ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает преимущество по предварительной заявке на патент США № 62/755905, поданной 5 ноября 2018 г.; предварительной заявке на патент США № 62/675846, поданной 24 мая 2018 г.; предварительной заявке на патент США № 62/667406, поданной 4 мая 2018 г.; предварительной заявке на патент США № 62/663206, поданной 26 апреля 2018 г.; предварительной заявке на патент США № 62/663219, поданной 26 апреля 2018 г.; и предварительной заявке на патент США № 62/609504, поданной 22 декабря 2017 г.; содержание которых в полном объеме включено в настоящий документ путем ссылки.
ОБЛАСТЬ ПРИМЕНЕНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к фармацевтическим продуктам и способам лечения депрессии (например, большого депрессивного расстройства). В некоторых вариантах осуществления указанные способы применимы для лечения терапевтически рефрактерной или терапевтически резистентной депрессии. В других вариантах осуществления указанные способы применимы для лечения суицидальной идеации. Данное изобретение включает введение нуждающемуся в этом пациенту имеющего клинически доказанную безопасность и терапевтически эффективного количества эскетамина в виде монотерапии или в виде комбинированной терапии по меньшей мере с одним антидепрессантом.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Большое депрессивное расстройство (БДР) поражает около 7-15% общей совокупности населения. БДР связано со значительной заболеваемостью и смертностью и является основной причиной недееспособности во всем мире. Приблизительно у одной трети пациентов не удается достичь ремиссии, несмотря на лечение несколькими антидепрессантами, и считается, что они имеют терапевтически резистентную депрессию (ТРД). Те пациенты, которым приносят пользу пероральные АД, имеют высокие уровни рецидивов даже при продолжении лечения.
Влияние ТРД на жизнь пациента трудно описать с достаточной точностью. У многих пациентов случаются депрессивные эпизоды, длящиеся годами. Пациенты с сильной депрессией теряют волю к жизни, при этом наблюдается увеличение числа попыток самоубийства в 7 раз. Средняя продолжительность жизни снижается на 10 лет. В крайних случаях они не могут даже осуществлять элементарный уход за собой, такой, как гигиенические процедуры или питание, или заботиться о себе сами, предоставляя самим себе тех, кто находится на их попечении, например, родителей, супругов и т. д. Это влияет не только на самих пациентов, но также и на семью и тех, кто зависим от них. Они также теряют способность испытать удовольствие, делая те вещи, которые им нравились, что лишает людей смысла жизни и того, что является ее движущей силой. В результате ТРД лишает их жизни. Теоретически эти эффекты связывают с дисрегуляцией глутаматного пути.
Глутамат является основным возбуждающим нейротрансмиттером в головном мозге млекопитающих и играет важную роль в синаптической пластичности, обучении и памяти. При повышенных концентрациях глутамат представляет собой сильный нейронный эксайтотоксин, который может провоцировать быструю или отсроченную нейротоксичность. На протяжении многих лет наблюдается растущий интерес к роли глутамата в патофизиологии депрессии, поскольку аномальная активность глутаматергической системы, вероятно, способствует нарушению синаптической пластичности, наблюдаемой у пациентов с депрессией. Кетамин, классическое анестетическое лекарственное средство, продемонстрировало активность не только в животных моделях депрессии, но также в мелкомасштабных клинических исследованиях на пациентах с большим депрессивным расстройством, включая испытуемых с терапевтически резистентной депрессией. В низких субанестетических дозах, вводимых путем внутривенной инфузии, кетамин продемонстрировал сильный антидепрессивный эффект у пациентов, который длился в течение нескольких суток после однократной дозы и мог сохраняться в течение нескольких недель при помощи повторных инфузий.
Кетамин (рацемическая смесь соответствующих S- и R-энантиомеров) представляет собой неизбирательного антагониста в сайте связывания фенциклидина, связывающего N-метил-D-аспартат (NMDA) рецептора глутамата, хотя это не может быть основной причиной антидепрессивного эффекта. Энантиомер S-кетамин (эскетамин) проявляет приблизительно в 3-4 раза большую аффинность к глутаматному рецептору NMDA in vitro, чем R-кетамин. Серьезной проблемой, связанной с кетамином и эскетамином, является потенциальная нейротоксичность, связанная с длительным применением, и то, могут ли повторные дозы кетамина/эскетамина сохранять значимый антидепрессивный эффект в долгосрочной перспективе (Molero, et al., “Antidepressant Efficacy and Tolerability of Ketamine and Esketamine: A Critical Review,” CNS Drugs (2018) 32:411-420). В частности, предыдущие исследования показали, что эскетамин, в отличие от R-кетамина, не может вызывать стойкий антидепрессивный эффект в животной модели у грызунов (C. Yang et al., “R-Ketamine: a rapid onset and sustained antidepressant without psychotomimetic side effects,” Transl. Psychiatry (2015) 5:1-11). Более того, эскетамин продемонстрировал больше нежелательных психотомиметических побочных эффектов по сравнению с R-кетамином, включая значимое снижение количества PV-положительных клеток в головном мозге, что связано с психозом и нарушением когнитивных функций (id.). В литературе отсутствует руководство относительно кумулятивного эффекта или переносимости долгосрочного приема эскетамина.
Сохраняется потребность в обеспечении эффективного, долгосрочного и безопасного лечения депрессии, в частности, у пациентов, у которых диагностирована терапевтически рефрактерная или терапевтически резистентная депрессия.
ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам лечения депрессии (например, большого депрессивного расстройства), включающим введение нуждающемуся в этом пациенту клинически доказанного безопасного и терапевтически эффективного количества эскетамина.
Настоящее изобретение дополнительно относится к способу лечения депрессии (например, большого депрессивного расстройства), включающему применение к нуждающемуся в этом пациенту комбинированной терапии на основе имеющего клинически доказанную безопасность и терапевтически эффективного количества эскетамина и по меньшей мере одного антидепрессанта по определению в данном документе.
Настоящее изобретение также относится к способам поддержания стабильной ремиссии или стабильного ответа, достигаемых пациентом с депрессией, после введения терапевтически эффективного количества эскетамина во время начальной фазы введения, включающим продолжение введения терапевтически эффективного количества эскетамина в течение по меньшей мере пяти месяцев во время последующей фазы введения. В некоторых вариантах осуществления депрессия представляет собой большое депрессивное расстройство или терапевтические резистентную депрессию.
Настоящее изобретение дополнительно относится к способам долгосрочного лечения депрессии у пациента, включающим введение нуждающемуся в лечении пациенту имеющего клинически доказанную безопасность и/или клинически доказанную эффективность терапевтически эффективного количества эскетамина в течение по меньшей мере шести месяцев. В некоторых вариантах осуществления депрессия представляет собой большое депрессивное расстройство или терапевтические резистентную депрессию.
Указанный способ лечения включает долгосрочное лечение, в том числе продолжительностью по меньшей мере около шести месяцев. В некоторых вариантах осуществления лечение может длиться по меньшей мере около одного года, по меньшей мере около 18 месяцев или по меньшей мере около двух лет. Например, долгосрочное лечение может включать в себя продолжительность в диапазоне от около шести месяцев до около двух лет. Лечение может продолжаться в течение гораздо более длительных периодов времени при условии, что пациент получает пользу от такой терапии.
В некоторых вариантах осуществления по меньшей мере один антидепрессант независимо выбран из группы, состоящей из ингибиторов моноаминоксидазы, трициклических соединений, ингибиторов обратного захвата серотонина, ингибиторов обратного захвата серотонина-норэпинефрина, норадренергических и специфических серотонинергических агентов, ингибиторов обратного захвата норадреналина, натуральных продуктов, диетических добавок, нейропептидов, соединений, нацеленных на нейропептидные рецепторы, и гормонов.
В других вариантах осуществления предложены способы лечения депрессии (например, большого депрессивного расстройства), которые включают введение нуждающемуся в этом пациенту имеющего клинически доказанную безопасность и терапевтически эффективного количества эскетамина в комбинации с одним или более соединениями, выбранными из группы, состоящей из ингибиторов моноаминоксидазы (MAOI), таких как необратимые MAOI (фенелзин, транилципромин), обратимые MAOI (моклобемид) и т.п.; трициклических соединений, таких как имипрамин, амитриптилин, дезипрамин, нортриптилин, доксепин, протриптилин, тримипрамин, кломипрамин, амоксапин и т.п.; тетрациклических соединений, таких как мапротилин и т.п.; нециклических соединений, таких как номифензин и т.п.; триазолопиридинов, таких как тразодон и т.п.; антихолинергических средств, например, скополамина; ингибиторов обратного захвата серотонина, таких как флуоксетин, сертралин, пароксетин, циталопрам, флувоксамин и т.п.; антагонистов рецепторов серотонина, таких как нефазодон, тианептин и т.п.; норадренергических ингибиторов обратного захвата серотонина, таких как венлафаксин, дес-венлафаксин, милнаципран, лево-милнаципран и т.п.; норадренергических и специфических серотонинергических агентов, таких как миртазапин и т.п.; ингибиторов обратного захвата норадреналина, таких как ребоксетин и т.п.; атипических антипсихотических средств, таких как бупропион и т.п.; лития, ингибиторов обратного захвата тройного типа, натуральных продуктов, таких как кава-кава, зверобой и т.п.; диетических добавок, таких как S-аденозилметионин и т.п.; и нейропептидов, таких как тиреотропин-рилизинг гормон и т.п.; соединений, нацеленных на нейропептидные рецепторы, таких как антагонисты нейрокининовых рецепторов и т.п.; и гормонов, таких как трийодотиронин и т.п.
В других вариантах осуществления предложены способы лечения депрессии (например, большого депрессивного расстройства), которые включают введение нуждающемуся в этом пациенту имеющего клинически доказанную безопасность и терапевтически эффективного количества эскетамина в комбинации с одним или более соединениями, выбранными из группы, состоящей из ингибиторов моноаминоксидазы; трициклических соединений; тетрациклических соединений; нециклических соединений; триазолопиридинов; ингибиторов обратного захвата серотонина; антагонистов рецепторов серотонина; норадренергических ингибиторов обратного захвата серотонина; норадренергических ингибиторов обратного захвата серотонина; норадренергических и специфических серотонинергических агентов; ингибиторов обратного захвата норадреналина; атипичных антипсихотических средств; натуральных продуктов; диетических добавок; нейропептидов; соединений, нацеленных на нейропептидные рецепторы; и гормонов. Предпочтительно эскетамин вводят в комбинации с одним или более соединениями, выбранными из группы, состоящей из ингибиторов моноаминоксидазы, трициклических соединений, ингибиторов обратного захвата серотонина, норадренергических ингибиторов обратного захвата серотонина, норадренергических и специфических серотонинергических агентов, атипичных антипсихотических средств, и/или вспомогательной терапией антипсихотическим лекарственным средством (например, рисперидоном, оланзапином, кветиапином, арипипразолом и зипрасидоном). Более предпочтительно эскетамин вводят в комбинации с одним или более соединениями, выбранными из группы, состоящей из ингибиторов моноаминоксидазы, трициклических соединений, ингибиторов обратного захвата серотонина и ингибиторов обратного захвата серотонина-норэпинефрина. Более предпочтительно эскетамин вводят в комбинации с одним или более соединениями, выбранными из группы, состоящей из ингибиторов обратного захвата серотонина и ингибиторов обратного захвата серотонина-норэпинефрина.
В дополнительных вариантах осуществления предложены способы лечения депрессии (например, большого депрессивного расстройства), которые включают введение нуждающемуся в этом пациенту имеющего клинически доказанную безопасность и терапевтически эффективного количества эскетамина в комбинации с одним или более соединениями, выбранными из группы, состоящей из фенелзина, транилципромина, моклобемида, имипрамина, амитриптилина, дезипрамина, нортриптилина, доксепина, протриптилина, тримипрамина, кломипрамина, амоксапина, флуоксетина, сертралина, пароксетина, циталопрама, флувоксамина, венлафаксина, милнаципрана, миртазапина, бупропиона, тиреотропин-рилизинг гормона и трийодотиронина.
Предпочтительно эскетамин вводят в комбинации с одним или более соединениями, выбранными из группы, состоящей из лития, рилузола, фенелзина, транилципромина, моклобемида, имипрамина, амитриптилина, дезипрамина, нортриптилина, доксепина, протриптилина, тримипрамина, кломипрамина, амоксапина, флуоксетина, сертралина, пароксетина, циталопрама, флувоксамина, венлафаксина, милнаципрана, левомилнаципрана, миртазапина и бупропиона. Более предпочтительно эскетамин вводят в комбинации с одним или более соединениями, выбранными из группы, состоящей из фенелзина, транилципромина, моклобемида, имипрамина, амитриптилина, дезипрамина, нортриптилина, доксепина, протриптилина, тримипрамина, кломипрамина, амоксапина, флуоксетина, сертралина, пароксетина, циталопрама и флувоксамина. Более предпочтительно эскетамин вводят в комбинации с одним или более соединениями, выбранными из группы, состоящей из флуоксетина, сертралина, пароксетина, циталопрама, эсциталопрама и флувоксамина.
В дополнительных вариантах осуществления предложены способы лечения депрессии (например, большого депрессивного расстройства), которые включают введение нуждающемуся в этом пациенту имеющего клинически доказанную безопасность и терапевтически эффективного количества эскетамина в комбинации с одним или более соединениями, выбранными из группы, состоящей из нейропептидов, таких как тиреотропин-рилизинг гормон и т.п.; соединений, нацеленных на нейропептидные рецепторы, таких как антагонисты нейрокининовых рецепторов и т.п.; и гормонов, таких как трийодотиронин и т.п.
В других вариантах осуществления предложены способы лечения депрессии (например, большого депрессивного расстройства), которые включают применение к нуждающемуся в этом пациенту комбинированной терапии на основе имеющего клинически доказанную безопасность и терапевтически эффективного количества эскетамина, по меньшей мере одного антидепрессанта и по меньшей мере одного атипичного антипсихотического средства по определению в данном документе.
В дополнительных вариантах осуществления предложены способы лечения депрессии (например, большого депрессивного расстройства), которые включают применение к нуждающемуся в этом пациенту комбинированной терапии на основе имеющего клинически доказанную безопасность и терапевтически эффективного количества эскетамина, по меньшей мере одного антидепрессанта и по меньшей мере одного атипичного антипсихотического средства, выбранного из группы, состоящей из кветиапина, арипипразола, брекспипразола, оланзапина, луразидона, рисперидона и палиперидона.
В других вариантах осуществления способы лечения депрессии можно комбинировать со вспомогательной терапией, такой как антипсихотическая терапия, электрошоковая терапия (ЭШТ), транскраниальная магнитная стимуляция (ТМС) или их комбинации.
Настоящее изобретение дополнительно относится к применению эскетамина в приготовлении лекарственного средства для лечения депрессии (например, большого депрессивного расстройства) у нуждающегося в этом пациента. В некоторых вариантах осуществления указанное лекарственное средство предназначено для лечения терапевтически рефрактерной или терапевтически резистентной депрессии. В других вариантах осуществления указанное лекарственное средство предназначено для лечения суицидальной идеации.
Настоящее изобретение дополнительно относится к эскетамину для применения в способе лечения депрессии (например, большого депрессивного расстройства), предпочтительно терапевтически рефрактерной или терапевтически резистентной депрессии, у нуждающегося в этом субъекта.
В другом варианте осуществления предложены композиции, содержащие эскетамин для лечения депрессии (например, большого депрессивного расстройства). В некоторых вариантах осуществления указанные композиции предназначены для лечения терапевтически рефрактерной или терапевтически резистентной депрессии. В других вариантах осуществления указанное лекарственное средство предназначено для лечения суицидального поведения и/или суицидальной идеации.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На Фиг. 1 проиллюстрирован схематически дизайн клинического исследования фазы 3 ESKETINTRD3002.
На Фиг. 2 проиллюстрированы средние значения изменений, полученные методом наименьших квадратов (± СП), в общей оценке по шкале MADRS (шкала Монтгомери-Асберг для оценки депрессии, англ. «Montgomery-Asberg Depression Rating Scale») в динамике по времени, наблюдаемые в случае СМПИ во время двойной слепой индукционной фазы. Среднее по МНК и СП основаны на СМПИ с изменением относительно исходного уровня в качестве переменной ответа и условиями модели с фиксированными эффектами для лечения (эскетамин интраназально+АД перорально, АД перорально+плацебо интраназально), сутками, страной, классом перорального антидепрессанта (SNRI или SSRI), и лечением по суткам, а также исходным значением в качестве ковариаты. Отрицательное изменение оценки указывает на улучшение. *1-сторонний p<0,025.
На Фиг. 3 представлен столбчатый график скорости ответа на 2 сутки; ответ представляет собой улучшение ≥50% по шкале MADRS относительно исходного уровня для пациентов, принимающих эскетамин и пероральный антидепрессант.
На Фиг. 4 представлен столбчатый график скорости ответа на 28 сутки; ответ представляет собой улучшение ≥50% по шкале MADRS относительно исходного уровня для пациентов, принимающих эскетамин и пероральный антидепрессант.
На Фиг. 5 представлен столбчатый график скорости ремиссии на 28 сутки; ремиссия соответствует общей оценке по шкале MADRS ≤12.
На Фиг. 6 представлена гистограмма, иллюстрирующая процент субъектов, сообщивших о проблемах (уровни 2-5), связанных с двигательной активностью, уходом за собой, активностью, болью и тревогой/депрессией.
На Фиг. 7 проиллюстрировано среднее арифметическое значение (± СП) систолического кровяного давления в динамике по времени во время двойной слепой индукционной фазы с использованием выборки для анализа безопасности.
На Фиг. 8 проиллюстрировано среднее арифметическое значение (± СП) диастолического кровяного давления в динамике по времени; двойная слепая индукционная фаза с использованием выборки для анализа безопасности.
На Фиг. 9 проиллюстрирована общая оценка по оцениваемой клиницистом шкале диссоциативных симптомов (CADSS, англ. «clinician-assessed dissociative symptom scale») в динамике по времени во время двойной слепой индукционной фазы с использованием выборки для анализа безопасности.
На Фиг. 10 проиллюстрировано среднее арифметическое значение (± СП) оценки по шкале модифицированной оценки наблюдателем активности/седации (MOAA/S, англ. «modified observer’s assessment of alertness/sedation») в динамике по времени; двойная слепая индукционная фаза с использованием выборки для анализа безопасности.
На Фиг. 11 проиллюстрировано среднее значение изменения, полученное методом наименьших квадратов, в общей оценке по шкале MADRS в динамике по времени (наблюдаемые случаи) у пациентов с ТРД в США.
На Фиг. 12 проиллюстрирована оцененная пациентом степень тяжести депрессивного заболевания (наблюдаемые случаи) у пациентов с ТРД в США по оценке PHQ-9.
На Фиг. 13 проиллюстрировано функциональное нарушение (наблюдаемые случаи) у пациентов с ТРД в США по оценке SDS.
На Фиг. 14 проиллюстрирован процент пациентов с ТРД в США, достигших ответа в течение 4 недель после начальной дозы (наблюдаемый случай).
На Фиг. 15 проиллюстрирован процент пациентов с ТРД в США, достигших ремиссии по оценке клинициста в течение 4 недель после начальной дозы.
На Фиг. 16 проиллюстрировано частотное распределение категорий тяжести PHQ-9 (наблюдаемый случай) у пациентов с ТРД в США.
На Фиг. 17 проиллюстрирован процент пациентов с ТРД в США, у которых наблюдалось снижение CGI-S на ≥1 пункта (наблюдаемый случай) через 4 недели после начальной дозы.
На Фиг. 18 проиллюстрирован схематически дизайн клинического исследования фазы 3 ESKETINTRD3005.
На Фиг. 19 проиллюстрированы средние значения изменений, полученные методом наименьших квадратов (± СП), в общей оценке по шкале MADRS в динамике по времени, наблюдаемые в случае СМПИ; двойная слепая индукционная фаза (исследование ESKETINTRD3005: полная выборка для анализа).
На Фиг. 20 проиллюстрировано среднее арифметическое значение (± СП) систолического кровяного давления в динамике по времени во время двойной слепой индукционной фазы с использованием выборки для анализа безопасности ESKETINTRD3005.
На Фиг. 21 проиллюстрировано среднее арифметическое значение (± СП) диастолического кровяного давления в динамике по времени во время двойной слепой индукционной фазы с использованием выборки для анализа безопасности ESKETINTRD3005.
На Фиг. 22 представлен график общей оценки по шкале CADSS в динамике по времени во время двойной слепой индукционной фазы с использованием выборки для анализа безопасности.
На Фиг. 23 проиллюстрированы средние значения изменений, полученные методом наименьших квадратов (± СП), в общей оценке по шкале MADRS в динамике по времени LOCF ANCOVA во время двойной слепой индукционной фазы с использованием полной выборки для анализа. Среднее по МНК и СП основаны на модели ковариационного анализа (ANCOVA) с изменением относительно исходного уровня в качестве переменной ответа и факторами для лечения (эскетамин интраназально+АД перорально, АД перорально+плацебо интраназально), областью и классом перорального антидепрессанта (SNRI или SSRI), и исходным значением в качестве ковариаты. Результаты не корректируют для переоценки размера выборки. Отрицательное изменение оценки указывает на улучшение.
На Фиг. 24 проиллюстрирована форест-диаграмма для общей оценки по шкале MADRS, показывающая рассчитанную методом наименьших квадратов среднюю по лечению разницу изменения относительно исходного уровня (95% доверительный интервал) до 28 суток СМПИ по подгруппе во время двойной слепой индукционной фазы с использованием полной выборки для анализа. Подгруппы с менее чем 5 субъектами не представлены. Результаты не корректируют для переоценки размера выборки.
На Фиг. 25 проиллюстрированы средние арифметические значения изменений (± СП) в общей оценке по шкале MADRS в динамике по времени, наблюдаемые в возрастной группе 65-74 года, во время двойной слепой индукционной фазы с использованием полной выборки для анализа.
На Фиг. 26 проиллюстрированы средние арифметические значения изменений (± СП) в общей оценке по шкале MADRS в динамике по времени, наблюдаемые в возрастной группе ≥75 лет, во время двойной слепой индукционной фазы с использованием полной выборки для анализа.
На Фиг. 27 проиллюстрированы средние значения изменений, полученные методом наименьших квадратов (± СП), в общей оценке по шкале MADRS в динамике по времени (наблюдаемые случаи) в СМПИ для стадии 1 во время двойной слепой индукционной фазы с использованием полной выборки для анализа. Среднее по МНК и СП основаны на смешанной модели повторных измерений (СМПИ) с изменением относительно исходного уровня в качестве переменной ответа и условиями модели с фиксированными эффектами для лечения (эскетамин интраназально+АД перорально, АД перорально+плацебо интраназально), сутками, областью, классом перорального антидепрессанта (SNRI или SSRI) и лечением по суткам, а также исходным значением в качестве ковариаты. Результаты не корректируют для переоценки размера выборки. Отрицательное изменение оценки указывает на улучшение.
На Фиг. 28 проиллюстрированы средние значения изменений, полученные методом наименьших квадратов (± СП), в общей оценке по шкале MADRS в динамике по времени (наблюдаемые случаи) в СМПИ для стадии 2 во время двойной слепой индукционной фазы с использованием полной выборки для анализа. Среднее по МНК и СП основаны на смешанной модели повторных измерений (СМПИ) с изменением относительно исходного уровня в качестве переменной ответа и условиями модели с фиксированными эффектами для лечения (эскетамин интраназально+АД перорально, АД перорально+плацебо интраназально), сутками, областью, классом перорального антидепрессанта (SNRI или SSRI) и лечением по суткам, а также исходным значением в качестве ковариаты. Результаты не корректируют для переоценки размера выборки. Отрицательное изменение оценки указывает на улучшение.
На Фиг. 29 проиллюстрировано среднее значение изменения, полученное методом наименьших квадратов, в общей оценке по шкале MADRS в динамике по времени (наблюдаемые случаи) у пациентов с ТРД в США в возрасте ≥65 лет. Общая оценка MADRS соответствует диапазону 0-60; более высокая оценка указывает на более тяжелое состояние.
На Фиг. 30 проиллюстрировано частотное распределение тяжести заболевания на основании оценки CGI-S на исходном уровне и для конечной точки двойной слепой фазы (LOCF). Оценка CGI-S находится в диапазоне от 1 (норма, полное отсутствие заболевания) до 7 (среди наиболее тяжелобольных пациентов). Оценка CGI-S находится в диапазоне от 1 (норма, полное отсутствие заболевания) до 7 (среди наиболее тяжелобольных пациентов).
На Фиг. 31 проиллюстрирован процент пациентов в США в возрасте ≥65 лет с ТРД, достигших ответа (наблюдаемый случай) по оценке MADRS. Оцениваемый клиницистом ответ был определен как a ≥ 50% снижение относительно исходного уровня общей оценки по шкале MADRS.
На Фиг. 32 проиллюстрирован процент пациентов в США в возрасте ≥65 лет с ТРД, достигших ремиссии (наблюдаемый случай) по оценке MADRS. Оцениваемая клиницистом ремиссия была определена как общая оценка по шкале MADRS ≤12.
На Фиг. 33 проиллюстрирован процент пациентов в США в возрасте ≥65 лет с ТРД, достигших оцениваемой пациентом ремиссии (наблюдаемый случай) по оценке PHQ-9.
На Фиг. 34 проиллюстрирован процент пациентов в США в возрасте ≥65 лет с ТРД, которые имели клинически существенный ответ по оценке CGI-S.
На Фиг. 35 проиллюстрирован процент пациентов в США в возрасте ≥65 лет с ТРД, которые имели клинически значимый ответ по оценке CGI-S. Клинически существенный и клинически значимый ответы были определены как снижение в CGI-S на ≥1 пункт или на ≥2 пункта относительно исходного уровня соответственно.
На Фиг. 36 проиллюстрировано частотное распределение тяжести заболевания на основании оценки по шкале общего клинического впечатления о тяжести заболевания (CGI-S, англ. «clinical global impression-severity») на исходном уровне и для конечной точки двойной слепой фазы. Медианные оценки CGI-s повышались от исходного уровня до конечного момента времени в группе ЭСК + АД, медианное (диапазон) изменение относительно исходного уровня составляло -1,0 (-4,1), медианное (диапазон) изменение от исходною уровня до конечного момента времени в группе АД + ПБО составляло 0 (4,3). Отношение шансов для улучшения оценки CGI-5 составляло 5,3, что указывает на то, что пациенты, получавшие ЭСК + АД, имели в 5,3 раз большую вероятность по сравнению с получавшими АД + ПБО улучшения оценки CGI-S в двойной слепой индукционной фазе.
На Фиг. 37 показан дизайн исследования для оценки эффективности и безопасности интраназального введения эскетамина в целях быстрого снижения симптомов большого депрессивного расстройства, включая суицидальную идеацию, у субъектов с угрозой самоубийства. a) Рандомизация будет стратифицирована по результатам оценки врачом потребности субъекта в лечении антидепрессантами (монотерапия антидепрессантами или антидепрессант плюс усиливающая терапия) до рандомизации на 1 сутки; b) Будет начато стандартное лечение антидепрессантами или оптимизировано на 1 сутки; с) Фаза скрининга может быть продлена до 48 часов до введения интраназальной дозы на 1 сутки после консультации с медицинским наблюдателем спонсора.
На Фиг. 38 показаны средние значения изменений относительно исходного уровня, полученные методом наименьших квадратов (± СП), для общей оценки по шкале MADRS в динамике по времени в двойной слепой индукционной фазе с использованием данных последнего документированного значения. Среднее по МНК и СП основаны на модели ковариационного анализа (ANCOVA) с лечением (плацебо, 84 мг эскетамина), терапией антидепрессантами (монотерапия АД, АД плюс аугментационная терапия) и аналитическим центром в качестве факторов и исходным значением в качестве ковариаты.
На Фиг. 39 показаны средние изменения (СП) в CGJSR относительно исходного уровня до 4 и 24 часов. Среднее изменение и СП основано на степени изменения относительно данных исходного уровня (LOCF) и проанализировано с помощью модели ANCOVA с лечением, аналитическим центром и SoC в качестве фиксированных эффектов и исходного значения (не ранжированного) в качестве ковариаты.
На Фиг. 40 приведена корреляция процента пациентов с разрешением риска самоубийства в моменты времени 4 и 24 часов.
На Фиг. 41 показано частотное распределение оценок по шкале SIBAT на двойном слепом исходном уровне, сутки 1, 4 часа после приема дозы, двойной слепой конечный показатель и конечный показатель наблюдения. Клиническое глобальное заключение о риске самоубийств соответствует диапазону от 0-6. 0: Не суицидальный; 1: Присутствуют периодические идеи самоубийства, но никакого специального вмешательства не требуется; 2: Присутствуют некоторые очевидные идеи самоубийства; пациенту рекомендуется обращаться за профессиональной помощью по мере необходимости; 3: Риск суицида требует амбулаторного наблюдения; но более никакого незамедлительного вмешательства; 4: Риск суицида требует незамедлительного вмешательства, но не госпитализации (например, приема лекарства, срочного амбулаторного наблюдения); 5: Риск суицида требует незамедлительной госпитализации, но без предупреждения суицида; 6: Риск суицида требует госпитализации с предупреждением суицида.
На Фиг. 42 показаны средние значения изменений относительно исходного уровня в MADRS, полученные методом наименьших квадратов (СП), в моменты времени до 4 часов (первичный конечный результат) и около 24 часов.
На Фиг. 43 приведена корреляция процента пациентов с соответствующими ответом MADRS и ремиссией на 1, 2 сутки и в конечной точке.
На Фиг. 44 приведена корреляция процента пациентов с ремиссией в ДС конечной точке и во время последующего наблюдения.
На Фиг. 45 показаны средние значения изменений относительно исходного уровня, полученные методом наименьших квадратов (± СП), для общей оценки по шкале BSS в динамике по времени в двойной слепой индукционной фазе с использованием данных последнего документированного значения.
На Фиг. 46 и 47 представлены средние значения кровяного давления в динамике по времени по группе лечения в двойной слепой фазе.
На Фиг. 48 представлен график общей оценки по шкале CADSS в динамике по времени во время двойной слепой индукционной фазы (исследование ESKETINSUI2001: выборка для анализа безопасности).
На Фиг. 49 представлен дизайн исследования для примера 4.
На Фиг. 50 представлена блок-схема с обобщенной информацией по субъектам и лечению из примера 4. (a) 1 пациент со стабильным ответом был ошибочно рандомизирован как находящийся в стабильной ремиссии; (b) 1 субъект, не отвечающий критериям стабильной ремиссии и стабильного ответа в конце фазы оптимизации, был ошибочно рандомизирован как имеющий стабильный ответ.
На Фиг. 51 представлена совокупная доля субъектов, у которых не наблюдался рецидив; фаза поддержания дозы (расчет по методу Каплана - Мейера) (полная (пациенты со стабильной ремиссией) выборка для анализа) для примера 4.
На Фиг. 52 представлена совокупная доля субъектов, у которых не наблюдался рецидив; фаза поддержания дозы (расчет по методу Каплана - Мейера) (полная (пациенты с клиническим ответом) выборка для анализа) для примера 4.
На Фиг. 53 показано среднее арифметическое значение (± СП) систолического кровяного давления в динамике по времени; фаза поддержания дозы (выборка для анализа безопасности (MA)) для примера 4.
На Фиг. 54 показано среднее арифметическое значение (± СП) диастолического кровяного давления в динамике по времени; фаза поддержания дозы (выборка для анализа безопасности (MA)) для примера 4.
На Фиг. 55 показано среднее арифметическое значение (± СП) общей оценки по шкале CADSS в динамике по времени; фаза поддержания дозы (выборка для анализа безопасности (MA)) для примера 4.
На Фиг. 56 представлена форест-диаграмма отношения рисков по подгруппе: Регрессия Кокса (полная (пациенты со стабильной ремиссией) выборка для анализа) для примера 4. Оценки отношения рисков для подгрупп без событий в любой из ветвей не отображены. Подгруппы с менее чем 5 субъектами не представлены.
На Фиг. 57 представлен дизайн исследования для примера 5. На момент включения в исследование не демонстрирующие клинический ответ субъекты продолжали получать тот же пероральный антидепрессант, что и в исследовании ESKETINTRD3005. Новый пероральный АД предназначен только для напрямую включенных субъектов.
На Фиг. 58 показано частотное распределение частот для CGI-S из примера 5.
На Фиг. 59 показано среднее арифметическое значение (± СП) обнаружения - внимательности (время простой реакции) (все включенные выборки для анализа) для возрастной группы ≥ 65 лет из примера 5.
На Фиг. 60-62 показан уровень нарушения для EQ-5D-5L посредством определения тревожности/депрессии, обычной активности и боли/дискомфорта, соответственно.
На Фиг. 63 показано среднее арифметическое значение (± СП) систолического кровяного давления в динамике по времени; фазы индукции и оптимизации/поддержания дозы (все включенные выборки для анализа) для примера 5.
На Фиг. 64 показано среднее арифметическое значение (± СП) диастолического кровяного давления в динамике по времени; фазы индукции и оптимизации/поддержания дозы (все включенные выборки для анализа) для примера 5.
На Фиг. 65 представлен график общей оценки по шкале CADSS в динамике по времени во время фазы индукции и оптимизации/поддержания дозы (все включенные выборки для анализа) для примера 5.
На Фиг. 66 представлен график, показывающий среднее значение ( ± СП) для общей оценки по подшкале положительных симптомов краткой психиатрической оценочной шкалы в динамике по времени во время фаз индукции и оптимизации/поддержания дозы (выборка для анализа из всех включенных субъектов) для примера 5.
На Фиг. 67 показаны средние значения общей оценки по шкале MADRS в динамике по времени в фазах ИНД и ОП/ПО на основании данных по наблюдаемым случаям для примера 5.
На Фиг. 68 показан ответ для пациентов, имеющих ответ с ≥ 50% снижением относительно исходного уровня и ремиссией с MADRS ≤ 12.
На Фиг. 69 показаны средние значения общей оценки по шкале PHQ-9 в динамике по времени в фазах ИНД и ОП/ПО на основании данных по наблюдаемым случаям для примера 5.
На Фиг. 70 представлена иллюстрация снижения и повышения активности. MK-801-индуцированные изменения активности описаны в разделе 3.3 примера 6. Макропатология не выявила каких-либо изменений в ткани.
На Фиг. 71 показано исследование нейротоксичности с повторными дозами. Окрашенная гематоксилином-эозином (HE) ретроспленальная кора демонстрирует отсутствие нейронального некроза у обработанной эскетамином-HCl крысы (54 мг/сутки) и его присутствие у обработанной (+)MK-801 малеатом крысы, как описано в примере 6. На Фиг. 71A представлено изображение ретроспленальной коры обработанной эскетамином-HCl крысы (54 мг/сутки), демонстрирующее отсутствие нейронального некроза. На Фиг. 71B представлено изображение ретроспленальной коры от обработанного (+)MK-801 малеатом животного. Стрелками показаны некротические нейроны (сморщенная, эозинофильная цитоплазма с конденсированными ядрами). На Фиг. 71C представлено увеличенное изображение некротических нейронов (стрелки) в ретроспленальной коре от обработанного (+)MK-801 малеатом животного.
На Фиг. 72 проиллюстрировано исследование нейротоксичности с повторными дозами. Окрашенная Fluoro-Jade (FJ) ретроспленальная кора демонстрирует отсутствие нейронального некроза у обработанной эскетамином-HCl крысы (54 мг/сутки) и его присутствие у обработанной (+)MK-801 малеатом крысы, как описано в примере 6. На Фиг. 72A представлено изображение ретроспленальной коры обработанной эскетамином-HCl крысы (54 мг/сутки), демонстрирующее отсутствие нейронального некроза. На Фиг. 72B представлено изображение ретроспленальной коры от обработанного (+)MK-801 малеатом животного.
На Фиг. 73 представлена блок-схема, демонстрирующая диспозицию пациентов из примера 7. Семь участников вступили в фазу наблюдения раньше, чем 74 сутки, получая на протяжении 2 недель исследуемый препарат во время открытой фазы исследования. Пациенты могли иметь несколько причин неудачи при скрининге. Участники могли иметь несколько причин неудачи при скрининге. Участники вошли в фазу последующего наблюдения, если они не выбрали прекращение исследования.
На Фиг. 74A и 74B представлены линейные графики, показывающие среднее значение изменения (± СП) общей оценки по шкале MADRS в динамике по времени в двойной слепой фазе из примера 7. Изменения показаны в периоды 1(A) и 2(B). Период 2 состоял только из участников, получавших плацебо во время периода 1, и имел умеренные или тяжелые симптомы (n=28). Период 1 (сутки 1-8) и период 2 (сутки 8-15) обсуждаются в секции «Дизайн» раздела «Способы» и показаны по вертикальной оси на Фиг. 73. ИУ обозначает исходный уровень; 2H, 2 часа после введения дозы. Планки погрешностей указывают СП. Период 2 состоял только из участников, принимавших плацебо во время периода 1 и имевших умеренные или тяжелые симптомы (n=28).
На Фиг. 75 представлен линейный график, показывающий общую оценку по шкале MADRS, среднее значение изменения относительно исходного уровня до конечной точки последующего наблюдения для участников, которые были включены в открытую фазу примера 7. Период 1 (сутки 1-8), период 2 (сутки 8-15), открытый период (сутки 15-74) и период последующего наблюдения (сутки 74-130) обсуждаются в секции «Дизайн» раздела «Способы» и показаны по вертикальной оси на Фиг. 73. ИУ обозначает исходный уровень; планки погрешностей, СП.
На Фиг. 76A и 76B представлены линейные графики, показывающие среднее значение (± СП) общей оценки по шкале MADRS в динамике по времени в двойной слепой фазе из примера 7. Период 2 состоял только из участников, принимавших плацебо во время периода 1 и имевших умеренные или тяжелые симптомы (n=28). В момент времени 2 часа использовали модифицированную шкалу MADRS с переносом далее исходных оценок характеристик сна и аппетита.
На Фиг. 77 представлен график среднего значения систолического кровяного давления в динамике по времени для участников, которые получали одинаковое лечение в течение обоих периодов во время двойной слепой фазы в примере 7.
На Фиг. 78 представлен график среднего значения диастолического кровяного давления в динамике по времени для участников, которые получали одинаковое лечение в течение обоих периодов во время двойной слепой фазы в примере 7.
На Фиг. 79 представлен график среднего значения общей оценки по шкале CADSS в динамике по времени для участников, которые получали одинаковое лечение в течение обоих периодов в примере 7.
На Фиг. 80 представлен график профиля средняя плазменная концентрация - время для эскетамина.
На Фиг. 81 представлен график профиля средняя плазменная концентрация - время для норэскетамина.
На Фиг. 82A и 82B представлены значения Cмакс в зависимости от дозы и ППКпосл в зависимости от дозы, соответственно, для данных примера 10. Показана линия регрессии для Cмакс (r=0,53) и ППКпосл (r=0,70).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам лечения депрессии (например, большого депрессивного расстройства), включающим введение нуждающемуся в этом пациенту клинически доказанного безопасного и терапевтически эффективного количества эскетамина. В некоторых вариантах осуществления указанные способы предназначены для лечения терапевтически рефрактерной депрессии или терапевтически резистентной депрессии. В других вариантах осуществления указанное лекарственное средство предназначено для лечения суицидальной идеации.
Эти способы преимущественно позволяют адаптировать эффективную схему лечения для пациентов с депрессией. Такие пациенты включают тех, у кого уже диагностированы БДР, ТРД, кто имеет суицидальные наклонности или кто не проходил лечение депрессии.
Также описаны способы поддержания стабильной ремиссии или стабильного ответа, достигаемых пациентом с депрессией после введения терапевтически эффективного количества эскетамина во время фазы начального введения. Такие способы включают продолжение введения терапевтически эффективного количества эскетамина в течение по меньшей мере пяти месяцев во время фазы последующего введения.
Таким образом, также предложены способы долгосрочного лечения депрессии у пациента. Эти способы включают введение нуждающемуся в лечении пациенту имеющего клинически доказанную безопасность и клинически доказанную эффективность терапевтически эффективного количества эскетамина в течение по меньшей мере шести месяцев. Желательно, чтобы когнитивная деятельность пациента оставалась стабильной на основании исходного измерения после шести месяцев лечения. В некоторых вариантах осуществления лечение может длиться по меньшей мере около одного года, по меньшей мере около 18 месяцев или по меньшей мере около двух лет. Например, долгосрочное лечение может включать в себя продолжительность в диапазоне от около шести месяцев до около двух лет. Лечение также можно продолжать в течение более длительных периодов времени, в том числе, без ограничения, 4, 5, 6, 7, 8, 9, 10 или более лет, согласно определению лечащего врача. В некоторых вариантах осуществления дозу эскетамина вводят сначала дважды в неделю в течение до четырех недель во время индукционной фазы, а после этого вводят реже, чем два раза в неделю.
В некоторых вариантах осуществления настоящего изобретения эскетамин можно вводить в комбинации с одним или более антидепрессантами, как описано в данном документе, предпочтительно в комбинации с одним-тремя антидепрессантами, более предпочтительно в комбинации с одним-двумя антидепрессантами.
В некоторых вариантах осуществления настоящего изобретения эскетамин можно вводить в комбинации с одним или более антидепрессантами, и дополнительно в комбинации с одним или более антипсихотическими средствами, описанными в данном документе.
В одном варианте осуществления настоящее изобретение относится к комбинированной терапии, включающей эскетамин и один или более антидепрессантов; причем эскетамин вводят в виде неотложного лечения. В другом варианте осуществления настоящее изобретение относится к комбинированной терапии, включающей эскетамин и один или более антидепрессантов, причем эскетамин вводят в виде неотложного лечения, а один или более антидепрессантов вводят в виде постоянного лечения.
В других вариантах осуществления, например во время индукционной фазы, эскетамин можно использовать в качестве монотерапии, а не в комбинации с любыми другими активными соединениями.
Некоторые количественные выражения, приведенные в данном документе, не уточнены с помощью термина «около». Понятно, что, независимо от того, используется ли термин «около» явным образом или нет, предполагается, что каждое приведенное в данном документе численное значение относится к фактическому данному значению, и предполагается, что оно относится к приближению к такому данному значению, которое можно надлежащим образом оценить на основании обычной квалификации в данной области техники, включая приближения, связанные с условиями проведения эксперимента и/или измерения для такого данного значения.
В контексте данного документа, если не указано иное, термин «эскетамин» означает (S)-энантиомер кетамина, т.е. соединение формулы (I):
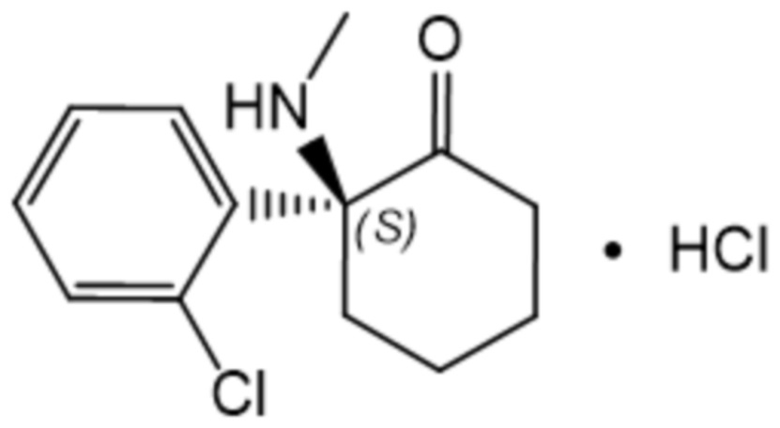 (I),
(I),
также известное как (S)-2-(2-хлорфенил)-2-(метиламино)циклогексанон. «Эскетамин» также означает соль, например, хлоридную соль, такую как гидрохлоридная соль, (S)-энантиомера кетамина, т.е. соединение формулы (II):
(II),
также известное как гидрохлорид (S)-2-(2-хлорфенил)-2-(метиламино)циклогексанона.
В некоторых вариантах осуществления эскетамин, по существу, не содержит (R)-энантиомер кетамина, т.е. соединение формулы (III):
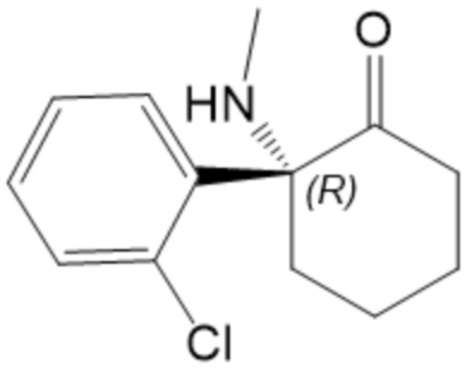 (III)
(III)
В других вариантах осуществления эскетамин содержит менее чем около 10% по массе на основании массы образца эскетамина (R)-энантиомера кетамина. В дополнительных вариантах осуществления эскетамин содержит менее чем около 10, 9, 8, 7, 6, 5, 4, 3, 2, 1, 0,5, 0,1, 0,005 или 0,001% по массе на основании массы образца эскетамина (R)-энантиомера кетамина. В других вариантах осуществления эскетамин содержит от около 0,001% до около 10% по массе на основании массы образца эскетамина (R)-энантиомера кетамина. В дополнительных вариантах осуществления эскетамин содержит от около 0,001 до около 10%, от около 0,001 до около 5%, от около 0,001 до около 1, от около 0,001 до около 0,5, от около 0,001 до около 0,1, от около 0,1 до около 5, от около 0,1 до около 1, от около 0,1 до около 5 или от около 0,5 до около 5% по массе на основании массы образца эскетамина (R)-энантиомера кетамина.
Термин «эскетамин» также может включать другие фармацевтически приемлемые соли, которые могут быть легко выбраны специалистами в данной области техники. Подразумевается, что «фармацевтически приемлемая соль» представляет собой соль эскетамина, которая является нетоксичной, биологически переносимой или иным образом биологически подходящей для введения субъекту. В общем случае смотрите G.S. Paulekuhn, «Trends in Active Pharmaceutical Ingredient Salt Selection based on Analysis of the Orange Book Database», J. Med. Chem., 2007, 50:6665-72, S.M. Berge, "Pharmaceutical Salts", J Pharm Sci., 1977, 66:1-19, и Handbook of Pharmaceutical Salts, Properties, Selection, and Use, Stahl and Wermuth, Eds., Wiley-VCH and VHCA, Zurich, 2002. Примерами фармацевтически приемлемых солей являются те, которые являются фармакологически эффективными и подходящими для введения пациентам без неспецифической токсичности, раздражения или аллергической реакции.
Примеры фармацевтически приемлемых солей включают сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, моногидрогенфосфаты, дигидрогенфосфаты, метафосфаты, пирофосфаты, бромиды (такие как гидробромиды), йодиды (такие как гидройодиды), ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изобутираты, капроаты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себацинаты, фумараты, малеаты, бутин-1,4-диоаты, гексин-1,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты, динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, сульфонаты, ксилолсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, γ-гидроксибутираты, гликоляты, тартраты, метансульфонаты, пропансульфонаты, нафтален-1-сульфонаты, нафтален-2-сульфонаты и манделаты. В частности, соль эскетамина представляет собой гидрохлоридную соль.
В определенных вариантах осуществления настоящего изобретения эскетамин вводят интраназально. В определенных вариантах осуществления настоящего изобретения эскетамин вводят интраназально в виде соответствующей гидрохлоридной соли. В определенных вариантах осуществления настоящего изобретения эскетамин вводят интраназально в виде соответствующей гидрохлоридной соли в 16,14% масса/объем растворе (эквивалент 14% масса/объем основания эскетамина).
В определенных вариантах осуществления настоящего изобретения эскетамин вводят интраназально в виде раствора, содержащего 161,4 мг/мл гидрохлорида эскетамина (эквивалент 140 мг/мл основания эскетамина), 0,12 мг/мл этилендиаминтетрауксусной кислоты (ЭДТУ) и 1,5 мг/мл лимонной кислоты при pH 4,5 в воде. В определенных вариантах осуществления настоящего изобретения эскетамин вводят интраназально, причем интраназальная доставка включает 100 мкл раствора, содержащего 161,4 мг/мл гидрохлорида эскетамина (эквивалент 140 мг/мл основания эскетамина), 0,12 мг/мл этилендиаминтетрауксусной кислоты (ЭДТУ) и 1,5 мг/мл лимонной кислоты при pH 4,5 в воде. В определенных вариантах осуществления эскетамин вводят интраназально, используя насос для назального впрыскивания, причем насос доставляет 100 мкл раствора, содержащего 161,4 мг/мл гидрохлорида эскетамина (эквивалент 140 мг/мл основания эскетамина), 0,12 мг/мл этилендиаминтетрауксусной кислоты (ЭДТУ) и 1,5 мг/мл лимонной кислоты при pH 4,5 в воде.
В целом один насос из назального распылительного устройства может быть выполнен с возможностью доставки от около 50 мкл до около 200 мкл раствора эскетамина в ноздрю пациента, в том числе около 60 мкл, около 70 мкл, около 80 мкл, около 90 мкл, около 100 мкл, около 110 мкл, около 120 мкл, около 130 мкл, около 140 мкл, около 150 мкл, около 160 мкл, около 170 мкл, около 180 мкл и около 200 мкл. Соответственно два насоса доставляют субъекту от около 100 мкл до около 400 мкл.
В определенных вариантах осуществления настоящего изобретения пациент, нуждающийся в лечении имеющим клинически доказанную безопасность и терапевтически эффективным количеством эскетамина, представляет собой пациента, страдающего от эпизода депрессии (например, большого депрессивного расстройства). В определенных вариантах осуществления настоящего изобретения нуждающийся пациент страдает от эпизода депрессии (например, большого депрессивного расстройства), причем эпизод депрессии (например, большое депрессивное расстройство) не отвечает на лечение по меньшей мере двумя пероральными антидепрессантами (т.е. пациент не отвечал на лечение по меньшей мере двумя пероральными антидепрессантами). В других вариантах осуществления нуждающийся пациент страдает от эпизода депрессии (например, большого депрессивного расстройства), причем эпизод депрессии (например, большое депрессивное расстройство) не отвечает на лечение двумя пероральными антидепрессантами (т.е. пациент старческого возраста не отвечал на лечение двумя пероральными антидепрессантами).
В определенных вариантах осуществления настоящего изобретения нуждающийся пациент страдает от депрессии (например, большого депрессивного расстройства). Например, это пациент, имеющий оценку 18 или более по шкале MADRS или оценку 4 или более по шкале CGI.
В контексте данного документа термин «депрессия» включает большое депрессивное расстройство, устойчивое депрессивное расстройство, сезонное аффективное расстройство, послеродовую депрессию, предменструальное дисфорическое расстройство, психогенную депрессию, ангедонию, меланхолию, депрессию среднего возраста, депрессию старшего возраста, депрессию вследствие идентифицируемых факторов стресса, терапевтически резистентную депрессию или их комбинации. В определенных вариантах осуществления депрессия представляет собой большое депрессивное расстройство. В других вариантах осуществления большое депрессивное расстройство сопровождается характеристиками меланхолии или тревожным расстройством. В дополнительных вариантах осуществления депрессия представляет собой терапевтически резистентную депрессию.
В контексте данного документа термин «не демонстрирующий ответ» означает пациентов, которые полностью не восстанавливаются на антидепрессантах (например, изменение в 25% или менее относительно исходного уровня общей оценки MADRS).
В контексте данного документа термин «эпизод большого депрессивного расстройства» означает непрерывный период (например, около 2 недель или более), в течение которого пациент демонстрирует симптомы большого депрессивного расстройства, достаточные для соответствия критериям большой депрессии, определенным в Diagnostic and statistical Manual of Mental Disorders, 5th Edition: DSM 5.
В контексте данного документа термин «самоубийство» означает «акт лишения себя жизни». Смотрите http://en.wikipedia.org/wiki/Suicide - cite_note-7. Самоубийство включает попытку самоубийства или нефатальное суицидальное поведение, которое представляет собой нанесение себе повреждений с целью лишения себя жизни, которое не приводит к смерти. Попытка самоубийства - это последовательность действий, совершаемых по собственной инициативе индивида, который изначально ожидал, что этот комплекс действий приведет к его или ее собственной смерти.
В контексте данного документа «суицидальная идеация» относится к мыслям о самоубийстве или необычном зацикливании на нем, или мыслям о прекращении жизни или нежелании более жить дальше, необязательно сопровождающимся активными попытками воплотить их. Диапазон суицидальной идеации сильно варьируется от кратковременной до постоянной и может прогрессировать до подробного планирования, ролевого исполнения и неудачных попыток, которые могут предприниматься намеренно с целью неудачи или обнаружения, либо могут быть полностью направлены на достижение летального исхода. В некоторых вариантах осуществления пациента классифицируют как «суицидального», если пациент имеет среднюю исходную общую оценку по шкале MADRS около 38 или выше. В других вариантах осуществления пациента классифицируют как суицидального, если пациент имеет среднюю исходную оценку по шкале BBSS около 22 или выше. В дополнительных вариантах осуществления пациента классифицируют как суицидального, если пациент имеет оценку 6 или выше по клинической глобальной оценке риска суицида SIBAT. В других вариантах осуществления пациент имеет одну или более комбинаций этих оценок.
В контексте данного документа термины «совместная терапия», «комбинированная терапия», «дополнительное лечение», «дополнительная терапия», «комбинированное лечение» и «совместное введение» означают лечение нуждающегося в этом пациента путем введения эскетамина в комбинации с одним или более антидепрессантами, причем эскетамин и антидепрессант(ы) вводят любыми подходящими способами. В некоторых вариантах осуществления эскетамин вводят по схеме с одним - пятью антидепрессантами. В других вариантах осуществления эскетамин вводят по схеме с одним, двумя, тремя, четырьмя или пятью антидепрессантами. В других вариантах осуществления эскетамин вводят по схеме с одним или двумя антидепрессантами. В дополнительных вариантах осуществления эскетамин вводят по схеме с антидепрессантом, который в данный момент вводят пациенту. В других вариантах осуществления эскетамин вводят по схеме с отличным антидепрессантом. В дополнительных вариантах осуществления эскетамин вводят по схеме с антидепрессантом, который ранее не вводили пациенту. В других вариантах осуществления эскетамин вводят по схеме с антидепрессантом, который ранее вводили пациенту. Если эскетамин и антидепрессант(ы) вводят в отдельных дозированных формах, число вводимых суточных доз каждого из соединений могут быть одинаковыми или разными, и, как правило, разными. Дозу антидепрессанта можно применять в соответствии с предписаниями лечащего врача и/или согласно информации на этикетке, а дозу эскетамина применяют, как описано в данном документе. Как правило, пациент находится на одновременном лечении как антидепрессантом, так и эскетамином, причем оба вводят в соответствии с предписанными схемами дозирования.
Эскетамин и антидепрессант(ы) можно вводить одним путем или разными путями введения. Примеры подходящих способов введения включают, но не ограничиваются этим, пероральный, внутривенный (в/в), интраназальный (и/н), внутримышечный (в/м), подкожный (п/к), трансдермальный, буккальный или ректальный. В некоторых вариантах осуществления эскетамин вводят интраназально. В контексте данного документа, если не указано иное, термин «антидепрессант» означает любой фармацевтический агент, который можно использовать для лечения депрессии. Соответствующие примеры включают, без ограничения, ингибитор моноаминоксидазы, трициклическое соединение, ингибитор обратного захвата серотонина, норадренергический ингибитор обратного захвата серотонина, норадренергический и специфический серотонинергический агент или атипичное антипсихотическое средство. Другие примеры включают, но не ограничиваются этим, ингибиторы моноаминоксидазы, такие как фенелзин, транилципромин, моклобемид и т. п; трициклические соединения, такие как имипрамин, амитриптилин, дезипрамин, нортриптилин, доксепин, протриптилин, тримипрамин, кломипрамин, амоксапин и т.п.; тетрациклические соединения, такие как мапротилин и т.п.; нециклические соединения, такие как номифензин и т.п.; триазолопиридины, такие как тразодон и т.п.; ингибиторы обратного захвата серотонина, таких как флуоксетин, сертралин, пароксетин, циталопрам, эсциталопрам, флувоксамин и т.п.; антагонисты рецепторов серотонина, такие как нефазодон и т.п.; норадренергические ингибиторы обратного захвата серотонина, такие как венлафаксин, милнаципран, дес-венлафаксин, дулоксетин, лево-милнаципран и т.п.; норадренергические и специфические серотонинергические агенты, такие как миртазапин и т.п.; ингибиторы обратного захвата норадреналина, такие как ребоксетин, эдивоксетин и т.п.; атипические антипсихотические средства, такие как бупропион и т.п.; натуральные продукты, такие как кава-кава, зверобой и т.п.; диетические добавки, такие как S-аденозилметионин и т.п.; и нейропептиды, такие как тиреотропин-рилизинг гормон и т.п.; соединения, нацеленные на нейропептидные рецепторы, такие как антагонисты нейрокининовых рецепторов и т.п.; и гормоны, такие как трийодотиронин и т.п. В некоторых вариантах осуществления антидепрессант представляет собой имипрамин, амитриптилин, дезипрамин, нортриптилин, доксепин, протриптилин, тримипрамин, мапротилин, амоксапин, тразодон, бупропион, кломипрамин, флуоксетин, дулоксетин, эсциталопрам, циталопрам, сертралин, пароксетин, флувоксамин, нефазодон, венлафаксин, милнаципран, ребоксетин, миртазапин, фенелзин, транилципромин, моклобемид, кава-кава, зверобой, S-аденозилметионин, тиреотропин-рилизинг гормон, антагонист нейрокининовых рецепторов или трийодотиронин. Предпочтительно антидепрессант выбран из группы, состоящей из флуоксетина, имипрамина, бупропиона, венлафаксина и сертралина.
Специалист в данной области техники может легко определить терапевтически эффективные количества/уровни дозировок и схемы приема для антидепрессантов (например, ингибиторов моноаминоксидазы, трициклических соединений, ингибиторов обратного захвата серотонина, норадренергических ингибиторов обратного захвата серотонина, норадренергических и специфических серотонинергических агентов, ингибиторов обратного захвата норадреналина, натуральных продуктов, диетических добавок, нейропептидов, соединений, нацеленных на нейропептидные рецепторы, гормонов и других фармацевтических агентов, описанных в данном документе). Например, количества терапевтических дозировок и схемы приема фармацевтических агентов, одобренных для продажи, находятся в открытом доступе, например, указаны на этикетках упаковок, в руководствах по стандартным дозировкам, в справочниках по стандартным дозировкам, таких как Physician’s Desk Reference (Medical Economics Company или онлайн на http:///www.pdrel.com), или доступны из других источников.
В контексте данного документа термин «антипсихотическое средство» включает, но не ограничивается этим:
(a) типичные или традиционные антипсихотические средства, такие как фенотиазины (например, хлорпромазин, тиоридазин, флуфеназин, перфеназин, трифлуоперазин, левомепромазин), тиоксантены (например, тиотиксен, флупентиксол), бутирофеноны (например, галоперидол), дибензоксазепины (например, локсапин), дигидроиндолоны (например, молиндон), замещенные бензамиды (например, сульприд, амисульприд) и т.п.; и
(b) атипичные антипсихотические средства и стабилизаторы настроения, такие как палиперидон, клозапин, рисперидон, оланзапин, кветиапин, зотепин, зипрасидон, илоперидон, пероспирон, блонансерин, сертиндол, ORG-5222 (Organon) и т.п.; и другие, такие как сонепипразол, арипипразол, немонаприд, SR-31742 (Sanofi), CX-516 (Cortex), SC-111 (Scotia), NE-100 (Taisho), дивалпроат (стабилизатор настроения) и т.п.
В одном из вариантов осуществления «атипичное антипсихотическое средство» выбрано из группы, состоящей из арипипразола, кветиапина, оланзапина, рисперидона и палиперидона. В другом варианте осуществления атипичное антипсихотическое средство выбрано из группы, состоящей из арипипразола, кветиапина, оланзапина и рисперидона; предпочтительно атипичное антипсихотическое средство выбрано из группы, состоящей из арипипразола, кветиапина и оланзапина.
В контексте данного документа термин «терапевтически рефрактерная или терапевтически резистентная депрессия» и сокращение «ТРД» определены как большое депрессивное расстройство у пациента, который в текущем депрессивном эпизоде не отвечает должным образом на по меньшей мере два разных антидепрессанта, предпочтительно от двух до пяти антидепрессантов. В других вариантах осуществления ТРД определена как большое депрессивное расстройство у пациента, который в текущем депрессивном эпизоде не отвечал по меньшей мере на два пероральных антидепрессанта при адекватных дозе и длительности.
Специалисту в данной области техники будет понятно, что отсутствие ответа на адекватный курс данного антидепрессанта можно определить ретроспективно или перспективно. В одном варианте осуществления по меньшей мере один из случаев отсутствия ответа на адекватный курс антидепрессанта определяют перспективно. В другом варианте осуществления по меньшей мере два случая отсутствия ответа на адекватный курс антидепрессанта определяют перспективно. В другом варианте осуществления по меньшей мере один из случаев отсутствия ответа на адекватный курс антидепрессанта определяют ретроспективно. В другом варианте осуществления по меньшей мере два случая отсутствия ответа на адекватный курс антидепрессанта определяют ретроспективно в текущем депрессивном эпизоде.
«По меньшей мере два пероральных антидепрессанта» или «по меньшей мере два разных пероральных антидепрессанта» вводили пациенту в адекватной дозе, которая может быть определена лечащим врачом. Аналогично, антидепрессант вводили в течение необходимого времени, определенного лечащим врачом.
В контексте данного документа, если не указано иное, термины «терапия», «лечение» и т.п. включают ведение субъекта или пациента (предпочтительно млекопитающего, более предпочтительно человека) и уход за ним для противостояния заболеванию, патологическому состоянию или расстройству и включает введение соединения по настоящему изобретению для предотвращения возникновения симптомов или осложнений, для облегчения симптомов или осложнений, или для устранения заболевания, патологического состояния или расстройства.
В контексте данного документа, если не указано иное, термин «клинически доказанный» (используемый независимо или для модификации терминов «безопасный» и/или «эффективный») означает, что доказательство было доказано клиническим испытанием фазы III, чего достаточно для соответствия стандартам одобрения Управлением США по контролю качества продуктов питания и лекарственных средств, или аналогичным исследованием для допуска на рынок ЕАЛС. Предпочтительно для клинического доказательства эффектов эскетамина используют рандомизированное двойное слепое контролируемое исследование с надлежащей выборкой для исследования эскетамина. Наиболее предпочтительно, чтобы для того, чтобы клинически доказать эффекты эскетамина при лечении большого депрессивного расстройства, например, терапевтически резистентной депрессии, это было бы рандомизированное двойное слепое активно контролируемое исследование интраназального эскетамина в гибкой дозе (28 мг, 56 мг или 84 мг ± 20%), вводимого вместе с новым или недавно появившимся пероральным антидепрессантом, по сравнению с новым или недавно появившимся пероральным антидепрессантом (активный препарат сравнения) плюс плацебо, причем состояние пациента оценивается с помощью описанных в данном документе методик, таких как MADRS, шкала Гамильтона, CGI, шкала Бека для депрессии, QIDS или PHQ-9, включая оценки, начиная с 1 суток до 28 суток, а также оценки во время периодов последующего введения, как описано в данном документе.
В контексте данного документа, если не указано иное, термин «имеющий клинически доказанную эффективность» означает, что эффективность лечения была доказана клиническим исследованием фазы III как статистически значимая, т.е. результаты клинического исследования маловероятно получены случайно с уровнем значимости менее 0,05, или результаты клинической эффективности достаточны для соответствия стандартам одобрения Управлением США по контролю качества продуктов питания и лекарственных средств, или аналогичным исследованием для допуска на рынок ЕАЛС. Например, было клинически доказано, что эскетамин является эффективным для лечения пациентов с большим депрессивным расстройством, например, терапевтически резистентной депрессией, при введении гибких интраназальных доз в терапевтически эффективной дозе 28 мг, 56 мг или 84 мг (± 25%) и совместном введении с новым или недавно появившимся пероральным антидепрессантом, со снижением оценки по шкале MADRS по меньшей мере на около 50% по сравнению с определенной исходной оценкой по шкале MADRS пациента, в качестве части схемы дозирования, включая фазы индукции и поддержания, описанные в данном документе, и как конкретным образом описано в примерах.
В контексте данного документа, если не указано иное, термин «безопасный» применительно к фармацевтическому лечению (терапии) или комбинированной терапии означает отсутствие нежелательных побочных эффектов (таких как токсичность, раздражение или аллергическая реакция), соответствие с приемлемым соотношением польза/риск при применении способом по данному изобретению.
В контексте данного документа, если не указано иное, термин «имеющий клинически доказанную безопасность» означает, что безопасность лечения была доказана клиническим исследованием фазы III с помощью анализа данных исследования и результатов, позволяющих установить, что лечение не сопровождается нежелательными побочными эффектами и соответствует статистически значимой клинической пользе (например, эффективности), достаточной для соответствия стандартам одобрения Управлением США по контролю качества продуктов питания и лекарственных средств, или аналогичным исследованием для допуска на рынок ЕАЛС. Например, было клинически доказано, что эскетамин является безопасным для лечения пациентов с большим депрессивным расстройством, например, терапевтически резистентной депрессией, при введении гибких интраназальных доз в терапевтически эффективной дозе 28 мг, 56 мг или 84 мг (± 25%) и совместном введении с новым или недавно появившимся пероральным антидепрессантом в качестве части схемы дозирования, включая фазы индукции и поддержания, описанные в данном документе, и как конкретным образом описано в примерах.
В контексте данного документа термин «терапевтически эффективное количество» означает количество активного соединения или фармацевтического агента, которое вызывает биологически или медицински значимый ответ со стороны системы тканей животного или человека, подразумеваемый исследователем, ветеринаром, врачом или другим клиницистом, и который включает облегчение симптомов заболевания или расстройства, подлежащего лечению. Желательно, чтобы терапевтически эффективное количество представляло собой количество с клинически доказанной безопасностью и клинически доказанной эффективностью. В некоторых вариантах осуществления антидепрессант применяют в терапевтически эффективном количестве, определенном лечащим врачом. В других вариантах осуществления эскетамин применяют в терапевтически эффективном количестве.
Терапевтически эффективное количество эскетамина и/или антидепрессанта можно вводить во время фазы (фаз) инициации и/или последующей(их) фазы (фаз), как описано в данном документе. В некоторых вариантах осуществления терапевтически эффективное количество эскетамина составляет от около 20 до около 100 мг. В других вариантах осуществления терапевтически эффективное количество эскетамина составляет от около 30 до около 90 мг. В дополнительных вариантах осуществления терапевтически эффективное количество эскетамина составляет от около 40 до около 80 мг. В других вариантах осуществления терапевтически эффективное количество эскетамина составляет около 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99 или 100 мг. В дополнительных вариантах осуществления терапевтически эффективное количество составляет около 28 мг, около 56 мг или около 84 мг. В других вариантах осуществления терапевтически эффективное количество составляет около 56 мг или около 84 мг. В дополнительных вариантах осуществления терапевтически эффективное количество эскетамина составляет около 28 мг. В других вариантах осуществления терапевтически эффективное количество эскетамина составляет около 56 мг. В дополнительных вариантах осуществления терапевтически эффективное количество эскетамина составляет около 84 мг.
В контексте данного документа, если не указано иное, термины «субъект» и «пациент» относятся к животному, предпочтительно млекопитающему, наиболее предпочтительно человеку, которые являются объектом лечения, наблюдения или эксперимента. Предпочтительно, субъект или пациент испытывал и/или демонстрировал по меньшей мере один симптом заболевания или расстройства, подлежащего лечению и/или предотвращению.
В некоторых вариантах осуществления субъект или пациент является взрослым. В контексте данного документа термин «взрослый» относится к человеку возрастом от около 18 лет до около 65 лет.
В других вариантах осуществления субъект или пациент является человеком пожилого или старческого возраста. В контексте данного документа термины «пожилой» и «старческий» взаимозаменяемо используются для обозначения субъекта-человека в возрасте около 65 лет или старше. Пожилые пациенты в возрасте от ≥65 до ≤75 более восприимчивы к лечение, чем пациенты ≥75.
В дополнительных вариантах осуществления субъект или пациент является педиатрическим субъектом. В контексте данного документа термин «педиатрический» относится к субъекту-человеку в возрасте менее чем около 18 лет.
В контексте данного документа подразумевается, что термин «композиция» включает продукт, содержащий конкретные ингредиенты в конкретных количествах, а также к любому продукту, который можно получать, прямо или косвенно, из комбинаций конкретных ингредиентов в конкретных количествах.
В контексте данного документа термин «стабильная ремиссия» относится к пациенту, имеющему общую оценку по шкале MADRS 12 или менее в течение по меньшей мере 3 из последних 4 недель после достижения пациентом, по существу, полного ответа на эскетамин во время индукционной фазы. В определенных примерах вариантов осуществления пациенты, находящиеся на стадии «стабильной ремиссии», включают пациентов с одним изменением общей оценки по шкале MADRS более 12 или без оценки MADRS на 13 или 14 неделе после индукционной фазы. В других вариантах осуществления пациенты, находящиеся на стадии «стабильной ремиссии», включают пациентов с общей оценкой по шкале MADRS на 15 и 16 неделях, составляющей 12 или менее, после индукционной фазы.
В контексте данного документа термин «стабильный ответ» относится к пациенту со снижением общей оценки по шкале MADRS на 50% или более от исходного уровня (сутки 1 индукционной фазы; предварительная рандомизация/перед первой интраназальной дозой) на каждую из последних 2 недель после достижения пациентом, по существу, полного ответа на эскетамин во время индукционной фазы, но не отвечающему критериям стабильной ремиссии.
Как указано выше, описаны способы лечения депрессии у пациента. Указанные способы включают введение эскетамина в одной, двух или, необязательно, трех фазах, т.е. начальной и последующих фазах введения. В некоторых вариантах осуществления фазы включают начальную индукционную фазу, продленную индукционную фазу, фазу поддержания или любую их комбинацию. Соответственно, в каждой фазе вводят эффективное количество эскетамина. Врач может оценивать состояние пациента для определения наиболее подходящих начальной/индукционной и поддерживающей доз для пациента из диапазона дозировок и частот введения, определенных в данном документе. Эффективное количество эскетамина может быть одинаковым для каждой фазы или может отличаться.
Описанные в данном документе способы позволяют оптимизировать дозировки эскетамина для введения пациенту, у которого наблюдается депрессия или который к ней предрасположен, во время «фазы оптимизации». Оптимизацию может рассматривать как часть фазы поддержания, которая следует за индукционной фазой. В некоторых вариантах осуществления описанные в данном документе способы не требуют корректировки дозировки эскетамина. Фактически, эскетамин можно вводить во время обсуждаемых в данном документе фаз (например, индукции и поддержания) при самой низкой частоте введения, при которой у пациента наблюдается и сохраняется ответ на эскетамин. Было установлено, что эффективное количество эскетамина составляет от около 28 до около 84 мг.
В контексте данного документа термин «индукционная фаза» или «фаза однократного введения» относится к периоду времени, в течение которого эскетамин изначально вводят пациенту. В некоторых вариантах осуществления индукционная фаза является достаточно длинной, чтобы обеспечить надежное, стабильное снижение депрессивных симптомов. Индукционная фаза может зависеть от таких факторов, как, без ограничения, конкретный пациент и/или пол, возраст, вес, время введения, частота введения и сопутствующие заболевания конкретного пациента. Индукционная фаза может включать начальную индукционную фазу и продленную индукционную фазу. В целом индукционная фаза (начальная и продленная фазы вместе) может составлять период от около 4 до около 12 недель, от около 4 до около 11 недель, от около 4 до около 10 недель, от около 4 до около 9 недель, от около 4 до около 8 недель, от около 4 до около 7 недель, от около 4 до около 6 недель, от около 5 до около 12 недель, от около 5 до около 11 недель, от около 5 до около 10 недель, от около 5 до около 9 недель, от около 5 до около 8 недель, от около 5 до около 7 недель, от около 5 до около 6 недель, от около 6 до около 12 недель, от около 6 до около 11 недель, от около 6 до около 10 недель, от около 6 до около 9 недель, от около 6 до около 8 недель, от около 7 до около 12 недель, от около 7 до около 11 недель, от около 7 до около 10 недель, от около 7 до около 9 недель, от около 8 до около 12 недель, от около 8 до около 11 недель или от около 8 до около 10 недель. В некоторых вариантах осуществления весь индукционный период составляет от около 4 до около 8 недель.
В начальный индукционный период пациенту вводят терапевтически эффективное количество эскетамина с заданной частотой по меньшей мере два раза в неделю. В некоторых вариантах осуществления пациенту вводят терапевтически эффективное количество эскетамина с заданной частотой 3 раза в неделю. В том случае, когда введение происходит 3 раза в неделю, введение осуществляют на 1, 3 и 5 сутки недели ± 1 сутки. Начальная индукционная фаза, как правило, составляет период времени, в течение которого пациент отвечает на лечение, но не готов к переходу к фазе поддержания. В соответствующие моменты времени ответ пациента оценивает специалист в данной области техники. В некоторых вариантах осуществления ответ пациента оценивают ежесуточно. В некоторых вариантах осуществления ответ пациента оценивают дважды в неделю. В некоторых вариантах осуществления ответ пациента оценивают через сутки. В других вариантах осуществления ответ пациента оценивают в конце начальной индукционной фазы. Как правило, ответ пациента можно оценивать с помощью методик и тестов, известных специалистам в данной области техники. В некоторых вариантах осуществления определяют оценку пациента по шкале MADRS и используют в качестве определения, завершена ли начальная индукционная фаза. Начальная индукционная фаза желательно является длинной, чтобы обеспечить снижение депрессивных симптомов. В некоторых вариантах осуществления начальная индукционная фаза составляет период от около 1 до около 4 недель. В других вариантах осуществления индукционная фаза составляет до около 1 недели, до около 2 недель, до около 3 недель или до около 4 недель. В дополнительных вариантах осуществления начальный индукционный период составляет от около 1 до около 3 недель, от около 1 до около 2 недель, от около 2 до около 4 недель, от около 2 до около 3 недель, от около 3 до около 4 недель, 1 неделю, 2 недели, 3 недели, 4 недели, до 1 недели, до 2 недель, до 3 недель или до 4 недель. Эффективное количество эскетамина, вводимое во время начальной индукционной фазы, может быть определено лечащим врачом. В некоторых вариантах осуществления эффективное количество эскетамина, вводимое во время начальной индукционной фазы, составляет около 28 мг. В некоторых вариантах осуществления эффективное количество эскетамина, вводимое во время начальной индукционной фазы, составляет около 56 мг. В других вариантах осуществления эффективное количество эскетамина, вводимое во время начальной индукционной фазы, составляет около 84 мг.
В контексте данного документа термин «дважды в неделю» относится к частоте, которая составляет два раза в недельный (7-суточный) период. Например, в данном документе «дважды в неделю» может относиться к введению эскетамина. «Дважды в неделю» также может относиться к частоте мониторинга пациента во время одной или более фаз, обсуждаемых в данном документе. В некоторых вариантах осуществления дважды в неделю относится к частоте введения, которое происходит на 1 сутки и 2 сутки недели. В других вариантах осуществления дважды в неделю относится к частоте введения, которое происходит на 1 сутки и 3 сутки недели. В дополнительных вариантах осуществления дважды в неделю относится к частоте введения, которое происходит на 1 сутки и 4 сутки недели. В других вариантах осуществления дважды в неделю относится к частоте введения, которое происходит на 1 сутки и 5 сутки недели. «1 сутки» могут приходиться на любой день недели, включая, воскресенье, понедельник, вторник, среду, четверг, пятницу или субботу. Как правило, в отношении введения эскетамина, дважды в неделю относится к частоте введения, которое происходит на 1 сутки и 4 сутки недели. В случае пропуска дозы дозу следует принять как можно быстрее и после этого продолжить следовать предписанной схеме.
Среди некоторых популяциях пациентов (таких как пожилые люди) снижение депрессивных симптомов во время начальной индукционной фазы является недостаточным, и необходима продленная индукционная фаза. Во время продленной индукционной фазы продолжают введение терапевтически эффективного количества эскетамина с заданной частотой по меньшей мере два раза в неделю. В соответствующие моменты времени ответ пациента снова оценивает специалист в данной области техники. В некоторых вариантах осуществления ответ пациента оценивают ежесуточно. В некоторых вариантах осуществления ответ пациента оценивают дважды в неделю. В некоторых вариантах осуществления ответ пациента оценивают через сутки. Как правило, ответ пациента можно оценивать с помощью методик и тестов, известных специалистам в данной области техники. В некоторых вариантах осуществления определяют оценку пациента по шкале MADRS и используют в качестве определения, завершена ли продленная индукционная фаза. Продленная индукционная фаза желательно является длинной для достижения существенного снижения депрессивных симптомов с достижением, таким образом, по существу, полного ответа на эскетамин.
В контексте данного документа термин «по существу полный ответ на эскетамин» относится к пациенту со снижением оценки по шкале MADRS относительно исходного уровня до по меньшей мере 50% улучшения относительно исходного уровня. В некоторых вариантах осуществления, по существу, полный ответ на эскетамин относится к пациенту с оценкой по шкале MADRS, соответствующей по меньшей мере 50% улучшению относительно исходного уровня или на около -20 ниже исходной оценки пациента. В других вариантах осуществления, по существу, полный ответ включает снижение оценки по шкале MADRS, составляющее около -20 или менее, -19, или менее, -18 или менее, -17 или менее, -16 или менее, -15 или менее, -14 или менее, -13 или менее, -12 или менее, -11 или менее или -10 или менее. В дополнительных вариантах осуществления, по существу, полный ответ приводит к снижению исходной оценки пациента по шкале MADRS, составляющему от около -15 до около -20. По существу полный ответ на эскетамин также может быть получен, если оценки пациента по шкале MADRS снижаются на около 50% относительно оценки по шкале MADRS в начале лечения. Такой по существу полный ответ можно наблюдать в любой момент времени лечения эскетамином. В некоторых вариантах осуществления, по существу, полный ответ наблюдается, когда пациент имеет снижение общей оценки по шкале MADRS относительно исходного уровня через 4 часа после лечения. В других вариантах осуществления, по существу, полный ответ наблюдается, когда пациент имеет снижение общей оценки по шкале MADRS относительно исходного уровня через 2 суток после лечения.
Продленная индукционная фаза составляет период времени, в течение которого достигается, по существу, полный ответ на эскетамин. В некоторых вариантах осуществления продленная индукционная фаза составляет от около 1 до около 8 недель. В других вариантах осуществления продленная индукционная фаза составляет период до около 1 недели, до около 2 недель, до около 3 недель, до около 4 недель, до около 5 недель, до около 6 недель, до около 7 недель или до около 8 недель. В дополнительных вариантах осуществления продленный индукционный период составляет от около 1 до около 8 недель, от около 1 до около 7 недель, от около 1 до около 6 недель, от около 1 до около 5 недель, от около 1 до около 4 недель, от около 1 до около 3 недель, от около 2 до около 8 недель, от около 2 до около 7 недель, от около 2 до около 7 недель, от около 2 до около 6 недель, от около 2 до около 5 недель, от около 2 до около 4 недель, от около 3 до около 8 недель, от около 3 до около 7 недель, от около 3 до около 6 недель, от около 3 до около 5 недель, от около 4 до около 8 недель, от около 4 до около 7 недель, от около 4 до около 6 недель, от около 5 до около 8 недель, от около 5 до около 7 недель, около 1 недели, около 2 недель, около 3 недель, около 4 недель, около 5 недель, около 6 недель, около 7 недель или около 8 недель. Эффективное количество эскетамина, вводимое во время продленной индукционной фазы, может быть определено лечащим врачом. В некоторых вариантах осуществления эффективное количество эскетамина, вводимое во время продленной индукционной фазы, составляет около 56 мг. В других вариантах осуществления эффективное количество эскетамина, вводимое во время продленной индукционной фазы, составляет около 84 мг.
Введение может дополнительно включать фазу оптимизации/поддержания, которая следует за индукционной фазой, и при этом после достижения пациентом, по существу, полного ответа на эскетамин во время индукционной фазы эскетамин вводят с частотой менее двух раз в неделю во время фазы оптимизации/поддержания. В некоторых вариантах осуществления частота введения во время фазы оптимизации/поддержания составляет один раз в неделю, один раз в две недели, один раз в месяц или их комбинацию.
На любой стадии во время одной или более из индукционной фазы, фазы оптимизации или фазы поддержания ответ пациента на лечение можно оценивать с помощью методик, описанных в данном документе. Такое оценивание можно проводить до тех пор, пока специалист в данной области техники не решит, что пациент достиг подходящего ответа на схему лечения. В некоторых вариантах осуществления индукционный период может считаться завершенным, когда оценка пациента по шкале MADRS снижается на ≥50% относительно исходного уровня или на от около 20 до около 13. В других вариантах осуществления оценка пациента по шкале MADRS может составлять около 19, около 18, около 17, около 16, около 15, около 14 или около 13. Пациентов с оценками по шкале MADRS ≤12 считают достигшими ремиссии и, если они стабильны в течение четырех недель, их следует переводить в фазу поддержания или оставлять на ней.
В конце индукционной фазы или продленной индукционной фазы лечащий врач должен оценить пациента, чтобы оптимизировать частоту и количество дозы для любых последующих фаз введения, таких как «фаза поддержания» или «фаза длительной терапии». Предполагается, что частота интраназального лечения во время последующего введения, например, фазы поддержания, будет снижена по сравнению с индукционной фазой или продленной индукционной фазой (по меньшей мере дважды в неделю) до введения один раз в неделю в течение по меньшей мере 4 недель. В некоторых вариантах осуществления последующее введение, такое как фаза поддержания, происходит в течение по меньшей мере около 4 недель, около 5 недель, около 6 недель, около 7 недель, около 8 недель, около 9 недель, около 10 недель, около 11 недель, около 12 недель, около 13 недель, около 17 недель, около 18 недель, около 19 недель, около 20 недель, около 6 месяцев, около 7 месяцев, около 8 месяцев, около 9 месяцев, около 10 месяцев, около 11 месяцев, 1 года или около 2 лет. В некоторых вариантах осуществления введение эскетамина во время последующей фазы введения продолжают в течение по меньшей мере шести месяцев. В других вариантах осуществления введение эскетамина во время последующей фазы введения продолжают в течение по меньшей мере одного года. В дополнительных вариантах осуществления частота введения во время последующей фазы введения составляет один раз в неделю, или один раз в две недели, или их комбинацию. В других вариантах осуществления частота дозирования и эффективное количество эскетамина во время последующей фазы введения соответствуют минимальным частоте и количеству для поддержания стабильной ремиссии или стабильного ответа.
Последующее введение, например, во время периода поддержания, может включать более длительные периоды времени в зависимости от состояния пациента. В некоторых вариантах осуществления эти более длительные периоды могут составлять по меньшей мере около 3 лет, около 4 лет, около 5 лет, около 6 лет, около 7 лет, около 8 лет, около 9 лет, около 10 лет или более чем около 10 лет, включая неограниченное время. Например, у пациентов с диагнозом ТРД лечение может продолжаться неограниченное время. В других вариантах осуществления частоту лечения снижают до одного раза в две недели. В дополнительных вариантах осуществления частоту лечения снижают до одного раза в три недели. В других вариантах осуществления частоту лечения снижают до одного раза в месяц. Пациентов продолжают лечить в соответствии с графиком до тех пор, пока пациент не достигает ремиссии, пока у пациента не сохраняется ответ или пока лечение не оказывается безрезультатным. Если пациент достигает ремиссии или у него сохраняется ответ при лечении один раз неделю в течение по меньшей мере 4 недель, частоту интраназальных сеансов лечения можно снизить до поддерживающей дозы один раз в две недели с учетом тяжести депрессивных симптомов, а для некоторых популяций пациентов частоту лечения можно снизить приблизительно до одного раза в три или четыре недели, как обсуждается выше.
Специалисту в данной области техники понятно, что описанная в настоящем документе фаза поддержания может продолжаться, пока не потребуется дополнительное лечение, и определяться, например, длительной ремиссией депрессии (включая, например, ремиссию одного или более симптомов, связанных с депрессией), социальным и/или профессиональным функциональным улучшением до нормального или преклинического уровней, либо другими известными показателями при депрессии.
Во время фазы поддержания пациенту вводят эффективное количество эскетамина. Как указано выше, количество эскетамина, вводимое во время фазы поддержания представляет количество, которое вызывает биологический или медицинский ответ со стороны системы тканей, что обсуждалось выше для индукционной фазы. В некоторых вариантах осуществления эффективное количество эскетамина представляет количество, при котором поддерживается фармакодинамическое равновесное состояние эскетамина, достигнутое во время индукционной фазы. В других вариантах осуществления, если симптомы депрессии начинают ухудшаться при лечении раз в две недели, в три недели или в четыре недели, введение эскетамина повышают для стабилизации пациента. Например, если пациент принимает дозу раз в две недели, а симптомы начинают ухудшаться, эскетамин можно вводить раз в неделю для поддержания ответа во время фазы поддержания. Аналогично вышеуказанному в любое время в течение фазы поддержания можно проводить повторную оценку ответа пациента.
Для пожилых пациентов рекомендуемая доза эскетамина составляет от около 28 до около 84 мг. Рекомендуемая начальная доза (во время первого курса лечения) составляет около 28 мг эскетамина. В зависимости от эффективности и переносимости дозы около 28 мг во время следующего курса лечения можно сохранять дозу около 28 мг или повышать ее до около 56 мг. В зависимости от эффективности и переносимости дозы около 56 мг во время последующего курса лечения можно сохранять дозу около 56 мг или повышать ее до около 84 мг, или снижать до около 28 мг. В зависимости от переносимости дозы около 84 мг во время последующих курсов лечения можно сохранять дозу около 84 мг или снижать ее до около 56 мг.
Для пациентов с печеночной недостаточностью рекомендуемая доза эскетамина составляет от около 28 до около 56 мг. Рекомендуемая начальная доза (во время первого курса лечения) составляет около 28 мг эскетамина. В зависимости от эффективности и переносимости дозы около 28 мг во время следующего курса лечения можно сохранять дозу около 28 мг или повышать ее до около 56 мг. Врачи должны регулярно следить за пациентами с печеночной недостаточностью в отношении переносимости лекарства, поскольку эскетамин обширно метаболизируется в печени.
В случае лечения пациентов с большим депрессивным расстройством с суицидальной идеацией и с высоким риском суицида введение является более агрессивным из-за тяжести состояния. Способы включают введение эскетамина в течение одной или двух фаз, т.е. начальной индукционной фазы и, необязательно и в определенных обстоятельствах, фазы поддержания. Вследствие высокого риска для жизни пациента начальную дозу эскетамина вводят в наиболее эффективном количестве эскетамина, переносимом пациентом, дважды в неделю во время индукционной фазы. В некоторых вариантах осуществления пациент продолжает терапию текущим (т.е. прием которого уже начат) антидепрессантом одновременно с началом терапии эскетамином во время индукционной фазы. В других вариантах осуществления пациент начинает прием нового антидепрессанта одновременно с началом терапии эскетамином во время индукционной фазы. В некоторых вариантах осуществления пациент продолжает терапию ранее вводимым антидепрессантом одновременно с началом терапии эскетамином во время индукционной фазы. Антидепрессант следует вводить, как показано при лечении БДР, способом, соответствующим состоянию/здоровью пациента. Индукционная фаза должна составлять от около 4 до около 8 недель, от около 4 до около 7 недель, от около 4 до около 6 недель, наиболее предпочтительно около 4 недель. В конце индукционной фазы введение эскетамина следует прекратить, если пациент демонстрирует адекватный ответ на лечение или находится в состоянии ремиссии. Необходимо осуществлять мониторинг пациента, чтобы убедиться в том, что пациент остается стабильным или находится в состоянии ремиссии на одном антидепрессанте. Если состояние пациента не стабилизируется при первой комбинации эскетамина и антидепрессанта или лечение антидепрессантом, начатое вместе с эскетамином, не дает результата после прекращения введения эскетамина, можно начать вторую индукционную фазу.
Во время второй индукционной фазы пациенту повторно назначают эскетамин в самой высокой переносимой дозе одновременно со вторым новым антидепрессантом. В альтернативном варианте пациенту повторно назначают эскетамин в самой высокой переносимой дозе одновременно с тем же антидепрессантом, который использовали во время предыдущей индукционной фазы. Эскетамин вводят дважды в неделю. Антидепрессант следует вводить, как показано при лечении БДР, способом, соответствующим состоянию/здоровью пациента. Вторая индукционная фаза должна составлять от около 4 до около 8 недель, от около 4 до около 7 недель, от около 4 до около 6 недель, наиболее предпочтительно около 4 недель. В конце второй индукционной фазы, если пациент демонстрирует адекватный ответ на лечение или находится в состоянии ремиссии, введение эскетамина следует прекратить, и необходимо осуществлять мониторинг пациента, чтобы убедиться в том, что пациент остается стабильным/ или находится в состоянии стабильной ремиссии на одном антидепрессанте. Если состояние пациента не стабилизируется или лечение антидепрессантом, начатое вместе с эскетамином, не дает результата после прекращения введения эскетамина, можно начать третью индукционную фазу.
Во время третьей индукционной фазы пациенту повторно назначают эскетамин в самой высокой переносимой дозе одновременно с третьим новым антидепрессантом. В альтернативном варианте пациенту повторно назначают эскетамин в самой высокой переносимой дозе одновременно с тем же антидепрессантом, который использовали во время второй индукционной фазы. Эскетамин вводят дважды в неделю. Антидепрессант следует вводить, как показано при лечении БДР, способом, соответствующим состоянию/здоровью пациента. Третья индукционная фаза должна составлять от около 4 до около 8 недель, от около 4 до около 7 недель, от около 4 до около 6 недель, наиболее предпочтительно около 4 недель. В конце третьей индукционной фазы пациент переходит к фазе поддержания, показанной при ТРД, поскольку теперь пациент классифицируется как пациент с ТРД. Описанные в данном документе способы позволяют оптимизировать дозировки эскетамина для введения пациенту, у которого наблюдается депрессия или который к ней предрасположен. В некоторых вариантах осуществления описанные в данном документе способы не требуют корректировки дозировки эскетамина.
В общем случае пациенту может быть повторно назначен эскетамин в самой высокой переносимой дозе одновременно с тем же антидепрессантом, который использовали во время любой предыдущей индукционной фазы, в том числе антидепрессантом, с которым состояние пациента не смогло стабилизироваться или лечение каким-либо иным образом оказалось безрезультатным. Например, в способе лечения терапевтически резистентной депрессии у пациента, если пациент не отвечал по меньшей мере на два пероральных антидепрессанта во время текущего депрессивного эпизода, пациенту можно вводить эскетамин по меньшей мере дважды в неделю по схеме только эскетамин или вместе с первым пероральным антидепрессантом, который является таким же или отличным от ранее неэффективного перорального антидепрессанта во время первой индукционной фазы. В случае, если пациент не смог достичь по существу полного ответа на эскетамин, пациенту может быть повторно назначен эскетамин в самой высокой переносимой дозе одного эскетамина или одновременно со вторым пероральным антидепрессантом, который является таким же или отличным от первого перорального антидепрессанта во время второй индукционной фазы. В случае, если пациент достигает, по существу, полного ответа на эскетамин во время второй индукционной фазы, пациенту после этого можно вводить терапевтически эффективное количество эскетамина реже, два раза в неделю, во время последующей фазы поддержания.
В случае пропуска одной или более (например, двух) доз эскетамина во время любой из фаз, описанных в данном документе, следующую дозу планируют по мере возможности с учетом схемы введения. Если пропущено более 2 доз, в соответствии с клинической оценкой, может потребоваться корректировка дозы или частоты введения эскетамина.
В предпочтительной фармацевтической композиции по настоящему изобретению S-кетамина гидрохлорид в качестве активного ингредиента тщательно смешивают с фармацевтическим носителем, предпочтительно водой, в соответствии с традиционными фармацевтическими методами приготовления смесей, при этом такой носитель может принимать ряд форм в зависимости от формы препарата, необходимой для введения. Подходящие фармацевтически приемлемые носители хорошо известны в данной области техники. Описания некоторых из этих фармацевтически приемлемых носителей можно найти в The Handbook of Pharmaceutical Excipients, опубликованной Американской фармацевтической ассоциацией и Фармацевтическим обществом Великобритании.
Способы составления фармацевтических композиций описаны в многочисленных публикациях, таких как Pharmaceutical Dosage Forms: Tablets, Second Edition, Revised and Expanded, Volumes 1-3, edited by Lieberman et al.; Pharmaceutical Dosage Forms: Parenteral Medications, Volumes 1-2, edited by Avis et al.; и Pharmaceutical Dosage Forms: Disperse Systems, Volumes 1-2, edited by Lieberman et al.; опубликованных издательством Marcel Dekker, Inc.
Один подходящий водный состав S-кетамина содержит воду и S-кетамин; причем S-кетамин присутствует в количестве в диапазоне от около 25 мг/мл до около 250 мг/мл, предпочтительно от около 55 мг/мл до около 250 мг/мл или от около 100 мг/мл до около 250 мг/мл, или любом количестве или диапазоне в этих пределах в расчете на общий объем фармацевтической композиции. Предпочтительно S-кетамин присутствует в количестве в диапазоне от около 150 мг/мл до около 200 мг/мл или в любом количестве или диапазоне в этих пределах. Более предпочтительно S-кетамин присутствует в количестве в диапазоне от около 150 мг/мл до около 175 мг/мл или в любом количестве или диапазоне в этих пределах. Более предпочтительно S-кетамин присутствует в количестве в диапазоне от около 160 мг/мл до около 163 мг/мл, например, в количестве около 161,4 мг/мл.
Другой подходящий водный состав S-кетамина содержит воду и S-кетамин; причем S-кетамин присутствует в количестве в диапазоне от около экв. 100 мг/мл до около экв. 250 мг/мл или любом количестве или диапазоне в этих пределах в расчете на общий объем фармацевтической композиции. Предпочтительно S-кетамин присутствует в количестве в диапазоне от около экв. 125 мг/мл до около экв. 180 мг/мл или любом количестве или диапазоне в этих пределах. Более предпочтительно S-кетамин присутствует в количестве в диапазоне от около экв. 140 мг/мл до около экв. 160 мг/мл или любом количестве или диапазоне в этих пределах, например, в количестве около экв. 140 мг/мл.
Подходящие фармацевтические композиции для применения в настоящем изобретении предпочтительно представляют собой водный состав. В контексте данного документе, если не указано иное, термин «водный» означает, что основным жидким компонентом состава является вода. Предпочтительно вода составляет более чем около 80 масс. % жидкого компонента фармацевтической композиции, более предпочтительно - более чем около 90 масс. %, более предпочтительно - более чем около 95 масс. %, более предпочтительно - около 98 масс. %.
В подходящих фармацевтических композициях для применения в настоящем изобретении содержание воды в композиции находится в диапазоне 85 ± 14 масс. %, более предпочтительно 85 ± 12 масс. %, еще более предпочтительно 85 ± 10 масс. %, наиболее предпочтительно 85 ± 7,5 масс. %, и, в частности, 85 ± 5 масс. %, в расчете на общую массу композиции.
В подходящих фармацевтических композициях для применения в настоящем изобретении предпочтительно содержание воды в композиции находится в диапазоне 90 ± 14 масс. %, более предпочтительно 90 ± 12 масс. %, еще более предпочтительно 90 ± 10 масс. %, наиболее предпочтительно 80 ± 7,5 масс. %, и, в частности, 90 ± 5 масс. %, в расчете на общую массу композиции.
В другой фармацевтической композиции для применения в настоящем изобретении содержание воды в композиции находится в диапазоне 95 ± 4,75 масс. %, более предпочтительно 95 ± 4,5 масс. %, еще более предпочтительно 95 ± 4 масс. %, еще более предпочтительно 95 ± 3,5 масс. %, наиболее предпочтительно 95 ± 3 масс. %, и, в частности, 95 ± 2,5 масс. %, в расчете на общую массу композиции.
В другой фармацевтической композиции для применения в настоящем изобретении содержание воды в композиции находится в диапазоне от 75 до 99,99 масс. %, более предпочтительно от 80 до 99,98 масс. %, еще более предпочтительно от 85 до 99,95 масс. %, более предпочтительно от 90 до 99,9 масс. %, наиболее предпочтительно от 95 до 99,7 масс. % и, в частности, от 96,5 до 99,5 масс. %, в расчете на общую массу композиции.
В другой фармацевтической композиции для применения в настоящем изобретении композиция дополнительно содержит один или более буферов и/или одну или более буферных систем (т.е. комбинаций пар кислота - основание).
В контексте данного документа термин «буфер» означает любую твердую или жидкую композицию (предпочтительно водную, жидкую композицию), которая при добавлении в водный состав регулирует значение pH указанного состава. Специалисту в данной области техники понятно, что буфер может регулировать pH водного состава в любом направлении (до более кислотного, более основного или более нейтрального pH). Предпочтительно буфер является фармацевтически приемлемым.
Подходящие примеры буферов, которые можно использовать в водных составах по настоящему изобретению, включают, не ограничиваясь этим, лимонную кислоту, первичный кислый фосфат натрия, вторичный кислый фосфат натрия, уксусную кислоту, борную кислоту, борат натрия, янтарную кислоту, винную кислоту, яблочную кислоту, молочную кислоту, фумаровую кислоту и т.п. Предпочтительно буфер или буферную систему выбраны из группы, состоящей из NaOH, лимонной кислоты, первичного кислого фосфата натрия или вторичного кислого фосфата натрия.
В одном варианте осуществления буфер выбран так, чтобы регулировать pH фармацевтических композиций S-кетамина гидрохлорида по настоящему изобретению (например, водных составов, описанных в данном документе) до pH в диапазоне от около pH 3,5 до около pH 6,5 или любом количестве или диапазоне в этих пределах. Предпочтительно буфер выбран так, чтобы регулировать pH композиций S-кетамина гидрохлорида по настоящему изобретению приблизительно в диапазоне от около pH 4,0 до около pH 5,5 или любом количестве или диапазоне в этих пределах, более предпочтительно в диапазоне от около pH 4,5 до около pH 5,0 или любом количестве или диапазоне в этих пределах.
Предпочтительно концентрация буфера и буферной системы, соответственно, предпочтительно NaOH, отрегулирована для обеспечения достаточной буферной емкости.
В одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей S-кетамина гидрохлорид, воду и буфер или буферную систему, предпочтительно NaOH; причем буфер или буферная система присутствуют в количестве, достаточном для получения состава с pH в диапазоне от около pH 4,0 до около pH 6,0 или в любом количестве или диапазоне в этих пределах.
Необязательно фармацевтические композиции по настоящему изобретению могут содержать консервант.
В контексте данного документа, если не указано иное, термины «противомикробный консервант» и «консервант» предпочтительно относятся к любому веществу, которое обычно добавляют в фармацевтические композиции для предотвращения микробиологического разложения или роста микроорганизмов. В связи с этим рост микроорганизмов, как правило, играет важную роль, т.е. консервант служит основной цели предотвращения микробного заражения. В качестве дополнительного аспекта также может быть желательно предотвратить любое влияние микроорганизмов на активные ингредиенты и наполнители, соответственно, т.е. чтобы предотвратить микробиологическое разложение.
Типовые примеры консервантов включают, но не ограничиваются этим, бензалкония хлорид, бензетония хлорид, бензойную кислоту, бензоат натрия, бензиловый спирт, бронопол, цетримид, цетилпиридиния хлорид, хлоргексидин, хлорбутанол, хлорокрезол, хлороксиленол, крезол, этиловый спирт, глицерин, гексетидин, имидомочевина, фенол, феноксиэтанол, фенилэтиловый спирт, нитрат фенилртути, пропиленгликоль, пропионат натрия, тимеросал, метилпарабен, этилпарабен, пропилпарабен, бутилпарабен, изобутилпарабен, бензилпарабен, сорбиновую кислоту и сорбат калия.
Полное отсутствие консервантов в фармацевтических композициях, используемых в настоящем изобретении, является предпочтительным, когда содержание S-кетамина гидрохлорида является достаточно высоким, так что вследствие его консервирующего свойства необходимые срок хранения или стабильность могут быть обеспечены за счет наличия самого лекарственного препарата. Предпочтительно в таких случаях концентрация S-кетамина гидрохлорида составляет по меньшей мере экв. 120 мг/мл, предпочтительно в диапазоне от около экв. 120 мг/мл до около экв. 175 мг/мл или любом количестве или диапазоне в этих пределах, более предпочтительно в количестве в диапазоне от около экв. 125 мг/мл до около экв. 150 мг/мл или любом количестве или диапазоне в этих пределах, например, около экв. 126 мг/мл или около экв. 140 мг/мл.
В контексте данного документа термины «агент, улучшающий проникновение», «усилитель проникновения» и «вещество, обеспечивающее проникновение» относятся к любому веществу, которое повышает или облегчает всасывание и/или биодоступность активного ингредиента (например, S-кетамина гидрохлорида) фармацевтической композиции. Предпочтительно агенты, улучшающие проникновение, повышают или облегчают всасывание и/или биодоступность активного ингредиента (например, S-кетамина гидрохлорида) фармацевтической композиции после назального введения в (т.е. повышают или облегчают всасывание и/или биодоступность активного ингредиента через слизистую оболочку).
Соответствующие примеры включают, но не ограничиваются этим, тетрадецилмальтозид, гликохолат натрия, тауроурсодеоксихолевую кислоту (TUDCA), лецитины и т.п.; и хитозан (и соли), а также поверхностно-активные ингредиенты, такие как бензалкония хлорид, додецилсульфат натрия, докузат натрия, полисорбаты, лаурет-9, октоксинол, дезоксихолат натрия, полиаргинин и т.п. Предпочтительно агент, улучшающий проникновение, представляет собой тауроурсодеоксихолевую кислоту (TUDCA).
Агент, улучшающий проникновение, может действовать за счет любого механизма, включая, например, увеличение подвижности мембраны, создание временных гидрофильных пор в эпителиальных клетках, снижение вязкости слизистого слоя или открытие плотных контактов. Некоторые агенты, улучшающие проникновение (например, соли желчных кислот и производные фузидовой кислоты), также могут ингибировать ферментативную активность в мембране, таким образом улучшая биодоступность активного ингредиента.
Предпочтительно агент, улучшающий проникновение, выбран так, чтобы соответствовать одному или более, более предпочтительно всем, из следующих общих требований:
(a) Он эффективен в отношении повышения всасывания (предпочтительно всасывания в полости носа) активного ингредиента, предпочтительно временным и/или обратимым образом;
(b) Он является фармакологически инертным;
(c) Он не вызывает аллергию, нетоксичен и/или не вызывает раздражение;
(d) Он очень эффективен (эффективен в небольших количествах);
(e) Он совместим с другими компонентами фармацевтической композиции;
(f) Он не имеет запаха, цвета и/или вкуса;
(g) Он одобрен регуляторными органами; и
(h) Он является недорогим и доступен в форме с высокой степенью чистоты.
В одном варианте осуществления настоящего изобретения агент, улучшающий проникновение, выбран так, чтобы повышать проникновение (всасывание и/или биодоступность S-кетамина гидрохлорида) без раздражения тканей носа. В другом варианте осуществления настоящего изобретения агент, улучшающий проникновение, выбран так, чтобы улучшать всасывание и/или биодоступность S-кетамина гидрохлорида; и дополнительно выбран так, чтобы повышать эффективность равномерного введения.
В одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей S-кетамин и воду; в данном случае фармацевтическая композиция не содержит противомикробный консервант; и при этом фармацевтическая композиция дополнительно содержит усилитель проникновения, предпочтительно TUDCA.
В другом варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей S-кетамин и воду; в данном случае фармацевтическая композиция не содержит противомикробный консервант; и при этом фармацевтическая композиция дополнительно содержит тауроурсодеоксихолевую кислоту (TUDCA); причем TUDCA присутствует в концентрации в диапазоне от около 1,0 мг/мл до около 25,0 мг/мл или любом количестве или диапазоне в этих пределах, предпочтительно в концентрации в диапазоне от около 2,5 мг/мл до около 15 мг/мл или любом количестве или диапазоне в этих пределах, предпочтительно в концентрации в диапазоне от около 5 мг/мл до около 10 мг/мл или любом количестве или диапазоне в этих пределах. В другом варианте осуществления настоящее изобретение относится к фармацевтической композиции, в которой TUDCA присутствует в концентрации около 5 мг/мл. В другом варианте осуществления настоящее изобретение относится к фармацевтической композиции, в которой TUDCA присутствует в концентрации около 10 мг/мл.
Фармацевтические композиции для применения в настоящем изобретении могут дополнительно содержать один или более дополнительных наполнителей, например смачивающих агентов, поверхностно-активных компонентов, солюбилизирующих агентов, загустителей, красителей, антиоксидантов и т.п.
Примеры подходящих антиоксидантов, в случае их применения, включают, но не ограничиваются этим, одно или более из следующих веществ: сульфиты; аскорбиновая кислота; аскорбаты, такие как аскорбат натрия, аскорбат кальция или аскорбат калия; аскорбилпальмитат; фумаровая кислота; этилендиаминтетрауксусная кислота (ЭДТА) или ее натриевые или кальциевые соли; токоферол; галлаты, такие как пропилгаллат, октилгаллат или додецилгалат; витамин E; и их смеси. Антиоксидант обеспечивает долгосрочную стабильность жидких композиций. Добавление антиоксиданта может способствовать повышению и гарантии стабильности композиций и делать композиции стабильными даже после шести месяцев при 40 °C. Подходящее количество антиоксиданта, в случае его наличия, составляет от около 0,01 масс. % до около 3 масс. %, предпочтительно от около 0,05 масс. % до около 2 масс. % от общей массы композиции.
Для облегчения более равномерной дисперсии активного ингредиента или другого наполнителя, который в целом не растворим в жидком носителе, можно включать солюбилизирующие и эмульгирующие агенты. Примеры приемлемого эмульгирующего агента, в случае его применения, включают, но не ограничиваются этим, желатин, холестерин, аравийскую камедь, трагакант, пектин, метилцеллюлозу, карбомер и их смеси. Примеры подходящего солюбилизирующего агента включают полиэтиленгликоль, глицерин, D-маннит, трегалозу, бензилбензоат, этанол, трисаминометан, холестерин, триэтаноламин, карбонат натрия, цитрат натрия, салицилат натрия, ацетат натрия и их смеси.
Предпочтительно солюбилизирующий агент включает глицерин. Солюбилизирующий или эмульгирующий агент в общем случае присутствуют в количестве, достаточном для растворения или диспергирования активного ингредиента, т.е. S-кетамина, в носителе. Типовые количества при включении солюбилизатора или эмульгатора составляют от около 1 масс. % до около 80 масс. %, предпочтительно от около 20 масс. % до около 65 масс. % и более предпочтительно от около 25 масс. % до около 55 масс. % от общей массы композиции.
Подходящий изотонизирующий агент, в случае его применения, включает хлорид натрия, глицерин, D-маннит, D-сорбит, глюкозу и их смеси. Подходящее количество изотонизирующего агента, в случае его включения, как правило, составляют от около 0,01 масс. % до около 15 масс. %, более предпочтительно от около 0,3 масс. % до около 4 масс. % и более предпочтительно от около 0,5 масс. % до около 3 масс. % от общей массы композиции.
В фармацевтические композиции по настоящему изобретению можно добавлять суспендирующий агент или повышающий вязкость агент, например, для увеличения времени пребывания в носу. Подходящие примеры включают, но не ограничиваются этим, гидроксипропилметилцеллюлозу, кармеллозу натрия, микрокристаллическую целлюлозу, карбомер, пектин, альгинат натрия, соли хитозана, геллановую камедь, полоксамер, поливинилпирролидон, ксантановую камедь и т.п.
Преимущественно эскетамин можно вводить в одной суточной дозе, или же общую суточную дозу можно вводить разделенными дозами два, три или четыре раза в сутки, предпочтительно два раза в сутки. Как правило, разделенные дозы следует вводить с небольшой разницей во времени. В некоторых вариантах осуществления разделенные дозы вводят в течение около 20 минут, около 15 минут, около 10 минут, около 5 минут, около 4 минут, около 3 минут, около 2 минут, около 1 минуты или менее друг за другом. Кроме того, по гибкой схеме введения пациенту можно вводить дозу ежесуточно, два раза в неделю, один раз в неделю, один раз в две недели или один раз в месяц. Например, одну дозу эскетамина вводят на 1 сутки, а другую дозу эскетамина вводят на 2 сутки, или одну дозу эскетамина вводят на 1 сутки, а другую дозу эскетамина вводят на 3 сутки, одну дозу эскетамина вводят на 1 сутки, а другую дозу эскетамина вводят на 4 сутки, одну дозу эскетамина вводят на 1 сутки, а другую дозу эскетамина вводят на 5 сутки. Кроме того, эскетамин предпочтительно вводят в интраназальной форме путем местного применения подходящих интраназальных средств, таких как назальный распылительный насос.
Как описано, способы введения эскетамина пациенту приводят к фармакокинетическому профилю с достижением максимальной концентрации в плазме (Cмакс) эскетамина от около 45 до около 165 нг/мл. Специалисту в данной области техники понятно, что любой из диапазонов или отдельных значений Cмакс могут варьироваться на ± 30%. В некоторых вариантах осуществления Cмакс составляет около 45, около 50, около 55, около 60, около 65, около 70, около 75, около 80, около 85, около 90, около 95, около 100, около 105, около 110, около 115, около 120, около 125, около 130, около 135, около 140, около 145, около 150, около 155, около 160 или около 165 нг/мл. В других вариантах осуществления Cмакс составляет от около 50 до около 150, от около 50 до около 125, от около 50 до около 100, от около 50 до около 75, от около 75 до около 150, от около 75 до около 125 или от около 75 до около 100 нг/мл. В дополнительных вариантах осуществления Cмакс составляет от около 45 до около 75, от около 50 до около 70, от около 55 до около 65, от около 45 до около 70, от около 45 до около 65, от около 45 до около 60, от около 45 до около 55, от около 55 до около 75 или от около 60 до около 70 нг/мл при введении около 28 мг эскетамина. В других вариантах осуществления Cмакс составляет от около 65 до около 120, от около 70 до около 120, от около 70 до около 110, от около 70 до около 100, от около 70 до около 90, от около 70 до около 80, от около 80 до около 120, от около 80 до около 110, от около 80 до около 90, от около 90 до около 120 или от около 90 до около 110 нг/мл при введении около 56 мг эскетамина. В дополнительных вариантах осуществления Cмакс составляет от около 90 до около 165, от около 95 до около 165, от около 95 до около 155, от около 95 до около 145, от около 95 до около 135, от около 95 до около 125, от около 95 до около 115, от около 105 до около 165, от около 105 до около 155, от около 105 до около 145, от около 105 до около 135, от около 105 до около 125, от около 105 до около 115, от около 115 до около 165, от около 115 до около 155, от около 115 до около 145, от около 115 до около 135, от около 115 до около 125, от около 125 до около 165, от около 125 до около 155, от около 125 до около 145, от около 125 до около 135, от около 135 до около 165, от около 135 до около 155, от около 135 до около 145 или от около 145 до около 165 нг/мл при введении около 84 мг эскетамина.
Аналогично, способы введения эскетамина пациенту приводят к фармакокинетическому профилю с достижением площади под кривой плазменная концентрация - время, начиная с момента времени 0 до времени последней количественно определяемой концентрации (ППКпосл) от около 125 до около 490 нг*ч/мл. В контексте данного документа термин «ППКпосл» относится к площади под кривой плазменная концентрация - время, начиная с нулевого момента времени до времени последней измеряемой концентрации. Термин «нулевой момент времени» в общем контексте относится к началу введения предполагаемой дозы. Например, в примере 1 в отношении интраназального введения момент времени 0 определяется как время первого интраназального впрыскивания в одну ноздрю из первого интраназального устройства. В случае, если предполагаемая доза требует введения двух пероральных таблеток, момент времени 0 соответствует времени введения первой таблетки. Специалисту в данной области техники понятно, что любые из диапазонов или отдельных значений ППКпосл могут варьироваться на ± 30%. В некоторых вариантах осуществления ППКпосл составляет около 125, около 130, около 135, около 140, около 145, около 150, около 160, около 170, около 180, около 190, около 200, около 210, около 220, около 230, около 240, около 250, около 260, около 270, около 280, около 290, около 300, около 310, около 320, около 330, около 340, около 350, около 360, около 370, около 380, около 390, около 400, около 410, около 420, около 430, около 440, около 450, около 460, около 470, около 480 или около 490 нг*ч/мл. В других вариантах осуществления ППКпосл составляет от около 150 до около 450, от около 200 до около 400, от около 250 до около 350, от около 150 до около 350 или от около 200 до около 300 нг*ч/мл. В дополнительных вариантах осуществления ППКпосл составляет от около 125 до около 185, от около 130 до около 180, от около 135 до около 175, от около 140 до около 170, от около 145 до около 165 или от около 150 до около 160 нг*ч/мл при введении около 28 мг эскетамина. В других вариантах осуществления ППКпосл составляет от около 210 до около 320, от около 220 до около 310, от около 230 до около 300, от около 240 до около 290, от около 250 до около 280 или от около 260 до около 270 нг*ч/мл при введении около 56 мг эскетамина. В дополнительных вариантах осуществления ППКпосл составляет от около 305 до около 490, от около 310 до около 480, от около 320 до около 470, от около 330 до около 460, от около 340 до около 450, от около 350 до около 450, от около 360 до около 440, от около 370 до около 430, от около 380, от около 420 или от около 390 до около 410 нг*ч/мл при введении около 84 мг эскетамина.
Способы введения эскетамина также могут приводить к фармакокинетическому профилю, который позволяет получить комбинации отдельных значений и диапазонов Cмакс и ППКпосл, описанных выше.
Типовое устройство для назального распыления раскрыто в патенте США № 6321942, включенном в данный документ посредством ссылки. Например, для осуществления способов, раскрытых в данном документе, можно использовать одноразовый распылитель для впрыскивания частичного впрыскиваемого количества в виде спрея. Как правило, такие устройства позволяют впрыскивать лекарственное средство в обе ноздри пациента за два последовательных нажима. Такое устройство может быть готовым к использованию, когда лекарственное средство высвобождается из контейнера со средой. Такое устройство, как правило, позволяет разделять первый нажим для впрыскивания и второй нажим для впрыскивания, чтобы предотвратить опустошение контейнера со средой за один раз. Такое устройство может иметь форму одноразового насоса двойного нажима, который утилизируется после однократного применения и обеспечивает осуществление отдельного частичного впрыскивания с высокой точностью дозы и надежностью.
В одном варианте осуществления назальное распылительное устройство представляет собой одноразовое устройство, которое доставляет в общей сложности 28 мг эскетамина за два впрыскивания, по одному впрыскиванию на ноздрю. Пациент может использовать такое устройство под наблюдением медицинского работника. Что касается дозировок, одно устройство можно использовать для дозы 28 мг, два устройства - для дозы 56 мг или три устройства - для дозы 84 мг. Также предпочтительно наличие 5-минутного интервала между использованием каждого устройства. Как описано в примере 1, момент времени 0 определяется как время первого интраназального впрыскивания в одну ноздрю из первого интраназального устройства.
Аспекты данного раскрытия
Настоящее раскрытие относится и включает по меньшей мере следующие аспекты.
1. Способ лечения большого депрессивного расстройства, включающий интраназальное введение нуждающемуся в этом пациенту имеющего клинически доказанную безопасность и клинически доказанную эффективность терапевтически эффективного количества эскетамина;
причем нуждающийся пациент представляет собой пациента-человека, переживающего большой депрессивный эпизод, и при этом пациент не отвечал по меньшей мере на два пероральных антидепрессанта во время текущего депрессивного эпизода.
2. Способ лечения большого депрессивного расстройства, включающий введение нуждающемуся в этом пациенту эскетамина;
причем нуждающийся пациент переживает большой депрессивный эпизод и при этом пациент не отвечал по меньшей мере на два пероральных антидепрессанта во время текущего депрессивного эпизода;
при этом эскетамин вводят интраназально;
и при этом терапевтически эффективное количество эскетамина, вводимое пациенту, имеет клинически доказанные безопасность и эффективность.
3. Способ лечения большого депрессивного расстройства у пациента-человека, включающий такие этапы:
(a) диагностирование указанного пациента-человека путем определения исходной оценки по шкале MADRS указанного пациента-человека;
(b) интраназальное введение указанному пациенту-человеку терапевтически эффективного количества эскетамина, которое имеет клинически доказанные безопасность и эффективность;
причем терапевтически эффективное количество улучшает указанную оценку по шкале MADRS по меньшей мере на 50% относительно определенной исходной оценки по шкале MADRS;
и при этом эскетамин вводят через заранее определенные интервалы; и
(c) повторная оценка указанного пациента-человека через регулярные интервалы после этапа (b) для определения относительной эффективности;
причем повторная оценка включает определения оценки по шкале MADRS указанного пациента-человека.
4. Способ по аспектам 1, 2 или 3, в котором большое депрессивное расстройство представляет собой терапевтически рефрактерную депрессию или терапевтически резистентную депрессию.
5. Способ по аспектам 1, 2, 3 или 4, в котором терапевтически эффективное количество по меньшей мере одного антидепрессанта вводят совместно с эскетамином.
6. Способ по аспекту 5, в котором комбинированная терапия включает применение эскетамина и одного или двух антидепрессантов.
7. Способ по аспекту 5, в котором каждый антидепрессант независимо выбран из группы, состоящей из имипрамина, амитриптилина, дезипрамина, нортриптилина, доксепина, протриптилина, тримипрамина, мапротилина, амоксапина, тразодона, бупропиона, кломипрамина, флуоксетина, дулоксетина, эсциталопрама, циталопрама, сертралина, пароксетина, флувоксамина, нефазодона, венлафаксина, милнаципрана, ребоксетина, миртазапина, фенелзина, транилципромина, моклобемида, кава-кава, зверобоя, S-аденозилметионина, тиреотропин-рилизинг гормона, антагонист нейрокининовых рецепторов и трийодотиронина.
8. Способ по аспекту 5, в котором каждый антидепрессант независимо выбран из группы, состоящей из ингибиторов моноаминоксидазы, трициклических соединений, ингибиторов обратного захвата серотонина, ингибиторов обратного захвата серотонина-норэпинефрина; норадренергических и специфических серотонинергических агентов и атипичных антидепрессанов.
9. Способ по аспекту 5, в котором каждый антидепрессант независимо выбран из группы, состоящей из фенелзина, транилципромина, моклобемида, имипрамина, амитриптилина, дезипрамина, нортриптилина, доксепина, протриптилина, тримипрамина, кломипрамина, амоксапина, флуоксетина, сертралина, пароксетина, циталопрама, флувоксамина, венлафаксина, милнаципрана, миртазапина и бупропиона.
10. Способ по аспекту 5, в котором комбинированная терапия включает применение эскетамина и одного или двух антидепрессантов, независимо выбранных из группы, состоящей из флуоксетина, имипрамина, бупропиона, венлафаксина и сертралина.
11. Способ по аспекту 5, в котором комбинированная терапия, включающая применение эскетамина и по меньшей мере одного антидепрессанта, дополнительно включает применение атипичного антидепрессанта.
12. Способ по аспекту 11, в котором атипичный антидепрессант выбран из группы, состоящей из арипипразола, кветиапина, оланзапина, рисперидона и палиперидона.
13. Способ по аспекту 12, в котором атипичный антидепрессант выбран из группы, состоящей из арипипразола, кветиапина и оланзапина.
14. Фармацевтическая композиция для лечения терапевтически рефрактерной или терапевтически резистентной депрессии, содержащая эскетамин, необязательно по меньшей мере один антидепрессант, и по меньшей мере один фармацевтически приемлемый носитель.
15. Применение эскетамина в приготовлении лекарственного средства для лечения терапевтически рефрактерной или терапевтически резистентной депрессии у нуждающегося в этом пациента.
16. Эскетамин для применения в способе лечения терапевтически рефрактерной или терапевтически резистентной депрессии у нуждающегося в этом пациента.
17. Композиция, содержащая эскетамин для лечения терапевтически рефрактерной или терапевтически резистентной депрессии.
18. Фармацевтический продукт, содержащий эскетамин, для введения пациенту, страдающему от терапевтически резистентной депрессии, причем эскетамин вводят указанному пациенту интраназально в количестве с клинически доказанными безопасностью и эффективностью.
19. Способ поддержания стабильной ремиссии или стабильного ответа, достигаемых пациентом с депрессией, после введения терапевтически эффективного количества эскетамина во время начальной фазы введения, включающий продолжение введения терапевтически эффективного количества эскетамина в течение по меньшей мере пяти месяцев во время последующей фазы введения.
20. Способ по аспекту 19, в котором депрессия представляет собой терапевтически резистентную депрессию.
21. Способ по аспекту 19 или 20, в котором терапевтически эффективное количество эскетамина вводят интраназально, внутримышечно, подкожно, трансдермально, буккально или ректально на начальной и последующей стадиях введения.
22. Способ по аспекту 21, в котором введение является интраназальным.
23. Способ по любому одному из аспектов 19-22, в котором терапевтически эффективное количество по меньшей мере одного антидепрессанта вводят совместно с эскетамином на начальной и последующей стадиях введения.
24. Способ по аспекту 23, в котором эскетамин вводят совместно с одним или двумя антидепрессантами.
25. Способ по аспекту 24, в котором каждый антидепрессант независимо представляет собой имипрамин, амитриптилин, дезипрамин, нортриптилин, доксепин, протриптилин, тримипрамин, мапротилин, амоксапин, тразодон, бупропион, кломипрамин, флуоксетин, дулоксетин, эсциталопрам, циталопрам, сертралин, пароксетин, флувоксамин, нефазодон, венлафаксин, милнаципран, ребоксетин, миртазапин, фенелзин, транилципромин, моклобемид, кава-кава, зверобой, S-аденозилметионин, тиреотропин-рилизинг гормон, антагонист нейрокининовых рецепторов или трийодотиронин.
26. Способ по любому одному из аспектов 23-25, в котором каждый антидепрессант независимо представляет собой ингибитор моноаминоксидазы, триклическое соединение, ингибитор обратного захвата серотонина, ингибитор обратного захвата серотонина-норэпинефрина, норадренергический и специфический серотонинергический агент или атипичный антидепрессант.
27. Способ по любому одному из аспектов 23-26, в котором каждый антидепрессант независимо представляет собой фенелзин, транилципромин, моклобемид, имипрамин, амитриптилин, дезипрамин, нортриптилин, доксепин, протриптилин, тримипрамин, кломипрамин, амоксапин, флуоксетин, сертралин, пароксетин, циталопрам, флувоксамин, венлафаксин, милнаципран, миртазапин или бупропион.
28. Способ по любому одному из аспектов 23-27, в котором каждый антидепрессант независимо представляет собой флуоксетин, имипрамин, бупропион, венлафаксин или сертралин.
29. Способ по аспекту 23, в котором по меньшей мере один антидепрессант представляет собой атипичный антидепрессант.
30. Способ по аспекту 29, в котором атипичный антидепрессант представляет собой арипипразол, кветиапин, оланзапин, рисперидон или палиперидон.
31. Способ по аспекту 30, в котором атипичный антидепрессант представляет собой арипипразол, кветиапин или оланзапин.
32. Способ по любому одному из аспектов 19-31, в котором начальная фаза введения включает индукционную фазу, причем эскетамин вводят с частотой по меньшей мере два раза в неделю.
33. Способ по аспекту 32, в котором частота составляет два раза в неделю.
34. Способ по аспекту 32 или 33, дополнительно включающий оценку ответа пациента во время индукционной фазы.
35. Способ по любому одному из аспектов 32-34, в котором начальная фаза введения дополнительно включает фазу оптимизации, которая следует за индукционной фазой, и при этом после достижения пациентом, по существу, полного ответа на эскетамин во время индукционной фазы эскетамин вводят с частотой менее двух раз в неделю во время фазы оптимизации.
36. Способ по аспекту 35, дополнительно включающий оценку ответа пациента во время фазы оптимизации и корректировку частоты введения во время фазы оптимизации на основании ответа для достижения стабильной ремиссии или стабильного ответа.
37. Способ по аспекту 36, в котором частота введения во время фазы оптимизации составляет один раз в неделю, один раз в две недели или их комбинацию.
38. Способ по любому одному из аспектов 19-37, в котором эффективное количество эскетамина составляет 28 мг, 56 мг или 84 мг во время начальной и последующей фаз введения.
39. Способ по любому одному из аспектов 32-38, в котором продолжение введения эскетамина во время последующей фазы введения происходит в течение по меньшей мере шести месяцев.
40. Способ по любому одному из аспектов 32-39, в котором продолжение введения эскетамина во время последующей фазы введения происходит в течение по меньшей мере одного года.
41. Способ по любому одному из аспектов 32-40, в котором частота введения во время последующей фазы введения составляет один раз в неделю, один раз в две недели или их комбинацию.
42. Способ по любому одному из аспектов 32-41, в котором эффективное количество эскетамина во время последующей фазы введения составляет 56 мг или 84 мг.
43. Способ по любому одному из аспектов 32-42, в котором частота введения и эффективное количество эскетамина во время последующей фазы введения соответствуют минимальным частоте и количеству для поддержания стабильной ремиссии или стабильного ответа.
44. Способ по любому одному из аспектов 19-43, в котором терапевтически эффективное количество эскетамина представляет собой количество с клинически доказанной безопасностью и клинически доказанной эффективностью.
45. Способ долгосрочного лечения депрессии у пациента, включающий введение нуждающемуся в лечении пациенту имеющего клинически доказанную безопасность и клинически доказанную эффективность терапевтически эффективного количества эскетамина в течение по меньшей мере шести месяцев.
46. Способ по аспекту 45, в котором эскетамин вводят в течение по меньшей мере одного года.
47. Способ по аспекту 45 или 46, в котором эскетамин вводят в течение периода, составляющего до двух лет.
48. Способ по любому одному из аспектов 45-47, в котором депрессия представляет собой терапевтически резистентную депрессию.
49. Способ по любому одному из аспектов 45-47, в котором эскетамин вводят интраназально.
50. Способ по любому одному из аспектов 45-48, в котором терапевтически эффективное количество по меньшей мере одного антидепрессанта вводят совместно с эскетамином.
51. Способ по аспекту 50, в котором эскетамин вводят совместно с одним или двумя антидепрессантами.
52. Способ по аспекту 51, в котором каждый антидепрессант независимо представляет собой имипрамин, амитриптилин, дезипрамин, нортриптилин, доксепин, протриптилин, тримипрамин, мапротилин, амоксапин, тразодон, бупропион, кломипрамин, флуоксетин, дулоксетин, эсциталопрам, циталопрам, сертралин, пароксетин, флувоксамин, нефазодон, венлафаксин, милнаципран, ребоксетин, миртазапин, фенелзин, транилципромин, моклобемид, кава-кава, зверобой, S-аденозилметионин, тиреотропин-рилизинг гормон, антагонист нейрокининовых рецепторов или трийодотиронин.
53. Способ по любому одному из аспектов 50-52, в котором каждый антидепрессант независимо представляет собой ингибитор моноаминоксидазы, триклическое соединение, ингибитор обратного захвата серотонина, ингибитор обратного захвата серотонина-норэпинефрина, норадренергический и специфический серотонинергический агент или атипичный антидепрессант.
54. Способ по любому одному из аспектов 50-53, в котором каждый антидепрессант независимо представляет собой фенелзин, транилципромин, моклобемид, имипрамин, амитриптилин, дезипрамин, нортриптилин, доксепин, протриптилин, тримипрамин, кломипрамин, амоксапин, флуоксетин, сертралин, пароксетин, циталопрам, флувоксамин, венлафаксин, милнаципран, миртазапин или бупропион.
55. Способ по любому одному из аспектов 50-54, в котором каждый антидепрессант независимо представляет собой флуоксетин, имипрамин, бупропион, венлафаксин или сертралин.
56. Способ по аспекту 55, в котором по меньшей мере один антидепрессант представляет собой атипичный антидепрессант.
57. Способ по аспекту 56, в котором атипичный антидепрессант представляет собой арипипразол, кветиапин, оланзапин, рисперидон или палиперидон.
58. Способ по аспекту 57, в котором атипичный антидепрессант представляет собой арипипразол, кветиапин или оланзапин.
59. Способ по любому одному из аспектов 45-58, в котором дозу эскетамина вводят сначала дважды в неделю в течение до четырех недель во время индукционной фазы, а после этого вводят реже, чем два раза в неделю.
60. Способ по аспекту 59, в котором дозу эскетамина вводят один раз в неделю или один раз в две недели после индукционной фазы.
61. Способ по любому одному из аспектов 45-60, в котором терапевтически эффективное количество эскетамина составляет 28 мг, 56 мг или 84 мг.
62. Способ по любому одному из аспектов 45-61, в котором когнитивная деятельность пациента остается стабильной на основании данных исходного измерения после шести месяцев лечения.
63. Способ лечения большого депрессивного расстройства у пожилого пациента, содержащий этапы, на которых:
пациенту, нуждающемуся в лечении большого депрессивного расстройства, вводят терапевтически эффективное количество эскетамина с частотой по меньшей мере два раза в неделю во время начальной индукционной фазы определенной длительности;
ответ пациента оценивают после начальной индукционной фазы; и
продолжают введение с частотой по меньшей мере два раза в неделю во время продленной индукционной фазы на основании оценки того, достиг ли пациент, по существу, полного ответа на эскетамин.
64. Способ по аспекту 63, в котором пожилой пациент не демонстрировал ответ по меньшей мере на два пероральных антидепрессанта во время текущего депрессивного эпизода.
65. Способ по аспекту 64, в котором терапевтически эффективное количество эскетамина вводят интраназально, внутримышечно, подкожно, трансдермально, буккально или ректально.
66. Способ по любому одному из аспектов 63-65, в котором введение является интраназальным.
67. Способ по любому одному из аспектов 63-66, в котором начальная индукционная фаза составляет до 2 недель.
68. Способ по любому одному из аспектов 63-66, в котором начальная индукционная фаза составляет до 3 недель.
69. Способ по любому одному из аспектов 63-66, в котором начальная индукционная фаза составляет до 4 недель.
70. Способ по любому одному из аспектов 63-66, в котором продленная индукционная фаза составляет до 8 недель.
71. Способ по любому одному из аспектов 63-70, в котором эффективное количество составляет 28 мг, 56 мг или 84 мг.
72. Способ по любому одному из аспектов 63-71, в котором после достижения пожилым пациентом, по существу, полного ответа на эскетамин введение эскетамина осуществляют с частотой не более одного раза в неделю во время фазы оптимизации.
73. Способ по аспекту 72, дополнительно включающий периодическое оценивание ответа пациента во время фазы оптимизации.
74. Способ по любому одному из аспектов 63-73, в котором частота во время начальной индукционной фазы, продленной индукционной фазы или их комбинации составляет два раза в неделю.
75. Способ по любому одному из аспектов 63-74, в котором большое депрессивное расстройство представляет собой терапевтически рефрактерную депрессию или терапевтически резистентную депрессию.
76. Способ по любому одному из аспектов 63-75, в котором терапевтически эффективное количество по меньшей мере одного антидепрессанта вводят совместно с эскетамином.
77. Способ по любому одному из аспектов 63-76, в котором комбинированная терапия включает применение эскетамина и одного или двух антидепрессантов.
78. Способ по аспекту 77, в котором каждый антидепрессант независимо представляет собой имипрамин, амитриптилин, дезипрамин, нортриптилин, доксепин, протриптилин, тримипрамин, мапротилин, амоксапин, тразодон, бупропион, кломипрамин, флуоксетин, дулоксетин, эсциталопрам, циталопрам, сертралин, пароксетин, флувоксамин, нефазодон, венлафаксин, милнаципран, ребоксетин, миртазапин, фенелзин, транилципромин, моклобемид, кава-кава, зверобой, S-аденозилметионин, тиреотропин-рилизинг гормон, антагонист нейрокининовых рецепторов или трийодотиронин.
79. Способ по аспекту 77 или 78, в котором каждый антидепрессант независимо представляет собой ингибитор моноаминоксидазы, триклическое соединение, ингибитор обратного захвата серотонина, ингибитор обратного захвата серотонина-норэпинефрина, норадренергический и специфический серотонинергический агент или атипичный антидепрессант.
80. Способ по любому одному из аспектов 77-79, в котором каждый антидепрессант независимо представляет собой фенелзин, транилципромин, моклобемид, имипрамин, амитриптилин, дезипрамин, нортриптилин, доксепин, протриптилин, тримипрамин, кломипрамин, амоксапин, флуоксетин, сертралин, пароксетин, циталопрам, флувоксамин, венлафаксин, милнаципран, миртазапин или бупропион.
81. Способ по любому одному из аспектов 77-80, включающий применение одного или двух антидепрессантов, которые независимо представляют собой флуоксетин, имипрамин, бупропион, венлафаксин или сертралин.
82. Способ по аспекту 79, в котором по меньшей мере один антидепрессант представляет собой атипичный антидепрессант.
83. Способ по аспекту 82, в котором атипичный антидепрессант представляет собой арипипразол, кветиапин, оланзапин, рисперидон или палиперидон.
84. Способ по аспекту 82 или 83, в котором атипичный антидепрессант представляет собой арипипразол, кветиапин или оланзапин.
85. Способ по любому одному из аспектов 63-84, в котором возраст пациента составляет по меньшей мере 65 лет.
86. Способ лечения пациента с большим депрессивным расстройством, включающий введение пациенту, нуждающемуся в лечении большого депрессивного расстройства, имеющего клинически доказанную безопасность и клинически доказанную эффективность терапевтически эффективного количества эскетамина.
87. Способ по аспекту 86, в котором пациент не демонстрировал ответ по меньшей мере на два пероральных антидепрессанта при адекватных дозе и длительности во время текущего депрессивного эпизода.
88. Способ по аспекту 86 или 87, в котором у пациента была диагностирована терапевтически рефрактерная депрессия или терапевтически резистентная депрессия.
89. Способ по аспекту 86, в котором симптомом большого депрессивного расстройства является суицидальная идеация у пациента.
90. Способ по аспекту 89, в котором пациент имеет высокий риск суицида.
91. Способ по любому одному из аспектов 86-90, в котором пациент является взрослым.
92. Способ по любому одному из аспектов 86-91, в котором пациент является пожилым пациентом.
93. Способ по любому одному из аспектов 86-92, в котором эскетамин вводят интраназально, внутримышечно, подкожно, трансдермально, буккально или ректально.
94. Способ по любому одному из аспектов 86-93, в котором эскетамин вводят интраназально.
95. Способ по любому одному из аспектов 86-94, в котором терапевтически эффективное количество по меньшей мере одного антидепрессанта вводят совместно с эскетамином.
96. Способ по аспекту 95, в котором эскетамин вводят совместно с одним или двумя антидепрессантами.
97. Способ по аспекту 95 или 96, в котором каждый антидепрессант независимо представляет собой имипрамин, амитриптилин, дезипрамин, нортриптилин, доксепин, протриптилин, тримипрамин, мапротилин, амоксапин, тразодон, бупропион, кломипрамин, флуоксетин, дулоксетин, эсциталопрам, циталопрам, сертралин, пароксетин, флувоксамин, нефазодон, венлафаксин, милнаципран, ребоксетин, миртазапин, фенелзин, транилципромин, моклобемид, кава-кава, зверобой, S-аденозилметионин, тиреотропин-рилизинг гормон, антагонист нейрокининовых рецепторов или трийодотиронин.
98. Способ по аспекту 95 или 96, в котором каждый антидепрессант независимо представляет собой ингибитор моноаминоксидазы, триклическое соединение, ингибитор обратного захвата серотонина, ингибитор обратного захвата серотонина-норэпинефрина, норадренергический и специфический серотонинергический агент или атипичный антидепрессант.
99. Способ по любому одному из аспектов 95-98, в котором каждый антидепрессант независимо представляет собой фенелзин, транилципромин, моклобемид, имипрамин, амитриптилин, дезипрамин, нортриптилин, доксепин, протриптилин, тримипрамин, кломипрамин, амоксапин, флуоксетин, сертралин, пароксетин, циталопрам, флувоксамин, венлафаксин, милнаципран, миртазапин или бупропион.
100. Способ по любому одному из аспектов 96-99, в котором каждый антидепрессант независимо представляет собой флуоксетин, имипрамин, бупропион, венлафаксин или сертралин.
101. Способ по аспекту 95, в котором по меньшей мере один антидепрессант представляет собой атипичный антидепрессант.
102. Способ по аспекту 101, в котором атипичный антидепрессант представляет собой арипипразол, кветиапин, оланзапин, рисперидон или палиперидон.
103. Способ по аспекту 101 или 102, в котором атипичный антидепрессант представляет собой арипипразол, кветиапин или оланзапин.
104. Фармацевтическая композиция для лечения большого депрессивного расстройства, содержащая эскетамин, необязательно по меньшей мере один антидепрессант, и по меньшей мере один фармацевтически приемлемый носитель.
105. Фармацевтическая композиция для лечения терапевтически рефрактерной депрессии или терапевтически резистентной депрессии, содержащая эскетамин, необязательно по меньшей мере один антидепрессант, и по меньшей мере один фармацевтически приемлемый носитель.
106. Фармацевтическая композиция для лечения суицидальной идеации, содержащая эскетамин, необязательно по меньшей мере один антидепрессант, и по меньшей мере один фармацевтически приемлемый носитель.
107. Применение эскетамина в приготовлении лекарственного средства для лечения большого депрессивного расстройства у нуждающегося в этом пациента.
108. Применение по аспекту 107, отличающееся тем, что пациент страдает от терапевтически рефрактерной депрессии или терапевтически резистентной депрессии.
109. Применение по аспекту 107, отличающееся тем, что пациент страдает от суицидальной идеации.
110. Эскетамин для применения в способе лечения большого депрессивного расстройства у нуждающегося в этом пациента.
111. Эскетамин по аспекту 110, в котором пациент страдает от терапевтически рефрактерной депрессии или терапевтически резистентной депрессии.
112. Эскетамин по аспекту 110, в котором пациент страдает от суицидальной идеации.
113. Композиция, содержащая эскетамин, для лечения большого депрессивного расстройства.
114. Композиция, содержащая эскетамин, для лечения терапевтически рефрактерной депрессии или терапевтически резистентной депрессии.
115. Композиция, содержащая эскетамин, для лечения суицидальной идеации.
116. Фармацевтический продукт, содержащий эскетамин, для введения пациенту, страдающему от большого депрессивного расстройства, причем эскетамин вводят указанному пациенту интраназально в количестве с клинически доказанными безопасностью и эффективностью.
117. Фармацевтический продукт по аспекту 116, в котором пациент страдает от терапевтически рефрактерной депрессии или терапевтически резистентной депрессии.
118. Фармацевтический продукт по аспекту 116, в котором пациент страдает от суицидальной идеации.
119. Способ введения эскетамина пациенту, включающий
первую фазу, длительность которой составляет от около одной недели до около четырех недель, причем пациенту вводят от около 28 мг до около 84 мг эскетамина с частотой два раза в неделю, а способ имеет клинически доказанную безопасность.
120. Способ по аспекту 119, в котором вводят около 28 мг эскетамина.
121. Способ по аспекту 119 или 120, в котором введение эскетамина позволяет достигать максимальной плазменной концентрации (Cмакс) эскетамина от около 45 до около 75 нг/мл, площади под кривой плазменная концентрация - время, начиная от момента времени 0 до времени последней количественно определяемой концентрации (ППКпосл), от около 125 до около 185 нг*ч/мл, или их комбинации.
122. Способ по любому одному из аспектов 119-121, в котором эскетамин вводят интраназально.
123. Способ по аспекту 122, в котором около 28 мг эскетамина вводят по меньшей мере за два отдельных впрыскивания.
124. Способ по аспекту 123, в котором около 28 мг эскетамина вводят посредством одного впрыскивания в каждую ноздрю.
125. Способ по аспекту 119, в котором вводят около 56 мг эскетамина.
126. Способ по аспекту 119 или 125, в котором введение эскетамина позволяет достигать максимальной плазменной концентрации (Cмакс) эскетамина от около 65 до около 120 нг/мл, площади под кривой плазменная концентрация - время, начиная от момента времени 0 до времени последней количественно определяемой концентрации (ППКпосл), от около 210 до около 320 нг*ч/мл, или их комбинации.
127. Способ по аспекту 125 или 126, в котором эскетамин вводят интраназально.
128. Способ по аспекту 127, в котором около 56 мг эскетамина вводят по меньшей мере за 4 впрыскивания.
129. Способ по аспекту 128, в котором эскетамин вводят посредством одного впрыскивания в каждую ноздрю в момент времени 0 в общем количестве около 28 мг и повторяют через около 5 минут в общем количестве около 56 мг.
130. Способ по аспекту 119, в котором вводят около 84 мг эскетамина.
131. Способ по аспекту 119 или 130, в котором введение эскетамина позволяет достигать максимальной плазменной концентрации (Cмакс) эскетамина от около 90 до около 165 нг/мл, площади под кривой плазменная концентрация - время, начиная от момента времени 0 до времени последней количественно определяемой концентрации (ППКпосл), от около 305 до около 490 нг*ч/мл, или их комбинации.
132. Способ по аспекту 130 или 131, в котором эскетамин вводят интраназально.
133. Способ по аспекту 132, в котором около 84 мг эскетамина вводят по меньшей мере за 6 впрыскиваний.
134. Способ по аспекту 133, в котором эскетамин вводят посредством одного впрыскивания в каждую ноздрю в момент времени 0 в общем количестве около 28 мг, повторяют через около 5 минут в общем количестве около 56 мг и снова повторяют через около 5 минут в общем количестве около 84 мг, всего за около 10 минут.
135. Способ по любому одному из аспектов 119-134, в котором длительность первой фазы составляет около 4 недель.
136. Способ по любому одному из аспектов 119-135, дополнительно включающий вторую фазу, длительность которой составляет от около 1 до около 4 недель, следующую за первой фазой, причем пациенту вводят от около 56 мг до около 84 мг эскетамина с частотой один раз в неделю.
137. Способ по аспекту 136, в котором пациенту вводят около 56 мг эскетамина с частотой один раз в неделю во время второй фазы.
138. Способ по аспекту 136, в котором пациенту вводят около 84 мг эскетамина с частотой один раз в неделю во время второй фазы.
139. Способ по любому одному из аспектов 136-138, в котором эскетамин вводят интраназально во время второй фазы.
140. Способ по любому одному из аспектов 136-139, в котором длительность второй фазы составляет около 4 недель.
141. Способ по любому одному из аспектов 119-140, дополнительно включающий третью фазу, длительность которой составляет по меньшей мере одну неделю, следующую за второй фазой, причем пациенту вводят от около 56 мг до около 84 мг эскетамина с частотой один раз в 2 недели или один раз в неделю.
142. Способ по аспекту 141, в котором пациенту вводят около 56 мг эскетамина с частотой один раз в 2 недели или один раз в неделю во время третьей фазы.
143. Способ по аспекту 141, в котором пациенту вводят около 84 мг эскетамина с частотой один раз в 2 недели или один раз в неделю во время третьей фазы.
144. Способ по любому одному из аспектов 141-143, в котором эскетамин вводят интраназально во время третьей фазы.
145. Способ по любому одному из аспектов 141-144, в котором длительность третьей фазы составляет по меньшей мере около одного месяца.
146. Способ по любому одному из аспектов 141-144, в котором длительность третьей фазы составляет по меньшей мере около двух месяцев.
147. Способ по любому одному из аспектов 141-144, в котором длительность третьей фазы составляет по меньшей мере около трех месяцев.
148. Способ по любому одному из аспектов 141-144, в котором длительность третьей фазы составляет по меньшей мере около четырех месяцев.
149. Способ по любому одному из аспектов 141-144, в котором длительность третьей фазы составляет по меньшей мере около пяти месяцев.
150. Способ по любому одному из аспектов 141-144, в котором длительность третьей фазы составляет по меньшей мере около шести месяцев.
151. Способ по любому одному из аспектов 141-144, в котором длительность третьей фазы составляет по меньшей мере около года.
152. Способ по любому одному из аспектов 141-144, в котором длительность третьей фазы составляет по меньшей мере около двух лет.
153. Способ по любому одному из аспектов 119-152 в котором способ дополнительно включает совместное введение антидепрессанта, причем способ имеет клинически доказанную эффективность в отношении лечения большого депрессивного расстройства.
154. Способ по аспекту 153, в котором антидепрессант вводят перорально.
155. Способ по аспектам 153 или 154, в котором большое депрессивное расстройство представляет собой терапевтически резистентную депрессию.
156. Фармацевтический продукт, содержащий одно или более интраназальных распылительных устройств, причем одно или более устройств содержат композицию эскетамина и одно или более устройств выполнены с возможностью введения от около 28 до около 84 мг эскетамина, и при этом фармацевтический продукт имеет клинически доказанную безопасность и/или клинически доказанную эффективность в отношении лечения большого депрессивного расстройства.
157. Фармацевтический продукт по аспекту 156, в котором большое депрессивное расстройство представляет собой терапевтически резистентную депрессию.
158. Фармацевтический продукт по аспекту 156 или 157, в котором продукт содержит одно устройство.
159. Фармацевтический продукт по аспекту 156, в котором устройство выполнено с возможностью введения эскетамина за два или более впрыскиваний.
160. Фармацевтический продукт по аспекту 158 или 159, в котором устройство содержит около 28 мг эскетамина.
161. Фармацевтический продукт по аспекту 156, в котором продукт содержит более одного устройства, а каждое устройство содержит около 28 мг эскетамина.
162. Фармацевтический продукт по аспекту 161, в котором каждое устройство является одноразовым устройством.
163. Фармацевтический продукт по аспекту 162, содержащий три устройства.
164. Фармацевтический продукт по любому одному из аспектов 156-163, дополнительно содержащий инструкции по осуществлению любого из способов по аспектам 119-155.
165. Способ лечения большого депрессивного расстройства с суицидальной идеацией, включающий
введение эскетамина в наибольшей переносимой дозе два раза в неделю во время первой индукционной фазы определенной длительности;
введение первого перорального антидепрессанта одновременно с эскетамином; и
оценку состояния пациента для определения, достигнут ли по существу полный ответ на эскетамин.
166. Способ по аспекту 165, в котором лечение прекращают, если пациент достигает, по существу, полного ответа на эскетамин.
167. Способ по аспекту 166, в котором осуществляют мониторинг пациента, чтобы убедиться, что пациент остается стабильным или находится в состоянии ремиссии только на первом пероральном антидепрессанте.
168. Способ по аспекту 165, в котором в случае недостижения, по существу, полного ответа во время первой индукционной фазы, инициируют вторую индукционную фазу.
169. Способ по аспекту 168, в котором во время второй индукционной фазы пациенту повторно назначают эскетамин в наибольшей переносимой дозе одновременно со вторым пероральным антидепрессантом.
170. Способ по аспекту 169, в котором второй пероральный антидепрессант является таким же, как и первый пероральный антидепрессант.
171. Способ по аспекту 169, в котором второй пероральный антидепрессант отличается от первого перорального антидепрессанта.
172. Способ по любому одному из аспектов 169-171, в котором осуществляют мониторинг пациента, чтобы убедиться, что пациент остается стабильным или находится в состоянии ремиссии только на втором пероральном антидепрессанте.
173. Способ по любому одному из аспектов 169-172, в котором в случае недостижения, по существу, полного ответа во время второй индукционной фазы, инициируют третью индукционную фазу.
174. Способ по аспекту 173, в котором во время третьей индукционной фазы пациенту повторно назначают эскетамин в наибольшей переносимой дозе одновременно с третьим пероральным антидепрессантом.
175. Способ по аспекту 174, в котором третий пероральный антидепрессант является таким же, как и второй пероральный антидепрессант.
176. Способ по аспекту 174, в котором третий пероральный антидепрессант отличается от второго перорального антидепрессанта.
177. Способ по любому одному из аспектов 165-176, дополнительно включающий введение пациенту терапевтически эффективного количества эскетамина реже чем два раза в неделю во время последующей фазы поддержания.
178. Способ по любому одному из аспектов 165-177, в котором длительность первой, второй и третьей индукционной фазы независимо составляет по меньшей мере 4 недели.
179. Способ лечения терапевтически резистентной депрессии у пациента, в котором пациент не отвечал по меньшей мере на два пероральных антидепрессанта во время текущего депрессивного эпизода, содержащий этапы, на которых:
пациенту вводят первый пероральный антидепрессант, и
пациенту вводят эскетамин по меньшей мере два раза в неделю во время первой индукционной фазы определенной длительности;
пациента оценивают во время первой индукционной фазы; и
если пациент не достигает, по существу, полного ответа на эскетамин, пациенту повторно назначают наибольшую переносимую дозу эскетамина одновременно со вторым пероральным антидепрессантом во время второй индукционной фазы определенной длительности.
180. Способ по аспекту 179, в котором первый пероральный антидепрессант является таким же, как по меньшей мере один из по меньшей мере двух пероральных антидепрессантов.
181. Способ по аспекту 179, в котором первый пероральный антидепрессант отличается от по меньшей мере одного из по меньшей мере двух пероральных антидепрессантов.
182. Способ по аспекту 179, в котором первый пероральный антидепрессант отличается от по меньшей мере двух пероральных антидепрессантов.
183. Способ по любому одному из аспектов 179-182, в котором если пациент не достигает, по существу, полного ответа на эскетамин во время второй индукционной фазы, пациенту повторно назначают эскетамин одновременно с третьим пероральным антидепрессантом во время третьей индукционной фазы определенной длительности.
184. Способ по аспекту 183, в котором третий пероральный антидепрессант является таким же, как и второй пероральный антидепрессант.
185. Способ по аспекту 183, в котором третий пероральный антидепрессант отличается от второго перорального антидепрессанта.
186. Способ по любому одному из аспектов 179-185, дополнительно включающий то, что при достижении пациентом, по существу, полного ответа на эскетамин пациенту вводят терапевтически эффективное количество эскетамина не чаще одного раза в неделю во время последующей фазы поддержания.
187. Способ по любому одному из аспектов 179-186, в котором длительность первой, второй и третьей индукционной фазы независимо составляет по меньшей мере 4 недели.
188. Способ лечения терапевтически резистентной депрессии у пациента, содержащий этапы, на которых:
указанному пациенту вводят терапевтически эффективное количество перорального антидепрессанта; и
указанному пациенту интраназально вводят терапевтически эффективное количество эскетамина по меньшей мере два раза в неделю во время индукционной фазы длительностью по меньшей мере 4 недели; и
пациенту интраназально вводят терапевтически эффективное количество эскетамина не чаще одного раза в неделю во время последующей фазы поддержания,
причем способ имеет клинически доказанную безопасность и/или клинически доказанную эффективность.
189. Способ по аспекту 188, в котором эскетамин вводят один раз в две недели во время последующей фазы поддержания.
190. Способ по аспекту 188, в котором частоту введения можно корректировать во время индукционной фазы и/или фазы поддержания.
191. Способ по аспекту 188, в котором терапевтически эффективное количество эскетамина, вводимое во время индукционной фазы, составляет от около 28 мг до около 84 мг.
192. Способ по аспекту 191, в котором терапевтически эффективное количество эскетамина составляет около 28 мг.
193. Способ по аспекту 191, в котором терапевтически эффективное количество эскетамина составляет около 56 мг.
194. Способ по аспекту 191, в котором терапевтически эффективное количество эскетамина составляет около 84 мг.
195. Способ по аспекту 191, в котором терапевтически эффективное количество эскетамина составляет около 56 мг в начале индукционной фазы и корректируется до около 84 мг во время индукционной фазы.
196. Способ по аспекту 192, в котором возраст пациента составляет 65 лет или более.
197. Способ по аспекту 188, в котором терапевтически эффективное количество эскетамина, вводимое во время фазы поддержания, составляет около 56 мг или около 84 мг.
198. Способ по любому одному из аспектов 188-197, в котором терапевтически эффективное количество эскетамина во время индукционной фазы и фазы поддержания доставляют из интраназального устройства введения за 2 или более впрыскиваний.
199. Способ по любому одному из аспектов 188-198, в котором лечение продолжают в течение по меньшей мере шести месяцев.
200. Способ по любому одному из аспектов 188-198, в котором лечение продолжают в течение периода до двух лет.
В контексте данного документа АД=антидепрессант; НЯ=нежелательное явление; ЭСК=назальное впрыскивание эскетамина; ПБО=назальное впрыскивание плацебо; PHQ-9=Опросник по ответственности пациента; SDS=Шкала нетрудоспособности Шихана; CGI-S=Общее клиническое впечатление о тяжести заболевания; MADRS=Шкала Монтгомери - Асберг для оценки депрессии; СО=стандартное отклонение; SNRI=ингибиторы обратного захвата серотонина и норэпинефрина; SSRI=селективные ингибиторы обратного захвата серотонина; МНК=метод наименьших квадратов; СП=стандартная погрешность; ИМТ=индекс массы тела; BPIC-SS=Оценка симптомов боль в мочевом пузыре/интерстициального цистита; BPRS+ = подшкала положительных симптомов из 4 пунктов краткой психиатрической оценочной шкалы; К=клинический визит; CADSS=Применяемая клиницистами шкала диссоциативных состояний; CGADR=Глобальная клиническая оценка готовности к выписке; C-SSRS=Шкала оценки выраженности суицидальных тенденций Колумбийского университета; ДНК=дезоксирибонуклеиновая кислота; ЭКГ=электрокардиограмма; EQ-5D-5L=Европейский опросник для оценки качества жизни по 5 направлениям, 5-уровневая шкала; ДП=досрочное прекращение (участия в исследовании); GAD-7=Генерализированное тревожное расстройство, шкала из 7 пунктов; HE=окрашивание гематоксилином и эозином; HbA1c-тест, тест на гликированный гемоглобин; HRUQ=Опросник по использованию ресурсов в здравоохранении; HVLT-R=Пересмотренный тест на заучивание слов Хопкинса; IDS-C30=Опросник депрессивной симптоматики - клиническая оценка, шкала из 30 пунктов; ОЭ=отсутствие эффективности; БДР=большое депрессивное расстройство; НПН=невозможность последующего наблюдения; MGH-ATRQ=Опросник по истории лечения антидепрессантами Центральной больницы штата Массачусетс; MGH-Female RLHQ=Опросник по женскому репродуктивному жизненному цинку и гормонам Центральной больницы штата Массачусетс; MINI=Краткое международное нейропсихиатрическое интервью; MOAA/S=Модифицированная оценка наблюдателем активности/седации; Н/З=статистически незначимо; ОИ=открытое исследование; ДР=другая причина прекращения (участия в исследовании); PAQ=Опросник по ответственности пациента; PHQ-9=Опросник по состоянию здоровья пациента - 9; PWC-20=Врачебный контрольный список по прекращению, шкала из 20 пунктов; QIDS=Краткое описание депрессивных симптомов из 16 пунктов - самоотчет; РНК=рибонуклеиновая кислота; SDS=Шкала нетрудоспособности Шихана; SAFER=ситуативная vs. личностная (тревожность), возможность оценки, внешняя достоверность, экологическая достоверность, правило трех «P»; STOP-Bang=храп, усталость, наблюдаемая одышка, высокое кровяное давление, индекс массы тела, возраст, обхват шеи, пол (опросник); ТРД=терапевтически резистентная депрессия; TSH=тиреостимулирующий гормон; RA=только дистанционная оценка; LOCF=использование последнего документированного значения; ПП=прекращение пациентом; ПР=прекращение.
Следующие примеры приведены, чтобы помочь понять данное изобретение, и их не следует рассматривать как ограничивающие каким-либо образом данное изобретение, определяемое приведенной ниже формулой изобретения.
Пример 1:
Эффективность интраназального эскетамина для лечения терапевтически резистентной депрессии (ТРД), клиническое исследование фазы 3
Способность эскетамина лечить терапевтически рефрактерную или терапевтически резистентную депрессию (ТРД) оценивали с помощью описанного ниже клинического исследования, которое проводили для оценки эффективности, безопасности и переносимости вводимого в гибких дозах интраназального эскетамина и нового назначенного перорального антидепрессанта у взрослых субъектов с ТРД. Исследование служило в качестве ключевого краткосрочного исследования эффективности и безопасности фазы 3 для обеспечения выполнения требований регуляторных органов для регистрации интраназального эскетамина для лечения ТРД.
Предположение для данного исследования состояло в том, что у взрослых субъектов с ТРД перевод с неудачного лечения антидепрессантами на интраназальный эскетамин плюс новый назначенный пероральный антидепрессант имел бы преимущество перед переводом на лечение новым назначенным пероральным антидепрессантом (действующий препарат сравнения) плюс интраназальное плацебо для уменьшения депрессивных симптомов.
Первичной целью данного исследования была оценка эффективности перевода взрослых субъектов с ТРД с предыдущего лечения антидепрессантами (на которое они не отвечали) на вводимый в гибких дозах интраназальный эскетамин (28 мг, 56 мг или 84 мг) плюс новый назначенный пероральный антидепрессант по сравнению с переводом на новый назначенный пероральный антидепрессант (действующий препарат сравнения) плюс интраназальное плацебо в отношении уменьшения депрессивных симптомов, оцениваемого по изменению относительно исходного уровня общей оценки по шкале MADRS от 1 суток (до рандомизации) до конца 4-недельной двойной слепой индукционной фазы.
Ключевыми вторичными целями было оценивание эффекта интраназального эскетамина плюс новый назначенный пероральный антидепрессант по сравнению с новым назначенным пероральным антидепрессантом (действующий препарат сравнения) плюс интраназальное плацебо на следующие параметры у взрослых субъектов с ТРД: (a) депрессивные симптомы (сообщаемые субъектами), (b) начало клинического ответа на 2 сутки и (с) функционирование и связанная с ним недееспособность. Дополнительные вторичные цели включали (а) частоту ответа депрессии, (b) частоту ремиссии депрессии, (с) общую тяжесть депрессивного заболевания, (d) тревожные симптомы и (e) связанное со здоровьем качество жизни и состояние здоровья.
Для изучения безопасности и переносимости интраназального эскетамина плюс новый назначенный пероральный антидепрессант по сравнению с новым назначенным пероральным антидепрессантом (действующий препарат сравнения) плюс интраназальное плацебо у взрослых субъектов с ТРД также определяли следующие параметры: (a) НЯВЛ, включая НЯ, представляющие особый интерес, (b) локальную назальную переносимость, (c) воздействие на частоту сердечных сокращений, кровяное давление, частоту дыхания и насыщение крови кислородом, (d) воздействие на активность и седацию, (e) потенциальные психозоподобные эффекты, (f) диссоциативные симптомы, (g) потенциальное воздействие на когнитивную функцию, (h) потенциальное воздействие на суицидальные идеацию/поведение, (i) потенциальные возникшие при лечении симптомы цистита и/или симптомы со стороны нижних мочевыводящих путей, (j) потенциальные синдром отмены и/или симптомы отдачи после завершения лечения интраназальным эскетамином и (k) потенциальное воздействие на ощущение запаха.
ФК интраназального эскетамина у взрослых субъектов с ТРД, получавших интраназальный эскетамин плюс новый назначенный пероральный антидепрессант также оценивали в качестве части вторичных целей.
ИНФОРМАЦИЯ ПО ИССЛЕДУЕМОМУ ЛЕКАРСТВЕННОМУ ПРЕПАРАТУ
Эскетамин был предоставлен в виде прозрачного бесцветного интраназального раствора гидрохлорида эскетамина (16,14% масса/объем [масс./об.]; эквивалент 14% масс./об. основания эскетамина) в насосе для назального впрыскивания. Раствор состоял из 161,4 мг/мл гидрохлорида эскетамина (эквивалент 140 мг основания эскетамина), приготовленного в 0,12 мг/мл этилендиаминтетрауксусной кислоты (ЭДТА) и 1,5 мг/мл лимонной кислоты при pH 4,5 в воде для инъекций. Он был предоставлен в насосе для назального впрыскивания, который доставлял 16,14 мг гидрохлорида эскетамина (14 мг основания эскетамина) на 100 мкл спрея. Каждый отдельный насос для назального впрыскивания (устройство) содержал в общей сложности 28 мг (т.е. на 2 впрыскивания).
Раствор плацебо был предоставлен в виде прозрачного бесцветного интраназального раствора воды для инъекций, с горьким агентом (денатониум бензоат [Bitrex®] в конечной концентрации 0,001 мг/мл), добавленным для имитации вкуса интраназального раствора с активным лекарственным препаратом. Раствор плацебо был предоставлен в аналогичных устройствах типа насоса для назального впрыскивания. Хлорид бензалкония добавляли в качестве консерванта в концентрации 0,3 мг/мл. Каждый отдельный насос для назального впрыскивания (устройство) содержал раствор на 2 впрыскивания.
Медикаментозные пероральные антидепрессанты
Дулоксетин (30 мг) был получен из коммерческого источника и предоставлен под ответственность спонсора. Физическое описание и перечень вспомогательных веществ смотрите на вкладыше упаковки/ОХЛП.
Эсциталопрам (10 мг) был получен из коммерческого источника и предоставлен под ответственность спонсора. Физическое описание и перечень вспомогательных веществ смотрите на вкладыше упаковки/ОХЛП.
Сертралин (50 мг и 25 мг) (в случае применения) был получен из коммерческого источника и предоставлен под ответственность спонсора. Физическое описание и перечень вспомогательных веществ смотрите на вкладыше упаковки/ОХЛП.
Венлафаксин (75 мг и 37,5 мг) (в случае применения) был получен из коммерческого источника и предоставлен под ответственность спонсора. Физическое описание и перечень вспомогательных веществ смотрите на вкладыше упаковки/ОХЛП.
Обзор дизайна исследования
Это было рандомизированное, двойное слепое, контролируемое по действующему препарату, многоцентровое исследование на взрослых субъектах мужского и женского пола с ТРД для оценки эффективности, безопасности и переносимости вводимого в гибких дозах интраназального эскетамина (28 мг, 56 мг или 84 мг) плюс новый назначенный пероральный антидепрессант по сравнению с новым назначенным пероральным антидепрессантом (действующий препарат сравнения) плюс интраназальное плацебо. Данное исследование включало 3 фазы, которые кратко описаны ниже. Схема дизайна исследования представлена на Фиг. 1.
Фаза скрининга/проспективного наблюдения (длительность 4 недели)
Во время этой фазы проспективно оценивали ответ на лечение по текущей схеме лечения субъекта пероральными антидепрессантами. Через 4 недели следования одной схеме лечения (при одной дозировке) субъекты, которые не отвечали на текущее лечение пероральными антидепрессантами (по оценке независимыми удаленными экспертами), допускались к переходу к двойной слепой индукционной фазе. Исследователи центра работали с обезличенными данными в отношении критериев исследования по отсутствию ответа.
Отвечающие критериям субъекты, которые попали в двойную слепою индукционную фазу, прекращали прием своих текущих пероральных антидепрессантов. При наличии клинических показаний прием текущих пероральных антидепрессантов субъекта можно было уменьшать и прекращать в течение дополнительного необязательного периода до 3 недель согласно локальным инструкциям по применению или клиническому заключению.
Так как прием нового перорального антидепрессанта начинался на 1 сутки двойной слепой индукционной фазы, допущенные субъекты, для которых не требовалось постепенное прекращение приема их текущих антидепрессантов, незамедлительно переходили к двойной слепой индукционной фазе.
Двойная слепая индукционная фаза (длительность 4 недели)
В исследование вошли 227 рандомизированных субъектов (4 из которых не получали интраназальный и/или пероральный АД исследуемый препарат и, следовательно, не были включены в выборки для анализа), которые были случайным образом распределены в соотношении 1:1 (n=98 субъектов на группу лечения) для прохождения двойного слепого лечения интраназальным эскетамином или интраназальным плацебо. Курс интраназального лечения (эскетамином или плацебо) проводили дважды в неделю. Кроме того, все субъекты на 1 сутки начали принимать новый немаскированный пероральный антидепрессант, который они принимали ежесуточно в течение всей фазы. Назначенным пероральным антидепрессантом был 1 из 4 пероральных антидепрессантов (дулоксетин, эсциталопрам, сертралин или венлафаксин с продленным высвобождением [XR]), на который у субъекта ранее не наблюдалось отсутствие ответа во время текущего депрессивного эпизода, непереносимости (в течение жизни) и который был доступен в стране-участнице.
В конце индукционной фазы субъекты, которые демонстрировали ответ (определяемый как ≥ 50% снижение общей оценки по шкале MADRS относительно исходного уровня [1 сутки до рандомизации] до конца 4-недельной двойной слепой индукционной фазы), допускались к участию в последующем исследовании ESKETINTRD3003, если они соответствовали всем остальным критериям включения в исследование (ESKETINTRD3003 представляет собой долгосрочное исследования поддержания эффективности, включающее повторяемые курсы лечения интраназальным эскетамином).
Если субъект выбыл из исследования до окончания двойной слепой индукционной фазы по причинам, отличным от отзыва согласия, то в течение 1 недели с даты прекращения проводили визит досрочного отстранения, за которым следовала фаза последующего наблюдения.
Фаза последующего наблюдения (длительность 24 недели)
В эту фазу были включены все субъекты, которые не были допущены или которые предпочли не участвовать в фазе поддержании эффекта ESKETINTRD3003 и получили по меньшей мере 1 дозу интраназального исследуемого лекарственного средства в двойной слепой индукционной фазе. Во время этой фазы курс интраназального лечения не проводили.
В начале фазы последующего наблюдения исследователь и/или лечащий врач субъекта обеспечивали дополнительное клиническое/стандартное лечение депрессии. Решение о продолжении приема перорального антидепрессанта на этой фазе принимал исследователь, однако, чтобы лучше оценить потенциальные симптомы отказа от интраназального исследуемого препарата, рекомендовалось продолжать прием перорального антидепрессанта в течение по меньшей мере первых 2 недель фазы последующего наблюдения, если это не считали клинически неприемлемым.
Фаза последующего наблюдения также позволила собрать дополнительные информативные данные для оценки течения большого депрессивного эпизода субъекта в продолжении 6-месячного периода.
Принимая во внимание необязательный период постепенного уменьшения дозы длительностью до 3 недель, продолжительность участия субъекта в исследовании составляла 11 недель (для субъектов, которые перешли к ESKETINTRD3003) или 35 недель (для субъектов, завершивших этап последующего наблюдения).
Исследуемая популяция
Критерии включения для набора субъектов в данное исследование были следующими. Каждый потенциальный субъект удовлетворял всем из перечисленных ниже критериев для включения в исследование.
1. На момент подписания формы информированного согласия (ФИС) субъект представлял собой мужчину или женщину возрастом от 18 лет (или старше, если минимальный установленный законом возраст согласия в стране, в которой проводится исследование, составляет > 18) до 64 лет включительно.
2. В начале фазы скрининга/проспективного наблюдения субъект соответствовал диагностическим критериям DSM-5 в отношении однократного эпизода БДР (в случае однократного эпизода БДР его длительность составляла ≥ 2 лет) или рецидива БДР без психотических признаков на основании клинической оценки и с подтверждением MINI.
3. В начале фазы скрининга/проспективного наблюдения у субъекта в анамнезе было отсутствие ответа на лечение ≥ 2, но ≤ 5 пероральными антидепрессантами во время текущего эпизода депрессии, оцененное с помощью MGH-ATRQ и подтвержденное задокументированной историей болезни и фармацевтическими/рецептурными записями. Субъект принимал пероральные антидепрессанты с отсутствием ответа в начале фазы скрининга/проспективного наблюдения. Субъекты соблюдали схему непрерывного лечения антидепрессантами (без коррекции дозировки) во время фазы скрининга/проспективного наблюдения, что задокументировано в PAQ. Пропуск приема антидепрессанта в течение ≥ 4 суток в течение предшествующего 2-недельного периода рассматривали как неадекватное соблюдение схемы приема. Субъектов, которые не демонстрировали ответ на текущие пероральные антидепрессанты во время фазы скрининга/проспективного наблюдения (по оценке независимыми удаленными экспертами), допускали к рандомизации при условии соответствия всем остальным критериям.
4. В начале фазы скрининга/проспективного наблюдения субъект имел общую оценку IDS-C30 ≥34.
5. Текущий большой депрессивный эпизод субъекта и ответ на лечение антидепрессантами в текущем депрессивном эпизоде были подтверждены с помощью независимой от исследовательского центра квалификационной оценки.
6. Субъект был стабильным с медицинской точки зрения на основании физического осмотра, истории болезни, основных физиологических показателей (в том числе кровяного давления), пульсоксиметрии и ЭКГ в 12-ти отведениях, проведенных во время фазы скрининга/проспективного наблюдения. Если имелись какие-либо отклонения, не указанные в критериях включения и исключения, определение их клинической значимости выполнялось исследователем и записывалось в первичной документации субъекта, и инициировалось исследователем.
7. Субъект был стабильным с медицинской точки зрения на основании клинических лабораторных тестов, проведенных во время фазы скрининга/проспективного наблюдения. Если результаты панели биохимического анализа сыворотки, гематологии или анализа мочи находились за пределами нормальных референсных диапазонов, субъекта включали только в том случае, если исследователь оценил аномалии или отклонения от нормы, как не имеющие клинической значимости или соответствующие и целесообразные для исследуемой популяции. Это определение записывалось в первичной документации субъекта и инициировалось исследователем. Субъекты с анамнезом заболеваний/расстройств щитовидной железы, которые проходили лечение тиреоидными гормонами, принимали постоянную дозу в течение 3 месяцев перед началом фазы скрининга/проспективного наблюдения и имели уровни тиреостимулирующего гормона (TSH) в пределах нормального диапазона во время фазы скрининга/проспективного наблюдения.
8. Субъекты не испытывали затруднений с самостоятельным введением интраназального препарата и были способны выполнять инструкции по интраназальному введению.
9. До начала фазы скрининга/проспективного наблюдения субъекты-женщины (a) находились в состоянии отсутствия детородного потенциала: после менопаузы (в возрасте > 45 лет с аменореей в течение по меньшей мере 12 месяцев или в любом возрасте с аменореей в течение по меньшей мере 6 месяцев, и с уровнем фолликулостимулирующего гормона (FSH) в сыворотке > 40 МЕ/л); имели необратимую стерильность (например, перевязка маточных труб, гистерэктомия, двусторонняя сальпингэктомия); или иным образом не были способными к беременности; или (b) имели детородный потенциал и использовали высокоэффективный способ контрацепции, соответствующий местным правовым нормам в отношении применения способов контрацепции для субъектов, участвующих в клинических исследованиях, например, использовали утвержденные для применения пероральные, инъекционные или имплантируемые гормональные средства контрацепции; размещение внутриматочных спиралей (IUD) или внутриматочных систем (IUS); барьерные способы (например, презерватив со спермицидной пенкой/гелем/пленкой/кремом/суппозиторием или окклюзионный колпачок [диафрагма или цервикальный/вагинальный колпачок] со спермицидной пенкой/гелем/пленкой/кремом/суппозиторием); стерилизацию мужчины-партнера (вазэктомированный партнер должен быть единственным партнером для данного субъекта); или полное воздержание (если это соответствует предпочтительному и обычному образу жизни субъекта). Если детородный потенциал менялся после начала исследования (например, женщина, которая ранее не был гетеросексуально активной, становилась активной), женщина-субъект начинала применять высокоэффективный способ контрацепции, как описано выше. Женщины давали согласие на продолжение использования этих способов контрацепции в течение всего исследования и в течение по меньшей мере 6 недель после последней дозы исследуемого интраназального препарата.
10. Женщина с детородным потенциалом имела негативный сывороточный результат (хорионический гонадотропин человека β [β-hCG]) в начале фазы скрининга/проспективного наблюдения и отрицательный результат теста мочи на беременность на 1 сутки двойной слепой индукционной фазы перед рандомизацией.
11. Мужчина, который был сексуально активен с женщиной с детородным потенциалом и не подвергавшийся вазэктомии, давал согласие на использование барьерного способа контрацепции, например, презерватива со спермицидной пенкой/гелем/пленкой/кремом/суппозиторием или использование партнершей окклюзионного колпачка (диафрагмы или цервикального/вагинального колпачка) со спермицидной пенкой/гелем/пленкой/кремом/суппозиторием, начиная с 1 суток двойной слепой индукционной фазы (перед рандомизацией) и в течение 3 месяцев после последней дозы исследуемого интраназального препарата. В альтернативном варианте женщины-партнерши с детородным потенциалом могли использовать высокоэффективный способ контрацепции, например, использовали утвержденные для применения пероральные, инъекционные или имплантируемые гормональные средства контрацепции; размещение внутриматочных спиралей (IUD) или внутриматочных систем (IUS); стерилизацию мужчины-партнера. Если детородный потенциал менялся после начала исследования, женщина-партнерша исследуемого субъекта-мужчины начинала применять высокоэффективный способ контрацепции, как описано выше.
12. Субъект имел готовность и способность к соблюдению указанных в протоколе клинического исследования запретов и ограничений.
13. Каждый субъект подписывал ФИС с указание, что он/она понимают цели и процедуры, необходимые для исследования, и готовы принимать участие в исследовании.
Критерии исключения для набора субъектов в данное исследование были следующими. Любой потенциальный субъект, который соответствовал любому одному из нижеприведенных критериев, был исключен из участия в исследовании.
1. Депрессивные симптомы субъекта ранее демонстрировали отсутствие ответа на: (a) эскетамин или кетамин во время текущего большого депрессивного эпизода согласно клиническому заключению или (b) все варианты лечения пероральными антидепрессантами, доступными в соответствующей стране, для двойной слепой индукционной фазы (т.е. дулоксетин, эсциталопрам, сертралин и венлафаксин XR) во время текущего большого депрессивного эпизода (на основании MGH-ATRQ), или (c) адекватный курс лечения электрошоковой терапией (ЭШТ) во время текущего большого депрессивного эпизода, определяемый как по меньшей мере 7 сеансов лечения односторонней ЭШК.
2. Субъект имеет имплантат для стимуляции блуждающего нерва (VNS) или проходил глубокую стимуляция головного мозга (DBS) во время текущего эпизода депрессии.
3. Субъект имел текущий или предшествующий диагноз по DSM-5 психотического расстройства или БДР с психозом, биполярных или схожих расстройства (подтвержденных MINI), сопутствующего обсессивно-компульсивного расстройства, умственной отсталости (только диагностический код 319 DSM-5), пограничного расстройства личности, антисоциального расстройства личности, истерического расстройства личности или нарциссического расстройства личности.
4. Субъект имел гомицидальные идеи/намерения согласно клиническому заключению исследователя или имел суицидальные идеи с некоторым намерением действовать в течение 6 месяцев перед началом фазы скрининга/проспективного наблюдения согласно клиническому заключению исследователя или на основании C-SSRS в соответствии с ответом «да» на пункт 4 (активная суицидальная идеация с некоторым намерением действовать без конкретного плана) или на пункт 5 (активная суицидальная идеация со специфическим планом или намерением) в отношении суицидальной идеации по C-SSRS, или анамнез суицидального поведения в течение прошедшего года перед началом фазы скрининга/проспективного наблюдения. Субъектов, сообщивших о суицидальной идеации с намерением действовать или суицидальным поведением перед началом двойной слепой индукционной фазы, исключали.
5. Субъект имел в анамнезе расстройство, связанное с употреблением наркотиков или алкоголя, в соответствии с критериями DSM-5, за исключением никотина или кофеина, в течение 6 месяцев перед началом фазы скрининга/проспективного наблюдения. Анамнез (на протяжении жизни) расстройства, связанного с употреблением галлюциногенов кетамина, фенциклидина (PCP), диэтиламида лизергиновой кислоты (ЛСД) или 3,4-метилендиокси-метамфетамина (MDMA), был исключающим критерием.
6. Субъект в настоящем или в прошлом имел в анамнезе судорожные припадки (перенесенные в детстве фебрильные судороги без осложнений, не имевшие последствий, не являются исключающим критерием).
7. Субъект имел общую оценку по шкале UPSIT ≤ 18, что указывает на отсутствие обоняния, во время фазы скрининга/проспективного наблюдения.
8. Субъект имел одно из следующих сердечно-сосудистых патологических состояний: (a) цереброваскулярное заболевание с анамнезом инсульта или временной ишемической атаки, (b) аневризматическое сосудистое заболевание (включая внутричерепную, грудную или брюшную аорту или периферические артериальные сосуды), (c) ишемическую болезнь сердца с инфарктом миокарда, нестабильную стенокардию, процедуру восстановления сосудов (например, коронарную ангиопластику или операцию по установке обходного сосудистого шунта) в течение 12 месяцев перед началом фазы скрининга/проспективного наблюдения или запланированную процедуру восстановления сосудов, (d) гемодинамически значимый порок клапана сердца, такой как недостаточность митрального клапана, аортальный стеноз или недостаточность аортального клапана, или (e) сердечную недостаточность класса III-IV по классификации Нью-Йоркской кардиологической ассоциации (NYHA) любой этиологии.
9. Субъект имел в анамнезе неконтролируемую гипертензию несмотря на диету, физические упражнения или антигипертензивную терапию в начале фазы скрининга/проспективного наблюдения или гипертонический криз в анамнезе в прошлом, или текущие признаки неконтролируемой гипертензии, определяемые как систолическое кровяное давление (СКД) в положении лежа на спине > 140 мм рт.ст. или диастолическое кровяное давление (ДКД) > 90 мм рт.ст. На 1 сутки двойной слепой индукционной фазы перед рандомизацией исключающими критериями также являются СКД в положении лежа на спине > 140 мм рт.ст. или ДКД > 90 мм рт.ст.
Текущая схема антигипертензивной терапии потенциального субъекта может быть скорректирована во время фазы скрининга/проспективного наблюдения, после чего проводят повторное оценивание субъектов для оценки контроля кровяного давления. Субъект придерживался постоянной схемы в течение по меньшей мере 2 недель до 1 суток двойной слепой индукционной фазы.
10. Субъект в настоящем или в прошлом имел в анамнезе существенные легочную недостаточность/легочное заболевание или насыщенность артериальной крови кислородом (SpO2) < 93% в начале фазы скрининга/проспективного наблюдения или на 1 сутки перед рандомизацией.
11. Субъект имел клинически значимые аномалии ЭКГ в начале фазы скрининга/проспективного наблюдения или на 1 сутки двойной слепой индукционной фазы перед рандомизацией, определяемые как: (a) интервал QT с коррекцией по формуле Фридеричиа (QTcF): ≥450 мс, (b) признаки AV-блокады 2-ой и 3-ей степени или AV-блокада 1-ой степени с PR-интервалом >200 мс, блокада левой ножки пучка (LBBB) или блокада правой ножки пучка (RBBB), (c) признаки новой ишемии, (d) аритмия (за исключением преждевременных сокращений предсердия [PAC] и преждевременных сокращений желудочка [PVC]).
12. Субъект имел в анамнезе дополнительные факторы риска в отношении двунаправленной тахикардии (например, сердечную недостаточность, гипокалиемию, семейный анамнез синдрома удлинения интервала QT) или использования сопутствующих препаратов, которые продлевают интервал QT/корректируют интервал QT (QTc).
13. Субъект имел в анамнезе симптомы и признаки, позволяющие предположить наличие цирроза печени (например, расширение вен пищевода, асциты и повышенное протромбиновое время), или значения аланинаминотрансферазы (АЛТ) или аспартатаминотрансферазы (АСТ), в ≥2x раз превышающие верхнюю границу нормы, или общий билирубин, в > 1,5 раза превышающий ВГН, во время фазы скрининга/проспективного наблюдения. В случае повышения уровня билирубина, если, по мнению исследователя и по согласию медицинского инспектора спонсора, повышение уровня билирубина было связано с заболеванием Гилберта, субъект имел возможность участвовать в исследовании.
14. Субъект имел положительный(-е) результат(-ы) теста на препараты, вызывающие лекарственную зависимость (включая барбитураты, метадон, опиаты, кокаин, фенциклидин и амфетамин/метамфетамин), в начале фазы скрининга/проспективного наблюдения или на 1 сутки двойной слепой индукционной фазы перед рандомизацией. Субъекты, которые имели положительный результат теста при скрининге из-за предписанных/отпускаемых без рецепта опиатов, барбитуратов или амфетаминов, были допущены к продолжению фазы скрининга/проспективного наблюдения, если они прекращали прием препарата по меньшей мере за 1 неделю или 5 времен полувыведения, в зависимости от того, что занимало больше времени, до 1 суток двойной слепой индукционной фазы (перед рандомизацией) в соответствии с ограничениями, представленными исследователю и воспроизведенными ниже в Таблице 6. Чтобы субъект участвовал в рандомизации, результат теста на 1 сутки (перед рандомизацией) на препараты, вызывающие лекарственную зависимость, должен был быть отрицательным. Повторное тестирование для получения положительного(ых) результата(ов) теста не допускалось, за исключением указанных выше причин. Предшествующее периодическое применение каннабиноидов перед началом фазы скрининга/проспективного наблюдения не являлось исключающим критерием, при условии, что субъект не соответствовал критериям расстройства, связанного с употреблением наркотических веществ. Однако положительный результат теста на каннабиноиды перед введением дозы на 1 сутки двойной слепой индукционной фазы был исключающим критерием.
15. Субъект имел неконтролируемый сахарный диабет или вторичный диабет, о чем свидетельствует HbA1c > 9% во время фазы скрининга/проспективного наблюдения, или анамнез в течение 3 месяцев перед началом фазы скрининга/проспективного наблюдения диабетического кетоацидоза, гипергликемической комы или тяжелой гипогликемии с потерей сознания.
16. Субъект имел не подвергнутую лечению глаукому, текущее проникающее или перфоративное повреждение глаза, повреждение головного мозга, гипертензивную энцефалопатию, интратекальную терапию с желудочными шунтами или любое другое состояние, связанное с повышенным внутричерепным давлением или повышенным внутриглазным давлением, или запланированную операцию на глазах.
17. Субъект имел любое анатомическое или медицинское состояние, которое может препятствовать доставке или всасыванию исследуемого интраназального препарата (например, существенные структурные или функциональные аномалии носа или верхних дыхательных путей; закупорку или поражения слизистой оболочки ноздрей или носовых проходов; подвергался хирургии околоносовых пазух в течение 2 предшествующих лет).
18. Субъект имел аномальную или неисправленную искривленную носовую перегородку с любым 1 или более следующими симптомами: (a) закупорка 1 или обеих ноздрей за последние несколько месяцев, которая может повлиять на участие в исследовании, (b) заложенность носа (особенно односторонняя), (c) частые носовые кровотечения, (d) частые инфекции пазух носа или (e) шумное дыхание во время сна.
19. Субъект имел в анамнезе злокачественные новообразования в течение 5 лет до начала фазы скрининга/проспективного наблюдения (исключениями были плоскоклеточная и базальноклеточная карцинома кожи и карцинома шейки матки in situ, или злокачественное новообразование, которое, по мнению исследователя и по согласованию с медицинским наблюдателем спонсора, считалось излеченным с минимальным риском рецидива).
20. Субъект имел известные аллергические реакции, гиперчувствительность, непереносимость или противопоказания к эскетамину/кетамину и/или вспомогательным веществам, или всем доступным вариантам лечения пероральными антидепрессантами для двойной слепой индукционной фазы.
21. Субъект принимал любые запрещенные варианты терапии, которые не позволили бы введение дозы 1 сутки, как описано в разделе под названием «Предварительное исследование и сопутствующая терапия», а также в Таблице 6.
22. Субъект принимал суммарную суточную дозу бензодиазепинов, превышающую эквивалент 6 мг/день лоразепама, в начале фазы скрининга/проспективного наблюдения.
23. Субъект имел оценку ≥5 по опроснику STOP-Bang, в случае чего следовало решать проблему синдрома обструктивного апноэ во сне (например, индекс апноэ-гипопноэ [AHI] < 30). Субъект с синдромом обструктивного апноэ во сне мог быть включен, если он или она использовали аппарат поддержания положительного давления в дыхательных путях или другой вариант лечения/терапии, который эффективно помогал при апноэ во сне.
24. Субъект получал экспериментальный препарат (включая экспериментальные вакцины) или использовал инвазивное экспериментальное медицинское устройство в течение 60 суток перед началом фазы скрининга/проспективного наблюдения, или участвовал в 2 или более клинических интервенционных исследованиях, связанных с БДР или другими психиатрическими состояниями, в течение 1 года до начала фазы скрининга/проспективного наблюдения, или на текущий момент был включен в экспериментальное исследование.
25. Субъект являлся женщиной, которая была беременна, кормила грудью или планировала беременность во время участия в данном исследовании или в пределах 6 недель после введения последней дозы исследуемого интраназального препарата.
26. Субъект имел диагноз синдрома приобретенного иммунодефицита (СПИД). Для данного исследования не требовалось проведение тестирования на вирус иммунодефицита человека (ВИЧ).
27. Субъект имел любые состояние или ситуацию/обстоятельство, по которым, по мнению исследователя, участие не было бы в интересах субъекта (например, поставило бы под угрозу благополучие), или которые могли бы помешать, ограничить или исказить определенные протоколом оценки.
28. Субъект подвергся обширному хирургическому вмешательству (например, требующему общей анестезии) в течение 12 недель до начала фазы скрининга/проспективного наблюдения или полностью не восстановился после операции, или имел запланированное хирургическое вмешательство в то время, когда предполагалось, что субъект должен принимать участие в исследовании. К участию допускались субъекты с запланированными хирургическими процедурами, которые проводятся под местной анестезией.
29. Субъект являлся сотрудником исследователя или исследовательского центра, непосредственно участвующим в намеченном исследовании или в других исследованиях под руководством данного исследователя или исследовательского центра, а также членом семей сотрудников или исследователя.
Исследователи гарантировали соответствие всем критериям включения в исследование. Если статус субъекта менялся (включая лабораторные результаты или получение дополнительных медицинских записей) до приема первой дозы исследуемого препарата так, что он или она больше не соответствовали всем критериям включения, то субъекта исключали из участия в исследовании.
Кроме того, потенциальные субъекты должны были проявлять готовность и способность придерживаться следующих запретов и ограничений в ходе исследования для того, чтобы иметь право на участие в нем:
1. Критерии включения и исключения;
2. Ограничения на предварительное исследование и сопутствующую терапию, включая перечень запрещенных сопутствующих препаратов для интраназального исследуемого препарата.
3. Положительный анализ мочи на запрещенные вещества в отношении применения фенциклидина (PCP), 3,4-метилендиокси-метамфетамина (MDMA) или кокаина от 1 суток индукционной фазы до последнего визита в двойной слепой индукционной фазе приведет к прекращению участия.
4. Субъекты должны воздерживаться от употребления алкоголя в течение 24 часов до и после каждого сеанса интраназального лечения. При наличии у субъекта интоксикации введение дозы происходить не должно.
5. Во все дни введения интраназального исследуемого препарата все субъекты должны были оставаться в центре клинического исследования до завершения процедур исследования и готовности субъектов к выходу, и должны были сопровождаться ответственным взрослым, покидая центр клинического исследования. Субъекты не должны были водить машину или работать с машинами в течение 24 часов после введения дозы исследуемого препарата.
6. Субъекты не должны были употреблять грейпфрутовый сок, горький апельсин или хинин в течение 24 часов до введения интраназальной дозы исследуемого препарата.
7. ЭШТ, СКК, транскраниальная магнитная стимуляция (ТМС) и СБН были запрещены, начиная с момента включения в исследование и до окончания двойной слепой индукционной фазы.
8. Субъекты, проходящие психотерапию, могли продолжать проходить психотерапию при условии, что эта терапия была постоянной в контексте частоты в течение последних 6 месяцев до фазы скрининга/проспективного наблюдения и оставалась неизменной до окончания двойной слепой индукционной фазы.
Распределение, рандомизация и маскировка лечения
В данном исследовании использовали центральную рандомизацию. Субъектов случайным образом распределяли в 1 из 2 групп лечения в соотношении 1:1 на основании сгенерированного компьютером графика рандомизации, подготовленного перед исследованием спонсором или под его руководством. Рандомизацию балансировали, используя случайно переставленные блоки, и стратифицировали по стране и классу пероральных антидепрессантов (SNRI или SSRI) для инициации в двойной слепой индукционной фазе. Интерактивная система веб-связи (IWRS) присваивала уникальный код лечения, который определял назначение лечения и соответствующие наборы исследуемых препаратов для субъекта. После того как исследователь выбрал вариант лечения пероральными антидепрессантами для двойной слепой индукционной фазы, центр вводил эту информацию в IWRS. Запросчик использовал свой собственный идентификационный номер пользователя и персональный идентификационный номер при обращении к IWRS, а затем получал релевантные сведения о субъекте для уникальной идентификации субъекта.
Исследователю не были предоставлены коды рандомизации. Эти коды хранились в IWRS, которая имела функциональные возможности, позволяющие исследователю раскрыть сведения по отдельному субъекту.
Данные, которые потенциально могли раскрыть сведения по назначению лечения (например, плазменные концентрации исследуемого интраназального препарата, распределение лечения), обрабатывали с особой осторожностью, чтобы гарантировать сохранение маскировки и свести к минимуму возможность ошибки. Это могло включать соблюдение специальных норм, таких как скрытие представляющих интерес данных от исследователей, клинической команды или других, в случае необходимости, до времени блокировки базы данных и раскрытия сведений.
При нормальных обстоятельствах сведения не должны раскрываться до тех пор, пока все субъекты не завершат исследование и база данных не будет финализирована. В противном случае сведения могут быть раскрыты только тогда, когда конкретные неотложное лечение/порядок действий продиктованы информацией о состоянии лечения субъекта. В таких случаях исследователь может в экстренном порядке определить идентичность лечения, связавшись с IWRS. Было рекомендовано, чтобы исследователь, по возможности, связывался со спонсором или его представителем для обсуждения конкретной ситуации, прежде чем раскрывать сведения. Телефонный контакт со спонсором или его представителем был доступен 24 часа в сутки, 7 дней в неделю. В случае раскрытия сведений спонсора информировали как можно быстрее. Дата и время раскрытия сведений документировались в IWRS, а причина раскрытия сведений документировалась в электронной истории болезни (eCRF) и в первичном документе. Документация, полученная от IWRS, указывающая на взламывание кода, хранилась вместе с первичными документами субъекта в защищенном виде.
Субъекты с раскрытыми сведениями о назначении лечения должны были вернуться к запланированному досрочному прекращению участия и визитам последующего наблюдения.
В целом коды рандомизации полностью раскрывали только в том случае, если исследование было завершено, а клиническая база данных была закрыта. Для промежуточного анализа коды рандомизации и, при необходимости, перенос кодов рандомизации на группы лечения и контроля, раскрывали для уполномоченных сотрудников и только для субъектов, включенных в промежуточный анализ.
В конце двойной слепой индукционной фазы базу данных блокировали для анализа и отчетности по этой фазе. Назначение лечения субъекта было видимым только для исследовательского персонала спонсора. Исследователи и персонал центра были заслеплены в отношении назначения лечения до тех пор, пока все субъекты не завершали участие в исследовании после фазы последующего наблюдения.
Для сохранения маскировки исследуемого интраназального препарата интраназальные устройства для эскетамина и плацебо были неразличимы.
В данном исследовании в целом было рандомизировано 227 субъектов. Из них 3 субъекта не получали любой из препаратов исследования (интраназальный или пероральный АД) и 1 субъект не получал ни интраназальный, ни пероральный АД препарат исследования.
Демографические и исходные характеристики для субъектов в данном исследовании приведены ниже в Таблице 1. В целом группы лечения были аналогичными в отношении исходных характеристик. Большинство субъектов, попавших в исследование, составляли женщины со средним возрастом всех субъектов 45,7 лет, в диапазоне от 19 до 64 лет.
(N=223)
b Статус гипертензии классифицируется как «Да», если в истории болезни указана гипертензия.
Из 227 рандомизированных субъектов 197 завершили 28-суточную двойную слепую индукционную фазу. Наиболее частой причиной прекращения участия становилось нежелательное явление. После этого 86 субъектов перешли в фазу последующего наблюдения, а 118 субъектов продолжили участие в клиническом исследовании ESKETINTRD3003. В Таблице 2 ниже представлены количества и причины прекращения участия в исследовании.
(N=227)
Исходный анамнез психиатрических расстройств соответствовал представленному в Таблице 3 ниже. Средняя (СО) исходная общая оценка по шкале MADRS составляла 37,1, в диапазоне от 21 до 52.
(N=223)
b Число антидепрессантов с отсутствием ответа (определяемым как ≤ 25% улучшение), взятое за по меньшей мере 6 недель во время текущего эпизода, полученное из MGH-ATRQ.
СПОСОБ ПРИМЕНЕНИЯ И ДОЗЫ
Фаза скрининга/проспективного наблюдения
В начале фазы скрининга/проспективного наблюдения субъекты проходили лечение пероральными антидепрессантами без ответа на момент начала фазы скрининга/проспективного наблюдения и продолжали такое же лечение в течение этой фазы для подтверждения отсутствия ответа. Центр и исследователи работали с обезличенными данными в отношении критериев исследования по отсутствию ответа. Во время этой фазы соблюдение схемы лечения антидепрессантами оценивали, используя PAQ.
По завершении 4 недель проспективного лечения антидепрессантами и оценки ответа на лечение антидепрессантами дозу антидепрессантов могли постепенно уменьшать и прекращать прием в течение периода до 3 недель в соответствии с местными инструкциями по применению или клиническим заключением (например, лечение антидепрессантами с коротким периодом полувыведения, такими как пароксетин и венлафаксин XR; или проблемы переносимости).
Двойная слепая индукционная фаза
Во время этой фазы субъекты самостоятельно проводили двойное слепое интраназальное лечение эскетамином (56 мг или 84 мг) или плацебо дважды в неделю в течение 4 недель по схеме с гибкой дозой в исследовательском центре. Кроме того, субъекты на 1 сутки одновременно начали принимать новый немаскированный пероральный антидепрессант (т.е. дулоксетин, эсциталопрам, сертралин или венлафаксин XR), который они продолжали принимать в течение этой фазы.
Исследуемый интраназальный препарат
Во время всех сеансов интраназального лечения с субъектом во время сеанса интраназального лечения и периода наблюдения после введения дозы присутствовали врач, медсестра или другой соответствующий представитель персонала центра, недавно (т.е. в течение 1 года) прошедшие тренинг по направлению сердечно-легочной реанимации. Кроме того, было доступно оборудование для поддерживающей вентиляции и реанимации. Ниже в Таблице 4 описано, каким образом следовало проводить каждый сеанс интраназального лечения во время двойной слепой индукционной фазы.
b В каждый момент времени использовали одно устройство. Каждое отдельное устройство для интраназального введения содержало раствор на 2 впрыскивания. Устройства для интраназального введения, содержащие эскетамин, доставляли по 14 мг при каждом впрыскивании, что в целом составляло 28 мг на отдельное устройство (т.е. 2 впрыскивания).
Перед введением первой интраназальной дозы на 1 сутки субъекты практиковались в распылении (в воздух, а не интраназально) с помощью демонстрационного интраназального устройства, которое было заполнено раствором плацебо.
Все субъекты самостоятельно вводили исследуемый интраназальный препарат (эскетамин или плацебо) во время сеансов лечения в исследовательском центре два раза в неделю в течение 4 недель. Первый сеанс лечения проводили на 1 сутки. Сеансы интраназального лечения не проводили по последовательным дням.
На 1 сутки субъекты, рандомизированные в группу приема интраназального эскетамина, начинали с дозы 56 мг. На 4 сутки дозу повышали до 84 мг или оставляли на уровне 56 мг по решению исследователя на основании эффективности и переносимости. На 8 сутки дозу повышали до 84 мг (если доза на 4 сутки составляла 56 мг), оставляли такой же или снижали до 56 мг (если доза на 4 сутки составляла 84 мг) по решению исследователя на основании эффективности и переносимости. На 11 сутки дозу повышали до 84 мг (если доза на 8 сутки составляла 56 мг), оставляли такой же или снижали до 56 мг (если доза на 8 сутки составляла 84 мг) по решению исследователя на основании эффективности и переносимости. На 15 сутки допускалось снижение дозы с 84 мг до 56 мг в случае необходимости по соображениям переносимости; повышение дозы на 15 сутки не допускалось. После 15 суток доза оставалась стабильной (неизменной).
Перед каждым введением исследуемого препарата ограничивали прием пищи в течение по меньшей мере 2 часов. Употребление любых жидкостей ограничивали по меньшей мере за 30 минут до первого назального впрыскивания.
Если у пациента был заложен нос в день приема дозы, рекомендовалось перенести день приема дозы (в пределах допустимого окна для проведения визита). Дозы не следовало принимать по последовательным дням. Если для уменьшения заложенности использовался интраназальное противоотечное средство, его не следовало использовать в течение 1 часа перед введением исследуемого интраназального препарата.
Во время всех сеансов интраназального лечения субъекты оставались в центре клинического исследования до завершения процедур исследования и готовности субъектов к выходу, и сопровождались ответственным взрослым, покидая центр клинического исследования. Субъекты не должны были водить машину или работать с машинами в течение 24 часов после введения последней дозы исследуемого препарата на каждые сутки введения дозы.
Пероральный антидепрессант
Начиная с 1 суток все субъекты начинали лечение новым немаскированным пероральным антидепрессантом и продолжали его в течение этой фазы. Пероральный антидепрессант представляет собой 1 из 4 пероральных антидепрессантов (дулоксетин, эсциталопрам, сертралин или венлафаксин XR). Антидепрессант назначался исследователем на основании рассмотрения MGH-ATRQ и соответствующей информации, касающейся предшествующего лечения антидепрессантами, и был таким, на который субъект ранее не демонстрировал отсутствие ответа в ходе текущего депрессивного эпизода, ранее не демонстрировал непереносимости (в течение жизни), и который был доступен в участвующей стране.
Прием дозы перорального антидепрессанта начинали на 1 сутки в соответствии с местными инструкциями по применению соответствующего продукта с принудительным титрованием до максимально переносимой дозы. Указанная в протоколе схема титрования соответствовала представленной в Таблице 5 ниже.
В случае непереносимости более высоких доз допускалось снижение дозировки на основании клинического заключения. Однако максимальная переносимая субъектом доза не должна была быть ниже, чем следующие минимальные терапевтические дозы: Сертралин (50 мг/сутки), венлафаксин XR (150 мг/сутки), эсциталопрам (10 мг/сутки) и дулоксетин (60 мг/сутки). Хотя субъекты, которым требовались более низкие дозы, могли продолжить участие в исследовании и завершить двойную слепую индукционную фазу, такие субъекты не допускались к участию в исследовании поддержания эффекта ESKETINTRD3003 и переходили к фазе последующего наблюдения после завершения двойной слепой индукционной фазы.
Все субъекты получили дополнительный 4-недельный запас перорального антидепрессанта, чтобы исключить прерывание терапии антидепрессантами во время перехода к дальнейшему клиническому/стандартному лечению.
Персонал исследовательского центра инструктировал субъектов в отношении введения и хранения пероральных антидепрессантов, применяемых во время двойной слепой индукционной фазы, для домашнего использования.
Во время сеансов интраназального лечения рекомендовалось принимать пероральный антидепрессантам вечером и в одно и то же время суток во время двойной слепой индукционной фазы. Кроме того, в дни интраназального введения дозы, если частота приема перорального антидепрессанта превышала один раз в сутки (например, два раза в сутки), рекомендовалось не принимать дозу в течение по меньшей мере 3 часов после сеанса интраназального лечения.
Руководство по мониторингу кровяного давления в дни интраназального введения дозы:
Учитывая потенциальную возможность связанного с лечением повышения систолического и диастолического кровяного давления, в дни интраназального введения дозы придерживались следующего руководства:
Если после выполнения критериев включения и исключения на 1 сутки систолическое кровяное давление субъекта (СКД) перед введением дозы составляло ≥ 160 мм рт.ст. и/или диастолическое кровяное давление (ДКД) составляло ≥ 100 мм рт.ст., рекомендовалось повторить измерение кровяного давления в течение 10 минут в сидящем или горизонтальном положении. Если повторно измеренное перед введением дозы СКД составляло ≥ 160 мм рт.ст. и/или ДКД составляло ≥ 100 мм рт.ст., то введение дозы откладывали и планировали возвращение субъекта на следующий день или в пределах заданного окна проведения визита. Если повышение кровяного давления сохранялось во время следующего визита, перед дальнейшим введением дозы планировали консультацию субъекта с кардиологом или врачом первичной медицинской помощи.
Если в любой момент времени после введения дозы в день введения СКД составляло ≥ 180 мм рт.ст., но < 200 мм рт.ст., и/или ДКД составляло ≥ 110 мм рт.ст., но < 120 мм рт.ст., дальнейшее интраназальное введение дозы прерывали, а субъект обращался к кардиологу или врачу первичной медицинской помощи за последующей оценкой.
После оценки, проведенной кардиологом или врачом первичной медицинской помощи, и при условии, что субъекту было разрешено продолжить участие в исследовании, субъект мог продолжить интраназальное введение дозы, если измеренное перед введением дозы кровяное давление во время следующего запланированного визита находилось в приемлемом диапазоне.
Если в любой момент времени после введения дозы в день введения СКД составляло ≥ 200 мм рт.ст. и/или ДКД составляло ≥ 120 мм рт.ст., субъект должен был прекратить дальнейшее введение дозы и обратиться к кардиологу или врачу первичной медицинской помощи за последующей оценкой.
Во время двойной слепой индукционной фазы, через 1,5 часа после введения дозы, если СКД составляло ≥ 160 мм рт.ст. и/или ДКД составляло ≥ 100 мм рт.ст., измерения следовало продолжать каждые 30 минут до тех пор, пока кровяное давление не соответствовало СКД < 160 мм рт.ст. и ДКД составляло < 100 мм рт.ст., или пока субъект не обращался за соответствующей медицинской помощью в случае клинических показаний.
Фаза последующего наблюдения
Субъекты, получившие по меньшей мере 1 дозу интраназального исследуемого препарата во время двойной слепой индукционной фазы, но не вошедшие в последующее клиническое исследование поддержания эффекта ESKETINTRD3003, переходили к 24-недельной фазе последующего наблюдения. Во время этой фазы исследуемый интраназальный препарат не вводили.
В начале фазы последующего наблюдения исследователь и/или лечащий врач субъекта обеспечивали дополнительное клиническое/стандартное лечение депрессии. Решение о продолжении приема перорального антидепрессанта на этой фазе принимал исследователь; однако, чтобы лучше оценить потенциальные симптомы отказа от интраназального исследуемого препарата, рекомендовалось продолжать прием перорального антидепрессанта в течение по меньшей мере первых 2 недель фазы последующего наблюдения, если это не считали клинически неприемлемым.
Соблюдение режима лечения
Исследователь или назначенный персонал исследовательского цента должны были вести журнал выдачи и возврата всех исследуемых интраназальных препаратов и пероральных антидепрессантов. На протяжении всего исследования регистрировали и учитывали выдачу лекарственных препаратов для каждого субъекта.
Субъекты получили инструкции по соблюдению режима лечения пероральным антидепрессантом. В ходе исследования исследователь или назначенный персонал исследовательского цента отвечали за предоставление дополнительных инструкций для повторного обучения любого субъекта, чтобы обеспечить соблюдение прием перорального антидепрессанта.
Соблюдение схемы лечения антидепрессантами во время фазы скрининга/проспективного наблюдения оценивали, используя PAQ. Пропуск приема антидепрессанта в течение ≥ 4 суток в течение предшествующего 2-недельного периода рассматривали как неадекватное соблюдение схемы приема.
Соблюдение режима лечения антидепрессантами во время двойной слепой индукционной фазы оценивали путем подсчета таблеток (т.е. контроль соблюдения) и учета препарата.
Субъекты самостоятельно вводили все дозы исследуемого интраназального препарата самостоятельно в исследовательском центре под непосредственным наблюдением исследователя или его представителя, при этом все дозы подлежали регистрации.
Терапия до исследования и сопутствующая терапия
Данные о не включающей антидепрессанты терапии, проводимой до исследования вплоть до 30 суток перед началом фазы скрининга/проспективного наблюдения, записывали в начале этой фазы.
Все антидепрессанты, включая вспомогательное лечение БДР, принимаемые во время текущего депрессивного эпизода (т.е. включая принимаемые более чем за 30 суток до начала фазы скрининга/проспективного наблюдения), записывали в начале фазы скрининга/проспективного наблюдения. Кроме того, также получали информацию в отношении анамнеза непереносимости любого из 4 вариантов антидепрессантов (т.е. дулоксетина, эсциталопрама, сертралина или венлафаксина XR).
В течение всего исследования записывали данные о сопутствующей терапии, начиная с подписания информированного согласия и до последнего визита фазы последующего наблюдения. За пределами этого времени информацию о сопутствующей терапии получали только в связи с новыми нежелательными явлениями или их ухудшением до разрешения проблемы этих явлений.
Субъекты продолжали принимать разрешенные сопутствующие препараты (например, антигипертензивные препараты) по стандартной схеме; однако с учетом ограничений и нижеприведенной Таблицы 6. Следует отметить, что, если пероральные антигипертензивные препараты принимали утром, утреннюю дозу следовало принимать и в дни интраназального введения дозы.
Субъекты, проходящие психотерапию, могли продолжать проходить психотерапию при условии, что эта терапия была постоянной в контексте частоты в течение последних 6 месяцев до начала фазы скрининга/проспективного наблюдения и оставалась неизменной до завершения двойной слепой индукционной фазы.
Записывали данные обо всех видах терапии (отпускаемые по рецепту или без рецепта препараты, включая вакцины, витамины, травяные добавки; нефармакологические виды терапии, такие как психотерапия, электростимуляция, акупунктура, специальные диеты и комплексы физических упражнений), отличных от исследуемого препарата. Модификацию эффективной предшествующей терапии не следовало проводить с конкретной целью включения субъекта в исследование, если это не допускалось протоколом (например, корректировка препаратов для давления).
Препараты неотложной помощи
Препараты неотложной помощи не были предоставлены спонсором. В случае возникновения связанных с лечением нежелательных явлений, которые невозможно устранить путем прекращения дальнейшего введения интраназального эскетамина/плацебо, можно рассмотреть применение следующих препаратов неотложной помощи:
При возбужденном или тревожном состоянии: при необходимости, мидазолам (максимальная доза 2,5 мг, перорально или в/м) или краткосрочный прием бензодиазепина
При тошноте: при необходимости, ондансетрон, 8 мг под язык, метоклопрамид (10 мг перорально, в/в или в/м) или дименгидринат (25-50 мг, в/в или в/м)
Рекомендовалось не лечить временное повышение кровяного давления, так как кровяное давление, как правило, возвращается к уровням до введения дозы в течение 2 часов. Эффект любого лечения может привести к гипотензии.
Запрещенные препараты
Перечень запрещенных препаратов (не полный) был предоставлен в качестве общего руководства для исследователя и приведен ниже в Таблице 6. Спонсора уведомляли заранее (или как можно быстрее после этого) о любых случаях применения запрещенных вариантов терапии.
- Если субъект принимает ингибитор моноаминоксидазы (MAOI) во время фазы скрининга/проспективного наблюдения, минимальный интервал отмывки должен составлять 2 недели перед первой дозой интраназального исследуемого препарата.
Снотворные препараты, не содержащие бензодиазепин, запрещены в течение 12 часов перед началом исследования когнитивной функции, но допускаются за ночь до введения дозы, если исследование когнитивной функции не запланировано.
Примеры (не исчерпывающие): Индинавир, нелфинавир, ритонавир, кларитромицин, итраконазол, кетоконазол, нефазодон, саквинавир и телитромицин
Примеры (не исчерпывающие): Эфавиренц, невирапин, барбитураты, карбамазепин, глюкокортикоиды, модафинил, окскарбазепин, фенобарбитал, фенитоин, рифабутин, рифампицин и зверобой
Число доз интраназального исследуемого препарата приведено ниже в Таблице 7.
Обобщенные данные по средней, режимной и конечной дозе интраназального исследуемого препарата приведены ниже в Таблице 8.
На 25 сутки двойной слепой индукционной фазы 66/99 (66/7%) субъектов принимали дозу эскетамина, составляющую 84 мг. Из 115 субъектов, проходивших лечение интраназальным эскетамином, 11 (9,6%) субъектов снижали дозу во время двойной слепой фазы. Длительность приема исследуемого перорального антидепрессанта приведена ниже в Таблице 9.
ОЦЕНКИ В РАМКАХ ИССЛЕДОВАНИЯ
План времени и событий соответствовал представленным ниже в Таблице 10 и Таблице 11 и содержит обобщенные данные по частоте и срокам эффективности, ФК, биомаркерам, фармакогеномике, использовании медицинских ресурсов, состоянии здоровья и безопасности, применимых в случае данного исследования.
(исходный уровень)
процедуры
a Допускался дополнительный необязательный период длительностью до 3 недель для постепенного уменьшения дозы и прекращения приема текущего(их) антидепрессанта(ов) после завершения оценок на 4 неделе (визит 1.3) в соответствии с местными инструкциями по применению или клиническим заключением. Для субъектов, которые не нуждались в постепенном уменьшении дозы и которые, таким образом, допускались к незамедлительному переходу к двойной слепой индукционной фазе, визит 1.3 и визит 2.1 происходят в один и тот же день.
b Если субъект выбыл из исследования до окончания двойной слепой индукционной фазы (т.е. до завершения визита 2.10/сутки 28) по причинам, отличным от отзыва согласия, то в течение 1 недели с даты прекращения проводился визит досрочного отстранения, за которым следовала фаза последующего наблюдения. Если визит досрочного отстранения проводился в тот же день, что и запланированный визит, то дублирование оценок не требовалось.
c До введения дозы (если/когда проводилось в дни интраназального дозирования). За исключением оценок после введения дозы, сообщенные субъектами оценки результатов обрабатывали до всех других относящихся к исследованию процедур во время клинического визита.
d Проводили только для субъектов, которым требовался период постепенного уменьшения дозы во время фазы скрининга/проспективного наблюдения; результат считали исходной оценкой MADRS пациента для двойной слепой индукционной фазы. Для всех остальных субъектов исходная оценка MADRS для двойной слепой индукционной фазы представляла собой оценку MADRS, выполненную в конце 4 недели фазы скрининга/проспективного наблюдения.
e Оценку основных физиологических показателей проводили через 40 минут, 1 час и 1,5 часа после введения дозы.
f ЭКГ в двенадцати отведениях проводили перед введением дозы и в момент t=1 час после введения дозы во время визита 2.1. ЭКГ в двенадцати отведениях проводили в момент t=1 час после введения дозы во время визитов 2.3-2.9, но во время визитов 2.3-2.9 ЭКГ до введения дозы не требовалась. Допускалось временное окно ± 15 минут.
g Оценку MOAA/S не проводили во время визита Visit 1.1 (только пульсоксиметрия). Оценку MOAA/S проводили каждые 15 минут от момента времени до введения дозы до t = +1,5 часа после введения дозы. Пульсоксиметрию проводили каждые 15 минут от момента времени до введения дозы до t = +1,5 часа после введения дозы.
h BPRS+ и CADSS проводили до введения дозы и через 40 минут и через 1,5 часа после введения дозы.
I CGADR проводили через 1 час и через 1,5 часа после введения дозы; если ответ не был «Да» через 1,5 часа после введения дозы, оценку повторяли каждые 15 минут до тех пор, пока не получали ответ «Да», или пока субъект не обращался за соответствующей медицинской помощью в случае клинических показаний. Субъект не должен был покидать центр ранее, чем через 1,5 часа.
j PWC-20 проводили только в том случае, если субъект не продолжал участие в исследовании ESKETINTRD3003.
k Опросник по назальным симптомам использовали перед введением дозы и через 1 час после введения дозы.
l Взятие крови для ФК проводили в моменты t=40 минут и t=2 часа после введения дозы (где момент времени=0 определялся как момент времени первого интраназального впрыскивания).
m Образцы крови брали перед введением дозы. Было предпочтительно, чтобы субъекты придерживались диеты с низким содержанием жиров в день сбора проб.
a Чтобы лучше оценить потенциальные симптомы отказа от интраназального исследуемого препарата, рекомендовалось продолжать прием перорального антидепрессанта в течение по меньшей мере первых 2 недель фазы последующего наблюдения, если это не считали клинически неприемлемым.
b Для HRUQ требовалась проводимая клиницистом оценка (на основании ответов субъекта).
c Проводилось в телефонном режиме квалифицированным персоналом центра.
d Было предпочтительно, чтобы субъекты придерживались диеты с низким содержанием жиров в день сбора проб.
За исключением оценок после введения дозы, выполняемые во время визитов оценки сообщаемых субъектом результатов проводили или завершали до любых тестов, процедур или других консультаций для этого клинического визита, чтобы предотвратить влияние на восприятие субъекта. Был предоставлен рекомендованный порядок процедур исследований. Фактические даты и время проведения оценок были записаны в первичной документации и в eCRF
Общий объем крови, который нужно было взять у каждого субъекта, составлял приблизительно 123,5 мл (смотрите Таблицу 12 ниже). Повторные или незапланированные образцы можно было брать из соображений безопасности или из-за технических проблем с образцами. Можно было проводить дополнительные тесты сыворотки или мочи на беременность, определенные как необходимые исследователем или требуемые в соответствии с местным законодательством, для определения отсутствия беременности в любое время во время участия субъекта в исследовании.
b Биохимия сыворотки включает тесты на сывороточный β-hCG на беременность (для женщин детородного потенциала) и липидограмму.
c При необходимости измеряют HbA1c в образцах, собранных для гематологии.
d Объем крови, приведенный под белковыми биомаркерами, представляет объединенный объем нескольких разных пробирок для сбора проб.
Для сбора проб крови можно использовать вживленную в/в канюлю.
Повторные или незапланированные образцы можно брать из соображений безопасности или из-за технических проблем с образцами.
Фаза скрининга/проспективного наблюдения
До проведения любой исследовательской процедуры исследователь (или назначенный исследовательский персонал) проводил ознакомление с письменным ФИС и его разъяснение для каждого субъекта. После подписания ФИС проводили скрининг субъектов, которым было от 18 (или больше, если минимальный установленный законом возраст согласия в стране, в которой проводится исследование, составляет >18) до 64 лет (включительно), чтобы определить допустимость участия в исследовании.
Субъекты должны были соответствовал диагностическим критериям DSM-5 однократного эпизода БДР (в случае однократного эпизода БДР его продолжительность должна составлять ≥2 лет) или рецидива БДР без психотических признаков на основании клинической оценки и с подтверждением MINI. Кроме того, в начале фазы скрининга/проспективного наблюдения субъект должен был иметь общую оценку IDS-C30 ≥34.
В начале этой фазы у субъектов в анамнезе должно быть отсутствие ответа на лечение ≥2, но ≤5 пероральными антидепрессантами во время текущего эпизода депрессии, оцененное с помощью MGH-ATRQ и подтвержденное задокументированной историей болезни и фармацевтическими/рецептурными записями. Субъект проходил лечение пероральными антидепрессантами с отсутствием ответа на момент включения и продолжал это лечение в такой же дозе на протяжении этой фазы для проспективного подтверждения отсутствия ответа. Соблюдение схемы лечения антидепрессантами оценивали, используя PAQ. Пропуск приема антидепрессанта в течение ≥4 суток в течение предшествующего 2-недельного периода рассматривали как неадекватное соблюдение схемы приема.
Текущий большой депрессивный эпизод субъекта и ответ на лечение антидепрессантами, используемыми в текущем депрессивном эпизоде, были подтверждены с помощью независимой от исследовательского центра квалификационной оценки.
Независимый работающий с обезличенными данными эксперт проводил дистанционную оценку MADRS для оценки депрессивных симптомов во время этой фазы. От исследователя и исследовательского центра были скрыты конкретные подробности, касающиеся критериев ответа для включения в двойную слепою индукционную фазу. Допущенные не демонстрирующие ответ субъекты (определенные удаленным работающим с обезличенными данными экспертом) прекращали прием своих текущих антидепрессантов и любых других запрещенных психотропных препаратов, включая вспомогательные атипичные антипсихотические средства. Позволялось продолжение приема бензодиазепинов или не содержащих бензодиазепины снотворных препаратов, но имелись конкретные ограничения в отношении времени введения относительно сеансов интраназального лечения.
Все остальные субъекты, которые не перешли в двойную слепую индукционную фазу, заканчивали участие в исследовании в этот момент времени. Никаких дополнительных визитов исследования или последующего наблюдения не требовалось.
Необязательный период постепенного уменьшения дозы антидепрессанта
Так как все не демонстрирующие ответ субъекты начинали прием нового перорального антидепрессанта во время двойной слепой индукционной фазы, после прекращения текущего лечения антидепрессантами не требовалось периода отмывки или воздержания от приема препарата. При этом допускался дополнительный необязательный период длительностью до 3 недель для постепенного уменьшения дозы и прекращения приема текущего перорального антидепрессанта в соответствии с местными инструкциями по применению или клиническим заключением.
Период постепенного уменьшения дозы не начинался до завершения 4 недель проспективного лечения антидепрессантами и оценки ответа на лечение антидепрессантами.
Двойная слепая индукционная фаза
Во время этой фазы субъекты самостоятельно проводили двойное слепое интраназальное лечение эскетамином (56 мг или 84 мг) или плацебо дважды в неделю в течение 4 недель по схеме с гибкой дозой. Кроме того, субъекты одновременно начинали принимать новый немаскированный пероральный антидепрессант.
Субъектов исследования (с ТРД) случайным образом распределяли в 1 из следующих 2 групп двойного слепого лечения в соотношении 1:1 (приблизительно 98 субъектов на группу): 1. Интраназальное плацебо или 2. Интраназальный эскетамин (56 мг или 84 мг). На те же сутки (т.е. сутки 1) субъекты переходили на лечение новым немаскированным пероральным антидепрессантом. Пероральный антидепрессант представляет собой 1 из 4 пероральных антидепрессантов (дулоксетин, эсциталопрам, сертралин или венлафаксин XR). Антидепрессант назначался исследователем (на основании рассмотрения MGH-ATRQ и соответствующей информации, касающейся предшествующего лечения антидепрессантами) и был таким, на который субъект ранее не демонстрировал отсутствие ответа в ходе текущего депрессивного эпизода, ранее не демонстрировал непереносимости (в течение жизни), и который был доступен в участвующей стране. Введение дозы перорального антидепрессанта начинали на 1 сутки в соответствии с местными инструкциями по применению соответствующего продукта с принудительным титрованием до максимально переносимой дозы. План титрования для выбранного перорального антидепрессанта соответствовал представленному в Таблице 5 выше.
В случае информации, полученной в телефонном режиме, разговор письменно документировали для внесения в первичные документы. Во время визитов субъектов персоналом центра в телефонном режиме получали информацию о нежелательных явлениях и сопутствующей терапии. Кроме того, указанные проводимые клиницистом оценки проводил надлежащим образом квалифицированный персонал.
В конце двойной слепой индукционной фазы субъекты, которые демонстрировали ответ (определяемый как ≥50% снижение общей оценки по шкале MADRS относительно исходного уровня [1 сутки до рандомизации] до конца 4-недельной двойной слепой индукционной фазы), допускались к участию в последующем клиническом исследовании поддержания эффективности (исследовании ESKETINTRD3003). Для сохранения маскировки исследования все демонстрирующие ответ субъекты, включая субъектов, демонстрирующих ответ на действующий препарат сравнения (т.е. пероральный антидепрессант плюс интраназальное плацебо), были допущены к включению в исследование ESKETINTRD3003. Участие в ESKETINTRD3003 начиналось сразу же после завершения двойной слепой индукционной фазы. Субъекты получали пероральный антидепрессант и были проинструктированы, чтобы продолжать принимать пероральный антидепрессант до своего следующего визита исследования (т.е. визита исследования фазы стабилизации в исследовании ESKETINTRD3003).
Те субъекты, которые не вошли в исследование ESKETINTRD3003, перешли в фазу последующего наблюдения.
Досрочное прекращение (участия)
Если субъект выбывал до окончания двойной слепой индукционной фазы по причинам, отличным от отзыва согласия, то в течение 1 недели с даты прекращения проводили визит досрочного отстранения, за которым следовала фаза последующего наблюдения. Если визит досрочного отстранения происходил в тот же день, что и запланированный визит, визит досрочного отстранения проводили в тот же день и дублирование оценок не требовалось.
Исследователь и/или лечащий врач субъекта обеспечивали дополнительное клиническое/стандартное лечение депрессии. Исследователь и/или лечащий врач определяли, стоит или нет продолжать лечение текущими пероральными антидепрессантами.
При необходимости досрочно выбывшие субъекты получали дополнительный пероральный антидепрессант, и им рекомендовалось продолжать принимать пероральный антидепрессант в течение по меньшей мере первых 2 недель фазы последующего наблюдения, если это не считали клинически неприемлемым.
Фаза последующего наблюдения
Субъекты, получившие по меньшей мере 1 дозу интраназального исследуемого препарата во время двойной слепой индукционной фазы, но не участвующие в следующем исследовании ESKETINTRD3003, переходили к 24-недельной фазе последующего наблюдения. Клинические визиты и визиты для дистанционной оценки проводили согласно плану времени и событий. Во время этой фазы оценивали безопасность и переносимость, включая потенциальные симптомы выбывания, после прекращения приема интраназального эскетамина. Кроме того, собирали данные для оценки течения большого депрессивного эпизода субъекта в продолжении 6-месячного периода.
Исследователь и/или лечащий врач субъекта обеспечивали дополнительное клиническое/стандартное лечение депрессии. Во время этой фазы исследуемый интраназальный препарат не вводили. Чтобы лучше оценить потенциальные симптомы отказа от интраназального исследуемого препарата, рекомендовалось продолжать прием перорального антидепрессанта в течение по меньшей мере первых 2 недель фазы последующего наблюдения, если это не считали клинически неприемлемым. Решение о продолжении приема антидепрессанта принимал исследователь.
В случае информации, полученной в телефонном режиме, разговор письменно документировали, чтобы он был доступен при изучении первичных документов.
Любые клинически значимые отклонения от нормы, которые сохранялись к концу исследования, наблюдались исследователем до их устранения или до достижения клинически стабильного состояния. Все нежелательные явления и особые сообщаемые ситуации, серьезные или несерьезные, сообщались до завершения субъектом последней связанной с исследованием процедуры.
ОЦЕНКИ ЭФФЕКТИВНОСТИ
Рекомендовалось, чтобы различные оценки сообщаемых субъектом результатов были завершены до проведения других процедур.
Первичная оценка эффективности
Первичная оценка эффективности представляла собой общую оценку по шкале MADRS. Оценку MADRS проводили независимые удаленные эксперты во время исследования. Применяемая клиницистами, оцениваемая клиницистами шкала MADRS из 10 пунктов была разработана для использования в случае субъектов с БДР для определения общей тяжести депрессивных симптомов, включая тяжесть депрессии, и для обнаружения изменений, обусловленных лечением антидепрессантами. Шкалу MADRS использовали в качестве первичной меры эффективности для данного исследования, поскольку она является проверенной, надежной и приемлемой для регуляторных органов здравоохранения в качестве первичной шкалы для определения эффективности при большой депрессии.
Шкала MADRS состоит из 10 пунктов, каждый из которых оценивается от 0 (параметр отсутствует или соответствует норме) до 6 (тяжелое или постоянное наличие симптомов) с общей возможной оценкой 60. Более высокая оценка свидетельствует о более тяжелом состоянии. Шкала MADRS позволяет оценить объективные признаки подавленности, субъективные признаки подавленности, внутреннее напряжение, сон, аппетит, уровень концентрации, утомляемость, уровень интереса, пессимистичные мысли и суицидальные мысли. Этот тест демонстрирует высокую межэкспертную надежность.
Первичным ожидаемым результатом эффективности было изменение общей оценки по шкале MADRS от исходного уровня (1 сутки перед рандомизацией) до конца 4-недельной двойной слепой индукционной фазы.
В данном исследовании субъекты в любой из 2 групп лечения, которые демонстрировали ответ на исследуемый препарат (т.е. пациенты с ответом), были определены как субъекты, соответствующие критерию ответа, определяемому как снижение ≥50% общей оценки MADRS от исходного уровня (1 сутки перед рандомизацией) до конца 4-недельной двойной слепой индукционной фазы.
Помимо того, что она является основной меры эффективности, MADRS также использовали для оценки ключевого вторичного ожидаемого результата эффективности, начала клинического ответа (т.е. эффекта антидепрессанта) на 2 сутки, который сохранялся в течение двойной слепой индукционной фазы. Начало клинического ответа определялось как ≥50% улучшение общей оценки MADRS на 2 сутки (т.е. через сутки после приема первой дозы двойного слепого интраназального препарата), которое сохранялось до конца двойной слепой фазы.
MADRS также использовали, чтобы оценить вторичную цель оценки доли субъектов с ответом и субъектов в состоянии ремиссии (определяемых как субъекты с общей оценкой по шкале MADRS ≤12) в конце 4-недельной двойной слепой индукционной фазы.
Ключевая вторичная оценка эффективности (выполняемая клиницистом)
MADRS применяли, используя модифицированный период оценки длительностью 24 часа, для ключевой вторичной оценки эффективности, связанной с началом клинического ответа на 2 сутки, который сохранялся в течение двойной слепой индукционной фазы.
MADRS с 24-часовым периодом оценки использовали на 2 сутки. Применимость такого укороченного периода оценки была подтверждена пациентами и врачами, и имеются данные, подтверждающие психометрические свойства этого укороченного периода оценки. Для всех последующих оценок MADRS, используемых для ключевой вторичной оценки эффективности (сохранение клинического ответа, достигнутого на 2 сутки, в течение двойной слепой индукционной фазы), использовали MADRS с 7-дневной оценкой.
Ключевая вторичная оценка эффективности (сообщаемые пациентом результаты)
Опросник здоровья пациента (PHQ-9) представляет собой критерий на основании сообщаемых пациентом результатов из 9 пунктов, который использовали для оценки депрессивных симптомов. Данная шкала учитывает каждый из 9 доменов симптомов критериев DSM-5 для БДР, и ее использовали как в качестве инструмента скрининга, так и в качестве меры ответа на лечение депрессии. Каждый пункт оценивался по 4-балльной шкале (0=никогда, 1=несколько дней, 2=более половины из всех дней и 3=практически каждый день). Ответы субъекта по всем пунктам суммировали для получения общей оценки (в диапазоне от 0 до 27), причем более высокие оценки указывают на большую тяжесть депрессивных симптомов. Период оценки составлял 2 недели.
Шкалу нетрудоспособности Шихана (SDS) использовали для оценки вторичной цели функционального влияния и связанной с ним нетрудоспособности. SDS представляет собой критерий на основании сообщаемых пациентом результатов и является опросником из 5 пунктов, который широко используется и является приемлемым для оценки функционального нарушения и связанной с ним нетрудоспособности. Первые три пункта оценивают прекращение (1) посещения работы/школы, (2) социальной жизни и (3) выполнения семейных обязанностей/домашних обязанностей с использованием оценочной шкалы 0-10. Оценки по первым трем пунктам суммировали для получения общей оценки 0-30, причем более высокая оценка указывает на большее нарушение. В SDS также есть один пункт по пропущенным дням школы или работы и один пункт по непродуктивным дням. Период оценки для данного исследования составлял 7 дней.
Общее клиническое впечатление о степени тяжести (CGI-S) обеспечивает общую определяемую клиницистом суммарную меру тяжести заболевания субъекта с учетом всей доступной информации, включая знание анамнеза субъекта, психосоциальных обстоятельств, симптомов, поведения и влияния симптомов на способность субъекта функционировать. CGI-S позволяет оценить степень тяжести психопатологии по шкале от 0 до 7. С учетом общего клинического опыта субъекта оценивали по тяжести психического заболевания на момент проведения оценки в соответствии с: 0=оценка отсутствует; 1=норма (полное отсутствие заболевания); 2=на грани психического заболевания; 3=легкая степень заболевания; 4=умеренная степень заболевания; 5=заметная степень заболевания; 6=тяжелая степень заболевания; 7=среди наиболее больных пациентов. CGI-S позволяет проводить глобальную оценку состояния субъекта в данный момент времени.
Для определения вторичной цели симптомов тревожности использовали сообщаемую субъектом оценку по шкале генерализированного тревожного расстройства из 7 пунктов (GAD-7). GAD-7 является краткой и проверенной мерой общей степени беспокойства. Каждый пункт оценивался по 4-балльной шкале (0=никогда; 1=несколько дней; 2=более половины из всех дней; и 3=практически каждый день). Ответы по пунктам суммировали для получения общей оценки в диапазоне от 0 до 21, причем более высокие оценки указывают на большую тревожность. Период оценки составлял 2 недели.
Euro-Qol-5 Dimention-5 Level (EQ-5D-5L) представляет собой стандартизированный инструмент для применения в качестве оценки эффекта на общее состояние здоровья, разработанный главным образом для самостоятельного выполнения респондентами. Он состоит из описательной системы EQ-5D-5L и визуальной аналоговой шкалы EQ (EQ-VAS). Описательная система EQ-5D-5L содержит следующие 5 параметров: Мобильность, самообслуживание, повседневная деятельность, боль/дискомфорт и тревожность/депрессия. Каждый из 5 параметров разделен на 5 уровней восприятия проблем (уровень 1 указывает на отсутствие проблем, уровень 2 указывает на небольшие проблемы, уровень 3 указывает на умеренные проблемы, уровень 4 указывает на серьезные проблемы, а уровень 5 указывает на экстремальные проблемы).
Субъект выбирал ответ для каждого из 5 параметров с учетом ответа, который лучше всего соответствует его здоровью «на сегодня». Описательную систему использовали для представления состояния здоровья. В самооценке EQ-VAS записывалась собственная оценка респондентом состояния его/ее здоровья на момент завершения по шкале от 0 до 100.
Первичный ожидаемый результат
Первичным ожидаемым результатом эффективности было изменение общей оценки по шкале MADRS, определяемое по изменению от исходного уровня (1 сутки перед рандомизацией) до конца 4-недельной двойной слепой индукционной фазы.
Итоги по первичному ожидаемому результату:
Для учета множественности и строгого контроля ошибок I рода для первичного и 3 ключевых вторичных ожидаемых результатов эффективности (начало клинического ответа, изменение общей оценки SDS и изменение общей оценки PHQ-9) использовали подход серийного фильтрования (фиксированная последовательность). Последовательно анализировали 3 ключевых вторичных ожидаемых результата и считали их статистически значимыми при 1-стороннем уровне 0,025, только если ожидаемый результат был в индивидуальном порядке значимым при 1-стороннем уровне 0,025, а предыдущие ожидаемые результаты в иерархии были значимыми при 1-стороннем уровне 0,025, включая первичный ожидаемый результат. Если первичный ожидаемый результат был статистически значимым, выбранные вторичные ожидаемые результаты оценивали в следующем порядке: начало клинического ответа, изменение общей оценки SDS, изменение общей оценки PHQ-9.
Первичным ожидаемым результатом эффективности было изменение общей оценки по шкале MADRS относительно исходного уровня на 28 сутки. Общая оценка MADRS соответствует диапазону 0-60. Первичный анализ эффективности проводили на полной выборке для анализа, которая включала всех рандомизированных субъектов, которые получили по меньшей мере 1 дозу интраназального исследуемого препарата и 1 дозу перорального исследуемого антидепрессанта. Как показано ниже в Таблице 13, результаты изменения общей оценки по шкале MADRS свидетельствовали в пользу интраназального эскетамина+пероральный АД в сравнении с пероральным АД+интраназальное плацебо. (На Фиг. 2 представлены средние значения изменений относительно исходного уровня, полученные методом наименьших квадратов (± СП), для общей оценки по шкале MADRS в динамике по времени в двойной слепой фазе на основании анализа СМПИ.) Среднее изменение относительно исходного уровня (СО) на 28 сутки составляло -21,4 (12,32) для эскетамина+пероральный АД и -17,0 (13,88) для действующего препарата сравнения. На основании модели СМПИ с лечением, сутками, страной, классом перорального антидепрессанта и посуточным лечением в качестве факторов и исходным значением в качестве ковариаты, среднеквадратическая разница (СП) между эскетамином+пероральный АД и действующим препаратом сравнения составляла -4,0 (1,69). Разница между группами лечения была статистически значимой (одностороннее p=0,010). Анализ СМПИ рассматривали в качестве первичного анализа для всех досье, за исключением EU.
Результаты на основании модели ANCOVA для изменения общей оценки MADRS от исходного уровня до конечной точки (ДС) с учетом факторов лечения, страны и класса перорального антидепрессанта и исходного значения в качестве ковариаты согласовались с анализом СМПИ (среднеквадратическая разница (СП) между эскетамином+пероральный АД и действующим препаратом сравнения составляла -3,5 (1,63), одностороннее p=0,017).
интраназальное плацебо (N=109)
Отрицательное изменение оценки указывает на улучшение.
Вторичные ожидаемые результаты
Первый ключевой вторичный ожидаемый результат представлял собой изменение от исходного уровня (1 сутки перед рандомизацией) до конца 4-недельной двойной слепой индукционной фазы депрессивных симптомов, сообщенное субъектом с использованием общей оценки PHQ-9.
Второй ключевой вторичный ожидаемый результат представлял собой долю субъектов, демонстрирующих начало клинического ответа на 2 сутки, который сохранялся до конца 4-недельной двойной слепой индукционной фазы. Начало клинического ответа определялось как ≥ 50% улучшение общей оценки MADRS на через сутки после приема первой дозы двойного слепого препарата [2 сутки], которое сохранялось до конца 4-недельной двойной слепой фазы. Субъектов, прекративших участие в исследовании до окончания двойной слепой индукционной фазы, не учитывали как сохранивших клинический ответ.
Третий ключевой вторичный ожидаемый результат представлял собой изменение общей оценки SDS, определяемое по изменению от исходного уровня (1 сутки перед рандомизацией) до конца 4-недельной двойной слепой индукционной фазы.
Другие вторичные ожидаемые результаты эффективности включали: (a) Долю субъектов с ответом (≥ 50% снижение общей оценки MADRS относительно исходного уровня) в конце 4-недельной двойной слепой индукционной фазы, (b) долю субъектов в состоянии ремиссии (MADRS ≤12) в конце 4-недельной двойной слепой индукционной фазы, и (c) изменение относительно исходного уровня (1 сутки перед рандомизацией) к концу 4-недельной двойной слепой индукционной фазы: тяжести депрессивного заболевания с помощью CGI-S, симптомов тревожности, определяемых по GAD-7, обусловленного здоровьем качества жизни и состояния здоровья по оценке EQ-5D-5L.
Итоги по вторичным ожидаемым результатам:
Начало клинического ответа
Субъекта определяли как имеющего клинический ответ при наличии по меньшей мере 50% улучшения относительно исходной общей оценки MADRS с началом на 2 сутки, которое сохранялось до 28 суток при каждом визите. Допускалось одно отклонение (отсутствие ответа) у субъектов на 8, 15 или 22 сутки, при этом оценка должна демонстрировать улучшение по меньшей мере на 25%. Субъектов, которые по какой-либо причине не соответствовали такому критерию или прекратили прием во время исследования до 28 суток, считали не демонстрирующими ответ и присваивали им значение «нет», что означает, что они не соответствовали критериям начала клинического ответа.
Как показано в Таблице 14 ниже, 7,9% субъектов в группе, получавшей эскетамин+пероральный АД, достигали клинического ответа по сравнению с 4,6% субъектов в активной группе, получавшей действующий препарат сравнения. Разница между группами лечения не была статистически значимой при 1-стороннем уровне 0,025. Таким образом, на основании предопределенной последовательности тестирования ключевых вторичных ожидаемых результатов формально невозможно было оценить общую оценку по SDS и общую оценку по PHQ-9.
b Обобщенный тест Кохрана - Мантеля - Хензеля (CMH) позволяет определить среднюю разницу в оценках между вариантами лечения с корректировкой в отношении страны и класса перорального антидепрессанта (SNRI или SSRI).
c Результат анализа считали статистически значимым при 1-стороннем уровне 0,025, только если результаты анализа общей оценки MADRS также был значимым.
d Шансы достижения начала клинического ответа при приеме интраназального эскетамина+пероральный АД, разделенные на шансы достижения начала клинического ответа при приеме перорального АД+интраназальное плацебо.
Частота ответа и ремиссии на основании общей оценки по шкале MADRS
Частоты ответа (≥50% улучшение относительно исходного уровня общей оценки по шкале MADRS) и ремиссии (общая оценка по шкале MADRS ≤12) соответствовали представленным в Таблице 15 и на Фиг. 3-5.
Шкала нетрудоспособности Шихана (SDS)
SDS представляет собой критерий на основании сообщаемых пациентом результатов и является опросником из 5 пунктов, который широко используется и является приемлемым для оценки функционального нарушения и связанной с ним нетрудоспособности. Первые три пункта оценивают прекращение (1) посещения работы/школы, (2) социальной жизни и (3) выполнения семейных обязанностей/домашних обязанностей с использованием оценочной шкалы 0-10. Оценки по первым трем пунктам суммируют для получения общей оценки 0-30, причем более высокая оценка указывает на большее нарушение.
Как показано ниже в Таблице 16, результаты изменения общей оценки по шкале SDS свидетельствовали в пользу интраназального эскетамина+пероральный АД в сравнении с пероральным АД+интраназальное плацебо. Среднее изменение относительно исходного уровня (СО) на 28 сутки составляло -13,3 (8,22) для эскетамина+пероральный АД и -9,5 (8,38) для действующего препарата сравнения. На основании модели СМПИ с лечением, сутками, страной, классом перорального антидепрессанта и посуточным лечением в качестве факторов и исходным значением в качестве ковариаты, среднеквадратическая разница (СП) между эскетамином+пероральный АД и действующим препаратом сравнения составляла -3,6 (1,18). На основании предопределенной последовательности тестирования ключевых вторичных ожидаемых результатов общую оценку по шкале SDS формально невозможно было оценить из-за отсутствия статистически значимой разницы между группами лечения для начала клинического ответа. Номинальное одностороннее p-значение=0,001.
Результаты на основании модели ANCOVA для изменения общей оценки SDS от исходного уровня до конечной точки (ДС) с учетом факторов лечения, страны и класса перорального антидепрессанта и исходного значения в качестве ковариаты согласовались с анализом СМПИ.
b Результат анализа считали статистически значимым при 1-стороннем уровне 0,025, только если результаты анализа общей оценки MADRS и начала клинического ответа также были значимыми.
Отрицательное изменение оценки SDS указывает на улучшение.
Опросник по состоянию здоровья пациента - 9 пунктов (PHQ-9)
PHQ-9 представляет собой шкалу самоанализа из 9 пунктов для оценки депрессивных симптомов. Каждый пункт оценивается по 4-балльной шкале (0=никогда, 1=несколько дней, 2=более половины из всех дней и 3=практически каждый день) с общей оценкой в диапазоне 0-27. Более высокая оценка указывает на более тяжелую депрессию.
Как показано ниже в Таблице 17, результаты изменения общей оценки по шкале PHQ-9 свидетельствовали в пользу интраназального эскетамина+пероральный АД в сравнении с пероральным АД+интраназальное плацебо. Среднее изменение относительно исходного уровня (СО) на 28 сутки составляло -12,8 (6,43) для эскетамина+пероральный АД и -10,2 (7,84) для действующего препарата сравнения. На основании модели СМПИ с лечением, сутками, страной, классом перорального антидепрессанта и посуточным лечением в качестве факторов и исходным значением в качестве ковариаты, среднеквадратическая разница (СП) между эскетамином+пероральный АД и действующим препаратом сравнения составляла -2,2 (0,89). На основании предопределенной последовательности тестирования ключевых вторичных ожидаемых результатов общую оценку по шкале PHQ-9 формально невозможно было оценить из-за отсутствия статистически значимой разницы между группами лечения для начала клинического ответа. Номинальное одностороннее p-значение=0,006.
Результаты на основании модели ANCOVA для изменения общей оценки PHQ-9 от исходного уровня до конечной точки (ДС) с учетом факторов лечения, страны и класса перорального антидепрессанта и исходного значения в качестве ковариаты согласовались с анализом СМПИ (смотрите дополнение 3).
пероральный АД (N=114)
b Результат анализа считали статистически значимым при 1-стороннем уровне 0,025, только если результаты анализа общей оценки MADRS, начала клинического ответа и общей оценки SDS также были значимыми.
Отрицательное изменение оценки PHQ-9 указывает на улучшение.
Оценки безопасности
Любые клинически релевантные изменения, происходящие во время исследования, записывали в разделе «Нежелательные явления» в eCRF. Любые клинически значимые отклонения от нормы, которые сохранялись к концу/досрочному прекращению исследования, наблюдались исследователем до их устранения или до достижения клинически стабильного состояния. Данное исследование включало следующие оценки безопасности и переносимости в соответствии с моментами времени, указанными в плане времени и событий.
Нежелательные явления
Во время исследования о нежелательных явлениях сообщал субъект (или, при необходимости, лицо, осуществляющее уход, замещающее лицо или законный представитель субъекта). Нежелательные явления отслеживались исследователем. НЯВЛ, представляющие особый интерес, исследовали отдельно.
Клинические лабораторные тесты
Собирали образцы крови для биохимического анализа сыворотки и гематологии, и образцы мочи для общего анализа мочи. Исследователь изучал лабораторный отчет, документировал результат изучения и записывал любые клинически релевантные изменения, возникшие в ходе исследования, в разделе «Нежелательные явления» в eCRF. Лабораторные отчеты были поданы вместе с первичными документами. Пользоваться услугами местных лабораторий было разрешено в тех случаях, когда начало лечения или последующего наблюдения для оценки безопасности являлось критическим по времени и ожидалось, что результаты центральной лаборатории не будут доступны до необходимости начинать введение дозы, или в случае необходимости принятия мер по соображениям безопасности.
Если не указано иное, центральной лабораторией проводились следующие тесты:
Следующие тесты проводили в моменты времени, указанные в плане времени и событий:
1. Липидограмма: Общий холестерин, липопротеины низкой плотности (ЛПНП)-холестерин, липопротеины высокой плотности (ЛПВП)-холестерин и триглицериды
2. Тест сыворотки и мочи на беременность (только для женщин детородного потенциала)
3. Анализ мочи на наличие наркотических веществ: Барбитуаты, метадон, опиаты, кокаин, каннабиноиды (тестирование на каннабиноиды проводится только на 1 сутки перед введением дозы), фенциклидин и амфетамин/метамфетамин
4. Тест на алкоголь в выдыхаемом воздухе
5. Тиреостимулирующий гормон (TSH)
6. Тест на гликированный гемоглобин (HbA1c)
7. Тест на уровень фолликулостимулирующего гормона (FSH) в сыворотке, только в случае необходимости для подтверждения, что субъект женского пола не имеет детородный потенциал (смотрите критерии включения № 0)
ЭКГ в одном и 12-ти отведениях
Во время проведения ЭКГ субъекты должны пребывать в спокойном состоянии без отвлекающих факторов (например, телевидение, мобильные телефоны). Субъекты должны находиться в положении лежа на спине в течение по меньшей мере 5 минут перед проведением ЭКГ и должны воздерживаться от разговоров или движений руками или ногами.
Все электрокардиограммы направляли в центральную лабораторию ЭКГ. ЭКГ считывали в определенные моменты времени и обобщали в центральной лаборатории ЭКГ. Исследователь или младший исследователь должен был изучить все ЭКГ во время визита исследования для оценки любых потенциальных проблем безопасности или свидетельств наличия исключающих условий.
Основные физиологические показатели (температура, пульс/частота сердечных сокращений, частота дыхания, кровяное давление)
Измерения кровяного давления и пульса/частоты сердечных сокращений проводили в положении лежа на спине с помощью полностью автоматизированного устройства. Ручные методы использовали только в случае недоступности автоматизированного устройства. Измерениям кровяного давления и пульса/частоты сердечных сокращений предшествовало пребывание в спокойном состоянии без отвлекающих факторов (например, телевидение, мобильные телефоны) по меньшей мере в течение 5 минут.
Рекомендовалось измерение тимпанальной температуры. Для измерения частоты дыхания использовали автоматизированное устройство.
Пульсоксиметрия
Пульсоксиметрию использовали для определения степени насыщения артериальной крови кислородом. В каждый день введения дозы устройство подсоединяли к пальцу на руке, пальцу на ноге или уху перед первым назальным впрыскиванием, а затем после первого впрыскивания отслеживали и документировали его показатели. Любая степень насыщения артериальной крови кислородом (SpO2) < 93%, длящаяся более 2 минут и подтвержденная дополнительным проведенным вручную измерением или измерением на другой части тела, регистрировалась как нежелательное явление.
В дни сеансов интраназального лечения пульсоксиметрию проводили каждые 15 минут от момента времени до введения дозы до t=1,5 часа после введения дозы. Если результат был ≤ 93% в течение интервала времени 1,5 часа после введения дозы, пульсоксиметрию повторяли каждые 5 минут до возвращения результата к ≥ 93%, или пока субъект не обращался за соответствующей медицинской помощью в случае клинических показаний.
Физический осмотр, рост, масса тела и окружность шеи
Физический осмотр, измерения массы тела и высоты проводили в соответствии с планом событий и времени. Кроме того, рассчитывали индекс массы тела (ИМТ) и измеряли окружность шеи в качестве части информации, требуемой для опросника STOP-Bang.
Осмотр полости носа
Осмотр полости носа (в том числе верхних дыхательных путей/горла) проводил квалифицированный медицинский работник. Цель осмотра при скрининге заключалась в исключении любых субъектов с анатомическими или медицинскими состояниями, которые могут препятствовать доставке или всасыванию препарата.
Последующий осмотр включал визуальный осмотр ноздрей, слизистой оболочки носа и горла в отношении наличия покраснения полости носа, ринореи, ринита, повреждения капилляров/кровеносных сосудов и носового кровотечения с оценкой их как отсутствующих, слабых, умеренных или тяжелых. Любое связанное с лечением изменение или ухудшение относительно исходного уровня регистрировали как нежелательное явление.
Опросник по назальным симптомам
Субъекты заполняли опросник по назальным симптомам. Опросник по назальным симптомам был разработан для оценки назальной переносимости после интраназального введения исследуемого препарата. Опросник содержит вопросы по назальным симптомам, которые были оценены субъектом как отсутствующие, легкие, умеренные или тяжелые, на основании ощущений на момент оценки.
C-SSRS
C-SSRS проводили для оценки потенциальных суицидальных идеации и поведения. C-SSRS представляет собой создающую низкую нагрузку меру определения спектра суицидальных идеации и поведения, которая была разработана в исследовании подросткового суицида Национального института психического здоровья для оценки тяжести и отслеживания суицидальных событий в ходе любого лечения. Она представляет собой клиническое интервью, включающее обобщенную информацию как по суицидальной идеации, так и по поведению, которое можно использовать во время проведения любых оценок или оценок риска для определения уровня и типа присутствующей суицидальности. C-SSRS также можно использовать во время лечения, чтобы следить за клиническим ухудшением.
В данном исследовании использовали две версии C-SSRS, исходную/скрининговую версию и версию «с момента последнего визита». Исходную/скрининговую версию C-SSRS использовали во время фазы скрининга/проспективного наблюдения. В этой версии суицидальную идеацию оценивали в 2 момента времени («на протяжении жизни» и «за последние 6 месяцев»), а суицидальное поведение оценивали в 2 момента времени («на протяжении жизни» и «за последний год»). Во всех последующих оценках C-SSRS в этом исследовании использовали версию «с момента последнего визита», в которой оценивались суицидальные идеация и поведение с момента последнего визита субъекта.
CADSS
CADSS представляет собой инструмент для определения текущих диссоциативных симптомов и применяется для оценки возникающих в ходе лечения диссоциативных симптомов. CADSS состоит из 23 субъективных пунктов, разделенных на 3 компоненты: Деперсонализация (пункты, 3-7, 20 и 23), дереализация (пункты 1, 2, 8-13, 16-19 и 21) и амнезия (пункты 14, 15 и 22). Ответы участников оценивались по 5-балльной шкале (от 0=полное отсутствие до 4=крайняя степень). CADSS обладает превосходной межэкспертной надежностью и внутренней согласованностью.
BPRS+
Для оценки потенциальных возникших в ходе лечения психотических симптомов применяли четыре пункта шкалы BPRS. BPRS представляет собой оценочную шкалу из 18 пунктов, используемую для оценки диапазона психотических и аффективных симптомов, оцениваемых как по наблюдениям за субъектом, так и по отчету самого субъекта. Считается, что она обеспечивает быструю и эффективную оценку ответа на лечение в клинических исследованиях лекарственных препаратов и в клинических условиях. В данном исследовании использовали только подшкалу положительных симптомов из 4 пунктов (т.е. подозрительность, галлюцинации, необычное содержание мыслей и концептуальная дезорганизация). Она очень чувствительна к изменениям, и с помощью подготовки и стандартной процедуры интервью можно достичь превосходной межэкспертной надежности.
MOAA/S
MOAA/S использовали для определения возникающей в ходе лечения седации с корреляцией с уровнями седации, определенными континуумом Американского общества анестезиологов (ASA). Оценки MOAA/S находятся в диапазоне от 0 = «отсутствие ответа на болевой стимул» (соответствует континууму ASA для общей анестезии) до 5 = «легко отвечает на имя, произнесенное нормальным тоном» (бодрствование; соответствует континууму ASA для минимальной седации).
В каждый день интраназального введения дозы MOAA/S проводили каждые 15 минут от момента времени до введения дозы до t = +1,5 часа после введения дозы. Если оценка составляла ≤ 3 в любое время в течение интервала 1,5 часа после введения дозы, MOAA/S проводили каждые 5 минут до достижения оценки 4 (в этот момент можно возобновлять частоту каждые 15 минут до t = +1,5 часа после введения дозы). Если у субъектов не было оценки по меньшей мере 5 в момент t = +1,5 часа после введения дозы, за ними дополнительно наблюдали. Для субъектов с оценкой 4 оценивание повторяли каждые 15 минут. И для субъектов с оценкой ≤ 3 оценивание повторяли каждые 5 минут до возвращения оценки к 5, или пока субъект не обращался за соответствующей медицинской помощью в случае клинических показаний.
CGADR
CGADR использовалась для определения текущего клинического статуса субъекта и представляла собой оценку клиницистом готовности покинуть исследовательский центр. Клиницист отвечал «Да» или «Нет» на вопрос «Считается ли субъект готовым покинуть центр на основании общего клинического статуса (например, седации, кровяного давления и других нежелательных явлений)?»
В каждый день интраназального введения дозы CGADR проводили через 1 час и 1,5 часа после введения дозы; если ответ не был «Да» через 1,5 часа после введения дозы, оценку повторяли каждые 15 минут до тех пор, пока не получали ответ «Да», или пока субъект не обращался за соответствующей медицинской помощью в случае клинических показаний. Субъект не должен был покидать центр ранее, чем через 1,5 часа. В дни всех сеансов интраназального лечения субъекты оставались на клиническом центре до завершения процедур исследования и готовности субъекта покинуть центр.
PWC-20
PWC-20 использовали для оценки потенциальных симптомов отказа после прекращения интраназального лечения эскетамином. Оценку проводили на 25 сутки для определения исходного уровня перед прекращением интраназального лечения эскетамином. Чтобы лучше оценить потенциальные симптомы отказа от интраназального исследуемого препарата, рекомендовалось продолжать прием перорального антидепрессанта в течение по меньшей мере первых 2 недель фазы последующего наблюдения, если это не считали клинически неприемлемым.
PWC-20 представляет собой состоящий из 20 пунктов простой и точный способ оценки потенциального развития симптомов отмены после прекращения приема исследуемого препарата. PWC-20 представляет собой надежный и чувствительный инструмент для оценки симптомов отмены. Симптомы отмены возникают рано и исчезают довольно быстро, в зависимости от скорости уменьшения дозы, суточной дозы препарата и периода полувыведения препарата.
BPIC-SS
BPIC-SS представляет собой меру оценки сообщаемых субъектом результатов, которая была разработана для выявления соответствующей популяции с синдромом болезненного мочевого пузыря/интерстициальным циститом для клинических исследований, в которых оценивали новые варианты лечения синдрома болезненного мочевого пузыря.
BPIC-SS использовали для наблюдения за потенциальными симптомами цистита, болезненности мочевого пузыря и интерстициального цистита. BPIC-SS содержит 8 вопросов с периодом оценки, составляющим последние 7 суток, и затрагивает ключевые симптомы, определяемые субъектами с СБМП, включая симптомы боли и/или давления в мочевом пузыре и частоту мочеиспускания. Субъекты отвечали по пунктам, используя 5-балльную шкалу (0=никогда, 1=редко, 2=иногда, 3=большую часть времени, 4=всегда в отношении вопросов о частоте и 0=вовсе нет, 1=немного, 2=в некоторой степени, 3=умеренно и 4=сильно в отношении пунктов, касающихся беспокойства, связанного с симптомами). Вопрос 8 отражает самую сильную боль в мочевом пузыре за последние 7 суток с использованием числовой оценочной шкалы 0-10. Общую оценку рассчитывали, суммируя числа рядом с вариантами ответа, выбранными субъектом. Диапазон возможных оценок для данной шкалы составляет от 0 до 38. Общая оценка 19 или более демонстрировала хорошую чувствительность/специфичность и считалась релевантным пороговым значением для отделения пациентов с существенными симптомами в мочевом пузыре или циститом.
В случае отсутствия каких-либо пунктов общую оценку рассчитать невозможно.
В текущем исследовании, если субъект имел оценку > 18 по шкале BPIC-SS и отсутствовали признаки инфекции мочевыводящих путей на основании общего анализа мочи и микроскопии, его или ее передавали специалисту для дополнительной оценки. Следовательно, помимо общего анализа мочи в соответствующий день исследования проводили бакпосев мочи, если оценка BPIC-SS составляла > 18.
Тестирование когнитивной функции: Компьютеризованный комплект когнитивных тестов и HVLT-R
Компьютеризованный комплект когнитивных тестов обеспечивает оценку нескольких когнитивных доменов, включая внимание, визуальное обучение и память, а также исполнительную функцию. В этих тестах используются нейтральные в отношении культуры стимулы, позволяющие использовать их в условиях многоязычной/многокультурной выборки. Компьютеризованный комплект включает:
Тесты на время простой реакции и время реакции выбора; оцениваемые в отношении скорости ответа (среднее по log 10-преобразованному времени реакции для правильных ответов)
Визуальную эпизодическую память; тест на визуальное запоминание, оцениваемый с помощью арксинус-преобразования доли правильных ответов
Кратковременную память (n-назад); оцениваемую в отношении скорости правильного ответа (среднее по log 10-преобразованному времени реакции для правильных ответов)
Исполнительную функцию; тест на лабиринт/последовательность, оцениваемый в отношении общего числа ошибок
Все измерения были проверены на соответствие традиционным нейропсихологическим тестам и являются чувствительными в отношении эффектов различных препаратов на когнитивную деятельность, включая алкоголь и бензодиазепины. Для выполнения комплекта когнитивных тестов требуется приблизительно 25 минут.
HVLT-R, мера вербального научения и памяти, представляет собой тест на воспроизведение списка слов из 12 пунктов. Его прохождение включает 3 испытаний на научение, перечень из 24 слов для распознавания (включая 12 целевых и 12 контрастных слов) и испытание на отсроченное (20 минут) воспроизведение. Прохождение осуществляется с помощью компьютера; инструкции и списки слов отображаются на экране. Тест-администратор записывает каждое правильно воспроизведенное слово, а оценки за научение, краткосрочное и отсроченное воспроизведение генерируются с помощью программного обеспечения теста. HVLT-R является хорошо проверенной и широко используемой мерой оценки вербальной эпизодической памяти.
Тесты проводили в следующем порядке: HVLT-R, компьютеризованный комплект когнитивных тестов и отсроченная HVLT-R.
UPSIT определение порога обонятельных ощущений
Для оценки любых потенциальных связанных с лечением эффектов на чувство запаха, количественно и количественно оценивали обонятельную функцию, используя проверенные стандартизованные обонятельные тесты, до и в определенные моменты времени в ходе исследования. Применяемыми 2 тестами были:
UPSIT позволяет оценить способность субъекта определять запахи. Этот стандартизированный тест, наиболее широко используемый в мире обонятельный тест, основан на теории проведения базовых психологических тестов и сфокусирован на сравнительной способности субъектов определять пахнущие вещества на сверхпороговом уровне. UPSIT состоит из 4 буклетов размера конверта, каждый из которых содержит 10 пахнущих веществ типа «потереть и понюхать», заключенных в 10-50-мкм полимерных микрокапсулах, размещенных на коричневых полосках в нижней части страниц буклетов. Коэффициенты внутренней согласованности и стабильности результатов повторного тестирования составляют > 0,90. Многочисленные исследования показали, что этот и схожие тесты чувствительны к незначительным изменениям в функции обоняния, связанным с разнообразной этиологией, включая вирусы, травму головы и ряд нейродегенеративных заболеваний.
Тест на определение порога обонятельных ощущений позволяет оценить порог обонятельных ощущений, используя процедуру вынужденного выбора со ступенеобразным порогом. Этот тест позволяет количественно определить порог обнаружения в случае вещества с запахом розы, фенилэтилового спирта (ФЭС). Это пахнущее вещество используют, так как оно обладает слабой способностью стимулировать тройничный нерв в носу. Этот тест чувствителен к обонятельной недостаточности вследствие широкого спектра расстройств.
Эти тесты проводили двусторонним путем (т.е. для обеих ноздрей одновременно). Тестирование проводили во время фазы скрининга/проспективного наблюдения для определения исходной чувствительности субъекта. Затем с течением времени определяли степень изменения от этого исходного уровня. Процентное изменение относительно исходного уровня служило в качестве зависимого показателя для каждого субъекта для каждого теста.
MINI
Субъекты проходили MINI (краткое, структурированное диагностическое интервью) для подтверждения диагноза БДР и определения наличия других психиатрических состояний. Время прохождения составляет приблизительно 15 минут.
MGH-ATRQ
Для определения терапевтической резистентности при БДР использовали MGH-ATRQ. MGH-ATRQ позволяет оценить адекватность длительности и дозировки всех антидепрессантов, используемых для текущего большого депрессивного эпизода. Кроме того, MGH-ATRQ позволяет оценить степень улучшения по шкале от 0% (отсутствие улучшения) до 100% (полное улучшение). MGH-ATRQ проводил клиницист в сотрудничестве с субъектом.
Опросник STOP-Bang
Опросник STOP-Bang представляет собой краткий, простой в использовании, проверенный и чувствительный инструмент скрининга для синдрома обструктивного апноэ во сне (ОАС). Этот опросник содержит 8 пунктов, которые затрагивают ключевые факторы риска для синдрома обструктивного апноэ во сне: храп, утомляемость, наблюдаемое прерывистое дыхание во время сна, высокое кровяное давление, индекс массы тела, возраст, обхват шеи и пол. В вопросах STOP-Bang не уточнен период оценки. Субъекты отвечают да или нет на вопросы, касающиеся храпа, утомляемости, наблюдаемого прерывистого дыхания и высокого кровяного давления (эти пункты относятся к «STOP» в акрониме STOP-BANG); это занимает приблизительно 1 минуту.
Персона исследовательского центра отвечает да или нет на вопросы, касающиеся индекса массы тела (более 35 кг/м²?), возраста (старше 50 лет?), окружности шеи (более 17 дюймов [43 см] у мужчин или более 16 дюймов [41 см] у женщин?) и пола (мужской?).
Общую оценку STOP-BANG рассчитывали, суммируя число положительных ответов с получением диапазона оценок от 0 до 8. Оценка ≥5 по STOP-Bang указывает на умеренный или высокий риск синдрома обструктивного апноэ во сне (индекс апноэ-гипопноэ >30).
Независимая квалификационная оценка
Независимые психиатры/психологи проводили для всех субъектов независимую квалификационную оценку в телефонном режиме во время фазы скрининга/проспективного наблюдения с целью подтверждения диагноза депрессии и пригодности к участию в исследовании.
IDS-C30
IDS-C30 из 30 пунктов разработана для оценки тяжести депрессивных симптомов. IDS позволяет оценить все определяющие домены симптомов, разработанные DSM-5, для диагностирования большого депрессивного эпизода. Эти оценки можно использовать для выявления депрессии, хотя преимущественно их используют в качестве меры тяжести симптомов. 7-суточный период перед оценкой представляет собой обычные временные рамки для оценки тяжести симптомов. Психометрический свойства IDS-C30 были установлены в различных примерах исследований.
Опросник по женскому репродуктивному жизненному цинку и гормонам Центральной больницы штата Массачусетс (MGH-Female RLHQ): Модуль I и отслеживание менструального цикла
Модуль I MGH-Female RLHQ (детородный потенциал, статут менопаузы и менструальный цикл) представляет собой краткий опросник, предназначенный для стандартизации минимального объема релевантной информации о репродуктивном статусе и гормонах. Он заполнялся клиницистом. Эта информация может облегчить исследовательский анализ воздействия эндогенных и экзогенных репродуктивных гормонов на курс лечения БДР и потенциально обеспечить информацию об уходе за женщинами с БДР в будущем.
Отслеживание менструального цикла (дата начала последнего менструального периода) документировали во время визитов исследования, указанных в плане времени и событий.
PAQ
Соблюдение субъектами схемы лечения пероральными антидепрессантами во время фазы скрининга/проспективного наблюдения оценивали, используя PAQ. Она представляет собой краткую меру оценки сообщаемых субъектом результатов из 2 пунктов, которая была разработана в Юго-Западном медицинском центре Университета Техаса для оценки того, как часто субъект принимал препарат и вносил ли он какие-либо изменения в схему лечения антидепрессантами в течение последних 2 недель. Общую оценку рассчитывали путем добавления выбранных ответов на вопросы 1c-1f, где 0=соблюдение, а 1 или более=несоблюдение.
Сбор и обработка проб
Фактические даты и время сбора проб записывали в eCRF или бланке заявки на проведение лабораторного анализа. Если образцы крови брали с помощью постоянной канюли, соответствующее количество (1 мл) сукровицы, немного большее объема мертвого пространства наконечника, удаляли из канюли и выливали перед взятием каждого образца крови. После сбора проб крови канюлю продували 0,9% раствором хлорида натрия фармакопеи США (USP) (или эквивалентным), и заполняли объемом, равным объему мертвого пространства наконечника.
ЗАВЕРШЕНИЕ/ПРЕКРАЩЕНИЕ УЧАСТИЯ В ИССЛЕДОВАНИИ ПАЦИЕНТОМ
Завершение участия
Считалось, что субъект завершил участие в двойной слепой индукционной фазе исследования, если он или она завершили оценивание по шкале MADRS в конце 4-недельной двойной слепой индукционной фазы (т.е. MADRS на 28 сутки). Субъектов, которые преждевременно прекратили исследуемое лечение по какой-либо причине до завершения двойной слепой индукционной фазы, не считали завершившими участие в двойной слепой индукционной фазе исследования. Субъектов, которые перешли в фазу последующего наблюдения, считали завершившими эту фазу исследования, если он или она завершили оценивание по шкале MADRS на 24 неделе фазы последующего наблюдения.
Прекращение участия в исследовании
Субъект выбывал из исследования по любой из следующих причин:
1. Невозможность последующего наблюдения
2. Отзыв согласия
3. Нарушение процедур протокола (определяемое в каждом конкретном случае)
4. Раскрытие данных (двойной слепой индукционной фазы)
5. Отсутствие эффективности
6. Исследователь или спонсор посчитал (например, что по причинам безопасности или переносимости, таким как нежелательные явления), что прекращение участия в исследовании было в интересах данного субъекта.
7. Беременность субъекта
8. Исследование прервано спонсором по причинам нецелесообразности
9. Смерть
В случае невозможности последующего наблюдения какого-либо субъекта персонал исследовательского центра прилагал все разумные усилия к тому, чтобы связаться с субъектом и определить причину прекращения/прерывания участия.
Если субъект прекращал участие до завершения исследования, причину прекращения документировали. Исследуемый препарат, назначенный прекратившему участие субъекту, не назначали другому субъекту. Если субъект выбыл из исследования до окончания двойной слепой индукционной фазы по причинам, отличным от отзыва согласия, то в течение 1 недели с даты прекращения проводили визит досрочного отстранения, за которым следовала фаза последующего наблюдения.
Анализ безопасности
Данные о безопасности анализировали для двойной слепой индукционной фазы, используя выборку для анализа безопасности.
Нежелательные явления
Дословные термины, используемые в eCRF исследователями для определения нежелательных явлений, были закодированы с помощью MedDRA. В анализ были включены все сообщенные нежелательные явления, начавшиеся во время двойной слепой индукционной фазы (т.е. НЯВЛ и нежелательные явления, которые ухудшились относительно исходного уровня). Для каждого нежелательного явления суммировали процент субъектов, у которых наблюдался по меньшей мере 1 эпизод данного явления, по группе лечения. Нежелательные явления, возникшие во время фазы последующего наблюдения, суммировали отдельно.
НЯВЛ, представляющие особый интерес, исследовали отдельно. НЯ, представляющие особый интерес, перечислены в SAP. Умерших субъектов, которые прекратили лечение по причине нежелательного явления или которые испытали тяжелое или серьезное нежелательное явление, учитывали отдельно.
Клинические лабораторные тесты
Лабораторные данные обобщали по типу лабораторных тестов. В обобщенных лабораторных данных использовали референсные диапазоны и заметно отклоняющиеся от нормы результаты (указанные в плане статистического анализа). Описательную статистику рассчитывали для каждого лабораторного аналита на исходном уровне и в каждый запланированный момент времени на каждой фазе исследования. Представлены изменения относительно исходных результатов. Приведены сводные таблицы о частоте аномалий. Приведены списки субъектов с лабораторными результатами за пределами референсных диапазонов и заметно отклоняющимися от нормы результатами.
ЭКГ
Эффекты на сердечно-сосудистые показатели оценивали с помощью описательной статистики и сводных таблиц частот. Эти таблицы включают наблюдаемые значения и изменения относительно исходных значений.
Данные электрокардиограмм обобщали с помощью параметра ЭКГ. Описательную статистику рассчитывали на исходном уровне и для наблюдаемых значений и изменений относительно исходного уровня в каждый запланированный момент времени. Были составлены сводные таблицы о частоте аномалий.
Анализируемыми показателями ЭКГ были частота сердечных сокращений, интервал PR, интервал QRS, интервал QT и интервал QTc с использованием следующих методов коррекции: QT корректировали по формуле Базетта (QTcB) и QTcF.
Описательные статистические данные для интервалов QTc и изменений относительно двойного слепого исходного уровня обобщали в каждый запланированный момент времени. Суммировали процент субъектов с интервалом QTc > 450 мс, > 480 мс или > 500 мс, а также процент субъектов с повышением интервала QTc относительно исходного уровня < 30 мс, 30-60 мс или > 60 мс.
Были представлены все важные отклонения от нормы в форме сигнала ЭКГ, которые являлись изменениями относительно исходных показателей (например, изменения в морфологии T-волн или появление U-волн).
Основные физиологические показатели
В каждый запланированный момент времени обобщали описательные статистические данные по температуре, пульсу/частоте сердечных сокращений, частоте дыхания, пульсоксиметрии и кровяному давлению (систолическому и диастолическому) (в положении лежа на спине) и изменениям относительно исходного уровня. Обобщали процент субъектов со значениями за пределами клинически значимых границ.
Осмотр полости носа
Изменения в результатах относительно исходного осмотра полости носа (включая верхние дыхательные пути/горло) были перечислены для каждой группы лечения. Результатом осмотра были оценки (отсутствуют, слабые, умеренные или тяжелые), основанные на визуальном осмотре ноздрей, слизистой оболочки носа и горла в отношении покраснения полости носа, ринореи, ринита, повреждения капилляров/кровеносных сосудов и носового кровотечения. Для каждой группы лечения была представлена таблица сдвигов для изменений относительно двойного слепого исходного уровня в оценках каждого типа осмотра.
Опросник по назальным симптомам
Оценку из опросника по назальным симптомам обобщали описательным образом для каждого запланированного момента времени для каждой группы лечения.
C-SSRS
Связанные с суицидом мысли и поведение на основании C-SSRS обобщали по группам лечения в таблицах частоты возникновения и сдвигов. Были определены отдельные ожидаемые результаты для суицидальной идеации и суицидального поведения и обобщены по группе лечения. Отсутствующие оценки не учитывали.
CADSS, BPRS+ и MOAA/S
Описательные статистические данные для каждой оценки и изменений относительно момента до введения дозы обобщали в каждый запланированный момент времени.
Глобальная клиническая оценка готовности к выписке, PWC-20, BPIC-SS, UPSIT и тест на определение порога обонятельных ощущений
Описательную статистику по каждой оценке и изменениям и/или процентным изменениям относительно исходного уровня обобщали в каждый запланированный момент времени.
Тестирование когнитивной функции
Описательную статистику по оценкам и изменениям относительно исходного уровня по доменам когнитивных функций обобщали в каждый запланированный момент времени.
Определения и классификация нежелательных явлений
Нежелательным явлением является любое неблагоприятное медицинское явление у субъекта клинического исследования, которому вводят лекарственный (исследуемый или не исследуемый) продукт. Нежелательное явление необязательно имеет причинно-следственную связь с лечением. Таким образом, нежелательное явление может представлять собой любые неблагоприятные и непреднамеренные признак (включая аномальный результат), симптом или заболевание, связанные по времени с применением лекарственного (исследуемого или не исследуемого) продукта, независимо от того, связаны они с данным лекарственным (исследуемым или не исследуемым) продуктом или нет (по определению на Международной конференции по гармонизации [ICH]). Это включает любое событие, которое является новым в отношении начала или ухудшенным в отношении тяжести или частоты относительно исходного состояния, или представляет собой аномальные результаты диагностических процедур, включая аномалии в лабораторных тестах.
Серьезным нежелательным явлением, согласно руководствам ICH и EU по фармакологическому надзору за лекарственными средствами для медицинского применения, является любое неблагоприятное медицинское явление, которое при любой дозе:
• Приводит к смерти
• Представляет угрозу для жизни (например, субъект подвергался риску смерти во время события. Термин «представляет угрозу для жизни» не относится к событию, которое гипотетически могло бы привести к смерти, если бы оно было более тяжелым.)
• Требует стационарной госпитализации или продления текущей госпитализации
• Приводит к постоянной или значительной нетрудоспособности/недееспособности
• Является врожденной аномалией/дефектом при рождении
• Подозревается в переносе любых возбудителей инфекции посредством лекарственного продукта
• Является важным с медицинской точки зрения*
*Медицинское и научное заключение следует использовать при решении, является ли экспресс-отчетность целесообразной также в других ситуациях, например, важных медицинских событиях, которые могут непосредственно не представлять угрозу для жизни или не приводить к смерти, но могут подвергать субъекта опасности или могут требовать вмешательства для предотвращения одного из других результатов, перечисленных в определении выше. Их, как правило, следует рассматривать как серьезные.
В случае возникновения серьезного и неожиданного нежелательного явления, предполагающего наличие причинно-следственной связи между исследуемым препаратом и событием (например, смерть от анафилактического шока), это событие регистрировали как серьезную и неожиданную нежелательную реакцию, даже если оно являлось компонентом ожидаемого результата исследования (например, смертность по всем причинам).
Нежелательное явление не вносили в список, если природа или тяжесть не соответствовали применимой справочной информации по безопасности продукта. В случае эскетамина ожидаемость нежелательного явления определялась тем, было оно перечислено в разделе справочной информации по безопасности Брошюры исследователя или нет.
В случае дулоксетина, эсциталопрама, сертралина и венлафаксина XR ожидаемость нежелательного явления определялась тем, было оно перечислено в SmPC или инструкции США по применению препарата или нет.
Нежелательное явление считалось связанным с применением препарата, если отнесение было возможным, вероятным или очень вероятным согласно определениям отнесения, перечисленным ниже.
Не связано: Нежелательное явление, которое не было связано с применением препарата.
Сомнительно: Нежелательное явление, для которого было более вероятным альтернативное объяснение, например, сопутствующие препараты, сопутствующие заболевания или временная взаимосвязь позволяют предположить, что причинно-следственная связь маловероятна.
Возможно: Нежелательное явление, которое может быть связано с применением препарата. Альтернативное объяснение, например, сопутствующие препараты, сопутствующие заболевания, было неубедительным. Временная взаимосвязь была обоснованной; следовательно, причинно-следственная связь не может быть исключена.
Вероятно: Нежелательное явление, которое может быть связано с применением препарата. Временная взаимосвязь позволяла сделать такое предположение (например, подтверждена пробной отменой). Альтернативное объяснение было маловероятным, например, сопутствующие препараты, сопутствующие заболевания.
Очень вероятно: Нежелательное явление, которое было указано как возможная нежелательная реакция и не может быть надлежащим образом объяснено альтернативным объяснением, например, сопутствующими препаратами, сопутствующими заболеваниями. Временная взаимосвязь позволяла сделать очень вероятное предположение (например, подтверждена пробными отменой и возобновлением).
Оценку степени тяжести осуществляли, используя следующие общие категориальные дескрипторы:
Легкая: Ощущение симптомов, которые легко переносились, вызывая минимальный дискомфорт и не мешая повседневной деятельности.
Средняя: Наличие достаточного дискомфорта, чтобы мешать повседневной деятельности.
Тяжелая: Максимальный дистресс, приводящий к существенному нарушению функционирования или недееспособности. Мешал нормальной повседневной деятельности.
Исследователь использовал клиническое заключение при оценке тяжести событий, не испытываемых субъектом непосредственно (например, отклонения от нормы в лабораторных результатах).
Ситуации, требующие специальной отчетности
Представляющие интерес события безопасности в связи с исследуемым препаратом спонсора, которые могут требовать экспресс-отчетности и/или оценки безопасности, включали, но не ограничивались этим:
Передозировку исследуемого препарата спонсора
Подозрение в злоупотреблении/неправильном применении исследуемого препарата спонсора
Неумышленное или случайное применение исследуемого препарата спонсора
Ошибка в применении лекарства, связанная с продуктом спонсора (с применением субъектом/пациентом исследуемого препарата спонсора или без, например, перепутанные имена)
Ситуации, требующие специальной отчетности, записывали в eCRF. Любую ситуацию, требующую специальной отчетности, которая соответствовала критериям серьезного нежелательного явления, записывали на странице серьезных нежелательных явлений в eCRF.
Процедуры: Все нежелательные явления
Все нежелательные явления и ситуации, требующие специальной отчетности, серьезные или несерьезные, записывали с момента получения подписанного и датированного ФИС и до завершения последней связанной с исследованием процедуры субъекта (которая может включать связь для осуществления последующего наблюдения в вопросах безопасности). Серьезные нежелательные явления, включая спонтанно сообщенные исследователю в течение 30 дней после последней дозы исследуемого препарата, регистрировали, используя форму для серьезных нежелательных явлений. Спонсор оценивал любую информацию по безопасности, которая была спонтанно сообщена исследователем в сроки, указанные в протоколе.
Все явления, которые соответствуют определению серьезного нежелательного явления, регистрировали как серьезные нежелательные явления, независимо от того, являлись ли они определенными в протоколе оценками. Ожидаемые явления записывали и регистрировали.
Все нежелательные явления, независимо от серьезности, тяжести или предполагаемой взаимосвязи с исследуемым препаратом, записывали, используя медицинскую терминологию в первичном документе и eCRF. По возможности диагноз ставили тогда, когда признаки и симптомы возникали в результате общей этиологии (например, кашель, насморк, чиханье, боль в горле и приливы крови к голове следовало регистрировать как «инфекцию верхних дыхательных путей»). Исследователи записывали в eCRF свое мнение относительно взаимосвязи нежелательного явления с исследуемой терапией. Все меры, необходимые для борьбы с нежелательными явлениями, были записаны в первичной документации и зарегистрированы.
Спонсор взял на себя ответственность за надлежащее информирование регуляторных органов о нежелательных явлениях.
Для всех исследований с амбулаторной фазой, включая открытые исследования, субъект получал «карточку-памятку (исследования)» и был проинструктирован носить эту карточку с собой в течение всего исследования, при этом в карточке были указаны:
• Номер исследования
• Указание на местном(ых) языке(ах), что субъект участвует в клиническом исследовании
• Имя исследователя и 24-часовой контактный номер телефона
• Имя местного спонсора и 24-часовой контактный номер телефона (только для медперсонала)
• Номер центра
• Номер субъекта
• Любая другая информация, необходимая для неотложного раскрытия данных
Серьезные нежелательные явления
О всех серьезных нежелательных явлениях, происходящих во время исследования, сообщали соответствующему контактному лицу спонсора в течение 24 часов с момента получения информации о явлении.
Все серьезные нежелательные явления, которые не были устранены к концу исследования или которые не были устранены после прекращения участия субъекта в исследовании, подлежали наблюдению до тех пор, пока не происходило одно из следующего:
• Явление было устранено
• Явление было стабилизировано
• Явление вернулось к исходному уровню, если был известен исходный уровень/статус
• Явление могло было связано с агентами, отличными от исследуемого препарата, или с факторами, не относящимися к проведению исследования
• Становилось маловероятно, что можно получить какую-либо дополнительную информацию (отказ субъекта или медработника предоставить дополнительную информацию, невозможность последующего наблюдения после проведения комплексной проверки усилий, связанных с последующим наблюдением)
Подозрение передачи возбудителя инфекции через лекарственный продукт регистрировали как серьезное нежелательное явление. Любое явление, требующее госпитализации (или продления госпитализации), которое произошло в ходе участия субъекта в исследовании, регистрировали как серьезное нежелательное явление, за исключением госпитализации по следующим причинам:
• Госпитализация, не связанная с лечением острого заболевания или нежелательного явления (например, социальные причины, такие, как ожидание размещения в учреждении долговременного ухода)
• Хирургическая операция или процедура, запланированная до включения в исследование (должна быть задокументирована в eCRF). Госпитализацию, запланированную до подписания ФИС, при условии, что первопричинное патологическое состояние, в связи с которым планировалась госпитализация, не ухудшились, не считали серьезным нежелательным явлением. Любое нежелательное явление, которое привело к продлению первоначально запланированной госпитализации, нужно было регистрировать как новое серьезное нежелательное явление.
Для удобства исследователь мог принимать решение о госпитализации субъекта в течение периода лечения.
Случай смерти субъекта в ходе исследования, вне зависимости от того, была ли она ожидаема или связана с исследуемым препаратом, считали серьезным нежелательным явлением.
Беременность
Персонал исследовательского центра должен был сообщать спонсору о всех первоначальных сообщениях о беременности в течение 24 часов с момента получения информации об этом событии, используя соответствующую форму уведомления о беременности. Аномальные варианты разрешения беременности (например, спонтанный аборт, мертворождение и врожденная аномалия) считали серьезными нежелательными явлениями, и о них следовало сообщать, используя форму для серьезных нежелательных явлений. Любой субъект, который забеременел в ходе исследования, должен был незамедлительно выбыть из исследования и прекратить дальнейшее лечение.
Поскольку эффект исследуемого препарата на сперму неизвестен, о беременности партнерш субъектов-мужчин, включенных в исследование, следовало сообщать персоналу исследовательского центра в течение 24 часов с момента получения информации об этом событии, используя соответствующую форму уведомления о беременности.
Необходимо было предоставлять информацию по периоду последующего наблюдения, касающуюся разрешения беременности и любых постнатальных последствий для новорожденного.
ОБОБЩЕННАЯ ИНФОРМАЦИЯ ПО ВСЕМ НЕЖЕЛАТЕЛЬНЫМ ЯВЛЕНИЯМ
Обобщенная информация по всем возникшим в ходе лечения нежелательным явлениям (НЯВЛ) во время двойной слепой фазы представлена в Таблице 18. В целом, у 84,3% субъектов в группе, получавшей эскетамин+пероральный АД, и у 60,6% субъектов в группе, получавшей действующий препарат сравнения, наблюдалось по меньшей мере одно НЯВЛ во время двойной слепой фазы.
b Нежелательное явление, которое началось во время двойной слепой индукционной фазы и привело к прекращению приема во время фазы последующего наблюдения, считали возникшим в ходе лечения во время двойной слепой индукционной фазы.
Частота определялась числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений.
На Фиг. 6 представлен процент субъектов, сообщивших о проблемах на исходном уровне и в конечный момент времени, определенных по индивидуальным параметрам EQ-5D-%L.
Нежелательные явления, возникшие в ходе лечения во время двойной слепой фазы (> 5% субъектов в каждой группе лечения), обобщены по группе лечения для выборки для анализа безопасности и приведены в Таблице 19 ниже. Наиболее распространенными (> 20%) НЯВЛ в группе, получавшей эскетамин+пероральный АД во время двойной слепой фазы, были тошнота (26,1%), вертиго (26,1%), дисгевзия (24,3%) и головокружение (20,9%). Наиболее распространенным НЯВЛ в группе, получавшей действующий препарат сравнения, была головная боль (17,4%).
Нежелательные явления, приводящие к прекращению приема исследуемого препарата
9 субъектов (8 субъектов в группе, получавшей эскетамин+пероральный АД, и 1 субъект в группе, получавшей действующий препарат сравнения), прекратили прием интраназального исследуемого препарата во время двойной слепой индукционной фазы из-за нежелательных явлений, возникших в ходе лечения (Таблица 20). 4 субъекта в группе, получавшей эскетамин+пероральный АД, прекратили прием перорального антидепрессанта во время двойной слепой индукционной фазы из-за нежелательных явлений, возникших в ходе лечения (Таблица 21). Три субъекта в группе, получавшей эскетамин+пероральный АД, прекратили участие в двойной слепой индукционной фазе как из-за интраназального препарата, так и перорального АД. (Данные обобщены в Таблицах 20 и 21).
Частота определялась числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений.
Частота определялась числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений.
Серьезные нежелательные явления
Два субъекта испытали серьезное возникшее в ходе лечения нежелательное явление во время двойной слепой фазы. Один субъект в группе, получавшей действующий препарат сравнения, испытывал постуральное головокружение, которое посчитали маловероятно связанным как с интраназальным плацебо, так и с пероральным АД. Один субъект в группе, получавшей эскетамин+пероральный АД, получил множественные повреждения в мототранспортном происшествии (и впоследствии умер после формальной блокировки базы данных). Это событие посчитали не связанным с эскетамином и маловероятно связанным с пероральным АД.
У одного субъекта в группе, получавшей эскетамин+пероральный АД, произошло внутримозговое кровоизлияние во время фазы последующего наблюдения через 83 дней после последнего интраназального введения эскетамина. Это посчитали маловероятно связанным с эскетамином и не связанным с пероральным АД.
Кровяное давление
Временное кровяное давление увеличивается до пика для группы, получавшей эскетамин, приблизительно через 40 минут после введения дозы и возвращается к нормальному диапазону через 90 минут. Максимальное среднее повышение (по всем дням введения дозы) систолического КД составило 11,6 в группе, получавшей эскетамин+пероральный АД, и 5,0 в группе, получавшей действующий препарат сравнения. Максимальное среднее повышение (по всем дням введения дозы) диастолического КД составило 8,1 в группе, получавшей эскетамин, и 4,5 в группе, получавшей действующий препарат сравнения. На Фиг. 7 и 8 представлены средние значения измеренного кровяного давления в динамике по времени по группе лечения во время двойной слепой фазы.
Применяемая клиницистами шкала диссоциативных симптомов (CADSS)
Оценку по применяемой клиницистами шкале диссоциативных состояний (CADSS) определяли перед началом введения каждой дозы, через 40 минут и 1,5 часа после введения дозы. Шкалу CADSS использовали для оценки возникших в ходе лечения диссоциативных симптомов и изменений восприятия, а общая оценка находилась в диапазоне от 0 до 92, причем более высокая оценка представляла более тяжелое состояние. Диссоциативные симптомы и изменения восприятия, оцененные по шкале CADSS, позволяют предположить, что эти симптомы появились вскоре после начала введения дозы и они разрешились через 1,5 часа после введения дозы (как показано на Фиг. 9).
Модифицированная оценка наблюдателем активности/седации (MOAA/S)
Модифицированную оценку наблюдателем активности/седации (MOAA/S) использовали для определения возникающей в ходе лечения седации с корреляцией с уровнями седации, определенными континуумом Американского общества анестезиологов (ASA). Оценки MOAA/S находились в диапазоне от 0 (отсутствие ответа на болевой стимул; соответствует континууму ASA для общей анестезии) до 5 (легко отвечает на имя, произнесенное нормальным тоном [бодрствование]; соответствует континууму ASA для минимальной седации). Седация, оцененная с помощью MOAA/S, позволяет предположить, что седация была разрешена через 1,5 часа после введения дозы (как показано на Фиг. 10).
ФАРМАКОКИНЕТИКА
Образцы венозной крови объемом приблизительно 2 мл собирали для измерения концентраций эскетамина, норэскетамина и других метаболитов в плазме (в случае обоснованности) в моменты времени, указанные в плане времени и событий. Записывали точные даты и время сбора образцов крови для ФК.
Анализ образцов плазмы для определения концентраций эскетамина (и норэскетамина в случае обоснованности) проверенным, специфическим, ахиральным и чувствительным методом жидкостной хроматографии в сочетании с тандемной масс-спектрометрией (ЖХ-МС/МС) проводился спонсором или под наблюдением спонсора. При необходимости некоторые образцы плазмы анализировали, чтобы задокументировать наличие других аналитов (например, циркулирующих метаболитов или денатония), используя подходящий исследовательский метод. Кроме того, образцы плазмы для ФК могли хранить для анализа метаболического профиля в будущем.
Фармакокинетические параметры
Данные зависимости плазменной концентрации эскетамина (и норэскетамина в случае обоснованности) от времени анализировали, используя популяционную ФК-модель. Типичные популяционные значения основных ФК-параметров (например, объем распределения выведения эскетамина) оценивали вместе с межиндивидуальной вариабельностью. Исследовали эффекты демографии, значений лабораторных параметров и других ковариат субъектов на ФК эскетамина.
Оценки фармакокинетики/фармакодинамики
Оценивали взаимосвязь между общей оценкой по шкале MADRS (и, возможно, выбранными нежелательными явлениями в качестве дополнительных ФД-параметров) и ФК-показателями эскетамина. При наличии какой-либо видимой тенденции в графическом анализе применяли подходящие модели для описания взаимосвязи воздействие - эффект.
Оценки биомаркеров, фармакогеномики (ДНК) и экспрессии (РНК)
Во время исследования собирали кровь для оценки биомаркеров в моменты времени, указанные в плане времени и событий. Образцы крови для анализа биомаркеров брали перед введением дозы. Было предпочтительно, чтобы субъекты придерживались диеты с низким содержанием жиров в день сбора образцов.
В крови исследовали биомаркеры (белки, метаболиты и рибонуклеиновые кислоты [РНК]), связанные с (но без ограничения этим) активностью иммунной системы, активацией гипоталамо-гипофизарно-надпочечниковой (ГГН) оси, нейротрофическими факторами и метаболическими факторами. Биомаркеры добавляли или удаляли на основании научной информации или технических инноваций при условии, что общий объем собранной крови не увеличивался.
Образцы крови для анализа ДНК собирали в моменты времени, указанные в плане времени и событий, для оценки генетической и эпигенетической вариации в генах в случае путей, связанных с депрессией (например, ось ГГН, воспаление, факторы роста, переносчики моноаминов, ионные каналы и суточный ритм). Генотипирование проводили только для скринингового образца; фармакогеномные и эпигенетические оценки можно было проводить для любых/всех собранных образцов.
Образцы ДНК использовали для исследования, связанного с эскетамином, пероральными антидепрессантами, ТРД или БДР. Их также можно было использовать для разработки тестов/анализов, связанных с эскетамином, пероральными антидепрессантами, ТРД или БДР. Фармакогеномные исследования состояли из анализа 1 или более кандидатных генов или анализа генетических маркеров по всему геному (при необходимости), связанных склиническими ожидаемыми результатами для эскетамина, пероральных антидепрессантов, ТРД или БДР.
Использование медицинских ресурсов
Во время фазы последующего наблюдения исследования собирали данные об использовании медицинских ресурсов, связанных с обращением за медицинской помощью. Процедуры, тесты и обращения, предусмотренные протоколом, были исключены. Собранные данные могут использоваться для проведения исследовательского экономического анализа и включают: (a) число и длительность обращений за медицинской помощью, включая хирургические операции и другие выбранные процедуры (стационарные и амбулаторные), (b) длительность госпитализации (общее количество суток пребывания, включая длительность пребывания в больничном отделении; например, отделении интенсивной терапии), (c) число и характер диагностических и терапевтических тестов и процедур и/или (d) амбулаторные обращения за медицинской помощью и лечение (включая визиты к врачу или в кабинет неотложной помощи, тесты и процедуры, а также лекарственные препараты).
Фармакокинетический анализ
Значения плазменной концентрации эскетамина (и норэскетамина в случае обоснованности) перечислены для всех субъектов. Данные зависимости плазменной концентрации эскетамина (и норэскетамина в случае обоснованности) от времени анализировали, используя популяционную ФК-модель. Данные могли объединять с данными других выбранных исследований для подтверждения релевантной структурной модели. Типичные популяционные значения основных ФК-параметров оценивали вместе с межиндивидуальной вариабельностью. Исследовали эффекты демографии, значений лабораторных параметров и других ковариат субъектов на ФК эскетамина.
Фармакокинетический/фармакодинамический анализ
Оценивали взаимосвязь между общей оценкой по шкале MADRS (и, возможно, выбранными нежелательными явлениями в качестве дополнительных ФД-параметров) и ФК-показателями эскетамина. При наличии какой-либо видимой тенденции в графическом анализе применяли подходящие модели для описания взаимосвязи воздействие - эффект.
Анализ биомаркеров и фармакогеномики
Обобщали исходные значения биомаркеров и изменения от исходных значений биомаркеров до моментов времени, указанных в плане времени и событий. Исследовательский анализ мог включать сравнение показателей биомаркеров между группами лечения и корреляцию с исходным уровнем и изменениями значений биомаркеров относительно исходного уровня в случае эффективности и других показателей. Дополнительный исследовательский анализ мог также включать взаимосвязь между исходным уровнем и изменением относительно исходного уровня в показателях биомаркеров и клиническим ответом, поддержанием/стабилизацией ответа, рецидивом и отсутствием ответа.
Фармакогеномный анализ также мог включать анализ кандидатных генов или полногеномный анализ ассоциаций, связанных с ответом на лечение, поддержанием/стабилизацией ответа, рецидивом, отсутствием ответа и БДР/ТРД. Анализ экспрессии мог включать известных транскриптов матричной РНК/микроРНК (мРНК/микроРНК) или полный транскриптомный анализ в отношении ответа на лечение антидепрессантами и БДР/ТРД.
СТАТИСТИЧЕСКИЕ МЕТОДЫ, ИСПОЛЬЗУЕМЫЕ ПРИ АНАЛИЗЕ
Ниже приведено общее описание статистических методов, используемых для анализа данных по эффективности и безопасности. В конце двойной слепой индукционной фазы базу данных блокировали для анализа и отчетности по этой фазе. Назначение лечения субъекта было видимым только для исследовательского персонала спонсора. Исследователи и персонал центра были заслеплены в отношении назначения лечения до тех пор, пока все субъекты не завершали участие в исследовании после фазы последующего наблюдения.
Первичные выборки для анализа эффективности и безопасности были следующими:
Полная выборка для анализа: Все рандомизированные субъекты, которые получили по меньшей мере 1 дозу интраназального исследуемого препарата и 1 дозу перорального антидепрессанта в двойной слепой индукционной фазе.
Выборка для анализа безопасности: Все рандомизированные субъекты, которые получили по меньшей мере 1 дозу интраназального исследуемого препарата или 1 дозу перорального антидепрессанта в двойной слепой индукционной фазе.
Максимальный размер выборки, запланированный для данного исследования, рассчитывали исходя из разницы в лечении для двойной слепой индукционной фазы, соответствующей 6,5 баллам общей оценки MADRS между эскетамином и действующим препаратом сравнения, стандартному отклонению 12, 1-стороннему уровню значимости 0,0125 и частоте исключения 25%. Для достижения 90% мощности с использованием фиксированного дизайна без промежуточного анализа необходимо рандомизировать максимум около 98 субъектов в каждую группу лечения. Разница в лечении и стандартное отклонение, используемые в данном расчете, были основаны на результатах панели A исследования ESKETINTRD2003 и клиническом заключении.
Промежуточный анализ для повторной оценки размера выборки или приостановки из-за нецелесообразности
Проводили один немаскированный промежуточный анализ через 4 недели после рандомизации 66 субъектов в исследование (приблизительно по 33 субъекта на группу лечения). Предполагалось, что в это время приблизительно 50 субъектов в полной выборке для анализа завершат двойную слепою индукционную фазу исследования (приблизительно по 25 субъектов на группу лечения). Отслеживали частоту исключения, чтобы гарантировать, что в промежуточный анализ включено достаточное количество субъектов. Цель промежуточного анализа заключалась либо в переоценке размера выборки, либо в приостановке исследования из-за нецелесообразности. Размер выборки можно было корректировать для получения необходимой мощности при сохранении контроля общей вероятности ошибки I рода. Максимальный размер выборки, запланированный для данного исследования, составлял 98 человек на группу лечения.
Были разработаны строгий план промежуточного статистического анализа (SAP, англ. «statistical analysis plan») и концепция с подробным описанием алгоритма для повторной оценки размера выборки на основании промежуточных данных и способа выполнения анализа. IDMC провел промежуточный анализ и вынес рекомендации в отношении любой корректировки размера выборки на основании правил, определенных в промежуточном SAP. Любые изменения размера выборки были переданы IDMC (или статистиком из группы статистической поддержки) поставщику IWRS для гарантии включения в исследование соответствующего количества субъектов. Ни один из членов команды разработчиков эскетамина или членов персонала исследовательских центров, проводивших клиническое исследование, не был проинформирован о результатах промежуточного анализа и о любых корректировках, внесенных в размер выборки.
Были приняты меры, чтобы гарантировать, что результаты промежуточного анализа не повлияют на проведение исследования, исследователей или субъектов.
Анализ эффективности
Анализ эффективности проводили на полной выборке для анализа, которая включала всех рандомизированных субъектов, которые получили по меньшей мере 1 дозу интраназального исследуемого препарата и 1 дозу перорального антидепрессанта во время двойной слепой индукционной фазы.
Первичную переменную эффективности, изменение общей оценки MADRS относительно исходного уровня на 4 неделе двойной слепой индукционной фазы, анализировали, используя СМПИ. Данная модель включала исходную общую оценку MADRS в качестве ковариаты и лечение, страну, класс антидепрессанта (SNRI или SSRI), сутки и зависимость сутки - лечение в качестве фиксированных эффектов, а также эффект случайного субъекта. Сравнение группы эскетамина плюс пероральный антидепрессант с группой перорального антидепрессанта плюс интраназальное плацебо проводили, используя соответствующий контраст.
В случае досье EU первичный анализ эффективности был основан на модели ковариационного анализа (ANCOVA) с использованием данных последнего документированного значения (LOCF). Данная модель включала факторы лечения, страны и класса перорального антидепрессанта (SNRI или SSRI) и исходную общую оценку MADRS в качестве ковариаты. Сравнение группы эскетамина плюс пероральный антидепрессант с группой интраназального плацебо плюс пероральный антидепрессант проводили, используя соответствующий контраст.
При условии нормативно-правового признания PHQ-9 в качестве ключевого вторичного ожидаемого результата, первые из 3 ключевых вторичных ожидаемых результатов эффективности, изменение общей оценки PHQ-9 относительно исходного уровня на 4 неделе двойной слепой индукционной фазы, анализировали, используя те же модели, что были описаны выше для общей оценки MADRS.
Для анализа вторых ключевых вторичных ожидаемых результатов эффективности долю субъектов, демонстрировавших на 2 сутки начало клинического ответа, который сохранялся в течение двойной слепой индукционной фазы, в группе эскетамина плюс пероральный антидепрессант, сравнивали с группой перорального антидепрессанта плюс интраназальное плацебо, используя критерий хи-квадрат Кохрана - Мантеля - Хензеля с корректировкой в отношении страны и класса антидепрессанта (SNRI или SSRI). Клинический ответ определялся как ≥ 50% улучшение общей оценки MADRS на 2 сутки (т.е. через сутки после приема первой дозы двойного слепого интраназального препарата), которое сохранялось до конца двойной слепой фазы. Субъектов, прекративших участие в исследовании до окончания двойной слепой индукционной фазы, не учитывали как сохранивших клинический ответ.
Третий ключевой вторичный ожидаемый результат эффективности, изменение общей оценки SDS относительно исходного уровня на 4 неделе двойной слепой индукционной фазы, анализировали, используя ANCOVA. Данная модель включала факторы лечения, страны и класса перорального антидепрессанта (SNRI или SSRI) и исходную общую оценку SDS в качестве ковариаты. Сравнение каждой группы интраназального эскетамина плюс пероральный антидепрессант с группой перорального антидепрессанта плюс интраназальное плацебо проводили, используя соответствующий контраст. Ответы на вопросы H1-H3 суммировали отдельно.
Для учета множественности и строгого контроля ошибок I рода для первичного и 3 ключевых вторичных ожидаемых результатов эффективности (изменение общей оценки PHQ-9, начало клинического ответа и изменение общей оценки SDS) использовали подход серийного фильтрования (фиксированная последовательность).
Значения частоты ответа и ремиссии обобщали при каждом визите.
Изменение относительно исходного уровня общих оценок GAD-7 и ранг изменения относительно исходного уровня оценок CGI-S в конце двойной слепой индукционной фазы анализировали на основании данных LOCF, используя модель ANCOVA, со страной и классом антидепрессанта (SNRI или SSRI) в качестве факторов и соответствующей исходной оценкой (неранжированная оценка в случае CGI-S) в качестве ковариаты.
Оценки показателей данных EQ-5D-5L, индекса состояния здоровья и оценку общего состояния здоровья обобщали с течением времени.
Кроме того, суммировали оценки всех ожидаемых результатов эффективности для всех визитов во время двойной слепой индукционной фазы. Сводные данные представлены, чтобы продемонстрировать соответствие эффекта среди релевантных подгрупп (например, антидепрессантов класса SNRI и SSRI).
Анализ субпопуляции США - Клиническая эффективность и безопасность
В общем анализе ЭСК+АД демонстрировали статистически значимое и клинически существенное превосходство по сравнению с АД+ПБО в отношении первичного ожидаемого результата эффективности (т.е. изменения относительно исходного уровня общей оценки по шкале MADRS (Montgomery, British Journal of Psychiatry. 1979; 134:382-389)). В этом анализе эффективность и безопасность этих групп лечения анализировали только среди пациентов США и для оценки различий в эффективности и безопасности между популяцией США и общей исследуемой популяцией.
A. Клинические результаты
В случае проводимых клиницистом оценок шкалу MADRS использовали на исходном уровне; и на 2 (~ 24 часа после введения дозы), 8, 15, 22 и 28 сутки. Аналогично, шкалу общего клинического впечатления о тяжести заболевания (CGI-S) (Guy W. ECDEU Assessment Manual for Psychopharmacology (028 Clinical Global Impressions [CGI]). 1976:218-222) использовали на исходном уровне; на 4, 8, 11, 15, 22 сутки; и в конечный момент времени 4-недельной двойной слепой индукционной фазы.
В случае определяемых пациентом оценок Опросник по состоянию здоровья пациента - 9 из 9 пунктов (PHQ-9) (Spitzer, JAMA. 1999; 282(18):1737-1744) и шкалу нетрудоспособности Шихана (SDS) (Sheehan DV. The Anxiety Disease. A Leading Psychiatrist Offers New Hope for Victims of Severe Anxiety. New York, NY: Charles Scribner & Sons; 1983) использовали на исходном уровне, на 15 сутки и на 28 сутки.
B. Демография пациентов/Характеристики заболевания
Критерии включения включали взрослых в возрасте от 18 до 64 лет (включительно), которые соответствовали диагностическим критериям БДР-5, подтвержденным Кратким международным нейропсихиатрическим интервью и общей оценкой по Опроснику депрессивной симптоматики с клинической оценкой из 30 пунктов, составляющей ≥ 34 (умеренная или тяжелая депрессия).
У пациентов должны были быть ТРД и отсутствие ответа в конце фазы скрининга, определяемое как ≤ 25% улучшение общей оценки MADRS, начиная с 1 недели и до 4 недели, и общая оценка MADRS ≥ 28 на 2 неделе и 4 неделе.
Из 91 пациента США 46 получали ЭСК+АД, 44 получали АД+ПБО и один не получал дозу. Исходные демографические показатели и характеристики заболевания пациентов в целом были сходными у 2 групп лечения. Смотрите Таблицу 22. Общий средний возраст составлял 44,1 года, приблизительно две трети (61,1%) пациентов были женщинами и большинство (83,3%) пациентов были европеоидной расы. Средний возраст диагноза БДР составлял 27,5 лет, что указывает в среднем на > 15-летнюю историю депрессии. Исходные оценки MADRS, CGI-S и PHQ-9 соответствовали популяции с ТРД.
Женский
29 (63,0)
26 (59,1)
Представители белой европеоидной расы
Черные или афроамериканцы
Азиаты
Комбинированная
6 (13,0)
1 (2,2)
1 (2,2)
5 (11,4)
1 (2,3)
1 (2,3)
SNRI
SSRI
19 (41,3)
18 (40,9)
bОценка по шкале CGI-S находится в диапазоне от 1 (норма, полное отсутствие заболевания) до 7 (среди наиболее тяжело больных пациентов).
cОбщая оценка по шкале SDS находится в диапазоне 0-30, где 0=отсутствие нарушения и 30=сильное нарушение. dОбщая оценка по шкале PHQ-9 находится в диапазоне 0-27; более высокая оценка указывает на большую депрессию.
C. Эффективность
Эффективность определяли, измеряя общие оценки MADRS, оценки SDS, оценки PHQ-9 и оценки CGI-S. В случае общих оценок MADRS, оценок SDS и оценок PHQ-9 исследование эффекта лечения было основано на смешанной модели для повторных измерений (СМПИ) с изменением относительно исходного уровня в качестве переменной ответа и условиями модели с фиксированными эффектами для лечения (ЭСК+АД, АД+ПБО), сутками, классом перорального антидепрессанта (ингибитор обратного захвата серотонина и норэпинефрина [SNRI] или селективный ингибитор обратного захвата серотонина [SSRI]), посуточным лечением, а также исходным значением в качестве ковариаты. В случае оценок CGI-S исследование эффекта лечения было основано на модели ковариационного анализа (ANCOVA) с использованием данных последнего документированного значения (LOCF) с рангами изменения относительно исходного уровня в качестве переменной ответа и факторами для лечения (ЭСК+АД, АД+ПБО) и классом перорального антидепрессанта (SNRI или SSRI), а также исходным (неранжированным) значением в качестве ковариаты. В случае каждого анализа отрицательная разница свидетельствует в пользу назального впрыскивания эскетамина плюс новый пероральный АД.
Результаты показывают, что среднеквадратичные (МНК) изменения общей оценки MADS снижались в обеих группах лечения во время 4-недельной двойной слепой индукционной фазы. Смотрите Фиг. 11.
Эффект лечения был более выражен в группе ЭСК+АД приблизительно через 24 часа после введения дозы (2 сутки) и на 28 сутки, причем разница достигала статистической значимости на 28 сутки. Смотрите Таблицу 23. Средняя по МНК разница (СП) составляла -1,6 (2,15; P=0,225) приблизительно через 24 часа после введения дозы (2 сутки) и -5,5 (2,58; P=0,017) на 28 сутки.
Статистически значимую разницу в уменьшении тяжести депрессивного заболевания, определяемую по CGI-S, наблюдали между 2 группами лечения на 4 сутки (P=0,015). Разница приближалась к значимости через 4 недели после начальной дозы (P=0,070). Смотрите Таблицу 24.
Определяемая пациентом тяжесть депрессивного заболевания снижалась в обеих группах лечения, но степень снижения была большей в группе ЭСК+АД на 28 сутки. Смотрите Фиг. 12. Средние оценки PHQ-9 на исходном уровне составляли 20,2 в группе ЭСК+АД и 20,9 в группе АД+ПБО. На 28 сутки средние общие оценки PHQ-9 составляли 8,0 и 11,7 соответственно. Средняя по МНК разница (СП) в PHQ-9 составляла -3,1 (1,52; P=0,024).
Функциональное нарушение снижалось в обеих группах лечения, но степень улучшения была большей в группе ЭСК+АД на 28 сутки. Смотрите Фиг. 13. Средние оценки SDS на исходном уровне составляли 23,4 в группе ЭСК+АД и 24,1 в группе АД+ПБО. На 28 сутки средние оценки, полученные по SDS, составляли 9,7 и 16,7, соответственно. Средняя по МНК разница (СП) в общей оценке SDS составляла -5,2 (2,13; P=0,009).
D. Безопасность
Безопасность оценивали по возникшим в ходе лечения нежелательным явлениям (НЯВЛ), серьезным НЯ, основным физиологическим показателям, психиатрическим симптомам, оцениваемым по краткой психиатрической оценочной шкале (BPRS), диссоциации, оцениваемой по применяемой клиницистами шкале диссоциативных состояний (CADSS) и готовности покинуть центр.
В целом НЯВЛ наблюдались у 91,3% пациентов в группе ЭСК+АД и у 77,3% пациентов в группе АД+ПБО. Смотрите Таблицу 25A. Летальные исходы отсутствовали. Один пациент в группе ЭСК+АД испытал СНЯ во время фазы последующего наблюдения (кровоизлияние в мозг на 98 сутки). Четыре пациента прекратили прием назального впрыскиваемого препарата (n=3 ЭСК; n=1 ПБО); ни один пациент не прекратил прием нового перорального АД.
В Таблице 4 приведены наиболее распространенные НЯВЛ (≥ 5% в каждой группе лечения). Частота возникновения НЯВЛ была сходной между пациентами США и общей исследуемой популяцией. НЯ, наблюдаемые во время исследования, были в основном легкими или умеренными по тяжести и временными по своей природе.
Согласно наблюдениям в общей популяции текущие диссоциативные симптомы и временные эффекты восприятия, определяемые по общей оценке CADSS, спонтанно разрешались во время периода наблюдения после введения дозы перед уходом из центра (в течение 60-90 минут после введения дозы). Большинство (> 90%) пациентов в каждой экспериментальной группе были готовы покинуть центр через 1,5 часа после введения дозы. Основные физиологические показатели и результаты BPRS согласовались с общей популяцией.
E. Краткие итоги
Эти результаты продемонстрировали, что ЭСК+АД обеспечивали быстрое начало действия, которое продолжалось в течение 4 недель, и в целом хорошо переносились пациентами с ТРД в США. Эти наблюдения согласуются с данными общей исследуемой популяции, что свидетельствует о том, что популяция США не имела значимой разницы в эффективности. На 3 уровне STAR-D (т.е. пациенты с БДР, которым не помогло лечение 1 уровня или 2 уровня) достижение первичного результата (оценка ≤7 по шкале Гамильтона для оценки депрессии из 17 пунктов) происходило у 8-12% пациентов за время ~6 недель. Смотрите, Rush, CNS Drugs. 2009; 23(8):627-647. Для сравнения, через 4 недели после введения начальной дозы ЭСК+АД приводили к улучшению среднего по МНК изменения общей оценки MADRS, определяемой пациентом тяжести депрессивного заболевания и функционального нарушения.
Более того, статистически значимое улучшение определяемой клиницистом тяжести депрессивного заболевания наблюдали через 24 часа после введения дозы ЭСК+АД. Улучшения оцениваемых клиницистами и пациентами показателей эффективности были отмечены в группах лечения ЭСК+АД и АД+ПБО.
ЭСК+АД по сравнению с АД+ПБО (действующий препарат сравнения) у пациентов с ТРД в США обеспечили клинически существенное, статистически значимое и быстрое снижение депрессивных симптомов. У некоторых пациентов значительное улучшение определяемой клиницистом тяжести депрессивного заболевания наблюдали уже через 24 часа после введения дозы. Улучшение среднего по МНК изменения общей оценки MADRS, определяемой пациентом тяжести депрессивного заболевания и функционального нарушения наблюдали через 4 недели после введения начальной дозы.
В целом результаты по безопасности и ответу/ремиссии у пациентов в США были сходными с результатами общей популяции.
Финальный анализ субпопуляции США - Ответ, ремиссия и безопасность
Как обсуждалось выше для общего анализа, ЭСК+АД демонстрировали статистически значимое и клинически существенное превосходство по сравнению с АД+ПБО в отношении первичного ожидаемого результата эффективности (т.е. изменения относительно исходного уровня общей оценки по шкале MADRS. Смотрите приведенную выше ссылку на Montgomery. В этом анализе ответ, ремиссию и безопасность в этих группах лечения анализировали только среди пациентов США и для оценки различий в эффективности и безопасности между популяцией США и общей исследуемой популяцией.
A. Клинические результаты
В случае проводимых клиницистом оценок шкалу MADRS использовали на исходном уровне; и на 2 (~ 24 часа после введения дозы), 8, 15, 22 и 28 сутки. Аналогично, шкалу общего клинического впечатления о тяжести заболевания (CGI-S) (Guy W. ECDEU Assessment Manual for Psychopharmacology (028 Clinical Global Impressions [CGI]). 1976:218-222) использовали на исходном уровне; на 4, 8, 11, 15, 22 сутки; и в конечный момент времени 4-недельной двойной слепой индукционной фазы.
В случае определяемых пациентом оценок Опросник по состоянию здоровья пациента - 9 из 9 пунктов (PHQ-9) (Spitzer, JAMA. 1999; 282(18):1737-1744) и шкалу нетрудоспособности Шихана (SDS) (Sheehan DV. The Anxiety Disease. A Leading Psychiatrist Offers New Hope for Victims of Severe Anxiety. New York, NY: Charles Scribner & Sons; 1983) использовали на исходном уровне, на 15 сутки и на 28 сутки.
B. Демография пациентов/Характеристики заболевания
Критерии включения включали взрослых в возрасте от 18 до 64 лет (включительно), которые соответствовали диагностическим критериям БДР-5, подтвержденным Кратким международным нейропсихиатрическим интервью и общей оценкой по Опроснику депрессивной симптоматики с клинической оценкой из 30 пунктов, составляющей ≥ 34 (умеренная или тяжелая депрессия).
У пациентов должны были быть ТРД и отсутствие ответа в конце фазы скрининга, определяемое как ≤ 25% улучшение общей оценки MADRS, начиная с 1 недели и до 4 недели, и общая оценка MADRS ≥ 28 на 2 неделе и 4 неделе.
Из 91 пациента США 46 получали ЭСК+АД, 44 получали АД+ПБО и один не получал дозу. Исходные демографические показатели и характеристики заболевания пациентов в целом были сходными у 2 групп лечения. Смотрите Таблицу 22. Общий средний возраст составлял 44,1 года, приблизительно две трети (61,1%) пациентов были женщинами и большинство (83,3%) пациентов были европеоидной расы. Средний возраст диагноза БДР составлял 27,5 лет, что указывает в среднем на > 15-летнюю историю депрессии. Исходные оценки MADRS, CGI-S и PHQ-9 соответствовали популяции с ТРД.
C. Эффективность
Эффективность оценивали путем определения ответа, ремиссии и изменения клинически оцениваемой тяжести симптомов. Пациента считали демонстрирующим ответ при наличии снижения ≥ 50% исходной оценки MADRS. Пациента классифицирован как находящегося «в состоянии ремиссии», если определенная клиницистом оценка MADRS составляла ≤ 12, а определенная пациентом оценка PHQ-9 составляла < 5. И наконец, считали, что у пациента наблюдалось изменение в клинически оцениваемой тяжести симптомов при наличии снижения CGI-S на ≥ 1 балл и снижения CGI-S на ≥ 2 балла.
Приблизительно через 24 часа после введения дозы (2 сутки) 11/43 (25,6%) пациентов в группе ЭСК+АД и 9/40 (22,5%) пациентов в группе АД+ПБО достигали ответа. Ответы на 28 сутки наблюдали у 26/40 (65,0%) пациентов в группе ЭСК+АД по сравнению с 15/38 (39,5%) в группе АД+ПБО. Смотрите Фиг. 14. Аналогично, приблизительно через 24 часа после введения дозы (2 сутки) 6/43 (14,0%) пациентов в группе ЭСК+АД и 4/40 (10,0%) пациентов в группе АД+ПБО достигали клинически определяемой ремиссии.
Частота клинически определяемой ремиссии на 28 сутки составляла 18/40 (45,0%) пациентов в группе ЭСК+АД и 9/38 (23,7%) пациентов в группе АД+ПБО. Смотрите Фиг. 15.
Распределение частот категорий тяжести PHQ-9 на 15 сутки и 28 сутки приведено на Фиг. 16. На 15 сутки процент пациентов в состоянии ремиссии (т.е. с оценкой < 5) составлял 11,4% в группе ЭСК+АД и 19,5% в группе АД+ПБО. Через 4 недели после введения начальной дозы процент пациентов в состоянии ремиссии (т.е. с оценкой < 5) составлял 23,8% в группе ЭСК+АД и 18,4% в группе АД+ПБО. Кроме того, через 4 недели после введения начальной дозы процент пациентов с тяжелой депрессией (т.е. с оценкой 20-27) был более чем в 10 раз выше в группе АД+ПБО (26,3%), чем в группе ЭСК+АД (2,4%).
Кроме того, на 4 сутки снижение в CGI-S на ≥ 1 балл наблюдалось у практически в два раза большего числа пациентов в группе ЭСК+АД по сравнению с группой ПБО+АД (47,6 и 26,3%); На 28 сутки этот процент составил 77,5 и 63,9%, соответственно. Смотрите Фиг. 17. Процент пациентов с ≥ 2-балльным снижением в CGI-S был практически в 2 раза больше в группе ЭСК+АД по сравнению с группой ПБО+АД (14,3 и 7,9%) на 4 сутки; на 28 сутки этот процент составил 52,5 и 44,4%, соответственно.
D. Безопасность
Безопасность оценивали по возникшим в ходе лечения нежелательным явлениям (НЯВЛ), серьезным НЯ, основным физиологическим показателям, психиатрическим симптомам, оцениваемым по краткой психиатрической оценочной шкале (BPRS), диссоциации, оцениваемой по применяемой клиницистами шкале диссоциативных состояний (CADSS) и готовности покинуть центр. Смотрите Таблицу 25B, в которой представлена глобальная клиническая оценка готовности покинуть центр, которую оценивали на основании общего клинического состояния (включая седацию, изменения восприятия, кровяное давление и другие нежелательные явления).
В целом НЯВЛ наблюдались у 91,3% пациентов в группе ЭСК+АД и у 77,3% пациентов в группе АД+ПБО. Смотрите Таблицу 23. Летальные исходы отсутствовали. Один пациент в группе ЭСК+АД испытал СНЯ во время фазы последующего наблюдения (кровоизлияние в мозг на 98 сутки). Четыре пациента прекратили прием назального впрыскиваемого препарата (n=3 ЭСК; (n =1 ПБО), но ни один пациент не прекратил прием нового перорального АД.
В Таблице 23 приведены наиболее распространенные НЯВЛ (≥ 5% в каждой группе лечения). Частота возникновения НЯВЛ была сходной между пациентами США и общей исследуемой популяцией. НЯ, наблюдаемые во время исследования, были в основном легкими или умеренными по тяжести и временными по своей природе.
Согласно наблюдениям в общей популяции текущие диссоциативные симптомы и временные эффекты восприятия, определяемые по общей оценке CADSS, спонтанно разрешались во время периода наблюдения после введения дозы перед уходом из центра (в течение 60-90 минут после введения дозы). Большинство (>90%) пациентов в каждой экспериментальной группе были готовы покинуть центр через 1,5 часа после введения дозы. Основные физиологические показатели и результаты BPRS согласовались с общей популяцией.
E. Заключение
Эти результаты продемонстрировали, что ЭСК+АД в сравнении с АД+ПБО обеспечивали быстрое начало действия, которое продолжалось в течение 4 недель, и в целом хорошо переносились пациентами с ТРД в США. Эти результаты также показали, что ЭСК+АД демонстрировали клинически существенные улучшения в ответе и ремиссии в отношении депрессивных симптомов и имели благоприятный профиль безопасности у пациентов с ТРД в США. В частности, ЭСК+АД продемонстрировали улучшение определяемых клиницистами (CGI-S) и определяемых пациентами (PHQ-9) оценок. Снова, эти наблюдения согласуются с данными общей исследуемой популяции, что свидетельствует о том, что популяция США не имела значимой разницы в эффективности.
Однако на 3 уровне STAR-D, который включал пациентов с БДР, которым не помогло лечение 1 уровня или 2 уровня, общая частота острого ответа (на основании Краткого описания депрессивных симптомов (самоотчет), которое использовали при каждом клиническом визите острого лечения) составляла 16,8%, а общая частота острой ремиссии составляла 13,7%. Смотрите Rush, American Journal of Psychiatry. 2006; 163(11):1905-1917 и Howland RH. Journal of Psychosocial Nursing and Mental Health Services. 2008; 46(10):21-24. Для сравнения, частоты ответа и ремиссии, наблюдаемые в группе ЭСК+АД через 4 недели после введения начальной дозы, были значительно выше (45 и 65,0%, соответственно, по оценке MADRS).
Пример 2.
Эффективность интраназального эскетамина для лечения терапевтически резистентной депрессии (ТРД) у пожилых пациентов, клиническое исследование фазы 3
Способность эскетамина лечить терапевтически рефрактерную или терапевтически резистентную депрессию (ТРД) оценивали с помощью описанного ниже клинического исследования, которое проводили для оценки эффективности, безопасности и переносимости вводимого в гибких дозах интраназального эскетамина и нового назначенного перорального антидепрессанта у пожилых субъектов с ТРД. Исследование служило в качестве ключевого краткосрочного исследования эффективности и безопасности фазы 3 для обеспечения выполнения требований регуляторных органов для регистрации интраназального эскетамина для лечения ТРД. Схема дизайна исследования представлена на Фиг. 18.
Предположение для данного исследования состояло в том, что у пожилых субъектов с ТРД перевод с неудачного лечения антидепрессантами на интраназальный эскетамин плюс новый назначенный пероральный антидепрессант имел бы преимущество перед переводом на лечение новым назначенным пероральным антидепрессантом (действующий препарат сравнения) плюс интраназальное плацебо для уменьшения депрессивных симптомов.
Первичной целью данного исследования была оценка эффективности перевода пожилых субъектов с терапевтически резистентной депрессией (ТРД) с предыдущего лечения антидепрессантами (на которое они не отвечали) на вводимый в гибких дозах интраназальный эскетамин (28 мг, 56 мг или 84 мг) плюс новый назначенный пероральный антидепрессант по сравнению с переводом на новый назначенный пероральный антидепрессант плюс интраназальное плацебо в отношении уменьшения депрессивных симптомов, оцениваемого по изменению относительно исходного уровня общей оценки по шкале Монтгомери-Асберг для оценки депрессии (MADRS) от 1 суток (до рандомизации) до конца 4-недельной двойной слепой индукционной фазы.
Ключевыми вторичными целями было оценивание эффекта интраназального эскетамина плюс новый назначенный пероральный антидепрессант по сравнению с новым назначенным пероральным антидепрессантом (действующий препарат сравнения) плюс интраназальное плацебо на следующие параметры у пожилых субъектов с ТРД: (a) депрессивные симптомы (сообщаемые субъектами), (b) начало клинического ответа на 2 сутки и (с) функционирование и связанная с ним недееспособность. Дополнительные вторичные цели включали (а) частоту ответа депрессии, (b) частоту ремиссии депрессии, (с) общую тяжесть депрессивного заболевания, (d) тревожные симптомы и (e) связанное со здоровьем качество жизни и состояние здоровья.
Для изучения безопасности и переносимости интраназального эскетамина плюс новый назначенный пероральный антидепрессант по сравнению с новым назначенным пероральным антидепрессантом (действующий препарат сравнения) плюс интраназальное плацебо у пожилых субъектов с ТРД также определяли следующие параметры: (a) НЯВЛ, включая НЯ, представляющие особый интерес, (b) локальную назальную переносимость, (c) воздействие на частоту сердечных сокращений, кровяное давление, частоту дыхания и насыщение крови кислородом, (d) воздействие на активность и седацию, (e) потенциальные психозоподобные эффекты, (f) диссоциативные симптомы, (g) потенциальное воздействие на когнитивную функцию, (h) потенциальное воздействие на суицидальные идеацию/поведение, (i) потенциальные возникшие при лечении симптомы цистита и/или симптомы со стороны нижних мочевыводящих путей, (j) потенциальные синдром отмены и/или симптомы отдачи после завершения лечения интраназальным эскетамином и (k) потенциальное воздействие на ощущение запаха.
Эскетамин, растворы плацебо и пероральные антидепрессанты были предоставлены как описано в примере 1, в разделе «Информация о исследуемом препарате».
Обзор дизайна исследования
Это было рандомизированное, двойное слепое, активно контролируемое, многоцентровое исследование, которое включало 138 рандомизированных пожилых субъектов с ТРД. Данное исследование включало 3 фазы, которые кратко описаны ниже.
Фаза скрининга/проспективного наблюдения (длительностью 4 недели) была такой же, как описано в примере 1.
Двойная слепая индукционная фаза (длительность 4 недели)
Данное исследование включало 138 рандомизированных субъектов (один субъект не получал никакого исследуемого препарата (интраназального или перорального АД) и поэтому не был включен в выборки для анализа безопасности и полного анализа). Остальные 137 субъектов получали как интраназальный исследуемый препарат, так и пероральный АД и были включены в полную выборку для анализа (ПВА). Курс интраназального лечения (эскетамином или плацебо) проводили дважды в неделю. Кроме того, все субъекты на 1 сутки начали принимать новый немаскированный пероральный антидепрессант, который они принимали ежесуточно в течение всей фазы. Назначенным пероральным антидепрессантом был 1 из 4 пероральных антидепрессантов (дулоксетин, эсциталопрам, сертралин или венлафаксин с продленным высвобождением [XR]), на который у субъекта ранее не наблюдалось отсутствие ответа во время текущего депрессивного эпизода, непереносимости (в течение жизни) и который был доступен в стране-участнице.
В конце индукционной фазы субъекты, которые демонстрировали ответ (определяемый как ≥ 50% снижение общей оценки по шкале MADRS относительно исходного уровня [1 сутки до рандомизации] до конца 4-недельной двойной слепой индукционной фазы), допускались к участию в последующем исследовании ESKETINTRD3003, если они соответствовали всем остальным критериям включения в исследование (ESKETINTRD3003 представляет собой долгосрочное исследования поддержания эффективности, включающее повторяемые курсы лечения интраназальным эскетамином).
Если субъект выбыл из исследования до окончания двойной слепой индукционной фазы по причинам, отличным от отзыва согласия, то в течение 1 недели с даты прекращения проводили визит досрочного отстранения, за которым следовала фаза последующего наблюдения.
Фаза последующего наблюдения (длительностью 24 недели) была такой же, как описано в примере 1.
Исследуемая популяция
Критерии включения и исключения для включения субъектов в данное исследование были такими, как описано в примере 1 в разделе «Исследуемая популяция», за исключением того, что во время подписания формы информированного согласия (ФИС) субъект являлся мужчиной или женщиной в возрасте ≥ 65 лет включительно. Каждый потенциальный субъект удовлетворял всем критериям для включения в исследование.
Кроме того, потенциальные субъекты должны были проявлять готовность и способность придерживаться запретов и ограничений, описанных в примере 1 в разделе «Исследуемая популяция».
Распределение, рандомизация и маскировка лечения
Распределение, рандомизацию и маскировку проводили так, как описано в примере 1.
В ПВА 130/137 (94,9%) субъектов были белыми, а 85/137 (62,0%) субъектов были женщинами. Средний возраст составлял 70,0 лет, в диапазоне от 65 до 86 лет. Из 138 субъектов во всей рандомизированной выборке для анализа 122 (88,4%) завершили двойную слепую фазу, а 16 выбыли раньше, из которых 6 выбыли из-за «нежелательных явлений», 3 - из-за «прекращения участия субъектом», 4 - из-за «отсутствия эффективности», 1 - из-за «невозможности последующего наблюдения», 1 - из-за нарушения протокола и 1 - по «другим» причинам. После этого 15 субъектов перешли в фазу последующего наблюдения, 111 субъектов продолжили участие в исследовании ESKETINTRD3004, а 2 субъекта продолжили участие в 54135419TRD3008.
Информация о субъектах и лечении
Всего проводили исследование на 302 субъектах в 57 локациях в 13 странах (Бельгия, Бразилия, Болгария, Финляндия, Франция, Италия, Литва, Польша, Южная Африка, Испания, Швеция, Соединенное Королевство и США). За исключением 3 субъектов из США из-за проблем с GCP, 138 субъектов с диагнозом БДР по DSM-5 (Диагностико-статистическое руководство по психическим расстройствам, 5-ое издание) (в возрасте 65 лет или старше) рандомизировали в две группы в соотношении 1:1 (72 в группу интраназального эскетамина плюс пероральный АД и 66 в группу перорального АД плюс интраназальное плацебо).
Из 138 рандомизированных субъектов 1 субъект не получал никакого исследуемого препарата (интраназального или перорального АД) и поэтому не был включен в выборки для анализа безопасности и полного анализа. Остальные 137 субъектов получали как интраназальный исследуемый препарат, так и пероральный АД и были включены в полную выборку для анализа.
интраназальное плацебо (N=66)
Из 138 рандомизированных субъектов 122 (88,4%) завершили 28-суточную двойную слепою индукционную фазу. Результаты представлены в Таблице 27. Наиболее частой причиной прекращения участия становилось нежелательное явление. После этого 15 субъектов перешли в фазу последующего наблюдения, 111 субъектов продолжили участие в исследовании ESKETINTRD3004, а 2 субъекта продолжили участие в 54135419TRD3008 после закрытия ESKETINTRD3004.
Данные по демографическим и исходным характеристикам приведены в Таблице 28 для полной выборки для анализа. В целом группы лечения были аналогичными в отношении исходных характеристик. Большинство субъектов, включенных в исследование, составляли женщины (62,0%). Средний (СО) возраст всех субъектов составлял 70,0 (4,52) лет, в диапазоне от 65 до 86 лет. Смотрите Фиг. 26.
b Статус гипертензии классифицируется как «Да», если в истории болезни указана гипертензия.
Исходная психиатрическая история для полной выборки для анализа представлена в Таблице 29. Средняя (СО) исходная общая оценка по шкале MADRS составляла 35,2 (6,16), в диапазоне от 19 до 51. На момент скрининга 84,7% субъектов имели задокументированное отсутствие ответа на 2 или более антидепрессантов, принимаемых в течение по меньшей мере 6 недель, по MGH-ATRQ. Оставшиеся 15,3% субъектов на момент скрининга имели задокументированное отсутствие ответа на 1 антидепрессант, а отсутствие ответа на второй антидепрессант было подтверждено во время фазы скрининга/проспективного наблюдения.
жизни a, n (%)
b Число антидепрессантов с отсутствием ответа (определяемым как ≤ 25% улучшение), взятое за по меньшей мере 6 недель во время текущего эпизода, полученное из MGH-ATRQ.
СПОСОБ ПРИМЕНЕНИЯ И ДОЗЫ
Фаза скрининга/проспективного наблюдения: Фаза скрининга/проспективного наблюдения была такой же, как описана в примере 1.
Двойная слепая индукционная фаза: Двойная слепая индукционная фаза была такой же, как описана в примере 1.
Исследуемый интраназальный препарат: Исследуемый интраназальный препарат был таким же, как описан в примере 1. Смотрите Таблицу 4.
Пероральный антидепрессант: Лечение пероральным антидепрессантом было таким же, как описано в примере 1. Смотрите Таблицу 5.
Руководство по мониторингу кровяного давления в дни интраназального введения дозы: Руководство по мониторингу кровяного давления в дни интраназального введения дозы было таким же, как описано в примере 1.
Фаза последующего наблюдения: Фаза последующего наблюдения была такой же, как описана в примере 1.
Соблюдение режима лечения: Соблюдение режима лечения было таким же, как описано в примере 1.
Терапия до исследования и сопутствующая терапия: Терапия до исследования и сопутствующая терапия были такими же, как описаны в примере 1.
Препараты неотложной помощи: Применение препаратов неотложной помощи описано в примере 1.
Запрещенные препараты: Перечень запрещенных препаратов совпадает с препаратами, перечисленными в Таблице 6 примера 1.
Число доз интраназального исследуемого препарата было таким же, как описано в Таблице 7 примера 1.
Обобщенные данные по средней, режимной и конечной дозе интраназального исследуемого препарата приведены в Таблице 30. На 15 сутки двойной слепой индукционной фазы 49/65 (75,4%) субъектов принимали дозу эскетамина, составляющую 84 мг. Из 72 субъектов, проходивших лечение интраназальным эскетамином, 17 (23,6%) субъектов снижали дозу во время двойной слепой фазы.
Обобщенные данные по средней, режимной и конечной дозе перорального АД для каждого типа перорального АД приведены в Таблице 31.
Длительность приема исследуемого перорального антидепрессанта приведена в Таблице 32 и 33.
ОЦЕНКИ В РАМКАХ ИССЛЕДОВАНИЯ
Оценки в рамках исследования проводили, как описано в примере 1. План времени и событий был таким же, как описан в примере 1 в Таблицах 10 и 11. Приблизительный общий объем крови, который нужно было взять у каждого субъекта, был таким же, как описан в примере 1. Смотрите Таблицу 12.
Фаза скрининга/проспективного наблюдения
Фаза скрининга/проспективного наблюдения была такой же, как описана в примере 1. После подписания ФИС субъекты, возраст которых составлял ≥65 лет (включительно), проходили скрининг для определения пригодности для участия в исследовании.
Необязательный период постепенного уменьшения дозы антидепрессанта: Необязательный период постепенного уменьшения дозы антидепрессанта проводили так, как описано в примере 1.
Двойная слепая индукционная фаза: Двойную слепую индукционную фазу проводили так, как описано в примере 1.
Досрочное прекращение (участия): Досрочное прекращение участия пациента отслеживали в соответствии с процедурой, описанной в примере 1.
Фаза последующего наблюдения: Фазу последующего наблюдения проводили так, как описано в примере 1.
ОЦЕНКИ ЭФФЕКТИВНОСТИ
Оценки эффективности проводили, как описано в примере 1.
Первичная оценка эффективности: Первичная оценка эффективности описана в примере 1.
Ключевая вторичная оценка эффективности (выполняемая клиницистом): Ключевая вторичная оценка эффективности (выполняемая клиницистом) описана в примере 1.
Ключевая вторичная оценка эффективности (сообщаемые пациентом результаты): Ключевая вторичная оценка эффективности (сообщаемые пациентом результаты) описана в примере 1.
Первичный ожидаемый результат: Первичный ожидаемый результат эффективности описан в примере 1.
Итоги по первичному ожидаемому результату: Первичные ожидаемые результаты являются такими же, как описаны в примере 1. Смотрите Таблицу 34 и Фиг. 19.
b Отличие от плацебо представляет собой медианную несмещенную оценку, которая является взвешенной комбинацией определенных методом наименьших квадратов средних значений отличия от плацебо;
c 2-сторонний гибкий доверительный интервал;
d P-значение основано на взвешенной комбинации тестовой статистики.
Общая оценка MADRS соответствует диапазону 0-60; более высокая оценка указывает на более тяжелое состояние. Отрицательное изменение оценки указывает на улучшение.
На Фиг. 23 представлены средние значения изменений (±) относительно исходного уровня, полученные методом наименьших квадратов, для общей оценки по шкале MADRS в динамике по времени в двойной слепой фазе на основании анализа СМПИ. Эти данные показывают, что по сравнению с более молодой популяцией для достижения необходимого ответа требуется больший индукционный период. Смотрите Фиг. 2, на которой показан ответ для соответствующей более молодой популяции (-▲-).
Вторичные ожидаемые результаты: Вторичные ожидаемые результаты были такими же, как описаны в примере 1.
Анализ в подгруппах
На Фиг. 24 представлена форест-диаграмма, на которой показана разница в лечения на основании анализа СМПИ для предварительно спланированных подгрупп. Наблюдалась заметная разница в зависимости от возрастной подгруппы. Среднеарифметические изменения в течение времени для общей оценки по шкале MADRS представлены по возрастным группам на Фиг. 25 и 26. Разница в среднем по МНК изменении (СП) на основании анализа СМПИ на 28 сутки составляла -4,9 (2,04) для субъектов в возрасте 65-74 и -0,4 (5,02) для субъектов в возрасте 75 лет и старше. Однако число субъектов в более возрастной группе было низким.
Ретроспективный анализ для первичного ожидаемого результата
Как указано в SAP, взвешенный комбинированный тест был основным анализом для первичного ожидаемого результата эффективности, поскольку проводили промежуточный анализ для повторной оценки размера выборки. Проводили ретроспективный анализ, используя невзвешенный анализ СМПИ (в целом не учитывая данный промежуточного анализа), а одностороннее p-значение в этом подходе составляло 0,018. Ретроспективное одностороннее p-значение составляло 0,017 при использовании невзвешенного анализа ANCOVA.
Кроме того, исследовали лечение в зависимости от стадии (до и после проведения ПА). Эффект дифференциального лечения наблюдали для стадии 1 (субъекты, включенные до проведения ПА) и стадии 2 (субъекты, включенные после проведения ПА). Средняя по МНК (СП) разница в лечении составляла -1,6 (2,62) для стадии 1 и -5,6 (2,63) для стадии 2. Смотрите Фиг. 27 (стадия 1) и Фиг. 28 (стадия 2) для средних по МНК значений изменения с течением времени для каждой группы лечения.
Частота ответа и ремиссии на основании общей оценки по шкале MADRS
Частоты ответа (≥ 50% улучшение относительно исходного уровня общей оценки по шкале MADRS) и ремиссии (общая оценка по шкале MADRS ≤ 12) представлены в Таблице 35.
Частота ответа (≥ 50% улучшение относительно исходного уровня) на 28 сутки на основании общей оценки MADRS составляла 17/63 (27,0%) и 8/60 (13,3%) для групп, получавших интраназальный эскетамин+пероральный АД и пероральный АД+интраназальное плацебо, соответственно. Частота ремиссии (общая оценка по шкале MADRS ≤ 12) на 28 сутки составляла 11/63 (17,5%) и 4/60 (6,7%) для групп, получавших интраназальный эскетамин+пероральный АД и пероральный АД+интраназальное плацебо, соответственно.
Оценки безопасности: Оценки безопасности проводили, как описано в примере 1.
Нежелательные явления: Нежелательные явления отслеживали, как описано в примере 1.
Клинические лабораторные тесты: Клинические лабораторные тесты проводили, как описано в примере 1.
ЗАВЕРШЕНИЕ/ПРЕКРАЩЕНИЕ УЧАСТИЯ В ИССЛЕДОВАНИИ ПАЦИЕНТОМ
Завершение участия
Считалось, что субъект завершил участие в двойной слепой индукционной фазе исследования, если он или она завершили оценивание по шкале MADRS в конце 4-недельной двойной слепой индукционной фазы (т.е. MADRS на 28 сутки), как описано в примере 1.
Прекращение участия в исследовании: Субъект выбывал из исследования по любой из девяти причин, приведенных в примере 1.
Анализ безопасности: Данные о безопасности анализировали для двойной слепой индукционной фазы, используя выборку для анализа безопасности.
Нежелательные явления: Дословные термины, используемые в eCRF исследователями для определения нежелательных явлений, были закодированы с помощью MedDRA, как описано в примере 1.
Клинические лабораторные тесты: Клинические лабораторные тесты проводили, как описано в примере 1.
ЭКГ: Эффекты на сердечно-сосудистые показатели оценивали с помощью описательной статистики и сводных таблиц частот. Эти таблицы включают наблюдаемые значения и изменения относительно исходных значений. Данные электрокардиограмм обобщали с помощью параметра ЭКГ, как описано в примере 1.
Основные физиологические показатели: Основные физиологические показатели получали, как описано в примере 1.
Осмотр полости носа: Осмотр полости носа проводили, как описано в примере 1.
Опросник по назальным симптомам: Оценку из опросника по назальным симптомам обобщали описательным образом для каждого запланированного момента времени для каждой группы лечения, как описано в примере 1.
C-SSRS: Связанные с суицидом мысли и поведение на основании C-SSRS обобщали по группам лечения в таблицах частоты возникновения и сдвигов, как описано в примере 1. Были определены отдельные ожидаемые результаты для суицидальной идеации и суицидального поведения и обобщены по группе лечения. Отсутствующие оценки не учитывали.
CADSS, BPRS+ и MOAA/S: Описательные статистические данные для каждой оценки и изменений относительно момента до введения дозы обобщали в каждый запланированный момент времени, как описано в примере 1.
Глобальная клиническая оценка готовности к выписке, PWC-20, BPIC-SS, UPSIT и тест на определение порога обонятельных ощущений
Описательную статистику по каждой оценке и изменениям и/или процентным изменениям относительно исходного уровня обобщали в каждый запланированный момент времени, как описано в примере 1.
Тестирование когнитивной функции: Описательную статистику по оценкам и изменениям относительно исходного уровня по доменам когнитивных функций обобщали в каждый запланированный момент времени.
Определения и классификация нежелательных явлений: Определения и классификацию нежелательных явлений осуществляли, как описано в примере 1.
Ситуации, требующие специальной отчетности: Ситуации, требующие специальной отчетности, обсуждались в примере 1.
Процедуры: Все нежелательные явления
Все нежелательные явления и ситуации, требующие специальной отчетности, серьезные или несерьезные, записывали с момента получения подписанного и датированного ФИС и до завершения последней связанной с исследованием процедуры субъекта (которая может включать связь для осуществления последующего наблюдения в вопросах безопасности), как описано в примере 1.
Серьезные нежелательные явления: Исследования серьезных нежелательных явлений проводили, как описано в примере 1.
Беременность: Беременность оценивали, как описано в примере 1.
ОБОБЩЕННАЯ ИНФОРМАЦИЯ ПО ВСЕМ НЕЖЕЛАТЕЛЬНЫМ ЯВЛЕНИЯМ
Обобщенная информация по всем возникшим в ходе лечения нежелательным явлениям (НЯВЛ) во время двойной слепой фазы представлена в Таблице 36. В целом, у 70,8% субъектов в группе, получавшей эскетамин+пероральный АД, и у 60,0% субъектов в группе, получавшей пероральный АД+плацебо, наблюдалось по меньшей мере одно НЯВЛ во время двойной слепой фазы.
b Нежелательное явление, которое началось во время двойной слепой индукционной фазы и привело к прекращению приема во время фазы последующего наблюдения, считается возникшим в ходе лечения во время двойной слепой индукционной фазы.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений. Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Нежелательные явления, возникшие в ходе лечения во время двойной слепой фазы (> 5% субъектов в каждой группе лечения), обобщены по группе лечения для выборки для анализа безопасности и приведены в Таблице 37. Наиболее распространенными (> 10%) НЯВЛ в группе, получавшей эскетамин+пероральный АД во время двойной слепой фазы, были головокружение (20,8%), тошнота (18,1%), головная боль (12,5%), утомляемость (12,5%), повышенное кровяное давление (12,5%), вертиго (11,1%) и диссоциация (11,1%). Наиболее распространенным НЯВЛ в группе, получавшей пероральный АД+плацебо, были тревожность (7,7%), головокружение (7,7%) и утомляемость (7,7%). Летальные исходы отсутствовали.
Нежелательные явления, приводящие к прекращению приема исследуемого препарата
6 субъектов (4 субъектов в группе, получавшей эскетамин+пероральный АД, и 2 субъекта в группе, получавшей пероральный АД+плацебо), прекратили прием интраназального исследуемого препарата во время двойной слепой индукционной фазы из-за нежелательных явлений, возникших в ходе лечения. Смотрите Таблицу 19. 2 субъекта (1 субъектов в группе, получавшей эскетамин+пероральный АД, и 1 субъект в группе, получавшей пероральный АД+плацебо), прекратили участие во время двойной слепой индукционной фазы из-за НЯВЛ. Смотрите Таблицы 38 и 39.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений. Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Серьезные нежелательные явления
Пять субъектов испытали серьезные возникшие в ходе лечения нежелательные явления во время двойной слепой фазы. Один субъект в группе, получавшей эскетамин+пероральный АД, испытал тревожное расстройство, которое посчитали возможно связанным как с интраназальным эскетамином, так и с пероральным АД. Один субъект в группе, получавшей эскетамин+пероральный АД, испытал повышение кровяного давления, которое посчитали вероятно связанным с интраназальным эскетамином и не связанным с пероральным АД. Кроме того, один субъект в группе, получавшей эскетамин+пероральный АД, получил перелом шейки бедра, который посчитали не связанным как с интраназальным эскетамином, так и с пероральным АД. Один субъект в группе, получавшей пероральный АД+плацебо, испытывал чувство отчаяния и нарушение походки. Первое явление посчитали возможно связанным с интраназальным плацебо и не связанным с пероральным АД. Второе явление посчитали возможно связанным с интраназальным плацебо и с большой вероятностью связанным с пероральным АД. Один субъект в группе, получавшей пероральный АД+плацебо, испытывал головокружение, которое посчитали маловероятно связанным как с интраназальным плацебо, так и с пероральным АД.
Кровяное давление
Временное кровяное давление увеличивается до пика для группы, получавшей эскетамин+пероральный АД, приблизительно через 40 минут после введения дозы и возвращается к нормальному диапазону через 90 минут. Максимальное среднее повышение (по всем дням введения дозы) систолического КД составило 16,0 мм рт.ст. в группе, получавшей эскетамин+пероральный АД, и 11,1 мм рт.ст. в группе, получавшей пероральный АД+плацебо. Максимальное среднее повышение (по всем дням введения дозы) диастолического КД составило 9,5 мм рт.ст. в группе, получавшей эскетамин+пероральный АД, и 6,8 мм рт.ст. в группе, получавшей пероральный АД+плацебо. На Фиг. 20 и 21 представлены средние значения кровяного давления в динамике по времени по группе лечения в двойной слепой фазе.
Применяемая клиницистами шкала диссоциативных симптомов (CADSS)
Оценку по применяемой клиницистами шкале диссоциативных состояний (CADSS) определяли перед началом введения каждой дозы, через 40 минут и 1,5 часа после введения дозы. Шкалу CADSS используют для оценки возникших в ходе лечения диссоциативных симптомов и изменений восприятия, а общая оценка находится в диапазоне от 0 до 92, причем более высокая оценка представляет более тяжелое состояние.
Диссоциативные симптомы и изменения восприятия, оцененные по шкале CADSS, позволяют предположить, что эти симптомы появились вскоре после начала введения дозы и они разрешились через 1,5 часа после введения дозы. Смотрите Фиг. 22. Доля субъектов с седацией (оценка MOAA/S после введения дозы ≤ 3) составляла < 4% для группы, получавшей эскетамин, в каждый день введения дозы.
Модифицированная оценка наблюдателем активности/седации (MOAA/S)
Модифицированную оценку наблюдателем активности/седации (MOAA/S) использовали для определения возникающей в ходе лечения седации с корреляцией с уровнями седации, определенными континуумом Американского общества анестезиологов (ASA). Оценки MOAA/S находились в диапазоне от 0 (отсутствие ответа на болевой стимул; соответствует континууму ASA для общей анестезии) до 5 (легко отвечает на имя, произнесенное нормальным тоном [бодрствование]; соответствует континууму ASA для минимальной седации). Доля субъектов, имевших оценку MOAA/S ≤ 3 после введения дозы в день введения дозы, представлена в Таблице 40. Доля субъектов с седацией составляла < 4% для группы, получавшей эскетамин, в каждый день введения дозы.
ФАРМАКОКИНЕТИКА
Образцы венозной крови объемом приблизительно 2 мл собирали для измерения концентраций эскетамина, норэскетамина и других метаболитов в плазме (в случае обоснованности) в моменты времени, указанные в плане времени и событий, как описано в примере 1. Образцы плазмы анализировали, как описано в примере 1.
Фармакокинетические параметры: Данные зависимости плазменной концентрации эскетамина (и норэскетамина в случае обоснованности) от времени анализировали, как описано в примере 1.
Оценки фармакокинетики/фармакодинамики: Взаимосвязь между общей оценкой по шкале MADRS (и, возможно, выбранными нежелательными явлениями в качестве дополнительных ФД-параметров) и ФК-показателями эскетамина оценивали, как описано в примере 1.
Оценки биомаркеров, фармакогеномики (ДНК) и экспрессии (РНК)
Во время исследования собирали кровь для оценки биомаркеров в моменты времени, указанные в плане времени и событий, как описано в примере 1.
Биомаркеры (белки, метаболиты и рибонуклеиновые кислоты [РНК]), связанные с (но без ограничения этим) активностью иммунной системы, активацией гипоталамо-гипофизарно-надпочечниковой (ГГН) оси, нейротрофическими факторами и метаболическими факторами, в крови оценивали, как описано в примере 1.
Образцы крови для анализа ДНК собирали в моменты времени, указанные в плане времени и событий, для оценки генетической и эпигенетической вариации в генах в случае путей, связанных с депрессией (например, ось ГГН, воспаление, факторы роста, переносчики моноаминов, ионные каналы и суточный ритм), как описано в примере 1.
Использование медицинских ресурсов: во время фазы последующего наблюдения исследования собирали данные об использовании медицинских ресурсов, связанных с обращением за медицинской помощью, как описано в примере 1.
Фармакокинетический анализ: Фармакокинетический анализ проводили, как описано в примере 1.
Фармакокинетический/фармакодинамический анализ: Взаимосвязь между общей оценкой по шкале MADRS (и, возможно, выбранными нежелательными явлениями в качестве дополнительных ФД-параметров) и ФК-показателями эскетамина оценивали, как описано в примере 1.
Анализ биомаркеров и фармакогеномики: Анализ биомаркеров и фармакогеномики проводили, как описано в примере 1.
СТАТИСТИЧЕСКИЕ МЕТОДЫ, ИСПОЛЬЗУЕМЫЕ ПРИ АНАЛИЗЕ
Общее описание статистических методов, используемых для анализа данных по эффективности и безопасности, приведено в примере 1.
Промежуточный анализ для повторной оценки размера выборки или приостановки из-за нецелесообразности: промежуточный анализ для повторной оценки размера выборки или приостановки из-за нецелесообразности проводили, как описано в примере 1.
Анализ эффективности: Анализ эффективности проводили, как описано в примере 1.
Анализ субпопуляции США - Клиническая эффективность и безопасность
Хотя анализ первичной эффективности с использованием взвешенного комбинированного теста не был статистически значимым, разница в лечении была клинически существенной для изменения ЭСК+АД по сравнению с АД+ПБО в отношении улучшения депрессивных симптомов, оцениваемого по изменению общей оценки MADRS через 28 суток у пожилых субъектов с ТРД. На основании анализа СМПИ медианная несмещенная оценка разницы разности между ЭСК+АД и АД+ПБО составляла -3,6 (95% ДИ: -7,20, 0,07). Разница была сходной с наблюдаемой в исследованиях, в которых оценивали пероральные SSRI/SNRI АД в сравнении с плацебо у взрослых с депрессией.
Большее численное улучшение ответа при депрессии (приблизительно в 2 раза большее) и частоты ремиссии (приблизительно в 3 раза большее) для ЭСК+АД по сравнению с АД+ПБО на 28 сутки позволяет предположить наличие действительной клинической пользы от лечения назальным впрыскиванием эскетамина плюс пероральный антидепрессант у пожилых субъектов с ТРД.
Также наблюдалась клинически существенная разница в лечении в отношении снижения общей тяжести депрессивного заболевания на основании оценки CGI-S для ЭСК+АД по сравнению с АД+ПБО.
A. Клинические результаты
В случае определяемых клиницистом оценок шкалу MADRS использовали на исходном уровне и на 8, 15, 22 и 28 сутки. Оценки MADRS были получены дистанционно в телефонном режиме независимыми экспертами, от которых были скрыты данные по ответу субъекта на лечение. Аналогично, шкалу Общего клинического впечатления о тяжести заболевания (CGI-S) использовали на исходном уровне; на 4, 8, 11, 15, 18, 22, 25 сутки; и в конечный момент времени 4-недельной двойной слепой индукционной фазы.
В случае определяемых пациентами оценок Опросник по соблюдению пациентом режима лечения - 9 из 9 пунктов (PHQ-9) и шкалу нетрудоспособности Шихана (SDS) использовали на исходном уровне, на 15 сутки и на 28 сутки. Хотя оценки PHQ-9 и SDS были удалены при изменении исследования, журналы цента не были модифицированы, поэтому данные были получены.
B. Демография пациентов/Характеристики заболевания
Критерии включения включали взрослых в возрасте ≥ 65 лет, которые соответствовали диагностическим критериям DSM-5 для рецидивирующего БДР без психотических характеристик или однократного эпизода БДР (с длительностью эпизода > 2 лет) и имели оценку по Опроснику по симптомам депрессии, оцениваемым клиницистом, из 30 пунктов ≥ 31.
Критерии включения также включали отсутствие ответа (≤ 25% улучшение по MADRS) на ≥ 1, но ≤ 8 АД во время текущего эпизода депрессии, принимаемых в течение по меньшей мере 6 недель в терапевтической дозе (на основании Опросника по истории лечения антидепрессантами Центральной больницы штата Массачусетс, версии для пожилых людей).
Пациенты должны переживать текущий большой депрессивный эпизод, иметь серьезные симптомы депрессии (общая оценка по шкале MADRS на 1 неделе ≥ 24) и ответ на лечение АД во время текущего депрессивного эпизода, подтвержденные с помощью независимой квалификационной оценки.
Из 70 пациентов США в возрасте ≥ 65 лет 34 получали ЭСК+АД и 36 получали АД+ПБО. Исходные демографические показатели и характеристики заболевания пациентов в целом были сходными у 2 групп лечения (Таблица 41). Общий средний возраст составлял 70,0 лет, 57,1% были женщинами и большинство пациентов были белыми (98,6%). Средний возраст при диагнозе БДР составлял 42,5 лет, что указывает в среднем на > 27-летнюю историю депрессии в этой популяции. Исходные оценки MADRS, CGI-S и PHQ-9 соответствовали взрослой популяции с ТРД.
Мужской
Женский
19 (55,9)
21 (58,3)
Белые
Комбинированная
1 (2,9)
0
SNRI
SSRI
18 (52,9)
19 (52,8)
C. Эффективность
Эффективность определяли, измеряя общие оценки MADRS, оценки SDS, оценки PHQ-9 и оценки CGI-S. Первичным ожидаемым результатом эффективности, сравниваемым между группами лечения, было изменение общей оценки по шкале MADRS относительно исходного уровня на 28 сутки. Другими показателями эффективности были изменения оценок CGI-S, общих оценок SDS и общих оценок PHQ-9, которые определяли изменения в общем клиническом состоянии и функционировании. Анализ эффективности проводили при одностороннем уровне значимости 0,025.
В случае общих оценок MADRS, PHQ-9 и SDS исследование эффекта лечения было основано на смешанной модели для повторных измерений (СМПИ), на данных наблюдаемых случаев с изменением относительно исходного уровня в качестве переменной ответа и условиями модели с фиксированными эффектами для лечения (ЭСК+АД, АД+ПБО), сутками, классом перорального АД (SNRI или SSRI) и посуточным лечением, а также исходным значением в качестве ковариаты. В случае оценок CGI-S исследование эффекта лечения было основано на модели ковариационного анализа (ANCOVA) с использованием данных последнего документированного значения (LOCF) с рангами изменения относительно исходного уровня в качестве переменной ответа и факторами для лечения (ЭСК+АД, АД+ПБО) и классом перорального АД (SNRI или SSRI), а также исходным (неранжированным) значением в качестве ковариаты.
Результаты показывают, что среднеквадратичные (МНК) изменения общей оценки MADS снижались в обеих группах лечения во время 4-недельной двойной слепой индукционной фазы. Смотрите Фиг. 29. Статистически значимое улучшение общей оценки по шкале MADRS наблюдали при применении ЭСК+АД по сравнению с АД+ПБО на 28 сутки (средняя по МНК разница [СП]: -5,4 [2,48]; 1-стороннее P=0,016). Смотрите Таблицу 42.
В целом, ЭСК+АД по сравнению с АД+ПБО (действующий препарат сравнения) продемонстрировали клинически существенное, статистически значимое снижение депрессивных симптомов и уменьшение общей тяжести депрессивного заболевания, а также улучшение качества жизни и функционирования у пациентов в США в возрасте ≥ 65 лет с ТРД через 4 недели.
Определяемая клиницистом тяжесть депрессивного заболевания по оценке CGI-S была сходной через 4 суток после введения начальной дозы. При этом наблюдали статистически значимую разницу в уменьшении тяжести депрессивного заболевания, определяемую по CGI-S, между двумя группами лечения через 4 недели после введения начальной дозы (1-стороннее P=0,005). Смотрите Таблицу 43.
Частотное распределение тяжести заболевания на основании оценок CGI-S на исходном уровне и в конечный момент времени двойной слепой фазы приведено на Фиг. 30. На исходном уровне процент пациентов с нормальным/пограничным/легким заболеванием был сходным в группах ЭСК+АД и АД+ПБО (2,9% и 2,8%, соответственно). В конечный момент времени двойной слепой фазы процент пациентов с нормальным/пограничным/легким заболеванием был в 3,8 раза выше в группе ЭСК+АД по сравнению с группой АД+ПБО (42,4% и 11,2%, соответственно).
Определяемая пациентами тяжесть депрессивного заболевания по общим оценкам PHQ-9, и функционального нарушения по общим оценкам SDS снижалась в обеих группах лечения, но величина разницы была существенно большей в группе ЭСК+АД на 28 сутки. Смотрите Таблицу 24. В случае PHQ-9 средняя по МНК разница (СП) составляла -4,4 (1,68; 1-стороннее P=0,006). В случае SDS средняя по МНК разница (СП) составляла -7,6 [2,68; 1-стороннее P=0,004].
D. Безопасность
Оценка безопасности включала сообщения о нежелательных явлениях, результаты клинических лабораторных тестов, измерения основных физиологических показателей, физические осмотры, электрокардиограммы и осмотры полости носа. Безопасность оценивали по возникшим в ходе лечения НЯ (НЯВЛ). В целом НЯВЛ наблюдались у 64,7% пациентов из США в группе ЭСК+АД и у 58,3% пациентов в группе АД+ПБО. Смотрите Таблицу 25. Летальные исходы отсутствовали. В каждой группе наблюдали одно серьезное НЯ. Три пациента прекратили назальное впрыскивание (n=2 ЭСК [повышение систолического КД > 180; перелом шейки бедра, n=1 ПБО) и один пациент в группе АД+ПБО прекратил прием перорального АД (повышенное КД, головокружение и периферический отек). Большинство НЯВЛ были слабыми или умеренными по тяжести, и никаких новых или неожиданных сигналов безопасности не наблюдалось.
В Таблице 44 приведены наиболее распространенные (≥ 5% в каждой группе лечения) НЯВЛ. НЯВЛ, как правило, были слабыми или умеренными по тяжести и временными по своей природе. Частота возникновения НЯ у пациентов из США была сходной с наблюдаемой в общей популяции исследования. Результаты показывают, что ЭСК+АД демонстрировали статистически значимую, клинически существенную эффективность АД у пациентов в возрасте ≥ 65 лет с профилем безопасности, сходным с наблюдаемым у более молодых пациентов. Эти наблюдения сходны с глобальным анализом, который демонстрирует клинически существенную эффективность АД, и были сходны с наблюдаемыми для более молодой популяции в исследовании ЭСК фазы 3 и в исследованиях фазы 2.
Анализ субпопуляции США - Ответ, ремиссия и безопасность
Как обсуждалось выше для общего анализа, ЭСК+АД демонстрировали статистически значимое и клинически существенное превосходство по сравнению с АД+ПБО в отношении первичного ожидаемого результата эффективности (т.е. изменения относительно исходного уровня общей оценки по шкале MADRS у пожилых пациентов. Смотрите приведенную выше ссылку на Montgomery. В этом анализе ответ, ремиссию и безопасность в этих группах лечения анализировали только среди пожилых пациентов из США и для оценки различий в эффективности и безопасности между популяцией из США и общей исследуемой популяцией.
A. Клинические результаты
В случае определяемых клиницистом оценок шкалу MADRS использовали на исходном уровне и на 8, 15, 22 и 28 сутки. Аналогичным образом шкалу Общего клинического впечатления о тяжести заболевания (CGI-S) использовали на исходном уровне и на 4, 8, 11, 15, 18, 22, 25 и 28 сутки.
В случае определяемой пациентами оценки Опросник по соблюдению пациентом режима лечения - 9 из 9 пунктов (PHQ-9) использовали на исходном уровне, на 15 сутки и на 28 сутки.
B. Демография пациентов/Характеристики заболевания
Критерии включения включали взрослых в возрасте ≥ 65 лет, которые соответствовали диагностическим критериям Диагностико-статистического руководства по психическим расстройствам, пятое издание (DSM-5) для рецидивирующего БДР без психотических характеристик или однократного эпизода БДР (с длительностью эпизода > 2 лет). Отсутствие ответа (≤ 25% улучшение по шкале Монтгомери - Асберг для оценки депрессии [MADRS]) на ≥ 1, но ≤ 8 АД во время текущего эпизода депрессии, принимаемых в течение по меньшей мере 6 недель в терапевтической дозе (на основании Опросника по истории лечения антидепрессантами Центральной больницы штата Массачусетс, версии для пожилых людей). Оценка по опроснику по симптомам депрессии, оцениваемым клиницистом, из 30 пунктов ≥ 31. Текущий большой депрессивный эпизод, серьезные симптомы депрессии (общая оценка по шкале MADRS на 1 неделе ≥ 24) и ответ на лечение АД во время текущего депрессивного эпизода, подтвержденные с помощью независимой квалификационной оценки.
Из 70 пациентов из США в возрасте ≥ 65 лет 34 получали ЭСК+АД и 36 получали АД+ПБО. Исходные демографические показатели и характеристики заболевания пациентов в целом были сходными у 2 групп лечения. Смотрите Таблицу 45. Общий средний возраст составлял 70,0 лет, 57,1% были женщинами и большинство пациентов были белыми (98,6%). Средний возраст при диагнозе БДР составлял 42,5 лет, что указывает в среднем на > 27-летнюю историю депрессии в этой популяции. Исходные оценки MADRS, CGI-S и PHQ-9 соответствовали взрослой популяции с ТРД.
Мужской
Женский
19 (55,9)
21 (58,3)
Белые
Комбинированная
1 (2,9)
0
SNRI
SSRI
18 (52,9)
19 (52,8)
C. Эффективность
Эффективность определяли, измеряя общие оценки MADRS, оценки SDS, оценки PHQ-9 и оценки CGI-S. Пациента считали демонстрирующим ответ при наличии снижения ≥ 50% исходной оценки MADRS. Пациента классифицировали как находящегося в «состоянии ремиссии», если определенная клиницистом: оценка MADRS составляла ≤ 12, а определенная пациентом оценка PHQ-9 составляла < 5. Считали, что у пациента наблюдалось изменение в клинически оцениваемой тяжести симптомов при наличии клинически существенного ответа с ≥ 1-балльным снижением в CGI-S и клинически значимого ответа с ≥ 2-балльным снижением по CGI-S.
Приблизительно через восемь дней после введения начальной дозы частота ответа на основании MADRS составляла 6,3% (2/32) и 0% (0/35) в ЭСК+АД и АД+ПБО, соответственно.
Через двадцать восемь дней после введения начальной дозы частота ответа на основании MADRS была практически в 2 раза выше у пациентов, которых лечили ЭСК+АД, по сравнению с пациентами, которых лечили АД+ПБО (8/30 [26,7%] и 5/34 [14,7%]). Смотрите Фиг. 31. Через двадцать восемь дней после введения начальной дозы частота ремиссии на основании MADRS была приблизительно в 5 раз больше у пациентов, которых лечили ЭСК+АД, по сравнению с пациентами, которых лечили АД+ПБО (5/30 [16,7%] и 1/34 [2,9%]). Смотрите Фиг. 32. На 28 сутки после введения начальной дозы оцененная пациентами ремиссия была почти в 2,5 раза выше в группе ЭСК+АД по сравнению с группой АД+ПБО (22,6% [7/31] и 9,4% [3/32]). Смотрите Фиг. 33.
На 15 сутки после введения начальной дозы оцененная пациентами частота ремиссии на основании PHQ-9 была сходной между группами лечения (8,0% [2/25] и 8,7% [2/23]).
Через 4 недели после введения начальной дозы клинически существенный ответ был почти в 2 раза выше, а клинически значимый ответ был почти в 4 раза выше в группе ЭСК+АД по сравнению с группой АД+ПБО (63,3% и 29,4% и 43,2% и 11,8%, соответственно). Смотрите Фиг. 34 и 35.
D. Безопасность
Безопасность оценивали по возникшим в ходе лечения НЯ (НЯВЛ). В целом НЯВЛ наблюдались у 64,7% пациентов из США в группе ЭСК+АД и у 58,3% пациентов в группе АД+ПБО. Смотрите Таблицу 27. Летальные исходы отсутствовали; в каждой группе наблюдали одно серьезное НЯ. Три пациента прекратили назальное впрыскивание (n=2 ЭСК [повышение систолического КД > 180; перелом шейки бедра, n=1 ПБО) и один пациент в группе АД+ПБО прекратил прием перорального АД (повышенное КД, головокружение и периферический отек).
В Таблице 46 приведены наиболее распространенные (≥ 5% в каждой группе лечения) НЯВЛ. НЯВЛ, как правило, были слабыми или умеренными по тяжести и временными по своей природе. Частота возникновения НЯВЛ у пациентов из США была сходной с наблюдаемой в общей популяции исследования.
E. Заключение
Эти результаты продемонстрировали, что в этой субпопуляции пациентов с ТРД из США в возрасте ≥ 65 лет из более крупного, многонационального исследования, почти в два раза больше пациентов достигли ответа (т.е. ≥ 50% снижения исходной оценки MADRS) при лечении ЭСК+АД по сравнению теми, кого лечили АД+ПБО. Кроме того, частота ремиссии (т.е. оценка MADRS ≤ 12) была приблизительно в 5 раз выше у пациентов, которых лечили ЭСК+АД, по сравнению с пациентами, которых лечили АД+ПБО.
Таким образом, ЭСК+АД продемонстрировали клинически существенное и клинически значимое улучшение определяемой клиницистами (CGI-S) и определяемой пациентами (PHQ-9) ремиссии. Результаты по безопасности, ответу и ремиссии пациентов из США были сходными с результатами общей популяции исследования.
Пример 3
Это было рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое исследование. Смотрите Canuso, “Efficacy and Safety of Intranasal Esketamine for the Rapid Reduction of Symptoms of Depression and Suicidality in Patients at Imminent Risk for Suicide: Results of a Double-Blind, Randomized Placebo-Controlled Study,” Am. J. Psych., 2018, 1-11, которая включена в данный документ посредством ссылки. Были включены приблизительно 70 человек мужского и женского пола в возрасте от 19 до 64 лет с БДР с высоким риском суицида, попавшие в кабинет неотложной помощи (КНП) или другое соответствующее учреждение, и по оценкам имеющие высокий риск суицида. Большинство субъектов были женщинами, а средний возраст всех субъектов составлял приблизительно 36 лет. Средняя исходная общая оценка по шкале Монтгомери - Асберг для оценки депрессии (MADRS) составляла более 38 (что соответствует тяжелой депрессии), а средняя исходная оценка по шкале Бека для суицидальной идеации (BSS) составляла более 22. Более половины субъектов имели оценку 6 согласно Глобальной клинической оценке риска суицида на основе инструмента оценки суицидальных идеаций и поведения (SIBAT), что соответствует риску суицида, требующему госпитализации с предупреждением суицида. Схема дизайна исследования представлена на Фиг. 37.
Данное исследование состояло из скрининговой оценки, проводимой в течение 24 часов (или до 48 часов после консультации с медицинским наблюдателем, назначенным спонсором) до 1 суток введения дозы, сразу после 25-суточной двойной слепой фазы лечения (сутки 1-25) с введением дозы дважды в неделю и 56-суточной фазой последующего наблюдения (от 26 суток до 81 суток). Исследование было разработано как POC-исследование (исследование обоснованности концепции) и, следовательно, в нем использовали двусторонний уровень значимости 0,20.
Рандомизация: 68 субъектов рандомизировали в соотношении 1:1 для лечения 1 из 2 препаратов: интраназальный эскетамин 84 мг (N=36) или интраназальное плацебо (N=32). Рандомизацию стратифицировали по исследовательскому центру и по оценке врачом потребности субъекта в лечении стандартными антидепрессантами перед рандомизацией на 1 сутки (т.е. монотерапия антидепрессантом или антидепрессант плюс усиливающая терапия). Кроме того, всем субъектам была оказана активная клиническая помощь, включающая госпитализацию и инициацию или оптимизацию приема стандартного антидепрессанта (определяемого лечащим врачом на основании клинического заключения и практических руководств).
Первичная выборка для анализа эффективности: Первичный анализ эффективности был основан на выборке для анализа «по намеченному лечению» (ПНЛ), которая по определению включала всех рандомизированных субъектов, которые получают по меньшей мере 1 дозу исследуемого препарата во время двойной слепой фазы и имеют общую оценку MADRS как на исходном уровне, так и на 1 сутки через 4 часа после введения дозы.
Первичные параметры эффективности/первичный момент времени: Изменение общей оценки MADRS от исходного уровня (1 сутки, до введения дозы) до 1 суток, через 4 часа после введения дозы. Шкала MADRS состоит из 10 пунктов, которые охватывают все основные депрессивные симптомы, при этом каждый пункт оценивается от 0 (отсутствует или в пределах нормы) до 6 (тяжелое или постоянное присутствие симптома). Более высокая оценка представляет более тяжелое состояние.
Вторичные параметры эффективности:
a. Изменение общей оценки MADRS от исходного уровня до 2 суток и 25 суток
b. Изменения для пункта MADRS, касающегося суицида, от исходного уровня до 1 суток, через 4 часа после введения дозы, 2 суток и 25 суток
c. Изменения от исходного уровня до 1 суток, через 4 часа после введения дозы, 2 суток и 25 суток для глобальной клинической оценки риска суицида на основе инструмента оценки суицидальных идеации и поведения (SIBAT)
d. Устойчивый ответ (начало клинического ответа), определяемый как по меньшей мере 50% снижение общей оценки MADRS от исходного уровня с началом на 1 сутки, которое сохраняется до конца двойной слепой фазы (сутки 25)
e. Изменения от исходного уровня до 1 суток, 4 часа после введения дозы, 2 суток и 25 суток по шкале Бека для суицидальной идеации (BSS)
f. Изменения от исходного уровня до 1 суток, 4 часа после введения дозы, и 25 суток по шкале Бека для безнадежности.
Ожидаемый размер эффекта и планируемый размер выборки: Размер выборки был основан на допущении, что разница в лечении составляет по меньшей мере 6 пунктов в отношении среднего изменения общей оценки MADRS от исходного уровня до 1 суток (4 часа после введения дозы) между группами, получавшими эскетамин и плацебо. Для обеих групп использовали стандартное отклонение 9. Требовалось, чтобы при использовании 2-выборочного t-критерия 32 субъекта в каждой группе обнаружили разницу в лечении в 6 пунктов с мощностью 91% при общем 1-стороннем уровне значимости 0,10 (что соответствует 2-стороннему уровню значимости 0,20). Принимая во внимание, что 8% рандомизированных субъектов выбывают до проведения измерений эффективности после исходного уровня, общее количество субъектов, необходимое для каждой группы лечения, составляет 35. Цель выбора размера выборки для этого исследования обоснованности концепции фазы 2a заключалась в повышении чувствительности к обнаружению терапевтического сигнала, с сохранением при этом небольшого размера выборки. Таким образом, мощность была установлена на высокое значение (≥ 90%; β ≤ 0,1), но частота ошибок 1 рода была указана при 1-стороннем α=0,10.
Первичная цель: Первичная цель заключается в оценке эффективности интраназального эскетамина в дозе 84 мг по сравнению с интраназальным плацебо в отношении уменьшения симптомов БДР, включая суицидальную идеацию, у субъектов, которые по оценкам имеют высокий риск суицида, оцениваемой по изменению общей оценки по шкале Монтгомери - Асберг для оценки депрессии (MADRS) от исходного уровня до 4 часов после введения дозы на 1 сутки.
Информация о субъектах и лечении:
Это было рандомизированное, двойное слепое, плацебо-контролируемое исследование, которое включало 68 рандомизированных субъектов с диагнозом БДР с высоким риском суицида без психотических характеристик, поставленным на основании клинической оценки (DSM-IV 296.22, 296.23, 296.32 или 296.33) и подтвержденным Кратким международным нейропсихиатрическим интервью (MINI). Субъекты должны иметь в настоящем суицидальную идеацию с намерением, подтвержденным ответом «Да» на вопрос B5 [Думаете о суициде (самоубийстве)?] и вопрос B9 [Намереваетесь действовать согласно мыслям о самоубийстве?], полученным в MINI; и субъекты должны иметь общую оценку по шкале MADRS ≥ 22 перед введением дозы на 1 сутки. Из-а меньшей, чем предполагалось, частоты выбывания в момент времени, соответствующий первичному ожидаемому результату (1 сутки: 4 часа после введения дозы), набор в исследование прекратили на 68 субъектах, что является достаточным количеством подходящих для оценки субъектов. Из 68 рандомизированных субъектов 2 субъекта не получали исследуемого препарата и поэтому не были включены в выборки для анализа безопасности или ПНЛ. В выборке для анализа ПНЛ 35/66 (53,0%) субъектов были белыми, а 43/66 (65,2%) субъектов были женщинами. Средний возраст составлял 35,8 лет, в диапазоне от 19 до 64 лет. Из 68 субъектов во всей рандомизированной выборке для анализа 49 (72,1%) завершили двойную слепую фазу, а 19 выбыли раньше, из которых 6 выбыли из-за нежелательных явлений, 5 - из-за отсутствия эффективности, 2 - из-за невозможности последующего наблюдения, 2 - из-за отзыва согласия и 4 - по другим причинам. После этого 49 субъектов перешли в 56-суточную фазу последующего наблюдения.
Эффективность:
Первичный ожидаемый результат эффективности
На основании модели ANCOVA, результаты по изменению общей оценки MADRS от исходного уровня до 1 суток, 4 часа после введения дозы, свидетельствовали в пользу эскетамина в дозе 84 мг с определенной методом наименьших квадратов средней разницей (СП) с плацебо -5,3 (2,10). Разница между группами лечения была статистически значимой (двустороннее p=0,015) при двустороннем уровне значимости 0,20.
Вторичные ожидаемые результаты эффективности
В Таблице 47 ниже обобщены итоги для вторичных ожидаемых результатов эффективности.
Безопасность
Наиболее распространенными (> 20%) НЯВЛ в группе, получавшей 84 мг эскетамина во время двойной слепой фазы, были тошнота (37,1%), головокружение (34,3%), дисгевзия (31,4%), головная боль (31,4%), диссоциация (31,4%) и рвота (20,0%). Наиболее распространенным (> 20%) НЯВ в группе плацебо была головная боль (25,8%).
Во время двойной слепой фазы четыре субъекта испытали серьезное возникшее во время лечения нежелательное явление, и все они находились в группе, получавшей 84 мг эскетамина. У двух субъектов наблюдалась суицидальная идеация, у 1 субъекта наблюдалось возбуждение и у 1 субъекта наблюдались депрессивные симптомы. Шесть субъектов испытали серьезное нежелательное явление во время фазы последующего наблюдения (5 в группе, получавшей плацебо, и 1 в группе, получавшей 84 мг эскетамина). СНЯ в группе плацебо включали 3 попытки самоубийства (не фатальные), 1 субъекта, у которого наблюдалась суицидальная идеация, и 1 субъекта с целлюлитом. СНЯ у субъекта из группы, получавшей эскетамин, была суицидальная идеация.
6 субъектов (1 субъектов в группе плацебо и 5 субъектов в группе, получавшей 84 мг эскетамина) выбыли из двойной слепой фазы из-за нежелательных явлений, возникших в ходе лечения.
Временное увеличение кровяного давления для группы, получавшей 84 мг эскетамина, достигало пикового значения через 40 минут после введения дозы, при этом максимальное среднее повышение (во все дни введения дозы) систолического КД составляло 8,7 и 16,7 в группах, получавших плацебо и 84 мг эскетамина, соответственно. Максимальное среднее повышение (во все дни введения дозы) диастолического КД составляло 7,6 и 11,9 в группах, получавших плацебо и 84 мг эскетамина, соответственно.
Диссоциативные симптомы и симптомы изменения восприятия, оцененные по шкале CADSS, позволяют предположить, что эти симптомы появились вскоре после начала введения дозы и они разрешились через 2 часа после введения дозы.
РЕЗУЛЬТАТЫ
Информация о субъектах и лечении
Всего 68 субъектов с диагнозом БДР по DSM-IV-TR (Диагностико-статистическое руководство, 4-ое издание, пересмотренное) (в возрасте 19-64 лет) рандомизировали в две группы в соотношении 1:1 (32 в группе плацебо и 36 в группе 84 мг эскетамина). Количество субъектов, включенных в каждую выборку для анализа, приведено в Таблице 48. Все включенные в исследование субъекты были из США.
Среди 68 рандомизированных субъектов 66 были включены в выборку для анализа безопасности (по определению, это субъекты, получающие по меньшей мере одну дозу исследуемого препарата в двойной слепой фазе). Два субъекта были рандомизированы, но не получали исследуемого препарата. Все субъекты из выборки для анализа безопасности (N=66) были включены в выборку для анализа «по намеченному лечению» (ПНЛ) (по определению, это субъекты, получающие по меньшей мере одну дозу исследуемого препарата во время двойной слепой фазы и имеющие общую оценку MADRS как на исходном уровне, так и на 1 сутки через 4 часа после введения дозы). Сорок девять субъектов были включены в выборку для анализа безопасности (ПН) (по определению, это все субъекты, которые во время фазы последующего наблюдения проведи по меньшей мере 1 визит).
Информация о завершении исследования/прекращении участия в исследовании
Из 68 субъектов во всей рандомизированной выборке для анализа 19 (27,9%) субъектов выбыли из двойной слепой фазы. Информация о завершении и прекращении участия субъектов в двойной слепой фазе приведена в Таблице 49. Больше субъектов в группе, получавшей 84 мг эскетамина, прекратили участие из-за нежелательных явлений (5 субъектов в группе, получавшей 84 мг эскетамина, по сравнению с 1 субъектом в группе, получавшей плацебо), тогда как больше субъектов в группе, получавшей плацебо, прекратили участие из-за отсутствия эффективности (4 субъекта в группе, получавшей плацебо, по сравнению с 1 субъектом в группе, получавшей 84 мг эскетамина).
Четыре субъекта выбыли во время двойной слепой фазы по другим причинам. Подробная информация приведена ниже.
1) Один субъект (в группе, получавшей 84 мг эскетамина) имел повышенное кровяное давление после рандомизации, но до введения дозы, и, таким образом, выбыл.
2) Один субъект (в группе, получавшей 84 мг эскетамина) выбыл из-за отсутствия транспорта.
3) Один субъект (в группе плацебо) передумала и решила не участвовать в исследовании. Однако координатор исследования ошибочно рандомизировал ее в исследование.
4) Один субъект (в группе плацебо) не получил дозу на 22 сутки вследствие недоступности клинициста, затем этот субъект не показывался до 25 суток. Этот субъект вернулся для осуществления визита досрочного прекращения участия, но не возвращался во время фазы последующего наблюдения.
Демографические и исходные характеристики
Данные по демографическим и исходным характеристикам приведены в Таблице 50 для выборки для анализа ПНЛ. В целом группы лечения были аналогичными в отношении исходных характеристик. Большинство субъектов, попавших в двойную слепую фазу, составляли женщины (65,2%). Средний (СО) возраст всех субъектов составлял 35,8 (13,03) лет, в диапазоне от 19 до 64 лет. 75,8% субъектов должны были получать монотерапию антидепрессантами; 24,2% субъектов должны были получать антидепрессант плюс усиливающую терапию;
Исходная психиатрическая история для выборки для анализа ПНЛ представлена в Таблице 51. Средняя (СО) исходная общая оценка по шкале MADRS составляла 38,6 (6,53), в диапазоне от 20 до 52. Большинство субъектов имели оценку 6 согласно клинической глобальной оценке риска суицида, определяемой по SIBAT, модуль 8 (51,5%). Значение 6 соответствует риску суицида, требующему госпитализации с предупреждением суицида.
Оценки SIBAT (модуль 8) были следующими: 0=отсутствие риска суицида, 1=присутствуют нерегулярные суицидальные идеи, но никакого специального вмешательства не требуется, 2=присутствуют некоторые очевидные идеи самоубийства, пациенту рекомендуется обратиться за профессиональной помощью при необходимости; 3=риск суицида требует планового амбулаторного наблюдения; но без какого-либо иного немедленного вмешательства, 4=риск суицида требует немедленного вмешательства, но не госпитализации (например, приема лекарственных средств, неотложного амбулаторного наблюдения), 5=риск суицида требует немедленной госпитализации, но без предупреждения суицида, 6=риск суицида требует госпитализации с предупреждением суицида.
Степень воздействия
Число дней введения дозы представлено в Таблице 52. Больше субъектов в группе, получавшей 84 мг эскетамина, чем в группе, получавшей плацебо, прошли все 8 сеансов введения дозы (74,3% и 64,5%). Длительность приема препарата во время двойной слепой фазы представлена в Таблице 53. Пять субъектов уменьшили дозу эскетамина с 84 мг до 56 мг. Из них 3 уменьшили дозу из-за нежелательного явления, а 2 уменьшили дозу по ошибке и считались основными отклонениями протокола. Кроме того, у 1 субъекта был записан неправильный номер медицинского набора для устройства 2 на 15 сутки, поэтому получилось, что доза была снижена, но в действительности это было не так (Таблица 54).
Анализ первичного ожидаемого результата - изменение общей оценки MADRS от исходного уровня до 1 суток, 4 часа после введения дозы.
Первичным ожидаемым результатом эффективности было изменение общей оценки по шкале MADRS относительно исходного уровня на 1 сутки: 4 часа после введения дозы. Общая оценка MADRS соответствует диапазону 0-60. Первичный анализ эффективности проводили на выборке для анализа «по намеченному лечению» (ПНЛ), которая включала всех рандомизированных субъектов, которые получали по меньшей мере 1 дозу исследуемого препарата во время двойной слепой фазы и имели общую оценку MADRS как на исходном уровне, так и на 1 сутки через 4 часа после введения дозы. Так как это исследование обоснованности концепции фазы 2a, статистическую значимость определяли по двустороннему альфа-уровню 0,20. Все p-значения, представленные в данном документе, являются двусторонними.
Как показано ниже в Таблице 55, результаты изменения общей оценки по шкале MADRS свидетельствовали в пользу эскетамина в дозе 84 мг в сравнении с плацебо. Среднее изменение относительно исходного уровня (СО) на 1 сутки через 4 часа после введения дозы составляло -13,4 (9,03) для 84 мг эскетамина и -9,1 (8,38) для плацебо. На основании модели ANCOVA с лечением, терапией антидепрессантами и аналитическим центром в качестве факторов и исходным значением в качестве ковариаты, среднеквадратическая разница (СП) между эскетамином в дозе 84 мг и плацебо составляла -5,3 (2,10). Разница между группами лечения была статистически значимой (двустороннее p=0,015) при двустороннем уровне значимости 0,20.
Отрицательное изменение оценки указывает на улучшение. Исходный уровень соответствует значению до введения доза на 1 сутки.
Анализ вторичных ожидаемых результатов
Общая оценка по шкале MADRS: Изменение от исходного уровня до 2 суток (ДС) и до конечного момента времени (ДС)
Результаты изменения общей оценки MADRS на 2 сутки (ДС) приведены в Таблице 56. Среднее изменение относительно исходного уровня (СО) составляло -19,3 (12,02) для эскетамина в дозе 84 мг и -12,8 (9,77) для плацебо. На основании модели ANCOVA с лечением, терапией антидепрессантами и аналитическим центром в качестве факторов и исходным значением в качестве ковариаты группа эскетамина в дозе 84 мг статистически превосходила группу плацебо (двустороннее p-значение=0,015) с использованием двустороннего уровня значимости 0,20. Таким образом, изменения общей оценки MADRS на 2 сутки и 25 сутки конечного момента времени ДС были статистически большими в группе эскетамина по сравнению с группой плацебо (двустороннее p=0,015 и p=0,159, соответственно).
Отрицательное изменение оценки указывает на улучшение. Исходный уровень соответствует значению до введения доза на 1 сутки.
Как показано ниже в Таблице 57 ниже, результаты изменения общей оценки по шкале MADRS в конечный момент времени (ДС) свидетельствовали в пользу эскетамина в дозе 84 мг в сравнении с плацебо. Среднее изменение относительно исходного уровня (СО) составляло -26,4 (14,52) для эскетамина в дозе 84 мг и -23,0 (10,83) для плацебо. На основании той же модели ANCOVA, упомянутой выше, группа эскетамина в дозе 84 мг статистически превосходила группу плацебо (двустороннее p-значение=0,159) с использованием двустороннего уровня значимости 0,20. Смотрите Фиг. 38.
Отрицательное изменение оценки указывает на улучшение. Исходный уровень соответствует значению до введения доза на 1 сутки.
Пункт суицида в MADRS: Изменение относительно исходного уровня с течением времени
Результаты изменения относительно исходного уровня с течением времени для пункта суицида из оценки MADRS можно найти в Приложении 1. Статистически значимая разница в пользу эскетамина в дозе 84 мг была обнаружена на 1 сутки через 4 часа после введения дозы (двустороннее p=0,002), 2 сутки (ДС) (двустороннее p=0,129) и в конечный момент времени (ДС) (двустороннее p=0,143).
Клиническая глобальная оценка риска суицида SIBAT: Изменение от исходного уровня до 1 суток, через 4 часа после введения дозы, 2 суток и конечного момента времени (ДС)
Клиническая глобальная оценка риска суицида (модуль 8) обобщает общее клиническое заключение относительно риска суицида, полученное на основе информации, собранной с помощью всего инструмента SIBAT. Она работает подобно многочисленным другим шкалам тяжести CGI, которые использовали в других психиатрических исследованиях. Изменение клинической глобальной оценки риска суицида предназначено для непосредственного определения клинические значимых изменений в суицидальной идеации и возможности классификации риска суицида.
Анализ изменения оценки SIBAT был основан на модели ANCOVA на рангах изменения в SIBAT с лечением, терапией антидепрессантами и аналитическим центром в качестве факторов и исходным значением (неранжированным) в качестве ковариаты. Наблюдалась значимая разница (двустороннее p-значение=0,112) между двумя группами лечения при сравнении среднего ранга изменения относительно исходного уровня на 1 сутки, через 4 часа после введения дозы, в пользу 84 мг эскетамина. Смотрите Таблицу 58. Смотрите Фиг. 39. В частности, изменения клинической глобальной оценки риска суицида SIBAT были статистически превосходящими (т.е. двухстороннее p < 0,2) на 1 сутки, через 4 часа после введения дозы, и на 2 сутки в группе эскетамина по сравнению с группой плацебо. Эта разница не была очевидна в конечный момент времени ДС на 25 сутки. Смотрите Фиг. 40, на которой приведены данные LOCF и анализ с применением контрольного теста Кохрана-Мантеля-Хензеля для аналитического центра и терапии антидепрессантами.
Отрицательное изменение оценки указывает на улучшение. Исходный уровень соответствует значению до введения доза на 1 сутки.
Как показано в Таблице 59, сходные результаты были получены для изменения от исходного уровня до 2 суток (ДС). Хотя данные не приведены, в конечный момент времени (ДС) отсутствовала статистически значимая разница между группами лечения не наблюдалось (двустороннее p=0,922).
Отрицательное изменение оценки указывает на улучшение. Исходный уровень соответствует значению до введения доза на 1 сутки.
Для субъектов ПНЛ процент субъектов с исходной оценкой SIBAT 5 (риск суицида требует немедленной госпитализации, но без предупреждения суицида) или 6 (риск суицида требует госпитализации с предупреждением суицида) составлял 96,8% и 100% для групп плацебо и эскетамина в дозе 84 мг, соответственно. На 1 сутки через 4 часа после введения дозы процент субъектов с оценкой 5 или 6 составлял 80,6% для группы плацебо и 63,6% для группы эскетамина в дозе 84 мг. На гистограмме Фиг. 41 показано частотное распределение оценок по шкале SIBAT на исходном уровне двойной слепой фазы, на 1 сутки, 4 часа после введения дозы, в конечный момент времени двойной слепой фазы и в конечный момент времени фазы последующего наблюдения. На гистограмме Фиг. 42 показаны полученные методом наименьших квадратов (СП) средние значения изменений оценки MADRS от исходного уровня до 4 часов (первичный ожидаемый результат) и около 24 часов.
Устойчивый ответ (начало клинического ответа) по общей оценке MADRS
Результаты для устойчивого ответа приведена в Таблице 60. Устойчивый ответ определяется как по меньшей мере 50% снижение общей оценки MADRS относительно исходного уровня с началом на 1 сутки, 4 часа после введения дозы, которое сохраняется до конца двойной слепой фазы (25 сутки). У четырех субъектов в группе, получавшей 84 мг эскетамина, и у 2 субъектов в группе, получавшей плацебо, наблюдался устойчивый ответ в течение всей двойной слепой фазы. Статистически значимая разница между группами лечения отсутствовала (двустороннее p=0,608).
Устойчивый ответ определяется как по меньшей мере 50% снижение общей оценки MADRS от исходного уровня с началом на 1 сутки, которое сохраняется до конца двойной слепой фазы (сутки 25).
a Обобщенный тест Кохрана - Мантеля - Хензеля для разницы в средней оценки наличия устойчивого ответа с контролем в отношении аналитического цента и терапии антидепрессантами (монотерапия АД, АД плюс усиливающая терапия).
На Фиг. 43 приведена гистограмма с корреляцией процента пациентов с соответствующими ответом MADRS и ремиссией на 1, 2 сутки и в конечный момент времени.
На Фиг. 44 приведена гистограмма с корреляцией процента пациентов с ремиссией в конечный момент времени ДС и во время последующего наблюдения на 53 и 81 сутки.
Шкала Бека суицидальной идеации (BSS): Изменение от исходного уровня до 1 суток, 4 часа после введения дозы, 2 суток (ДС) и конечного момента времени (ДС)
BSS представляет собой инструмент самооценки из 21 пункта для обнаружения и определения степени тяжести суицидальной идеации у взрослых и подростков в возрасте 17 лет и старше. Общая оценка по шкале BSS представляет степень тяжести суицидальной идеации и рассчитывается путем суммирования оценок по первым 19 пунктам; общая оценка находится в диапазоне от 0 до 38, причем более высокая оценка представляет более выраженную суицидальную идеацию. Повышение оценок отражает повышение риска суицида.
Как показано в Таблицах 61-63, отсутствовала статистически значимая разница между эскетамином в дозе 84 мг и плацебо в отношении изменения общей оценки BSS на 1 сутки, 4 часа после введения дозы, 2 сутки (ДС) и в конечный момент времени (ДС), с использованием той же модели ANCOVA, что описана выше для общей оценки MADRS. Смотрите Фиг. 45.
Отрицательное изменение оценки указывает на улучшение. Исходный уровень соответствует значению до введения доза на 1 сутки.
Отрицательное изменение оценки указывает на улучшение. Исходный уровень соответствует значению до введения доза на 1 сутки.
Отрицательное изменение оценки указывает на улучшение. Исходный уровень соответствует значению до введения доза на 1 сутки.
Шкала безнадежности Бека (BHS): Изменение от исходного уровня до 1 суток, 4 часа после введения дозы, и конечного момента времени (ДС)
BHS представляет собой средство самооценки для оценки чьего-либо уровня негативных ожиданий или пессимизма в отношении будущего. Она состоит из 20 пунктов типа истина - ложь, которые позволяют определить отношение отвечающего за последнюю неделю путем подтверждения пессимистического утверждения или отрицания оптимистического утверждения. В случае каждого утверждения каждому ответу присваивается оценка 0 или 1. Общий оценка BHS представляет собой сумму ответов элемента и находится в диапазоне от 0 до 20, причем более высокая оценка представляет более высокий уровень безнадежности.
Как показано в Таблице 64, отсутствовала статистически значимая разница между эскетамином в дозе 84 мг и плацебо в отношении изменения общей оценки BHS на 1 сутки, 4 часа после введения дозы (двустороннее p=0,297). При этом в конечный момент времени (ДС) эскетамин в дозе 84 мг статистически превосходил (двустороннее p=0,165) плацебо при двустороннем уровне значимости 0,20. (Таблица 65).
Отрицательное изменение оценки указывает на улучшение. Исходный уровень соответствует значению до введения доза на 1 сутки.
Отрицательное изменение оценки указывает на улучшение. Исходный уровень соответствует значению до введения доза на 1 сутки.
БЕЗОПАСНОСТЬ
Обобщенная информация по всем нежелательным явлениям
Обобщенная информация по всем возникшим в ходе лечения нежелательным явлениям (НЯВЛ) во время двойной слепой фазы представлена в Таблице 66. В целом, у 80,6% субъектов в группе, получавшей плацебо, и у 94,3% субъектов в группе, получавшей эскетамин в дозе 84 мг, наблюдалось по меньшей мере одно НЯВЛ во время двойной слепой фазы.
В целом у 33/35 (94,3%) субъектов, получавших эскетамин в дозе 84 мг, и у 25/31 (80,6%) субъектов, получавших плацебо, наблюдалось по меньшей мере одно возникшее в ходе лечения нежелательное явление (НЯВЛ) во время двойной слепой фазы.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений. Нежелательные явления закодированы с помощью MedDRA, версия 18.0.
Нежелательные явления, возникшие в ходе лечения во время двойной слепой фазы (> 5% субъектов в любой группе лечения), обобщены по группе лечения для выборки для анализа безопасности и приведены в Таблице 67. Наиболее распространенными (> 20%) НЯВЛ в группе, получавшей 84 мг эскетамина во время двойной слепой фазы, были тошнота (37,1%), головокружение (34,3%), дисгевзия (31,4%), головная боль (31,4%), диссоциация (31,4%) и рвота (20,0%). Наиболее распространенным НЯВЛ в группе плацебо была головная боль (25,8%).
Нежелательные явления, возникшие во время фазы последующего наблюдения, обобщены в Таблице 68. В целом, у 77,3% субъектов в группе, получавшей плацебо, и у 48,1% субъектов в группе, получавшей эскетамин в дозе 84 мг, наблюдалось по меньшей мере одно нежелательное явление во время фазы последующего наблюдения.
Смерть
Летальные исходы отсутствовали.
Нежелательные явления, приводящие к прекращению приема исследуемого препарата
6 субъектов (1 субъект в группе плацебо и 5 субъектов в группе, получавшей 84 мг эскетамина) выбыли из двойной слепой фазы из-за нежелательных явлений, возникших в ходе лечения. Смотрите Таблицу 69.
Серьезные нежелательные явления
Во время двойной слепой фазы четыре субъекта испытали серьезное возникшее в ходе лечения нежелательное явление, и все они находились в группе, получавшей 84 мг эскетамина (Таблица 70). У двух субъектов наблюдалась суицидальная идеация, у 1 субъекта наблюдалось возбуждение и у 1 субъекта наблюдались депрессивные симптомы. Один субъект из группы плацебо имел серьезное большое депрессивное расстройство, ухудшившееся после двойной слепой фазы после прекращения участия в исследовании. Этот субъект не участвовал в фазе последующего наблюдения.
Как показано в Таблице 71, 6 субъектов испытали серьезное нежелательное явление во время фазы последующего наблюдения (5 в группе, получавшей плацебо, и 1 в группе, получавшей 84 мг эскетамина).
Основные физиологические показатели
Временное увеличение кровяного давления для группы эскетамина достигало пикового значения приблизительно через 40 минут после введения дозы, при этом максимальное среднее повышение (во все дни введения дозы) систолического КД составляло 8,7 в группе плацебо и 16,7 в группе, получавшей 84 мг эскетамина. Максимальное среднее повышение (по всем дням введения дозы) диастолического КД составило 7,6 в группе плацебо и 11,9 в группе, получавшей 84 мг эскетамина. В целом, в группе, получавшей эскетамин, наблюдали повышение временного кровяного давления, которое обычно возвращалось к нормальному диапазону через 2 часа после введения дозы. Смотрите Фиг. 46 и 47.
Другие наблюдения безопасности
Применяемая клиницистами шкала диссоциативных симптомов (CADSS)
Оценку CADSS определяли перед началом введения каждой дозы, через 40 минут, 2 часа и 4 часа после введения дозы. Шкалу CADSS используют для оценки возникших в ходе лечения диссоциативных симптомов и изменений восприятия, а общая оценка находится в диапазоне от 0 до 92, причем более высокая оценка представляет более тяжелое состояние.
Диссоциативные симптомы и изменения восприятия, оцененные по шкале CADSS, позволяют предположить, что эти симптомы появились вскоре после начала введения дозы и разрешились через 2 часа после введения дозы (Фиг. 55). Смотрите Таблицы 72A и 72B.
В целом диссоциативные симптомы, определенные по CADSS, наблюдаемые в группе, получавшей эскетамин, соответствовали предшествующим исследованиям. Эти симптомы были временными (разрешались в течение 2 часов) и становились слабее при повторном введении дозы.
Сводная информация
Интраназальный эскетамин в дозе 84 мг по сравнению с плацебо продемонстрировал клинически существенное и статистически значимое быстрое уменьшение депрессивных симптомов у пациентов с БДР, которые по оценке имели высокий риск суицида, о чем свидетельствует изменение общей оценки MADRS относительно исходного уровня как через 4 часа, так и на 2 сутки. Также наблюдалось значительное улучшение суицидальности через 4 часа и на 2 сутки по определению по пункту суицида в MADRS клинической глобальной оценке риска суицида SIBAT. В любой из этих моментов времени не было обнаружено разницы по BSS. Как наблюдалось в предыдущих исследованиях эскетамина при ТРД, перцептивные (диссоциативные) симптомы, определяемые по CADSS и повышению КД, по-видимому, происходят вскоре после начала введения и разрешаются через 2 часа после введения. Кроме того, перцептивные симптомы становились слабее при повторном введении дозы. Следует отметить, что во время периода последующего наблюдения 3 субъекта в группе плацебо, но ни один в группе эскетамина, предприняли попытки самоубийства (не фатальные).
Результаты исследования обоснованности концепции фазы 2a подтверждают гипотезу о том, что интраназальный эскетамин является эффективным лечением для быстрого уменьшения симптомов БДР, включая суицидальную идеацию, у пациентов, которые по оценке имеют высокий риск суицида. Субъекты, включенные в это исследование, имели тяжелую депрессию и суицидальные намерения, о чем свидетельствуют их высокие исходные оценки MADRS и BSS. Всем субъектам было оказано активное лечение с начальной госпитализацией и оптимизацией стандартных антидепрессантов. Таким образом, неудивительно, что субъекты в обеих группах лечения испытали клинически существенное улучшение по всем критериям эффективности в течение двойного слепого периода. Несмотря на неспецифическое улучшение в группе плацебо, благоприятный эффект эскетамина на симптомы БДР, определяемый по общей оценке MADRS и пункту суицида в MADRS, можно выделить в ранние моменты времени и в конечный момент времени двойной слепой фазы.
Пример 4
Первичная цель данного исследования заключается в оценке эффективности интраназального эскетамина плюс пероральный антидепрессант по сравнению с пероральным антидепрессантом плюс интраназальное плацебо в отношении задержки рецидива депрессивных симптомов у пациентов с ТРД, которые достигли стабильной ремиссии (первичной) или стабильного ответа (вторичного) после индукции и оптимизации курса интраназального эскетамина плюс пероральный антидепрессант (АД), для оценки эффективности эскетамина плюс пероральный АД по сравнению с пероральным АД плюс интраназальное плацебо в отношении задержки рецидива депрессивных симптомов. Ключевым являлся вопрос о том, можно ли прекратить прием эскетамина в группах со стабильной ремиссией/ответом и достичь долгосрочного поддержания эффекта только с пероральным АД. Экспертный комитет по рецидивам рассмотрел события, которые были сочтены клинически релевантными для определения того, произошел ли рецидив.
Наряду с 3 краткосрочными исследованиями по эффективности и безопасности и долгосрочным открытым исследованием по безопасности это исследование направлено на подтверждение соблюдения требований регуляторных органов по регистрации назального спрея эскетамина для лечения ТРД.
Информация о субъектах и лечении
Это было рандомизированное, двойное слепое, активно контролируемое, многоцентровое исследование в параллельных группах, которое включало 705 субъектов с ТРД. В этом исследовании оценивали эффективность, безопасность и переносимость интраназального эскетамина плюс пероральный антидепрессант по сравнению с пероральным антидепрессантом плюс интраназальное плацебо в отношении задержки рецидива депрессивных симптомов у взрослых мужчин и женщин с БДР, которые находятся в состоянии стабильной ремиссии после фазы индукции и оптимизации лечения интраназальным эскетамином плюс пероральный антидепрессант. Смотрите Фиг. 49 в отношении дизайна исследования.
Из 705 включенных в исследование субъектов 437 (62,0%) были напрямую включены в исследование 3003, 150 (21,3%) были переведены из исследования ESKETINTRD3001, а 118 (16,7%) были переведены из исследования ESKETINTRD3002. Смотрите Фиг. 50. В полной выборке для анализа 635 (90,1%) субъектов были белыми, а 457 (64,8%) субъектов были женщинами. Средний возраст составлял 46,1 лет, в диапазоне от 18 до 64 лет. Из 437 субъектов из выборки для анализа безопасности (ИНД) (только напрямую включенные субъекты), 273 (62,5%) субъектов завершили 28-суточную ИНД-фазу, а 164 (37,5%) выбыли. Большинство субъектов прекратили участие в ИНД-фазе из-за субъектов, которые не соответствовали критериям для продолжения участия в следующей фазе (114 субъектов).
Из 455 субъектов, проходивших лечение эскетамином, переходящих к фазе ОП (включая 182 проходивших лечение эскетамином субъектов, переведенных из исследования TRD3001 или TRD3002), 297 (65,3%) субъектов завершили 12-недельную фазу ОП, а 158 (34,7%) субъектов выбыли. Наиболее частые причины прекращения участия были связаны с тем, что субъект не соответствовал критериям для продолжения участия в следующей фазе (107 субъектов).
Из 176 субъектов в полной выборке для анализа (в состоянии стабильной ремиссии) 159 (90,3%) субъектов завершили фазу ПО (из них 63 (35,8%) имели рецидив, а 96 (54,5%) не имели рецидивов на момент прекращения исследования). Наиболее частой причиной прекращения участия было «прочее» (8 субъектов).
Из 121 субъектов в полной выборке для анализа (демонстрирующие стабильный ответ) 113 (93,4%) субъектов завершили фазу ПО (из них 50 (41,3%) имели рецидив, а 63 (52,1%) не имели рецидивов на момент прекращения исследования). Наиболее частой причиной прекращения участия было «прекращение участия субъектом» (3 субъекта). В общей сложности 545 субъектов перешли в фазу последующего наблюдения, а 532 (97,6%) завершили фазу последующего наблюдения.
На исходном уровне (ИНД) субъекты имели медианную общую оценку MADRS, составляющую 38 (тяжелая депрессия), и медианную длительность текущего депрессивного эпизода, составляющую 64 недели, при этом 27,4% в течение жизни имели в анамнезе суицидальную идеацию (29,1% в течение последних 6 месяцев) и 14,9% в течение жизни имели в анамнезе суицидальное поведение. Более 89,0% субъектов получали 3 или более АД с отсутствием ответа (определяемым как улучшение ≤ 25%) перед началом индукционной фазы. Субъекты сообщили о семейной истории депрессии (45,1%), тревожного расстройства (9,1%) и злоупотребления алкоголем (13,5%).
Длительность лечения/длительность исследования: Каждый субъект участвовал максимум в 5 фазах: фазе скрининга и проспективного наблюдения (только напрямую включенные субъекты) длительностью 4 недели+необязательный период постепенного уменьшения дозы до 3 недель, 4-недельной открытой индукционной фазе (только напрямую включенные субъекты), 12-недельной фазе оптимизации (открытой для напрямую включенных субъектов и двойной слепой для переведенных субъектов), двойной слепой фазе поддержания вариабельной длительности и 2-недельной фазе последующего наблюдения. Максимальная продолжительность участия субъекта была вариабельной в зависимости от того, непосредственно попали ли он или она в исследование напрямую или были переведены из одного из двойных скрытых краткосрочных исследований, и соответствовали ли он или она критериям, специфичным для фазы (например, соответствие критериям ответа в конце индукционной фазы, нахождение в состоянии стабильных ремиссии/ответа в конце фазы оптимизации, и время рецидива в фазе поддержания). Напрямую включенные субъекты участвовали максимум в 5 фазах, а переведенные субъекты участвовали максимум в 3 фазах текущего исследования после участия в фазе скрининга и проспективного наблюдения и индукционной фазе в исследованиях, из которых они были переведены. Критерии включения/исключения были одинаковыми для напрямую включенных и переведенных субъектов.
Эффективность
Уровень значимости
Запланированный промежуточный анализ данных эффективности провели, когда среди находящихся в состоянии стабильной ремиссии произошло 33 случая рецидива, причем по меньшей мере 30 рецидивов (в действительности в промежуточный анализ было включено 31) произошли у рандомизированных находящихся в состоянии стабильной ремиссии пациентов, проходивших лечение интраназальным эскетамином плюс пероральный антидепрессант во время фазы оптимизации (перечень 1). Цели промежуточного анализа заключались в переоценке размера выборки или в приостановке исследования из-за нецелесообразности. Независимая внешняя статистическая группа поддержки (Cytel) провела анализ, а IDMC изучила раскрытые результаты и рекомендовала продолжить исследование. На основании заранее определенных правил конечный размер выборки, определенный на основании повторной оценки размера выборки, составил 59. Группа Janssen и центры продолжали работать с маскированными данными в отношении рекомендации по размеру выборки IDMC до тех пор, пока в перечне 1 не появилось 59 рецидивов.
Промежуточный анализ эффективности проводили при уровне значимости 0,0097 (двусторонний). Поскольку исследование не было остановлено из-за эффективности при проведении промежуточного анализа, окончательный анализ эффективности следовало проводить при уровне значимости 0,046 (двусторонний).
Первичный ожидаемый результат
Первичный анализ эффективности проводили на полной (находящиеся в состоянии стабильной ремиссии пациенты) выборке для анализа, которая включая 175 находящихся в состоянии стабильной ремиссии пациентов и 1 пациента со стабильным ответом (который был ошибочно рандомизирован как находящийся в состоянии стабильной ремиссии), определяемых как рандомизированные субъекты, которые находились в состоянии стабильной ремиссии в конце фазы оптимизации после лечения интраназальным эскетамином плюс пероральный антидепрессант. Субъектов рандомизировали как находящихся в состоянии стабильной ремиссии (перечень 1) в соотношении 1:1 для продолжения приема интраназального эскетамина плюс пероральный антидепрессант (N=90) или для прекращения приема эскетамина и получения перорального антидепрессанта плюс интраназальное плацебо (N=86); Эти субъекты получили по меньшей мере 1 дозу интраназального исследуемого препарата и 1 дозу перорального антидепрессанта во время фазы поддержания.
Результаты по времени до рецидива во время фазы поддержания свидетельствовали в пользу интраназального эскетамина+пероральный АД в отношении задержки рецидива по сравнению с пероральным АД+интраназальное плацебо. В целом 24 (26,7%) субъекта в группе интраназального эскетамина+пероральный АД и 39 (45,3%) субъектов в группе перорального АД+интраназальное плацебо имели рецидив во время фазы поддержания. На основании взвешенного комбинированного теста разница между группами лечения была статистически значимой (двустороннее p=0,003), что было ниже порога статистической значимости (0,046). Расчетное отношение рисков для интраназального эскетамина+пероральный АД по сравнению с пероральным АД+интраназальное плацебо на основании взвешенных оценок составляло 0,49 (95% ДИ: 0,29, 0,84) с использованием ADDPLAN. Наиболее распространенной причиной рецидива была общая оценка по шкале MADRS ≥ 22 для 2 последовательных определений, разделенных 5-15 сутками.
Не наблюдали существенных различий по региону, полу, прямому включению или переводу или группе пероральных АД (SNRI и SSRI).
Другие вторичные ожидаемые результаты эффективности
Вторичные параметры эффективности включали:
- Время между рандомизацией субъекта и первой документацией (самая ранняя дата) рецидива в фазе поддержания для субъектов со стабильным ответом (но которые не находились в состоянии ремиссии) в конце фазы оптимизации после лечения интраназальным эскетамином плюс пероральный антидепрессант.
- Изменение MADRS от исходного уровня (ПО) до конечного момента времени (ПО)
- Доля субъектов с ответом и ремиссией на основании MADRS
- Изменение PHQ-9 от исходного уровня (ПО) до конечного момента времени (ПО)
- Изменение CGI-S от исходного уровня (ПО) до конечного момента времени (ПО)
- Изменение GAD-7 от исходного уровня (ПО) до конечного момента времени (ПО)
- Изменение EQ-5D-5L от исходного уровня (ПО) до конечного момента времени (ПО)
- Изменение SDS от исходного уровня (ПО) до конечного момента времени (ПО)
Результаты для времени между рандомизацией субъекта и первой документацией (самая ранняя дата) рецидива в фазе поддержания для субъектов в полной (демонстрирующих стабильный ответ) выборке для анализа (включая 121 субъектов: 120 пациентов со стабильным ответом и 1 субъект, не соответствующий критериям стабильной ремиссии или стабильного ответа в конце фазы оптимизации) после лечения интраназальным эскетамином плюс пероральный антидепрессант, свидетельствовали в пользу интраназального эскетамина плюс пероральный АД в отношении задержки рецидива по сравнению с пероральным АД+интраназальное плацебо. В целом 16 (25,8%) субъектов в группе интраназального эскетамина+пероральный АД и 34 (57,6%) субъекта в группе перорального АД+интраназальное плацебо имели рецидив во время фазы поддержания. Разница между группами лечения была статистически значимой (двустороннее p < 0,001) при двустороннем логранговом критерии. Расчетное отношение рисков для интраназального эскетамина+пероральный АД по сравнению с пероральным АД+интраназальное плацебо на основании модели пропорциональных рисков Кокса с лечением в качестве фактора составляло 0,30 (95% ДИ: 0,16, 0,55).
Субъектов рандомизировали как находящихся в состоянии стабильной ремиссии (перечень 2) в соотношении 1:1 для продолжения приема интраназального эскетамина плюс пероральный АД (N=62) или для прекращения приема эскетамина и получения перорального АД плюс интраназальное плацебо (N=59); Оба перечня рандомизации были стратифицированы по стране.
Безопасность
В целом 76,9% субъектов испытали по меньшей мере одно НЯВЛ во время ИНД-фазы. В выборке для анализа безопасности (ОП) (выборки для анализа безопасности (ОП) и безопасности (ПО) не включали переведенных субъектов, которые продолжали получать пероральный АД+плацебо (TEP) во время последующих фаз), 73,6% субъектов испытали по меньшей мере одно НЯВЛ во время ОП-фазы. В выборке для анализа безопасности (ПО) 82,2% субъектов в группе, получавшей эскетамин+пероральный АД, и 45,5% субъектов в группе, получавшей пероральный АД+плацебо, испытали по меньшей мере одно НЯВЛ во время ПО-фазы В случае субъектов TEP 61,6% субъектов испытали по меньшей мере одно НЯВЛ во время ОП-фазы, а 68,5% испытали по меньшей мере одно НЯВЛ во время ПО-фазы.
Наиболее распространенными НЯВЛ (> 10%) во время ИНД-фазы были вертиго (22,7%), головокружение (22,2%), тошнота (21,5%), дисгевзия (20,6%), сонливость (14,9%), головная боль (13,7%), параанестезия (11,0%), диссоциация (11,0%), ненормальное ощущение (10,8%), размытое зрение (10,3%) и седация (10,1%). В выборке для анализа безопасности (ОП) наиболее распространенными НЯВЛ во время ОП-фазы были вертиго (20,0%), дисгевзия (17,4%), сонливость (13,8%), головокружение (13,4%), головная боль (12,5%) и тошнота (10,5%). В выборке для анализа безопасности (ПО) наиболее распространенными НЯВЛ в группе эскетамина+пероральный АД во время двойной слепой ПО-фазы были дисгевзия (26,3%), вертиго (25,0%), сонливость (21,1%), головокружение (20,4%), головная боль (17,8%), тошнота (16,4%), размытое зрение (15,8%), диссоциация (13,8%) и пероральная гипостезия (13,2%). У >10% субъектов с пероральным АД+плацебо в выборке для анализа безопасности (ПО) не наблюдалось ни одного НЯВЛ. В случае субъектов TEP наиболее распространенным НЯВЛ была головная боль (18,6%) во время ОП-фазы и вирусная инфекция верхних дыхательных путей (24,1%), головная боль (22,2%) и дисгевзия (14,8%) во время ПО-фазы.
В этом исследовании летальных случаев зарегистрировано не было.
В этом исследовании было зарегистрировано 32 субъекта с 39 серьезными нежелательными явлениями (СНЯ). В общей сложности 13 субъектов испытывали серьезные возникшие в ходе лечения нежелательные явления (НЯВЛ) во время ИНД-фазы в выборке для анализа безопасности (ИНД). Три субъекта имели серьезные НЯВЛ, которые исследователи посчитали с большой вероятностью связанными с интраназальным эскетамином: дезориентация (1 сутки), суицидальная идеация (8 сутки), седация (22 сутки), а один субъект имел два серьезных НЯВЛ в одни сутки (5 сутки), которые посчитали с большой вероятностью связанными с интраназальным эскетамином: дисбаланс автономной нервной системы и простой парциальный припадок. Один субъект имел серьезное НЯВЛ, которое исследователь посчитал вероятно связанным с интраназальным эскетамином: лакунарный инсульт (1 сутки). Один субъект имел серьезное НЯВЛ, которое посчитали возможно связанным с интраназальным эскетамином: гипотермия (10 сутки). Одиннадцать субъектов испытывали серьезные возникшие в ходе лечения нежелательные явления во время ОП-фазы в выборке для анализа безопасности (ОП). Ни одно НЯВЛ не посчитали возможно, вероятно или с большой вероятностью связанным с эскетамином. Пять субъектов (4 в группе эскетамина+пероральный АД, 1 в группе пероральный АД+плацебо) в выборке для анализа безопасности (ПО) испытывали серьезные возникшие в ходе лечения нежелательные явления во время ПО-фазы. Все явления посчитали не связанными с интраназальным препаратом или пероральным АД. Два субъекта имели серьезные НЯ, которые посчитали не связанными с пероральным АД в фазе последующего наблюдения. Один субъект испытал два серьезных НЯ, которые посчитали возможно связанными с пероральным АД. Субъект TEP испытал серьезное возникшее в ходе лечения нежелательное явление во время ОП-фазы. Один субъект TEP испытал серьезное НЯВЛ во время ПО-фазы, которое посчитали не связанным с интраназальным препаратом или пероральным АД.
22 субъекта прекратили прием интраназального исследуемого препарата в ИНД-фазе из-за возникших в ходе лечения нежелательных явлений в выборке для анализа безопасности (ИНД), а 5 субъектов прекратили прием интраназального исследуемого препарата в ОП-фазе из-за возникших в ходе лечения нежелательных явлений в выборке для анализа безопасности (ОП). Согласно дизайну исследования эти субъекты могли продолжать принимать пероральный АД во время фазы последующего наблюдения при необходимости. Восемь субъектов прекратили прием перорального АД в ИНД-фазе, а 2 субъекта прекратили прием перорального АД в ОП-фазе из-за возникших в ходе лечения нежелательных явлений. 7 субъектов (4 субъекта в группе, получавшей эскетамин+пероральный АД, и 3 субъекта в группе, получавшей пероральный АД+плацебо), прекратили прием интраназального исследуемого препарата в ПО-фазе из-за возникших в ходе лечения нежелательных явлений в выборке для анализа безопасности (ПО). Согласно дизайну исследования эти субъекты могли продолжать принимать пероральный АД во время фазы последующего наблюдения при необходимости. Три субъекта в группе, получавшей эскетамин+пероральный АД, прекратили прием перорального исследуемого АД в ПО-фазе из-за возникших в ходе лечения нежелательных явлений; ни один субъект в группе, получавшей пероральный АД+интраназальное плацебо, не прекратил прием перорального исследуемого АД в ПО-фазе из-за возникших в ходе лечения нежелательных явлений. Ни один субъект TEP не прекратил прием интраназального исследуемого препарата или перорального АД в ОП-фазе из-за возникших в ходе лечения нежелательных явлений в выборке для анализа безопасности (ОП_TEP). 2 субъекта TEP прекратили прием интраназального исследуемого препарата в ПО-фазе в выборке для анализа безопасности (ПО_TEP). Один из этих субъектов прекратил участие в ПО-фазе как из-за интраназального препарата, так и из-за и перорального АД.
Другие наблюдения безопасности
Временное кровяное давление увеличивается до пика для группы, получавшей эскетамин+пероральный АД, приблизительно через 40 минут после введения дозы и возвращается ближе к уровням до введения дозы через 1,5 часа после введения дозы.
Диссоциативные симптомы/симптомы изменения восприятия, оцененные по шкале CADSS, позволяют предположить, что эти симптомы появились вскоре после начала сеанса интраназального введения дозы и разрешились через 1,5 часа после введения дозы.
Доля субъектов с седацией (определяемой по шкале MOAA/S ≤ 3) составляла ≤ 3,9% для группы эскетамина+пероральный АД для каждых суток введения дозы во всех фазах.
Информация о субъектах и лечении
Всего было включено 1097 субъектов из 164 центров в 16 странах (Бельгия, Бразилия, Канада, Чехия, Эстония, Франция, Германия, Венгрия, Италия, Мексика, Польша, Словакия, Испания, Швеция, Турция и Соединенные Штаты). За исключением 378 случаев неудачного скрининга и 14 субъектов из центра PL10002 из-за проблем с GCP, в исследование были включены 705 субъектов с диагнозом БДР по DSM-5 (Диагностико-статистическое руководство по психическим расстройствам, 5-ое издание).
Четыреста тридцать семь субъектов были напрямую включены в исследование 3003, 150 субъектов были переведены из исследования ESKETINTRD3001, а 118 субъектов были переведены из исследования ESKETINTRD3002. Результаты представлены в Таблице 73.
Число субъектов в каждой фазе и выборке для анализа представлено в Таблице 74.
b Полная (стабильная ремиссия) выборка для анализа во время промежуточного анализа. Стабильная ремиссия: Общая оценка MADRS ≤ 12 по меньшей мере на 3 из последних 4 недель фазы оптимизации, но допускается одна общая оценка MADRS > 12 или одна пропущенная оценка MADRS только на 13 или 14 неделе фазы оптимизации. Общая оценка MADRS на 15 и 16 неделях должна составлять ≤ 12. Стабильный ответ: ≥50% снижение общей оценки MADRS относительно исходного уровня (1 сутки индукционной фазы; до рандомизации/перед первой интраназальной дозой) на каждую из последних 2 недель фазы оптимизации, но несоответствие критериям стабильной ремиссии. В случае переведенных субъектов 1 сутки открытой индукционной фазы будут приходиться на исследования ESKETINTRD3001 или ESKETINTRD3002.
Информация о завершении исследования/прекращении участия в исследовании
Из 437 субъектов из выборки для анализа безопасности (ИНД) (только напрямую включенные субъекты), 273 (62,5%) субъектов завершили 28-суточную ИНД-фазу, а 164 (37,5%) выбыли. Результаты представлены в Таблице 75. Большинство субъектов прекратили участие в ИНД-фазе из-за того, что субъект не соответствовал критериям для продолжения участия в следующей фазе (114 субъектов).
Из 455 субъектов, попавших в ОП-фазу (включая 182 проходивших лечение эскетамином субъектов, переведенных из исследования TRD3001 или TRD3002), в выборке для анализа безопасности (ОП) 297 (65,3%) субъектов завершили 12-недельную ОП-фазу, а 158 (34,7%) субъектов выбыли. Результаты представлены в Таблице 76.
b Этот критерий применим к субъектам до принятия поправки к Протоколу 4.
Наиболее частые причины прекращения участия были связаны с тем, что субъект не соответствовал критериям для продолжения участия в следующей фазе (107 субъектов). Следует отметить, что выборки для анализа безопасности (ОП) и безопасности (ПО) не включают переведенных субъектов, которые продолжали получать пероральный АД+плацебо (TEP) во время последующих фаз, смотрите Таблицы 77 и 78 в отношении информации по завершению/прекращению участия для этих выборок для анализа.
b Этот критерий применим к субъектам до принятия поправки к Протоколу 4.
Из 176 субъектов в полной выборке для анализа (в состоянии стабильной ремиссии) 159 (90,3%) субъектов завершили фазу ПО (из них 63 (35,8%) имели рецидив, а 96 (54,5%) не имели рецидивов на момент прекращения исследования). Результаты представлены в Таблице 79. Наиболее частой причиной прекращения участия было «прочее» (8 субъектов).
(N=176)
Из 121 субъектов в полной выборке для анализа (демонстрирующие стабильный ответ) 113 (93,4%) субъектов завершили фазу ПО (из них 50 (41,3%) имели рецидив, а 63 (52,1%) не имели рецидивов на момент прекращения исследования). Результаты представлены в Таблице 80. Наиболее частой причиной прекращения участия было «прекращение участия субъектом» (3 субъекта).
(N=121)
Субъекты могли перейти в фазу последующего наблюдения из ИНД-фазы, ОП-фазы или ПО-фазы. В общей сложности 545 субъектов перешли в фазу последующего наблюдения, а 532 (97,6%) завершили фазу последующего наблюдения.
Демографические и исходные характеристики
Данные по демографическим и исходным характеристикам приведены в Таблице 81 для выборки для анализа из всех включенных субъектов. Большинство субъектов, включенных в исследование, составляли женщины (64,8%).
b Статус гипертензии классифицируется как «Да», если в истории болезни указана гипертензия.
Средний возраст (СО) составлял 46,1 (11,10) лет, в диапазоне от 18 до 64 лет. Исходная психиатрическая история для выборки для анализа из всех включенных субъектов представлена в Таблице 82. Средняя (СО) исходная (ИНД) общая оценка по шкале MADRS составляла 37,9 (5,50), в диапазоне от 4 до 53.
b Число антидепрессантов с отсутствием ответа (определяемым как ≤ 25% улучшение), принятое за по меньшей мере 6 недель во время текущего эпизода, полученное из MGH-ATRQ во время первого скринингового визита.
Демографические и исходные характеристики и исходный анамнез психиатрических расстройств для выборки для анализа безопасности (ИНД) представлены в Таблицах 83 и 84. Большинство субъектов, включенных в исследование, составляли женщины (61,3%). Средний возраст (СО) составлял 46,5 (10,96) лет, в диапазоне от 19 до 64 лет. Средняя (СО) исходная (ИНД) общая оценка по шкале MADRS составляла 37,8 (5,51), в диапазоне от 4 до 53.
b Статус гипертензии классифицируется как «Да», если в истории болезни указана гипертензия.
b Число антидепрессантов с отсутствием ответа (определяемым как ≤ 25% улучшение), принятое за по меньшей мере 6 недель во время текущего эпизода, полученное из MGH-ATRQ во время первого скринингового визита.
Демографические и исходные характеристики и исходный анамнез психиатрических расстройств для полной (стабильная ремиссия) выборки для анализа представлены в Таблицах 85 и 86. Большинство находящихся в состоянии стабильной ремиссии субъектов, рандомизированных в ПО-фазу, составляли женщины (66,5%). Средний возраст (СО) составлял 45,8 (11,64) лет, в диапазоне от 19 до 64 лет. Средняя (СО) исходная (ИНД) общая оценка по шкале MADRS составляла 37,5 (4,93), в диапазоне от 26 до 49.
(N=176)
b Статус гипертензии классифицируется как «Да», если в истории болезни указана гипертензия.
(N=176)
b Число антидепрессантов с отсутствием ответа (определяемым как ≤ 25% улучшение), принятое за по меньшей мере 6 недель во время текущего эпизода, полученное из MGH-ATRQ во время первого скринингового визита.
Степень воздействия
Степень воздействия интраназального исследуемого препарата во время ПО-фазы для полной (стабильная ремиссия) выборки для анализа и полной (стабильный ответ) выборки для анализа представлена в Таблицах 87 и 88.
На 1 сутки ПО-фазы 40/90 (44,4%) субъектов, получавших интраназальный эскетамин в полной (стабильная ремиссия) выборке для анализа, получали эскетамин в дозе 56 мг, а 50/90 (55,6%) получали эскетамин в дозе 84 мг. В полной (стабильный ответ) выборке для анализа 20/61 (32,8%) субъектов получали эскетамин в дозе 56 мг, а 41/61 (67,2%) субъектов получали эскетамин в дозе 84 мг. Начиная с недели 4 (ПО) частоту сеансов интраназального лечения можно было корректировать (при необходимости) с фиксированными 4-недельными интервалами. В Таблицах 89 и 90 приведены схемы введения дозы, которых субъекты придерживались в течение по меньшей мере 50% времени ПО-фазы, для полной (стабильная ремиссия) выборки для анализа и полной (стабильный ответ) выборки для анализа. Из 90 рандомизированных находящихся в состоянии стабильной ремиссии субъектов, проходивших лечение интраназальным эскетамином во время ПО-фазы, 62 (68,9%) субъектов придерживались планового введения дозы «один раз в две недели» в течение большей части времени. Из 62 рандомизированных демонстрирующих стабильный ответ субъектов, проходивших лечение интраназальным эскетамином во время ПО-фазы, 21 (33,9%) субъектов придерживались планового введения дозы «один раз в две недели» в течение большей части времени.
АНАЛИЗ ПЕРВИЧНОЙ КОНЕЧНОЙ ТОЧКИ
Промежуточный анализ эффективности проводили при уровне значимости 0,0097 (двусторонний). Поскольку исследование не было остановлено из-за эффективности при проведении промежуточного анализа, окончательный анализ эффективности проводили при уровне значимости 0,046 (двусторонний).
Первичный анализ эффективности основан на полной (стабильная ремиссия) выборке для анализа, определяемой как рандомизированные субъекты, находящиеся в состоянии стабильной ремиссии в конце фазы оптимизации и получившие по меньшей мере 1 дозу интраназального исследуемого препарата и 1 дозу перорального антидепрессанта во время фазы поддержания. Один субъект со стабильным ответом, который был неправильно рандомизирован как находящийся в состоянии стабильной ремиссии, был включен в эту выборку для анализа. Первичным ожидаемым результатом эффективности является время от рандомизации до первой документации (самая ранняя дата) рецидива во время фазы поддержания у проходивших лечение эскетамином субъектов, которые достигли стабильной ремиссии в конце фазы оптимизации.
Рецидив определяется как любое из следующего:
• Общая оценка по шкале MADRS ≥ 22 для 2 последовательных оценок, разделенных 5-15 сутками. Дату второй оценки по шкале MADRS использовали как дату рецидива.
• Госпитализация при ухудшении депрессии или любом другом клинически релевантном событии, определяемом по клиническому заключению как предполагающее рецидив депрессивного заболевания, таком как попытка суицида, завершенный суицид, или госпитализация для предупреждения суицида. При госпитализации вследствие любого из этих событий дату госпитализации использовали как дату рецидива. В ином случае использовали дату события, если субъект не был госпитализирован.
• При соответствии обоим критериям рецидива более раннюю дату определяли как дату рецидива для этого субъекта.
Один субъект был рандомизирован рано (на 12 неделе ОП-фазы), но не начинал участие в ПО-фазе до следующей недели. Время до рецидива для этого субъекта рассчитывали по дате начала фазы поддержания. Первичный анализ эффективности проводили на полной (находящиеся в состоянии стабильной ремиссии пациенты) выборке для анализа, которая включая 175 находящихся в состоянии стабильной ремиссии пациентов и 1 пациента со стабильным ответом (который был ошибочно рандомизирован как находящийся в состоянии стабильной ремиссии), в конце фазы оптимизации после лечения интраназальным эскетамином плюс пероральный антидепрессант. Эти субъекты получили по меньшей мере 1 дозу интраназального исследуемого препарата и 1 дозу перорального антидепрессанта во время фазы поддержания. Как показано в Таблице 91 ниже, результаты свидетельствовали в пользу интраназального эскетамина+пероральный АД в отношении задержки рецидива по сравнению с пероральным АД+интраназальное плацебо. В целом 24 (26,7%) субъекта в группе интраназального эскетамина+пероральный АД и 39 (45,3%) субъектов в группе перорального АД+интраназальное плацебо имели рецидив во время фазы поддержания. На основании взвешенного комбинированного теста разница между группами лечения была статистически значимой (двустороннее p=0,003) и была ниже 0,046 (порог статистической значимости). Расчетное отношение рисков для интраназального эскетамина+пероральный АД по сравнению с пероральным АД+интраназальное плацебо на основании взвешенных оценок составляло 0,49 (95% ДИ: 0,29, 0,84) при использовании R. Расчет отношения рисков с помощью ADDPLAN был очень схожим 0,49 (0,29; 0,83).
b Отношение рисков и ДИ являются взвешенными оценками по Wassmer (2006) и были рассчитаны, используя R.
c Двустороннее P-значение учитывает конечную статистику критерия, которая представляет собой взвешенную комбинацию логранговой статистики критерия, рассчитанной по полной выборке для промежуточного анализа и по полной выборке для анализа для находящихся в состоянии стабильной ремиссии.
Н/О означает «не поддается оценке».
Кривые Каплана-Мейера для времени до рецидива для двух групп лечения представлены на Фиг. 51. Причины случаев рецидива у субъектов, имеющих рецидив, обобщены в Таблице 92. Наиболее распространенной причиной рецидива была общая оценка по шкале MADRS ≥ 22 для 2 последовательных определений, разделенных 5-15 сутками.
(N=176)
Анализ в подгруппах
На Фиг. 56 представлена форест-диаграмма, на которой показано отношение рисков на основании модели пропорциональных рисков Кокса для предварительно спланированных подгрупп. В целом, в случае подгрупп результаты свидетельствовали в пользу групп лечения эскетамином+пероральный АД.
Анализ чувствительности
Проводили два анализа чувствительности на полной (стабильная ремиссия) выборке для анализа, используя невзвешенный логранговый критерий и модель пропорциональных рисков Кокса с накоплением 63 событий и с учетом даты прекращения сбора данных на 59-м событии. Следует отметить, что анализ чувствительности на момент 59-ом события в действительности был выполнен при 61 рецидиве, так как 3 рецидива произошли в тот же день, что и 59-ое событие. Результаты представлены в Таблицах 93 и 94. Расчетное отношение рисков для интраназального эскетамина+пероральный АД по сравнению с пероральным АД+интраназальное плацебо составляло 0,47 (95% ДИ: 0,28, 0,78) с учетом 63 событий и 0,46 (0,27, 0,77) с учетом 61 события. Результаты согласуются с первичным анализом эффективности.
b Регрессионный анализ данных выживаемости на основании модели пропорциональных рисков Кокса с лечением в качестве фактора.
c Логранговый критерий.
Н/О означает «не поддается оценке».
b Регрессионный анализ данных выживаемости на основании модели пропорциональных рисков Кокса с лечением в качестве фактора.
c Логранговый критерий.
Н/О означает «не поддается оценке».
АНАЛИЗ ДРУГОГО ВТОРИЧНОГО ОЖИДАЕМОГО РЕЗУЛЬТАТА ЭФФЕКТИВНОСТИ
Время до рецидива у демонстрирующих стабильный ответ (но не ремиссию) пациентов
Время между рандомизацией субъекта и первой документацией (самая ранняя дата) рецидива в фазе поддержания сравнивали между группами лечения для субъектов в полной (стабильный ответ) выборке для анализа. Один субъект был рандомизирован рано (на 12 неделе ОП-фазы), но не начинал участие в ПО-фазе до следующей недели, а один субъект пропусти 1 неделю ПО-фазы. Время до рецидива для этих двух субъектов рассчитывали по дате начала фазы поддержания. Как показано в Таблице 95 ниже, результаты свидетельствовали в пользу интраназального эскетамина+пероральный АД в отношении задержки рецидива по сравнению с пероральным АД+интраназальное плацебо. В целом 16 (25,8%) субъектов в группе интраназального эскетамина+пероральный АД и 34 (57,6%) субъектов в группе перорального АД+интраназальное плацебо имели рецидив во время фазы поддержания. Разница между группами лечения была статистически значимой (двустороннее p < 0,001) при двустороннем логранговом критерии. Расчетное отношение рисков для интраназального эскетамина+пероральный АД по сравнению с пероральным АД+интраназальное плацебо на основании модели пропорциональных рисков Кокса с лечением в качестве фактора составляло 0,30 (95% ДИ: 0,16, 0,55). Кривые Каплана - Мейера для времени до рецидива для двух групп лечения представлены на Фиг. 52.
Медианное время до рецидива (95% ДИ) для группы интраназального эскетамина+пероральный АД составляло 635,0 (264,0; 635,0) суток; медианное время до рецидива (95% ДИ) для группы перорального АД+интраназальное впрыскивание плацебо составляло 88,0 (46,0; 196,0) суток на основании оценок Каплана - Мейера. Как отмечалось, оценку медианного времени до рецидива для группы эскетамина плюс пероральный АД следует интерпретировать с осторожностью, поскольку на нее сильно влияет один субъект, у которого до рецидива прошло много времени.
b Регрессионный анализ данных выживаемости на основании модели пропорциональных рисков Кокса с лечением в качестве фактора.
c Логранговый критерий.
Н/О означает «не поддается оценке».
БЕЗОПАСНОСТЬ
Обобщенная информация по всем нежелательным явлениям
Общая сводная информация по всем возникшим в ходе лечения нежелательным явлениям (НЯВЛ) во время фаз ИНД, ОП и ПО для выборок для анализа безопасности (ИНД), безопасности (ОП) и безопасности (ПО) (выборки для анализа безопасности (ОП) и безопасности (ПО) не включают переведенных субъектов, которые продолжали получать пероральный АД+плацебо (TEP) во время последующих фаз) представлены в Таблицах 96-98. В целом 76,9% субъектов испытали по меньшей мере одно НЯВЛ во время ИНД-фазы; 73,6% субъектов испытали по меньшей мере одно НЯВЛ во время ОП-фазы в выборке для анализа безопасности (ОП); 82,2% субъектов в группе, получавшей эскетамин+пероральный АД, и 45,5% субъектов в группе, получавшей пероральный АД+плацебо, испытали по меньшей мере одно НЯВЛ во время ПО-фазы в выборке для анализа безопасности (ПО).
b Нежелательное явление, которое началось во время индукционной фазы и привело к прекращению приема во время следующей фазы, считается возникшим в ходе лечения во время индукционной фазы.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
b Нежелательное явление, которое началось во время фазы оптимизации и привело к прекращению приема во время следующей фазы, считается возникшим в ходе лечения во время фазы оптимизации.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
b Нежелательное явление, которое началось во время фазы поддержания и привело к прекращению приема во время фазы последующего наблюдения, считается возникшим в ходе лечения во время фазы поддержания.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
НЯВЛ для переведенных субъектов, которые продолжали получать пероральный АД+плацебо во время фаз ОП и ПО, обобщены в Таблицах 99 и 100. В целом 61,6% субъектов TEP испытали по меньшей мере одно НЯВЛ во время ОП-фазы; 68,5% субъектов TEP испытали по меньшей мере одно НЯВЛ во время ПО-фазы;
b Нежелательное явление, которое началось во время фазы оптимизации и привело к прекращению приема во время следующей фазы, считается возникшим в ходе лечения во время фазы оптимизации.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений. Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
b Нежелательное явление, которое началось во время фазы поддержания и привело к прекращению приема во время фазы последующего наблюдения, считается возникшим в ходе лечения во время фазы поддержания.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений. Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Нежелательные явления, возникшие в ходе лечения во время фаз ИНД, ОП и ПО (> 5% субъектов в любой группе лечения), обобщены по группе лечения для выборки для анализа безопасности (ИНД), безопасности (ОП) и безопасности (ПО) и приведены в Таблицах 101-103. Наиболее распространенными НЯВЛ (> 10%) во время ИНД-фазы были вертиго (22,7%), головокружение (22,2%), тошнота (21,5%), дисгевзия (20,6%), сонливость (14,9%), головная боль (13,7%), параанестезия (11,0%), диссоциация (11,0%), ненормальное ощущение (10,8%), размытое зрение (10,3%) и седация (10,1%) в выборке для анализа безопасности (ИНД). Наиболее распространенными НЯВЛ во время ОП-фазы были вертиго (20,0%), дисгевзия (17,4%), сонливость (13,8%), головокружение (13,4%), головная боль (12,5%) и тошнота (10,5%) в выборке для анализа безопасности (ОП). Наиболее распространенными НЯВЛ в группе эскетамина+пероральный АД во время двойной слепой ПО-фазы были дисгевзия (26,3%), вертиго (25,0%), сонливость (21,1%), головокружение (20,4%), головная боль (17,8%), тошнота (16,4%), размытое зрение (15,8%), диссоциация (13,8%) и пероральная гипостезия (13,2%) в выборке для анализа безопасности (ПО). У >10% субъектов в группе перорального АД+плацебо в выборке для анализа безопасности (ПО) не наблюдалось ни одного НЯВЛ. Большинство НЯ наблюдались после введения дозы в дни введения дозы и разрешились в тот же день.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Нежелательные явления, возникшие в ходе лечения во время фаз ОП и ПО (> 5% субъектов) для субъектов TEP, обобщены для выборки для анализа безопасности (ОП_TEP) и безопасности (ПО_TEP) и приведены в Таблицах 104 и 105. Наиболее распространенным НЯВЛ для субъектов TEP во время ОП-фазы была головная боль (18,6%). Наиболее распространенными НЯВЛ для субъектов TEP во время ПО-фазы были инфекция верхних дыхательных путей (24,1%), головная боль (22,2%) и дисгевзия (14,8%).
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Нежелательные явления, приводящие к прекращению приема исследуемого препарата
22 субъекта прекратили прием интраназального исследуемого препарата в ИНД-фазе из-за возникших в ходе лечения нежелательных явлений (Таблица 106), а 8 субъектов прекратили прием перорального АД из-за НЯВЛ (Таблица 107) в выборке для анализа безопасности (ИНД). 5 субъектов прекратили прием интраназального исследуемого препарата в ОП-фазе из-за возникших в ходе лечения нежелательных явлений (Таблица 108), а 2 субъекта прекратили прием перорального АД из-за НЯВЛ (Таблица 109) в выборке для анализа безопасности (ОП). Субъекты, которые прекратили прием интраназального исследуемого препарата, могли продолжать принимать пероральный АД во время фазы последующего наблюдения при необходимости.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
1 субъект завершил прием интраназального препарата на 25 сутки и не соответствовал критериям для продолжения участия в следующей фазе. Действие, предпринятое в отношении нежелательного явления (депрессия), было указано как прекращение приема перорального АД. Этого субъекта посчитали завершившим исследование в сводной информации по завершению исследования/прекращению участия в исследовании для индукционной фазы.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
7 субъектов (4 субъекта в группе, получавшей эскетамин+пероральный АД, 3 субъекта в группе, получавшей пероральный АД+плацебо), прекратили прием интраназального исследуемого препарата в ПО-фазе из-за возникших в ходе лечения нежелательных явлений в выборке для анализа безопасности (ПО) (Таблица 110). Четыре из этих субъектов (3 субъекта в группе, получавшей эскетамин+пероральный АД, 1 субъект в группе, получавшей пероральный АД+плацебо) имели рецидив во время фазы поддержания и сообщили о нежелательном явлении, приведшем к рецидиву, поэтому прием интраназального исследуемого препарата был прекращен из-за этого явления. Эти субъекты могли продолжать принимать пероральный АД во время фазы последующего наблюдения при необходимости. Три субъекта в группе, получавшей эскетамин+пероральный АД, прекратили прием перорального исследуемого антидепрессанта в ПО-фазе из-за возникших в ходе лечения нежелательных явлений в выборке для анализа безопасности (ПО) (Таблица 111). Из них три субъекта имели рецидив во время фазы поддержания и сообщили о нежелательном явлении, приведшем к рецидиву, поэтому прием перорального АД был прекращен из-за этого явления. Ни один субъект в группе, получавшей пероральный АД+интраназальное плацебо, не прекратил прием перорального исследуемого АД в ПО-фазе из-за возникших в ходе лечения нежелательных явлений.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
4 субъекта (3 в группе эскетамина+пероральный АД, 1 в группе перорального АД+плацебо), которые имели рецидив (депрессия), включены в эту сводную информацию. Действие, предпринятое в отношении нежелательного явления рецидива, было указано как прекращение приема интраназального препарата. Их посчитали завершившими исследование в сводной информации по завершению исследования/прекращению участия в исследовании для фазы поддержания.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
3 субъекта, которые имели рецидив, включены в эту сводную информацию. Действие, предпринятое в отношении нежелательного явления рецидива, было указано как прекращение приема перорального АД. Их посчитали завершившими исследование в сводной информации по завершению исследования/прекращению участия в исследовании для фазы поддержания.
Ни один переведенный субъект, получавший плацебо, не прекратил прием интраназального исследуемого препарата или перорального АД в ОП-фазе из-за возникших в ходе лечения нежелательных явлений в выборке для анализа безопасности (ОП_TEP) (Таблицы 112 и 113).
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений. Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
2 переведенных субъекта, получавших плацебо, прекратили прием интраназального исследуемого препарата в ПО-фазе в выборке для анализа безопасности (ПО_TEP). Один из этих субъектов прекратил участие в ПО-фазе как из-за интраназального препарата, так и из-за и перорального АД, и включен в обе Таблицы 114 и 115.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Серьезные нежелательные явления
В этом исследовании летальных случаев зарегистрировано не было.
В этом исследовании было зарегистрировано 32 субъекта с 39 серьезными нежелательными явлениями (СНЯ). В общей сложности 13 субъектов испытывали серьезные возникшие в ходе лечения нежелательные явления (НЯВЛ) во время ИНД-фазы в выборке для анализа безопасности (ИНД) (Таблица 116).
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Три субъекта имели серьезные НЯВЛ, которые исследователи посчитали с большой вероятностью связанными с интраназальным эскетамином: дезориентация (1 сутки), суицидальная идеация (8 сутки), седация (22 сутки), а один субъект имел два серьезных НЯВЛ, которые посчитали с большой вероятностью связанными с интраназальным эскетамином: дисбаланс автономной нервной системы (5 сутки) и простой парциальный припадок (5 сутки). Один субъект имел серьезное НЯВЛ, которое исследователь посчитал вероятно связанным с интраназальным эскетамином: лакунарный инсульт (1 сутки). Один субъект имел серьезное НЯВЛ, которое посчитали возможно связанным с интраназальным эскетамином: гипотермия (10 сутки). Одиннадцать субъектов испытывали серьезные возникшие в ходе лечения нежелательные явления во время ОП-фазы в выборке для анализа безопасности (ОП) (Таблица 117). Ни одно НЯВЛ не посчитали возможно, вероятно или с большой вероятностью связанным с эскетамином.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Пять субъектов (4 в группе эскетамина+пероральный АД, 1 в группе пероральный АД+плацебо) в выборке для анализа безопасности (ПО) испытывали серьезные возникшие в ходе лечения нежелательные явления во время ПО-фазы (Таблица 118). Все явления посчитали не связанными с интраназальным препаратом или пероральным АД.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Два субъекта имели серьезные НЯ, которые посчитали не связанными с пероральным АД в фазе последующего наблюдения (Таблица 119). Один субъект испытал два серьезных НЯ, которые посчитали возможно связанными с пероральным АД.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Ни один из переведенных субъектов, получавших плацебо, не испытывал серьезные возникшие в ходе лечения нежелательные явления во время ОП-фазы в выборке для анализа безопасности (ОП_TEP) (Таблица 120).
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Один субъект TEP испытал серьезное НЯВЛ во время ПО-фазы, которое посчитали не связанным с интраназальным препаратом или пероральным АД (Таблица 121).
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Основные физиологические показатели
На Фиг. 53 и 54 представлены средние значения кровяного давления в динамике по времени по группе лечения в фазе поддержания.
Временное кровяное давление увеличивается до пика для группы, получавшей эскетамин, приблизительно через 40 минут после введения дозы и возвращается ближе к уровням до введения дозы через 1,5 часа после введения дозы.
Другие наблюдения безопасности
Применяемая клиницистами шкала диссоциативных симптомов (CADSS)
Оценку по применяемой клиницистами шкале диссоциативных состояний (CADSS) определяли перед началом введения каждой дозы, через 40 минут и 1,5 часа после введения дозы. Шкалу CADSS используют для оценки возникших в ходе лечения диссоциативных симптомов и изменений восприятия, а общая оценка находится в диапазоне от 0 до 92, причем более высокая оценка представляет более тяжелое состояние.
Диссоциативные симптомы и изменения восприятия, оцененные по шкале CADSS, позволяют предположить, что эти симптомы появились вскоре после начала введения дозы и разрешились через 1,5 часа после введения дозы (Фиг. 55).
Модифицированная оценка наблюдателем активности/седации (MOAA/S)
Модифицированную оценку наблюдателем активности/седации (MOAA/S) использовали для определения возникающей в ходе лечения седации с корреляцией с уровнями седации, определенными континуумом Американского общества анестезиологов (ASA). Оценки MOAA/S находились в диапазоне от 0 (отсутствие ответа на болевой стимул; соответствует континууму ASA для общей анестезии) до 5 (легко отвечает на имя, произнесенное нормальным тоном [бодрствование]; соответствует континууму ASA для минимальной седации).
Доля субъектов с седацией (определяемой по шкале MOAA/S ≤ 3) составляла ≤ 3,9% для группы эскетамина+пероральный АД для каждых суток введения дозы во всех фазах.
ЗАКЛЮЧЕНИЕ
Продолжение лечения эскетамином плюс пероральный АД продемонстрировало статистически значимое превосходство над лечением пероральным АД плюс интраназальное плацебо в отношении увеличения времени до рецидива у тех пациентов, кто находился в состоянии стабильной ремиссии через 16 недель лечения эскетамином плюс пероральный АД.
У субъектов, которые находились в состоянии стабильной ремиссии через 16 недель лечения эскетамином+пероральный АД, продолжение лечения эскетамином плюс пероральный АД продемонстрировало клинически существенное и статистически значимое (двустороннее p=0,003) превосходство над лечением пероральным АД+назальное впрыскивание плацебо согласно определению по увеличению времени до рецидива.
В целом 26,7% субъектов в группе эскетамина+пероральный АД и 45,3% субъектов в группе пероральный АД+назальное впрыскивание плацебо испытали рецидив; уровни рецидива за 6 месяцев на основании оценок Каплана - Мейера составляли 34,5% и 48,6%, соответственно. Рассчитанное отношение рисков (95% ДИ) эскетамина+пероральный АД по сравнению с пероральным АД+плацебо на основании взвешенных оценок составляло 0,49 (0,29, 0,84), что указывает на то, что в любой момент времени в течение исследования субъекты, которые находились в состоянии стабильной ремиссии и продолжали лечение эскетамином+пероральный АД, имели в среднем на 51% меньшую вероятность рецидива, чем субъекты, которые перешли на пероральный АД плюс плацебо. На основании оценок Каплана - Мейера медианное время до рецидива (момент времени, в который кумулятивная функция выживаемости равна 0,5 [или 50%]) для группы эскетамина плюс пероральный АД не поддавалось оценке (Н/О), так как эта группа никогда не достигала 50%. Медианное время до рецидива (95% ДИ) для группы перорального АД+назальное впрыскивание плацебо составляло 273 (97,0; Н/О) суток.
У субъектов, которые демонстрировали стабильный ответ (но не находились в состоянии стабильной ремиссии), через 16 недель лечения эскетамином+пероральный АД, продолжение лечения эскетамином плюс пероральный АД продемонстрировало клинически существенное и статистически значимое превосходство над лечением пероральным АД+назальное впрыскивание плацебо в отношении увеличения времени до рецидива (двустороннее p < 0,001).
Уровни рецидива за 6 месяцев на основании оценок Каплана - Мейера составляли 24,4% и 59,4%, соответственно. Расчетное отношение рисков для интраназального эскетамина+пероральный АД по сравнению с пероральным АД+назальное впрыскивание плацебо на основании модели пропорциональных рисков Кокса составляло 0,30 (95% ДИ: 0,16, 0,55), что указывает на то, что в любой момент времени в течение исследования субъекты, которые демонстрировали стабильный ответ и продолжали лечение эскетамином+пероральный АД, имели в среднем на 70% меньшую вероятность рецидива, чем субъекты, которые перешли на пероральный АД плюс назальное впрыскивание плацебо. Следует отметить, что оценку медианного времени до рецидива для группы эскетамина плюс пероральный АД следует интерпретировать с осторожностью, поскольку на нее сильно влияет один субъект, у которого до рецидива прошло много времени (т.е. 635 суток).
Как показано, продолжение лечения эскетамином плюс пероральный АД продемонстрировало статистически значимое превосходство над лечением одним пероральным АД в отношении увеличения времени до рецидива у тех пациентов, кто демонстрировал стабильный ответ (но не ремиссию), через 16 недель лечения эскетамином плюс пероральный АД.
Пример 5
Длительность лечения/длительность исследования
Каждый субъект участвовал максимум в 4 фазах: фазе скрининга длительностью до 4 недель (только напрямую включенные субъекты), 4-недельной открытой индукционной (ИНД) фазе (напрямую включенные субъекты и переведенные не демонстрирующие ответ субъекты), 48-недельной открытой фазе оптимизации/поддержания (ОП/ПО) (все демонстрирующие ответ субъекты из открытой ИНД-фазы текущего исследования и переведенные демонстрирующие ответ субъекты) и 4-недельной фазе последующего наблюдения. Максимальная продолжительность участия субъекта в исследовании ESKETINTRD3004 составляла 60 недель для напрямую включенных субъектов; 56 недель для переведенных не демонстрирующих ответ субъектов и 52 недели для переведенных демонстрирующих ответ субъектов. По оценкам размер выборки 750 включал по меньшей мере 300 субъектов, получавших лечение интраназальным эскетамином в течение 6 месяцев, и по меньшей мере 100 субъектов - в течение 12 месяцев. Кроме того, были включены переведенные субъекты из исследования 3005 для получения 100 пожилых субъектов, принимающих эскетамин. Смотрите Фиг. 57 в отношении дизайна исследования.
Выборки для анализа эффективности и безопасности
Анализ эффективности и безопасности основан на полной (ИНД) выборке для анализа и полной (ОП/ПО) выборке для анализа. Полная (ИНД) выборка для анализа определяется, как все субъекты, который получают по меньшей мере 1 дозу интраназального исследуемого препарата или 1 дозу перорального антидепрессанта в открытой ИНД-фазе (для напрямую включенных и переведенных не демонстрирующих ответ субъектов). Полная (ОП/ПО) выборка для анализа определяется, как все субъекты, который получают по меньшей мере 1 дозу интраназального исследуемого препарата или 1 дозу перорального антидепрессанта в ОП/ПО-фазе. Показатели безопасности включают когнитивную функцию в динамике по времени, возникшие в ходе лечения нежелательные явления (НЯВЛ), включая НЯВЛ, представляющие особый интерес, основные физиологические показатели в динамике по времени, Применяемую клиницистами шкалу диссоциативных состояний (CADDS) в динамике по времени и оценку по шкале Модифицированной оценки наблюдателем активности/седации (MOAA/S) ≤ 3. Показатели безопасности включают шкалу MADRS, которая состоит из 10 пунктов, которые охватывают все основные депрессивные симптомы, при этом каждый пункт оценивается от 0 (симптом отсутствует или в пределах нормы) до 6 (тяжелое или постоянное присутствие симптома). Общую оценку (от 0 до 60) рассчитывали путем суммирования оценок по каждому из всех 10 пунктов. Более высокая оценка представляет более тяжелое состояние.
Первичная цель
Первичная цель этого исследования заключается в оценке долгосрочной безопасности и переносимости интраназального эскетамина плюс новый назначенный пероральный антидепрессант у субъектов с ТРД с особым вниманием к потенциальному воздействию на когнитивную функцию, потенциальным связанным с лечением симптомам цистита и/или симптомов нижних мочевыводящих путей, а также потенциальным симптомам отмены и/или отдачи после прекращения лечения интраназальным эскетамином.
Вторичные цели
Оценка эффекта интраназального эскетамина плюс новый назначенный пероральный антидепрессант у субъектов с ТРД на:
• Безопасность и переносимость с особым вниманием к следующему:
- Возникшие в ходе лечения нежелательные явления (НЯВЛ), включая НЯВЛ, представляющие особый интерес
- Локальная назальная переносимость
- Эффект на частоту сердечных сокращений, кровяное давление, частоту дыхания и насыщенность крови кислородом
- Эффект на активность и седацию
- Потенциальные психореспираторные симптомы
- sis-подобный эффекты
- Диссоциативные симптомы
• Потенциальный эффект на суицидальные идеацию/поведение.
• Долгосрочная эффективность, включая эффект на:
Депрессивные симптомы (оцениваемые врачом и самостоятельно), общую тяжесть депрессивного заболевания, функциональное нарушение и связанную с ними недееспособность, симптомы беспокойства, а также связанное со здоровьем качество жизни и состояние здоровья
Частота ответа в динамике по времени, определяемая как:
- процент субъектов с ≥50% снижением относительно исходного уровня (ИНД-фаза) общей оценки по шкале для оценки депрессии Монтгомери - Асберг (MADRS),
- процент субъектов с ≥50% снижением относительно исходного уровня (ИНД-фаза) общей оценки по Опроснику по состоянию здоровья пациента из 9 пунктов (PHQ-9),
Частота ремиссии в динамике по времени, определяемая как:
- процент субъектов с общей оценкой MADRS ≤12,
- процент субъектов с общей оценкой PHQ-9 ≤5
ИНФОРМАЦИЯ О СУБЪЕКТАХ И ЛЕЧЕНИИ
Были проведены скрининг и включение всего 1161 субъектов в 123 центрах в 21 стране (Аргентина, Австралия, Австрия, Бельгия, Бразилия, Болгария, Финляндия, Франция, Германия, Италия, Республика Корея, Малайзия, Мексика, Польша, Южная Африка, Испания, Швеция, Тайвань, Турция, Соединенное Королевство и Соединенные Штаты). За исключением 338 случаев неудачного скрининга и 21 субъектов из центра US10025 из-за проблем с GCP, в исследование были включены 802 субъекта с диагнозом БДР по DSM-5 (Диагностико-статистическое руководство по психическим расстройствам, 5-ое издание).
Шестьсот девяносто один субъект был напрямую включен в исследование 3004, а 111 субъектов были переведены из исследования TRD3005 (88 не демонстрирующих ответ субъектов и 23 демонстрирующих ответ субъекта).
Это было открытое, многоцентровое, долгосрочное исследование для оценки безопасности и эффективности интраназального эскетамина плюс новый назначенный пероральный антидепрессант у субъектов с ТРД. Исследование включало 802 взрослых субъектов мужского и женского пола с ТРД. Из 802 субъектов, включенных в исследование, 691 (86,2%) были напрямую включены, а 111 (13,8%) были переведены из исследования ESKETINTRD3005 (88 не демонстрирующих ответ при вступлении в ИНД-фазу и 23 демонстрирующих ответ при вступлении в исследование в ОП/ПО-фазе). В полной выборке для анализа 686 (85,5%) субъектов были белыми, а 502 (62,6%) субъектов были женщинами. Средний возраст составлял 52,2 лет, в диапазоне от 18 до 86 лет. Гендерное распределение было аналогичным с исследованиями острой фазы 3 (преимущественно женщины), в то время как медианный возраст был несколько выше, что отражает включение пожилых субъектов. С 178 пожилыми субъектами, на долю которых приходится 22,2% выборки для анализа из включенных субъектов, это исследование удовлетворяло нормативным требованиям в отношении включения минимум 100 пожилых субъектов.
Из 779 напрямую включенных или переведенных из исследования TRD3005 не демонстрирующих ответ субъектов в полной (ИНД) выборке для анализа 580 (74,5%) завершили ИНД-фазу, а 198 (25,4%) выбыли досрочно. Большинство субъектов прекратили участие в ИНД-фазе из-за того, что субъект не соответствовал критериям для продолжения участия в следующей фазе (84 субъекта), и из-за нежелательного явления (52 субъекта). Из 603 субъектов, вошедших в ОП/ПО-фазу (включая 23 переведенных из исследования TRD3005 демонстрирующих ответ субъектов), 150 (24,9%) завершили ОП/ПО-фазу. Из 453 субъектов, выбывших до конца 48-недельной ОП/ПО-фазы, 331 выбыли из-за прекращения исследования спонсором (необходимое количество субъектов соответствовало достаточному уровню воздействия лечения). Другие наиболее частые причины прекращения участия были обусловлены «прекращением участия субъектом» (30 субъектов) и прекращением участия из-за «нежелательного явления» и «недостатка эффективности» (по 25 субъектов в каждом случае). Субъекты могли перейти в фазу последующего наблюдения из ИНД-фазы или ОП/ПО-фазы. В общей сложности 357 субъектов перешли в фазу последующего наблюдения, а 326 (91,3%) завершили фазу последующего наблюдения.
Из 802 включенных в исследование субъектов 1 субъект не получал интраназальный исследуемый препарат, но получал пероральный АД, а 1 субъект получал интраназальный исследуемый препарат, но не получал пероральный АД. Эти субъекты включены во все выборки для анализа из включенных субъектов. Смотрите Таблицы 122 и 123.
Субъекты получали гибкие дозы интраназального ЭСК в течение 1-ых 2 недель, затем фиксированные дозы (28 мг - только для пожилых субъектов, 56 мг или 84 мг во всех возрастных группах) плюс новый назначенный пероральный антидепрессант (один из следующих: сертралин, эсциталопрам, венлафаксин XR или флуоксетин). Дозу эскетамина вводили два раза в неделю во время ИНД-фазы. Во время ОП/ПО-фазы введение раз в неделю происходило на неделях 5-8. На 9-52 неделях ОП/ПО-фазы дозу эскетамина вводили один раз в неделю или один раз в две недели в зависимости от оценки MADRS с целью достижения наименьшей частоты для поддержания ремиссии. Переход на лечение один раз в две недели (если общая оценка MADRS составлял ≤ 12) или обратно к лечению один раз в неделю (если общая оценка MADRS составляла > 12) был возможен с 4-недельными интервалами, начиная с 8 недели. С 15 суток (пациенты < 65 лет) или 18 суток (пациенты ≥ 65 лет) доза назального впрыскивания эскетамина оставалась одинаковой. После начального периода титрования с повышением дозы доза пероральных антидепрессантов оставалась одинаковой. Для обоих препаратов допускалось уменьшение дозы в связи с переносимостью.
Готовность субъекта покинуть центр оценивали с учетом общих нежелательных явлений (включая головокружение, седацию, изменения восприятия, кровяное давление): Приблизительно 60-65% из субъектов были готовы покинуть центр через 1 час после введения дозы, и более 95% субъектов были готовы покинуть центр через 1,5 ч после введения дозы во время визитов ИНД-фазы; процент субъектов, готовых покинуть центр, составлял приблизительно 65-70% через 1 час после введения дозы и 97-99% через 1,5 ч после введения дозы в ОП/ПО-фазе.
Информация о завершении исследования/прекращении участия в исследовании
Из 779 субъектов из полной (ИНД) выборки для анализа (напрямую включенные и переведенные не демонстрирующие ответ субъекты), 580 (74,5%) субъектов завершили 28-суточную ИНД-фазу, а 198 (25,4%) выбыли досрочно. Результаты представлены в Таблице 124. Большинство субъектов прекратили участие в ИНД-фазе из-за того, что субъект не соответствовал критериям для продолжения участия в следующей фазе (< 50% улучшение общей оценки MADRS) (84 субъекта), и из-за нежелательного явления (52 субъекта).
Из 603 субъектов, вошедших в ОП/ПО-фазу (включая 23 переведенных из исследования TRD3005 демонстрирующих ответ субъектов), 150 (24,9%) субъектов завершили 48-недельную ОП/ПО-фазу. Из 453 субъектов, выбывших до конца 48-недельной ОП/ПО-фазы, 331 выбыли из-за прекращения исследования спонсором (необходимое количество субъектов соответствовало достаточному уровню воздействия лечения). Результаты представлены в Таблице 125. Наиболее частые причины прекращения участия были обусловлены «прекращением исследования спонсором» (331 субъект), «прекращением участия субъектом» (30 субъектов) и прекращением участия из-за «нежелательного явления» и «недостатка эффективности» (по 25 субъектов в каждом случае). (Примечание: исследование было прекращено после достижения целевых показателей воздействия эскетамина (по меньшей мере 300 субъектов, проходящих лечение в течение 6 месяцев, и 100 субъектов, проходящих лечение в течение 12 месяцев).
Субъекты могли перейти в фазу последующего наблюдения из ИНД-фазы или ОП/ПО-фазы. В общей сложности 357 субъектов перешли в фазу последующего наблюдения, а 326 (91,3%) завершили фазу последующего наблюдения.
2 субъекта прекратили лечение во время ОП/ПО-фазы. После последней менструации одна субъект дважды приняла ЭСК в дозе 56 мг, а 2-ая субъект один раз приняла ЭСК в дозе 84 мг. В случае обеих беременностей во время первого триместра происходил спонтанный аборт, оценка исследователем связи с эскетамином была не применима. Во время исследования наблюдался один случай воздействия эскетамина на ребенка через отца. Беременность партнерши субъекта протекала без осложнений, и она родила нормального доношенного ребенка в ходе спонтанных родов.
Демографические и исходные характеристики
Данные по демографическим и исходным характеристикам приведены в Таблице 126 для выборки для анализа из всех включенных субъектов. Большинство субъектов, включенных в исследование, составляли женщины (62,6%) и белые (85,5%). Средний (СО) возраст всех субъектов составлял 52,2 (13,69) лет, в диапазоне от 18 до 86 лет.
b Статус гипертензии классифицируется как «Да», если в истории болезни указана гипертензия.
Исходная психиатрическая история для выборки для анализа из всех включенных субъектов представлена в Таблице 127. Средняя (СО) исходная общая оценка по шкале MADRS составляла 31,4 (5,39), в диапазоне от 19 до 49.
2=58,0% ≥ 4 =16,6%
a категория C-SSRS: Отсутствие событий=0; Суицидальная идеация=1, 2, 3, 4, 5; Суицидальное поведение=6, 7, 8, 9, 10
b Число антидепрессантов с отсутствием ответа (определяемым как ≤ 25% улучшение или 26%-< 50% улучшение для напрямую включенных субъектов и определяемым как ≤ 25% улучшение для переведенных субъектов), взятое за по меньшей мере 6 недель во время текущего эпизода, полученное из MGH-ATRQ во время первого скринингового визита.
c Напрямую включенные субъекты должны принимать ≥ 2 пероральных антидепрессантов во время текущего эпизода депрессии, а переведенные субъекты должны принимать ≥ 1 пероральных антидепрессантов во время текущего эпизода.
d Субъекты из 3005, которые не демонстрировали ответ на 1 АД и продемонстрировали проспективное отсутствие ответа на 2-ой АД во время скрининга исследования 3005.
Степень воздействия
Число доз интраназального исследуемого препарата во время ИНД-фазы приведено в Таблице 128.
Обобщенные данные по средней, режимной и конечной дозе интраназального исследуемого препарата во время ИНД-фазы приведены в Таблице 129. На 25 сутки ИНД-фазы 28/675 (4,1%) субъектов получали дозу 28 мг эскетамина, 298/675 (44,1%) получали дозу 56 мг эскетамина и 349/675 (51,7%) получали дозу 84 мг эскетамина.
Конечная доза представляет собой последнюю ненулевую дозу, принятую во время индукционной фазы.
Степень воздействия интраназального исследуемого препарата во время комбинированный ИНД- и ОП/ПО-фаз приведено в Таблице 130.
Частота субъектов с 6-месячным и 12-месячным приемом эскетамина представлена в таблице 131.
Обобщенные данные по средней, режимной и конечной дозе интраназального исследуемого препарата во время ОП/ПО-фазы приведены в Таблице 132.
Конечная доза представляет собой последнюю ненулевую дозу, принятую во время фазы оптимизации/поддержания.
На 48 неделе ОП/ПО-фазы 7/143 (4,9%), 69/143 (48,3%), 1/143 (0,7%) и 66/143 (46,2%) субъектов получали дозу 28 мг, дозу 56 мг, дозу 70 мг и дозу 84 мг эскетамина, соответственно. Начиная с недели 4 (ОП/ПО) частоту сеансов интраназального лечения можно было корректировать (при необходимости) с фиксированными 4-недельными интервалами. Из 603 субъектов, проходивших лечение интраназальным эскетамином во время ОП/ПО-фазы 275 (47,6%) субъектов перешли с введения дозы один раз в неделю на один раз в две недели на 4 неделе (ОП/ПО). Большинство субъектов не меняли схему введения дозы в течение оставшихся недель ОП/ПО-фазы. Смотрите Таблицу 133.
В Таблице 134A показаны изменения схемы введения дозы во время ОП/ПО-фазы.
Схема: введение дозы эскетамина один раз в неделю или введение дозы эскетамина один раз в две недели.
Число изменений: 0=доза один раз в неделю в течение всей ОП/ПО-фазы; 1=1 одно изменение с одного раза в неделю на один раз в две недели (РДН); 2=изменение с РДН обратно на один раз в неделю; 3=2 изменения с одного раза в неделю на РДН, остановка на РДН; 4=2 изменения с одного раза в неделю на РДН и обратно на один раз в неделю; 5=3 изменения с одного раза в неделю на РДН, остановка на РДН; 6=3 изменения с одного раза в неделю на РДН и обратно на один раз в неделю.
a Приблизительно 22% возвращались к одному разу в неделю, всего 46% на схеме один раз в неделю, и 16% возвращались к одному разу в две недели (всего 54% на схеме один раз в две недели).
БЕЗОПАСНОСТЬ
Когнитивная функция
Первичная цель исследования заключалась в оценке потенциальных эффектов эскетамина на когнитивную функцию. Потенциальное воздействие эскетамина на когнитивную функцию оценивали с помощью компьютеризированной когнитивной батареи Cogstate.
Число проанализированных субъектов
Полная выборка для анализа из включенных субъектов: Всего проанализировали 796 субъектов.
Выборка для анализа последующего наблюдения, включающая только субъектов, которые были включены в фазу последующего наблюдения: Всего проанализировали 356 субъектов.
Проанализированные моменты времени
Полная выборка для анализа из включенных субъектов:
Открытая индукционная фаза
• Исходный уровень
• Сутки 28
Фаза оптимизации/поддержания
• Неделя 20
• Неделя 32
• Неделя 44
• Конечный момент времени (последний момент времени в фазе оптимизации/поддержания получения данных от субъекта)
Выборка для анализа последующего наблюдения:
• Исходный уровень
• Конечный момент времени (предыдущая фаза - последний момент времени в последней фазе лечения, в которой участвовали субъекты перед переходом в фазу последующего наблюдения)
• Неделя 4
Критерии оценки
Батарея Cogstate
• Теста на обнаружение (DET; для определения уровня внимания)
• Тест на идентификацию (IDN; для определения уровня внимания)
• Тест на запоминание с одной картой (OCL; для определения зрительной памяти)
• Тест n-назад(ONB; для определения рабочей памяти)
• Тест на обучение в лабиринте Гротона (GML; для определения исполнительной функции)
Тест на заучивание слов Хопкинса, пересмотренный (HVLT-R)
• Общее запоминание (для определения вербального научения)
• Отсроченное запоминание (для определения вербальной памяти)
• Истинно положительные значения (для определения памяти распознавания)
• Индекс различения распознавания (для определения памяти распознавания)
В целом, средние по группе показатели испытаний когнитивной функции для оценки внимания [тесты на обнаружение (DET) и идентификацию (IDN), оценивающие время простой реакции и реакции выбора, соответственно], зрительной памяти, рабочей памяти, исполнительной функции и отсроченной вербальной памяти, а также памяти распознавания для выборки для анализа из всех включенных субъектов и отдельно для субъектов < 65, либо демонстрировали улучшение относительно исходного уровня, либо субъекты оставались на исходном уровне во время ИНД-фазы и ОП/ПО-фазы. Такой же профиль когнитивной деятельности наблюдался у субъектов в возрасте ≥ 65 лет, за исключением тестов для оценки внимания/скорости обработки информации (DET и IDN), в случае которых наблюдалось снижение от исходного уровня, начиная с 20 недели, с наибольшим снижением на 44 неделе исследования. Несмотря на то, что размер выборки для пожилых субъектов снизился на 44 неделе, снижение показателей внимания в этой возрастной группе также наблюдалось в анализе завершивших исследование.
Наибольшее среднее снижение скорости выполнения для DET и IDN (-0,1032 и -0,0587, соответственно), наблюдаемое на 44 неделе исследования, было меньшим по сравнению со стандартным отклонением (СО) измерений на исходном уровне (0,15955 и 0,09465, для ATN и IDN, соответственно). Для справки, наблюдаемая разница была ниже, чем снижение внимания, наблюдаемое при приеме 1 мг алпразолама три раза в сутки. В предыдущем исследовании на здоровых субъектах среднее по группе снижение показателей внимания порядка 1 СО после введения 1 мг алпразолама три раза в сутки было сочтено клинически значимым (Maruff et al. 2006).
Исходная оценка данных об уровне пациента показывает, что у 17 из 28 пожилых субъектов, завершивших исследование, наблюдалось постоянное снижение показателей внимания (достоверный индекс изменений [RCI) < -1,65 по меньшей мере на 2 измерений]. Снижение показателей в тестах на обнаружение и идентификацию согласуется в терминах величины и направления среди субъектов в возрасте от 65 до < 75 и субъектов в возрасте ≥ 75 лет, хотя размер выборки для возрастной группы ≥ 75 является очень маленьким и не позволяет делать достоверные выводы об изменении.
Показатели когнитивной функции, включая показатели внимания/времени реакции, оставались стабильными во время периода последующего наблюдения как у пожилых, так и у более молодых субъектов. Не наблюдалось постоянного снижения когнитивной функции, о котором сообщалось в популяциях, злоупотребляющих применением больших (часто ежедневных) доз кетамина.
2 субъекта испытали нежелательное явление нарушения памяти в ИНД-фазе и 2 субъекта - в ОП/ПО-фазе. Два субъекта испытали возникшее в ходе лечение когнитивное нарушение во время ИНД-фазы. Ни один субъект не испытал возникшее в ходе лечение когнитивное нарушение во время ОП/ПО-фазы.
В целом 723/802 (90,1%) субъектов испытали по меньшей мере одно НЯВЛ во время ИНД- и ОП/ПО-фаз. Наиболее распространенными (≥ 10%) НЯВЛ во время ИНД- и ОП/ПО-фаз были головокружение (33,0%), тошнота (25,1%), головная боль (24,9%), диссоциация (22,4%), сонливость (16,7%), дисгевзия и гипостезия (11,8% в каждом случае), рвота и вертиго (10,8% в каждом случае) и вирусная инфекция верхних дыхательных путей (10,2%).
76 (9,5%) субъектов прекратили прием интраназального исследуемого препарата во время ИНД- или ОП/ПО-фазы из-за возникших в ходе лечения нежелательных явлений. Наиболее распространенными НЯ, приведшими к прекращению приема интраназального исследуемого препарата, были тревожность (1,1%), суицидальная идеация (0,9%), депрессия, головокружение и повышение кровяного давления (0,7% в каждом случае), диссоциация (0,6%) и мышечная слабость (0,5%). Эти субъекты могли продолжать принимать пероральный АД во время фазы последующего наблюдения при необходимости. 33 (4,1%) субъекта прекратили прием перорального исследуемого антидепрессанта во время ИНД- или ОП/ПО-фазы из-за возникших в ходе лечения нежелательных явлений. Наиболее распространенными НЯ, приведшими к прекращению приема перорального исследуемого антидепрессанта, были тревожность (0,9%) и суицидальная идеация (0,6%).
В общей сложности 423 (52,7%) субъектов испытали нежелательные явления, указывающие на потенциальное злоупотребление, зависимость и синдром отмены препарата, во время ИНД- и ОП/ПО-фаз.
Временное увеличение кровяного давления для группы, получавшей эскетамин+пероральный АД, достигало пикового значения через 40 минут после введения дозы, при этом максимальное среднее повышение (во все дни введения дозы во время соответствующей фазы) систолического/диастолического КД составляло 9,6/5,6 мм рт.ст. и 8,6/5,2 мм рт.ст. во время ИНД- и ОП/ПО-фаз, соответственно. Повышение систолического и диастолического кровяного давления после введения дозы наблюдали во время обеих фаз исследования. Субъекты с гипертензией и без нее имели схожую величину среднего повышения СКД и ДКД.
Во время исследования наблюдались временные изменения систолического и диастолического кровяного давления, что согласуется с исследованиями острых доз эскетамина. Большинство субъектов, которые выбыли из-за повышения кровяного давления, сделали это после первых 2 сеансов введения дозы.
Частота возникновения в ходе лечения острой гипертензии у субъектов приведена в Таблице 134B. Частота возникновения острой гипертензии была почти в 3 раза выше у субъектов с анамнезом гипертензии по сравнению с субъектами без гипертензии.
В целом диссоциативные симптомы и симптомы изменения восприятия, оцененные по шкале CADSS, позволяют предположить, что эти симптомы появились вскоре после начала введения дозы и разрешились через 1,5 часа после введения дозы. Эти симптомы становились слабее при повторном введении дозы в течение времени. Интенсивность симптомов, наблюдаемых по CADSS после введения дозы, снижалась при введении повторных доз и оставалась низкой в течение всей ОП/ПО-фазы.
Эффективность: Наблюдалось клинически существенное улучшение депрессивных симптомов: среднее изменение (СО) общей оценки MADRS от исходного уровня (ИНД) до конечного момента времени (ИНД) составляло -16,4 (8,76) для эскетамина+пероральный АД с исходным значением (ИНД) (СО) 31,2 (5,27). Среднее изменение (СО) от исходного уровня (ОП/ПО) до конечного момента времени (ОП/ПО) составляло 0,3 (8,12) для эскетамина+пероральный АД с исходным значением (ОП/ПО) (СО) 11,0 (4,52), что указывает на сохранение эффекта антидепрессанта. Аналогично, медианное изменение (диапазон) от исходного уровня (ИНД) до конечного момента времени (ИНД) составляло -18,0, а от исходного уровня (ОП/ПО) до конечного момента времени (ОП/ПО) составляло 0 для эскетамина+пероральный АД.
Среднее изменение (СО) от исходного уровня (ИНД) общей оценки PHQ-9 до конечного момента времени (ИНД) составляло -8,9 (6,67) для эскетамина+пероральный АД. Среднее изменение (СО) от исходного уровня (ОП/ПО) общей оценки PHQ-9 до конечного момента времени (ОП/ПО) составляло -0,2 (5,65) для эскетамина+пероральный АД.
Эти результаты согласуются с MADRS в направлении изменений в конце обеих фаз исследования. Результаты общей эффективности в ИНД-фазе согласуются с результатами, полученными в исследованиях острой эффективности (3001 и 3002). Такое улучшение подтверждалось сдвигами в общей степени тяжести заболевания по оценкам CGI-S: процент субъектов, которые были в норме/на грани/имели легкое заболевание, увеличился с 2,7% на исходном уровне до 63,8% в конце ИНД-фазы. Смотрите Фиг. 58.
В Таблицах 134-137 представлены обобщенные данные о средних значениях и средних изменениях в течение времени в показателях обнаружения и внимания (время простой реакции) для субъектов в возрасте < 65 лет и ≥ 65 лет, соответственно.
b Число ошибок, меньшая оценка=лучшее выполнение
c Точность выполнения, большая оценка=лучшее выполнение
d Большее изменение относительно исходного уровня соответствует лучшему выполнению.
b Число ошибок, меньшая оценка=лучшее выполнение
c Точность выполнения, большая оценка=лучшее выполнение
d Большее изменение относительно исходного уровня соответствует лучшему выполнению.
b Число ошибок, меньшая оценка=лучшее выполнение
c Точность выполнения, большая оценка=лучшее выполнение
d Большее изменение относительно исходного уровня соответствует лучшему выполнению.
b Число ошибок, меньшая оценка=лучшее выполнение
c Точность выполнения, большая оценка=лучшее выполнение
d Большее изменение относительно исходного уровня соответствует лучшему выполнению.
В Таблицах 138-141 представлены обобщенные данные о средних значениях и средних изменениях в течение времени в показателях идентификации и внимания (время простой реакции) для субъектов в возрасте < 65 лет и ≥ 65 лет, соответственно. Смотрите также Фиг. 59.
b Число ошибок, меньшая оценка=лучшее выполнение
c Точность выполнения, большая оценка=лучшее выполнение
d Большее изменение относительно исходного уровня соответствует лучшему выполнению.
b Число ошибок, меньшая оценка=лучшее выполнение
c Точность выполнения, большая оценка=лучшее выполнение
d Большее изменение относительно исходного уровня соответствует лучшему выполнению.
b Число ошибок, меньшая оценка=лучшее выполнение
c Точность выполнения, большая оценка=лучшее выполнение
d Большее изменение относительно исходного уровня соответствует лучшему выполнению.
b Число ошибок, меньшая оценка=лучшее выполнение
c Точность выполнения, большая оценка=лучшее выполнение
d Большее изменение относительно исходного уровня соответствует лучшему выполнению.
2 субъекта испытали нарушения памяти в ИНД-фазе и 2 субъекта - в ОП/ПО-фазе. Два субъекта испытали возникшее в ходе лечение когнитивное нарушение во время ИНД-фазы. Ни один субъект не испытал возникшее в ходе лечение когнитивное нарушение во время ОП/ПО-фазы.
В целом, проведенные перед введением дозы когнитивные оценки продемонстрировали общее сохранение или улучшение когнитивной функции относительно исходного уровня у субъектов в возрасте < 65 лет. Данные по когнитивной функции у субъектов ≥ 65 лет также продемонстрировали сохранение или улучшение в большинстве исследуемых доменов, но наблюдались относительно небольшие снижения двух показателей внимания. Данные об уровне субъекта, лежащие в основе этих видимых изменений, дополнительно оценивают для определения того, в какой степени они могут быть клинически существенными у некоторых субъектов.
Обобщенная информация по всем нежелательным явлениям
Обобщенная информация по всем возникшим в ходе лечения нежелательным явлениям (НЯВЛ) во время ИНД- и ОП/ПО-фаз представлена в Таблицах 142A и 142B. В целом 90,1% субъектов испытали по меньшей мере одно НЯВЛ во время ИНД- и ОП/ПО-фаз.
b Нежелательное явление, которое началось во время индукционной фазы или фазы оптимизации/поддержания и привело к прекращению приема во время фазы последующего наблюдения, считается возникшим в ходе лечения в этой таблице.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Нежелательные явления, возникшие во время ИНД- и ОП/ПО-фаз (> 5% субъектов) обобщены для выборки для анализа из всех включенных субъектов в Таблице 143. Наиболее распространенными (≥ 10%) НЯВЛ во время ИНД- и ОП/ПО-фаз были головокружение (33,0%), тошнота (25,1%), головная боль (24,9%), диссоциация (22,4%), сонливость (16,7%), дисгевзия и гипостезия (11,8% в каждом случае), рвота и вертиго (10,8% в каждом случае) и вирусная инфекция верхних дыхательных путей (10,2%). Частота возникновения тошноты была наибольшей на 1 сутки введения дозы (10,7%) и снижалась в последующие сутки введения дозы в диапазоне от 0,0 до 4,4%. Частота возникновения рвоты была наибольшей на 1 сутки введения дозы (3,3%) и снижалась в последующие сутки введения дозы в диапазоне от 0,0 до 1,9%.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
На Фиг. 60-62 показан уровень нарушения для EQ-5D-5L посредством определения тревожности/депрессии, обычной активности и боли/дискомфорта, соответственно. Оценки находились в диапазоне 1-5: 1 (отсутствует), 2 (легкое), 3 (умеренное), 4 (тяжелое) и 5 (экстремальное).
Серьезные нежелательные явления
Во время ОП/ПО-фазы этого исследования было зарегистрировано два летальных исхода.
Один 60-летний мужчина с анамнезом гипертензии и операции на венах умер вследствие острой сердечной и дыхательной недостаточности на 113 сутки лечения эскетамином, что посчитали маловероятно связанным с эскетамином. Этот субъект ранее не испытывал каких-либо сердечных нежелательных явлений во время лечения эскетамином и пероральным АД и имел нормальное кровяное давление во время исследования. Субъект получал эскетамин в дозе 56 мг, причем последнюю дозу он принял за 5 суток до смерти.
Одна 55-летняя женщина умерла вследствие суицида на 188 сутки исследования. Этот субъект демонстрировал ремиссию депрессивных симптомов, на что указывает оценка MADRS 7 и 9, полученная во время 2 последних клинических визитов за 13 суток и 6 суток до происшествия, соответственно. Это событие посчитали не связанным с эскетамином. Субъект получал эскетамин в дозе 84 мг, причем последнюю дозу она приняла за 13 суток до смерти.
Всего 55 (6,9%) субъектов испытали в общей сложности 68 серьезных возникших в ходе лечения нежелательных явлений во время ИНД- и ОП/ПО-фаз для выборки для анализа из всех включенных субъектов. Четыре субъекта имели серьезные НЯВЛ, оцененные исследователями как связанные (возможно, вероятно или с большой вероятностью) с интраназальным эскетамином: делирий, тревожность и бред, суицидальная идеация и попытка суицида. Следующие СНЯ привели к прекращению лечения эскетамином: 5 случаев суицидальной идеации; 2 случая попыток суицида, депрессии, тревожности и токсичного воздействия нескольких агентов (указанных как «интоксикация золпидемом и оксазепамом» у одного участника, и «интоксикация лекарственным препаратом» у другого); и по 1 событию каждого из следующего: алкоголизм, суицидальная депрессия (депрессия с суицидальными мыслями), бред, делирий и гепатит B.
СНЯ делирия было зарегистрировано на 127 сутки для субъекта, который продолжал лечения для предупреждения алкоголизма в течение исследования (хотя на тот момент не злоупотреблял), в течение нескольких минут после введения дозы 56 мг эскетамина. Это СНЯ оценили как с большой вероятностью связанное с ЭСК и не связанное с пероральным АД. В течение этого события у субъекта наблюдался период возбуждения со случайными движениями конечностей с последующим 10-минутным периодом невосприимчивости к стимулам. До этого события этот субъект хорошо переносил эскетамин. Во время этого визита не проводили никаких тестов на наркотики или алкоголь. Субъект был госпитализирован, выведен из исследования и восстановился после события за 18 суток. Результаты КТ-сканирования, МРТ и ЭЭГ были нормальными. Точная причина делирия остается неизвестной, нельзя исключить употребление других веществ.
Сообщалось о СНЯ тревожности и бреда до введения дозы ЭСК на 5 сутки вместе с СНЯ злоупотребления алкоголем (которое было оценено исследователем как не связанное с эскетамином). Этот субъект имел в анамнезе психоз и бред, а также отрицательные скрининговые пробы на алкоголь. Впоследствии субъект признался в избыточном употреблении алкоголя. Субъект был госпитализирован и выведен из исследования.
СНЯ, связанные с почечной функцией, включали пиелонефрит, острый пиелонефрит и 1 случай тубулоинтерстициального нефрита. Все эти СНЯ разрешались после соответствующего лечения без последствий, а субъекты продолжили участие в исследовании.
Три субъекта имели серьезные НЯВЛ, которые исследователи посчитали связанными с пероральным антидепрессантом: гастроэнтерит, микроскопический колит и суицидальная идеация. Смотрите Таблицы 144A и 144B.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
(N=604)
Случай суицидальной идеации (умеренной интенсивности) начался на 207 сутки исследования и привел к прекращению участия. Также у этого субъекта была зарегистрирована акатизия на 206 сутки (легкая). Субъект восстановился от суицидальной идеации в течение 30 суток.
Нежелательные явления, приводящие к прекращению приема исследуемого препарата
Число возникших в ходе лечения нежелательных явлений (НЯВЛ), приведших к прекращению исследуемого лечения, было низким для данного исследования и популяции пациентов с ТРД. Прекращение участия из-за НЯ во время ИНД-фазы происходило в большинстве после начальных сеансов лечения ЭСК.
76 (9,5%) субъектов прекратили прием интраназального исследуемого препарата во время ИНД- или ОП/ПО-фазы из-за возникших в ходе лечения нежелательных явлений (Таблицы 145A и 145B). В целом 53/779 (6,8%) субъектов прекратили прием интраназального исследуемого препарата во время ИНД-фазы и 23/603 (3,8%) субъектов прекратили прием интраназального исследуемого препарата во время ОП/ПО-фазы из-за возникших в ходе лечения нежелательных явлений. Это намного меньше, чем в случае других препаратов для БДР, например, уровня 23,7% в 52-недельном исследовании кветиапина у субъектов с БДР/ТРД (Berman et al., 2011) и 24,5% в течение 76 недель лечения симбиаксом субъектов с БДР/ТРД (Corya, Long-term antidepressant efficacy and safety of olanzapine/fluoxetine combination: a 76-week open-label study. Journal of Clinical Psychiatry: 64 (11) p.1349-56. 2003).
Наиболее распространенными НЯ, приведшими к прекращению приема интраназального исследуемого препарата, были тревожность (1,1%), суицидальная идеация (0,9%), депрессия, головокружение и повышение кровяного давления (0,7% в каждом случае), диссоциация (0,6%) и мышечная слабость (0,5%). Эти субъекты могли продолжать принимать пероральный АД во время фазы последующего наблюдения при необходимости. 33 (4,1%) субъекта прекратили прием перорального исследуемого антидепрессанта во время ИНД- или ОП/ПО-фазы из-за возникших в ходе лечения нежелательных явлений (Таблица 146). В целом 20/779 (2,6%) субъектов прекратили прием перорального антидепрессанта во время ИНД-фазы и 14/603 (2,3%) субъектов прекратили прием перорального исследуемого антидепрессанта во время ОП/ПО-фазы из-за возникших в ходе лечения нежелательных явлений. Один субъект прекратил прием перорального антидепрессанта во время ИНД-фазы и перешел на другой пероральный антидепрессант. Наиболее распространенными НЯ, приведшими к прекращению приема перорального исследуемого антидепрессанта, были тревожность (0,9%) и суицидальная идеация (0,6%). Двадцать шесть субъектов прекратили участие во время ИНД- или ОП/ПО-фазы из-за нежелательных явлений, связанных как с интраназальным препаратом, так и с пероральным АД, и включены в обе таблицы.
У одного субъекта диагностировали СНЯ тяжелого гепатита B и рака яичников умеренной степени тяжести, которые исследователи посчитали не связанными с ЭСК. У этого субъекта наблюдались клинически значимые повышения печеночных проб, что изначально подозревали как проявление лекарственного поражения печени. При этом данный субъект продемонстрировал улучшение лабораторных анализов и клинической картины в ответ на начатое противовирусное лечение, что вместе с положительной серологией на гепатит подтвердило диагноз гепатита B.
Среди 76 субъектов с НЯВЛ, приведшими к прекращению приема интраназального эскетамина во время ИНД- или ОП/ПО-фаз, 51 субъект имел НЯ, которые посчитали связанными (возможно, вероятно или с большой вероятностью) с эскетамином. Среди 33 субъектов с НЯВЛ, приведшими к прекращению приема перорального АД во время ИНД- или ОП/ПО-фаз, 14 субъектов имели НЯ, которые посчитали связанными с пероральным АД.
Девять субъектов выбыли из-за повышения кровяного давления или гипертензии. Шесть из этих субъектов досрочно выбыли в ИНД-фазе (на 1, 2, 4, 4, 8 и 9 сутки исследования). Четыре из этих субъектов соответствовали критериям повышенного кровяного давления (СКД ≥ 200 мм рт.ст. и/или ДКД ≥ 120 мм рт.ст. для субъектов в возрасте < 65 лет и СКД ≥ 190 и ДКД ≥ 110).
В общей сложности 7 субъектов имели суицидальную идеацию, а 2 предприняли попытки суицида во время ИНД- или ОП/ПО-фаз, что привело к прекращению приема исследуемого препарата эскетамина. В случае одной попытки суицида у 46-летней женщины на 44 сутки была передозировка лекарственного препарата и интоксикация монооксидом углерода. Это СНЯ оценили как представляющее угрозу для жизни, вероятно связанное с эскетамином и не связанное с пероральным АД. Этот субъект демонстрировал ответ, но при этом имел хронические и острые жизненные факторы стресса. Субъект восстановился от этого события в течение 11 суток.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Частота определяется числом субъектов, испытавших по меньшей мере одно нежелательное явление, а не количеством явлений.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Цистит, инфекции мочевыводящих путей и другие нарушения со стороны почек и мочевыводящей системы
Случаев интерстициального цистита или язвенного цистита не наблюдалось. Пять (0,6%) субъектов испытали возникший во время лечения цистит во время ИНД- и ОП/ПО-фаз. У трех субъектов наблюдался умеренный цистит длительностью 2, 4 и 9 суток, и у одного субъекта наблюдался умеренный цистит в течение 7 суток во время ИНД-фазы. У одного субъекта наблюдался умеренный цистит в течение 7 суток во время ОП/ПО-фазы. В случае этих событий не проводили изменение дозы или прекращение приема интраназального эскетамина или перорального антидепрессанта. Смотрите Таблицу 147.
Нежелательные явления закодированы с помощью MedDRA, версия 20.0.
Всего у 65 субъектов (8,1%) наблюдались инфекции мочевыводящих путей, а у 84 субъектов (10,5%) наблюдались нарушения со стороны почек и мочевыводящей системы во время ИНД- и ОП/ПО-фаз.
НЯВЛ цистита имели преимущественно легкую степень тяжести и возникали преимущественно во время ИНД-фазы (13, 15, 20, 40 и 78 сутки), были временными и приводили к самоограничению, что указывает на инфекционную этиологию случаев ИМП. Четыре субъекта сообщили о недержание мочи, которое оценили как связанное с ЭСК (т.е. возможно, вероятно или с большой вероятностью).
Нежелательные явления, связанные со злоупотреблением, зависимостью и отменой препарата
В Таблице 148 представлены нежелательные явления, связанные со злоупотреблением, зависимостью и отменой препарата, во время ИНД- и ОП/ПО-фаз. В общей сложности 423 (52,7%) субъектов испытали нежелательные явления, связанные со злоупотреблением, зависимостью и отменой препарата, во время ИНД- и ОП/ПО-фаз.
Основные физиологические показатели и масса тела
На Фиг. 63 и 64 представлены средние значения кровяного давления в динамике по времени во время ИНД- и ОП/ПО-фаз.
Временное увеличение кровяного давления для группы, получавшей эскетамин+пероральный АД, достигало пикового значения через 40 минут после введения дозы, при этом максимальное среднее повышение (во все дни введения дозы во время соответствующей фазы) систолического КД составляло 9,6 и 8,6 во время ИНД- и ОП/ПО-фаз, соответственно. Максимальное среднее повышение (во все дни введения дозы) диастолического КД составляло 5,6 и 5,2 во время ИНД- и ОП/ПО-фаз, соответственно.
Всего 18 субъектов имели систолическое кровяное давление ≥ 180 и 18 имели диастолическое кровяное давление ≥ 110 в любое время во время ИНД- и ОП/ПО-фаз, соответственно.
Шесть субъектов прекратили прием интраназального эскетамина в связи с повышенным кровяным давлением и 1 субъект прекратил прием перорального АД в связи с повышенным кровяным давлением. Эти субъекты могли продолжать принимать пероральный АД во время фазы последующего наблюдения при необходимости.
Средняя масса тела на исходном уровне ИНД-фазы составляла 78,53 кг, а на 28 сутки ИНД-фазы составляла 78,24 кг. Средняя масса тела на исходном уровне ОП/ПО-фазы составляла 79,01 кг, а в конечный момент времени составляла 79,16 кг.
В целом назальная переносимость была благоприятной, о чем свидетельствуют результаты осмотра полости носа, проведенного исследователем перед введением дозы, и опросника по назальной безопасности, заполняемого субъектами перед введением дозы и через 1 час после введения дозы.
Никаких очевидных, клинически значимых, связанных с препаратом изменений относительно исходного уровня в средних параметрах лабораторной гематологии и/или биохимии не наблюдалось во время ИНД- и ОП/ПО-фазы. У 13 субъектов (1,6%) было зарегистрировано бессимптомное повышение АЛТ > 3 x ВПН (верхний предел нормы), большинство случаев которого произошли в первые 1-3 месяца лечения. У большинства субъектов эти повышения нормализовались в течение лечения. Постоянных изменений в АЛТ не наблюдалось. Был зарегистрирован один случай предполагаемого ЛПП с повышением АЛТ/АСТ > 5 x и билирубином > 2 x ВПН, при этом была зафиксирована альтернативная этиология (гепатит B).
Другие наблюдения безопасности
Применяемая клиницистами шкала диссоциативных симптомов (CADSS)
Оценку по применяемой клиницистами шкале диссоциативных состояний (CADSS) определяли перед началом введения каждой дозы, через 40 минут и 1,5 часа после введения дозы. Шкалу CADSS используют для оценки возникших в ходе лечения диссоциативных симптомов и изменений восприятия, а общая оценка находится в диапазоне от 0 до 92, причем более высокая оценка представляет более тяжелое состояние.
Диссоциативные симптомы и изменения восприятия, оцененные по шкале CADSS, позволяют предположить, что эти симптомы появились вскоре после начала введения дозы и разрешились через 1,5 часа после введения дозы (Фиг. 65). Наибольшее увеличение оценки CADSS через 40 минут наблюдалось после первой дозы эскетамина во время индукционной фазы.
Модифицированная оценка наблюдателем активности/седации (MOAA/S)
MOAA/S использовали для определения возникающей в ходе лечения седации с корреляцией с уровнями седации, определенными континуумом Американского общества анестезиологов (ASA). Оценки MOAA/S находились в диапазоне от 0 (отсутствие ответа на болевой стимул; соответствует континууму ASA для общей анестезии) до 5 (легко отвечает на имя, произнесенное нормальным тоном [бодрствование]; соответствует континууму ASA для минимальной седации).
65/777 (8,4%) субъектов имели оценку MOAA/S ≤ 3 в любое время в течение ИНД-фазы, а 42/603 (7,0%) субъектов имели оценку MOAA/S ≤ 3 в любое время в течение ОП/ПО-фазы.
У двух субъектов наблюдалась глубокая седация, эквивалентная оценке 0 по шкале MOAA/S, во время одного из визитов исследования. У одного из этих субъектов было зарегистрировано снижение оценки MOAA/S на 8 сутки (ИНД) как нежелательное явление седации умеренной интенсивности с длительностью 1 час 30 минут. Субъект прекратил прием эскетамина в связи с тошнотой и дискомфортом ЖКТ, возникшими в один день. У другого субъекта зарегистрировали НЯ нечувствительности к стимулам на 15 сутки (ИНД) сильной интенсивности, а дозу эскетамина снизили с 84 мг до 56 мг.
Кроме того, у 3 субъектов наблюдалось временное снижение MOAA/S до оценки 1 во время одного визита и в один момент времени после введения дозы (4 сутки (ИНД), 11 сутки (ИНД) и 10 неделя (ОП/ПО)).
Не одному субъекту в исследовании не требовалась сердечно-сосудистая реанимация. Субъекты с оценкой 0 по шкале MOAA/S имели нормальную пульсоксиметрию и не имели снижение кровяного давления или частоты дыхания. В целом, частота дыхания и пульсоксиметрия оставались стабильными после введения ЭСК во время ИНД- и ОП/ПО-фазы. Хотя у отдельных субъектов наблюдались снижения пульсоксиметрии < 93%, эти снижения были бессимптомными, субъекты оставались в сознании, и не наблюдалось случаев угнетения дыхательной функции.
Подшкала положительных симптомов краткой психиатрической оценочной шкалы (BPRS+)
На Фиг. 66 представлен график, показывающий среднее значение ( ± СП) для общей оценки по подшкале положительных симптомов краткой психиатрической оценочной шкалы в динамике по времени во время фаз индукции и оптимизации/поддержания дозы (выборка для анализа из всех включенных субъектов) для примера 5.
Анализ эффективности
Анализ эффективности проводили на полной выборке для анализа для ИНД- и ОП/ПО-фаз, которая включала всех включенных субъектов, которые получили по меньшей мере 1 дозу интраназального исследуемого препарата и 1 дозу перорального исследуемого антидепрессанта в соответствующих фазах.
Шкала Монтгомери-Асберг для оценки депрессии (MADRS)
MADRS состоит из 10 пунктов, каждый из которых оценивается от 0 (симптом отсутствует или в пределах нормы) до 6 (тяжелая степень или постоянное присутствие симптома). Общую оценку (от 0 до 60) рассчитывали путем суммирования оценок по каждому из всех 10 пунктов. Более высокая оценка представляет более тяжелое состояние.
Среднее изменение (СО) от исходного уровня (ИНД) общей оценки MADRS до конечного момента времени (ИНД) составляло -16,4 (8,76) для эскетамина+пероральный АД. Среднее изменение (СО) от исходного уровня (ОП/ПО) общей оценки MADRS до конечного момента времени (ОП/ПО) составляло 0,3 (8,12) для эскетамина+пероральный АД. Смотрите Фиг. 67.
Частоты ответа (≥ 50% улучшение относительно исходного уровня (ИНД) общей оценки по шкале MADRS) и ремиссии (общая оценка по шкале MADRS ≤ 12) представлены для ИНД- и ОП/ПО-фаз в Таблицах 149 и 150, соответственно.
В конечный момент времени ИНД-фазы частота ответа составляла 78,4%, а частота ремиссии составляла 47,2%; среди пациентов, перешедших в фазу ОП/ПО, 76,5% демонстрировали ответ, а 58,2% находились в состоянии ремиссии в конечный момент времени. Функциональное восстановление, определяемое по SDS, сопровождалось некоторым запаздыванием после улучшения настроения. В конечный момент времени ИНД-фазы частота ремиссии, определяемая по SDS, в конечный момент времени ИНД-фазы (21,1%, наблюдаемых случаев). Частота ремиссии удвоилась в течение ОП/ПО-фазы (от 25,2% на 4 неделе до 51,1% на 48 неделе, наблюдаемый случай). Смотрите Фиг. 68.
Общая оценка по опроснику по состоянию здоровья пациента (PHQ-9)
PHQ-9 представляет собой шкалу самоанализа из 9 пунктов для оценки депрессивных симптомов. Каждый пункт оценивается по 4-балльной шкале (0=никогда, 1=несколько дней, 2=более половины из всех дней и 3=практически каждый день) с общей оценкой в диапазоне 0-27. Более высокая оценка указывает на более тяжелую депрессию.
Среднее изменение (СО) от исходного уровня (ИНД) общей оценки PHQ-9 до конечного момента времени (ИНД) составляло -8,9 (6,67) для эскетамина+пероральный АД. Среднее изменение (СО) от исходного уровня (ОП/ПО) общей оценки PHQ-9 до конечного момента времени (ОП/ПО) составляло -0,2 (5,65) для эскетамина+пероральный АД. Смотрите Фиг. 69.
Пример 6
Хотя кетамин хорошо известен своим нейротоксическим потенциалом у крыс, эскетамин в этом отношении не исследовали. Исследование нейротоксичности с однократным и повторным введением дозы проводили на самках крыс линии Спрег - Доули в возрасте 12-14 недель, чтобы исследовать, приводит ли интраназальная инстилляция эскетамина-HCl в однократной дозе до 72 мг или в течение 14 последовательных суток в дозах до 54 мг/сутки к гистопатологическим свидетельствам нейродегенерации (некроза) в головном мозге. Выраженные клинические признаки, связанные с центральной нервной системой, отмечались у крыс, обработанных эскетамином-HCl, включая связанное с дозой слюноотделение, атаксию, снижение двигательной активности, сопровождаемое лежанием на боку и каталепсией, повышение двигательной активности, брадипноэ и слышимое дыхание. Масштабные гистопатологические исследования головного мозга не показали морфологических признаков нейрональной дегенерации. У крыс, получивших одну подкожную инъекцию положительного контроля (+)MK-801 малеата, наблюдали нейрональный некроз, как и ожидалось, в задней поясной извилине и ретроспленальной коре. Терапевтический интервал эскетамина на основании Cмакс и ППК по сравнению с максимальной дозой 84 мг для людей был приблизительно 60- и 86-кратным в исследовании нейротоксичности с однократной дозой и приблизительно 17- и 11-кратным в 14-суточном в исследовании нейротоксичности, соответственно.
1. Введение
Это исследование включает исследования нейротоксичности с применением однократной дозы и повторяемой в течение 14 суток дозы интраназально вводимого эскетамина на самках крыс в возрасте 12-14 недель. Эти исследования проводили для исследования, индуцировали ли однократные или повторные интраназальные введения эскетамина нейродегенеративные изменения в головном мозге крысы. Результаты исследования использовали для оценки безопасного терапевтического интервала воздействия эскетамина по сравнению с максимальной дозой эскетамина, вводимого интраназально взрослым пациентам с депрессией в клинических исследованиях.
2. Материалы и способы
2.1. Испытательный центр
Исследования нейротоксичности с применением однократной дозы и повторяемой в течение 14 суток дозы проводили в Janssen Research & Development (JRD), отделении Janssen Pharmaceutica NV, в Берсе, Бельгия. Janssen Pharmaceutica NV является фармацевтической компанией Johnson & Johnson. Испытательный центр был утвержден Международной ассоциацией оценки и аккредитации лабораторных исследований на животных (AAALAC). Все животные получали гуманную обработку и уход в соответствии с Европейской конвенцией (Европейская конвенция (ETS № 123) для защиты позвоночных животных, используемых в экспериментальных и других научных целях. Директива Совета от 24 ноября 1986 г. (86/609/EEG) о сближении законодательств, правил и административных положений государств-членов в отношении защиты животных, используемых для экспериментальных и других научных целей, дополненная Рекомендацией комиссии от 18 июня 2007 г. (2007/526/EC) по руководствам в отношении размещения и ухода за животными, используемыми для экспериментальных и других научных целей) и бельгийскими (Закон Бельгии от 18 октября 1991 г.): Защита позвоночных животных, используемых в экспериментальных и других научных целях. Королевский указ от 14 ноября 1993 г. о защите лабораторных животных) руководствами, а также принципами эвтаназии, изложенными в Докладе Американской ветеринарной медицинской ассоциации (Американская ветеринарная медицинская ассоциация (AVMA), 2013 г. Руководство по AVMA по эвтаназии животных, издание 2013 г. Американская ветеринарная медицинская ассоциация (AVMA), Шаумбург). Исследования проводили в соответствии с этическими протоколами JRD, которые были одобрены местным комитетом по этике.
2.2. Нормативные положения
Все виды деятельности осуществляли в соответствии с современными требованиями к лабораторным исследованиям (ТЛИ) ОЭСР (Организация экономического сотрудничества и развития; ОЭСР, 1998 г. Серия ОЭСР о принципах проведения лабораторных исследований и контроля за их соблюдением. № 1, Принципы проведения лабораторных исследований. Доступ с: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=env/mc/chem(98)17&doclanguage=en). Принципы ТЛИ ОЭСР признаются регуляторными органами во всех странах-членах организации ОЭСР, как описано в документе о взаимном признании данных (12 мая 1981 г. - C(81)30/окончательный вариант, с поправками от 26 ноября 1997 г. - C(97) 186/окончательный вариант). Аутсорсинг и мониторинг биоанализа эскетамина осуществлялся в соответствии с принципами ТЛИ ОЭСР в отношении организации и управления многоцентровыми исследованиями (ОЭСР, 2002 г. Серия ОЭСР о принципах проведения лабораторных исследований и контроля за их соблюдением. № 13, Применение принципов ТЛИ ОЭСР для организации и управления многоцентровыми исследованиями. Доступ с: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?doclanguage=en&cote=env/jm/mono(2002)9). Биоаналитическую часть исследования проводили в соответствии с нормами ТЛИ FDA США для неклинических лабораторных исследований, 21 CFR часть 58 (FDA, 2014. FDA 21 CFR часть 58, Требования к лабораторным исследованиям для неклинических лабораторных исследований. Управление по контролю за качеством пищевых продуктов и лекарственных средств США (FDA)), и в соответствии с надлежащими стандартными процедурами JRD. Дизайн этого исследования основывался на международных руководствах (Европейский союз (2001). Директива 2001/83/EC Европейского парламента и Совета от 6 ноября 2001 г. да о кодексе Сообщества, касающемся лекарственных продуктов для использования человеком; FDA, 2007 г. Токсикологические принципы оценки безопасности прямых пищевых добавок и цветовых добавок в пищевых продуктах. Redbook 2000. FDA (Управление по контролю за качеством пищевых продуктов и лекарственных средств США); Руководство ICH S3A, 1994 г. Примечание для руководства по токсикокинетикие: оценка системного воздействия в исследованиях токсичности; Руководство ICH M3 (Rs), 2009 г. Руководство по неклиническим исследованиям безопасности для проведения клинических испытаний на людях и регистрации фармацевтических препаратов; JMHW, 1995 г. Руководство по неклиническим исследованиям лекарственных препаратов на японском языке, 1995 г. Nippo Yakuji, Токио. JMHW (Министерство здравоохранения и социального обеспечения Японии); ОЭСР, 2008 г. Тест № 407: 28-Суточное исследование пероральной токсичности в повторной дозой на грызунах, Руководство ОЭСР по тестированию химических веществ, раздел 4, OECD Publishing, Париж. DOI: http://dx.doi.org/10.1787/9789264070684-en).
2.3. Животные и содержание
Для этих исследований использовали самок беспатогенных (БП) крыс линии Спрег - Доули, которые на момент начала введения дозы были в возрасте приблизительно 12-14 недель и весили 227-293 граммов в исследовании однократной дозы и 235-296 граммов в 14-суточном исследовании с повторной дозой. Крысы поставлялись компанией Charles River (Зульцфельд, Германия). Группу животных содержали в полисульфоновых клетках с площадью пола 3000 см2, а в качестве материала подстилки предоставлялись кукурузные початки (размер 12, Eurocob, France). Обогащение среды состояло из отходов грызунов (Bio Serv, США) и осиновых деревянных блоков (Datesand, Соединенное Королевство). В помещение для животных кондиционировали воздухом (собственная подача фильтрованного свежего воздуха) и обеспечивали 12-часовой световой цикл (300 люкс на высоте 1 м). Животным кормили ad libitum облученным пеллетированным диетическим рационом R/M-H от компании Ssniff (Германия). Гидрогельtm (поставляемый компанией Bio-services, Уден, Нидерланды) был предусмотрен для крыс, которым вводили дозу MK-801, в исследовании с однократной дозой.
2.4. Составы
2.4.1. Эскетамин-HCl
Исследуемый клинический продукт представляет собой водный раствор лекарственного вещества, эскетамина гидрохлорида (HCl). Этот состав интраназально вводят пациентам, используя двойное устройство для назального впрыскивания, которое доставляет по одному впрыскиванию в каждую ноздрю. При комнатной температуре максимальная растворимость эскетамина-HCl при pH 4,5 составляет 207 мг/мл (т.е. 180 мг основания эскетамина/мл при применении фактора 1,15 для преобразования соли эскетамина-HCl в концентрацию основания эскетамина). Состав хранили при 37°C в целях обеспечения стабильности до введения дозы.
2.4.2. (+)MK-801 малеат
В качестве положительного контроля использовали водный раствор (+)MK-801 малеата с апирогенной водой, содержащей NaOH/HCl для pH 4,5 и маннит для изотоничности. Для преобразования соли малеата в основные уровни дозы использовали фактор 1,52.
2.4.3. Базовый раствор
В качестве базового раствора использовали клинический исследуемый состав без эскетамина-HCl.
2.5. Экспериментальный дизайн исследования нейротоксичности с однократной дозой
Эскетамин-HCl интраназально закапывали в дозах 0 (базовый раствор), 36, 54 или 72 мг (выраженных в виде основания эскетамина) самкам крыс в один день. Уровни дозы получали с помощью 2, 3 или 4 последовательных интраназальных закапываний, соответственно, в обе ноздри в объеме 50 мкл на ноздрю. При каждом закапывании дозу во вторую ноздрю вводили сразу после первой ноздри. Интервал между последовательными закапываниями в обе ноздри составлял 5 минут.
Контрольная группа, получавшая базовый раствор, получала 4 последовательных интраназальных закапывания базового раствора в обе ноздри (по 50 мкл на ноздрю) с 5-минутными интервалами.
Положительный контроль (+)MK-801 малеат инъецировали подкожно в дозе 1 мг/кг массы тела (выраженной в виде основания). Инъецируемый объем составлял 5 мл/кг.
Группы, которым вводили базовый раствор и дозу 36 и 54 мг, состояли из 24 животных на группу, тогда как группы, получавшие дозу 72 мг, и группы положительного контроля включали по 30 животных на группу. В группы животных основного исследования, которым вводили дозу эскетамина-HCl, было добавлено по четыре сателлитных животных в целях токсикокинетики (ТК). У этих животных плазменное воздействие эскетамина измеряли в день введения дозы в различные моменты времени вплоть до 24 часов после первого закапывания.
Для подготовки к исследованию гистопатологии головного мозга, проводили перфузию всех животных основного исследования при вскрытии. Перед перфузией крыс анестезировали изофлураном. После инъекции 0,1 мл гепарина кровь крыс продували физиологическим раствором путем внутриартериального введения иглы в брюшную аорту. После этого проводили перфузию крысы глутаральдегидом, 3% в фосфате калия, 0,09 M, и 1,4% сахарозы. Из-за логистических ограничений максимум 30 животных могли подвергнуть полной процедуре перфузии всего в заданный день. Таким образом, для всех 5 групп животных основного исследования применяли ступенчатое начало обработки. Следовательно, подгруппам из 4 животных основного исследования из групп, получавших базовый раствор и дозы 36 и 54 мг, и 5 животных основного исследования из групп, получавших дозу 72 мг и групп положительного контроля, дозу вводили в течение 6 последовательных дней соответственно. В Таблице 151 обобщена информация по дизайну исследования нейротоксичности с применением однократной дозы.
2 Однократная подкожная инъекция
Животных, получавших дозу контрольного базового раствора, положительного контроля и эскетамина-HCl, умерщвляли через 48 часов (т.е. первых 12 или 15 крыс на группу) или через 96 часов после времени введения дозы (т.е. последних 12 или 15 крыс на группу). Ткань головного мозга крыс, получавших дозу контрольного базового раствора, положительного контроля и эскетамина-HCl, обрабатывали в одно время и одинаковым образом в партиях из 4 или 5 животных на группу.
2.6. Экспериментальный дизайн 14-суточного исследования нейротоксичности с повторной дозой
Эскетамин-HCl вводили в каждую ноздрю самок крыс в объеме 50 мкл на ноздрю один, два или три раза в сутки путем интраназального закапывания в течение 14 последовательных суток в дозах 0 (базовый раствор), 18, 36 или 54 мг/сутки. При каждом закапывании дозу во вторую ноздрю вводили сразу после первой ноздри. Интервал между последовательными закапываниями в обе ноздри составлял 5 минут при введении дозы 2 или 3 раза в сутки.
Контрольная группа, получавшая базовый раствор, получала 3 последовательных интраназальных закапывания базового раствора в обе ноздри (по 50 мкл на ноздрю) с 5-минутными интервалами в течение 14 последовательных суток.
Группа положительного контроля получала одну подкожную инъекцию (+)MK-801 за 48 часов до вскрытия животных, обработанных контрольным базовым раствором и эскетамином-HCl. Уровень дозы (+)MK-801 малеата составлял 1 мг/кг массы тела.
Каждая группа животных основного исследования состояла из 12 самок крыс. В группы животных основного исследования, которым вводили дозу эскетамина-HCl, было добавлено по четыре сателлитных животных в целях ТК. Плазменное воздействие эскетамина измеряли у этих животных на первые и последние сутки введения дозы в различные моменты времени вплоть до 24 часов после первого закапывания.
Из-за логистических ограничений, связанных со процедурой полной перфузии животных при вскрытии, в исследовании использовали ступенчатое начало. Следовательно, все 5 групп животных основного исследования были разделены на три подгруппы по 4 животных в каждой. Для каждой из этих подгрупп обработку начинали в другие сутки. Процедура перфузии являлась такой же, как описана для исследования с однократной дозой. В Таблице 152 обобщена информация по дизайну 14-суточного исследования нейротоксичности с применением повторной дозы.
Всех животных, обработанных базовым раствором и эскетамином-HCl, умерщвляли через 48 часов после последнего закапывания. Всех животных, обработанных (+)MK-801 малеатом, умерщвляли через 48 часов после введения дозы. Ткань головного мозга крыс, получавших дозу контрольного базового раствора, положительного контроля и эскетамина-HCl, обрабатывали в одно время и одинаковым образом в партиях из 4 животных на группу.
2.7. Исследуемые параметры в исследованиях нейротоксичности с однократной дозой и повторной дозой
Всех животных по меньшей мере один раз в сутки проверяли в отношении плохого здоровья, аномального поведения или необычного внешнего вида, клинических признаков, токсичных или фармакологических реакций, агонии или смерти. Кроме того, проводили клинические наблюдения для крыс, получавших дозу эскетамина-HCl, в день введения дозы в моменты от 0 до 5 минут, 15 и 30 минут и через 1, 2, 4, 6 часов после введения дозы, в конце рабочего дня и через 24 часа после последнего закапывания в исследовании с однократной дозой, и в первый день введения дозы, через одну неделю и вплоть до конца периода введения дозы в исследовании с повторной дозой. Ежедневно измеряли массу тела и прирост массы тела. Потребление пищи измеряли еженедельно в исследовании с повторной дозой.
В день умерщвления проводили полный физический осмотр крыс в состоянии натощак и записывали массу тела. Выживших животных анестезировали путем ингаляционного воздействия смеси изофлуран (IsoFlo® Zoetis, Бельгия)/кислород и умерщвляли путем обескровливания через сонную артерию. Для всех животных проводили полное вскрытие и регистрировали все макроскопические изменения. Всех умерщвленных в конце исследования крыс или тех, которые были умерщвлены до конца, перфузировали 4% раствором параформальдегида в исследовании с однократной дозой и глутаральдегидом, 3% в калий-фосфатном буфере, 0,09 М, с 1,4% сахарозы, в исследовании с повторной дозой.
Получали образцы ткани головного мозга и обрабатывали в обычном режиме. Получали срезы вырезанной и заключенной ткани и окрашивали гематоксилином и эозином (HE). Гистообработку и окрашивание проводили в трех партиях из 4 животных на группу (каждая партия, таким образом, содержала 4 животных из обработанных контрольным базовым раствором, положительным контролем и трех обработанных эскетамином-HCl групп). В исследовании с однократной дозой дубликатные срезы головного мозга окрашивали Fluoro-Jade (FJ).
Все ткани, имеющие выраженные аномалии, исследовали макроскопически. Гистопатологическое исследование проводили на 7 уровнях ткани головного мозга в соответствии с Bolon (2013. Toxicol. Pathol. 41(7), 1028-1048) от обработанных контрольным базовым раствором, положительным контролем и высокой дозой эскетамина-HCl групп в исследовании с однократной дозой и во всех группах в исследовании с повторной дозой.
Микроскопическое обнаружение нейронального некроза в PCG/RSC головного мозга оценивали односторонне в зависимости от количества некротических нейронов, наблюдаемых на протяжении всей длины структуры PCG/RSC, видимой в срезе ткани (как правило, приблизительно 3-5 мм), используя следующие критерии:
Степень 1: минимальное гистологическое изменение (< 10 некротических нейронов),
Степень 2: небольшое гистологическое изменение (10-20 некротических нейронов),
Степень 3: умеренное гистологическое изменение (20-30 некротических нейронов) или
Степень 4: выраженное гистологическое изменение (> 30 некротических нейронов)
2.8. Статистический анализ
Значимость разницы между группой каждой дозы и группой базового раствора оценивали с помощью одностороннего критерия точной вероятности Фишера для летальности и гистопатологии, двустороннего критерия точной вероятности Фишера для клинических наблюдений и двустроннего U-критерия Манна - Уитни для массы тела и прироста массы. Не проводили статистический анализ для потребления пищи и выраженной патологии. Все тесты проводили при уровне значимости пять процентов (α=0,05).
3. Результаты
3.1 Исследование с однократной дозой
3.1.1. Эскетамин-HCl
Когда самкам крыс вводили однократную дозу эскетамина-HCl до 54 мг, никаких связанных с исследуемым препаратом изменений летальности или выраженной патологии не наблюдали. Не наблюдали никаких релевантных эффектов на массу тела при дозе до 72 мг.
При дозе 72 мг два животных основного исследования и два сателлитных TK животных умерли в течение 30 минут после получения 4 закапываний эскетамина-HCl в обе ноздри. Клинические признаки, отмеченные непосредственно перед смертью, включали атаксию, брадипноэ, сильное снижение общей активности, лежание на боку, каталепсию (ригидность позы) и слюноотделение. У одной из этих крыс также наблюдалось сужение глазных щелей. Провели вскрытие двух животных основного исследования и наблюдали выраженные изменения в легких (обесцвечивание [у одной крысы] и набухание) и плевральной полости (кровоизлияние). Причину смерти определить не удалось. Тем не менее, посчитали, что смерть была связана с комбинацией большого объема дозы и высокой дозы эскетамина-HCl.
В группах всех доз эскетамина-HCl были отмечены заметные клинические наблюдения. Непосредственно после введения дозы наблюдали слюноотделение, слышимое дыхание, атаксию и небольшое повышение общей активности. После этого наблюдали сильное снижение активности, сопровождаемое лежанием на боку, брадипноэ, птозом и каталепсией, начиная с < 15 минут после введения дозы и позже. Продолжительность этих симптомов зависела от дозы эскетамина-HCl. При дозе 36 мг эти симптомы наблюдались в основном до 15-30 минут, при дозе 54 мг - от 30 минут до одного часа и при дозе 72 мг - до 1-2 часов после последнего закапывания. Во время этого периода сужение глазных щелей происходило дозозависимым образом и поражало от приблизительно одной трети животных при дозе 36 мг до приблизительно двух третей животных при дозе 72 мг. После того, как снижение активности постепенно сходило на нет, у большинства животных снова наблюдалась атаксия, длящаяся приблизительно 1,5 часа при дозе 36 мг, 1,5-3 часа при дозе 54 мг и до 3 часов или дольше при дозе 72 мг. Атаксия сопровождалась небольшим (или иногда умеренным) повышением общей активности у приблизительно половины крыс. Слышимое дыхание было отмечено после введения дозы у приблизительно половины животных, получавших дозы 36 мг, и у 2 или 3 крыс при дозах 54 и 72 мг, соответственно. Все симптомы, за исключением слышимого дыхания, полностью сходили на нет в течение 24 часов. У одной получившей высокую дозу крысы слышимое дыхание все еще наблюдалось на 2 сутки. Дополнительную информацию можно найти в Таблице 153. Временная динамика снижения и повышения общей активности проиллюстрирована на Фиг. 70.
Несмотря на то, что наблюдались летальность и выраженные ЦНС-симптомы (например, каталепсия), гистопатологические поражения отсутствовали в мозгу крыс, обработанных эскетамином-HCl, умерщвленных через 48 или 96 часов после введения одной дозы 72 мг. В частности, не были обнаружены поражения Олни в PCG/RSC любого из обработанных эскетамином-HCl животных. Дополнительную информацию можно найти в Таблице 155 (смотрите Раздел 3.3).
Терапевтический интервал на основании Cмакс и ППК для эскетамина по сравнению с максимальной клинической дозой 84 мг был приблизительно 60- и 86-кратным, соответственно.
3.2. Исследование с повторной дозой
3.2.1. Эскетамин-HCl
Не наблюдали связанной с исследуемым препаратом смертности. Массу тела, прирост массы и потребление пищи оставались неизменными, когда самкам крыс вводили эскетамин-HCl в дозе до 54 мг/сутки в течение 14 суток.
При всех уровнях дозы эскетамина-HCl регистрировали дозозависимое увеличение продолжительности атаксии и слюноотделения. Несколько повышенная общая активность отмечалась у всех получавших дозу эскетамина-HCl животных, начиная с 5 минут после введения и продолжаясь до 1 или 2 часа после введения последней суточной дозы. Эта повышенная двигательная активность наблюдалась ежедневно при дозе 18 мг/сутки, от 3-5 суток до завершения исследования при дозе 36 мг/сутки и, главным образом, в течение последней недели исследования при дозе 54 мг/сутки. Также наблюдалось небольшое или резкое снижение общей активности у всех животных с дозозависимым повышением тяжести и длительности (от 15 или 30 минут до 1 или 2 часов после введения дозы). Иногда замечали лежание на боку в течение первых суток исследования при дозе 36 и 54 мг/сутки. После того как снижение активности постепенно сходило на нет, снова наблюдалось повышение общей активности, сопровождаемое атаксией (Фиг. 71). При дозе 36 или 54 мг/сутки почти у всех животных были отмечены периоды дыхательной аномалии (брадипноэ, слышимое дыхание), которые наблюдались несколько дней в течение всего исследования, от 5 до 30 минут или от 1 до 4 часов после введения дозы. Дополнительную информацию можно найти в Таблице 154.
Через 48 часов после последнего интраназального закапывания в головном мозге животных, получавших дозы эскетамина-HCl до 54 мг/сутки, не наблюдали никаких гистопатологических поражений. В частности, в PCG/RSC любого из обработанных эскетамином-HCl животных, не было обнаружено поражений Олни. Дополнительную информацию можно найти в Таблице 6 и на Фиг. 72 и 73 (смотрите Раздел 3.3).
В 14-суточном исследовании терапевтический интервал на основании Cмакс и ППК для эскетамина был приблизительно 17- и 11-кратным, соответственно.
3.3. Результаты для (+)MK-801 малеата в исследованиях нейротоксичности с однократной дозой и повторной дозой эскетамина-HCl
В исследовании с однократной дозой или повторной дозой не наблюдали смертельных исходов, когда самки крыс получали однократную подкожную дозу (+)MK-801 малеата при 1 мг/кг массы тела.
У большинства животных наблюдались умеренное или сильное снижение общей активности и лежание на боку. Кроме того, были отмечены каталепсия, брадипноэ, тремор и слюноотделение. У одного животного проявились клонические судороги.
В течение периода последующего наблюдения до вскрытия, сохранялись снижение активности и атаксия, но они постепенно становились менее сильными, а каталепсия и брадипноэ наблюдались только иногда. Кроме того, почти у всех животных проявилась хромодакриорея. Иногда у некоторых животных наблюдались и другие признаки общего дискомфорта, такие как пилоэрекция, сгорбленная спина, выделения из глаз и сужение глазной щели. Иногда наблюдались небольшие повышения общей активности. Временная динамика снижения и повышения общей активности проиллюстрирована на Фиг. 70.
Крысы, которым вводили дозу (+)MK-801 малеата демонстрировали снижение массы тела приблизительно на 13-14% в течение первых 48 часов после обработки. В исследовании с однократной дозой, где половину животных оставляли до 96 часов после введения дозы, животные восстановили массу в период от 48 до 96 часов после введения дозы.
Через 48 и 96 часов после введения дозы в исследовании с однократной дозой и через 48 часов после введения последней дозы в исследовании с повторной дозой (+)MK-801 малеат индуцировал появление поражений Олни, о чем свидетельствовало появление нейронального некроза в PCG/RSC всех животных, обработанных этим препаратом положительного контроля. В обоих исследованиях большинство поражений были оценены как умеренные или заметные (степень 3 или 4). Заметной разницы в степени тяжести нейронального некроза между животными, вскрытыми через 48 или 96 часов после введения дозы в исследовании с однократной дозой, не наблюдалось. В этом исследовании некротические нейроны, как правило, были несколько более многочисленными при окрашивании на FJ по сравнению с окрашиванием HE. В обоих исследованиях поражения были соответственно более тяжелыми в RSC по сравнению с PCG.
В Таблицах 155 и 156 ниже приведен обзор количества случаев и степени тяжести.
4. Обсуждение
Кетамин представляет собой препарат с приписываемыми ему противоречивыми свойствами. С одной стороны, он может быть нейропротективным (Hudetz, 2010. J. Cardiothor. Vasc. Anesth. 24, 131-142), как, например, было показано снижением содержания катехоламинов в плазме и улучшением результата при неполной ишемии головного мозга у крыс (Hoffman, 1992. J. Anesthesiology 76(5), 755-762). Снижение нейрональной дегенерации и уровней тревожности наблюдалось при введении кетамина во время индуцированной в раннем возрасте эпилепсии у крыс (Loss, 2012. Brain Research 1474, 110-117). В крысиной модели глобальной ишемии переднего мозга, индуцированной гипобарической гипотензией, однократное В/Б введение эскетамина в дозе 60 или 90 мг/кг массы тела через 15 минут после ишемии головного мозга значительно снижало потерю нейрональных клеток в коре головного мозга, тогда как другие структуры головного мозга, такие как гиппокамп, были менее защищены (Proescholdt, 2001. Brain Res. 904, 245-251). С другой стороны, известно, что кетамин провоцирует появление поражений Олни в мозге крыс после введения однократной (Olney 1989; Jevtovic-Todorovic 2000; Jevtovic-Todorovic 2005) или повторной дозы ( , 1997. Brain Res. 753(2), 181-195). По-прежнему необходимо установить точные пороговые значения для дозы и продолжительности воздействия, вызывающие нейротоксичность у животных. Релевантность нейротоксического действия кетамина у животных для человека неизвестна.
, 1997. Brain Res. 753(2), 181-195). По-прежнему необходимо установить точные пороговые значения для дозы и продолжительности воздействия, вызывающие нейротоксичность у животных. Релевантность нейротоксического действия кетамина у животных для человека неизвестна.
Потенциальную индукцию нейронального поражения после острого интраназального введения эскетамина-HCl изучали в исследовании нейротоксичности с однократной дозой, в котором взрослые самки крыс линии Спрег - Доули (в возрасте 12-14 недель) получали дозы 36, 54 или 72 мг. Эти дозы обеспечивали за один сеанс однократной обработки, состоящей из 2, 3 или 4 последовательных интраназальных закапываний, соответственно, в каждую ноздрю по 50 μл водного раствора, содержащего 180 мг/мл эскетамина-HCl. Интервал между последовательными закапываниями в обе ноздри составлял 5 минут. Наибольшая доза 72 мг представляла максимальную возможную дозу для острого исследования, учитывающую максимальный объем дозы, который можно было вводить за несколько раз через 5-минутные интервалы (50 μл на ноздрю), и максимальную растворимость эскетамина-HCl в воде, составляющую 180 мг/мл. Следовательно, наибольшую дозу 72 мг в каждую ноздрю закапывали в общем количестве 200 μл эскетамина-HCl в течение 15 минут. Больший объем дозы не представляется возможным с учетом того, что крыса обладает облигатным носовым дыханием с носовой полостью, объем которой составляет приблизительно 200 μл на ноздрю (Gizurarson, 1990. Acta Pharm. Nord. 2(2), 105-122). Гистопатологию головного мозга проводили через 48 часов и через 96 часов после введения дозы. Выбор этих моментов времени был обусловлен опубликованными результатами экспериментов, в которых говорилось, что острое воздействие MK-801 или (+)MK-801 в зависимости от дозы индуцирует некроз в PCG/RSC крыс через 1-14 суток после введения однократной дозы (Auer 1996; Bender, 2010a. Neuroscience 169, 720-732; Colbourne 1999; De Olmos, 2009. Neuroscience164(3), 1347-1359; Fix 1993; Fix 1994; Fix 1995; Fix 1996; Fix 2000; Jevtovic-Todorovic 2001; Willis, 2007. NeuroToxicology 28, 161-167). В эти два момента времени не было обнаружено никаких признаков нейронального некроза у любого из животных, обработанных эскетамином-HCl, изучаемых в исследовании с однократной дозой. Терапевтический интервал на основании Cмакс и ППК для эскетамина по сравнению с максимальной клинической дозой 84 мг был приблизительно 60- и 86-кратным, соответственно.
На данный момент в имеющейся литературе отсутствуют данные о потенциальной индукции нейронального некроза в PCG/RSC взрослых самок крыс линии Спрег-Доули, получивших острую обработку эскетамином или кетамином. Однако имеются достаточные доказательства того, что кетамин индуцирует нейрональную вакуолизацию в этих областях головного мозга самок крыс вскоре после обработки однократной дозой. В этом отношении кетамин напоминает MK-801 или (+)MK-801 (Auer and Coulter 1994; Farber 1995; Fix 1993; Fix 1994; Fix 1996; Fix 2000; Jevtovic-Todorovic, 1997. Journal of Cerebral Blood Flow and Metabolism 17,168-174; Jevtovic-Todorovic 2001). Нейрональную вакуолизацию наблюдали в PCG/RSC взрослых крыс линии Спрег-Доули через 4 часа после однократной подкожной инъекции кетамина в дозе 40 мг/кг массы тела, но не при 10 или 20 мг/кг (Olney 1989). RSC самок крыс линии Спрег-Доули в возрасте 2 месяцев, которые получали однократную дозу кетамина 60 мг/кг массы тела, демонстрировала нейрональную вакуолизацию через 3 часа после введения дозы. Реакция была более выраженной в возрасте 3 месяцев (Jevtovic-Todorovic 2001). ED50-значение для индукции нейрональной вакуолизации в PCG/RSC взрослых самок крыс линии Спрег-Доули через 3 часа после однократной внутрибрюшинной инъекции кетамина составляла 47,5 мг/кг массы тела (Jevtovic-Todorovic 2000). Такие ранние моменты времени после введения дозы не исследовали в исследовании нейротоксичности с однократной дозой, поскольку нейрональная вакуолизация считается обратимой. Более позднее нейродегенеративное поражение считается более серьезным, чем нейрональная вакуолизация, и необратимым (Bender 2010a; Zhang 1996).
В текущем исследовании с однократной дозой с интраназальным введением эскетамина-HCl крысам линии Спрег - Доули средняя масса тела составляла 255 г. Таким образом, наибольшая доза 72 мг соответствует приблизительно 282 мг/кг массы тела. Эта доза существенно больше, чем уровни доз кетамина, которые, как было описано ранее, стимулировали вакуолированные нейроны в PCG/RSC (Olney 1989; Jevtovic-Todorovic 2000; Jevtovic-Todorovic 2005). Неясно, почему после однократной интраназальной дозы 282 мг/кг массы тела эскетамина отсутствовали гистопатологические нейрональные поражения, тогда как нейрональную вакуолизацию наблюдали, начиная с однократной внутрибрюшинной инъекции 47,5 мг/кг массы тела кетамина и выше. Возможной причиной может быть то, что в текущем исследовании нейрональный некроз исследовали через 48 и 96 часов после острой дозы эскетамина, тогда как в литературе с кетамином описана только нейрональная вакуолизация через несколько часов после острого введения. Возможно, нейрональную вакуолизацию не наблюдали из-за того, что за 48 часов происходило ее восстановление (как показано Auer and Coulter 1994 для MK-801). Другая причина может заключаться в том, что интраназально вводимый эскетамин и парентерально вводимый кетамин отличаются по биодоступности и, следовательно, приводят к разному содержанию в плазме и/или разной кинетике в головном мозге.
Не сообщалось о поражениях Олни или других нейропатологических изменениях в головном мозге крыс после повторного введения доз эскетамина или кетамина. Кроме того, в открытой литературе не хватает нейропатологических исследований головного мозга крыс после повторного введения доз MK-801. В головном мозге крыс, обработанных П/К 0,3 мг/кг массы тела MK-801 в течение 4 последовательных дней не наблюдали нейрональную вакуолизацию через 4 часа после введения последней дозы (Olney 1989). Когда крыс ежесуточно обрабатывали MK-801 в течение 4 дней при более высоких дозах или по схеме с более резким увеличением дозы, а головной мозг исследовали через 4 часа после последней дозы, наблюдали нейрональную вакуолизацию. Отсутствовали данные о кумулятивном эффекте или прогрессировании реакции до необратимого состояния (Olney 1989). У крыс, обрабатываемых 3 раза в сутки (+)MK-801 в течение двух дней, нейродегенерация наблюдалась не только в RSC, но и в других областях головного мозга, хотя и менее выраженная  1997).
1997).
Чтобы исследовать потенциальное образование нейродегенеративных поражений после повторного интраназального введения эскетамина-HCl, проводили 14-суточное исследование нейротоксичности с повторной дозой на крысах линии Спрег - Доули того же пола и возраста, что и в исследовании с однократной дозой. Наибольшую дозу эскетамина-HCl 54 мг/сутки выбрали на основании 14-суточного исследования диапазона дозы, где после повторного интраназального введения этот уровень дозы посчитали максимально переносимой дозой (МПД) на основании сильного снижения общей активности, лежания на боку и дыхательных аномалий (внутреннее исследование). В 14-суточном исследовании нейротоксичности животных, получавших базовый раствор и дозу эскетамина-HCl, умерщвляли через 48 часов после введения 14-ой дозы эскетамина, а животных, получавших (+)MK-801 - через 48 часов после острой дозы (+)MK-801-малеата, которую вводили в последний день введения дозы исследования. В Fix 1996 сообщается о нейрональном некрозе в PCG/RSC крысиного головного мозга после однократной дозы MK-801 через время, составляющее от 24 часов до 14 суток после введения дозы. Через 24 часа эффект наблюдался лишь в малой степени и иногда, тогда как через 72 часа после введения дозы он был выраженным. В Fix 1993 сообщается о нейрональном некрозе в RSC крыс через время, составляющее от 48 часов до 14 суток, после однократной дозы (+)MK-801. Момент времени умерщвления 48 часов был выбран, поскольку основная цель исследования заключалась в изучении потенциального возникновения нейронального некроза в головном мозге. Четырнадцать суток повторного введения эскетамина-HCl посчитали приемлемой продолжительностью обработки, чтобы обнаружить дегенеративные нейроны через 48 часов после введения последней дозы. Ожидалось, что после 14 суток обработки не будет обнаруживаться нейрональная вакуолизация, поскольку она существует в течение короткого промежутка времени и считается обратимой (Auer and Coulter 1994; Bender 2010a; Farber 1995; Fix 1993; Fix 1994; Fix 1996; Fix 2000; Jevtovic-Todorovic 1997; Jevtovic-Todorovic 2000; Jevtovic-Todorovic 2001; Olney 1989; Zhang 1996). Следовательно, нейрональную вакуолизацию не считали представляющим интерес ожидаемым результатом. Нейрональный некроз в PCG/RSC наблюдали через 48 часов после однократной дозы (+)MK-801 при 1 мг/кг массы тела, было принято решение вводить группе положительного контроля дозу только один раз на сутки, когда обработанные эскетамином-HCl крысы получали последнее введение дозы, и оценивать все образцы тканей головного мозга через 48 часов после последней дозы. Хотя, как и ожидалось, у обработанных (+)MK-801 крыс отмечался нейрональный некроз, у обработанных эскетамином-HCl животных он отсутствовал. При наибольшей исследуемой дозе 54 мг/кг эскетамина-HCl терапевтический интервал на основании Cмакс и ППК для эскетамина по сравнению с максимальной клинической дозой 84 мг был приблизительно 17- и 11-кратным.
При интраназальном введении эскетамина-HCl взрослым самкам крыс в дозе до 72 мг (что соответствует приблизительно 282 мг/кг массы тела) или повторно в дозе до 54 мг/сутки (что соответствует приблизительно 196 мг/кг массы тела) были отмечены схожие результаты клинических наблюдений. Продолжительность анестезии составляла 15-30 минут при 36 мг, от 30 минут до часа при 54 мг и от одного до 2 часов при дозе 72 мг для крыс. Во время анестезии иногда наблюдалось брадипноэ. Хотя кетамин рассматривается как агент, вызывающий минимальное угнетение дыхания, некоторое угнетение дыхания может проявляться при более высоких уровнях дозы, что подтверждается более высокими значениями pCO2 для кетамина в дозе 80 мг/кг массы тела (Hoffmann, 2003. Pharmacology, Biochemistry and Behavior 74, 933-941). Кетамин является широко используемым анестетиком для крыс, но в основном он используется в комбинации с другими агентами (например, ксилазином) для индукции диссоциативной анестезии с анальгезией и неподвижностью. Начало анестезии после внутримышечного введения дозы 50 мг/кг кетамина крысе происходит быстро (в течение 5 минут) с потерей рефлекса выпрямления через 7 минут и длительностью полной анестезии в течение 35 минут. Через 45 минут рефлекс выпрямления снова появляется. Внутримышечные дозы до 150 мг/кг массы тела вызывали пиковый эффект в течение 10 минут, который мог сохраняться в течение 30-40 минут. Общее восстановление от анестезии наблюдалось через 1,5 часа (Green 1981. Lab. Anim. 1981, 15: 163).
Анестезии, индуцированной эскетамином-HCl, в основном предшествовали атаксия и небольшое повышение активности крыс в течение первых минут после введения дозы, до того, как через 15 минут отмечались лежание на боку и каталепсия. Гиперактивность и атаксию также отмечались после восстановления от анестезии и длились приблизительно до 1,5 часа при 36 мг, до 1-3 часов при 54 мг и до 3 часов или дольше при 72 мг. Известно, что субанестетические дозы кетамина вызывают гиперактивность и атаксию. Внутрибрюшинная доза кетамина 10 мг/кг массы тела приводит к повышению активности в открытом поле, что считается связанным с изменениями в активности дофамина (Wilson, 2005. Pharmacology, Biochemistry and Behavior 81 (2005) 530-534). Внутривенное введение кетамина в дозе от 5 до 80 мг/кг массы тела продемонстрировало повышение продолжительности атаксии и гиперактивности при увеличении дозы. Продолжительность гиперактивности была немного больше, чем продолжительность атаксии (Wilson 2005; Cohen 1973. Anesthesiol. 39: 370-376). Аналогичные результаты были получены Compton (Compton, 2013. International Journal of Life Science and Medical Research Vol. 3 Iss. 5, 179-192), так как крысы, которым внутрибрюшинно инъецировали кетамин в дозе 5 мг/кг, демонстрировали повышенную общую активность, тогда как крысы, которым вводили дозу 40 мг/кг, нет.
Положительным контролем в обоих исследованиях был (+)MK-801-малеат. Этот неизбирательный антагонист HMDA-рецепторов хорошо известен как вызывающий нейрональную вакуолизацию и дегенерацию (некроз) в PCG/RSG головного мозга крыс (Auer and Coulter 1994; De Olmos 2009; Fix 1993; Fix 1994; Fix 1995; Fix 1996; Fix 2000; Olney 1989; Olney 1991). (+)-энантиомер в 7 раз более активен, чем (-)-энантиомер. В данных исследованиях его вводили путем одной подкожной инъекции в дозе 1 мг/кг. В исследовании для определения диапазона дозы эту дозу 1 мг/кг массы тела считали МПД для однократной подкожной инъекции (+)MK-801-малеата самкам крыс линии Спрег - Доули в возрасте 13-14 недель. Сообщалось, что однократное или внутрибрюшинное (В/Б) введение MK-801 в дозе 5 мг/кг массы тела приводило к сильному нарушению координации, повышенной двигательной активности, атаксии, мотанию головой перед лежачим периодом от 5,5 до 6 часов у самцов крыс и от 24 до 40 часов у самок крыс. Этот длительный лежачий период приводил к серьезным потерям массы тела. Также сообщалось о смерти крыс в состоянии крайней неподвижности с неглубоким дыханием (Colbourne 1999; De Olmos 2008; Fix 1995; Auer 1996; Hur, 1999. Environmental Toxicology and Pharmacology 7, 143-146). Самки крыс, которым вводили В/Б дозу 5 мг/кг массы тела (+)MK-801, демонстрировали лежание, тяжелую гипотермию и потерю массы тела, длящиеся до 3-7 суток после введения дозы (Zajaczkowski, 2000. Neurotox. Res. 1(4), 299-310). Сообщалось также о поведенческом расстройстве и лежании у крыс, однократно обработанных 10 мг/кг В/Б (Bender, 2010b. Neurotoxicology and Teratology 32, 542-550). Однократная П/К доза 1 мг/кг MK-801 индуцировала лежание в течение по меньшей мере 7 часов после введения дозы (Fix 1995). При более низких уровнях дозы (от 0,05 до 0,2 мг/кг П/К или В/Б) MK-801 индуцировал повышенную локомоцию и атаксию у крыс  1999. J. Pharmacol. Exp. Ther. 290(3), 1393-408;
1999. J. Pharmacol. Exp. Ther. 290(3), 1393-408;  1999. Neuropsychopharmacol. 21, 414-426). У самок крыс локомоция была максимальной при 0,1-0,2 мг/кг, тогда как о стереотипическом сопении сообщалось при 0,1-0,5 мг/кг, и атаксии при 0,2-1 мг/кг MK-801 1999). Клинические результаты, приведенные в литературе, также наблюдались в исследованиях после однократной дозы (+)MK-801 при 1 мг/кг массы тела. В результате длительного периода лежания животные не принимали пищу или не пили, что приводило к большой потере массы тела на 13-14% в течение первых 48 часов после введения дозы. Было заявлено, что любая доза MK-801, равная или превышающая 0,2 мг/кг, вероятно, считается вызывающей сильную интоксикацию у крыс (Wozniak, 1990. Psychopharmacology 101(1), 47-56). Однако примечательно, что многие другие исследователи вообще не упоминали никаких клинических наблюдений даже после однократной дозы, равной 10 мг/кг (Wozniak, 1998. Neurobiology of Disease. 5(5), 305-322; Farber 1995; Auer and Coulter 1994; Fix 1993; Fix 1996; Willis and Ray 2007; Bueno, 2003. Experimental and Toxicologic Pathology 54, 319-334; 1997; De Olmos 2009; Fix 2000).
1999. Neuropsychopharmacol. 21, 414-426). У самок крыс локомоция была максимальной при 0,1-0,2 мг/кг, тогда как о стереотипическом сопении сообщалось при 0,1-0,5 мг/кг, и атаксии при 0,2-1 мг/кг MK-801 1999). Клинические результаты, приведенные в литературе, также наблюдались в исследованиях после однократной дозы (+)MK-801 при 1 мг/кг массы тела. В результате длительного периода лежания животные не принимали пищу или не пили, что приводило к большой потере массы тела на 13-14% в течение первых 48 часов после введения дозы. Было заявлено, что любая доза MK-801, равная или превышающая 0,2 мг/кг, вероятно, считается вызывающей сильную интоксикацию у крыс (Wozniak, 1990. Psychopharmacology 101(1), 47-56). Однако примечательно, что многие другие исследователи вообще не упоминали никаких клинических наблюдений даже после однократной дозы, равной 10 мг/кг (Wozniak, 1998. Neurobiology of Disease. 5(5), 305-322; Farber 1995; Auer and Coulter 1994; Fix 1993; Fix 1996; Willis and Ray 2007; Bueno, 2003. Experimental and Toxicologic Pathology 54, 319-334; 1997; De Olmos 2009; Fix 2000).
Наличие нейронального некроза в PCG/RSC оценивали в зависимости от количества некротических нейронов, наблюдаемых на протяжении всей длины структуры, видимой в срезе (как правило, около 3-5 мм). Использовали четыре степени, которые охватывали изменения в диапазоне от минимального (<10 некротических нейронов) до выраженного (> 30 некротических нейронов). С помощью этой системы оценки было показано, что (+)MK-801 малеат после одной П/К инъекции при 1 мг/кг массы тела вызвал типичный нейрональный некроз, связанный с нейрональным некрозом в PCG/RSC, у всех обработанных животных. Степень тяжести поражений была оценена как выраженная у большинства животных (т.е. 25 из 30 самок в исследовании с однократной дозой и 7 из 12 животных в исследовании с повторной дозой). Не наблюдалось никакой релевантной разницы между исследованиями с однократной дозой и повторной дозой или между 48- и 96-часовыми моментами времени после введения дозы в исследовании с однократной дозой. Таким образом, можно сделать вывод, что крысы, которым однократно вводили (+)MK-801 малеат в дозе 1 мг/кг, служили в качестве проверенной группы положительного контроля для обоих исследований нейротоксичности, и что ответ был сильным и согласующимся.
4.1. Заключение
Несмотря на то, что при интраназальном введении эскетамина-HCl в исследовании нейротоксичности с однократной дозой были отмечены случаи смерти и выраженные связанные с ЦНС клинические наблюдения (например, каталепсия), никаких нейропатологических поражений не наблюдали в головном мозге взрослых самок крыс вплоть до самой высокой исследованной дозы 72 мг при оценке через 48 и 96 часов после введения дозы. Терапевтический интервал на основании Cмакс и ППК для эскетамина по сравнению с максимальной клинической дозой 84 мг был приблизительно 60- и 86-кратным, соответственно. Гистопатологические поражения в мозге животных, обработанных эскетамином-HCl, включенных в 14-суточное исследовании нейротоксичности с повторной дозой, обнаружены не были. В последнем исследовании наибольшая исследованная доза составляла 54 мг/сутки. При этой дозе терапевтический интервал на основании Cмакс и ППК для эскетамина по сравнению с максимальной клинической дозой 84 мг был приблизительно 17- и 11-кратным. Как и ожидалось, положительный контроль (+)MK801 малеат индуцировал нейродегенеративные поражения, о чем свидетельствует типичный нейрональный некроз в PCG/RSC головного мозга у всех крыс, обработанных этим положительным контрольным соединением.
Пример 7
В исследованиях данного примера оценивали эффективность, безопасность и зависимость доза-ответ интраназального эскетамина у пациентов с терапевтически резистентной депрессией (ТРД).
Материалы и способы
Соблюдение этических норм
Независимый обзорный совет (Соединенные Штаты)/Независимый комитет по этике (Бельгия) утвердил протокол исследования и поправки. Исследование проводилось в соответствии с этическими принципами, которые изложены в Хельсинкской декларации, в соответствии с надлежащей клинической практикой и применимыми нормативными требованиями. Все пациенты предоставили письменное информированное согласие перед участием в исследовании. Это исследование зарегистрировано на clinicaltrials.gov, NCT01998958.
Исследуемая популяция
Это исследование включало стабильных с медицинской точки зрения (на основе физического осмотра, анамнеза, основных физиологических показателей и ЭКГ в 12-ти отведениях, проводимых при скрининге) взрослых (от 20 до 64 лет) с диагнозом БДР, согласно Диагностико-статистическому руководству по психическим расстройствам, четвертое издание, пересмотренный текст (DSM-IV-TR). Смотрите American Psychiatric Association. Diagnostic and statistical manual of mental disorders (DSM-IV-TR). 4th ed, text revised. Washington, DC: American Psychiatric Association, 2000.
Все участники имели ТРД, определяемую как неадекватный ответ на ≥ 2 антидепрессантов (оцениваемый по Опроснику по истории лечения антидепрессантами Центральной больницы штата Массачусетс; Rush, “The Inventory of Depressive Symptomatology (IDS): Psychometric properties”, Psychol. Med., 1996, 26(3):477-486), причем по меньшей мере 1 во время текущего эпизода депрессии. В ином случае допускался неудачный прием антидепрессанта во время предыдущего эпизода. Все участники продолжали принимать антидепрессанты, которые они принимали во время включения в исследование, во время исследования. При скрининге и перед введением дозы на 1 сутки пригодные участники имели оценку ≥ 34 по состоящему из 30 пунктов Опроснику депрессивной симптоматики с клинической оценкой (IDS-C30) (Rush 1996 and Trivedi, “The Inventory of Depressive Symptomatology, Clinician Rating (IDS-C) and Self-Report (IDS-SR), and the Quick Inventory of Depressive Symptomatology, Clinician Rating (QIDS-C) and Self-Report (QIDS-SR) in public sector patients with mood disorders: a psychometric evaluation”, Psychol. Med., 2004, 34(1):73-82), что соответствует умеренной или сильной депрессии. Основные критерии исключения включали недавнюю или текущую суицидальную идеацию с намерением действовать, суицидальное поведение или гомицидальные идеацию/намерения, диагноз биполярных или родственных расстройств, умственную инвалидность, психотическое расстройство, БДР с психозом, посттравматическое стрессовое расстройство, обсессивно-компульсивное расстройство, расстройства, связанные с употреблением наркотиков/алкоголя за последний год, и недавнее употребление каннабиса.
Это исследование включало стабильных с медицинской точки зрения (на основе физического осмотра, анамнеза, основных физиологических показателей и ЭКГ в 12-ти отведениях, проводимых при скрининге) взрослых (от 20 до 64 лет) с диагнозом БДР, согласно Диагностико-статистическому руководству по психическим расстройствам, четвертое издание, пересмотренный текст (DSM-IV-TR). Основной депрессивный эпизод участника и ответ на лечение оценивали для подтверждения соответствия участника критериям, используя интервью в отношении критериев ситуативной vs. личностной (тревожности), возможности оценки, внешней достоверности, экологической достоверности, правила трех «P» (SAFER) [Targum, “Redefining affective disorders: relevance for drug Development”, CNS Neurosci. Ther., 2008, 14(1):2-9], проводимое удаленными независимыми экспертами.
Основные критерии исключения включали недавнюю или текущую суицидальную идеацию с намерением действовать, суицидальное поведение или гомицидальные идеацию/намерения, диагноз биполярных или родственных расстройств, умственную инвалидность, психотическое расстройство, большое депрессивное расстройство (БДР) с психозом, расстройство личности кластера B (на основании клинической оценки исследователем), посттравматическое стрессовое расстройство, обсессивно-компульсивное расстройство, анамнез отсутствия ответа на электрошоковую терапию. Были исключены лица, злоупотребляющие наркотиками или алкоголем или имеющие зависимость в течение последнего года, а также имеющие положительный результат теста на каннабис при скрининге.
Дизайн исследования
Это 2-панельное, двойное слепое, дважды рандомизированное, плацебо-контролируемое исследование фазы 2 с отсроченным началом (вариант дизайна последовательного параллельного сравнения) проводилось с 28 января 2014 г. по 25 сентября 2015 г. Смотрите, например, Chi, “On clinical trials with a high placebo rate. Contemporary Clinical Trials Communications”, 2016, 2:34-53; Fava, “The problem of the placebo response in clinical trials for psychiatric disorders: culprits, possible remedies, and a novel study design approach”, Psychother. Psychosom. 2003, 72(3):115-27; Chen, “Evaluation of performance of some enrichment designs dealing with high placebo response in psychiatric clinical trials”, Contemp. Clin. Trials, 2011, 32(4):592-604; Fava, “A double-blind, placebo-controlled study of aripiprazole adjunctive to antidepressant therapy among depressed outpatients with inadequate response to prior antidepressant therapy (ADAPT-A Study)”, Psychother. Psychosom., 2012, 81(2):87-97); Chen, “A sequential enriched design for target patient population in psychiatric clinical trials” Stat. Med., 2014, 33(17):2953-2967; Doros, “A repeated measures model for analysis of continuous outcomes in sequential parallel comparison design studies”, Stat. Med., 2013, 32(16):2767-2789; Huang, “Comparison of test statistics for the sequential parallel design”, Statistics in Biopharmaceutical Research, 2010, 2(1):42-50; Ivanova, “Optimality, sample size, and power calculations for the sequential parallel comparison design”, Stat. Med., 2011, 30(23):2793-803; Papakostas, “L-methylfolate as adjunctive therapy for SSRI-resistant major depression: results of two randomized, double-blind, parallel-sequential trials”, Am. J. Psychiatry, 2012, 169(12):1267-1274; Roy, “An examination of the efficiency of the sequential parallel design in psychiatric clinical trials”, Clinical Trials. 2007, 4:309-317; Rybin, “Placebo non-response measure in sequential parallel comparison design studies”, Stat. Med., 2015, 34(15):2281-2293; Tamura, “An examination of the efficiency of the sequential parallel design in psychiatric clinical trials”, Clin. Trials. 2007, 4(4):309-317; и Tamura, “Estimation of treatment effect for the sequential parallel design”, Stat. Med., 2011, 30(30):3496-506.
В панели A, представленной в данном документе, 14 исследовательских центров (13 в США, 1 в Бельгии) проводили набор участников в это исследование. Исследование состояло из 4 фаз: 1) скрининг, 2) двойное слепое лечение (сутки 1-15), состоящее из двух 1-недельных периодов (период 1, период 2), 3) необязательно дополнительное лечение (сутки 15-74) с постепенным уменьшением частоты интраназального введения дозы и 4) период последующего наблюдения после лечения (8 недель). На основании предыдущих исследований кетамина, в которых эффективность отмечалась после 1-2 доз, продолжительность каждого периода в двойной слепой фазе составляла 1 неделю, и в течение этого времени ожидалось возможное достижение эффективности. Такой дизайн позволял оценить дозу(ы), необходимую(ые) для проведения оценки в фазе 3. Целью открытой фазы с гибкой дозой была оценка влияния менее частого введения дозы на сохранение эффективности.
В начале двойного слепого периода 1 допущенные участники были рандомизированы (3:1:1:1) в группу интраназального плацебо или эскетамина в дозе 28, 56 или 84 мг два раза в неделю в неделю (сутки 1 и 4) на основании первого из двух сгенерированных компьютером планов рандомизации (период 1 и период 2). Рандомизация была сбалансирована при помощи рандомизации внутри блоков и стратифицирована по исследовательскому центру. В конце периода 1 пациентов, рандомизированных в группу плацебо, которые имели умеренные или тяжелые симптомы (оцениваемые по Краткому опроснику депрессивной симптоматики с самоотчетом (Trivedi 2016 and Rush, “The 16-item Quick Inventory of Depressive Symptomatology (QIDS) Clinician Rating (QIDS-C) and Self-Report (QIDS-SR): A psychometric evaluation in patients with chronic major depression”, Biol. Psychiatry, 2003, 54(5):573-583) [QIDS-SR16] с общей оценкой: умеренный, 11-16; тяжелые, > 16) повторно рандомизировали (1:1:1:1) в группу интраназального эскетамина в дозе 28, 56 или 84 мг или плацебо, два раза в неделю (сутки 8 и 11), тогда как пациенты с легкими симптомами или без симптомов продолжали принимать плацебо. Для сохранения маскировки все участники выполнили идентичный процесс перед вступлением в период 2, независимо от того, были ли они повторно рандомизированы. Независимо от ответа в двойной слепой фазе, все участники имели право перейти в необязательную открытую фазу. Эскетамин (56 мг) вводили на первые сутки открытой фазы (15 сутки исследования); последующие дозы можно было корректировать (диапазон: 28-84 мг) на основании клинического заключения исследователя с введением два раза в неделю в течение первых 2 недель, один раз в неделю в течение следующих 3 недель, затем один раз в 2 недели.
Исследуемый препарат и введение
Исследуемый препарат был предоставлен в виде одноразового устройства для назального впрыскивания, содержащего 200 мкл раствора (т.е. на 2 впрыскивания). Каждое устройство доставляло либо 16,14 мг эскетамина гидрохлорида (14 мг основания эскетамина) на 100 мкл спрея, либо плацебо. Для сохранения маскировки раствор плацебо (интраназальный раствор воды для инъекций) содержал горький агент (денатония бензоат), добавленный для имитации вкуса интраназального раствора эскетамина. Как описано выше, антидепрессант, принимаемый участниками непосредственно перед включением в исследование, оставался неизменным.
На каждые сутки введения дозы во время двойной слепой фазы участники самостоятельно проводили 1 впрыскивание исследуемого препарата (эскетамина или плацебо) в каждую ноздрю в 3 момента времени, которые разделяло 5 минут. В открытой фазе, в зависимости от выбранной дозы, участники самостоятельно проводили 1 впрыскивание эскетамина в каждую ноздрю в 1, 2 или 3 момента времени (соответствующих 28, 56 или 84 мг, соответственно), которые разделяло 5 минут.
Оценки эффективности
Эффективность оценивали с помощью Шкалы Монтгомери - Асберг для оценки депрессии (MADRS; Montgomery, “A new depression scale designed to be sensitive to change”, Br. J. Psychiatry, 1979, 134:382-389; Williams, “Development and reliability of a structured interview guide for the Montgomery Asberg Depression Rating Scale (SIGMA)”, Br. J. Psychiatry, 2008, 192(1):52-58) на сутки 1 (перед введением дозы и через 2 часа после введения дозы), 2, 8 (перед введением дозы), 9 и 15, используя руководство по проведению структурированного интервью (SIGMA). Смотрите Williams 2008.
Общую тяжесть заболевания оценивали по шкале общего клинического впечатления о тяжести заболевания (CGI-S) (Guy, “ECDEU Assessment Manual for Psychopharmacology -Revised (DHEW Publ No ADM 76-338)”, Rockville, MD: U.S. Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, NIMH Psychopharmacology Research Branch, Division of Extramural Research Programs; 1976, pp 218-222). Участники оценивали тяжесть своих тревожных симптомов по шкале оценки генерализованного тревожного расстройства из 7 пунктов (GADS-7) (смотрите Таблицы 157 и 15). Смотрите Spitzer, “A brief measure for assessing generalized anxiety disorder - the GAD-7”, Arch. Intern. Med., 2006, 166(10):1092-1097.
Оценки безопасности
В течение всего исследования отслеживали нежелательные явления. Другие оценки безопасности (т.е. лабораторные исследования, основные физиологические показатели, физический осмотр) проводили в заранее определенные моменты времени. Основные физиологические показатели, используя Применяемую клиницистами шкалу диссоциативных состояний (CADSS; Bremner, “Measurement of dissociative states with the clinician-administered dissociative states scale (CADSS)”, J. Traumatic Stress, 1998, 11(1):125-136) и подшкалу положительных симптомов из 4 пунктов краткой психиатрической оценочной шкалы (BPRS; Overall, “The Brief Psychiatric Rating Scale. Psychological Reports”, 1962, 10:799-812), оценивали до введения дозы, через 40 минут и 2 часа после введения дозы.
Статистические методы
Данные эффективности анализировали в выборках для анализа «по намеченному лечению» (ПНЛ) для каждых периода и фазы. Выборки для анализа ПНЛ включали всех участников, которые получили по меньшей мере 1 дозу исследуемого препарата в течение этих периода или фазы и имели исходную и по меньшей мере одну последующую общую оценку MADRS в течение этих периода или фазы.
Данные по безопасности анализировали в наборах данных за период 1, период 2, двойную слепую и открытую фазы для всех субъектов, получивших по меньшей мере 1 дозу исследуемого препарата.
Ожидаемые результаты эффективности и анализ
Первичный ожидаемый результат эффективности - изменение от исходного уровня (до введения дозы, 1 сутки в каждом периоде) до конечного момента времени (8 сутки в каждом периоде) общей оценки MADRS - анализировали, используя модель ковариационного анализа (ANCOVA). Для периода 1 данная модель в качестве факторов включала лечение и страну, и исходную общую оценку MADRS в качестве ковариаты. Для периода 2 данная модель в качестве факторов включала лечение и страну, исходную оценку QIDS-SR16 (умеренное или тяжелое) для периода 2 и исходную общую оценку MADRS для периода 2 в качестве ковариаты.
Учитывая согласованность между периодами 1 и 2 (Chi 2016), группы доз эскетамина сравнивали с плацебо, используя комбинированный тест со взвешенной статистикой критериев для каждого периода в двойной слепой фазе лечения. Анализ зависимости доза-ответ для первичного ожидаемого результата эффективности проводили, используя данные, объединенные по двум периодам. Использовали метод множественных сравнений с моделированием (MCP-Mod) (Williams 2008; Bretz, “Combining multiple comparisons and modeling techniques in dose-response studies”, Biometrics, 2005, 61:738-748).
Определение размера выборки
Размер выборки определяли на основании следующих различий между интраназальным эскетамином и плацебо для среднего изменения относительно исходного уровня общей оценки MADRS: 9-точечная разница в лечении была принята для периода 1 (сутки 8), 7-точечная разница для периода 2 (сутки 15) была принята для пациентов с «умеренной» оценкой QIDS-SR16 и 9-точечная разница для периода 2 (сутки 15) была принята для пациентов с «тяжелой» оценкой QIDS-SR16.
На основании результатов исследования эскетамина (Singh 2016), было установлено, что 40% участников, принимающих плацебо, будут иметь умеренную оценку QIDS-SR16, а 55% - тяжелую оценку QIDS-SR16 в конце периода 1 (сутки 8 до введения дозы). Дополнительные допущения для расчета размера выборки включали стандартное отклонение 10, 92,5% мощность для объединенных данных по 8 суткам и 15 суткам (Liu, “Doubly-randomized delayed-start design for enrichment studies with responders or nonresponders”, J. Biopharm. Stat., 2012, 22(4):737-757), общий 1-сторонний уровень значимости 0,05 и 5% частоту исключения для периода 1. Было рассчитано, что для этой панели дважды рандомизированного дизайна на основании результатов требовалось случайное назначение 60 пациентам лечения на 1 сутки в соотношении 3:1:1:1 (30 в группу плацебо и 10 в группу интраназальной дозы эскетамина).
Результаты исследования
Участники
Проводили скрининг в общей сложности 126 человек, из которых 67 соответствовали критериям приемлемости и были рандомизированы. Из 33 участников, рандомизированных в группу плацебо в период 1, 28 имели оценку QIDS-SR16 ≥ 11 в конце периода 1 и, таким образом, им были в случайном порядке назначены эскетамин или плацебо в период 2 (Фиг. 73). Большинство рандомизированных участников (63/67, 94%) завершили период 1 и 2-недельную двойную слепую фазу (т.е. объединенные периоды 1 и 2 60/67, 90%; далее они называются «завершившими». Из них 57 попали в открытую фазу, причем 51 позднее перешли в фазу последующего наблюдения, из которых 41 выполнили визит последующего наблюдения на 8 неделе.
Группы лечения были схожи по демографическим и исходным клиническим характеристикам (Таблица 159). Шестьдесят четыре процента участников сообщили лишь об 1 неуспешном лечении антидепрессантами в текущем эпизоде (в дополнение к 1 в предыдущие эпизоды), приблизительно 22% имели 2, а остальные сообщили о ≥ 3 случаях неуспешного лечения антидепрессантов. Следует отметить, что 39% участников сообщили об использовании атипических антипсихотических средств в качестве вспомогательной терапии БДР перед включением в исследование.
N=33
N=11
N=11
N=12
N=67
Результаты оценки эффективности
Средняя оценка по шкале MADRS снизилась от исходного уровня до 8 суток в период 1 и от 8 суток до 15 суток в период 2 во всех группах с большим улучшением во всех группах доз эскетамина по сравнению с плацебо (средняя по МНК разница в диапазоне от -5,0 до -10,5 в период 1 и от -3,1 до -6,9 в период 2; Таблица 160). Изменение общей оценки по шкале MADRS было больше во всех 3 группах эскетамина, чем в группе плацебо, через 1 неделю лечения (p=0,02, p=0,001 и p < 0,001 для эскетамина в дозе 28 мг, 56 мг и 84 мг, соответственно); зависимость увеличение дозы-ответ была значимой (p < 0,001). Ответ был быстрым в начале (Фиг. 74A И 74B; Фиг. 76A и 76B) и имел тенденцию к повышению с течением времени во при повторном введении, о чем свидетельствует снижение средней общей оценки MADRS по сравнению с открытой фазой (среднее [СП] изменение от открытого исходного уровня до 74 суток): -7,2 [1,84]), несмотря на снижение частоты дозирования в открытой фазе. Кроме того, улучшение средних оценок MADRS сохранялось в течение 8-недельной фазы последующего наблюдения (без дополнительных доз эскетамина) у тех участников, которые оставались в исследовании (Фиг. 75).
В случае завершивших участников, которые 2 недели получали такое же лечение в двойной слепой фазе, среднее снижение общей оценки MADRS было больше в каждой группе доз эскетамина по сравнению с плацебо на 15 сутки, причем величина снижения непосредственно связана с дозой (разница в лечении по сравнению с плацебо -12,5, -8,3 и -6,0 для эскетамина в дозе 84 мг, 56 мг и 28 мг, соответственно). Эффективность лучше сохранялась между введениями препарата в 2 более высоких дозах (Фиг. 73).
Среди тех, кто получал одинаковое лечение в течение обоих периодов и завершил двойную слепую фазу, доля пациентов с ответом (определяемым как ≥ 50% улучшение от исходного уровня общей оценке MADRS) в каждой группе доз эскетамина была численно выше, чем в группе плацебо в конечный момент времени периода 2 (28 мг: 37,5% [3/8], 56 мг: 36,4% [4/11], 84 мг: 50,0% [5/10], плацебо 10% [1/10]). Аналогичную тенденцию в отношении ремиссии (определяемой как общая оценка MADRS ≤ 10) наблюдали между группами. Среди завершивших, получавших одинаковое лечение в течение обоих периодов, больше участников, получавших 2 более высокие дозы эскетамина, по сравнению с плацебо, имели ремиссию через 2 недели лечения (12,5%, 27,3% и 40,0% в группах доз 28 мг, 56 мг и 84 мг, соответственно, и 10,0% в группе плацебо). Частоты ответа и ремиссии в конце открытой фазы и фазы последующего наблюдения представлены по типу обработки в двойной слепой и открытой фазах в Таблице 161.
ОФ эскетамин, N=10
эскетамин/ОФ эскетамин, N=20
эскетамин/ОФ эскетамин, N=27
N=57
Результаты оценки безопасности
Три из 56 (5%) участников, получавших эскетамин, во время двойной слепой фазы (по сравнению с 0 в группе плацебо) и 1 из 57 (2%) во время открытой фазы имели нежелательные явления, приведшие к прекращению приема исследуемого препарата (по 1 событию, включая обморок, головную боль, диссоциативный синдром и внематочную беременность). Во время двойной слепой фазы 3 наиболее распространенными возникшими в ходе лечения нежелательными явлениями, наблюдаемыми у участников, получавших эскетамин, были головокружение, головная боль и изменения восприятия/диссоциативные симптомы; частота каждого из них была в > 2 раза выше для эскетамина, чем для плацебо (Таблица 162). Была отмечена зависимость доза-ответ для головокружения и тошноты, но не для других нежелательных явлений. Тип и частота нежелательных явлений, о которых сообщалось в ходе открытой фазы, были аналогичны двойной слепой фазе; явления, зарегистрированные для > 10% участников открытой фазы включали головокружение (38,6%), дисгевзию (22,8%), тошноту (15,8%), головную боль (14,0%) и седацию (10,5%). В целом 24,6% (14/57) участников сообщили о временных диссоциативных симптомах. Большинство нежелательных явлений, происходящих в дни введения дозы, были временными и либо легкими, либо умеренными по степени тяжести. Не сообщалось о летальных исходах.
N=33
N=19
N=20
N=17
N=56
bУчастники сообщили о диссоциативной реакции или о диссоциативных симптомах, и, в зависимости от словесного термина, эти события были закодированы как диссоциативное расстройство или диссоциативные симптомы, соответственно.
cВсе нежелательные явления, закодированные как диссоциация или диссоциативное расстройство, разрешались в те же сутки, когда проводили введение дозы.
Большинство участников, получавших эскетамин, демонстрировали временное повышение кровяного давления (максимальное среднее изменение: систолическое, 19,0 мм рт.ст.; диастолическое, 10,3 мм рт.ст.) и частоты сердечных сокращений (максимальное среднее изменение: 9,4 уд/мин) в сутки введения дозы. Максимальные значения кровяного давления наблюдались в большинстве случаев через 10 или 40 минут после введения дозы (систолическое: 199 мм рт.ст.; диастолическое: 115 мм рт.ст.); повышенные значения, как правило, возвращались к нормальному диапазону через 2 часа после введения дозы (Фиг. 77 и 78). Эффект дозы не наблюдался в случае частоты сердечных сокращений, хотя наибольшее среднее повышение от исходного уровня в течение обоих периодов наблюдали в группе, получавшей 84 мг эскетамина.
Изменения восприятия/диссоциативные симптомы, определенные по CADSS, начинались вскоре после начала интраназального введения дозы, достигали пиковых значения в течение 30-40 минут и разрешались через 2 часа (Фиг. 79). Изменения восприятия/диссоциативные симптомы становились слабее в группах всех доз при повторном введении дозы.
Ни у одного участника не проявились симптомы, свидетельствующие о психозе, на основании положительной оценки BPRS.
Обсуждение
Наблюдался значимый и клинически существенный эффект лечения (по сравнению с плацебо) дозами эскетамина 28 мг, 56 мг и 84 мг, о чем свидетельствует изменение общей оценки MADRS со значительной зависимостью между ответом на дозу эскетамина и антидепрессанта, наблюдаемым через 1 неделю лечения. Длительность сохранения эффективности была меньше, когда дозу 28 мг вводили два раза в неделю. Результаты открытой фазы свидетельствуют о том, что улучшение депрессивных симптомов можно поддерживать при более низкой частоте (один раз в неделю/один раз в 2 недели) введения эскетамина. Следует отметить, что величина разницы препарат-плацебо была существенной от исходного уровня до одной недели и была больше, чем средняя разница по сравнению с плацебо, наблюдаемая через 6-8 недель в исследованиях антидепрессантов в базе данных FDA (Khan, "Has the rising placebo response impacted antidepressant clinical trial outcome? Data from the US Food and Drug Administration 1987-2013”, World Psychiatry. 2017, 16(2):181-192). У большинства участников сохранялось улучшение в течение 2-месячной фазы последующего наблюдения.
Следует отметить, что интраназальные дозы эскетамина 56 и 84 мг приводили к уровням эскетамина в плазме, которые находятся в фармакокинетическом диапазоне, достигаемом В/В введением эскетамина в дозе 0,2 мг/кг, которое приводило к аналогичным клиническим результатам, что и кетамин в дозе 0,5 мг/кг В/В (в соответствии с более высокой аффинностью к рецепторам NMDA по сравнению с аркетамином (White, “Comparative pharmacology of the ketamine isomers. Studies in volunteers”, Br. J. Anaesth., 1985, 57(2):197-2030). Смотрите Singh 2016.
В этом первом исследовании интраназального эскетамина для ТРД эффективность и безопасность сравнивали с плацебо, используя двойной слепой, дважды рандомизированный дизайн с отложенным началом (Chi 2016), что позволяет получить меньший размер выборки для оценки эффективности, зависимости доза-ответ и безопасности, чем стандартный дизайн с параллельными группами, с сохранением при этом низкого риска возникновения ошибок 2 рода с целью не пропустить сигнал эффективности. Ключевая цель дизайна заключалась в том, чтобы включить только участников, принимавших плацебо, из периода 1, которым требовалось лечение в период 2, и повторно рандомизировать их для получения 1 из 3 интраназальных доз эскетамина или интраназального плацебо. В конце испытания данные об эффективности по обеим рандомизациям (сутки 1 и сутки 8) объединяли в интегрированном анализе. Поскольку ожидалось, что у повторно рандомизированных пациентов без ответа, получавших плацебо, будет более низкий ответ на плацебо, этот подход использовали для снижения высоких ответов на плацебо, наблюдаемых в психиатрических клинических исследованиях (Chi 2016). Согласованность результатов, полученных за период 1 и период 2, обосновывает их комбинацию с использованием весовых коэффициентов (Chi 2016), хотя при интерпретации необходимо соблюдать осторожность из-за небольшого размера выборки.
В целом дозы эскетамина, оцененные в данном исследовании (28, 56 и 84 мг), оказались безопасными, и не наблюдалось никаких новых или неожиданных проблем с безопасностью. Анализ симптомов изменения восприятия (определяемых по оценке CADSS) предполагает их начало вскоре после начала приема эскетамина и разрешение через 2 часа после введения. Эти симптомы были дозозависимыми и становились слабее при повторном введении. Напротив, эффективность антидепрессантов не становилась слабее при всех вариантах введения.
В целом временное повышение кровяного давления после введения дозы, в частности повышение систолического кровяного давления, обосновывает предположение, что повышение сердечного выброса является основным механизмом, что согласуется с предыдущими сообщениями о кетамине (Murrough 2013).
Обобщаемость результатов исследования ограничена малым размером выборки и критериями включения, которые исключили лиц с анамнезом психотических симптомов, расстройствами, связанными с употреблением наркотиков/алкоголя, недавним употреблением каннабиса или значительными медицинскими сопутствующими патологиями. Также были исключены субъекты, имеющие текущую суицидальную идеацию с намерением, группа, которую оценивали в отдельном исследовании (Murrough 2013). Еще одним ограничением являются затруднения с маскировкой эскетамина, несмотря на добавление горького агента в плацебо для имитации вкуса эскетамина.
Заключение
В целом интраназальный эскетамин, вводимый в дозах 28, 56 и 84 мг, оказался эффективным при лечении ТРД. Наблюдались свидетельства надежной и долговременной эффективности в фазе двойного слепого лечения (56 и 84 мг). Улучшение депрессивных симптомов сохранялось во время открытой фазе, несмотря на снижение частоты введения дозы, и в течение до 2 месяцев после прекращения введения эскетамина.
Пример 8
Валидированный фармакокинетический способ
В данном примере предложен способ ЖХ-МС/МС для определения кетамина и норкетамина в смешанной с гепарином натрия плазме крови человека с использованием кетамина-d4 и норкетамина-d4 в качестве соответствующих внутренних стандартов (ВС).
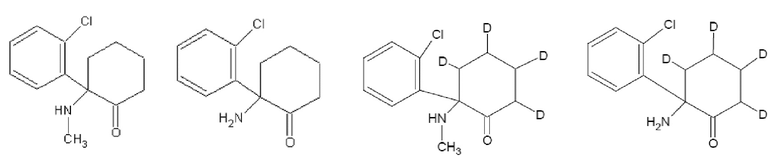
Кетамин Норкетамин Кетамин-d4 Норкетамин-d4
Все представленные в данном документе данные соответствовали критериям приемлемости валидации способа, определенным в протоколе валидации, и удовлетворяли требованиям и рекомендациям, изложенные в руководстве FDA для валидации бианалитических способов в отношении исследуемых параметров.
В целом этот способ был валидирован для определения кетамина и норкетамина в смешанной с гепарином натрия плазме крови человека. На основании объема образца, равного 25 мкл, нижний предел количественного определения (НПКО) составляет 0,500/0,500 нг/мл для кетамина/норкетамина. Динамический диапазон способа составляет 0,500/0,500-500/500 нг/мл для кетамина/норкетамина. Повторно введенные образцы были воспроизводимы при комнатной температуре в течение по меньшей мере 161,5 часов после начального ввода. Существенного переноса вещества из предыдущих проб обнаружено не было.
В исследовании валидации успешно оценивали точность и достоверность внутри серии и между сериями, селективность, чувствительность, линейность, восстановление, эффект матрицы, максимальную оценку размера партии, воспроизводимость при повторном вводе и оценку переноса вещества из предыдущих проб. Данный способ был определен как подходящий для определения кетамина и норкетамина в человеческой плазме с помощью твердофазной экстракции (ТФЭ). Смотрите Таблицу 163.
610 суток при -20°C для растворов кетамина/норкетамина с добавкой
3 цикла замораживания (-20 °C)/размораживания (комнатная температура)
ВС-нормализованный матричный множитель=1,02 ± 0,06 при 375 нг/мл с % CV=5,9% для кетамина
ВС-нормализованный матричный множитель=1,05 ± 0,03 при 1,50 нг/мл с % CV=2,9% для норкетамина
ВС-нормализованный матричный множитель=1,04 ± 0,05 при 375 нг/мл с % CV=4,8% для норкетамина
1. Экспериментальная часть
1.1. Биоаналитический способ
Этот биоаналитический способ для определения кетамина и норкетамина в смешанной с гепарином натрия плазме крови человека с использованием автоматической ТФЭ был валидирован.
1.2. Оборудование
• ЖХ-МС/МС, Sciex API 4000 (система №№ 220 и 222) с насосом для ВЭЖХ Shimadzu (система №№ 219 и 223) и автосэмплером
• Программное обеспечение для получения данных Analyst (1.4.2)
• Колонка: Phenomenex Synergi Polar-RP, 50× 2,0 мм, 4 микрон
• Многоходовой клапан, Valco Instruments
• Система очистки воды ELGA (Система 56-2), модель PL5231
• Аналитические весы, Sartorius CP22P, способные взвешивать 0,00001 г
• Микровесы, Mettler Toledo MX5, способные взвешивать 0,000001 г
• Центрифуга, Beckman GS-6R, (система № 694)
• Импульсный вихревой смеситель, Glas Col® кат. № 099A PVM12
• Шейкер для титровальных планшетов, Thermo Scientific, Barnstead/Lab-Line, модель 4625
• 1,40 мл некодированные пробирки с U-образным дном Pushcap, изделие № MP32022, NOVA
• Микротитровальный планшет с глубокими полипропиленовыми квадратными лунками/коническим дном, 2,0 мл, Microliter Analytical Supplies, Inc., изделие № 07-7400
• 96-луночный планшет с квадратными лунками, с прокалываемой крышкой, EVA, Microliter Analytical Supplies, Inc., продукт № 07-0017N
• Система ТФЭ DryTM 96, Biotage
• Автоматическая система Tomtec, Tomtec Quadra 3 (система № 114)
• 96-луночный планшет Oasis MCX для ТФЭ, 30 мкм, 10 мг
1.3. Стандарты и реагенты
• Кетамина гидрохлорид (Фармакопея США, чистота 99,9% (коэффициент превращения соли 237,73/274,19 применяли в отношении чистоты (99,9%) при использовании)
• Кетамина-d4 гидрохлорид (Cerilliant, 100,0 ± 0,5 мкг/мл в метаноле (в виде свободного основания))
• Норетамина гидрохлорид (Tocris Bioscience, чистота 100% (коэффициент превращения соли 223,70/260,16 применяли в отношении чистоты (100%) при использовании)
• (±)-норкетамина-d4 гидрохлорид (Cerilliant, 100,0 ± 0,5 мкг/мл в метаноле (в виде свободного основания))
• Вода, очищенная
• Метанол (Fisher Scientific, степень чистоты для ВЭЖХ)
• Формиат аммония (ACROS, степень чистоты GR)
• Ацетат аммония (Fisher Scientific, степень чистоты реагента ACS)
• Гидроксид аммония, 28-30% (Fisher Scientific, степень чистоты реагента ACS)
• Муравьиная кислота (ACROS, степень чистоты реагента ACS)
1.4. Биологическая матрица
Контрольную смешанную с гепарином натрия плазму человека покупали в Bioreclamation IVT. Объединенную плазму использовали для получения калибровочных стандартов, проб QC, проб валидации, холостых проб и двойных холостых проб. Плазму (объединенные и отдельные партии) хранили при температуре -20 °C.
1.5. Концентрации рабочих стандартов
Свежие рабочие стандарты готовили в каждый день проведения анализа в объединенной контрольной плазме с уровнями концентрации, перечисленными в Таблице 164.
1.6. Концентрации проб КК
Пробы КК готовили в каждый день проведения анализа в объединенной человеческой плазме с уровнями концентрации, перечисленными в Таблице 165. Пробы КК хранили при -20 °C.
2. Оценка данных
Время удержания и площадь пика определяли с помощью программного обеспечения для получения/обработки данных Analyst® (версия 1.4.2). Концентрации аналита получали на основании калибровочной кривой, полученной путем построения зависимости площади пика от концентрации с использованием Watson LIMS (версия 7.3). Для статистических расчетов использовались Watson LIMS и Microsoft Office Excel. При использовании Office Excel вычисления проверялись на 100%. Концентрации рассчитывали, используя линейную регрессию в соответствии со следующим уравнением:
y=ax+b
где:
y =отношение площади пика аналита/внутреннего стандарта
a=наклон соответствующей стандартной кривой
x=концентрация аналита (нг/мл)
b=отсечение соответствующей стандартной кривой
Применение 1/x2 в качестве весового коэффициента
Для расчета точности и достоверности использовали следующие формулы:
Точность:
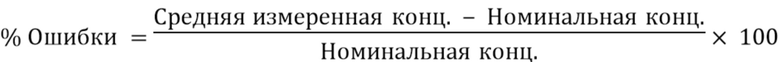
Достоверность:
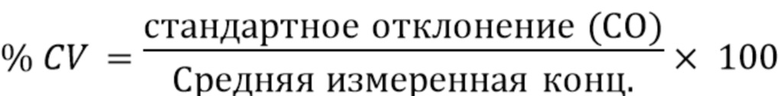
Точность и достоверность представляли до десятых. Все данные по концентрации представляли до трех значимых цифр.
3. Селективность матрицы
Селективность определяется как способность хроматографического способа измерять ответ от аналита без влияния со стороны биологической матрицы. Этого достигали путем оценки шести отдельных партий человеческой плазмы, полученных в качестве холостых проб и на нижнем пределе количественного определения (НПКО, 0,500 нг/мл).
3.1. Оценка на основании проб НПКО
Пробы селективности НПКО были приемлемы, если точность находилась в пределах ± 20,0% для по меньшей мере 5 из 6 проб, а достоверность составляла ≤ 20,0% для всех проб.
3.2. Оценка на основании холостых проб
Площади пика аналита в шести холостых пробах сравнивали со средней площадью пика аналита в пробах селективности НПКО. Оценка была приемлемой, если площадь пика в 5 из 6 холостых проб во время удержания аналита находилась в пределах ≤ 20,0% средней площади пика аналита проб селективности НПКО. Кроме того, площадь пика в 5 из 6 холостых проб во время удержания ВС должна была находиться в пределах ≤ 5,0% средней площади пика ВС проб селективности НПКО. Результаты для кетамина и норкетамина соответствовали критериям приемлемости.
Время удержания кетамина и кетамина-d4 (ВС) составляло приблизительно 2,3 минуты. Время удержания норкетамина и норкетамина-d4 (ВС) составляло приблизительно 2,0 минуты.
3.3. Перенос вводимого вещества из предыдущих проб
Целью исследования переноса вводимого вещества из предыдущих проб является оценка степени переноса представляющего интерес аналита из одной пробы в другую в каждом аналитическом эксперименте. Во время валидационных экспериментов двойную холостую пробу вводили после высокого стандарта из набора калибраторов. Перенос вводимого вещества для аналита составлял менее 20% площади пика НПКО (стандарт 1) для всех двойных холостых проб, что соответствовало критериям приемлемости. Кроме того, площадь пика ВС составляла 0,0% от среднего значения площади пика ВС из принятых калибровочных стандартов партии и проб КК, что хорошо соответствует критериям приемлемости 5,0%.
3.4. Эффект матрицы
Эффект матрицы определяется как подавление или повышение ионизации аналитов присутствием компонентов матрицы в биологических образцах. Смотрите, например, C.T. Viswanathan, “Quantitative Bioanalytical Methods Validation and Implementation: Best Practices for Chromatographic and Ligand Binding Assays,” Pharmaceutical Research, Vol. 24, No. 10, October 2007, p. 1969. Эффект матрицы оценивали путем экстракции единичных реплик шести партий контрольной человеческой плазмы и добавления в каждую партию КК при низких и высоких уровнях концентрации (1,50 нг/мл и 375 нг/мл) после экстракции. Соотношение площади шести партий дополненных проб плазмы после экстракции сравнивали со средним соотношением площади, полученным по трем репликам чистого раствора, приготовленного при том же уровне концентрации в очищенной воде.
ВС-нормализованный матричный коэффициент рассчитывали в соответствии со следующей формулой:

Вариабельность ВС-нормализованных матричных коэффициентов (% CV) для 6 партий проб плазмы составила ≤ 15%, что было приемлемо.
Диапазон количественного определения составлял от 0,5 до 500 нг/мл как для эскетамина, так и для норэскетамина. Все критерии приемлемости для анализа были выполнены.
3.5. Обратно рассчитанные концентрации калибровочных стандартов
Были определены обратно рассчитанные концентрации калибровочных стандартов для кетамина и норкетамина. Средние обратно рассчитанные концентрации не отличались более чем на 15% от номинальных концентраций (20,0% при НПКО), а % CV для каждого уровня концентрации составлял не более 15,0% (20,0% на НПКО).
3.6. Регрессионная модель
Линейность способа оценивали в линейном диапазоне 0,500/0,500-500/500 нг/мл для кетамина/норкетамина в человеческой плазме. Линейную регрессию (с весовым коэффициентом 1/x2) использовали для получения наилучшей аппроксимации зависимости показания детектора от концентрации для кетамина и норкетамина в человеческой плазме. Все калибровочные кривые для кетамина и норкетамина имели коэффициент детерминации (R2) ≥ 0,98, что соответствует приемлемости.
3.7. Чувствительность
Валидацию проводили с целевым НПКО 0,500 нг/мл для кетамина и норкетамина в человеческой плазме. Для оценки чувствительности шесть проб КК, приготовленных на НПКО, анализировали в трех экспериментах с отдельными партиями в качестве части точности и достоверности способа внутри серии и между сериями. Концентрации рассчитывали с помощью калибровочной кривой. Результаты показали, что данный способ соответствует критериям приемлемости для чувствительности (точность в пределах ± 20,0% и % CV не более 20,0%).
Следовательно, способ был достаточно чувствительным для определения кетамина и норкетамина в плазме в концентрации 0,500 нг/мл.
3.8. Точность и достоверность внутри серии и между сериями
Точность и достоверность способа внутри серии и между сериями исследовали при четырех разных уровнях концентрации КК (0,500 нг/мл (НПКО), 1,50 нг/мл, 80,0 нг/мл и 375 нг/мл). Результаты продемонстрировали, что точность и достоверность способа внутри серии и между сериями соответствуют критериям приемлемости (точность в пределах ± 15,0% (в пределах ± 20,0% для НПКО) и % CV не более 15,0% (20,0% для НПКО)).
3.9. Восстановление
Восстановление образца препарата оценивали путем сравнения среднего соотношения площади проб КК со средним соотношением площади проб кетамина/норкетамина с непосредственным добавлением (в тех же концентрациях) в экстрагированной объединенной плазме. Восстановление рассчитывали в соответствии со следующей формулой:

Восстановление было определено для кетамина и норкетамина при трех уровнях концентрации КК (низкий, средний и высокий). Для каждой концентрации проводили шесть измерений. Вариабельность (% CV) соотношения пиковой площади для каждого уровня КК должна составлять ≤ 15%. Результаты были приемлемыми. В соответствии с руководством BIO-201 при использовании меченного стабильным изотопом ВС, восстановление, установленное для немеченого аналита, будет достаточным, и восстановление для меченного стабильным изотопом ВС, не потребуется.
3.10. Воспроизводимость повторного ввода
Для оценки воспроизводимости повторного ввода после выдерживания при комнатной температуре в течение 161,5 часов проводили ввод партии аналита, содержащей пробы стандартной кривой и КК. Калибровочные стандарты и пробы КК (низкая, средняя и высокая концентрация) соответствовали общим критериям приемлемости партии, что свидетельствует о том, что образцы можно повторно вводить в течение до 161,5 часов после начального ввода.
3.11. Оценка размера партии
При валидации аналитическую серию № 3 использовали для имитации размера партии для анализа проб. В партию было включено в общей сложности 214 проб, которые включали как пробы стандартной кривой, так и пробы КК. Калибровочные стандарты и образцы КК внутри серии соответствовали общим критериям приемлемости партии для выполнения анализа образца.
Пример 9
Фармакокинетические исследования
Первичная цель данного исследования заключалась в оценке фармакокинетики (ФК) интраназального введения эскетамина у здоровых субъектов.
1. Способы
1.1. Обзор дизайна исследования
1.1.1. Общий дизайн
Это была открытое, одноцентровое исследование. Субъекты представляли собой здоровых мужчин и женщин белой европеоидной расы в возрасте от 20 до 55 лет включительно. Как описано в примере 1, эскетамин был предоставлен в виде прозрачного бесцветного интраназального раствора гидрохлорида эскетамина (16,14% масса/объем [масс./об.]; эквивалент 14% масс./об. основания эскетамина) в насосе для назального впрыскивания. Раствор состоял из:
161,4 мг/мл гидрохлорида эскетамина;
0.12 мг/мл этилендиаминтетрауксусной кислоты (ЭДТА);
1,5 мг/мл лимонной кислоты;
при pH 4,5 в воде для инъекций.
Раствор был предоставлен в насосе для назального впрыскивания, который доставлял 16,14 мг гидрохлорида эскетамина (14 мг основания эскетамина) на 100 мкл спрея.
Все субъекты самостоятельно применяли каждую из 3 разных схем введения единичной дозы интраназального эскетамина (варианты лечения A, B и C) в течение 3 периодов лечения (т.е. 1 лечения на период) открытым образом под непосредственным наблюдением исследователя или его представителя.
• Вариант лечения А: 1 впрыскивание 14% раствора эскетамина в каждую ноздрю в момент времени 0 (общая доза 28 мг);
• Вариант лечения B: 1 впрыскивание 14% раствора эскетамина в каждую ноздрю в момент времени 0 и повтор через 5 минут (общая доза 56 мг);
• Вариант лечения C: 1 впрыскивание 14% раствора эскетамина в каждую ноздрю в момент времени 0 и повтор каждые 5 минут ×2 (общая доза 84 мг);
Субъектам случайным образом назначили вариант лечения A и вариант лечения B в первые 2 периода (т.е. вариант лечения A в период 1 и вариант лечения B в период 2, или в обратном порядке). Все субъекты получали вариант лечения C в период 3 (Таблица 166). Схемы отличались по количеству впрыскиваний для достижения общей дозы и общей вводимой дозой эскетамина. Каждую схему введения интраназального эскетамина отделял период отмывки от 5 до 14 суток.
A. 1 впрыскивание 14% раствора эскетамина в каждую ноздрю в момент времени 0 (общая доза 28 мг)
B. 1 впрыскивание 14% раствора эскетамина в каждую ноздрю в момент времени 0 и повтор через 5 минут (общая доза 56 мг)
C. 1 впрыскивание 14% раствора эскетамина в каждую ноздрю в момент времени 0 и повтор каждые 5 минут ×2 (общая доза 84 мг)
После предоставления письменного информированного согласия проводили оценку субъектов в период с -21 по -2 сутки во время фазы скрининга для определения пригодности для участия, что включало обзор критериев включения и исключения. Все субъекты должны были соответствовать критериям включения/исключения перед допуском в исследовательский центр для каждого периода лечения. Субъектов допускали в исследовательский центр на -1 сутки каждого периода и выпускали из исследовательского центра после сбора последнего 24-часового образца ФК на 2 сутки каждого периода лечения. На -1 сутки первого периода лечения пациенты, имеющие право на участие, практиковались самостоятельно вводить прозрачный бесцветный интраназальный раствор плацебо для введения (вода для инъекций 0,001 мг/мл, [0,0001%] денатония бензоата) в положении полулежа, используя устройства, идентичные используемым для введения эскетамина. На 1 сутки каждого периода лечения субъекты самостоятельно применяли каждую интраназальную схему введения эскетамина (смотрите раздел 1.5, Дозировка и введение).
Образцы крови для фармакокинетики для измерения концентраций эскетамина и норэскетамина в плазме собирали от момента времени до введения дозы до 24 часов после каждого интраназального введения эскетамина на 1 сутки каждого периода.
Субъектов возвращали в исследовательский центр через 11 ( ± 2) суток после последней дозы исследуемого препарата для оценок окончания исследования. В альтернативном варианте оценки окончания исследования проводили в момент времени досрочного прекращения участия. Окончанием исследования была дата последнего посещения последнего субъекта, участвующего в исследовании. Общая длительность исследования от фазы скрининга до последующего наблюдения составляла до 63 суток.
Обоснование дизайна исследования
• Исследуемый агент: Эскетамин имеет более высокую аффинность к рецепторам NMDA, тем самым снижая необходимую нагрузку препарата и потенциально приводя к более быстрому восстановлению функций головного мозга и менее неприятным психотомиметическим эффектам, чем рацемический R-кетамин и кетамин, соответственно;
• Исследуемая популяция: Ожидалось, что размера выборки, составляющего 12 субъектов, будет достаточно для адекватного изучения характеристик ФК каждой схемы введения интраназального эскетамина на основании вариабельности, указанной в предыдущих исследованиях с интраназальным кетамином, и ее посчитали представляющей профиль у соответствующих субъектов с ТРД, которые будут включены в будущие клинические исследования;
• Дизайн исследования: Этап скрининга длительностью до 21 суток обеспечивал достаточное время для оценки пригодности субъекта в соответствии с критериями включения/исключения для исследования, а визит последующего наблюдения после лечения через 11 (± 2) суток облегчал оценку для оценки безопасности и переносимости субъектов. Рандомизацию использовали для того, чтобы избежать ошибок при отнесении субъектов к группе последовательности лечения и повысить вероятность того, что известные и неизвестные свойства субъекта (например, демографические и исходные характеристики) будут равномерно распределены между последовательностями лечения. Дизайн перекрестного исследования уменьшал общее количество субъектов, которые должны были быть включены в исследование, и позволяла проводить сравнения для одного субъекта;
• Доза и введение: В данном исследовании для инициации и поддержания анестезии использовали схемы приема доз интраназального эскетамина, которые ниже по сравнению с В/В схемами эскетамина, обычно применяемыми для индукции и поддержания анестезии. На основании опубликованных данных исследований предполагалось, что дозы хорошо переносятся;
• Фармакокинетика: Для оценки ФК при однократной дозе как эскетамина, так и его метаболита норкетамина, было достаточно 24-часового интервала для сбора крови после введения дозы.
1.2. Исследуемая популяция
В исследование были включены четырнадцать здоровых взрослых субъектов белой европеоидной расы, с одинаковым числом субъектов обоих полов для равного отношения мужчин к женщинам. В это исследование были включены здоровые мужчины и женщины в возрасте от 20 до 55 лет, включительно, с индексом массы тела (ИМТ) от 18 до 28 кг/м2, включительно, и массой тела не менее 50 кг. Субъекты имели систолическое кровяное давление от 90 мм рт.ст. до 145 мм рт.ст., включительно, и диастолическое кровяное давление не выше 90 мм рт.ст., нормальный синусовый ритм, пульс от 45 до 90 ударов в минуту, интервал QTc ≤ 450 миллисекунд (мс), интервал QRS ≤ 120 мс, интервал PR < 210 мс и морфологию ЭКГ, соответствующую здоровым состоянию и функции сердца. Согласно протоколу субъекты не должны были иметь в анамнезе суицидальной или гомицидальной идеации, существенного первичного расстройства сна или каких-либо противопоказаний к применению кетамина или эскетамина.
Удаление субъектов по причинам терапии или оценки для прекращения субъектом участия в исследовании могло включать следующее:
• невозможность последующего наблюдения;
• отзыв согласия;
• субъект не соблюдает требования исследования, в том числе критерии включения, критерии исключения, запреты и ограничения;
• прекращение прохождения исследуемого лечения (с получением конечных оценок). Субъект должен быть отстранен от исследуемого лечения, если:
• исследователь посчитал, что по соображениям безопасности (например, НЯ) прекращение лечения находится интересах субъекта
• субъект забеременел.
В случае невозможности последующего наблюдения субъекта персонал исследовательского центра должен был приложить все разумные усилия к тому, чтобы связаться с субъектом и определить причину прекращения участия. Меры, предпринятые для осуществления последующего наблюдения, следовало документировать.
Если субъект выбывал до завершения исследования, то причина прекращения должна быть отражена в индивидуальной регистрационной форме (CRF) и в первичном документе. Исследуемый агент, назначенный прекратившему участие субъекту, не подлежал назначению другому субъекту. По меньшей мере 12 субъектов (включая 4 каждого пола) должны были завершить процедуры исследования всех периодов лечения, включая 24-часовой сбор проб крови для ФК, и оценки завершения исследования. Субъектов, которые прекратили участие, следовало предпочтительно заменять субъектом того же пола, чтобы получить необходимых 12 субъектов для лечения.
Соблюдение режима лечения
Исследуемый агент самостоятельно вводили в контролируемой среде клинического исследовательского центра, и непосредственное наблюдение за введением исследуемого препарата персоналом исследования обеспечивало соблюдение требований исследования. Дату и время введения каждого исследуемого препарата регистрировали в CRF.
Предшествующая и сопутствующая терапия
В течение всего исследования были запрещены рецептурные или безрецептурные препараты, отличные от исследуемого агента (включая витамины и травяные добавки; сосудосуживающие средства и противозастойные средства, вводимые офтальмологическими или интраназальными путями), за исключением ацетаминофена, пероральных противозачаточных средств и заместительной гормональной терапии. Применение ацетаминофена допускалось в течение 3 суток перед введением каждого исследуемого агента.
В течение всего исследования для лечения головной боли или другого вида боли было разрешено принимать максимум 3 мг в сутки 500 мг ацетаминофена и не более 3 г в неделю. При использовании ацетаминофена дозу и схему применения дозы, а также причину применения, необходимо было зафиксировать в ОФД.
На протяжении всего исследования женщины, использующие гормональные противозачаточные средства в качестве контрацепции, продолжали использовать те же гормональные противозачаточные средства. В течение всего исследования женщины, использующие гормональную заместительную терапию, продолжали использовать ту же гормональную заместительную терапию.
Спонсора необходимо было немедленно уведомлять о применении запрещенной терапии. Все лекарственные препараты, принимаемые субъектом (рецептурные или безрецептурные), которые не являлись исследуемым агентом, были задокументированы в разделе сопутствующей терапии CRF. Они включали препараты, взятые за 30 суток до, во время и до завершения визита окончания исследования (от 9 до 13 суток после последнего введения исследуемого агента).
1.3. Информация по исследуемому лекарственному препарату
Интраназальный эскетамин и плацебо для практического введения были предоставлены спонсором в двойном устройстве для назального впрыскивания. Каждое устройство содержало 200 мкл и доставляло 16,14 мг гидрохлорида эскетамина (14 мг основания эскетамина) или 0,1 мкг бензоата денатония на 100 мкл спрея, соответственно. Информация о исследуемом агенте приведена в Таблице 167.
1.4. Рандомизация и маскировка
Это была открытое исследование; таким образом, маскировку лечения не проводили. Каждая схема введения интраназальной дозы была помечена кодом рандомизации персоналом центра и применялась открытым образом.
При замене субъектов заменяющим субъектам назначали ту же последовательность лечения, что и субъектам, которых они заменяли. Замена субъектов начиналась с периода 1.
1.5. Способ применения и дозы
Субъекты самостоятельно вводили интраназальный эскетамин открытым перекрестным образом. Они получали схемы эскетамина в течение 3 периодов лечения (т.е. 1 лечение на период, таблица 168), как указано в последовательностях лечения (Таблица C). Схемы отличались по количеству впрыскиваний для достижения общей дозы и общей вводимой дозой эскетамина.
aВремя 0 определяется как время впрыскивания первых 100 мкл. Впрыскивание на каждую ноздрю следует осуществлять быстро в запланированные моменты времени (т.е. не должно быть промедления между впрыскиванием в каждую ноздрю в каждый момент времени). Субъекты должны находиться в полулежащем положении при осуществлении впрыскивания и оставаться в полулежащем положении в течение по меньшей мере 10 минут после последнего впрыскивания.
b Концентрация эскетамина (процент раствора эскетамина) и общая доза выражены в виде основания эскетамина.
Применение схем введения интраназального эскетамина осуществляли самостоятельно под непосредственным наблюдением исследователя или его представителя, используя модифицированные инструкции, предоставленные в центре (т.е. в полулежащем положении в течение по меньшей мере 10 минут после последнего впрыскивания; после введения дозы могло начинаться чиханье).
Пищу ограничивали в течение по меньшей мере 8 часов, начиная с вечера до введения дозы, до 2 часов после каждого введения эскетамина. Употребление воды или любого другого разрешенного напитка было ограничено за 30 минут до первого назального впрыскивания и до 30 минут после последнего назального впрыскивания по заданной схеме. Приблизительно через 2 часа после впрыскивания последней назальной дозы субъекты во всех 3 периодах лечения должны были выпить от 180 до 240 мл воды.
1.6. Оценки исследования и статистические методы
1.6.1. Оценки фармакокинетики
1.6.1.1. Сбор и обработка проб
Образцы крови (по 4 мл каждый) для определения концентраций эскетамина и норэскетамина в плазме собирали в соответствующую пробирку для сбора (например, в пробирки Vacutainer®) в моменты времени 0,00, 0,12, 0,20, 0,37, 0,53, 0,67, 0,83, 1,00, 1,25, 1,50, 2,00, 3,00, 4,00, 6,00, 9,00, 12,00, 18,00 и 24,00 ч. Общее количество крови для проведения клинических лабораторных анализов и оценок ФК составляет приблизительно 327 мл.
Точную дату и время сбора проб записывали в CRF при необходимости. Перед обработкой пробирки осторожно переворачивали от 8 до 10 раз для обеспечения перемешивания и помещали в криоблок (в вертикальном положении) или в смесь ледяной воды приблизительно до высоты, соответствующей уровню крови в пробирке. Образцы крови центрифугировали в течение 60 минут после сбора в клинической центрифуге при 1300 g (около 2500-3000 об/мин) в течение 10 минут при 5°C с получением приблизительно 1,8 мл плазмы из каждого 4 мл образца цельной крови. Отделенную плазму немедленно переносили (равномерно разделенную) в 2 предварительно помеченные полипропиленовые пробирки для хранения с помощью чистой одноразовой стеклянной или полиэтиленовой пипеткой, используя новую пипетку для каждого образца. Одну пробирку метили «эскетамин, основной», а вторую «эскетамин, резерв». Образцы плазмы хранили в вертикальном положении при -20°C или ниже до их переноса в биоаналитический блок. Время между сбором крови и замораживанием плазмы не должно было превышать 2 часа.
1.6.1.2. Биоаналитические процедуры
Образцы плазмы анализировали в отношении концентраций эскетамина и норэскетамина, используя проверенный, специфический и чувствительный ахиральный метод жидкостной хроматографии в сочетании с тандемной масс-спектрометрией (ЖХ-МС/МС) из примера 8.
1.6.1.3. Фармакокинетические параметры
Серийно разведенные образцы крови для ФК (по 4 мл каждый) брали в течение 24-часового периода у каждого субъекта (периоды 1, 2 и 3). Некомпартментные ФК-параметры эскетамина и его метаболита норэскетамина, оцениваемые в плазме, включали:
• Cмакс - максимальная концентрация в плазме в течение интервала введения дозы tмакс
• tмакс - время до достижения максимальной концентрации в плазме
• ППКпосл - площадь под кривой плазменная концентрация - время, начиная с момента времени 0 до времени последней количественно определяемой концентрации
• ППК∞ - площадь под кривой плазменная концентрация - время от момента времени 0 до бесконечности, вычисляемая как сумма ППКпосл и Cпосл/λz, где Cпосл - последняя наблюдаемая количественно определяемая концентрация
• t1/2,λ - период полувыведения, связанный с наклоном в конечной стадии (λz) полулогарифмической кривой концентрация препарата - время, рассчитываемый как 0,693/ λz
• λz - константа скорости первого порядка, связанная с конечной частью кривой, определяемая как отрицательный наклон конечной log-линейной фазы кривой концентрация препарата – время.
1.6.4. Статистические методы
1.6.4.1. Размер выборки
По меньшей мере 12 субъектов (включая 4 каждого пола) должны были завершить процедуры исследования всех периодов лечения, включая 24-часовой сбор проб крови для ФК, и оценки завершения исследования. Субъектов, которые прекратили участие, предпочтительно заменяли субъектом того же пола, чтобы получить необходимых 12 субъектов для лечения. На основании ранее завершенного исследования коэффициент межиндивидуальной вариабельности для Cмакс и ППК интраназального рацемического кетамина, по оценкам, составлял по меньшей мере 55%. Принимая коэффициент межиндивидуальной вариабельности 55% для ФК-параметров эскетамина, размер выборки из 12 субъектов был достаточным для обеспечения того, чтобы оценка средних ФК-параметров эскетамина оказалась в пределах 71% и 142% от истинного значения с доверительным интервалом 95%.
1.6.4.2. Исходные характеристики субъектов
Для всех субъектов, получивших по меньшей мере 1 дозу исследуемого агента, описательную статистику (среднее значение, стандартное отклонение [СО], медиана, минимум, максимум) проводили для возраста, ИМТ, массы и высоты. Пол и раса были записаны и занесены в таблицу.
1.6.4.3. Фармакокинетический анализ
Для эскетамина и норэскетамина были записаны данные для всех субъектов с доступными концентрациями в плазме. Все концентрации в плазме, находящиеся ниже наименьшей количественно определяемой концентрации в образце, или отсутствующие данные, были помечены как таковые в приведенных данных о концентрации.
Концентрации ниже наименьшей количественно определяемой концентрации обрабатывали как нулевые при расчете ФК-параметров и суммарной статистики. Все субъекты и образцы, исключенные из анализа, должны быть четко задокументированы.
Описательную статистику использовали для обобщения плазменных концентраций эскетамина и норэскетамина в каждый момент сбора проб. Анализ включал данные по всем субъектам с доступными данными для по меньшей мере 1 дозы исследуемого агента. Данные о концентрации в плазме в каждый момент времени обобщали с получением среднего, медианы, минимума, максимума, СО и процентного коэффициента вариабельности для всех субъектов, получивших по меньшей мере 1 дозу исследуемого агента.
Следующие ключевые параметры эскетамина и норэскетамина в плазме рассчитывали, используя некомпартментные методы и действительно время сбора проб: Cмакс, tмакс, AUCпосл, AUC∞, t1/2,λ и λz.
Все оцененные ФК-параметры эскетамина и норэскетамина обобщали для каждого варианта лечения с получением среднего, медианы, минимума, максимума, СО и процентного коэффициента вариабельности для каждого предоставленного варианта лечения.
2. Информация о субъектах и лечении
2.1. Распределение субъектов и информация о завершении исследования/прекращении участия в исследовании
Первоначально в исследование включили 14 субъектов белой европеоидной расы и лечили эскетамином, причем для каждой последовательности лечения были рандомизированы по 7 субъектов. Из этих 14 субъектов 13 субъектов завершили исследование, по 7 субъектов в каждой последовательности лечения (ABC и BAC). Один субъект прекратил участие в исследовании; субъект не соответствовал критерию включения для периода 2 из-за положительного результата анализа мочи на наличие наркотических веществ при регистрации в период 2.
2.2. Демографические и исходные характеристики
Демографические и исходные характеристики субъектов, получавших дозу эскетамина, представлены в (Таблице 169).
ABC=эскетамин, 28 мг/эскетамин, 56 мг/эскетамин, 84 мг.
BAC=эскетамин, 56 мг/эскетамин, 28 мг/эскетамин, 84 мг.
2.6. Степень воздействия
Субъекты использовали все 3 разные схемы введения единичной дозы интраназального эскетамина (варианты лечения A, B и C) в периоды 1, 2 и 3 согласно рандомизированной последовательности. Впрыскивание проводили следующим образом:
• вариант лечения A, 28 мг в момент времени 0;
• вариант лечения B, 28 мг в каждый момент времени 0 и 5 минут (всего 56 мг); и
• вариант лечения C, 28 мг в каждый момент времени 0, 5 и 10 минут (всего 84 мг).
Выбывший субъект получал исследуемый агент таким образом:
• Субъект последовательности лечения 2 получал эскетамин, 28 мг в момент времени 0 и 5 минут (всего 56 мг) во время периода 1, сутки 1.
3. Результаты исследования фармакокинетики
Образцы плазмы анализировали в отношении концентраций эскетамина и норэскетамина проверенным, специфическим и чувствительным методом жидкостной хроматографии в сочетании с тандемной масс-спектрометрией (ЖХ-МС/МС), как описано в примере 8. В это исследование были включены в общей сложности 14 субъектов белой европеоидной расы (9 мужчин, 5 женщин), которые получили по меньшей мере 1 дозу 28 мг, 56 мг или 84 мг эскетамина. Один субъект выбыл из исследования после завершения первого периода лечения, в течение которого субъект самостоятельно вводил вариант лечения B (эскетамин, 56 мг).
Кроме того, один образец варианта лечения A (эскетамин, 28 мг, до введения дозы) был исключен из ФК-анализа эскетамина и норэскетамина, так как он был получен во время прекращения исследования.
Терминальную фазу профиля концентрация - время эскетамина невозможно было надежным образом оценить со значением R2adj < 0,900 и/или экстраполяцией ППК∞ > 20%. В результате ППК∞, ППК∞/доза, t1/2 и λz для эскетамина были исключены из описательной статистики для 3 субъектов, т.е. варианта лечения A (эскетамин, 28 мг), варианта лечения B (эскетамин, 56 мг) и варианта лечения C (эскетамин, 84 мг).
Терминальную фазу профиля концентрация - время норэскетамина невозможно было надежным образом оценить со значением R2adj < 0,900 и/или экстраполяцией ППК∞ > 20%. В результате ППК∞, ППК∞/доза, t1/2 и λz для норэскетамина были исключены из описательной статистики для 2 субъектов, т.е. варианта лечения B (эскетамин, 56 мг) и варианта лечения C (эскетамин, 84 мг).
Кроме того, соотношение метаболита и родительского соединения для ППК∞ было исключено из описательной статистики, поскольку значение ППК∞ было исключено из описательной статистики для эскетамина или норэскетамина для 5 субъектов, т.е. варианта лечения A (эскетамин, 28 мг), варианта лечения B (2 субъекта; эскетамин 56 мг) и варианта лечения C (2 субъекта; эскетамин, 84 мг).
Все ФК-параметры рассчитывали, используя фактическое время получения проб крови.
Результаты исследования фармакокинетики
Средние профили плазменная концентрация - время эскетамина и норэскетамина представлены на Фиг. 80 и 81, соответственно.
После интраназального введения получали максимальные концентрации (Cмакс) эскетамина с медианным tмакс в диапазоне от 0,67 до 0,83 часа у здоровых субъектов, соответственно, для 3 интраназальных схем введения эскетамина (28, 56 и 84 мг) (Таблица G). Максимальные концентрации метаболита норэскетамина наблюдали позже с медианным tмакс в диапазоне от 1,25 до 1,5 часа у здоровых субъектов, соответственно (Таблица H).
Для Cмакс эскетамина, вариабельность (выраженная в виде процентного коэффициента вариабельности) для 3 групп лечения находилась в диапазоне от 35,7% до 36,7%. Для ППК эскетамина (как ППКпосл, так и ППК∞) вариабельность находилась в диапазоне от 25,0% до 30,4%. Межиндивидуальная вариабельность для Cмакс норэскетамина находилась в диапазоне от 25,8% до 33,7% у субъектов. Для ППК норэскетамина (как ППКпосл, так и ППК∞) вариабельность находилась в диапазоне от 17,0% до 21,3%.
Для каждого уровня дозы строили графики Cмакс или ППК эскетамина в зависимости от общей массы тела. Аналогичные графики получали для метаболита. Результаты указывают на тенденцию к снижению Cмакс и ППК эскетамина и норэскетамина с увеличением массы тела. Сила этих тенденций варьировалась среди групп доз.
На основании визуального осмотра средние Cмакс, ППКпосл и ППК∞ для эскетамина и Cмакс для норэскетамина увеличиваются с повышением доз не совсем дозозависимым образом (Фиг. 80 и 81 и Таблицы G и H). Средние значения ППКпосл и ППК∞ для норэскетамина увеличивались образом, который был пропорционален дозе эскетамина.
Среднее значение t1/2 эскетамина находилось в диапазоне от 7,11 до 7,25 часа у субъектов (Таблица 170).
aМедиана (Мин-Макс), bn=13, cn=11, dn=12.
Среднее значение t1/2 норэскетамина находилось в диапазоне от 7,48 до 7,74 часа у субъектов (Таблица 171).
мл)
/мл)
мл)
aМедианное значение (Мин-Макс); bn=12; cn=13; dn=11; en=10
Концентрации в плазме норэскетамина в целом были выше по сравнению с родительским соединением. В случае Cмакс средние соотношения норэскетамина и эскетамина находились в диапазоне от 1,96 до 2,55. В случае ППКпосл средние соотношения находились в диапазоне от 3,62 до 4,32. В случае ППК∞ средние соотношения находились в диапазоне от 3,80 до 4,46 (Таблица H).
Заключение:
Средние значения Cмакс и ППК для эскетамина повышались не совсем дозозависимым образом для схем интраназального введения эскетамина в дозе 28 мг, 56 мг и 84 мг.
Пример 10
Данные, представленные в данном документе, были собраны из 14 исследований фазы 1; каждое исследование фазы 1 проводили аналогично исследованию, приведенному в примере 9, в котором оценивали ФК эскетамина.
ФК-параметры эскетамина, включая Tмакс, Cмакс, ППКпосл, и терминальное t1/2, после введения 28 мг, 56 мг или 84 мг назального эскетамина приведены в Таблице 172. tмакс эскетамина, как правило, наблюдали через 20-40 минут, 30-45 минут или 30-50 минут после первого назального впрыскивания 28 мг, 56 мг или 84 мг эскетамина, соответственно (т.е. приблизительно через 20-40 минут после последнего впрыскивания заданной дозы). Дозозависимое линейное увеличение средних значений Cмакс и ППКпосл является очевидным (Фиг. 82A и 82B).
Диапазон медианных значений приведен для Tмакс; диапазон средних значений приведен для Cмакс и ППКпосл.
После достижения Cмакс эскетамина после назального введения снижение концентраций в плазме происходит быстро в течение первых 2-4 часов, а затем более плавно. Среднее терминальное t1/2 эскетамина находился в диапазоне от 7 до 12 часов. Медианное терминальное t1/2 в данном исследовании составляло 10,7 часа.
Хотя приведенное выше описание содержит сведения о принципах настоящего изобретения с примерами, приведенными для иллюстрации, следует понимать, что практическое применение изобретения охватывает все обычные вариации, адаптации и/или модификации, входящие в объем приведенной ниже формулы изобретения и ее эквивалентов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНТРАНАЗАЛЬНОЕ ВВЕДЕНИЕ ЭСКЕТАМИНА | 2020 |
|
RU2830614C1 |
| Способы лечения биполярного расстройства | 2011 |
|
RU2653408C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ, ИХ КОМПОЗИЦИИ И ПРИМЕНЕНИЯ | 2017 |
|
RU2766155C2 |
| 9-АМИНОМЕТИЛМИНОЦИКЛИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2760186C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ДГЛК, И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2714323C2 |
| ЛИКСИСЕНАТИД И МЕТФОРМИН ДЛЯ ЛЕЧЕНИЯ ДИАБЕТА ТИПА 2 | 2012 |
|
RU2623023C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ЭОЗИНОФИЛЬНОГО ЭЗОФАГИТА | 2017 |
|
RU2766577C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ДЕКСТРОМЕТОРФАНОВОЕ СОЕДИНЕНИЕ И ХИНИДИН ДЛЯ ЛЕЧЕНИЯ ВОЗБУЖДЕНИЯ ПРИ ДЕМЕНЦИИ | 2020 |
|
RU2838188C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С РЕЦЕПТОРОМ S1P1 | 2021 |
|
RU2821032C1 |
| ЛЕЧЕНИЕ ВОССТАНОВЛЕННЫМ ЛИПОПРОТЕИНОМ ВЫСОКОЙ ПЛОТНОСТИ ИНФАРКТА МИОКАРДА | 2017 |
|
RU2798830C2 |
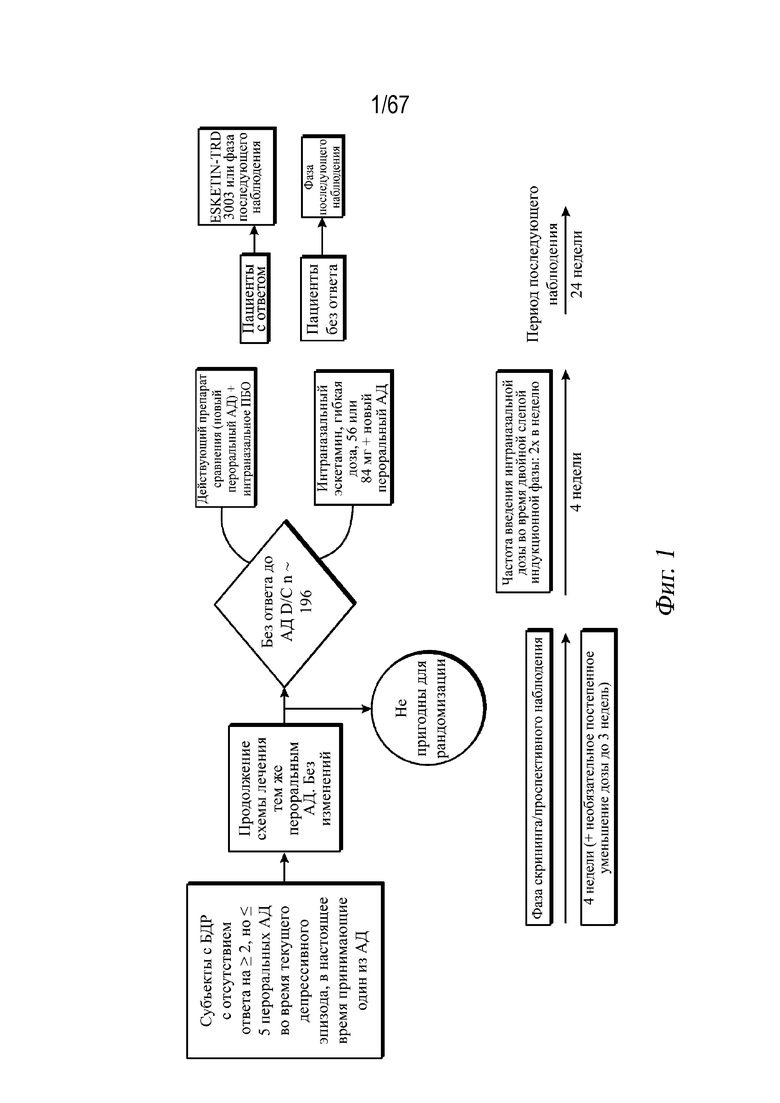
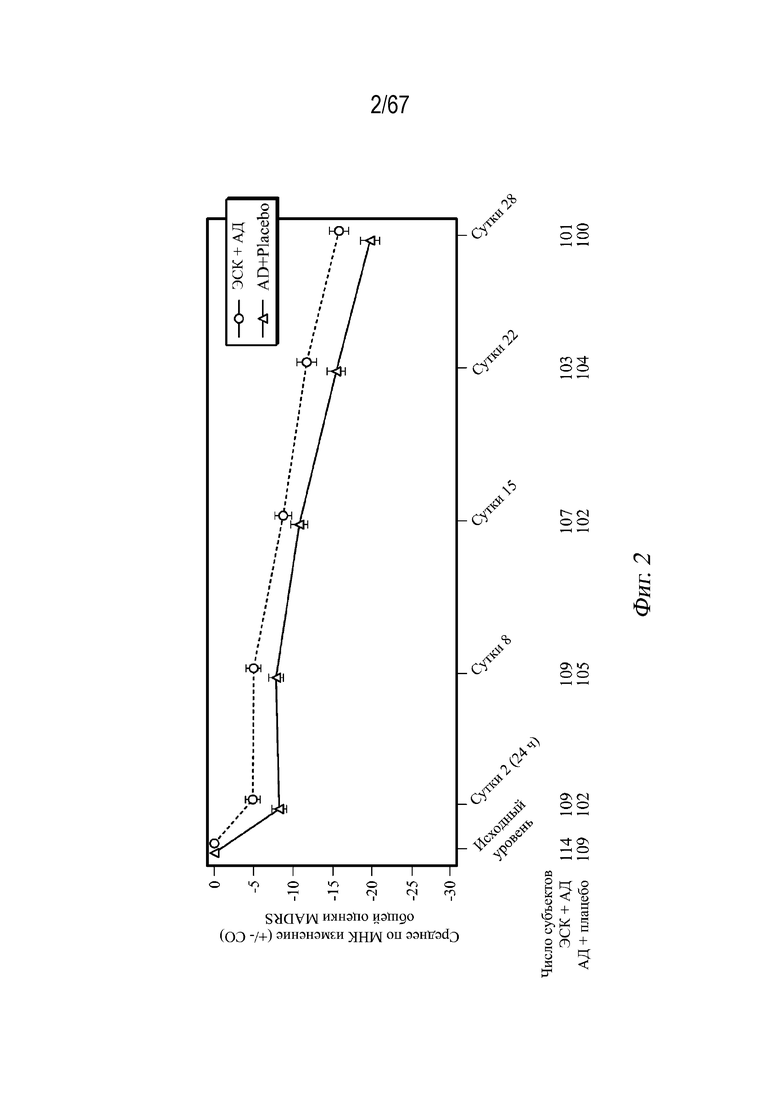
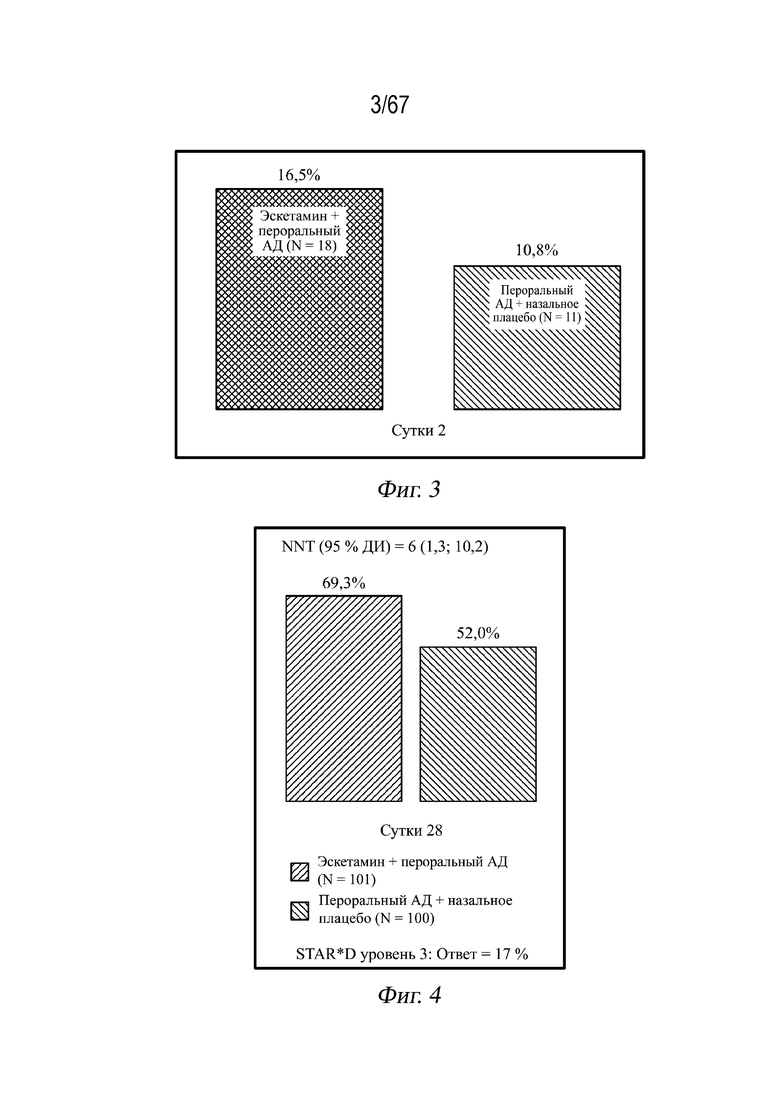









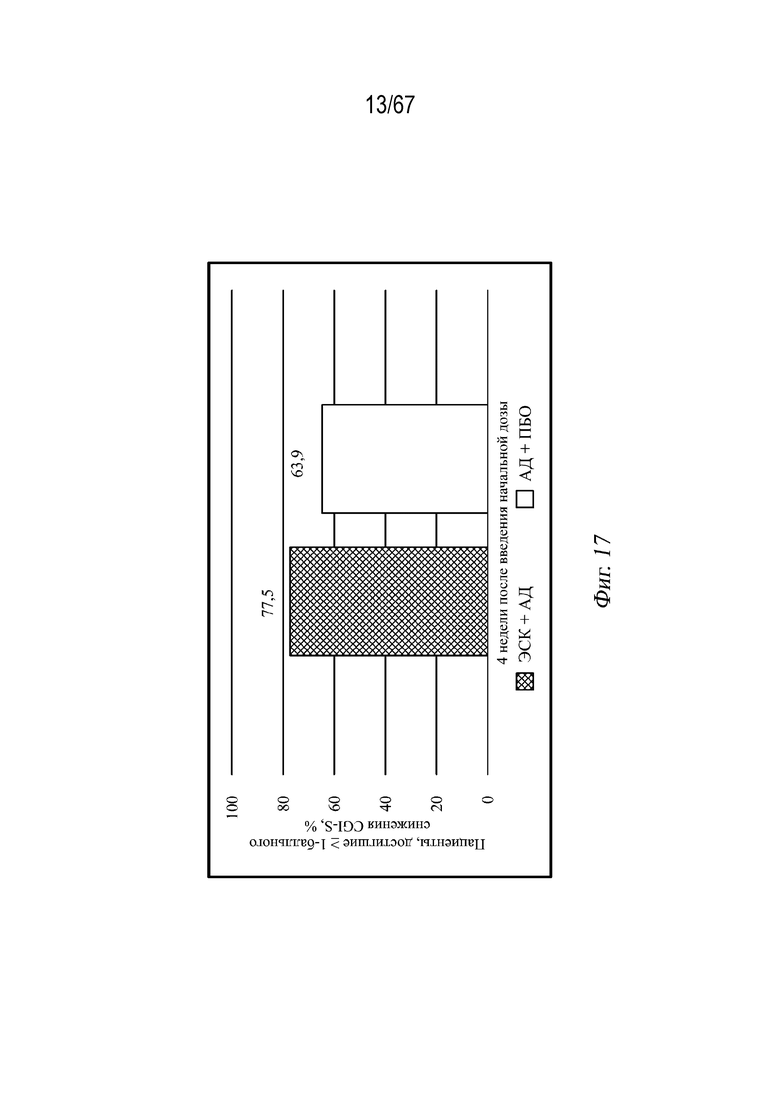

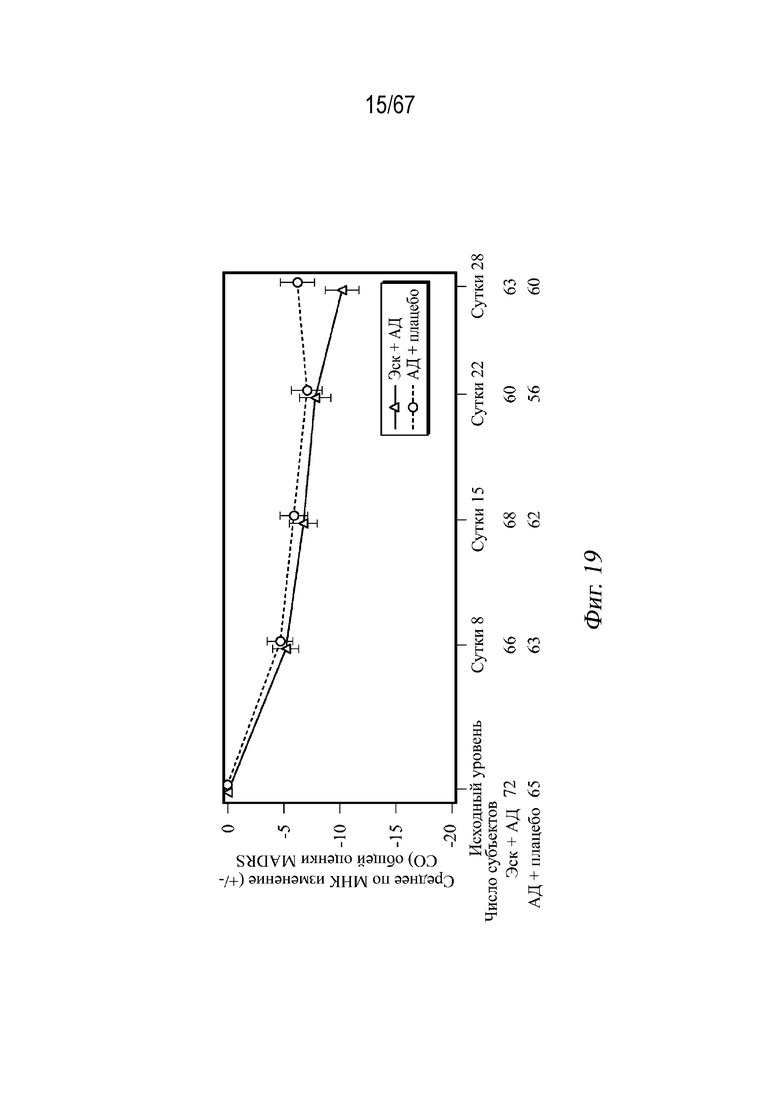
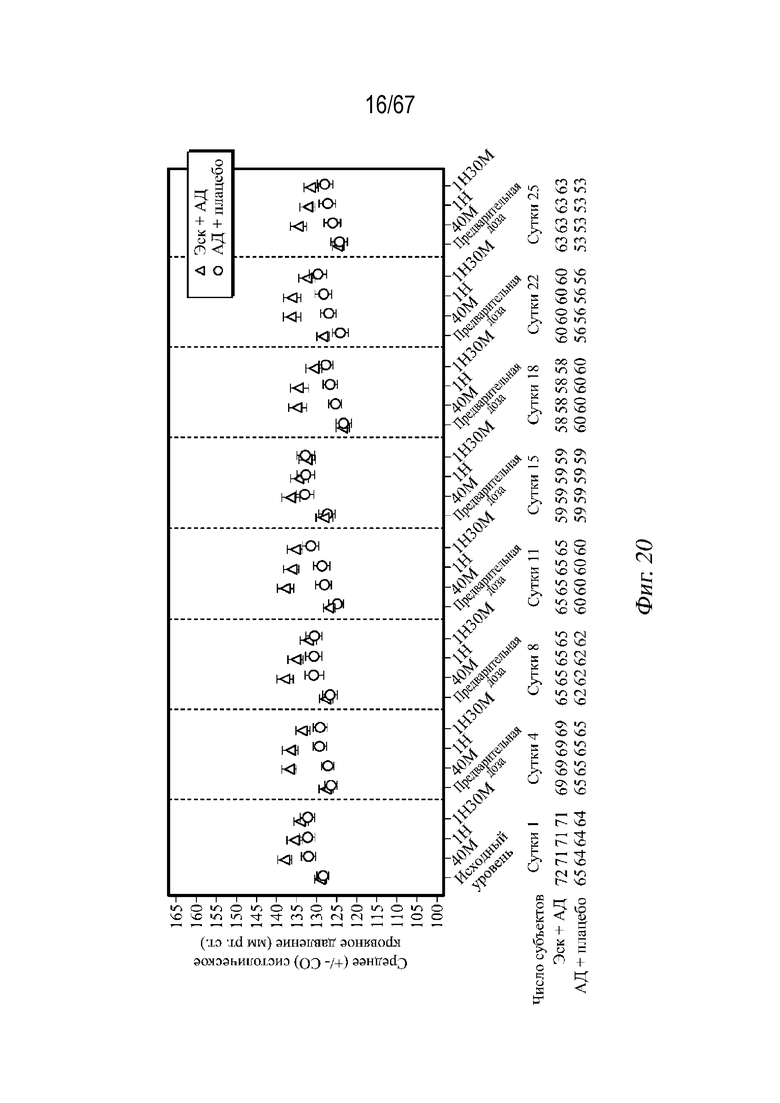
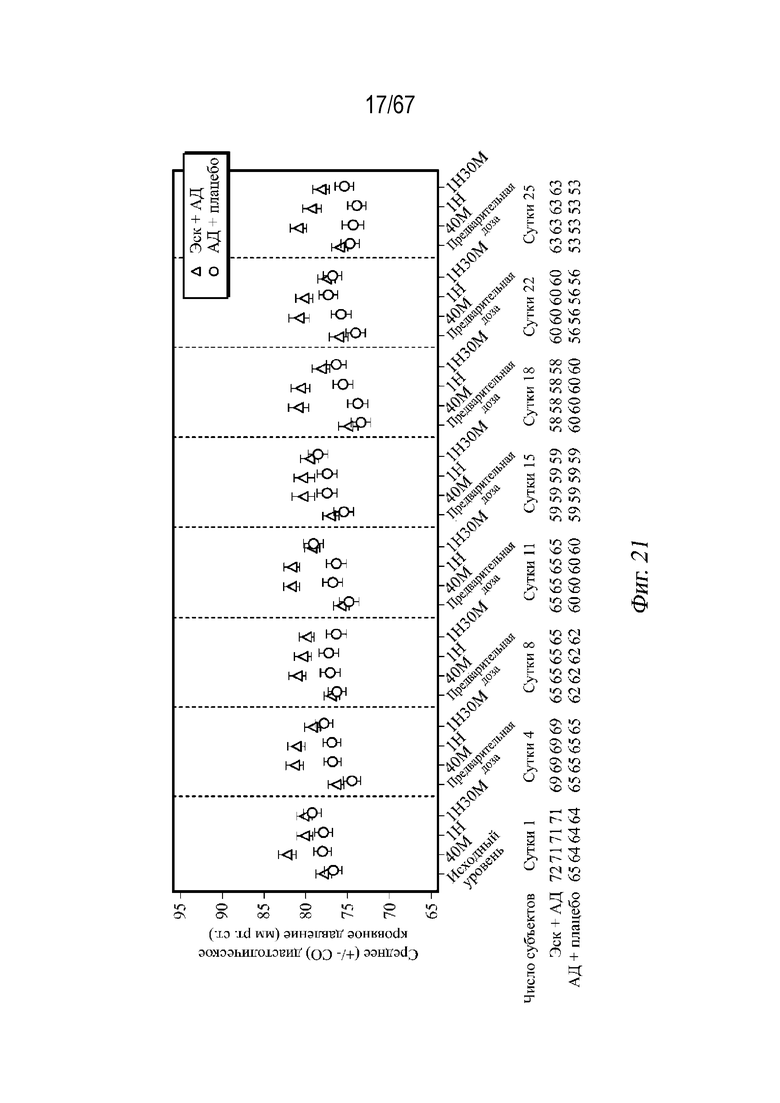



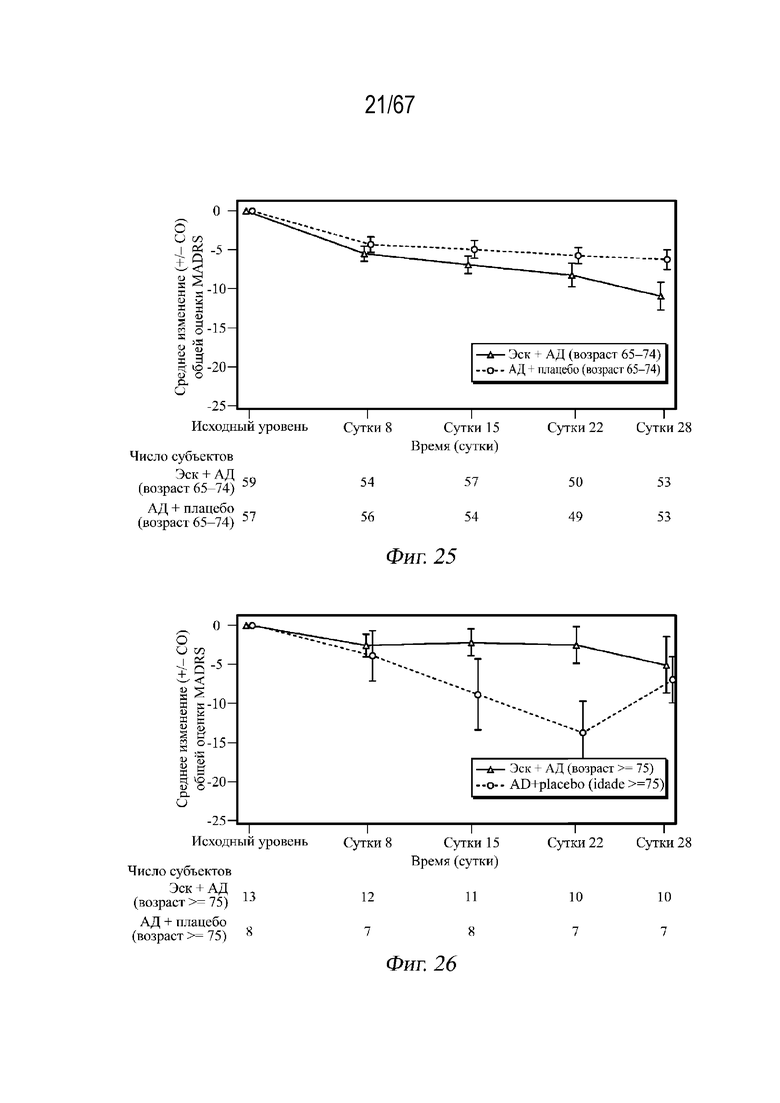
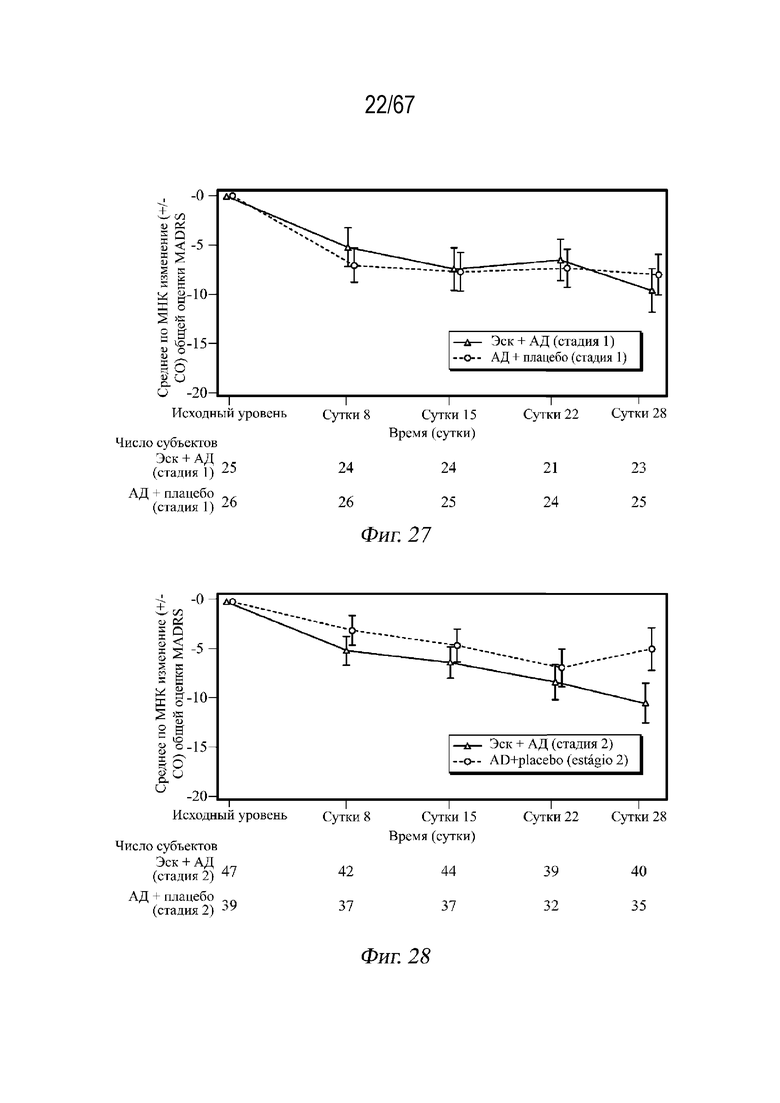






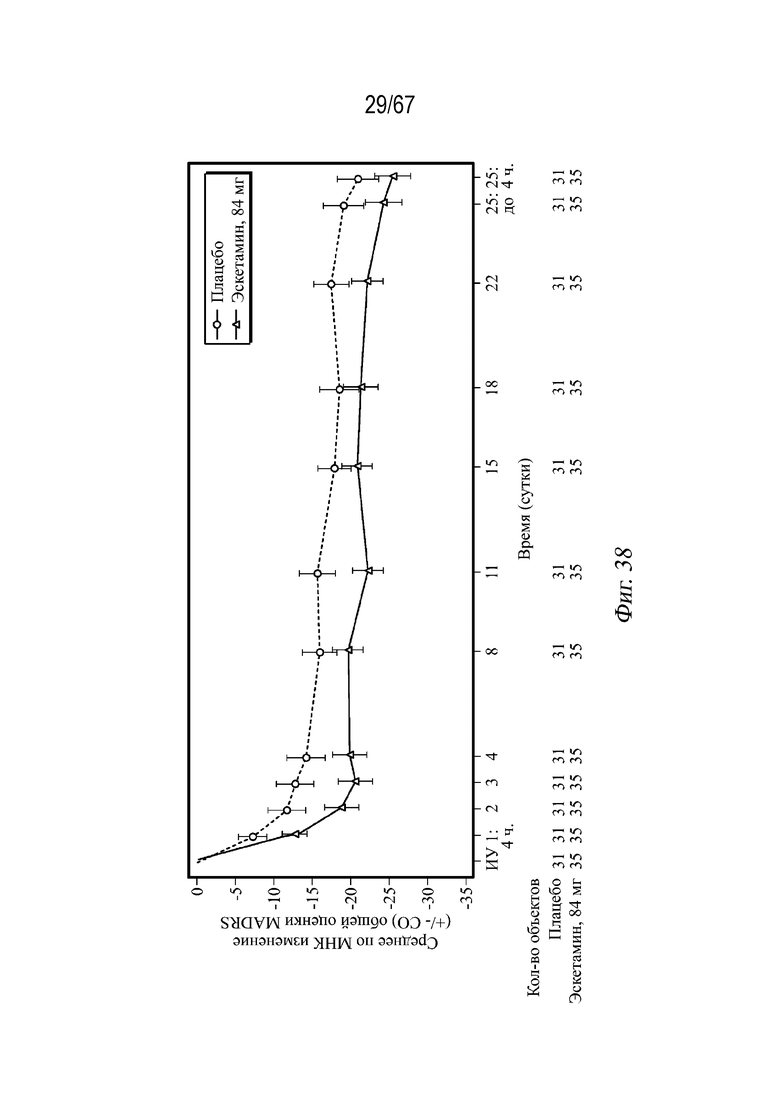





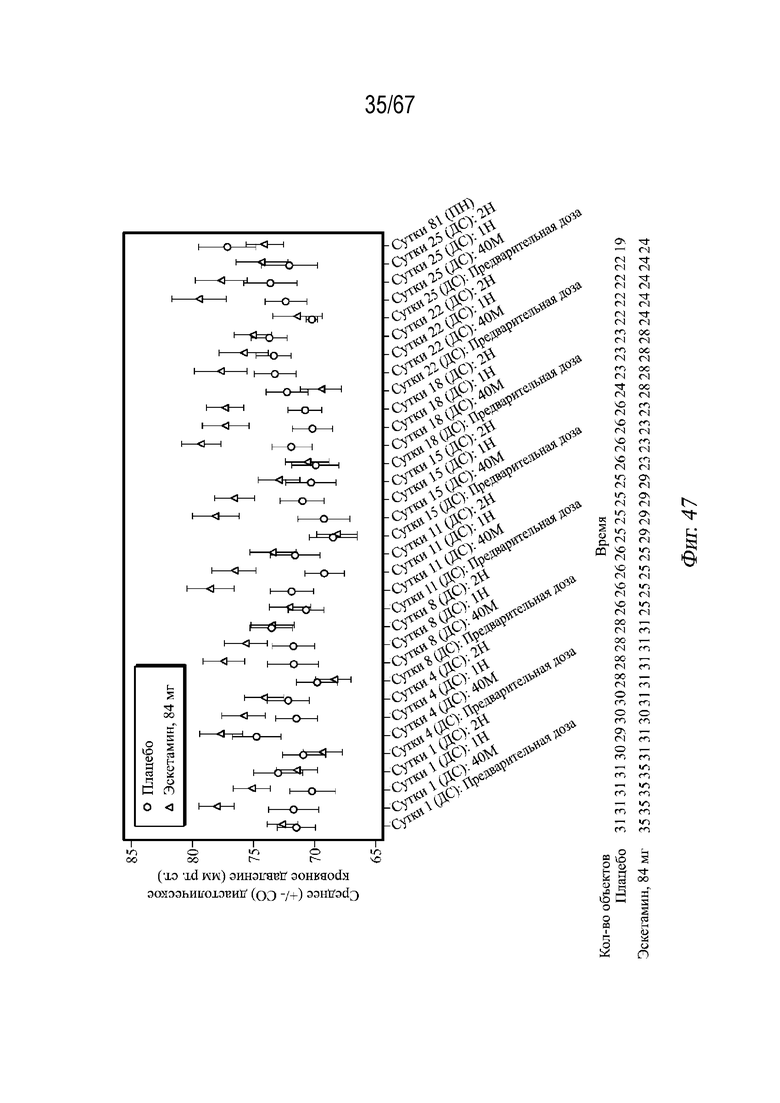

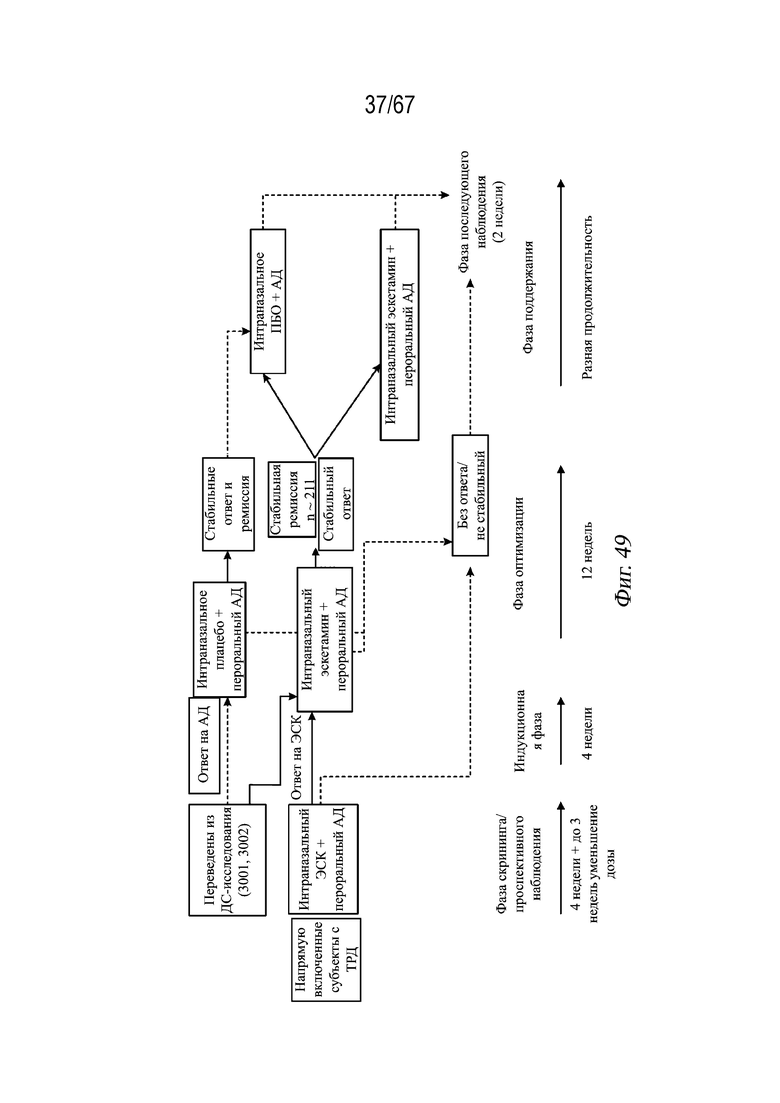

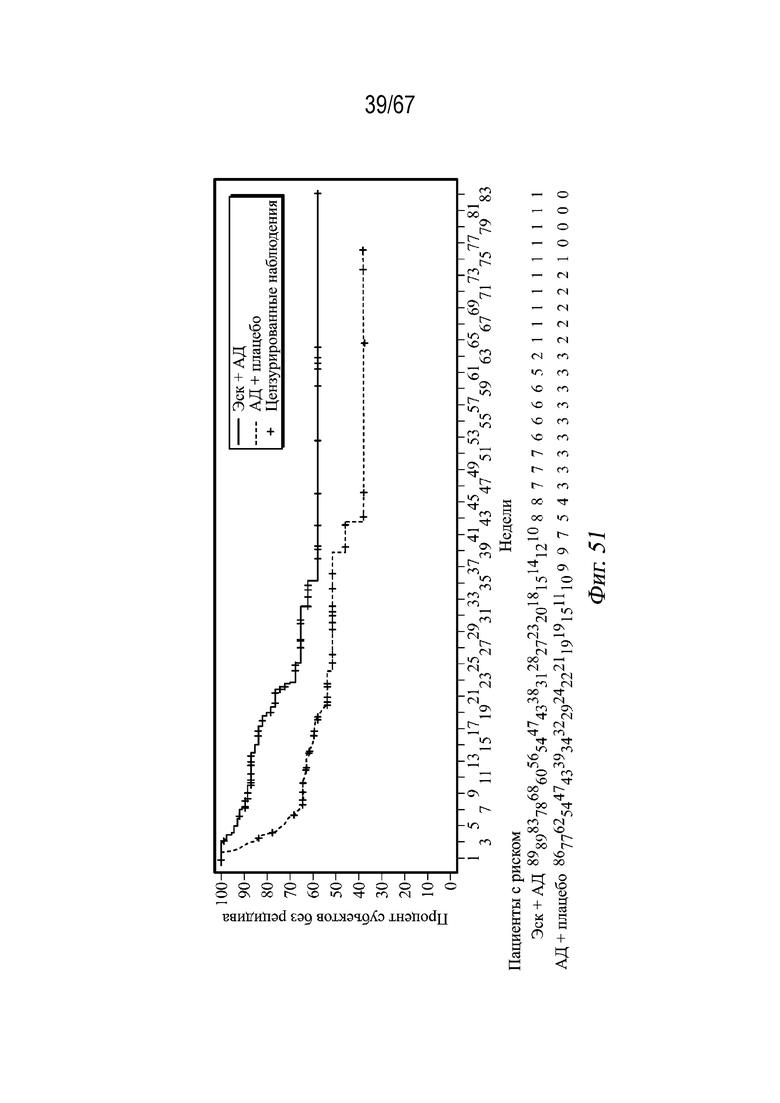



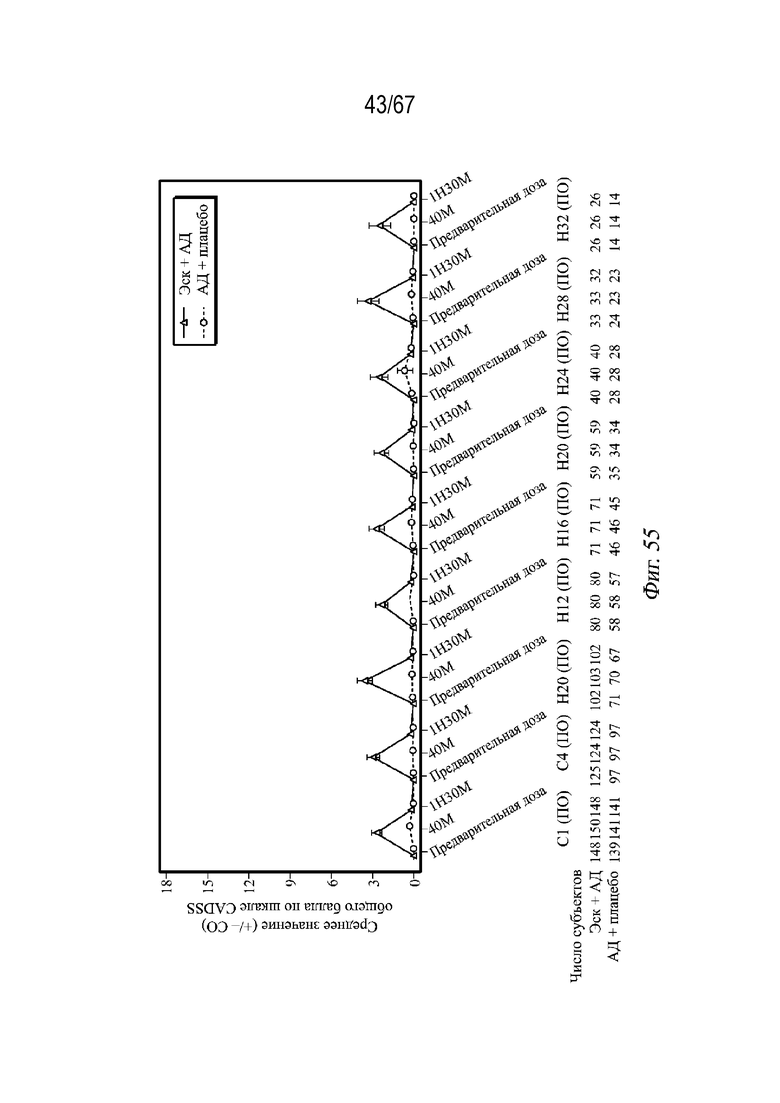






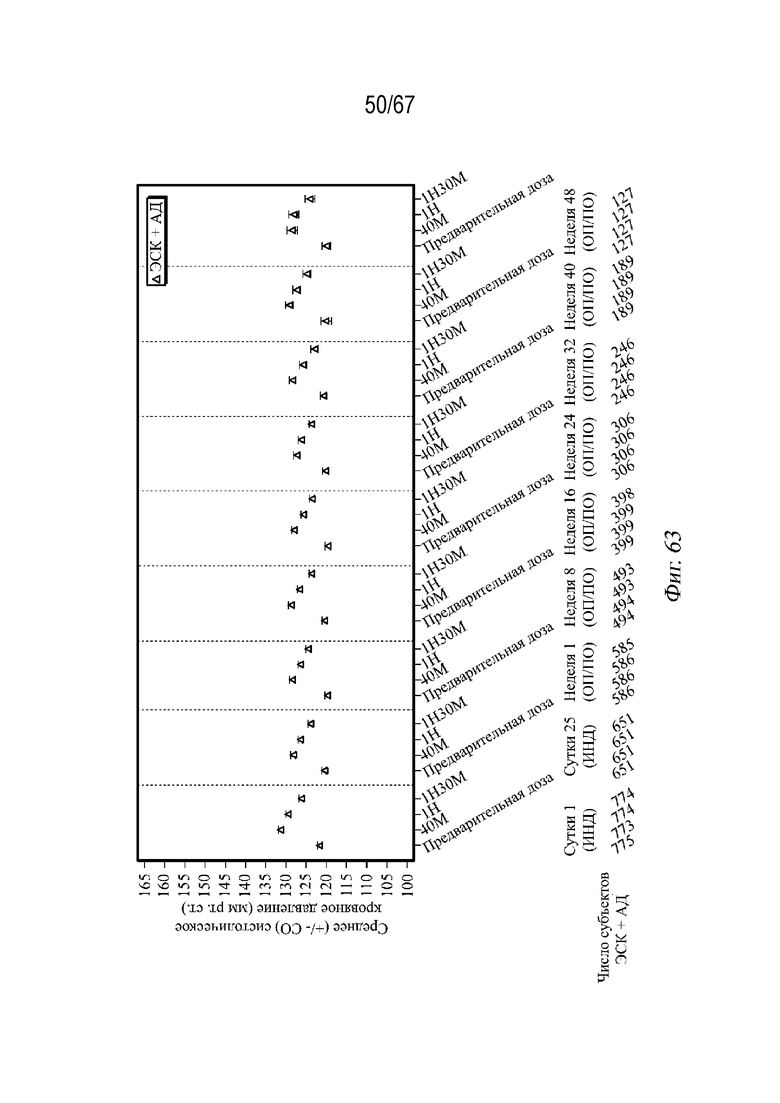




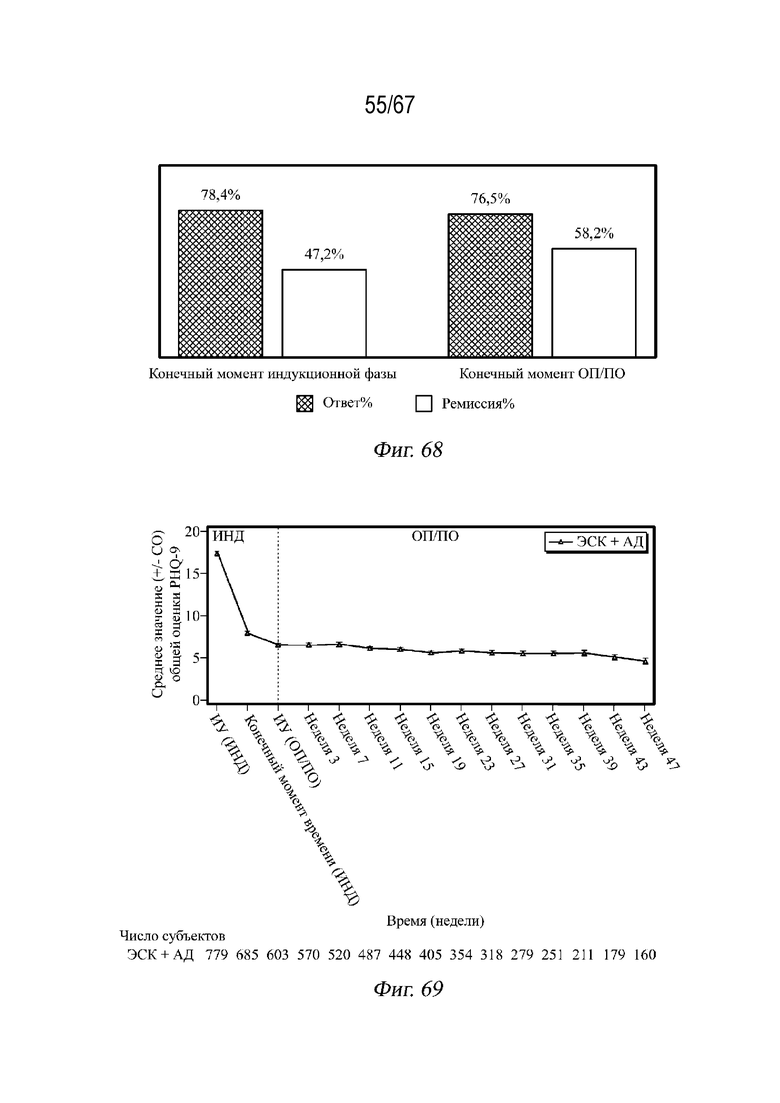



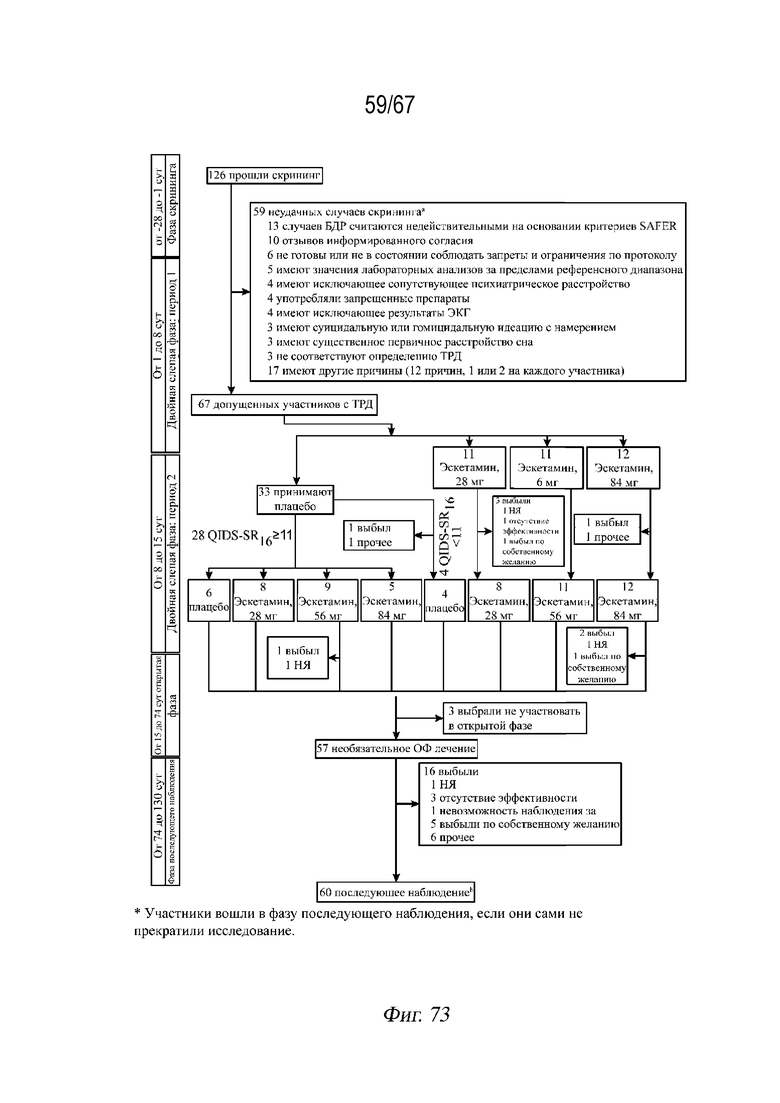





Изобретение относится к медицине, а именно к психиатрии, и может быть использовано при лечении депрессии. Способ по изобретению включает интраназальное введение во время первой индукционной фазы, составляющей не менее 4 недель, от 28 мг до 84 мг эскетамина пациенту, который не ответил, как минимум, на два пероральных антидепрессанта в текущий депрессивный эпизод. Проводят оценку пациента во время первой индукционной фазы, если пациент не может достичь практически полного ответа на эскетамин, повторно вводят пациенту от 28 мг до 84 мг эскетамина одновременно со вторым пероральным антидепрессантом во второй индукционной фазе, составляющей не менее 4 недель. Первый пероральный антидепрессант и второй пероральный антидепрессант независимо представляют собой сертралин, эсциталопрам, венлафаксин, дулоксетин или флуоксетин. Использование изобретения позволяет лечить терапевтически резистентную депрессию. 14 з.п. ф-лы, 172 табл., 82 ил., 10 пр.
1. Способ лечения устойчивой к лечению депрессии у пациента, включающий введение эскетамина, где пациент не ответил как минимум на два пероральных антидепрессанта в текущий депрессивный эпизод, где способ включает: введение пациенту первого перорального антидепрессанта, и интраназальное введение пациенту от 28 мг до 84 мг эскетамина не менее двух раз в неделю во время первой индукционной фазы, составляющей не менее 4 недель; оценку пациента во время первой индукционной фазы; и, где, если пациент не может достичь практически полного ответа на эскетамин, повторное назначение пациенту от 28 мг до 84 мг эскетамина и одновременно со вторым пероральным антидепрессантом во второй индукционной фазе, составляющей не менее 4 недель; где первый пероральный антидепрессант и второй пероральный антидепрессант независимо представляют собой сертралин, эсциталопрам, венлафаксин, дулоксетин или флуоксетин.
2. Способ по п.1, где первый пероральный антидепрессант представляет собой то же самое, что по меньшей мере один из по меньшей мере двух пероральных антидепрессантов.
3. Способ по п.1, где первый пероральный антидепрессант отличается от по меньшей мере одного из по меньшей мере двух пероральных антидепрессантов.
4. Способ по п.1, где первый пероральный антидепрессант отличается от по меньшей мере двух пероральных антидепрессантов.
5. Способ по п.1, где первый пероральный антидепрессант и второй пероральный антидепрессант представляют собой то же самое.
6. Способ по любому из пп.1-5, где в случае, если пациент не достигает практически полного ответа на эскетамин во время второй индукционной фазы, способ включает повторное назначение пациенту эскетамина и одновременно с третьим пероральным антидепрессантом в третьей индукционной фазе, составляющей не менее 4 недель.
7. Способ по п.6, где третий пероральный антидепрессант является тем же, что и второй пероральный антидепрессант.
8. Способ по п.6, где третий пероральный антидепрессант отличается от второго перорального антидепрессанта.
9. Способ по любому из пп.1-8, где, когда пациент достигает практически полного ответа на эскетамин, способ дополнительно включает введение пациенту терапевтически эффективного количества эскетамина в самое частое один раз в неделю во время последующей поддерживающей фазы.
10. Способ по любому из пп.1-9, где эскетамин вводится интраназально с частотой один раз каждую неделю или один раз каждые две недели или их комбинацию во время последующей поддерживающей фазы.
11. Способ по любому из пп.1-10, где количество вводимого эскетамина составляет:
(а) 28 мг, 56 мг или 84 мг,
(b) 56 мг или 84 мг,
(c) 28 мг,
(d) 56 мг, или
(e) 84 мг.
12. Способ по любому из пп.1-11, где пациент 65 лет и старше.
13. Способ по любому из пп.1-12, где эскетамин вводится с помощью устройства, представляющего собой одноразовый назальный спрей, который обеспечивает в общей сложности 28 мг эскетамина за два впрыскивания, по одному в каждую ноздрю.
14. Способ по п.13, где:
(а) одно устройство используется для дозы 28 мг,
(b) два устройства используются для дозы 56 мг, или
(c) три устройства используются для дозы 84 мг.
15. Способ по п.14, где существует 5-минутный интервал между использованием каждого устройства.
| WO2016187491 A1, 24.11.2016 | |||
| SG11201405530S A, 27.11.2014 | |||
| WO2017041112 A1, 09.03.2017 | |||
| ДРОБИЖЕВ М.Ю | |||
| и др., "Патогенетический и фармакологический подход при выборе антидепрессантов" Социальная и клиническая психиатрия, vol | |||
| Пишущая машина для тюркско-арабского шрифта | 1922 |
|
SU24A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Пюпитр для работы на пишущих машинах | 1922 |
|
SU86A1 |

