ОБЛАСТЬ ТЕХНИКИ
[1] Настоящее изобретение относится к области биомедицины и, в частности, относится к нацеливающемуся на TROP2 конюгату антитело-лекарственное средство, способу его получения и его применению.
УРОВЕНЬ ТЕХНИКИ
[2] Опухолеассоциированный трансдуктор кальциевого сигнала 2 (Trop2, TROP2) представляет собой трансмембранный гликопротеин I типа, кодируемый геном TACSTD2, являющийся трансдуктором кальциевого сигнала, связанным с регуляцией внутриклеточных кальциевых сигналов. Экспрессия Trop2 может активировать ERK. В нормальных условиях Trop2 в основном экспрессируется в трофобластных клетках и играет важную роль в развитии эмбриональных органов. Trop2 является сверхэкспрессированным во множестве эпителиальных опухолевых тканей человека, включая рак молочной железы, рак легкого, рак желудка, колоректальный рак, рак поджелудочной железы, рак предстательной железы, рак шейки матки, рак яичника и т. д., и уровень экспрессии связан с ухудшением и прогнозированием в отношении опухоли. Геномный анализ пациентов с раком молочной железы и результаты современных клинических исследований показывают, что Trop2 является клинически подтвержденной терапевтической мишенью. Trop2 привлек широко внимание вследствие своей высокой экспрессии на поверхности различных опухолевых клеток. Уровень Trop2 на поверхности одиночной клетки составляет от десятков тысяч до сотен тысяч. Эпидемиологические исследования доказали, что Trop2 связан с развитием различных опухолей и прогнозом в их отношении, и Trop2 стал мишенью для разработки лекарственных средств на основе антитела. После связывания антитела к Trop2 с белком Trop2 его эндоцитозная эффективность становится очень высокой, что является подходящим для разработки конъюгатов антитело-лекарственное средство.
[3] Конъюгат антитело-лекарственное средство (ADC) представляет собой новое поколение нацеленных терапевтических лекарственных средств, в основном применяемых для лечения раковых опухолей. Лекарственные средства на основе ADC состоят из трех частей: низкомолекулярное цитотоксическое лекарственное средство (лекарственное средство), антитело (антитело) и линкер (линкер), который соединяет антитело с цитотоксическим лекарственным средством. Низкомолекулярное цитотоксическое лекарственное средство соединено с белком антитела путем химического сочетания. Лекарственные средства на основе ADC предусматривают использование антител для обеспечения специфического распознавания и направления низкомолекулярных лекарственных средств на раковые клетки-мишени, экспрессирующие специфические для рака антигены, и проникновения в раковые клетки посредством эндоцитоза. Линкерная часть разрушается под действием внутриклеточной среды с низким значением pH или лизосомальной протеазы с высвобождением низкомолекулярных цитотоксических лекарственных средств, за счет чего обеспечивается эффект специфического уничтожения раковых клеток без повреждения клеток нормальной ткани. Следовательно, лекарственные средства на основе ADC имеют одновременно характеристики нацеливаемой специфичности антител и высокой токсичности низкомолекулярных токсинов в отношении раковых клеток, в значительной степени расширяя терапевтическое окно эффективности лекарственного средства. Клинические исследования доказали, что лекарственные средства на основе ADC обладают высокой эффективностью и являются относительно стабильными в крови, и могут эффективно снижать токсичность низкомолекулярных цитотоксических лекарственных средств (химиотерапевтические лекарственные средства) в системе кровообращения и здоровых тканях, и в настоящее время они представляют собой горячую точку в разработке противораковых лекарственных средств во всем мире.
СОДЕРЖАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
[4] Техническая задача, решаемая настоящим изобретением, заключается в том, чтобы преодолеть существующий в настоящее время дефицит ограниченных типов конъюгатов антитело-лекарственное средство, и в настоящем изобретении предусмотрен нацеливающийся на TROP2 конъюгат антитело-лекарственное средство, способ его получения и его применение.
[5] В настоящем изобретении разработан конъюгат антитело-лекарственное средство с хорошей нацеливающей способностью, сильным ингибирующим эффектом в отношении опухолевых клеток, которые являются положительными в отношении экспрессии TROP2, и хорошей лекарственностью и высокой безопасностью. Конъюгат антитело-лекарственное средство характеризуется ингибирующим эффектом в отношении TROP2, а также характеризуется хорошим подавляющим эффектом в отношении по меньшей мере одного из клеток NCI-N87, MDA-MB-468 и BXPC3.
[6] В настоящем изобретении решается вышеупомянутая техническая проблема посредством следующего технического решения.
[7] В настоящем изобретении предусмотрен конъюгат антитело-лекарственное средство формулы I, его фармацевтически приемлемая соль, его сольват или сольват его фармацевтически приемлемой соли;
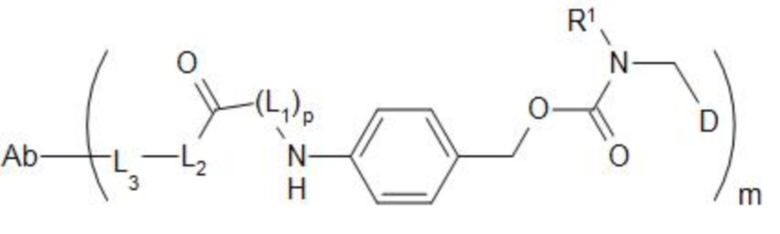 ;
;
I
[8] где Ab представляет собой антитело к TROP2 или вариант антитела к TROP2;
[9] аминокислотная последовательность легкой цепи в антителе к TROP2 представлена под SEQ ID NO: 1, и аминокислотная последовательность тяжелой цепи представлена под SEQ ID NO: 2;
[10] вариант антитела к TROP2 на по меньшей мере 70%, 75%, 80%, 85%, 90%, 95%, 98% или 99% идентичен антителу к TROP2;
[11] m составляет от 2 до 8;
[12] D представляет собой цитотоксическое лекарственное средство, являющееся ингибитором топоизомеразы;
[13] R1 представляет собой C1-C6алкил, замещенный одним или более чем одним -NR1-1R1-2, C1-C6алкил, замещенный одним или более чем одним R1-3S(O)2-, C1-C6алкил, C3-C10циклоалкил, C6-C14арил или 5-14-членный гетероарил; при этом гетероатом в 5-14-членном гетероариле выбран из одного или более чем одного из N, O и S, и число гетероатомов равняется 1, 2, 3 или 4; при этом каждый из R1-1, R1-2 и R1-3 независимо представляет собой C1-C6алкил;
[14] L1 независимо представляет собой одно или более чем одно из остатка фенилаланина, остатка аланина, остатка глицина, остатка глутаминовой кислоты, остатка аспарагиновой кислоты, остатка цистеина, остатка глутаминовой кислоты, остатка гистидина, остатка изолейцина, остатка лейцина, остатка лизина, остатка метионина, остатка пролина, остатка серина, остатка треонина, остатка триптофана, остатка тирозина и остатка валина; p составляет от 2 до 4;
[15] L2 представляет собой  ,
, 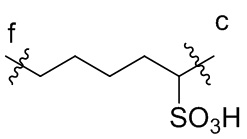 ,
, 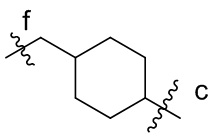 ,
, 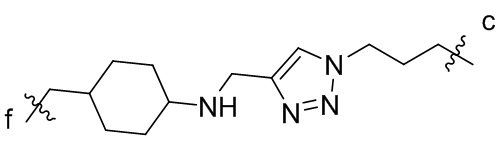 ,
, 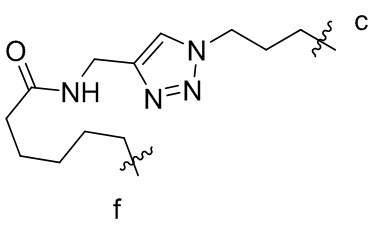 или
или 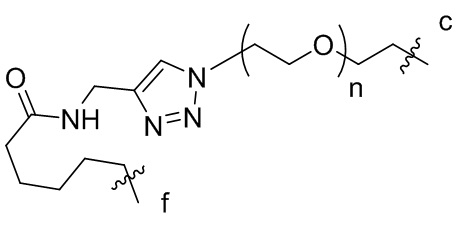 , где n независимо составляет от 1 до 12, c-конец присоединен к L1 посредством карбонильной группы, и f-конец присоединен к d-концу L3;
, где n независимо составляет от 1 до 12, c-конец присоединен к L1 посредством карбонильной группы, и f-конец присоединен к d-концу L3;
[16] L3 представляет собой 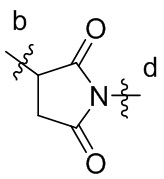 , где b-конец присоединен к Ab, и d-конец присоединен к f-концу L2.
, где b-конец присоединен к Ab, и d-конец присоединен к f-концу L2.
[17] В предпочтительном варианте осуществления настоящего изобретения некоторые группы конъюгата антитело-лекарственное средство определены следующим образом, и определение любой из неуказанных групп является таким, как описано в любом из приведенных выше вариантов осуществления (данный абзац далее в данном документе передается как «в предпочтительном варианте осуществления настоящего изобретения»):
[18] b-конец L3 предпочтительно присоединен к сульфгидрильной группе антитела в форме тиоэфирной связи. Принимая в качестве примера, форма связи с остатком цистеина в антителе представляет собой 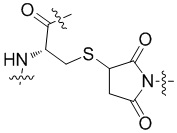 .
.
[19] В предпочтительном варианте осуществления настоящего изобретения, если D представляет собой цитотоксическое лекарственное средство, являющееся ингибитором топоизомеразы, цитотоксическое лекарственное средство, являющееся ингибитором топоизомеразы, предпочтительно представляет собой 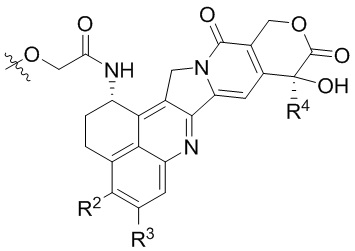 или
или 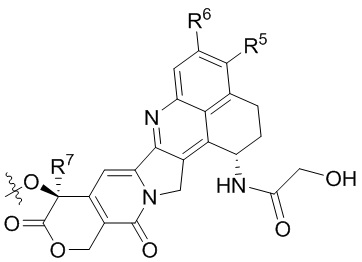 ; при этом каждый из R2 и R5 независимо представляет собой H, C1-C6алкил или галоген; каждый из R3 и R6 независимо представляет собой H, C1-C6алкил или галоген; каждый из R4 и R7 независимо представляет собой C1-C6алкил.
; при этом каждый из R2 и R5 независимо представляет собой H, C1-C6алкил или галоген; каждый из R3 и R6 независимо представляет собой H, C1-C6алкил или галоген; каждый из R4 и R7 независимо представляет собой C1-C6алкил.
[20] В предпочтительном варианте осуществления настоящего изобретения, если каждый из R2 и R5 независимо представляет собой C1-C6алкил, C1-C6алкил предпочтительно представляет собой C1-C4алкил, дополнительно предпочтительно метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, наиболее предпочтительно метил.
[21] В предпочтительном варианте осуществления настоящего изобретения, если каждый из R2 и R5 независимо представляет собой галоген, галоген предпочтительно представляет собой фтор, хлор, бром или йод, дополнительно предпочтительно фтор.
[22] В предпочтительном варианте осуществления настоящего изобретения, если каждый из R3 и R6 независимо представляет собой C1-C6алкил, C1-C6алкил предпочтительно представляет собой C1-C4алкил, дополнительно предпочтительно метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, наиболее предпочтительно метил.
[23] В предпочтительном варианте осуществления настоящего изобретения, если каждый из R3 и R6 независимо представляет собой галоген, галоген предпочтительно представляет собой фтор, хлор, бром или йод, дополнительно предпочтительно фтор.
[24] В предпочтительном варианте осуществления настоящего изобретения, если каждый из R4 и R7 независимо представляет собой C1-C6алкил, C1-C6алкил предпочтительно представляет собой C1-C4алкил, дополнительно предпочтительно метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, наиболее предпочтительно этил.
[25] В предпочтительном варианте осуществления настоящего изобретения каждый из R2 и R5 независимо представляет собой C1-C6алкил.
[26] В предпочтительном варианте осуществления настоящего изобретения каждый из R3 и R6 независимо представляет собой галоген.
[27] В предпочтительном варианте осуществления настоящего изобретения R4 и R7 представляют собой этил.
[28] В предпочтительном варианте осуществления настоящего изобретения D представляет собой 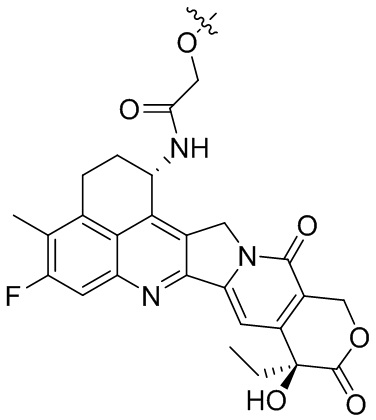 или
или 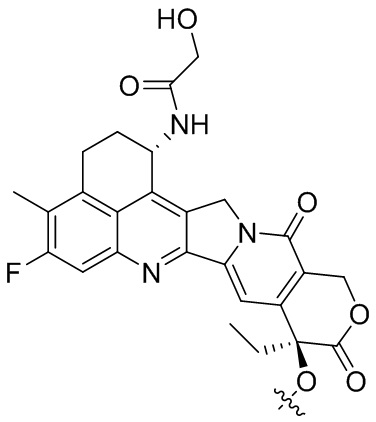 .
.
[29] В предпочтительном варианте осуществления настоящего изобретения, если D представляет собой или , конъюгат антитело-лекарственное средство может представлять собой 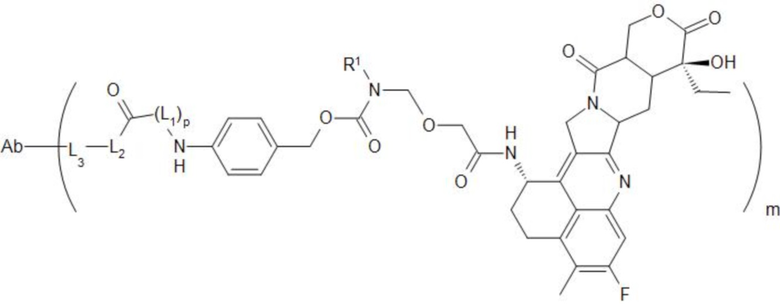 или
или 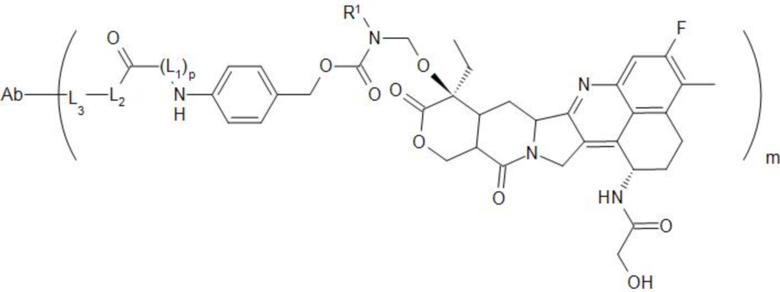 .
.
[30] В предпочтительном варианте осуществления настоящего изобретения, если R1 представляет собой C1-C6алкил, замещенный одним или более чем одним -NR1-1R1-2, C1-C6алкил предпочтительно представляет собой метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, наиболее предпочтительно этил.
[31] В предпочтительном варианте осуществления настоящего изобретения, если R1 представляет собой C1-C6алкил, замещенный более чем одним -NR1-1R1-2, «более чем один» составляет два или три.
[32] В предпочтительном варианте осуществления настоящего изобретения, если каждый из R1-1 и R1-2 независимо представляет собой C1-C6алкил, C1-C6алкил предпочтительно представляет собой метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, наиболее предпочтительно метил.
[33] В предпочтительном варианте осуществления настоящего изобретения, если R1 представляет собой C1-C6алкил, замещенный одним или более чем одним -NR1-1R1-2, -NR1-1R1-2 предпочтительно представляет собой -N(CH3)2.
[34] В предпочтительном варианте осуществления настоящего изобретения, если R1 представляет собой C1-C6алкил, замещенный одним -NR1-1R1-2, C1-C6алкил, замещенный одним -NR1-1R1-2, представляет собой 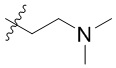 .
.
[35] В предпочтительном варианте осуществления настоящего изобретения, если R1 представляет собой C1-C6алкил, замещенный одним или более чем одним R1-3S(O)2-, C1-C6алкил предпочтительно представляет собой C1-C4алкил, дополнительно предпочтительно метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, наиболее предпочтительно этил.
[36] В предпочтительном варианте осуществления настоящего изобретения, если R1 представляет собой C1-C6алкил, замещенный более чем одним R1-3S(O)2-, «более чем один» составляет два или три.
[37] В предпочтительном варианте осуществления настоящего изобретения, если R1-3 представляет собой C1-C6алкил, C1-C6алкил предпочтительно представляет собой метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, наиболее предпочтительно метил.
[38] В предпочтительном варианте осуществления настоящего изобретения, если R1 представляет собой C1-C6алкил, замещенный одним R1-3S(O)2-, C1-C6алкил, замещенный одним R1-3S(O)2-, представляет собой 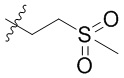 .
.
[39] В предпочтительном варианте осуществления настоящего изобретения, если R1 представляет собой C1-C6алкил, C1-C6алкил предпочтительно представляет собой метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, наиболее предпочтительно метил.
[40] В предпочтительном варианте осуществления настоящего изобретения m представляет собой целое число (такое как 2, 3, 4, 5, 6, 7 или 8) или нецелое число, предпочтительно от 3 до 5, дополнительно предпочтительно от 3,5 до 4,5, еще более предпочтительно от 3,8 до 4,2, такое как 3,88, 3,90, 3,96, 3,97, 3,98, 4,00, 4,03, 4,05, 4,10, 4,12 или 4,13.
[41] В предпочтительном варианте осуществления настоящего изобретения L1 предпочтительно представляет собой одно или более чем одно из остатка фенилаланина, остатка аланина, остатка глицина, остатка изолейцина, остатка лейцина, остатка пролина и остатка валина; более предпочтительно одно или более чем одно из остатка фенилаланина, остатка аланина, остатка глицина или остатка валина; дополнительно предпочтительно остатка валина и/или остатка аланина; «более чем один» предпочтительно равняется двум или трем; p предпочтительно равняется 2.
[42] В предпочтительном варианте осуществления настоящего изобретения (L1)p дополнительно предпочтительно представляет собой 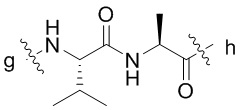 , где g-конец присоединен к c-концу L2 посредством карбонильной группы.
, где g-конец присоединен к c-концу L2 посредством карбонильной группы.
[43] В предпочтительном варианте осуществления настоящего изобретения n предпочтительно составляет от 8 до 12, как например 8, 9, 10, 11 и 12, дополнительно как например 8 или 12.
[44] В предпочтительном варианте осуществления настоящего изобретения, если каждый из R1-1, R1-2 и R1-3 независимо представляет собой C1-C6алкил, C1-C6алкил предпочтительно представляет собой C1-C4алкил, дополнительно предпочтительно метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, наиболее предпочтительно метил.
[45] В предпочтительном варианте осуществления настоящего изобретения R1 предпочтительно представляет собой C1-C6алкил, замещенный одним или более чем одним -NR1-1R1-2, C1-C6алкил, замещенный одним или более чем одним R1-3S(O)2-, C1-C6алкил или C3-C10циклоалкил; более предпочтительно C1-C6алкил, замещенный одним или более чем одним -NR1-1R1-2, C1-C6алкил, замещенный одним или более чем одним R1-3S(O)2-, или C1-C6алкил; дополнительно предпочтительно C1-C6алкил, замещенный одним или более чем одним -NR1-1R1-2, или C1-C6алкил, замещенный одним или более чем одним R1-3S(O)2-; наиболее предпочтительно C1-C6алкил, замещенный одним или более чем одним R1-3S(O)2-.
[46] В настоящем изобретении, если Ab представляет собой антитело к TROP2 или вариант антитела к TROP2, антитело к TROP2 или вариант антитела к TROP2 представляет собой остаток антитела к TROP2 (группа, образованная путем замены водорода в одной из сульфгидрильных групп в антителе к TROP2) или остаток варианта антитела к TROP2 (группа, образованная путем замены водорода в одной из сульфгидрильных групп в варианте антитела к TROP2).
[47] В предпочтительном варианте осуществления настоящего изобретения соединение формулы I представлено любой из следующих схем:
[48] схема I:
[49] Ab представляет собой антитело к TROP2 или вариант антитела к TROP2;
[50] D представляет собой или , каждый из R2 и R5 независимо представляет собой H, C1-C6алкил или галоген; каждый из R3 и R6 независимо представляет собой H, C1-C6алкил или галоген; каждый из R4 и R7 независимо представляет собой C1-C6алкил; D предпочтительно представляет собой или ;
[51] L1 независимо представляет собой одно или более чем одно из остатка фенилаланина, остатка аланина, остатка глицина, остатка изолейцина, остатка лейцина, остатка пролина и остатка валина;
[52] схема II:
[53] Ab представляет собой антитело к TROP2 или вариант антитела к TROP2;
[54] D представляет собой или , каждый из R2 и R5 независимо представляет собой C1-C6алкил; каждый из R3 и R6 независимо представляет собой галоген, D предпочтительно представляет собой или ;
[55] R1 представляет собой C1-C6алкил, замещенный одним или более чем одним -NR1-1R1-2, C1-C6алкил, замещенный одним или более чем одним R1-3S(O)2-, или C1-C6алкил;
[56] L1 независимо представляет собой одно или более чем одно из остатка фенилаланина, остатка аланина, остатка глицина и остатка валина;
[57] схема III:
[58] Ab представляет собой антитело к TROP2;
[59] D представляет собой или , каждый из R2 и R5 независимо представляет собой C1-C6алкил; каждый из R3 и R6 независимо представляет собой галоген, D предпочтительно представляет собой или ;
[60] m составляет от 3,5 до 4,5,
[61] R1 представляет собой C1-C6алкил, замещенный одним или более чем одним -NR1-1R1-2, C1-C6алкил, замещенный одним или более чем одним R1-3S(O)2-, или C1-C6алкил;
[62] L1 независимо представляет собой остаток валина и/или остаток аланина;
[63] схема IV:
[64] Ab представляет собой антитело к TROP2;
[65] D представляет собой или ,
[66] R1 представляет собой C1-C6алкил, замещенный одним или более чем одним -NR1-1R1-2, C1-C6алкил, замещенный одним или более чем одним R1-3S(O)2-, или C1-C6алкил;
[67] L1 независимо представляет собой остаток валина и/или остаток аланина;
[68] схема V:
[69] Ab представляет собой антитело к TROP2;
[70] D представляет собой или ,
[71] m составляет от 3,5 до 4,5,
[72] R1 представляет собой C1-C6алкил, замещенный одним или более чем одним -NR1-1R1-2, C1-C6алкил, замещенный одним или более чем одним R1-3S(O)2-, или C1-C6алкил;
[73] L1 независимо представляет собой остаток валина и/или остаток аланина.
[74] В предпочтительном варианте осуществления настоящего изобретения конъюгат антитело-лекарственное средство предпочтительно представляет собой 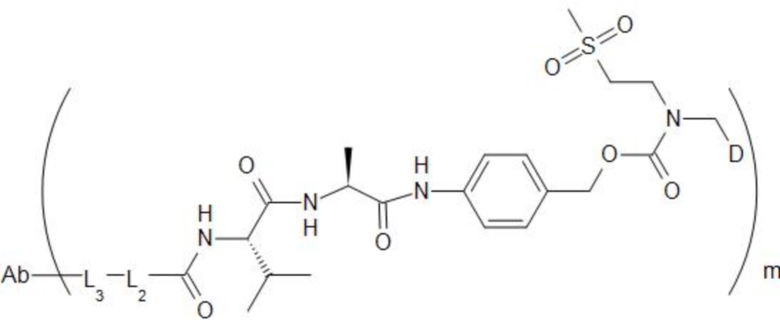 ,
, 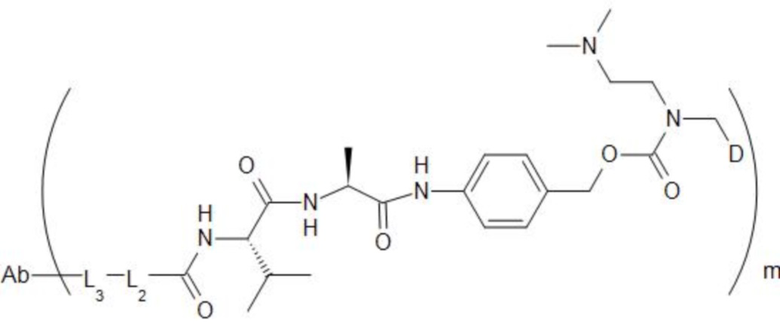 или
или
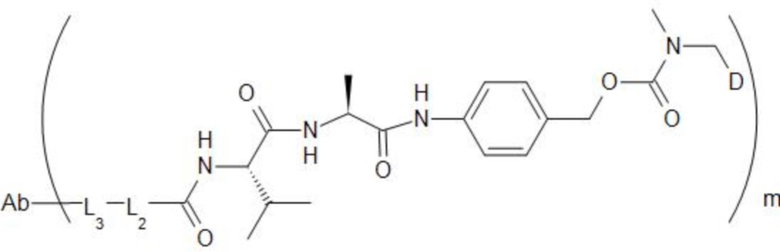 .
.
[75] В предпочтительном варианте осуществления настоящего изобретения L2 предпочтительно представляет собой .
[76] В предпочтительном варианте осуществления настоящего изобретения Ab представляет собой антитело к TROP2 или вариант антитела к TROP2, где аминокислотная последовательность легкой цепи в антителе к TROP2 предпочтительно представлена под SEQ ID NO: 1; аминокислотная последовательность тяжелой цепи в антителе к TROP2 представлена под SEQ ID NO: 2; D представляет собой или ; L1 представляет собой остаток валина и/или остаток аланина, p равняется 2, (L1)p предпочтительно представляет собой ; R1 представляет собой C1-C6алкил, замещенный одним или более чем одним -NR1-1R1-2, C1-C6алкил, замещенный одним или более чем одним R1-3S(O)2-, или C1-C6алкил, предпочтительно C1-C6алкил, замещенный одним или более чем одним -NR1-1R1-2, или C1-C6алкил, замещенный одним или более чем одним R1-3S(O)2-, дополнительно предпочтительно C1-C6алкил, замещенный одним или более чем одним R1-3S(O)2-; R1-1, R1-2 и R1-3 независимо представляют собой C1-C4алкил, предпочтительно метил; C1-C6алкил, замещенный одним или более чем одним -NR1-1, R1-2 предпочтительно представляет собой 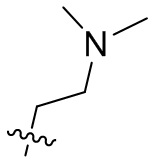 ; C1-C6алкил, замещенный одним или более чем одним R1-3S(O)2- предпочтительно представляет собой
; C1-C6алкил, замещенный одним или более чем одним R1-3S(O)2- предпочтительно представляет собой 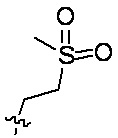 ; L2 представляет собой ; L3 представляет собой .
; L2 представляет собой ; L3 представляет собой .
[77] В предпочтительном варианте осуществления настоящего изобретения конъюгат антитело-лекарственное средство предпочтительно представляет собой любое из следующих соединений:
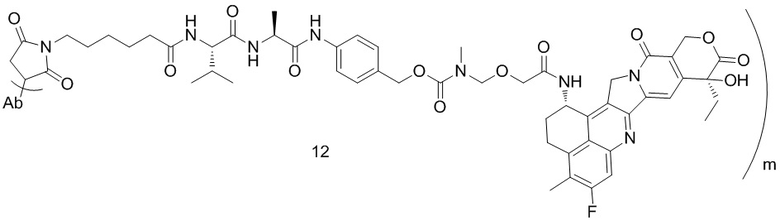 ,
, 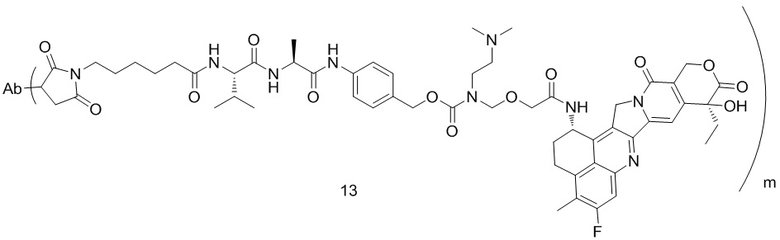 ,
, 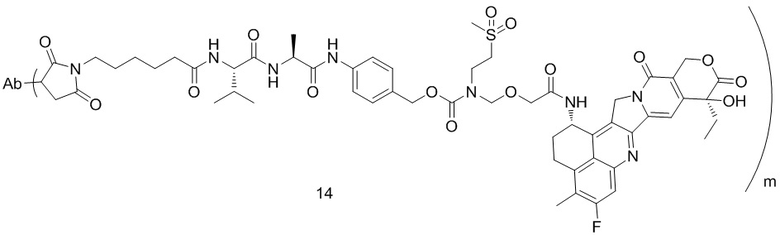 ,
, 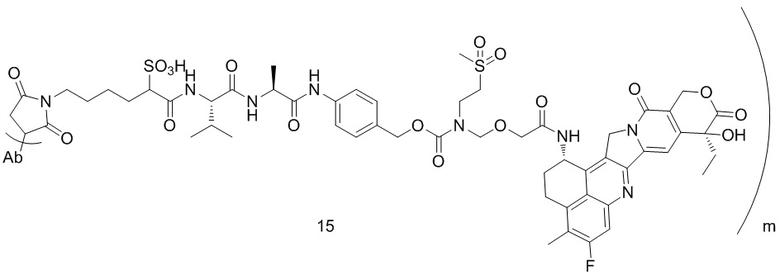 ,
, 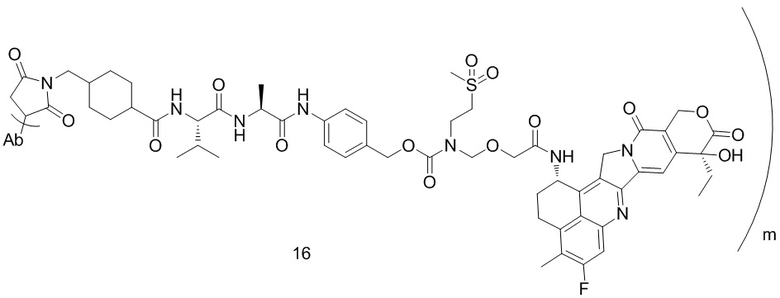 ,
, 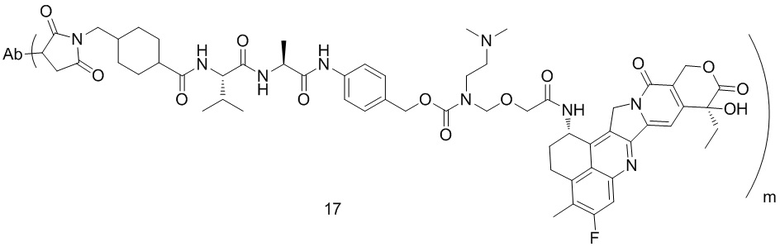 ,
, 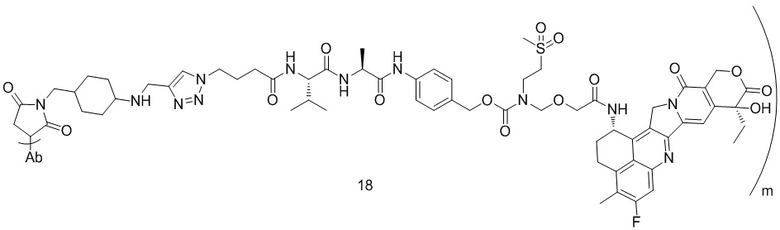 ,
, 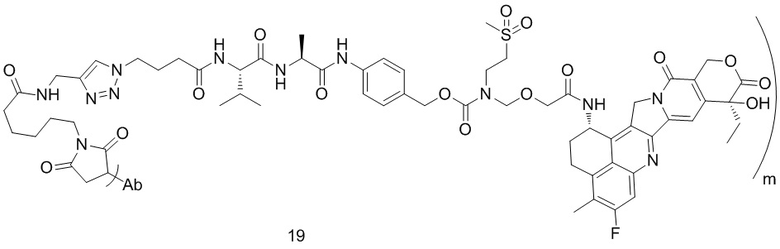 ,
, 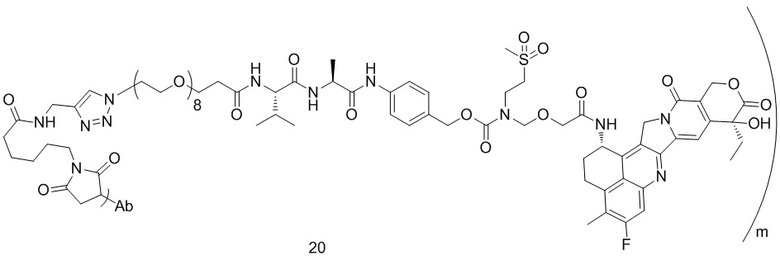 ,
, 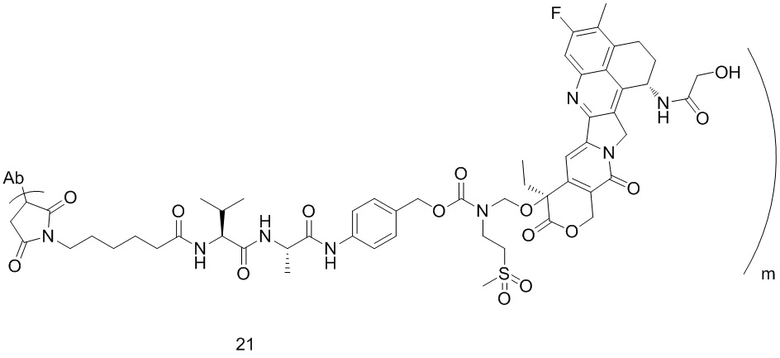 или
или 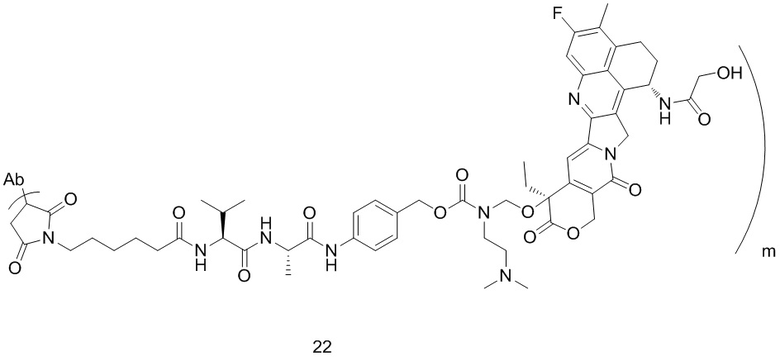 ;
;
[78] где Ab представляет собой антитело к TROP2 или вариант антитела к TROP2, и m составляет от 3,8 до 4,2, предпочтительно 3,88, 3,90, 3,96, 3,97, 3,98, 4,00, 4,03, 4,05, 4,10, 4,12 или 4,13; аминокислотная последовательность легкой цепи в варианте антитела к TROP2 предпочтительно представлена под SEQ ID NO: 1, аминокислотная последовательность тяжелой цепи предпочтительно представлена под SEQ ID NO: 2.
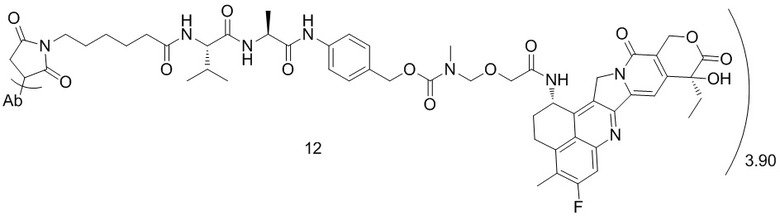
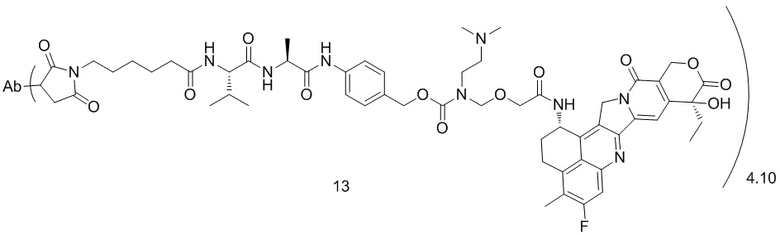
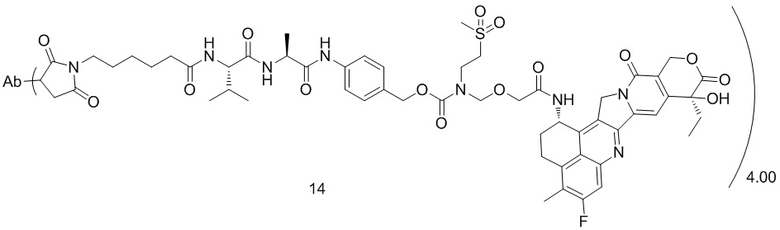
[79] В предпочтительном варианте осуществления настоящего изобретения конъюгат антитело-лекарственное средство предпочтительно представляет собой любое из следующих соединений:
 ,
,  ,
,  ,
, 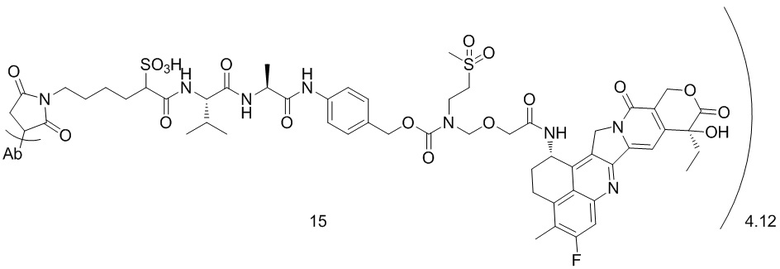 ,
, 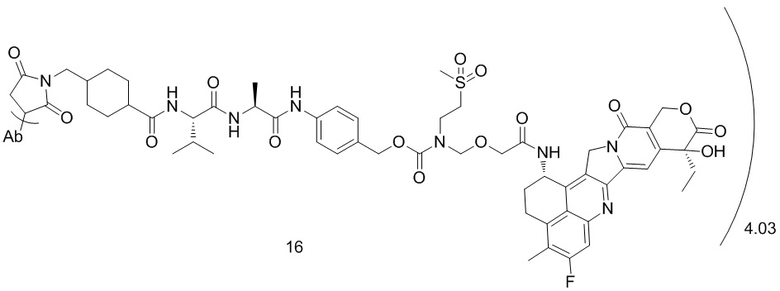 ,
, 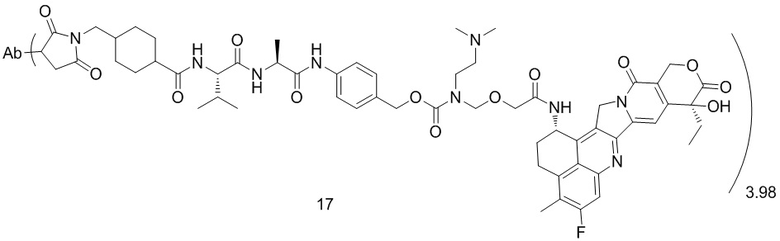 ,
, 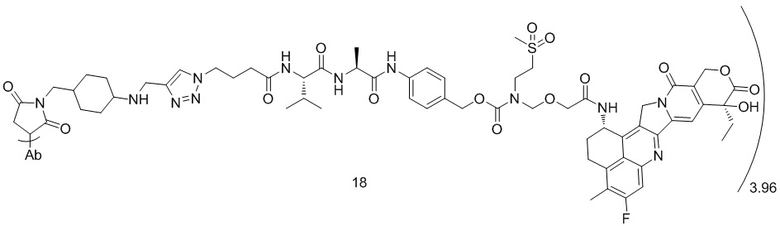 ,
, 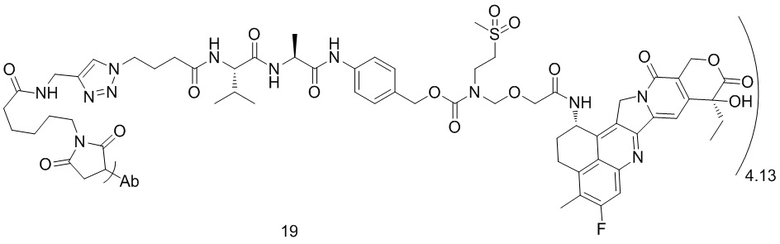 ,
, 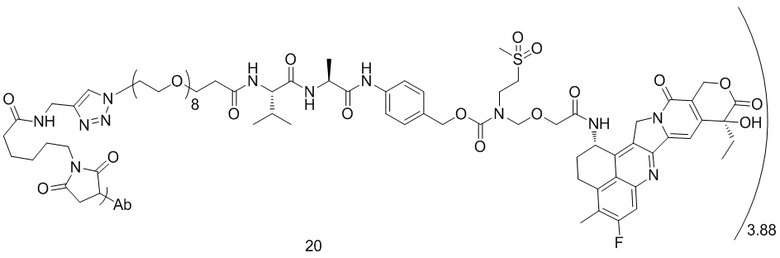 ,
, 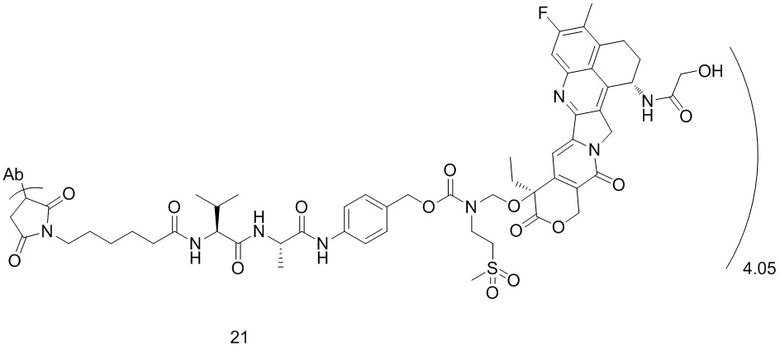 или
или 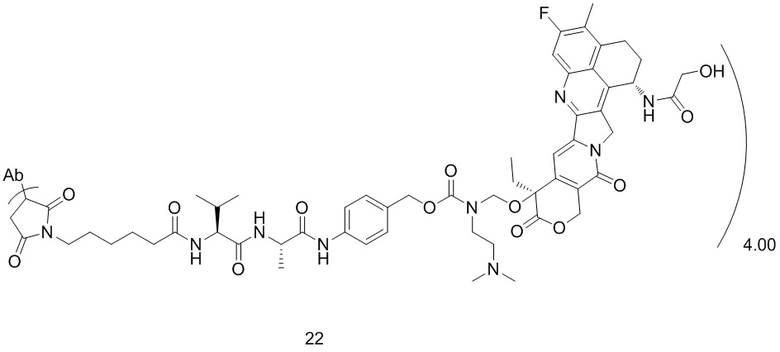 ;
;
[80] где Ab представляет собой антитело к TROP2 или вариант антитела к TROP2, предпочтительно антитело к TROP2; аминокислотная последовательность легкой цепи в варианте антитела к TROP2 предпочтительно представлена под SEQ ID NO: 1, и аминокислотная последовательность тяжелой цепи предпочтительно представлена под SEQ ID NO: 2.
[81] В предпочтительном варианте осуществления настоящего изобретения конъюгат антитело-лекарственное средство предпочтительно представляет собой любое из следующих соединений:
, или ;
[82] где Ab представляет собой антитело к TROP2 или вариант антитела к TROP2; m предпочтительно равняется 3,90, 4,00 или 4,10; аминокислотная последовательность легкой цепи в варианте антитела к TROP2 предпочтительно представлена под SEQ ID NO: 1, и аминокислотная последовательность тяжелой цепи предпочтительно представлена под SEQ ID NO: 2.
[83] В предпочтительном варианте осуществления настоящего изобретения конъюгат антитело-лекарственное средство предпочтительно представляет собой любое из следующих соединений:
, или ;
[84] где Ab представляет собой антитело к TROP2 или вариант антитела к TROP2.
[85] В предпочтительном варианте осуществления настоящего изобретения конъюгат антитело-лекарственное средство предпочтительно представляет собой любое из следующих соединений:
 , при этом Ab представляет собой антитело к TROP2, и m предпочтительно равняется 3,90;
, при этом Ab представляет собой антитело к TROP2, и m предпочтительно равняется 3,90;
, при этом Ab представляет собой антитело к TROP2, и m предпочтительно равняется 4,10;
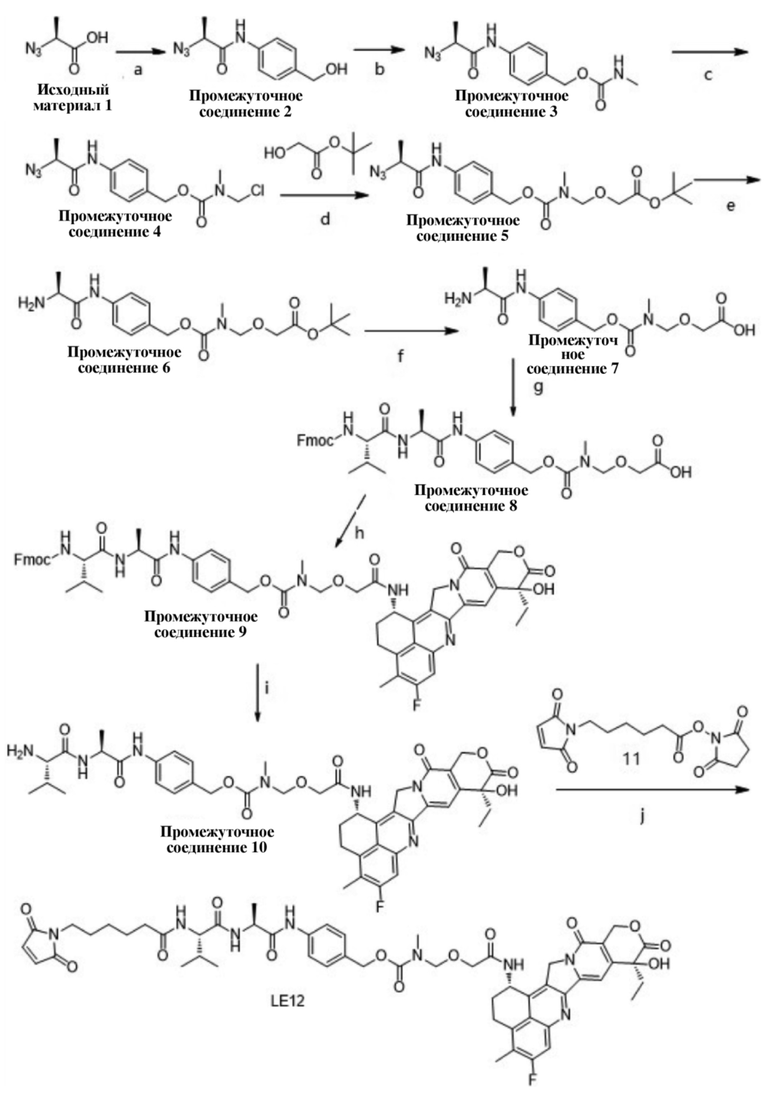
, при этом Ab представляет собой антитело к TROP2, и m предпочтительно равняется 4,00;
, при этом Ab представляет собой антитело к TROP2, и m предпочтительно равняется 4,12;
, при этом Ab представляет собой антитело к TROP2, и m предпочтительно равняется 4,03;
, при этом Ab представляет собой антитело к TROP2, и m предпочтительно равняется 3,98;
, при этом Ab представляет собой антитело к TROP2, и m предпочтительно равняется 3,96;
, при этом Ab представляет собой антитело к TROP2, и m предпочтительно равняется 4,13;
, при этом Ab представляет собой антитело к TROP2, и m предпочтительно равняется 3,88;
, при этом Ab представляет собой антитело к TROP2, и m предпочтительно равняется 4,05;
, при этом Ab представляет собой антитело к TROP2, и m предпочтительно равняется 4,00.
[86] В настоящем изобретении также предусмотрен способ получения конъюгата антитело-лекарственное средство, включающий следующую стадию: осуществление реакции сочетания между соединением формулы II и Ab-водородом, как показано ниже;
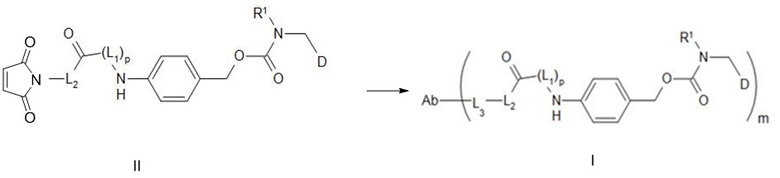
[87] где L1, L2, L3, R1, p и Ab определены выше.
[88] В настоящем изобретении условия проведения реакции сочетания и процедуры для ее проведения могут представлять собой общепринятые в уровне техники условия и процедуры для проведения реакции сочетания.
[89] В настоящем изобретении также предусмотрена фармацевтическая композиция, содержащая вещество X и фармацевтически приемлемое вспомогательное вещество, при этом вещество X представляет собой конъюгат антитело-лекарственное средство, его фармацевтически приемлемую соль, его сольват или сольват его фармацевтически приемлемой соли.
[90] В фармацевтической композиции вещество X может применяться в терапевтически эффективном количестве.
[91] В настоящем изобретении также предусмотрено применение вещества X или фармацевтической композиции в изготовлении ингибитора белка TROP2. В настоящем изобретении также предусмотрено применение вещества X или фармацевтической композиции в изготовлении лекарственного средства для лечения и/или предупреждения опухоли, и опухоль предпочтительно представляет собой TROP2-положительную опухоль.
[92] При применении TROP2-положительная опухоль предпочтительно представляет собой одно или более чем одно из TROP2-положительного рака желудка, трижды отрицательного рака молочной железы и рака поджелудочной железы человека.
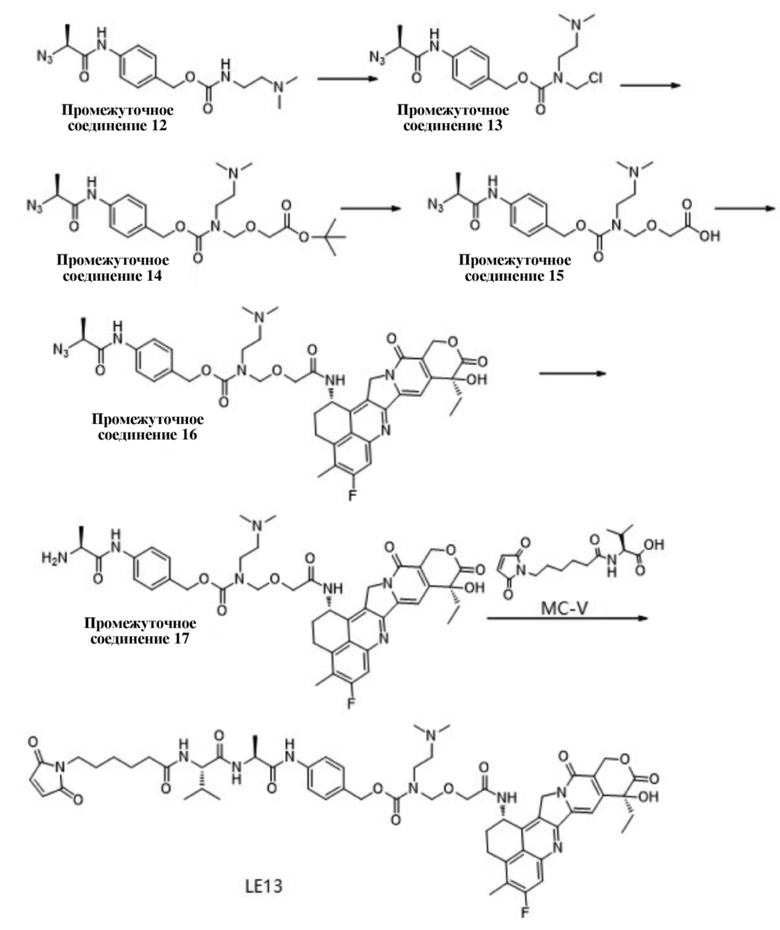
[93] В некоторых вариантах осуществления настоящего изобретения, в случае рака желудка, клетки рака желудка представляют собой клетки NCI-N87;
[94] в некоторых вариантах осуществления настоящего изобретения, в случае трижды отрицательного рака молочной железы, клетки трижды отрицательного рака молочной железы представляют собой клетки MDA-MB-468;
[95] в некоторых вариантах осуществления настоящего изобретения, в случае рака поджелудочной железы, клетки рака поджелудочной железы представляют собой клетки BXPC3.
[96] Если не указано иное, следующие термины при нахождении в описании и формуле настоящего изобретения имеют следующие значения.
[97] Фармацевтическое вспомогательное вещество может представлять собой вспомогательное вещество, широко используемое в области фармацевтического производства. Вспомогательное вещество в основном применяется для обеспечения безопасной, стабильной и функциональной фармацевтической композиции, и может также обеспечивать способ растворения активного ингредиента с требуемой скоростью после введения или облегчения эффективного всасывания активного ингредиента у субъекта после того, как субъект подвергается процедуре введения композиции. Фармацевтическое вспомогательное вещество может представлять собой инертный наполнитель или может обеспечивать определенную функцию, такую как стабилизация общего значения pH композиции или предупреждение разрушения активного ингредиента композиции. Фармацевтические вспомогательные вещества могут включать одно или более из следующих вспомогательных веществ: буферы, хелатирующие средства, консерванты, солюбилизаторы, стабилизаторы, среды-носители, поверхностно-активные вещества, красящие вещества, ароматизирующие средства и подсластители.
[98] Природная или естественная последовательность TROP2 может быть выделена из природной среды или получена посредством методики рекомбинантной ДНК, химического синтеза или комбинации указанных выше и аналогичных методик.
[99] Антитело в данном документе трактуется в самом широком смысле, которое может специфично связываться с мишенью посредством по меньшей мере одной области распознавания антигена, расположенной в вариабельной области молекулы иммуноглобулина, такого как углевод, полинуклеотид, жир, полипептид и т. д. В частности, оно включает полные моноклональные антитела, поликлональные антитела, биспецифические антитела и фрагменты антитела, в том случае, если они обладают требуемой биологической активностью. Варианты антител по настоящему изобретению относятся к мутантным аминокислотным последовательностям и ковалентным производным природных полипептидов, при условии, что сохраняется биологическая активность, эквивалентная таковой природных полипептидов. Отличие между мутантными аминокислотными последовательностями и природными аминокислотными последовательностями обычно заключается в том, что одна или более аминокислот в природной аминокислотной последовательности заменены или одна или более аминокислот удалены из полипептидной последовательности и/или вставлены в нее. Мутанты с делецией включают фрагменты природных полипептидов и мутанты с усечением N-концевой и/или C-концевой части. Обычно мутантная аминокислотная последовательность на по меньшей мере 70% (например, 70%, 75%, 80%, 85%, 90%, 95%, 98% или 99%) идентична природной последовательности.
[100] Антитела по настоящему изобретению можно получить посредством хорошо известных в уровне техники методик, таких как гибридомные технологии, методики рекомбинантной ДНК, методики фагового дисплея, методики синтеза, или их комбинаций, или посредством других методик, известных из уровня техники.
[101] Описание термина «соотношение лекарственное средство-антитело» (DAR). L-D представляет собой реакционноспособную группу с участком конъюгации на антителе, при этом L представляет собой линкер, D представляет собой цитотоксическое средство, дополнительно связанное с антителом, присоединенным к L. В настоящем изобретении D представляет собой Dxd. Числовое значение DAR для каждого антитела, в конечном итоге связанного с D, представлено m, или m также может представлять собой число D, связанных с одним антителом. В некоторых вариантах осуществления m фактически представляет собой среднее значение от 2 до 8, от 3 до 7 или от 3 до 5, или m представляет собой целое число, составляющее 2, 3, 4, 5, 6, 7 или 8; в некоторых вариантах осуществления m представляет собой среднее значение от 3,5 до 4,5 (например, 3,88, 3,90, 3,96, 3,97, 3,98, 4,00, 4,03, 4,05, 4,10, 4,12 или 4,13); в других вариантах осуществления m представляет собой среднее значение 2, 3, 4, 5, 6, 7 или 8.
[102] Линкер относится к прямому или непрямому соединению между антителом и лекарственным средством. Линкер может быть присоединен к mAb множеством способов, таких как посредством поверхностного лизина, восстановительного связывания с окисленными углеводами и посредством остатков цистеина, высвобождаемых посредством восстановления межцепочечных дисульфидных связей. Множество систем связывания ADC известны из уровня техники, включая варианты связывания на основе гидразонов, дисульфидов и пептидов.
[103] Термин «фармацевтически приемлемая соль» относится к соли, полученной из соединения по настоящему изобретению и относительно нетоксичной и фармацевтически приемлемой кислоты или основания. В случае если соединение по настоящему изобретению содержит относительно кислотные функциональные группы, соль присоединения основания можно получить путем приведения в контакт достаточного количества фармацевтически приемлемого основания с нейтральной формой соединения в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемая соль присоединения основания включает без ограничения соль лития, соль натрия, соль калия, соль кальция, соль алюминия, соль магния, соль цинка, соль висмута, соль аммония и соль диэтаноламина. Если соединение по настоящему изобретению содержит относительно основные функциональные группы, то соль присоединения кислоты можно получить посредством приведения в контакт достаточного количества фармацевтически приемлемой кислоты с нейтральной формой соединения в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемая кислота включает неорганическую кислоту, и неорганическая кислота включает без ограничения хлористоводородную кислоту, бромистоводородную кислоту, йодистоводородную кислоту, азотную кислоту, угольную кислоту, ортофосфорную кислоту, фосфорную кислоту, серную кислоту и т. д. Фармацевтически приемлемая кислота включает органическую кислоту, и органическая кислота включает без ограничения уксусную кислоту, пропионовую кислоту, щавелевую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, транс-бутендиовую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, лимонную кислоту, салициловую кислоту, винную кислоту, метансульфоновую кислоту, изоникотиновую кислоту, лимонную кислоту, олеиновую кислоту, дубильную кислоту, пантотеновую кислоту, битартрат, аскорбиновую кислоту, гентизиновую кислоту, фумаровую кислоту, глюконовую кислоту, сахарную кислоту, муравьиную кислоту, этансульфоновую кислоту, памоевую кислоту (т. е. 4,4′-метилен-бис(3-гидрокси-2-нафтoйную кислоту)), аминокислоту (например, глутаминовую кислоту и аргинин) и т. д. Если соединение по настоящему изобретению содержит относительно кислотные функциональные группы и относительно основные функциональные группы, его можно преобразовать в соль присоединения основания или соль присоединения кислоты. Подробнее см. Berge et al., «Pharmaceutical Salts», Journal of Pharmaceutical Science 66: 1-19 (1977) или Handbook of Pharmaceutical Salts: Properties, Selection, and Use (P. Heinrich Stahl and Camille G. Wermuth, ed., Wiley-VCH, 2002).
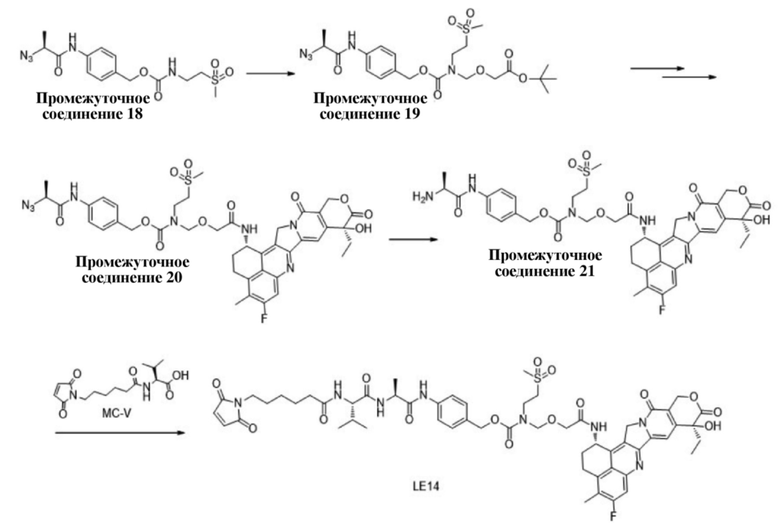
[104] Термин «сольват» относится к веществу, образующемуся при объединении соединения по настоящему изобретению со стехиометрическим или нестехиометрическим количеством растворителя. Молекулы растворителя в сольвате могут существовать в упорядоченном или неупорядоченном вариантах расположения. Растворитель включает без ограничения воду, метанол, этанол и т.п.
[105] Термин «лечение» или эквивалентное ему выражение при его использовании применительно, например, к раку относится к процедуре или процессу для уменьшения или устранения некоторого количества раковых клеток в организме пациента или облегчения симптомов рака. «Лечение» рака или другого пролиферативного нарушения не обязательно означает, что раковые клетки или другие нарушения будут фактически устранены, но количество клеток или нарушений будет фактически уменьшено, или симптомы видов рака или других нарушений будут фактически облегчены. В общем, способ лечения рака будет осуществляться, даже если существует лишь низкая вероятность успеха, но все еще считается, что индуцируется общий полезный ход действий, принимая во внимание историю болезни пациента и расчетную прогнозируемую выживаемость.
[106] Термин «предупреждение» относится к снижению риска приобретения или развития заболевания или нарушения.
[107] Термин «циклоалкил» относится к насыщенной циклической углеводородной группе, содержащей от трех до двадцати атомов углерода (например, C3-C6циклоалкил), в том числе моноциклический и полициклический циклоалкил. Циклоалкил содержит от 3 до 20 атомов углерода, предпочтительно от 3 до 10 атомов углерода и более предпочтительно от 3 до 6 атомов углерода. Примеры циклоалкила включают без ограничения циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил и циклодецил.
[108] Термин «алкил» относится к алкильной группе с прямой или разветвленной цепью с указанным числом атомов углерода. Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, т-бутил, изобутил, втор-бутил, н-пентил, изопентил, н-гексил, н-гептил, н-октил и подобные алкильные группы.
[109] Термин «галоген» относится к фтору, хлору, брому или йоду.
[110] Термин «гетероарил» относится к арильной группе (или ароматическому кольцу), содержащей 1, 2, 3 или 4 гетероатома, независимо выбранных из N, O и S, который может быть моноциклической, бициклической или трициклической ароматической системой, такой как фурил, пиридинил, пиридазинил, пиримидинил, пиразинил, тиофенил, изоксазолил, оксазолил, диазолил, имидазолил, пирролил, пиразолил, триазолил, тетразолил, тиазолил, изотиазолил, тиадиазолил, бензимидазолил, индолил, индазолил, бензотиазолил, бензизотиазолил, бензоксазолил, бензизоксазолил, хинолил, изохинолил и т. д.
[111] Термин «арил» относится к любому устойчивому моноциклическому или бициклическому карбоциклу, где все кольца представляют собой ароматические кольца. Примеры арильной структурной единицы включают фенил или нафтил.
[112] Вышеуказанные предпочтительные условия можно произвольно объединять для получения предпочтительных вариантов осуществления настоящего изобретения без нарушения общеизвестных сведений в уровне техники.
[113] Если не указано иное, комнатная температура в настоящем изобретении относится к температуре от 20 до 30°C.
[114] Все реагенты и исходные материалы, используемые в настоящем изобретении, являются коммерчески доступными.
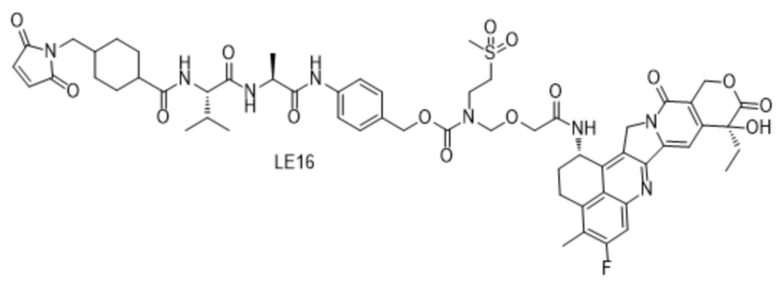
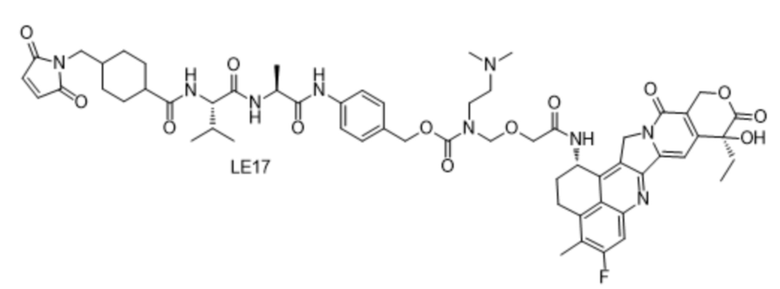
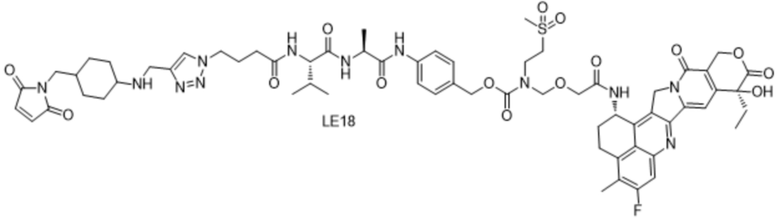
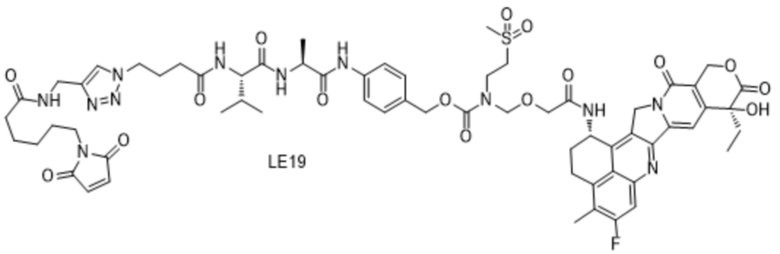
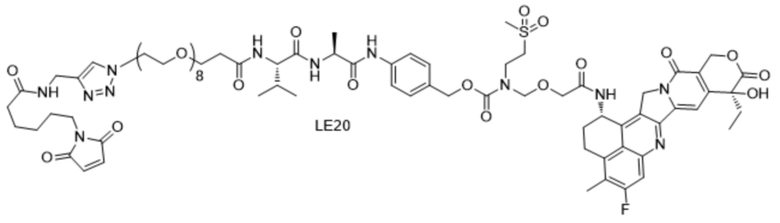
[115] Положительные прогрессивные эффекты, обеспечиваемые настоящим изобретением, являются следующими.
[116] 1. Конъюгат антитело-лекарственное средство, содержащий антитело к TROP2, предусмотренный настоящим изобретением, характеризуется хорошей нацеливающей способностью и обладает хорошим подавляющим эффектом в отношении различных опухолевых клеток, экспрессирующих TROP2, и т. д.
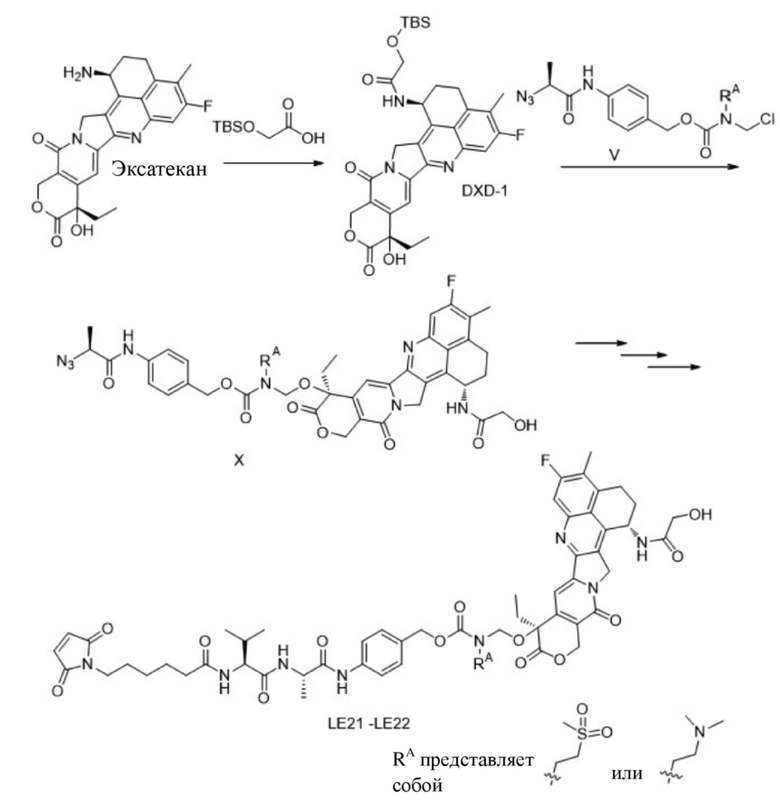
[117] 2. Исследования in vivo показали, что конъюгат антитело-лекарственное средство по настоящему изобретению характеризуется лучшей цитотоксичностью in vitro и противоопухолевой активностью in vivo.
[118] 3. Конъюгат антитело-лекарственное средство по настоящему изобретению характеризуется хорошей растворимостью и хорошей лекарственностью. Аномальных явлений, таких как осаждение, во время процедуры обеспечения сочетания, не наблюдалось, что является очень благоприятным для получения конъюгата антитело-лекарственное средство.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[119] На фигуре 1 показана конструкция векторов экспрессии для легких и тяжелых цепей антитела FDA018; где Ab-L представляет собой легкую цепь антитела, и Ab-H представляет собой тяжелую цепь антитела.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНОГО ВАРИАНТА ОСУЩЕСТВЛЕНИЯ
[120] Настоящее изобретение будет дополнительно описано ниже со ссылкой на примеры, но при этом настоящее изобретение не ограничивается объемом примеров. Экспериментальные способы без конкретных условий в следующих примерах выбраны в соответствии с общепринятыми способами и условиями или в соответствии с коммерческой спецификацией.
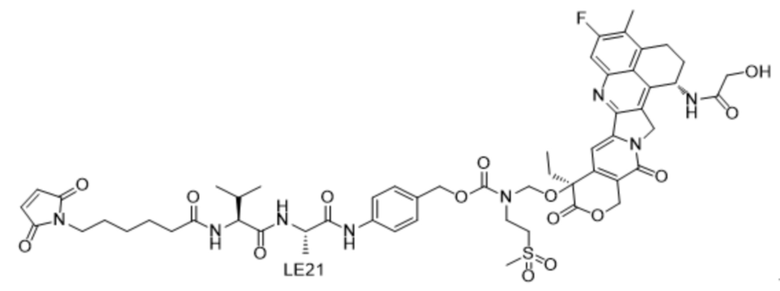
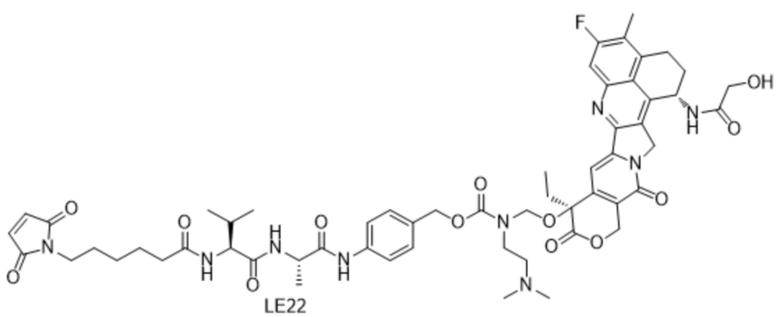
[121] Описание сокращений
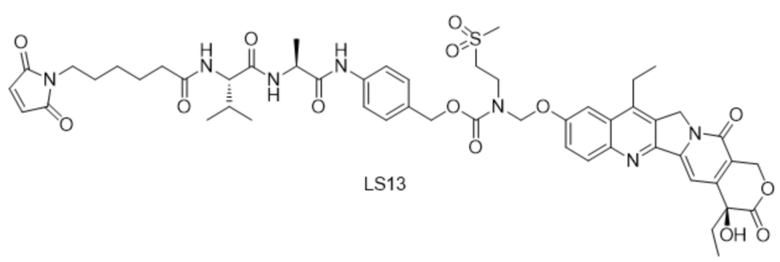
[122] ПЦР - полимеразная цепная реакция
[123] CHO - клетки яичника китайского хомячка
[124] HTRF - гомогенная флуоресценция с временным разрешением
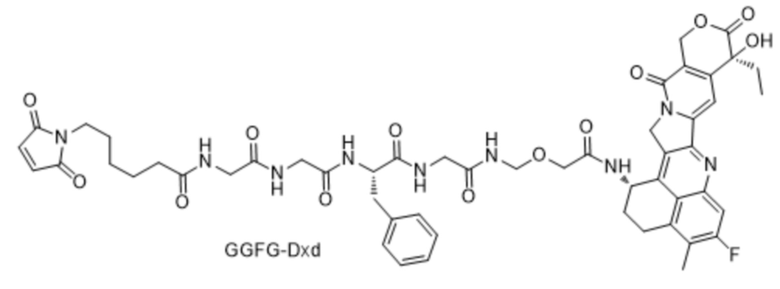
[125] PB - фосфатный буфер
[126] EDTA - этилендиаминтетрауксусная кислота
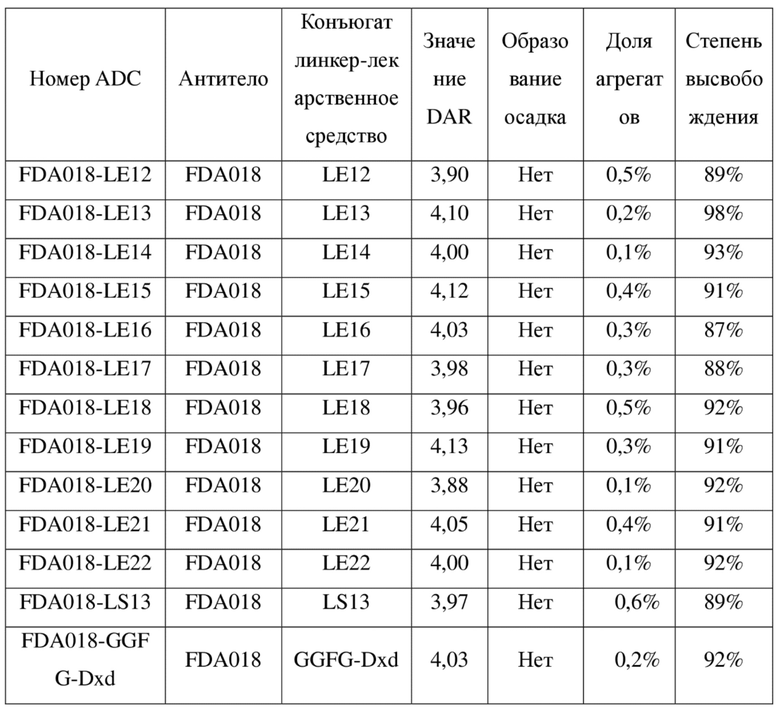
[127] TECP - трис(2-карбоксиэтил)фосфин
[128] DMSO - диметилсульфоксид
[129] DMF - N,N-диметилформамид
[130] His - гистидин
[131] HATU - 2-(7-азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат
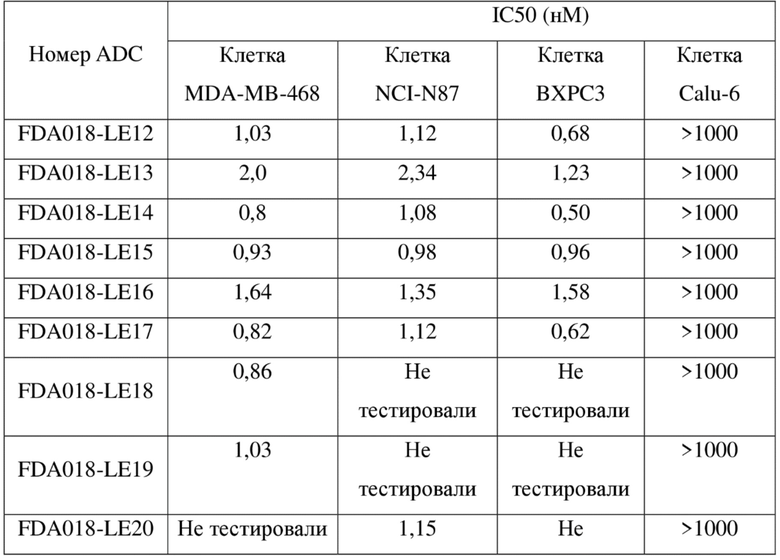
[132] об./об. - объем/объем, объемное соотношение
[133] UV - ультрафиолетовый свет
[134] ELISA - иммуноферментный твердофазный анализ
[135] BSA - бычий сывороточный альбумин
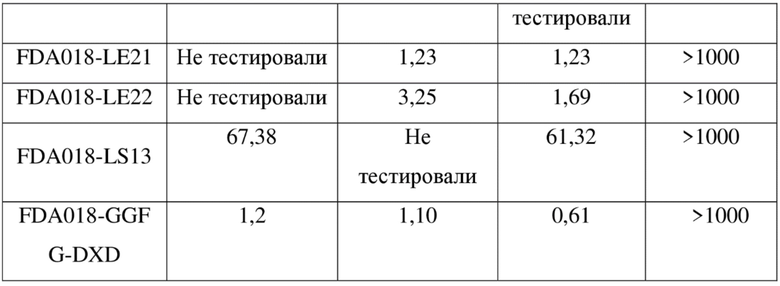
[136] rpm - обороты в минуту
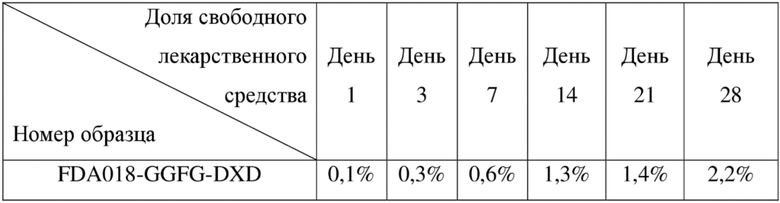
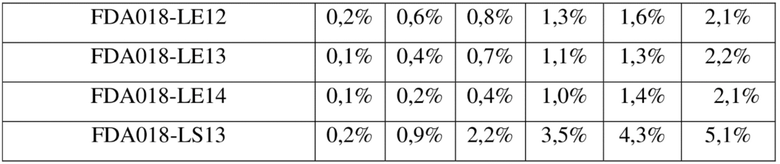
[137] FBS - фетальная бычья сыворотка
[138] Пример 1. Получение антитела к TROP2
[139] В настоящем изобретении выбирали моноклональное антитело FDA018 с высокой аффинностью и специфическим нацеливанием на TROP2, аминокислотная последовательность легкой цепи которого представлена под SEQ ID NO: 1, и аминокислотная последовательность тяжелой цепи которого представлена под SEQ ID NO: 2. Нуклеотидные последовательности легкой и тяжелой цепей FDA018 получали путем полного генного синтеза (Suzhou Genewiz). Их по отдельности конструировали с включением в вектор pV81 (как показано на фигуре 1) путем двойного расщепления EcoR I и Hind III (приобретенных у TAKARA), а затем трансформировали в компетентные клетки Trans 1-T1 (приобретенные у Beijing TransGen Biotech, номер продукта: CD501-03) путем лигирования, которые собирали для клонирования, идентификации с помощью ПЦР и отсылали для подтверждения секвенирования. Положительные клоны культивировали и размножали для экстрагирования плазмид с получением таким образом эукариотической плазмиды экспрессии легкой цепи антитела FDA018-L/pV81 и эукариотической плазмиды экспрессии тяжелой цепи антитела FDA018-H/pV81. Эти две плазмиды линеаризовывали путем расщепления с помощью XbaⅠ (приобретенной от Takara, номер продукта: 1093S). Эукариотические плазмиды экспрессии легкой и тяжелой цепей трансформировали в клетки CHO, адаптированные для роста в суспензии (приобретенные у ATCC) при соотношении 1,5/1 путем электропорации. После электропорации клетки высевали при 2000-5000 клеток/лунка в 96-луночный планшет. Через 3 недели культивирования уровень экспрессии измеряли посредством способа HTRF (гомогенная флуоресценция с временным разрешением). Десять наилучших клеточных пулов в отношении уровня экспрессии выбирали для размножения и криоконсервации. Клетки восстанавливали в 125 мл колбе для встряхивания (объем культивирования 30 мл) и культивировали при 37°C, 5,0% CO2 и 130 об./мин с применением вибрации в течение 3 дней, затем размножали в колба для встряхивания объемом 1000 мл (при объеме культуры 300 мл) и культивировали при 37°C, 5,0% CO2 и 130 об./мин с применением вибрации. Начиная с 4-го дня, каждый день добавляли свежую культуральную среду в количестве от 5 до 8% от исходного объема культуры. Культивирование завершали в период от 10-го до 12-го дня и собранную жидкость центрифугировали при 9500 об./мин в течение 15 минут с удалением клеточного осадка. Супернатант собирали и фильтровали через мембранный фильтр на 0,22 мкм. Обработанный образец очищали с применением аффинной хроматографической колонки MabSelect (приобретенной у GE) с получением антитела FDA018.
[140] Аминокислотная последовательность легкой цепи FDA018 показана ниже:
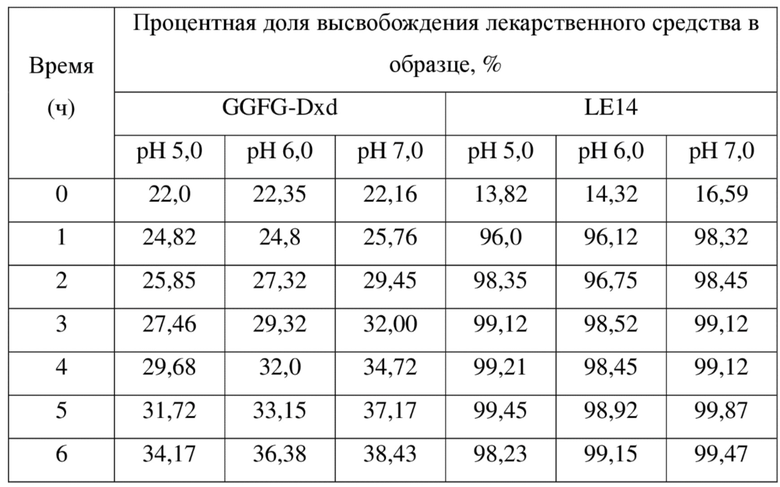
SEQ ID NO: 1:
DIQLTQSPSS LSASVGDRVS ITCKASQDVS IAVAWYQQKP GKAPKLLIYS 50
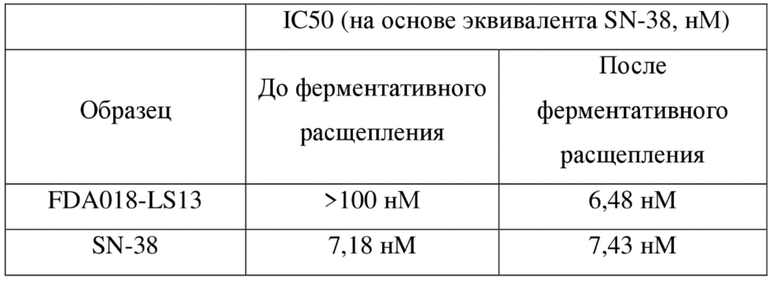
ASYRYTGVPD RFSGSGSGTD FTLTISSLQP EDFAVYYCQQ HYITPLTFGA 100
GTKVEIKRTV AAPSVFIFPP SDEQLKSGTA SVVCLLNNFY PREAKVQWKV 150
DNALQSGNSQ ESVTEQDSKD STYSLSSTLT LSKADYEKHK VYACEVTHQG 200
LSSPVTKSFN RGEC 214
[141] Аминокислотная последовательность тяжелой цепи FDA018 показана ниже:
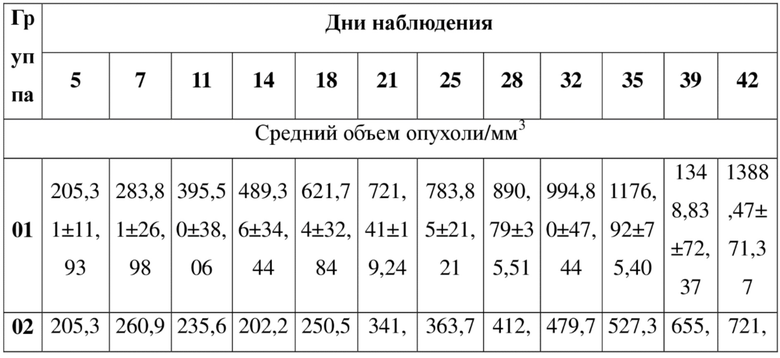
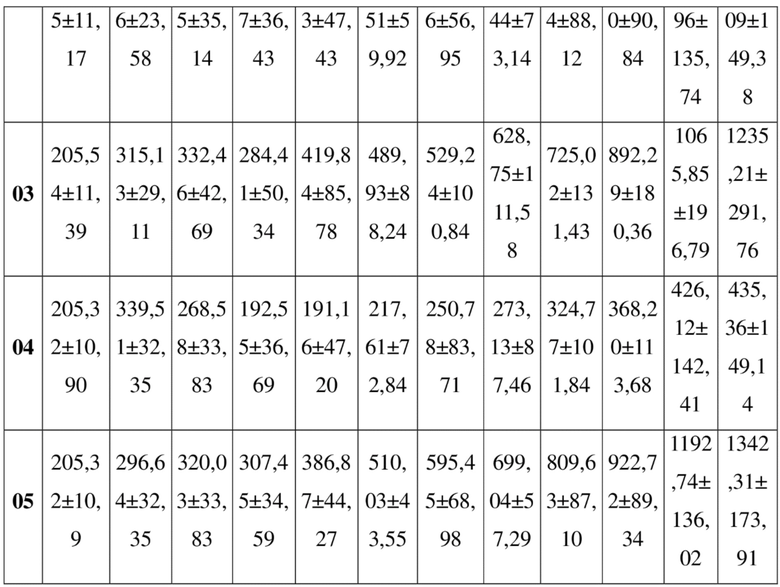
SEQ ID NO: 2:
QVQLQQSGSE LKKPGASVKV SCKASGYTFT NYGMNWVKQA PGQGLKWMGW 50
INTYTGEPTY TDDFKGRFAF SLDTSVSTAY LQISSLKADD TAVYFCARGG 100
FGSSYWYFDV WGQGSLVTVS SASTKGPSVF PLAPSSKSTS GGTAALGCLV 150
KDYFPEPVTV SWNSGALTSG VHTFPAVLQS SGLYSLSSVV TVPSSSLGTQ 200
TYICNVNHKP SNTKVDKRVE PKSCDKTHTC PPCPAPELLG GPSVFLFPPK 250
PKDTLMISRT PEVTCVVVDV SHEDPEVKFN WYVDGVEVHN AKTKPREEQY 300
NSTYRVVSVL TVLHQDWLNG KEYKCKVSNK ALPAPIEKTI SKAKGQPREP 350
QVYTLPPSRE EMTKNQVSLT CLVKGFYPSD IAVEWESNGQ PENNYKTTPP 400
VLDSDGSFFL YSKLTVDKSR WQQGNVFSCS VMHEALHNHY TQKSLSLSPG 450
K 451
[142] Пример 2. Синтез конъюгатов линкер-лекарственное средство
[143] Пример 2-1. Синтез LE12
[144] Синтез промежуточного соединения 2
[145] Растворяли (S)-2-азидопропионовую кислоту (10 г, 86,9 ммоль) и 4-аминобензиловый спирт (21,40 г, 173,8 ммоль) в смешанном растворителе из 300 мл дихлорметана и метанола (в объемном соотношении 2:1) и затем к полученному добавляли 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин (21,49 г, 86,9 ммоль). Реакцию проводили при комнатной температуре в течение 5 часов. Растворитель затем выпаривали при пониженном давлении и полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле [дихлорметан:этилацетат = 1:1 (об./об.)] с получением промежуточного соединения 2 (16,3 г, выход 85%), ESI-MS масса/заряд: 221 (M+H).
[146] Синтез промежуточного соединения 3
[147] Промежуточное соединение 2 (15 г, 68,2 ммоль) смешивали с бис(п-нитрофенил)карбонатом (22,82 г, 75,02 ммоль) и растворяли в 200 мл безводного N,N-диметилформамида, и затем к полученному добавляли 25 мл триэтиламина и проводили реакцию при комнатной температуре в течение 2 часов. После завершения реакции исходные материалы контролировали с помощью жидкостной хроматографии-масс-спектрометрии, к полученному добавляли метиламина гидрохлорид (6,91 г, 102,3 ммоль) и обеспечивали продолжение реакции при комнатной температуре в течение 1 часа. После завершения реакции большую часть растворителя удаляли путем перегонки при пониженном давлении, затем к полученному добавляли 200 мл воды и 200 мл этилацетата. Органическую фазу собирали после того, как фазы разделяли, и органическую фазу высушивали и концентрировали, и затем полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле [дихлорметан:этилацетат = 10:1 (об./об.)] с получением промежуточного соединения 3 (18,9 г, выход 100%), ESI-MS масса/заряд: 278 (M+H).
[148] Синтез промежуточного соединения 5
[149] Промежуточное соединение 3 (10 г, 36,1 ммоль) смешивали с полиформальдегидом (1,63 г, 54,2 ммоль) и растворяли в 150 мл безводного дихлорметана. К полученному медленно добавляли триметилхлорсилан (6,28 г, 57,76 ммоль) и реакцию проводили при комнатной температуре в течение 2 часов с получением неочищенного раствора промежуточного соединения 4. Реакцию контролировали с помощью жидкостной хроматографии-масс-спектрометрии после отбора образцов и гашения с помощью метанола. После завершения реакции реакционную смесь фильтровали и затем добавляли трет-бутилгидроксиацетат (9,54 г, 72,2 ммоль) и триэтиламин (10 мл, 72,2 ммоль) в фильтрат и обеспечивали продолжение реакции при комнатной температуре в течение 2 часов. После завершения реакции большинство растворителя удаляли путем перегонки при пониженном давлении, и затем полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле [петролейный эфир:этилацетат = 3:1 (об./об.)] с получением промежуточного соединения 5 (11,2 г, выход 74%), ESI-MS масса/заряд: 422 (M+H).
[150] Синтез промежуточного соединения 6
[151] Промежуточное соединение 5 (10 г, 23,8 ммоль) растворяли в 80 мл безводного тетрагидрофурана и к полученному добавляли 80 мл воды, а затем к полученному добавляли трис(2-карбоксиэтилфосфин)гидрохлорид (13,6 г, 47,6 ммоль) и реакцию проводили в течение 4 часов при комнатной температуре. После завершения реакции тетрагидрофуран удаляли путем перегонки при пониженном давлении и затем смесь экстрагировали этилацетатом. Полученную органическую фазу высушивали и выпаривали с удалением растворителя при пониженном давлении и очищали с помощью колоночной хроматографии на силикагеле [дихлорметан:метанол = 10:1 (об./об.)] с получением промежуточного соединения 6 (8,1 г, выход 86%), ESI-MS масса/заряд: 396 (M+H).
[152] Синтез промежуточного соединения 8
[153] Промежуточное соединение 6 (5 г, 12,7 ммоль) растворяли в 60 мл смешанного растворителя из дихлорметана и метанола (об./об. = 2:1) и к полученному медленно добавляли 3 мл трифторуксусной кислоты, и реакцию проводили при комнатной температуре в течение 30 мин. После завершения реакции к полученному добавляли равный объем воды и этилацетата и органическую фазу высушивали и концентрировали, и полученный неочищенный продукт непосредственно применяли на следующей стадии.
[154] Неочищенный продукт, полученный на предыдущей стадии, растворяли в 50 мл безводного N,N-диметилформамида и затем к полученному добавляли сложный гидроксисукцинимидный эфир Fmoc-L-валина (8,3 г, 19,1 ммоль) и триэтиламин (5 мл) и реакцию проводили при комнатной температуре в течение 2 часов. После завершения реакции большинство растворителя удаляли путем перегонки при пониженном давлении, и затем полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле [дихлорметан:метанол = 10:1 (об./об.)] с получением промежуточного соединения 8 (5,4 г, выход 64%), ESI-MS масса/заряд: 661 (M+H).
[155] Синтез промежуточного соединения 9
[156] Промежуточное соединение 8 (1 г, 1,5 ммоль) смешивали с эксатекана метансульфонатом (0,568 г, 1 ммоль) в 30 мл безводного N,N-диметилформамида, а затем к полученному добавляли 2-(7-азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (1,14 г, 3,0 ммоль) и 2 мл триэтиламина и реакцию проводили при комнатной температуре в течение 2 часов. После завершения реакции растворитель удаляли путем перегонки при пониженном давлении, и затем полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле [хлороформ:метанол = 10:1 (об./об.)] с получением промежуточного соединения 9 (0,94 г, выход 87%), ESI-MS масса/заряд: 1078 (M+H).
[157] Синтез соединения LE12
[158] Промежуточное соединение 9 (1 г, 0,929 ммоль) растворяли в 20 мл безводного DMF, затем к полученному добавляли 0,5 мл 1,8-диазабицикло[5.4.0]ундец-7-ена и реакцию проводили при комнатной температуре в течение 1 часа. После завершения реакции исходных материалов непосредственно добавляли N-сукцинимидил-6-малеимидогексаноат (428,5 мг, 1,39 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли путем перегонки при пониженном давлении и затем полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле [хлороформ:метанол = 8:1 (об./об.)] с получением указанного в заголовке соединения (0,7 г, выход: 73%), ESI-MS масса/заряд: 1035 (M+H).
[159] Пример 2-2. Синтез соединения LE13
[160] Синтез промежуточного соединения 14
[161] Коммерчески доступное промежуточное соединение 12 (267 мг, 0,8 ммоль) смешивали с параформальдегидом (50 мг, 1,6 ммоль) и растворяли в 20 мл безводного дихлорметана. Затем медленно добавляли триметилхлорсилан (0,3 мл, 3,4 ммоль). После завершения добавления реакцию осуществляли при комнатной температуре в течение 2 часов. Затем реакцию контролировали с помощью жидкостной хроматографии-масс-спектрометрии после отбора образцов и гашения с помощью метанола. После завершения реакции реакционную смесь фильтровали и затем в фильтрат добавляли трет-бутил-2-гидроксиацетат (211 мг, 1,6 ммоль) и пемпидин (0,5 мл) и обеспечивали продолжение реакции при комнатной температуре в течение приблизительно 2 часов. После завершения реакции большинство растворителя удаляли путем перегонки при пониженном давлении и полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле [дихлорметан:метанол = 20:1 (об./об.)] с получением промежуточного соединения 14 (260 мг, выход 68%), ESI-MS масса/заряд: 479 (M+H).
[162] Синтез промежуточного соединения 15
[163] Промежуточное соединение 14 (238 мг, 0,50 ммоль) растворяли в 6 мл смешанного растворителя из дихлорметана и метанола (об./об. = 2:1) и к полученному медленно добавляли 0,3 мл трифторуксусной кислоты, и реакцию проводили при комнатной температуре в течение 30 мин. После завершения реакции к полученному добавляли равный объем воды и этилацетата и органическую фазу высушивали и концентрировали, и полученный неочищенный продукт непосредственно применяли на следующей стадии.
[164] Синтез промежуточного соединения 16
[165] Неочищенный продукт, полученный на предыдущей стадии, смешивали с эксатекана метансульфонатом (170 мг, 0,30 ммоль) в 5 мл безводного N,N-диметилформамида и затем к полученному добавляли 2-(7-азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (341 мг, 0,90 ммоль) и 0,60 мл триэтиламина и реакцию проводили при комнатной температуре в течение 2 часов. После завершения реакции растворитель удаляли путем перегонки при пониженном давлении, и затем полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле [хлороформ:метанол = 10:1 (об./об.)] с получением промежуточного соединения 16 (210 мг, 83%), ESI-MS масса/заряд: 840 (M+H).
[166] Синтез промежуточного соединения 17
[167] Промежуточное соединение 16 (100 мг, 0,12 ммоль) растворяли в 15 мл безводного тетрагидрофурана и к полученному добавляли 3 мл воды, затем к полученному добавляли 0,3 мл водного раствора 1 моль/л триэтилфосфина, и реакцию проводили при комнатной температуре в течение 4 часов. После контроля реакции на предмет завершения реакционную смесь перегоняли при пониженном давлении с удалением тетрагидрофурана. К оставшемуся водному раствору добавляли бикарбонат натрия для регулирования pH до нейтрального значения и затем добавляли дихлорметан для экстрагирования. Полученную органическую фазу высушивали и выпаривали при пониженном давлении с удалением растворителя. Полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле [дихлорметан:метанол = 10:1 (об./об.)] с получением промежуточного соединения 17 (69 мг, выход 71%), ESI-MS масса/заряд: 814 (M+H).
[168] Синтез соединения LE13
[169] Промежуточное соединение 17 (120 мг, 0,15 ммоль), полученное в соответствии с предыдущим способом синтеза, смешивали с коммерчески доступным исходным материалом MC-V (102 мг, 0,33 ммоль) в 40 мл дихлорметана и добавляли конденсирующее средство 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин (82 мг, 0,33 ммоль) с обеспечением реакции в течение ночи при комнатной температуре. После завершения реакции растворитель выпаривали при пониженном давлении и полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле [дихлорметан:метанол = 10:1 (об./об.)] с получением соединения LE13 (116 мг, выход 70%), ESI-MS масса/заряд: 1106,5 (M+H).
[170] Пример 2-3. Синтез соединения LE14
[171] Синтез промежуточного соединения 19
[172] Коммерчески доступное промежуточное соединение 18 (300 мг, 0,8 ммоль) смешивали с полиформальдегидом (50 мг, 1,6 ммоль) и растворяли в 20 мл безводного дихлорметана. Затем к полученному медленно добавляли триметилхлорсилан (0,3 мл, 3,4 ммоль) и реакцию проводили при комнатной температуре в течение 2 часов. Реакцию контролировали с помощью жидкостной хроматографии-масс-спектрометрии после отбора образцов и гашения с помощью метанола. После завершения реакции реакционную смесь фильтровали и затем добавляли трет-бутил-2-гидроксиацетат (211 мг, 1,6 ммоль) и триэтиламин (0,22 м, 1,6 ммоль) в фильтрат. Обеспечивали продолжение реакции при комнатной температуре в течение приблизительно 2 часов. После завершения реакции большую часть растворителя удаляли путем перегонки при пониженном давлении и полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле [дихлорметан:метанол = 20:1 (об./об.)] с получением промежуточного соединения 19 (349 мг, выход 85%), ESI-MS масса/заряд: 514 (M+H), 1H ЯМР (400 MГц, CDCl3) δ 8,13 (s, 1H), 7,56 (d, J = 7,5 Гц, 2H), 7,35 (s, 2H), 5,14 (s, 2H), 4,91 (s, 2H), 4,25 (q, J = 7,1 Гц, 1H), 3,99 (d, J = 42,5 Гц, 2H), 3,85 (t, J = 6,2 Гц, 2H), 3,40 (dd, J = 18,5, 7,6 Гц, 2H), 2,89 (d, J = 48,6 Гц, 3H), 1,65 (d, J = 6,8 Гц, 3H), 1,46 (s, 9H).
[173] Синтез промежуточного соединения 20
[174] Промежуточное соединение 19 (257 мг, 0,50 ммоль) растворяли в 6 мл смешанного растворителя из дихлорметана и метанола (об./об. = 2:1) и к полученному медленно добавляли 0,3 мл трифторуксусной кислоты, и реакцию проводили при комнатной температуре в течение 30 мин. После завершения реакции к полученному добавляли равный объем воды и этилацетата и органическую фазу высушивали и концентрировали, и полученный неочищенный продукт непосредственно применяли на следующей стадии.
[175] Полученный неочищенный продукт смешивали с эксатекана метансульфонатом (170 мг, 0,30 ммоль) в 5 мл безводного N,N-диметилформамида и затем к полученному добавляли 2-(7-азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (341 мг, 0,90 ммоль) и 0,60 мл триэтиламина и реакцию проводили при комнатной температуре в течение 2 часов. После завершения реакции растворитель удаляли путем перегонки при пониженном давлении, и затем полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле [дихлорметан:метанол = 20:1 (об./об.)] с получением промежуточного соединения 20 (212 мг, выход 81%), ESI-MS масса/заряд: 875 (M+H). 1H ЯМР (400 MГц, CDCl3) δ 8,27 (d, J = 34,7 Гц, 1H), 7,63-7,35 (m, 5H), 7,21-7,10 (m, 1H), 5,71-5,48 (m, 2H), 5,24-4,95 (m, 3H), 4,95-4,72 (m, 4H), 4,45 (s, 1H), 4,33-3,97 (m, 3H), 3,75 (s, 2H), 3,39-2,99 (m, 4H), 2,76 (d, J = 15,3 Гц, 3H), 2,43-2,15 (m, 5H), 2,04 (s, 1H), 1,94-1,75 (m, 2H), 1,62 (d, J = 6,6 Гц, 3H), 1,11-0,89 (m, 3H).
[176] Синтез промежуточного соединения 21
[177] Промежуточное соединение 20 (77 мг, 0,09 ммоль) растворяли в 12 мл безводного тетрагидрофурана и к полученному добавляли 3 мл воды, затем к полученному добавляли 0,3 мл водного раствора 1 моль/л триэтилфосфина, и реакцию проводили при комнатной температуре в течение 4 часов. После завершения реакции реакционную смесь перегоняли при пониженном давлении с удалением тетрагидрофурана. К оставшемуся водному раствору добавляли бикарбонат натрия для регулирования pH до нейтрального значения и затем добавляли дихлорметан для экстрагирования. Полученную органическую фазу высушивали и выпаривали при пониженном давлении с удалением растворителя. Полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле [дихлорметан:метанол = 10:1 (об./об.)] с получением промежуточного соединения 21 (53 мг, выход 69%), ESI-MS масса/заряд: 849 (M+H). 1H ЯМР (400 MГц, DMSO) δ 8,52 (s, 1H), 7,79 (d, J = 10,8 Гц, 1H), 7,67-7,55 (m, 2H), 7,47-7,21 (m, 3H), 6,51 (s, 1H), 5,60 (s, 1H), 5,52-5,32 (m, 2H), 5,30-5,11 (m, 2H), 5,11-4,94 (m, 2H), 4,94-4,74 (m, 2H), 4,02 (s, 2H), 3,81-3,66 (m, 2H), 3,60-3,35 (m, 4H), 3,24-3,08 (m, 2H), 2,94 (d, J = 30,8 Гц, 3H), 2,39 (s, 3H), 2,28-2,04 (m,2H), 2,00-1,73 (m, 2H), 1,22 (d, J = 6,6 Гц, 3H), 0,96-0,70 (m, 3H).
[178] Синтез соединения LE14
[179] Промежуточное соединение 21 (134 мг, 0,16 ммоль) смешивали с коммерчески доступным исходным материалом MC-V (102 мг, 0,33 ммоль) в 40 мл дихлорметана и добавляли конденсирующее средство 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин (82 мг, 0,33 ммоль) с обеспечением реакции в течение ночи при комнатной температуре. После завершения реакции растворитель выпаривали при пониженном давлении и полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле [дихлорметан:метанол = 10:1 (об./об.)] с получением соединения LE14 (137 мг, выход 75%), ESI-MS масса/заряд: 1141,4 (M+H). 1H ЯМР (400 MГц, DMSO) δ 9,97 (s, 1H), 8,52 (s, 1H), 8,27-8,09 (m, 1H), 7,88-7,70 (m, 2H), 7,63-7,51 (m, 2H), 7,28 (s, 3H), 6,99 (s, 2H), 6,51 (s, 1H), 5,59 (s, 1H), 5,50-5,32 (m, 2H), 5,17 (s, 2H), 4,98 (s, 2H), 4,85 (d, J = 17,3 Гц, 2H), 4,43-4,33 (m, 1H), 4,21-4,12 (m, 1H), 4,03 (s, 2H), 3,74-3,64 (m, 2H), 3,20-3,03 (m, 3H), 3,02-2,84 (m, 4H), 2,36 (s, 3H), 2,23-2,09 (m, 4H), 2,01-1,90 (m, 1H), 1,90-1,78 (m, 2H), 1,55-1,39 (m, 4H), 1,30 (d, J = 6,7 Гц, 3H), 1,23-1,11 (m, 2H), 0,93-0,77 (m, 9H).
[180] Пример 2-4. Синтез соединений LE15-LE20
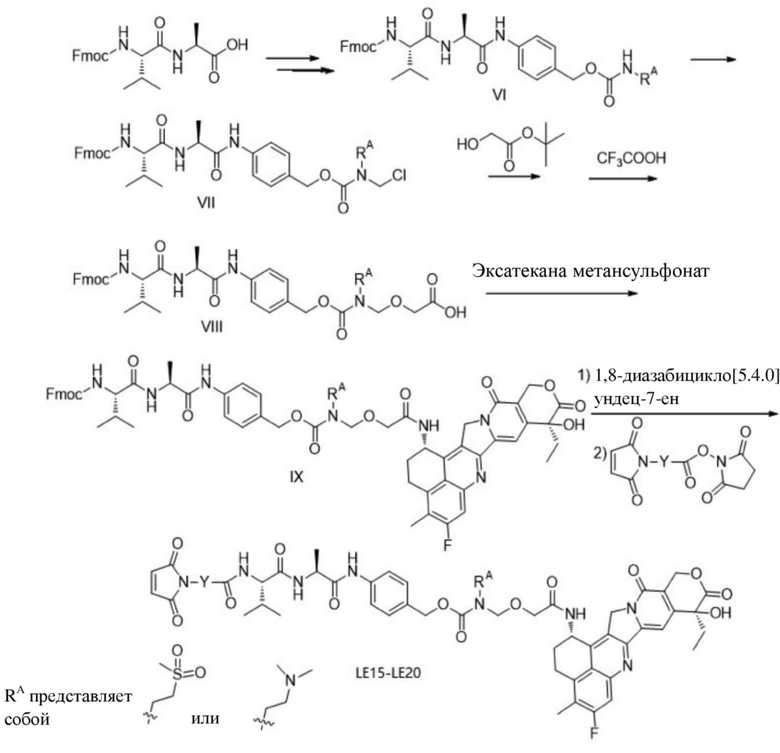
[181] Промежуточное соединение VI может быть получено путем замены гидрохлорида метиламина на стадии b на соответствующее коммерчески доступное аминосоединение с применением Fmoc-L-валил-L-аланина в качестве исходного материала и со ссылкой на стадии a и b в способе синтеза промежуточного соединения 3 в примере 2-1. Последующие стадии проводили, начиная с промежуточного соединения VI, и с применением тех же способов, что на стадиях c, d, f и h примера 2-1, и получали промежуточное соединение IX, сходное с промежуточным соединением 9. Затем, в соответствии с теми же стадиями i и j, что в примере 6, удаляли защитную аминогруппу и конденсировали с применением разных коммерчески доступных малеимидных соединений с получением конечного продукта. Структуры используемых аминосоединений и малеимидов показаны в таблице 1. Соединение LE15: серо-белое твердое вещество, ESI-MS масса/заряд: 1121,2 (M+H); соединение LE16: светло-желтое твердое вещество, ESI-MS масса/заряд: 1167,1 (M+H); соединение LE17: желтое твердое вещество, ESI-MS масса/заряд: 1132,3 (M+H); соединение LE18: светло-желтое твердое вещество, ESI-MS масса/заряд: 1305,4 (M+H); соединение LE19: светло-желтое твердое вещество, ESI-MS масса/заряд: 1307,4 (M+H); соединение LE20: светло-желтое твердое вещество, ESI-MS масса/заряд: 1337,6 (M+H).
[182] Таблица 1. Промежуточные соединения, используемые для синтеза LE15-LE20
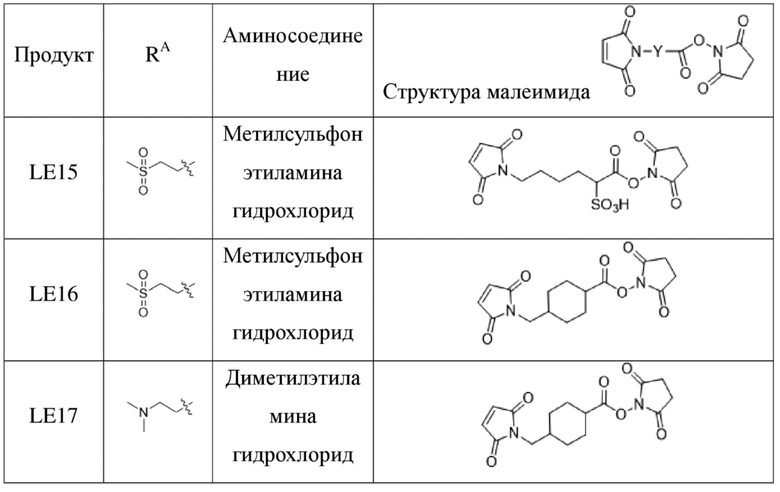
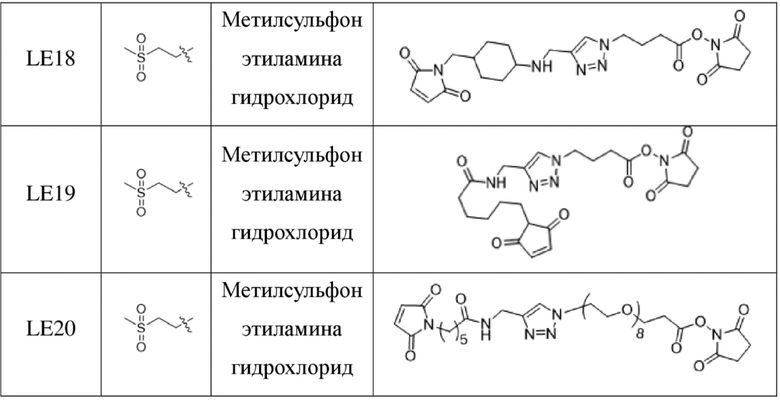
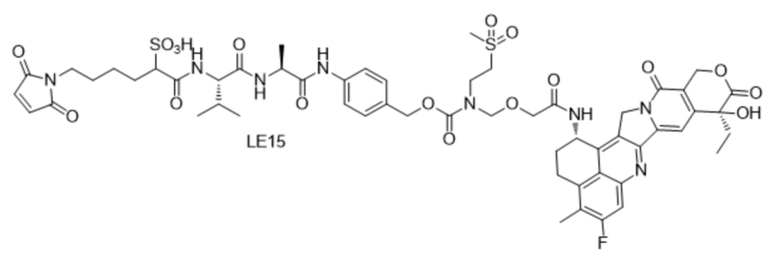 ,
,  ,
,  ,
,  ,
,  ,
, 
[183] Пример 2-5. Синтез соединений LE21 и LE22
[184] Синтез соединения DXD-1
[185] Коммерчески доступный эксатекана метансульфонат (0,568 г, 1 ммоль) смешивали с коммерчески доступной 2-(трет-бутилдиметилсилилокси)уксусной кислотой (CAS: 105459-05-0, 0,38 г, 2 ммоль) в 20 мл безводного дихлорметана. К полученному добавляли конденсирующее средство HATU (0,76 г, 2 ммоль) и 1 мл пиридина и перемешивали при комнатной температуре в течение 2 часов. После завершения реакции растворитель выпаривали до сухого состояния при пониженном давлении и полученный неочищенный продукт очищали с помощью колоночной хроматографии [дихлорметан:метанол = 50:1 (об./об.)] с получением указанного в заголовке соединения DXD-1 (0,55 г, выход 90%), ESI-MS масса/заряд: 608,1 (M+H). 1H ЯМР (400 MГц, CDCl3) δ 7,73 (d, J = 10,5 Гц, 1H), 7,64 (s, 1H), 7,05 (d, J = 9,2 Гц, 1H), 5,80-5,62 (m, 2H), 5,41-5,14 (m, 4H), 4,29-4,15 (m, 2H), 4,08-4,03 (m, 1H), 3,27-3,07 (m, 2H), 2,45 (s, 3H), 2,38-2,28 (m, 2H), 1,96-1,81 (m, 2H), 1,04 (t, J = 7,4 Гц, 3H), 0,80 (s, 9H), 0,11 (s, 3H), 0,03 (s, 3H).
[186] Получение промежуточного соединения V
[187] Промежуточное соединение V может быть получено путем замены гидрохлорида метиламина на стадии b на соответствующее коммерчески доступное аминосоединение в соответствии со способом получения соединения 4 в примере 2-1.
[188] Синтез LE21-LE22
[189] Промежуточное соединение V вводили в реакцию с DXD-1 и затем обрабатывали с помощью 10% раствора трифторуксусной кислоты/дихлорметана с получением промежуточного соединения X. Затем промежуточное соединение X вводили в реакцию в соответствии с последующими стадиями e, g, i и j для соединения 5 в примере 2-1: Промежуточное соединение X восстанавливали с получением аминосоединения и аминосоединение затем конденсировали со сложным гидроксисукцинимидным эфиром Fmoc-L-валина. Затем защитную группу Fmoc аминогруппы удаляли из полученного продукта и полученный аминопродукт затем вводили в реакцию с N-сукцинимидил-6-малеимидогексанoатом с получением конечного продукта. Соединение LE21: желтое твердое вещество, ESI-MS масса/заряд: 1141,2 (M+H); соединение LE22: желтое твердое вещество, ESI-MS масса/заряд: 1106,6 (M+H).
 ,
,
[190] Пример 2-6. Синтез соединения LS13
[191] Ссылаясь на способ синтеза LE15 в примерах 2-4, SN-38 (7-этил-10-гидроксикамптотецин) вводили в реакцию с промежуточным соединением VII (R1 представляет собой метилсульфонилэтил) с получением соединения LS13 после удаления защитной группы, конденсации и других стадий: 1H ЯМР (400 MГц, DMSO) δ 9,92 (d, J = 22,4 Гц, 1H), 8,14 (s, 1H), 8,08 (d, J = 9,1 Гц, 1H), 7,81 (d, J = 8,0 Гц, 1H), 7,70-7,50 (m, 3H), 7,47 (d, J = 7,2 Гц, 1H), 7,34 (d, J = 7,2 Гц, 1H), 7,27 (s, 1H), 7,20 (s, 1H), 6,98 (s, 2H), 6,51 (s, 1H), 5,61 (s, 2H), 5,48-5,35 (m, 2H), 5,27 (s, 2H), 5,10 (d, J = 20,6 Гц, 2H), 4,36 (s, 1H), 4,21-4,07 (m, 1H), 3,84 (s, 2H), 3,48 (s, 2H), 3,21-2,92 (m, 6H), 2,25-2,04 (m, 2H), 2,04-1,78 (m, 3H), 1,55-1,36 (m, 4H), 1,36-1,10 (m, 9H), 0,95-0,71 (m, 10H).
[192] Пример 2-7. Синтез соединения GGFG-Dxd
[193] Соединение GGFG-Dxd получали в соответствии с известным способом синтеза, описанным в WO2015146132A1. ESI-MS масса/заряд: 1034,5 (M+H), 1H-ЯМР (400 MГц, DMSO-d6) δ 8,61 (t, J = 6,4 Гц, 1H), 8,50 (d, J = 8,5 Гц, 1H), 8,28 (t, J = 5,1 Гц, 1H), 8,11 (d, J = 7,5 Гц, 1H), 8,05 (t, J = 5,7 Гц, 1H), 7,99 (t, J = 5,9 Гц, 1H), 7,77 (d, J = 11,0 Гц, 1H), 7,31 (s, 1H), 7,25-7,16 (m, 5H), 6,98 (s, 2H), 6,51 (s, 1H), 5,59 (dt, J = 7,4, 4,1 Гц, 1H), 5,41 (s, 2H), 5,20 (s, 2H), 4,64 (d, J = 6,1 Гц, 2H), 4,53-4,40 (m, 1H), 4,02 (s, 2H), 3,74-3,37 (m, 8H), 3,18-3,00 (m, 2H), 3,04-2,97 (m, 1H), 2,77 (dd, J = 13,5, 9,4 Гц, 1H), 2,38 (s, 3H), 2,19 (dd, J = 14,9, 8,5 Гц, 2H), 2,11-2,05 (m, 2H), 1,86 (dd, J = 14,0, 6,7 Гц, 2H), 1,45 (s, 4H), 1,20-1,14 (m, 2H), 0,87 (t, J = 7,1 Гц, 3H).
[194] Пример 3. Получение конъюгатов антитело-лекарственное средство
[195] Антитело FDA018 к TROP2 получали в соответствии со способом из примера 1, и при этом осуществляли замену на буфер 50 мМ PB/1,0 мМ EDTA (pH 7,0) с применением обессоливающей колонки G25. К полученному добавляли 5 эквивалентов TECP и смесь перемешивали при 25°C в течение 1 часа с полным раскрытием дисульфидных межцепочечных связей антитела. Затем применяли лимонную кислоту для регулирования pH восстановленного раствора антитела до 5,0 и для образца осуществляли замену на 20 мМ цитратный буфер и 1 мМ буфер EDTA (pH 5,0) с применением обессоливающей колонки G25. Температуру водяной бани поддерживали при 25°C в течение реакции сочетания. Конъюгаты линкер-лекарственное средство LE12-LE22, LS13 и GGFG-Dxd, полученные в соответствии с указанным выше примером 2, растворяли в DMSO соответственно и добавляли по каплям 4,5 эквивалента конъюгатов линкер-лекарственное средство в восстановленный раствор антитела. Добавляли дополнительный DMSO до конечной концентрации 5% (об./об.) и реакционную смесь перемешивали при 25°C в течение 0,5 часа. После завершения реакции образец фильтровали посредством мембраны с порами 0,22 мкм. Для очистки и удаления неконъюгированных малых молекул применяли систему фильтрации с тангенциальным потоком. Буфер представлял собой 25 мМ His, 6% раствор сахарозы (pH 6,0). После очистки образец хранили в холодильнике при -20°C. Значения поглощения измеряли при 280 нм и 370 нм с применением УФ-способа соответственно и рассчитывали значение DAR. Результаты представлены в таблице 2 ниже.
[196] Реакцию сочетания проводили тем же образом, что в данном примере, и все образцы получали в соответствии с наибольшим DAR (т. е. избыточное сочетание). Наблюдали образование осадка во время каждой реакции сочетания и рассчитывали полимерное соотношение и степень высвобождения после каждой реакции сочетания. Результаты также показаны в таблице 2.
[197] Таблица 2. Условия сочетания для получение разных конъюгатов антитело-лекарственное средство (ADC)
[198] «/» указывает на то, что степень высвобождения не рассчитывалась
[199] При процедуре получения конъюгата антитело-лекарственное средство по настоящему изобретению образование осадка не происходило, и доля агрегатов находилась в пределах нормального диапазона, что указывает на то, что конъюгаты линкер-лекарственное средство, предусмотренные настоящим изобретением, обладают хорошими физико-химическими свойствами.
[200] Пример 1 применительно к эффекту. Оценка in vitro уничтожающей активности конъюгатов антитело-лекарственное средство
[201] Клетки NCI-N87 (ATCC) выбирали в качестве клеточной линии для определения активности in vitro. 2000 клеток на лунку высевали в 96-луночный клеточный культуральный планшет и культивировали в течение периода от 20 до 24 часов. Конъюгаты антитело-лекарственное средство, полученные в соответствии со способом из примера 3, составляли в тестовых растворах с 11 градиентными концентрациями 1000, 166,7, 55,6, 18,6, 6,17, 2,06, 0,69, 0,23, 0,08, 0,008 и 0 нМ с применением среды для культивирования клеток L15, содержащей 10% FBS. Разбавленные тестовые растворы добавляли в культуральный планшет, содержащий высеянные клетки при 100 мкл/лунка, и инкубировали в течение 144 часов при 37°C в инкубаторе с 5% CO2. Добавляли реагент для анализа жизнеспособности клеток CellTiter-Glo® Luminescent (50 мкл/лунка) и планшет встряхивали при 500 об./мин при комнатной температуре в течение 10 минут для тщательного перемешивания. Данные считывали с применением микропланшет-ридера SpectraMaxL (OD 570 нм, считывание с интервалами 2 с) и рассчитывали результаты IC50, как показано в таблице 3.
[202] С применением того же способа, что описан выше, тестировали цитотоксическую активность каждого конъюгата антитело-лекарственное средство против опухолевых клеток MDA-MB-468 и BXPC3, приобретенных у ATCC. Результаты показаны в таблице 3. Из результатов в таблице 3 можно увидеть, что конъюгаты антитело-лекарственное средство, предусмотренные настоящим изобретением, характеризуются превосходной уничтожающей активностью in vitro против клеток, таких как NCI-N87, MDA-MB-468 и BXPC3. Однако конъюгат антитело-лекарственное средство не обладает уничтожающей активностью в отношении отрицательных клеток Calu-6, что указывает на то, что полученный ADC характеризуется специфичной нацеленной уничтожающей активностью.
[203] Таблица 3. Уничтожающая активность in vitro конъюгатов антитело-лекарственное средство
[204] Пример 2 применительно к эффекту. Анализ in vitro стабильности в плазме крови
[205] В данном примере оценивают стабильность конъюгата антитело-лекарственное средство, полученного в соответствии со способом из примера 3, в плазме крови человека. Конкретно в данном примере конъюгат антитело-лекарственное средство из примера 3 добавляли в плазму крови человека и помещали в водяную баню при 37°C на период продолжительностью 1, 3, 7, 14, 21 и 28 дней. Добавляли внутренний стандарт (эксатекан в качестве вещества внутреннего стандарта) и экстрагировали, а затем подвергали определению посредством высокоэффективной жидкостной хроматографии с выявлением высвобождения свободных лекарственных средств. Результаты показаны в таблице 4.
[206] Таблица 4. Оценка стабильности разных ADC в плазме крови человека
Результаты стабильности в плазме крови демонстрируют, что стабильность ADC, полученного с помощью нового технического решения, не хуже, чем у FDA018-GGFG-DXD, и иногда даже превосходит. В то же время приведенные выше результаты теста активности также доказывают, что некоторые из новых полученных ADC характеризуются лучшей активностью, чем FDA018-GGFG-DXD.
[208] Пример 3 применительно к эффекту. Эксперимент в отношении ферментативного расщепления in vitro конъюгатов линкер-лекарственное средство
[209] Конъюгат линкер-лекарственное средство (LE14 и GGFG-Dxd) совместно инкубировали с катепсином B в трех буферах с разным pH (5,0, 6,0, 7,0). Образцы брали в разные моменты времени и вводили в прибор для высокоэффективной жидкостной хроматографии-масс-спектрометрии. Применяли способ внешнего стандарта (с DXD в качестве внешнего стандарта) для определения процентной доли высвобождения лекарственного средства. Результаты эксперимента (показанные в таблице 5) показали, что GGFG-Dxd характеризуется медленной скоростью ферментативного расщепления в пределах применяемого диапазона pH, при этом LE14 по настоящему изобретению может быть быстро ферментативно расщеплен при pH в пределах диапазона от 5,0 до 7,0.
[210] Таблица 5. Ферментативное расщепление LE14 и GGFG-Dxd при разных значениях pH in vitro
[211] Пример 4 применительно к эффекту. Эксперимент в отношении ферментативного расщепления in vitro FDA018-LS13
[212] В качестве экспериментальных клеточных линий выбирали клеточную линию NCI-N87. После инкубирования образца в системе на основе катепсина B (100 мМ буфер на основе ацетата натрия-уксусной кислоты, 4 мМ дитиотреитол, pH 5,0) при 37°C в течение 4 часов полученный образец разбавляли с помощью культуральной среды до различных концентраций. Устанавливали 8 концентраций (от 1,5 до 10-кратного разбавления) с концентрацией от 70 нМ до 0,003 нМ SN-38. Уничтожающую (подавляющую) способность клеточной линии наблюдали в течение 144 часов. Значение IC50 рассчитывали путем считывания данных флуоресценции после химического люминесцентного окрашивания с помощью люминесцентного анализа жизнеспособности клеток CellTiter-Glo®.
[213] Указанные выше образцы ферментативного расщепления, полученные путем инкубирования в системе на основе катепсина B при 37°C в течение 4 часов, осаждали с помощью подходящего количества этанола с удалением белка и подвергали обнаружению посредством высокоэффективной жидкостной хроматографии в отношении высвобождения низкомолекулярных соединений. Степень высвобождения в течение 4 часов измеряли с применением эквивалентного количества SN-38 в качестве эталона, и результаты демонстрировали, что степень высвобождения достигала 99%.
[214] Результаты эксперимента (показанные в таблице 6) демонстрируют, что после обработки в виде ферментативного расщепления цитотоксическая активность FDA018-LS13 является почти такой же, как активность SN-38 в эквивалентной дозе, что также указывает на то, что FDA018-LS13 обеспечил почти полное высвобождение SN-38 при действии катепсина B и имел значение. Однако FDA018-LS13 мог претерпеть непредсказуемые изменения при поглощении эндоцитозом в лизосомы, в результате чего SN-38 не способен функционировать эффективно.
[215] Таблица 6. Изменения активности FDA018-LS13 в отношении уничтожения клеточной линии NCI-N87 перед ферментативным расщеплением и после него посредством системы на основе катепсина B
[216] Пример 5 применительно к эффекту. Тестирование противоопухолевой активности FDA018-LE14 в модели рака желудка человека NCI-N87
[217] Самкам «голых» мышей Balb/c возрастом от 6 до 8 недель подкожно путем инъекции в правую сторону шеи и спины вводили 5×106 клеток рака желудка человека (NCI-N87), растворенных в 100 мкл раствора PBS. Когда опухоль вырастала до среднего объема от 150 до 200 мМ3, мышей произвольным образом разделяли на 5 групп в соответствии с размером опухоли и весом мышей с 6 животными в каждой группе. Группы представляли собой контрольную группу холостого раствора и две группы дозы конъюгатов антитело-лекарственное средство FDA018-GGFG-DXD и FDA018-LE14 соответственно. Конкретно группа 01 представляла собой контрольную группу холостого раствора; группа 02 представляла собой группу FDA018-GGFG-DXD при 4,0 мг/кг; группа 03 представляла собой группу FDA018-GGFG-DXD при 2,0 мг/кг; группа 04 представляла собой группу FDA018-LE14 при 4,0 мг/кг; группа 05 представляла собой группу FDA018-LE14 при 2,0 мг/кг; при этом введение осуществляли внутрибрюшинно один раз в неделю. Дважды в неделю измеряли вес тела и объем опухоли животных, при этом в ходе эксперимента наблюдали за жизненным статусом экспериментальных животных. Как показано в таблице 7, средний объем опухоли мышей в контрольной группе холостого раствора составлял 1388,47 мм3 в конце обработки. Средний объем опухоли в группе обработки FDA018-GGFG-DXD при 2,0 мг/кг составлял 1235,21 мм3 на 14-й день после завершения обработки, и средний объем опухоли в группе обработки FDA018-GGFG-DXD при 4,0 мг/кг составлял 721,09 мм3 на 14-й день после завершения обработки. Средний объем опухоли в группе обработки FDA018-LE14 при 2,0 мг/кг составлял 1342,31 мм3 на 14-й день после завершения обработки, и средний объем опухоли в группе обработки FDA018-LE14 при 4,0 мг/кг составлял 435,36 мм3 на 14-й день после завершения обработки. Результаты экспериментов демонстрировали, что FDA018-LE14 обладает хорошей противоопухолевой активностью in vivo, и все экспериментальные мыши не имели случаев смерти или потери веса, что указывает на то, что FDA018-LE14 характеризуется хорошей безопасностью.
[218] Таблица 7. Противоопухолевая активность FDA018-LE14 в модели рака желудка человека NCI-N87
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Шанхай Фудань-Чжанцзян Био-Фармасьютикал, Ко., Лтд.
<120> КОНЪЮГАТ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО,
НАЦЕЛИВАЮЩИЙСЯ НА TROP2, И СПОСОБ ЕГО ПОЛУЧЕНИЯ
И ЕГО ПРИМЕНЕНИЕ
<130> P23412149RU
<160> 2
<170> PatentIn версии 3.5
<210> 1
<211> 214
<212> БЕЛОК
<213> Homo sapiens
<400> 1
Asp Ile Gln Leu Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Ser Ile Thr Cys Lys Ala Ser Gln Asp Val Ser Ile Ala
20 25 30
Val Ala Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Ser Ala Ser Tyr Arg Tyr Thr Gly Val Pro Asp Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Val Tyr Tyr Cys Gln Gln His Tyr Ile Thr Pro Leu
85 90 95
Thr Phe Gly Ala Gly Thr Lys Val Glu Ile Lys Arg Thr Val Ala Ala
100 105 110
Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125
Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala
130 135 140
Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln
145 150 155 160
Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser
165 170 175
Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr
180 185 190
Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205
Phe Asn Arg Gly Glu Cys
210
<210> 2
<211> 451
<212> БЕЛОК
<213> Homo sapiens
<400> 2
Gln Val Gln Leu Gln Gln Ser Gly Ser Glu Leu Lys Lys Pro Gly Ala
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly Tyr Thr Phe Thr Asn Tyr
20 25 30
Gly Met Asn Trp Val Lys Gln Ala Pro Gly Gln Gly Leu Lys Trp Met
35 40 45
Gly Trp Ile Asn Thr Tyr Thr Gly Glu Pro Thr Tyr Thr Asp Asp Phe
50 55 60
Lys Gly Arg Phe Ala Phe Ser Leu Asp Thr Ser Val Ser Thr Ala Tyr
65 70 75 80
Leu Gln Ile Ser Ser Leu Lys Ala Asp Asp Thr Ala Val Tyr Phe Cys
85 90 95
Ala Arg Gly Gly Phe Gly Ser Ser Tyr Trp Tyr Phe Asp Val Trp Gly
100 105 110
Gln Gly Ser Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser
115 120 125
Val Phe Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala
130 135 140
Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val
145 150 155 160
Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala
165 170 175
Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val
180 185 190
Pro Ser Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His
195 200 205
Lys Pro Ser Asn Thr Lys Val Asp Lys Arg Val Glu Pro Lys Ser Cys
210 215 220
Asp Lys Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Leu Leu Gly
225 230 235 240
Gly Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met
245 250 255
Ile Ser Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser His
260 265 270
Glu Asp Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val
275 280 285
His Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr
290 295 300
Arg Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly
305 310 315 320
Lys Glu Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile
325 330 335
Glu Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val
340 345 350
Tyr Thr Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser
355 360 365
Leu Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu
370 375 380
Trp Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro
385 390 395 400
Val Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val
405 410 415
Asp Lys Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met
420 425 430
His Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser
435 440 445
Pro Gly Lys
450
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ ДЛЯ ЕГО ПОЛУЧЕНИЯ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2800137C1 |
| КОНЪЮГАТЫ АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА ДЛЯ РАЗРУШЕНИЯ ГЕМОПОЭТИЧЕСКИХ СТВОЛОВЫХ КЛЕТОК | 2017 |
|
RU2781444C2 |
| КОНЪЮГАТ АНТИ-TROP2 АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2021 |
|
RU2840864C2 |
| ПРОИЗВОДНЫЕ КАЛИХЕАМИЦИНА И ИХ КОНЪЮГАТЫ "АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО" | 2018 |
|
RU2732568C1 |
| КОНЪЮГАТ АНТИ-TROP2 АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2014 |
|
RU2743077C2 |
| ПРОИЗВОДНЫЕ МАЙТАНЗИНОИДА С САМОРАСЩЕПЛЯЮЩИМИСЯ ПЕПТИДНЫМИ ЛИНКЕРАМИ И ИХ КОНЪЮГАТЫ | 2018 |
|
RU2765098C2 |
| КОНЪЮГАТ АНТИТЕЛА К TROP-2 И АНАЛОГА ЭКЗАТЕКАНА И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2830167C1 |
| КОНЪЮГАТ АНТИТЕЛО-ПРОИЗВОДНОЕ ПИРРОЛОБЕНЗОДИАЗЕПИНА | 2018 |
|
RU2820928C2 |
| КОНЪЮГАТЫ АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА, СОДЕРЖАЩИЕ МОДУЛЯТОР СПЛАЙСИНГА НА ОСНОВЕ ГЕРБОКСИДИЕНА, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2820607C2 |
| КОНЪЮГАТ АНТИТЕЛО К B7H3-АНАЛОГ ЭКЗАТЕКАНА И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2019 |
|
RU2785664C2 |
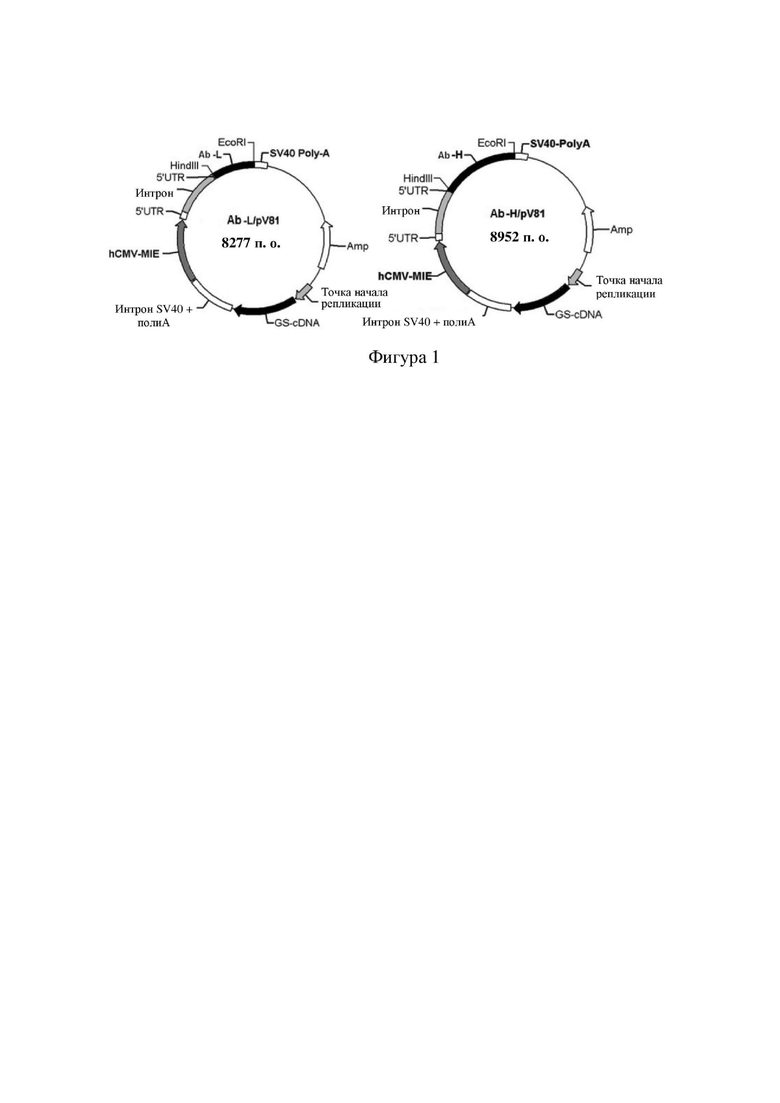
Изобретение относится к области биотехнологии. Описана группа изобретений, включающая конъюгат антитело-лекарственное средство к TROP2, способ получения конъюгата антитело-лекарственное средство к TROP2, фармацевтическую композицию для лечения и/или предупреждения TROP2-положительного рака желудка, применение конъюгата или фармацевтической композиции в изготовлении ингибитора белка TROP2, применение конъюгата или фармацевтической композиции для лечения и/или предупреждения TROP2-положительного рака желудка. Изобретение расширяет арсенал средств, связывающихся с TROP2. 5 н. и 7 з.п. ф-лы, 1 ил., 7 табл., 5 пр.
1. Конъюгат антитело-лекарственное средство формулы I к TROP2, его фармацевтически приемлемая соль;
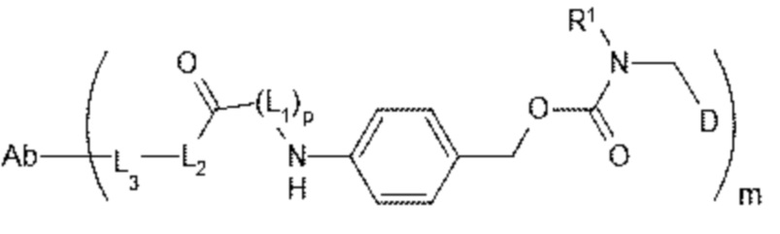 ;
;
I
где Ab представляет собой антитело к TROP2;
аминокислотная последовательность легкой цепи в антителе к TROP2 представлена под SEQ ID NO: 1 и аминокислотная последовательность тяжелой цепи представлена под SEQ ID NO: 2;
m составляет от 3,5 до 4,5;
D представляет собой 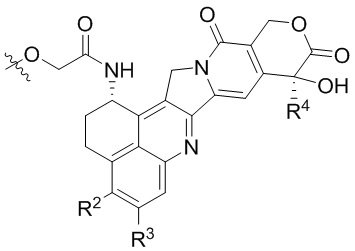 или
или 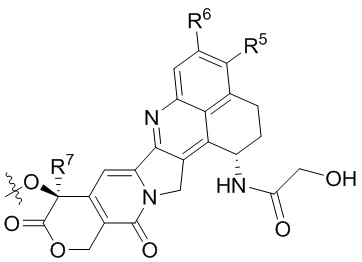 ; каждый из R2 и R5 независимо представляет собой H, C1-C6алкил или галоген; каждый из R3 и R6 независимо представляет собой H, C1-C6алкил или галоген; каждый из R4 и R7 независимо представляет собой C1-C6алкил;
; каждый из R2 и R5 независимо представляет собой H, C1-C6алкил или галоген; каждый из R3 и R6 независимо представляет собой H, C1-C6алкил или галоген; каждый из R4 и R7 независимо представляет собой C1-C6алкил;
R1 представляет собой C1-C6алкил, замещенный одним -NR1-1R1-2, C1-C6алкил, замещенный R1-3S(O)2-, или C1-C6алкил; при этом каждый из R1-1, R1-2 и R1-3 независимо представляет собой C1-C6алкил;
L1 независимо представляет собой одно из остатка аланина и остатка валина; p составляет 2;
L2 представляет собой 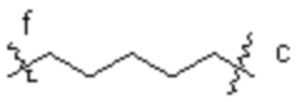 ,
,  ,
,  ,
,  ,
,  или
или 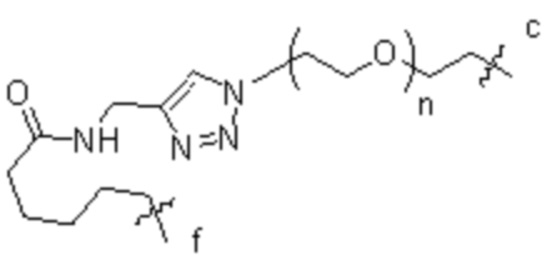 , где n независимо составляет от 8 до 12, c-конец присоединен к L1 посредством карбонильной группы и f-конец присоединен к d-концу L3;
, где n независимо составляет от 8 до 12, c-конец присоединен к L1 посредством карбонильной группы и f-конец присоединен к d-концу L3;
L3 представляет собой 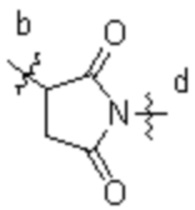 , где b-конец присоединен к Ab и d-конец присоединен к f-концу L2.
, где b-конец присоединен к Ab и d-конец присоединен к f-концу L2.
2. Конъюгат антитело-лекарственное средство к TROP2, его фармацевтически приемлемая соль по п. 1, где
b-конец L3 присоединен к сульфгидрильной группе антитела в форме тиоэфирной связи;
и/или, если R1 представляет собой C1-C6алкил, замещенный одним -NR1-1R1-2, C1-C6алкил представляет собой метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, предпочтительно этил;
и/или, если каждый из R1-1 и R1-2 независимо представляет собой C1-C6алкил, C1-C6алкил представляет собой метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, предпочтительно метил;
и/или, если R1 представляет собой C1-C6алкил, замещенный одним R1-3S(O)2-, C1-C6алкил представляет собой C1-C4алкил, предпочтительно метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, наиболее предпочтительно этил;
и/или, если R1-3 представляет собой C1-C6алкил, C1-C6алкил представляет собой метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, предпочтительно метил;
и/или, если R1 представляет собой C1-C6алкил, C1-C6алкил представляет собой метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, предпочтительно метил;
и/или m представляет собой целое число или нецелое число;
и/или n составляет 8;
и/или, если R1-1, R1-2 и R1-3 независимо представляют собой C1-C6алкил, C1-C6алкил представляет собой C1-C4алкил, предпочтительно метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, наиболее предпочтительно метил.
3. Конъюгат антитело-лекарственное средство к TROP2, его фармацевтически приемлемая соль по п. 1, где,
если каждый из R2 и R5 независимо представляет собой C1-C6алкил, C1-C6алкил представляет собой C1-C4алкил, дополнительно предпочтительно метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, наиболее предпочтительно метил;
и/или, если каждый из R2 и R5 независимо представляет собой галоген, галоген представляет собой фтор, хлор, бром или йод, предпочтительно фтор;
и/или, если каждый из R3 и R6 независимо представляет собой C1-C6алкил, C1-C6алкил представляет собой C1-C4алкил, дополнительно предпочтительно метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, наиболее предпочтительно метил;
и/или, если каждый из R3 и R6 независимо представляет собой галоген, галоген представляет собой фтор, хлор, бром или йод, дополнительно предпочтительно фтор;
и/или, если каждый из R4 и R7 независимо представляет собой C1-C6алкил, C1-C6алкил представляет собой C1-C4алкил, дополнительно предпочтительно метил, этил, н-пропил, изопропил, н-бутил, изобутил или трет-бутил, наиболее предпочтительно этил;
и/или, если R1 представляет собой C1-C6алкил, замещенный одним -NR1-1R1-2, -NR1-1R1-2 представляет собой -N(CH3)2;
и/или, если R1 представляет собой C1-C6алкил, замещенный одним -NR1-1R1-2, C1-C6алкил, замещенный одним -NR1-1R1-2, представляет собой  ;
;
и/или, если R1 представляет собой C1-C6алкил, замещенный одним R1-3S(O)2-, C1-C6алкил, замещенный одним R1-3S(O)2-, представляет собой  ;
;
и/или m составляет от 3,8 до 4,2, как например 3,88, 3,90, 3,96, 3,97, 3,98, 4,00, 4,03, 4,05, 4,10, 4,12 или 4,13;
и/или (L1)p представляет собой 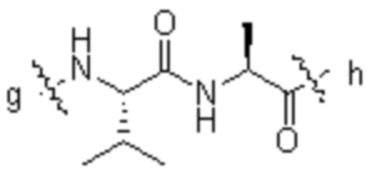 , где g-конец присоединен к c-концу L2 посредством карбонильной группы.
, где g-конец присоединен к c-концу L2 посредством карбонильной группы.
4. Конъюгат антитело-лекарственное средство к TROP2, его фармацевтически приемлемая соль по п. 1, где
каждый из R2 и R5 независимо представляет собой C1-C6алкил;
и/или каждый из R3 и R6 независимо представляет собой галоген;
и/или R4 и R7 представляют собой этил;
и/или L1 представляет собой одно из остатка аланина и остатка валина;
и/или R1 представляет собой C1-C6алкил, замещенный одним -NR1-1R1-2, или C1-C6алкил, замещенный одним R1-3S(O)2-, наиболее предпочтительно C1-C6алкил, замещенный одним R1-3S(O)2-;
предпочтительно D представляет собой 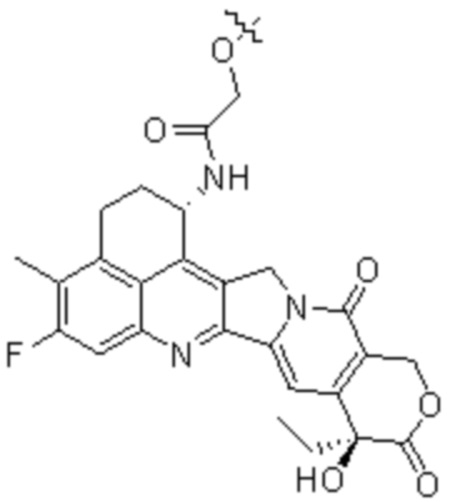 или
или 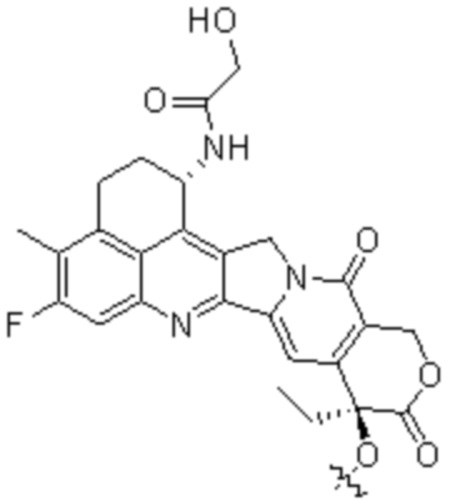 .
.
5. Конъюгат антитело-лекарственное средство к TROP2, его фармацевтически приемлемая соль по любому из пп. 1-4, где конъюгат антитело-лекарственное средство представлен любой из следующих схем:
схема I:
D представляет собой или ;
L1 независимо представляет собой одно из остатка аланина и остатка валина;
схема II:
каждый из R2 и R5 независимо представляет собой C1-C6алкил; каждый из R3 и R6 независимо представляет собой галоген; D предпочтительно представляет собой или ;
L1 независимо представляет собой одно из остатка аланина и остатка валина;
схема III:
Ab представляет собой антитело к TROP2;
каждый из R2 и R5 независимо представляет собой C1-C6алкил; каждый из R3 и R6 независимо представляет собой галоген; D предпочтительно представляет собой или ;
m составляет от 3,5 до 4,5;
L1 независимо представляет собой остаток валина или остаток аланина;
схема IV:
Ab представляет собой антитело к TROP2;
D представляет собой или ;
L1 независимо представляет собой остаток валина или остаток аланина;
схема V:
Ab представляет собой антитело к TROP2;
D представляет собой или ;
m составляет от 3,5 до 4,5;
L1 независимо представляет собой остаток валина или остаток аланина.
6. Конъюгат антитело-лекарственное средство к TROP2, его фармацевтически приемлемая соль по п. 1, где конъюгат антитело-лекарственное средство к TROP2 представляет собой любое из следующих соединений:
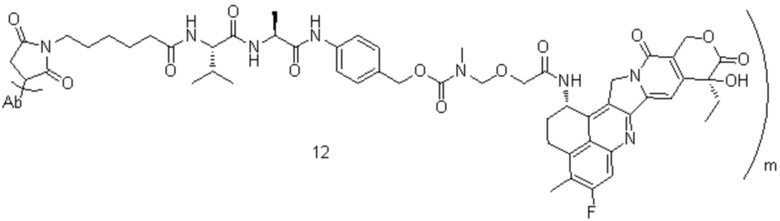 ,
, 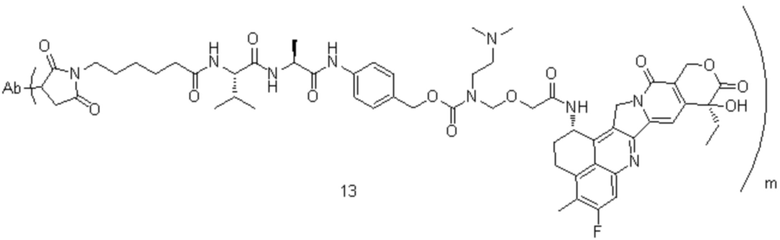 ,
, 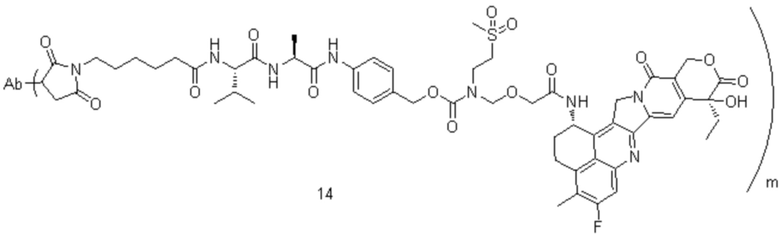 ,
, 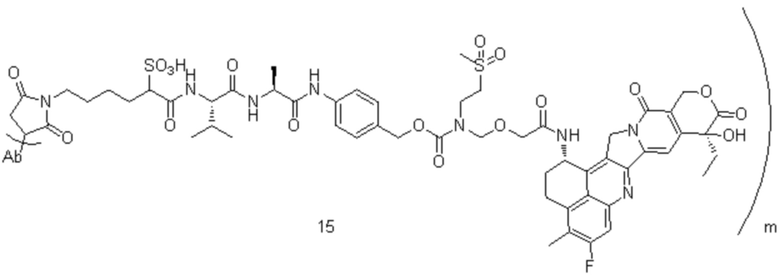 ,
, 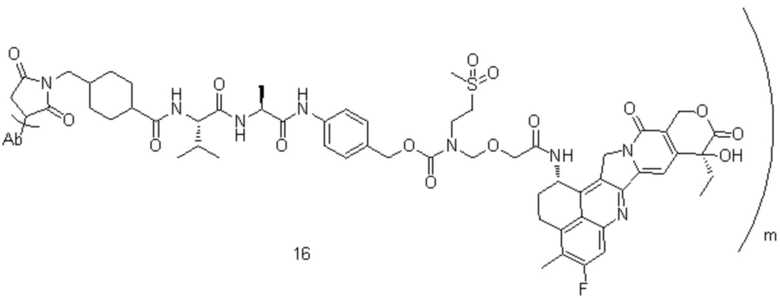 ,
, 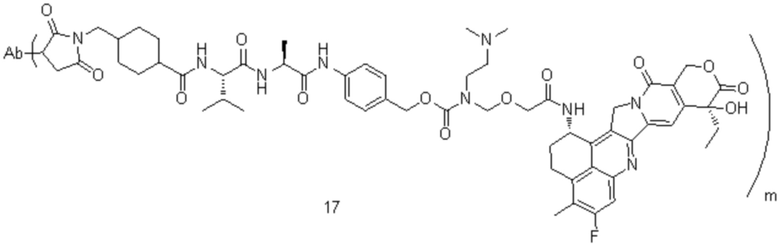 ,
, 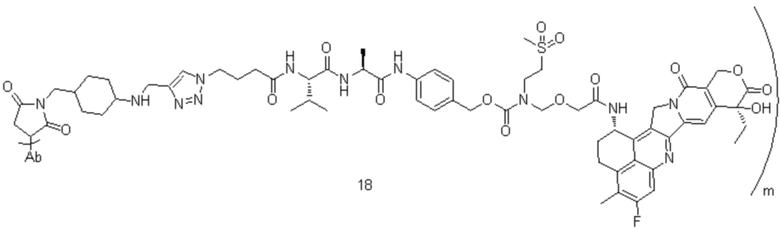 ,
, 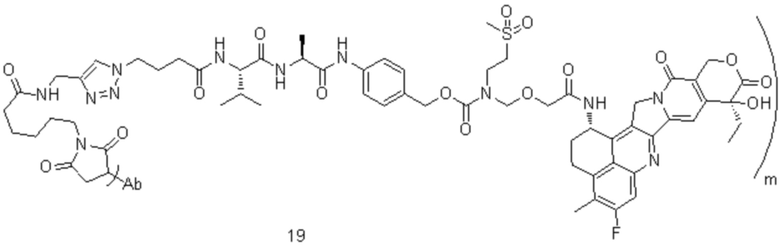 ,
, 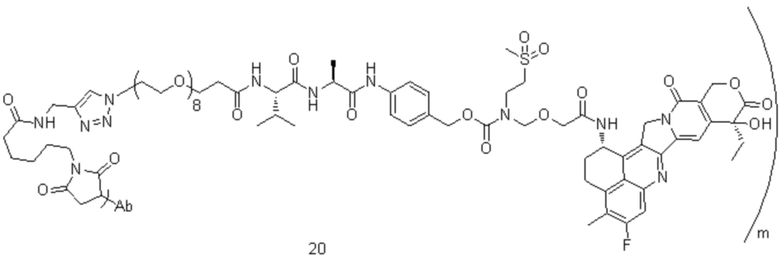 ,
, 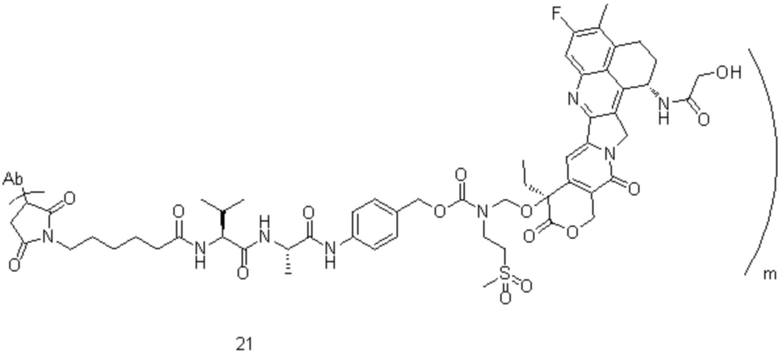 или
или 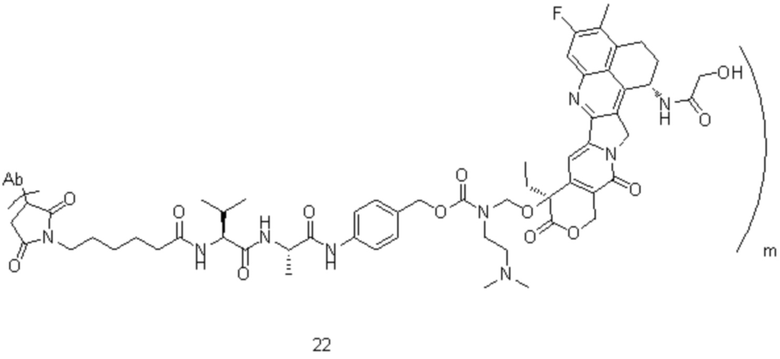 ;
;
где Ab представляет собой антитело к TROP2 и m составляет от 3,8 до 4,2, предпочтительно 3,88, 3,90, 3,96, 3,97, 3,98, 4,00, 4,03, 4,05, 4,10, 4,12 или 4,13; аминокислотная последовательность легкой цепи в антителе к TROP2 предпочтительно представлена под SEQ ID NO: 1 и аминокислотная последовательность тяжелой цепи предпочтительно представлена под SEQ ID NO: 2.
7. Конъюгат антитело-лекарственное средство к TROP2, его фармацевтически приемлемая соль по п. 1, где конъюгат антитело-лекарственное средство представлен любой из следующих схем:
схема A:
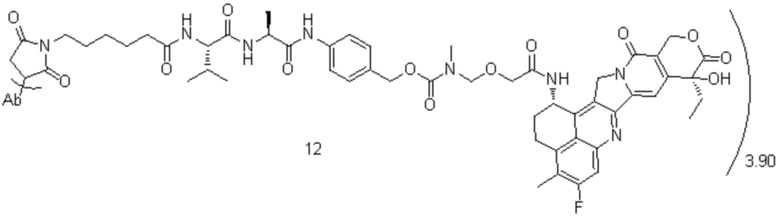 ,
, 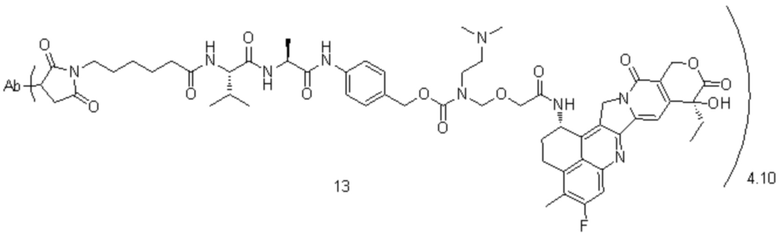 ,
, 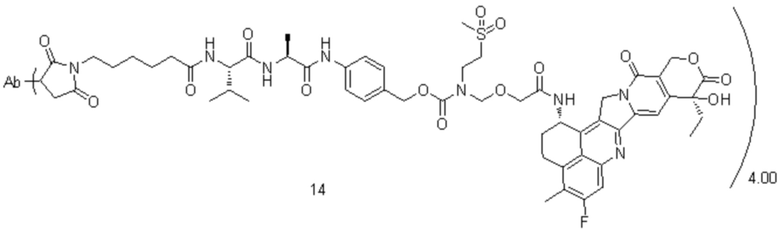 ,
, 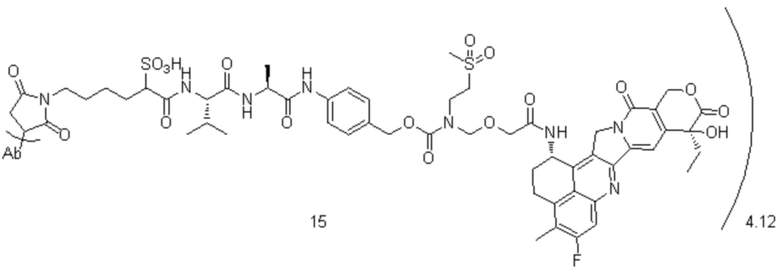 ,
, 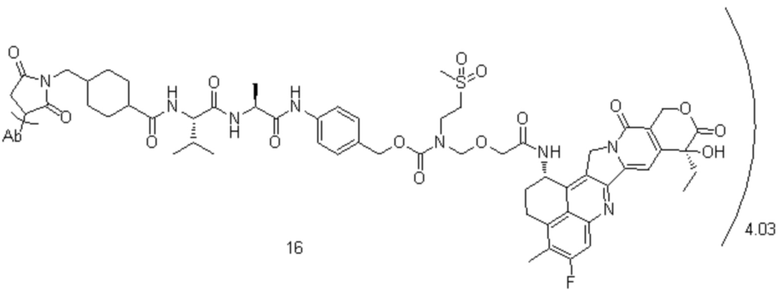 ,
, 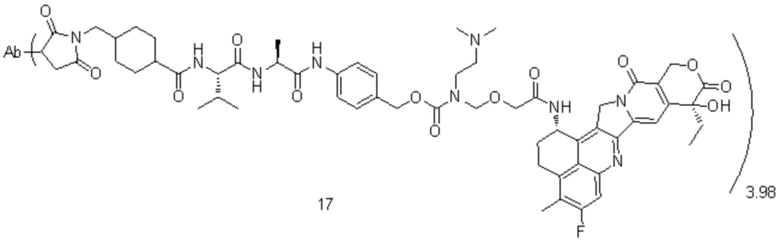 ,
, 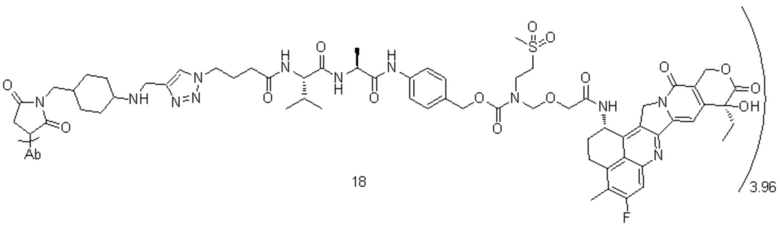 ,
, 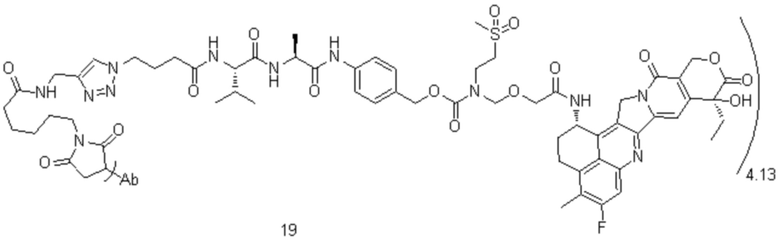 ,
, 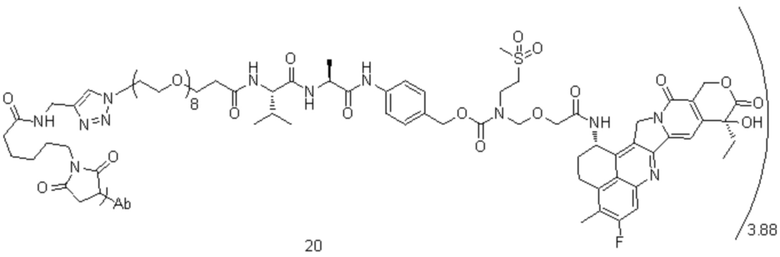 ,
, 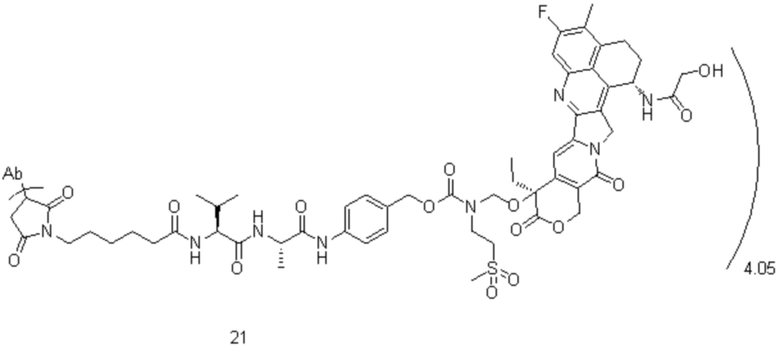 или
или 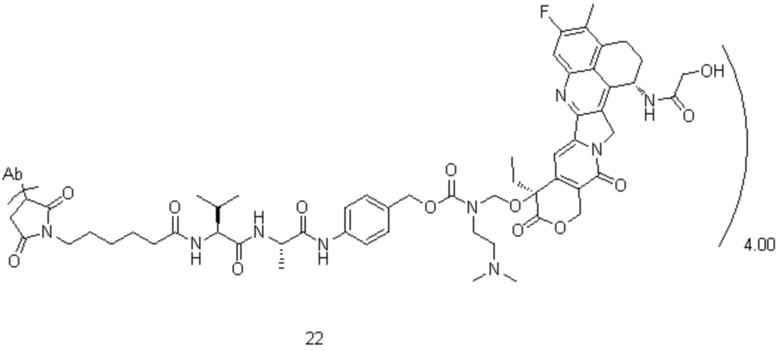 ;
;
где Ab представляет собой антитело к TROP2, предпочтительно антитело к TROP2; аминокислотная последовательность легкой цепи в антителе к TROP2 предпочтительно представлена под SEQ ID NO: 1 и аминокислотная последовательность тяжелой цепи предпочтительно представлена под SEQ ID NO: 2;
схема B:
, или ,
где Ab представляет собой антитело к TROP2; m предпочтительно равняется 3,90, 4,00 или 4,10; аминокислотная последовательность легкой цепи в антителе к TROP2 предпочтительно представлена под SEQ ID NO: 1 и аминокислотная последовательность тяжелой цепи предпочтительно представлена под SEQ ID NO: 2;
схема C:
, или ;
где Ab представляет собой антитело к TROP2;
схема D:
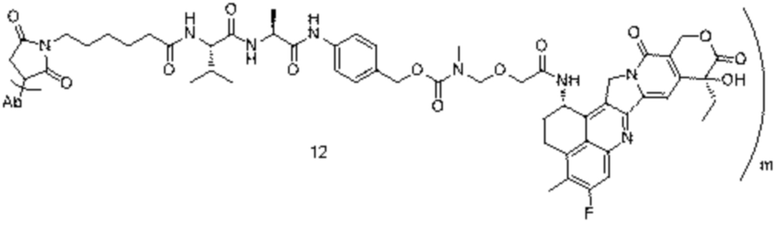 ,
,
при этом Ab представляет собой антитело к TROP2 и m предпочтительно равняется 3,90;
,
при этом Ab представляет собой антитело к TROP2 и m предпочтительно равняется 4,10;
,
при этом Ab представляет собой антитело к TROP2 и m предпочтительно равняется 4,00;
,
при этом Ab представляет собой антитело к TROP2 и m предпочтительно равняется 4,12;
,
при этом Ab представляет собой антитело к TROP2 и m предпочтительно равняется 4,03;
,
при этом Ab представляет собой антитело к TROP2 и m предпочтительно равняется 3,98;
,
при этом Ab представляет собой антитело к TROP2 и m предпочтительно равняется 3,96;
,
при этом Ab представляет собой антитело к TROP2 и m предпочтительно равняется 4,13;
,
при этом Ab представляет собой антитело к TROP2 и m предпочтительно равняется 3,88;
,
при этом Ab представляет собой антитело к TROP2 и m предпочтительно равняется 4,05;
,
при этом Ab представляет собой антитело к TROP2 и m предпочтительно равняется 4,00.
8. Способ получения конъюгата антитело-лекарственное средство к TROP2 по любому из пп. 1-7, включающий следующие стадии: осуществление реакции сочетания между соединением формулы II и Ab-водородом, как показано ниже;
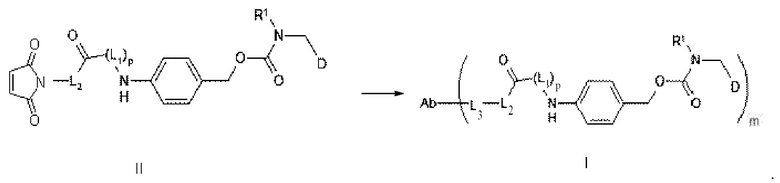
9. Фармацевтическая композиция, содержащая вещество X и фармацевтически приемлемое вспомогательное вещество; при этом вещество X представляет собой конъюгат антитело-лекарственное средство к TROP2, его фармацевтически приемлемую соль по любому из пп. 1-7, при этом количество вещества X представляет собой терапевтически эффективное количество, при этом фармацевтическую композицию используют для лечения и/или предупреждения TROP2-положительного рака желудка.
10. Применение вещества X или фармацевтической композиции по п. 9 в изготовлении ингибитора белка TROP2; при этом вещество X представляет собой конъюгат антитело-лекарственное средство к TROP2, его фармацевтически приемлемую соль по любому из пп. 1-7.
11. Применение вещества X или фармацевтической композиции по п. 9 в изготовлении лекарственного средства для лечения и/или предупреждения TROP2-положительного рака желудка.
12. Применение вещества X или фармацевтической композиции по п. 11, при этом клетки рака желудка представляют собой клетки NCI-N87.
| КОНЪЮГАТ АНТИ-TROP2 АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2014 |
|
RU2705367C2 |
| EP 3464381 A4, 11.12.2019 | |||
| Gray J | |||
| E | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Устройство для автоматического тушения огнеопасных жидкостей | 1926 |
|
SU5711A1 |

