477,1 [M+H]+.
Пример 16. Синтез 1-(3-хлор-2-фторбензил)-4-((3-фтор-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 16)
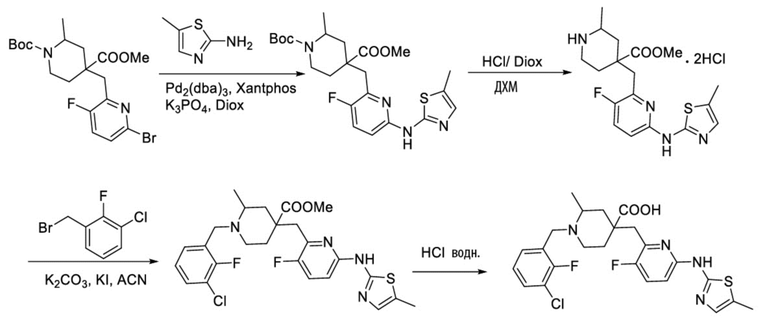
Используя 1-(трет-бутил)-4-метил-4-((6-бром-3-фторпиридин-2-ил)метил)-2-метилпиперидин-1,4-дикарбоксилат и 5-метилтиазол-2-амин в качестве исходных материалов, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, ДМСО-d6) δ: 11,66 (с, 1H), 10,59 (с, 1H), 7,76 (т, J = 7,2 Гц, 1H), 7,72-7,66 (м, 2H), 7,55 (т, J = 9,2 Гц, 1H), 7,34 (т, J = 7,9 Гц, 1H), 7,01 (м, 1H), 4,72 (д, J = 13,3 Гц, 1H), 4,36 (дд, J = 13,6, 8,3 Гц, 1H), 3,88 (с, 2H), 3,26-3,21 (м, 2H), 3,09 (д, J = 12,9 Гц, 1H), 2,27 (с, 3H), 2,16-1,95 (м, 4H), 1,50 (д, J = 6,0 Гц, 3H); ИЭР-МС m/z: 508,1 [M+H]+.
Пример 17. Синтез 1-((3-хлор-2-фторфенил)метил-d2)-4-((3-фтор-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 17)
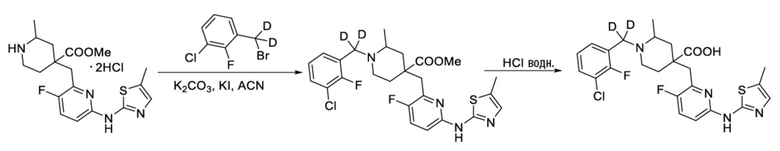
Используя гидрохлорид метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилата и 1-(бромметил-d2)-3-хлор-2-фторбензол в качестве исходных материалов, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, CD3OD) δ: 7,75 (т, J = 8,7 Гц, 1H), 7,72-7,56 (м, 3H), 7,28 (с, 1H), 7,20 (дд, J = 8,8, 2,9 Гц, 1H), 3,96 (с, 2H), 3,57-3,44 (м, 2H), 3,36 (с, 1H), 2,57 (с, 3H), 2,41-1,96 (м, 4H), 1,53 (м, 3H); ИЭР-МС m/z: 510,0 [M+H]+.
Пример 18. Синтез 1-(3-хлор-2-фторбензил)-4-((3-фтор-6-((4-метилтиазол-2-ил)амино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 18)
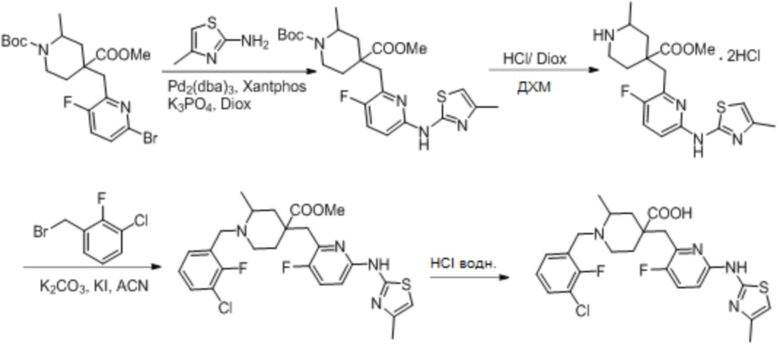
Используя 1-(трет-бутил)-4-метил-4-((6-бром-3-фторпиридин-2-ил)метил)-2-метилпиперидин-1,4-дикарбоксилат и 4-метилтиазол-2-амин в качестве исходных материалов, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, ДМСО-d6) δ: 11,64 (с, 1H), 10,53 (с, 1H), 7,73 (т, J = 7,2 Гц, 1H), 7,70-7,66 (м, 2H), 7,55 (т, J = 9,2 Гц, 1H), 7,34 (т, J = 7,9 Гц, 1H), 6,56 (с, 1H), 4,68 (д, J = 13,3 Гц, 1H), 4,37 (дд, J = 13,6, 8,3 Гц, 1H), 3,83 (с, 2H), 3,26-3,21 (м, 2H), 3,06 (д, J = 12,9 Гц, 1H), 2,17 (с, 3H), 2,14-1,93 (м, 4H), 1,51 (д, J = 6,0 Гц, 3H); ИЭР-МС m/z: 508,1 [M+H]+.
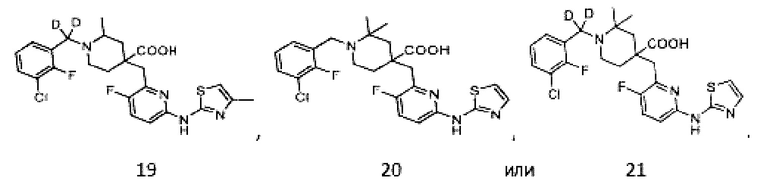
Пример 19. Синтез 1-((3-хлор-2-фторфенил)метил-d2)-4-((3-фтор-6-((4-метилтиазол-2-ил)амино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 19)
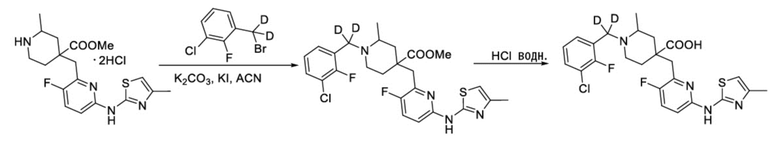
Используя гидрохлорид метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилат и 1-(бромметил-d2)-3-хлор-2-фторбензол в качестве исходных материалов, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, CD3OD) δ: 7,76 (т, J = 8,7 Гц, 1H), 7,73-7,57 (м, 3H), 7,22 (дд, J = 8,8, 2,9 Гц, 1H), 6,58 (с, 1H), 3,92 (с, 2H), 3,44-3,31 (м, 2H), 3,16 (м, 1H), 2,19 (с, 3H), 2,15-1,86 (м, 4H), 1,51 (м, 3H); ИЭР-МС m/z: 510,0 [M+H]+.
Пример 20. Синтез 1-(3-хлор-2-фторбензил)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2,2-диметилпиперидин-4-карбоновой кислоты (соединение 20)
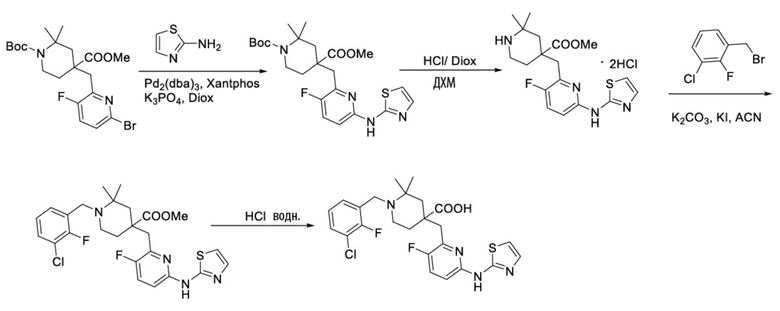
Используя 1-(трет-бутил)-4-метил-4-((6-бром-3-фторпиридин-2-ил)метил)-2,2-диметилпиперидин-1,4-дикарбоксилат и тиазол-2-амин в качестве исходных материалов, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, CD3OD) δ: 7,76 (т, J = 8,7 Гц, 1H), 7,70-7,51 (м, 3H), 7,35-7,24 (м, 2H), 7,18 (дд, J = 8,8, 2,9 Гц, 1H), 4,61 (д, J = 13,3 Гц, 1H), 4,32 (дд, J = 13,6, 8,3 Гц, 1H), 3,94 (с, 2H), 3,34-3,22 (м, 2H), 2,31 (s, 2H), 2,02-1,81 (м, 2H), 1,45 (с, 6H); ИЭР-МС m/z: 508,2 [M+H]+.
Пример 21. Синтез 1-((3-хлор-2-фторфенил)метил-d2)-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2,2-диметилпиперидин-4-карбоновой кислоты (соединение 21)
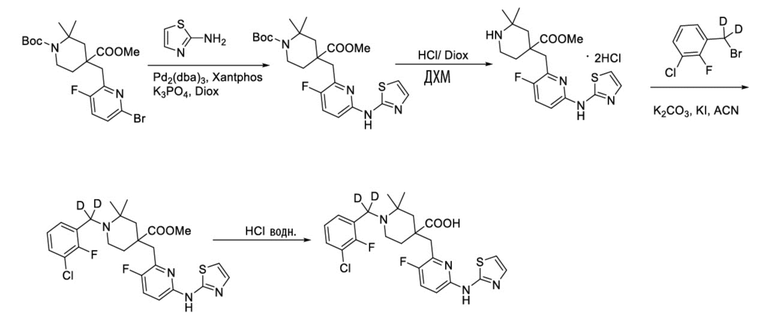
Используя 1-(трет-бутил)-4-метил-4-((6-бром-3-фторпиридин-2-ил)метил)-2,2-диметилпиперидин-1,4-дикарбоксилат и тиазол-2-амин в качестве исходных материалов, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, CD3OD) δ: 7,75 (т, J = 8,7 Гц, 1H), 7,70-7,55 (м, 3H), 7,37-7,26 (м, 2H), 7,20 (дд, J = 8,8, 2,9 Гц, 1H), 3,96 (с, 2H), 3,57-3,44 (м, 2H), 2,32 (с, 2H), 2,02-1,85 (м, 2H), 1,43 (с, 6H); ИЭР-МС m/z: 509,2 [M+H]+.
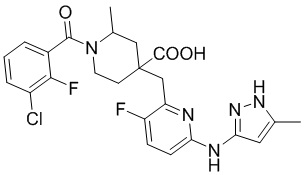
Пример 22. Синтез 1-(3-хлор-2-фторбензоил)-4-((3-фтор-6-((5-метил-1H-пиразол-3-ил)амино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (пример 22)
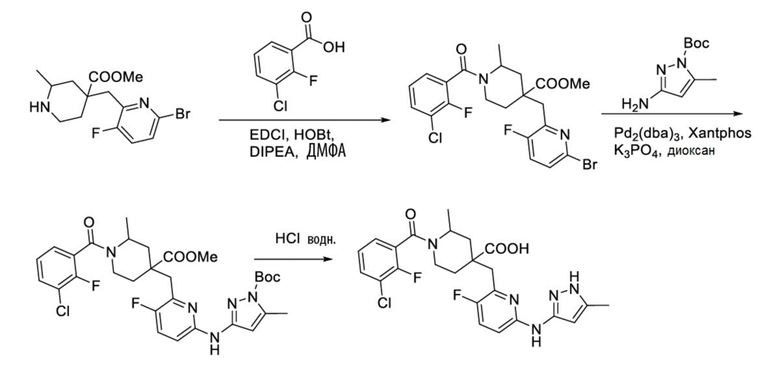
Метил-4-((6-бром-3-фторпиридин-2-ил)метил)-1-(3-хлор-2-фторбензоил)-2-метилпиперидин-4-карбоксилат
3-Хлор-2-фторбензойную кислоту (262 мг, 1,50 ммоль), ДМФА (20 мл), EDCI (431 мг, 2,25 ммоль), HOBt (304 мг, 2,25 ммоль) и DIPEA (970 мг, 7,52 ммоль) добавляли в колбу объемом 100 мл, перемешивали смесь при комнатной температуре в течение 30 минут под защитным слоем Ar, добавляли метил-4-((3-фтор-6-(тиазол-2-иламино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилат (345 мг, 1,0 ммоль), затем перемешивали при комнатной температуре в течение 20 часов. После завершения реакции, по данным ЖХ-МС, смесь гасили водой (40 мл), экстрагировали EA (50 мл* 2), объединяли с органической фазой и промывали насыщенным раствором NaCl, концентрировали, очищали остаток колоночной хроматографией с получением требуемого промежуточного соединения (426 мг, выход 85%), ИЭР-МС m/z: 501,1/503,1 [M+H]+.
1-(3-Хлор-2-фторбензоил)-4-((3-фтор-6-((5-метил-1H-пиразол-3-ил)амино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновая кислота
Метил-4-((6-бром-3-фторпиридин-2-ил)метил)-1-(3-хлор-2-фторбензоил)-2-метилпиперидин-4-карбоксилат (426 мг, 0,85 ммоль), трет-бутил-3-амино-5-метил-1H-пиразол-1-карбоксилат (201 мг, 1,02 ммоль), Pd2(dba)3 (92 мг, 0,10 ммоль), Xantphos (116 мг, 0,20 ммоль), K3PO4 (96 мг, 0,45 ммоль) и 1,4-диоксан (10 мл) добавляли в колбу объемом 100 мл, нагревали до 100°С и дополнительно проводили реакцию около 5 часов под защитным слоем Ar. После завершения реакции, установленного по ЖХ-МС, концентрировали смесь при пониженном давлении и затем очищали колоночной хроматографией (ДХМ/MeOH = от 10/0 до 5/1) с получением требуемого продукта в виде желтого пенистого вещества (394 мг, выход 75%), ИЭР-МС m/z: 618,1 [M+H]+.
Используя вышеуказанное промежуточное соединение в качестве исходного материала, получали требуемое соединение таким же способом синтеза, как описан в примере 1.
1H ЯМР (400 МГц, CD3OD) δ: 7,65 (дт, J = 16,0, 8,4 Гц, 3H), 7,35 (т, J = 7,9 Гц, 1H), 6,92 (дд, J = 9,0, 3,1 Гц, 1H), 6,14-6,05 (м, 1H), 3,93 (дд, J = 11,5, 6,1 Гц, 1H), 2,57-2,49 (м, 3H), 2,40 (с, 3H), 2,23-2,00 (м, 4H), 1,61-1,48 (м, 3H); ИЭР-МС m/z: 504,2 [M+H]+.
Пример 23. Синтез 1-((3-хлор-2-фторфенил)метил-d2)-4-((3-фтор-6-((5-метил-1H-пиразол-3-ил)амино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (пример 23)
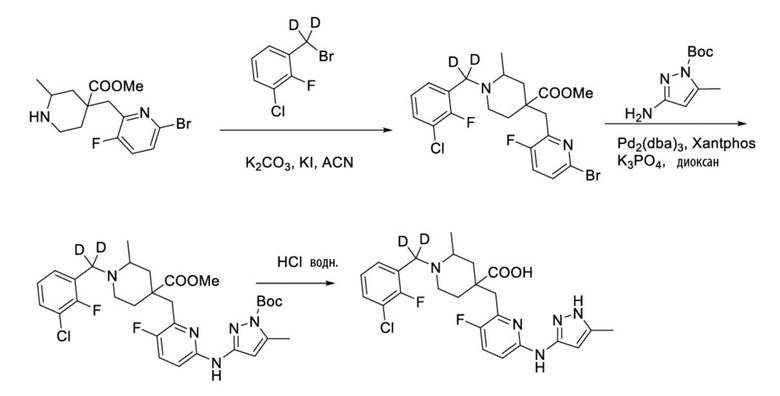
Метил-4-((6-бром-3-фторпиридин-2-ил)метил)-1-((3-хлор-2-фторфенил)метил-d2)-2-метилпиперидин-4-карбоксилат
1-(Трет-бутил)-4-метил-4-((6-бром-3-фторпиридин-2-ил)метил)-2-метилпиперидин-1,4-дикарбоксилат (1 г, 2,62 ммоль, его синтезировали со ссылкой на способ, описанный в патенте WO2016077161), 1-(бромметил-d2)-3-хлор-2-фторбензол (650 мг, 2,88 ммоль), K2CO3 (1,811 г, 13,1 ммоль), KI (10 мг) и ACN (20 мл) добавляли в колбу объемом 100 мл, перемешивали при комнатной температуре около 2 часов. После завершения реакции, установленного по ЖХ-МС, концентрировали смесь и затем очищали колоночной хроматографией (PE/EA = от 20/1 до 8/1) с получением метил-4-((6-бром-3-фторпиридин-2-ил)метил)-1-((3-хлор-2-фторфенил)-метил-d2)-2-метилпиперидин-4-карбоксилата (985 мг, выход 77%) в виде бесцветного маслянистого вещества, ИЭР-МС m/z: 489,1/491,1 [M+H]+.
Метил-4-((6-((1-(трет-бутоксикарбонил)-5-метил-1H-пиразол-3-ил)амино)-3-фторпиридин-2-ил)метил)-1-((3-хлор-2-фторфенил)метил-d2)-2-метилпиперидин-4-карбоксилат
Метил-4-((6-бром-3-фторпиридин-2-ил)метил)-1-((3-хлор-2-фторфенил)-метил-d2)-2-метилпиперидин-4-карбоксилат (985 мг, 2,01 ммоль), трет-бутил-3-амино-5-метил-1H-пиразол-1-карбоксилат (476 мг, 2,41 ммоль), Pd2(dba)3 (92 мг, 0,10 ммоль), Xantphos (116 мг, 0,20 ммоль), K3PO4 (1,067 ммоль) и 1,4-диоксан (20 мл) добавляли в колбу объемом 100 мл, нагревали до 100°С и дополнительно проводили реакцию в течение 5 часов под защитным слоем Ar. После завершения реакции, установленного по ЖХ-МС, концентрировали смесь и затем очищали колоночной хроматографией (PE/EA = от 10/1 до 5/1) с получением требуемого промежуточного соединения в виде желтого вещества (1,03 г, выход 84%), ИЭР-МС m/z: 605,3 [M+H]+.
1-((3-Хлор-2-фторфенил)метил-d2)-4-((3-фтор-6-((5-метил-1H-пиразол-3-ил)амино)пиридин-2-ил)метил)-2-метилпиперидин-4-карбоновая кислота
Метил-4-((6-((1-(трет-бутоксикарбонил)-5-метил-1H-пиразол-3-ил)амино)-3-фторпиридин-2-ил)метил)-1-((3-хлор-2-фторфенил)метил-d2)-2-метилпиперидин-4-карбоксилат (1,03 г, 1,70 ммоль) добавляли в колбу объемом 100 мл, затем добавляли воду (15 мл) и конц. HCl (15 мл), нагревали до 105°С и кипятили с обратным холодильником в течение 5 часов. После завершения реакции, установленного по ЖХ-МС, концентрировали смесь и суспендировали остаток с ACN (30 мл) при комнатной температуре. После фильтрования промывали осадок на фильтре ACN (5 мл*2) и сушили с получением требуемого желтого порошка (844 мг, выход 88%).
1H ЯМР (400 МГц, CD3OD) δ: 7,63 (дт, J = 16,0, 8,4 Гц, 3H), 7,32 (т, J = 7,9 Гц, 1H), 6,90 (дд, J = 9,0, 3,1 Гц, 1H), 6,04-6,01 (м, 1H), 3,96 (дд, J = 11,5, 6,1 Гц, 1H), 3,58-3,30 (м, 4H), 2,40 (с, 3H), 2,23-2,00 (м, 5H), 1,61-1,48 (м, 3H); ИЭР-МС m/z: 492,2 [M+H]+.
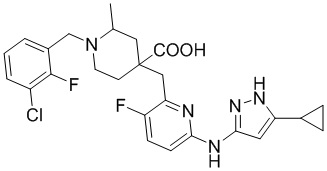
Пример 24. Синтез 1-(3-хлор-2-фторбензил)-4-((6-((5-циклопропил-1H-пиразол-3-ил)амино)-3-фторпиридин-2-ил)метил)-2-метилпиперидин-4-карбоновой кислоты (соединение 24)
Используя метил-4-((6-бром-3-фторпиридин-2-ил)метил)-2-метилпиперидин-4-карбоксилат и 1-(бромметил)-3-хлор-2-фторбензол в качестве исходных материалов, получали требуемое соединение таким же способом синтеза, как описан в примере 23.
1H ЯМР (400 МГц, CD3OD) δ: 7,61 (дт, J = 16,0, 8,4 Гц, 3H), 7,34 (т, J = 7,9 Гц, 1H), 6,85 (дд, J = 9,0, 3,1 Гц, 1H), 6,01-5,95 (м, 1H), 4,56 (д, J = 13,3 Гц, 1H), 4,13 (дд, J = 13,6, 8,3 Гц, 1H), 3,93 (дд, J = 11,5, 6,1 Гц, 1H), 3,68-3,41 (м, 4H), 2,23-2,00 (м, 4H), 1,61-1,48 (м, 4H), 0,85-0,67 (м, 4H); ИЭР-МС m/z: 516,2 [M+H]+.
Пример 25. Анализ активности аврора-киназы
Использовали анализ изменения подвижности Caliper для измерения активности соединений в отношении аврора-киназы. Получали 10 различных концентраций соединений посредством трехкратных серийных разбавлений. Рекомбинантные аврора-киназы в киназном буфере (20 мМ HEPES, рН 7,5, 0,01% Triton X-100) инкубировали с соединениями в течение 10 минут при комнатной температуре. Затем добавляли FAM-меченные пептидные субстраты для инициации реакции при 25°С. Степень превращения измеряли с помощью Caliper после остановки реакций. В качестве положительного контроля использовали LY-3295668. Данные нормализовали по данным, полученным для контрольного образца с носителем, и рассчитывали процент ингибирования и IC50. Результаты представлены в таблице 2.
Пример 26. Анализ пролиферации H1975
Клетки H1975, растущие на логарифмической фазе, трипсинизировали, растирали до одноклеточных суспензий и высевали в 384-луночные планшеты с плотностью 5×103/лунка в 50 мкл. Оставляли клетки прикрепляться в течение ночи и добавляли к клеткам предложенные соединения для дополнительной инкубации в течение 72 часов. Затем добавляли 50 мкл CTL для измерения выживания клеток посредством количественного определения АТФ. Рассчитывали IC50 в программе GRAPHPAD, результаты представлены в таблице 2.
Таблица 2. Значения IC50 ингибирования аврора-киназы и антипролиферативного действия в отношении H1975
Как указано выше, соединения общей формулы (1), где L имеет соответствующий размер, такой как CD2 вместо CH2, и/или W представляет собой , демонстрируют улучшенную эффективность против аврора-A, аврора-B и H1975. Оптические изомеры в данном описании демонстрируют различную активность относительно рацемических смесей. Например, оптический изомер, соединение 1-1, является более эффективным, чем его рацемическая смесь, соединение 1. Таким же образом может быть измерена активность других оптических изомеров по данному описанию, и она может превышать активность соединения 1-1.
Пример 27. In vivo действие против роста опухоли в ксенотрансплантатах H1975 у мышей
Клетки H1975 выращивали в среде 1640, содержащей 10% FBS, при 37°С и с 5% CO2. Клетки пересевали и собирали посредством трипсинизации. 8×106 клеток имплантировали в левую подмышку «голых» мышей. Мышей случайным образом разделяли на четыре группы по 6 мышей в каждой, когда объем опухоли достигал около 80 мм3, и вводили дозу носителя и 6 мг/мл LY-3295668, соединение 13 и соединение 23 в дозе 0,1 мг/10 г, соответственно, через желудочный зонд. Объем опухоли и массу тела проверяли через день. Мышей усыпляли на 21 день лечения. Рассчитывали и анализировали относительный объем опухоли (RTV) и рост опухоли (T/C), а также ингибирование роста опухоли (TGI). Результаты представлены в таблице 3.
Таблица 3. In vivo эффективность в моделях ксенотрансплантата H1975 у мышей
* : P < 0,05 относительно контрольного образца, ** : P < 0,01 относительно контрольного образца, *** : P < 0,001 относительно контрольного образца, **** : P < 0,0001 относительно контрольного образца; D1: первый день лечения; D21: последний день лечения; qd*21: один раз в сутки в течение 21 дня; RTV: относительный объем опухоли: RTV = Vt / V0; T/C (%) = TRTV / CRTV X 100; TRTV : относительный объем опухоли (RTV) в экспериментальной группе; CRTV: относительный объем опухоли (RTV) в контрольной группе с носителем. TGI: ингибирование роста опухоли (%); T/C (%) > 60%: неэффективно; T/C (%) ≤ 60% и P < 0,05: эффективно.
Как показано в таблице 3, по сравнению с LY-3295668, соединение 13 демонстрирует более высокую активность in vivo, что позволяет предположить, что соединения общей формулы (1), где L имеет соответствующий размер, такой как CD2, и/или W представляет собой , демонстрируют заметно улучшенную эффективность in vivo, что имеет большое значение для прицельного воздействия на рак ингибиторами аврора-киназы.
Пример 28. In vivo действие против роста опухоли в ксенотрансплантатах H69 у мышей
Клетки H69 выращивали в среде 1640, содержащей 10% FBS, при 37°С и с 5% CO2. Клетки пересевали и собирали посредством трипсинизации. 1×107 клеток H69 имплантировали в левую подмышку «голых» мышей. Мышей случайным образом разделяли на четыре группы по 8 мышей в каждой, когда объем опухоли достигал около 290 мм3, и перорально вводили носитель (0,5% МЦ) и 2,5 мг/кг, 5 мг/кг и 10 мг/кг соединения 1-1 два раза в сутки, соответственно. Объем опухоли и массу тела измеряли через день. Мышей усыпляли на 21 день лечения. Рассчитывали и статистически анализировали относительный объем опухоли (RTV) и рост опухоли (T/C), а также ингибирование роста опухоли (TGI). Результаты представлены ниже.
Таблица 4. In vivo эффективность в моделях ксенотрансплантата H69 у мышей
* : P < 0,05 относительно контрольного образца, ** : P < 0,01 относительно контрольного образца, *** : P < 0,001 относительно контрольного образца, **** : P < 0,0001 относительно контрольного образца; D1: первый день лечения; D21: последний день лечения; qd*21: один раз в сутки в течение 21 дня; RTV: относительный объем опухоли: RTV = Vt / V0; T/C (%) = TRTV / CRTV X 100; TRTV : относительный объем опухоли (RTV) в экспериментальной группе; CRTV: относительный объем опухоли (RTV) в контрольной группе с носителем; TGI: ингибирование роста опухоли (%); T/C (%) > 60%: неэффективно; T/C (%) ≤ 60% и P < 0,05: эффективно.
Как показано в таблице 4, соединение 1-1 блокирует рост опухоли H69 дозозависимым образом с заметно улучшенной эффективностью по сравнению с LY-3295668.
Несмотря на то, что выше описаны конкретные варианты реализации данного изобретения, специалистам в данной области техники понятно, что они являются лишь примерами, и в отношении приведенных вариантов реализации могут быть сделаны различные изменения или модификации без отступления от принципов и сущности данного изобретения. Соответственно, объем данного изобретения определяется прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ПИПЕРИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2013 |
|
RU2581834C1 |
| ПРОИЗВОДНЫЕ 1,5-НАФТИРИДИНА И ИНГИБИТОРЫ MELK, СОДЕРЖАЩИЕ ИХ | 2012 |
|
RU2645339C1 |
| ДИГИДРОНАФТИРИДИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ, ПОДХОДЯЩИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2012 |
|
RU2664055C2 |
| Соединения триазоло-пиримидина и их применение | 2019 |
|
RU2802866C2 |
| СОЕДИНЕНИЯ БЕНЗОЛСУЛЬФОНАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ТЕРАПЕВТИЧЕСКИХ СРЕДСТВ | 2017 |
|
RU2769827C2 |
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ | 2021 |
|
RU2817736C1 |
| ИНГИБИТОРЫ ФОСФОИНОЗИТИД-3-КИНАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2007 |
|
RU2468027C2 |
| ПИРИМИДИНГИДРАЗИДНЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ PGDS | 2008 |
|
RU2464262C2 |
| ДИГИДРОНАФТИРИДИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ, ПОДХОДЯЩИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2804468C2 |
| ИНГИБИТОРЫ РЕПЛИКАЦИИ ВИРУСА ГРИППА, СПОСОБЫ ИХ ПРИМЕНЕНИЯ И ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2737190C2 |
Изобретение относится к новому типу пиридиновых соединений, а именно к соединению формулы (1) или его фармацевтически приемлемой соли, где R1 представляет собой арил, гетероарил, 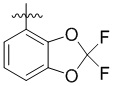 или
или 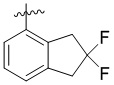 , где арил и гетероарил необязательно замещены 1-3 группами, выбранными из группы, состоящей из галогена, С1-С3 алкила, С1-С3 алкоксила, галогензамещенного С1-С3 алкила или галогензамещенного С1-С3 алкоксила; R2 представляет собой Н или метил; R3 представляет собой Н или F; W представляет собой
, где арил и гетероарил необязательно замещены 1-3 группами, выбранными из группы, состоящей из галогена, С1-С3 алкила, С1-С3 алкоксила, галогензамещенного С1-С3 алкила или галогензамещенного С1-С3 алкоксила; R2 представляет собой Н или метил; R3 представляет собой Н или F; W представляет собой  и Ra представляет собой Н, С1-С3 алкил или С3-С6 циклоалкил; и L представляет собой СН2, СО, CD2, СН(Ме), С(Ме)2,
и Ra представляет собой Н, С1-С3 алкил или С3-С6 циклоалкил; и L представляет собой СН2, СО, CD2, СН(Ме), С(Ме)2,  или
или 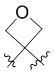 . Технический результат изобретения заключается в разработке новых ингибиторов аврора-киназы с улучшенной активностью in vitro и in vivo. 4 з.п. ф-лы, 4 табл., 28 пр.
. Технический результат изобретения заключается в разработке новых ингибиторов аврора-киназы с улучшенной активностью in vitro и in vivo. 4 з.п. ф-лы, 4 табл., 28 пр.
1. Соединение формулы (1) или фармацевтически приемлемая соль:
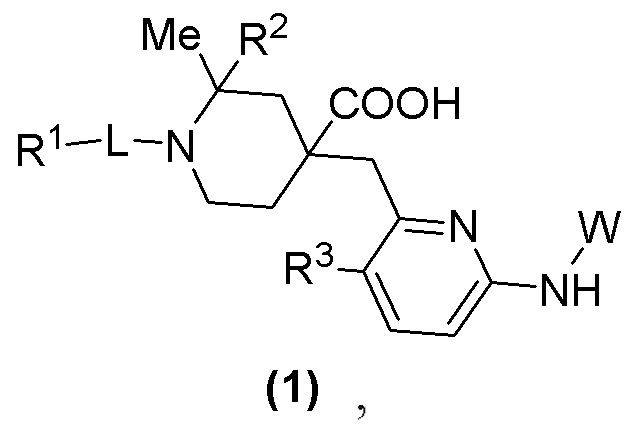
в формуле (1):
R1 представляет собой арил, гетероарил,  или
или 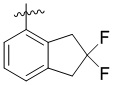 , где арил и гетероарил необязательно замещены 1-3 группами, выбранными из группы, состоящей из галогена, С1-С3 алкила, С1-С3 алкоксила, галогензамещенного С1-С3 алкила или галогензамещенного С1-С3 алкоксила;
, где арил и гетероарил необязательно замещены 1-3 группами, выбранными из группы, состоящей из галогена, С1-С3 алкила, С1-С3 алкоксила, галогензамещенного С1-С3 алкила или галогензамещенного С1-С3 алкоксила;
R2 представляет собой Н или метил;
R3 представляет собой Н или F;
W представляет собой 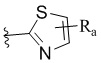 и Ra представляет собой Н, С1-С3 алкил или С3-С6 циклоалкил; и
и Ra представляет собой Н, С1-С3 алкил или С3-С6 циклоалкил; и
L представляет собой СН2, СО, CD2, СН(Ме), С(Ме)2,  или
или  .
.
2. Соединение по п. 1, отличающееся тем, что в формуле (1) R1 представляет собой  ,
,  ,
, 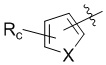 ,
, 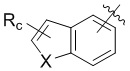 , или , где X представляет собой NH, О или S, Rc и Rd независимо представляют собой Н, галоген, С1-С3 алкил, С1-С3 алкоксил, галогензамещенный С1-С3 алкил или галогензамещенный С1-С3 алкоксил.
, или , где X представляет собой NH, О или S, Rc и Rd независимо представляют собой Н, галоген, С1-С3 алкил, С1-С3 алкоксил, галогензамещенный С1-С3 алкил или галогензамещенный С1-С3 алкоксил.
3. Соединение по п. 2, отличающееся тем, что в формуле (1) R1 представляет собой  ,
, ,
, 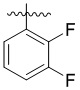 ,
, 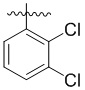 ,
, 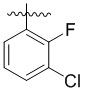 ,
, 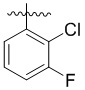 ,
, 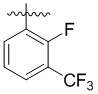 ,
, 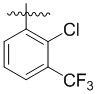 ,
, 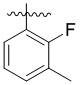 ,
, 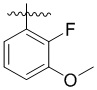 ,
, 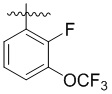 ,
, 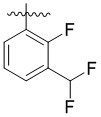 ,
, 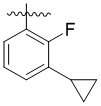 ,
, 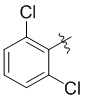 ,
, 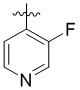 ,
, 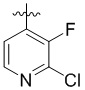 ,
, 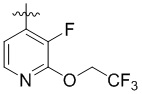 ,
, 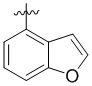 ,
, 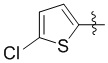 , или .
, или .
4. Соединение по п. 1, отличающееся тем, что в формуле (1)
W представляет собой: 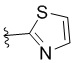 ,
, 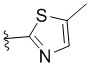 ,
, 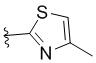 ,
, 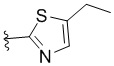 ,
, 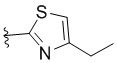 ,
, 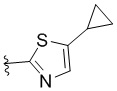 или
или 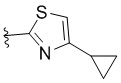 .
.
5. Соединение по пп. 1-4 или его фармацевтически приемлемая соль, отличающееся тем, что указанное соединение представляет собой:
| WO 2009104802 A1, 27.08.2009 | |||
| CN 107108567 A, 29.08.2017 | |||
| VASILEVICH N.I | |||
| et al, General Ser/Thr Kinases Pharmacophore Approach for Selective Kinase Inhibitors Search as Exemplified by Design of Potent and Selective Aurora A Inhibitors, Chem Biol Drug Des, 2016, vol.88, p.54-65 | |||
| VASILEVICH N.I | |||
| et al, Search for Potent and Selective Aurora A |

