ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к соединениям и производным замещенного гидроксистильбена, в частности, к соединениям и производным 2-замещенного гидроксистильбена, к синтезу таких соединений и их применению в терапии.
УРОВЕНЬ ТЕХНИКИ
В патентном документе WO2012/149608 раскрыты новые производные пренилированного полигидроксистильбена и способы многостадийного получения таких соединений. Одной из стадий является стадия перегруппировки O- в C-пренил, изображенной в общем виде ниже:
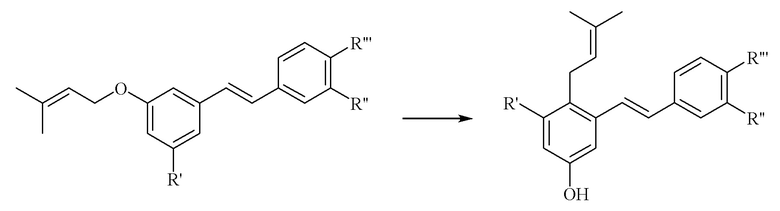
Способы, раскрытые в патентном документе WO2012/149608, имеют ряд недостатков. Синтез конечных соединений пренилированного полигидроксистильбена из общих промежуточных соединений требует проведения большого количества стадий. Проведение стадии перегруппировки осложняется тем, что она является специфичной главным образом для пренильной группы и для относительно узкого диапазона других типов заместителей. Метод перегруппировки требует использования хроматографического разделения и, как правило, дает низкий выход. Таким образом, способы предшествующего уровня техники не подходят для получения широкого спектра синтетических производных соединений пренилированного полигидроксистильбена и не подходят для синтеза в промышленном масштабе. Полное содержание патентного документа WO2012/149608 включено в настоящее изобретение путем ссылки на него.
Существует потребность в альтернативных способах получения соединений и производных замещенного гидроксистильбена, в частности, соединений и производных 2-замещенного гидроксистильбена.
Любое обсуждение документов, актов, материалов, устройств, статей и других подобных источников информации, которые включены в описание настоящего изобретения, не следует рассматривать как признание того, что какие-либо или все эти предметы обсуждения образуют часть базы предшествующего уровня техники или являются общеизвестными в области, относящейся к настоящему изобретению, в силу того, что они существовали до даты приоритета каждого из прилагаемых пунктов формулы изобретения.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В процессе создания настоящего изобретения авторы синтезировали широкий ряд соединений 2-замещенного гидроксистильбена, используя новый процесс, обеспечивающий эффективное и применимое в промышленности получение ряда соединений, имеющих важное значение при проведении лекарственной терапии. Новый процесс также позволил получать ряд новых соединений, которые, как было продемонстрировано, оказались перспективными для применения в терапии, например, при лечении рака и заболеваний или нарушений кожи.
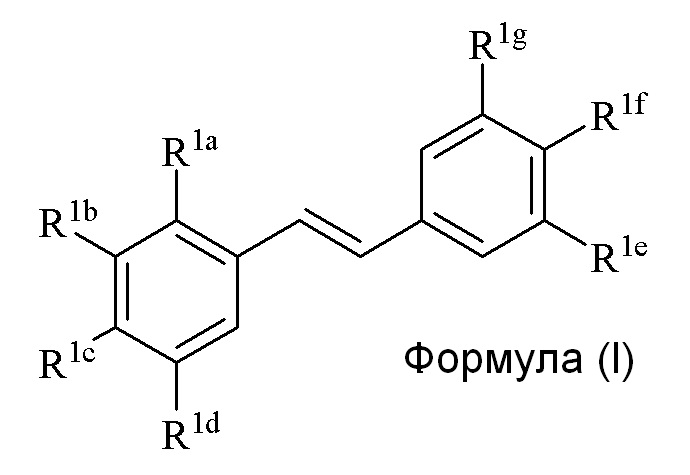
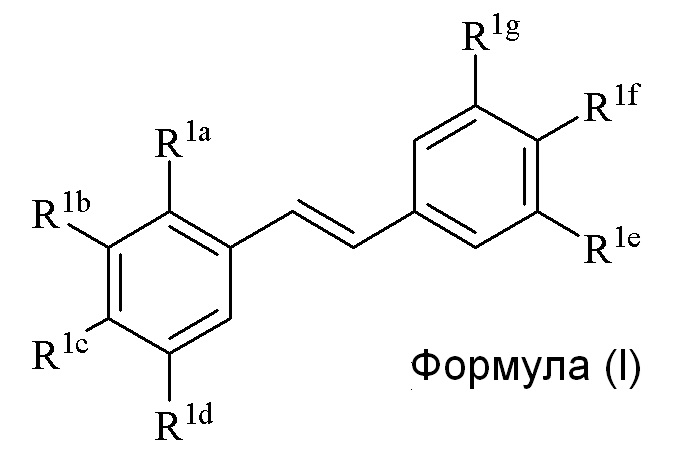
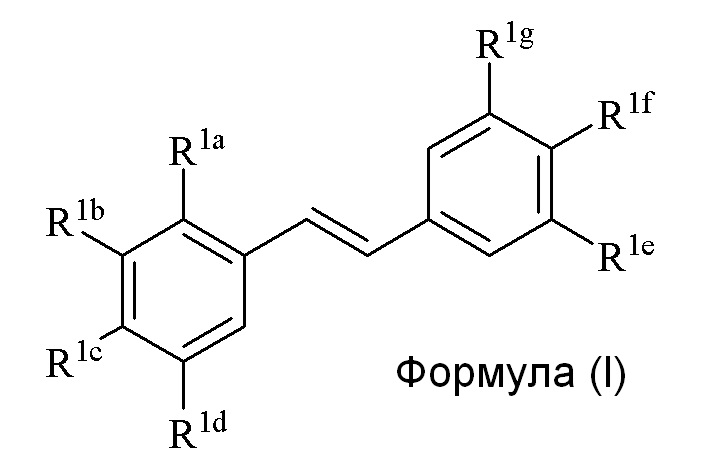
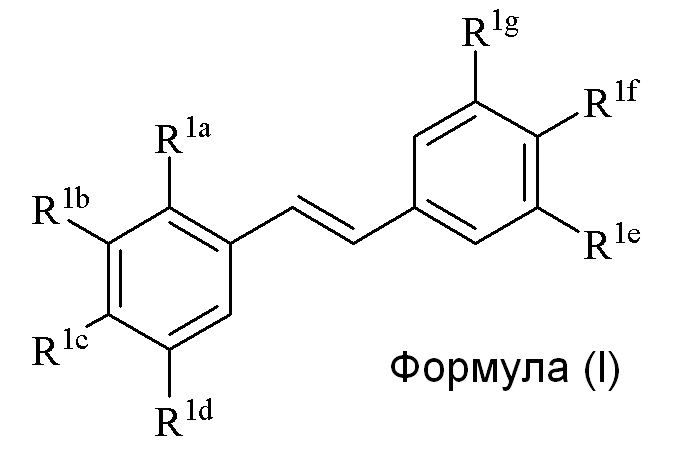
Настоящее изобретение относится к синтезу соединения формулы (I):
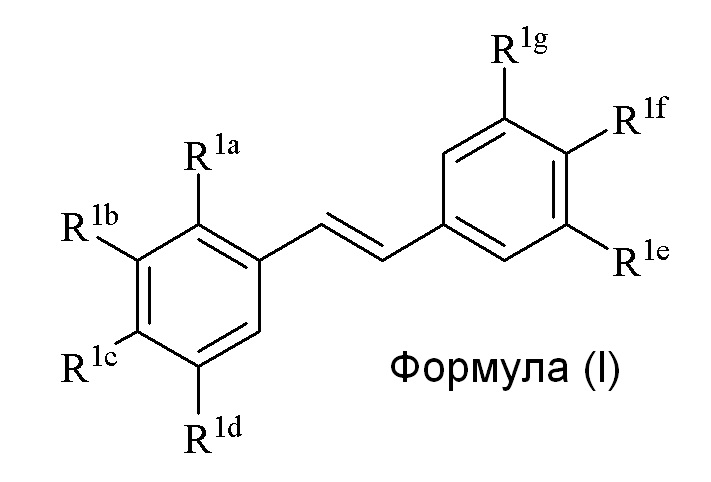
где
R1a представляет собой независимо аллил, кротил, пренил, геранил, фарнезил, бензил или 2-алкенил, 2-алкинил;
R1b представляет собой независимо СF3, OH или OR2, NO2, NHEt, NMe2, NMeEt, NHR3, NMeR3, NHC=NH(NH2) или COOR2;
R1c представляет собой независимо H, алкил, алкенил, алкинил, алкандиенил, пренил, геранил, фарнезил или бензил;
R1d представляет собой независимо СF3, OH или OR2, NHR3, NO2, NMeR3, NHC=NH(NH2) или COOR2;
R1e представляет собой независимо H, C1-C6 алкил, OH, OR2, NO2, NMe2, NHR3, NMeR3, NHC(O)H или SR2;
R1f представляет собой независимо H, OH, OR2, NO2, NHR3, NHC=NH(NH2), COOR2, COOH;
R1g представляет собой независимо H, алкил, CF3, OH или OR2;
и не более чем один из R1e, R1f или R1g может представлять собой H;
или R1e и R1f образуют пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу или (CO)CH2 группу, когда R1e независимо представляет собой OH, NH2 или NHMe, и R1f представляет собой NH2, так что азот в R1f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R1e;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил, COCOOR4 или O-защитная группа, когда R2 присоединен к O, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этокси]метила (SEM), CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой независимо H, CH3, OH, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, CO(CH2)nNH2, CONH2, CO(CH2)nCOOH или COCOOR4, где n=0, 1 или 2; и
R4 представляет собой C1-C6 алкил.
Описанное в изобретении соединение 2-замещенного гидроксистильбена формулы (I) представляет собой два бензольных кольца, которые обозначены как кольцо A и кольцо B. Описанное в изобретении кольцо A представляет бензольное кольцо, несущее заместители R1a, R1b, R1c и R1d, и описанное в изобретении кольцо B представляет бензольное кольцо, несущее заместители R1e, R1f и R1g.
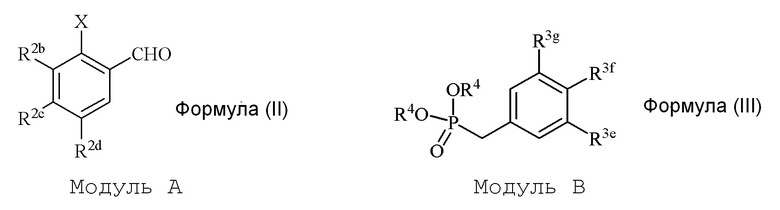
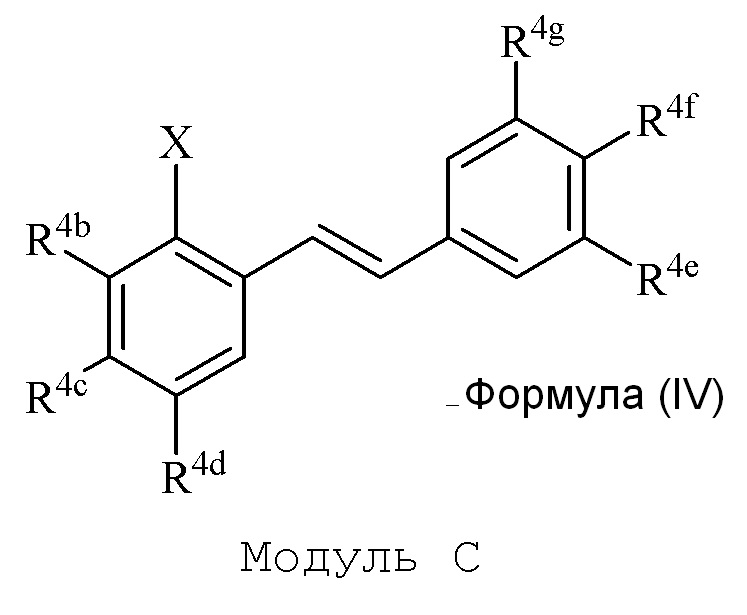
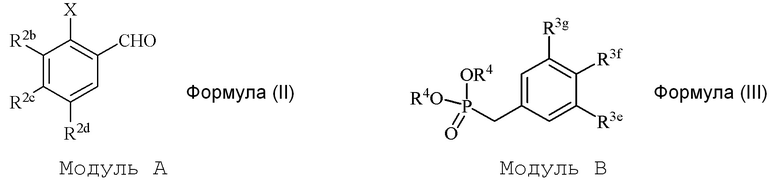
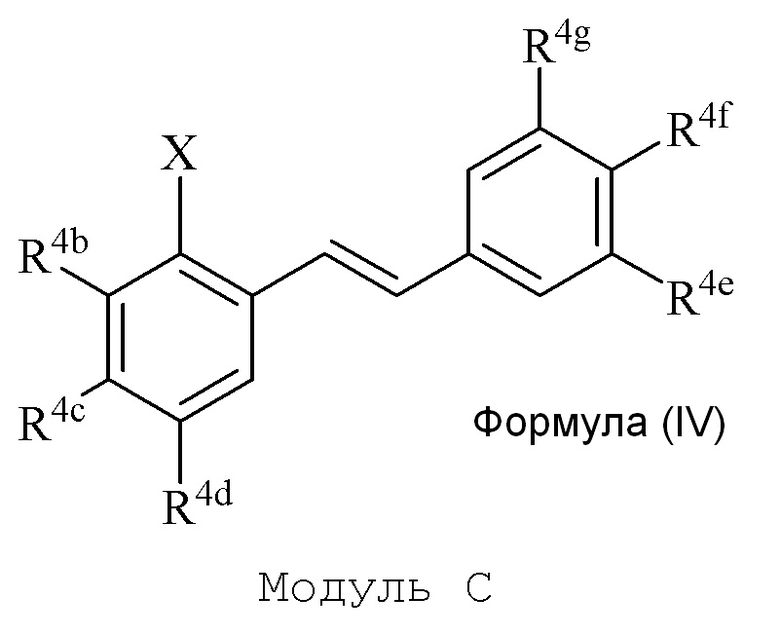
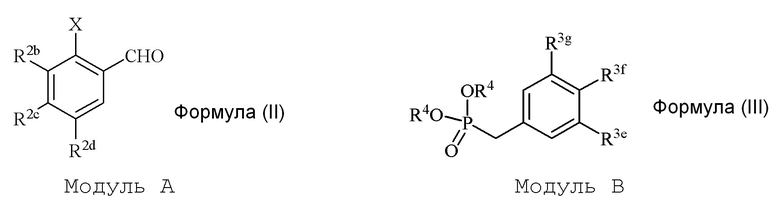
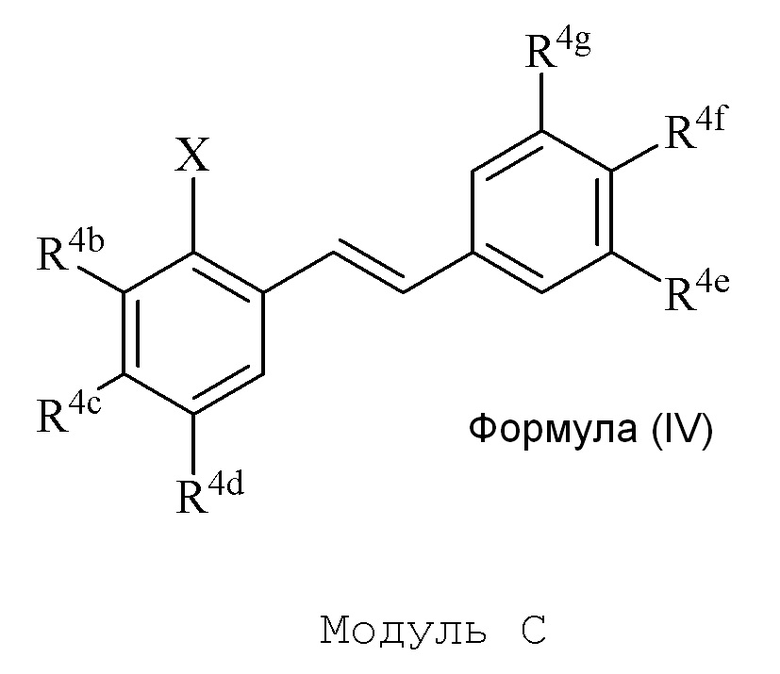
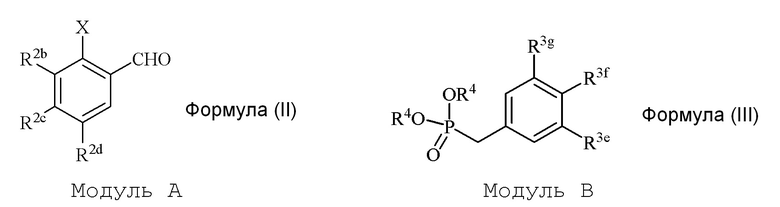
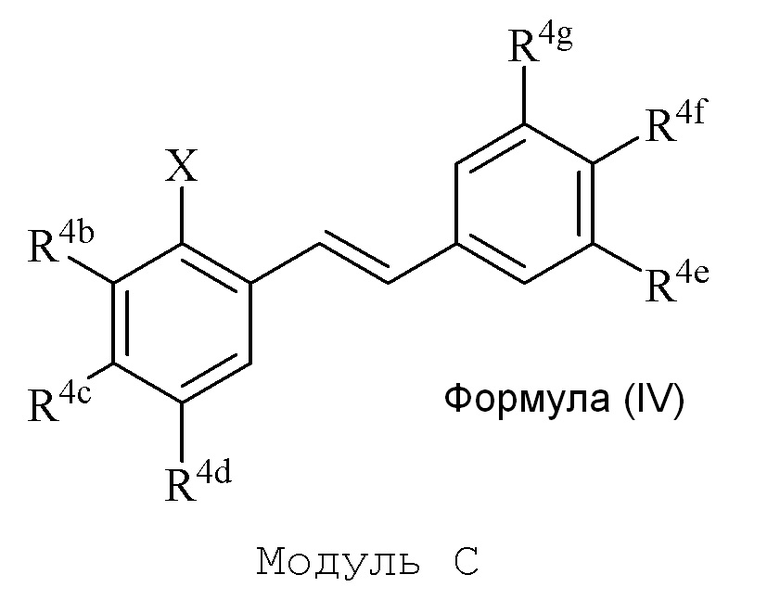
В настоящем изобретении используется модульный подход для синтеза соединения формулы (I). Кольцо A и кольцо B соединения 2-замещенного гидроксистильбена образуют из двух раздельных модулей, модуля A и модуля B, соответственно. Модуль A представляет собой галогенидное или гидроксильное производное бензальдегида, а модуль B представляет собой ароматическое фосфонатное производное. Согласно настоящему изобретению, реакция сочетания галоген/гидроксибензальдегида (модуля A) с ароматическим фосфонатом (модулем B) дает описанное выше производное 2-галоген- или 2-гидроксизамещенного гидроксистильбена (модуль C).
В описании изобретения, производное 2-галоген или 2-гидроксизамещенного гидроксистильбена (модуль C) может называться как производное 2-галоген/гидроксилзамещенного гидроксистильбена, производное 2-галоген/гидроксилгидроксистильбена, 2-галоген/гидроксилгидроксистильбен или модуль C, и может включать как производные 2-галогензамещенного гидроксистильбена, так и производные 2-гидроксизамещенного гидроксистильбена, и упомянутые выше варианты эквивалентных названий.
Авторы настоящего изобретения неожиданно обнаружили, что этот метод позволяет проводить альтернативный синтез соединений 2-замещенного гидроксистильбена и использование различных заместителей в 2-положении соединений 2-замещенного гидроксистильбена, благодаря чему группа, замещенная 2-положении, может быть не только пренильной группой. Соответственно, в настоящем изобретении предлагается способ получения описанного выше соединения 2-замещенного гидроксистильбена формулы (I), при котором введение 2-алкенильной или бензильной группы в положение R1a происходит в результате непосредственного сочетания 2-алкенильной или бензильной группы бороновой кислоты/производного или оловоорганического соединения, содержащего эти группы, с производным 2-галоген/гидроксизамещенным гидроксистильбена.
В одном варианте осуществления, синтез упомянутого выше соединения (I) характеризуется высокой эффективностью с точки зрения небольшого количества стадий синтеза из приготовленных исходных модулей и универсальности и гибкости применения при получении широкого разнообразия синтетических производных. В еще одном варианте осуществления, каждая стадия синтеза, начиная с приготовленных модулей, характеризуется высоким выходом.
В одном варианте осуществления, способ получения описанного выше соединения формулы (I), когда R1a представляет собой пренильную группу, позволяет достигать повышения выхода на стадиях, описанных в патентном документе WO2012/149608, которые продуцируют смесь продуктов.
В одном варианте осуществления настоящего изобретения предлагается метод синтеза, в котором стадия перегруппировки O- в C-пренил, описанная в патентном документе WO2012/149608, заменена на непосредственное сочетание пренильной группы с ароматическим кольцом путем проведения реакции сочетания пренилбороновых кислот/эфиров или пренилтрибутилстаннана с производным 2-галоген- или 2-гидроксилзамещенного гидроксистильбена. Преимуществом метода является то, что в R1a положение структуры гидроксистильбена могут быть введены группы, не являющиеся пренилом.
Преимуществом является и то, что авторы изобретения могут применять раскрытый в изобретении универсальный метод для эффективного получения в промышленных масштабах ранее выявленных соединений 2-пренилгидроксистильбена, которые являются важными при проведении лекарственной терапии. Благодаря универсальности описанного в изобретении метода, авторы изобретения смогли получить широкий ряд соединений 2-замещенных гидроксистильбена формулы (I), которые они ранее не могли получить с использованием метода перегруппировки. Авторы изобретения неожиданно обнаружили, что соединения 2-замещенного гидроксистильбена формулы (I), полученные описанным в изобретении методом, являются чрезвычайно перспективными для разработки новых терапевтических средств, применяемых при лечении таких заболеваний, как рак, и заболеваний или нарушений кожи, таких как атопический дерматит и псориаз.
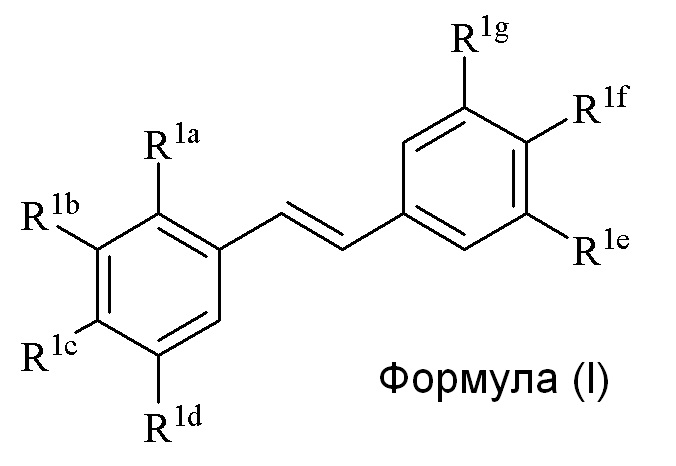
В первом аспекте, в настоящем изобретении предлагается способ синтеза соединения формулы (I),
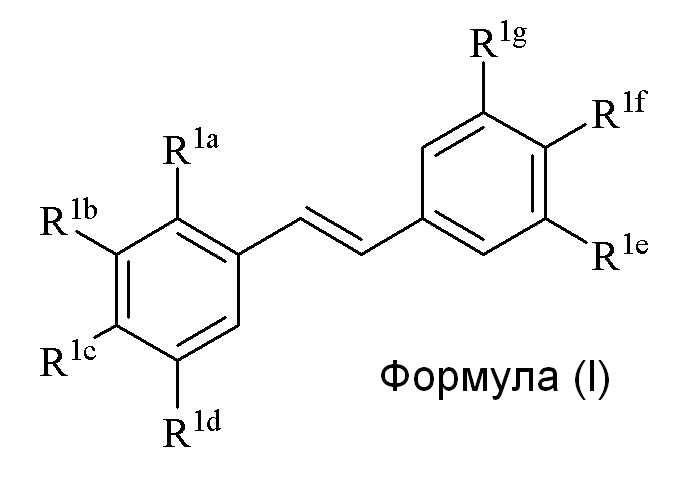
где
R1a представляет собой независимо аллил, кротил, пренил, геранил, фарнезил, бензил или 2-алкенил, 2-алкинил;
R1b представляет собой независимо СF3, OH или OR2, NO2, NHEt, NMe2, NMeEt, NHR3, NMeR3, NHC=NH(NH2) или COOR2;
R1c представляет собой независимо H, алкил, алкенил, алкинил, алкандиенил, пренил, геранил, фарнезил или бензил;
R1d представляет собой независимо СF3, OH или OR2, NHR3, NO2, NMeR3, NHC=NH(NH2) или COOR2;
R1e представляет собой независимо H, C1-C6 алкил, OH, OR2, NO2, NMe2, NHR3, NMeR3 , NHC(O)H или SR2;
R1f представляет собой независимо H, OH, OR2, NO2, NHR3, NHC=NH(NH2), COOR2, COOH;
R1g представляет собой независимо H, алкил, CF3, OH или OR2;
и не более чем один из R1e, R1f или R1g может представлять собой H;
или R1e и R1f образуют пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу или (CO)CH2 группу, когда R1e независимо представляет собой OH, NH2 или NHMe, и R1f представляет собой NH2, так что азот в R1f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R1e;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил, COCOOR4 или O-защитную группу, когда R2 присоединен к O;
R3 представляет собой независимо H, CH3, OH, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, CO(CH2)nNH2, CONH2, CO(CH2)nCOOH или COCOOR4, где n представляет собой 0, 1 или 2; и
R4 представляет собой C1-C6 алкил;
где способ включает:
i) стадию сочетания соединения формулы (II) (модуля A) с соединением формулы (III) (модулем B) с получением соединения формулы (IV) (модуля C):
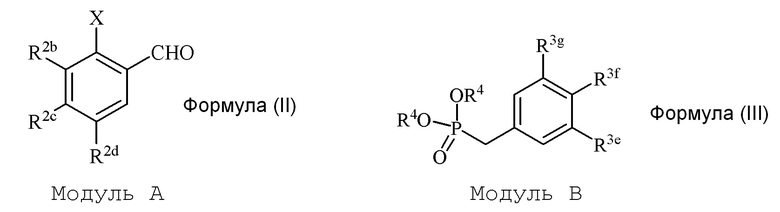
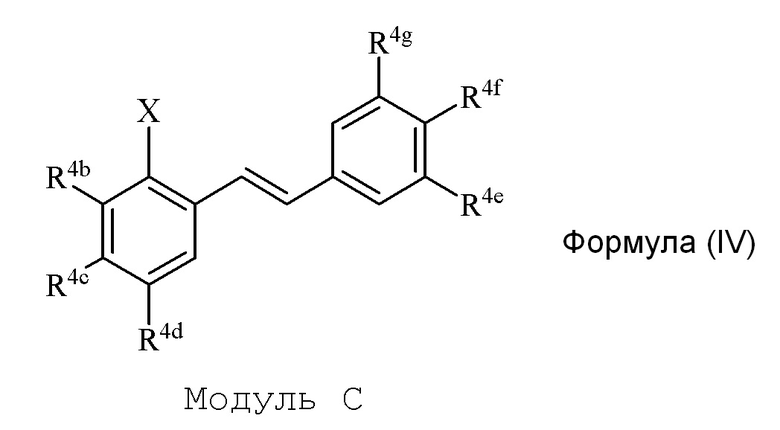
где
в формуле II (модуле A):
R2b представляет собой независимо СF3, OH, OR2, NO2, NMe2, NHEt, NMeEt, NHR3 или NMeR3;
R2c представляет собой независимо H, алкил, алкенил, алкинил, алкандиенил, пренил, геранил, фарнезил или бензил;
R2d представляет собой независимо СF3, OH или OR2, NO2,NHR3, NMeR3, NHC=NH(NH2) или COOR2;
X представляет собой галогенид или гидроксильную группу, где галогенид выбирают из группы, состоящей из F, Cl, Br, I и At;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил, или O-защитную группу, когда R2 присоединен к O; и
R3 представляет собой независимо H, OH, CH3, (CO)H, COMe, SO2Me, COCH2NH2 или CH2COOR2; и
в формуле III (модуле B):
R3e представляет собой независимо H, C1-C6 алкил, OH, OR2, NO2, NHMe, NMe2, NHR3 или NMeR3, SR2;
R3f представляет собой независимо H, OH, OR2, NO2, NHR3, NHC=NH(NH2), COOH или COOR2;
R3g представляет собой независимо H, алкил, CF3, OH или OR2;
и не более чем один из R3e, R3f или R3g может представлять собой H;
или R3e и R3f образуют пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу или (CO)CH2 группу, когда R3e представляет собой OH, NH2 или NHMe, и R3f представляет собой NH2, так что азот в R3f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R3e;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 присоединен к O,
R3 представляет собой независимо H, CH3, OH, (CO)H, COMe, SO2Me, COCH2NH2, CH2COOH или CH2COOR2; и
в формуле IV (Модуль C):
R4b представляет собой независимо СF3, OH или OR2, NO2, NMe2, NHEt, NMeEt, NHR3 или NMeR3;
R4c представляет собой независимо H, алкил, алкенил, алкинил, алкандиенил, пренил, геранил, фарнезил или бензил;
R4d представляет собой независимо СF3, OH или OR2, OR3, NO2 или NHR3, NMeR3, NHC=NH(NH2) или COOR2;
R4e представляет собой независимо H, C1-C6 алкил, OH, OR2, NO2, NMe2, NHR3 или NMeR3, SR2;
R4f представляет собой независимо H, OH, OR2, NO2, NHR3, NHC=NH(NH2), COOH или COOR2;
R4g представляет собой независимо H, алкил, CF3, OH или OR2;
и не более чем один из R4e, R4F или R4g может представлять собой H;
или R4e и R4f образуют пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу или (CO)CH2 группу, когда R4e представляет собой OH, NH2 или NHMe, и R4f представляет собой NH2, так что азот в R4f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R4e;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 присоединен к O;
R3 представляет собой независимо H, CH3, OH, (CO)H, COMe, SO2Me, COCH2NH2, CH2COOH или CH2COOR2; и
X представляет собой галогенид или гидроксильную группу, где галогенид выбирают из группы, состоящей из F, Cl, Br, I и At.
Соединения 2-замещенного галоген/гидрокси гидроксистильбена формулы (IV) (модуль C) подвергают последующей реакции с получением описанного в изобретении соединения формулы (I).
В одном варианте осуществления, соединение 2-замещенного галогенгидроксистильбена формулы (IV) (модуль C) подвергают последующей реакции сочетания с 2-алкенил- или бензилбороновой кислотой или эфиром, с соединением 2-алкенил- или бензилтрифторбората или с соединением 2-алкенил- или бензилорганостаннана с получением соединения формулы (I).
В еще одном варианте осуществления, когда X представляет собой гидроксил в формуле (IV), требуется активация гидроксила в трифлатную группу (OTf, трифторметилсульфонат) перед сочетание с 2-алкенил- или бензилбороновой кислотой или эфиром, с соединением 2-алкенил- или бензилтрифторбората или с соединением 2-алкенил- или бензилорганостаннана с получением соединения формулы (I).
Соединение формулы (IV) или (I) может быть необязательно подвергнуто одной или более или последующим реакциям, включающим, но этим не ограничивая, реакцию с одним или более -NH2, NO2 и/или -OH заместителями в формуле (IV) или формуле (I).
Во втором аспекте, в настоящем изобретении предлагается соединение формулы (I)
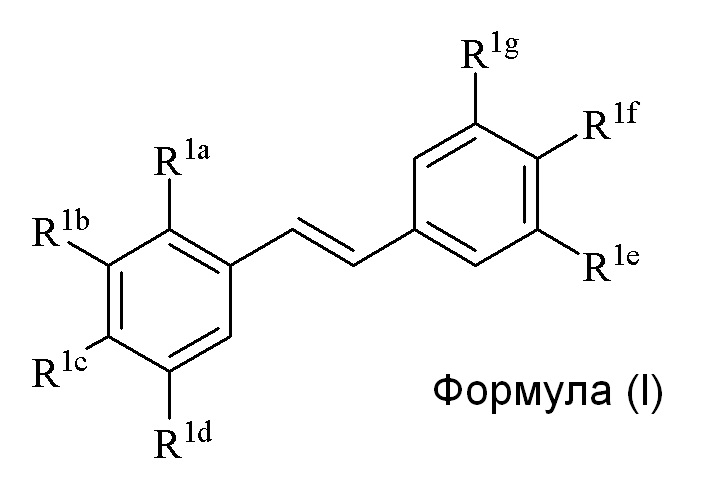
где
R1a представляет собой независимо аллил, кротил, пренил, геранил, фарнезил, бензил, 2-алкенил или 2-алкинил;
R1b представляет собой независимо СF3, OH или OR2, NO2, NHEt, NMe2, NMeEt, NHR3, NMeR3, NHC=NH(NH2) или COOR2;
R1c представляет собой независимо H, алкил, алкенил, алкинил, алкандиенил, пренил, геранил, фарнезил или бензил;
R1d представляет собой независимо СF3, OH или OR2, NHR3, NO2, NMeR3, NHC=NH(NH2) или COOR2;
R1e представляет собой независимо H, C1-C6 алкил, OH, OR2, NO2, NMe2, NHR3 или NMeR3, NHC(O)H или SR2;
R1f представляет собой независимо H, OH, OR2, NO2, NHR3, NHC=NH(NH2), COOR2 или COOH;
R1g представляет собой независимо H, алкил, CF3, OH или OR2;
и только один из R1e, R1f или R1g может представлять собой H;
или R1e и R1f образуют пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу или (CO)CH2 группу, когда R1e представляет собой OH, NH2 или NHMe, и R1f представляет собой NH2, так что азот в R1f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R1e;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил, COCOOR4 или O-защитную группу, когда R2 присоединен к O, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этокси]метила (SEM), CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой независимо H, CH3, OH, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, CO(CH2)nNH2, CONH2, CO(CH2)nCOOH или COCOOR4, где n=0, 1 или 2;
R4 представляет собой C1-C6 алкил; и
галогенид выбирают из группы, состоящей из F, Cl, Br, I и At;
где соединение формулы (I) получают способом в соответствии с описанным в изобретении первым аспектом.
В третьем аспекте, в настоящем изобретении предлагается соединение формулы (I)
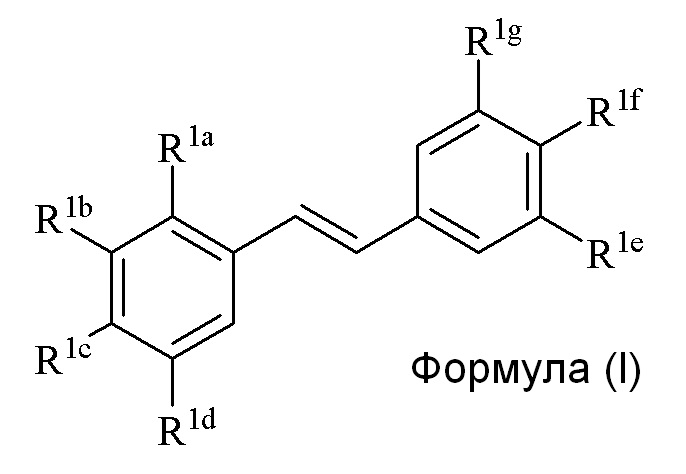
где
R1a представляет собой независимо аллил, кротил, пренил, геранил, фарнезил, бензил, 2-алкенил или 2-алкинил;
R1b представляет собой независимо СF3, OH, OR2, NO2, NHEt, NMe2, NMeEt, NHR3, NMeR3, NHC=NH(NH2) или COOR2;
R1c представляет собой независимо H, алкил, алкенил, алкинил, алкандиенил, пренил, геранил, фарнезил или бензил;
R1d представляет собой независимо СF3, OH, OR2, NO2, NHR3, NMeR3, NHC=NH(NH2) или COOR2;
R1e представляет собой независимо H, C1-C6 алкил, OH, OR2, NO2, NMe2, NHR3 или NMeR3, NHC(O)H и SR2;
R1f представляет собой независимо H, OH, OR2, NO2, NHR3, NHC=NH(NH2), COOR2, COOH;
R1g представляет собой независимо H, алкил, CF3, OH или OR2;
и только один из R1e, R1f или R1g может представлять собой H;
или R1e и R1f образуют пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу или (CO)CH2 группу, когда R1e представляет собой OH, NH2 или NHMe, и R1f представляет собой NH2, так что азот в R1f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R1e;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил, COCOOR4 или O-защитную группу, когда R2 присоединен к O, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этокси]метил (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой независимо H, CH3, OH, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, CO(CH2)nNH2, CONH2, CO(CH2)nCOOH или COCOOR4, где n представляет собой 0, 1 или 2;
R4 представляет собой C1-C6 алкил;
галогенид выбирают из группы, состоящей из F, Cl, Br, I и At; и
только один из R1e, R1f или R1g может представлять собой H,
при условии, что
когда R1a и/или R1c представляет собой пренил, R1d представляет собой OH, R1f представляет собой OH, R1g представляет собой H, и R1e представляет собой OH, OMe или OEt, то тогда R1b не может представлять собой OH или OR2, где R представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил или бензил.
В четвертом аспекте, в настоящем изобретении предлагается способ лечения рака, включающий введение терапевтически эффективного количества соединения по второму или третьему аспекту настоящего изобретения или фармацевтически приемлемой соли, сольвата или фармацевтической композиции, включающей указанные соединения, пациенту, нуждающемуся в этом.
В пятом аспекте, в настоящем изобретении предлагается способ лечения заболевания или нарушения кожи, включающий введение терапевтически эффективного количества соединения по второму или третьему аспекту настоящего изобретения или фармацевтически приемлемой соли, сольвата или фармацевтической композиции, включающей указанные соединения, пациенту, нуждающемуся в этом.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
В изобретении предлагается способ синтеза соединения формулы (I):
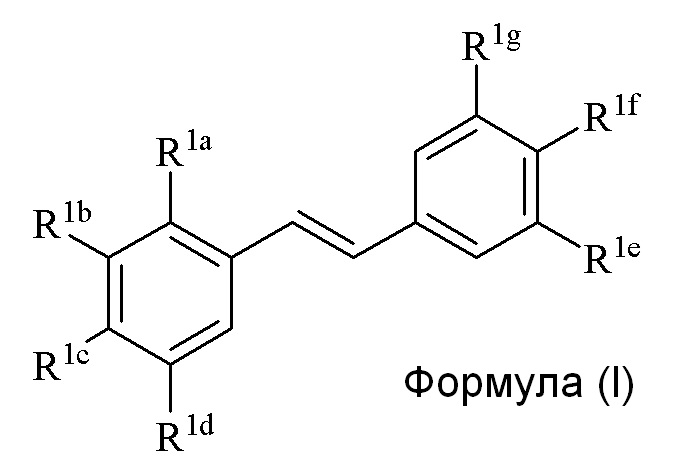
где
R1a представляет собой независимо аллил, кротил, пренил, геранил, фарнезил, бензил, 2-алкенил или 2-алкинил;
R1b представляет собой независимо СF3, OH, OR2, NO2, NHEt, NMe2, NMeEt, NHR3, NMeR3, NHC=NH(NH2) или COOR2;
R1c представляет собой независимо H, алкил, алкенил, алкинил, алкандиенил, пренил, геранил, фарнезил или бензил;
R1d представляет собой независимо СF3, OH, OR2, NHR3, NO2, NMeR3, NHC=NH(NH2) или COOR2;
R1e представляет собой независимо H, C1-C6 алкил, OH, OR2, NO2, NMe2, NHR3 или NMeR3, NHC(O)H, SR2;
R1f представляет собой независимо H, OH, OR2, NO2, NHR3, NHC=NH(NH2), COOR2, COOH;
R1g представляет собой независимо H, алкил, CF3, OH или OR2;
и не более чем один из R1e, R1f или R1g может представлять собой H;
или R1e и R1f образуют пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу или (CO)CH2 группу, когда R1e независимо представляет собой OH, NH2 или NHMe, и R1f представляет собой NH2, так что азот в R1f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R1e;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил, COCOOR4 или O-защитную группу, когда R2 присоединен к O;
R3 представляет собой независимо H, CH3, OH, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, CO(CH2)nNH2, CONH2, CO(CH2)nCOOH или COCOOR4, где n представляет собой 0, 1 или 2; и
R4 представляет собой C1-C6 алкил;
где способ включает:
i) стадию сочетания соединения формулы (II) (модуля A) с соединением формулы (III) (модулем B) с получением соединения формулы (IV) (модуля C):
где
в формуле II (модуле A):
R2b представляет собой независимо СF3, OH, OR2, NO2, NMe2, NHEt, NMeEt, NHR3 или NMeR3;
R2c представляет собой независимо H, алкил, алкенил, алкинил, алкандиенил, пренил, геранил, фарнезил или бензил;
R2d представляет собой независимо СF3, OH или OR2, NO2, NHR3, NMeR3, NHC=NH(NH2) или COOR2;
X представляет собой галогенид или гидроксильную группу, где галогенид выбирают из группы, состоящей из F, Cl, Br, I и At;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу;
R3 представляет собой независимо H, OH, CH3, (CO)H, COMe, SO2Me, COCH2NH2 или CH2COOR2; и
в формуле III (модуле B):
R3e представляет собой независимо H, C1-C6 алкил, OH, OR2, NO2, NHMe, NMe2, NHR3 или NMeR3, SR2;
R3f представляет собой независимо H, OH, OR2, NO2, NHR3, NHC=NH(NH2), COOH или COOR2;
R3g представляет собой независимо H, алкил, CF3, OH, OR2;
и не более чем один из R3e, R3f или R3g может представлять собой H
или R3e и R3f образуют пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу или (CO)CH2 группу, когда R3e представляет собой OH, NH2 или NHMe, и R3f представляет собой NH2, так что азот в R3f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R3e;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 присоединен к O,
R3 представляет собой H, CH3, OH, (CO)H, COMe, SO2Me, COCH2NH2, CH2COOH и CH2COOR2; и
в формуле IV (модуле C):
R4b представляет собой независимо СF3, OH или OR2, NO2, NMe2, NHEt, NMeEt, NHR3 или NMeR3;
R4c представляет собой независимо H, алкил, алкенил, алкинил, алкандиенил, пренил, геранил, фарнезил или бензил;
R4d представляет собой независимо СF3, OH, OR2, OR3, NO2 или NHR3, NMeR3, NHC=NH(NH2) или COOR2;
R4e представляет собой независимо H, C1-C6 алкил, OH, OR2, NO2, NMe2, NHR3 или NMeR3, NHC(O)H или SR2;
R4f представляет собой независимо H, OH, OR2, NO2, NHR3, NHC=NH(NH2), COOH или COOR2;
R4g представляет собой независимо H, алкил, CF3, OH или OR2;
или R4e и R4f образуют пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу или (CO)CH2 группу, когда R4e представляет собой OH, NH2 или NHMe, и R4f представляет собой NH2, так что азот в R4f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R4e;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 присоединен к O;
R3 представляет собой независимо H, CH3, OH, (CO)H, COMe, SO2Me, COCH2NH2, CH2COOH или CH2COOR2; и
X представляет собой галогенид или гидроксильную группу, где галогенид выбирают из группы, состоящей из F, Cl, Br, I и At.
O-защитная группа может быть выбрана из известных в данной области защитных групп. Подходящие O-защитные группы, которые могут применяться в настоящем изобретении, хорошо известны специалисту в данной области.
O-защитная группа может представлять собой COMe, t-BuSi(CH3)2, 2-(триметилсилил)этокси]метил (SEM), CH(OEt)CH3, тетрагидропиранил или C(OEt)(CH3)2.
В соответствии с соединениями, определяемыми в изобретении формулой (I), формулой (III) и формулой (IV), R1e, 3e, 4e и R1f, 3f, 4f могут образовывать пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу или (CO)CH2 группу, когда R1e, 3e, 4e представляет собой OH, NH2 или NHMe, и R1f, 3f, 4f представляет собой NH2, так что азот в R1f, 3f, 4f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R1e, 3e, 4e. В одном варианте осуществления, азот в R1f, 3f, 4f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R1e, 3e, 4e с образованием группы, выбранной из любой одной из групп: -NH -C(O)- O -, -NH-C(O)-CH2- O -, - NH -CH2C(O)-O-, -NH-C(O)-NH-, -NH-C(O) CH2-NH-, -NH-CH2C(O)-NH-, -NH -C(O)-N(Me)-, -NH-C(O) CH2-NMe- или -NH -CH2C(O)-N(Me)-. В одном варианте осуществления R1f, 3f, 4f представляет собой NH2, и R1e, 3e, 4e представляет собой NHMe, и азот в R1f, 3f, 4f соединен мостиковой связью через карбонильную группу с азотом в R1e, 3e, 4e с образованием -NH -C(O)-N(Me)-, так что R1e, 3e, 4e и R1f, 3f, 4f образуют пятичленное гетероциклическое кольцо. В еще одном варианте осуществления, R1f, 3f, 4f представляет собой NH2, и R1e, 3e, 4e представляет собой OH, и азот в R1f, 3f, 4f соединен мостиковой связью через карбонильную группу с кислородом R1e, 3e, 4e с образованием -NH-C(O)-O-, так что R1e, 3e, 4e и R1f, 3f, 4f образуют пятичленное гетероциклическое кольцо.
Соединение 2-замещенного галоген/гидрокси гидроксистильбена формулы (IV) (модуль C) подвергают последующей реакции с получением описанного в изобретении соединения формулы (I).
В модуле A и C, группа X представляет собой галогенид или гидроксильную группу. В одном варианте осуществления, X представляет собой галогенид. Когда X представляет собой галогенид, формула (II) и (IV) может быть названа как галогенид формулы (II) и галогенид формулы (IV). В еще одном варианте осуществления, X представляет собой гидроксильную группу. Когда X представляет собой гидроксильную группу, формула (II) и (IV) может быть названа как гидроксил формулы (II) и гидроксил формулы (IV). Гидроксил формулы (IV) может быть активирован в соответствующую трифлатную (трифторметилсульфонатную) группу и может быть назван как трифлат формулы (IV) или 2-замещенный трифлат гидроксистильбена формулы (IV).
Превращение гидроксильной группы в трифлатную группу может быть проведено известными в данной области методами. В одном варианте осуществления, когда X представляет собой гидроксил, гидроксил формулы (IV) обрабатывают с помощью трифторметансульфонового ангидрида в присутствии N-метилморфина с получением трифлата формулы (IV).
В одном варианте осуществления, когда X представляет собой галогенид в формуле (IV), галогенид формулы (IV) подвергают реакции сочетания с одним из следующих соединений:
a) с соединением R1a-замещенной бороновой кислоты или эфира;
b) с соединением R1a-замещенного трифторбората; или
c) с соединением R1a-замещенного органостаннана;
с образованием соединения формулы (I).
В одном варианте осуществления, соединение 2-замещенного галогенгидроксистильбена формулы (IV) (модуль C) подвергают последующей реакции сочетания с соединением 2-алкенил- или бензилбороновой кислоты или эфира с получением соединения формулы (I). В одном варианте осуществления, соединение 2-замещенного галогенгидроксистильбена формулы (IV) (модуль C) подвергают последующей реакции сочетания с соединением 2-алкенил- или бензилтрифторбората с получением соединения формулы (I). В еще одном варианте осуществления, соединение 2-замещенного галогенгидроксистильбена формулы (IV) (модуль C) подвергают последующей реакции сочетания с соединением 2-алкенил- или бензилорганостаннана с получением соединения формулы (I).
В одном варианте осуществления, когда X представляет собой гидроксил в формуле (IV) (гидроксил формулы (IV)), требуется активация в трифлатную группу (OTf, трифторметилсульфонат) перед тем как проводить реакцию сочетания. В одном варианте осуществления, гидроксильную группу формулы (IV) превращают в трифлатную (трифторметилсульфонатную) группу с образованием трифлата формулы (IV), и трифлат формулы (I) подвергают реакции сочетания с одним из следующих соединений:
a) с соединением R1a-замещенной бороновой кислоты или эфира;
b) с соединением R1a-замещенного трифторбората; или
c) с соединением R1a-замещенного органостаннана
с образованием соединения формулы (I).
В одном варианте осуществления, соединение 2-замещенного гидроксигидроксистильбена формулы (IV) (модуль C) активируют в 2-замещенный трифлатгидроксистильбен, который может быть затем подвергнут реакции сочетания с 2-алкенил- или бензилбороновой кислотой или эфиром с получением соединения формулы (I). В еще одном варианте осуществления, трифлат формулы (IV) подвергают последующей реакции сочетания с соединением 2-алкенил- или бензилтрифторбората с получением соединения формулы (I). В еще одном варианте осуществления, трифлат формулы (IV) подвергают последующей реакции сочетания с соединением 2-алкенил- или бензилорганостаннана с получением соединения формулы (I).
В одном варианте осуществления, описанные в изобретении соединение галогенида формулы (IV) или трифлата формулы (IV) подвергают реакции сочетания с соединением R1a-замещенной бороновой кислоты с образованием соединения формулы (I), где R1a выбирают из группы, состоящей из аллила, кротила, пренила, геранила, фарнезила, бензила, 2-алкенила и 2-алкинила.
В еще одном варианте осуществления, описанные в изобретении соединение галогенида формулы (IV) или трифлата формулы (IV) подвергают реакции сочетания с соединением R1a-замещенного органостаннана с образованием соединения формулы (I), где R1a выбирают из группы, состоящей из аллила, кротила, пренила, геранила, фарнезила, бензила, 2-алкенила и 2-алкинила.
В еще одном варианте осуществления, описанные в изобретении соединение галогенида формулы (IV) или трифлата формулы (IV) подвергают реакции сочетания с R1a-замещенным трифторборатом с образованием соединения формулы (I), где R1a выбирают из группы, состоящей из аллила, кротила, пренила, геранила, фарнезила, бензила, 2-алкенила и 2-алкинила.
В еще одном варианте осуществления, заместитель R1a в формуле (II) и (IV) выбирают из группы, состоящей из аллила, кротила, пренила, бензила или 2-алкенила. В еще одном варианте осуществления, R1a выбирают из группы, состоящей из аллила, кротила, пренила или бензила.
Реакция сочетания описанных в изобретении соединения формулы (IV) или трифлата формулы (IV) с соединением R1a-замещенной бороновой кислоты, с соединением R1a-замещенного органостаннана или с соединением R1a-замещенного трифторбората может быть катализирована с помощью подходящего катализатора, включающего соединение палладия. Палладиевый катализатор включает, но этим не ограничивая, тетракис(трифенилфосфин)палладий(0) и 1,1′-бис(ди-фенилфосфино)ферроцен]дихлорпалладий(II). Описанная в изобретении реакция сочетания может быть также проведена в присутствии основания, включающего, но этим не ограничивая, карбонаты щелочных металлов, такие как карбонат калия (K2CO3), гидрокарбонаты, карбонат кальция, карбонат магния и алкиламины, в том числе триэтиламин.
В одном варианте осуществления, соединение R1a-замещенной бороновой кислоты представляет собой пинаколовый эфир аллил-, кротил-, пренил-, бензил-, 2-алкенил-бороновой кислоты. Пинаколовый эфир пренил-бороновой кислоты включает, но этим не ограничивая, пинаколовый эфир 3-метилбут-2-енилбороновой кислоты, пинаколовый эфир кротилбороновой кислоты, пинаколовый эфир аллилбороновой кислоты, пинаколовый эфир бензилбороновой кислоты.
В одном варианте осуществления, описанные в изобретении соединение галогенида формулы (IV) или трифлата формулы (IV) подвергают реакции сочетания с пинаколовым эфиром R1a-замещенной бороновой кислоты, катализируемой с помощью палладиевого катализатора в присутствии основания. В еще одном варианте осуществления, соединение галогенида формулы (IV) подвергают реакции сочетания с R1a-замещенным трибутилстаннаном, катализируемой с помощью тетракис(трифенилфосфин)палладия(0).
В еще одном варианте осуществления, описанные в изобретении соединение галогенида формулы (IV) или трифлата формулы (IV) подвергают реакции сочетания с R1a-замещенным трифторборатом. В одном конкретном варианте осуществления, соединение галогенида или трифлата формулы (IV) подвергают реакции сочетания с аллилтрифторборатом калия, катализируемой с помощью 1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) и триэтиламина. Этот метод сочетания позволяет вводить аллильную группу в R1a положении в кольце A.
В одном варианте осуществления, предлагается способ синтеза соединения формулы (I),
где в формуле (I):
R1a представляет собой независимо аллил, кротил, пренил, бензил или 2-алкенил;
R1b независимо представляет собой OH, OR2 или NHC(O)Me;
R1c представляет собой H;
R1d независимо представляет собой OH, NHR3, NH(CO)H или NO2;
R1e независимо представляет собой OH, OR2, NMe2, NHR3, NMeR3, NHC(O)H или SR2;
R1f представляет собой независимо H, OH, NO2, OR2, NHR3, NHC=NH(NH2) или COOH;
R1g представляет собой независимо H, алкил, OH или OR2;
и не более чем один из R1e, R1f или R1g может представлять собой H;
или R1e и R1f образуют пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу, когда R1e представляет собой OH, NH2 или NHMe, и R1f представляет собой NH2, так что азот в R1f соединен мостиковой связью через карбонильную группу с кислородом или азотом в R1e;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил или O-защитную группу, когда R2 присоединен к O, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этокси]метила (SEM), CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой независимо H, CH3, OH, (CO)H, (CO)Me, SO2Me, CO(CH2)nNH2, CH2COOR2, CO(CH2)nCOOH или COCOOR4, где n представляет собой 0, 1 или 2; и
R4 представляет собой C1-C6 алкил;
в формуле II (модуле A):
R2b представляет собой OH или OR2;
R2c представляет собой H;
R2d независимо представляет собой OH или OR2, NO2, NHR3 или COOR2;
X представляет собой галогенид (Hal) или гидроксильную группу, где галогенид выбирают из группы, состоящей из F, Cl, Br, I и At;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил, или O-защитную группу;
R3 представляет собой независимо H, OH, CH3, (CO)H, COMe, SO2Me, COCH2NH2 или CH2COOR2; и
в формуле III (модуле B):
R3e независимо представляет собой OH, OR2, NO2, NHMe, NMe2, NHR3 NMeR3 или SR2;
R3f представляет собой независимо H, OH, OR2, NO2, NHR3, NHC=NH(NH2), COOH, COOR2;
R3g представляет собой независимо H, алкил, OH, OR2;
и не более чем один из R3e, R3f или R3g может представлять собой H;
или R3e и R3f образуют пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу или (CO)CH2 группу, когда R3e представляет собой OH, NH2 или NHMe, и R3f представляет собой NH2, так что азот в R3f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R3e;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 присоединен к O,
R3 представляет собой независимо H, CH3, OH, (CO)H, COMe, SO2Me, COCH2NH2, CH2COOH или CH2COOR2; и
в формуле IV (модуле C):
R4b представляет собой OH или OR2, NHC(O)Me;
R4c представляет собой H;
R4d независимо представляет собой OH или OR2, NO2 или NHR3, NMeR3;
R4e независимо представляет собой OH, OR2, NO2, NMe2, NHR3 или NMeR3, NHC(O)H или SR2;
R4f представляет собой независимо H, OH, OR2, NO2, NHR3, NHC=NH(NH2), COOH или COOR2;
R4g представляет собой независимо H, алкил, CF3, OH или OR2;
и не более чем один из R4e, R4f или R4g может представлять собой H;
или R4e и R4f образуют пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу или (CO)CH2 группу, когда R4e представляет собой OH, NH2 или NHMe, и R4f представляет собой NH2, так что азот в R4f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R4e;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 присоединен к O;
R3 представляет собой независимо H, CH3, OH, (CO)H, COMe, SO2Me, COCH2NH2, CH2COOH или CH2COOR2; и
X представляет собой галогенид или гидроксильную группу, где галогенид выбирают из группы, состоящей из F, Cl, Br, I и At.
В одном варианте осуществления, предлагается синтез соединения формулы (I):
где в формуле (I):
R1a независимо представляет собой кротил, пренил, бензил или 2-алкенил;
R1b представляет собой CF3, OR2 или NHC(O)Me;
R1c представляет собой H;
R1d независимо представляет собой OH, NHR3 или NO2;
R1e представляет собой независимо OR2, NMe2, NHR3 или SR2;
R1f представляет собой независимо H, OH, NO2, NHR3, NHC=NH(NH2) или COOH;
R1g представляет собой независимо H, алкил, OH или OR2;
или R1e и R1f образуют пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу, когда R1e представляет собой OH, NH2 или NHMe, и R1f представляет собой NH2, так что азот в R1f соединен мостиковой связью через карбонильную группу с кислородом или азотом в R1e;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил;
R3 представляет собой независимо H, Me, (CO)H, (CO)Me, SO2Me, CO(CH2)nNH2, CO(CH2)nCOOH или COCOOR4 (где n=0, 1 или 2); и
R4 представляет собой C1-C6 алкил;
где синтез включает:
i) стадию сочетания соединения формулы (II) (модуля A) с соединением формулы (III) (модулем B) с получением соединения формулы (IV) (модуля C):
где
в формуле II (модуле A):
R2b представляет собой CF3, OH, OR2, NO2, NMe2, NHEt, NMeEt, NHR3 или NMeR3;
R2c представляет собой H, алкил, алкенил, алкинил, алкандиенил, пренил, геранил, фарнезил или бензил;
R2d представляет собой CF3, OH или OR2, NO2, NHR3, NMeR3, NHC=NH(NH2) или COOR2;
X представляет собой галогенид (Hal) или гидроксильную группу, где галогенид выбирают из группы, состоящей из F, Cl, Br, I и At;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу;
R3 представляет собой H, OH, CH3, (CO)H, COMe, SO2Me, COCH2NH2, или CH2COOR2; и
в формуле III (модуле B):
R3e представляет собой независимо OH, OR2, NO2, NHMe, NMe2, NHR3 или NMeR3, (CO)H, NHC(O)H или SR2;
R3f представляет собой независимо H, OH, OR2, NO2, NHR3;
R3g представляет собой независимо H, алкил, OH, OR2;
и не более чем один из R3e, R3f или R3g может представлять собой H;
или R3e и R3f образуют пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу или (CO)CH2 группу, когда R3e представляет собой OH, NH2 или NHMe, и R3f представляет собой NH2, так что азот в R3f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R3e;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 присоединен к O,
R3 представляет собой H, CH3, (CO)H, COMe, SO2Me, COCH2NH2 или CH2COOR2; и
в формуле IV (модуле C):
R4b представляет собой независимо СF3, OH или OR2, NHC(O)Me;
R4c представляет собой H;
R4d представляет собой OH, NH2, N(CO)H;
R4e независимо представляет собой OH, OR2, NO2, NHR3 или NMeR3, SR2;
R4f представляет собой независимо H, OH, OR2, NO2, NHR3;
R4g представляет собой независимо H, алкил, CF3, OH, OR2;
и только один из R4e, R4f или R4g может представлять собой H;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 присоединен к O;
R3 представляет собой независимо H, (CO)H, COMe, SO2Me или CH2COOR2; и
X представляет собой галогенид или гидроксильную группу, где галогенид выбирают из группы, состоящей из F, Cl, Br, I и At.
В одном варианте осуществления, предлагается способ синтеза соединения формулы (I):
где в формуле (I):
R1a представляет собой независимо аллил, кротил, пренил, бензил;
R1b представляет собой независимо OR2 или NHC(O)Me;
R1c представляет собой H;
R1d независимо представляет собой OH, NH2, N(CO)H;
R1e независимо представляет собой OH, OR2, NO2, NHR3 или NMeR3, (CO)H, SMe;
R1f представляет собой независимо H, OH, OR2, NO2, NHR3, NHC=NH(NH2), COOH;
R1g представляет собой независимо H, алкил, OH, OR2;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 присоединен к O, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этоксиметила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой независимо H, CH3, OH, (CO)H, (CO)Me, SO2Me, CH2COOR2, COCH2NH2, CO(CH2)nCOOH (где n=0, 1 или 2);
где синтез включает:
i) стадию сочетания соединения формулы (II) (модуля A) с соединением формулы (III) (модулем B) с получением соединения формулы (IV) (модуля C):
где
в формуле II:
R2b представляет собой CF3, OH или OR2;
R2c представляет собой H,
R2d представляет собой OH или NH2, N(CO)H;
X представляет собой галогенид (Hal) или гидроксильную группу, где галогенид выбирают из группы, состоящей из F, Cl, Br, I и At;
галогенид, выбранный из группы, состоящей из F, Cl, Br, I и At;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 присоединен к O, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этоксиметила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2; и
в формуле III:
R3e представляет собой OH, OR2, NO2, NHR3 или NMeR3, SR2;
R3f представляет собой H, OH, OR2, NO2, NHR3;
R3g представляет собой H, алкил, OH, OR2;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 представляет собой защитную группу для O, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этоксиметила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2; и
R3 представляет собой H, CH3, (CO)H, COMe, SO2Me или CH2COOR2, COCH2NH2; и
в формуле IV:
R4b представляет собой CF3, OH или OR2;
R4c представляет собой H;
R4d представляет собой OH, NH2, N(CO)H;
R4e представляет собой OH, OR2, NO2, NHR3 или NMeR3, SR2;
R4f представляет собой H, OH, OR2, NO2, NHR3;
R4g представляет собой H, алкил, OH, OR2;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 присоединен к O, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этоксиметила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой H, (CO)H, COMe, SO2Me, или CH2COOR2; и
X представляет собой галогенид (Hal) или гидроксильную группу, где галогенид выбирают из группы, состоящей из F, Cl, Br, I и At;
В одном варианте осуществления, предлагается способ синтеза соединения формулы (I):
где
в формуле (I):
R1a представляет собой аллил, кротил, пренил, бензил;
R1b представляет собой OR2, NHC(O)Me
R1c представляет собой H;
R1d представляет собой OH;
R1e представляет собой OR2, NO2;
R1f представляет собой NO2, NHR3, NHC=NH(NH2);
R1g представляет собой H, алкил, OH, OR2;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил;
R3 представляет собой H, (CO)H, (CO)Me, SO2Me, CH2COOR2, COCH2NH2, CO(CH2)nCOOH (где n=0, 1 или 2);
где синтез включает:
i) стадию сочетания соединения формулы (II) (модуля A) с соединением формулы (III) (модулем B) с получением соединения формулы (IV) (модуля C):
где
в формуле II:
R2b представляет собой CF3, OH или OR2;
R2c представляет собой H,
R2d представляет собой OH или NH2, N(CO)H;
Hal представляет собой галогенид, выбранный из группы, состоящей из F, Cl, Br, I и At;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 присоединен к O, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этоксиметила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2; и
в формуле III:
R3e представляет собой OH, OR2, NO2, NHR3 или NMeR3, SR2;
R3f представляет собой H, OH, OR2, NO2, NHR3;
R3g представляет собой H, алкил, OH, OR2;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 представляет собой защитную группу для O, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этоксиметила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2; и
R3 представляет собой H, CH3, (CO)H, COMe, SO2Me или CH2COOR2, COCH2NH2; и
в формуле IV:
R4b представляет собой CF3, OH или OR2;
R4c представляет собой H;
R4d представляет собой OH, NH2, N(CO)H;
R4e представляет собой OH, OR2, NO2, NHR3 или NMeR3, SR2;
R4f представляет собой H, OH, OR2, NO2, NHR3;
R4g представляет собой H, алкил, OH, OR2;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 присоединен к O, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этоксиметила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой H, (CO)H, COMe, SO2Me или CH2COOR2; и
Hal представляет собой галогенид, выбранный из группы, состоящей из F, Cl, Br, I и At;
где соединение формулы IV подвергают реакции сочетания с пинаколовым эфиром R1a-бороновой кислоты, выбранным из группы, состоящей из пинаколового эфира 3-метилбут-2-енилбороновой кислоты, пинаколового эфира кротилбороновой кислоты, пинаколового эфира аллилбороновой кислоты и пинаколового эфира бензилбороновой кислоты, катализируемой с помощью палладиевого катализатора в присутствии основания, с получением соединения формулы (I).
В одном варианте осуществления, предлагается способ синтеза соединения формулы (I):
где
в формуле (I):
R1a представляет собой кротил, пренил;
R1b представляет собой OR2,
R1c представляет собой H;
R1d представляет собой OH;
R1e представляет собой OR2;
R1f представляет собой NO2, NHR3, NHC=NH(NH2);
R1g представляет собой H;
R2 представляет собой метил, этил;
R3 представляет собой H, (CO)H, (CO)Me, SO2Me, CH2COOR2, COCH2NH2, CO(CH2)nCOOH (где n=0, 1 или 2); и
в формуле II:
R2b представляет собой OH или OR2;
R2c представляет собой H,
R2d представляет собой OH, OSEM;
галогенид представляет собой Br;
R2 представляет собой метил или этил; и
в формуле III:
R3e представляет собой OH, OR2;
R3f представляет собой NO2, NHR3, NHC=NH(NH2);
R3g представляет собой H;
R2 представляет собой метил, этил, и
R3 представляет собой H, (CO)H, COMe, SO2Me или CH2COOR2, COCH2NH2 или CO(CH2)nCOOH, где n представляет собой 0, 1 или 2; и
в формуле IV:
R4b представляет собой OR2;
R4c представляет собой H;
R4d представляет собой OH;
R4e представляет собой OR2;
R4f представляет собой NO2, NHR3, NHC=NH(NH2);
R4g представляет собой H;
R2 представляет собой метил, этил;
R3 представляет собой H, (CO)H, COMe, SO2Me или CH2COOR2, COCH2NH2, CO(CH2)nCOOH и
галогенид представляет собой Br;
где соединение формулы IV подвергают реакции сочетания с пинаколовым эфиром R1a-бороновой кислоты, выбранным из группы, состоящей из пинаколового эфира 3-метилбут-2-енилбороновой кислоты, пинаколового эфира кротилбороновой кислоты, пинаколового эфира аллилбороновой кислоты и пинаколового эфира бензилбороновой кислоты, катализируемой с помощью палладиевого катализатора в присутствии основания, с получением соединения формулы (I).
В одном варианте осуществления описанного выше способа, R1f в формуле (I) представляет собой NO2. Соединение формулы (I) может быть подвергнуто последующей реакции с восстановителем для превращения группы NO2 в группу NH2. Это позволяет получать аминные соединения, такие как соединения 23 и 24. В одном варианте осуществления, соединение формулы (I), где R1f представляет собой NH2, подвергают последующей реакции с метансульфонилхлоридом в присутствии основания, такого как триэтиламин, с получением соединения формулы (I), где R1f представляет собой NHSO2Me. Это позволяет получать аминные соединения, такие как соединения 25 and 24. В еще одном варианте осуществления, соединение формулы (I), где R1f представляет собой NH2, подвергают последующей реакции с этилформиатом с получением соединения формулы (I), где R1f представляет собой NH(CO)H. Это позволяет получать аминные соединения, такие как соединения 27 and 28. Предварительные результаты показали, что соединения 27 и 28 являются очень перспективными для терапевтического применения при лечении атопического дерматита и псориаза.
В одном варианте осуществления, предлагается способ синтеза соединения формулы (I):
где
в формуле (I):
R1a представляет собой кротил, пренил;
R1b представляет собой OR2,
R1c представляет собой H;
R1d представляет собой OH;
R4e и R4f образуют пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу, когда R4e представляет собой OH или NH2, и R4f представляет собой NH2, так что азот в R4f соединен мостиковой связью через карбонильную группу с кислородом или азотом в R4e;
R1g представляет собой H;
R2 представляет собой метил, этил;
R3 представляет собой H, (CO)H, (CO)Me, SO2Me, CH2COOR2, COCH2NH2 или CO(CH2)nCOOH (где n=0, 1 или 2);
в формуле II:
R2b представляет собой OR2;
R2c представляет собой H,
R2d представляет собой OSEM;
Hal представляет собой Br;
R2 представляет собой метил или этил, и
в формуле III:
R3e и R3f образуют пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу, когда R3e представляет собой OH или NH2, и R3f представляет собой NH2, так что азот в R3f соединен мостиковой связью через карбонильную группу с кислородом или азотом в R3e;
R3g представляет собой H; и
в формуле IV:
R4b представляет собой OR2;
R4c представляет собой H;
R4d представляет собой OSEM;
R4e и R4f образуют пяти- или шестичленное гетероциклическое кольцо, содержащее карбонильную группу, когда R4e представляет собой OH или NH2, и R4f представляет собой NH2, так что азот в R4f соединен мостиковой связью через карбонильную группу с кислородом или азотом в R4e;
R4g представляет собой H;
R2 представляет собой метил, этил,
и
галогенид представляет собой Br;
где соединение формулы (IV) подвергают реакции сочетания с пинаколовым эфиром R1a-бороновой кислоты, выбранным из группы, состоящей из пинаколового эфира 3-метилбут-2-енилбороновой кислоты, пинаколового эфира кротилбороновой кислоты, пинаколового эфира аллилбороновой кислоты и пинаколового эфира бензилбороновой кислоты, катализируемой с помощью палладиевого катализатора в присутствии основания, с получением соединения формулы (I).
В одном конкретном варианте осуществления описанного выше способа, соединение формулы (IV) подвергают реакции сочетания с пинаколовым эфиром 3-метилбут-2-енилбороновой кислоты, катализируемой с помощью тетракис(трифенилфосфин)палладия(0) в присутствии карбоната калия. В одном варианте осуществления, соединение формулы (I) выбирают из соединений 51 и 52.
В одном варианте осуществления способа синтеза соединения формулы (I), соединение формулы (I) выбирают из группы, состоящей из:
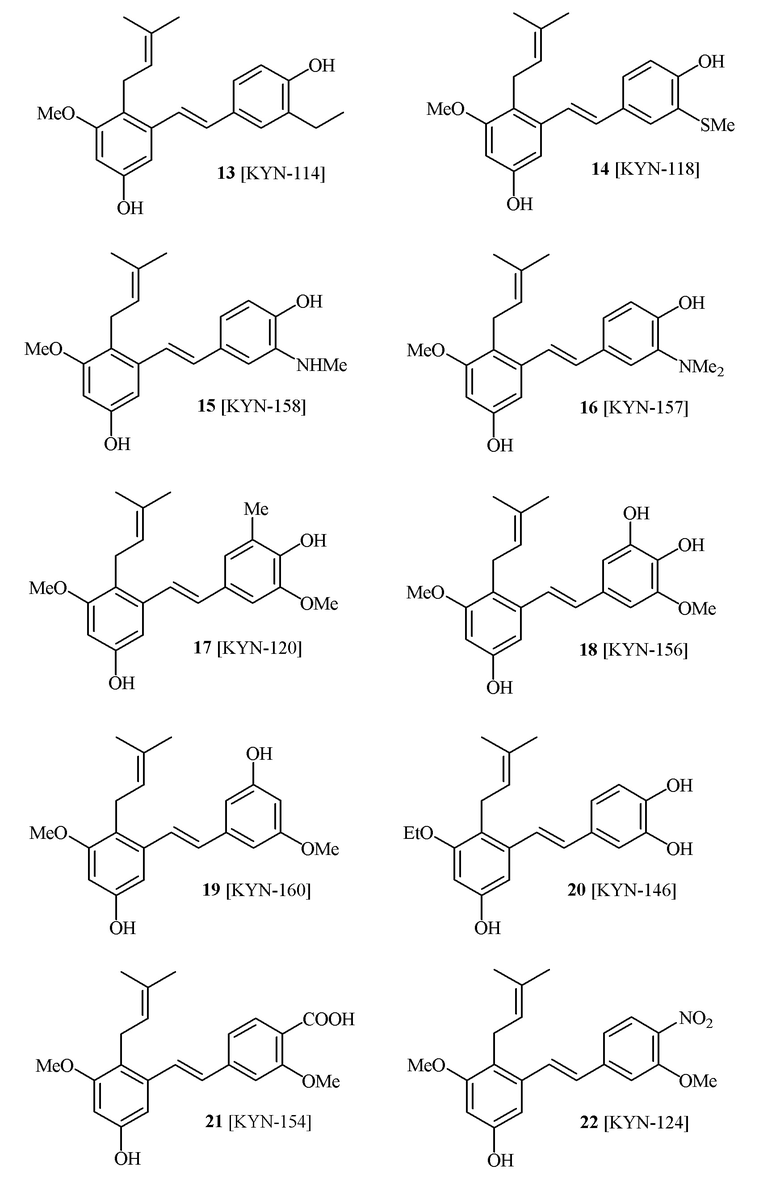
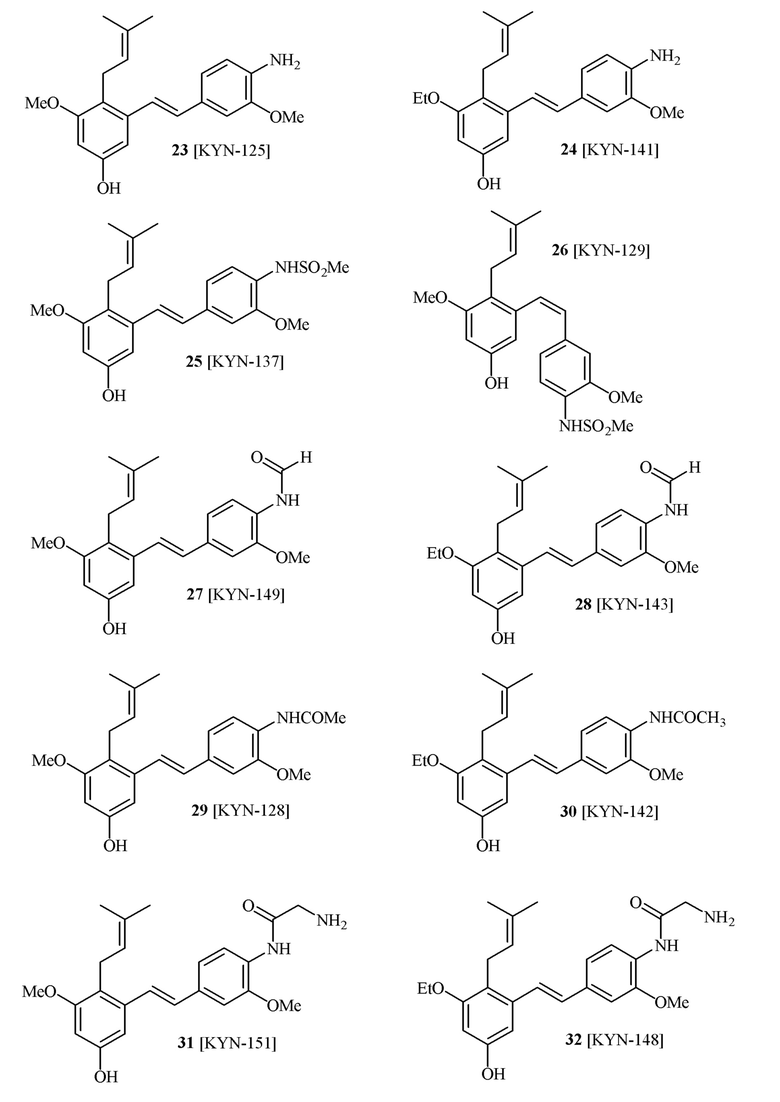
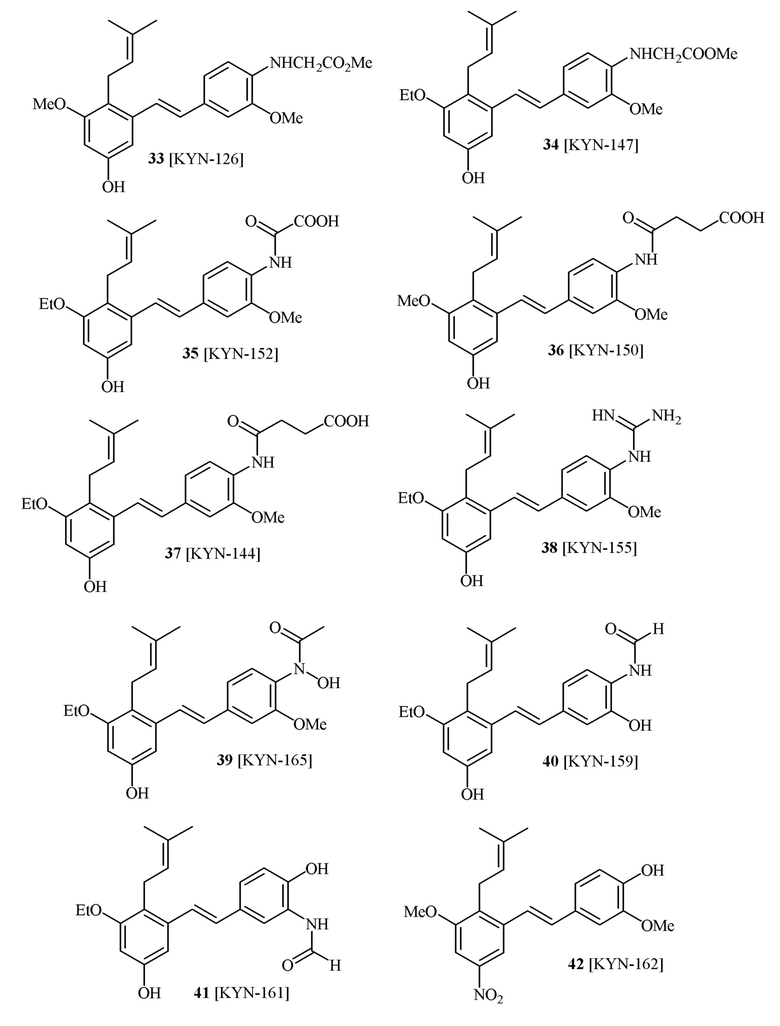
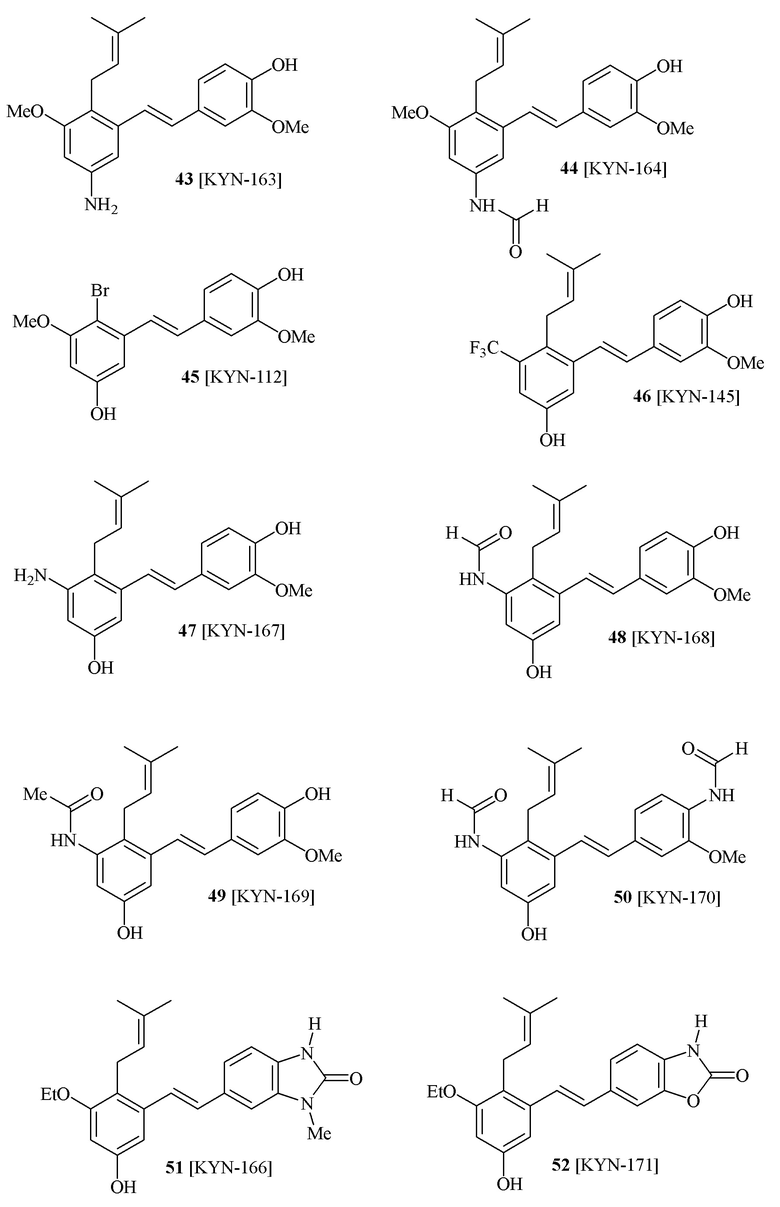
В одном варианте осуществления способа синтеза соединения формулы (I), соединение формулы (I) выбирают из группы, состоящей из:
соединений 22, 23, 24, 25, 26 27, 28 , 29, 30, 31, 36, 24, 33, 34, 35, 36, 37, 38, 39, 40.
Соединение формулы (IV) или (I) может быть необязательно подвергнуто одной или более последующих реакций, включающих, но этим не ограничивая, реакцию с одним или более заместителями -NH2, NO2 и/или -OH в соединении формулы (IV) или формулы (I). В одном варианте осуществления, эта последующая реакция протекает на заместителе R1f соединения формулы (I). В одном варианте осуществления, один или более заместителей -OH в соединении формулы (IV) или формулы (I) могут быть превращены в заместители метоксиоксоацетамидо (-NHC(O)C(O)OCH3) путем проведения реакции соединения, содержащего заместитель OH, с метилхлороксоацетатом в присутствии основания, такого как триэтиламин. В одном варианте осуществления, эту последующую реакции проводят по заместителю R1f в соединении формулы (I).
В одном варианте осуществления, один или более заместителей -NH2 могут быть превращены в заместители метилоксалаты путем проведения реакции соединения, содержащего заместитель NH2, с метилхлороксоацетатом в присутствии основания, такого как триэтиламин. В одном варианте осуществления, эту последующую реакцию проводят по заместителю R1f в соединении формулы (I).
В одном варианте осуществления, один или более заместителей метоксиоксоацетамидо подвергают гидролизу с образованием заместителей -NHC(O)C(O)OH путем проведения реакции соединения, содержащего заместитель N-метоксиоксоацетамидо, с основанием, например, LiOH. В одном варианте осуществления, эту последующую реакцию проводят по заместителю R1f соединения формулы (I).
В одном варианте осуществления, один или более заместителей метилоксалатов могут быть подвергнуты гидролизу с образованием заместителей -OH путем проведения реакции соединения, содержащего заместитель метилоксалат, с основанием, например, LiOH. В одном варианте осуществления, эту последующую реакцию проводят по заместителю R1f соединения формулы (I).
В одном варианте осуществления, один или более заместителей -NH2 могут быть превращены в заместители гуанидин (-NHC(NH)NH2) путем проведения реакции соединения, содержащего заместитель NH2, с цианамидом в присутствии кислоты, такой как п-толуолсульфоновая кислота. В одном варианте осуществления, эту последующую реакцию проводят по заместителю R1f соединения формулы (I).
В одном варианте осуществления, один или более заместителей -NH2 могут быть превращены в заместители формамид (-NHC(O)H) путем проведения реакции соединения, содержащего заместитель NH2, с этилформиатом. В одном варианте осуществления, эту последующую реакцию проводят по заместителю R1f соединения формулы (I).
В одном варианте осуществления, один или более заместителей -NH2 могут быть превращены в заместители ацетамидо (-NHC(O)CH3) путем проведения реакции соединения, содержащего заместитель NH2, с уксусным ангидридом в присутствии основания, такого как DIPEA (диизопропилэтиламин). В одном варианте осуществления, эту последующую реакцию проводят по заместителю R1f соединения формулы (I).
В одном варианте осуществления, один или более заместителей -NH2 могут быть превращены в заместители амидные производные (-NHC(O)CH2CH2C(O)OH) путем проведения реакции соединения, содержащего заместитель NH2, с янтарным ангидридом в присутствии основания, такого как DIPEA (диизопропилэтиламин). В одном варианте осуществления, эту последующую реакцию проводят по заместителю R1f соединения формулы (I).
В одном варианте осуществления, один или более заместителей -NH2 могут быть превращены в заместители метиламины (-NHMe) путем проведения реакции соединения, содержащего заместитель NH2, с формальдегидом в присутствии триацетоксиборгидрида. В одном варианте осуществления, эту последующую реакцию проводят по заместителю R1f соединения формулы (I).
В одном варианте осуществления, один или более заместителей -OH могут быть превращены в заместители ацетаты (-OC(O)CH3) путем проведения реакции соединения, содержащего заместитель -OH, с уксусным ангидридом в присутствии основания, такого как DIPEA (диизопропилэтиламин). В одном варианте осуществления, эту последующую реакцию проводят по заместителю R1f соединения формулы (I).
В одном варианте осуществления, один или более заместителей ацетаты могут быть подвергнуты гидролизу с образованием заместителей -OH путем проведения реакции соединения, содержащего заместитель ацетат, с основанием, например, LiOH. В одном варианте осуществления, эту последующую реакцию проводят по заместителю R1f соединения формулы (I).
В одном варианте осуществления, один или более заместителей -NO2 могут быть восстановлены до заместителей аминов путем проведения реакции, например, соединения, содержащего заместитель NO2, с Zn в присутствии хлорида аммония. В одном варианте осуществления, эту последующую реакцию проводят по заместителю R1f соединения формулы (I).
Модуль A
В одном варианте осуществления описанного в изобретении способа, соединение формулы (II) представляет собой
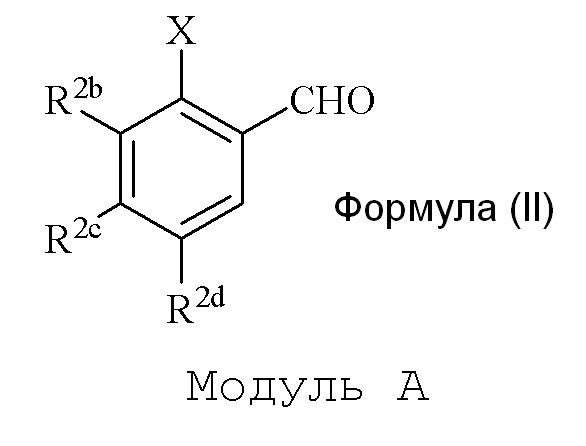
где
R2b=OMe
R2d=OH или OSEM
R2c=H
Hal представляет собой Br
В одном варианте осуществления описанного в изобретении способа, соединение формулы (II) может быть получено путем проведения стадий, включающих i) бромирование соединения моно- или дигидроксилзамещенного толуола; ii) защиту одной гидроксильной группы и/или алкилирование другой гидроксильной группы; iii) удаление защитной группы; iv) ацилирование гидроксильной группы; v) дибромирование метила в ароматическом соединении; vi) гидролиз дибромметила с образованием альдегида; и vii) защиту гидроксильной группы. В одном варианте осуществления, предлагается на стадии i) бромирование соединения дигидроксилзамещенного толуола. В одном варианте осуществления описанного в изобретении способа, соединение формулы (II) может быть получено путем проведения стадий, включающих i) бромирование соединения дигидроксилзамещенного толуола в положении, находящимся рядом с C-связанным заместителем, с помощью бромирующего реагента; ii) защиту гидроксильной группы с помощью O-защитной группы с последующим алкилированием с помощью галоген-C1-C3 алкила; iii) удаление O-защитной группы в присутствии основания; iv) ацилирование гидроксильной группы с использованием уксусного ангидрида в присутствии основания; v) дибромирование метильной группы ароматического соединения с использованием бромирующего реагента с образованием дибромметильной группы; vi) гидролиз дибромметильной группы с образованием альдегида; и vii) защиту гидроксильной группы путем проведения реакции с подходящим соединением, способным образовывать O-защитную группу, включающим но этим не ограничивая, COMe, t-BuSi(CH3)2, 2-(триметилсилил)этоксиметил (SEM), CH(OEt)CH3, тетрагидропиранил или C(OEt)(CH3). В одном варианте осуществления, предлагается на стадии vii) защиту гидроксильной группы путем проведения реакции с 2-(триметилсилил)этоксиметила (SEM) хлоридом с образованием защитной группы 2-(триметилсилил)этоксиметила (SEM) ацеталя. В одном конкретном варианте осуществления, соединение формулы (II) представляет собой модуль A и его получают следующим способом:
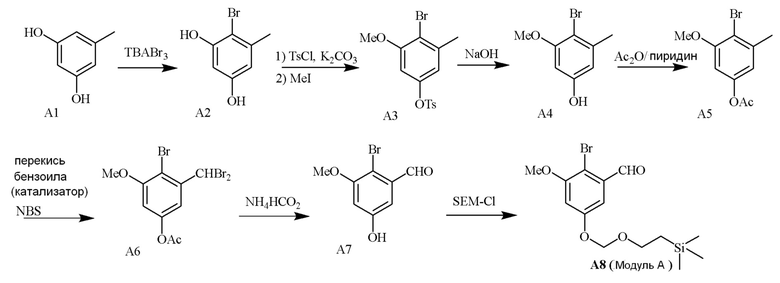
Модуль B
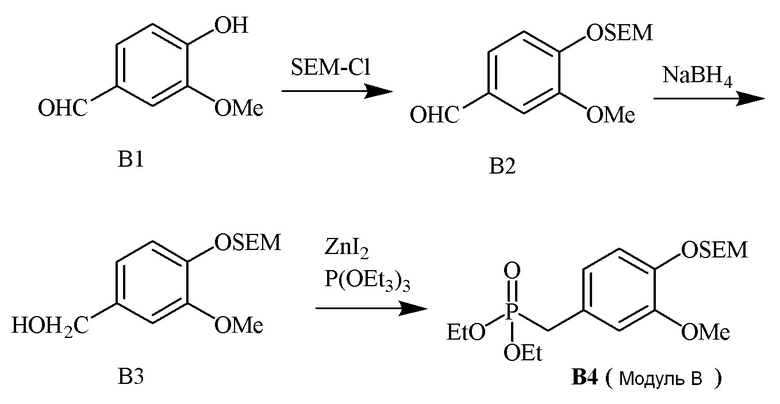
В одном варианте осуществления описанного в изобретении способа, соединение формулы (III) может быть получено исходя из соединения ди- или тризамещенного гидроксил- и/или алкоксил- бензальдегида путем проведения стадий, включающих i) защиту гидроксильной группы; ii) восстановление ароматического альдегида в бензиловый спирт; iii) сочетание бензилового спирта с триалкилфосфонатом и йодидом цинка(II) с образованием диалкилбензилфосфоната.
В одном варианте осуществления, соединение формулы (III) представляет собой модуль B:
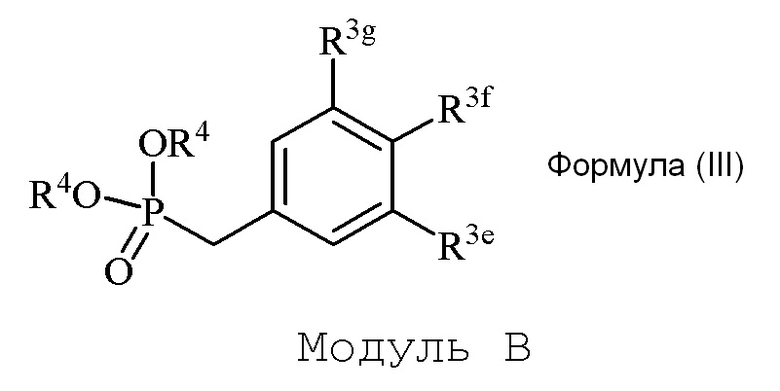
где
R3g=H
R3f=OSEM
R3e=OMe
R4=Et
В одном варианте осуществления описанного в изобретении способа, соединение формулы (III) представляет собой модуль B и его получают следующим способом:
Модуль C
В одном варианте осуществления, соединение формулы (IV) представляет собой модуль C:
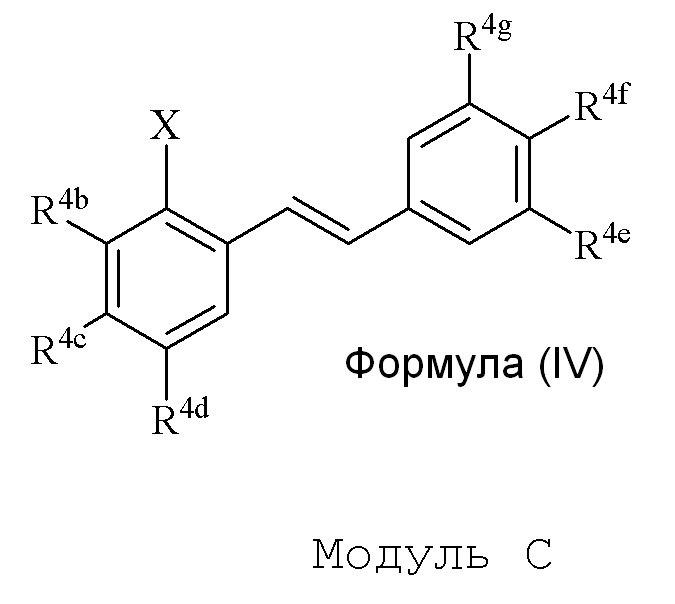
где
R4b=OMe
R4c=H
R4d=OSEM, OH
R4g=H
R4e=OMe
R4f=OSEM
Hal=Br
В одном варианте осуществления описанного в изобретении способа, соединение формулы (IV) получают следующим способом:
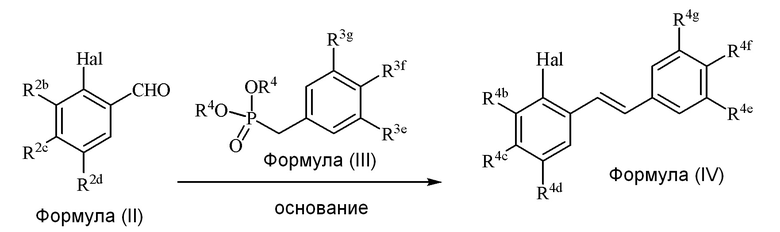
где основание может представлять собой, но этим не ограничивая, NaH, и где формулы (II), (III) и (IV) определены в изобретении.
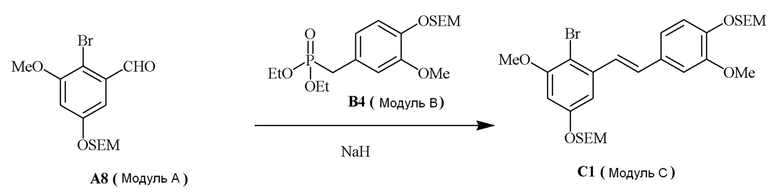
В одном варианте осуществления описанного в изобретении способа, соединение формулы (II) представляет собой модуль A, соединение формулы (III) представляет собой модуль B, и соединение формулы (IV) представляет собой модуль C в соответствии со следующей схемой реакции:
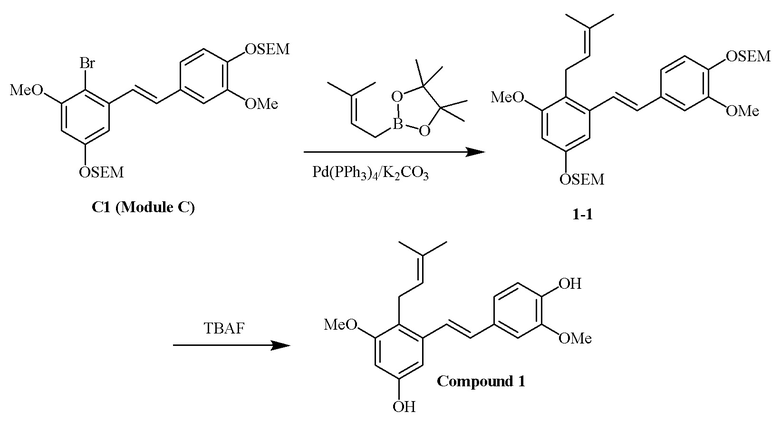
Перекрестное алкилирование арилгалогенида, модуля C, может быть достигнуто, например, с использованием алкилбороновой кислота. В качестве примера описанного в изобретении способа предлагается следующий вариант осуществления:
i) Модуль C подвергают реакции сочетания с пинаколовым эфиром 3-метилбут-2-енилбороновой кислоты в присутствии тетракис-(трифенилфосфин)палладия(0) и карбонат калия с получением соединения 1-1;
ii) соединение 1-1 подвергают реакции с тетрабутиламмония фторидом с получением соединения 1,
в соответствии со следующей схемой реакции:
Соединения и композиции
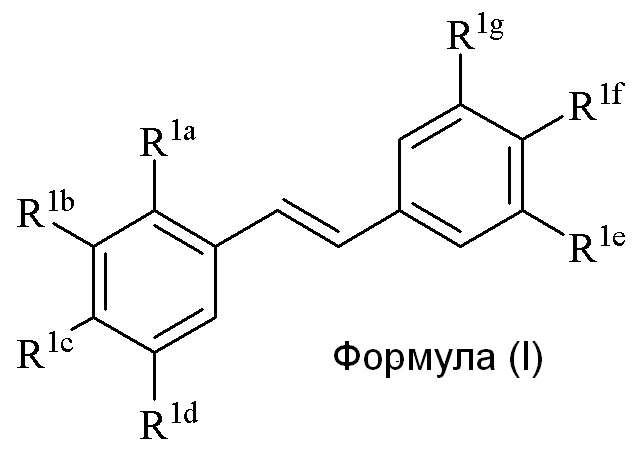
В изобретении описано соединение формулы (I)
 ,
,
полученное описанным в изобретении способом, где
R1a представляет собой Hal, аллил, кротил, пренил, геранил, фарнезил, бензил, 2-алкенил, 2-алкинил;
R1b представляет собой CF3, OH или OR2, NO2, NHEt, NMe2, NMeEt, NHR3, NMeR3, NHC=NH(NH2) или COOR2;
R1c представляет собой H, алкил, алкенил, алкинил, алкандиенил, пренил, геранил, фарнезил или бензил;
R1d представляет собой CF3, OH или OR2, NHR3, NO2, NMeR3, NHC=NH(NH2) или COOR2;
R1e представляет собой H, C1-C6 алкил, OH, OR2, NO2, NMe2, NHR3 или NMeR3, SR2;
R1f представляет собой H, OH, OR2, NO2, NHR3, NHC=NH(NH2), COOR2, COOH;
R1g представляет собой H, алкил, CF3, OH или OR2;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил, COCOOR4 или O-защитную группу, когда присоединен к O, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этокси]метила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой H, CH3, OH, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, CO(CH2)nNH2, CONH2, CO(CH2)nCOOH или COCOOR4 (где n=0, 1 или 2);
R4 представляет собой C1-C6 алкил; и
Hal представляет собой галогенид, выбранный из группы, состоящей из F, Cl, Br, I и At;
только один из R1e, R1f или R1g может представлять собой H; и
когда R1e представляет собой OH, NH2 или NHMe, и R1f представляет собой NH2, азот в R1f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R1e с образованием пяти- или шестичленного гетероциклического кольца.
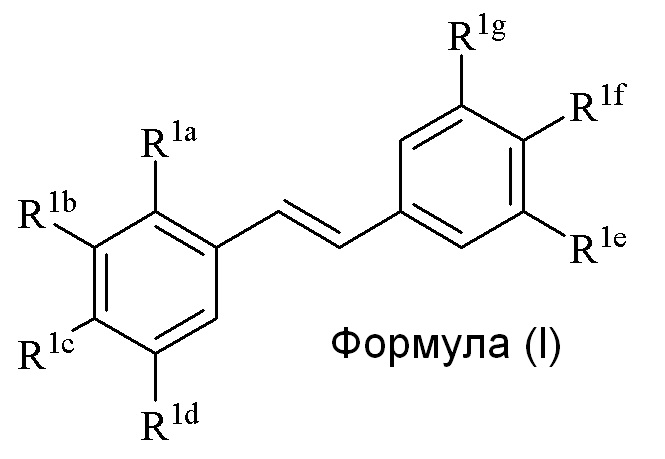
Кроме того, в изобретении описано соединение формулы (I):
где
R1a представляет собой Hal, аллил, кротил, пренил, геранил, фарнезил, бензил, 2-алкенил, 2-алкинил;
R1b представляет собой CF3, OH или OR2, NO2, NHEt, NMe2, NMeEt, NHR3, NMeR3, NHC=NH(NH2) или COOR2;
R1c представляет собой H, алкил, алкенил, алкинил, алкандиенил, пренил, геранил, фарнезил или бензил;
R1d представляет собой CF3, OH или OR2, NO2, NHR3, NMeR3, NHC=NH(NH2) или COOR2;
R1e представляет собой H, C1-C6 алкил, OH, OR2, NO2, NMe2, NHR3 или NMeR3, SR2;
R1f представляет собой H, OH, OR2, NO2, NHR3, NHC=NH(NH2), COOR2, COOH;
R1g представляет собой H, алкил, CF3, OH или OR2;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил, COCOOR4 или O-защитную группу, которая присоединена к O, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этокси]метила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой H, CH3, OH, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, CO(CH2)nNH2, CONH2, CO(CH2)nCOOH или COCOOR4 (где n=0, 1 или 2);
R4 представляет собой C1-C6 алкил; и
Hal представляет собой галогенид, выбранный из группы, состоящей из F, Cl, Br, I и At;
только один из R1e, R1f или R1g может представлять собой H,
когда R1e представляет собой OH, NH2 или NHMe и R1f представляет собой NH2, азот в R1f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R1e с образованием пяти- или шестичленного гетероциклического кольца;
при условии, что
когда R1a и/или R1c представляет собой пренил, R1d представляет собой OH, R1f представляет собой OH, R1g представляет собой H, и R1e представляет собой OH, OMe или OEt, то тогда R1b не может представлять собой OH, OR, где R=метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, или бензил.
В одном конкретном варианте осуществления, предлагается соединение формулы (I) где
R1a представляет собой аллил, пренил, бензил, 2-алкенил;
R1b представляет собой CF3, OH или OR2;
R1c представляет собой H;
R1d представляет собой OH, NH2, N(CO)H;
R1e представляет собой OH, OR2, NO2, NHR3 или NMeR3, (CO)H, SMe;
R1f представляет собой H, OH, OR2, NO2, NHR3, NHC=NH(NH2), COOH;
R1g представляет собой H, алкил, OH;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил;
R3 представляет собой H, CH3, OH, (CO)H, (CO)Me, SO2Me, CH2COOR2, COCH2NH2, CO(CH2)nCOOH (где n=0, 1 или 2).
при условии, что
когда R1a или R1c представляет собой пренил, R1d представляет собой OH, R1f представляет собой OH, R1g представляет собой H, и R1e представляет собой OH, OMe или OEt, то тогда R1b не может представлять собой OR, где R=метил, этил, изопропил, пропил, бутил, изобутил, трет-бутил или бензил.
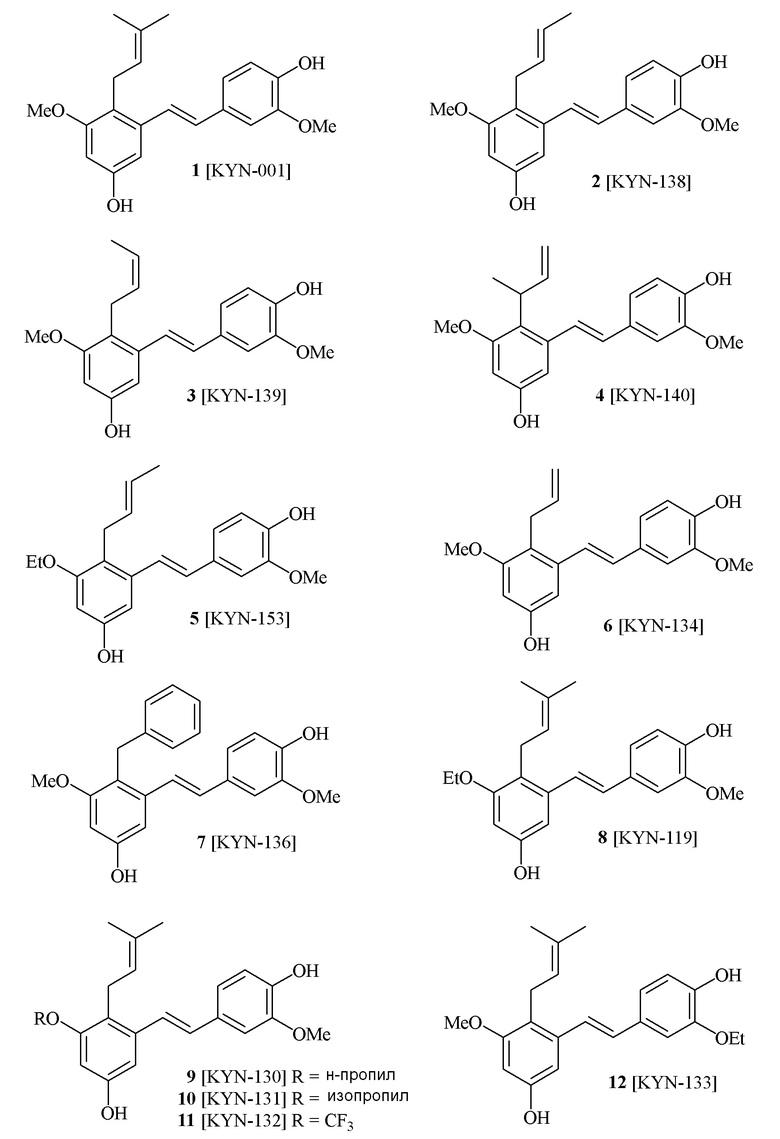
В одном варианте осуществления, соединение формулы (I) представляет собой соединение, выбранное из группы, состоящей из описанных в изобретении соединений 2-7, 11, 13 to 19, 21-44, 46-49, 50, 51, 52 [KYN 138, 139, 140, 153, 134, 136, 132, 114, 118, 158, 157, 120, 156, 160, 154, 124, 125, 141, 137, 129, 149, 143, 128, 142, 151, 148, 126, 147, 152, 150, 144, 155, 165, 159, 161, 162, 163, 164, 145, 167, 168, 169, 166, 171, KYN-170].
Кроме того, в изобретении описаны варианты осуществления, где один из R1b и R1d формулы (I) представляет собой заместитель, не являющийся OH или OR2. В одном варианте осуществления, по меньшей мере, один из R1b или R1d представляет собой CF3, NO2, NHR3, NMeR3, NHC=NH(NH2) или COOR. Соответственно, предлагается также соединение формулы (I):
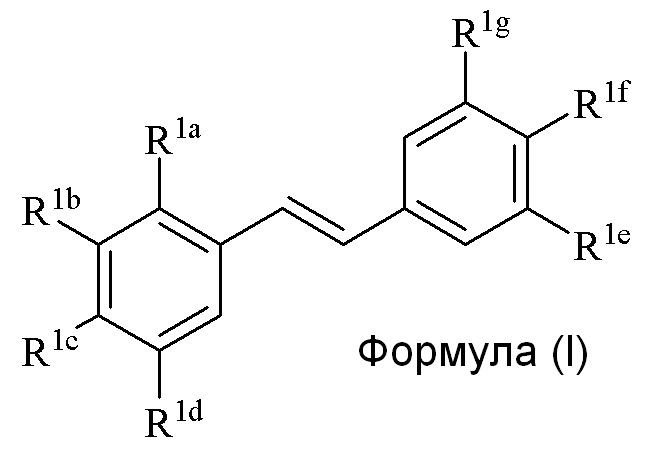
где
R1a представляет собой Hal, аллил, кротил, пренил, геранил, фарнезил, бензил, 2-алкенил, 2-алкинил;
R1b представляет собой CF3, OH или OR2, NO2, NHEt, NMe2, NMeEt, NHR3, NMeR3, NHC=NH(NH2) или COOR2;
R1c представляет собой H, алкил, алкенил, алкинил, алкандиенил, пренил, геранил, фарнезил или бензил;
R1d представляет собой CF3, OH или OR2, NO2, NHR3, NMeR3, NHC=NH(NH2) или COOR2;
R1e представляет собой H, C1-C6 алкил, OH, OR2, NO2, NMe2, NHR3 или NMeR3, SR2;
R1f представляет собой H, OH, OR2, NO2, NHR3, NHC=NH(NH2), COOR2, COOH;
R1g представляет собой H, алкил, CF3, OH или OR2;
где по меньшей мере один из R1b или R1d представляет собой CF3, NO2, NHR3, NMeR3, NHC=NH(NH2) или COOR2;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил, COCOOR4 или O-защитную группу, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этокси]метила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой H, CH3, OH, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, CO(CH2)nNH2, CONH2, CO(CH2)nCOOH или COCOOR4 (где n=0, 1 или 2);
R4 представляет собой C1-C6 алкил; и
Hal представляет собой галогенид, выбранный из группы, состоящей из F, Cl, Br, I и At;
только один из R1e, R1f или R1g может представлять собой H,
когда R1e представляет собой OH, NH2 или NHMe, и R1f представляет собой NH2, азот в R1f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R1e с образованием пяти- или шестичленного гетероциклического кольца.
В одном варианте осуществления, соединение формулы (I) представляет собой соединение, выбранное из описанных в изобретении соединений 11, 43, 44, 46, 47, 48, 49, 50 [KYN-132, 163, 164, 145, 167, 168, 169 [KYN-170].
Кроме того, в изобретении описаны варианты осуществления где заместитель R1f в кольце B не представляет собой H, OH или OR2, и R1e не представляет собой H. В одном варианте осуществления, предлагается соединение формулы (I)
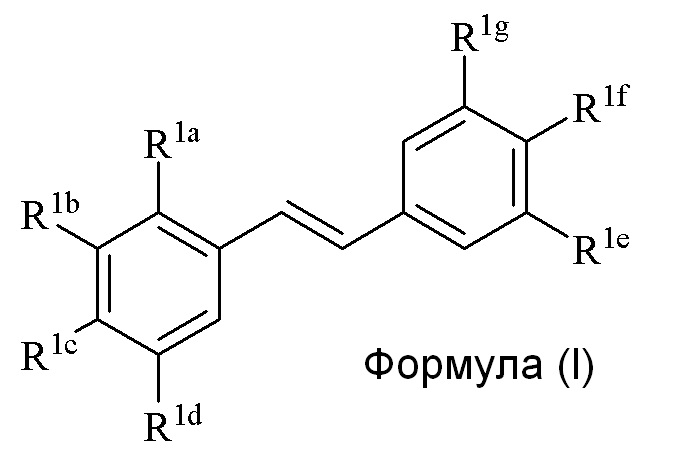
где
R1a представляет собой Hal, аллил, кротил, пренил, геранил, фарнезил, бензил, 2-алкенил, 2-алкинил;
R1b представляет собой CF3, OH или OR2, NO2, NHMe, NHEt, NMe2, NMeEt, NHR3, NMeR3, NHC=NH(NH2) или COOR2, NHR3, NMeR3, NHC=NH(NH2) или COOR2;
R1c представляет собой H, алкил, алкенил, алкинил, алкандиенил, пренил, геранил, фарнезил или бензил;
R1d представляет собой CF3, OH или OR2, NHR3, NO2, NH2, NHMe, NHC=NH(NH2) или COOR2;
R1e представляет собой OH, OR2;
R1f представляет собой NO2, NHR3, NHC=NH(NH2), COOR2, COOH, NHSO2Me, NHC(O)(CH2)COOH, NHC(O)COOH, NHC(O)(CH2)NNH2, NHC(O)CH2NH2, NHCH2COOMe;NHC(O)H, NHC(O)CH3, NHC(O)(CH2)2COOH, NHC(NH)NH2;
R1g представляет собой H;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этокси]метила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой H, CH3, OH, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, COCH2NH2, CONH2, CO(CH2)nCOOH, CO(CH2)nCOOR4 (где n=0, 1 или 2);
R4 представляет собой C1-C6 алкил;
Hal представляет собой галогенид, выбранный из группы, состоящей из F, Cl, Br, I и At;
только один из R1e, R1f или R1g может представлять собой H, и
когда R1e представляет собой OH, NH2 или NHMe, и R1f представляет собой NH2, азот в R1f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R1e с образованием пяти- или шестичленного гетероциклического кольца.
В одном варианте осуществления, соединение формулы (I) представляет собой соединение, выбранное из описанных в изобретении соединений 21-40, 50, 51, 52 [KYN-154, 124, 125, 141, 137, 129, 149, 143, 128, 142, 151, 148, 126, 147, 152, 150, 144, 155, 165, 159, 166, 170, 171].
Кроме того, в изобретении описаны варианты осуществления, где заместитель R1e в кольце B не представляет собой H, OH или OR2, и R1fe не представляет собой H. В одном варианте осуществления, предлагается соединение формулы (I):
где
R1a представляет собой Hal, аллил, кротил, пренил, геранил, фарнезил, бензил, 2-алкенил, 2-алкинил;
R1b представляет собой CF3, OH или OR2, NO2, NHMe, NHEt, NMe2, NMeEt, NHR3, NMeR3, NHC=NH(NH2) или COOR2, NHR3, NMeR3, NHC=NH(NH2) или COOR2;
R1c представляет собой H, алкил, алкенил, алкинил, алкандиенил, пренил, геранил, фарнезил или бензил;
R1d представляет собой CF3, OH или OR2, NHR3, NO2, NH2, NHMe, NHC=NH(NH2) или COOR2;
R1e представляет собой C1-C6 алкил, NO2, NH2, NHMe, NMe2, NHR3 или NMeR3, SR2;
R1f представляет собой OH, OR2;
R1g представляет собой H;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этокси]метила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой H, CH3, OH, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, COCH2NH2, CONH2, CO(CH2)nCOOH (где n=0, 1 или 2); и
Hal представляет собой галогенид, выбранный из группы, состоящей из F, Cl, Br, I и At;
только один из R1e, R1f или R1g может представлять собой H,
когда R1e представляет собой OH, NH2 или NHMe, и R1f представляет собой NH2, азот в R1f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R1e с образованием пяти- или шестичленного гетероциклического кольца.
В одном варианте осуществления, соединение формулы (I) представляет собой соединение, выбранное из описанных в изобретении соединений 13-16, 41 [KYN-114, 118, 158, 157, 161].
Кроме того, в изобретении описаны варианты осуществления, где отсутствует пренильная группа на кольце A или кольце B. В одном варианте осуществления, предлагается соединение формулы (I):
где
R1a представляет собой Hal, аллил, кротил, геранил, фарнезил, бензил, 2-алкенил, 2-алкинил;
R1b представляет собой CF3, OH или OR2, NO2, NHMe, NHEt, NMe2, NMeEt, NHR3, NMeR3, NHC=NH(NH2) или COOR2;
R1c представляет собой H, алкил, алкенил, алкинил, алкандиенил, геранил, фарнезил или бензил;
R1d представляет собой CF3, OH или OR2, NHR3, NO2, NH2, NHMe, NHC=NH(NH2) или COOR2;
R1e представляет собой H, OH, OR2, NO2, NH2, NHMe, NMe2, NHR3 или NMeR3, SR2;
R1f представляет собой H, OH, OR2, NO2, NH2, NHMe, NHR3, NHC=NH(NH2), COOR2, COOH;
R1g представляет собой H, алкил, CF3, OH или OR2;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил, C(O)C(O)OR4 или O-защитную группу, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этокси]метила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой H, CH3, OH, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, COCH2NH2, CONH2, CO(CH2)nCOOH (где n=0, 1 или 2);
R4 представляет собой C1-C6 алкил; и
Hal представляет собой галогенид, выбранный из группы, состоящей из F, Cl, Br, I и At;
только один из R1e, R1f или R1g может представлять собой H, и
когда R1e представляет собой OH, NH2 или NHMe, и R1f представляет собой NH2, азот в R1f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R1e с образованием пяти- или шестичленного гетероциклического кольца.
В конкретном варианте осуществления, предлагается соединение формулы (I), где
R1a представляет собой аллил, кротил, геранил, фарнезил, бензил, 2-алкенил, 2-алкинил;
R1b представляет собой CF3, OH, OR2;
R1c представляет собой H;
R1d представляет собой OH или OR2, NH2, NH(CO)H;
R1e представляет собой OR2, R4, NO2, NH2, NHMe, NMe2, SR2;
R1f представляет собой OH, OR2, NO2, NH2, NHMe, NHR3, NHC=NH(NH2), COOR2, COOH;
R1g представляет собой H;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил, C(O)C(O)OR4 или O-защитную группу, где защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этокси]метила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой H, CH3, OH, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, COCH2NH2, CONH2, CO(CH2)nCOOH (где n=0, 1 или 2);
R4 представляет собой C1-C6 алкил,
и
Hal представляет собой галогенид, выбранный из группы, состоящей из F, Cl, Br, I и At;
только один из R1e, R1f или R1g может представлять собой H,
когда R1e представляет собой OH, NH2 или NHMe, и R1f представляет собой NH2, азот в R1f соединен мостиковой связью через карбонильную группу или (CO)CH2 группу с кислородом или азотом в R1e с образованием пяти- или шестичленного гетероциклического кольца.
В одном варианте осуществления, соединение формулы (I) представляет собой соединение, выбранное из описанных в изобретении соединений 2, 3, 4, 5, 6, 7 [KYN-138, 139, 140, 153, 134, 136].
В еще одном варианте осуществления, предлагается соединение формулы (I), где
R1a представляет собой пренил;
R1b представляет собой OR2;
R1c представляет собой H;
R1d представляет собой OH;
R1e представляет собой OR2;
R1f представляет собой NHR3, NHC=NH(NH2);
R1g представляет собой H;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил; и
R3 представляет собой H, CH3, OH, (CO)H, (CO)Me, SO2Me, CH2COOR2, COCH2NH2, CO(CH2)nCOOH (где n=0, 1 или 2).
В еще одном варианте осуществления, предлагается соединение формулы (I), где
R1a представляет собой пренил;
R1b представляет собой OR2;
R1c представляет собой H;
R1d представляет собой OH:
R1e представляет собой OR2;
R1f представляет собой OH;
R1g представляет собой алкил, OH;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил; и
алкил представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил.
Кроме того, в изобретении описано соединение формулы (I), имеющее, по меньшей мере, одну формамидную группу и/или этоксидную группу на кольце A и/или кольце B. А одном варианте осуществления, кольцо A имеет, по меньшей мере, один этоксидный заместитель. Этоксидный заместитель может находиться в положении R1b или R1d на кольце A. В еще одном варианте осуществления, кольцо A имеет формамидный заместитель. В еще одном варианте осуществления, кольцо B имеет формамидный заместитель. В еще одном варианте осуществления, кольцо A имеет, по меньшей мере, один этоксидный заместитель и кольцо B имеет формамидный заместитель. В конкретном варианте осуществления, соединение формулы (I) могут быть выбраны из соединений 8, 20, 24, 27, 28, 30, 32, 34, 35, 37 to 41, 50, 51 [KYN-119, 146, 141, 149, 143, 142, 148, 147, 152, 144, 155, 165, 159, 161, 170]
Способы лечения
Предлагается способ лечения рака, включающий введение терапевтически эффективного количества описанного в изобретении соединения формулы (I) или фармацевтически приемлемой соли, сольвата, или фармацевтической композиции, включающей указанные соединения, пациенту, нуждающемуся в этом.
Предварительные результаты
Обсуждение
В патентном документе WO2012/149608 описана противораковая активность конкретных производных пренилированного полигидроксистильбена. В частности, было обнаружено, что описанное в настоящем изобретении соединение 1 [KYN-001] обладает высокой ингибирующей активностью в отношении раковых клеток. Активность соединений по настоящему изобретению сравнивали с активностью соединения 1.
Результаты предварительных исследований показывают, что соединения, в которых R1b группу OEt и/или, по меньшей мере, одну фенольную группу OH заменяют на формамидную группу, являются очень эффективными ингибиторами рака. Как видно в таблице 6, в которой представлены величины IC50 в случае 42 линий раковых клеток (средняя активность), ряд активности представлен как 28 > 27 > 8 > 20 > 1. Соединение 28 с формамидом и OEt характеризовалось самой высокой активностью, затем следовало соединение 27 с R1b=OMe и формамидом.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фигуре 1 приведена хроматограмма LM-6-44 (смеси соединений 2, 3 и 4) перед разделением.
На фигуре 2 приведена хроматограмма LM-6-44-P1 (соединения 4) после разделения.
На фигуре 3 приведена хроматограмма LM-6-44-P2 (соединения 2) после разделения.
На фигуре 4 приведена хроматограмма LM-6-44-P3 (соединения 3) после разделения.
На фигуре 5 приведена хроматограмма LM-6-85 (смеси соединения 5, E-5-4 и E-5-5) перед разделением.
На фигуре 6 приведена хроматограмма LM-6-85-P2 (соединения 5) после разделения.
На фигуре 7 приведена хроматограмма LM-6-85-P1 (соединения 5-5) после разделения.
На фигуре 8 приведена хроматограмма LM-6-85-P3 (соединения 5-4) после разделения.
На фигуре 9 представлена в графическом виде зависимость % воздействия на ингибирование высвобождения CCL2 и подавление жизнеспособности клеток от изменяющихся концентраций соединения 28.
На фигуре 10 представлена в графическом виде зависимость % воздействия на ингибирование высвобождения IL-8 и подавление жизнеспособности клеток от изменяющихся концентраций соединения 28.
На фигуре 11 представлена в графическом виде зависимость % воздействия на ингибирование высвобождения IL-17A и подавление жизнеспособности клеток от изменяющихся концентраций соединения 28.
На фигуре 12 представлена в графическом виде зависимость % воздействия на ингибирование высвобождения IL-2 и IL-4 и подавление жизнеспособности клеток от изменяющихся концентраций соединения 28.
На фигуре 13 приведены кривые концентрация-эффект для соединений 1, 8, 20, 27, 28, 31 и 35.
На фигуре 13,1 в графическом виде представлены данные по воздействию соединения 1 на: A. высвобождение CCL2 при воспалении, индуцированном IL-4, IL-13, IL-22 и IFN-γ, в кератиноцитах человека; B. высвобождение IL-8 при воспалении, индуцированном IL-17 и TNF-α, в кератиноцитах человека; C. высвобождение IL-17A при воспалении, индуцированном CD3, CD28 и IL-23, в мононуклеарных клетках периферической крови (PBMC) человека; D. высвобождение IL-4 и IL-2 при воспалении, индуцированном CD2, CD3 и CD28, в T-клетках. Параллельно в каждом исследовании оценивали воздействия соединения 1 на жизнеспособность клеток.
На фигуре 13,2 в графическом виде представлены данные по воздействию соединения 8 на: A. высвобождение CCL2 при воспалении, индуцированном IL-4, IL-13, IL-22 и IFN-γ, в кератиноцитах человека; B. высвобождение IL-8 при воспалении, индуцированном IL-17 и TNF-α, в кератиноцитах человека; C. высвобождение IL-17A при воспалении, индуцированном CD3, CD28 и IL-23, в мононуклеарных клетках периферической крови (PBMC) человека; D. высвобождение IL-4 и IL-2 при воспалении, индуцированном CD2, CD3 и CD28, в T-клетках. Параллельно в каждом исследовании оценивали воздействия соединения 8 на жизнеспособность клеток.
На фигуре 13,3 в графическом виде представлены данные по воздействию соединения 20 на: A. высвобождение CCL2 при воспалении, индуцированном IL-4, IL-13, IL-22 и IFN-γ, в кератиноцитах человека; B. высвобождение IL-8 при воспалении, индуцированном IL-17 и TNF-α, в кератиноцитах человека; C. высвобождение IL-17A при воспалении, индуцированном CD3, CD28 и IL-23, в мононуклеарных клетках периферической крови (PBMC) человека; D. высвобождение IL-4 и IL-2 при воспалении, индуцированном CD2, CD3 и CD28, в T-клетках. Параллельно в каждом исследовании оценивали воздействия соединения 20 на жизнеспособность клеток.
На фигуре 13,4 в графическом виде представлены данные по воздействию соединения 27 на: A. высвобождение CCL2 при воспалении, индуцированном IL-4, IL-13, IL-22 и IFN-γ, в кератиноцитах человека; B. высвобождение IL-8 при воспалении, индуцированном IL-17 и TNF-α, в кератиноцитах человека; C. высвобождение IL-17A при воспалении, индуцированном CD3, CD28 и IL-23, в мононуклеарных клетках периферической крови (PBMC) человека; D. высвобождение IL-4 и IL-2 при воспалении, индуцированном CD2, CD3 и CD28, в T-клетках. Параллельно в каждом исследовании оценивали воздействия соединения 27 на жизнеспособность клеток.
На фигуре 13,5 в графическом виде представлены данные по воздействию соединения 28 на: A. высвобождение CCL2 при воспалении, индуцированном IL-4, IL-13, IL-22 и IFN-γ, в кератиноцитах человека; B. высвобождение IL-8 при воспалении, индуцированном IL-17 и TNF-α, в кератиноцитах человека; C. высвобождение IL-17A при воспалении, индуцированном CD3, CD28 и IL-23, в мононуклеарных клетках периферической крови (PBMC) человека; D. высвобождение IL-4 и IL-2 при воспалении, индуцированном CD2, CD3 и CD28, в T-клетках. Параллельно в каждом исследовании оценивали воздействия соединения 28 на жизнеспособность клеток.
На фигуре 13,6 в графическом виде представлены данные по воздействию соединения 31 на: A. высвобождение CCL2 при воспалении, индуцированном IL-4, IL-13, IL-22 и IFN-γ, в кератиноцитах человека; B. высвобождение IL-8 при воспалении, индуцированном IL-17 и TNF-α, в кератиноцитах человека; C. высвобождение IL-17A при воспалении, индуцированном CD3, CD28 и IL-23, в мононуклеарных клетках периферической крови (PBMC) человека; D. высвобождение IL-4 и IL-2 при воспалении, индуцированном CD2, CD3 и CD28, в T-клетках. Параллельно в каждом исследовании оценивали воздействия соединения 31 на жизнеспособность клеток.
На фигуре 13,7 в графическом виде представлены данные по воздействию соединения 35 на: A. высвобождение CCL2 при воспалении, индуцированном IL-4, IL-13, IL-22 и IFN-γ, в кератиноцитах человека; B. высвобождение IL-8 при воспалении, индуцированном IL-17 и TNF-α, в кератиноцитах человека; C. высвобождение IL-17A при воспалении, индуцированном CD3, CD28 и IL-23, в мононуклеарных клетках периферической крови (PBMC) человека; D. высвобождение IL-4 и IL-2 при воспалении, индуцированном CD2, CD3 и CD28, в T-клетках. Параллельно в каждом исследовании оценивали воздействия соединения 35 на жизнеспособность клеток.
На фигуре 14 в графическом виде представлены данные по зависимости от концентрации ингибирующего действия соединения 1 [KYN-001], 20 [KYN-146] и 27 [KYN-149] в отношении полученных у трех пациентов образцов клеток мультиформной глиобластомы (GBM), обозначенных как A. BNA99, B. BNB04 и C. BNB18.
На фигуре 15 представлены данные по концентрациям соединения 27 в образцах различных тканей, взятых у подвергнутых лечению мышей.
На фигуре 16 представлены данные гистохимического анализа образцов опухоли у подвергнутых лечению мышей группы 1 и 2.
На фигуре 17 представлены данные гистохимического анализа образцов у подвергнутых лечению мышей группы 3, 4 и 5.
На фигуре 18 в графическом виде представлены данные по воздействию соединения 27 на меланому B16F10, ксенотрансплантированную мышам линии BALB/c.
Определения
Если специально не указано иное, то все технические и научные термины, используемые в настоящем изобретении, будут иметь такие же значения, которые является общепринятыми для специалиста в данной области (например, в химии, биохимии, медицинской химии, микробиология и в других областях). Несмотря на то, что при практическом применении или испытании настоящего изобретения могут использоваться способы и материалы, аналогичные или эквивалентные тем, что описаны в настоящем изобретении, тем не менее, далее описаны подходящие способы и материалы. В случае возникновения противоречий, описание настоящего изобретения, включая определения, будет иметь преимущественную силу. Кроме того, материалы, методы и примеры носят исключительно иллюстративный характер и не являются ограничениями.
Следует иметь в виду, что используемый в изобретении термин "и/или", например, "X и/или Y", означает либо "X и Y", либо "X или Y", и его следует рассматривать как относящемуся к обоим значениям, так и к любому одному из значений, например, A и/или B включает в себя варианты i) A, ii) B или iii) A и B.
Если не указано иное, то используемый в изобретении термин приблизительно, относится к отклонению +/- 20%, как правило, +/- 10%, как правило +/- 5%, от обозначенной величины.
Используемые в изобретении формы единственного числа включают как форму единственного числа, так и форму множественного числа, если из контекста не очевидно иное.
Следует иметь в виду, что некоторые признаки, которые для ясности описаны в изобретении на примере отдельных вариантов осуществления, могут быть также предоставлены в комбинации в одном варианте осуществления. И наоборот, различные признаки, которые для краткости описаны на примере одного варианта осуществления, могут быть также предоставлены отдельно или в любой субкомбинации.
В настоящем описании изобретения, различные аспекты и компоненты изобретения могут быть представлены в формате диапазона. Формат диапазона используют для удобства и его не следует интерпретировать в качестве жесткого ограничения объема изобретения. Соответственно, следует иметь в виду, что описание диапазона конкретно раскрывает все возможные поддиапазоны, а также отдельные числовые значения в пределах этого диапазона, если не указано иное. Например, описание диапазона, например, от 1 до 5, следует рассматривать как включающее конкретно раскрытые поддиапазоны, такие как от 1 до 3, от 1 до 4, от 1 до 5, от 2 до 4, от 2 до 5, от 3 до 5 и так далее, а также целые и десятичные числа в указанном диапазоне, например, 1, 2, 3, 4, 5, 5,5 и 6, за исключением тех случаев, когда в контексте однозначно определены или подразумеваются целые числа. Это применимо независимо от широты раскрываемого диапазона. Если речь идет о конкретных значениях, то они будут указаны в описании изобретения.
Следует иметь в виду, что, в описании это изобретения, слово "содержать" или его варианты, такие как "содержит" или "содержащий", подразумевает включение указанного элемента, целого числа или стадии, или группы элементов, целых чисел или стадий, но не исключение любого другого элемента, целого числа или стадии, или группы элементов, целых чисел или стадий.
Используемый в изобретении термин "галоген" обозначает фтор, хлор, бром, йод или астат.
Используемый в изобретении термин "углеводород" включает химическое соединение, состоящее из атомов водорода и углерода.
Используемый в изобретении термин "алкил" включает как неразветвленные (то есть, линейные), так и разветвленные углеводородные группы. Примеры алкильных групп включают, но этим не ограничивая, метильную, этильную, н-пропильную, изопропильную, н-бутильную, трет-бутильную, изобутильную, вторбутильную, пентильную и гексильную группы. В одном примере, алкильная группа содержит от одного до шести углеродных атомов (то есть C1-6 алкил).
Используемый в изобретении термин "алкокси" относится к группе -O-алкил, где "алкил" описан выше. Примеры алкоксильных групп включают, но этим не ограничивая, метоксильную, этоксильную, пропоксильную и бутоксильную группы. В одном примере, алкоксильная группа содержит от одного до шести углеродных атомов (то есть -O-C1-6 алкил).
Используемый в изобретении термин "алкенил" относится как к линейным, так и к разветвленным ненасыщенным углеводородным группам, по меньшей мере, с одной двойной связью углерод-углерод. Примеры алкенильных групп включают, но этим не ограничивая, этенильную, пропенильную, бутенильную, пентенильную и гексенильную группы. В одном примере, алкенильная группа содержит от двух до шести углеродных атомов (то есть C2-6 алкенил). Термин "2-алкенил" относится к алкеновому заместителю, который имеет двойную связь в положении 2 от точки его присоединения. Он включает аллильную, кротильную, пренильную, геранильную и фарнезильную группы
Используемый в изобретении термин "алкинил" относится как к линейным, так и к разветвленным ненасыщенным углеводородным группам, по меньшей мере, с одной тройной связью углерод-углерод. Примеры алкинильных групп включают, но этим не ограничивая, этинильную, пропинильную, бутинильную, пентинильную и гексинильную группы. В одном примере, алкинильная группа содержит от двух до шести углеродных атомов (то есть C2-6 алкинил).
Используемый в изобретении термин "галогеналкил" относится к алкильной группе, имеющей, по меньшей мере, один галогенный заместитель, где "алкил" и "галоген" описаны выше. Аналогично, термин "дигалогеналкил" обозначает алкильную группу, имеющую два галогенных заместителя, и термин "тригалогеналкил" обозначает алкильную группу, имеющую три галогенных заместителя. Примеры галогеналкильных групп включают фторметильную, хлорметильную, бромметильную, йодметильную, фторпропильную и фторбутильную группы. Примеры дигалогеналкильных групп включают дифторметильную и дифторэтильную группы. Примеры тригалогеналкильных групп включают трифторметильную и трифторэтильную группы. В одном примере, галогеналкильная группа содержит от одного до шести углеродных атомов (то есть C1-6 галогеналкил).
Используемый в изобретении термин "гетероциклил" относится к ароматической или неароматической циклической группе, которая аналогична карбоциклической группе, но в которой от одного до трех углеродных атомов заменены на один или более гетероатомов, независимо выбранных из азота, кислорода или серы. Гетероциклильная группа может быть, например, моноциклической или полициклической (например, бициклической). Гетероатом может представлять собой N, O или S.
Хотя при применении в терапии и возможно введение терапевтически эффективного количества описанных в изобретении соединений или их фармацевтически приемлемой соли или сольвата в форме не подвергавшегося обработке химического вещества, тем не менее, в одном аспекте настоящего изобретения, активный ингредиент представлен в форме фармацевтической композиции. Поэтому, в дополнительном варианте осуществления изобретение предлагается фармацевтическая композиция, содержащая раскрытое в изобретение соединение формулы (I) или его фармацевтически приемлемую соль или сольват в смеси с одним или более фармацевтически приемлемыми носителями, разбавителями или вспомогательными веществами. Носитель (носители), разбавитель (разбавители) или вспомогательное вещество (вспомогательные вещества) должны быть приемлемыми с точки зрения их совместимости с другими ингредиентами композиции и отсутствия вредного воздействия на реципиента.
В случае, когда это применимо, соединения по настоящему изобретению, включающие соединения формулы (I), могут находиться в форме фармацевтически приемлемой соли и/или могут быть введены в форме фармацевтически приемлемой соли.
Используемый в изобретении термин "фармацевтически приемлемая соль" относится к солям, которые являются безопасными с токсикологической точки зрения при системном введении. Фармацевтически приемлемые соли могут быть выбраны из солей щелочных или щелочноземельных металлов, включающих, натрий, литий, калий, кальций, магний и другие подобные металлы, а также из солей нетоксичных катионов аммония, четвертичного аммония и амина, включающих, но этим не ограничивая, аммоний, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин, этиламин, триэтаноламин и другие подобные катионы.
Используемый в изобретении термин "фармацевтически приемлемое вспомогательное вещество" относится к твердому или жидкому наполнителю, разбавителю или инкапсулирующему веществу, которое может быть безопасно использовано при системном введении. В зависимости от конкретного способа введения, может быть использован целый ряд хорошо известных в данной области носителей. Эти носители или вспомогательные вещества могут быть выбраны из группы, включающей сахара, крахмалы, целлюлозу и ее производные, солод, желатин, тальк, сульфат кальция, растительные масла, синтетические масла, полиолы, альгиновую кислоту, забуференные фосфатом растворы, эмульгаторы, изотонический раствор и апирогенную воду.
Используемый в изобретении термин "сольват" относится к комплексу переменной стехиометрии, образованному растворенным веществом (в этом изобретении, соединение формулы (I) или (Ia) или его соль или физиологически функциональное производное) и растворителем. Такие растворители для осуществления решаемых в изобретении задач не должны отрицательно влиять на биологическую активность растворенного вещества. Примеры подходящих растворителей включают, но этим не ограничивая, воду, метанол, этанол и уксусную кислоту. В частность, используемый растворитель представляет собой фармацевтически приемлемый растворитель. Примеры подходящих фармацевтически приемлемых растворителей включают, без ограничения, воду, этанол, уксусную кислоту, глицерин, жидкие полиэтиленгликоли и их смеси. Конкретный растворитель представляет собой воду.
Введение соединений формулы (I) может быть осуществлено в форме "пролекарства". Пролекарство представляет собой неактивную форму соединения, которая превращается in vivo в активную форму. Подходящие пролекарства включают эфиры, фосфонатные эфиры и другие эфиры активной формы соединения.
Следует иметь в виду, что помимо конкретно указанных выше ингредиентов, композиции могут включать другие традиционные для данной области средства с учетом типа рассматриваемой композиции.
Соединения по настоящему изобретению могут применяться для лечения заболеваний у человека или животного. В одном варианте осуществления, пациентом является млекопитающее, в том числе человек, лошадь, собака, кошка, овца, корова или примат. В одном варианте осуществления, пациентом является человек. В еще одном варианте осуществления, пациентом является не человек.
Используемый в изобретении термин "эффективное количество" обозначает то количество лекарственного средства или фармацевтического средства, которое позволяет достигать биологического или лечебного ответа ткани, системы, животного или человека, которого стремится достигнуть, например, исследователь или клиницист. Кроме того, термин "терапевтически эффективное количество " обозначает любое количество, которое, по сравнению с соответствующим субъектом, который не получал такое количество, приводит к улучшению результатов лечения, выздоровлению, предотвращению или облегчению заболевания, расстройства или побочного эффекта, или к снижению скорости развития заболевания или расстройства. Термин также включает в себя количества, эффективные для усиления нормальной физиологической функции.
Используемый в изобретении термин "лечение" относится к защите от проявления симптома или к подавлению симптома, лечению симптома, отсрочке появления симптома, снижению тяжести развития симптома и/или уменьшению количества или типов симптомов, от которых страдает индивидуум, по сравнению с ситуацией, когда не применяют фармацевтическую композицию, содержащую соединение по изобретению. Термин "лечение" включает в себя и паллиативное лечение.
Для специалистов в данной области является очевидным, что при получении соединений по изобретению может быть необходимой или желательной защита одной или более реакционно способных групп в молекуле с целью предотвращения протекания нежелательных побочных реакций. Подходящие защитные группы для применения в соответствии с настоящим изобретением хорошо известны специалистам в данной области и могут быть использованы традиционным способом. Смотрите, например, монографию "Protective groups in organic synthesis" by T. W. Greene and P. G. M. Wuts (John Wiley & sons 1991 ) или монографию "Protecting Groups" by PJ. Kocienski (Georg Thieme Verlag 1994). Примеры подходящих защитных групп для аминогрупп включают ацильный тип защитных групп (например формил, трифторацетил, ацетил), тип ароматических уретановых защитных групп (например бензилоксикарбонил (Cbz) и замещенный Cbz), алифатические уретановые защитные группа (например 9-флуоренилметоксикарбонил (Fmoc), трет-бутилоксикарбонил (Boc), изопропилоксикарбонил, циклогексилоксикарбонил) и алкильный тип защитных групп (например бензил, тритил, хлортритил).
Согласно четвертому аспекту изобретения, соединения формулы (I) могут применять при лечении рака. Типы рака, которые могут быть подвергнуты лечению, включают лейкоз, немелкоклеточный рак легкого, рак толстой кишки, опухоль центральной нервной системы (CNS), меланому, рак яичников, рак почки, рак предстательной железы или рак молочной железы. Более предпочтительно, когда подвергаемый лечению рак представляет собой лейкоз или меланому.
Противоопухолевое действие соединений по настоящему изобретению может быть использовано в форме монотерапии или может использоваться вместе с одним или более другими веществами и/или методами лечения. Такое совместное лечение может быть достигнуто в результате одновременного, последовательного или раздельного применения индивидуальных используемых при лечении компонентов. В области терапевтической онкологии, обычной практикой является использование комбинации различных форм терапии при лечении каждого пациента, страдающего раком, такие как комбинация хирургического вмешательства, лучевой терапии и/или химиотерапии. В частности, известно, что лучевая терапия или лечение с помощью антиангиогенных и/или снижающих сосудистую проницаемость средств может приводить к увеличению количества гипоксической ткани внутри опухоли. Поэтому, эффективность соединений по настоящему изобретению может быть повышена путем проведения совместного лечения с использованием лучевой терапии и/или с помощью антиангиогенного средства.
Индивидуальные компоненты таких комбинаций могут быть применены раздельно в разные моменты времени в течение курса терапии или одновременно в раздельных или единых комбинированных формах. Поэтому, следует иметь в виду, что настоящее изобретение охватывает все такие режимы одновременного или чередующегося лечения, и термин "введение (применение)" следует интерпретировать соответствующим образом. Следует иметь в виду, что объем комбинаций соединений по настоящему изобретению с другими противоопухолевыми средствами включает, в принципе, любую комбинацию с любой фармацевтической композицией, применяемой для лечения рака.
При комбинировании в одной лекарственной форме, следует иметь в виду, что два соединения должны быть стабильными и совместимыми друг с другом и другими компонентами лекарственной формы, и они могут быть приготовлены для введения. При приготовлении раздельных лекарственных форм, соединения могут быть приготовлены в любой удобной форме с помощью такого метода, который известен для таких соединений в данной области техники.
Фармацевтические композиции по изобретению могут быть приготовлены для введения пациентам любым подходящим способом, например пероральным (в том числе буккальным или сублингвальным), ректальным, назальным, местным (в том числе буккальным, сублингвальным или трансдермальным), вагинальным или парентеральным (в том числе подкожным, внутримышечным, внутривенным или интрадермальным) способом. Поэтому, фармацевтические композиции по изобретению могут быть приготовлены, например, в форме таблеток, капсул, порошков, гранул, пастилок, кремов или жидких препаратов, таких как пероральные или стерильные парентеральные растворы или суспензии. Такие фармацевтические препараты могут быть приготовлены любым способом, известным в области фармацевтики, например, путем связывания активного ингредиента с носителем (носителями) или вспомогательным веществом (вспомогательными веществами). Такие фармацевтические препараты могут быть приготовлены в форме гранул, таблеток или капсул с энтеросолюбильным покрытием, применяемых для перорального введения, и в форме препаратов с отсроченным высвобождением.
В случае использования соединения в комбинации со вторым терапевтическим средством, обладающим активностью в отношении того же заболевания, доза каждого соединения может отличаться от дозы, которую используют в случае применения соединений в форме монотерапии. Соответствующие дозы могут быть определены без особых затруднений специалистами в данной области.
ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Для более глубокого понимания сущности изобретения, далее описан ряд примеров.
Получение модуля A
Модуль A представлен описанным в изобретении соединением формулы (II) по настоящему изобретению.
В одном варианте осуществления, модуль A представлен соединением A7 и A8, и его получают следующим способом:
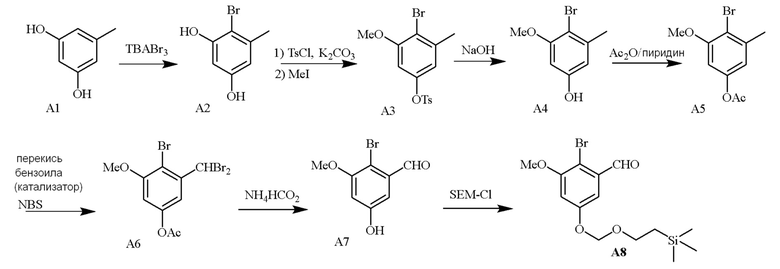
4-Бром-5-метилбензол-1,3-диол (A2).
Тетрабутиламмония трибромид (120 г, 248 ммоль, 1,0 экв) в смеси 3 : 2 дихлорметана и метанола (300 мл) добавляли по каплям к раствору соединения A1 (30 г, 242 ммоль, 1,0 экв) в смеси 3 : 2 дихлорметана и метанола (200 мл) при 15°C в течение 3 часов. Полученную смесь медленно подогревали до комнатной температуры и перемешивали в течение 16 часов. Растворители удаляли при пониженном давлении. К остатку добавляли воду (200 мл). Смесь экстрагировали этилацетатом (2×250 мл) и метилтрет-бутиловым эфиром (500 мл). Объединенные органические слои промывали 2,5% водным раствором бикарбоната натрия (3×500 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на силикагеле (2,5 кг), элюируя с градиентом от 0 до 50% этилацетата в гептанах, с получением соединения A2 (36,83 г, 75% выход) в виде желтовато-белого твердого вещества. (LM-1-96).
Дополнительное соединение A1 (270 г, 2175 ммоль, 1,0 экв) подвергали обработке таким же образом. Реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в дихлорметане (3 л) и очищали на силикагеле (6 кг), элюируя с градиентом от 0 до 100% этилацетата в гептанах, с получением частично очищенного соединения A2 (407 г, ~70% чистота). Этот неочищенный продукт перекристаллизовывали два раза в смеси 1 : 3 метанола и воды (1,2 л). Твердое вещество фильтровали, ополаскивали водой (500 мл) и сушили под высоким вакуумом при 45°C в течение 16 часов с получением соединения A2 (163,78 г 37% выход) в виде желтовато-белого твердого вещества. (LM-1-100).
4-Бром-3-метокси-5-метилфенил 4-метилбензолсульфонат (A3).
Карбонат калия (886 г, 6402 ммоль, 6,5 экв) добавляли к раствору соединения A2 (200 г, 985 ммоль, 1,0 экв) в ацетоне (15 л). После перемешивания в течение 30 минут, добавляли п-толуолсульфонилхлорид (187,8 г, 985 ммоль, 1,0 экв), и полученную смесь кипятили с обратным холодильником в течение 16 часов. К реакционной смеси добавляли метилйодид (166 мл, 2660 ммоль, 2,7 экв) и продолжали кипячение с обратным холодильником в течение еще 24 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали, и твердые вещества споласкивали ацетоном (4 л). Фильтрат концентрировали при пониженном давлении. Остаток растворяли в дихлорметане (2 л) и пропускали через силикагель (6 кг), элюируя с градиентом от 0 до 15% этилацетата в гептанах (16 л) с получением соединения A3 (350 г, ~60% чистота), которое использовали на следующей стадии. (LM-6-1).
4-Бром-3-метокси-5-метилфенол (A4).
6M водный раствор гидроксида натрия (108 мл, 8,0 экв) добавляли к раствору неочищенного соединения A3 (30 г, 1,0 экв) в смеси 1 : 1 тетрагидрофурана и метанола (400 мл). После кипячения с обратным холодильником в течение 16 часов, анализ методом LCMS указывал на завершение реакции. Другую партию неочищенного соединения A3 (300 г) подвергали обработке аналогичным способом, и обе партии объединяли для выделения продукта. Затем смесь охлаждали до комнатной температуры, добавляли ацетонитрил (4 л), и смесь промывали гептанами (2×4 л). Гептановые слои отбрасывали. Смесь тетрагидрофуран/метанол/ацетонитрил/вода подкисляли 1N раствором хлористоводородной кислоты (~6,2 л) до pH=7, насыщали хлоридом натрия (3 кг) и экстрагировали этилацетатом (2×4 л). Объединенные органические слои концентрировали при пониженном давлении. Остаток разбавляли в воде (500 мл) и экстрагировали этилацетатом (3×1 л). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток разбавляли ацетонитрилом (1 л) и гептанами (500 мл), и слои разделяли. Гептановый слой отбрасывали. Ацетонитрильный слой пропускали через слой силикагеля (300 г), который элюировали этилацетатом (2 л). Фильтрат концентрировали при пониженном давлении. Полученное твердое вещество растирали с 10% этилацетата в гептанах (1,1 л) с получением соединения A4 (96,6 г, 45% выход за 2 стадии) в виде желтовато-белого твердого вещества. (LM-6-12, LM-6-14)
4-Бром-3-метокси-5-метилфенилацетат (A5).
Уксусный ангидрид (63,1 мл, 668 ммоль, 1,5 экв) добавляли к раствору соединения A4 (96,6 г, 445 ммоль, 1,0 экв) в смеси пиридина (377 мл) и дихлорметана (1000 мл) при комнатной температуре. Полученную смесь перемешивали в течение 16 часов. Добавляли ледяную воду (500 мл) для прерывания реакции. Органический слой промывали ледяным 1N раствором HCl (3×500 мл), насыщенным раствором бикарбоната натрия (500 мл), насыщенным солевым раствором (500 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток затем сушили под высоким вакуумом при 45°C в течение 16 часов с получением соединения A5 (114,53 г, 99% выход) в виде желтовато-белого твердого вещества, которое использовали на следующей стадии. (LM-6-15).
4-Бром-3-(дибромметил)-5-метоксифенилацетат (A6).
N-Бромсукцинимид (82,6 г, 464 ммоль, 2,1 экв) и пероксид бензоила (5,35 г, 22,1 ммоль, 0,1 экв) последовательно добавляли к раствору соединения A5 (57,27 г, 221 ммоль, 1,0 экв) в четыреххлористом углероде (1 л). Полученную смесь кипятили с обратным холодильником в течение 24 часов, после чего анализ методом LCMS указывал на завершение реакции. Другую партию соединения A5 (57,27 г) подвергали обработке аналогичным способом, и обе партии объединяли для выделения продукта. Затем смесь охлаждали до комнатной температуры, добавляли бисульфит натрия (250 г), и реакционную смесь перемешивали в течение 24 часов при комнатной температуре (тест на влажной реактивной крахмальной бумаге показывал отсутствие пероксида). Полученную смесь фильтровали через целит (200 г), который споласкивали дихлорметаном (2 л). Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения A6 в виде желтого твердого вещества, которое использовали на следующей стадии. (LM-6-17)
2-Бром-5-гидрокси-3-метоксибензальдегид (A7):
Формиат аммония (153 г, 2431 ммоль, 5,5 экв) и смесь 1 : 1 этанола и воды (600 мл) добавляли к раствору соединения A6 (~442 ммоль, 1,0 экв) в этаноле (800 мл) при 50°C. Полученную смесь кипятили с обратным холодильником в течение 70 часов. Анализ методом LCMS указывал на завершение реакции. Реакционную смесь охлаждали до комнатной температуры и добавляли концентрированную хлористоводородную кислоту (15 мл). Растворители удаляли при пониженном давлении. Остаток разбавляли в воде (500 мл), экстрагировали этилацетатом (2×1 л) и метилтрет-бутиловым эфиром (2×1 л). Водный слой фильтровали через слой целита (50 г), который споласкивали этилацетатом (2 л). Органический слой отделяли. Все органические слои объединяли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток пропускали через слой силикагеля (300 г), который элюировали тетрагидрофураном (3 л) и этилацетатом (4 л). Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения A7 (141 г) в виде черного масла, которое использовали на следующей стадии. (LM-6-18)
2-Бром-3-метокси-5-((2-(триметилсилил)этокси)метокси)-бензальдегид (A8 типичное для модуля A).
Диизопропилэтиламин (16,5 мл, 94,7 ммоль, 1,5 экв) и 2-(триметилсилил)этоксиметилхлорид (13,4 мл, 75,6 ммоль, 1,2 экв) добавляли последовательно к раствору неочищенного соединения A7 (20 г, ~63 ммоль, 1,0 экв) в смеси 1 : 1 безводного тетрагидрофурана и дихлорметана (200 мл) при комнатной температуре. После перемешивания при комнатной температуре в течение 16 часов, анализ методом LCMS указывал на ~65% превращение исходного реагента. Добавляли дополнительное количество диизопропилэтиламина (16,5 мл, 94,7 ммоль, 1,5 экв) и 2-(триметилсилил)этоксиметилхлорида (13,4 мл, 75,6 ммоль, 1,2 экв), и смесь перемешивали в течение еще 24 часов. Добавляли насыщенный раствор бикарбоната натрия (250 мл) для прерывания реакции, и органический растворитель удаляли при пониженном давлении. Оставшийся водный слой экстрагировали метилтрет-бутиловым эфиром (500 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной системе Interchim (колонка 330 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением A8 (типичного модуля A) (8,44 г) в виде белого твердого вещества. (LM-6-20) Дополнительное количество неочищенного соединения 7 (120 г) подвергали обработке аналогичным образом. Добавляли насыщенный раствор бикарбоната натрия (250 мл) для прерывания реакции, и растворитель удаляли при пониженном давлении. Оставшийся водный слой разбавляли водой (250 мл), насыщенным солевым раствором (250 мл) и экстрагировали метилтрет-бутиловым эфиром (2×1 л). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток фильтровали через слой силикагеля (1 кг), который элюировали смесью 20% этилацетата в гептанах. Фильтрат концентрировали при пониженном давлении. Остаток разделяли на 6 равных порций. Каждую порцию очищали на автоматизированной системе Interchim (колонка 330 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах. Все количество выделенного промежуточного соединения A затем очищали путем растирания с 5% дихлорметана в гексанах (300 мл) с получением A8 (типичного соединения для модуля A) (26,73 г, 99% чистота и 42,72 г, ~90% чистота) в виде белого твердого вещества после сушки под вакуумом при 45°C в течение 16 часов. Суммарный выход составлял 43% за 3 стадии. (LM-6-21)
Белое твердое вещество, температура плавления 58,3-59,1°C; анализ методом HPLC: 99,6% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 10,80 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=378,0 (M+H2O)+; C14H21BrO4Si; 1H ЯМР (400 МГц, CDCl3) δ=10,40 (с, 1H), 7,20 (д, J=2,8 Гц,1H), 6,84 (д, J=2,7 Гц, 1H), 5,25 (с, 2H), 3,92 (с, 3H), 3,78-3,73 (м, 2H), 0,97-0,93 (м, 2H), 0,00 (с, 9H); 13C ЯМР (100 МГц, CDCl3) δ=191,82, 157,76, 157,12, 134,85, 109,31, 107,49, 106,62, 93,06, 66,72, 56,65, 18,02, -1,43.
В еще одном варианте осуществления, модуль A представлен соединением A9, которое получают следующим способом:
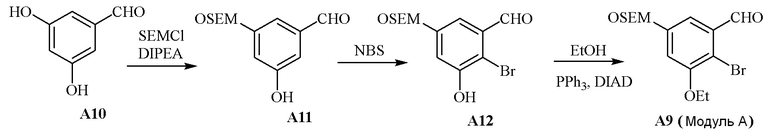
3-Гидрокси-5-((2-(триметилсилил)этокси)метокси)бензальдегид (A11).
N, N-Диизопропилэтиламин (122 мл, 700 ммоль, 1,4 экв) добавляли к раствору соединения A10 (69,06 г, 500 ммоль, 1,0 экв) в безводном диметилформамиде (400 мл) при комнатной температуре. Добавляли по каплям 2-(триметилсилил)этоксиметилхлорид (106,2 мл, 600 ммоль, 1,2 экв) в течение 30 минут. Полученную смесь перемешивали при комнатной температуре в течение 70 часов. Добавляли воду (1,5 л), и смесь экстрагировали метилтрет-бутиловым эфиром (2×2,5 л). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток разделяли на 3 равных порции. Каждую порцию очищали на автоматизированной системе Interchim (колонка 330 g), элюируя с градиентом от 0 до 50% этилацетата в гептанах, с получением соединения A11 (49,64 г, 26% выход, 71% чистота по количественному анализу методом ЯМР) в виде желтого масла. (LM-6-84)
2-Бром-3-гидрокси-5-((2-(триметилсилил)этокси)метокси)-бензальдегид (A12).
Соединение A11 (49,64 г, 71% чистота по количественному анализу методом ЯМР, 131,3 ммоль, 1,0 экв) помещали в глубокий вакуум (66,7 Па) при 50°C в течение 2 часов для удаления остаточного 2-(триметилсилил)этанола. После охлаждения до комнатной температуры, добавляли безводный дихлорметан (700 мл), и смесь охлаждали до -17°C. Добавляли по каплям N-бромсукцинимид (25,12 г, 141,1 ммоль, 1,07 экв) в безводном дихлорметане (1 л) в течение 1 часа при поддержании температуры в диапазоне от -15°C до -20°C. После добавления, анализ методом ЯМР показывал, что реакция прошла не до конца (оставалось непрореагировавшим 16% соединения A11). Добавляли по каплям дополнительное количество N-бромсукцинимида (4,62 г, 26 ммоль, 0,2 экв) в безводном дихлорметане (200 мл) в течение 10 минут. Полученную смесь перемешивали при -17°C в течение еще 30 минут. Добавляли воду (800 мл), и смесь подогревали до комнатной температуры. Органический слой промывали насыщенным солевым раствором (1 л), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток разделяли на 5 равных порций. Каждую порцию очищали на автоматизированной системе Interchim (ряд колонок 2×120 g), элюируя с градиентом от 0 до 12% этилацетата в гептанах, с получением соединения A12 (35,84 г, 79% выход) в виде желтого масла. (LM-6-87)
2-Бром-3-этокси-5-((2-(триметилсилил)этокси)метокси)-бензальдегид (A9, модуль A):
Трифенилфосфин (40,6 г, 154,8 ммоль, 1,5 экв) и этанол (9 мл, 154,8 ммоль, 1,5 экв) добавляли к раствору соединения A12 (35,84 г, 103,2 ммоль, 1,0 экв) в безводном тетрагидрофуране (1 л). После охлаждения до 0°C в течение 10 минут, добавляли по каплям диизопропилазодикарбоксилат (31 мл, 154,8 ммоль, 1,5 экв) в течение 15 минут. Полученную смесь медленно подогревали до комнатной температуры и перемешивали в течение 16 часов. Растворитель удаляли при пониженном давлении. Остаток разделяли на 5 равных порций. Каждую порцию очищали на автоматизированной системе Interchim (ряд колонок 2×120 g), элюируя смесью 5% этилацетата в гептанах, с получением A9 (17,5 г, 45% выход) в виде желтого твердого вещества. (LM-6-88)
Получение модуля B
Модуль B представлен описанным в изобретении соединением формулы (III) по настоящему изобретению. В одном варианте осуществления, модуль B представлен соединением B4, которое получают следующим способом:
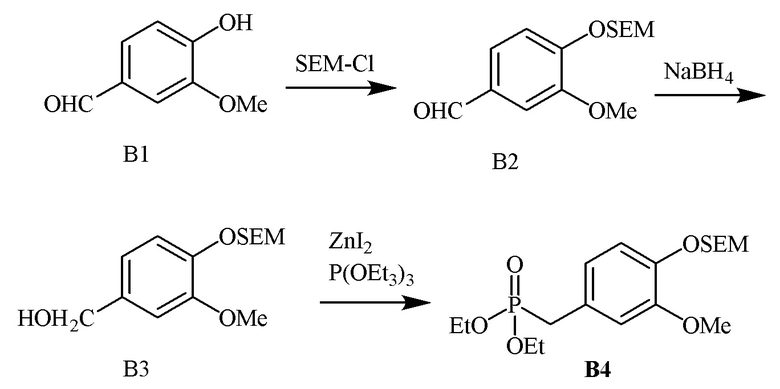
3-Метокси-4-((2-(триметилсилил)этокси)метокси)бензальдегид (B2).
N, N-Диизопропилэтиламин (1224 г, 9,47 моль, 1,2 экв) и 2-(триметилсилил)этоксиметилхлорид (1315 г, 7,89 моль, 1,0 экв) последовательно добавляли к раствору соединения B1 (1200 г, 7,84 моль, 1,0 экв) в безводном дихлорметане (12 л). После перемешивания при комнатной температуре в течение ночи, смесь выливали в делительную воронку, содержащую насыщенный водный раствор бикарбоната натрия (20 л). Слои разделяли, и водный слой экстрагировали дихлорметаном (2×5 л). Объединенные органические слои концентрировали при пониженном давлении. Остаток объединяли с 2 остатками, полученных при проведении реакций при равной загрузке, и очищали на силикагеле (2 кг), элюируя с градиентом от 0 до 50% MTBE в гептанах, с получением соединения B2 (2200 г, 99% выход) в виде бледно-желтого масла. (EK-22-137)
(3-Метокси-4-((2-(триметилсилил)этокси)метокси)фенил)метанол (B3).
К раствору соединения B2 (2000 г, 7,10 моль) в метаноле (8 л) добавляли порциями боргидрид натрия (250 г, 6,61 моль) в течение 20 минут при поддержании температуры ниже 20ºC. После перемешивания в течение ночи, медленно добавляли воду (1 л) в течение 20 минут. Смесь объединяли со смесью, полученной при проведении реакции с равной загрузкой, и концентрировали при пониженном давлении с удалением большой части метанола. Суспензию выливали в делительную воронку и разбавляли с помощью MTBE (8 л). Слои разделяли, и водный слой снова экстрагировали с помощью MTBE (2×4 л). Объединенные органические слои промывали насыщенным солевым раствором (4 л), сушили над сульфатом натрия (200 г) и концентрировали при пониженном давлении с получением соединения B3 (1970g, 98% выход) в виде желтого масла, которое использовали на следующей стадии. (EK-22-153)
Диэтил (3-метокси-4-((2-(триметилсилил)этокси)метокси)-бензил)фосфонат (B4, модуль B):
В колбу объемом 22 л загружали THF (7 л) и йодид цинка(II) (2,24 кг, 2,03 моль, 2 экв), добавление приводило к повышению температуры до 40ºC. Последовательно добавляли триэтилфосфит (1,17 кг, 7,03 моль, 2 экв) и соединение B3 (1000 г, 3,52 моль, 1 экв) в THF (1 л). После нагревания при 65ºC в течение 16 часов, реакционную смесь охлаждали до комнатной температуры и разбавляли водой (5 л). Добавляли порциями карбонат калия (1,2 кг, 8,68 моль, 2,47 экв) в течение 5 минут, доводя величину pH до 9. В процессе добавления, образования пены не наблюдалось. Соли фильтровали, и осадок на фильтре промывали с помощью MTBE (2×4 л). Объединенные фильтраты промывали насыщенным солевым раствором (4 л), сушили над сульфатом натрия (500 г) и концентрировали при пониженном давлении. Полученное масло очищали на силикагеле (2,0 кг), элюируя с градиентом от 25 до 100% этилацетата в гептанах, с получением B4 (1035 г, 72% выход) в виде желтого масла. (EK-22-154) C18H33PSiO6
1H ЯМР (400 МГц, CDCl3) δ=7,11 (д, J=8,2 Гц, 1H), 6,89 (м, 1H), 6,80 (дм, J=8,2 Гц, 1H), 5,26 (с, 2H), 4,08-3,98 (м, 4H), 3,88 (с, 3H), 3,82 (м, 2H), 3,10 (д, J=21,1 Гц, 2H), 1,26 (т, J=7,1 Гц, 6H), 0,98-0,93 (м, 2H), 0,00 (с, 9H).
Получение модуля C
Модуль C представлен описанным в изобретении соединением формулы (IV) по настоящему изобретению.
В одном варианте осуществления, модуль C представлен соединением C1, которое получают следующим способом:
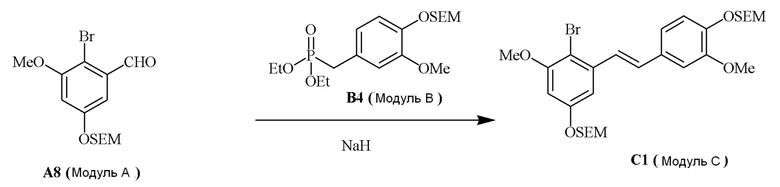
(E)-(2-((4-(2-бром-3-метокси-5-((2-(триметилсилил)этокси)- метокси)стирил)-2-метоксифенокси)метокси)этил)триметилсилан (C1):
60% дисперсию гидрида натрия в минеральном масле (1,07 г, 26,7 ммоль, 2 экв) добавляли одной порцией к раствору соединения B4 (5,4 г, 13,4 ммоль, 1,0 экв) в безводном тетрагидрофуране (120 мл) при 0°C. Смесь подогревали до комнатной температуры и перемешивали 30 минут. Затем добавляли по каплям раствор соединения A8 (4,82 г, 13,4 ммоль, 1 экв) в безводном тетрагидрофуране (30 мл), и смесь перемешивали при комнатной температуре в течение 16 часов, после чего анализ методом LCMS показывал, что достигалась ~30% конверсия в модуль C. Добавляли дополнительное количество 60% дисперсии гидрида натрия в минеральном масле (1,07 г, 26,7 ммоль, 2 экв), что в результате давало ~50% конверсию через 8 часов, что определялось путем проведения анализа методом LCMS. Добавляли дополнительное количество 60% дисперсии гидрида натрия в минеральном масле (2,14 г, 53,4 ммоль, 4 экв), в результате чего достигалась полная конверсия чем 16 часов, что определялось путем проведения анализа методом LCMS. Реакцию осторожно прерывали насыщенным солевым раствором (100 мл, 1 капля в минуту для первых 5 мл солевого раствора) при 0°C. Смесь экстрагировали этилацетатом (2×500 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 220 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением C1 (5,2 г, 64% выход) в виде желтого масла. (LM-6-27)
C28H43BrSiO6 1H ЯМР (400 МГц, CDCl3) δ=7,39 (д, J=16,1 Гц, 1H), 7,17 (д, J=8,3 Гц, 1H), 7,09 (д, J=1,7 Гц, 1H), 7,07 (дд, J=2,0, 8,6 Гц, 1H), 6,99 (д, J=2,4 Гц, 1H), 6,96 (д, J=16,2 Гц, 1H), 6,56 (д, J=2,7 Гц, 1H), 5,29 (с, 2H), 5,25 (с, 2H), 3,94 (с, 3H), 3,88 (с, 3H), 3,83-3,77 (м, 4H), 1,00-0,94 (м, 4H), 0,02 (с, 9H), 0,00 (с, 9H).
В еще одном варианте осуществления, модуль C представлен соединением C2, которое получают следующим способом:
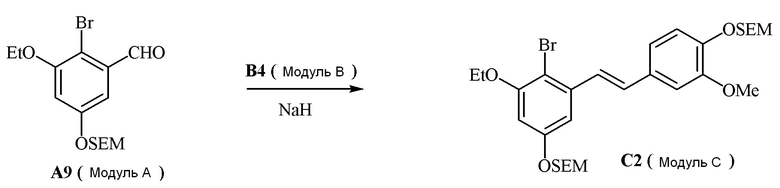
(E)-(2-((4-(2-Бром-3-этокси-5-((2-(триметилсилил)этокси)-метокси)стирил)-2-метоксифенокси)метокси)этил)триметилсилан (C2):
К раствору соединения B4 (3,8 г, 9,4 ммоль, 1,0 экв) в безводном тетрагидрофуране (150 мл) добавляли одной порцией 60% дисперсию гидрида натрия в минеральном масле (750 мг, 18,8 ммоль, 2 экв) при 0°C. Смесь подогревали до комнатной температуры и перемешивали 30 минут. Затем добавляли по каплям раствор соединения D4 (3,52 г, 9,4 ммоль, 1 экв) в безводном тетрагидрофуране (50 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию осторожно прерывали насыщенным солевым раствором (150 мл, 1 капля в минуту для первых 5 мл) при 0°C. Смесь экстрагировали метилтрет-бутиловым эфиром (2×150 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 120 g), элюируя с градиентом от 0-15% этилацетата в гептанах, с получением C2 (1,69 г, 29% выход) в виде желтого масла. (LM-6-80).
Получение соединения 1 [KYN-001]
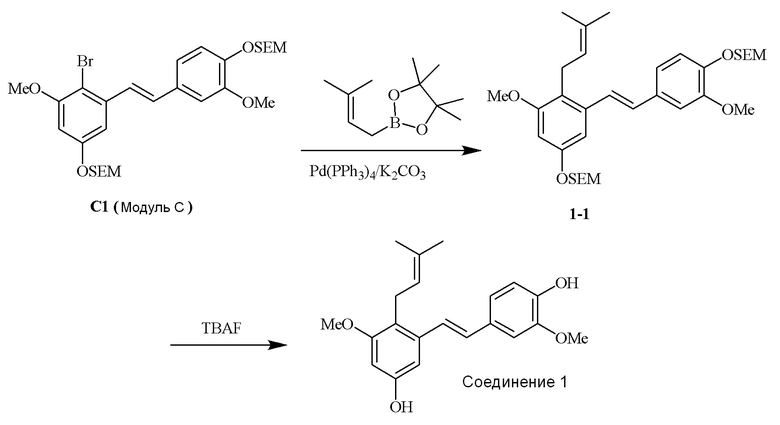
(E)-(2-((2-метокси-4-(3-метокси-2-(3-метилбут-2-ен-1-ил)-5-((2-(триметилсилил)этокси)метокси)стирил)фенокси)метокси)этил)-триметилсилан (1-1).
Тетракис(трифенилфосфин)палладий(0) (29 мг, 0,025 ммоль, 0,05 экв), карбонат калия (138 мг, 1 ммоль, 2 экв), пинаколовый эфир 3-метилбут-2-енилбороновой кислоты (147 мг, 0,75 ммоль, 1,5 экв) и воду (5 мл) добавляли последовательно к раствору соединения C1 (306 мг, 0,5 ммоль, 1 экв) в тетрагидрофуране (5 мл). После продувки азотом в течение 10 минут, реакционную смесь нагревали при 50°C в течение 20 часов. После охлаждения до комнатной температуры, смесь экстрагировали этилацетатом (15 мл), и органический слой концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 40 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 1-1 (190 мг, 63% выход) в виде желтого масла. (LM-6-29)
(E)-3-(4-гидрокси-3-метоксистирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенол (1).
1M раствор тетрабутиламмония фторида в тетрагидрофуране (4,43 мл, 4,43 ммоль, 14 экв) добавляли к соединению 1-1 (190 мг, 0,32 ммоль, 1 экв) при комнатной температуре. После нагревания при 67°C в течение 16 часов, смесь охлаждали до комнатной температуры и разбавляли водой (4 мл). Смесь экстрагировали этилацетатом (15 мл), и органический слой концентрировали при пониженном давлении. Остаток очищали два раза на автоматизированной хроматографической системе Interchim (2 колонки 25 g), элюируя каждый раз с градиентом от 0 до 60% этилацетата в гептанах, с получением соединения 1 (60 мг, 56% выход) в виде желтовато-белого твердого вещества после сушки под вакуумом при 40°C в течение 16 часов. (LM-6-30)
Белое твердое вещество, температура плавления 132,3-134,0°C; анализ методом HPLC: 96,3% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 8,4 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=341,2 (M+H)+; C21H24O4; 1H ЯМР (400 МГц, CDCl3) δ=7,15 (д, J=16,0 Гц, 1H), 7,00 (уш.с, 1H), 6,99 (дд, J=2,0, 8,2 Гц, 1H), 6,90 (д, J=8,2 Гц, 1H), 6,85 (д, J=16,1 Гц, 1H), 6,66 (д, J=2,6 Гц, 1H), 6,36 (д, J=2,4 Гц, 1H), 5,12 (тм, J=6,8 Гц, 1H), 3,94 (с, 3H), 3,80 (с, 3H), 3,41 (уш.д, J=6,7 Гц, 2H), 1,81 (с, 3H), 1,68 (д, J=1,1 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=158,52, 154,37, 146,65, 145,54, 138,13, 130,59, 130,40, 130,24, 124,24, 123,63, 120,65, 120,55, 114,53, 108,22, 103,85, 98,17, 55,86, 55,69, 25,78, 24,45, 17,97.
Получение соединения 2, 3 и 4.
4-((E)-бут-2-ен-1-ил)-3-((E)-4-гидрокси-3-метоксистирил)-5-метоксифенол (2) [KYN-138]
4-((Z)-бут-2-ен-1-ил)-3-((E)-4-гидрокси-3-метоксистирил)-5-метоксифенол (3) [KYN-139]
(E)-4-(бут-3-ен-2-ил)-3-(4-гидрокси-3-метоксистирил)-5-метоксифенол (4) [KYN-140]
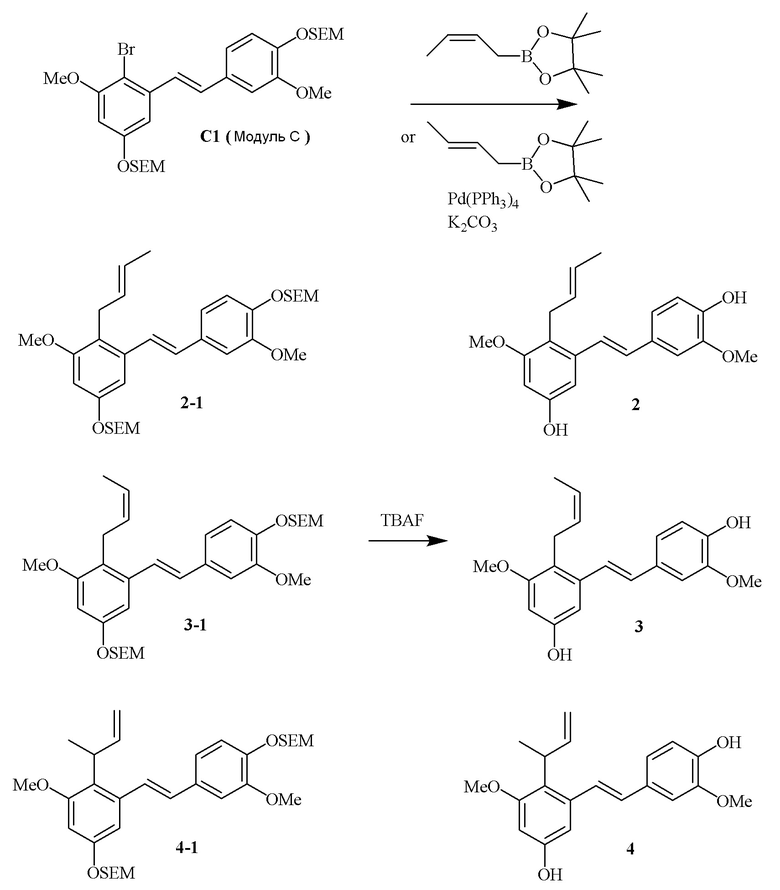
(2-((4-((E)-2-((E)-бут-2-ен-1-ил)-3-метокси-5-((2-(триметил-силил)этокси)метокси)стирил)-2-метоксифенокси)метокси)этил)-триметилсилан (2-1)
(2-((4-((E)-2-((Z)-бут-2-ен-1-ил)-3-метокси-5-((2-(триметил-силил)этокси)метокси)стирил)-2-метоксифенокси)метокси)этил)-триметилсилан (3-1)
(E)-(2-((4-(2-(бут-3-ен-2-ил)-3-метокси-5-((2-(триметил-силил)этокси)метокси)стирил)-2-метоксифенокси)метокси)этил)-триметилсилан (4-1).
Тетракис(трифенилфосфин)палладий(0) (116 мг, 0,1 ммоль, 0,05 экв), карбонат калия (553 мг, 4 ммоль, 2 экв), пинаколовый эфир транс-кротилбороновой кислоты (728 мг, 4 ммоль, 2 экв) и воду (10 мл) добавляли последовательно к раствору соединения C1 (1,22 г, 2 ммоль, 1 экв) в тетрагидрофуране (10 мл). После продувки азотом в течение 10 минут, реакционную смесь нагревали при 77°C в течение 20 часов. Еще одну партию соединения C1 (1,22 г) и пинаколового эфира цис-кротилбороновой кислоты (2 экв) подвергали обработке таким же образом, и обе партии объединяли для выделения продукта реакции. После охлаждения до комнатной температуры, смесь экстрагировали этилацетатом (2×50 мл), и органический слой концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 80 g), элюируя с градиентом от 0 до 15% этилацетата в гептанах, с получением смеси соединений 2-1, 3-1 и 4-1 (2,2 г, 94% выход) в виде желтого масла. (LM-6-42, LM-6-43)
4-((E)-бут-2-ен-1-ил)-3-((E)-4-гидрокси-3-метоксистирил)-5-метоксифенол (2) / 4-((Z)-бут-2-ен-1-ил)-3-((E)-4-гидрокси-3-метоксистирил)-5-метоксифенол 3) / (E)-4-(бут-3-ен-2-ил)-3-(4-гидрокси-3-метоксистирил)-5-метоксифенол (4).
1M раствор тетрабутиламмония фторида в тетрагидрофуране (52,5 мл, 52,5 ммоль, 14 экв) добавляли к смеси соединений 2, 3 и 4 (2,2 г, 3,75 ммоль, 1 экв) при комнатной температуре. После нагревания при 67°C в течение 16 часов, смесь охлаждали до комнатной температуры и разбавляли водой (50 мл). Смесь экстрагировали дихлорметаном (2×150 мл), и органический слой концентрировали при пониженном давлении. Остаток очищали три раза на автоматизированной хроматографической системе Interchim (3 колонки 80 g), элюируя каждый раз с градиентом от 0 до 60% этилацетата в гептанах, с получением смеси соединений 2, 3 и 4 (760 мг) в виде желтого масла. Эту маслообразную смесь подвергали сверхкритической жидкостной хроматографии (Lotus Separations) с получением трех соединений, каждое из которых выглядело как черные остатки маслообразные остатки. Эти остатки по отдельности очищали повторно очищали на автоматизированной хроматографической системе Interchim (колонка 25 g), элюируя с градиентом от 0 до 60% этилацетата в гептанах, с получением соединений 2 (90 мг, 7% выход), 3 (60 мг, 5% выход) и 4 (290 мг, 24% выход), соответственно, в виде желтовато-белых твердых веществ после сушки под вакуумом при 40°C в течение 16 часов. (LM-6-44)
Проводимое далее разделение методом SFC (условия приведены ниже) давало 400 мг пика-1 (4, LM-6-44-P1), 108 мг пика-2 (2, LM-6-44-P2), и 110 мг пика-3 (3, LM-6-44-P3). Хроматограммы включены в этот отчет.
На фигуре 1 представлена хроматограмма LM-6-44 (смесь соединений 2, 3 и 4) перед разделением
На фигуре 2 представлена хроматограмма LM-6-44-P1 (соединение 4) после разделения.
На фигуре 3 представлена хроматограмма LM-6-44-P2 (соединение 2) после разделения.
На фигуре 4 представлена хроматограмма LM-6-44-P3 (соединение 3) после разделения:
Структурные данные
4-((E)-бут-2-ен-1-ил)-3-((E)-4-гидрокси-3-метоксистирил)-5-метоксифенол (соединение 2) [KYN-138]:
желтовато-белое твердое вещество; анализ методом HPLC: 96,3% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 8,1 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=327,2 (M+H)+; C20H22O4; 1H ЯМР (400 МГц, CDCl3) δ=7,17 (д, J=16,1 Гц, 1H), 7,03-6,98 (м, 2H), 6,93-6,89 (м, 1H), 6,86 (д, J=16,1 Гц, 1H), 6,67 (д, J=2,3 Гц, 1H), 6,36 (д, J=2,4 Гц, 1H), 5,67 (с, 1H), 5,59-5,49 (м, 1H), 5,47-5,36 (м, 1H), 4,73 (с, 1H), 3,94 (с, 3H), 3,80 (с, 3H), 3,41 (тд, J=1,3, 5,9 Гц, 2H), 1,63 (квд, J=1,4, 6,2 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=158,63, 154,50, 146,67, 145,59, 138,42, 130,41, 130,31, 129,67, 124,93, 124,34, 120,35, 119,57, 114,59, 108,56, 103,91, 98,24, 55,89, 55,78, 28,29, 17,91.
4-((Z)-бут-2-ен-1-ил)-3-((E)-4-гидрокси-3-метоксистирил)-5-метоксифенол (соединение 3) [KYN-139]:
желтовато-белое твердое вещество; анализ методом HPLC: 98,8% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 8,2 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=327,2 (M+H)+; C20H22O4; 1H ЯМР (400 МГц, CDCl3) δ=7,15 (д, J=16,0 Гц, 1H), 7,01-6,97 (м, 2H), 6,90 (д, J=7,9 Гц, 1H), 6,85 (д, J=16,0 Гц, 1H), 6,66 (д, J=2,3 Гц, 1H), 6,36 (д, J=2,4 Гц, 1H), 5,68 (с, 1H), 5,53-5,32 (м, 2H), 4,87 (с, 1H), 3,93 (с, 3H), 3,80 (с, 3H), 3,48 (д, J=6,7 Гц, 2H), 1,81 (квд, J=1,2, 6,4 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=158,60, 154,49, 146,68, 145,58, 138,29, 130,56, 130,24, 129,71, 124,20, 122,99, 120,59, 120,16, 114,56, 108,29, 104,00, 98,23, 55,86, 55,69, 23,37, 12,99.
(E)-4-(бут-3-ен-2-ил)-3-(4-гидрокси-3-метоксистирил)-5-метоксифенол (соединение 4) [KYN-140]:
желтовато-белое твердое вещество; анализ методом HPLC: 96,1% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 8,2 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=327,2 (M+H)+; C20H22O4; 1H ЯМР (400 МГц, CDCl3) δ=7,32 (д, J=16,0 Гц, 1H), 6,99-6,95 (м, 2H), 6,92-6,89 (м, 1H), 6,74 (д, J=16,0 Гц, 1H), 6,61 (д, J=2,4 Гц, 1H), 6,37 (д, J=2,4 Гц, 1H), 6,22 (ддд, J=4,8, 10,5, 17,4 Гц, 1H), 5,67 (с, 1H), 5,10-5,03 (м, 2H), 4,85 (с, 1H), 4,15 тдкв, J=2,1, 4,6, 7,0 Гц, 1H), 3,94 (с, 3H), 3,78 (с, 3H), 1,39 (д, J=7,1 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=158,72, 154,52, 146,69, 145,51, 143,90, 138,85, 130,37, 130,14, 125,60, 124,11, 120,38, 114,56, 111,76, 108,30, 105,18, 98,73, 55,86, 55,70, 34,49, 18,55.
4-((E)-Бут-2-ен-1-ил)-3-этокси-5-((E)-4-гидрокси-3-метокси-стирил)фенол (соединение 5) [KYN-153]
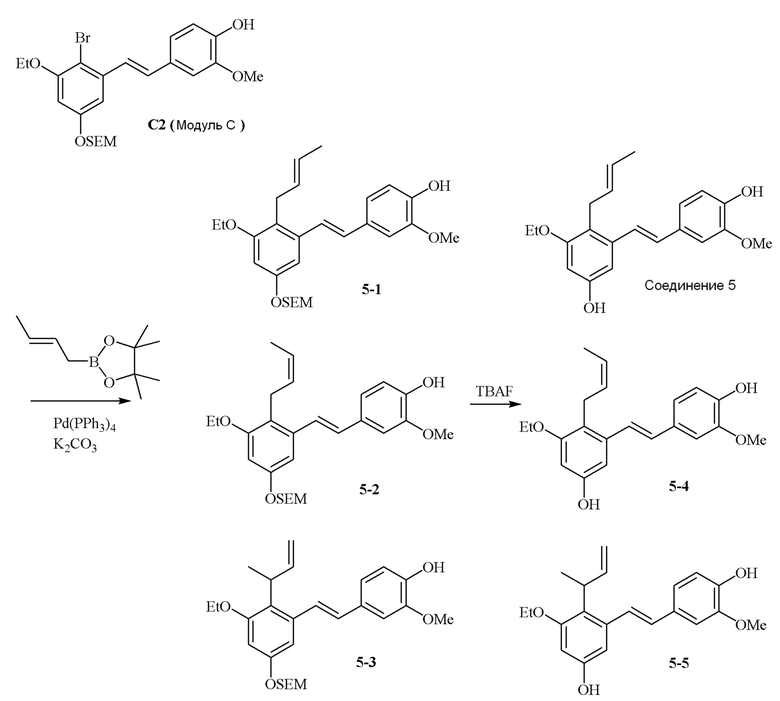
(2-((4-((E)-2-((E)-бут-2-ен-1-ил)-3-этокси-5-((2-(триметил-силил)этокси)метокси)стирил)-2-метоксифенокси)метокси)этил)-триметилсилан (5-1).
Тетракис(трифенилфосфин)палладий(0) (156 мг, 0,14 ммоль, 0,05 экв), карбонат калия (1,12 г, 8,1 ммоль, 3 экв), пинаколовый эфир транс-кротилбороновой кислоты (1,48 г, 8,1 ммоль, 3 экв) и воду (10 мл) добавляли последовательно к раствору соединения C2 (1,69 г, 2,7 ммоль, 1 экв) в 1,4-диоксане (15 мл). После продувки азотом в течение 10 минут, реакционную смесь нагревали при 95°C в течение 16 часов. После охлаждения до комнатной температуры, смесь экстрагировали метилтрет-бутиловым эфиром (2×20 мл), и объединенные органические слои концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 40 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением смеси соединения 5-1, 5-2 и 5-3 (1,54 г, 89% выход) в виде желтого масла (LM-6-82)
4-((E)-Бут-2-ен-1-ил)-3-этокси-5-((E)-4-гидрокси-3-метокси-стирил)фенол (5).
1M раствор тетрабутиламмония фторида в тетрагидрофуране (33,5 мл, 33,5 ммоль, 14 экв) добавляли к смеси соединения 5-1, 5-2 и 5-3 (1,44 г, 2,4 ммоль, 1 экв) в тетрагидрофуране (50 мл) при комнатной температуре. После нагревания при 67°C в течение 16 часов, смесь охлаждали до комнатной температуры и разбавляли этилацетатом (200 мл). Смесь промывали насыщенным солевым раствором (50 мл), и органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали два раза на автоматизированной хроматографической системе Interchim (2 колонки 40 g, 2 колонки 25 g), элюируя каждый раз с градиентом от 0 до 100% этилацетата в гептанах, с получением смеси соединения 5, 5-4 и 5-5 (610 мг) в виде желтого масла. Эту маслоподобную смесь подвергали сверхкритической жидкостной хроматографии (Lotus Separations) с получением трех соединений, которые имели вид черных масляных остатков. Остаток LM-6-85-P2 повторно очищали на автоматизированной хроматографической системе Interchim (колонка 25 g), элюируя с градиентом от 0 до 60% этилацетата в гексанах, с получением соединения 5 (60 мг, 7% выход) в виде желтовато-белого твердого вещества после сушки под вакуумом при 40°C в течение 16 часов (LM-6-85). Проводимое далее разделение методом SFC (условия приведены ниже) давало 425 мг пика-1 (5-5, LM-6-85-P1), 88 мг пика-2 (5, LM-6-85-P2) и 70 мг пика-3 (5-4, LM-6-85-P3). Пик-2 был подвергнут повторной обработке для повышения частоты.
Хроматограммы включены в этот отчет.
На фигуре 5 представлена хроматограмма LM-6-85 (смесь соединения 5, E-5-4 и E-5-5) перед разделением.
На фигуре 6 представлена хроматограмма LM-6-85-P2 (соединение 5) после разделения.
На фигуре 7 представлена хроматограмма (соединение 5-5) после разделения.
На фигуре 8 представлена хроматограмма LM-6-85-P3 (соединение 5-4) после разделения.
Структурные данные
Соединение 5 [KYN-153]
Желтовато-белое твердое вещество; анализ методом HPLC: 97,7% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 8,6 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=341,2 (M+H)+; C21H24O4; ЯМР (400 МГц, CDCl3) δ=7,19 (д, J=16,1 Гц, 1H), 7,03-6,98 (м, 2H), 6,92 (д, J=8,2 Гц, 1H), 6,85 (д, J=16,1 Гц, 1H), 6,65 (д, J=2,4 Гц, 1H), 6,34 (д, J=2,4 Гц, 1H), 5,66 (с, 1H), 5,59-5,40 (м, 2H), 4,70 (с, 1H), 4,00 (кв, J=7,0 Гц, 2H), 3,95 (с, 3H), 3,42 (уш.д, J=5,9 Гц, 2H), 1,65-1,61 (м, 3H), 1,41 (т, J=7,0 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=157,95, 154,40, 146,68, 145,57, 138,31, 130,35, 130,29, 129,79, 124,88, 124,43, 120,34, 119,89, 114,59, 108,54, 103,84, 99,17, 63,99, 55,89, 28,49, 17,90, 14,87.
Получение (E)-4-аллил-3-(4-гидрокси-3-метоксистирил)-5-метоксифенола (6) [KYN-134]
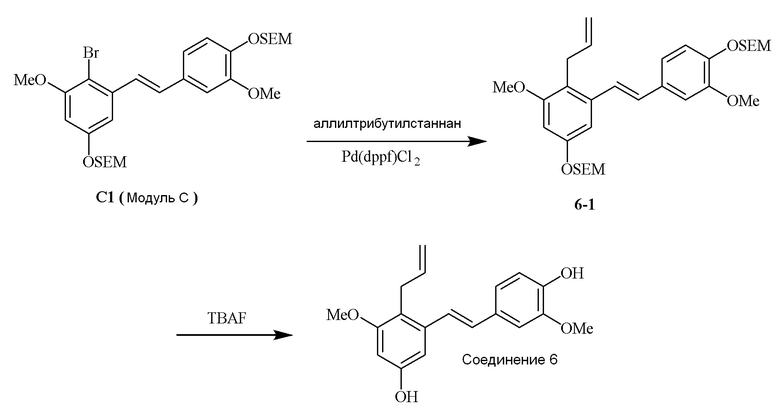
(E)-(2-((4-(2-аллил-3-метокси-5-((2-(триметилсилил)этокси)-метокси)стирил)-2-метоксифенокси)метокси)этил)триметилсилан (6-1):
[1,1′-Бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (46 мг, 0,063 ммоль, 0,25 экв) и аллилтрибутилстаннан (248 мг, 0,75 ммоль, 3 экв) добавляли к раствору соединения C1 (Модуль C)(153 мг, 0,25 ммоль, 1 экв) в безводном диметилформамиде (3 мл). После продувки азотом в течение 10 минут, реакционную смесь нагревали при 100°C в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, загружали в сухой картридж 12 g c силикагелем (10 г) и очищали на автоматизированной хроматографической системе Interchim (колонка 25 g), элюируя с градиентом от 0 до 40% этилацетата в гептанах, с получением соединения 6-1 (140 мг, 98% выход) в виде желтого масла. (LM-6-33)
(E)-4-аллил-3-(4-гидрокси-3-метоксистирил)-5-метоксифенол (6) [KYN-134]:
1M раствор тетрабутиламмония фторида в тетрагидрофуране (3,5 мл, 3,5 ммоль, 14 экв) добавляли к соединению 6-1 (140 мг, 0,25 ммоль, 1 экв) при комнатной температуре, и смесь нагревали при 67°C в течение 16 часов. Затем смесь охлаждали до комнатной температуры, добавляли воду (4 мл) для прерывания реакции. Смесь экстрагировали этилацетатом (15 мл), и органический слой концентрировали при пониженном давлении. Остаток очищали два раза на автоматизированной хроматографической системе Interchim (2 колонки 25 g), элюируя каждый раз с градиентом от 0 до 60% этилацетата в гептанах, с получением соединения 6 (34 мг, 45% выход) в виде желтовато-белого твердого вещества после сушки под вакуумом при 35°C в течение 16 часов. (LM-6-34)
Белое твердое вещество; анализ методом HPLC: 96,3% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 7,48 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=313,1 (M+H)+; C19H20O4
1H ЯМР (400 МГц, CDCl3) δ=7,14 (д, J=16,2 Гц, 1H), 7,00 (дд, J=1,5, 7,7 Гц, 1H), 6,98 (с, 1H), 6,90 (д, J=7,8 Гц, 1H), 6,85 (д, J=16,1 Гц, 1H), 6,67 (д, J=2,5 Гц, 1H), 6,36 (д, J=2,2 Гц, 1H), 5,94 (ддт, J=5,9, 10,2, 17,0 Гц, 1H), 5,66 (уш.с, 1H), 4,99 (дкв, J=1,7, 8,5 Гц, 1H), 4,96 (дкв, J=1,7, 17,1 Гц, 1H), 4,76 (уш.с, 1H), 3,93 (с, 3H), 3,79 (с, 3H), 3,48 (дт, J=1,7, 5,8 Гц, 2H).
13C ЯМР (100 МГц, CDCl3) δ=158,73, 154,70, 146,70, 145,64, 138,71, 137,25, 130,63, 130,26, 124,22, 120,43, 118,57, 114,61, 114,40, 108,56, 104,03, 98,25, 55,92, 55,80, 29,50.
Получение (E)-4-бензил-3-(4-гидрокси-3-метоксистирил)-5-метоксифенола (7) [KYN-136]
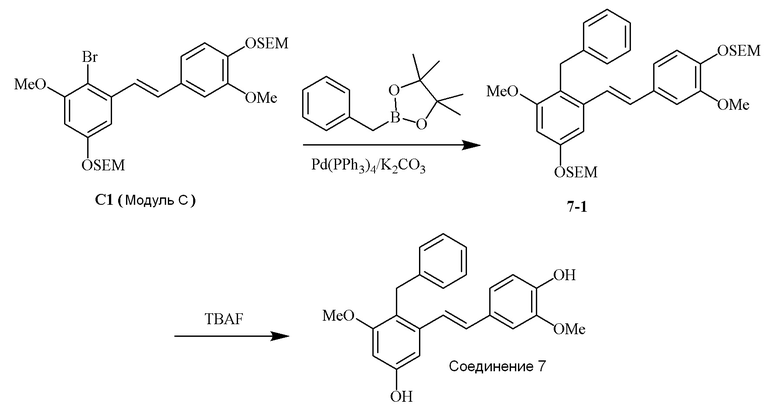
(E)-(2-((4-(2-бензил-3-метокси-5-((2-(триметилсилил)этокси)-метокси)стирил)-2-метоксифенокси)метокси)этил)триметилсилан (Соединение 7-1).
Тетракис(трифенилфосфин)палладий(0) (16 мг, 0,014 ммоль, 0,05 экв), карбонат калия (77 мг, 0,56 ммоль, 2 экв), пинаколовый эфир бензилбороновой кислоты (122 мг, 0,56 ммоль, 2 экв) и воду (5 мл) добавляли последовательно к раствору соединения C1 (170 мг, 0,28 ммоль, 1 экв) в тетрагидрофуране (5 мл). После продувки азотом в течение 10 минут, реакционную смесь нагревали при 77°C в течение 16 часов. После охлаждения до комнатной температуры, смесь экстрагировали этилацетатом (15 мл), и органический слой концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 40 g), элюируя с градиентом от 0 до 50% этилацетата в гептанах, с получением соединения 7-1 (172 мг, 99% выход) в виде желтого масла. (LM-6-35)
(E)-4-бензил-3-(4-гидрокси-3-метоксистирил)-5-метоксифенол (7) [KYN-136].
1M раствор тетрабутиламмония фторида в тетрагидрофуране (3,92 мл, 3,92 ммоль, 14 экв) добавляли к соединению 7-1 (172 мг, 0,28 ммоль, 1 экв) при комнатной температуре. После нагревания при 67°C в течение 16 часов, смесь охлаждали до комнатной температуры и разбавляли водой (4 мл). Смесь экстрагировали этилацетатом (15 мл), и органический слой концентрировали при пониженном давлении. Остаток очищали два раза на автоматизированной хроматографической системе Interchim (25 колонок 40 g), элюируя каждый раз с градиентом от 0 до 60% этилацетата в гептанах, с получением соединения 7 (37 мг, 37% выход) в виде желтовато-белого твердого вещества после сушки под вакуумом при 35°C в течение 16 часов. (LM-6-36)
Желтовато-белое твердое вещество; анализ методом HPLC: 96,4% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 8,09 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=363,1 (M+H)+; C23H22O4;
1H ЯМР (400 МГц, CDCl3) δ=7,24-7,00 (м, 2H), 7,18-7,10 (м, 3H), 7,09 (д, J=16,1 Гц, 1H), 6,89 (дд, J=1,7, 8,6 Гц, 1H), 6,85 (д, J=8,1 Гц, 1H), 6,85 (д, J= 1,7 Гц), 6,82 (д, J=15,9 Гц), 6,68 (д, J=2,5 Гц, 1H), 6,39 (д, J=2,5 Гц, 1H), 5,65 (уш.с, 1H), 4,87 (уш.с, 1H), 4,10 (с, 2H), 3,87 (с, 3H), 3,77 (с, 3H).
13C ЯМР (100 МГц, CDCl3) δ=159,03, 154,85, 146,63, 145,58, 141,66, 138,83, 130,62, 130,12, 128,22, 128,16, 125,55, 124,31, 120,60, 119,60, 114,48, 108,23, 104,06, 98,21, 55,83, 55,72, 30,92.
Получение (E)-3-Этокси-5-(4-гидрокси-3-метоксистирил)-4-(3-метилбут-2-ен-1-ил)фенола (8) [KYN-119]
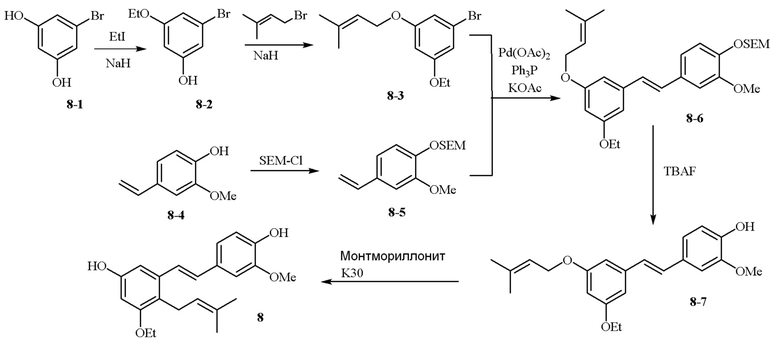
3-Бром-5-этоксифенол (8-2).
60% дисперсию гидрида натрия (2,9 г, 73,62 ммоль, 1,1 экв) в минеральном масле добавляли несколькими порциями к раствору соединения 8-1 (12,65 г, 66,93 ммоль, 1 экв) в безводном тетрагидрофуране (700 мл) при комнатной температуре. После перемешивания при комнатной температуре в течение 15 минут, добавляли этилйодид (5,38 мл, 66,93 ммоль, 1 экв). Полученный раствор нагревали при 50°C в течение 8 часов. Реакционную смесь охлаждали до комнатной температуры и переносили в делительную воронку, содержащую насыщенный водный раствор хлорида аммония (500 мл). Добавляли этилацетат (1 л), и органический слой отделяли. Водный слой экстрагировали этилацетатом (3×400 мл), и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech 120 g), элюируя с градиентом от 0 до 50% этилацетата в гептанах, с получением соединения 8-2 (4,37 г, 31% выход) в виде желтого масла. (TA-1-107)
1-Бром-3-этокси-5-((3-метилбут-2-ен-1-ил)оксибензол (8-3).
60% дисперсию гидрида натрия (1,1 г, 28,64 ммоль, 1,1 экв) в минеральном масле и 3,3-диметилаллилбромид (4,27 г, 28,6 ммоль, 1,1 экв) последовательно добавляли к раствору соединения 8-2 (5,65 г, 26,03 ммоль, 1 экв) в безводном THF (500 мл). Реакционную смесь нагревали при 50°C и перемешивали в течение ночи. Реакционную смесь охлаждали до комнатной температуры и переносили в делительную воронку, содержащую насыщенный водный раствор хлорида аммония (400 мл). Добавляли этилацетат (700 мл), и органический слой отделяли. Водный слой экстрагировали этилацетатом (3×200 мл), и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech 120 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 8-3 (6,8 г, 88% выход) в виде желтого масла. (TA-1-102).
(2-((2-Метокси-4-винилфенокси)метокси)этил)триметилсилан (8-5).
N, N-Диизопропилэтиламин (8,8 мл, 50 ммоль, 1,5 экв) и 2-(триметилсилил)этоксиметил хлорид (7,1 мл, 40 ммоль, 1,2 экв) последовательно добавляли к раствору соединения 8-4 (5 г, 33,33 ммоль, 1,0 экв) в безводном дихлорметане (50 мл) при комнатной температуре. После перемешивания при комнатной температуре в течение ночи, добавляли насыщенный водный раствор хлорида аммония (50 мл) для прерывания реакции. Водный слой экстрагировали дихлорметаном (2×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 10% этилацетата в гептанах, с получением соединения 8-5 (6,5 г, 70% выход) в виде прозрачного масла. (NK-1-45)
(E)-(2-((4-(3-Этокси-5-((3-метилбут-2-ен-1-ил)оксистирил)-2-метоксифенокси)метокси)этил)триметилсилан (8-6).
Соединение 8-5 (1,49 г, 5,32 ммоль, 1,2 экв), трифенилфосфин (110 мг, 0,443 ммоль, 0,1 экв), ацетат калия (868 мг, 8,86 ммоль, 2 экв) и ацетат палладия (50 мг, 0,221 ммоль, 0,05 экв) добавляли к раствору соединения 8-3 (1,26 г, 4,43 ммоль, 1,0 экв) в безводном толуоле (30 мл). Реакционную смесь продували азотом в течение 15 минут. После кипячения с обратным холодильником (110°C) в течение 16 часов, реакционную смесь охлаждали до комнатной температуры и фильтровали через целит (~ 5 г). Фильтрат переносили в делительную воронку, содержащую насыщенный раствор бикарбоната натрия (30 мл). Органический слой отделяли, и водный слой экстрагировали этилацетатом (2 X 20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Еще одну партию, используя соединение 3 (1,26 г), подвергали обработке аналогичным способом. Объединенный остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 8-6 (960 мг, 22% выход) в виде бледно-желтого масла. (NK-1-49)
(E)-4-(3-Этокси-5-((3-метилбут-2-ен-1-ил)оксистирил)-2-метоксифенол (8-7).
1M раствор тетрабутиламмония фторида в THF (14,0 мл, 13,88 ммоль, 7 экв) добавляли к раствору соединения 8-6 (960 мг, 1,98 ммоль, 1 экв) в безводном THF (40 мл) при комнатной температуре. Смесь кипятили с обратным холодильником в течение ночи (66°C), после чего анализ методом LCMS указывал на завершение реакции. Затем смесь охлаждали до комнатной температуры, добавляли воду (10 мл) для прерывания реакции. Летучие компоненты удаляли при пониженном давлении, затем добавляли этилацетат (15 мл). Органический слой отделяли, и водный слой экстрагировали этилацетатом (2×15 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech 12 g), элюируя с градиентом от 0 до 50% этилацетата в гептанах, с получением соединения 8-7 (503 мг, 71% выход) в виде желтого масла. (NK-1-44)
(E)-3-Этокси-5-(4-гидрокси-3-метоксистирил)-4-(3-метилбут-2-ен-1-ил)фенол (8) [KYN-119].
Порошок монтмориллонита K30 (500 мг) добавляли к раствору соединения 8-7 (500 мг, 1,41 ммоль) в безводном дихлорметане (20 мл) при комнатной температуре, и смесь перемешивали при комнатной температуре в течение ночи. Анализ методом LCMS не указывал на образование продукта. Смесь фильтровали и добавляли свежий порошок монтмориллонита K30 (500 мг), и смесь перемешивали при комнатной температуре в течение ночи, после чего анализ методом LCMS указывал на образование 30-40% требуемого соединения 8 наряду с двумя региоизомерами и непрореагировавшим исходным соединением 7 в качестве побочных продуктов. Смесь фильтровали, и твердое вещество промывали дихлорметаном (2×15 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 50% этилацетата в гептанах. Растирание с 30% дихлорметана в гептанах (10 мл) давало чистое соединение 8 [KYN-119] (60 мг, 12% выход) в виде белого твердого вещества. (NK-1-52-C)
Бежевое твердое вещество, температура плавления 133,9-134,7°C; анализ методом HPLC: 96,7% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 9,10 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=355,2 (M+H)+; C22H26O4; 1H ЯМР (400 МГц, CDCl3) δ=7,16 (д, J=16,1 Гц, 1H), 7,00 (уш.с, 1H), 6,99 (дд, J=1,9, 9,0 Гц, 1H), 6,90 (д, J=8,4 Гц, 1H), 6,84 (д, J=16,1 Гц, 1H), 6,64 (д, J=2,4 Гц, 1H), 6,33 (д, J=2,4 Гц, 1H), 5,13 (тм, J=1,4, 7,0 Гц, 1H), 3,98 (кв, J=7,0 Гц, 2H), 3,93 (с, 3H), 3,43 (д, J=7,0 Гц, 2H), 1,81 (д, J=1,0 Гц, 3H), 1,67 (д, J=1,2 Гц, 3H), 1,41 (т, J=7,0 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=157,88, 154,27, 146,68, 145,53, 138,12, 130,40, 130,34, 130,32, 124,39, 123,74, 120,89, 120,55, 114,56, 108,32, 103,86, 99,12, 63,94, 55,88, 25,78, 24,54, 18,01, 14,89.
Соединение 8 (KYN-119) может быть также синтезировано с использованием модульного метода по настоящему изобретению, применяя эквиваленты для модуля A, B и C и последующее проведение реакции перекрестного алкилирования арилгалогенида.
Получение (E)-3-(4-Гидрокси-3-метоксистирил)-4-(3-метилбут-2-ен-1-ил)-5-пропоксифенола (9) [KYN-130]
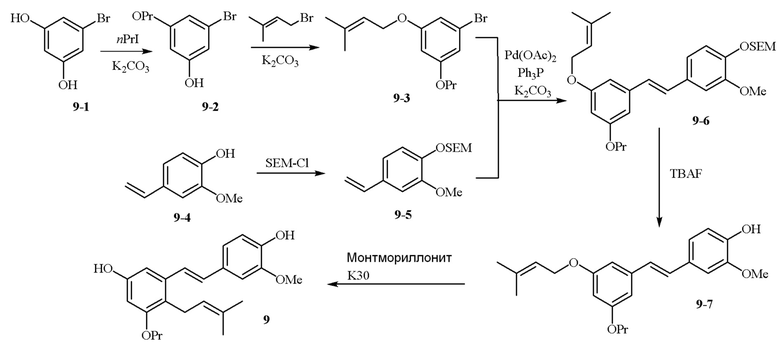
3-Бром-5-пропоксифенол (9-2).
Карбонат калия (21,9 г, 158,73 ммоль, 1,2 экв) и пропилйодид (15 мл, 158,73 ммоль, 1,2 экв) добавляли последовательно к раствору соединения 9-1 (10,0 г, 52,91 ммоль, 1 экв) в ацетонитриле (300 мл) при комнатной температуре. Полученную суспензию кипятили с обратным холодильником (80°C) в течение 16 часов. После охлаждения до комнатной температуры, реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток растворяли в этилацетате (150 мл) и переносили в делительную воронку, содержащую 1M раствор хлористоводородной кислоты (100 мл). Слои разделяли, и водный слой экстрагировали этилацетатом (2×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 40 g), элюируя с градиентом от 0 до 30% этилацетата в гептанах, с получением соединения 9-2 (11,3 г, 37% выход) в виде светло-коричневого масла (NRK-1-89).
1-Бром-3-((3-метилбут-2-ен-1-ил)окси-5-пропоксибензол (9-3).
Карбонат калия (10,1 г, 73,36 ммоль, 1,5 экв) и 3,3-диметилаллилбромид (6,8 мл, 58,70 ммоль, 1,2 экв) добавляли последовательно к раствору соединения 9-2 (11,3 г, 48,91 ммоль, 1 экв) в ацетонитриле (200 мл) при комнатной температуре. Полученную суспензию кипятили с обратным холодильником (80°C) в течение 16 часов. После охлаждения до комнатной температуры, реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением соединения 9-3 (16,0 г, 99% выход) в виде светло-коричневого масла (NRK-1-90), которое использовали на следующей стадии,
(E)-(2-((2-метокси-4-(3-((3-метилбут-2-ен-1-ил)окси-5-пропоксистирил)фенокси)метокси)этил)триметилсилан (9-6).
(2-((2-Метокси-4-винилфенокси)метокси)этил)триметилсилан (9-5, смотрите 8-5 для получения) (12,3 г, 4,15 ммоль, 1,1 экв), трифенилфосфин (2,1 г, 8,02 ммоль, 0,2 экв), карбонат калия (11 г, 80,26 ммоль, 2 экв) и ацетат палладия (910 мг, 4,01 ммоль, 0,1 экв) добавляли к раствору соединения 9-3 (12,0 г, 40,13 ммоль, 1,0 экв) в безводном толуоле (300 мл) при комнатной температуре. Реакционную смесь продували азотом в течение 15 минут. После кипячения с обратным холодильником (110°C) в течение 16 часов, реакционную смесь охлаждали до комнатной температуры и фильтровали через целит (~ 30 г). Фильтрат переносили в делительную воронку, содержащую насыщенный раствор бикарбоната натрия (200 мл). Слои разделяли, и водный слой экстрагировали этилацетатом (2×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 80 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 9-6 (6,25 г, 31% выход) в виде светло-коричневого масла. (NRK-1-91)
(E)-2-метокси-4-(3-((3-метилбут-2-ен-1-ил)окси-5-пропокси-стирил)фенол (8-7).
1M раствор тетрабутиламмония фторида в THF (139 мл, 138,59 ммоль, 7 экв) добавляли к раствору соединения 9-6 (9,86 г, 19,8 ммоль, 1 экв) в безводном THF (200 мл) при комнатной температуре. Смесь кипятили с обратным холодильником (66°C) в течение 16 часов, после чего анализ методом LCMS указывал на завершение реакции. Затем смесь охлаждали до комнатной температуры, добавляли воду (20 мл). Летучие компоненты удаляли при пониженном давлении, затем добавляли этилацетат (150 мл). Слои разделяли, и водный слой экстрагировали этилацетатом (2×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 80 g), элюируя с градиентом от 0 до 50% этилацетата в гептанах, с получением соединения 9-7 (6,66 г, 92% выход) в виде желтого твердого вещества. (NRK-1-92)
(E)-3-(4-гидрокси-3-метоксистирил)-4-(3-метилбут-2-ен-1-ил)-5-пропоксифенол (9) [KYN-130].
Порошок монтмориллонита K30 (6,66 г) добавляли к раствору соединения 8-7 (6,66 г, 18,09 ммоль) в безводном дихлорметане (200 мл) при комнатной температуре. После перемешивания при комнатной температуре в течение ночи, анализ методом LCMS указывал на образование 30-40% требуемого соединения 9 наряду с двумя региоизомерами и непрореагировавшим исходным соединением 9-7 в качестве побочных продуктов. Смесь фильтровали через целит, который промывали дихлорметаном (2×50 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 80 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах. Продукт растирали с 30% дихлорметана в гептанах (20 мл) с получением чистого соединения 9 [KYN-130] (1,02 г, 15% выход) в виде белого твердого вещества. (NRK-1-95-C)
Белое твердое вещество, температура плавления 141,6-142,8°C; анализ методом HPLC: 99,48% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 9,4 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации отрицательных ионов) m/z=367,2 (M-H)-; C23H28O4; 1H ЯМР (400 МГц, CDCl3) δ=7,16 (д, J=16,1 Гц, 1H), 7,00 (уш.с, 1H), 6,99 (дд, J=2,0, 8,2 Гц, 1H), 6,90 (д, J=8,3 Гц, 1H), 6,84 (д, J=15,9 Гц, 1H), 6,64 (д, J=2,5 Гц, 1H), 6,33 (д, J=2,4 Гц, 1H), 5,14 (тм, J=7,0 Гц, 1H), 3,93 (с, 3H), 3,88 (т, 6,4 Гц, 2H), 3,43 (д, J=6,8 Гц, 2H), 1,83 (м, J=6,4, 7,6 Гц, 2H), 1,81 (д, J=1,0 Гц, 3H), 1,67 (д, J=1,2 Гц, 3H), 1,04 (т, J=7,5 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=158,02, 154,27, 146,68, 145,57, 138,14, 130,36, 130,33, 130,31, 124,42, 123,84, 120,88, 120,55, 114,56, 108,32, 103,75, 98,97, 69,90, 55,90, 25,78, 24,57, 22,68, 18,01, 10,68.
Соединение 9 (KYN-130) может быть также синтезировано с использованием модульного метода по настоящему изобретению, применяя эквиваленты для модуля A, B и C и последующее проведение реакции перекрестного алкилирования арилгалогенида.
Получение 3-(4-Гидрокси-3-метоксистирил)-5-изопропокси-4-(3-метилбут-2-ен-1-ил)фенола (10) [KYN-131]
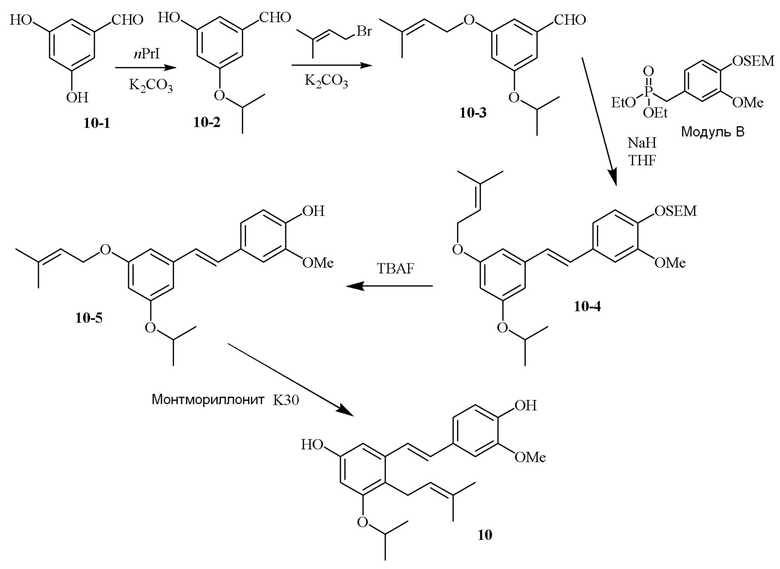
3-Гидрокси-5-изопропоксибензальдегид (10-2):
Смесь соединения 10-1 (3 г, 21,7 ммоль, 1 экв), карбоната калия (4,5 г, 32,6 ммоль, 1,5 экв) и изопропилйодида (2,1 мл, 21,7 ммоль, 1 экв) в N, N’-диметилформамиде (60 мл) нагревали при 70°C в течение 16 часов. Добавляли дополнительное количество изопропилйодида (1,05 мл, 10,9 ммоль, 0,5 экв), и продолжали проведение реакции в течение 2 часов. После охлаждения до комнатной температуры, величину pH доводили до 4 с помощью 6 N HCl. Оставшееся твердое вещество удаляли фильтрацией. Фильтрат экстрагировали этилацетатом (4×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (40 г SorbTech колонка), элюируя с градиентом от 0 до 70% этилацетата в гептанах, с получением соединения 10-2 (1,13 г, 29% выход) в виде коричневого твердого вещества. (QZH-RCI-4).
3-Изопропокси-5-((3-метилбут-2-ен-1-ил)оксибензальдегид (10-3).
Смесь соединения 10-2 (1,13 г, 6,27 ммоль, 1 экв), карбоната калия (1,73 г, 12,5 ммоль, 2 экв) и 1-бром-3-метилбут-2-ена (0,87 мл, 7,53 ммоль, 1,2 экв) в ацетоне (35 мл) кипятили с обратным холодильником в течение 16 часов. После охлаждения до комнатной температуры, реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (2×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (30 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной автоматической системе Büchi (колонка SorbTech 24 g), элюируя с градиентом от 0 до 50% этилацетата в гептанах, с получением соединения 10-3 (1,33, 86% выход) в виде желтого масла. (QZH-RCI-6).
2-((4-(3-Изопропокси-5-((3-метилбут-2-ен-1-ил)оксистирил)-2-метоксифенокси)метокси)этил)триметилсилан (10-4).
Раствор модуля B (2,17 г, 5,36 ммоль, 1 экв) в тетрагидрофуране (10 мл) добавляли к суспензии 60% дисперсии гидрида натрия в минеральном масле (0,43 г, 10,7 ммоль, 2 экв) в тетрагидрофуране (30 мл) при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 часа. Добавляли раствор соединения 10-3 (1,33 г, 5,36 ммоль, 1 экв) в тетрагидрофуране (10 мл), и реакционную смесь подогревали до комнатной температуры в течение ночи. Реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали насыщенным солевым раствором (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной автоматической системе Büchi (колонка RediSep 24 g), элюируя с градиентом от 0 до 10% этилацетата в гептанах, с получением соединения 10-4 (1,65 г, 62% выход) в виде желтого масла. (QZH-RCI-9).
4-(3-Изопропокси-5-((3-метилбут-2-ен-1-ил)оксистирил)-2-метоксифенол (10-5).
Раствор соединения 10-4 (1,65 г, 3,3 ммоль, 1 экв) и 1,0 M раствора тетрабутиламмония фторида в тетрагидрофуране (16,5 мл, 16,5 ммоль, 5 экв) в тетрагидрофуране (20 мл) кипятили с обратным холодильником в течение ночи. После охлаждения до комнатной температуры, реакционную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (50 мл), сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали на автоматизированной автоматической системе Büchi (колонка RediSep 12 g), элюируя с градиентом от 0 до 30% этилацетата в гептанах, с получением соединения 10-5 (0,85 г, 70% выход) в виде коричневого масла. (QZH-RCI-10)
3-(4-Гидрокси-3-метоксистирил)-5-изопропокси-4-(3-метилбут-2-ен-1-ил)фенол (10) [KYN-131].
Порошок монтмориллонита К10 (1 г, Sigma-Aldrich, # по каталогу 69866) добавляли к раствору соединения 10-5 (0,85 г, 2,3 ммоль, 1 экв) в безводном дихлорметане (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, после чего анализ методом LC/MS указывал на >90% конверсию исходного материала. Твердое вещество удаляли фильтрацией, и осадок на фильтре споласкивали дихлорметаном (100 мл). Фильтрат концентрировали при пониженном давлении. Остаток изначально очищали на автоматизированной автоматической системе Büchi (колонка SorbTech 25 g), элюируя с градиентом от 0 до 30% этилацетата в гептанах. Фракции, которые содержали продукт (>85% чистота, определенная методом LC/MS), собирали и снова очищали на автоматизированной хроматографической системе InterChim (колонка InterChim 25 g c силикагелем с размером частиц от 20 до 45 микрон), элюируя с градиентом от 0 до 30% этилацетата в гептанах, с получением соединения 10 [KYN-131] (75 мг, 9% выход, 99,2% чистота by HPLC) в виде желтовато-белого твердого вещества. (QZH-RCI-12).
Желтовато-белое твердое вещество; анализ методом HPLC: 99,2% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 9,2 мин; подвижная фаза: ACN/муравьиная кислота/вода; C23H28O4; 1H ЯМР (400 МГц, CDCl3) δ=7,15 (д, J=16,1 Гц, 1H), 7,00 (уш.с, 1H), 6,99 (дд, J=2,0, 8,0 Гц, 1H), 6,90 (д, J=8,3 Гц, 1H), 6,85 (д, J=16,1 Гц, 1H), 6,64 (д, J=2,4 Гц, 1H), 6,35 (д, J=2,2 Гц, 1H), 5,12 (тм, J=1,4, 7,0 Гц, 1H), 4,49 (септет, J=6,0 Гц, 1H), 3,94 (с, 3H), 3,41 (д, J=6,9 Гц, 2H), 1,82 (д, J=1,0 Гц, 3H), 1,67 (д, J=1,2 Гц, 3H), 1,34 (д, J=6,1 Гц, 6H); 13C ЯМР (100 МГц, CDCl3) δ=156,75, 154,19, 146,68, 145,57, 138,37, 130,34, 130,27, 130,23, 124,52, 123,87, 121,87, 120,55, 114,55, 108,32, 103,95, 100,46, 70,35, 55,90, 25,78, 24,69, 22,17, 18,10.
Соединение 10 (KYN-131) может быть также синтезировано с использованием модульного метода по настоящему изобретению, применяя эквиваленты для модуля A, B и C и последующее проведение реакции перекрестного алкилирования арилгалогенида.
Получение (E)-3-(4-Гидрокси-3-метоксистирил)-4-(3-метилбут-2-ен-1-ил)-5-(трифторметокси)фенола (11) [KYN-132]
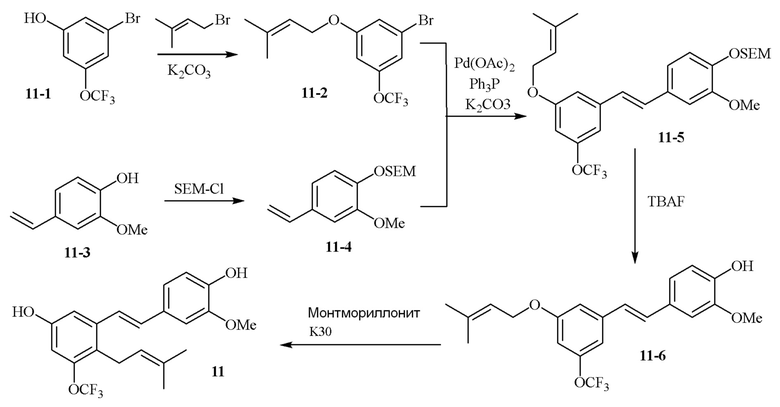
1-Бром-3-((3-метилбут-2-ен-1-ил)окси-5-(трифторметокси)-бензол (11-2).
Карбонат калия (5,36 г, 38,9 ммоль, 2 экв) и 3,3-диметил-аллилбромид (2,9 мл, 23,34 ммоль, 1,2 экв) добавляли последовательно к раствору соединения 11-1 (5,0 г, 19,45 ммоль, 1 экв) в ацетонитриле (100 мл) при комнатной температуре. Полученную суспензию кипятили с обратным холодильником (80°C) в течение 16 часов. После охлаждения до комнатной температуры, реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении. Неочищенный остаток растворяли в этилацетате (100 мл) и промывали 1M раствором хлористоводородной кислоты (2×50 мл). Органический слой сушили над сульфатом натрия и испаряли при пониженном давлении с получением соединения 11-2 (5,0 г, 79% выход) в виде бледно-желтого масла (NRK-1-96), которое использовали на следующей стадии.
(E)-(2-((2-метокси-4-(3-((3-метилбут-2-ен-1-ил)окси-5-(трифторметокси)стирил)фенокси)метокси)этил)триметилсилан (11-5).
(2-((2-Метокси-4-винилфенокси)метокси)этил)триметилсилан (11-4, смотрите 8-5 для приготовления) (4,30 г, 15,38 ммоль, 1,0 экв), трифенилфосфин (800 мг, 3,07 ммоль, 0,2 экв), карбонат калия (4,25 г, 30,76 ммоль, 2 экв) и ацетат палладия (350 мг, 1,6 ммоль, 0,1 экв) добавляли к раствору соединения 11-2 (5,0 г, 15,38 ммоль, 1,0 экв) в безводном толуоле (150 мл) при комнатной температуре. Реакционную смесь продували азотом в течение 15 минут. После кипячения с обратным холодильником (110°C) в течение 16 часов, реакционную смесь охлаждали до комнатной температуры и фильтровали через целит (~ 20 г). Фильтрат переносили в делительную воронку, содержащую насыщенный раствор бикарбоната натрия (100 мл). Слои разделяли, и водный слой экстрагировали этилацетатом (2×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 80 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 11-5 (4,89 г, 61% выход) в виде светло-коричневого масла. (NRK-1-97)
(E)-2-метокси-4-(3-((3-метилбут-2-ен-1-ил)окси-5-(трифтор-метокси)стирил)фенол (11-6).
1M раствор тетрабутиламмония фторида раствор в THF (66 мл, 65,24 ммоль, 7 экв) добавляли к раствору соединения 11-5 (4,89 г, 9,32 ммоль, 1 экв) в безводном THF (200 мл) при комнатной температуре. После кипячения с обратным холодильником (66°C) в течение 16 часов, анализ методом LCMS показывал, что реакция завершена. Смесь охлаждали до комнатной температуры и разбавляли водой (20 мл). Летучие компоненты удаляли при пониженном давлении, и остаток разбавляли этилацетатом (150 мл). Слои разделяли, и водный слой экстрагировали этилацетатом (2×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 80 g), элюируя с градиентом от 0 до 50% этилацетата в гептанах, с получением соединения 11-6 (3,2 г, 87% выход) в виде бледно-желтого масла. (NRK-1-98)
(E)-3-(4-гидрокси-3-метоксистирил)-4-(3-метилбут-2-ен-1-ил)-5-(трифторметокси)фенол (11) [KYN-132].
Порошок монтмориллонита K30 (3,2 г) добавляли к раствору соединения 11-6 (3,2 г, 8,89 ммоль) в безводном дихлорметане (100 мл) при комнатной температуре, и смесь перемешивали при комнатной температуре в течение ночи. Анализ методом LCMS указывал на образование 10-15% требуемого соединения 11 наряду с двумя региоизомерами и непрореагировавшим исходным соединением 11-6 в качестве побочных продуктов. Смесь фильтровали и обрабатывали свежим порошком монтмориллонита K30 (3,2 г) при комнатной температуре в течение еще 24 часов. Реакционную смесь фильтровали, и твердое вещество промывали дихлорметаном (2×50 мл). Объединенный фильтрат концентрировали при пониженном давлении. Остаток изначально очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 80 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением нескольких смешанных фракций, содержащих соединение 11. Фракции собирали вместе, и полученный материал очищали на автоматизированной хроматографической системе Reveleris (колонка Redisep Rf Gold HP C18, 100 g), элюируя с градиентом от 0 до 80% ацетонитрила в воде. Продукт растирали с помощью 30% дихлорметана в гептанах (10 мл) с получением чистого соединения 11 (60 мг, 1,7% выход) в виде белого твердого вещества. (NRK-1-99-C)
Белое твердое вещество, температура плавления 141,6-142,6°C; анализ методом HPLC: 98,81% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 9,8 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации отрицательных ионов) m/z=393,0 (M-H)-; C21H21F3O4; 1H ЯМР (400 МГц, CDCl3) δ=7,09 (д, J=15,9 Гц, 1H), 7,00 (дд, J=1,8, 7,1 Гц, 1H), 6,99 (уш.с, 1H), 6,99 (м, 1H), 6,92 (д, J=8,9 Гц, 1H), 6,87 (д, J=16,1 Гц, 1H), 6,69 (м, 1H), 5,07 (тм, J=6,9 Гц, 1H), 3,94 (с, 3H), 3,42 (д, J=6,9 Гц, 2H), 1,80 (с, 3H), 1,69 (с, 3H); 19F ЯМР (376 МГц, CDCl3) δ=-56,88 (с, 3F); 13C ЯМР (100 МГц, CDCl3) δ=153,95, 148,13, 146,73, 145,92, 139,81, 131,69, 131,66, 129,80, 124,31, 123,25, 122,29, 120,77, 114,64, 110,74, 108,43, 106,97, 55,90, 25,68, 25,02, 17,93.
Соединение 11 (KYN-132) может быть также синтезировано с использованием модульного метода по настоящему изобретению, применяя эквиваленты для модуля A, B и C и последующее проведение реакции перекрестного алкилирования арилгалогенида.
Получение 3-(3-Этокси-4-гидроксистирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенол (12) [KYN-133]:
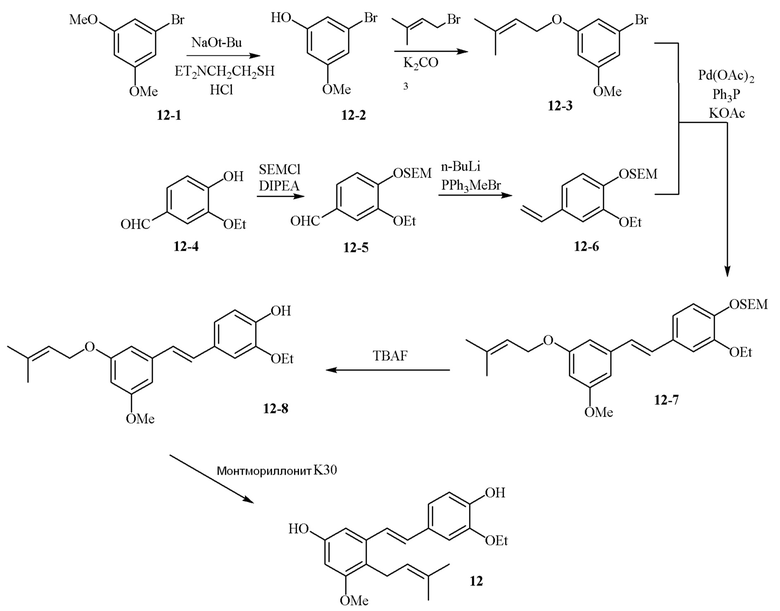
3-Бром-5-метоксифенол (12-2).
Смесь 2-(диэтиламино)этантиола гидрохлорида (46,92 г, 276,5 ммоль, 1,2 экв) в DMF (1000 мл) охлаждали на ледяной бане, и добавляли порциями третбутоксид натрия (55,3 г, 576 ммоль, 2,5 экв) при поддержании температуры ниже 25°C. После перемешивания в течение 5 минут, реакционную смесь подогревали до комнатной температуры. Через 5 минут, добавляли соединение 12-1 (50 г, 230,4 ммоль, 1,0 экв), и смесь кипятили с обратным холодильником (138°C) в течение 3 часов, после чего анализ методом LCMS показывал, что реакция завершена. Реакционную смесь охлаждали до 0°C и доводили до pH 1 с помощью 1M HCl (~1000 мл), при поддержании температуры ниже 20°C. Смесь экстрагировали этилацетатом (4×1 л), и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Дополнительное соединение 1 (25 г) обрабатывали таким же образом и объединяли с полученным выше остатком. Объединенный остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech 330 g), элюируя с градиентом от 10 до 20% этилацетата в гептанах, с получением соединения 12-2 (60 г, 86% выход) в виде светло-коричневого твердого вещества. (NK-1-5)
1-Бром-3-метокси-5-((3-метилбут-2-ен-1-ил)оксибензол (12-3).
Порошок карбоната калия (82 г, 594,20 ммоль, 2 экв) и 3,3-диметилаллилбромид (53 г, 355,7 ммоль, 1,2 экв) добавляли последовательно к раствору соединения 12-2 (60 г, 297,02 ммоль, 1,0 экв) в ацетоне (500 мл) при комнатной температуре. После кипячения с обратным холодильником (55°C) в течение ночи, реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech 220 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 12-3 (60 г, 75% выход) в виде светло-коричневого масла. (NK-1-8)
3-Этокси-4-((2-(триметилсилил)этокси)метокси)бензальдегид (12-5).
2-(Триметилсилил)этоксиметилхлорид (3,4 мл, 19 ммоль, 1,05 экв) добавляли к раствору соединения 12-4 (3 г, 18,1 ммоль, 1 экв) и N, N’-диизопропилэтиламина (9,5 мл, 54,3 ммоль, 3 экв) в дихлорметане (60 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 16 часов, после чего анализ методом LC/MS указывал на полное завершение реакции. Реакционную смесь промывали последовательно водой (100 мл), насыщенным водным раствором хлорида аммония (100 мл) и насыщенным солевым раствором (50 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 12-5 (6 г, > теоретического выхода) в виде оранжевого масла, которое использовали на следующей стадии. (QZH-RCI-34).
(2-((2-Этокси-4-винилфенокси)метокси)этил)триметилсилан (12-6).
2,5 M раствор н-бутиллития в гексане (8 мл, 20 ммоль, 1,1 экв) добавляли к смеси метилтрифенилфосфония бромида (7,1 г, 20 ммоль, 1,1 экв) в тетрагидрофуране (90 мл) при 0°C. После перемешивания в течение 30 минут, добавляли раствор соединения 12-5 (6 г, 18,1 ммоль, 1 экв) в тетрагидрофуране (20 мл) с такой скоростью, чтобы поддерживать внутреннюю температуру реакционной смеси ниже 5°C. Добавляли гексан (150 мл), и образовавшееся твердое вещество фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали на автоматизированной автоматической системе Büchi (колонка 80 g), элюируя с градиентом от 0 до 30% этилацетата в гептанах, с получением соединения 12-6 (4,43 г, 84% выход) в виде прозрачного масла. (QZH-RCI-35).
((2-((2-Этокси-4-(3-метокси-5-((3-метилбут-2-ен-1-ил)окси-стирил)фенокси)метокси)этил)триметилсилан (12-7).
Смесь соединения 12-6 (4,43 г, 15 ммоль, 1 экв), 1-бром-3-метокси-5-((3-метилбут-2-ен-1-ил)оксибензола (12-3) (4,3 г, 15,8 ммоль, 1,05 экв), ацетат палладия(II) (0,34 г, 1,5 ммоль, 0,1 экв), трифенилфосфина (0,79 г, 3 ммоль, 0,2 экв) и карбоната калия (4,14 г, 30 ммоль, 2 экв) в толуоле (130 мл) продували азотом в течение десяти минут. После кипячения с обратным холодильником в течение 16 часов, реакционную смесь охлаждали до комнатной температуры и разбавляли гептанами (150 мл). Твердые вещества удаляли фильтрацией и отбрасывали. Фильтрат концентрировали при пониженном давлении, и остаток очищали на автоматизированной автоматической системе Büchi (колонка 120 g), элюируя с градиентом от 0 до 25% этилацетата в гептанах, с получением соединения 12-7 (3,18 г, 44% выход) в виде коричневого масла. (QZH-RCI-37).
2-Этокси-4-(3-метокси-5-((3-метилбут-2-ен-1-ил)оксистирил)-фенол (12-8).
Раствор соединения 12-7 (3,18 г, 6,56 ммоль, 1 экв) и 1,0 M раствора тетрабутиламмония фторида в тетрагидрофуране (45,9 мл, 45,9 ммоль, 7 экв) в тетрагидрофуране (32 мл) кипятили с обратным холодильником в течение ночи. После охлаждения до комнатной температуры, добавляли воду (100 мл) и насыщенный водный раствор хлорида аммония (200 мл). Реакционную смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (50 мл), сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали на автоматизированной автоматической системе Büchi (колонка 24 g), элюируя с градиентом от 0 до 35% этилацетата в гептанах, с получением соединения 12-8 (1,76 г, 76% выход) в виде желтого масла. (QZH-RCI-38).
3-(3-Этокси-4-гидроксистирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенол (12) (KYN-133).
Порошок монтмориллонита К10 (1,76 г, Sigma-Aldrich, # по каталогу 69866) добавляли к раствору соединения 12-8 (1,76 г, 4,96 ммоль, 1 экв) в безводном дихлорметане (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, после чего анализ методом LC/MS указывал на >90% конверсию исходного материала. Реакционную смесь непосредственно абсорбировали на целите (25 г) и очищали на автоматизированной автоматической системе Büchi (колонка 80 g), элюируя с градиентом от 0 до 35% этилацетата в гептанах, с получением соединения 12 [KYN-133] (0,17 г, 10% выход, 99,4% чистота by HPLC%) в виде желтовато-белого твердого вещества.(QZH-RCI-38).
Желтовато-белое твердое вещество, температура плавления 126,6-129,1°C; анализ методом HPLC: 99,4% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 9,1 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=355,2 (M+H)+; C22H26O4; 1H ЯМР (400 МГц, CDCl3) δ=7,12 (д, J=16,2 Гц, 1H), 6,98 (с, 1H), 6,97 (дд, J=2,0, 8,5 Гц, 1H), 6,90 (дм, J=8,6 Гц, 1H), 6,82 (д, J=15,9 Гц, 1H), 6,64 (д, J=2,4 Гц, 1H), 6,34 (д, J=2,4 Гц, 1H), 5,74 (с, 1H), 5,13 (тм, J=1,4, 6,9 Гц, 1H), 4,85 (с, 1H), 4,14 (кв, J=6,9 Гц, 2H), 3,78 (с, 3H), 3,40 (д, J=6,9 Гц, 2H), 1,80 (д, J=0,8 Гц, 3H), 1,67 (д, J=1,2 Гц, 3H), 1,46 (т, J=7,1 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=158,54, 154,34, 145,93, 145,65, 138,17, 130,58, 130,46, 130,24, 124,19, 123,66, 120,71, 120,44, 114,50, 109,28, 103,94, 98,23, 64,49, 55,68, 25,74, 24,45, 17,93, 14,87.
Соединение 12 (KYN-133) может быть также синтезировано с использованием модульного метода по настоящему изобретению, применяя эквиваленты для модуля A, B и C и последующее проведение реакции перекрестного алкилирования арилгалогенида.
Получение (E)-3-(3-Этил-4-гидроксистирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенола (13) [KYN-114]
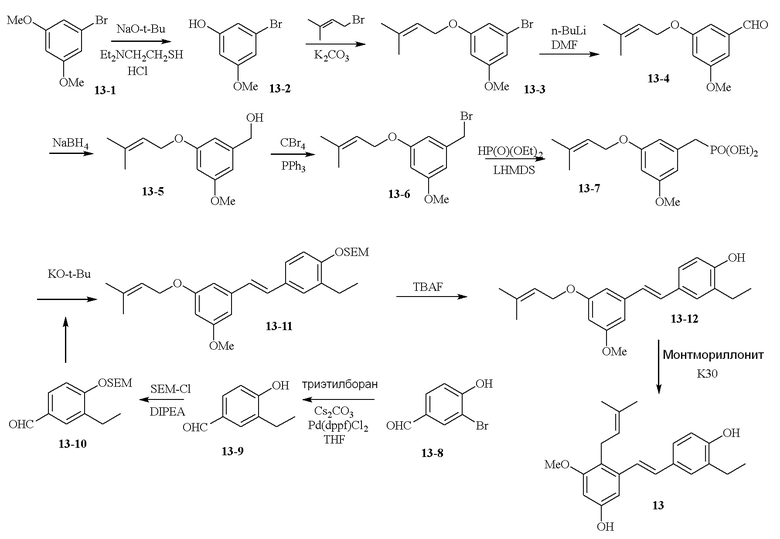
3-Бром-5-метоксифенол (13-2).
Третбутоксид натрия (332,05 г, 3455,3 ммоль, 2,5 экв) добавляли порциями при поддержании температуры ниже 25°C к смеси 2-(диэтиламино)этантиола гидрохлорида (281,48 г, 1658,5 ммоль, 1,2 экв) в DMF (3000 мл) на ледяной бане. Через 5 минут, реакционную смесь подогревали до комнатной температуры и перемешивали в течение еще 5 минут. Добавляли соединение 13-1 (300 г, 1382,1 ммоль, 1,0 экв), и смесь кипятили с обратным холодильником (138°C) в течение 3 часов, после чего анализ методом LCMS указывал на полное завершение реакции. После охлаждения до комнатной температуры, смесь охлаждали на ледяной бане и добавляли 1M HCl (~3000 мл) для доведения до pH 1. Смесь экстрагировали этилацетатом (3×5 л), и объединенные органические слои промывали насыщенным солевым раствором (4 л), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на силикагеле (2,6 кг), элюируя с градиентом от 0 до 25% этилацетата в гептанах, с получением соединения 13-2 (257,1 г, 92% выход) в виде коричневого твердого вещества. (GB-27-172)
1-Бром-3-метокси-5-((3-метилбут-2-ен-1-ил)оксибензол (13-3).
Порошок карбоната калия (350,0 г, 2532,5 ммоль, 2 экв) и 3,3-диметилаллилбромид (226,45 г, 1519,5 ммоль, 1,2 экв) добавляли последовательно к раствору соединения 13-2 (257,1 г, 1266,2 ммоль, 1,0 экв) в ацетоне (7500 мл) при комнатной температуре. После кипячения с обратным холодильником в течение ночи, реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали на колонке Biotage-150, элюируя с градиентом от 0 до 10% этилацетата в гептанах, с получением соединения 13-3 (358,0 г, >100% выход) в виде коричневого масла. (GB-27-174)
3-Метокси-5-((3-метилбут-2-ен-1-ил)оксибензальдегид (13-4). 2,5 M раствор н-бутиллития в гексанах (557 мл, 1392,6 ммоль, 1,1 экв) добавляли по каплям, при поддержании температуры ниже -70°C, к раствору соединения 13-3 (358,0 г, ~1266,2 ммоль, 1,0 экв) в безводном THF (7000 мл) при -75°C. После перемешивания при -75°C в течение 30 минут, добавляли по каплям безводный DMF (147 мл, 1899,0 ммоль, 1,5 экв) при поддержании температуры ниже -70°C. Полученную смесь медленно подогревали до комнатной температуры в течение ночи. Добавляли ледяную воду (500 мл) для прерывания реакции. Смесь разбавляли этилацетатом (5 л), и слои разделяли. Органический слой промывали насыщенным солевым раствором (500 мл). Объединенные водные слои экстрагировали этилацетатом (2×500 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на колонке Biotage-150, элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 13-4 (221,5 г, 79% суммарный выход за две стадии) в виде желтого масла. (GB-27-175)
(3-Метокси-5-((3-метилбут-2-ен-1-ил)оксифенил)метанол (13-5).
Боргидрид натрия (38,04 г, 1005,6 ммоль, 1,0 экв) добавляли порциями, при поддержании температуры ниже 10°C, к раствору соединения 13-4 (221,5 г, 1005,6 ммоль, 1,0 экв) в метаноле (3500 мл) при 0°C. После перемешивания при 5-10°C в течение 1 часа, добавляли воду (200 мл) для прерывания реакции. Смесь концентрировали при пониженном давлении с удалением большей части метанола. Остаток разбавляли этилацетатом (2,5 л) и промывали насыщенным солевым раствором (600 мл). Водный слой экстрагировали этилацетатом (2×500 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 13-5 (218,61 г, 98% выход) в виде желтого масла, которое использовали на следующей стадии. (GB-27-178).
1-(Бромметил)-3-метокси-5-((3-метилбут-2-ен-1-ил)оксибензол (13-6).
Тетрабромид углерода (358,76 г, 1081,84 ммоль, 1,1 экв) добавляли к раствору соединения 13-5 (218,61 г, 983,49 ммоль, 1,0 экв) в дихлорметане (4400 мл) при комнатной температуре. Полученную смесь охлаждали до 0°C, и добавляли порциями трифенилфосфин (283,76 г, 1081,84 ммоль, 1,1 экв). После перемешивания при комнатной температуре в течение 4 часов, смесь концентрировали при пониженном давлении. Остаток очищали на колонке Biotage-150, элюируя с градиентом от 0 до 10% этилацетата в гептанах, с получением соединения 13-6 (273,76 г, 97% выход) в виде желтого масла. (GB-27-181) Примечание: соединение 13-6 проявляет нестабильность при очистке на некоторых предварительно загруженных силикагелем колонках.
Диэтил (3-метокси-5-((3-метилбут-2-ен-1-ил)оксибензил)-фосфонат (13-7).
1M раствор гексаметилдисилилазида лития в THF (6,57 мл, 6,57 ммоль, 1,8 экв) добавляли к раствору диэтилфосфита (0,94 мл, 7,3 ммоль, 2,0 экв) в безводном THF (20 мл) при 0°C. После перемешивания при 0°C в течение 30 минут, добавляли раствор соединения 13-6 (1,04 г, 3,65 ммоль, 1,0 экв) в безводном THF (10 мл), и смесь перемешивали при комнатной температуре в течение ночи. Реакцию прерывали водой (30 мл), и смесь промывали насыщенным солевым раствором (30 мл). Водный слой экстрагировали этилацетатом (2×60 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 400 g), элюируя с градиентом от 0 до 10% метанола в дихлорметане, с получением соединения 13-7 (1,10 г, 88% выход) в виде коричневого масла. (LM-1-41)
3-Этил-4-гидроксибензальдегид (13-9).
[1,1′-Бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (2,18 г, 2,98 ммоль, 0,05 экв) и карбонат цезия (38,9 г, 119,4 ммоль, 2 экв) добавляли к раствору 3-бром-4-гидроксилбензальдегида (13-8) (12 г, 59,7 ммоль, 1 экв) в безводном THF (150 мл). Добавляли 1M раствор триэтилборана в THF (119,4 мл, 119,4 ммоль, 1,8 экв), и смесь кипятили с обратным холодильником (66°C) в течение ночи. После охлаждения до комнатной температуры, реакционную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (2×250 мл). Объединенные органические слои промывали насыщенным солевым раствором (200 мл), сушили над сульфатом натрия и концентрировали при пониженном давлении. Неочищенный остаток очищали первоначально на автоматизированной хроматографической системе InterChim (колонка 220 g), элюируя с градиентом от 0 до 40% этилацетата в гептанах. Вторая очистка на автоматизированной хроматографической системе InterChim (колонка 80 g), с элюированием с градиентом от 0 до 40% этилацетата в гептанах, давала соединение 13-9 (5,21 г, 58% выход) в виде коричневого масла. (LM-1-2).
3-Этил-4-((2-(триметилсилил)этокси)метокси)бензальдегид (13-10).
2-(Триметилсилил)этоксиметилхлорид (6,1 мл, 34,52 ммоль, 1,1 экв) добавляли к раствору соединения 13-9 (4,71 г, 31,38 ммоль, 1 экв) в безводном дихлорметане (120 мл). Добавляли диизопропилэтиламин (8,2 мл, 47,07 ммоль, 1,5 экв), и смесь перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь гасили насыщенным раствором бикарбоната натрия (50 мл) и экстрагировали этилацетатом (2×100 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении. Неочищенный остаток очищали изначальна на автоматизированной хроматографической системе InterChim (колонка 220 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах. Вторая очистка на автоматизированной хроматографической системе InterChim (колонка 120 g), c элюированием с градиентом от 0 до 20% этилацетата в гептанах, давала соединение 13-10 (6,77 г, 77% выход) в виде прозрачного масла. (LM-1-4)
(E)-(2-((2-Этил-4-(3-метокси-5-((3-метилбут-2-ен-1-ил)оксистирил)фенокси)метокси)этил)триметилсилан (13-11).
1,0 M раствор третбутоксида калия в THF (11,7 мл, 11,68 ммоль, 2 экв) добавляли по каплям к охлажденному раствору диэтил (3-метокси-5-((3-метилбут-2-ен-1-ил)оксибензил)фосфоната (13-7) (2 г, 5,84 ммоль, 1 экв) в безводном THF (20 мл) при 0°C. После перемешивания в течение 15 минут при комнатной температуре, добавляли по каплям раствор соединения 9 (2,5 г, 8,76 ммоль, 1,5 экв) в безводном THF (5 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем гасили насыщенным солевым раствором (50 мл) и экстрагировали этилацетатом (200 мл). Органический слой сушили над сульфатом натрия и концентрировали при пониженном давлении. Неочищенный остаток очищали на автоматизированной хроматографической системе InterChim (колонка 80 g), элюируя с градиентом от 0 до 30% этилацетата в гептанах, с получением соединения 13-11 (1,32 г, 48% выход) в виде прозрачного масла. (CSK-2-97)
(E)-2-Этил-4-(3-метокси-5-((3-метилбут-2-ен-1-ил)оксистирил)фенол (13-12).
1,0 M раствор тетрабутиламмония фторида в THF (19,7 мл, 19,7 ммоль, 7 экв) добавляли к раствору соединения 13-11 (1,32 г, 2,81 ммоль, 1 экв) в безводном THF (20 мл), и полученную смесь кипятили с обратным холодильником в течение ночи. Реакционную смесь охлаждали до комнатной температуры и разбавляли водой (25 мл). Смесь экстрагировали этилацетатом (100 мл). Органический слой промывали насыщенным солевым раствором (25 мл), сушили над сульфатом натрия и концентрировали при пониженном давлении. Неочищенный остаток очищали изначально на автоматизированной хроматографической системе InterChim (колонка 80 g), элюируя с градиентом от 0 до 100% этилацетата в гептанах. Вторая очистка на автоматизированной хроматографической системе InterChim (колонка 40 g), c элюированием с градиентом от 0 до 30% этилацетата в гептанах, давала соединение 13-12 (770 мг, 80% выход) в виде вязкого прозрачного масла. (CSK-2-98)
(E)-3-(3-Этил-4-гидроксистирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенол (13) [KYN-114].
Монтмориллонит K30 (620 мг) добавляли к раствору соединения 13-12 (620 мг, 1,83 ммоль, 1 экв) в безводном дихлорметане (12 мл), и полученную смесь перемешивали при комнатной температуре в течение ночи. Смесь фильтровали через слой целита, который споласкивали дихлорметаном (50 мл). Фильтрат концентрировали при пониженном давлении. Неочищенный остаток очищали на автоматизированной хроматографической системе InterChim (колонка 25 g), элюируя с градиентом от 0 до 30% этилацетата в гептанах, с получением соединения 13 [KYN-114] (120 мг, 19% выход) в виде желтовато-белого твердого вещества. (CSK-2-99)
1H ЯМР (400 МГц, метанол-d3) δ=7,22 (д, J=2,2 Гц, 1H), 7,13 (дд, J=2,0, 8,3 Гц, 1H), 7,11 (д, J=16,1 Гц, 1H), 6,83 (д, J=16,1 Гц, 1H), 6,72 (д, J=8,3 Гц, 1H), 6,63 (д, J=2,2 Гц, 1H), 6,33 (д, J=2,2 Гц, 1H), 5,06 (септет, J=1,4, 6,8 Гц, 1H), 3,77 (с, 3H), 3,37 (уш.д, J=6,6 Гц, 2H), 2,62 (кв, J=7,5 Гц, 2H), 1,80 (д, J=1,0 Гц, 3H), 1,67 (д, J=1,2 Гц, 3H), 1,21 (т, J=7,5 Гц, 3H); 13C ЯМР (100 МГц, метанол-d3) δ=159,69, 157,12, 156,09, 139,51, 131,92, 131,12, 130,70, 128,30, 126,20, 125,49, 124,54, 115,95, 104,67, 98,95, 55,96, 25,87, 25,09, 24,26, 18,06, 14,68.
Соединение KYN-114 может быть также синтезировано с использованием модульного метода по настоящему изобретению, применяя эквиваленты для модуля A, B и C и последующее проведение реакции перекрестного алкилирования арилгалогенида.
Получение (E)-3-(4-гидрокси-3-(метилтио)стирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенола (14) [KYN-118]
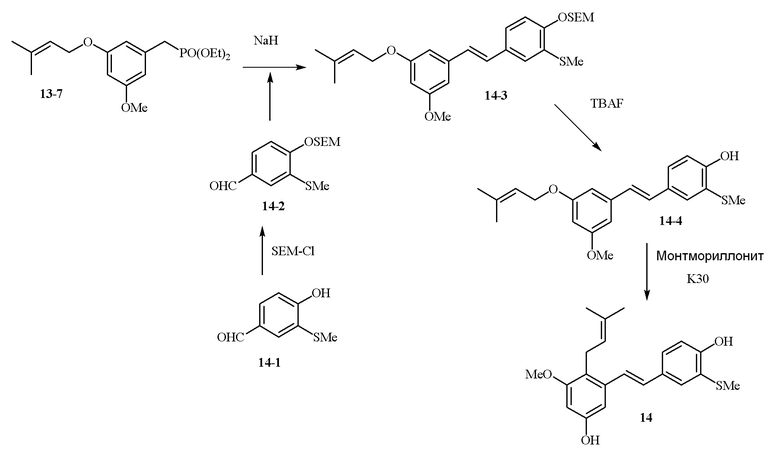
3-(Метилтио)-4-((2-(триметилсилил)этокси)метокси)-бензальдегид (14-2).
N, N-Диизопропилэтиламин (0,78 мл, 4,46 ммоль, 1,5 экв) и 2-(триметилсилил)этоксиметилхлорид (0,63 мл, 3,57 ммоль, 1,2 экв) добавляли последовательно к раствору соединения 14-1 (0,50 г, 2,97 ммоль, 1,0 экв) в безводном дихлорметане (30 мл) при комнатной температуре и перемешивали при комнатной температуре в течение ночи. Объединяли с другой реакционной смесью, полученной реакцией с равной загрузкой, и гасили насыщенным раствором бикарбоната натрия (50 мл). Слои разделяли, и водный слой экстрагировали дихлорметаном (2×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 80 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 14-2 (1,67 г, 94% выход) в виде бледно-желтого масла. (LM-1-39 and LM-1-40)
(E)-(2-((4-(3-Метокси-5-((3-метилбут-2-ен-1-ил)оксистирил)-2-(метилтио)фенокси)метокси)этил)триметилсилан (14-3).
Раствор соединения 13-7 (1,1 г, 3,2 ммоль, 1,0 экв) в безводном THF (10 мл) добавляли по каплям к 60% дисперсии гидрида натрия в минеральном масле (257 мг, 6,4 ммоль, 2,0 экв) в безводном THF (20 мл) при 0°C. Смесь подогревали до комнатной температуры и перемешивали в течение 30 минут. Затем добавляли по каплям раствор соединения 14,2 (0,955 г, 3,2 ммоль, 1,0 экв) в безводном THF (10 мл), и смесь перемешивали при комнатной температуре в течение ночи. Реакцию прерывали водой (30 мл), и смесь промывали насыщенным солевым раствором (70 мл). Водный слой экстрагировали этилацетатом (2×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 40 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 14-3 (1,01 г, 79% выход в расчете на 0,17 г извлеченного исходного соединения 14,2) в виде желтого масла. (LM-1-42)
(E)-4-(3-Метокси-5-((3-метилбут-2-ен-1-ил)оксистирил)-2-(метилтио)фенол (14,4).
1 M раствор тетрабутиламмония фторида в THF (7 мл, 7 ммоль, 7 экв) добавляли к раствору соединения 14,3 (0,486 г, 1,0 ммоль, 1,0 экв) в безводном THF (10 мл) при комнатной температуре. После кипячения с обратным холодильником в течение ночи, смесь охлаждали до комнатной температуры и разбавляли водой (20 мл) и этилацетатом (50 мл). Слои разделяли, и органический слой промывали насыщенным солевым раствором (30 мл). Объединенные водные слои экстрагировали этилацетатом (2×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 25 g), элюируя с градиентом от 0 до 30% этилацетата в гептанах, с получением соединения 14,4 (0,25 г, 70% выход) в виде коричневого масла. (LM-1-43)
(E)-3-(4-Гидрокси-3-(метилтио)стирил)-5-метокси-4-(3-метил-бут-2-ен-1-ил)фенол (14).
Порошок монтмориллонита K30 (Aldrich # 69866, 0,25 г) добавляли к раствору соединения 14,4 (0,25 г, 0,7 ммоль) в безводном дихлорметане (15 мл) при комнатной температуре, и смесь перемешивали при комнатной температуре в течение ночи, после чего анализ методом LCMS указывал на завершение реакции. Смесь фильтровали, и твердое вещество споласкивали дихлорметаном (2×15 мл). Объединенный фильтрат концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 25 g, 20-45 мкм), элюируя с градиентом от 0 до 40% этилацетата в гептанах, с получением смеси соединений 14A, 14B и14 (0,18 г, 70% суммарный выход). (LM-1-44)
Дополнительное соединение 14-4 (0,27 г) из 11 использованных обрабатывали таким же образом и получали смесь соединений 14A, 14B и 14 (0,22 г, 81% суммарный выход). (LM-1-46)
Неочищенный соединения 14A, 14B и 14 (LM-1-44 и LM-1-46) затем очищали на автоматизированной хроматографической системе Interchim (колонка 25 g, 20-45 мкм), элюируя с градиентом от 0 до 40% этилацетата в гептанах, с получением соединения 14B (50 мг, 10% выход) в виде бледно-желтого твердого вещества, соединения 14B (100 мг, 19% выход) в виде бледно-желтого твердого вещества и соединения 14 (170 мг, 33% выход) в виде белого твердого вещества (LM-1-54A, LM-1-54B и LM-1-54C). Примечание: структуры этих трех соединений определяли методом ЯМР в Университете штата Флорида США (University of Florida).
Соединение 14 может быть также синтезировано с использованием модульного метода по настоящему изобретению, применяя эквиваленты для модуля A, B и C и последующее проведение реакции перекрестного алкилирования арилгалогенида.
(E)-3-(4-Гидрокси-3-(метилтио)стирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенол (14) [KYN-118].
Белое твердое вещество, температура плавления 124,5-125,2°C; анализ методом HPLC: 98,9% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 9,00 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=357,1 (M+H)+; C21H24O3S; 1H ЯМР (400 МГц, CDCl3) δ=7,60 (д, J=2,2 Гц, 1H), 7,37 (дд, J=2,2, 8,3 Гц, 1H), 7,17 (д, J=16,2 Гц, 1H), 6,98 (д, J=8,3 Гц, 1H), 6,82 (д, J=16,1 Гц, 1H), 6,66 (с, 1H), 6,65 (д, J=2,5 Гц, 1H), 6,36 (д, J=2,5 Гц, 1H), 5,11 (септет, J=1,3, 6,8 Гц, 1H), 4,71 (уш.с, 1H), 3,80 (с, 3H), 3,41 (уш.д, J=6,8 Гц, 2H), 2,36 (с, 3H), 1,81 (д, J=1,0 Гц, 3H), 1,68 (д, J=1,2 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=158,61, 155,89, 154,39, 138,04, 132,98, 131,00, 130,83, 129,29, 128,98, 125,01, 123,58, 121,37, 120,92, 115,13, 103,93, 98,39, 55,74, 25,79, 24,47, 19,90, 17,98.
(E)-3-(4-Гидрокси-3-(метиламино)стирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенол (15) [KYN-158]
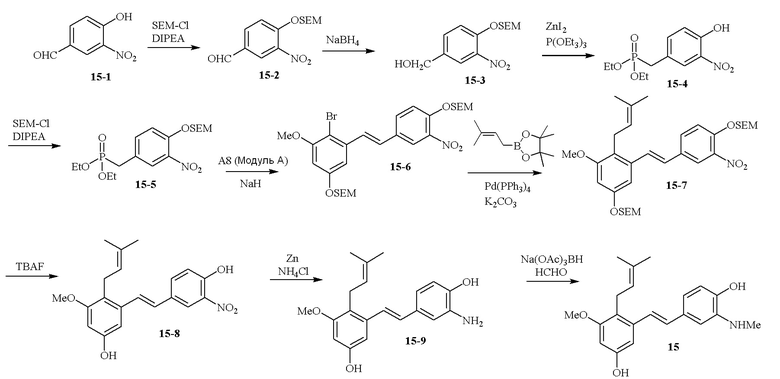
3-Нитро-4-((2-(триметилсилил)этокси)метокси)бензальдегид (15-2).
2-(Триметилсилил)этоксиметил хлорид (17,9 г,166,70 ммоль, 1,2 экв) и N, N-диизопропилэтиламин (24,0 мл, 134,73 ммоль, 1,5 экв) последовательно добавляли к раствору соединения 15-1 (15,0 г, 89,82 ммоль, 1,0 экв) в дихлорметане (150 мл) при комнатной температуре. После перемешивания в течение 16 часов, реакционную смесь разбавляли насыщенным раствором бикарбоната натрия (150 мл). Слои разделяли, и водный слой экстрагировали дихлорметаном (2×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 80 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 15-2 (23,5 г, 88% выход) в виде желтого масла (NRK-1-179).
(3-Нитро-4-((2-(триметилсилил)этокси)метокси)фенил)метанол (15-3).
Боргидрид натрия (2,92 г, 79,12 ммоль, 1,0 экв) добавляли двумя порциями к раствору соединения 15-2 (23,5 г, 79,12 ммоль, 1,0 экв) в метаноле при 0°C. Полученный раствор перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь гасили водой (20 мл). Летучие компоненты удаляли при пониженном давлении, и полученный неочищенный остаток растворяли в этилацетате (150 мл) и промывали насыщенным водным раствором хлорида аммония (100 мл). Водный слой экстрагировали этилацетатом (2×100 мл). Объединенные органические слои сушили над сульфатом натрия и испаряли при пониженном давлении с получением соединения 15-3 (23,7 г, 99% выход) в виде оранжевого масла, которое использовали на следующей стадии. (NRK-1-120)
Диэтил (4-гидрокси-3-нитробензил)фосфонат (15-4).
Йодид цинка (21,31 г, 66,80 ммоль, 2,0 экв) добавляли к триэтилфосфиту (11,5 мл, 66,80 ммоль, 2,0 экв). После перемешивания в течение 15 минут при комнатной температуре, добавляли раствор соединения 15-3 (10,0 г, 33,40 ммоль, 1,0 экв) в безводном тетрагидрофуране (100 мл). Полученный раствор кипятили с обратным холодильником (66°C) в течение 4 часов. После охлаждения до комнатной температуры, летучие компоненты удаляли при пониженном давлении. Неочищенный остаток растворяли в этилацетате (150 мл) и промывали 1M раствором гидроксида натрия (150 мл). Водный слой нейтрализовывали с помощью 1M HCL (250 мл). Полученный водный слой экстрагировали этилацетатом (3×200 мл). Объединенные органические слои сушили над сульфатом натрия и испаряли при пониженном давлении с получением соединения 15-4 (7,12 г, 76% выход) в виде желтого масла, которое использовали на следующей стадии. (NRK-1-187)
Диэтил (3-нитро-4-((2-(триметилсилил)этокси)метокси)бензил)-фосфонат (15-5).
2-(Триметилсилил)этоксиметилхлорид (4,1 г,24,63 ммоль, 1,0 экв) и N, N-диизопропилэтиламин (6,5 мл, 36,94 ммоль, 1,5 экв) последовательно добавляли к раствору соединения 15-4 (7,12 г, 24,63 ммоль, 1,0 экв) в дихлорметане (100 мл) при комнатной температуре. После перемешивания в течение 20 часов, реакционную смесь разбавляли насыщенным раствором бикарбоната натрия (100 мл). Слои разделяли, и водный слой экстрагировали дихлорметаном (2×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 15-5 (9,45 г, 92% выход) в виде желтого масла, которое использовали на следующей стадии. (NRK-1-189)
(E)-(2-((4-(2-Бром-3-метокси-5-((2-(триметилсилил)этокси)-метокси)стирил)-2-нитрофенокси)метокси)этил)триметилсилан (15-6).
60% дисперсию гидрида натрия в минеральном масле (3,6 г, 90,24 ммоль, 4 экв) добавляли 5 порциями к раствору соединения 15-5 (9,45 г, 22,56 ммоль, 1,0 экв) в безводном тетрагидрофуране (100 мл) при 0°C. Смесь подогревали до комнатной температуры и перемешивали в течение 30 минут. Добавляли по каплям раствор A8 (модуль A) (8,15 г, 22,56 ммоль, 1 экв) в безводном тетрагидрофуране (20 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию осторожно прерывали насыщенным солевым раствором (20 мл, 1 капля в минуту для первых 5 мл солевого раствора) при 0°C. Летучие компоненты удаляли при пониженном давлении. Добавляли насыщенный водный раствор хлорида аммония (100 мл). Смесь экстрагировали этилацетатом (2×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток сначала очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 330 g), элюируя с градиентом от 0 до 10% этилацетата в гептанах. Продукт затем очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 330 g), элюируя с градиентом от 0 до 10% этилацетата в гептанах, с получением соединения 15-6 (1,63 г, 12% выход) в виде желтого масла (NRK-1-190).
(E)-(2-((4-(3-Метокси-2-(3-метилбут-2-ен-1-ил)-5-((2-(три-метилсилил)этокси)метокси)стирил)-2-нитрофенокси)метокси)этил)-триметилсилан (15-7).
Тетракис(трифенилфосфин)палладий(0) (300 мг, 0,26 ммоль, 0,1 экв), карбонат калия (710 мг, 5,2 ммоль, 2 экв), пинаколовый эфир 3-метилбут-2-енилбороновой кислоты (1,01 г, 5,2 ммоль, 2 экв) и воду (2 мл) добавляли последовательно к раствору соединения 15-6 (1,6 г, 2,60 ммоль, 1 экв) в 1,4-диоксане (20 мл) в герметизированной пробирке. После продувки азотом в течение 10 минут, реакционную смесь нагревали при 100°C в течение 16 часов. После охлаждения до комнатной температуры, реакционную смесь фильтровали через целит. Фильтрат испаряли при пониженном давлении. Остаток суспендировали в насыщенном растворе бикарбоната натрия (100 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 80 g), элюируя с градиентом от 0 до 10% этилацетата в гептанах, с получением соединения 15-7 (1,8 г, >100% выход) в виде оранжевого масла. (NRK-6-25)
(E)-3-(4-Гидрокси-3-нитростирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенол (15-8).
1M раствор тетрабутиламмония фторида в тетрагидрофуране (41 мл, 40,92 ммоль, 14 экв) добавляли к раствору соединения 15-7 (1,8 г, 2,92 ммоль, 1 экв) в тетрагидрофуране (20 мл) при комнатной температуре. После нагревания при 67°C в течение 16 часов, смесь охлаждали до комнатной температуры и разбавляли водой (20 мл) и насыщенным водным раствором хлорида аммония (100 мл). Летучие компоненты удаляли при пониженном давлении, и оставшийся водный слой экстрагировали этилацетатом (2×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 80 g), элюируя с градиентом от 0 до 80% этилацетата в гептанах, с получением соединения 15-8 (370 мг, 36% выход) в виде желтого твердого вещества (NRK-6-26).
(E)-3-(3-Амино-4-гидроксистирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенол (15-9).
Порошок активированного цинка (680 мг, 10,4 ммоль, 10,0 экв), хлорид аммония (560 мг, 10,4 ммоль, 10,0 экв) и воду (6 мл) добавляли последовательно к раствору соединения 15-8 (370 мг, 1,04 ммоль, 1,0 экв) в тетрагидрофуране (20 мл) при комнатной температуре. Полученную суспензию перемешивали при комнатной температуре в течение 16 часов. Суспензию фильтровали через целит, и твердые вещества промывали этилацетатом (20 мл). Фильтрат испаряли досуха при пониженном давлении. Остаток суспендировали в насыщенном водном растворе хлорида аммония (50 мл) и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 15-9 (270 мг, 81% выход) в виде желтовато-белого твердого вещества, которое использовали на следующей стадии (NRK-6-32).
(E)-3-(4-Гидрокси-3-(метиламино)стирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенол (15).
37 масс.% водный раствор формальдегида (52 мкл, 0,62 ммоль. 2,0 экв) и триацетоксиборгидрид натрия (262 мг, 1,24 ммоль, 4,0 экв) последовательно добавляли к раствору соединения 15-9 (100 мг, 0,31 ммоль, 1,0 экв) в дихлорметане (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли дихлорметаном (20 мл) и промывали насыщенным раствором бикарбоната натрия (15 мл). Водный слой экстрагировали дихлорметаном (2×10 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток первоначально очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 100% этилацетата в гептанах, с получением соединения 15 [KYN-158] (52 мг, 52% выход) в виде желтовато-белого твердого вещества (NRK-6-40).
Примечание: в пробном синтезе, соединение 15-9 (50 мг) превращали в соединение 15, используя описанные выше условия реакции. Очистка неочищенного соединения 15, полученного в этой реакции, на автоматизированной хроматографической системе Reveleris (колонка Redisep Rf Gold HP C18, 50 g), c элюированием с градиентом от 0 до 80% ацетонитрила в воде, приводила к образованию примеси (с массой m/z=2M-1). Анализ методом LCMS показывал, что относительное содержание этой примеси превышает 15%, увеличиваясь с течением времени. Аналогичный результат был получен при выдерживании раствор соединения 15 в метаноле в течение 16 часов, что позволяет предположить, что соединение 15 может быть нестабильным в растворе.
(E)-3-(3-(Диметиламино)-4-гидроксистирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенол (16) [KYN-157]
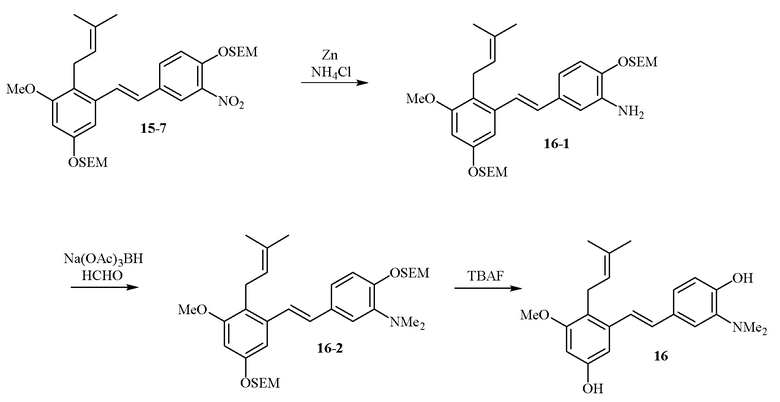
(E)-5-(3-Метокси-2-(3-метилбут-2-ен-1-ил)-5-((2-(триметил-силил)этокси)метокси)стирил)-2-((2-(триметилсилил)этокси)-метокси)анилин) (16-1).
Порошок активированного цинка (1,89 г, 29,1 ммоль, 10,0 экв), хлорид аммония (1,57 г, 29,1 ммоль, 10,0 экв) и воду (3 мл) добавляли последовательно к раствору соединения 15-7 (1,79 г, 2,91 ммоль, 1,0 экв) в тетрагидрофуране (50 мл) при комнатной температуре. Полученную суспензию перемешивали при комнатной температуре в течение 16 часов. Суспензию фильтровали, и твердые вещества промывали этилацетатом (20 мл). Фильтрат испаряли досуха при пониженном давлении. Остаток суспендировали в насыщенном водном растворе хлорида аммония (30 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 16-1 (1,7 г, 99% выход) в виде светло-желтого масла, которое использовали на следующей стадии. (NRK-1-199)
(E)-5-(3-Метокси-2-(3-метилбут-2-ен-1-ил)-5-((2-(триметил-силил)этокси)метокси)стирил)-N, N-диметил-2-((2-(триметилсилил)-этокси)метокси)анилин (16-2).
37 масс.% водный раствор формальдегида (37 мкл, 0,46 ммоль. 1,0 экв) и триацетоксиборгидрид натрия (196 мг, 0,92 ммоль, 2,0 экв) последовательно добавляли к раствору соединения 16-1 (267 мг, 0,46 ммоль, 1,0 экв) в дихлорметане (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Анализ методом LCMS указывал на образование как моно-, так и ди-N-метилированных продуктов. К реакционной смеси добавляли дополнительное количество водного раствора формальдегида (0,5 мл, в избытке) и перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли дихлорметаном (20 мл) и промывали насыщенным раствором бикарбоната натрия (20 мл). Водный слой экстрагировали дихлорметаном (2×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток изначально очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 15% этилацетата в гептанах. Полученный продукт дополнительно очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 15% этилацетата в гептанах, с получением соединения 16-2 (194 мг, 69% выход) в виде светло-желтого масла. (NRK-1-198)
(E)-3-(3-(Диметиламино)-4-гидроксистирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенол (16) [KYN-157].
1M раствор тетрабутиламмония фторида в тетрагидрофуране (4,5 мл, 4,42 ммоль, 14 экв) добавляли к раствору соединения 16-2 (194 мг, 0,32 ммоль, 1 экв) в тетрагидрофуране (10 мл) при комнатной температуре. После нагревания при 67°C в течение 8 часов, смесь охлаждали до комнатной температуры и разбавляли водой (20 мл) и насыщенным водным раствором хлорида аммония (20 мл). Летучие компоненты удаляли при пониженном давлении, и оставшийся водный слой экстрагировали этилацетатом (3×30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (Redisep Rf Gold HP C18, колонка 50 g), элюируя с градиентом от 0 до 60% ацетонитрил в воде, с получением соединения 16 [KYN-157] (40 мг, 36% выход) в виде бледно-желтого твердого вещества (NRK-1-201).
Бледно-желтое твердое вещество, температура плавления 45-53°C; анализ методом HPLC: 97,0% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: Water Atlantis T3, 2,1×50 мм, 3 мкм; время удерживания: 5,75 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=354,2 (M+H)+; C22H27NO3; 1H ЯМР (400 МГц, CDCl3) δ=7,31 (д, J=2,0 Гц, 1H), 7,17 (дд, J=2,0, 8,3 Гц, 1H), 7,14 (д, J=16,1 Гц, 1H), 6,92 (д, J=8,3 Гц, 1H), 6,84 (д, J=16,0 Гц, 1H), 6,67 (д, J=2,3 Гц, 1H), 6,37 (д, J=2,3 Гц, 1H), 5,12 (септет, J=1,3, 6,8 Гц, 1H), 3,79 (с, 3H), 3,41 (уш.д, J=6,8 Гц, 2H), 2,69 (с, 6H), 1,82 (с, 3H), 1,69 (д, J=1,0 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=158,55, 154,55, 151,23, 140,76, 138,25, 130,52, 130,27, 130,07, 124,75, 124,06, 123,76, 120,54, 118,70, 114,21, 103,93, 98,24, 55,69. 45,14 (2C), 25,77, 24,47, 17,98.
Получение соединения 17 [KYN-120]
(E)-4-(5-Гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)стирил)-2-метокси-6-метилфенол (17)
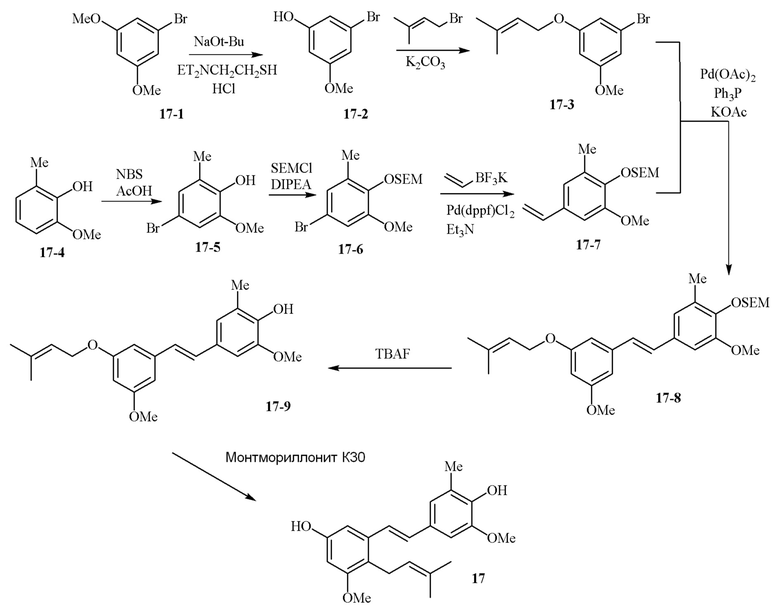
4-Бром-2-метокси-6-метилфенол (17-5).
N-Бромсукцинимид (5,15 г, 28,98 ммоль, 1 экв) добавляли к раствору соединения 17-4 (4 г, 28,98 ммоль, 1,0 экв) в уксусной кислоте (60 мл) при комнатной температуре. После перемешивания при комнатной температуре в течение ночи, летучие компоненты удаляли при пониженном давлении. Остаток разбавляли водой (200 мл) и экстрагировали этилацетатом (3×60 мл). Объединенный органический слой промывали насыщенным раствором бикарбоната натрия (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech 12 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 17-5 (7 г, 99% выход) в виде бесцветного масла (NK-1-35).
(2-((4-Бром-2-метокси-6-метилфенокси)метокси)этил)триметил-силан (17-6).
N, N-Диизопропилэтиламин (3,6 мл, 20,73 ммоль, 1,5 экв) и 2-(триметилсилил)этоксиметилхлорид (2,9 мл, 16,58 ммоль, 1,2 экв) последовательно добавляли к раствору соединения 17-5 (3 г, 13,82 ммоль, 1,0 экв) в безводном дихлорметане (60 мл) при комнатной температуре. После перемешивания в течение ночи, добавляли насыщенный водный раствор хлорида аммония (100 мл). Водный слой экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech 12 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 17-6 (2,37 г, 88% выход) в виде бледно-желтого масла (NK-1-29).
(2-((2-Метокси-6-метил-4-винилфенокси)метокси)этил)триметил-силан (17-7).
Винилтрифторборат калия (1,08 г, 8,06 ммоль, 1,4 экв), триэтиламин (1,3 мл, 9,2 ммоль, 1,6 экв) и Pd(dppf)Cl2 (210 мг, 0,029 ммоль, 0,05 экв) добавляли к раствору соединения 17-6 (2,0 г, 5,75 ммоль, 1,0 экв) в 2-пропаноле (40 мл). После продувки азотом в течение 10 минут, смесь кипятили с обратным холодильником (80°C) в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через целит (~ 5 г). Фильтрат концентрировали при пониженном давлении, разбавляли насыщенным раствором бикарбоната натрия (20 мл) и экстрагировали этилацетатом (3×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 17-7 (1,06 г, 65% выход) в виде бесцветного масла. (NK-1-34)
(E)-(2-((2-Метокси-4-(3-метокси-5-((3-метилбут-2-ен-1-ил)-оксистирил)-6-метилфенокси)метокси)этил)триметилсилан (17-8).
1-Бром-3-метокси-5-((3-метилбут-2-ен-1-ил)оксибензол (17-3) (1,49 г, 5,26 ммоль, 1,2 экв), трифенилфосфин (114 мг, 0,438 ммоль, 0,1 экв), ацетат калия (850 мг, 8,76 ммоль, 2 экв) и ацетат палладия (50 мг, 0,219 ммоль, 0,05 экв) добавляли к раствору соединения 17-7 (1,29 г, 4,38 ммоль, 1,0 экв) в безводном толуоле (60 мл). После продувки азотом в течение 15 минут, смесь кипятили с обратным холодильником (110°C) в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через целит (~ 5 г). Фильтрат переносили в делительную воронку, содержащую насыщенный раствор бикарбоната натрия 20 мл). Органический слой отделяли, и водный слой экстрагировали этилацетатом (2×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 30% этилацетата в гептанах, с получением соединения 17-8 (420 мг, 20% выход) в виде бесцветного масла (NK-1-40).
1M раствор тетрабутиламмония фторида в THF (6,1 мл, 6,06 ммоль, 7 экв) добавляли к раствору соединения 17-8 (420 мг, 0,86 ммоль, 1 экв) в безводном THF (40 мл) при комнатной температуре. После кипячения с обратным холодильником в течение ночи (66°C), анализ методом LCMS указывал на завершение реакции. Смесь охлаждали до комнатной температуры и разбавляли водой (10 мл). Органические летучие компоненты удаляли при пониженном давлении и разбавляли этилацетатом (20 мл). Органический слой отделяли, и водный слой экстрагировали этилацетатом (2×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech 14 g), элюируя с градиентом от 0 до 50% этилацетата в гептанах, с получением соединения 17-9 (220 мг, 73% выход) в виде желтого масла. (NK-1-41)
(E)-4-(5-Гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)стирил)-2-метокси-6-метилфенол (17) [KYN-120].
Порошок монтмориллонита K30 (220 мг) добавляли к раствору соединения 17-9 (220 мг, 0,45 ммоль) в безводном дихлорметане (20 мл) при комнатной температуре. После перемешивания при комнатной температуре в течение ночи, анализ методом LCMS указывал на образование 30-40% требуемого соединения 10, наряду с двумя региоизомерами и непрореагировавшим исходным соединением 9. Смесь фильтровали, промывали дихлорметаном (2×15 мл), и фильтрат концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (Sorbtech 12 г колонка), элюируя с градиентом от 0 до 50% этилацетата в гептанах. Растирание с 30% дихлорметан в гептанах (10 мл) давало чистое соединение 17 [KYN-120] (38 мг, 17% выход) в виде белого твердого вещества. (NK-1-42-S3)
Белое твердое вещество, температура плавления 140,3-140,4°C; анализ методом HPLC: 97,6% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 9,29 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=355 (M+H)+; C22H26O4; 1H ЯМР (400 МГц, CDCl3) δ=7,14 (д, J=16,1 Гц, 1H), 6,88 (м, 1H), 6,87 (м, 1H), 6,83 (д, J=16,1 Гц, 1H), 6,65 (д, J=2,5 Гц, 1H), 6,35 (д, J=2,5 Гц, 1H), 5,72 (д, J=0,5 Гц, 1H), 5,13 (тм, J=1,4, 6,9 Гц, 1H), 4,60 (с, 1H), 3,92 (с, 3H), 3,80 (с, 3H), 3,42 (уш.д, J=6,9 Гц, 2H), 2,27 (с, 3H), 1,82 (д, J=1,0 Гц, 3H), 1,69 (д, J=1,2 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=158,58, 154,34, 146,37, 143,77, 138,32, 130,62, 130,53, 129,17, 124,00, 123,87, 123,73, 122,35, 120,71, 105,96, 103,87, 98,13, 56,00, 55,71, 25,78, 24,49, 17,97, 15,45.
Соединение 17 может быть также синтезировано с использованием модульного метода по настоящему изобретению, применяя эквиваленты для модуля A, B и C и последующее проведение реакции перекрестного алкилирования арилгалогенида.
(E)-5-(5-Гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)стирил)-3-метоксибензол-1,2-диол (18) [KYN-156]
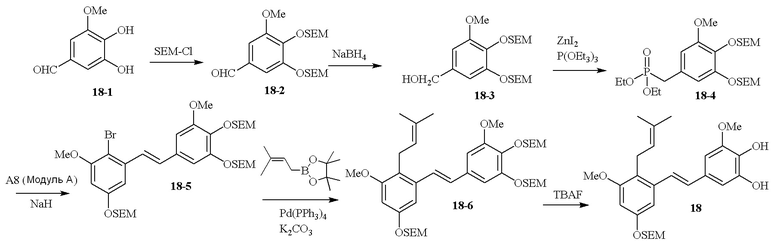
3-Метокси-4,5-бис((2-(триметилсилил)этокси)метокси)-бензальдегид (18-2).
N, N-Диизопропилэтиламин (3,14 мл, 18 ммоль, 3,0 экв) и 2-(триметилсилил)этоксиметилхлорид (2,53 мл, 14,3 ммоль, 2,4 экв) добавляли последовательно к раствору соединения 18-1 (1,0 г, 6 ммоль, 1,0 экв) в смеси 1:1:2 безводный тетрагидрофуран/диметилформамид/дихлорметан (80 мл) при комнатной температуре. После перемешивания при комнатной температуре в течение 16 часов, добавляли насыщенный раствор бикарбоната натрия (100 мл) для прерывания реакции. Слои разделяли, и водный слой экстрагировали метилтрет-бутиловым эфиром (2×250 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 80 g), элюируя с градиентом от 0 до 35% этилацетата в гептанах, с получением соединения 18-2 (2 г, 78% выход) в виде бесцветного масла. (LM-6-96)
(3-Метокси-4,5-бис((2-(триметилсилил)этокси)метокси)фенил)-метанол (18-3).
Боргидрид натрия (176 мг, 4,66 ммоль, 1,0 экв) добавляли одной порцией в раствор соединения 18-2 (2 г, 4,66 ммоль, 1,0 экв) в метаноле (50 мл) при 0°C. После перемешивания при 0°C в течение 1 часа, добавляли воду (50 мл) для прерывания реакции. Смесь концентрировали при пониженном давлении с удалением большей части метанола. Остаток разбавляли насыщенным солевым раствором (50 мл) и экстрагировали этилацетатом (2×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 18-3 (2 г) в виде бесцветного масла, которое использовали на следующей стадии. (LM-6-104).
Диэтил (3-метокси-4,5-бис((2-(триметилсилил)этокси)метокси)-бензил)фосфонат (18-4).
Йодид цинка (2,97 г, 9,32 ммоль, 2,0 экв) и триэтилфосфит (1,6 мл, 9,32 ммоль, 2,0 экв) добавляли последовательно к раствору соединения 18-3 (2 г, 4,66 ммоль, 1,0 экв) в безводном тетрагидрофуране (100 мл) при комнатной температуре. Смесь кипятили с обратным холодильником (68°C) в течение 16 часов. После охлаждения до комнатной температуры, добавляли последовательно воду (50 мл), карбонат калия (1,61 г, 2,5 экв) и метилтрет-бутиловый эфир (200 мл). Смесь фильтровали через слой целита (20 г), который споласкивали этилацетатом (300 мл). Слои разделяли, и органический слой промывали насыщенным солевым раствором (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 80 g), элюируя с градиентом от 0 до 100% этилацетата в гептанах, с получением смеси соединения 4A и 4B (1 г). К раствору соединений 4A и 4B (1 г, 2,4 ммоль, 1,0 экв) в безводном дихлорметане (100 мл) добавляли successively при комнатной температуре N, N-диизопропилэтиламин (2,52 мл, 14,4 ммоль, 6 экв) и 2-(триметил-силил)этоксиметилхлорид (2,04 мл, 11,2 ммоль, 4,8 экв). После перемешивания при комнатной температуре в течение 16 часов, добавляли насыщенный раствор бикарбоната натрия (100 мл) для прерывания реакции. Слои разделяли, и водный слой экстрагировали дихлорметаном (2×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 80 g), элюируя с градиентом от 0 до 100% этилацетата в гептанах, с получением соединения 18-4 (1,15 г, 45% выход за три стадии) в виде бесцветного масла. (LM-8-4, LM-8-5).
(E)-(((((5-(2-Бром-3-метокси-5-((2-(триметилсилил)этокси)-метокси)стирил)-3-метокси-1,2-фенилен)бис(окси)бис(метилен))-бис(окси)бис(этан-2,1-диил))бис(триметилсилан) (18-5).
60% дисперсию гидрида натрия в минеральном масле (168 мг, 4,2 ммоль, 2 экв) добавляли одной порцией к раствору соединения 18-4 (1,15 г, 2,1 ммоль, 1,0 экв) в безводном тетрагидрофуране (50 мл) при 0°C. Смесь подогревали до комнатной температуры и перемешивали 30 минут. Добавляли по каплям раствор промежуточного соединения A (754 мг, 2,1 ммоль, 1 экв) в безводном тетрагидрофуране (20 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию осторожно прерывали насыщенным солевым раствором (20 мл, 1 капля в минуту для первых 5 мл солевого раствора) при 0°C. Смесь экстрагировали метилтрет-бутиловым эфиром (2×150 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 80 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 18-5 (1,18 г, 75% выход) в виде бесцветного масла. (LM-8-6)
(E)-(((((3-Метокси-5-(3-метокси-2-(3-метилбут-2-ен-1-ил)-5-((2-(триметилсилил)этокси)метокси)стирил)-1,2-фенилен)бис(окси)-бис(метилен))бис(окси)бис(этан-2,1-диил))бис(триметилсилан) (18-6).
Тетракис(трифенилфосфин)палладий(0) (90 мг, 0,078 ммоль, 0,05 экв), карбонат калия (428 мг, 3,1 ммоль, 2 экв), пинаколовый эфир 3-метилбут-2-енилбороновой кислоты (611 мг, 3,1 ммоль, 2 экв) и воду (4 мл) добавляли последовательно к раствору соединения 18-5 (1,18 г, 1,56 ммоль, 1 экв) в 1,4-диоксане (12 мл). После продувки азотом в течение 10 минут, реакционную смесь нагревали при 95°C в течение 16 часов. После охлаждения до комнатной температуры, смесь экстрагировали метилтрет-бутиловым эфиром (2×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 80 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 18-6 (1 г, 86% выход) в виде бесцветного масла. (LM-8-7)
(E)-5-(5-Гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)стирил)-3-метоксибензол-1,2-диол (18) [KYN-156].
1M раствор тетрабутиламмония фторида в тетрагидрофуране (25,3 мл, 25,3 ммоль, 21 экв) добавляли к раствору соединения 18-6 (900 мг, 1,2 ммоль, 1 экв) в тетрагидрофуране (25 мл) при комнатной температуре. После нагревания при 67°C в течение 16 часов, смесь охлаждали до комнатной температуры и разбавляли водой (20 мл) и насыщенным солевым раствором (20 мл). Смесь экстрагировали этилацетатом (300 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали три раза на автоматизированной хроматографической системе Interchim (колонка 80 g, 2 колонки 40 g, 2 колонки 25 g), элюируя каждый раз с градиентом от 0 до 100% этилацетата в гептанах, с получением соединения 18 (70 мг, ~85% чистота). Это неочищенное соединение 18 разделяли на 7 равных порций. Каждую порцию очищали на автоматизированной хроматографической системе ACCQPrep HP125 с обращенной фазой (колонка SunFire Prep C18 OBD 5 мкм 19×250 мм), элюируя с градиентом от 0 до 100% ацетонитрила в воде, с получением соединения 18 (50 мг). Этот материал дополнительно очищали на автоматизированной хроматографической системе Interchim (колонка 25 g), элюируя с градиентом от 0 до 100% этилацетата в гексанах, с получением соединения 18 [KYN-156] (27 мг, 6,3% выход) в виде желтовато-белого твердого вещества после сушки под вакуумом при 40°C в течение 16 часов. (LM-8-9).
Желтовато-белое твердое вещество; анализ методом HPLC: 96,0% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: Luna C18(2), 2,0×20 мм, 3 мкм; время удерживания: 7,7 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=357,1 (M+H)+; C21H24O5; 1H ЯМР (400 МГц, ацетон-d6) δ=8,08 (с, 1H), 7,72-7,41 (м, 2H), 7,19 (д, J=16,1 Гц, 1H), 6,84 (д, J=16,0 Гц, 1H), 6,75-6,72 (м, 2H), 6,72 (д, J=2,3 Гц, 1H), 6,40 (д, J=2,2 Гц, 1H), 5,09 (септет, J=1,3, 7,0 Гц, 1H), 3,86 (с, 3H), 3,78 (с, 3H), 3,41 (уш.д, J=7,0 Гц, 2H), 1,82 (д, J=0,7 Гц, 3H), 1,64 (д, J=1,1 Гц, 3H); 13C ЯМР (100 МГц, Ацетон-d6) δ=158,88, 156,66, 148,63, 145,92, 138,33, 134,46, 130,72, 129,84, 129,52, 124,69, 124,46, 119,36, 107,91, 104,14, 102,28, 98,62, 55,95. 55,38, 25,38, 24,37, 17,58.
(E)-3-(3-Гидрокси-5-метоксистирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенол (19) [KYN-160]
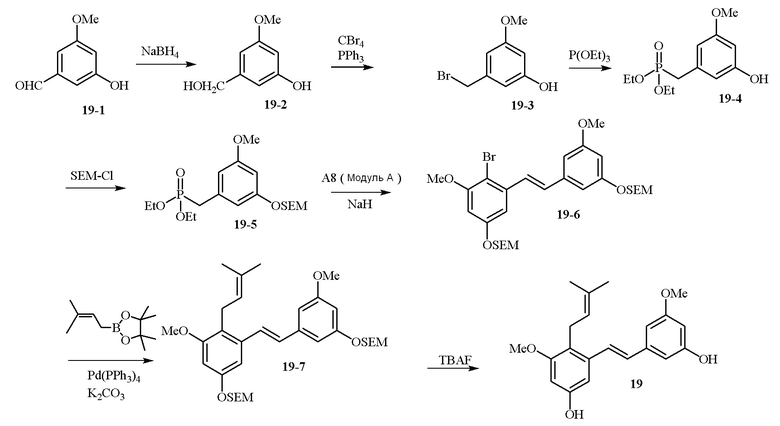
3-(Гидроксиметил)-5-метоксифенол (19-2).
Боргидрид натрия (263 мг, 4,66 ммоль, 1,0 экв) добавляли одной порцией к раствору соединения 19-1 (1,06 г, 7 ммоль, 1,0 экв) в метаноле (70 мл) при 0°C. После перемешивания при 0°C в течение 1 часа, добавляли воду (50 мл) для прерывания реакции. Смесь концентрировали при пониженном давлении с удалением большей части метанола. Остаток разбавляли насыщенным солевым раствором (50 мл) и экстрагировали этилацетатом (2×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (колонка 40 g), элюируя с градиентом от 0 до 100% этилацетата в гептанах, с получением соединения 19-2 (1,07 г, 99% выход) в виде белого твердого вещества. (LM-8-17)
3-(Бромметил)-5-метоксифенол (19-3).
Тетрабромид углерода (2,55 г, 7,7 ммоль, 1,1 экв) добавляли к раствору соединения 19-2 (1,06 г, 7 ммоль, 1,0 экв) в смеси 1 : 2 безводного тетрагидрофурана и дихлорметана (75 мл) при комнатной температуре. Смесь охлаждали на ледяной бане и добавляли одной порцией трифенилфосфин (2,02 г, 7,7 ммоль, 1,1 экв). Смесь затем подогревали до комнатной температуры и перемешивали в течение 2 часов. Растворитель удаляли при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (колонка 80 g), элюируя с градиентом от 0 до 50% этилацетата в гептанах, с получением соединения 19-3 (1,18 г, 79% выход) в виде желтовато-белого твердого вещества. (LM-8-18)
Диэтил (3-гидрокси-5-метоксибензил)фосфонат (19-4).
Триэтилфосфит (2,8 мл, 16,3 ммоль, 3 экв) добавляли к раствору соединения 19-3 (1,18 г, 5,4 ммоль, 1,0 экв) в безводном толуоле (100 мл) при комнатной температуре. После кипячения с обратным холодильником (110°C) в течение 16 часов, реакционную смесь охлаждали до комнатной температуры, и растворитель удаляли при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (колонка 80 g), элюируя с градиентом от 0 до 100% этилацетата в гептанах, с получением соединения 19-4 (1,32 г, 89% выход) в виде бесцветного масла (LM-8-19).
Диэтил (3-метокси-5-((2-(триметилсилил)этокси)метокси)-бензил)фосфонат (19-5).
N, N-Диизопропилэтиламин (3,1 мл, 24 ммоль, 5 экв) и 2-(триметилсилил)этоксиметилхлорид (3,4 мл, 19,3 ммоль, 4 экв) добавляли последовательно к раствору соединения 19-4 (1,32 г, 4,8 ммоль, 1,0 экв) в безводном дихлорметане (50 мл) при комнатной температуре. После перемешивания при комнатной температуре в течение 16 часов, добавляли насыщенный раствор бикарбоната натрия (50 мл) для прерывания реакции. Слои разделяли, и водный слой экстрагировали дихлорметаном (2×200 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (колонка 80 g), элюируя с градиентом от 0 до 100% этилацетата в гептанах, с получением соединения 19-5 (1,27 г, 65% выход) в виде бесцветного масла (LM-8-20).
(E)-(2-((3-(2-Бром-3-метокси-5-((2-(триметилсилил)этокси)-метокси)стирил)-5-метоксифенокси)метокси)этил)триметилсилан (19-6).
60% дисперсию гидрида натрия в минеральном масле (204 мг, 5,1 ммоль, 2 экв) добавляли одной порцией к раствору соединения 19-5 (1,03 г, 2,55 ммоль, 1,0 экв) в безводном тетрагидрофуране (50 мл) при 0°C. Смесь подогревали до комнатной температуры и перемешивали 30 минут. Добавляли по каплям раствор соединения A8 (модуль A) (0,92 мг, 2,55 ммоль, 1 экв) в безводном тетрагидрофуране (20 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию осторожно прерывали насыщенным солевым раствором (20 мл, 1 капля в минуту для первых 5 мл солевого раствора) при 0°C. Смесь экстрагировали метилтрет-бутиловым эфиром (2×150 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (колонка 80 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 19-6 (1,23 г, 79% выход) в виде бесцветного масла (LM-8-21).
(E)-(2-((3-Метокси-5-(3-метокси-2-(3-метилбут-2-ен-1-ил)-5-((2-(триметилсилил)этокси)метокси)стирил)фенокси)метокси)этил)-триметилсилан (19-7).
Тетракис(трифенилфосфин)палладий(0) (116 мг, 0,1 ммоль, 0,05 экв), карбонат калия (553 мг, 4 ммоль, 2 экв), пинаколовый эфир 3-метилбут-2-енилбороновой кислоты (785 мг, 4 ммоль, 2 экв) и воду (5 мл) добавляли последовательно к раствору соединения 19-6 (1,23 г, 2 ммоль, 1 экв) в 1,4-диоксане (15 мл). После продувки азотом в течение 10 минут, реакционную смесь нагревали при 95°C в течение 16 часов. После охлаждения до комнатной температуры, смесь экстрагировали метилтрет-бутиловым эфиром (2×20 мл), и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (колонка 80 g), элюируя с градиентом от 0 до 15% этилацетата в гептанах, с получением соединения 19-7 (1,1 г, 91% выход) в виде бесцветного масла (LM-8-22).
(E)-3-(3-Гидрокси-5-метоксистирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенол (19) [KYN-160].
1M раствор тетрабутиламмония фторида в тетрагидрофуране (25,6 мл, 25,6 ммоль, 14 экв) добавляли к раствору соединения 19-7 (1,1 г, 1,83 ммоль, 1 экв) в тетрагидрофуране (25 мл) при комнатной температуре. После нагревания при 67°C в течение 16 часов, смесь охлаждали до комнатной температуры и разбавляли водой (20 мл) и насыщенным солевым раствором (20 мл). Смесь экстрагировали этилацетатом (300 мл), и органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали два раза на автоматизированной хроматографической системе InterChim (колонка 120 g, 2 колонки 25 g), элюируя каждый раз с градиентом от 0 до 100% этилацетата в гептанах, с получением соединения 19 (110 мг, 17% выход) в виде желтовато-белого твердого вещества после сушки под вакуумом при 40°C в течение 16 часов. (LM-8-23).
E)-4-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)стирил)-бензол-1,2-диол (20) [KYN-146]
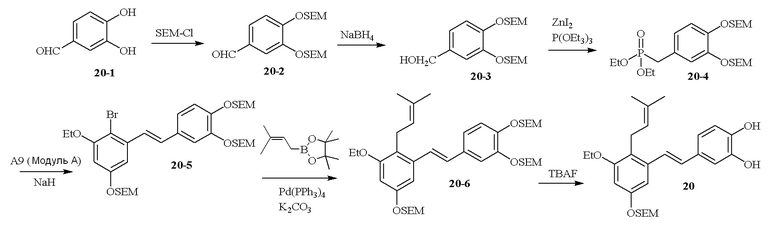
3,4-Бис((2-(триметилсилил)этокси)метокси)бензальдегид (20-2).
N, N-Диизопропилэтиламин (37,8 мл, 217,2 ммоль, 3,0 экв) и 2-(триметилсилил)этоксиметилхлорид (29,0 г, 173,8 ммоль, 2,4 экв) добавляли последовательно к раствору соединения 20-1 (10,0 г, 72,4 ммоль, 1,0 экв) в безводном дихлорметане (200 мл) при комнатной температуре. После перемешивания при комнатной температуре в течение 16 часов, добавляли насыщенный раствор бикарбоната натрия (150 мл). Слои разделяли, и водный слой экстрагировали дихлорметаном (2×150 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 120 g), элюируя с градиентом от 0 до 50% этилацетата в гептанах, с получением соединения 20-2 (28,03 г, 97% выход) в виде желтого масла. (LM-1-75)
(3,4-Бис((2-(триметилсилил)этокси)метокси)фенил)метанол (20-3).
Боргидрид натрия (2,66 г, 70,3 ммоль, 1,0 экв) добавляли одной порцией к раствору соединения 20-2 (28,03 г, 70,3 ммоль, 1,0 экв) в метаноле (200 мл) при 0°C. После перемешивания при 0°C в течение 1 часа, добавляли воду (75 мл) для прерывания реакции. Смесь концентрировали при пониженном давлении с удалением большей части метанола. Остаток разбавляли насыщенным солевым раствором (75 мл) и экстрагировали этилацетатом (2×200 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 120 g), элюируя с градиентом от 0 до 100% этилацетата в гептанах, с получением соединения 20-3 (26,84 г, 95% выход) в виде желтого масла. (LM-1-80)
Диэтил (3,4-бис((2-(триметилсилил)этокси)метокси)бензил)-фосфонат (20-4).
Йодид цинка (42,77 г, 134,0 ммоль, 2,0 экв) и триэтилфосфит (23 мл, 134,0 ммоль, 2,0 экв) добавляли последовательно к раствору соединения 20-3 (26,84 г, 67,0 ммоль, 1,0 экв) в безводном тетрагидрофуране (400 мл) при комнатной температуре. Смесь кипятили с обратным холодильником (68°C) в течение 4 часов, после чего анализ методом LCMS показывал, что реакция завершена. После охлаждения до комнатной температуры, смесь концентрировали при пониженном давлении с удалением большей части тетрагидрофурана. Остаток разбавляли насыщенным раствором бикарбоната натрия (150 мл) и экстрагировали метилтрет-бутиловым эфиром (2×400 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 220 g), элюируя с градиентом от 0 до 100% этилацетата в гептанах, с получением соединения 20-4 (24,18 г, 69% выход) в виде желтого масла. (LM-1-82)
(E)-(((((4-(2-Бром-3-этокси-5-((2-(триметилсилил)этокси)-метокси)стирил)-1,2-фенилен)бис(окси)бис(метилен))бис(окси)-бис(этан-2,1-диил))бис(триметилсилан) (20-5).
60% дисперсию гидрида натрия в минеральном масле (2,43 г, 60,6 ммоль, 2 экв) добавляли одной порцией к раствору соединения 20-4 (15,76 г, 30,3 ммоль, 1,0 экв) в безводном тетрагидрофуране (300 мл) при 0°C. Смесь подогревали до комнатной температуры и перемешивали 30 минут. Добавляли по каплям раствор, содержащий соединение A9 (модуль A) (11,36 г, 30,3 ммоль, 1 экв) в безводном тетрагидрофуране (100 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию осторожно прерывали насыщенным солевым раствором (250 мл, 1 капля в минуту для первых 5 мл солевого раствора) при 0°C. Смесь экстрагировали метилтрет-бутиловым эфиром (2×500 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 330 g), элюируя с градиентом от 0 до 15% этилацетата в гептанах, с получением соединения 20-5 (15,5 г, 69% выход) в виде желтого масла. (LM-6-89)
(E)-(((((4-(3-этокси-2-(3-метилбут-2-ен-1-ил)-5-((2-(три-метилсилил)этокси)метокси)стирил)-1,2-фенилен)бис(окси)бис-(метилен))бис(окси)бис(этан-2,1-диил))бис(триметилсилан) (20-6).
Тетракис(трифенилфосфин)палладий(0) (1,21 г, 1,05 ммоль, 0,05 экв), карбонат калия (5,78 г, 41,8 ммоль, 2 экв), пинаколовый эфир 3-метилбут-2-енилбороновой кислоты (8,2 г, 41,8 ммоль, 2 экв) и воду (50 мл) добавляли последовательно к раствору соединения 20-5 (15,5 г, 20,9 ммоль, 1 экв) в 1,4-диоксане (150 мл). После продувки азотом в течение 10 минут, реакционную смесь нагревали при 95°C в течение 16 часов. После охлаждения до комнатной температуры, смесь экстрагировали метилтрет-бутиловым эфиром (2×300 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 330 g), элюируя с градиентом от 0 до 15% этилацетата в гептанах, с получением соединения 20-6 (15,28 г, 99% выход) в виде желтого масла (LM-6-90).
(E)-4-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)стирил)-бензол-1,2-диол (20) [KYN-146].
1M раствор тетрабутиламмония фторида в тетрагидрофуране (513 мл, 513 ммоль, 21 экв) добавляли к смеси соединения 20-6 (17,86 г, 24,4 ммоль, 1 экв) в тетрагидрофуране (500 мл) при комнатной температуре. После нагревания при 67°C в течение 16 часов, смесь охлаждали до комнатной температуры и разбавляли водой (350 мл) и насыщенным солевым раствором (350 мл). Смесь экстрагировали метилтрет-бутиловым эфиром (1,5 л) и этилацетатом (1,5 л). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали три раза на автоматизированной хроматографической системе Interchim (2 колонки 40 g, 2 колонки 80 g, 2 колонки 40 g), элюируя каждый раз с градиентом от 0 до 100% этилацетата в гептанах, с получением cмеси SEM-защищенных соединений (3,94 г) и соединения 20 (1,5 г, ~70% чистота). Это неочищенное соединение 20 дополнительно очищали на автоматизированной хроматографической системе Interchim с обращенной фазой (колонка 300 g), элюируя с градиентом от 0 до 100% ацетонитрила в воде, с получением соединения 20 (480 мг). Смесь SEM-защищенных соединений (3,94 г) обрабатывали 1M раствором тетрабутиламмония фторида в тетрагидрофуране (58,7 мл, 58,7 ммоль, 7,0 экв) и обрабатывали таким же образом с получением соединения 20 (450 мг). Весь SEM-защищенный материал объединяли (480 мг, 450 мг и 130 мг, LM-6-69) и очищали на автоматизированной хроматографической системе Interchim (колонка 40 g), элюируя с градиентом от 0 до 55% этилацетата в гексанах, с получением соединения 20 [KYN-146] (1,0 г, 10% суммарный выход) в виде желтовато-белого твердого вещества после сушки под вакуумом при 40°C в течение 16 часов. (LM-6-91).
Структурные данные
Соединение 20 [KYN-146]
Желтовато-белое твердое вещество; анализ методом HPLC: 96,3% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 8,3 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=341,2 (M+H)+; C21H24O4; 1H ЯМР (400 МГц, Ацетон-d6) δ=7,98 (уш.с, 2H), 7,18 (д, J=16,1 Гц, 1H), 7,08 (д, J=2,1 Гц, 1H), 6,90 (ддд, J=0,4, 2,1, 8,1 Гц, 1H), 6,87-6,80 (м, 2H), 6,70 (д, J=2,3 Гц, 1H), 6,37 (д, J=2,3 Гц, 1H), 5,11 (септет, J=1,3, 7,0 Гц, 1H), 3,99 (кв, J=7,0 Гц, 2H), 3,42 (д, J=7,0 Гц, 2H), 1,81 (д, J=0,9 Гц, 3H), 1,64 (д, J=1,1 Гц, 3H), 1,38 (т, J=7,0 Гц, 3H); 13C ЯМР (100 МГц, Ацетон-d6) δ=158,18, 156,56, 145,65, 145,59, 138,34, 130,60, 130,33, 129,87, 124,65, 124,23, 119,52, 119,50, 115,75, 113,29, 104,08, 99,43, 63,86, 25,40, 24,44, 17,65, 14,75.
(E)-4-(5-Гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)стирил)-2-метоксибензойная кислота (21) [KYN-154]
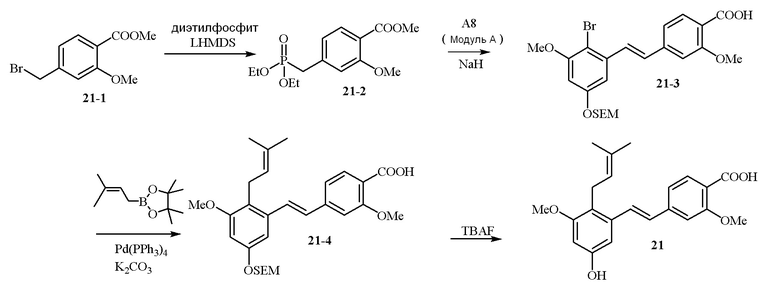
Метил 4-((диэтоксифосфорил)метил)-2-метоксибензоат (21-2).
1M раствор бис(триметилсилил)амида лития в тетрагидрофуране (7 мл, 7 ммоль, 1,8 экв) добавляли к раствору диэтилфосфита (1 мл, 7,7 ммоль, 2,0 экв) в безводном тетрагидрофуране (30 мл) при 0°C. После перемешивания при 0°C в течение 30 минут, добавляли по каплям раствор соединения 21-1 (1 г, 3,9 ммоль, 1 экв) в безводном тетрагидрофуране (10 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли насыщенным солевым раствором (50 мл) и экстрагировали этилацетатом (2×300 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 40 g), элюируя с градиентом от 0 до 100% этилацетата в гептанах, с получением соединения 21-2 (1,22 г, 99% выход) в виде бесцветного масла. (LM-6-92)
(E)-4-(2-Бром-3-метокси-5-((2-(триметилсилил)этокси)-метокси)стирил)-2-метоксибензойная кислота (21-3).
60% дисперсию гидрида натрия в минеральном масле (160 мг, 4 ммоль, 2 экв) добавляли одной порцией к раствору соединения 21-2 (0,63 г, 2 ммоль, 1,0 экв) в безводном тетрагидрофуране (30 мл) при 0°C. Смесь подогревали до комнатной температуры и перемешивали 30 минут. Добавляли по каплям раствор соединения A8 (модуль A) (0,72 г, 2 ммоль, 1 экв) в безводном тетрагидрофуране (10 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли насыщенным солевым раствором (20 мл, 1 капля в минуту для первых 5 мл солевого раствора) при 0°C. Смесь экстрагировали метилтрет-бутиловым эфиром (2×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 40 g), элюируя с градиентом от 0 до 100% этилацетата в гептанах, с получением соединения 21-3 (0,55 г, 55% выход) в виде желтого твердого вещества. (LM-6-93)
(E)-2-Метокси-4-(3-метокси-2-(3-метилбут-2-ен-1-ил)-5-((2-(триметилсилил)этокси)метокси)стирил)бензойная кислота (21-4).
Тетракис (трифенилфосфин)палладий(0) (58 мг, 0,05 ммоль, 0,05 экв), карбонат калия (415 мг, 3 ммоль, 3 экв), пинаколовый эфир 3-метилбут-2-енилбороновой кислоты (392 мг, 2 ммоль, 2 экв) и воду (4 мл) добавляли последовательно к раствору соединения 21-3 (509 мг, 1 ммоль, 1 экв) в 1,4-диоксане (12 мл). После продувки азотом в течение 10 минут, реакционную смесь нагревали при 95°C в течение 16 часов. После охлаждения до комнатной температуры, смесь экстрагировали этилацетатом (2×20 мл), и объединенные органические слои концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 25 g), элюируя с градиентом от 0 до 80% этилацетата в гептанах, с получением соединения 21-4 (490 мг, 98% выход) в виде желтого масла. (LM-6-94)
(E)-4-(5-Гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)стирил)-2-метоксибензойная кислота (21) [KYN-154]:
1M раствор тетрабутиламмония фторида в тетрагидрофуране (7 мл, 7 ммоль, 7 экв) добавляли к смеси соединения 21-4 (490 мг, 1 ммоль, 1 экв) в тетрагидрофуране (20 мл) при комнатной температуре. После нагревания при 67°C в течение 16 часов, смесь охлаждали до комнатной температуры и разбавляли этилацетатом (200 мл). Смесь промывали насыщенным солевым раствором (50 мл), и органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали два раза на автоматизированной хроматографической системе Interchim (2 колонки 25 g), элюируя каждый раз с градиентом от 0 до 100% этилацетата в гептанах, и два раза на автоматизированной хроматографической системе Interchim с обращенной фазой (колонка 50 g), элюируя каждый раз с градиентом от 0 до 100% ацетонитрила в воде, с получением соединения S10 (70 мг, ~70% чистота). Этот материал разделяли на 8 равных порций. Каждую порцию очищали на автоматизированной хроматографической системе ACCQPrep HP125 с обращенной фазой (колонка SunFire Prep C18 OBD 5 мкм 19×250 мм), элюируя с градиентом от 0 до 100% ацетонитрила в воде, с получением соединения 21 [KYN-154] (30 мг, 8% выход) в виде желтовато-белого твердого вещества после лиофилизации в течение 16 часов (LM-6-95).
Желтовато-белое твердое вещество; анализ методом HPLC: 98,8% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 8,4 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=369,2 (M+H)+; C22H24O5; 1H ЯМР (400 МГц, Ацетон-d6) δ=7,92 (д, J=8,1 Гц, 1H), 7,58 (д, J=16,1 Гц, 1H), 7,38 (д, J=1,1 Гц, 1H), 7,30 (дд, J=1,2, 8,2 Гц, 1H), 7,05 (д, J=16,3 Гц, 1H), 6,78 (д, J=2,3 Гц, 1H), 6,48 (д, J=2,2 Гц, 1H), 5,08 (септет, J=1,3, 7,0 Гц, 1H), 4,09 (с, 3H), 3,80 (с, 3H), 3,45 (уш.д, J=6,8 Гц, 2H), 1,81 (с, 3H), 1,64 (д, J=1,0 Гц, 3H); 13C ЯМР (100 МГц, ацетон-d6) δ=165,55, 159,48, 158,97, 156,82, 144,29, 137,40, 133,08, 130,20, 130,14, 129,15, 124,52, 120,27, 119,38, 118,33, 110,57, 104,49, 99,64, 56,36. 55,46, 25,36, 24,36, 17,57.
Получение соединения 22 [KYN-124] и соединение 23 [KYN-125]
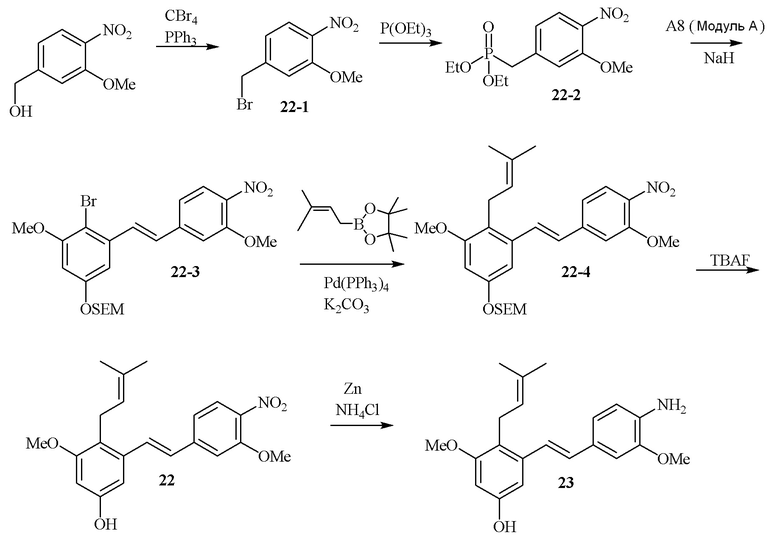
4-(Бромметил)-2-метокси-1-нитробензол (22-1).
Тетрабромид углерода (21,7 г, 65,5 ммоль, 1,2 экв) добавляли 5 порциями к раствору 3-метокси-4-нитробензилового спирта (10 г, 54,5 ммоль, 1,0 экв) и трифенилфосфина (17,2 г, 65,5 ммоль, 1,2 экв) в дихлорметане (300 мл) при 0°C. После перемешивания при комнатной температуре в течение 16 часов, реакционную смесь концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (колонка 330 g), элюируя с градиентом от 0 до 50% этилацетата в гептанах, с получением соединения 22-1 (12,8 г, 95% выход) в виде желтого твердого вещества (CH-LM-02-26)
Диэтил (3-метокси-4-нитробензил)фосфонат (22-2).
Триэтилфосфит (26,8 мл, 156 ммоль, 3,0 экв) добавляли к раствору соединения 22-1 (12,8 г, 52 ммоль, 1,0 экв) в толуоле (400 мл). После кипячения с обратным холодильником (110°C) в течение 40 часов, анализ методом ЯМР указывал на завершение реакции. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (колонка 330 g), элюируя с градиентом от 0 до 100% этилацетата в гептанах, затем 10% метанола в дихлорметане, с получением соединения 22-2 (15,5 г, 98% выход) в виде бледно-желтого масла (CH-LM-02-27).
(E)-(2-((4-Бром-3-метокси-5-(3-метокси-4-нитростирил)-фенокси)метокси)этил)триметилсилан (22-3).
60% дисперсию гидрида натрия в минеральном масле (6,1 г, 153,3 ммоль, 3,0 экв) добавляли 3 порциями к раствору соединения 22-2 (15,5 г, 51,1 ммоль, 1,0 экв) в безводном тетрагидрофуране (450 мл) при 0°C. Смесь подогревали до комнатной температуры и перемешивали в течение 30 минут. Добавляли по каплям раствор промежуточного соединения A (18,5 г, 51,1 ммоль, 1,0 экв) в безводном тетрагидрофуране (150 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию осторожно прерывали водой (500 мл, 1 капля в минуту для первых 5 мл воды) при 0°C и экстрагировали метилтрет-бутиловым эфиром (1,5 л) и этилацетатом (1 л). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали тремя равными порциями на автоматизированной хроматографической системе InterChim (3 колонки 330 g), элюируя с градиентом от 10 до 50% этилацетата в гептанах, с получением соединения 22-3 (9,9 г, 38% выход) в виде желтого твердого вещества (CH-LM-02-28) .
(E)-(2-((3-Метокси-5-(3-метокси-4-нитростирил)-4-(3-метил-бут-2-ен-1-ил) фенокси)метокси)этил)триметилсилан (22-4).
Тетракис(трифенилфосфин)палладий(0) (2,1 г, 1,8 ммоль, 0,05 экв), карбонат калия (10 г, 72 ммоль, 2,0 экв), пинаколовый эфир 3-метилбут-2-енилбороновой кислоты (14,1 г, 72 ммоль, 2,0 экв) и воду (90 мл) добавляли последовательно к раствору соединения 22-3 (18,3 г, 36 ммоль, 1,0 экв) в 1,4-диоксане (270 мл) в герметизированной пробирке. После продувки азотом в течение 10 минут, реакционную смесь нагревали при 100°C в течение 16 часов. После охлаждения до комнатной температуры, реакционную смесь экстрагировали метилтрет-бутиловым эфиром (500 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали двумя равными порциями на автоматизированной хроматографической системе InterChim (2 колонки 220 g), элюируя с градиентом от 0 до 12% этилацетата в гептанах, с получением соединения 22-4 (15,1 г, 84% выход) в виде желтого твердого вещества. (CH-LM-02-29)
(E)-3-Метокси-5-(3-метокси-4-нитростирил)-4-(3-метилбут-2-ен-1-ил)фенол (22).
1M раствор тетрабутиламмония фторида в тетрагидрофуране (366 мл, 366 ммоль, 7,0 экв) добавляли к раствору соединения 22-4 (26,1 г, 52,3 ммоль, 1,0 экв) в тетрагидрофуране (400 мл) при комнатной температуре. После нагревания при 68°C в течение 16 часов, смесь охлаждали до комнатной температуры и разбавляли водой (100 мл). Летучие компоненты удаляли при пониженном давлении. Остаток разбавляли насыщенным водным раствором хлорида аммония (250 мл) и экстрагировали этилацетатом (2×500 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали тремя равными порциями на автоматизированной хроматографической системе InterChim (3 колонки 330 g), элюируя с градиентом от 30 до 100% этилацетата в гептанах, с получением соединения 22 (17,1 г, 89% выход) в виде желтого твердого вещества (CH-LM-02-30).
Желтое твердое вещество, температура плавления 167,1-169,0°C; анализ методом HPLC: 99,2% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 9,77 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=370,1 (M+H)+; C21H23NO5; 1H ЯМР (400 МГц, CDCl3) δ=7,91 (д, J=8,3 Гц, 1H), 7,43 (д, J=16,1 Гц, 1H), 7,12 (ддд, J=0,5, 1,7, 8,3 Гц, 1H), 7,11 (уш.д, J=1,5 Гц, 1H), 6,91 (д, J=16,1 Гц, 1H), 6,68 (д, J=2,4 Гц, 1H), 6,42 (д, J=2,4 Гц, 1H), 5,09 (тм, J=6,8 Гц, 1H), 4,76 (с, 1H), 4,01 (с, 3H), 3,82 (с, 3H), 3,43 (д, J=6,8 Гц, 2H), 1,80 (д, J=0,7 Гц, 3H), 1,68 (д, J=1,2 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=158,67, 154,55, 153,70, 144,15, 138,12, 136,81, 130,97, 130,89, 128,38, 126,57, 123,42, 121,71, 118,09, 111,20, 104,15, 99,44, 56,45, 55,76, 25,74, 24,46, 17,99.
(E)-3-(4-Амино-3-метоксистирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенол (23) [KYN-125].
Порошок цинка (30,3 г, 463 ммоль, 10,0 экв), хлорид аммония (25 г, 463 ммоль, 10,0 экв) и воду (70 мл) добавляли последовательно к раствору соединения 22 (17,1 г, 46,3 ммоль, 1,0 экв) в тетрагидрофуране (700 мл) при комнатной температуре. Полученную суспензию перемешивали при комнатной температуре в течение 4 часов. Суспензию фильтровали, и твердые вещества промывали этилацетатом (1 л). Фильтрат испаряли досуха при пониженном давлении. Остаток очищали пятью равными порциями на автоматизированной хроматографической системе InterChim (5 колонок 120 g), элюируя каждый раз с градиентом от 0 до 40% этилацетата в гептанах, с получением соединения 23 (10,5 г, 67% выход) в виде желтовато-белого твердого вещества (CH-LM-02-31).
Температура плавления 159,3-163,3°C; анализ методом HPLC: 94,8% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 7,80 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=340,2 (M+H)+; C21H25NO3; 1H ЯМР (400 МГц, CDCl3) δ=7,10 (д, J=16,1 Гц, 1H), 6,95 (д, J=1,7 Гц, 1H), 6,92 (дд, J=1,9, 7,9 Гц, 1H), 6,81 (д, J=15,9 Гц, 1H), 6,69 (д, J=7,8 Гц, 1H), 6,62 (д, J=2,5 Гц, 1H), 6,34 (д, J=2,4 Гц, 1H), 5,12 (тм, J=1,5, 6,9 Гц, 1H), 3,89 (с, 3H), 3,80 (с, 3H), 3,41 (д, J=6,6 Гц, 2H), 1,81 (д, J=1,0 Гц, 3H), 1,67 (д, J=0,9 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=158,56, 154,40, 147,42, 138,48, 136,08, 130,88, 130,49, 128,67, 123,77, 122,88, 120,54, 120,50, 114,86, 108,07, 103,83, 97,97, 55,69. 55,45, 25,77, 24,48, 17,97.
(E)-3-(4-Амино-3-метоксистирил)-5-этокси-4-(3-метилбут-2-ен-1-ил)фенол (24) [KYN-141]
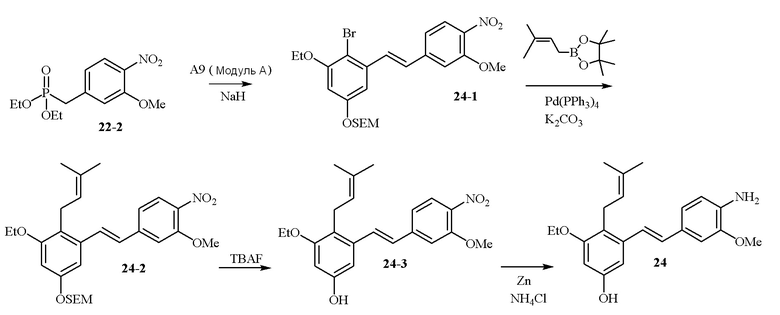
(E)-(2-((4-Бром-3-этокси-5-(3-метокси-4-нитростирил)-фенокси)метокси)этил)триметилсилан (24-1).
60% дисперсию гидрида натрия в минеральном масле (2,7 г, 67,3 ммоль, 3,0 экв) добавляли 3 порциями к раствору соединения 22-2 (6,8 г, 22,4 ммоль, 1,0 экв) в безводном тетрагидрофуране (200 мл) при 0°C. Смесь подогревали до комнатной температуры и перемешивали в течение 30 минут. Добавляли по каплям раствор соединение A9 (модуль A) (8,4 г, 22,4 ммоль, 1,0 экв) в безводном тетрагидрофуране (50 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию осторожно прерывали водой (200 мл, 1 капля в минуту для первых 5 мл воды) при 0°C и экстрагировали метилтрет-бутиловым эфиром (1 л) и этилацетатом (500 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (колонка 220 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 24-1 (9,04 г, 77% выход) в виде желтого твердого вещества (LM-8-98).
(E)-(2-((3-Этокси-5-(3-метокси-4-нитростирил)-4-(3-метилбут-2-ен-1-ил)фенокси)метокси)этил)триметилсилан (24-2).
Тетракис (трифенилфосфин) палладий(0) (1 г, 0,86 ммоль, 0,05 экв), карбонат калия (4,76 г, 34,47 ммоль, 2,0 экв), пинаколовый эфир 3-метилбут-2-енилбороновой кислоты (6,76 г, 34,47 ммоль, 2,0 экв) и воду (30 мл) добавляли последовательно к раствору соединения 24-1 (9,04 г, 17,24 ммоль, 1,0 экв) в 1,4-диоксане (120 мл) в герметизированной пробирке. После продувки азотом в течение 10 минут, реакционную смесь нагревали при 100°C в течение 16 часов. После охлаждения до комнатной температуры, реакционную смесь экстрагировали метилтрет-бутиловым эфиром (200 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (2 колонки 200 g), элюируя с градиентом от 0 до 7% этилацетата в гептанах, с получением соединения 24-2 (8,84 г, 99% выход) в виде желтого масла. (LM-8-99).
(E)-3-Этокси-5-(3-метокси-4-нитростирил)-4-(3-метилбут-2-ен-1-ил)фенол (24-3).
1M раствор тетрабутиламмония фторида в тетрагидрофуране (130 мл, 130 ммоль, 7,0 экв) добавляли к раствору соединения 24-2 (9,53 г, 18,55 ммоль, 1,0 экв) в тетрагидрофуране (200 мл) при комнатной температуре. После нагревания при 68°C в течение 16 часов, смесь охлаждали до комнатной температуры и разбавляли водой (50 мл). Летучие компоненты удаляли при пониженном давлении. Остаток разбавляли насыщенным водным раствором хлорида аммония (250 мл) и экстрагировали этилацетатом (2×400 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (колонка 330 g), элюируя с градиентом от 0 до 60% этилацетата в гептанах, с получением соединения 24-3 (5,05 г, 71% выход) в виде желтого твердого вещества. (LM-8-100).
(E)-3-(4-Амино-3-метоксистирил)-5-этокси-4-(3-метилбут-2-ен-1-ил)фенол (24).
Порошок цинка (8,61 г, 131,7 ммоль, 10,0 экв), хлорид аммония (7,1 г, 131,7 ммоль, 10,0 экв) и воду (20 мл) добавляли последовательно к раствору соединения 24-3 (5,05 г, 13,17 ммоль, 1,0 экв) в тетрагидрофуране (200 мл) при комнатной температуре. Полученную суспензию перемешивали при комнатной температуре в течение 16 часов. Суспензию фильтровали через слой целита (50 г), который споласкивали этилацетатом (400 мл). Фильтрат испаряли досуха при пониженном давлении. Остаток очищали два раза на автоматизированной хроматографической системе InterChim (2 колонки 120 g), элюируя каждый раз с градиентом от 0 до 100% этилацетата в гептанах, с получением соединения 24 (4,56 г, 98% выход) в виде желтовато-белого твердого вещества. (LM-8-101).
Желтовато-белое твердое вещество, температура плавления 187-196°C; анализ методом HPLC: 97,9% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 8,3 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=354,2 (M+H)+; C22H27NO3; 1H ЯМР (400 МГц, CDCl3) δ=7,11 (уш.д, J=16,0 Гц, 1H), 6,95 (уш.с, 1H), 6,92 (уш.д, J=7,7 Гц, 1H), 6,80 (уш.д, J=16,0 Гц, 1H), 6,70 (уш.д, J=7,9 Гц, 1H), 6,59 (уш.с, 1H), 6,31 (уш.с, 1H), 5,14 (уш.т, J=5,9 Гц, 1H), 4,90 (уш.с, 1H), 3,99 (кв, J=6,6 Гц, 2H), 3,90 (с, 5H), 3,42 (уш.д, J=6,1 Гц, 2H), 1,91-1,74 (м, 3H), 1,74-1,61 (м, 3H), 1,41 (уш.т, J=6,8 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=157,88, 154,28, 147,40, 138,44, 136,10, 130,80, 130,30, 128,66, 123,84, 122,96, 120,67, 120,51, 114,83, 108,10, 103,74, 98,82, 63,90, 55,46, 25,79, 24,55, 18,03, 14,93.
(E)-N-(4-(5-гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)метансульфонамид (25) [KYN-137] и (Z)-N-(4-(5-гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)стирил)-2-метоксифенил)метансульфонамид (26) [KYN-129]
(E*Z*)-3-метокси-5-(3-метокси-4-(метилсульфонамидо)стирил)-4-(3-метилбут-2-ен-1-ил)фенилметансульфонат (25 и 26).
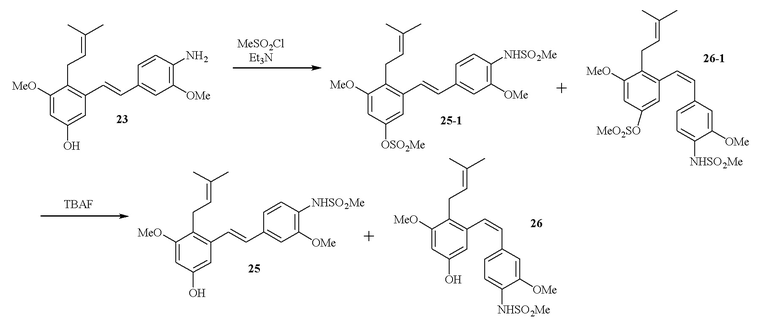
Триэтиламин (0,15 мл, 1,08 ммоль, 3 экв) и метансульфонил-хлорид (0,1 мл, 0,70 ммоль, 2 экв) последовательно добавляли к раствору соединения 23 (120 мг, 0,36 ммоль, 1,0 экв) в безводном THF (10 мл) при комнатной температуре. После кипячения с обратным холодильником (66°C) в течение 16 часов, реакционную смесь охлаждали до комнатной температуры, и летучие компоненты удаляли при пониженном давлении. Остаток растворяли в этилацетате (20 мл) и промывали насыщенным раствором бикарбоната натрия (20 мл). Водный слой экстрагировали этилацетатом (2 X 15 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 30% этилацетата в гептанах, с получением соединений 25-1 и 26-1 (100 мг, 66% выход) в виде белого твердого вещества. (NRK-1-74)
1M раствор тетрабутиламмония фторида в THF (2 мл, 1,63 ммоль, 7 экв) добавляли к раствору соединений 25-1 и 26-1 (100 мг, 0,234 ммоль, 1 экв) в безводном THF (10 мл) при комнатной температуре. После кипячения с обратным холодильником (66°C) в течение 16 часов, анализ методом LCMS показывал, что реакция завершена. Затем смесь охлаждали до комнатной температуры и разбавляли насыщенным водным раствором хлорида аммония (10 мл). Летучие компоненты удаляли при пониженном давлении, и остаток разбавляли этилацетатом (20 мл). Слои разделяли, и водный слой экстрагировали этилацетатом (2×15 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка RediSep Rf Gold HP C18, 50 g), элюируя с градиентом от 0 до 40% ацетонитрила в воде, с получением соединения 25 [KYN-137] (15,0 мг) в виде желтовато-белого твердого вещества и соединения 26 [KYN-129] (19 мг, 22% выход) в виде светло-коричневого масла.
(E)-N-(4-(5-гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)метансульфонамид (25) [KYN-137]
Желтовато-белое твердое вещество; температура плавления 170,7-173,5°C; анализ методом HPLC: 92,4% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 8,4 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=418,1 (M+H)+; C22H27NO5S; 1H ЯМР (400 МГц, CDCl3) δ=7,50 (д, J=8,2 Гц, 1H), 7,27 (с, 1H), 7,26 (с, 1H), 7,25-7,22 (м, 1H), 7,02 (д, J=1,6 Гц, 1H), 6,88 (д, J=16,0 Гц, 1H), 6,68 (д, J=2,3 Гц, 1H), 6,39 (д, J=2,3 Гц, 1H), 5,11 (септет, J=1,3, 6,8 Гц, 1H), 4,92 (уш.с, 1H), 3,93 (с, 3H), 3,81 (с, 3H), 3,42 (уш.д, J=6,7 Гц, 2H), 2,97 (с, 3H), 1,81 (с, 3H), 1,68 (д, J=1,0 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=158,60, 154,52, 149,51, 137,63, 135,35, 130,67, 129,64, 126,58, 125,48, 123,62, 120,99, 120,75, 120,06, 108,33, 104,01, 98,68, 55,77, 55,72, 39,14, 25,76, 24,47, 17,99.
(Z)-N-(4-(5-гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)метансульфонамид (26) [KYN-129]
Светло-коричневое масло; анализ методом HPLC: 97,8% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 9,2 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=418,1 (M+H)+; C22H27NO5S; 1H ЯМР (400 МГц, CDCl3) δ=7,31 (д, J=8,3 Гц, 1H), 6,77 (дд, J=1,7, 8,4 Гц, 1H), 6,66 (д, J=1,7 Гц, 1H), 6,63 (д, J=12,3 Гц, 1H), 6,50 (д, J=12,2 Гц, 1H), 6,35 (д, J=2,5 Гц, 1H), 6,19 (д, J=2,2 Гц, 1H), 5,08 (тм, J=1,4, 6,9 Гц, 1H), 3,79 (с, 3H), 3,50 (с, 3H), 3,27 (уш.д, J=7,1 Гц, 2H), 2,90 (с, 3H), 1,68 (с, 3H), 1,58 (д, J=0,9 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=158,87, 154,74, 148,69, 138,86, 134,39, 130,97, 129,90, 129,71, 124,78, 122,90, 122,67, 120,75, 120,01, 111,24, 107,31, 98,13, 55,65, 55,32, 39,14, 25,75, 25,66, 17,87.
(E)-N-(4-(5-Гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)формамид (27) [KYN-149]
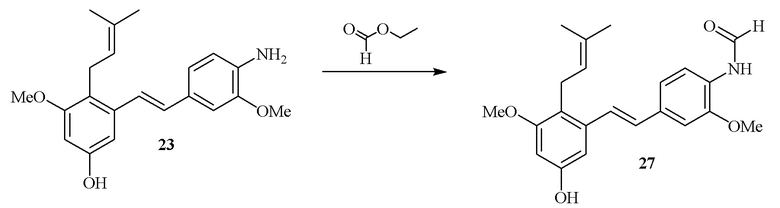
Соединение 23 [KYN-125] (120 мг, 0,35 ммоль, 1,0 экв) добавляли к этилформиату (15 мл) с получением мутного раствора, который нагревали при 55°C в течение 20 часов (раствор становился прозрачным при нагревании). Реакционную смесь охлаждали до комнатной температуры и испаряли досуха при пониженном давлении. Остаток растворяли в этилацетате (20 мл) и промывали насыщенным водным раствором хлорида аммония (20 мл). Водный слой экстрагировали этилацетатом (2×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток изначально очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 60% этилацетата в гептанах. Продукт растирали с 10% этилацетата в гептанах (10 мл) с получением соединения 27 (50 мг, 38% выход) в виде желтовато-белого твердого вещества (NRK-1-159).
(E)-N-(4-(5-гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)формамид (27) [KYN-149]
Белое твердое вещество, температура плавления 181,2-181,7°C; анализ методом HPLC: 99,6% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 8,4 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации отрицательных ионов) m/z=733,3 (2M-H)-; C22H25NO4; 1H ЯМР (400 МГц, DMSO-d6) δ=9,69 (с, 1H), 9,24 (с, 1H), 8,30 (д, J=1,8 Гц, 1H), 8,16 (д, J=8,2 Гц, 1H) 7,31-7,22 м, 2H), 7,09 (д, J=8,6 Гц, 1H), 6,90 (д, J=16,1 Гц, 1H), 6,64 (д, J=2,1 Гц, 1H), 6,35 (д, J=2,2 Гц, 1H), 5,00 (септет, J=1,4, 6,8 Гц, 1H), 3,91 (с, 3H), 3,73 (с, 3H), 3,36 (уш.д, J=6,7 Гц, 2H), 1,76 (с, 3H), 1,60 (с, 3H); 13C ЯМР (100 МГц, DMSO-d6) δ=160,45, 158,45, 156,69, 149,10, 137,43, 133,78, 129,85, 127,03, 125,99, 124,50, 120,63, 119,75, 118,74, 109,00, 104,13, 99,16, 56,25. 55,91, 25,96, 24,28, 18,21.
E)-N-(4-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)формамид (28) [KYN-143]
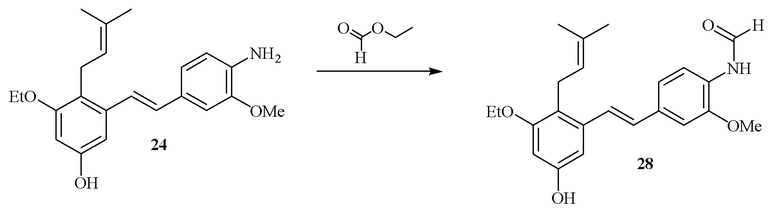
(E)-N-(4-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)формамид (28) [KYN-143].
Соединение 24 (100 мг, 0,29 ммоль, 1,0 экв) добавляли к этилформиату (10 мл) с получением мутного раствора, который нагревали при 55°C в течение 20 часов (раствор становился прозрачным при нагревании). Реакционную смесь охлаждали до комнатной температуры и испаряли досуха при пониженном давлении. Остаток растворяли в этилацетате (20 мл) и промывали насыщенным водным раствором хлорида аммония (20 мл). Водный слой экстрагировали этилацетатом (2×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток изначально очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 12 g), элюируя с градиентом от 0 до 50% этилацетата в гептанах. Продукт растирали с 10% этилацетата в гептанах (10 мл) с получением соединения 28 [KYN-143] (45 мг, 64% выход) в виде желтовато-белого твердого вещества (NRK-1-145).
Желтовато-белое твердое вещество, температура плавления 173-176°C; анализ методом HPLC: 98,9% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 9,0 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации отрицательных ионов) m/z=761,3 (M-H)-; C23H27NO4; 1H ЯМР (400 МГц, DMSO-d6) δ=9,69 (с, 1H), 9,20 (с, 1H), 8,30 (д, J=1,7 Гц, 1H), 8,16 (д, J=8,3 Гц, 1H), 7,29 (д, J=16,1 Гц, 1H), 7,25 (д, J=1,1 Гц, 1H), 7,10 (уш.д, J=8,3 Гц, 1H), 6,90 (д, J=16,0 Гц, 1H), 6,62 (д, J=2,1 Гц, 1H), 6,32 (д, J=2,0 Гц, 1H), 5,01 (уш.т, J=7,0 Гц, 1H), 3,95 (кв, J=7,0 Гц, 2H), 3,91 (с, 3H), 3,37 (уш.д, J=6,7 Гц, 2H), 1,76 (с, 3H), 1,60 (с, 3H), 1,33 (т, J=6,9 Гц, 3H); 13C ЯМР (100 МГц, DMSO-d6) δ=160,44, 157,71, 156,59, 149,09, 137,40, 133,79, 129,74 (2C), 127,01, 126,04, 124,52, 120,62, 119,72, 118,90, 109,03, 104,1, 99,96, 63,74, 56,27, 25,99, 24,34, 18,25, 15,25.
(E)-N-(4-(5-гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)ацетамид (29) [KYN-128]:
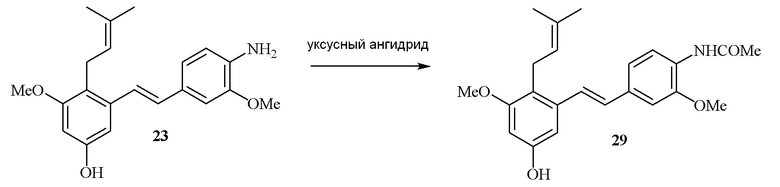
N, N-Диизопропилэтиламин (0,1 мл, 0,38 ммоль, 2 экв) и уксусный ангидрид (0,1 мл, 0,38 ммоль, 2 экв) последовательно добавляли к раствору соединения 23 [KYN-125] (65 мг, 0,19 ммоль, 1,0 экв) в безводном THF (10 мл) при комнатной температуре. После кипячения с обратным холодильником (66°C) в течение 16 часов, реакционную смесь охлаждали до комнатной температуры, и летучие компоненты удаляли при пониженном давлении. Остаток растворяли в этилацетате (20 мл) и промывали насыщенным раствором бикарбоната натрия (20 мл). Водный слой экстрагировали этилацетатом (2×10 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка RediSep Rf Gold HP C18, 50 g), элюируя с градиентом от 0 до 80% ацетонитрила в воде, с получением относительно чистого соединения 29, которое дополнительно очищали растиранием с 30% дихлорметана в гептанах (10 мл) с получением чистого соединения 29 [KYN-128] (17 мг, 23% выход) в виде белого твердого вещества (NRK-1-66) .
Желтовато-белое серое твердое вещество, температура плавления 205,7-205,9°C; анализ методом HPLC: 97,4% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 8,4 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=382,2 (M+H)+; C23H27NO4; 1H ЯМР (400 МГц, CDCl3) δ=8,40 (д, J=8,8 Гц, 1H), 7,84 (с, 1H), 7,23 (д, J=16,1 Гц, 1H), 7,04 (д, J=1,8 Гц, 1H), 7,03 (дд, J=1,7, 6,0 Гц, 1H), 6,88 (д, J=16,1 Гц, 1H), 6,79 (д, J=2,3 Гц, 1H), 6,39 (д, J=2,3 Гц, 1H), 6,24 (с, 1H), 5,12 (тм, J=1,4, 6,8 Гц, 1H), 3,93 (с, 3H), 3,81 (с, 3H), 3,42 (д, J=6,7 Гц, 2H), 2,24 (с, 3H), 1,81 (д, J=0,6 Гц, 3H), 1,67 (д, J=1,0 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=168,65, 158,58, 155,07, 147,91, 137,68, 133,65, 130,42, 129,84, 127,11, 125,77, 123,86, 120,71, 120,44, 119,67, 106,87, 103,98, 98,62, 55,68, 55,65, 25,75, 24,88, 24,45, 17,98.
(E)-N-(4-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)ацетамид (30) [KYN-142]
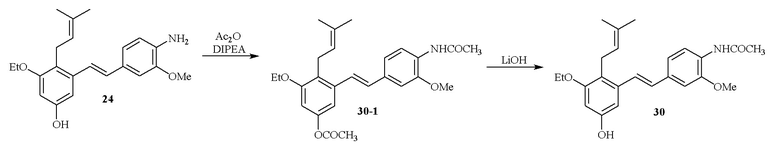
(E)-3-(4-Ацетамидо-3-метоксистирил)-5-этокси-4-(3-метилбут-2-ен-1-ил)фенилацетат (30-1).
N, N-Диизопропилэтиламин (0,25 мл, 1,36 ммоль, 4 экв) и уксусный ангидрид (0,1 мл, 0,75 ммоль, 2,2 экв) последовательно добавляли к раствору соединения 24 (120 мг, 0,34 ммоль, 1,0 экв) в безводном THF (20 мл) при комнатной температуре. После кипячения с обратным холодильником (66°C) в течение 16 часов, реакционную смесь охлаждали до комнатной температуры, и летучие компоненты удаляли при пониженном давлении. Остаток растворяли в этилацетате (20 мл) и промывали насыщенным раствором бикарбоната натрия (20 мл). Водный слой экстрагировали этилацетатом (2×10 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 30-1 (136 мг, 92% выход) в виде бледно-желтого твердого вещества, которое использовали на следующей стадии (NRK-1-133).
(E)-N-(4-(3-этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)ацетамид (30) [KYN-142].
1M раствор гидроксида лития (1,6 мл, 1,55 ммоль, 5,0 экв) добавляли к раствору соединения 30-1 (136 мг, 0,31 ммоль, 1,0 экв) в тетрагидрофуране (10 мл) при комнатной температуре. Полученный раствор перемешивали при комнатной температуре в течение 4 часов. Летучие компоненты удаляли при пониженном давлении. Остаток растворяли в этилацетате (20 мл) и промывали насыщенным водным раствором хлорида аммония (10 мл). Водный слой экстрагировали этилацетатом (2×10 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 12 g), элюируя с градиентом от 0 до 50% этилацетата в гептанах. Продукт растирали с диэтиловым эфиром (5 мл) с получением чистого соединения 30 [KYN-142](20 мг, 17% выход) в виде белого твердого вещества (NRK-1-135).
Желтовато-белое твердое вещество, температура плавления 188-189°C; анализ методом HPLC: 99,3% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 9,0 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=396,2 (M+H)+; C24H29NO4; 1H ЯМР (400 МГц, CDCl3) δ=8,41 (д, J=8,1 Гц, 1H), 7,85 (с, 1H), 7,26-7,22 (м, 1H), 7,06-7,00 m, 2H), 6,88 (д, J=16,0 Гц, 1H), 6,79 (д, J=2,1 Гц, 1H), 6,42-6,31 (м, 2H), 5,14 (септет, J=1,2, 6,8 Гц, 1H), 4,00 (кв, J=6,9 Гц, 2H), 3,93 (с, 3H), 3,44 (уш.д, J=6,7 Гц, 2H), 2,24 (с, 3H), 1,82 (с, 3H), 1,68 (с, 3H), 1,42 (т, J=7,0 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=168,71, 157,91, 155,05, 147,92, 137,60, 133,73, 130,22, 129,71, 127,06, 125,90, 123,96, 120,78, 120,55, 119,67, 106,82, 103,91, 99,52, 63,88, 55,68, 25,79, 24,88, 24,53, 18,04, 14,93.
(E)-2-Амино-N-(4-(5-гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)стирил)-2-метоксифенил)ацетамид (31) [KYN-151]
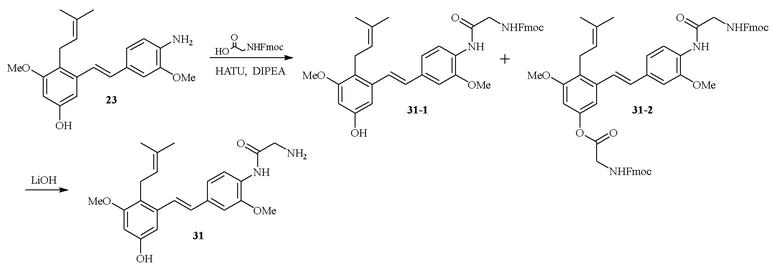
(9H-Флуорен-9-ил)метил (E)-(2-((4-(5-гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)стирил)-2-метоксифенил)амино)-2-оксоэтил)-карбамат.
Fmoc-глицин (520 мг, 1,76 ммоль, 3,0 экв), N, N-диизопропилэтиламин (0,5 мл, 2,95 ммоль, 5,0 экв) и 1-[бис(ди-метиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния 3-оксида гексафторфосфат (HATU) (900 мг, 2,36 ммоль, 4,0 экв) последовательно добавляли к раствору (E)-3-(4-амино-3-метокси-стирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенола (соединение 23) [KYN-125] (200 мг, 0,59 ммоль, 1,0 экв) в дихлорметане (10 мл). Полученный раствор перемешивали при комнатной температуре в течение 16 часов. Анализ методом LC-MS указывал на образование соединения 11 и соединения 12. Реакционную смесь разбавляли дихлорметаном (20 мл) и промывали насыщенным раствором бикарбоната натрия (30 мл). Водный слой экстрагировали дихлорметаном (2×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 60% этилацетата в гептанах, с получением смесь соединения 31-1 и соединения 31-2 (330 мг) в виде желтовато-белого твердого вещества (NRK-1-161).
(E)-2-амино-N-(4-(5-гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)стирил)-2-метоксифенил)ацетамид (31).
1M раствор гидроксида лития (5 мл) добавляли к раствору соединения 31-1 и соединения 31-2 (330 мг) в тетрагидрофуране при комнатной температуре. После перемешивания в течение 16 часов, летучие компоненты удаляли при пониженном давлении. Остаток растворяли в этилацетате (30 мл) и промывали насыщенным водным раствором хлорида аммония (30 мл). Водный слой экстрагировали этилацетатом (2×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 5% метанола в дихлорметане, содержащим 1% 7M раствора аммиака в метаноле, с получением соединения 31 (25,0 мг, 23% выход за 2 стадии) в виде желтовато-белого твердого вещества (NRK-1-164).
(E)-2-амино-N-(4-(5-гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)стирил)-2-метоксифенил)ацетамид (31) [KYN-151]
Желтовато-белое твердое вещество, температура плавления 196,0-201,9°C; анализ методом HPLC: 98,7% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 5,78 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=397,2 (M+H)+; C23H28N2O4; 1H ЯМР (400 МГц, DMSO-d6) δ=10,10 (уш.с, 1H), 9,24 (с, 1H), 8,29 (д, J=8,3 Гц, 1H), 7,30-7,23 (м, 2H), 7,10 (дд, J=1,7, 8,4 Гц, 1H), 6,90 (д, J=16,0 Гц, 1H), 6,63 (д, J=2,2 Гц, 1H), 6,35 (д, J=2,2 Гц, 1H), 5,00 (септет, J=1,2, 7,0 Гц, 1H), 3,92 (с, 3H), 3,73 (с, 3H), 3,38-3,33 (м, 2H), 3,28-3,24 (м, 2H), 2,33 (уш.с, 2H), 1,76 (с, 3H), 1,60 (с, 3H); 13C ЯМР (100 МГц, DMSO-d6) δ=171,88, 158,45, 156,68, 148,71, 137,47, 133,12, 129,92, 129,83, 127,42, 125,67, 124,51, 119,96, 118,87, 118,69, 108,71, 104,09, 99,11, 56,30. 55,91, 45,67, 25,95, 24,28, 18,20.
(E)-2-Амино-N-(4-(3-этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)стирил)-2-метоксифенил)ацетамид (32) [KYN-148]
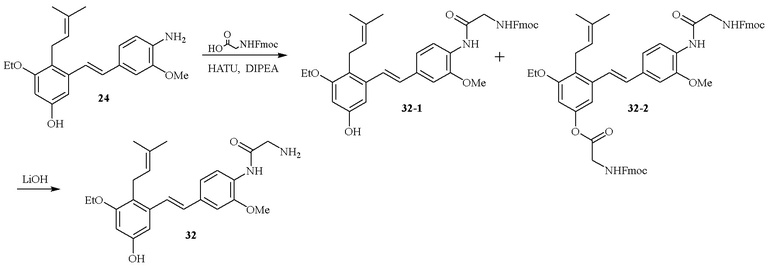
(9H-Флуорен-9-ил)метил (E)-(2-((4-(3-этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)стирил)-2-метоксифенил)амино)-2-оксоэтил)-карбамат (12).
Fmoc-глицин (500 мг, 1,71 ммоль, 3,0 экв), N, N-диизопропилэтиламин (0,5 мл, 2,85 ммоль, 5,0 экв) и 1-[бис(ди-метиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния 3-оксида гексафторфосфат (HATU) (860 мг, 2,28 ммоль, 4,0 экв) последовательно добавляли к раствору соединения 24 (200 мг, 0,57 ммоль, 1,0 экв) в дихлорметане (10 мл). Полученный раствор перемешивали при комнатной температуре в течение 16 часов. Анализ методом LC-MS указывал на образование соединения 32-1 и соединения 32-2. Реакционную смесь разбавляли дихлорметаном (20 мл) и промывали насыщенным раствором бикарбоната натрия (30 мл). Водный слой экстрагировали дихлорметаном (2×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 60% этилацетата в гептанах, с получением смеси соединения 32-1 и соединения 32-2 (550 мг) в виде желтовато-белого твердого вещества (NRK-1-158).
(E)-2-Амино-N-(4-(3-этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)стирил)-2-метоксифенил)ацетамид (32) [KYN-148].
1M раствор гидроксида лития (5 мл) добавляли к раствору соединения 32-1 и соединения 32-2 (550 мг) в тетрагидрофуране (20 мл) при комнатной температуре. После перемешивания в течение 16 часов, летучие компоненты удаляли при пониженном давлении. Неочищенное вещество растворяли в этилацетате (30 мл) и промывали насыщенным водным раствором хлорида аммония (30 мл). Водный слой экстрагировали этилацетатом (2×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток сначала растирали с 30% этилацетата в гептанах (30 мл) с получением бежевого твердого вещества, которое дополнительно очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 5% метанола в дихлорметане, содержащим 1% 7M раствора аммиака в метаноле, с получением соединения 32 [KYN-148] (30,0 мг, 13% выход за 2 стадии) в виде желтовато-белого твердого вещества (NRK-1-162).
Желтовато-белое твердое вещество, температура плавления 200,0-203,7°C; анализ методом HPLC: 99,5% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 8,25 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=411,2 (M+H)+; C24H30N2O4; 1H ЯМР (400 МГц, DMSO-d6) δ=10,08 (уш.с, 1H), 9,20 (с, 1H), 8,29 (д, J=8,3 Гц, 1H), 7,32-7,23 (м, 2H), 7,11 (дд, J=1,5, 8,4 Гц, 1H), 6,90 (д, J=16,0 Гц, 1H), 6,63 (д, J=2,1 Гц, 1H), 6,32 (д, J=2,1 Гц, 1H), 5,01 (уш.т, J=7,0 Гц, 1H), 3,95 (кв, J=6,8 Гц, 2H), 3,92 (с, 3H), 3,40-3,34 (м, 2H), 3,29-3,24 (м, 2H), 2,33 (уш.с, 2H), 1,77 (с, 3H), 1,61 (с, 3H), 1,33 (т, J=6,9 Гц, 3H); 13C ЯМР (100 МГц, DMSO-d6) δ=171,88, 157,72, 156,59, 148,71, 137,46, 133,14, 129,87, 129,73, 127,41, 125,74, 124,54, 119,93, 118,86, 108,74, 104,08, 99,92, 63,74, 56,32, 45,67, 25,99, 24,35, 18,25, 15,25.
Метил (E)-(4-(5-гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)глицинат (33) [KYN-126].
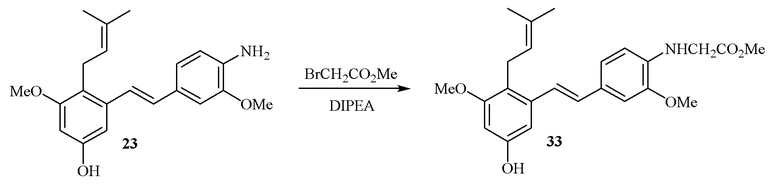
N, N-Диизопропилэтиламин (0,5 мл, 2,92 ммоль, 4 экв) и метилбромацетат (0,2 мл, 2,21 ммоль, 3 экв) последовательно добавляли к раствору соединения 23 [KYN-125] в безводном THF (10 мл) при комнатной температуре. После кипячения с обратным холодильником (66°C) в течение 16 часов, реакционную смесь охлаждали до комнатной температуры, и летучие компоненты удаляли при пониженном давлении. Остаток растворяли в этилацетате (30 мл) и промывали насыщенным раствором бикарбоната натрия (20 мл). Водный слой экстрагировали этилацетатом (2 X 30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка RediSep Rf Gold HP C18, 50 g), элюируя с градиентом от 0 до 70% ацетонитрила в воде, с получением соединения 33 [KYN-126] (180 мг, 59% выход) в виде желтовато-белого твердого вещества (NRK-1-65-1).
Желтовато-белое серое твердое вещество, температура плавления 147,3-160,2°C; анализ методом HPLC: 98,1% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: Luna C18(2), 2,0×20 мм, 3 мкм; время удерживания: 3,84 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=412,2 (M+H)+; C24H29NO5; 1H ЯМР (400 МГц, CDCl3) δ=7,10 (д, J=16,2 Гц, 1H), 6,97 (дд, J=1,6, 6,7 Гц, 1H), 6,96 (уш.с, 1H), 6,84 (д, J=15,8 Гц, 1H), 6,65 (д, J=2,4 Гц, 1H), 6,45 (д, J=8,6 Гц, 1H), 6,34 (д, J=2,4 Гц, 1H), 5,13 (тм, J=1,5, 6,9 Гц, 1H), 4,92 (уш.т, J= 4,3 Гц, 1H), 4,75 (уш.с, 1H), 3,98 (д, J=5,1 Гц, 2H), 3,91 (с, 3H), 3,80 (с, 3H), 3,79 (с, 3H), 3,41 (д, J=6,6 Гц, 2H), 1,82 (д, J=0,7 Гц, 3H), 1,68 (д, J=1,2 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=171,44, 158,55, 154,37, 147,19, 138,52, 137,00, 130,88, 130,47, 127,54, 123,79, 122,63, 120,85, 120,50, 109,64, 107,10, 103,78, 97,92, 55,69, 55,47, 52,26, 45,43, 25,76, 24,47, 17,98.
Метил (E)-(4-(3-этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)глицинат (34) [KYN-147]
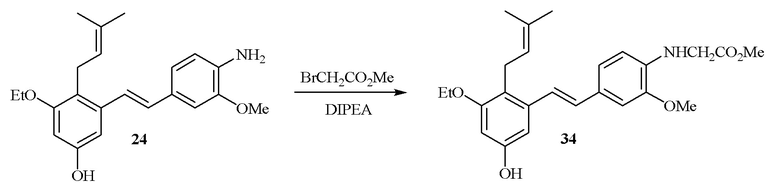
Метил (E)-(4-(3-этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)глицинат (34) [KYN-147].
N, N-Диизопропилэтиламин (0,4 мл, 2,24 ммоль, 4 экв) и метилбромацетат (0,21 мл, 1,68 ммоль, 3 экв) последовательно добавляли к раствору соединения 24 (200 мг, 0,56 ммоль, 1,0 экв) в безводном THF (20 мл) при комнатной температуре. После кипячения с обратным холодильником (66°C) в течение 16 часов, реакционную смесь охлаждали до комнатной температуры, и летучие компоненты удаляли при пониженном давлении. Остаток растворяли в этилацетате (30 мл) и переносили в делительную воронку, содержащую насыщенный раствор бикарбоната натрия раствор (20 мл). Слои разделяли, и водный слой экстрагировали этилацетатом (2×30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток изначально очищали на автоматизированной хроматографической системе Reveleris (колонка Redisep Rf Gold HP C18, 50 g), элюируя с градиентом от 0 до 60% ацетонитрила в воде. Продукт растирали с 10% этилацетата в гептанах (10 мл) с получением соединения 34 [KYN-147] (40 мг, 17% выход) в виде желтовато-белого твердого вещества (NRK-1-151).
Желтовато-белое твердое вещество, температура плавления 144-169°C; анализ методом HPLC: 97,3% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 10,06 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=426,2 (M+H)+; C25H31NO5; 1H ЯМР (400 МГц, CDCl3) δ=7,11 (д, J=16,0 Гц, 1H), 6,99-6,94 (м, 2H), 6,84 (уш.д, J=16,0 Гц, 1H), 6,63 (д, J=2,3 Гц, 1H), 6,45 (д, J=8,6 Гц, 1H), 6,31 (д, J=2,4 Гц, 1H), 5,14 (септет, J=1,3, 7,0 Гц, 1H), 4,92 (уш.с, 1H), 4,73 (уш.с, 1H), 3,99 (кв, J=7,0 Гц, 2H), 3,97 (с, 2H), 3,91 (с, 3H), 3,79 (с, 3H), 3,43 (уш.д, J=6,8 Гц, 2H), 1,82 (с, 3H), 1,68 (д, J=0,9 Гц, 3H), 1,41 (т, J=7,0 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=171,44, 157,88, 154,27, 147,20, 138,45, 136,96, 130,77, 130,28, 127,59, 123,86, 122,74, 120,82, 120,65, 109,66, 107,14, 103,69, 98,79, 63,91, 55,48, 52,25, 45,44, 25,79, 24,54, 18,04, 14,92.
(E)-2-((4-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)амино)-2-оксоуксусная кислота (35) [KYN-152]
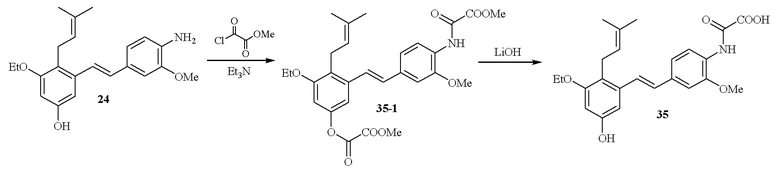
(E)-3-Этокси-5-(3-метокси-4-(2-метокси-2-оксоацетамидо)-стирил)-4-(3-метилбут-2-ен-1-ил)фенилметилоксалат (35-1).
Метилхлороксоацетат (0,1 мл, 0,85 ммоль, 2,5 экв) добавляли к раствору триэтиламина (0,25 мл, 1,7 ммоль, 5,0 экв) и соединения 24 [KYN-141] (120 мг, 0,34 ммоль, 1,0 экв) в тетрагидрофуране (20 мл) при 5°C. Реакционную смесь подогревали до комнатной температуры и перемешивали в течение 16 часов. Реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток растворяли в этилацетате (20 мл) и промывали насыщенным водным раствором хлорида аммония (20 мл). Водный слой экстрагировали этилацетатом (2×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 60% этилацетата в гептанах, с получением соединения 35-1 (130,0 мг, 73%) в виде бежевого твердого вещества (NRK-1-168).
(E)-2-((4-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)амино)-2-оксоуксусная кислота (35) [KYN-152]
1M раствор гидроксида лития (0,75 мл, 0,75 ммоль, 3,0 экв) добавляли к раствору соединения 35-1 (130 мг, 0,25 ммоль, 1,0 экв) в тетрагидрофуране (20 мл) при комнатной температуре. После перемешивания в течение 16 часов, летучие компоненты удаляли при пониженном давлении. Остаток растворяли в этилацетате (30 мл) и промывали с помощью 1M HCl (10 мл). Водный слой экстрагировали этилацетатом (2×30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток растирали с 10% этилацетата в гептанах (20 мл) с получением соединения 35 [KYN-152] (42,0 мг, 38% выход) в виде желтовато-белого твердого вещества (NRK-1-170).
Желтовато-белое твердое вещество, температура плавления 207,8-207,9°C; анализ методом HPLC: 99,1% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 8,24 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации отрицательных ионов) m/z=424,1 (M-H)-; C24H27NO6; 1H ЯМР (400 МГц, DMSO-d6) δ=9,63 (с, 1H), 9,21 (уш.с, 1H), 8,10 (д, J=8,3 Гц, 1H), 7,38-7,29 (м, 2H), 7,18 (уш.д, J=8,4 Гц, 1H), 6,93 (д, J=16,1 Гц, 1H), 6,63 (д, J=2,0 Гц, 1H), 6,33 (д, J=2,1 Гц, 1H), 5,01 (уш.т, J=7,0 Гц, 1H), 4,06-3,91 (м, 5H), 3,38 (уш.д, J=6,8 Гц, 2H), 1,76 (с, 3H), 1,60 (с, 3H), 1,33 (т, J=6,9 Гц, 3H); 13C ЯМР (100 МГц, DMSO-d6) δ=162,25, 157,73, 156,61,156,08, 149,59, 137,30, 135,11, 129,78, 129,62, 126,77, 125,77, 124,51, 120,32, 119,82, 119,04, 109,14, 104,17, 100,10, 63,75, 56,53, 25,99, 24,35, 18,26, 15,25.
(E)-4-((4-(5-Гидрокси-3-метокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)амино)-4-оксобутановая кислота (соединение 36) [KYN-150]
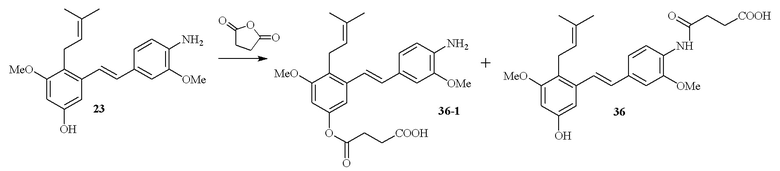
Янтарный ангидрид (140 мг, 1,4 ммоль, 4,0 экв) и N, N-диизопропилэтиламин (0,36 мл, 2,1 ммоль, 6,0 экв) последовательно добавляли к раствору (E)-3-(4-амино-3-метоксистирил)-5-метокси-4-(3-метилбут-2-ен-1-ил)фенола (соединение 23) [KYN-125] (120 мг, 0,35 ммоль, 1,0 экв) в толуоле (20 мл). Полученный мутный раствор кипятили с обратным холодильником (110°C) в течение 16 часов (при нагревании раствор становился прозрачным). Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом (20 мл). Органический слой промывали насыщенным водным раствором хлорида аммония (20 мл). Водный слой экстрагировали этилацетатом (2×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением смеси соединения 36-1 и соединения 36 (210 мг) в виде светло-коричневого масла. Остаток растворяли в тетрагидрофуране (10 мл) и обрабатывали 1M раствором гидроксида лития (1,0 мл) при комнатной температуре в течение 16 часов. Реакционную смесь испаряли досуха, и полученный остаток растворяли в этилацетате (20 мл). Органический слой промывали насыщенным водным раствором хлорида аммония (20 мл). Органический слой сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток растирали с 10% этилацетата в гептанах (10 мл). Продукт суспендировали в диэтиловом эфире (10 мл) и перемешивали при комнатной температуре в течение 16 часов. Суспензию фильтровали с получением соединения 36 (60 мг, 40% выход за 2 стадии) в виде светло-серого твердого вещества (NRK-1-163).
(E)-N-((4-(3-метокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)амино)-4-оксобутановая кислота (соединение 36) [KYN-150]
Светло-серое твердое вещество, температура плавления 169,4-175,7°C; анализ методом HPLC: 97,8% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 7,98 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации отрицательных ионов) m/z=438,2 (M-H)-; C25H29NO6; 1H ЯМР (400 МГц, Ацетонитрил-d3) δ=8,25 (уш.с, 1H), 8,21 (уш.д, J=8,3 Гц, 1H), 7,28 (д, J=16,1 Гц, 1H), 7,17 (д, J=1,7 Гц, 1H), 7,07 (дд, J=1,8, 8,4 Гц, 1H), 6,94 (д, J=16,1 Гц, 1H), 6,68 (д, J=2,3 Гц, 1H), 6,39 (д, J=2,3 Гц, 1H), 5,05 (септет, J=1,4, 6,8 Гц, 1H), 3,92 (с, 3H), 3,77 (с, 3H), 3,41 (уш.д, J=6,8 Гц, 2H), 2,70-2,65 (м, 2H), 2,64-2,60 (м, 2H), 1,80 (д, J=0,7 Гц, 3H), 1,64 (д, J=1,1 Гц, 3H); 13C ЯМР (100 МГц, ацетонитрил-d3) δ=174,42, 171,50, 159,61, 156,96, 138,79, 131,50, 130,93, 128,74, 126,52, 124,90, 120,85, 120,77, 120,69, 109,17, 104,80, 99,68, 56,67. 56,39, 32,51, 29,64, 25,88, 24,99, 18,23.
(E)-4-((4-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)амино)-4-оксобутановая кислота (37) [KYN-144]
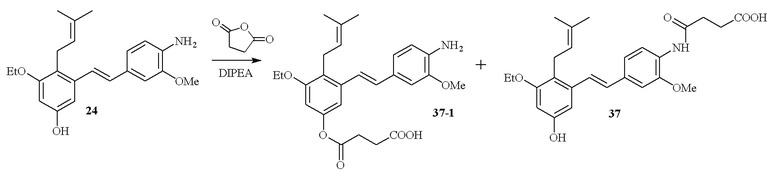
(E)-4-((4-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)амино)-4-оксобутановая кислота (соединение 37) [KYN-144]:
Янтарный ангидрид (58 мг, 0,58 ммоль, 2,0 экв) и N, N-диизопропилэтиламин (0,15 мл, 0,87 ммоль, 3,0 экв) последовательно добавляли к раствору соединения 24 (100 мг, 0,29 ммоль, 1,0 экв) в толуоле (20 мл). Полученный мутный раствор кипятили с обратным холодильником (110°C) в течение 16 часов (при нагревании раствор становился прозрачным). Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом (20 мл). Органический слой промывали насыщенным водным раствором хлорида аммония (20 мл). Водный слой экстрагировали этилацетатом (2×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением смеси соединения 37-1 и соединения 37 (165 мг) в виде светло-коричневого твердого вещества. Остаток растворяли в тетрагидрофуране (10 мл) и обрабатывали 1M раствором гидроксида лития (1,0 мл) при комнатной температуре в течение 16 часов. Реакционную смесь испаряли досуха, и полученный остаток растворяли в этилацетате (20 мл). Органический слой промывали насыщенным водным раствором хлорида аммония (20 мл). Органический слой собирали, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток растирали с 10% этилацетата в гептанах (10 мл). Продукт суспендировали в диэтиловом эфире (10 мл) и перемешивали при комнатной температуре в течение 16 часов. Суспензию фильтровали с получением соединения 37 [KYN-144] (40 мг, 31% выход за 2 стадии) в виде светло-серого твердого вещества (NRK-1-148).
Светло-серое твердое вещество, температура плавления 173-180°C; анализ методом HPLC: 96,4% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 8,52 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации отрицательных ионов) m/z=452,2 (M-H)-; C26H31NO6; 1H ЯМР (400 МГц, Ацетонитрил-d3) δ=8,25 (уш.с, 1H), 8,21 (уш.д, J=8,3 Гц, 1H), 7,29 (д, J=16,1 Гц, 1H), 7,17 (д, J=1,7 Гц, 1H), 7,07 (дд, J=1,7, 8,3 Гц, 1H), 6,94 (д, J=16,1 Гц, 1H), 6,67 (д, J=2,3 Гц, 1H), 6,36 (д, J=2,3 Гц, 1H), 5,07 (септет, J=1,5, 7,0 Гц, 1H), 3,99 (кв, J=7,0 Гц, 2H), 3,92 (с, 3H), 3,43 (уш.д, J=7,0 Гц, 2H), 2,70-2,65 (м, 2H), 2,64-2,60 (м, 2H), 1,80 (с, 3H), 1,64 (д, J=1,0 Гц, 3H), 1,37 (т, J=7,0 Гц, 3H); 13C ЯМР (100 МГц, ацетонитрил-d3) δ=174,41, 171,48, 158,91, 156,86, 149,80, 138,77, 134,49, 131,38, 130,85, 128,72, 126,61, 124,95, 120,95, 120,84, 120,68, 109,18, 104,74, 100,55, 64,93, 55,67, 32,52, 29,64, 25,90, 25,07, 18,32, 15,30.
(E)-1-(4-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)гуанидин (38) [KYN-155]
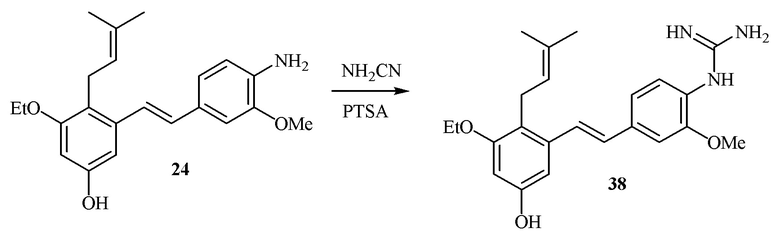
(E)-1-(4-(3-этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)гуанидин (38) [KYN-155].
Цианамид (237 мг, 5,66 ммоль, 20,0 экв) и п-толуолсульфоновую кислоту (1,07 г, 5,66 ммоль, 20,0 экв) добавляли к раствору соединения 24 (100 мг, 0,28 ммоль, 1,0 экв) в этаноле (50 мл). После кипячения с обратным холодильником (79°C) в течение 16 часов, к реакционной смеси добавляли дополнительное количество цианамида (237 мг, 5,66 ммоль, 20,0 экв) и п-толуолсульфоновой кислоты (1,07 г, 5,66 ммоль, 20,0 экв) и кипятили с обратным холодильником в течение еще 16 часов. Реакционную смесь охлаждали до комнатной температуры, и летучие компоненты удаляли при пониженном давлении. Остаток растворяли в этилацетате (50 мл) и промывали насыщенным раствором бикарбоната натрия (50 мл). Водный слой экстрагировали этилацетатом (2×30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Дополнительное соединение 24 (100 мг) превращали в соединение 38 аналогичным образом. Объединенный остаток сначала очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 15% метанола в дихлорметане, содержащем 5% 7M раствора аммиака в метаноле, с получением относительно чистого соединения 38. Продукт дополнительно очищали на автоматизированной хроматографической системе Reveleris (колонка Redisep Rf Gold HP C18, 50 g), элюируя с градиентом от 0 до 45% ацетонитрила в воде, содержащей 10 мM бикарбоната аммония и 5% метанола, с получением соединения 38 [KYN-155] (37,0 мг, 16% выход) в виде белого твердого вещества (NRK-1-177).
Белое твердое вещество, температура плавления 130,5-130,6°C; анализ методом HPLC: 99,7% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 6,45 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=396,2 (M+H)+; C23H29N3O3; 1H ЯМР (400 МГц, DMSO-d6) δ=7,20 (д, J=16,0 Гц, 1H), 7,11 (с, 1H), 7,01 (д, J=7,5 Гц, 1H), 6,96-6,80 (м, 2H), 6,62 (д, J=2,2 Гц, 1H), 6,30 (д, J=2,2 Гц, 1H), 5,06-4,98 (м, 1H), 3,94 (кв, J=6,9 Гц, 2H), 3,82-3,72 (м, 3H), 3,35 (уш.д, J=6,8 Гц, 2H), 1,77 (с, 3H), 1,61 (с, 3H), 1,33 (т, J=6,9 Гц, 3H); 13C ЯМР (100 МГц, DMSO-d6) δ=157,70, 156,60, 153,04, 137,68, 130,36, 129,71, 124,83, 124,59, 124,37, 120,26, 118,60, 110,05, 103,97, 99,72, 63,74, 55,76, 25,99, 24,34, 18,27, 15,27.
(E)-N-(4-(3-этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-метоксифенил)-N-гидроксиацетамид (соединение 39) [KYN-165]
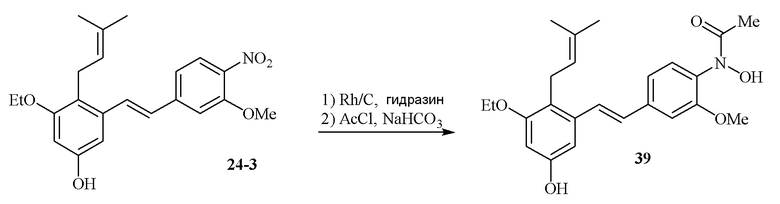
5% родия на активированном угле (5 мг, 0,672 мкмоль, 0,003 экв) и моногидрат гидразина (27 мг, 0,27 ммоль, 1,2 экв) добавляли к раствору соединения 24-3 (86 мг, 0,224 ммоль, 1,0 экв) в тетрагидрофуране (8 мл) при 0°C. После перемешивания при 0°C в течение 1 часа, к реакционной смеси последовательно добавляли бикарбонат натрия (75 мг, 0,896 ммоль, 4 экв) и ацетилхлорид (53 мг, 0,672 ммоль, 3 экв). Смесь подогревали до комнатной температуры и перемешивали в течение 1 часа. Суспензию разбавляли этилацетатом (30 мл) и промывали насыщенным водным раствором хлорида аммония (10 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (колонка 25 g), элюируя с градиентом от 0 до 100% этилацетата в гептанах, с получением соединения 39 (44 мг, 44% выход) в виде желтовато-белого масла. (LM-8-65)
Анализ методом HPLC: 97,2% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: Waters Atlantis T3, 2,1×50 мм, 3 мкм; время удерживания: 8,3 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=412,2 (M+H)+; C24H29NO5; 1H ЯМР (400 МГц, CDCl3) δ=9,69-7,44 (м, 1H), 7,40-7,28 (м, 2H), 7,08 (уш.д, J=6,6 Гц, 1H), 7,02 (с, 1H), 6,87 (уш.д, J=15,4 Гц, 1H), 6,70 (уш.с, 1H), 6,41 (уш.с, 1H), 5,12 (уш.т, J=6,7 Гц, 1H), 3,98 (кв, J=6,9 Гц, 2H), 3,86 (с, 3H), 3,43 (уш.д, J=6,5 Гц, 2H), 1,97 (уш.с, 3H), 1,80 (с, 3H), 1,67 (с, 3H), 1,40 (т, J=6,9 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=167,75, 157,86, 155,23, 154,91, 141,36, 136,98, 130,47, 130,07, 128,96, 126,01, 123,73, 120,97, 119,27, 109,83, 104,13, 100,11, 63,93, 55,75, 25,75, 24,48, 19,41, 18,01, 14,87.
(E)-N-(4-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-гидроксифенил)формамид (40) [KYN-159]
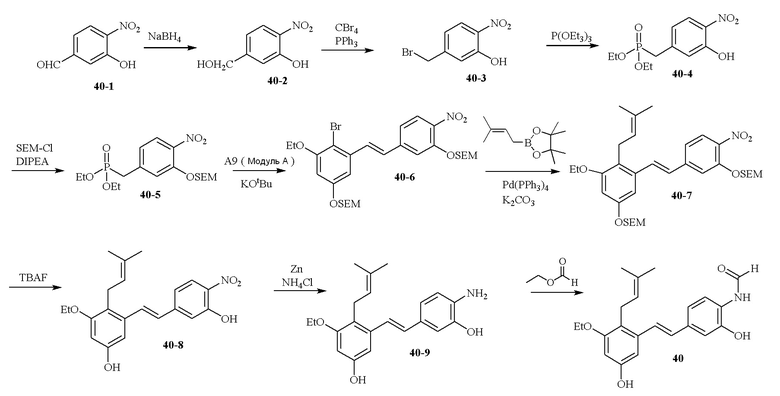
5-(гидроксиметил)-2-нитрофенол (40-2).
Боргидрид натрия (2,27 г, 59,88 ммоль, 1,0 экв) добавляли двумя порциями к раствору соединения 40-1 (10,0 г, 59,88 ммоль, 1,0 экв) в метаноле при 0°C. Полученный раствор перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь гасили путем добавления воды (20 мл). Летучие компоненты удаляли при пониженном давлении, и полученный неочищенный остаток растворяли в этилацетате (150 мл) и промывали с помощью 1M HCl (100 мл). Водный слой экстрагировали этилацетатом (2×100 мл). Объединенные органические слои сушили над сульфатом натрия и испаряли при пониженном давлении с получением соединения 40-2 (9,98 г, 98% выход) в виде желтого твердого вещества (NRK-1-202), которое использовали на следующей стадии.
5-(Бромметил)-2-нитрофенол (40-3).
Тетрабромид углерода (4,05 г, 12,18 ммоль, 2,0 экв) добавляли 4 порциями к раствору соединения 40-2 (1,0 г, 6,09 ммоль, 1,0 экв) и трифенилфосфина (3,19 г, 12,18 ммоль, 2,0 экв) в дихлорметане (50 мл) при 0°C. Полученный раствор перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь гасили путем добавления 1M HCl (40 мл). Слои разделяли, и водный слой экстрагировали дихлорметаном (2×20 мл). Объединенные органические слои сушили над сульфатом натрия и испаряли при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 40 g), элюируя с градиентом от 0 до 40% этилацетата в гептанах, с получением соединения 40-3 (1,5 г, 92% выход) в виде желтого твердого вещества (NRK-6-23).
Диэтил (3-гидрокси-4-нитробензил)фосфонат (40-4).
Триэтилфосфит (3,3 мл, 19,39 ммоль, 3,0 экв) добавляли к раствору соединения 40-3 (1,5 г, 6,46 ммоль, 1,0 экв) в толуоле (30 мл). Полученный раствор кипятили с обратным холодильником (110°C) в течение 20 часов. После охлаждения до комнатной температуры, летучие компоненты удаляли при пониженном давлении. Неочищенный остаток растворяли в дихлорметане (50 мл) и промывали 1M раствором гидроксида натрия (100 мл). Водный слой нейтрализовывали 1M раствором хлористоводородной кислоты (150 мл). Полученный водный слой экстрагировали этилацетатом (3×100 мл). Объединенные органические слои сушили над сульфатом натрия и испаряли при пониженном давлении с получением соединения 40-4 (1,0 г, 56% выход) в виде желтого твердого вещества, которое использовали на следующей стадии (NRK-6-24).
Диэтил (4-нитро-3-((2-(триметилсилил)этокси)метокси)бензил)-фосфонат (40-5).
2-(Триметилсилил)этоксиметилхлорид (0,6 мл,3,46 ммоль, 1,0 экв) и N, N-диизопропилэтиламин (0,9 мл, 5,19 ммоль, 1,5 экв) последовательно добавляли к раствору соединения 40-4 (1,0 г, 3,46 ммоль, 1,0 экв) в дихлорметане (50 мл) при комнатной температуре. После перемешивания в течение 20 часов, реакционную смесь разбавляли насыщенным раствором бикарбоната натрия (30 мл). Слои разделяли, и водный слой экстрагировали дихлорметаном (2×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 5 (1,02 г, 71% выход) в виде желтого масла, которое использовали на следующей стадии. (NRK-6-29)
(E)-(2-((5-(2-Бром-3-этокси-5-((2-(триметилсилил)этокси)-метокси)стирил)-2-нитрофенокси)метокси)этил)триметилсилан (40-6).
Третбутоксид калия (800 мг, 7,06 ммоль, 2 экв) добавляли 3 порциями к раствору соединения 5 (1,02 г, 3,53 ммоль, 1,0 экв) в безводном тетрагидрофуране (50 мл) при 0°C. Смесь подогревали до комнатной температуры и перемешивали в течение 30 минут. Добавляли по каплям раствор модуля D (1,32 г, 3,53 ммоль, 1 экв) в безводном тетрагидрофуране (20 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию осторожно прерывали насыщенным солевым раствором (20 мл, 1 капля в минуту для первых 5 мл солевого раствора) при 0°C. Летучие компоненты удаляли при пониженном давлении. Добавляли насыщенный водный раствор хлорида аммония (50 мл), и смесь экстрагировали этилацетатом (2×60 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 40 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 40-6 (867 мг, 39% выход) в виде желтого масла. (NRK-6-31)
(E)-(2-((5-(3-Этокси-2-(3-метилбут-2-ен-1-ил)-5-((2-(три-метилсилил)этокси)метокси)стирил)-2-нитрофенокси)метокси)этил)-триметилсилан (40-7).
Тетракис (трифенилфосфин)палладий(0) (156 мг, 0,14 ммоль, 0,1 экв), карбонат калия (373 мг, 2,7 ммоль, 2 экв), пинаколовый эфир 3-метилбут-2-енилбороновой кислоты (530 мг, 2,7 ммоль, 2 экв) и воду (3 мл) добавляли последовательно к раствору соединения 40-6 (867 мг, 1,4 ммоль, 1 экв) в 1,4-диоксане (30 мл) в герметизированной колбе. После продувки азотом в течение 10 минут, реакционную смесь нагревали при 100°C в течение 16 часов. Анализ методом LCMS указывал на образование требуемого соединения 40-7 (70% конверсия). Добавляли дополнительное количество пинаколового эфира 3-метилбут-2-енилбороновой кислоты (530 мг, 2,7 ммоль, 2 экв), и реакционную смесь нагревали при 100°C в течение 16 часов. После охлаждения до комнатной температуры, реакционную смесь фильтровали через целит, и фильтрат испаряли при пониженном давлении. Остаток суспендировали в насыщенном водном растворе хлорида аммония (20 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 40 g), элюируя с градиентом от 0 до 30% этилацетата в гептанах, с получением соединения 40-7 (852 мг, 99% выход) в виде желтого масла. (NRK-6-37)
(E)-3-Этокси-5-(3-гидрокси-4-нитростирил)-4-(3-метилбут-2-ен-1-ил)фенол (40-8).
1M раствор тетрабутиламмония фторида в тетрагидрофуране (19 мл, 18,90 ммоль, 14 экв) добавляли к раствору соединения 40-7 (852 мг, 1,36 ммоль, 1 экв) в тетрагидрофуране (20 мл) при комнатной температуре. После нагревания при 67°C в течение 16 часов, смесь охлаждали до комнатной температуры и разбавляли водой (10 мл) и насыщенным водным раствором хлорида аммония (30 мл). Летучие компоненты удаляли при пониженном давлении, и оставшийся водный слой экстрагировали этилацетатом (3×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 40 g), элюируя с градиентом от 0 до 80% этилацетата в гептанах, с получением соединения 40-8 (410 мг, 83% выход) в виде желтого твердого вещества. (NRK-6-26)
(E)-3-(4-Амино-3-гидроксистирил)-5-этокси-4-(3-метилбут-2-ен-1-ил)фенол (40-9).
Порошок активированного цинка (730 мг, 11,1 ммоль, 10,0 экв), хлорид аммония (590 мг, 11,1 ммоль, 10,0 экв) и воду (5 мл) добавляли последовательно к раствору соединения 40-8 (410 мг, 1,11 ммоль, 1,0 экв) в тетрагидрофуране (20 мл) при комнатной температуре. Полученную суспензию перемешивали при комнатной температуре в течение 16 часов. Суспензию фильтровали, и твердые вещества промывали этилацетатом (20 мл). Фильтрат испаряли досуха при пониженном давлении. Остаток суспендировали в насыщенном водном растворе хлорида аммония (20 мл) и экстрагировали этилацетатом (3×20 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 80% этилацетата в гептанах, с получением соединения 40-9 (234 мг, 63% выход ) в виде бледно-желтого твердого вещества (NRK-6-39).
(E)-N-(4-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-гидроксифенил)формамид (40) [KYN-159].
Соединение 40-9 (100 мг, 0,3 ммоль, 1,0 экв) добавляли к этилформиату (10 мл), и реакционную смесь нагревали при 55°C в течение 20 часов. Реакционную смесь охлаждали до комнатной температуры и испаряли досуха при пониженном давлении. Остаток растворяли в этилацетате (20 мл) и промывали насыщенным водным раствором хлорида аммония (20 мл). Водный слой экстрагировали этилацетатом (2×15 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 80% этилацетата в гептанах. Продукт растирали с 10% этилацетата в гептанах (10 мл) с получением соединения 40 (74 мг, 69% выход) в виде желтовато-белого твердого вещества (NRK-6-41).
Желтовато-белое твердое вещество; температура плавления 157,9-162,9°; анализ методом HPLC: 97,0% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: Waters Atlantis T3, 2,1×50 мм, 3 мкм; время удерживания: 8,4 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=368,2 (M+H)+; C22H25NO4; 1H ЯМР (400 МГц, DMSO-d6) δ=10,04 (уш.с, 1H), 9,63 (с, 1H), 9,19 (уш.с, 1H), 8,28 (д, J=1,7 Гц, 1H), 8,04 (д, J=8,3 Гц, 1H), 7,16 (д, J=16,1 Гц, 1H), 7,05 (д, J=1,7 Гц, 1H), 6,97(дд, J=1,7, 8,3 Гц, 1H), 6,83 (д, J=16,1 Гц, 1H), 6,62 (д, J=2,2 Гц, 1H), 6,31 (д, J=2,1 Гц, 1H), 5,02 (уш.т, J=7,0 Гц, 1H), 3,94 (кв, J=6,9 Гц, 2H), 3,34 (уш.с, 2H), 1,75 (с, 3H), 1,62 (с, 3H), 1,33 (т, J=6,9 Гц, 3H); 13C ЯМР (100 МГц, DMSO-d6) δ=160,41, 157,69, 156,59, 147,23, 137,32, 133,69, 130,08, 129,78, 126,32, 125,48, 124,32, 121,06, 118,76, 118,45, 112,80, 104,10, 99,93, 63,75, 25,99, 24,29, 18,28, 15,25.
(E)-N-(5-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-гидроксифенил)формамид (41) [KYN-161]
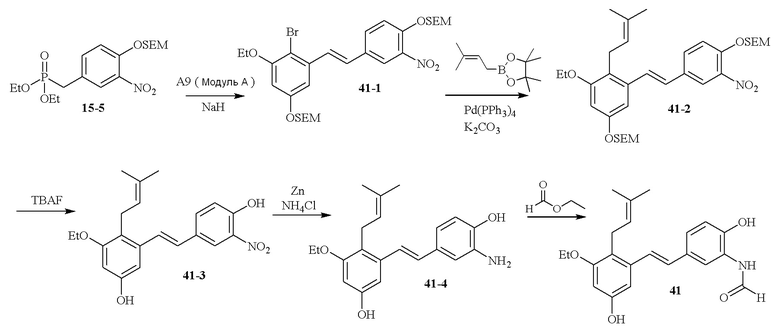
(E)-(2-((4-(2-Бром-3-этокси-5-((2-(триметилсилил)этокси) метокси)стирил)-2-нитрофенокси)метокси)этил)триметилсилан (41-1).
60% дисперсию гидрида натрия в минеральном масле (1,09 г, 45,52 ммоль, 4 экв) добавляли 2 порциями к раствору соединения 15-5 (3,29 г, 11,38 ммоль, 1,0 экв) в безводном тетрагидрофуране (60 мл) при 0°C. Смесь подогревали до комнатной температуры и перемешивали в течение 30 минут. Добавляли по каплям раствор модуля D (4,3 г, 11,38 ммоль, 1 экв) в безводном тетрагидрофуране (20 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию осторожно прерывали насыщенным солевым раствором (20 мл, 1 капля в минуту для первых 5 мл солевого раствора) при 0°C. Летучие компоненты удаляли при пониженном давлении. Добавляли насыщенный водный раствор хлорида аммония (60 мл), и смесь экстрагировали этилацетатом (3×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 80 g), элюируя с градиентом от 0 до 10% этилацетата в гептанах, с получением соединения 41-1 (740 мг, 16% выход) в виде желтого масла (NRK-6-48).
(E)-(2-((4-(3-Этокси-2-(3-метилбут-2-ен-1-ил)-5-((2-(три-метилсилил)этокси)метокси)стирил)-2-нитрофенокси)метокси)этил)-триметилсилан (41-2).
Тетракис(трифенилфосфин)палладий(0) (120 мг, 0,1 ммоль, 0,1 экв), карбонат калия (276 мг, 2,0 ммоль, 2 экв), пинаколовый эфир 3-метилбут-2-енилбороновой кислоты (400 мг, 2,0 ммоль, 2 экв) и воду (2 мл) добавляли последовательно к раствору соединения 41-1 (640 мг, 1,0 ммоль, 1 экв) в 1,4-диоксане (10 мл) в герметизированной колбе. После продувки азотом в течение 10 минут, реакционную смесь нагревали при 100°C в течение 16 часов. После охлаждения до комнатной температуры, реакционную смесь фильтровали через целит. Фильтрат концентрировали при пониженном давлении. Остаток суспендировали в насыщенном растворе бикарбоната натрия (30 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 40 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 41-2 (525 мг, 71% выход) в виде желтого масла (NRK-6-53).
(E)-3-Этокси-5-(4-гидрокси-3-нитростирил)-4-(3-метилбут-2-ен-1-ил)фенол (41-3).
1M раствор тетрабутиламмония фторида в тетрагидрофуране (12 мл, 11,68 ммоль, 14 экв) добавляли к раствору соединения 41-2 (525 мг, 0,84 ммоль, 1 экв) в тетрагидрофуране (12 мл) при комнатной температуре. После нагревания при 67°C в течение 16 часов, смесь охлаждали до комнатной температуры и разбавляли водой (20 мл) и 1M HCl (30 мл). Летучие компоненты удаляли при пониженном давлении, и оставшийся водный слой экстрагировали этилацетатом (3×30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 80 g), элюируя с градиентом от 0 до 60% этилацетата в гептанах, с получением соединения 41-3 (120 мг, 40% выход) в виде желтого твердого вещества. (NRK-6-54)
(E)-3-(3-Амино-4-гидроксистирил)-5-этокси-4-(3-метилбут-2-ен-1-ил)фенол (41-4).
Порошок активированного цинка (210 мг, 3,26 ммоль, 10,0 экв), хлорид аммония (176 мг, 3,26 ммоль, 10,0 экв) и воду (2 мл) добавляли последовательно к раствору соединения 41-3 (120 мг, 0,326 ммоль, 1,0 экв) в тетрагидрофуране (10 мл) при комнатной температуре. Полученную суспензию перемешивали при комнатной температуре в течение 16 часов. Суспензию фильтровали через целит, который промывали этилацетатом (20 мл). Фильтрат концентрировали досуха при пониженном давлении. Остаток суспендировали в насыщенном водном растворе хлорида аммония (20 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 60% этилацетата в гептанах, с получением соединения 41-4 (100 мг, 91% выход ) в виде бледно-желтого твердого вещества (NRK-6-55).
(E)-N-(5-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)-стирил)-2-гидроксифенил)формамид (41) [KYN-161].
Раствор соединения 41-4 (100 мг, 0,3 ммоль, 1,0 экв) в этилформиате (10 мл) нагревали при 55°C в течение 20 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали досуха при пониженном давлении. Остаток растворяли в этилацетате (30 мл) и промывали насыщенным водным раствором хлорида аммония (30 мл). Водный слой экстрагировали этилацетатом (2×30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 60% этилацетата в гептанах, с получением соединения 41 (80 мг, 73% выход) в виде бледно-желтого твердого вещества (NRK-6-57).
Бледно-желтое твердое вещество; анализ методом HPLC: 97,5% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: Waters Atlantis T3, 2,1×50 мм, 3 мкм; время удерживания: 8,3 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=368,2 (M+H)+; C22H25NO4; 1H ЯМР (400 МГц, DMSO-d6) δ=10,08 (уш.с, 1H), 9,58 (с, 1H), 9,16 (уш.с, 1H), 8,31 (д, J=1,7 Гц, 1H), 8,28 (д, J=2,1 Гц, 1H), 7,20-7,02 (м, 2H), 6,91-6,78 (м, 2H), 6,63-6,58 (м, 1H), 6,30 (д, J=2,2 Гц, 1H), 5,00 (уш.т, J=7,0 Гц, 1H), 3,94 (кв, J=6,9 Гц, 2H), 3,35 (уш.с, 1H), 3,31-3,28 (м, 1H), 1,77-1,70 (м, 3H), 1,67-1,47 (м, 3H), 1,33 (т, J=7,0 Гц, 3H); 13C ЯМР (100 МГц, DMSO-d6) δ=160,55, 157,67, 156,57, 147,20, 137,62, 130,08, 128,90, 126,77, 124,51, 124,29, 123,97, 123,49, 118,83, 118,52, 115,61, 103,97, 99,69, 63,74, 25,96, 24,34, 18,24, 15,26.
(E)-2-Метокси-4-(3-метокси-2-(3-метилбут-2-ен-1-ил)-5-нитро-стирил)фенол (42) [KYN-162]
(E)-4-(5-Амино-3-метокси-2-(3-метилбут-2-ен-1-ил)стирил)-2-метоксифенол (S16) (43) [KYN-163]
(E)-N-(3-(4-Гидрокси-3-метоксистирил)-5-метокси-4-(3-метил-бут-2-ен-1-ил)фенил)формамид (S17) (44) [KYN-164]
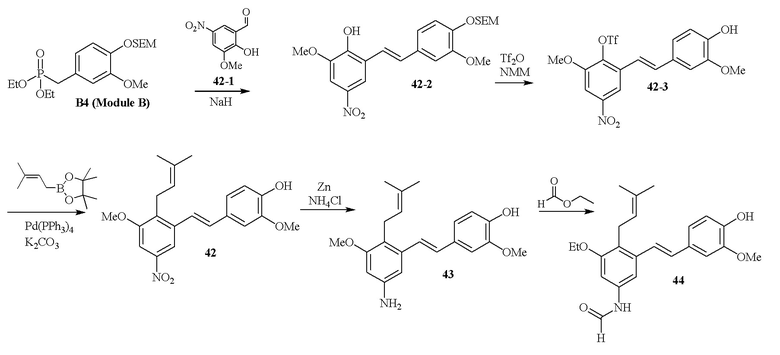
(E)-2-Метокси-6-(3-метокси-4-((2-(триметилсилил)этокси)-метокси)стирил)-4-нитрофенол (42-2).
60% дисперсию гидрида натрия в минеральном масле (14,2 г, 356 ммоль, 9 экв) добавляли одной порцией к раствору соединения B4 (модуля B) (16 г, 39,5 ммоль, 1 экв) в безводном тетрагидрофуране (400 мл) при 0°C. Смесь подогревали до комнатной температуры и перемешивали 30 минут. Добавляли по каплям раствор соединения 42-1 (11,7 г, 59,3 ммоль, 1,5 экв) в безводном тетрагидрофуране (100 мл), и смесь кипятили с обратным холодильником (68°C) в течение 16 часов. Анализ методом LCMS указывал на ~15% конверсию в продукт. Добавляли дополнительное количество 60% дисперсии гидрида натрия в минеральном масле (4,7 г, 119 ммоль, 3 экв), и смесь кипятили с обратным холодильником (68°C) в течение еще 24 часов. Реакционную смесь охлаждали до 0°C и осторожно гасили водой (40 мл, 1 капля в минуту для первых 10 мл) и затем разбавленным насыщенным водным раствором хлорида аммония (2 л). Смесь экстрагировали метилтрет-бутиловым эфиром (2 л) и этилацетатом (3 л). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали два раза на автоматизированной хроматографической системе Interchim (2 колонки 120 g), элюируя каждый раз с градиентом от 0 до 100% этилацетата в гептанах, с получением соединения 42-2 (2,8 г, 16% выход) в виде желтого твердого вещества (LM-8-46).
(E)-2-(4-Гидрокси-3-метоксистирил)-6-метокси-4-нитрофенил- трифторметансульфонат (42-3).
Трифторметансульфоновый ангидрид (1,9 г, 6,7 ммоль, 1,07 экв) добавляли по каплям в течение 5 минут, поддерживая температуру реакции при 0°C, к раствору соединения 42-2 (2,8 г, 6,26 моль, 1,0 экв) и N-метилморфолина (1,03 мл, 9,4 ммоль, 1,5 экв) в дихлорметане (100 мл) при 0°C. После перемешивания при 0°C в течение 3 часов, реакционную смесь гасили путем добавления 10% лимонной кислоты (100 мл) в течение 15 минут, поддерживая температуру реакции ниже 5°C. Слои разделяли, и водный слой экстрагировали дихлорметаном (2×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 80 g), элюируя с градиентом от 0 до 60% этилацетата в гептанах, с получением соединения 42-3 (2,26 г, 80% выход) в виде желтого твердого вещества (LM-8-47).
(E)-2-Метокси-4-(3-метокси-2-(3-метилбут-2-ен-1-ил)-5-нитро-стирил)фенол (42) [KYN-162].
Тетракис(трифенилфосфин)палладий(0) (237 мг, 0,2 ммоль, 0,05 экв), карбонат калия (1,2 г, 8,2 ммоль, 2 экв), пинаколовый эфир 3-метилбут-2-енилбороновой кислоты (1,6 г, 8,2 ммоль, 2 экв) и воду (11 мл) добавляли последовательно к раствору соединения 42-3 (1,83 г, 4,1 ммоль, 1 экв) в 1,4-диоксане (33 мл). После продувки азотом в течение 10 минут, реакционную смесь нагревали при 95°C в течение 16 часов. После охлаждения до комнатной температуры, смесь разбавляли насыщенным водным раствором хлорида аммония раствор (30 мл) и экстрагировали метилтрет-бутиловым эфиром (2×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 120 g), элюируя с градиентом от 0 до 60% этилацетата в гептанах, с получением соединения 42 (0,82 г, 54% выход) в виде желтого масла.
Анализ методом HPLC: 97,3% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: Waters Atlantis T3, 2,1×50 мм, 3 мкм; время удерживания: 10,7 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=370,2 (M+H)+; C21H23NO5; 1H ЯМР (400 МГц, CDCl3) δ=8,10 (д, J=2,2 Гц, 1H), 7,59 (д, J=2,2 Гц, 1H), 7,18-7,13 (м, 1H), 7,08-7,01 (м, 3H), 6,94 (д, J=8,2 Гц, 1H), 5,71 (с, 1H), 5,07 (септет, J=1,3, 6,8 Гц, 1H), 3,95 (с, 3H), 3,93 (с, 3H), 3,55 (уш.д, J=6,8 Гц, 2H), 1,84 (д, J=0,9 Гц, 3H), 1,70 (д, J=1,1 Гц, 3H); 13C ЯМР (100 МГц, CDCl3) δ=157,84, 147,03, 146,73, 146,17, 138,48, 135,17, 132,87, 132,61, 129,45, 122,31, 121,10, 120,63, 114,73, 113,29, 108,83, 103,38, 56,10, 55,91, 25,75, 25,52, 18,09.
(E)-4-(5-Амино-3-метокси-2-(3-метилбут-2-ен-1-ил)стирил)-2-метоксифенол (43) [KYN-163].
Воду (6 мл), хлорид аммония (613 мг, 11,4 ммоль, 10 экв) и порошок цинка (743 мг, 11,4 ммоль, 10 экв) добавляли последовательно к раствору соединения 42 (420 мг, 1,14 ммоль, 1 экв) в тетрагидрофуране (60 мл) при комнатной температуре. Полученную суспензию перемешивали при комнатной температуре в течение 3 часов. Суспензию фильтровали через целит, который промывали этилацетатом (50 мл). Фильтрат испаряли досуха при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 80 g), элюируя с градиентом от 0 до 100% этилацетата в гептанах, с получением соединения 43 (60 мг, >95% чистота и 180 мг, ~90% чистота; 62% выход) в виде желтовато-белого твердого вещества после сушки под вакуумом при 40°C в течение 16 часов (LM-8-53).
Желтовато-белое твердое вещество; анализ методом HPLC: 97,6% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: Waters Atlantis T3, 2,1×50 мм, 3 мкм; время удерживания: 6,7 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=340,2 (M+H)+; C21H25NO3; 1H ЯМР (400 МГц, DMSO-d6) δ=9,08 (с, 1H), 7,12-7,04 (м, 2H), 6,91 (дд, J=1,9, 8,3 Гц, 1H), 6,81-6,73 (м, 2H), 6,43 (д, J=2,0 Гц), 6,17 (д, J=2,0 Гц), 4,99 (септет, J=1,3, 6,8 Гц, 1H), 4,88 (с, 2H), 3,81 (с, 3H), 3,69 (с, 3H), 3,28 (уш.д, J=7,0 Гц, 2H), 1,75 (д, J=0,6 Гц, 3H), 1,60 (д, J=0,7 Гц, 3H); 13C ЯМР (100 МГц, DMSO-d6) δ=158,21, 148,28, 147,79, 146,99, 137,40, 129,49, 129,22, 125,10, 124,35, 120,35, 116,08, 115,59, 110,09, 103,11, 97,74, 55,99, 55,68, 55,36, 25,97, 24,18, 18,18.
(E)-N-(3-(4-Гидрокси-3-метоксистирил)-5-метокси-4-(3-метил-бут-2-ен-1-ил)фенил)формамид (44) [KYN-164].
Раствор соединения 43 (90 мг, 0,27 ммоль, 1,0 экв) в этилформиате (10 мл) нагревали при 55°C в течение 20 часов. Реакционную смесь охлаждали до комнатной температуры и испаряли досуха при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка 25 g), элюируя с градиентом от 0 до 60% этилацетата в гептанах, с получением соединения 44 (80 мг, 82% выход) в виде желтовато-белого твердого вещества после сушки под вакуумом при 40°C в течение 16 часов (LM-8-57).
Желтовато-белое твердое вещество; анализ методом HPLC: 97,3% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: Waters Atlantis T3, 2,1×50 мм, 3 мкм; время удерживания: 6,7 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=368,2 (M+H)+; C22H25NO4; 1H ЯМР (400 МГц, DMSO-d6) δ=10,08 (с, 1H), 9,98 (д, J=11,1 Гц, 1H), 9,15 (с, 1H), 8,90-8,80 (м, 1H), 8,27 (д, J=1,8 Гц, 1H), 7,42 (д, J=1,8 Гц), 7,22-7,11 (м, 2H), 7,07-6,91 (м, 2H), 6,87-6,70 (м, 2H), 5,04-4,96 (м, 1H), 3,82 (с, 3H), 3,78-3,74 (м, 3H), 3,40 (уш.д, J=6,7 Гц, 2H), 1,77 (с, 3H), 1,61 (с, 3H); 13C ЯМР (100 МГц, DMSO-d6) δ=163,17, 159,99, 157,64, 148,30, 147,31, 137,64, 137,56, 137,52, 130,91, 130,48, 129,07, 123,75, 123,72, 123,52, 123,08, 120,66, 120,60, 116,09, 110,38, 108,53, 101,90, 56,19, 56,03, 56,00, 25,94, 24,56, 18,24.
(E)-4-Бром-3-(4-гидрокси-3-метоксистирил)-5-метоксифенол (45) [KYN-112].
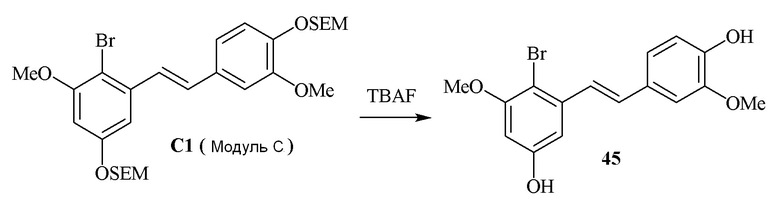
1,0M раствор тетрабутиламмония фторида в THF (2,3 мл, 2,29 ммоль, 7 экв) добавляли к раствору (E)-(2-((4-(2-Бром-3-метокси-5-((2-(триметилсилил)этокси)метокси)стирил)-2-метоксифенокси)-метокси)этил)триметилсилана (C1) (200 мг, 0,327 ммоль, 1 экв) в безводном THF (5 мл) и кипятили с обратным холодильником в течение ночи. После охлаждения до комнатной температуры, реакционную смесь гасили водой (25 мл) и экстрагировали этилацетатом (100 мл). Органический слой промывали насыщенным солевым раствором (25 мл), сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали два раза на автоматизированной системе AnaLogixs (колонка 12 g), элюируя каждый раз с градиентом от 0 до 100% этилацетата в гептанах, с получением соединения 45 [KYN-112] (21 мг, 18% выход) в виде коричневой пленки (CSK-2-95).
Анализ методом HPLC: 94,3% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 7,0 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации отрицательных ионов) m/z=349/351 (M-H)-; C16H15BrO4; 1H ЯМР (400 МГц, Метанол-d3) δ=7,31 (д, J=16,2 Гц, 1H), 7,10 (д, J=2 Гц, 1H), 7,00 (дд, J=2,0, 8,1 Гц, 1H), 6,93 (д, J=16,1 Гц, 1H), 6,79 (д, J=8,0 Гц, 1H), 6,75 (д, J=2,7 Гц, 1H), 6,40 (д, J=2,7 Гц, 1H), 3,90 (с, 3H), 3,82 (с, 3H); 13C ЯМР (100 МГц, Метанол-d3) δ=158,69, 158,32, 149,15, 148,08, 140,14, 132,49, 130,67, 126,01, 121,41, 116,38, 110,75, 105,77, 103,89, 100,20, 56,57, 56,37.
Получение соединения 46 [KYN-145] - (E)-3-(4-гидрокси-3-метоксистирил)-4-(3-метилбут-2-ен-1-ил)-5-(трифторметил)фенола (46) [KYN-145]
Предлагаемый модульный синтез
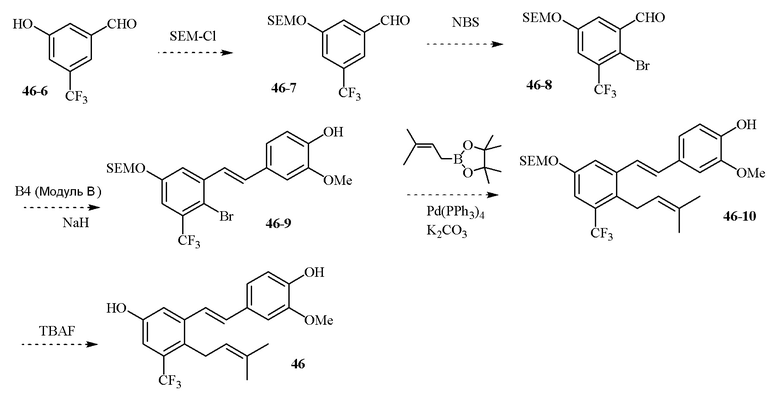
Предлагаются следующие ключевые стадии для модульного синтеза. Реакция 2-бром-3-трифторметил-5-((2-триметилсилил)-этокси)метокси)бензальдегида (модуля A) с соединением B4 (модулем B) в присутствии гидрида натрия с получением продукта модуля C, который может быть превращен в соединение 46 за 2 стадии.
Получение методом перегруппировки
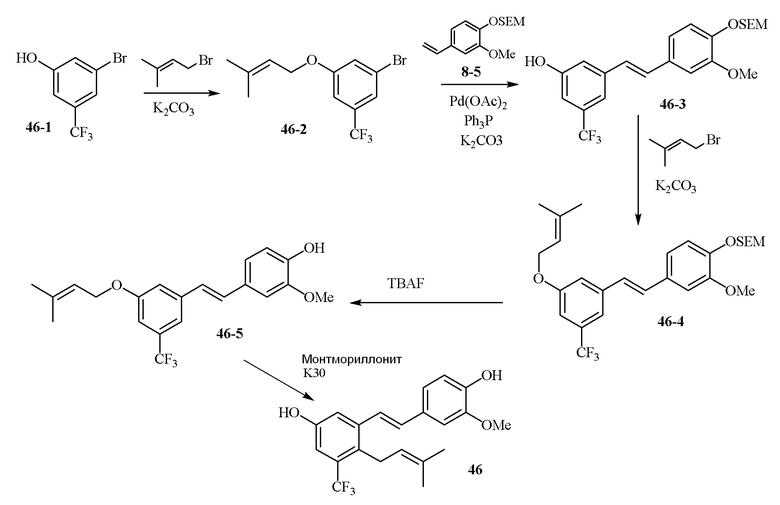
1-Бром-3-((3-метилбут-2-ен-1-ил)окси-5-(трифторметил)бензол (46-2).
Карбонат калия (28 г, 206 ммоль, 2 экв) и 3,3-диметилаллил- бромид (18,5 г, 124,48 ммоль, 1,2 экв) добавляли последовательно к раствору соединения 46-1 (25,0 г, 107,73 ммоль, 1 экв) в ацетонитриле (100 мл) при комнатной температуре. Полученную суспензию кипятили с обратным холодильником (80°C) в течение 16 часов. После охлаждения до комнатной температуры, реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении. Неочищенный остаток растворяли в этилацетате (300 мл) и промывали 1M раствором хлористоводородной кислоты (2×70 мл). Органический слой сушили над сульфатом натрия и испаряли при пониженном давлении с получением соединения 46-2 (31,0 г, 99% выход) в виде бледно-желтого масла (NRK-1-106), которое использовали на следующей стадии.
(E)-3-(3-метокси-4-((2-(триметилсилил)этокси)метокси)-стирил)-5-(трифторметил)фенол (46-3).
(2-((2-Метокси-4-винилфенокси)метокси)этил)триметилсилан (8-5) (3,97 г, 14,20 ммоль, 1,0 экв), трифенилфосфин (750 мг, 2,84 ммоль, 0,2 экв), карбонат калия (4,25 г, 30,76 ммоль, 2 экв) и ацетат палладия (320 мг, 1,42 ммоль, 0,1 экв) добавляли к раствору соединения 46-2 (5,0 г, 14,20 ммоль, 1,0 экв) в безводном толуоле (100 мл) при комнатной температуре. Реакционную смесь продували азотом в течение 15 минут. После кипячения с обратным холодильником (110°C) в течение 16 часов, реакционную смесь охлаждали до комнатной температуры и фильтровали через целит (~ 20 г). Фильтрат переносили в делительную воронку, содержащую насыщенный раствор бикарбоната натрия (150 мл). Слои разделяли, и водный слой экстрагировали этилацетатом (2×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 40 g) элюируя с градиентом от 0 до 50% этилацетата в гептанах, с получением соединения 46-3 (5,25 г, 85% выход) в виде бледно-желтого масла (NRK-1-107).
(E)-(2-((2-метокси-4-(3-((3-метилбут-2-ен-1-ил)окси-5-(три-фторметил)стирил)фенокси)метокси)этил)триметилсилан (46-4).
Карбонат калия (3,36 г, 24,40 ммоль, 2 экв) и 3,3-диметил-аллилбромид (2,18 г, 14,64 ммоль, 1,2 экв) добавляли последовательно к раствору соединения 46-3 (5,25 г, 12,20 ммоль, 1 экв) в ацетонитриле (150 мл) при комнатной температуре. После кипячения с обратным холодильником (80°C) в течение 16 часов, реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Неочищенный остаток растворяли в этилацетате (100 мл) и промывали 1M раствором хлористоводородной кислоты (2×50 мл). Органический слой сушили над сульфатом натрия и испаряли при пониженном давлении с получением соединения 46-4 (5,0 г, 80% выход) в виде бледно-желтого масла, которое использовали на следующей стадии. (NRK-1-111).
(E)-2-метокси-4-(3-((3-метилбут-2-ен-1-ил)окси-5-(трифтор-метил)стирил)фенол (46-5).
1M раствор тетрабутиламмония фторида в THF (69 мл, 68,78 ммоль, 7 экв) добавляли к раствору соединения 46-4 (5,0 г, 9,82 ммоль, 1 экв) в безводном THF (200 мл) при комнатной температуре. После кипячения с обратным холодильником (66°C) в течение 16 часов, анализ методом LCMS указывал на завершение реакции. Смесь охлаждали до комнатной температуры и разбавляли водой (20 мл). Летучие компоненты удаляли при пониженном давлении. Смесь разбавляли этилацетатом (150 мл). Слои разделяли, и водный слой экстрагировали этилацетатом (2×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 80 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 46-5 (3,25 г, 87% выход) в виде бледно-желтого твердого вещества (NRK-1-113).
(E)-3-(4-гидрокси-3-метоксистирил)-4-(3-метилбут-2-ен-1-ил)-5-(трифторметил)фенол (46) [KYN-145].
Порошок монтмориллонита K30 (3,25 г) добавляли к раствору соединения 46-7 (3,2 г, 8,89 ммоль) в безводном дихлорметане (200 мл) при комнатной температуре. После перемешивания в течение ночи, анализ методом LCMS указывал на образование и 10-15% требуемого соединения 46 наряду с двумя региоизомерами и непрореагировавшим исходным соединением 46-5 в качестве побочных продуктов. Смесь фильтровали и обрабатывали свежим порошком монтмориллонита K30 (3,2 г) при комнатной температуре в течение еще 24 часов. Реакционную смесь фильтровали, и твердые вещества промывали дихлорметаном (2×50 мл). Объединенные фильтраты концентрировали при пониженном давлении. Остаток сначала очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 80 g), элюируя с градиентом от 0 до 50% этилацетата в гептанах, с получением нескольких смешанных фракций, содержащих соединение 46. Фракции собирали и снова очищали на автоматизированной хроматографической системе Reveleris (колонка Sorbtech 24 g), элюируя с градиентом от 0 до 40% этилацетата в гептанах. Все фракции, содержащие соединение 46 (наряду с другим изомером), собирали и очищали снова на автоматизированной хроматографической системе Reveleris (колонка Redisep C-18 100 g), элюируя с градиентом от 0 до 100% ацетонитрила в воде, с получением соединения 46 (118 мг, 85% чистота методом HPLC, 3% выход) в виде желтого твердого вещества. Этот материал растворяли в этилацетате (3 мл) и гептанах (7 мл), и раствор медленно испаряли до объема ~ 3 мл в течение ночи при комнатной температуре с получением слоя кристаллов на дне раствора. Твердое вещество собирали с получением соединения 46 [KYN-145] (34 мг, 96,5% чистота методом HPLC) в виде желтовато-белого твердого вещества (NRK-1-115) и (QZH-RCI-47).
Желтовато-белое твердое вещество; анализ методом HPLC: 96,5% чистота; длина волны 210 нм, диапазон пропускания 4; колонка: SorbTech C18AQ, 2,1×50 мм, 3 мкм; время удерживания: 11,9 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=379,1 (M+H)+; C21H21F3O3; 1H ЯМР (400 МГц, CD3OD) δ=7,33 (д, J=16,1 Гц, 1H), 7,21 (уш.м, 1H), 7,19 (с, 1H), 7,12 (уш.м, 1H), 6,88 (д, J=16,2 Гц, 1H), 6,88 (уш.м, 1H), 6,64 (с, 1H), 5,21 (тм, J=1,5, 7,1 Гц, 1H), 3,90 (с, 3H), 3,36 (уш.д, J=7,1 Гц, 2H), 1,78 (д, J=1,0 Гц, 3H), 1,73 (д, J=0,9 Гц, 3H); 19F ЯМР (376 МГц, CD3OD) δ=-64,35 (с, 3F); 13C ЯМР (100 МГц, CD3OD) δ=159,66, 148,18, 147,81, 142,37, 134,79, 133,34, 133,01, 132,66, 129,41, 128,29, 127,12, 126,81, 125,22, 117,72, 117,28, 114,99, 114,95, 111,53, 111,49, 110,19, 56,78, 32,91, 25,99, 18,15.
The соединение 46 KYN-145 может быть также синтезировано с использованием модульного метода по настоящему изобретению, применяя эквиваленты для модуля A, B и C и последующее проведение реакции перекрестного алкилирования арилгалогенида.
(E)-3-Амино-5-(4-гидрокси-3-метоксистирил)-4-(3-метилбут-2-ен-1-ил)фенол (47)
(E)-N-(5-Гидрокси-3-(4-гидрокси-3-метоксистирил)-2-(3-метил-бут-2-ен-1-ил)фенил)формамид (48)
(E)-N-(5-Гидрокси-3-(4-гидрокси-3-метоксистирил)-2-(3-метил-бут-2-ен-1-ил)фенил)ацетамид (49)
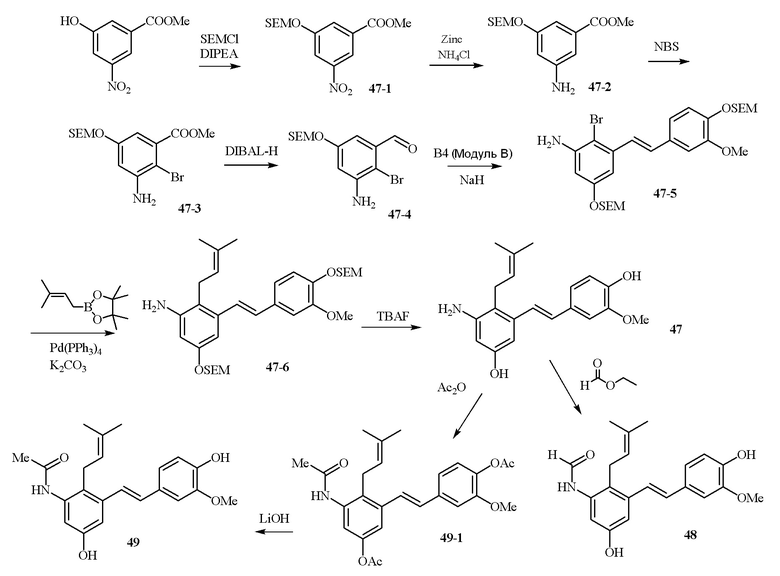
Метил 3-нитро-5-((2-(триметилсилил)этокси)метокси)бензоат (47-1).
N, N-Диизопропилэтиламин (10,3 мл, 59 ммоль, 3 экв) и 2-(три-метилсилил)этоксиметилхлорид (5,2 мл, 29,6 ммоль, 1,5 экв) последовательно добавляли к раствору метил 3-нитро-5-гидрокси-бензоата (3,88 г, 19,7 моль, 1,0 экв) в безводном дихлорметане (200 мл). После перемешивания при комнатной температуре в течение 16 часов, добавляли насыщенный раствор бикарбоната натрия (250 мл). Слои разделяли, и водный слой экстрагировали дихлорметаном (250 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (колонка 120 g), элюируя с градиентом от 0 до 30% этилацетата в гептанах, с получением соединения 47-1 (6,12 г, 95% выход) в виде бесцветного масла (CHLM-02-9).
Метил 3-амино-5-((2-(триметилсилил)этокси)метокси)бензоат (47-2).
Порошок цинка (18,33 г, 280 ммоль, 15,0 экв), хлорид аммония (15,1 г, 280 ммоль, 15,0 экв) и воду (20 мл) добавляли последовательно к раствору соединения 47-1 (6,12 г, 18,7 ммоль, 1,0 экв) в тетрагидрофуране (200 мл) при комнатной температуре. Полученную суспензию кипятили с обратным холодильником (68°C) в течение 5 часов. Суспензию охлаждали до комнатной температуры, разбавляли этилацетатом (500 мл), фильтровали через целит, который промывали этилацетатом (500 мл). Фильтрат испаряли досуха при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (2 колонки 120 g), элюируя с градиентом от 0 до 60% этилацетата в гептанах, с получением соединения 47-2 (4,76 г, 86% выход) в виде бледно-желтого масла (CH-LM-02-10).
Метил 3-амино-2-бром-5-((2-(триметилсилил)этокси)метокси)-бензоат (47-3).
N-Бромсукцинимид (2,85 г, 16 ммоль, 1,0 экв) добавляли порционно к раствору соединения 47-2 (4,76 г, 16 ммоль, 1,0 экв) в безводном дихлорметане (500 мл) при 0°C. После перемешивания при 0°C в течение 1 часа, добавляли ледяную воду (500 мл), и смесь подогревали до комнатной температуры. Слои разделяли, и органический слой промывали насыщенным раствором бикарбоната натрия (500 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (2 колонки 120 g), элюируя с градиентом от 0 до 20% этилацетата в гептанах, с получением соединения 47-3 (5,05 г, 84% выход) в виде бледно-желтого масла (CH-LM-02-14).
3-Амино-2-бром-5-((2-(триметилсилил)этокси)метокси)-бензальдегид (47-4).
1M раствор гидрида диизобутилалюминия в толуоле (26,84 мл, 26,84 ммоль, 2,0 экв) добавляли по каплям к раствору соединения 47-3 (5,05 г, 13,42 ммоль, 1,0 экв) в толуоле (250 мл) при -78°C. Полученную смесь перемешивали при -78°C в течение 3 часов. Добавляли по каплям насыщенный раствор тартрата калия/натрия (120 мл), и смесь медленно подогревали до комнатной температуры. После перемешивания в течение 16 часов, добавляли метилтрет-бутиловый эфир (300 мл). Слои разделяли, и органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (2 колонки 120 g), элюируя с градиентом от 0 до 60% этилацетата в гептанах, с получением соединения 47-4 (3,25 г, 70% выход) в виде желтого масла (CH-LM-02-16).
(E)-2-Бром-3-(3-метокси-4-((2-(триметилсилил)этокси)-метокси)стирил)-5-((2-(триметилсилил)этокси)метокси)анилин (47-5).
60% дисперсию гидрида натрия в минеральном масле (1,34 г, 33,6 ммоль, 4,0 экв) добавляли одной порцией к раствору модуля B (3,4 г, 8,4 ммоль, 1,0 экв) в безводном тетрагидрофуране (150 мл) при 0°C. Смесь подогревали до комнатной температуры и перемешивали в течение 30 минут. Добавляли по каплям раствор соединения 47-4 (3,25 г, 9,4 ммоль, 1,12 экв) в безводном тетрагидрофуране (50 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию осторожно прерывали водой (250 мл, 1 капля в минуту для первых 5 мл воды) при 0°C и экстрагировали метилтрет-бутиловым эфиром (400 мл) и этилацетатом (400 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (2 колонки 120 g), элюируя с градиентом от 0 до 30% этилацетата в гептанах, с получением соединения 47-5 (2,6 г, 52% выход) в виде желтого твердого вещества (CH-LM-02-17).
(E)-3-(3-Метокси-4-((2-(триметилсилил)этокси)метокси)-стирил)-2-(3-метилбут-2-ен-1-ил)-5-((2-(триметилсилил)этокси)-метокси)анилин (47-6).
Тетракис(трифенилфосфин)палладий(0) (252 мг, 0,22 ммоль, 0,05 экв), карбонат калия (1,2 г, 8,71 ммоль, 2,0 экв), пинаколовый эфир 3-метилбут-2-енилбороновой кислоты (1,71 г, 8,71 ммоль, 2,0 экв) и воду (15 мл) добавляли последовательно к раствору соединения 47-5 (2,6 г, 4,36 ммоль, 1,0 экв) в 1,4-диоксане (45 мл) в герметизированной колбе. После продувки азотом в течение 10 минут, реакционную смесь нагревали при 100°C в течение 16 часов. После охлаждения до комнатной температуры, реакционную смесь экстрагировали метилтрет-бутиловым эфиром (100 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (колонка 120 g), элюируя с градиентом от 0 до 35% этилацетата в гептанах, с получением соединения 47-6 (2,07 г, 81% выход) в виде желтого твердого вещества (CH-LM-02-18).
(E)-3-Амино-5-(4-гидрокси-3-метоксистирил)-4-(3-метилбут-2-ен-1-ил)фенол (47) [KYN-167].
1M раствор тетрабутиламмония фторида в тетрагидрофуране (35 мл, 35 ммоль, 14,0 экв) добавляли к раствору соединения 47-6 (1,47 г, 2,51 ммоль, 1,0 экв) в тетрагидрофуране (120 мл) при комнатной температуре. После нагревания при 68°C в течение 16 часов, смесь охлаждали до комнатной температуры и разбавляли водой (50 мл). Летучие компоненты удаляли при пониженном давлении. Остаток разбавляли насыщенным водным раствором хлорида аммония (150 мл) и экстрагировали этилацетатом (2×300 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (колонка 80 g), элюируя с градиентом от 0 до 100% этилацетата в гептанах. Продуктовые фракции объединяли, концентрировали при пониженном давлении и затем очищали на автоматизированной хроматографической системе InterChim (колонка RediSep Rf GOLD HP C18 100 g), элюируя с градиентом от 0 до 100% ацетонитрила в воде, с получением соединения 47 (148 мг, 18% выход) в виде желтовато-белого твердого вещества (CH-LM-02-22). Еще одну партию соединения 11 (400 мг) подвергали обработке таким же образом с получением соединения 47 (15 мг, 7% выход) в виде желтовато-белого твердого вещества (CH-LM-02-19).
Анализ методом HPLC: 99,2% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: Waters Atlantis T3, 2,1×50 мм, 3 мкм; время удерживания: 6,1 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=326,2 (M+H)+; C20H23NO3; 1H ЯМР (400 МГц, DMSO-d6) δ=9,06 (уш.с, 1H), 8,72 (уш.с, 1H), 7,12-7,05 (м, 2H), 6,91 (дд, J=1,9, 8,3 Гц, 1H), 6,77-6,70 (м, 2H), 6,27 (д, J=2,3 Гц), 6,06 (д, J=2,3 Гц), 4,99 (септет, J=1,3, 6,7 Гц, 1H), 4,62 (с, 2H), 3,81 (с, 3H), 3,24-3,17 (м, 2H), 1,77 (с, 3H), 1,64 (с, 3H); 13C ЯМР (100 МГц, DMSO-d6) δ=156,14, 148,25, 147,75, 146,90, 137,67, 129,92, 129,60, 129,53, 124,97, 124,23, 120,36, 116,04, 114,60, 110,16, 101,93, 101,72, 56,03, 25,98, 25,57, 18,26.
(E)-N-(5-Гидрокси-3-(4-гидрокси-3-метоксистирил)-2-(3-метил-бут-2-ен-1-ил)фенил)формамид (48).
Соединение 47 (49 мг, 0,15 ммоль, 1,0 экв) добавляли к этилформиату (10 мл) с получением мутного раствора, который нагревали при 55°C (при нагревании раствор становился прозрачным). Через 24 часа, реакционную смесь охлаждали до комнатной температуры и испаряли досуха при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (колонка RediSep Rf GOLD HP C18 100 g), элюируя с градиентом от 0 до 100% ацетонитрила в воде, с получением соединения 48 (25 мг, 47% выход) в виде желтовато-белого твердого вещества после лиофилизации (CH-LM-02-23).
Анализ методом HPLC: 99,8% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: Waters Atlantis T3, 2,1×50 мм, 3 мкм; время удерживания: 6,6 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=354,2 (M+H)+; C21H23NO4; 1H ЯМР (400 МГц, DMSO-d6) δ=9,66-9,45 (м, 1H), 9,23 (уш.с, 2H), 8,26-8,19 (м, 1H), 7,17-7,06 (м, 3H), 6,99-6,85 (м, 2H), 6,83 (д, J=2,7 Гц, 1H), 6,76 (д, J=8,1 Гц), 6,49 (д, J=2,3 Гц, 1H), 4,95 (уш.с, 1H), 3,81 (с, 3H), 3,39-3,33 (м, 2H), 1,78-1,73 (м, 3H), 1,64-1,59 (м, 3H); 13C ЯМР (100 МГц, DMSO-d6) δ=163,08, 159,42, 155,17, 154,60, 147,22, 146,16, 137,35, 135,29, 130,02, 129,59, 129,53, 129,34, 128,21, 128,11, 122,94, 122,89, 122,79, 122,64, 122,56, 121,68, 119,68, 119,63, 115,00, 110,26, 109,37, 109,21, 108,56, 55,00, 54,96, 24,88, 17,32.
(E)-3-Ацетамидо-5-(4-ацетокси-3-метоксистирил)-4-(3-метил-бут-2-ен-1-ил)фенилацетат (49-1).
N, N-Диизопропилэтиламин (0,32 мл, 1,8 ммоль, 6 экв) и уксусный ангидрид (0,1 мл, 1 ммоль, 3,3 экв) последовательно добавляли к раствору соединения 47 (98 мг, 0,3 ммоль, 1,0 экв) в безводном тетрагидрофуране (50 мл) при комнатной температуре. После кипячения с обратным холодильником (66°C) в течение 16 часов, реакционную смесь охлаждали до комнатной температуры, и летучие компоненты удаляли при пониженном давлении. Остаток растворяли в этилацетате (20 мл) и промывали насыщенным раствором бикарбоната натрия (20 мл). Водный слой экстрагировали этилацетатом (2×10 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 49-1 (135 мг, 99% выход) в виде бледно-желтого твердого вещества, которое использовали на следующей стадии (CH-LM-02-24).
(E)-N-(5-гидрокси-3-(4-гидрокси-3-метоксистирил)-2-(3-метил-бут-2-ен-1-ил)фенил)ацетамид (49) [KYN-169].
1M раствор гидроксида лития (1,27 мл, 1,27 ммоль, 5,0 экв) добавляли к раствору соединения 49-1 (115 мг, 0,255 ммоль, 1,0 экв) в смеси 5 : 1 тетрагидрофурана и метанола (30 мл) при комнатной температуре. Полученный раствор перемешивали при комнатной температуре в течение 1 часа. Еще одну партию соединения 49-1 (20 мг) подвергали обработке таким же образом. Две партии объединяли, и летучие компоненты удаляли при пониженном давлении. Остаток растворяли в этилацетате (50 мл) и промывали насыщенным водным раствором хлорида аммония (30 мл). Водный слой экстрагировали этилацетатом (2×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе InterChim (колонка RediSep Rf GOLD HP C18 100 g), элюируя с градиентом от 0 до 100% ацетонитрила в воде, с получением соединения 49 (50 мг, 46% выход) в виде желтовато-белого твердого вещества после лиофилизации (CH-LM-02-25).
Желтовато-белое твердое вещество температура плавления 87,3-193,8°C (разложение); анализ методом HPLC: 99,3% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: Waters Atlantis T3, 2,1×50 мм, 3 мкм; время удерживания: 6,6 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=368,2 (M+H)+; C22H25NO4; 1H ЯМР (400 МГц, DMSO-d6) δ=9,19 (с, 2H), 9,14 (уш.с, 1H), 7,18-7,09 (м, 2H), 6,95 (дд, J=1,8, 8,2 Гц, 1H), 6,91-6,79 (м, 2H), 6,76 (д, J=8,1 Гц, 1H), 6,70 (д, J=2,2 Гц, 1H), 4,94 (септет, J=1,2, 6,8 Гц, 1H), 3,81 (с, 3H), 3,37-3,32 (м, 2H), 2,01 (с, 3H), 1,75 (с, 3H), 1,60 (с, 3H); 13C ЯМР (100 МГц, DMSO-d6) δ=168,82, 155,54, 148,27, 147,17, 138,26, 137,47, 130,43, 129,96, 129,34, 124,94, 124,37, 123,98, 120,64, 116,05, 113,45, 110,34, 109,99, 56,06, 26,29, 25,98, 23,67, 18,29.
(E)-N-(5-Гидрокси-3-(4-формамидо-3-метоксистирил)-2-(3-метилбут-2-ен-1-ил)фенил)формамид (50) [KYN-170].
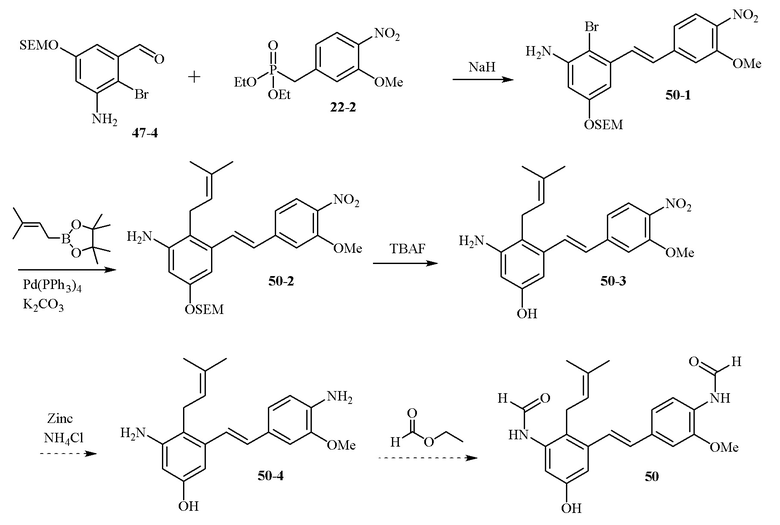
Соединение 47-4 (3,8 г) обрабатывали гидридом натрия (7,2 экв) и соединением 22-2 (1,8 экв) при комнатной температуре в тетрагидрофуране в течение 16 часов с получением соединения 50-1 (4,6 г, 85% выход).
Соединение 50-1 (4,6 г) обрабатывали тетракис (трифенилфосфин)палладием(0) (0,05 экв), карбонатом калия (2 экв и пинаколовым эфиром 3-метилбут-2-енилбороновой кислоты (2 экв) в смеси 3 : 1 диоксан/вода при 100°C с получением соединения 50-2 (3,99 г, 89% выход). Соединение 50-2 (0,79 г) обрабатывали 1M раствором тетрабутиламмония фторида в тетрагидрофуране (7 экв) при 68°C в течение 16 часов с получением соединения 50-3.
(E)-6-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)стирил)-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (51) [KYN-166].
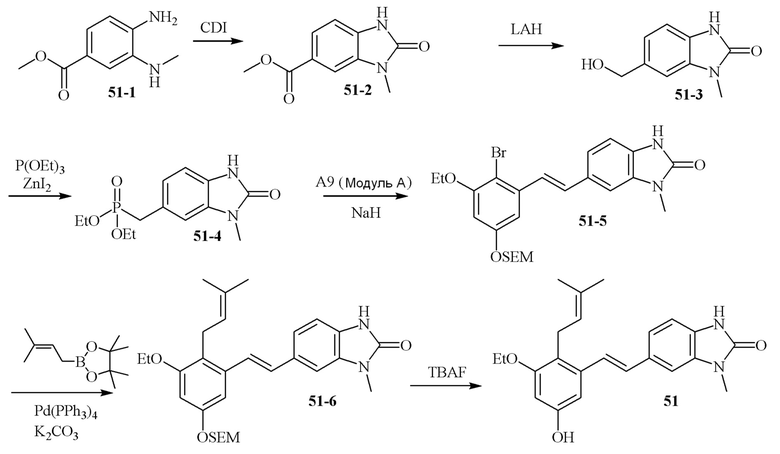
Метил 3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-карбоксилат (51-2).
Смесь соединения 51-1 (10,4 г, 57,7 ммоль, 1 экв), карбонилдиимидазола (14,0 г, 86,5 ммоль, 1,5 экв) в N, N’-диметилформамиде (104 мл) нагревали до 80°C в течение 2 часов, после чего анализ методом LC/MS указывал на завершение реакции. После охлаждения до комнатной температуры, растворитель удаляли при пониженном давлении. Добавляли к остатку 1N HCl (100 мл) и воду (80 мл), и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Твердое вещество фильтровали и сушили под вакуумом при 40°C в течение 16 часов с получением соединения 51-2 (11,5 г, 97% выход) в виде белого твердого вещества (YZ-3-165).
6-(Гидроксиметил)-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (51-3).
1M раствор алюмогидрида лития в тетрагидрофуране (83,7 мл, 83,7 ммоль, 1,5 экв) добавляли медленно к раствору соединения 51-2 (11,5 г, 55,8 ммоль, 1 экв) в безводном тетрагидрофуране (230 мл) при -79°C. Реакционную смесь постепенно подогревали до комнатной температуры в течение 4 часов и перемешивали в течение ночи, после чего анализ методом LC/MS указывал на завершение реакции. К реакционной смеси добавляли последовательно воду (10 мл), 1N водный раствор гидроксида натрия (5 мл). Смесь перемешивали при комнатной температуре в течение 30 минут и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech с силикагелем 330 g), элюируя с градиентом от 0 до 10% метанола в дихлорметане, с получением соединения 51-3 (7,2 г, 72% выход) в виде белого твердого вещества (YZ-3-166).
Диэтил ((3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)метил)фосфонат (51-4).
Триэтилфосфит (13,8 мл, 13,4 г, 40,4 ммоль, 2 экв) добавляли к раствору соединения 51-3 (7,2 г, 40,4 ммоль, 1 экв) и йодида цинка (25,8 г, 80,8 ммоль, 2 экв) в безводном тетрагидрофуране (72 мл). Реакционную смесь перемешивали при 70°C в течение 4 часов. Анализ методом 1H-ЯМР указывал на завершение реакции. После охлаждения до комнатной температуры, реакционную смесь разбавляли метилтрет-бутиловым эфиром (200 мл) и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech с силикагелем 330 g), элюируя с градиентом от 0 до 6% метанола в дихлорметане, с получением соединения 51-4 (2,7 г, 22% выход) в виде белого твердого вещества (YZ-3-167).
(E)-6-(2-Бром-3-этокси-5-((2-(триметилсилил)этокси)метокси)-стирил)-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (51-5).
Третбутоксид калия (3,0 г, 27,2 ммоль, 3 экв) добавляли к раствору соединения 51-4 (2,7 г, 9,0 ммоль, 1 экв) в безводном диметоксиэтане (35 мл) при 0°C. После перемешивания в течение 30 минут при 0°C, добавляли порциями раствор модуля D (3,7 г, 10,0 ммоль, 1,1 экв). Реакционную смесь постепенно подогревали до комнатной температуры и перемешивали в течение 20 часов. Анализ методом LC/MS указывал на завершение реакции. Реакционную смесь разбавляли метилтрет-бутиловым эфиром (200 мл) и насыщенным водным раствором хлорида аммония (200 мл). Слои разделяли, и водный слой экстрагировали дополнительным количеством метилтрет-бутилового эфира (200 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали на автоматизированной хроматографической системе Biotage (колонка Sorbtech с силикагелем 330 g), элюируя с градиентом от 25 до 60% этилацетата в гептане, с получением соединения 51-5 (300 мг, 6% выход) в виде желтовато-белого твердого вещества (YZ-3-168).
(E)-6-(3-Этокси-2-(3-метилбут-2-ен-1-ил)-5-((2-(триметил-силил)этокси)метокси)стирил)-1-метил-1,3-дигидро-2H-бензо[d]-имидазол-2-он (51-6).
Смесь 3 : 1 диоксана и воды (16 мл) продували азотом в течение 30 минут. Добавляли последовательно соединение 51-5 (290 мг, 0,56 ммоль, 1 экв), карбонат калия (32 мг, 1,12 ммоль, 2 экв), тетракис(трифенилфосфин)палладий(0) (32 мг, 0,03 ммоль, 0,05 экв) и 4,4,5,5-тетраметил-2-(3-метилбут-2-ен-1-ил)-1,3,2-диоксаборолан (220 г, 1,12 ммоль, 2 экв). Реакционную смесь перемешивали при 100°C в течение 7 часов. Анализ методом LC/MS указывал на завершения реакции. Реакционную смесь разбавляли метилтрет-бутиловым эфиром (100 мл) и водой (100 мл). Слои разделяли, и водный слой экстрагировали дополнительным количеством метилтрет-бутилового эфира (100 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали на автоматизированной хроматографической системе Biotage (колонка Sorbtech с силикагелем 40 g), элюируя с градиентом от 10 до 40% этилацетата в гептане, с получением соединения 51-6 (160 мг, 56% выход) в виде желтовато-белого твердого вещества (YZ-3-170).
(E)-6-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)стирил)-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (51) [KYN-166].
1M раствор тетрабутиламмония фторида в тетрагидрофуране (2,0 мл, 2,0 ммоль, 7 экв) добавляли к раствору соединения 51-6 (145 мг, 0,29 ммоль, 1 экв) в тетрагидрофуране (3 мл). После перемешивания при 70°C в течение 24 часов, анализ методом LC/MS указывал на завершение реакции. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом (100 мл) и насыщенным солевым раствором (100 мл). Слои разделяли, и водный слой экстрагировали дополнительным количеством этилацетата (100 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech с силикагелем 80 g), элюируя с градиентом от 0 до 5% метанола в дихлорметане, с получением соединения 51 (29 мг, 27% выход, 98,4% чистота) в виде желтовато-белого твердого вещества (YZ-3-172).
Анализ методом HPLC: 97,3% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: Waters Atlantis T3, 2,1×50 мм, 3 мкм; время удерживания: 8,4 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=379,2 (M+H)+; C23H26N2O3; 1H ЯМР (400 МГц, DMSO-d6) δ=7,32 (с, 1H), 7,27 (д, J=16,1 Гц, 1H), 7,16 (д, J=7,9 Гц, 1H), 6,97 (с, 1H), 6,94 (д, J=6,6 Гц, 1H), 6,63 (д, J=2,1 Гц, 1H), 6,31 (д, J=2,0 Гц, 1H), 5,02 (уш.т, J=6,8 Гц, 1H), 4,25-3,69 (м, 4H), 3,42-3,34 (м, 2H), 1,77 (с, 3H), 1,61 (с, 3H), 1,33 (т, J=6,9 Гц, 3H); 13C ЯМР (100 МГц, DMSO-d6) δ=157,77, 156,49, 155,02, 137,69, 132,04, 130,90, 130,62, 129,79, 128,52, 124,59, 121,14, 118,77, 109,10, 105,26, 103,95, 99,67, 63,77, 26,89, 26,03, 24,37, 18,27, 18,28.
(E)-6-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)стирил)-бензо[d]оксазол-2(3H)-он (52) [KYN-171].
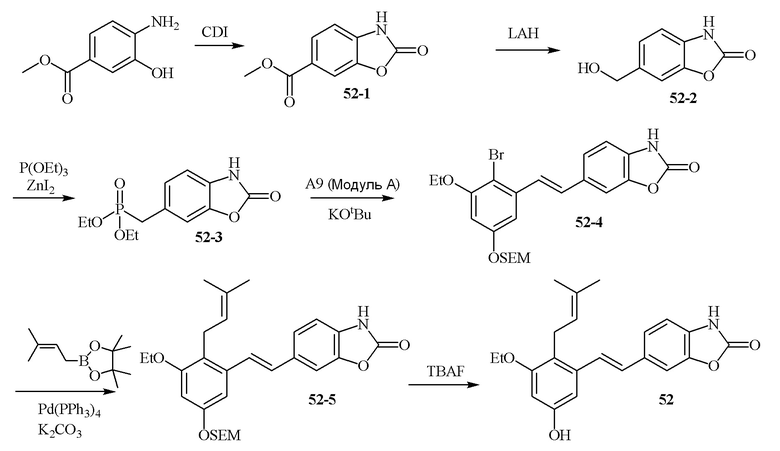
Метил 2-оксо-2,3-дигидробензо[d]оксазол-6-карбоксилат (52-1).
Смесь метил 4-амино-3-гидроксибензоата (10 г, 59,8 ммоль, 1 экв), карбонилдиимидазола (14,5 г, 89,7 ммоль, 1,5 экв) в N, N’-диметилформамиде (100 мл) нагревали до 80°C в течение 2 часов, после чего анализ методом LC/MS указывал на завершение реакции. После охлаждения до комнатной температуры, растворитель удаляли при пониженном давлении. К остатку добавляли 1N HCl (120 мл), и полученную смесь перемешивали в течение 3 часов при комнатной температуре. Твердое вещество фильтровали и сушили под вакуумом при 40°C в течение 16 часов с получением соединения 52-1 (11,6 г, 99% выход, 97% чистота) в виде желтовато-белого твердого вещества (CH-QZH-01-17).
6-(Гидроксиметил)бензо[d]оксазол-2(3H)-он (52-2).
1M раствор алюмогидрида лития в тетрагидрофуране (39 мл, 39 ммоль, 1,5 экв) добавляли, при скорости, позволяющей поддерживать температуру реакции ниже 5°C, к смеси соединения 52-1 (5 г, 25,6 ммоль, 1 экв) в тетрагидрофуране (120 мл) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение ночи, после чего анализ методом LC/MS указывал на завершение реакции. Реакционную смесь охлаждали до 5°C, добавляли последовательно воду (1,5 мл), 4N раствор гидроксида натрия (3 мл) и воду (6 мл). Смесь перемешивали при комнатной температуре в течение 30 минут и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech с силикагелем 220 g), элюируя с градиентом от 0 до 10% метанола в дихлорметане с получением соединения 52-2 (1,03 г, 24% выход) в виде желтого твердого вещества (CH-QZH-01-021).
Диэтил ((2-оксо-2,3-дигидробензо[d]оксазол-6-ил)метил)-фосфонат (52-3).
Триэтилфосфит (3,8 мл, 3,73 г, 22,48 ммоль, 2 экв) добавляли к раствору соединения 52-2 (1,86 г, 11,24 ммоль, 1 экв) и йодида цинка (7,18 г, 22,48 ммоль, 2 экв) в тетрагидрофуране (55 мл). Реакционную смесь перемешивали при 70°C в течение 3 часов. Анализ методом 1H-ЯМР указывал на завершение реакции. После охлаждения до комнатной температуры, реакционную смесь разбавляли метилтрет-бутиловым эфиром (200 мл) и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech с силикагелем 220 g), элюируя с градиентом от 0 до 5% метанола в дихлорметане, с получением соединения 52-3 (1,9 г, 59% выход) в виде желтовато-белого твердого вещества (YZ-3-151).
(E)-6-(2-Бром-3-этокси-5-((2-(триметилсилил)этокси)метокси)-стирил)бензо[d]оксазол-2(3H)-он (52-4).
Третбутоксид калия (2,10 г, 18,96 ммоль, 3 экв) добавляли к раствору соединения 52-3 (1,8 г, 6,30 ммоль, 1 экв) в безводном диметоксиэтане (53 мл) при 0°C. После перемешивания в течение 1 часа при 0°C, добавляли медленно раствор модуля D (2,61 г, 6,96 ммоль, 1,1 экв) в безводном диметоксиэтане (10 мл). Реакционную смесь постепенно подогревали до комнатной температуры и перемешивали в течение 2 дней. Анализ методом LC/MS указывал на завершение реакции. Реакционную смесь разбавляли метилтрет-бутиловым эфиром (200 мл) и насыщенным водным раствором хлорида аммония (200 мл). Слои разделяли, и водный слой экстрагировали дополнительным количеством метилтрет-бутилового эфира (200 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали на автоматизированной хроматографической системе Biotage (колонка Sorbtech с силикагелем 220 g), элюируя с градиентом от 5 до 30% этилацетата в гептане, с получением соединения 52-4 (0,94 г, 29% выход) в виде желтовато-белого твердого вещества (YZ-3-155).
(E)-6-(3-Этокси-2-(3-метилбут-2-ен-1-ил)-5-((2-(триметил-силил)этокси)метокси)стирил)бензо[d]оксазол-2(3H)-он (52-5).
Смесь диоксан/вода (45 мл/15 мл) продували азотом в течение 30 минут. Добавляли последовательно соединение 52-4 (0,94 г, 1,86 ммоль, 1 экв), карбонат калия (0,51 г, 3,72 ммоль, 2 экв), тетракис(трифенилфосфин)палладий(0) (0,11 г, 0,08 ммоль, 0,05 экв) и 4,4,5,5-тетраметил-2-(3-метилбут-2-ен-1-ил)-1,3,2-диоксаборолан (0,73 г, 3,72 ммоль, 2 экв). Реакционную смесь перемешивали при 100°C в течение 20 часов. Анализ методом LC/MS указывал на завершение реакции. Реакционную смесь разбавляли метилтрет-бутиловым эфиром (200 мл) и водой (200 мл). Слои разделяли, и водный слой экстрагировали дополнительным количеством метилтрет-бутилового эфира (200 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали на автоматизированной хроматографической системе Biotage (колонка Sorbtech с силикагелем 220 g), элюируя с градиентом от 5 до 30% этилацетата в гептане, с получением соединения 52-5 (0,54 г, 59% выход) в виде белого твердого вещества (YZ-3-156).
(E)-6-(3-Этокси-5-гидрокси-2-(3-метилбут-2-ен-1-ил)стирил)-бензо[d]оксазол-2(3H)-он (52).
1M раствор тетрабутиламмония фторида в тетрагидрофуране (4,9 мл, 4,94 ммоль, 7 экв) добавляли к раствору соединения 52-5 (0,35 г, 0,71 ммоль, 1 экв) в безводном тетрагидрофуране (5 мл). Реакционную смесь перемешивали при 70°C в течение 24 часов. Анализ методом LC/MS указывал на образования следовых количеств продукта, но большая часть исходного материала оставалась непрореагировавшей. Объем реакционной смеси концентрировали при пониженном давлении до ~ 1 мл и перемешивали при 70°C в течение еще 24 часов. Анализ методом LC/MS указывал на завершение реакции. Реакционную смесь разбавляли этилацетатом (100 мл) и насыщенным солевым раствором (100 мл). Слои разделяли, и водный слой экстрагировали дополнительным количеством этилацетата (100 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали на автоматизированной хроматографической системе Interchim (колонка Sorbtech с силикагелем 80 g), элюируя с градиентом от 0 до 2% метанола в дихлорметане, с получением соединения 52 (58 мг, >95% чистота, и 78 мг, ~70% чистота, 53% суммарный выход) в виде белого твердого вещества. (YZ-3-158).
Анализ методом HPLC: 97,0% чистота; длина волны 254 нм, диапазон пропускания 4; колонка: Waters Atlantis T3, 2,1×50 мм, 3 мкм; время удерживания: 8,9 мин; подвижная фаза: ACN/муравьиная кислота/вода; масс-спектр (в режиме регистрации положительных ионов) m/z=366,2 (M+H)+; C22H23NO4; 1H ЯМР (400 МГц, DMSO-d6) δ=11,65 (уш.с, 1H), 9,20 (уш.с, 1H), 7,56 (с, 1H), 7,38-7,23 (м, 2H), 7,07 (д, J=8,1 Гц, 1H), 6,95 (д, J=16,0 Гц, 1H), 6,66-6,58 (м, 1H), 6,36-6,27 (м, 1H), 5,01 (уш.т, J=6,4 Гц, 1H), 3,94 (кв, J=6,8 Гц, 2H), 3,41-3,34 (м, 2H), 1,74 (с, 3H), 1,60 (с, 3H), 1,33 (т, J=6,8 Гц, 3H); 13C ЯМР (100 МГц, DMSO-d6) δ=157,70, 156,57, 154,94, 144,44, 137,36, 132,39, 130,35, 129,81, 129,71, 125,91, 124,51, 123,41, 118,94, 110,22, 107,20, 104,16, 99,96, 63,74, 25,98, 24,27, 18,25, 15,25.
ИССЛЕДОВАНИЯ БИОЛОГИЧЕСКОЙ АКТИВНОСТИ СОЕДИНЕНИЙ ПО ИЗОБРЕТЕНИЮ НА КЛЕТОЧНЫХ КУЛЬТУРАХ
In vitro исследования, относящиеся к соединениям 1-15, в линиях раковых клеток человека
Введение
Оценивали соединения 1-15 и их воздействие на рост раковых клеток при проведении исследования выживаемости клеток многих типов рака (таблица 1-5). Раковые клетки, включающие клетки рака поджелудочной железы, мочевого пузыря, почки, толстой кишки, молочной железы, легкого, печени и остеобластических сарком, полученные из ATCC (Американской коллекции типовых культур), аутентифицировали перед использованием с помощью анализа коротких концевых повторов в геноме (Reid Y, Storts D, Riss T and Minor L. Authentication of Human Cell Lines by STR DNA Profiling Analysis. 2013 May 1. In: Markossian S, Sittampalam GS, Grossman A, et al., editors. Assay Guidance Manual. Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences; 2004.). Протокол эксперимента для каждой линии раковых клеток перед каждым использованием верифицировали и оптимизировали в соответствии с опубликованными в научной литературе рекомендациями (таблицы 1-3).
Культивирование клеток/Протоколы молекулярной биологии
Криосохранение линий клеток млекопитающих
Раковые клетки выращивали в чашке диаметром 100 мм в соответствии с рекомендациями фирмы-производителя. После удаления культуральной среды, клетки разделяли путем промывания в PBS (1х), затем добавляли трипсин (1 мл) и инкубировали в течение 2-5 мин при 37°С. Клетки ресуспендировали в среде для культивирования, затем переносили в пробирку фирмы Falcon объемом 15 мл и затем центрифугировали в течение 5 мин при 1000 об/мин при комнатной температуре. Надосадочную жидкость удаляли, а осадок клеток ресуспендировали в фетальной бычьей сыворотке (900 мкл). Суспензию клеток (900 мкл) переносили в криопробирку объемом 1 мл, содержащую DMSO (100 мкл). Принимали меры для соблюдения требования по поводу того, чтобы клетки в среде для замораживания подвергались минимальному воздействию комнатной температуры (<10 мин). Затем криопробирки переносили в содержащий изопропанол штатив для замораживания CoolCell и хранили при -80°C. Штатив CoolCell обеспечивает стабильное снижение температуры на 1°C в минуту. Криопробирки извлекали из штатива CoolCell через 24 часа, затем переносили в жидкий азот для длительного хранения. В жидком азоте хранили минимум 4 аликвоты для каждой клеточной линии.
Размораживание и пассирование раковых клеток
Криопробирку извлекали из емкости с жидким азотом и выдерживали при комнатной температуре до тех пор, пока стенки пробирки не оттаивали, но центр остался замороженным. К частично замороженным клеткам добавляли по каплям подогретую среду (9 мл). Суспензию клеток центрифугировали в течение 5 мин при 1000 об/мин при комнатной температуре. Надосадочную жидкость удаляли, а осадок клеток ресуспендировали в 1 мл среды. Суспензию переносили в культуральную чашку диаметром 100 мм и инкубировали при 37°С в течение 24 часов. После прочного сцепления, к клеткам добавляли свежую среду. Смену сред повторяли каждые 3 дня до тех пор, пока плотность клеток не достигала конфлюентности. При достижении клетками приблизительно 80% конфлюэнтности (80% поверхности колбы покрыто клеточным монослоем), проводили субкультивирование. Готовность использования клеток в эксперименте определяли по величине индекса разведения от 1:3 до 1:10. Субкультирование проводили до 10 пассажей и менее.
Анализ жизнеспособности клеток
1,7 х 103 клеток/лунка высевали в четырех экземплярах в 96-луночном планшете после культивирования. Добавляли лекарственные средства при концентрации 5 мкМ и при 18 концентрациях, полученных путем последовательного 2-кратного разведения (диапазон концентраций 0,000038146-5 мкМ) для построения кривой зависимости "доза-эффект". Рост клеток оценивали на день 1, 3 и 5 после обработки лекарственными средствами, используя люминесцентный анализ жизнеспособности клеток CellTiter Glo Luminescence Cell Viability Assay (Promega) в соответствии с протоколом фирмы-производителя. Для считывания люминесцентного сигнала с планшета использовали многорежимный планшет-ридер Thermo Scientific Varioskan LUX. Данные представлены как среднее величина интенсивности люминесценции (в произвольных единицах) ± S.D. (среднеквадратическое отклонение).
Расчет величины IC50
Анализ жизнеспособности клеток использовали для определения концентрации соединения 1 (KYN-001) и его аналогов, при которой достигалось снижение ответной реакции наполовину. В каждом случае обработки раковых клеток, использовали растворы соединений с концентрацией 1, 2, 5, 10 и 20 мкМ. Для аппроксимации кривых зависимости "доза-эффект" использовали программное обеспечение GraphPad Prism 7. Строили графические зависимости относительных единиц люминесценции от log применяемых величин доз, и для анализа использовали модель нелинейной регрессии.
Результаты
Воздействие соединения 1 (KYN-001) и его аналогов на жизнеспособность раковых клеток
Оценивали зависимое от времени и дозы воздействие экзогенно добавляемых соединений на пролиферацию раковых клеток, используя люминесцентный анализ жизнеспособности клеток CellTiter Glo Luminescence Cell Viability Assay. Во всех случаях рассчитывали величину концентрации лекарственного средства, при которой ответная реакция снижалась наполовину (IC50). Ингибирующее действие аналогов соединения 1 (KYN-001) в сравнении с ингибирующим действием исходных соединений 1 (KYN-001), 8 (KYN-119) и 27 (KYN-149) представлено в таблицах 1-5.
Соединение 8 [KYN-119] обладало самой высокой активностью (наименьшей величиной IC50) среди оцениваемых соединений в отношении всех испытуемых клеточных линий, представленных в таблицах 1 и 2. Соединения 17 [KYN-120], 23 (KYN-125), 29 (KYN-128) и 33 (KYN-126) проявляли существенно меньшую активность, чем исходное соединение 1. В случае соединения 22 (KYN-124) наблюдалось потеря антипролиферативного действия.
Протокол исследования клеточной пролиферации оптимизировали в случае соединение 8 (KYN-119), для того чтобы можно было более точно оценить его низкую величину IC50. В этом протоколе, применяли 18 доз (2-х кратные разбавления в диапазоне от 0,000038146 до 5 мкМ) на клетках, посеянных в 96-луночных планшетах, и через 1, 3 и 5 дней после обработки измеряли люминесценцию. Следует отметить, что ни соединение 1, ни соединение 8 не оказывали воздействия на жизнеспособность клеток на день 1 после обработки при дозах <1 мкМ. Это наблюдение дает основание предположить, что при дозах < 1 мкМ, соединения 1 (KYN-001) и 8 (KYN-119) не оказывают токсического действия на клетки.
Проводили сравнительное исследование соединения 1 [KYN-001] и 8 [KYN-119] на 83 линиях раковых клеток (таблица 3). Высокая активность была продемонстрирована соединением 1 [KYN-001] с величинами IC50 в диапазоне от 0,01 до 0,1 мкМ в отношении следующих клеточных линий: рака языка SCC-15, CAL 27, UPCI:SCC090, SCC-4; лейомиосаркомы SK-UT-1B, SK-LMS-1; рака мочевого пузыря UMUC_3, SCABER, 5637, RT-112; рака молочной железы MDA-MB-231; рака печени SNU-423, SNU-398, Huh7; саркомы соединительной ткани TE 381.T, HT-1080; остеосарком MG3, KHOS_N/P; рака легкого NCI-H661, NCI-H1299, NCI-H2126, BEAS-2B, NCI-H1048; рака гортанной части глотки UPCI:SCC152; фибросарком SW 684; липосаркомы 93T449, 94T778; рака глотки FADU; рабдомиосаркомы SJCRH30, CCL 166; рака почки CCL 166. В среднем, активность соединения 8 [KYN-119] было в 10 раз выше активности соединения 1 [KYN-001]. Очень высокая активность была продемонстрирована соединением 8 [KYN-119] с величинами IC50 в диапазоне от 0,001 до 0,01 мкМ в отношении следующих клеточных линий: рака легкого NCI-H2126, NCI-H1299; рака мочевого пузыря RT-112, 5637, UMUC_3; остеосарком T1-73, MG3; фибросарком SW 684; рабдомиосаркомы CCL 166; рака печени SNU-398; рака молочной железы MDA-MB-231; рака почки Caki-1; рака поджелудочной железы CAPAN-2. Высокая активность была продемонстрирована соединением 8 [KYN-119] с величинами IC50 в диапазоне от 0,01 до 0,1 мкМ в отношении следующих клеточных линий: рака мочевого пузыря SCABER, HT-1197, T24, J82; рака толстой кишки SW_480, HT_116; саркомы соединительной ткани HT-1080; саркомы Юинга HTB-166; рака гортанной части глотки UPCI:SCC152; рака почки ACHN, SK-NEP-1, 769-P, 786-O; лейомиосаркомы SK-LMS-1; липосаркомы 93T449, 94T778; Liver HepG2, Huh7, SNU-475, JHH-6; рака легкого NCI-H661, NCI-H1048, BEAS-2B, NCI-H1437, NCI-H1563, NCI-H1573, A549; остеосарком KHOS_N/P, SAOS-2, Hs888.T, HOS_CRL_1543, KHOS_312H, U2OS; рака поджелудочной железы PANC-1, MIA PaCa-2, CFPAC; рака глотки FADU; рабдомиосаркомы TE 159.T, A-204, SJCRH30, Hs 729; рака языка SCC-15, SCC-4, UPCI:SCC090, CAL 27, SCC-9.
Проводили испытания аналогов соединения 1 [KYN-001], включающих соединения 8 [KYN-129], 12 [KYN-130] и 13 [KYN-131], на их воздействие на жизнеспособность раковых клеток, используя оптимизированный протокол анализа пролиферации клеток, в месте с соединениями 1 [KYN-001] и 9 [KYN-119], и результаты представлены в таблице 4. Ни одно из этих соединений не является более эффективным, чем соединение 9 [KYN-119]. Соединения 12 [KYN-130] и 13 [KYN-131] проявляли эффективность, аналогичную эффективности соединения 1 [KYN-001], при ингибировании роста раковых клеток на день 3 обработки. Полная потеря антипролиферативного действия наблюдалась в случае соединения 8 [KYN-129].
Было проведена сравнительная оценка воздействия соединений 1 [KYN_001] и 8 [KYN-119], и их аналогов, величин IC50 (мкМ), в отношении линий раковых клеток A498 и MIA PaCa-2 в 8 экспериментах (таблица 5A и 5B).
Активность в отношении клеток A498 в диапазоне активности величины IC50 менее, чем 0,1 мкМ, наблюдалась только для соединения 30 [KYN-142]. Ряд активности (от высокой к низкой) для соединений в диапазоне умеренной активности от 0,1 до 1 мкМ является следующим: соединения 2, 3, 4, 7, 8, 9, 10, 11, 12, 17, 20, 23, 27, 28, 29, 33, 34, 38 [KYN-155, 131, 138, 120, 130, 143, 136, 132, 125, 128, 119, 139, 146, 140, 133, 147, 149, 126]. Активность в отношении клеток MIA PaCa-2 в диапазоне высокой активности величины IC50, меньшей чем 0,001 мкМ, наблюдалась только для соединения 28 [KYN-143]. Порядок активности для соединений с величинами IC50 в диапазоне очень высокой активности от 0,01 до 0,1 мкМ является следующим: 1, 5, 8, 10, 20, 27, 30 [KYN-149, 119, 153, 146, 142, 001, 131]. Ряд соединений с величинами IC50 в диапазоне активности от 0,1 до 1 мкМ является следующим: 2, 3, 4, 9, 11, 23, 29, 32, 34 [KYN-130, 138, 147, 132, 125, 128, 139, 148, 140].
Зависимости активности подавления жизнеспособности раковых клеток от структуры (SARs) для соединений 1-49 и 51 [KYN-001, KYN-119, KYN-120, KYN-124, KYN-125, KYN-126, KYN-128 - KYN-134, KYN-136 - KYN-169] на основе величин IC50 в таблицах 1-5.
Синтезировали на основе ключевого соединения 1 [KYN-001] аналоги с различными вариантами химической структуры с изменением заместителей на стильбеновом ядре с целью улучшения и оптимизации биологической активности, биодоступности и растворимости.
Изменение пренильного заместителя в соединении 1 [KYN-001] (R1a=пренил) осуществляли путем проведения органического синтеза с получением следующих аналогов: 45 [KYN-112] (R1a=Br), 7 [KYN-136] (R1a=бензил), 6 [KYN-134] (R1a=аллил), 2 [KYN-138] (R1a=E-кротил ((E)-бут-2-ен-1-ил)), 3 [KYN-139] (R1a=Z-кротил ((Z)-бут-2-ен-1-ил)), 4 [KYN-140] (R1a=изокротил (бут-3-ен-2-ил)). В целом, все изменения приводили к снижению биологической активности (таблицы 1, 4 и 5 ) за исключением соединения 2 [KYN-138] с E-кротиловой группой, которое демонстрировало биологическую активность, сопоставимую с активностью соединения 1 [KYN-001]. Рассчитано, что соединение 2 [KYN-138] является менее липофильным и приблизительно в 3 раза лучше растворяется в воде, чем соединение 1 [KYN-001], указывая на то, что оно может иметь более высокую биодоступность и более высокие фармакокинетические свойства.
Были получены следующие аналоги путем замены R1b заместителя в соединении 1 [KYN-001] (R=OMe): 8 [KYN-119] (R=OEt), 9 [KYN-130] (R=пропокси, 10 [KYN-131] (R=изопропокси, 11 [KYN-132] (R=трифторметокси), 46 [KYN-145] (R=трифторметил). Замена метоксильной группы на трифторметильную группу приводила к полной потере биологической активности в отношении испытуемых клеточных линий. Соединение 8 [KYN-119] с этоксильной группой демонстрировало в среднем усиление в 10 раз подавление жизнеспособности раковых клеток в случае 83 клеточных линий (таблица 3). Дополнительное увеличение размера алкильной группы, как в случае соединения 9 [KYN-130] (R=пропокси) и соединения 10 [KYN-131] (R=изопропокси) приводило к сопоставимой или более слабой биологической активности и, кроме того, было рассчитано, что эти соединения являются значительно более липофильными, чем соединений 1 [KYN-001], и значительно менее водорастворимыми.
Аналог соединения 1 [KYN-001] (R1e=OMe), соединение 12 [KYN-133] (R1e=OEt), при испытаниях на клеточных линиях, продемонстрировало снижение приблизительно в 10 раз подавления жизнеспособности раковых клеток. Это представляет резкий контраст с усилением в 10 раз подавления жизнеспособности раковых клеток, наблюдаемое в случае соединения 8 [KYN-119], в которой изменена этоксильная группа в соединении 1 [KYN-001]. Другие аналоги соединения 1 [KYN-001] (R1e=OMe), соединения 15 [KYN-158] (R1e=NHMe), 16 [KYN-157] (R1e=NHMe), 41 [KYN-161] (R1e=NHMe) при испытаниях проявляли отсутствие активности или слабую активность.
Аналоги соединения 1 [KYN-001] получали путем изменения заместитель R1f в соединении 1 [KYN-001] (R1f=OH). Замена гидроксильной группы на аминогруппу дает дополнительную валентность для введения дополнительной химической структуры. Аналог с аминогруппой, соединение 23 [KYN-125], проявляло активность, подобную, но в целом более низкую, в отношении испытуемых клеточных линий, по сравнению с соединением 1 [KYN-001]. С дополнительной валентностью, доступной через азот, были синтезированы ряд аминопроизводных соединения 23 [KYN-125] в виде соединений 25, 26, 27, 29, 31, 33, 36 [KYN-126, 128, 129, 137, 149, 150, 151]. Ацетамидное производное 29 [KYN-128] (R1f=-(CO)CH3) демонстрировало активность, подробную активности соединения 1 [KYN-001], и формамидное производное 27 [KYN-149] (R1f=-(CO)H), испытуемое на двух клеточных линиях, характеризовалось величиной IC50 0,015 мкМ в отношении клеток MIA PaCa-2, приблизительно в 5 раз более высокой активностью, чем активность соединения 1 [KYN-001]. Все аминопроизводные, более крупные чем ацетамид, в целом, характеризовались более слабым подавлением жизнеспособности раковых клеток или отсутствием активности. Замена гидроксильной группы R1f в соединении 1 [KYN-001] на карбоксильную группу давало аналог 21 [KYN-154] (R1f=COOH) с величиной IC50 более 3 мкМ на двух клеточных линиях, A498 и MIA PaCa-2. Замена атома водорода R1g в соединении 1 [KYN-001] на метильную группу с получением аналога 17 [KYN-120] (R1g=CH3) приводило к снижению приблизительно в 3 раза ингибирующего действия в отношении подвергаемых испытанию шести клеточных линий (таблица 2). Аналогичная потеря активности наблюдалась для аналога 18 [KYN-156] (R1g=OH) с дополнительной гидроксильной группой (таблица 5A). Перемещение гидроксильной группы R1f в соединение 1 [KYN-001], как в аналоге 19 [KYN-160] (R1g=OH), также приводило к сильному ослаблению активности в отношении испытуемых клеточных линий.
В случае, когда было сделано единственное изменение заместителя в соединении 1 [KYN-001], было обнаружено, что химическая структура с двумя изменениями заместителей независимо усиливает подавление жизнеспособности раковых клеток, R1d=OEt, как наблюдалось в случае соединения 8 [KYN-119] (R1b=OEt), и R1f=NH(CO)H, как наблюдалось в случае соединения 27 [KYN-149] (R1f=-NH(CO)H). Синтезировали соединение с изменениями обоих этих заместителей, 28 [KYN-143] (R1b=OEt; R1f=NH(CO)H), и было обнаружено, что оно значительно усиливает подавление клеточных линий A498 (IC50 0,18 мкМ) и MIA PaCa-2 (IC50 0,00094 мкМ). Эти величины подавления выгодно отличаться от соединения 8 [KYN-119], которое демонстрировало более низкую величину подавления клеточных линий A498 (IC50 0,36 мкМ) и MIA PaCa-2 (IC50 0,025 мкМ). Замена метоксильной группы R1e в соединении 28 [KYN-143] на гидроксильную группу группа, как в аналоге 40 [KYN-159] (R1e=OH), сильно понижала активность в отношении двух испытуемых клеточных линий (таблица 5A). N-гидроксипроизводное гидроксильной группы соединения 28 [KYN-143], как в аналоге 39 [KYN-165] (R1f=N(OH)(CO)H), характеризовалось практически полной потерей противораковой активности в отношении двух испытуемых клеточных линий (таблица 5B).
Замена гидроксильной группы R1d в соединении 1 [KYN-001] на нитрогруппу давала аналог 42 [KYN-162] (R1d=NO2), который характеризовался практически полной потерей противораковой активности в отношении двух испытуемых клеточных линий (таблица 5A). Не наблюдалось существенного усиления активности в случае, когда нитрогруппу R1d превращали в аминогруппу, как в соединении 43 [KYN-163] (R1d=NH2), и затем в формамидную группу, как в соединении 44 [KYN-164] (R1d=NH(CO)H) (таблица 5A).
Замена гидроксильной группы R1b в соединении 1 [KYN-001] на аминогруппу давала аналог 47 [KYN-167] (R1b=NH2), который вызывал неопределенную ответную реакцию на противораковое действие в отношении двух клеточных линий (таблица 5B). Синтезировали формамидное и ацетамидное производное в качестве соединения 48 [KYN-168] (R1b=NH(CO)H) и соединения 49 [KYN-169] (R1b=NH(CO)Me), соответственно, которые, как было определено, обладали противораковым действием, сопоставимым с противораковым действием соединения 1 [KYN-001], в отношении двух линий раковых клеток (таблица 5A).
Аналог 8 [KYN-119] с 5-членным гетероциклическим кольцом, конденсированным через присоединение к R1e и R1f, соединение 51 [KYN-166] и соединение 52 [KYN-171], не проявляли значительной активности в отношении двух испытуемых клеточных линий (таблица 5A).
Таблица 1. Величины IC50 (мкМ) соединений 1, 13, 14, 45 [KYN-001, 112, 114, 118] на день 3 после обработки лекарственным средством (на основе 10 доз в диапазоне 0,0001-20 мкМ [31 октября 2018 года]
Таблица 2. Величины IC50 (мкМ) для соединений 1, 17, 22, 23, 29, 33 на день 5 после обработки лекарственным средством (на основе 10 доз в диапазоне 0,0001-20 мкМ) и для соединение 8 (на основе 18 доз в диапазоне 0,000038146-5 мкМ [31 октября 2018, скорректированные 9 июня 2019 года]
Таблица 3. Воздействие соединений 1 [KYN-001] и 8 [KYN-119] на жизнеспособность раковых клеток, оцененное люминесцентным анализом CellGlow Titer Assay, представленное в виде величины IC50 мкМ. Клетки обрабатывали соединениями в 18 (2-кратных) разбавлениях (диапазон концентраций: 5-0,00003 мкМ) через 24 часа после посева, и измерения люминесценции проводили через 3 дня после обработки. Приведены объединенные результаты 7 экспериментов от 31 октября 2018 года, 19 декабря 2018 года, 15 мая 2019 года, 4 июня 2019 года, 6 июня 2019 года, 8 июня 2019 года, 19 июня 2019 года.
IC50
мкМ
IC50
мкМ
IC50
Концентрации DMSO 0,005% - 4,88×10-6% - 3×10-8% служили в качестве отрицательных контролей при проведении анализа, так как эти разведения соответствуют самой высокой, медианной и самой низкой концентрации DMSO в диапазоне 18 испытуемых концентраций лекарственного средства
Величины IC50 (мкМ) соответствуют зависимости log(ингибитор) против ответной реакции (четыре параметра), и кривые зависимости "доза-эффект" строили, используя программное обеспечение GraphPad Prism 8.
Таблица 4. Воздействие соединений на жизнеспособность раковых клеток, оцененное люминесцентным анализом CellGlow Titer Assay, представленное в виде величины IC50 мкМ. Клетки обрабатывали соединениями в 18 (2-кратных) разбавлениях (диапазон концентраций: 5-0,00003 мкМ) через 24 часа после посева, и измерения люминесценции проводили через 3 дня после обработки. Приведены объединенные результаты экспериментов от 4 июня 2019 года, 6 июня 2019 года, 8 июня 2019 года.
Таблица 5A. Воздействие соединений на жизнеспособность раковых клеток, оцененное люминесцентным анализом CellGlow Titer Assay. Сравнительная оценка величин IC50s (мкМ) для соединений 1 [KYN-001] и 27 [KYN-149], и их аналогов. Клетки обрабатывали соединениями в 11 разбавлениях (диапазон концентраций: 3-0,00001 мкМ). Приведены объединенные результаты семи экспериментов
Таблица 5B. Воздействие соединений на жизнеспособность раковых клеток, оцененное люминесцентным анализом CellGlow Titer Assay. Сравнительная оценка величин IC50 (мкМ) для соединений 1 [KYN-001] и 27 [KYN-149], и их аналогов. Клетки обрабатывали соединениями в 11 разбавлениях (диапазон концентраций: 3-0,00001 мкМ). Приведены объединенные результаты трех экспериментов
CRL-1420
HTB-14
Таблица 6. Воздействие соединений на жизнеспособность раковых клеток, оцененное люминесцентным анализом CellGlow Titer Assay. Сравнительная оценка величин IC50 (мкМ) для соединения 1 [KYN-001] и соединение 8 [KYN-119], и их аналогов. Клетки обрабатывали соединениями в 11 разбавлениях (диапазон концентраций: 3-0,00001 мкМ)
На 3 день после обработки клеток рака поджелудочной железы MIA PaCa_2, через 24 часа после посева (11 разбавлений), оценено люминесцентным анализом CellGlow Titer Assay.
Величины IC50 (мкМ) соответствуют зависимости log(ингибитор) против ответной реакции (четыре параметра), и кривые зависимости "доза-эффект" строили, используя программное обеспечение GraphPad Prism 8.
Соединения 23, 29 и 33 синтезировали для того, чтобы получить соединения с улучшенной биодоступностью и улучшенными фармакокинетическими свойствами (более оптимальное значение logP и растворимость в воде) по сравнению с ключевыми противораковыми соединениями 1 и 8.
Для специалистов в данной области является очевидным, что в отношении описанных выше вариантов осуществления могут быть предложены многочисленные изменения и/или модификации без отступления от объема настоящего изобретения. Поэтому, варианты осуществления настоящего изобретения следует считать во всех аспектах иллюстративными, и не ограничивающими объем изобретения.
Оценка соединения 1 при лечении заболеваний кожи
Цель исследования
Оценить возможность применения соединений 1, 8, 20, 27, 28, 31 и 35 при заболевании кожи с проведением in vitro исследований. Оценку проводила компания LEO Pharma Open Innovation, USA.
Конкретные задачи
1. Определение ингибирования высвобождения CCL2 из IL-4, IL-13, IL-22 и IFN-γ-стимулированных первичных кератиноцитов человека [исследование номер 1280] и определение воздействия на выживаемость клеток [исследование номер 1243].
2. Определение ингибирования высвобождения IL-8 из IL-17A и TNFα-индуцированных первичных кератиноцитов человека [исследование номер 1250] и определение воздействия на выживаемость клеток [исследование номер 1244].
3. Определение ингибирования секреции IL-17A из мононуклеарных клеток периферической крови (PBMC) человека, стимулированной с помощью antiCD3/antiCD28-покрытых сфер [исследование номер 1321] и определение воздействия на выживаемость клеток [исследование номер 1322].
4. Определение ингибирования секреции IL-4 и IL-2 из CD4-положительных T-клеток человека, стимулированной с помощью antiCD3/antiCD28-покрытых сфер [исследование номер 1297, 1298] и определение воздействия на выживаемость клеток [исследование номер 1299].
Краткое описание возможности применения соединений при заболевании
1. Можно ожидать, что соединения, которые ингибируют секрецию CCL2 из кератиноцитов, обладают лечебным эффектом при атопическом дерматите.
2. Можно ожидать, что соединения, которые ингибируют секрецию IL-8 из кератиноцитов, обладают лечебным эффектом при псориазе.
3. Можно ожидать, что соединения, которые ингибируют секрецию IL-17A, обладают лечебным эффектом при псориазе.
4. Можно ожидать, что соединения, которые ингибируют секрецию IL-4, обладают лечебным эффектом при атопическом дерматите. Соединения, которые также ингибируют секрецию IL-2, обладают более широким иммунодепрессивным эффектом, так как IL-2 является аутокринным фактором роста для T-клеток.
Результаты
Пример кривых "концентрация-эффект" для соединения 28 [KYN-143].
1. Высвобождение CCL2
На фигуре 9 в графическом виде представлены данные по % воздействия изменяющихся концентраций соединения 28 на ингибирование высвобождения CCL2 и подавление жизнеспособности клеток.
Обсуждение: соединение 28 проявляло умеренную активность по снижению высвобождения CCL2 с аналогичным снижением жизнеспособности клеток. Не приводя в качестве подтверждения какую-либо теорию, тем не менее, авторы изобретения считают, что горизонтальный участок кривой, соответствующий частичному уровню подавления жизнеспособности клеток от максимального, по-видимому, не обусловлен цитотоксичностью, а на самом деле может быть частью механизма воздействия (MoA) на молекулу/мишень.
2. Высвобождение IL-8
На фигуре 10 в графическом виде представлены данные по % воздействия изменяющихся концентраций соединения 28 на ингибирование высвобождения IL-8 и подавление жизнеспособности клеток.
Обсуждение: соединение 28 проявляло умеренную активность по снижению высвобождения IL-8, аналогичную активности, полученной по снижению высвобождения CCL2. Однако, в этом исследовании, соединение 28 продемонстрировало одинаковое воздействие как на снижение высвобождения IL-8, так и на подавление жизнеспособности клеток. Не приводя в качестве подтверждения какую-либо теорию, тем не менее, авторы изобретения считают, что это указывает на кератиноцит-связанный механизм, но с меньшим эффектом по высвобождению IL-8, чем в случае высвобождения CCL2, описанного выше и представленного на фигуре 9.
3. Высвобождение IL-17A
На фигуре 11 в графическом виде представлены данные по % воздействия изменяющихся концентраций соединения 28 на ингибирование высвобождения IL-17A и подавление жизнеспособности клеток.
Обсуждение: соединение 28 проявляло низкую активность по снижению высвобождения IL-17A.
4. Высвобождение IL-2 и IL-4
На фигуре 12 в графическом виде представлены данные по % воздействия изменяющихся концентраций соединения 28 на ингибирование высвобождения IL-2 и IL-4 и подавление жизнеспособности клеток.
Обсуждение: Соединение 28 проявляло умеренную активность по снижению высвобождения конкретно IL-4 и низкую активность по высвобождению IL-2 и подавлению пролиферации клеток. Не приводя в качестве подтверждения какую-либо теорию, тем не менее, авторы изобретения считают, что, по-видимому, имеет место специфическое воздействие по высвобождению IL-4 в T-клетках.
На фигуре 13 представлены кривые зависимости "концентрация-эффект" для соединений 1, 8, 20, 27, 28, 31 и 35.
Обсуждение: соединения 1 (KYN-001), 8 (KYN-119), 20 (KYN-146), 27 (KYN-149), 28 (KYN-143), 31 (KYN-151) и 35 (KYN-152) характеризуются аналогичным профилем, при котором более резко выраженный эффект с меньшими величинами EC50 может быть отмечен при проведении исследования по высвобождению CCL2, затем в случае высвобождения IL-8 и IL-4; менее чем 100% снижение выживаемости клеток, за исключением соединения 8 и соединения 31 (таблица 7, фигура 13). Наличие генерализованного цитотоксического эффекта в испытуемых молекулах является типичным, и он может достигать 100%, но это характерно не для всех соединений. Это может указывать на специфический механизм действия, который подавляет жизнеспособность клеток без проявления цитотоксичности. Кроме того, по-видимому, имеет место специфическое действие по высвобождению IL-4 в T-клетках, но с более низкой активностью. Соединения 28 (KYN-143) и 27 (KYN-149) отличаются от остальных соединений в этих исследованиях как более активные молекулы, и в соответствии с их активностью они следует в ряду сразу после соединения 8 (KYN-119) (таблица 7).
Соединение 28 [KYN-143] ингибировало секрецию CCL2 в кератиноцитах при величине EC50 40 нM с 100% максимальной эффективностью и подавляло жизнеспособность клеток при величине EC50 124 нM с 68% максимальным снижением жизнеспособности клеток. Соединение 27 [KYN-149] ингибировало секрецию CCL2 в кератиноцитах при величине EC50 32 нМ с 100% максимальной эффективностью и подавляло жизнеспособность клеток при величине EC50 41 нМ с 67% максимальным снижением жизнеспособности клеток.
Соединение 28 [KYN-143] ингибировало высвобождение IL-8 из IL-17A и TNFα-индуцированных первичных кератиноцитов человека при величине EC50 44 нМ с 85% максимальной эффективностью и подавляло жизнеспособность клеток при величине EC50 35 нМ с 81% максимальным снижением жизнеспособности клеток. Соединение 27 [KYN-149] ингибировало высвобождение IL-8 из IL-17A и TNFα-индуцированных первичных кератиноцитов человека при величине EC50 46 нМ с 75% максимальной эффективностью и подавляло жизнеспособность клеток при величине EC50 32 нМ с 83% максимальным снижением жизнеспособности клеток.
Соединение 28 [KYN-143] ингибировало секрецию IL-17A из PBMC человека, стимулированную с помощью antiCD3/antiCD28-покрытых сфер при величине EC50 526 нМ с 73% максимальной эффективностью и подавляло жизнеспособность клеток при величине EC50 659 нМ с 54% максимальным снижением жизнеспособности клеток. Соединение 27 [KYN-149] и другие представляющие интерес соединения были значительно менее эффективными.
Для ингибирования секреции IL-2 из CD4-положительных T-клеток человека, стимулированных с помощью antiCD2/antiCD3/antiCD28-покрытых сфер, ни одно из представляющих интерес соединений не проявляло эффективности в этом исследовании.
Соединение 28 [KYN-143] ингибировало секрецию IL-4 из CD4-положительных T-клеток человека, стимулированных с помощью antiCD2/antiCD3/antiCD28-покрытых сфер при величине EC50 278 нМ с 80% максимальной эффективностью и подавляло жизнеспособность клеток при величине EC50 2360 нМ с 60% максимальным снижением жизнеспособности клеток. Соединение 27 [KYN-149] ингибировало секрецию IL-4 из CD4-положительных T-клеток человека, стимулированных с помощью antiCD2/antiCD3/antiCD28-покрытых сфер при величине EC50 507 нМ с 79% максимальной эффективностью и подавляло жизнеспособность клеток при величине EC50 4350 нМ с 52% максимальным снижением жизнеспособности клеток.
В целом, результаты указывают, что соединения 28 [KYN-143] и 27 [KYN-149] являются наиболее перспективными соединениями для эффективного лечения атопического дерматита и псориаза.
Таблица 7. Результаты исследований соединений 1, 8, 20, 27, 28, 31 и 35 по ингибированию высвобождения/секреции CCl2, IL-8, IL-17, IL-2 и IL-4 из клеток и воздействия соединений на жизнеспособность клеток.
Abs EC50: концентрация соединения, при которой достигается половина максимального эффекта (нМ) по сравнению с контролем
Max Eff.: проявляемый молекулой наивысший эффект в виде процента от контрольного максимального эффекта
Abs EC50: концентрация соединения, при которой достигается половина максимального эффекта (нМ) по сравнению с контролем
Max Eff.: проявляемый молекулой наивысший эффект в виде процента от контрольного максимального эффекта
Методы
1. Высвобождение CCL2 из кератиноцитов
Ингибирование высвобождение CCL2 из IL-4, IL-13, IL-22 и IFN-γ-стимулированных первичных кератиноцитов человека, LEO Pharma Open Innovation исследование номер 1280
Применимость для лечения заболевания
Воспалительное заболевание кожи атопический дерматит (AD) характеризуется высвобождаемыми T-клетками цитокинами, включающими IL-4, IL-13, IL-22 и IFN-γ. В настоящем исследовании, кератиноциты стимулируют смесью этих цитокинов, и высвобождение CCL2 (также называемого моноцитарным хемоаттрактантным белком 1 (MCP-1)) измеряют в культуральной надосадочной жидкости бесконтактным методом гомогенной флуоресценции с временным разрешением (HTRF). Целью исследования является измерение ингибирования уровней высвобождаемого кератиноцитами CCL2 при воздействии испытуемых соединений. Можно предполагать, что соединение, которое ингибирует секрецию CCL2 кератиноцитами, обладает лечебным эффектом при атопическом дерматите.
Известный стероид, бетаметазон, ингибирует высвобождение CCL2 в этом исследовании с величиной EC50 приблизительно 15 нМ и с величиной Emax (горизонтальный участок аппроксимирующей кривой) приблизительно 60%. Высокая величина Emax указывает на то, что соединение ингибирует большую часть секреции CCL2. Низкая величина EC50 указывает на то, что соединение является активным и оказывает ингибирующее действие при низких концентрациях.
Описание метода
Культура клеток
HEKa представляют собой эпидермальные кератиноциты человека, выделенные из кожи взрослого человека. Клетки криозамораживали в конце первичной стадии культивирования в среде, содержащей 10% DMSO. Применяется стерилизация культуры клеток.
Инициирование культур из криозамороженных клеток HEKa
Разморозить флакон с исходными замороженными клетками HEKa на водяной бане/шариковой бане с температурой 37°C.
Перенести суспензию клеток в колбу для культивирования клеток T75 объемом 30.
Отобрать 15 мл и распределить в двух колбах T75 (по 15 мл в каждой).
Приготовить к исследованию через 5-6 дней или сразу переходить к исследованию.
Сохранение исходных культур
Заменить культуральную среду на свежую среду с добавками через период времени от 24 до 36 часов после формирования вторичной культуры из криосохраненных клеток. Для последующих субкультур, заменять среду через 48-72 часов после формирования субкультуры.
В исследовании провести только 3-5 пассажей.
Субкультура HEKa
Визуально оценить и подтвердить 80% конфлюэнтность.
Удалить из колбы всю культуральную среду from.
Добавить в колбу 3 мл раствора TrypLE Express раствор. Покачать колбу, для того чтобы убедиться в том, что вся поверхность покрыта раствором.
Немедленно удалить из колбы полностью 3 мл раствор TrypLE Express.
Добавить в колбу 1½ мл (T75) (3 мл T175) свежего раствора TrypLE Express.
Визуально оценить культуру под микроскопом. Инкубировать колбу при комнатной температуре до тех пор, пока клетки не станут полностью сферическими, приблизительно через 8-10 минут.
Очень осторожно постучать по колбе для отделения клеток от поверхности колбы.
Добавить в колбу 3-4 мл (T75) (7 мл T175) раствора Trypsin Neutralizer и перенести отделенные клетки в стерильную коническую пробирку.
Центрифугировать клетки при 170 x g в течение 7-10 минут. Получить осадок клеток.
Удалить надосадочную жидкость из пробирки, проявляя осторожность, чтобы не удалить осадок клеток.
Ресуспендировать осадок клеток в 15 мл среды, дополненной HKGS (cat no S-001-K), но без гидрокортизона (при применении для исследования). Набирать клетки в пипетку и выпускать из пипетки, используя пипетку объемом 10 мл, для того чтобы убедиться в гомогенности суспензии клеток.
Определить концентрацию клеток в суспензии.
Разбавить клетки в среде с добавками и посеять новую культуру в колбе T175. Инкубировать культуры при 37°C в увлажненной атмосфере 5% CO2/95% воздух в инкубаторе для клеточных культур.
День 0 исследования
Трипсинизировать клетки, как описано выше
Ресуспендировать осадок клеток в 10 мл среды для исследования (набор среды EpiLife с HKGS, но без гидрокортизона)
Отфильтровать клетки через фильтр с размером пор 37 мкм
Подсчитать клетки на цитометре
Посеять клетки HEKa в 384-луночных прозрачных планшетах при 3500 клеток/лунка/40 мкл.
Поместить планшеты с крышками в увлажнительную камеру с крышкой (бокс 24,5×24,5 см с добавляемой на дно H2O).
Инкубировать планшеты в течение ночи при 37°C в атмосфере 5% CO2/95% воздух.
День 1 исследования
Приготовить исходный планшет, содержащий соединение, то есть в планшет для исследований должны быть перенесены титрации в DMSO.
Добавить 80 нл соединений и контроли из исходного планшета в планшет для исследования (с клетками).
Добавить 40 мкл стимулятора (рекомбинантного интерлейкина человека (IL) 4, 13, 22 при 10 нг/мл и интерферона-гамма при 1 нг/мл) в среде с добавкой, но без гидрокортизона.
Инкубировать планшеты при 37°C в атмосфере 5% CO2/95% воздух в течение 2 дней.
День 3 исследования
Удалить планшеты из инкубатора и привести в равновесие с комнатной температурой в течение 30 минут.
Перенести 8 мкл надосадочной жидкости в белые 384-луночные планшеты (ProxiPlate-384 Plus, White) для детекции CCL2.
Добавить 8 мкл среды в планшет для детекции.
Добавить 4 мкл реагента HTRF для детекции CCL2 (в белый 384-луночный планшет (ProxiPlate-384 Plus, White)).
Герметизировать и инкубировать в течение 4 часов, и провести считывание флуоресценции на планшет-ридере.
Анализ данных и вычисления
Концентрацию CCL2 в надосадочных жидкостях измеряют, используя метод гомогенного резонансного переноса энергии флюоресценции с временным разрешением (TR-FRET). Количественные данные получают путем измерения флуоресценции при 665 нм (пропорциональной концентрации CCL2) и при 620 нм (контроль). Рассчитывают отношение 665/620*1000.
% Воздействия
Способность испытуемого соединения ингибировать высвобождение CCL2 нормализуют к величине сигнала в контрольных лунках с кератиноцитами, инкубированными с 0,1% DMSO (0%) и 10 мкМ терфенадина (100%), которые полностью ингибируют высвобождение CCL2.
2. Высвобождение IL-8 из кератиноцитов
Ингибирование высвобождения IL-8 из IL-17A и TNFα-индуцированных первичных кератиноцитов человека, LEO Pharma Open Innovation исследование номер 1250 (openinnovation.leo-pharma.com)
Применимость для лечения заболевания
Воспалительное заболевание кожи псориаз характеризуется цитокинами IL-17A и TNFα. В настоящем исследовании, первичные кератиноциты человека стимулируют смесью этих цитокинов, и высвобождение IL-8 (также называемых CXCL8) измеряют в культуральной надосадочной жидкости бесконтактным методом гомогенной флуоресценции с временным разрешением (HTRF). Целью исследования является измерение ингибирования уровней высвобождаемого кератиноцитами IL-8 при воздействии испытуемого соединения, что может указывать на способность соединения подавлять часть воспаления, возникающего в клетках.
Можно предполагать, что соединения, которые ингибирует секрецию IL-8 кератиноцитами, обладает лечебным эффектом при псориазе.
Известный стероид, бетаметазон, ингибирует высвобождение IL-8 в этом исследовании с величиной EC50 приблизительно 10 нМ, и с величиной Emax (горизонтальный участок аппроксимирующей кривой) приблизительно 50%. Высокая величина Emax указывает на то, что соединение ингибирует большую часть секреции IL-8. Низкая величина EC50 указывает на то, что соединение является активным и оказывает ингибирующее действие при низких концентрациях.
Описание метода
Культура клеток
HEKa представляют собой эпидермальные кератиноциты человека, выделенные из кожи взрослого человека (смотрите метод культивирования, описанный выше для высвобождения CCL2 из кератиноцитов).
День 0 исследования
Отделить клетки, как описано выше.
Ресуспендировать осадок клеток в 10 мл среды EpiLife, дополненной HKGS, но без гидрокортизона
Отфильтровать клетки через фильтр с размером пор 37 мкм
Подсчитать клетки на цитометре
Посеять клетки эпидермальных кератиноцитов взрослого человека (HEKa) в 384-луночных прозрачных планшетах при 3500 клеток/лунка/40 мкл. Использовать для клеток среду с добавкой, но без гидрокортизона, в качестве среды для исследования.
Поместить планшеты с крышками в увлажнительную камеру с крышкой (бокс 24,5×24,5 см с добавляемой на дно H2O).
Инкубировать планшеты в течение ночи при 37°C в атмосфере 5% CO2/95% воздух.
День 1 исследования
Приготовить исходный планшет, содержащий соединение, то есть в планшет для исследований должны быть перенесены титрации в DMSO.
Опорожнить планшет для исследования и повторно добавить 25 мкл среды для исследования.
Добавить 75 нл соединений и контролей из исходного планшета в планшет для исследования (с клетками).
Добавить 25 мкл среды для исследования и инкубировать в течение 2 часов.
Добавить 25 мкл стимулирующей смеси в среду для исследования (рекомбинантного интерлейкина человека 17A и фактор некроза опухолей альфа) с получением конечной концентрации 10 нг/мл обоих цитокинов.
Инкубировать планшеты при 37°C в атмосфере 5% CO2/95% воздух в течение 3 дней.
День 4 исследования
Удалить планшеты из инкубатора и привести в равновесие с комнатной температурой в течение 30 минут.
Перенести 2 мкл надосадочной жидкости в белые 384-луночные планшеты (ProxiPlate-384 Plus, White) для детекции IL-8.
Добавить 2 мкл среды для исследования
Приготовить IL-8 HTRF смесь (в соответствии с протоколом фирмы-производителя) и добавить по 2 мкл в лунку.
Герметизировать и инкубировать в течение 3 часов, и провести считывание флуоресценции на планшет-ридере.
Анализ данных и вычисления
Концентрацию IL-8 в надосадочной жидкости измеряют, используя метод гомогенного резонансного переноса энергии флюоресценции с временным разрешением (TR-FRET). Количественные данные получают путем измерения флуоресценции при 665 нм (пропорциональной концентрации IL8) и при 620 нм (контроль). Рассчитывают отношение 665/620*1000 для определения количества IL-8 в надосадочной жидкости.
% Воздействия
Способность испытуемого соединения ингибировать высвобождение IL-8 нормализуют к величине сигнала в лунках с отрицательным контролем с кератиноцитами, инкубированными с 0,1% DMSO (0%), и с 10 мкМ терфенадина (100%), которые полностью ингибируют высвобождение IL-8.
3. Исследование секреции IL-17 в PBMC
Ингибирование секреции IL-17A из PBMC человека, стимулированных с помощью сфер, покрытых антителами antiCD3/antiCD28, LEO Pharma Open Innovation исследование номер 1321
Применимость для лечения заболевания
Воспалительное заболевание кожи псориаз характеризуется цитокинами IL-17A и TNFα. В настоящем исследовании, мононуклеарные клетки периферической крови человека (PBMC) стимулируют с помощью сфер, покрытых антителами против CD3 и CD28 для активации рецептора T-клеток, и стимулируют с помощью IL-23 для промотирования активности T-helper17. Клетки инкубируют в течение трех дней, затем измеряют в культуральной надосадочной жидкости секретируемый уровень IL-17A, используя AlphaLISA, и измеряют количество живых клеток путем добавления резазурина (PrestoBlue®) для выявления соединений, которые ингибируют пролиферацию клеток или подавляют жизнеспособность клеток. Целью исследования является измерение ингибирования секреции IL-17A мононуклеарными клетками периферической крови человека при воздействии испытуемого соединения. Можно предполагать, что соединения, которые ингибируют секрецию IL-17A, обладает лечебным эффектом при псориазе.
Референсным соединением в этом исследовании является ингибитор кальциневрина такролимус, который ингибирует высвобождение IL-17A с величиной EC50 приблизительно 0,34 нМ, и с величиной Emax (горизонтальный участок аппроксимирующей кривой) приблизительно 90%. Высокая величина Emax указывает на то, что соединение ингибирует большую часть секреции IL-17. Низкая величина EC50 указывает на то, что соединение является активным и оказывает ингибирующее действие при низких концентрациях.
(набор для активации/ размножения T-клеток фирмы Miltenyi Biotec номер по каталогу 130-091-441). Одна сфера/клетка.
Описание метода
Выделение PBMC
PBMC выделяют из свежих лейкоцитарных пленок человека путем центрифугирования в среде с градиентом плотности Lymphoprep® в соответствии с инструкциями Sebmate®
Клетки сушат в центрифуге и ресуспендируют в замораживающей среде (RPMI1640+20% FBS+5% DMSO)
Клетки замораживают порциями по 8×107 клеток/флакон в количестве, соответствующем для использования в одном 384-луночном планшете. Клетки помещают в штатив для охлаждения клеток Coolcell box и помещают в морозильную камеру с температурой -80°C на один день. Флаконы переносят в морозильную камеру с температурой -150°C в предварительно охлажденном штативе.
День 1 исследования
Приготовить среду для исследования: RPMI1640+10% термоинактивированная FBS и 1% пенициллин/стрептомицин, дополненную человеческим IL-23, 20 нг/мл (R&D systems, номер по каталогу 1290-IL-010)
Испытуемые соединения: приготовить исходный планшет, содержащий соединение, то есть титрации в DMSO следует перенести в планшет для исследования.
Добавить 70 нл соединений и контролей из исходного планшета в планшет для исследования.
Разморозить клетки путем добавления 1 флакона в 20 мл подогретой среды для исследования
Центрифугировать клетки при 170 х g в течение 10 минут, удалить надосадочную жидкость и ресуспендировать осадок клеток в новой среде.
Отфильтровать клетки через фильтр с размером пор 37 мкм.
Подсчитать клетки на цитометре.
Сферы (набор для активации/размножения T-клеток фирмы Miltenyi Biotec номер по каталогу 130-091-441): нанести покрытие из антител (antiCD3 и antiCD28).
Добавить сферы к клеткам, одну сферу на клетку, 2,1 х 106 клеток/мл. Использовать пипетку для осторожного смешения клеток и сфер.
Посеять смесь клеток/сфер в планшете для исследования, 70 мкл/лунка (1,3 х 105 клеток/лунка)
Поместить планшеты в увлажнительную камеру с крышкой (бокс 24,5×24,5 см с добавляемой на дно H2O).
Инкубировать планшеты в течение 3 дней при 37°C в атмосфере 5% CO2/95% воздух.
День 4 исследования
Измеряют концентрацию IL-17A в надосадочных жидкостях, используя набор alphaLISA (Perkin Elmer, номер по каталогу AL219F).
Предварительное испытание для определения оптимального разбавления образцов: отобрать 5 мкл надосадочной жидкости из контрольных лунок и провести анализ IL-17A набором alphaLISA при инкубации только в течение 2 часов.
После того, как данные будут получены, удалить планшеты для исследования из инкубатора и привести в равновесие с комнатной температурой в течение 30 минут.
Перенести 5 мкл или скорректированные количества надосадочной жидкости в белые 384-луночные планшеты ProxiPlate-384 Plus
Добавить 5 мкл сфер акцепторов alphaLISA.
Инкубировать 1 час при комнатной температуре
Добавить 5 мкл сфер доноров
Инкубировать в течение ночи при комнатной температуре при защите от воздействия света.
Добавить 7 мкл реагента PrestoBlue (резазурина)/лунка в планшет для исследования и инкубировать в течение 2-4 часов при 37°C/5% CO2. Измерить флуоресценцию в лунках (длина волны возбуждения 615 нм/ длина волны излучения 535 нм).
День 5 исследования
Считать планшет alphaLISA в планшет-ридере
Анализ данных и вычисления
Концентрацию IL-17A в надосадочной жидкости измеряют, используя набор alphaLISA (Perkin Elmer, номер по каталогу AL219F).
% Воздействия на IL-17.
Способность испытуемого соединения ингибировать высвобождение IL-17A нормализуют к величине сигнала в лунках с отрицательным контролем, обработанных токсичным соединением терфенадином.
Уровень живых клеток в лунках измеряют путем добавления PrestoBlue® и измерения превращения резазурина в резоруфин (флуоресцирующий) в живых клетках.
% Воздействия на жизнеспособность/пролиферацию.
Воздействие испытуемых соединений на жизнеспособность/пролиферацию нормализуют к сигналу PrestoBlue в лунках с отрицательным контролем, обработанных токсичным соединением терфенадином.
4 и 5. Исследование секреции IL-2 и IL-4 T-клетками
Ингибирование секреции IL-4 и IL-2 из CD4-положительных T-клеток человека, стимулированных с помощью сфер, покрытых с помощью антител antiCD2/antiCD3/antiCD28. LEO Pharma Open Innovation исследования номер 1297, 1298, 1299.
Применимость для лечения заболевания
Воспалительное заболевание кожи атопический дерматит характеризуется цитокинами IL-4, IL-13 и IL-22, секретируемыми T-клетками. В настоящем исследовании, T-клетки человека стимулируют с помощью сфер, покрытых антителами против CD2, CD3 и CD28, для активации рецептора T-клетки. Затем измеряют уровень секретируемого IL-4 в надосадочной культуральной жидкости, используя наборы для электрохемилюминесценции (наборы MSD, Meso Scale Discovery), измеряют уровень IL-2 бесконтактным методом гомогенной флуоресценции с временным разрешением (HTRF) и измеряют количество живых клеток путем добавления резазурина (PrestoBlue®), что позволяет выявить соединения, которые ингибируют пролиферацию T-клеток или подавляют их жизнеспособность. Целью исследования является измерение способности испытуемого соединения ингибировать секрецию IL-4 T-клетками, что указывает на способность соединения ингибировать часть воспаления, возникающего на коже.
Можно предполагать, что соединения, которые ингибируют секрецию IL-4 обладают лечебным действием при атопическом дерматите. Соединения, которые также ингибируют секрецию IL-2, имеют более широкий спектр иммуносупрессорного действия, так как IL-2 является аутокринным фактором роста для T-клеток. Известный стероид, бетаметазон, ингибирует высвобождение IL-4 в этом исследовании с величиной EC50 приблизительно 15 нМ и с величиной Emax (горизонтальный участок аппроксимирующей кривой) приблизительно 80%. Высокая величина Emax указывает на то, что соединение ингибирует большую часть секреции IL-4. Низкая величина EC50 указывает на то, что соединение является активным и оказывает ингибирующее действие при низких концентрациях.
Ключевые параметры исследования
(набор для активации/размножения T-клеток фирмы Miltenyi Biotec номер по каталогу 130-091- 441). Одна сфера/лунка.
Описание метода
Выделение CD4+ T-клеток
Мононуклеарные клетки периферической крови (PBMC) человека выделяют из свежих лейкоцитарных пленок человека путем центрифугирования в среде с градиентом плотности Lymphoprep® в соответствии с инструкциями Sebmate™ (Stemcell).
Клетки ресуспендируют в 30 мл PBS+2% сыворотка и подсчитывают.
CD4 положительные T-клетки выделяют из свежих выделенных PBMC, используя набор для выделения CD4+ T-клеток человека EasySep™ humane Isolation Kit (каталог # 17952) и следуя протоколу фирмы-производителя.
Клетки сушат в центрифуге и ресуспендируют в замораживающей среде (RPMI1640+20% FBS+5% DMSO)
Клетки замораживают порциями по 3×107 клеток/флакон в количестве, соответствующем для использования в одном 384-луночном планшете. Клетки помещают в штатив для охлаждения клеток Coolcell box и помещают в морозильную камеру с температурой -80°C на один день, затем переносят в морозильную камеру с температурой -150°C в предварительно охлажденном штативе.
День 1 исследования
Приготовить среду для исследования: 500 мл X-Vivo 15+5 мл Glutamax (конечная концентрация 2 мM) + 5 мл пенициллин/стрептомицин (конечная концентрация 100 Е/мл).
Добавить 5 мкл среды в лунки 384-луночного планшета (Greiner номер по каталогу 788986).
CD4+ клетки: разморозить замороженные клетки из морозильной камеры с температурой -150°C в бане с температурой 37°C, и добавить клетки с помощью пипетки в 10 мл среды. Центрифугировать в течение 10 минут при 170 х g.
Ресуспендировать осадок клеток в 3,5 мл в количестве 2,5×107 клеток и добавить 10 мкл на лунку в планшет (40000 клеток/лунка)
Испытуемые соединения: приготовить исходный планшет, содержащий соединение, то есть должны быть перенесены в планшет для исследования титрации в DMSO.
Добавить 25 нл соединений и контролей из исходного планшета в планшет для исследования (с клетками).
Сферы (набор для активации/размножения T-клеток фирмы Miltenyi Biotec номер по каталогу 130-091-441): нанести покрытие из антител на сферы в соответствии с инструкцией, приложенной к набору.
Добавить 10 мкл сфер в каждую лунку, за исключением лунок с отрицательными контролями. Одна сфера на лунку. Добавить 10 мкл среды в лунки с отрицательными контролями.
Инкубировать планшеты в течение 2 дней при 37°C в атмосфере 5% CO2/95% воздух.
День 3 исследования
Удалить планшеты из инкубатора и привести в равновесие с комнатной температурой в течение 30 минут.
Измеряют концентрацию IL-4 в 10 мкл надосадочной жидкости, используя набор MSD IL-4 384-plate kit (изготовленный на заказ) в соответствии с протоколом фирмы-производителя.
Измеряют концентрации IL-2 в надосадочной жидкости с использованием набора IL-2 HTRF kit, CisBio, номер по каталогу 62HIL02PEH.
Добавить 4 мкл среды в белые 384-луночные планшеты ProxiPlate-384 Plus
Добавить 1 мкл надосадочной жидкости.
Добавить 4 мкл смеси mHTRF (каждая разбавлена 1:300 в 2/3 буфера для разбавления и 1/3 среды для исследования).
Центрифугировать планшеты в течение 1 минуты при 170 х g
Герметизировать и инкубировать в течение ночи и считать флуоресценцию в планшет-ридере
Количество жизнеспособных клеток измеряют с использованием реагента PrestoBlue®
Добавить 15 мкл разбавленного реагента PrestoBlue® (резазурина), что соответствует 2,5 мкл Prestoblue/лунка, в планшет для исследования.
Инкубировать в течение ночи при 37°C/5% CO2. Измеряют флуоресценцию в лунках (длина волны возбуждения 615 нм/ длина волны излучения 535 нм).
Анализ данных и вычисления
Концентрацию IL-4 в надосадочных жидкостях измеряют, используя электрохемилюминесценцию (наборы MSD, Meso Scale Discovery). Концентрацию IL-2 в надосадочных жидкостях измеряют, используя метод гомогенного резонансного переноса энергии флюоресценции с временным разрешением (TR-FRET). Количественные данные получают путем измерения флуоресценции при 665 нм (пропорциональной концентрации IL-2) и 620 нм (контроль). Рассчитывают отношение 665/620*1000.
% Воздействия на IL-2 и IL-4
Способность испытуемого соединения ингибировать высвобождение нормализуют к величине сигнала в лунках с отрицательным контролем с нестимулированными T-клетками.
Уровень живых клеток в лунках измеряют путем добавления PrestoBlue® и превращения резазурина в резоруфин (флуоресцирующий) в живые клетки.
% Воздействия на жизнеспособность/пролиферацию
Воздействие испытуемых соединений на жизнеспособность/пролиферацию нормализуют к сигналу PrestoBlue в лунках с отрицательным контролем, обработанных токсическим соединением терфенадином.
Ex vivo исследование: прогноз ответной реакции пациентов на воздействие противоопухолевых лекарственных средств
ROK-DRPS-001 - Краткие выводы об ответной реакции на воздействие лекарственных средств (19 июня 2020 года)
Пилотное исследование ответной реакции на воздействие лекарственных средств при мультиформной глиобластоме (GBM)
KIYATEC, Inc., Greenville, South Carolina, USA
1. Описание/аннотация проекта
Этот проект включает скрининговое ex vivo исследование фирмы ROKiT Pharma ответной реакции при воздействии 3 испытуемых лекарственных средств и темозоломида в 3 предварительно разложенных и криосохраненных образцов GBM.
2. Техническое осуществление
2.1 Для использования в этом исследовании были взяты три предварительно обработанных образца у пациентов, страдающих мультиформной глиобластомой (GBM).
IDH1
IDH1: изоцитратдегидрогеназа-1; MGMT: O-6-метилгуанин-ДНК метилтрансфераза
2.2 Размораживали 1-2 флакона с каждым образцом, и клетки в форме 3D сфероидов высевали в одном 384-луночном планшете в случае каждого образца.
2.3 Через 24 часа после посева, получали изображения всех лунок и добавляли в лунки лекарственные средства/контроли.
Испытуемые соединения: KYN-001, KYN-146, KYN-149, кривая доза - эффект, построенная по 10 точках при разбавлении 1:3, начиная с 100 мкМ и заканчивая с 0,005 мкМ
Среды для лекарственных средств: среда+0,5% DMSO, среда+1% DMSO
Положительные контроли: 10% DMSO и 2% DMSO
Лунки, содержащие только среду и не содержащие клеток, использовали в качестве холостых проб
2.4 Жизнеспособность клеток измеряли через 4 дня после обработки лекарственными средствами, используя анализ CellTiter-Glo 3D.
Регистрировали изображения всех лунок перед добавлением реагента CellTiter Glo.
2.5 После очистки первичных данных и удаления резко отклоняющихся значений (определяемых с помощью анализа с использованием программного обеспечения и выделенных красным цветом в файлах первичных данных), определяли величины IC50 с использованием нелинейной регрессии (нормализованной к не подвергавшемуся обработке контролю) в программе GraphPad Prism.
Величины IC50 выражаются в мкМ и определяются как концентрация лекарственного средства, при которой жизнеспособность клеток снижается наполовину.
3. Краткое изложение результатов эксперимента
3.1. Ответная реакция на воздействие испытуемых соединений и темозоломида (TMZ) представлена в таблице ниже:
3.2. Значения RLU (относительных световых единиц) были высокими для клеток из каждого образца GBM, указывая на пролиферацию/жизнеспособность в течение всего времени проведения исследования.
3.3. Факторы Z отражали успешность исследований в случае всех образцов и лекарственных средств.
3.4. Ответная реакция на действие TMZ:
NR: отсутствие ответа
MR: умеренный ответ
R: ответ
Обсуждение
В этом исследовании использовали три образца клеток GBM, а именно BNA99, BNB04 и BNB18, демонстрирующих три четко различимых особенностей роста при их выращивании в 3D сфероиды, как показано на фигуре 14. Эти клетки располагаются в ряд от резистентных (BNA99) до умеренно резистентных (BNB04) и восприимчивых (BNB18) к воздействию используемого в клинике лекарственного средства темозоломида (TMZ). В присутствии испытуемых соединений 1, 20 и 27 сфероиды всех трех клеточных линий разрушались, что вызывало гибель клеток и распространение в культуральную среду. Эти явления были наглядно продемонстрированы изображениями (фигура 14), полученными перед добавлением реагента CellTiter Glo, который использовали для количественной оценки жизнеспособности клеток путем измерения уровня АТФ живых клеток.
Испытуемые соединения демонстрировали ингибирование этих образцов 3D сфероидов клеток GBM в диапазоне от умеренного до высокого с величиной IC50 27 в субмикромолярном диапазоне в случае восприимчивой линии клеток BNB18 (IC50=0,63 мкM). Как и ожидалось, испытуемые соединения были активными в отношении восприимчивой линии клеток BNB18 мультиформной глиобластомы (GBM), но менее активными в отношении сфероидов BNB04 и значительно менее активными в отношении сфероидов BNA99. Несмотря на это, было показано, что испытуемые соединения, в частности, соединение 27, является относительно активным в отношении TMZ-резистентных клеток (BNA99) с величиной IC50 8,86 мкM, в то время как темозоломид (TMZ) не вызывал ответной реакции в этой клеточной линии.
Результаты этого исследования показывают, что испытуемые соединения, в частности, соединение 27, являются весьма перспективными для разработки новых противораковых лекарственных средств.
Краткие выводы
Результаты исследования были положительными для всех лекарственных средств и образцов, исходя из факторов z и сигналов RLU
Величины IC50 указывали на различную восприимчивость к воздействию KYN-001, KYN-146 и KYN-149 для всех 3 образцов.
Ответная реакция на воздействие TMZ изменялась для всех 3 образцов по всем 3 категориям ответной реакции.
Яркие поля изображения сфероидов соответствуют результатам анализа CellTiterGlo (данные не представлены).
Соединение 27 [KYN-149] является более эффективным, чем соединение 1 [KYN-001] и 20 [KYN-146] в отношении всех трех типов 3D-сфероидных образцов мультиформной глиобластомы (NR, MR и R).
Ссылка на литературный источник
Shuford, S., et al., 2019. Prospective Validation of an Ex Vivo, Patient-Derived 3D Spheroid Model for Response Predictions in Newly Diagnosed Ovarian Cancer, Nature Scientific Reports, 9:11153 | https://doi.org/10,1038/s41598-019-47578-7
Максимально переносимая доза соединения 27 [KYN-149] - результаты по оценке величины LD/50 при пероральном введении разовой дозы
Оценку проводила фирма Kynan Pharma, USA
Цель исследования
Получить исходную информацию относительно токсичности и переносимости соединения 27.
Дизайн исследования
Приготовленную для перорального введения максимальную разовую дозу 2000 мг/кг вводили 5 мышам линии BalbC. Наблюдение проводили в течение 5 дней.
Экспериментаторы: Tiziana Palumbo, Matt Kiosea
Дата проведения испытаний: 8-12 июня 2020 года
Метод
Соединение 27 взвешивали и разбавляли в той же самой среде SEDDS, которую использовали и для S1. Среда SEDDS состоит из следующих компонентов:
Лабразол (30%)
PEG-400 (20%)
Коллифор RH-40 (30%)
Лабрафил (20%)
Концентрация соединения 27 в среде SEDDS составляла 200 мг/мл, которая в пересчете для мыши массой 20 г, которой вводят 200 мкл, составляла пероральную дозу 2000 мг/кг. Пероральное введение проводили 5 мышам, и животных тщательно обследовали на наличие признаков токсичности в течение первого часа, а затем эпизодически в течение 5 дней.
Результаты
Все пять мышей выжили и не обнаруживали прямых признаков тяжелой формы недомогания. В течение оставшихся дней наблюдения, не было зарегистрировано ни случаев недомогания, ни каких-либо других случаев дискомфорта.
Выводы
Результаты исследования порядка величины LD/50 указывают, что соединение 27 характеризуется высокой степенью пероральной переносимостью в данной лекарственной форме. 2000 мг/кг представляет собой максимальную дозу, рекомендуемую для испытания токсикологических воздействий Управлением по контролю за качеством пищевых продуктов и лекарственных препаратов США.
Предварительное изучение воздействия соединения 27 [KYN-149] на мышах с ксенотрансплантатом клеток B16F10.
Оценку проводила фирма Kynan Pharma, USA
Цели
Наблюдение и оценка воздействия перорального введения соединения 27 на самочувствие животного.
Количественное определение соединения 27 в тканях (плазме, опухоли, печени, селезенки и головного мозга) для оценки биологической аккумуляции и распределения соединения.
Метод
Клетки B16F10 (25×105 клеток/мышь) инъецировали подкожно в правую боковую поверхность самок мышей линии BALB/c в возрасте 7-8 недель. Мышей (n=10) разделяли на 2 группы и ежедневно вводили перорально суспензию соединения 27 (подвергавшаяся обработке группа) или среду (контрольная группа) в течение 15 дней. Подвергавшейся обработке группе перорально вводили суспензию соединения 27 при 900 мг/кг. Контрольной группе вводили суспензию среды (SEDDS). Измерения проводили только в дни 0, 9 и 15. Собирали образцы тканей (плазмы, опухоли, печени, селезенки и головного мозга) на протяжении 3 часов после финальной обработки. Затем брали на биопсию ткани опухоли для гистопатологического исследования.
Образцы тканей опухолей, как подвергавшихся обработке мышей, так и контрольных мышей, фиксировали формалином. Гистопатологическое исследование этих образцов опухоли проводила Lore Laboratory, CA, USA.
Концентрации соединения 27 в различных тканях определяли методом жидкостной хроматографии с тандемной масс-спектрометрией (LC/MS/MS).
Результаты
У мышей не обнаруживалось признаков токсического воздействия при дозе 900 мг/кг соединения 27. Определенные концентрации соединения 27 тканях представлены в таблице 1 и в графическом виде на фигуре 15A и B.
Таблица 8. Измерение концентраций соединения 27 в тканях мыши методом масс-спектрометрии
NA=не определяли
Фигура 15. Концентрации соединения 27 в образцах различных тканей, взятых от обработанных мышей. Мышам перорально вводили 900 мг/кг соединения 27 в виде суспензии в среде SEDDS или среду (контроль) ежедневно в течение 15 дней.
Было обнаружено, что концентрация соединения 27 по-разному распределялась в различных тканях, как показано на фигуре 15. Низкое содержание соединения 27 в плазме может быть объяснено тем, что соединение связывается с белками плазмы, такими как альбумин.
Гистохимический анализ образцов опухоли
Предварительное исследование показало, что два из пяти взятых на биопсию образцов у пяти мышей, подвергавшихся обработке с помощью соединения 27, выявляли меланому, как подробно описывается ниже.
Подвергнутая обработке мышь 1.
Было обнаружено, что очаг поражения располагался глубоко в коже и поверхностном слое кожи. Обнаруживался слой плеоморфных круглых клеток с эпизодически пигментированной цитоплазмой. Эти клетки преимущественно были вырожденными или омертвевшими с прилегающими областями лизирующего некроза (фигура 16). Отмечались смешанные воспалительные клетки нам периферии.
Подвергнутая обработке мышь 2.
Было обнаружено, что очаг поражения располагался в поверхностным и срединном слое кожи. Обнаруживался слой плеоморфных круглых клеток с эпизодически пигментированной цитоплазмой (фигура 16). Отмечались смешанные воспалительные клетки на периферии.
Гистохимический анализ образцов опухоли подвергнутых обработке мышей 1 и 2.
Эти две опухоли содержали меланому, которая быстра росла в течение периода времени от дня 0 до дня 9. Опухоли последовательно уменьшались после дня 10, что указывало, по-видимому, на воздействие соединения 27 (фигура 16).
Гистохимический анализ образцов опухоли подвергнутых обработке мышей 3, 4 и 5.
Гистохимический анализ этих образцов выявил наличие преимущественно меланоцитов, которые распространялись в подкожном и поверхностном слое со слабовыраженным мононуклеарным воспалением (фигура 17). Эти результаты совпадают с отсутствием роста опухоли, как показано в таблице 8 и на фигуре 18.
Таблица 8. Объем опухоли (мм3) у мышей, подвергнутых обработке с помощью соединения 27
Фигура 18. Воздействие соединения 27 на клетки меланомы B16F10, ксенотрансплантированные мышам линии BALB/c.
Заключения гистопатологического исследования
За исключением двух опухолей, в случае которых вводили лекарственное средство (подвергнутая обработке мышь 1 и подвергнутая обработке мышь 2), другие образцы представляли собой сильно пигментированные меланоциты с круглым удлиненным ядром и без митотической активности.
Возможно, что в тех образцах, в которых гистологически не наблюдали злокачественного новообразования, либо не было возникшего изначально злокачественного новообразования, либо не были отобраны и собраны образцы злокачественной ткани.
Несмотря на то, что большинство подвергнутых анализу тканей состояло из меланоцитов, тем не менее, две подвергнутые обработке ткани содержали меланому, которая была некротической и дегенеративной, что, по-видимому, свидетельствует об эффекте лечения. Отсутствие меланомы в контрольных необработанных тканях ставит под сомнение эти выводы.
Общее заключение
В заключение, исследование показало, что соединение 27 имеет низкую токсичность при пероральном введении и характеризуется сильной тенденцией к накоплению в головном мозге. Это может быть объяснено высокой липофильностью соединения.
Гистохимический анализ образцов опухоли показал, что ксенотрансплантированные мышам клетки меланомы, в случае двух из пяти мышей, подвергавшихся обработке с помощью соединения 27, лизировались и/или подвергались некрозу, что указывает на возможную эффективность соединения 27 при лечении меланомы in vivo.
Однако необходимы дальнейшие исследования для определения дополнительных фармакокинетических характеристик соединения 27 и оценки противоопухолевой эффективности in vivo.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЦИТОТОКСИЧЕСКИЕ ПЕПТИДЫ И ИХ КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2012 |
|
RU2586885C2 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА RIP-1 КИНАЗЫ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2800652C2 |
| ЗАМЕЩЕННЫЕ ХРОМАНЫ | 2015 |
|
RU2718060C2 |
| ФОСФОИНДОЛЫ КАК ИНГИБИТОРЫ ВИЧ | 2005 |
|
RU2393163C2 |
| ИНГИБИРУЮЩИЕ JAK СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИМИДИНА И СПОСОБЫ | 2010 |
|
RU2675857C2 |
| ИНГИБИРУЮЩИЕ JAK СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИМИДИНА И СПОСОБЫ | 2010 |
|
RU2567238C2 |
| СОЕДИНЕНИЯ ГЕТЕРОАРИЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ТКБ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2742122C2 |
| ХИНОЛИНКАРБОКСАМИДНЫЕ И ХИНОЛИНКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ mGLuR2-НЕГАТИВНЫХ АЛЛОСТЕРИЧЕСКИХ МОДУЛЯТОРОВ, КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2610262C2 |
| ГЕТЕРОАРОМАТИЧЕСКИЕ МОДУЛЯТОРЫ РЕТИНОЛ-СВЯЗАННОГО ОРФАННОГО РЕЦЕПТОРА ГАММА | 2017 |
|
RU2771280C2 |
| ПРОИЗВОДНЫЕ БЕНЗИЗОКСАЗОЛСУЛЬФОНАМИДА | 2020 |
|
RU2813356C2 |
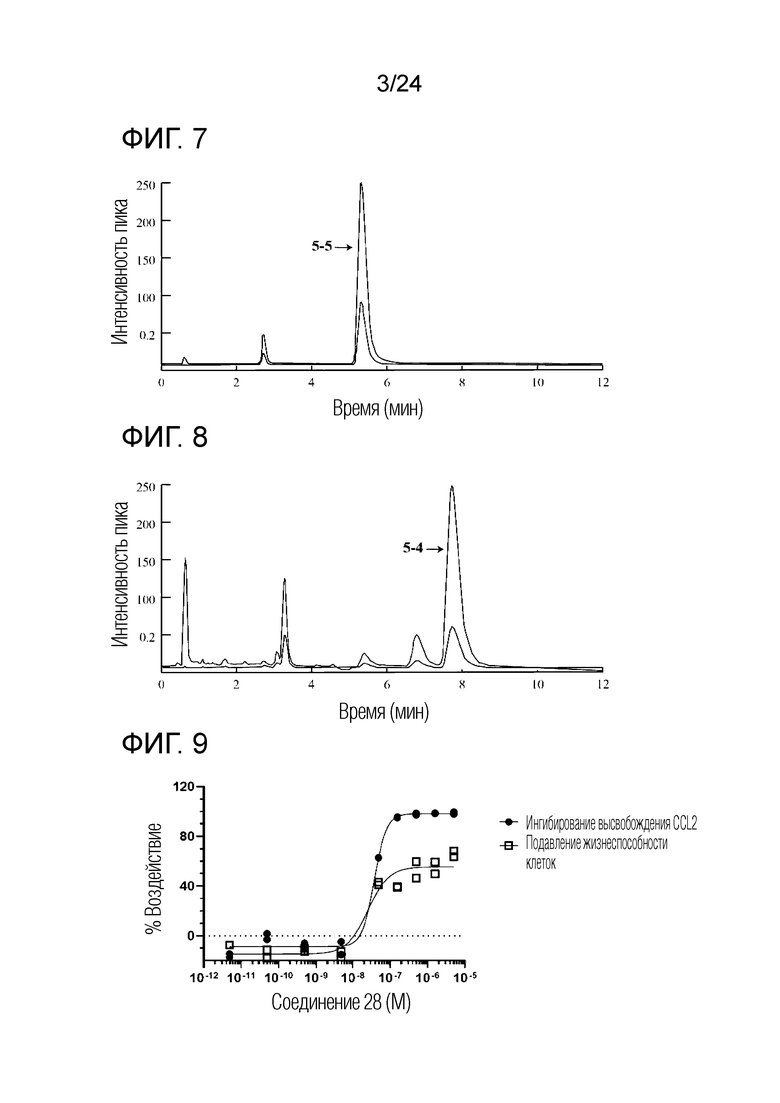
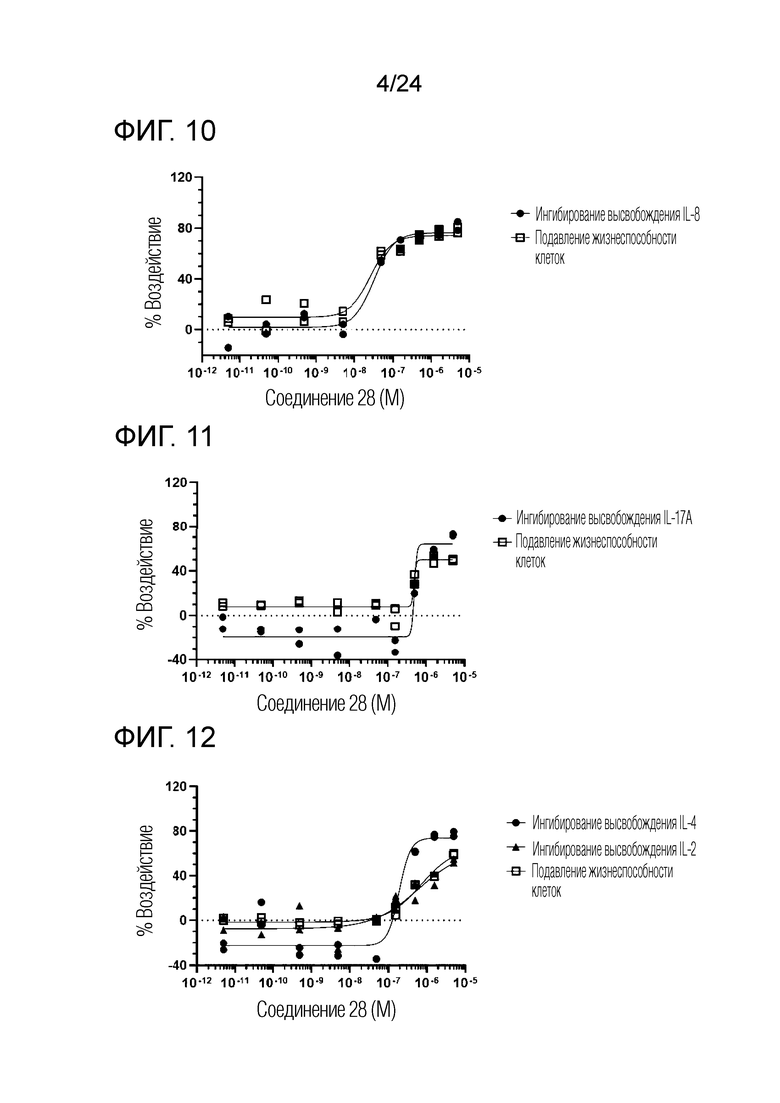
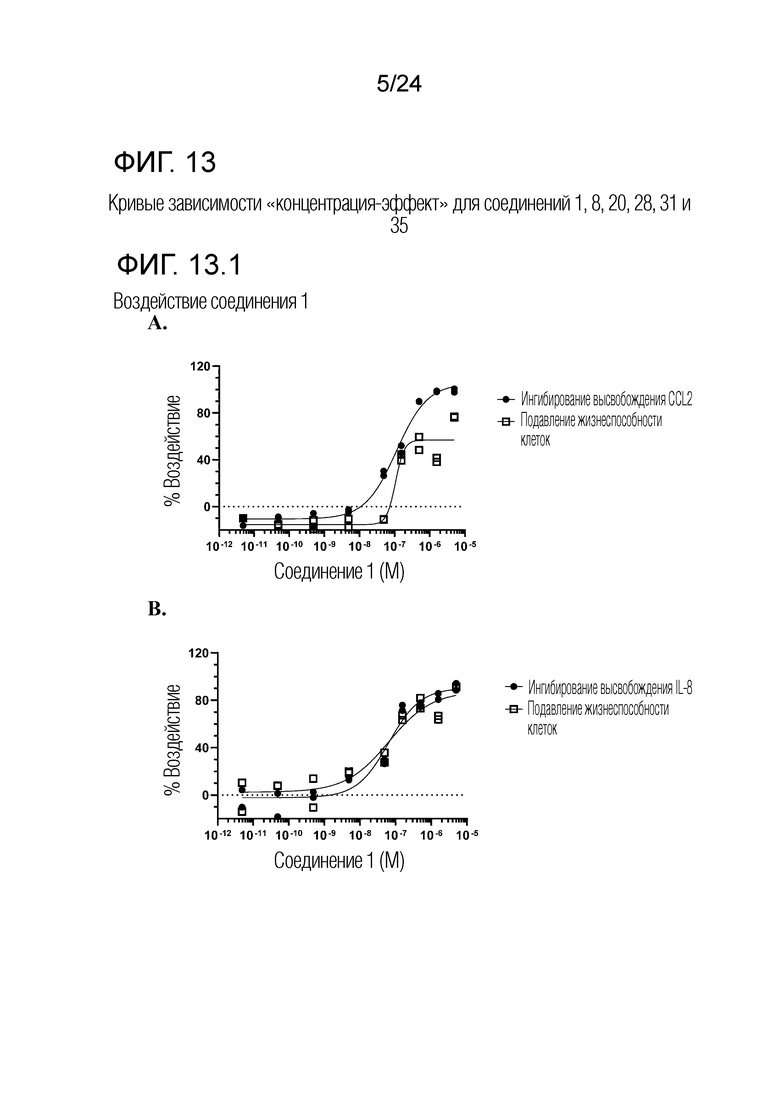
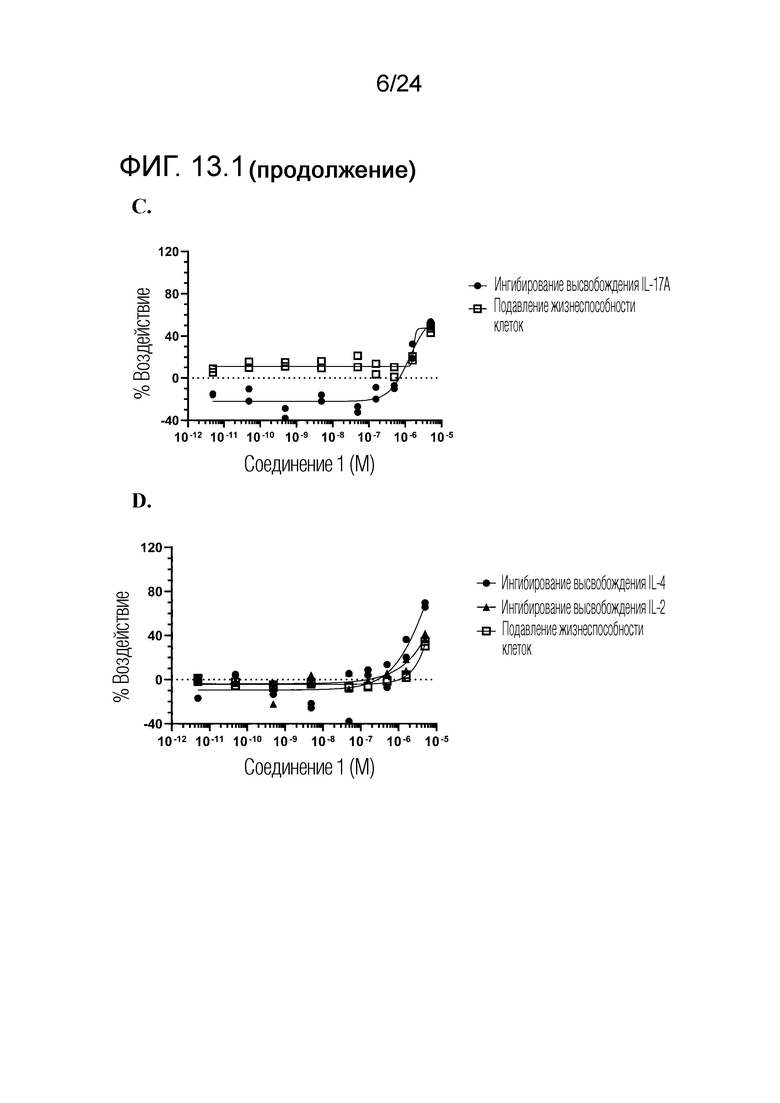
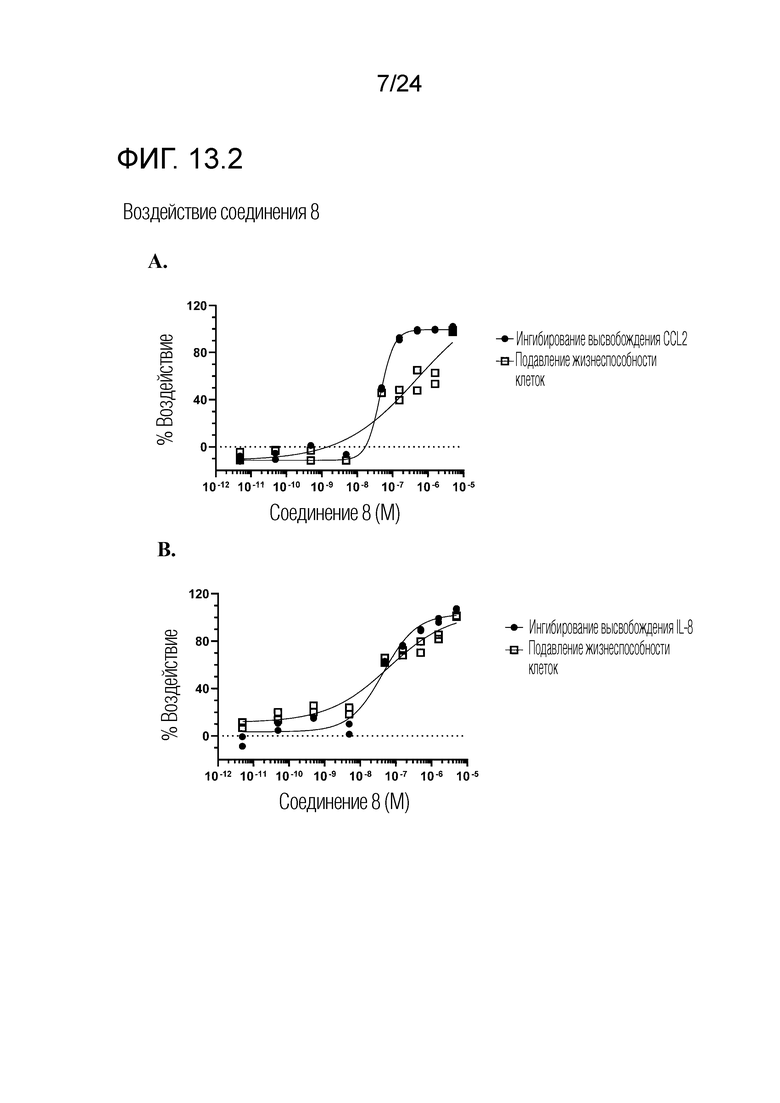
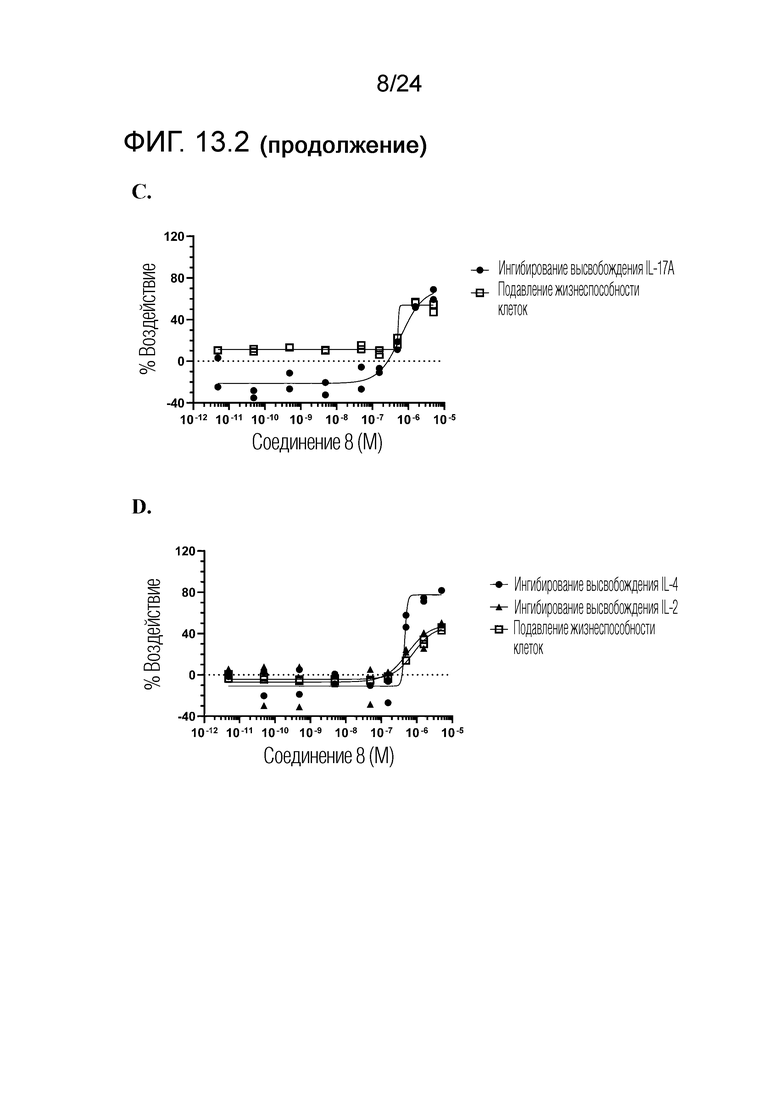
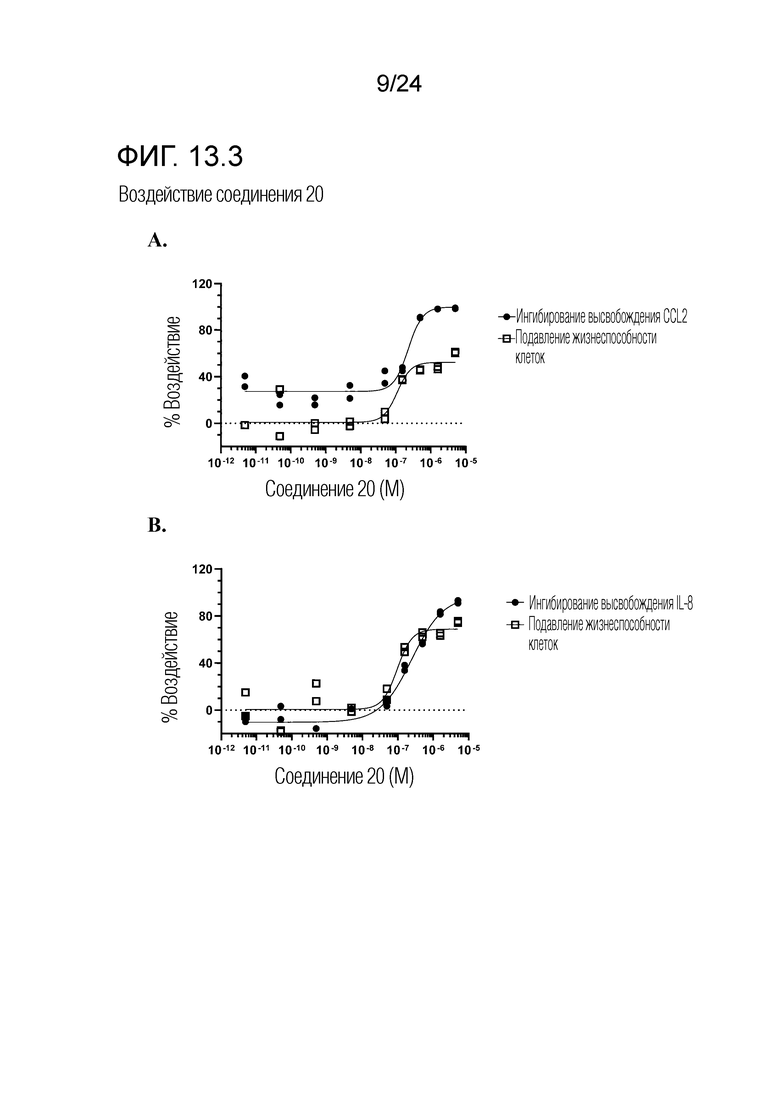
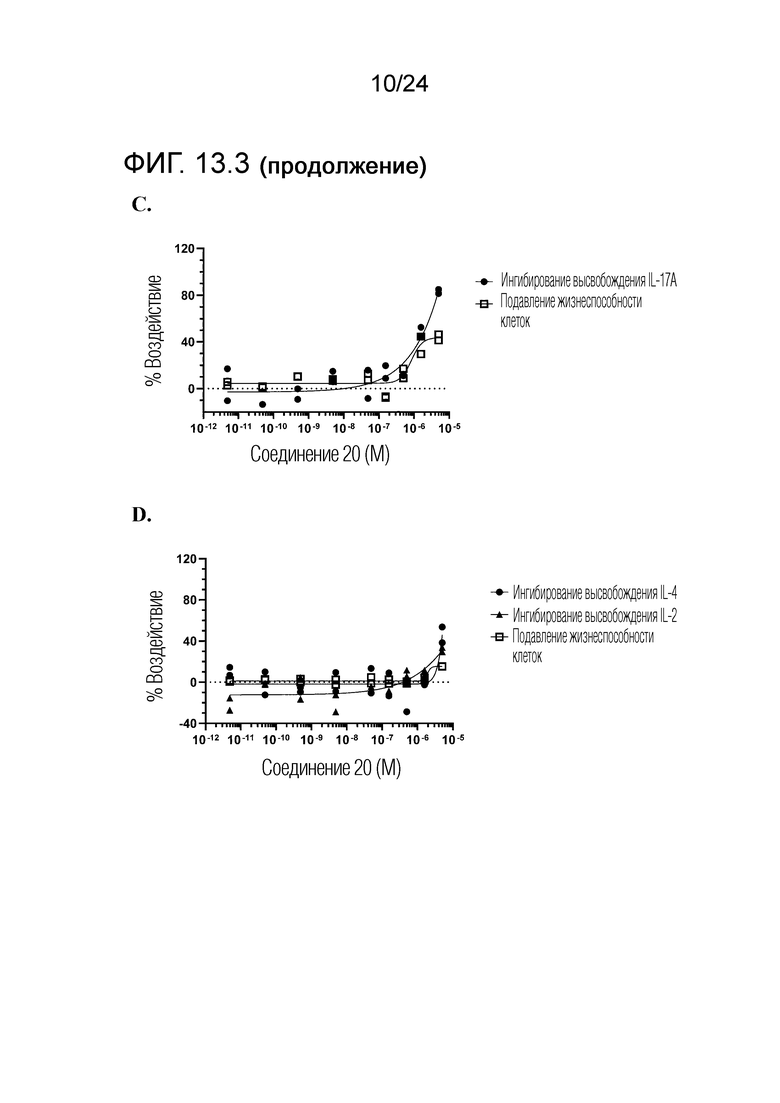
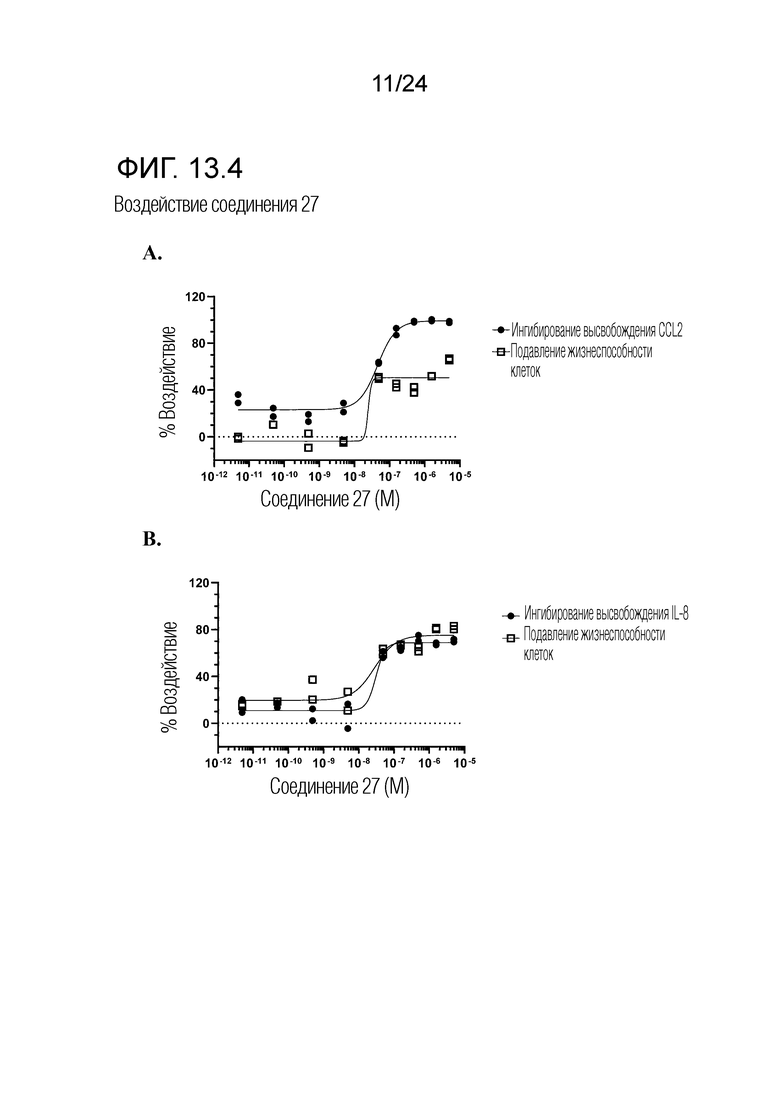
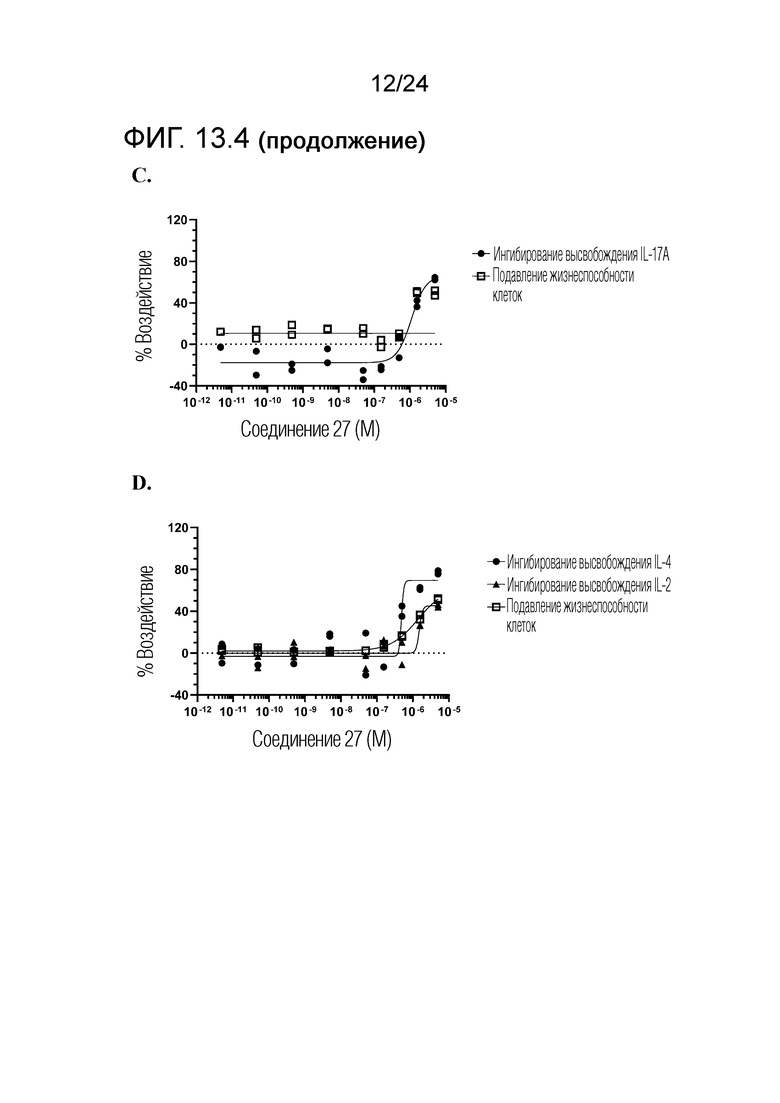
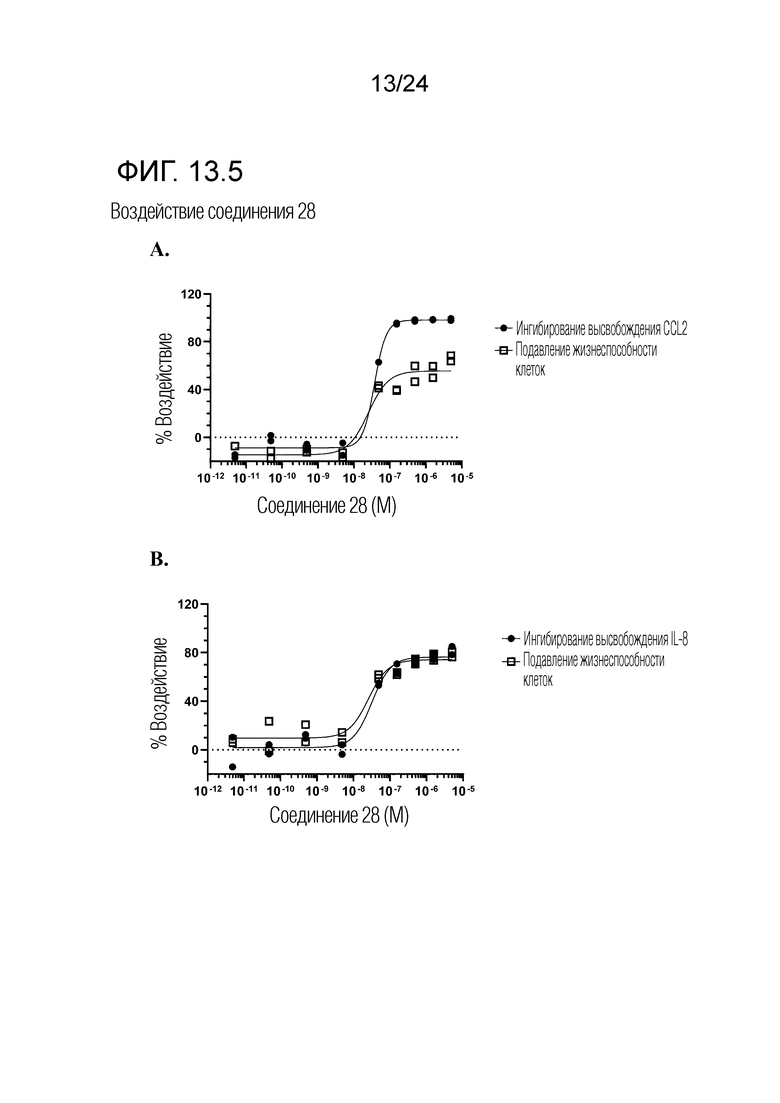
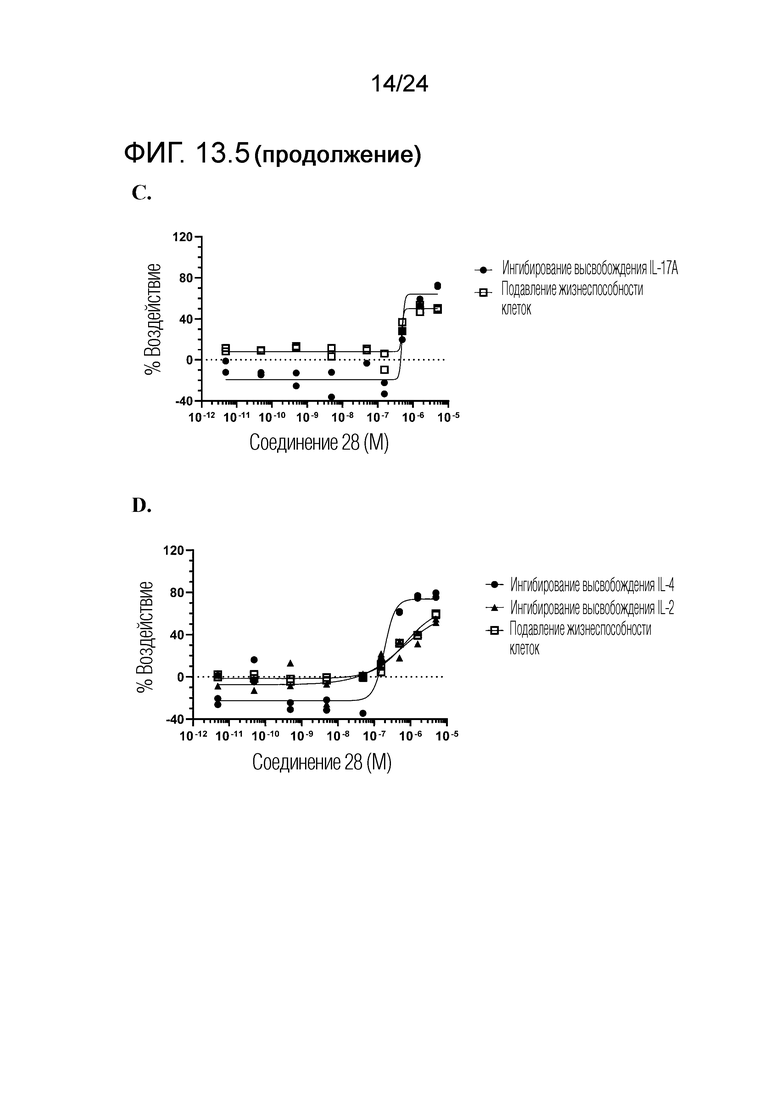
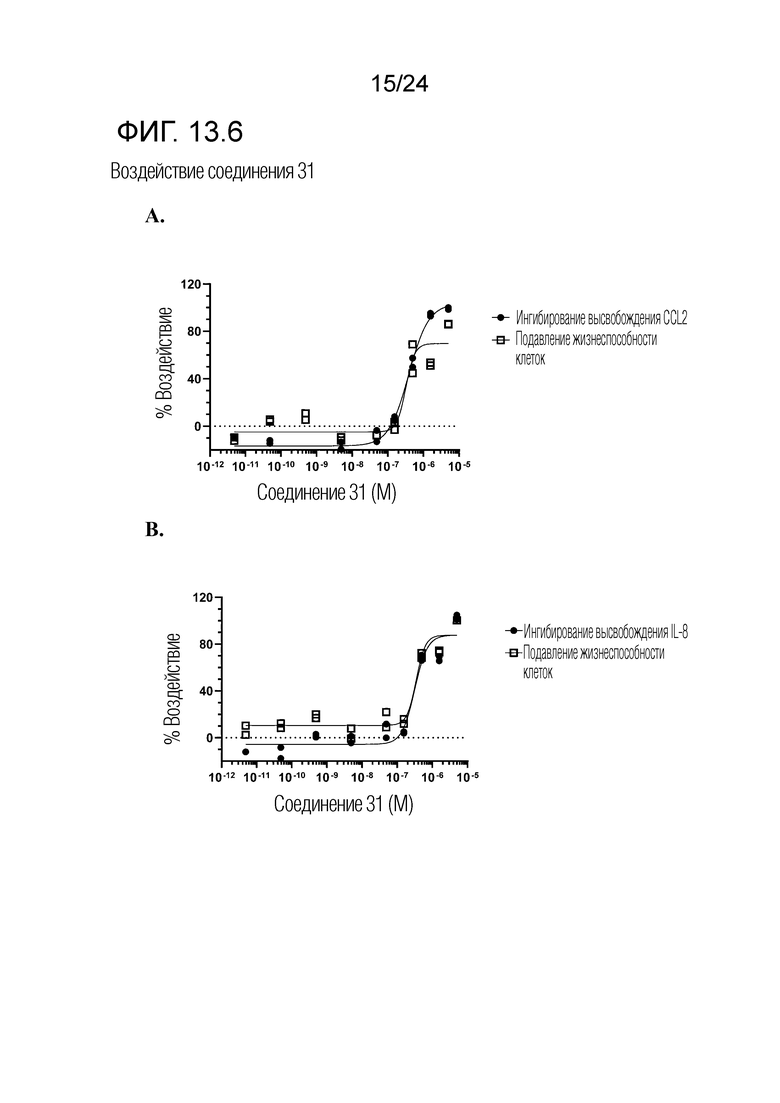
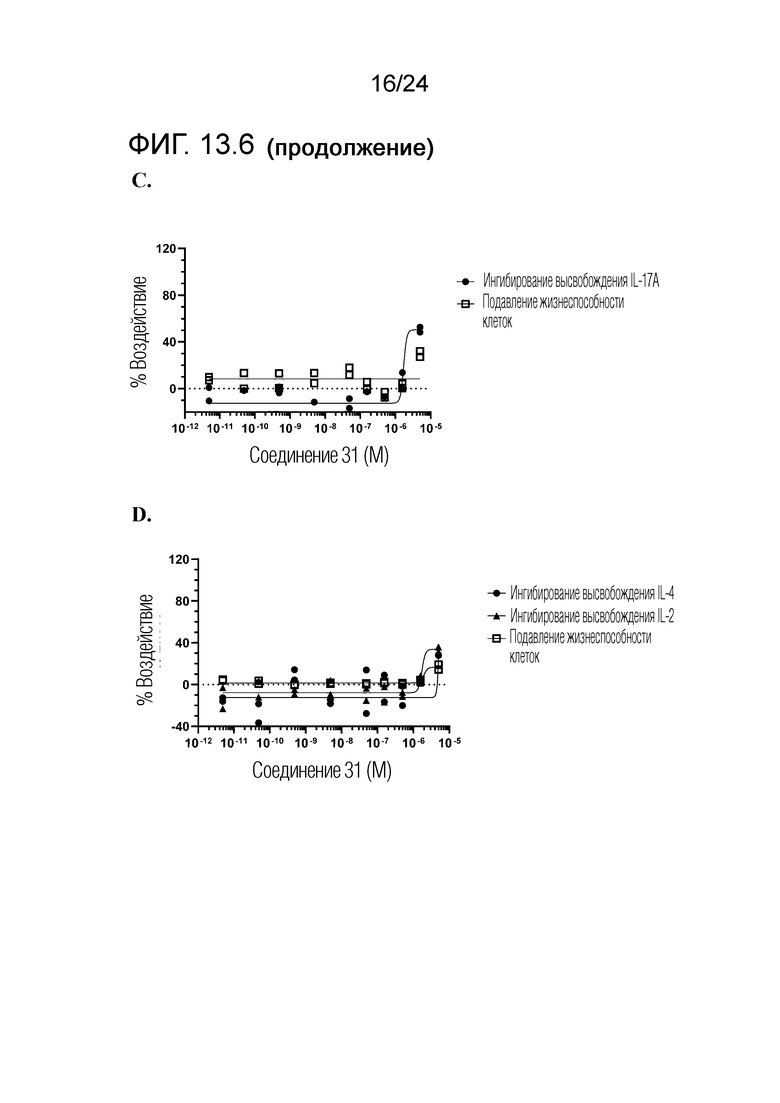
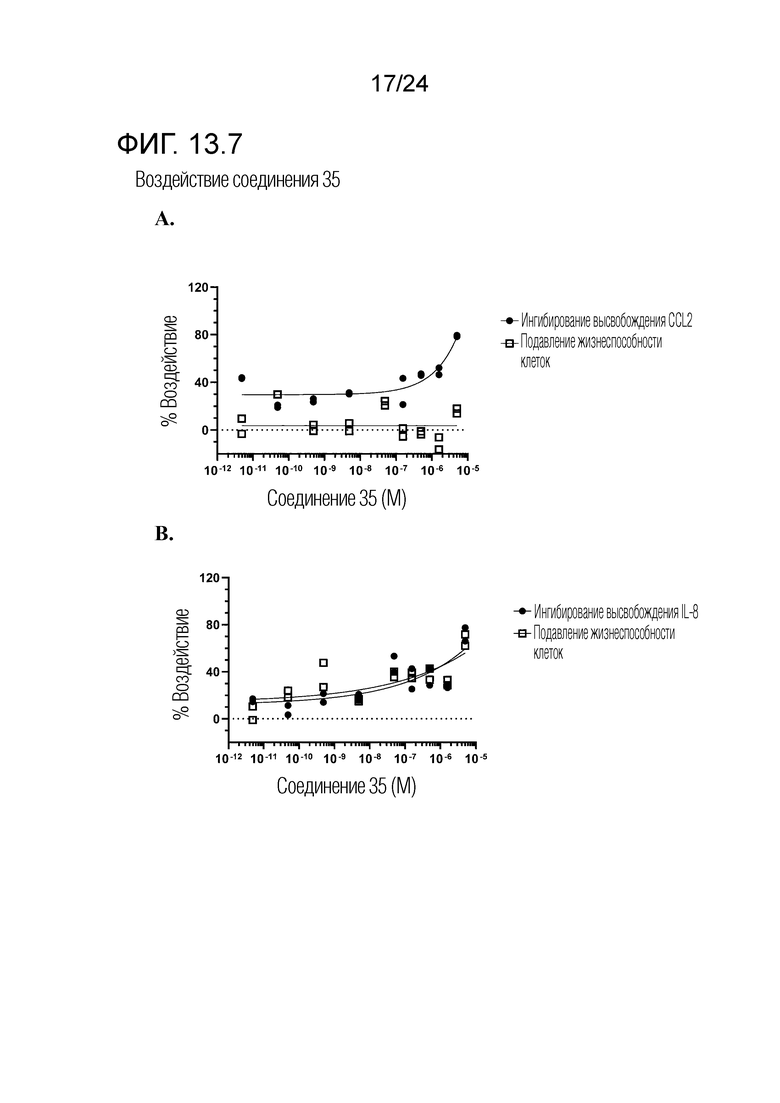
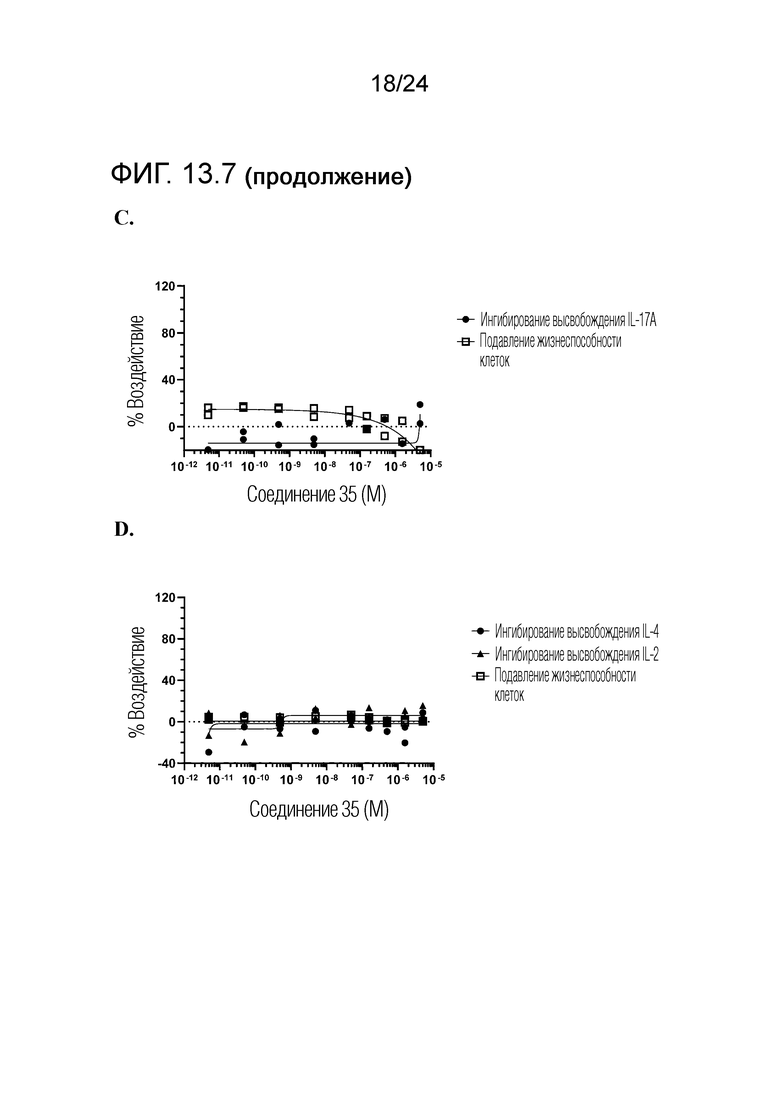
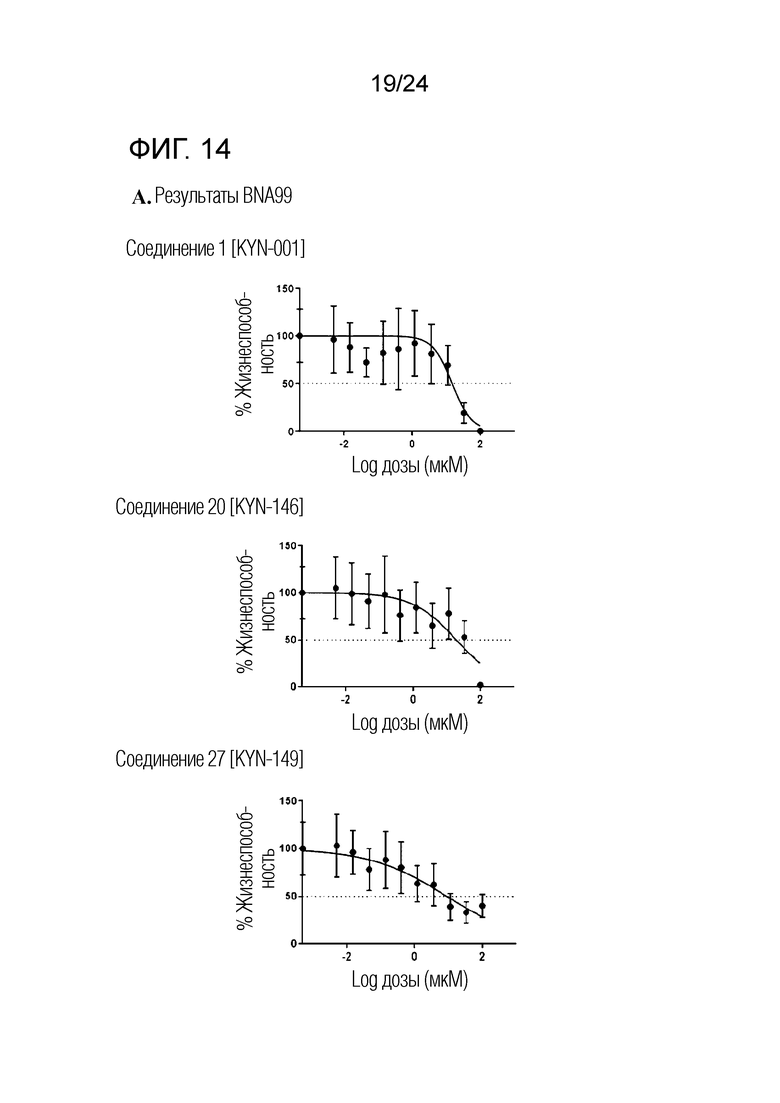
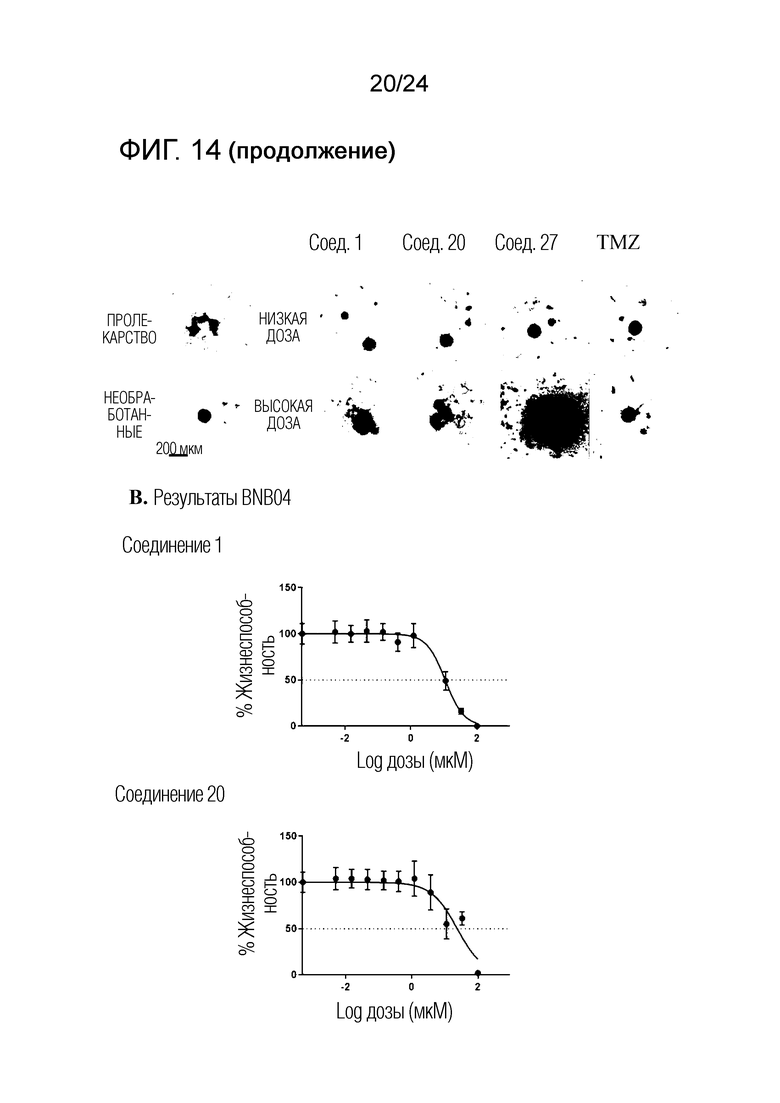
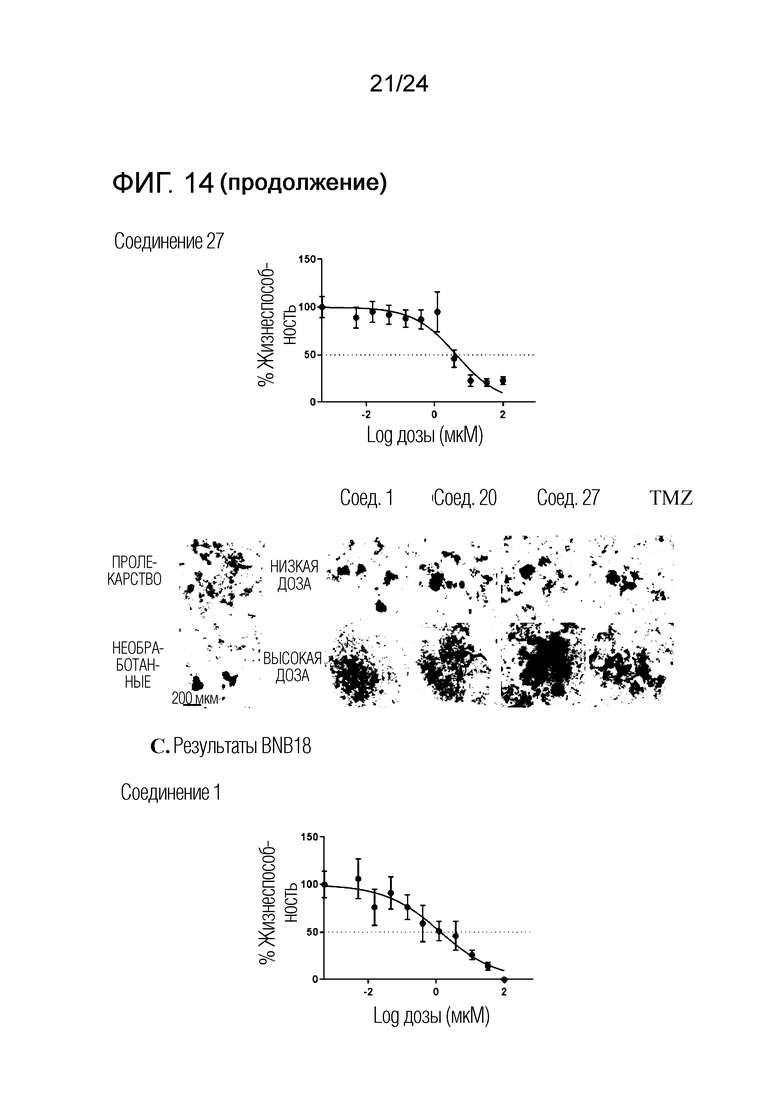
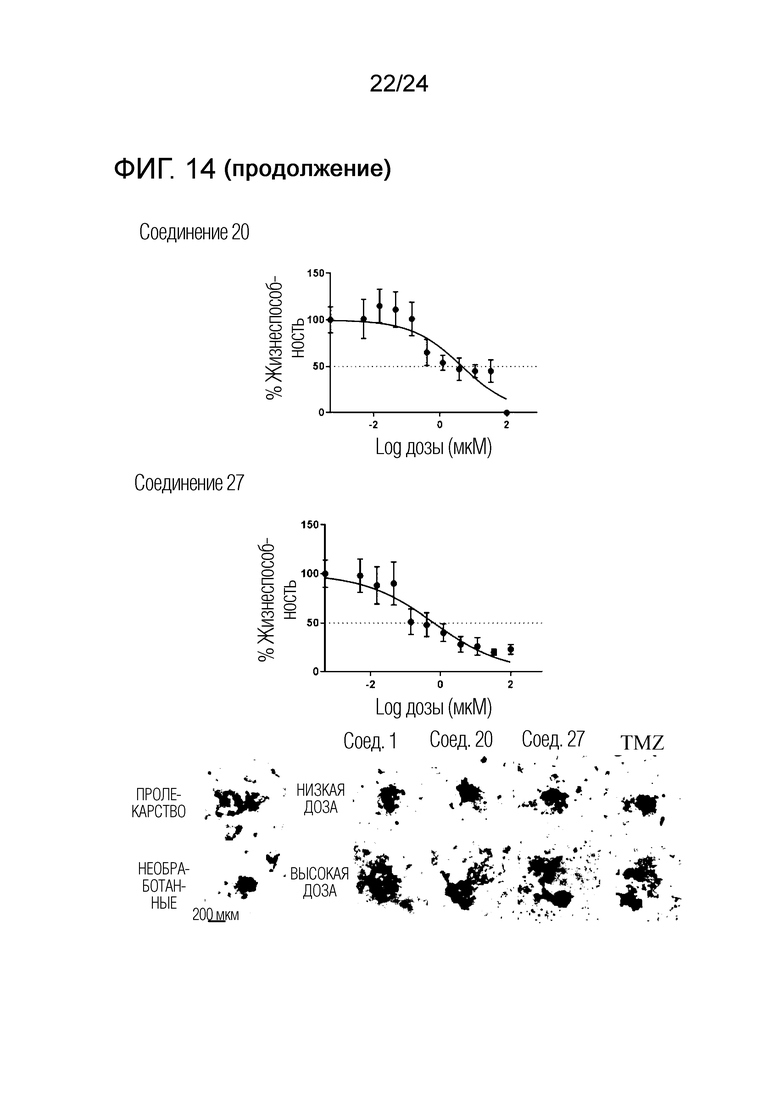
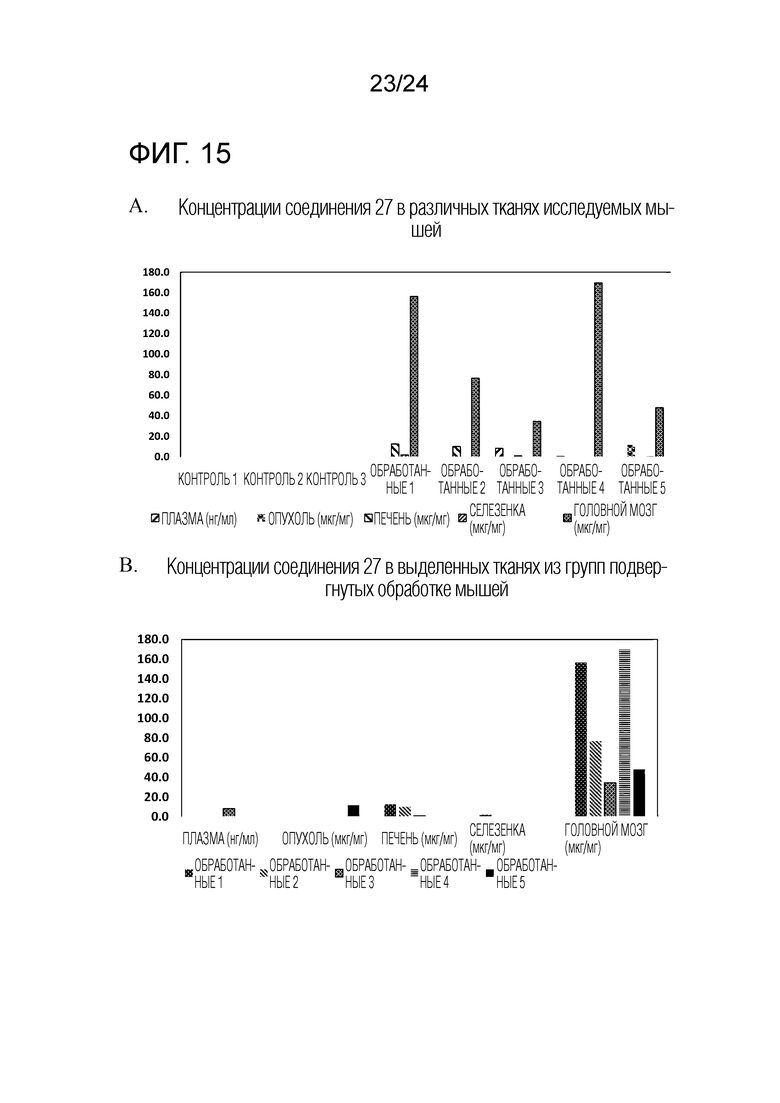
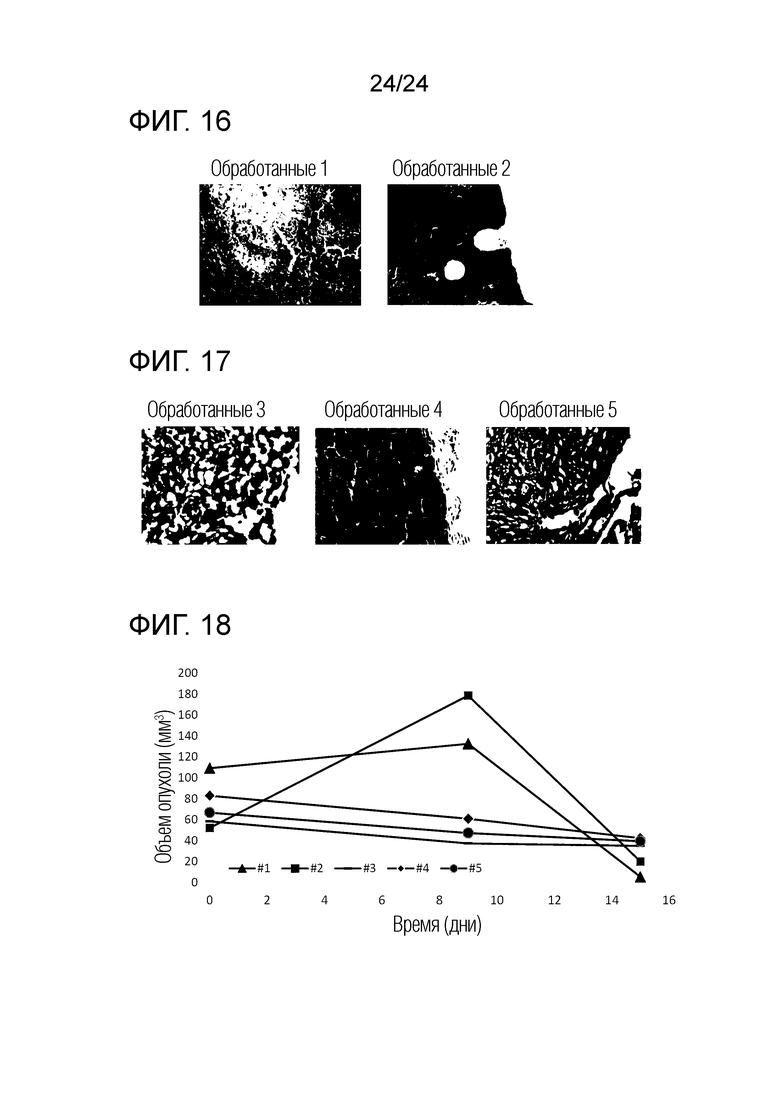
Настоящее изобретение относится к способу синтеза соединения формулы (I):
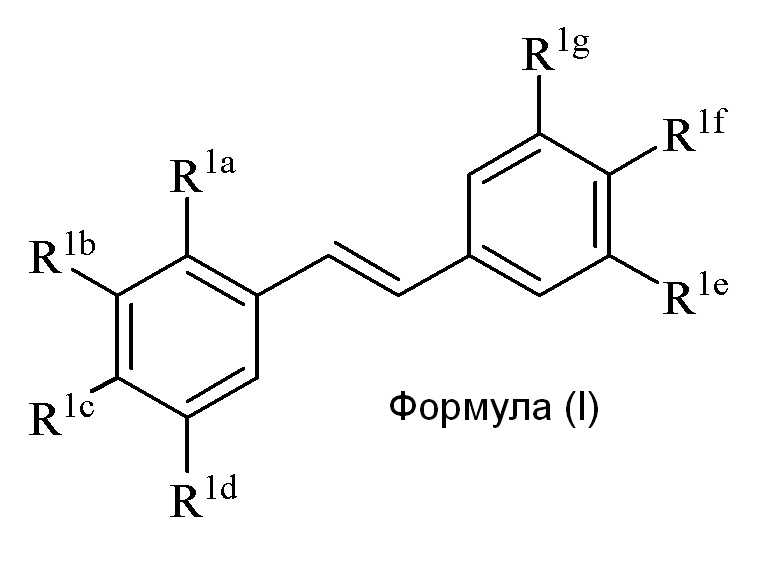
где R1a представляет собой независимо аллил, кротил, пренил или бензил; R1b представляет собой независимо СF3, OR2 или NHR3; R1c представляет собой H; R1d представляет собой независимо OH, O-защитную группу, NO2 или NHR3; R1b и R1d не являются одинаковыми; R1e представляет собой независимо C1-C6 алкил, OH, OR2, NO2, NHR3, NMeR3 или SR2; R1f представляет собой независимо H, OH, O-защитную группу, NO2, NHR3, NHC=NH(NH2), N(OH)(CO)Me или COOH; R1g представляет собой независимо H, C1-C6 алкил или OH; и не более чем один из R1f или R1g может представлять собой H; R3 представляет собой независимо H, CH3, (CO)H, (CO)Me, SO2Me, CH2COOR2, CO(CH2)nNH2 или CO(CH2)nCOOH, где n равен 0, 1 или 2; или
R1e и R1f образуют пятичленное гетероциклическое кольцо, содержащее карбонильную группу, когда R1e независимо представляет собой OH, NH2 или NHMe, и R1f представляет собой NH2, так что азот в R1f соединен мостиковой связью через карбонильную группу с кислородом или азотом в R1e, и R1g представляет собой Н;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, третбутил, трифторметил или O-защитную группу, когда R2 присоединен к O. Данный способ включает стадию сочетания соединения формулы (II) (модуля A) с соединением формулы (III) (модулем B) с получением соединения формулы (IV) (модуля C):
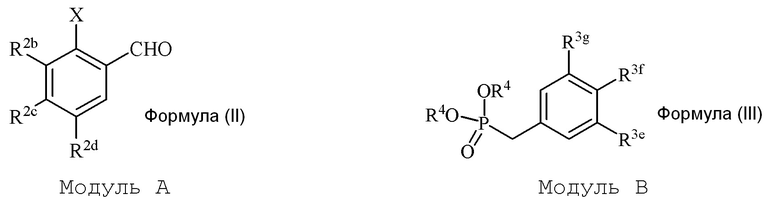
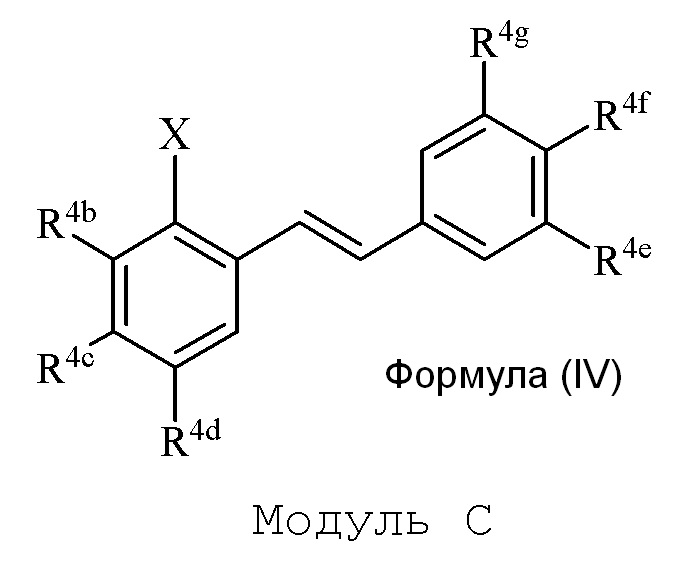
В соединении формулы (II) X представляет собой галоген или гидроксильную группу, где галоген выбирают из группы, состоящей из F, Cl, Br, I и At, в соединении формулы (IV) R4 представляет собой C1-C6алкил. Также изобретение относится к соединению формулы (I) и к способам лечения рака, атопического дерматита и псориаза с использование данного соединения или содержащей данное соединение фармацевтической композиции. Технический результат - создание нового процесса, обеспечивающего эффективное и применимое в промышленности получение ряда соединений 2-замещенного гидроксистильбена, являются перспективными для применения при лечении рака и заболеваний или нарушений кожи. 7 н. и 26 з.п. ф-лы, 18 ил., 8 табл.
1. Способ синтеза соединения формулы (I):
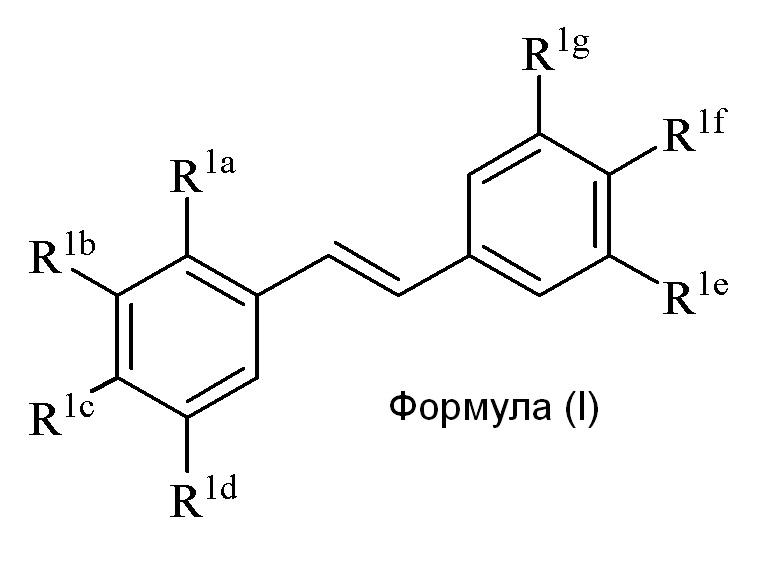
где
R1a представляет собой независимо аллил, кротил, пренил или бензил;
R1b представляет собой независимо СF3, OR2 или NHR3;
R1c представляет собой H;
R1d представляет собой независимо OH, O-защитную группу, NO2 или NHR3;
R1b и R1d не являются одинаковыми;
R1e представляет собой независимо C1-C6 алкил, OH, OR2, NO2, NHR3, NMeR3 или SR2;
R1f представляет собой независимо H, OH, O-защитную группу, NO2, NHR3, NHC=NH(NH2), N(OH)(CO)Me или COOH;
R1g представляет собой независимо H, C1-C6 алкил или OH;
и не более чем один из R1f или R1g может представлять собой H;
R3 представляет собой независимо H, CH3, (CO)H, (CO)Me, SO2Me, CH2COOR2, CO(CH2)nNH2 или CO(CH2)nCOOH, где n равен 0, 1 или 2;
или
R1e и R1f образуют пятичленное гетероциклическое кольцо, содержащее карбонильную группу, когда R1e независимо представляет собой OH, NH2 или NHMe, и R1f представляет собой NH2, так что азот в R1f соединен мостиковой связью через карбонильную группу с кислородом или азотом в R1e, и R1g представляет собой Н;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, третбутил, трифторметил или O-защитную группу, когда R2 присоединен к O;
способ включает:
i) стадию сочетания соединения формулы (II) (модуля A) с соединением формулы (III) (модулем B) с получением соединения формулы (IV) (модуля C):
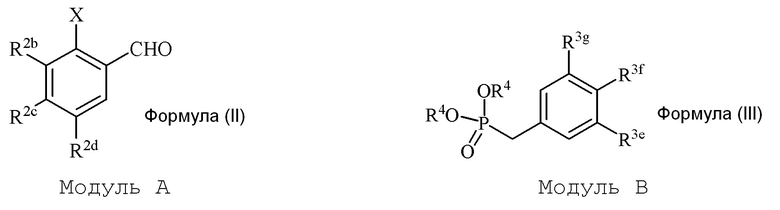
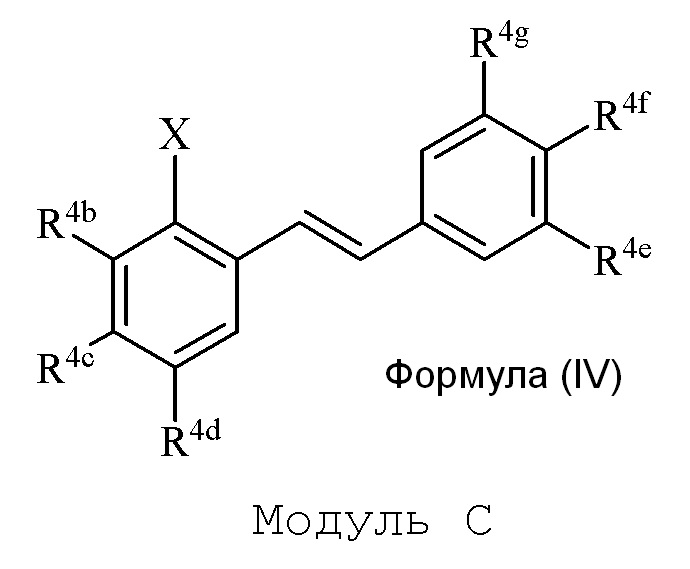
где
в формуле II (модуле A):
R2b представляет собой независимо СF3, OR2 или NH2;
R2c представляет собой H;
R2d представляет собой независимо O-защитную группу или NO2;
X представляет собой галоген или гидроксильную группу, где галоген выбирают из группы, состоящей из F, Cl, Br, I и At;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил, или O-защитную группу, когда R2 присоединен к O;
и
в формуле III (модуле B):
R3e представляет собой независимо C1-C6 алкил, OR2, NO2 или SR2;
R3f представляет собой независимо H, O-защитную группу, NO2 или COOR2;
R3g представляет собой независимо H, C1-C6 алкил, OH или OR2;
и не более чем один из R3f или R3g может представлять собой H;
или R3e и R3f образуют пятичленное гетероциклическое кольцо, содержащее карбонильную группу, когда R3e представляет собой OH, NH2 или NHMe, и R3f представляет собой NH2, так что азот в R3f соединен мостиковой связью через карбонильную группу с кислородом или азотом в R3e, и R3g представляет собой Н;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 присоединен к O; и
R4 представляет собой C1-C6алкил;
в формуле IV (модуле C):
R4b представляет собой независимо СF3, OR2 или NH2;
R4c представляет собой независимо H;
R4d представляет собой независимо O-защитную группу или NO2;
R4e представляет собой независимо C1-C6 алкил, OR2, NO2, SR2;
R4f представляет собой независимо H, O-защитную группу, NO2 или COOH;
R4g представляет собой независимо H, C1-C6 алкил, ОН или OR2;
и не более чем один из R4f или R4g может представлять собой H;
или
R4e и R4f образуют пятичленное гетероциклическое кольцо, содержащее карбонильную группу, когда R4e представляет собой OH, NH2 или NHMe, и R4f представляет собой NH2, так что азот в R4f соединен мостиковой связью через карбонильную группу с кислородом или азотом в R4e, и R4g представляет собой Н;
R2 представляет собой независимо метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 присоединен к O;
и
X представляет собой галоген или гидроксильную группу, где галоген выбирают из группы, состоящей из F, Cl, Br, I и At;
где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этокси]метила (SEM), CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2.
2. Способ по п. 1, где, если
i) X в формуле (II) и (IV) представляет собой галоген,
галогенид формулы (IV) подвергают реакции сочетания с одним из следующих соединений:
a) соединением R1a-замещенной бороновой кислоты или эфира, включая пинаколовый эфир R1a-замещенной бороновой кислоты;
b) соединением R1a-замещенного трифторбората, включая R1a-замещенный трифторборат калия; или
c) соединением R1a-замещенного органостаннана, включая R1a-замещенный трибутилстаннан;
с образованием соединения формулы (I),
где R1a выбирают из группы, состоящей из аллила, кротила, пренила и бензила;
или
ii) X в формуле (II) и (IV) представляет собой гидроксил,
гидроксильную группу формулы (IV) превращают в трифлатную (трифторметилсульфонатную) группу с образованием трифлата формулы (IV), и трифлат формулы (IV) подвергают реакции сочетания с одним из следующих соединений:
a) соединением R1a-замещенной бороновой кислоты или эфира, включая пинаколовый эфир R1a-замещенной бороновой кислоты;
b) соединением R1a-замещенного трифторбората, включая R1a-замещенный трифторборат калия; или
c) соединением R1a-замещенного органостаннана, включая R1a-замещенный трибутилстаннан
с образованием соединения формулы (I),
где R1a выбирают из группы, состоящей из аллила, кротила, пренила и бензила; и
необязательно где реакция сочетания катализируется соединением палладия в присутствии основания.
3. Способ по п. 1 или 2, где соединение Формулы (I) подвергают последующей реакции, выбранной из одной или более следующих реакций:
- превращение заместителя NH2 в положении R1b в заместитель NH(CO)H или NHCOMe;
- превращение заместителя NO2 в положении R1d в заместитель NHR3;
- превращение заместителя NO2 в положении R1e в заместитель NHR3 или NMeR3;
- в заместитель NO2 в положении R1f в заместитель NHR3, NHC(NH)NH2 или N(OH)C(O)Me;
- где R3 представляет собой независимо H, CH3, (CO)H, (CO)Me, SO2Me, CH2COOR2, CO(CH2)nNH2 или CO(CH2)nCOOH, где n равен 0, 1 или 2.
4. Способ по п. 1, где соединение формулы (I) выбирают из группы, состоящей из:
9 [KYN-130] R = н-пропил
10 [KYN-131] R = изопропил
11 [KYN-132] R = CF3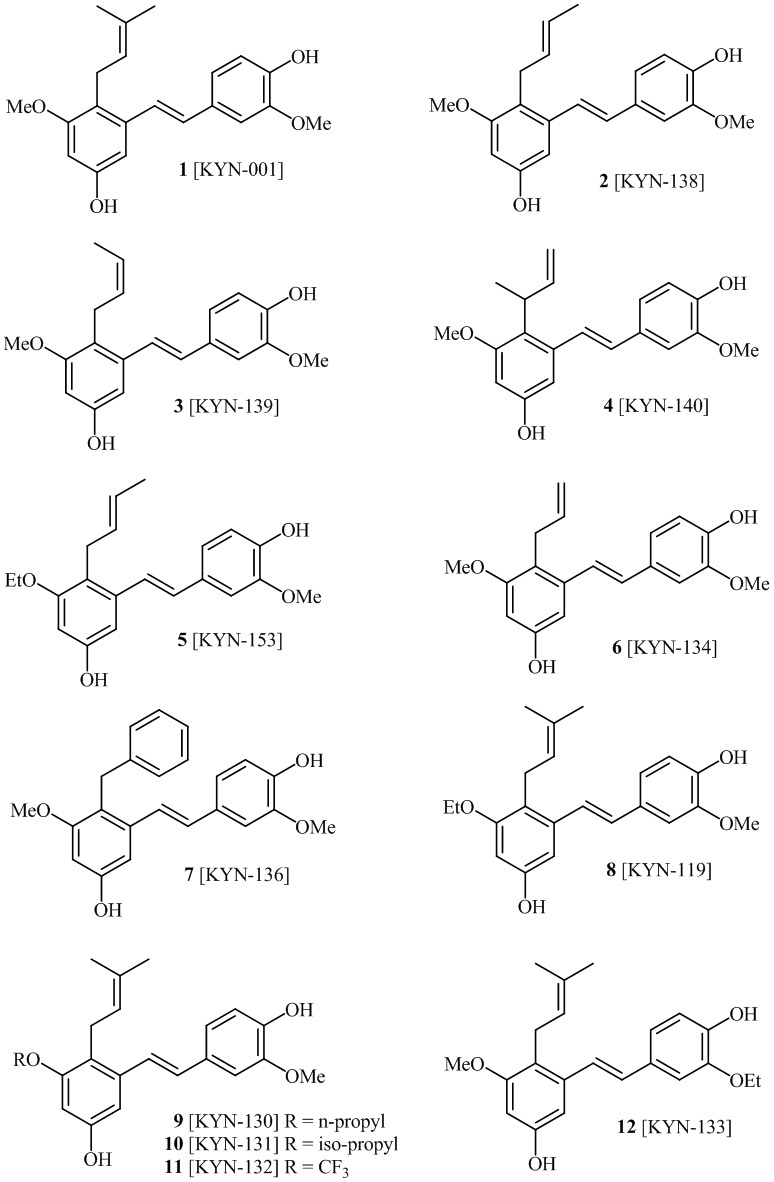
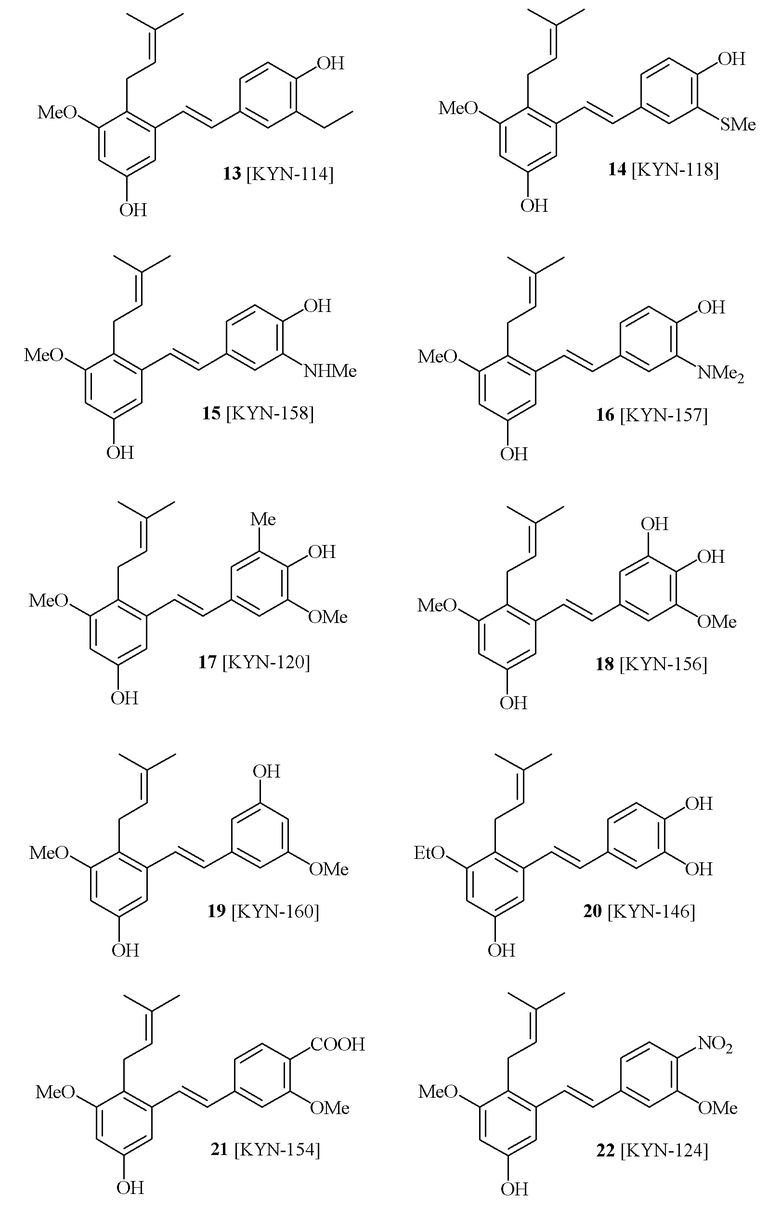
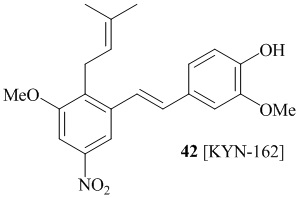
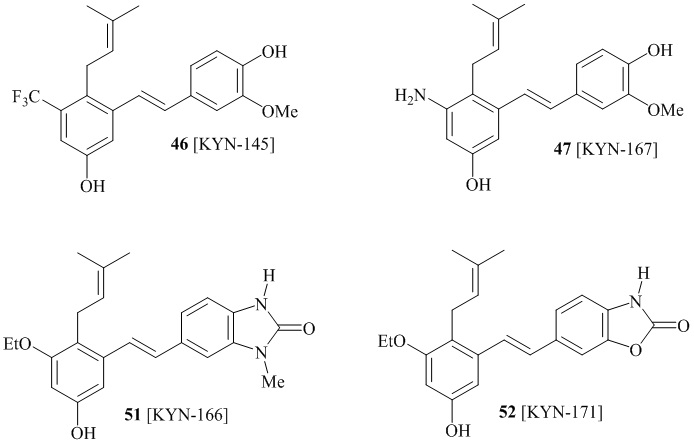
5. Способ по п. 3, где в результате реакции соединения Формулы (I) образуется соединение, выбранное из группы, состоящей из:
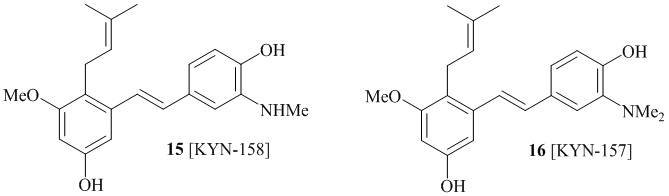
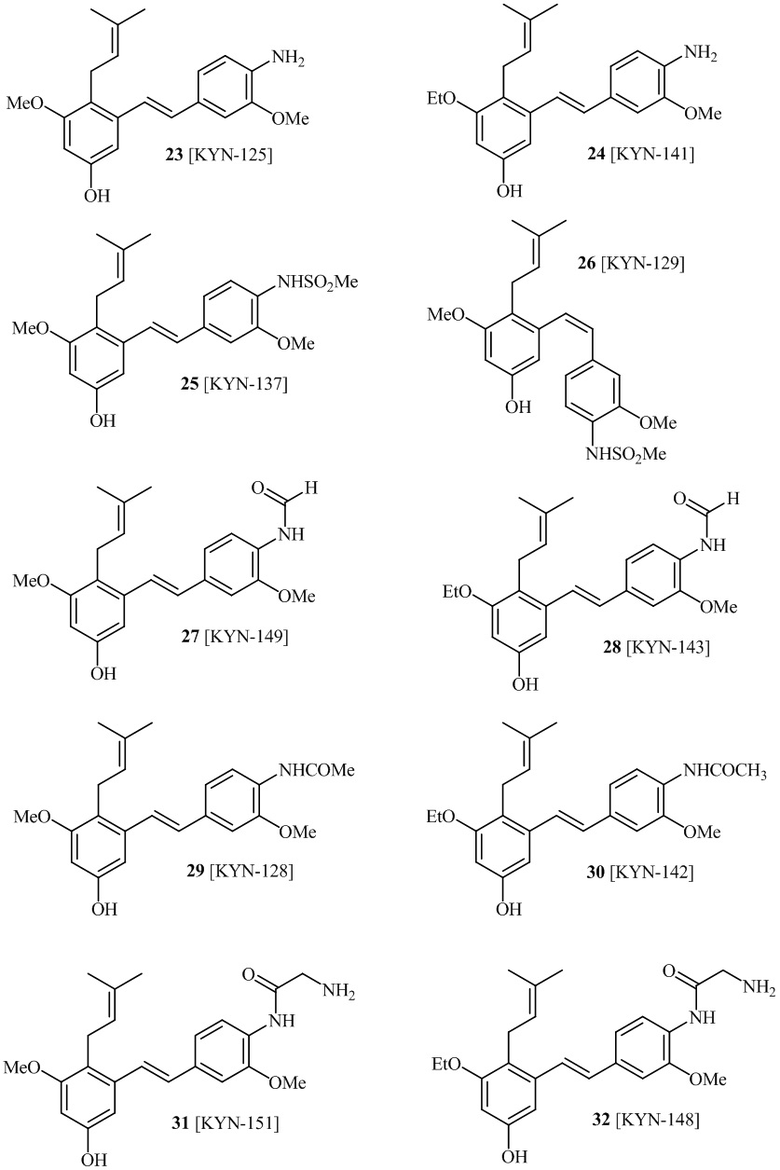
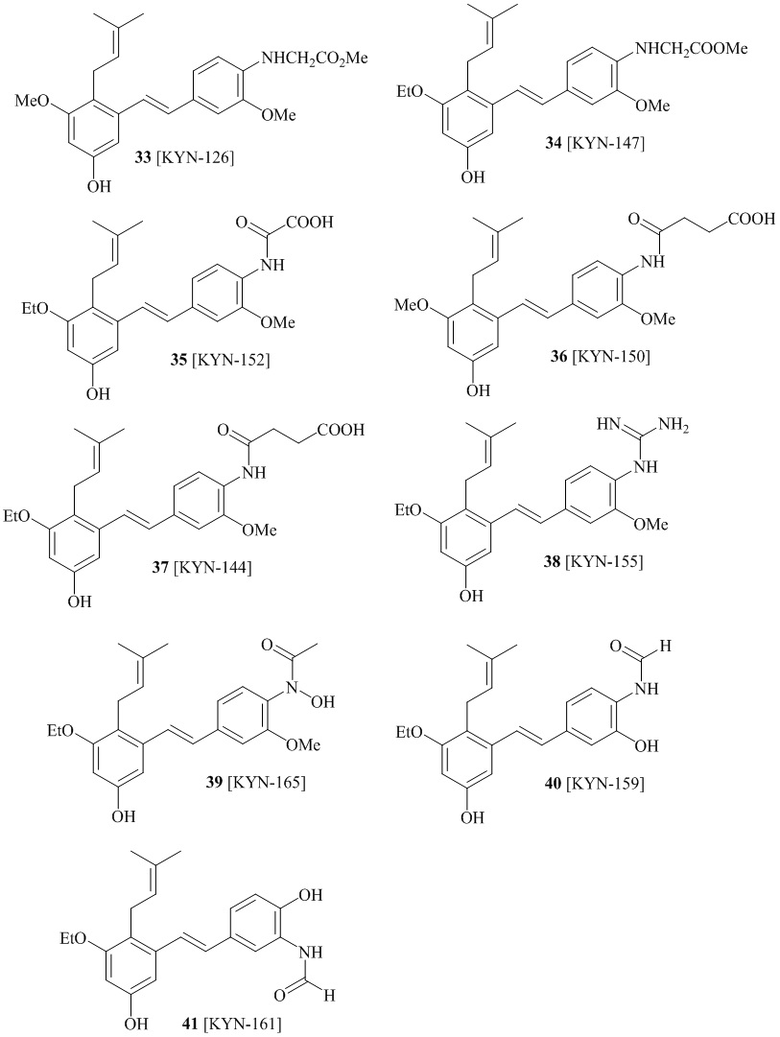
6. Способ по п. 1 или 2,
где в формуле (I):
R1a представляет собой аллил, кротил, пренил или бензил,
R1b представляет собой OR2 или NH2,
R1c представляет собой H;
R1d представляет собой OH;
R1e представляет собой OH или OR2;
R1f представляет собой NO2;
R1g представляет собой H;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил или трет-бутил;
R3 представляет собой H, (CO)H, (CO)Me, SO2Me, CH2COOR2, COCH2NH2 или CO(CH2)nCOOH (где n=0, 1 или 2);
в формуле (II):
R2b представляет собой OR2 или NH2;
R2c представляет собой H,
R2d представляет собой O-защитную группу;
Х представляет собой Hal, представляющий собой галоген, выбранный из группы, состоящей из F, Cl, Br, I и At;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, когда R2 присоединен к O,
где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этоксиметила (SEM), CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2; и
в формуле (III):
R3e представляет собой OR2;
R3f представляет собой NO2;
R3g представляет собой H;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил или трет-бутил, и
в формуле (IV):
R4b представляет собой OR2 или NH2;
R4c представляет собой H;
R4d представляет собой O-защитную группу;
R4e представляет собой OR2;
R4f представляет собой NO2;
R4g представляет собой H;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил или трет-бутил,
и
X представляет собой галоген, выбранный из группы, состоящей из F, Cl, Br, I и At;
где соединение формулы IV подвергают реакции сочетания с пинаколовым эфиром R1a-бороновой кислоты, выбранным из группы, состоящей из пинаколового эфира 3-метилбут-2-енилбороновой кислоты, пинаколового эфира кротилбороновой кислоты, пинаколового эфира аллилбороновой кислоты и пинаколового эфира бензилбороновой кислоты, катализируемой с помощью палладиевого катализатора в присутствии основания, с получением соединения формулы (I).
7. Способ по п. 6, где соединение Формулы (I) подвергают последующей реакции, выбранной из одной или более следующих реакций:
превращение заместителя NH2 в положении R1b в заместитель NH(CO)H или NHCOMe;
превращение заместителя NO2 в положении R1f в заместитель NH2, NHSO2Me, NHC(O)H, NHC(O)Me, NHCO(CH2)2NH2, NHCH2COOR2 или NHC=NH(NH2).
8. Способ по п. 7, где последующая реакция соединения формулы (I) приводит к получению соединения, выбранного из группы, состоящей из:
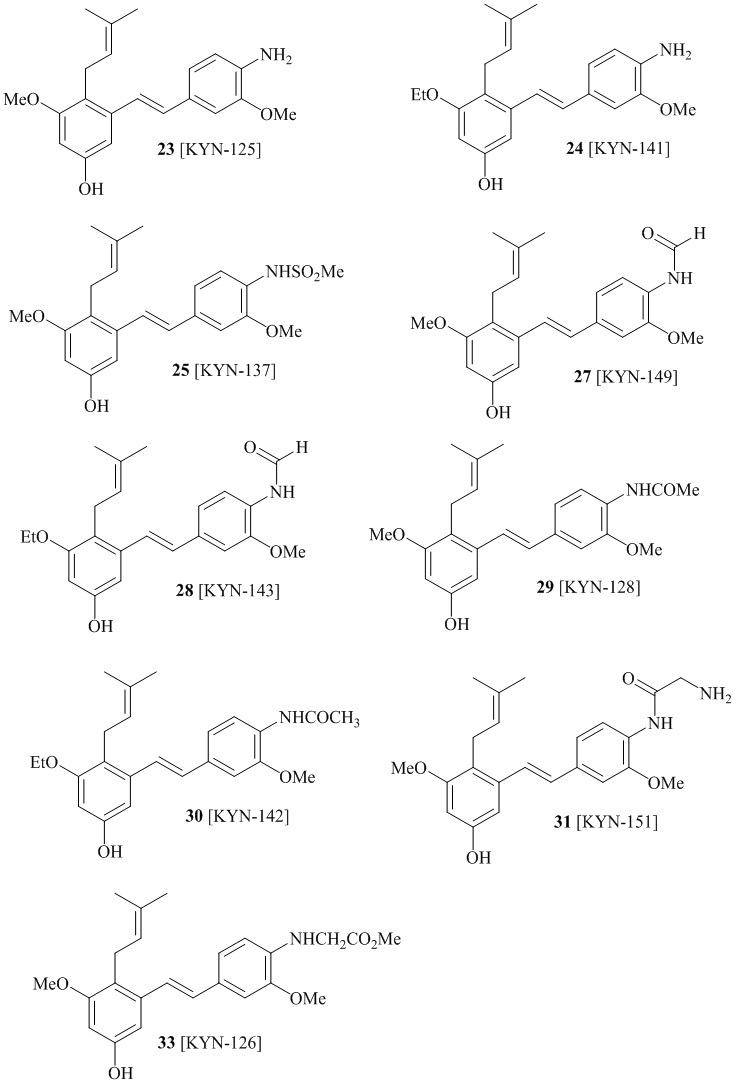
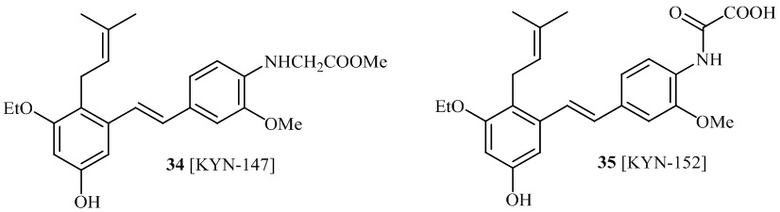
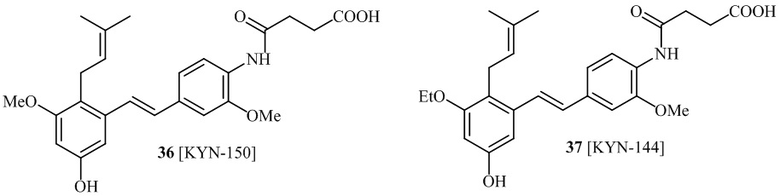
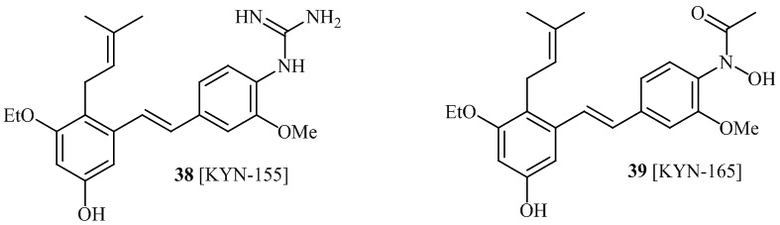
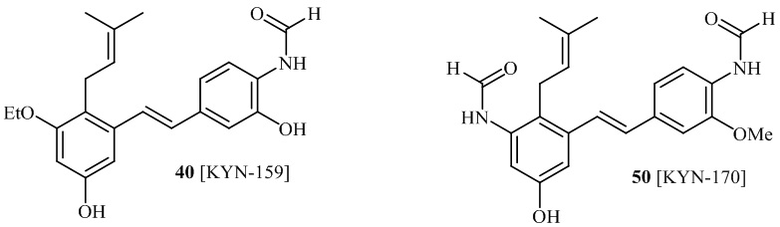 .
.
9. Способ по п. 1, где соединение формулы (I) подвергают последующей реакции, выбранной из одной или более следующих реакций:
- превращения одного или более заместителей -NH2 в метилоксалатные заместители путем реакции соединения, содержащего заместитель NH2, с метилхлороксоацетатом в присутствии основания, такого как триэтиламин, с последующим гидролизом с образованием заместителя -ОН;
- превращения одного или более заместителей -NH2 в гуанидиновые (-NHC(NH)NH2) заместители путем реакции соединения, содержащего заместитель NH2 с цианамидом в присутствии кислоты, такой как п-толуолсульфоновая кислота;
- превращения одного или более заместителей -NH2 в формамидные (-NHC(O)H) заместители путем реакции соединения, содержащего заместитель NH2, с этилформиатом;
- превращения одного или более заместителей -NH2 в заместители ацетамидо (-NHC(O)CH3) путем реакции соединения, содержащего заместитель NH2, с уксусным ангидридом в присутствии основания, такого как DIPEA (диизопропилэтиламин);
- превращения одного или более заместителей -NH2 в заместители амидного производного (-NHC(O)CH2CH2C(O)OH) путем реакции соединения, содержащего заместитель NH2, с янтарным ангидридом в присутствии основания, такого как DIPEA (диизопропилэтиламин);
- превращения одного или более заместителей -NH2 в метиламинные (-NHMe) заместители путем реакции соединения, содержащего заместитель NH2, с формальдегидом в присутствии триацетоксиборгидрида;
- восстановления одного или более заместителей -NO2 в аминные заместители, например, реакцией соединения, содержащего заместитель NO2, с Zn в присутствии хлорид аммония.
10. Способ по п. 1, где соединение формулы (II) представляет собой Модуль А
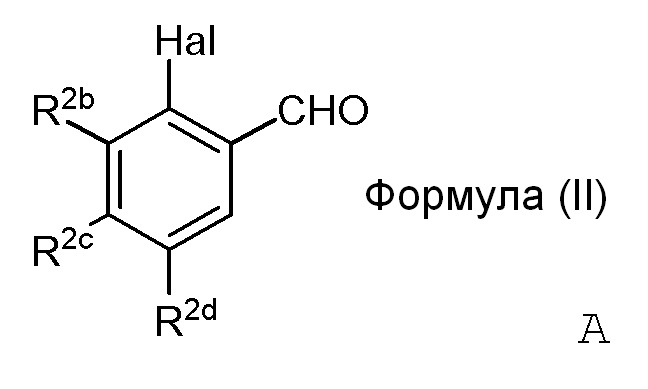
Модуль А
где
R2b = OMe;
R2c = H;
R2d = OSEM;
и
Х представляет собой Hal, представляющий собой Br;
который получают следующим способом:
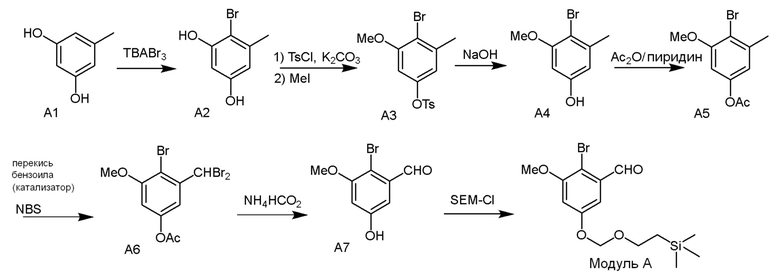
11. Способ по п. 1, где соединение формулы (III) представляет собой Модуль В
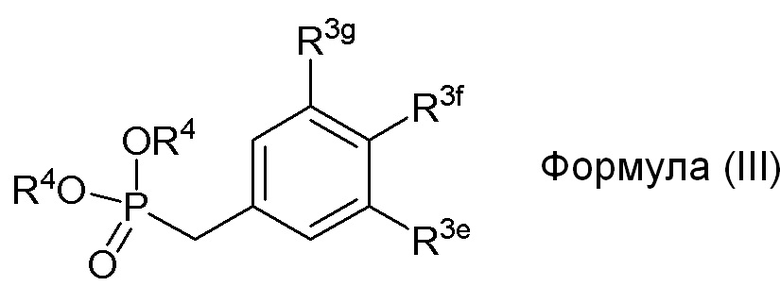
Модуль В
где
R3g = H;
R3f = OSEM;
R3e = OMe;
R4 = Et;
который получают следующим способом:
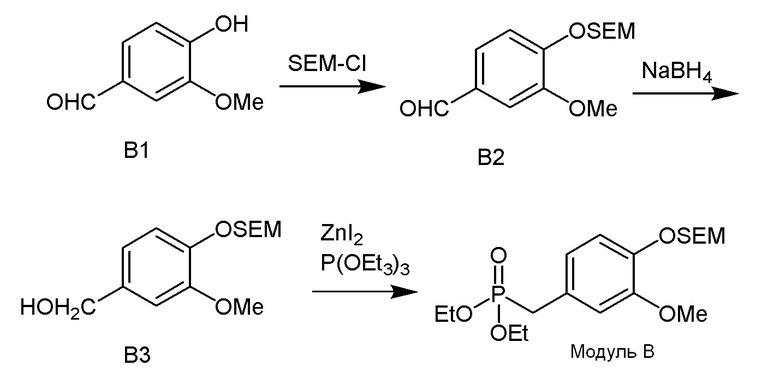
12. Способ по п.1, где соединение формулы (II) представляет собой модуль A, соединение формулы (III) представляет собой модуль B и соединение формулы (IV) представляет собой модуль C, в соответствии со следующей схемой реакции:
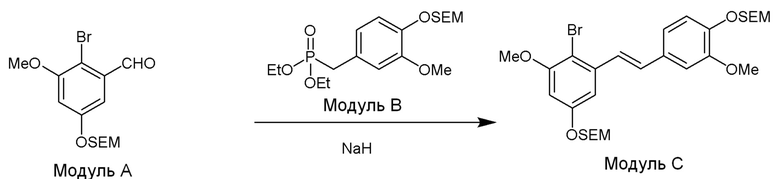
где
i) модуль C подвергают реакции сочетания с пинаколовым эфиром 3-метилбут-2-енилбороновой кислоты в присутствии тетракис-(трифенилфосфин)палладия(0) и карбонат калия с получением соединения 1-1; и
ii) соединение 1-1 обрабатывают тетрабутиламмония фторидом с получением соединения 1,
в соответствии со следующей схемой реакции:
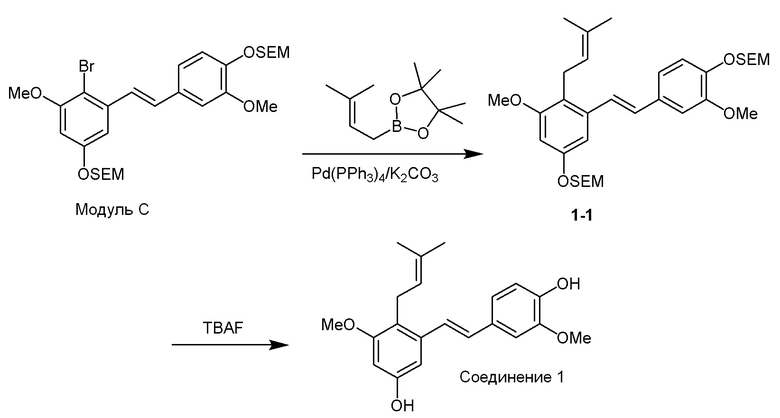
13. Соединение формулы (I)
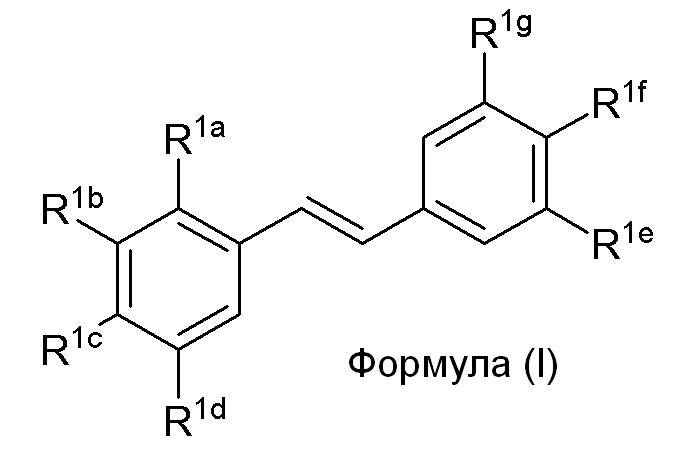
где:
R1a представляет собой аллил, кротил, пренил или бензил;
R1b представляет собой CF3, OR2 или NHR3;
R1c представляет собой H;
R1d представляет собой OH, NO2 или NHR3;
R1e представляет собой C1-C6 алкил, OH, OR2, NO2, NHR3, NMeR3 или SR2;
R1f представляет собой H, OH, NO2, NHR3, NHC=NH(NH2), COOH или N(OH)(CO)CH3;
R1g представляет собой H, C1-C6 алкил, OH или OR2;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил, COCOOR4 или O-защитную группу, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этоксиметила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой H, CH3, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, CO(CH2)nNH2, CONH2, CO(CH2)nCOOH или COCOOR4 (где n=0, 1 или 2);
R4 представляет собой C1-C6 алкил; и
только один из R1f или R1g может представлять собой H; или
R1e и R1f вместе образуют пятичленное гетероциклическое кольцо, содержащее карбонильную группу, когда R1e представляет собой OH, NH2 или NHMe, и R1f представляет собой NH2, так что азот в R1f соединен мостиковой связью через карбонильную группу с кислородом или азотом в R1e, и R1g представляет собой Н;
при условии, что:
i) когда R1a представляет собой пренил, R1d представляет собой OH, R1f представляет собой OH, R1g представляет собой H и R1e представляет собой OH или OR2, то тогда R1b не может представлять собой OR2, где R2=метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил или бензил; и
ii) R1b и R1d не являются одинаковыми.
14. Соединение формулы (I) по п. 13, где
R1a представляет собой аллил, пренил или бензил;
R1b представляет собой CF3 или OR2;
R1c представляет собой H;
R1d представляет собой OH, NH2 или NH(CO)H;
R1e представляет собой OH, OR2, NO2, NHR3 или SMe;
R1f представляет собой H, OH, NO2, NHR3, NHC=NH(NH2) или COOH;
R1g представляет собой H, C1-C6 алкил или OH;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил или трет-бутил; и
R3 представляет собой H, CH3, (CO)H, (CO)Me, SO2Me, CH2COOR2, COCH2NH2 или CO(CH2)nCOOH (где n=0, 1 или 2);
только один из R1f и R1g представляет собой H;
при условии, что
i) когда R1a представляет собой пренил, R1d представляет собой OH, R1f представляет собой OH, R1g представляет собой H, и R1e представляет собой OH или OR2, то тогда R1b не может представлять собой OR2, где R2=метил, этил, изопропил, пропил, бутил, изобутил или трет-бутил.
15. Соединение по п. 13, где соединение формулы (I) представляет собой соединение, выбранное из группы, состоящей из:
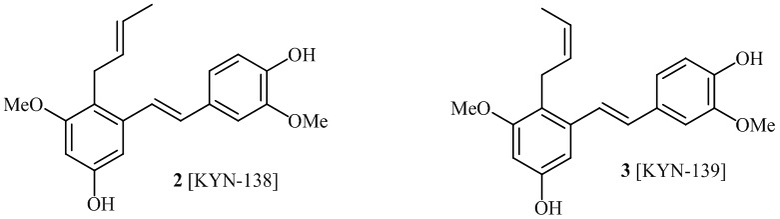
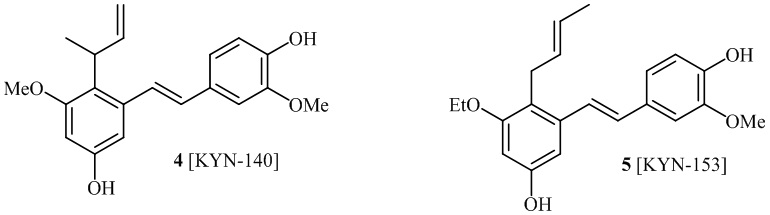
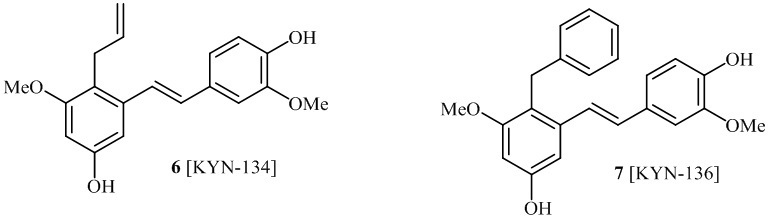
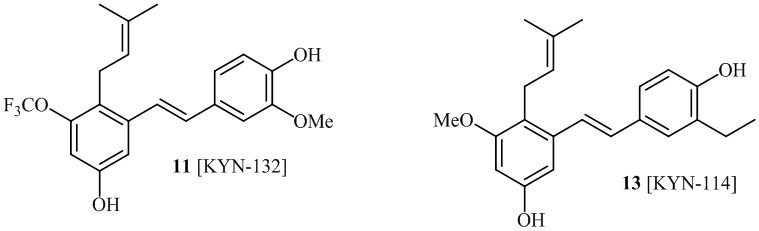
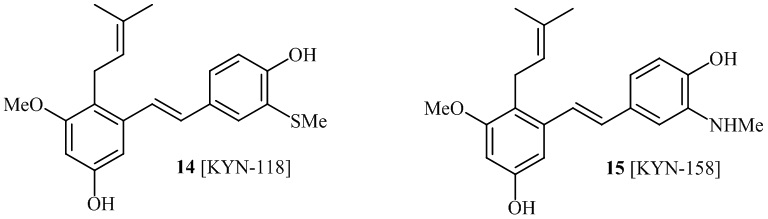
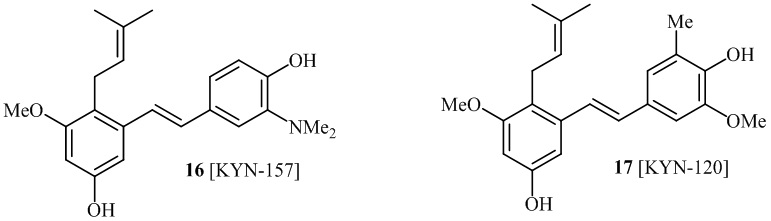
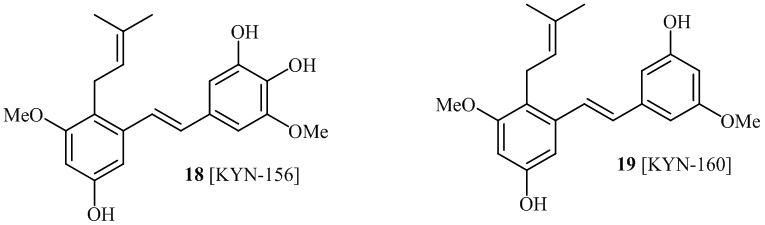
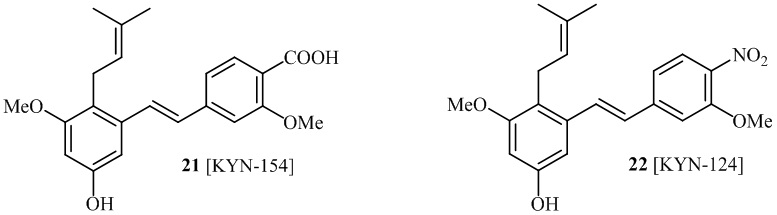
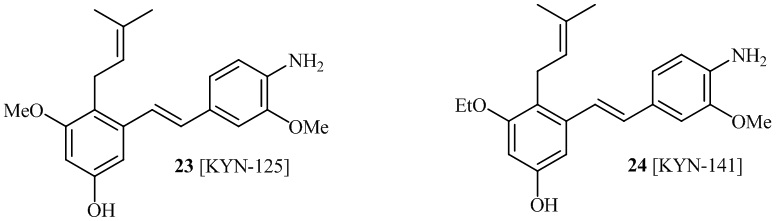
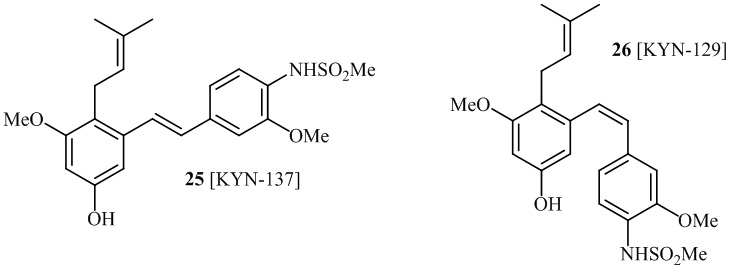
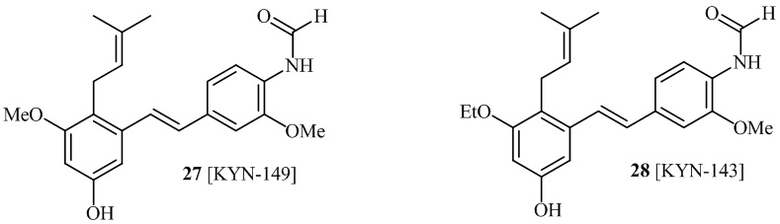
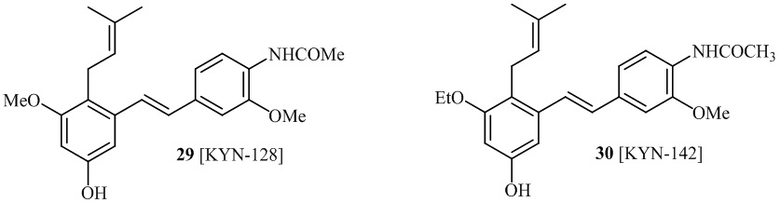
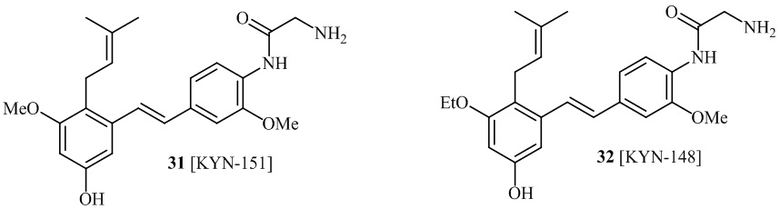
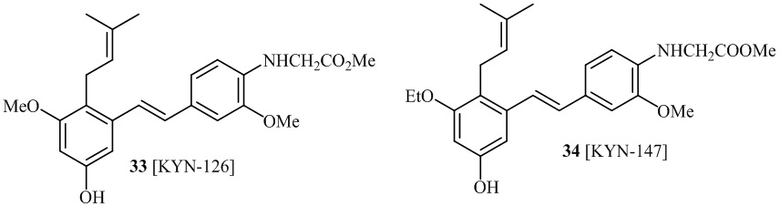
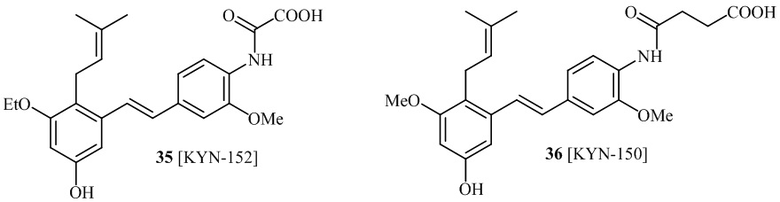
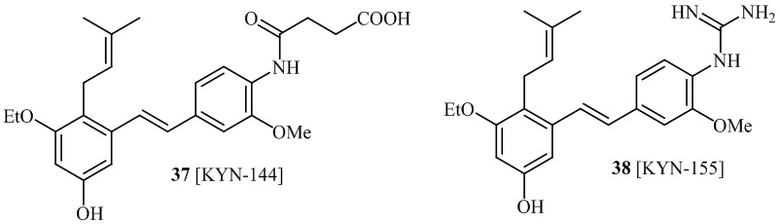
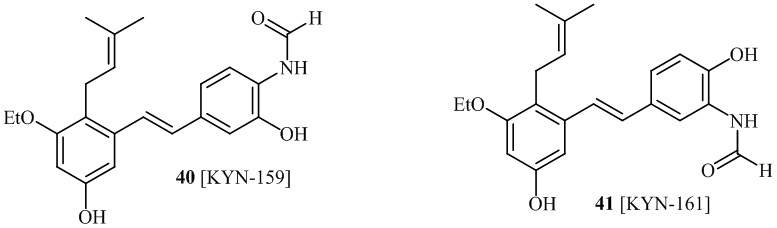
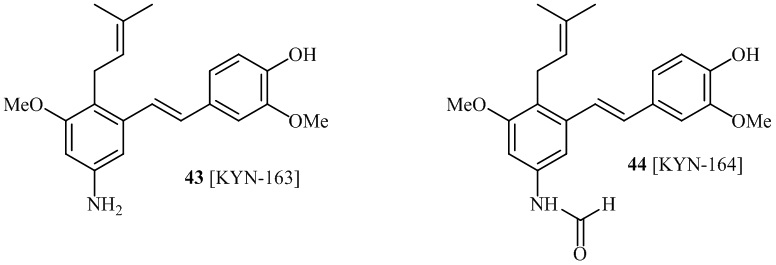
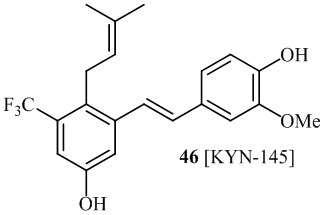 .
.
16. Соединение по п. 13, где в формуле (I)
R1a представляет собой, аллил, кротил, пренил или бензил;
R1b представляет собой CF3, OR2 или NHR3;
R1c представляет собой H;
R1d представляет собой OH, NO2 или NHR3;
R1e представляет собой C1-C6 алкил, OH, OR2, NO2, NHR3 или SR2;
R1f представляет собой H, OH, NO2, NHR3, NHC=NH(NH2) или COOH;
R1g представляет собой H, C1-C6 алкил, OH или OR2;
где по меньшей мере один из R1b или R1d представляет собой NHR3;
при этом R1b и R1d не являются одинаковыми;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, третбутил, трифторметил, COCOOR4 или O-защитную группу, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этоксиметила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой H, CH3, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, CO(CH2)nNH2, CONH2, CO(CH2)nCOOH или COCOOR4 (где n=0, 1 или 2);
R4 представляет собой C1-C6 алкил; и
только один из R1f или R1g может представлять собой H.
17. Соединение по п. 16, где соединение формулы (I) представляет собой соединение, выбранное из группы, состоящей из:
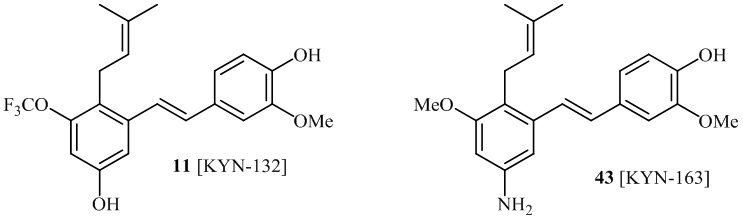
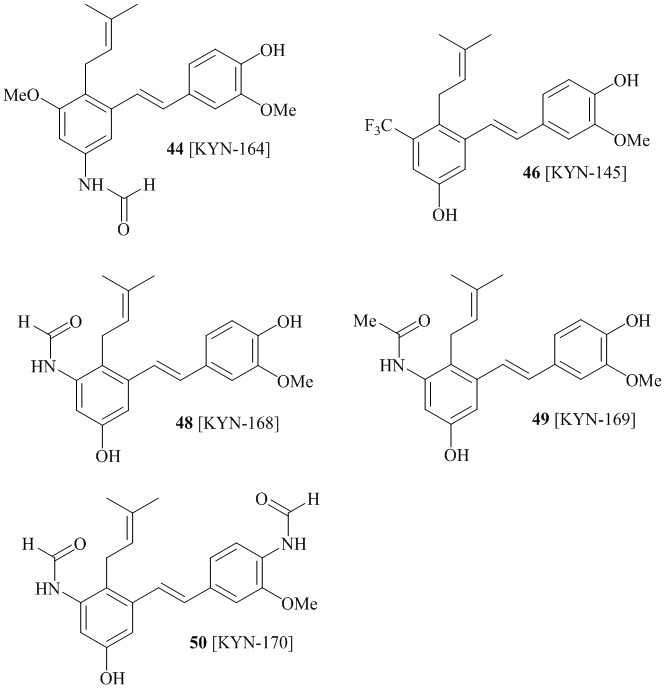
18. Соединение по п. 13, где в формуле (I)
R1a представляет собой аллил, кротил, пренил или бензил;
R1b представляет собой CF3, OR2 или NHR3;
R1c представляет собой H;
R1d представляет собой OH, NHR3, NO2;
R1e представляет собой OH или OR2;
R1f представляет собой NO2, NHR3, NHC=NH(NH2) или COOH;
R1g представляет собой H;
при этом R1b и R1d не являются одинаковыми;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этоксиметила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой H, CH3, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, COCH2NH2, CONH2, CO(CH2)nCOOH или CO(CH2)nCOOR4 (где n=0, 1 или 2); и
R4 представляет собой C1-C6 алкил.
19. Соединение по п. 18, где соединение формулы (I) представляет собой соединение, выбранное из группы, состоящей из:
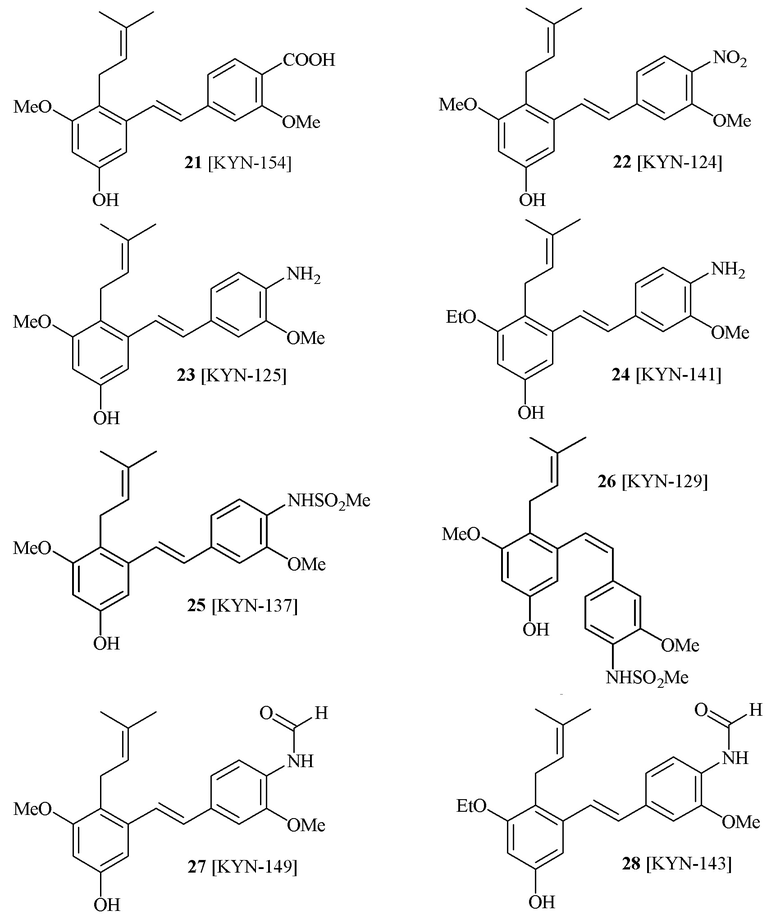
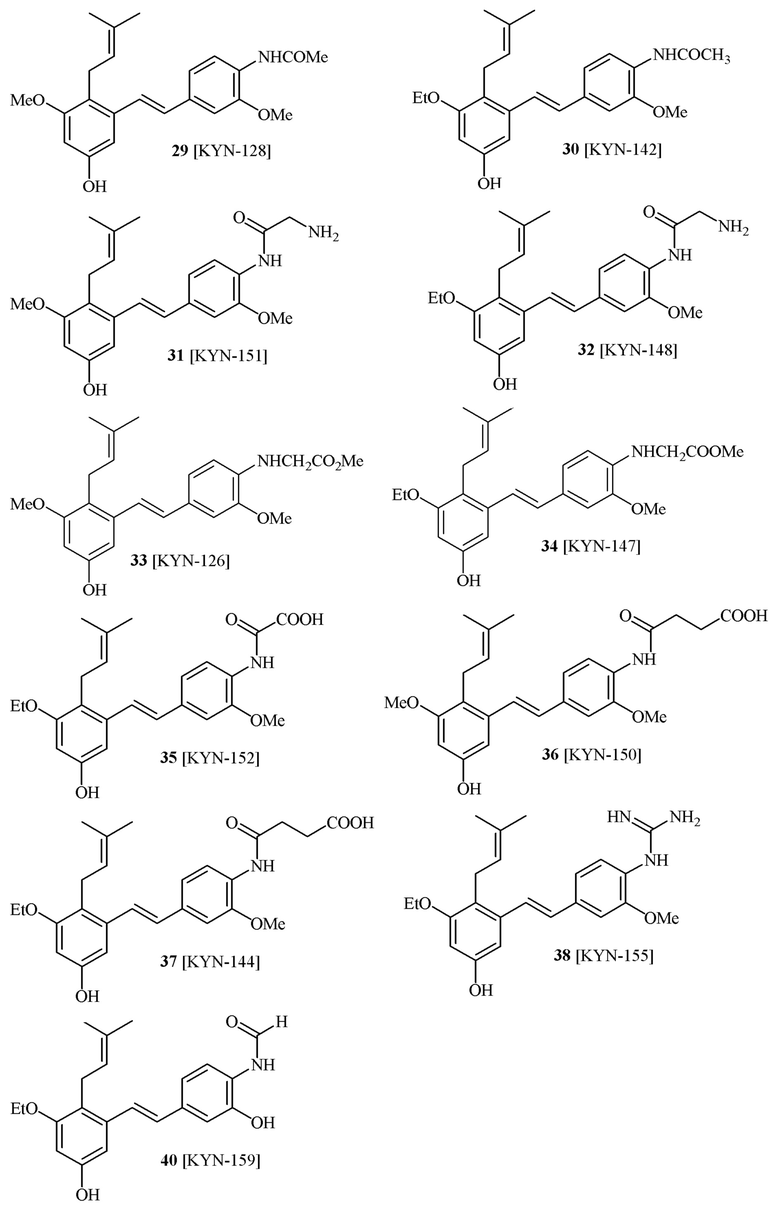
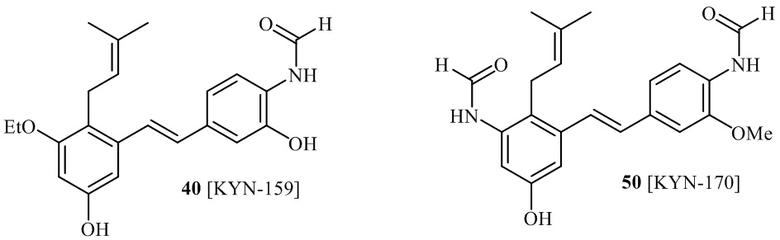
20. Соединение по п. 13, где в формуле (I)
R1a представляет собой аллил, кротил, пренил или бензил;
R1b представляет собой CF3, OR2 или NHR3;
R1c представляет собой H;
R1d представляет собой OH, NHR3 или NO2;
R1e представляет собой C1-C6 алкил, NO2, NHR3 или SR2;
R1f представляет собой OH;
R1g представляет собой H;
при этом R1b и R1d не являются одинаковыми;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил или O-защитную группу, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этокси]метила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой H, CH3, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, COCH2NH2, CONH2 или CO(CH2)nCOOH (где n=0, 1 или 2).
21. Соединение по п. 20, где соединение формулы (I) представляет собой соединение, выбранное из группы, состоящей из:
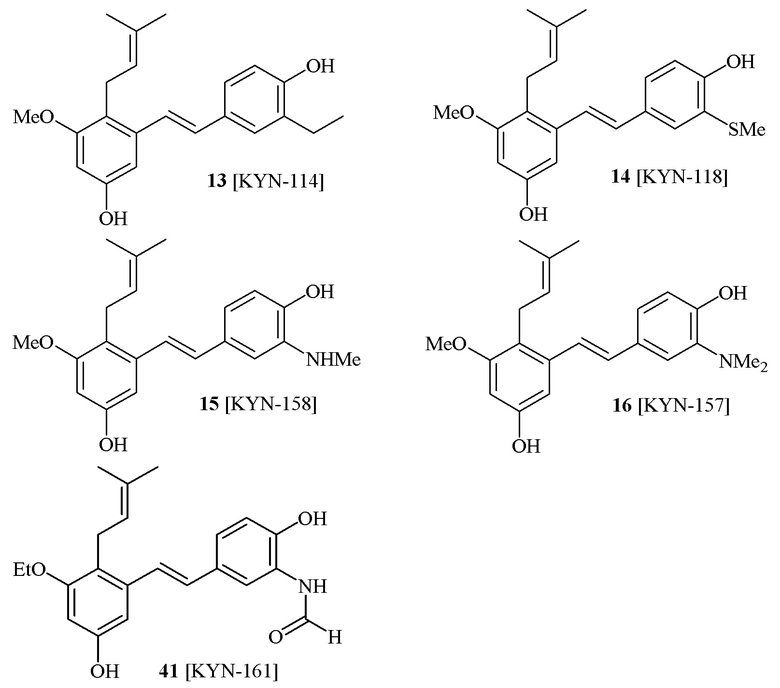
22. Соединение по п. 13, где в формуле (I)
R1a представляет собой аллил, кротил или бензил;
R1b представляет собой CF3, OR2 или NHR3;
R1c представляет собой H;
R1d представляет собой OH, NHR3 или NO2;
R1e представляет собой OH, OR2, NO2, NHR3 или SR2;
R1f представляет собой H, OH, NO2, NHR3, NHC=NH(NH2) или COOH;
R1g представляет собой H, C1-C6 алкил, OH или OR2;
при этом R1b и R1d не являются одинаковыми;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил, C(O)C(O)OR4 или O-защитную группу, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этокси]метила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой H, CH3, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, COCH2NH2, CONH2 или CO(CH2)nCOOH (где n=0, 1 или 2);
R4 представляет собой C1-C6 алкил; и
только один из R1f или R1g может представлять собой H.
23. Соединение формулы (I) по п. 13
R1a представляет собой аллил, кротил или бензил;
R1b представляет собой OR2;
R1c представляет собой H;
R1d представляет собой OH;
R1e представляет собой OR2;
R1f представляет собой OH;
R1g представляет собой H;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил, C(O)C(O)OR4 или O-защитную группу, где защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этокси]метила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2.
24. Соединение по п. 23, где соединение формулы (I) представляет собой соединение, выбранное из группы, состоящей из:
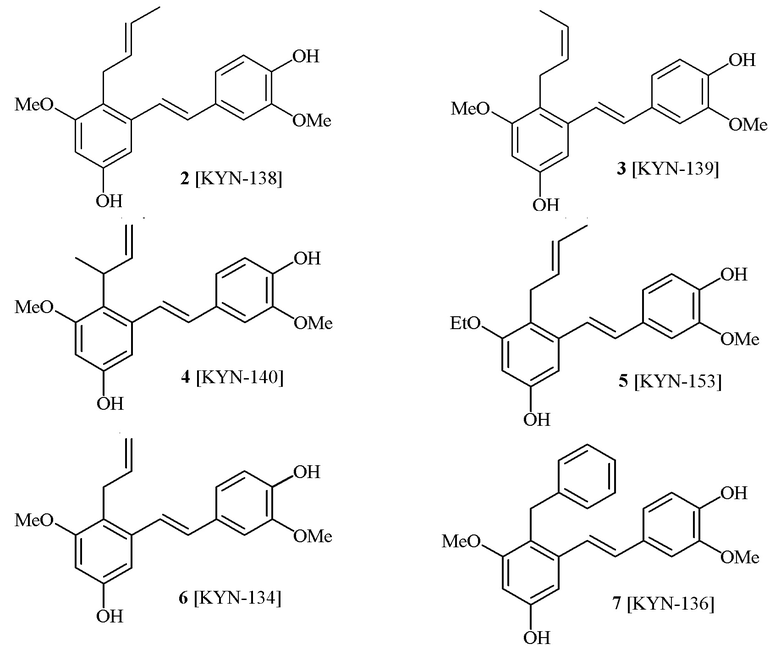
25. Соединение формулы (I) по п. 13, где соединение выбирают из группы, состоящей из:
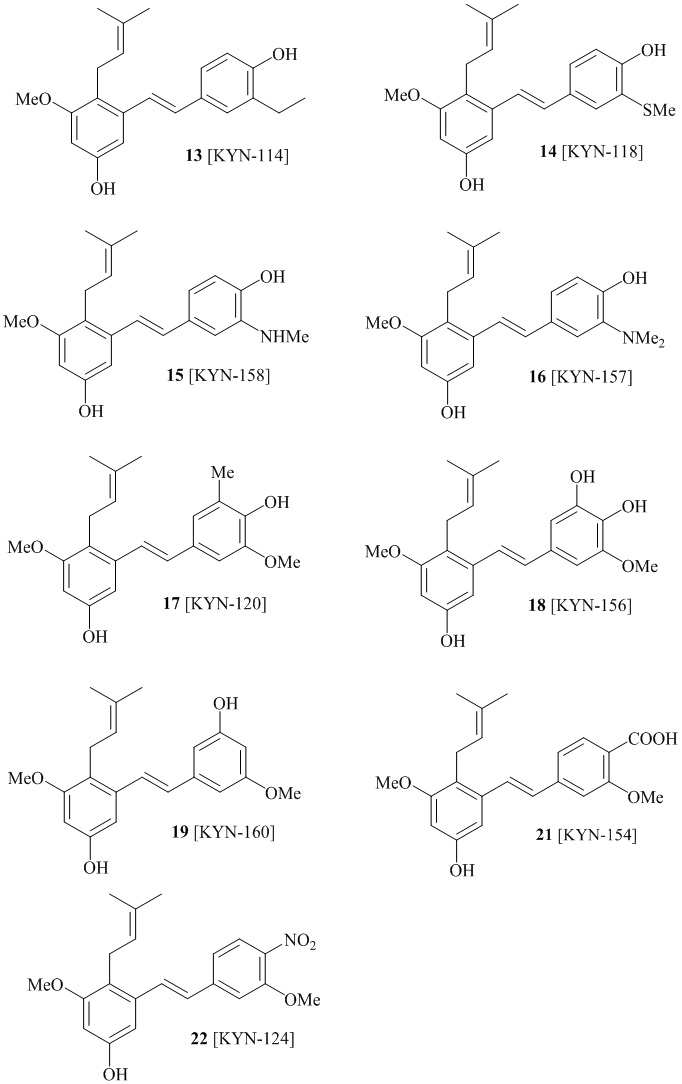
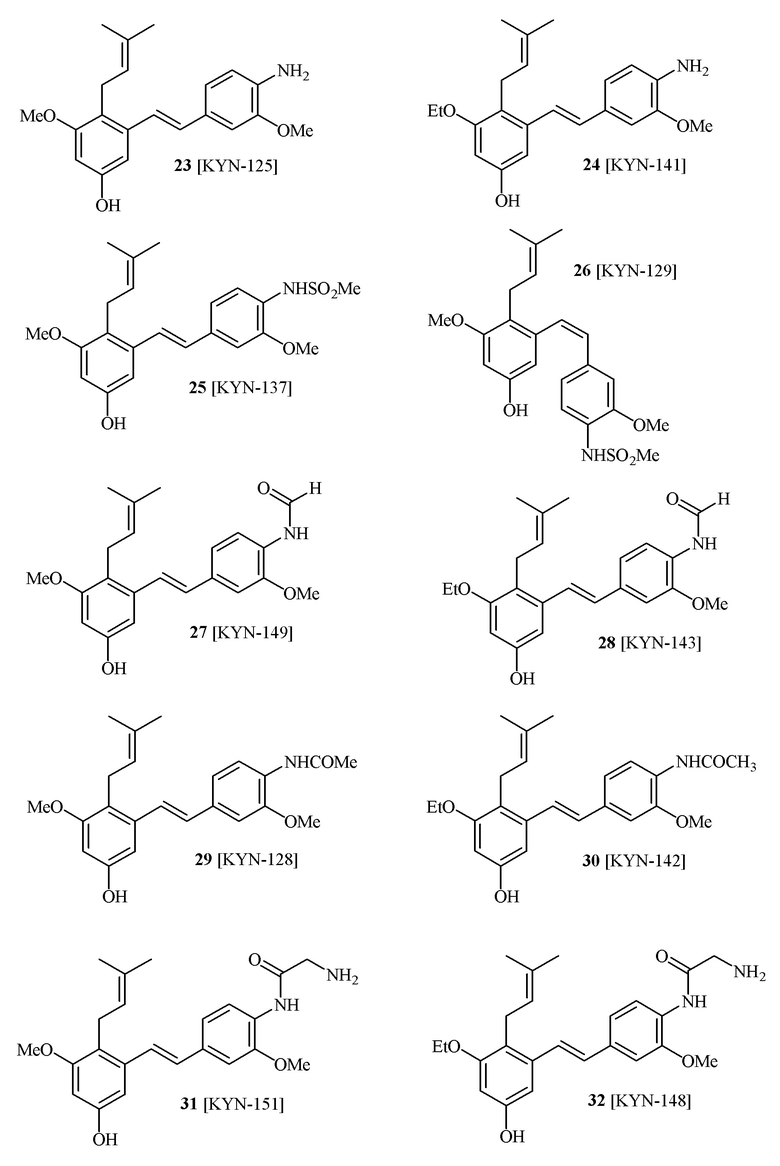
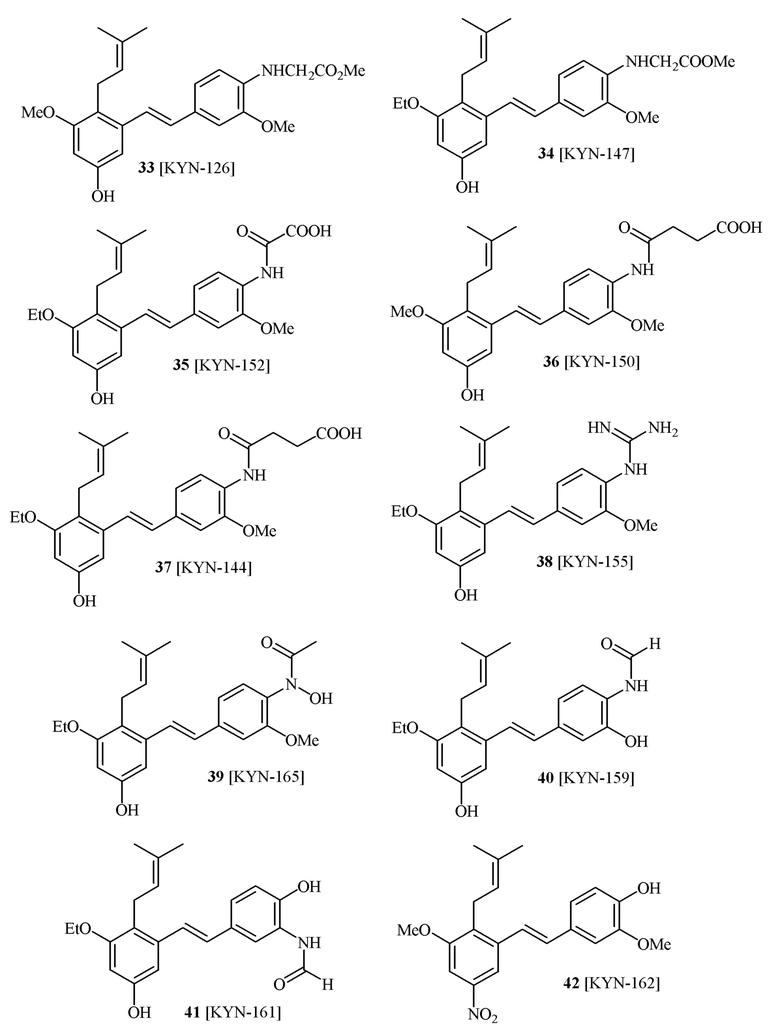
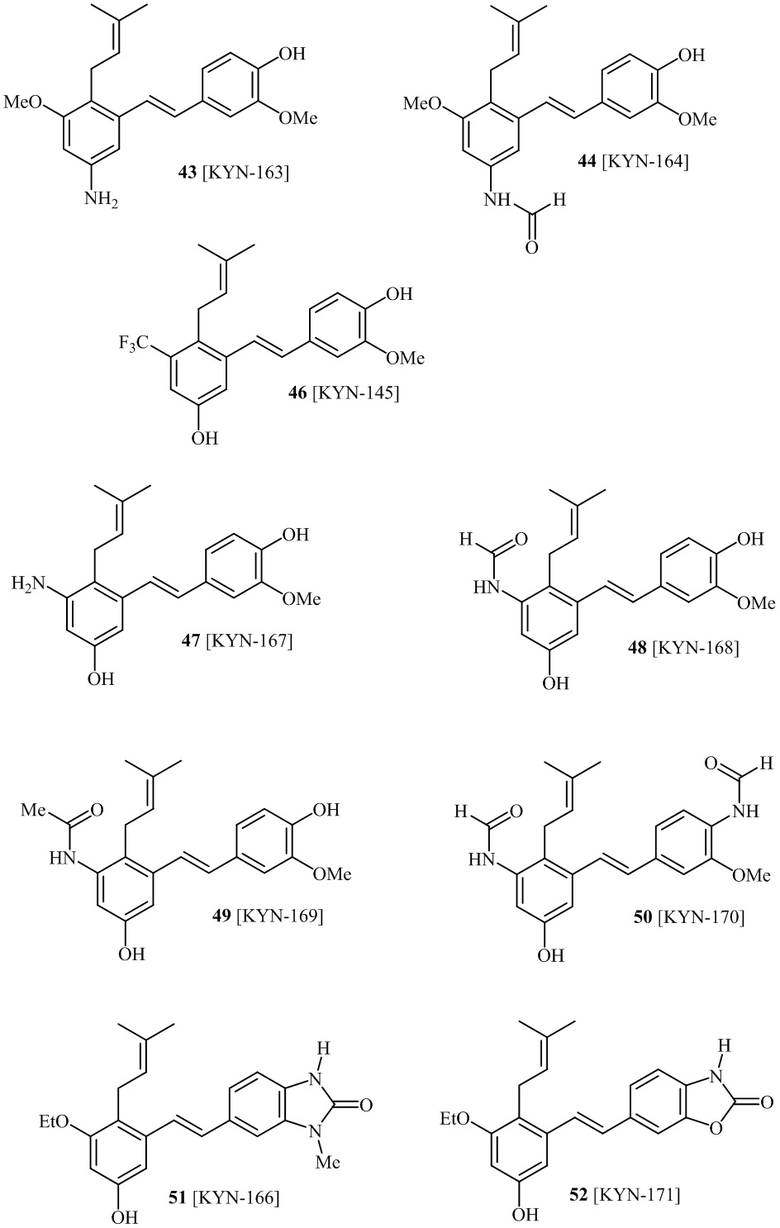
26. Соединение формулы (I) по п. 13, где
R1a представляет собой пренил;
R1b представляет собой OR2;
R1c представляет собой H;
R1d представляет собой OH;
R1e представляет собой OR2;
R1f представляет собой NHR3 или NHC=NH(NH2);
R1g представляет собой H;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил; и
R3 представляет собой H, CH3, (CO)H, (CO)Me, SO2Me, CH2COOR2, COCH2NH2 или CO(CH2)nCOOH (где n=0, 1 или 2).
27. Соединение формулы (I) по п. 13, где
R1a представляет собой пренил;
R1b представляет собой OR2;
R1c представляет собой H;
R1d представляет собой OH;
R1e представляет собой OR2;
R1f представляет собой OH;
R1g представляет собой C1-C6 алкил или OH;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил;
С1-С6 алкил представляет собой метил, этил, пропил, изопропил, бутил, изобутил или трет-бутил.
28. Соединение по п. 13, где в формуле (I)
R1a представляет собой аллил, кротил, пренил или бензил;
R1b представляет собой CF3, OR2 или NHR3;
R1c представляет собой H;
R1d представляет собой OH, NO2 или NHR3;
R1e представляет собой C1-C6 алкил, OH, OR2, NO2, NHR3 или SR2;
R1f представляет собой H, OH, NO2, NHR3, NHC=NH(NH2) или COOH;
R1g представляет собой H, С1-С6 алкил, CF3, OH или OR2;
при этом R1b и R1d не являются одинаковыми;
R2 представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, трифторметил, COCOOR4 или O-защитную группу, где O-защитную группу выбирают из группы, состоящей из COMe, t-BuSi(CH3)2, 2-(триметилсилил)этокси]метила (SEM) ацеталя, CH(OEt)CH3, тетрагидропиранила или C(OEt)(CH3)2;
R3 представляет собой H, CH3, (CO)H, (CO)Me, SO2Me, CH2COOH, CH2COOR2, CO(CH2)nNH2, CONH2, CO(CH2)nCOOH или COCOOR4 (где n=0, 1 или 2);
R4 представляет собой C1-C6 алкил; и
только один из R1f или R1g может представлять собой H;
где, по меньшей мере, один из R1e или R1f представляет собой - NHC(O)H или NHC(O)Me.
29. Соединение формулы:
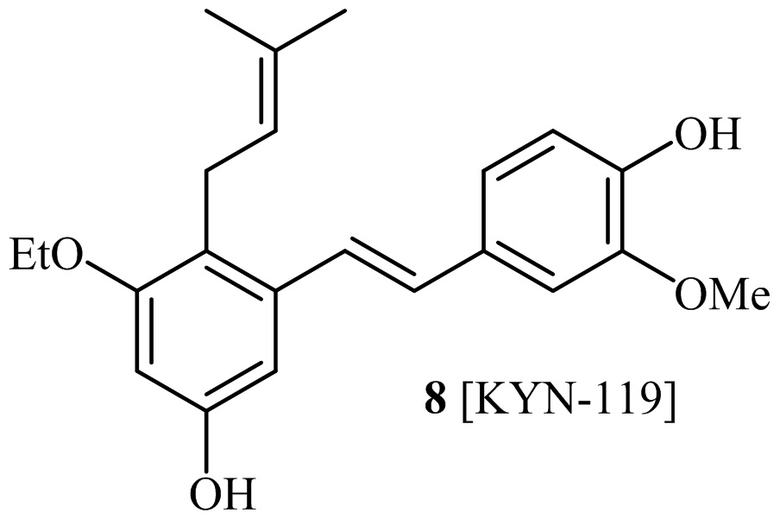 .
.
30. Соединение формулы:
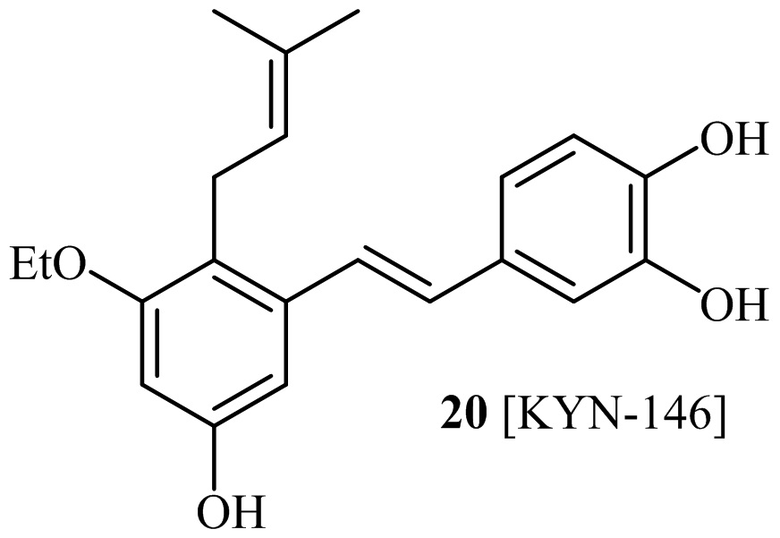 .
.
31. Соединение формулы:
 .
.
32. Способ лечения рака, включающий введение терапевтически эффективного количества соединения по любому одному из пп. 13-31 или фармацевтической композиции, включающей указанные соединения, пациенту, нуждающемуся в этом, где рак выбирают из группы, состоящей из рака кожи, включая меланому; рака поджелудочной железы, рака мочевого пузыря, рака почек, рака толстой кишки, рака молочной железы, рака легкого, рака печени и клеточного рака остеобластической саркомы.
33. Способ лечения заболеваний и нарушений кожи, включающий введение терапевтически эффективного количества соединения по любому одному из пп. 13-31 или фармацевтической композиции, включающей указанные соединения, пациенту, нуждающемуся в этом, где заболевания и нарушения кожи выбраны из атопического дерматита и псориаза.
| Jan P | |||
| Steynberg et al | |||
| Synthesis of Condensed Tannins | |||
| Способ использования делительного аппарата ровничных (чесальных) машин, предназначенных для мериносовой шерсти, с целью переработки на них грубых шерстей | 1921 |
|
SU18A1 |
| J | |||
| CHEM | |||
| SOC | |||
| PERKIN TRANS., 1978, 1, 1705-1712 | |||
| Alyssa M | |||
| Hartung et al | |||
| Stilbenes as κ Selective, Non-nitrogenous Opioid Receptor | |||

