Область техники, к которой относится изобретение
Настоящее изобретение относится к новым соединениям для использования при лечении или профилактике раковых заболеваний и других пролиферативных состояний, которые, например, характеризуются клетками, экспрессирующими цитохром P450 1В1 (CYP1B1) и его аллельные варианты. Настоящее изобретение также относится к фармацевтическим композициям, включающим одно или более таких соединений, для применения при консервативном лечении, например, при лечении или профилактике раковых заболеваний или других пролиферативных состояний, а также к способам лечения раковых заболеваний или других состояний у людей или животных. Настоящее изобретение также относится к способам идентификации новых соединений для использования при лечении или профилактике раковых заболеваний и других пролиферативных состояний, которые, например, характеризуются клетками, экспрессирующими CYP1B1 и его аллельные варианты. Настоящее изобретение также относится к способу определения эффективности соединения согласно изобретению в лечении рака.
Предпосылки создания изобретения
CYP1B1 является членом семейства диоксин-индуцируемых CYP1-генов, которое также включает CYP1A1 и CYP1A2, как описывается Sutter и др. (J. Biol. Chem., 269(18), 13092-9 (1994, 6 мая)). CYP1B1 представляет собой фермент гем-тиолатмонооксигеназу, который способен метаболизировать и активировать множество субстратов, включая стероиды, ксенобиотики, лекарственные средства и/или пролекарства. CYP1B1-белок экспрессируется с высокой частотой в случае большого ряда первичных и метастатических раковых заболеваний человека различных гистогенных типов и не экспрессируется или экспрессируется в незначительных количествах в нормальной ткани (см., например: McFadyen M.C., Melvin W.T. и Murray G.I., «Cytochrome P450 Enzymes: Novel Options for Cancer Therapeutics», Mol. Cancer Ther., 3(3), 363-71 (2004); McFadyen M.C. и Murray G.I., «Cytochrome P450 1B1: a Novel Anticancer Therapeutic Target», Future Oncol., 1(2), 259-63 (2005); Sissung T.M., Price D.K., Sparreboom A. и Figg W.D., «Pharmacogenetics and Regulation of Human Cytochrome Р450 1B1: Implications in Hormone-Mediated Tumor Metabolism and a Novel Target for Therapeutic Intervention», Mol. Cancer Res., 4(3), 135-50 (2006)).
Более конкретно, показано, что CYP1B1 экспрессируется в случае раковых заболеваний мочевого пузыря, головного мозга, молочной железы, ободочной кишки, головы и шеи, почки, легкого, печени, яичника, простаты и кожи, но не экспрессируется в соответствующей нормальной ткани. Например, авторами Barnet и др., в Clin. Cancer Res., 13(12), 3559-67 (2007), сообщалось, что CYP1B1 сверхэкспрессируется в глиальных опухолях, включая глиобластомы, анапластические астроцитомы, олигодендроглиомы и анапластические олигодендроглиомы, однако, ткань головного мозга не затрагивается; авторами Carnell и др., в Int. J. Radiat. Oncol. Biol. Phys., 58(2), 500-9 (2004), сообщалось, что CYP1B1 сверхэкспрессируется в аденокарциномах простаты, но не в соответствующей нормальной ткани простаты; авторами Carnell и др., (2004) (там же), также показано, что CYP1B1 экспрессируется в (n=22, 100%) карциномах мочевого пузыря; авторами Downie и др., в Clin. Cancer Res., 11(20), 7369-75 (2005), и McFadyen и др., в Br. J. Cancer, 85(2), 242-6 (2001), сообщалось об увеличивающейся экспрессии CYP1B1 в случае первичного и метастатического овариального рака, но не в нормальной ткани яичника; и авторами Gibson и др., в Mol. Cancer Ther., 2(6), 527-34 (2003), и Kumarakulasingham и др., в Clin. Cancer Res., 11(10), 3758-65 (2005), сообщалось, что CYP1B1 сверхэкспрессируется в аденокарциномах ободочной кишки по сравнению с соответствующей нормальной тканью.
В некоторых исследованиях показано, что CYP1B1 сверхэкспрессируется в случае рака молочной железы по сравнению с соответствующей нормальной тканью (см., например: Murray G.I., Taylor M.C., McFadyen M.C., McKay J.A., Greenlee W.F., Burke M.D. и Melvin W.T., «Tumor-Specific Expression of Cytochrome P450 CYP1B1», Cancer Res., 57(14), 3026-31 (1997); Haas S., Piert C., Harth V., Pesch B., Rabstein S., Bruning T., Ko Y., Hamann U., Justenhoven C., Brauch H. и Fischer H.P., «Expression of Xenobiotic and Steroid Hormone Metabolizing Enzymes in Human Breast Carcinomas», Int. J. Cancer, 119(8), 1785-91 (2006); McKay J.A., Murray G.I., Ah-See A.K., Greenlee W.F., Marcus C.B., Burke M.D. и Melvin W.T., «Differential Expression of CYP1A1 и CYP1B1 in Human Breast Cancer», Biochem. Soc. Trans., 24(2), 327S (1996)).
Авторами Everett и др., в J. Clin. Oncology, 25, 18S (2007), сообщалось, что CYP1B1 сверхэкспрессируется в случае злокачественной меланомы и диссеминированного заболевания, но не в случае нормальной кожи. Авторами Chang и др., в Toxicol. Sci., 71(1), 11-9 (2003), сообщалось, что CYP1B1-белок не присутствует в нормальной печени; с другой стороны, Everett и др., (2007) (там же), подтвердили сверхэкспрессию CYP1B1 в случае метастаза меланомы стадии IV в печени, но не в примыкающей нормальной печеночной ткани.
Авторами Greer и др., в Proc. Am. Assoc. Cancer Res., 45, 3701 (2004), сообщалось, что CYP1B1 сверхэкспрессируется во время злокачественной прогрессии плоскоклеточного рака головы и шеи, но не в нормальном эпителии.
Авторами McFadyen и др., в Br. J. Cancer, 91(5), 966-71 (2004), определен CYP1B1 в почечных карциномах, но не в соответствующей нормальной ткани.
Murray и др., (2004) (там же), использовали иммуногистохимию для обнаружения сверхэкспрессии CYP1B1 в случае клеток рака легкого по сравнению с нормальной тканью легкого. Su и др., в Anti-Cancer Res., 2, 509-15 (2009), использовали иммуногистохимию для обнаружения сверхэкспрессии CYP1B1 в случае продвинутой стадии IV немелкоклеточного рака легкого по сравнению с более ранними стадиями заболевания.
Из цитированных выше многочисленных раскрытий очевидно, что экспрессия CYP1B1 является характерной в случае ряда различных раковых заболеваний и других пролиферативных состояний и что экспрессия CYP1B1 может быть использована для определения такого ряда раковых заболеваний и других состояний. Так как нормальные (не являющиеся раковыми) клетки не экспрессируют значительные уровни CYP1B1, можно также обоснованно ожидать, что соединения, которые проявляют цитотоксичность в клетках, экспрессирующих CYP1B1, но являются посуществу не цитотоксическими в случае нормальных клеток, могут быть полезными в качестве целенаправленных противораковых агентов в случае раковых заболеваний, характеризующихся экспрессией CYP1B1. Под термином «целенаправленный» понимают, что такие соединения могут быть системно доставляемыми и могут быть активированы только в присутствии раковых клеток, экспрессирующих CYP1B1, оставаясь посуществу нетоксичными в отношении остальной части организма.
Кроме того, целый ряд ферментов цитохромов Р450 известен в отношении метаболизации и детоксификации множества противораковых лекарственных средств. Авторами McFadyen и др. (Biochem. Pharmacol., 62(2), 207-12 (2001, 15 июля)) продемонстрировано значительное уменьшение в отношении чувствительности доцетаксела в клетках, экспрессирующих CYP1B1, по сравнению с не экспрессирующими CYP1B1 клетками. Это обнаружение указывает на то, что наличие CYP1B1 в клетках может уменьшать их чувствительность к некоторым цитотоксическим лекарственным средствам. CYP1B1-активируемые пролекарства, следовательно, могут быть пригодны для лечения раковых заболеваний, лекарственная резистентность которых опосредуется CYP1B1.
Кроме того, CYP1B1-ген является высокополиморфным в случае рака и идентифицированы некоторые отдельные нуклеотидные полиморфизмы, содержащиеся в CYP1B1-гене, которые изменяют экспрессию и/или активность кодируемого белка. Из них аллель CYP1B1*3 (4326C>G; L432V) характеризуется как увеличивающейся экспрессией, так и кинетиками фермента CYP1B1 по отношению к некоторым субстратам, как описывается Sissung и др. в Mol. Cancer Ther., 7(1), 19-26 (2008) и в приведенных там ссылках. Это обнаружение указывает на то, что не только CYP1B1, но и аллельные варианты фермента также могут способствовать активации пролекарства и целенаправленности на рак.
Пролекарства исследовали в качестве средства для уменьшения нежелательной токсичности или какого-либо другого нежелательного свойства лекарственного средства без потери эффективности. Пролекарство представляет собой лекарственное средство, которое химически модифицировано для превращения его в неактивную форму, но которое после введения метаболизируется или другим образом превращается в активную форму лекарственного средства в организме. Сверхэкспрессия CYP1B1 в первичных опухолях и при метастатическом заболевании по сравнению с нормальной тканью является великолепной возможностью для создания активируемых CYP1B1 пролекарств в отношении целенаправленной раковой терапии, как рассматривается McFadyen и др., Mol. Cancer Ther., 3(3), 363-71 (2004). В самом деле, обнаружение и создание активируемых CYP1B1 пролекарств для целенаправленной раковой терапии, вероятно, представляет собой значительные фармакологические преимущества над существующими нецеленаправленными, активируемыми цитохромом Р450, клинически используемыми пролекарствами, как, например, алкилирующие пролекарство агенты циклофосфамид, ифосфамид, дакарбазин, прокарбазин, которые активируются цитохромами Р450, экспрессируемыми в нормальной ткани, как рассмотрено Patterson L.H. и Murray G.I. в Curr. Pharm. Des., 8(15), 1335-47 (2002).
Семейство цитохромов Р450 человека содержит 57 активных изоферментов, которые функционируют при нормальном метаболизме, фармакокинетиках влияния лекарственного средства и отрицательных результатах эффекта у пациентов через посредство взаимодействий лекарственное средство-лекарственное средство. Цитохром-Р450-изоферменты метаболизируют приблизительно две трети известных лекарственных средств у людей, с 80% из них, приписываемых пяти изоферментам, а именно CYP1A2, CYP2C9, CYP2C19, CYP2D6 и CYP3A4, как описывается в книге Ortiz de Montellano P.R. (ред.) «Cytochrome P450: structure, mechanism and biochemistry», Kluwer Academic/Plenum Publichers, New York, 2005.
Среди генов, обнаруженных в плане человеческого генома, имеются CYP2R1, CYP2W1, CYP2S1, CYP2S1, CYP2U1, но функция, полиморфизм и регуляция этих генов еще должны быть полностью выяснены, как рассматривается Ingelman-Sundberg M., «Toxicol. Appl. Pharmacol., 207, 52-6 (2005). В дополнение к CYP1B1, некоторое количество этих цитохром-Р450-оксидоредуктаз, включая CYP1B1, CYP2A/2B, CYP2F1, CYP2R1, CYP2U1, Cyp3A5, CYP3A7, CYP4Z1, CYP26A1 и CYP51, присутствуют на значительно более высоком уровне интенсивности, чем в случае нормального яичника, как определено путем иммуногистохимии и оптической микроскопии, как описано Downie и др., Clin. Cancer Res., 11(20), 7369-75 (2005). Кроме того, используя подобные методы определения в случае первичного колоректального рака, некоторые цитохромы Р450, включая CYP1B1, CYP2S1, CYP2U1, CYP3A5 и CYP51, часто сверхэкспрессируются по сравнению с нормальной ободочной кишкой, как описывается Kumarakulasingham и др., Clin. Cancer Res., 11(10), 3758-65 (2005). При том же самом исследовании, некоторые цитохромы Р450, включая CYP1B1, CYP2A/2B, CYP2F1, CYP4V2 и CYP39, коррелируют с их присутствием в первичной опухоли. Также показано, что CYP2W1 сверхэкспрессируется в случае колоректального рака, согласно Elder и др., Eur. J. Cancer, 45(4), 705-12. CYP4Z1 сверхэкспрессируется в случае рака молочной железы, представляет собой ген, ассоциированный с промотированием и прогрессированием немелкоклеточного рака легкого, как описывается Reiger и др., Cancer Res., 64(7), 2357-64 (2004), и Bankovic и др., Lung Cancer, 67(2), 151-9 (2010), соответственно.
Главной проблемой в данной области является выяснение функции человеческих цитохромов Р450, так называемого «сиротского» статуса, в отношении специфичности к неизвестному субстрату, как рассматривается Stark K. Guengerich F.P. в Drug. Metab. Rev., 39(2-3), 627-37 (2007). Некоторое количество субстратов известно для CYP1B1, немногие из которых специфически метаболизируются ферментом, например, 7-этоксирезоруфин подвергается окислительному деэтилированию, когда активируется всеми членами семейства CYP1, включая CYP1A1, CYP1A2 и CYP1B1, как описывается Chang T.K. и Waxman D.J. в Method Mol. Biol., 320, 85-90 (2006). Некоторое количество флюорогенных и люминогенных субстратов-образцов доступно для оценки активности цитохрома Р450 с высокой чувствительностью, но они проявляют широкую специфичность и как таковые метаболизируются рядом цитохромов Р450 в случае семейств CYP1, CYP2 и CYP3. Например, авторами Cali и др., Expert Opin. Drug. Toxicol., 2(4), 62-45 (2006), описывается использование люминогенных субстратов, которые ассоциируются с люминесценцией люциферазы светляка, по технологии, называемой Р450-Glo. Другим примером является 7-этоксикумарин, который подвергается катализируемому цитохромом Р450 О-деэтилированию с высвобождением высокофлуоресцентного аниона, как описывается Waxman D.J. и Change T.K.H. в «The use 7-ethoxycoumarin to minitor multiple enzymes in the human CYP1, CYP2, CYP3 families» в Methods in Molecular Biology, том 320, Cytochrome P450 Protocols, второе издание, под редакцией Phillips I.R. и Shephard E.A., 2006.
Everett и др., Biochem. Pharmacol., 63, 1629-39 (2002), описывают восстановительную фрагментацию модельных индолхиноновых пролекарств с помощью цитохром-Р450-редуктазы (не путать с цитохромами Р450) при гипоксии с высвобождением 7-гидрокси-4-метилкумарин-аниона. Модельное пролекарство является не флуоресцентным при предварительно выбранной длине волны эмиссии и восстановительная фрагментация может быть точно определена путем мониторинга продуцирования кумарин-аниона (λ=380 нм/λem=450 нм), используя кинетическую спектрофлуориметрию.
Взаимодействия между ограниченным количеством соединений (типично <100) и изоферментами цитохромами Р450 описаны, но результаты таких исследований затруднительны для сравнения из-за различий в технологиях, условиях анализа и методах анализа данных, как описывается Rendic S. «Summary of information on human CYP enzymes: human P450 mttabolism data» в Drug Metab. Rev., 34, 83-448 (2002). Развиты главным образом вычислительные стратегии для создания моделей предиктивной активности субстрата к изоферменту цитохрому Р450, но они ограничены за счет отсутствия единого большого набора различных данных в отношении активностей изофермента цитохрома Р450, как описывается Veith и др., Nature Biotechnology, 27, 1050-55 (2009). Авторы описывают создание базы данных по биоактивности цитохрома 450, используя количественный высокопроизводительный скрининг (HTS) с биолюминесцентным анализом в отношении ингибирования субстрата фермента, для скрининга 17143 химических соединений по отношению к пяти изоферментам цитохрома Р450 (CYP1A2, CYP2C9, CYP2C19, CYP2D6 и CYP3A4), экспрессируемым в нормальных тканях главным образом печени и ответственным за так называемый метаболизм фазы 1 лекарственных средств. Сделан вывод, что база данных должна содействовать созданию и тестированию новых предиктивных моделей в отношении активности цитохрома Р450 для способствования достижениям ранней стадии обнаружения лекарственного средства.
Jensen и др., J. Med. Chem., 50, 501-11 (2007), описывают способы для in silico прогнозирования ингибирования CYP2D6 и CYP3A4, базирующиеся на новом Gaussion Kernel позиционном алгоритме «k-ближайшего соседа (k-NN)», в свою очередь, базирующемся на поисках подобия по Tanimoto согласно расширенному методу «отпечатков пальцев». Набор данных включает моделирование 1153 и 1182 лекарственных «кандидатов», тестируемых в отношении ингибирования CYP2D6 и CYP3A4 в случае человеческих печеночных микросом. В случае CYP2D6, 82% классифицированных тест-соединений предсказаны в отношении точного класса и, в случае CYP3A4, 88% классифицированных тест-соединений точно классифицированы.
Теоретически может быть возможным использование цитохрома Р450 HTS для создания большой базы данных в отношении биоактивностей цитохромов Р450 в случае опухоли и нормальной ткани и затем для разработки модели предсказания субстрата в качестве базиса для дизайна и синтеза селективных CYP1B1-активируемых пролекарств, тогда как скрининг в отношении фармакологических «склонностей» ассоциирован с метаболизмом фазы 1 за счет цитохромов Р450 нормальной ткани. Однако доведение до практики не является очевидным из уровня техники и должно быть рационалистически объяснено в отношении структуры пролекарства и механизма конверсии в активное лекарственное средство как только происходит активация за счет экспрессирующихся опухолью цитохромов Р450.
Использование так называемой «триггер-линкер-эффектор»-химии при дизайне лекарственного средства требует активации триггера для инициации фрагментации линкера в целях высвобождения эффектора (типично активное лекарственное средство), биологическая активность которого замаскирована в пролекарственной форме. Модульный дизайн селективных пролекарств, направленных на экспрессирующиеся опухолью цитохромы Р450, как например CYP1B1, требует (1) идентификации селективных триггерных составляющих, (2) использования биостабильных линкеров, фрагмент которых рационально следует за активацией триггера (обычно путем ароматического гидроксилирования), и (3) подходящих эффекторов или лекарственных средств, которые не интерферируют с эффективностью процесса триггерирования.
CYP1B1 мРНК экспрессируется конститутивно во всех нормальных экстрагепатических человеческих тканях, хотя белок является недетектируемым. В противоположность, CYP1B1-белок экспрессируется до высоких уровней в опухолях. Предполагают, что в случае большого ряда установленных или иммортилизованных, происходящих от людей, опухолевых клеточных линий (как например клетки MCF-7 рака молочной железы), которые подвергались значительному пассированию in vitro, но не констутитивно, экспрессируется активный CYP1B1-белок. Хотя CYP1B1 не конститутивно экспрессируется в опухолевых клетках MCF-7 молочной железы, он способен индуцировать экспрессию CYP1-фермента, как в случае мРНК, так и в случае уровня белка, путем обработки арилуглеводородными агонистами, такими как диоксин TCDD.
В международной заявке WO 99/40944 описываются пролекарства, которые включают лекарственную составляющую, связанную с основой-носителем, причем описано, что пролекарство активируется даже во время гидроксилирования с помощью CYP1B1 при высвобождении лекарственной составляющей.
Краткое изложение сущности изобретения
Авторами настоящей заявки неожиданно найдено, что описанные в данном контексте соединения, отличные от соединений, описанных в международной заявке WO 99/40944, разрушаются в некоторых клетках, в частности, в клетках, которые экспрессируют цитохром Р450 1В1 (ниже обозначается как CYP1B1), но не в нормальных клетках, как результат соединений, разрушающихся при гидроксилировании (например, осуществляемым экспрессирующими CYP1B1 клетками) и, в особенности, раковыми клетками.
В соответствии с первым аспектом, следовательно, настоящее изобретение относится к соединению формулы (I):
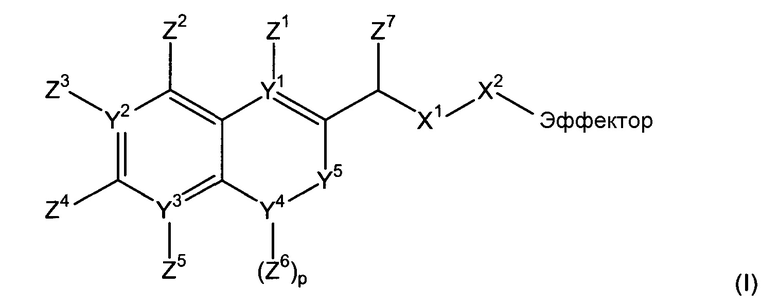
(где
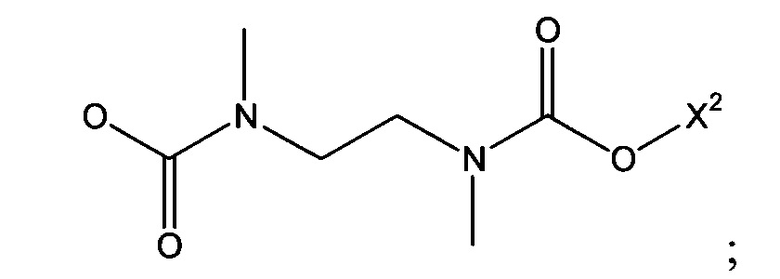
Х1 является таким, что -Х1-Х2 означает -О-Х2-, -S-X2, -SO2-O-X2, -SO2NZ10-X2, конъюгированный алкенметилокси, конъюгированный алкенметилтио, конъюгированный алкенметил-SO2-O, конъюгированный алкенметил-SO2NZ10 или формулу:
-Х2 отсутствует или является таким, что Х1-Х2-эффектор означает один из:
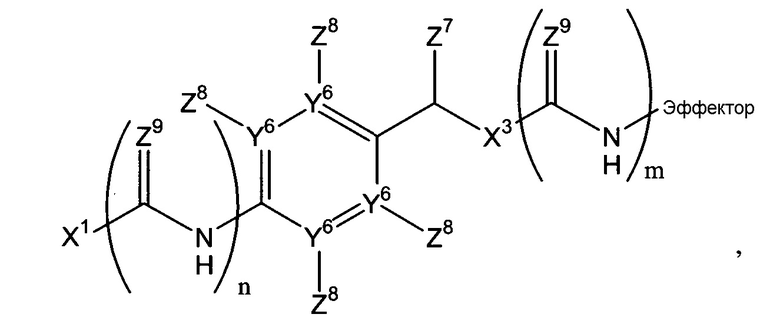
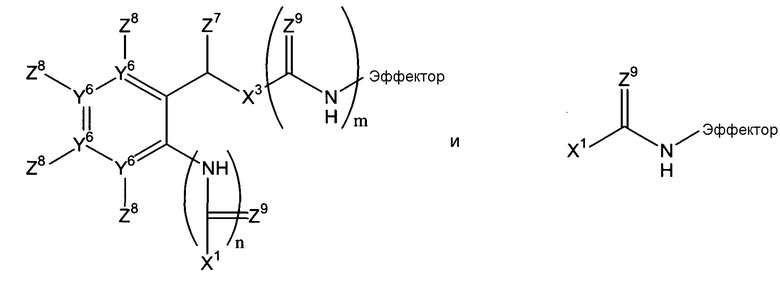
каждый n и m независимо означает 0 или 1;
р означает 0, 1 или 2;
Х3 означает кислород или серу и, дополнительно, когда m=0, может представлять собой SO2-O, SO2NZ10, конъюгированный алкенметилокси, конъюгированный алкенметилтио, конъюгированный алкенметил-SO2-O или конъюгированный алкенметил-SO2NZ10;
каждый из Y1, Y2 и Y3 независимо означает углерод или азот, где, если Y1 означает азот, Z1 отсутствует, если Y2 означает азот, Z3 отсутствует, и, если Y3 означает азот, Z5 отсутствует;
Y4 означает атом кислорода, углерода или азота, сульфоксид или сульфон;
-Y5- означает или (i) одинарную связь, (ii) =СН-, где двойная связь = в =СН- связана с Y4, или (iii) -СН2- или -СН2СН2-, или один из (ii)-(iii), где атом водорода в (ii) или один или более атомов водорода в (iii) заменены заместителем Z11, где Z11 независимо выбирают из группы, состоящей из алкила, алкенила, алкинила, арила, аралкила, алкилокси, алкенилокси, алкинилокси, арилокси, аралкилокси, алкилтиокси, алкенилтиокси, алкинилтиокси, арилтиокси, аралкилтиокси, амино, гидрокси, тио, галогена, карбокси, формила, нитро и циано;
каждый из Z1-Z4, когда присутствует, независимо выбирают из группы, состоящей из водорода, алкила, алкенила, алкинила, арила, аралкила, алкилокси, алкенилокси, алкинилокси, арилокси, аралкилокси, алкилтиокси, алкенилтиокси, алкинилтиокси, арилтиокси, аралкилтиокси, амино, гидрокси, тио, галогена, карбокси, формила, нитро и циано; и Z5, когда присутствует, независимо выбирают из группы, состоящей из водорода, алкила, алкенила, алкинила, арила, аралкила, алкилокси, алкенилокси, алкинилокси, арилокси, аралкилокси, алкилтиокси, алкенилтиокси, алкинилтиокси, арилтиокси, аралкилтиокси, амино, гидрокси, тио, карбокси, формила, нитро и циано, или один из Z2 и Z3, Z3 и Z4 и Z4 и Z5, вместе с атомами, с которыми они связаны, образуют ароматический цикл, конденсированный с остатком соединения, при условии, что по меньшей мере один из Z1, Z2 и Z4 означает водород;
Z6 выбирают из группы, состоящей из водорода, алкила, алкенила, алкинила, арила и аралкила;
ни один, один или два из Y6 могут означать атомы азота с остаточными атомами углерода;
каждый Z7 независимо означает водород, алкил или арил;
каждый Z8 независимо выбирают из группы, состоящей из водорода, электроноакцепторной группы, незамещенного С1-С6-алкила, замещенного С1-С6-алкила, незамещенного С1-С6-алкокси и замещенного С1-С6-алкокси, где замещенный алкил или алкокси замещен одной или более группами, выбираемыми из группы, состоящей из простого эфира, амино, моно- или дизамещенного амино, циклического С1-С5-алкиламино, имидазолила, С1-С6-алкилпиперазинила, морфолино, тиола, простого тиоэфира, тетразола, карбоновой кислоты, сложного эфира, амидо, моно- или дизамещенного амидо, N-связанного амида, N-связанного сульфонамида, сульфокси, сульфоната, сульфонила, сульфокси, сульфината, сульфинила, фосфоноокси, фосфата и сульфонамида;
каждый Z9 независимо означает кислород или серу;
Z10 означает водород или алкил, например, С1-4-алкил;
эффектор представляет собой молекулу, обладающую фармакологической, диагностической или скрининговой функцией),
или его фармацевтически приемлемой соли, сложному эфиру, амиду или сольвату.
Согласно второму аспекту, данное изобретение относится к композиции, содержащей соединение в соответствии с первым аспектом данного изобретения или его фармацевтически приемлемую соль, сложный эфир, амид или сольват, вместе с фармацевтически приемлемым носителем.
Согласно третьему аспекту, данное изобретение относится к соединению согласно первому аспекту данного изобретения или его фармацевтически приемлемой соли, сложному эфиру, амиду или сольвату для применения в качестве лекарственного средства.
Согласно четвертому аспекту, данное изобретение относится к соединению согласно первому аспекту данного изобретения или его фармацевтически приемлемой соли, сложному эфиру, амиду или сольвату для применения в способе лечения или профилактики пролиферативного состояния.
Согласно пятому аспекту, данное изобретение относится к способу лечения или профилактики пролиферативного состояния, причем вышеуказанный способ включает введение терапевтически или профилактически пригодного количества соединения согласно первому аспекту данного изобретения или его фармацевтически приемлемой соли, сложного эфира, амида или сольвата, субъекту, нуждающемуся в этом.
Согласно шестому аспекту, данное изобретение относится к применению соединения согласно первому аспекту данного изобретения или его фармацевтически приемлемой соли, сложного эфира, амида или сольвата для получения лекарственного средства, пригодного для лечения или профилактики пролиферативного состояния.
Согласно седьмому аспекту, данное изобретение относится к способу идентификации соединения, которое специфически активируется ферментом цитохромом Р450, причем вышеуказанный способ включает стадии:
(а) введение в контакт ряда соединений, в соответствии с первым аспектом данного изобретения, в которых эффектор представляет собой флуорофор, с вышеуказанным ферментом цитохромом Р450, и определение, приводит ли вышеуказанный контакт к высвобождению вышеуказанного флуорофора из одного или более соединений вышеуказанного ряда;
(b) введение в контакт вышеуказанного ряда соединений с контрольной тканью, тканевым или клеточным экстрактом, или с ферментом, и определение, приводит ли вышеуказанный контакт к высвобождению вышеуказанного флуорофора из одного или более соединений вышеуказанного ряда; и
(с) идентификация вышеуказанного соединения, специфически активируемого вышеуказанным цитохромом Р450, в качестве любого соединения из вышеуказанного ряда соединений, которое высвобождает вышеуказанный флуорофор на стадии (а), но не высвобождает или только в гораздо меньшей степени высвобождает на стадии (b).
Согласно восьмому аспекту, данное изобретение относится к способу определения, является ли соединение согласно данному изобретению, где эффектор представляет собой молекулу, обладающую фармакологической функцией, эффективным при лечении ракового заболевания, причем вышеуказанный способ включает введение вышеуказанного соединения животному с раковым заболеванием, где вышеуказанное раковое заболевание является проистекающим от имплантации любой рекомбинантной клетки, модифицированной таким образом, чтобы конститутивно экспрессировать фермент цитохром Р450, ткани, взятой непосредственно из опухоли или карциномы, или клетки из клеточной линии раннего пассирования, происходящей из ткани, взятой непосредственно из опухоли или карциномы, которая экспрессирует вышеуказанный фермент цитохром Р450 на уровнях, подобных таковым в случае опухоли или карциномы, из которых она происходит.
Дальнейшие аспекты и воплощение данного изобретения следуют из обсуждения, которое следует ниже.
Краткое описание фигур
Фиг.1 представляет собой вестерн-блоттинг, показывающий детектирование экспрессии CYP1B1 в трансфицированной клеточной линии СНО/CYP1B1/CPR (панель А) и экспрессии CYP1A1 в трансфицированной клеточной линии СНО/CYP1A1/CPR (панель В). Подробное описание приведено в нижеприводимой экспериментальной части.
Фиг.2а демонстрирует механизм индуцируемого CYP1B1 3-гидроксилирования соединения согласно данному изобретению (ссылаются в данном контексте как на SU025-04), с последующим спонтанным высвобождением цитотоксической молекулы эффектора (N,N’-бис(2-хлорэтил)фосфордиамидат (также называемый как IPM-хлорид)) путем 1,4-элиминирования.
Фиг.2b демонстрирует механизм индуцируемого CYP1B1 4-гидроксилирования соединения согласно данному изобретению (ссылаются в данном контексте как на SU025-04), с последующим спонтанным высвобождением цитотоксической молекулы эффектора (N,N’-бис(2-хлорэтил)фосфордиамидат (также называемый как IPM-хлорид)) путем 1,6-элиминирования.
Фиг.2с демонстрирует механизм индуцируемого CYP1B1 6-гидроксилирования соединения согласно данному изобретению (ссылаются в данном контексте как на SU025-04), с последующим спонтанным высвобождением цитотоксической молекулы эффектора (N,N’-бис(2-хлорэтил)фосфордиамидат (также называемый как IPM-хлорид)) путем 1,8-элиминирования.
Фиг.3 демонстрирует механизм индуцируемого CYP1B1 6-гидроксилирования соединения согласно данному изобретению (ссылаются в данном контексте как на SU024-1-03), с последующим спонтанным высвобождением молекулы эффектора путем 1,8-элиминирования.
Подробное описание данного изобретения
Настоящее изобретение является результатом получения пролекарств, в которых так называемая молекула эффектора, которая может быть цитостатической, цитотоксической, диагностической или скрининговой молекулой, как описано более подробно в дальнейшем, химически модифицирована путем введения ее во взаимодействие, посредством чего образуется соединение формулы (I). Авторами данного изобретения найдено, что гидроксилирование соединений формулы (I), в особенности, гидроксилирование, индуцируемое CYP1B1, дает возможность высвобождать молекулы эффектора путем разрушения соединений формулы (I), которое происходит спонтанно при непосредственном гидроксилировании или гидроксилировании через образование эпоксида.
В общих чертах, структуру соединений формулы (I) можно рассматривать как включающую три части: триггерная область, линкер и молекула эффектора. Триггер служит в качестве субстрата для типичного гидроксилирования, индуцируемого CYP1B1, и может вообще подразумевать включение бициклической составляющей, изображенной с левой стороны формулы (I), и ее заместителей, т.е. включение части соединений, содержащей Y1-Y5, Z1-Z6, и остающихся атомов углерода, к которым некоторые из этих составляющих присоединены. Триггерная область соединений присоединена через связывающую область, включающую элемент С(Z7)-X1-X2, к молекуле эффектора, которая помечена, как таковая.
Состав и вариабельность этих трех областей - триггера, линкера и эффектора - соединений формулы (I) теперь описываются.
При последующем обсуждении ссылку делают на некоторое количество терминов, под которыми нужно понимать, что они имеют нижеприводимое значение, за исключением противоположного предписанному согласно данному контексту.
Под алкилом подразумевают, согласно данному контексту, насыщенный углеводородный радикал, который может иметь линейную, циклическую или разветвленную цепь (обычно, линейную цепь, за исключением противоположного предписанному согласно данному контексту). Когда алкильная группа имеет один или более участков ненасыщенности, то они могут быть образованы двойными углерод-углеродными связями или тройными углерод-углеродными связями. Когда алкильная группа включает двойную углерод-углеродную связь, это приводит к образованию алкенильной группы; присутствие тройной углерод-углеродной связи приводит к образованию алкинильной группы. Обычно алкильные, алкенильные и алкинильные группы включают 1-25 атомов углерода, более конкретно, 1-10 атомов углерода, еще более конкретно, 1-6 атомов углерода, при этом, конечно, подразумевают, что нижний предел в случае алкенильных и алкинильных групп составляет 2 атома углерода и в случае циклоалкильных групп - 3 атома углерода.
Алкильные, алкенильные или алкинильные группы могут быть замещены, например, одно-, двух- или трехкратно, например, однократно, т.е. формально заменены один или более атомов водорода алкильной группы. Примерами таких заместителей являются галоген (например, фтор, хлор, бром и йод), арил, гидрокси, нитро, амино, алкокси, алкилтио, карбокси, циано, тио, формил, сложный эфир, ацил, тиоацил, амидо, сульфонамидо, карбамат и т.п.
Под карбокси подразумевают, согласно данному контексту, функциональную группу СО2Н, которая может быть в депротонированной форме (СО2-).
Галоген означает фтор, бром, хлор или йод.
Под ацилом и тиоацилом подразумевают функциональные группы формул -С(О)-алкил или -С(S)-алкил, соответственно, где алкил имеет значение, как описано выше.
Под сложным эфиром подразумевают функциональную группу, содержащую остаток -ОС(=О)-.
Под амидо подразумевают функциональную группу, содержащую остаток -N(H)C(=O)-; под карбаматом подразумевают функциональную группу, содержащую остаток -N(H)C(=O)О-; и под сульфонамидо подразумевают функциональную группу, содержащую остаток -SO2N(H)2-, в которой каждый указанный атом водорода может быть заменен (независимо, в сульфонамидо) алкилом или арилом.
Алкилокси (синоним алкокси) и алкилтио имеют формулы -О-алкил и -S-алкил, соответственно, где алкил имеет значение, как описано выше.
Также алкенилокси, алкинилокси, алкенилтио и алкинилтио являются таковыми формул -О-алкенил, -О-алкинил, -S-алкенил и -S-алкинил, где алкенил и алкинил имеют значения, как описано выше.
Под аминогруппой подразумевают, согласно данному контексту, группу формулы -N(R)2, в которой каждый R независимо означает водород, алкил или арил, например, ненасыщенный, незамещенный С1-6-алкил, такой как метил или этил, или в котором два R, присоединенные к атому азота N, связаны. Одним примером этого является то, каким образом -R-R- образует алкиленовый дирадикал, формально происходящий от алкана, из которого удалены два атома водорода, типично, от концевых атомов углерода, с образованием цикла вместе с атомом азота амина. Как известно, дирадикал в циклических аминах необязательно должен быть алкиленом: морфолин (в котором -R-R- означает -(СН2)2О(СН2)2-) представляет собой один такой пример, согласно которому может быть получен циклический аминозаместитель.
Ссылки на аминогруппы, согласно данному контексту, также должны подразумеваться как охватывающие в их пределах кватернизированные или протонированные производные аминов, проистекающие от соединений, содержащих такие аминогруппы. Под примерами последних могут подразумеваться соли, такие как гидрохлоридные соли.
Под арилом подразумевают, согласно данному контексту, радикал, формально образованный за счет удаления атома водорода из ароматического соединения.
Ариленовые дирадикалы происходят от ароматических остатков, формально, путем удаления двух атомов водорода, и могут быть и типично являются, за исключением противоположного предписанному согласно данному контексту, моноциклическими, например, фенилен. Как известно квалифицированному специалисту в данной области, гетероароматические составляющие являются подмножеством ароматических составляющих, которые содержат один или более гетероатомов, обычно О, N или S, вместо одного или более атомов углерода, и туда присоединены любые атомы водорода. Примеры гетероароматических составляющих, например, включают пиридин, фуран, пиррол и пиримидин. Дальнейшие примеры гетероароматических циклов включают пиридил, пиридазин (в котором 2 атома азота являются смежными в ароматическом 6-членном цикле); пиразин (в котором 2 атома азота находятся в 1,4-положении в 6-членном ароматическом цикле); пиримидин (в котором 2 атома азота находятся в 1,3-положении в 6-членном цикле); или 1,3,5-триазин (в котором 3 атома азота находятся в 1,3,5-положении в 6-членном ароматическом цикле).
Арил или арилен могут быть замещены один или более раз электроноакцепторной группой (например, группой, выбираемой из группы, состоящей из галогена, циано (-CN), галогеналкила, амида, нитро, кето (-COR), алкенила, алкинила, четвертичного амино (-N+R3), сложного эфира, амидо (-CONR2), N-связанного амидо (-NR-C(=O)-R), N-связанного сульфонамидо (-NR-S(=O)2-R), сульфокси (-S(=O)2-ОН), сульфоната (S(=O)2OR), сульфонила (S(=O)2R) и сульфонамида (S(=O)2-NR2), где (каждый) R независимо выбирают из группы, состоящей из С1-С6-алкильной группы, С3-С20-гетероциклической группы или С3-С20-арильной группы, обычно, С1-С6-алкильной группы, незамещенной С1-С6-алкоксильной группы и замещенной С1-С6-алкоксильной группы, где замещенный алкил или алкокси замещены одной или более группами, выбираемыми из группы, состоящей из простого эфира, амино, моно- или дизамещенного амино, циклического С1-С5-алкиламино, имидазолила, С1-С6-алкилпиперазинила, морфолино, тиола, тиоэфира, тетразола, карбоновой кислоты, сложного эфира, амида, моно- или дизамещенного амида, N-связанного амида (-NR-C(=O)-R), N-связанного сульфонамида (-NR-S(=O)2-R), сульфокси (-S(=O)2-ОН), сульфоната (S(=O)2OR), сульфонила (S(=O)2R), сульфокси (S(=O)OH), сульфината (S(=O)OR), сульфинила (S(=O)R), фосфоноокси (-ОР(=О)(ОН)2), фосфата (ОР(=О)(OR)2) и сульфонамида (-S(=O)2-NR2), где (каждый) R независимо выбирают из группы, состоящей из С1-С6-алкильной группы, С3-С20-гетероциклической группы или С3-С20-арильной группы.
Триггерная область соединений формулы (I) обычно включает бициклическую составляющую, содержащую ароматический цикл (который включает Y2 и Y3, как указано), конденсированную со вторым циклом (который включает Y1, Y4 и Y5, которые могут быть ароматическими или неароматическими).
Без связывания с теорией, полагают, что активность соединений формулы (I) в качестве субстратов для гидроксилирования, например, производимого за счет CYP1B1, достигается частично за счет структуры триггерной составляющей, чувствительной к гидроксилированию, когда Z2 или Z4 означает водород или когда Y1-Z1 означает С-Н, причем гидроксилирование, таким образом, имеет место по одному из их трех атомов углерода, с которыми связаны Z2 и Z4, и Y1, где Y1 означает углерод. Как изображено на фиг.2, гидроксилирование, в случае любого из этих положений в типичном соединении согласно данному изобретению, меченном SU025-04, приводит к спонтанному разрушению соединения путем процесса элиминирования, или 1,4-, 1,6- или 1,8-элиминирование, в зависимости от того, в случае какого из этих положений имеет место гидроксилирование.
В отношении структуры соединений формулы (I) следует заметить, что, благодаря конъюгации атомов углерода, к которым присоединены Z2 и Z4, через Y1, для линкерной составляющей, может иметь место любой из трех механизмов спонтанного разрушения соединения, независимо от природы области Z6-Y4-Y5 соединений. Таким образом, может быть допустимо большое разнообразие природы этой области соединений формулы (I), как обсуждается ниже. Также, продолжение области конъюгации достигается, между прочим, за счет использования конъюгированных составляющих Х1, как описывается в данном контексте.
В соединениях формулы (I) каждый из атомов, указанных как Y1, Y2 и Y3, может независимо представлять собой атом углерода или атом азота. Когда связанный атом представляет собой атом азота, то должен отсутствовать соответствующий заместитель (Z1, Z3 или Z5, соответственно). Согласно некоторым воплощениям данного изобретения, Y2 или Y3 означает атом углерода. Согласно конкретным воплощениям данного изобретения, Y2 и Y3 означают атомы углерода. Согласно любому из этих воплощений - в которых оба Y2 или Y3 означает атом углерода или в которых Y2 и Y3 означают атомы углерода - или в которых ни тот, ни другой из Y2 или Y3 не означает атом углерода, Y1 может быть атомом углерода.
Заместители Z1, Z2 и Z4 могут обычно представлять собой такие, как описанные в п.1 формулы изобретения. Однако по меньшей мере одна из этих составляющих означает атом водорода, для того, чтобы иметь участок для гидроксилирования соединения. Согласно некоторым воплощениям данного изобретения, или Z2 или Z4 означает водород. Согласно другим воплощениям, Z2 и Z4 означают водород. Согласно любому из этих воплощений - в которых Z2 или Z4 означает атом водорода или в которых как Z2, так и Z4 означают атомы водорода - или в которых ни тот, ни другой из Z2 или Z4 не означает атом водорода, Z1 может означать водород. Согласно некоторым воплощениям данного изобретения, каждый из Z1, Z2 и Z4 означает атом водорода.
Как Z3, так и Z4, вместе со смежным заместителем у ароматического цикла (т.е. Z2 или Z4, или Z3 или Z5, соответственно), вместе с атомами ароматического цикла, с которыми эти заместители связаны, могут образовывать ароматический цикл, конденсированный с остатком соединения. Таким образом, Z2 и Z3, вместе с атомом углерода, с которым связан Z2, и Y2 могут образовывать ароматический цикл. Подобным образом, например, Z4, Z5 и атом углерода, с которым связан Z4, и Y3 вместе могут образовывать ароматический цикл.
Согласно некоторым воплощениям данного изобретения, ни одна или только две из пары заместителей Z2 и Z3, Z3 и Z4 и Z4 и Z5, вместе образуют конденсированный ароматический цикл. Таким образом, согласно некоторым воплощениям, нет ароматических циклов, конденсированных с ароматическим циклом, содержащим Y2 и Y3.
Конкретно, заместители Z3 и Z5 обычно не являются частью ароматического цикла, конденсированного с остатком соединения формулы (I). Там, где это имеет место, т.е. где эти составляющие представляют собой индивидуальные заместители, Z3 может означать алкил, алкенил, алкинил, арил, аралкил, алкилокси, алкенилокси, алкинилокси, арилокси, аралкилокси, алкилтиокси, алкенилтиокси, алкинилтиокси, арилтиокси, аралкилтиокси, амино, гидрокси, тио, галоген, карбокси, формил, нитро и циано, и Z5 может означать алкил, алкенил, алкинил, арил, аралкил, алкилокси, алкенилокси, алкинилокси, арилокси, аралкилокси, алкилтиокси, алкенилтиокси, алкинилтиокси, арилтиокси, аралкилтиокси, амино, гидрокси, тио, карбокси, формил, нитро и циано. В отдельных воплощениях данного изобретения, Z3 может означать алкил, алкенил, алкинил, арил, аралкил, алкилокси, алкенилокси, алкинилокси, арилокси, аралкилокси, алкилтиокси, алкенилтиокси, алкинилтиокси, арилтиокси, аралкилтиокси, амино, гидрокси, тио, галоген, карбокси, формил, нитро и циано.
Согласно некоторым воплощениям данного изобретения, Z3 и Z5 представляют собой индивидуальные заместители, другие, чем атомы водорода. Когда Z3 и Z5 являются одними и теми же заместителями или другими, Z3 и Z5, в соответствии с некоторыми воплощениями данного изобретения, являются электронодонорными группами, такими как алкокси, алкилтиокси, арилокси, арилтиокси. Согласно конкретным воплощениям данного изобретения, Z3 или Z5, оба, означают амино или алкокси, например, С1-С6-алкокси. Примеры таких алкоксильных групп включают метокси, этокси, изопропокси, н-пропокси и т.п. Согласно некоторым воплощениям данного изобретения, или Z3 или Z5, или Z3 и Z5, означают метокси. Согласно некоторым воплощениям данного изобретения, Z3 и Z5 являются одинаковыми и представляют собой любой из непосредственно вышеуказанных заместителей, или классов заместителей. Как указано выше, соединения формулы (I) могут значительно изменяться в отношении их структуры, в части, которая включают Z6-Y4-Y5. Таким образом, Y4 может означать кислород, серу, сульфоксид или сульфон, вследствие чего не присутствует заместитель Z6 (р=0), атом азота (где р=0 или 1) или атом углерода, вследствие чего р=1 или 2. Согласно некоторым воплощениям данного изобретения, р=0 и Y4 означает кислород, серу, сульфон или сульфоксид. Согласно конкретным воплощениям данного изобретения, р=0 и Y4 означает кислород или серу. Согласно некоторым воплощениям данного изобретения, р=0 и Y4 означает кислород.
-Y5- может представлять собой одну из (i) одинарной связи, в случае которой триггерная составляющая базируется на 6-членном ароматическом цикле, содержащем Y2- и Y3-, конденсированном с 5-членным циклом, так как, согласно этому воплощению, Y5 действительно отсутствует; или (ii) =СН-, где двойная связь = связана с Y4. Согласно этим воплощениям данного изобретения, триггерная составляющая, таким образом, образована из двух конденсированных ароматических циклов и квалифицированному специалисту понятно, что, когда -Y5- означает =СН-, тогда Y4 означает или атом азота и р=0, или атом углерода и р=1. Наконец, -Y5- может представлять собой (iii) -СН2- или -СН2СН2-, в случае которых триггерная составляющая включает бициклическую систему, содержащую 6- или 7-членный цикл, конденсированный с ароматическим 6-членным циклом, замещенным Y2 и Y3. Согласно некоторым воплощениям данного изобретения, или один или более водородов или атомов водорода, указанных в пунктах (ii) и (iii) для -Y5-, могут быть заменены Z11, например, алкилом или галогеном. Согласно некоторым воплощениям данного изобретения, Z11 не присутствует. Согласно конкретным воплощениям данного изобретения, -Y5- представляет собой одинарную связь, например, когда р=0 и Y4 означает кислород, серу, сульфон или сульфоксид, р=0 и Y4 означает кислород или серу, и, в особенности, когда р=0 и Y4 означает кислород.
Теперь описывается связующая составляющая СН(Z7)-X1-X2.
Z7 означает водород или алкильную или арильную группу, которая, согласно некоторым воплощениям данного изобретения, является незамещенной. Согласно некоторым воплощениям данного изобретения, Z7 или каждый Z7 означает алкильную группу, например, незамещенную алкильную группу, такую как незамещенная С1-С6-алкильная группа. Примеры Z7 включают метил и этил. Согласно конкретным воплощениям данного изобретения, Z7 означает водород, так что -СН(Z7)- означает метилен. Согласно другим воплощениям, Z7 или каждый Z7 представляет собой замещенную алкильную группу, например, замещенную метильную или этильную группу. Примеры таких воплощений включают аминозамещенные алкильные группы, например, морфолино- или пиперидинилалкильные группы, или другие группы, которые позволяют повышаться растворимости в воде. Альтернативно, Z7, каждый, или по меньшей мере один Z7, может быть необязательно замещен гетероарилом, таким как пиридил.
Х1 может представлять собой различные связующие атомы или двухвалентные связующие составляющие, например, Х1 может означать кислород, серу, сульфонамид или сульфонатный эфир. В дополнение, Х1 может означать этан-1,2-диилбис(метилкарбамат) или конъюгированный алкенметилокси остаток.
Под конъюгированным алкенметилокси остатком подразумевают остаток формулы (=СН-СН)q=CH-CH2-O-, где q представляет собой целое число от 0 до 6, например, от 0 до 3, например, 0 или 1. Квалифицированному специалисту понятно, что атом кислорода, указанный в алкенметилокси остатках, может быть замещен атомом серы, SO2-O или SO2NZ10, для образования конъюгированных алкенметилсульфонатных остатков или конъюгированных алкенметилсульфонамидных остатков, как изложено выше, где атомы кислорода или серы, или сульфонатные или сульфонамидные остатки (SO2-O или SO2-NZ10) присоединены к Х2 или, если он отсутствует, к эффектору.
В соответствии с некоторыми воплощениями данного изобретения, Х1 означает кислород или серу. Во множестве воплощений данного изобретения Х1 означает кислород.
Х2 представляет собой необязательную дополнительную связующую составляющую, которая или отсутствует или введена между Х1 и эффектором.
Х2 может включать множество остатков, как описано в данном контексте, или может отсутствовать. Согласно некоторым воплощениям данного изобретения, Х2 отсутствует, или Х1-Х2-эффектор является одним из:
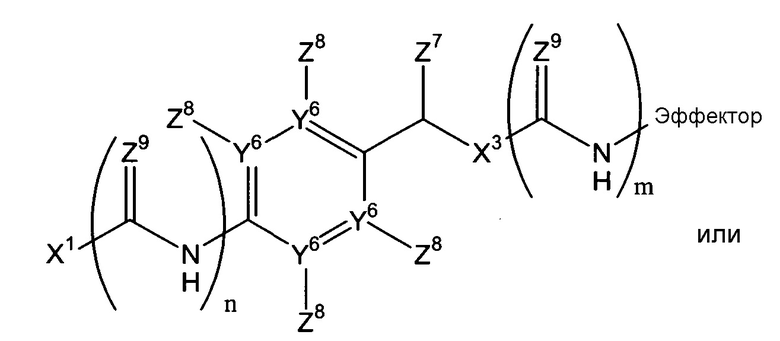
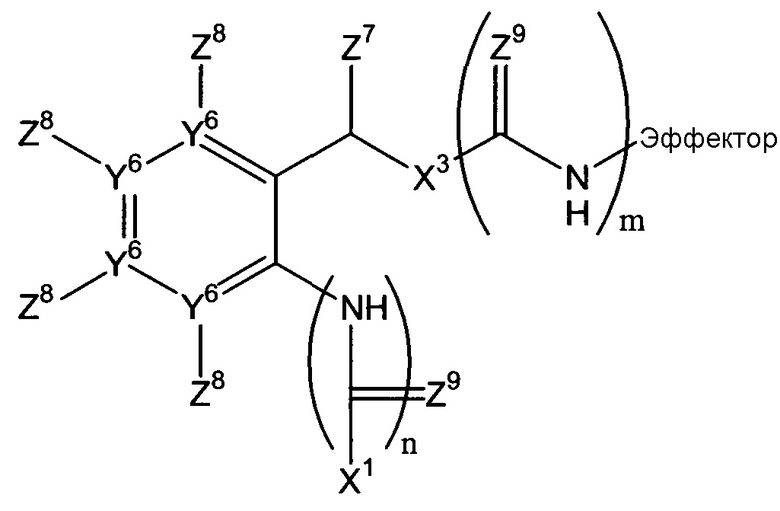
Например, Х2 может включать остаток арилен-СН(Z7)Х3 (в дальнейшем, остаток -ArCH(Z7)Х3-) и/или амид. Когда присутствует, остаток -Ar-CH(Z7)Х3- может быть фланкирован одним или двумя амидными или тиоамидными группами (С(Z9)NH). Если он фланкирован одной амидной или тиоамидной группой, он может быть расположен непосредственно между Х1 и ароматическим циклом остатка -Ar-CH(Z7)Х3 (где n=1), или введен между Х3 и эффекторной составляющей (где m=1). Альтернативно, амидная или тиоамидная группа могут присутствовать в обоих положениях или ни в одном из этих положений. Согласно некоторым воплощениям данного изобретения, n=0 и m=1. Когда Х2 включает остаток -Ar-CH(Z7)Х3-, фланкирован ли он или нет одним или двумя амидными или тиоамидными остатками, остаток Х1, который присоединен к ароматическому циклу или прямо или непрямо через амидный или тиоамидный остаток, может быть присоединен в любом из двух положений в ароматическом цикле, которые находится в орто-положении к остатку СН(Z7)X3 системы -Ar-CH(Z7)Х3-, или в пара-положении. Создание этих точек присоединения в ароматических циклах в случае остатков Х2, которые включают остатки Ar-CH(Z7)Х3-, позволяющие осуществляться 1,4-, 1,6- или 1,8-элиминированию молекулы эффектора. Должно быть понятно, что ариленовая группа, присутствующая, согласно некоторым воплощениям, в случае Х2, может быть гетероароматической, т.е. говорят о том, что один или два или атомы Y6 могут означать атомы азота с остаточными атомами углерода. Примером такого гетероариленового остатка является пиридилен, в котором один Y6 означает атом азота. В случае множества воплощений данного изобретения, каждый Y6, где присутствует, означает атом углерода.
Когда ариленовая группа присутствует в остатке Х2, она может быть замещена, как указано, в одном из четырех положений можно замещать, как указано в любом из четырех положений (не связывая ариленовую группу с эффекторным и триггерным концами соединений формулы (I), т.е. за счет заместителей Z8, которые могут быть независимо выбраны, как указано в п.1 формулы изобретения.
Когда Х2 включает один или более амидных или тиоамидных остатков -СН(Z9)NH, типично, где присутствует, (каждый) Z9 означает кислород, посредством чего получается один или более амидных остатков, хотя, где присутствует более чем один Z9, каждый Z9 может быть выбран независимо.
В заключение, эффекторная часть соединений формулы (I) представляет собой остаток, который обеспечивает желательное целенаправленное действие в клетках, обычно таких, в которых экспрессируется CYP1B1. Эффекторный компонент может быть любой молекулой, обладающей фармакологической, диагностической или скрининговой функцией, когда высвобождается из соединения формулы (I). Под фармакологической или диагностической функцией подразумевают, что эффекторный компонент, когда высвобождается, оказывает распознаваемое фармакологическое или диагностическое действие на клетки, в которых он высвобождается.
Специалисту в данной области должно быть понятно, что эффекторный компонент (эффектор) в соединениях формулы (I), когда высвобождается, может включать атом, описываемый в данном случае как часть Х1 - например, как атом кислорода или серы, или часть Х2, например, Х3, например, атом кислорода или серы. Однако, должно быть понятно, что различия между триггерной, линкерной и эффекторной частями соединений формулы (I) сделаны просто для содействия описанию соединений согласно изобретению; специалисту в данной области должно быть известно, что эффекторная часть в соединениях согласно изобретению составляет большую часть эффекторной молекулы, которая высвобождается после индуцируемого гидроксилированием разрушения, но что один или несколько из атомов в эффекторной молекуле, которая высвобождается, могут быть обеспечены атомами, описанными в данном контексте, как в случае Х1, части Х1 или Х2, и, безусловно, где-нибудь в другом месте (например, атомы водорода, происходящие от молекул воды). Альтернативно, эффекторная молекула может быть присоединена к остальной части соединений формулы (I), например, через посредство кетогрупп или формильных групп.
Эффекторная молекула, там, где она проявляет фармакологическое действие, может быть, например, любым химическим продуктом, который оказывает цитостатическое или цитотоксическое действие на клетку, которая служит для осуществления его высвобождения (например, CYP1B1-экспрессирующие клетки). Как известно, цитотоксической молекулой является молекула, которая является токсичной для клеток, тогда как цитостатический агент представляет собой агент, который подавляет рост и/или репликацию клеток.
Согласно некоторым воплощениям изобретения, эффекторная молекула представляет собой цитотоксический агент. Примеры цитотоксических агентов, которые могут быть использованы, включают, но не исчерпывающим образом, алкилирующие агенты, антимитотические агенты, антифолаты, антиметаболиты, ДНК-повреждающие агенты и ингибиторы ферментов (например, ингибиторы тирозинкиназы). Конкретные примеры возможных цитотоксических составляющих лекарственных средств включают, но не исчерпывающим образом, бис(галогенэтил)фосфороамидаты, циклофосфамиды, гемцитабин, цитарабин, 5-фторурацил, 6-меркаптопурин, камптотецин, топотекан, доксорубицин, даунорубицин, дуокармицин, этопозид, диетопозид, комбретастатин А-4, винбластин, винкристин, AQ4N, гидроксимочевину, маитансины, энедийены, эпотилоны, таксаны, блеомицины, калихеамицины, колхицин, дакарбазин, дактиномицин, эпирубицин, производные эпирубицина, флударабин, гидроксимочевинопентатостатин, метотраксат, митомицин, митоксантрон, карбоплатин, цисплатин, такселы, 6-тиогуанин, винка-алкалоиды, координационные комплексы платины, антрацендионы, замещенные мочевины, производные метилгидразина и хлорметины.
Согласно некоторым воплощениям изобретения, эффекторная молекула представляет собой фосфорамидиприт, т.е. производное фосфорной кислоты, в котором одна или две, обычно две, гидроксильные группы фосфорной кислоты заменены на хлорметин, или его кислород- или серосодержащий аналог, и необязательно группа Р(=О) заменена на Р(=S). Хлорметин, согласно данному контексту, определяется как неспецифически алкилирующий амин, структурно родственный горчичному газу (1,5-дихлор-3-тиапентан), в котором атом серы заменен атомом азота и, необязательно, одна хлорэтильная боковая цепь заменена атомом водорода или алкильной группой, или один или оба концевых хлорзаместителей заменены удаляемой группой, такой как бром, йод или мезилат (-OSO2CH3). Примеры фосфорамидипритов включают соединения, известные как фосфорамидиприт (РМ) и изофосфорамидиприт (IPM):
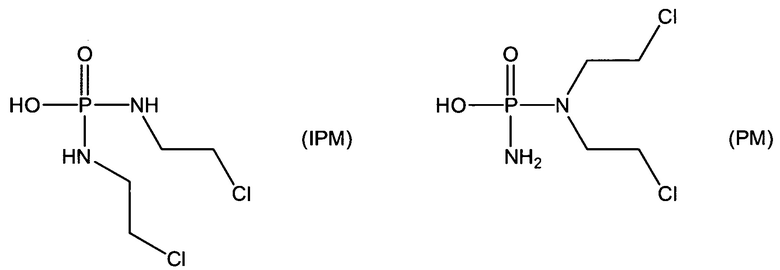
Таким образом, нужно заметить, что соединение РМ представляет собой пример, а также классическое название соединений, известных как фосфорамидиприты, так как его можно рассматривать как производное фосфорной кислоты, в котором одна или две гидроксильные группы заменены хлорметином (причем другая гидроксильная группа заменена на аминогруппу (NH2)).
Согласно этим воплощениям изобретения, в случае которых эффекторная молекула представляет собой фосфорамидиприт, в котором одна или две, обычно две, гидроксильные группы производного фосфорной кислоты заменены на кислородсодержащий или серосодержащий аналог хлорметина, под которым подразумевают аналоги фосфорамидипритов, в которых хлорметин заменен на аналог, в котором одна хлорэтильная боковая группа отсутствует и атом азота заменен на атом серы или атом кислорода.
Согласно конкретному воплощению настоящего изобретения, эффекторная молекула связана с остальной частью соединения через атом кислорода или серы и -эффектор отвечает формуле (II):
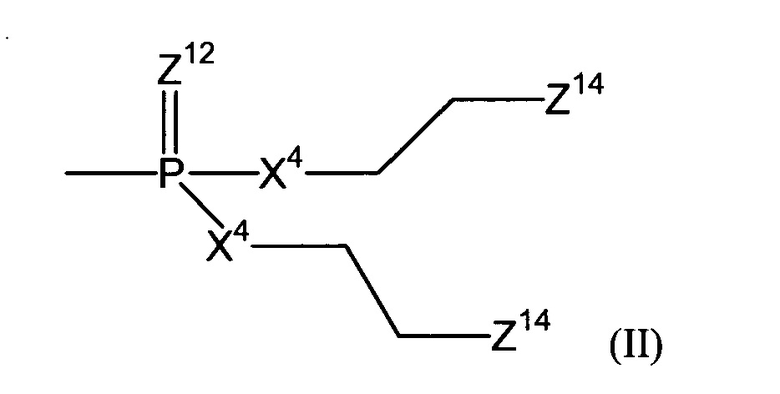
(где
Z12 означает кислород или серу;
каждый Х4 независимо означает кислород, серу или NZ13, где каждый -Z13 независимо означает -(СН2)2-Z14, -алкил или -водород; и
каждый Z14 независимо означает хлор, бром, йод или мезилат).
Согласно некоторым воплощениям изобретения, Z12 означает кислород. Согласно этим и другим конкретным воплощениям, каждый Х4 имеет одно и то же значение. Согласно этим и другим конкретным воплощениям, каждый Х4 означает NZ13. Согласно этим и другим конкретным воплощениям, каждый Z13 означает водород. Согласно этим и другим конкретным воплощениям настоящего изобретения, каждый имеющийся Z14 такой же и/или означает бром или хлор. В частности, согласно воплощениям изобретения каждый Z14 (который может представлять собой два, три или четыре Z14-остатка) означает бром.
Альтернативно, эффекторная молекула может представлять собой молекулу, которая выполняет диагностическую функцию, например, позволяющую осуществлять идентификацию или полное понимание природы, опухоли, в которой, например, экспрессируется CYP1B1. Примером класса эффекторных молекул, которые являются диагностическими молекулами, являются флуорофорные молекулы. Они могут быть пригодны в случае диагноза раковых клеток. Примеры флуорофорных соединений включают кумарины, резорцины, флуоресцеины и родамины и, на самом деле, через посредство ряда экспериментов, проводимых при использовании соединений согласно настоящему изобретению, включающих кумарины в качестве эффекторной молекулы, продемонстрирована «жизнеспособность» настоящего изобретения (см. примеры ниже).
Таким образом, должно быть понятно, что соединения формулы (I), где эффектор выполняет диагностическую функцию, могут быть использованы в методах диагноза и такие методы составляют дальнейшие аспекты настоящего изобретения. Следовательно, настоящее изобретение относится к соединению формулы (I), или его фармацевтически приемлемой соли, сложному эфиру, амиду или сольвату, для применения в методе диагноза пролиферативного состояния, как например, предраковая или злокачественная клеточная пролиферация, рак, лейкоз, псориаз, костное заболевание, фибропролиферативное нарушение или артеросклероз, например, пролиферативное состояние, выбираемое из ракового заболевания мочевого пузыря, головного мозга, молочной железы, ободочной кишки, головы и шеи, почки, легкого, печени, яичника, простаты и кожи, причем вышеуказанный метод включает введение количества соединения, или его фармацевтически приемлемой соли, сложного эфира, амида или сольвата формулы (I) субъекту, имеющему или у которого подозревают, что имеет, такое пролиферативное состояние, и мониторинге в отношении распределения высвобождающихся эффекторных молекул у субъекта, на основании чего может быть сделан диагноз.
Альтернативно, эффектор может представлять собой эффектор, который выполняет скрининговую функцию, например, в качестве части коллекции библиотеки модельных пролекарств, чтобы идентифицировать комбинации триггера и линкера, фрагмент которых затем активируется с помощью CYP1B1 и его аллельных вариантов. Примером класса эффекторных молекул являются флуорофорные молекулы. Примеры флуорофорных соединений включают хорошо известные кумарины, резоруфины, флуоресцеины и родамины. На самом деле, через посредство ряда экспериментов, проводимых при использовании соединений согласно изобретению, включающих кумарины в качестве эффекторной молекулы, продемонстрирована «жизнеспособность» настоящего изобретения (см. пример 1 в нижеприводимом разделе). Таким образом, должно быть понятно, что соединения формулы (I), в которых эффектор выполняет скрининговую функцию, могут быть использованы в случае идентификации комбинаций триггера и линкера для дизайна и синтеза пролекарств, активируемых с помощью CYP1B1, и такие методы составляют дальнейшие аспекты настоящего изобретения.
Таким образом, может быть признано, что соединения формулы (I), в которых эффектор выполняет скрининговую функцию, в качестве части коллекции библиотеки модельных пролекарств, могут быть использованы в комбинации с моделями прогнозирования субстрата цитохрома Р450 для руководствования в отношении дизайна и синтеза пролекарств с селективностью к, например, CYP1B1, и его аллельным вариантам, как, например, CYP1B1*3. В целях ясности, комбинация пролекарства из библиотеки модельного пролекарства с моделью прогнозирования субстрата специфически связывает субстрат с активацией пролекарства и фрагментацией с помощью CYP1B1, что представляет собой фундаментальный принцип дизайна. Кроме того, таким образом может быть признано, что соединения формулы (I), в которых эффектор выполняет скрининговую функцию, могут быть использованы в комбинации с моделями прогнозирования субстрата цитохрома Р450 для руководствования в отношении дизайна и синтеза пролекарств, которые не активируются цитохромами Р450 нормальной ткани, например, CYP1A1, CYP1A2, CYP2C9, CYP2C19, CYP2D6 и CYP3A4. Примером модели прогнозирования субстрата является позиционированный Gaussian Kernel k-NN алгоритм, базирующийся на поисках подобия согласно Tanimoto по, но не ограничиваясь этим, дескрипторам, как, например, расширенная связность по методу «отпечатков пальцев». Модели прогнозирования субстрата цитохрома Р450 для дизайна пролекарства могут быть созданы при использовании баз данных по биоактивности, происходящих от цитохрома Р450 HTS из коллекций структурно различных соединений. На самом деле, через посредство ряда экспериментов, осуществляемых при использовании соединений согласно изобретению, включающих кумарины в качестве эффекторной молекулы, используемых в комбинации с моделью прогнозирования CYP1B1-субстрата, продемонстрирована «жизнеспособность» настоящего изобретения (см. примеры 1 и 2).
Альтернативно, эффектор может быть эффектором, который выполняет скрининговую функцию, в качестве части коллекции библиотеки модельных пролекарств, в целях идентификации комбинаций триггера и линкера, фрагмент которых, когда активируется с помощью CYP1B1 и/или других цитохромов Р450 и его аллельных вариантов, сверхэкспрессируется в случае рака и других пролиферативных состояний. Примером класса эффекторных молекул являются флуорофорные молекулы. Примеры флуорофорных соединений включают кумарины, резоруфины, флуоресцеины и родамины. Примеры цитохромов Р450, других, чем CYP1B1, которые сверхэкспрессируются в случае рака, включают CYP2A/2B, CYP2F1, CYP2R1, CYP2S1, CYP2U1, CYP2W1, CYP3A5, CYP3A7, CYP4Z1, CYP26A1 и CYP51.
Таким образом, может быть признано, что соединения формулы (I), в которых эффектор выполняет скрининговую функцию, в качестве части коллекции библиотеки модельных пролекарств, могут быть использованы в комбинации с моделями прогнозирования субстрата цитохрома Р450 для руководствования в отношении дизайна и синтеза пролекарств с селективностью к CYP1B1 и/или другим цитохромам Р450 и его аллельным вариантам, сверхэкспрессируемым в случае рака и других пролиферативных состояний. Примером класса эффекторных молекул являются флуорофорные молекулы. Примеры флуорофорных соединений включают кумарины, резоруфины, флуоресцеины и родамины. Примеры цитохромов Р450, других, чем CYP1B1, которые сверхэкспрессируются в случае рака, включают CYP2A/2B, CYP2F1, CYP2R1, CYP2S1, CYP2U1, CYP2W1, CYP3A5, CYP3A7, CYP4Z1, CYP26A1 и CYP51. Примером модели прогнозирования субстрата является позиционированный Gaussian Kernel k-NN алгоритм, базирующийся на поисках подобия согласно Tanimoto по, но не ограничиваясь этим, дескрипторам, как, например, расширенная связность по методу «отпечатков пальцев». Модели прогнозирования субстрата цитохрома Р450 для дизайна пролекарства могут быть созданы при использовании баз данных по биоактивности, происходящих от цитохрома Р450 HTS, из коллекций структурно различных соединений.
Согласно аспектам и воплощениям настоящего изобретения, когда эффектор выполняет скрининговую функцию, например, в соответствии с седьмым аспектом настоящего изобретения, набор соединений типично включает множество соединений, например, включает по меньшей мере 10, например по меньшей мере 20 соединений. В случае некоторых воплощений, набор может включать вплоть до 100, 1000, 10000 или даже 100000 соединений. Такие наборы соединений, т.е. множества соединений согласно первому аспекту изобретения, где эффектор представляет собой флуорофор, а также другие множества соединений, в случае которых эффектор не является таким образом ограниченным и/или соединения могут быть фармацевтически приемлемыми солями, сложными эфирами, амидами или сольватами, составляют еще дальнейший аспект настоящего изобретения.
Согласно воплощениям седьмого аспекта настоящего изобретения, где соединение высвобождает флуорофор на стадии (а), но не высвобождает или высвобождает только в гораздо меньшей степени на стадии (b), под этим понимают, что фермент Р450 обычно высвобождает по меньшей мере в 10 раз, например по меньшей мере в 20 раз, больше вышеуказанного флуорофора на стадии (а) по сравнению со стадией (b).
Где действует скрининг, например, согласно воплощениям седьмого аспекта настоящего изобретения, например и обычно, соединение, которое высвобождает флуорофор на стадии (а), но не высвобождает или высвобождает только в гораздо меньшей степени на стадии (b), метод согласно седьмому аспекту настоящего изобретения необязательно включает дополнительно стадии:
(d) моделирование соединений, идентичных по структуре таковым, идентифицированным на стадии (с), за исключением того, что флуорофор заменяют молекулой, имеющей фармакологическую функцию в отношении связывания с активным участком вышеуказанного фермента цитохрома Р450; и
(е) синтез соединений, моделированных на стадии (d), которые должны прогнозировать субстраты для вышеуказанного фермента цитохрома Р450.
Альтернативно, эти стадии ((d) и (e)) могут быть использованы независимо от обязательных стадий согласно седьмому аспекту данного изобретения (т.е. (а)-(с)) и, следовательно, составляют еще дальнейшее воплощение настоящего изобретения.
Типично, фермент цитохром Р450 выбирают из группы, состоящей из CYP1B1, CYP2S1, CYP2W1, CYP4Z1 и их аллельных вариантов, например, CYP1B1 и его аллельного варианта, например, CYP1B1.
Аспектом настоящего изобретения является использование первичных человеческих опухолевых клеточных линий с числом раннего пассирования <20 in vitro, получаемых из резицированных раковых препаратов. Первичные клеточные линии плоскоклеточного рака головы и шеи UT-SCC, описанные в нижеприводимых примерах 4 и 5, конститутивно экспрессируют CYP1B1 на уровне мРНК и белка и могут быть подкожно трансплантированы мышам с иммунодефицитом (например, «голые» или с сильным комбинированным иммунодефицитом SCID мыши) с высокими скоростями приживления трансплантата для генерирования первичных человеческих опухолевых ксенотрансплантатов, где конститутивная экспрессия белка цитохрома Р450 соответствует экспрессии его в возникающей опухоли у пациента. Эти модели кесенотрансплантата первичной человеческой опухоли, путем поддерживания экспрессии цитохром Р450 мРНК/белок, подобной в возникающей у пациента опухоли, следовательно, могут быть использованы для оценки эффективности соединения согласно изобретению, где эффекторным остатком является агент, обладающий фармакологической активностью, при лечении рака. Кроме того, в клиническом контексте, эти модели первичного человеческого опухолевого ксенотрансплантата могут быть использованы для контроля, если ответные реакции на соединение согласно изобретению, где эффекторным остатком является обладающий фармакологической активностью агент, коррелируют с клиническими ответными реакциями и последствиями, что указывает на пригодность для персональной химиотерапии. Модели первичной человеческой опухоли также могут быть использованы для сравнения эффективности соединения по п.1 формулы изобретения, где эффекторный остаток представляет собой обладающий фармакологической активностью агент со стандартными химиотерапевтическими режимами и, следовательно, для идентификации наиболее эффективных режимов для соединений по п.1 формулы изобретения, индивидуально или в комбинации с другими химиотерапевтическими агентами.
Кроме того, в качестве части настоящего изобретения можно получать ксенотрансплантаты первичной человеческой опухоли путем осуществления прямой имплантации опухолевой ткани, получаемой прямой резекцией от пациентов, имплантации подкожно, например, «голым», SCID и «nonobese» диабетическим/с тяжелым комбинированным иммунодефицитом (NOD/SCID) мышам. Можно генерировать сначала человеческие опухолевые ксенотрансплантаты первого поколения в случае ряда различных раковых заболеваний, которые сохраняют гистологические и генетические характеристики возникающей опухоли и в качестве таковых конститутивно экспрессируют CYP1B1 мРНК/белок на уровне, подобном возникающей опухоли. Эти модели первичного человеческого опухолевого ксенотрансплантата, за счет сохранения экспрессии CYP1B1 мРНК/белок, подобно как в случае возникающей у пациента опухоли, следовательно, могут быть использованы для оценки эффективности соединения согласно изобретению, где эффекторным остатком является обладающий фармакологической активностью агент, при лечении рака. Кроме того, в клиническом контексте, эти модели первичного человеческого опухолевого ксенотрансплантата могут быть использованы для контроля, если ответные реакции на соединение по п.1 формулы изобретения, где эффекторным остатком является обладающий фармакологической активностью агент, коррелируют с клиническими ответными реакциями и последствиями, что указывает на пригодность для персональной химиотерапии. Модели первичной человеческой опухоли также могут быть использованы для сравнения эффективности соединения согласно изобретению, где эффекторный остаток представляет собой обладающий фармакологической активностью агент, со стандартными химиотерапевтическими режимами и, следовательно, для идентификации наиболее эффективных режимов для соединений согласно изобретению, индивидуально или в комбинации с другими химиотерапевтическими агентами.
Согласно восьмому аспекту настоящего изобретения, рак возникает в результате имплантации клетки из клеточной линии раннего пассирования, получаемой из ткани, которую берут прямо из опухоли или рака, которые экспрессируют вышеуказанный фермент цитохром Р450 при уровнях, подобных таковым в случае опухоли или карциномы, в которых он возникает, уровни могут быть рассмотрены как подобные, если они составляют в пределах 10% от таковых в случае опухоли или карциномы, от которой он происходит, например, в пределах 5%.
Для использования согласно настоящему изобретению, соединения или их физиологически приемлемая соль, сольват, сложный эфир или амид, описанные в данном контексте, могут быть представлены в виде фармацевтической композиции, включающей соединение или его физиологически приемлемую соль, сложный эфир, амид или его иное физиологически функциональное производное, вместе с одним или более фармацевтически приемлемыми носителями для этой цели и необязательно с другими терапевтическими и/или профилактическими ингредиентами. Любые носители приемлемы в том смысле, что являются совместимыми с другими ингредиентами композиции и не вредными для их реципиента.
Примеры физиологически приемлемых солей соединений согласно изобретению включают соли, образованные с органическими карбоновыми кислотами, такими как уксусная кислота, молочная кислота, винная кислота, малеиновая кислота, лимонная кислота, пирувиновая кислота, щавелевая кислота, фумаровая кислота, щавелевоуксусная кислота, изетионовая кислота, лактобионовая кислота и янтарная кислота; органическими сульфокислотами, такими как метансульфокислота, этансульфокислота, бензолсульфокислота и п-толуолсульфокислота, и неорганическими кислотами, такими как соляная кислота, серная кислота, фосфорная кислота и сульфаминовая кислота.
Определение физиологически приемлемых сложных эфиров или амидов, особенно сложных эфиров, хорошо известно квалифицированным специалистам в данной области.
Может быть подходящим или желательным получение, очистка и/или манипулирование в отношении соответствующих сольватов соединений, описанных в данном контексте, которые могут быть использованы в любом из описанных применений/методов. Термин «сольват» используется в данном контексте в отношении комплекса растворенного вещества, такого как соединение или соль соединения, и растворителя. Если растворителем является вода, сольват может быть назван гидратом, таким как, например, моногидрат, дигидрат, тригидрат и т.д., в зависимости от количества молекул воды, имеющихся на молекулу субстрата.
Должно быть понятно, что соединения согласно настоящему изобретению могут существовать в различных стереоизомерных формах и соединения согласно настоящему изобретению, как указано выше, включают все стереоизомерные формы и их смеси, включая энантиомеры и рацемические смеси. Настоящее изобретение включает, в пределах его объема, использование любой такой стереоизомерной формы или смеси стереоизомеров, включая индивидуальные энантиомеры соединений формулы (I) или (II), а также полностью или частично рацемические смеси таких энантиомеров.
Специалисту в данной области также должно быть понятно, что противораковые пролекарства, как например таковые, описанные в данном контексте, могут быть направлены на конкретные опухоли путем присоединения направленной на опухоль составляющей, такой как нацеливаемый на опухоль пептид, например, малые пептиды, идентифицируемые через посредство развития фагообнаруживаемых пептидных библиотек. Такие пептиды или другие составляющие могут содействовать в нацеливании конъюгатов, которые включают их, на конкретную карциному, особенно, на солидную опухоль. Таким образом, обеспечение такими конъюгатами, т.е. соединением согласно изобретению, конъюгированным с нацеленной на опухоль составляющей, составляет дальнейший аспект данного изобретения, как и получение композиций, применения и методы, описанные в данном контексте, которые включают или влекут за собой использование таких конъюгатов.
Соединения согласно настоящему изобретению могут быть получены при использовании реагентов и способов, без труда доступных согласно уровню техники, и/или типичных способов, таких как описанные в дальнейшем. Найдено, что соединения согласно настоящему изобретению проявляют цитотоксичность в случае клеток, экспрессирующих фермент CYP1B1, но по существу являются нетоксичными в случае нормальных клеток, которые не экспрессируют CYP1B1. Соединения согласно изобретению также проявляют цитотоксичность в случае клеток, экспрессирующих фермент CYP1A1. На практике, следовательно, соединения согласно изобретению являются нетоксическими пролекарствами, которые превращаются (обычно за счет CYP1B1) в цитотоксические агенты.
Соответственно, соединения согласно изобретению имеют значение цитотоксичности IC50, как определено ниже, или менее чем 10 мкМ, преимущественно менее чем 5 мкМ, например, менее чем 1,0 мкМ, или 0,5 мкМ.
Согласно некоторым воплощениям, цитотоксичность соединения согласно изобретению может быть определена путем инкубации соединения, при различных последовательных разведениях, с клетками, сконструированными путем генной инженерии для экспрессирования CYP1B1. Соответственно, вышеуказанные клетки могут быть клетками яичника китайского хомячка (СНО), которые могут содержать рекомбинантный CYP1B1 и цитохром-Р450-редуктазу (CPR). Высокие уровни функционального фермента, когда происходит коэкспрессия с человеческой Р-450-редуктазой, могут быть достигнуты при использовании амплификации гена дигидрофолатредуктазы (DHFR). Типично, генноинженерные клетки могут быть инкубированы с соединением и, спустя соответствующий период времени (например, 96 часов), инкубированы далее (например, в течение 1,5 часов) с подходящим реагентом для анализа в целях обеспечения указания на число живых клеток в культуре. Подходящим реагентом для анализа является MTS (см. ниже), который биовосстанавливается клетками до формазана, продукта, который растворим в тканевой культуральной среде. Абсорбция формазана может быть прямо измерена при длине волны 510 нм и количественный формазановый продукт, как определенный по величине оптической плотности при длине волны 490 нм или 510 нм, прямо пропорционален числу живых клеток в культуре. Подробные способы определения значения IC50 соединения согласно изобретению описываются ниже в примере 3.
С целью сравнения, значения IC50 соединений согласно изобретению также могут быть определены в клетках (например, клетки яичника китайского хомячка), которые не содержат CYP1B1, например, СНО-клетки дикого типа. Соединения согласно изобретению, соответственно, могут иметь кратную селективность в случае CYP1B1-экспрессирующих клеток по меньшей мере 200, где термин «кратная селективность» определяют как частное от деления значения IC50 данного соединения в не экспрессирующих CYP1 клетках на значение IC50 того же самого соединения в экспрессирующих CYP1B1 клетках.
Согласно некоторым воплощениям, цитотоксичность соединения согласно изобретению может быть также определена путем инкубации соединения, при различных последовательных разведениях, с клетками первичной опухоли головы и шеи, происходящими от пациентов с плоскоклеточным раком головы и шеи, как описывается в примере 4.
Согласно некоторым воплощениям, in vivo эффективность соединения согласно изобретению может быть определена путем имплантации клеток первичной опухоли плоскоклеточного рака головы и шеи, которые конститутивно экспрессируют CYP1B1, подкожно, в бок «голой» мыши для создания моделей первичного человеческого опухолевого ксенотрансплантата и определения действия обработки пролекарством на рост опухоли, как описывается в примере 5.
Как таковое, настоящее изобретение также относится к применению одного или более из соединений согласно изобретению, включая вышеуказанные фармацевтически приемлемые сложные эфиры, амиды, соли, сольваты и пролекарства, для использования при обработке человеческого или животного организма путем терапии, особенно, для лечения или профилактики пролиферативных состояний, таких, как, например, пролиферативные нарушения или заболевания, у людей и животных, включая пролиферативные состояния, которые указаны в случае некоторых воплощений данного изобретения, характеризующиеся клетками, которые экспрессируют CYP1B1. Более конкретно, настоящее изобретение относится к применению одного или более из соединений согласно изобретению для лечения раковых заболеваний, характеризующихся, согласно некоторым воплощениям настоящего изобретения, экспрессией CYP1B1.
Под «пролиферативным состоянием», согласно данному контексту, понимают заболевание или нарушение, которое характеризуется нежелательной или неконтролируемой клеточной пролиферацией избыточных или анормальных клеток, которые нежелательны, как например непластический или гиперпластический рост, происходящий или in vivo, или in vitro. Примерами пролиферативных состояний являются предраковая или злокачественная клеточная пролиферация, включая злокачественные неоплазмы и опухоли, карциномы, лейкозы, псориаз, костные заболевания, фибропролиферативные нарушения (например, соединительных тканей) и атеросклероз.
Вышеуказанное пролиферативное состояние может характеризоваться, согласно некоторым воплощениям данного изобретения, клетками, которые экспрессируют CYP1B1.
Вышеуказанное пролиферативное состояние может быть выбрано из рака мочевого пузыря, головного мозга, молочной железы, ободочной кишки, головы и шеи, почки, легкого, печени, яичника, простаты и кожи. Согласно некоторым воплощениям, вышеуказанное пролиферативное состояние может включать солидную опухоль.
Под термином «лечение», согласно данному контексту, понимают обработку путем терапии, осуществляется ли она в случае человека или животного (например, при применениях в ветеринарии), при которой достигают некоторого желательного терапевтического воздействия на пролиферативное состояние, например, ингибирование прогрессирования нарушения, включая снижение скорости прогрессирования, прекращение прогрессирования, уменьшение интенсивности нарушения или излечивание состояния. Также включается лечение как профилактическая мера. Ссылки, согласно данному контексту, на предотвращение или профилактику, как указывается в данном контексте, не обозначают или не свидетельствуют о необходимом условии полного предотвращения состояния; его проявление, взамен, может быть ослаблено или замедлено за счет профилактики или предотвращения согласно настоящему изобретению. Под термином «терапевтически эффективное количество», согласно данному контексту, понимают количество одного или более соединений согласно изобретению или фармацевтической композиции, включающей такое одно или более соединений, которое является эффективным для продуцирования такого терапевтического эффекта, соразмерного с допустимым соотношение польза/риск.
Соединения согласно настоящему изобретению, кроме того, могут быть использованы в качестве противораковых агентов. Под термином «противораковый агент», согласно данному контексту, понимают соединение, которое лечит рак (т.е. соединение, которое пригодно в терапии рака). Противораковый эффект соединений согласно изобретению может возникать через посредство одного или более механизмов, включая регуляцию клеточной пролиферации, ингибирование ангиогенеза, ингибирование метастаз, ингибирование инвазии или промотирование апоптоза.
Должно быть понятно, что соответствующие дозы соединений согласно изобретению могут изменяться от пациента к пациенту. Определение оптимальной дозы обычно предполагает балансировку уровня терапевтической пользы против любого риска или вредных побочных эффектов лечений согласно настоящему изобретению. Выбираемый уровень дозы зависит от множества факторов, включая активность конкретного соединения, путь введения, время введения, скорость экскреции соединения, продолжительность лечения, другие лекарственные средства, соединения или материалы, используемые в комбинации, и возраст, пол, массу, состояние, общее состояние здоровья и предшествующую историю болезни пациента. Количество соединения(ий) и путь введения, в конечном счете, должны быть по усмотрению врача, хотя обычно доза должна достигать локальных концентраций в месте действия с тем, чтобы достигать желательного эффекта.
Введение in vivo может быть осуществлено в виде одной дозы, непрерывно или периодически, в продолжение курса лечения. Способы определения наиболее эффективного средства и дозы введения хорошо известны специалисту в данной области и могут изменяться в зависимости от готовой лекарственной формы, используемой для терапии, цели терапии, подвергаемой обработке клетки-мишени и подвергаемого лечению субъекта. Одинарные или многократные введения могут быть осуществлены в соответствии с уровнем дозы и схемами приема, выбираемыми лечащим врачом.
Фармацевтические композиции включают таковые, подходящие для перорального, местного (включая дермальное, буккальное и сублингвальное), ректальное и парентеральное (включая подкожное, интрадермальное, внутримышечное и внутривенное), назальное и пульмонарное введение, например, путем ингаляции. Композиция, где соответствует, может быть подходящим образом представлена в виде дискретных дозированных единиц и может быть получена любым из способов, хорошо известных в фармацевтике. Способы обычно включают стадию введения в ассоциацию активного соединения с жидкими носителями или тонко измельченными твердыми носителями или обоими и затем, если необходимо, формирования продукта до получения желательной готовой лекарственной формы.
Фармацевтические композиции, подходящие для перорального введения, где носителем является твердое вещество, наиболее предпочтительно представлены в виде композиций в унифицированной дозе, таких как болюсы, капсулы или таблетки, содержащие предопределенное количество активного соединения. Таблетка может быть получена путем прессования или литья под давлением, необязательно с одним или более добавочными ингредиентами. Прессованные таблетки могут быть получены путем прессования в подходящей машине активного соединения в свободнотекучей форме, такой как порошок или гранулы, необязательно смешанного со связующим, смазкой, инертным разбавителем, смазывающим веществом, поверхностно-активным веществом или диспергатором. Отлитые под давлением таблетки могут быть получены путем отливки в форме активного соединения с инертным жидким разбавителем. Таблетки могут быть необязательно снабжены покрытием и, если не имеют покрытия, необязательно могут быть снабжены насечкой. Капсулы могут быть получены путем насыпания активного соединения, либо индивидуально, либо в смеси с одним или более добавочными ингредиентами, в капсульные оболочки и затем герметизации капсулы обычным образом. Крахмальные облатки аналогичны капсулам, где активное вещество вместе с любым(и) добавочным(и) ингредиентом(ами) герметизировано в обертке из рисовой бумаги. Активное соединение также может быть представлено в виде диспергируемых гранул, которые могут быть, например, суспендированы в воде перед введением или насыпаны на пищу. Гранулы могут быть упакованы, например, в саше. Готовые лекарственные формы для перорального введения, где носителем является жидкость, могут быть представлены в виде раствора или суспензии в водной или неводной жидкости или в виде жидкой эмульсии масло-в-воде.
Готовые лекарственные формы для перорального введения включают лекарственные формы с контролируемым высвобождением, например, таблетки, где активное соединение находится в соответствующей, контролирующей высвобождение, матрице или покрыто подходящей, контролирующей высвобождение, пленкой. Такие готовые лекарственные формы могут быть особенно подходящими для профилактического использования.
Фармацевтические композиции, подходящие для ректального введения, где носителем является твердое вещество, наиболее предпочтительно представлены в виде суппозиториев с разовой дозой. Подходящие носители включают масло какао и другие вещества, обычно используемые в уровне техники. Суппозитории могут быть подходящим образом получены путем смешения активного соединения с размягченным или расплавленным носителем(ями) с последующим охлаждением и формированием в формах.
Фармацевтические композиции, подходящие для парентерального введения, включают стерильные растворы или суспензии активного соединения в водных или маслянистых эксципиентах.
Инъецируемые препараты могут быть адаптированы для болюсной инъекции или непрерывной инфузии. Такие препараты пригодным образом представлены в виде контейнеров с разовой дозой или с многократной дозой, которые герметически закрыты после введения композиции вплоть до необходимости для использования. Альтернативно, активное соединение может находиться в порошкообразной форме, которая перед использованием структурируется с помощью подходящего эксципиента, такого как, например, стерильная непирогенная вода.
Активное соединение также может быть представлено в виде пролонгированных депо-препаратов, которые могут быть введены путем внутримышечной инъекции или путем имплантации, например, подкожно или внутримышечно. Депо-препараты могут включать, например, подходящие полимерные или гидрофобные вещества или ионобменные смолы. Такие готовые лекарственные формы пролонгированного действия особенно пригодны для профилактического использования.
Готовые лекарственные формы, подходящие для пульмонарного введения через щечный карман, представляют собой такие, частицы которых, содержащие активное соединение и желательно имеющие диаметр в пределах 0,5-7 микрон, доставляются по бронхиальному дереву реципиента.
В качестве одной возможности, такие готовые лекарственные формы находятся в виде тонко измельченных порошков, которые могут быть подходящим образом представлены либо в прокалываемой капсуле, соответственно, из, например, желатина, для использования в устройстве для ингаляции, или, альтернативно, в виде самоприводящейся в движение композиции, включающей активное соединение, подходящий жидкий или газообразный пропеллент и, необязательно, другие ингредиенты, как, например, поверхностно-активное вещество и/или твердый разбавитель. Подходящие жидкие пропелленты включают пропан и хлорфторуглеводороды и подходящие газообразные пропелленты включают диоксид углерода. Самоприводящиеся в движение композиции также могут быть применены там, где активное соединение распределяется в форме капель раствора или суспензии.
Такие самоприводящиеся в движение композиции аналогичны таковым, известным в уровне техники, и могут быть получены известными способами. Подходящим образом, они находятся в контейнере, снабженном либо управляемым вручную или функционирующим автоматически клапаном, имеющем желательные характеристики получения спрея; преимущественно клапан представляет собой таковой измерительного типа, доставляющий фиксированный объем, например, от 25 до 100 микролитров, во время каждого его действия.
В качестве дальнейшей возможности, активное соединение может быть в форме раствора или суспензии для использования в распыливателе или аэрозольном аппарате, где используют ускоренный ток воздуха или ультразвуковое перемешивание для получения мелкокапельного тумана для ингаляции.
Готовые лекарственные формы, подходящие для назального введения, включают препараты, обычно подобные таковым, описанным выше для пульмонарного введения. Когда приготавливают, такие готовые лекарственные формы должны иметь желательно диаметр частицы в пределах от 10 микрон до 200 микрон для создания возможности удерживания в полости носа; они могут быть успешно получены путем, как соответствует, использования порошка с подходящим размером частиц или выбора соответствующего клапана. Другие подходящие готовые лекарственные формы включают «крупные» порошки, имеющие диаметр частицы в пределах от 20 микрон до 500 микрон, для введения путем быстрой ингаляции через носовой вход из контейнера, удерживаемого закрытым вплоть до носа, и капли в нос включают 0,2-5% масс./об. активного соединения в водном или масляном растворе или суспензии.
Должно быть понятно, что в дополнение к вышеуказанным ингредиентам носителей, вышеописанные фармацевтические композиции могут включать, подходящие, один или более, дополнительных ингредиентов носителей, как например разбавители, буферы, ароматизаторы, связующие, поверхностно-активные вещества, загустители, смазочные вещества, консерванты (включая антиоксиданты) и т.п., и вещества, включаемые для цели получения композиции, изотонической с кровью подразумеваемого реципиента.
Фармацевтически приемлемые носители хорошо известны специалисту в данной области и включают, но не исчерпывающим образом, 0,1 М и, предпочтительно, 0,05 М фосфатный буфер или 0,8% солевой раствор. Дополнительно, фармацевтически приемлемые носители могут быть водными или неводными растворами, суспензиями и эмульсиями. Примерами неводных растворителей являются пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло, и инъецируемые сложные органические эфиры, такие как этилолеат. Водные носители включают воду, спирто/водные растворы, эмульсии или суспензии, включая солевой раствор и забуферивающие среды. Парентеральные наполнители включают раствор хлорида натрия, декстрозу Рингера, декстрозу и хлорид натрия, лактат Рингера или нелетучие масла. Консерванты и другие добавки также могут присутствовать, такие как, например, антимикробные средства, антиоксиданты, хелатирующие агенты, инертные газы и т.п.
Композиции, подходящие для локальной готовой лекарственной формы, могут быть предусмотрены в виде гелей, кремов или мазей.
Также могут быть предусмотрены жидкие или порошкообразные композиции, которые могут наноситься путем пульверизации или посыпания прямо на обрабатываемое место, например, рану или язву. Альтернативно, носитель, такой как повязка, марля, сетка или т.п., может быть опрыскан или посыпан композицией и затем нанесен на подвергаемое лечению место.
Терапевтические композиции для использования в ветеринарии могут быть подходящим образом в форме либо порошкообразного, либо жидкого концентрата. В соответствии со стандартной ветеринарной практикой получения готовой лекарственной формы, подходящие водорастворимые эксципиенты, такие как лактоза или сахароза, могут быть включены в порошки для улучшения их физических свойств. Так, особенно подходящие порошки согласно настоящему изобретению включают 50-100% масс./масс. и, предпочтительно, 60-80% масс./масс., активного(ых) ингредиента(ов) и 0-50% масс./масс. и, предпочтительно, 20-40% масс./масс. обычных эксципиентов в случае ветеринарии. Эти порошки могут быть добавлены либо к животному корму, например, путем промежуточного предварительного смешивания, либо разведены в воде для питья животного.
Жидкие концентраты согласно настоящему изобретению подходящим образом содержат соединение, или его производное, или его соль и, необязательно, могут включать приемлемый в ветеринарии смешивающийся с водой растворитель, например, полиэтиленгликоль, пропиленгликоль, глицерин, глицеринформаль или такой растворитель, смешанный с количеством вплоть до 30% об./об. этанола. Жидкие концентраты могут быть введены в воду для питья животных.
Обычно подходящая доза одного или более соединений согласно изобретению может находиться в диапазоне от 1 мкг до примерно 5000 мкг/кг массы тела субъекта в сутки, например, 1, 5, 10, 25, 50, 100, 250, 1000, 2500 или 5000 мкг/кг в сутки. Когда соединение(я) представляет(ют) собой соль, сольват, пролекарство или т.п., вводимое количество может быть рассчитано на основе исходного соединения и таким образом используемая действительная масса может быть пропорционально увеличена.
Согласно некоторым воплощениям, одно или более соединений согласно настоящему изобретению могут быть использованы в комбинированных терапиях для лечения пролиферативных состояний рода, описанного выше, т.е. в сочетании с другими терапевтическими агентами. Примеры таких других терапевтических агентов включают, но не исчерпывающим образом, ингибиторы топоизомеразы, алкилирующие агенты, антиметаболиты, ДНК-связующие и ингибиторы микротрубочек (нацеленные на тубулин агенты), такие как цисплатин, циклофосфамид, доксорубицин, этопозид, иринотекан, флударабин, 5FU, таксаны и митомицин С. Другие терапевтические агенты должны быть очевидны специалисту в данной области. В случае активных соединений, комбинируемых с другими терапиями, две или более обработок можно осуществлять по графикам индивидуально изменяющейся дозы и посредством различных путей.
Комбинация вышеперечисленных агентов с соединением согласно настоящему изобретению должна быть определена по усмотрению врача, который обычно выбирает дозировки, используя свое общее знание и режимы дозировки, известные квалифицированному практику.
Когда соединение согласно данному изобретению вводят согласно комбинированной терапии с одним, двумя, тремя, четырьмя или более, предпочтительно одним или двумя, предпочтительно одним другим, терапевтическими агентами, то соединения можно вводить одновременно или последовательно. Когда вводят последовательно, то их можно вводить через близко расположенные интервалы (например, через период времени 5-10 минут) или через более длительные интервалы (например, 1, 2, 3, 4 или более часов, раздельно, или даже более длительный период, раздельно, когда необходимо), причем точная схема приема соразмерна со свойствами терапевтического(их) агента(ов).
Соединения согласно данному изобретению также можно вводить в сочетании с нехимиотерапевтическими видами лечения, такими как радиотерапия, фотодинамическая терапия, генная терапия, хирургия и контролируемые диеты.
Данное изобретение теперь поясняется со ссылкой на следующие, не ограничивающие объема охраны изобретения, примеры.
Получение соединений
Общее
Спектры 1Н-, 13С- и 31Р-ядерного магнитного резонанса (ЯМР) регистрировали в указанном растворителе при использовании или спектрометра Bruker Avance DPX 500 МГц или спектрометра Bruker Avance 300 МГц. Химические сдвиги выражали в миллионных долях (м.д.). Изображение расщепления сигнала описывали как синглет (s), уширенный синглет (bs), дублет (d), триплет (t), квартет (q), мультиплет (m) или их комбинация. Масс-спектры низкого разрешения с ионизацией электронным распылением (ES) регистрировали на масс-спектрометре Bruker MicroTof, работая по методу образования положительных ионов с использованием или смеси метанол/вода (95:5) или смеси вода/ацетонитрил (1:1) + 0,1% муравьиной кислоты в качестве подвижной фазы. Измерения при использовании прибора высокого разрешения с ионизацией электронным распылением осуществляли на масс-спектрометре Bruker Microtof. Анализы LC-MS осуществляли с помощью Agilent HPLC 1100 (колонка Phenomenex Gemini, 5 мкм, С18, 110 Å, размером 50×3,0 мм, элюируя с помощью (0-20% МеОН/Н2О), и последовательно с помощью масс-спектрометра Bruker Microtof, при использовании детектора с диодной матрицей. Колоночную хроматографию осуществляли при использовании колонок, предварительно заполненных силикагелем (230-400 меш) или 4, 12, 40 или 80 г кремнезема RediSep®. Все исходные вещества являлись коммерчески доступными и использовались без дальнейшей очистки. Все взаимодействия осуществляли в безводных или инертных условиях, за исключением иначе указанного.
[Соединения, указанные ниже с указанным в скобках крестиком (†), не являются примерами соединений согласно данному изобретению, но включены для лучшего его понимания].
1. Фосфороамидатмустиновые пролекарства
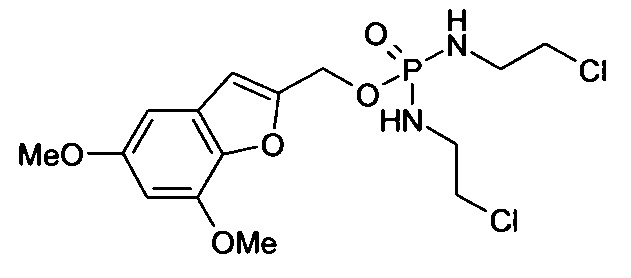
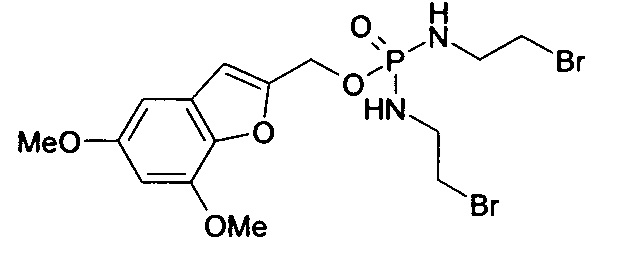
Синтез фосфороамидатных пролекарств SU025-04 и SU046-04
1. Синтез триггерного компонента пролекарств
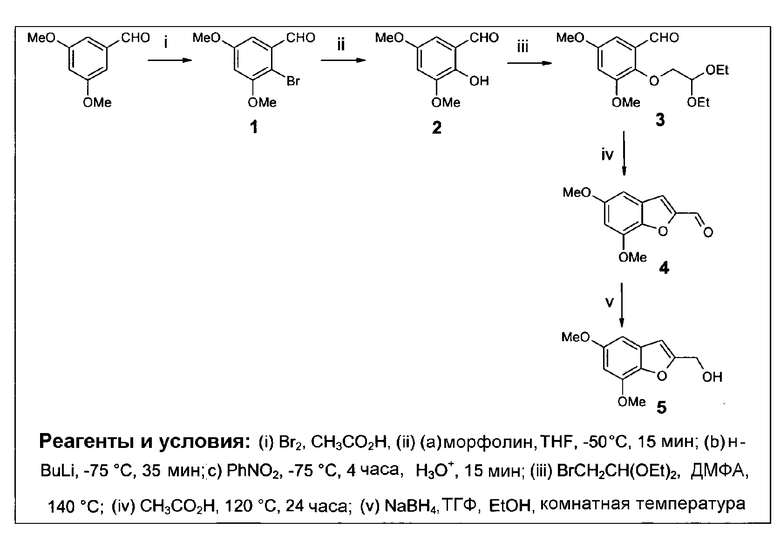
2-Бром-3,5-диметоксибензальдегид (1)
3,5-Диметоксибензальдегид (12,6 г, 76 ммоль) растворяют в уксусной кислоте (350 мл). Полученный бесцветный раствор охлаждают до температуры 0°С. По каплям добавляют раствор брома (3,9 мл), в уксусной кислоте (50 мл) в течение 1 часа. После завершения добавления убирают ледяную баню и полученный раствор бледно-зеленого цвета перемешивают в течение ночи при комнатной температуре. К раствору добавляют холодную воду. Полученное твердое вещество белого цвета собирают путем вакуумной фильтрации и промывают водой. Твердое вещество затем повторно растворяют в EtOAc и адсорбируют на силикагеле. Продукт очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (4:1), получая соединение 1 (12,5 г, выход 66%) в виде твердого вещества белого цвета. m/z = 345,98 (М+Н).
1H ЯМР (500 МГц, CDCl3): δ: 10,43 (1H, с, CHO), 7,06 (1H, с, ArH), 6,73 (1H, с, ArH), 3,93 (3H, с, CH3O), 3,86 (3H, с, CH3O).
13С-ЯМР (500 МГц, CDCl3): δ: 192,09 (СНО), 159,92 (С-5), 157,02 (С-3), 134,67 (С-1), 109,12 (С-2), 105,83, 103,37 (С-4 и С-6), 56,60 (ОМе), 55,82 (ОМе).
2-Гидрокси-3,5-диметоксибензальдегид (2)
Морфолин (2,05 г, 24 ммоль) и ТГФ (40 мл) вводят в трехгорлую круглодонную колбу, оснащенную мешалкой, мембранным колпачком, капельной воронкой, термометром и вводом для аргона. Колбу охлаждают на бане со смесью сухой лед-ацетон до температуры -50°С и добавляют весь раствор n-BuLi в гексане (1,6 М раствор, 15 мл, 24 ммоль), за один раз. Спустя 10 минут, по каплям, добавляют раствор соединения 1 (4,9 г, 20 ммоль) в ТГФ (30 мл) посредством шприца, в течение периода времени 4 минуты, и смесь охлаждают до температуры ~ -75°С в течение 20 минут. Затем по каплям добавляют н-BuLi в гексане (1,6 М раствор, 20 мл, 32 ммоль) в течение 45 минут, поддерживая температуру равной -75°С. После завершения добавления н-BuLi, раствор перемешивают в течение 35 минут. Из капельной воронки добавляют раствор нитробензола (6,90 г, 46 ммоль) в 10 мл ТГФ, поддерживая температуру равной -75°С. Полученную смесь темного цвета перемешивают при температуре -75°С в течение 4 часов и затем оставляют нагреваться до комнатной температуры. Эту смесь подкисляют до рН=1 с помощью 6 н HCl и перемешивают в течение 15 минут. После разбавления насыщенным солевым раствором (100 мл), ТГФ удаляют в вакууме. Водный раствор экстрагируют диэтиловым эфиром (4×40 мл). Объединенные органические слои экстрагируют 2 н раствором NaOH (3×40 мл). Объединенные NaOH-экстракты промывают диэтиловым эфиром (3×20 мл) и затем подкисляют до рН=1 с помощью концентрированной HCl. Полученную смесь экстрагируют CH2Cl2 (3×20 мл), и объединенные органические экстракты промывают насыщенным солевым раствором, сушат (MgSO4) и адсорбируют на силикагеле. Продукт очищают с помощью флэш-хроматографии, элюируя смесью EtOAc/гексан (1:2). Чистое соединение 2 получают (2,0 г, выход 55%) в виде твердого вещества желтого цвета. m/z = 183,06 (М+Н).
1H ЯМР (500 МГц, CDCl3): δ: 10,71 (1H, с, OH), 9,91 (1H, с, CHO), 6,77 (1H, д, J4,6=2,8 Гц, H-6), 6,61 (1H, д, J4,6=2,8 Гц, Н-4), 3,92 (3H, с, OMe), 3,84 (3H, с, OMe).
13С-ЯМР CDEPT135 (500 МГц, CDCl3): δ: 196,11 (СНО), 107,93 (С-6), 103,90 (С-4), 56,29 (ОМе), 55,83 (ОМе).
2-(2,2-Диэтоксиэтокси)-3,5-диметоксибензальдегид (3)
К перемешиваемой суспензии, содержащей соединение 2 (1,1 г, 6,0 ммоль) и К2СО3 (1,0 г, 7,2 ммоль), в ДМФА (100 мл), по каплям, добавляют бромацетальдегиддиэтилацеталь (0,93 мл, 6,0 ммоль). Смесь кипятят с обратным холодильником в течение 4 часов. После охлаждения осадок отфильтровывают и растворитель выпаривают в вакууме. Сырой остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (4:1), получая соединение 3 (1,2 г, выход 67%) в виде прозрачного масла.
1H ЯМР (500 МГц, CDCl3): δ: 10,50 (1H, с, CHO), 6,88 (1H, д, J4,6=2,9 Гц, H-6), 6,74 (1H, д, J4,6=2,9 Гц, H-4), 4,83 (1H, т, J4,6=5,3 Гц, CH), 4,14 (2H, д, J4,6=5,3 Гц, CH2), 3,88 (3H, с, OMe), 3,83 (3H, с, OMe), 3,77-3,71 (2H, м, CH2CH3), 3,63-3,58 (2H, м, CH2CH3), 1,24 (6H, т, J= 7,1 Гц, 2 ×·CH3).
5,7-Диметоксибензофуран-2-карбальдегид (4)
Перемешиваемый раствор соединения 3 (1,2 г, 4,0 ммоль) в уксусной кислоте (35 мл) кипятят с обратным холодильником в течение 16 часов. После охлаждения раствор выпаривают досуха. Сырой продукт адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (2:1), получая соединение 4 (230 мг, выход 28%) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, CDCl3): δ: 9,89 (1H, с, CHO), 7,50 (1H, с, H-3), 6,69 (1H, д, J4,6=2,2 Гц, H-6), 6,64 (1H, д, J4,6=2,2 Гц, H-4), 4,01 (3H, с, OMe), 3,87 (3H, с, OMe).
13С-ЯМР CDEPT 135 (500 МГц, CDCl3): δ: 179,91 (СНО), 153,37 (С-2), 116,10 (С-3), 101,80 (С-6), 94,90 (С-4), 56,20 (ОМе), 55,88 (ОМе).
(5,7-Диметоксибензофуран-2-ил)метанол (5)
Соединение 4 (460 мг, 2,23 ммоль) растворяют в ТГФ (5 мл) и EtOH (1 мл). Порциями добавляют NaBH4 (102 мг, 2,68 ммоль) при температуре 0°С при энергичном перемешивании. Суспензию перемешивают при температуре 0°С в течение 15 минут, и затем при комнатной температуре в течение 1 часа. Растворители выпаривают в вакууме. Сырой остаток обрабатывают EtOAc и промывают водой, насыщенным солевым раствором и сушат (MgSO4). Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (1:1), получая соединение 5 (388 мг, выход 82%) в виде масла. m/z = 209,08 (М+Н).
1H ЯМР (500 МГц, CDCl3): δ: 6,62 (1H, с, H-3), 6,60 (1H, с, H-6), 6,46 (1H, с, H-4), 4,76 (2H, с, 2-CH2), 3,99 (3H, с, OMe), 3,85 (3H, с, OMe).
13С-ЯМР (500 МГц, CDCl3): δ: 157,23 (С-5), 156,72 (С-1), 145,36 (С-7), 139,50 (С-1а), 129,38 (С-4а), 104,62 (С-3), 96,96 (С-6), 94,58 (С-4), 57,98 (2-СН2), 55,95 (ОМе), 55,83 (ОМе).
2. Синтез эффекторных компонентов пролекарств
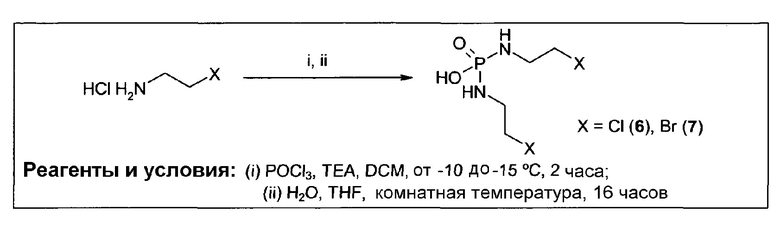
N,N-бис(2-Хлорэтил)фосфонамидокислота (6)
К суспензии 2-хлорэтиламингидрохлорида (7,2 г, 62 ммоль) в CH2Cl2 (110 мл) добавляют POCl3 (2,84 мл, 31 ммоль) в течение 15 минут при температуре -78°С, при энергичном перемешивании, затем добавляют раствор ТЕА (17,5 мл, 124 ммоль) в CH2Cl2 (30 мл) в течение 4 часов. Реакционную смесь перемешивают при температуре -78°С в течение 1 часа и потом оставляют нагреваться до комнатной температуры и перемешивают в течение 2 часов. Полученное твердое вещество отфильтровывают и промывают холодным EtOAc. Твердое вещество удаляют. Фильтрат концентрируют в вакууме до объема примерно 5 мл и добавляют EtOAc (10 мл). Полученную суспензию отфильтровывают и промывают EtOAc (2×10 мл). Твердое вещество снова удаляют. Фильтрат концентрируют в вакууме досуха. Остаток затем растворяют в ТГФ (7 мл), потом добавляют водный раствор NaBr (NaBr (5 г) в 100 мл воды) при температуре 0°С в течение 20 мин. Смесь затем нагревают до комнатной температуры и перемешивают в течение 15 часов на водяной бане. Из реакционной смеси осаждается твердое вещество белого цвета. Смесь затем выдерживают в морозильной камере при температуре -20°С в течение 2 часов. Кристаллическое твердое вещество отфильтровывают и промывают холодной водой (2×50 мл, температура 0°С) и холодным EtOAc (2×50 мл, температура 0°С). После высушивания при комнатной температуре в вакууме в течение ночи получают продукт 6 (1,8 г, выход 26%) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, ДМСО-d6): δ: 5,29 (3H, ушир., OH и NH), 3,55 (4H, т, J=7,0 Гц, 2 × CH2), 3,01 (4H, дт, J=12,2, 7,0 Гц, 2 × CH2).
31P ЯМР (500 МГц, ДМСО-d6) δ: 12,28 ppm.
N,N-бис(2-бромэтил)фосфонамидная кислота (7)
Соединение 7 синтезируют, используя подобную методику, как описано выше. Получают соединение с выходом 18% (1,64 г).
1H ЯМР (500 МГц, ДМСО-d6): δ: 6,08 (3H, с, OH и NH), 3,46 (4H, т, J=7,0 Гц, 2 × CH2), 3,01 (4H, дт, J=12,2, 7,0 Гц, 2 × CH2).
31P ЯМР (500 МГц, ДМСО-d6) δ: 12,23 ppm.
3. Реакция связывания для синтеза SU025-04 и SU046-04
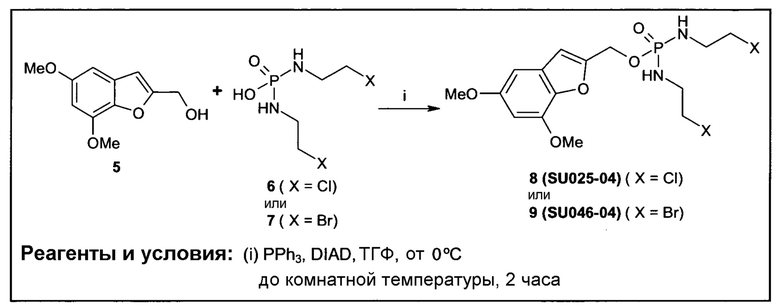
5,7-Диметоксибензофуран-2-ил)метил-N,N’-бис(2-хлорэтил)фосфордиамидат (8) SU025-04
К суспензии соединения 5 (300 мг, 1,44 ммоль), соединения 6 (479 мг, 2,16 ммоль) и PPh3 (565 мг, 2,16 ммоль) в ТГФ (20 мл) по каплям добавляют DIAD (0,426 мл, 2,16 ммоль), при температуре 0°С. Полученную суспензию нагревают до комнатной температуры и перемешивают в течение 2 часов. Растворитель удаляют и остаток очищают с помощью флэш-хроматографии (70% ацетона в толуоле), получая соединение 8 (250 мг, выход 42%) в виде масла. m/z = 823,08 (2М+Н).
1H ЯМР (500 МГц, ДМСО-d6): δ 7,27 (2H, с, 2 × NH), 6,75 (1H, с, ArH-3), 6,62 (1H, д, J4,6=2,3 Гц, ArH-4), 6,49 (1H, д, J4,6=2,3 Гц, ArH-6), 5,13 (2H, д, J=9,3 Гц, 2H), 3,99 (3H, с, OMe), 3,86 (3H, с, OMe), 3,29 (4H, м, 2 × CH2).
31Р-ЯМР (500 МГц, ДМСО-d6), δ: 14,76 м.д.
HRMS: рассчитано для C15H21N2O5PCl2Na, 433,0463; найдено 433,0471.
5,7-Диметоксибензофуран-2-ил)метил-N,N’-бис(2-бромэтил)фосфордиамидат (9) SU046-04
Соединение 9 (SU046-04) синтезируют, используя подобную методику, как описано выше. Получают соединение с выходом 25% (10 мг). m/z = 500,96 (М+Н), 1000,93 (2М+Н).
1H ЯМР (500 МГц, CDCl3): δ 6,76 (1H, с, ArH-3), 6,61 (1H, д, J4,6=2,2 Гц, ArH-4), 6,48 (1H, д, J4,6=2,2 Гц, ArH-6), 5,13 (2H, д, J=9,4 Гц, 2H), 3,99 (3H, с, OMe), 3,86 (3H, с, OMe), 3,49-3,45 (4H, м, 2 × CH2), 3,40-3,33 (4H, м, 2 × CH2), 3,23 (2H, ушир.с, NH).
13С-ЯМР (500 МГц, CDCl3): δ: 156,98, 152,91, 145,58, 139,97, 129,06, 128,24, 107,45, 97,70, 94,56, 59,73, 56,00, 42,90, 34,76, 30,98.
HRMS: рассчитано для C15H21N2O5PBr2Na, 520,9453; найдено 520,9454.
2. Модельные, связанные через образование простого эфира и тиоэфира пролекарства
Синтез связанных через образование простого эфира и тиоэфира пролекарств
7-(Бензофуран-2-илметокси)-4-метил-2Н-хромен-2-он (10) TLE-M2-SU010A
2-(Бромметил)бензофуран (10)
Бензофуран-2-илметанол (1,0 г, 6,7 ммоль) растворяют в толуоле (50 мл) и добавляют пиридин (653 мкл, 8,1 ммоль). Раствор охлаждают до температуры 0°С. По каплям добавляют PBr3 (760 мкл, 8,1 ммоль) в течение 15 минут. Реакционную смесь затем доводят до комнатной температуры и перемешивают в течение 1 часа. Смесь промывают раствором К2СО3 и экстрагируют EtOAc (3×30 мл). Этилацетатный слой промывают насыщенным солевым раствором и сушат (MgSO4). Растворитель выпаривают в вакууме и продукт очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (4:1), получая соединение 10 (780 мг, выход 55%) в виде масла.
1H ЯМР (500 МГц, CDCl3): δ: 7,57 (1H, д, J=7,75 Гц, H-4), 7,52 (1H, д, J=8,40 Гц, H-7), 7,35 (1H, т, J=8,90 Гц, H-4), 7,27 (1H, т, J=8,9 Гц, H-5), 6,79 (1H, с, H-3), 4,64 (2H, с, 2-CH2).
13С-ЯМР CDEPT 135 (500 МГц, CDCl3): δ: 155,34 (С-2), 152,65 (С-7а), 129,08 (С-3а), 125,20 (С-6), 123,16 (С-5), 121,34 (С-С-4), 111,46 (С-7), 106,30 (С-3), 23,61 (СН2-Br).
7-(Бензофуран-2-илметокси)-4-метил-2Н-хромен-2-он (11) TLE-M2-SU010A
Этоксид натрия (77 мг, 1,13 ммоль) добавляют к ДМФА (10 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (200 мг, 1,13 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси порциями добавляют соединение 10 (200 мг, 0,94 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 2 часов. ДМФА выпаривают в вакууме и остаток обрабатывают EtOAc и промывают насыщенным солевым раствором, водой и 1 М раствором NaOH (2×30 мл). Органический слой сушат (MgSO4) и продукт очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (2:1), получая соединение 11 (35 мг, выход 12%) в виде твердого вещества белого цвета. m/z = 307 (М+Н).
1H ЯМР (500 МГц, ДМСО-d6): δ=7,75-7,60 (2H, м, ArH), 7,40-7,25 (2H, м, ArH), 7,23 (1H, с, ArH), 7,12 (2H, д, CH), 6,25 (1H, с, CH), 5,41 (2H, с, CH2), 2,38 (3H, с, CH3).
7-((5-Фторбензофуран-2-ил)метокси)-4-метил-2Н-хромен-2-он (16) VG015-05
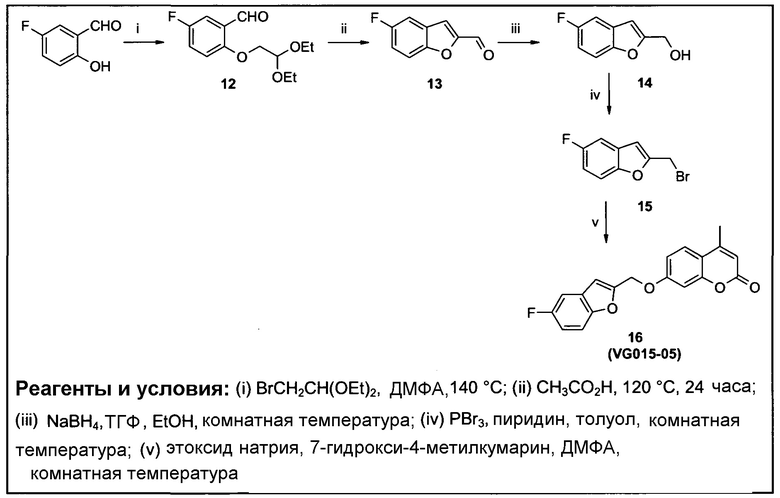
2-(2,2-Диэтоксиэтокси)-5-фторбензальдегид (12)
К перемешиваемой суспензии, содержащей 2-гидрокси-5-фторбензальдегид (500 мг, 3,57 ммоль) и К2СО3 (524 мг, 13,79 ммоль), в ДМФА (10 мл) по каплям добавляют бромацетальдегид-диэтилацеталь (0,6 мл, 3,93 ммоль). Смесь кипятят с обратным холодильником в течение 4 часов. После охлаждения осадок отфильтровывают и растворитель выпаривают в вакууме. Сырой остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (4:1), получая соединение 12 (300 мг, выход 33%) в виде масла.
1H ЯМР (500 МГц, CDCl3): δ: 10,30 (1H, с, CHO), 7,31 (1H, д, J=7,85 Гц), 7,10 (1H, т, J=7,80 Гц), 6,87 (1H, д, J=8,86 Гц), 4,75 (1H, с, CH), 3,97 (2H, д, J=2,35 Гц, CH2), 3,67-3,64 (2H, м, CH2CH3), 3,53-3,50 (2H, м, CH2CH3), 1,11 (6H, т, J=6,00 Гц, 2 × CH3).
5-Фторбензофуран-2-карбальдегид (13)
Перемешиваемый раствор соединения 12 (300 мг, 4,0 ммоль) в уксусной кислоте (10 мл) кипятят с обратным холодильником в течение 24 часов. После охлаждения раствор выпаривают досуха. Сырой продукт адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (4:1), получая соединение 13 (180 мг, выход 94%) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, CDCl3): δ: 9,89 (1H, с, CHO), 7,57 (2H, м, ArH), 7,41 (1H, д, J=7,20 Гц), 7,26 (1H, д, J=6,94 Гц, H-4).
13С-ЯМР CDEPT 135 (500 МГц, CDCl3): δ: 179,74 (СНО), 117,73 (С-3), 117,25 (С-7), 113,81 (С-6), 108,68 (С-4).
(5-Фторбензофуран-2-ил)метанол (14)
Соединение 13 (180 мг, 1,10 ммоль) растворяют в EtOH (12 мл). Порциями добавляют NaBH4 (45 мг, 1,21 ммоль) при температуре 0°С при энергичном перемешивании. Суспензию перемешивают при температуре 0°С в течение 15 минут и затем при комнатной температуре в течение 1,5 часов. Растворители выпаривают в вакууме. Сырой остаток обрабатывают EtOAc и промывают водой, насыщенным солевым раствором и сушат (MgSO4). Растворитель выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (3:1), получая соединение 14 (150 мг, выход 91%) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, CDCl3): δ: 7,36 (1H, д, J=8,35 Гц, H-7), 7,19 (1H, д, J=7,80 Гц, H-4), 7,00 (1H, т, J=8,75 Гц, H-6), 6,60 (1H, с, H-3), 4,75 (2H, с, CH2).
2-(Бромметил)-5-фторбензофуран (15)
Соединение 14 (150 мг, 0,90 ммоль) растворяют в толуоле (10 мл) и раствор охлаждают до температуры 0°С. По каплям добавляют PBr3 (102 мкл, 1,08 ммоль) в течение 15 минут. Реакционную смесь затем доводят до комнатной температуры и перемешивают в течение 1 часа. Растворитель выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (4:1), получая соединение 15 (150 мг, выход 72%) в виде масла.
1H ЯМР (500 МГц, CDCl3): δ: 7,43 (1H, д, J=7,60 Гц, H-7), 7,21 (1H, т, J=8,10 Гц, H-4), 7,06 (1H, т, J=8,90 Гц, H-4), 6,75 (1H, с, H-3), 4,60 (2H, с, 2-CH2).
13С-ЯМР CDEPT 135 (500 МГц, CDCl3): δ: 113,08 (С-7), 112,08 (С-6), 106,87 (С-4), 106,34 (С-3), 60,42 (СН2-Br).
7-((5-Фторбензофуран-2-ил)метокси)-4-метил-2Н-хромен-2-он (16) VG015-05
Этоксид натрия (8,9 мг, 0,13 ммоль) добавляют к ДМФА (3 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (25,4 мг, 0,14 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси порциями добавляют соединение 15 (30 мг, 0,13 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 2 часов. ДМФА выпаривают в вакууме и остаток очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (3:1), получая соединение 16 (9,0 мг, выход 21%) в виде твердого вещества белого цвета. m/z = 325,20 (М+Н).
7-((5,7-Дифторбензофуран-2-ил)метокси)-4-метил-2Н-хромен-2-он (19) VG016-05
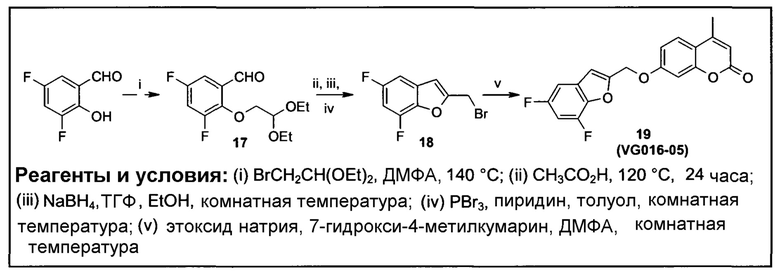
2-(2,2-Диэтоксиэтокси)-3,5-дифторбензальдегид (17)
К перемешиваемой суспензии, содержащей 2-гидрокси-3,5-фторбензальдегид (1,0 г, 6,32 ммоль) и К2СО3 (960 мг, 6,95 ммоль) в ДМФА (10 мл), по каплям добавляют бромацетальдегиддиэтилацеталь (1,07 мл, 6,95 ммоль). Смесь кипятят с обратным холодильником в течение 4 часов. После охлаждения осадок отфильтровывают и растворитель выпаривают в вакууме. Сырой остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (4:1), получая соединение 17 (380 мг, выход 22%) в виде масла.
1H ЯМР (500 МГц, CDCl3): δ: 10,41 (1H, с, CHO), 7,29 (1H, д, J=6,80 Гц), 7,10 (1H, т, J=8,30 Гц), 4,80 (1H, с, CH), 4,20 (2H, д, J=3,25 Гц, CH2), 3,71 (2H, т, J=7,10 Гц, CH2CH3), 3,57 (2H, т, J=7,45 Гц, CH2CH3), 1,19 (6H, т, J=6,25 Гц, 2 × CH3).
2-(Бромметил)-5,7-дифторбензофуран (18)
Перемешиваемый раствор соединения 17 (380 мг, 1,39 ммоль) в уксусной кислоте (10 мл) кипятят с обратным холодильником в течение 24 часов. После охлаждения, раствор выпаривают досуха. Сырой продукт (300 мг) растворяют в EtOH (5 мл). Порциями, добавляют NaBH4 (73 мг, 1,98 ммоль), при температуре 0°С, при энергичном перемешивании. Суспензию перемешивают при температуре 0°С в течение 15 минут и затем при комнатной температуре в течение 1,5 часов. Растворители выпаривают в вакууме. Сырой остаток (280 мг) растворяют в толуоле (20 мл) и раствор охлаждают до температуры 0°С. По каплям, добавляют PBr3 (142 мкл, 1,52 ммоль) в течение 15 минут. Реакционную смесь затем доводят до комнатной температуры и перемешивают в течение 1 часа. Растворитель выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (4:1), получая соединение 18 (210 мг, выход 61%).
1H ЯМР (500 МГц, CDCl3): δ: 7,03 (1H, д, J=7,50 Гц, H-4), 6,87 (1H, т, J=9,80 Гц, H-5), 6,79 (1H, с, H-3), 4,59 (2H, с, 2-CH2).
7-((5,7-Дифторбензофуран-2-ил)метокси)-4-метил-2Н-хромен-2-он (19) VG016-05
Этоксид натрия (8,9 мг, 0,13 ммоль) добавляют к ДМФА (3 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (25,4 мг, 0,14 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси порциями добавляют 2-(бромметил)-5-фторбензофуран (30 мг, 0,12 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 2 часов. ДМФА выпаривают в вакууме и остаток очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (3:1), получая соединение 19 (8,8 мг, выход 21%) в виде твердого вещества белого цвета. m/z = 343,12 (М+Н).
7-((5,7-Дифторбензофуран-2-ил)метокси)-4-метил-2Н-хромен-2-он (22) VG017-05
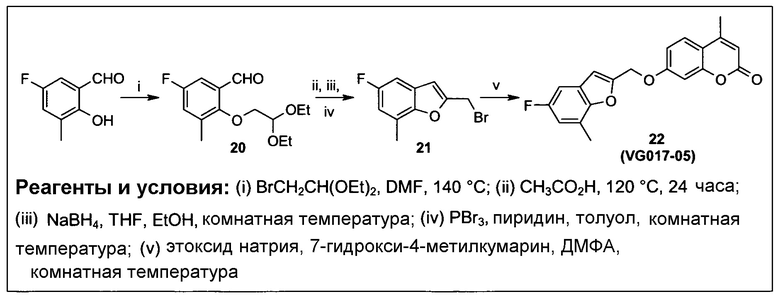
2-(2,2-Диэтоксиэтокси)-5-фтор-3-метилбензальдегид (20)
К перемешиваемой суспензии, содержащей 5-фтор-2-гидрокси-3-метилбензальдегид (1,0 г, 6,49 ммоль) и К2СО3 (980 мг, 7,10 ммоль), в ДМФА (8 мл), по каплям добавляют бромацетальдегид-диэтилацеталь (1,10 мл, 7,15 ммоль). Смесь кипятят с обратным холодильником в течение 4 часов. После охлаждения, осадок отфильтровывают и растворитель выпаривают в вакууме. Сырой остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии. Продукт элюируют смесью гексан/EtOAc (4:1), получая соединение 20 (350 мг, выход 20%) в виде масла.
1H ЯМР (500 МГц, CDCl3): δ: 10,40 (1H, с, CHO), 7,32 (1H, д, J=7,30 Гц, ArH), 7,14 (1H, д, J=7,75 Гц, ArH), 4,85 (1H, с, CH), 3,95 (2H, с, CH2), 3,75 (2H, т, J=7,15 Гц, CH2CH3), 3,61 (2H, т, J=7,20 Гц, CH2CH3), 2,36 (3H, с, CH3), 1,24 (6H, т, J=5,65 Гц, 2 × CH3).
2-(Бромметил)-5-фтор-7-метилбензофуран (21)
Перемешиваемый раствор соединения 20 (350 мг, 1,30 ммоль) в уксусной кислоте (10 мл) кипятят с обратным холодильником в течение 24 часов. После охлаждения, раствор выпаривают досуха. Сырой продукт (300 мг) растворяют в ТГФ (5 мл). Порциями добавляют NaBH4 (78 мг, 2,02 ммоль) при температуре 0°С при энергичном перемешивании. Суспензию перемешивают при температуре 0°С в течение 15 минут и затем при комнатной температуре в течение 1,5 часов. Растворители выпаривают в вакууме. Сырой остаток (260 мг) растворяют в толуоле (20 мл) и раствор охлаждают до температуры 0°С. По каплям, добавляют PBr3 (135 мкл, 1,44 ммоль) в течение 15 минут. Реакционную смесь затем доводят до комнатной температуры и перемешивают в течение 1 часа. Растворитель выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (4:1), получая соединение 21 (200 мг, выход 36%).
1H ЯМР (500 МГц, CDCl3): δ: 7,03 (1H, д, J=7,50 Гц, H-4), 6,88 (1H, т, J=9,80 Гц, H-5), 6,75 (1H, с, H-3), 4,69 (2H, с, 2-CH2), 2,54 (3H, с, CH3).
7-((5,7-Дифторбензофуран-2-ил)метокси)-4-метил-2Н-хромен-2-он (22) VG017-05
Этоксид натрия (8,9 мг, 0,13 ммоль) добавляют к ДМФА (3 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (25,4 мг, 0,14 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси порциями добавляют соединение 21 (30 мг, 0,12 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 2 часов. ДМФА выпаривают в вакууме и остаток очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (3:1), получая соединение 22 в виде твердого вещества белого цвета (11 мг, выход 26%). m/z = 33,20 (М+Н).
7-((5-Метоксибензофуран-2-ил)метокси)-4-метил-2Н-хромен-2-он (27) VG027-05
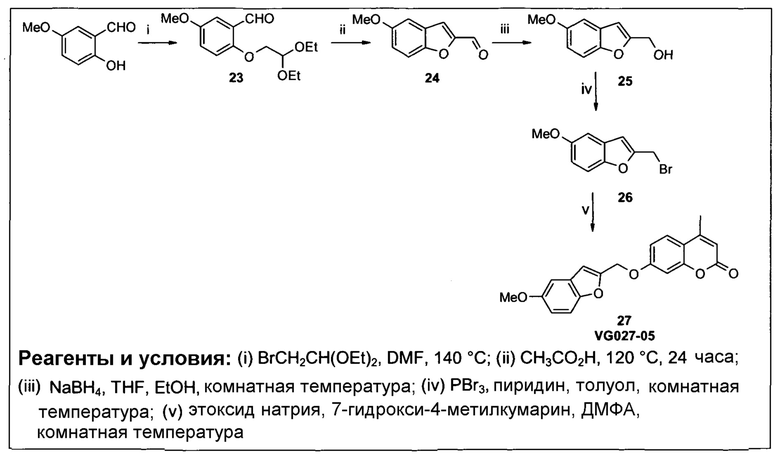
2-(2,2-Диэтоксиэтокси)-5-метоксибензальдегид (23)
К перемешиваемой суспензии, содержащей 2-гидрокси-5-метоксибензальдегид (2,0 г, 13,16 ммоль) и К2СО3 (2,18 г, 15,79 ммоль), в ДМФА (20 мл), по каплям, добавляют бромацетальдегид-диэтилацеталь (2,43 мл, 15,79 ммоль). Смесь кипятят с обратным холодильником в течение 4 часов. После охлаждения осадок отфильтровывают и растворитель выпаривают в вакууме. Сырой остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии. Продукт элюируют смесью гексан/EtOAc (4:1), получая целевое соединение 23 (1,10 г, выход 31%) в виде масла.
1H ЯМР (500 МГц, CDCl3): δ: 10,49 (1H, с, CHO), 7,33 (1H, д, J=3,30 Гц, ArH), 7,12 (1H, дд, J=5,75 и 3,30 Гц, ArH), 6,97 (1H, д, J=9,05 Гц), 4,87 (1H, т, J=5,25 Гц, CH), 4,09 (2H, д, J=5,25 Гц, CH2), 3,81-3,78 (2H, м, CH2CH3), 3,67-3,64 (2H, м, CH2CH3), 1,26 (6H, т, J=7,05 Гц, 2 × CH3).
5-Метоксибензофуран-2-карбальдегид (24)
Перемешиваемый раствор соединения 23 (1,0 г, 3,74 ммоль) в уксусной кислоте (10 мл) кипятят с обратным холодильником в течение 16 часов. После охлаждения раствор выпаривают досуха. Сырой продукт адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (4:1), получая соединение 24 (160 мг, выход 24%) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, CDCl3): δ: 9,80 (1H, с, CHO), 7,49-7,45 (2H, м, ArH), 7,12-7,09 (2H, м, ArH), 3,85 (3H, с, OCH3).
(5-Метоксибензофуран-2-ил)метанол (25)
Соединение 24 (3,5 г, 19,9 ммоль) растворяют в EtOH (20 мл). Порциями добавляют NaBH4 (957 мг, 25,87 ммоль) при температуре 0°С при энергичном перемешивании. Суспензию перемешивают при температуре 0°С в течение 15 минут и затем при комнатной температуре в течение 1,5 часов. Растворитель выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (2:1), получая соединение 25 (3,0 г, выход 85%), в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, CDCl3): δ: 7,36 (1H, д, J=8,90 Гц, H-7), 7,02 (1H, д, J=2,6 Гц, H-4), 6,90 (1H, дд, J=6,30 и 2,60 Гц, H-6), 6,61 (1H, с, H-3), 4,76 (2H, с, 2-CH2), 3,86 (3H, с, OCH3), 2,16 (1H, ушир.с, OH).
13С-ЯМР CDEPT 135 (500 МГц, CDCl3): δ: 113,07 (С-7), 111,69 (С-6), 104,34 (С-4), 103,60 (С-3), 58,24 (СН2), 55,92 (ОСН3).
2-(Бромметил)-5-метоксибензофуран (26)
Соединение 25 (40 мг, 0,22 ммоль) растворяют в толуоле (5 мл) и раствор охлаждают до температуры 0°С. По каплям, добавляют PBr3 (21 мкл, 0,22 ммоль) в течение 10 минут. Реакционную смесь затем доводят до комнатной температуры и перемешивают в течение 1 часа. Растворитель выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (4:1), получая соединение 26 (40 мг, выход 74%) в виде масла.
1H ЯМР (500 МГц, CDCl3): δ: 7,39 (1H, д, J=8,90 Гц, H-7), 7,01 (1H, д, J=2,55 Гц, H-4), 6,94 (1H, дд, J=6,35 и 2,60 Гц, H-6), 6,72 (1H, с, H-3), 4,61 (2H, с, 2-CH2), 3,86 (3H, с, OCH3).
7-((5-Метоксибензофуран-2-ил)метокси)-4-метил-2Н-хромен-2-он (27) VG027-05
Этоксид натрия (12 мг, 0,18 ммоль) добавляют к ДМФА (5 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (32 мг, 0,17 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси порциями добавляют соединение 26 (40 мг, 0,17 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре, в течение 2 часов. ДМФА выпаривают в вакууме и остаток очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (3:1), получая соединение 27 (17 мг, выход 30%) в виде твердого вещества белого цвета. m/z = 337,04 (М+Н), 673,13 (2М+Н).
7-((7-Метоксибензофуран-2-ил)метокси)-4-метил-2Н-хромен-2-он (31) VG029-05
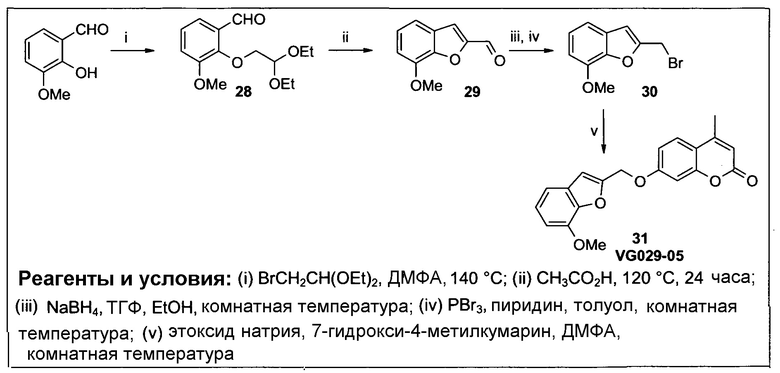
2-(2,2-Диэтоксиэтокси)-3-метоксибензальдегид (28)
К перемешиваемой суспензии, содержащей 2-гидрокси-3-метоксибензальдегид (4,0 г, 26,3 ммоль) и К2СО3 (4,36 г, 31,60 ммоль) в ДМФА (15 мл), по каплям, добавляют бромацетальдегиддиэтилацеталь (4,86 мл, 31,60 ммоль). Смесь кипятят с обратным холодильником в течение 4 часов. После охлаждения осадок отфильтровывают и растворитель выпаривают в вакууме. Сырой остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (4:1), получая соединение 28 (2,60 г, выход 36%).
1H ЯМР (500 МГц, CDCl3): δ: 10,53 (1H, с, CHO), 7,42 (1H, м, ArH), 7,14-7,12 (2H, м, ArH), 4,83 (1H, т, J=5,30 Гц, CH), 4,21 (2H, д, J=5,35 Гц, CH2), 3,90 (3H, с, OCH3), 3,74-3,71 (2H, м, CH2CH3), 3,60-3,57 (2H, м, CH2CH3), 1,22 (6H, т, J=7,05 Гц, 2 × CH3).
7-Метоксибензофуран-2-карбальдегид (29)
Перемешиваемый раствор соединения 28 (2,0 г, 7,46 ммоль) в уксусной кислоте (10 мл) кипятят с обратным холодильником в течение 24 часов. После охлаждения раствор выпаривают досуха. Сырой продукт адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (4:1), получая соединение 29 (450 мг, выход 34%) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, CDCl3): δ: 9,89 (1H, с, CHO), 7,55 (1H, с, ArH), 7,30 (1H, д, J=6,95 Гц, ArH), 7,24 (1H, т, J=7,85 Гц, ArH), 6,97 (1H, д, J=6,90 Гц), 4,02 (3H, с, OCH3).
2-(Бромметил)-7-метоксибензофуран (30)
Соединение 29 (450 мг, 2,56 ммоль) растворяют в EtOH (10 мл). Порциями добавляют NaBH4 (104 мг, 2,81 ммоль) при температуре 0°С при энергичном перемешивании. Суспензию перемешивают при температуре 0°С в течение 15 минут и затем при комнатной температуре в течение 1,5 часов. Растворитель выпаривают в вакууме. Полученный сырой спиртовой остаток растворяют в толуоле (5 мл) и раствор охлаждают до температуры 0°С. По каплям, добавляют PBr3 (240 мкл, 2,56 ммоль) в течение 10 минут. Реакционную смесь затем доводят до комнатной температуры и перемешивают в течение 1 часа. Растворитель выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (4:1), получая соединение 30 (150 мг, выход 24%) в виде масла.
1H ЯМР (500 МГц, CDCl3): δ: 7,19-7,17 (2H, м, ArH), 6,85 (1H, д, J=5,60 Гц, ArH), 6,78 (1H, с, ArH), 4,62 (2H, с, 2-CH2), 4,04 (3H, с, OCH3).
7-((7-Метоксибензофуран-2-ил)метокси)-4-метил-2Н-хромен-2-он (31) VG029-05
Этоксид натрия (12 мг, 0,18 ммоль) добавляют к ДМФА (5 мл), при температуре 0°С, и суспензию перемешивают в течение 10 мин. Медленно добавляют 7-гидрокси-4-метилкумарин (32 мг, 0,17 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси порциями добавляют соединение 30 (40 мг, 0,17 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 2 часов. ДМФА выпаривают в вакууме и остаток очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (3:1), получая соединение 31 (14 мг, выход 25%) в виде твердого вещества белого цвета. m/z = 337,04 (М+Н), 673,13 (2М+Н).
7-((5-Бромбензофуран-2-ил)метокси)-4-метил-2Н-хромен-2-он (32) VG035-04
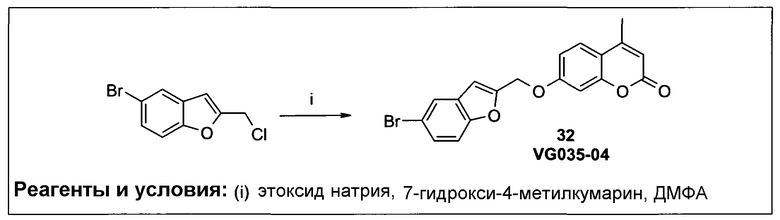
7-((5-Бромбензофуран-2-ил)метокси)-4-метил-2Н-хромен-2-он (VG035-04)
Этоксид натрия (76 мг, 1,10 ммоль) добавляют к ДМФА (10 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (215 мг, 1,22 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси порциями добавляют 5-бром-2-(хлорметил)бензофуран (250 мг, 1,02 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 2 часов. ДМФА выпаривают в вакууме и остаток очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (3:1), получая соединение 32 (120 мг, выход 31%) в виде твердого вещества белого цвета. m/z = 386 (М+Н).
1H ЯМР (500 МГц, ДМСО-d6): δ=7,91 (1H, д, J=2,0 Гц, ArH), 7,71 (1H, д, J=8,80 Гц, ArH), 7,61 (1H, д, J=8,75 Гц, ArH), 7,49 (1H, дд, J=6,70 и 2,05 Гц, ArH), 7,20 (1H, д, J=2,45 Гц, ArH), 7,13 (1H, с, ArH), 7,09 (1H, дд, J=6,30 и 2,50 Гц, ArH), 6,25 (1H, с, CH), 5,43 (2H, с, CH2), 2,41 (3H, с, CH3).
13С-ЯМР CDEPT 135 (500 МГц, CDCl3): δ: 127,55 (ArCH), 126,59 (ArCH), 124,04 (ArCH), 113,32 (ArCH), 112,56 (ArCH), 111,44 (ArCH), 106,87 (ArCH), 101,66 (ArCH), 62,40 (СН2), 16,11 (CH3).
7-((5-Хлорбензофуран-2-ил)метокси)-4-метил-2Н-хромен-2-он (36) VG028-05
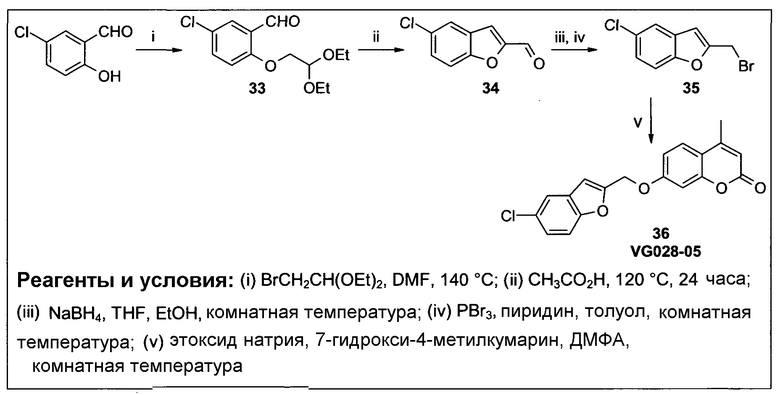
5-Хлор-2-(2,2-диэтоксиэтокси)бензальдегид (33)
К перемешиваемой суспензии 5-хлор-2-гидроксибензальдегида (5,0 г, 32,1 ммоль) и К2СО3 (4,87 г, 35,3 ммоль) в ДМФА (20 мл), по каплям, добавляют бромацетальдегиддиэтилацеталь (5,43 мл, 35,3 ммоль). Смесь кипятят с обратным холодильником в течение 4 часов. После охлаждения осадок отфильтровывают и растворитель выпаривают в вакууме. Сырой остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии. Продукт элюируют смесью гексан/EtOAc (4:1), получая соединение 33 (4,10 г, выход 38%) в виде масла.
1H ЯМР (500 МГц, CDCl3): δ: 10,42 (1H, с, CHO), 7,76 (1H, д, J=2,8 Гц, ArH), 7,46 (1H, дд, J=6,15 и 2,75 Гц, ArH), 6,96 (1H, д, J=8,90 Гц, ArH), 4,87 (1H, т, J=5,25 Гц, CH), 4,10 (2H, д, J=5,25 Гц, CH2), 3,80-3,77 (2H, м, CH2CH3), 3,67-3,62 (2H, м, CH2CH3), 1,24 (6H, т, J=7,05 Гц, 2 × CH3).
5-Хлорбензофуран-2-карбальдегид (34)
Перемешиваемый раствор соединения 33 (4,10 г, 15,07 ммоль) в уксусной кислоте (20 мл) кипятят с обратным холодильником в течение 24 часов. После охлаждения раствор выпаривают досуха. Сырой продукт адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (4:1), получая соединение 34 (550 мг, выход 20%) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, CDCl3): δ: 9,91 (1H, с, CHO), 7,76 (1H, д, J=1,85 Гц, ArH), 7,57 (1H, д, J=8,90 Гц, ArH), 7,53 (1H, с, ArH), 7,50 (1H, дд, J=8,90 и 2,10 Гц, ArH).
2-(Бромметил)-5-хлорбензофуран (35)
Соединение 34 (160 мг, 0,89 ммоль) растворяют в EtOH (5 мл). Порциями добавляют NaBH4 (36 мг, 0,98 ммоль) при температуре 0°С при энергичном перемешивании. Суспензию перемешивают при температуре 0°С в течение 15 минут и затем при комнатной температуре в течение 1,5 часов. Растворитель выпаривают в вакууме. Полученный сырой спиртовой остаток растворяют в толуоле (5 мл) и раствор охлаждают до температуры 0°С. По каплям, добавляют PBr3 (92 мкл, 0,98 ммоль) в течение 10 минут. Смесь затем доводят до комнатной температуры и перемешивают в течение 1 часа. Растворитель выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (4:1), получая соединение 35 (128 мг, выход 57%) в виде масла.
1H ЯМР (500 МГц, CDCl3): δ: 7,48 (1H, д, J=2,05 Гц, ArH), 7,37 (1H, д, J=8,70 Гц, ArH), 7,25 (1H, дд, J=8,80 и 2,05 Гц, ArH), 6,68 (1H, с, ArH), 4,55 (2H, с, 2-CH2).
7-((5-Хлорбензофуран-2-ил)метокси)-4-метил-2Н-хромен-2-он (36) VG028-05
Этоксид натрия (60 мг, 0,24 ммоль) добавляют к ДМФА (5 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (47 мг, 0,27 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси порциями добавляют 2-(бромметил)-5-метоксибензофуран (40 мг, 0,17 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 2 часов. ДМФА выпаривают в вакууме и остаток очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (3:1), получая целевое соединение в виде твердого вещества белого цвета (2,7 мг, выход 3%). m/z = 341,10 (М+Н).
7-((5,7-Диметоксибензофуран-2-ил)метокси)-4-метил-2Н-хромен-2-он (42) VG035-05
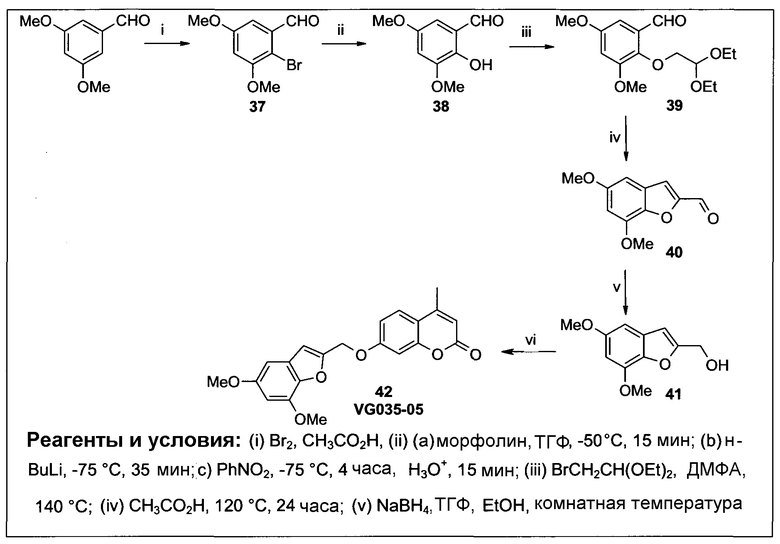
2-Бром-3,5-диметоксибензальдегид (37)
3,5-Диметоксибензальдегид (12,6 г, 76 ммоль) растворяют в уксусной кислоте (350 мл). Полученный бесцветный раствор охлаждают до температуры 0°С. По каплям, добавляют раствор брома (3,9 мл) в уксусной кислоте (50 мл) в течение 1 часа. После завершения добавления ледяную баню убирают и полученный раствор бледно-зеленого цвета перемешивают в течение ночи при комнатной температуре. К раствору добавляют холодную воду. Полученное твердое вещество белого цвета собирают путем вакуумной фильтрации и промывают водой. Твердое вещество затем повторно растворяют в EtOAc и адсорбируют на силикагеле. Продукт очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (4:1), получая соединение 37 (12,5 г, выход 66%) в виде твердого вещества белого цвета. m/z = 344,98 (М+Н).
1H ЯМР (500 МГц, CDCl3): δ: 10,43 (1H, с, CHO), 7,06 (1H, с, ArH), 6,73 (1H, с, ArH), 3,93 (3H, с, CH3O), 3,86 (3H, с, CH3O).
13С-ЯМР (500 МГц, CDCl3): δ: 192,09 (СНО), 159,92 (С-5), 157,02 (С-3), 134,67 (С-1), 109,12 (С-2), 105,83, 103,37 (С-4 и С-6), 56,60 (ОМе), 55,82 (ОМе).
2-Гидрокси-3,5-диметоксибензальдегид (38)
Морфолин (2,05 г, 24 ммоль) и ТГФ (40 мл) вводят в сухую, трехгорлую, круглодонную колбу, оснащенную мешалкой, мембранным колпачком, капельной воронкой, термометром и вводом для аргона. Колбу охлаждают на бане со смесью сухой лед-ацетон до температуры -50°С и добавляют весь раствор н-BuLi в гексане (1,6 М раствор, 15 мл, 24 ммоль), за один раз. Спустя 10 минут, по каплям, добавляют раствор 2-бром-3,5-диметоксибензальдегида, соединения 37 (4,9 г, 20 ммоль) в ТГФ (30 мл) посредством шприца в течение периода времени 4 минуты и смесь охлаждают при температуре ~ -75°С в течение 20 минут. Затем по каплям добавляют н-BuLi в гексане (1,6 М раствор, 20 мл, 32 ммоль) в течение 45 минут, поддерживая температуру равной -75°С. После завершения добавления н-BuLi, раствор перемешивают в течение 35 минут. Из капельной воронки добавляют раствор нитробензола (6,90 г, 46 ммоль) в 10 мл ТГФ, поддерживая температуру равной -75°С. Полученную смесь темного цвета перемешивают при температуре -75°С в течение 4 часов и затем оставляют нагреваться до комнатной температуры. Смесь подкисляют до рН=1 с помощью 6 н HCl и перемешивают в течение 15 минут. После разбавления насыщенным солевым раствором (100 мл), ТГФ удаляют в вакууме. Водный раствор экстрагируют диэтиловым эфиром (4×40 мл). Объединенные органические слои экстрагируют 2 н раствором NaOH (3×40 мл). Объединенные NaOH-экстракты промывают диэтиловым эфиром (3×20 мл) и затем подкисляют до рН=1 с помощью концентрированной HCl. Полученную смесь экстрагируют CH2Cl2 (3×20 мл) и объединенные органические экстракты промывают насыщенным солевым раствором, сушат (MgSO4) и адсорбируют на силикагеле. Продукт очищают с помощью флэш-хроматографии, элюируя смесью EtOAc/гексан (1:2), получая соединение 38 (2,0 г, выход 55%) в виде твердого вещества желтого цвета. m/z = 183,06 (М+Н).
1H ЯМР (500 МГц, CDCl3): δ: 10,71 (1H, с, OH), 9,91 (1H, с, CHO), 6,77 (1H, д, J4,6=2,8 Гц, H-6), 6,61 (1H, д, J4,6=2,8 Гц, H-4), 3,92 (3H, с, OMe), 3,84 (3H, с, OMe).
13С-ЯМР CDEPT 135 (500 МГц, CDCl3): δ: 196,11 (СНО), 107,93 (С-6), 103,90 (С-4), 56,29 (ОМе), 55,83 (ОМе).
2-(2,2-Диэтоксиэтокси)-3,5-диметоксибензальдегид (39)
К перемешиваемой суспензии, содержащей соединение 38 (1,1 г, 6,0 ммоль) и К2СО3 (1,0 г, 7,2 ммоль) в ДМФА (100 мл), по каплям, добавляют бромацетальдегиддиэтилацеталь (0,93 мл, 6,0 ммоль). Смесь кипятят с обратным холодильником в течение 4 часов. После охлаждения осадок отфильтровывают и растворитель выпаривают в вакууме. Сырой остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии. Продукт элюируют смесью гексан/EtOAc (4:1), получая соединение 39 (1,2 г, выход 67%) в виде масла.
1H ЯМР (500 МГц, CDCl3): δ: 10,50 (1H, с, CHO), 6,88 (1H, д, J4,6=2,9 Гц, H-6), 6,74 (1H, д, J4,6=2,9 Гц, H-4), 4,83 (1H, т, J4,6=5,3 Гц, CH), 4,14 (2H, д, J4,6=5,3 Гц, CH2), 3,88 (3H, с, OMe), 3,83 (3H, с, OMe), 3,77-3,71 (2H, м, CH2CH3), 3,63-3,58 (2H, м, CH2CH3), 1,24 (6H, т, J=7,1 Гц, 2 × CH3).
5,7-Диметоксибензофуран-2-карбальдегид (40)
Перемешиваемый раствор соединения 39 (1,2 г, 4,0 ммоль) в уксусной кислоте (35 мл) кипятят с обратным холодильником в течение 16 часов. После охлаждения раствор выпаривают досуха. Сырой продукт адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (2:1), получая соединение 40 (230 мг, выход 28%) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, CDCl3): δ: 9,89 (1H, с, CHO), 7,50 (1H, с, H-3), 6,69 (1H, д, J4,6=2,2 Гц, H-6), 6,64 (1H, д, J4,6=2,2 Гц, H-4), 4,01 (3H, с, OMe), 3,87 (3H, с, OMe).
13С-ЯМР CDEPT 135 (500 МГц, CDCl3): δ: 179,91 (СНО), 153,37 (С-2), 116,10 (С-3), 101,80 (С-6), 94,90 (С-4), 56,20 (ОМе), 55,88 (ОМе).
(5,7-Диметоксибензофуран-2-ил)метанол (41)
Соединение 39 (460 мг, 2,23 ммоль) растворяют в ТГФ (5 мл) и EtOH (1 мл). Порциями добавляют NaBH4 (102 мг, 2,68 ммоль), при температуре 0°С, при энергичном перемешивании. Суспензию перемешивают при температуре 0оС в течение 15 минут и затем при комнатной температуре в течение 1 часа. Растворители выпаривают в вакууме. Сырой остаток обрабатывают EtOAc и промывают водой, насыщенным солевым раствором, и сушат (MgSO4). Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (1:1), получая соединение 41 (388 мг, выход 82%) в виде твердого вещества белого цвета. m/z = 209,08 (М+Н).
1H ЯМР (500 МГц, CDCl3): δ: 6,62 (1H, с, H-3), 6,60 (1H, с, H-6), 6,46 (1H, с, H-4), 4,76 (2H, с, 2-CH2), 3,99 (3H, с, OMe), 3,85 (3H, с, OMe).
13С-ЯМР (500 МГц, CDCl3): δ: 157,23 (С-5), 156,72 (С-1), 145,36 (С-7), 139,50 (С-1а), 129,38 (С-4а), 104,62 (С-3), 96,96 (С-6), 94,58 (С-4), 57,98 (2-СН2), 55,95 (ОМе), 55,83 (ОМе).
7-((5,7-Диметоксибензофуран-2-ил)метокси)-4-метил-2Н-хромен-2-он (42) VG035-05
Соединение 41 (130 мг, 0,63 ммоль) растворяют в толуоле (5 мл) и раствор охлаждают до температуры 0°С. По каплям, добавляют PBr3 (64 мкл, 0,69 ммоль) в течение 10 минут. Реакционную смесь затем доводят до комнатной температуры и перемешивают в течение 1 часа. Растворитель выпаривают в вакууме. Сырой остаток используют на следующей стадии.
Этоксид натрия (80 мг, 0,24 ммоль) добавляют к ДМФА (5 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (47 мг, 0,27 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси порциями добавляют 2-(бромметил)-5-метоксибензофуран (40 мг, 0,17 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 2 часов. ДМФА выпаривают в вакууме и остаток очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (3:1), получая соединение 42 (2,7 мг, выход 3%) в виде твердого вещества белого цвета. m/z = 367,05 (М+Н), 733,15 (2М+Н).
4-Метил-7-(нафталин-1-илметокси)-2Н-хромен-2-он (43)
TLE-M1-SU001A
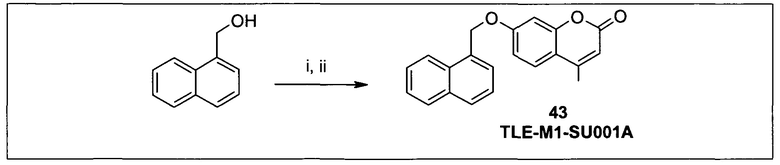

1-Нафталинметанол (2,0 г, 12,7 ммоль) растворяют в толуоле (30 мл) и добавляют пиридин (1,02 мл, 12,7 ммоль). Раствор охлаждают до температуры 0°С. По каплям, добавляют PBr3 (1,19 мл, 12,7 ммоль) в течение 15 минут. Реакционную смесь затем доводят до комнатной температуры и перемешивают в течение 1 часа. Смесь промывают раствором К2СО3 и экстрагируют EtOAc (3×30 мл). Слой EtOAc промывают насыщенным солевым раствором и сушат (MgSO4). Растворитель выпаривают в вакууме, получая 1-(бромметил)нафталин (1,5 г, выход 53%) в виде бесцветного масла. Этот промежуточный продукт используют в следующей реакции.
Этоксид натрия (169 мг, 2,49 ммоль) добавляют к ДМФА (5 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (438 мг, 2,49 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа, затем оставляют достигать комнатной температуры. К этой смеси порциями добавляют 1-(бромметил)нафталин (500 мг, 2,26 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 16 часов. ДМФА выпаривают в вакууме и остаток обрабатывают EtOAc и промывают насыщенным солевым раствором (2×50 мл), водой (2×50 мл) и 1 М раствором NaOH (2×30 мл). Органический слой сушат (MgSO4) и продукт очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (2:1), получая соединение 43 (200 мг, выход 28%) в виде твердого вещества белого цвета. Температура плавления: 181-183°С.
1H ЯМР (500 МГц, ацетон-d6): δ = 8,10 (1H, д, ArH), 8,00-7,99 (2H, м, Ar), 7,97-7,71 (2H, м, ArH), 7,70-7,53 (3H, м, ArH), 7,24 (1H, с, ArH), 7,09 (1H, д, CH), 6,23 (1H, с, CH), 5,68 (2H, с, CH2), 2,40 (3H, с, CH3).
4-Метил-7-(нафталин-2-илметокси)-2Н-хромен-2-он (44)
VG040-03
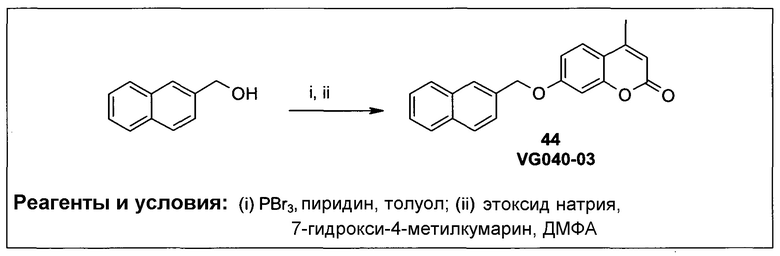
Нафталин-2-илметанол (2,0 г, 12,7 ммоль) растворяют в толуоле (30 мл) и добавляют пиридин (1,02 мл, 12,7 ммоль). Раствор охлаждают до температуры 0°С. По каплям, добавляют PBr3 (1,19 мл, 12,7 ммоль) в течение 15 минут. Смесь затем доводят до комнатной температуры и перемешивают в течение 1 часа. Смесь промывают раствором К2СО3 и экстрагируют EtOAc (3×30 мл). Слой EtOAc промывают насыщенным солевым раствором и сушат (MgSO4). Растворитель выпаривают в вакууме, получая сырой 1-(бромметил)нафталин. Этот промежуточный продукт используют в следующих реакциях.
Этоксид натрия (169 мг, 2,49 ммоль) добавляют к ДМФА (5 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (438 мг, 2,49 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа, затем оставляют достигать комнатной температуры. К этой смеси порциями добавляют 1-(бромметил)нафталин (500 мг, 2,26 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 16 часов. ДМФА выпаривают в вакууме и остаток обрабатывают EtOAc и промывают насыщенным солевым раствором (2×50 мл), водой (2×50 мл) и 1 М раствором NaOH (2×30 мл). Органический слой сушат (MgSO4) и продукт очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (2:1), получая соединение 44 (1,44 г, выход 36%) в виде твердого вещества белого цвета. m/z = 317,12 (М+Н), 633,24 (2М+Н).
1H ЯМР (500 МГц, CDCl3): δ = 7,93-7,87 (4H, м, ArH), 7,58-7,52 (4H, м, Ar), 6,97 (1H, т, J=2,43 Гц, ArH), 6,16 (1H, с, ArH), 5,32 (2H, с, CH2), 2,41 (3H, с, CH3).
7-(Бензгидрилокси)-4-метил-2Н-хромен-2-он (45) TLE-M1-SU004A
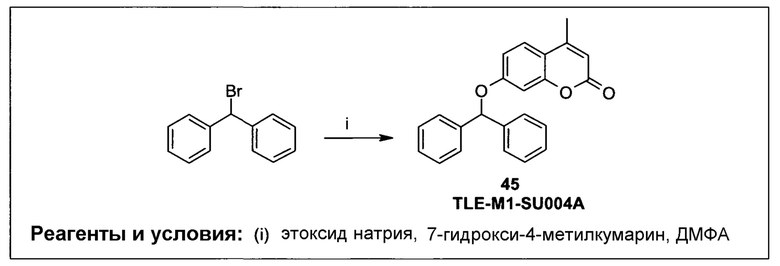
Этоксид натрия (165 мг, 2,43 ммоль) добавляют к ДМФА, при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (428 мг, 2,43 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа, затем оставляют достигать комнатной температуры. К этой смеси порциями добавляют дифенилметилбромид (500 мг, 2,02 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 16 часов. ДМФА выпаривают в вакууме и остаток обрабатывают EtOAc и промывают насыщенным солевым раствором (2×30 мл), водой (2×30 мл) и 1 М раствором NaOH (2×30 мл). Органический слой сушат (MgSO4) и продукт очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (2:1), получая соединение 45 (200 мг, выход 29%) в виде твердого вещества белого цвета. Температура плавления: 146-148ºС.
1H ЯМР (500 МГц, ДМСО-d6): δ = 7,63 (1H, д, ArH), 7,53 (4H, д, ArH), 7,38 (4H, т, ArH), 7,29 (2H, т, ArH), 7,09 (2H, д, ArH), 7,04 (1H, д, ArH), 6,75 (1H, с, ArH), 6,18 (1H, с, CH), 2,37 (3H, с, CH3).
13С-ЯМР (500 МГц, ДМСО-d6, DEPT135): δ = 160,2, 154,4, 153,2, 140,8, 129,90, 128,0, 126,8, 113,7, 111,4, 111,2, 102,9, 80,2, 18,2.
4-Метил-7-(1-(нафталин-2-ил)этокси)-2Н-хромен-2-он (46) VG039-03
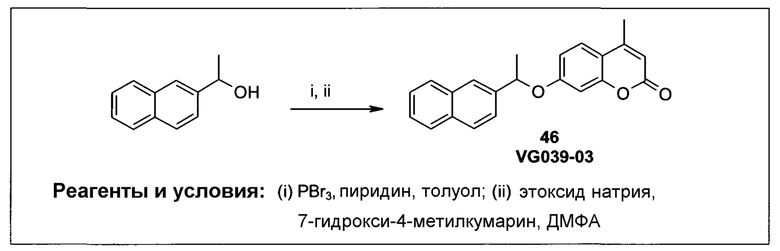
1-(Нафталин-2-ил)этанол (2,0 г, 11,6 ммоль) растворяют в толуоле (30 мл). Раствор охлаждают до температуры 0°С. По каплям, добавляют PBr3 (1,09 мл, 11,6 ммоль) в течение 15 минут. Реакционную смесь затем доводят до комнатной температуры и перемешивают в течение 1 часа. Смесь промывают раствором К2СО3 и экстрагируют EtOAc (3×30 мл). Слой EtOAc промывают насыщенным солевым раствором и сушат (MgSO4). Растворитель выпаривают в вакууме, получая сырой 2-(1-бромэтил)нафталин. Этот промежуточный продукт используют на следующей стадии.
Этоксид натрия (63,4 мг, 0,93 ммоль) добавляют к ДМФА (5 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (147 мг, 0,84 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа, затем оставляют достигать комнатной температуры. К этой смеси порциями добавляют 2-(1-бромэтил)нафталин (200 мг, 0,85 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 16 часов. ДМФА выпаривают в вакууме и остаток обрабатывают EtOAc и промывают насыщенным солевым раствором (2×50 мл), водой (2×50 мл) и 1 М раствором NaOH (2×30 мл). Органический слой сушат (MgSO4) и продукт очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (2:1), получая соединение 46 (60 мг, выход 21%) в виде твердого вещества белого цвета. m/z = 331,15 (М+Н), 661,29 (2М+Н).
1H ЯМР (500 МГц, ДМСО-d6): δ = 7,97 (1H, с, ArH), 7,93-7,86 (3H, м, ArH), 7,60-7,50 (4H, м, Ar), 6,99 (1H, кв., J=6,45 и 2,35 Гц, ArH), 6,96 (1H, д, J=2,40 Гц, ArH), 6,13 (1H, с, ArH), 5,84 (1H, кв., J=6,35 Гц, CH), 2,26 (3H, с, CH3), 1,67 (3H, д, J=6,35 Гц, CH3).
7-(Антрацен-9-илметокси)-4-метил-2Н-хромен-2-он (48) SU06-02
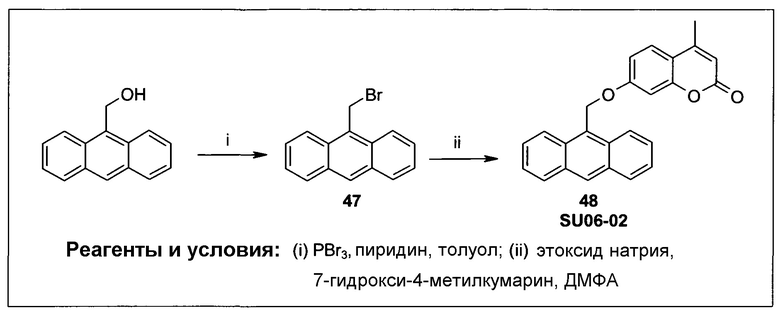
9-(Бромметил)антрацен (47)
К перемешиваемой суспензии 9-антраценметанола (2,0 г, 9,6 ммоль), при температуре 0°С, в толуоле (100 мл), добавляют PBr3 (1,2 мл, 12,51 ммоль) и суспензию перемешивают при температуре 0ºС в течение 1 часа. Реакционную смесь затем доводят до комнатной температуры и оставляют перемешиваться в течение следующего часа. Смесь превращается в раствор желтого цвета. Добавляют К2СО3 (10 мл), для того, чтобы погасить реакцию. Толуол выпаривают в вакууме. Остаток обрабатывают EtOAc и промывают насыщенным водным раствором К2СО3, водой и насыщенным солевым раствором, и сушат (MgSO4). Растворитель выпаривают в вакууме и сырой остаток очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (2:1), получая соединение 47 (1,4 г, выход 54%) в виде твердого вещества желтого цвета.
1H ЯМР (500 МГц, CDCl3): δ = 8,45 (1H, с, Ar-10H), 8,27 (2H, д, Ar-1, 8H), 8,00 (2H, д, Ar-4, 6H), 7,62 (2H, д, Ar-2, 7H), 7,48 (2H, д, Ar-3, H), 5,50 (2H, с, CH2).
7-(Антрацен-9-илметокси)-4-метил-2Н-хромен-2-он (48) SU06-02
Этоксид натрия (151 мг, 2,21 ммоль) добавляют к ДМФА (5 мл), при температуре 0°С, и суспензию перемешивают в течение 10 мин. Медленно добавляют 7-гидрокси-4-метилкумарин (390 мг, 2,21 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа, затем оставляют достигать комнатной температуры. К этой смеси порциями добавляют соединение 47 (500 мг, 1,85 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 16 часов. ДМФА выпаривают в вакууме и остаток обрабатывают EtOAc и промывают насыщенным солевым раствором, водой и 1 М раствором NaOH (2×30 мл). Органический слой сушат (MgSO4) и продукт очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (2:1), получая соединение 48 (200 мг, выход 30%) в виде твердого вещества желтого цвета. Температура плавления: 216-218°С.
1H ЯМР (500 МГц, ДМСО-d6): δ = 8,10 (1H, д, ArH), 8,00-7,99 (2H, м, Ar), 7,97-7,71 (2H, м, ArH), 7,70-7,53 (3H, м, ArH), 7,24 (1H, с, ArH), 7,09 (1H, д, CH), 6,23 (1H, с, CH), 5,68 (2H, с, CH2), 2,40 (3H, с, CH3).
13С-ЯМР (500 МГц, ДМСО-d6, DEPT135): δ = 160,8 (qC), 152,0 (2 × qC), 129,0 (2 × CH), 128,9 (2 × CH), 126,8 (2 × CH), 126,8 (Ar, CH), 126,5 (Ar, CH), 125,3 (Ar, CH), 124,1 (Ar, CH), 112,9 (кумарин 3-СН), 111,2 (кумарин 6-СН), 101,7 (кумарин 8-CH), 62,9 (СН2), 18,2 (СН3).
7-(бис(4-Метоксифенил)метокси)-4-метил-2Н-хромен-2-он (49) SU010-02
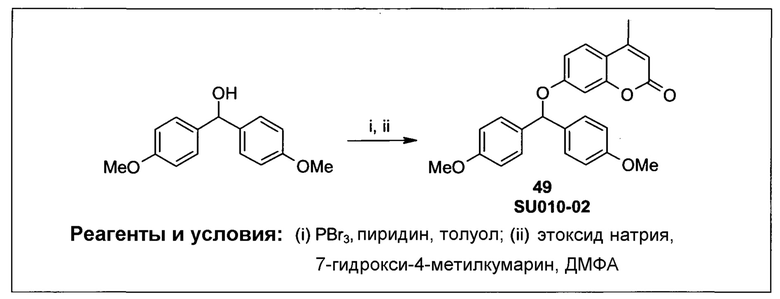
бис(4-Метоксифенил)метанол (2,0 г, 8,2 ммоль) растворяют в толуоле (60 мл) и добавляют пиридин (661 мкл, 8,2 ммоль). Раствор охлаждают до температуры 0°С. По каплям, добавляют PBr3 (768 мкл, 8,2 ммоль) в течение 15 минут. Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 1 часа. Смесь промывают раствором К2СО3 и экстрагируют EtOAc (3×30 мл). Слой EtOAc промывают насыщенным солевым раствором и сушат (MgSO4). Растворитель выпаривают в вакууме, получая сырой продукт, 4,4’-(бромметилен)бис(метоксибензол) (780 мг, выход 31%), в виде бесцветного масла. Этот продукт используют на следующей реакционной стадии без дальнейшей очистки.
Этоксид натрия (133 мг, 1,96 ммоль) добавляют к ДМФА (5 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (345 мг, 1,96 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси порциями добавляют 4,4’-(бромметилен)бис(метоксибензол) (500 мг, 1,63 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 16 часов. ДМФА выпаривают в вакууме и остаток обрабатывают EtOAc и промывают насыщенным солевым раствором, водой и 1 М раствором NaOH (2×30 мл). Органический слой сушат (MgSO4) и продукт очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (2:1), получая соединение 49 (100 мг, выход 15%) в виде твердого вещества белого цвета. Температура плавления: 142-145°С. m/z = 403 (М+Н).
1H ЯМР (500 МГц, ацетон-d6): δ = 7,60 (1H, д, ArH), 7,45 (4H, д, ArH), 7,04 (1H, д, ArH), 6,94 (5H, д, ArH), 6,56 (1H, с, ArH), 6,10 (1H, с, qCH), 3,78 (6H, с, 2 × CH3O), 2,39 (3H, с, CH3).
13С-ЯМР (500 МГц, ацетон-d6, DEPT135): δ = 206,3 (qC), 134,1 (2 × qC), 129,3 (2 × CH), 129,2 (2 × CH), 129,0 (2 × CH), 126,9 (Ar, CH), 114,4 (Ar, CH), 114,7 (Ar, CH), 115,1 (Ar, CH), 112,5 (Ar, СН), 104,0 (Ar, СН), 81,7 (CH), 55,6 (2 × СН3), 18,2 (СН3).
7-((1Н-Бензо[d]имидазол-2-ил)метокси)-4-метил-2Н-хромен-2-он (50) VG033-03
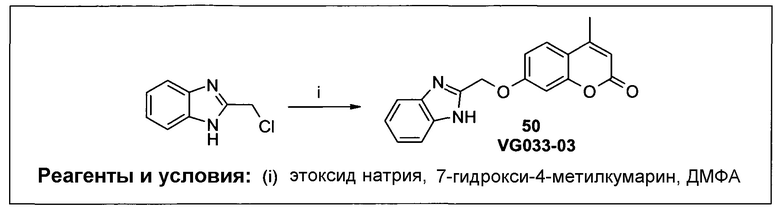
Этоксид натрия (82 мг, 1,20 ммоль) добавляют к ДМФА (10 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (253 мг, 1,44 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси порциями добавляют 2-(хлорметил)-1Н-бензо[d]имидазол (200 мг, 1,20 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 2 часов. ДМФА выпаривают в вакууме и остаток очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (3:1), получая соединение 50 (200 мг, выход 54%) в виде твердого вещества белого цвета. m/z = 307,11 (М+Н), 613,22 (2М+Н).
1H ЯМР (500 МГц, ДМСО-d6): δ = 12,75 (1H, ушир.с, NH), 7,74 (1H, д, J=8,85 Гц, ArH), 7,60-7,59 (2H, м, ArH), 7,23-7,12 (4H, м, ArH), 6,25 (1H, с, ArH), 5,48 (2H, с, CH2), 2,40 (3H, с, CH3).
13С-ЯМР CDEPT 135 (500 МГц, CDCl3): δ: 206,52 (qC), 160,80, 160,03, 154,52, 153,33, 149,27, 126,57, 126,28, 122,04, 119,42, 113,64, 112,50, 111,47, 101,86, 101,77, 64,23, 30,67, 18,10.
7-(Бензо[d]тиазол-2-илметокси)-4-метил-2Н-хромен-2-он (51) VG014-04
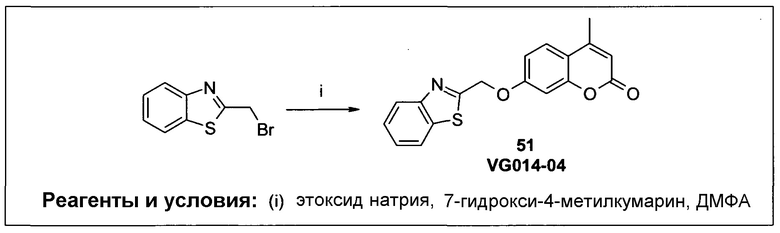
Этоксид натрия (30 мг, 0,44 ммоль) добавляют к ДМФА (10 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (77 мг, 0,44 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси порциями добавляют 2-(хлорметил)-1Н-бензо[d]имидазол (100 мг, 0,44 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 2 часов. ДМФА выпаривают в вакууме и остаток очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (3:1), получая соединение 51 (25 мг, выход 18%) в виде твердого вещества белого цвета. m/z = 324,06 (М+Н), 647,12 (2М+Н).
4-Метил-7-(4-(тиофен-2-ил)бензилокси)-2Н-хромен-2-он (52)
VG015-04
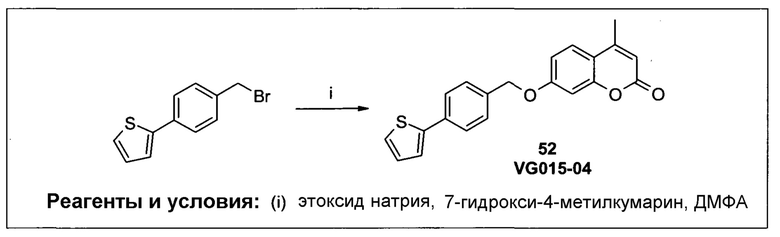
Этоксид натрия (27 мг, 0,40 ммоль) добавляют к ДМФА (10 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (70 мг, 0,40 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси порциями добавляют 2-(хлорметил)-1Н-бензо[d]имидазол (100 мг, 0,40 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 2 часов. ДМФА выпаривают в вакууме и остаток очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (3:1), получая соединение 52 (30 мг, выход 18%) в виде твердого вещества белого цвета. m/z = 349,09 (М+Н), 697,16 (2М+Н).
6-(Бензгидрилтио)-9Н-пурин (53) (VG015-02)
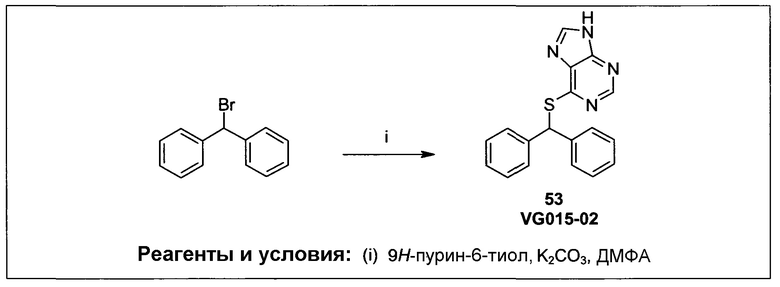
6-Меркаптопурин (151 мг, 0,88 ммоль) растворяют в ДМФА (5 мл). К полученной суспензии добавляют К2СО3 (122 мг, 1,2 ммоль) и дифенилметилбромид (200 мг, 0,8 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 4 часов. Смесь выливают на лед и полученный осадок отделяют путем фильтрации, промывают диэтиловым эфиром и сушат в вакууме, получая соединение 53 (35 мг, выход 14%) в виде твердого вещества белого цвета. m/z = 319 (М+Н).
1Н ЯМР (500 МГц, ацетон): δ = 8,46 (1Н, с, СН), 8,2 (1Н, с, СН), 7,4 (4Н, м, СН, J=3 Гц), 7,2 (4Н, м, СН, J=3,83 Гц), 7,1 (2Н, м, СН, J=2,12 Гц), 6,7 (1Н, с, СН).
3. Карбамат-связанные нуклеозидные аналогичные пролекарства
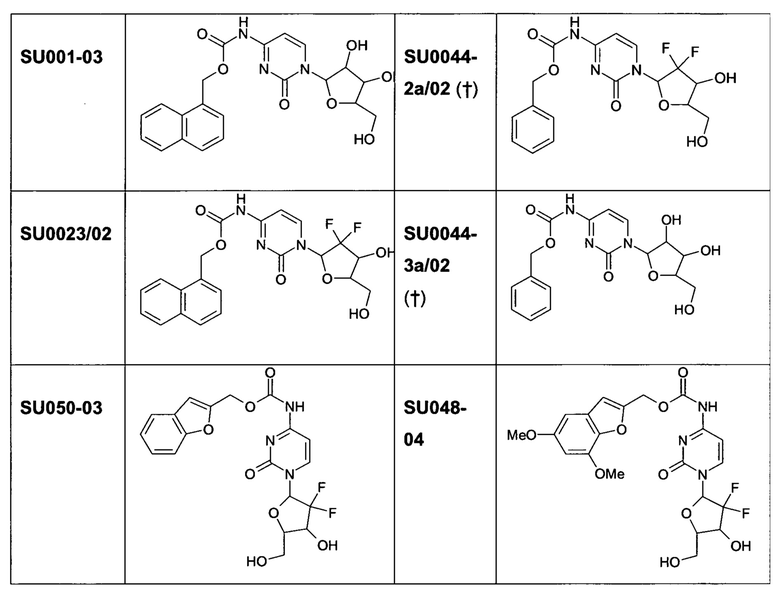
Нафталин-1-илметил-1-(3,4-дигидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)-2-оксо-1,2-дигидропиримидин-4-илкарбамат (55) SU001-03
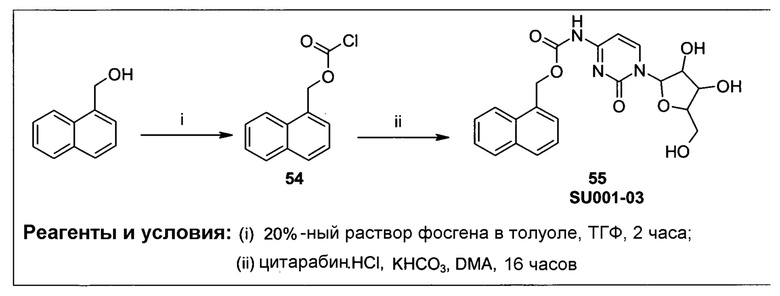
Нафталин-1-илметанол (3,0 г, 19,0 ммоль) добавляют, в виде одной порции, к COCl2 (13,3 мл, в виде 20%-ного раствора COCl2 в толуоле), в ТГФ (30 мл). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Избыток COCl2 и ТГФ удаляют при пониженном давлении. Твердый остаток растворяют в горячем гексане и отфильтровывают. Гексан затем медленно выпаривают в вакууме, получая промежуточный хлорформиат 54 в виде твердого вещества белого цвета. Это вещество непосредственно используют на следующей стадии.
Соединение 54 (330 мг, 1,5 ммоль) и КНСО3 (252 мг, 2,52 ммоль) добавляют к раствору цитарабин⋅HCl (243 мг, 0,87 ммоль) в диметилацетамиде (5 мл) и смесь перемешивают в течение 16 часов при комнатной температуре. Растворитель выпаривают в вакууме и продукт очищают с помощью флэш-хроматографии, элюируя градиентом 2,5-12% МеОН в DCM, получая соединение 55 (38 мг, выход 10%) в виде твердого вещества белого цвета. m/z = 428,15 (М+Н).
Нафталин-1-илметил-1-(3,3-дифтор-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)-2-оксо-1,2-дигидропиримидин-4-илкарбамат (56) SU0023-02
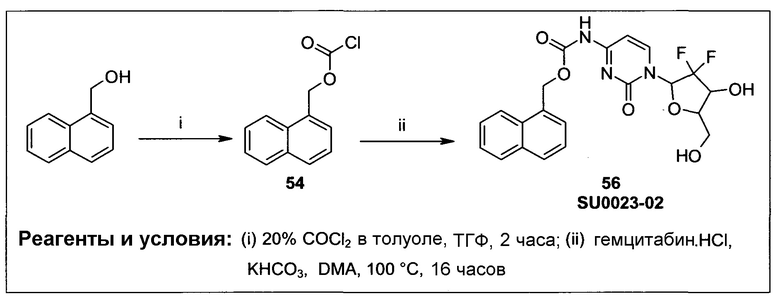
Нафталин-1-илметанол (1,0 г, 6,3 ммоль) добавляют, в виде одной порции, к COCl2 (4,4 мл, в виде 20%-ного раствора COCl2 в толуоле), в ТГФ (20 мл). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Избыток COCl2 и ТГФ удаляют при пониженном давлении. Твердый остаток растворяют в горячем гексане и отфильтровывают. Растворитель, гексан, затем медленно выпаривают в вакууме, получая промежуточный хлорформиат 54 в виде твердого вещества белого цвета. Это вещество непосредственно используют на следующей стадии.
Гемцитабин⋅HCl (200 мг, 0,67 ммоль) растворяют в Н2О (2 мл). К этому раствору добавляют КНСО3 (67 мг, 0,67 ммоль) и соединение 54 (147 мг, 0,67 ммоль), предварительно растворенное в этилацетате (5 мл). Смесь перемешивают при температуре 100°С в течение 16 часов. Растворитель выпаривают в вакууме и продукт очищают с помощью флэш-хроматографии, элюируя 3% МеОН в этилацетате, получая соединение 56 (15 мг, выход 5%) в виде масла. m/z = 448,13 (М+Н).
Бензил-1-(3,3-дифтор-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)-2-оксо-1,2-дигидропиримидин-4-илкарбамат (57) SU0044-2а/02
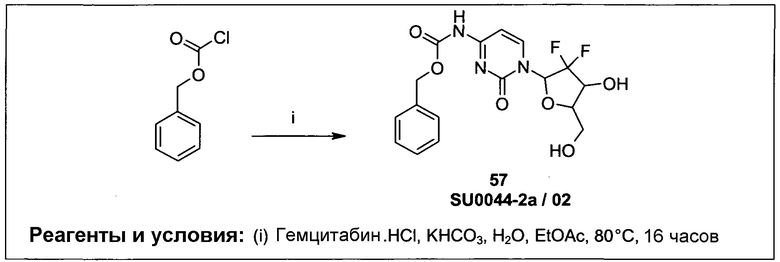
Гемцитабин⋅HCl (200 мг, 0,67 ммоль) растворяют в Н2О (2 мл). К этому раствору добавляют КНСО3 (67 мг, 0,67 ммоль) и бензилкарбонохлоридат (95 мкл, 0,67 ммоль), предварительно растворенный в этилацетате (5 мл). Смесь перемешивают при температуре 80оС в течение 16 часов. Растворитель выпаривают в вакууме и продукт очищают с помощью флэш-хроматографии, элюируя 3% МеОН в этилацетате, получая соединение 57 (40 мг, выход 15%) в виде масла. m/z = 398,12 (М+Н).
Бензил-1-(3,4-дигидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)-2-оксо-1,2-дигидропиримидин-4-илкарбамат (58) SU0044-3а/02
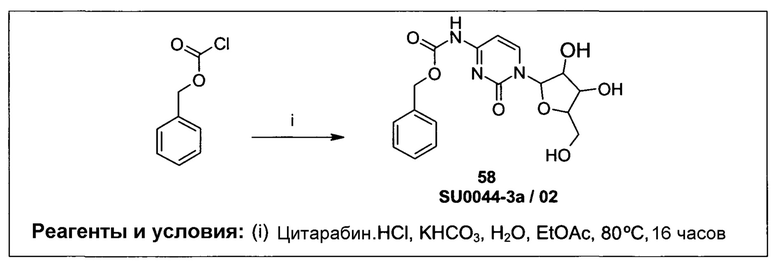
Цитарабин⋅HCl (200 мг, 0,72 ммоль) растворяют в Н2О (2 мл). К этому раствору добавляют КНСО3 (72 мг, 0,72 ммоль) и бензилкарбонохлоридат (107 мкл, 0,72 ммоль), предварительно растворенный в этилацетате (5 мл). Смесь перемешивают при температуре 80°С в течение 16 часов. Растворитель выпаривают в вакууме и продукт очищают с помощью флэш-хроматографии, элюируя 3% МеОН в этилацетате, получая соединение 58 (40 мг, выход 15%) в виде масла. m/z = 378,13 (М+Н).
Бензофуран-2-илметил-4-нитрофенилкарбонат (60)
и
(5,7-Диметоксибензофуран-2-ил)метил-4-нитрофенилкарбонат (61)
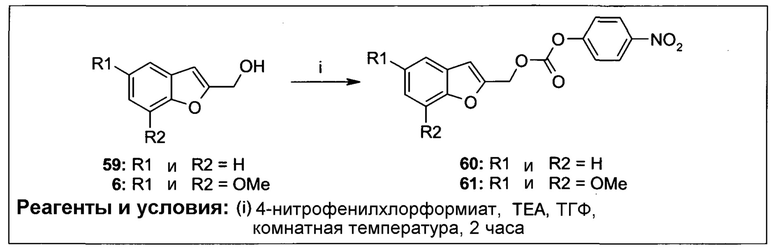
Бензофуран-2-илметил-4-нитрофенилкарбонат (60)
Раствор бензофуран-2-илметанола 59 (300 мг, 2,03 ммоль) в ТГФ (5 мл) охлаждают до температуры 0°С. По каплям, добавляют ТЕА (280 мкл, 2,03 ммоль), затем порциями добавляют п-нитрофенилхлорформиат (282 мг, 3,05 ммоль). Полученный раствор перемешивают при комнатной температуре в течение 2 часов. Растворитель выпаривают в вакууме и сырой остаток очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (3:1), получая соединение 60 (350 мг, выход 54%) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, CDCl3): δ = 8,31 (2H, м, ArH), 7,62 (1H, д, J=7,70 Гц, ArH), 7,54 (1H, д, J=7,70 Гц, ArH), 7,43-7,27 (3H, м, ArH), 7,29 (1H, т, J=7,72 Гц, ArH), 6,93 (1H, с, ArH), 5,43 (2H, с, CH2).
13С-ЯМР CDEPT 135 (500 МГц, CDCl3): δ = 125,47, 125,37, 123,23, 121,80, 121,66, 111,60, 108,53, 62,95.
(5,7-Диметоксибензофуран-2-ил)метил-4-нитрофенилкарбонат (61)
Раствор (5,7-диметоксибензофуран-2-ил)метанола 6 (100 мг, 0,48 ммоль) в ТГФ (3 мл) охлаждают до температуры 0°С. По каплям, добавляют ТЕА (69 мкл, 0,48 ммоль), затем порциями п-нитрофенилхлорформиат (100 мг, 0,72 ммоль). Полученный раствор перемешивают при комнатной температуре в течение 2 часов. Растворитель выпаривают в вакууме и сырой остаток очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (3:1), получая соединение 61 (120 мг, выход 67%) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, CDCl3): δ = 8,29 (2H, д, J=9,0 Гц, ArH), 7,40 (2H, д, J=9,0 Гц, ArH), 6,82 (1H, с, ArH), 6,63 (1H, с, ArH), 6,52 (1H, с, ArH), 5,39 (2H, с, CH2), 4,00 (3H, с, OCH3), 3,85 (3H, с, OCH3).
Бензофуран-2-илметил-1-(3,3-дифтор-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)-2-оксо-1,2-дигидропиримидин-4-илкарбамат (65) SU050-03
и
(5,7-Диметоксибензофуран-2-ил)метил-1-(3,3-дифтор-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)-2-оксо-1,2-дигидропиримидин-4-илкарбамат (66) SU048-04
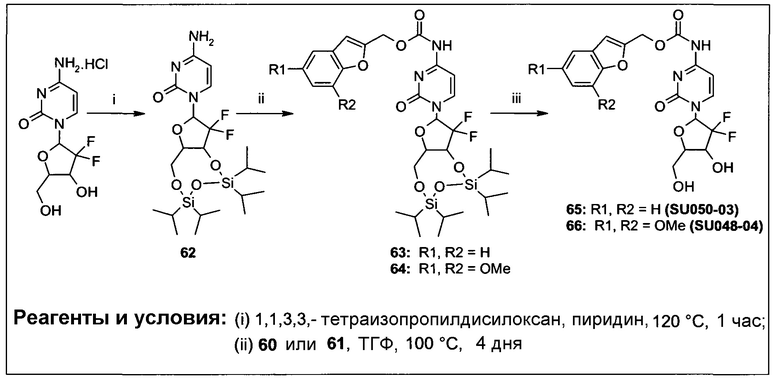
4-Амино-1-(9,9-дифтор-2,2,4,4-тетраизопропилтетрагидро-6Н-фуро[3,2-f][1,3,5,2,4]триоксадисилоцин-8-ил)пиримидин-2(1Н)-он (62)
Гемцитабин⋅HCl (1,0 г, 3,3 ммоль) перемешивают в пиридине (10 мл) в течение 10 минут (2×5 мл). Пиридин выпаривают. Добавляют пиридин (10 мл) и, по каплям, 1,1,3,3-тетраизопропилдисилоксан (1,17 мл, 3,63 ммоль). Полученную смесь перемешивают при температуре 100оС в течение 16 часов. Добавляют еще порцию 1,1,3,3-тетраизопропилдисилоксана (1 мл) и смесь перемешивают при температуре 120°С в течение 1 часа. Реакционную смесь охлаждают до комнатной температуры и растворитель выпаривают в вакууме. Полученное сырое твердое вещество перекристаллизуют из смеси EtOAc/диэтиловый эфир (1:1), получая соединение 62 (600 мг, выход 36%) в виде твердого вещества белого цвета. m/z = 506,23 (М+Н).
Бензофуран-2-илметил-1-(9,9-дифтор-2,2,4,4-тетраизопропилтетрагидро-6Н-фуро[3,2-f][1,3,5,2,4]триоксадисилоцин-8-ил)-2-оксо-1,2-дигидропиримидин-4-илкарбамат (63)
К перемешиваемому раствору соединения 62 (300 мг, 0,59 ммоль) в ТГФ (5 мл) добавляют бензофуран-2-илметил-4-нитрофенилкарбонат (223 мг, 0,71 ммоль). Полученный раствор перемешивают при температуре 100°С в течение 4 дней. Растворитель выпаривают в вакууме и продукт очищают с помощью препаративной ВЭЖХ, получая соединение 63 (350 мг, выход 87%) в виде масла. m/z = 680,0 (М+Н), 1359,49 (2М+Н).
Бензофуран-2-илметил-1-(3,3-дифтор-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)-2-оксо-1,2-дигидропиримидин-4-илкарбамат (65) SU050-03
Соединение 63 (200 мг, 0,29 ммоль) растворяют в ТГФ (1,5 мл). К этому раствору добавляют тетра-н-бутиламмонийфторид и полученный раствор перемешивают при комнатной температуре в течение 15 минут. Растворитель выпаривают в вакууме. Продукт очищают с помощью флэш-хроматографии, элюируя 5% МеОН в EtOAc, получая соединение 65 (30 мг, выход 23%) в виде масла. m/z = 438,14 (М+Н), 874,24 (2М+Н).
(5,7-Диметоксибензофуран-2-ил)метил-1-(3,3-дифтор-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)-2-оксо-1,2-дигидропиримидин-4-илкарбамат (66) SU048-04
К перемешиваемому раствору соединения 62 (108 мг, 0,21 ммоль) в ТГФ (5 мл) добавляют соединение 61 (100 мг, 0,27 ммоль). Полученный раствор перемешивают при температуре 100°С в течение 4 дней. Растворитель выпаривают в вакууме, получая соединение 64 в виде масла. Это соединение используют на следующей стадии, без дальнейшей очистки.
Соединение 64 (100 мг, 0,14 ммоль) растворяют в ТГФ (1,5 мл). К этому раствору добавляют тетра-н-бутиламмонийфторид и полученный раствор перемешивают при комнатной температуре в течение 15 минут. Растворитель выпаривают в вакууме. Продукт очищают с помощью флэш-хроматографии, элюируя 5% МеОН в EtOAc, получая соединение 66 (18 мг, выход 26%) в виде масла. m/z = 498,14 (М+Н), 995,29 (2М+Н).
1H ЯМР (500 МГц, ацетон-d6): δ = 9,60 (1H, ушир.с, NH), 8,34 (1H, д, J=7,62 Гц, ArH), 7,26 (1H, д, J=9,00 Гц, ArH), 6,92 (1H, с, ArH), 6,71 (1H, д, J=2,20 Гц, ArH), 6,56 (1H, д, J=2,20 Гц, ArH), 6,26 (1H, т, J=7,56 Гц, CH), 5,64 (2H, с, CH2), 4,55-4,45 (1H, м, CH), 4,05-4,02 (2H, м, CH2), 3,97 (3H, с, OCH3), 3,91-3,3,87 (1H, м, CH), 3,82 (3H, с, OCH3), 2,92 (2H, ушир.с, OH).
4. Карбамат-связанные азот- и анилинмустиновые пролекарства
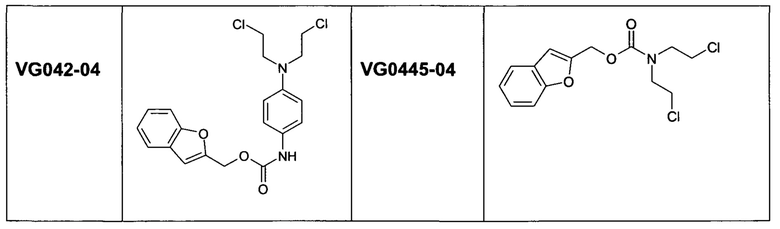
Бензофуран-2-илметил-4-(бис(2-хлорэтил)амино)фенилкарбамат (VG042-04)
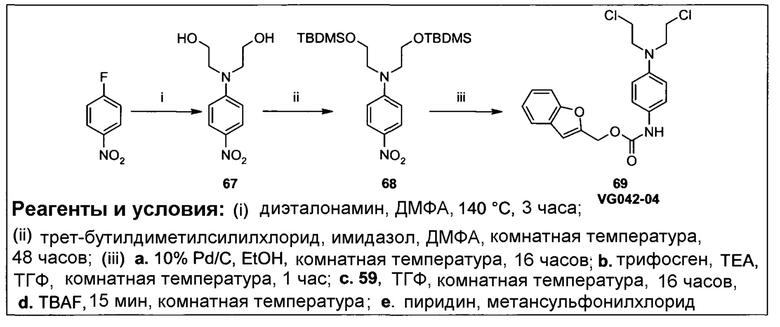
2,2’-(4-Нитрофенилазанедиил)диэтанол (67)
Диэтаноламин (2,70 мл, 2,5 ммоль) добавляют к 1-фтор-4-нитробензолу (1,0 г, 7,09 ммоль) в ДМФА (30 мл). Полученную смесь перемешивают при температуре 140°С в течение 3,5 часов. Раствор охлаждают до комнатной температуры и растворитель выпаривают в вакууме. Остаток растворяют в EtOAc (30 мл) и промывают водой (3×10 мл) и насыщенным солевым раствором (3×20 мл) и сушат (MgSO4). Растворитель выпаривают в вакууме и продукт очищают с помощью флэш-хроматографии, элюируя EtOAc, получая соединение 67 (400 мг, выход 25%) в виде твердого вещества желтого цвета.
1H ЯМР (500 МГц, CDCl3): δ = 8,06 (2H, д, J=9,50 Гц, ArH), 6,87 (2H, д, J=9,50 Гц, ArH), 4,27 (2H, т, J=5,35 Гц, 2 × OH), 3,83 (4H, кв., J=5,55 и 5,45 Гц, 2 × CH2), 3,74 (4H, т, J=5,62 Гц, 2 × CH2).
N,N-бис(2-(трет-Бутилдиметилсилилокси)этил)-4-нитроанилин (68)
К охлажденному раствору соединения 67 (400 мг, 1,77 ммоль) и имидазола (481 мг, 7,08 ммоль) в ДМФА (10 мл), по каплям, добавляют трет-бутилдиметилсилилхлорид (2,72 мг, 3,54 ммоль). Смесь оставляют достигать комнатной температуры и перемешивают в течение 48 часов. Растворитель выпаривают в вакууме и продукт очищают с помощью флэш-хроматографии, элюируя 10% EtOAc в диэтиловом эфире, получая соединение 68 (200 мг, выход 25%) в виде твердого вещества желтого цвета.
1H ЯМР (500 МГц, CDCl3): δ = 8,06 (2H, д, J=9,45 Гц, ArH), 6,65 (2H, д, J=9,45 Гц, ArH), 3,80 (4H, т, J=5,80 Гц, 2 × CH2), 3,62 (4H, т, J=5,80 Гц, 2 × CH2), 0,85 (18H, с, 6 × CH3), -0,01 (12H, с, 4 × CH3).
Бензофуран-2-илметил-4-(бис(2-хлорэтил)амино)фенилкарбамат (69) VG042-04
Соединение 68 (200 мг, 0,44 ммоль) обрабатывают водородом в присутствии 10% палладия-на-угле (20 мг). После перемешивания в течение 16 часов смесь отфильтровывают через целит и растворитель выпаривают в вакууме, получая промежуточный аминоанилиновый продукт. Его затем вводят во взаимодействие с трифосгеном (195 мг, 0,70 ммоль) в присутствии триэтиламина (260 мкл, 0,70 ммоль) в ТГФ (15 мл). После перемешивания в течение 1 часа при комнатной температуре, осадок белого цвета отфильтровывают и растворитель выпаривают в вакууме, получая сырой остаток анилинизоцианата. Этот остаток непосредственно используют на следующей стадии.
Промежуточный изоцианат растворяют в ТГФ (10 мл). Раствор охлаждают до температуры 0°С. Добавляют бензофуран-2-илметанол 59 (100 мг, 1,35 ммоль) и полученную смесь перемешивают при комнатной температуре в течение 16 часов. Смесь охлаждают на льду и, по каплям, добавляют TBAF (996 мкл, 3,38 ммоль) в течение 5 минут. Полученную смесь оставляют нагреваться до комнатной температуры и затем перемешивают в течение 20 минут. ТГФ выпаривают в вакууме. Промежуточный продукт растворяют в пиридине (5 мл) и к этому раствору добавляют метансульфонилхлорид (12,5 мкл, 0,16 ммоль). Смесь перемешивают при комнатной температуре в течение 1 часа. Пиридин выпаривают в вакууме и сырой продукт очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (3:1), получая соединение 69 (5 мг, выход 2%) в виде твердого вещества белого цвета. m/z = 408,07 (М+Н), 837,16 (2М+Н).
Бензофуран-2-илметилбис(2-хлорэтил)карбамат (70) VG045-04
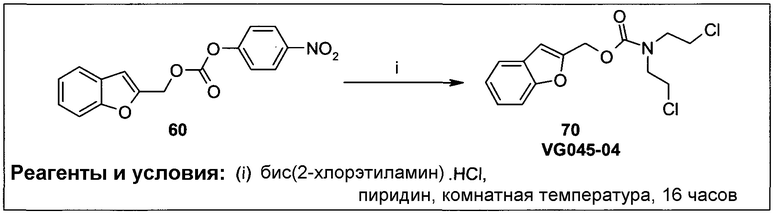
Раствор соединения 60 (200 мг, 0,64 ммоль) в пиридине (3 мл) добавляют к раствору бис(2-хлорэтиламин)гидрохлорида (227 мг, 1,28 ммоль) в пиридине (25 мл). Смесь перемешивают при комнатной температуре в течение 16 часов. Добавляют DCM (10 мл) и смесь промывают 2% раствором лимонной кислоты (2×50 мл), водой (50 мл), насыщенным солевым раствором (50 мл) и сушат (MgSO4). Растворитель выпаривают в вакууме и продукт очищают с помощью флэш-хроматографии, элюируя смесью CH2Cl2:гексан (2:1), получая соединение 70 (125 мг, выход 62%) в виде масла. m/z = 338,05 (М+Na).
1H ЯМР (500 МГц, CDCl3): δ = 7,60 (1H, д, J=7,70 Гц, ArH), 7,51 (1H, д, J=8,05 Гц, ArH), 7,33 (1H, т, J=6,80 Гц, ArH), 7,26 (1H, т, J=6,80 Гц, ArH), 6,79 (1H, с, ArH), 5,28 (2H, с, CH2), 3,72-3,63 (8H, м, 4 × CH2).
5. Связанное через образование простого эфира пролекарство, ингибитор топоизомеразы I
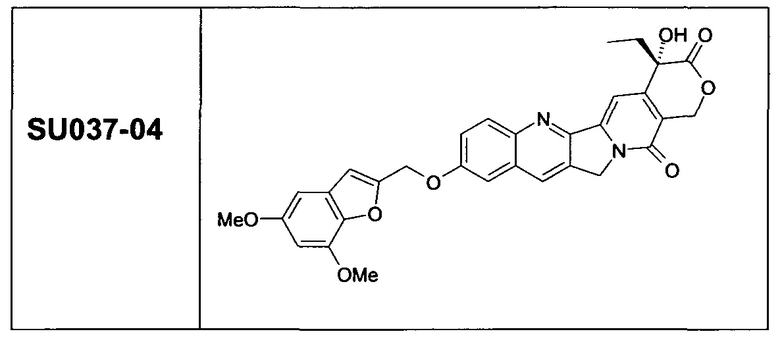
(5,7-Диметоксибензофуран-2-ил)метилкамптотецин (71) SU037-04
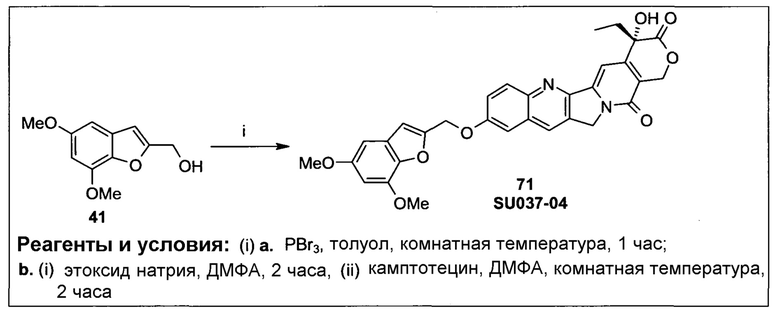
Соединение 41 (100 мг, 0,48 ммоль) растворяют в толуоле (5 мл) и раствор охлаждают до температуры 0°С. По каплям, добавляют PBr3 (46 мкл, 0,48 ммоль) в течение 10 минут. Реакционную смесь затем доводят до комнатной температуры и перемешивают в течение 1 часа. Растворитель выпаривают в вакууме. Сырой остаток используют на следующей стадии.
Этоксид натрия (15 мг, 0,22 ммоль) добавляют к ДМФА (5 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют камптотецин (81 мг, 0,22 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси порциями добавляют сырой остаток с предыдущей стадии, 2-(бромметил)-5,7-диметоксибензофуран (50 мг, 0,18 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 2 часов. ДМФА выпаривают в вакууме и остаток очищают с помощью флэш-хроматографии, элюируя смесью DCM:EtOAc (2:1), получая целевое соединение в виде твердого вещества белого цвета (10 мг, выход 10%). m/z = 555,19 (М+Н).
6. Связанное через образование простого эфира пролекарство, ингибитор тирозинкиназы
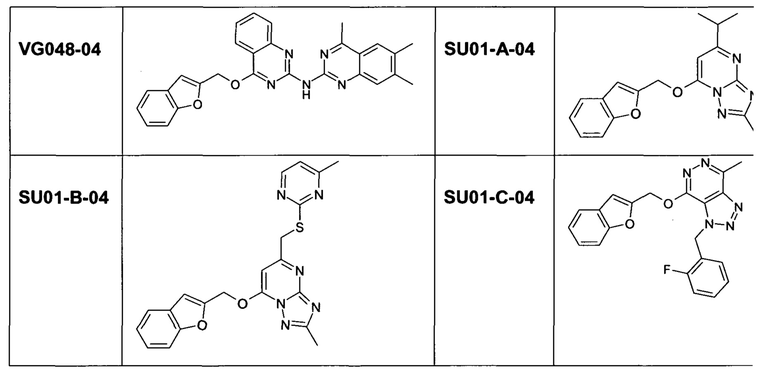
N-(4-(Бензофуран-2-илметокси)хиназолин-2-ил)-4,6,7-триметилхиназолин-2-амин (72) VG048-04
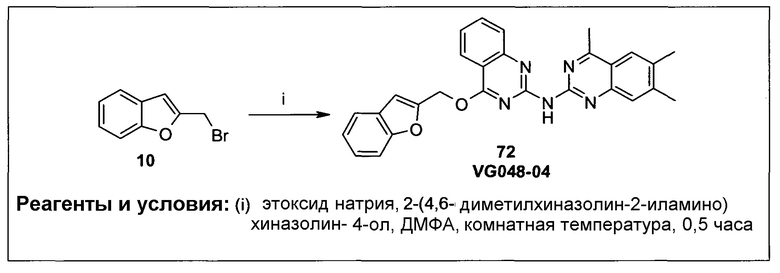
Этоксид натрия (3 мг, 0,05 ммоль) добавляют к ДМФА (2 мл), при температуре 0°С, и суспензию перемешивают в течение 5 минут. Медленно добавляют 2-(4,6-диметилхиназолин-2-иламино)хиназолин-4-ол (15 мг, 0,05 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси добавляют 2-(бромметил)бензофуран (16 мг, 0,08 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 1 часа. ДМФА выпаривают в вакууме, получая сырое твердое вещество белого цвета. Это вещество очищают путем промывки холодным диэтиловым эфиром и EtOAc, получая соединение 72 (3 мг, выход 11%) в виде твердого вещества белого цвета. m/z = 462,2 (М+Н).
7-(Бензофуран-2-илметокси)-5-изопропил-2-метил-[1,2,4]триазоло[1,5-a]пиримидин (73) SU01-А-04
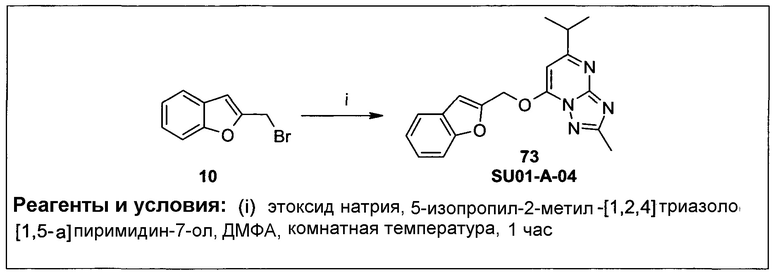
Этоксид натрия (7,2 мг, 0,10 ммоль) добавляют к ДМФА (2 мл), при температуре 0°С, и суспензию перемешивают в течение 5 минут. Медленно добавляют 5-изопропил-2-метил-[1,2,4]триазоло[1,5-a]пиримидин-7-ол (20 мг, 0,10 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси добавляют 2-(бромметил)бензофуран (16 мг, 0,08 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 1 часа. ДМФА выпаривают в вакууме, получая сырое твердое вещество белого цвета. Это вещество очищают с помощью полупрепаративной ВЭЖХ, получая соединение 73 (6,3 мг, выход 19%) в виде твердого вещества белого цвета. m/z = 323,13 (М+Н), 645,27 (2М+Н).
7-(Бензофуран-2-илметокси)-2-метил-5-((4-метилпиримидин-2-илтио)метил)-[1,2,4]триазоло[1,5-a]пиримидин (74) SU01-В-04
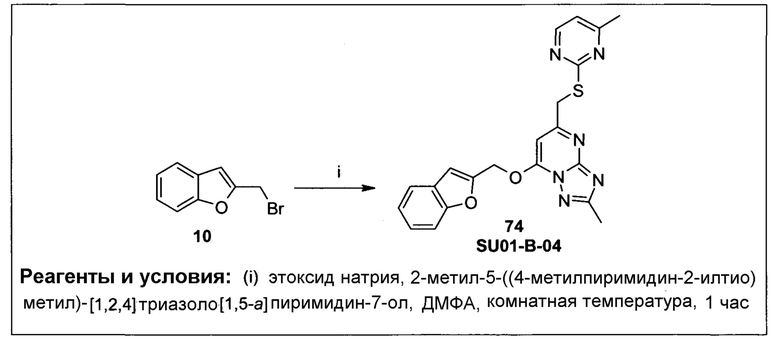
Этоксид натрия (4,7 мг, 0,07 ммоль) добавляют к ДМФА (2 мл), при температуре 0°С, и суспензию перемешивают в течение 5 минут. Медленно добавляют 2-метил-5-((4-метилпиримидин-2-илтио)метил)-[1,2,4]триазоло[1,5-a]пиримидин-7-ол (20 мг, 0,07 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси добавляют соединение 10 (16 мг, 0,08 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 1 часа. ДМФА выпаривают в вакууме, получая сырое твердое вещество белого цвета. Это вещество очищают с помощью полупрепаративной ВЭЖХ, получая соединение 74 (5,2 мг, выход 18%) в виде твердого вещества белого цвета. m/z = 419,09 (М+Н), 837,23 (2М+Н).
7-(Бензофуран-2-илметокси)-1-(2-фторбензил)-4-метил-1Н-[1,2,3]триазоло[4,5-d]пиридазин (75) SU01-С-04
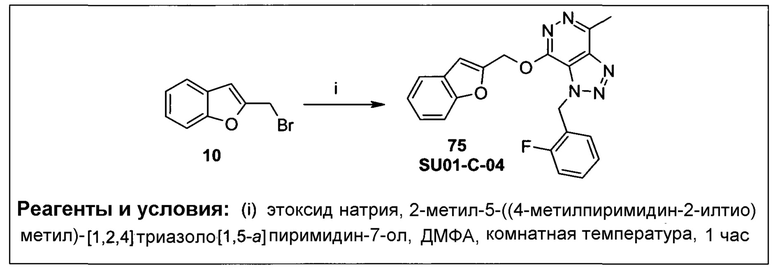
Этоксид натрия (5,2 мг, 0,08 ммоль) добавляют к ДМФА (2 мл), при температуре 0°С, и суспензию перемешивают в течение 5 минут. Медленно добавляют 1-(2-фторбензил)-4-метил-1Н-[1,2,3]триазоло[4,5-d]пиридазин-7-ол (20 мг, 0,08 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси добавляют 2-(бромметил)бензофуран (16 мг, 0,08 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 1 часа. ДМФА выпаривают в вакууме, получая сырое твердое вещество белого цвета. Это вещество очищают с помощью полупрепаративной ВЭЖХ, получая соединение 75 (3 мг, выход 10%) в виде твердого вещества белого цвета. m/z = 390,07 (М+Н), 801,12 (2М+Na).
7. Карбамат-связанные модельные кумариновые пролекарства
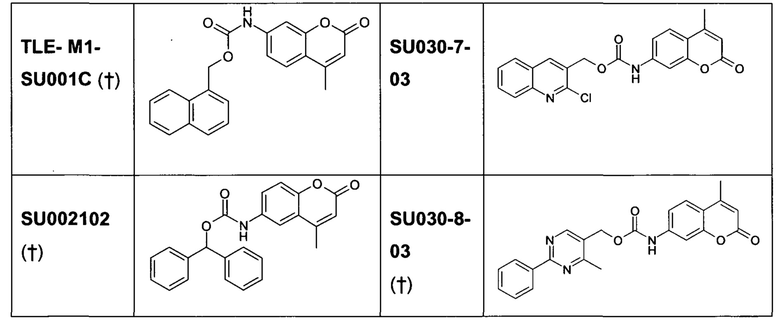
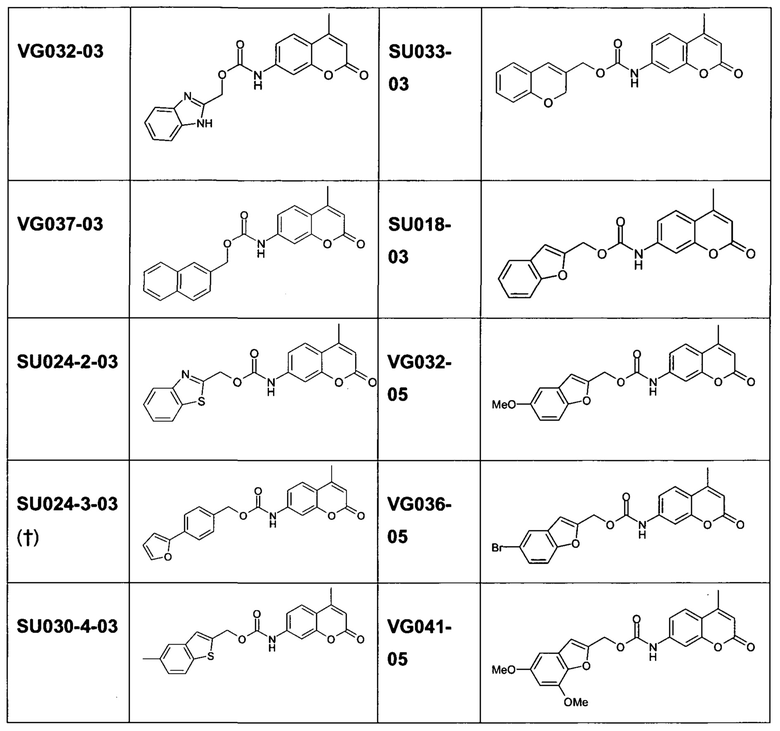
7-Изоцианато-4-метилкумарин (76)
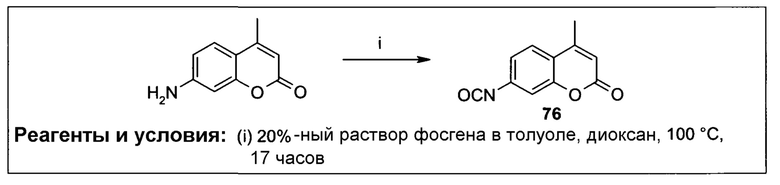
В трехгорлую колбу емкостью 200 мл, оснащенную конденсатором с охлаждением сухим льдом и магнитной мешалкой, вводят раствор 20%-ного фосгена в толуоле (2,0 мл) и диоксан (80 мл). К этой смеси добавляют 7-амино-4-метил-2Н-хромен-2-он (2,00 г, 11,4 ммоль). Смесь перемешивают при температуре 100°С в течение 12 часов. Первоначальный желтый цвет исчезает и осаждается твердое вещество белого цвета. Добавляют еще раствор 20%-ного фосгена в толуоле (7,0 мл) и смесь нагревают в течение дополнительных 5 часов, после чего раствор осветляется. Избыток фосгена и следы HCl удаляют путем барботирования газообразного азота через раствор. Мутный раствор отфильтровывают, удаляя непрореагировавший 7-амино-4-метил-2Н-хромен-2-он, и концентрируют, получая соединение 76 (0,5 г, выход 25%) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, CDCl3): δ = 7,50 (1H, д, J=7,40 Гц, ArH), 7,46 (2H, с, ArH), 6,20 (1H, с, ArH), 2,35 (3H, с, CH3): ir (CH2Cl2) 2314 (N=C=O), 1726 и 1615.
Нафталин-1-илметил-4-метил-2-оксо-2Н-хромен-7-илкарбамат (77) VG020-02
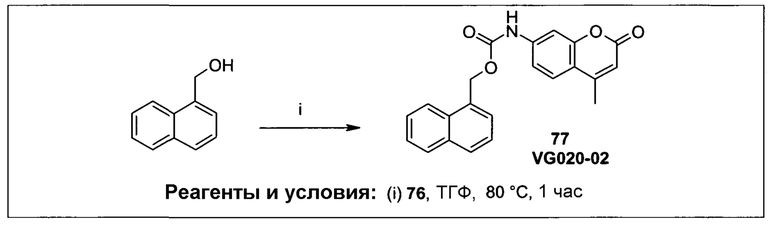
Нафталин-1-илметанол (56 мг, 0,28 ммоль) и соединение 76 (200 мг, 1,27 ммоль) растворяют в ТГФ (2 мл). Полученную смесь перемешивают при комнатной температуре в течение 15 минут и затем при температуре 80°С в течение 1 часа. ТГФ выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью CH2Cl2/гексан/EtOAc (1:1:1), получая соединение 77 (3 мг, выход 5%) в виде твердого вещества белого цвета. m/z = 360,14 (М+Н), 719,27 (2М+Н).
(2-Хлорхинолин-3-ил)метил-4-метил-2-оксо-2Н-хромен-7-илкарбамат (78) SU030-7-03
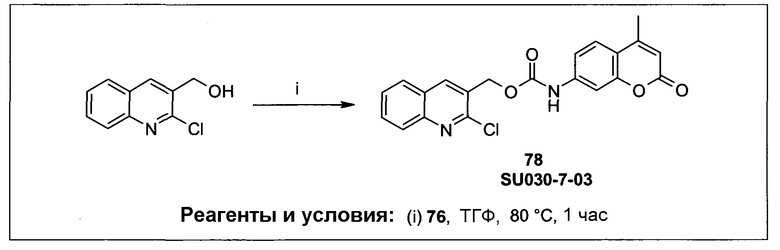
(2-Хлорхинолин-3-ил)метанол (100 мг, 0,52 ммоль) и соединение 76 (155 мг, 0,77 ммоль) растворяют в ТГФ (2 мл). Полученную смесь перемешивают при комнатной температуре в течение 15 минут и затем при температуре 80°С в течение 1 часа. ТГФ выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью CH2Cl2/EtOAc (1:1), получая соединение 78 (38 мг, выход 19%) в виде твердого вещества белого цвета. m/z = 395,08 (М+Н).
Бензгидрил-4-метил-2-оксо-2Н-хромен-7-илкарбамат (79) SU0021-02
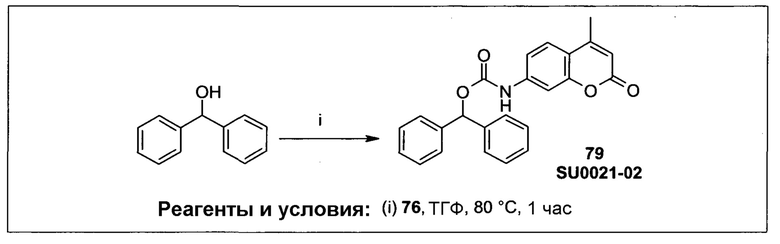
(2-Хлорхинолин-3-ил)метанол (119 мг, 0,65 ммоль) и соединение 76 (70 мг, 0,35 ммоль) растворяют в ТГФ (2 мл). Полученную смесь перемешивают при комнатной температуре в течение 15 минут и затем при температуре 80°С в течение 1 часа. ТГФ выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью CH2Cl2/EtOAc (1:1), получая соединение 79 (50 мг, выход 37%) в виде твердого вещества белого цвета. m/z = 386,16 (М+Н).
Бензгидрил-4-метил-2-оксо-2Н-хромен-7-илкарбамат (80) SU0021-02
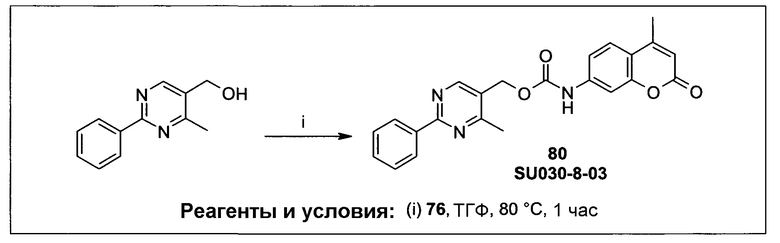
(4-Метил-2-фенилпиримидин-5-ил)метанол (100 мг, 0,50 ммоль) и соединение 76 (151 мг, 0,75 ммоль) растворяют в ТГФ (2 мл). Полученную смесь перемешивают при комнатной температуре в течение 15 минут и затем при температуре 80°С в течение 1 часа. ТГФ выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью CH2Cl2/EtOAc (1:1), получая соединение 80 (70 мг, выход 35%) в виде твердого вещества белого цвета. m/z = 402,15 (М+Н).
(1Н-Бензо[d]имидазол-2-ил)метил-4-метил-2-оксо-2Н-хромен-7-илкарбамат (81) VG032-03
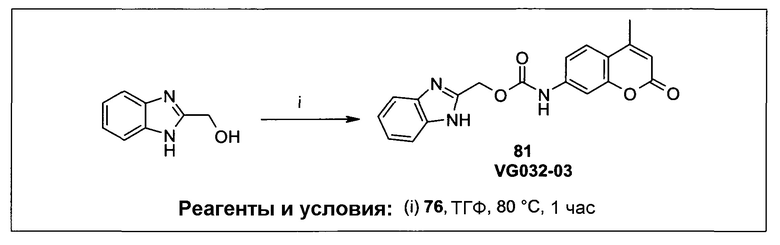
(1Н-Бензо[d]имидазол-2-ил)метанол (200 мг, 1,35 ммоль) и соединение 76 (272 мг, 1,35 ммоль) растворяют в ТГФ (2 мл). Полученную смесь перемешивают при комнатной температуре, в течение 15 минут и затем при температуре 80°С в течение 1 часа. ТГФ выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью CH2Cl2/EtOAc (1:1), получая соединение 81 (80 мг, выход 17%) в виде твердого вещества белого цвета. m/z = 350,12 (М+Н).
(2Н-Хромен-3-ил)метил-4-метил-2-оксо-2Н-хромен-7-илкарбамат (82) SU033-03
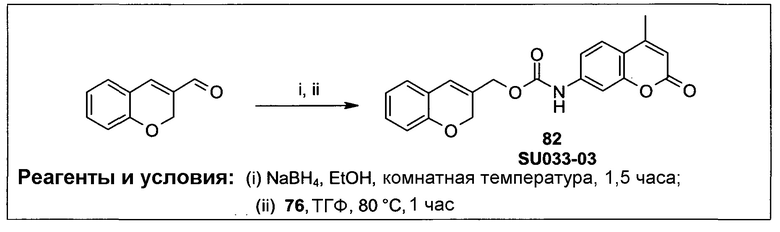
2Н-Хромен-3-карбальдегид (500 мг, 3,13 ммоль) растворяют в EtOH (10 мл). Порциями добавляют NaBH4 (119 мг, 3,13 ммоль), при температуре 0°С, при энергичном перемешивании. Суспензию перемешивают при температуре 0°С в течение 15 минут и затем при комнатной температуре в течение 1,5 часов. Растворитель выпаривают в вакууме, получая промежуточный спирт в виде масла. Этот спирт растворяют в ТГФ (5 мл) и добавляют соединение 76 (155 мг, 0,77 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 15 минут и затем при температуре 80оС в течение 1 часа. ТГФ выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью CH2Cl2/EtOAc (1:1), получая соединение 82 (80 мг, выход 8%) в виде твердого вещества белого цвета. m/z = 364,12 (М+Н), 727,23 (2М+Н).
Нафталин-2-илметил-4-метил-2-оксо-2Н-хромен-7-илкарбамат (83) VG037-03
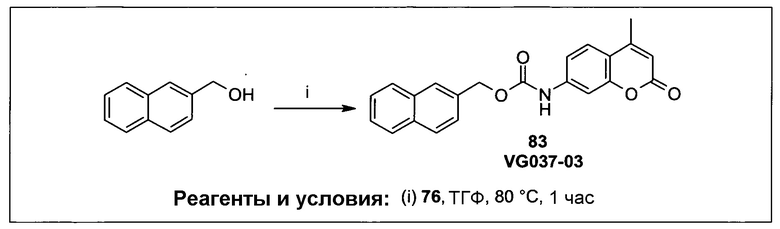
Нафталин-2-илметанол (200 мг, 1,27 ммоль) и соединение 76 (279 мг, 1,39 ммоль) растворяют в ТГФ (2 мл). Полученную смесь перемешивают при комнатной температуре в течение 15 минут и затем при температуре 80°С в течение 1 часа. ТГФ выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью CH2Cl2/EtOAc (1:1), получая соединение 83 (26 мг, выход 6%) в виде твердого вещества белого цвета. m/z = 360,13 (М+Н), 719,25 (2М+Н).
Бензофуран-2-илметил-4-метил-2-оксо-2Н-хромен-7-илкарбамат (84) SU018-03
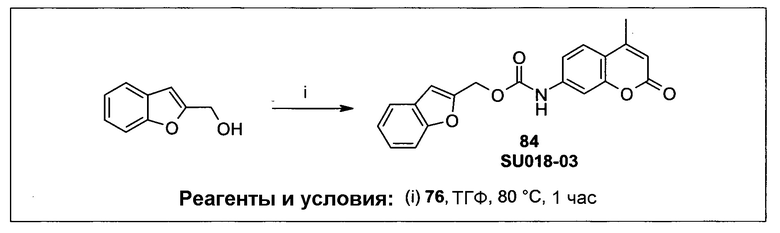
Бензофуран-2-илметанол (300 мг, 2,03 ммоль) и соединение 76 (407 мг, 2,03 ммоль) растворяют в ТГФ (2 мл). Полученную смесь перемешивают при комнатной температуре, в течение 15 минут и затем при температуре 80°С в течение 1 часа. ТГФ выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью CH2Cl2/EtOAc (1:1), получая соединение 84 (130 мг, выход 18%) в виде твердого вещества белого цвета. m/z = 350,09 (М+Н), 699,17 (2М+Н).
Бензо[d]тиазол-2-илметил-4-метил-2-оксо-2Н-хромен-7-илкарбамат (85) SU024-3-03
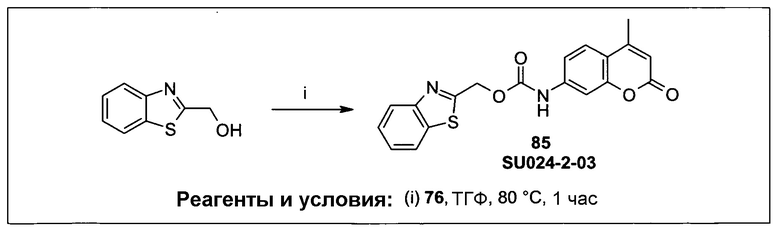
Бензо[d]тиазол-2-илметанол (200 мг, 1,21 ммоль) и соединение 76 (365 мг, 1,8 ммоль) растворяют в ТГФ (2 мл). Полученную смесь перемешивают при комнатной температуре в течение 15 минут и затем при температуре 80°С в течение 1 часа. ТГФ выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью CH2Cl2/EtOAc (1:1), получая соединение 85 (90 мг, выход 20%) в виде твердого вещества белого цвета. m/z = 367,02 (М+Н), 733,13 (2М+Н).
4-(Фуран-2-ил)бензил-4-метил-2-оксо-2Н-хромен-7-илкарбамат (86) SU024-3-03
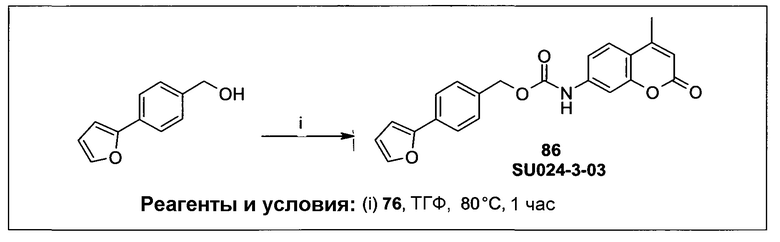
(4-(Фуран-2-ил)фенил)метанол (100 мг, 0,57 ммоль) и соединение 76 (139 мг, 0,69 ммоль) растворяют в ТГФ (10 мл). Полученную смесь перемешивают при комнатной температуре, в течение 15 минут и затем при температуре 80°С в течение 1 часа. ТГФ выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью CH2Cl2/EtOAc (1:1), получая целевое соединение (20 мг, выход 9%) в виде твердого вещества белого цвета. m/z = 376,11 (М+Н), 751,22 (2М+Н).
(5-Метилбензо[d]тиофен-2-ил)метил-4-метил-2-оксо-2Н-хромен-7-илкарбамат (87) SU030-4-03
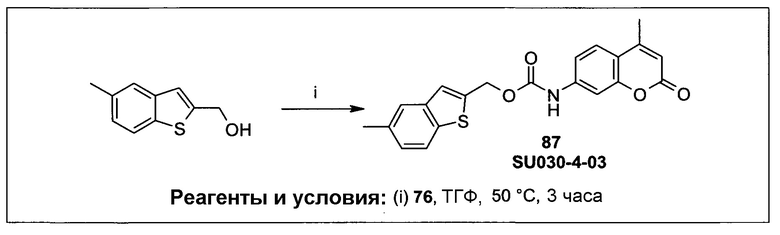
(5-Метилбензо[d]тиофен-2-ил)метанол (100 мг, 0,56 ммоль) и соединение 76 (136 мг, 0,67 ммоль) растворяют в ТГФ (2 мл). Полученную смесь перемешивают при комнатной температуре в течение 15 минут и затем при температуре 50°С в течение 3 часов. ТГФ выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью CH2Cl2/EtOAc (1:1), получая соединение 87 (38 мг, выход 18%) в виде твердого вещества белого цвета. m/z = 380,09 (М+Н), 759,17 (2М+Н).
(5-Метоксибензофуран-2-ил)метил-4-метил-2-оксо-2Н-хромен-7-илкарбамат (88) VG032-05
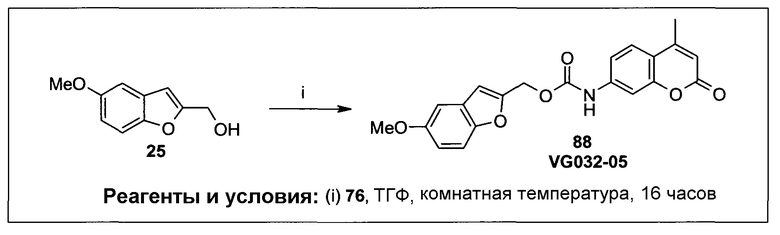
(5-Метоксибензофуран-2-ил)метанол 25 (200 мг, 1,12 ммоль) и соединение 76 (190 мг, 0,95 ммоль) растворяют в ТГФ (10 мл). Полученную смесь перемешивают при комнатной температуре в течение 15 минут и затем при комнатной температуре в течение 16 часов. ТГФ выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью гексан/EtOAc (1:1), получая соединение 88 (20 мг, выход 5%) в виде твердого вещества белого цвета. m/z = 380,13 (М+Н), 759,26 (2М+Н).
(5-Бромбензофуран-2-ил)метил-4-метил-2-оксо-2Н-хромен-7-илкарбамат (89) VG036-05
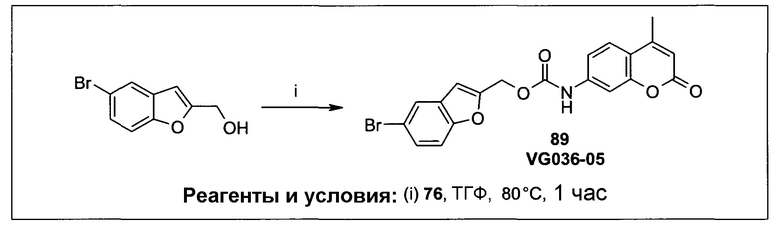
(5-Бромбензофуран-2-ил)метанол (100 мг, 0,44 ммоль) и соединение 76 (106 мг, 0,52 ммоль) растворяют в ТГФ (2 мл). Полученную смесь перемешивают при комнатной температуре в течение 15 минут и затем при температуре 80°С в течение 1 часа. ТГФ выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью CH2Cl2/EtOAc (1:1), получая соединение 89 (5,0 мг, выход 3%) в виде твердого вещества белого цвета. m/z = 429,10 (М+Н).
(5,7-Диметоксибензофуран-2-ил)метил-4-метил-2-оксо-2Н-хромен-7-илкарбамат (90) VG041-05
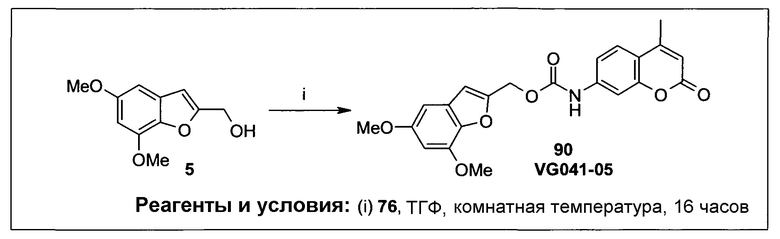
(5,7-Диметоксибензофуран-2-ил)метанол 5 (50 мг, 0,24 ммоль) и 7-изоцианато-4-метилкумарин (58 мг, 0,29 ммоль) растворяют в ТГФ (10 мл). Полученную смесь перемешивают при комнатной температуре в течение 16 часов. ТГФ выпаривают в вакууме. Остаток адсорбируют на силикагеле и очищают с помощью флэш-хроматографии, элюируя смесью CH2Cl2/EtOAc (1:1), получая соединение 90 (5,0 мг, выход 3%) в виде твердого вещества белого цвета. m/z = 410,04 (М+Н).
8. Удлиненные линкеры: простой оксибензиловый эфир, простой карбаматбензиловый эфир, оксибензилкарбамат
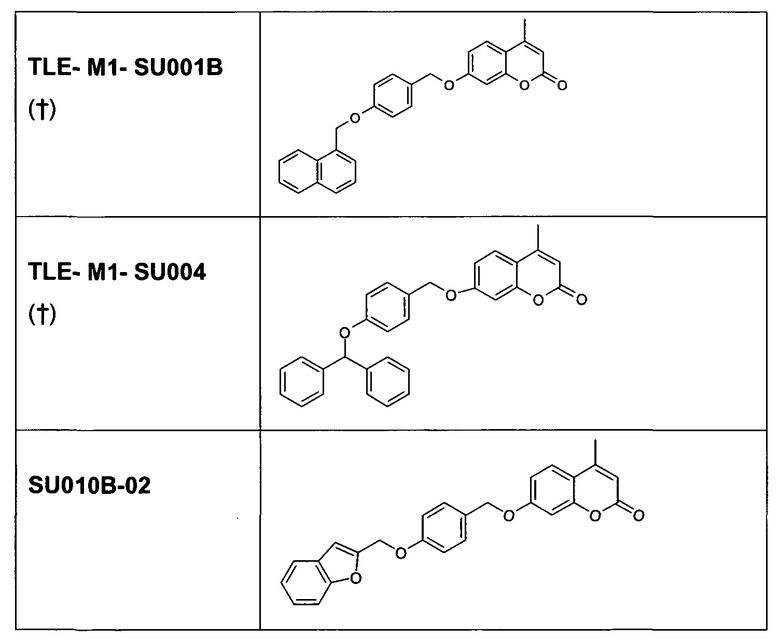
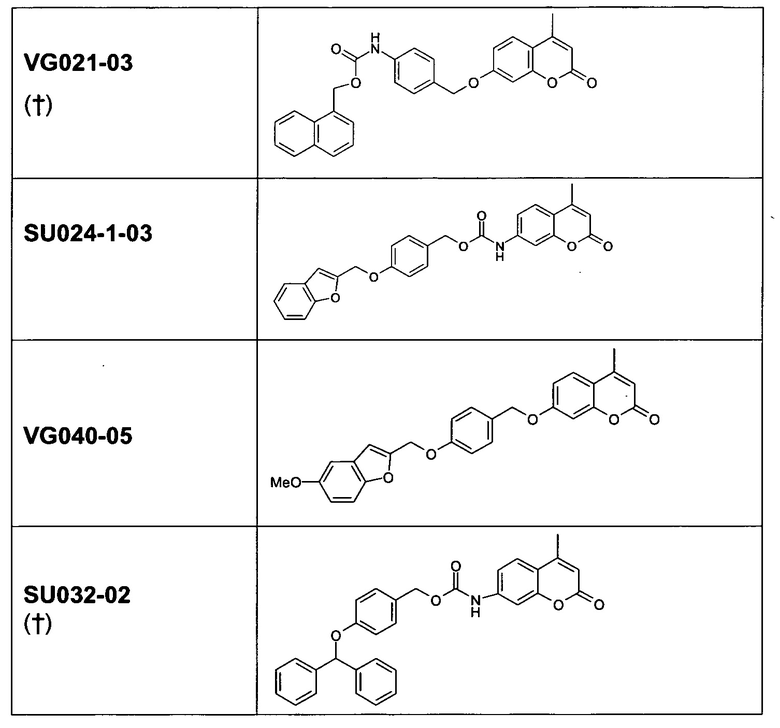
4-Метил-7-(4-нафталин-1-илметокси)бензилокси)-2Н-хромен-2-он (91) TLE-M1-SU001B
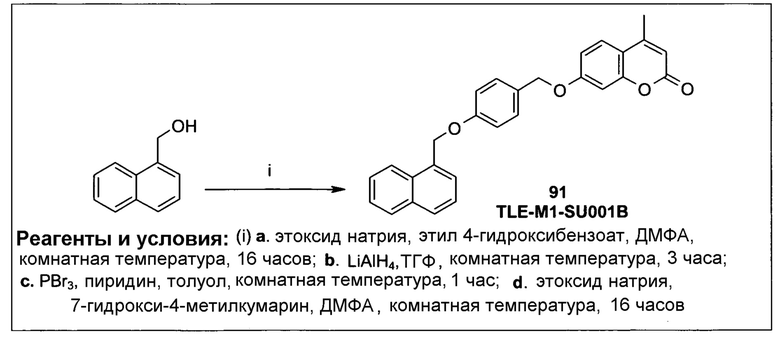
Суспензию этоксида натрия (924 мг, 13,6 ммоль) в ДМФА перемешивают при температуре 0°С в течение 10 минут. Медленно добавляют этил-4-гидроксибензоат (2,26 г, 13,6 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа, затем оставляют достигать комнатной температуры. К этой смеси по каплям добавляют 1-(бромметил)нафталин (2,0 г, 9,0 ммоль) [(предварительно растворенный в ДМФА (5 мл)]. Полученную смесь перемешивают при комнатной температуре в течение 16 часов. ДМФА выпаривают в вакууме и остаток обрабатывают EtOAc и промывают насыщенным солевым раствором, водой и 1 М раствором NaOH (2×30 мл). Органический слой сушат (MgSO4) и растворитель выпаривают в вакууме, получая этил-4-(нафталин-1-илметокси)бензоат (1,7 г, 5,55 ммоль). Этот продукт затем растворяют в ТГФ и порциями добавляют LiAlH4 (211 мг, 5,55 ммоль), при энергичном перемешивании. Суспензию перемешивают при комнатной температуре в течение 3 часов. ТГФ выпаривают в вакууме. Сырой остаток обрабатывают EtOAc и промывают водой, насыщенным солевым раствором и сушат (MgSO4). EtOAc выпаривают в вакууме, получая (4-(нафталин-1-илметокси)фенил)метанол (1,3 г, 4,9 ммоль) в виде сырого продукта. Этот продукт используют на следующей реакционной стадии без дальнейшей очистки.
(4-(Нафталин-1-илметокси)фенил)метанол (1,0 г, 3,8 ммоль) растворяют в толуоле (30 мл) и добавляют пиридин (305 мкл, 3,8 ммоль). Раствор охлаждают до температуры 0°С. По каплям добавляют PBr3 (359 мкл, 3,8 ммоль) в течение 15 минут. Смесь доводят до комнатной температуры и перемешивают в течение 1 часа. Эту смесь промывают раствором К2СО3 и экстрагируют EtOAc (3×30 мл). Этилацетатный слой промывают насыщенным солевым раствором и сушат (MgSO4). Растворитель выпаривают в вакууме, получая 1-((4-(бромметил)фенокси)метил)нафталин (660 мг, выход 53%). Этот промежуточный продукт используют в следующей реакции.
Этоксид натрия (156 мг, 2,29 ммоль) добавляют к ДМФА, при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (403 мг, 2,29 ммоль) и полученную смесь перемешивают в течение 0,5 часа и затем оставляют достигать комнатной температуры. К этой смеси порциями добавляют 1-((4-(бромметил)фенокси)метил)нафталин (500 мг, 1,53 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 16 часов. ДМФА выпаривают в вакууме и остаток обрабатывают EtOAc и промывают насыщенным солевым раствором (2×50 мл), водой (2×50 мл) и 1 М раствором NaOH (2×30 мл). Органический слой сушат (MgSO4) и очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (2:1), получая соединение 91 (200 мг, выход 31%) в виде твердого вещества белого цвета. Температура плавления = 154-156°С.
1H ЯМР (500 МГц, ацетон-d6): δ = 8,10 (1H, д, ArH), 8,00-7,99 (2H, м, Ar), 7,97-7,71 (2H, м, ArH), 7,70-7,53 (3H, м, ArH), 7,24 (1H, с, ArH), 7,09 (1H, д, CH), 6,23 (1H, с, CH), 5,68 (2H, с, CH2), 2,40 (3H, с, CH3).
13С-ЯМР (500 МГц, ацетон-d6, DEPT135): δ = 154,6 (qC), 153,4 (qC), 133,3 (qC), 131,1, 129,8, 128,9, 128,7, 128,5, 126,9, 126,7, 126,5, 126,4, 126,0, 125,9 (11 × Ar, CH), 125,3 (Ar, CH), 123,8 (Ar, CH), 114,8 (Ar, CH), 113,1 (Ar, CH), 112,7 (Ar, CH), 111,2 (Ar, CH), 111,1 (Ar, CH), 101,7 (СН), 69,6 и 67,9 (2 × СН2), 18,1 (СН3).
7-(4-(Бензгидрилокси)бензилокси)-4-метил-2Н-хромен-2-он (92) TLE-M1-SU004B
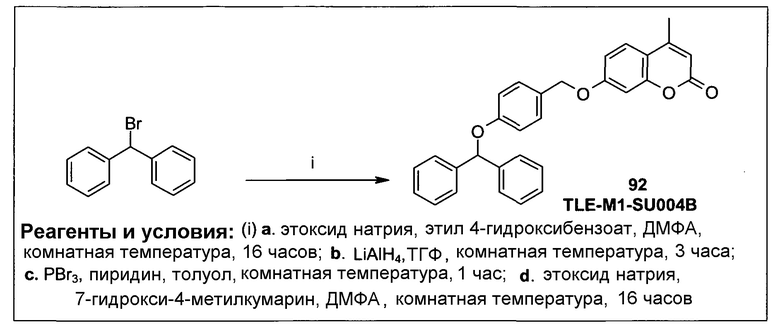
Этоксид натрия (661 мг, 9,7 ммоль) добавляют к ДМФА (5 мл), при температуре 0°С. Полученную суспензию перемешивают в течение 15 минут. Медленно добавляют этил-4-гидроксибензоат (1,61 г, 9,7 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа, затем оставляют достигать комнатной температуры. К этой смеси порциями добавляют дифенилметилбромид (2,0 г, 8,1 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 16 часов. ДМФА выпаривают в вакууме и остаток обрабатывают EtOAc и промывают насыщенным солевым раствором, водой и 1 М раствором NaOH (2×30 мл). Органический слой сушат (MgSO4) и растворитель выпаривают в вакууме, получая этил-4-(бензгидрилокси)бензоат (1,0 г, 3,0 ммоль). Это соединение затем растворяют в ТГФ (5 мл) и порциями добавляют LiAlH4 (114 мг, 3,0 ммоль), при энергичном перемешивании. Суспензию перемешивают при комнатной температуре в течение 3 часов. ТГФ выпаривают в вакууме. Сырой остаток обрабатывают EtOAc и промывают водой, насыщенным солевым раствором и сушат (MgSO4). EtOAc выпаривают в вакууме, получая (4-(бензгидрилокси)фенил)метанол (760 мг, 2,62 ммоль) в виде сырого продукта. Этот продукт используют на следующей реакционной стадии без дальнейшей очистки.
(4-(Бензгидрилокси)фенил)метанол (500 мг, 1,72 ммоль) растворяют в толуоле (20 мл) и добавляют пиридин (139 мкл, 1,72 ммоль). Раствор охлаждают до температуры 0°С. По каплям добавляют PBr3 (163 мкл, 1,72 ммоль) в течение 15 минут. Полученную смесь перемешивают при комнатной температуре в течение 1 часа. Эту смесь затем промывают насыщенным раствором К2СО3 и экстрагируют EtOAc (3×30 мл). Этилацетатный слой промывают насыщенным солевым раствором и сушат (MgSO4). Растворитель выпаривают в вакууме, получая ((4-(бромметил)фенокси)метилен)дибензол в виде масла (450 мг, выход 74%). Этот промежуточный продукт используют в следующей реакции.
Этоксид натрия (87 мг, 1,28 ммоль) добавляют к ДМФА (3 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (225 мг, 1,28 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа, затем оставляют достигать комнатной температуры. К этой смеси порциями добавляют ((4-(бромметил)фенокси)метилен)дибензол (300 мг, 1,53 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 16 часов. ДМФА выпаривают в вакууме и остаток обрабатывают EtOAc и промывают насыщенным солевым раствором (2×50 мл), водой (2×50 мл) и 1 М раствором NaOH (2×30 мл). Органический слой сушат (MgSO4) и очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (2:1), получая соединение 92 (80 мг, выход 21%) в виде твердого вещества белого цвета. Температура плавления = 179-181°С.
1H ЯМР (500 МГц, ацетон-d6): δ = 7,66 (1H, д, ArH), 7,56 (4H, д, ArH), 7,39-7,34 (6H, м, ArH), 7,08 (2H, д, ArH), 6,97 (2H, д, ArH), 6,93 (2H, д, ArH), 6,51 (1H, с, CH), 6,12 (1H, с, CH), 5,12 (2H, с, CH2), 2,42 (3H, с, CH3).
7-(4-(Бензофуран-2-илметокси)бензилокси)-4-метил-2Н-хромен-2-он (94) SU010B-02
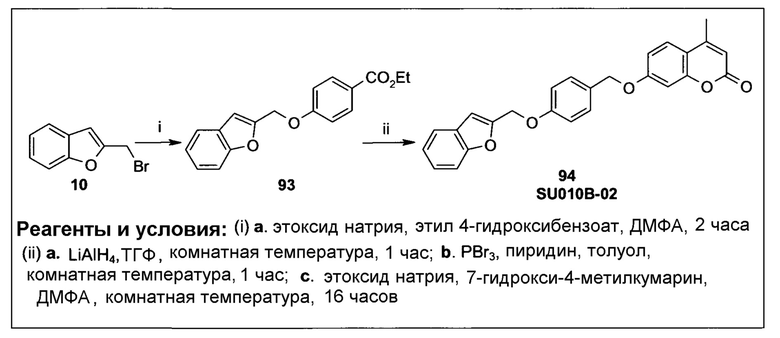
Этил-4-(бензофуран-2-илметокси)бензоат (93)
Этоксид натрия (580 мг, 8,5 ммоль) добавляют к ДМФА (10 мл), при температуре 0°С. Полученную суспензию перемешивают в течение 15 минут. Медленно добавляют этил-4-гидроксибензоат (1,4 г, 8,5 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа, затем оставляют достигать комнатной температуры. К этой смеси, по каплям, добавляют соединение 10 (1,5 г, 7,1 ммоль), предварительно растворенное в ДМФА (5 мл). Полученную реакционную смесь перемешивают при комнатной температуре в течение 2 часов. ДМФА выпаривают в вакууме и остаток обрабатывают EtOAc и промывают насыщенным солевым раствором, водой и 1 М раствором NaOH (2×30 мл). Органический слой сушат (MgSO4) и растворитель выпаривают в вакууме, получая соединение 93 в виде твердого вещества белого цвета (920 мг, выход 44%). m/z = 423,18 (М+Н).
1H ЯМР (500 МГц, ацетон-d6): δ = 8,0 (2H, д, ArH), 7,60 (1H, д, ArH), 7,30-7,15 (2H, м, ArH), 7,27-7,19 (1H, м, CH), 7,00 (2H, д, ArH), 6,80 (1H, с, CH), 5,20 (2H, с, CH2), 4,10 (2H, кв., CH2), 1,25 (3H, т, CH3).
7-(4-(Бензофуран-2-илметокси)бензилокси)-4-метил-2Н-хромен-2-он (94) SU010B-02
Соединение 93 (400 мг, 1,29 ммоль) растворяют в ТГФ (15 мл) и порциями добавляют LiAlH4 (49 мг, 1,29 ммоль), при энергичном перемешивании. Суспензию перемешивают при комнатной температуре в течение 1 часа. ТГФ выпаривают в вакууме. Сырой остаток обрабатывают EtOAc и промывают водой, насыщенным солевым раствором и сушат (MgSO4). EtOAc выпаривают в вакууме, получая (4-(бензофуран-2-илметокси)фенил)метанол (220 мг, выход 67%) в виде сырого продукта. Этот продукт растворяют в толуоле (10 мл). Раствор охлаждают до температуры 0°С. По каплям добавляют PBr3 (98 мкл, 1,04 ммоль) в течение 15 минут. Полученную смесь перемешивают при комнатной температуре в течение 1 часа. Эту смесь затем промывают насыщенным раствором К2СО3 и экстрагируют EtOAc (3×30 мл). Этилацетатный слой промывают насыщенным солевым раствором и сушат (MgSO4). Растворитель выпаривают в вакууме, получая 2-((4-(бромметил)фенокси)метил)бензофуран в виде бесцветного масла (132 мг). Этот промежуточный продукт используют в следующей реакции.
Этоксид натрия (43 мг, 0,63 ммоль) добавляют к ДМФА, при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (110 мг, 0,63 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа, затем оставляют достигать комнатной температуры. К этой смеси порциями добавляют соединение 93 (132 мг, 0,42 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 16 часов. ДМФА выпаривают в вакууме и остаток обрабатывают EtOAc и промывают насыщенным солевым раствором (2×50 мл), водой (2×50 мл) и 1 М раствором NaOH (2×30 мл). Органический слой сушат (MgSO4) и очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (2:1), получая соединение 94 (80 мг, выход 47%) в виде твердого вещества белого цвета. Температура плавления = 153-155°С. m/z = 413 (М+Н).
1H ЯМР (500 МГц, ацетон-d6): δ = 7,56-7,46 (2H, м, CH), 7,40 (1H, т, J=8,2 Гц, CH), 7,34 (2H, д, J=8,50 Гц, CH), 7,27-7,19 (1H, м, CH), 7,14-7,09 (1H, м, CH), 7,00-6,98 (2H, д, CH, 7=11,8 Гц), 6,86-6,80 (3H, м, CH), 5,99 (1H, с, CH), 5,14 (2H, с, CH2), 5,03 (2H, с, CH2), 2,28 (3H, с, CH3).
Нафталин-1-илметил-4-((4-метил-2-оксо-2Н-хромен-7-илокси)метил)фенилкарбамат (95) VG021-03
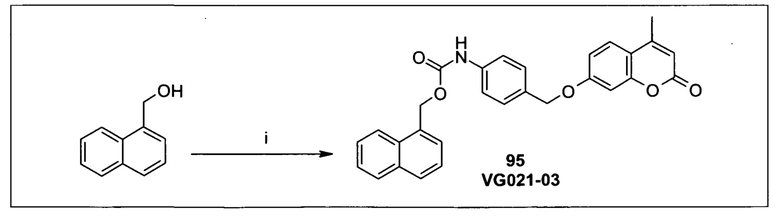

К перемешиваемому раствору нафталинметанола (4,0 г, 25,3 ммоль) в ТГФ (30 мл) добавляют ТЕА (100 мкл). К этому раствору, по каплям, добавляют этилцианобензоат (4,0 г, 21,0 ммоль), предварительно растворенный в ТГФ (10 мл). Полученный раствор перемешивают при комнатной температуре в течение 16 часов. Растворитель выпаривают, получая сырой промежуточный продукт, этил-4-((нафталин-1-илметокси)карбониламино)бензоат (1,3 г). Этот продукт затем растворяют в ТГФ (15 мл) и порциями добавляют LiAlH4 (141 мг, 3,75 ммоль), при энергичном перемешивании. Суспензию перемешивают при комнатной температуре в течение 1 часа. ТГФ выпаривают в вакууме. Сырой остаток обрабатывают EtOAc и промывают водой, насыщенным солевым раствором и сушат (MgSO4). EtOAc выпаривают в вакууме, получая (нафталин-1-илметил-4-(гидроксиметил)фенилкарбамат (500 мг, 1,62 ммоль) в виде сырого продукта. Этот продукт растворяют в толуоле (10 мл). Раствор охлаждают до температуры 0°С. По каплям добавляют PBr3 (154 мкл, 1,62 ммоль) в течение 15 минут. Полученную смесь перемешивают при комнатной температуре в течение 1 часа. Растворитель выпаривают в вакууме, получая нафталин-1-илметил-4-(бромметил)фенилкарбамат в виде масла (300 мг). Этот промежуточный продукт используют в следующей реакции без дальнейшей очистки.
Этоксид натрия (44 мг, 0,65 ммоль) добавляют к ДМФА, при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (114 мг, 0,70 ммоль) и смесь перемешивают при этой же температуре в течение 0,5 часа, затем оставляют достигать комнатной температуры. К этой смеси порциями добавляют 2-((4-(бромметил)фенокси)метил)бензофуран (200 мг, 0,42 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 16 часов. ДМФА выпаривают в вакууме и остаток обрабатывают EtOAc и промывают насыщенным солевым раствором (2×50 мл), водой (2×50 мл) и 1 М раствором NaOH (2×30 мл). Органический слой сушат (MgSO4) и очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (2:1), получая соединение 95 (160 мг, выход 63%) в виде твердого вещества белого цвета. Температура плавления = 153-155°С. m/z = 488,2 (М+Н).
1H ЯМР (500 МГц, ацетон-d6): δ = 7,56-7,46 (2H, м, CH), 7,40 (1H, т, J=8,2 Гц, CH), 7,34 (2H, д, J=8,50 Гц, CH), 7,27-7,19 (1H, м, CH), 7,14-7,09 (1H, м, CH), 7,00-6,98 (2H, д, CH, J=11,8 Гц), 6,86-6,80 (3H, м, CH), 5,99 (1H, с, CH), 5,14 (2H, с, CH2), 5,03 (2H, с, CH2), 2,28 (3H, с, CH3).
4-(Бензофуран-2-илметокси)бензил-4-метил-2-оксо-2Н-хромен-7-илкарбамат (96) (SU024-1-03)
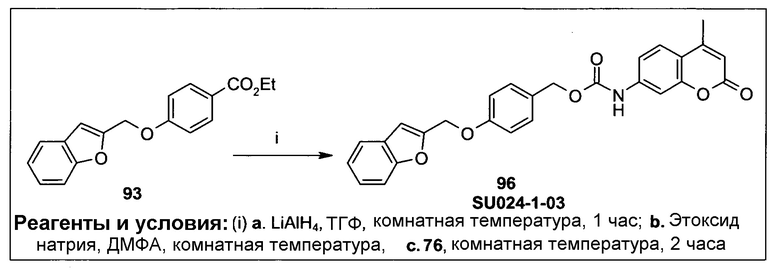
Соединение 93 (300 мг, 1,01 ммоль) растворяют в ТГФ (15 мл) и порциями добавляют LiAlH4 (38 мг, 1,01 ммоль), при энергичном перемешивании. Суспензию перемешивают при комнатной температуре в течение 1 часа. ТГФ выпаривают в вакууме. Сырой остаток обрабатывают EtOAc и промывают водой, насыщенным солевым раствором и сушат (MgSO4). EtOAc выпаривают в вакууме, получая (4-(бензофуран-2-илметокси)фенил)метанол (150 мг, 0,59 ммоль) в виде сырого продукта. Этот продукт используют на следующей стадии без дальнейшей очистки.
Этоксид натрия (40 мг, 0,59 ммоль) добавляют к ДМФА, при температуре 0°С, и суспензию перемешивают в течение 10 минут. Добавляют (4-(бензофуран-2-илметокси)фенил)метанол (150 мг, 0,59 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа, затем оставляют достигать комнатной температуры. К этой смеси порциями добавляют соединение 76 (130 мг, 0,65 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 2 часов. ДМФА выпаривают в вакууме. Сырой остаток очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (2:1), получая целевое соединение (20 мг, выход 8%) в виде твердого вещества белого цвета. m/z=456,10 (М+Н).
1H ЯМР (500 МГц, ДМСО-d6): δ = 10,22 (1H, ушир.с, NH), 7,69-7,64 (2H, м, ArH), 7,58 (1H, д, J=8,15 Гц, ArH), 7,55 (1H, с, ArH), 7,42 (2H, д, J=8,00 Гц, ArH), 7,33 (1H, т, J=7,70 Гц, ArH), 7,26 (1H, т, J=7,5 Гц, ArH), 7,11 (2H, д, J=8,28 Гц, ArH), 7,06 (1H, с, ArH), 6,23 (1H, с, ArH), 5,29 (2H, с, CH2), 5,12 (2H, с, CH2), 2,37 (3H, с, CH3).
7-(4-((5-Метоксибензофуран-2-ил)метокси)бензилокси)-4-метил-2Н-хромен-2-он (97) VG040-05
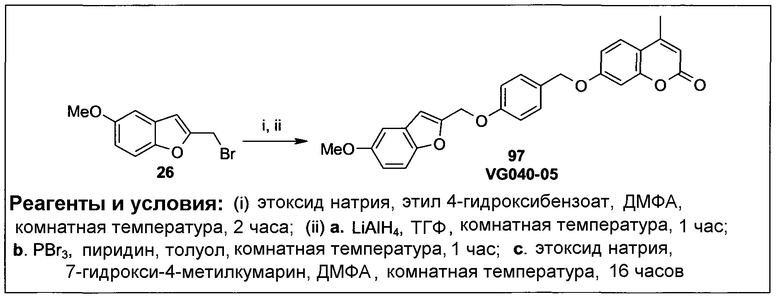
Этоксид натрия (62 мг, 0,90 ммоль) добавляют к ДМФА (3 мл), при температуре 0°С. Полученную суспензию перемешивают в течение 15 минут. Медленно добавляют этил-4-гидроксибензоат (148 мг, 0,90 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа, затем оставляют достигать комнатной температуры. К этой смеси добавляют соединение 26 (180 мг, 0,75 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 1 часа. ДМФА выпаривают в вакууме, получая сырой промежуточный продукт (150 мг). Этот продукт затем растворяют в ТГФ (5 мл) и порциями добавляют LiAlH4 (34 мг, 0,90 ммоль), при энергичном перемешивании. Суспензию перемешивают при комнатной температуре в течение 1 часа. ТГФ выпаривают в вакууме. Сырой остаток обрабатывают EtOAc и промывают водой, насыщенным солевым раствором и сушат (MgSO4). EtOAc выпаривают в вакууме, получая (4-((5-метоксибензофуран-2-ил)метокси)фенил)метанол (120 мг) в виде сырого продукта. Этот продукт растворяют в толуоле (4 мл). Раствор охлаждают до температуры 0°С. По каплям добавляют PBr3 (84 мкл, 1,04 ммоль) в течение 5 минут. Полученную смесь перемешивают при комнатной температуре в течение 1 часа. Растворитель выпаривают в вакууме, получая 2-((4-(бромметил)фенокси)метил)-5-метоксибензофуран в виде бесцветного масла (90 мг). Этот промежуточный продукт используют в следующей реакции.
Этоксид натрия (22 мг, 0,31 ммоль) добавляют к ДМФА (2 мл), при температуре 0°С, и суспензию перемешивают в течение 10 минут. Медленно добавляют 7-гидрокси-4-метилкумарин (54 мг, 0,31 ммоль) и полученную смесь перемешивают при этой же температуре в течение 0,5 часа. К этой смеси порциями добавляют 2-((4-(бромметил)фенокси)метил)-5-метоксибензофуран (90 мг, 0,23 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 16 часов. ДМФА выпаривают в вакууме и остаток очищают с помощью флэш-хроматографии, элюируя смесью гексан:EtOAc (1:1), получая соединение 94 (22 мг, выход 7%), в виде твердого вещества белого цвета. m/z = 443 (М+Н).
4-(Бензгидрилокси)бензил-4-метил-2-оксо-2Н-хромен-7-илкарбамат (98) SU032-02
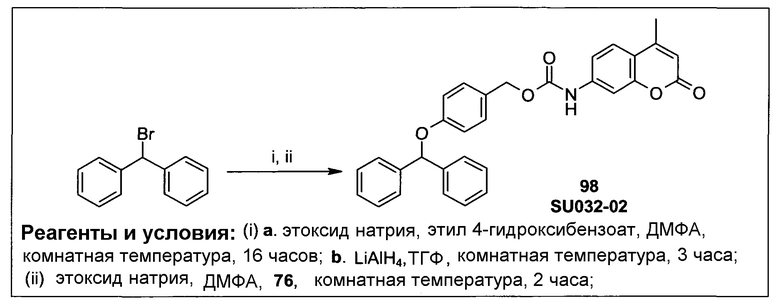
Этоксид натрия (300 мг, 4,05 ммоль) добавляют к ДМФА (10 мл), при температуре 0°С. Полученную суспензию перемешивают в течение 10 минут. Медленно добавляют этил-4-гидроксибензоат (739 мг, 4,05 ммоль) и полученную смесь перемешивают при этой температуре в течение 20 минут. К этой смеси порциями добавляют дифенилметилбромид (1,0 г, 4,05 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 16 часов. ДМФА выпаривают в вакууме и остаток обрабатывают этилацетатом и промывают водой и солевым раствором. Органический слой сушат (MgSO4) и растворитель выпаривают в вакууме, получая 4-(бензгидрилокси)бензоат (830 мг). Его затем растворяют в ТГФ (5 мл) и порциями при интенсивном перемешивании добавляют LiAlH4 (114 мг, 3,0 ммоль). Суспензию перемешивают при комнатной температуре в течение 3 часов. ТГФ выпаривают в вакууме. Сырой остаток обрабатывают этилацетатом и промывают водой, солевым раствором и сушат (MgSO4). Этилацетат выпаривают в вакууме, получая (4-(бензгидрилокси)фенил)метанол (760 мг, 2,62 ммоль) в виде сырого продукта. Его используют на ближайшей реакционной стадии без дальнейшей очистки.
Раствор (4-(бензгидрилокси)фенил)метанола (100 мг, 0,34 ммоль) и соединения 76 в толуоле (10 мл) кипятят с обратным холодильником в течение 2 часов. Реакционную смесь охлаждают до комнатной температуры и выпавший в результате осадок отфильтровывают и промывают холодным диэтиловым эфиром и этилацетатом, получая соединение 98 (100 мг, 60%) в виде твердого вещества белого цвета. m/z = 491,55 (М+Н).
Биологическая активность
Пример 1: CYP1B1-метаболизм пролекарств
Воздействие заместителя на фрагментацию связанных через посредство бензофуран-простой эфир-карбамат кумаринов с помощью CYP1-изоферментов и человеческих печеночных микросом (HLM)
Коммерчески доступный SupersomalTM CYP1A1, CYP1A2, CYP1B1 и объединенные в общий фонд человеческие печеночные микросомы (выпускаются фирмой BD Gentest, Оксфорд, Великобритания) включены в ферментативный скрининг в отношении идентификации взаимосвязей активности со структурой (SAR), лежащих в основе структурных свойств, которые контролируют эффективность и селективность пролекарственной фрагментации с помощью CYP1B1, экспрессирующегося в карциноме, по сравнению с ферментами цитохром Р450, экспрессирующимися в нормальных тканях, включая печень. HLM происходят от печени человека и, согласно поставщику, содержат комплект цитохромов Р450, включая CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2D6, CYP2E1, CYP3A4 и CYP4A, но не CYP1A1 или CYP1B1.
В случае типичных исследований метаболизма фермента SupersomalTM CYP1A1, CYP1A2, CYP1B1 использут 10 пмоль фермента, 100 мкмоль/дм3 NADPH в 10 ммоль/дм3 калийфосфатного буфера с рН 7,4 и температуру 37°С. Метаболизм фермента SupersomalTM инициируют добавлением исходного раствора пролекарства, растворенного в ДМСО, с получением конечной концентрации 10 мкмоль/дм3 пролекарства и 0,5% ДМСО. Для скрининга HLM используют 60 микролитров микросом, 100 мкмоль/дм3 NADPH в 10 ммоль/дм3 калийфосфатного буфера с рН 7,4 и температуру 37°С, в общем реакционном объеме 1,5 мл.
Соединения согласно изобретению включают ряд гетероароматических триггеров, связанных простыми эфирными и карбаматными линкерами с гидроксильной группой 7-гидрокси-4-метилкумарина и 7-амино-4-метилкумарина, соответственно. Дальнейшие примеры согласно изобретению включают соединения, где гетероароматические триггеры связаны через так называемый расширенный оксибензил-простой эфир - линкер (-Ar-CH(Z7)X3- = -фенил-СН2О-) с гидроксильной группой 7-гидрокси-4-метилкумарина. Дальнейшие примеры согласно изобретению включают соединения, где гетероароматические триггеры связаны через так называемый расширенный оксибензилкарбаматный линкер с аминогруппой 7-амино-4-метилкумарина. Дальнейшие примеры согласно изобретению включают соединения, где гетероароматические триггеры связаны через карбамат-бензил-простой эфир - линкер с гидроксильной группой 7-гидрокси-4-метилкумарина.
Как 7-гидрокси-4-метилкумарин, так и 7-амино-4-метилкумарин частично депротонированы при физиологическом значении рН 7,4 и оба кумаринаниона являются высокофлуоресцентными с эмиссией флуоресценции с максимумами при длине волны 450 и 445 нм, соответственно. Когда кумарины связаны с гетероароматическими триггерами через линкеры, описанные согласно данному изобретению, флуоресценция кумарин-аниона гасится. Следовательно, ферментативное гидроксилирование гетероароматического триггера и получаемая фрагментация линкера могут быть контролируемы в реальное время за счет высвобождения кумарин-аниона путем кинетической флуориметрии. Эту стратегию дизайна пролекарства удачно использовали для мониторинга фрагментации так называемых биовосстановительных, активируемых гипоксией, пролекарств с помощью Р450-редуктазы, не смешиваемой с CYP1B1, которая представляет собой фермент монооксигеназу (см., например, Everett S.A. и др. «Modifying rates of reductive elimination of leaving groups from indolequinone prodrugs: a key factor in controlling hypoxia-selective drug release», Biochem. Pharmacol., 63, 1629-39 (2002)).
Высвобождение кумарин-аниона, указывающее на фрагментацию линкера, контролируют, используя флуоресцентную ячейку с длиной пути 1 см, в кинетическом флуориметре Cary Eclipse c набором щелей возбуждения и эмиссии по 5 нм. Высвобождение кумарин-аниона из соединений согласно изобретению детектировали при длине волны возбуждения λех=350 нм и длине волны эмиссии λem=450 нм. Изменение интенсивности флуоресценции количественно определяли по линейной калибровочной кривой интенсивности флуоресценции в отношении концентрации кумарина (0-3,5 мкмоль/дм3) в 10 ммоль/дм3 калийфосфатном буфере с рН 7,4, используя те же самые установочные параметры, как в случае метаболизма фермента.
Удельные активности в отношении фрагментации (на пмоль кумарина мин-1 пмоль цитохрома Р450-1) для CYP1-изофермента и HLM-активируемой фрагментации и высвобождение кумарина из бензофуран-простой эфир-карбамат-связанных кумаринов представлены в таблице 3. Электронодонорный заместитель (Ме, МеО) или электроноакцепторные заместители (Cl, Br, F) в одном или обоих из положений 5 и 7 бензофурана оказывают значительное воздействие как на специфичность, так и эффективность фрагментации. Положения 4 и 6 в бензофуране остаются незамещенными (R4 и R6 = Н), так как они, вероятно, представляют собой положения для ферментативного гидроксилирования, необходимого для индуцирования фрагментации простого эфирного или карбаматного линкера согласно предлагаемому механизму. Взаимосвязь активности со структурой (SAR), определяющая влияние заместителя в положениях 5 и 7 бензофурана на эффективность и селективность индуцируемой CYP1-изоферментом фрагментации, не прогнозируема для фрагментации как простого эфирного, так и карбаматного линкера.
SU10A (см. нижеприводимую таблицу 3), где Z3 = H, Z5 = H, которые несет связанный через образование простого эфира кумарин, фрагментируется с помощью CYP1A1, CYP1A2 и CYP1B1, а также HLM. В случае HLM, включение 10 мкмоль/дм3 α-нафтофлавона (CYP1-селективный ингибитор фермента) ингибирует фрагментацию, что указывает на то, что CYP1A2 является ответственным за опосредуемое HLM высвобождение кумарина. Следовательно, бензофуран является генетической триггерной составляющей, которая может облегчать фрагментацию связанных простой эфирной связью пролекарств с помощью CYP1-изоферментов.
Электроноакцепторные заместители у бензофурана в VGO16-04 (см. также нижеприводимую таблицу 3), где Z3 = F, Z5 = F, ингибируют индуцируемую CYP1-изоферментом и HLM фрагментацию простого эфирного линкера. Однако, электронодонорные заместители в VG035-05 (см. также нижеприводимую таблицу 3), где Z3 = МеО, Z5 = МеО, приводят к CYP1B1-специфической фрагментации простой эфирной связи, так как не обнаруживают фрагментации линкера в случае CYP1A или HLM. Удельная активность в отношении фрагментации в случае VG035-05 с помощью CYP1B1 составляет 13,65±1,00 пмоль кумарина мин-1 на пмоль цитохрома Р450-1, представляет собой самую высокую эффективность различных связанных с бензофураном простой эфирной связью кумаринов, которая исследована (см. нижеприводимую таблицу 3, ниже). Следовательно, 5,7-диметоксибензофурановый остаток может быть использован для специфического триггера фрагментации связанных через посредство образования простого эфира пролекарств с помощью CYP1B1-фермента, который сверхэкспрессируется в карциноме.
В противоположность VG035-04 (который содержит связанный через посредство образования простого эфира кумарин), VG041-05 (который содержит соответствующий, связанный через посредство карбамата, кумарин), где Z3 = MeO, Z5 = MeO, селективно фрагментируется с помощью CYP1A1 (но не CYP1A2 или CYP1B1) с удельной активностью в отношении фрагментации, составляющей 5,51±0,06 пмоль кумарина мин-1 на пмоль цитохрома Р450-1. Согласно нижеприводимой таблице 3, все приведенные в качестве примеров соединений структуры В, содержащие карбаматный линкер, фрагментируются с помощью CYP1A1, но не CYP1B1. Единственное исключение составляет VG032-05, где Х3 = МеО, Z5 = МеО, показывающий удельную активность в отношении фрагментации за счет CYP1B1, составляющую 1,53±0,09 пмоль кумарина мин-1 на пмоль цитохрома Р450-1, которая приблизительно в 6 раз ниже, чем в случае VG027-05, который содержит линкер в виде простого эфира.
Пример 2: Комбинация библиотеки модельного пролекарства с моделью прогнозирования CYP1B1-субстрата соединяет специфичность субстрата с активацией и фрагментацией пролекарства
Выполнение высокоспецифичного исследования для построения базы данных для CYP1B1
Фермент-мишень CYP1B1 подвергали скринингу при использовании двух коммерчески доступных библиотек, включая коллекцию 50000 тест-соединений ChemDiv Diversity и коллекцию 10000 тест-соединений ChemDiv Kinase Targeted, c целью идентификации активности дифференцирующих субструктур и большого набора данных по биоактивности, из которых создается модель специфичности субстрата. HTS выполняли при использовании миниатюризованного 384-луночного планшета, используя жидкостной манипулятор (Beckman FXp), дозаторы массы (Matrix Wellmate) и многоканальный люминесцентный спектрофотометр для прочтения планшетов (Molecular Devices Analyst AD plate Reader). Р450-GloTM-анализы выполняются люминесцентным методом для измерения активности цитохрома Р450. Стандартную реакцию осуществляли путем инкубации рекомбинантного фермента человеческого суперсомального CYP1B1 плюс редуктаза (BD GentestTM, Великобритания) вместе с люминогенным субстратом цитохрома Р450, а именно 6’-хлорэтиловый простой эфир люциферина (люциферин-CEE), который представляет собой субстрат для CYP1B1, но не для люциферазы. Люциферин-СЕЕ превращается в продукт люциферин, который детектируют по второй реакции с реагентом для детектирования люциферина (набор для люминесцентного анализа CYP1B1, P450-GloTM, выпускается фирмой Promega, Мэдисон, США). Реагент одновременно прекращает реакцию цитохрома Р450 и инициирует люминесцентный сигнал с периодом полупревращения >2 часов. Количество свечения, продуцируемое во второй реакции, пропорционально активности CYP1B1. Биохимической конечной точкой являлось ингибирование субстрата фермента (0,5 пмоль/лунка), достигаемое при кажущейся Km для люциферина-СЕЕ (20 мкмоль/дм3). Анализ характеризуется превосходными Z’-факторами, обычно больше, чем 0,6 (где Z’=1,0 означает вполне надежный высоковоспроизводимый анализ), когда работают с использованием 384-луночного планшета. Отрицательным контролем являлся уровень активности, который определяет немодифицированное состояние мишени-фермента, тогда как положительный контроль представлял собой уровень активности, который определяет попадание. Отрицательный контроль содержит реакционную смесь CYP1B1/KPO4/NADPH/субстрат и эквивалентную концентрацию 1% ДМСО, используемую для солюбилизации тест-соединений. Положительный контроль в случае анализа содержит реакционную смесь CYP1B1/KPO4/NADPH/субстрат с α-нафтофлавоном, который полностью ингибирует активность CYP1B1-фермента при конечной концентрации 5 мкмоль/дм3. Положительный и отрицательный контроли помещают в наружные колонки каждого 384-луночного планшета с тест-соединениями, помещенными в остающиеся 320 лунок. Определением попадания является тест-соединение, которое представляет собой субстратный ингибитор активности CYP1B1 на 80-100% при концентрации 0,5 мкмоль/дм3.
Конвейерный пилот (Scitegic, Сан-Диего, США) использовали для упрощения и интеграции большого количества данных по идентичности SAR из CYP1B1 HTS, подтверждаемых специалистами в области вычислительной техники в случае UCF SMDC. Программное обеспечение использовали для идентификации (1) предварительных SAR результатов попаданий против непопаданий, (2) определения физико-химических свойств, как, например, молекулярная масса, вычисленный log P, взаимодействия донор/акцептор популяции попаданий, (3) определения частоты циклических фрагментов и функциональных групп и (4) определения in silico модели для прогнозирования ингибирования CYP1B1-субстрата в качестве базиса дизайна будущего пролекарства. Важным является то, что s, значительное число попаданий, ~10% допускает молекулярную массу 400-500, последняя является максимальной молекулярной массой тест-соединений, пригодной в обеих коллекциях соединений. Эта информация определяет максимальную молекулярную массу пролекарства, допустимую, пока сохраняется специфичность CYP1B1-субстрата. Структурный анализ клеточных каркасов попадания в SARvision v2 из CHEMAPPSTM (La Jolla, CA, США) подтверждает, что тест-соединения не содержат соответствующую функциональную группу (например, триггерный гироксиметильный заместитель) для прямой интеграции в химию связывания, указанную на схеме 1. Однако, в случае идентификации циклических фрагментов, с высокой частотой в случае попаданий против непопаданий, можно идентифицировать образцы для триггерных частей, которые затем могут быть функционализированы подходящим образом для реакций связывания.
Модель in silico для прогнозирования ингибирования субстрата цитохрома Р450 в подтверждение дизайна пролекарства
Главной проблемой в дизайне пролекарства является определение стратегии интеграции с точки зрения химии триггера, линкера и эффектора, пока поддерживается специфичность субстрата к ферменту-мишени. CYP1B1 HTS является в высшей степени ценным в случае идентификации потенциальных триггерных составляющих, но при последующей химии «управляемого попадания», включая линкерное и эффекторное лекарственное средство, следует иметь в виду, что конечная пролекарственная структура не должна быть оптимальной для активации фермента-мишени. Оптимальное использование большого количества структурных данных из скринингов HTS (равняющегося 60000 тест-соединений) достигнуто за счет разработки in silico модели прогнозирования ингибирования субстрата цитохрома P4501B1, используя Gaussian Kernel позиционный алгоритм «k-ближайшего соседа (k-NN)», базирующийся на поисках подобия согласно Tanimoto по расширенному методу «отпечатков пальцев». Оптимальные параметры позиционной k-NN ядра CYP1B1 модели выбирали, используя подтверждение правильности «leave-one-out-cross» в случае специальной совокупности, выбираемой из 45000 и 9000 тест-соединений из библиотек «ChemDiv Diverse and Kinase». Остальную часть тест-соединений, в целом 6000, использовали в качестве набора для внутреннего теста в целях подтверждения правильности модели для прогнозируемого субстратного ингибирования. Любые тест-соединения, проявляющие >20%, но <80% ингибирования, обозначали неклассифицированными. Правильная модель прогнозировала 89% неклассифицированных субстратных ингибиторов и 95% классифицированных субстратных ингибиторов. Протокол модели прогнозирования CYP1B1-субстрата загружали в главную систему «Scitegic Web Port» для облегчения стыковки предполагаемых пролекарственных структур через интерфейс с пакетом чертежей стандартной химии, таких как, например, ChemDraw/IsisDraw.
Подтверждение правильности модели прогнозирования CYP1B1-субстрата, используя набор для внутреннего теста соединений
384-луночный исходный планшет, предназначаемый для набора в отношении внутреннего теста для модели прогнозирования CYP1B1-субстрата, конструировали и включали (1) известные ингибиторы CYP1B1-субстрата, включающие, например, тетраметоксистильбен, β-эстрадиол, α-нафтофлавон, этоксирезоруфин, ресвератрол, (2) соединения, которые не являются ингибиторами CYP1B1-субстрата, включая, например, хинидин (эффективный специфический ингибитор CYP2D6), сульфафеназол (эффективный специфический ингибитор CYP2C9), и (3) модельные пролекарства VG016-05 и VG035-05 и (4) мустинфосфорамидатные пролекарства SU025-04 и SU046-04. Исходная концентрация набора для внутреннего теста составляла 10 ммоль/дм3 в ДМСО и процент ингибирования CYP1B1-субстрата в конечной концентрации 0,5 ммоль/дм3 определяли, используя те же самые методы, как описанные для основного CYP1B1 HTS. Эксперименты осуществляли в тройном повторении для получения среднего % субстратного ингибирования активности CYP1B1 ± стандартное отклонение. Все структуры набора для внутреннего теста представляли на рассмотрение в виде запросов в отношении модели прогнозирования CYP1B1-субстрата через Scitegic Web Port для генерирования прогнозируемых значений % субстратного ингибирования CYP1B1 для сравнения с действительным биохимическим измерением % ингибирования субстрата.
Сравнительные действительные и прогнозированные значения % ингибирования субстрата для CYP1B1 были следующими:
Модель прогнозирования CYP1B1-субстрата являлась правильной в случае прогнозируемого % субстратного ингибирования CYP1B1 многочисленными классами соединений с широким диапазоном активности, что подтверждает правильность модели при использовании набора для внутреннего теста соединений. Является важным, с точки зрения уровня изобретения, что модель точно способна прогнозировать активность двух моделей пролекарств VG016-05 и VG035-05 с точки зрения ингибирования CYP1B1-субстрата, которое может быть прямо связано с эффективностью фрагментации пролекарства и высвобождения аниона 7-гидрокси-4-метилкумарина. Согласно таблице 3, электронодонорные или электроноакцепторные заместители в случае R5 и R7 бензофуранового триггера активируют эти модельные пролекарства до ароматического гидроксилирования и фрагментации. Когда R5 и R7 означают F, т.е. электроноакцепторные заместители, как в VG016-05, модельное пролекарство не активируется CYP1B1, как правильно прогнозировано, и, как следствие, не происходит фрагментации линкера. Заметной противоположностью является то, что, когда R5 и R7 означают МеО, т.е. электронодонорные заместители, как в случае VG035-05, модельное пролекарство активируется CYP1B1, как точно прогнозировано, приводя к фрагментации линкера с высокой эффективностью. Включение диметоксибензофурановой триггерной составляющей в мустинфосфорамидатные пролекарства SU025-04 и SU046-04 генерирует соединения, которые должны быть точно прогнозированы как хорошие ингибиторы субстрата CYP1B1. В заключение, комбинация библиотек модельных пролекарств и моделей прогнозирования CYP1B1-субстрата, базирующаяся на базе данных по биоактивности CYP1B1, облегчает дизайн специфически активируемых CYP1B1 пролекарств.
Пример 3: Цитотоксичность пролекарства в случае клеток СНО дикого типа и генноинженерных клеток СНО для экспрессирования изоферментов CYP1A1 и CYP1B1
Генноинженерные СНО-клетки использовали для демонстрации селективного клеточного киллинга, опосредуемого экспрессией CYP1. В нижеописываемых экспериментах соединения подвергали воздействию на генноинженерные клетки СНО дикого типа для экспрессирования либо CYP1A1 (CHO/CYP1A1), либо CYP1B1 (CHO/CYP1B1) ферментов.
СНО-клетки: клетки яичника китайского хомячка (СНО) DUKXB11 выращивали в стандартных условиях культивирования клеток в α-МЕМ, дополненной 10% FCS, 1 Ед/мл каждого из гипоксантина и тимидина, и пенициллином (100 Межд.Ед./мл) и стрептомицином (100 мкг/мл), в соответствии с известными из литературы методами (Ding S. и др., Arch. Biochem. Biophys., 348, 403-410 (1997), содержание которых включено в данный контекст путем ссылки). Клетки выращивали при температуре 37°С в увлажненной атмосфере плюс 5% СО2.
Клетки CHO/CYP1A1 и CHO/CYP1B1: СНО-клетки, содержащие рекомбинантный CYP1A1 и рекомбинантный CYP1B1, коэкспрессирующие Р450-редуктазу, а именно (CHO/CYP1A1) и (CHO/CYP1B1), соответственно, культивировали, используя стандартную культуральную среду для СНО-клеток, дополненную с помощью 0,4 мг/мл G418-дисульфата и 0,3 мкМ метотрексата (Sigma/Aldrich Co., Gillingham, Dorset, Великобритания), согласно описанным в литературе методам (см. выше). Клетки выращивали при температуре 37°С в увлажненной атмосфере плюс 5% СО2.
Экспрессия рекомбинантного CYP1A1 и CYP1B1
Амплификацию гена дигидрофолатредуктазы (DHFR) либо человеческого кДНК-CYP1A1, либо кДНК-CYP1B1 в СНО-клетках использовали для достижения высоких уровней функционального фермента, когда коэкспрессируют с человеческой Р450-редуктазой (там же; Ding S. и др., Biochem. J., 356 (часть 2), 613-619 (2001)). Модифицированную CYP1A1- или CYP1B1-кДНК рестриктировали и лигировали с вектором экспрессии млекопитающего pDHFR с генерированием плазмид pDHFR/1A1 и pDHFR/1B1, соответственно (там же). Трансфекцию клеточной культуры и ДНК до CHO-DUKXB11 осуществляли согласно методам, описанным в литературе, и трансфицированные клетки селекционировали для фенотипа DHFR+ путем культивирования в среде с дефицитом нуклеозида (там же). Клоны DHFR+ группировали и культивировали при возрастающих концентрациях МТХ (0,02-1 мкМ) для амплификации тренсфицированной CYP1A1- или CYP1B1-кДНК. Клеточные клоны, которые пережили селекцию в 0,1 МТХ, выделяли, затем селекционировали далее с помощью 0,3 мкМ МТХ. Полученные клеточные линии анализировали в отношении экспрессии CYP1A1 и CYP1B1 путем иммуноблоттинга. Клеточные линии, экспрессирующие высокий уровень каждого фермента, стабильно трансфицировали с помощью плазмиды pcDNA/HR, содержащей полной длины человеческий цитохром-Р450-редуктаза(CPR)-кДНК, и селекционировали с помощью G418 (0,8 мг/мл) и МТХ (0,3 мкМ) согласно методам, описанным в литературе (там же). После выделения резистентных клонов концентрацию G418 уменьшали до 0,4 мг/мл и гомогенность клеточных линий обеспечивали путем повторяющегося клонирования. СНО-клеточную линию трансфицировали с помощью несущего плазмиду кДНК-CYP1A1, затем трансфицировали с помощью CPR-кДНК, обозначали СНО/CYP1A1 и СНО-клеточную линию трансфицировали с помощью несущего плазмиду кДНК-CYP1B1, затем трансфицировали с помощью CPR-кДНК, обозначали СНО/CYP1B1.
Иммунохимическое детектирование CYP1A1 и CYP1B1
Клетки собирали и лизировали путем обработки ультразвуком, используя стандартные методы, известные в литературе (Ding S. и др., 1997, содержание которых включено в данный контекст путем ссылки). Белки (обычно 50 мкг лизата) подвергали разделению путем электрофореза в полиакриламидном геле с додецилсульфатом натрия (SDS/PAGE), переносили на нитроцеллюлозную мембрану и зондировали, используя стандартные методы (Paine M.J. и др., Arch. Biochem. Biophys., 328, 380-388 (1996), содержания которых включены в данный контекст путем ссылки). Человеческий CYP1A1 плюс редуктаза SupersomesTM, человеческий CYP1A2 плюс редуктаза SupersomesTM и CYP1B1 плюс редуктаза SupersomesTM (BD Biosciences, Оксфорд, Великобритания) использовали в качестве положительных контролей (типично 0,03-0,3 пмоль) для иммунохимического детектирования экспрессии фермента в клеточных линиях. Первичное антитело WB-1B1 (разведение 1:1500, BD Biosciences, Оксфорд, Великобритания) и анти-CYP1A2-антитело, которое перекрестно реагирует с CYP1A1 (разведение 1:2000, Cancer Research Technology, Лондон, Великобритания) использовали для детектирования экспрессии CYP1B1 и CYP1A1, соответственно. Вторичное антитело представляло собой козье антитело против кроличьего IgG, используемое при разведении 1:500. Иммуноблоты обнаруживали, используя набор для детектирования по методу Вестерн-блоттинга с использованием усиленной хемилюминесценции (ECL) (GE Healthcare Life Sciences, Amersham, Buckinghamshire, Великобритания).
Характеристика путем Вестерн-блоттинга экспрессии CYP1A1 и CYP1B1 в случае генноинженерных СНО-клеток
Фиг.1а из сопровождаемых чертежей представляет собой типичный вестерн-блоттинг, показывающий детектирование экспрессии CYP1B1-белка в лизате из СНО/CYP1B1-клеточной линии, которая не детектируема ни в каких нетрансфицированных СНО DUKXB11-клетках, а также не в клеточной линии СНО/CYP1A1. Полоса соответствует молекулярной массе 56 кДа и подходит полосе человеческого фермента CYP1B1 SupersomalTM. Полоса 1b представляет собой типичный вестерн-блоттинг, показывающий детектирование экспрессии CYP1A1-белка в лизате из СНО/CYP1A1-клеточной линии, которая не детектируема ни в каких нетрансфицированных СНО DUKXB11-клетках, а также не в клеточной линии СНО/CYP1B1. Полоса соответствует молекулярной массе 60 кДа и подходит полосе человеческого фермента CYP1A1 SupersomalTM, детектируемого путем перекрестной реактивности анти-CYP1A2 антитела.
Функциональная активность фермента CYP1
Анализ в отношении О-деэтилирования этоксирезоруфина (EROD) широко используется для подтверждения функциональной активности CYP1 (Chang T.K. and Waxman D.J. «Enzymatic Analysis of cDNA-Expressed Human CYP1A1, CYP1A2 and CYP1B1 with 7-Ethoxyresorufin as Substrate», Methods Mol. Biol., 320, 85-90 (2006), содержания которого включены в данный контекст путем ссылки). Анализ определяет О-деалкилирование 7-этоксирезоруфина за счет CYP1A1, CYP1A2 и CYP1B1 с образованием ферментативного продукта резоруфина, который непрерывно контролируют по эмиссии флуоресценции при длине волны 580 нм. Альтернативный анализ для определения активности фермента представляет собой коммерчески доступный анализ Promega P450-GloTM, при котором используют люциферин-СЕЕ в качестве люминогенного субстрата для CYP1-ферментов, согласно Cali J.J. и др., Expert. Opin. Drug Metabolism Toxicol., 2(4), 629-45 (2006), содержания которого включены в данный контекст путем ссылки. EROD-анализ и Promega P450-GloTM-анализ с селективным и неселективным ингибиторами CYP1 использовали для подтверждения того, что СНО-клеточные линии, упомянутые выше, экспрессируют ожидаемые CYP1-ферменты в функциональной форме.
В отсутствие ингибиторов, СНО/CYP1A1 и СНО/CYP1B1 (но не клетки СНО дикого типа) превращают 7-этоксирезофурин в резофурин или люциферин-СЕЕ в люциферин, таким образом подтверждая функциональную экспрессию CYP1 в этих клетках (см. нижеприводимую таблицу 1).
Как ожидалось, добавление ингибитора CYP1 широкого спектра, α-нафтофлавона, уничтожает активность в случае обеих CYP1-экспрессирующих клеточных линий (см. нижеприводимую таблицу 1). Селективный ингибитор, тетраметоксистильбен, является в 30 раз селективнее для CYP1B1 относительно CYP1A1 (Chun Y.J., Kim S., Kim D., Lee S.K. и Guengerich F.P. «A New Selective and Potent Inhibitor of Human Cytochrome P450 1B1 and its Application to Antimutagenesis», Cancer Res., 61(22), 8164-70 (2001)). Тетраметоксистильбен уничтожает активность при высоких концентрациях в случае обеих клеточных линий, экспрессирующих CYP1, и, предпочтительно, уменьшает активность в случае экспрессирующих CYP1B1 клеток (по сравнению с CYP1A1-экспрессирующими клетками) при низких концентрациях (см. нижеприводимую таблицу 1).
Эти результаты независимо обеспечивают подтверждение того, что уровни экспрессии CYP1A1 и CYP1B1 являются такими, как ожидаемые.
Определяющие цитотоксичность значения IC50 в случае клеточных линий CHO, CHO/CYP1A1 и CHO/CYP1B1
Единую клеточную суспензию CHO, CHO/CYP1A1 и CHO/CYP1B1 в 100 мкл необходимой клеточной культуральной среды высевали на 96-луночные планшеты при клеточной плотности 1500 клеток на лунку и помещали в инкубатор на 24 часа при температуре 37°С. Затем добавляли исходный раствор тест-соединения в ДМСО для получения концентрационного ряда 100, 30, 10, 3, 1, 0,3, 0,1, 0,03, 0,01, 0,003, 0,001, 0 мкМ. Найдено, что конечная концентрация 0,2% ДМСО не влияет на характеристики роста различных СНО-клеточных линий. Клетки инкубировали вместе с тест-соединением в течение 72 часов или 96 часов, спустя которые всю среду удаляли и заменяли с помощью 100 мкл свежей среды для возмещения потери среды вследствие испарения. Клетки инкубировали с 20 мкл реагента для MTS-анализа в течение 1,5 часов и оптическую плотность на лунку определяли при длине волны 510 нм, используя планшет-ридер. Среднюю оптическую плотность и стандартное отклонение для каждой концентрации тест-соединения рассчитывали в сравнении с серией контролей, включающих (а) клетки плюс среда, (b) клетка плюс среда, содержащая 0,2% ДМСО, (с) одна среда и (d) среда, содержащая 0,2% ДМСО, и рядом концентраций соединения от 0 мкмоль/дм3 до 100 мкмоль/дм3. Значение цитотоксичности IC50 рассчитывали из графика зависимости процента клеточного роста (где 100% клеточного роста соответствует необработанным контрольным клеткам) от концентрации тест-соединения.
Значения цитотоксичности IC50 согласно данному контексту определяли в виде концентрации соединения, которая ликвидирует 50% клеток, и общую селективность рассчитывали путем деления IC50 в случае неэкспрессирующих CYP1 клеткок на IC50 в случае экспрессирующих CYP1A1 или CYP1B1 клеток. Соотношения дифференциальной цитотоксичности IC50 рассчитывали, исходя из IC50 соединения в случае нормальных клеток, деленной на IC50 в случае трансфицированных CYP1A1 или CYP1B1 СНО-клеток.
PromegaTM CellTiter 96® Aqueous нерадиоактивный (MTS)-анализ клеточной пролиферации
Коммерчески доступный MTS-анализ представляет собой гомогенный колориметрический метод определения количества жизнеспособных клеток в случае анализов в отношении пролиферации, цитотоксичности или чувствительности к химиотерапии. Анализ заключается в использовании растворов тетразолиевого соединения [3-(4,5-диметилтиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2Н-тетразолий, внутренняя соль; MTS] и электроносвязывающего реагента (феназинметосульфат) PMS. MTS биовосстанавливается клетками до продукта формазана, который растворим в тканевой культуральной среде. Оптическая плотность формазана при длине волны 510 нм может быть прямо определена из 96-луночных аналитических планшетов. Количество формазана, которое измеряется по величине оптической плотности при 490 нм или 510 нм, прямо пропорционально количеству живых клеток в культуре.
Два соединения согласно изобретению (SU025-04 и SU046-04) предназначены для высвобождения мустинфосфорамидатов, N,N-бис(2-хлорэтил)фосфорамида (CI-IPM) и N,N-бис(2-бромэтил)фосфорамида (Br-IPM), соответственно, когда активируются CYP1B1. Высокие токсичности двух мустинфосфорамидатов CI-IPM и Br-IPM значительно снижены, когда включены в пролекарства SU025-04 и SU046-04, соответственно. Как SU025-04, так и SU046-04 имеют значения цитотоксичности IC50 не менее чем 10 мкмоль/дм3 в случае СНО-клеток дикого типа при экспонировании в течение 72 часов или 96 часов в заметной противоположности CI-IPM и Br-IPM, которые имеют значения цитотоксичности IC50 ниже 0,007 мкмоль/дм3 в случае СНО-клеток дикого типа после экспонирования в течение 72 часов (см. нижеприводимую таблицу 2). Механизм активации обоих пролекарств может быть выведен из их сравнительных значений цитотоксичности IC50 в случае СНО-клеток дикого типа (в которых отсутствует экспрессия CYP1-фермента), клеток СНО/1А1 и СНО/CYP1B1. Например, SUO25 и SUO46 проявляют низкую токсичность в случае СНО-клеток дикого типа, но являются высокотоксичными в отношении CHO/1B1-клеток, давая дифференциальные соотношения цитотоксичности IC50 1689 и 5075, соответственно, при экспонировании в течение 72 часов. При более продолжительном времени экспонирования 96 часов, CYP1B1-селективные пролекарства SU025-04 и SU046-04 являются в 3367 и 5400 раз более токсичными по отношению к экспрессирующим CYP1B1 клеткам, чем в случае не экспрессирующих CYP1B1 клеток (см. нижеприводимую таблицу 2). Соединения SU025-04 и SU046-04 поэтому являются доказуемо CYP1B1-активируемыми пролекарствами. SU025-04 и SU046-04 проявляют подобным образом низкую цитотоксичность в отношении СНО-клеток дикого типа и СНО/CYP1A1-клеток с соотношениями дифференциальной цитотоксичности IC50<1 при экспонировании в течение 72 часов, что указывает на то, что высокотоксичные мустинфосфорамидаты не высвобождаются CYP1A1-активацией (см. нижеприводимую таблицу 2). Как следует из литературы, два клинически используемых пролекарства, ифосамид и циклофосфамид, которые также генерируют алкилирующие мустинизофосфамидаты, когда активируются ферментами CYP2B6 и CYP3A4, но не CYP1 (например, McFadyen M.C., Melvin W.T. и Murray G.I. «Cytochrome P450 Enzymes: Novel Options for Cancer Therapeutics», Mol. Cancer Ther., 3(3), 363-71 (2004)), оба являются нетоксичными при самой высокой концентрации 100 мкмоль/дм3 и самых длительных временах экспонирования 96 часов, используемых в этом анализе по цитотоксичности (см. снова нижеприводимую таблицу 2).
Пример 4: Цитотоксичность пролекарства в случае человеческих первичных опухолевых клеточных линий
Цитотоксичность пролекарства в случае человеческой первичной опухолевой клеточной линии плоскоклеточного рака головы и шеи (UT-SCC-14), которая конститутивно экспрессирует CYP1B1
Авторами Greer и др., в Proc. Am. Assoc. Cancer Res., 45, 3701 (2004), сообщалось, что CYP1B1 сверхэкспрессируется во время злокачественной прогрессии плоскоклеточного рака головы и шеи (HNSCC), но не в случае нормального эпителия. Первичную UT-SCC-14 опухолевую клеточную линию получали от ракового больного с HNSCC (см., например, Yaromina и др., Radiother. Oncol., 83, 304-10 (2007), и Hessel и др., Int. J. Radiat. Biol., 80, 719-727 (2004)). Пациентом являлся мужчина в возрасте 25 лет с HNSCC, который характеризовался следующими клиникопатологическими параметрами: локация, пораженный плоскоклеточным раком язык; T3 N1, M0; место, язык; патологическое изменение, первичность; стадия G2. Клеточная линия UT-SCC-14 конститутивно экспрессирует CYP1B1 на уровне мРНК и белка и была использована для демонстрирования цитотоксичности соединения в случае раковой клетки, происходящей от рака человека, характеризующегося сверхэкспрессией CYP1B1 (Greer и др., в Proc. Am. Assoc. Cancer Res., 45, 3701 (2004)).
Опухолевые клетки UT-SCC-14: клеточную линию HNSCC культивировали в условиях стандартного клеточного культивирования в ЕМЕМ (500 мл), дополненной фетальной телячьей сывороткой (50 мл), не являющимися незаменимыми аминокислотами (100Х, 5 мл), пируватом натрия (100 ммоль/дм3, 5 мл), L-глутамином (200 ммоль/дм3, 5 мл), вместе с пенициллином 100 Межд.Ед./мл/стрептомицином (100 мкг/мл, 5 мл), согласно известным из литературы методам (Hessel и др., Int. J. Radiat. Biol., 80, 719-27 (2004), содержания которых включены в данный контекст путем ссылки).
Определение значений цитотоксичности пролекарства IC50 в первичных опухолевых клеточных линиях головы и шеи
Использовали суспензию опухолевых клеток UT-SCC-14 по 2000 клеток на лунку на 96-луночном планшете и, если необходимо, добавляли свежую среду для получения общего объема на лунку 100 мкл. Клетки оставляли фиксироваться в течение 4 часов в инкубаторе. Спустя 4 часа, подтверждали, при использовании микроскопа, что клетки прилипли ко дну 96-луночного планшета, затем среду удаляли и заменяли свежей средой, содержащей исходный раствор тест-соединения в этаноле для получения следующих конечных концентраций: 0, 0,001, 0,003, 0,01, 0,03, 0,1, 0,3, 1, 3, 10, 30, 100 мкмоль/дм3 при конечном объеме 100 мкл на лунку. Обнаружено, что конечная концентрация этанола 0,2% не действует на характеристики роста клеточной линии UT-SCC-14. Клетки UT-SCC-14 инкубировали с тест-соединением в течение 72 часов, спустя которые всю среду удаляли и заменяли 100 мкл свежей среды для компенсации потери среды вследствие испарения. Клетки инкубировали с 20 мкл реагента для MTS-анализа в течение 1,5 часов и измеряли оптическую плотность на лунку при 510 нм, используя планшет-ридер. Среднюю оптическую плотность и стандартное отклонение для каждой концентрации тест-соединения рассчитывали при использовании серии контролей, включая (а) клетки плюс среда, (b) клетка плюс среда, содержащая 0,2% этанола, (с) одна среда и (d) среда, содержащая 0,2% этанола, и ряд концентраций тест-соединения от 0 мкмоль/дм3 до 100 мкмоль/дм3. Значение цитотоксичности IC50 рассчитывали из графика зависимости роста клеток в процентах (где рост клеток 100% соответствует необработанным контрольным клеткам) от концентрации тест-соединения.
Значения цитотоксичности IC50 определяли, согласно данному контексту, в виде концентрации соединения, которая ликсидирует 50% опухолевых клеток UT-SCC-14. Коммерчески доступный MTS-анализ представляет собой гомогенный колориметрический метод определения количества жизнеспособных клеток в случае анализов по пролиферации, цитотоксичности и чувствительности к химиотерапии и использовали, как описано ранее в вышеприведенном примере 3.
Два соединения согласно изобретению (SU025-04 и SU046-04) предназначены для высвобождения мустинфосфорамидатов, N,N-бис(2-хлорэтил)фосфорамида (CI-IPM) и N,N-бис(2-бромэтил)фосфорамида (Br-IPM), соответственно, когда активируются CYP1B1. Значения цитотоксичности IC50 для SU025-04 и SU046-04 в случае опухолевых клеток UT-SCC14 после экспонирования в течение 72 часов составляли 0,05±0,01 мкмоль/дм3 и 0,02±0,01 мкмоль/дм3, соответственно. Данные показывают сильную цитотоксичность SU025-04 и SU046-04 в случае клеточной линии UT-SCC-14 от ракового пациента с HNSCC, которая сверхэкспрессирует CYP1B1.
SU025-04 и SU046-04 оценивали в 3 дополнительных первичных клеточных линиях головы и шеи, включая UT-SCC-8, UT-SCC-9 и UT-SCC-10, культивируемых при тех же самых условиях, как в случае UT-SCC-14. В случае SU025-04 цитотоксичность IC50 в мкмоль/дм3 составляла для UT-SCC-8 (0,31±0,06), UT-SCC-9 (0,43±0,07), UT-SCC-10 (0,22±0,03) после экспонирования в течение 72 часов. В случае SU046-04 цитотоксичность IC50 в мкмоль/дм3 составляла для UT-SCC-8 (0,06±0,02), UT-SCC-9 (0,15±0,02), UT-SCC-10 (0,09±0,03) после экспонирования в течение 72 часов. Данные показывают, что SU046-04 является более сильным цитотоксином, чем SU025-04, в случае ряда первичных клеточных линий головы и шеи, которые конститутивно экспрессируют CYP1B1.
Одно соединение согласно изобретению, SU037-04, предназначено для высвобождения камптотецина, когда активируется CYP1B1. В случае SU037-04 цитотоксичность IC50 в мкмоль/дм3 для каждой первичной опухолевой клеточной линии составляла UT-SCC-8 (0,56±0,04), UT-SCC-9 (0,22±0,08), UT-SCC-10 (0,21±0,04), UT-SCC-14 (0,12±0,07) после экспонирования в течение 72 часов.
Одно соединение согласно изобретению, SU048-04, предназначено для высвобождения гемцитабина, когда активируется CYP1B1. В случае SU048-04 цитотоксичность IC50 в отношении опухолевой клеточной линии UT-SCC-14 составляла 0,94±0,02 мкмоль/дм3 после экспонирования в течение 72 часов. Совместная инкубация с α-нафтофлавоном (сильный ингибитор CYP1B1) в количестве 10 мкмоль/дм3 значительно снижала токсичность SU048-04 до 12,2±0,2 мкмоль/дм3, посредством этого обеспечивая непрямо доказательство активации пролекарства за счет CYP1B1, конститутивно экспрессирующегося в клетках.
Пример 5: Противоопухолевая активность SU046-04 в случае модели первичного человеческого опухолевого ксенотрансплантата, который конститутивно экспрессирует CYP1B1
Первичные клеточные линии UTSCC-14 3×106 имплантировали подкожно в бок «голых» мышей. Мышей рандомизировали по 10 животных на группу, когда объем опухоли составлял от 100 мм3 до 150 мм3. SU046-04 вводили интраперитонеально по 12, 25 и 50 мг/кг в PBS против только эксципиента в 2 цикла: ежедневно в течение 5 дней/2 дня без. Объем опухоли измеряли каждые 4 дня, используя штангенциркули. Значительное ингибирование роста опухоли наблюдали в случае всех трех обработанных передних лап по сравнению с использованием только эксципиента. Замедление роста опухоли на 28 дней составляло 31% при 12 мг/кг, 56% при 25 мг/кг и 90% при 50 мг/кг с 4/10 полными ответами. Не наблюдали вредных эффектов или значительного уменьшения массы тела после самого высокого воздействия 250 мг/кг.
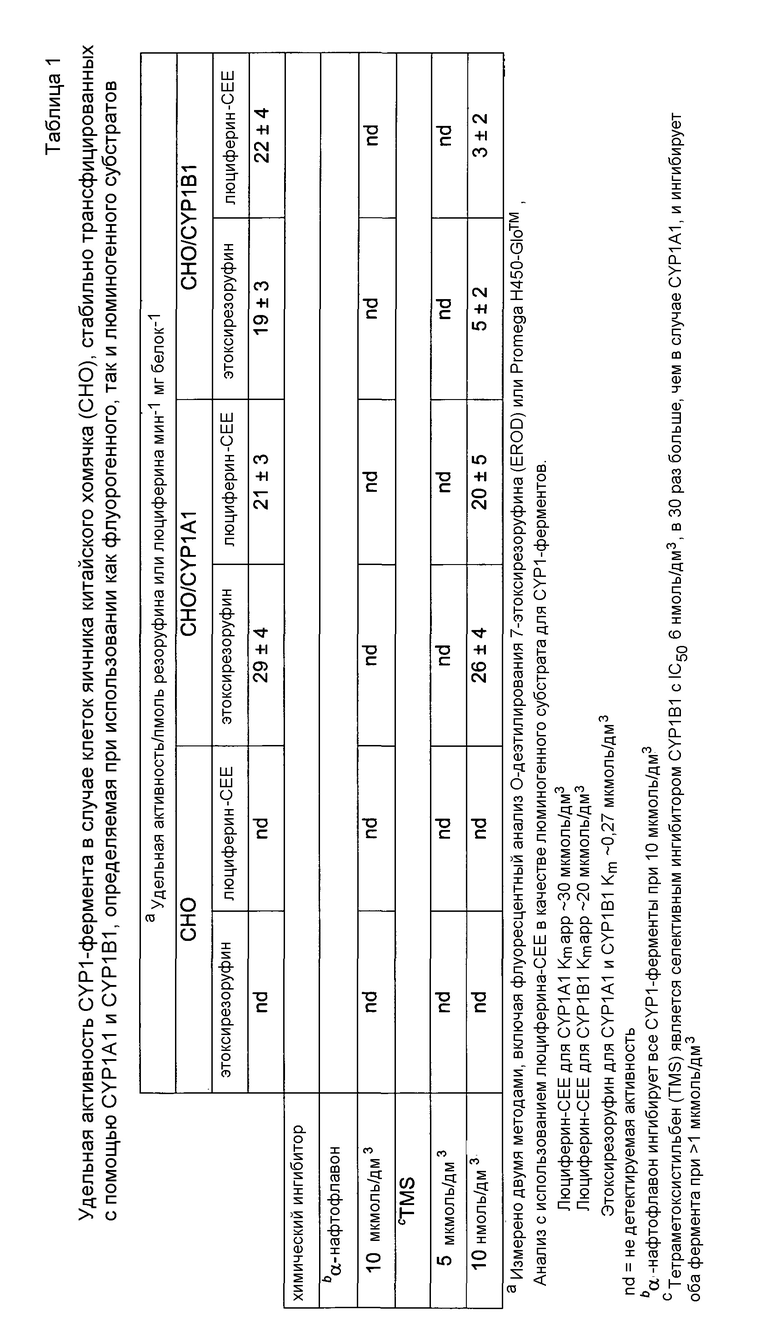
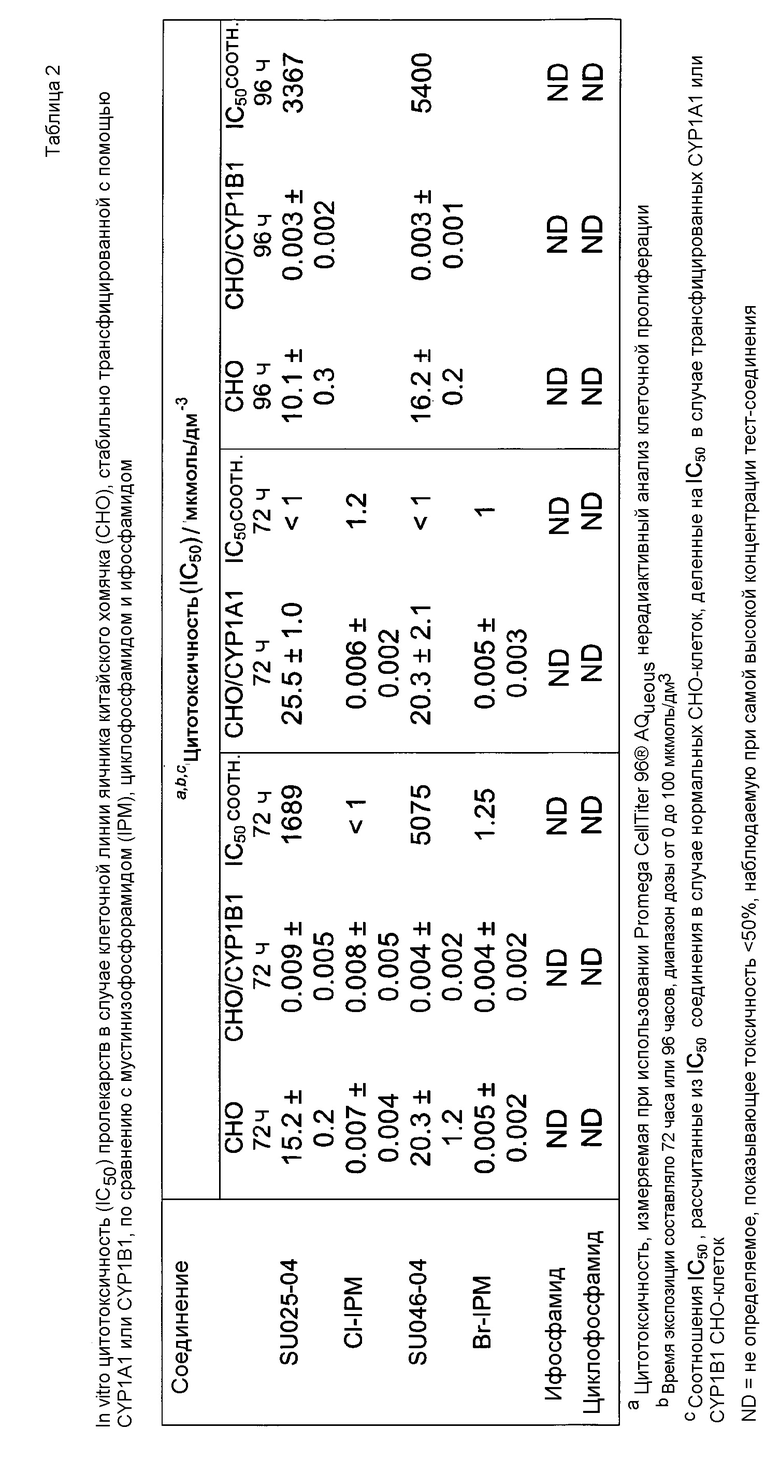
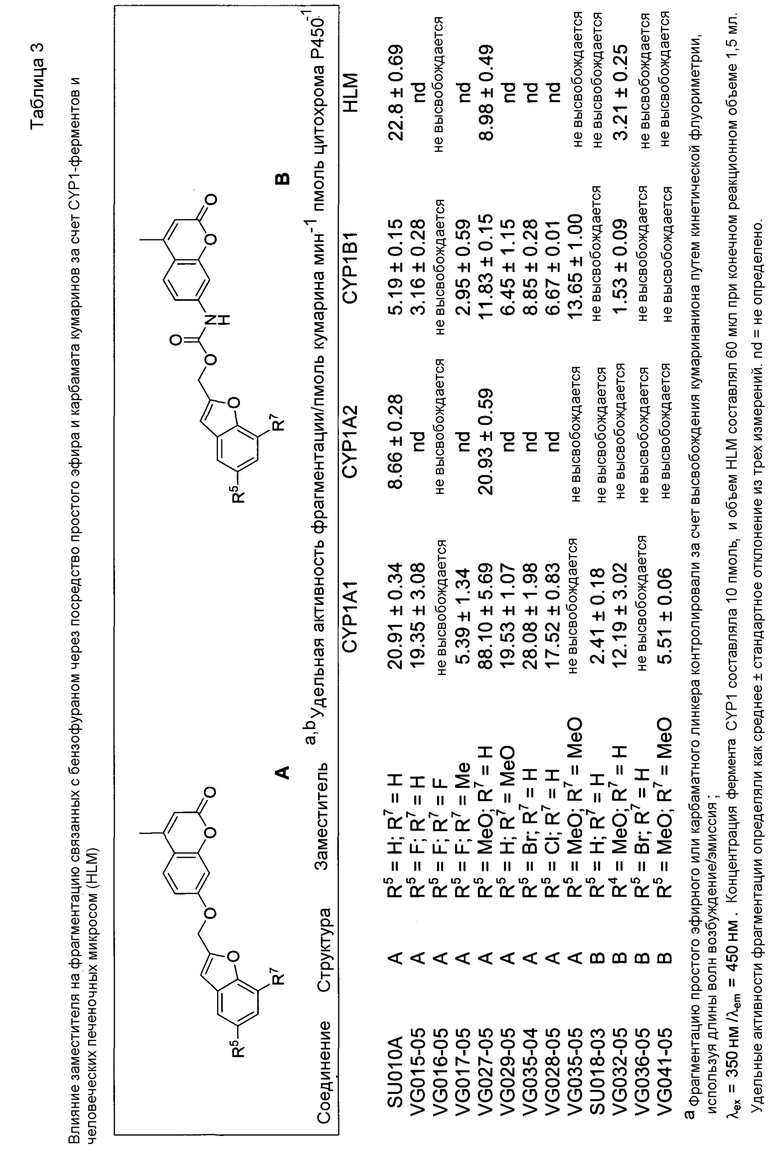
| название | год | авторы | номер документа |
|---|---|---|---|
| МЕТАБОЛИЧЕСКИ УСТОЙЧИВЫЕ АНАЛОГИ CYP-ЭЙКОЗАНОИДОВ ДЛЯ ЛЕЧЕНИЯ КАРДИОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2016 |
|
RU2761438C2 |
| L-НУКЛЕОЗИДЫ, ОБЛАДАЮЩИЕ АНТИ-HBV ИЛИ АНТИ-EBV АКТИВНОСТЬЮ, СПОСОБ ИНГИБИРОВАНИЯ HBV ИЛИ EBV ИНФЕКЦИИ | 1995 |
|
RU2171809C2 |
| СОЕДИНЕНИЯ | 2008 |
|
RU2461559C2 |
| ЛИГАНДЫ, СЕЛЕКТИВНО РАЗРУШАЮЩИЕ АНДРОГЕННЫЕ РЕЦЕПТОРЫ (SARD), И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2017 |
|
RU2795431C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ АТИПИЧНОГО АНТИПСИХОТИКА И МОДУЛЯТОРА NMDA ДЛЯ ЛЕЧЕНИЯ ШИЗОФРЕНИИ, БИПОЛЯРНОГО РАССТРОЙСТВА, КОГНИТИВНОГО НАРУШЕНИЯ И КЛИНИЧЕСКОЙ ДЕПРЕССИИ | 2016 |
|
RU2802972C2 |
| СОЕДИНЕНИЯ, ОБРАЗУЮЩИЕ ПРОЛЕКАРСТВА | 2014 |
|
RU2667942C2 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ИЗОХИНОЛИНЫ В КАЧЕСТВЕ ЛИГАНДОВ ДЛЯ ДОПАМИНОВЫХ РЕЦЕПТОРОВ | 1996 |
|
RU2177001C2 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ ИМАТИНИБА, НЕОБЯЗАТЕЛЬНО МЕЧЕННОГО РАДИОАКТИВНЫМ ИЗОТОПОМ | 2006 |
|
RU2440999C2 |
| Амидные соединения, содержащие их фармацевтические композиции и способы их применения | 2017 |
|
RU2759913C2 |
| ИНГИБИТОРЫ Р38 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2004 |
|
RU2357957C2 |
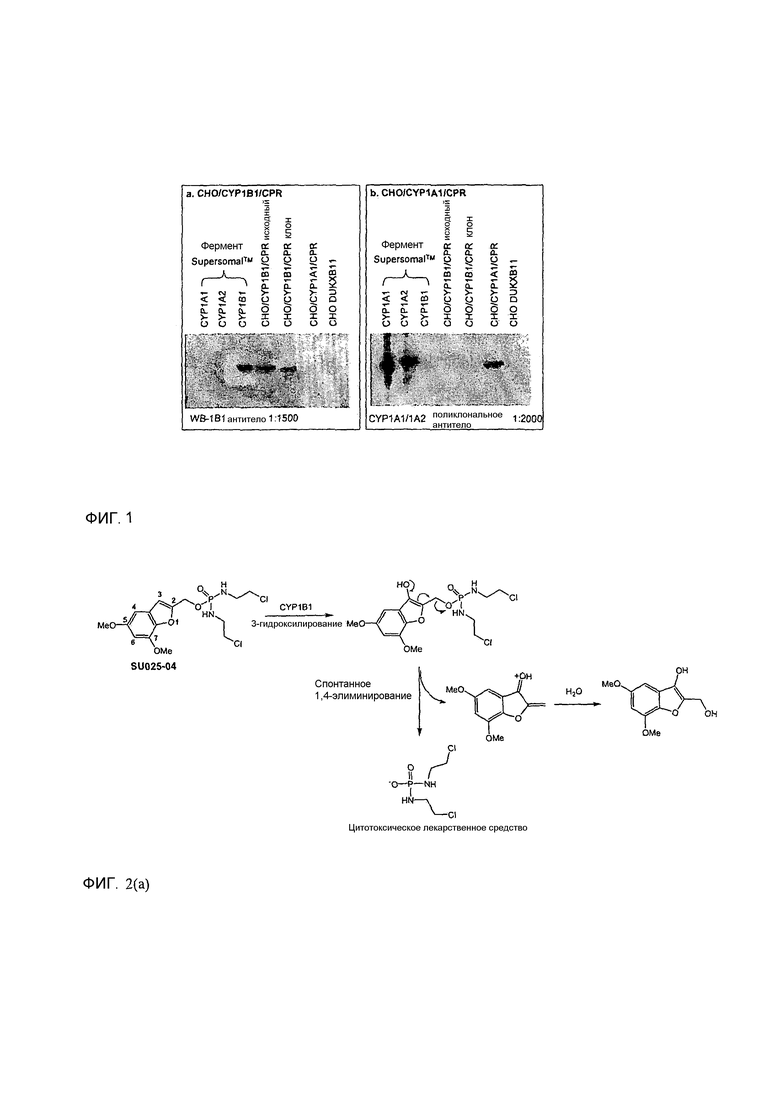
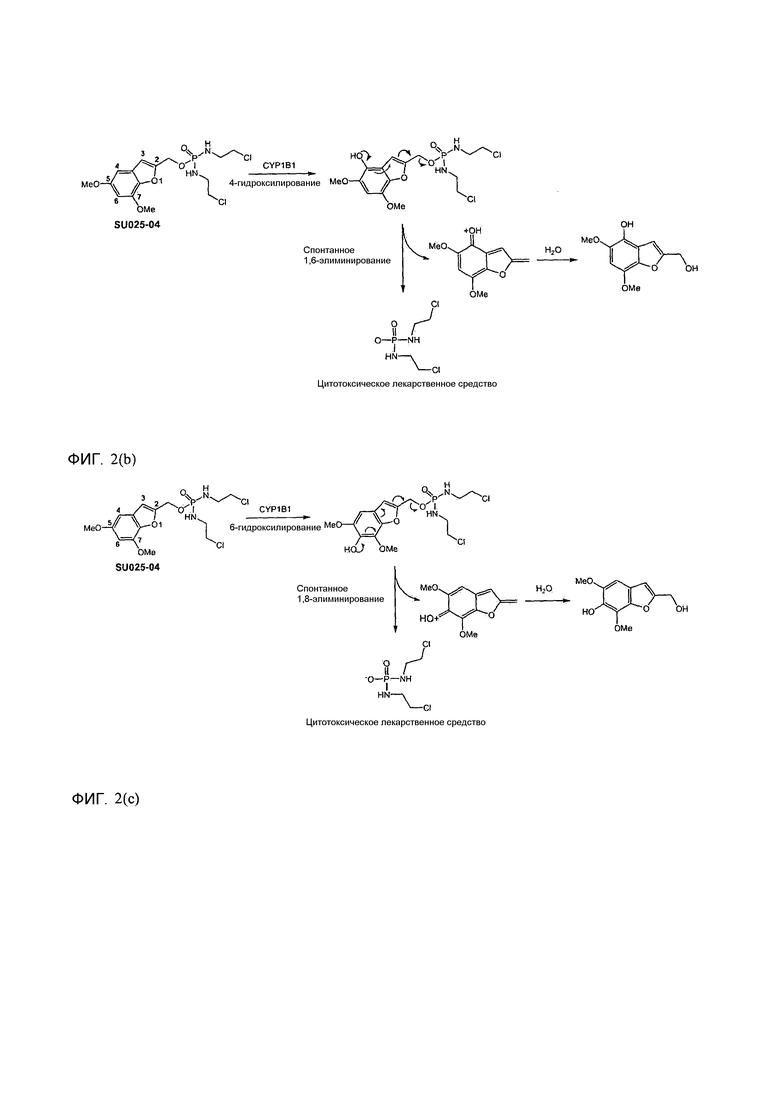
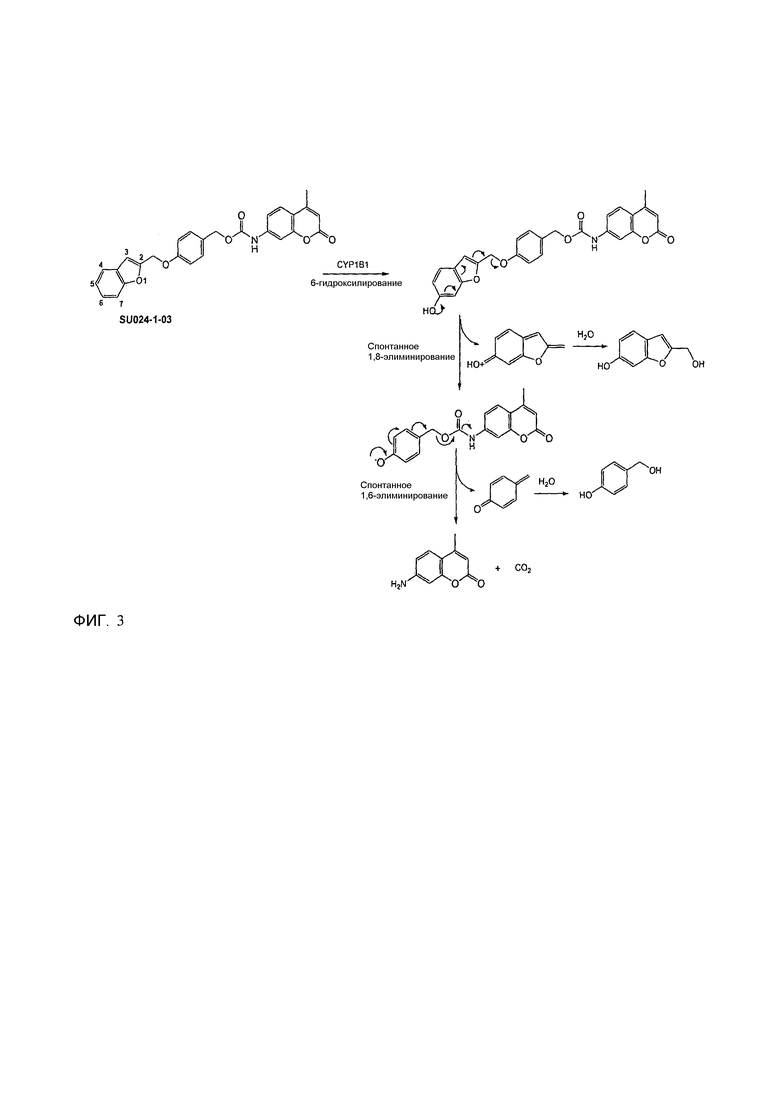
Изобретение относится к новым соединению формулы (I) или его фармацевтически приемлемой соли, где Х1 представляет собой связующий атом или двухвалентную связующую составляющую, выбранные из группы, состоящей из –О-, -S-, -SO2-O- и SO2NZ10; -Х2 отсутствует или является таким, что Х1-Х2-эффектор имеет формулу, выбранную из группы, состоящей из структур (приведенных ниже); каждый n и m независимо означает 0 или 1; р означает 0, 1 или 2; Х3 означает кислород или серу и, дополнительно, когда m=0, может представлять собой SO2-O или SO2NZ10; каждый Y1 представляет собой углерод или азот, и каждый Y2 и Y3 представляют собой углерод, где, если Y1 означает азот, Z1 отсутствует; Y4 означает атом кислорода или углерода; -Y5- означает или (i) одинарную связь, (ii) =СН-, где двойная связь = в =СН- связана с Y4; каждый из Z1, Z2 и Z4 означает водород; Z3 выбирают из группы, состоящей из C1-C6алкила, C1-C6алкилокси и галогена; Z5 выбирают из группы, состоящей из водорода, C1-C6алкила, C1-C6алкилокси; или Z3 и Z4 совместно с атомами, с которыми они связаны, образуют ароматический 6-членный цикл, конденсированный с остатком соединения, при условии, что по меньшей мере один из Z1, Z2 и Z4 означает водород; Z6 выбирают из группы, состоящей из водорода и C1-C6алкила; Y6 означает атом углерода; каждый Z7 независимо означает водород или C1-C6алкил; каждый Z8 независимо означает водород или C1-C6алкил; каждый Z9 независимо означает кислород или серу; Z10 означает водород или алкил, например, С1-4-алкил; и эффектор представляет собой фрагмент, который при высвобождении из соединения формулы (I) обеспечивает флуорофор, выбранный из кумаринов, резофуринов, флуоресцеинов и родаминов; или обеспечивает цитотоксический агент, выбранный из бис(галогенэтил)фосфороамидатов, циклофосфамидов, гемцитабина, цитарабина, 5-фторурацила, 6-меркаптопурина, камптотецина, топотекана, и др. Соединения формулы (I) предназначены для лечения пролиферативного состояния, характеризующегося клетками, экспрессирующими цитохром Р450. 2 н. и 13 з.п. ф-лы, 3 табл., 5 пр., 5 ил.
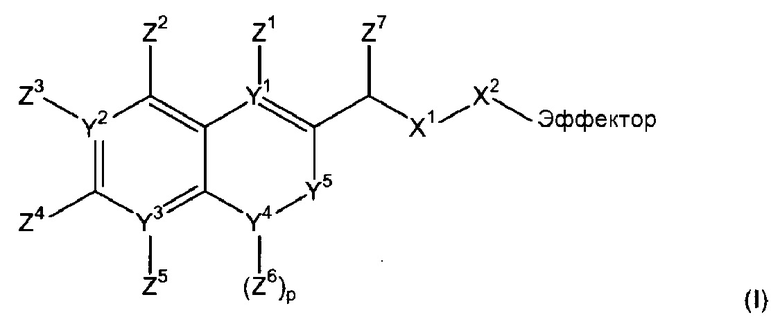 ,
,
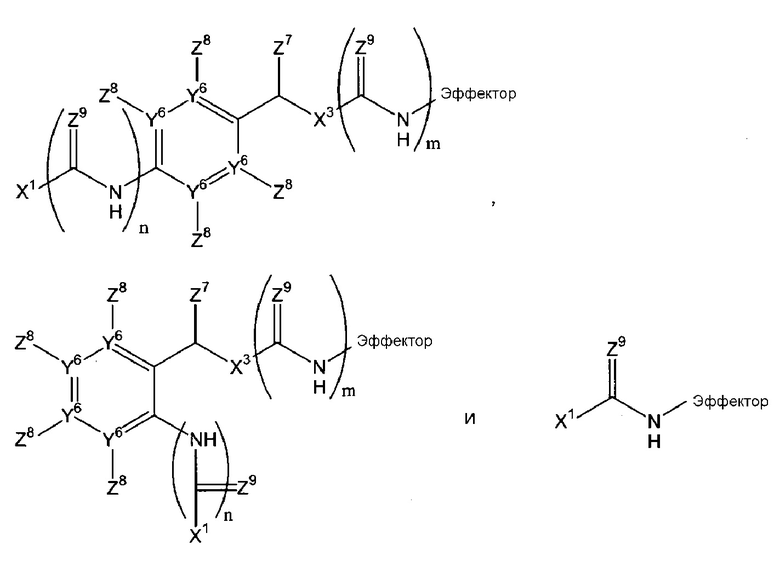
1. Соединение формулы (I):
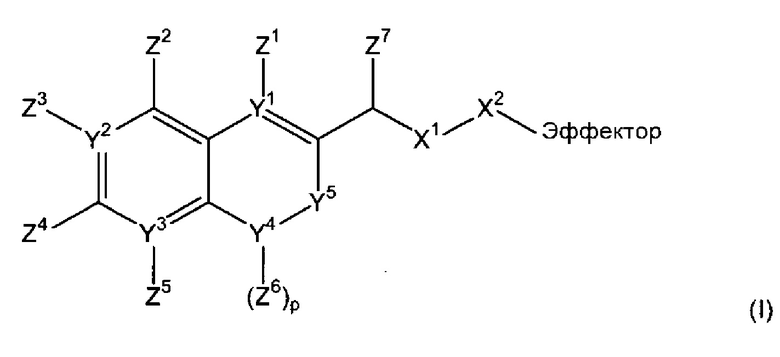
или его фармацевтически приемлемая соль,
где
Х1 представляет собой связующий атом или двухвалентную связующую составляющую, выбранные из группы, состоящей из –О-, -S-, -SO2-O- и SO2NZ10;
-Х2 отсутствует или является таким, что Х1-Х2-эффектор имеет формулу, выбранную из группы, состоящей из:
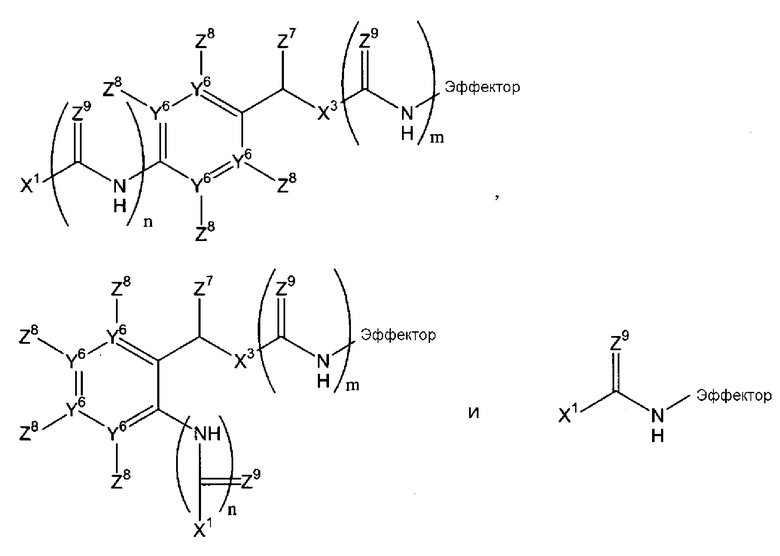
каждый n и m независимо означает 0 или 1;
р означает 0, 1 или 2;
Х3 означает кислород или серу и, дополнительно, когда m=0, может представлять собой SO2-O или SO2NZ10;
каждый Y1 представляет собой углерод или азот, и каждый Y2 и Y3 представляют собой углерод, где, если Y1 означает азот, Z1 отсутствует;
Y4 означает атом кислорода или углерода;
-Y5- означает или (i) одинарную связь, (ii) =СН-, где двойная связь = в =СН- связана с Y4;
каждый из Z1, Z2 и Z4 означает водород;
Z3 выбирают из группы, состоящей из C1-C6алкила, C1-C6алкилокси и галогена;
Z5 выбирают из группы, состоящей из водорода, C1-C6алкила, C1-C6алкилокси; или
Z3 и Z4 совместно с атомами, с которыми они связаны, образуют ароматический 6-членный цикл, конденсированный с остатком соединения, при условии, что по меньшей мере один из Z1, Z2 и Z4 означает водород;
Z6 выбирают из группы, состоящей из водорода и C1-C6алкила;
Y6 означает атом углерода;
каждый Z7 независимо означает водород или C1-C6алкил;
каждый Z8 независимо означает водород или C1-C6алкил;
каждый Z9 независимо означает кислород или серу;
Z10 означает водород или алкил, например, С1-4-алкил; и
эффектор представляет собой фрагмент, который при высвобождении из соединения формулы (I) обеспечивает флуорофор, выбранный из кумаринов, резофуринов, флуоресцеинов и родаминов; или обеспечивает цитотоксический агент, выбранный из бис(галогенэтил)фосфороамидатов, циклофосфамидов, гемцитабина, цитарабина, 5-фторурацила, 6-меркаптопурина, камптотецина, топотекана, доксорубицина, даунорубицина, дуокармицина, этопозида, диетопозида, комбретастатина А-4, винбластина, винкристина, AQ4N, гидроксимочевины, маитансинов, энедийенов, эпотилонов, таксанов, блеомицинов, калихеамицинов, колхицина, дакарбазина, дактиномицина, эпирубицина, производных эпирубицина, флударабина, гидроксимочевинопентатостатина, метотраксата, митомицина, митоксантрона, карбоплатина, цисплатина, такселов, 6-тиогуанина, винка-алкалоидов, координационных комплексов платины, антрацендионов, гидроксимочевины, метилгидразина и хлорметинов.
2. Соединение или фармацевтически приемлемая соль по п.1, где каждый Z7 означает водород и Х1 означает кислород.
3. Соединение или фармацевтически приемлемая соль по п.1 или 2, где Y1 означает углерод, Z1 означает водород.
4. Соединение или фармацевтически приемлемая соль по пп.1-3, где Z2 и/или Z4 означает водород.
5. Соединение или фармацевтически приемлемая соль по пп.1-4, где Y4 означает кислород и р=0.
6. Соединение или фармацевтически приемлемая соль пп.1-5, где Y4 означает кислород и -Y5- представляет собой одинарную связь.
7. Соединение или фармацевтически приемлемая соль по пп.1-6, где Z3 и Z5, каждый, означают метокси.
8. Соединение или фармацевтически приемлемая соль по пп.1-7, где Х1 представляет собой О.
9. Соединение или фармацевтически приемлемая соль по пп.1-8, где Х2 отсутствует или -Х1-Х2-эффектор отвечает формуле:
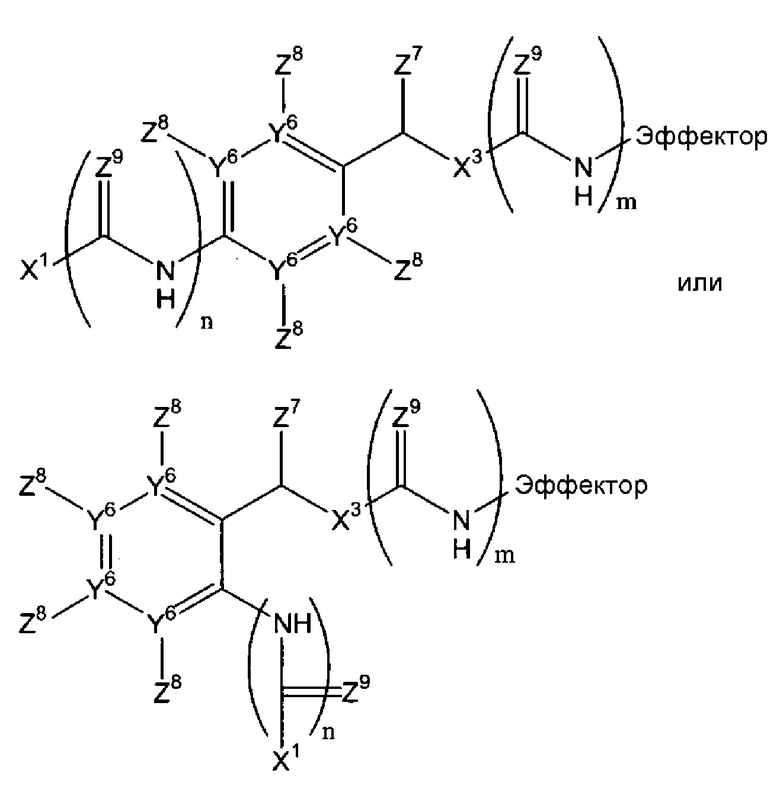
10. Соединение или фармацевтически приемлемая соль по п. 9, где один из n и m представляет собой 0, или оба n и m представляют собой 0 и где каждый Z9 означает кислород.
11. Соединение или фармацевтически приемлемая соль по п.1, где эффектор представляет собой цитотоксический или цитостатический агент, где цитотоксический агент выбран из бис(галогенэтил)фосфороамидата, циклофосфамида, гемцитабина, цитарабина, 5-фторурацила, 6-меркаптопурина, камптотецина, топотекана, доксорубицина, даунорубицина, дуокармицина, этопозида, диетопозида, комбретастатина А-4, винбластина, винкристина, AQ4N, гидроксимочевины, маитансина, энедийена, эпотилона, таксана, блеомициныа калихеамицина, колхицина, дакарбазина, дактиномицина, эпирубицина, производных эпирубицина, флударабина, гидроксимочевинопентатостатина, метотраксата, митомицина, митоксантрона, карбоплатина, цисплатина, таксела, 6-тиогуанина, винка-алкалоидов, координационных комплексов платины, антрацендиона, гидроксимочевины, метилгидразина и хлорметинов.
12. Соединение или фармацевтически приемлемая соль по пп.1-10, где эффектор связан с остатком соединения через атом кислорода или серы и эффектор представляет собой бис(галогенэтил)фосфорамидат формулы (II):
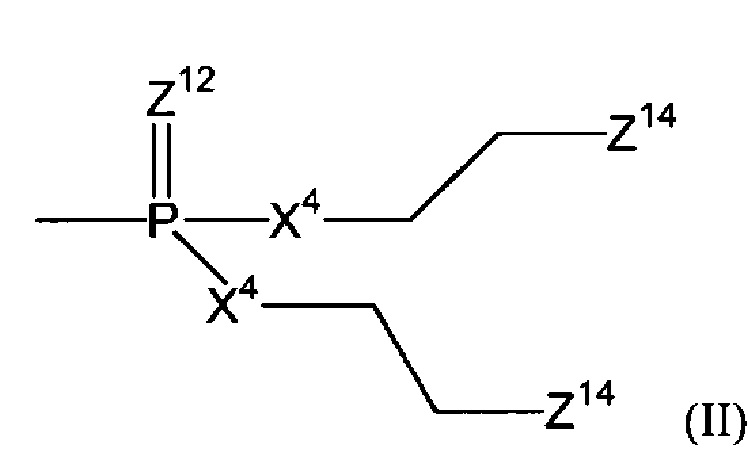
где
Z12 означает кислород или серу;
каждый Х4 независимо означает кислород, серу или NZ13, где каждый -Z13 независимо означает -(СН2)2-Z14, алкил или водород; и
каждый Z14 независимо означает хлор, бром, йод или мезилат.
13. Фармацевтическая композиция, предназначенная для лечения пролиферативного состояния, характеризующегося клетками, экспрессирующими цитохром Р450, содержащая эффективное количество соединения или фармацевтически приемлемой соли по пп.1-12, вместе с фармацевтически приемлемым носителем.
14. Соединение или фармацевтически приемлемая соль по пп.1-12 для применения в способе лечения или профилактики пролиферативного состояния, где пролиферативное состояние представляет собой предраковую или злокачественную клеточную пролиферацию или рак, и где рак выбран из рака мочевого пузыря, головного мозга, молочной железы, ободочной кишки, головы и шеи, почки, легкого, печени, яичника, простаты и кожи.
15.Соединение по п.1, выбранное из
 ;
; ;
; ;
;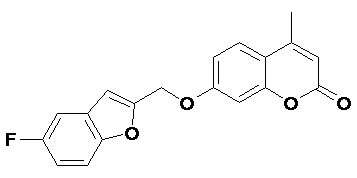 ;
;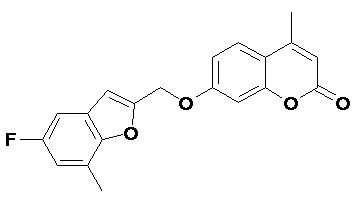 ;
;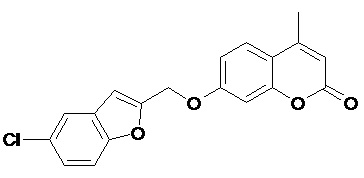 ;
;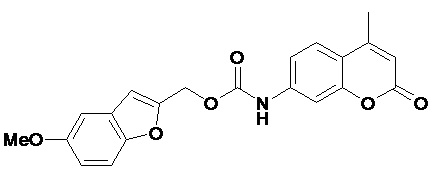 ;
;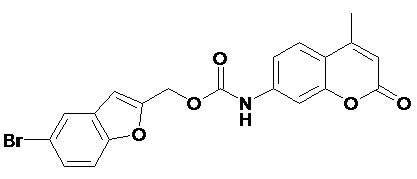 ;
;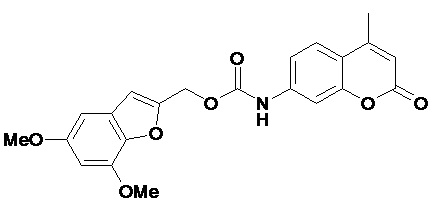 ; или
; или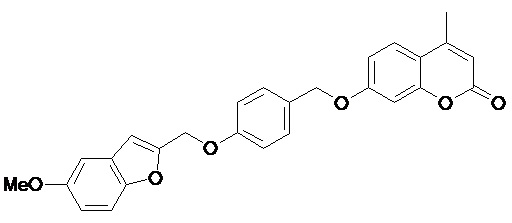 ;
;
или его фармацевтически приемлемой соли.
| Способ изготовления волокнистого модифицированного материала | 1985 |
|
SU1676834A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| РЕЛЕЙНАЯ СЛЕДЯЩАЯ СИСТЕМА | 0 |
|
SU247381A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| 0 |
|
SU363796A1 | |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| FLADER C | |||
| et al.: "Development of novel quinone phosphorodiamidate prodrugs targeted to DT-diaphorase" Journal of Medicinal Chemistry, 2000, vol.43, p.3157-3167 | |||
| HERNICK MARCY | |||

