Область техники, к которой относится изобретение
Настоящее изобретение относится к новому пиперидиновому соединению, обладающему ингибирующей киназу aurora A активностью, или его соли, и применению соединения или его соли.
Предпосылки создания изобретения
Aurora A является членом серин-треониновых киназ, и она в значительной степени связана, например, с образованием и созреванием центросом, динамикой веретена и выравниванием хромосом в митотической фазе (M фаза) клеточного цикла, посредством этого, регулируя протекание митоза (непатентный документ 1). К настоящему времени, сверхэкспрессия и/или амплификация aurora A подтверждена при большом разнообразии карцином (непатентный документ 2). Кроме того, поскольку ингибирование киназы aurora A в опухолевых клетках вызывает не только остановку митоза, но также и апоптоз, aurora A является одной из важных молекул-мишеней в терапии рака.
Между тем, средства, воздействующие на микротрубочки, как представлено таксаном и алкалоидом барвинка, широко применяются в качестве ключевого лекарственного средства в химиотерапии рака. Однако устойчивые и достаточные терапевтические эффекты не всегда получают из-за потери восприимчивости к лекарственным средствам или возникновения устойчивости к лекарственным средствам. Следовательно, есть клиническая необходимость в разработке лекарственного средства, способного усиливать противоопухолевый эффект таксановых лекарственных средств, поскольку данное лекарственное средство обещает обеспечить более эффективными терапевтическими возможностями. Разрушающий клетки эффект таксановых противораковых средств требует активации контрольной точки сборки веретена в клеточном цикле, при этом сообщают, что опухолевые клетки, имеющие пониженную активность контрольной точки сборки веретена, обладают пониженной чувствительностью к таксановым противораковым лекарственным средствам (непатентный документ 3). Кроме того, известно, что клеточная линия, сверхэкспрессирующая aurora A, становится устойчивой к паклитакселу (непатентный документ 4), и ингибирование aurora A усиливает активность паклитаксела или доцетаксела (непатентный документ 5). Между тем, сообщалось, что хотя aurora B, которая представляет собой ее подтип, обладает активностью на митотической фазе (M фаза) клеточного цикла с aurora A, при этом ингибирование aurora B снижает активность контрольной точки сборки веретена (непатентный документ 6). Следовательно, предполагается, что ингибирование aurora B сможет смягчить эффект таксановых лекарственных средств. Кроме того, aurora C сильно экспрессируется, например, в семеннике или сперматозоидах, и результаты анализа генома человека показали, что aurora C является важной в сперматогенезе (непатентный документ 7). Известно, что aurora C функционирует в качестве дополнения к функционированию aurora B при делении клеток (непатентный документ 8). Аналогично ингибированию aurora B, ингибирование aurora C вызывает анеуплоидию в клетках, приводящую к проявлению фенотипа, который сильно отличается от проявляемого при ингибировании aurora A и, вероятно, можно ожидать усиления эффекта таксановых лекарственных средств. Более того, нельзя не обратить внимания на влияние на репродуктивную систему и, следовательно, желательно, чтобы лекарственное средство не обладало ингибирующей aurora C активностью.
Согласно указанному выше, ожидают, что при введении лекарственного средства, которое селективно ингибирует киназу aurora A, в комбинации с таксановым противораковым средством лекарственное средство будет эффективно усиливать противоопухолевую активность таксанового противоракового средства, обеспечивая посредством этого больший терапевтический эффект.
Кроме того, сообщалось, что активность, прекращающая клеточный цикл, вызванная паклитакселом, поддерживается в течение нескольких дней на модели опухоли мышей, которым трансплантирована клеточная линия рака человека (непатентный документ 9). Следовательно, средство для перорального введения считают подходящим, когда одновременно вводят ингибитор aurora A, поскольку обеспечивается непрерывное воздействие.
К настоящему времени, сообщалось, что аминопиридиновое производное, обладающее ингибирующей aurora A активностью, можно вводить перорально (патентный документ 1). Однако хотя в патентном документе 1 описана ингибирующая активность aurora A и активность клеточной пролиферации in vitro, не было обнаружено какого-либо описания, относящегося к оценке перорального введения указанного выше соединения.
[Список цитируемых документов]
[Патентный документ]:
[патентный документ 1] WO2009/104802
[Непатентный документ]:
[непатентный документ 1] Nat. Rev. Drug Discov., 8, стр. 547-566 (2009)
[непатентный документ 2] Cancer Treat. Rev., 34, стр. 175-182 (2008)
[непатентный документ 3] Mol. Cancer Ther., 5, стр. 2963-2969 (2006)
[непатентный документ 4] Cancer Cell, 3, стр. 51-62 (2003)
[непатентный документ 5] Cancer Res., 65, стр. 2899-2905 (2005)
[непатентный документ 6] Mol. Cancer Ther., 8, стр. 2046-2056 (2009)
[непатентный документ 7] Nature Genet. 39: стр. 661-665, 2007
[непатентный документ 8] Genes Клетки 10: стр. 617-626, 2005
[непатентный документ 9] Cancer Res., 71, стр. 4608-4616 (2011)
Сущность изобретения
Задача настоящего изобретения
Соответственно, задача настоящего изобретения состоит в предоставлении нового соединения, которое обладает превосходной селективной ингибирующей aurora A активностью и является пригодным в качестве перорально вводимого противоракового лекарственного средства. Кроме того, другая задача настоящего изобретения состоит в предоставлении нового средства для усиления противоопухолевого эффекта лекарственных средств, воздействующих на микротрубочки, включающих таксановое противораковое средство, и комбинированной терапии.
Способы решения задачи
Авторы настоящего изобретения провели тщательное исследование для того, чтобы решить указанные выше задачи. Вследствие этого, было обнаружено, что пиперидиновое соединение, имеющее конкретный заместитель в пиридиновом кольце, обладает превосходной селективной ингибирующей aurora A активностью и активностью, ингибирующей пролиферацию раковых клеток, и является вводимым перорально. Кроме того, было обнаружено, что данное пиперидиновое соединение заметно усиливает противоопухолевые эффекты лекарственных средств, воздействующих на микротрубочки, включающих таксановое противораковое средство, что составило настоящее изобретение.
Конкретно, настоящее изобретение относится к пиперидиновому соединению, представленному общей формулой (I), или его соли:
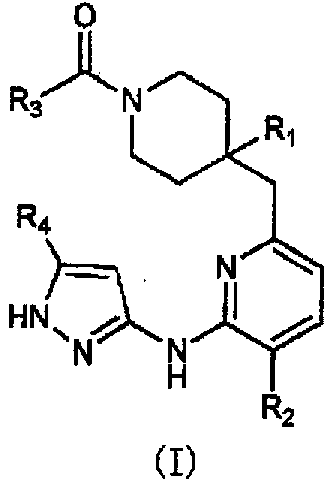
где R1 представляет собой карбоксильную группу, -C(=O)NR5R6 или оксадиазолильную группу, необязательно содержащую C1-C6алкильную группу или трифторметильную группу в качестве заместителя;
R2 представляет собой атом галогена или C1-C6алкоксигруппу;
R3 представляет собой фенильную группу, необязательно содержащую 1-3 одинаковые или различные группы, выбранные из атома галогена, C1-C6алкильной группы, C1-C6алкоксигруппы и трифторметильной группы в качестве заместителя;
R4 представляет собой атом водорода или C1-C6алкильную группу; и
R5 и R6 представляют собой одинаковые или различные группы, и каждый представляет собой атом водорода, C1-C6алкильную группу или C3-C6циклоалкильную группу, или R5 и R6 необязательно образуют 3-6-членную содержащую азот насыщенную гетероциклическую группу вместе с атомом азота, с которым связаны R5 и R6.
Настоящее изобретение также относится к лекарственному средству, содержащему пиперидиновое соединение, представленное приведенной выше общей формулой (I), или его соли в качестве активного ингредиента.
Настоящее изобретение также относится к селективному ингибитору aurora A, противоопухолевому средству или средству для усиления противоопухолевого эффекта лекарственного средства, воздействующего на микротрубочки, содержащему пиперидиновое соединение, представленное приведенной выше общей формулой (I), или его соль в качестве активного ингредиента.
Настоящее изобретение также относится к применению пиперидинового соединения, представленного приведенной выше общей формулой (I), или его соли для получения селективного ингибитора aurora A, противоопухолевого средства или средства для усиления противоопухолевого эффекта лекарственного средства, воздействующего на микротрубочки.
Настоящее изобретение также относится к пиперидиновому соединению, представленному приведенной выше общая формула (I), или его соли для селективного ингибирования aurora A, лечения рака или усиления противоопухолевого эффекта лекарственного средства, воздействующего на микротрубочки.
Настоящее изобретение также относится к способу селективного ингибирования aurora A, лечения рака или усиления противоопухолевого эффекта лекарственного средства, воздействующего на микротрубочки, включающему введение эффективной дозы пиперидинового соединения, представленного приведенной выше общей формулой (I), или его соли.
Настоящее изобретение также относится к терапевтическому средству для лечения рака, содержащему пиперидиновое соединение, представленное приведенной выше общей формулой (I), или его соль и лекарственное средство, воздействующее на микротрубочки; композиции, содержащей пиперидиновое соединение, представленное приведенной выше общей формулой (I), или его соль и лекарственное средство, воздействующее на микротрубочки, для лечения рака; применению композиции, содержащей пиперидиновое соединение, представленное приведенной выше общей формулой (I), или его соль и средство, воздействующее на микротрубочки, для получения лекарственного средства для лечения рака; и способу лечения рака, включающему введение эффективной дозы пиперидинового соединения, представленного приведенной выше общей формулой (I), или его соли одновременно с эффективной дозой лекарственного средства, воздействующего на микротрубочки.
Кроме того, настоящее изобретение относится к приведенному выше лекарственному средству, селективному ингибитору aurora A, противоопухолевому средству или средству для усиления противоопухолевого эффекта агониста микротрубочек для перорального введения; применению приведенного выше селективного ингибитора aurora A, противоопухолевого средства или средства, воздействующего на микротрубочки, для перорального введения для получения усилителя противоопухолевого эффекта; приведенному выше соединению или его соли для приведенного выше селективного ингибирования aurora A, лечения рака или усиления противоопухолевого эффекта лекарственного средства, воздействующего на микротрубочки, пероральным введением; и способу приведенного выше селективного ингибирования aurora A, лечения рака или усиления противоопухолевого эффекта лекарственного средства, воздействующего на микротрубочки, где введение осуществляют посредством перорального введения.
Эффекты изобретения
Соединение (I) настоящего изобретения или его соль обладает превосходной селективной ингибирующей aurora A активностью и ингибирующей пролиферацию раковых клеток активностью, и является перорально вводимым. Соединение (I) настоящего изобретения или его соль является пригодной не только в качестве противоопухолевого средства, но также и для введения в комбинации со средством, воздействующим на микротрубочки, включающим таксановое противораковое средство.
Подробное описание изобретения
В соединении (I) настоящего изобретения выбор R2 является важным с точки зрения селективной ингибирующей aurora A активности, перорального поглощения, противоопухолевой активности пероральным введением и усиления противоопухолевого эффекта лекарственных средств, воздействующих на микротрубочки, включающих таксановое противораковое средство. Соединение настоящего изобретения характеризуется тем, что R2 представляет собой атом галогена или C1-C6алкоксигруппу.
В описании настоящего изобретения "C1-C6алкильная группа" представляет собой линейную или разветвленную алкильную группу, содержащую 1-6 атомов углерода, и ее конкретные примеры включают метильную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, пентильную группу и гексильную группу. C1-C6алкильная группа предпочтительно представляет собой линейную или разветвленную алкильную группу, содержащую 1-4 атома углерода (C1-C4алкильная группа).
В описании настоящего изобретения "оксадиазолильная группа" относится к 1,2,4-оксадиазолильной группе или 1,3,4-оксадиазолильной группе. Оксадиазолильное кольцо является предпочтительно незамещенным или замещено C1-C4алкильной группой или трифторметильной группой, и оксадиазолильное кольцо является более предпочтительно незамещенным или замещено метильной группой или трифторметильной группой.
В описании настоящего изобретения примеры "атома галогена" включают атом фтора, атом хлора, атом брома и атом йода.
В описании настоящего изобретения "C1-C6алкоксигруппа" представляет собой линейную или разветвленную алкоксигруппу, содержащую 1-6 атомов углерода, и ее конкретные примеры включают метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу, н-бутоксигруппу, изобутоксигруппу, трет-бутоксигруппу, пентоксигруппу и гексоксигруппу. C1-C6алкоксигруппа предпочтительно представляет собой линейную или разветвленную алкоксигруппу, содержащую 1-4 атома углерода (C1-C4алкоксигруппа).
В описании настоящего изобретения "C3-C6циклоалкильная группа" относится к моноциклической циклоалкильной группе, содержащей 3-6 атомов углерода, и ее конкретные примеры включают циклопропильную группу, циклобутильную группу, циклопентильную группу и циклогексильную группу. C3-C6циклоалкильная группа предпочтительно представляет собой циклопропильную группу или циклобутильную группу.
В описании настоящего изобретения фраза "R5 и R6 необязательно образуют 3-6-членную, содержащую азот насыщенную гетероциклическую группу вместе с атомом азота, с котором соединены R5 и R6" означает, что R5 и R6 необязательно образуют, вместе с атомом азота, с которым связаны R5 и R6 (то есть как -NR5R6), 3-6-членную насыщенную гетероциклическую группу, дополнительно содержащую 0-2 атома азота и/или атома кислорода в кольце. Конкретные примеры 3-6-членной, содержащей азот насыщенной гетероциклической группы, которая необязательно образуется, включают азетидинильную группу, пирролидинильную группу, пиперидинильную группу, пиперазинильную группу, морфолинильную группу и изоксазолидинильную группу.
В общей формуле (I) R1 предпочтительно представляет собой карбоксильную группу, -C(=O)NR5R6 (где R5 и R6 являются одинаковыми или различными, и каждый представляет собой атом водорода, C1-C6алкильную группу или C3-C6циклоалкильную группу, или R5 и R6 необязательно образуют азетидинильную группу, пирролидинильную группу или изоксазолидинильную группу вместе с атомом азота, с которым связаны R5 и R6) или оксадиазолильную группу, необязательно содержащую C1-C6алкильную группу или трифторметильную группу в качестве заместителя.
В общей формуле (I) R1 более предпочтительно представляет собой карбоксильную группу, -C(=O)NR5R6 (где R5 и R6 являются одинаковыми или различными, и каждый представляет собой атом водорода, метильную группу, циклопропильную группу или циклобутильную группу, или R5 и R6 представляют собой азетидинильную группу, пирролидинильную группу или изоксазолидинильную группу вместе с атомом азота, с которым связаны R5 и R6) или оксадиазолильную группу, необязательно содержащую метильную группу или трифторметильную группу в качестве заместителя.
Как показано в примерах ниже, является важным, чтобы R2 в общей формуле (I) представлял собой атом галогена или C1-C6алкоксигруппу с точки зрения селективной ингибирующей aurora A активности, перорального поглощения, противоопухолевой активности пероральным введением и усиления противоопухолевого эффекта лекарственных средств, воздействующих на микротрубочки, включающих таксановое противораковое средство. R2 предпочтительно представляет собой атом фтора, атом хлора или C1-C4алкоксигруппу, более предпочтительно атом фтора, атом хлора или метоксигруппу.
В общей формуле (I) R3 предпочтительно представляет собой фенильную группу, необязательно содержащую 1-3 одинаковые или различные группы, выбранные из атома галогена, C1-C4алкильной группы, C1-C4алкоксигруппы и трифторметильной группы в качестве заместителя, более предпочтительно фенильную группу, содержащую 1-2 одинаковые или различные группы, выбранные из приведенных выше заместителей. R3 даже более предпочтительно представляет собой фенильную группу, содержащую 1-2 одинаковые или различные группы, выбранные из атома фтора, атома хлора, метильной группы, метоксигруппы и трифторметильной группы в качестве заместителя.
В общей формуле (I) R4 предпочтительно представляет собой атом водорода или C1-C4алкильную группу, и более предпочтительно атом водорода или метильную группу.
В соединении настоящего изобретения предпочтительно, чтобы R1 представлял собой карбоксильную группу, -C(=O)NR5R6 (где R5 и R6 являются одинаковыми или различными, и каждый представляет собой атом водорода, C1-C4алкильную группу или C3-C6циклоалкильную группу, или R5 и R6 необязательно образуют азетидинильную группу, пирролидинильную группу или изоксазолидинильную группу вместе с атомом азота, с которым связаны R5 и R6) или оксадиазолильную группу, необязательно содержащую C1-C4алкильную группу или трифторметильную группу в качестве заместителя; R2 представлял собой атом фтора, атом хлора или C1-C4алкоксигруппу; R3 представлял собой фенильную группу, необязательно содержащую 1-2 одинаковые или различные группы, выбранные из атома галогена, C1-C4алкильной группы, C1-C4алкоксигруппы и трифторметильной группы в качестве заместителя; и R4 представлял собой атом водорода или C1-C4алкильную группу.
Кроме того, в качестве соединения настоящего изобретения, соединение, где в общей формуле (I) R1 представляет собой карбоксильную группу, -C(=O)NR5R6 (где R5 и R6 являются одинаковыми или различными, и каждый представляет собой атом водорода, метильную группу, циклопропильную группу или циклобутильную группу, или R5 и R6 представляют собой азетидинильную группу, пирролидинильную группу или изоксазолидинильную группу вместе с атомом азота, с которым связаны R5 и R6) или оксадиазолильную группу, необязательно содержащую метильную группу или трифторметильную группу в качестве заместителя; R2 представляет собой атом фтора, атом хлора или метоксигруппу; R3 представляет собой фенильную группу, необязательно содержащую 1-2 одинаковые или различные группы, выбранные из атома фтора, атома хлора, метильной группы, метоксигруппы и трифторметильной группы в качестве заместителя; и R4 представляет собой атом водорода или метильную группу, является предпочтительным.
Кроме того, соединение, где R1 представляет собой карбоксильную группу, -C(=O)NR5R6 (где R5 и R6 являются одинаковыми или различными, и каждый представляет собой атом водорода или метильную группу, или R5 и R6 представляют собой изоксазолидинильную группу вместе с атомом азота, с которым связаны R5 и R6) или 1,2,4-оксадиазолильную группу или 1,3,4-оксадиазолильную группу, необязательно содержащую метильную группу в качестве заместителя; R2 представляет собой атом фтора, атом хлора или метоксигруппу; R3 представляет собой фенильную группу, содержащую 1-2 одинаковые или различные группы, выбранные из атома фтора, атома хлора, метильной группы, метоксигруппы и трифторметильной группы в качестве заместителя; и R4 представляет собой атом водорода или метильную группу, является более предпочтительным.
Кроме того, соединение, где R1 представляет собой карбоксильную группу или 1,2,4-оксадиазолильную группу, необязательно содержащую метильную группу в качестве заместителя; R2 представляет собой атом фтора; R3 представляет собой фенильную группу, содержащую 1-2 одинаковые или различные группы, выбранные из атома фтора и атома хлора в качестве заместителя; и R4 представляет собой метильную группу, является особенно предпочтительным.
Следующие соединения могут быть приведены в качестве примеров особенно предпочтительных соединений настоящего изобретения.
1-(2,3-дихлорбензоил)-4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновая кислота (соединение 1),
1-(2-фтор-3-трифторметилбензоил)-4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновая кислота (соединение 2),
1-(3-хлор-2-фторбензоил)-4-((5-фтор-6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновая кислота (соединение 10),
1-(3-хлор-2-фторбензоил)-4-((5-метокси-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновая кислота (соединение 11),
1-(3-хлор-2-фторбензоил)-4-((5-хлор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновая кислота (соединение 12),
1-(3-хлор-2-фторбензоил)-4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновая кислота (соединение 13),
1-(3-хлор-2-фторбензоил)-4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)-N-метилпиперидин-4-карбоксамид (соединение 14),
1-(3-хлор-2-фторбензоил)-4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)-N,N-диметилпиперидин-4-карбоксамид (соединение 16),
азетидин-1-ил(1-(3-хлор-2-фторбензоил)-4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-ил)метанон (соединение 19),
(1-(3-хлор-2-фторбензоил)-4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-ил)(изоксазолидин-2-ил)метанон (соединение 21),
(3-хлор-2-фторфенил)(4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)-4-(5-метил-1,2,4-оксадиазол-3-ил)пиперидин-1-ил)метанон (соединение 22),
(2,3-дихлорфенил)(4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)-4-(5-метил-1,2,4-оксадиазол-3-ил)пиперидин-1-ил)метанон (соединение 23),
(3-хлор-2-фторфенил)(4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)-4-(1,2,4-оксадиазол-3-ил)пиперидин-1-ил)метанон (соединение 24),
(3-хлор-2-фторфенил)(4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)-4-(5-метил-1,3,4-оксадиазол-2-ил)пиперидин-1-ил)метанон (соединение 28),
(3-хлор-2-фторфенил)(4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)-4-(3-метил-1,2,4-оксадиазол-5-ил)пиперидин-1-ил)метанон (соединение 29).
Далее, репрезентативный способ получения соединения (I) настоящего изобретения показан в настоящем описании ниже.
Соединение (I) настоящего изобретения можно получить, например, следующим способом получения, показанным в примерах. Однако способ получения соединения (I) настоящего изобретения не ограничивается данными примерами реакций. Исходные соединения, необходимые для получения соединения настоящего изобретения, можно получить в виде коммерческих продуктов или легко получить способом получения, описанным, например, в документах предшествующего уровня техники.
Среди соединений (I) настоящего изобретения, соединение (I-1), где R1 представляет собой карбоксильную группу, можно получить, например, следующим способом получения 1.
Способ получения 1


где X и Y, каждый представляет собой уходящую группу; P представляет собой атом водорода или защитную группу; и Z представляет собой общую формулу (a) или (b):
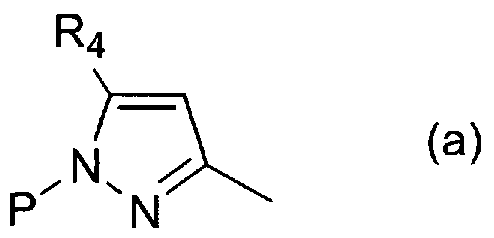
 ,
,
и R2, R3 и R4 являются такими, как определено выше.
В приведенном выше способе получения 1 примеры уходящей группы, представленной X или Y, включают атом галогена, и она предпочтительно является атомом брома. Примеры защитной группы, представленной P, включают трет-бутильную группу, метоксиметильную группу, [(2-триметилсилил)этокси]метильную группу и бензильную группу, и предпочтительно трет-бутильную группу.
(Стадия 1)
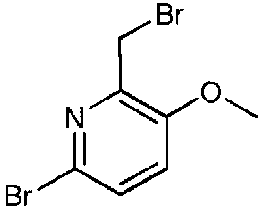
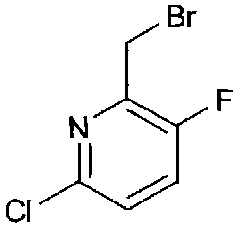
Данная стадия представляет собой способ получения соединения (IV) взаимодействием соединения (II) с основанием, и затем с соединением (III). Примеры соединения (III), которые будут использовать на данной стадии, включают 6-бром-2-бромметил-5-фторпиридин, 6-бром-2-хлорметил-5-фторпиридин, 2-бромметил-6-хлор-5-фторпиридин, 2-бромметил-5,6-дихлорпиридин и 6-бром-2-бромметил-5-метоксипиридин, и предпочтительно 6-бром-2-бромметил-5-фторпиридин. Соединение (III) можно получить в виде коммерческого продукта или получить согласно общеизвестному способу.
Количество соединения (III), которое будут использовать на данной стадии, составляет 0,1-10 эквивалентов, предпочтительно 0,8-2 эквивалентов на один эквивалент соединения (II). Температура реакции составляет от -90 до 100°C, предпочтительно от -78 до 0°C. Продолжительность реакции составляет 0,1-100 часов, предпочтительно 0,5-10 часов. Примеры основания включают диизопропиламин лития и гексаметилдисилазид лития, и основание можно использовать в количестве 0,5-10 эквивалентов, предпочтительно 1-1,5 эквивалентов. Растворитель, который будут использовать в данной реакции, конкретно не ограничен, при условии, что он не мешает протеканию реакции, и его примеры включают тетрагидрофуран, 2-метилтетрагидрофуран, диэтиловый эфир, 1,2-диметоксиэтан и толуол. Данные растворители можно использовать отдельно или в виде смеси.
Соединение (IV), полученное, как описано выше, может быть подвергнуто последующей стадии с или без выделения и очистки общеизвестными способами выделения и очистки, такими как концентрирование, концентрирование при пониженном давлении, кристаллизация, экстракция растворителем, переосаждение и хроматография.
(Стадия 2)
Данная стадия представляет собой способ получения соединения (VI) реакцией сочетания соединения (IV) и соединения (V). Примеры соединения (V) для использования на данной стадии (соединение (V-1) или соединение (V-2)) включают 1-трет-бутил-3-метил-1H-пиразол-5-амин, 1-трет-бутил-1H-пиразол-5-амин, 1-{[2-(триметилсилил)этокси]метил}-1H-пиразол-3-амин и 5-метил-1-{[2-(триметилсилил)этокси]метил}-1H-пиразол-3-амин. Соединение (V) можно получить в виде коммерческого продукта или получить согласно общеизвестному способу.
Количество соединения (V) для использования на данной стадии составляет 0,5-10 эквивалентов, предпочтительно 0,8-2 эквивалента на один эквивалент соединения (IV). Примеры катализатора для использования на данной стадии включают металлический катализатор, такой как трис(бензилиденацетон)дипалладий и ацетат палладия, и катализатор можно использовать в количестве 0,001-5 эквивалентов, предпочтительно 0,005-0,1 эквивалента на один эквивалент соединения (IV). Примеры лиганда приведенного выше металлического катализатора включают 4,5-бис(дифенилфосфино)-9,9-диметилксантен и 2,2'-бисдифенилфосфино-1,1'-бинафтил, и данные лиганды можно использовать в количестве 0,001-5 эквивалентов, предпочтительно 0,005-0,2 эквивалента на один эквивалент соединения (IV). Температура реакции составляет 0-200°C, предпочтительно от комнатной температуры до 130°C. Продолжительность реакции составляет 0,1-100 часов, предпочтительно 0,5-20 часов. Примеры основания включают неорганическое основание, такое как фосфат калия, карбонат натрия, карбонат калия, карбонат цезия и трет-бутоксид натрия, и органические амины, такие как триметиламин, диизопропилэтиламин и пиридин, и данные основания можно использовать в количестве 0,5-10 эквивалентов, предпочтительно 1-3 эквивалента. Растворитель для использования в данной реакции конкретно не ограничен, при условии, что он не препятствует протеканию реакции, и его примеры включают толуол, тетрагидрофуран, диоксан, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидин-2-он, трет-бутанол и трет-амиловый спирт. Данные растворители можно использовать отдельно или в виде смеси.
Соединение (VI), полученное, как описано выше, может быть подвергнуто последующей стадии с или без выделения и очистки общеизвестными способами выделения и очистки, такими как концентрирование, концентрирование при пониженном давлении, кристаллизация, экстракция растворителем, переосаждение и хроматография.
(Стадия 3)
Данная стадия представляет собой способ получения соединения (VII) удалением трет-бутоксикарбонильной группы, которая представляет собой защитную группу соединения (VI), в присутствии кислоты. Что касается условий реакции, применяемых на данной стадии, данную стадию можно осуществлять согласно способу, описанному в документе (Protective Groups in Organic Synthesis, written by T. W. Greene, John Wiley & Sons, Inc. (1981)), или способу, эквивалентному приведенному выше способу. Примеры кислоты для использования включают трифторуксусную кислоту, хлористоводородную кислоту, серную кислоту, метансульфоновую кислоту и толуолсульфоновую кислоту, и кислоту можно использовать в количестве 0,1-100 эквивалентов, предпочтительно 1-10 эквивалентов. Температура реакции составляет 0-200°C, предпочтительно от комнатной температуры до 100°C. Продолжительность реакции составляет 0,1-100 часов, предпочтительно 0,5-20 часов. Растворитель для использования в данной реакции конкретно не ограничен, при условии, что он не препятствует протеканию реакции, и его примеры включают хлороформ, ацетонитрил, толуол, тетрагидрофуран, диоксан, воду и уксусную кислоту. Данные растворители можно использовать отдельно или в виде смеси.
Соединение (VII), полученное, как описано выше, может быть подвергнуто последующей стадии с или без выделения и очистки общеизвестными способами выделения и очистки, такими как концентрирование, концентрирование при пониженном давлении, кристаллизация, экстракция растворителем, переосаждение и хроматография.
(Стадия 4)
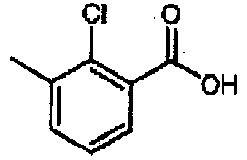
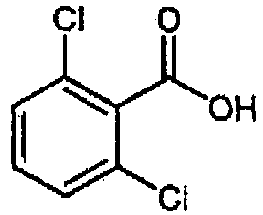
Данная стадия представляет собой реакцию получения соединения (IX) реакцией конденсации с дегидратацией между соединением (VII) и соединением (VIII). Примеры соединения (VIII) для использования на данной стадии включают 2-фтор-3-хлорбензойную кислоту и 2,3-дихлорбензойную кислоту. Соединение (VIII) может быть получено в виде коммерческого продукта или получено согласно общеизвестному способу. На данной стадии, используя широко применяемый конденсирующий агент, соединение (IX) может быть получено согласно общеизвестному способу. Примеры конденсирующего агента включают N,N'-дициклогексилкарбодиимид (DCC), N,N'-диизопропилкарбодиимид (DIC), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (WSC), дифенилфосфорилазид (DPPA), гексафторфосфат (бензотриазол-1-илокси)трисдиметиламинофосфония (BOP), гексафторфосфат (бензотриазол-1-илокси)трипирролидинфосфония (PyBOP), фосфат (7-азабензотриазол-1-илокси)триспирролидинфосфония (PyAOP), гексафторфосфат бромтриспирролидинфосфония (BroP), гексафторфосфат хлортрис(пирролидин-1-ил)фосфония (PyCroP), 3-(диэтоксифосфорилокси)-1,2,3-бензотриазин-4(3H)-он (DEPBT), гексафторфосфат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU) и гидрохлорид 4-(5,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолина (DMTMM). Примеры добавки для использования на данной стадии включают 1-гидроксибензотриазол (HOBt), 1-гидрокси-7-азабензотриазол (HOAt) и N-гидроксисукцинимид (HOSu). Данные добавки можно использовать в количестве 0,1-100 эквивалентов, предпочтительно 1-10 эквивалентов. При желании, основание, такое как триметиламин, триэтиламин, трипропиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин, лутидин и коллидин, можно использовать в количестве 0,1-100 эквивалентов, предпочтительно 1-10 эквивалентов. Растворитель конкретно не ограничен и можно использовать, например, воду, метанол, этанол, 2-пропанол, тетрагидрофуран, 1,4-диоксан, толуол, метиленхлорид, хлороформ, ацетонитрил, N,N-диметилформамид и N,N-диметилацетамид, диметилсульфоксид. Температура реакции составляет от -30 до 200°C, предпочтительно 0-50°C. Продолжительность реакции составляет 0,1-100 часов, предпочтительно 0,5-24 часов.
Соединение (IX), полученное, как описано выше, может быть подвергнуто последующей стадии с или без выделения и очистки общеизвестными способами выделения и очистки, такой как концентрирование, концентрирование при пониженном давлении, кристаллизация, экстракция растворителем, переосаждение и хроматография.
(Стадия 5)
Данная стадия представляет собой способ получения соединения (I-1) одновременным проведением гидролиза цианогруппы соединения (IX) и удалением защитной группы (P) заместителя Z в кислых условиях. Примеры кислоты для использования включают хлористоводородную кислоту, серную кислоту, метансульфоновую кислоту, толуолсульфоновую кислоту и трифторуксусную кислоту. Данные кислоты можно использовать в количестве 0,1-100 эквивалентов, предпочтительно 1-10 эквивалентов. Температура реакции составляет от комнатной температуры до 200°C, предпочтительно 60-130°C. Продолжительность реакции составляет 0,1-100 часов, предпочтительно 0,5-20 часов. Растворитель для использования в данной реакции конкретно не ограничен, при условии, что он не препятствует протеканию реакции, и его примеры включают диоксан, воду, уксусную кислоту, толуол, тетрагидрофуран и 2-пропанол. Данные растворители можно использовать отдельно или в виде смеси. Соединение (I-1), полученное, как описано выше, можно выделить и очистить общеизвестными способами выделения и очистки, такими как концентрирование, концентрирование при пониженном давлении, кристаллизация, экстракция растворителем, переосаждение и хроматография.
Среди соединений общей формулы (I), соединение (I-2), в котором R1 представляет собой -C(=O)NR5R6, может быть получено, например, следующим способом получения 2.
Способ получения 2

где R2, R3, R4, R5 и R6 являются такими, как указано выше.
(Стадия 6)
Данная стадия представляет собой способ получения соединения (I-2) реакцией конденсации с дегидратацией между соединением (I-1), полученным способом получения 1, и соединением (X). Примеры соединения (X) для использования на данной стадии включают амин, такой как метиламин, диметиламин, хлорид аммония, циклопропиламин и пирролидин, а также их соли. Соединение (X) может быть получено в виде коммерческого продукта или получено согласно общеизвестному способу. На данной стадии, используя широко применяемый конденсирующий агент, соединение (I-2) может быть получено согласно общеизвестному способу. Примеры конденсирующего агента включают N,N'-дициклогексилкарбодиимид (DCC), N,N'-диизопропилкарбодиимид (DIC), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (WSC), дифенилфосфорилазид (DPPA), гексафторфосфат (бензотриазол-1-илокси)трисдиметиламинофосфония (BOP), гексафторфосфат (бензотриазол-1-илокси)трипирролидинфосфония (PyBOP), фосфат (7-азабензотриазол-1-илокси)триспирролидинфосфония (PyAOP), гексафторфосфат бромтриспирролидинфосфония (BroP), гексафторфосфат хлортрис(пирролидин-1-ил)фосфония (PyCroP), 3-(диэтоксифосфорилокси)-1,2,3-бензотриазин-4(3H)-он (DEPBT), гексафторфосфат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU) и гидрохлорид 4-(5,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолина (DMTMM). Примеры добавки для использования на данной стадии включают 1-гидроксибензотриазол (HOBt), 1-гидрокси-7-азабензотриазол (HOAt), и N-гидроксисукцинимид (HOSu). Данные добавки можно использовать в количестве 0,1-100 эквивалентов, предпочтительно 1-10 эквивалентов. При желании, можно использовать основание, такое как триметиламин, триэтиламин, трипропиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин, лутидин и коллидин, в количестве 0,1-100 эквивалентов, предпочтительно 1-10 эквивалентов. Растворитель конкретно не ограничен, и можно использовать, например, воду, метанол, этанол, 2-пропанол, тетрагидрофуран, 1,4-диоксан, толуол, метиленхлорид, хлороформ, ацетонитрил, N,N-диметилформамид, N,N-диметилацетамид и диметилсульфоксид. Температура реакции составляет от -30 до 200°C, предпочтительно 0-50°C. Продолжительность реакции составляет 0,1-100 часов, предпочтительно 0,5-24 часов.
Соединение (I-2), полученное, как описано выше, можно выделить и очистить общеизвестными способами выделения и очистки, такими как концентрирование, концентрирование при пониженном давлении, кристаллизация, экстракция растворителем, переосаждение и хроматография.
Среди соединений общей формулы (I), соединение (I-3), в котором R1 представляет собой 1,2,4-оксадиазол, замещенный R7, может быть получено, например, следующим способом получения 3.
Способ получения 3
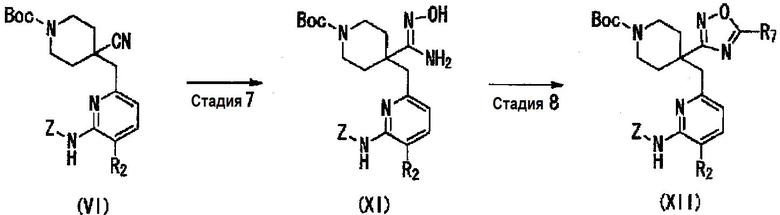

где R7 представляет собой C1-C6алкильную группу или трифторметильную группу; R2, R3, R4, R5, R6 и Z являются такими, как указано выше.
(Стадия 7)
Данная стадия представляет собой способ получения соединения (XI) преобразованием цианогруппы в соединении (VI), которое получают в качестве промежуточного продукта при получении в способе получения 1, в амидоксим. Реакции, применяемые на данной стадии, можно осуществлять, например, способами, описанными в международной публикации No. WO2005/026123, международной публикации No. WO2008/117175, международной публикации No. WO2008/156721, или согласно способу, эквивалентному данным способам. Например, реакции можно осуществлять взаимодействием соединения (VI) с гидроксиламином в спиртовом растворителе, таком как этанол и 2-пропанол. При желании, можно использовать основание, и примеры основания включают органические амины, такие как триэтиламин, диизопропилэтиламин, N-метилморфолин и пиридин, и неорганические соли, такие как бикарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия, метоксид натрия, этоксид натрия и трет-бутоксид калия. Гидроксиламин можно использовать в количестве 1-100 эквивалентов, предпочтительно 1-10 эквивалентов. Температура реакции составляет от комнатной температуры до 150°C, предпочтительно 50-100°C. Продолжительность реакции составляет 0,1-100 часов, предпочтительно 0,5-20 часов.
Соединение (XI), полученное, как описано выше, может быть подвергнуто последующей стадии с или без выделения и очистки общеизвестными способами выделения и очистки, такими как концентрирование, концентрирование при пониженном давлении, кристаллизация, экстракция растворителем, переосаждение и хроматография.
(Стадия 8)
Данная стадия представляет собой способ получения соединения (XII) преобразованием амидоксимной группы в соединении (XI) в 1,2,4-оксадиазольное кольцо. Реакции, применяемые на данной стадии, можно осуществлять, например, способами, описанными в международной публикации No. WO2005/026123, международной публикации No. WO2008/117175, международной публикации No. WO2008/156721, или согласно способу, эквивалентному данным способам. Например, реакции можно осуществлять взаимодействием соединения (XI) с уксусным ангидридом, ацетилхлоридом, триэтилортоформиатом и триэтилортоацетатом в растворителе, таком как толуол, хлороформ, уксусная кислота, N,N-диметилформамид, N-метилпирролидин-2-он и пиридин. При желании, можно использовать основание, и примеры основания включают триэтиламин, диизопропилэтиламин, N-метилморфолин и пиридин. Температура реакции составляет от комнатной температуры до 150°C, предпочтительно 50-100°C. Продолжительность реакции составляет 0,1-100 часов, предпочтительно 0,5-20 часов.
Соединение (XII), полученное, как описано выше, может быть подвергнуто последующей стадии с или без выделения и очистки общеизвестными способами выделения и очистки, такими как концентрирование, концентрирование при пониженном давлении, кристаллизация, экстракция растворителем, переосаждение и хроматография.
(Стадия 9)
Данная стадия представляет собой способ получения соединения (XIII) удалением трет-бутоксикарбонильной группы, которая представляет собой защитную группу соединения (XII), в присутствии кислоты. Данную стадию можно осуществлять способом, аналогично применяемому на приведенной выше стадии 3, или способом, эквивалентным способу, применяемому на стадии 3.
Соединение (XIII), полученное, как описано выше, может быть подвергнуто последующей стадии с или без выделения и очистки общеизвестными способами выделения и очистки, такими как концентрирование, концентрирование при пониженном давлении, кристаллизация, экстракция растворителем, переосаждение и хроматография.
(Стадия 10)
Данная стадия представляет собой способ получения соединения (XIV) реакцией конденсации с дегидратацией между соединением (XIII) и соединением (VIII). Данную стадию можно осуществлять способом, аналогично применяемому на приведенной выше стадии 4, или способом, эквивалентным способу, применяемому на стадии 4.
Соединение (XIV), полученное, как описано выше, может быть подвергнуто последующей стадии с или без выделения и очистки общеизвестными способами выделения и очистки, такими как концентрирование, концентрирование при пониженном давлении, кристаллизация, экстракция растворителем, переосаждение и хроматография.
(Стадия 11)
Данная стадия представляет собой способ получения соединения (I-3) удалением защитной группы (P) заместителя Z в соединении (XIV) в присутствии кислоты. Данную стадию можно осуществлять способом, аналогично применяемому на приведенной выше стадии 5, или способом, эквивалентным способу, применяемому на стадии 5.
Соединение (I-3), полученное, как описано выше, можно выделить и очистить общеизвестными способами выделения и очистки, такими как концентрирование, концентрирование при пониженном давлении, кристаллизация, экстракция растворителем, переосаждение и хроматография.
Среди соединений общей формулы (I), соединение (I-4), в котором R1 представляет собой 1,3,4-оксадиазол, замещенный R7, может быть получено, например, следующим способом получения 4.
Способ получения 4
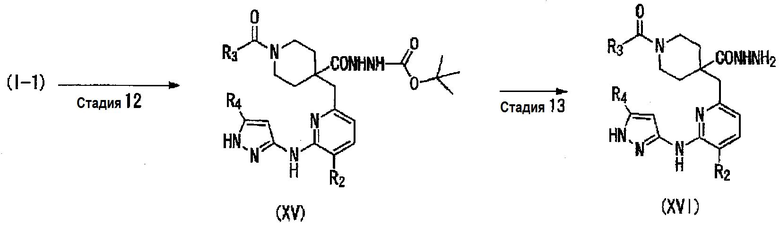

где R7 представляет собой C1-C6алкильную группу или трифторметильную группу; R2, R3 и R4 являются такими, как указано выше.
(Стадия 12)
Данная стадия представляет собой способ получения соединения (XV) реакцией конденсации с дегидратацией между соединением (I-1), полученным способом получения 1, и трет-бутоксикарбонилгидразидом. Данную стадию можно осуществлять способом, аналогично применяемому на приведенной выше стадии 4, или способом, эквивалентным способу, применяемому на стадии 4.
Соединение (XV), полученное, как описано выше, может быть подвергнуто последующей стадии с или без выделения и очистки общеизвестными способами выделения и очистки, такими как концентрирование, концентрирование при пониженном давлении, кристаллизация, экстракция растворителем, переосаждение и хроматография.
(Стадия 13)
Данная стадия представляет собой способ получения соединения (XVI) удалением трет-бутоксикарбонильной группы, которая представляет собой защитную группу соединения (XV), в присутствии кислоты. Данную стадию можно осуществлять способом, аналогично применяемому на приведенной выше стадии 3, или способом, эквивалентным способу, применяемому на стадии 3.
Соединение (XVI), полученное, как описано выше, может быть подвергнуто последующей стадии с или без выделения и очистки общеизвестными способами выделения и очистки, такими как концентрирование, концентрирование при пониженном давлении, кристаллизация, экстракция растворителем, переосаждение и хроматография.
(Стадия 14)
Данная стадия представляет собой способ получения соединения (I-4) преобразованием ацилгидразидной группы в соединении (XVI) в 1,3,4-оксадиазольное кольцо. Данную стадию можно осуществлять способом, аналогично применяемому на приведенной выше стадии 8, или способом, эквивалентным способу, применяемому на стадии 8.
Соединение (I-4), полученное, как описано выше, может быть подвергнуто последующей стадии с или без выделения и очистки общеизвестными способами выделения и очистки, такими как концентрирование, концентрирование при пониженном давлении, кристаллизация, экстракция растворителем, переосаждение и хроматография.
Когда соединение настоящего изобретения включает изомеры, такие как оптические изомеры, стереоизомеры, изомеры положения и ротационные изомеры, смесь любых изомеров также включена соединением настоящего изобретения. Например, когда оптические изомеры существуют для соединения настоящего изобретения, оптические изомеры, которые выделяют из рацемических форм, также включены соединением настоящего изобретения. Данные изомеры можно получить индивидуально в виде отдельного соединения способом получения или способом разделения, известными per se (такими как концентрирование, экстракция растворителем, колоночная хроматография и перекристаллизация).
Соединение настоящего изобретения или его соль могут представлять собой кристалл, и отдельная кристаллическая форма, а также полиморфная смесь включены соединением настоящего изобретения или его солью. Кристалл можно получить проведением кристаллизации, применяя способ кристаллизации, известный per se. Соединение настоящего изобретения или его соль могут представлять собой сольват (такой как гидрат) или несольват, и обе формы включены соединением настоящего изобретения или его солью. Соединение, меченное, например, изотопом (например, 2H, 3H, 13C, 14C, 18F, 35S и 125I), также включено соединением настоящего изобретения или его солью.
Соль соединения настоящего изобретения относится к общепринятой соли, используемой в области органической химии, и ее примеры включают соли, такие как, когда соединение содержит карбоксильную группу, аддитивную соль основания карбоксильной группы, и когда соединение содержит аминогруппу или основную гетероциклическую группу, аддитивную соль кислоты аминогруппы или основной гетероциклической группы.
Примеры аддитивной соли основания включают соль щелочного металла, такую как натриевая соль и калиевая соль; соль щелочноземельного металла, такую как кальциевая соль и магниевая соль; аммониевую соль; и соль органического амина, такую как триметиламмониевая соль, триэтиламмониевая соль, соль дициклогексиламина, соль этаноламина, соль диэтаноламина, соль триэтаноламина, соль прокаина и соль N,N'-дибензилэтилендиамина.
Примеры аддитивной соли кислоты включают соль неорганической кислоты, такую как гидрохлорид, сульфат, нитрат, фосфат и перхлорат; соль органической кислоты, такую как ацетат, формиат, малеат, фумарат, тартрат, цитрат, аскорбат и трифторацетат; и сульфонат, такой как метансульфонат, изетионат, бензолсульфонат и п-толуолсульфонат.
Соединение настоящего изобретения или его соль обладают превосходной селективной ингибирующей aurora A активностью, и в частности, обладают наиболее сильной селективной ингибирующей aurora A активностью, по сравнению с ингибирующей aurora B и aurora C активность и, таким образом, является пригодным в качестве селективного ингибитора aurora A. Кроме того, соединение настоящего изобретения или его соль обладают превосходным противоопухолевым эффектом и, таким образом, является пригодным в качестве противоопухолевого средства. Хотя рак, который будут лечить, конкретно не ограничен, его примеры включают рак головы и шеи, рак пищевода, рак желудка, рак двенадцатиперстной кишки, рак толстой кишки, рак прямой кишки, рак печени, рак желчного пузыря и желчных протоков, рак желчевыводящих путей, рак поджелудочной железы, рак легких, рак молочной железы, рак яичников, рак шейки матки, рак матки, рак почки, рак мочевого пузыря, рак предстательной железы, рак яичка, саркому кости и мягких тканей, гематологический рак, множественную миелому, рак кожи, опухоль головного мозга, мезотелиому и гематологический рак. Предпочтительно, рак, который будут лечить, представляет собой гематологический рак, такой как В-клеточная лимфома, хронический лимфоцитарный лейкоз, периферическая Т-клеточная лимфома, миелодиспластический синдром, острый миелолейкоз, острый лимфоцитарный лейкоз и множественная миелома, рак желудка, рак молочной железы, рак предстательной железы, рак яичника, рак легкого, рак толстой кишки. Кроме того, лекарственное средство настоящего изобретения можно применять на людях и животных, отличных от людей.
Кроме того, принимая во внимание, что соединение настоящего изобретения или его соль обладает превосходной селективной ингибирующей aurora A активностью, когда его применяют в комбинации со средством, воздействующим на микротрубочки, он усиливает противоопухолевый эффект лекарственного средства, воздействующего на микротрубочки. Следовательно, соединение настоящего изобретения или его соль является пригодным в качестве усилителя противоопухолевого эффекта для лекарственного средства, воздействующего на микротрубочки. Композиция, содержащая соединение настоящего изобретения или его соль и агонист микротрубочек, является пригодной в качестве противоопухолевого средства (лекарственного средства для лечения рака). Комбинированное введение соединения настоящего изобретения или его соли и средства, воздействующего на микротрубочки, является пригодным в качестве способа лечения рака. Примеры лекарственного средства, воздействующего на микротрубочки, включают лекарственное средство, стабилизирующее микротрубочки, такое как таксановое противораковое средство и эпотилоновое противораковое средство, и предпочтительно средством, воздействующим на микротрубочки, является таксановое противораковое лекарственное средство. Примеры таксанового противоракового лекарственного средства включают паклитаксел, доцетаксел и кабазитаксел, и предпочтительно таксановым противораковым лекарственным средством является паклитаксел. Примеры эпотилонового противоракового лекарственного средства включают эпотилон B и эпотилон D. Усилитель противоопухолевого эффекта настоящего изобретения можно вводить в любое время, то есть, перед или после, или одновременно с введением средства, воздействующего на микротрубочки. Предпочтительно, усилитель противоопухолевого эффекта настоящего изобретения можно вводить в то же время или в пределах четырех часов после или перед введением средства, воздействующего на микротрубочки. Когда усилитель противоопухолевого эффекта настоящего изобретения вводят отдельно или одновременно с агонистом микротрубочек, например, усилитель противоопухолевого эффекта можно вводить в таком количестве, что количество, по меньшей мере, одного компонента, выбранного из соединений настоящего изобретения или их солей, находится в диапазоне 0,01-100 моль, предпочтительно 0,05-50 моль, более предпочтительно 0,1-20 моль на один моль средства, воздействующего на микротрубочки. Хотя рак, который будут лечить, конкретно не ограничен, его примеры включают рак головы и шеи, рак пищевода, рак желудка, рак двенадцатиперстной кишки, рак толстой кишки, рак прямой кишки, рак печени, рак желчного пузыря и желчных протоков, рак желчевыводящих путей, рак поджелудочной железы, рак легких, рак молочной железы, рак яичников, рак шейки матки, рак матки, рак почки, рак мочевого пузыря, рак предстательной железы, рак яичка, саркому кости и мягких тканей, гематологический рак, множественную миелому, рак кожи, опухоль головного мозга, мезотелиому и гематологический рак. Предпочтительно, рак, который будут лечить, представляет собой гематологический рак, такой как В-клеточная лимфома, хронический лимфоцитарный лейкоз, периферическая Т-клеточная лимфома, миелодиспластический синдром, острый миелолейкоз, острый лимфоцитарный лейкоз и множественная миелома, рак желудка, рак молочной железы, рак предстательной железы, рак яичника, рак легкого, рак толстой кишки. Кроме того, лекарственное средство настоящего изобретения можно применять на людях и животных, отличных от людей.
Кроме того, согласно настоящему изобретению, можно комбинировать компонент, выбранный из группы, состоящей из соединений настоящего изобретения или их солей, которые являются активным ингредиентом приведенного выше усилителя противоопухолевого эффекта, с лекарственным средством, воздействующим на микротрубочки, с получением противоракового лекарственного средства, сформулированного с усилителем противоопухолевого эффекта. В данном случае, противораковое средство можно применять в форме смешанной композиции, содержащей суммарный активный ингредиент, состоящий из средства, воздействующего на микротрубочки, и компонента, выбранного из группы, состоящей из соединений настоящего изобретения или их солей, в одном препарате, альтернативно, противораковое средство можно получить в виде отдельного препарата, причем каждый отдельно содержит активные ингредиенты, альтернативно, противораковое средство можно получить в виде набора.
Когда соединение настоящего изобретения или его соль применяют в качестве лекарственного средства, фармацевтический носитель можно смешивать, при необходимости, с получением фармацевтической композиции. Можно применять различные дозированные формы в зависимости от цели предотвращения или лечения. В качестве дозированной формы возможна, например, любая из перорального средства, инъекции, суппозитория, мази и пластыря. Соединение настоящего изобретения или его соль обладает превосходным пероральным поглощением и проявляет превосходную противоопухолевую активность при пероральном введении. В свете приведенного выше, предпочтительно применять пероральное средство. Каждую из данных дозированных форм можно получить способами получения лекарственных средств, которые являются общеизвестными и широко применяемыми специалистами в данной области техники.
В качестве фармацевтического носителя используют любой тип органических или неорганических веществ-носителей, широко применяемых в качестве фармацевтических материалов, и фармацевтический носитель вмешивают в твердый препарат, такой как, например, эксципиент, связующее, дезинтегрант, лубрикант и краситель, или в жидкий препарат, такой как, например, растворитель, агент, способствующий растворению, суспендирующий агент, изотонический агент, буфер и смягчитель. Кроме того, можно также применять, при необходимости, добавку для лекарственного препарата, такую как консервант, антиоксидант, краситель, подсластитель и стабилизирующий агент.
Когда получают пероральный твердый препарат, эксципиент и, при желании, например, эксципиент, связующее, дезинтегрант, лубрикант, краситель и корригент, добавляют к соединению настоящего изобретения, и затем можно получить обычным способом, например, таблетку, таблетку с покрытием, гранулу, порошок и капсулу. Когда получают инъекцию, например, агент, регулирующий pH, буфер, стабилизатор, изотонический агент и местный анестетик, можно добавлять к соединению настоящего изобретения, и подкожную, внутримышечную и внутривенную инъекцию можно получить обычным способом.
Количество соединения настоящего изобретения, которое будут вводить в каждую из приведенных выше единичных дозированных форм, не является постоянным, но зависит, например, от симптомов у пациента, для которого применяют соединение, и от лекарственной формы соединения. Однако, обычно, количество для единичной дозированной формы желательно составляет 0,05-1000 мг для перорального средства, желательно 0,01-500 мг для инъекции и желательно 1-1000 мг для суппозитория.
Кроме того, дневная доза лекарственного средства приведенной выше дозированной формы изменяется в зависимости от, например, симптомов, массы тела, возраста или пола пациента и, таким образом, не может быть конкретно определена. Однако дневная доза соединения настоящего изобретения для нормального взрослого (массой 50 кг) может составлять 0,05-5000 мг, предпочтительно 0,1-1000 мг, и лекарственное средство предпочтительно вводят один раз в день или приблизительно два или три раза в день в виде разделенных доз.
[Примеры]
Далее, настоящее изобретение конкретно описано примерами и примерами испытаний. Однако настоящее изобретение не ограничивается данными примерами.
Что касается различных реагентов, использованных в примерах, используют коммерческие продукты, если не указано иное. Что касается колоночной хроматографии на силикагеле, использовали Purif-Pack (R) SI, полученную у Schott Moritex Corporation, KP-Sil (R) колонку с силикагелем, полученную у Biotage, или HP-Sil (R) колонку с силикагелем, полученную у Biotage. Что касается основной колоночной хроматографии на силикагеле, использовали Purif-Pack (R) NH, полученную Schott Moritex Corporation, или KP-NH (R) колонку с силикагелем, полученную Biotage. Что касается препаративной тонкослойной хроматографии, использовали Kieselgel TM60F254, Art. 5744, полученную Merck или NH2 силикагельную 60F254 пластину, полученную Wako Pure Chemical Industries, Ltd. ЯМР спектры регистрировали на спектрометре типа AL400 (400 МГц; JEOL, Ltd.), Mercury 400 (400 МГц; Agilent Technologies, Inc.) или спектрометре типа Inova 400 (400MHz; Agilent Technologies, Inc.), снабженном OMNMR зондом (Protasis), используя в качестве внутреннего стандарта тетраметилсилан, когда он содержался в дейтерированном растворителе, или используя в качестве внутреннего стандарта ЯМР растворитель в любом другом случае, и все δ значения выражены в м.д. Микроволновые реакции осуществляли, используя Initiator 8, полученный у Biotage.
Кроме того, что касается спектров ЖХМС, использовали ACQUITY SQD (квадруполь), полученный у Waters Corporation.
Сокращения имеют следующие значения:
с: синглет
д: дуплет
т: триплет
дд: дуплет дуплетов
м: мультиплет
ушир.: уширенный
ушир.с: уширенный синглет
ДМСО-d6: дейтерированный диметилсульфоксид
CDCl3: дейтерированный хлороформ
CD3OD: дейтерированный метанол
Xantphos: 4,5-бис(дифенилфосфино)-9,9-диметилксантен
Pd2(dba)3: трис(дибензилиденацетон)дипалладий (0)
K3PO4: фосфат трикалия
MsOH: метансульфоновая кислота
AIBN: азобисизобутиронитрил
HPMC: гидроксипропилметилцеллюлоза
Пример 1
Получение 1-(2,3-дихлорбензоил)-4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновой кислоты (соединение 1)
(Стадия a) получение трет-бутил 4-((6-бром-5-фторпиридин-2-ил)метил)-4-цианопиперидин-1-карбоксилата
N-Boc-4-цианопиперидин (5,35 г, 25,4 ммоль) растворяли в 100 мл тетрагидрофурана. После охлаждения полученной в результате смеси до -78°C, добавляли раствор комплекса диизопропиламид лития/тетрагидрофуран в циклогексане (1,5M, 16,5 мл, 24,8 ммоль), поддерживая внутреннюю температуру равную -70°C или ниже. Полученную в результате реакционную смесь перемешивали при -78°C в течение 20 минут. К полученной таким образом реакционной смеси добавляли 10 мл раствора 2-бром-6-(бромметил)-3-фторпиридина в ТГФ (6,28 г, 23,4 ммоль), поддерживая внутреннюю температуру равную -70°C или ниже, с последующим перемешиванием при -78°C в течение 20 минут. К данному реакционному раствору добавляли смесь хлористоводородной кислоты (5M, 4,95 мл, 24,8 ммоль) и 95 мл насыщенного водного раствора хлорида аммония, с последующим перемешиванием при комнатной температуре и затем экстракцией этилацетатом. Полученный таким образом экстракт промывали насыщенным раствором соли и сушили над безводным сульфатом натрия, затем фильтровали и концентрировали. Смолистый остаток растворяли в 6 мл этилацетата, и по каплям добавляли 50 мл гептана при перемешивании. Затем добавляли затравочные кристаллы, с последующим перемешиванием при комнатной температуре в течение одного часа. К полученной в результате светло-желтой суспензии дополнительно по каплям добавляли 50 мл гептана, с последующим перемешиванием в течение ночи. Полученное таким образом твердое вещество собирали фильтрованием, промывали раствором этилацетата в гептане и затем сушили при пониженном давлении, с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (7,10 г, 17,8 ммоль) (выход 76%). Значения физических свойств показаны ниже.
1H-ЯМР (CDCl3) δ: 7,45 (1H, т, J=8,1 Гц), 7,31 (1H, дд, J=8,1, 3,5 Гц), 4,16 (2H, ушир.), 3,09-2,93 (2H, м), 3,04 (2H, с), 1,95-1,84 (2H, м), 1,68-1,57 (2H, м), 1,48 (9H, с); ESI-МС m/z 298, 300 (MH+).
(Стадия b) получение трет-бутил 4-((6-(1-трет-бутил-3-метил-1H-пиразол-5-иламино)-5-фторпиридин-2-ил)метил)-4-цианопиперидин-1-карбоксилата
Соединение (6,37 г, 16,0 ммоль), полученное на приведенной выше стадии a, 5-амино-1-трет-бутил-3-метилпиразол (2,42 г, 15,8 ммоль), xantphos (65,9 мг, 114 мкмоль), Pd2(dba)3 (51,1 мг, 55,8 мкмоль) и K3PO4 (3,63 г, 17,1 ммоль) помещали в реакционный контейнер, и добавляли в конце 50 мл толуола, с последующей дегазацией и замещением аргоном. Полученную таким образом смесь перемешивали при 110°C в течение восьми часов, с последующим добавлением 200 мл этилацетата при комнатной температуре. Полученную таким образом смесь промывали водой и насыщенным раствором соли, сушили над сульфатом натрия, затем фильтровали и концентрировали. Остаток растворяли в 10 мл этилацетата, и добавляли 40 мл гептана при перемешивании при 75°C, с последующим перемешиванием при комнатной температуре в течение ночи. Полученное в результате твердое вещество собирали фильтрованием и промывали 15% смесью этилацетат/гептан, и затем сушили при пониженном давлении, с получением указанного в заголовке соединения (4,15 г, 8,81 ммоль) в виде белого твердого вещества (выход 56%). Значения физических свойств показаны ниже.
1H-ЯМР (CDCl3) δ: 7,26 (1H, дд, J=10,7, 8,0 Гц), 6,74 (1H, дд, J=8,0, 3,2 Гц), 6,23-6,15 (2H, м), 4,19-3,92 (2H, м), 3,09-2,92 (2H, м), 2,85 (2H, с), 2,26 (3H, с), 1,95-1,86 (2H, м), 1,64 (9H, с), 1,58-1,48 (2H, м), 1,46 (9H, с); ESI-МС m/z 471 (MH+).
(Стадия c) получение 4-((6-(1-трет-бутил-3-метил-1H-пиразол-5-иламино)-5-фторпиридин-2-ил)метил)пиперидин-4-карбонитрила
Соединение (4,11 г, 8,73 ммоль), полученное на приведенной выше стадии b, растворяли в ТГФ (33 мл), к которому добавляли MsOH (7,0 мл) на водяной бане. Полученный таким образом раствор перемешивали при комнатной температуре в течение двух часов, и полученное в результате содержимое выливали в 160 мл воды. Полученный таким образом водный раствор промывали 50 мл изопропилового эфира и добавляли 21,5 мл 5М гидроксида натрия, с последующей экстракцией этилацетатом. Полученный таким образом этилацетатный раствор промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали, с получением указанного в заголовке соединения (3,09 г, 8,34 ммоль) (выход 96%). Значения физических свойств показаны ниже.
1H-ЯМР (CDCl3) δ: 7,22 (1H, дд, J=10,6, 8,0 Гц), 6,71 (1H, дд, J=8,0, 3,2 Гц), 6,25-6,16 (2H, м), 3,02-2,95 (2H, м), 2,91-2,84 (2H, м), 2,83 (2H, с), 2,21 (3H, с), 1,90-1,83 (2H, м), 1,61 (9H, с), 1,59-1,49 (2H, м); ESI-МС m/z 371 (MH+).
(Стадия d) получение 4-((6-(1-трет-бутил-3-метил-1H-пиразол-5-иламино)-5-фторпиридин-2-ил)метил)-1-(2,3-дихлорбензоил)пиперидин-4-карбонитрила
К смеси соединения (3,65 г, 9,85 ммоль), полученного на приведенной выше стадии c, добавляли 2,3-дихлорбензойную кислоту (2,05 г, 10,8 ммоль) и моногидрат 1-гидроксибензотриазола (1,80 г, 13,3 ммоль), 25 мл ацетонитрила, и затем добавляли гидрохлорид WSC (2,05 г, 10,7 ммоль). Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем добавляли 30 мл 1М гидроксида натрия, с последующим перемешиванием в течение 15 минут. Полученную таким образом смесь экстрагировали этилацетатом. Полученный в результате этилацетатный слой промывали последовательно водой, 1М хлористоводородной кислотой, водой и насыщенным раствором соли. Полученный таким образом этилацетатный раствор промывали безводным сульфатом натрия, затем фильтровали и концентрировали, с получением указанного в заголовке соединения (5,55 г) в виде белого твердого вещества (выход 100%). Значения физических свойств показаны ниже.
ESI-МС m/z 543, 545 (MH+).
(Стадия e) получение соединения 1
Соединение (524 мг, 0,964 ммоль), полученное на приведенной выше стадии d, растворяли в 3 мл 1,4-диоксана, и затем добавляли 3 мл 5М хлористоводородной кислоты. Полученный в результате раствор нагревали при 150°C в течение 10 минут в аппарате для микроволновых реакций. Полученную в результате реакционную смесь концентрировали при пониженном давлении, и полученный таким образом остаток растворяли в хлороформе, с последующей промывкой насыщенным раствором соли. Полученный таким образом хлороформный раствор сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле (хлороформ/метанол=100/0-90/10), и полученное таким образом твердое вещество переосаждали из смеси этанол-этилацетат, с получением указанного в заголовке соединения (290 мг, 0,573 ммоль) в виде белого твердого вещества (выход 59%). Значения физических свойств показаны в таблице 9.
Примеры 2-13
Используя исходные соединения, перечисленные в таблицах 1-3, соединения примеров 2-13 получали согласно способу примера 1. Значения физических свойств показаны в таблицах 9-17.














Пример 14
Получение 1-(3-хлор-2-фторбензоил)-4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)-N-метилпиперидин-4-карбоксамида (соединение 14)
К смеси соединения 13 (50 мг, 0,1 ммоль), полученного в примере 13, добавляли гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (40 мг, 0,21 ммоль), моногидрат 1-гидроксибензотриазола (30 мг, 0,22 ммоль), гидрохлорид метиламина (25 мг, 0,37 ммоль) и 1 мл диметилформамида, 0,05 мл триэтиламина, с последующим перемешиванием при комнатной температуре в течение 13 часов. К полученной в результате реакционной смеси добавляли воду, с последующей экстракцией этилацетатом. Полученный в результате экстракт сушили над безводным сульфатом магния, фильтровали и затем концентрировали. Полученный таким образом остаток очищали ВЭЖХ, с получением указанного в заголовке соединения (41 мг, 0,082 ммоль) в виде белого твердого вещества (выход 82%). Значения физических свойств показаны в таблицах 9-17.
Примеры 15-21
В примерах 15-21, используя исходные соединения, указанные в таблицах 4-5, соединения получали способом согласно примеру 14. Значения физических свойств показаны в таблицах 9-17.
Пример 22
Получение (3-хлор-2-фторфенил)(4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)-4-(5-метил-1,2,4-оксадиазол-3-ил)пиперидин-1-ил)метанона (соединение 22)
(Стадия a) получение трет-бутил 4-((6-(1-трет-бутил-3-метил-1H-пиразол-5-иламино)-5-фторпиридин-2-ил)метил)-4-(N'-гидроксикарбамимидоил)пиперидин-1-карбоксилата
Соединение (50 г), полученное в примере 1 (стадия b), растворяли в 530 мл этанола при 60°C. Полученный в результате раствор охлаждали до комнатной температуры и добавляли 65 мл 50% водного раствора гидроксиламина, с последующим перемешиванием при 60°C в течение 46 часов. Полученный в результате реакционный раствор добавляли к дистиллированной воде, с последующей экстракцией этилацетатом. Полученный в результате органический слой промывали дистиллированной водой и насыщенным раствором соли. Полученный в результате раствор сушили над сульфатом натрия и затем концентрировали при пониженном давлении, с получением указанного в заголовке соединения (53 г, 106 ммоль) (выход 100%). Значения физических свойств показаны ниже.
ESI-МС m/z 504 (MH+).
(Стадия b) получение трет-бутил 4-((6-(1-трет-бутил-3-метил-1H-пиразол-5-иламино)-5-фторпиридин-2-ил)метил)-4-(5-метил-1,2,4-оксадиазол-3-ил)пиперидин-1-карбоксилата
Соединение (53 г, 106 ммоль), полученное на приведенной выше стадии a, суспендировали в 525 мл толуола и добавляли 10 мл уксусного ангидрида, с последующим перемешиванием при комнатной температуре в течение одного часа и 20 минут, и затем при 100°C в течение 16 часов. К полученному в результате реакционному раствору последовательно на бане со льдом добавляли 175 мл водного аммиака, 500 мл дистиллированной воды и 500 мл этилацетата, с последующей промывкой насыщенным раствором соли. Полученный в результате водный слой экстрагировали этилацетатом, полученный в результате органический слой промывали насыщенным раствором соли и сушили над сульфатом натрия, затем концентрировали при пониженном давлении, с получением указанного в заголовке соединения (58 g) в виде грубо очищенного продукта. Значения физических свойств показаны ниже.
ESI-МС m/z 528(MH+).
(Стадия c) получение N-(1-трет-бутил-3-метил-1H-пиразол-5-ил)-3-фтор-6-((4-(5-метил-1,2,4-оксадиазол-3-ил)пиперидин-4-ил)метил)пиридинe-2-амина
Соединение (57 г), полученное на приведенной выше стадии b, растворяли в 210 мл ацетонитрила и на бане со льдом добавляли 27 мл метансульфоновой кислоты, с последующим перемешиванием на бане со льдом в течение одного часа и затем при комнатной температуре в течение 17 часов. Полученный в результате реакционный раствор на бане со льдом добавляли к 500 мл дистиллированной воды, с последующей промывкой 500 мл диизопропилового эфира. К полученному в результате водному слою на бане со льдом добавляли 100 мл 5M гидроксида натрия, и водный слой экстрагировали этилацетатом. Полученный в результате органический слой промывали насыщенным раствором соли, сушили над сульфатом натрия и затем концентрировали при пониженном давлении, с получением указанного в заголовке соединения (44 г, 102 ммоль) (выход 91%). Значения физических свойств показаны ниже.
ESI-МС m/z 428(MH+).
(Стадия d) получение 4-((6-(1-трет-бутил-3-метил-1H-пиразол-5-иламино)-5-фторпиридин-2-ил)метил)-4-(5-метил-1,2,4-оксадиазол-3-ил)пиперидин-1-ил)(3-хлор-2-фторфенил)метанона
Соединение (44 г), полученное на приведенной выше стадии c, 3-хлор-2-фторбензойную кислоту (20 г) и моногидрат 1-гидроксибензотриазола (21 г) растворяли в 343 мл ацетонитрила, и на бане со льдом добавляли гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (22 г), с последующим перемешиванием при комнатной температуре в течение 15 часов. К полученному в результате реакционному раствору добавляли 1М гидроксид натрия (500 мл), с последующей экстракцией этилацетатом. Полученный в результате органический слой промывали дистиллированной водой, 1М хлористоводородной кислотой, дистиллированной водой и насыщенным раствором соли, затем сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный в результате остаток кристаллизовали из смеси гептан-этилацетат, с получением указанного в заголовке соединения (52 г, 89 ммоль) (выход 86%). Значения физических свойств показаны ниже.
ESI-МС m/z 584,586 (MH+).
(Стадия e) получение соединения 22
Соединение (2,94 г, 5,03 ммоль), полученное на приведенной выше стадии d, растворяли в 30 мл 5М хлористоводородной кислоты и 20 мл 2-пропанола, с последующим нагреванием при 100°C в течение двух часов. Полученный в результате реакционный раствор охлаждали на льду, затем добавляли воду и 5М гидроксид натрия, доводя pH до приблизительно 8, с последующей экстракцией этилацетатом. Полученный таким образом экстракт промывали водой и насыщенным раствором соли, затем сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле (хлороформ/метанол=100/0-95/5), с получением указанного в заголовке соединения (2,26 г, 4,28 ммоль) (выход 85%). Значения физических свойств показаны в таблицах 9-17.
Примеры 23-27
В примерах 23-27, используя исходные соединения, указанные в таблицах 6-7, соединения получали способом согласно примеру 22. Значения физических свойств показаны в таблицах 9-17.
Пример 28
Получение (3-хлор-2-фторфенил)(4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)-4-(5-метил-1,3,4-оксадиазол-2-ил)пиперидин-1-ил)метанона (соединение 28)
(Стадия a) получение трет-бутил 2-(1-(3-хлор-2-фторбензоил)-4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-карбонил)гидразинкарбоксилата
Соединение 13 (62 мг, 0,13 ммоль), полученное в примере 13, трет-бутоксикарбонилгидразид (25 мг, 0,19 ммоль) и моногидрат 1-гидроксибензотриазола (30 мг, 0,22 ммоль) растворяли в 3 мл диметилформамида и добавляли гидрохлорид (1-этил-3-(3-диметиламинопропил)карбодиимида (41 мг, 0,22 ммоль), с последующим перемешиванием при комнатной температуре в течение трех часов. К полученной в результате реакционной смеси добавляли воду, полученную в результате смесь экстрагировали этилацетатом и затем сушили над безводным сульфатом магния. Полученную в результате смесь фильтровали и концентрировали, и полученный таким образом остаток очищали колоночной хроматографией на силикагеле (хлороформ/метанол=100/0-95/5), с получением указанного в заголовке соединения (70 мг, 0,12 ммоль) (выход 92%). Значения физических свойств показаны ниже.
ESI-МС m/z 604, 606(MH+).
(Стадия b) получение соединения 28
Соединение (70 мг, 0,12 ммоль), полученное на приведенной выше стадии a, растворяли в 4 мл хлороформа и добавляли 2 мл трифторуксусной кислоты, с последующим перемешиванием при комнатной температуре в течение трех часов. Полученную в результате реакционную смесь концентрировали, и к остатку добавляли хлороформ и насыщенный водный раствор бикарбоната натрия для разделения фаз. Хлороформный слой сушили над безводным сульфатом магния, фильтровали и концентрировали. К полученному таким образом остатку добавляли 4 мл толуола и 0,5 мл этилацетата, с последующим перемешиванием при нагревании при 110°C в течение двух часов. К полученному в результате реакционному раствору добавляли воду при комнатной температуре, с последующей экстракцией этилацетатом. Полученный таким образом экстракт сушили над безводным сульфатом магния, затем фильтровали и концентрировали. Полученный таким образом остаток очищали колоночной хроматографией на силикагеле (хлороформ/метанол=100/0-90/10), с получением указанного в заголовке соединения (39 мг, 0,077 ммоль) (выход 64%). Значения физических свойств показаны в таблицах 9-17.
Пример 29
Получение (3-хлор-2-фторфенил)(4-((5-фтор-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)-4-(3-метил-1,2,4-оксадиазол-5-ил)пиперидин-1-ил)метанона (соединение 29)
К смеси соединения 13 (49 мг, 0,10 ммоль), полученного в примере 13, добавляли гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (38 мг, 0,20 ммоль), моногидрат 1-гидроксибензотриазола (27 мг, 0,20 ммоль), ацетамидоксим (15 мг, 0,20 ммоль), диметилформамид (1 мл) и диизопропилэтиламин (0,07 мл), с последующим перемешиванием при комнатной температуре в течение семи часов. К полученной в результате реакционной смеси добавляли воду, с последующей экстракцией этилацетатом. Полученный таким образом экстракт сушили над безводным сульфатом натрия, затем фильтровали и концентрировали. К полученному таким образом неочищенному продукту добавляли 1,4-диоксан (1 мл), и полученную в результате смесь облучали при 120°C в течение шести часов при перемешивании, применяя аппарат для микроволновых реакций (Biotage Initiator 8). После концентрирования, полученный таким образом остаток очищали ВЭЖХ, с получением указанного в заголовке соединения (29 мг, 0,055 ммоль) в виде светло-оранжевого твердого вещества (выход 55%). Значения физических свойств показаны в таблицах 9-17.
Сравнительный пример 1
Получение 5-(1-(3-хлор-2-фторбензоил)-4-((6-(1H-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-ил)-1,3,4-оксадиазол-2(3H)-она (сравнительное соединение 1)
Сравнительное соединение 1 получали следующим образом согласно способу, описанному в международной публикации No. WO2009/104802. К 5-(4-((6-((1-трет-бутил-1H-пиразол-5-ил)амино)пиридин-2-ил)метил)-1-(3-хлор-2-фторбензоил)пиперидин-4-ил)-1,3,4-оксадиазол-2-(3H)-ону (4,45 г, 8,03 ммоль) добавляли 5М хлористоводородную кислоту (40 мл) и 2-пропанол (40 мл), с последующим перемешиванием при 100°C в течение четырех часов. К полученной в результате реакционной смеси добавляли 5М гидроксид натрия (40 мл), полученный в результате раствор отделяли и экстрагировали хлороформом. Полученный в результате хлороформный экстракт сушили над безводным сульфатом магния, затем фильтровали и концентрировали. Полученный таким образом остаток очищали колоночной хроматографией на силикагеле (хлороформ/метанол=100/0-95/5), и затем промывали в этилацетате при перемешивании, с получением указанного в заголовке соединения (1,63 г, 3,29 ммоль) в виде светло-оранжевого твердого вещества (выход 41%). Значения физических свойств показаны в таблице 18.
Сравнительные примеры 2-5 и 7
В сравнительных примерах 2-5 и 7, используя исходные соединения, указанные в таблице 8, соединения получали согласно способу, эквивалентно применяемому в примере 1. Значения физических свойств показаны в таблице 18.




Сравнительный пример 6
Получение 1-(3-хлор-2-фторбензоил)-4-((5-циано-6-(5-метил-1H-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновой кислоты (сравнительное соединение 6)
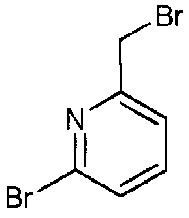
(Стадия a) получение трет-бутил 4-этил 4-((5-бром-6-хлорпиридин-2-ил)метил)пиперидин-1,4-дикарбоксилата
В 21 мл тетрахлорида углерода растворяли 3-бром-2-хлор-6-метилпиридин (880 мг, 4,26 ммоль) и N-бромсукцинимид (682 мг, 3,83 ммоль) и добавляли AIBN (70 мг, 0,426 ммоль), с последующим перемешиванием при 90°C в течение одного часа. Полученный в результате реакционный раствор концентрировали, и полученный таким образом остаток очищали колоночной хроматографией на силикагеле (хлороформ/метанол=100/0-95/5), с получением 3-бром-6-(бромметил)-2-хлорпиридина в виде грубо очищенного продукта.
В 18 мл тетрагидрофурана растворяли этил-N-Boc-пиперидинкарбоксилат (1,16 мл, 4,72 ммоль) и добавляли раствор комплекса диизопропиламид лития/тетрагидрофуран в циклогексане (1,5M, 3,3 мл, 4,96 ммоль) при -78°C, с последующим перемешиванием в течение 40 минут. Затем по каплям добавляли 2 мл раствора 3-бром-6-(бромметил)-2-хлорпиридина в тетрагидрофуране, полученного выше, с последующим дополнительным перемешиванием в течение 10 минут. К полученному в результате реакционному раствору добавляли насыщенный водный раствор хлорида аммония, температуру повышали, и раствор распределяли между водой и этилацетатом. Полученный в результате этилацетатный слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния, затем фильтровали и концентрировали. Полученный таким образом остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат=95/5-65/35), с получением указанного в заголовке соединения (592 мг, 1,28 ммоль) (выход 27%). Значения физических свойств показаны ниже.
ESI-МС m/z 461, 463, 465 (MH+).
(Стадия b) получение этил 4-((5-бром-6-хлорпиридин-2-ил)метил)-1-(3-хлор-2-фторбензоил)пиперидин-4-карбоксилата
Соединение (590 мг, 1,28 ммоль), полученное на приведенной выше стадии a, растворяли в 5 мл хлороформа и добавляли 2 мл трифторуксусной кислоты, с последующим перемешиванием при комнатной температуре в течение одного часа. Полученный в результате реакционный раствор концентрировали и растворяли в 5 мл ДМФА. Затем добавляли 1H-бензо[b][1,2,3]триазол-1-ил-3-хлор-2-фторбензоат (411 мг, 1,41 ммоль) и N,N-диизопропилэтиламин (0,45 мл, 2,56 ммоль) при 0°C, с последующим перемешиванием в течение 15 минут. Добавляли воду, и полученную в результате смесь экстрагировали этилацетатом. Полученный в результате этилацетатный слой промывали водой и насыщенным раствором соли, сушили над безводным сульфатом магния, затем фильтровали и концентрировали. Полученный таким образом остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат=90/10-50/50), с получением указанного в заголовке соединения (581 мг, 1,13 ммоль) (выход 88%). Значения физических свойств показаны ниже.
ESI-МС m/z 517, 519, 521 (MH+).
(Стадия c) получение 4-((5-бром-6-хлорпиридин-2-ил)метил)-1-(3-хлор-2-фторбензоил)пиперидин-4-карбоновой кислоты
Соединение (310 мг, 0,598 ммоль), полученное на приведенной выше стадии b, растворяли в 6 мл этанола и добавляли 5М гидроксид натрия (0,96 мл), с последующим перемешиванием при 80°C в течение 1,5 часов. Полученный в результате реакционный раствор разбавляли водой и добавляли 5М хлористоводородную кислоту, доводя pH до 1, с последующей экстракцией этилацетатом. Полученный в результате этилацетатный слой промывали водой и насыщенным раствором соли, сушили над безводным сульфатом магния, затем фильтровали и концентрировали. Полученный таким образом остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат=30/70-0/100→хлороформ/метанол=100/0-90/10), с получением указанного в заголовке соединения (259 мг, 0,526 ммоль) (выход 88%). Значения физических свойств показаны ниже.
ESI-МС m/z 489, 491, 493 (MH+).
(Стадия d) получение сравнительного соединения 6
К соединению (80 мг, 0,163 ммоль), полученному на приведенной выше стадии c, и цианиду меди (16 мг, 0,180 ммоль) добавляли 1 мл N-метилпирролидона, и полученную в результате смесь облучали при 195°C в течение 40 минут при перемешивании, применяя аппарат для микроволновых реакций (Biotage Initiator 8). Полученный в результате реакционный раствор разбавляли водой и добавляли 1М хлористоводородную кислоту, с последующей экстракцией этилацетатом. Полученный в результате этилацетатный слой промывали водой и насыщенным раствором соли, сушили над безводным сульфатом магния, затем фильтровали и концентрировали. Полученный таким образом остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат=50/50-0/100→хлороформ/метанол=100/0-90/10), с получением, таким образом, 4-((5-циано-6-хлорпиридин-2-ил)метил)-1-(3-хлор-2-фторбензоил)пиперидин-4-карбоновой кислоты (9 мг) в виде грубо очищенного продукта. К грубо очищенному продукту, полученному на данной стадии (9 мг), добавляли 5-амино-1-трет-бутил-3-метилпиразол (3,5 мг, 0,022 ммоль), xantphos (2,4 мг, 0,0041 ммоль), Pd2(dba)3 (2,0 мг, 0,0023 ммоль), фосфат калия (8,7 мг, 0,041 ммоль) и 0,2 мл диоксана, с последующим перемешиванием при 100°C в течение 3,5 часов. Полученный в результате реакционный раствор разбавляли водой и экстрагировали этилацетатом. Полученный в результате этилацетатный слой промывали водой и насыщенным раствором соли, сушили над безводным сульфатом магния, затем фильтровали и концентрировали. Полученный таким образом остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат=25/75-0/100→хлороформ/метанол=100/0-90/10), с получением 4-((6-(1-трет-бутил-5-метил-1H-пиразол-3-иламино)-5-цианопиридин-2-ил)метил)-1-ил)-1-(3-хлор-2-фторбензоил)пиперидин-4-карбоновой кислоты (4 мг) в виде неочищенного продукта. Полученный таким образом неочищенный продукт (4 мг) растворяли в 0,5 мл трифторуксусной кислоты и 0,05 мл анизола, с последующим перемешиванием при нагревании при 85°C в течение одного часа. После концентрирования, полученный таким образом остаток очищали ВЭЖХ с обращенной фазой, с получением указанного в заголовке соединения (1 мг, 0,002 ммоль) (выход 1%). Значения физических свойств показаны в таблице 18.
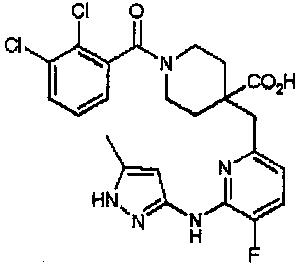



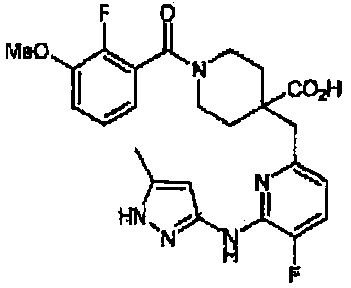





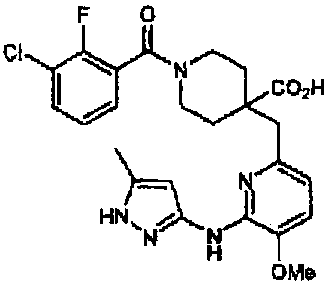


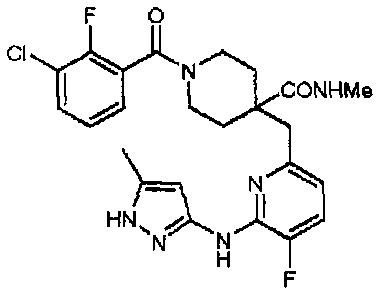


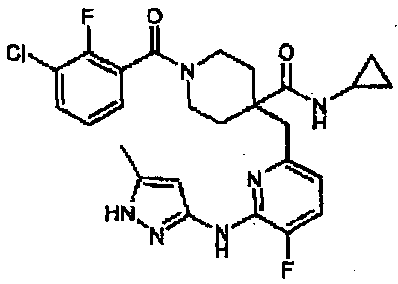
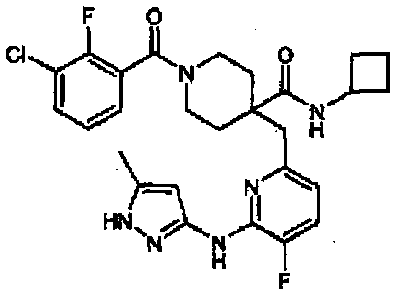

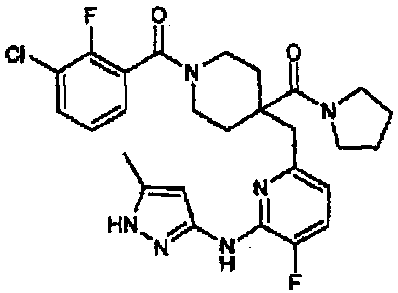
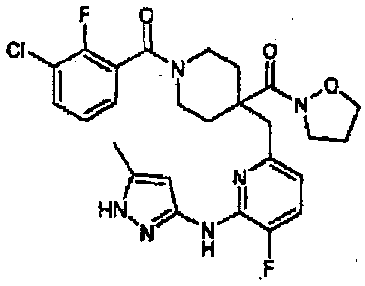

(MH+).

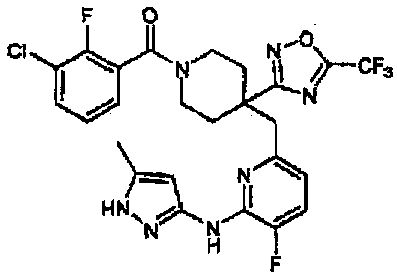










Пример испытания 1: оценка ингибирующих aurora A и aurora B активностей
Ингибирующую активность тестируемого соединения на aurora A и aurora B измеряли согласно следующему способу. В качестве контрольного соединения использовали MLN8237, который проходит клинические испытания в качестве селективного ингибитора aurora A.
1) Очистка белка aurora A
кДНК, кодирующую aurora A человека, содержащую присоединенную по N-концу гистидиновую метку, вставляли в экспрессирующий вектор, и данный белок экспрессировали с высокой концентрацией в штамм E. coli BL21-CodonPlus (DE3)-RIL. E. coli собирали и солюбилизировали, и белок aurora A человека с присоединенной гистидиновой меткой экстрагировали адсорбированием на никелевой хелатной колонке, и затем элюировали с колонки имидазолом. Активную фракцию обессоливали на колонке для обессоливания, с получением, таким образом, очищенного фермента.
2) Измерение ингибирующей активности на aurora A
Способ in vitro измерения ингибирующей активности приведенных выше соединений на киназную активность aurora A осуществляли, ссылаясь на способ, описанный в JP-A-2008-81492. На первой стадии измерения ингибирующей активности соединения, тестируемое соединение последовательно разводили диметилсульфоксидом (ДМСО). Затем очищенный белок aurora A человека, FL-Peptide 21 (Caliper Life Sciences, Inc., конечная концентрация 100 нм), АТФ (конечная концентрация 5 мкм) и раствор соединения настоящего изобретения в ДМСО (конечная концентрация в ДМСО 5%) добавляли к реакционному буферу [50 мМ буфер трис-хлористоводородная кислота (pH 7,4), 15 мМ ацетат магния и 0,2 мМ этилендиамин-N,N,N',N'-тетрауксусная кислота (EDTA)]. Затем полученную в результате смесь инкубировали при 25°C в течение 50 минут, осуществляя киназные реакции. Затем добавляли к ней IMAP (R) Progressive Binding Reagent, разведенный в 500 раз IMAP (R) Progressive Binding Buffer A (продукт Molecular Devices, LLC.), для завершения киназной реакции. После выдерживания полученного в результате продукта в темноте при комнатной температуре в течение 120 минут, степень фосфорилирования определяли из степени флуоресцентной поляризации, как измерено PHERAstar (BMG LABTECH, длина волны возбуждения 485 нм, длина волны детектирования 520 нм). Затем концентрацию соединения, при которой реакция фосфорилирования ингибируется на 50%, определяли как значение IC50 (нМ), и полученные результаты показаны в таблице 19.
3) Измерение киназной активности aurora B
Способ in vitro измерения ингибирующей активности тестируемого соединения на киназную активность aurora B осуществляли аналогичным способом, как приведено выше для aurora A, и очищенный рекомбинантный белок aurora B человека получали у Carna Biosciences, Inc. Реакционный буфер имел следующий состав: 20 мМ HEPES (pH 7,4), 2 мМ DTT, 0,01% Tween-20, хлорид магния при конечной концентрации 1 мМ и АТФ при конечной концентрации 40 мкМ, и время инкубирования составляло 60 минут. Концентрацию соединения, при которой реакция фосфорилирования ингибируется на 50%, определяли как значение IC50 (нМ), и полученные результаты показаны в таблице 19.
Вследствие этого, было подтверждено, что соединения настоящего изобретения обладают большей ингибирующей активностью в отношении aurora A и меньшей ингибирующей активностью в отношении aurora B даже по сравнению с MLN8237, которое являлось контрольным соединением, посредством этого проявляя селективность в отношении aurora A. Наоборот, соединение сравнительного примера 6 не обладало ни ингибирующей активностью в отношении aurora A, ни селективностью в отношении aurora A. Из приведенных выше результатов, было сделано предположение, что введение специфического заместителя в специфическое положение (атом галогена или C1-C6алкоксигруппа при R2) в пиридиновом кольце в структуре соединения настоящего изобретения, представленной общей формулой (I), будет придавать не только высокую ингибирующую активность в отношении aurora A, но также селективность в отношении aurora A.
Пример испытания 2: Оценка ингибирующего эффекта на клеточную пролиферацию
Клетки клеточной линии рака желудка человека SNU-16 стандартно пересевали в RPMI-1640 среде, содержащей 10% фетальную бычью сыворотку (FBS), и среде Игла, модифицированной по способу Дульбекко, содержащей 10% FBS (DMEM), для поддержания клеточной плотности не более чем 80%. Для того чтобы начать испытание на ингибирующую клеточную пролиферацию активность, клетки суспендировали в приведенной выше среде и высевали в каждой лунке 96-луночного планшета с плоским дном (черный планшет с прозрачным дном) при 2500 или 3000 клеток на лунку. Затем клетки выращивали в течение одного дня при 37°C в инкубаторе, содержащем 5% газообразный диоксид углерода. На следующей день, соединение настоящего изобретения и сравнительное соединение растворяли в ДМСО и, применяя ДМСО, тестируемые соединения последовательно разводили до концентрации 200 кратной конечной концентрации. Раствор тестируемых соединений в ДМСО разводили средой, применяемой для выращивания, и затем добавляли к каждой лунке планшета для клеточных культур при конечной концентрации в ДМСО 0,5%. Клетки выращивали в течение 72 часов при 37°C в инкубаторе, содержащем 5% газообразный диоксид углерода. Клетки считали во время добавления тестируемых соединений и через 72 часа после выращивания, используя набор для люминесцентного анализа жизнеспособности клеток CellTiter-Glo (продукт Promega) на основе протокола, рекомендованного Promega. Реагент, включенный в набор, добавляли к каждому планшету, с последующим перемешиванием, и планшеты оставляли при комнатной температуре на 10 минут. После завершения реакции, люминесцентный сигнал измеряли, используя микропланшет-ридер.
Скорость ингибирования клеточной пролиферации рассчитывали по следующей формуле, и определяли концентрацию тестируемого соединения, при которой клеточная пролиферация ингибировалась на 50% (GI50(нМ)). Результаты показаны в таблице 20.
Скорость ингибирования клеточной пролиферации (%)=(C-T)/(C-C0)×100
T: люминесцентный сигнал в лунке с добавлением тестируемого соединения
C: люминесцентный сигнал в лунке без добавления тестируемого соединения
C0: люминесцентный сигнал в лунке, измеренный перед добавлением соединения
Вследствие этого, соединение настоящего изобретения проявляло ингибирующий клеточную пролиферацию эффект и, таким образом, оно предполагается пригодным в качестве противоопухолевого средства.
Пример испытания 3: оценка перорального поглощения
Тестируемое соединение суспендировали или растворяли в 0,5% HPMC и перорально вводили BALB/cA мышам. Кровь отбирали из ретроорбитального венозного сплетения через 0,5, 1, 2, 4 и 6 часов после перорального введения, получая плазму. Полученную таким образом концентрацию соединения в плазме измеряли ЖХМС, и оценивали пероральное поглощение.
Вследствие этого, после перорального введения, приемлемые концентрации в плазме наблюдали для соединений примеров 1, 11, 12, 13 и 22, которые являются соединениями настоящего изобретения, обладающими подходящим пероральным поглощением. Между тем, соединения сравнительных примеров 2-5 и 7 не обладали подходящим пероральным поглощением (AUC0-6час была меньше чем 1/8 AUC0-6час соединений настоящего изобретения). Следовательно, включение данных соединений в перорально вводимые препараты в качестве активного ингредиента считается сложным и, таким образом, не ожидают клинического эффекта после перорального введения. На основе приведенных выше результатов, было обнаружено, что введение атома галогена или C1-C6алкоксигруппы при R2 в пиридиновом кольце в структуре соединения настоящего изобретения, представленной общей формулой (I), придает хорошее пероральное поглощение.
Пример испытания 4: оценка противоопухолевого эффекта (1)
Клеточную линию рака шейки матки человека с введенной люциферазой (Hela-Luc) трансплантировали подкожно безтимусным мышам, и когда объем трансплантированной опухоли достигал 100-200 мм3, распределяли по пять мышей в одной группе в группу с введением одного лекарственного средства и группу с комбинированным введением способом случайного распределения так, чтобы получить равномерный объем опухоли в каждой группе (1 день). В группах с введением одного лекарственного средства, группа 1: паклитаксел (30 мг/кг) вводили внутривенно в 1 день, группа 2: соединение настоящего изобретения (соединение 1) (60 мг/кг) вводили перорально дважды в день на 2 и 3 день, и группа 3: сравнительное соединение 1 (60 мг/кг) вводили перорально дважды в день на 2 и 3 день. В группах с комбинированным введением, группа 4: паклитаксел (30 мг/кг) вводили внутривенно в 1 день, и соединение 1 (60 мг/кг) вводили перорально дважды в день на 2 и 3 день, и группа 5: паклитаксел (30 мг/кг) вводили внутривенно в 1 день, и сравнительное соединение 1 (60 мг/кг) вводили перорально дважды в день на 2 и 3 день. Для того чтобы сравнить противоопухолевый эффект, обусловленный введением лекарственного средства, задавая объем опухоли в момент распределения на группы равный 1, относительный объем опухоли (RTV) определяли согласно следующей формуле как скорость пролиферации опухоли:
RTV=(объем опухоли в день измерения объема опухоли)/(объем опухоли в момент распределения на группы)
В таблице 21 показаны средние значения RTV контроля и групп с введением одного лекарственного средства (группы 2 и 3) на день 23 после распределения на группы, и показаны средние значения RTV группы с введение только паклитаксела (группа 1) и групп с комбинированным введением (группы 4 и 5) на день 23 и 46 после распределения на группы.
Кроме того, частоту контроля заболевания (DCR), описанную, например, в J. Clin. Oncol., 29 (31), стр. 4129-4136, (2010), также применяли в качестве показателя противоопухолевого эффекта, вызываемого введением лекарственного средства. DCR определяли как отношение индивидов, у которых RTV не превышал 1 в последний день измерения объема опухоли (день 46). DCR получали согласно следующей формуле, и результаты показаны в таблице 21:
DRC (%)=[(количество индивидов, у которых RTV не превышал 1 в последний день измерения объема опухоли)/(количество мышей, доживших до последнего дня)]×100
Между тем, в качестве показателя общей токсичности, вызванной введением лекарственного средства, применяли изменение массы тела (BWC). BWC рассчитывали согласно следующей формуле, и средние значения BWC показаны в таблице 21:
BWC (%)=([(масса тела мыши в день измерения массы тела)-(масса тела мыши в момент распределения на группы)]/(масса тела мыши в момент распределения на группы))×100
Вследствие этого, по сравнению с группой с введением только паклитаксела (группа 1), противоопухолевый эффект заметно усиливался в группе, для которой осуществляли комбинированное введение соединения настоящего изобретения (соединение 1) и паклитаксела (группа 4) без значительного увеличения токсичности, которая проявлялась, например, в виде снижения массы тела. Кроме того, на основе величин RTV и DCR на день 46 введения, было обнаружено, что непрерывный эффект уменьшения опухоли можно ожидать в результате введения соединения 1. Между тем, как очевидно из величин RTV и DCR на день 23 и 46 введения, сравнительное соединение 1 не усиливало явно противоопухолевый эффект при введении в комбинации с паклитакселом (группа 5), по сравнению с группой с введением только паклитаксела (группа 1).
Пример испытания 5: оценка противоопухолевого эффекта (2)
Способом, аналогично применяемому в примере испытания 4, клеточную линию рака шейки матки человека с введенной люциферазой (Hela-Luc) трансплантировали подкожно безтимусным мышам, и распределяли по пять мышей в одной группе в группу с введением одного лекарственного средства и группу с комбинированным введением способом случайного распределения (день 1). В группах с введением одного лекарственного средства, паклитаксел (20 мг/кг) вводили внутривенно в день 1. Кроме того, соединение 13 (30 мг/кг) или соединение 22 (100 мг/кг) перорально вводили дважды в день на 2 и 3 день. В группах с комбинированным введением паклитаксел (20 мг/кг) вводили внутривенно в 1 день, и соединение 13 (30 мг/кг) или соединение 22 (60 мг/кг) вводили перорально дважды в день на 2 и 3 день. Средние значения RTV и BWC на день 11 после распределения на группы показаны в таблицах 22 и 23.
Вследствие этого, по сравнению с группой с введением только паклитаксела, противоопухолевый эффект заметно усиливался в группе, для которой осуществляли комбинированное введение соединения 13 или соединения 22, оба из которых представляют собой соединения настоящего изобретения, и паклитаксел, без сильно повышенной токсичности, которая проявляется, например, в виде снижения веса тела.
Пример испытания 6: Измерение киназной активности aurora C
Ингибирующую активность соединения настоящего изобретения на киназную активность aurora C измеряли in vitro. Конкретно, применяя «офф-чип» анализ подвижности пятна, реакции осуществляли следующим способом.
К реакционному буферу (20 мМ HEPES, 0,01% Triton X-100, 2 мМ DTT, pH 7,5) добавляли соединение настоящего изобретения, АТФ (конечная концентрация 25 мкМ), субстраты (кемптид, конечная концентрация 1000 нМ), магний (конечная концентрация 5 мМ) и очищенную киназу aurora C человека, с последующим перемешиванием. Затем проводили реакции при комнатной температуре в течение одного часа. Для получения очищенной киназы aurora C человека, GST белок сливали с N-концом полноразмерной киназы aurora C, и полученный в результате белок экспрессировали бакуловирусом. Слитый белок GST-aurora C очищали глютатионсефарозной хроматографией. После завершения реакции, добавляли раствор для прекращения реакции (QuickScout Screening Assist MSA; Carna Biosciences, Inc.), и пептид, являющийся субстратом, и фосфорилированный пептид в реакционном растворе разделяли и количественно определяли LabChip 3000 системой (Carna Biosciences, Inc.). Способ in vitro для измерения ингибирующей активности соединения настоящего изобретения на киназную активность aurora C проводили согласно способу измерения ингибирующей aurora A активности, показанному в примере испытания 1-2) выше, и определяли концентрацию соединения, при которой реакция фосфорилирования может ингибироваться на 50%, в виде значения IC50 (нМ), и полученную в результате ингибирующую активность на киназу aurora C сравнивали с ингибирующей активностью на aurora A, полученной в приведенном выше примере испытания 1-2).
Вследствие этого, ингибирующая aurora C активность соединения настоящего изобретения была в 80-100 раз меньше ингибирующей aurora A активности, предполагая значительную селективную ингибирующую aurora A активность соединения настоящего изобретения.
Пример испытания 7: способность усиливать эффект лекарственных средств, воздействующих на микротрубочки (in vitro)
Клеточную линию рака желудка OCUM-2M человека и клеточную линию рака шейки матки человека с введенной люциферазой HeLa стандартным способом пересевали в среду Игла, модифицированную по способу Дульбекко (DMEM), содержащую 10% фетальную бычью сыворотку (FBS), при плотности клеток не больше 80%. Для того чтобы начать испытание на ингибирующую клеточную пролиферацию активность, клетки суспендировали в приведенной выше среде и высевали в каждой лунке 96-луночного планшета с плоским дном (черный планшет с прозрачным дном) при 2500 или 3000 клеток на лунку. Затем клетки выращивали в течение одного дня при 37°C в инкубаторе, содержащем 5% газообразный диоксид углерода. На следующий день, последовательно разведенные растворы лекарственных средств, воздействующих на микротрубочки (доцетаксел, кабазитаксел и эпотилон B), получали с ДМСО (10 доз получали для каждого лекарственного средства при тестируемой концентрации в диапазоне 0,03 нМ-1000 нМ), и растворы разводили средой. Затем полученные в результате растворы добавляли к каждой лунке планшета для клеточных культур при конечной концентрации в ДМСО 0,1%. Кроме того, для того чтобы проверить комбинированный эффект лекарственного средства, воздействующего на микротрубочки, и соединения настоящего изобретения, приведенное выше соединение разводили ДМСО до конечной концентрации 300 нМ (конечная концентрация в ДМСО 0,1%) и затем добавляли к каждой лунке планшета для клеточных культур. В качестве группы сравнительного контроля, лунки, каждая из которых содержала только средство, воздействующее на микротрубочки, или только соединение настоящего изобретения, получали отдельно, и клетки в них выращивали в инкубаторе, содержащем 5% газообразный диоксид углерода при 37°C в течение 72 часов. Клетки считали, используя набор для люминесцентного анализа жизнеспособности клеток CellTiter-Glo (продукт Promega) на основе протокола, рекомендованного Promega. Реагент, включенный в набор, добавляли к каждой лунке, с последующим перемешиванием, и планшеты оставляли при комнатной температуре на 10 минут. После завершения реакции, люминесцентный сигнал измеряли, используя микропланшет-ридер.
Скорость клеточной пролиферации рассчитывали по следующей формуле, и определяли концентрацию тестируемого соединения, при которой клеточная пролиферация ингибировалась на 50% (IC50(мкМ)).
Скорость клеточной пролиферации (%)=T/C×100
T: люминесцентный сигнал в лунке с добавлением тестируемого соединения
C: люминесцентный сигнал в лунке без добавления тестируемого соединения
Кроме того, определяли значения IC50 только средства, воздействующего на микротрубочки (IC50 - средство, воздействующее на микротрубочки), и значения IC50 средства, воздействующего на микротрубочки, при комбинированном введении агониста микротрубочек и соединения настоящего изобретения (IC50 - комбинированное введение). Последнее значение IC50 (значение IC50 - комбинированное введение) рассчитывали из скорости ингибирования клеточной пролиферации (переведенное значение) в группе с комбинированным введением путем определения скорости клеточной пролиферации только соединения настоящего изобретения как 100%. Степень усиления эффекта лекарственного средства, воздействующего на микротрубочки, при добавлении соединения настоящего изобретения оценивали порядком величины, рассчитанной по следующей формуле:
(Усиливающий эффект комбинированного введения)=(значение IC50_комбинированное введение)/(значение IC50_средство, воздействующее на микротрубочки)
Оценку проводили согласно критерию, по которому величину, большую 1, оценивали как проявляющую сильный усиливающий эффект, тогда как величину, меньшую или равную 1, оценивали как проявляющую слабый усиливающий эффект.
Вследствие этого, путем добавления соединения настоящего изобретения к средствам, воздействующим на микротрубочки, таким как доцетаксел, кабазитаксел и эпотилон B, величину, большую 2, получали по приведенной выше формуле, что указывает на то, что соединение настоящего изобретения усиливало ингибирующий клеточную пролиферацию эффект данных агонистов микротрубочек.
Как показано выше, соединения настоящего изобретения селективно и отлично ингибировали aurora A, показывая превосходный противоопухолевый эффект с подходящим пероральным поглощением. Кроме того, было показано, что соединение настоящего изобретения сильно усиливает противоопухолевый эффект паклитаксела в испытаниях in vivo при использовании безтимусных мышей. Кроме того, было показано, что соединение настоящего изобретения усиливает также ингибирующий клеточную пролиферацию эффект лекарственных средств, воздействующих на микротрубочки.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДУЛИРУЮЩИЕ JAK КИНАЗУ ХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2529019C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2340611C2 |
| ИНГИБИТОРЫ АВРОРА-КИНАЗЫ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2822464C2 |
| ИНГИБИТОРЫ КИНАЗ, ПРИМЕНИМЫЕ ДЛЯ ЛЕЧЕНИЯ МИЕЛОПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ И ДРУГИХ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2007 |
|
RU2482112C2 |
| ДИАМИНОГЕТЕРОЦИКЛИЧЕСКОЕ КАРБОКСАМИДНОЕ СОЕДИНЕНИЕ | 2010 |
|
RU2526253C2 |
| НОВЫЙ ИНГИБИТОР AURORA КИНАЗЫ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2835080C1 |
| НОВОЕ ПРОИЗВОДНОЕ НИКОТИНАМИДА ИЛИ ЕГО СОЛЬ | 2011 |
|
RU2560163C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНОНА И ПИРИДАЗИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ПОЛИ(ADP-РИБОЗА) ПОЛИМЕРАЗЫ (PARP) | 2007 |
|
RU2472782C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА И СОДЕРЖАЩИЕ ИХ ИНГИБИТОРЫ MELK | 2011 |
|
RU2582610C2 |
| НОВОЕ 2-ГЕТЕРОАРИЛ-ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ БЕНЗИМИДАЗОЛА | 2004 |
|
RU2329261C2 |
Настоящее изобретение относится к новому пиперидиновому соединению, представленному общей формулой (I), или к его фармацевтически приемлемой соли. Данное соединение обладает превосходным селективным ингибирующим aurora A действием и является пригодным в качестве перорально вводимого противоракового лекарственного средства. Кроме того, настоящее изобретение относится к новому средству для усиления противоопухолевого эффекта агонистов микротрубочек, которые включают таксановое противораковое средство для комбинированной терапии. В соединении формулы (I) R1 представляет собой карбоксильную группу -C(=O)NR5R6 или оксадиазолильную группу, необязательно содержащую C1-C6алкильную группу или трифторметильную группу в качестве заместителя; R2 представляет собой атом галогена или C1-C6алкоксигруппу; R3 представляет собой фенильную группу, необязательно содержащую 1-3 одинаковые или различные группы, выбранные из атома галогена, C1-C6алкильной группы, C1-C6алкоксигруппы и трифторметильной группы в качестве заместителя; R4 представляет собой атом водорода или C1-C6алкильную группу; и R5 и R6 являются одинаковыми или различными, и каждый представляет собой атом водорода, C1-C6алкильную группу или C3-C6циклоалкильную группу, или R5 и R6 необязательно образуют 3-6-членную содержащую азот насыщенную гетероциклическую группу вместе с атомом азота, с которым связаны R5 и R6. 10 н. и 14 з.п. ф-лы, 23 табл., 28 пр.
1. Пиперидиновое соединение, представленное общей формулой (I), или его фармацевтически приемлемая соль

где R1 представляет собой карбоксильную группу, -C(=O)NR5R6 или оксадиазолильную группу, необязательно содержащую С1-С6алкильную группу или трифторметильную группу в качестве заместителя;
R2 представляет собой атом галогена или С1-С6алкоксигруппу;
R3 представляет собой фенильную группу, необязательно содержащую 1-3 одинаковые или различные группы, выбранные из атома галогена, С1-С6алкильной группы, С1-С6алкоксигруппы и трифторметильной группы в качестве заместителя;
R4 представляет собой атом водорода или С1-С6алкильную группу;
R5 и R6 являются одинаковыми или различными, и каждый представляет собой атом водорода, С1-С6алкильную группу или С3-С6циклоалкильную группу, или R5 и R6 необязательно образуют 3-6-членную содержащую азот насыщенную гетероциклическую группу вместе с атомом азота, с которым связаны R5 и R6.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где в общей формуле (I) R1 представляет собой карбоксильную группу, -C(=O)NR5R6 (где R5 и R6 являются одинаковыми или различными, и каждый представляет собой атом водорода, С1-С6алкильную группу или С3-С6циклоалкильную группу, или R5 и R6 необязательно образуют азетидинильную группу, пирролидинильную группу или изоксазолидинильную группу вместе с атомом азота, с которым связаны R5 и R6) или оксадиазолильную группу, необязательно содержащую С1-С6алкильную группу или трифторметильную группу в качестве заместителя.
3. Соединение или его фармацевтически приемлемая соль по п. 1, где в общей формуле (I) R1 представляет собой карбоксильную группу, -C(=O)NR5R6 (где R5 и R6 являются одинаковыми или различными, и каждый представляет собой атом водорода, метильную группу, циклопропильную группу или циклобутильную группу, или R5 и R6 представляют собой азетидинильную группу, пирролидинильную группу или изоксазолидинильную группу вместе с атомом азота, с которым связаны R5 и R6) или оксадиазолильную группу, необязательно содержащую метильную группу или трифторметильную группу в качестве заместителя.
4. Соединение или его фармацевтически приемлемая соль по п. 1, где в общей формуле (I) R2 представляет собой атом фтора, атом хлора или С1-С4алкоксигруппу.
5. Соединение или его фармацевтически приемлемая соль по п. 1, где в общей формуле (I) R2 представляет собой атом фтора, атом хлора или метоксигруппу.
6. Соединение или его фармацевтически приемлемая соль по п. 1, где в общей формуле (I) R3 представляет собой фенильную группу, необязательно содержащую 1-2 одинаковые или различные группы, выбранные из атома галогена, С1-С4алкильной группы, С1-С4алкоксигруппы и трифторметильной группы в качестве заместителя.
7. Соединение или его фармацевтически приемлемая соль по п. 1, где в общей формуле (I) R3 представляет собой фенильную группу, необязательно содержащую 1-2 одинаковые или различные группы, выбранные из атома фтора, атома хлора, метильной группы, метоксигруппы и трифторметильной группы в качестве заместителя.
8. Соединение или его фармацевтически приемлемая соль по п. 1, где в общей формуле (I) R4 представляет собой атом водорода или С1-С4алкильную группу.
9. Соединение или его фармацевтически приемлемая соль по п. 1, где в общей формуле (I) R1 представляет собой карбоксильную группу, -C(=O)NR5R6 (где R5 и R6 являются одинаковыми или различными, и каждый представляет собой атом водорода, С1-С4алкильную группу или С3-С6циклоалкильную группу, или R5 и R6 необязательно образуют азетидинильную группу, пирролидинильную группу или изоксазолидинильную группу вместе с атомом азота, с которым связаны R5 и R6) или оксадиазолильную группу, необязательно содержащую С1-С4алкильную группу или трифторметильную группу в качестве заместителя; R2 представляет собой атом фтора, атом хлора или С1-С4алкоксигруппу; R3 представляет собой фенильную группу, необязательно содержащую 1-2 одинаковые или различные группы, выбранные из атома галогена, С1-С4алкильной группы, С1-С4алкоксигруппы и трифторметильной группы в качестве заместителя; и R4 представляет собой атом водорода или С1-С4алкильную группу.
10. Соединение или его фармацевтически приемлемая соль по п. 9, где в общей формуле (I) R1 представляет собой карбоксильную группу, -C(=O)NR5R6 (где R5 и R6 являются одинаковыми или различными, и каждый представляет собой атом водорода, метильную группу, циклопропильную группу или циклобутильную группу, или R5 и R6 представляют собой азетидинильную группу, пирролидинильную группу или изоксазолидинильную группу вместе с атомом азота, с которым связаны R5 и R6) или оксадиазолильную группу, необязательно содержащую метильную группу или трифторметильную группу в качестве заместителя; R2 представляет собой атом фтора, атом хлора или метоксигруппу; R3 представляет собой фенильную группу, необязательно содержащую 1-2 одинаковые или различные группы, выбранные из атома фтора, атома хлора, метильной группы, метоксигруппы и трифторметильной группы в качестве заместителя; и R4 представляет собой атом водорода или метильную группу.
11. Соединение или его фармацевтически приемлемая соль по п. 10, где в общей формуле (I) R1 представляет собой карбоксильную группу, -C(=O)NR5R6 (где R5 и R6 являются одинаковыми или различными, и каждый представляет собой атом водорода или метильную группу, или R5 и R6 представляет собой изоксазолидинильную группу вместе с атомом азота, с которым связаны R5 и R6) или 1,2,4-оксадиазолильную группу или 1,3,4-оксадиазолильную группу, необязательно содержащую метильную группу в качестве заместителя; R2 представляет собой атом фтора, атом хлора или метоксигруппу; R3 представляет собой фенильную группу, содержащую 1-2 одинаковые или различные группы, выбранные из атома фтора, атома хлора, метильной группы, метоксигруппы и трифторметильной группы в качестве заместителя; и R4 представляет собой атом водорода или метильную группу.
12. Соединение или его фармацевтически приемлемая соль по п. 11, где в общей формуле (I) R1 представляет собой карбоксильную группу или 1,2,4-оксадиазолильную группу, необязательно содержащую метильную группу в качестве заместителя; R2 представляет собой атом фтора; R3 представляет собой фенильную группу, содержащую 1-2 одинаковые или различные группы, выбранные из атома фтора и атома хлора в качестве заместителя; и R4 представляет собой метильную группу.
13. Соединение или его фармацевтически приемлемая соль по п. 1, где соединение представляет собой соединение, выбранное из следующей группы соединений:
1-(2,3-дихлорбензоил)-4-((5-фтор-6-(5-метил-1Н-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновой кислоты;
1-(2-фтор-3-трифторметилбензоил)-4-((5-фтор-6-(5-метил-1Н-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновой кислоты;
1-(3-хлор-2-фторбензоил)-4-((5-фтор-6-(1Н-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновой кислоты;
1-(3-хлор-2-фторбензоил)-4-((5-метокси-6-(5-метил-1Н-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновой кислоты;
1-(3-хлор-2-фторбензоил)-4-((5-хлор-6-(5-метил-1Н-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновой кислоты;
1-(3-хлор-2-фторбензоил)-4-((5-фтор-6-(5-метил-1Н-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-карбоновой кислоты;
1-(3-хлор-2-фторбензоил)-4-((5-фтор-6-(5-метил-1Н-пиразол-3-иламино)пиридин-2-ил)метил)-N-метилпиперидин-4-карбоксамида;
1-(3-хлор-2-фторбензоил)-4-((5-фтор-6-(5-метил-1Н-пиразол-3-иламино)пиридин-2-ил)метил)-N,N-диметилпиперидин-4-карбоксамида;
азетидин-1-ил(1-(3-хлор-2-фторбензоил)-4-((5-фтор-6-(5-метил-1Н-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-ил)метанона;
(1-(3-хлор-2-фторбензоил)-4-((5-фтор-6-(5-метил-1Н-пиразол-3-иламино)пиридин-2-ил)метил)пиперидин-4-ил)(изоксазолидин-2-ил)метанона;
(3-хлор-2-фторфенил)(4-((5-фтор-6-(5-метил-1Н-пиразол-3-иламино)пиридин-2-ил)метил)-4-(5-метил-1,2,4-оксадиазол-3-ил)пиперидин-1-ил)метанона;
(2,3-дихлорфенил)(4-((5-фтор-6-(5-метил-1Н-пиразол-3-иламино)пиридин-2-ил)метил)-4-(5-метил-1,2,4-оксадиазол-3-ил)пиперидин-1-ил)метанона;
(3-хлор-2-фторфенил)(4-((5-фтор-6-(5-метил-1Н-пиразол-3-иламино)пиридин-2-ил)метил)-4-(1,2,4-оксадиазол-3-ил)пиперидин-1-ил)метанона;
(3-хлор-2-фторфенил)(4-((5-фтор-6-(5-метил-1Н-пиразол-3-иламино)пиридин-2-ил)метил)-4-(5-метил-1,3,4-оксадиазол-2-ил)пиперидин-1-ил)метанона;
(3-хлор-2-фторфенил)(4-((5-фтор-6-(5-метил-1Н-пиразол-3-иламино)пиридин-2-ил)метил)-4-(3-метил-1,2,4-оксадиазол-5-ил)пиперидин-1-ил)метанона.
14. Селективный ингибитор aurora А, содержащий соединение или его фармацевтически приемлемую соль по любому из пп. 1-13 в качестве активного ингредиента.
15. Фармацевтическая композиция, предназначенная для лечения заболеваний, связанных с aurora А, содержащая соединение или его фармацевтически приемлемую соль по любому из пп. 1-13 и фармацевтический носитель.
16. Противоопухолевое средство, содержащее соединение или его фармацевтически приемлемую соль по любому из пп. 1-13 в качестве активного ингредиента.
17. Средство для усиления противоопухолевого эффекта агониста микротрубочек, содержащее соединение или его фармацевтически приемлемую соль по любому из пп. 1-13 в качестве активного ингредиента.
18. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-13 для получения противоопухолевого средства.
19. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-13 для получения средства для усиления противоопухолевого эффекта агониста микротрубочек.
20. Соединение или его фармацевтически приемлемая соль по п. 1 для лечения рака.
21. Соединение или его фармацевтически приемлемая соль по п. 1 для усиления противоопухолевого эффекта агониста микротрубочек.
22. Способ лечения рака, включающий введение эффективной дозы соединения или его фармацевтически приемлемой соли по п. 1.
23. Способ усиления противоопухолевого эффекта агониста микротрубочек, включающий введение эффективной дозы соединения или его фармацевтически приемлемой соли по п. 1.
24. Способ лечения рака, включающий комбинирование эффективной дозы соединения или его фармацевтически приемлемой соли по п. 1 и эффективной дозы агониста микротрубочек.
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| НОВЫЕ АМИНОПИРИДИНОВЫЕ ИЛИ АМИНОПИРАЗИНОВЫЕ ПРОИЗВОДНЫЕ С СЕЛЕКТИВНОЙ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ АВРОРЫ А | 2007 |
|
RU2437880C2 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |

