Изобретение относится к медицине, а именно к фармацевтической химии и медицинской биохимии. Аминокислотный анализ занимает ключевое место при исследовании объектов белковой или пептидной природы. Методы аминокислотного анализа востребованы для объектов животного, растительного, микробного происхождения. Широта исследуемых объектов, их матричный эффект, определяют значимость и сложность аналитических подходов при пробоподготовке и собственно анализе аминокислот в объектах природного происхождения и фармацевтических препаратах, по критериям: воспроизводимость, точность и чувствительность [1]. Используемые варианты кислотного гидролиза подразумевают длительное термическое воздействие на образец в условиях 6 М хлористоводородной кислоты. При этом для каждого образца требуется подбирать оптимальные временные параметры. Так, для оценки степени гидролиза рекомендуется выполнять гидролиз в течение 24, 48, 72 часов при температуре 110°С [2, 3]. Условия гидролиза критически важны, поскольку они всегда представляют собой компромисс между сохранением наиболее чувствительных аминокислот (в частности цистина, серила, треонина, и тирозина) и расщеплением наиболее стабильных пептидных связей с участием таких аминокислот, как изолейцин, лейцин и валин [4].
В отечественной медицинской и фармацевтической практике востребованы препараты, представляющие собой белковые, пептидные вещества, а также их комбинацию с иными структурами. Все это обуславливает необходимость разработки воспроизводимых экспрессных методов, позволяющих осуществить пробоподготовку образца в воспроизводимых условиях. Одним из вариантов решения является применение микроволнового излучения.
Микроволновый нагрев обладает определенными преимуществами в сравнении с обычным вариантом кислотного гидролиза и нагреванием. Так, при микроволновом излучении передача энергии происходит за счет поляризации, а не столкновения молекул. Величина эффекта поляризации зависит от дипольного характера участвующих молекул и обладает одним из наиболее высоких показателей для молекул воды. Гидролиз, происходящий в водно-кислотной среде, обуславливает мгновенное поглощение энергии излучения и приводит к сокращению общего времени гидролиза уменьшая время пробоподготовки от нескольких десятков часов (от 24 до 72-80), необходимых для обычного гидролиза, до нескольких минут. Поэтому образцы белка могут быть подвергнуты гидролизу и проанализированы в течение нескольких часов, одних суток, что представляет собой основное преимущество.
Из литературы известны варианты использования сверхвысокочастотного (СВЧ) излучения для осуществления кислотного гидролиза. Так, в условиях опубликованного исследования [5], для проведения гидролиза белков проводили следующую процедуру. Около 40 мкл 0,25 мг/мл раствора белка смешивали с 0,5 мкл 500 мМ дитиотриетола (ДТТ) и 40 мкл 6 М HCl в полипропиленовом флаконе. Флакон закрывали и облучали в бытовой микроволновой печи (Panasonic, London Drugs, Эдмонтон) мощностью 1200 Вт при частоте 2450 МГц. Контейнер со 100 мл воды также помещали в микроволновую печь для поглощения избыточной микроволновой энергии. Образец подвергали 60-секундному микроволновому воздействию, затем охлаждали и сушили в вакуумной центрифуге SpeedVac для удаления всей кислоты. Для восстановления образца во флакон добавляли 30 мкл 250 мМ NH4HCO3. Затем добавляли 1 мкл 500 мМ ДТТ и проводили восстановление дисульфидом путем инкубации в течение 60 мин при 37°С. Однако в данном варианте используется полипропиленовый сосуд, который при локальных высоких температурах может деформироваться, загрязняя образец продуктами температурных изменений пластика. Вместе с этим, указанные временные параметры для обработки не всегда приводят к полному гидролизу образца, а сам вариант не предназначен для получения гидролизата из образцов с большими массами навески.
Близким к заявляемому является способ определения аминогликозидных антибиотиков методом обращено-фазовой высокоэффективной жидкостной хроматографии [6], включающий изократический режим элюирования с использованием хроматографической колонки с применением ультрафиолетового детектора и объеме вводимой пробы 10 мкл, при этом заранее проводят дериватизацию гентамицина, к 100 мкл образца добавляют 25 мкл раствора карбоната натрия (5% в воде) и 25 мкл раствора фенилизотиоцианата (5% в ацетонитриле), смесь интенсивно перемешивают на вортексе в течение 30 секунд и нагревают в течение 15 минут при 50°С в твердотельном термостате, затем реакционную смесь разбавляют 850 мкл смеси ацетонитрил/20 мМ ацетат натрия, рН 4,70 в соотношении 20:80, длина волны ультрафиолетового детектора 250 нм. В данном способе для дериватизации с фенилизотиоцианатом используется раствор в ацетонитриле. Однако недостатком метода является высокая селективность, ограниченная преимущественно определением аминогликозидов. При этом ацетонитрил, как компонент растворителя ключевого реактива, обладает рядом недостатков. Так, его заявленная концентрация относительно мала для обеспечения межмолекулярного взаимодействия липофильного фенилизотиоцианата с аминокислотами.
Рассматривая ацетонитрил как апротоный растворитель, следует учесть его недостаток - низкую устойчивость к кислотам с возможностью образования побочных продуктов. Все эти факторы снижают привлекательность ацетонитрила как растворителя для фенилизотиоцианата.
Из уровня техники [7] известно, что гидролиз осуществляют в среде 6 М хлористоводородной кислоты в течение 17 часов. Для проведения гидролиза в виалы, снабженные плотно завинчивающимися крышками, помещали 1 мл солянокислого экстракта предстательной (поджелудочной) железы или 100 мг гомогенизированного комбикорма (мяса). Затем добавляли 10 мл 6 М раствора соляной кислоты. Смесь тщательно перемешивали и обдували током азота в течение 2 мин. Виалы плотно закрывали крышками и помещали в термостат. Гидролиз проводили при температуре 110°С в течение 17 ч, а фенилизотиоцианат растворяется в изопропиловом спирте. Однако, известный способ не рассматривает особенностей проведения гидролиза для различных типов образцов, (корректировка параметров гидролиза по времени, температуре), а применение дериватизирующего реактива не учитывает наличия липофильных веществ (в том числе, собственно липидов) анализируемых образцов, что затрудняет воспроизводимость метода при индивидуальных особенностях образца.
Недостатками известных способов являются ограниченные возможности по универсальности метода, позволяющего анализировать образцы различных морфологических образований, а также белковые образования микроорганизмов, растительные объекты. Сопутствующие компоненты образцов способны влиять на конечный результат, тем самым снижая воспроизводимость метода за счет «матричного» эффекта образца. Подбор условий для преодоления индивидуальных особенностей образца формирует длительность исследования затягивая получение конечного результата.
Задачей изобретения является повышение чувствительности дериватизирующего реактива при упрощении пробоподготовки.
Технический результат достигается с помощью способа пробоподготовки образцов для аминокислотного анализа в условиях обращено-фазовой высокоэффективной жидкостной хроматографии, в котором кислотный гидролиз образца белковой или пептидной природы проводят при 6 М растворе хлористоводородной кислоты в условиях микроволнового облучения СВЧ-диапазона с мощностью микроволнового излучения 1800 Вт при частоте 2450 МГц в температурном интервале 150-160°С при давлении 2,8-3,5 атм с временем экспозиции в пределах 25-55 мин. Полученный гидролизат высушивают в условиях, исключающих контакт с кислородом воздуха. Защелачивают полученный гидролизат натрия гидроксидом 1 М до рН=7,5-7,8 и проводят дериватизацию с применением раствора фенилизотиоцианата, приготовленного в смеси изопропиловый спирт-диметилсульфоксид, взятых в объемном соотношении 1:1.
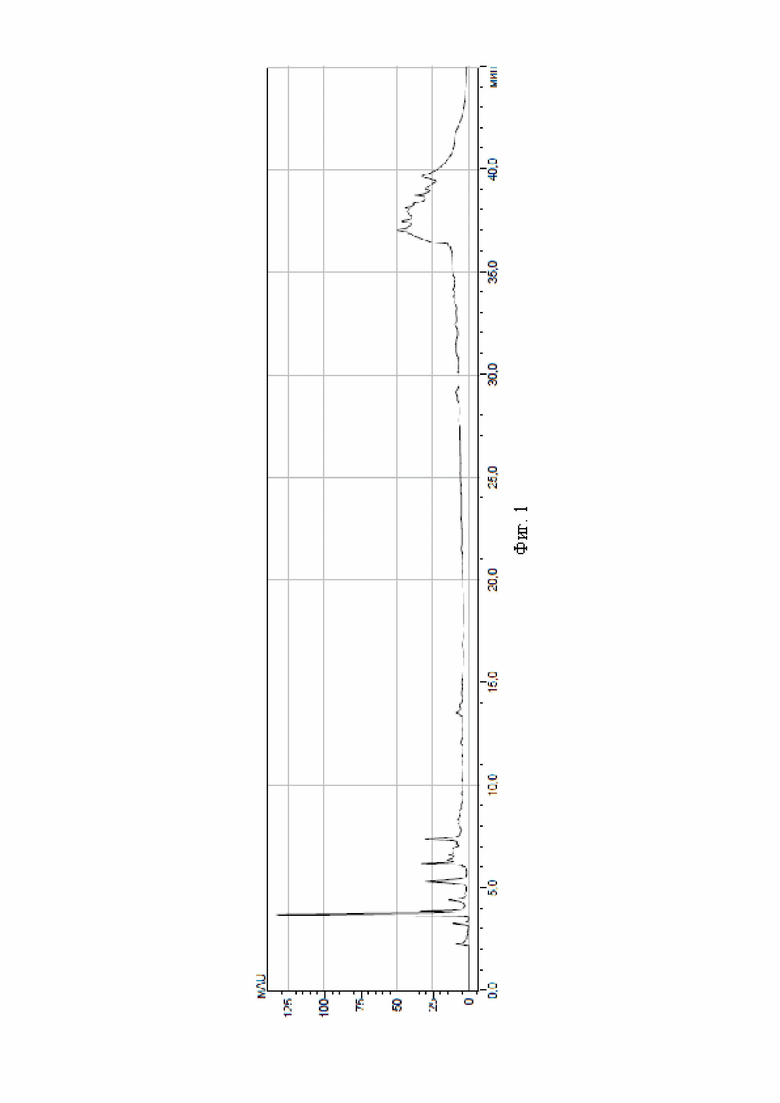
На фиг. 1 приведена хроматограмма аминокислот мышечной ткани (образец 1 ВФ) в условиях обращенно-фазной высокоэффективной жидкостной хроматографии после дериватизации фенилизотиоцианатом в изопропиловом спирте.
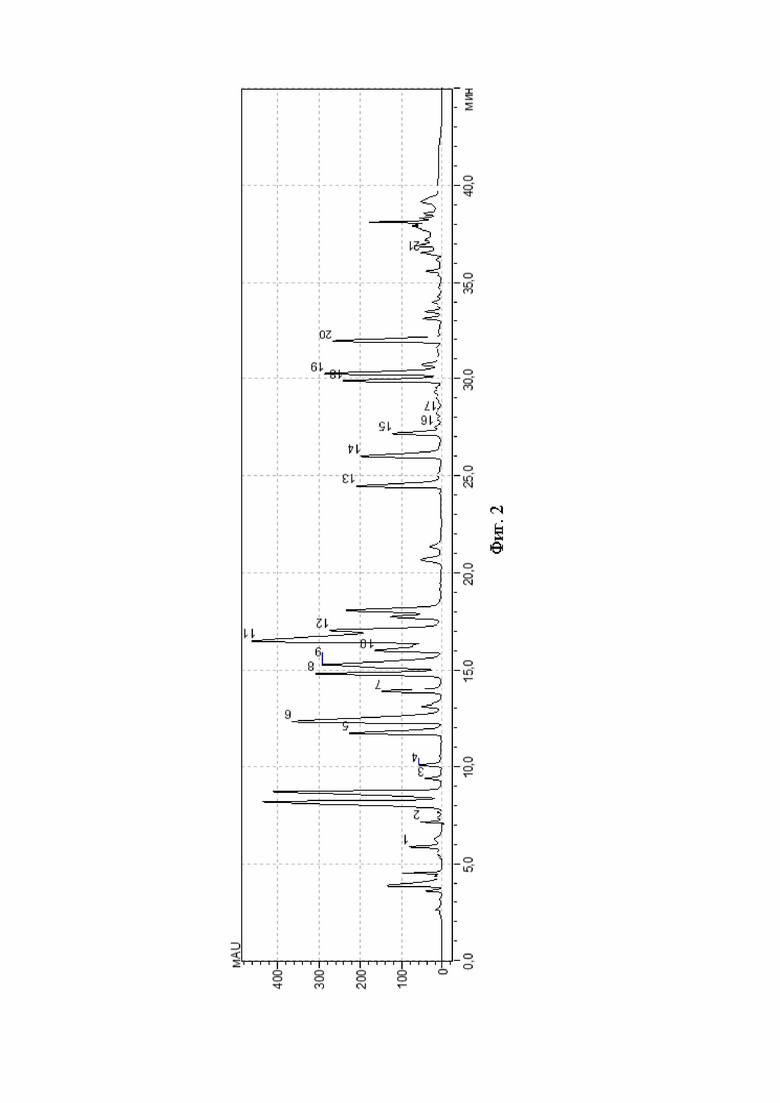
На фиг. 2 показана хроматограмма аминокислот мышечной ткани (образец 1 ВФ) в условиях обращенно-фазной высокоэффективной жидкостной хроматографии после дериватизации фенилизотиоцианатом в растворе изопропиловый спирт-диметилсульфоксид в объемном соотношении 1:1. Детекция 254 нм. Подписи пиков: Пик 1 - Асп, пик 2 - Глу, пик 3 - о-Про, пик 4 - Сер, пик 5 - Гли, пик 6 - Гис, пик 7 - Арг, пик 8 - Трп, пик 9 - Тре, пик 10 - Ала, пик 11, 12 - Про, пик 13 - Тир, пик 14 - Мет, пик 15 - Вал, пик 16 - Цис, пик 17 - Цис, пик 18 - Иле, пик 19 - Лей, пик 20 - Фен, пик 21 - Лиз.
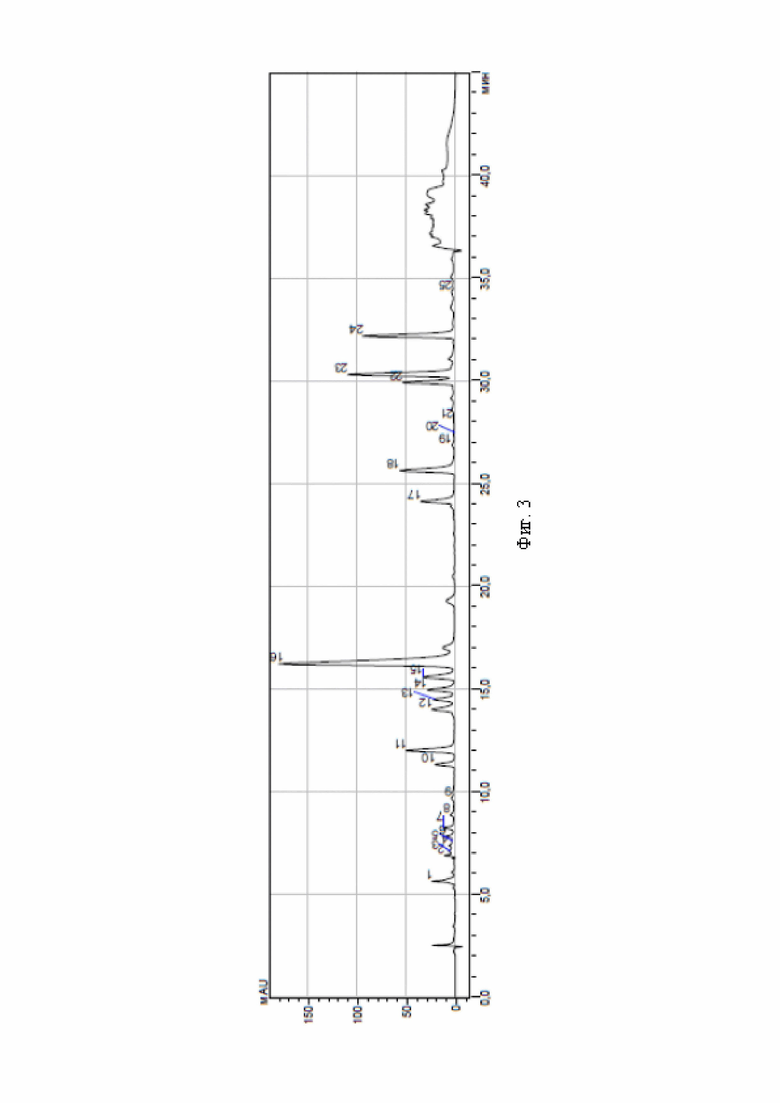
На фиг. 3 приведена хроматограмма аминокислот опытного образца апротинина в условиях обращенно-фазной высокоэффективной жидкостной хроматографии после дериватизации фенилизотиоцианатом в растворе изопропиловый спирт-диметилсульфоксид в объемном соотношении 1:1.
Примеры конкретного выполнения способа пробоподготовки образцов для аминокислотного анализа в условиях обращенно-фазовой высокоэффективной жидкостной хроматографии
Пример 1. Образец мышечной ткани массой 500,0 мг подвергли кислотному гидролизу. Для этого, точную навеску поместили в кислотоустойчивый контейнер, добавили 8 мл хлористоводородной кислоты 6 М и подвергли микроволновому облучению при 150°С в течение 55 мин с мощностью микроволнового излучения 1800 Вт при частоте 2450 МГц и давлении 3,2+0,2 атм.
Полученный гидролизат высушили в вакууме.
К высушенному гидролизату добавили раствор натрия гидроксида 1 М до рН 7,5 и тщательно перемешали.
Затем добавили 0,5 мл раствора фенилизотиоционата. Дериватизацию проводили в течение 30 мин при температуре 25°С, оставив пробу в темном месте.
Раствора фенилизотиоционата приготовили заранее растворением 1,6 мл фенилизотиоцианата в 100 мл смеси изопропиловый спирт-диметилсульфоксид в объемном соотношении 1:1.
После дериватизации смесь упарили, а полученный остаток растворили в 1 мл дистиллированной воды, отфильтровали через мембранный фильтр с диаметром пор 0,45 мкм и ввели в хроматограф для анализа.
Условия выполнения хроматографического анализа были следующими. Разделение аминокислотных дериватов проводили на хроматографе Shimadzu LC-20 Prominence с диодно-матричным детектором Shimadzu SPD20MA (Shimadzu, Япония). Хроматографическая колонка Kromasil 5 мкм С18, 110 А, 250×4,6 мм, объем инжекции 20 мкл. Температура термостата колонки 40°С. Условия подачи элюента 1,2 мл/мин в градиентном режиме, подвижная фаза представляла смесь 6,0 мМ раствора ацетата натрия с рН 5,5 (компонент А), 1% раствор изопропилового спирта в ацетонитриле (компонент В) и 6,0 мМ раствора ацетата натрия с рН 4,05 (компонент С). Программа подачи градиента согласно условиям [7].
Пример 2. Точную навеску образца фермента апротинин массой 100,0 мг поместили в кислотоустойчивый контейнер, добавилиляли 8 мл хлористоводородной кислоты 6 М, и подвергли микроволновому облучению при 160°С в течение 25 мин с мощностью микроволнового излучения 1800 Вт при частоте 2450 МГц и давлении 2,8+0,2 атм.
Полученный гидролизат высушили в вакууме.
К высушенному образцу добавили раствор натрия гидроксида 1 М до рН 7,8 и тщательно перемешали. Затем добавили 0,5 мл раствора фенилизотиоционата. Раствор приготавливали аналогично примеру 1. Дериватизацию проводили аналогично примеру 1.
После этого смесь упарили, а полученный остаток растворили в 1 мл дистиллированной воды, отфильтровали через мембранный фильтр с диаметром пор 0,45 мкм и ввели в хроматограф для анализа. Условия проведения хроматографического анализа согласно аналогично примеру 1.
Результаты хроматографического анализа по приведенным примерам, а также контрольного образца - мышечной ткани после дериватизации фенилизотиоцианатом в изопропиловом спирте - приведены на фиг. 1-3. Из хроматограмм видно, что дериватизация раствором фенилизотиоционата в смеси изопропиловый спирт-диметилсульфоксид, взятых в объемном соотношении 1:1, повышает чувствительность дериватизирующего реактива - на фиг. 2, 3 отражено и идентифицировано значительно большее количество пиков, чем для контрольного образца.
Литература:
1. Considerations for amino acid analysis by liquid chromatography-tandem mass spectrometry: A tutorial review / J.P. Violi, D.P. Bishop, M.P. Padula, J.R. Steele, K.J. Rodgers // TrAC Trends in Analytical Chemistry. 2020. Vol. 131, P. 116018 doi.org/10.1016/j.trac.2020.116018.
2. HPAEC-PAD analysis for determination of the amino acid profiles in protein fractions from oat flour combined with correction of amino acid loss during hydrolysis / R.R. Sardari, A. Jasilionis, N.T. Renhuldt, P. Adlercreutz, E.N. Karlsson // J. Cereal Science. 2023. Vol. 109, P. 103589. doi.org/10.1016/j.jcs. 2022.103589.
3. Estimation of correction factors to determine the true amino acid concentration of protein after a 24-hour hydrolysis / H. Lapierre, S. Binggeli, M. Sok, D. Pellerin, D.R. Ouellet // Dairy Science. 2019. Vol. 102, Issue 2, P. 1205-1212, doi.org/10.3168/jds. 2018-15392.
4. Correction for amino acid loss during acid hydrolysis of a purified protein / A.J. Darragh, D.J. Garrick, P.J. Moughan, W.H. Hendriks // Analytical Biochemistry. 1996. Vol. 236, Issue 2. P. 199-207. doi.org/10.1006/abio. 1996.0157.
5. Microwave-assisted acid hydrolysis of proteins combined with peptide fractionation and mass spectrometry analysis for characterizing protein terminal sequences / L. Chen, N. Wang, D. Sun, L. Li // J. Proteomics. 2014. Vol. 100 P. 68-78.
6. Способ определения аминогликозидных антибиотиков методом обращенно-фазной высокоэффективной жидкостной хроматографии Патент на изобретение Российской Федерации №2786839. Заявка: 2022129592, 15.11.2022. Опубл. 26.12.2022 Бюл. №36.
7. Методика определения важнейших аминокислот в сложных объектах биологического происхождения методом обращенно-фазовой ВЭЖХ с получением фенилтиогидантоинов аминокислот / Руденко А.О., Карцова Л.А., Снарский С.И. // Сорбционные и хроматографические процессы. 2010. Т. 10. №2. С. 223-230.
Изобретение относится к медицине. Раскрыт способ пробоподготовки образцов для аминокислотного анализа в условиях обращенно-фазовой высокоэффективной жидкостной хроматографии, включающий получение гидролизата аминокислот с использованием 6 М раствора хлористоводородной кислоты, дериватизацию фенилизотиоцианатом с использованием хроматографической колонки, где гидролиз образца белковой или пептидной природы осуществляют при микроволновом облучении при температуре 150-160°С в течение 25-55 мин с мощностью микроволнового излучения 1800 Вт при частоте 2450 МГц и давлении 2,8-3,5 атм, гидролизат высушивают в вакууме, добавляют раствор натрия гидроксида 1 М до рН=7,5-7,8, перемешивают, дериватизацию проводят в течение 30 мин при температуре 25°С раствором фенилизотиоционата в смеси изопропиловый спирт-диметилсульфоксид, взятых в объемном соотношении 1:1. Изобретение обеспечивает повышение чувствительности дериватизирующего реактива при упрощении пробоподготовки. 1 з.п. ф-лы, 3 ил., 2 пр.
1. Способ пробоподготовки образцов для аминокислотного анализа в условиях обращенно-фазовой высокоэффективной жидкостной хроматографии, включающий получение гидролизата аминокислот с использованием 6 М раствора хлористоводородной кислоты, дериватизацию фенилизотиоцианатом с использованием хроматографической колонки, отличающийся тем, что гидролиз образца белковой или пептидной природы осуществляют при микроволновом облучении при температуре 150-160°С в течение 25-55 мин с мощностью микроволнового излучения 1800 Вт при частоте 2450 МГц и давлении 2,8-3,5 атм, гидролизат высушивают в вакууме, добавляют раствор натрия гидроксида 1 М до рН=7,5-7,8, перемешивают, дериватизацию проводят в течение 30 мин при температуре 25°С раствором фенилизотиоционата в смеси изопропиловый спирт-диметилсульфоксид, взятых в объемном соотношении 1:1.
2. Способ по п.1, отличающийся тем, что раствор фенилизотиоционата готовят растворением 1,6 мл фенилизотиоцианата в 100 мл смеси изопропиловый спирт-диметилсульфоксид в объемном соотношении 1:1.
| CHEN L | |||
| et al | |||
| Microwave-assisted acid hydrolysis of proteins combined with peptide fractionation and mass spectrometry analysis for characterizing protein terminal sequences // Journal of Proteomics, 2014, V | |||
| Облицовка комнатных печей | 1918 |
|
SU100A1 |
| Способ получения смеси хлоргидратов опийных алкалоидов (пантопона) из опийных вытяжек с любым содержанием морфия | 1921 |
|
SU68A1 |
| РАЗОРЕНОВА К.Н | |||
| Аминокислотный состав надземной части Geranium pratense L., Geranium sylvaticum L., Geranium palustre L | |||

