Область техники, к которой относится изобретение
Настоящее изобретение относится к производному соединению, в котором в аминоалкановую кислоту введена бифенильная группа, его стереоизомеру или его фармацевтически приемлемой соли, и к фармацевтической композиции для профилактики и/или лечения инфекционного грибкового заболевания, которая включает это соединение в качестве активного ингредиента.
Предпосылки создания изобретения
Чем дольше живут современные люди, тем больше увеличивается число условно-патогенных грибковых инфекций, особенно среди пожилых людей, из-за снижения у них иммунных функций. Кроме того, инфекция, вызываемая условно-патогенными инфекционными грибами, растет во всем мире, в частности, среди пациентов с ослабленным иммунитетом, которых лечат иммунодепрессантами для уменьшения реакции отторжения трансплантата, или среди пациентов, перенесших трансплантацию органов, с нарушенными иммунными функциями, или среди пациентов с ослабленным иммунитетом из-за химиотерапии или синдрома приобретенного иммунодефицита (СПИДа). В прошлом из-за грибковых инфекций обычно возникали местные грибковые инфекции, такие как микоз стопы, опоясывающий лишай и молочница, но в последнее время системные грибковые инфекции, как правило, возникают настолько часто, что они занимают четвертое место среди всех типов инфекций в больницах. В качестве репрезентативных условно-патогенных грибов сообщалось о Candida albicans, Candida glabrata, Candida krusei, Cryptococcus neoformans и т.п. Патогенный гриб Cryptococcus neoformans, вызывающий системные инфекции, обычно встречается в почве по всему миру, и его базидиоспоры могут попадать из окружающей среды в легкие через органы дыхания человека. В случае пациентов с ослабленным иммунитетом, таких как пациенты с трансплантацией органов или пациенты со СПИДом, грибы, оказавшиеся в легких, могут вызывать легочные инфекции и проникать в центральную нервную систему через гематоэнцефалический барьер (ГЭБ), вызывая опасный для жизни менингит. В частности, менингит, вызываемый Cryptococcus, приводит к самому высокому уровню смертности среди менингитов: ежегодно во всем мире умирает более 600000 человек. Однако, поскольку грибы состоят из эукариотических клеток, таких же как клетки животных, биохимические метаболические пути грибов и млекопитающих настолько похожи, что трудно найти специфичную для грибов мишень для лекарств. Таким образом, обычные противогрибковые средства для лечения криптококкоза имеют ряд ограничений при их клиническом применении. Противогрибковые средства, разработанные к настоящему времени для подавления грибов Cryptococcus, включают класс полиенов, включающий амфотерицин B; класс азолов, включающий кетоконазол, флуконазол, итраконазол и вориконазол; и не-азольный класс, такой как тербинафин и флуцитозин; класс эхинокандинов, такой как каспофунгин. Амфотерицин B, одно из полиеновых противогрибковых средств, связывается с эргостеролом в клеточной мембране Cryptococcus, вызывая окислительное повреждение, приводя к гибели клеток грибов. Однако амфотерицин B вызывает побочные эффекты из-за его сильной токсичности для человеческого организма. Как известно, противогрибковые средства класса азолов ингибируют биосинтез эргостерина, одного из основных элементов клеточной мембраны грибов, путем ингибирования 14-α-деметилазы, которая участвует в превращении ланостерина в эргостерин, ослабляя тем самым клеточную мембрану и вызывая гибель клеток грибов. Однако сообщалось о росте устойчивости грибов к азолам. Тербинафин подавляет синтез эргостерина, ингибируя превращение сквалена в эпоксидный сквален. Флуцитозин, который является метаболическим антагонистом, ингибирующим синтез нуклеиновых кислот, проявляет противогрибковые эффекты, вызывая неправильное кодирование РНК грибов и противодействуя синтезу ДНК грибов. Противогрибковые средства класса эхинокандинов проявляют противогрибковые эффекты, ингибируя синтез клеточной стенки грибов, в то время как другие противогрибковые средства, упомянутые выше, действуют на мембраны клеток грибов. Как указано выше, обычные противогрибковые средства, как и другие лекарственные средства, имеют ряд проблем, связанных с побочными эффектами, такими как сильная токсичность, развитие лекарственной устойчивости и т.п. Следовательно, крайне необходима разработка противогрибкового средства нового класса, которое способно проявлять повышенные противогрибковые эффекты действия, при минимуме побочных эффектов.
Раскрытие изобретения
Техническая проблема
Настоящее изобретение предлагает новое производное аминоалкановой кислоты, содержащее бифенильную группу, его соль и/или сольват.
Кроме того, настоящее изобретение предлагает противогрибковую фармацевтическую композицию, содержащую указанное производное аминоалкановой кислоты, его соль и/или сольват в качестве активного ингредиента.
Кроме того, настоящее изобретение предлагает сельскохозяйственное противогрибковое средство, содержащее указанное производное аминоалкановой кислоты, его соль и/или сольват в качестве активного ингредиента.
Кроме того, настоящее изобретение предлагает противогрибковое средство для животных, содержащее указанное производное аминоалкановой кислоты, его соль и/или сольват в качестве активного ингредиента.
Кроме того, настоящее изобретение предлагает противогрибковую композицию, содержащую указанное производное аминоалкановой кислоты, его соль и/или сольват в качестве активного ингредиента.
Кроме того, настоящее изобретение предлагает очищающую композицию для тела человека, косметическую композицию или композицию шампуня, содержащую указанное производное аминоалкановой кислоты, его соль и/или сольват в качестве активного ингредиента.
Кроме того, целью настоящего изобретения является создание способа получения бензилоксибензиламинилового аминокислотного производного по изобретению.
Техническое решение
В качестве одного из аспектов для достижения вышеуказанных целей, настоящее изобретение предлагает соединение, представленное формулой 1, его стереоизомер или его фармацевтически приемлемую соль:
Формула 1
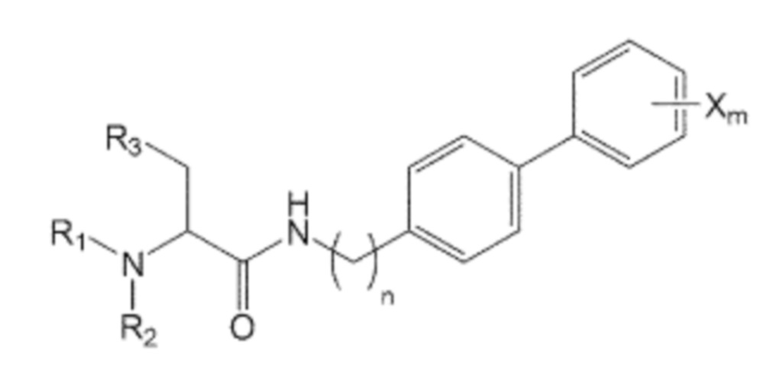
где в указанной формуле 1:
n равно 0, 1, 2, 3, 4 или 5,
R1, R2 и R3 независимо друг от друга являются одинаковыми или отличающимися друг от друга, и каждый независимо выбран из водорода, C1-7 алкила, гидроксила, галогена, галогенированного C1-7 алкила, C1-7 алкилокси и галогенированного C1-7 алкилокси, и
X представляет собой m заместителей (где m представляет собой целое число от 1 до 5), которые являются одинаковыми или отличающимися друг от друга, выбранными из группы, состоящей из группы галогена, галогенированной C1-7 алкильной группы и галогенированной C1-7 алкоксигруппы.
Кроме того, соединение, представленное формулой 1, представляет собой соединение, не имеющее ограничений в отношении трехмерной структуры расположения заместителя, присоединенного к хиральному углероду, и оно может включать все структурно доступные энантиомеры или оптические изомеры. В частности, соединение, представленное формулой 1, может быть предоставлено, но без ограничения указанным, в форме одного (R) или (S) изомера, или их смеси, например, их рацемической смеси или рацемата.
В настоящем изобретении галоген может быть выбран из группы, состоящей из фтора, хлора, брома и йода.
C1-7 алкил может быть линейным, разветвленным или циклическим алкилом, и он может быть выбран из группы, состоящей из метила, этила, пропила, изопропила, циклопропила, бутила, изобутила, втор-бутила, трет-бутила, пентила, гексила, гептила и октила.
C1-7 алкилокси группа может быть выбрана из группы, состоящей из метокси, этокси, пропокси, бутокси, пентокси, гексилокси, гептилокси и октилокси.
Галогенированный C1-7 алкил может быть выбран из группы, состоящей из дифторметила, трифторметила, дифторэтила, трифторэтила, трифторпропила, трифторпентила, трифторгексила и трифторгептила.
Галогенированный C1-7 алкилокси может быть выбран из группы, состоящей из дифторметилокси, трифторметилокси, дифторэтилокси, трифторэтилокси, трифторпропилокси, трифторпентилокси, трифторгексилокси и трифторгептилокси.
Настоящее изобретение может включать не только соединение формулы 1 или его фармацевтически приемлемую соль, но также сольват или гидрат, обладающие такими же эффектами действия, которые могут быть получены при их использовании в соответствии с настоящим изобретением.
Соединение по изобретению основано на аминоалкановой кислоте и оно может представлять собой производное, в котором в аминоалкановую кислоту введена бифенильная группа.
В частности, аминоалкановая кислота может представлять собой производное α-аминокислоты, содержащее прямую углеводородную цепь C2-4 в боковой цепи, например α-аминомасляную кислоту, норвалин или норлейцин.
Используемый в данном документе термин «α-аминомасляная кислота (AABA)» относится к соединению, представленному приведенной ниже формулой 6, которое имеет название по IUPAC 2-аминомасляная кислота, представляющую собой непротеиногенную α-аминокислоту формулы C4H9NO2, также известную в биохимии как гомоаланин. Эта α-аминомасляная кислота включает прямую углеводородную цепь C2 в боковой цепи, которая содержит дополнительный C1 по сравнению с аланином.
Формула 6
Используемый здесь термин «норвалин (Nva)» относится к соединению, представленному формулой 7, которое имеет название по IUPAC 2-аминопентановая кислота, представляющую собой водорастворимую аминокислоту, являющуюся изомером валина, и представляет собой аминокислоту с разветвленной цепью (BCAA) формулы CH3(CH2)2CH(NH2)CO2H.
Формула 7

Используемый здесь термин «норлейцин (Nle)» относится к соединению, представленному следующей формулой 8, которое имеет название по IUPAC 2-аминогексановая кислота, и представляет собой аминокислоту, имеющую формулу CH3(CH2)3CH(NH2)CO2H.
Формула 8
Например, соединение по изобретению может быть соединением, в котором каждый R1 и R2 независимо представляют собой H или метил, этил, н-пропил, изопропил, циклопропил, н-бутил, изобутил, циклобутил, н-пентил, циклопентил, н-гексил или циклогексил, и R3 представляет собой метил, этил, н-пропил, изопропил, циклопропил, н-бутил, изобутил, втор-бутил или трет-бутил.
Кроме того, соединение по изобретению может быть соединением, в котором каждый из R1 и R2 независимо представляет собой H или метил, и R3 представляет собой метил, этил или н-пропил, но без ограничения указанным.
Например, в соединении по изобретению X может быть одним или двумя одинаковыми или разными заместителями, выбранными из группы, состоящей из фтора, хлора, трифторметила и трифторметокси. Например, X может быть одним, двумя или более заместителями, которые идентичны друг другу, или быть разными.
Например, X в соединении по изобретению может представлять собой фтор, хлор, трифторметил или трифторметокси, и, в частности, X может представлять собой п-фтор, м-фтор, п, м-дифтор, п-хлор, м-хлор, п, м-дихлор, п-трифторметил или п-трифторметокси, но без ограничения указанным.
В частности, соединение по изобретению может представлять собой следующие соединения:
1) 2-амино-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)бутанамид;
2) 2-амино-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)пентанамид;
3) 2-амино-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)гексанамид;
4) 2-амино-N-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)пентанамид;
5) N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)-2-(метиламино)бутанамид;
6) N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)-2-(метиламино)пентанамид;
7) N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)-2-(метиламино)гексанамид;
8) N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)-2-(диметиламино)пентанамид;
9) 2-амино-N-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)пентанамид;
10) 2-амино-N-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)гексанамид;
11) 2-амино-N-((4’-(трифторметил)-[1,1’-бифенил]-4-ил)метил)пентанамид;
12) 2-амино-N-((4’-(трифторметил)-[1,1’-бифенил]-4-ил)метил)гексанамид;
13) 2-амино-N-((4’-(трифторметокси)-[1,1’-бифенил]-4-ил)метил)пентанамид;
14) 2-амино-N-((4’-(трифторметокси)-[1,1’-бифенил]-4-ил)метил)гексанамид;
15) N-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)-2-(метиламино)пентанамид;
16) 2-(метиламино)-N-((4’-(трифторметил)-[1,1’-бифенил]-4-ил)метил)пентанамид;
17) N-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)-2-(диметиламино)пентанамид;
18) 2-амино-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)бутанамид;
19) 2-амино-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)пентанамид;
20) 2-амино-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)гексанамид;
21) 2-амино-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)бутанамид;
22) 2-амино-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)пентанамид;
23) 2-амино-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)гексанамид;
24) 2-амино-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)бутанамид;
25) 2-амино-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)пентанамид;
26) 2-амино-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)гексанамид;
27) 2-амино-N-(2-(3’,4’-дифтор-[1,1’-бифенил]-4-ил)этил)пентанамид;
28) N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)-2-(метиламино)бутанамид;
29) N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)-2-(метиламино)пентанамид;
30) N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)-2-(метиламино)гексанамид;
31) 2-(метиламино)-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)бутанамид;
32) 2-(метиламино)-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)пентанамид;
33) 2-(метиламино)-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)гексанамид;
34) 2-(метиламино)-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)бутанамид;
35) 2-(метиламино)-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)пентанамид;
36) 2-(метиламино)-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)гексанамид; или
37) N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)-2-(диметиламино)пентанамид;
но не ограничиваясь указанными.
Соединение по изобретению может быть в форме фармацевтически приемлемой соли. Солью может быть кислотно-аддитивная соль, образованная фармацевтически приемлемой свободной кислотой. Используемый здесь термин «фармацевтически приемлемая соль» относится к органической или неорганической аддитивной соли соединения, представленного формулой 1, которая переносится пациентами и которая нетоксична в степени, достаточной для использования ее в концентрации, проявляющей фармакологические эффекты соединения.
Кислотно-аддитивную соль получают обычными способами, например растворением соединения в избытке водного раствора кислоты, с осаждением полученной соли с использованием смешивающегося с водой органического растворителя, например метанола, этанола, ацетона или ацетонитрила. Такое же молярное количество соединения и кислоты или спирта (например, монометилового эфира гликоля) в воде нагревают, а затем полученную смесь можно упарить и высушить, или осажденную соль можно отфильтровать отсасыванием.
В этом случае в качестве свободной кислоты можно использовать органические кислоты и неорганические кислоты. В качестве неорганических кислот можно использовать хлористоводородную кислоту, фосфорную кислоту, серную кислоту, азотную кислоту, винную кислоту и тому подобное, а в качестве органических кислот можно использовать метансульфоновую кислоту, п-толуолсульфоновую кислоту, уксусную кислоту, трифторуксусную кислоту, малеиновую кислоту, янтарную кислоту, щавелевую кислоту, бензойную кислоту, винную кислоту, фумаровую кислоту, миндальную кислоту, пропионовую кислоту, лимонную кислоту, молочную кислоту, гликолевую кислоту, глюконовую кислоту, галактуроновую кислоту, глутаминовую кислоту, глутаровую кислоту, глюкуроновую кислоту, аспарагиновую кисло, аскорбиновую кислоту, угольную кислоту, ванилиновую кислоту, иодистоводородную кислоту и т.п., при этом свободная кислота не ограничивается указанными.
Кроме того, с использованием основания может быть получена фармацевтически приемлемая соль металла. Соль щелочного металла или соль щелочноземельного металла получают, например, растворением соединения в избыточном количестве раствора гидроксида щелочного металла или гидроксида щелочноземельного металла, с последующим фильтрованием нерастворенной соли соединения, выпариванием и сушкой фильтрата. В этом случае в качестве фармацевтически приемлемой соли металла предпочтительно получать, в частности, соли натрия, калия или кальция, при этом соль металла не ограничивается перечисленными. Кроме того, может быть получена соответствующая соль серебра путем взаимодействия соли щелочного или щелочноземельного металла с подходящей солью серебра (например, с нитратом серебра).
Фармацевтически приемлемая соль соединения по изобретению включает соль, образованную с кислотной или основной группой, которая может присутствовать в соединении формулы 1, если не указано иное. Например, фармацевтически приемлемая соль может содержать соли натрия, кальция, калия и т.п. гидроксильной группы. Примеры других фармацевтически приемлемых солей аминогруппы включают гидробромид, сульфат, гидросульфат, фосфат, гидрофосфат, дигидрофосфат, ацетат, сукцинат, цитрат, тартрат, лактат, манделат, метансульфонат (мезилат), п-толуолсульфонат (тозилат) и т.п., и они могут быть получены способом получения солей, известным в данной области.
Соль соединения формулы 1 по изобретению представляет собой фармацевтически приемлемую соль, хотя может быть использована без ограничения любая соль при условии, что эта соль обладает фармакологической активностью, эквивалентной активности соединения формулы 1. Например, когда соль соединения формулы 1 проявляет противогрибковую активность.
В качестве другого аспекта настоящее изобретение предлагает способ получения производного соединения, его стереоизомера или его фармацевтически приемлемой соли, в котором в аминоалкановую кислоту введена бифенильная группа, при этом способ включает первую стадию образования пептидной связи посредством взаимодействия производного аминоалкановой кислоты, защищенного бутоксикарбонильной (Boc) защитной группой, представленного формулой 2, с производным бифенила, включающим первичную аминогруппу, представленным следующей формулой 3; и вторую стадию удаления защитной группы Boc путем взаимодействия соединения, полученного на первой стадии, с кислотой:
Формула 1
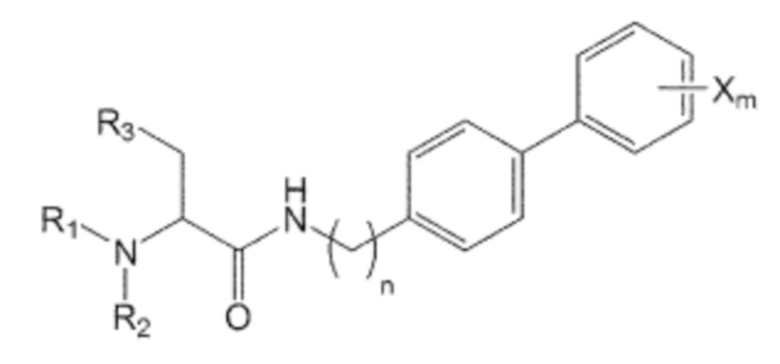
Формула 2
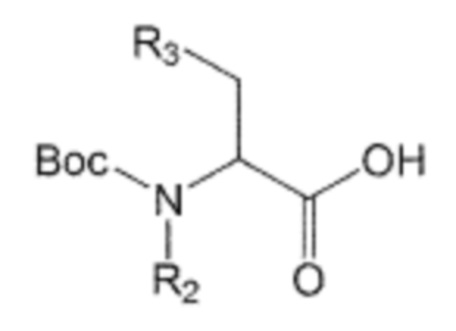
Формула 3
где в формуле 1:
n равно 0, 1, 2, 3, 4 или 5,
R1, R2 и R3 независимо друг от друга являются одинаковыми или отличающимися друг от друга, и каждый независимо выбран из группы, состоящей из водорода, C1-7 алкила, гидроксила, галогена, галогенированного C1-7 алкила, C1-7 алкилокси и галогенированного C1-7 алкилокси, и
X представляет собой m заместителей (где m представляет собой целое число от 1 до 5), которые являются одинаковыми или отличающимися друг от друга, выбранными из группы, состоящей из группы галогена, галогенированной C1-7 алкильной группы и галогенированной C1-7 алкоксигруппы.
В способе получения по изобретению производное аминоалкановой кислоты, защищенное защитной группой Boc, которое представлено формулой 2, может быть получено взаимодействием производного аминокислоты, представленного формулой 4, с ди-трет-бутилдикарбонатом (также известным как ангидрид Boc):
Формула 4
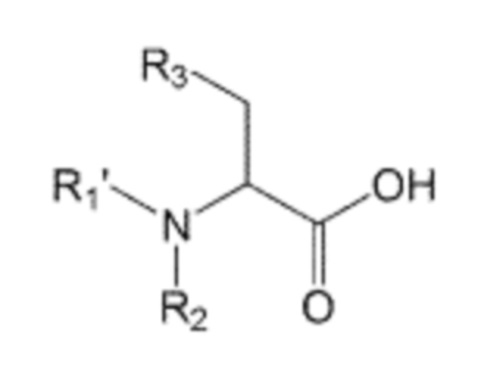
где в формуле 4:
R1’, R2 и R3 независимо друг от друга являются одинаковыми или отличающимися друг от друга, и каждый независимо выбран из группы, состоящей из водорода, C1-7 алкила, гидроксила, галогена, галогенированного C1-7 алкила, C1-7 алкилокси и галогенированного C1-7 алкилокси.
В этом случае, когда R2 полученного соединения представляет собой алкил, то после указанной выше реакции может быть проведена дополнительная стадия алкилирования соединения путем взаимодействия соединения с галогеналканом в присутствии основания. Например, алкилирование можно проводить путем растворения соединения, представленного формулой 4, и галогеналканового соединения, соответствующего 5-20 эквивалентам соединения, например, йодистого алкана, в органическом растворителе, например, в тетрагидрофуране, добавляя гидрид натрия в качестве основание при низкой температуре, например, при 0°C, а затем проводя взаимодействие реагентов при температуре от 15 до 30°C в течение от 12 до 48 часов, но без ограничения указанным. Известная в данной области реакция алкилирования аминов может использоваться без ограничения, или она может осуществляться с модификациями.
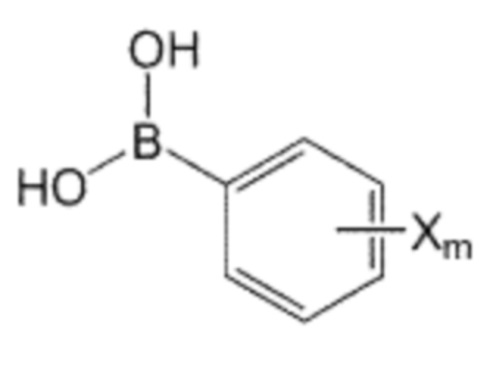
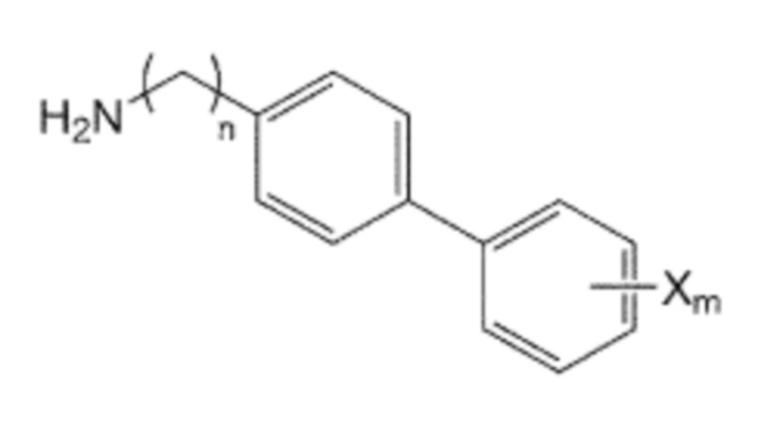
Помимо прочего, производное бифенила, содержащее первичную аминогруппу, которая представлена формулой 3, используемое в способе получения по изобретению, может быть получено взаимодействием производного C0-2 алкиламина, в котором галогенфенильная группа на одном конце замещена, представленного формулой 5, с ди-трет-бутилдикарбонатом для введения защитной группы Boc в аминогруппу, с последующим взаимодействием полученного производного алкиламина с производным фенилбороновой кислоты, представленного формулой 6, и с последующим взаимодействием реагентов с кислотой для удаления защитной группы Boc:
Формула 5
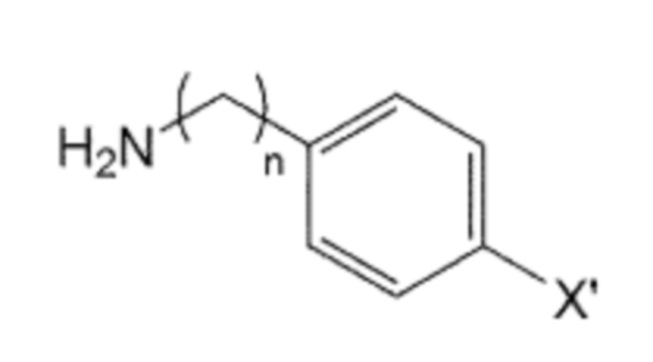
Формула 6
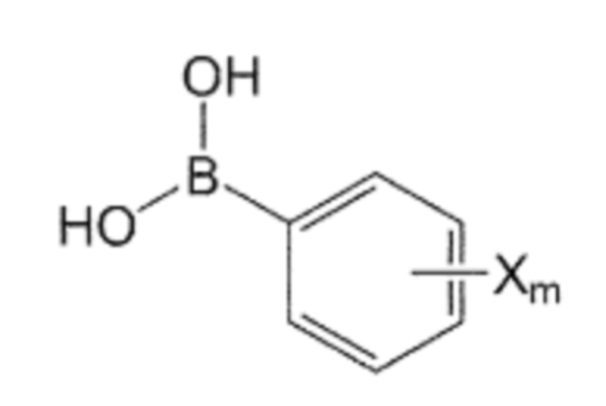
где в указанных формулах:
X’ представляет собой галоген, и
X представляет собой m заместителей (где m представляет собой целое число от 1 до 5), которые являются одинаковыми или отличающимися друг от друга, выбранными из группы, состоящей из группы галогена, галогенированной C1-7 алкильной группы и галогенированной C1-7 алкоксигруппы.
В этом случае реакция с производным фенилбороновой кислоты может быть выполнена путем реакции перекрестного сочетания (кросс-сочетания) с использованием металлического катализатора в присутствии основания. Например, реакцию можно проводить в основных условиях с использованием металлического катализатора, такого как палладий или никель. Металлический катализатор может быть катализатором, в котором фосфиновый лиганд связан с металлом. Например, реакция может быть реакцией кросс-сочетания Сузуки-Мияуры, с использованием Pd(PPh3)4 в присутствии Na2CO3, но не ограничиваясь указанным.
Например, в способе получения по изобретению первая стадия может быть выполнена путем реакции ангидридного сочетания, проводимой в органическом растворителе в присутствии N-метилморфолина (NMM) или изобутилхлорформиата (IBCF). В качестве органического растворителя можно использовать тетрагидрофуран, но не ограничиваясь указанным органическим растворителем.
Например, в способе получения по изобретению вторая стадия удаления защитной группы Boc может быть выполнена путем проведения реакции с хлористоводородной кислотой, но не ограничиваясь указанным.
Кроме того, способ получения по изобретению может дополнительно включать третью стадию образования вторичного амина путем алкилирования амина после второй стадии, когда каждый из R1 и R2 полученного соединения представляет собой алкил. Аминирование может быть выполнено путем взаимодействия с формальдегидом при подаче газообразного водорода в качестве восстановителя в присутствии Pd/C. Например, реакцию можно проводить при температуре от 15 до 30°С в течение от 6 до 24 часов, но без ограничения указанным, и реакция алкилирования аминов, известная в данной области, может использоваться как таковая или с модификациями.
В качестве еще одного аспекта настоящее изобретение обеспечивает противогрибковую композицию, содержащую в качестве активного ингредиента производное соединение, в котором в аминоалкановую кислоту введена бифенильная группа, его стереоизомер или его фармацевтически приемлемую соль.
В качестве еще одного аспекта настоящее изобретение обеспечивает фармацевтическую композицию для лечения или профилактики грибкового инфекционного заболевания, где композиция включает в качестве активного ингредиента производное аминоалкановой кислоты, в которую введена бифенильная группа, его стереоизомер или фармацевтически его приемлемая соль.
Например, новое производное аминоалкановой кислоты, в которую введена бифенильная группа, его стереоизомер или его фармацевтически приемлемая соль по изобретению может проявлять противогрибковую активность в отношении условно-патогенных инфекционных грибов, и, таким образом, оно может использоваться в качестве активного ингредиента в противогрибковой композиции, и кроме того, его можно использовать для предотвращения или лечения грибкового инфекционного заболевания.
Используемый здесь термин «профилактика» (или «предотвращение») относится к любым действиям, которые подавляют, ингибируют или задерживают начало, развитие или рецидив любого рассматриваемого заболевания при введении указанной фармацевтической композиции. Термин «лечение» относится к любым действиям, при которых симптомы любого рассматриваемого заболевания облегчаются или благоприятно улучшаются при введении указанной фармацевтической композиции.
Например, грибковое инфекционное заболевание, которое можно предотвратить или лечить фармацевтической композицией по изобретению, может включать, например, инфекционные заболевания, вызываемые Cryptococcus neoformans, Candida albicans, Candida auris, Candida glabrata и Aspergillus fumigatus. Инфекционным грибковым заболеванием также может быть менингит, вызванный Cryptococcus, но без ограничения указанным.
Фармацевтическая композиция по изобретению может содержать в качестве активного ингредиента соединение, представленное формулой 1, его стереоизомер или его фармацевтически приемлемую соль, и дополнительно может включать фармацевтически приемлемый носитель, разбавитель или эксципиент. Например, фармацевтическая композиция по изобретению может быть получена и использована в различных формах, таких как пероральная лекарственная форма, такая как порошок, гранулы, таблетка, капсула, суспензия, эмульсия, сироп, или аэрозоль, или стерильный раствор для инъекций, при этом введение лекарственной формы осуществляют обычным способом в соответствии с каждым предполагаемым применением, и лекарственная форма может вводиться перорально или другими различными путями, включая внутривенное, внутрибрюшинное, подкожное, ректальное, местное введение и т.п. Примеры подходящего носителя, разбавителя или эксципиента, которые могут быть включены в такую композицию, включают лактозу, декстрозу, сахарозу, сорбит, маннит, ксилит, эритрит, мальтит, крахмал, гуммиарабик, альгинат, желатин, фосфат кальция, силикат кальция, целлюлозу, метилцеллюлозу, микрокристаллическую целлюлозу, поливинилпирролидон, воду, метилгидроксибензоат, пропилгидроксибензоат, тальк, стеарат магния, минеральное масло и т.п. Кроме того, композиция по изобретению может дополнительно включать наполнитель, антикоагулянт, лубрикант, смачивающий агент, ароматизатор, эмульгирующий агент, консервант и т.п.
Твердый препарат для перорального введения включает таблетку, пилюлю, порошок, гранулы, капсулу и т.п., и твердый препарат готовят путем смешивания по меньшей мере одного эксципиента, например, крахмала, карбоната кальция, сахарозы, лактозы, желатина и т.п. с композицией. Кроме того, помимо простого эксципиента, можно использовать стеарат магния и тальк в качестве лубриканта.
В качестве жидкого препарата для перорального введения можно использовать суспензию, жидкость для приема во внутрь, эмульсию, сироп и т.п., а в дополнение к воде и жидкому парафину, которые являются простыми и обычно используемыми разбавителями, можно использовать различные вспомогательные вещества, например, можно использовать смачивающий агент, подсластитель, ароматизатор, консервант или тому подобное.
Примеры препарата для парентерального введения включают водный стерильный раствор, раствор в неводном растворителе, суспензию, эмульсию, лиофилизированный препарат и суппозиторий. В качестве неводного растворителя для раствора и суспензии можно использовать пропиленгликоль, полиэтиленгликоль, растительное масло, такое как оливковое масло, сложный эфир, используемый для инъекций, такой как этилолеат, и т.п. В качестве основы суппозитория можно использовать витепсол®, макрогол, твин 61, масло какао, лауриновый жир, глицерожелатин и т.п. Помимо прочего, препараты для инъекции могут включать добавки, известные из уровня техники, такие как солюбилизатор, изотонический агент, суспендирующий агент, эмульгатор, стабилизатор и консервант.
Композицию по изобретению вводят в фармацевтически эффективном количестве. Используемый здесь термин «фармацевтически эффективное количество» относится к количеству, которое достаточно для лечения заболеваний с разумным соотношением польза/риск, применимым к медицинскому лечению, не вызывая побочных эффектов, а эффективный уровень дозировки может быть определен в соответствии с различными факторами, включающими состояние здоровья пациента, тип заболевания, тяжесть заболевания, активность лекарства, чувствительность к лекарствам, способ введения, время введения, путь введения, скорость выведения, продолжительность лечения, а также с учетом лекарств, используемых в комбинации или одновременно, и с учетом других факторов, известных в данной области медицины. Композицию по изобретению можно вводить в виде индивидуального терапевтического средства или в комбинации с другими терапевтическими средствами, и ее можно вводить последовательно или одновременно с некоторыми обычными терапевтическими средствами, при этом ее можно вводить в виде одной дозы или нескольких доз. Важно вводить композицию в минимальном количестве, чтобы можно было получить максимальные эффекты без каких-либо побочных эффектов, принимая во внимание все вышеупомянутые факторы, и это количество может быть легко определено специалистами в данной области.
Например, поскольку количество может быть увеличено или уменьшено в зависимости от пути введения, тяжести заболевания, пола, веса тела, возраста и т.п., конкретная дозировка никоим образом не предназначена для ограничения объема настоящего изобретения.
Кроме того, настоящее изобретение обеспечивает способ лечения грибкового инфекционного заболевания, где способ включает введение фармацевтической композиции нуждающемуся в этом индивидууму.
Используемый здесь термин «индивидуум» относится к животным, включая обезьяну, корову, лошадь, овцу, свинью, курицу, индейку, перепела, кошку, собаку, мышь, крысу, кролика и морскую свинку, а также он включает человека, у которого развилось грибковое инфекционное заболевание или у которого возможно развитие грибкового инфекционного заболевания, и такое заболевание можно эффективно предотвратить или лечить путем введения индивидууму фармацевтической композиции по изобретению. Кроме того, фармацевтическая композиция по изобретению проявляет терапевтический эффект в отношении заболевания, вызванного грибковой инфекцией, благодаря ее противогрибковой активности, и она может проявлять синергический эффект при введении ее в комбинации с известным терапевтическим средством.
Используемый здесь термин «введение» относится к предоставлению пациенту заранее определенного материала любым подходящим способом. Что касается пути введения композиции по изобретению, то композицию по изобретению можно вводить любым известным путем, который может обеспечить достижение ткани-мишени. Путь введения может представлять собой внутрибрюшинное введение, внутривенное введение, внутримышечное введение, подкожное введение, внутрикожное введение, пероральное введение, местное введение, интраназальное введение, внутрилегочное введение и ректальное введение, но не ограничиваясь указанными. Кроме того, фармацевтическая композиция по изобретению также можно вводить с помощью любого устройства, которое может позволить активному материалу или ингредиенту перемещаться к клетке-мишени. Предпочтительные способы введения и препараты представляют собой внутривенную инъекцию, подкожную инъекцию, внутрикожную инъекцию, внутримышечную инъекцию, капельную инъекцию и т.п. Препарат для инъекций может быть приготовлен с использованием водного растворителя, такого как физиологический раствор и раствор Рингера, неводного растворителя, такого как растительное масло, сложный эфир высших жирных кислот (например, этилолеат и т.п.) и спирт (например, этанол, бензиловый спирт, пропиленгликоль, глицерин и т.п.), и такой препарат может включать фармацевтический носитель, стабилизатор для предотвращения порчи продукта (например, аскорбиновая кислота, бисульфит натрия, пиросульфит натрия, ВНА, токоферол, EDTA и т.п.), эмульгатор, буфер для поддержания pH и консервант для ингибирования роста микробов (например, нитрат фенилртути, тимероcал, бензалкония хлорид, фенол, крезол, бензиловый спирт и т.п.).
Термин «терапевтически эффективное количество», используемый в настоящем изобретении в отношении активного ингредиента, относится к количеству соединения производного аминоалкановой кислоты, где в аминоалкановую кислоту введена бифенильная группа, где такое производное эффективно для предотвращения или лечения целевого заболевания, или к количеству стереоизомера соединения или его фармацевтически приемлемой соли.
Технические результаты и эффекты
Согласно различным иллюстративным вариантам осуществления изобретения, недостатки обычных лекарств, используемых в качестве противогрибковых средств, можно преодолеть с помощью соединения на основе аминоалкановой кислоты, включающего бифенильную группу, например, соединения, содержащего в качестве основного скелета альфа-аминомасляную кислоту, норвалин или норлейцин. В частности, настоящее изобретение предоставляет противогрибковые соединения, обладающие повышенной безопасностью и эффективностью, для облегчения или устранения побочных эффектов обычных противогрибковых средств и для усиления терапевтических эффектов действия. Следовательно, настоящее изобретение предоставляет фармацевтическую композицию для лечения и/или предотвращения различных грибковых инфекционных заболеваний. Кроме того, соединение по изобретению можно использовать для получения антибактериальной композиции против грамположительных, грамотрицательных и устойчивых к MRSA бактерий. Кроме того, соединение по изобретению также можно использовать для изготовления противовоспалительного терапевтического средства.
Описание фигур
ФИГ. 1 отражает результаты сравнения противогрибковой активности соединения 74 по изобретению с активностью коммерчески доступных сравнительных лекарственных средств.
ФИГ. 2 отражает результаты сравнения фунгицидной активности соединения 74 по изобретению с активностью коммерчески доступных сравнительных лекарственных средств.
ФИГ. 3 отражает результаты сравнения эффектов в отношении удаления биопленки под действием соединения 74 по изобретению с действием коммерчески доступных сравнительных лекарственных средств.
Лучший вариант выполнения изобретения
Далее настоящее изобретение будет описано более подробно со ссылкой на конкретные примеры получения соединений и примеры выполнения изобретения. Следует иметь ввиду, что эти примеры приведены только для иллюстрации настоящего изобретения, и объем изобретения не ограничивается этими примерами.
Во-первых, реакции, используемые в синтезе соединения по изобретению, могут быть обобщены и представлены в общем виде следующим образом.
Схема реакции а - Введение защитной группы Boc
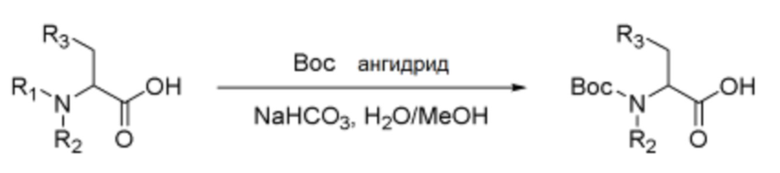
Норлейцин (1,0 экв.), ангидрид Boc (1,5 экв.) и бикарбонат натрия (1,5 экв.) растворяли в смешанном растворителе, состоящем из дистиллированной воды и метанола в соотношении 1:1, и реакцию проводили при комнатной температуре в течение 36-48 часов. После концентрирования смеси в вакууме pH водного слоя доводили до 2 с помощью 1,0 M хлористоводородной кислоты. Затем сульфатом натрия удаляли влагу из органического слоя, полученного экстракцией этилацетатом, и растворитель выпаривали в вакууме, получая указанное в заголовке соединение.
Схема реакции b - Метилирование аминогруппы

Соединение (1,0 экв.), полученное по схеме реакции а, и йодметан (10 экв.) растворяли в тетрагидрофурановом растворителе, и к нему при 0°C очень медленно по каплям добавляли гидрид натрия (10 экв.). Реагенты взаимодействовали при комнатной температуре в течение 24 часов. После завершения реакции полученный продукт разбавляли эфирным растворителем и добавляли к нему дистиллированную воду. Величину pH водного слоя доводили до 2 с помощью 20% раствора лимонной кислоты. Затем сульфатом натрия удаляли влагу из органического слоя, полученного экстракцией этилацетатом, и растворитель выпаривали в вакууме. Полученный остаток отделяли и очищали хроматографией на силикагеле, получая указанное в заголовке соединение.
Схема реакции c - Введение защитной группы Boc в группу первичного амина
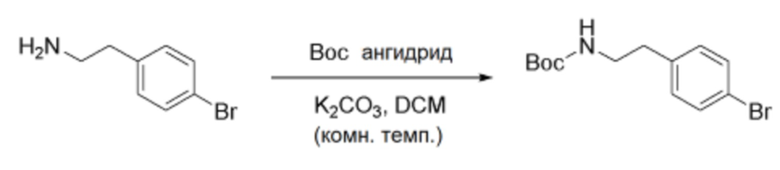
После растворения 4-бромфенетиламина (1,0 экв.) в растворителе метиленхлориде, к полученному раствору добавляли карбонат калия (1,5 экв.) и ангидрид Boc (1,05 экв.). Реакция в полученной смеси протекала при комнатной температуре в течение примерно 12-18 часов. Реакционную смесь разбавляли хлористым метиленом и дважды промывали дистиллированной водой. Органический слой сушили сульфатом натрия, а затем концентрировали в вакууме. Полученный остаток промывали гексаном, а затем упаривали в вакууме, получая указанное в заголовке соединение.
Схема реакции d - Синтез гидрохлорида производного бифениламина
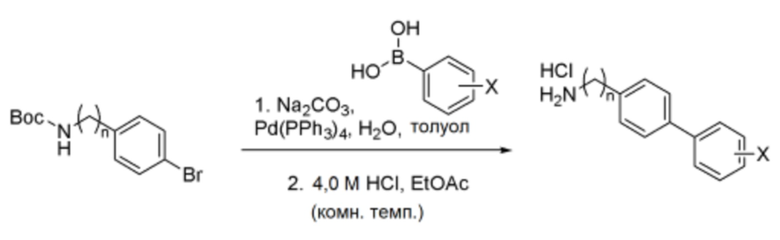
Соединение, полученное по схеме реакции c, трет-бутил(4-бромбензил)карбамат или трет-бутил(4-бромфенил)карбамат (1,0 экв.), бензолбороновую кислоту (1,5 экв.), карбонат натрия (5,0 экв.) и тетракис(трифенилфосфин)палладий (0,04 экв.) растворяли в смешанном растворителе из дегазированного толуола и дистиллированной воды в соотношении от 2:1 до 2,5:1, и реакцию проводили при кипячении с обратным холодильником при температуре 140°C в течение 12-18 часов. После завершения реакции катализатор удаляли фильтрованием через целит, и растворитель выпаривали из отфильтрованного органического слоя под вакуумом. Полученный остаток отделяли и очищали хроматографией на силикагеле. После растворения очищенного продукта в этилацетатном растворителе, полученный раствор перемешивали при комнатной температуре, добавляя к нему 4,0 М хлористоводородную кислоту (6,0-10,0 экв.). Полученное белое твердое вещество в форме соли промывали этилацетатом, а затем полностью сушили в вакууме, получая указанное в заголовке соединение.
Схема реакции e - Реакция смешанного ангидридного сочетания (MAC)
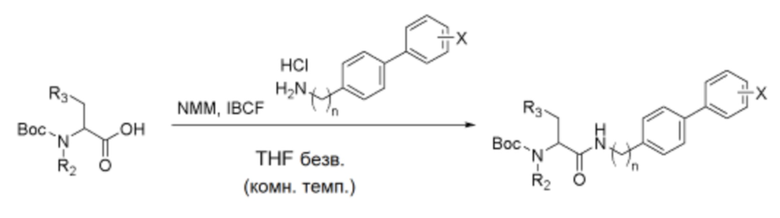
Соединение, синтезированное по схеме реакции a, или соединение, синтезированное по схеме реакции b (1,0 экв.), и N-метилморфолин (NMM, 2,5-2,8 экв.) помещали в дистиллированный тетрагидрофурановый растворитель, и полученную смесь перемешивали в течение 15 минут. Затем к ней добавляли изобутилхлорформиат (IBCF, 1,3 экв.), и полученную смесь дополнительно перемешивали в течение 15 минут. Затем к ней добавляли соединение (1,05 экв.), полученное по схеме реакции d. Реакционной смеси давали возможность прореагировать при комнатной температуре в течение примерно 3-5 часов. Смесь фильтровали, и растворитель выпаривали в вакууме. Полученный остаток отделяли и очищали хроматографией на силикагеле, получая указанное в заголовке соединение.
Схема реакции f - Удаление защитной группы Boc
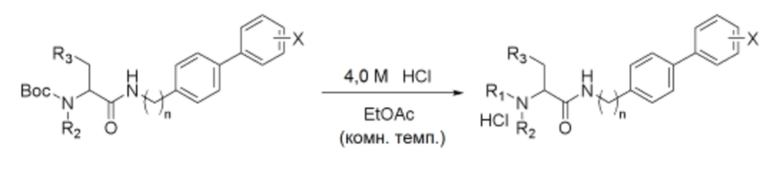
После растворения производного соединения (1,0 экв.), полученного по схеме реакции е, в этилацетатном растворителе, полученный раствор перемешивали при комнатной температуре, добавляя к нему 4,0 М хлористоводородную кислоту (6,0-10,0 экв.). Полученное белое твердое вещество в форме соли промывали этилацетатом, а затем полностью сушили в вакууме, получая указанное в заголовке соединение.
Схема реакции g - Диметилирование аминогруппы
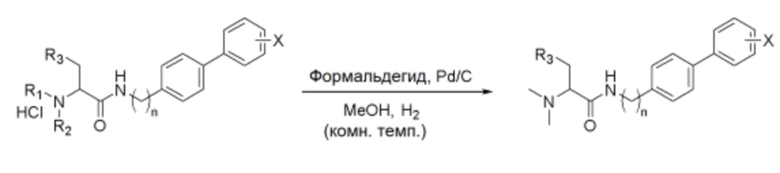
Соединение (1,0 экв.), полученное по схеме реакции f, растворяли в метаноле, и к нему добавляли триэтиламин (6,0 экв.). Затем последовательно добавляли формальдегид (37 масс.% раствор, от 1,0 до 2,5 экв.) и 10% палладиевый катализатор (от 0,1 до 0,5 экв.). Реагенты взаимодействовали при комнатной температуре в течение 18 часов. После завершения реакции катализатор удаляли фильтрованием через целит, и отфильтрованный органический слой упаривали в вакууме, получая белое твердое вещество. Полученный продукт перекристаллизовывали из метанола и диэтилового эфира, получая указанное в заголовке соединение.
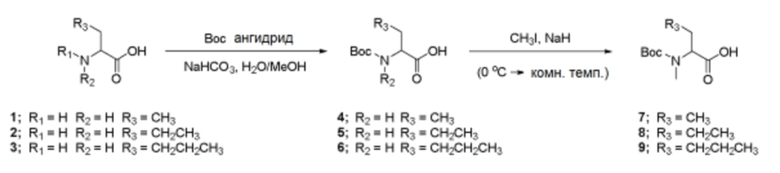
Ниже представлены следующие Примеры получения для синтеза соединения по изобретению.
Примеры получения
Пример получения 1: Получение (R)/(S)-2-((трет-бутоксикарбонил)амино)бутановой кислоты (4)
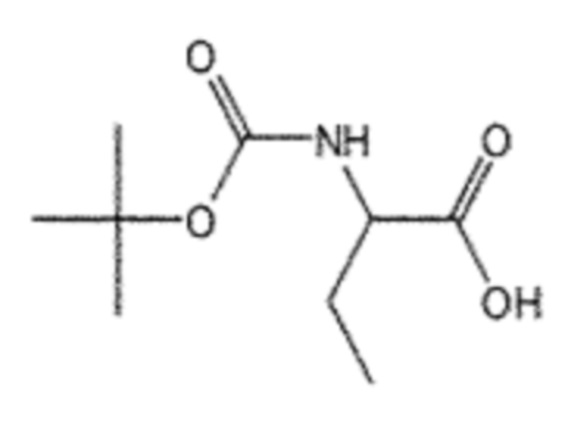
Соединение 1 (2-аминобутановая кислота, 5,00 г, 48,5 ммоль), ангидрид Boc (19,9 мл, 72,7 ммоль) и NaHCO3 (6,11 г, 72,7 ммоль) взаимодействовали между собой по схеме реакции a для синтеза соединения 4, (R)/(S)-2-((трет-бутоксикарбонил)амино)бутановой кислоты (8,25 г, 83%), получаемого в виде белого порошка.
Rf=0,00 (DCM:метанол 9,5:0,5, и несколько капель уксусной кислоты);
1H ЯМР (DMSO-d6, 300 МГц) 12,40 (C(O)OH), 7,02 (д, J=7,9 Гц, Boc-NH), 3,69-3,82 (м, хиральный-H), 1,48-1,72 (м, CH2CH3), 1,38 (с, Boc), 0,87 (т, J=7,3 Гц, CH2CH3).
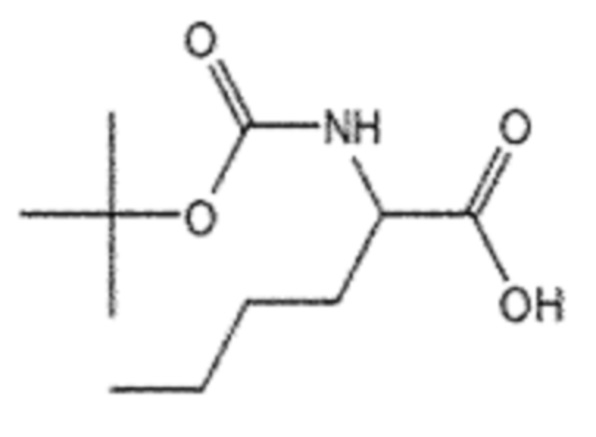
Пример получения 2: Получение (R)/(S)-2-((трет-бутоксикарбонил)амино)пентановой кислоты (5)
Соединение 2 (2-аминопентановая кислота, 10,00 г, 25,6 ммоль), ангидрид Boc (35,1 мл, 128,0 ммоль) и NaHCO3 (10,8 г, 128,0 ммоль) взаимодействовали между собой по схеме реакции a для синтеза соединения 5, (R)/(S)-2-((трет-бутоксикарбонил)амино)пентановой кислоты (13,40 г, 83%), получаемого в виде белого порошка.
Rf=0,85 (DCM:метанол 3:17);
1H ЯМР (DMSO-d6, 400 МГц) 12,40 (C(O)OH), 7,03 (д, J=8,0 Гц, Boc-NH), 3,75-3,89 (м, хиральный-H), 1,50-1,65 (м, CH2CH2CH3), 1,20-1,38 (м, CH2CH2CH3, Boc), 0,85 (т, J=7,4 Гц, CH2CH2CH3).
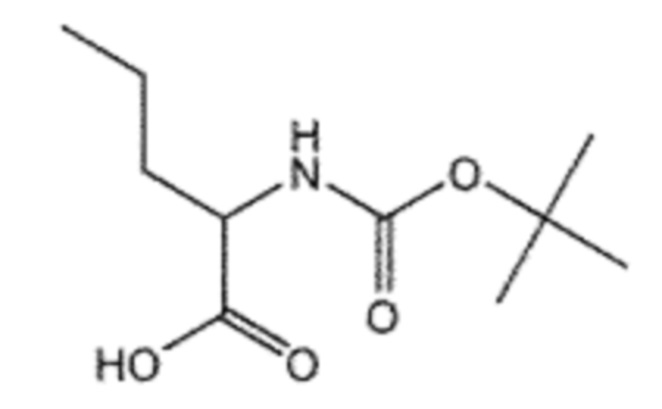
Пример получения 3: Получение (R)/(S)-2-((трет-бутоксикарбонил)амино)гексановой кислоты (6)
Соединение 3 (2-аминогексановая кислота, 5,00 г, 38,1 ммоль), ангидрид Boc (15,7 мл, 57,2 ммоль) и NaHCO3 (4,80 г, 57,2 ммоль) взаимодействовали между собой по схеме реакции a для синтеза соединения 6, (R)/(S)-2-((трет-бутоксикарбонил)амино)гексановой кислоты (7,14 г, 81%), получаемого в виде белого порошка.
Rf=0,40 (DCM:метанол 9:1);
1H ЯМР (CDCl3, 400 МГц) 10,26 (C(O)OH), 5,00 (д, J=7,6 Гц, Boc-NH), 4,32-4,33 (м, хиральный-H), 1,63-1,87 (м, CH2CH2CH2CH3), 1,47 (с, Boc), 1,31-1,38 (м, CH2CH2CH2CH3), 0,93 (т, J=7,0 Гц, CH2CH2CH2CH3).
Пример получения 4: Получение (R)/(S)-2-((трет-бутоксикарбонил)метиламино)бутановой кислоты (7)
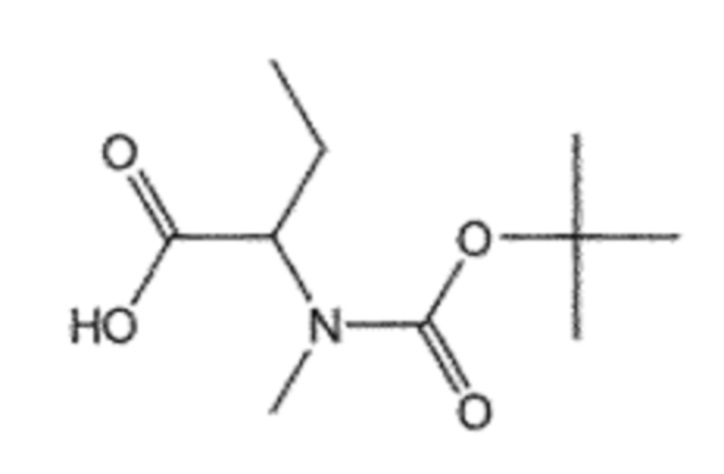
Соединение 4 (3,00 г, 14,8 ммоль), CH3I (9,2 мл, 147,6 ммоль) и NaH (3,54 г, 147,6 ммоль) взаимодействовали между собой по схеме реакции b, для синтеза соединения 7, (R)/(S)-2-((трет-бутоксикарбонил)метиламино)бутановой кислоты (2,84 г, 88%), получаемого в виде желтого масла.
Rf=0,45 (DCM:метанол 9:1, и несколько капель уксусной кислоты);
1H ЯМР (DMSO-d6, 300 МГц) 12,7 (C(O)OH), 4,14-4,43 (м, хиральный-H), 2,71 (с, NCH3), 1,50-1,73 (м, CH2CH3), Boc), 0,79-0,87 (м, CH2CH3).
Пример получения 5: Получение (R)/(S)-2-((трет-бутоксикарбонил)метиламино)пентановой кислоты (8)
Соединение 5 (1,50 г, 6,90 ммоль), CH3I (4,3 мл, 69,0 ммоль) и NaH (1,66 г, 69,0 ммоль) взаимодействовали между собой по схеме реакции b, для синтеза соединения 8, (R)/(S)-2-((трет-бутоксикарбонил)метиламино)пентановой кислоты (1,34 г, 83%), получаемого в виде желтого масла.
Rf=0,45 (DCM:метанол 9:1, и несколько капель уксусной кислоты);
1H ЯМР (DMSO-d6, 300 МГц) 12,7 (C(O)OH), 4,54-4,28 (м, хиральный-H), 2,70 (с, NCH3), 1,79-1,64 (м, CH2CH2CH3), 1,41-1,37 (м, CH2CH2CH3, Boc), 1,37-1,29 (м, CH2CH2CH3).
Пример получения 6: Получение (R)/(S)-2-((трет-бутоксикарбонил)метиламино)гексановой кислоты (9)
Соединение 6 (3,00 г, 13,0 ммоль), CH3I (8,1 мл, 129,7 ммоль) и NaH (5,19 г, 129,7 ммоль) взаимодействовали между собой по схеме реакции b для синтеза соединения 9, (R)/(S)-2-((трет-бутоксикарбонил)метиламино)гексановой кислоты (3,18 г, 100%), получаемого в виде желтого масла.
Rf=0,38 (DCM:метанол 9:1);
1H ЯМР (CDCl3, 400 МГц) 12,6 (C(O)OH), 4,25-4,52 (м, хиральный-H), 2,70 (с, NCH3), 1,66-1,79 (м, CH2CH2CH2CH3), 1,18-1,40 (м, CH2CH2CH2CH3, Boc), 0,86-0,89 (м, CH2CH2CH2CH3).
Пример получения 7: Получение трет-бутил(4-бромфенэтил)карбамата (12)
4-бромфенэтиламин (3,9 мл, 25,1 ммоль), K2CO3 (5,21 г, 37,7 ммоль) и ангидрид Boc (7,2 мл, 26,4 ммоль) взаимодействовали между собой по схеме реакции c для синтеза соединения 12, трет-бутил(4-бромфенэтил)карбамата (6,23 г, 83%), получаемого в виде белого порошка.
Rf=0,36 (EtOAc:н-гексан 1:5);
1H ЯМР (DMSO-d6, 400 МГц) 7,46 (д, J=8,6 Гц, ArH), 7,15 (д, J=8,2 Гц, ArH), 6,87 (с, NH), 3,09-3,14 (м, NHCH2CH2), 2,64-2,67 (м, NHCH2CH2), 1,35 (с, Boc).
Пример получения 8: Получение гидрохлорида 3’,4’-дихлор-[1,1’-бифенил]-4-амина (13)
После получения соединения взаимодействием соединения 10 (трет-бутил-4-бромфенилкарбамат, 4,00 г, 14,7 ммоль), 3,4-дихлорфенилбороновой кислоты (3,37 г, 17,6 ммоль), тетракис(трифенилфосфин)палладия (0,68 г, 0,59 ммоль) и Na2CO3 (7,80 г, 73,5 ммоль) по схеме реакции d, Boc-группу удаляли с использованием 4,0 M HCl (7,9 мл, 31,5 ммоль в диоксане) для синтеза соединения 13, гидрохлорида 3’,4’-дихлор-[1,1’-бифенил]-4-амина (1,27 г, 34%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 9,94 (с, NH3), 7,95 (д, J=2,0 Гц, ArH), 7,40-7,80 (м, ArH), 7,39 (д, J=8,5 Гц, ArH).
Пример получения 9: Получение гидрохлорида 4’-(трифторметокси)-[1,1’-бифенил]-4-амина (14)
После получения соединения взаимодействием соединения 10 (3,99 г, 14,7 ммоль), 4-(трифторметокси)фенилбороновой кислоты (7,77 г, 22,0 ммоль), тетракис(трифенилфосфин)палладия (0,68 г, 0,59 ммоль) и Na2CO3 (7,77 г, 73,3 ммоль) по схеме реакции d, Boc-группу удаляли с использованием 4,0 M HCl (12,8 мл, 51,1 ммоль в диоксане) для синтеза соединения 14, гидрохлорида 4’-(трифторметокси)-[1,1’-бифенил]-4-амина (1,99 г, 48%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 9,45 (шир. с, NH3), 7,77 (д, J=8,7 Гц, ArH), 7,71 (д, J=8,4 Гц, ArH), 7,45 (д, J=8,4 Гц, ArH), 7,28 (д, J=8,2 Гц, ArH).
Пример получения 10: Получение гидрохлорида 2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)метан-1-амина (15)
После получения соединения взаимодействием соединения 11 (трет-бутил-4-бромбензилкарбамат, 6,00 г, 21,0 ммоль), 3,4-дихлорфенилбороновой кислоты (4,80 г, 25,2 ммоль), тетракис(трифенилфосфин)палладия (0,97 г, 0,84 ммоль) и Na2CO3 (111,1 г, 104,8 ммоль) по схеме реакции d, Boc-группу удаляли с использованием 4,0 M HCl (3,1 мл, 12,3 ммоль в диоксане) для синтеза соединения 15, гидрохлорида 2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)метан-1-амина (1,08 г, 17%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,71 (с, NH3), 7,97 (с, ArH), 7,63-7,83 (м, ArH), 4,07 (с, NH3CH2).
Пример получения 11: Получение гидрохлорида (4’-(трифторметил)-[1,1’-бифенил]-4-ил)метанамина (16)
После получения соединения взаимодействием соединения 11 (6,00 г, 21,0 ммоль), 4-(трифторметил)фенилбороновой кислоты (5,97 г, 31,5 ммоль), тетракис(трифенилфосфин)палладия (0,97 г, 0,84 ммоль) и Na2CO3 (11,1 г, 104,8 ммоль) по схеме реакции d, Boc-группу удаляли, используя 4,0 M HCl (17,9 мл, 71,7 ммоль в диоксане), для синтеза соединения 16, гидрохлорида (4’-(трифторметил)-[1,1’-бифенил]-4-ил)метанамина (1,08 г, 66%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,49 (с, NH3), 7,93 (д, J=8,2 Гц, ArH), 7,83 (т, J=9,0 Гц, ArH), 7,64 (д, J=8,2 Гц, ArH), 4,09 (с, NH3CH2).
Пример получения 12: Получение гидрохлорида (4’-(трифторметокси)-[1,1’-бифенил]-4-ил)метанамина (17)
После получения соединения взаимодействием соединения 11 (4,00 г, 14,0 ммоль), 4-(трифторметокси)фенилбороновой кислоты (4,32 г, 21,0 ммоль), тетракис(трифенилфосфин)палладия (0,65 г, 0,56 ммоль) и Na2CO3 (7,41 г, 69,9 ммоль) по схеме реакции d, Boc-группу удаляли, используя 4,0 M HCl (13,9 мл, 55,6 ммоль в диоксане), для синтеза соединения 17, гидрохлорида (4’-(трифторметокси)-[1,1’-бифенил]-4-ил)метанамина (2,73 г, 65%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,33 (с, NH3), 7,81-7,83 (м, ArH), 7,75 (д, J=8,2 Гц, ArH), 7,59 (д, J=8,2 Гц, ArH), 7,48 (д, J=8,3 Гц, ArH), 4,08 (с, NH3CH2).
Пример получения 13: Получение гидрохлорида 2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этан-1-амина (18)
После получения соединения взаимодействием соединения 12 (трет-бутил(4-бромфенэтил)карбамат, 1,00 г, 3,33 ммоль), 3,4-дихлорфенилбороновой кислоты (0,76 г, 4,00 ммоль), тетракис(трифенилфосфин)палладия (0,15 г, 0,15 ммоль) и Na2CO3 (1,77 г, 16,7 ммоль) по схеме реакции d, Boc-группу удаляли с использованием 4,0 M HCl (2,50 мл, 10,0 ммоль в диоксане) для синтеза соединения 18, гидрохлорида 2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этан-1-амина (2,73 г, 65%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,33 (с, NH3), 7,93 (д, J=1,9 Гц, ArH), 7,66-7,72 (м, ArH), 7,39 (д, J=8,2 Гц, ArH), 2,98-3,07 (м, NH3CH2CH2).
Пример получения 14: Получение гидрохлорида 2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этан-1-амина (19)
После получения соединения взаимодействием соединения 12 (0,50 г, 1,67 ммоль), 4-(трифторметил)фенилбороновой кислоты (0,38 г, 2,00 ммоль), тетракис(трифенилфосфин)палладия (0,08 г, 0,07 ммоль) и Na2CO3 (0,88 г, 8,33 ммоль) по схеме реакции d, Boc-группу удаляли с использованием 4,0 M HCl (1,25 мл, 5,00 ммоль в диоксане) для синтеза соединения 19, гидрохлорида 2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этан-1-амина (0,28 г, 56%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,37 (с, NH3), 7,71-7,91 (м, ArH), 7,44 (д, J=8,1 Гц, ArH), 3,01-3,11 (м, NH3CH2CH2).
Пример получения 15: Получение гидрохлорида 2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этан-1-амина (20)
После получения соединения взаимодействием соединения 12 (1,50 г, 5,00 ммоль), 4-(трифторметокси)фенилбороновой кислоты (1,23 г, 6,00 ммоль), тетракис(трифенилфосфин)палладия (0,23 г, 0,20 ммоль) и Na2CO3 (2,65 г, 25,0 ммоль) по схеме реакции d, Boc-группу удаляли с использованием 4,0 M HCl (3,75 мл, 15,0 ммоль в диоксане) для синтеза соединения 20, гидрохлорида 2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этан-1-амина (0,88 г, 55%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,31 (с, NH3), 7,79 (д, J=8,7 Гц, ArH), 7,66 (д, J=8,1 Гц, ArH), 7,45 (д, J=8,2 Гц, ArH), 7,40 (д, J=8,1 Гц, ArH), 2,97-3,10 (м, NH3CH2CH2).
Пример получения 16: Получение гидрохлорида 2-(3’,4’-дифтор[1,1’-бифенил]-4-ил)этан-1-амина (21)
После получения соединения взаимодействием соединения 12 (1,00 г, 3,33 ммоль), 3,4-дихлорфенилбороновой кислоты (0,76 г, 4,00 ммоль), тетракис(трифенилфосфин)палладия (0,15 г, 0,15 ммоль) и Na2CO3 (1,77 г, 16,7 ммоль) по схеме реакции d, Boc-группу удаляли с использованием 4,0 M HCl (2,50 мл, 10,0 ммоль в диоксане) для синтеза соединения 21, гидрохлорида 2-(3’,4’-дифтор-[1,1’-бифенил]-4-ил)этан-1-амина (2,73 г, 65%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 7,95 (с, NH3), 7,74-7,79 (м, ArH), 7,67 (д, J=8,1 Гц, ArH), 7,48-7,54 (м, ArH), 7,37 (д, J=8,1 Гц, ArH), 2,90-3,09 (м, NH3CH2CH2).
Пример получения 17: Получение (R)/(S)-трет-бутил(1-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)амино)-1-оксобутан-2-ил)карбамата (22)
Соединение 4 (0,63 г, 3,12 ммоль), NMM (0,96 мл, 8,74 ммоль), IBCF (0,53 мл, 4,06 ммоль) и соединение 13 (0,90 г, 3,28 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 22, (R)/(S)-трет-бутил(1-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)амино)-1-оксобутан-2-ил)карбамата (1,09 г, 82%), получаемого в виде бледно-желтого порошка.
Rf=0,33 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 8,55 (с, C(O)NH), 7,58 (д, J=7,3 Гц, ArH), 7,44-7,47 (м, ArH), 7,34 (дд, J=1,8 Гц, 8,3 Гц, ArH), 5,12 (с, Boc-NH), 4,18 (с, хиральный-H), 1,67-2,05 (м, CH2CH3), 1,47 (с, Boc), 1,03 (т, J=7,4 Гц, CH2CH3).
Пример получения 18: Получение (R)/(S)-трет-бутил(1-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)амино)-1-оксопентан-2-ил)карбамата (23)
Соединение 5 (0,30 г, 1,52 ммоль), NMM (0,42 мл, 3,80 ммоль), IBCF (0,26 мл, 1,98 ммоль) и соединение 13 (0,44 г, 1,60 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 23, (R)/(S)-трет-бутил(1-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)амино)-1-оксопентан-2-ил)карбамата (0,61 г, 92%), получаемого в виде белого порошка.
Rf=0,37 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 8,53 (с, C(O)NH), 7,62 (д, J=8,6 Гц, ArH), 7,48-7,50 (м, ArH), 7,38 (дд, J=2,0 Гц, 8,3 Гц, ArH), 5,08 (с, Boc-NH), 4,24 (с, хиральный-H), 1,63-1,99 (м, CH2CH2CH3), 1,47-1,50 (м, Boc, CH2CH2CH3), 0,99 (т, J=7,3 Гц, CH2CH2CH3).
Пример получения 19: Получение (R)/(S)-трет-бутил(1-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)амино)-1-оксогексан-2-ил)карбамата (24)
Соединение 6 (0,80 г, 3,46 ммоль), NMM (0,95 мл, 8,69 ммоль), IBCF (0,58 мл, 4,50 ммоль) и соединение 13 (1,00 г, 3,64 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 24, (R)/(S)-трет-бутил(1-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)амино)-1-оксогексан-2-ил)карбамата (1,11 г, 71%), получаемого в виде белого порошка.
Rf=0,50 (EtOAc:н-гексан 1:3);
1H ЯМР (DMSO-d6, 400 МГц) 10,10 (с, NH), 7,91 (д, J=1,9 Гц, ArH), 7,64-7,74 (м, ArH), 7,04 (д, J=7,8 Гц, NH), 4,02-4,07 (м, NHCHCH2), 1,57-1,64 (м, CH2CH2CH2CH3), 1,39 (с, Boc), 1,26-1,32 (м, CH2CH2CH2CH3)0,86 (т, J=6,8 Гц, CH2CH2CH2CH3).
Пример получения 20: Получение (R)/(S)-трет-бутил(1-оксо-1-((4’-(трифторметокси)-[1,1’-бифенил]-4-ил)амино)пентан-2-ил)карбамата (25)
Соединение 5 (0,43 г, 1,97 ммоль), NMM (0,61 мл, 5,52 ммоль), IBCF (0,33 мл, 2,56 ммоль) и соединение 14 (0,60 г, 2,07 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 25, (R)/(S)-трет-бутил(1-оксо-1-((4’-(трифторметокси)-[1,1’-бифенил]-4-ил)амино)пентан-2-ил)карбамата (0,66 г, 73%), получаемого в виде белого порошка.
Rf=0,30 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 8,62 (с, C(O)NH), 7,61 (д, J=7,3 Гц, ArH), 7,54 (д, J=7,6 Гц, ArH), 7,49 (д, J=7,9 Гц, ArH), 7,26-7,29 (м, ArH), 5,19 (д, J=7,4 Гц, Boc-NH), 4,29 (с, хиральный-H), 1,65-1,98 (м, CH2CH2CH3), 1,43-1,56 (м, Boc, CH2CH2CH3), 0,99 (т, J=7,1 Гц, CH2CH2CH3).
Пример получения 21: Получение (R)/(S)-трет-бутил(1-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)амино)-1-оксобутан-2-ил)метилкарбамата (26)
Соединение 7 (0,68 г, 3,12 ммоль), NMM (0,96 мл, 8,74 ммоль), IBCF (0,53 мл, 4,06 ммоль) и соединение 13 (0,90 г, 3,28 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 26, (R)/(S)-трет-бутил(1-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)амино)-1-оксобутан-2-ил)метилкарбамата (0,74 г, 54%), получаемого в виде масла.
Rf=0,50 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 8,50 (с, C(O)NH), 7,58-7,63 (м, ArH), 7,46-7,50 (м, ArH), 7,38 (дд, J=1,8 Гц, 8,2 Гц, ArH), 4,57 (с, хиральный-H), 2,83 (с, NCH3), 1,71-2,04 (м, CH2CH3), 1,51 (д, J=6,8 Гц, Boc), 0,97 (т, J=7,3 Гц, CH2CH3).
Пример получения 22: Получение (R)/(S)-трет-бутил(1-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)амино)-1-оксопентан-2-ил)метилкарбамата (27)
Соединение 8 (0,86 г, 3,72 ммоль), NMM (1,14 мл, 10,4 ммоль), IBCF (0,63 мл, 4,83 ммоль) и соединение 13 (1,07 г, 3,90 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 27, (R)/(S)-трет-бутил(1-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)амино)-1-оксопентан-2-ил)метилкарбамата (0,79 г, 47%), получаемого в виде желтого порошка.
Rf=0,48 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 8,49 (с, C(O)NH), 7,58-7,64 (м, ArH), 7,47-7,51 (м, ArH), 7,38 (д, J=8,3 Гц, ArH), 4,66 (с, хиральный-H), 2,82 (с, NCH3), 1,67-2,04 (м, CH2CH2CH3), 1,51 (с, Boc), 1,33-1,39 (м, CH2CH2CH3), 0,99 (т, J=7,3 Гц, CH2CH2CH3).
Пример получения 23: Получение (R)/(S)-трет-бутил(1-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)амино)-1-оксогексан-2-ил)метилкарбамата (28)
Соединение 9 (R)/(S)-2-((трет-бутоксикарбонил)метиламино)гексановая кислота (0,84 г, 3,47 ммоль), NMM (1,10 мл, 9,71 ммоль), IBCF (0,58 мл, 4,51 ммоль) и соединение 13 (1,00 г, 3,64 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 28, (R)/(S)-трет-бутил(1-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)амино)-1-оксогексан-2-ил)метилкарбамата (0,86 г, 53%), получаемого в виде масла.
Rf=0,55 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 8,49 (с, C(O)NH), 7,58-7,64 (м, ArH), 7,47-7,51 (м, ArH), 7,38 (дд, J=2,1 Гц, 8,4 Гц, ArH), 4,64 (с, хиральный-H), 2,82 (с, NCH3), 1,67-2,01 (м, CH2CH2CH2CH3), 1,52 (с, Boc), 1,24-1,44 (м, CH2CH2CH2CH3), 0,93 (т, J=7,1 Гц, CH2CH2CH3).
Пример получения 24: Получение гидрохлорида (R)/(S)-2-амино-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)бутанамида (29)
Соединение 22 (1,06 г, 2,50 ммоль) и 4,0 M HCl (3,80 мл, 15,0 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 29, гидрохлорида (R)/(S)-2-амино-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)бутанамида (0,87 г, 97%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (CDCl3, 400 МГц) 11,05 (с, C(O)NH), 8,38 (с, NH3), 7,93 (д, J=1,9 Гц, ArH), 7,66-7,79 (м, ArH), 4,01-4,04 (м, хиральный-H), 1,86-1,91 (м, CH2CH3), 0,96 (т, J=7,5 Гц, CH2CH3).
Пример получения 25: Получение гидрохлорида (R)/(S)-2-амино-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)пентанамида (30)
Соединение 23 (0,58 г, 1,33 ммоль) и 4,0 M HCl (2,00 мл, 7,95 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 30, гидрохлорида (R)/(S)-2-амино-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)пентанамида (0,40 г, 81%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 11,05 (с, C(O)NH), 8,40 (с, NH3), 7,93 (д, J=1,5 Гц, ArH), 7,66-7,79 (м, ArH), 4,06 (с, хиральный-H), 1,79-1,85 (м, CH2CH2CH3), 1,36-1,43 (м, CH2CH2CH3), 0,91 (т, J=7,3 Гц, CH2CH2CH3).
Пример получения 26: Получение гидрохлорида (R)/(S)-2-амино-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)гексанамида (31)
Соединение 24 (1,10 г, 2,44 ммоль) и 4,0 M HCl (3,66 мл, 14,6 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 31, гидрохлорида (R)/(S)-2-амино-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)гексанамида (0,81 г, 86%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 10,97 (с, C(O)NH), 8,38 (с, NH3), 7,93 (д, J=2,0 Гц, ArH), 7,66-7,78 (м, ArH), 4,03 (с, хиральный-H), 1,81-1,87 (м, CH2CH2CH2CH3), 1,33-1,39 (м, CH2CH2CH2CH3), 0,87 (т, J=6,9 Гц, CH2CH2CH2CH3).
Пример получения 27: Получение гидрохлорида (R)/(S)-2-амино-N-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)пентанамида (32)
Соединение 25 (0,64 г, 1,41 ммоль) и 4,0 M HCl (2,12 мл, 8,46 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 32, гидрохлорида (R)/(S)-2-амино-N-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)пентанамида (0,51 г, 93%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 10,90 (с, C(O)NH), 8,35 (с, NH3), 7,76-7,79 (м, ArH), 7,70 (д, J=8,8 Гц, ArH), 7,45 (д, J=8,2 Гц, ArH), 4,03 (с, хиральный-H), 1,82 (кв, J=6,9 Гц, 7,9 Гц, CH2CH2CH3), 1,34-1,47 (м, CH2CH2CH3), 0,92 (т, J=7,3 Гц, CH2CH2CH3).
Пример получения 28: Получение гидрохлорида (R)/(S)-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)-2-(метиламино)бутанамида (33)
Соединение 26 (0,69 г, 1,58 ммоль) и 4,0 M HCl (2,40 мл, 9,50 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 33, гидрохлорида (R)/(S)-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)-2-(метиламино)бутанамида (0,55 г, 93%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 11,00 (с, C(O)NH), 9,13 (с, NH2), 7,94 (д, J=1,7 Гц, ArH), 7,66-7,78 (м, ArH), 3,96 (т, J=5,5 Гц, хиральный-H), 2,57 (с, NCH3), 1,87-2,05 (м, CH2CH3), 0,94 (т, J=7,5 Гц, CH2CH3).
Пример получения 29: Получение гидрохлорида (R)/(S)-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)-2-(метиламино)пентанамида (34)
Соединение 27 (0,38 г, 0,83 ммоль) и 4,0 M HCl (1,25 мл, 4,98 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 34, гидрохлорида (R)/(S)-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)-2-(метиламино)пентанамида (0,23 г, 71%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 10,98 (с, C(O)NH), 9,09 (с, NH2), 7,94 (с, ArH), 7,66-7,76 (м, ArH), 3,96 (т, J=6,1 Гц, хиральный-H), 2,57 (с, NCH3), 1,80-1,93 (м, CH2CH2CH3), 1,31-1,39 (м, CH2CH2CH3), 0,91 (т, J=7,3 Гц, CH2CH2CH3).
Пример получения 30: Получение гидрохлорида (R)/(S)-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)-2-(метиламино)гексанамида (35)
Соединение 28 (0,82 г, 1,77 ммоль) и 4,0 M HCl (2,65 мл, 10,6 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 35, гидрохлорида (R)/(S)-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)-2-(метиламино)гексанамида (0,60 г, 84%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 10,81 (с, C(O)NH), 9,05 (с, NH2), 7,94 (д, J=2,0 Гц, ArH), 7,66-7,77 (м, ArH), 3,92 (с, хиральный-H), 2,57 (с, NCH3), 1,87-1,99 (м, CH2CH2CH2CH3), 1,29-1,33 (м, CH2CH2CH2CH3)0,86 (т, J=6,8 Гц, CH2CH2CH2CH3).
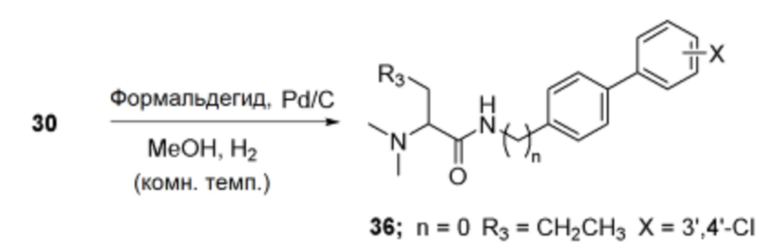
Пример получения 31: Получение (R)/(S)-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)-2-(диметиламино)пентанамида (36)
Соединение 30 (1,0 экв.), триэтиламин (6,0 экв.), формальдегид (2,0 экв.) и палладиевый катализатор (0,4 экв.) взаимодействовали между собой по схеме реакции g для синтеза соединения 36, (R)/(S)-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)-2-(диметиламино)пентанамида.
1H ЯМР (DMSO-d6, 400 МГц) 10,98 (с, C(O)NH), 7,94 (с, ArH), 7,66-7,76 (м, ArH), 3,96 (т, J=6,1 Гц, хиральный-H), 2,57 (с, N(CH3)2), 1,80-1,93 (м, CH2CH2CH3), 1,31-1,39 (м, CH2CH2CH3), 0,91 (т, J=7,3 Гц, CH2CH2CH3).
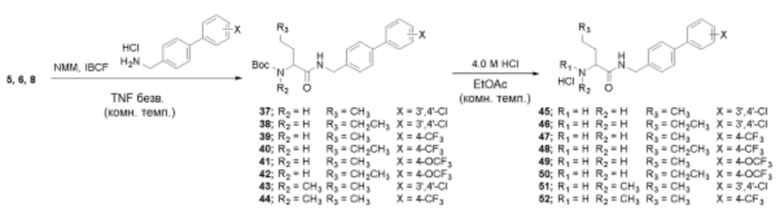
Пример получения 32: Получение (R)/(S)-трет-бутил(1-(((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)амино)-1-оксопентан-2-ил)карбамата (37)
Соединение 5 (0,57 г, 2,64 ммоль), NMM (0,73 мл, 6,60 ммоль), IBCF (0,45 мл, 3,43 ммоль) и соединение 15 (0,80 г, 2,77 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 37, (R)/(S)-трет-бутил(1-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)амино)-1-оксопентан-2-ил)карбамата (1,20 г, 100%), получаемого в виде белого порошка.
Rf=0,04 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 7,64 (д, J=2,0 Гц, ArH), 7,49 (дд, J=2,6 Гц, 8,5 Гц, ArH), 7,33-7,40 (м, ArH), 6,48 (с, C(O)NH), 4,93 (с, Boc-NH), 4,49 (д, J=4,2 Гц, NHCH2), 4,07-4,09 (м, хиральный-H), 1,36-1,43 (м, CH2CH2CH3, Boc), 0,95 (т, J=7,3 Гц, CH2CH2CH3).
Пример получения 33: Получение (R)/(S)-трет-бутил(1-(((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)амино)-1-оксогексан-2-ил)карбамата (38)
Соединение 6 (0,38 г, 1,65 ммоль), NMM (0,45 мл, 4,13 ммоль), IBCF (0,28 мл, 2,15 ммоль) и соединение 15 (0,50 г, 1,74 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 38, (R)/(S)-трет-бутил(1-((((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)амино)-1-оксогексан-2-ил)карбамата (0,46 г, 60%), получаемого в виде белого порошка.
Rf=0,19 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 7,63 (д, J=2,0 Гц, ArH), 7,49 (дд, J=4,3 Гц, 8,2 Гц, ArH), 7,33-7,39 (м, ArH), 6,55 (с, C(O)NH), 4,98 (д, J=3,8 Гц, Boc-NH), 4,49 (д, J=5,5 Гц, NHCH2), 4,07-4,11 (м, хиральный-H), 1,58-1,90 (м, CH2CH2CH2CH3), 1,42 (с, Boc), 1,34 (д, J=2,2 Гц, CH2CH2CH2CH3), 0,88-0,94 (м, CH2CH2CH2CH3).
Пример получения 34: Получение (R)/(S)-трет-бутил(1-оксо-1(((4’-(трифторметил)-[1,1’-бифенил]-4-ил)метил)амино)пентан-2-ил)карбамата (39)
Соединение 5 (0,36 г, 1,66 ммоль), NMM (0,46 мл, 4,14 ммоль), IBCF (0,28 мл, 2,15 ммоль) и соединение 16 (0,50 г, 1,74 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 39, (R)/(S)-трет-бутил(1-оксо-1-((((4’-(трифторметил)-[1,1’-бифенил]-4-ил)метил)амино)пентан-2-ил)карбамата (0,72 г, 96%), получаемого в виде белого порошка.
Rf=0,17 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 300 МГц) 7,62-7,70 (м, ArH), 7,44 (дд, J=8,1 Гц, 44,7 Гц, ArH), 6,85 (с, C(O)NH), 5,16-5,18 (м, Boc-NH), 4,47-4,49 (м, ArCH2), 4,11-4,15 (м, хиральный-H), 1,54-1,89 (м, CH2CH2CH3), 1,41 (с, Boc, CH2CH2CH3), 0,93 (т, J=7,2 Гц, CH2CH2CH3).
Пример получения 35: Получение (R)/(S)-трет-бутил(1-оксо-1(((4’-(трифторметил)-[1,1’-бифенил]-4-ил)метил)амино)гексан-2-ил)карбамата (40)
Соединение 6 (0,54 г, 2,32 ммоль), NMM (0,71 мл, 6,49 ммоль), IBCF (0,39 мл, 3,01 ммоль) и соединение 16 (0,70 г, 2,43 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 40, (R)/(S)-трет-бутил(1-оксо-1-((((4’-(трифторметил)-[1,1’-бифенил]-4-ил)метил)амино)гексан-2-ил)карбамата (0,87 г, 81%), получаемого в виде белого порошка.
Rf=0,16 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 7,70 (кв, J=4,6 Гц, 8,6 Гц, ArH), 7,57 (д, J=8,2 Гц, ArH), 7,39 (д, J=8,2 Гц, ArH), 6,50 (с, C(O)NH), 4,96 (с, Boc-NH), 4,53 (д, J=4,7 Гц, NHCH2), 4,09-4,10 (м, хиральный-H), 1,59-1,94 (м, CH2CH2CH2CH3), 1,45 (с, Boc), 1,37-1,38 (м, CH2CH2CH2CH3), 0,93 (т, J=7,0 Гц, CH2CH2CH2CH3).
Пример получения 36: Получение (R)/(S)-трет-бутил(1-оксо-1(((4’-(трифторметокси)-[1,1’-бифенил]-4-ил)метил)амино)пентан-2-ил)карбамата (41)
Соединение 5 (0,55 г, 2,51 ммоль), NMM (0,77 мл, 7,02 ммоль), IBCF (0,42 мл, 3,26 ммоль) и соединение 17 (0,80 г, 2,63 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 41, (R)/(S)-трет-бутил(1-оксо-1-((((4’-(трифторметокси)-[1,1’-бифенил]-4-ил)метил)амино)пентан-2-ил)карбамата (1,05 г, 90%), получаемого в виде белого порошка.
Rf=0,09 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 7,57-7,61 (м, ArH), 7,53 (д, J=8,2 Гц, ArH), 7,37 (д, J=8,2 Гц, ArH), 7,30 (д, J=8,2 Гц, ArH), 6,47 (с, C(O)NH), 4,96 (с, Boc-NH), 4,52 (с, NHCH2), 4,10-4,11 (м, хиральный-H), 1,59-1,94 (м, CH2CH2CH3), 1,37-1,45 (м, CH2CH2CH3, Boc), 0,97 (т, J=7,3 Гц, CH2CH2CH3).
Пример получения 37: Получение (R)/(S)-трет-бутил(1-оксо-1(((4’-(трифторметокси)-[1,1’-бифенил]-4-ил)метил)амино)гексан-2-ил)карбамата (42)
Соединение 6 (0,58 г, 2,51 ммоль), NMM (0,77 мл, 7,02 ммоль), IBCF (0,42 мл, 3,26 ммоль) и соединение 17 (0,80 г, 2,63 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 42, (R)/(S)-трет-бутил(1-оксо-1-((((4’-(трифторметокси)-[1,1’-бифенил]-4-ил)метил)амино)гексан-2-ил)карбамата (1,06 г, 88%), получаемого в виде белого порошка.
Rf=0,18 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 7,58-7,61 (м, ArH), 7,53 (д, J=8,1 Гц, ArH), 7,37 (д, J=8,1 Гц, ArH), 7,30 (д, J=8,5 Гц, ArH), 6,52 (с, C(O)NH), 4,99 (с, Boc-NH), 4,53 (д, J=5,1 Гц, NHCH2), 4,09-4,11 (м, хиральный-H), 1,62-1,94 (м, CH2CH2CH2CH3), 1,45 (с, Boc), 1,36-1,37 (м, CH2CH2CH2CH3), 0,92 (т, J=6,9 Гц, CH2CH2СН2СН3).
Пример получения 38: Получение (R)/(S)-трет-бутил(1-((((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)амино)-1-оксопентан-2-ил)метилкарбамата (43)
Соединение 8 (0,38 г, 1,65 ммоль), NMM (0,46 мл, 4,13 ммоль), IBCF (0,28 мл, 2,15 ммоль) и соединение 15 (0,50 г, 1,73 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 43, (R)/(S)-трет-бутил(1-(((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)амино)-1-оксопентан-2-ил)метилкарбамата (0,57 г, 73%), получаемого в виде масла.
Rf=0,18 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 300 МГц) 7,30-7,68 (м, ArH), 6,27-6,64 (м, C(O)NH), 4,41-4,59 (м, NHCH2, хиральный-H), 2,78 (с, NCH3), 2,04-1,63 (м, CH2CH2CH3), 1,44 (с, Boc), 1,32-1,26 (м, CH2CH2CH3), 0,96 (т, J=7,3 Гц, CH2CH2CH3).
Пример получения 39: Получение (R)/(S)-трет-бутилметил(1-оксо-1(((4’-(трифторметил)-[1,1’-бифенил]-4-ил)метил)амино)пентан-2-ил)карбамата (44)
Соединение 8 (0,38 г, 1,66 ммоль), NMM (0,46 мл, 4,14 ммоль), IBCF (0,28 мл, 2,15 ммоль) и соединение 16 (0,50 г, 1,74 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 44, (R)/(S)-трет-бутилметил(1-оксо-1-(((4’-(трифторметил)-[1,1’-бифенил]-4-ил)метил)амино)пентан-2-ил)карбамата (0,49 г, 64%), получаемого в виде масла.
Rf=0,22 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 300 МГц) 7,64-7,71 (м, ArH), 7,44 (дд, J=7,9 Гц, 53,4 Гц, ArH), 6,28-6,64 (м, C(O)NH), 4,43-4,61 (м, NHCH2, хиральный-H), 2,78 (с, NCH3), 1,63-2,04 (м, CH2CH2CH3), 1,44 (с, Boc), 1,26-1,35 (м, CH2CH2CH3), 0,96 (т, J=7,3 Гц, CH2CH2CH3).
Пример получения 40: Получение гидрохлорида (R)/(S)-2-амино-N-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)пентанамида (45)
Соединение 37 (1,19 г, 2,64 ммоль) и 4,0 M HCl (3,95 мл, 15,8 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 45, гидрохлорида (R)/(S)-2-амино-N-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)пентанамида (0,73 г, 71%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 300 МГц) 9,04 (с, C(O)NH), 8,21 (с, NH3), 7,94 (с, ArH), 7,66-7,73 (м, ArH), 7,40 (д, J=8,1 Гц, ArH), 4,39-4,41 (м, NHCH2), 3,78-3,82 (т, J=6,3 Гц, хиральный-H), 1,68-1,76 (м, CH2CH2CH3), 1,29-1,39 (м, CH2CH2CH3), 0,89 (т, J=7,2 Гц, CH2CH2CH3).
Пример получения 41: Получение гидрохлорида (R)/(S)-2-амино-N-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)гексанамида (46)
Соединение 38 (0,46 г, 0,99 ммоль) и 4,0 M HCl (1,48 мл, 1,48 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 46, гидрохлорида (R)/(S)-2-амино-N-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)гексанамида (0,27 г, 85%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 9,17 (м, C(O)NH), 8,32 (с, NH3), 7,94 (д, J=2,0 Гц, ArH), 7,66-7,73 (м, ArH), 7,41 (д, J=8,2 Гц, ArH), 4,34-4,45 (м, NHCH2), 3,81 (т, J=6,1 Гц, хиральный-H), 1,75-1,76 (м, CH2CH2CH2CH3), 1,27-1,28 (м, CH2CH2CH2CH3), 0,85 (т, J=6,6 Гц, CH2CH2CH2CH3).
Пример получения 42: Получение гидрохлорида (R)/(S)-2-амино-N-((4’-(трифторметил)-[1,1’-бифенил]-4-ил)метил)пентанамида (47)
Соединение 39 (0,70 г, 1,55 ммоль) и 4,0 M HCl (2,33 мл, 9,32 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 47, гидрохлорида (R)/(S)-2-амино-N-((4’-(трифторметил)-[1,1’-бифенил]-4-ил)метил)пентанамида (0,60 г, 99%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 300 МГц) 9,37 (т, J=5,7 Гц, C(O)NH), 8,46 (с, NH3), 7,71-7,92 (м, ArH), 7,45-7,51 (м, ArH), 4,40-4,43 (м, NHCH2), 3,89 (с, хиральный-H), 1,74-1,82 (м, CH2CH2CH3), 1,16-1,42 (м, CH2CH2CH3)0,89 (т, J=7,2 Гц, CH2CH2CH3).
Пример получения 43: Получение гидрохлорида (R)/(S)-2-амино-N-((4’-(трифторметил)-[1,1’-бифенил]-4-ил)метил)гексанамида (48)
Соединение 40 (0,85 г, 1,83 ммоль) и 4,0 M HCl (2,75 мл, 11,0 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 48, гидрохлорида (R)/(S)-2-амино-N-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)метил)гексанамида (0,72 г, 98%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 9,09 (с, C(O)NH), 8,23 (с, NH3), 7,89 (д, J=8,2 Гц, ArH), 7,82 (д, J=8,4 Гц, ArH), 7,73 (д, J=8,1 Гц, ArH), 7,44 (д, J=8,1 Гц, ArH), 4,36-4,46 (м, NHCH2), 3,80 (т, J=6,4 Гц, хиральный-H), 1,74-1,76 (м, CH2CH2CH2CH3), 1,27-1,29 (м, CH2CH2CH2CH3), 0,85 (т, J=6,5 Гц, CH2CH2CH2CH3).
Пример получения 44: Получение гидрохлорида (R)/(S)-2-амино-N-((4’-(трифторметокси)-[1,1’-бифенил]-4-ил)метил)пентанамида (49)
Соединение 41 (1,04 г, 2,22 ммоль) и 4,0 M HCl (3,34 мл, 13,3 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 49, гидрохлорида (R)/(S)-2-амино-N-((4’-(трифторметокси)-[1,1’-бифенил]-4-ил)метил)пентанамида (0,82 г, 92%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 9,14 (т, J=5,7 Гц, C(O)NH), 8,30 (с, NH3), 7,77-7,81 (м, ArH), 7,67 (д, J=8,2 Гц, ArH), 7,46 (д, J=8,1 Гц, ArH), 7,41 (д, J=8,2 Гц, ArH), 4,40 (д, J=5,8 Гц, NHCH2), 3,82 (т, J=6,5 Гц, хиральный-H), 1,71-1,76 (м, CH2CH2CH3), 1,28-1,38 (м, CH2CH2CH3), 0,89 (т, J=7,3 Гц, CH2CH2CH3).
Пример получения 45: Получение гидрохлорида (R)/(S)-2-амино-N-((4’-(трифторметокси)-[1,1’-бифенил]-4-ил)метил)гексанамида (50)
Соединение 42 (1,04 г, 2,17 ммоль) и 4,0 M HCl (3,26 мл, 13,0 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 50, гидрохлорида (R)/(S)-2-амино-N-((4’-(трифторметокси)-[1,1’-бифенил]-4-ил)метил)гексанамида (0,88 г, 97%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 9,09 (т, J=5,8 Гц, C(O)NH), 8,25 (с, NH3), 7,78 (д, J=8,7 Гц, ArH), 7,66 (д, J=8,2 Гц, ArH), 7,45 (д, J=8,3 Гц, ArH), 7,41 (д, J=8,2 Гц, ArH), 4,35-4,44 (м, NHCH2), 3,80 (т, J=6,4 Гц, хиральный-H), 1,72-1,75 (м, CH2CH2CH2CH3), 1,27-1,28 (м, CH2CH2CH2CH3), 0,85 (т, J=6,5 Гц, CH2СН2СН2СН3).
Пример получения 46: Получение гидрохлорида (R)/(S)-N-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)-2-(метиламино)пентанамида (51)
Соединение 43 (0,84 г, 1,81 ммоль) и 4,0 M HCl (2,80 мл, 10,9 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 51, гидрохлорида (R)/(S)-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)-2-(метиламино)пентанамида (0,64 г, 88%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 300 МГц) 9,10-9,78 (м, NH2), 9,57 (т, J=5,5 Гц, C(O)NH), 7,96 (с, ArH), 7,69-7,72 (м, ArH), 7,43 (д, J=10,7 Гц, ArH), 4,42-4,44 (м, NHCH2), 3,85-3,90 (м, хиральный-H), 2,48 (с, NCH3), 1,76-1,90 (м, CH2CH2CH3), 1,26-1,38 (м, CH2CH2CH3), 0,90 (т, J=7,1 Гц, CH2CH2CH3).
Пример получения 47: Получение гидрохлорида (R)/(S)-2-(метиламино)-N-((4’-(трифторметил)-[1,1’-бифенил]-4-ил)метил)пентанамида (52)
Соединение 44 (0,45 г, 0,97 ммоль) и 4,0 M HCl (1,45 мл, 5,81 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 52, гидрохлорида (R)/(S)-2-(метиламино)-N-((4’-(трифторметил)-[1,1’-бифенил]-4-ил)метил)пентанамида (0,32 г, 82%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 300 МГц) 9,03-9,81 (м, NH2), 9,52 (т, J=5,5 Гц, C(O)NH), 7,72-7,92 (м, ArH), 7,46 (д, J=8,2 Гц, ArH), 4,43-4,45 (м, NHCH2), 3,86 (с, хиральный-H), 2,48 (с, NCH3), 1,77-1,89 (м, CH2CH2CH3), 1,25-1,38 (м, CH2CH2CH3), 0,90 (т, J=7,2 Гц, CH2CH2CH3).
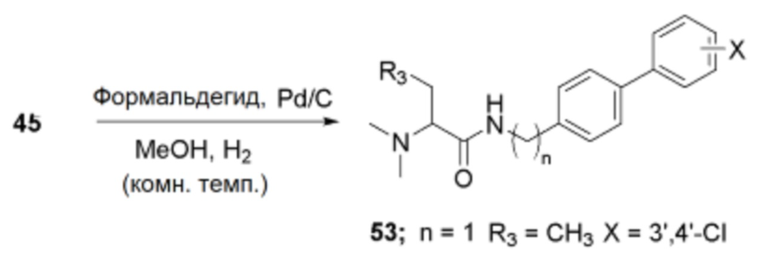
Пример получения 48: Получение (R)/(S)-N-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)-2-(диметиламино)пентанамида (53)
Соединение 45 (1,0 экв.), триэтиламин (6,0 экв.), формальдегид (1,05 экв.) и палладиевый катализатор (0,2 экв.) взаимодействовали между собой по схеме реакции g для синтеза соединения 53, (R)/(S)-N-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)-2-(диметиламино)пентанамида.
1H ЯМР (DMSO-d6, 300 МГц) 9,57 (т, J=5,5 Гц, C(O)NH), 7,96 (с, ArH), 7,69-7,72 (м, ArH), 7,43 (д, J=10,7 Гц, ArH), 4,42-4,44 (м, NHCH2), 3,85-3,90 (м, хиральный-H), 2,48 (с, N (CH3)2), 1,76-1,90 (м, CH2CH2CH3), 1,26-1,38 (м, CH2CH2CH3), 0,90 (т, J=7,1 Гц, CH2CH2CH3).
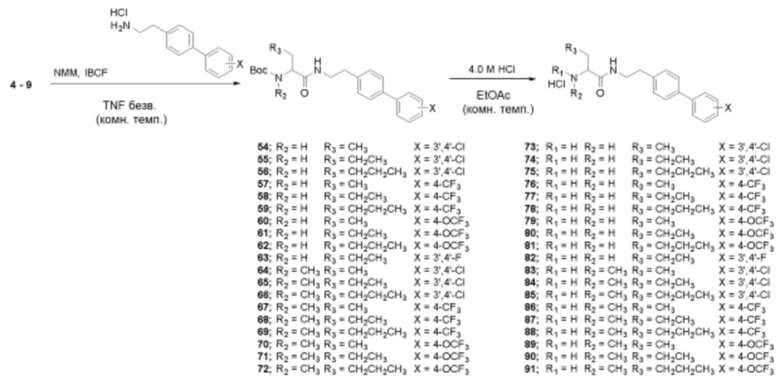
Пример получения 49: Получение (R)/(S)-трет-бутил(1-((2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)амино)-1-оксобутан-2-ил)карбамата (54)
Соединение 4 (0,32 г, 1,57 ммоль), NMM (0,43 мл, 3,93 ммоль), IBCF (0,27 мл, 2,05 ммоль) и соединение 18 (0,50 г, 1,65 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 54, (R)/(S)-трет-бутил(1-((2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)амино)-1-оксобутан-2-ил)карбамата (0,66 г, 93%), получаемого в виде белого порошка.
Rf=0,05 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 7,65 (с, ArH), 7,47-7,50 (м, ArH), 7,39 (д, J=8,2 Гц, ArH), 7,26-7,28 (м, ArH), 6,07 (с, C(O)NH), 4,94 (с, Boc-NH), 3,93 (д, J=6,6 Гц, хиральный-H), 3,50-3,94 (м, NHCH2CH2), 2,86 (т, J=6,9 Гц, NHCH2CH2), 1,55-2,88 (м, CH2CH3), 1,43 (с, Boc), 0,91 (т, J=7,4 Гц, CH2CH3).
Пример получения 50: Получение (R)/(S)-трет-бутил(1-((2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)амино)-1-оксопентан-2-ил)карбамата (55)
Соединение 5 (0,50 г, 2,30 ммоль), NMM (0,51 мл, 4,60 ммоль), IBCF (0,39 мл, 2,99 ммоль) и соединение 18 (0,73 г, 2,42 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 55, (R)/(S)-трет-бутил(1-((2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)амино)-1-оксопентан-2-ил)карбамата (0,96 г, 89%), получаемого в виде белого порошка.
Rf=0,26 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 7,46-7,64 (м, ArH), 7,26-7,40 (м, ArH), 6,36 (с, C(O)NH), 5,07 (с, Boc-NH), 4,01- 4,03 (м, хиральный-H), 3,47-3,61 (м, NHCH2CH2), 2,85 (т, J=7,0 Гц, NHCH2CH2), 2,67 (с, NCH3), 1,48-1,80 (м, CH2CH2CH3), 1,42 (с, Boc), 1,26-1,36 (м, CH2CH2CH3), 0,90 (т, J=7,2 Гц, CH2CH2CH3).
Пример получения 51: Получение (R)/(S)-трет-бутил(1-((2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)амино)-1-оксогексан-2-ил)карбамата (56)
Соединение 6 (0,36 г, 1,57 ммоль), NMM (0,43 мл, 3,93 ммоль), IBCF (0,27 г, 2,05 ммоль) и соединение 18 (0,50 г, 16,5 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 56, (R)/(S)-трет-бутил(1-((2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)амино)-1-оксогексан-2-ил)карбамата (0,76 г, 100%), получаемого в виде белого порошка.
Rf=0,07 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 7,65 (с, ArH), 7,48 (д, J=6,3 Гц, ArH), 7,39 (д, J=8,0 Гц, ArH), 7,26-7,28 (м, ArH), 6,07 (с, C(O)NH), 4,91 (с, Boc-NH), 3,97 (д, J=5,6 Гц, хиральный-H), 3,51-3,62 (м, NHCH2CH2), 2,86 (т, J=6,1 Гц, NHCH2CH2), 1,51-1,81 (м, CH2CH2CH2CH3), 1,42 (с, Boc), 1,27 (д, J=6,7 Гц, CH2CH2CH2CH3), 0,87 (д, J=5,5 Гц, CH2CH2CH2CH3).
Пример получения 52: Получение (R)/(S)-трет-бутил(1-оксо-1-((2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)амино)бутан-2-ил)карбамата (57)
Соединение 4 (0,42 г, 2,08 ммоль), NMM (0,57 мл, 5,21 ммоль), IBCF (0,35 мл, 2,71 ммоль) и соединение 19 (0,66 г, 2,19 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 57, (R)/(S)-трет-бутил(1-оксо-1-((2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)амино)бутан-2-ил)карбамата (0,92 г, 98%), получаемого в виде белого порошка.
Rf=0,16 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 7,70 (с, ArH), 7,57 (д, J=8,0 Гц, ArH), 7,29-7,33 (м, ArH), 6,15 (с, C(O)NH), 5,00 (s, Boc-NH), 3,94-3,99 (м, хиральный-H), 3,53-3,66 (м, NHCH2CH2), 2,90 (т, J=7,0 Гц, NHCH2CH2), 1,58-1,90 (м, CH2CH3), 1,45 (с, Boc), 0,93 (т, J=7,5 Гц, CH2CH3).
Пример получения 53: Получение (R)/(S)-трет-бутил(1-оксо-1-((2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)амино)пентан-2-ил)карбамата (58)
Соединение 5 (0,55 г, 2,53 ммоль), NMM (0,69 мл, 6,31 ммоль), IBCF (0,43 мл, 3,28 ммоль) и соединение 19 (0,80 г, 2,65 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 58, (R)/(S)-трет-бутил(1-оксо-1-((2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)амино)пентан-2-ил)карбамата (1,05 г, 89%), получаемого в виде белого порошка.
Rf=0,20 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 300 МГц) 7,67 (с, ArH), 7,41 (дд, J=8,1 Гц, 63,6 Гц, ArH), 6,36 (с, C(O)NH), 5,05-5,07 (м, Boc-NH), 4,00-4,05 (м, хиральный-H), 3,45-3,66 (м, NHCH2CH2), 2,87 (т, J=7,1 Гц, NHCH2CH2), 1,50-1,83 (м, CH2CH2CH3), 1,42 (с, Boc), 1,28-1,39 (м, CH2CH2CH3), 0,90 (т, J=7,2 Гц, CH2CH2CH3).
Пример получения 54: Получение (R)/(S)-трет-бутил(1-оксо-1-((2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил).)амино)карбамата (59)
Соединение 6 (0,76 г, 3,16 ммоль), NMM (0,87 мл, 7,89 ммоль), IBCF (0,53 мл, 4,10 ммоль) и соединение 19 (1,00 г, 3,31 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 59, (R)/(S)-трет-бутил(1-оксо-1-((2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)амино)карбамата (1,34 г, 89%), получаемого в виде белого порошка.
Rf=0,13 (EtOAc:н-гексан 1:3);
1H ЯМР (DMSO-d6, 400 МГц) 7,87 (д, J=8,2 Гц, ArH, C(O)NH), 7,80 (д, J=8,5 Гц, ArH), 7,66 (д, J=8,1 Гц, ArH), 7,35 (д, J=8,1 Гц, ArH), 6,73 (д, J=8,2 Гц, Boc-NH), 3,83 (кв, J=5,6 Гц, 8,4 Гц, хиральный-H), 3,24-3,43 (м, NHCH2CH2), 2,77 (т, J=7,0 Гц, NHCH2CH2), 1,37-1,52 (м, CH2CH2CH2CH3, Boc), 1,15-1,20 (м, CH2CH2CH2CH3), 0,80 (т, J=6,8 Гц, CH2CH2CH2CH3).
Пример получения 55: Получение (R)/(S)-трет-бутил(1-оксо-1-((2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)амино)бутан-2-ил)карбамата (60)
Соединение 4 (0,49 г, 2,40 ммоль), NMM (0,66 мл, 6,00 ммоль), IBCF (0,41 мл, 3,12 ммоль) и соединение 20 (0,80 г, 2,52 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 60, (R)/(S)-трет-бутил(1-оксо-1-((2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)амино)бутан-2-ил)карбамата (0,94 г, 84%), получаемого в виде белого порошка.
Rf=0,14 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 7,68-7,73 (м, ArH), 7,51-7,62 (м, ArH), 7,29-7,33 (м, ArH), 6,17 (д, J=5,6 Гц, C(O)NH), 5,01 (с, NH), 3,95-3,98 (м, хиральный-H), 3,51-3,67 (м, NHCH2CH2), 2,87-2,92 (м, NHCH2CH2), 1,56-1,92 (м, CH2CH3), 1,45 (с, Boc), 0,93 (т, J=7,4 Гц, CH2CH3).
Пример получения 56: Получение (R)/(S)-трет-бутил(1-оксо-1-((2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил))амино)пентан-2-ил)карбамата (61)
Соединение 5 (0,33 г, 1,50 ммоль), NMM (0,41 мл, 3,75 ммоль), IBCF (0,25 мл, 1,95 ммоль) и соединение 20 (0,50 г, 1,57 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 61, (R)/(S)-трет-бутил(1-оксо-1-((2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)амино)пентан-2-ил)карбамата (0,70 г, 98%), получаемого в виде белого порошка.
Rf=0,12 (EtOAc:н-гексан 1:3);
1H ЯМР (DMSO-d6, 400 МГц) 7,88 (т, J=5,6 Гц, C(O)NH), 7,74-7,77 (м, ArH), 7,59 (д, J=8,2 Гц, ArH), 7,44 (д, J=8,1 Гц, ArH), 7,32 (д, J=8,2 Гц, ArH), 6,74 (д, J=8,2 Гц, Boc-NH), 4,86 (кв, J=5,6 Гц, 8,2 Гц, хиральный-H), 3,26-3,40 (м, NHCH2CH2), 2,76 (т, J=7,0 Гц, NHCH2CH2), 1,37-1,52 (м, CH2CH2CH3, Boc), 1,17-1,25 (м, CH2CH2CH3), 0,81 (т, J=7,3 Гц, CH2CH2CH3).
Пример получения 57: Получение (R)/(S)-трет-бутил(1-оксо-1-((2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил))амино)гексан-2-ил)карбамата (62)
Соединение 6 (0,50 г, 2,18 ммоль), NMM (0,60 мл, 5,45 ммоль), IBCF (0,37 мл, 2,83 ммоль) и соединение 20 (0,72 г, 2,29 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 62, (R)/(S)-трет-бутил(1-оксо-1-((2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)амино)гексан-2-ил)карбамата (0,95 г, 88%), получаемого в виде белого порошка.
Rf=0,13 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 7,70-7,73 (м, ArH), 7,57 (д, J=8,1 Гц, ArH), 7,32 (д, J=8,1 Гц, ArH), 6,15 (с, C(O)NH), 4,95 (s, Boc-NH), 4,00 (q, J=6,4 Гц, 7,2 Гц, хиральный-H), 3,53-3,65 (м, NHCH2CH2), 2,90 (т, J=7,0 Гц, NHCH2CH2), 1,52-1,85 (м, CH2CH2CH2CH3), 1,45 (с, Boc), 1,31-1,34 (м, CH2CH2CH2CH3), 0,90 (т, J=6,9 Гц, CH2CH2CH2CH3).
Пример получения 58: Получение (R)/(S)-трет-бутил(1-((2-(3’,4’-дифтор-[1,1’-бифенил]-4-ил)этил)амино)-1-оксопентан-2-ил)карбамата (63)
Соединение 5 (0,50 г, 2,29 ммоль), NMM (0,70 мл, 6,40 ммоль), IBCF (0,39 мл, 2,97 ммоль) и соединение 21 (0,80 г, 2,40 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 63, (R)/(S)-трет-бутил(1-((2-(3’,4’-дифтор-[1,1’-бифенил]-4-ил)этил)амино)-1-оксопентан-2-ил)карбамата (0,97 г, 98%), получаемого в виде белого порошка.
Rf=0,10 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 7,46 (д, J=7,9 Гц, ArH), 7,33-7,38 (м, ArH), 7,17-7,27 (м, ArH), 6,11 (с, C(O)NH), 4,92 (с, Boc-NH), 3,96-4,01 (м, хиральный-H), 3,47-3,63 (м, NHCH2CH2), 2,85 (т, J=7,0 Гц, NHCH2CH2), 1,48- 1,82 (м, CH2CH2CH3), 1,42 (с, Boc), 1,26-1,36 (м, CH2CH2CH3), 0,90 (т, J=7,3 Гц, CH2CH2CH3).
Пример получения 59: Получение (R)/(S)-трет-бутил(1-((2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)амино)-1-оксобутан-2-ил)метилкарбамата (64)
Соединение 7 (0,82 г, 3,78 ммоль), NMM (1,04 мл, 9,44 ммоль), IBCF (0,64 мл, 4,91 ммоль) и соединение 18 (1,20 г, 3,97 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 64, (R)/(S)-трет-бутил(1-((2-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)амино)-1-оксобутан-2-ил)метилкарбамата (1,38 г, 79%), получаемого в виде масла.
Rf=0,22 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 7,66 (д, J=2,0 Гц, ArH), 7,49-7,53 (м, ArH), 7,41 (дд, J=2,1 Гц, 6,3 Гц, ArH), 7,27-7,29 (м ArH), 6,26 (с, C(O)NH), 4,44 (с, хиральный-H), 3,55-3,64 (м, NHCH2CH2), 2,87 (т, J=6,7 Гц, NHCH2CH2), 2,70 (с, NCH3), 1,61-1,98 (м, CH2CH3), 1,45 (с, Boc), 0,88 (т, J=7,4 Гц, CH2CH3).
Пример получения 60: Получение (R)/(S)-трет-бутил(1-((2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)амино)-1-оксопентан-2-ил)метилкарбамата (65)
Соединение 8 (0,35 г, 1,51 ммоль), NMM (0,33 мл, 3,03 ммоль), IBCF (0,26 мл, 1,97 ммоль) и соединение 18 (0,48 г, 1,59 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 65, (R)/(S)-трет-бутил(1-((2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)амино)-1-оксопентан-2-ил)метилкарбамата (0,69 г, 95%), получаемого в виде масла.
Rf=0,33 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 7,46-7,64 (м, ArH), 7,35-7,40 (м, ArH), 7,25-7,27 (м, ArH), 5,95-6,25 (м, C(O)NH), 4,51 (с, хиральный-H), 3,53-3,60 (м, NHCH2CH2), 2,84 (т, J=6,6 Гц, NHCH2CH2), 2,67 (с, NCH3), 1,57-1,90 (м, CH2CH2CH3), 1,42 (с, Boc), 1,22-1,32 (м, CH2CH2CH3), 0,95 (т, J=7,3 Гц, CH2CH2CH3).
Пример получения 61: Получение (R)/(S)-трет-бутил(1-((2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)амино)-1-оксогексан-2-ил)метилкарбамата (66)
Соединение 9 (0,29 г, 1,20 ммоль), NMM (0,33 мл, 2,99 ммоль), IBCF (0,20 мл, 0,16 ммоль) и соединение 18 (0,38 г, 1,26 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 66, (R)/(S)-трет-бутил(1-((2-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)амино)-1-оксогексан-2-ил)метилкарбамата (0,60 г, 97%), получаемого в виде масла.
Rf=0,30 (EtOAc:н-гексан 1:3);
1H ЯМР (DMSO-d6, 400 МГц) 7,89 (д, J=1,9 Гц, ArH, C(O)NH), 7,62-7,71 (м, ArH), 7,30 (д, J=8,1 Гц, ArH), 4,27-4,47 (м, хиральный-H), 3,32-3,35 (м, NHCH2CH2), 2,77 (т, J=6,5 Гц, NHCH2CH2), 2,67 (с, NCH3), 1,53-1,71 (м, CH2CH2CH2CH3), 1,39 (с, Boc), 1,12-1,28 (м, CH2CH2CH2CH3), 0,84 (с, CH2CH2CH2CH3).
Пример получения 62: Получение (R)/(S)-трет-бутилметил(1-оксо-1((2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)амино)бутан-2-ил)карбамата (67)
Соединение 7 (0,62 г, 2,84 ммоль), NMM (0,87 мл, 7,95 ммоль), IBCF (0,48 мл, 3,69 ммоль) и соединение 19 (0,90 г, 2,98 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 67, (R)/(S)-трет-бутилметил(1-оксо-1-((2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)амино)бутан-2-ил)карбамата (0,89 г, 67%), получаемого в виде белого порошка.
Rf=0,15 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 7,70 (т, J=9,7 Гц, ArH), 7,56 (д, J=7,9 Гц, ArH), 7,28-7,32 (м, ArH), 5,94-6,22 (м, C(O)NH), 4,45 (с, хиральный-H), 3,57-3,64 (м, NHCH2CH2), 2,89 (д, J=6,1 Гц, NHCH2CH2), 2,70 (с, NCH3), 1,61-1,99 (м, CH2CH3), 1,45 (с, Boc), 0,89 (т, J=7,3 Гц, CH2CH3).
Пример получения 63: Получение (R)/(S)-трет-бутилметил(1-оксо-1((2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)амино)пентан-2-ил)карбамата (68)
Соединение 8 (0,58 г, 2,53 ммоль), NMM (0,69 мл, 6,31 ммоль), IBCF (0,43 мл, 3,28 ммоль) и соединение 19 (0,80 г, 2,65 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 68, (R)/(S)-трет-бутилметил(1-оксо-1-((2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)амино)пентан-2-ил)карбамата (1,19 г, 98%), получаемого в виде масла.
Rf=0,26 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 300 МГц) 7,64-7,70 (м, ArH), 7,41 (дд, J=8,1 Гц, 67,4 Гц, ArH), 5,95-6,21 (с, C(O)NH), 4,52 (с, хиральный-H), 3,54-3,63 (м, NHCH2CH2), 2,86 (т, J=6,8 Гц, NHCH2CH2), 2,67 (с, NCH3), 1,55-1,91 (м, CH2CH2CH3), 1,42 (с, Boc), 1,23-1,27 (м, CH2CH2CH3), 0,88-0,95 (м, CH2CH2CH3).
Пример получения 64: Получение (R)/(S)-трет-бутилметила ((1-оксо-1((2-(4’-(трифторметил)-[1,1’-бифенил]-4-ила)этил)амино)гексан-2-ил)карбамата (69)
Соединение 9 (0,70 г, 2,84 ммоль), NMM (0,87 мл, 7,95 ммоль), IBCF (0,48 мл, 3,69 ммоль) и соединение 19 (0,90 г, 2,98 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 69, (R)/(S)-трет-бутилметил(1-оксо-1-((2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)амино)гексан-2-ил)карбамата (0,88 г, 63%), получаемого в виде желтого порошка.
Rf=0,32 (EtOAc:н-гексан 1:3);
1H ЯМР (DMSO-d6, 400 МГц) 7,87 (д, J=8,3 Гц, ArH, C(O)NH), 7,80 (д, J=8,4 Гц, ArH), 7,66 (д, J=8,2 Гц, ArH), 7,33 (д, J=8,1 Гц, ArH), 4,27-4,47 (м, хиральный-H), 3,32-3,33 (м, NHCH2CH2), 2,78 (т, J=6,8 Гц, NHCH2CH2), 2,66 (с, NCH3), 1,53-1,71 (м, CH2CH2CH2CH3), 1,39 (с, Boc), 1,12-1,28 (м, CH2CH2CH2CH3)0,84-0,89 (м, CH2CH2CH2CH3).
Пример получения 65: Получение (R)/(S)-трет-бутилметил(1-оксо-1((2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)амино)бутан-2-ил)карбамата (70)
Соединение 7 (0,65 г, 3,00 ммоль), NMM (0,92 мл, 8,99 ммоль), IBCF (0,51 мл, 3,90 ммоль) и соединение 20 (1,00 г, 3,15 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 70, (R)/(S)-трет-бутилметил(1-оксо-1-((2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)амино)бутан-2-ил)карбамата (1,00 г, 69%), получаемого в виде масла.
Rf=0,19 (EtOAc:н-гексан 1:3);
1H ЯМР (CDCl3, 400 МГц) 7,51-7,70 (м, ArH), 7,29 (т, J=3,7 Гц, ArH), 5,98-6,26 (м, C(O)NH), 4,45 (с, хиральный-H), 3,59-3,60 (м, NHCH2CH2), 2,87 (с, NHCH2CH2), 2,70 (д, J=3,2 Гц, NCH3), 1,62-2,07 (м, CH2CH3), 1,45 (с, Boc), 0,90 (м, CH2CH3).
Пример получения 66: Получение (R)/(S)-трет-бутилметил(1-оксо-1((2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил))амино)пентан-2-ил)карбамата (71)
Соединение 8 (0,25 г, 1,08 ммоль), NMM (0,30 мл, 2,69 ммоль), IBCF (0,18 мл, 1,40 ммоль) и соединение 20 (0,36 г, 1,13 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 71, (R)/(S)-трет-бутилметил(1-оксо-1-((2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)амино)пентан-2-ил)карбамата (0,32 г, 60%), получаемого в виде белого порошка.
Rf=0,18 (EtOAc:н-гексан 1:3);
1H ЯМР (DMSO-d6, 400 МГц) 7,89 (с, C(O)NH), 7,76 (д, J=8,4 Гц, ArH), 7,59 (д, J=7,8 Гц, ArH), 7,44 (д, J=8,2 Гц, ArH), 7,30 (д, J=7,8 Гц, ArH), 4,30-4,49 (м, хиральный-H), 3,28-3,40 (м, NHCH2CH2), 2,77 (с, NHCH2CH2), 2,66 (с, NCH3), 1,53-1,90 (м, CH2CH2CH3), 1,39 (с, Boc), 1,16-1,23 (м, CH2CH2CH3), 0,88-0,89 (м, CH2CH2CH3).
Пример получения 67: Получение (R)/(S)-трет-бутилметил(1-оксо-1((2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил))амино)гексан-2-ил)карбамата (72)
Соединение 9 (0,74 г, 3,00 ммоль), NMM (0,92 мл, 8,99 ммоль), IBCF (0,51 мл, 3,90 ммоль) и соединение 20 (1,00 г, 3,15 ммоль) взаимодействовали между собой по схеме реакции e для синтеза соединения 72, (R)/(S)-трет-бутилметил(1-оксо-1-((2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)амино)гексан-2-ил)карбамата (0,92 г, 60%), получаемого в виде масла.
Rf=0,29 (EtOAc:н-гексан 1:3);
1H ЯМР (DMSO-d6, 400 МГц) 7,88 (с, C(O)NH), 7,74-7,77 (м, ArH), 7,59 (д, J=8,2 Гц, ArH), 7,44 (д, J=8,0 Гц, ArH), 7,30 (д, J=8,1 Гц, ArH), 4,28-4,47 (м, хиральный-H), 3,30-3,34 (м, NHCH2CH2), 2,77 (т, J=6,7 Гц, NHCH2CH2), 2,66 (с, NCH3), 1,53-1,72 (м, CH2CH2CH2CH3), 1,39 (с, Boc), 1,12-1,29 (м, CH2CH2CH2CH3), 0,84-0,90 (м, CH2CH2CH2CH3).
Пример получения 68: Получение гидрохлорида (R)/(S)-2-амино-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)бутанамида (73)
Соединение 54 (0,66 г, 1,47 ммоль) и 4,0 M HCl (2,20 мл, 8,81 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 73, гидрохлорида (R)/(S)-2-амино-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)бутанамида (0,52 г, 91%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,66 (т, J=5,3 Гц, C(O)NH), 8,23 (с, NH3), 7,92 (д, J=1,9 Гц, ArH), 7,65- 7,72 (м, ArH), 7,35 (д, J=8,1 Гц, ArH), 3,67 (т, J=6,0 Гц, хиральный-H), 3,31-3,55 (м, NHCH2CH2), 2,79-2,83 (м, NHCH2CH2), 1,67-1,74 (м, CH2CH3), 0,79 (т, J=7,4 Гц, CH2CH3).
Пример получения 69: Получение гидрохлорида (R)/(S)-2-амино-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)пентанамида (74)
Соединение 55 (0,95 г, 2,04 ммоль) и 4,0 M HCl (3,06 мл, 12,2 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 74, гидрохлорида (R)/(S)-2-амино-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)пентанамида (0,66 г, 80%), получаемого в виде желтого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,82 (т, J=5,3 Гц, C(O)NH), 8,34 (с, NH3), 7,64-7,90 (м, ArH), 7,35-7,38 (д, J=8,1 Гц, ArH), 3,73 (т, J=6,3 Гц, хиральный-H), 3,29-3,57 (м, NHCH2CH2), 2,82-2,85 (м, NHCH2CH2), 1,60-1,67 (м, CH2CH2CH3), 1,14-1,22 (м, CH2CH2CH3), 0,80 (т, J=7,1 Гц, CH2CH2CH3).
Пример получения 70: Получение гидрохлорида (R)/(S)-2-амино-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)гексанамида (75)
Соединение 56 (0,75 г, 1,56 ммоль) и 4,0 M HCl (2,35 мл, 9,39 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 75, гидрохлорида (R)/(S)-2-амино-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)гексанамида (0,60 г, 92%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,60 (т, J=5,5 Гц, C(O)NH), 8,18 (с, NH3), 7,91 (д, J=2,0 Гц, ArH), 7,65- 7,72 (м, ArH), 7,35 (д, J=8,2 Гц, ArH), 3,66 (т, J=6,2 Гц, хиральный-H), 3,28-3,58 (м, NHCH2CH2), 2,81-2,85 (м, NHCH2CH2), 1,59-1,64 (м, CH2CH2CH2CH3), 1,14-1,23 (м, CH2CH2CH2CH3), 0,79 (т, J=6,8 Гц, CH2СН2СН2СН3).
Пример получения 71: Получение гидрохлорида (R)/(S)-2-амино-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)бутанамида (76)
Соединение 57 (0,90 г, 2,00 ммоль) и 4,0 M HCl (3,00 мл, 12,0 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 76, гидрохлорида (R)/(S)-2-амино-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)бутанамида (0,75 г, 96%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,62 (с, C(O)NH), 8,17 (с, NH3), 7,88 (д, J=8,2 Гц, ArH), 7,81 (д, J=8,4 Гц, ArH), 7,68 (д, J=8,3 Гц, ArH), 7,38 (д, J=8,1 Гц, ArH), 3,66 (т, J=6,1 Гц, хиральный-H), 3,30-3,57 (м, NHCH2CH2), 2,82 (т, J=7,0 Гц, NHCH2CH2), 1,67-1,74 (м, CH2CH3), 0,79 (т, J=7,5 Гц, CH2CH3).
Пример получения 72: Получение гидрохлорида (R)/(S)-2-амино-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)пентанамида (77)
Соединение 58 (1,03 г, 2,22 ммоль) и 4,0 M HCl (5,56 мл, 22,2 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 77, гидрохлорида (R)/(S)-2-амино-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)пентанамида (0,75 г, 85%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 300 МГц) 8,81 (т, J=5,3 Гц, C(O)NH), 8,35 (с, NH3), 7,84 (дд, J=8,2 Гц, 15,7 Гц, ArH), 7,54 (дд, J=8,1 Гц, 73,4 Гц, ArH), 3,71-3,75 (т, J=6,3 Гц, хиральный-H), 3,30-3,60 (м, NH2 CH2CH2), 2,85 (т, J=6,3 Гц, NHCH2CH2), 1,61-1,68 (м, CH2CH2CH3), 1,15-1,22 (м, CH2CH2CH3), 0,80 (т, J=7,2 Гц, CH2CH2CH3).
Пример получения 73: Получение гидрохлорида (R)/(S)-2-амино-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)гексанамида (78)
Соединение 59 (1,32 г, 2,76 ммоль) и 4,0 M HCl (4,14 мл, 16,6 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 78, гидрохлорида (R)/(S)-2-амино-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)гексанамида (0,82 г, 72%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,69 (т, J=4,9 Гц, C(O)NH), 8,26 (с, NH3), 7,88 (д, J=8,2 Гц, ArH), 7,81 (д, J=8,3 Гц, ArH), 7,68 (д, J=8,1 Гц, ArH), 7,39 (д, J=8,0 Гц, ArH), 3,69 (т, J=6,2 Гц, хиральный-H), 3,29-3,59 (м, NHCH2CH2), 2,80-2,89 (м, NHCH2CH2), 1,61-1,67 (м, CH2CH2CH2CH3), 1,14-1,23 (м, CH2CH2CH2CH3), 0,79 (т, J=6,8 Гц, CH2CH2CH2CH3).
Пример получения 74: Получение гидрохлорида (R)/(S)-2-амино-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)бутанамида (79)
Соединение 60 (0,91 г, 1,95 ммоль) и 4,0 M HCl (2,92 мл, 11,7 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 79, гидрохлорида (R)/(S)-2-амино-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)бутанамида (0,43 г, 54%), получаемого в виде желтого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,67 (с, C(O)NH), 8,24 (с, NH3), 7,35-7,89 (м, ArH), 3,67 (д, J=4,3 Гц, хиральный-H), 3,30-3,55 (м, NHCH2CH2), 2,80-2,85 (м, NHCH2CH2), 1,68-1,75 (м, CH2CH3), 0,80 (т, J=7,5 Гц, CH2СН3).
Пример получения 75: Получение гидрохлорида (R)/(S)-2-амино-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)пентанамида (80)
Соединение 61 (0,70 г, 1,45 ммоль) и 4,0 M HCl (2,18 мл, 8,70 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 80, гидрохлорида (R)/(S)-2-амино-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)пентанамида (0,55 г, 91%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,71 (т, J=5,4 Гц, C(O)NH), 8,28 (с, NH3), 7,77 (д, J=8,8 Гц, ArH), 7,62 (д, J=8,2 Гц, ArH), 7,35-7,45 (м, ArH), 3,70 (т, J=6,4 Гц, хиральный-H), 3,28-3,57 (м, NHCH2CH2), 2,80-2,86 (м, NHCH2CH2), 1,61-1,65 (м, CH2CH2CH3), 1,13-1,24 (м, CH2CH2CH3), 0,80 (т, J=7,4 Гц, CH2CH2CH3).
Пример получения 76: Получение гидрохлорида (R)/(S)-2-амино-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)гексанамида (81)
Соединение 62 (0,79 г, 1,59 ммоль) и 4,0 M HCl (2,38 мл, 9,54 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 81, гидрохлорида (R)/(S)-2-амино-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)гексанамида (0,62 г, 91%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,66 (т, J=5,4 Гц, C(O)NH), 8,23 (с, NH3), 7,88 (д, J=8,2 Гц, ArH), 7,81 (д, J=8,4 Гц, ArH), 7,68 (д, J=8,2 Гц, ArH), 7,39 (д, J=8,2 Гц, ArH), 3,68 (т, J=6,4 Гц, хиральный-H), 3,29- 3,59 (м, NHCH2CH2), 2,81-2,87 (м, NHCH2CH2), 1,60-1,66 (м, CH2CH2CH2CH3), 1,13-1,23 (м, CH2CH2CH2CH3), 0,79 (т, J=7,0 Гц, CH2CH2CH2CH3).
Пример получения 77: Получение гидрохлорида (R)/(S)-2-амино-N-(2-(3’,4’-дифтор-[1,1’-бифенил]-4-ил)этил)пентанамида (82)
Соединение 62 (0,94 г, 2,17 ммоль) и 4,0 M HCl (3,26 мл, 13,0 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 82, гидрохлорида (R)/(S)-2-амино-N-(2-(3’,4’-дифтор-[1,1’-бифенил]-4-ил)этил)пентанамида (0,68 г, 85%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,55 (с, C(O)NH), 8,10 (с, NH3), 7,72-7,77 (м, ArH), 7,62 (д, J=8,1 Гц, ArH), 7,50-7,53 (м, ArH), 7,33 (д, J=8,1 Гц, ArH), 3,65 (т, J=5,9 Гц, хиральный-H), 3,29-3,57 (м, NHCH2CH2), 2,78-2,84 (м, NHCH2CH2), 1,58 (кв, J=6,9 Гц, 8,8 Гц, CH2CH2CH3), 1,14-1,20 (м, CH2CH2CH3), 0,80 (т, J=7,3 Гц, CH2CH2CH3).
Пример получения 78: Получение гидрохлорида (R)/(S)-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)-2-(метиламино)бутанамида (83)
Соединение 64 (1,31 г, 2,81 ммоль) и 4,0 M HCl (4,20 мл, 16,9 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 83, гидрохлорида (R)/(S)-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)-2-(метиламино)бутанамида (1,07 г, 94%), получаемого в виде желтого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,79 (с, C(O)NH, NH2), 7,92 (д, J=2,0 Гц, ArH), 7,65-7,72 (м, ArH), 7,36 (д, J=8,1 Гц, ArH), 3,59 (т, J=6,4 Гц, хиральный-H), 3,39-3,53 (м, NHCH2CH2), 2,83 (т, J=6,8 Гц, NHCH2CH2), 2,38 (т, J=6,1 Гц, NCH3), 1,68-1,80 (м, CH2CH3), 0,77 (т, J=7,5 Гц, CH2CH3).
Пример получения 79: Получение гидрохлорида (R)/(S)-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)-2-(метиламино)пентанамида (84)
Соединение 65 (0,65 г, 1,36 ммоль) и 4,0 M HCl (2,03 мл, 8,13 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 84, гидрохлорида (R)/(S)-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)-2-(метиламино)пентанамида (0,33 г, 57%), получаемого в виде желтого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 9,26 (с, NH2), 8,96 (т, J=5,3 Гц, C(O)NH), 7,90-7,91 (м, ArH), 7,64-7,71 (м, ArH), 7,36 (д, J=8,1 Гц, ArH), 3,64-3,68 (м, хиральный-H), 3,42-3,56 (м, NHCH2CH2), 2,84 (т, J=6,8 Гц, NHCH2CH2), 2,35 (с, NCH3), 1,60-1,77 (м, CH2CH2CH3), 1,09-1,16 (м, CH2CH2CH3), 0,80 (т, J=7,2 Гц, CH2СН2СН3).
Пример получения 80: Получение гидрохлорида (R)/(S)-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)-2-(метиламино)гексанамида (85)
Соединение 66 (0,57 г, 1,16 ммоль) и 4,0 M HCl (1,74 мл, 6,95 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 85, гидрохлорида (R)/(S)-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)-2-(метиламино)гексанамида (0,38 г, 75%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,70-9,36 (с, NH2), 8,80 (т, J=5,4 Гц, C(O)NH), 7,91 (д, J=2,0 Гц, ArH), 7,65-7,72 (м, ArH), 7,35 (д, J=8,2 Гц, ArH), 3,55 (т, J=6,6 Гц, хиральный-H), 3,38-3,52 (м, NHCH2CH2), 2,83 (т, J=6,8 Гц, NHCH2CH2), 2,35 (с, NCH3), 1,60-1,76 (м, CH2CH2CH2CH3), 1,05-1,23 (м, CH2CH2CH2CH3), 0,78 (т, J=7,2 Гц, CH2CH2CH2CH3).
Пример получения 81: Получение гидрохлорида (R)/(S)-2-(метиламино)-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)бутанамида (86)
Соединение 67 (0,86 г, 1,84 ммоль) и 4,0 M HCl (2,77 мл, 11,1 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 86, гидрохлорида (R)/(S)-2-(метиламино)-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)бутанамида (0,65 г, 88%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 9,04 (с, NH2), 8,79 (с, C(O)NH), 7,87 (д, J=8,3 Гц, ArH), 7,80 (д, J=8,4 Гц, ArH), 7,68 (д, J=8,2 Гц, ArH), 7,39 (д, J=8,2 Гц, ArH), 3,59 (т, J=2,5 Гц, 4,9 Гц, хиральный-H), 3,40-3,56 (м, NHCH2CH2), 2,84 (т, J=6,9 Гц, NHCH2CH2), 2,37 (с, NCH3), 1,67-1,85 (м, CH2CH3), 0,77 (т, J=7,5 Гц, CH2CH3).
Пример получения 82: Получение гидрохлорида (R)/(S)-2-(метиламино)-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)пентанамида (87)
Соединение 68 (1,17 г, 2,45 ммоль) и 4,0 М HCl (3,67 мл, 14,7 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 87, гидрохлорида (R)/(S)-2-(метиламино)-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)пентанамида (81%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,93-9,79 (м, NH2), 9,04 (т, J=5,2 Гц, C(O)NH), 7,84 (дд, J=8,3 Гц, 15,1 Гц, ArH), 7,54 (дд, J=8,0 Гц, 73,4 Гц, ArH), 3,68-3,72 (м, хиральный-H), 3,42-3,58 (м, NHCH2CH2), 2,87 (т, J=6,7 Гц, NHCH2CH2), 2,36 (с, NCH3), 1,64-1,82 (м, CH2CH2CH3), 1,09-1,22 (м, CH2CH2CH3), 0,81 (т, J=7,1 Гц, СН2СН2СН3).
Пример получения 83: Получение гидрохлорида (R)/(S)-2-(метиламино)-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)гексанамида (88)
Соединение 69 (0,85 г, 1,72 ммоль) и 4,0 M HCl (2,60 мл, 10,3 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 88, гидрохлорида (R)/(S)-2-(метиламино)-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)гексанамида (0,53 г, 71%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 9,00 (с, NH2), 8,75 (т, J=5,4 Гц, C(O)NH), 7,87 (д, J=8,3 Гц, ArH), 7,81 (д, J=8,4 Гц, ArH), 7,68 (д, J=8,2 Гц, ArH), 7,38 (д, J=8,2 Гц, ArH), 3,57-3,61 (м, хиральный-H), 3,40-3,55 (м, NHCH2CH2), 2,84 (т, J=6,8 Гц, NHCH2CH2), 2,36 (с, NCH3), 1,60-1,75 (м, CH2CH2CH2CH3), 1,06-1,24 (м, CH2CH2CH2CH3), 0,78 (т, J=7,2 Гц, CH2CH2CH2CH3).
Пример получения 84: Получение гидрохлорида (R)/(S)-2-(метиламино)-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)бутанамида (89)
Соединение 70 (0,97 г, 2,01 ммоль) и 4,0 M HCl (3,00 мл, 12,1 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 89, гидрохлорида (R)/(S)-2-(метиламино)-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)бутанамида (0,73 г, 88%), получаемого в виде желтого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 9,00 (с, NH2), 8,77 (с, C(O)NH), 7,77 (д, J=8,7 Гц, ArH), 7,61 (д, J=8,2 Гц, ArH), 7,44 (д, J=8,2 Гц, ArH), 7,35 (д, J=8,1 Гц, ArH), 3,57-3,60 (м, хиральный-H), 3,39-3,55 (м, NHCH2CH2), 2,82 (т, J=6,9 Гц, NHCH2CH2), 2,37 (с, NCH3), 1,67-1,83 (м, CH2CH3), 0,77 (т, J=7,5 Гц, CH2CH3).
Пример получения 85: Получение гидрохлорида (R)/(S)-2-(метиламино)-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)пентанамида (90)
Соединение 71 (0,83 г, 1,67 ммоль) и 4,0 M HCl (2,51 мл, 10,0 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 90, гидрохлорида (R)/(S)-2-(метиламино)-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)пентанамида (0,40 г, 61%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,85-9,19 (м, NH2), 8,75 (с, C(O)NH), 7,76 (д, J=7,7 Гц, ArH), 7,61 (д, J=7,5 Гц, ArH), 7,44 (д, J=8,0 Гц, ArH), 7,35 (д, J=7,4 Гц, ArH), 3,60 (с, хиральный-H), 3,40-3,56 (м, NHCH2CH2), 2,83 (т, J=6,5 Гц, NHCH2CH2), 2,36 (с, NCH3), 1,60-1,66 (м, CH2CH2CH3), 1,09-1,17 (м, CH2CH2CH3), 0,80 (т, J=7,0 Гц, CH2CH2CH3).
Пример получения 86: Получение гидрохлорида (R)/(S)-2-(метиламино)-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)гексанамида (91)
Соединение 72 (0,89 г, 1,74 ммоль) и 4,0 M HCl (2,61 мл, 10,4 ммоль в диоксане) взаимодействовали между собой по схеме реакции f для синтеза соединения 91, гидрохлорида (R)/(S)-2-(метиламино)-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)гексанамида (0,66 г, 86%), получаемого в виде белого порошка.
Rf=0,00 (EtOAc:ацетон 9:1);
1H ЯМР (DMSO-d6, 400 МГц) 8,95 (с, NH2), 8,72 (с, C(O)NH), 7,76 (д, J=8,7 Гц, ArH), 7,61 (д, J=8,1 Гц, ArH), 7,44 (д, J=8,4 Гц, ArH), 7,35 (д, J=8,1 Гц, ArH), 3,57-3,60 (м, хиральный-H), 3,29-3,56 (м, NHCH2CH2), 2,83 (т, J=6,8 Гц, NHCH2CH2), 2,37 (с, NCH3), 1,59-1,74 (м, CH2CH2CH2CH3), 1,08-1,24 (м, CH2CH2CH2CH3), 0,78 (т, J=7,2 Гц, CH2CH2CH2CH3).
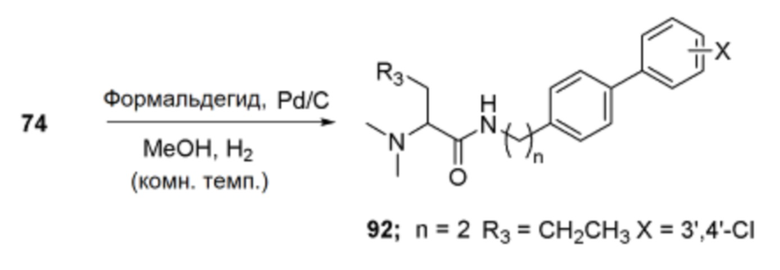
Пример получения 87: Получение (R)/(S)-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)-2-(диметиламино)пентанамида (92)
Соединение 74 (1,0 экв.), триэтиламин (6,0 экв.), формальдегид (1,05 экв.) и палладиевый катализатор (0,2 экв.) взаимодействовали между собой по схеме реакции g для синтеза соединения 92, (R)/(S)-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)-2-(диметиламино)пентанамида.
1H ЯМР (DMSO-d6, 400 МГц) 8,96 (т, J=5,3 Гц, C(O)NH), 7,90-7,91 (м, ArH), 7,64-7,71 (м, ArH), 7,36 (д, J=8,1 Гц, ArH), 3,64-3,68 (м, хиральный-H), 3,42-3,56 (м, NHCH2CH2), 2,82-2,85 (м, NHCH2CH2), 2,35 (с, N (CH3)2), 1,60-1,77 (м, CH2CH2CH3), 1,09-1,16 (м, CH2CH2CH3), 0,80 (т, J=7,2 Гц, CH2CH2CH3).
Варианты выполнения изобретения
Примеры
Пример 1: Анализ противогрибковой активности в отношении грибов, патогенных для человека
Тест на чувствительность к противогрибковым препаратам in vitro проводили в соответствии с руководящими принципами Института клинических и лабораторных стандартов США (CLSI) для измерения степени противогрибковой активности соединений, синтезированных в рамках Примеров получения, против условно-патогенных грибов. В настоящем примере определяли минимальные ингибирующие концентрации (MIC), при которых соединения могут подавлять рост патогенных грибов Cryptococcus neoformans, Candida albicans, Candida glabrata и Aspergillus fumigatus, как показано в Таблице 1. Значение MIC (мкг/мл) выражали в виде диапазона значений. В частности, грибы видов Candida и Cryptococcus neoformans инкубировали на твердой агаровой среде Сабуро с декстрозой (SDA) (Sigma-Aldrich) в течение 24 и 48 часов, соответственно, для получения отдельных колоний. Полученные одиночные колонии суспендировали в 0,85% физиологическом растворе с получением суспензии клеток грибов (от 1 до 5 х 10 6 клеток/л). Клетки грибов, разбавленные в 2000 раз в бульоне RPMI1640 (Sigma-Aldrich), высевали в 96-луночные планшеты (от 5 х 102 до 2,5 х 103 клеток/мл, из расчета 195 мкл на лунку). Последовательные разведения соединений с двукратным шагом (128, 64, 32, 16, 8, 4, 2, 1 и 0,5 мкг/мл) готовили путем разбавления соединений, синтезированных в соответствии с приведенными выше Примерами получения. Затем 5 мкл каждого разведения добавляли в лунку 96-луночного планшета, содержащего суспензию клеток грибов, с получением конечной суспензии из расчета 200 мкл на лунку. Обработанные соединениями виды Candida и C. neoformans инкубировали при 35°C соответственно в течение 48 часов и 72 часов, а затем определяли MIC путем визуального контроля дна 96-луночного планшета для определения факта развития и роста грибов. В качестве положительного контроля использовали амфотерицин B (AMB), который очень токсичен для человеческого организма, хотя он считается типичным противогрибковым агентом. Результаты представлены ниже в Таблицах 1-3.
Пример 2: Анализ противогрибковой активности соединения 74
Для оценки противогрибковой активности соединения 74 измеряли минимальную ингибирующую концентрацию (MIC), способную ингибировать рост грибов в соответствии с руководящими принципами Института клинических и лабораторных стандартов (CLSI). Клетки грибов распределяли на твердой среде YPD (Sigma-Aldrich), а затем аликвоты переносили в лунки 96-луночных планшетов (2,5 х 10 3 микроконидий/мл для видов T. rubrum и T. mentagrophytes или 2,5 х 10 3 КОЕ/мл для видов Candida albicans, Candida glabrata и Cryptococcus neoformans, из расчета 195 мкл на лунку). Готовили серийные разведения соединения 74 с двукратным шагом в пределах концентраций от 128 до 0,5 мкг/мл (128, 64, 32, 16, 8, 4, 2, 1 и 0,5 мкг/мл). Затем 5 мкл каждого разведения соединения 74 добавляли в лунки 96-луночного планшета, содержащие клетки грибов, с получением конечной суспензии объемом 200 мкл на лунку. Обработанные соединением клетки грибов инкубировали при 35°C в течение 48 часов (виды Candida) и 72 часов (виды Trichophyton и C. neoformans), соответственно. После инкубации определяли MIC (мкг/мл) путем визуального контроля дна 96-луночного планшета для определения факта роста грибов. Результаты представлены ниже в Таблице 4.
Пример 3: Сравнение быстроты действия соединения 74 с коммерчески доступными сравнительными препаратами
Для оценки быстроты действия соединения 74 изучали рост грибов Candida albicans при обработке их соединением 74 или коммерчески доступными противогрибковыми лекарственными препаратами, такими как эфинаконазол, таваборол, тербинафин и циклопирокс, в той же концентрации и в течение такого же периода времени. В частности, получали клетки Candida albicans в концентрации 2,5 х 10 3 КОЕ/мл и аликвоты, из расчета 195 мкл на лунку, переносили в 96-луночные планшеты. Затем клетки Candida Albicans в 96-луночном планшете обрабатывали соединением 74 или каждым из коммерчески доступных сравнительных лекарственных препаратов (50 мкг/мл или 100 мкг/мл) в течение 30 минут, соответственно. Затем клетки грибов, обработанные соединением 74, высевали на среду YPD (Sigma-Aldrich) и подсчитывали количество выросших одиночных колоний. Результаты представлены на Фиг. 1. Как следует из Фиг. 1, подтверждено, что соединение 74 обладает превосходной быстротой действия по сравнению с коммерчески доступными сравнительными лекарственными препаратами.
Пример 4: Сравнение фунгицидного действия соединения 74 с коммерчески доступными сравнительными препаратами
Для оценки фунгицидного действия соединения 74 изучали рост T. rubrum после обработки его соединением 74 или коммерчески доступными лекарственными противогрибковыми препаратами, такими как эфинаконазол, аморолфин, тербинафин, циклопирокс и амфотерицин B (при 0,5-, 1-, 2-, 4-, 8- или 16-кратном превышении соответствующих величин MIC для соединения 74 или других коммерчески доступных противогрибковых лекарственных препаратов) в течение заданного времени. В частности, получали клетки T. rubrum (6 х 10 3 КОЕ/мл) и аликвоты, из расчета 195 мкл на лунку, переносили в 96-луночные планшеты. Затем 5 мкл соединения 74 или каждого из других коммерчески доступных лекарственных препаратов добавляли в лунки 96-луночного планшета, содержащие клетки T. rubrum, с получением конечного объема 200 мкл на лунку. Через 0, 2, 4, 6, 12, 24, 48, 72 или 120 часов после добавления, клетки грибов, обработанные соединением 74 или другими противогрибковыми препаратами, наносили на твердую среду YPD, а затем подсчитывали количество выросших одиночных колоний. Результаты представлены на Фиг. 2. Как следует из Фиг. 2, соединение 74 проявляет фунгицидное действие при концентрации, соответствующей MIC, что позволяет предположить, что соединение 74 обладает фунгицидным действием при более низкой концентрации, чем другие коммерчески доступные сравнительные препараты.
Пример 5: Сравнение активности соединения 74 с коммерчески доступными сравнительными препаратами в отношении разрушения биопленки
Изучали активность соединения 74 в отношении разрушения биопленки. Для этого формировали биопленку Candida albicans. Затем к полученной биопленке добавляли соединение 74 или коммерчески доступный противогрибковый препарат, такой как каспофунгин, амфотерицин В или эфинаконазол, в соответствующих концентрациях 4, 8, 16 и 32 мкг/мл, и оценивали разрушение биопленки. В частности, клетки Candida albicans (2,5 х 103 КОЕ/л) высевали в 96-луночный планшет и инкубировали в течение 90 минут для образования биопленки, а затем биопленку обрабатывали соединением 74 или каждым из указанных выше сравнительных препаратов, а затем через 24 часа после обработки оценивали рост Candida albicans. Результаты представлены на Фиг. 3. Как следует из Фиг. 3, установлено, что соединение 74 по изобретению проявляет превосходные эффекты в отношении ингибирования роста Candida albicans, разрушая биопленку, особенно по сравнению с коммерчески доступными сравнительными лекарственными препаратами.
Пример 6: Анализ антибактериальной активности
Для соединений по изобретению также оценивали антибактериальную активность в отношении грамположительных бактерий, грамотрицательных бактерий или бактерий с множественной лекарственной устойчивостью. Исследование по оценке MIC проводили в соответствии с рекомендациями CLSI. При этом использовали E. coli(-), P. aeruginosa(-) и S. enterica(-) в качестве типовых грамотрицательных бактерий, B. cereus(+), B. subtilis(+), B. coagulans(+), L. monocytogenes(+), M. luteus(+), P. acnes(+), S. epidermidis(+) и S. aureus(+)в качестве типовых грамположительных бактерий. Метициллин-устойчивый золотистый стафилококк Staphylococcus aureus (MRSA) использовали в качестве типовой бактерии с множественной лекарственной устойчивостью. Бактериальные клетки инкубировали до тех пор, пока величина OD600 отдельных колоний не достигала значения 0,1. Клетки, разбавленные в 100 раз жидкой средой MHB (Sigma-Aldrich), высевали на 96-луночные планшеты, с получением суспензии (100 мкл на лунку) с концентрацией, соответствующей величине OD600, равной 0,001. Затем последовательно разводили соединения по изобретению с двукратным шагом (64, 32, 16, 8, 4, 2, 1 и 0,5 мкг/мл), и добавляли в лунки, чтобы получить конечную суспензию объемом 200 мкл на лунку. Затем бактериальные клетки инкубировали при 37°C в течение 24 часов, и определяли MIC (мкг/мл) путем визуального контроля и определения оптической плотности при 600 нм (OD600). В качестве положительного контроля использовали нофлоксацин и ванкомицин. Результаты показаны в ниже в Таблицах 5-8.
Пример 7: Анализ противовоспалительной активности
Противовоспалительное действие соединений по изобретению оценивали с использованием клеток RAW264.7, стимулированных для секреции интерлейкина 6 (IL-6). В частности, клетки мышиных макрофагов RAW264.7 (полученные из Korean Cell Line Bank) вносили в 12-луночный планшет из расчета 5 х 105 клеток на лунку, и затем инкубировали при 37°C в течение 16-24 часов. Соединения по изобретению добавляли к клеточной культуральной среде, содержащей клетки RAW264.7, и инкубировали, как указано выше, в конечной концентрации 0 или 8 мкг/мл, а затем инкубировали при 37°C в течение 1 часа. После этого дополнительно добавляли в среду липополисахарид (LPS, Sigma-Aldrich, США) в конечной концентрации 1 мкг/мл, и среду инкубировали при 37°C в течение 24 часов, чтобы индуцировать экспрессию IL-6. Затем измеряли уровни IL-6, продуцируемого клетками RAW264.7, с использованием набора для ELISA (Komabiotech Inc., Корея) в соответствии с инструкциями производителя. Результаты показаны в Таблице 9. В Таблице 9 уровни IL-6 выражены как относительный процент (%) между уровнями LPS-индуцированного IL-6 после обработки соединением по изобретению и уровнями LPS-индуцированного IL-6 без обработки соединением по изобретению.
(%)
Промышленная применимость
Настоящее изобретение относится к новому соединению и его применению, и это соединение может быть использовано для приготовления противогрибковых и антибактериальных композиций, а также может быть использовано для получения фармацевтической композиции для профилактики и лечения грибковых инфекционных заболеваний.
| название | год | авторы | номер документа |
|---|---|---|---|
| С-ГЛИКОЗИДНЫЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ КОНДЕНСИРОВАННОЕ ФЕНИЛЬНОЕ КОЛЬЦО, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ ТАКОВЫХ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ТАКОВЫЕ | 2017 |
|
RU2739024C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2734390C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2681849C2 |
| ПРОИЗВОДНОЕ БЕНЗАМИДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ РАКА, СОДЕРЖАЩАЯ ЕГО В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2022 |
|
RU2841260C2 |
| Новое соединение в качестве ингибитора протеинкиназы и содержащая его фармацевтическая композиция | 2019 |
|
RU2785734C1 |
| ГЕТЕРОАРИЛЬНОЕ ПРОИЗВОДНОЕ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С PI3 КИНАЗАМИ, СОДЕРЖАЩАЯ ДАННОЕ ДЕЙСТВУЮЩЕЕ ВЕЩЕСТВО | 2016 |
|
RU2719367C2 |
| Гетероциклическое соединение в качестве ингибитора протеинкиназы | 2018 |
|
RU2783723C2 |
| ПРОИЗВОДНЫЕ 1,3-ДИОКСОИНДЕНА, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ АНТИВИРУСНАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2012 |
|
RU2566761C2 |
| ПРОИЗВОДНОЕ ТИОФЕНКАРБОКСАМИДА И СРЕДСТВО ДЛЯ КОНТРОЛЯ ЗАБОЛЕВАНИЯ РАСТЕНИЙ, СОДЕРЖАЩЕЕ ЕГО | 2019 |
|
RU2776177C1 |
| НОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2814103C1 |
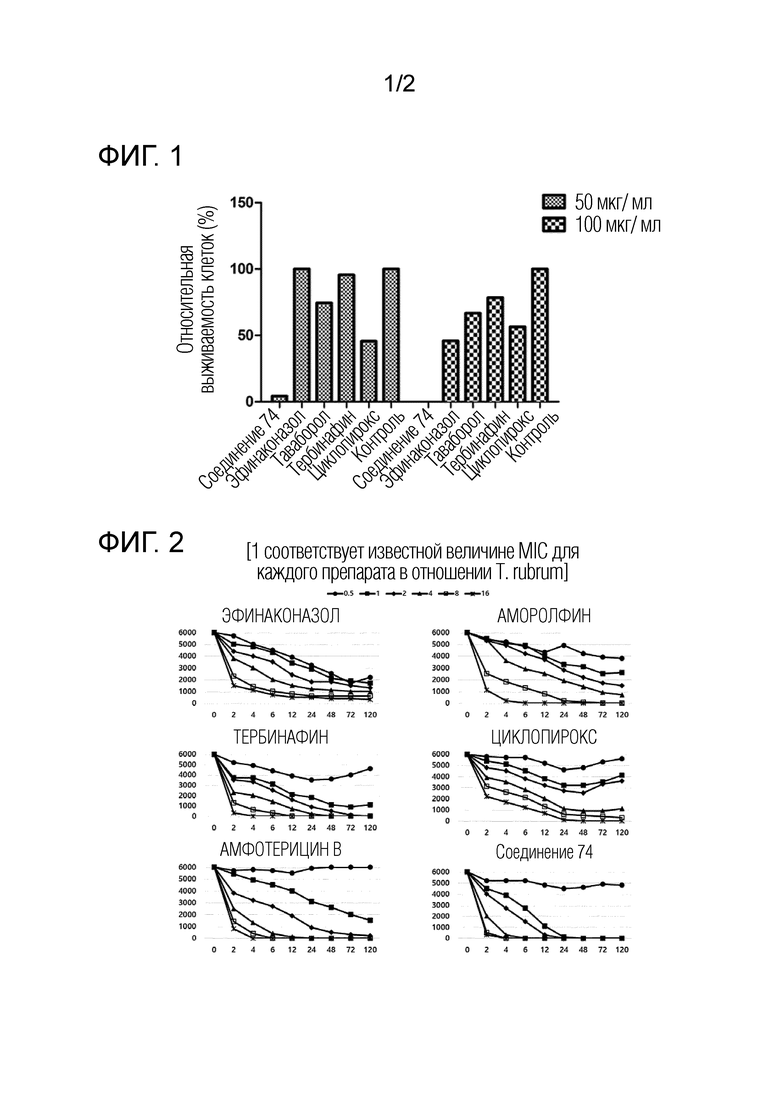
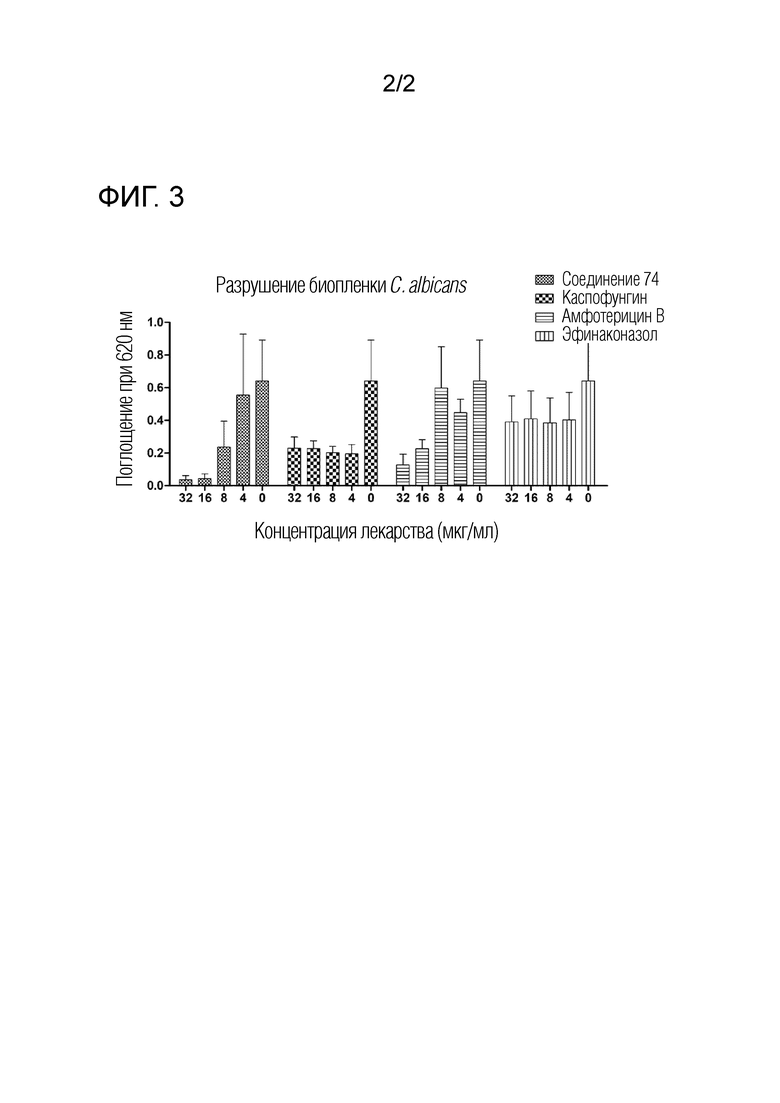
Изобретение относится к соединению формулы 1, где R1-R3, n и m определены в формуле изобретения, его стереоизомеру или его фармацевтически приемлемой соли. Соединение по изобретению проявляет превосходные противогрибковые и фунгицидные эффекты. Кроме того, соединение по изобретению проявляет синергический эффект при использовании его в сочетании с обычным противогрибковым средством, имеет широкий спектр противогрибковой активности в отношении большого числа грибковых патогенов и может широко использоваться в областях, где необходимо использовать противогрибковые или фунгицидные средства против патогенных грибов человека и патогенных грибов животных, а также фитопатогенных грибов. 7 н. и 12 з.п. ф-лы, 3 ил., 9 табл., 94 пр.
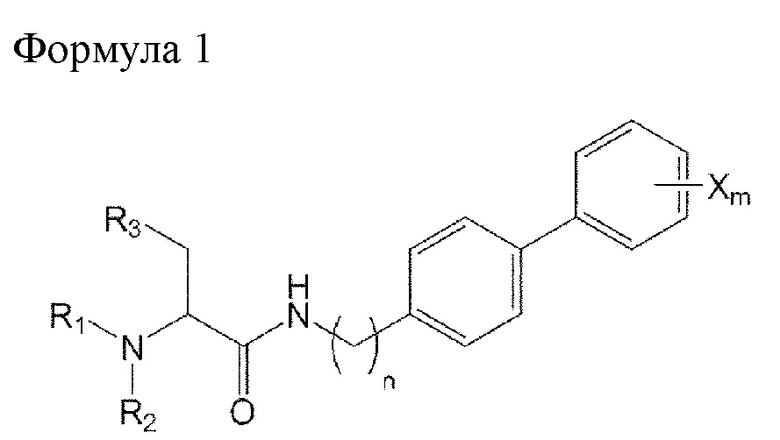
1. Соединение, представленное следующей химической формулой 1, его стереоизомер или его фармацевтически приемлемая соль:
Формула 1
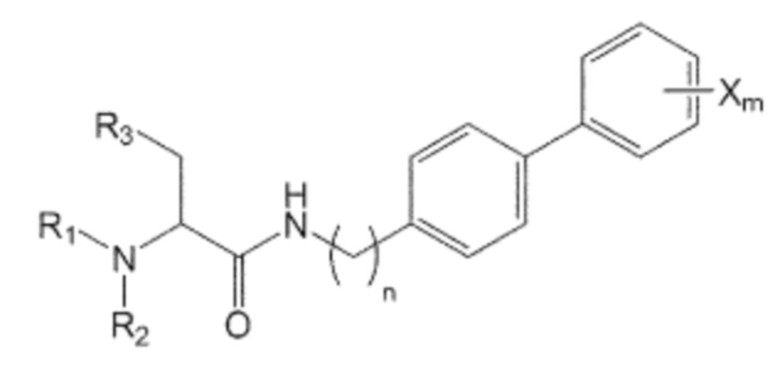
где n равно 0, 1, 2, 3, 4 или 5, m представляет собой целое число от 1 до 5;
R1, R2 и R3 независимо друг от друга являются одинаковыми или отличающимися друг от друга и каждый из них независимо выбран из группы, состоящей из водорода и C1-7 алкила, и
X представляет собой m заместителей, которые являются одинаковыми или отличающимися друг от друга, выбранными из группы, состоящей из группы галогена, галогенированной C1-7 алкильной группы и галогенированной C1-7 алкоксигруппы;
при условии, что, когда n равно 0 или 1, и R1 и R2 представляют собой водород, R3 является C1-3 алкилом; и когда n равно 0, R3 не является водородом.
2. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п.1, где R1 и R2 независимо друг от друга представляют собой Н или метил, этил, н-пропил, изопропил, н-бутил, изобутил, н-пентил или н-гексил, и
R3 представляет собой метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил или трет-бутил.
3. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п.1, где X представляет собой любой один или два одинаковых или разных заместителя, выбранных из группы, состоящей из фтора, хлора, трифторметила и трифторметокси.
4. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п.3, где X представляет собой п-фтор, м-фтор, п,м-дифтор, п-хлор, м-хлор, п,м-дихлор, п-трифторметил или п-трифторметокси.
5. Соединение, его стереоизомер или его фармацевтически приемлемая соль по п.1, где соединение представляет собой
3) 2-амино-N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)гексанамид;
5) N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)-2-(метиламино)бутанамид;
6) N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)-2-(метиламино)пентанамид;
7) N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)-2-(метиламино)гексанамид;
8) N-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)-2-(диметиламино)пентанамид;
10) 2-амино-N-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)гексанамид;
12) 2-амино-N-((4’-(трифторметил)-[1,1’-бифенил]-4-ил)метил)гексанамид;
14) 2-амино-N-((4’-(трифторметокси)-[1,1’-бифенил]-4-ил)метил)гексанамид;
15) N-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)-2-(метиламино)пентанамид;
16) 2-(метиламино)-N-((4’-(трифторметил)-[1,1’-бифенил]-4-ил)метил)пентанамид;
17) N-((3’,4’-дихлор-[1,1’-бифенил]-4-ил)метил)-2-(диметиламино)пентанамид;
18) 2-амино-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)бутанамид;
19) 2-амино-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)пентанамид;
20) 2-амино-N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)гексанамид;
21) 2-амино-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)бутанамид;
22) 2-амино-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)пентанамид;
23) 2-амино-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)гексанамид;
24) 2-амино-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)бутанамид;
25) 2-амино-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)пентанамид;
26) 2-амино-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)гексанамид;
27) 2-амино-N-(2-(3’,4’-дифтор-[1,1’-бифенил]-4-ил)этил)пентанамид;
28) N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)-2-(метиламино)бутанамид;
29) N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)-2-(метиламино)пентанамид;
30) N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)-2-(метиламино)гексанамид;
31) 2-(метиламино)-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)бутанамид;
32) 2-(метиламино)-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)пентанамид;
33) 2-(метиламино)-N-(2-(4’-(трифторметил)-[1,1’-бифенил]-4-ил)этил)гексанамид;
34) 2-(метиламино)-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)бутанамид;
35) 2-(метиламино)-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)пентанамид;
36) 2-(метиламино)-N-(2-(4’-(трифторметокси)-[1,1’-бифенил]-4-ил)этил)гексанамид или
37) N-(2-(3’,4’-дихлор-[1,1’-бифенил]-4-ил)этил)-2-(диметиламино)пентанамид.
6. Способ получения соединения, его стереоизомера или его фармацевтически приемлемой соли по любому из пп.1-5, включающий
первую стадию образования пептидной связи путем взаимодействия производного соединения аминоалкановой кислоты, защищенного бутоксикарбонильной (Boc) защитной группой, которая представлена формулой 2, с производным бифенила, содержащим первичную аминогруппу, представленным формулой 3; и
вторую стадию удаления защитной группы Boc путем взаимодействия соединения, полученного на первой стадии, с кислотой:
Формула 1
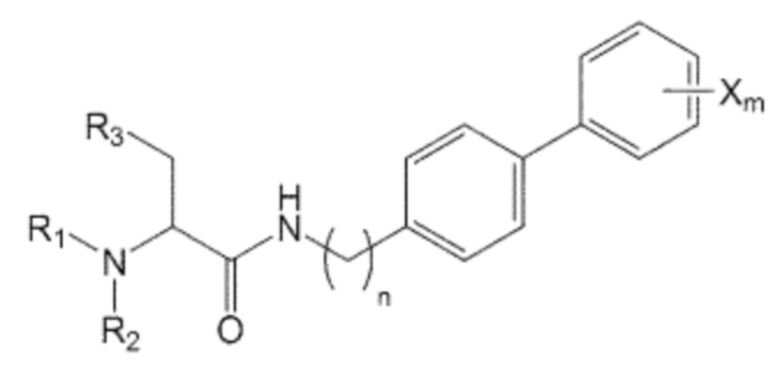
Формула 2
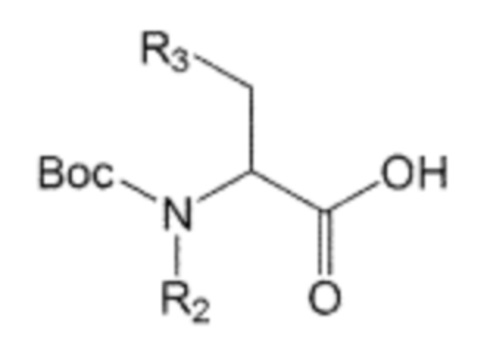
Формула 3
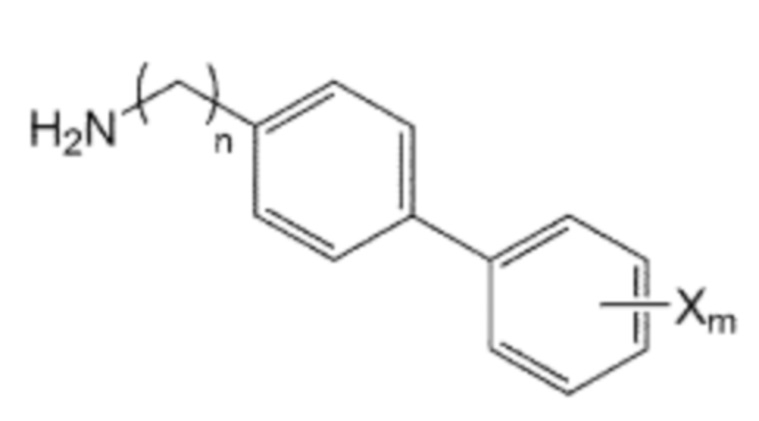
где n равно 0, 1, 2, 3, 4 или 5, m представляет собой целое число от 1 до 5;
R1, R2 и R3 независимо друг от друга являются одинаковыми или отличающимися друг от друга и каждый независимо выбран из группы, состоящей из водорода и C1-7 алкила, и
X представляет собой m заместителей, которые являются одинаковыми или отличающимися друг от друга, выбранными из группы, состоящей из группы галогена, галогенированной C1-7 алкильной группы и галогенированной C1-7 алкоксигруппы.
7. Способ по п.6, где производное аминоалкановой кислоты, защищенное Boc-защитной группой, которое представлено формулой 2, получают путем взаимодействия производного аминокислоты, представленного формулой 4, с ди-трет-бутилдикарбонатом (ангидрид Boc):
Формула 4

где в формуле:
R1’ представляет собой водород, и каждый R2 и R3 независимо друг от друга являются одинаковыми или отличающимися друг от друга, и каждый из них независимо выбран из группы, состоящей из водорода и C1-7 алкила,
причем когда R2 полученного соединения формулы 2 представляет собой алкил, то после указанной выше стадии получения соединения формулы 2 проводят стадию алкилирования указанного соединения путем взаимодействия соединения с галогеналканом в присутствии основания.
8. Способ по п.6, где соединение производного бифенила, содержащего группу первичного амина, которое представлено формулой 3, получают реакцией взаимодействия производного C0-2 алкиламина, в котором галогенфенильная группа на одном конце замещена, как представлено формулой 5, с ди-трет-бутилдикарбонатом для введения защитной группы Boc в аминогруппу с последующим взаимодействием полученного производного алкиламина с производным фенилбороновой кислоты, представленным формулой 6, и затем взаимодействием реагентов с кислотой для удаления защитной группы Boc:
Формула 5
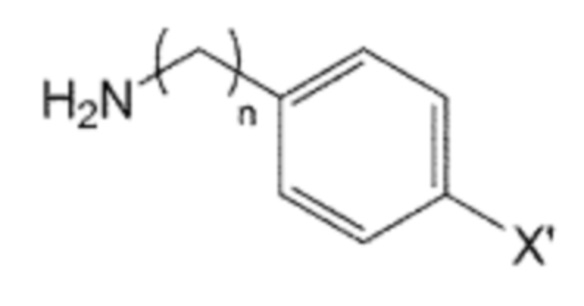
Формула 6
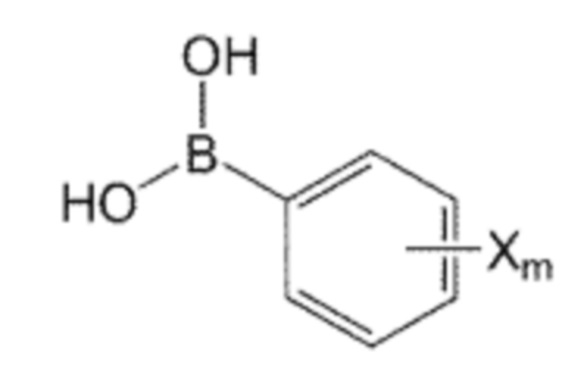
где в формулах:
X’ представляет собой галоген и
X представляет собой m заместителей, где m представляет собой целое число от 1 до 5, которые являются одинаковыми или отличающимися друг от друга, выбранными из группы, состоящей из группы галогена, галогенированной C1-7 алкильной группы и галогенированной C1-7 алкоксигруппы.
9. Способ по п.8, где реакцию с производным фенилбороновой кислоты осуществляют посредством реакции кросс-сочетания в присутствии основания с использованием металлического катализатора.
10. Способ по п.6, где первую стадии осуществляют посредством реакции сочетания ангидрида, проводимой в органическом растворителе в присутствии N-метилморфолина (NMM) и изобутилхлорформиата (IBCF).
11. Способ по п.6, дополнительно включающий после второй стадии третью стадию алкилирования амина с получением соединения, представленного формулой 1, в котором R1 и R2 оба представляют собой алкилы.
12. Противогрибковая композиция, содержащая в качестве активного ингредиента соединение, его стереоизомер или его фармацевтически приемлемую соль по любому из пп.1-5 и фармацевтически приемлемый носитель, разбавитель или эксципиент.
13. Противогрибковая композиция по п.12, где противогрибковая композиция обладает противогрибковой активностью в отношении Cryptococcus neoformans, Candida albicans, Candida glabrata или Aspergillus fumigatus.
14. Фармацевтическая композиция для лечения или профилактики грибкового инфекционного заболевания, содержащая в качестве активного ингредиента соединение, его стереоизомер или его фармацевтически приемлемую соль по любому из пп.1-5 и фармацевтически приемлемый носитель, разбавитель или эксципиент.
15. Фармацевтическая композиция по п.14, где грибковое инфекционное заболевание вызывается инфекциями Cryptococcus neoformans, Candida albicans, Candida glabrata или Aspergillus fumigatus.
16. Антибактериальная композиция, содержащая в качестве активного ингредиента соединение, его стереоизомер или его фармацевтически приемлемую соль по любому из пп.1-5 и фармацевтически приемлемый носитель, разбавитель или эксципиент.
17. Антибактериальная композиция по п.16, где антибактериальная композиция обладает антибактериальной активностью в отношении грамотрицательных бактерий, грамположительных бактерий или бактерий с множественной лекарственной устойчивостью.
18. Фармацевтическая композиция для лечения или профилактики бактериального инфекционного заболевания, содержащая в качестве активного ингредиента соединение, его стереоизомер или его фармацевтически приемлемую соль по любому из пп.1-5 и фармацевтически приемлемый носитель, разбавитель или эксципиент.
19. Фармацевтическая композиция для лечения или профилактики воспалительного заболевания, содержащая соединение, его стереоизомер или его фармацевтически приемлемую соль по любому из пп.1-5 и фармацевтически приемлемый носитель, разбавитель или эксципиент.
| WO 2007088571 A2, 09.08.2007 | |||
| WO 2012125832 A2, 20.09.2012 | |||
| WO 2013131018 A1, 06.09.2013 | |||
| RU 2019100054 A, 24.01.2019 | |||
| Cai Zhan et al, "Design, synthesis, and SAR study of 3-(benzo[d][1,3]dioxol-5 -yl)-N-benzylpropanamide as novel potent synergists against fluconazole resistant Candida albicans", Bioorganic & Medicinal Chemistry Letters, 2017, |

