Область изобретения
Настоящее изобретение относится к новым производным бензизоксазолсульфонамида, которые действуют как ингибиторы лизинацетилтрансферазы (KAT) семейства MYST и могут быть использованы для лечения у пациентов аномального роста клеток, такого как рак. Настоящее изобретение относится также к фармацевтическим композициям, содержащим соединения, и к способам применения соединений и композиций для лечения аномального роста клеток у пациентов.
Предпосылки изобретения
Семейство MYST является самым большим семейством KAT и названо в честь членов-основателей дрожжей и млекопитающих: MOZ, Ybf2/ Sas3, Sas2 и TIP60 (Dekker 2014). Белки MYST опосредуют многие биологические функции, включая регуляцию генов, репарацию ДНК, регуляцию и развитие клеточного цикла (Avvakumov 2007; Voss 2009). Белки KAT семейства MYST играют ключевую роль в посттрансляционной модификации гистонов и, таким образом, оказывают глубокое влияние на структуру хроматина в ядре эукариот (Avvakumov 2007). В настоящее время семейство включает пять KAT млекопитающих: TIP60 (KAT5; HTATIP; MIM 601409), MOZ (KAT6A; MIM 601408; MYST3), MORF (KAT6b; QKF; MYST4), HBO (KAT8; HBO1; MYST2) и MOF (KAT8; MYST1) (Voss 2009). Эти пять членов семейства MYST присутствуют у людей, и известно, что нарушение функционирования белков MYST связано с раком (Avvakumov 2007). Наиболее часто используемыми названиями членов семейства MYST являются следующие:
Функциональные домены MYS
Белки MYST функционируют в составе многосубъединичных белковых комплексов, включая адаптеры, такие как белки ING, которые опосредуют связывание ДНК (Avvakumov 2007). Например, TIP60 связан с мультипротеиновым комплексом NuA4 (который включает более 16 членов) (Zhang 2017). Однако были также некоторые сообщения о ДНК-связывающем мотиве спираль-поворот-спираль в структуре самого белка MOZ (Holbert 2007), что указывает на способность связываться непосредственно с ДНК.
На ацетилтрансферазную активность белков MYST влияет домен MYST (каталитический домен). Домен MYST содержит мотив связывания ацетил-кофермента А, который структурно консервативен с другими HAT, и необычный цинковый палец C2HC-типа (Voss 2009). Высококонсервативный домен MYST, включая ацетил-CoA-связывающий мотив и цинковый палец, считается определяющим признаком этого семейства ферментов (Avvakumov 2007).
Роль белков MYST
Ацетилирование остатков гистонов обычно связано с активацией транскрипции. Однако в некоторых случаях репрессия транскрипции также приписывается белкам MYST (Voss 2009). Известно, что отдельные члены семейства MYST участвуют в широком спектре важных биохимических взаимодействий:
HBO1 положительно регулирует инициацию репликации ДНК (Avvakumov 2007; Aggarwal 2004; Doyon 2006; Iizuka 2006) посредством ацетилирования гистоновых субстратов, что предположительно приводит к более доступной конформации хроматина (Avvakumov 2007, Iizuka 2006). Также известно, что HBO1 играет роль в патогенезе рака молочной железы, способствуя обогащению раковых стволовых клеток (Duong 2013) и дестабилизируя рецептор эстрогена α (ERα) посредством убихинициации, которая происходит за счет гистон-ацетилирующей активности HBO1 (Iizuka 2013). HBO1 также участвует в развитии острого миелоидного лейкоза (AML) (Shi 2015).
TIP60 (KAT5) является наиболее изученным членом семейства MYST. TIP60 играет важную роль не только в регуляции транскрипции, но и в процессе репарации повреждений ДНК, особенно в двухцепочечных разрывах ДНК (DSB) (Gil 2017). TIP60 может ацетилировать p53, ATM и c-Myc. TIP60 и MOF специфически ацетилируют лизин 120 (K120) р53 при повреждении ДНК (Avvakumov 2007). TIP60 также играет важную роль в биологии регуляторных Т-клеток (Treg). FOXP3 является главным регулятором в развитии и функции Treg, и было показано, что ацетилирование FOXP3 с помощью TIP60 важно для активности FOXP3 (Li 2007, Xiao 2014). В подтверждение этого, условная делеция TIP60 у мышей ведет к фатальному аутоиммунному заболеванию, подобному налету, имитирующему фенотип, наблюдаемый у мышей, нокаутированных по FOXP3 (Xiao 2014). При раке Treg-клетки могут способствовать прогрессированию опухоли, подавляя адаптивный иммунитет против опухоли.
MOF («самцы, отсутствующие на первом») первоначально был идентифицирован как один из компонентов дозовой компенсации у дрозофилы и был классифицирован как член семейства MYST на основании функциональных исследований и анализа последовательности (Su 2016). Ортолог человека обнаруживает значительное сходство с MOF дрозофилы; содержащий ацетил-CoA-связывающий сайт, хромодомен (который связывает гистоны) и цинковый палец C2HC-типа (Su 2016). MOF является ключевым ферментом для ацетилирования гистона H4K16, а комплексы, содержащие MOF, участвуют в различных основных клеточных функциях, связанных с раком (Su 2016). Помимо глобального снижения ацетилирования гистонов, истощение MOF в клетках млекопитающих может привести к аномальной транскрипции генов, в частности, вызывая аномальную экспрессию определенных генов-супрессоров опухолей или онкогенов, что указывает на критическую роль MOF в онкогенезе (Su 2016). Например, было показано, что KAT-активность MOF необходима для поддержания лейкемии MLL-AF9 и может быть важна для нескольких подтипов AML (Valerio 2017).
KAT6B (Querkopf) впервые был идентифицирован при скрининге мутаций генов, регулирующих баланс между пролиферацией и дифференцировкой во время эмбрионального развития (Thomas 2000). Мыши, гомозиготные по мутантному аллелю KAT6B, имеют серьезные дефекты в развитии коры головного мозга, возникающие в результате серьезного снижения как пролиферации, так и дифференцировки, в частности, популяции корковых предшественников во время эмбрионального развития. KAT6B необходим для поддержания популяции взрослых нервных стволовых клеток и является частью системы, регулирующей дифференцировку стволовых клеток в нейроны (Merson 2006). KAT6B также мутирует при редких формах лейкемии (Vizmanos 2003).
Локус MOZ занимает 12-е место среди наиболее часто амплифицируемых областей среди всех типов рака (Zack 2013). MOZ находится внутри ампликона 8p11-p12, что наблюдается с частотой около 10-15% при различных формах рака, особенно рака молочной железы и яичников (Turner-Ivey 2014). MOZ был впервые идентифицирован как партнер слияния CREB-связывающего белка (CBP) во время исследования специфической хромосомной транслокации при остром миелоидном лейкозе (AML) (Avvakumov 2007; Borrow 1996). Активность MOZ KAT необходима для стимулирования экспрессии MEIS1 и HOXa9, белков, которые обычно сверхэкспрессируются при некоторых лимфомах и лейкозах. Наблюдается повышенная выживаемость MOZ+/- гетерозиготных мышей в трансгенной модели Eμ-Myc B-клеточной лимфомы, где потеря одного аллеля MOZ приводит к биологически значимому снижению уровней Meis1 и Hoxa9 в пре-B-клетках (Sheikh 2015).
Ингибиторы некоторых MYST являются известными. Например, сообщается о следующем производном анакардовой кислоты (Ghizzoni 2012) как ингибирующее TIP60 (IC50=74 мкМ) и MOF (IC50=47 мкМ):
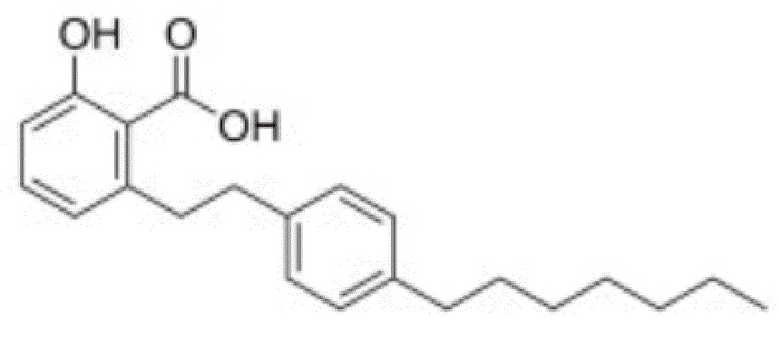
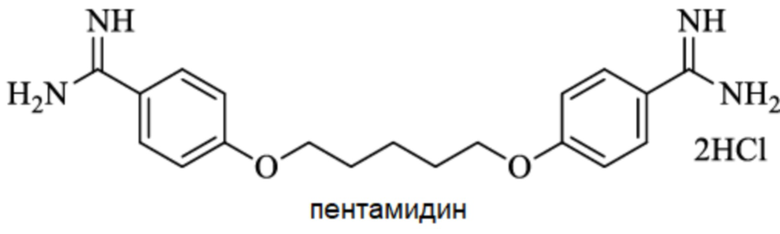
Другие известные ингибиторы включают (Zhang 2017):
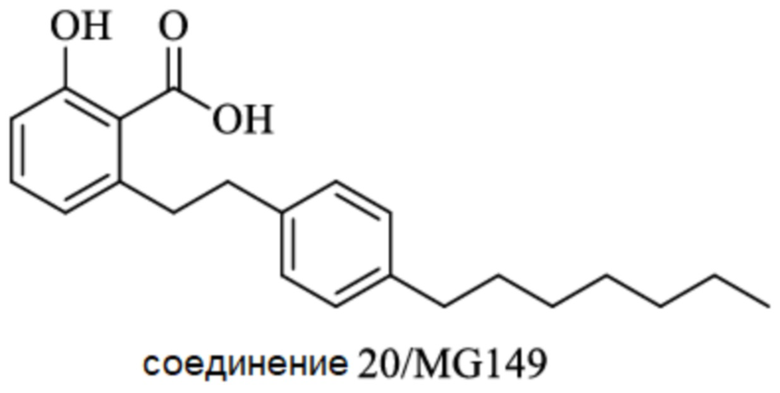
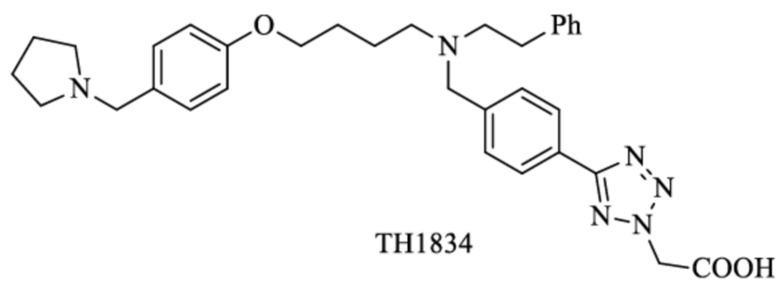
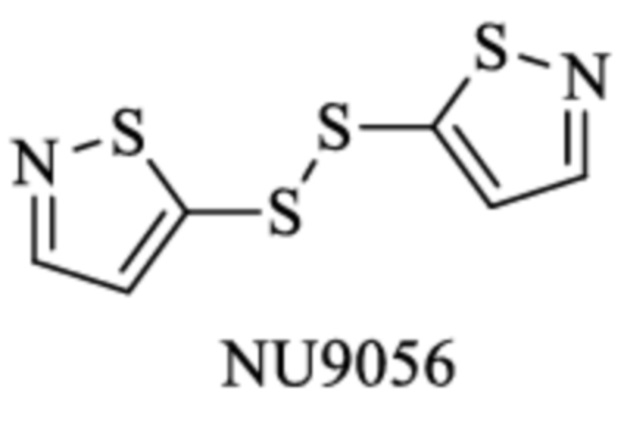
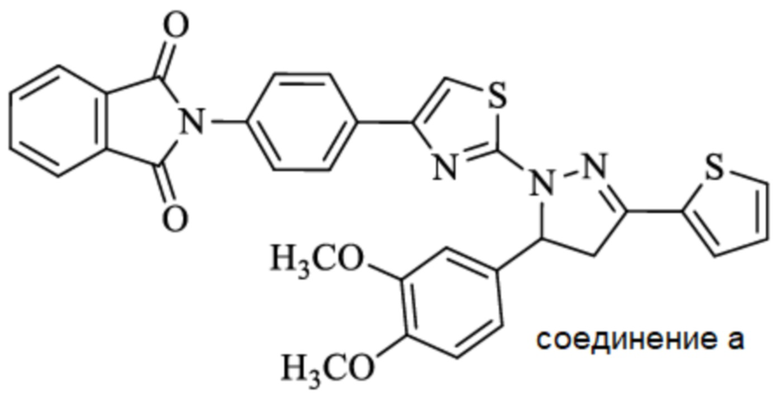
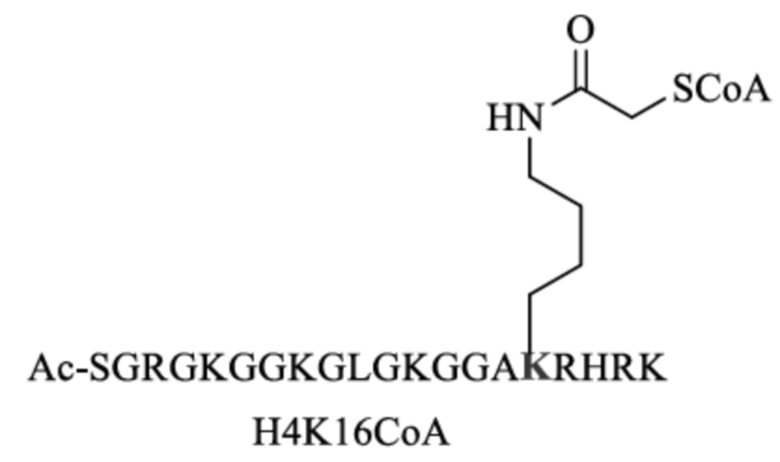
SGRGKGGKGLGKGGAKRHRK,
SEQ ID NO:1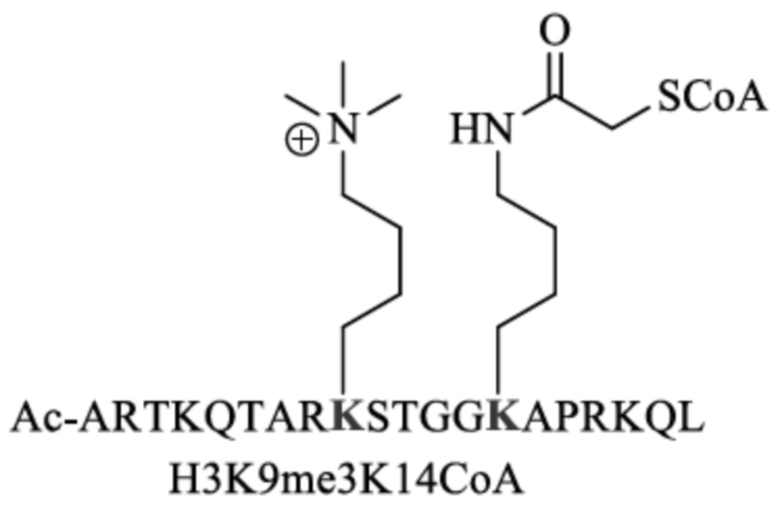
ARTKQTARKSTGGKAPRKQL,
SEQ ID NO:2
В свете установленной роли KAT в целом и MYST в частности при таких заболеваниях, как рак, существует потребность в новых ингибиторах этих белков.
Сущность изобретения
Каждый из вариантов осуществления настоящего изобретения, описанных ниже, может быть объединен с одним или несколькими другими вариантами осуществления настоящего изобретения, описанными в настоящем документе, что не противоречит вариантам осуществления, с которыми оно объединено. Кроме того, каждый из приведенных ниже вариантов осуществления, описывающих изобретение, предусматривает в пределах своего объема фармацевтически приемлемые соли соединений по изобретению. Соответственно, фраза «или его фармацевтически приемлемая соль» подразумевается при характеристике всех соединений, описанных в настоящем документе.
Данное изобретение относится к соединению формулы (I)
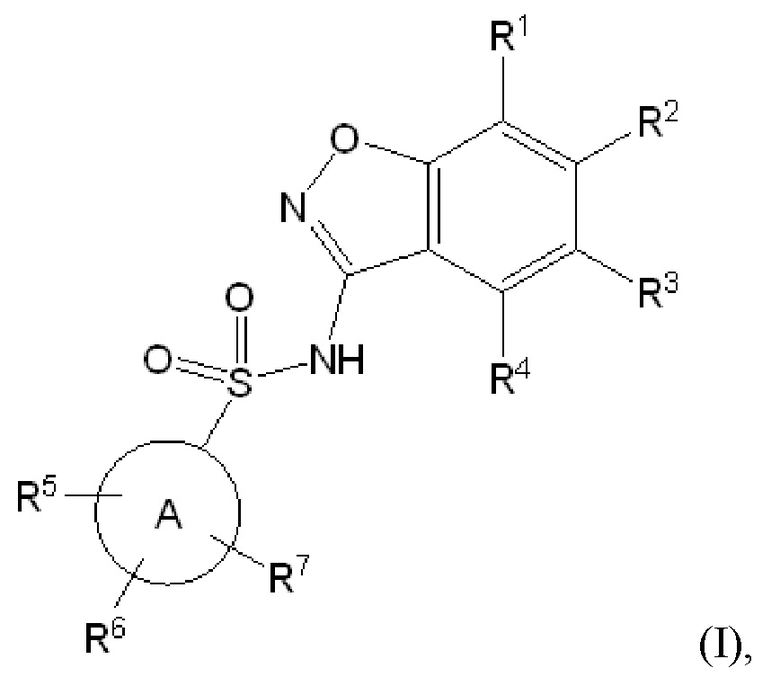
или его фармацевтически приемлемой соли,
где
R1 представляет собой водород или 5-6 членный гетероарил, необязательно замещенный метилом;
R2 представляет собой водород или -(CHR8)n-(5-9 членный гетероарил), необязательно замещенный галогеном, C1-C3 алкилом, -CH2OH или -OH,
при условии что один из R1 и R2 представляет собой водород,
и еще при условии, что R1 и R2 оба не представляют собой водород;
R3 представляет собой водород, галоген, C1-C3 алкил, циклопропил, -CHF2, -CF3, C1-C4 алкокси, -OCHF2 или -OCF3;
R4 представляет собой водород, галоген, C1-C3 алкил, циклопропил, C1-C4 алкокси или -O-циклопропил,
кольцо A представляет собой C6-C10 арил или 9-10 членный гетероарил;
R5 представляет собой водород, фтор, циано, C1-C3 алкил, -CHF2, -CF3, циклопропил, C1-C3 алкокси, -OCHF2, -OCF3, -O-циклопропил, -CH2-O-CH3, -C(O)OCH3 или -C(O)N(H)CH3;
R6 представляет собой водород, фтор, метил, -OH или метокси;
R7 представляет собой водород, бром, хлор, фтор или метокси;
R8 представляет собой водород или -OH; и
n обозначает 0 или 1.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где R1 представляет собой 5-6 членный гетероарил и R2 представляет собой водород; R1 представляет собой 5 членный гетероарил и R2 представляет собой водород; или R1 представляет собой пиразолил и R2 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где R1 представляет собой водород и R2 представляет собой 5-6 членный гетероарил; R1 представляет собой водород и R2 представляет собой 5 членный гетероарил; R1 представляет собой водород и R2 представляет собой пиразолил; R1 представляет собой водород, R2 представляет собой -(CHR8)-(5-6 членный гетероарил), необязательно замещенный галогеном, C1-C3 алкилом, -CH2OH или -OH, и R8 представляет собой -OH; R1 представляет собой водород и R2 представляет собой -(CH2)-(5-6 членный гетероарил), необязательно замещенный галогеном, C1-C3 алкилом, -CH2OH или -OH; R1 представляет собой водород, R2 представляет собой -(CHR8)-(5-6 членный гетероарил), и R8 представляет собой -OH; R1 представляет собой водород и R2 представляет собой -(CH2)-(5-6 членный гетероарил); R1 представляет собой водород, R2 представляет собой -(CHR8)-(5 членный гетероарил), необязательно замещенный галогеном, C1-C3 алкилом, -CH2OH или -OH, и R8 представляет собой -OH; R1 представляет собой водород и R2 представляет собой -(CH2)-(5 членный гетероарил), необязательно замещенный галогеном, C1-C3 алкилом, -CH2OH или -OH; R1 представляет собой водород, R2 представляет собой -(CHR8)-(5 членный гетероарил), и R8 представляет собой -OH; R1 представляет собой водород и R2 представляет собой -(CH2)-(5 членный гетероарил); R1 представляет собой водород и R2 представляет собой -(CH2)-триазолил; R1 представляет собой водород и R2 представляет собой -(CH2)-пиразолил, необязательно замещенный галогеном или C1-C3 алкил; R1 представляет собой водород и R2 представляет собой -(CH2)-пиразолил, необязательно замещенный галогеном; R1 представляет собой водород и R2 представляет собой -(CH2)-пиразолил, необязательно замещенный C1-C3 алкилом; R1 представляет собой водород и R2 представляет собой -(CH2)-пиразолил, замещенный метилом; R1 представляет собой водород и R2 представляет собой -(CH2)-пиразолил; R1 представляет собой водород и R2 представляет собой -(CH2)-(6 членный гетероарил); R1 представляет собой водород и R2 представляет собой -(CHR8)-(6 членный гетероарил), и R8 представляет собой -OH; R1 представляет собой водород и R2 представляет собой -(CH2)-пиридин, -(CH2)-пиразин или -(CH2)-пиримидин; R1 представляет собой водород и R2 представляет собой -(CH2)-(5-9 членный гетероарил); или R1 представляет собой водород и R2 представляет собой -(CH2)-индазолил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где R3 представляет собой галоген, C1-C3 алкил, циклопропил, -CHF2, -CF3, C1-C4 алкокси, -OCHF2 или -OCF3; и R4 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где R3 представляет собой водород и R4 представляет собой галоген, C1-C3 алкил, циклопропил, C1-C4 алкокси или -O-циклопропил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где R3 представляет собой водород, галоген или C1-C3 алкил; R3 представляет собой водород, фтор, бром или метил; R3 представляет собой фтор: R3 представляет собой метил; R3 представляет собой водород; R4 представляет собой водород, фтор, метил, этил, циклопропил, -O-циклопропил или C1-C4 алкокси; R4 представляет собой водород; R4 представляет собой C1-C3 алкокси; или R4 представляет собой метокси, и любую комбинацию R3 и R4.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где по меньшей мере один из R3 и R4 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где R3 представляет собой водород, галоген или C1-C3 алкил и R4 представляет собой водород; R3 представляет собой водород, фтор, бром или метил и R4 представляет собой водород; R3 представляет собой метил и R4 представляет собой водород; R3 представляет собой водород и R4 представляет собой водород, фтор, метил, этил, циклопропил, -O-циклопропил или C1-C4 алкокси; R3 представляет собой водород и R4 представляет собой водород; R3 представляет собой водород и R4 представляет собой C1-C3 алкокси; R3 представляет собой водород и R4 представляет собой метокси; или R3 представляет собой фтор и R4 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил, хинолинил, бензоксазолил, инданил или тетрагидронафтил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил и R5 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил и R6 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил и R6 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил и R7 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил, R5 представляет собой метокси и R6 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил, R5 представляет собой метокси, R6 представляет собой метокси и R7 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил, R5 представляет собой метокси, R6 представляет собой водород и R7 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли где кольцо A представляет собой инданил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где кольцо A представляет собой тетрагидронафтил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где кольцо A представляет собой инданил или тетрагидронафтил, R5 представляет собой метокси, R6 представляет собой водород и R7 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где кольцо A представляет собой хинолинил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где кольцо A представляет собой бензоксазолил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где кольцо A представляет собой хинолинил или бензоксазолил, R5 представляет собой метил или этил, R6 представляет собой водород и R7 представляет собой водород.
Следует понимать, что любой из вышеупомянутых вариантов осуществления формулы (I) может быть объединен с любыми другими вариантами осуществления, указанными выше, в той степени, в которой они не являются несовместимыми.
Данное изобретение относится к соединению формулы (Ia)
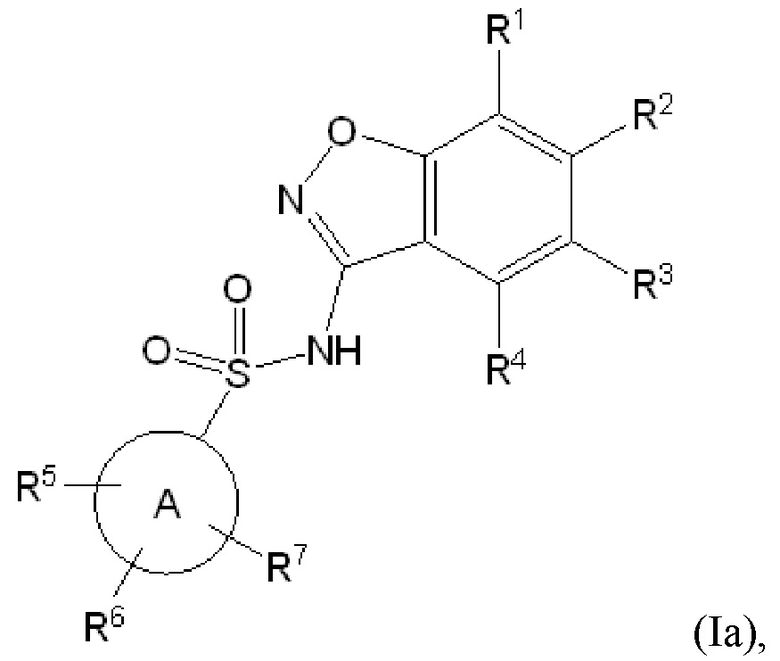
или его фармацевтически приемлемой соли,
где
R1 представляет собой водород или 5-6 членный гетероарил, необязательно замещенный метилом;
R2 представляет собой водород или -(CH2)n-(5-6 членный гетероарил), необязательно замещенный галогеном, C1-C3 алкилом, -CH2OH или -OH,
при условии что один из R1 и R2 представляет собой водород,
и еще при условии, что R1 и R2 оба не представляют собой водород;
R3 представляет собой водород, галоген, C1-C3 алкил, -CF2H, -CF3, C1-C4 алкокси, -OCHF2 или -OCF3;
R4 представляет собой водород, галоген, C1-C3 алкил, циклопропил, C1-C4 алкокси или -O-циклопропил,
при условии, что по меньшей мере один из R3 и R4 представляет собой водород;
кольцо A представляет собой C6-C10 арил или 9-10 членный гетероарил;
R5 представляет собой водород, фтор, циано, C1-C3 алкил, -CHF2, -CF3, циклопропил, C1-C3 алкокси, -OCHF2, -OCF3, -O-циклопропил, -CH2-O-CH3, -C(O)OCH3 или -C(O)N(H)CH3;
R6 представляет собой водород, фтор, метил, -OH или метокси;
R7 представляет собой водород, бром, хлор, фтор или метокси; и
n обозначает 0 или 1.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где R1 представляет собой 5-6 членный гетероарил и R2 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где R1 представляет собой 5 членный гетероарил и R2 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где R1 представляет собой пиразолил и R2 представляет собой водород; R1 представляет собой водород и R2 представляет собой 5-6 членный гетероарил; R1 представляет собой водород и R2 представляет собой 5 членный гетероарил; R1 представляет собой водород и R2 представляет собой пиразолил; R1 представляет собой водород и R2 представляет собой -(CH2)-(5-6 членный гетероарил), необязательно замещенный галогеном, C1-C3 алкилом, -CH2OH или -OH; R1 представляет собой водород и R2 представляет собой -(CH2)-(5-6 членный гетероарил); R1 представляет собой водород и R2 представляет собой -(CH2)-(5 членный гетероарил), необязательно замещенный галогеном, C1-C3 алкилом, -CH2OH или -OH; R1 представляет собой водород и R2 представляет собой -(CH2)-(5 членный гетероарил); R1 представляет собой водород и R2 представляет собой -(CH2)-триазолил; R1 представляет собой водород и R2 представляет собой -(CH2)-пиразолил, необязательно замещенный галогеном или C1-C3 алкил; R1 представляет собой водород и R2 представляет собой -(CH2)-пиразолил, необязательно замещенный галогеном; R1 представляет собой водород и R2 представляет собой -(CH2)-пиразолил, необязательно замещенный C1-C3 алкилом; R1 представляет собой водород и R2 представляет собой -(CH2)-пиразолил, замещенный метилом; или R1 представляет собой водород и R2 представляет собой -(CH2)-пиразолил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где R3 представляет собой галоген, C1-C3 алкил, -CF2H, -CF3, C1-C4 алкокси, -OCHF2 или -OCF3; и R4 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где R3 представляет собой водород и R4 представляет собой галоген, C1-C3 алкил, циклопропил, C1-C4 алкокси или -O-циклопропил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли, где по меньшей мере один из R3 и R4 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где R3 представляет собой водород, галоген или C1-C3 алкил и R4 представляет собой водород; R3 представляет собой водород, фтор, бром или метил и R4 представляет собой водород; R3 представляет собой метил и R4 представляет собой водород; R3 представляет собой водород и R4 представляет собой водород, фтор, метил, этил, циклопропил, -O-циклопропил или C1-C4 алкокси; R3 представляет собой водород и R4 представляет собой водород; R3 представляет собой водород и R4 представляет собой C1-C3 алкокси; или R3 представляет собой водород и R4 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил, хинолинил, бензоксазолил, инданил или тетрагидронафтил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил и R5 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил и R6 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил и R6 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил и R7 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил, R5 представляет собой метокси и R6 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил, R5 представляет собой метокси и R6 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил, R5 представляет собой метокси, R6 представляет собой метокси и R7 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил, R5 представляет собой метокси, R6 представляет собой водород и R7 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где кольцо A представляет собой инданил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где кольцо A представляет собой тетрагидронафтил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где кольцо A представляет собой инданил или тетрагидронафтил, R5 представляет собой метокси, R6 представляет собой водород и R7 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где кольцо A представляет собой хинолинил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где кольцо A представляет собой бензоксазолил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (Ia) или его фармацевтически приемлемой соли, где кольцо A представляет собой хинолинил или бензоксазолил, R5 представляет собой метил или этил, R6 представляет собой водород и R7 представляет собой водород.
Следует понимать, что любой из вышеупомянутых вариантов осуществления формулы (Ia) может быть объединен с любыми другими вариантами осуществления, указанными выше, в той степени, в которой они не являются несовместимыми.
Данное изобретение относится к соединению формулы (II)
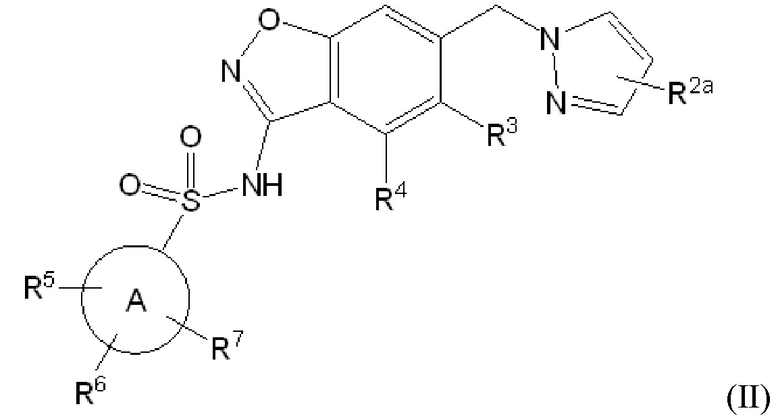
или его фармацевтически приемлемой соли,
где
R2a отсутствует или представляет собой галоген, C1-C3 алкил, -CH2OH или -OH;
R3 представляет собой водород, галоген, C1-C3 алкил, циклопропил, -CHF2, -CF3, C1-C4 алкокси, -OCHF2 или -OCF3;
R4 представляет собой водород, галоген, C1-C3 алкил, циклопропил, C1-C4 алкокси или -O-циклопропил;
кольцо A представляет собой C6-C10 арил или 9-10 членный гетероарил;
R5 представляет собой водород, фтор, циано, C1-C3 алкил, -CHF2, -CF3, циклопропил, C1-C3 алкокси, -OCHF2, -OCF3, -O-циклопропил, -CH2-O-CH3, -C(O)OCH3 или -C(O)N(H)CH3;
R6 представляет собой водород, фтор, метил, -OH или метокси; и
R7 представляет собой водород, бром, хлор, фтор или метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где R2a отсутствует или представляет собой фтор, метил, -CH2OH или -OH; R2a отсутствует или представляет собой фтор или метил; R2a отсутствует; R2a представляет собой фтор; или R2a представляет собой метил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где R3 представляет собой водород, галоген, C1-C3 алкил, -CHF2, -CF3, C1-C4 алкокси, -OCHF2 или -OCF3.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где R3 представляет собой водород, галоген или C1-C3 алкил; R3 представляет собой водород, фтор, бром или метил; R3 представляет собой фтор; R3 представляет собой метил; R4 представляет собой водород, фтор, метил, этил, циклопропил, -O-циклопропил или C1-C4 алкокси; R4 представляет собой водород: R4 представляет собой C1-C3 алкокси; или R4 представляет собой метокси, и любую комбинацию R3 и R4.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где R3 представляет собой фтор и R4 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где по меньшей мере один из R3 и R4 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где R3 представляет собой водород, галоген или C1-C3 алкил и R4 представляет собой водород; R3 представляет собой водород, фтор, бром или метил и R4 представляет собой водород; R3 представляет собой метил и R4 представляет собой водород; R3 представляет собой водород и R4 представляет собой водород, фтор, метил, этил, циклопропил, -O-циклопропил или C1-C4 алкокси; R3 представляет собой водород и R4 представляет собой водород; R3 представляет собой водород и R4 представляет собой C1-C3 алкокси; R3 представляет собой водород и R4 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил, хинолинил, бензоксазолил, инданил или тетрагидронафтил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил и R5 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил и R6 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил и R6 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил и R7 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил, R5 представляет собой метокси и R6 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил, R5 представляет собой метокси, R6 представляет собой метокси и R7 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где кольцо A представляет собой фенил, R5 представляет собой метокси, R6 представляет собой водород и R7 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где кольцо A представляет собой инданил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где кольцо A представляет собой тетрагидронафтил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где кольцо A представляет собой инданил или тетрагидронафтил, R5 представляет собой метокси, R6 представляет собой водород и R7 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где кольцо A представляет собой хинолинил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где кольцо A представляет собой бензоксазолил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (II) или его фармацевтически приемлемой соли, где кольцо A представляет собой хинолинил, R5 представляет собой метил или этил, R6 представляет собой водород и R7 представляет собой водород.
Следует понимать, что любой из вышеупомянутых вариантов осуществления формулы (II) может быть объединен с любыми другими вариантами осуществления, указанными выше, в той степени, в которой они не являются несовместимыми.
Данное изобретение относится к соединению формулы (III)
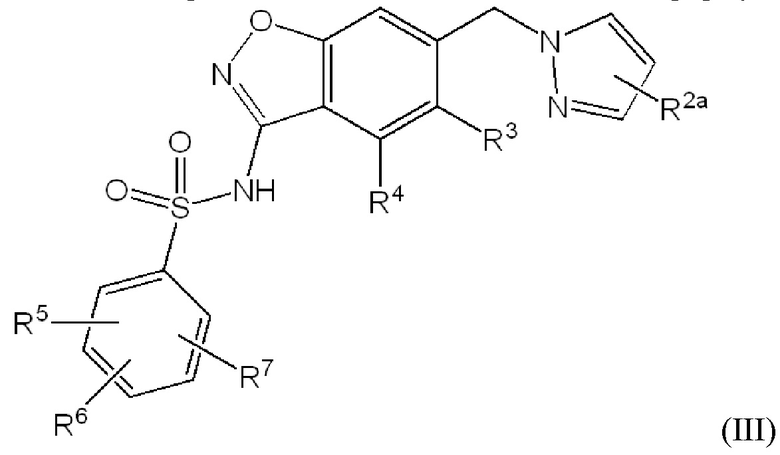
или его фармацевтически приемлемой соли,
где
R2a отсутствует или представляет собой галоген, C1-C3 алкил, -CH2OH или -OH;
R3 представляет собой водород, галоген, C1-C3 алкил, циклопропил, -CHF2, -CF3, C1-C4 алкокси, -OCHF2 или -OCF3;
R4 представляет собой водород, галоген, C1-C3 алкил, циклопропил, C1-C4 алкокси или -O-циклопропил;
R5 представляет собой водород, фтор, циано, C1-C3 алкил, -CHF2, -CF3, циклопропил, C1-C3 алкокси, -OCHF2, -OCF3, -O-циклопропил, -CH2-O-CH3, -C(O)OCH3 или -C(O)N(H)CH3;
R6 представляет собой водород, фтор, метил, -OH или метокси; и
R7 представляет собой водород, бром, хлор, фтор или метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (III) или его фармацевтически приемлемой соли, где R2a отсутствует или представляет собой фтор, метил, -CH2OH или -OH; R2a отсутствует или представляет собой фтор или метил; R2a отсутствует; R2a представляет собой фтор; или R2a представляет собой метил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (III) или его фармацевтически приемлемой соли, где R3 представляет собой водород, галоген или C1-C3 алкил; R3 представляет собой водород, фтор, бром или метил; R3 представляет собой фтор; R3 представляет собой метил; R4 представляет собой водород, фтор, метил, этил, циклопропил, -O-циклопропил или C1-C4 алкокси; R4 представляет собой водород; R4 представляет собой C1-C3 алкокси; R4 представляет собой метокси; или R3 представляет собой фтор и R4 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (III) или его фармацевтически приемлемой соли, где по меньшей мере один из R3 и R4 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (III) или его фармацевтически приемлемой соли, где R3 представляет собой водород, галоген или C1-C3 алкил и R4 представляет собой водород; R3 представляет собой водород, фтор, бром или метил и R4 представляет собой водород; R3 представляет собой метил и R4 представляет собой водород; R3 представляет собой водород и R4 представляет собой водород, фтор, метил, этил, циклопропил, -O-циклопропил или C1-C4 алкокси; R3 представляет собой водород и R4 представляет собой водород; R3 представляет собой водород и R4 представляет собой C1-C3 алкокси; или R3 представляет собой водород и R4 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (III) или его фармацевтически приемлемой соли, где R5 представляет собой метокси, R6 представляет собой метокси и R7 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (III) или его фармацевтически приемлемой соли, где R5 представляет собой метокси, R6 представляет собой водород и R7 представляет собой водород.
Следует понимать, что любой из вышеупомянутых вариантов осуществления формулы (III) может быть объединен с любыми другими вариантами осуществления, указанными выше, в той степени, в которой они не являются несовместимыми.
Данное изобретение относится к соединению формулы (IV)
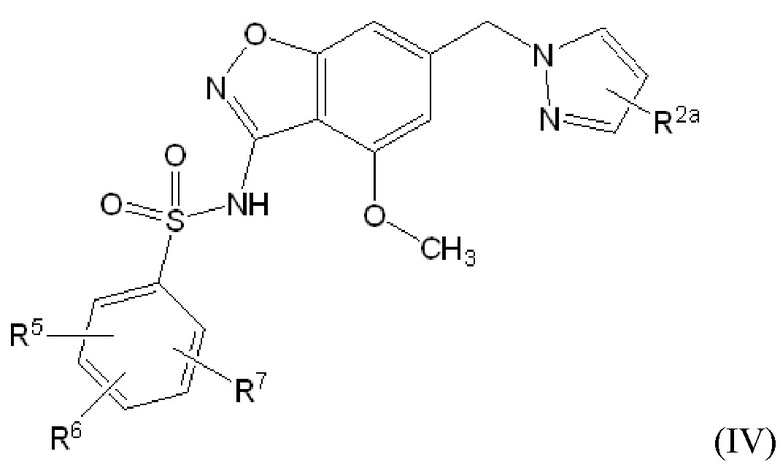
или его фармацевтически приемлемой соли,
где
R2a отсутствует или представляет собой галоген, C1-C3 алкил, -CH2OH или -OH;
R5 представляет собой водород, фтор, циано, C1-C3 алкил, -CHF2, -CF3, циклопропил, C1-C3 алкокси, -OCHF2, -OCF3, -O-циклопропил, -CH2-O-CH3, -C(O)OCH3 или -C(O)N(H)CH3;
R6 представляет собой водород, фтор, метил, -OH или метокси; и
R7 представляет собой водород, бром, хлор, фтор или метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (IV) или его фармацевтически приемлемой соли, где R2a отсутствует или представляет собой фтор, метил, -CH2OH или -OH; R2a отсутствует или представляет собой фтор или метил; R2a отсутствует; R2a представляет собой фтор; или R2a представляет собой метил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (IV) или его фармацевтически приемлемой соли, где R5 представляет собой метокси, R6 представляет собой метокси и R7 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (IV) или его фармацевтически приемлемой соли, где R5 представляет собой метокси, R6 представляет собой водород и R7 представляет собой водород.
Следует понимать, что любой из вышеупомянутых вариантов осуществления формулы (IV) может быть объединен с любыми другими вариантами осуществления, указанными выше, в той степени, в которой они не являются несовместимыми.
Данное изобретение относится к соединению формулы (V)
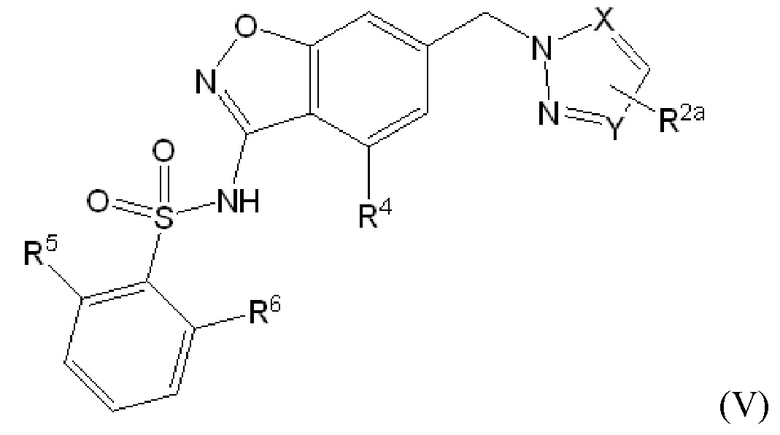
или его фармацевтически приемлемой соли,
где
X представляет собой N или -C(H)-;
Y представляет собой N или -C(H)-,
при условии, что по меньшей мере один из X и Y представляет собой -C(H)-;
R2a отсутствует или представляет собой галоген, C1-C3 алкил, -CH2OH или -OH;
R4 представляет собой водород, галоген, C1-C3 алкил, циклопропил, C1-C4 алкокси или -O-циклопропил;
R5 представляет собой водород, метил, -CF3, C1-C3 алкокси, -CH2-OCH3 или -C(O)OCH3; и
R6 представляет собой водород, фтор, метил, -OH или метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (V) или его фармацевтически приемлемой соли, где X представляет собой N и Y представляет собой -C(H)-.
Один вариант осуществления настоящего изобретения относится к соединению формулы (V) или его фармацевтически приемлемой соли, где X представляет собой -C(H)- и Y представляет собой N.
Один вариант осуществления настоящего изобретения относится к соединению формулы (V) или его фармацевтически приемлемой соли, где X представляет собой -C(H)- и Y представляет собой -C(H)-.
Один вариант осуществления настоящего изобретения относится к соединению формулы (V) или его фармацевтически приемлемой соли, где R2a отсутствует или представляет собой фтор, метил, -CH2OH или -OH; R2a отсутствует или представляет собой фтор или метил; R2a отсутствует; R2a представляет собой фтор; или R2a представляет собой метил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (V) или его фармацевтически приемлемой соли, где R4 представляет собой водород, фтор, этил, циклопропил, C1-C4 алкокси или -O-циклопропил; R4 представляет собой C1-C4 алкокси; R4 представляет собой метокси; или R4 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (V) или его фармацевтически приемлемой соли, где R5 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (V) или его фармацевтически приемлемой соли, где R6 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (V) или его фармацевтически приемлемой соли, где R6 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (V) или его фармацевтически приемлемой соли, где R5 представляет собой метокси и R6 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (V) или его фармацевтически приемлемой соли, где R5 представляет собой метокси и R6 представляет собой водород.
Следует понимать, что любой из вышеупомянутых вариантов осуществления формулы (V) может быть объединен с любыми другими вариантами осуществления, указанными выше, в той степени, в которой они не являются несовместимыми.
Данное изобретение относится к соединению формулы (VI)
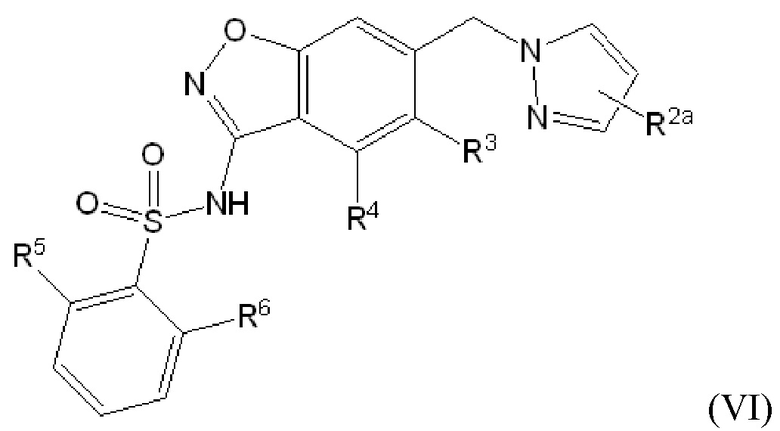
или его фармацевтически приемлемой соли,
где
R2a отсутствует или представляет собой галоген, C1-C3 алкил, -CH2OH или -OH;
R3 представляет собой водород, галоген или C1-C3 алкил;
R4 представляет собой водород, галоген, C1-C3 алкил, циклопропил, C1-C4 алкокси или -O-циклопропил,
при условии, что по меньшей мере один из R3 и R4 представляет собой водород;
R5 представляет собой водород, метил, -CH2-OCH3, -CF3, C1-C3 алкокси или -C(O)OCH3; и
R6 представляет собой водород, фтор, метил, -OH или метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (VI) или его фармацевтически приемлемой соли, где R2a отсутствует или представляет собой фтор, метил, -CH2OH или -OH; R2a отсутствует или представляет собой фтор или метил; R2a отсутствует; R2a представляет собой фтор; или R2a представляет собой метил.
Один вариант осуществления настоящего изобретения относится к соединению формулы (VI) или его фармацевтически приемлемой соли, где R3 представляет собой водород, фтор или метил и R4 представляет собой водород; R3 представляет собой водород и R4 представляет собой водород; R3 представляет собой метил и R4 представляет собой водород; R3 представляет собой водород и R4 представляет собой водород, фтор, этил, циклопропил, C1-C4 алкокси или -O-циклопропил; R3 представляет собой водород и R4 представляет собой C1-C4 алкокси; или R3 представляет собой водород и R4 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (VI) или его фармацевтически приемлемой соли, где R5 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (VI) или его фармацевтически приемлемой соли, где R6 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (VI) или его фармацевтически приемлемой соли, где R6 представляет собой водород.
Один вариант осуществления настоящего изобретения относится к соединению формулы (VI) или его фармацевтически приемлемой соли, где R5 представляет собой метокси и R6 представляет собой метокси.
Один вариант осуществления настоящего изобретения относится к соединению формулы (VI) или его фармацевтически приемлемой соли, где R5 представляет собой метокси и R6 представляет собой водород.
Следует понимать, что любой из вышеупомянутых вариантов осуществления формулы (VI) может быть объединен с любыми другими вариантами осуществления, указанными выше, в той степени, в которой они не являются несовместимыми.
Один вариант осуществления настоящего изобретения относится к соединению, выбранному из группы, включающий соединения, представленные в примерах 1-133, включительно, или его фармацевтически приемлемой соли.
Данное изобретение относится к соединению по любому из вариантов соединений формулы (I), формулы (Ia), формулы (II), формулы (III), формулы (IV), формулы (V) или формулы (VI) или его фармацевтически приемлемой соли, которое мечено дейтерием.
Данное изобретение относится к фармацевтической композиции, содержащей соединение по любому из вариантов соединений формулы (I), формулы (Ia), формулы (II), формулы (III), формулы (IV), формулы (V) или формулы (VI) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или разбавитель.
Данное изобретение относится к фармацевтической композиции, содержащей соединение по любому из вариантов соединений формулы (I), формулы (Ia), формулы (II), формулы (III), формулы (IV), формулы (V) или формулы (VI) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или разбавитель для лечения рака.
Данное изобретение относится к способу лечения рака у пациента, включающий введение пациенту такого количества соединения по любому из вариантов соединений формулы (I), формулы (Ia), формулы (II), формулы (III), формулы (IV), формулы (V) или формулы (VI) или формулы (V) или его фармацевтически приемлемой соли, которое является эффективным при лечении рака.
Данное изобретение относится к соединению по любому из вариантов соединений формулы (I), формулы (Ia), формулы (II), формулы (III), формулы (IV), формулы (V) или формулы (VI) или его фармацевтически приемлемой соли для применения при лечении рака у пациента.
Данное изобретение относится к применению соединения по любому из вариантов соединений формулы (I), формулы (Ia), формулы (II), формулы (III), формулы (IV), формулы (V) или формулы (VI) или его фармацевтически приемлемой соли при изготовлении лекарственного препарата для лечения рака.
Данное изобретение относится к комбинация соединения по любому из вариантов соединений формулы (I), формулы (Ia), формулы (II), формулы (III), формулы (IV), формулы (V) или формулы (VI) или его фармацевтически приемлемой соли с противоопухолевым средством или с лучевой терапией для лечения рака.
Данное изобретение относится к комбинация соединения по любому из вариантов соединений формулы (I), формулы (Ia), формулы (II), формулы (III), формулы (IV), формулы (V) или формулы (VI) или его фармацевтически приемлемой соли с противоопухолевым средством для лечения рака.
В одном варианте осуществления настоящего изобретения рак представляет собой рак молочной железы.
В одном варианте осуществления настоящего изобретения рак представляет собой рак молочной железы, где рак молочной железы представляет собой ER положительный рак молочной железы.
Краткое описание фигур
На ФИГ. 1 показан спектр порошковой рентгеновской дифракции безводного 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (форма 1).
Подробное описание изобретения
Настоящее изобретение может быть легче понято при обращении к нижеследующему подробному описанию предпочтительных вариантов осуществления изобретения и включенных в него примеров. Следует учесть, что терминология, используемая в настоящем документе, предназначена только для описания конкретных вариантов осуществления и не предназначена для ограничения. Кроме того, следует понимать, что, если в настоящем документе не указано иное, терминология, используемая в настоящем документе, должна иметь ее традиционное значение, известное в соответствующей области.
Как используется в настоящем документе, форма единственного числа включает ссылку на множественное число, если не указано иное. Например, термин «заместитель» включает один или несколько заместителей.
Изобретение, описанное в настоящем документе, подходящим образом может быть осуществлено на практике в отсутствие какого-либо элемента(ов), конкретно не раскрытого в настоящем документе. Таким образом, например, в настоящем документе в каждом случае любой из терминов «содержащий», «состоящий по существу из» и «состоящий из» может быть заменен любым из двух других терминов.
Для удобства многие химические фрагменты и соединения представлены с использованием хорошо известных сокращений, включая, помимо прочего, Ac (ацетил), AcOH (уксусная кислота), AIBN (азобисизобутиронитрил), н-BuLi (н-бутиллитий), CN (циано), CPME (циклопентилметиловый эфир), DCM (дихлорметан или метиленхлорид), ацетон-d6 (дейтерированный ацетон), CDCl3 (дейтерированный хлороформ), ДМСО-d6 (дейтерированный диметилсульфоксид), метанол-d4 (дейтерированный метанол), D2O (дейтерированная вода), DIAD (диизопропилазодикарбоксилат), DMAP (N, N-диметилпиридин-4-амин), ДМФ (N, N-диметилформамид), ДМСО (диметилсульфоксид), dppf (1,1ʼ-бис(дифенилфосфино)ферроцен), dppp (1,3-бис(дифенилфосфино)пропан), Et (этил), этилацетат (EtOAc), EtOH (этанол), LDA (диизопропиламид лития), Me (метил), MeOH (метанол), MeCN (ацетонитрил), MeOAc (метилацетат), Ms (метансульфонил), MsCl (метансульфонилхлорид), MTBE (метил трет- бутиловый эфир), NADPH (никотинамидадениндинуклеотидфосфат), N/D (не определено); NaOMe (метоксид натрия), NaOtPn (трет-пентоксид натрия), Pd(OAc)2 (ацетат палладия(II)), PdCl2(dppf) или Pd(dppf)Cl2 (1,1ʼ-бис(дифенилфосфино)ферроцендихлорпалладий (II)), Pd(PPh3)4 (тетракис(трифенилфосфин)палладий(0)), пет. эфир (петролейный эфир), Ph (фенил), 2-PrOH (изопропанол, 2-пропанил), t-Bu (трет-бутил), TBAF (фторид тетра-н-бутиламмония), TBS (трет-бутилдиметилсилил), TMG (тетраметилгуанидин), TBSCl (трет-бутилдиметилсилил хлорид), TEA (триэтиламин), ТФУ (трифторуксусная кислота), ТГФ (тетрагидрофуран), TMEDA (тетраметилэтилендиамин) и X-Phos (2-дициклогексилфосфино-2ʼ,4ʼ,6ʼ-триизопропилбифенил).
Кроме того, ТСХ относится к тонкослойной хроматографии, ВЭЖХ относится к высокоэффективной жидкостной хроматографии, ЖХМС относится к жидкостной хроматографии-масс-спектрометрии и SFC (сверхкритическая жидкостная хроматография).
Другие сокращения: rt или Rt (время удерживания), мин (минута или минуты), ч (час или часы), КТ (комнатная температура), водн. (водный), насыщ. (насыщенный), экв или экв. (эквивалент(ы)).
Термин «галоген», как используется в настоящем документе, относится к атому фтора, хлора, брома или йода или обозначает фтор (F), хлор (Cl), бром (Br) или йод (I).
Термин «алкил», как используется в настоящем документе, относится к насыщенным одновалентным углеводородным радикалам, содержащим в некоторых вариантах от одного до шести или от одного до трех атомов углерода, имеющих линейные или разветвленные фрагменты. Термин «C1-C4 алкил» относится к алкильному радикалу, содержащему от одного до четырех атомов углерода, имеющему прямые или разветвленные фрагменты. Термин «C1-C4 алкил» включает в свое определение термин «C1-C3 алкил». Примеры алкильных групп включают, но не ограничиваются ими, метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил.
Термин «алкокси», как используется в настоящем документе, относится к алкильному радикалу, который связан прямой связью с атомом кислорода. Точка присоединения алкокси радикала к молекуле осуществляется через атом кислорода. Алкокси радикал может быть обозначен как алкил-O-. Термины «C1-C4 алкокси» и «C1-C3 алкокси», относятся к алкокси радикалу, содержащему от одного до четырех атомов углерода и от одного до трех атомов углерода, соответственно, с линейными или разветвленными фрагментами. Алкокси группы включают, но не ограничиваются ими, метокси, этокси, пропокси, изопропокси, бутокси и тому подобное.
Термин «арил», как используется в настоящем документе, относится к циклической группе, производной от ароматического углеводорода. Термин «C6-C10 арил» включает от шести до десяти атомов углерода. Примеры таких групп включают, но не ограничиваются ими, фенил и нафтил. Термин «арил» также включает конденсированные полициклические ароматические кольцевые системы, в которых ароматическое кольцо конденсировано с одним или несколькими кольцами. Примеры включают, но не ограничиваются ими, 1-нафтил, 2-нафтил, 1-антрацил и 2-антрацил. Также в объем термина «арил», как он используется в настоящем документе, входит группа, в которой ароматическое кольцо конденсировано с одним или несколькими неароматическими кольцами, такими как инданил (2,3-дигидро-1H-инден) или тетрагидронафтил (также известный как 1,2,3,4-тетрагидронафтил), где радикал или точка присоединения находятся на ароматическом кольце.
Термин «гетероцикл», как используется в настоящем документе, относится к группе, производной от арильной группы, в которой по меньшей мере один из кольцевых атомов углерода заменен гетероатомом, выбранным из кислорода, азота и серы.
Термин «гетероарил», используемый в настоящем документе, относится к группе, производной от ароматического моноциклического или бициклического гетероцикла, и, в частности, в отношении бициклического гетероцикла, к бензоконденсированной гетероциклической группе, в которой ароматический или неароматический гетероцикл конденсирован с фенильной группой. Как используется в настоящем документе, термин «5 членный гетероарил» обозначает всего 5 атомов в своей кольцевой системе, термин «5-6-членный гетероарил» обозначает всего 5 или 6 атомов в своей кольцевой системе, а термин «5-9-членный гетероарил» обозначает всего 5, 6, 7, 8 или 9 атомов в своей кольцевой системе. Кроме того, каждая из групп «5-членный гетероарил», «5-6-членный гетероарил» и «5-9-членный гетероарил» имеет один, два или три гетероатома, независимо выбранных из азота и кислорода, при условии, что кольцевая система не содержит два соседних атома кислорода. Примеры включают, но не ограничиваются ими, пиразолил и триазолил. Как используется в настоящем документе, термин «9-10 членный гетероарил», обозначает всего 9 или 10 атомов в своей кольцевой системе и один или два гетероатома, каждый из которых независимо выбран из азота и кислорода, при условии, что кольцевая система не содержит два соседних атома кислорода.
Примеры «9-10-членного гетероарила» согласно настоящему изобретению включают, но не ограничиваются ими,
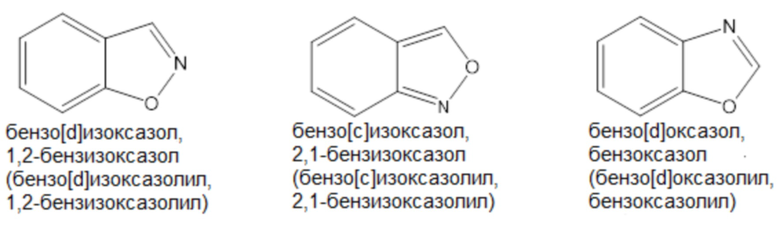
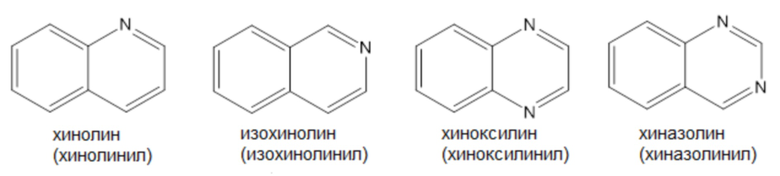
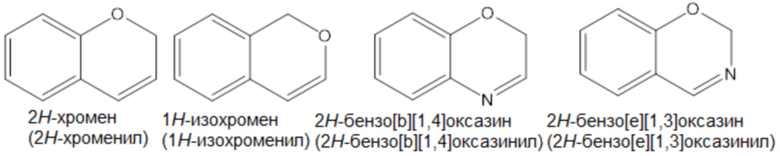
Термин «лечение», как используется в настоящем документе, если не указано иное, означает обращение, облегчение, подавление прогрессирования или предотвращение заболевания, расстройства или состояния, к которому применяется такой термин, или одного или нескольких симптомов такого заболевания, расстройства или состояния. Термин «лечение», как используется в настоящем документе, если не указано иное, относится к акту лечения, поскольку «лечение» определено непосредственно выше.
Термин «комбинация», как используется в настоящем документе, если не указано иное, означает комбинацию с фиксированной дозой или комбинацию агентов, которую вводят периодически, одновременно или последовательно в соответствии с тем же или другим путем введения. Как используется в настоящем документе, «эффективное» количество относится к количеству вещества, агента, соединения или композиции, которое является достаточным для уменьшения тяжести симптомов заболевания, увеличения частоты и продолжительности бессимптомных периодов заболевания или предотвращения ухудшения состояния или инвалидности из-за заболевания - либо в виде разовой дозы, либо в соответствии с режимом многократного приема, отдельно или в комбинации с другими агентами или веществами. Специалист в данной области сможет определить эти количества на основе таких факторов, как размер пациента, тяжесть симптомов пациента и выбранная конкретная комбинация, композиция или путь введения. Пациент или субъект может быть человеком или млекопитающим, не являющимся человеком, нуждающимся в лечении. В одном из вариантов осуществления изобретения пациентом является человек.
Если не указано иное, все ссылки в настоящем документе на соединения по изобретению включают ссылки на их соли, сольваты, гидраты и комплексы, а также на сольваты, гидраты и комплексы их солей, включая их полиморфы, стереоизомеры и их меченные изотопами версии.
Описанные в настоящем документе варианты осуществления включают меченные изотопами соединения, которые идентичны соединениям, указанным в формулах (I), (Ia) (II), (III), (IV), (V) или (VI), но с учетом того, что один или несколько атомов заменены атомом с атомной массой или массовым числом, отличным от атомной массы или массового числа, обычно встречающихся в природе. Примеры изотопов, которые могут быть включены в соединения описанных в настоящем документе вариантов осуществления изобретения, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как, но не ограничиваясь ими, 2H, 3H, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F и 36Cl, соответственно. В одном варианте изотоп, включенный в соединения формул (I), (Ia) (II), (III), (IV), (V) или (VI), представляет собой 2H. Соединения, описанные в настоящем документе, и фармацевтически приемлемые соли указанных соединений, которые содержат вышеупомянутые изотопы и/или другие изотопы других атомов, входят в объем настоящих вариантов осуществления изобретения. Некоторые меченные изотопами соединения вариантов осуществления изобретения, описанные в настоящем документе, например, те, в которые включены радиоактивные изотопы, такие как 3H и 14C, могут быть использованы в анализах распределения лекарственного средства и/или субстрата в тканях. Изотопы тритий, то есть 3H, и углерод-14, то есть 14C, особенно предпочтительны из-за простоты их получения и обнаружения. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, то есть 2H, может дать определенные терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличенным периодом полувыведения Inc vivo или уменьшенными требованиями к дозировке, и, следовательно, может быть предпочтительным в некоторых обстоятельствах. Меченные изотопами соединения вариантов осуществления изобретения, описанные в настоящем документе, обычно могут быть получены путем осуществления способов, представленных на схемах и/или в примерах ниже, путем замены легко доступного неизотопно-меченного реагента на изотопно-меченный реагент. В одном варианте осуществления изобретения соединения формул (I), (Ia) (II), (III), (IV), (V) или (VI) мечены дейтерием.
Некоторые варианты осуществления изобретения относятся к фармацевтически приемлемым солям соединений, описанным в настоящем документе. Соединения, описанные в настоящем документе, которые являются основными по природе, способны образовывать широкий спектр солей с различными неорганическими и органическими кислотами. Кислоты, которые можно использовать для получения фармацевтически приемлемых кислотно-аддитивных солей таких основных соединений, описанных в настоящем документе, являются такими, которые образуют нетоксичные кислотно-аддитивные соли, например, соли, содержащие фармакологически приемлемые анионы, такие как соли гидрохлорид, гидробромид, гидройодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, изоникотинат, ацетат, лактат, салицилат, цитрат, кислый цитрат, тартрат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат и памоат [то есть 1,1ʼ-метилен-бис-(2-гидрокси-3-нафтоат)]. Соединения, описанные в настоящем документе, которые включают основную составляющую, такую как аминогруппа, могут образовывать фармацевтически приемлемые соли с различными аминокислотами в дополнение к кислотам, упомянутым выше.
Также могут быть образованы полусоли кислот и оснований, например, гемисульфатные и гемикальциевые соли.
Обзор подходящих солей см. в справочнике Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley-VCH, 2002). Способы получения фармацевтически приемлемых солей соединений, описанных в настоящем документе, известны специалисту в данной области.
Термин «сольват» используется в настоящем документе для описания молекулярного комплекса, содержащего соединение, описанное в настоящем документе, и одну или несколько молекул фармацевтически приемлемого растворителя, например этанола.
Соединения, описанные в настоящем документе, также могут существовать в несольватированной и сольватированной формах. Соответственно, некоторые варианты осуществления изобретения относятся к гидратам и сольватам соединений, описанных в настоящем документе. Когда растворитель или вода связаны прочно, комплекс будет иметь четко определенную стехиометрию независимо от влажности. Однако, когда растворитель или вода связаны слабо, как в канальных сольватах и гигроскопичных соединениях, содержание воды/растворителя будет зависеть от влажности и условий сушки. В таких случаях нестехиометрия будет нормой. Термин «сольват» используется в настоящем документе для описания молекулярного комплекса, содержащего соединение по настоящему изобретению и одну или несколько фармацевтически приемлемых молекул растворителя, например этанола. Термин «гидрат» используется, когда растворителем является вода. Фармацевтически приемлемые сольваты в соответствии с изобретением включают гидраты и сольваты, в которых кристаллизационный растворитель может быть изотопно замещенным, например D2O, d6-ацетон, d6-ДМСО.
В объем изобретения также входят комплексы, такие как клатраты, комплексы включения лекарственного вещества-хозяина, в которых, в отличие от вышеупомянутых сольватов, лекарственное вещество и хозяин присутствуют в стехиометрических или нестехиометрических количествах. Также включены комплексы лекарственного вещества, содержащие два или более органических и/или неорганических компонента, которые могут находиться в стехиометрических или нестехиометрических количествах. Полученные комплексы могут быть ионизированными, частично ионизированными или неионизированными. Для обзора таких комплексов см. J Pharm Sci, 64 (8), 1269-1288 by Haleblian (August 1975), описание которого полностью включено в настоящий документ посредством ссылки.
Изобретение также относится к пролекарствам соединений приведенных в настоящем документе формул. Таким образом, некоторые производные соединений по настоящему изобретению, которые сами по себе могут обладать небольшой фармакологической активностью или не иметь ее, при введении пациенту могут быть преобразованы в соединения по настоящему изобретению, например, путем гидролитического расщепления. Такие производные называются «пролекарствами». Дополнительную информацию об использовании пролекарств можно найти в «Pro Drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) и «Bioreversible Carriers in Drug Design», Pergamon Press, 1987 (ed. E B Roche, American Pharmaceutical Association), описания которых полностью включены в настоящий документ посредством ссылок.
Пролекарства в соответствии с изобретением могут быть получены, например, путем замены соответствующих функциональных групп, присутствующих в соединениях по изобретению, определенными фрагментами, известными специалистам в данной области как «пролекарства», как описано, например, в «Design of Prodrugs» by H Bundgaard (Elsevier, 1985), описание которого полностью включено в настоящий документ посредством ссылки.
Некоторые неограничивающие примеры пролекарств в соответствии с изобретением включают следующие:
(i) когда соединение содержит функциональную группу карбоновой кислоты (COOH), ее сложный эфир, например, замена водорода на (C1-C8)алкил;
(ii) когда соединение содержит функциональную группу спирта (ОН), ее простой эфир, например, замена водорода на (C1-C6) алканоилоксиметил или на группу простого фосфатного эфира; и
(iii) если соединение содержит первичную или вторичную амино-функциональную группу (-NH2 или -NHR, где R≠H), ее амид, например, замена одного или обоих атомов водорода подходящей метаболически лабильной группой, такой как амид, карбамат, мочевина, фосфонат, сульфонат и т.д.
Дополнительные примеры замещающих групп в соответствии с приведенными выше примерами и примеры других типов пролекарств можно найти в вышеупомянутых ссылках. Наконец, некоторые соединения изобретения могут сами действовать как пролекарства других соединений изобретения.
В объем изобретения также входят метаболиты соединений описанных в настоящем документе формул, то есть соединения, образующиеся in vivo при введении лекарственного средства.
Соединения, описанные в настоящем документе, содержащие один или несколько асимметричных атомов углерода, могут существовать в виде двух или более стереоизомеров. Если соединение, описанное в настоящем документе, содержит алкенильную или алкениленовую группу, возможны геометрические цис/транс (или Z/E) изомеры. Если структурные изомеры взаимно преобразуются через низкий энергетический барьер, может возникать таутомерная изомерия («таутомерия»). Она может принимать форму протонной таутомерии в соединениях, описанных в настоящем документе, содержащих, например, имино-, кето- или оксимную группу, или так называемой валентной таутомерии в соединениях, которые содержат ароматический фрагмент. Одно соединение может проявлять более одного типа изомерии.
Соединения описанных в настоящем документе вариантов осуществления включают все стереоизомеры (например, цис- и транс-изомеры) и все оптические изомеры соединений, описанных в настоящем документе (например, R и S энантиомеры), а также рацемические, диастереомерные и другие смеси таких изомеров. Хотя все стереоизомеры входят в объем нашей формулы изобретения, специалист в данной области поймет, что конкретные стереоизомеры могут быть предпочтительными.
В некоторых вариантах осуществления изобретения соединения, описанные в настоящем документе, могут существовать в нескольких таутомерных формах, включая енольную и иминную форму, а также кето- и енаминную форму и их геометрические изомеры и их смеси. Все такие таутомерные формы включены в объем настоящих вариантов осуществления изобретения. Таутомеры существуют в виде смесей таутомеров в растворе. В твердой форме обычно преобладает один таутомер. Даже несмотря на то, что может быть описан один таутомер, настоящие варианты осуществления изобретения включают все таутомеры настоящих соединений.
Настоящие варианты осуществления изобретения также включают атропоизомеры описанных в настоящем документе соединений. Атропоизомеры относятся к соединениям, которые можно разделить на изомеры с ограничением вращения.
В объем настоящих вариантов осуществления изобретения включены все стереоизомеры, геометрические изомеры и таутомерные формы соединений, описанные в настоящем документе, включая соединения, проявляющие более одного типа изомерии, и смеси одного или нескольких из них.
Цис/транс-изомеры можно разделить обычными методами, хорошо известными специалистам в данной области, например, хроматографией и фракционной кристаллизацией.
Обычные способы получения/выделения индивидуальных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата (или рацемата соли или производного) с использованием, например, хиральной высокоэффективной жидкостной хроматографии (ВЭЖХ) или SFC.
В качестве альтернативы, рацемат (или рацемический предшественник) может взаимодействовать с подходящим оптически активным соединением, например, спиртом, или, в случае, когда соединение, описанное в настоящем документе, содержит кислотный или основной фрагмент, основанием или кислотой, такими как 1-фенилэтиламин или винная кислота. Полученная диастереомерная смесь может быть разделена хроматографией и/или фракционной кристаллизацией, и один или оба диастереоизомера могут быть преобразованы в соответствующий чистый энантиомер(ы) способами, хорошо известными квалифицированному специалисту.
«Аномальный рост клеток» или «рак» в контексте настоящего описания, если не указано иное, относится к росту клеток, который не зависит от нормальных регуляторных механизмов (например, потеря контактного ингибирования). Это включает аномальный рост следующего: (1) опухолевые клетки (опухоли), которые пролиферируют путем экспрессии мутированной тирозинкиназы или сверхэкспрессии рецепторной тирозинкиназы; (2) доброкачественные и злокачественные клетки других пролиферативных заболеваний, при которых происходит аберрантная активация тирозинкиназы; (3) любые опухоли, которые размножаются рецепторными тирозинкиназами; (4) любые опухоли, которые пролиферируют за счет аберрантной активации серин/треонинкиназы; (5) доброкачественные и злокачественные клетки других пролиферативных заболеваний, при которых происходит аберрантная активация серин/треонинкиназы; (6) любые опухоли, которые размножаются по аберрантным сигнальным, метаболическим, эпигенетическим и транскрипционным механизмам; и (7) доброкачественные и злокачественные клетки других пролиферативных заболеваний, при которых нарушены механизмы передачи сигналов, метаболизма, эпигенетики и транскрипции.
Для удобства в настоящем документе могут быть использованы некоторые хорошо известные сокращения, включая: положительный по рецептору эстрогена (ER+), отрицательный по рецептору 2 эпидермального фактора роста человека (HER2-), немелкоклеточный рак легкого (NSCLC) и устойчивый к кастрации рак простаты (CRPC).
Дополнительные варианты осуществления изобретения относятся к способам лечения аномального роста клеток у пациента. Дополнительные варианты осуществления относятся к способу лечения аномального роста клеток у пациента, включающему введение пациенту такого количества соединения, описанного в настоящем документе, которое эффективно при лечении аномального роста клеток.
В других вариантах осуществления изобретения аномальный рост клеток представляет собой рак.
В некоторых вариантах осуществления изобретения рак выбран из группы, включающей рак легких, мезотелиому, рак костей, рак поджелудочной железы, рак кожи, рак головы или шеи, кожную или внутриглазную меланому, рак матки, рак яичников, рак прямой кишки, рак анальной области, рак желудка, рак печени, рак толстой кишки, рак молочной железы, рак матки, рак маточных труб, рак эндометрия, рак шейки матки, рак влагалища, рак вульвы, болезнь Ходжкина, рак пищевода, рак тонкой кишки, рак эндокринной системы, рак щитовидной железы, рак паращитовидной железы, рак надпочечника, саркому мягких тканей, рак уретры, рак половой член, рак простаты, гематологические злокачественные новообразования, хронический или острый лейкоз, лимфоцитарные лимфомы, рак мочевого пузыря, рак почки или мочеточника, почечно-клеточную карциному, карциному почечной лоханки, новообразования центральной нервной системы (ЦНС), первичную лимфому ЦНС, опухоли оси позвоночника, глиобластому, глиому ствола головного мозга, аденому гипофиза или комбинацию двух или более из вышеперечисленных видов рака.
Дополнительные варианты осуществления относятся к способам лечения солидных опухолей у пациента. Некоторые варианты осуществления изобретения относятся к лечению солидных опухолей у пациента, включающему введение пациенту такого количества соединения, описанного в настоящем документе, которое эффективно при лечении солидной опухоли.
В одном варианте осуществления изобретения солидная опухоль представляет собой молочную железу, легкое, толстую кишку, мозг, простату, желудок, поджелудочную железу, яичник, меланому, эндокринную опухоль, матку, яичко или мочевой пузырь.
В одном варианте осуществления изобретения солидная опухоль представляет собой молочную железу, легкое, простату, поджелудочную железу или яичник.
В одном варианте осуществления изобретения рак представляет собой рак молочной железы.
В одном варианте осуществления изобретения рак молочной железы представляет собой ER+ рак молочной железы.
В одном варианте осуществления изобретения рак молочной железы представляет собой ER+ HER2- рак молочной железы.
В одном варианте осуществления изобретения рак молочной железы представляет собой местно-распространенный или метастатический рак молочной железы ER+ HER2-.
В одном варианте осуществления изобретения рак легкого представляет собой немелкоклеточный рак легкого.
В одном варианте осуществления изобретения рак легкого представляет собой местнораспространенный или метастатический немелкоклеточный рак легкого.
В одном варианте осуществления изобретения рак простаты представляет собой устойчивый к кастрации рак простаты.
В одном варианте осуществления изобретения рак простаты представляет собой местно-распространенный или метастатический устойчивый к кастрации рак простаты.
Дополнительные варианты осуществления относятся к способам лечения гематологических опухолей у пациента. Некоторые варианты осуществления относятся к лечению гематологических опухолей у пациента, включающему введение пациенту такого количества соединения, описанного в настоящем документе, которое эффективно при лечении гематологической опухоли.
В одном варианте осуществления изобретения гематологическая опухоль представляет собой лейкоз, лимфому или множественную миелому.
В одном варианте осуществления изобретения гематологическая опухоль представляет собой лейкоз или лимфому.
Дополнительные варианты осуществления относятся к способам лечения рака у пациента, включающим введение пациенту такого количества соединения, описанного в настоящем документе, которое является эффективным при лечении рака.
В одном варианте осуществления изобретения рак представляет собой рак молочной железы, легкого, толстой кишки, мозга, простаты, желудка, поджелудочной железы, яичников, меланомы, эндокринный, маточный, тестикулярный, мочевого пузыря или гематологический.
В одном варианте осуществления изобретения рак представляет собой рак молочной железы, легкого, простаты, поджелудочной железы, яичников или гематологический.
В одном варианте осуществления изобретения рак представляет собой рак молочной железы, легкого, простаты, поджелудочной железы или яичников.
В одном варианте осуществления изобретения рак представляет собой рак молочной железы.
В одном варианте осуществления изобретения рак молочной железы представляет собой ER+ рак молочной железы.
В одном варианте осуществления изобретения рак молочной железы представляет собой ER+ HER2- рак молочной железы.
В одном варианте осуществления изобретения рак молочной железы представляет собой местно-распространенный или метастатический рак молочной железы ER+ HER2-.
В одном варианте осуществления изобретения рак легкого представляет собой немелкоклеточный рак легкого.
В одном варианте осуществления изобретения рак легкого представляет собой местнораспространенный или метастатический немелкоклеточный рак легкого.
В одном варианте осуществления изобретения рак простаты представляет собой устойчивый к кастрации рак простаты.
В одном варианте осуществления изобретения рак простаты представляет собой местно-распространенный или метастатический устойчивый к кастрации рак простаты.
В одном варианте осуществления изобретения рак является гематологическим.
В одном варианте осуществления изобретения гематологическая опухоль представляет собой лейкоз или лимфому.
Дополнительные варианты осуществления изобретения относятся к способам лечения рака у пациента, которые включают введение пациенту такого количества соединения, описанного в настоящем документе, которое эффективно для лечения рака в комбинации с противоопухолевым агентом, выбранным из группы, включающей митотические ингибиторы, алкилирующие агенты, антиметаболиты, интеркалирующие антибиотики, ингибиторы фактора роста, облучение, ингибиторы клеточного цикла, ферменты, ингибиторы топоизомеразы, модификаторы биологического ответа, антитела, цитотоксические средства, антигормоны и антиандрогены.
Другие варианты осуществления изобретения относятся к фармацевтическим композициям для лечения рака у пациента, содержащим соединение, описанное в настоящем документе, которое является эффективным при лечении рака, и фармацевтически приемлемый носитель.
Дополнительные варианты осуществления относятся к способу лечения рака у пациента, в частности у человека, включающему введение пациенту такого количества соединения, описанного в настоящем документе, или его фармацевтически приемлемой соли, сольвата, гидрата или пролекарства, которые эффективны при лечении рака. В одном варианте осуществления этого способа рак включает, но не ограничивается ими, рак легких, рак костей, рак поджелудочной железы, рак кожи, рак головы или шеи, кожную или внутриглазную меланому, рак матки, рак яичников, рак яичников, рак прямой кишки, рак анальной области, рак желудка, рак толстой кишки, рак молочной железы, рак матки, рак маточных труб, рак эндометрия, рак шейки матки, рак влагалища, рак вульвы, болезнь Ходжкина, рак пищевода, рак тонкой кишки, рак эндокринной системы, рак щитовидной железы, рак паращитовидной железы, рак надпочечника, саркому мягких тканей, рак уретры, рак половой член, рак простаты, хронический или острый лейкоз, лимфоцитарные лимфомы, рак мочевого пузыря, рак почки или мочеточника, почечно-клеточный рак, рак почечной лоханки, новообразования центральной нервной системы (ЦНС), первичную лимфому ЦНС, опухоли оси позвоночника, глиому ствола головного мозга, аденому гипофиза или комбинацию одного или нескольких из вышеупомянутых видов рака. В одном варианте осуществления изобретения способ включает введение пациенту такого количества соединения, описанного в настоящем документе, которое эффективно при лечении указанной раковой солидной опухоли. В одном предпочтительном воплощении солидная опухоль представляет собой рак молочной железы, легких, толстой кишки, мозга, простаты, желудка, поджелудочной железы, яичников, кожи (меланома), эндокринный рак, рак матки, яичек и мочевого пузыря.
В другом варианте осуществления этого способа указанный рак представляет собой доброкачественное пролиферативное заболевание, включая, помимо прочего, псориаз, доброкачественную гипертрофию предстательной железы или рестеноз.
Некоторые варианты осуществления изобретения относятся к способу лечения рака у пациента, который включает введение указанному пациенту такого количества соединения, описанного в настоящем документе, или его фармацевтически приемлемой соли, сольвата, гидрата или пролекарства, которое эффективно при лечении рака в комбинации с противоопухолевым агентом, выбранным из группы, включающей ингибиторы митоза, алкилирующие агенты, антиметаболиты, интеркалирующие антибиотики, ингибиторы фактора роста, ингибиторы клеточного цикла, ферменты, ингибиторы топоизомеразы, модификаторы биологического ответа, антитела, цитотоксические средства, антигормоны и антиандрогены.
Дополнительные варианты осуществления изобретения относятся к фармацевтической композиции для лечения рака у пациента, в частности, у человека, содержащей такое количество соединения, описанного в настоящем документе, или его фармацевтически приемлемой соли, сольвата, гидрата или пролекарства, которое эффективно при лечении рака, и фармацевтически приемлемый носитель. В одном варианте осуществления указанной композиции рак включает, помимо прочего, рак легких, рак костей, рак поджелудочной железы, рак кожи, рак головы или шеи, кожную или внутриглазную меланому, рак матки, рак яичников, рак прямой кишки, рак анальной области, рак желудка, рак толстой кишки, рак молочной железы, рак матки, рак маточных труб, рак эндометрия, рак шейки матки, рак влагалища, рак вульвы, болезнь Ходжкина, рак пищевода, рак тонкой кишки, рак эндокринной системы, рак щитовидной железы, рак паращитовидной железы, рак надпочечников, саркому мягких тканей, рак уретры, рак полового члена, рак простаты, хронический или острый лейкоз, лимфоцитарные лимфомы, рак мочевого пузыря, рак почки или мочеточника, почечно-клеточный рак, рак почечной лоханки, новообразования центральной нервной системы (ЦНС), первичную лимфому ЦНС, опухоли оси позвоночника, глиому ствола головного мозга, аденому гипофиза или комбинация одного или нескольких из вышеперечисленных видов рака. В другом варианте осуществления указанной фармацевтической композиции указанный аномальный рост клеток представляет собой доброкачественное пролиферативное заболевание, включая, помимо прочего, псориаз, доброкачественную гипертрофию предстательной железы или рестиноз.
Дополнительные варианты осуществления изобретения относятся к способу лечения рака у пациента, который включает введение указанному пациенту такого количества соединения, описанного в настоящем документе, или его фармацевтически приемлемой соли, сольвата или гидрата, которое эффективно при лечении рака в комбинации с другим противоопухолевым агентом, выбранным из группы, включающей ингибиторы митоза, алкилирующие агенты, антиметаболиты, интеркалирующие антибиотики, ингибиторы фактора роста, ингибиторы клеточного цикла, ферменты, ингибиторы топоизомеразы, модификаторы биологического ответа, антитела, цитотоксические средства, антигормоны и антиандрогены. В некоторых вариантах осуществления изобретения рассматривается фармацевтическая композиция для лечения аномального роста клеток, где композиция включает соединение, описанное в настоящем документе, или его фармацевтически приемлемую соль, сольват или гидрат, которые эффективны при лечении аномального роста клеток, и другой противоопухолевый агент, выбранный из группа, включающей ингибиторы митоза, алкилирующие агенты, антиметаболиты, интеркалирующие антибиотики, ингибиторы фактора роста, ингибиторы клеточного цикла, ферменты, ингибиторы топоизомеразы, модификаторы биологического ответа, антитела, цитотоксические средства, антигормоны и антиандрогены.
Еще больше вариантов осуществления изобретения относятся к способу лечения расстройства, связанного с ангиогенезом у пациента, в том числе человека, включающему введение указанному пациенту такого количества соединения, описанного в настоящем документе, как определено выше, или его фармацевтически приемлемой соли, сольвата, гидрата или пролекарства, которое эффективно для лечения указанного нарушения, в комбинации с одним или несколькими противоопухолевыми агентами, перечисленными выше. Такие нарушения включают раковые опухоли, такие как меланома; глазные расстройства, такие как возрастная дегенерация желтого пятна, предполагаемый синдром глазного гистоплазмоза и неоваскуляризация сетчатки в результате пролиферативной диабетической ретинопатии; ревматоидный артрит; нарушения потери костной массы, такие как остеопороз, болезнь Педжета, гуморальная гиперкальциемия злокачественных новообразований, гиперкальциемия от опухолей, метастазирующих в кости, и остеопороз, вызванный лечением глюкокортикоидами; коронарный рестеноз; и некоторые микробные инфекции, включая инфекции, связанные с микробными патогенами, выбранными из аденовируса, хантавирусов, Borrelia burgdorferi, Yersinia spp., Bordetella pertussis, и Streptococcus группы А.
Некоторые варианты осуществления изобретения относятся к способу (и к фармацевтической композиции для) лечения рака у пациента, который включает некоторое количество соединения, описанного в настоящем документе, или его фармацевтически приемлемой соли, сольвата или гидрата в сочетании с некоторым количеством одного или большее веществ, выбранных из агентов против ангиогенеза, ингибиторов сигнальной трансдукции (например, ингибирующих средства, с помощью которых регуляторные молекулы, которые регулируют фундаментальные процессы роста, дифференциации и выживания клеток, передаются внутри клетки) и антипролиферативных агентов, где вместе количество которых является эффективным при лечении указанного аномального роста клеток.
в способах и фармацевтических композициях, описанных в настоящем документе, в комбинации с соединением, описанным в настоящем документе, могут использоваться агенты против ангиогенеза, такие как ингибиторы MMP-2 (матрикс-металлопротиеназа 2), ингибиторы MMP-9 (матрикс-металлопротиеназа 9) и ингибиторы COX-II (циклооксигеназы II).
С соединением, описанным в настоящем документе, также можно комбинировать ингибиторы тирозинкиназы.
С соединением, описанным в настоящем документе, также можно комбинировать ингибиторы VEGF, например супент и акситиниб.
В комбинации с соединением, описанным в настоящем документе, могут быть введены ингибиторы рецептора ErbB2. Также было показано, что различные другие соединения, такие как производные стирола, обладают свойствами ингибирования тирозинкиназы, и некоторые из ингибиторов тирозинкиназы были идентифицированы как ингибиторы рецептора erbB2.
В комбинации с соединением по настоящему изобретению могут быть введены ингибиторы рецептора эпидермального фактора роста (EGFR).
В комбинации с соединением по настоящему изобретению могут быть введены ингибиторы PI3K, такие как ингибиторы PI3K альфа или PI3K бета.
В комбинации с соединением по настоящему изобретению могут быть введены ингибиторы мишени для рапамицина у млекопитающих (mTOR).
В комбинации с соединением по настоящему изобретению могут быть введены c-Met ингибиторы.
В комбинации с соединением по настоящему изобретению могут быть введены CDK ингибиторы.
В комбинации с соединением по настоящему изобретению могут быть введены MEK ингибиторы.
В комбинации с соединением по настоящему изобретению могут быть введены PARP ингибиторы.
В комбинации с соединением по настоящему изобретению могут быть введены JAK ингибиторы.
В комбинации с соединением по настоящему изобретению может быть введен антагонист белка запрограммированной клеточной смерти 1 (PD-1).
В комбинации с соединением по настоящему изобретению может быть введен антагонист лиганда 1 запрограммированной клеточной гибели (PD-L1).
Другие антипролиферативные агенты, которые можно использовать с соединениями, описанными в настоящем документе, включают ингибиторы фермента фарнезил-протеинтрансферазы и ингибиторы рецепторной тирозинкиназы PDGFr.
Соединение, описанное в настоящем документе, может использоваться с другими агентами, применимыми для лечения аномального роста клеток или рака, включая, помимо прочего, агенты, способные усиливать противоопухолевые иммунные ответы, такие как антитела CTLA4 (цитотоксический лимфоцитарный антиген 4), и другие агенты, способные к блокировке CTLA4; и антипролиферативные агенты, такие как другие ингибиторы фарнезилпротеинтрансферазы, например, фарнезилпротеинтрансфераза.
Соединение, описанное в настоящем документе, может применяться в качестве единственной терапии или может включать, например, одно или несколько других противоопухолевых веществ, выбранных, например, из ингибиторов митоза, алкилирующих агентов, антиметаболитов, ингибиторов фактора роста, ингибиторов клеточного цикла, интеркалирующих антибиотиков, ферментов и антигормонов.
Соединения, описанные в настоящем документе, можно использовать отдельно или в комбинации с одним или несколькими противоопухолевыми средствами или средствами поддерживающего ухода. Например, соединения, описанные в настоящем документе, можно использовать с цитотоксическими агентами. Некоторые варианты осуществления также предполагают использование соединений, описанных в настоящем документе, вместе с гормональной терапией. Кроме того, в некоторых вариантах осуществления изобретения предложено соединение, описанное в настоящем документе, отдельно или в комбинации с одним или несколькими продуктами для поддерживающей терапии, например, продукт, выбранный из группы, включающей филграстим (нейпоген), ондансетрон (зофрана), фрагмин, прокрит, алокси, эменда или их комбинации. Такое совместное лечение может быть достигнуто путем одновременного, последовательного или раздельного дозирования отдельных компонентов лечения.
Соединения, описанные в настоящем документе, можно использовать с противоопухолевыми агентами, алкилирующими агентами, антиметаболитами, антибиотиками, противоопухолевыми агентами растительного происхождения, производными камптотецина, ингибиторами тирозинкиназы, антителами, интерферонами и/или модификаторами биологического ответа. В этом отношении далее приводится неограничивающий список примеров вторичных агентов, которые можно использовать с соединениями, описанными в настоящем документе.
Некоторые варианты осуществления изобретения также относятся к фармацевтической композиции, содержащей соединение формул (I), (Ia) (II), (III), (IV), (V) или (VI) или фармацевтически приемлемую соль или сольват, определенные в настоящем документе выше, вместе с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Дополнительные варианты осуществления изобретения относятся к фармацевтической композиции, которая включает смешивание соединения формул (I), (Ia) (II), (III), (IV), (V) или (VI) или фармацевтически приемлемой соли или сольвата, определенных в настоящем документе выше, с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Для вышеупомянутых терапевтических применений вводимая доза, конечно, будет варьироваться в зависимости от применяемого соединения, способа введения, желаемого лечения и указанного нарушения. Суточная доза соединения формул (I), (Ia) (II), (III), (IV), (V) или (VI) или его фармацевтически приемлемой соли может находиться в диапазоне от 1 мг до 1 грамма, от 1 мг до 250 мг, от 1 мг до 100 мг, от 1 мг до 50 мг, от 1 мг до 25 мг и от 1 мг до 10 мг.
Настоящие варианты осуществления изобретения включают также композиции с замедленным высвобождением.
На введение описанных в настоящем документе соединений (далее «активное(ые) соединение(ия)») можно воздействовать любым способом, который обеспечивает доставку соединений к месту действия. Эти методы включают пероральные пути, интрадуоденальные пути, парентеральную инъекцию (включая внутривенную, подкожную, внутримышечную, внутрисосудистую или инфузию), местное и ректальное введение.
Активное соединение может применяться в качестве единственной терапии или может включать одно или несколько других противоопухолевых веществ, например, выбранных, например, из ингибиторов митоза, например винбластина; алкилирующих агентов, например цисплатина, карбоплатина и циклофосфамида; антиметаболитов, например, 5-фторурацила, цитозинарабинозида и гидроксимочевина, или, например, одного из предпочтительных антиметаболитов, описанных в Европейской патентной заявке № 239362, такой как N-(5-[N-(3,4-дигидро-2-метил-4-оксохиназолин-6-илметил)-N-метиламино]-2-теноил)-L-глутаминовая кислота; ингибиторов фактора роста; ингибиторов клеточного цикла; интеркалирующих антибиотиков, например, адриамицина и блеомицина; ферментов, например интерферона; и антигормонов, например, антиэстрогенов, таких как Nolvadex® (тамоксифен), или, например, антиандрогенов, таких как Casodex® (4Θ-циано-3-(4-фторфенилсульфонил)-2-гидрокси-2-метил-3ʼ-(трифторметил)пропионанилид). Такое совместное лечение может быть достигнуто путем одновременного, последовательного или раздельного дозирования отдельных компонентов лечения.
Фармацевтическая композиция может быть представлена в подходящей форме, например, для перорального введения в виде таблетки, капсулы, пилюли, порошка, составов с замедленным высвобождением, раствора, суспензии; для парентеральной инъекции в виде стерильного раствора, суспензии или эмульсии; для местного применения в виде мази или крема; или для ректального введения в виде суппозитория. Фармацевтическая композиция может быть в виде единичных дозированных форм, подходящих для разового введения точных доз. Фармацевтическая композиция будет включать обычный фармацевтический носитель или наполнитель и соединение, описанное в настоящем документе, в качестве активного ингредиента. Кроме того, она может включать другие лекарственные или фармацевтические агенты, носители, адъюванты и т.д.
Примеры форм для парентерального введения включают растворы или суспензии активных соединений в стерильных водных растворах, например, водных растворах пропиленгликоля или декстрозы. При желании такие лекарственные формы могут быть соответствующим образом забуферены.
Подходящие фармацевтические носители включают инертные разбавители или наполнители, воду и различные органические растворители. При желании фармацевтические композиции могут содержать дополнительные ингредиенты, такие как ароматизаторы, связующие вещества, наполнители и тому подобное. Таким образом, для перорального введения таблетки, содержащие различные вспомогательные вещества, такие как лимонная кислота, могут использоваться вместе с различными разрыхлителями, такими как крахмал, альгиновая кислота и некоторые сложные силикаты, и со связующими агентами, такими как сахароза, желатин и аравийская камедь. Кроме того, для целей таблетирования часто используются смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции подобного типа также могут использоваться в мягких и твердых желатиновых капсулах. Поэтому предпочтительные материалы включают лактозу или молочный сахар и высокомолекулярные полиэтиленгликоли. Когда для перорального введения желательны водные суспензии или эликсиры, активное соединение в них можно комбинировать с различными подсластителями или ароматизаторами, красящими веществами или красителями и, при желании, с эмульгаторами или суспендирующими агентами вместе с разбавителями, такими как вода, этанол, пропиленгликоль, глицерин или их комбинации.
Примеры и способы получения, представленные далее, дополнительно иллюстрируют и предоставляют примеры соединений, описанных в настоящем документе, и способы получения таких соединений. Объем описанных в настоящем документе вариантов осуществления никоим образом не ограничивается следующими примерами и способами получения. В следующих примерах молекулы с одним хиральным центром, если не указано иное, существуют в виде рацемической смеси. Если не указано иное, молекулы с двумя или более хиральными центрами существуют в виде рацемической смеси диастереомеров. Отдельные энантиомеры/диастереомеры могут быть получены способами, известными специалистам в данной области.
В показанных примерах солевые формы иногда выделялись как следствие добавок подвижной фазы в процессе хроматографической очистки на основе ВЭЖХ. В этих случаях соли, такие как формиат, трифторацетат и ацетат, выделяли и исследовали без дальнейшей обработки. Следует понимать, что средний специалист в данной области сможет получить форму свободного основания с помощью стандартного метода (такого как использование ионообменных колонок или проведение простых основных экстракций с использованием мягкого водного основания).
В общем, соединения, описанные в настоящем документе, могут быть получены способами, известными в области химии, особенно в свете приведенного в настоящем документе описания. Некоторые способы получения описанных в настоящем документе соединений представлены в качестве дополнительных признаков вариантов осуществления изобретения и проиллюстрированы на схемах реакций, приведенных ниже, и в экспериментальном разделе.
ПРИМЕРЫ
Следующие далее примеры предоставлены исключительно для иллюстрации настоящего изобретения и не предназначены для ограничения объема изобретения, описанного в настоящем документе.
Общие экспериментальные детали
Если не указано иное, применяются следующие обобщения. Спектры 1H ЯМР регистрировали на Bruker Ultrashield Plus (400 МГц) или Bruker AVANCE III (400 МГц). Кратность сигнала обозначается следующими сокращениями: с, синглет; д, дублет; т, триплет; кв, квартет; п, пентет; септ, септет; дд, дуплет дублетов; дт, дуплет триплетов; тт, триплет триплетов; шир., широкий; м, мультиплет. Все наблюдаемые константы взаимодействия, J, выражены в герцах (Гц). Способные к обмену протоны наблюдаются не всегда. Данные ЖХМС были получены с использованием Agilent 6100 Series Single Quad, Agilent 1260 Infinity Series UPLC/MS, Agilent 1200 (ЖХМС-A), Waters 2695 alliance, Agilent 6120 Single Quad или масс-направленной ВЭЖХ-МС. Изотопы хлора указаны как 35Cl, изотопы брома указаны либо как 79Br, либо как 81Br, или как оба 79Br/81Br.
Характерный способ ЖХМС представлен ниже:
Прибор: одноквадрупольный ВЭЖХ Agilent серии 6100, ВЭЖХ Agilent серии 1200, насос: четвертичный насос G1311A серии 1200, автосамплер: термостатированный автосамплер серии 1200 G1329A, детектор: детектор с переменной длиной волны G1314B серии 1200
Условия ЖХ: анализ ВЭЖХ с обращенной фазой, колонка: Luna C8 (2) 5 мкм 50×4,6 мм 100 Å, температура колонки: 30ºC, объем впрыска: 5 мкл, растворитель A: вода 0,1%-ная муравьиная кислота, растворитель B: MeCN 0,1%-ная муравьиная кислота, градиент: 5-100% растворитель B в течение 10 мин, детекция: 254 нм или 214 нм
Условия МС: источник ионов: квадруполь, ионный режим: многомодовый ES, температура сушильного газа: 300ºC, температура испарителя: 200ºC, капиллярное напряжение (В): 2000 (положительное), капиллярное напряжение (В): 4000 (отрицательное), диапазон сканирования: 100-1000, размер шага: 0,1 с, время сбора данных: 10 мин.
ЖХМС способ A (ЖХМС-A): модель ЖХ: Agilent 1200, тип насоса: бинарный насос, тип детектора: DAD, модель МС: Agilent G6110A Quadrupole, условия ЖХ: колонка: Xbridge-C18, 2,5 мкм, 2,1×30 мм, температура колонки: 30ºC, длина волны: 214 нм, 254 нм, подвижная фаза: A: 0,07% водный раствор HCOOH, B: MeOH; условия МС: МС: источник ионов: ES+ (или ES-) диапазон МС: 50-900 m/z, фрагментатор: 60, расход осушающего газа: 10 л/мин, давление в распылителе: 35 фунтов на кв. дюйм, температура осушающего газа: 350ºC, Vcap: 3,5 кВ
Таблица градиентов:
Приготовление образца:
Образец растворяли в метаноле, концентрация около 0,11-1 мг/мл, затем фильтровали через шприцевой фильтр с размером пор 0,22 мкм. (Объем впрыска: 1-10 мкл)
Общие способы:
Если не указано иное, переменные на схемах I-VI имеют такие же значения, какие определены в настоящем документе.
Схема I:
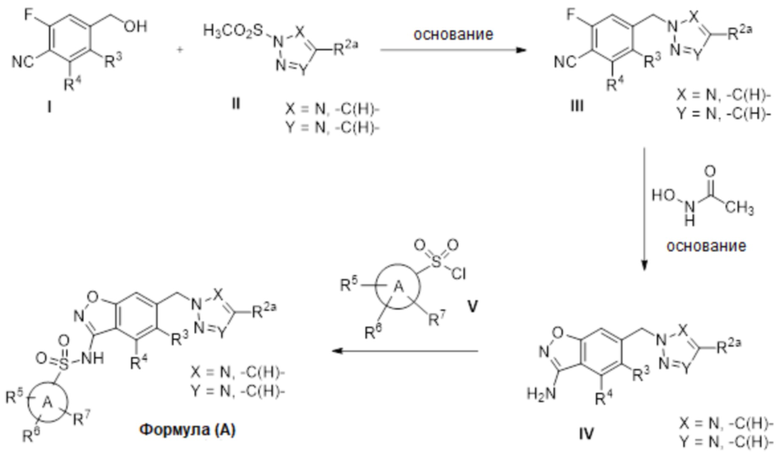
Как показано на схеме I, соединение типа I можно обработать соединением типа II в присутствии эффективного основания (такого как Cs2CO3) в подходящем растворителе (таком как MeCN) с получением соединения типа III. Соединение типа III можно преобразовать в соединение типа IV обработкой N-гидроксиацетамидом в присутствии эффективного основания (такого как K2CO3 или 1,1,3,3-тетраметилгуанидин) в подходящей смеси растворителей (такой как ДМФ/H2O). Соединение типа IV можно обработать соединением типа V в присутствии эффективного основания (такого как пиридин, NaH или NaOtPn), в чистом виде или в подходящем растворителе (таком как ТГФ или ДМФ) с получением соединения формулы (A). В некоторых случаях соединения типа II, III и IV или формулы (A) могут содержать защитные группы, которые могут быть добавлены или удалены дополнительными стадиями в последовательности синтеза с использованием условий, известных в данной области (Protective Groups in Organic Synthesis, A. Wiley-Interscience Publication, 1981 или Protecting Groups, 10 Georg Thieme Verlag, 1994). Соединения на каждой стадии можно очищать стандартными методами, такими как колоночная хроматография, кристаллизация или обращенно-фазовая SFC или ВЭЖХ. Переменные R2a, R3, R4, R5, R6, R7 и R8 определены в вариантах осуществления, схемах, примерах и формуле изобретения в настоящем документе.
Схема II:
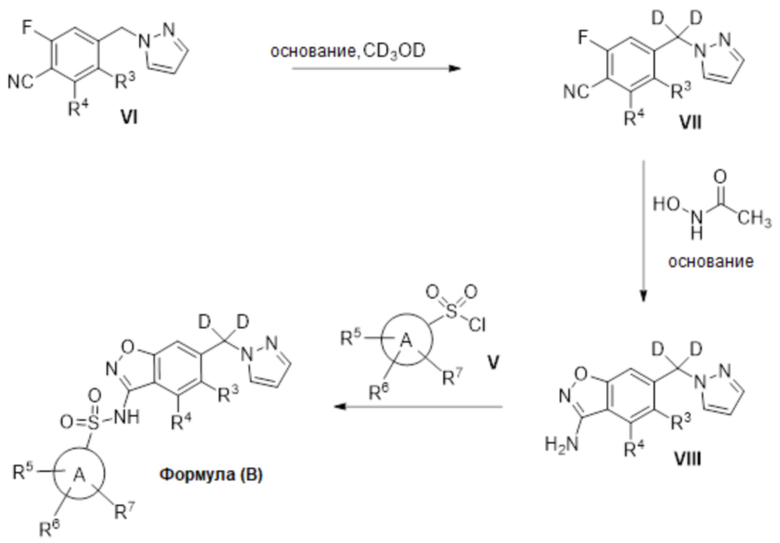
Как показано на схеме II, соединение типа VI может быть дейтерировано обработкой эффективным основанием (таким как Cs2CO3) в подходящем растворителе (таком как CD3OD) с получением соединения типа VII. Соединение типа VII может быть преобразовано в соединение типа VIII обработкой N-гидроксиацетамидом в присутствии эффективного основания (такого как 1,1,3,3-тетраметилгуанидин) в подходящей смеси растворителей (такой как MeCN/D2O). Соединение типа VIII можно обработать соединением типа V в присутствии эффективного основания (такого как пиридин), в чистом виде, с получением соединения формулы (B). Соединения на каждой стадии можно очищать стандартными методами, такими как колоночная хроматография, кристаллизация или обращенно-фазовая SFC или ВЭЖХ. Переменные R3, R4, R5, R6 и R7 определены в вариантах осуществления, схемах, примерах и формуле изобретения в настоящем документе.
Схема III:
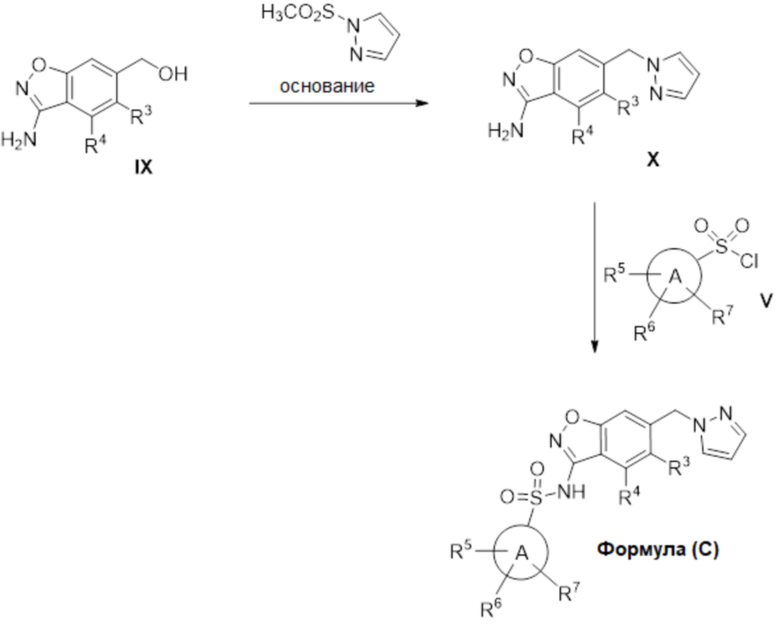
Как показано на схеме III, соединение типа IX может быть преобразовано в соединение типа X обработкой 1-(метансульфонил)-1H-пиразолом в присутствии эффективного основания (такого как Cs2CO3) в подходящем растворителе (таком как MeCN). Соединение типа X можно обработать соединением типа V в присутствии эффективного основания (такого как пиридин), в чистом виде, с получением соединения формулы (C). Соединения на каждой стадии можно очищать стандартными методами, такими как колоночная хроматография, кристаллизация или обращенно-фазовая SFC или ВЭЖХ. Переменные R3, R4, R5, R6 и R7 определены в вариантах осуществления, схемах, примерах и формуле изобретения в настоящем документе.
Схема IV:
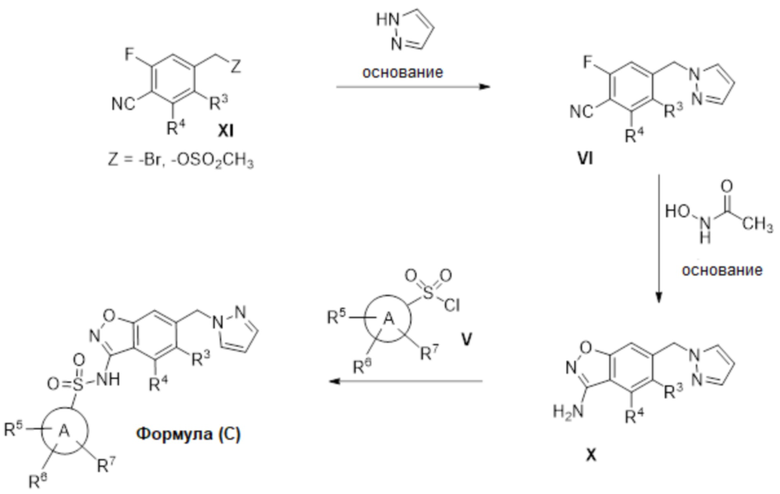
Как показано на схеме IV, соединение типа XI, с подходящей удаляемой группой (такой как -Br или -SO2CH3) может быть преобразовано в соединение типа VI обработкой 1H-пиразолом в присутствии эффективного основания (такого как Cs2CO3) в подходящем растворителе (таком как MeCN). Соединение типа VI может быть преобразовано в соединение типа X обработкой N-гидроксиацетамидом в присутствии эффективного основания (такого как 1,1,3,3-тетраметилгуанидин) в подходящей смеси растворителей (такой как ДМФ/H2O). Соединение типа X можно обработать соединением типа V в присутствии подходящего основания (такого как пиридин), в чистом виде, с получением соединения формулы (C). Соединения на каждой стадии можно очищать стандартными методами, такими как колоночная хроматография, кристаллизация или обращенно-фазовая SFC или ВЭЖХ. Переменные R3, R4, R5, R6 и R7 определены в вариантах осуществления, схемах, примерах и формуле изобретения в настоящем документе.
Схема V
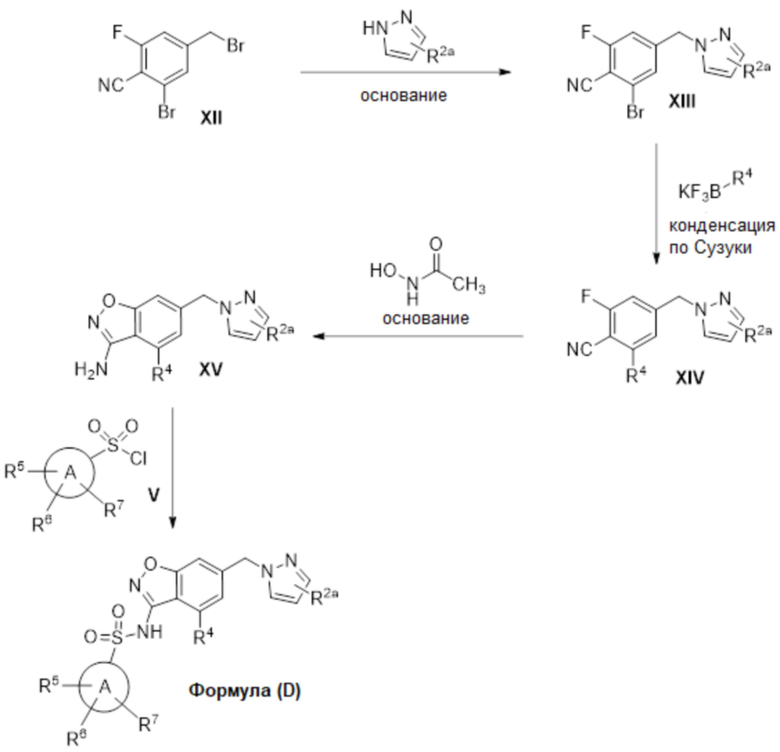
Как показано на схеме V, соединение типа XII может быть преобразовано в соединение типа XIII обработкой соответствующим образом необязательно замещенным 1H-пиразолом в присутствии эффективного основания (такого как Cs2CO3) в подходящем растворителе (таком как MeCN). Соединение типа XIII может быть преобразовано в соединение типа XIV в условиях кросс-сочетания Сузуки в присутствии эффективного катализатора (такого как метансульфонато(три-трет-бутилфосфино)(2Θ-амино-1,1-бифенил-2-ил)палладий(II)) в подходящей смеси растворителей (такой как PhMe/H2O). Соединение типа XIV может быть преобразовано в соединение типа XV обработкой N-гидроксиацетамидом в присутствии эффективного основания (такого как 1,1,3,3-тетраметилгуанидин) в подходящей смеси растворителей (такой как ДМФ/H2O). Соединение типа XV можно обработать соединением типа V в присутствии подходящего основания (такого как пиридин), в чистом виде, с получением соединения формулы (D). Соединения на каждой стадии можно очищать стандартными методами, такими как колоночная хроматография, кристаллизация или обращенно-фазовая SFC или ВЭЖХ. Переменные R2a, R4, R5, R6 и R7 определены в вариантах осуществления, схемах, примерах и формуле изобретения в настоящем документе.
Схема VI:
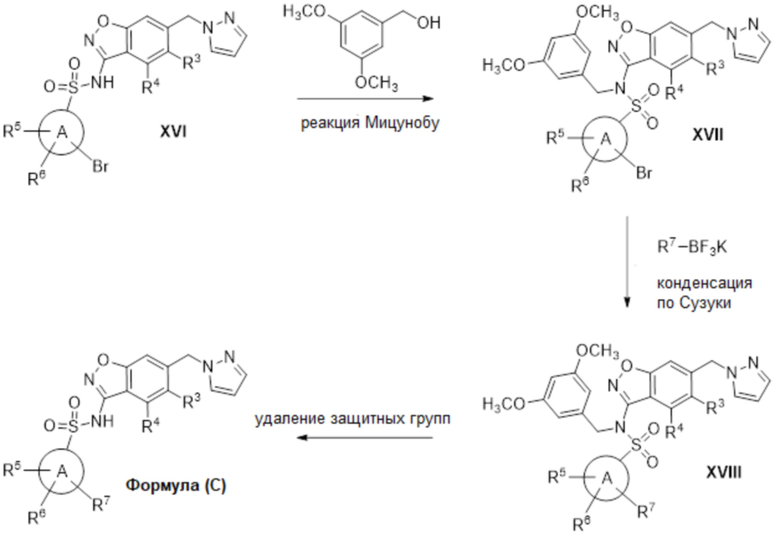
Как показано на схеме VI, соединение типа XVI может быть преобразовано в соединение типа XVII обработкой (3,5-диметоксифенил)метанолом в условиях Мицунобу (PPh3, DIAD) в подходящем растворителе (таком как 2-Me-ТГФ). Соединение типа XVII может быть преобразовано в соединение типа XVIII в условиях кросс-сочетания Сузуки в присутствии в присутствии эффективной комбинации катализатор/лиганд (такой как Pd(OAc)2/X-Phos) в подходящем растворителе (таком как CPME/H2O). Соединение типа XVII может быть преобразовано в соединение формулы (C) обработкой эффективной кислотой (такой как ТФУ) в подходящем растворителе (таком как DCM). Соединения на каждой стадии можно очищать стандартными методами, такими как колоночная хроматография, кристаллизация или обращенно-фазовая SFC или ВЭЖХ. Переменные R3, R4, R5, R6, и R7 определены в вариантах осуществления, схемах, примерах и формуле изобретения в настоящем документе.
Синтез промежуточных соединений:
Получение 2-фтор-4-(гидроксиметил)-6-метоксибензонитрила (Int-01) в соответствии со схемой 1.
Схема 1:
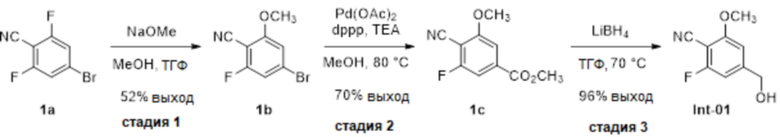
Стадия 1: Синтез 4-бром-2-фтор-6-метоксибензонитрила (1b).
К раствору 4-бром-2,6-дифторбензонитрила (1a) (40,0 г, 183,5 ммоль) в ТГФ (210,0 мл) и MeOH (30,0 мл) по частям при температуре 0ºC добавляли NaOMe (11,9 г, 220 ммоль). Смесь перемешивали при комнатной температуре в течение 16 ч. ТСХ анализ (1:4 EtOAc/петролейный эфир) показывал, что исходное вещество расходовалось. Смесь переносили в делительную воронку и промывали H2O (150 мл). Водный слой экстрагировали EtOAc (300 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали с помощью флеш хроматографии (330 г, SiO2, 10% EtOAc/петролейный эфир) с получением 4-бром-2-фтор-6-метоксибензонитрила (1b) (15,7 г, 52%-ный выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,49 (дд, J=1,5, 8,8 Гц, 1H), 7,41 (с, 1H), 3,98 (с, 3H).
Стадия 2: Синтез метил 4-циано-3-фтор-5-метоксибензоата (1c).
Раствор 4-бром-2-фтор-6-метоксибензонитрила (1b) (15,7 г, 68,2 ммоль), TEA (20,7 г, 205 ммоль), dppp (2,8 г, 6,8 ммоль) и Pd(OAc)2 (766 мг, 3,4 ммоль) в MeOH (150 мл) перемешивали при температуре 80ºC в атмосфере CO в течение 16 ч. ТСХ анализ (1:4 EtOAc/петролейный эфир) показывал, что исходное вещество расходовалось. Реакционную смесь концентрировали досуха. Остаток очищали с помощью флеш хроматографии (120 г, SiO2, 1:4 EtOAc/петролейный эфир) с получением метил 4-циано-3-фтор-5-метоксибензоата (1c) (10,0 г, 70%-ный выход) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,53-7,47 (м, 2H), 4,03 (с, 3H), 3,91 (с, 3H).
Стадия 3: Синтез 2-фтор-4-(гидроксиметил)-6-метоксибензонитрила (Int-01).
К раствору метил 4-циано-3-фтор-5-метоксибензоата (1c) (9,5 г, 45,4 ммоль) в ТГФ (50 мл) по частям при температуре 0ºC добавляли LiBH4 (2,0 г, 90,8 ммоль). Смесь перемешивали при температуре 70ºC в течение 2 ч. ЖХМС анализ показывал, что исходное вещество расходовалось с образованием желаемой массы продукта. Реакцию гасили путем добавления H2O (100 мл). Смесь переносили в делительную воронку и экстрагировали EtOAc (2×150 мл). Объединенные органические экстракты промывали насыщенным солевым раствором и насыщенным водным раствором NaHCO3, сушили над Na2SO4, фильтровали и концентрировали с получением 2-фтор-4-(гидроксиметил)-6-метоксибензонитрила (Int-01) (7,9 г, 96%-ный выход) в виде масла коричневого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,05 (с, 1H), 6,98 (д, J=10,0 Гц, 1H), 4,58 (д, J=5,7 Гц, 2H), 3,95 (с, 3H).
Промежуточные соединения, приведенные в таблице ниже, получали в соответствии со способами, используемыми для синтеза 2-фтор-4-(гидроксиметил)-6-метоксибензонитрила (Int-01). Следующие промежуточные соединения синтезировали с некритическими изменениями или заменами в приведенных в качестве примеров способах, которые мог бы осуществить специалист в данной области.
Таблица 1:
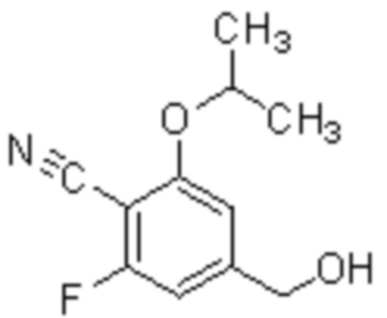
2-фтор-4-(гидроксиметил)-6-[(пропан-2-ил)окси]бензонитрил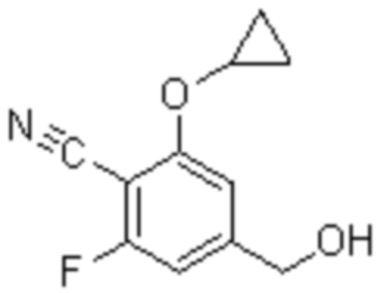
2-(циклопропилокси)-6-фтор-4-(гидроксиметил)бензонитрил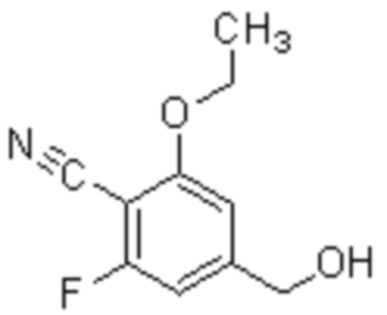
2-этокси-6-фтор-4-(гидроксиметил)бензонитрил
Альтернативное получение 2-фтор-4-(гидроксиметил)-6-метоксибензонитрила (Int-01) в соответствии со схемой 2.
Схема 2:
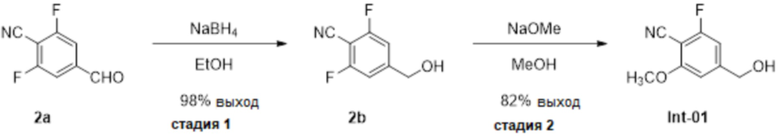
Стадия 1: Синтез 2,6-дифтор-4-(гидроксиметил)бензонитрила (2b).
Раствор 2,6-дифтор-4-формилбензонитрила (2a) (21,5 г, 129 ммоль) в абсолютном EtOH (400 мл) охлаждали на ледяной бане до температуры ~3ºC (внутренняя). Добавляли твердый NaBH4 (5×1 г, гранулы, 5,0 г, 130 ммоль), что вызывало небольшое выделение газа. Смесь перемешивали при охлаждении на ледяной бане в течение 2 ч и затем гасили при той же температуре путем добавления по каплям деионизированной H2O (100 мл в течение 5 мин). Медленно добавляли водную HCl (2,0 н, 50 мл в течение 30 мин), поддерживая температуру <10ºC (внутренняя). Раствор концентрировали в вакууме для удаления EtOH. Водный остаток переносили в делительную воронку, оставляя после себя липкое белое твердое вещество Водную фазу экстрагировали EtOAc (2×). Объединенные органические экстракты промывали насыщенным солевым раствором (2×), сушили над MgSO4, фильтровали и концентрировали. Сырое вещество растирали в гептане, фильтровали и сушили в вакууме с получением 2,6-дифтор-4-(гидроксиметил)бензонитрила (2b) (21,3 г, 98%) в виде сыпучего твердого вещества белого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,34 (д, J=9,3 Гц, 2H), 5,69 (шир. с, 1H), 4,60 (шир. с, 2H).
Стадия 2: Синтез 2-фтор-4-(гидроксиметил)-6-метоксибензонитрила (Int-01).
Раствор 2,6-дифтор-4-(гидроксиметил)бензонитрила (2b) (21,3 г, 126 ммоль) в безводном MeOH (400 мл) охлаждали до температуры −40ºC (внутренняя) на бане сухой лед/ацетонитрил. В течение периода 10 мин с помощью капельной воронки добавляли раствор NaOMe (5,0 M в MeOH, 100 мл, 500 ммоль). После завершения добавления охлаждающую баню удаляли. Смеси давали нагреться естественным образом до комнатной температуры и перемешивали в течение еще 8 ч. Реакционную смесь охлаждали до температуры 0ºC (внутренняя) и добавляли по каплям HCl (2,0 N, 200 мл) с получением раствора с pH ~5-6. Смесь концентрировали в вакууме для удаления MeOH. Водный раствор экстрагировали EtOAc (3×). Объединенные органические экстракты промывали насыщенным солевым раствором (2×), сушили над MgSO4 и фильтровали. Смесь концентрировали до ~150 мл на роторном испарителе (температура бани ~35ºC) и образовавшейся взвеси давали охладиться до комнатной температуры. Твердые продукты собирали путем фильтрации. Слой продукта на фильтре промывали гептаном (2×). Промывки фильтрата и гептана дополнительно концентрировали с получением второй порции твердых продуктов, которые собирали путем фильтрации. Объединенные твердые продукты сушили в вакууме с получением 2-фтор-4-(гидроксиметил)-6-метоксибензонитрила (Int-01) (18,6 г, 82%) в виде твердого вещества бледно-желтого цвета. 1H ЯМР (400 МГц, CDCl3) δ 6,82 (с, 1H), 6,79 (д, J=9,2 Гц, 1H), 4,76 (с, 2H), 3,97 (с, 3H).
Промежуточное соединение, представленные в таблице ниже, получали в соответствии со способами, используемыми для синтеза 2-фтор-4-(гидроксиметил)-6-метоксибензонитрила (Int-01). Следующие промежуточное соединение синтезировали с некритическими изменениями или заменами в приведенных в качестве примеров способах, которые мог бы осуществить специалист в данной области.
Таблица 2:
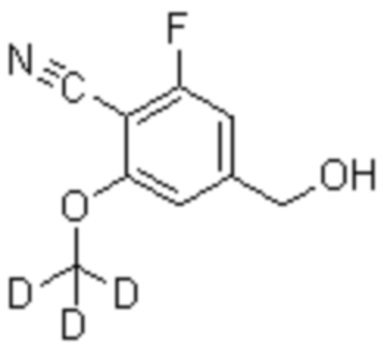
2-фтор-4-(гидроксиметил)-6-[(2H3)метилокси]-бензонитрил
Получение 2-фтор-4-(гидроксиметил)бензонитрила (Int-06) в соответствии со схемой 3.
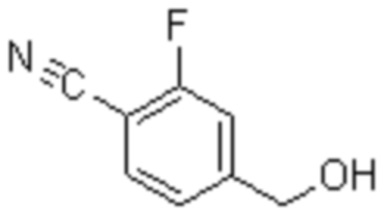
Схема 3:
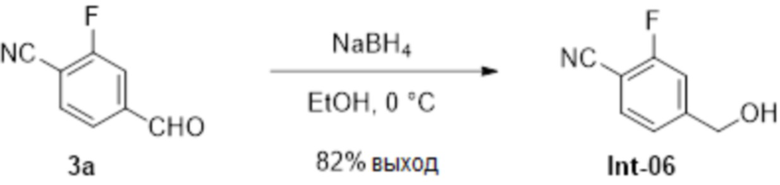
Раствор (5,0 г, 33,5 ммоль) 2-фтор-4-формилбензонитрила (3a) в EtOH (100 мл) охлаждали до температуры 0ºC. Добавляли NaBH4 (1,3 г, 33 ммоль) и реакционную смесь перемешивали при температуре 0ºC в течение 2 ч. Смесь гасили добавлением по каплям H2O (25 мл в течение 5 мин). Добавляли разбавленную HCl (2 н, 13 мл), поддерживая внутреннюю температуру <10ºC. Раствор концентрировали в вакууме для удаления EtOH. Водную смесь экстрагировали EtOAc (2×). Объединенные органические продукты промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Остаток растирали в гептане и сушили с получением 2-фтор-4-(гидроксиметил)бензонитрила (Int-06) (4,2 г, 82%-ный выход) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,87 (дд, J=7,9, 6,8 Гц, 1H), 7,42 (дд, J=10,6, 1,3 Гц, 1H), 7,35 (дд, J=8,0, 1,3 Гц, 1H), 5,55 (т, J=5,7 Гц, 1H), 4,60 (д, J=5,7 Гц, 2H).
Получение 5-бром-4-(бромметил)-2-фторбензонитрила (Int-07) в соответствии со схемой 4.
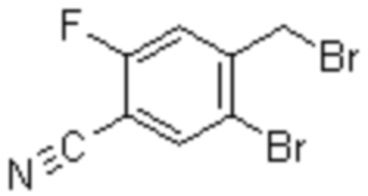
Схема 4:
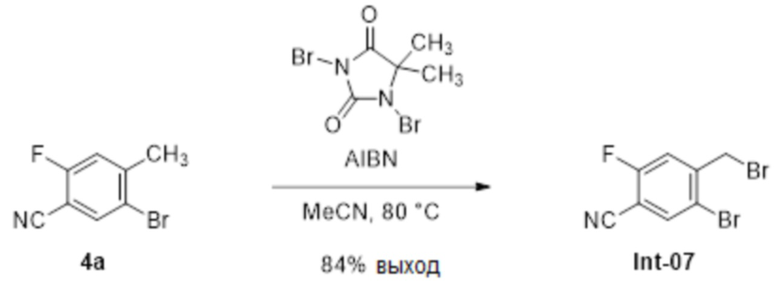
К раствору 5-бром-2-фтор-4-метилбензонитрила (4a) (1,01 г, 4,72 ммоль) в MeCN (10,0 мл) добавляли 1,3-дибром-5,5-диметилимидазолидин-2,4-дион (701 мг, 2,45 ммоль) и AIBN (101 мг, 0,613 ммоль). Смесь перемешивали при температуре 80ºC в течение ночи и затем концентрировали досуха. Остаток очищали с помощью флеш хроматографии (80 г, SiO2, 0-30% EtOAc/гептан) с получением (Int-07) (847 мг, 84%-ный выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3) δ 7,84 (д, J=6,0 Гц, 1H), 7,38 (д, J=8,9 Гц, 1H), 4,54 (с, 2H).
Промежуточное соединение, представленное в таблице ниже, получали в соответствии со способами, используемыми для синтеза 5-бром-4-(бромметил)-2-фторбензонитрила (Int-07). Следующие промежуточное соединение синтезировали с некритическими изменениями или заменами в приведенных в качестве примеров способах, которые мог бы осуществить специалист в данной области.
Таблица 3:
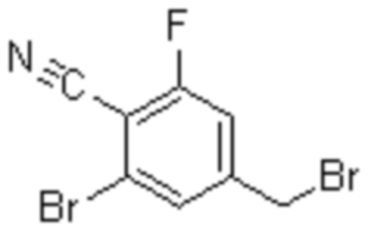
2-бром-4-(бромметил)-6-фторбензонитрил
Получение (4-циано-2,5-дифторфенил)метил метансульфоната (Int-09) в соответствии со схемой 5.
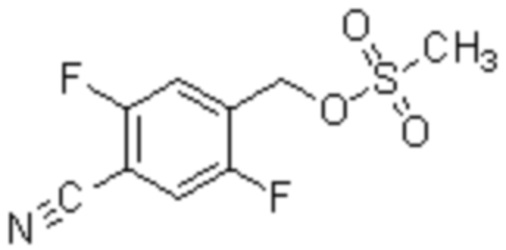
Схема 5:
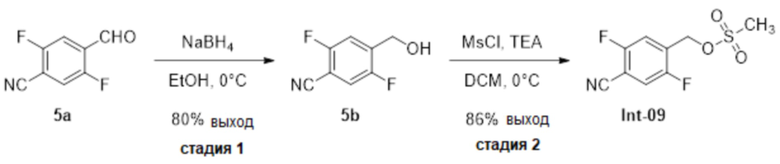
Стадия 1: Синтез 2,5-дифтор-4-(гидроксиметил)бензонитрила (5b)
Раствор 2,5-дифтор-4-формилбензонитрила (5a) (250 мг, 1,5 ммоль) в EtOH (5,0 мл) охлаждали до температуры 0ºC на ледяной бане и затем добавляли NaBH4 (60 мг, 1,6 ммоль). Смесь перемешивали при температуре 0ºC в течение 30 мин. Реакцию гасили при той же температуре путем добавления H2O (0,5 мл) и HCl (6,0 н, 0,32 мл). Смесь экстрагировали EtOAc. Органический слой промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали с получением 2,5-дифтор-4-(гидроксиметил)бензонитрила (5b) (202 мг, 80%-ный выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3) δ 7,44 (дд, J=5,62, 8,80 Гц, 1H), 7,30 (дд, J=4,89, 8,56 Гц, 1H), 4,85 (с, 2H).
Стадия 2: Синтез (4-циано-2,5-дифторфенил)метил метансульфоната (Int-09)
Раствор 2,5-дифтор-4-(гидроксиметил)бензонитрила (5b) (915 мг, 5,41 ммоль) в DCM (25,0 мл) охлаждали до температуры 0ºC и затем добавляли TEA (871 мг, 5,84 ммоль) и MsCl (649 г, 5,66 ммоль). Через 2 ч реакционную смесь непосредственно помещали на SiO2 и очищали с помощью флеш хроматографии (40 г, SiO2, 0-75% EtOAc/гептан) с получением (4-циано-2,5-дифторфенил)метил метансульфоната (Int-09) (1,15 г, 86%-ный выход) в виде прозрачного масла, которое затвердевало при стоянии. 1H ЯМР (400 МГц, CDCl3) δ 7,44-7,39 (м, 2H), 5,34-5,32 (м, 2H), 3,14 (с, 3H).
Промежуточное соединение, представленные в таблице ниже, получали в соответствии со способами, используемыми для синтеза (4-циано-2,5-дифторфенил)метил метансульфоната (Int-09). Следующие промежуточное соединение синтезировали с некритическими изменениями или замены приведенных в качестве примеров способов, которые сможет осуществить специалист в данной области техники.
Таблица 4:
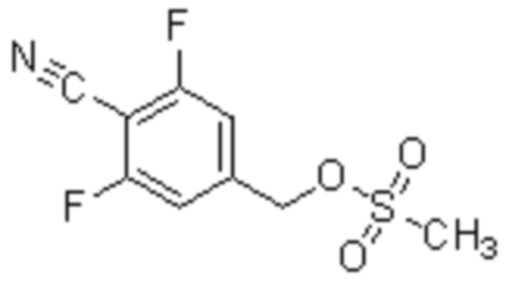
(4-циано-3,5-дифторфенил)метил метансульфонат
Получение 2-фтор-4-(гидроксиметил)-5-метилбензонитрила (Int-11) в соответствии со схемой 6.
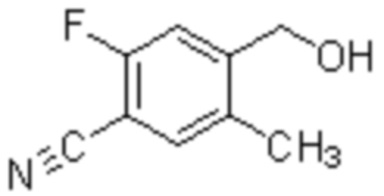
Схема 6:
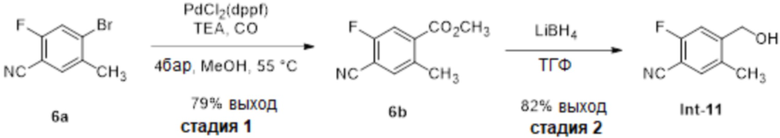
Стадия 1: Синтез метил 4-циано-5-фтор-2-метилбензоата (6b)
К раствору 4-бром-2-фтор-5-метилбензонитрила (6a) (1,0 г, 4,67 ммоль) и TEA (1,7 г, 17 ммоль) в MeOH (30,0 мл) в сосуде из нержавеющей стали емкостью 100 мл добавляли PdCl2(dppf) (247 мг, 0,327 ммоль). В сосуде создавали давление CO до 4 бар и перемешивали при температуре 55ºC в течение 20 ч. Реакционную смесь фильтровали и концентрировали. Остаток очищали с помощью флеш хроматографии (40 г, SiO2, 0-55% EtOAc/гептан) с получением метил 4-циано-5-фтор-2-метилбензоата (6b) (716 мг, 79%-ный выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3) δ 7,74 (д, J=9,4 Гц, 1H), 7,52 (д, J=6,1 Гц, 1H), 3,95 (с, 3H), 2,59 (с, 3H).
Стадия 2: Синтез 2-фтор-4-(гидроксиметил)-5-метилбензонитрила (Int-11)
К раствору метил 4-циано-5-фтор-2-метилбензоата (6b) (710 мг, 3,68 ммоль) в ТГФ (18,4 мл) добавляли LiBH4 (120 мг, 5,51 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. Реакцию гасили H2O (3 мл). Смесь перемешивали в течение 30 мин и затем охлаждали на ледяной бане. Смесь осторожно гасили HCl (6,0 н, 0,60 мл). ТГФ удаляли в вакууме и остаток распределяли между EtOAc и 1:1 H2O/насыщенным водным раствором NaHCO3. Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью флеш хроматографии (24 г, SiO2, 0-80% EtOAc/гептан) с получением 2-фтор-4-(гидроксиметил)-5-метилбензонитрила (Int-11) (495 мг, 82%-ный выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3) δ 7,44-7,35 (м, 2H), 4,74 (д, J=5,5 Гц, 2H), 2,26 (с, 3H), 1,84 (т, J=5,5 Гц, 1H); 19F ЯМР (376 МГц, CDCl3) δ -110,29 (дд, J=5,7, 10,3 Гц).
Получение (3-амино-5-фтор-4-метокси-1,2-бензоксазол-6-ил)метанола (Int-12) в соответствии со схемой 7.
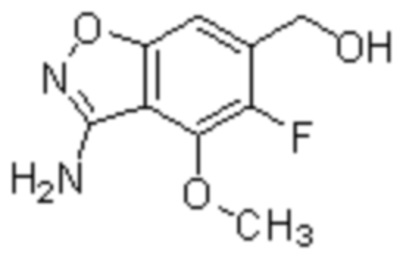
Схема 7:
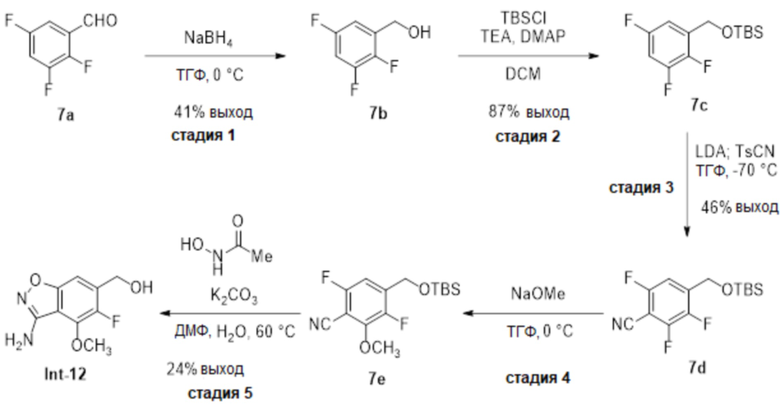
Стадия 1: Синтез (2,3,5-трифторфенил)метанола (7b)
К раствору 2,3,5-трифторбензальдегида (7a) (1,8 г, 11 ммоль) в ТГФ (30 мл) по частям при температуре 0ºC добавляли NaBH4 (468 мг, 12,4 ммоль). Смесь перемешивали при температуре 0ºC в течение 2 ч. ЖХМС анализ показывал, что исходное вещество расходовалось. Реакцию гасили путем добавления H2O (10 мл) и концентрировали досуха. Остаток очищали с помощью флеш хроматографии (40 г, SiO2, 0-50% EtOAc/гептан) с получением (2,3,5-трифторфенил)метанола (7b) (740 мг, 41%-ный выход) в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 7,10-6,96 (м, 1H), 6,94-6,78 (м, 1H), 4,81 (д, J=5,9 Гц, 2H), 1,91 (т, J=6,1 Гц, 1H).
Стадия 2: Синтез трет-бутил(диметил)[(2,3,5-трифторфенил)метокси]силана (7c)
К раствору (2,3,5-трифторфенил)метанола (7b) (740 мг, 4,56 ммоль) в DCM (20 мл) добавляли DMAP (27,9 мг, 0,228 ммоль), TEA (639 мг, 6,85 ммоль) и раствор TBSCl (894 мг, 5,93 ммоль) в DCM (5 мл). Смесь перемешивали при температуре окружающей среды в течение 18 ч. ЖХМС показывал, что исходное вещество расходовалось. Смесь концентрировали досуха и остаток очищали с помощью флеш хроматографии (40 г, SiO2, 0-10% EtOAc/петролейный эфир) с получением трет-бутил(диметил)[(2,3,5-трифторфенил)метокси]силана (7c) (1,1 г, 87%-ный выход) в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 7,06-6,95 (м, 1H), 6,87-6,70 (м, 1H), 4,79 (с, 2H), 0,95 (с, 9H), 0,13 (с, 6H).
Стадия 3: Синтез 4-({[трет-бутил(диметил)силил]окси}метил)-2,3,6-трифторбензонитрила (7d)
Раствор LDA (0,07 M в ТГФ, 20,0 мл, 1,41 ммоль ) в ТГФ (20 мл) при охлаждении до температуры —70ºC и затем обрабатывали по каплям раствором трет-бутил(диметил)[(2,3,5-трифторфенил)метокси]силана (7c) (300 мг, 1,09) в ТГФ (5 мл) в течение 5 мин. Смесь перемешивали в течение 2 ч при температуре —70ºC и затем обрабатывали по каплям раствором п-толилсульфонил цианида (216 мг, 1,19 ммоль) в ТГФ (5 мл) в течение 10 мин. Смесь перемешивали при температуре —70ºC в течение 1 ч. Смесь гасили путем добавления насыщенного водного раствора NH4Cl и распределяли между EtOAc (60 мл) и H2O (60 мл). Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью флеш хроматографии (SiO2, 3:1-10:1 EtOAc/петролейный эфир). Объединенные фракции, содержащие продукт, опять очищали путем препаративной ВЭЖХ с колонкой Agela DuraShell C18 (150×25 мм, размер частиц 5 мкм), которые элюировали 70-100% MeCN/H2O (+0,04% NH4OH, +10 мм NH4HCO3) со скоростью потока 25 мл/мин с получением 4-({[трет-бутил(диметил)силил]окси}метил)-2,3,6-трифторбензонитрила (7d) (150 мг, 46%-ный выход) в виде масла желтого цвета. 1H ЯМР (400 МГц, CDCl3) δ 7,10-7,02 (м, 1H), 4,69 (с, 2H), 0,81 (с, 9H), 0,00 (с, 6H).
Стадия 4: Синтез 4-({[трет-бутил(диметил)силил]окси}метил)-3,6-дифтор-2-метоксибензонитрила (7e)
К раствору 4-({[трет-бутил(диметил)силил]окси}метил)-2,3,6-трифторбензонитрила (7d) (150 мг, 0,498 ммоль) в ТГФ (20 мл) добавляли NaOMe (71,7 мг, 0,398 ммоль) при температуре 0ºC. Смесь перемешивали при температуре 0ºC в течение 1 ч. Смесь гасили H2O и распределяли между EtOAc и H2O. Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали с получением сырого 4-({[трет-бутил(диметил)силил]окси}метил)-3,6-дифтор-2-метоксибензонитрила (7e) (150 мг, 96%-ный выход) в виде масла желтого цвета, которое использовали без дополнительной очистки.
Стадия 5: Синтез (3-амино-5-фтор-4-метокси-1,2-бензоксазол-6-ил)метанола (Int-12)
К раствору сырого 4-({[трет-бутил(диметил)силил]окси}метил)-3,6-дифтор-2-метоксибензонитрила (7e) (150 мг,0,479 ммоль) и N-гидроксиацетамида (108 мг, 1,33 ммоль) в ДМФ (10 мл) и H2O (2 мл) добавляли K2CO3 (397 мг, 2,87 ммоль). Смесь перемешивали при температуре 60ºC в течение 16 ч. Смесь фильтровали и фильтрат концентрировали досуха. Остаток очищали путем препаративной ВЭЖХ колонкой Agela DuraShell C18 (150×25 мм, размер частиц 5 мкм), которую элюировали 5-35% MeCN/H2O H2O (+0,04% NH4OH, +10 мм NH4HCO3) со скоростью потока 25 мл/мин с получением (3-амино-5-фтор-4-метокси-1,2-бензоксазол-6-ил)метанола (Int-12) (25 мг, 24%-ный выход за две стадии) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,12 (д, J=4,3 Гц, 1H), 6,03 (с, 2H), 5,47 (т, J=5,8 Гц, 1H), 4,62 (д, J=5,6 Гц, 2H), 4,05 (с, 3H); m/z (ESI+) 213,1 (M+H)+.
Получение 1-(метансульфонил)-1H-пиразола (Int-13) в соответствии со схемой 8.
Схема 8:
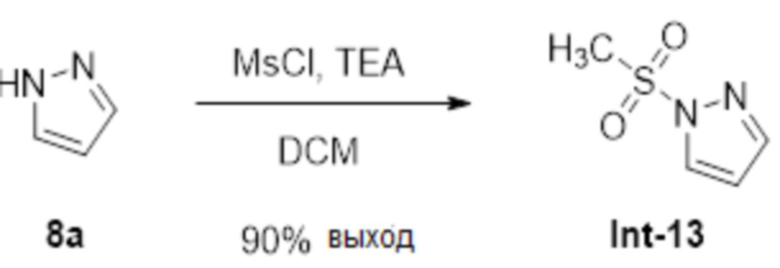
К раствору 1H-пиразола (8a) (33,0 г, 485 ммоль) и TEA (73,6 мг, 727 ммоль) в DCM медленно при температуре 0ºC добавляли MsCl (73,9 г, 645 ммоль). Смесь перемешивали при температуре 0ºC в течение 10 мин и затем при комнатной температуре в течение 1 ч. ТСХ анализ (1:1 EtOAc/петролейный эфир) показывал, что исходное вещество расходовалось. Реакционную смесь разбавляли насыщенным водным раствором NH4Cl (200 мл) и смесь разделяли. Водный слой экстрагировали DCM (200 мл). Объединенные органические слои промывали насыщенным солевым раствором (300 мл) и насыщенным водным раствором Na2CO3 (300 мл), сушили над безводным Na2SO4, фильтровали и концентрировали с получением 1-(метансульфонил)-1H-пиразола (Int-13) (64 г, 90%-ный выход) в виде масла бледно-желтого цвета. 1H ЯМР (400 МГц, CDCl3) δ 8,04 (д, J=2,6 Гц, 1H), 7,86-7,79 (м, 1H), 6,46 (дд, J=1,6, 2,7 Гц, 1H), 3,33 (с, 3H).
Промежуточные соединения, приведенные в таблице ниже, получали в соответствии со способами, используемыми для синтеза 1-(метансульфонил)-1H-пиразола (Int-13). Следующие промежуточные соединения синтезировали с некритическими изменениями или заменами в приведенных в качестве примеров способах, которые мог бы осуществить специалист в данной области. Если указано, региоизомерные смеси выделяли без дальнейшего разделения.
Таблица 5:
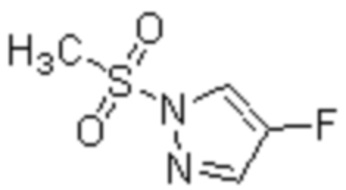
4-фтор-1-(метансульфонил)-1H-пиразола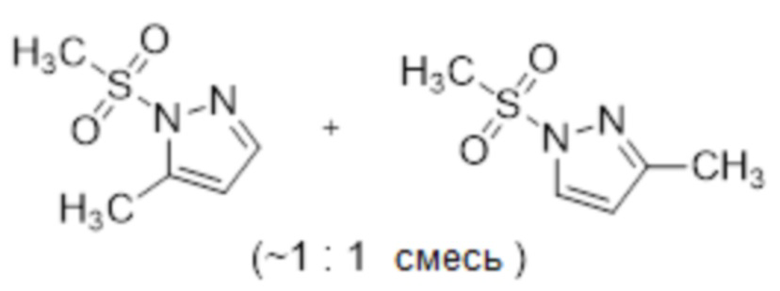
1-(метансульфонил)-5-метил-1H-пиразола и 1-(метансульфонил)-3-метил-1H-пиразола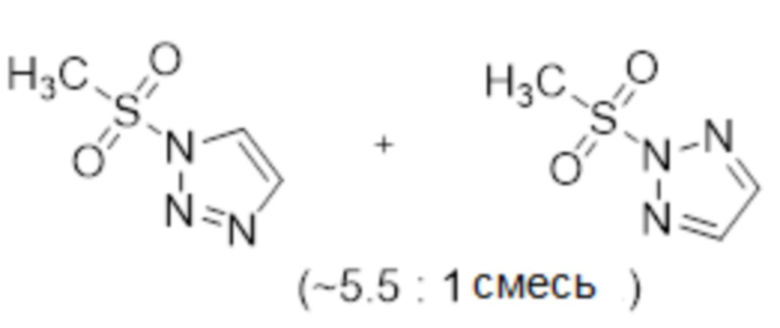
1-(метансульфонил)-1H-1,2,3-триазол и 2-(метансульфонил)-2H-1,2,3-триазол
Получение 4-({[трет-бутил(диметил)силил]окси}метил)-1-(метансульфонил)-1H-пиразола (Int-17) в соответствии со схемой 9.
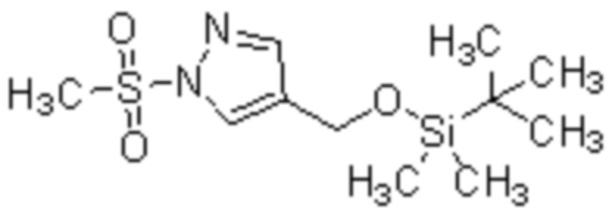
Схема 9:
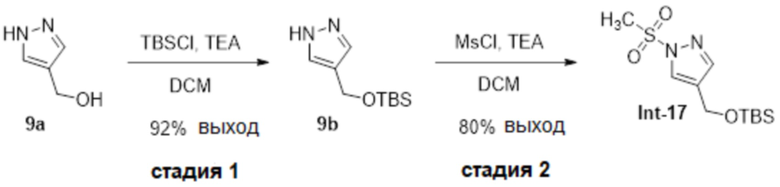
Стадия 1: Синтез 4-({[трет-бутил(диметил)силил]окси}метил)-1H-пиразола (9b).
К раствору (1H-пиразол-4-ил)метанола (9a) (500 мг, 5,1 ммоль) в DCM (10,0 мл) добавляли TBSCl (845 мг, 5,6 ммоль), TEA (774 мг, 7,7 ммоль) и DMAP (31,1 мг, 0,26 ммоль). Раствор перемешивали при комнатной температуре в течение 16 ч. ЖХМС анализ показывал, что исходное вещество расходовалось с образованием желаемой массы продукта. Смесь разбавляли DCM (20 мл) и последовательно промывали H2O (20 мл), насыщенным водным раствором NaHCO3 (20 мл) и насыщенным солевым раствором (20 мл). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением 4-({[трет-бутил(диметил)силил]окси}метил)-1H-пиразола (9b) (1,0 г, 92%-ный выход), которое использовали без дополнительной очистки. m/z (ESI+) 212,8 (M+H)+.
Стадия 2: Синтез 4-({[трет-бутил(диметил)силил]окси}метил)-1-(метансульфонил)-1H-пиразола (Int-17).
К раствору 4-({[трет-бутил(диметил)силил]окси}метил)-1H-пиразола (9b) (1,0 г, 4,7 ммоль) в DCM (15,0 мл) добавляли TEA (619 мг, 6,1 ммоль). Смесь охлаждали до температуры 0ºC на ледяной бане. Добавляли по каплям MsCl (3,8 г, 33,0 ммоль). Смесь перемешивали в течение 2 ч при температуре 0ºC и при комнатной температуре в течение 16 ч. ЖХМС анализ показывал, что исходное вещество расходовалось с образованием желаемой массы продукта. Смесь разбавляли DCM (100 мл) и последовательно промывали H2O (50 мл), насыщенным водным раствором NaHCO3 (50 мл) и насыщенным солевым раствором (50 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением 4-({[трет-бутил(диметил)силил]окси}метил)-1-(метансульфонил)-1H-пиразола (Int-17) (1,1 г, 80%-ный выход), которое использовали без дополнительной очистки. m/z (ESI+) 291,1 (M+H)+.
Промежуточное соединение, представленное в таблице ниже, получали в соответствии со способами, используемыми для синтеза 4-({[трет-бутил(диметил)силил]окси}метил)-1-(метансульфонил)-1H-пиразола (Int-17). Следующие промежуточное соединение синтезировали с некритическими изменениями или заменами в приведенных в качестве примеров способах, которые мог бы осуществить специалист в данной области. Если указано, региоизомерные смеси выделяли без дальнейшего разделения.
Таблица 6:
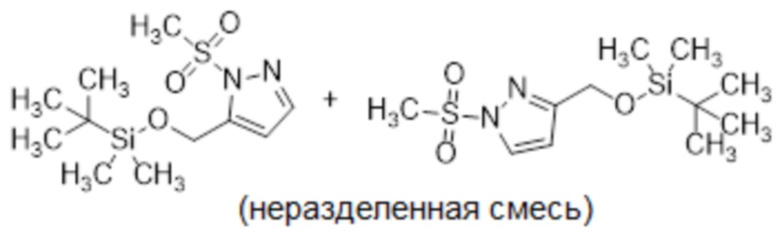
5-({[трет-бутил(диметил)силил]окси}метил)-1-(метансульфонил)-1H-пиразола и 3-({[трет-бутил(диметил)силил]окси}метил)-1-(метансульфонил)-1H-пиразола
Получение 2,4,6-триметоксибензол-1-сульфонил хлорида (Int-19) в соответствии со схемой 10.
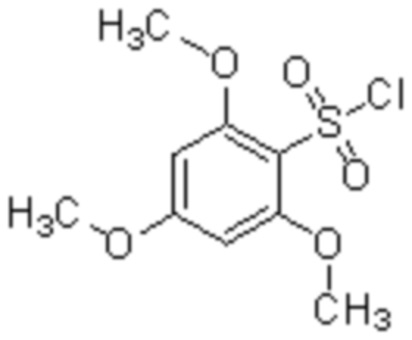
Схема 10:
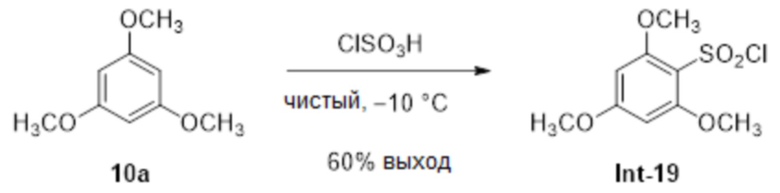
Хлорсульфоновую кислоту (15,0 мл) охлаждали до температуры —10ºC и одной порцией добавляли 1,3,5-триметоксибензол (10a) (1,4 г, 8,4 ммоль). Смесь перемешивали при температуре —10ºC в течение 15 мин. ТСХ анализ (1:1 EtOAc/петролейный эфир) указывал на расходование исходного вещества. Реакцию гасили осторожным выливанием в ледяную воду. Смесь экстрагировали DCM (3×100 мл). Объединенные органические экстракты промывали насыщенным водным раствором NaHCO3 (50 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью флеш хроматографии (20 г, SiO2, 0-50% EtOAc/петролейный эфир) с получением 2,4,6-триметоксибензол-1-сульфонил хлорида (Int-19) (1,8 г, 60%-ный выход) в виде твердого вещества, которое использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 6,12 (с, 2H), 3,96 (с, 6H), 3,89 (с, 3H).
Промежуточное соединение, представленное в таблице ниже, получали в соответствии со способами, используемыми для синтеза 2,4,6-триметоксибензол-1-сульфонил хлорида (Int-19). Следующие промежуточное соединение синтезировали с некритическими изменениями или заменами в приведенных в качестве примеров способах, которые мог бы осуществить специалист в данной области.
Таблица 7:
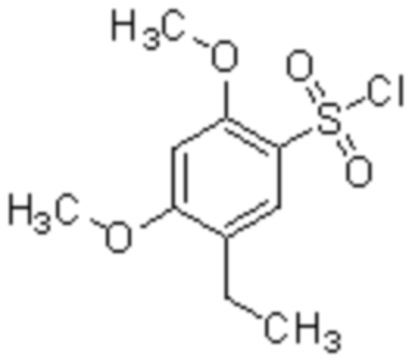
5-этил-2,4-диметоксибензол-1-сульфонил хлорид
Получение 2-метокси-5-(трифторметокси)бензол-1-сульфонил хлорида (Int-21) в соответствии со схемой 11.
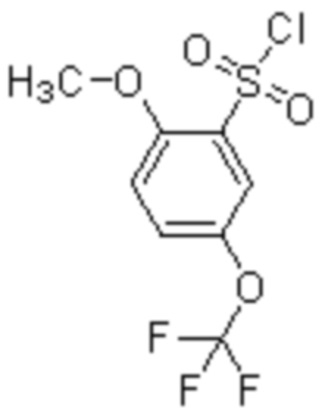
Схема 11:
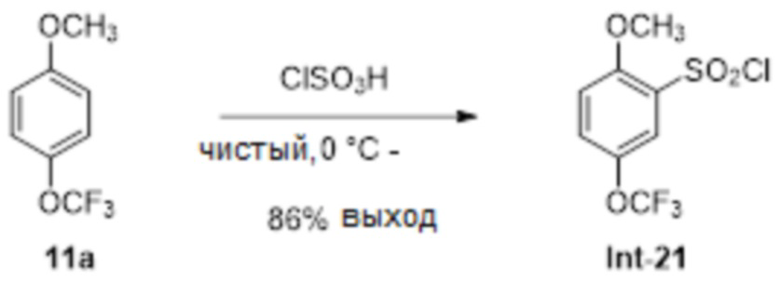
Хлорсульфоновую кислоту (26,0 мл) охлаждали до температуры 0ºC и одной порцией добавляли 1-метокси-4-(трифторметокси)бензол (11a) (2,0 г, 10,4 ммоль). Смесь перемешивали при комнатной температуре в течение 18 ч. Реакцию гасили осторожным выливанием в ледяную воду. Смесь экстрагировали EtOAc (2×60 мл). Объединенные органические экстракты промывали насыщенным водным раствором Na2CO3 (50 мл), сушили над Na2SO4, фильтровали и концентрировали с получением 2-метокси-5-(трифторметокси)бензол-1-сульфонил хлорида (Int-21) (2,6 г, 86%-ный выход) в виде масла желтого цвета, которое использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 7,85 (д, J=2,8 Гц, 1H), 7,56 (дд, J=2,4, 9,1 Гц, 1H), 7,16 (д, J=9,2 Гц, 1H), 4,08 (с, 3H).
Получение 2-метокси-5,6,7,8-тетрагидронафталин-1-сульфонил хлорида и 3-метокси-5,6,7,8-тетрагидронафталин-2-сульфонил хлорида (Int-22) в соответствии со схемой 12.
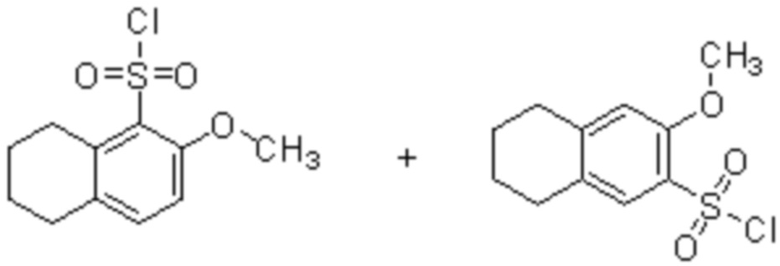
Схема 12:
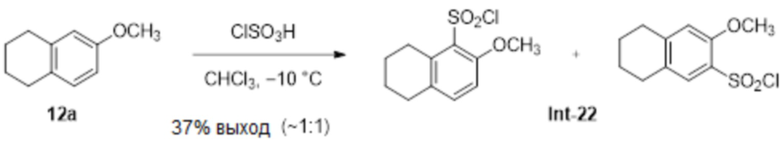
Смесь CHCl3 (10,0 мл) и хлорсульфоновую кислоту (1,0 мл) охлаждали до температуры —10ºC и добавляли 6-метокси-1,2,3,4-тетрагидронафталин (12a) (1,0 г, 6,1 ммоль). Смесь перемешивали при температуре —10ºC в течение 15 мин. ТСХ анализ (1:1 EtOAc/петролейный эфир) указывал на расходование исходного вещества. Реакцию гасили осторожным выливанием в ледяную воду. Смесь экстрагировали DCM (3×50 мл). Объединенные органические экстракты промывали насыщенным водным раствором NaHCO3 (50 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью флеш хроматографии (40 г, SiO2, 0-50% EtOAc/петролейный эфир) с получением 2-метокси-5,6,7,8-тетрагидронафталин-1-сульфонил хлорида и 3-метокси-5,6,7,8-тетрагидронафталин-2-сульфонил хлорида (Int-22) (~1:1 смесь, 600 мг, 37%-ный выход) в виде смолы бледно-желтого цвета. 1H ЯМР (400 МГц, CDCl3) δ 7,65 (с, 1H), 7,35 (д, J=8,5 Гц, 1H), 6,92 (д, J=8,5 Гц, 1H), 6,79 (с, 1H), 4,01 (с, 6H), 3,23 (т, J=6,0 Гц, 2H), 2,91-2,63 (м, 6H), 1,88-1,69 (м, 8H).
Промежуточное соединение, представленное в таблице ниже, получали в соответствии со способами, используемыми для синтеза 2-метокси-5,6,7,8-тетрагидронафталин-1-сульфонил хлорида и 3-метокси-5,6,7,8-тетрагидронафталин-2-сульфонил хлорида (Int-22). Следующие промежуточные соединения синтезировали с некритическими изменениями или замены приведенных в качестве примеров способов, которые сможет осуществить специалист в данной области техники.
Таблица 8:
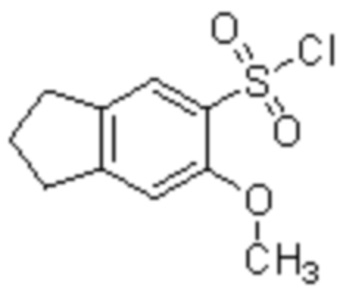
6-метокси-2,3-дигидро-1H-инден-5-сульфонил хлорид
Получение 4-циклопропил-2,6-диметоксибензол-1-сульфонил хлорида (Int-24) в соответствии со схемой 13.
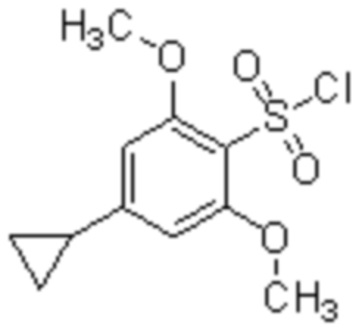
Схема 13:

Раствор (13a) (1,0 г, 5,61 ммоль) (J. Org. Chem. 2008, 7481-7485) и TMEDA (717 мг, 6,17 ммоль) в петролейном эфире (15,0 мл) охлаждали на ледяной бане и затем обрабатывали по каплям н-BuLi (2,5 M в гексане, 2,5 мл, 6,17 ммоль) с помощью капельной воронки, поддерживая температуру <5ºC (внутренняя). Смесь перемешивали при температуре 0ºC в течение 20 мин и затем охлаждали до температуры —70ºC на бане сухой лед/ацетон. Медленно добавляли предварительно охлажденный раствор (—65ºC) SO2 (5,4 г, 84,2 ммоль) в Et2O (100 мл), поддерживая температуру <—60ºC (внутренняя). Реакционную смесь бледно-желтого цвета медленно нагревали до температуры 10ºC. Полученные твердые продукты собирали путем фильтрации и промывали сухим Et2O. Слой продукта на фильтре суспендировали в гексане (30 мл) и смесь охлаждали до температуры 0ºC. К холодной суспензии медленно добавляли раствор SOCl2 (757 мг, 5,61 ммоль) в гексане (20 мл), поддерживая температуру <3ºC (внутренняя). Полученную смесь перемешивали при температуре 0ºC в течение 18 ч. Раствор фильтровали и слой продукта на фильтре промывали холодным гексаном (20 мл). Твердые продукты обрабатывали EtOAc (50 мл) и промывали H2O (50 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением 4-циклопропил-2,6-диметоксибензол-1-сульфонил хлорида (Int-24) (856 мг, 55%-ный выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3) δ 6,32 (с, 2H), 3,97 (с, 6H), 1,93 (тт, J=5,0, 8,3 Гц, 1H), 1,18-1,11 (м, 2H), 0,86-0,80 (м, 2H).
Получение 4-метокси-6-((4-метил-1H-пиразол-1-ил)метил)бензо[d]изоксазол-3-амина (Int-25) в соответствии со схемой 14.
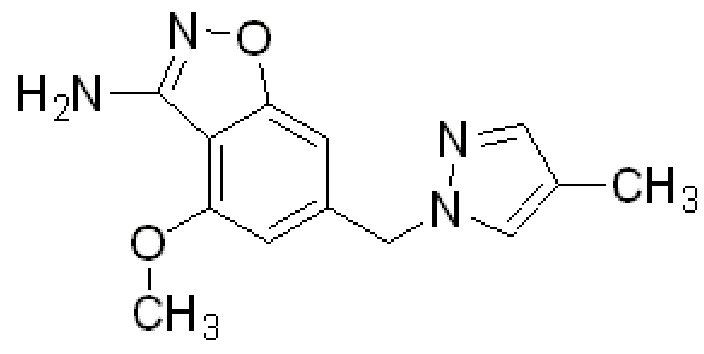
Схема 14:
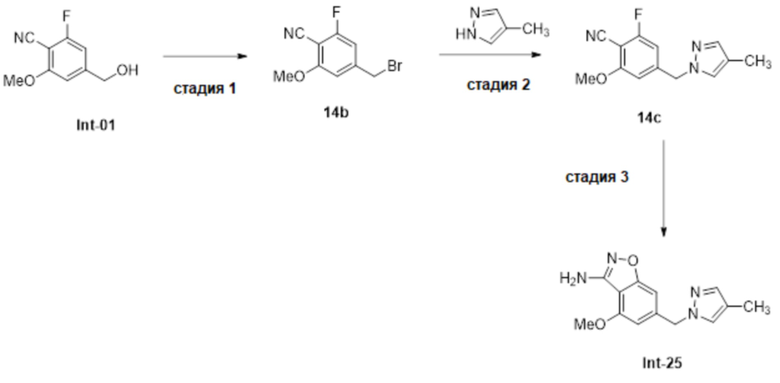
Стадия 1: Синтез 4-(бромметил)-2-фтор-6-метоксибензонитрила (14b).
К раствору 2-фтор-4-(гидроксиметил)-6-метоксибензонитрила (Int-01) (8,0 г, 44,2 ммоль) и PPh3 (18,7 г, 71,2 ммоль) в ацетонитриле (400 мл) добавляли Br2 (11,8 г, 73,8 ммоль) и смесь нагревали при температуре 55ºC в течение 2 ч. Добавляли воду и избыток Na2SO3 и смесь экстрагировали EtOAc. Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии (петролейный эфир/EtOAc=10/1) с получением указанного в заголовке соединения (9,7 г, 91%) в виде твердого вещества белого цвета, которое напрямую использовали на следующей стадии.
Стадия 2: Синтез 2-фтор-6-метокси-4-((4-метил-1H-пиразол-1-ил)метил)бензонитрила (14c).
Смесь 4-(бромметил)-2-фтор-6-метоксибензонитрила (14b) (100 мг, 0,41 ммоль), 4-метил-1H-пиразола (40 мг, 0,49 ммоль) и K2CO3 (113 мг, 0,82 ммоль) в ДМФ (5 мл) нагревали при температуре 60ºC в течение ночи. Смесь разбавляли водой, экстрагировали EtOAc и органический экстракт промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Масштаб реакции был увеличен соответственно с использованием 4-(бромметил)-2-фтор-6-метоксибензонитрила (14b) (500 мг, 2,05 ммоль) и две партии объединяли и очищали путем колоночной хроматографии (DCM/MeOH=10/1) с получением указанного в заголовке соединения (380 мг, 63%) в виде твердого вещества желтого цвета. m/z 246,0 [M+H]+.
Стадия 3: Синтез 4-метокси-6-((4-метил-1H-пиразол-1-ил)метил)бензо[d]изоксазол-3-амина (Int-25)
К раствору N-гидроксиацетамида (238 мг, 3,18 ммоль) в безводном ДМФ (13 мл) при температуре 0ºC добавляли t-BuOK (357 мг, 3,18 ммоль) и смесь перемешивали в течение 30 мин. Затем добавляли 2-Фтор-6-метокси-4-((4-метил-1H-пиразол-1-ил)метил)бензонитрил (14c) (260 мг, 1,06 ммоль) и смеси давали нагреться до комнатной температуры и перемешивали в течение ночи. Добавляли воду и смесь экстрагировали EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии (DCM/MeOH=50/1) с получением указанного в заголовке соединения (150 мг, 55%) в виде твердого вещества желтого цвета. m/z 259,1 [M+H]+. 1H ЯМР (400 МГц, CDCl3) δ 7,37 (с, 1H), 7,20 (с, 1H), 6,76 (с, 1H), 6,43 (с, 1H), 5,31 (с, 2H), 3,90 (с, 3H), 2,07 (с, 3H).
Получение 2,6-диметоксибензолсульфонил хлорида (Int-26) в соответствии со схемой 15.
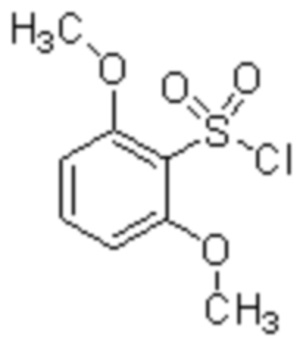
Схема 15:
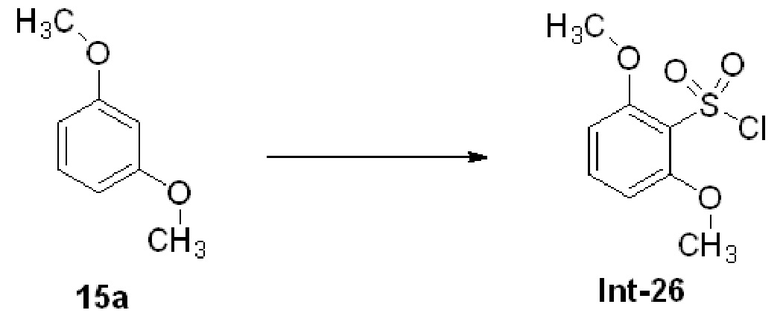
К раствору 1,3-диметоксибензола (5,0 г, 36 ммоль) и TMEDA (4,6 г, 39,8 ммоль) в н-гексане (100 мл) при температуре 0ºC в атмосфере N2 добавляли по каплям н-BuLi (2,5 M раствор в гексане, 16,0 мл, 39,8 ммоль), поддерживая внутреннюю температуру реакционной смеси ниже 5ºC. Смесь перемешивали при температуре 0ºC в течение 20 мин, затем охлаждали до температуры -78ºC и барботировали газообразный SO2 в течение 20 мин. Затем смеси давали медленно нагревали до температуры 10ºC и полученный осадок собирали путем фильтрации и промывали сухим диэтиловым эфиром. Твердый продукт суспендировали в н-гексане (100 мл), охлаждали до температуры 0ºC и добавляли по каплям раствор SO2Cl2 (4,9 г, 36 ммоль) в н-гексане (20 мл), поддерживая внутреннюю температуру ниже 3ºC. Затем смесь перемешивали при температуре 0ºC в течение 1 ч и твердые продукты собирали путем фильтрации и промывали холодным н-гексаном. Твердые продукты затем распределяли между диэтиловым эфиром и водой, слои разделяли и водный слой затем экстрагировали диэтиловым эфиром. Объединенные органические экстракты сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (4,0 г, 47%) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3) δ 7,54 (т, J=8,4 Гц, 1H), 6,66 (д, J=8,4 Гц, 2H), 3,97 (с, 6H).
Получение N-(6-бром-4-метоксибензо[d]изоксазол-3-ил)-2,6-диметоксибензолсульфонамида (Int-27) в соответствии со способом AA.
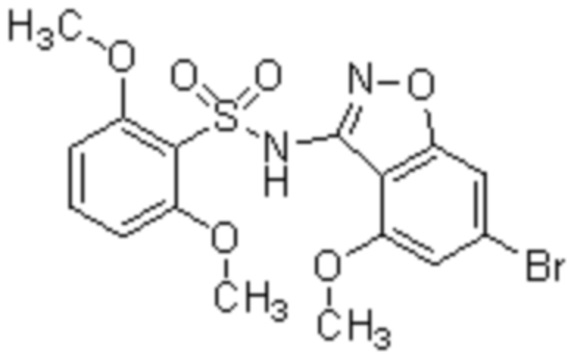
Способ AA:
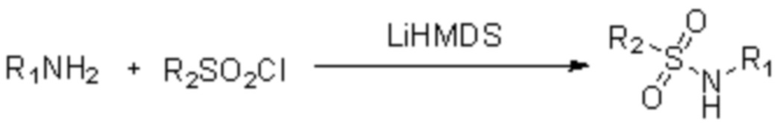
К раствору амина (0,5 ммоль, 1,0 экв.) в безводном ТГФ (10 мл) при температуре -78ºC в атмосфере N2 добавляли по каплям LiHMDS (1 M раствор в ТГФ, 3 экв.) и смесь перемешивали при температуре -78ºC в течение 30 мин. Затем добавляли по каплям раствор сульфонилхлорида (1,5 экв.) в безводном ТГФ (2,0 мл) и смеси давали нагреться до комнатной температуры и перемешивали в течение ночи. Добавляли воду и смесь экстрагировали EtOAc. Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии или преп. ТСХ с получением указанного в заголовке соединения. Варианты вышеуказанных условий отмечены в таблице ниже.
Таблица 9:
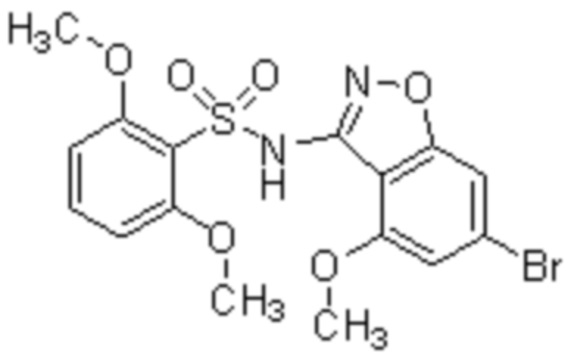
N-(6-бром-4-метоксибензо[d]-изоксазол-3-ил)-2,6-диметокси-бензолсульфонамид Int-27
6-бром-4-метоксибензо[d]изоксазол-3-амин (14b)
Преп. ТСХ (DCM/MeOH=100/1)
Получение 7-бром-5-метилбензо[d]изоксазол-3-амина (Int-28) в соответствии со схемой 16.
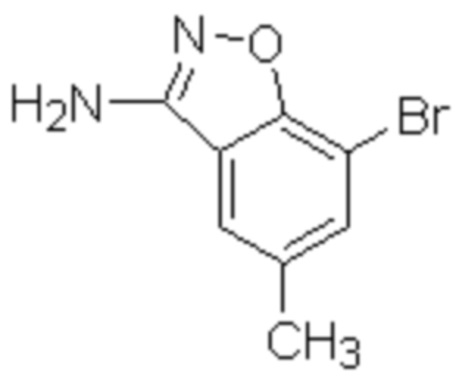
Схема 16:
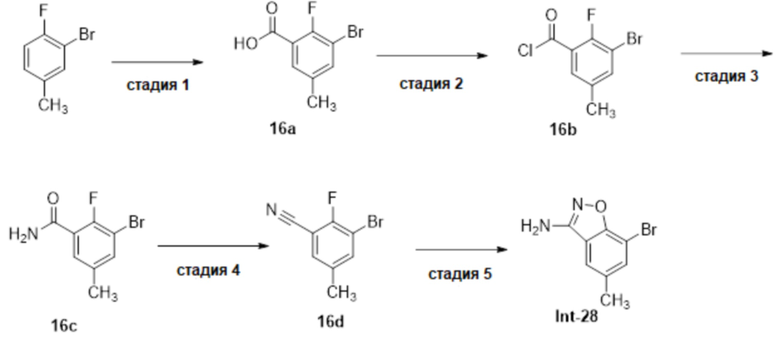
Стадия 1: Синтез 3-бром-2-фтор-5-метилбензойной кислоты (16a)
К раствору 2-бром-1-фтор-4-метилбензола (10,0 г, 53 ммоль) и диизопропиламина (5,9 г, 58 ммоль) в безводном ТГФ (200 мл) при температуре -78ºC в атмосфере N2 добавляли по каплям н-BuLi (2,5 M раствор в гексане, 25,6 мл, 64,0 ммоль) и смесь перемешивали при температуре -78ºC в течение 1 ч. Добавляли избыток твердого CO2 (сухой лед) добавляли и перемешивание продолжали при температуре -78ºC в течение 3 ч. Смесь разбавляли водой (500 мл) и экстрагировали EtOAc (500 мл). Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (12,3 г, 100%) в виде твердого вещества коричневого цвета, которое использовали на следующей стадии без дополнительной очистки. m/z 232,8 [M+H]+.
Стадия 2: Синтез 3-бром-2-фтор-5-метилбензоил хлорида (16b)
К раствору 3-бром-2-фтор-5-метилбензойной кислоты (16a) (12,3 г, 53 ммоль) и ДМФ (4 капли) в DCM (100 мл) при комнатной температуре в атмосфере N2 добавляли по каплям оксалил хлорид (13,0 г, 106 ммоль) и смесь перемешивали в течение 2 ч. Смесь концентрировали при пониженном давлении с получением указанного в заголовке соединения (14,0 г, 100%) в виде твердого вещества коричневого цвета, которое использовали на следующей стадии без дополнительной очистки.
Стадия 3: Синтез 3-бром-2-фтор-5-метилбензамида (16c)
Раствор 3-бром-2-фтор-5-метилбензоил хлорида (16b) (14,0 г, 53 ммоль) в DCM (100 мл) добавляли по каплям к 30%-ному водному раствору гидроксида аммония (100 мл) и смесь перемешивали в течение 2 ч. Смесь разбавляли EtOAc (200 мл), промывали водой (200 мл×3), насыщенным солевым раствором, и органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (12,0 г, 97%) в виде твердого вещества коричневого цвета, которое использовали на следующей стадии без дополнительной очистки. m/z 231,9 [M+H]+.
Стадия 4: Синтез 3-бром-2-фтор-5-метилбензонитрила (16d)
Раствор 3-бром-2-фтор-5-метилбензамида (16c) (10,0 г, 43,0 ммоль) и тионил хлорида (15,4 г, 129 ммоль) в ДМФ (100 мл) нагревали при температуре 100ºC в течение 3 ч. Смесь разбавляли EtOAc (200 мл) и промывали водой (400 мл×5), насыщенным солевым раствором и органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (5,0 г, 54%) в виде твердого вещества коричневого цвета, которое использовали на следующей стадии без дополнительной очистки. m/z 213,9 [M+H]+.
Стадия 5: Синтез 7-бром-5-метилбензо[d]изоксазол-3-амина (Int-28)
Суспензию N-гидроксиацетамида (5,27 г, 70,2 ммоль) и t-BuOK (7,88 г, 70,2 ммоль) в безводном ДМФ (200 мл) перемешивали при температуре 0ºC в течение 1 ч. Затем добавляли 3-бром-2-фтор-5-метилбензонитрил (16d) (5,0 г, 23,4 ммоль) и смеси давали нагреться до комнатной температуры и перемешивали в течение ночи. Смесь разбавляли EtOAc (300 мл), промывали водой (600 мл×4), насыщенным солевым раствором и органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии (петролейный эфир/EtOAc=10/1) с получением указанного в заголовке соединения (2,8 г, 52%) в виде твердого вещества желтого цвета. m/z 226,9 [M+H]+.
Получение N-(7-бром-5-метилбензо[d]изоксазол-3-ил)-2,6-диметоксибензолсульфонамида (Int-29) в соответствии со способом AB.
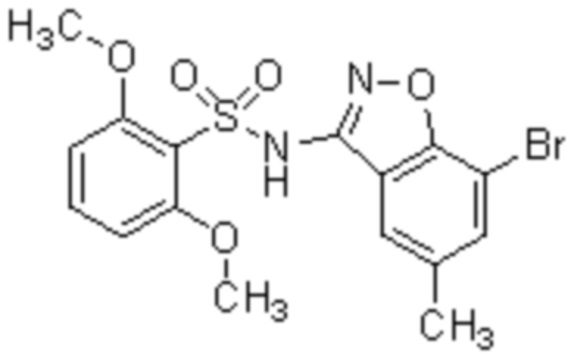
Способ AB:
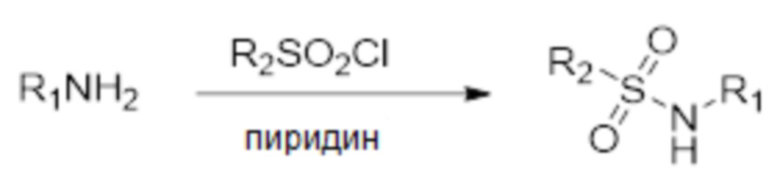
К раствору амина (0,2 ммоль, 1,0 экв.) в пиридине (2 мл) добавляли сульфонил хлорид (1,5 экв.) и смесь нагревали при температуре 120ºC при воздействии микроволнового облучения в течение 2 ч. Смесь распределяли между водой и EtOAc, слои разделяли и органический слой промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали путем преп. ТСХ с получением указанного в заголовке соединения. Варианты вышеуказанных условий отмечены в таблице ниже.
Таблица 10:
N-(7-бром-5-метилбензо[d]изоксазол-3-ил)-2,6-диметоксибензолсульфонамид, Int-29
бензолсульфонил хлорид (Int-26)
7-бром-5-метилбензо[d]изоксазол-3-амин (Int-28)
В процессе обработки органический слой промывали 0,1 M водн. HCl.
Преп. ТСХ (DCM/MeOH, 20/1)
Способ получения сульфонамидов:
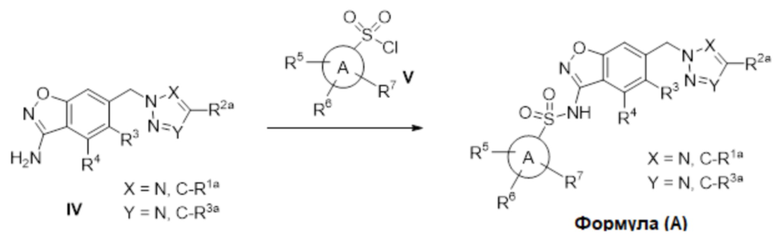
Способ A:
К раствору соединения типа IV (1,0 экв.) в пиридине (c=0,1 M) добавляли соединение типа V (1,2 экв.). Смесь перемешивали при нагревании при температуре от 80 до 120ºC в течение ~3-16 ч. Реакционную смесь охлаждали до комнатной температуры, концентрировали досуха и очищали стандартными способами, известными специалистам в данной области с получением соединения формулы (A).
Способ B:
К суспензии NaH (60%-ная дисперсия в минеральном масле, 3,0 экв.) в ТГФ (c=0,15 M) при температуре 0ºC добавляли по каплям раствор соединения типа IV (1,0 экв.) в смеси 1:1 ТГФ/ДМФ (c=0,15 M) или ТГФ (c=0,15 M). При той же температуре добавляли раствор соединения типа V (1,3 экв.) в смеси 2:1 ТГФ/ДМФ (c=0,15 M) или ТГФ (c=0,15 M). Реакционную смесь перемешивали при температуре 60ºC в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры, концентрировали досуха и очищали стандартными способами, известными специалистам в данной области с получением соединения формулы (A).
Способ C:
К раствору соединения типа IV (1,0 экв.) в ТГФ (c=0,3 M) добавляли NaOtPn (40% в PhMe, 1,0 экв.) и раствор соединения типа V (1,0 экв.) в ТГФ (c=0,3 M). Смесь перемешивали при температуре 60ºC в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры, концентрировали досуха и очищали стандартными способами, известными специалистам в данной области с получением соединения формулы (A).
Способ D:
К раствору соединения типа IV (1,0 экв.) и соединения типа V (1,2 экв.) в ACN (c=0,2 M) добавляли 0,05 M раствор ДМСО в ACN (1,0 мл/ммоль соединения типа IV, 0,05 экв. ДМСО), затем 3,5-лутидин (3,0 экв.). Смесь перемешивали при комнатной температуре в течение 16 часов, концентрировали досуха и очищали стандартными способами, известными специалистам в данной области с получением соединения формулы (A).
Примеры получения
Пример 01: Получение 5-этил-2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида в соответствии со схемой A.
Схема A:
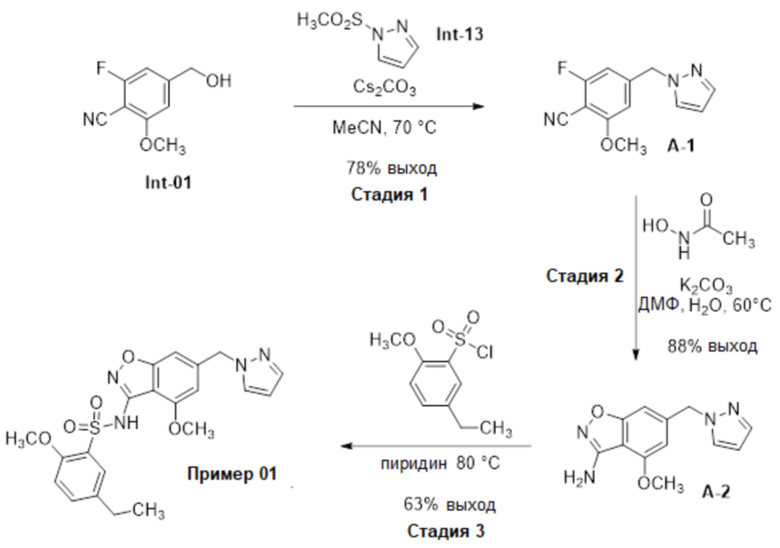
Стадия 1: Синтез 2-фтор-6-метокси-4-[(1H-пиразол-1-ил)метил]бензонитрила (A-1).
К раствору 2-фтор-4-(гидроксиметил)-6-метоксибензонитрила (Int-01) (7,0 г, 38,6 ммоль) и 1-(метансульфонил)-1H-пиразола (Int-13) (6,2 г, 42,5 ммоль) в MeCN (150 мл) добавляли Cs2CO3 (18,9 г, 58 ммоль). Смесь перемешивали при температуре 70ºC в течение 2 ч. ЖХМС анализ показывал, что исходное вещество расходовалось. Реакционную смесь фильтровали и фильтрат концентрировали досуха. Сырой остаток очищали с помощью флеш хроматографии (40 г, SiO2, 1:1 EtOAc/петролейный эфир) с получением 2-фтор-6-метокси-4-[(1H-пиразол-1-ил)метил]бензонитрила (A-1) (7,0 г, 78%-ный выход) в виде твердого вещества желтого цвета. m/z (ESI+) 231,8 (M+H)+.
Стадия 2: Синтез 4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (A-2).
К раствору 2-фтор-6-метокси-4-[(1H-пиразол-1-ил)метил]бензонитрила (A-1) (7,0 г, 30,3 ммоль) и N-гидроксиацетамида (6,8 г, 90,8 ммоль) в ДМФ (200 мл) и H2O (30 мл) добавляли K2CO3 (25,1 г, 182 ммоль). Смесь перемешивали при температуре 60ºC в течение 16 ч. ТСХ анализ (EtOAc) показывал, что исходное вещество расходовалось. Реакционную смесь концентрировали для удаления основного количества ДМФ и затем разбавляли H2O (100 мл). Полученный осадок собирали путем фильтрации. Слой продукта на фильтре промывали H2O (3×20 мл) и сушили в вакууме с получением 4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (A-2) (6,0 г). Вышеуказанный фильтрат экстрагировали EtOAc (2×30 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью флеш хроматографии (SiO2, EtOAc) с получением дополнительной партии 4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (A-2) (0,5 г). Две партии продукта объединяли и сушили в вакууме с получением 4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (A-2) (6,5 г, 88%-ный выход) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,88 (д, J=2,0 Гц, 1H), 7,51 (д, J=1,3 Гц, 1H), 6,70 (с, 1H), 6,63 (с, 1H), 6,31 (т, J=2,0 Гц, 1H), 6,08-5,78 (м, 2H), 5,52-5,31 (м, 2H), 3,93-3,73 (м, 3H). m/z (ESI+) 244,8 (M+H)+.
Стадия 3: Синтез 5-этил-2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 01) в соответствии со способом получения сульфонамида A.
К раствору 4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (A-2) (1,23 г, 5,032 ммоль) в пиридине (2,5 мл) добавляли 5-этил-2-метоксибензол-1-сульфонил хлорид (1,54 г, 6,54 ммоль). Реакционную смесь перемешивали при температуре 80ºC в течение 3,5 ч. ЖХМС анализ показывал, что исходное вещество расходовалось с образованием желаемой массы продукта. Реакционная смесь затвердевала при охлаждении. Твердый продукт растворяли в DCM и AcOH (1,4 мл) с минимальным количеством MeOH. Смесь очищали с помощью флеш хроматографии (40 г, SiO2, 10-70% MeOAc/гептан). Собирали чистые фракции, содержащие указанное в заголовке соединение. Неочищенные фракции опять очищали с помощью флеш хроматографии (40 г, SiO2, 10-70% MeOAc/гептан). Чистые фракции объединяли с ранее выделенными чистыми фракциями и концентрировали с получением твердого продукта белого цвета. Твердый продукт суспендировали в MeOAc, кипятили с обратным холодильником в течение 1 ч, и давали охладиться до комнатной температуры. Полученный твердый продукт собирали путем фильтрации и сушили в вакууме с получением 5-этил-2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 01) (1,4 г, 63%-ный выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,98 (с, 1H), 7,87 (д, J=2,0 Гц, 1H), 7,62 (д, J=2,3 Гц, 1H), 7,49 (д, J=1,5 Гц, 1H), 7,46 (дд, J=2,0, 8,5 Гц, 1H), 7,10 (д, J=8,5 Гц, 1H), 6,83 (с, 1H), 6,74 (с, 1H), 6,30 (т, J=2,0 Гц, 1H), 5,43 (с, 2H), 3,81 (с, 3H), 3,74 (с, 3H), 2,59 (кв, J=7,5 Гц, 2H), 1,13 (т, J=7,5 Гц, 3H); m/z (ESI+) 443,1 (M+H)+.
Пример 02: Получение 2,6-диметокси-N-{4-метокси-6-[(3-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида в соответствии со схемой B.
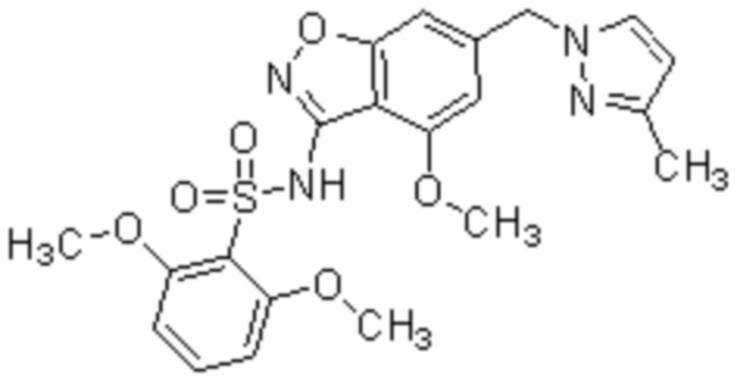
Пример 03: Получение 2,6-диметокси-N-{4-метокси-6-[(5-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида в соответствии со схемой B.
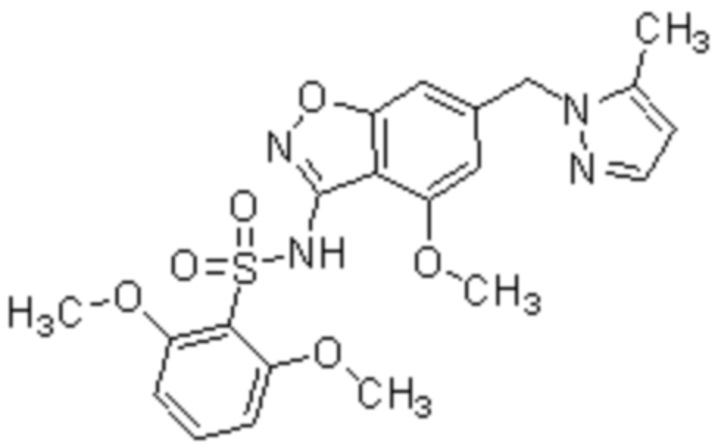
Схема B:
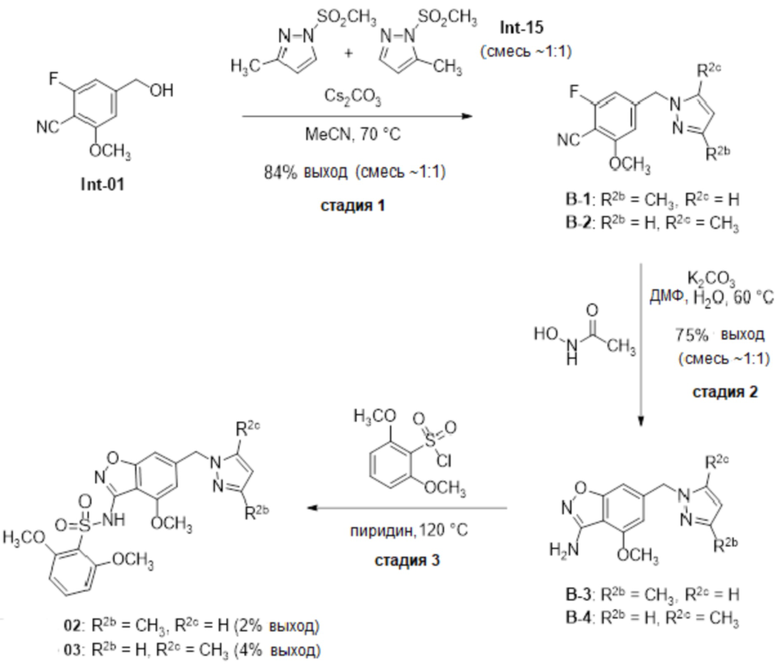
Стадия 1: Синтез 2-фтор-6-метокси-4-[(3-метил-1H-пиразол-1-ил)метил]бензонитрила (B-1) и 2-фтор-6-метокси-4-[(5-метил-1H-пиразол-1-ил)метил]бензонитрила (B-2).
К смеси (~1:1) 1-(метансульфонил)-3-метил-1H-пиразола и 1-(метансульфонил)-5-метил-1H-пиразола (Int-15) в MeCN (25 мл) добавляли 2-фтор-4-(гидроксиметил)-6-метоксибензонитрил (Int-01) (1,0 г, 5,5 ммоль) и Cs2CO3 (2,3 г, 7,2 ммоль). Смесь перемешивали при температуре 70ºC в течение 1 ч. ЖХМС анализ показывал, что исходное вещество расходовалось с образованием желаемой массы продукта. Реакционную смесь охлаждали до комнатной температуры и концентрировали досуха. Остаток очищали с помощью флеш хроматографии (20 г, SiO2, 100% EtOAc) с получением смеси (~1:1) 2-фтор-6-метокси-4-[(3-метил-1H-пиразол-1-ил)метил]бензонитрила (B-1) и 2-фтор-6-метокси-4-[(5-метил-1H-пиразол-1-ил)метил]бензонитрила (B-2) (1,13 г, 84%-ный выход) в виде смолы желтого цвета. m/z (ESI+) 245,8 (M+H)+.
Стадия 2: Синтез 4-метокси-6-[(3-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (B-3) и 4-метокси-6-[(5-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (B-4).
К смеси (~1:1) 2-фтор-6-метокси-4-[(3-метил-1H-пиразол-1-ил)метил]бензонитрила (B-1) и 2-фтор-6-метокси-4-[(5-метил-1H-пиразол-1-ил)метил]бензонитрила (B-2) (1,13 г, 4,73 ммоль) в ДМФ (20 мл) и H2O (3 мл) добавляли N-гидроксиацетамид (1,07 г, 14,2 ммоль) и K2CO3 (3,9 г, 28,4 ммоль). Смесь перемешивали при температуре 60ºC в течение 16 ч. ЖХМС анализ показывал, что исходное вещество расходовалось с образованием желаемой массы продукта. Смесь концентрировали досуха. Остаток обрабатывали EtOAc (30 мл) и промывали H2O (30 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью флеш хроматографии (20 г, SiO2, 100% EtOAc) с получением смеси (~1:1) 4-метокси-6-[(3-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (B-3) и 4-метокси-6-[(5-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (B-4) (916 мг, 75%-ный выход) в виде твердого вещества. m/z (ESI+) 258,8 (M+H)+.
Стадия 3: Синтез 2,6-диметокси-N-{4-метокси-6-[(3-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 02) и 2,6-диметокси-N-{4-метокси-6-[(5-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 03) в соответствии со способом получения сульфонамида A.
К смеси (~1:1) 4-метокси-6-[(3-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (B-3) и 4-метокси-6-[(5-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (B-4) (800 мг, 3,1 ммоль) в пиридине (10,0 мл) добавляли 2,6-диметоксибензол-1-сульфонил хлорид (Int-26) (1,1 г, 4,65 ммоль). Смесь перемешивали при температуре 120ºC в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали досуха. Остаток очищали с помощью флеш хроматографии (20 г, SiO2, 1:4 MeOH/EtOAc). Продукт опять очищали путем препаративной ВЭЖХ с колонкой YMC Triart (20×150 мм, размер частиц 7 мкм), которые элюировали смесью 23-63% MeCN/H2O (+0,225%-ная муравьиная кислота) со скоростью потока 25 мл/мин. Продукт опять очищали с помощью препаративной SFC с колонкой Diacel CHIRALCEL OD-H (30×250 мм, размер частиц 5 мкм), который элюировали 45% EtOH/CO2 (+0,1% NH4OH) со скоростью потока 60 мл/мин с получением 2,6-диметокси-N-{4-метокси-6-[(3-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 02) (63 мг, 4%-ный выход) в виде первого элюируемого пика в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,62 (шир. с, 1H), 7,74 (д, J=2,1 Гц, 1H), 7,49 (т, J=8,4 Гц, 1H), 6,81 (с, 1H), 6,77 (д, J=8,3 Гц, 3H), 6,07 (д, J=2,1 Гц, 1H), 5,33 (с, 2H), 3,93-3,84 (м, 3H), 3,77 (с, 6H), 2,15 (с, 3H); m/z (ESI+) 458,8 (M+H)+. 2,6-Диметокси-N-{4-метокси-6-[(5-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид (пример 03) (33 мг, 2%-ный выход) получали в виде второго элюируемого пика в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,64 (шир. с, 1H), 7,49 (т, J=8,6 Гц, 1H), 7,40 (д, J=1,7 Гц, 1H), 6,78 (с, 1H), 6,76 (с, 1H), 6,66 (с, 1H), 6,61 (с, 1H), 6,11 (дд, J=1,8, 0,9 Гц, 1H), 5,41 (с, 2H), 3,86 (с, 3H), 3,76 (с, 6H), 2,21 (с, 3H); m/z (ESI+) 458,8 (M+H)+.
Соединения по примерам, приведенные в таблице ниже, синтезировали в соответствии со способами, используемыми для синтеза 5-этил-2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 01), 2,6-диметокси-N-{4-метокси-6-[(3-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 02) и 2,6-диметокси-N-{4-метокси-6-[(5-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 03), и общим способом получения сульфонамидов A-D. Следующие соединения по примерам синтезировали с некритическими изменениями или замены приведенных в качестве примеров способов, которые сможет осуществить специалист в данной области техники. При необходимости разделение смесей региоизомеров проводили стандартными методами, известными в данной области, такими как SFC или ВЭЖХ, и проводили на любой подходящей стадии в последовательности синтеза.
Таблица 11:
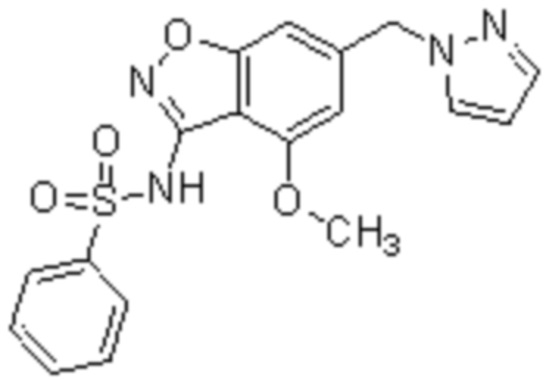
N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензолсульфонамид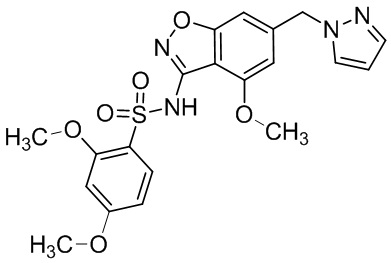
2,4-диметокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид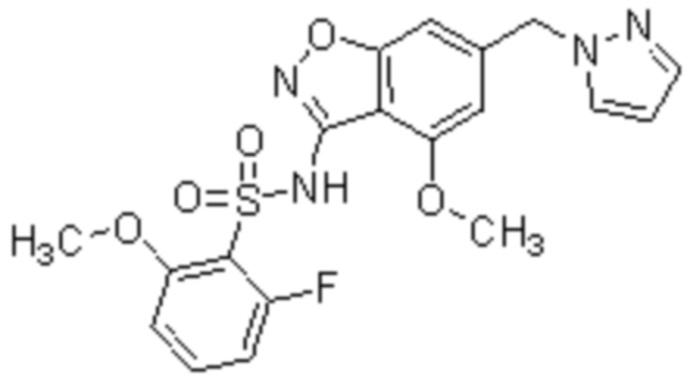
2-фтор-6-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид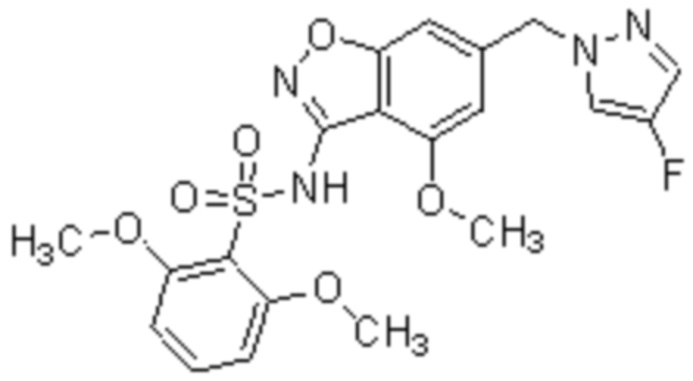
N-{6-[(4-фтор-1H-пиразол-1-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамид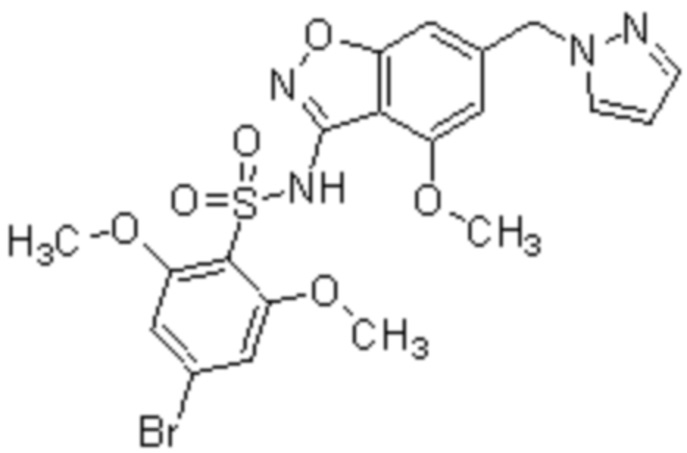
4-бром-2,6-диметокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид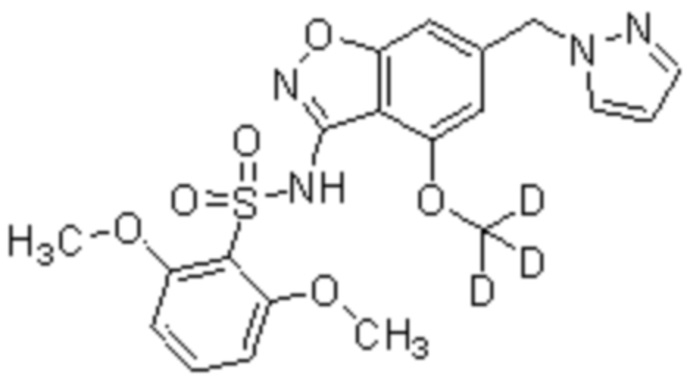
2,6-диметокси-N-{4-[(2H3)метилокси]-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид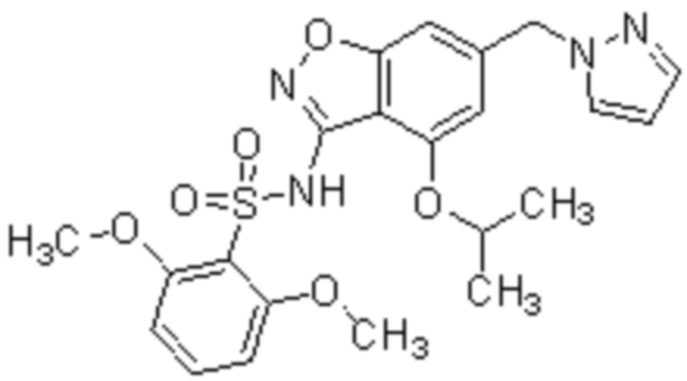
2,6-диметокси-N-{4-[(пропан-2-ил)окси]-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид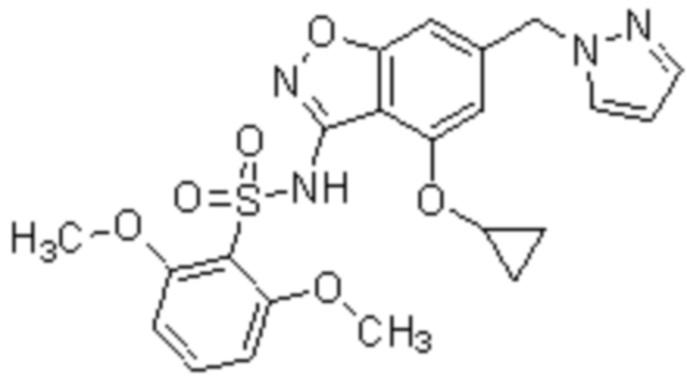
N-{4-(циклопропилокси)-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамид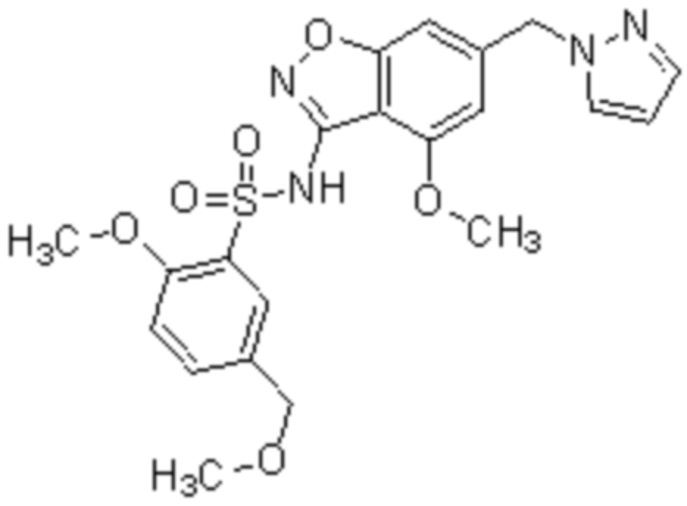
2-метокси-5-(метоксиметил)-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид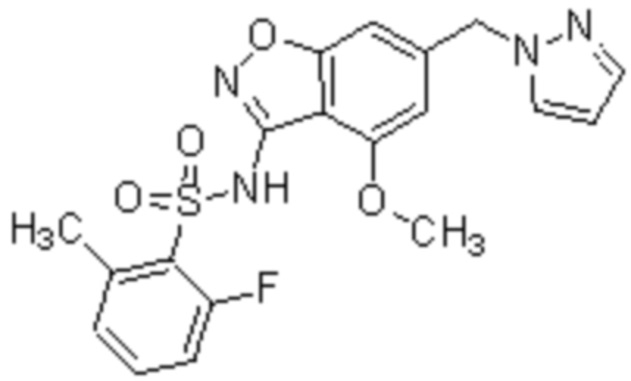
2-фтор-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-6-метилбензол-1-сульфонамид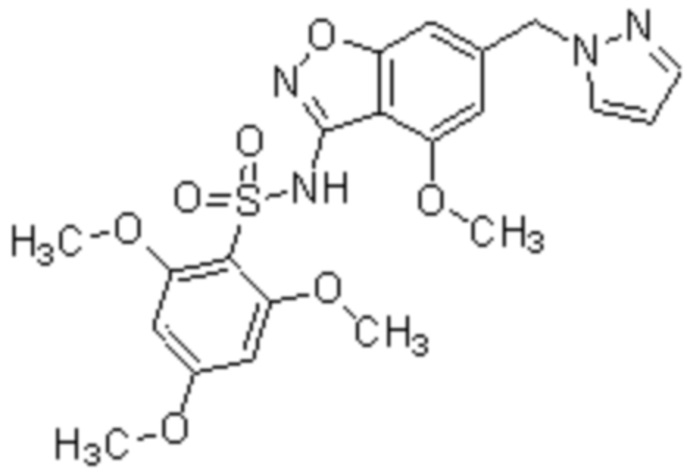
2,4,6-триметокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид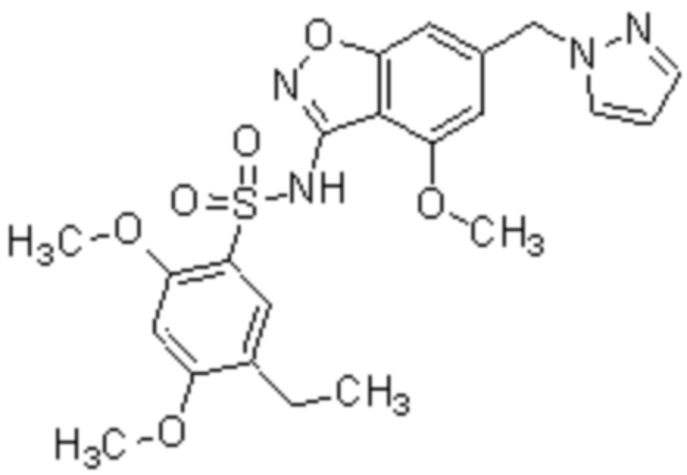
5-этил-2,4-диметокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид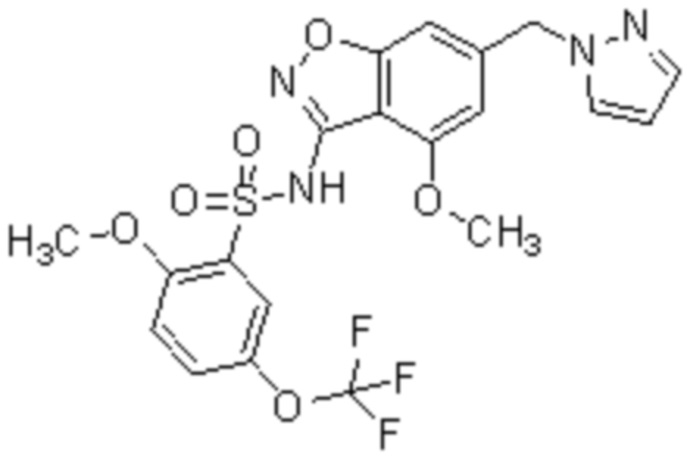
2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-5-(трифторметокси)бензол-1-сульфонамид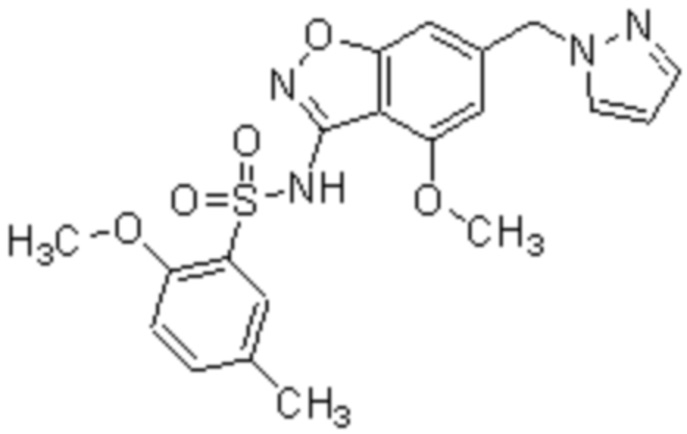
2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-5-метилбензол-1-сульфонамид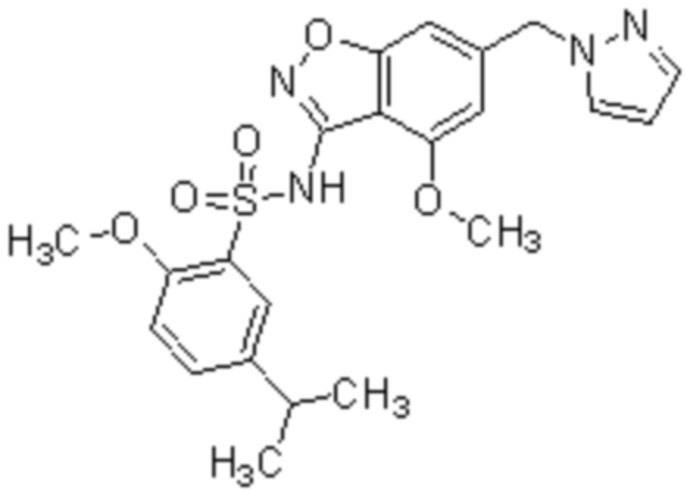
2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-5-(пропан-2-ил)бензол-1-сульфонамид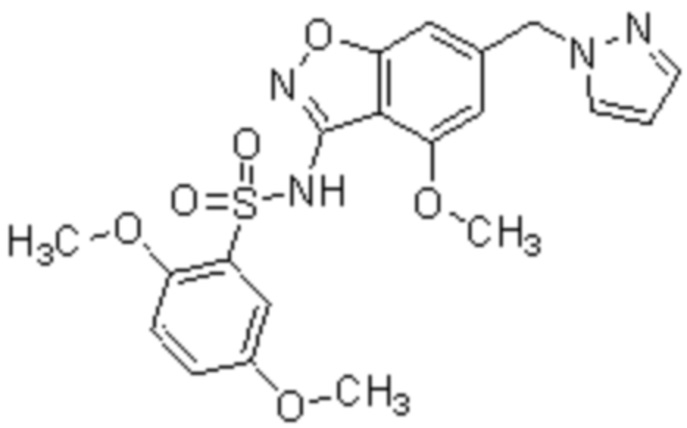
2,5-диметокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид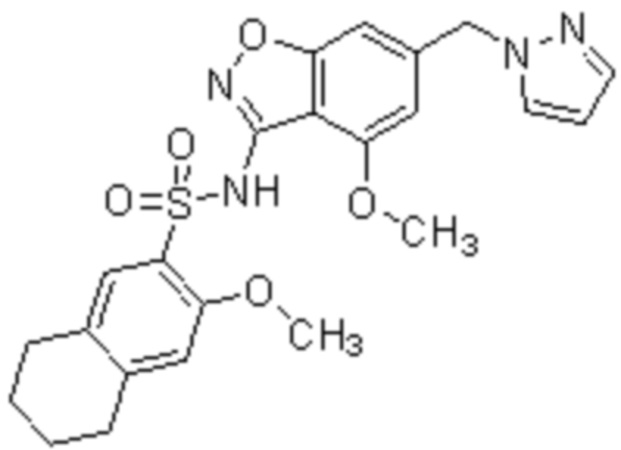
3-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-5,6,7,8-тетрагидронафталин-2-сульфонамид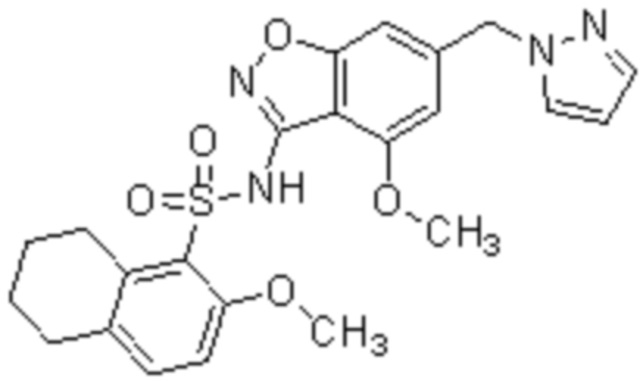
2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-5,6,7,8-тетрагидронафталин-1-сульфонамид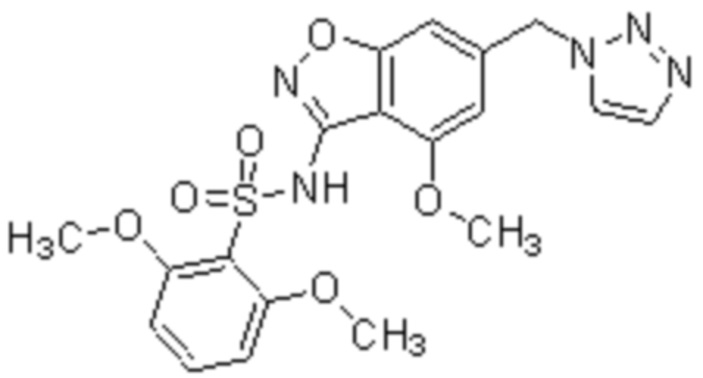
2,6-диметокси-N-{4-метокси-6-[(1H-1,2,3-триазол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид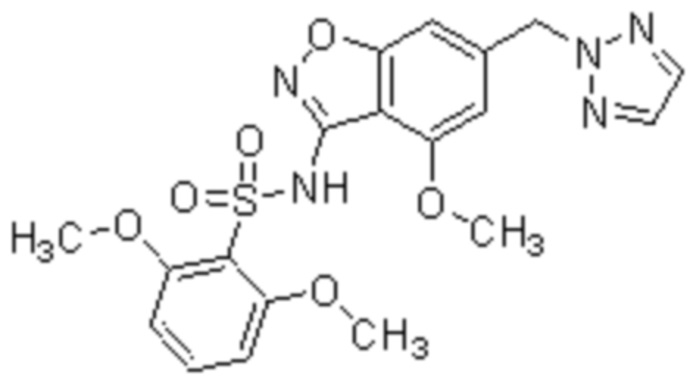
2,6-диметокси-N-{4-метокси-6-[(2H-1,2,3-триазол-2-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид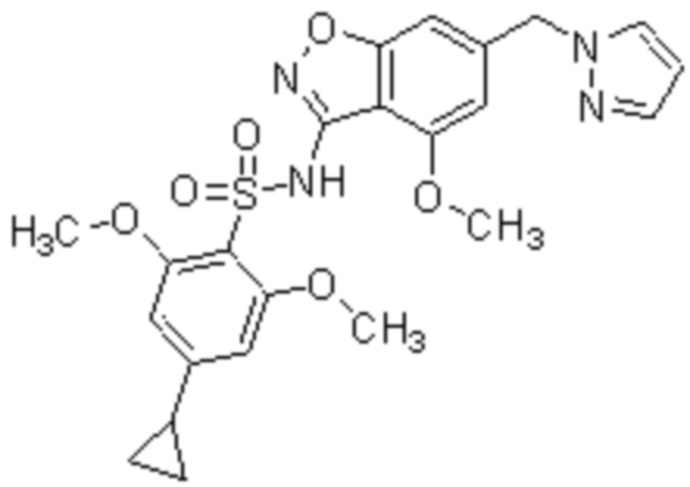
4-циклопропил-2,6-диметокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид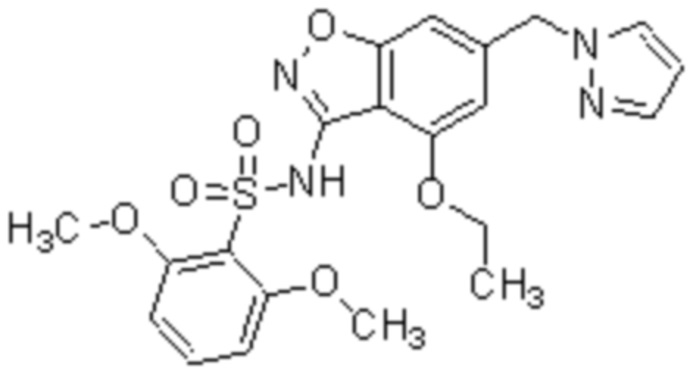
N-{4-этокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамид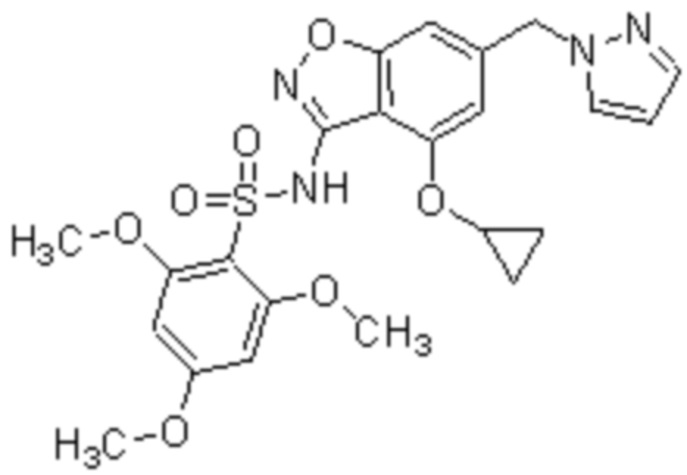
N-{4-(циклопропилокси)-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,4,6-триметоксибензол-1-сульфонамид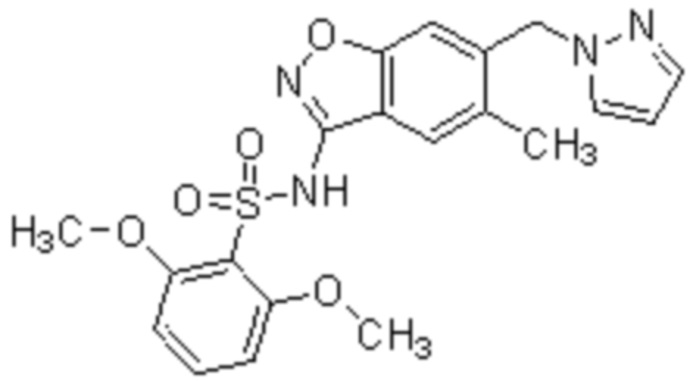
2,6-диметокси-N-{5-метил-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид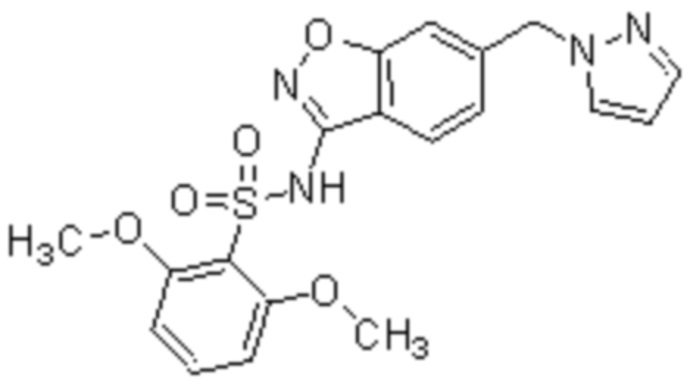
2,6-диметокси-N-{6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид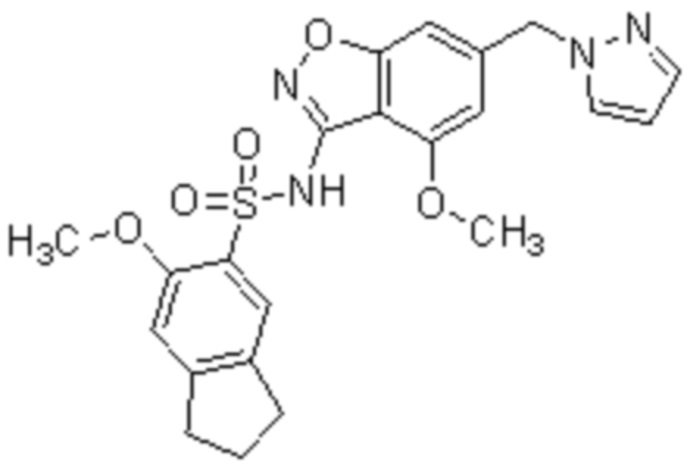
6-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,3-дигидро-1H-инден-5-сульфонамид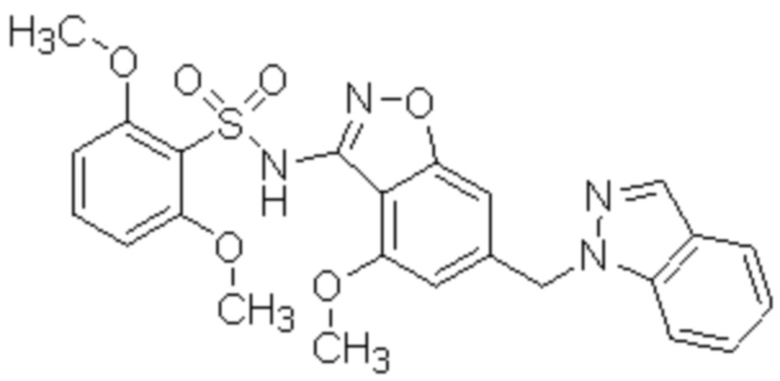
N-(6-((1H-индазол-1-ил)метил)-4-метоксибензо[d]изоксазол-3-ил)-2,6-диметоксибензолсульфонамид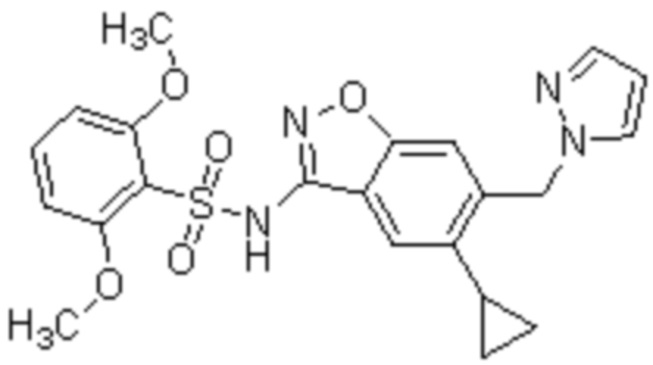
N-[5-циклопропил-6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-ил]-2,6-диметоксибензолсульфонамид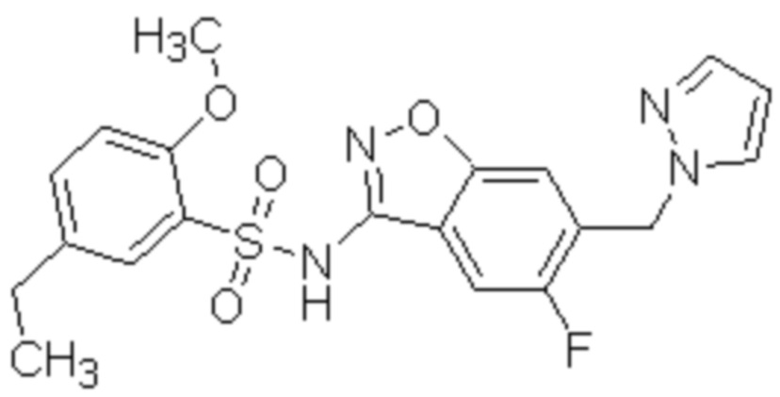
5-этил-N-[5-фтор-6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-ил]-2-метоксибензолсульфонамид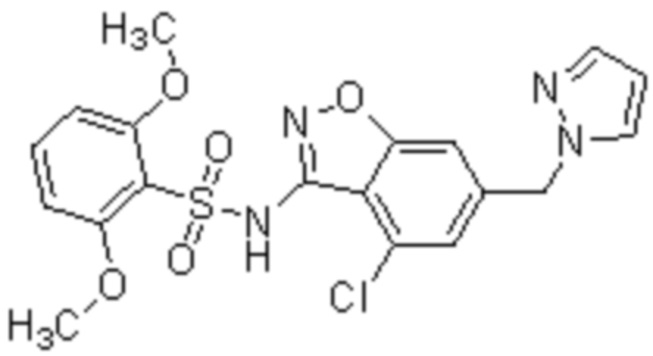
N-[4-хлор-6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-ил]-2,6-диметоксибензолсульфонамид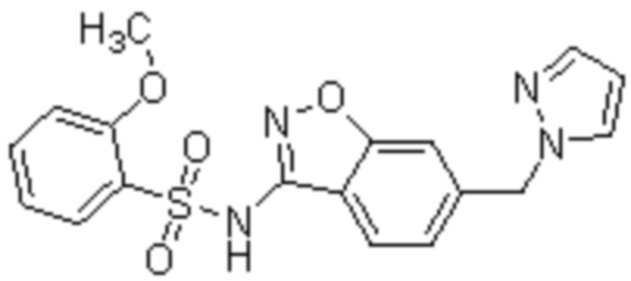
2-метокси-N-[6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамид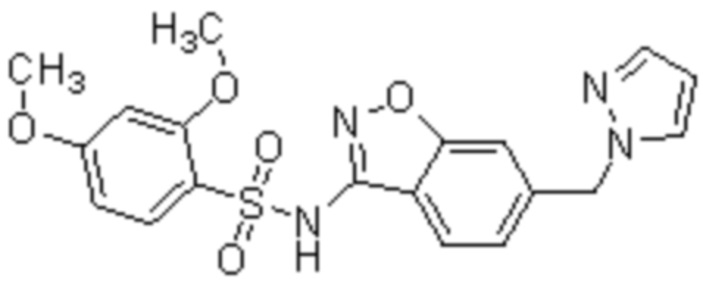
2,4-диметокси-N-[6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамид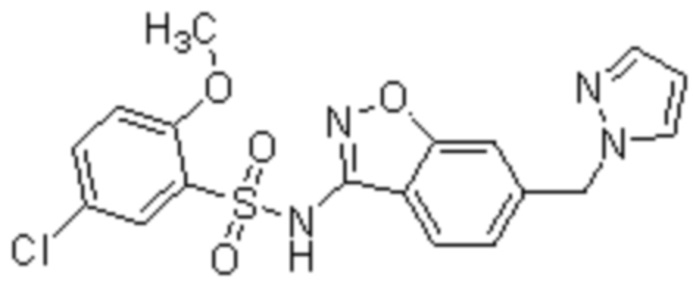
5-хлор-2-метокси-N-[6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамид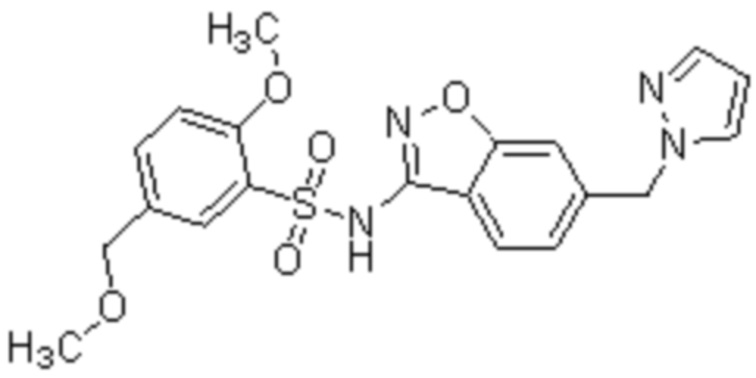
2-метокси-5-(метоксиметил)-N-[6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамид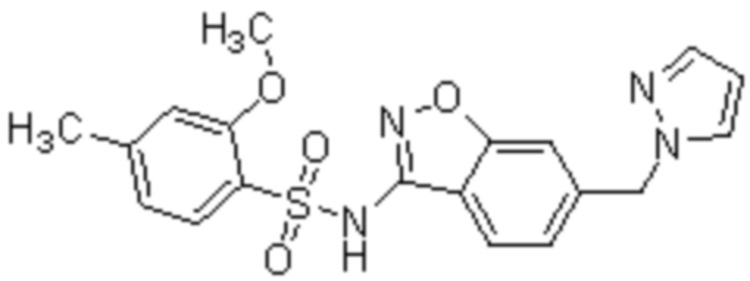
2-метокси-4-метил-N-[6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамид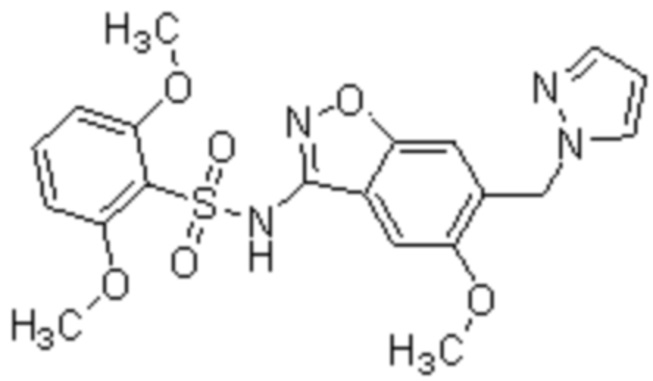
2,6-диметокси-N-[5-метокси-6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамид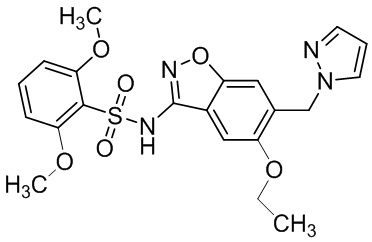
N-[5-этокси-6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-ил]-2,6-диметоксибензолсульфонамид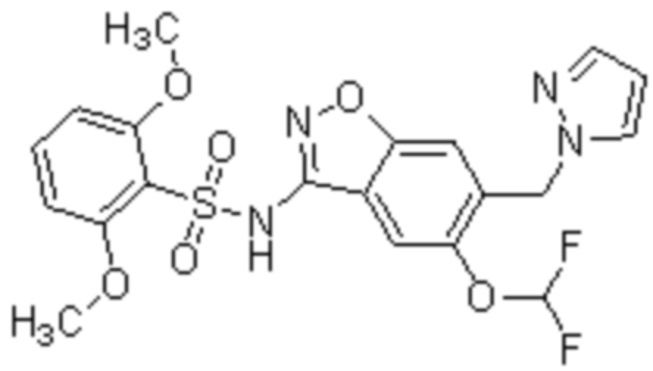
N-[5-(дифторметокси)-6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-ил]-2,6-диметоксибензолсульфонамид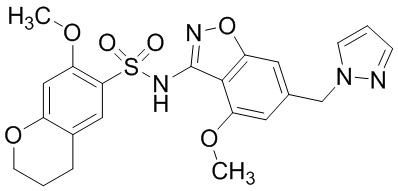
7-метокси-N-[4-метокси-6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-ил]-3,4-дигидро-2H-chromene-6-сульфонамид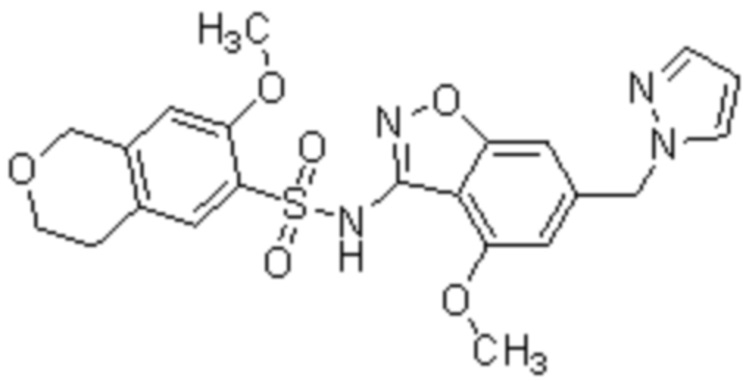
7-метокси-N-[4-метокси-6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-ил]-3,4-дигидро-1H-isochromene-6-сульфонамид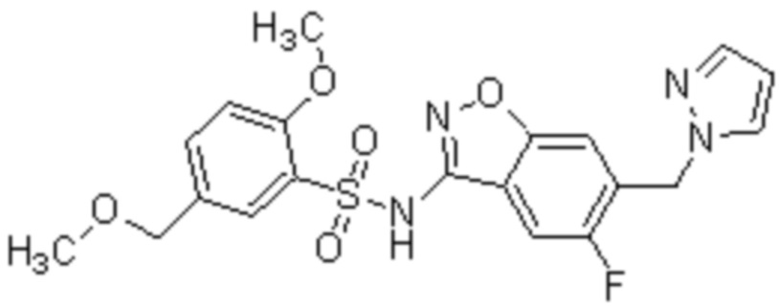
N-[5-фтор-6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-ил]-2-метокси-5-(метоксиметил)бензолсульфонамид
Пример 45: Получение 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида в соответствии со схемой C (путь A).
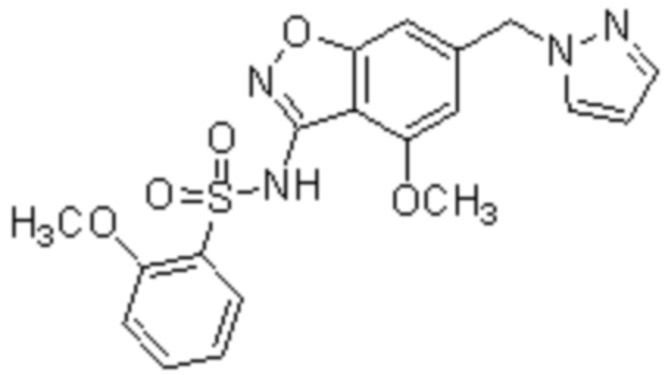
Схема C:
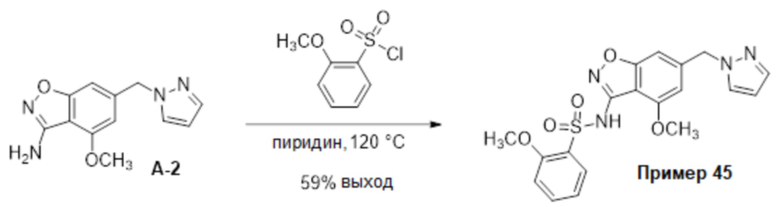
К суспензии 4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (A-2) (2,5 г, 10 ммоль) в пиридине (8,0 мл) добавляли 2-метоксибензол-1-сульфонил хлорид (3,17 г, 15,4 ммоль). Реакционную смесь перемешивали при температуре 120ºC в течение 1,5 ч. Смесь охлаждали до комнатной температуры и разбавляли MeOH. Полученную суспензию фильтровали. и слой продукта на фильтре промывали MeOH (30 мл). Твердые продукты растворяли в DCM (50 мл) и MeOH (30 мл) добавляли. The DCM удаляли в вакууме и осадок собирали путем фильтрации. Слой продукта на фильтре сушили путем лиофмлмзации с получением 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 45) (2,5 г, 59%-ный выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 10,18 (с, 1H), 7,87 (д, J=2,0 Гц, 1H), 7,80 (дд, J=1,6, 7,9 Гц, 1H), 7,66-7,59 (м, 1H), 7,49 (д, J=1,5 Гц, 1H), 7,19 (д, J=8,3 Гц, 1H), 7,09 (т, J=7,7 Гц, 1H), 6,83 (с, 1H), 6,74 (с, 1H), 6,30 (т, J=2,0 Гц, 1H), 5,44 (с, 2H), 3,82 (с, 3H), 3,78 (с, 3H); m/z (ESI+) 415,0 (M+H)+.
Пример 45: Альтернативное получение 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида в соответствии со схемой D.
Схема D:
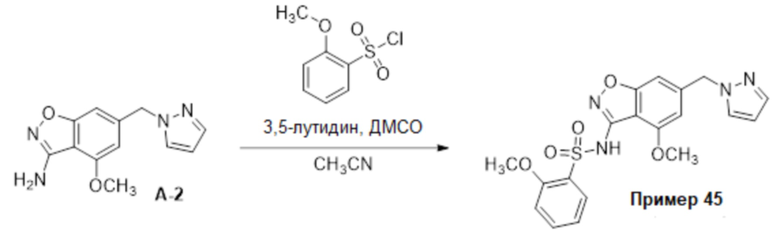
В реактор объемом 100 мл, оборудованный верхней мешалкой, загружали 4-метокси-6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-амин (A-2) (10,00 г, 40,94 ммоль), 2-метоксибензолсульфонил хлорид (10,15 г, 49,13 ммоль) и ацетонитрил (100 мл). Полученную суспензию перемешивали при температуре 25ºC в течение 55 минут. Одной порцией с помощью пипетки добавляли диметилсульфоксид (0,36 мл, 4,09 ммоль). С помощью пипетки в течение 15 минут добавляли по каплям 3,5-лутидин (14,8 мл, 122,82 ммоль). Полученную суспензию светло-желтого цвета перемешивали при температуре 25ºC в течение 18 часов до достижения >98% конверсии по данным ЖХМС. Реакционную смесь подкисляли 1 M водн. HCl (100 мл), затем концентрировали до ~80 мл (роторный испаритель, 40ºC, 85 мбар). Взвесь обрабатывали дополнительным количеством 1 M водн. HCl (40 мл) для промывки стенок сосуда, затем перемешивали при температуре 20ºC в течение 2,5 часов. Полученный осадок собирали фильтрованием с отсасыванием. Слой продукта на фильтре промывали водой (2×50 мл), затем сушили в вакууме при температуре 35ºC в течение 48 часов, получая сырой 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид (пример 45) (15,2 г, 90%-ный выход, 98%-ная чистота по данным ЖХМС) в виде твердого вещества. m/z 415,1 (M+H)+.
Для очистки сырого продукта суспензию сырого 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 45) (14,00 г, 33,78 ммоль) в дихлорметане (210 мл) нагревали на бане с температурой 40ºC до получения прозрачного раствора (10 минут). Смесь фильтровали, и фильтрат возвращали в чистый реакционный сосуд, используя дополнительное количество дихлорметана (70 мл) для количественного определения переноса. К раствору в течение 2 минут добавляли этилацетат (140 мл), затем смесь перемешивали в течение 2,5 часов. Кристаллизации не наблюдалось, поэтому раствор концентрировали при пониженном давлении (200 мбар) для удаления дихлорметана (объем уменьшался примерно на 70 мл). К остатку добавляли еще этилацетат (140 мл) и смесь перемешивали при комнатной температуре в течение 21 часов. Полученную суспензию концентрировали при пониженном давлении (40ºC, 200 мбар) до примерно 280 мл, затем перемешивали при комнатной температуре в течение 3 часов. Твердые продукты собирали путем фильтрации с использованием дополнительного количества этилацетата (70 мл) для промывки реакционного сосуда и слоя на фильтре. Слой продукта на фильтре сушили в вакуумной печи при температуре 35ºC в течение 23 часов, получая 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид (пример 45) (12,0 г, 85%-ный выход, 97,9%-ной чистоты по данным UPLC, никаких примесей более 0,5%) в виде твердого вещества. m/z 415,1 (M+H)+.
Для дальнейшей очистки суспензию 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 45) (2,0 г, 4,73 ммоль) в ацетоне (80 мл) нагревали до кипячения (температура бани 55ºC) при перемешивании в течение 2 часов. Пока смесь все еще нагревалась, медленно добавляли этилацетат (30 мл), так что внутренняя температура остается выше 45ºC. Образовавшуюся взвесь концентрировали до около 30 мл в мягком вакууме (температура бани 65ºC), затем охлаждали медленно со скоростью 1ºC/мин до 20ºC (~31 минут). Полученный осадок собирали фильтрованием с отсасыванием. Слой продукта на фильтре сушили в вакууме при температуре 50ºC в течение 22 часов, с получением 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 45) (1,825 г, 93%-ный выход, 99,5%-ная чистота по данным UPLC) в виде твердого кристаллического продукта. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 8,14 (дд, J=1,7, 7,8 Гц, 1H), 8,04 (с, 1H), 7,59-7,51 (м, 2H), 7,44 (д, J=2,2 Гц, 1H), 7,14-7,06 (м, 1H), 6,95 (д, J=8,3 Гц, 1H), 6,78 (д, J=0,6 Гц, 1H), 6,45 (с, 1H), 6,32 (т, J=2,1 Гц, 1H), 5,38 (с, 2H), 3,97 (с, 3H), 3,91 (с, 3H).
Пример 45b: Получение безводного свободного основания 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (форма 1) в соответствии со схемой C-1 (путь B).
Схема C-1:
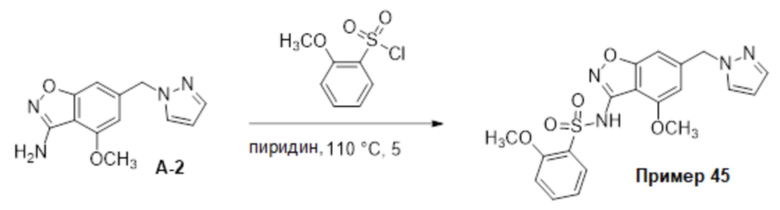
2-Метоксибензол-1-сульфонил хлорид (7,6 г, 37 ммоль) помещали в двухгорлую круглодонную колбу, снабженную внутренним термометром. Добавляли 4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амин (A-2) (8,18 г, 33,5 ммоль) и содержимое растворяли в пиридине (55 мл, 0,6 M) при слабом нагревании. Нагревание инициировали при температуре масляной бани 110ºC и внутренней температуре 101ºC. После 5 ч нагревания реакция была завершена, что было определено ЖХМС анализом. Реакционную смесь охлаждали до комнатной температуры и распределяли между DCM (200 мл), 6 н HCl (100 мл) и ледяной водой (100 мл). Продукт экстрагировали DCM (×3) и объединенный экстракт DCM промывали 1 н HCl (×3) для удаления следов пиридина. Экстракт DCM сушили над MgSO4 и концентрировали до темного масла. Масло очищали с помощью флэш-хроматографии, элюируя градиентом 40-100% EtOAc в гептане, с получением 4,6 г, продукта, что подтверждалось данными ЯМР. 4,6 г, продукта перекристаллизовывали, сначала растворяя в CH3CN (60 мл) при кипячении с обратным холодильником до растворения большей части твердых веществ. Этот горячий раствор фильтровали с использованием предварительно нагретой/горячей стеклянной воронки, снабженной гофрированной фильтровальной бумагой. На этом этапе удалялись все неорганические примеси или примеси силикагеля. Фильтровальную бумагу промывали небольшими порциями CH3CN, доводя общий объем промывки до 10 мл. Фильтрат собирали в химический стакан объемом 250 мл, оборудованный стержнем для перемешивания. К горячему фильтрату добавляли МТВЕ (45 мл) и начинали перемешивание. После 30 секунд перемешивания начинал образовываться белый осадок. Перемешивание продолжали со скоростью 400 об/мин, в то время как в верхнюю часть раствора подавался легкий поток газообразного N2 чтобы ускорить процесс испарения. Принудительное испарение N2 продолжали в течение 3 ч до достижения общего объема 50 мл. Белое твердое вещество фильтровали, промывая МТВЕ (×2) и гептаном (×2). Белый порошок помещали в кристаллизатор диаметром 3 дюйма, накрывали куском фильтровальной бумаги и нагревали в вакуумной печи при температуре 70ºC в течение 48 часов, используя медленный поток N2 в сушильную печь и из нее для облегчения процесса сушки. После сушки получали 3,9 г кристаллического продукта, что подтверждалось данными ЯМР. Точка плавления=203-204ºC. Анал. Вычисл. для C19H18N4O5S: C, 55,06; H, 4,38; N, 13,52. Найдено: C, 55,09; H, 4,41; N, 13,57.
Кристаллическое твердое вещество, полученное выше как безводное (форма 1), дополнительно охарактеризовали порошковой дифракцией рентгеновских лучей (PXRD). Порошковый рентгеноструктурный анализ проводили на порошковом рентгеновском дифрактометре Bruker A25 D8 Advance, оснащенном тета-2-тета-гониометром и детектором Lynxeye с размером окна PSD 3,3º, первичной щелью Соллера, установленной на 2,5º и щелями расхождения, установленными на постоянное освещение 0,6 мм. Напряжение и сила тока рентгеновской трубки были установлены на 40 кВ и 40 мА, соответственно. Данные собирали на длине волны меди с использованием шага 0,02 градуса, времени шага 0,3 с от 3,0 до 40,0º 2-тета. Образец готовили путем помещения порошка в держатель полости Si с низким уровнем фона. Порошок образца прессовали с помощью шпателя, чтобы гарантировать достижение нужной высоты образца. Данные собирали с использованием программного обеспечения Bruker DIFFRAC, а анализ выполняли с помощью программного обеспечения DIFFRAC EVA. Собранные рентгенограммы PXRD были импортированы в программное обеспечение Bruker DIFFRAC EVA. Выбор пика выполнялся с использованием «функции поиска пика» программного обеспечения, а затем был тщательно проверен и исправлен, чтобы гарантировать, что все положения пиков были точно назначены. Выбирали пики с относительной интенсивностью ≥4,0%. К этим данным относится типичная ошибка ±0,2º 2-тета в положениях пиков. Незначительная ошибка, связанная с этим измерением, может возникать из-за множества факторов, включая: (a) подготовка образца (например, высота образца), (b) прибор, (c) калибровка, (d) оператор (включая те ошибки, которые присутствуют при определении местоположения пиков) и (e) характер материала (например, предпочтительная ориентация и ошибки прозрачности). Поэтому считается, что пики имеют типичную связанную ошибку ±0,2º 2-тета. Когда два пика в списке считаются перекрывающимися, менее интенсивный пик удаляется из списка. Пики, существующие в виде плеч на соседнем пике с более высокой интенсивностью, также были удалены из списка пиков. Хотя плечи могут быть >0,2º 2-тета от положения соседнего пика, они не считаются отличимыми от соседнего пика.
Чтобы получить абсолютные положения пиков, порошковый образец следует выровнять относительно эталона. Это может быть либо смоделированный порошковый узор из кристаллической структуры той же формы, решенной при комнатной температуре, либо внутренний стандарт, например кремнезем или корунд. Смоделированную порошковую картину безводного 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (форма 1) получали из монокристаллической структуры. Для приготовления монокристалла 200 мг материала примера 45b растворяли в CH3CN (3 мл) при кипячении с обратным холодильником. Добавляли МТВЕ (2 мл) и смесь оставляли на 48 часов в пробирке, открытой для воздуха, что приводило к медленному испарению растворителей. Образовывались крупные кристаллы, которые фильтровали и промывали МТВЕ (×2) и гептаном (×2) и сушили в вакууме. Получали 116 мг (выход 58%) материала примера 45b в виде кристаллического белого твердого вещества, что подтверждалось данными 1H ЯМР. Кристаллы, визуализированные с помощью микроскопии в поляризованном свете, показывали крупный размер частиц и триклинную форму. Смоделированный порошковый узор из монокристаллической структуры получали путем расчета с использованием Mercury 4.1.0, который является частью CCDC Software Suite.
Диаграмма PXRD безводной формы 1 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 45a), показана на ФИГ. 1. Список пиков PXRD и данные относительной интенсивности для безводной формы 1 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 45a) (2-Thetaº) представлены в таблице 12 ниже. Положения характерных пиков PXRD обозначены звездочкой.
Таблица 12: Перечень пиков порошковой рентгеновской дифракции для формы 1 безводного свободного основания по примеру 45.
º2-тета
Один вариант осуществления настоящего изобретения относится к кристаллической форме безводного свободного основания 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида, имеющей порошковую рентгеновскую дифрактограмму, содержащую пики при значениях 2Θ: 13,4 и 18,1 º2Θ±0,2 º2Θ.
Один вариант осуществления настоящего изобретения относится к кристаллической форме безводного свободного основания 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида, имеющей порошковую рентгеновскую дифрактограмму, содержащую пики при значениях 2Θ: 13,4 и 18,1 º2Θ±0,2 º2Θ, и дополнительно содержащую по меньшей мере один пик, выбранный из значений 2Θ: 11,4, 14,1, и 17,5 º2Θ±0,2 º2Θ.
Один вариант осуществления настоящего изобретения относится к кристаллической форме безводного свободного основания 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида, имеющей порошковую рентгеновскую дифрактограмму, содержащую пики при значениях 2Θ: 13,4 и 18,1 º2Θ±0,2 º2Θ, и дополнительно содержащую пики при значениях 2Θ: 11,4, 14,1, и 17,5 º2Θ±0,2 º2Θ.
Один вариант осуществления настоящего изобретения относится к кристаллической форме безводного свободного основания 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида, имеющей порошковую рентгеновскую дифрактограмму, содержащую пики при значениях 2Θ: 11,4, 13,4, 14,1, 17,5 и 18,1 º2Θ±0,2 º2Θ.
Соединения по примерам, приведенным в таблице ниже, синтезировали в соответствии со способами, используемыми для синтеза 5-этил-2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 01), 2,6-диметокси-N-{4-метокси-6-[(3-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 02) и 2,6-диметокси-N-{4-метокси-6-[(5-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 03), и общим способом получения сульфонамида C в формате библиотеки с высокой пропускной способностью. Следующие соединения по примерам синтезировали с некритическими изменениями или заменами в приведенных в качестве примеров способах, которые мог бы осуществить специалист в данной области.
Таблица 13:
примера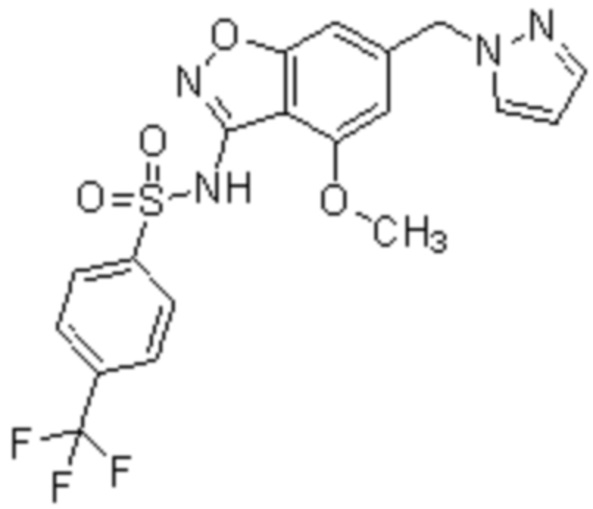
N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-4-(трифторметил)бензол-1-сульфонамид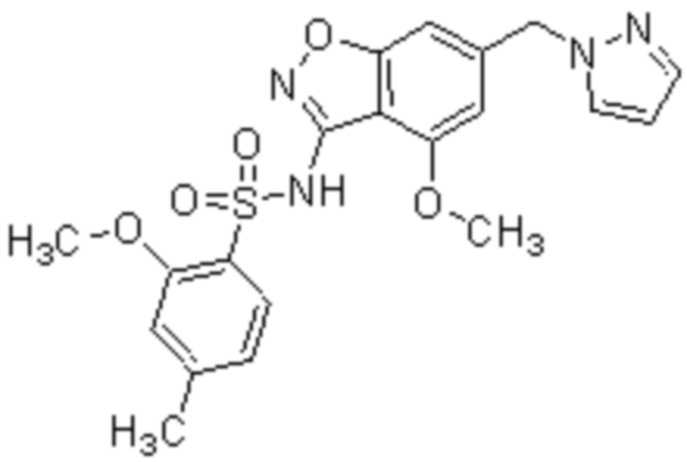
2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-4-метилбензол-1-сульфонамид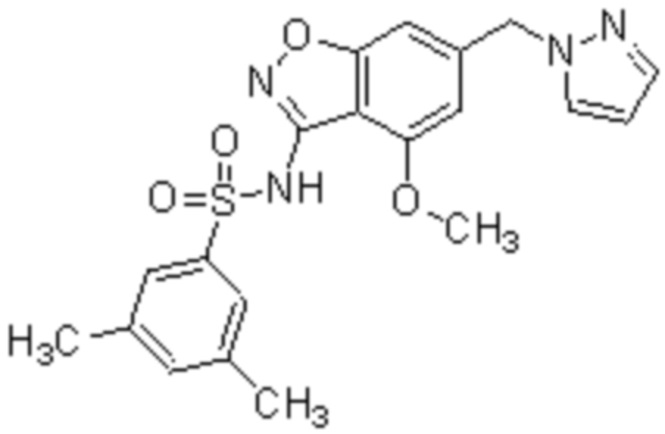
N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-3,5-диметилбензол-1-сульфонамид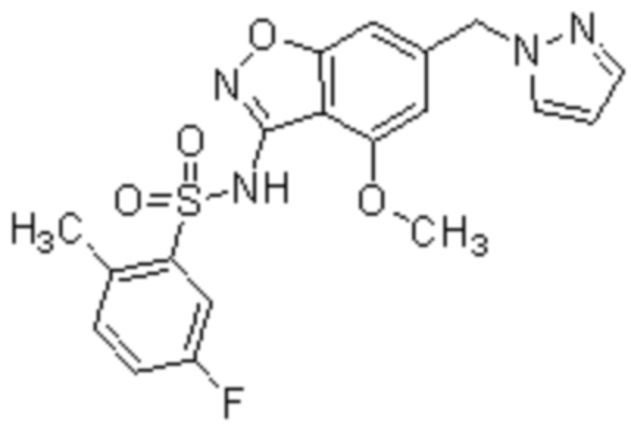
5-фтор-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2-метилбензол-1-сульфонамид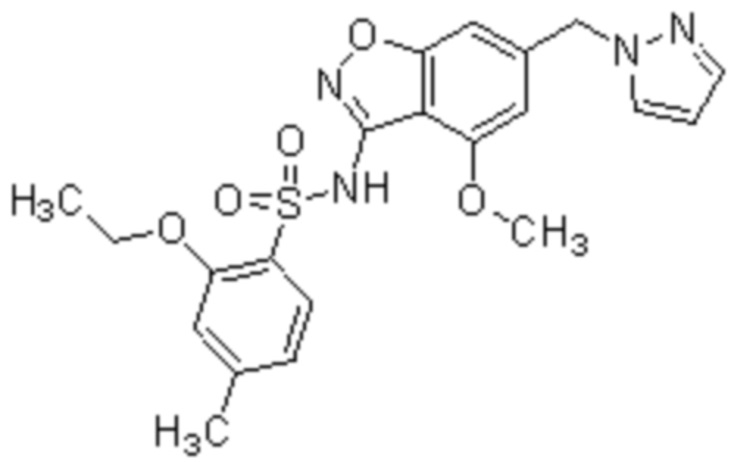
2-этокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-4-метилбензол-1-сульфонамид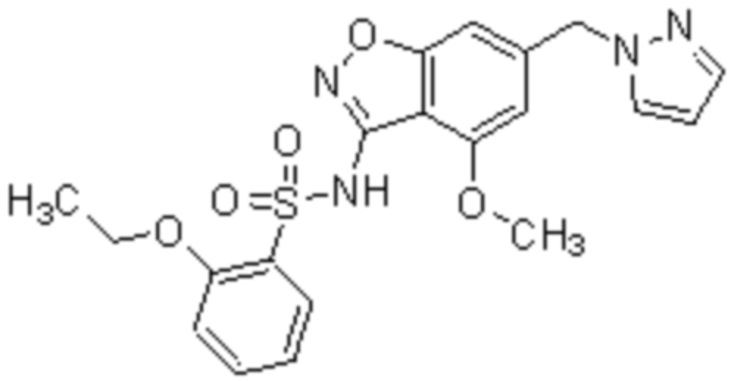
2-этокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид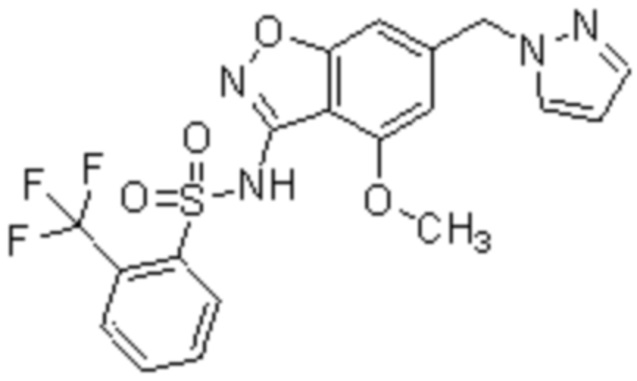
N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2-(трифторметил)бензол-1-сульфонамид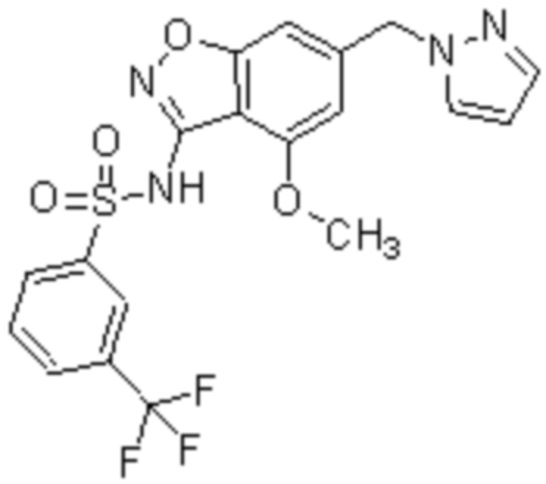
N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-3-(трифторметил)бензол-1-сульфонамид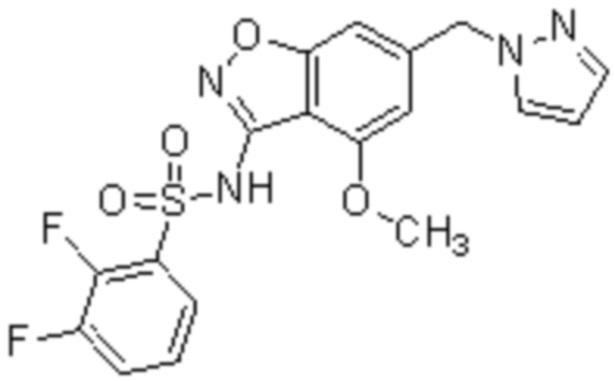
2,3-дифтор-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид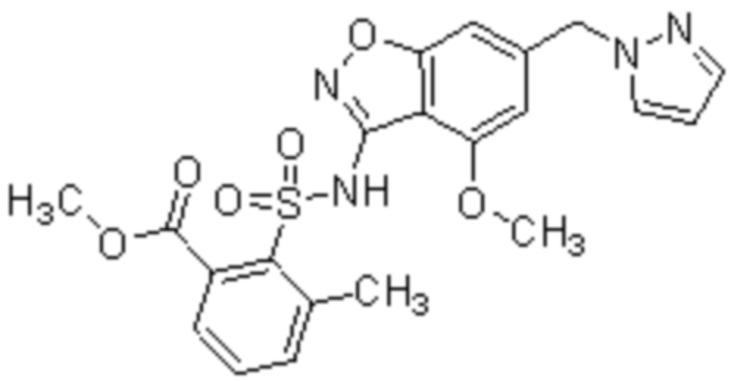
метил 2-({4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}сульфамоил)-3-метилбензоат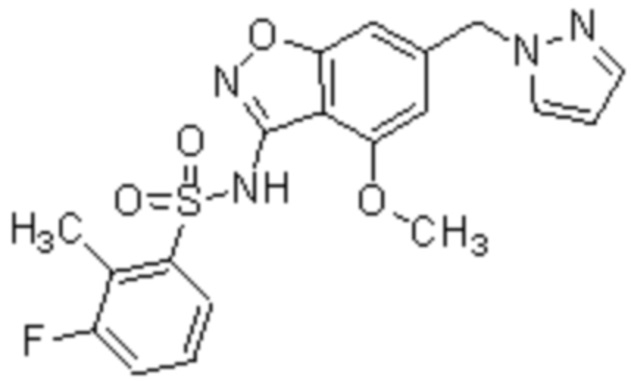
3-фтор-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2-метилбензол-1-сульфонамид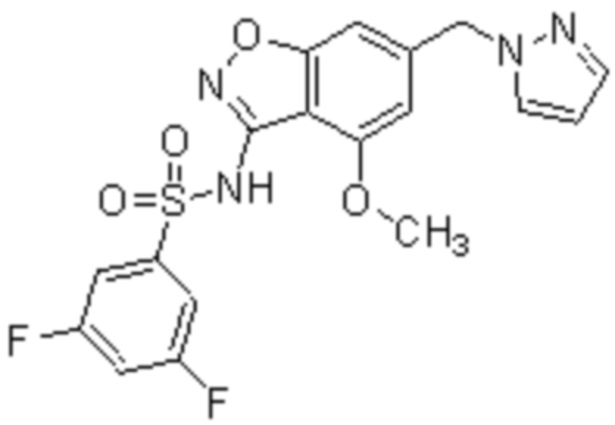
3,5-дифтор-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид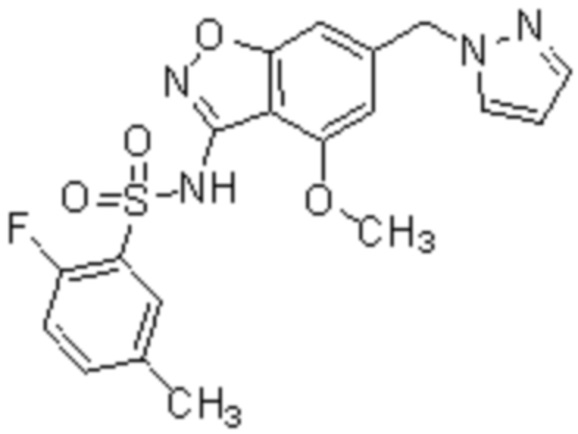
2-фтор-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-5-метилбензол-1-сульфонамид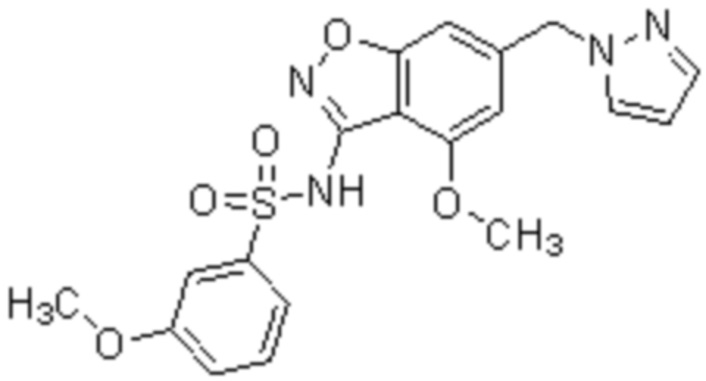
3-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид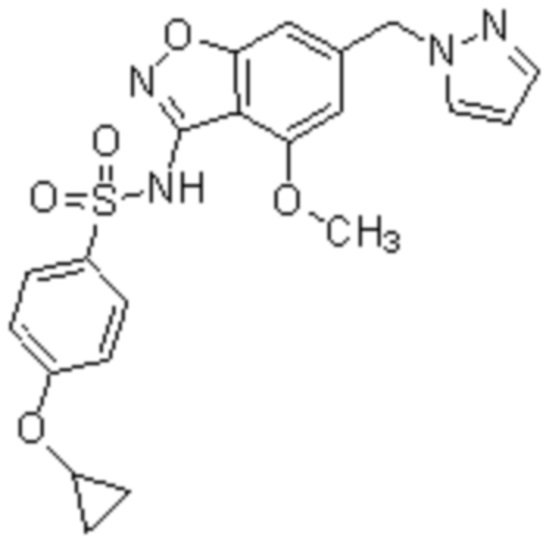
4-(циклопропилокси)-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид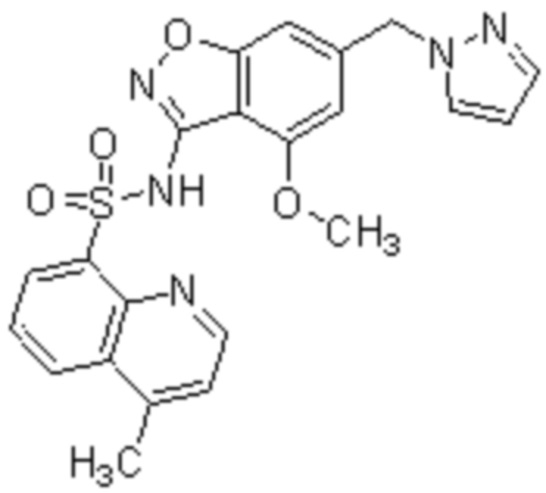
N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-4-метилхинолин-8-сульфонамид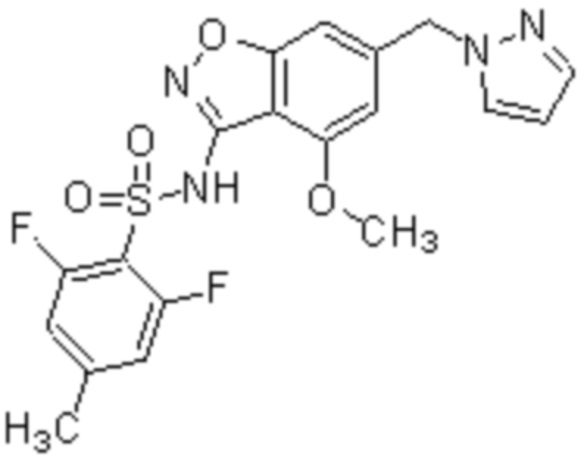
2,6-дифтор-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-4-метилбензол-1-сульфонамид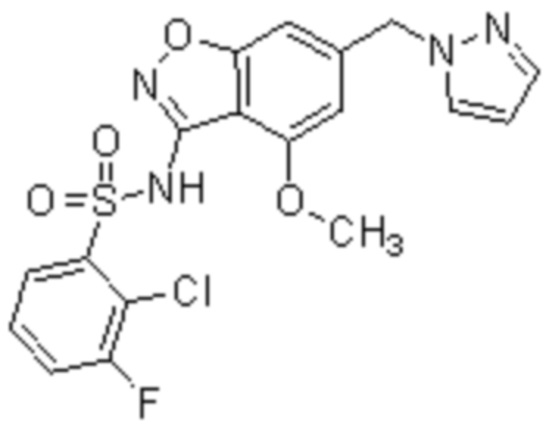
2-хлор-3-фтор-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид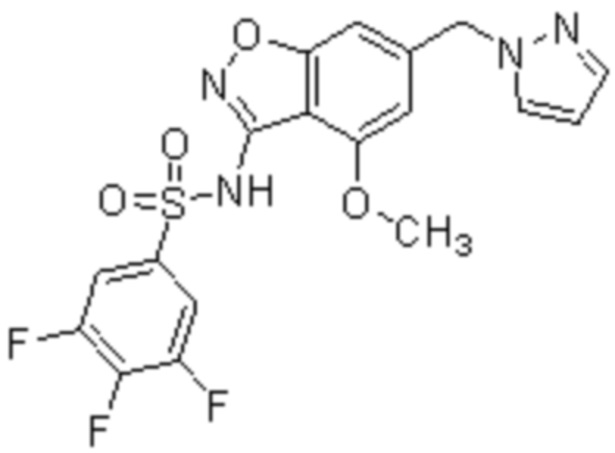
3,4,5-трифтор-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид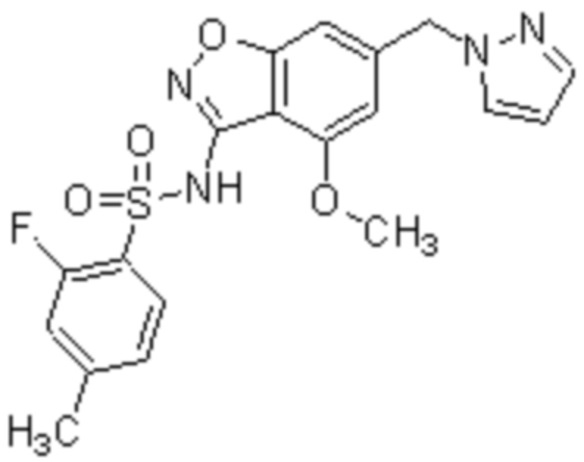
2-фтор-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-4-метилбензол-1-сульфонамид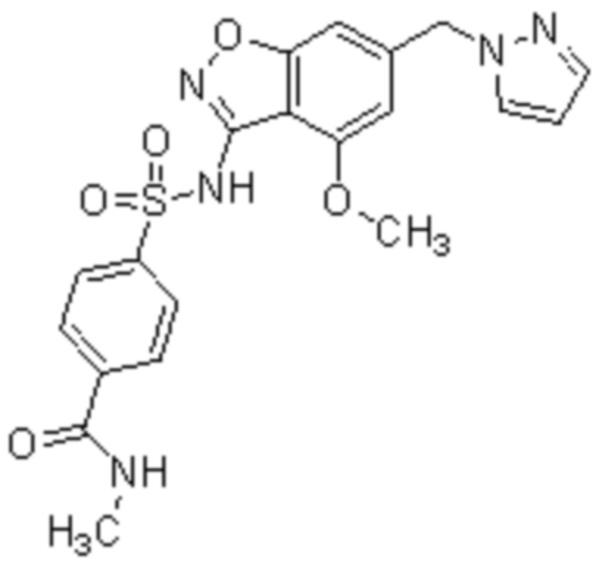
4-({4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}сульфамоил)-N-метилбензамид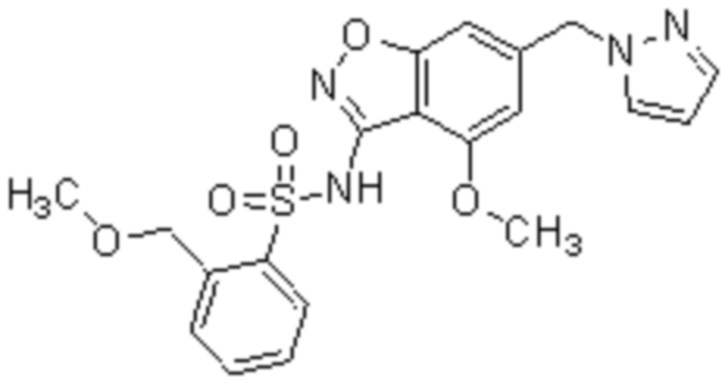
2-(метоксиметил)-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид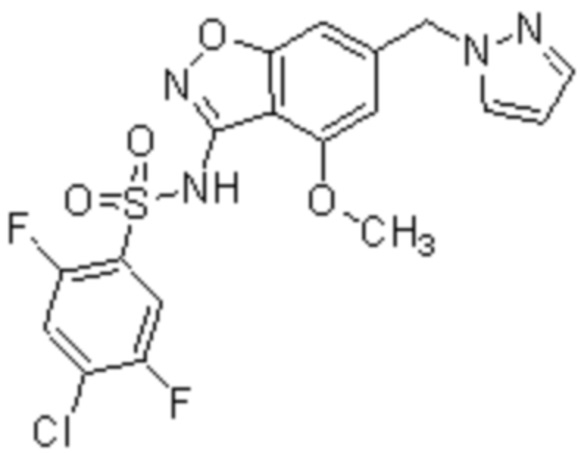
4-хлор-2,5-дифтор-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид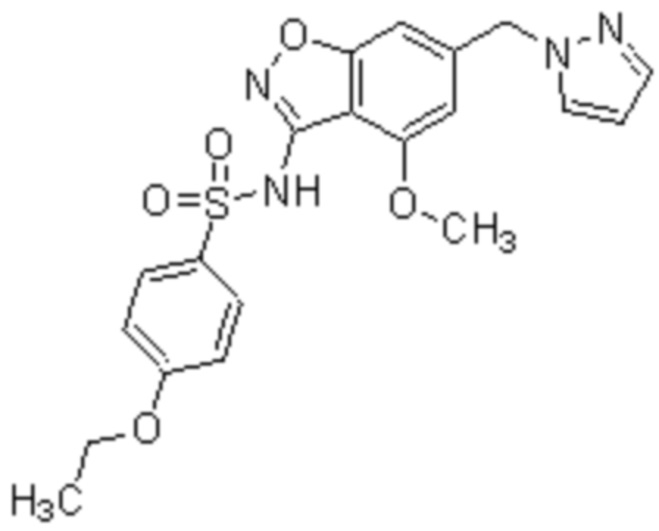
4-этокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид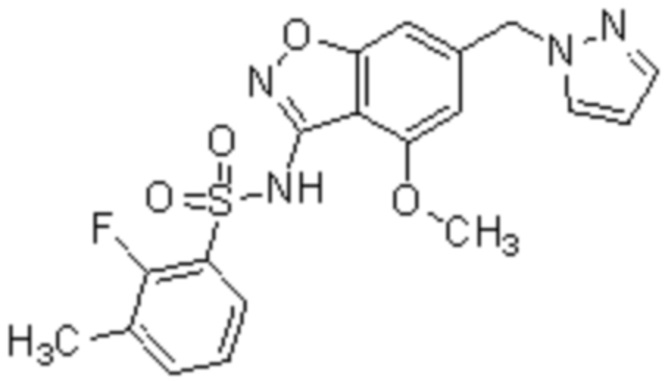
2-фтор-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-3-метилбензол-1-сульфонамид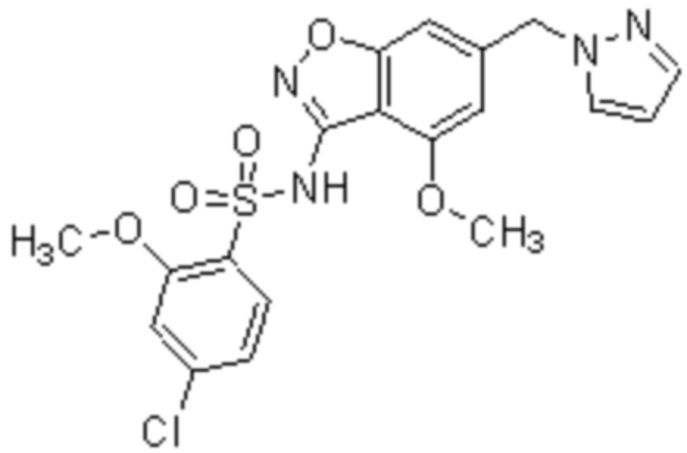
4-хлор-2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид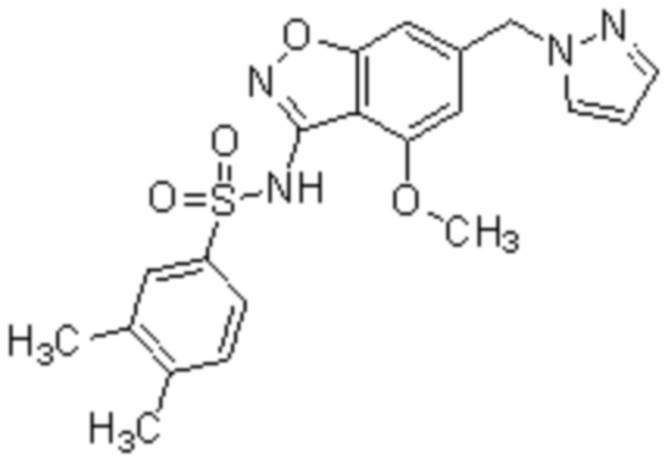
N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-3,4-диметилбензол-1-сульфонамид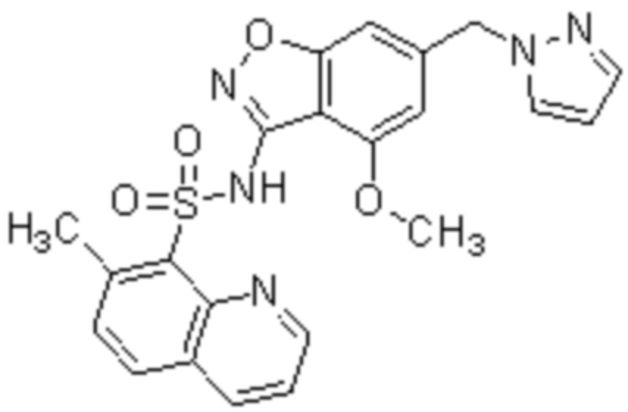
N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-7-метилхинолин-8-сульфонамид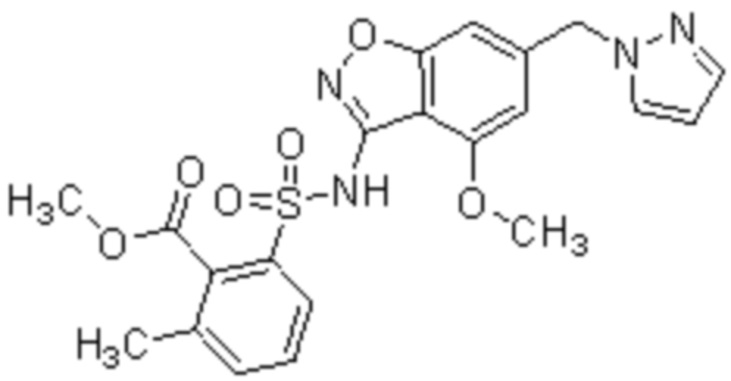
метил 2-({4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}сульфамоил)-6-метилбензоат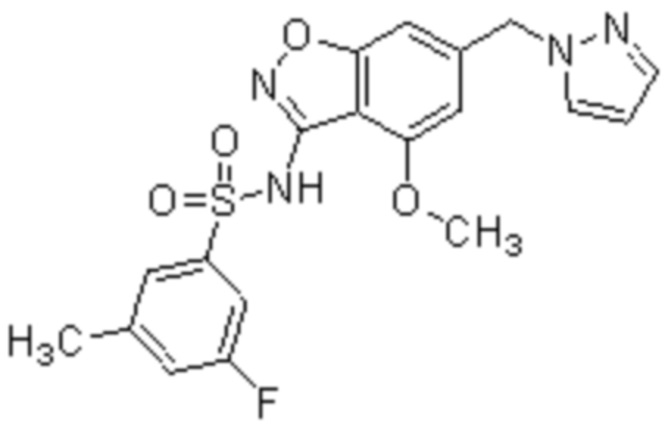
3-фтор-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-5-метилбензол-1-сульфонамид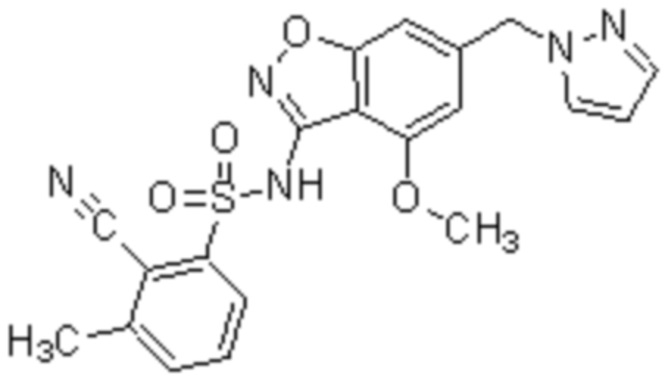
2-циано-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-3-метилбензол-1-сульфонамид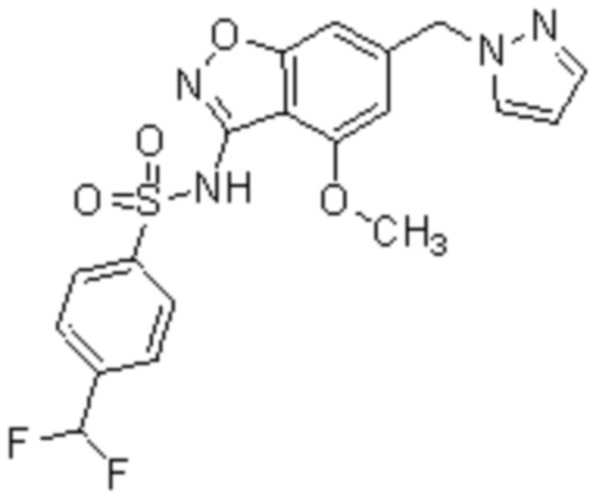
4-(дифторметил)-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид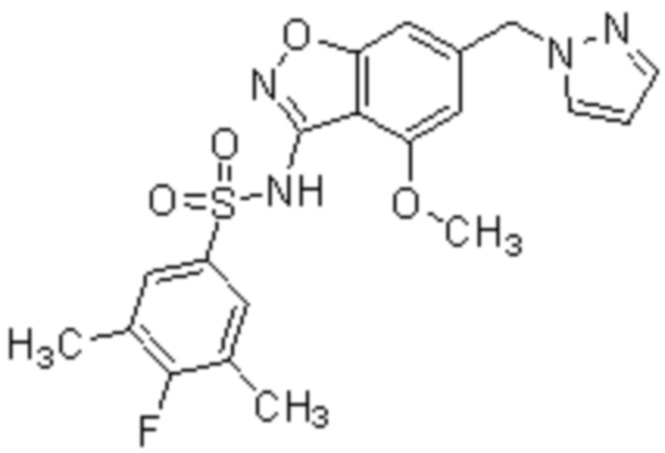
4-фтор-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-3,5-диметилбензол-1-сульфонамид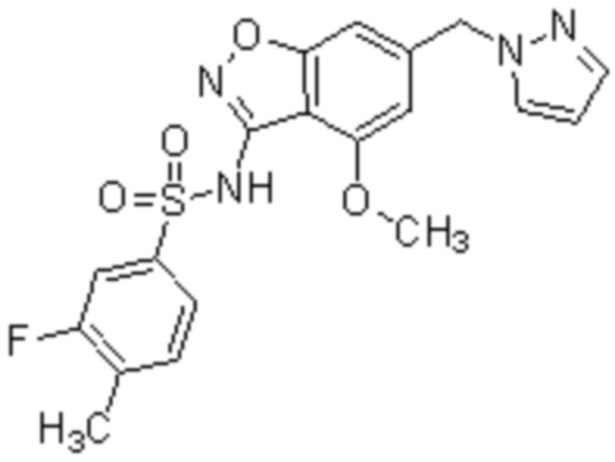
3-фтор-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-4-метилбензол-1-сульфонамид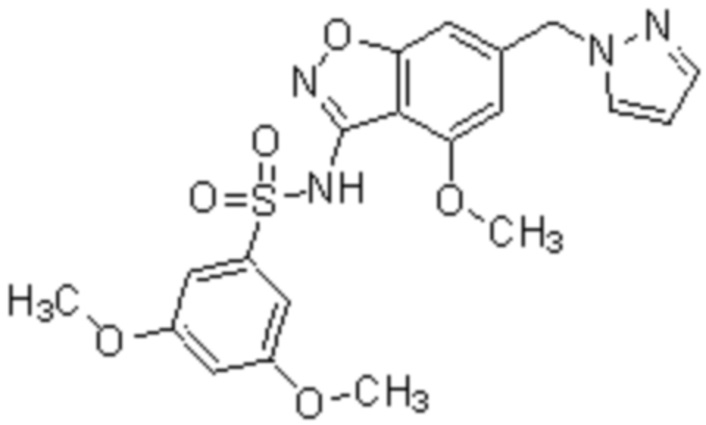
3,5-диметокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид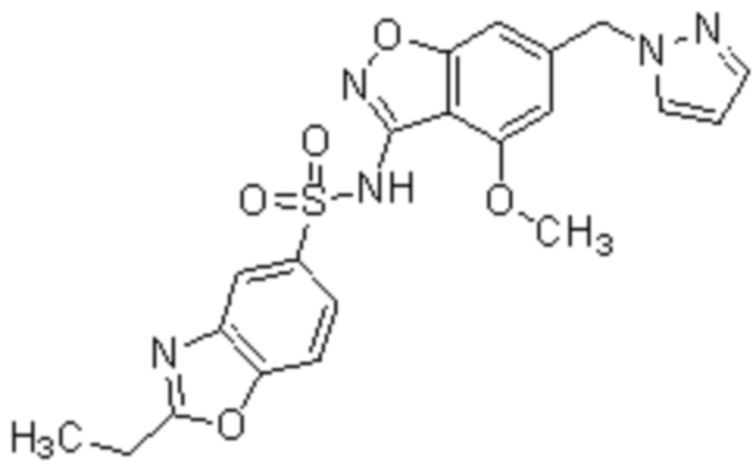
2-этил-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-1,3-бензоксазоле-5-сульфонамид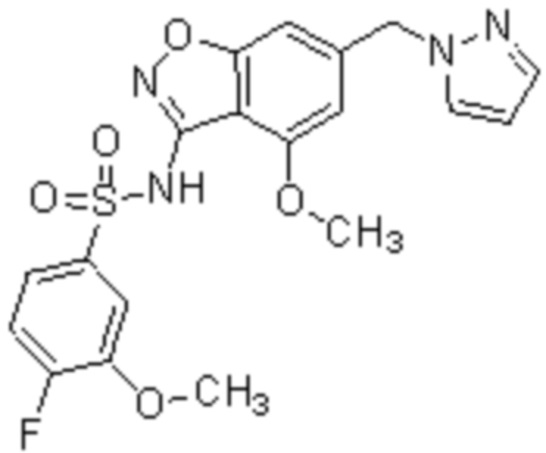
4-фтор-3-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид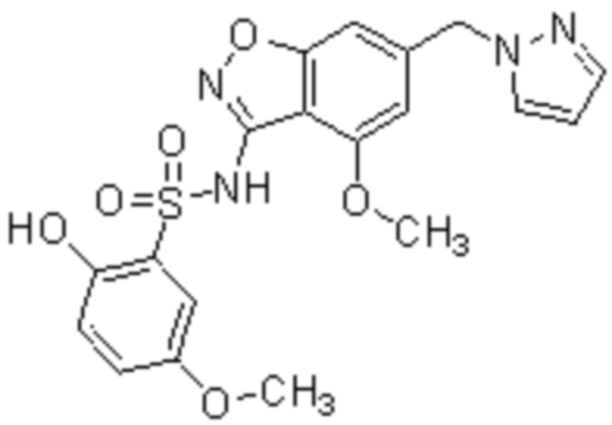
2-гидрокси-5-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид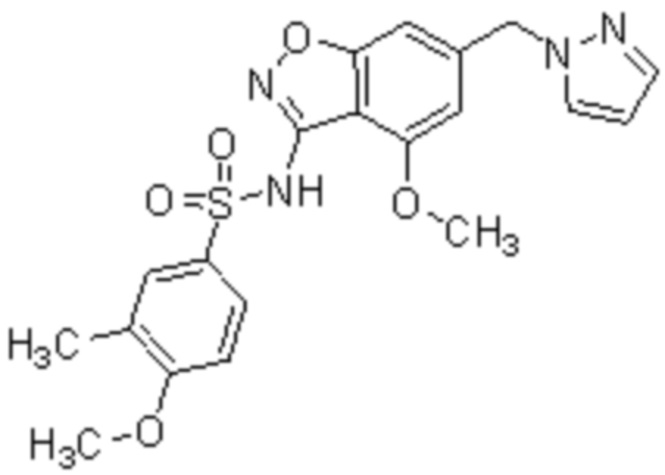
4-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-3-метилбензол-1-сульфонамид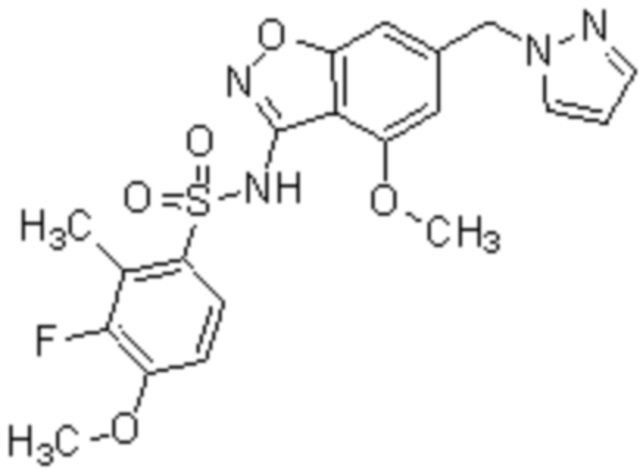
3-фтор-4-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2-метилбензол-1-сульфонамид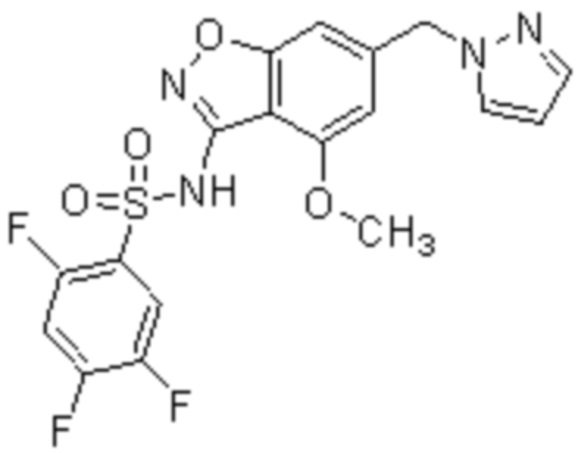
2,4,5-трифтор-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид
Пример 87: Получение N-(6-{[4-(гидроксиметил)-1H-пиразол-1-ил]метил}-4-метокси-1,2-бензоксазол-3-ил)-2,6-диметоксибензол-1-сульфонамида в соответствии со схемой E.
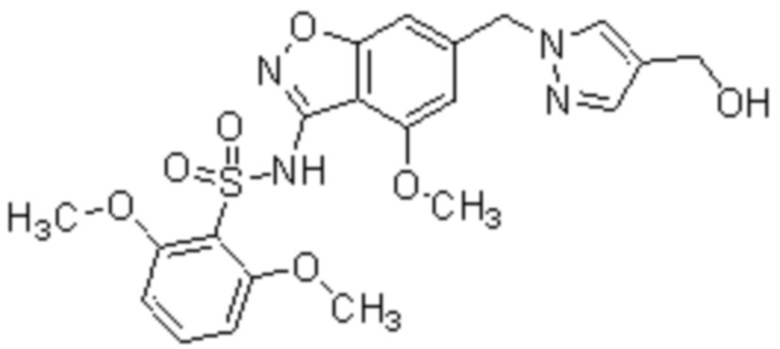
Схема E:

Стадия 1: Синтез 6-{[4-({[трет-бутил(диметил)силил]окси}метил)-1H-пиразол-1-ил]метил}-4-метокси-1,2-бензоксазол-3-амина (E-2).
К раствору 4-{[4-({[трет-бутил(диметил)силил]окси}метил)-1H-пиразол-1-ил]метил}-2-фтор-6-метоксибензонитрила (E-1) (получен как в примере 01, 500 мг, 1,33 ммоль) и N-гидроксиацетамида (300 мг, 3,99 ммоль) в ДМФ (10,0 мл) и H2O (2,0 мл) добавляли K2CO3 (1,1 г, 7,99 ммоль). Смесь перемешивали при температуре 60ºC в течение 16 ч. ЖХМС анализ показывал, что исходное вещество расходовалось. Реакционную смесь концентрировали для удаления ДМФ и разбавляли H2O. Полученный осадок собирали путем фильтрации. Слой продукта на фильтре сушили в вакууме. ЖХМС анализ показывал смесь желаемого продукт и des-TBS побочного продукта. Сырые твердые продукты объединяли с параллельным запуском реакции с 200 мг 4-{[4-({[трет-бутил(диметил)силил]окси}метил)-1H-пиразол-1-ил]метил}-2-фтор-6-метоксибензонитрила. Объединенные твердые продукты обрабатывали DCM (10,0 мл). Добавляли TBSCl (178 мг, 1,18 ммоль), TEA (149 мг, 1,48 ммоль) и DMAP (6,0 мг, 0,49 моль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Смесь разбавляли DCM (100 мл) и последовательно промывали H2O (50 мл), насыщенным раствором NaHCO3 (50 мл) и насыщенным солевым раствором (50 мл). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью флеш хроматографии (20 г, SiO2, 60-70% EtOAc/петролейный эфир) с получением 6-{[4-({[трет-бутил(диметил)силил]окси}метил)-1H-пиразол-1-ил]метил}-4-метокси-1,2-бензоксазол-3-амина (E-2) (280 мг, 34%-ный выход за 2 стадии) в виде твердого вещества белого цвета. m/z (ESI+) 388,9 (M+H)+.
Стадия 2: Синтез N-(6-{[4-({[трет-бутил(диметил)силил]окси}метил)-1H-пиразол-1-ил]метил}-4-метокси-1,2-бензоксазол-3-ил)-2,6-диметоксибензол-1-сульфонамида (E-3) в соответствии со способом получения сульфонамида B.
К суспензии NaH (60%-ная дисперсия в минеральном масле, 40,1 мг, 1,00 ммоль) в ТГФ (2,0 мл) добавляли раствор 6-{[4-({[трет-бутил(диметил)силил]окси}метил)-1H-пиразол-1-ил]метил}-4-метокси-1,2-бензоксазол-3-амина (E-2) (130 мг, 0,335 ммоль) в ТГФ (2,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 15 мин и затем добавляли раствор 2,6-диметоксибензол-1-сульфонил хлорида (Int-26) (95,0 мг, 0,402 ммоль) в ТГФ (2,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 17 ч. Суспензию фильтровали и концентрировали досуха. Остаток очищали с помощью флеш хроматографии (12 г, SiO2, 1:1 EtOAc/петролейный эфир) с получением N-(6-{[4-({[трет-бутил(диметил)силил]окси}метил)-1H-пиразол-1-ил]метил}-4-метокси-1,2-бензоксазол-3-ил)-2,6-диметоксибензол-1-сульфонамида (E3) (80 мг, 41%-ный выход) в виде смолы бледно-желтого цвета. m/z (ESI+) 589,1 (M+H)+
Стадия 3: Синтез N-(6-{[4-(гидроксиметил)-1H-пиразол-1-ил]метил}-4-метокси-1,2-бензоксазол-3-ил)-2,6-диметоксибензол-1-сульфонамида (пример 87).
К раствору N-(6-{[4-({[трет-бутил(диметил)силил]окси}метил)-1H-пиразол-1-ил]метил}-4-метокси-1,2-бензоксазол-3-ил)-2,6-диметоксибензол-1-сульфонамида (E-3) (80,0 мг, 0,16 ммоль) в ТГФ (2,0 мл) добавляли TBAF (76,3 мг, 0,32 ммоль). Реакционный раствор перемешивали в течение 1 ч. ЖХМС анализ показывал, что исходное вещество расходовалось с образованием желаемой массы продукта. Реакционную смесь концентрировали досуха. Остаток обрабатывали EtOAc (15 мл) и промывали H2O (10 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали досуха. Остаток очищали путем препаративной ВЭЖХ с колонкой YMC-Actus Triart C-18 (30×150 мм, размер частиц 5 мкм), который элюировали 5-25% MeCN/H2O (0,05% NH4OH) со скоростью потока 35 мл/мин с получением N-(6-{[4-(гидроксиметил)-1H-пиразол-1-ил]метил}-4-метокси-1,2-бензоксазол-3-ил)-2,6-диметоксибензол-1-сульфонамида (пример 87) (4,5 мг, 6%-ный выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,73 (шир. с, 1H), 7,40 (шир. с, 2H), 6,74 (шир. с, 4H), 5,35 (шир. с, 2H), 4,81 (т, J=5,4 Гц, 1H), 4,34 (д, J=5,4 Гц, 2H), 3,97-3,59 (м, 9H); m/z (ESI+) 475,0 (M+H)+.
Соединения по примерам, приведенным в таблице ниже, синтезировали в соответствии со способами, используемыми для синтеза N-(6-{[4-(гидроксиметил)-1H-пиразол-1-ил]метил}-4-метокси-1,2-бензоксазол-3-ил)-2,6-диметоксибензол-1-сульфонамида (пример 87). Следующие соединения по примерам синтезировали с некритическими изменениями или заменами в приведенных в качестве примеров способах, которые мог бы осуществить специалист в данной области. При необходимости разделение смесей региоизомеров проводили стандартными методами, известными в данной области, такими как SFC или ВЭЖХ, и проводили на любой подходящей стадии в последовательности синтеза.
Таблица 14:
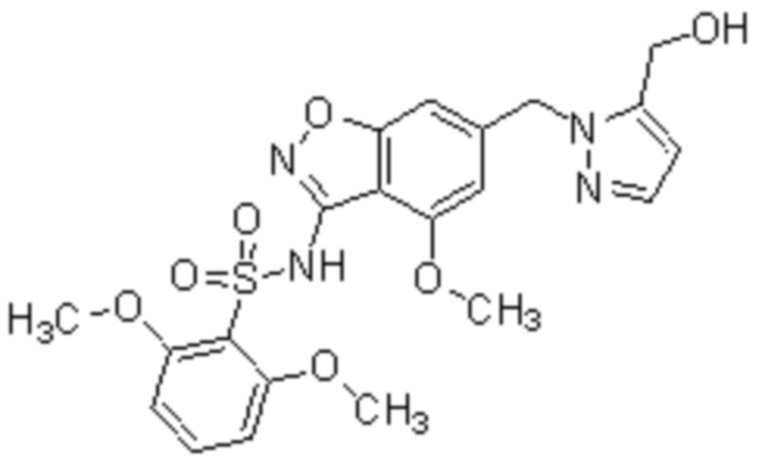
N-(6-{[5-(гидроксиметил)-1H-пиразол-1-ил]метил}-4-метокси-1,2-бензоксазол-3-ил)-2,6-диметоксибензол-1-сульфонамид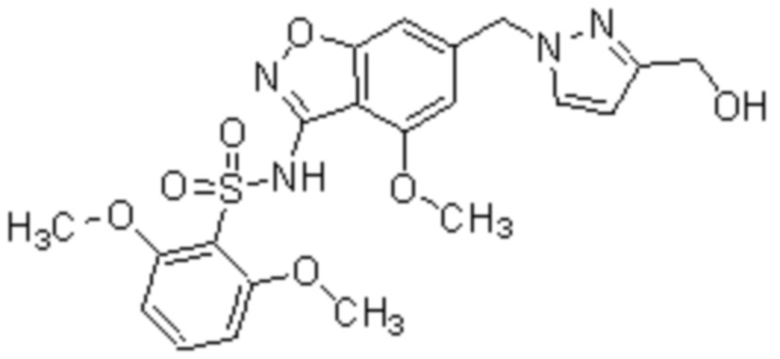
N-(6-{[3-(гидроксиметил)-1H-пиразол-1-ил]метил}-4-метокси-1,2-бензоксазол-3-ил)-2,6-диметоксибензол-1-сульфонамид
Пример 90: Получение 2,6-диметокси-N-{4-метокси-6-[(1H-пиразол-1-ил)(2H2)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида в соответствии со схемой F.
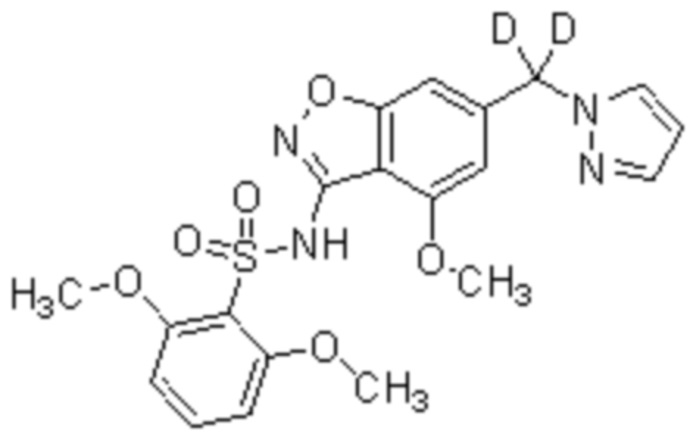
Схема F:
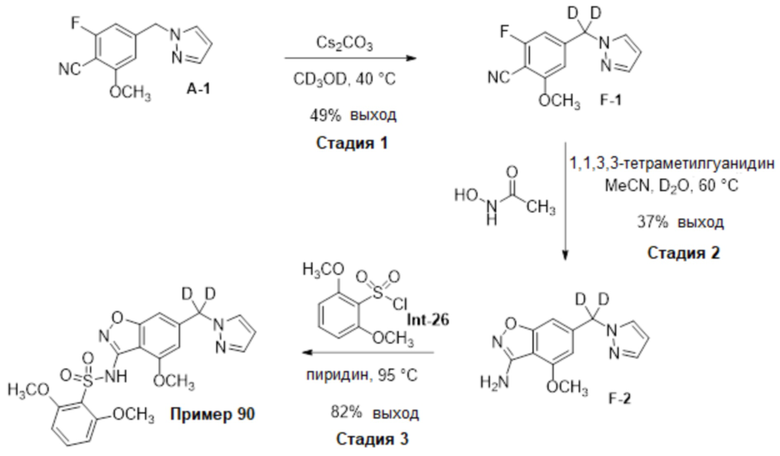
Стадия 1: Синтез 2-фтор-6-метокси-4-[(1H-пиразол-1-ил)(2H2)метил]бензонитрила (F-1).
К раствору 2-фтор-6-метокси-4-[(1H-пиразол-1-ил)метил]бензонитрила (164 мг, 0,703 ммоль) (A-1) в CD3OD (4,0 мл) добавляли Cs2CO3 (229 мг, 0,703 ммоль). Смесь перемешивали при температуре 40ºC в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали досуха. Остаток очищали с помощью флеш хроматографии (24 г, SiO2, 0-40% EtOAc/DCM) с получением 2-фтор-6-метокси-4-[(1H-пиразол-1-ил)(2H2)метил]бензонитрила (F-1) (81,0 мг, 49%-ный выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3) δ 7,61 (д, J=1,71 Гц, 1H), 7,47 (д, J=2,32 Гц, 1H), 6,52-6,57 (м, 2H), 6,37 (т, J=2,08 Гц, 1H), 3,88-3,91 (м, 3H); m/z (ESI+) 234,2 (M+H)+.
Стадия 2: Синтез 4-метокси-6-[(1H-пиразол-1-ил)(2H2)метил]-1,2-бензоксазол-3-амина (F-2).
К суспензии 2-фтор-6-метокси-4-[(1H-пиразол-1-ил)(2H2)метил]бензонитрила (F-1) (81,0 мг, 0,35 ммоль) и N-гидроксиацетамида (78,2 мг, 1,04 ммоль) в MeCN (2,7 мл) и D2O (0,3 мл) добавляли 1,1,3,3-тетраметилгуанидин (240 мг, 2,08 ммоль). Смесь перемешивали при температуре 60ºC в течение 7 ч и 65ºC в течение дополнительных 2 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали досуха. Остаток очищали с помощью флеш хроматографии (24 г, SiO2, 60-100% EtOAc/DCM) с получением 4-метокси-6-[(1H-пиразол-1-ил)(2H2)метил]-1,2-бензоксазол-3-амина (F-2) (32 мг, 37%-ный выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,85-7,89 (м, 1H), 7,49 (д, J=1,83 Гц, 1H), 6,70 (д, J=0,86 Гц, 1H), 6,63 (д, J=0,73 Гц, 1H), 6,30 (т, J=2,08 Гц, 1H), 5,93 (с, 2H), 3,83-3,87 (м, 3H); m/z (ESI+) 247,2 (M+H)+.
Стадия 3: Синтез 2,6-диметокси-N-{4-метокси-6-[(1H-пиразол-1-ил)(2H2)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 90) в соответствии со способом получения сульфонамида A.
Смесь 4-метокси-6-[(1H-пиразол-1-ил)(2H2)метил]-1,2-бензоксазол-3-амина (F-2) (25,0 мг, 0,10 ммоль) и 2,6-диметоксибензол-1-сульфонил хлорида (Int-26) (36,0 мг, 0,152 ммоль) в пиридине перемешивали при температуре 95ºC в течение 2 ч. Полученную смолу разбавляли DCM и обрабатывали AcOH (46 мкл, 0,812 ммоль). Смесь напрямую очищали с помощью флеш хроматографии (24 г, SiO2, 70-100% EtOAc/гептан) с получением 2,6-диметокси-N-{4-метокси-6-[(1H-пиразол-1-ил)(2H2)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 90) (37,0 мг, 82%-ный выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,60 (с, 1H), 7,88 (д, J=2,08 Гц, 1H), 7,43-7,54 (м, 2H), 6,84 (с, 1H), 6,73-6,80 (м, 3H), 6,30 (т, J=2,08 Гц, 1H), 3,87 (с, 3H), 3,76 (с, 6H); m/z (ESI+) 448,1 (M+H)+.
Соединения по примерам, представленные в таблице ниже, были синтезированы в соответствии со способами, используемыми для синтеза 2,6-диметокси-N-{4-метокси-6-[(1H-пиразол-1-ил)(2H2)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 90). Следующие соединения по примерам синтезировали с некритическими изменениями или замены приведенных в качестве примеров способов, которые сможет осуществить специалист в данной области техники.
Таблица 15:
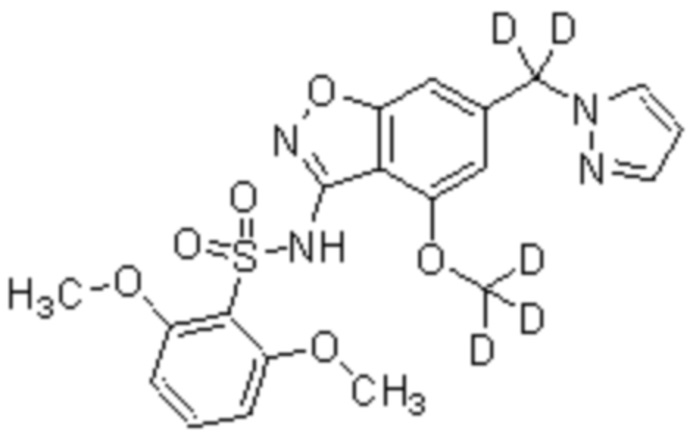
2,6-диметокси-N-{4-[(2H3)метилокси]-6-[(1H-пиразол-1-ил)(2H2)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид
Пример 92: Получение N-{5-бром-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида в соответствии со схемой G.
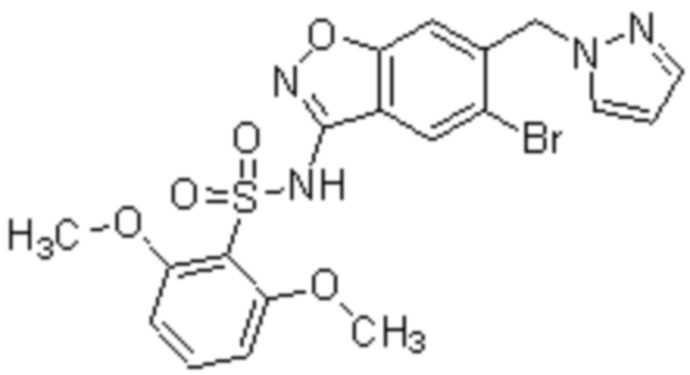
Схема G:
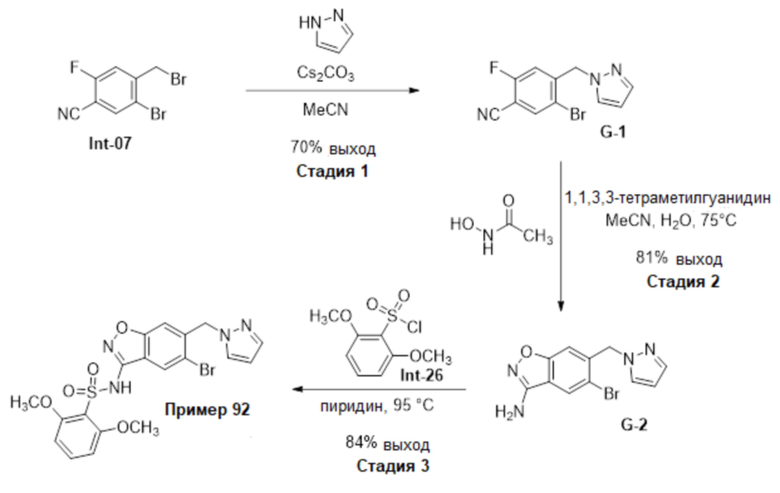
Стадия 1: Синтез 5-бром-2-фтор-4-[(1H-пиразол-1-ил)метил]бензонитрила (G-1)
К раствору 5-бром-4-(бромметил)-2-фторбензонитрила (Int-07) (280 мг, 0,956 ммоль) в MeCN (6,4 мл) добавляли 1H-пиразол (71,6 мг, 1,05 ммоль) и Cs2CO3 (0,374 мг, 1,15 ммоль). Смесь перемешивали при комнатной температуре в течение 6 ч. Смесь разбавляли EtOAc и фильтровали. Фильтрат концентрировали досуха. Остаток очищали с помощью флеш хроматографии (40 г, SiO2, 10-100% EtOAc/гептан) с получением 5-бром-2-фтор-4-[(1H-пиразол-1-ил)метил]бензонитрила (G-1) (188 мг, 70%-ный выход) в виде прозрачного масла, которое затвердевало при стоянии в виде твердого вещества бледно-желтого цвета. 1H ЯМР (400 МГц, CDCl3) δ 7,83 (д, J=5,7 Гц, 1H), 7,65 (д, J=1,7 Гц, 1H), 7,53 (д, J=2,2 Гц, 1H), 6,52 (д, J=9,3 Гц, 1H), 6,40 (т, J=2,1 Гц, 1H), 5,43 (с, 2H); m/z (ESI+) 280,0, 282,0 (M+H)+.
Стадия 2: Синтез 5-бром-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (G-2)
К раствору 5-бром-2-фтор-4-[(1H-пиразол-1-ил)метил]бензонитрила (G-1) (185 мг, 0,66 ммоль) и N-гидроксиацетамида (149 мг, 1,98 ммоль) в MeCN (3,5 мл) и H2O (0,35 мл) добавляли 1,1,3,3-тетраметилгуанидин (45,9 мг, 0,399 ммоль). Смесь перемешивали при температуре 75ºC в течение 4 ч. Реакционную смесь концентрировали досуха. Остаток очищали с помощью флеш хроматографии (40 г, SiO2, 30-90% EtOAc/гептан) с получением 5-бром-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (G-2) (157 мг, 81%-ный выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,18 (с, 1H), 7,87 (д, J=2,2 Гц, 1H), 7,55 (д, J=1,3 Гц, 1H), 6,80 (с, 1H), 6,50 (с, 2H), 6,34 (т, J=2,1 Гц, 1H), 5,51 (с, 2H); m/z (ESI+) 293,0, 295,0 (M+H)+.
Стадия 3: Синтез N-{5-бром-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида (пример 92)
Раствор 5-бром-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (G-2) (155 мг, 0,529 ммоль) и 2,6-диметоксибензол-1-сульфонил хлорида (Int-26) (188 мг, 0,793 ммоль) в пиридине (0,31 мл) нагревали до температуры 95ºC, после чего реакция становилась гомогенной. Реакционную смесь перемешивали при температуре 95ºC в течение 2 ч и затем концентрировали досуха. Остаток обрабатывали в минимальном количестве DCM и обрабатывали AcOH (0,10 мл, 1,75 ммоль). Смесь помещали непосредственно на SiO2 и очищали с помощью флеш хроматографии (40 г, SiO2, 35-100% MeOAc/гептан) с получением N-{5-бром-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида (пример 92) (221 мг, 84%-ный выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,55 (с, 1H), 8,39 (с, 1H), 7,87 (д, J=2,1 Гц, 1H), 7,54 (д, J=1,3 Гц, 1H), 7,47 (т, J=8,4 Гц, 1H), 6,88 (с, 1H), 6,74 (д, J=8,4 Гц, 2H), 6,34 (т, J=2,1 Гц, 1H), 5,52 (с, 2H), 3,75 (с, 6H); m/z (ESI+) 493,0, 495,0 (M+H)+.
Соединения по примерам, приведенным в таблице ниже, синтезировали в соответствии со способами, используемыми для синтеза N-{5-бром-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида (пример 92). Следующие соединения по примерам синтезировали с некритическими изменениями или заменами в приведенных в качестве примеров способах, которые мог бы осуществить специалист в данной области. При необходимости разделение смесей региоизомеров проводили стандартными методами, известными в данной области, такими как SFC или ВЭЖХ, и проводили на любой подходящей стадии в последовательности синтеза.
Таблица 16:
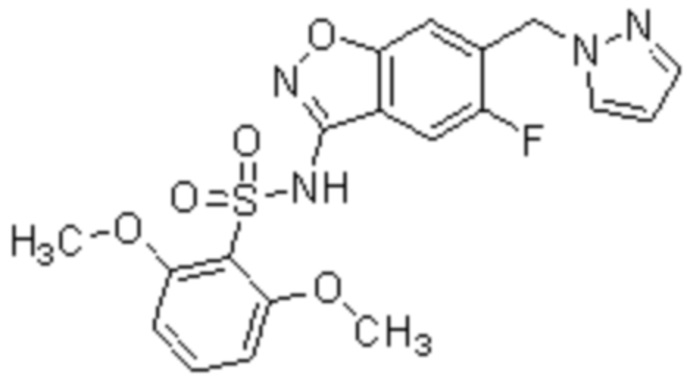
N-{5-фтор-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамид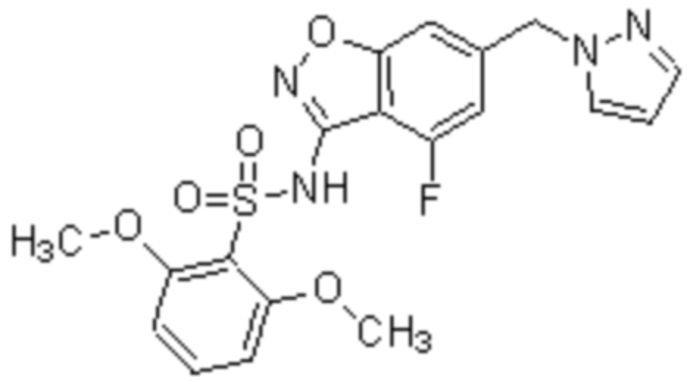
N-{4-фтор-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамид
Пример 95: Получение N-{4-этил-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида в соответствии со схемой H.
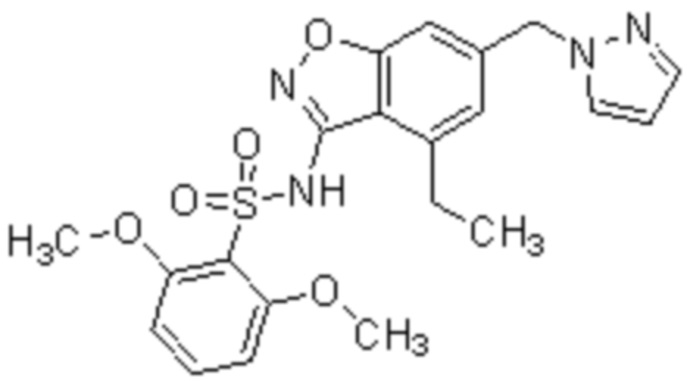
Схема H:
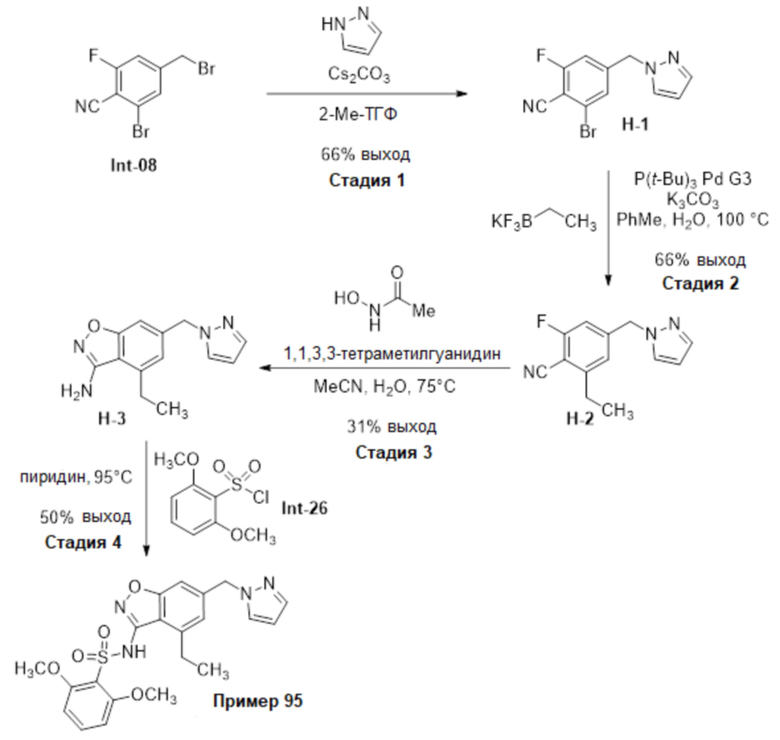
Стадия 1: Синтез 2-бром-6-фтор-4-[(1H-пиразол-1-ил)метил]бензонитрила (H-1)
Суспензию 2-бром-4-(бромметил)-6-фторбензонитрила (Int-08) (459 мг, 1,57 ммоль), 1H-пиразола (159 мг, 2,34 ммоль) и Cs2CO3 (767 мг) в 2-Me-ТГФ (3,1 мл) перемешивали при комнатной температуре в течение 5,5 ч. ЖХМС анализ показывал, что исходное вещество расходовалось. Смесь распределяли между H2O (5 мл) и EtOAc (20 мл). Водный слой экстрагировали EtOAc (20 мл). Объединенные органические слои промывали насыщенным солевым раствором (5 мл), сушили над MgSO4, фильтровали и концентрировали. Остаток очищали с помощью флеш хроматографии (40 г, SiO2, 0-100% EtOAc/гептан) с получением 2-бром-6-фтор-4-[(1H-пиразол-1-ил)метил]бензонитрила (H-1) (295 мг, 66%-ный выход) в виде масла желтого цвета. 1H ЯМР (400 МГц, CDCl3) δ 7,62 (д, J=1,7 Гц, 1H), 7,47 (д, J=2,3 Гц, 1H), 7,28 (с, 1H), 6,92 (д, J=8,9 Гц, 1H), 6,38 (т, J=2,1 Гц, 1H), 5,36 (с, 2H); m/z (ESI+) 280,0, 282,0 (M+H)+.
Стадия 2: Синтез 2-этил-6-фтор-4-[(1H-пиразол-1-ил)метил]бензонитрила (H-2)
В сосуд для микроволновой печи помещали 2-бром-6-фтор-4-[(1H-пиразол-1-ил)метил]бензонитрил (H-1) (79,3 мг, 0,283 ммоль), этилтрифторборат калия (60,0 мг, 0,441 ммоль), K2CO3 (108 мг, 0,778 ммоль) и метансульфонато(три-трет-бутилфосфино)(2"амино-1,1-бифенил-2-ил)палладий(II) (P(t-Bu)3 Pd (7,9 мг, 0,014 ммоль), герметично закрывали, вакуумировали и снова заполняли N2. Добавляли PhMe (0,60 мл) и деионизированную H2O (0,30 мл) и смесь перемешивали при температуре 100ºC в течение 5 ч. ЖХМС анализ показывал образование желаемой массы продукта. Смесь распределяли между насыщенным водным раствором NH4Cl (10 мл) и EtOAc (15 мл). Водный слой экстрагировали EtOAc (15 мл). Объединенные органические продукты промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Остаток очищали с помощью флеш хроматографии (12 г, SiO2, 0-100% EtOAc/гептан) с получением 2-этил-6-фтор-4-[(1H-пиразол-1-ил)метил]бензонитрила (H-2) (43,1 мг, 66%-ный выход) в виде твердого вещества не совсем белого цвета. 1H ЯМР (400 МГц, CDCl3) δ 7,60 (д, J=1,6 Гц, 1H), 7,46 (д, J=2,2 Гц, 1H), 6,92 (с, 1H), 6,76 (д, J=9,2 Гц, 1H), 6,36 (т, J=2,1 Гц, 1H), 5,36 (с, 2H), 2,85 (кв, J=7,6 Гц, 2H), 1,28 (т, J=7,6 Гц, 3H); m/z (APCI+) 230,1 (M+H)+.
Стадия 3: Синтез 4-этил-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (H-3)
К раствору 2-этил-6-фтор-4-[(1H-пиразол-1-ил)метил]бензонитрила (H-2) (40,6 мг, 0,177 ммоль) и N-гидроксиацетамида (43,9 мг, 0,585 мг) в MeCN (1,0 мл) и H2O (0,1 мл) добавляли 1,1,3,3-тетраметилгуанидин (120 мг, 1,0 ммоль). Смесь перемешивали при температуре 75ºC в течение 24 ч. Смесь концентрировали досуха и очищали с помощью флеш хроматографии (12 г, SiO2, 0-100% EtOAc/гептан) с получением 4-этил-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (H-3) (13,2 мг, 31%-ный выход). 1H ЯМР (400 МГц, CDCl3) δ 7,60 (д, J=1,6 Гц, 1H), 7,45 (д, J=2,0 Гц, 1H), 7,04 (с, 1H), 6,87 (с, 1H), 6,34 (т, J=2,0 Гц, 1H), 5,44 (с, 2H), 4,35 (шир. с, 2H), 2,93 (кв, J=7,6 Гц, 2H), 1,34 (т, J=7,6 Гц, 3H); m/z (APCI+) 243,1 (M+H)+.
Стадия 4: Синтез N-{4-этил-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида (пример 95)
Суспензию 4-этил-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (H-3) (13,2 мг, 0,055 ммоль) и 2,6-диметоксибензол-1-сульфонил хлорида (Int-26) (21,3 мг, 0,090 ммоль) в пиридине (0,15 мл) перемешивали при температуре 95ºC в течение 3 ч. ЖХМС анализ показывал, что исходное вещество расходовалось. Реакционную смесь концентрировали досуха и очищали с помощью флеш хроматографии (4 г, SiO2, 0-100% EtOAc/гептан) с получением N-{4-этил-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида (пример 95) (12,0 мг, 50%-ный выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ10,14 (с, 1H), 7,87 (д, J=2,2 Гц, 1H), 7,57-7,50 (м, 1H), 7,49 (д, J=1,5 Гц, 1H), 7,20 (шир. с, 1H), 7,06 (шир. с, 1H), 6,79 (шир. д, J=7,8 Гц, 2H), 6,29 (т, J=2,1 Гц, 1H), 5,46 (с, 2H), 3,77 (шир. с, 6H), 3,07 (кв, J=7,5 Гц, 2H), 1,21 (т, J=7,5 Гц, 3H); m/z (APCI+) 443,1 (M+H)+.
Соединения по примерам, представленные в таблице ниже, были синтезированы в соответствии со способами, используемыми для синтеза N-{4-этил-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида (пример 95). Следующее соединение по примеру синтезировали с некритическими изменениями или замены приведенных в качестве примеров способов, которые сможет осуществить специалист в данной области техники.
Таблица 17:
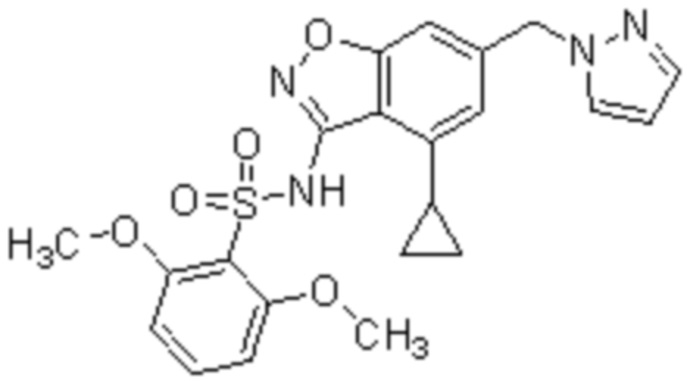
N-{4-циклопропил-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамид
Пример 97: Получение 5-циклопропил-2-метокси-N-{6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида в соответствии со схемой J.
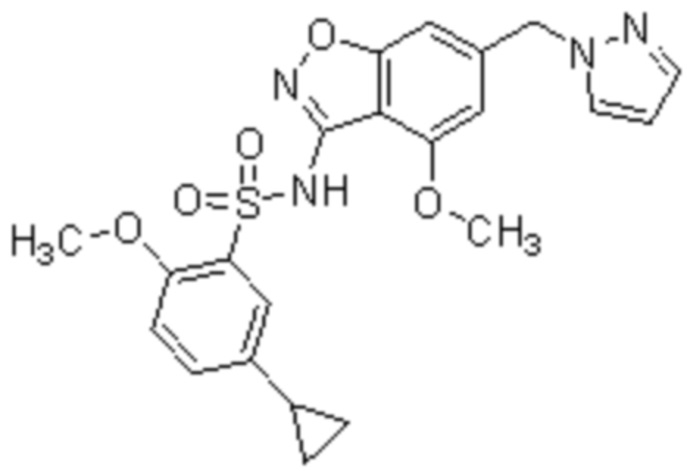
Схема J:
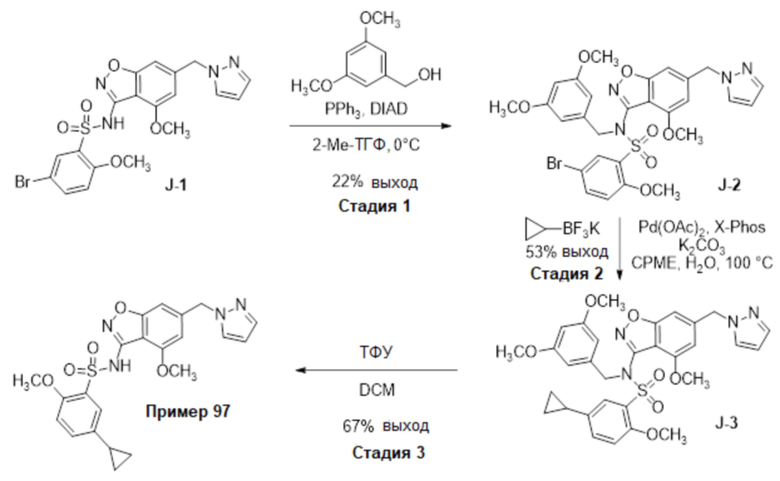
Стадия 1: Синтез 5-бром-N-[(3,5-диметоксифенил)метил]-2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (J-2)
К раствору 5-бром-2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (J-1) (получен как в примере 01, 700 мг, 1,42 ммоль), PPh3 (930 мг, 3,55 ммоль) и (3,5-диметоксифенил)метанола (358 мг, 2,13 ммоль) в 2-Me-ТГФ (30 мл) при температуре 0ºC добавляли по каплям DIAD (574 мг, 2,84 ммоль). Раствор перемешивали в течение 16 ч с получением суспензии бледно-желтого цвета. Суспензию фильтровали, и фильтрат концентрировали досуха. Остаток очищали с помощью флеш хроматографии (40 г, SiO2, 1:2 петролейный эфир/EtOAc) с получением 5-бром-N-[(3,5-диметоксифенил)метил]-2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (J-2) (200 мг, 22%-ный выход) в виде твердого вещества белого цвета.
Стадия 2: Синтез 5-циклопропил-N-[(3,5-диметоксифенил)метил]-2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (J-3)
К раствору 5-бром-N-[(3,5-диметоксифенил)метил]-2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (J-2) (200 мг, 0,311 ммоль) в CPME (10,0 мл) и H2O (1,0 мл) добавляли циклопропилтрифторборат калия (138 мг, 0,932 ммоль), Pd(OAc)2 (14,0 мг, 0,062 ммоль), K2CO3 (172 мг, 1,24 ммоль) и X-Phos (44,4 мг, 0,093 ммоль). Смесь откачивали и снова заполнял N2 (3×) и затем перемешивали при температуре 100ºC в атмосфере N2 в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли EtOAc (20 мл) и фильтровали. Фильтрат концентрировали досуха. Остаток очищали с помощью флеш хроматографии (20 г, SiO2, EtOAc) с получением 5-циклопропил-N-[(3,5-диметоксифенил)метил]-2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (J-3) (100 мг, 53%-ный выход) в виде твердого вещества белого цвета. m/z (ESI+) 605,3 (M+H)+.
Стадия 3: Синтез 5-циклопропил-2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 97)
К раствору 5-циклопропил-N-[(3,5-диметоксифенил)метил]-2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (J-3) (100 мг, 0,165 ммоль) в DCM (2,0 мл) добавляли ТФУ (2,0 мл). Смесь перемешивали в течение 1 ч и затем концентрировали досуха. Остаток очищали с помощью флеш хроматографии (12 г, SiO2, 1:10 MeOH/EtOAc). Материал повторно очищали путем препаративной ВЭЖХ с колонкой Phenomenex Gemini-NX (150×30 мм, размер частиц 5 мкм), которые элюировали 2-42% MeCN/H2O (+0,05% NH4OH) со скоростью потока 30 мл/мин с получением 5-циклопропил-2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 97) (50 мг, 67%-ный выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 10,10 (шир. с, 1H), 7,88 (д, J=1,9 Гц, 1H), 7,51 (дд, J=1,6, 11,0 Гц, 2H), 7,27 (шир. д, J=6,8 Гц, 1H), 7,05 (шир. д, J=8,9 Гц, 1H), 6,82 (шир. с, 1H), 6,72 (шир. с, 1H), 6,30 (т, J=1,9 Гц, 1H), 5,44 (с, 2H), 3,83 (с, 2H), 3,90 (шир. д, J=8,3 Гц, 1H), 3,73 (с, 2H), 3,76-3,67 (м, 1H), 2,01-1,89 (м, 1H), 1,04-0,78 (м, 2H), 0,70-0,42 (м, 2H). m/z (ESI+) 454,8 (M+H)+.
Пример 98: Получение N-(6-((1H-пиразол-1-ил)метил)-4-метоксибензо[d]изоксазол-3-ил)-2,6-диметоксибензолсульфонамида [или 2,6-диметокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида] в соответствии со схемой K.
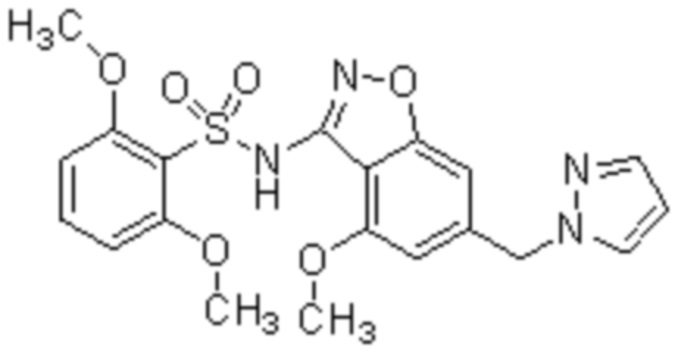
Схема K:
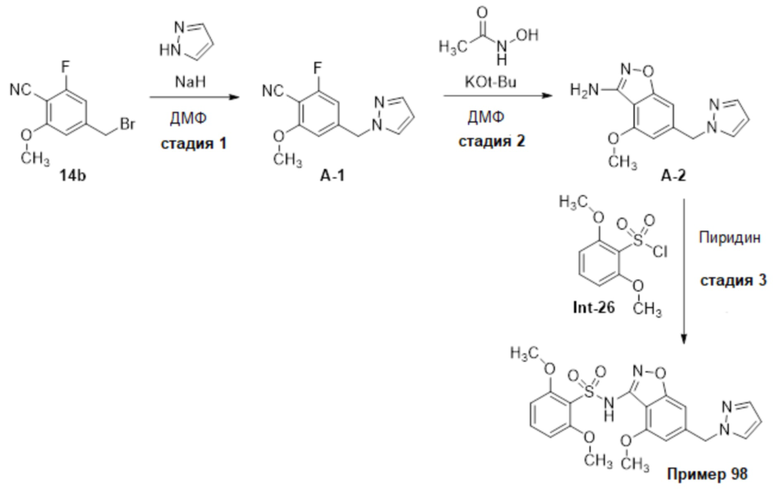
Стадия 1: Альтернативный синтез 4-((1H-пиразол-1-ил)метил)-2-фтор-6-метоксибензонитрила (A-1) из 14b
Раствор 1H-пиразола (2,0 г, 29,6 ммоль) и NaH (60% мас./мас. дисперсия в минеральном масле, 1,5 г, 37,1 ммоль) в ДМФ (520 мл) перемешивали при температуре 0ºC в течение 1 ч. Затем добавляли раствор 4-(бромметил)-2-фтор-6-метоксибензонитрила (14b) (6,0 г, 24,7 ммоль) в ДМФ (80 мл) и смесь перемешивали при комнатной температуре в течение ночи. Реакцию гасили водой и смесь экстрагировали EtOAc. Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии (петролейный эфир/EtOAc=6/1) с получением 4-((1H-пиразол-1-ил)метил)-2-фтор-6-метоксибензонитрила (A-1) (2,4 г, 42%) в виде твердого вещества желтого цвета. m/z 232,0 [M+H]+.
Стадия 2: Альтернативный синтез 6-((1H-пиразол-1-ил)метил)-4-метоксибензо[d]изоксазол-3-амина (A-2) с использованием трет-бутоксида калия
К раствору ацетогидроксамовой кислоты (3,7 г, 49,5 ммоль) в безводном ДМФ (150 мл) при комнатной температуре добавляли трет-бутоксид калия (5,6 г, 49,5 ммоль) и смесь перемешивали при комнатной температуре в течение 1 ч. Затем добавляли 4-((1H-пиразол-1-ил)метил)-2-фтор-6-метоксибензонитрил (A-1) (3,8 г, 16,5 ммоль) и перемешивание продолжали при температуре 60ºC в течение 4 ч. Добавляли воду и смесь экстрагировали EtOAc. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии (петролейный эфир/EtOAc=5/1) с получением 6-((1H-пиразол-1-ил)метил)-4-метоксибензо[d]изоксазол-3-амина (A-2) (2,1 г, 53%) в виде твердого вещества желтого цвета. m/z 245,0 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,87 (дд, J=1,6, 0,4 Гц, 1H), 7,50 (дд, J=1,6, 0,4 Гц, 1H), 6,69 (с, 1H), 6,62 (с, 1H), 6,30 (т, J=2,1 Гц, 1H), 5,93 (с, 2H), 5,41 (с, 2H), 3,86 (с, 3H).
Стадия 3: Синтез N-(6-((1H-пиразол-1-ил)метил)-4-метоксибензо[d]изоксазол-3-ил)-2,6-диметоксибензолсульфонамида (пример 98)
Смесь 6-((1H-пиразол-1-ил)метил)-4-метоксибензо[d]изоксазол-3-амина (A-2) (50 мг, 0,205 ммоль) и 2,6-диметоксибензолсульфонил хлорида (Int-26) (73 мг, 0,308 ммоль) в пиридине (1 мл) нагревали при температуре 120ºC в течение 2 ч при воздействии микроволнового облучения (партия 1).
Смесь 6-((1H-пиразол-1-ил)метил)-4-метоксибензо[d]изоксазол-3-амина (A-2)(500 мг, 2,1 ммоль) и 2,6-диметоксибензолсульфонил хлорида (Int-26) (746 мг, 3,2 ммоль) в пиридине (5 мл) нагревали при температуре 120ºC в течение 2 ч при воздействии микроволнового облучения (партия 2).
Эту реакцию повторяли еще раз в том же масштабе (партия 3).
Смесь 6-((1H-пиразол-1-ил)метил)-4-метоксибензо[d]изоксазол-3-амина (A-2)(350 мг, 1,4 ммоль) и 2,6-диметоксибензолсульфонил хлорида (Int-26) (509 мг, 2,2 ммоль) в пиридине (4 мл) нагревали при температуре 120ºC в течение 2 ч при воздействии микроволнового облучения (партия 4).
Четыре реакционные смеси объединяли, разбавляли водой, доводили до pH 5-6 с помощью 2 M водной HCl и экстрагировали EtOAc (300 мл×3). Объединенные органические экстракты сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии (петролейный эфир/EtOAc=2/1) с получением N-(6-((1H-пиразол-1-ил)метил)-4-метоксибензо[d]изоксазол-3-ил)-2,6-диметоксибензолсульфонамида (пример 98) (1,07 г, 43%) в виде твердого вещества белого цвета. m/z 445,0 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,58 (с, 1H), 7,87 (д, J=2,0 Гц, 1H), 7,50-7,46 (м, 2H), 6,83 (с, 1H), 6,76 (м, 3H), 6,30 (с, 1H), 5,44 (с, 2H), 3,87 (с, 3H), 3,76 (с, 6H).
Пример 98: Альтернативное получение 2,6-диметокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида в соответствии со схемой L.
Схема L:
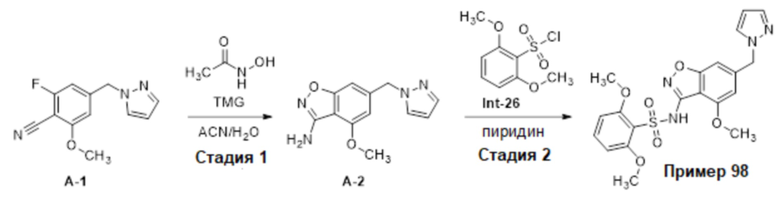
Стадия 1: Альтернативный синтез 4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (A-2), используя 1,1,3,3-тетраметилгуанидин.
Суспензию 2-фтор-6-метокси-4-(1H-пиразол-1-илметил)бензонитрила (A-1) (15,43 г, 66,7 ммоль), N-гидроксиацетамида (15,0 г, 200 ммоль) и 1,1,3,3-TMG (46,1 г, 400 ммоль) в ацетонитриле (270 мл) и деионизированной воде (30 мл) нагревали при температуре 60ºC в течение 7 часов. Ацетонитрил удаляли в вакууме, и оставшееся густое масло распределяли между этилацетатом (300 мл) и деионизированной водой (250 мл). Водный слой экстрагировали этилацетатом (2×150 мл). Все органические слои объединяли и промывали насыщ. водн. NaCl. Некоторые твердые вещества начали образовываться в органическом слое, поэтому добавляли метанол (~10 мл) и суспензию нагревали до гомогенности. После охлаждения до комнатной температуры органический слой сушили над сульфатом натрия, фильтровали и концентрировали. Полученную твердое вещество бледно-желтого цвета суспендировали в этилацетате (125 мл) и недолго нагревали до кипения с обратным холодильником. Суспензии давали охладиться до комнатной температуры, полученную твердые продукты собирали путем фильтрации и слой продукта на фильтре промывали в гептане. Фильтрат и промывку гептаном концентрировали досуха, остаточное твердое вещество суспендировали в этилацетате (15 мл), суспензию ненадолго нагревали до кипения с обратным холодильником и собирали вторую порцию осадка, как прежде. Объединенные фракции осадка сушили в вакууме с получением 4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (A-2) (11,86 г, 48,6 ммоль) в виде порошка бледно-желтого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,87 (д, J=1,8 Гц, 1H), 7,49 (д, J=1,2 Гц, 1H), 6,69 (с, 1H), 6,62 (с, 1H), 6,30 (т, J=2,1 Гц, 1H), 5,93 (с, 2H), 5,41 (с, 2H), 3,86 (с, 3H). ЖХМС: [M+H]+ 245.
Стадия 2: Синтез 22,6-диметокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 98)
Смесь 4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (A-2) (9,5 г, 39 ммоль) и 2,6-диметоксибензолсульфонил хлорида (Int-26) (12,1 г, 51,1 ммоль) в пиридине (20 мл) нагревали при внутренней температуре 97ºC в течение 1 часа. После охлаждения до температуры 50ºC, раствор выливали в колбу, содержащую колотый лед (200 г) и 6н HCl (100 мл). Реакционный сосуд промывали дихлорметаном для количественного определения переноса. Полученную водную смесь экстрагировали дихлорметаном (4×100 мл). Объединенные органические экстракты промывали деионизированной водой и насыщ. водн. NaCl, сушили над сульфатом магния, фильтровали и концентрировали до желтой пены. К пене добавляли метилацетат (50 мл) и суспензию перемешивали при комнатной температуре в течение 1 часа. Твердые продукты собирали фильтрованием с отсасыванием и промывали в гептане. После сушки в вакууме сырой 2,6-диметокси-N-[4-метокси-6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамида (пример 98)(16,1 г, 95%) получали в виде твердого вещества оранжево-коричневого цвета. твердого вещества с метилацетатом еще два раза не привело к исчезновению оранжевого цвета, поэтому неочищенный продукт растирали в теплом дихлорметане, давали ему охладиться до комнатной температуры и фильтровали с получением белого твердого вещества кремового цвета. Маточный раствор дихлорметана дополнительно очищали хроматографией (колонка с силикагелем 330 г, элюирование 60-100% этилацетатом в гептане) с получением белого твердого вещества. Твердые вещества от растирания с DCM и хроматографии фильтрата DCM объединяли, перемешивали в кипящем метилацетате и охлаждали до комнатной температуры в течение 2 часов. Полученное твердое вещество собирали фильтрованием с отсасыванием и сушили в вакуумном сушильном шкафу при 100ºC в течение ночи, получая очищенный 2,6-диметокси-N-[4-метокси-6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамид (пример 98) (15,3 г, 89%) в виде порошка не совсем белого цвета.
Три партии соединения по примеру 98 (всего 54,3 г), приготовленные, как описано выше, объединяли, суспендировали в метилацетате (250 мл) и нагревали с обратным холодильником в течение 1 часа. После удаления нагревающей бани перемешивание продолжали в течение 4 часов, пока смесь охлаждалась до комнатной температуры. Полученный осадок собирали путем фильтрации и промывали в гептане. Твердый продукт сушили в вакууме при комнатной температуре в течение 2 часов, затем еще сушили при 130яяC в вакуумной печи в течение 16 часов, получая 2,6-диметокси-N-[4-метокси-6-(1H-пиразол-1-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамид (пример 98) (53,55 г, 99%) в виде твердого вещества не совсем белого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,60 (с, 1H), 7,88 (д, J=1,7 Гц, 1H), 7,45-7,52 (м, 2H), 6,83 (с, 1H), 6,77 (д, J=8,4 Гц, 3H), 6,30 (т, J=2,1 Гц, 1H), 5,44 (с, 2H), 3,87 (с, 3H), 3,76 (с, 6H). ЖХМС: [M+H]+ 445. Анал. Вычисл. для C20H20N4O6S: C, 54,05; H, 4,54; N, 12,61; S, 7,21. Найдено: C, 53,91; H, 4,58; N, 12,51; S, 7,09.
Пример 99: Получение N-(6-((1H-пиразол-1-ил)метил)-4-метоксибензо[d]изоксазол-3-ил)-3-метилхинолин-8-сульфонамида в соответствии со способом AC.
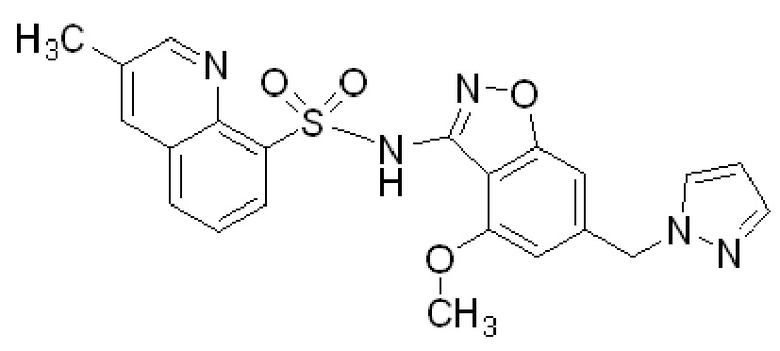
Пример 100: Получение 2,6-диметокси-N-(4-метокси-6-((4-метил-1H-пиразол-1-ил)метил)бензо[d]изоксазол-3-ил)бензолсульфонамида в соответствии со способом AC.
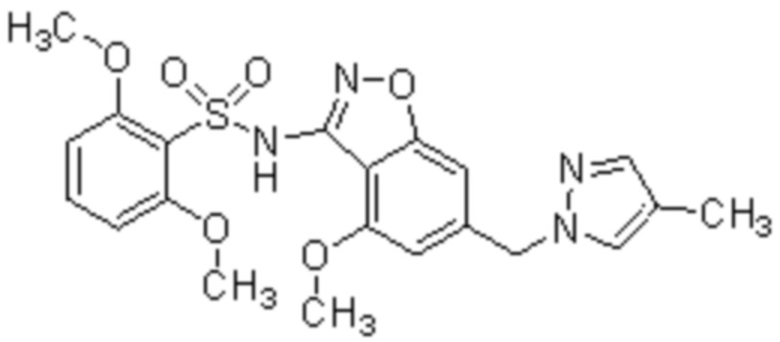
Способ AC:
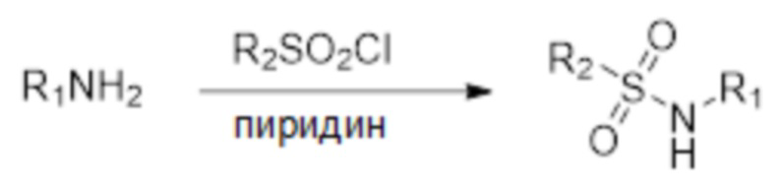
К раствору амина (0,2 ммоль, 1,0 экв.) в пиридине (2 мл) добавляли сульфонил хлорид (1,5 экв.) и смесь нагревали при температуре 120ºC при воздействии микроволнового облучения в течение 2 ч. Смесь распределяли между водой и EtOAc, слои разделяли, и органический слой промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали путем преп. ТСХ с получением указанного в заголовке соединения. Вариации вышеуказанных условий отмечены в таблице 18.
Таблица 18:
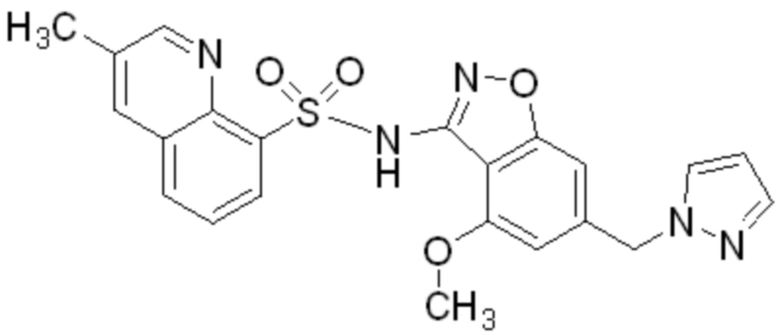
N-(6-((1H-пиразол-1-ил)метил)-4-метоксибензо[d]изоксазол-3-ил)-3-метилхинолин-8-сульфонамид
использовали 5 мл пиридин.
Перед обработкой доводили до pH 5 с помощью 1 M водн. HCl.
Преп. ТСХ (петролейный эфир/EtOAc=1/2)
2,6-диметокси-N-(4-метокси-6-((4-метил-1H-пиразол-1-ил)метил)бензо[d]изоксазол-3-ил)бензолсульфонамид
4-метокси-6-((4-метил-1H-пиразол-1-ил)метил)бензо-[d]изоксазол-3-амин (Int-25)
Преп. ТСХ (DCM/MeOH, 20/1)
Пример 101: Получение 2,6-диметокси-N-(4-метокси-6-(1-метил-1H-пиразол-4-ил)бензо[d]изоксазол-3-ил)бензолсульфонамида в соответствии со схемой M.
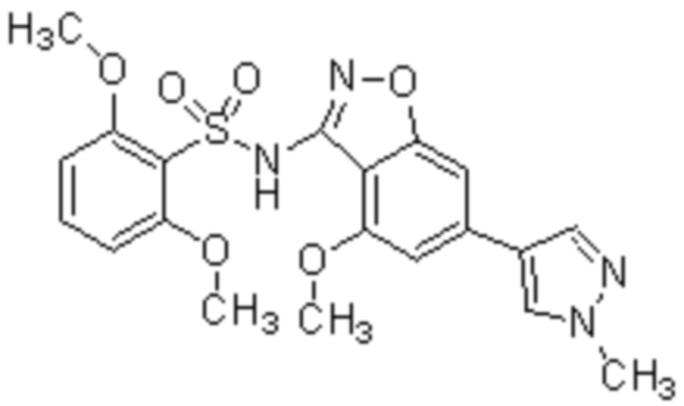
Схема M:
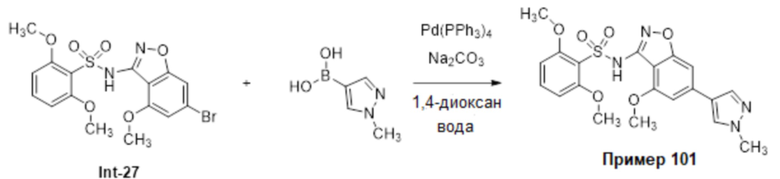
К раствору N-(6-бром-4-метоксибензо[d]изоксазол-3-ил)-2,6-диметоксибензолсульфонамида (Int-27) (40 мг, 0,09 ммоль) в 1,4-диоксане (8 мл) и воде (2 мл) добавляли (1-метил-1H-пиразол-4-ил)бороновую кислоту (80 мг, 0,631 ммоль), Na2CO3 (100 мг, 0,948 ммоль) и Pd(PPh3)4 (37 мг, 0,032 ммоль) и смесь нагревали при температуре кипения в атмосфере N2 в течение ночи. Смесь доводили до pH 4-5 с помощью 1 M водной HCl, разбавляли EtOAc и промывали водой, насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью преп. ТСХ (DCM/MeOH=50/1) с получением указанного в заголовке соединения (22 мг, 55%) в виде твердого вещества белого цвета. m/z 445,0 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,52 (с, 1H), 8,35 (с, 1H), 8,05 (с, 1H), 7,50 (т, J=8,5 Гц, 1H), 7,38 (с, 1H), 7,05 (с, 1H), 6,78 (д, J=8,5 Гц, 2H), 3,98 (с, 3H), 3,87 (с, 3H), 3,78 (с, 6H).
Пример 102: Получение 2,6-диметокси-N-(5-метил-7-(1-метил-1H-пиразол-4-ил)бензо[d]изоксазол-3-ил)бензолсульфонамида в соответствии со схемой N.
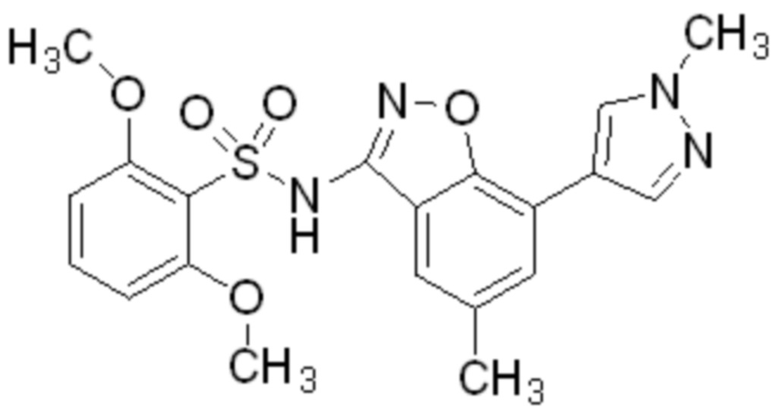
Схема N:
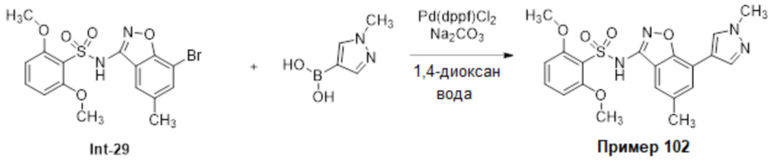
К раствору N-(7-бром-5-метилбензо[d]изоксазол-3-ил)-2,6-диметоксибензолсульфонамида (Int-29)(50 мг, 0,117 ммоль) в 1,4-диоксане (8 мл) и воде (2 мл) добавляли (1-метил-1H-пиразол-4-ил)бороновую кислоту (22 мг, 0,176 ммоль), Na2CO3 (50 мг, 0,468 ммоль) и Pd(dppf)Cl2 (9 мг, 0,012 ммоль) и смесь нагревали при температуре кипения в атмосфере N2 в течение ночи. Смесь доводили до pH 5 с помощью 1 M водной HCl, разбавляли водой и экстрагировали EtOAc (30 мл×3). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью преп. ТСХ (петролейный эфир/EtOAc=3/1) с получением указанного в заголовке соединения (24 мг, 48%) в виде твердого вещества белого цвета. m/z 429,0 [M+H]+. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,24 (с, 1H), 7,99 (с, 1H), 7,55 (с, 1H), 7,49-7,22 (м, 2H), 6,67 (д, J=8,6 Гц, 2H), 3,88 (с, 3H), 3,69 (с, 6H), 2,39 (с, 3H),38 (с, 1H), 7,05 (с, 1H), 6,78 (д, J=8,5 Гц, 2H), 3,98 (с, 3H), 3,87 (с, 3H), 3,78 (с, 6H).
Пример 103: Получение 3-гидрокси-2,6-диметокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида в соответствии со схемой O.
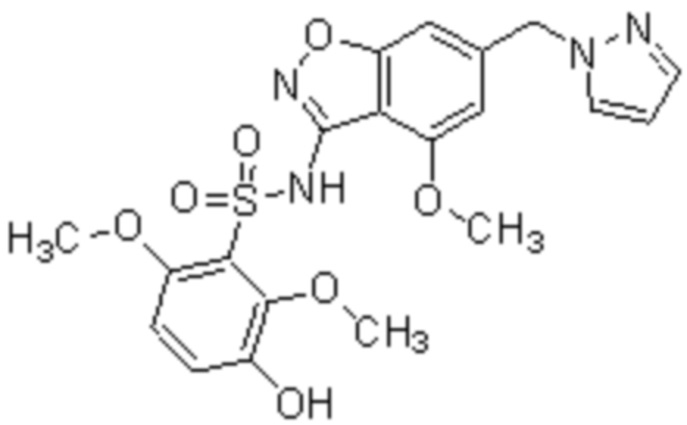
Пример 104: Получение 2-гидрокси-6-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида в соответствии со схемой O.
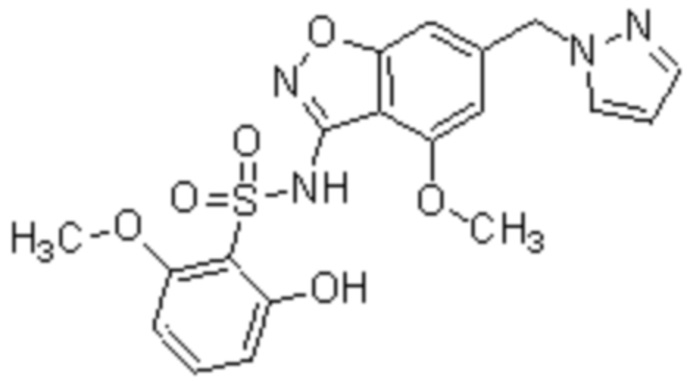
Пример 105: Получение N-{6-[(4-гидрокси-1H-пиразол-1-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида в соответствии со схемой O.
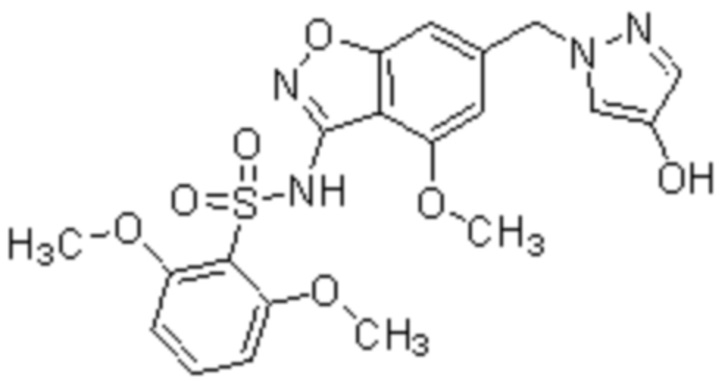
Схема O:
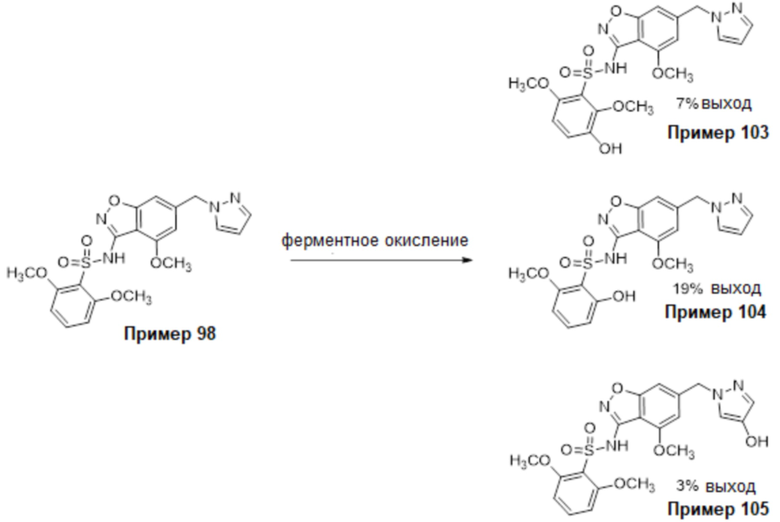
В колбу Эрленмейера на 500 мл добавляли H2O степени чистоты для ВЭЖХ (27,2 мл), водный калий-фосфатный буфер (1,0 M, 4,0 мл, pH 7,5), водный MgCl2 (165 мм, 0,8 мл), микросомы печени самцов крыс, индуцированные дексаметазоном (4,0 мл, 20 мг/мл, Xenotech) и раствор 2,6-диметокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (получен как в примере 98, 5 мм в MeCN, 0,20 мл, 1,0 мкмоль). Инкубацию начинали с добавления свежеприготовленного водного раствора NADPH (4,0 мл, 13 мм). Колбу Эрленмейера без крышки встряхивали на водяной бане, поддерживаемой при температуре 37ºC, в течение 1,5 часов. Инкубационную смесь гасили добавлением MeCN (40 мл) с последующим центрифугированием при ~1700 g в течение 5 мин. Супернатант частично упаривали на вакуумной центрифуге. Оставшийся раствор обрабатывали MeCN (0,5 мл), чистой муравьиной кислотой (0,5 мл) и деионизированной H2O до получения конечного объема ~ 50 мл. Раствор центрифугировали при ~ 40 000 g в течение 30 мин. Супернатант адсорбировали на колонке для ВЭЖХ Zorbax Polaris C18-A (250×4,6 мм, размер частиц 5 мкм) с использованием насоса для ВЭЖХ JASCO PU-1580 при скорости потока 0,8 мл/мин в течение ~60 мин. Колонку для ВЭЖХ переносили в масс-спектрометр Thermo LTQ Velos вместе с прибором Waters Acquity UВЭЖХ, состоящим из четвертичного насоса, автосэмплера и УФ/видимого детектора на матрице фотодиодов. Градиент MeCN/H2O (+0,1%-ная муравьиная кислота) применяли для разделения представляющих интерес продуктов. После прохождения через детектор PDA, элюент разделялся в соотношении приблизительно 15:1, причем большая часть направлялась в коллектор фракций, а меньшая часть в масс-спектрометр. Фракции собирали каждые 20 с, а те, которые содержали интересующие пики, анализировали с помощью UВЭЖХ-UV-HRMS с использованием масс-спектрометра с ионной ловушкой высокого разрешения Thermo Orbitrap Elite в сочетании с UВЭЖХ Thermo Accela и диодно-матричный УФ/видимый детектор с автоинжектором CTC Analytics Leap (Thermo-Fisher). Образцы вводили (10 мкл) в колонку Phenomenex Kinetex C18 UВЭЖХ (50×2,1 мм, размер частиц 1,7 мкм), выдерживаемая при 45ºC, которую элюировали градиентом MeCN/H2O (+0,1%-ная муравьиная кислота) со скоростью потока 0,4 мл/мин. После анализа UВЭЖХ-UV-HRMS фракции объединяли, и растворитель удаляли центрифугированием в вакууме. Высушенные образцы анализировали с помощью ЯМР-спектроскопии и количественно определяли с помощью внешней калибровки по спектру 1H ЯМР стандартного раствора 5,0 мМ бензойной кислоты в ДМСО-d6 с использованием функции ERETIC2 в Topspin V3.2. N-{6-[(4-гидрокси-1H-пиразол-1-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамид (пример 105) (0,028 мкмоль, 3%-ный выход) получали как пик первого элюирования. 1H ЯМР (600 МГц, ДМСО-d6) δ 8,45 (с, 1H), 7,38 (м, 1H), 7,32 (с, 1H), 7,06 (с, 1H), 6,70 (м, 3H), 6,63 (с, 1H), 5,22 (с, 2H), 3,86 (с, 3H), 3,69 (с, 6H). HRMS (ESI-TOF) рассчитано для (C20H21N4O7S)[M+H]+ m/z=461,1125, найдено 461,1121 (-0,45 м.д.). 3-Гидрокси-2,6-диметокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид (пример 103) (0,072 мкмоль, 7%-ный выход) получали как второй пик элюирования. 1H ЯМР (600 МГц, ДМСО-d6) δ 9,33 (с, 1H), 7,88 (д, J=2,4 Гц, 1H), 7,50 (с, 1H), 7,01 (д, J=9,0 Гц, 1H), 6,80 (с, 1H), 6,76-6,68 (м, 2H), 6,31 (т, J=2,3 Гц, 1H), 5,44 (с, 2H), 3,87 (с, 3H), 3,76 (с, 3H), 3,65 (с, 3H). HRMS (ESI-TOF) рассчитано для (C20H21N4O7S)[M+H]+ m/z=461,1125, найдено 461,1123 (-0,25 м.д.). 2-Гидрокси-6-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид (пример 104) (0,19 мкмоль, 19%-ный выход) получали как третий пик элюирования. 1H ЯМР (600 МГц, ДМСО-d6) δ 7,86 (с, 1H), 7,50 (с, 1H), 7,13 (м, 1H), 6,63 (с, 1H), 6,53 (м, 2H), 6,40 (д, J=8,1 Гц, 2H), 6,30 (с, 1H), 5,41 (с, 2H), 3,83 (с, 3H), 3,72 (с, 3H). HRMS (ESI-TOF) рассчитано для (C19H19N4O6S) [M+H]+ 431,1020, найдено 431,1017 (-0,28 м.д.).
Пример 106: Получение N-{6-[гидрокси(пиридин-2-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамида в соответствии со схемой P.
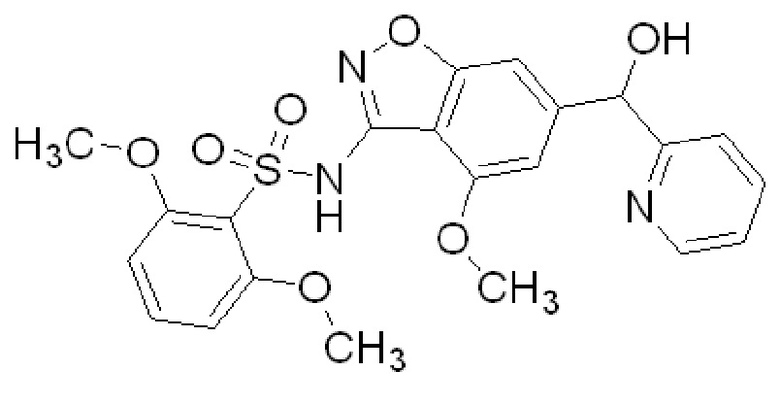
Пример 107: Получение 2,6-диметокси-N-[4-метокси-6-(пиридин-2-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамида в соответствии со схемой P.
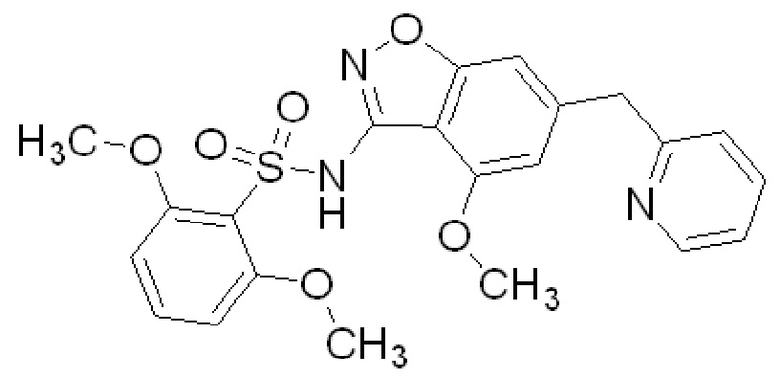
Схема P:
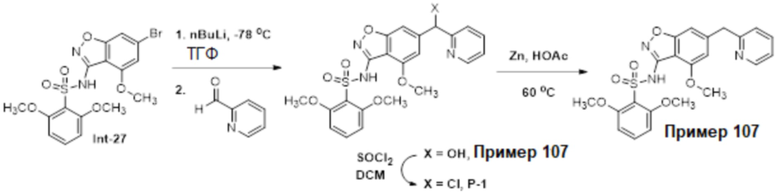
Стадия 1: Синтез N-{6-[гидрокси(пиридин-2-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамида (пример 106):
Раствор N-(6-бром-4-метоксибензо[d]изоксазол-3-ил)-2,6-диметоксибензолсульфонамида (Int-27) (628 мг, 1,42 ммоль) в безводном ТГФ (20 мл) охлаждали до температуры -78ºC. Добавляли по каплям н-BuLi (1,20 мл 2,5 в гексане, 3,00 ммоль). Образовавшуюся взвесь перемешивали при температуре -78ºC в течение 1 ч. Затем добавляли по каплям раствор пиридин-2-альдегида (187 мг, 1,75 ммоль) в безводном ТГФ (2 мл). Полученную реакционную смесь перемешивали при температуре -78ºC в течение 1 ч. Добавляли дополнительное количество пиридин-2-альдегида (75 мг, 0,71 ммоль) в 1 мл безводном ТГФ. Полученную реакционную смесь нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 18 ч. Реакцию гасили HOAc (0,5 мл). Погашенную реакционную смесь распределяли между EtOAc (50 мл) и воде (50 мл). Органическую фазу разделяли, промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и очищали флэш-хроматографией, элюируя градиентом 0-100% EtOAc в гептане, затем 0-20% 2-PrOH в EtOAc с получением N-{6-[гидрокси(пиридин-2-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамида (пример 106) в виде твердого вещества (247 мг, 37%). 1H ЯМР (400 МГц, CDCl3) δ 8,61 (д, J=4,9 Гц, 1H), 8,23 (с, 1H), 7,82 (т, J=7,7 Гц, 1H), 7,42-7,33 (м, 3H), 7,10 (с, 1H), 6,85 (с, 1H), 6,58 (д, J=8,4 Гц, 2H), 5,99 (с, 1H), 4,01 (с, 3H), 3,88 (с, 6H). m/z 472,2 [M+H].
Стадия 2: Синтез N-(6-(хлор(пиридин-2-ил)метил)-4-метоксибензо[d]изоксазол-3-ил)-2,6-диметоксибензолсульфонамида (P-1):
К раствору N-{6-[гидрокси(пиридин-2-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамида (пример 106) (113 мг, 0,240 ммоль) в безводном DCM (5 мл) добавляли SOCl2 (0,20 мл, 2,7 ммоль). Полученную реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Растворитель удаляли, и полученный остаток распределяли между EtOAc (50 мл) и насыщ. водный NaHCO3 (50 мл). Органическую фазу разделяли, сушили над сульфатом натрия и концентрировали с получением N-(6-(хлор(пиридин-2-ил)метил)-4-метоксибензо[d]изоксазол-3-ил)-2,6-диметоксибензолсульфонамида (P-1) (78 мг, 66%-ный выход), который использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 8,57 (шир. д, J=4,2 Гц, 1H), 8,25 (с, 1H), 7,75 (дт, J=1,7, 7,8 Гц, 1H), 7,57 (д, J=7,8 Гц, 1H), 7,37 (т, J=8,5 Гц, 1H), 7,27-7,22 (м, 1H), 7,10 (с, 1H), 6,82 (с, 1H), 6,57 (д, J=8,6 Гц, 2H), 6,17 (с, 1H), 4,00 (с, 3H), 3,87 (с, 6H), отсутствие сульфонамидного NH; m/z 490,1 [M+H]+.
Стадия 3: Синтез 2,6-диметокси-N-[4-метокси-6-(пиридин-2-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамида (пример 107):
К раствору N-(6-(хлор(пиридин-2-ил)метил)-4-метоксибензо[d]изоксазол-3-ил)-2,6-диметоксибензолсульфонамида (P-1) (75 мг, 0,153 ммоль) в HOAc (5 мл) добавляли порошок цинка (69 мг, 1,1 ммоль) и раствор нагревали до температуры 60ºC. После 3 ч при температуре 60ºC реакция завершалась. Реакционную смесь охлаждали до комнатной температуры и осторожно нейтрализовали насыщенным раствором NaHCO3. Органические продукты экстрагировали EtOAc (2×50 мл) и объединенный органический экстракт сушили над сульфатом натрия, концентрировали досуха и очищали с помощью флэш-хроматографии, элюируя градиентом 0-100% EtOAc в гептане затем 0-20% 2-PrOH в EtOAc. Концентрирование чистых фракций дает 2,6-диметокси-N-[4-метокси-6-(пиридин-2-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамид (пример 107) (38 мг, 55% за две стадии) в виде твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,57 (шир. с, 1H), 8,23 (с, 1H), 7,65 (т, J=7,6 Гц, 1H), 7,37 (т, J=8,5 Гц, 1H), 7,19 (шир. д, J=6,6 Гц, 2H), 6,89 (с, 1H), 6,64-6,53 (м, 3H), 4,23 (с, 2H), 3,98 (с, 3H), 3,87 (с, 6H). m/z 456,3 [M+H].
Пример 108: Получение N-{6-[(S*)-гидрокси(1,3-оксазол-2-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамида в соответствии со схемой Q.
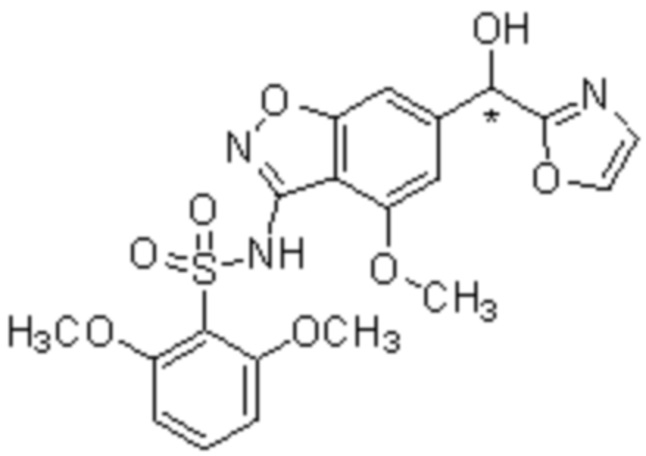
Пример 109: Получение N-{6-[(R*)-гидрокси(1,3-оксазол-2-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамида в соответствии со схемой Q.
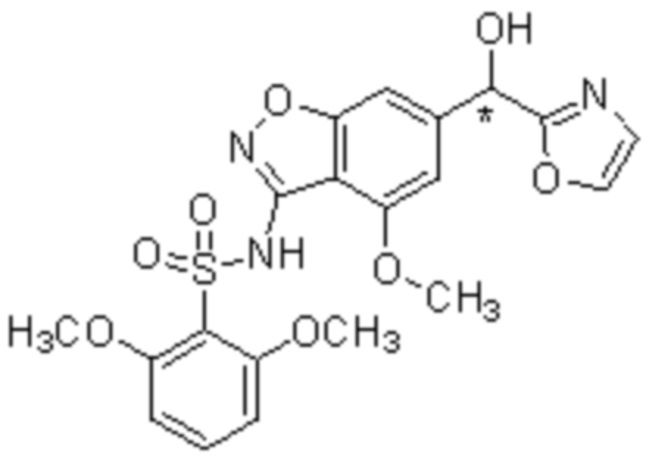
Пример 110: Получение 2,6-диметокси-N-[4-метокси-6-(1,3-оксазол-2-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамида в соответствии со схемой Q.
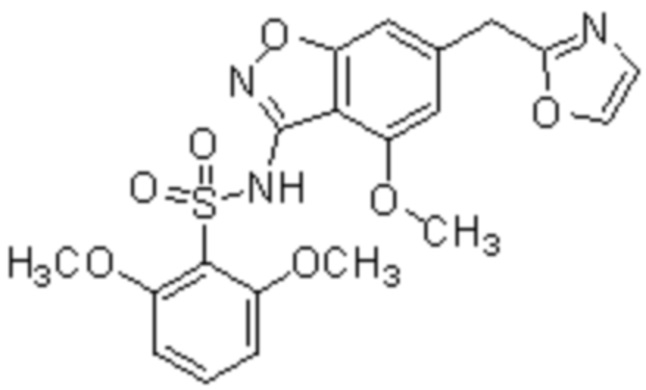
Схема Q:
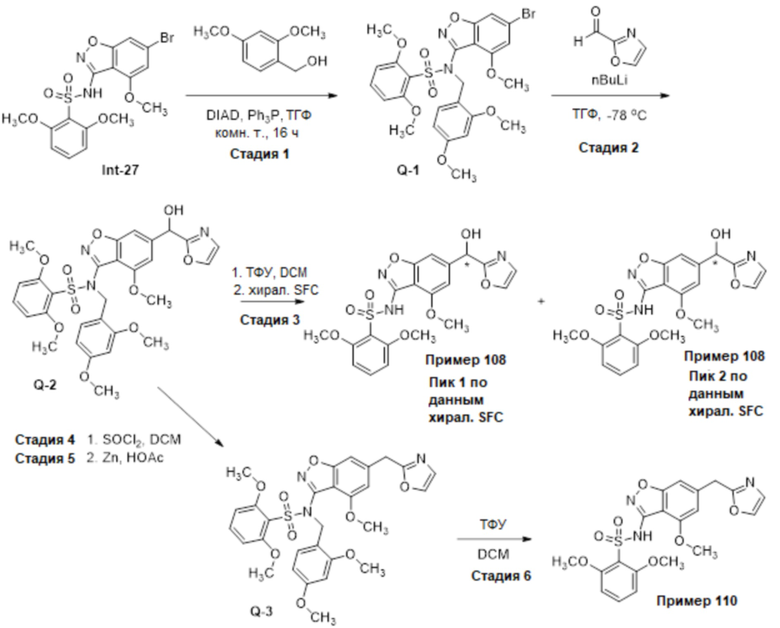
Стадия 1: Синтез N-(6-бром-4-метокси-1,2-бензоксазол-3-ил)-N-[(2,4-диметоксифенил)метил]-2,6-диметоксибензол-1-сульфонамида (Q-1)
К раствору N-(6-бром-4-метоксибензо[d]изоксазол-3-ил)-2,6-диметоксибензолсульфонамида (Int-27) (15 г, 34 ммоль) в ТГФ (300 мл) и 2,4-диметоксибензилового спирта (8,54 г, 50,8 ммоль), PPh3 (22,2 г, 84,6 ммоль) по каплям при температуре 0ºC добавляли DIAD (13,7 г, 67,7 ммоль). Реакционный раствор нагревали до комнатной температуры и оставляли перемешиваться в течение 16 ч. Реакционную смесь разбавляли EtOAc (300 мл), промывали водой (150 мл), насыщенным солевым раствором, насыщенным водн. раствором гидрокарбоната натрия и опять насыщенным солевым раствором. Органическую фазу сушили над сульфатом натрия и фильтровали. Растворитель удаляли при пониженном давлении с получением смеси, которую очищали с помощью флеш хроматографии, элюируя 60%-70% EtOAc в петролейном эфире с получением сырого продукта с небольшим количеством оставшегося трифенилфосфин оксида. Сырой твердый продукт перекристаллизовывали из MeOH с получением N-(6-бром-4-метокси-1,2-бензоксазол-3-ил)-N-[(2,4-диметоксифенил)метил]-2,6-диметоксибензол-1-сульфонамида (Q-1) (8,0 г, 40%-ный выход) в виде твердого вещества белого цвета.
Стадия 2: Синтез rac-N-[(2,4-диметоксифенил)метил]-N-{6-[гидрокси(1,3-оксазол-2-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида (Q-2)
К раствору N-(6-бром-4-метокси-1,2-бензоксазол-3-ил)-N-[(2,4-диметоксифенил)метил]-2,6-диметоксибензол-1-сульфонамида (Q-1) (500 мг, 0,843 ммоль) в ТГФ (9,5 мл) по каплям при температуре −78ºC в атмосфере аргона добавляли н-BuLi (0,506 мл 2,5 M раствора в гексане, 1,26 ммоль). Через 30 мин при температуре −78ºC добавляли оксазол-2-карбальдегид (123 мг, 1,26 ммоль) в виде раствора в ТГФ (0,5 мл). Реакционной смеси давали медленно нагреться до комнатной температуры и перемешивали 16 ч. Реакционную смесь выливали в насыщенный водн. раствор NH4Cl (20 мл). Водный слой экстрагировали двумя частями EtOAc (2×20 мл). Объединенные экстракт промывали насыщенным солевым раствором (20 мл), сушили над MgSO4 и концентрировали в вакууме. Остаток очищали с помощью флеш хроматографии, элюируя 100% EtOAc с получением rac-N-[(2,4-диметоксифенил)метил]-N-{6-[гидрокси(1,3-оксазол-2-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида (Q-2) (150 мг, 29%-ный выход) в виде смолы желтого цвета.
Стадия 3: Синтез N-{6-[(S*)-гидрокси(1,3-оксазол-2-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамида (пример 108) и N-{6-[(R*)-гидрокси(1,3-оксазол-2-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамида (пример 109)
Раствор rac-N-[(2,4-диметоксифенил)метил]-N-{6-[гидрокси(1,3-оксазол-2-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида (Q-2) (150 мг, 0,245 ммоль) в ТФУ (5 мл) перемешивали при комнатной температуре в течение 2 ч. Наблюдался раствор розового цвета, и реакционную смесь концентрировали в вакууме. Остаток предварительно очищали с помощью флеш хроматографии, элюируя EtOAc/MeOH 10:1 с получением рацемической смеси соединений по примерам 108 и 109, которые были подвергнуты хиральной очистке SFC. Соединения отделяли друг от друга с использованием колонки Chiralpak AS-3 100×4,6 мм внутр. диам., 3 мкм с подвижной фазой, состоящей из CO2 (A) и этанола с 0,05% DEA (B). Градиентное элюирование от 5% до 40% B в течение 4 мин и поддержание 40% B в течение 2,5 мин, затем 5% B в течение 1,5 мин. После разделения с помощью хиральной SFC получали 20 мг каждого продукта. Пик 1=соединение по примеру 108 1H ЯМР (400МГц, ДМСО-d6) δ 8,06 (д, J=0,6 Гц, 1H), 7,45 (шир. т, J=8,4 Гц, 1H), 7,18 (д, J=0,6 Гц, 1H), 7,15 (с, 1H), 6,86 (с, 1H), 6,74 (д, J=8,4 Гц, 2H), 6,67 (д, J=5,1 Гц, 1H), 5,93 (д, J=5,3 Гц, 1H), 3,88 (с, 3H), 3,74 (с, 6H), отсутствует пик сульфонамида NH; m/z 462,0 (M+H)+. Пик 2=соединение по примеру 109 1H ЯМР (400МГц, ДМСО-d6) δ 8,06 (д, J=0,6 Гц, 1H), 7,50 (т, J=8,5 Гц, 1H), 7,13-7,26 (м, 2H), 6,90 (с, 1H), 6,78 (д, J=8,4 Гц, 2H), 6,70 (д, J=5,4 Гц, 1H), 5,95 (д, J=5,3 Гц, 1H), 3,89 (с, 3H), 3,78 (с, 6H), отсутствует пик сульфонамида NH; m/z 462,0 (M+H)+.
Стадии 4 и 5: Синтез N-[(2,4-диметоксифенил)метил]-2,6-диметокси-N-{4-метокси-6-[(1,3-оксазол-2-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (Q-3)
К раствору rac-N-[(2,4-диметоксифенил)метил]-N-{6-[гидрокси(1,3-оксазол-2-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида (Q-2) (150 мг, 0,245 ммоль) в DCM (5 мл) при комнатной температуре добавляли тионил хлорид (290 мг, 2,44 ммоль). Раствор перемешивали в течение 1 ч, при этом образовывался раствор бледно-желтого цвета. По данным ТСХ реакция завершалась и смесь гасили водой (20 мл) и экстрагировали DCM (20 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением вторичного хлорида (150 мг, желтое масло), которое использовали на следующей стадии без дополнительной очистки. К раствору вторичного хлорида (150 мг, 0,238 ммоль) в HOAc (5 мл) ) при комнатной температуре добавляли порошок цинка (467 мг, 7,14 ммоль. Реакционной смеси давали перемешиваться при комнатной температуре в течение 1 ч. По данным ТСХ реакция завершалась и смесь разбавляли EtOAc (50 мл) и фильтровали. Фильтрат доводили до pH 7-8 с использованием насыщ. водн. Na2CO3. Органический слой сушили над Na2SO4 и концентрировали с получением остатка, который очищали с помощью флеш хроматографии с получением N-[(2,4-диметоксифенил)метил]-2,6-диметокси-N-{4-метокси-6-[(1,3-оксазол-2-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (Q-3) (80 мг) в виде желтого масла.
Стадия 6: Синтез 2,6-диметокси-N-[4-метокси-6-(1,3-оксазол-2-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамида (пример 110)
Раствор N-[(2,4-диметоксифенил)метил]-2,6-диметокси-N-{4-метокси-6-[(1,3-оксазол-2-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (Q-3) (80 мг, 0,13 ммоль) в ТФУ (5 мл) перемешивали в течение 1 ч при комнатной температуре. Реакционную смесь концентрировали и предварительно очищали с помощью флеш хроматографии, элюируя EtOAc/MeOH 10:1. Полученный сырой продукт затем очищали с помощью преп. ВЭЖХ и чистые фракции замораживали и лиофилизовали с получением 10 мг продукта, все еще загрязненного примесью, как определялось данными 1H ЯМР. Этот образец затем очищали посредством преп. ТСХ с получением 2,6-диметокси-N-[4-метокси-6-(1,3-оксазол-2-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамида (пример 110) (5 мг, 8,4%-ный выход) в виде твердого вещества белого цвета. m/z 446,0 (M+H)+; 1H ЯМР (400МГц, МЕТАНОЛ-d4) δ 7,87 (с, 1H), 7,43 (т, J=8,5 Гц, 1H), 7,14 (с, 1H), 6,92 (с, 1H), 6,72 (д, J=8,5 Гц, 3H), 4,34-4,11 (м, 2H), 4,00 (с, 3H), 3,81 (с, 7H).
Пример 111: Получение 2,6-диметокси-N-[4-метокси-6-(пиразин-2-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамида в соответствии со схемой R.
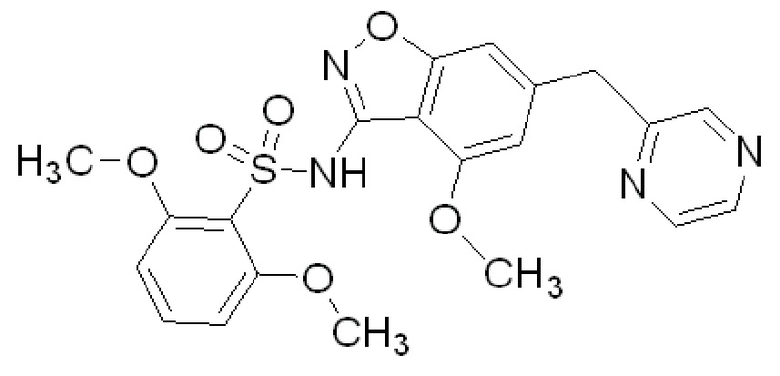
Схема R:
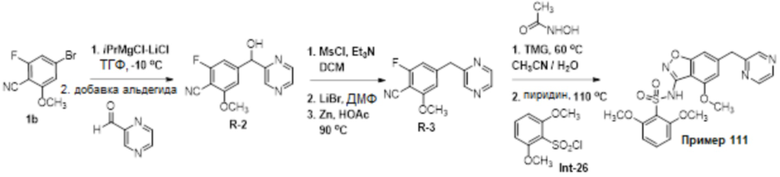
Стадия 1: Синтез 2-фтор-4-[гидрокси(пиразин-2-ил)метил]-6-метоксибензонитрила (R-2).
Трехгорлую круглодонную колбу на 250 мл оборудовали термометром, мешалкой и впускным отверстием для азота. В колбу помещали 4-бром-2-фтор-6-метоксибензонитрил (1b) (4,00 г, 17,4 ммоль) и 100 мл безводного ТГФ. Колбу закрывали пробкой с перегородкой, продували атмосферой азота и охлаждали до температуры -20ºC (баня лед/MeOH). Добавляли по каплям iPrMgCl-LiCl (17,5 мл 1,3 M, 22,8 ммоль), поддерживая при этом температуру ниже -15ºC. Полученную смесь перемешивали при температуре -20ºC в течение 1 ч. Затем добавляли пиразин-2-карбоксальдегид (2,95 г, 1,57 ммоль) в виде раствора в 20 мл безводного ТГФ, поддерживая при этом температуру ниже -10ºC. Полученную реакционную смесь перемешивали при температуре -10ºC в течение 1 ч и затем гасили 4 н HCl (10 мл). Погашенную реакционную смесь распределяли между EtOAc (200 мл) и водой (200 мл). Органическую фазу разделяли, и водную фазу опять экстрагировали EtOAc (1× 100 мл). Объединенные органические фазы сушили над Na2SO4, концентрировали досуха и очищали посредством флеш хроматографии с использованием градиента 20-100% EtOAc в гептане с получением 2-фтор-4-[гидрокси(пиразин-2-ил)метил]-6-метоксибензонитрила (R-2) (2,6 г, 58%-ный выход) в виде смолы. m/z 260,0 (M+H)+; 1H ЯМР (400 МГц, CDCl3) δ 8,67 (д, J=0,7 Гц, 1H), 8,56-8,50 (м, 2H), 6,93 (с, 1H), 6,87 (д, J=9,2 Гц, 1H), 5,88 (с, 1H), 4,57 (шир. с, 1H), 3,94 (с, 3H).
Стадии 2-4: Синтез 2-фтор-6-метокси-4-[(пиразин-2-ил)метил]бензонитрила (R-3).
К раствору 2-фтор-4-[гидрокси(пиразин-2-ил)метил]-6-метоксибензонитрила (R-2) (145 мг, 0,559 ммоль) и Et3N (0,120 мл, 0,861 ммоль) в безводном ТГФ (10 мл) при температуре 0ºC добавляли метансульфонил хлоридом (0,050 мл, 0,64 ммоль). Полученную смесь нагревали до комнатной температуры и перемешивали в течение 30 мин. Реакционную смесь разбавляли DCM (30 мл), промывали водой (1×30 мл) и насыщ. водн. NaHCO3 (1×30 мл). Экстракт сушили над Na2SO4 и концентрировали досуха. Продукт использовали на следующей стадии без дополнительной очистки. Смесь сырого мезилата (173 мг, 0,513 ммоль) и LiBr (137 мг, 1,58 ммоль) в безводном ДМФ (4 мл) перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь распределяли между EtOAc (50 мл) и воде (50 мл). Органическую фазу разделяли, промывали водой (1×50 мл) и насыщенным солевым раствором (1×50 мл), сушили над Na2SO4 и концентрировали. Осуществляли очистку посредством флеш хроматографии с использованием градиента 0-100% EtOAc в гептане с получением 72 мг, (44%) вторичного бромида. Вторичный бромид (43 мг, 0,13 ммоль) и порошок цинка (90 мг, 1,4 ммоль) перемешивали при температуре 80ºC в HOAc (2 мл) в течение 8 ч. После охлаждения до комнатной температуры реакционную смесь разбавляли EtOAc (50 мл) и осторожно промывали насыщ. водн. NaHCO3 (50 мл). Органический слой затем промывали насыщенным солевым раствором (1×50 мл) и сушили над Na2SO4. После концентрирования досуха сырой продукт очищали посредством флеш хроматографии, элюируя с градиентом 0-100% EtOAc в гептане с получением 10 мг (31%) 2-фтор-6-метокси-4-[(пиразин-2-ил)метил]бензонитрила (R-3) (10 мг, 31%-ный выход). 1H ЯМР (400 МГц, CDCl3) δ 8,56 (шир. с, 3H), 6,74-6,69 (м, 2H), 4,18 (с, 2H), 3,94 (с, 3H); m/z 244,0 (M+H)+.
Стадии 5-6: Синтез 2,6-диметокси-N-[4-метокси-6-(пиразин-2-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамида (пример 111).
К смеси 2-фтор-6-метокси-4-[(пиразин-2-ил)метил]бензонитрила (R-3) (178 мг, 0,732 ммоль) и N-гидроксиацетамида (165 мг, 2,20 ммоль) в CH3CN (5 мл) и воде (0,5 мл) добавляли 1,1,3,3-тетраметилгуанидин (0,55 мл, 4,4 ммоль). Полученную реакционную смесь перемешивали при температуре 60ºC в течение 16 ч и затем охлаждали до комнатной температуры. Растворитель удаляли, и полученный остаток распределяли между EtOAc и водой. Органическую фазу разделяли, и водную фазу экстрагировали второй раз EtOAc (50 мл). Объединенный органический экстракт сушили над Na2SO4, концентрировали досуха и очищали с помощью флэш-хроматографии, элюируя градиентом 40-100% EtOAc в гептане. Это давало 82 мг (44%) аминового промежуточное соединение в виде масла. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,68 (д, J=1,1 Гц, 1H), 8,60-8,55 (м, 1H), 8,51 (д, J=2,6 Гц, 1H), 6,91 (с, 1H), 6,69 (с, 1H), 5,87 (шир. с, 2H), 4,22 (с, 2H), 3,88 (с, 3H); m/z 257,1 (M+H)+. Амин (73 мг, 0,28 ммоль), полученный реакцией выше, обрабатывали 2,6-диметоксибензол-1-сульфонил хлоридом (Int-26) (100 мг, 0,43 ммоль) и пиридином (2 мл). Реакционную смесь перемешивали при температуре 110ºC в течение 3 ч. Затем добавляли дополнительное количество 2,6-диметоксибензол-1-сульфонил хлорида (Int-26) (50 мг, 0,21 ммоль) и продолжали нагревание при температуре 110ºC в течение еще 1 ч. После охлаждения до комнатной температуры реакционную смесь распределяли между этилацетатом (20 мл) и 2н HCl (20 мл). Органическую фазу разделяли, сушили над Na2SO4 и очищали с помощью флэш-хроматографии, элюируя градиентом 20-100% EtOAc в гептане, затем вторым градиентом 0-20% 2-PrOH в EtOAc с получением 2,6-диметокси-N-[4-метокси-6-(пиразин-2-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамида (пример 111) (44 мг, 34%-ный выход) в виде твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 9,50 (с, 1H), 8,68 (д, J=1,0 Гц, 1H), 8,58-8,53 (м, 1H), 8,50 (д, J=2,4 Гц, 1H), 7,49 (т, J=8,5 Гц, 1H), 7,07 (с, 1H), 6,83 (с, 1H), 6,77 (д, J=8,6 Гц, 2H), 4,25 (с, 2H), 3,89 (с, 3H), 3,77 (с, 6H); m/z 456,8 (M+H)+.
Соединения по примерам, приведенным в таблице ниже, синтезировали в соответствии со способами, используемыми для синтеза 2,6-диметокси-N-[4-метокси-6-(пиразин-2-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамида (пример 111). Следующие соединения по примерам синтезировали с некритическими изменениями или заменами в приведенных в качестве примеров способах, которые мог бы осуществить специалист в данной области. При необходимости разделение смесей региоизомеров проводили стандартными методами, известными в данной области, такими как SFC или ВЭЖХ, и проводили на любой подходящей стадии в последовательности синтеза.
Таблица 19:
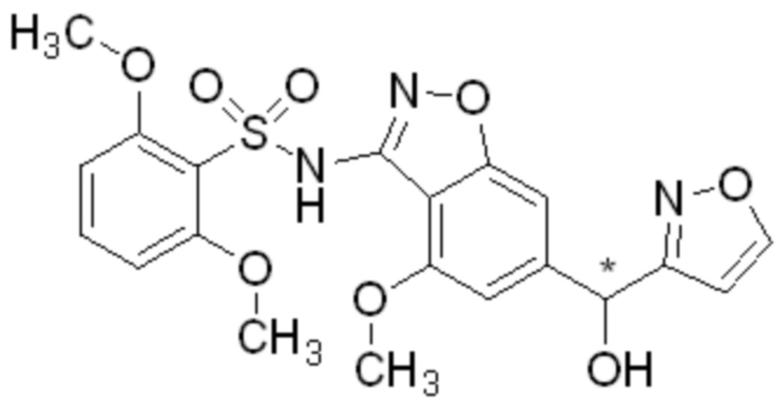
N-{6-[(S*)-гидрокси(1,2-оксазол-3-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамид, изомер-A
1-ый пик на колонке Chiralpak AS-3 150×4,6 мм внутр. диам., 3 мкм.
Подвижная фаза A: CO2 B:изопропанол (0,05% DEA); схема Q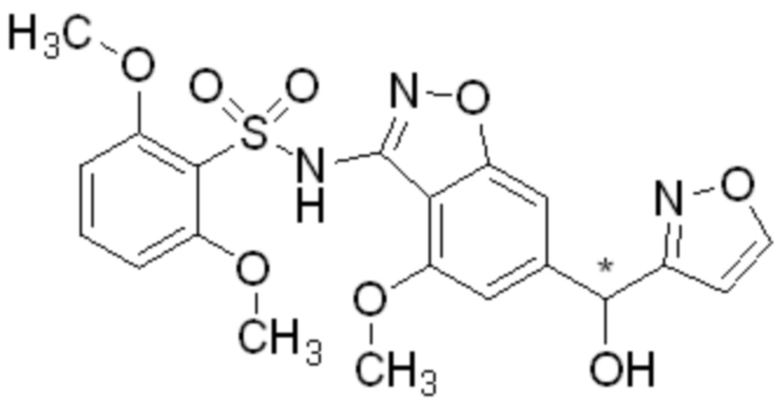
N-{6-[(R*)-гидрокси(1,2-оксазол-3-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамид, изомер-B
2-ой пик на колонке Chiralpak AS-3 150×4,6 мм внутр. диам., 3 мкм.
Подвижная фаза A: CO2 B:изопропанол (0,05% DEA); схема Q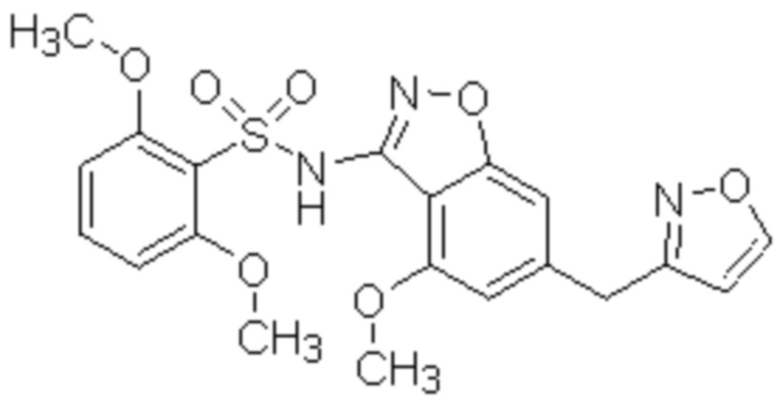
N-(6-(изоксазол-3-илметил)-4-метоксибензо[d]изоксазол-3-ил)-2,6-диметоксибензолсульфонамид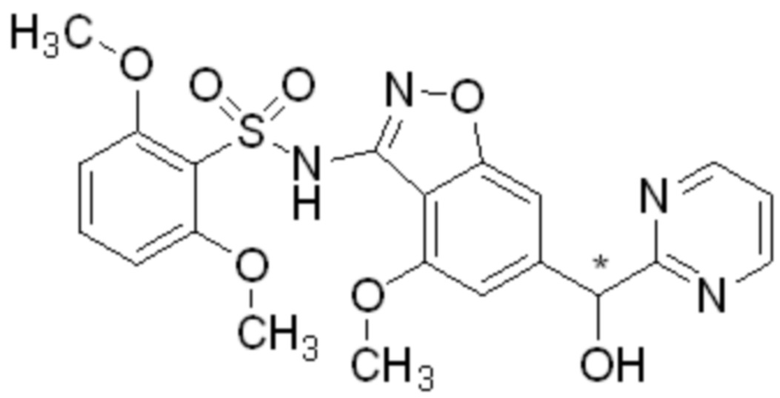
N-{6-[(S*)-гидрокси(пиримидин-2-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамид
Подвижная фаза: A: CO2 B: 2-PrOH (0,1% этаноламин);
схема Q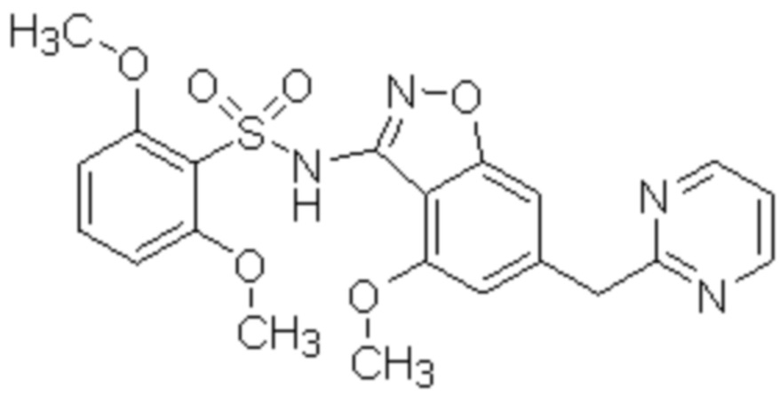
2,6-диметокси-N-[4-метокси-6-(пиримидин-2-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамид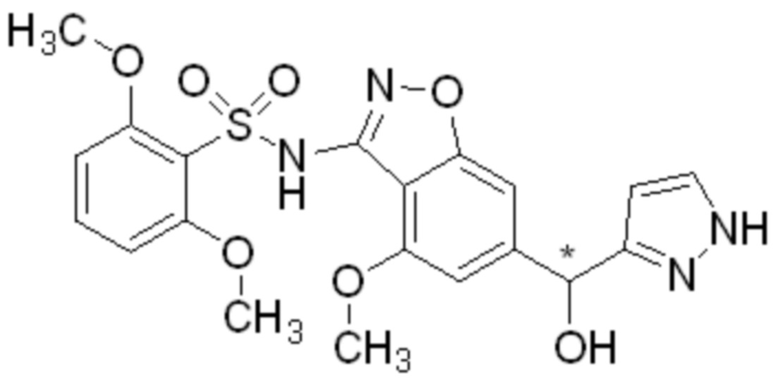
N-{6-[(S*)-гидрокси(1H-пиразол-3-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамид, изомер A
Подвижная фаза: A: CO2 B:изопропанол (0,05% DEA). Схема Q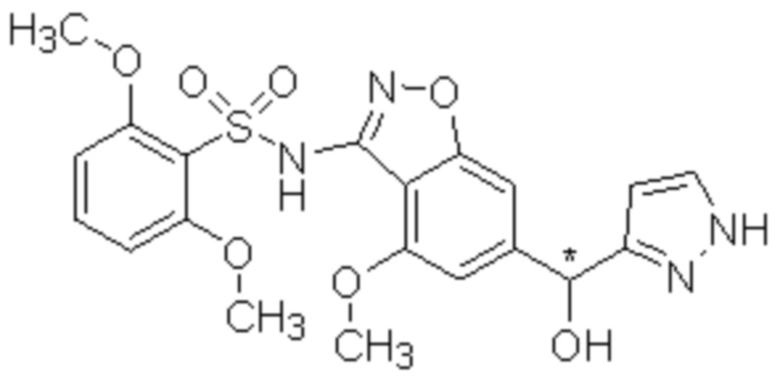
N-{6-[(R*)-гидрокси(1H-пиразол-3-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамид, изомер B
Подвижная фаза: A: CO2 B:изопропанол (0,05% DEA). Схема Q
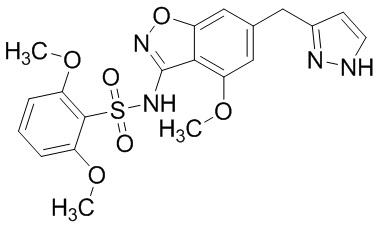
2,6-диметокси-N-[4-метокси-6-(1H-пиразол-3-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамид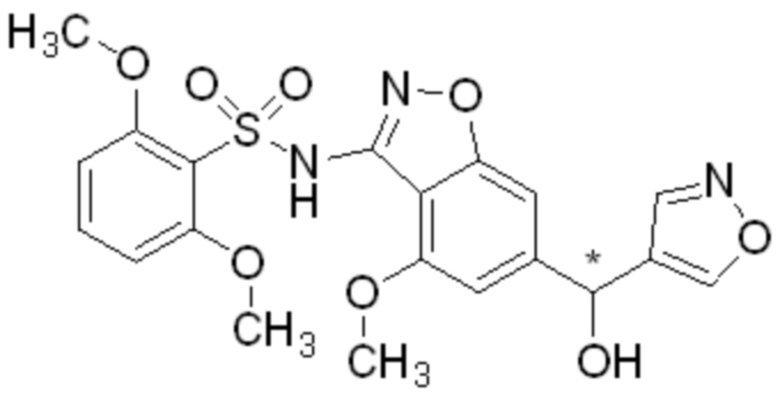
N-{6-[(S*)-гидрокси(1,2-оксазол-4-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамид, изомер A
Подвижная фаза: A: CO2 B: этанол (0,05% DEA).
Схема Q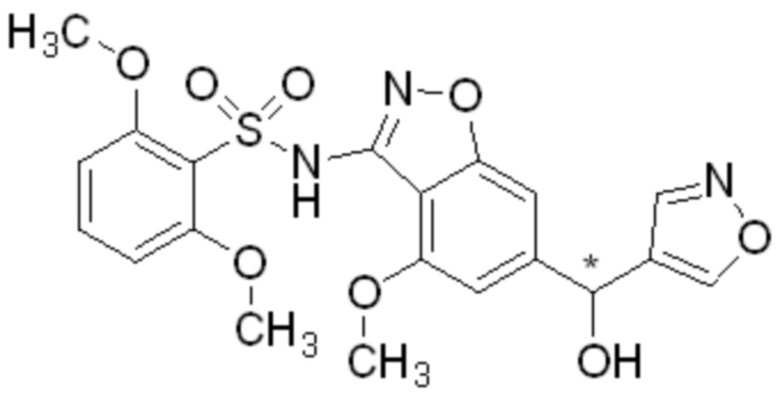
N-(6-(гидрокси(изоксазол-4-ил)метил)-4-метоксибензо[d]изоксазол-3- N-{6-[(R*)-гидрокси(1,2-оксазол-4-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамид, изомер B
Подвижная фаза: A: CO2 B: этанол (0,05% DEA).
Схема Q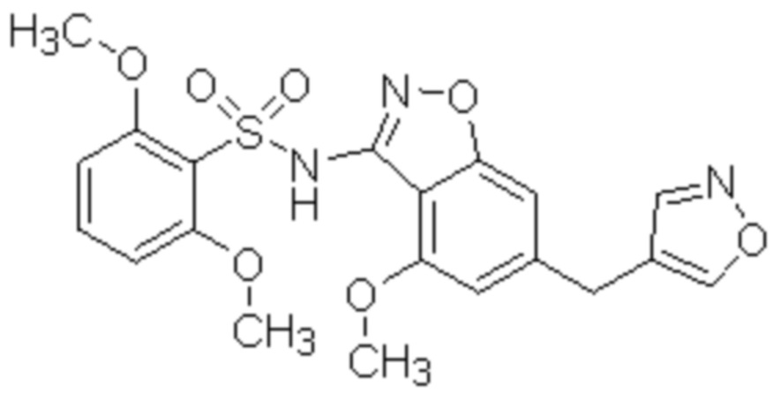
2,6-диметокси-N-[4-метокси-6-(1,2-оксазол-4-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамид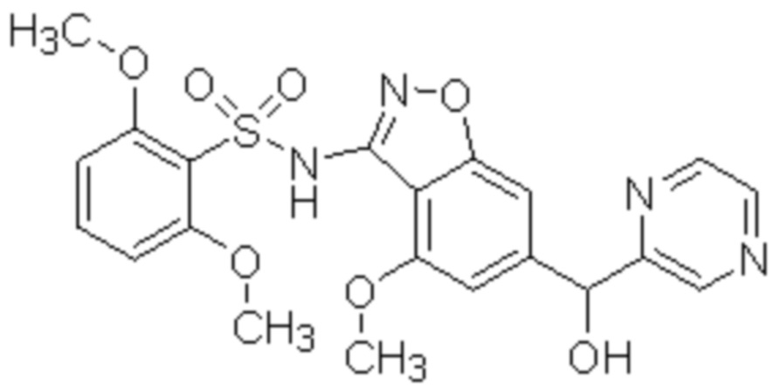
rac-N-{6-[гидрокси(пиразин-2-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамид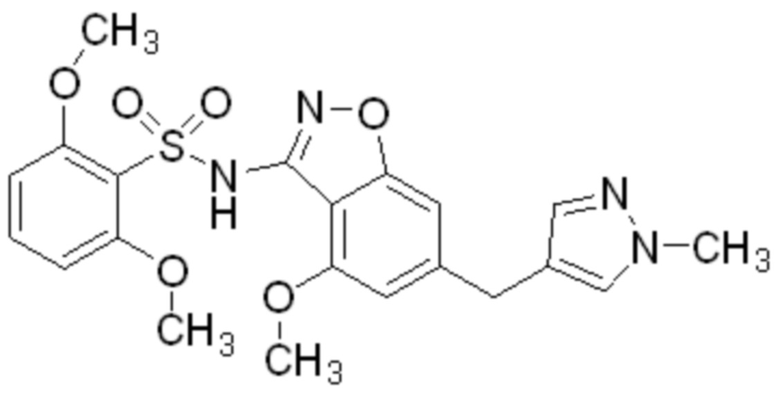
2,6-диметокси-N-{4-метокси-6-[(1-метил-1H-пиразол-4-ил)метил]-1,2-бензоксазол-3-ил}бензолсульфонамид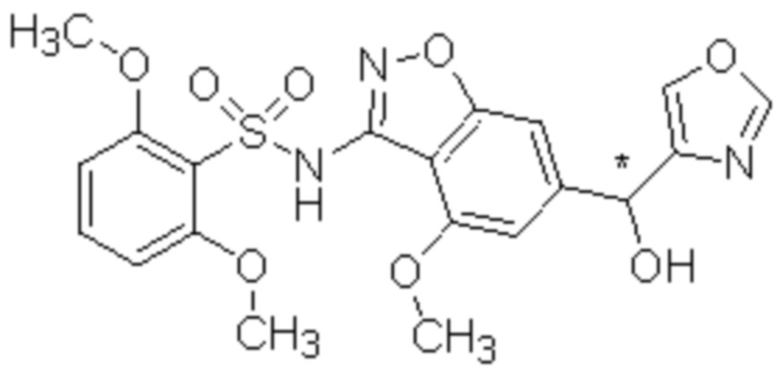
N-{6-[(S*)-гидрокси(1,3-оксазол-4-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамид
Подвижная фаза: A: CO2 B:метанол(0,05% DEA).
Изократическая: 40% B.
Схема Q
N-{6-[(R*)-гидрокси(1,3-оксазол-4-ил)метил]-4-метокси-1,2-бензоксазол-3-ил}-2,6-диметоксибензолсульфонамид
Подвижная фаза: A: CO2 B:метанол(0,05% DEA)
Изократическая: 40% B.
Схема Q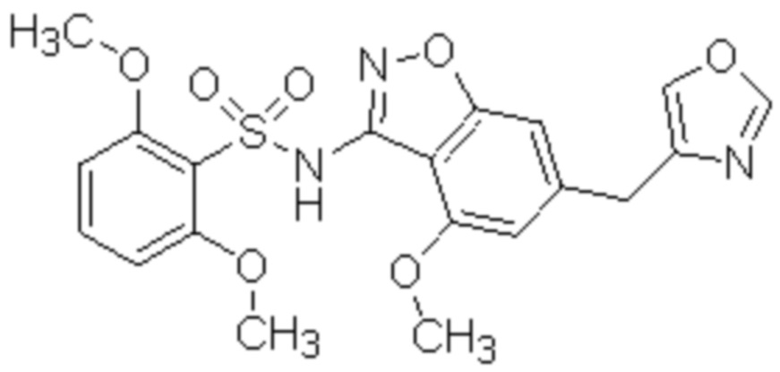
2,6-диметокси-N-[4-метокси-6-(1,3-оксазол-4-илметил)-1,2-бензоксазол-3-ил]бензолсульфонамид
Пример 128: Получение N-{5-фтор-4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида в соответствии со схемой S.
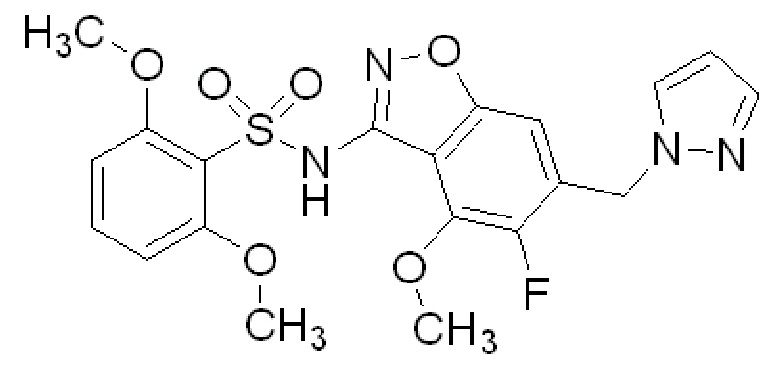
Схема S:
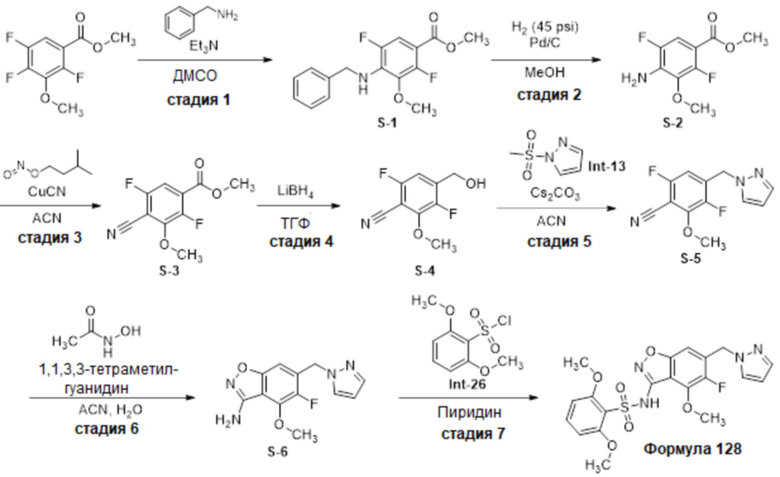
Стадия 1: Синтез метил 4-(бензиламино)-2,5-дифтор-3-метоксибензоата (S-1).
Раствор бензиламина (38,0 мл, 347 ммоль), метил 2,4,5-трифтор-3-метоксибензоата (51,0 г, 232 ммоль) и триэтиламина (161 мл, 1160 ммоль) в ДМСО (500 мл) нагревали при температуре 100ºC в течение 18 часов. После охлаждения до комнатной температуры смесь выливали в воду и экстрагировали этилацетатом. Органическую фазу промывали насыщ. водн. NaCl, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле (элюируя смесью петролейный эфир/этилацетат 5/1) с получением метил 4-(бензиламино)-2,5-дифтор-3-метоксибензоата (S−1) (41 г, 57%-ный выход) в виде масла светло-желтого цвета. ЖХМС m/z 308,1 [M+H]+; 1H ЯМР (400МГц, ХЛОРОФОРМ-d) δ 7,43-7,20 (м, 6H), 4,74 (шир. с, 1H), 4,63 (шир. с, 2H), 3,97-3,80 (м, 6H).
Стадия 2: Синтез метил 4-амино-2,5-дифтор-3-метоксибензоата (S-2).
Раствор метил 4-(бензиламино)-2,5-дифтор-3-метоксибензоата (S-1) (41 г, 133 ммоль) в метаноле (500 мл) обрабатывали Pd/C (7,0 г) и перемешивали при температуре 50ºC в атмосфере водорода (45 фунтов на кв. дюйм) в течение 48 часов. Суспензию фильтровали через слой из Целита®, и фильтрат концентрировали с получением метил 4-амино-2,5-дифтор-3-метоксибензоата (S-2) (28,0 г, 96%-ный выход) в виде твердого вещества не совсем белого цвета. ЖХМС m/z 217,9 [M+H]+; 1H ЯМР (400МГц, ДМСО-d6) δ 7,27 (дд, J=6,3, 11,6 Гц, 1H), 6,21 (с, 2H), 3,77 (д, J=1,1 Гц, 6H).
Стадия 3: Синтез метил 4-циано-2,5-дифтор-3-метоксибензоата (S-3)
Суспензию метил 4-амино-2,5-дифтор-3-метоксибензоата (S-2) (28,0 г, 129 ммоль) и цианид меди(I) (34,6 г, 387 ммоль) в CH3CN (1 л) нагревали до температуры 65ºC. По каплям добавляли изоамилнитрит (22,7 г, 193 ммоль) и реакционную смесь перемешивали при температуре 65ºC в течение 1 ч. Анализ ЖХМС показывал, что часть исходного материала оставалась и добавляли дополнительное количество изоамилнитрита (15,1 г, 129 ммоль). Реакционную смесь нагревали при температуре 65ºC в течение 18 ч. После охлаждения до комнатной температуры реакционную смесь разбавляли EtOAc (200 мл) и фильтровали. Фильтрат концентрировали и очищали с помощью флэш-хроматографии, элюируя градиентом 0-50% EtOAc в гептане с получением метил 4-циано-2,5-дифтор-3-метоксибензоата (S-3) (14,0 г, 47%-ный выход) в виде твердого вещества желтого цвета. 1H ЯМР (400МГц, ХЛОРОФОРМ-d) δ 7,36 (дд, J=4,8, 8,4 Гц, 1H), 4,21 (д, J=3,1 Гц, 3H), 3,97 (с, 3H).
Стадия 4: Синтез 3,6-дифтор-4-(гидроксиметил)-2-метоксибензонитрила (S-4)
К раствору метил 4-циано-2,5-дифтор-3-метоксибензоата (S-3) (14 г, 62 ммоль) в ТГФ (400 мл) медленно при температуре 0ºC добавляли LiBH4 (20 г, 92 ммоль). После завершения добавления реакционную смесь нагревали до комнатной температуры и затем нагревали до температуры 50ºC в течение 2 ч. После охлаждения до комнатной температуры реакционную смесь гасили медленным добавлением H2O (100 мл) и органические продукты экстрагировали EtOAc (2×300 мл). Объединенные органический экстракт промывали насыщенным солевым раствором и насыщ. NaHCO3, сушили над Na2SO4 и фильтровали. После удаления растворителя получали 3,6-дифтор-4-(гидроксиметил)-2-метоксибензонитрил (S-4) (10 г, 81%) в виде твердого вещества желтого цвета. Это вещество использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400МГц, ХЛОРОФОРМ-d) δ 7,06 (дд, J=4,8, 8,8 Гц, 1H), 4,81 (с, 2H), 4,17 (д, J=3,3 Гц, 3H), 2,48 (шир. с, 1H).
Стадия 5: Синтез 3,6-дифтор-2-метокси-4-[(1H-пиразол-1-ил)метил]бензонитрила (S-5)
К раствору 3,6-дифтор-4-(гидроксиметил)-2-метоксибензонитрила (S-4) (10 г, 50 ммоль) и 1-(метансульфонил)-1H-пиразола (Int-13) (8,8 г, 60 ммоль) в CH3CN (500 мл) добавляли Cs2CO3 (24,5 г, 75,3 ммоль) и перемешивали при температуре 70ºC в течение 2 ч. Анализ ЖХМС показывал, что исходное вещество расходовалось. Реакционную смесь охлаждали до комнатной температуры и фильтровали. После концентрирования фильтрата остаток очищали с помощью флеш хроматографии, элюируя градиентом 20-50% EtOAc в петролейном эфире с получением 3,6-дифтор-2-метокси-4-[(1H-пиразол-1-ил)метил]бензонитрила (S-5) (8,4 г, 67%-ный выход) в виде смолы желтого цвета. ЖХМС m/z 250,0 [M+H]+; 1H ЯМР (400МГц, ДМСО-d6) δ 7,89 (д, J=2,2 Гц, 1H), 7,62-7,40 (м, 1H), 6,76 (дд, J=5,0, 9,1 Гц, 1H), 6,33 (т, J=2,1 Гц, 1H), 5,49 (д, J=1,1 Гц, 2H), 4,13 (д, J=3,2 Гц, 3H).
Стадия 6: Синтез 5-фтор-4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амин (S-6)
К раствору 3,6-дифтор-2-метокси-4-[(1H-пиразол-1-ил)метил]бензонитрила (S-5) (8,4 г, 34 ммоль) и N-гидроксиацетамида (7,6 г, 100 ммоль) в CH3CN (400 мл) и воде (80 мл) добавляли 1,1,3,3-тетраметилгуанидин (23 г, 200 ммоль) медленно. Смесь нагревали при температуре 60ºC в течение 16 ч. Смесь охлаждали и концентрировали для удаления CH3CN. Из раствора выпадало в осадок желтое твердое вещество, которое промывали водой, а затем смесью 10% EtOAc в петролейном эфире. Полученное твердое вещество бледно-желтого цвета фильтровали с получением 5-фтор-4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (S-6) (6,1 г, 69%-ный выход) в виде твердого вещества бледно-желтого цвета. ЖХМС m/z 262,9 [M+H]+; 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 7,59 (д, J=1,7 Гц, 1H), 7,43 (д, J=2,2 Гц, 1H), 6,70 (д, J=9,2 Гц, 1H), 6,50 (с, 1H), 6,35 (т, J=2,1 Гц, 1H), 5,31 (с, 2H), 2,23 (тт, J=5,1, 8,4 Гц, 1H), 1,20-1,14 (м, 2H), 0,81-0,73 (м, 2H).
Стадия 7: Синтез N-{5-фтор-4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида (пример 128)
К раствору 5-фтор-4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-амина (S-6) (6,1 г, 23 ммоль) в пиридине (100 мл) добавляли 2,6-диметоксибензолсульфонил хлорид (Int-26) (6,1 г, 26 ммоль) и полученную смесь перемешивали при температуре 70ºC в течение 18 ч. Смесь концентрировали и очищали с помощью флеш хроматографии, элюируя 10% MeOH в DCM с получением сырого соединения по примеру 128 (8 г) в виде твердого вещества желтого цвета. Суспензию твердого вещества желтого в CH3CN (100 мл) кипятили с обратным холодильником в течение 10 мин. Осталась большая часть твердых частиц. Итак, добавляли дополнительное количество CH3CN (500 мл) порциями по 100 мл до полного растворения твердого вещества. Раствору давали остыть в течение 5 минут и добавляли МТВЕ (400 мл) при интенсивном перемешивании. Начиналось образование белого твердого вещества, смесь концентрировали до 1/3 объема и раствор интенсивно перемешивали при температуре 20ºC в течение 18 часов. Осадок собирали фильтрованием, промывали гептаном и сушили в вакууме с получением 3,2 г (30%) соединения по примеру 128 в виде твердого вещества белого цвета. Фильтрат концентрировали с получением 4,7 г сырого соединения по примеру 128 в виде твердого вещества желтого цвета, которое дополнительно очищали, как описано ниже. 4,7 г повторно очищали флэш-хроматографией, элюируя градиентом 0-20% EtOAc в DCM, с получением 3 граммов твердого продукта белого цвета, которое растворяли в CH3CN (10 мл) и MTBE (25 мл). Бесцветный раствор перемешивали до тех пор, пока он не станет мутным и не выпадет в осадок белое твердое вещество. Белое твердое вещество собирали фильтрацией и промывали MTBE (3×5 мл). Эту партию белого твердого вещества объединяли с загрузкой 3,2 г, и объединенное твердое вещество суспендировали в CH3CN (30 мл) и затем нагревали до растворения. Постепенно добавляли МТВЕ (60 мл), и из раствора выпадало в осадок белое твердое вещество. Смесь охлаждали до комнатной температуры и концентрировали до общего объема 30 мл. Полученное белое твердое вещество собирали фильтрованием и промывали МТВЕ (3×10 мл), сушили в вакуумном сушильном шкафу при температуре 60ºC в течение 6 часов с получением N-{5-фтор-4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида (пример 128) (5,3 г, 49%-ный выход) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, ДМСО-d6) δ 10,13 (шир. с, 1H), 7,86 (шир. с, 1H), 7,59-7,43 (м, 2H), 6,90 (шир. д, J=3,3 Гц, 1H), 6,78 (шир. д, J=8,5 Гц, 2H), 6,31 (шир. с, 1H), 5,50 (шир. с, 2H), 4,04 (шир. с, 3H), 3,76 (с, 6H); m/z 463,0 [M+H]+.
Соединения по примерам, приведенным в таблице ниже, синтезировали в соответствии со способами, используемыми для синтеза N-{5-фтор-4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида (пример 128), N-{4-этил-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2,6-диметоксибензол-1-сульфонамида (пример 95), 5-этил-2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 01), 2,6-диметокси-N-{4-метокси-6-[(3-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 02) и 2,6-диметокси-N-{4-метокси-6-[(5-метил-1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида (пример 03), и общим способом получения сульфонамидов A-D. Следующие соединения по примерам синтезировали с некритическими изменениями или замены приведенных в качестве примеров способов, которые сможет осуществить специалист в данной области техники.
Таблица 20:
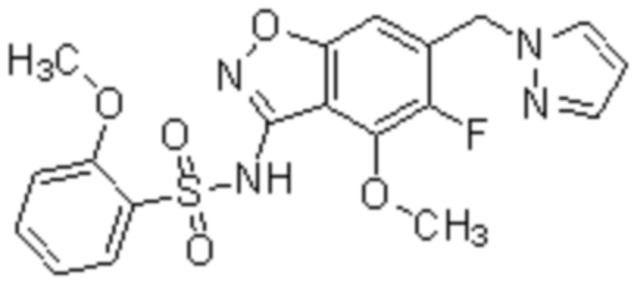
N-{5-фтор-4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2-метоксибензол-1-сульфонамид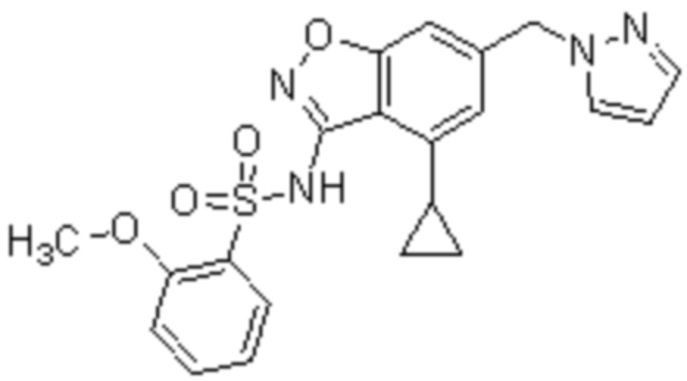
N-{4-циклопропил-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}-2-метоксибензол-1-сульфонамид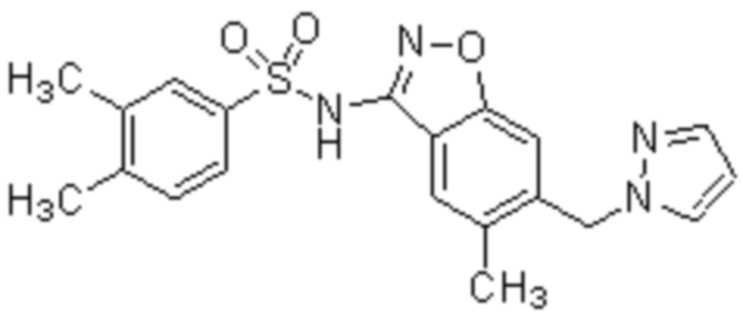
3,4-диметил-N-{5-метил-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид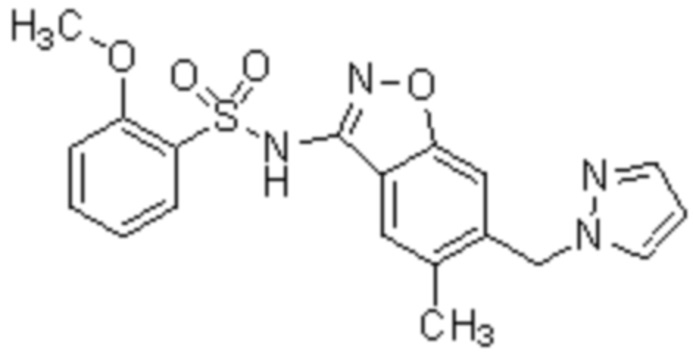
2-метокси-N-{5-метил-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид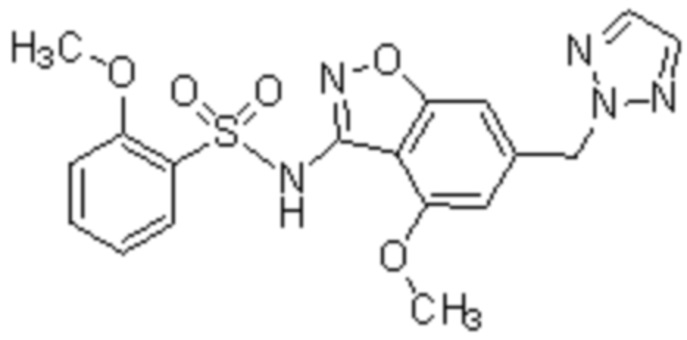
2-метокси-N-{4-метокси-6-[(2H-1,2,3-триазол-2-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид
Биологический анализ Раздел 1
Протокол анализа KAT:
Получение соединения
Приготовить 10 мМ исходных растворов в 100% ДМСО из твердого вещества
Серийное разведение 10 мм, 1 мм или 0,1 мм исходных соединений соединения 3-кратно в 100% ДМСО для 11-точечного ответа на дозу
Получение реагентов
Приготовить 1× буфер для анализа, содержащий 10 мм Tris HCl pH 8,0, 2,5 мМ NaCl, 0,5 мм EDTA, 0,005% BSG и 0,02% Tween-20
Развести пептид гистона (CPC Scientific) и AcCoA (Sigma) месте в буфере для анализа до 2×.
Развести фермент KAT в буфере для анализа до 2×.
Ферментная реакция
Конечные условия реакции для каждого анализа KAT в реакционном объеме анализа 20 мкл:
KAT5 25 нМ, 1 мкМ AcCoA, 2 мкМ H4 1-21 пептид, 30-минутное взаимодействие
KAT6A 15 нМ, 1 мкМ AcCoA, 2 мкМ H3 1-21 пептид, 45-минутное взаимодействие
KAT6B 25 нМ, 1 мкМ AcCoA, 2 мкМ H3 1-21 пептид, 60-минутное взаимодействие
KAT7 12,5 нМ, 1 мкМ AcCoA, 2 мкМ H3 1-21 пептид, 45-минутное взаимодействие
KAT8 15 нМ, 1 мкМ AcCoA, 2 мкМ H3 1-21 пептид, 45-минутное взаимодействие
Добавить 0,5 мкл разбавленного соединения в аналитический планшет (384-луночные полипропиленовые планшеты с V-образным дном) или 0,5 мкл ДМСО для контрольных лунок.
Добавить 10 мкл смеси 2× пептида гистона/2× AcCoA в планшет для анализа.
Добавить 10 мкл 2× фермента в планшет для анализа.
Остановить реакцию через указанное время добавлением 2 мкл 5% муравьиной кислоты.
Каждую реакцию анализировали с использованием времяпролетной масс-спектрометрии десорбции/ионизации самоорганизующегося монослоя (Mrksich, Milan (2008) Mass Spectrometry of Self-Assembled Monolayers: A New Tool for Molecular Surface Science ACS Nano 2008 2 (1), 7-18; SAMDI Tech, Inc. (Chicago, IL)).
Площадь под кривой (AUC) для пиков субстрата и продукта определяли для KAT5 при M.W. 2561 [субстрат+H]+ и 2603 [продукт+H]+ с допуском +/- 1 Да, соответственно
Площадь под кривой (AUC) для пиков субстрата и продукта была определена для KAT6A, KAT6B, KAT7 и KAT8 при M.W. 2723 [субстрат+H]+ и 2765 [продукт+H]+ с допуском +/- 1 Да, соответственно.
Процент превращения в продукт рассчитывали по формуле: AUCпродукт/(AUCсубстрат+AUCпродукт).
Анализ данных
Значения IC50 определяли путем подгонки % превращения при каждой концентрации ингибитора к 4-параметрическому уравнению IC50 с использованием запатентованного программного обеспечения Pfizer для построения кривых.
Значения Ki определяли путем подгонки% превращения при каждой концентрации ингибитора к уравнению Моррисона для конкурентных ингибиторов с сильным связыванием с использованием запатентованного программного обеспечения Pfizer для построения кривых.
Вещества
Ферменты KAT экспрессировали с использованием системы экспрессии бакуловируса и очищали по Pfizer, La Jolla. Пептид гистона H3 (1-21) (ARTKQTARKSTGGKAPRKQLA, SEQ ID NO:3) и пептид гистона H4 (1-21) (SGRGKGGKGLGKGGAKRHRKV, SEQ ID NO:4) были приобретены у компании CPC Scientific (Sunnyvale, CA). Ацетилкофермент А был приобретен у компании Sigma-Aldrich (St. Louis, MO). Все остальные биохимические реагенты были приобретены у компании Sigma-Aldrich или ThermoFisher Scientific (Waltham, MA).
Рекции KAT
KAT анализы проводили при комнатной температуре в буфере для анализа, содержащем 1 мкМ AcCoA, 2 мкМ пептид гистона, 10 мм Tris HCl pH 8,0, 2,5 мМ NaCl, 0,5 мм EDTA, 0,005% BSG и 0,02% Tween-20. 10 мкл смеси 2× пептида гистона/AcCoA добавляли в 384-луночный планшет для анализа полипропилена с V-образным дном, содержащий 0,5 мкл серийно разведенного тестируемого соединения в 100% диметилсульфоксиде (ДМСО). Для начала реакции в аналитический планшет добавляли 10 мкл 2х раствора фермента. Анализы KAT прекращали через 30-60 минут добавлением 2 мкл 5% муравьиной кислоты. Во всех анализах использовался пептид гистона H3 (1-21), за исключением анализа KAT5, в котором использовался пептид гистона H4 (1-21). Конечная концентрация фермента для каждого KAT была следующей: KAT5, 25 нМ; KAT6A, 15 нМ; KAT6B, 25 нМ; KAT7, 12,5 нМ; KAT8 15 нМ. Каждую реакцию анализировали с использованием времяпролетной масс-спектрометрии десорбции/ионизации самоорганизующегося монослоя (Mrksich, Milan (2008) Mass Spectrometry of Self-Assembled Monolayers: A New Tool for Molecular Surface Science ACS Nano 2008 2 (1), 7-18; SAMDI Tech, Inc. (Chicago, IL)).
Обработка и анализ данных
Площадь под кривой (AUC) для пиков субстрата и продукта определяли для KAT5 при M.W. 2561 [субстрат+H]+ и 2603 [продукт+H]+ с допуском +/- 1 Да, соответственно. Площадь под кривой (AUC) для пиков субстрата и продукта была определена для KAT6A, KAT6B, KAT7 и KAT8 при M.W. 2723 [субстрат+H]+ и 2765 [продукт+H]+ с допуском +/- 1 Да, соответственно. Процент превращения в продукт рассчитывали по формуле: AUCпродукт/(AUCсубстрат+AUCпродукт). Значения IC50 определяли путем подгонки % превращения при каждой концентрации ингибитора к 4-параметрическому уравнению IC50 с использованием запатентованного программного обеспечения Pfizer для построения кривых. Значения Ki определяли путем подгонки % превращения при каждой концентрации ингибитора к уравнению Моррисона для конкурентных ингибиторов с сильным связыванием с использованием запатентованного программного обеспечения Pfizer для построения кривых.
Ki KAT6a и KAT6b представлены в таблице 21 и Ki KAT5, KAT7, и KAT8 представлены в таблице 22 ниже.
Таблица 21:
Ki при 1 мкМ AcCoA (нМ)
Ki при 10 мкМ AcCoA (нМ)
Ki при 1 мкМ AcCoA (нМ)
Ki при 25 мкМ AcCoA (нМ)
Таблица 22:
Ki при 1 мкМ AcCoA (мкМ)
Ki при 1 мкМ AcCoA (мкМ)
Ki при 1 мкМ AcCoA (мкМ)
Биологический анализ Раздел 2
Получение белка
KAT5
Молекулярная биология: Оптимизированная по кодонам последовательность ДНК (для экспрессии в Escherichia coli), кодирующая аминокислотные остатки от 2 до 461 (Uniprot Q92993-2) KAT5 изоформы KAT5 человека, была синтезирована GenScript USA Inc (Piscataway, New Jersey, USA). Его лигировали в модифицированный экспрессионный вектор pET43a E. coli, сконструированный для кодирования N-концевой гексагистидиновой метки, за которой следует сайт расщепления протеазой вируса травления табака (TEV) и последовательность KAT5. Полученная последовательность белка приведена ниже (SEQ ID NO:5).
MGHHHHHHGTENLYFQGSAEVGEIIEGCRLPVLRRNQDNEDEWPLAEILSVKDISGRKLFYVHYIDFNKRLDEWVTHERLDLKKIQFPKKEAKTPTKNGLPGSRPGSPEREVKRKVEVVSPATPVPSETAPASVFPQNGAARRAVAAQPGRKRKSNCLGTDEDSQDSSDGIPSAPRMTGSLVSDRSHDDIVTRMKNIECIELGRHRLKPWYFSPYPQELTTLPVLYLCEFCLKYGRSLKCLQRHLTKCDLRHPPGNEIYRKGTISFFEIDGRKNKSYSQNLCLLAKCFLDHKTLYYDTDPFLFYVMTEYDCKGFHIVGYFSKEKESTEDYNVACILTLPPYQRRGYGKLLIEFSYELSKVEGKTGTPEKPLSDLGLLSYRSYWSQTILEILMGLKSESGERPQITINEISEITSIKKEDVISTLQYLNLINYYKGQYILTLSEDIVDGHERAMLKRLLRIDSKCLHFTPKDWSKRGKWAS*
Экспрессия белка: для получения рекомбинантного белка KAT5 экспрессионную плазмиду трансформировали в штамм E. coli BL21 DE3 и выращивали при встряхивании при температуре 37ºC в объеме 1 л бульона Terrific (TB) с добавлением 100 мкг/мл ампициллина и 50 мкМ цинка до достижения OD600 0,8. Культуры переносили при температуре 18ºC и экспрессию белка индуцировали добавлением изопропил-β-D-1-тиогалактопиранозида до конечной концентрации 0,5 мМ, и культуры встряхивали в течение ночи в течение следующих 16 часов. После экспрессии клеточные культуры центрифугировали при 5000xg в течение 20 мин и клеточный осадок хранили замороженными при температуре -20ºC.
Очистка белка: Очистку белка инициировали оттаиванием осадка клеток (25 г, сырой вес) в буфере для лизиса (50 мМ Hepes pH 7,4, 500 мМ NaCl, 5 мМ имидазола, 5% [об./об.] глицерина, 0,1% [мас./об.] CHAPS, 2 мМ 2-меркаптоэтанола, 3 мМ MgCl2, 0,5 мг/мл лизоцима, бензоназная эндонуклеаза [EMD Millipore], 1 мМ PMSF, таблетки полного ингибитора протеазы без EDTA [Roche]) с использованием соотношения 6 мл буфера на 1 г клеток. Клетки дополнительно лизировали обработкой ультразвуком с использованием процессора Misonix Liquid (6 импульсов по 30 секунд, амплитуда 60 [70 Вт]), а затем центрифугировали при 48000×g при температуре 4ºC. Супернатант (клеточный лизат) смешивали с 20 мл смолы Q-Sepharose FF (GE Healthcare), предварительно уравновешенной Q-буфером (20 мМ Hepes pH 7,4, 1 M NaCl). Несвязавшуюся фракцию Q-сефарозы FF затем инкубировали с 5 мл смолы cOmplete His-Tag Purification Resin (Roche), предварительно уравновешенной промывочным буфером IMAC (20 мМ Hepes pH 7,4, 500 мМ NaCl, 35 мМ имидазола). Смолу промывали промывочным буфером IMAC и связанный KAT5 элюировали буфером для элюции IMAC (20 мМ Hepes pH 7,4, 500 мМ NaCl, 300 мМ имидазола). Белок, элюированный IMAC, сразу обессоливали в буфере для хранения (50 мМ цитрата Na pH 6,5, 500 мМ NaCl, 5% [об./об.] глицерина), используя последовательно 2 колонки для обессоливания HiPrep 26/10 (GE Healthcare). Обессоленный белок дополнительно очищали пропусканием через колонку HiLoad 26/60 Superdex 75, предварительно уравновешенную буфером для хранения. Наконец, белок KAT5 концентрировали до 1,5 мг/мл с использованием блока центробежного фильтра Amicon Ultra (Utra-15 MWCO 10 кДа), мгновенно замораживали в жидком азоте и хранили в морозильной камере при температуре -70ºC.
KAT6A
Молекулярная биология: ДНК-последовательность, кодирующая аминокислотные остатки от 507 до 778 (Uniprot Q92794-1) человеческого KAT6A, амплифицировали с помощью PCR и лигировали в модифицированный вектор экспрессии pET E. coli, разработанный для кодирования метки растворимости NusA, за которой следует гексагистидиновая метка и сайт расщепления протеазой вируса травления табака (TEV) и последовательность KAT6A. Полученная последовательность белка приведена ниже (SEQ ID NO:6).
Mnkeilavveavsnekalprekifealesalatatkkkyeqeidvrvqidrksgdfdtfrrwlvvdevtqptkeitleaaryedeslnlgdyvedqiesvtfdrittqtakqvivqkvreaeramvvdqfrehegeiitgvvkkvnrdnisldlgnnaeavilredmlprenfrpgdrvrgvlysvrpeargaqlfvtrskpemlielfrievpeigeevieikaaardpgsrakiavktndkridpvgacvgmrgarvqavstelggeridivlwddnpaqfvinamapadvasivvdedkhtmdiaveagnlaqaigrngqnvrlasqlsgwelnvmtvddlqakhqaeahaaidtftkyldidedfatvlveegfstleelayvpmkelleiegldeptvealreraknalatiaqaqeeslgdnkpaddllnlegvdrdlafklaargvctledlaeqgiddladiegltdekagalimaarnicwfgdeatsgsghhhhhhsagenlyfqgamgrcpsviefgkyeihtwysspypqeysrlpklylcefclkymksrtilqqhmkkcgwfhppvneiyrknnisvfevdgnvstiycqnlcllaklfldhktlyydvepflfyvltqndvkgchlvgyfskekhcqqkynvscimilpqyqrkgygrflidfsyllskregqagspekplsdlgrlsymaywksvileclyhqndkqisikklskltgicpqditstlhhlrmldfrsdqfviirrekliqdhmaklqlnlrpvdvdpeclrwtp
Экспрессия белка: Для получения рекомбинантного белка KAT6A экспрессионную плазмиду трансформировали в штамм E. coli BL21 DE3 и выращивали при встряхивании при температуре 37ºC в объеме 1 л Terrific broth (TB) с добавлением 100 мкг/мл ампициллина до достижения OD600 0,8. Культуры переносили при температуре 18ºC и экспрессию белка индуцировали добавлением изопропил-β-D-1-тиогалактопиранозида до конечной концентрации 0,5 мМ, и культуры встряхивали в течение ночи в течение следующих 16 часов. После экспрессии клеточные культуры центрифугировали при 5000×g в течение 20 мин и клеточный осадок хранили замороженными при температуре -20ºC.
Очистка белка: Очистку белка инициировали оттаиванием осадка клеток (40 г, сырой вес) в буфере для лизиса (25 мМ Tris-HCl pH 7,8, 500 мМ NaCl, 5 мМ DTT, 0,01% [об./об.] Triton-X 100, 5% [об./об.] глицерина, 2 мМ MgCl2, 10 мМ имидазола, 0,5 мг/мл лизоцима, бензоназная эндонуклеаза [EMD Millipore], 1 мМ PMSF, таблетки полного ингибитора протеазы без EDTA [Roche]) с использованием соотношения 5 мл буфера на 1 г клеток. Клетки дополнительно лизировали за 3 прохода (при 15000 фунт/кв.дюйм) через дробилку клеток Avestin C5, охлаждаемую льдом, а затем центрифугировали при 48000×g, при температуре 4ºC. Супернатант (клеточный лизат) фильтровали через фильтр 5 мкм и наносили на 5 мл колонку HiTrap IMAC Sepharose FF (GE Healthcare), предварительно уравновешенную промывочным буфером IMAC (25 мМ Tris-HCl pH 7,8, 500 мМ NaCl, 5 мМ DTT, 0,01% [об./об.] Triton-X 100, 5% [об./об.] глицерина, 20 мМ имидазола) с использованием системы очистки для хроматографии Profinia Affinity (Bio-Rad). Затем колонку IMAC промывали промывочным буфером IMAC и связанный белок KAT6A, элюируя буфером для элюирования IMAC (25 мМ Tris-HCl pH 7,8, 500 мМ NaCl, 5% [об./об.] глицерина, 5 мМ DTT, 250 мМ имидазола). Белок, элюированный IMAC, дополнительно очищали пропусканием через колонку HiLoad 26/60 Superdex 200, предварительно уравновешенную в буфере для хранения (25 мМ Tris-HCl pH 7,8, 500 мМ NaCl, 5 мМ DTT, 5% [об./об.] глицерина). Наконец, белок KAT6A концентрировали до ≥1 мг/мл с использованием центробежного фильтра Amicon Ultra (Utra-15 MWCO 10 кДа), мгновенно замораживали в жидком азоте и хранили в морозильной камере при температуре -70ºC.
KAT7
Молекулярная биология: Оптимизированная по кодонам последовательность ДНК, кодирующая аминокислотные остатки от 325 до 611 (Uniprot O95251-1) человеческого KAT7, была синтезирована GenScript USA Inc (Piscataway, New Jersey, USA). Его лигировали в модифицированный экспрессионный вектор pET43a E. coli, разработанный для кодирования N-концевой гексагистидиновой метки, за которой следует сайт расщепления протеазой вируса травления табака (TEV) и последовательность KAT7. Полученная последовательность белка приведена ниже (SEQ ID NO:7).
MGHHHHHHGTENLYFQGSRLQGQITEGSNMIKTIAFGRYELDTWYHSPYPEEYARLGRLYMCEFCLKYMKSQTILRRHMAKCVWKHPPGDEIYRKGSISVFEVDGKKNKIYCQNLCLLAKLFLDHKTLYYDVEPFLFYVMTEADNTGCHLIGYFSKEKNSFLNYNVSCILTMPQYMRQGYGKMLIDFSYLLSKVEEKVGSPERPLSDLGLISYRSYWKEVLLRYLHNFQGKEISIKEISQETAVNPVDIVSTLQALQMLKYWKGKHLVLKRQDLIDEWIAKEAKRSNSNKTMDPSCLKWTPPKGTAS
Экспрессия белка: Для получения рекомбинантного белка KAT7 экспрессионную плазмиду трансформировали в штамм E. coli BL21 DE3 RIL и выращивали при встряхивании при температуре 37ºC в объеме 1 л Terrific broth (TB) с добавлением 100 мкг/мл ампициллина до достижения OD600 0,8. Культуры переносили при температуре 18ºC и экспрессию белка индуцировали добавлением изопропил-β-D-1-тиогалактопиранозида до конечной концентрации 0,5 мМ, и культуры встряхивали в течение ночи в течение следующих 16 часов. После экспрессии клеточные культуры центрифугировали при 5000×g в течение 20 мин и клеточный осадок хранили замороженными при температуре -20ºC.
Очистка белка: Очистку белка инициировали оттаиванием осадка клеток (10 г, сырой вес) в буфере для лизиса (50 мМ Hepes pH 7,5, 300 мМ NaCl, 5 мМ DTT, 5 мМ имидазола, 0,05% [об./об.] Brij 35, 10% [об./об.] глицерина, 3 мМ MgCl2, 0,5 мг/мл лизоцима, бензоназная эндонуклеаза [EMD Millipore], 1 мМ PMSF, таблетки полного ингибитора протеазы без EDTA [Roche]) с использованием соотношения 10 мл буфера на 1 г клеток. Клетки дополнительно лизировали обработкой ультразвуком с использованием процессора Misonix Liquid (6 импульсов по 30 секунд, амплитуда 60 [70 Вт]), а затем центрифугировали при 48000×g при температуре 4ºC. Супернатант (клеточный лизат) инкубировали с 1 мл cOmplete His-Tag Purification Resin (Roche), предварительно уравновешенным промывочным буфером 1 (25 мМ Hepes pH 7,5, 800 мМ NaCl, 5 мМ имидазола, 10% [об./об.] глицерина, 5 мМ DTT, 0,01% [об./об.] Brij 35, 50 мМ аргинина, 50 мМ глутаминовой кислоты). Смолу последовательно промывали промывочным буфером 1 IMAC и промывочным буфером 2 IMAC (25 мМ Hepes pH 7,5, 300 мМ NaCl, 20 мМ имидазола, 10% [об./об.] глицерина, 5 мМ DTT, 0,01% [об./об.] Brij 35, 50 мМ аргинина, 50 мМ глутаминовой кислоты). Связанный белок KAT7 элюировали буфером для элюирования IMAC (25 мМ Hepes pH 7,5, 200 мМ NaCl, 500 мМ имидазола, 10% [об./об.] глицерина, 5 мМ DTT 0,01% [об./об.] Brij 35, 50 мМ аргинина, 50 мМ глутаминовой кислоты). Элюирующий белок собирали непосредственно в 4 объема обессоливаемого буфера (50 мМ цитрата Na pH 6,5, 200 мМ NaCl, 0,01% [об./об.] Brij 35, 10% [об./об.] глицерина, 5 мМ DTT) для доведения конечной концентрации имидазола до 100 мМ. Белок, элюированный IMAC, сразу же обессоливали в обессоливающем буфере, используя последовательно две обессоливающие колонки HiPrep 26/10 (GE Healthcare). Обессоленный белок дополнительно очищали пропусканием через колонку HiLoad 26/60 Superdex 75, предварительно уравновешенную в буфере для хранения (50 мМ цитрата Na pH 6,5, 200 мМ NaCl, 10% [об./об.] глицерина, 5 мМ DTT). Наконец, белок KAT7 концентрировали до 3,5 мг/мл с использованием центробежного фильтра Amicon Ultra (Utra-15 MWCO 10 кДа), мгновенно замораживали в жидком азоте и хранили в морозильной камере при температуре -70ºC.
Биохимический анализ ацетилтрансферазы
Для определения ингибирования ферментативной активности KAT тестируемыми соединениями реакции анализа проводили в объеме 8 мкл в 384-луночных планшетах для анализа с низким объемом. Реакции проводили в буфере для анализа (100 мМ Tris-HCl, pH 7,8, 15 мМ NaCl, 1 мм EDTA, 0,01% Tween-20, 1 мМ дитиотреитола и 0,01% мас./об. альбумина куриного яичного белка)).
Реакции были установлены с 1 мкМ ацетилкофермента А, 100 нМ полноразмерного рекомбинантного гистона, меченного ограниченным биотинилированием (KAT6A, KAT7: H3,1, KAT5), 10/ 5/ 8/ 40/ 20 нМ фермента KAT5/KAT6A/KAT7, соответственно, и специфическое антитело к ацетил-лизину (H3,1: Cell Signaling Technology, H4: Abcam). Были приготовлены серии 11-точечных разведений тестируемых соединений в ДМСО; объем 100 нл переносили с помощью булавочного инструмента в аналитические планшеты, содержащие субстраты, перед добавлением фермента для начала реакции. Положительные (без соединения, только ДМСО) и отрицательные (без АсСоА) контрольные реакции включали в одни и те же планшеты и получали такое же количество ДМСО, что и лунки, обработанные соединением. После добавления всех реагентов планшеты герметично закрывали и инкубировали 90 мин при комнатной температуре. Затем добавляли дополнительные 4 мкл буфера для анализа, содержащего гранулы акцептора AlphaScreen® Protein A и гранулы донора стрептавидина (PerkinElmer, Waltham, MA) до конечной концентрации 8 мкг/мл. После инкубации в течение 2 часов планшеты считывали с помощью считывающего устройства для планшетов с несколькими метками EnVision 2103 multi label plate reader (PerkinElmer) в режиме HTS AlphaScreen®. Значения IC50 были получены из исходных данных путем расчета процента ингибирования (% I) для каждой реакции по сравнению с контролями на том же планшете (%I=(I-CN)/(CP-CN), где CN/CP средние значения отрицательные/положительные реакции, соответственно), затем подгонка данных % I к концентрации соединения [I] в %I=(A+((B-A)/(1+((C/[I])^D)))) где A обозначает нижнюю асимптоту, B обозначает верхнюю асимптоту, C обозначает значение IC50 и D обозначает наклон.
Результаты показаны в таблице 23 ниже:
Таблица 23:
Анализ биомаркера ацетилирования гистона H3, лизина 23
Соединения исследовали на их способность ингибировать ацетилирование маркера гистона H3K23 в следующем анализе:
Клеточную линию U2OS высевали с плотностью 9000 клеток на лунку в 96-луночные планшеты для культивирования тканей оптического качества в среде RPMI и 10% фетальной бычьей сыворотке и давали возможность прилипнуть в течение 24 часов в стандартных условиях культивирования (37 градусов Цельсия, 5% CO2). В конце этого периода среду отсасывали. Разведения соединений, приготовленные в ДМСО, были добавлены к среде, при этом лунки отрицательного контроля зарезервированы для обработки только ДМСО, а положительные контроли со 100% ингибированием, получавшие соединение с сильным ингибитором (например, cas 2055397-28-7, бензойная кислота, 3-фтор-5-(2-пиридиниил)-, 2-[(2-фторфенил)сульфонил]гидразид) (Baell, J., Nguyen, H.N., Leaver, D.J., Cleary, B.L., Lagiakos, H.R., Sheikh, B.N., Thomas. T.J., Aryl sulfonohydrazides, WO2016198507A1, 2016) в концентрации 10 мкМ и 200 мкл переносили в клетки. После инкубации в течение 24 часов клетки фиксировали 3,7% формальдегидом в PBS в течение 20 минут при комнатной температуре, промывали (5×5 минут) солевым раствором фосфатного буфера, содержащим 0,1% Твин 20, и блокировали блокирующим буфером Odyssey (LI-COR, Lincoln, NE), содержащим 0,1% TritonX100. Добавляли специфическое анти-H3K23ac антитело (Abcam ab177275) в блокирующем буфере Odyssey, содержащем 0,1% Tween 20, и инкубировали в течение 16 часов при 4 градусах Цельсия. После промывки (как указано выше) добавляли вторичное антитело, меченное красителем Alexa647 (LifeTechnologies) и Hoechst 33342 (1 мкн/мл, SigmaAldrich) для инкубации в течение 1 часа. Планшеты были промыты, как и ранее, и считаны на платформе для визуализации высокого содержания PerkinElmer Phenix. Используя конвейер анализа изображений Columbus, отдельные ядра были локализованы с помощью красителя Hoechst 33342, и уровень ацетилирования был рассчитан на основе интенсивности, связанной с Alexa647, в той же области. Полученную среднюю интенсивность на клетку непосредственно преобразовывали в процент ингибирования относительно контроля на том же планшете, и данные сравнивали с четырехпараметрической логистической моделью для определения 50% ингибирующей концентрации (IC50).
Результаты показаны в таблице 24 ниже:
Таблица 24:
IC50 (мкМ)
Анализ биомаркера ацетилирования гистона H3, лизина 14
Соединения исследовали на их способность ингибировать ацетилирование маркера гистона H3 лизина 14 в следующем анализе:
Клеточную линию U2OS высевали с плотностью 3000 клеток на лунку в 384-луночные планшеты для культивирования тканей оптического качества в среде RPMI с добавлением 10% фетальной бычьей сыворотки и 10 мМ Hepes. Клеткам давали возможность прикрепиться в течение 24 часов в стандартных условиях культивирования (37 градусов Цельсия, 5% CO2). В конце этого периода клетки промывали бессывороточной средой. Разведения соединений, приготовленные в ДМСО, добавляли к бессывороточной среде, при этом лунки отрицательного контроля зарезервированы для обработки только ДМСО, а положительные контроли со 100% ингибированием получали соединение с сильным ингибитором (например, (Z)-4-фтор-N-((3-гидроксифенил)сульфонил)-5-метил-[1,1Θ-бифенил]-3-карбогидразоновая кислота) в концентрации 10 мкМ. После инкубации в течение 24 часов клетки фиксировали 4% формальдегидом в PBS в течение 15 минут при комнатной температуре, промывали фосфатным буферным солевым раствором и блокировали блокирующим буфером, содержащим 0,2% TritonX100 и 2% BSA. Добавляли специфическое анти-H3K14ac антитело (Cell Signaling Technologies) в блокирующем буфере и инкубировали в течение ночи при 4 градусах Цельсия. После промывания добавляли вторичные антитела, меченные красителем AlexaFluor 488 (ThermoFisher) и Hoechst 33342 (1 мкг/мл, Life Technologies) для инкубации в течение 2 часов при комнатной температуре. Планшеты промывали и считывали на платформе визуализации высокого содержания PerkinElmer Opera HCS. Используя конвейер анализа изображений Columbus, отдельные ядра были локализованы с помощью красителя Hoechst 33342, и уровень ацетилирования был рассчитан на основе интенсивности, связанной с AlexaFluor 488, в той же области. Полученную среднюю интенсивность на клетку преобразовывали в процент ингибирования относительно контроля на той же планшете, и данные сравнивали с четырехпараметрической логистической моделью для определения 50% ингибирующей концентрации (IC50).
Результаты показаны в таблице 25 ниже:
Таблица 25:
Анализ биомаркера ацетилирования лизина 7 H2A.Z
Соединения исследовали на их способность ингибировать маркер ацетилирования гистона H2A.Z лизина 7 в следующем анализе:
Клеточную линию U2OS высевали с плотностью 3000 клеток на лунку в 384-луночные планшеты для культивирования тканей оптического качества в среде RPMI с добавлением 10% фетальной бычьей сыворотки и 10 мМ Hepes. Клеткам давали возможность прикрепиться в течение 24 часов в стандартных условиях культивирования (37 градусов Цельсия, 5% CO2). В конце этого периода клетки промывали бессывороточной средой. Разведения соединений, приготовленные в ДМСО, добавляли к бессывороточной среде, при этом лунки отрицательного контроля зарезервированы для обработки только ДМСО, а положительные контроли со 100% ингибированием получали мощный ингибитор энантиомера 1 соединения7-йод-N-(2-(оксазол-2-ил)-2-фенилэтил)-2H-бензо[e][1,2,4]тиадиазин-3-карбоксамид 1,1-диоксид, которое представляет собой соединение 146 одновременно рассматриваемой заявки GB1713962.7, поданной 31 августа 2018 г., в концентрации 30 мкМ. После инкубации в течение 24 часов клетки фиксировали 4% формальдегидом в PBS в течение 15 минут при комнатной температуре, промывали фосфатным буферным солевым раствором и блокировали блокирующим буфером, содержащим 0,2% TritonX100 и 2% BSA. Добавляли специфическое анти-H2A.ZK7ac антитело (Abcam) в блокирующем буфере и инкубировали в течение ночи при 4 градусах Цельсия. После промывания добавляли вторичные антитела, меченные красителем AlexaFluor 488 (ThermoFisher) и Hoechst 33342 (1 мкг/мл, Life Technologies), для инкубации в течение 2 часов при комнатной температуре. Планшеты промывали и считывали на платформе визуализации высокого содержания PerkinElmer Opera HCS. Используя конвейер анализа изображений Columbus, отдельные ядра были локализованы с помощью красителя Hoechst 33342, и уровень ацетилирования был рассчитан на основе интенсивности, связанной с AlexaFluor 488, в той же области. Полученную среднюю интенсивность на клетку преобразовывали в процент ингибирования относительно контроля на той же планшете, и данные сравнивали с четырехпараметрической логистической моделью для определения 50% ингибирующей концентрации (IC50).
Результаты показаны в таблице 26 ниже:
Таблица 26:
Ссылки
Aggarwal and Calvi, Nature, 2004, 430, 372-376 doi:10.1038/nature02694
Avvakumov et al., Oncogene, 2007, 26, 5395-5407 doi:10.1038/sj.onc.1210608
Berge et al., J. Pharm. Sci., 1977, 66, 1-19 doi:10.1002/jps.2600660104
Borrow et al., Nat. Genet., 1996, 14, 33-41 doi:10.1038/ng0996-33
Dekker et al., Drug, Discov. Today, 2014, 19, 654-660 doi:10.1016/j.drudis.2013.11.012
Doyon et al., Mol. Cell., 2006, 21, 51-64 doi:10.1016/j.molcel.2005.12.007
Dhuban et al., Sci. Immunol., 2017, 2, 9297 doi:10.1126/sciimmunol.aai9297
Duong et al., Cancer Res., 2013, 73, 5556-5568 doi:10.1158/0008-5472.CAN-13-0013
Ghizzoni et al., Eur. J. Med. Chem., 2012, 47, 337-344 doi:10.1016/j.ejmech.2011.11.001
Gil et al., J. Proteomics, 2017, 150, 297-309 doi:10.1016/j.jprot.2016.10.003
Gobert, M. et al., Cancer Research, 2009, 69, 2000-2009 doi:10.1158/0008-5472.CAN-08-2360
Holbert et al., J. Biol. Chem., 2007, 282, 36603-36613 doi:10.1074/jbc.M705812200
Iizuka et al., Mol. Cell. Biol., 2006, 26, 1098-1108 doi:10.1128/MCB.26.3.1098-1108.2006
Iizuka et al., Cancer Sci., 2013, 104, 1647-1655 doi:10.1111/cas.12303
Jeong, et al., Blood Res 2016 51(3), 152-154 doi:10.5045/br.2016.51.3.152
Joshi, et al., Immunity 2015, 43, 579-590 doi:10.1016/j.immuni.2015.08.006
Li, B. et al., PNAS, 2007, 104, 4571-4576 doi:10.1073/pnas.0700298104
Melero, et al. Nature Reviews Cancer, 2015, 15, 457-472 doi:10.1038/nrc3973
Merson et al., J. Neurosci., 2006, 26, 11359-11370 doi:10.1523/JNEUROSCI.2247-06.2006
Miller, A.M. et al. J. Immunol., 2006, 177, 7398-7405 doi:10.4049/jimmunol.177.10.7398
Persa, E. et al. Cancer Letters, 2015 368(2), 252-261 doi:10.1016/j.canlet.2015.03.003
Sheikh et al., Blood, 2015, 125(12), 1910-21 doi:10.1182/blood-2014-08-594655
Shi et al, Nature Biotech, 2015, 33, 661-667 doi:10.1038/nbt.3235
Su et al., Int. J. Mol. Sci., 2016, 17, 1-18 doi:10.3390/ijms17101594
Stern et al., Crit. Rev. Oncol. Hematol., 2005, 54, 11-29 doi:10.1016/j.critrevonc.2004.10.011
Thomas et al., Development, 2000, 127, 2537-2548 PMID:10821753
Tao, H. et al., Lung Cancer, 2012, 75, 95-101 doi:10.1016/j.lungcan.2011.06.002
Turner-Ivey et al., Neoplasia, 2014, 16(8): 644-655 doi:10.1016/j.neo.2014.07.007
Valerio et al., Cancer Research, 2017, 77(7), 1753-62 doi:10.1158/0008-5472.CAN-16-2374
Vizmanos et al., Genes Chromosomes Cancer, 2003, 36(4), 402-405 doi:10.1002/gcc.10174
Voss et al., BioEssays, 2009, 31(10), 1050-1061 doi:10.1002/bies.200900051
Wang, L., et al. EBioMedicine, 2016, 13, 99-112 doi:10.1016/j.ebiom.2016.10.018
Wang, X. et al., Oncogene, 2017, 36, 3048-3058 doi:10.1038/onc.2016.458
Xiao, Y. et al., Cell reports, 2014, 7, 1471-1480 doi:10.1016/j.celrep.2014.04.021
Yan, M. et al., Breast Cancer Research, 2011, 13, R47 doi:10.1186/bcr2869
Zack et al., Nature Genetics 2013 45, 1134-1140 doi:10.1038/ng.2760
Zhang et al., Mini. Rev. Med. Chem., 2017, 17, 1-8 doi:10.2174/1389557516666160923125031
--->
СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Pfizer Inc.
CTXT PTY LTD
<120> ПРОИЗВОДНЫЕ БЕНЗИЗОКСАЗОЛСУЛЬФОНАМИДА
<130> PC072525A
<150> US 62/863,199
<151> 2019-06-18
<150> US 62/953,223
<151> 2019-12-24
<150> US 63/025,278
<151> 2020-05-15
<160> 7
<170> FastSEQ for Windows Version 4.0
<210> 1
<211> 20
<212> PRT
<213> Homo sapiens
<400> 1
Ala Arg Thr Lys Gln Thr Ala Arg Lys Ser Thr Gly Gly Lys Ala Pro
1 5 10 15
Arg Lys Gln Leu
20
<210> 2
<211> 20
<212> PRT
<213> Homo sapiens
<400> 2
Ser Gly Arg Gly Lys Gly Gly Lys Gly Leu Gly Lys Gly Gly Ala Lys
1 5 10 15
Arg His Arg Lys
20
<210> 3
<211> 21
<212> PRT
<213> Homo sapiens
<400> 3
Ala Arg Thr Lys Gln Thr Ala Arg Lys Ser Thr Gly Gly Lys Ala Pro
1 5 10 15
Arg Lys Gln Leu Ala
20
<210> 4
<211> 21
<212> PRT
<213> Homo sapiens
<400> 4
Ser Gly Arg Gly Lys Gly Gly Lys Gly Leu Gly Lys Gly Gly Ala Lys
1 5 10 15
Arg His Arg Lys Val
20
<210> 5
<211> 480
<212> PRT
<213> Искусственная последовательность
<220>
<223> модифицированный белок KAT5
<400> 5
Met Gly His His His His His His Gly Thr Glu Asn Leu Tyr Phe Gln
1 5 10 15
Gly Ser Ala Glu Val Gly Glu Ile Ile Glu Gly Cys Arg Leu Pro Val
20 25 30
Leu Arg Arg Asn Gln Asp Asn Glu Asp Glu Trp Pro Leu Ala Glu Ile
35 40 45
Leu Ser Val Lys Asp Ile Ser Gly Arg Lys Leu Phe Tyr Val His Tyr
50 55 60
Ile Asp Phe Asn Lys Arg Leu Asp Glu Trp Val Thr His Glu Arg Leu
65 70 75 80
Asp Leu Lys Lys Ile Gln Phe Pro Lys Lys Glu Ala Lys Thr Pro Thr
85 90 95
Lys Asn Gly Leu Pro Gly Ser Arg Pro Gly Ser Pro Glu Arg Glu Val
100 105 110
Lys Arg Lys Val Glu Val Val Ser Pro Ala Thr Pro Val Pro Ser Glu
115 120 125
Thr Ala Pro Ala Ser Val Phe Pro Gln Asn Gly Ala Ala Arg Arg Ala
130 135 140
Val Ala Ala Gln Pro Gly Arg Lys Arg Lys Ser Asn Cys Leu Gly Thr
145 150 155 160
Asp Glu Asp Ser Gln Asp Ser Ser Asp Gly Ile Pro Ser Ala Pro Arg
165 170 175
Met Thr Gly Ser Leu Val Ser Asp Arg Ser His Asp Asp Ile Val Thr
180 185 190
Arg Met Lys Asn Ile Glu Cys Ile Glu Leu Gly Arg His Arg Leu Lys
195 200 205
Pro Trp Tyr Phe Ser Pro Tyr Pro Gln Glu Leu Thr Thr Leu Pro Val
210 215 220
Leu Tyr Leu Cys Glu Phe Cys Leu Lys Tyr Gly Arg Ser Leu Lys Cys
225 230 235 240
Leu Gln Arg His Leu Thr Lys Cys Asp Leu Arg His Pro Pro Gly Asn
245 250 255
Glu Ile Tyr Arg Lys Gly Thr Ile Ser Phe Phe Glu Ile Asp Gly Arg
260 265 270
Lys Asn Lys Ser Tyr Ser Gln Asn Leu Cys Leu Leu Ala Lys Cys Phe
275 280 285
Leu Asp His Lys Thr Leu Tyr Tyr Asp Thr Asp Pro Phe Leu Phe Tyr
290 295 300
Val Met Thr Glu Tyr Asp Cys Lys Gly Phe His Ile Val Gly Tyr Phe
305 310 315 320
Ser Lys Glu Lys Glu Ser Thr Glu Asp Tyr Asn Val Ala Cys Ile Leu
325 330 335
Thr Leu Pro Pro Tyr Gln Arg Arg Gly Tyr Gly Lys Leu Leu Ile Glu
340 345 350
Phe Ser Tyr Glu Leu Ser Lys Val Glu Gly Lys Thr Gly Thr Pro Glu
355 360 365
Lys Pro Leu Ser Asp Leu Gly Leu Leu Ser Tyr Arg Ser Tyr Trp Ser
370 375 380
Gln Thr Ile Leu Glu Ile Leu Met Gly Leu Lys Ser Glu Ser Gly Glu
385 390 395 400
Arg Pro Gln Ile Thr Ile Asn Glu Ile Ser Glu Ile Thr Ser Ile Lys
405 410 415
Lys Glu Asp Val Ile Ser Thr Leu Gln Tyr Leu Asn Leu Ile Asn Tyr
420 425 430
Tyr Lys Gly Gln Tyr Ile Leu Thr Leu Ser Glu Asp Ile Val Asp Gly
435 440 445
His Glu Arg Ala Met Leu Lys Arg Leu Leu Arg Ile Asp Ser Lys Cys
450 455 460
Leu His Phe Thr Pro Lys Asp Trp Ser Lys Arg Gly Lys Trp Ala Ser
465 470 475 480
<210> 6
<211> 791
<212> PRT
<213> Искусственная последовательность
<220>
<223> модифицированный белок KAT6A
<400> 6
Met Asn Lys Glu Ile Leu Ala Val Val Glu Ala Val Ser Asn Glu Lys
1 5 10 15
Ala Leu Pro Arg Glu Lys Ile Phe Glu Ala Leu Glu Ser Ala Leu Ala
20 25 30
Thr Ala Thr Lys Lys Lys Tyr Glu Gln Glu Ile Asp Val Arg Val Gln
35 40 45
Ile Asp Arg Lys Ser Gly Asp Phe Asp Thr Phe Arg Arg Trp Leu Val
50 55 60
Val Asp Glu Val Thr Gln Pro Thr Lys Glu Ile Thr Leu Glu Ala Ala
65 70 75 80
Arg Tyr Glu Asp Glu Ser Leu Asn Leu Gly Asp Tyr Val Glu Asp Gln
85 90 95
Ile Glu Ser Val Thr Phe Asp Arg Ile Thr Thr Gln Thr Ala Lys Gln
100 105 110
Val Ile Val Gln Lys Val Arg Glu Ala Glu Arg Ala Met Val Val Asp
115 120 125
Gln Phe Arg Glu His Glu Gly Glu Ile Ile Thr Gly Val Val Lys Lys
130 135 140
Val Asn Arg Asp Asn Ile Ser Leu Asp Leu Gly Asn Asn Ala Glu Ala
145 150 155 160
Val Ile Leu Arg Glu Asp Met Leu Pro Arg Glu Asn Phe Arg Pro Gly
165 170 175
Asp Arg Val Arg Gly Val Leu Tyr Ser Val Arg Pro Glu Ala Arg Gly
180 185 190
Ala Gln Leu Phe Val Thr Arg Ser Lys Pro Glu Met Leu Ile Glu Leu
195 200 205
Phe Arg Ile Glu Val Pro Glu Ile Gly Glu Glu Val Ile Glu Ile Lys
210 215 220
Ala Ala Ala Arg Asp Pro Gly Ser Arg Ala Lys Ile Ala Val Lys Thr
225 230 235 240
Asn Asp Lys Arg Ile Asp Pro Val Gly Ala Cys Val Gly Met Arg Gly
245 250 255
Ala Arg Val Gln Ala Val Ser Thr Glu Leu Gly Gly Glu Arg Ile Asp
260 265 270
Ile Val Leu Trp Asp Asp Asn Pro Ala Gln Phe Val Ile Asn Ala Met
275 280 285
Ala Pro Ala Asp Val Ala Ser Ile Val Val Asp Glu Asp Lys His Thr
290 295 300
Met Asp Ile Ala Val Glu Ala Gly Asn Leu Ala Gln Ala Ile Gly Arg
305 310 315 320
Asn Gly Gln Asn Val Arg Leu Ala Ser Gln Leu Ser Gly Trp Glu Leu
325 330 335
Asn Val Met Thr Val Asp Asp Leu Gln Ala Lys His Gln Ala Glu Ala
340 345 350
His Ala Ala Ile Asp Thr Phe Thr Lys Tyr Leu Asp Ile Asp Glu Asp
355 360 365
Phe Ala Thr Val Leu Val Glu Glu Gly Phe Ser Thr Leu Glu Glu Leu
370 375 380
Ala Tyr Val Pro Met Lys Glu Leu Leu Glu Ile Glu Gly Leu Asp Glu
385 390 395 400
Pro Thr Val Glu Ala Leu Arg Glu Arg Ala Lys Asn Ala Leu Ala Thr
405 410 415
Ile Ala Gln Ala Gln Glu Glu Ser Leu Gly Asp Asn Lys Pro Ala Asp
420 425 430
Asp Leu Leu Asn Leu Glu Gly Val Asp Arg Asp Leu Ala Phe Lys Leu
435 440 445
Ala Ala Arg Gly Val Cys Thr Leu Glu Asp Leu Ala Glu Gln Gly Ile
450 455 460
Asp Asp Leu Ala Asp Ile Glu Gly Leu Thr Asp Glu Lys Ala Gly Ala
465 470 475 480
Leu Ile Met Ala Ala Arg Asn Ile Cys Trp Phe Gly Asp Glu Ala Thr
485 490 495
Ser Gly Ser Gly His His His His His His Ser Ala Gly Glu Asn Leu
500 505 510
Tyr Phe Gln Gly Ala Met Gly Arg Cys Pro Ser Val Ile Glu Phe Gly
515 520 525
Lys Tyr Glu Ile His Thr Trp Tyr Ser Ser Pro Tyr Pro Gln Glu Tyr
530 535 540
Ser Arg Leu Pro Lys Leu Tyr Leu Cys Glu Phe Cys Leu Lys Tyr Met
545 550 555 560
Lys Ser Arg Thr Ile Leu Gln Gln His Met Lys Lys Cys Gly Trp Phe
565 570 575
His Pro Pro Val Asn Glu Ile Tyr Arg Lys Asn Asn Ile Ser Val Phe
580 585 590
Glu Val Asp Gly Asn Val Ser Thr Ile Tyr Cys Gln Asn Leu Cys Leu
595 600 605
Leu Ala Lys Leu Phe Leu Asp His Lys Thr Leu Tyr Tyr Asp Val Glu
610 615 620
Pro Phe Leu Phe Tyr Val Leu Thr Gln Asn Asp Val Lys Gly Cys His
625 630 635 640
Leu Val Gly Tyr Phe Ser Lys Glu Lys His Cys Gln Gln Lys Tyr Asn
645 650 655
Val Ser Cys Ile Met Ile Leu Pro Gln Tyr Gln Arg Lys Gly Tyr Gly
660 665 670
Arg Phe Leu Ile Asp Phe Ser Tyr Leu Leu Ser Lys Arg Glu Gly Gln
675 680 685
Ala Gly Ser Pro Glu Lys Pro Leu Ser Asp Leu Gly Arg Leu Ser Tyr
690 695 700
Met Ala Tyr Trp Lys Ser Val Ile Leu Glu Cys Leu Tyr His Gln Asn
705 710 715 720
Asp Lys Gln Ile Ser Ile Lys Lys Leu Ser Lys Leu Thr Gly Ile Cys
725 730 735
Pro Gln Asp Ile Thr Ser Thr Leu His His Leu Arg Met Leu Asp Phe
740 745 750
Arg Ser Asp Gln Phe Val Ile Ile Arg Arg Glu Lys Leu Ile Gln Asp
755 760 765
His Met Ala Lys Leu Gln Leu Asn Leu Arg Pro Val Asp Val Asp Pro
770 775 780
Glu Cys Leu Arg Trp Thr Pro
785 790
<210> 7
<211> 307
<212> PRT
<213> Искусственная последовательность
<220>
<223> модифицированный белок KAT7
<400> 7
Met Gly His His His His His His Gly Thr Glu Asn Leu Tyr Phe Gln
1 5 10 15
Gly Ser Arg Leu Gln Gly Gln Ile Thr Glu Gly Ser Asn Met Ile Lys
20 25 30
Thr Ile Ala Phe Gly Arg Tyr Glu Leu Asp Thr Trp Tyr His Ser Pro
35 40 45
Tyr Pro Glu Glu Tyr Ala Arg Leu Gly Arg Leu Tyr Met Cys Glu Phe
50 55 60
Cys Leu Lys Tyr Met Lys Ser Gln Thr Ile Leu Arg Arg His Met Ala
65 70 75 80
Lys Cys Val Trp Lys His Pro Pro Gly Asp Glu Ile Tyr Arg Lys Gly
85 90 95
Ser Ile Ser Val Phe Glu Val Asp Gly Lys Lys Asn Lys Ile Tyr Cys
100 105 110
Gln Asn Leu Cys Leu Leu Ala Lys Leu Phe Leu Asp His Lys Thr Leu
115 120 125
Tyr Tyr Asp Val Glu Pro Phe Leu Phe Tyr Val Met Thr Glu Ala Asp
130 135 140
Asn Thr Gly Cys His Leu Ile Gly Tyr Phe Ser Lys Glu Lys Asn Ser
145 150 155 160
Phe Leu Asn Tyr Asn Val Ser Cys Ile Leu Thr Met Pro Gln Tyr Met
165 170 175
Arg Gln Gly Tyr Gly Lys Met Leu Ile Asp Phe Ser Tyr Leu Leu Ser
180 185 190
Lys Val Glu Glu Lys Val Gly Ser Pro Glu Arg Pro Leu Ser Asp Leu
195 200 205
Gly Leu Ile Ser Tyr Arg Ser Tyr Trp Lys Glu Val Leu Leu Arg Tyr
210 215 220
Leu His Asn Phe Gln Gly Lys Glu Ile Ser Ile Lys Glu Ile Ser Gln
225 230 235 240
Glu Thr Ala Val Asn Pro Val Asp Ile Val Ser Thr Leu Gln Ala Leu
245 250 255
Gln Met Leu Lys Tyr Trp Lys Gly Lys His Leu Val Leu Lys Arg Gln
260 265 270
Asp Leu Ile Asp Glu Trp Ile Ala Lys Glu Ala Lys Arg Ser Asn Ser
275 280 285
Asn Lys Thr Met Asp Pro Ser Cys Leu Lys Trp Thr Pro Pro Lys Gly
290 295 300
Thr Ala Ser
305
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| N1/N2-ЛАКТАМНЫЕ ИНГИБИТОРЫ АЦЕТИЛ-КоА-КАРБОКСИЛАЗ | 2011 |
|
RU2540337C2 |
| СПОСОБЫ И КОМБИНАЦИИ ИНГИБИТОРОВ KAT6 ДЛЯ ЛЕЧЕНИЯ РАКА | 2021 |
|
RU2833058C1 |
| СОЕДИНЕНИЯ | 2019 |
|
RU2838187C2 |
| ДИГИДРОНАФТИРИДИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ, ПОДХОДЯЩИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2804468C2 |
| СОЕДИНЕНИЯ | 2016 |
|
RU2725616C2 |
| ПРОИЗВОДНЫЕ 3-(5-ГИДРОКСИ-1-ОКСОИЗОИНДОЛИН-2-ИЛ) ПИПЕРИДИН-2,6-ДИОНА И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С БЕЛКОМ С "ЦИНКОВЫМИ ПАЛЬЦАМИ" 2 (IKZF2) СЕМЕЙСТВА IKAROS | 2019 |
|
RU2797559C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОПИРИДИНИЛ-АМИНОПИРИДИНОВЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ РАКА | 2010 |
|
RU2619463C2 |
| ИНГИБИТОРЫ БЕЛКОВЫХ ТИРОЗИНФОСФАТАЗ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2833220C2 |
| КОНЪЮГАТ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО, НАЦЕЛИВАЮЩИЙСЯ НА TROP2, И СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2822663C1 |
| ИНГИБИТОРЫ ПРОТЕИН-ТИРОЗИНФОСФАТАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2799446C2 |
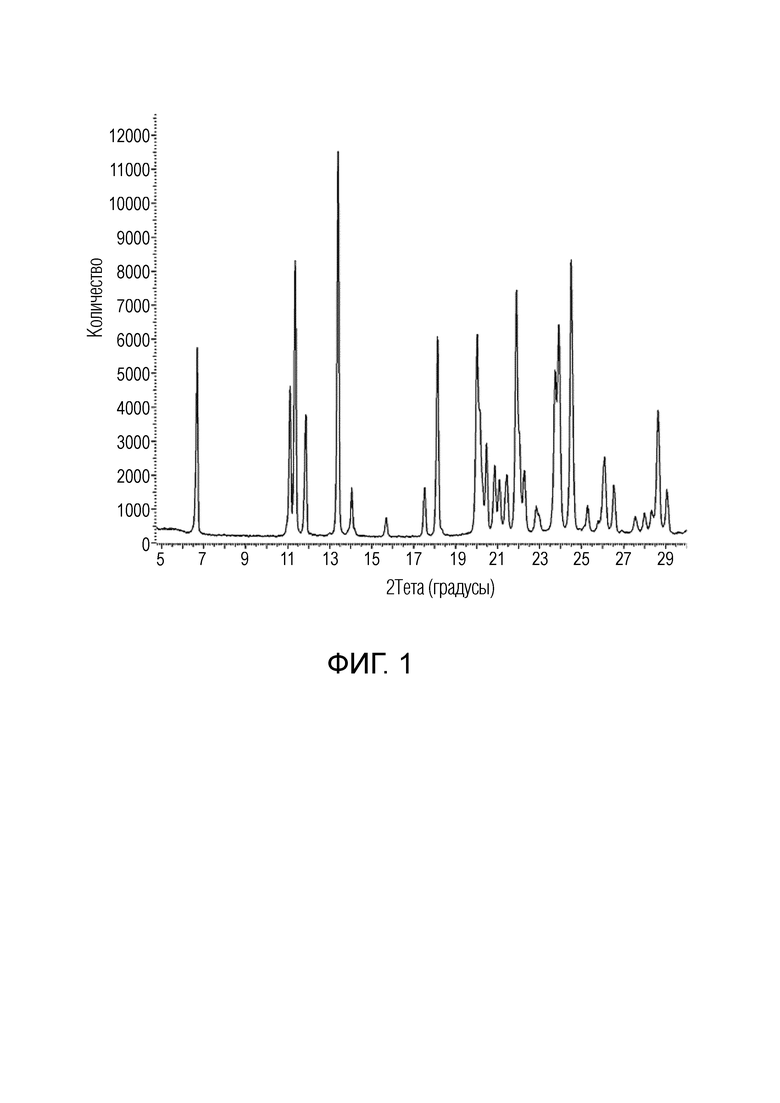
Изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, где кольцо A, R1-R8 и n определены в формуле изобретения. Новые производные бензизоксазолсульфонамида могут быть использованы для лечения аномального роста клеток, такого как рак, у пациентов. Также предложены комбинации, содержащие соединения, для лечения рака у пациента и способ лечения рака. 5 н. и 25 з.п. ф-лы, 1 ил., 26 табл., 128 пр.
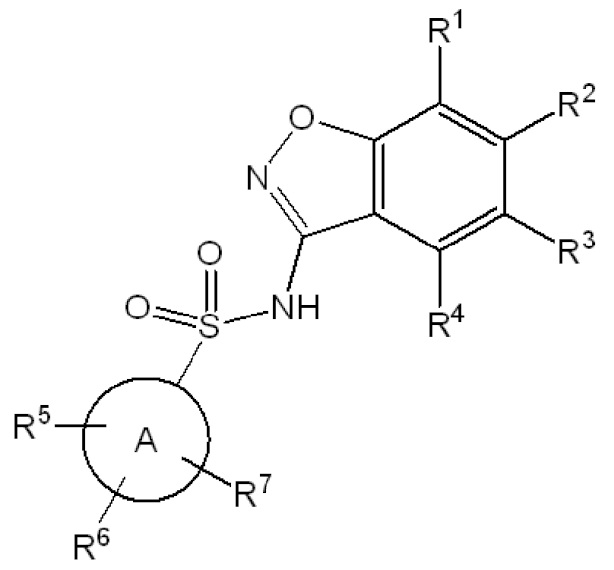
1. Соединение формулы (I)
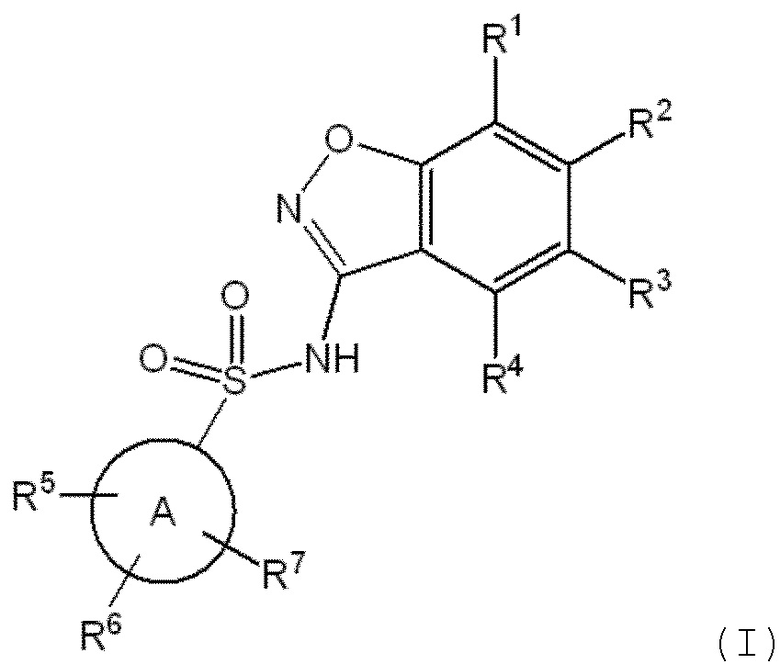
или его фармацевтически приемлемая соль, где
R1 представляет собой водород;
R2 представляет собой -(CHR8)-(5-9-членный гетероарил), необязательно замещенный галогеном, C1-C3 алкилом, -СН2ОН или -ОН,
R3 представляет собой водород, галоген, C1-C3 алкил, циклопропил, С1-С4 алкокси или -OCHF2;
R4 представляет собой водород, галоген, С1-С3 алкил, циклопропил, С1-С4 алкокси или -О-циклопропил;
кольцо А представляет собой С6-С10 арил или 9-10-членный гетероарил;
R5 представляет собой водород, фтор, циано, С1-С3 алкил, CHF2, -CF3, циклопропил, С1-С3 алкокси, -OCHF2, -О-циклопропил, -СН2-O-СН3, -С(O)ОСН3 или -С(О)N(Н)СН3;
R6 представляет собой водород, фтор, метил, -ОН или метокси;
R7 представляет собой водород, бром, хлор, фтор или метокси; и
R8 представляет собой водород или -ОН.
2. Соединение или соль по п. 1, имеющее формулу (II) 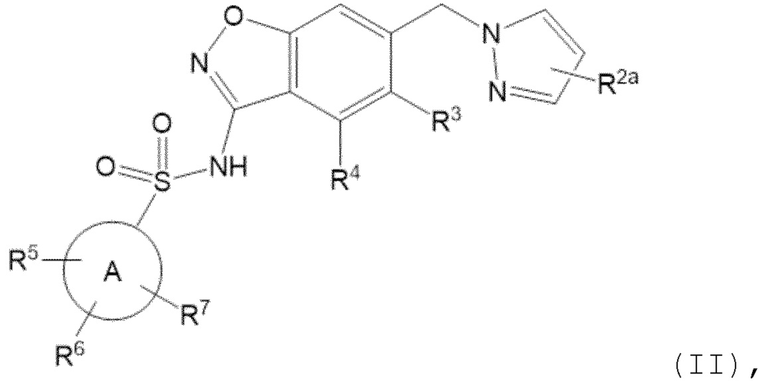
где R2a отсутствует или представляет собой галоген, C1-C3 алкил, -СН2ОН или -ОН;
R3 представляет собой водород, галоген, C1-C3 алкил, циклопропил, С1-С4 алкокси или -OCHF2;
R4 представляет собой водород, галоген, С1-С3 алкил, циклопропил, С1-С4 алкокси или -О-циклопропил;
кольцо А представляет собой С6-С10 арил или 9-10-членный гетероарил;
R5 представляет собой водород, фтор, циано, C1-C3 алкил, CHF2, -CF3, циклопропил, C1-C3 алкокси, -OCHF2, -О-циклопропил, -СН2-O-СН3, -С(O)ОСН3 или -С(О)N(Н)СН3;
R6 представляет собой водород, фтор, метил, -ОН или метокси; и
R7 представляет собой водород, бром, хлор, фтор или метокси.
3. Соединение или соль по п. 1 или 2, где кольцо А представляет собой фенил, хинолинил, бензоксазолил, инданил или тетрагидронафтил.
4. Соединение или соль по п.3, где кольцо А представляет собой фенил.
5. Соединение или соль по п. 1 или 2, имеющее формулу (III)
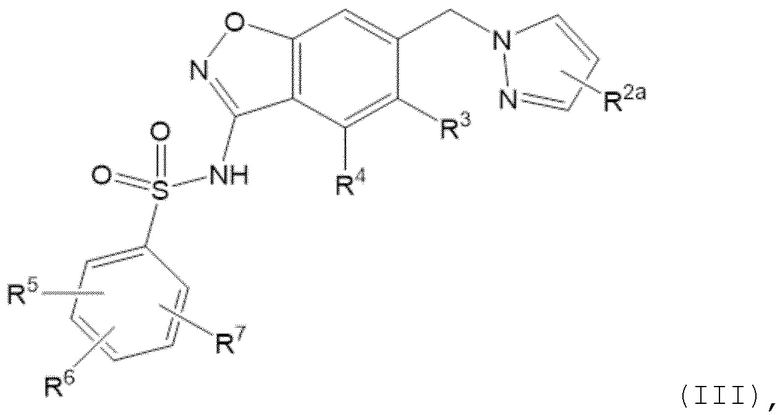
где R2a отсутствует или представляет собой галоген, C1-C3 алкил, -СН2ОН или -ОН;
R3 представляет собой водород, галоген, C1-C3 алкил, циклопропил, С1-С4 алкокси или -OCHF2;
R4 представляет собой водород, галоген, C1-C3 алкил, циклопропил, С1-С4 алкокси или -О-циклопропил;
R5 представляет собой водород, фтор, циано, C1-C3 алкил, CHF2, -CF3, циклопропил, C1-C3 алкокси, -OCHF2, -О-циклопропил, -СН2-O-СН3, -С(O)ОСН3 или -С(О)N(Н)СН3;
R6 представляет собой водород, фтор, метил, -ОН или метокси; и
R7 представляет собой водород, бром, хлор, фтор или метокси.
6. Соединение или соль по п. 1 или 2, имеющее формулу (IV)
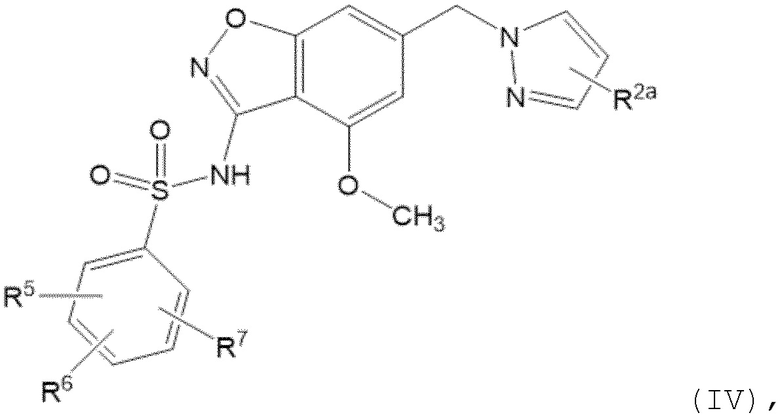
где R2a отсутствует или представляет собой галоген, C1-C3 алкил, -СН2ОН или -ОН;
R5 представляет собой водород, фтор, циано, C1-C3 алкил, CHF2, -CF3, циклопропил, С2-С3 алкокси, -OCHF2, -О-циклопропил, -СН2-O-СН3, -С(O)ОСН3 или -С(О)N(Н)СН3;
R6 представляет собой водород, фтор, метил, -ОН или метокси; и
R7 представляет собой водород, бром, хлор, фтор или метокси.
7. Соединение или соль по любому из пп. 1-6, где R5 представляет собой метокси, R6 представляет собой метокси и R7 представляет собой водород.
8. Соединение или соль по любому из пп. 1-6, где R5 представляет собой метокси, R6 представляет собой водород и R7 представляет собой водород.
9. Соединение или соль по любому из пп. 1, 2 или 5, имеющее формулу (VI)
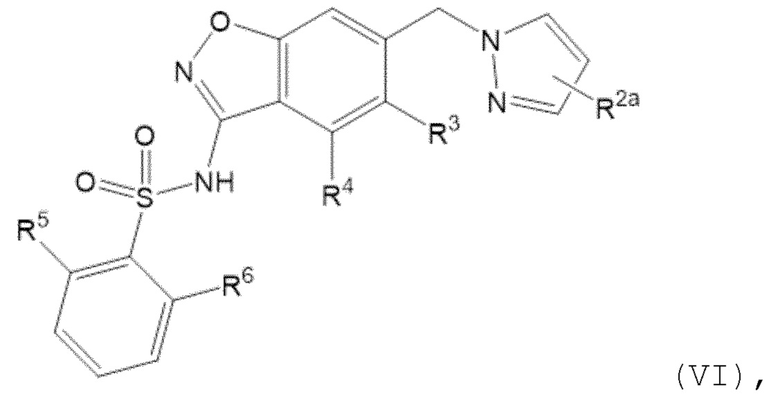
где R2a отсутствует или представляет собой галоген, C1-C3 алкил, -СН2ОН или -ОН;
R3 представляет собой водород, галоген или C1-C3 алкил;
R4 представляет собой водород, галоген, С1-С3 алкил, циклопропил, С1-С4 алкокси или -О-циклопропил,
при условии, что по меньшей мере один из R3 и R4 представляет собой водород;
R5 представляет собой водород, метил, -СН2-ОСН3, -CF3, C1-C3 алкокси или -С(O)ОСН3; и
R6 представляет собой водород, фтор, метил, -ОН или метокси.
10. Соединение или соль по любому из пп. 2-9, где R2a отсутствует или представляет собой фтор, метил, -CH2OH или -ОН.
11. Соединение или соль по любому из пп. 2-9, где R2a отсутствует.
12. Соединение или соль по любому из пп. 1-5 или 9-11, где R3 представляет собой водород, галоген или C1-C3 алкил.
13. Соединение или соль по любому из пп. 1-5 или 9-11, где R3 представляет собой водород, фтор, бром или метил.
14. Соединение или соль по любому из пп. 1-5 или 9-11, где R4 представляет собой водород, фтор, метил, этил, циклопропил, -O-циклопропил или С1-С4 алкокси.
15. Соединение или соль по любому из пп. 1-5 или 9-11, где R4 представляет собой водород.
16. Соединение или соль по любому из пп. 1-5 или 9-11, где R4 представляет собой C1-C3 алкокси.
17. Соединение или соль по любому из пп. 1-5 или 9-11, где R4 представляет собой метокси.
18. Соединение или соль по любому из пп. 1-5 или 9-11, где по меньшей мере один из R3 и R4 представляет собой водород.
19. Соединение или соль по любому из пп. 9-18, где R5 представляет собой метокси и R6 представляет собой метокси.
20. Соединение или соль по любому из пп. 9-18, где R5 представляет собой метокси и R6 представляет собой водород.
21. Соединение по п. 1, которое представляет собой
или его фармацевтически приемлемая соль.
22. Соединение по п. 1, которое представляет собой
или его фармацевтически приемлемая соль.
23. Соединение по п. 1, которое представляет собой
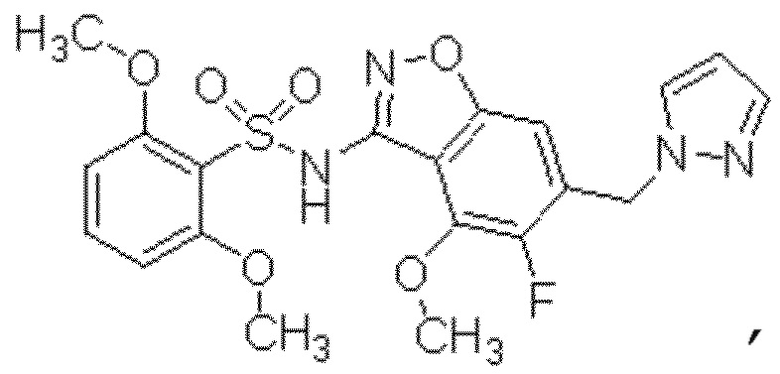
или его фармацевтически приемлемая соль.
24. Соединение по п. 1, которое представляет собой 2-метокси-N-{4-метокси-6-[(1Н-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамид.
25. Способ лечения рака у пациента, включающий введение пациенту такого количества соединения по любому из пп. 1-24 или его фармацевтически приемлемой соли, которое является эффективным при лечении рака молочной железы.
26. Способ по п. 25, где рак представляет собой ER положительный рак молочной железы.
27. Комбинация соединения по любому из пп. 1-24 или его фармацевтически приемлемой соли с противоопухолевым средством или с лучевой терапией для лечения рака молочной железы.
28. Комбинация соединения по любому из пп. 1-24 или его фармацевтически приемлемая соль с противоопухолевым средством для лечения рака молочной железы.
29. Комбинация по п. 28, где рак молочной железы представляет собой ER положительный рак молочной железы.
30. Кристаллическая форма безводного 2-метокси-N-{4-метокси-6-[(1H-пиразол-1-ил)метил]-1,2-бензоксазол-3-ил}бензол-1-сульфонамида, имеющая порошковую рентгеновскую дифрактограмму, содержащую пики при значениях 2θ: 13,4 и 18,1 °2θ ± 0,2 °2θ.
| WO 2018102419 A1, 07.06.2018 | |||
| RU 2015143834 A, 24.04.2017 | |||
| US 2007015787 A1, 18.01.2007. |
