Область техники, к которой относится изобретение
Настоящее изобретение относится к способам лечения синдрома Шегрена с применением ингибитора тирозинкиназы Брутона (BTK).
Предпосылки изобретения
Синдром Шегрена (SjS) представляет собой системное аутоиммунное заболевание неизвестной этиологии, характеризующееся лимфоидной инфильтрацией и прогрессирующим разрушением экзокринных желез (Brito-Zerón P., et al, (2016) Treating the Underlying Pathophysiology of Primary Sjögren Syndrome: Recent Advances and Future Prospects. Drugs p. 1601-1623).
Хотя заболевание в первую очередь поражает слезные и слюнные железы, воспалительный
процесс может затрагивать любой орган, при этом у приблизительно 15% пациентов наблюдаются тяжелые
экстрагландулярные проявления (Baldini C., et al (2014) Primary Sjögren's syndrome as a multi-organ disease: impact of the serological profile on the clinical presentation of the disease in a large cohort of Italian patients. Rheumatology (Oxford) p. 839-44). Клиническая картина чаще всего характеризуется в первую очередь экзокринопатией слюнных и слезных желез, проявляющейся сухостью во рту и глазах. Однако симптомы могут быть очень разнородными и помимо сухости они могут включать ряд симптомов от скелетно-мышечной боли и утомляемости, поражающих почти всех пациентов, до тяжелых, экстрагландулярных и системных поражений (характеризующихся периэпителиальной инфильтрацией лимфоцитов и отложением иммунных комплексов) у более ограниченной подгруппы пациентов. Механизм, лежащий в основе развития SjS, заключается в разрушении эпителия экзокринных желез вследствие действия аутореактивных В-клеток и Т-клеток. (Brito-Zerón P., et al, (2016) Treating the Underlying Pathophysiology of Primary Sjögren Syndrome: Recent Advances and Future Prospects. Drugs p. 1601-1623). Высокая распространенность аутоантител, в частности к Ro/SSA, даже на очень ранней стадии свидетельствует о том, что аутореактивные В-клетки участвуют в патологическом механизме SjS (Nocturne G., et al, (2018) B cells in the pathogenesis of primary Sjögren syndrome. Nat Rev Rheumatol p. 133-145).
Патология B-клеток также приводит к повышенному риску злокачественной трансформации, при этом B-клеточные лимфомы встречаются у 5% пациентов с SjS при 10-кратном повышении риска возникновения в течение жизни (Baldini C., et al, (2014) Primary Sjögren's syndrome as a multi-organ disease: impact of the serological profile on the clinical presentation of the disease in a large cohort of Italian patients. Rheumatology (Oxford) p. 839-44). Расчетная распространенность SjS составляет от 0,3 до 1 на 1000 человек (Qin B., et al. (2015) Epidemiology of primary Sjögren's syndrome: a systematic review and meta-analysis. Ann. Rheum. Dis. p. 1983-9), и в группе системных аутоиммунных заболеваний он уступает только ревматоидному артриту. Заболевание поражает в основном женщин, при этом соотношение женщины/мужчины составляет 9:1, и может возникать в любом возрасте. Основной эффект симптомов SjS заключается в серьезном воздействии на качество жизни и продуктивность, зачастую вызываемым ассоциированной с заболеванием утомляемостью, ограничивающей дееспособность (Mariette X., et al.(2018) Primary Sjögren's Syndrome. N. Engl. J. Med. p. 931-939). Также существует целый ряд потенциально серьезных системных осложнений, включая артрит, кожный васкулит, периферическую невропатию, гломерулонефрит, интерстициальный нефрит, билиарный холангит, обструктивный бронхиолит и другие, затрагивающие несколько систем органов и поражающие 20-40% пациентов (Seror R., et al (2014) Outcome measures for primary Sjögren's syndrome: a comprehensive review. J. Autoimmun. p. 51-6).
Клинические особенности синдрома Шегрена можно разделить на проявления, поддающиеся медицинской оценке, и симптоматические проявления у пациента. В настоящее время не существует единого инструмента оценки, который мог бы зафиксировать активность заболевание на основе обоих этих клинических проявлений SjS. Таким образом, для измерения симптоматических и системных проявлений SjS широко используются и являются подтвержденными "индекс оценки, сообщаемой пациентом с синдромом Шегрена, согласно Европейской лиге по борьбе с ревматизмом (EULAR)" (ESSPRI) и индекс активности заболевания при SS согласно EULAR (ESSDAI) (Franceschini F., et al, (2017), BMC Medicine, 15:69).
С точки зрения комплекса имеющихся в настоящее время средств и методов лечения не существует одобренных на международном уровне системных видов терапии SjS. Если рассматривать только сухость во рту и глазах, лечение пациентов с SjS ограничивается симптоматической терапией. Стероиды и типичные DMARD в большинстве своем неэффективны, а против тяжелой утомляемости, ограничивающей дееспособность, эффективного фармакологического вмешательства не существует. Отсутствие эффективных вариантов лечения подчеркивает необходимость в оценке более новых терапевтических подходов для лечения данного в высшей степени инвалидизирующего заболевания. Поскольку характер аутореактивности В-клеток в некоторой степени сходен с таковым при системной волчанке и ревматоидном артрите, в последнее время терапию путем истощения В-клеток с применением ритуксимаба, моноклонального антитела к CD20 (mAb), также оценивали в отношении как проявлений со стороны желез, так и экстрагландулярных проявлений SjS, а также в отношении контроля лимфомы с различной степенью успеха. Однако в настоящее время данный подход не является одобренным лечением SjS. Недостаточная эффективность ритуксимаба могла быть связана с неполным истощением В-клеток в пораженных тканях(Brito-Zerón P et al (2016) Treating the Underlying Pathophysiology of Primary Sjögren Syndrome: Recent Advances and Future Prospects. Drugs p. 1601-1623).
Несмотря на доступное лечение SjS, остается высокая медицинская потребность в новых вариантах лечения для субъектов с SjS.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является обеспечение нового способа лечения заболевания, представляющего собой синдром Шегрена, у нуждающегося в таком лечении субъекта, предусматривающего введение указанному субъекту терапевтически эффективного количества N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида или его фармацевтически приемлемой соли.
Следовательно, в данном документе раскрыты способы лечения синдрома Шегрена (SjS), предусматривающие введение нуждающемуся в таком лечении субъекту суточной дозы, составляющей от приблизительно 0,5 мг до приблизительно 600 мг, предпочтительно суточной дозы, составляющей от приблизительно 10 мг до приблизительно 200 мг, или более предпочтительно дозы, составляющей от приблизительно 10 мг до приблизительно 100 мг N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида или его фармацевтически приемлемой соли.
Также раскрыты N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид или его фармацевтически приемлемая соль для применения в лечении SjS, где N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид или его фармацевтически приемлемую соль вводят в суточной дозе, составляющей от приблизительно 0,5 мг до приблизительно 600 мг, предпочтительно в суточной дозе, составляющей от приблизительно 10 мг до приблизительно 200 мг, и наиболее предпочтительно в суточной дозе, составляющей от приблизительно 10 мг до приблизительно 100 мг.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фигура 1. Концентрация соединения (I) в крови в зависимости от времени после введения однократных нарастающих доз в диапазоне 0,5 мг - 600 мг.
Фигура 2. Концентрация соединения (I) в крови в зависимости от времени после введения многократных нарастающих доз в диапазоне 10 мг - 400 мг по схеме q.d.
Фигура 3. Концентрация соединения (I) в крови в зависимости от времени после введения многократных нарастающих доз от 100 мг по схеме b.i.d. до 200 мг по схеме b.i.d.
Фигура 4. Влияние приема пищи, наблюдаемое после введения однократной пероральной дозы, содержащей 60 мг соединения формулы (I).
Фигура 5. Среднее арифметическое (SD) процента занятости BTK в периферической крови после введения однократной дозы соединения формулы (I).
Фигура 6. Медиана процента ингибирования активации базофилов в зависимости от общей суточной дозы соединения формулы (I) в день 12 введения многократных нарастающих доз соединения формулы (I).
Фигура 7. Уменьшение размера папулы при кожной инъекционной пробе с многократными нарастающими дозами
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Тирозинкиназа Брутона (BTK) является цитоплазматической тирозинкиназой и представителем семейства киназ TEC. BTK экспрессируется в клетках как адаптивной, так и врожденной иммунной системы, включая В-клетки, макрофаги, базофилы, тучные клетки и тромбоциты. BTK необходима для передачи сигналов через Fc-эпсилон-рецептор (FcεR1 для IgE) и активирующие Fc-гамма-рецепторы (FcγR для IgG), а также через B-клеточный антигенраспознающий рецептор (BCR). Было показано, что ингибирование BTK является эффективным подходом для лечения B-клеточных злокачественных новообразований. Ковалентные ингибиторы BTK, ибрутиниб (Имбрувика®), акалабрутиниб (Калквенс®) и занубрутиниб (Брукинза®) одобрены для лечения определенных типов B-клеточных злокачественных новообразований (Thompson PA, et al, (2018) Bruton's tyrosine kinase inhibitors: first and second generation agents for patients with Chronic Lymphocytic Leukemia (CLL). Expert Opin Investig Drugs p. 31-42). Ингибирование BTK продемонстрировало перспективную эффективность в отношении B-клеточных аутоиммунных реакций в доклинических и клинических исследованиях (Tan SL., et al, (2013) Targeting the SYK-BTK axis for the treatment of immunological and hematological disorders: recent progress and therapeutic perspectives. Pharmacol. Ther. p. 294-309; Whang J.A., et al. (2014) Bruton's tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Drug Discov. Today p. 1200-4; Satterthwaite A.B. (2017) Bruton's Tyrosine Kinase, a Component of B Cell Signaling Pathways, Has Multiple Roles in the Pathogenesis of Lupus. Front Immunol p. 1986; Rip J., et al, (2018) The Role of Bruton's Tyrosine Kinase in Immune Cell Signaling and Systemic Autoimmunity. Crit. Rev. Immunol. p. 17-62). Следовательно, ингибирование BTK является привлекательным терапевтическим подходом для лечения различных аутоиммунных и хронических воспалительных заболеваний, включая ревматоидный артрит, рассеянный склероз, системную красную волчанку, хроническую крапивницу, атопический дерматит, астму и первичный синдром Шегрена (Tan SL, Liao C, Lucas MC, et al (2013) Targeting the SYK-BTK axis for the treatment of immunological and hematological disorders: recent progress and therapeutic perspectives. Pharmacol. Ther. p. 294-309; Whang JA, Chang BY (2014) Bruton's tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Drug Discov. Today p. 1200-4).
Кроме того, было показано, что уровни BTK повышены в циркулирующих B-клетках у значительной процентной доли пациентов с SjS в сочетании с высокими уровнями ревматоидного фактора (RF) в сыворотке крови (Corneth OBJ et al. (2017) Enhanced Bruton's Tyrosine Kinase Activity in Peripheral Blood B Lymphocytes From Patients With Autoimmune Disease. p. 1313-1324).
N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид или его фармацевтически приемлемая соль представляют собой ингибитор BTK, называемый в данном документе соединением формулы (I):
 (I),
(I),
или его фармацевтически приемлемой солью.
Данное соединение было описано в заявке WO2015/079417, поданной 4 июня 2015 г. (номер дела патентного поверенного PAT056021-WO-PCT). Данное соединение является селективным, активным, необратимым ковалентным ингибитором тирозинкиназы Брутона (BTK) и может применяться при заболевании или нарушении, опосредованных BTK.
Соответственно, на текущий момент авторы настоящего изобретения разработали схемы введения доз для лечения пациентов с SjS с помощью N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида или его фармацевтически приемлемой соли.
Определения
Для целей толкования данного описания будут применяться следующие определения, и при необходимости термины, используемые в единственном числе, будут также включать множественное число, и наоборот.
Используемая в данном документе фраза "фармацевтически приемлемый" относится к тем соединениям, материалам, композициям и/или лекарственным формам, которые в рамках здравого медицинского суждения являются подходящими для применения в контакте с тканями людей и животных, не сопровождаются чрезмерной токсичностью, раздражением, аллергической реакцией или другими проблемами или осложнениями, соответствуют приемлемому соотношению пользы и риска.
Подразумевается, что любая формула, приведенная в данном документе, также представляет немеченые формы, а также изотопно-меченные формы соединений. Изотопно-меченные соединения имеют структуры, изображенные посредством формул, приведенных в данном документе, за исключением того, что один или несколько атомов заменены атомом, характеризующимся выбранными атомной массой или массовым числом. Изотопы, которые могут быть введены в соединение по настоящему изобретению, включают, например, изотопы водорода, углерода, азота, кислорода, фтора и хлора, такие как 3H, 11C, 13C, 14C, 15N, 18F и 36Cl. Следовательно, следует понимать, что в настоящее изобретение включено соединение, в которое введены один или несколько любых из вышеуказанных изотопов, включая, например, радиоактивные изотопы, такие как 3H и 14C, или соединения, в которых присутствуют нерадиоактивные изотопы, такие как 2H и 13C. Такие изотопно-меченные соединения применимы в исследованиях метаболизма (с использованием 14C), исследованиях кинетики реакций (с использованием, например, 2H или 3H), методиках выявления или визуализации, таких как позитронно-эмиссионная томография (PET) или однофотонная эмиссионная компьютерная томография (SPECT), в том числе в анализах распределения лекарственного средства или субстрата в тканях, или в лечении пациентов с помощью радиоактивных препаратов. В частности, меченое 18F соединение может быть особенно пригодным для исследований с помощью PET или SPECT. Обычно изотопно-меченные соединения можно получать с помощью традиционных методик, известных специалистам в данной области техники, например с использованием подходящих изотопно-меченных реагентов вместо немеченого реагента, используемого ранее.
Используемый в данном документе термин "фармацевтическая комбинация" означает продукт, который получают путем применения или смешивания или объединения более чем одного активного ингредиента. Следует понимать, что используемая в данном документе фармацевтическая комбинация включает как фиксированные, так и нефиксированные комбинации активных ингредиентов. Термин "фиксированная комбинация" означает, что активные ингредиенты, например соединение формулы (I) или его фармацевтически приемлемая соль и один или несколько партнеров по комбинации, вводятся пациенту одновременно в виде одного препарата или лекарственной формы. Термин в таком случае относится к комбинации с фиксированной дозой в одной стандартной лекарственной форме (например, капсуле, таблетке или саше). Оба термина "нефиксированная комбинация" или "набор частей" означают, что активные ингредиенты, например соединение по настоящему изобретению и один или несколько партнеров по комбинации и/или одно или несколько дополнительных средств, вводятся или совместно вводятся пациенту независимо в виде отдельных единиц либо одновременно, либо параллельно, либо последовательно без конкретных временных ограничений, где такое введение обеспечивает терапевтически эффективные уровни двух соединений в организме пациента, особенно если эти временные интервалы позволяют партнерам по комбинации продемонстрировать совместный, например аддитивный или синергический, эффект. Термин "нефиксированная комбинация" также применяется в отношении "коктейльной терапии", например к введению трех или более активных ингредиентов. Таким образом, термин "нефиксированная комбинация" определяет, в частности, введение, применение, композицию или состав в том смысле, что дозы соединения, описанного в данном документе, можно вводить независимо от других соединений, т. е. одновременно или в разные моменты времени. Следует понимать, что термин "нефиксированная комбинация" также охватывает применение отдельного средства вместе с одним или несколькими продуктами на основе фиксированной комбинации, при этом каждый независимый состав имеет различные количества активных ингредиентов, содержащихся в нем. Дополнительно следует понимать, что продукты на основе комбинации, описанные в данном документе, а также термин "нефиксированные комбинации" охватывают активные ингредиенты (включая соединения, описанные в данном документе), где партнеры по комбинации вводятся в виде полностью отдельных фармацевтических лекарственных форм или в виде фармацевтических составов, которые также продаются независимо друг от друга. Инструкции по применению нефиксированной комбинации находятся или могут быть предоставлены в упаковке, например в виде листка-вкладыша и т. п., или в виде другой информации, которая предоставляется врачам и/или медицинскому персоналу. Затем независимые составы или части состава, продукты или композиции можно вводить одновременно или через определенные промежутки времени, что означает, что каждую отдельную часть из набора частей можно вводить в различные моменты времени и/или с одинаковыми или различными временными интервалами в случае любой части из набора частей. В частности, временные интервалы для введения доз выбирают таким образом, чтобы воздействие на заболевание, в отношении которого осуществляют лечение, при комбинированном применении частей было больше/сильнее, чем воздействие, наблюдаемое при применении только соединения формулы (I); таким образом, соединения, применяемые в фармацевтической комбинации, описанной в данном документе, являются совместно активными. Соотношение общих количеств соединения формулы I и второго средства, подлежащих введению в виде фармацевтической комбинации, можно варьировать или корректировать для обеспечения лучшего соответствия потребностям определенной субпопуляции пациентов, подлежащей лечению, или потребностям одного пациента, которые могут быть обусловлены, например, возрастом, полом, массой тела и т. д. пациентов.
Подразумевается, что используемые в данном документе термины "совместное введение" или "комбинированное введение" и т.п. охватывают введение одного или нескольких соединений, описанных в данном документе, вместе с выбранным партнером по комбинации одному нуждающемуся в этом субъекту (например, пациенту или субъекту), и предполагается, что они включают схемы лечения, в которых соединения необязательно вводятся одним и тем же путем введения и/или в одно и то же время.
Определяемый в данном документе термин "фармацевтическая композиция" относится к смеси (например, раствору или эмульсии), содержащей по меньшей мере один активный ингредиент или терапевтическое средство, подлежащие введению теплокровному животному, например млекопитающему или человеку, для того, чтобы предупредить или лечить конкретное заболевание или состояние, поражающее теплокровное животное.
Термин "терапевтически эффективное количество" соединения (т. е. соединения формулы (I) или его фармацевтически приемлемой соли) по настоящему изобретению относится к количеству соединения по настоящему изобретению, которое будет вызывать биологический или медицинский ответ у субъекта (пациента или субъекта), например снижение или ингибирование активности фермента или белка, или ослаблять симптомы, облегчать состояния, замедлять или сдерживать прогрессирование заболевания, или предупреждать заболевание и т. д. Терапевтически эффективная дозировка соединения, фармацевтической композиции или их комбинаций зависит от биологического вида пациента, массы тела, возраста, пола и индивидуального состояния, нарушения или заболевания, лечение которых осуществляют, или их тяжести. Врач, клиницист или ветеринар средней квалификации может легко определить эффективное количество каждого из активных ингредиентов, необходимое для предупреждения, лечения или подавления прогрессирования нарушения или заболевания.
Частоту введения дозы можно менять в зависимости от используемого соединения и конкретного состояния, подлежащего лечению или предупреждению. В целом, предпочтительным является применение минимальной дозы, достаточной для обеспечения эффективной терапии. Мониторинг терапевтической эффективности у пациентов обычно может осуществляться с применением анализов, подходящих для состояния, подлежащего лечению или предупреждению, что будет хорошо известно специалистам средней квалификации в данной области техники.
Используемые в данном документе термины "носитель" или "фармацевтически приемлемый носитель" включают все возможные растворители, дисперсионные среды, покрытия, поверхностно-активные вещества, антиоксиданты, консерванты (например, антибактериальные средства, противогрибковые средства), средства для обеспечения изотоничности, замедляющие всасывание средства, соли, консерванты, лекарственные средства, стабилизаторы лекарственных средств, связующие средства, вспомогательные вещества, разрыхляющие средства, смазывающие вещества, подслащивающие вещества, ароматизирующие средства, красители и т. п., а также их комбинации, которые будут известны специалистам в данной области техники (см., например, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289-1329). За исключением случаев, когда традиционный носитель несовместим с активным ингредиентом, предполагается его применение в терапевтических или фармацевтических композициях.
Используемый в данном документе термин "субъект" относится к животному. Как правило, животное является млекопитающим. Субъект также относится, например, к приматам (например, людям, мужчинам или женщинам), коровам, овцам, козам, лошадям, собакам, кошкам, кроликам, крысам, мышам, рыбам, птицам и т. п. В определенных вариантах осуществления субъектом является примат. В предпочтительном варианте осуществления субъектом является человек. Термин "субъект" используется взаимозаменяемо с "пациентом", если он относится к человеку.
Используемый в данном документе субъект "нуждается в" лечении, если в результате такого лечения такой субъект получит пользу с биологической, медицинской точки зрения, или улучшится качество его жизни.
Используемая в данном документе фраза "популяция пациентов" означает группу пациентов.
Термин "содержащий" охватывает "включающий", а также "состоящий", например композиция, "содержащая" X, может состоять исключительно из X или может включать что-либо дополнительное, например X+Y.
Термин "приблизительно" по отношению к числовому значению х означает, например, +/-10%. В случае использования перед числовым диапазоном или перечнем чисел термин "приблизительно" применяется к каждому числу в ряду чисел, например, фразу "приблизительно 1-5" следует интерпретировать как "приблизительно 1 - приблизительно 5", или, например, фразу "приблизительно 1, 2, 3, 4" следует интерпретировать как "приблизительно 1, приблизительно 2, приблизительно 3, приблизительно 4 и т. д.".
Термин "лечение" или "лечить" в данном документе определяется как применение или введение соединения в соответствии с настоящим изобретением (соединения формулы (I), или его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей указанное соединение, субъекту или в выделенную ткань или линию клеток от субъекта, где у субъекта имеется конкретное заболевание (например, SjS), симптом, ассоциированный с заболеванием (например, SjS), или предрасположенность к развитию заболевания (например, SjS) (если применимо), при этом целью является излечение (если применимо), задержка начала развития, уменьшение тяжести, облегчение, ослабление одного или нескольких симптомов заболевания, улучшение течения заболевания, уменьшение или снижение проявления любых ассоциированных симптомов заболевания или предрасположенности к развитию заболевания. Термин "лечение" или "лечить" включает лечение пациента с подозрением на наличие заболевания, а также пациентов, у которых имеется заболевание, или пациентов, которым был поставлен диагноз наличия заболевания или медицинского состояния, и включает подавление клинического рецидива.
Используемые в данном документе термины "осуществление отбора" и "отобранный", используемые в отношении пациента, означают, что конкретный пациент специально выбран из большей группы пациентов на основании того (вследствие того), что конкретный пациент отвечает заранее определенным критериям. Подобным образом, "селективное лечение" относится к обеспечению лечением пациента, имеющего конкретное заболевание, при этом данный пациент специально выбран из большей группы пациентов на основании того, что конкретный пациент отвечает заранее определенному критерию. Подобным образом, "селективное введение" относится к введению лекарственного средства пациенту, который специально выбран из большей группы пациентов на основании того (вследствие того), что конкретный пациент отвечает заранее определенному критерию. Под "осуществлением отбора", "осуществлением селективного лечения" и "осуществлением селективного введения" подразумевают, что пациенту обеспечивается доставка персонализированной терапии на основании личного анамнеза пациента (например, предшествующих терапевтических вмешательств, например, предшествующего лечения биологическими препаратами), биологических показателей пациента (например, конкретных генетических маркеров) и/или проявления у пациента (например, несоответствия определенным диагностическим критериям), вместо применения стандартной схемы лечения, основанной исключительно на принадлежности пациента к большей группе. Используемый в данном документе отбор, применительно к способу лечения, не относится к случайному лечению пациента, отвечающего конкретному критерию, а скорее относится к взвешенному решению, принимаемому в отношении введения лекарственного средства пациенту на основании того, что пациент отвечает конкретному критерию. Таким образом, селективное лечение/введение отличается от стандартного лечения/введения, в ходе которого доставка конкретного лекарственного средства обеспечивается всем пациентам, имеющим конкретное заболевание, независимо от их личного анамнеза, проявлений заболевания и/или биологических особенностей. В некоторых вариантах осуществления, пациент был выбран для лечения на основании наличия SjS.
Варианты осуществления настоящего изобретения
Синдром Шегрена и эффективность лечения в соответствии с настоящим изобретением
Раскрытый ингибитор BTK, т. е. соединение формулы (I) или его фармацевтически приемлемую соль, можно применять in vitro, ex vivo или включать в фармацевтические композиции и вводить in vivo для лечения пациентов с SjS (например, пациентов-людей).
Эффективность лечения пациента с синдромом Шегрена можно оценивать с применением различных известных способов и инструментов, которые позволяют измерить состояние при синдроме Шегрена и/или клинический ответ пациента с синдромом Шегрена. Некоторые примеры включают, например, индекс активности заболевания при синдроме Шегрена согласно EULAR (ESSDAI), шкалу общей оценки врача (PhGA), индекс оценки, сообщаемой пациентом с синдромом Шегрена, согласно EULAR (ESSPRI), опросник функциональной оценки терапии хронической болезни-шкала утомляемости (FACIT-утомляемость) и EQ5D.
Эффективность
Измерения клинической эффективности в отношении первичных и вторичных целей указаны ниже.
Индекс активности заболевания при синдроме Шегрена согласно EULAR (ESSDAI)
ESSDAI является подтвержденной мерой результата лечения заболевания в случае синдрома Шегрена и применяется к субъектам исследования (Seror R, et al (2015) Validation of EULAR primary Sjögren's syndrome disease activity (ESSDAI) and patient indexes (ESSPRI). Ann. Rheum. Dis. p. 859-66). Инструмент содержит 12 доменов, характеризующих определенные органы, вносящих вклад в оценку активности заболевания. В случае каждого домена признакам активности заболевания начисляются баллы на 3 или 4 уровнях в зависимости от их тяжести. Затем эти баллы взвешенным образом суммируют по всем 12 доменам с получением общего балла. Домены (взвешенные значения) являются следующими: общее состояние (3), лимфаденопатия (4), проявление со стороны желез (2), проявление со стороны суставов (2), проявление со стороны кожи (3), проявление со стороны легких (5), проявление со стороны почек (5), проявление со стороны мышц (6), проявление со стороны ПНС (5), проявление со стороны ЦНС (5), гематологические показатели (2) и биологические показатели (1). Максимальный возможный балл составляет 123.
В исследовании авторов настоящего изобретения для расчета ESSDAI все 12 органных доменов должны оцениваться отдельно в каждый запланированный момент времени (от скринингового визита вплоть до конца исследования). Оценки доменов вводятся в таблицу (предоставляемую центральным поставщиком), а балл ESSDAI рассчитывается программным обеспечением.
Что касается оценок, не перечисленных в протоколе в качестве обязательных тестов, но которые могут потребоваться для оценки ESSDAI, включая рентгенографию, компьютерную томографию высокого разрешения (HRCT), тест легочной функции (DLCO, FVC), расчетную скорость клубочковой фильтрации (eGFR), электромиографию (EMG), мышечную (или любую другую) биопсию, то их оценка проводится на усмотрение исследователя, исходя из признаков и симптомов пациента, чтобы обеспечить получение правильных результатов ESSDAI. Индекс активности заболевания при синдроме Шегрена согласно EULAR (ESSDAI), домен и определения пунктов, а так же взвешенные значения обобщены в таблице 1
За исключением лихорадки инфекционного происхождения и преднамеренного снижения массы тела
Низкий=1
Средний=2
Легкая или перемежающаяся лихорадка (37,5-38,5°C)/ночная потливость и/или непреднамеренное снижение массы тела на 5-10% от массы тела;
Средняя лихорадка (>38,5°C)/ночная потливость и/или непреднамеренное снижение массы тела на >10% от массы тела
За исключением инфекции
Низкий=1
Средний=2
Высокий=3
Лимфаденопатия ≥1 см в области любого узла или ≥2 см в паховой области
Лимфаденопатия ≥2 см в области любого узла или ≥3 см в паховой области, и/или спленомегалия (клинически пальпируемая или оцениваемая посредством визуализации)
Текущее злокачественное В-клеточное пролиферативное нарушение
За исключением камней или инфекции
Низкий =1
Средний=2
Небольшой отек желез с увеличением околоушных желез (≤3 см) или ограниченный отек подчелюстных или слезных желез
Большой отек желез с увеличением околоушных желез (>3 см) или значительный отек подчелюстных или слезных желез
За исключением остеоартрита
Низкий=1
Средний=2
Высокий=3
Артралгии в кистях рук, запястьях, лодыжках и стопах, сопровождаемые утренней скованностью (>30 мин)
Синовит у 1-5 (из общего числа 28) суставов
Синовит у ≥6 (из общего числа 28) суставов
В качестве ‘отсутствия активности’ рассматриваются стабильные долгосрочные признаки, связанные с повреждением
Низкий=1
Средний=2
Высокий=3
Мультиформная эритема
Ограниченный кожный васкулит, включая уртикарный васкулит, или пурпура, ограниченная стопой и лодыжкой, или подострая кожная волчанка
Диффузный кожный васкулит, включая уртикарный васкулит, или диффузная пурпура, или язвы, связанные с васкулитом
В качестве ‘отсутствия активности’ рассматриваются стабильные долгосрочные признаки, связанные с повреждением, или поражение дыхательных путей, не связанные с заболеванием (употребление табака и т. д.)
Низкий=1
Средний=2
Высокий=3
Постоянный кашель или поражение бронхов с отсутствием рентгенологических нарушений на рентгенограмме или рентгенологическое подтверждение или подтверждение с помощью HRCT интерстициального заболевания легких с отсутствием одышки и нормальным результатом теста легочной функции
Умеренно активное поражение легких, такое как интерстициальное заболевание легких, продемонстрированное с помощью HRCT, с одышкой при физической нагрузке (NYHA II) или аномальными результатами тестов легочной функции, находящимися в пределах 70%>DLCO≥40% или 80%>FVC≥60%
Высокоактивное поражение легких, такое как интерстициальное заболевание легких, продемонстрированное с помощью HRCT, с одышкой в состоянии покоя (NHYA III, IV) или аномальными результатами тестов легочной функции, составляющими DLCO<40% или FVC<60%
В качестве ‘отсутствия активности’ рассматриваются стабильные долгосрочные признаки, связанные с повреждением, и поражение почек, не связанное с заболеванием.
В случае если была выполнена биопсия, следует вначале рассматривать активность, исходя из гистологических признаков
Низкий=1
Средний=2
Высокий=3
Подтверждение легкого активного поражения почек, ограниченного канальцевым ацидозом без почечной недостаточности, или поражение клубочков с протеинурией (от 0,5 до 1 г/день) и без гематурии или почечной недостаточности (GFR ≥60 мл/мин)
Умеренно активное поражение почек, такое как канальцевый ацидоз с почечной недостаточностью (GFR <60 мл/мин), или поражение клубочков с протеинурией от 1 до 1,5 г/день и без гематурии или почечной недостаточности (GFR ≥60 мл/мин), или гистологическое подтверждение экстрамембранозного гломерулонефрита, или существенный интерстициальный лимфоидный инфильтрат
Высокоактивное поражение почек, такое как поражение клубочков с протеинурией >1,5 г/день или гематурией или почечной недостаточностью (GFR <60 мл/мин), или гистологическое подтверждение пролиферативного гломерулонефрита, или криоглобулинемия, связанная с поражением почек
За исключением слабости, обусловленной кортикостероидами
Низкий=1
Средний=2
Высокий=3
Легкий активный миозит, продемонстрированный с помощью аномальных результатов EMG или биопсии, при отсутствии слабости и уровне креатинкиназы (N<CK≤2N)
Умеренный активный миозит, подтвержденный с помощью аномальных результатов EMG или биопсии, со слабостью (максимальный дефицит со значением 4/5) или повышенным уровнем креатинкиназы (2N<CK≤4N)
Высокоактивный миозит, продемонстрированный с помощью аномальной EMG или биопсии, со слабостью (дефицит со значением ≤3/5) или повышенным уровнем креатинкиназы (>4N)
В качестве ‘отсутствия активности’ рассматриваются стабильные долгосрочные признаки, связанные с повреждением, и поражение ПНС, не связанное с заболеванием
Низкий=1
Средний=2
Высокий=3
Легкое активное поражение периферической нервной системы, такое как исключительно сенсорная аксональная полинейропатия, продемонстрированная с помощью NCS, или невралгия тройничного нерва (V)
Умеренное активное поражение периферической нервной системы, продемонстрированное с помощью NCS, такое как аксональная сенсомоторная нейропатия с максимальным двигательным дефицитом со значением 4/5, исключительно сенсорная нейропатия с присутствием криоглобулинемического васкулита, ганглиопатия с симптомами, ограниченными легкой/умеренной атаксией, воспалительная демиелинизирующая полинейропатия (CIDP) с легким нарушением функции (максимальный двигательный дефицит со значением 4/5 или легкая атаксия) или поражение черепных нервов в периферической части (за исключением невралгии тройничного нерва (V))
Высокоактивное поражение ПНС, продемонстрированное с помощью NCS, такое как аксональная сенсомоторная нейропатия с двигательным дефицитом со значением ≤3/5, поражение периферических нервов вследствие васкулита (множественный мононеврит и т. п.), тяжелая атаксия вследствие ганглиопатии, воспалительная демиелинизирующая полинейропатия (CIDP) с тяжелым нарушением функции: двигательный дефект со значением ≤3/5 или тяжелая атаксия
В качестве ‘отсутствия активности’ рассматриваются стабильные долгосрочные признаки, связанные с повреждением, и поражение ЦНС, не связанное с заболеванием
Высокий=3
Высокоактивные признаки поражения ЦНС, такие как церебральный васкулит с острым нарушением мозгового кровообращения или транзиторной ишемической атакой, судорожные припадки, поперечный миелит, лимфоцитарный менингит, подобный рассеянному склерозу синдром с двигательным дефицитом
В случае анемии, нейтропении и тромбопении следует учитывать только аутоиммунную цитопению. Исключение составляет цитопения, вызванная дефицитом витаминов или железа или приемом лекарственных средств.
Низкий=1
Средний=2
Высокий=3
Цитопения аутоиммунного происхождения с нейтропенией (1000<нейтрофилов<1500/мм3), и/или анемией (10<гемоглобина<12 г/дл), и/или тромбоцитопенией (100000<тромбоцитов<150000/мм3), или лимфопения (500<лимфоцитов<1000/мм3)
Цитопения аутоиммунного происхождения с нейтропенией (500≤нейтрофилов≤1000/мм3), и/или анемией (8≤гемоглобина≤10 г/дл), и/или тромбоцитопенией (50000≤тромбоцитов≤100000/мм3), или лимфопения (≤500/мм3)
Цитопения аутоиммунного происхождения с нейтропенией (нейтрофилов <500/мм3), и/или анемией (гемоглобина <8 г/дл), и/или тромбоцитопенией (тромбоцитов <50000/мм3)
Низкий=1
Средний=2
Клональный компонент и/или гипокомплементемия (низкий уровень C4 или C3 или CH50) и/или гипергаммаглобулинемия или высокий уровень IgG от 16 до 20 г/л.
Присутствие криоглобулинемии и/или гипергаммаглобулинемии или высокого уровня IgG >20 г/л и/или недавнее возникновение гипогаммаглобулинемии или недавнее снижение уровня IgG (<5 г/л)
Шкала общей оценки врача (PhGA)
Исследователь использует шкалу общей оценки врача для измерения активности заболевания своего пациента с применением 100 мм VAS, имеющей диапазон от "отсутствия активности заболевания" (0) до "максимальной активности заболевания" (100).
Для повышения объективности врач не должен знать об оценках результата лечения, сообщаемых конкретным пациентом, при проведении своей собственной оценки данного пациента. Поэтому данная оценка должна быть выполнена до просмотра общей оценки пациента, представляющей собой общий балл активности заболевания.
Индекс оценки, сообщаемой пациентом с синдромом Шегрена, согласно EULAR (ESSPRI)
ESSPRI представляет собой общепринятую меру результата лечения заболевания в случае синдрома Шегрена (Seror R, et al (2011) EULAR Sjögren 's Syndrome Patient Reported Index (ESSPRI): development of a consensus patient index for primary Sjögren 's syndrome. Ann. Rheum. Dis. p. 968-72). Он состоит из трех доменов: сухость, боль и утомляемость. Субъект может оценивать тяжесть симптомов, которые он испытывает, по единой числовой шкале 0-10 для каждого из трех доменов. Балл ESSPRI определяется как среднее значение баллов по трем шкалам: (сухость+боль+утомляемость) /3.
FACIT-утомляемость
Опросник функциональной оценки терапии хронической болезни-шкала утомляемости (FACIT-F, версия 4) представляет собой короткий простой в осуществлении инструмент, состоящий из 13 пунктов, с помощью которого измеряют уровень утомляемости индивидуума во время его обычной повседневной деятельности за последнюю неделю. Уровень утомляемости измеряют по 5-балльной шкале Ликерта (0=полное отсутствие, 1=небольшая, 2=в некоторой степени, 3=значительная, 4=очень сильная) (Webster K, et al. (2003) The Functional Assessment of Chronic Illness Therapy (FACIT) Measurement System: properties, applications, and interpretation. Health Qual Life Outcomes p. 79).
EQ5D
EQ-5D представляет собой стандартизированный инструмент, с помощью которого измеряют качество жизни, связанное со здоровьем. EQ-5D состоит из описательной системы и шкалы EQ VAS.
Описательная система содержит пять измерений: подвижность, самообслуживание, повседневная деятельность, боль/дискомфорт и тревога/депрессия. Ее можно применять в качестве количественной меры результата лечения, которая отражает собственное суждение пациента. Баллы по этим пяти измерениям могут быть представлены в виде профиля состояния здоровья или могут быть преобразованы в единый суммарный числовой индекс (индекс оценки общего состояния здоровья), являющийся предпочтительным по сравнению с другими профилями состояния здоровья.
С помощью EQ VAS регистрируют состояние здоровья согласно самооценке пациента на вертикальной визуальной аналоговой шкале, где 0 представляет 'наихудшее состояние здоровья, которое можно себе представить', а 100 представляет 'Наилучшее состояние здоровья, которое можно себе представить'.
Пригодность оценок эффективности
В данном исследовании меры эффективность в первую очередь основывались на ESSDAI (индекс активности заболевания при SS согласно EULAR), измеряющем органоспецифические критерии заболевания, и на ESSPRI (индекс оценки, сообщаемой пациентом с синдромом Шегрена [SS], согласно Европейской лиге по борьбе с ревматизмом [EULAR]), измеряющем субъективную оценку воздействия на пациента. Оба инструмента являются общепризнанными и подтвержденными, золотыми стандартами мер для системных и симптоматических проявлений SjS соответственно.
ESSDAI представляет собой системный индекс активности заболевания, с помощью которого активность заболевания классифицируют по 3-4 уровням в каждой из 12 доменов с дифференциальным образом взвешенными показателями (биологические показатели, гематологические показатели, проявление со стороны суставов, проявление со стороны желез, проявление со стороны кожи, общее состояние, лимфаденопатия, проявление со стороны почек, проявление со стороны легких, проявление со стороны ПНС, проявление со стороны ЦНС и проявление со стороны мышц). Сводный взвешенный балл обеспечивает точную оценку активности заболевания с надлежащей чувствительностью к изменениям, что подтверждено в исследованиях на нескольких когортах (Seror R et al (2015) Validation of EULAR primary Sjögren's syndrome disease activity (ESSDAI) and patient indexes (ESSPRI). Ann. Rheum. Dis. p. 859-66). С другой стороны инструмент ESSPRI представляет собой совокупный балл оценки, сообщаемой пациентом, которая оценивает симптомы сухости, боли в конечностях и утомляемости по визуальной аналоговой шкале 0-10 в течение предыдущих 2 недель (Seror R et al (2011) EULAR Sjögren 's Syndrome Patient Reported Index (ESSPRI): development of a consensus patient index for primary Sjögren 's syndrome. Ann. Rheum. Dis. p. 968-72). Баллы, сообщаемые пациентом, характеризуются неудовлетворительной чувствительностью к изменению активности заболевания, но среди доступных инструментов, отмечалось, что ESSPRI характеризуется значительно лучшей чувствительностью. В недавнем проспективном исследовании отмечали слабую корреляцию между системными баллами и баллами от пациентов, что позволяет предположить, что два индекса оценивают взаимодополняющие компоненты активности заболевания, следовательно подчеркивая важность оценки обоих параметров для получения точной оценки активности заболевания и ее изменения (Seror R et al (2015) Validation of EULAR primary Sjögren's syndrome disease activity (ESSDAI) and patient indexes (ESSPRI). Ann. Rheum. Dis. p. 859-66).
Фармацевтическая композиция
Ингибитор BTK, т. е. соединение формулы (I) или его фармацевтически приемлемую соль, можно использовать в качестве фармацевтической композиции в комбинации с фармацевтически приемлемым носителем. Такая композиция может содержать, в дополнение к соединению формулы (I), носители, различные разбавители, наполнители, соли, буферы, стабилизаторы, солюбилизаторы и другие материалы, известные из уровня техники. Характеристики носителя зависят от пути введения. Фармацевтические композиции для применения в раскрытых способах также могут содержать дополнительные терапевтические средства для лечения конкретного целевого нарушения. Например, фармацевтическая композиция может также включать противовоспалительные и противозудные средства. Такие дополнительные факторы и/или средства могут быть включены в фармацевтическую композицию для получения синергического эффекта с соединением формулы (I) или для сведению к минимуму побочных эффектов, вызванных соединением формулы (I). В предпочтительных вариантах осуществления фармацевтическая композиция для применения в раскрытых способах содержит соединение формулы (I) в дозе, составляющей 10 мг, 20 мг, 25 мг, 50 мг или приблизительно 100 мг.
Подходящие композиции для перорального введения включают эффективное количество соединения по настоящему изобретению, находящееся в форме таблеток, пастилок для рассасывания, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул, или сиропов, или настоек. Композиции, предназначенные для перорального применения, получают в соответствии с любым способом, известным в данной области техники для изготовления фармацевтических композиций, и такие композиции могут содержать одно или несколько средств, выбранных из группы, состоящей из подслащивающих веществ, ароматизирующих средств, красящих средств и консервантов, с целью получения препаратов, которые являются фармацевтически эстетичными и приятными на вкус. Таблетки могут содержать активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми вспомогательными веществами, которые являются подходящими для изготовления таблеток. Такие вспомогательные вещества представляют собой, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и разрыхляющие средства, например кукурузный крахмал или альгиновую кислоту; связующие средства, например крахмал, желатин или аравийскую камедь; и смазывающие средства, например стеарат магния, стеариновую кислоту или тальк. Таблетки являются непокрытыми или покрытыми посредством известных методик для замедления распада и всасывания в желудочно-кишечном тракте, и за счет чего обеспечивается устойчивое действие в течение более длительного периода. Например, для обеспечения замедленного действия можно использовать такой материал, как глицерилмоностеарат или глицерилдистеарат. Составы для перорального применения могут быть представлены в виде твердых желатиновых капсул, где активный ингредиент смешан с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где активный ингредиент смешан с водой или масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом.
Фармацевтические композиции для применения в раскрытых способах можно изготавливать обычным способом. В одном варианте осуществления фармацевтическая композиция предусмотрена для перорального введения. Например, фармацевтические композиции представляют собой таблетки или желатиновые капсулы, содержащие активный ингредиент вместе с
a) разбавителями, например лактозой, декстрозой, сахарозой, маннитом, сорбитом, целлюлозой и/или глицином;
b) смазывающими веществами, например диоксидом кремния, тальком, стеариновой кислотой, ее магниевой или кальциевой солью и/или полиэтиленгликолем; в случае таблеток также со
c) связующими средствами, например алюмосиликатом магния, крахмальной пастой, желатином, трагакантом, метилцеллюлозой, карбоксиметилцеллюлозой натрия и/или поливинилпирролидоном; при необходимости
d) разрыхлителями, например видами крахмала, агаром, альгиновой кислотой или ее натриевой солью или шипучими смесями; и/или
e) абсорбентами, красящими веществами, ароматизаторами и подсластителями.
Таблетки могут быть покрыты либо пленочной оболочкой, либо кишечнорастворимой оболочкой в соответствии со способами, известными в данной области техники.
Комбинации
При практическом применении некоторых способов лечения или вариантов применения настоящего изобретения терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли вводится пациенту, например млекопитающему (например, человеку). Хотя понятно, что раскрытые способы предусматривают лечение пациентов с синдромом Шегрена с применением соединения формулы (I) или его фармацевтически приемлемой соли, терапия не обязательно является монотерапией. Действительно, если пациент выбран для лечения с помощью соединения формулы (I), то соединение формулы (I) или его фармацевтически приемлемую соль можно вводить в соответствии со способами по настоящему изобретению либо отдельно, либо в комбинации с другими средствами и видами терапии для лечения пациентов с синдромом Шегрена, например, в комбинации с по меньшей мере одним дополнительным средством для лечения синдрома Шегрена. При совместном введении с одним или несколькими дополнительными средствами для лечения SjS соединение формулы (I) или его фармацевтически приемлемую соль можно вводить либо одновременно с другим средством, либо последовательно. При последовательном введении лечащий врач примет решение относительно соответствующей последовательности введения соединения формулы (I) или его фармацевтически приемлемой соли в комбинации с другими средствами и соответствующих дозировках для совместной доставки.
В ходе лечения SjS различные виды терапии можно объединять с раскрытым соединением формулы (I) или его фармацевтически приемлемой солью с обеспечением благоприятного эффекта. Такие виды терапии включают стероиды (кортикостероид, такой как преднизон или его эквивалент); DMARDS, такие как, например, гидроксихлорохин (Плаквенил), метотрексат (Трексалл), сульфасалазин (Азульфидин), миноциклин (Миноцин) или лефлуномид (Арава) или лекарственное средство, истощающее В-клетки, такое как ритуксимаб.
Квалифицированный специалист в данной области техники сможет определить соответствующие дозы вышеуказанных средств для лечения SjS для совместной доставки с раскрытыми соединением формулы (I) или его фармацевтически приемлемой солью.
Наборы по настоящему изобретению
Настоящее изобретение также охватывает наборы для лечения SjS. Такие наборы содержат ингибитор BTK, например N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид или фармацевтическую композицию на его основе. Дополнительно такие наборы могут содержать инструкции по применению.
В одном варианте осуществления набор содержит две или более отдельные фармацевтические композиции, по меньшей мере одна из которых содержит соединение формулы (I) или его фармацевтически приемлемую соль. В одном варианте осуществления набор содержит средства для раздельного содержания указанных композиций, такие как контейнер, разделенная бутылка или разделенный пакет из фольги. Примером такого набора является блистерная упаковка, как правило, применяемая для упаковывания таблеток, капсул и т. п.
Набор по настоящему изобретению можно применять для введения различных лекарственных форм, например для перорального и парентерального применения, для введения отдельных композиций с различными интервалами между введениями доз или для подбора доз отдельных композиций относительно друг друга. Для содействия соблюдению режима лечения набор по настоящему изобретению, как правило, содержит инструкции по введению.
В видах комбинированной терапии по настоящему изобретению соединение формулы (I) или его фармацевтически приемлемая соль и другое средство для лечения SjS (как определено в данном документе) могут быть изготовлены и/или составлены одним и тем же или различными производителями. Более того, соединение формулы (I) или его фармацевтически приемлемая соль и другое средство для лечения SjS могут быть объединены с получением средства комбинированной терапии: (i) до того, как комбинированный продукт попадет к врачам (например, в случае набора, содержащего соединение формулы (I) или его фармацевтически приемлемую соль и другое средство для лечения ); (ii) самими врачами (или под руководством врача) незадолго до введения; (iii) самими пациентами, например во время последовательного введения соединения формулы (I) или его фармацевтически приемлемой соли и другого средства для лечения SjS.
Дополнительные варианты осуществления
Соединение формулы (I) или его фармацевтически приемлемая соль в целях удобства вводятся пациенту (предпочтительно перорально) в дозе, составляющей от приблизительно 10 мг до приблизительно 200 мг в сутки.
Соединение формулы (I) или его фармацевтически приемлемая соль в целях удобства вводятся пациенту (предпочтительно перорально) в суточной дозе, составляющей от приблизительно 10 мг до приблизительно 200 мг в сутки.
В некоторых вариантах осуществления соединение формулы (I) или его фармацевтически приемлемая соль вводятся в суточной дозе, составляющей от приблизительно 10 мг до приблизительно 100 мг.
В других вариантах осуществления соединение формулы (I) или его фармацевтически приемлемая соль вводятся в суточной дозе, составляющей приблизительно 100 мг.
В других вариантах осуществления соединение формулы (I) или его фармацевтически приемлемая соль вводятся в суточной дозе, составляющей приблизительно 50 мг.
В других вариантах осуществления соединение формулы (I) или его фармацевтически приемлемая соль вводятся в суточной дозе, составляющей приблизительно 35 мг.
В других вариантах осуществления соединение формулы (I) или его фармацевтически приемлемая соль вводятся в суточной дозе, составляющей приблизительно 25 мг.
В других вариантах осуществления соединение формулы (I) или его фармацевтически приемлемая соль вводятся в суточной дозе, составляющей приблизительно 20 мг.
В одном варианте осуществления соединение формулы (I) или его фармацевтически приемлемая соль вводятся один раз в сутки в дозе, составляющей приблизительно 10 мг, приблизительно 35 мг, приблизительно 50 мг или приблизительно 100 мг.
В другом варианте осуществления соединение формулы (I) или его фармацевтически приемлемую соль вводят два раза в сутки в дозе, составляющей приблизительно 10 мг, приблизительно 25 мг, приблизительно 50 мг или приблизительно 100 мг.
Будет понятно, что для определенных пациентов может потребоваться повышение дозы, например для пациентов с синдромом Шегрена, у которых проявляется недостаточный ответ (например, как измерено с помощью любой из систем оценки синдрома Шегрена, раскрытых в данном документе). Также будет понятно, что для определенных пациентов может потребоваться снижение дозы, например для пациентов с синдромом Шегрена, у которых проявляются нежелательные явления или нежелательная реакция на лечение с помощью соединения формулы (I) или его фармацевтически приемлемой соли. Таким образом, дозировки соединения формулы (I) или его фармацевтически приемлемой соли могут составлять менее приблизительно 10 мг, приблизительно 20 мг, приблизительно 25 мг, приблизительно 50 мг или приблизительно 100 мг.
Сроки введения доз обычно отмеряют от дня введения первой дозы соединения формулы (I) или его фармацевтически приемлемой соли (который также известен как "день определения исходного уровня"). Сроки введения доз обычно отмеряют от дня введения первой дозы соединения формулы (I) или его фармацевтически приемлемой соли (который также известен как "день определения исходного уровня").
Однако медицинские работники зачастую используют разные правила наименования для идентификации графиков введения доз. Для обеспечения ясности, как раскрыто в данном документе, первый день введения доз называется день 1. Однако квалифицированному специалисту в данной области техники будет понятно, что этот способ наименования используется просто для обеспечения соответствия и не должен рассматриваться как ограничивающий, т. е. ежедневное введение доз представляет собой обеспечение ежедневной дозы соединения формулы (I) или его фармацевтически приемлемой соли, и врач может ссылаться на конкретный день как на "день 0" или "день 1".
В данном документе раскрыты способы лечения заболевания, представляющего собой синдром Шегрена (SjS), предусматривающие введение нуждающемуся в этом пациенту соединения формулы (I) или его фармацевтически приемлемой соли в дозе, составляющей от приблизительно 10 мг до приблизительно 200 мг.
В данном документе также раскрыты способы лечения заболевания, представляющего собой синдром Шегрена (SjS), предусматривающие введение нуждающемуся в этом пациенту соединения формулы (I) или его фармацевтически приемлемой соли в суточной дозе, составляющей от приблизительно 10 мг до приблизительно 200 мг.
В данном документе также раскрыто соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении SjS, где суточная доза соединения составляет от приблизительно 10 мг до приблизительно 200 мг.
В одном варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемая соль вводятся в суточной дозе, составляющей от приблизительно 10 мг до приблизительно 100 мг.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемая соль вводятся в суточной дозе, составляющей приблизительно 10 мг, приблизительно 20 мг, приблизительно 25 мг, приблизительно 35 мг, приблизительно 50 мг, приблизительно 100 мг или приблизительно 200 мг.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемая соль вводятся в суточной дозе, составляющей приблизительно 100 мг.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемая соль вводятся в суточной дозе, составляющей приблизительно 50 мг.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемая соль вводятся в суточной дозе, составляющей приблизительно 35 мг.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемая соль вводятся в суточной дозе, составляющей приблизительно 25 мг.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемая соль вводятся в суточной дозе, составляющей приблизительно 20 мг.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемая соль вводятся один раз в сутки в дозе, составляющей приблизительно 10 мг, приблизительно 35 мг, приблизительно 50 мг или приблизительно 100 мг.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов соединение формулы (I) или его фармацевтически приемлемая соль вводятся в дозе, составляющей приблизительно 10 мг, приблизительно 25 мг, приблизительно 50 мг или приблизительно 100 мг два раза в день.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов у пациента имеется SjS со степенью тяжести от средней до тяжелой. Пациента с SjS со степенью тяжести от средней до тяжелой определяют как пациента, у которого до лечения с помощью соединения формулы (I) или его фармацевтически приемлемой соли балл ESSDAI (на основе взвешенного балла, как показано в таблице 1) составлял ≥ 5 (т. е. по меньшей мере 5) на основе 8 определенных доменов (биологические показатели, гематологические показатели, проявление со стороны суставов, проявление со стороны кожи, проявление со стороны желез, лимфаденопатия, проявление со стороны почек, общее состояние), а балл ESSPRI составлял по меньшей мере 5.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов пациент является взрослым.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов к неделе 12 или к неделе 24 лечения у пациента достигается изменение от исходного уровня по меньшей мере одного из результатов лечения, сообщаемых пациентом и/или врачом (т. е. ESSPRI, FACIT-F, EQ-5D, PhGA).
В другом варианте осуществления раскрытых способов, вариантов применения и наборов к неделе 12 или к неделе 24 лечения у пациента достигается изменение от исходного уровня балла ESSPRI.
В другом варианте осуществления раскрытых способов, вариантов применений и наборов к неделе 12 или к неделе 24 лечения у пациента достигается снижение балла ESSPRI.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов к неделе 12 или к неделе 24 лечения у пациента достигается снижение балла ESSPRI по меньшей мере на один пункт, предпочтительно на по меньшей мере 2 пункта.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов к неделе 12 или к неделе 24 лечения у пациента достигается снижение балла ESSPRI. В другом варианте осуществления раскрытых способов, вариантов применения и наборов к неделе 12 или к неделе 24 лечения у пациента достигается снижение балла ESSPRI, составляющее по меньшей мере 15%, по меньшей мере 25%, по меньшей мере 35%, по меньшей мере 50% или по меньшей мере 60%. Снижение балла ESSPRI рассчитывается следующим образом:

В другом варианте осуществления раскрытых способов, вариантов применения и наборов к неделе 12 или к неделе 24 лечения у пациента достигается снижение балла ESSDAI.
В еще одном варианте осуществления раскрытых способов, вариантов применения и наборов к неделе 12 или к неделе 24 лечения у пациента достигается снижение балла ESSDAI на по меньшей мере 3 пункта.
В другом варианте осуществления раскрытых способов, вариантов применения и наборов к неделе 12 или к неделе 24 лечения у пациента достигается изменение от исходного уровня балла ESSDAI.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов пациент является взрослым. В некоторых вариантах осуществления раскрытых способов, вариантов применения и наборов пациент является подростком.
Дополнительные пронумерованные варианты осуществления
1. Способ лечения синдрома Шегрена (SjS), предусматривающий введение нуждающемуся в этом субъекту суточной дозы, составляющей от приблизительно 10 мг до приблизительно 200 мг N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида или его фармацевтически приемлемой соли.
2. Способ в соответствии с вариантом осуществления 1, где суточная доза N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида или его фармацевтически приемлемой соли составляет от приблизительно 10 мг до приблизительно 100 мг.
3. Способ в соответствии с вариантом осуществления 1, где суточная доза N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида или его фармацевтически приемлемой соли составляет приблизительно 100 мг.
4. Способ в соответствии с вариантом осуществления 1, где суточная доза N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида или его фармацевтически приемлемой соли составляет приблизительно 50 мг.
5. Способ в соответствии с вариантом осуществления 1, где суточная доза N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида или его фармацевтически приемлемой соли составляет приблизительно 35 мг.
5. Способ в соответствии с вариантом осуществления 1, где суточная доза N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида или его фармацевтически приемлемой соли составляет приблизительно 25 мг.
7. Способ в соответствии с вариантом осуществления 1, где суточная доза N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамида или его фармацевтически приемлемой соли составляет приблизительно 20 мг.
8. Способ в соответствии с вариантом осуществления 1, где N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид или его фармацевтически приемлемую соль вводят один раз в сутки в дозе, составляющей приблизительно 10 мг, приблизительно 35 мг, приблизительно 50 мг или приблизительно 100 мг.
9. Способ в соответствии с вариантом осуществления 1, где N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид или его фармацевтически приемлемую соль вводят в дозе, составляющей приблизительно 10 мг, приблизительно 25 мг, приблизительно 50 мг или приблизительно 100 мг, два раза в сутки.
10. Способ в соответствии с любым из предыдущих вариантов осуществления, где у субъекта имеется SjS со степенью тяжести от средней до тяжелой.
11. Способ в соответствии с любым из вариантов осуществления 1-10, где субъект выбран в соответствии с по меньшей мере одним из следующих критериев:
а) до лечения с помощью соединения формулы (I) или его фармацевтически приемлемой соли субъект имеет балл ESSPRI, составляющий 5 или больше;
b) до лечения с помощью соединения формулы (I) или его фармацевтически приемлемой соли субъект имеет балл ESSDAI, составляющий 5 или больше, на основе взвешенного балла по 8 определенным доменам, выбранным из биологических показателей, гематологических показателей, проявления со стороны суставов, проявления со стороны кожи, проявления со стороны желез, лимфаденопатии, проявления со стороны почек и общего состояния.
12. Способ в соответствии с любым из предыдущих вариантов осуществления, где субъект является взрослым.
13. Способ в соответствии с любым из предыдущих вариантов осуществления, где к неделе 12 или к неделе 24 лечения у указанного субъекта достигается по меньшей мере одно из следующего:
a) снижение балла ESSPRI и/или
b) снижение балла ESSDAI.
14. Способ в соответствии с любым из вышеперечисленных вариантов осуществления, где на неделе 5 после завершения лечения у указанного субъекта достигается устойчивый ответ, как измерено с помощью ESSPRI или ESSDAI.
15. Способ в соответствии с любым из предыдущих вариантов осуществления, где N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид или его фармацевтически приемлемая соль помещены в фармацевтический состав, где указанный фармацевтический состав дополнительно содержит фармацевтически приемлемые носители.
16. Способ в соответствии с любым из вариантов осуществления 1-15, где N-(3-(6-амино-5-(2-(N-метилакриламидо)этокси)пиримидин-4-ил)-5-фтор-2-метилфенил)-4-циклопропил-2-фторбензамид или его фармацевтически приемлемая соль или его фармацевтически приемлемая соль характеризуются Tmax, составляющей приблизительно 0,5-3 часа.
Подробности одного или нескольких вариантов осуществления настоящего изобретения приведены в сопутствующем вышеизложенном описании. Хотя при осуществлении на практике или тестировании настоящего изобретения могут применяться любые способы и материалы, аналогичные или эквивалентные тем, которые описаны в настоящем изобретении, в данный момент описаны предпочтительные способы и материалы. Другие признаки, объекты и преимущества настоящего изобретения будут очевидны из описания и из формулы изобретения. В описании и прилагаемой формуле изобретения формы единственного числа включают ссылки на множественное число, если контекст явно не предписывает иное. Если не указано иное, то все технические и научные термины, используемые в данном документе, имеют то же значение, которое обычно понятно специалисту средней квалификации в области техники, к которой относится настоящее изобретение. Все патенты и публикации, процитированные в данном описании, включены в него посредством ссылки. Следующие примеры представлены для того, чтобы более полно проиллюстрировать предпочтительные варианты осуществления настоящего изобретения. Данные примеры никоим образом не следует истолковывать как ограничивающие объем раскрытого объекта изобретения, определяемого в прилагаемой формуле изобретения.
Сокращения
AE нежелательное явление
AUC площадь под кривой
AUCinf площадь под кривой зависимости концентрации в плазме крови (или сыворотке крови, или крови) от времени, начиная с нулевого момента времени и до бесконечности (масса x время/объем)
AUClast площадь под кривой зависимости концентрации в плазме крови (или сыворотки крови, или крови) от времени,
начиная с нулевого момента времени и до момента времени, соответствующего последнему значению концентрации, поддающемуся количественному определению (масса x время/объем)
AUCtau площадь под кривой зависимости концентрации в плазме крови (или сыворотке крови, или крови) от времени, начиная с нулевого момента времени и до конца интервала введения доз тау (масса x время/объем)
BCR В-клеточный рецептор
Bid или b.i.d. два раза в день (с латыни: "bis in die")
BMI индекс массы тела
BTK тирозинкиназа Брутона
CBC общий клинический анализ крови
см сантиметр
CL/F кажущийся системный (или общий организменный) клиренс из плазмы крови (или сыворотки крови, или крови)
после введения (масса/объем)
ЦНС центральная нервная система
CV коэффициент вариации
DMARD болезнь-модифицирующие противоревматические лекарственные средства
ЭКГ электрокардиограмма
eGFR расчетная скорость клубочковой фильтрации
ELISA твердофазный иммуноферментный анализ
EMG электромиография
EQ-5D 5 измерений EuroQual (стандартный инструмент для измерения качества жизни, связанного со здоровьем)
ESSDAI индекс активности заболевания при синдроме Шегрена согласно EULAR
ESSPRI индекс, сообщаемый пациентами с синдромом Шегрена, согласно EULAR.
EULAR Европейская лига по борьбе с ревматизмом
FACIT-F опросник функциональной оценки терапии хронической болезни-шкала утомляемости
FIH первое применение у человека
ч. час
HRCT компьютерная томография высокого разрешения
i.v. внутривенный
IA промежуточный анализ
INR международный коэффициент нормализации
кг килограмм
LC-MS/MS жидкостная хроматография/масс-спектрометрия-масс-спектрометрия
mAb моноклональное антитело
MCP-Mod процедура множественных сравнений - моделирование
MMRM модель со смешанными эффектами для повторных измерений
MRT среднее время удержания
NOAC новый антикоагулянт для перорального введения
NSAID нестероидное противовоспалительное лекарственное средство
PD фармакодинамический(-ие) параметр(-ы)
PhGA шкала общей оценки врача
PK фармакокинетический(-ие) параметр(-ы)
ПНС периферическая нервная система
PT протромбиновое время
PTT частичное тромбопластиновое время
Qd или q.d. один раз в день (с латыни "quaque die")
QTcF интервал QT, скорректированный по формуле Фридеричиа
Racc коэффициент накопления лекарственного средства
SAE серьезное нежелательное явление
SjS синдром Шегрена
SOM руководство по организации работы исследовательского центра
SPT кожная инъекционная проба
SS выборка для оценки безопасности
TEC тирозиновая протеинкиназа
Vz/F кажущийся объем распределения во время терминальной фазы выведения
после введения (объем)
Пример 1. Доклинические исследования
Пример 1a. Взаимосвязь занятости BTK и PK/PD в доклинических исследованиях
PD-эффекты in vivo необратимого ингибитора BTK, такого как соединение (I), определяются степенью и продолжительностью ковалентной занятости BTK ингибитором. Занятость BTK после обработки соединением формулы (I) (также называемым соединением (I)) измеряли с помощью иммунологического анализа ex vivo. Фракцию незанятого белка BTK анализировали после инкубации in vitro с ковалентным биотинилированным зондом для BTK, поскольку соединение (I) и зонд связываются с BTK взаимоисключающим образом. Незанятый белок BTK, а также относительные уровни общего белка BTK определяли в лизатах выбранных тканей, и уровни незанятого белка BTK нормализовали относительно уровней общего белка BTK в тех же образцах.
У самок крыс однократная пероральная доза 3 мг/кг соединения (I) приводила к полной занятости BTK в селезенке, доза 1 мг/кг приводила к занятости 76%-81%, тогда как после однократной дозы 0,3 мг/кг достигалась только частичная занятость, составляющая 30%. Занятость BTK в крови достигла уровней, совпадающих с наблюдаемыми в селезенке уровнями. Из экспериментальных данных было очевидно, что кратковременное присутствие соединения (I) в системном кровотоке достаточно для достижения полной занятости BTK в нескольких тканях при низких пероральных дозах 1-3 мг/кг. Содержание соединения (I) в крови после введения дозы 1 мг/кг достигало 49,1 нМ через 0,5 часа и составляло 5,6 нМ через 5 часов после введения дозы. Такое кратковременное присутствие в системном кровотоке в очень низком количестве согласуется с типичной моделью PK/PD для необратимых ингибиторов.
Продолжительность занятости BTK для селезенки, крови, лимфатических узлов и легких определяли у крыс и мышей после введения однократной пероральной дозы соединения (I). У крыс для занятости BTK продемонстрирован длинный период полужизни в крови, составляющий примерно 87 часов. Расчетный период полужизни для занятости BTK в селезенке крысы значительно короче, чем в крови, и составляет всего примерно 5 часов. Различные скорости обновления может отражать тот факт, что В-клетки и моноциты, экспрессирующие BTK, находятся в периферической крови в состоянии покоя и метаболически относительно неактивны по сравнению с таковыми в селезенке. Ранее сообщалось о более длительной продолжительности занятости BTK в крови (Advani et al 2013, J Clin Onc; 31(1):88-94). Для всех остальных проанализированных тканей (легкие и лимфатические узлы) показано обновление BTK и период полужизни для занятости, подобные таковым в селезенке.
Поскольку уровни BTK-экспрессирующих клеток в коже были слишком низкими, чтобы обеспечить измерение занятости, продолжительность PD-эффекта в коже после однократной дозы оценивали на модели обратной пассивной реакции Артюса (RPA) с воспалением, опосредованным FcγRIII тучных клеток. В данной модели ингибирование отека кожи было максимальным, если соединение (I) вводили за 2 часа до индуцирования реакции Артюса. Эффект постепенно уменьшался и достигал исходного уровня, если реакцию Артюса запускали через 45 часов или позже после введения дозы соединения (I). Это указывает на то, что занятость BTK в коже характеризуется динамикой, подобной таковой в селезенке, легких и лимфатических узлах.
В данных доклинических фармакологических исследованиях занятость BTK и соответствующие фармакологические результаты продемонстрировали сильную корреляцию. Таким образом, занятость BTK является подходящим биомаркером PD для применения в клинических исследованиях и, следовательно, его использовали в клинических исследованиях фазы 1.
Пример 2. Клиническое испытание фазы 1
Исследование первого применения у человека проводили для оценки безопасности и переносимости, фармакокинетики (PK) и фармакодинамики (PD) однократной и многократных доз соединения (I) при пероральном введении как один раз в сутки (qd), так и два раза в сутки (bid) у здоровых добровольцев и пациентов с атопическим диатезом для поддержки дальнейшей клинической разработки соединения (I) при аутоиммунных заболеваниях. В данном исследовании также изучали влияние приема пищи.
Исследование первого применения у человека с участием до примерно 168 здоровых добровольцев (HV), из которых у 64 (в частях 2 и 4) имелся бессимптомный атопический диатез.
• Часть 1 представляла собой двойное слепое (для субъекта и исследователя данные были замаскированы, для спонсора - нет) плацебо-контролируемое исследование с повышением дозы с использованием однократных нарастающих доз (SAD) с участием 10 когорт (N=80).
• Часть 2 представляла собой двойное слепое (для субъекта и исследователя данные были замаскированы, для спонсора - нет), плацебо-контролируемое исследование с повышением дозы с использованием многократных нарастающих доз (MAD) (13 доз в течение 12 дней) с введением дозы один раз в сутки в 6 когортах здоровых добровольцев с бессимптомным атопическим диатезом (N=48).
• Часть 3 представляла собой открытое перекрестное исследование влияния приема пищи при введении однократной дозы у 12 HV.
• Часть 4 представляла собой двойное слепое (для субъекта и исследователя данные были замаскированы, для спонсора - нет), плацебо-контролируемое исследование с использованием многократных доз (25 доз в течение 12 дней) с введением дозы два раза в день с участием 2 когорт здоровых добровольцев с бессимптомным атопическим диатезом (N=16).
Часть SAD (часть 1) предусматривала десять уровней доз, а части MAD (части 2 и 4) предусматривали восемь уровней доз (6 когорт с введением дозы один раз в сутки в части 2 и 2 когорты с введением дозы два раза в сутки в части 4). В каждую когорту рандомизировали по восемь субъектов для получения либо соединения (I), либо соответствующего плацебо в соотношении 6:2 (активное вещество:плацебо) в частях SAD и MAD. В рамках части SAD, следовало подвергать оценке дозы, превышающие расчетную фармакологически активную дозу (PAD) не более чем в примерно 4 раза, до того, как была начата часть исследования MAD, при условии, что до того времени от части SAD не поступало сигнала безопасности. Общая суточная доза соединения (I), используемая в части 2 (схема MAD qd) и части 4 (схема многократных доз bid), не превышала наивысшего исследованного уровня дозы в части SAD. Более того, общая суточная доза в части 4 не превышала общую суточную дозу в части 2.
В части 1 (SAD) для первого введения на каждом уровне дозы должны были применять стратегию дозорного введения доз следующим образом. Первым двум субъектам дозу вводили в первый день (одному с активным лекарственным средством, одному с плацебо). После 48-часового периода наблюдения дозы вводили остальным 6 субъектам когорты (пяти с активным лекарственным средством, одному с плацебо).
На протяжении всех частей исследования использовали стандартный мониторинг безопасности. Была включена отдельная оценка возможных случаев возникновения синяков на коже. Для каждой когорты перед повышением дозы следовало слепым образом ознакомиться с всеми показателями жизненно важных функций, физикальным обследованием и анамнезом пациента, ЭКГ, нежелательными явлениями и лабораторными параметрами безопасности (биохимический анализ крови, гематологическое исследование и анализ мочи) до 96 часов после введения последней дозы, а также данными PK от группы предыдущей дозы (если таковые доступны) до 48 часов после введения последней дозы. Сводные отчеты о безопасности по сообщенным нежелательным явлениям, лабораторным параметрам клинической безопасности, QTc и частоте сердечных сокращений предоставляли после завершения каждого уровня дозы.
В частях 1, 2 и 4 каждый субъект принимал участие в 28-дневном периоде скрининга (от дня -29 до дня -2), периоде определения исходного уровня, периоде лечения и периоде последующего наблюдения, который включал оценку в конце исследования.
В части 1 субъекты поступали в исследовательский центр в день -2 или -1 для оценки безопасности на исходном уровне и подтверждения соответствия критериям пригодности для включения в исследование. Пригодные для включения в исследование субъекты получали однократную дозу соединения (I) или плацебо натощак в день 1. Их госпитализировали на срок от дня -1 до утра дня 5 (96 часов после введения последней дозы лекарственного средства).
В частях 2 и 4 субъекты поступали в день -2 или -1 для оценки безопасности на исходном уровне и подтверждения соответствия критериям пригодности для включения в исследование. Пригодные для включения в исследование субъекты получали первую дозу соединения (I) натощак в день 1 и продолжали принимать исследуемый препарат натощак до дня 12 включительно. Субъектов госпитализировали со дня -2 или -1 до утра дня 16, что соответствовало 96 часам после приема последней дозы соединения (I). В частях 2 и 4 исследуемый препарат давали один раз в сутки и два раза в сутки соответственно (подробности см. в графике проведения оценок).
Часть 3 представляла собой открытое рандомизированное двустороннее перекрестное исследование однократной дозы для оценки влияния приема пищи. В части 3 каждый субъект принимал участие в 28-дневном периоде скрининга (от дня -29 до дня -2), в 2 периодах определения исходного уровня (день -1) и 2 периодах лечения, каждый из которых состоял из введения однократной дозы в день 1 с последующей оценкой безопасности и PK до дня 5. Период лечения 2 состоял из визита последующего наблюдения и оценки в конце исследования в дни 22 и 40 соответственно. Два периода лечения были разделены периодом отмывки продолжительностью по меньшей мере 18 дней (+/- 1 день).
Первичная(-ые) цель(-и)
Включена специальная оценка частоты возникновения синяков на коже.
Вторичные цели
Поисковые цели
Реакции на известный аллерген при проведении кожной инъекционной пробы (части 2 и 4)
Ex vivo клеточные биомаркеры PD, которые обеспечивали дополнительные меры ответа на лекарственное средство, представляющее собой соединения (I), путем ингибирования дегрануляции базофилов периферической крови (FcεR-индуцированная экспрессия CD63 и CD203c)
Критерии включения:
1. Здоровые субъекты мужского и женского пола в возрасте от 18 до 65 лет (включительно), имеющие хорошее состояние здоровья, как определено на основании анамнеза, физикального обследования, показателей жизненно важных функций, электрокардиограммы и лабораторных тестов на момент скрининга. Здоровые субъекты принимали участие в части 2 или части 4 с атопическим диатезом согласно критериям пригодности для включения в исследование для данных конкретных частей исследования. У здоровых добровольцев с атопией на момент скрининга должен был присутствовать положительный результат кожной инъекционной пробы на известный аллерген (атопический диатез), но они были клинически бессимптомными и не нуждались в приеме каких-либо системных лекарственных препаратов.
2. Требовалось, чтобы масса тела субъектов составляла по меньшей мере 50 кг, а индекс массы тела (BMI) находился в диапазоне 18-30 кг/м2 (включительно). BMI=масса тела (кг)/[рост (м)]2
3. На момент скрининга и первого определения исходного уровня показатели жизненно важных функций (температура тела, систолическое и диастолическое кровяное давление и частота пульса) оценивали в положении сидя после пребывания субъекта в состоянии покоя в течение по меньшей мере 3 минут и еще раз (при необходимости) после 3 минут в положении стоя. Показатели жизненно важных функций в положении сидя должны находиться в пределах следующих диапазонов (включительно):
• температура тела при оральном измерении 35,0-37,5°C;
• систолическое кровяное давление 90-139 мм рт. ст.;
• диастолическое кровяное давление 50-89 мм рт. ст.;
• частота пульса 50-90 уд/мин.
Ключевые критерии исключения
1. В анамнезе повышенная чувствительность к любому из исследуемых лекарственных средств или к лекарственным средствам аналогичных химических классов.
2. В анамнезе клинически значимые отклонения ЭКГ или любые из следующих отклонений ЭКГ на момент скрининга и/или перед лечением:
• интервал PR > 200 мс;
• комплекс QRS > 120 мс;
• QTcF> 450 мс (мужчины);
• QTcF> 460 мс (женщины).
3. Уровни гемоглобина ниже 12,0 г/дл на момент скрининга или первого определения исходного уровня.
4. Число тромбоцитов выходит за пределы диапазона нормы (менее 150×109/л или более 450×109) на момент скрининга или первого определения исходного уровня.
5. Любые клинически значимые отклонения в любом из стандартных тестов коагуляции, включая протромбиновое время (PT), частичное тромбопластиновое время (PTT) или международное нормализованное отношение (INR) на момент скрининга и/или определения исходного уровня.
6. Тромботическое или тромбоэмболическое событие в анамнезе или их наличие, или повышенный риск развития тромботического или тромбоэмболического события.
Вводимые средства лечения
Часть 1 (SAD)
Субъектов распределяли в одну из следующих 10 когорт. В каждую когорту рандомизировали по 8 субъектов для получения либо соединения (I), либо соответствующего плацебо при общем соотношении 6:2. Первую субкогорту рандомизировали в соотношении 1:1: то есть один субъект получает соединение (I) и один субъект получает соответствующее плацебо. Остальных 6 субъектов из каждой когорты, которым вводили дозу после 48-часового периода наблюдения за 2 субъектами, получившими дозу изначально, рандомизировали в соотношении 5:1.
• Когорта 1: однократная пероральная доза 0,5 мг соединения (I) или соответствующего плацебо.
• Когорта 2: однократная пероральная доза 1,5 мг соединения (I) или соответствующего плацебо.
• Когорта 3: однократная пероральная доза 5 мг соединения (I) или соответствующего плацебо.
• Когорта 4: однократная пероральная доза 15 мг соединения (I) или соответствующего плацебо.
• Когорта 5: однократная пероральная доза 30 мг соединения (I) или соответствующего плацебо.
• Когорта 6: однократная пероральная доза 60 мг соединения (I) или соответствующего плацебо.
• Когорта 7: однократная пероральная доза 100 мг соединения (I) или соответствующего плацебо.
• Когорта 8: однократная пероральная доза 200 мг соединения (I) или соответствующего плацебо.
• Когорта 9: однократная пероральная доза 400 мг соединения (I) или соответствующего плацебо.
• Когорта 10: однократная пероральная доза 600 мг соединения (I) или соответствующего плацебо.
Часть 2 (MAD, схема qd)
Субъектов распределяли в одну из следующих 6 когорт. В каждой когорте по 8 субъектов рандомизировали для получения либо соединения (I), либо соответствующего плацебо в соотношении 6:2.
• Когорта 1: многократные пероральные дозы 10 мг соединения (I) или соответствующего плацебо.
• Когорта 2: многократные пероральные дозы 25 мг соединения (I) или соответствующего плацебо.
• Когорта 3: многократные пероральные дозы 50 мг соединения (I) или соответствующего плацебо.
• Когорта 4: многократные пероральные дозы 100 мг соединения (I) или соответствующего плацебо.
• Когорта 5: многократные пероральные дозы 400 мг соединения (I) или соответствующего плацебо.
• Когорта 6: многократные пероральные дозы до 600 мг соединения (I) или соответствующего плацебо.
Часть 3 (влияние приема пищи)
Субъектов рандомизировали в группу с одной из 2 последовательностей лечения в соотношении 1:1.
Часть 4 (MAD, схема bid)
Субъектов распределяли в одну из следующих когорт. В каждой когорте по 8 субъектов рандомизировали для получения либо соединения (I), либо соответствующего плацебо в соотношении 6:2.
• Когорта 1: многократные пероральные дозы 100 мг соединения (I) или соответствующего плацебо по схеме bid
• Когорта 2: многократные пероральные дозы 200 мг соединения (I) или соответствующего плацебо по схеме bid
Данные фармакокинетики
Биоаналитические методы
Фармакокинетические образцы получали из крови и оценивали при всех уровнях дозы у всех субъектов. Образцы от субъектов в группах плацебо не анализировали. Образцы для оценки PK от субъектов собирали в моменты времени, определенные в исследовании. Концентрации соединения (I) определяли в крови с помощью утвержденного метода LC-MS/MS.
Фармакокинетика однократных нарастающих доз, составляющих 0,5 мг - 600 мг
Средняя концентрация соединения (I) в крови в зависимости от времени после введения однократных нарастающих доз показана на фиг. 1.
Соединение (I) быстро всасывалось, при этом время до достижения Cmax составляло приблизительно 1-1,5 часа для всех доз. Фаза абсорбции характеризовалась единственным отчетливым пиком абсорбции у большинства субъектов. Распределение лекарственного средства демонстрировало двухфазное снижение. Большая часть лекарственного средства выводилась в фазе первоначального распределения, что позволяет предположить, что значительная часть клиренса лекарственного средства может происходить до достижения равновесия в тканях всего тела. Кажущаяся терминальная фаза выведения наступала только через 12 часов после введения дозы и была измеряемой только у субъектов, получавших дозы 100 мг и выше. Измеряемые терминальные периоды полувыведения находились в диапазоне от 4 часов (100 мг) до 18 часов (600 мг), что приводило к тому, что среднее время удержания (MRT) в кровотоке составляло от 1 часа до 5 часов (MRT≈ T1/2/ln2). Фаза распределения продемонстрировала доминирующий независимый от дозы T1/2, составляющий ~1 ч. Расчетное среднее геометрическое значение клиренса из крови после перорального введения однократной дозы (CL/F) находилось в диапазоне от 250 до 506 л/ч в когортах SAD, при этом оценочное значение составляло приблизительно 383 л/ч во всех когортах.
Фармакокинетика многократных пероральных доз
Средняя концентрация соединения (I) в крови в зависимости от времени после введения многократных нарастающих доз 10 г - 400 мг показана на фиг. 2.
Геометрическое среднее кажущегося клиренса в равновесном состоянии после перорального введения дозы (CLss/F, день 12 MAD, qd) находилось в диапазоне от 246 л/ ч до 414 л/ ч в разных когортах. В целом более низкий клиренс наблюдали в равновесном состоянии по сравнению с днем 1, но эта разница почти исчезала при дозах 100 мг и выше (таблица 2-1 (день 1) и таблица 2-2 (день 12)). Причиной такого поведения, вероятно, является ковалентное связывание мишени (BTK), вносящее вклад в первоначальный клиренс соединения (I). Данный эффект наиболее заметен в день 1, так как в последующие дни оставшаяся занятость мишень при минимальном уровне снижает долю вклада связывания мишени в клиренс (CLss/F). Естественно, данная разница уменьшается с увеличением дозы, когда занятость мишени при минимальном уровне является почти полной. Следовательно, обнаружили, что содержание лекарственного средства (AUC, Cmax) было выше в день 12 по сравнению с днем 1, что демонстрирует коэффициент накопления лекарственного средства (Racc) (в пределах субъекта), который находился в диапазоне от 5 (низкая доза) до 1,2 (высокая доза) и обычно был выше для AUC, чем для Cmax, что подтверждает возможность вовлечения влияния на системный клиренс.
Таблица 2-1. Сводная информация о PK-параметрах соединения (I) при введении многократных нарастающих доз, составляющих от 10 до 600 мг, введение дозы q.d.
Профиль: день 1
N=6
N=6
N=6
N=6
N=6
N=6
Медиана (мин.-макс.) [n]
CV%=коэффициент вариации (%)=SD/среднее *100
Для Tmax и T1/2 представлены только значения медианы (мин.-макс.) [n].
Таблица 2-2.
Профиль: день 12
N=6
N=6
N=6
N=6
N=6
N=6
Медиана (мин.-макс.) [n]
CV%=коэффициент вариации (%)=SD/среднее *100
Для Tmax и T1/2 представлены только значения медианы (мин.-макс.) [n].
В целом, концентрации в крови через 24 часа после введения последней дозы, как правило, были ниже 1 нг/мл, за исключением нескольких субъектов, принимавших дозы 100 мг и выше, что указывало на почти полное вымывание соединения (I) в пределах введения двух последовательных доз. Последнее также предполагает, что равновесное состояние достигается в пределах введения нескольких доз.
Из-за более быстрого обновления BTK в тканях также исследовали введение дозы по схеме b.i.d. Профили средней концентрации в крови с течением времени, полученные после введения многократных нарастающих доз 100 мг и 200 мг два раза в день, показаны на фигуре 3. Как и в случае результатов других когорт, быстрая абсорбция всасывание с Tmax, составляющим примерно 1 ч, наблюдалось для доз после схемы b.i.d. Наблюдаемый коэффициент накопления (Racc) составлял 1,5 (100 мг) и 2,0 (200 мг) для AUC и приблизительно 1,65 для Cmax (обе дозы). Дозопропорциональное увеличение AUCtau наблюдали в день 12, при этом для Cmax обнаружили лишь небольшое увеличение (в 1,33 раза). В заключение, введение дозы соединения (I) по схеме b.i.d обеспечивает возможность решения проблемы более быстрого ресинтеза мишени в тканях во время интервала между введением доз без негативного влияния на общий PK-профиль и необходимости лечения высокими дозами по схеме q.d.
Влияние приема пищи: результат части 3
Данные PK от когорты, в которой определяли влияние приема пищи, обобщенные в таблице 2-3 ниже, указывают на сниженную скорость абсорбции, о чем свидетельствует снижение Cmax в 1,25 раза, и более полную общую абсорбцию, о чем свидетельствует повышение значения AUC0-24 в 1,4 раза. Что наиболее важно, среднее значение Tmax смещалось с 1 ч (натощак) до >3 ч (после приема пищи) (фиг. 4)
Таблица 2-3 Сводные данные по PK-параметрам: влияние приема пищи на соединение (I) после введения однократной дозы, составляющей 60 мг

CV=коэффициент вариации (%)=SD/среднее значение*100
Фармакодинамика
Фармакодинамику (PD) характеризовали путем оценки занятости мишени и ингибирования дистальных элементов сигнального пути. Измерения занятости BTK в цельной крови человека (получаемой как соотношение свободной и общей BTK) служили в качестве прямого маркера терапевтического воздействия на мишень.
Взаимосвязь между занятостью BTK, дозой, системным воздействием соединения и действием на комплексный сигнальный путь in vivo и результатами по состоянию болезни была установлена на доклинических моделях для соединения формулы (I) (например, пример 1).
Соединение формулы (I) является необратимым ингибитором BTK, определяли степень и продолжительность занятости BTK. PD-эффект соединения (I) оценивали путем измерения содержания как свободной BTK (не связанной), так и общей BTK в цельной крови с помощью твердофазного иммуноферментного анализа (ELISA) на платформе Meso Scale Diagnostics (MSD) в двух отдельных анализах.
Взаимосвязь между дозой и фармакодинамикой характеризовали за счет измерений занятости BTK в крови человека (получаемой как соотношение свободной и общей BTK), прямого маркера терапевтического воздействия на мишень. Занятость BTK определяли для введения однократных нарастающих доз в диапазоне от 0,5 до 400 мг, для введения многократных нарастающих доз в диапазоне от 10 до 400 мг по схеме q.d и для введения многократных нарастающих доз в диапазоне от 100 до 200 мг по схеме b.i.d.
Соединение формулы (I) продемонстрировало явное дозозависимое увеличение как степени, так и продолжительности занятости BTK в периферической крови. Пиковую занятость мишени обычно наблюдали уже через 0,5 ч после введения дозы, что указывает на быстрое начало действия без значимого гистерезиса действия лекарственного средства по сравнению с пиковым содержанием лекарственного средства. Исходя из его способности ковалентно связывать BTK, занятость мишени сохранялась еще долго после его выведения из системного кровообращения, что указывает на неравновесную взаимосвязь PK-PD. Соответственно, установлено, что продолжительность занятости BTK определяется скоростью синтеза BTK de novo.
В отличие от когорт с более низкими дозами (0,5-1,5 мг) однократные дозы, составляющие 15 мг соединения (I) и выше, обеспечивали установление пиковой занятости мишени, приближающейся к 100%, почти у всех субъектов, при этом она оставалась выше 80% через 24 ч. В то время как при дозе 15 мг ответ среди субъектов значительно варьировался, дозы 30 мг и выше обеспечивали устойчивую (> 24 ч) и почти полную (>90%) занятость у всех субъектов при явно сниженной вариабельностью между субъектами. Время обновления пула белка BTK до уровней перед введением дозы составляло приблизительно 10 дней, что соответствует медианному T1/2 обновления, составляющему приблизительно 48 часов (фигура 5).
После введения многократных доз соединения (I) при использовании уже 10 мг соединения (I) по схеме q.d. достигалась остаточная занятость BTK в крови >96% перед введением дозы в день 12.
Кроме того, ингибирование активации базофилов ex vivo (отслеживаемое по поверхностной экспрессии CD63 и CD203c) использовали в качестве дистального биомаркера механизма действия для тестирования нижерасположенных PD-эффектов соединения (I). Для определения PD-эффекта соединения (I) в отношении активации базофилов цельную кровь стимулировали ex vivo с помощью антител к IgE. Дегрануляцию оценивали по процентной доле CD63+и CD203+базофилов с помощью проточной цитометрии.
После введения однократных нарастающих доз соединения (I) данные указывают на дозозависимое ингибирование FcεR1-опосредованной активации базофилов. Активация базофилов крови ex vivo, измеренная с помощью CD63, почти полностью подавлялась (>89%) при дозах, составляющих 60 мг, и достигала почти 100% ингибирования при более высоких дозах через 24 ч после введения дозы. Напротив, максимальное ингибирование CD203c через 24 ч после введения однократной дозы соединения (I) (примерно 50% ингибирование) достигалось только при использовании 200 мг соединения (I).
В день 12, через 8 ч после введения MAD соединения формулы (I) по схеме q.d. или b.i.d., уже самая низкая протестированная доза соединения (I) (10 мг по схеме q.d.) приводила к >90% ингибированию повышения экспрессии CD63, и остаточный уровень ингибирования CD63 составлял >90% при дозах соединения (I) ≥50 мг по схеме q.d. (фигура 6). Максимальное остаточное ингибирование активации CD203c в день 12 систематически было выше, чем после введения однократной дозы соединения (I), и достигалось только при введении 100 мг и 200 мг соединения (I) по схеме b.i.d.
Способность соединения (I) ингибировать ответ на определенный аллерген оценивали с помощью кожной инъекционной пробы (SPT) у здоровых субъектов с атопией в части исследования с использованием MAD в исследовании первого применения у человека. SPT выполняли перед введением дозы (на момент скрининга, при определении исходного уровня и перед введением дозы в день 1) и в различные моменты времени после введения первой дозы (день 1) и через 11 дней после введения дозы один раз в сутки (день 12).
Подобно ингибированию активации базофилов ex vivo дозозависимый эффект в отношении диаметра папулы был четко выражен в когортах многократных нарастающих доз, на что указывало уменьшение среднего размера папулы после введения дозы по сравнению с исходным уровнем (фигура 7). Эффект начал выходить на плато при введении примерно 100 мг соединения (I) по схеме q.d.
Обоснование выбора дозы/заключение
Здоровых добровольцев подвергали воздействию соединения (I) в клинических исследованиях фазы 1 с диапазоном доз от 0,5 мг до 600 мг, которые вводили в виде однократной дозы или вводили в течение не более 18 дней один или два раза в сутки. Соединение (I) хорошо переносилось, и отсутствовали серьезные или тяжелые нежелательные явления, связанные с приемом соединения (I). В клиническом исследовании наблюдаемые нежелательные явления (AE), по-видимому, не зависели от дозы, причем большинство из них были единичными и обычно носили умеренный характер. Таким образом, информация о клинической безопасности поддерживает дозы, выбранные для данного исследования фазы 2b.
Уровни доз по настоящему изобретению получали на основе следующих анализов: определение занятости BTK, ингибирование активации базофилов (отслеживаемой по повышению экспрессии CD63 и CD203c) у здоровых добровольцев и воздействие на кожные инъекционные пробы (SPT) у здоровых добровольцев с бессимптомной атопией - непрямой показатель ингибирования тучных клеток и базофилов в коже.
В данном вышеописанном клиническом испытании введение 10 мг соединения (I) по схеме q.d. приводило к почти полной занятости BTK в крови, >90% снижению повышения экспрессии CD63 (через 8 ч после введения СОЕДИНЕНИЯ (I) в равновесном состоянии) и минимальному ингибированию размера папулы при SPT. Таким образом, 10 мг соединения (I) по схеме q.d соответствует проявлению биологической активности. При дозе 100 мг соединения формулы (I) среднее уменьшение размера папулы при SPT начинало выходить на плато. Следовательно, 100 мг соединения соответствует максимальному эффекту СОЕДИНЕНИЯ (I). Средняя доза соединения (I), составляющая тридцать пять мг по схеме q.d., хорошо подходит для точного описания кривой доза-ответ для введения СОЕДИНЕНИЯ (I) по схеме q.d.
Соединение формулы (I) ингибирует BTK за счет ковалентного связывания. В то время как занятость BTK в крови составляет >24 часа (ч), из-за быстрого обновления BTK в ткани (например, примерно 5 часов в селезенке грызунов) для достижения максимальной эффективности может потребоваться введение соединения (I) по схеме b.i.d. Дозы 10, 25 и 100 мг соединения (I) по схеме b.i.d. соответственно точно описывают кривые доза-ответ для соединения (I) при приеме два раза в сутки.
Безопасность для человека
Для анализа нежелательных эффектов субъектов, получавших плацебо, из всех когорт с использованием SAD и MAD (по 2 на когорту) и разделенных на части по использованию SAD и MAD, объединяли в одну группу плацебо (n=20 для SAD и n=16 для MAD), чтобы сравнить с группой каждой отдельной дозы соединения (I) (по n=6 в каждой) и с общей группой соединения (I) (n=60 для SAD и n=48 для MAD). Не отмечены очевидные серьезные различия демографических данных между группой плацебо и группами активного соединения как для популяции SAD, так и для популяции MAD. В результате оценки безопасности в рамках исследования FIH на здоровых добровольцах не было выявлено значимых проблем с безопасностью при введении дозы вплоть до 600 мг.
Пример 3. Данные по эффективности и безопасности применения у пациентов с SjS со степенью тяжести от средней до тяжелой
Применение соединения формулы (I) осуществляют в исследовании фазы 2, разработанном для определения безопасности и эффективности соединения формулы (I), а также и описания зависимости доза-ответ у субъектов с SjS со степенью тяжести от средней до тяжелой, чтобы обеспечить возможность дальнейшей разработки соединения для лечения данного заболевания.
Краткий обзор протокола
Цель 2. Оценить эффективность соединения формулы (I) по сравнению с плацебо в отношении изменения результатов лечения, сообщаемых пациентом и врачом (ESSPRI, FACIT-F, EQ-5D, PhGA) по сравнению с исходным уровнем с течением времени.
Цель 3. Оценить эффективность соединения формулы (I) или его фармацевтически приемлемой соли по сравнению с плацебо в отношении изменения ESSDAI по сравнению с исходным уровнем с течением времени.
Цель 4. Оценить безопасность и переносимость соединения формулы (I) или его фармацевтически приемлемой соли посредством сообщения о возникновении AE, возникших после начала лечения (как серьезных, так и несерьезных), отклонении показателей жизненно важных функций, лабораторных показателей и значений ЭКГ в течение периода исследования.
Цель 5. Оценить PK-параметры соединения формулы (I) или его фармацевтически приемлемой соли (Cmax, AUC, Tmax и MRT и другие, при необходимости) в равновесном состоянии.
Классификация синдрома Шегрена согласно критериям ACR/EULAR 2016 на момент скрининга
Оценка ESSDAI на момент скрининга (на основе взвешенного балла) ≥5 на основе 8 определенных доменов (биологические показатели, гематологические показатели, проявление со стороны суставов, проявления со стороны кожи, проявления со стороны желез, лимфаденопатия, проявление со стороны почек, общее состояние). Пациенты с поражением, относящимся к одному или нескольким из оставшихся 4 доменов, пригодны к участию, но баллы по этим доменам не будут вносить вклад в оценку пригодности к участию, но являются частью общего балла ESSDAI для данного субъекта.
ESSPRI на момент скрининга ≥ 5
Серопозитивность по антителам к Ro/SSA на момент скрининга или в пределах 3 месяцев до него.
Скорость потока смешанной слюны без стимуляции составляет > 0 мл/мин на момент скрининга
Прием ритуксимаба или другого лекарственного средства, истощающего В-клетки, на протяжении 12 месяцев до момента скрининга. У субъектов, получавших такое лекарственное средство, число В-клеток должно быть в пределах нормы.
Предыдущее лечение на протяжении 6 месяцев до определения исходного уровня с помощью любого из следующих средств:
CTLA4-Fc Ig (абатацепт);
mAb к TNF-α;
внутривенного Ig;
применения плазмафереза;
циклофосфамида, вводимого внутривенно или перорально;
циклоспорина А, вводимого внутривенно или перорально.
Потребность в регулярном применении лекарственных препаратов, которые, как известно, в качестве основного побочного эффекта вызывают сухость во рту/глазах, и пациенты, которые не принимали стабильную дозу в течение по меньшей мере 30 дней перед моментом скрининга, или какое-либо ожидаемое изменение в схеме лечения во время периода исследования.
Значительный риск кровотечений или нарушения свертывания крови, включая без ограничения:
наличие в анамнезе или на момент исследования тромботического или тромбоэмболического события или повышенный риск возникновения тромботического или тромбоэмболического события;
необходимость приема антитромбоцитарных препаратов или антикоагулянтов (например, варфарина, клопидогрела или новых пероральных антикоагулянтов - NOAC), кроме ацетилсалициловой кислоты (до 100 мг/день);
желудочно-кишечные или внутримозговые или другие тяжелые предыдущие кровотечения в анамнезе, в том числе в ассоциации с применением нестероидного противовоспалительного лекарственного средства (NSAID).
Следующие значения лабораторного анализа CBC на момент скрининга:
уровень гемоглобина ниже 10 г/дл;
общее число лейкоцитов менее 3000/мкл;
количество тромбоцитов менее 100000/мкл;
число нейтрофилов ≤ 1500/мкл.
Капсулы плацебо
Физикальное обследование
Мониторинг лабораторных маркеров в крови и моче
Центральная оценка ЭКГ
Дополнительный мониторинг безопасности в отношении печени и почек
Кривые доза-ответ соединения формулы (I), основанные на улучшении ESSPRI во время визита на неделе 24, оценивают с использованием того же метода MCP-Mod, что и для первичного анализа. Изменение ESSDAI, ESSPRI, FACIT-F, EQ-5D и PhGA по сравнению с исходным уровнем анализируют с использованием MMRM, включая группу лечения, визит, взаимодействие факторов группа лечения и визит, фактор стратификации, учитывающий ESSDAI на исходном уровне (<10 или ≥10), и географический регион в качестве фиксированных факторов, а также исходное значение переменной, анализируемой как ковариата. Расчетные средние значения для дозы и визита и различия между группами лечения соединением формулы (I) и плацебо получают вместе с двухсторонними 95% доверительными интервалами.
Цели и конечные точки
Таблица 2-4 Цели и связанные с ними конечные точки
случаи нежелательных эффектов, возникших после начала лечения (как серьезных, так и несерьезных) в течение периода исследования
случаи отклонений показателей жизненно важных функций, лабораторных показателей и показателей ЭКГ, возникших после начала лечения, в течение периода исследования
Дизайн исследования
Исследование представляет собой адаптивное рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое,
интегрированное исследование фазы 2 с диапазоном доз для оценки безопасности и эффективности многократных доз соединения (I) у пациентов с синдромом Шегрена (SjS) со степенью тяжести от средней то тяжелой. В данном исследовании SjS со степенью тяжести от средней до тяжелой определяют как синдром Шегрена в соответствии с критериями ACR/EULAR, а также характеризующийся ESSDAI, составляющим по меньшей мере 5 (по 8 из 12 доменов), и ESSPRI, составляющим по меньшей мере 5. Если субъекты исследования получают определенную сопутствующую терапию для лечения своего первопричинного заболевания, но все еще соответствуют критериям включения, они будут продолжать получать данный вид терапии при условии, что она останется стабильной до конца исследования.
Исследование состоит из двух частей. В части 1 исследования самую высокую установленную
биологически активную однократную дозу (100 мг соединения формулы (I) или его фармацевтически приемлемой соли) испытывают в двух разных схемах введения доз: в виде введения дозы один раз в день (qd) или в виде введения дозы два раза в день (bid) и сравнивают с группой плацебо. В общей сложности примерно 72 субъектов исследования равным образом рандомизировали в данные 3 группы лечения с получением ожидаемого размера выборки, составляющего 24 субъекта на группу. В части 2 выбранную схему введения доз (qd или bid) расширяют до более низких доз, чтобы оценить безопасность и зависимость доза-ответ в данной схеме введения доз от установленных самой низкой (10 мг соединения формулы (I)) дозы до самой высокой (100 мг соединения формулы (I)) дозы. Это дает 4 группы лечения;а именно группу плацебо, а также группы соединения формулы (I) при трех уровнях дозы (100 мг bid/qd, 25 мг bid или 35 мг qd и 10 мг bid/qd). Ожидается, что в общей сложности примерно 180 субъектов равным образом рандомизируют в данные 4 группы лечения с получением размера выборки, составляющей 45 субъектов на группу.
Каждый отдельный субъект исследования сначала проходит период скрининга длительностью до 6 недель, лечение продолжительностью 24 недели и период последующего наблюдения, составляющий 30 дней после последнего введения исследуемого препарата, перед визитом в конце исследования. Общая продолжительность для каждого субъекта исследования,
включая скрининг, составляет до 35 недель.
В течение всего периода лечения (24 недели) субъекты два раза в день получают дозы соединения формулы (I) или плацебо, независимо от выбранной схемы введения доз, так что маскирование сохраняется на протяжении всего исследования.
Оценки безопасности включают физикальный осмотр, ЭКГ, оценку показателей жизненно важных функций, стандартные клинические лабораторные показатели (общий анализ крови, биохимический анализ, общий анализ мочи), а также мониторинг нежелательных явлений и серьезных нежелательных явлений.
Скрининг
После подписания информированного согласия субъектов оценивают по ESSDAI и ESSPRI, а также
проводят оценку безопасности и другие оценки, чтобы оценить пригодность к участию. Из соображений логистики
оценки выполняют в разные дни в течение 6-недельного периода скрининга, если исследователь
сочтет это целесообразным. Субъектов, не прошедших скрининг, могут повторно подвергнуть скринингу еще один
раз. Субъектам также выдают руководство по использованию носимого устройства (если они
выбрали его использование), которое им предоставляют в случае, если подтверждается пригодность субъекта к участию в исследовании.
Исходный уровень
Пригодные к участию субъекты возвращаются для визита оценки на исходном уровне в день 1. Субъекты могут остаться в исследовательском центре на ночь
из соображений логистики, хотя это не будет считаться госпитализацией.
Пригодность к участию подтверждают до рандомизации, и перед введением дозы в день 1 необходимо завершить требуемые оценки на исходном уровне. Для целей планирования на усмотрение исследовательского центра некоторые
оценки на исходном уровне проводят вечером перед днем 1.
Исследуемый препарат
Экспериментальное лекарственное средство поставляется в виде флаконов с этикеткой, которая надлежащим образом замаскирована. Флаконы содержат капсулы с активным веществом (соединение формулы (I)) в дозе 10 мг, или 25 мг, или 50 мг или соответствующим плацебо. Каждую дозу (2 капсулы) проглатывают, запивая водой, и их следует принимать натощак. Между утренними и вечерними дозами следует соблюдать интервал между введением доз, составляющий примерно 12 часов (от 10 до 14 часов). Подробная информация касательно требований к хранению и обращению с исследуемым препаратом и инструкции, которых необходимо придерживаться для нумерации субъектов, распределения и приема исследуемого препарата, изложены в SOM.
Группы лечения
В части 1 субъектов в день 1 распределяли в одну из следующих 3 групп лечения в соотношении 1:1:1.
• Соединение формулы (I), 100 мг bid
• Соединение формулы (I), 100 мг qd
• Плацебо
График введения доз и диапазон доз для части 2 основаны на данных IA. В части 2 субъектов в день 1 распределяли в одну из следующих 4 групп лечения в соотношении 1:1:1:1.
• Соединение формулы (I), 100 мг bid или qd
• Соединение формулы (I), 35 мг qd или 25 мг bid
• Соединение формулы (I), 10 мг bid или qd
• Плацебо
В качестве каждой дозы субъекты принимают по 2 капсулы. Для всех субъектов в обеих частях существуют утренняя и вечерняя дозы (по 2 капсулы).
Субъекты получают свою утреннюю дозу соединения формулы (I) или плацебо в исследовательском центре в дни 1, 29 (неделя 4), 57 (неделя 8), 85 (неделя 12), 113 (неделя 16), 141 (неделя 20) и 169 (неделя 24). Остальные утренние дозы и все вечерние дозы участвующие субъекты обычно принимают дома. Все субъекты получают соответствующий запас соединения формулы (I) или капсулы плацебо каждые 4 недели во время запланированных визитов в исследовательский центр.
Субъектов рандомизируют в соответствующие группы лечения для каждой части исследования. За исключением центров в Японии
рандомизация стратифицируется по ESSDAI на исходном уровне (< или ≥10 на основе взвешенных баллов).
Отдельные блоки номеров рандомизации генерируют для субъектов в Японии и других
странах, принимающих участие в исследовании, чтобы гарантировать, что субъекты из Японии равномерно распределены по всем
группам лечения в исследовании. Субъекты получают свою утреннюю дозу соединения формулы (I) или плацебо в исследовательском центре в дни 1 (неделя 1), 29 (неделя 4), 57 (неделя 8), 85 (неделя 12), 113 (неделя 16), 141 (неделя 20) и 169 (неделя 24). Затем субъектов обеспечивают исследуемым лекарственным средством, и они могут вернуться
домой, чтобы продолжить свою ежедневную схему введения доз (самостоятельное введение). Во время визитов на неделе 4 и неделе 24
субъектов также подвергают оценке безопасности и PK после введения дозы.
Субъекты возвращаются в исследовательский центр с интервалами примерно 4 недели, при этом имеется дополнительный визит
в конце недели 2 (день 15). В ходе визитов во время исследования субъектов подвергают оценкам ESSDAI и ESSPRI,
а также по другим шкалам/опросникам, и проводится сбор различных образцов для оценки безопасности, PK, PD и биомаркеров,
как указано в графике проведения оценок.
Каждую неделю субъектов просят заполнять дневники для регистрации своих симптомов SjS и
введения средства лечения.
Первичную конечную точку исследования оценивают после завершения 24-недельного лечения
(день 169; окончание визита на неделе 24) в конце части 2. В промежуточных анализах оценивают эффективность
и безопасность после 12 недель лечения как косвенный показатель результатов 24-недельного лечения в части 1.
Обоснование дозы/схемы и продолжительности лечения
Доза/схема для части 1
Самая высокая доза, запланированная для данного исследования (100 мг bid или qd), продемонстрировала максимальный эффект
для соединения формулы (I) на основе расчетной занятости BTK в крови (блокада B-клеток) и тканях, а также на основе ингибирования повышения экспрессии CD63 в базофилах (IB) (результаты в примере 2). Следовательно, установлено, что в случае SjS данная доза обеспечивает максимальный клинический эффект в ткани, включая лимфоидную ткань. В фазе 1 на людях-добровольцах дозы протестировали до 600 мг, как однократные, так и многократные, а также 200 мг в качестве доз, принимаемых два раза в день, и они доказали свою безопасность.
В части 1 дозу, которая, как установлено, обеспечивала максимальный эффект (100 мг), тестируют при схеме введения qd и bid и сравнивают с плацебо. Вследствие ковалентной природы связывания соединения формулы (I) с внутриклеточной BTK продолжительность эффекта лечения зависит от скорости обновления молекулы BTK. Имитационные модели показали, что схема введения 100 мг соединения формулы (I) qd в равновесном состоянии обеспечивает в среднем 83% занятость BTK через 24 часа после введения дозы (и непосредственно перед введением следующей дозы), тогда как схема введения bid такой же однократной дозы 100 мг обеспечивает в среднем 96% занятость BTK через 24 часа.
Более того, 70% ингибирование BTK в течение ~90% каждого периода после введения дозы в равновесном состоянии считается подходящим показателем для оптимальной клинической эффективности. Следовательно схемы введения доз, содержащих 100 мг соединения (I), как qd, так и bid, которые тестируют в части 1 данного исследования, обеспечивают эффективность.
Доза/схема для части 2
В части 2 будет оцениваться полный диапазон доз выбранной схемы введения доз с дозами от 10 мг до 100 мг. В исследовании первого применения у человека соединения формулы (I) введение 10 мг соединения формулы (I) q.d. приводило к почти полной занятости BTK в крови и снижению на >80% повышения экспрессии CD63, однако только к минимальному подавлению размера папулы при кожной инъекционной пробе (SPT). Следовательно, установлено, что 10 мг соединения формулы (I) q.d. будут соответствовать проявлению биологической активности с точки зрения фармакодинамической активности в тканях. Касательно средней дозы, считается, что суточная доза 35 мг соединения формулы (I) qd или 25 мг bid подходит для точного описания полной кривой доза-ответ для соединения формулы (I), вводимого qd или bid соответственно. Продолжение периода лечения с двойной маскировкой до 24 недель обеспечит непрерывные данные о безопасности и эффективности для соединения формулы (I).
Обоснование выбора контрольного лекарственного средства (плацебо)
Препарат сравнения представляет собой плацебо для обеспечения объективного подтверждения потенциальных AE и других данных о безопасности, а также данных о клинической эффективности и PD, полученных от субъектов, которых лечили с помощью соединения формулы (I) или его фармацевтически приемлемой соли на протяжении 24-недельного периода испытания. Поскольку одобренного системного лечения SjS не существует, применение плацебо является оправданным. Текущее стандарт лечения для пациентов с SjS ограничивается симптоматическим лечением признаков и симптомов (сухости) со стороны слизистых оболочек, а стероиды и традиционные DMARD зачастую являются неэффективными. Никакое фармакологическое воздействие не является эффективным при тяжелой ограничивающей дееспособность утомляемости, ассоциированной с SjS.

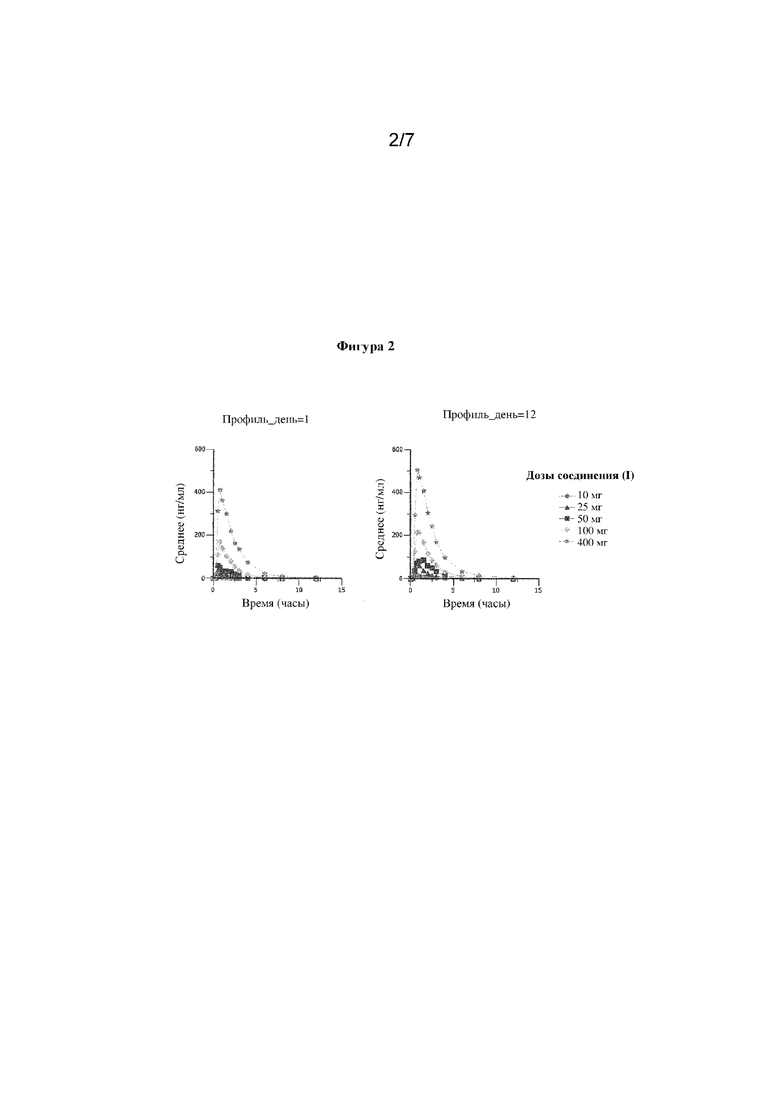




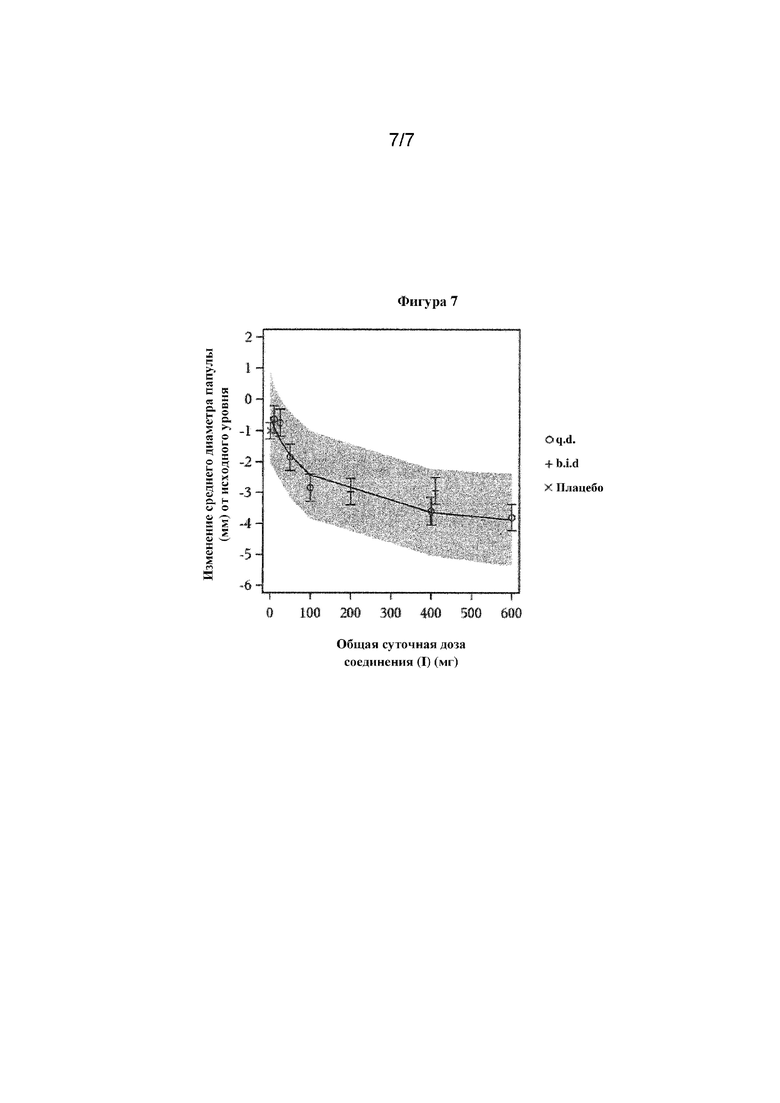
Изобретение относится к медицине. Раскрыт способ лечения синдрома Шегрена (SjS) у нуждающегося в таком лечении субъекта, включающий введение суточной дозы от приблизительно 10 мг до приблизительно 200 мг соединения формулы (I)
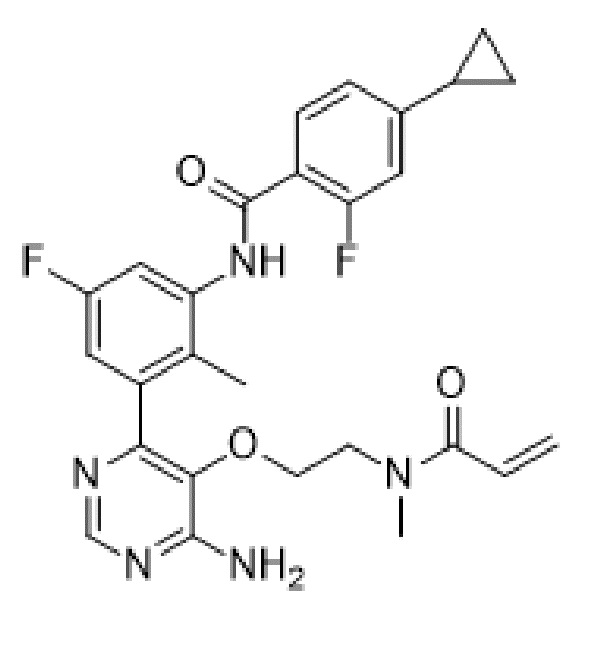 (I)
(I)
или его фармацевтически приемлемой соли, где термин «приблизительно» указывает на отклонение +/-10%. Изобретение обеспечивает лечение синдрома Шегрена соединением формулы (I) в эффективных и безопасных дозах. 15 з.п. ф-лы, 7 ил., 10 табл., 3 пр.
1. Способ лечения синдрома Шегрена (SjS) у нуждающегося в таком лечении субъекта, включающий введение суточной дозы от приблизительно 10 мг до приблизительно 200 мг соединения формулы (I)
 (I)
(I)
или его фармацевтически приемлемой соли,
где термин «приблизительно» указывает на отклонение +/-10%.
2. Способ по п. 1, где суточная доза составляет от приблизительно 10 мг до приблизительно 100 мг.
3. Способ по п. 1, где суточная доза составляет приблизительно 100 мг.
4. Способ по п. 1, где суточная доза составляет приблизительно 50 мг.
5. Способ по п. 1, где суточная доза составляет приблизительно 35 мг.
6. Способ по п. 1, где суточная доза составляет приблизительно 25 мг.
7. Способ по п. 1, где суточная доза составляет приблизительно 20 мг.
8. Способ по п. 1, где соединение формулы (I) вводится один раз в сутки в дозе, составляющей приблизительно 10 мг, приблизительно 35 мг, приблизительно 50 мг или приблизительно 100 мг.
9. Способ по п. 1, где соединение формулы (I) или его фармацевтически приемлемая соль вводятся в дозе, составляющей приблизительно 10 мг, приблизительно 25 мг, приблизительно 50 мг или приблизительно 100 мг, два раза в сутки.
10. Способ по любому из предыдущих пунктов, где у субъекта имеется SjS со степенью тяжести от средней до тяжелой.
11. Способ по любому из пп. 1-10, где субъект выбран в соответствии с по меньшей мере одним из следующих критериев:
а) до лечения с помощью соединения формулы (I) субъект имеет балл ESSPRI, составляющий 5 или больше;
b) до лечения с помощью соединения формулы (I) субъект имеет балл ESSDAI, составляющий 5 или больше, на основе взвешенного балла по 8 определенным доменам, выбранным из биологических показателей, гематологических показателей, проявления со стороны суставов, проявления со стороны кожи, проявления со стороны желез, лимфаденопатии, проявления со стороны почек и конституциональных симптомов.
12. Способ по любому из предыдущих пунктов, где субъект является взрослым.
13. Способ по любому из предыдущих пунктов, где у указанного субъекта к неделе 12 или к неделе 24 лечения достигается по меньшей мере одно из следующего:
a) снижение балла ESSPRI и/или
b) снижение балла ESSDAI.
14. Способ по любому из предыдущих пунктов, где у указанного субъекта достигается устойчивый ответ, измеренный с помощью ESSPRI или ESSDAI на неделе 5 после завершения лечения.
15. Способ по любому из предыдущих пунктов, где соединение формулы (I) или его фармацевтически приемлемая соль помещены в фармацевтический состав, и где указанный фармацевтический состав дополнительно содержит фармацевтически приемлемые носители.
16. Способ по любому из пп. 1-15, где соединение формулы (I) или его фармацевтически приемлемая соль характеризуются значением времени достижения максимальной концентрации (Tmax), составляющим приблизительно 0,5-3 ч.
| US 20150152068 A1, 04.06.2015 | |||
| КОМБИНАЦИИ ДЛЯ ЛЕЧЕНИЯ ИММУНОВОСПАЛИТЕЛЬНЫХ РАССТРОЙСТВ | 2002 |
|
RU2322984C2 |
| WO 2019087094 A1, 09.05.2019 | |||
| PAPAS A | |||
| et al | |||
| Приспособление с иглой для прочистки кухонь типа "Примус" | 1923 |
|
SU40A1 |
| Ударно-вращательная врубовая машина | 1922 |
|
SU126A1 |
| Эксцентричный фильтр-пресс для отжатия торфяной массы, подвергшейся коагулированию и т.п. работ | 1924 |
|
SU203A1 |

