В настоящей заявке испрашивается приоритет согласно китайской патентной заявке №201611174146.3 под названием "Способ синтеза ингибитора мультикиназ и его применение", поданной в Государственное ведомство по интеллектуальной собственности КНР 13 декабря 2016 года; китайской патентной заявке №201710426594.6 под названием "Ингибитор мультикиназ и его применение", поданной в Государственное ведомство по интеллектуальной собственности КНР 8 июня 2017 года; и китайской патентной заявке №201710593933.X под названием "Кристаллическая форма ингибитора мультикиназ и способ ее получения и ее применение", поданной в Государственное ведомство по интеллектуальной собственности КНР 20 июня 2017 года, содержание которых включено в настоящую заявку посредством ссылки во всей их полноте.
ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к области медицинской технологии, в частности относится к соединению - ингибитору мультикиназ, его кристаллической форме и его применению.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Нормальное клеточное деление необходимо для здоровья организма и выживаемости клеточных органов. Во время этого процесса внутриклеточное вещество полностью подвергается рекомбинации, и две идентичные копии хромосом распределяются в две дочерние клетки с помощью биполярного веретена. Когда в процессе митоза происходит ошибка, хромосомное число в клетке становится аномальным, что может приводить к клеточной смерти или способствовать превращению нормальных клеток в опухолевые клетки. Процесс митоза главным образом зависит от трех механизмов: 1) локализации белка; (2) протеолиза; (3) фосфорилирования. В эти процессы вовлечены некоторые серин/треонинкиназы, также известные как митотические киназы.
Киназа Aurora представляет собой одну разновидность митотических киназ и была открыта в 1995 году. Экспрессию киназы Aurora впервые наблюдали в опухолевой ткани человека в 1998 году. Сейчас она стала мишенью, представляющий интерес для противоопухолевых исследований. Семейство киназ Aurora включает три высокогомологичные киназы: Aurora A, Aurora В и Aurora С. Из них обнаружению поддаются Aurora А и Aurora В.
В настоящее время доказано, что Aurora А является онкогеном, сверхэкспрессия которого блокирует правильную сборку комплексов контрольных точек митотического цикла, приводя к генетической нестабильности и образованию опухоли. Aurora В является важной киназой, которая регулирует нормальный митоз клетки. Сверхэкспрессия Aurora В широко распространена в опухолях. Опухолевые клетки становятся более чувствительными, когда Aurora В ингибирована. Принимая во внимание ключевые роли Aurora А и Aurora В в процессе митоза клетки, исследование и разработка противоопухолевых лекарственных средств, направленных на киназу Aurora, привлекает все больше и больше внимания. Кроме того, киназы Aurora являются неэффективными в отношении непролиферирующих клеток, поскольку они экспрессируются и активируются при митозе. Поэтому ингибиторы киназ Aurora принадлежат к таргетным противоопухолевым лекарственным веществам и будут обладать большими преимуществами по сравнению с другими неспецифическими цитотоксическими лекарственными средствами.
В дополнение к связи со сверхэкспрессией митотических киназ, опухолевый рост и миграция также зависят от образования большого числа новых кровеносных сосудов, где сигнальный путь VEGF/VEGFR (фактор роста эндотелия сосудов/рецептор фактора роста эндотелия сосудов) играет ключевую роль в неоваскуляризации опухоли. Из них VEGFR представляет собой один тип тирозинкиназного трансмембранного гликопротеина, состоящего из внеклеточной части, содержащей 7 Ig(иммуноглобулин)-подобных доменов, одного трансмембранного домена и цитоплазматической тирозинкиназной структурной части. Существует три подтипа VEGFR, которые представляют собой VEGFR1, VEGFR2 и VEGFR3. Конформация VEGFR изменяется после связывания с VEGF, что приводит к димеризации рецептора, аутофосфорилированию тирозинового сайта во внутриклеточном сегменте и активации нисходящего пути сигнальной трансдукции. VEGFR2 (KDR) (рецептор, содержащий домен вставки киназы) распространен главным образом в клетках сосудистого эндотелия и гемопоэтических стволовых клетках. VEGFR2 (KDR) тесно связан с дисфункцией гемопоэтической системы перед злокачественными пролиферативными поражениями, такими как тромбоцитемия, первичная тромбоцитемия, миелофиброз (MF), хронический идиопатический миелофиброз (IMF), полицитемия (PV), предраковый миелодиспластический синдром и гемобластозы. Среди указанного, гемобластозы включают лейкоз (неходжкинскую лимфому), болезнь Ходжкина (также известную как лимфома Ходжкина) и миелому, например острый лимфобластный лейкоз (ALL), острый миелоидный лейкоз (AML), острый промиелоцитарный лейкоз (APL), хронический лимфоцитарный лейкоз (CLL), хронический миелоидный лейкоз (CML), хронический нейтрофильный лейкоз (CNL) и так далее, но не ограничены ими.
В настоящее время существуют клинические ингибиторы Aurora А и Aurora В соответственно, а также ингибиторы VEGFR. Однако нет никаких ингибиторов мультикиназ, которые являются одновременно эффективными в отношении указанных киназ. В WO 2013123840 A1 раскрыт класс производных азабензо[f]азулена, обладающих противоопухолевым действием, без какого-либо механизма их терапевтического воздействия.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложен класс соединений (ингибиторов мультикиназ), представленных формулами (I) и (II), или их фармацевтически приемлемых солей или стереоизомеров, способных ингибировать, регулировать и/или модулировать активность одной или более протеинкиназ, такой как киназа Aurora и VEGFR киназа; кристаллическая форма I соединения, представленного формулой (III); и фармацевтический препарат, и фармацевтическая композиция, содержащие вышеуказанные соединения и/или кристаллическую форму I, для применения в лечении заболеваний, опосредованных аномальностями этих киназ, в частности раковых заболеваний. Согласно изобретению также предложены способы получения вышеуказанных соединений и кристаллической формы, и способы применения соединений, кристаллической формы, фармацевтического препарата и/или фармацевтической композиции для лечения вышеописанных заболеваний у млекопитающих, в частности людей.
В вышеуказанных целях согласно настоящему изобретению прежде всего предложено соединение формулы (I) или его фармацевтически приемлемые соли или стереоизомеры:
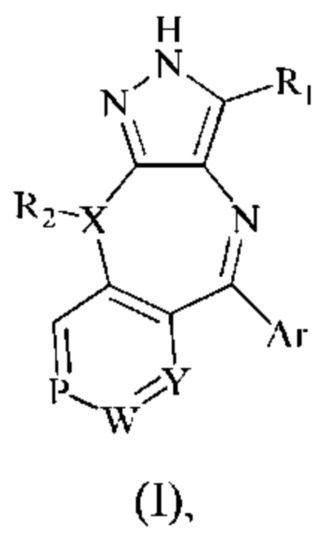
где
X выбран из СН или N;
R1 выбран из группы, состоящей из водорода, C1-6алкила, С3-6циклоалкила, C1-6алкокси, галогенированного C1-6алкила или галогенированного С1-6алкокси;
R2 выбран из группы, состоящей из водорода, С1-6алкила или С3-6циклоалкила;
Y выбран из CR3 или N;
Р выбран из CR4 или N;
W выбран из CR5 или N;
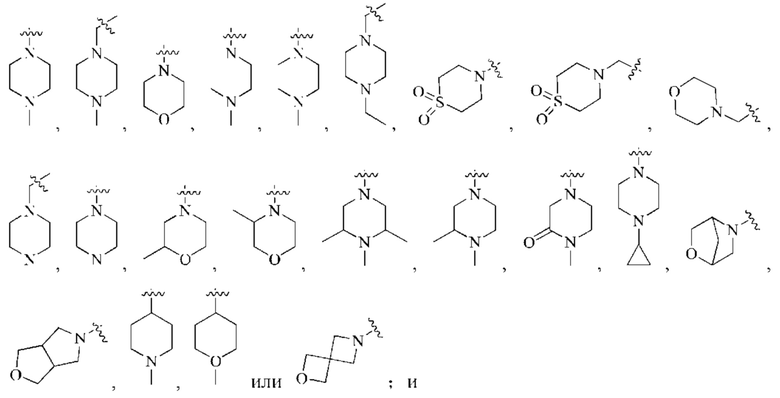
R3, R4 и R5 независимо выбраны из группы, состоящей из водорода, гидроксила, амино, карбоксила, циано, нитро, галогена, С1-6алкила, С1-6алкокси, С3-6циклоалкилокси, окса-С5-8циклоалкилокси, галогенированного С1-6алкокси, С2-8алкенила, С2-8алкинила, С3-6циклоалкиламино, С1-6алкилсульфонила, С3-8циклоалкилсульфонила, С1-6алкилкарбонила, С3-6циклоалкилкарбонила, -NR11-(CH2)n-N(R7)(R8), С1-6алкилтио-(СН2)n-, -(СН2)n-(3-14)-членного циклоалкила, -(СН2)n-(6-14)-членного арила, -(СН2)n-(5-14)-членного гетероциклила и -(СН2)n-(5-14)-членного гетероарила; где n равен 0-6; образующий кольцо атом S в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до S(O) или S(O)2, образующий кольцо атом С в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до С(О); и циклоалкил, арил, гетероарил или гетероциклил возможно могут быть замещены одним или более независимым С1-3алкилом или С3-6циклоалкилом;
R7 и R8 независимо выбраны из группы, состоящей из водорода, С1-6алкила, С3-6циклоалкила, С1-6алкокси, галогенированного С1-6алкила или галогенированного С1-6алкокси; и
Р, W и Y одновременно не являются N,
когда Y представляет собой CR3, Р представляет собой CR4, и W представляет собой N, R4 не может быть выбран из С1-6алкила,
когда Y представляет собой CR3, Р представляет собой CR4, и W представляет собой CR5, один из R4 и R5 должен представлять собой Н;
Ar выбран из группы, состоящей из 3-14-членного циклоалкила, 6-14-членного арила, 5-14-членного гетероциклила или 5-14-членного гетероарила; образующий кольцо атом S в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до S(O) или S(O)2, и образующий кольцо атом С в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до С(О); и Ar возможно может быть замещен 1-3 R6;
R6 каждый независимо выбран из группы, состоящей из водорода, гидроксила, амино, карбоксила, циано, нитро, галогена, С1-6алкила, С3-6циклоалкила, С1-6алкокси, С3-6циклоалкилокси, галогенированного С1-6алкокси, галогенированного С1-6алкила, С3-6циклоалкила, С2-8алкенила, С2-8алкинила, -NR11-(CH2)n-N(R9)(R10), аминоС1-6алкила, С3-6циклоалкиламино, С1-6алкилсульфонила, С3-8циклоалкилсульфонила, С1-6алкилкарбонила, С3-6циклоалкилкарбонила, С1-6алкилтио, -(СН2)n-(6-14)-членного циклоалкила, -(СН2)n-(6-14)-членного арила, -(СН2)n-(5-14)-членного гетероциклила или -(СН2)n-(5-14)-членного гетероарила; где n равен 0-6, и циклоалкил, арил, гетероарил или гетероциклил возможно могут быть замещены одним или более независимыми С1-3алкилами;
R9 и R10 независимо выбраны из группы, состоящей из водорода, С1-6алкила, С3-6циклоалкила, С1-6алкокси, галогенированного С1-6алкила или галогенированного С1-6алкокси;
R11 выбран из группы, состоящей из водорода, С1-6алкила или С3-6циклоалкила.
Одно воплощение настоящего изобретения относится к вышеуказанному соединению формулы (I), его фармацевтически приемлемым солям или его стереоизомерам, где
R1 выбран из С1-3алкила, предпочтительно метила или этила;
R2 выбран из группы, состоящей из водорода, метила и этила;
X выбран из N.
Одно воплощение настоящего изобретения относится к вышеуказанному соединению формулы (I), его фармацевтически приемлемым солям или его стереоизомерам, где
Ar выбран из группы, состоящей из 6-14-членного арила или 5-14-членного гетероарила; любой образующий кольцо атом S в ариле и гетероариле возможно может быть окислен до S(O) или S(O)2, образующий кольцо атом С в ариле и гетероариле возможно может быть окислен до С(О); и Ar возможно может быть замещен 1-3 R6;
R6 выбран из группы, состоящей из водорода, амино, циано, галогена, С1-4алкила, трифторметила, метилсульфонила, -(СН2)n-(5-14)-членного гетероциклила, -(СН2)n-(5-14)-членного гетероарила, где n равен 0-6, и гетероарильное кольцо и гетероциклил возможно могут быть замещены С1-3алкилом.
Одно воплощение настоящего изобретения относится к вышеуказанному соединению формулы (I), его фармацевтически приемлемым солям или его стереоизомерам, где
X представляет собой N;
R1 выбран из С1-3алкила, предпочтительно метила или этила;
R2 выбран из группы, состоящей из водорода, метила и этила;
Y выбран из CR3 или N;
Р выбран из CR4 или N;
W выбран из CR5 или N;
R3, R4 и R5 независимо выбраны из группы, состоящей из водорода, гидроксила, амино, карбоксила, циано, нитро, галогена, С1-6алкила, C1-6алкокси, С3-6циклоалкилокси, окса-С5-8циклоалкилокси, галогенированного С1-6алкокси, С2-8алкенила, С2-8алкинила, С3-6циклоалкиламино, С1-6алкилсульфонила, С3-8циклоалкилсульфонила, С1-6алкилкарбонила, С3-6циклоалкилкарбонила, -NR11-(CH2)n-N(R7)(R8), С1-6алкилтио-(СН2)n-, -(СН2)n-(3-14)-членного циклоалкила, -(СН2)n-(6-14)-членного арила, -(СН2)n-(5-14)-членного гетероциклила или -(СН2)n-(5-14)-членного гетероарила; где n равен 0-6; образующий кольцо атом S в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до S(O) или S(O)2, образующий кольцо атом С в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до С(О); и циклоалкил, арил, гетероарил или гетероциклил возможно могут быть замещены одним или более независимыми С1-3алкилами или С3-6циклоалкилами;
R7 и R8 независимо выбраны из группы, состоящей из водорода, С1-6алкила, С3-6циклоалкила, С1-6алкокси, галогенированного С1-6алкила или галогенированного C1-6алкокси; и
Р, W и Y одновременно не являются N,
когда Y представляет собой CR3, Р представляет собой CR4, и W представляет собой N, R4 не может быть выбран из С1-6алкила,
когда Y представляет собой CR3, Р представляет собой CR4, и W представляет собой CR5, один из R4 и R5 должен представлять собой Н;
R11 выбран из группы, состоящей из водорода, С1-6алкила и С3-6циклоалкила;
Ar выбран из группы, состоящей из 6-14-членного арила или 5-10-членного гетероарила; образующий кольцо атом S в ариле и гетероариле возможно может быть окислен до S(O) или S(O)2, образующий кольцо атом С в ариле и гетероариле возможно может быть окислен до С(О); и где Ar возможно может быть замещен 1-3 R6;
каждый R6 независимо выбран из группы, состоящей из водорода, амино, циано, галогена, С1-4алкила, трифторметила, метилсульфонила, -(СН2)n-(5-10)-членного гетероциклила или -(СН2)n-(5-10)-членного гетероарила; где n равен 0-6; и гетероарил и гетероциклил возможно могут быть замещены одним или более независимыми С1-3алкилами.
Одно воплощение настоящего изобретения относится к вышеуказанному соединению формулы (II), его фармацевтически приемлемым солям или его стереоизомерам:
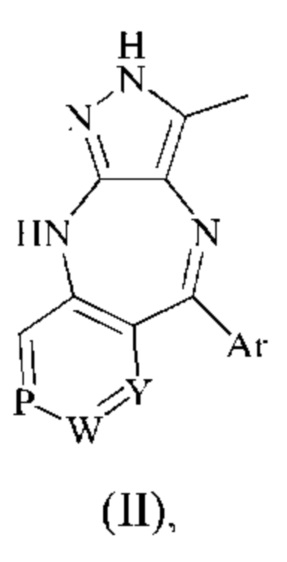
где
Ar выбран из группы, состоящей из 5-6-членного арила или 5-6-членного гетероарила; образующий кольцо атом S в ариле и гетероариле возможно может быть окислен до S(O) или S(O)2, и образующий кольцо атом С в ариле и гетероариле возможно может быть окислен до С(О); и где Ar возможно может быть замещен 1-3 R6;
каждый R6 независимо выбран из группы, состоящей из водорода, амино, циано, галогена, С1-4алкила, трифторметила или метилсульфонила, и галоген предпочтительно представляет собой хлор;
Y выбран из CR3 или N;
Р выбран из CR4 или N;
W выбран из CR5 или N;
R3, R4 и R5 независимо выбраны из группы, состоящей из водорода, гидроксила, амино, карбоксила, циано, нитро, галогена, С1-4алкила, С1-4алкокси, С3-6циклоалкилокси, окса-С5-8циклоалкилокси, галогенированного С1-4алкокси, С2-6алкенила, С2-6алкинила, С3-6циклоалкиламино, С1-4алкилсульфонила, С3-8циклоалкилсульфонила, С1-4алкилкарбонила, С3-6циклоалкилкарбонила, -NR11-(CH2)n-N(R7)(R8), -(СН2)n-(3-14)-членного циклоалкила, -(СН2)n-(5-11)-членного гетероциклила или -(СН2)n-(5-10)-членного гетероарила; где n равен 0-6; образующий кольцо атом S в циклоалкиле, гетероциклиле или гетероариле возможно может быть окислен до S(O) или S(O)2, образующий кольцо атом С в циклоалкиле, гетероциклиле или гетероариле возможно может быть окислен до С(О); и циклоалкил, гетероарил или гетероциклил возможно могут быть замещены одним или более независимыми C1-3алкилами или С3-6циклоалкилами;
R7 и R8 независимо выбраны из водорода, метила, этила, изопропила или циклопропила; и
Р, W и Y одновременно не являются N, и по меньшей мере один из Р, W и Y представляет собой N;
когда Y представляет собой CR3, Р представляет собой CR4, и W представляет собой N, R4 не может быть выбран из С1-4алкила;
R11 выбран из группы, состоящей из водорода, С1-6алкила и С3-6циклоалкила;
предпочтительно,
Y представляет собой CR3;
Р представляет собой CR4;
W представляет собой N;
R4 не может быть выбран из С1-4алкила.
Одно воплощение настоящего изобретения относится к вышеуказанному соединению формулы (I) или (II), его фармацевтически приемлемым солям или его стереоизомерам, где
Ar может быть выбран из группы, состоящей из:
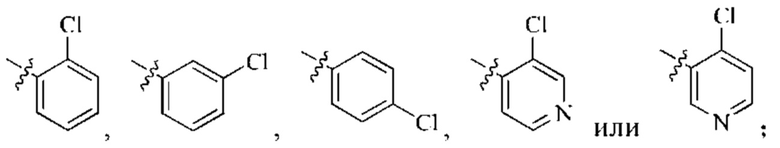
Y выбран из CR3 или N;
Р выбран из CR4 или N;
W выбран из CR5 или N;
R3, R4 и R5 независимо выбраны из группы, состоящей из: водорода, метила, этила, изопропила, 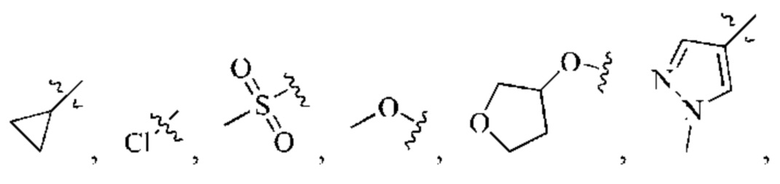
Р, W и Y одновременно не являются N, и по меньшей мере один из Р, W и Y представляет собой N;
когда Y представляет собой CR3, Р представляет собой CR4, и W представляет собой N, R4 не может являться метилом, этилом или изопропилом;
предпочтительно,
Y представляет собой CR3;
Р представляет собой CR4;
W представляет собой N.
Одно воплощение настоящего изобретения относится к вышеуказанному соединению формулы (I) или (II), его фармацевтически приемлемым солям или его стереоизомерам, где
Ar выбран из фенила или 5-6-членного гетероарила, Ar возможно может быть замещен 1-3 R6, и R6 каждый независимо выбран из группы, состоящей из водорода, амино, циано, галогена, С1-4алкила, трифторметила или метилсульфонила;
Y выбран из CR3;
Р выбран из CR4;
W выбран из N;
R3 выбран из водорода или С1-4алкила;
R4 выбран из группы, состоящей из водорода, гидроксила, амино, карбоксила, циано, нитро, галогена, С1-4алкокси, С3-6циклоалкилокси, окса-С5-8циклоалкилокси, галогенированного С1-4алкокси, С3-6циклоалкиламино, С1-4алкилсульфонила, С3-6циклоалкилсульфонила, С1-4алкилкарбонила, С3-6циклоалкилкарбонила, -NR11-(CH2)n-N(R7)(R8), -(СН2)n-С3-10циклоалкила, -(СН2)n-(5-11)-членного гетероциклила или -(СН2)n-(5-10)-членного гетероарила; где n равен 0-6; образующий кольцо атом S в циклоалкиле, гетероциклиле или гетероариле возможно может быть окислен до S(O) или S(O)2, образующий кольцо атом С в циклоалкиле, гетероциклиле или гетероариле возможно может быть окислен до С(О); и циклоалкил, гетероарил или гетероциклил возможно могут быть замещены одним или более независимыми C1-3алкилами или С3-6циклоалкилами.
Одно воплощение настоящего изобретения относится к вышеуказанному соединению формулы (I) или (II), его фармацевтически приемлемым солям или его стереоизомерам, где
Ar выбран из фенила или пиридила, Ar возможно может быть замещен 1-3 R6, каждый R6 независимо выбран из группы, состоящей из водорода, амино, циано, галогена, С1-4алкила, трифторметила или метилсульфонила;
Y представляет собой CR3;
Р представляет собой CR4;
W представляет собой N;
R3 выбран из водорода или С1-4алкила;
R4 выбран из группы, состоящей из водорода, гидроксила, амино, карбоксила, циано, нитро, галогена, С1-4алкокси, С3-6циклоалкилокси, галогенированного С1-4алкокси, С3-6циклоалкиламино, С1-4алкилсульфонила, С1-4алкилкарбонила, С3-6циклоалкилкарбонила, -NR11-(CH2)n-N(R7)(R8), -(СН2)n-С3-6циклоалкила, -(СН2)n-(5-6)-членного моногетероциклила, -(СН2)n-(7-11)-членного конденсированного гетероциклила, -(СН2)n-(5-6)-членного моногетероарила или -(СН2)n-(8-10)-членного конденсированного гетероарила, где n равен 0-6, образующий кольцо атом S в циклоалкиле, гетероциклиле или гетероариле возможно может быть окислен до S(O) или S(O)2, и образующий кольцо атом С в циклоалкиле, гетероциклиле или гетероариле возможно может быть окислен до С(О); циклоалкил, гетероарил, гетероциклил возможно могут быть замещены одним или более независимыми С1-3алкилами или С3-6циклоалкилами;
где (1) R4 предпочтительно выбран из группы, состоящей из галогена, С1-4алкокси, галогенированного С1-4алкокси, С1-4алкилсульфонила, С3-6циклоалкилкарбонила, -NR11-(CH2)n-N(R7)(R8), -(СН2)n-С3-6циклоалкила, -(СН2)n-(5-6)-членного моногетероциклила и -(СН2)n-(7-11)-членного конденсированного гетероциклила; где n равен 0-6; образующий кольцо атом S в циклоалкиле или гетероциклиле возможно может быть окислен до S(O) или S(O)2, и образующий кольцо атом С в циклоалкиле или гетероциклиле возможно может быть окислен до С(О); и циклоалкил или гетероциклил возможно могут быть замещены одним или более независимыми С1-3алкилами или С3-6циклоалкилами;
5-6-членный моно гетероциклил предпочтительно представляет собой 5-6-членный насыщенный моногетероциклил, и 7-11-членный конденсированный гетероциклил предпочтительно представляет собой 7-11-членный насыщенный конденсированный гетероциклил, более предпочтительно 7-11-членный насыщенный орто-конденсированный гетероциклил, 7-11-членный насыщенный спиро-гетероциклил или 7-11-членный насыщенный мостиковый гетероциклил;
(2) дополнительно R4 предпочтительно выбран из группы, состоящей из: галогена, С1-4алкокси, -NR11-(CH2)n-N(R7)(R8), С1-4алкилсульфонила, 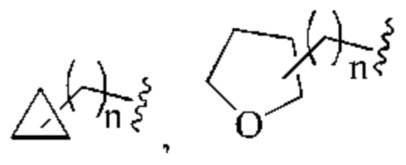
 n равен 0-3; где циклоалкил или гетероциклил возможно могут быть замещены одной или более независимыми С1-3алкильными группами или С3-6циклоалкильными группами;
n равен 0-3; где циклоалкил или гетероциклил возможно могут быть замещены одной или более независимыми С1-3алкильными группами или С3-6циклоалкильными группами;
(3) R4 более предпочтительно выбран из группы, состоящей из: галогена, С1-4алкокси, -NR11-(CH2)n-N(R7)(R8), С1-4алкилсульфонила, 
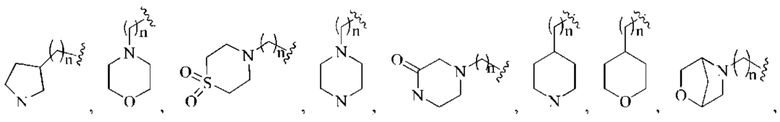
 n равен 0-3; где циклоалкил или гетероциклил возможно могут быть замещены одной или более независимыми С1-3алкильными группами или С3-6циклоалкильными группами;
n равен 0-3; где циклоалкил или гетероциклил возможно могут быть замещены одной или более независимыми С1-3алкильными группами или С3-6циклоалкильными группами;
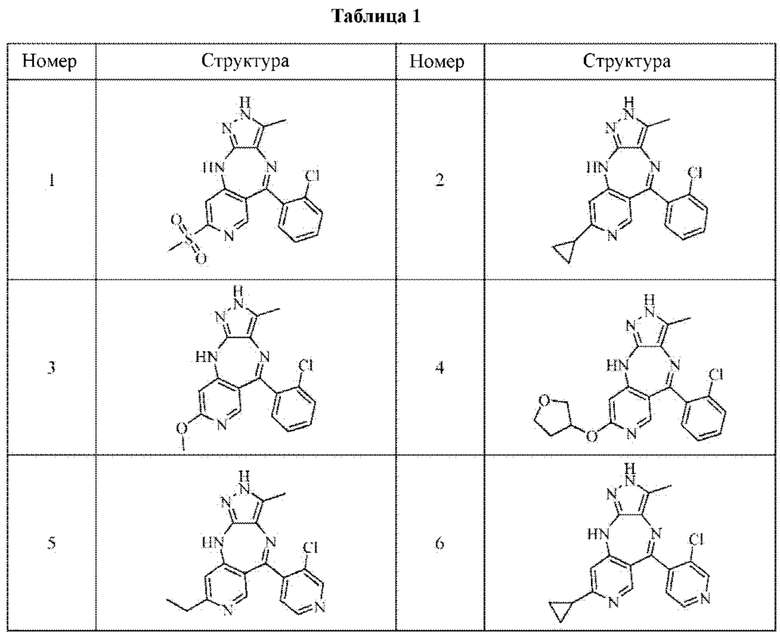
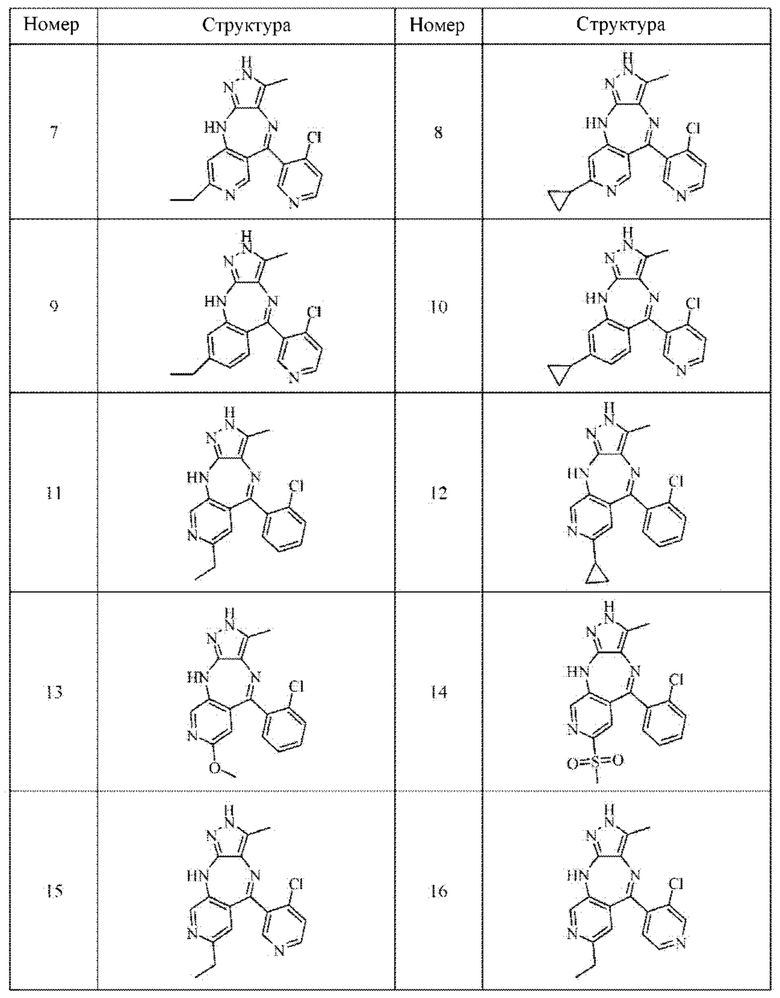
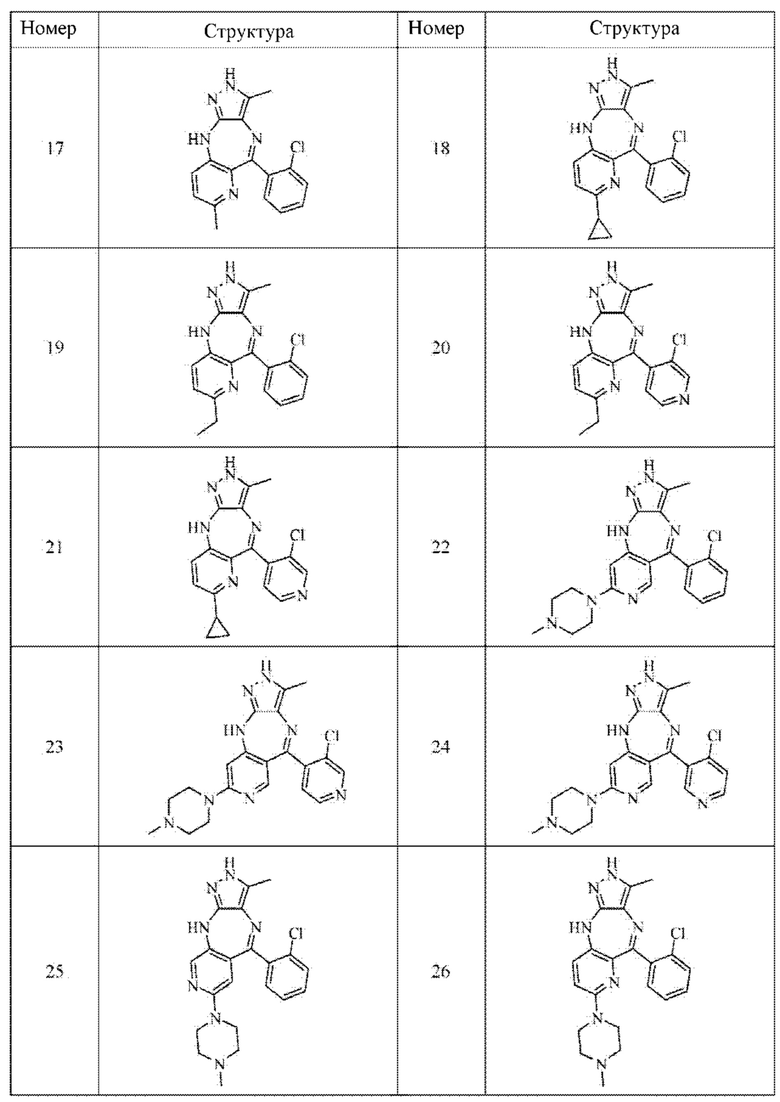
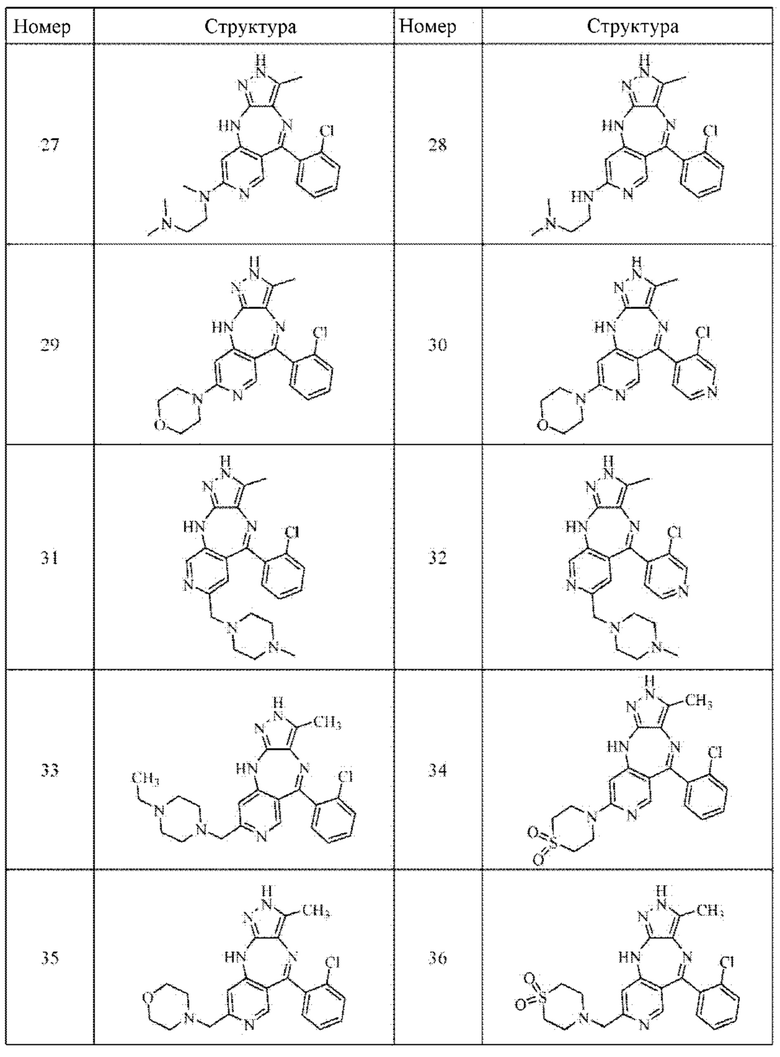
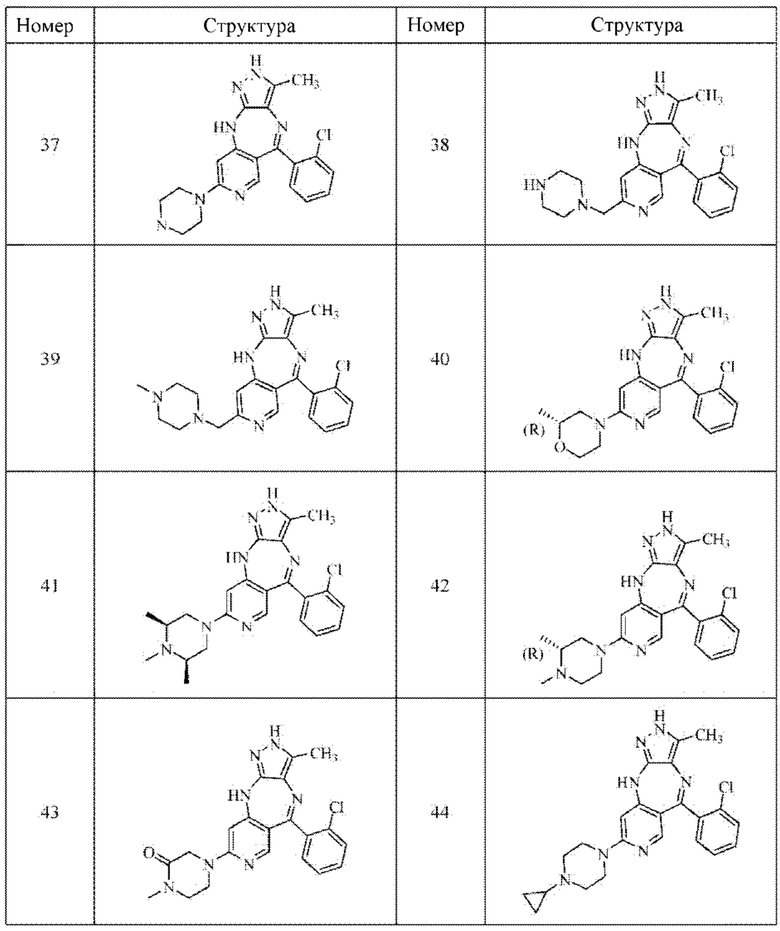
В одном воплощении настоящего изобретения вышеуказанное соединение формулы (I) или (II), его фармацевтически приемлемые соли или его стереоизомеры показаны в таблице 1:
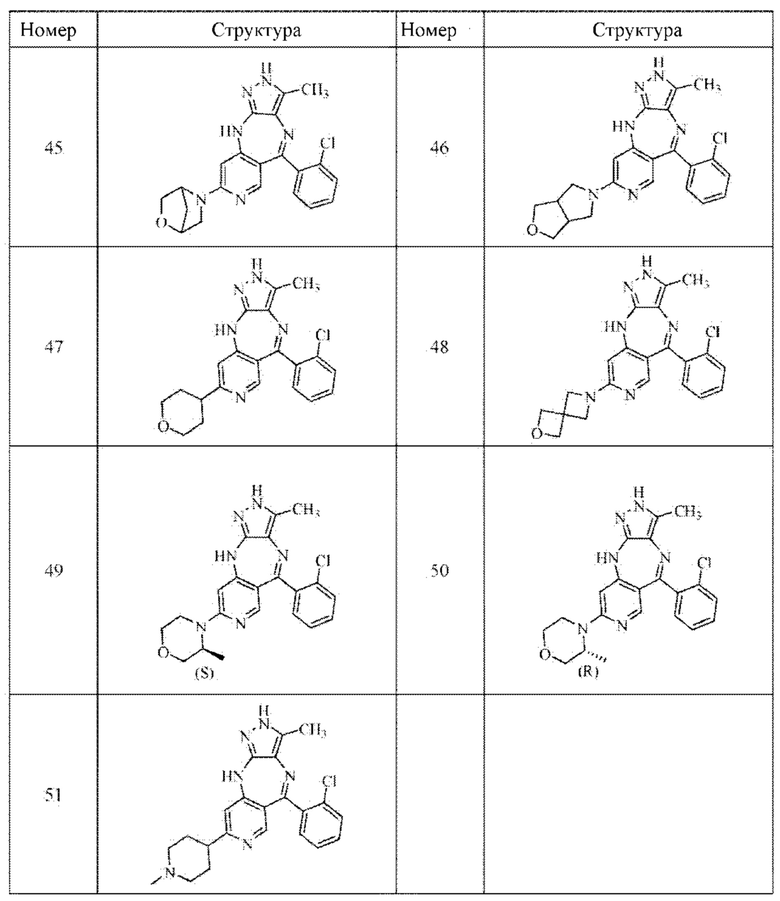
Поскольку кристаллическая форма соединения существенно отличается от других форм в отношении стабильности и растворимости, изучение кристаллической формы является очень важным при разработке лекарственного средства. Авторы настоящего изобретения изучили соединение формулы (III), как указано ниже, и получили кристаллическую форму соединения. На основании этого в настоящем изобретении также предложена кристаллическая форма I соединения формулы (III).
Кристаллическая форма I соединения формулы (III), 4-(5-(2-хлорфенил)-3-метил-2,10-дигидропиразоло[4,3-b]пиридо[4,3-е][1,4]диазепин-8-ил)-морфолина, имеет характеристические пики при 7,4±0,2°; 17,9±0,2°; 18,9±0,2°; 19,4±0,2°; 21,5±0,2° и 23,7±0,2° на картине дифракции рентгеновских лучей на порошке. В конкретном воплощении дифракции рентгеновских лучей на порошке по настоящему изобретению может быть использовано Cu-Kα-излучение, и характеристические пики представлены углами 2θ;
В одном воплощении изобретения, в дополнение к вышеописанным характеристическим пикам, кристаллическая форма I соединения формулы (III) также имеет характеристические пики при 14,0±0,2°; 15,0±0,2°; 20,7±0,2° и 25,4±0,2°.
В одном воплощении изобретения, в дополнение к вышеописанным характеристическим пикам, кристаллическая форма I соединения формулы (III) также имеет характеристические пики при 11,7±0,2°; 22,8±0,2° и 27,8±0,2°, представленные углами 2θ на картине дифракции рентгеновских лучей на порошке.
В изобретении также предложен способ получения кристаллической формы I соединения формулы (III), который может включать:
растворение соединения формулы (III) в отдельном растворителе или смешанном растворителе при нагревании и охлаждение для осаждения кристаллической формы I;
или
суспендирование соединения формулы (III) в отдельном растворителе или смешанном растворителе, перемешивание и фильтрование с получением кристаллической формы I;
или
растворение соединения формулы (III) в отдельном растворителе или смешанном растворителе и концентрирование под вакуумом с получением кристаллической формы I.
В одном воплощении настоящего изобретения отдельный растворитель или смешанный растворитель, используемый в вышеуказанном способе получения кристаллической формы I, может представлять собой один или более растворителей, выбранных из группы, состоящей из метанола, этанола, тетрагидрофурана, 2-метилтетрагидрофурана, дихлорметана, дихлорэтана, этилацетата, ацетонитрила, диметилсульфоксида, смеси диметилсульфоксид/вода, метанол/тетрагидрофуран, метанол/2-метилтетрагидрофуран, метанол/дихлорметан, этанол/2-метилтетрагидрофуран и дихлорметан/вода; предпочтительно метанола, этанола, тетрагидрофурана, 2-метилтетрагидрофурана, диметилсульфоксида, смеси диметилсульфоксид/вода, метанол/тетрагидрофуран, метанол/2-метилтетрагидрофуран, этанол/2-метилтетрагидрофуран и/или дихлорметан/вода.
Термин "диметилсульфоксид/вода" по настоящему изобретению означает смесь диметилсульфоксида и воды; "метанол/тетрагидрофуран" означает смесь метанола и тетрагидрофурана; "метанол/2-метилтетрагидрофуран" означает смесь метанола и 2-метилтетрагидрофурана, "метанол/дихлорметан" означает смесь метанола и дихлорметана, "этанол/2-метилтетрагидрофуран" означает смесь этанола и 2-метилтетрагидрофурана, и "дихлорметан/вода" означает смесь дихлорметана и воды.
В одном воплощении настоящего изобретения вышеуказанный отдельный или смешанный растворитель используют в количестве, необходимом для обеспечения растворения всех исходных веществ, например, объем отдельного или смешанного растворителя, необходимый для 1 г соединения формулы (III) составляет от 90 мл до 200 мл.
Объемное соотношение используемой смеси растворителей может находиться в диапазоне 0,1-20:1, предпочтительно в диапазоне 1-10:1, и более предпочтительно в диапазоне 1-5:1. Например, соотношение этанол/2-метилтетрагидрофуран составляет 5:1, соотношение дихлорметан/вода составляет 2:1, соотношение метанол/дихлорметан составляет 5:1, и тому подобное.
В изобретении также предложен способ получения кристаллической формы I соединения формулы (III), который может включать:
промывку соединения формулы (III) подходящим количеством отдельного или смешанного растворителя, перемешивание, фильтрование (предпочтительно фильтрование при пониженном давлении) и сушку с получением кристаллической формы I.
Одиночный или смешанный растворитель представляет собой один или более растворителей, выбранных из метанола, этанола, тетрагидрофурана, 2-метилтетрагидрофурана, дихлорметана, дихлорэтана, этилацетата, ацетонитрила, диметилсульфоксида, смеси диметилсульфоксид/вода, метанол/тетрагидрофуран, метанол/2-метилтетрагидрофуран, метанол/дихлорметан, этанол/2-метилтетрагидрофуран, дихлорметан/вода; предпочтительно метанола, этанола, тетрагидрофурана, 2-метилтетрагидрофурана, диметилсульфоксида, смеси диметилсульфоксид/вода, метанол/тетрагидрофуран, метанол/2-метилтетрагидрофуран, этанол/2-метилтетрагидрофуран и/или дихлорметан/вода.
В изобретении также предложен способ получения соединения формулы (III), включающий:
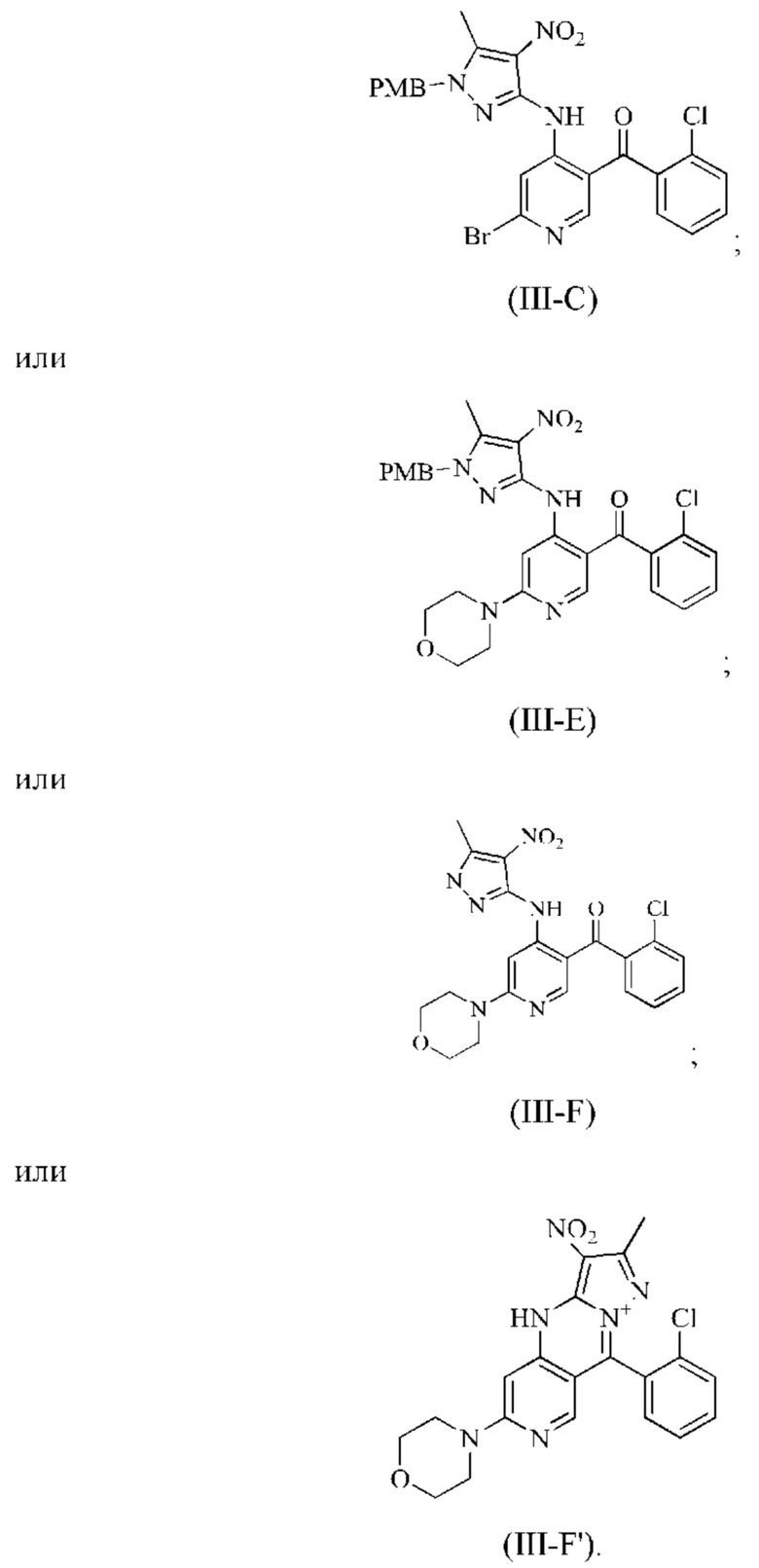
1) взаимодействие соединения формулы (III-А) с соединением формулы (III-В) с получением соединения формулы (III-С);
2) взаимодействие соединения формулы (III-С) с соединением формулы (III-D) с получением соединения формулы (III-Е);
3) снятие защиты с соединения формулы (III-Е) с получением соединения формулы (III-F) или (III-F') в качестве переходного состояния;
4) получение соединения формулы (III) из соединения формулы (III-F) или (III-F');
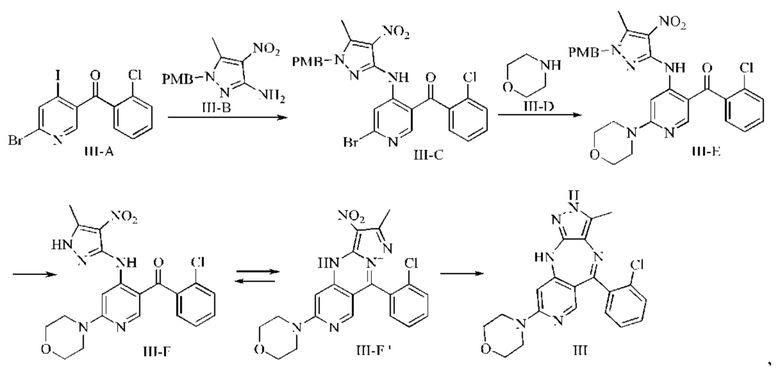
где аббревиатура "РМВ" означает пара-метоксибензил.
В настоящем изобретении также предложено промежуточное соединение для получения вышеуказанного соединения формулы (III), которое имеет следующую структурную формулу:
В изобретении также предложен фармацевтический препарат, содержащий вышеуказанные соединения формулы (I) или (II), их фармацевтически приемлемые соли или стереоизомеры, и/или содержащий вышеуказанную кристаллическую форму I соединения формулы (III).
В одном воплощении изобретения фармацевтический препарат может содержать один или более фармацевтически приемлемый носитель и может быть введен перорально, парентерально, ректально или посредством пульмонального введения пациенту или субъекту, нуждающемуся в этом. Для перорального введения фармацевтическая композиция может быть приготовлена в виде традиционного твердого препарата, такого как таблетка, капсула, пилюля и гранула и так далее; или перорального жидкого препарата, такого как пероральный раствор, пероральная суспензия и сироп, и так далее. При приготовлении перорального препарата может быть включен подходящий наполнитель, связующее вещество, разрыхлитель, смазывающее вещество и тому подобное. Для парентерального введения фармацевтическая композиция может быть приготовлена в виде инъекции, включающей инъекционную жидкость, стерильный порошок для инъекций и концентрированный раствор для инъекций. При приготовлении в виде инъекции препарат может быть получен традиционным в области медицины способом. Возможно приготовление инъекции с подходящими дополнительными агентами, в соответствии со свойствами лекарственного средства, или без дополнительных агентов. Для ректального введения фармацевтическая композиция может быть приготовлена в виде суппозитория или тому подобного. Для пульмонального введения фармацевтическая композиция может быть приготовлена в виде средства для ингаляции или спрея и так далее.
В конкретном воплощении по настоящему изобретению фармацевтический препарат может дополнительно содержать один или более чем один второй терапевтически активный агент, который представляет собой антиметаболит, ингибитор фактора роста, ингибитор митоза, противоопухолевый гормон, алкилирующий агент, металл, ингибитор топоизомеразы, гормональное лекарственное средство, иммуномодулятор, ген-супрессор опухолей, противораковую вакцину, иммунную контрольную точку или антитело для иммунотерапии опухолей, или низкомолекулярное лекарственное средство.
В настоящем изобретении также предложена фармацевтическая композиция, содержащая вышеуказанное соединение формулы (I) или (II), или его фармацевтически приемлемые соли или стереоизомеры, и/или фармацевтическая композиция, содержащая вышеуказанную кристаллическую форму I соединения формулы (III) и один или более чем один второй терапевтически активный агент.
В одном конкретном воплощении настоящего изобретения композиция может быть использована в комбинированном введении в "терапевтически эффективном количестве" вышеуказанного соединения формулы (I) или (II) или его фармацевтически приемлемых солей или стереоизомеров, и/или вышеуказанной кристаллической формы I соединения формулы (III), в сочетании с одним или более чем одним вторым терапевтически активным агентом, таком как последовательное введение, одновременное введение, альтернативно, терапевтически активные ингредиенты готовят в виде препарата сложного состава для введения.
Второй терапевтически активный агент представляет собой антиметаболит, ингибитор фактора роста, ингибиторы митоза, противоопухолевые гормоны, алкилирующие агенты, металлы, ингибиторы топоизомеразы, гормональные лекарственные средства, иммуномодуляторы, гены-супрессоры опухолей, противораковые вакцины, иммунные контрольные точки или антитела для иммунотерапии опухолей, или низкомолекулярные лекарственные средства.
В изобретении также предложено применение вышеуказанных соединений формулы (I) или (II) или их фармакологически приемлемых солей или стереоизомеров, вышеуказанной кристаллической формы I соединений формулы (III), или вышеуказанного фармацевтического препарата в изготовлении лекарственного средства для лечения опосредованного мультикиназами рака, такого как рак легкого, плоскоклеточная карцинома, рак мочевого пузыря, рак желудка, рак яичника, рак брюшины, рак молочной железы, протоковая карцинома молочной железы, рак головы и шеи, карцинома эндометрия, рак тела матки, рак прямой кишки, рак печени, рак почки, опухоль почечной лоханки, эзофагеальная карцинома, эзофагеальная аденокарцинома, глиома, рак предстательной железы, рак щитовидной железы, рак женской репродуктивной системы, карцинома in situ, лимфома, нейрофиброматоз, рак кости, рак кожи, рак головного мозга, рак толстой кишки, рак яичка, гастроинтестинальная стромальная опухоль, рак полости рта, фарингеальный рак, множественная миелома, лейкоз, неходжкинская лимфома, хорионаденома толстой кишки, меланома, цитома и саркома.
В изобретении также предложен способ лечения заболеваний. Способ включает введение в терапевтически эффективном количестве пациентам, нуждающимся в этом, вышеуказанного соединения формулы (I) или (II) или его фармакологически приемлемых солей или стереоизомеров, вышеуказанной кристаллической формы I соединения формулы (III), или вышеуказанных фармацевтических препаратов. Заболевания включают опосредованные мультикиназами раковые заболевания, такие как рак легкого, плоскоклеточная карцинома, рак мочевого пузыря, рак желудка, рак яичника, рак брюшины, рак молочной железы, протоковая карцинома молочной железы, рак головы и шеи, карцинома эндометрия, рак тела матки, рак прямой кишки, рак печени, рак почки, опухоль почечной лоханки, эзофагеальная карцинома, эзофагеальная аденокарцинома, глиома, рак предстательной железы, рак щитовидной железы, рак женской репродуктивной системы, карцинома in situ, лимфома, нейрофиброматоз, рак кости, рак кожи, рак головного мозга, рак толстой кишки, рак яичка, гастроинтестинальная стромальная опухоль, рак полости рта, фарингеальный рак, множественная миелома, лейкоз, неходжкинская лимфома, хорионаденома толстой кишки, меланома, цитома и саркома.
Термин "терапевтически эффективное количество", использованный здесь, относится к количеству вышеуказанного соединения, кристаллической формы I и/или фармацевтического препарата, которое способно по меньшей мере облегчить симптомы состояния у пациента при введении пациенту. Фактическое количество, включающее "терапевтически эффективное количество", будет изменяться в зависимости от различных обстоятельств, включая конкретное состояние, требующее лечения, тяжесть состояния, телосложение и здоровье пациента, и путь введения, без ограничения ими. Специалисты в области медицины могут легко определить подходящее количество, используя способы, известные в области терапевтического лечения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Используемый в настоящем изобретении термин "галоген" означает фтор, хлор, бром, йод и тому подобное, предпочтительно фтор и хлор.
Используемый здесь термин "оксо" означает, что любой атом С в заместителе может быть окислен до "-С(О)-"; если присутствует гетероатом, этот гетероатом может образовывать оксид, например 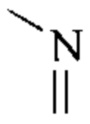 может быть окислен до
может быть окислен до 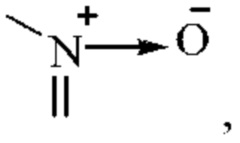 S возможно может быть окислен до S(O) или S(O)2.
S возможно может быть окислен до S(O) или S(O)2.
Используемый здесь термин "галогенированный" означает, что любой атом водорода в заместителе может быть замещен одним или более атомами галогена, одинаковым или разным. Термин "галоген" определен выше.
Используемый в настоящем изобретении термин "С1-6алкил" означает прямую или разветвленную алкильную группу, полученную посредством удаления одного атома водорода из углеводородной группировки, имеющей от 1 до 6 атомов углерода, такую как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, 2-метилбутил, неопентил, 1-этилпропил, н-гексил, изогексил, 4-метилпентил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил, 2-этилбутил и 1-метил-2-метилпропил и так далее. Термин "С1-4алкил" означает вышеуказанные примеры, имеющие от 1 до 4 атомов углерода.
Используемый в настоящем изобретении термин "С2-8алкенил" означает прямую или разветвленную алкенильную группу, полученную посредством удаления одного атома водорода из олефиновой группировки, имеющей от 2 до 8 атомов углерода, содержащей двойную углерод-углеродную связь, такую как винил, 1-пропенил, 2-пропенил, 1-бутенил, 2-бутенил, 1,3-бутадиенил, 1-пентенил, 2-пентенил, 3-пентенил, 1,3-пентадиенил, 1,4-пентадиенил, 1-гексенил и 1,4-гексадиенил и так далее.
"С2-8алкинильная группа" по настоящему изобретению означает прямую или разветвленную алкиновую группу, полученную посредством удаления одного атома водорода из алкиновой группировки, имеющей от 2 до 8 атомов углерода, содержащей тройную углерод-углеродную связь, такую как этинил, пропинил, 2-бутинил, 2-пентинил, 3-пентинил, 4-метил-2-пентинил, 2-гексинил, 3-гексинил и так далее. "С1-6алкилкарбониламино", "С1-6алкиламинокарбонил", "С1-6алкилсульфонил", "С1-6алкилкарбонил", "С1-6алкилтио" по настоящему изобретению означает С1-6алкил-С(O)-NH-, С1-6алкил-NH-С(О)-, С1-6алкил-S(O)2-, С1-6алкил-С(О)-, С1-6алкил-S- соответственно; "С1-6алкил" является таким, как определен выше, предпочтительно "С1-4алкил".
"С1-6алкокси" по настоящему изобретению означает группу, в которой "С1-6алкил", определенный выше, связан с атомом кислорода, который в свою очередь связан с исходной группировкой, то есть группу "С1-6алкил-О-", такую как метокси, этокси, пропокси, изопропокси, н-бутокси, трет-бутокси, н-пентилокси, неопентилокси и н-гексилокси и так далее. "С1-4алкокси" относится к вышеуказанным примерам, имеющим от 1 до 4 атомов углерода, то есть к группам "С1-4алкил-O-".
"Циклоалкил", "арил", "гетероциклил" и "гетероарил" по настоящему изобретению включают моноциклическую систему и конденсированную кольцевую систему (бициклическую систему или полициклическую систему). Моноциклическая система относится к группе в виде только одного кольца, и конденсированная кольцевая система относится к полициклической кольцевой структуре, образованной двумя или более циклическими структурами, соединенных в виде орто-конденсированных, спиро или мостиковых колец. Орто-конденсированное кольцо относится к конденсированной кольцевой структуре, образованной двумя или более циклическими структурами, имеющими друг с другом два общих смежных кольцевых атома (то есть имеющими одну общую связь). Мостиковое кольцо относится к конденсированной кольцевой структуре, образованной двумя или более циклическими структурами, имеющими друг с другом два общих несмежных кольцевых атома. Спирокольцо относится к конденсированной кольцевой структуре, образованной двумя или более циклическими структурами, имеющими друг с другом один общий кольцевой атом. Циклоалкил, арил, гетероциклил или гетероарил, определенные посредством числа атомов в настоящем изобретении, включают моноциклические и конденсированные кольцевые структуры, которые могут быть образованы, если не оговорено иное.
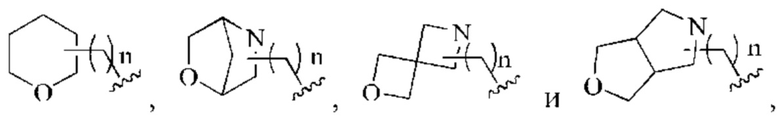
"Циклоалкил" по настоящему изобретению означает моноциклическую циклоалкильную, бициклическую циклоалкильную систему или полициклическую циклоалкильную систему. Эти группы могут быть насыщенными или ненасыщенными, но не ароматическими. Моноциклический циклоалкил может представлять собой С3-8циклоалкил, С3-6циклоалкил, С5-8циклоалкил и тому подобное. Примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклобутенил, циклопентенил, циклогексенил, 1,4-циклогексадиенил, циклогептенил, 1,4-циклогептадиенил, циклооктенил, 1,5-циклооктадиенил и тому подобное, но не ограничены ими. Орто-конденсированный циклоалкил может представлять собой 6-12-членный орто-конденсированный циклоалкил, 7-10-членный орто-конденсированный циклоалкил, и его типичные примеры включают бицикло[3.1.1]гептан, бицикло[2.2.1]гептан, бицикло[2.2.2]октан, бицикло[3.2.2]нонан, бицикло[3.3.1]нонан и бицикло[4.2.1]нонан, но не ограничены ими. Спироциклический циклоалкил может представлять собой 6-12-членные спироциклические группы, 7-11-членные спироциклические группы или подобное, и его примеры включают 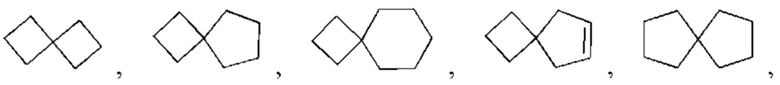
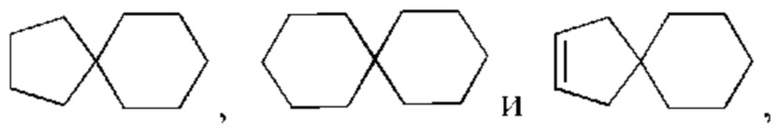 но не ограничены ими. Мостиковый циклоалкил может представлять собой 6-12-членные мостиковые кольцевые группы и 7-11-членные мостиковые кольцевые группы, и его примеры включают:
но не ограничены ими. Мостиковый циклоалкил может представлять собой 6-12-членные мостиковые кольцевые группы и 7-11-членные мостиковые кольцевые группы, и его примеры включают: 
 но не ограничены ими.
но не ограничены ими.
"3-14-Членный циклоалкил", "3-10-членный циклоалкил" и "3-6-членный циклоалкил" по настоящему изобретению включают моноциклические и конденсированные кольцевые структуры, которые могут быть образованы, если не оговорено иное.
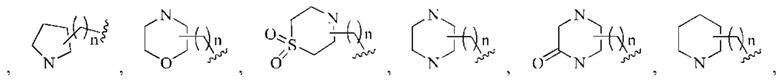
"Гетероциклил" по настоящему изобретению означает неароматическую циклическую группу, в которой по меньшей мере один кольцевой атом углерода замещен гетероатомом, выбранным из О, S и N, предпочтительно замещен 1-3 гетероатомами, и где атом углерода, атом азота и атом серы может быть окислен.
"Гетероциклил" означает моноциклическую гетероциклильную, бициклическую гетероциклильную или полициклическую гетероциклильную систему, включая насыщенный, частично насыщенный гетероциклил, но исключая ароматические кольца. Моногетероциклил может представлять собой 3-8-членный гетероциклил, 3-8-членный насыщенный гетероциклил, 3-6-членный гетероциклил, 4-7-членный гетероциклил, 5-7-членный гетероциклил, 5-6-членный гетероциклил, 5-6-членный кислородсодержащий гетероциклил, 5-6-членный азотсодержащий гетероциклил, 5-6-членный насыщенный гетероциклил или тому подобное. Примеры моногетероциклила включают азиридинил, оксиранил, тииранил, азетидинил, оксетанил, тиетанил, тетрагидрофуранил, тетрагидропирролил, тетрагидротиофенил, имидазолидинил, пиразолидинил, 1,2-оксазолидинил, 1,3-оксазолидинил, 1,2-тиазолидинил, 1,3-тиазолидинил, тетрагидро-2Н-пиранил, тетрагидро-2Н-тиопиранил, пиперидинил, пиперазинил, морфолинил, 1,4-диоксанил, 1,4-тиоксанил, но не ограничены ими; примеры частично насыщенного гетероциклила включают 4,5-дигидроизооксазолил, 4,5-дигидрооксазолил, 2,5-дигидрооксазолил, 2,3-дигидрооксазолил, 3,4-дигидро-2Н-пирролил, 2,3-дигидро-1H-пирролил, 2,5-дигидро-1Н-имидазолил, 4,5-дигидро-1Н-имидазолил, 4,5-дигидро-1Н-пиразолил, 4,5-дигидро-3Н-пиразолил, 4,5-дигидротиазолил, 2,5-дигидротиазолил, 2Н-пиранил, 4Н-пиранил, 2Н-тиопиранил, 4Н-тиопиранил, 2,3,4,5-тетрагидропиридил, 1,2-изооксазинил, 1,4-изооксазинил или 6Н-1,3-оксазинил и тому подобное, но не ограничены ими. Конденсированное гетероциклическое кольцо включает орто-конденсированный гетероциклил, спирогетероциклил, мостиковый гетероциклил, и может быть насыщенным, частично насыщенным или ненасыщенным, но не ароматическим. Конденсированный гетероциклил представляет собой 5- или 6-членное моноциклическое гетероциклическое кольцо, конденсированное с бензольным кольцом, то есть 5- или 6-членный моноциклический циклоалкил, 5- или 6-членный моноциклический циклоалкенил, 5- или 6-членный моноциклический гетероциклил, или 5- или 6-членный моноциклический гетероарил. Орто-конденсированный гетероциклил может представлять собой 6-12-членный орто-конденсированный гетероциклил, 7-11-членный орто-конденсированный гетероциклил, 6-10-членный орто-конденсированный гетероциклил, 6-12-членный насыщенный орто-конденсированный гетероциклил и 7-11-членный насыщенный орто-конденсированный гетероциклил, и его примеры включают 3-азабицикло[3.10.]гексил, 3,6-диазабицикло[3.2.0]гептил, 3,8-диазабицикло[4.2.0]октил, 3,7-диазабицикло[4.2.0]октил, октагидропирроло[3,4-с]пирролил, октагидропирроло[3,4-b]пирролил, октагидропирроло[3,4-b][1,4]оксазинил, октагидро-1Н-пирроло[3,4-с]пиридинил, 2,3-дигидробензофуран-2-ил, 2,3-дигидробензофуранил-3-ил, индолин-1-ил, индолин-2-ил, индолин-3-ил, 2,3-дигидробензотиофен-2-ил, октагидро-1H-индолил, октагидробензофуранил, но не ограничены ими.
Спирогетероциклил может представлять собой 6-12-членный спирогетероциклил, 7-11-членный спирогетероциклил, 7-11-членный насыщенный спирогетероциклил, 6-12-членный насыщенный спироциклил, и его примеры включают: 
 но не ограничены ими.
но не ограничены ими.
Мостиковый гетероциклил может представлять собой 6-12-членный мостиковый гетероциклил, 7-11-членный мостиковый гетероциклил, 6-12-членный насыщенный мостиковый гетероциклил и 7-11-членный насыщенный мостиковый гетероциклил, и его примеры включают: 
 но не ограничены ими.
но не ограничены ими.
5-14-Членный гетероциклил, 5-11-членный гетероциклил, 5-10-членный гетероциклил, 6-10-членный гетероциклил, 7-11-членный гетероциклил, 7-11-членный насыщенный гетероциклил по настоящему изобретению включают моноциклические и конденсированные кольцевые структуры, которые могут быть образованы, если не оговорено иное.
Арил по настоящему изобретению относится к ароматической циклической группе, включающей моноциклическую систему, бициклическую систему или поликлическую систему, и может представлять собой 6-14-членный арил, включая "6-8-членный моноциклический арил", например фенил, циклооктенил и так далее; и "8-14-членный конденсированный кольцевой арил", такой как пенталенил, нафтил, фенантрил и тому подобное.
Используемый здесь термин "гетероарил" относится к ароматической циклической группе, в которой по меньшей мере один кольцевой атом углерода замещен гетероатомом, выбранным из О, S и N, предпочтительно 1-3 гетероатомами, включая условие, что атом углерода или атом серы окислен, например атом углерода замещен С(О), S(O), S(O)2. Гетероарил включает моноциклический гетероарил и конденсированный гетероарил, который может представлять собой 5-14-членный гетероарил, 5-10-членный гетероарил, 5-7-членный гетероарил, 5-6-членный гетероарил, 8-10-членный гетероарил. Типичные примеры моноциклического гетероарила включают фуранил, имидазолил, изоксазолил, тиазолил, изотиазолил, оксадиазолил, оксазолил, изоксазолил, пиридил, пиридазинил, пиримидинил, пиразинил, пиразолил, пирролил, тетразолил, тиадиазолил, тиазолил, тиенил, триазолил и триазинил, но не ограничены ими. Конденсированный гетероарил относится к бициклической или поликлической кольцевой системе, конденсированной с фенилом, циклоалкилом, циклоалкенилом, гетероциклилом или гетероарилом. Конденсированный гетероарил может представлять собой 8-12-членный гетероарил, 8-10-членный гетероарил, 9-10-членный гетероарил. Типичные примеры конденсированного гетероарила включают бензимидазолил, бензофуранил, бензотиенил, бензотиенил, бензооксадиазолил, бензотиазолил, циннолинил, 5,6-дигидрохинолин-2-ил, 5,6-дигидроизохинолин-1-ил, индазолил, индолил, изохинолил, нафтиридинил, пуринил, хинолил, 5,6,7,8-тетрагидрохинол-2-ил, 5,6,7,8-тетрагидрохинолил, 5,6,7,8-тетрагидрохинол-4-ил, 5,6,7,8-тетрагидроизохинол-1-ил, 4,5,6,7-тетрагидро[с][1,2,5]оксадиазол и 6,7-дигидро[с][1,2,5]оксадиазол-4(5Н)кето, но не ограничены ими. В некоторых воплощениях конденсированный гетероарил представляет собой 5- или 6-членное моноциклическое гетероарильное кольцо, конденсированное с фенильным кольцом, то есть 5- или 6-членный моноциклический циклоалкил, 5- или 6-членный моноциклический циклоалкенил, 5- или 6-членный моноциклический гетероциклил или 5- или 6-членный моноциклический гетероарил.
5-14-Членный гетероарил, 5-10-членный гетероарил, 6-10-членный гетероарил, 5-6-членный гетероарил и 8-10-членный гетероарил по настоящему изобретению включают моноциклические и конденсированные кольцевые структуры, которые могут быть образованы, если не оговорено иное.
Используемый здесь термин "фармацевтически приемлемые соли" означает фармацевтически приемлемые соли присоединения и сольваты кислот и оснований. Такие фармацевтически приемлемые соли включают соли следующих кислот: например соляной кислоты, фосфорной кислоты, бромистоводородной кислоты, серной кислоты, сернистой кислоты, муравьиной кислоты, толуолсульфоновой кислоты, метансульфоновой кислоты, азотной кислоты, бензойной кислоты, лимонной кислоты, винной кислоты, малеиновой кислоты, йодистоводородной кислоты, карбоновой кислоты (такой как уксусная кислота, НООС-(СН2)n-СООН (где n составляет от 0 до 4)) и тому подобных. Такие фармацевтически приемлемые соли также включают соли оснований, такие как соли натрия, калия, кальция, аммония и тому подобные. Различные нетоксичные фармацевтически приемлемые соли присоединения известны специалистам в данной области техники.
Термин "стереоизомер" соединений формулы (I) или (II) по настоящему изобретению означает энантиомер в случае, когда соединение формулы (I) или (II) имеет ассиметрический атом углерода; цис-транс-изомер в случае, когда соединение имеет двойную углерод-углеродную связь или циклическую структуру; таутомеры в случае, когда в соединении присутствует кетон или оксим. Все энантиомеры, диастереомеры, рацематы, цис-транс-изомеры, таутомеры, геометрические изомеры, эпимеры и их смеси соединений формулы (I) или (II) включены в объем изобретения.
Положительные эффекты изобретения:
1) Соединения формулы (I) или (II) и кристаллическая форма I соединения формулы (III) по настоящему изобретению являются двойными ингибиторами в отношении митоза и ангиогенеза.
2) Лекарственные средства являются более эффективными благодаря координации мультикиназ, и демонстрируют лучшие фармакологические активности, такие как энзимология, цитология и фармакокинетику и так далее.
3) Соединение по настоящему изобретению имеет лучшие фармакокинетические свойства, физико-химические свойства и/или токсикологические свойства, а также возможность быть использованным в качестве лекарственного средства.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Чтобы проиллюстрировать воплощения настоящего изобретения и технические решения из предшествующего уровня техники более наглядно, графические материалы, используемые в воплощениях и предшествующем уровне техники, коротко представлены ниже. Очевидно, что графические материалы в следующем описании представляют собой лишь некоторые воплощения настоящего изобретения, и специалисты в данной области техники могут получить другие графические материалы на основании этих графических материалов без какой-либо творческой работы.
Фиг. 1 представляет собой картину дифракции рентгеновских лучей на порошке (XRPD) кристаллической формы I соединения формулы (III).
Фиг. 2 представляет собой картину дифференциального сканирующего термального анализа (калориметрии) (DSC) кристаллической формы I соединения формулы (III).
ВОПЛОЩЕНИЯ
Настоящее изобретение будет дополнительно подробно описано ниже со ссылкой на прилагаемые графические материалы, так чтобы проиллюстрировать задачи, технические решения и преимущества настоящего изобретения более наглядно. Очевидно, что описанные воплощения являются лишь частью воплощений изобретения, а не всеми из них. Все другие воплощения, полученные специалистами в данной области техники на основании воплощений по настоящему изобретению без творческих усилий, входят в объем настоящего изобретения.
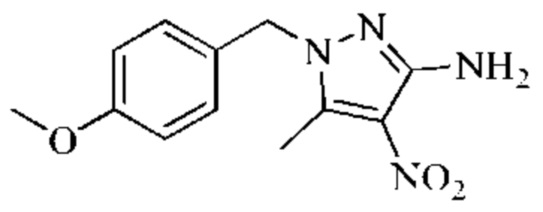
Подготовительный пример 1: синтез промежуточного соединения 1-(4-метоксибензил)-5-метил-4-нитро-1H-пиразол-3-амина
Стадия 1: синтез N-(5-метил-1H-пиразол-3-ил)ацетамида
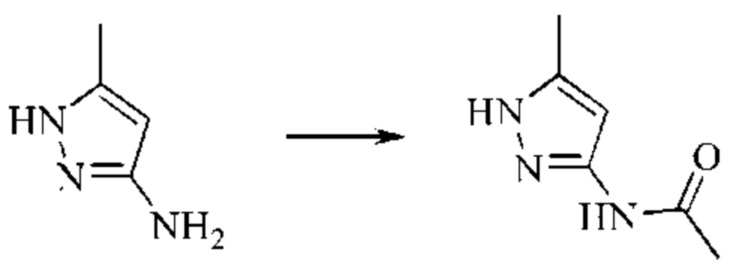
5-Метил-1H-пиразол-3-амин (300 г; 3,09 моль; 1,0 экв.) взвешивали в круглодонную колбу на 5 л, растворяли путем добавления воды (2800 мл) при механическом перемешивании при комнатной температуре. Порциями добавляли бикарбонат натрия (780 г; 9,28 моль; 3 экв.) с последующим перемешиванием в течение еще 30 минут после добавления. Затем медленно и по каплям в реакционную систему добавляли уксусный ангидрид (592 мл; 6,2 моль; 2 экв.) в течение примерно 1 часа при контролируемой скорости подачи капель. На данном этапе образовывалось большое количество пенистого белого твердого вещества. Температуру повышали до 100°С, и реакционную систему перемешивали в течение 2 часов. Твердое вещество постепенно растворялось, и система становилась прозрачной. Затем нагревание прекращали, и систему охлаждали до комнатной температуры. После перемешивания в течение ночи в осадок выпадало большое количество белого кристаллического твердого вещества. Другую партию 5-метил-1H-пиразол-3-амина (300 г) подвергали параллельному взаимодействию. После завершения взаимодействий эти две порции объединяли и фильтровали, и твердое вещество промывали водой (500 мл×2) и сушили с получением белого твердого вещества (554 г; выход 62%).
1Н ЯМР (ядерный магнитный резонанс) (400 МГц, DMSO (диметилсульфоксид)-d6) δ (м.д. (миллионная доля)): 10.32 (s, 1H), 6.21 (s, 1H), 2.18 (s, 3Н), 1.97 (s, 3Н).
Стадия 2: синтез N-(5-метил-4-нитро-1H-пиразол-3-ил)ацетамида
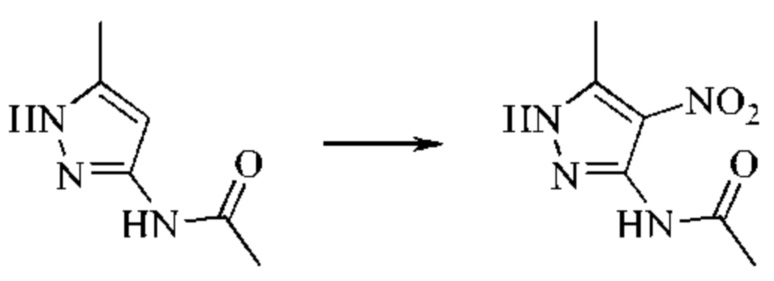
Концентрированную серную кислоту (2 л; примерно 36,8 моль; 9,2 экв.) загружали в 5 л круглодонную колбу и порциями добавляли N-(5-метил-1H-пиразол-3-ил)ацетамид (554 г; 3,98 моль; 1 экв.) в течение 1 часа при механическом перемешивании на бане с ледяной водой. Перемешивание продолжали до тех пор, пока твердое вещество полностью не растворялось. Затем в смесь добавляли дымящую азотную кислоту (250 мл; примерно 5,7 моль; 1,4 экв.) в течение 2 часов при контролируемой температуре ниже 15°С. Окончание взаимодействия контролировали посредством ЖХ-МС (жидкостная хроматография-масс-спектрометрия), после того как взаимодействие продолжалось в течение еще 15 минут. Осаждалось большое количество белого твердого вещества немедленно после того, как реакционную смесь медленно выливали в 5 л воды с колотым льдом при механическом перемешивании. Смесь оставляли отстаиваться в течение ночи и фильтровали. Полученное твердое вещество промывали водой (1000 мл×2) с получением белого твердого вещества (587 г; выход 80%) после сушки с помощью инфракрасного излучения.
1H ЯМР (400 МГц, DMSO-d6) δ (м.д.): 10.22 (s, 1H), 2.44 (s, 1H), 2.13 (s, 3Н).
Стадия 3: Синтез 5-метил-4-нитро-1H-пиразол-3-амина
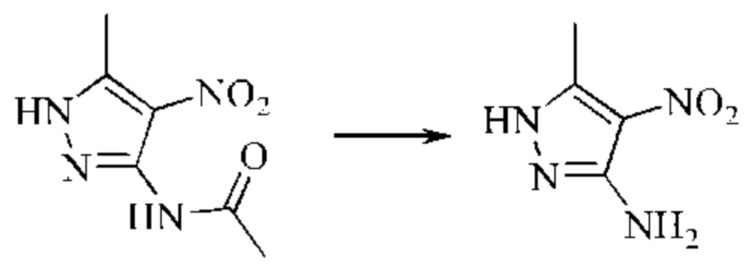
Воду (1,1 л) и концентрированную соляную кислоту (1 л; примерно 12 моль; 4 экв.) загружали в четырехгорлую круглодонную колбу на 5 л и постепенно нагревали до 80°С. Порциями добавляли N-(5-метил-4-нитро-1H-пиразол-3-ил)ацетамид (587 г; 3 моль; 1 экв.) в течение примерно 1 часа при механическом перемешивании. Смесь продолжали нагревать с обратным холодильником при 100°С в течение примерно 1 часа, пока смесь не становилась прозрачной. Смесь фильтровали после охлаждения для удаления нерастворимых веществ. Фильтрат концентрировали при пониженном давлении с получением неочищенного продукта, который растирали с метил-трет-бутиловым эфиром и фильтровали. Полученный осадок на фильтре сушили с получением оранжевого твердого вещества (610 г неочищенного продукта).
Стадия 4: Синтез 1-(4-метоксибензил)-5-метил-4-нитро-1H-пиразол-3-амина
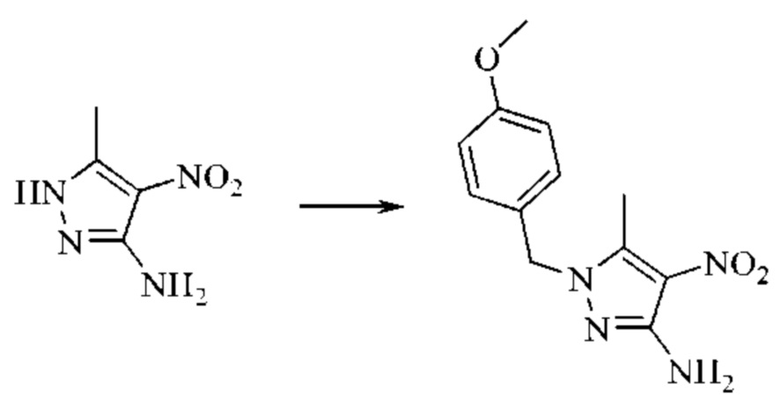
5-Метил-4-нитро-1H-пиразол-3-амина гидрохлорид (200 г; 1,12 моль; 1 экв.) загружали в 3 л круглодонную колбу, растворяли в DMF (диметилформамид) (1,8 л) при механическом перемешивании при комнатной температуре, медленно порциями добавляли карбонат калия (335 г; 2,42 моль; 2,1 экв.) в течение примерно 40 минут и затем по каплям добавляли 4-метоксибензилхлорид (177 г; 1,13 моль; 1 экв.) в течение 30 минут. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Посредством ТСХ (тонкослойная хроматография) и ЖХ-МС было установлено, что осталось небольшое количество исходного вещества. Две другие порции 5-метил-4-нитро-1H-пиразоле-3-амина гидрохлорида (200 г; 1,12 моль) подвергали параллельным взаимодействиям. После завершения взаимодействий смеси фильтровали. Фильтраты объединяли, концентрировали при пониженном давлении, пока не оставалась половина растворителя, выливали в ледяную воду (примерно 2,5 л) при перемешивании для осаждения коричневато-желтого твердого вещества и оставляли отстаиваться в течение ночи. Осадок на фильтре промывали DCM (дихлорметан) (1000 мл×3), концентрировали при пониженном давлении, выливали в ледяную воду (примерно 1000 мл) для осаждения коричневато-желтого твердого вещества и оставляли отстаиваться в течение ночи. Присоединяли осажденное ранее твердое вещество, промывали водой (500 мл×2). После сушки под вакуумом твердое вещество растирали с этилацетатом, фильтровали и сушили с получением желтого твердого вещества (268 г; выход 30%).
1Н ЯМР (400 МГц, DMSO-d6) δ (м.д.): 7.18 (d, J=8,6 Гц, 2Н), 6.90 (d, J=8,6 Гц, 2Н), 6.18 (s, 2Н), 5.09 (s, 2Н), 3.73 (s, 3Н), 2.56 (s, 3Н).
Подготовительный пример 2: Синтез промежуточного соединения (6-бром-4-иод-пиридин-3-ил)(2-хлорфенил)метанона
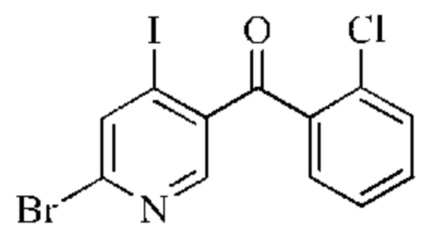
Стадия 1: Синтез (6-бромпиридин-3-ил)(2-хлорфенил)метанола
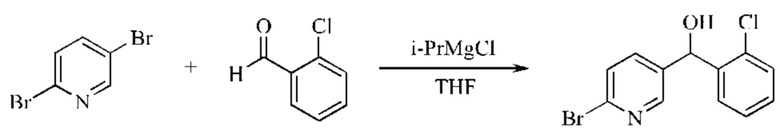
Безводный тетрагидрофуран (500 мл) и 2,5-дибромпиридин (100,0 г; 0,42 моль; 1,0 экв.) добавляли в четырехгорлую колбу на 2 л, и смесь охлаждали до 2°С при перемешивании на бане с ледяной водой. Затем по каплям добавляли хлорид изопропилмагния (210,5 мл; 2,0 М; 0,42 моль; 1,0 экв.) в течение примерно 0,5 часа при контролируемой температуре не выше 10°С. Реакционную смесь перемешивали при комнатной температуре (20°С) в течение 1 часа, затем охлаждали до 10°С на бане с ледяной водой. По каплям добавляли раствор 2-хлорбензальдегида (62,3 г; 0,443 моль; 1,05 экв.) в тетрагидрофуране (200 мл) в течение примерно 0,5 часа. После перемешивания при 10°С в течение 2 часов окончание взаимодействия контролировали посредством ТСХ. В реакционную смесь добавляли насыщенный водный раствор хлорида аммония (300 мл). После перемешивания в течение 10 минут органическую фазу отделяли от смеси и концентрировали до желтого масла. Водную фазу экстрагировали EtOAc (этилацетатом) (1,0 л×2). Полученное вещество затем объединяли с полученным ранее желтым маслом, промывали водой (500 мл), затем насыщенным рассолом (500 мл), сушили над безводным Na2SO4 и концентрировали с получением коричневого масла (140 г неочищенного продукта).
Стадия 2: Синтез (6-бромпиридин-3-ил)(2-хлорфенил)метанона
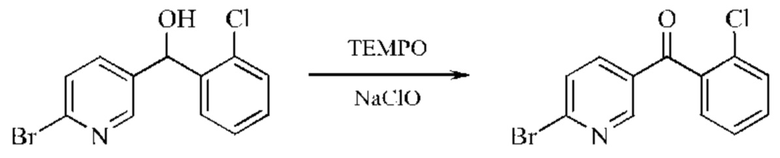
(6-Бромпиридин-3-ил)(2-хлорфенил)метанол (140 г неочищенного продукта) добавляли в DCM (1,3 л), затем добавляли TEMPO ((2,2,6,6-тетраметилпиперидин-1-ил)оксил) (1,51 г; 9,4 ммоль) и NaBr (1,92 г; 18,8 ммоль). Реакционную смесь охлаждали до 3°С на бане с ледяной водой и по каплям добавляли водный раствор NaClO (1,34 моль/л; 600 л; 0,71 моль), нейтрализованный NaHCO3 (45,0 г), при температуре не выше 20°С. После завершения добавления реакционную смесь перемешивали в течение 10 минут, и затем окончание взаимодействия контролировали посредством ТСХ. Водную фазу, отделенную от реакционной смеси, экстрагировали DCM (1,0 л). Органические фазы объединяли, промывали водой (1,0 л) затем насыщенным рассолом (1,0 л) и сушили над безводным Na2SO4. После концентрирования получали желтое масло, растирали его со смесью 150 мл метил-трет-бутилового эфира/500 мл петролейного эфира с получением желтого твердого вещества (50,3 г; выход: 39,7% за две стадии).
Стадия 3: Синтез (6-бром-4-иод-пиридин-3-ил)(2-хлорфенил)метанона
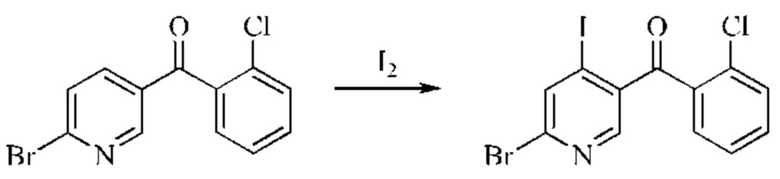
В атмосфере азота раствор лития тетраметилпиперидин/хлорида магния (281 мл; 1,5 моль/л; 0,43 моль; 2,5 экв.) добавляли в 2 л четырехгорлую колбу и охлаждали до -65°С на бане со смесью сухой лед/этанол. По каплям добавляли раствор (6-бромпиридин-3-ил)(2-хлорфенил)метанона (50,0 г; 0,17 моль; 1,0 экв.) в тетрагидрофуране (50 мл) в течение примерно 0,5 часа. Затем реакционную смесь нагревали до -45°С при перемешивании в течение 1 часа и затем снова охлаждали до -65°С. По каплям добавляли раствор I2 (129,3 г; 0,51 моль; 3,0 экв.) в тетрагидрофуране (400 мл) в течение примерно 1 часа. Реакционную систему перемешивали в течение 20 минут, и окончание взаимодействия контролировали посредством ТСХ. Затем в реакционную смесь добавляли насыщенный водный раствор хлорида аммония (500 мл) и насыщенный водный раствор NaHSO3 (500 мл) и перемешивали в течение 15 минут, затем фильтровали. Нерастворенные вещества промывали этилацетатом (500 мл×2). Фильтраты объединяли. Отделенную водную фазу экстрагировали EtOAc (1,0 л×2). Все органические фазы объединяли, промывали водой (800 мл), затем насыщенным рассолом (800 мл), сушили над безводным Na2SO4 и концентрировали с получением желтого твердого вещества. Желтое твердое вещество растирали со смесью метил-трет-бутиловый эфир (500 мл)/петролейный эфир (500 мл) и сушили с получением желтого твердого вещества (30 г; выход: 41,8%).
1H ЯМР (400 МГц, CDCl3) δ (м.д.): 7.38-7.50 (m, 2Н), 7.51-7.65 (m, 2Н), 8.17 (s, 1H), 8.24 (s, 1H).
Подготовительный пример 3: Синтез промежуточного соединения (6-бром-4-((1-(4-метоксибензил)-5-метил-4-нитро-1H-пиразол-3-ил)амино)пиридин-3-ил)(2-хлорфенил)метанона
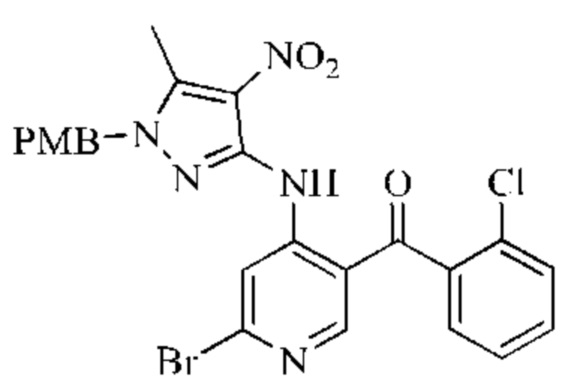
Стадия 1: Синтез (6-бром-4-((1-(4-метоксибензил)-5-метил-4-нитро-1H-пиразол-3-ил)амино)пиридин-3-ил)(2-хлорфенил)метанона
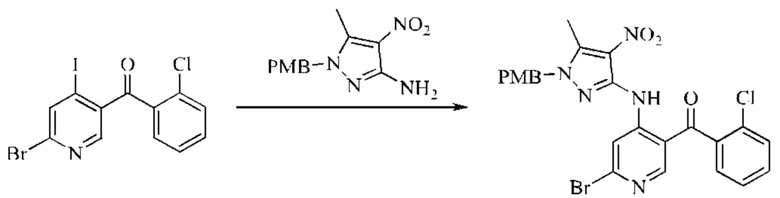
1-(4-Метоксибензил)-5-метил-4-нитро-1H-пиразол-3-амин (14,92 г; 56,9 ммоль) добавляли в безводный тетрагидрофуран (100 мл), растворяли при перемешивании в атмосфере азота. Порциями добавляли NaH (40% по массе; 4,82 г; 0,11 моль) на бане с ледяной водой и после добавления перемешивали в течение 1 часа, затем по каплям добавляли раствор (6-бром-4-иод-пиридин-3-ил)(2-хлорфенил)метанона (20 г; 47,4 ммоль) в тетрагидрофуране (100 мл). Взаимодействие проводили в течение 16 часов при комнатной температуре. Окончание взаимодействия контролировали посредством ТСХ. Для остановки реакции добавляли метанол (30 мл), затем насыщенный раствор хлорида аммония (50 мл). Полученную смесь фильтровали с получением желтого продукта (25 г; выход 80%).
1Н ЯМР (400 МГц, DMSO-d6): 12.36 (s, 1H), 8.77 (s, 1H), 8.11 (s, 1H), 7.66-7.53 (m, 4Н), 7.33 (d, J=8,8 Гц, 1H), 7.33 (d, J=8,8 Гц, 1H), 6.96 (d, J=8,8 Гц, 1H), 5.39 (s, 2Н), 3.75 (s, 3Н), 2.73(s, 3Н).
Пример 1: Синтез соединения 1
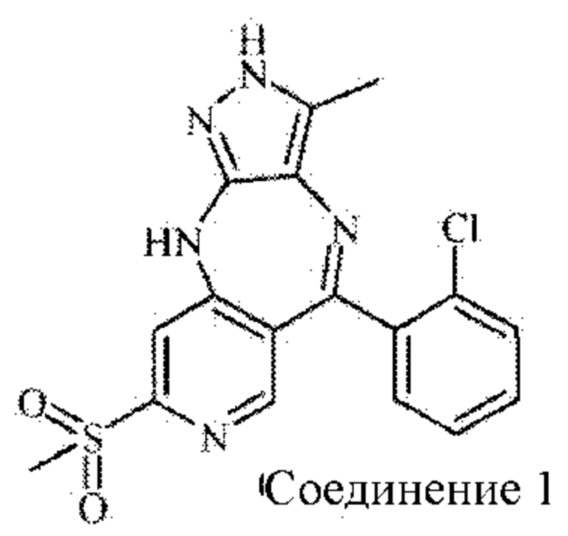
Путь синтеза:
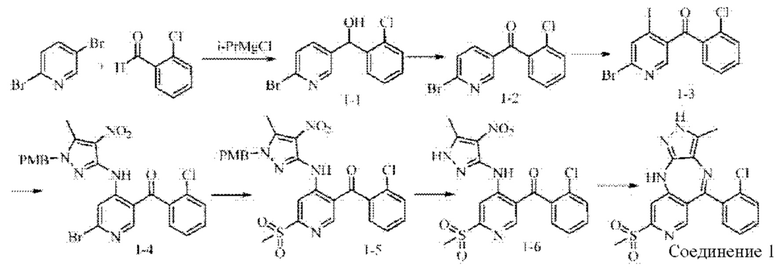
Стадия 1: Промежуточное соединение 1-3 получали согласно способу, описанному в Подготовительном примере 2.
Стадия 2: Промежуточное соединение 1-4 получали согласно способу, описанному в Подготовительном примере 3.
Стадия 3: Синтез промежуточного соединения 1-5
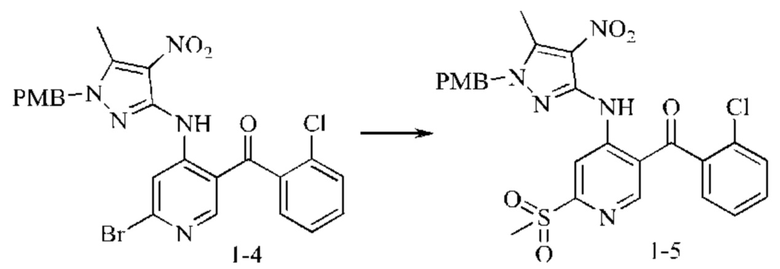
Промежуточное соединение 1-4 (5 г; 9 ммоль) добавляли в йодид меди (5,13 г; 27 ммоль) и метансульфинат натрия (2,76 г; 27 ммоль). Затем вводили DMSO (100 мл), после того как реактор трижды продували азотом. Реакционную систему нагревали до 130°С и подвергали взаимодействию в течение 5 часов. Окончание взаимодействия контролировали посредством ТСХ. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (элюировали дихлорметаном) с получением 1,1 г желтого твердого вещества с выходом 23%.
Стадия 4: Синтез промежуточного соединения 1-6
Промежуточное соединение 1-5 (1,1 г; 2,0 ммоль) добавляли в дихлорметан (3 мл) и растворяли. Медленно по каплям добавляли трифторуксусную кислоту (10 мл). Смесь нагревали до 70°С и подвергали взаимодействию в течение 8 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. Растворитель и трифторуксусную кислоту выпаривали на роторном испарителе с получением 1,5 г коричневого твердого вещества, которое непосредственно использовали на следующей стадии.
Стадия 5: синтез соединения 1
Промежуточное соединение 1-6 (1,5 г; 3,4 ммоль) добавляли в 2-метилтетрагидрофуран (15 мл) и растворяли. Дигидрат дихлорида олова (5,4 г; 24,1 ммоль) добавляли в атмосфере азота. Смесь нагревали до 100°С и подвергали взаимодействию в течение 16 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. рН смеси доводили до значения 10 путем добавления водного раствора гидроксида натрия, и смесь фильтровали через целит. Фильтрат экстрагировали 2-метилтетрагидрофураном и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (ЕА (этилацетат) : DCM, 1:3) с получением соединения 1 в виде желтого твердого вещества (60 мг; выход 10%).
1Н ЯМР (400 МГц, CDCl3) δ: 11.84 (s, 1H), 9.12 (s, 1H), 7.43-7.51 (m, 4Н), 7.17 (s, 1H), 7.11 (s, 1H), 3.11 (s, 3Н), 1.96 (s, 3Н).
Пример 2: Синтез соединения 2
Стадия 1: Синтез промежуточного соединения 2-1
Промежуточное соединение 1-4 (3 г; 5,4 ммоль) добавляли в толуол (90 мл), циклопропилбороновую кислоту (0,70 г; 8,1 ммоль), ацетат палладия (0,121 г; 0,54 ммоль), трициклогексилфосфин (0,310 г; 1,1 ммоль) и фосфат калия (4,0 г; 18,8 ммоль). Реактор трижды продували азотом. Реакционную систему нагревали до 120°С и подвергали взаимодействию в течение 16 часов. Окончание взаимодействия контролировали посредством ТСХ. После охлаждения смесь экстрагировали ЕА (100 мл), и отделенную водную фазу экстрагировали ЕА (20 мл). Органические фазы объединяли, промывали насыщенным водным хлоридом натрия, сушили над безводным сульфатом натрия, фильтровали, концентрировали и растирали со смесью этилацетат : метил-трет-бутиловый эфир (1:3; 10 мл). Смесь сушили с получением оранжевого твердого вещества (3,0 г; выход 86%).
Стадия 2: Синтез промежуточного соединения 2-2
Промежуточное соединение 2-1 (2,9 г; 5,6 ммоль) добавляли в толуол (60 мл) и растворяли. Медленно по каплям добавляли трифторуксусную кислоту (30 мл). После добавления смесь нагревали до 100°С и подвергали взаимодействию в течение 8 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. Растворитель и трифторуксусную кислоту выпаривали на роторном испарителе с получением 1,9 г коричневого твердого вещества, которое непосредственно использовали на следующей стадии.
Стадия 3: синтез соединения 2
Промежуточное соединение 2-2 (1,9 г; 3,7 ммоль) добавляли в 2-метилтетрагидрофуран (35 мл) и растворяли. Дигидрат дихлорида олова (5,4 г; 24,8 ммоль) медленно добавляли в атмосфере азота. Смесь нагревали до 100°С и подвергали взаимодействию в течение 16 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. рН смеси доводили до значения 10 путем добавления водного раствора гидроксида натрия, и смесь фильтровали через целит. Фильтрат экстрагировали 2-метилтетрагидрофураном и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (элюировали смесью EA : DCM, 1:3) с получением желтого твердого вещества (0,35 г; выход 27%).
1Н ЯМР (400 МГц, CDCl3) δ: 11.65 (s, 1H), 8.42 (s, 1H), 7.33-7.47 (m, 4Н), 7.01 (s, 1H), 6.40 (s, 1H), 1.96 (s, 3Н), 1.71-1.75 (m, 1H), 0.72-0.84 (m, 4Н).
Пример 3: Синтез соединения 3
Стадия 1: Синтез промежуточного соединения 3-1
Промежуточное соединение 1-4 (3 г; 5,4 ммоль) добавляли в THF (300 мл). В атмосфере азота по каплям добавляли раствор метилат натрия-метанол (полученный посредством взаимодействия гидрида натрия (1,4 г; 35 ммоль) и метанола (6 мл)). Смесь нагревали до 50°С и подвергали взаимодействию в течение 16 часов. Окончание взаимодействия контролировали посредством ТСХ. После охлаждения реакционную смесь добавляли в насыщенный водный раствор хлорида аммония (20 мл) и затем экстрагировали 2-метилтетрагидрофураном (50 мл). Органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт растирали со смесью метанол/метил-трет-бутиловый эфир (1:3) с получением желтого твердого вещества (2,1 г; выход 77%).
Стадия 2: Синтез промежуточного соединения 3-2
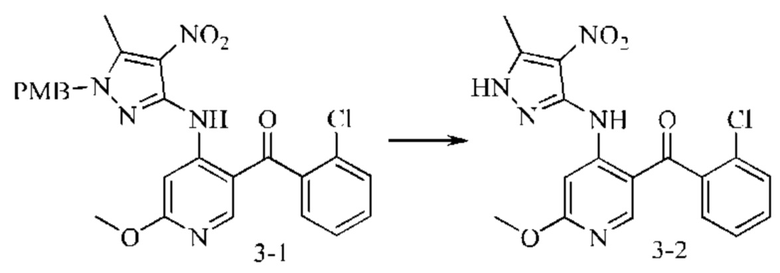
Промежуточное соединение 3-1 (1,8 г; 3,5 ммоль) добавляли в толуол (30 мл) и растворяли. Медленно по каплям добавляли трифторуксусную кислоту (20 мл; 0,268 моль). Смесь нагревали до 110°С и подвергали взаимодействию в течение 16 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. Растворитель и трифторуксусную кислоту выпаривали на роторном испарителе. Остаток растирали с метил-трет-бутиловым эфиром (20 мл) и фильтровали с получением красного твердого вещества (0,92 г; выход 52%).
Стадия 3: Синтез соединения 3
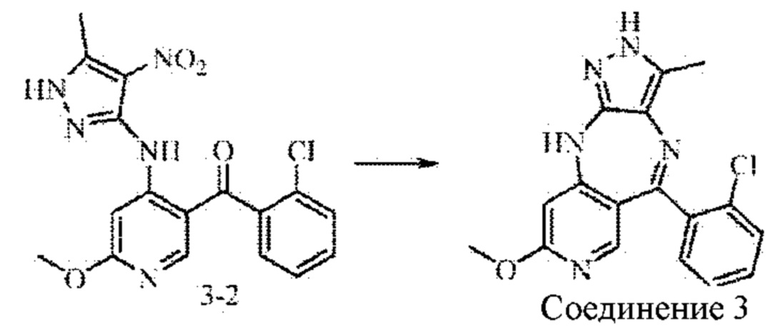
Промежуточное соединение 3-2 (0,92 г; 1,8 ммоль) добавляли в 2-метилтетрагидрофуран (15 мл) и растворяли. Дигидрат дихлорида олова (2,9 г; 12,8 ммоль) добавляли в атмосфере азота. Смесь нагревали до 100°С и подвергали взаимодействию в течение 16 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. рН смеси доводили до значения 10 путем добавления водного раствора гидроксида натрия, и смесь фильтровали через целит. Фильтрат экстрагировали 2-метилтетрагидрофураном и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (элюирование смесью EA : DCM, 1:3) с получением соединения 3 в виде желтого твердого вещества (0,32 г; выход 52%).
1Н ЯМР (400 МГц, CDCl3) δ: 11.69 (s, 1H), 8.60 (s, 1H), 7.36-7.49 (m, 4Н), 6.90 (s, 1H), 5.96 (s, 1H), 3.68 (s, 3Н), 1.99 (s, 3Н).
Пример 4: Синтез соединения 4
Стадия 1: синтез промежуточного соединения 4-1
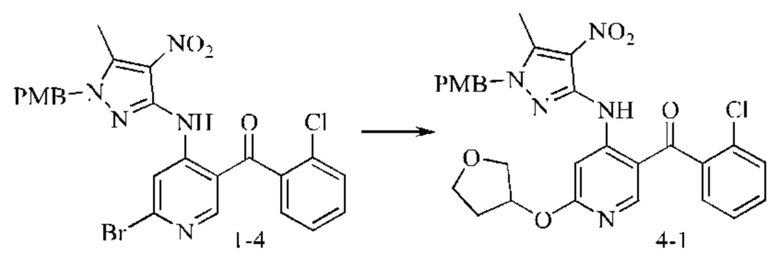
Промежуточное соединение 1-4 (3 г; 5,4 ммоль) добавляли в карбонат цезия (6,8 г; 26,5 ммоль), 3,4,7,8-тетраметил-1,10-фенантролин (0,127 г; 0,54 ммоль), йодид меди (0,080 г; 0,54 ммоль) и 3-гидрокситетрагидрофуран (1,0 г; 10,8 ммоль). Реактор трижды продували азотом и добавляли толуол (20 мл). Реакционную смесь нагревали до 100°С и подвергали взаимодействию в течение 16 часов. Окончание взаимодействия контролировали посредством ТСХ. После охлаждения реакционную смесь добавляли в насыщенный водный раствор хлорида аммония (20 мл). Отделенную водную фазу экстрагировали этилацетатом (50 мл). Органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт растирали со смесью метанол/метил-трет-бутиловый эфир (1:3) с получением желтого твердого вещества (0,7 г; выход 23%).
Стадия 2: Синтез промежуточного соединения 4-2
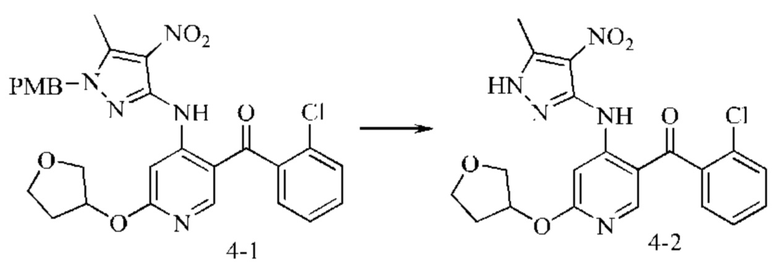
Промежуточное соединение 4-1 (0,7 г; 1,3 ммоль) добавляли в дихлорметан (3 мл) и растворяли. Медленно по каплям добавляли трифторуксусную кислоту (10 мл). После добавления смесь нагревали до 70°С и подвергали взаимодействию в течение 16 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. Растворитель и трифторуксусную кислоту выпаривали на роторном испарителе с получением красного твердого вещества (0,94 г), который использовали непосредственно на следующей стадии.
Стадия 3: синтез соединения 4
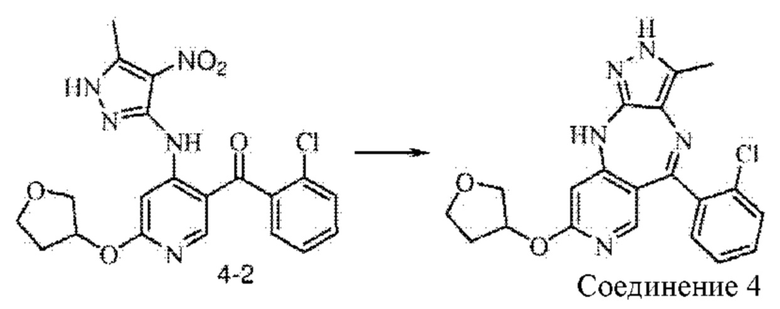
Промежуточное соединение 4-2 (0,94 г; 2,1 ммоль) добавляли в 2-метилтетрагидрофуран (15 мл) и растворяли. Дигидрат дихлорида олова (3,34 г; 14,8 ммоль) добавляли в атмосфере азота. Смесь нагревали до 100°С, и подвергали взаимодействию в течение 16 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. рН смеси доводили до значения 10 путем добавления водного раствора гидроксида натрия, и смесь фильтровали через целит. Фильтрат экстрагировали 2-метилтетрагидрофураном и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (элюирование смесью EA : DCM, 1:3) с получением соединения 4 в виде желтого твердого вещества (0,066 г; выход 10%).
1Н ЯМР (400 МГц, CDCl3) δ: 11.69 (s, 1H), 8.65 (s, 1H), 7.37-7.49 (m, 4Н), 6.88 (s, 1H), 5.92 (s, 1H), 5.29-5.30 (m, 1H), 3.61-3.78 (m, 4Н), 2.07-2.12 (m, 1H), 2.00 (s, 3Н), 1.86-1.90 (m, 1H).
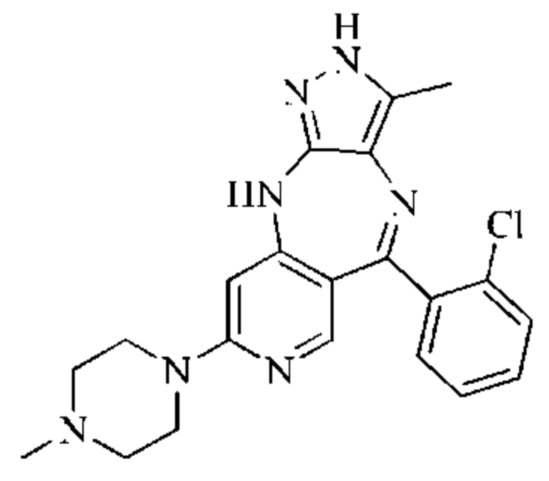
Пример 5: Синтез 5-(2-хлорфенил)-3-метил-8-(4-метилпиперазин-1-ил)-2,10-дигидропиразоло[4,3-b]пиридо[4,3-е][1,4]диазепина (соединение 22)
Стадия 1: Синтез (2-хлорфенил)(4-((1-(4-метоксибензил)-5-метил-4-нитро-1H-пиразол-3-ил)амино)-6-(4-метилпиперазин-1-ил)пиридин-3-ил)метанона
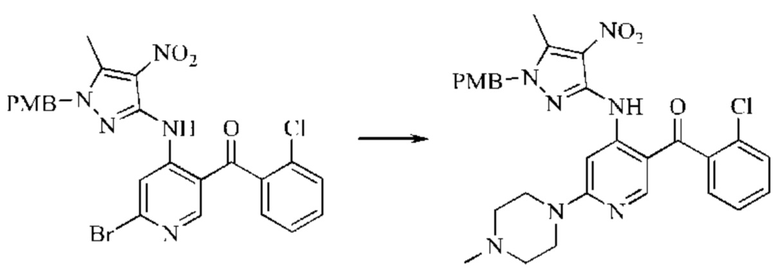
(6-Бром-4-((1-(4-метоксибензил)-5-метил-4-нитро-1Н-пиразол-3-ил)амино)пиридин-3-ил)(2-хлорфенил)метанон (промежуточное соединение 1-4) (0,80 г; 1,4 ммоль) растворяли в DMSO (10 мл), добавляли в метилпиперазин (0,431 г; 4,3 ммоль). Смесь нагревали до 110°С, подвергали взаимодействию в течение 4 часов. Анализ посредством ТСХ показал окончание взаимодействия. После охлаждения реакционную смесь выливали в воду (100 мл), и в осадок выпадало большое количество твердого вещества. Смесь фильтровали. Осадок на фильтре растворяли в дихлорметане, сушили и концентрировали под вакуумом с получением желтого твердого вещества (1,0 г неочищенного продукта).
Стадия 2: синтез (2-хлорфенил)(4-((5-метил-4-нитро-1H-пиразол-3-ил)амино)-6-(4-метилпиперазин-1-ил)пиридин-3-ил)метанона
(2-Хлорфенил-4-((1-(4-метоксибензил)-5-метил-4-нитро-1H-пиразол-3-ил)амино)-6-(4-метилпиперазин-1-ил)пиридин-3-ил)метанон (1,0 г; неочищенный продукт) растворяли в дихлорметане (3 мл), затем медленно по каплям добавляли в трифторуксусную кислоту (10 мл). Затем смесь нагревали до 70°С и подвергали взаимодействию в течение 16 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. Смесь концентрировали под вакуумом с получением красного твердого вещества (1,2 г; неочищенный продукт), которое использовали непосредственно на следующей стадии без очистки.
Стадия 3: Синтез 5-(2-хлорфенил)-3-метил-8-(4-метилпиперазин-1-ил)-2,10-дигидропиразоло[4,3-b]пиридо[4,3-е][1,4]диазепина
(2-Хлорфенил)(4-((5-метил-4-нитро-1H-пиразол-3-ил)амино)-6-(4-метилпиперазин-1-ил)пиридин-3-ил)метанон (1,2 г; неочищенный продукт) растворяли в 2-метилтетрагидрофуране (20 мл), добавляли в дигидрат дихлорида олова (4,2 г; 18,6 ммоль) в атмосфере азота. Смесь нагревали до 90°С и подвергали взаимодействию в течение 16 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. рН смеси доводили до значения 10 путем добавления водного раствора гидроксида натрия, и смесь фильтровали через целит. Фильтрат экстрагировали 2-метилтетрагидрофураном и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (дихлорметан : метанол, 10:1) с получением соединения 22 в виде желтого твердого вещества (0,033 г; выход: 5,8% за три стадии).
1Н ЯМР (400 МГц, DMSO-d6): 11.56 (s, 1H), 8.23 (s, 1H), 7.33-7.45 (m, 4Н), 6.88 (s, 1H), 5.96 (s, 1H), 3.37 (m, 4Н), 2.33 (m, 4Н), 2.19 (s, 3Н), 1.97 (s, 3Н).
Молекулярная формула: C21H22ClN7; молекулярная масса: 407,91; ЖХ-МС (Pos, m/z) составляет 408 [М+Н+].
Пример 6: Синтез 4-(5-(2-хлорфенил)-3-метил-2,10-дигидропиразоло[4,3-b]-пиридо[4,3-е][1,4]диазепин-8-ил)морфолина (соединение 29)
Стадия 1: Синтез (6-бром-4-((5-метил-4-нитро-1H-пиразол-3-ил)амино)пиридин-3-ил)(2-хлорфенил)метанона
(6-Бром-4-((1-(4-метоксибензил)-5-метил-4-нитро-1Н-пиразол-3-ил)амино)пиридин-3-ил)(2-хлорфенил)метанон (промежуточное соединение 1-4) (0,80 г; 1,4 ммоль) растворяли в DCM (2 мл), затем добавляли трифторуксусную кислоту (10 мл). Смесь нагревали до 70°С и подвергали взаимодействию в течение 4 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. Реакционную смесь охлаждали и концентрировали под вакуумом с получением желтого твердого вещества (1,0 г; неочищенный продукт).
Стадия 2: Синтез 8-бром-5-(2-хлорфенил)-3-метил-2,10-дигидропиразоло[4,3-b]-пиридо[4,3-е][1,4]диазепина
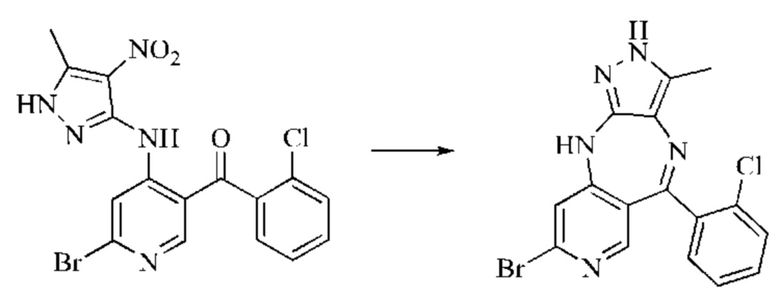
(6-Бром-4-((5-метил-4-нитро-1H-пиразол-3-ил)амино)пиридин-3-ил)(2-хлорфенил)-метанон (1,0 г; неочищенный продукт) растворяли в 2-метилтетрагидрофуране (15 мл), затем добавляли в дигидрат дихлорида олова (3,2 г; 14,2 ммоль). Смесь нагревали до 100°С и подвергали взаимодействию в течение 16 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. рН смеси доводили до значения 10 путем добавления водного раствора гидроксида натрия, и смесь фильтровали через целит. Фильтрат экстрагировали 2-метилтетрагидрофураном и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (дихлорметан : метанол, 30:1) с получением продукта (60 мг; выход 10,7% за две стадии).
Стадия 3: синтез 4-(5-(2-хлорфенил)-3-метил-2,10-дигидропиразоло[4,3-b]пиридо-[4,3-е][1,4]диазепин-8-ил)морфолина
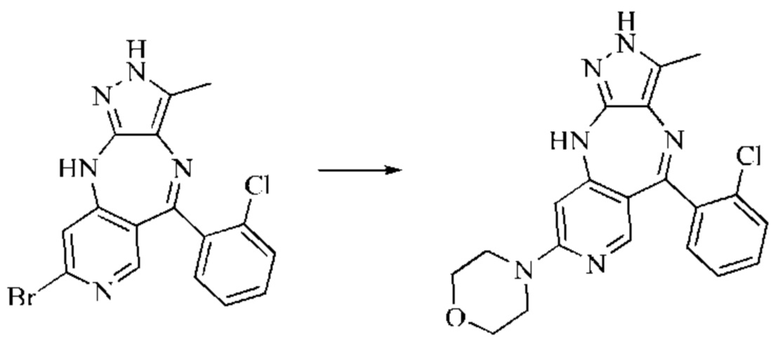
8-Бром-5-(2-хлорфенил)-3-метил-2,10-дигидропиразоло[4,3-b]пиридо[4,3-е][1,4]-диазепин (60 мг; 0,16 ммоль) растворяли в DMSO (2 мл), затем добавляли в морфолин (14 мг; 0,20 ммоль) в атмосфере азота. Смесь нагревали до 110°С и подвергали взаимодействию в течение 6 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. Реакционную смесь выливали в воду (10 мл), экстрагировали дихлорметаном (20 мл×2) и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (дихлорметан : метанол, 20:1) с получением продукта (16 мг; выход 25%).
1Н ЯМР (400 МГц, DMSO-d6): 11.56 (s, 1H), 8.29 (s, 1H), 7.33-7.47 (m, 4Н), 6.88 (s, 1H), 5.96 (s, 1H), 3.62 (m, 4Н), 3.33 (m, 4Н), 1.97 (s, 3Н).
Молекулярная формула: C20H19ClN6O, Молекулярная масса: 394,86, ЖХ-МС (Pos, m/z): 394,96 [М+Н+].
Пример 7: Синтез соединения 33
Стадия 1: Синтез промежуточного соединения 33-1
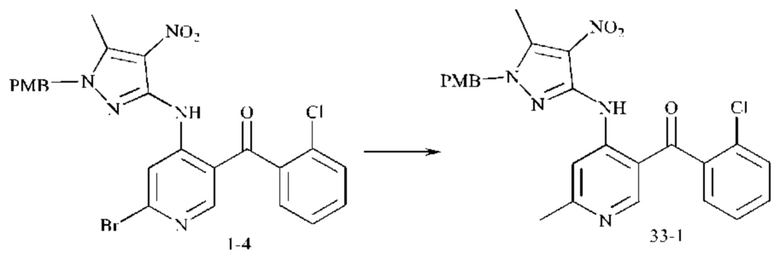
К промежуточному соединению 1-4 (3,5 г; 6,3 ммоль) добавляли 3,8 г (19,1 ммоль) карбоната калия и 1,06 г (0,63 ммоль) тетракис(трифенилфосфин)палладия. Реактор трижды продували азотом, затем в него по каплям добавляли 2 мл триметилбороксина (6,8 ммоль), затем добавляли 40 мл диоксана. Реакционную смесь нагревали до 110°С и подвергали взаимодействию в течение 16 часов. Окончание взаимодействия контролировали посредством ТСХ. После охлаждения реакционную смесь выливали в 100 мл воды, и большое количество твердого вещества выпадало в осадок. После фильтрации смесь добавляли в силикагель и подвергали колоночной хроматографии, элюируя смесью дихлорметан : метанол, 100:1, и роторному выпариванию досуха с получением 1,8 г промежуточного соединения 33-1 в виде желтого твердого вещества с выходом 58,8%.
Стадия 2: Синтез промежуточного соединения 33-2
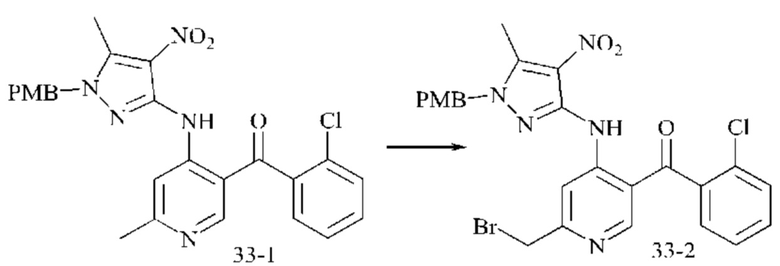
1,8 г (3,6 ммоль) промежуточного соединения 33-1 добавляли в 0,65 г NBS (N-бромсукцинимид) (3,6 ммоль) и 100 мл тетрахлорметана, перемешивали до растворения в течение 30 минут и затем добавляли 89 мг бензоилпероксида (0,36 ммоль). Реакционную смесь нагревали до 100°С и подвергали взаимодействию в течение 16 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. После выпаривания растворителя смесь растворяли в 20 мл метиленхлорида, фильтровали через силикагель и упаривали досуха с получением 2,0 г промежуточного соединения 33-2 в виде желтого твердого вещества, которое непосредственно использовали на следующей стадии.
Стадия 3: Синтез промежуточного соединения 33-3
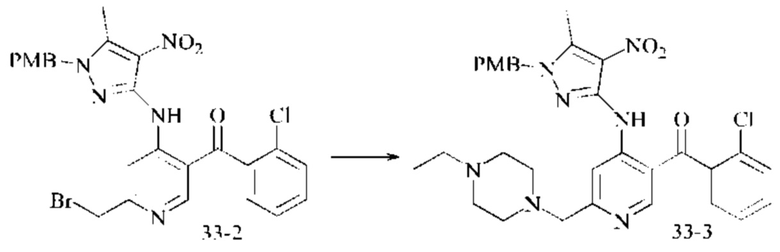
Промежуточное соединение 33-2 (2,0 г; 3,5 ммоль) добавляли в 0,55 г этилпиперазина (4,8 ммоль), 0,73 г карбоната калия (5,2 ммоль) и 20 мл ацетонитрила. Смесь нагревали до 80°С и подвергали взаимодействию в течение 8 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. Растворитель выпаривали и добавляли 100 мл DCM и 50 мл водного раствора хлорида аммония. После разделения жидкостей, сушки и концентрирования получали 1,9 г промежуточного соединения 33-3 в виде красного твердого вещества, которое непосредственно использовали на следующей стадии.
Стадия 4: Синтез промежуточного соединения 33-4
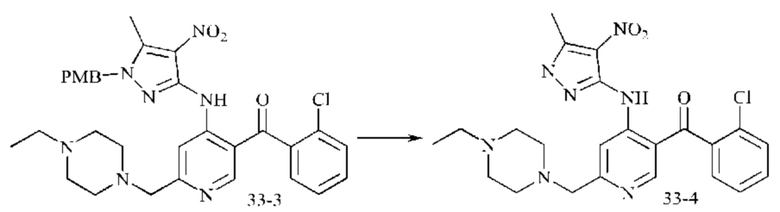
1,9 г Промежуточного соединения 33-3 (3,2 ммоль) растворяли в дихлорметане (3 мл) до растворения. Затем медленно по каплям добавляли трифторуксусную кислоту (10 мл). После добавления смесь нагревали до 70°С и подвергали взаимодействию в течение 16 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. Растворитель и трифторуксусную кислоту выпаривали с получением 2,4 г промежуточного соединения 33-4 в виде красного твердого вещества, которое непосредственно использовали на следующей стадии.
Стадия 4: синтез соединения 33
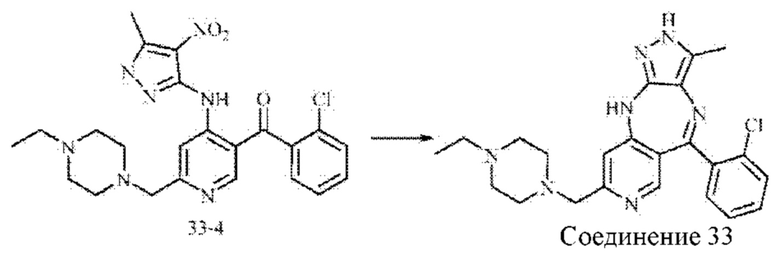
Промежуточное соединение 33-4 (2,4 г; 4,9 ммоль) растворяли в 2-метилтетрагидрофуране (25 мл). Затем добавляли дигидрат дихлорида олова (7,8 г; 34,8 ммоль) в атмосфере азота, и смесь нагревали до 90°С. Спустя 16 часов анализ посредством ЖХ-МС показал окончание взаимодействия. рН смеси доводили до значения 10 путем добавления водного раствора гидроксида натрия, и смесь фильтровали через целит. Фильтрат экстрагировали 2-метилтетрагидрофураном и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (дихлорметан : метанол, 10:1) с получением соединения 33 (0,047 г; выход 10%).
1Н ЯМР (400 МГц, DMSO): 11.50 (s, 1H), 8.72 (s, 1H), 7.43-7.47 (m, 4Н), 7.27 (s, 1H), 6.67 (s, 1H), 3.47 (s, 2Н), 3.06 (m, 4Н), 2.97 (m, 2Н), 2.71 (s, 4Н), 2.12 (s, 3Н), 1.26-1.29 (t, 3Н).
Пример 8: Синтез соединения 34
Стадия 1: Синтез промежуточного соединения 34-1
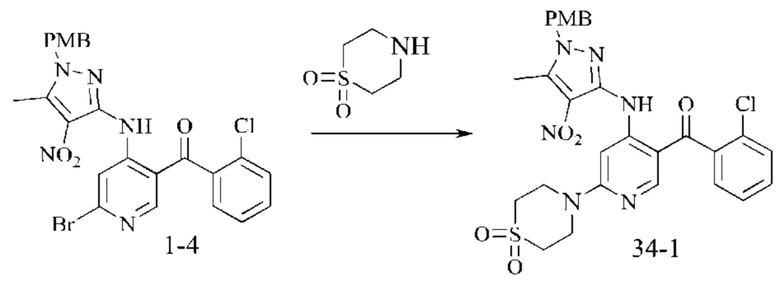
Промежуточное соединение 1-4 (120 мг; 0,216 ммоль; 1 экв.) добавляли в DMSO (2 мл), затем добавляли тиоморфолин 1,1-диоксид (58,1 мг; 0,43 ммоль; 2 экв.) и DIEA (диизопропилэтиламин) (83,6 мг; 0,65 ммоль; 3 экв.). Смесь нагревали до 80°С и подвергали взаимодействию в течение 3 часов. Окончание взаимодействия контролировали посредством ТСХ. В реакционную смесь по каплям добавляли воду (10 мл) и осаждали твердое промежуточное соединение 34-1 (70 мг; неочищенное), которое использовали непосредственно на следующей стадии.
Стадия 2: синтез промежуточного соединения 34-2
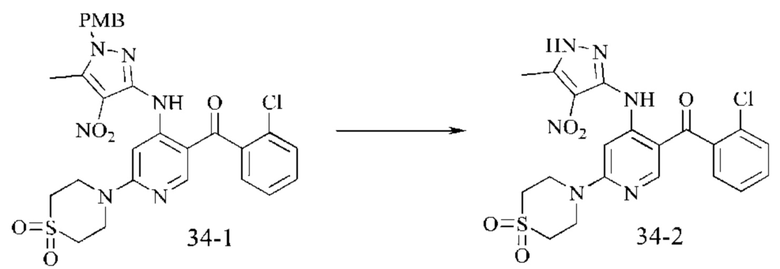
Промежуточное соединение 34-1 (70 мг; 0,11 ммоль; 1 экв.) добавляли в 2 мл трифторуксусной кислоты. Смесь нагревали до 80°С и подвергали взаимодействию в течение 5 часов. Окончание взаимодействия контролировали посредством ТСХ. Реакционную смесь выпаривали досуха и растирали с МТВЕ (метил-трет-бутиловый эфир) (10 мл) с получением промежуточного соединения 34-2 в виде желтого твердого вещества (30 мг; неочищенный продукт).
Стадия 3: Синтез соединения 34
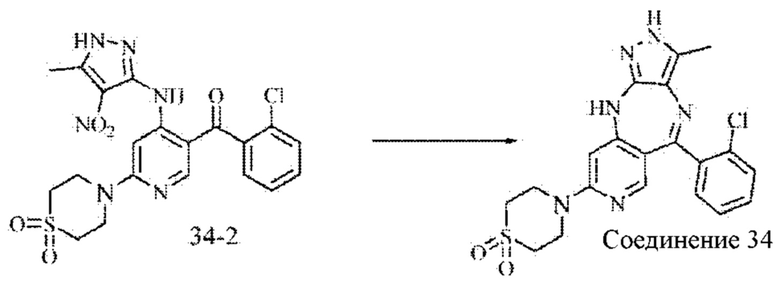
Промежуточное соединение 34-2 (300 мг; 0,62 ммоль; 1 экв.) добавляли в 25 мл одногорлую колбу, затем добавляли 2-метилтетрагидрофуран (20 мл) и хлорид олова (966 г; 4,28 ммоль; 7 экв.). Смесь нагревали до 90°С и перемешивали в течение 5 часов. Окончание взаимодействия контролировали посредством ТСХ. Реакционный раствор охлаждали, и доводили значение рН примерно до 8 гидрокарбонатом натрия. Смесь экстрагировали DCM (50 мл), и органическую фазу отделяли, сушили, объединяли и упаривали досуха с получением соединения 34 (21 мг; выход: 2,1% за три стадии) с применением силикагелевой пластины (DCM : MeOH, 30:1).
1Н ЯМР (400 МГц, DMSO-d6): 11.60 (s, 1H), 8.30 (s, 1H), 7.33-7.49 (m, 4Н), 6.92 (s, 1H), 6.11 (s, 1H), 3.88 (m, 4Н), 3.08-3.10 (m, 4Н), 1.98 (s, 3Н).
Пример 9: Синтез соединения 37
Стадия 1: взвешивали 500 мг промежуточного соединения 1-4 и растворяли в 15 мл ацетонитрила, затем добавляли 200 мг триэтиламина и 200 мг ВОС-пиперазина. Смесь нагревали до температуры дефлегмации в течение 3 часов. Окончание взаимодействия контролировали посредством ТСХ, и реакционный раствор упаривали досуха с получением промежуточного соединения 37-1 в виде желтого твердого вещества (400 мг; выход 80%).
Стадия 2: промежуточное соединение 37-1 растворяли в 15 мл TFA (трифторуксусная кислота), и смесь нагревали до температуры дефлегмации в течение 6 часов. Окончание взаимодействия контролировали посредством ЖХ-МС. Смесь упаривали досуха. После фильтрации неочищенный продукт растирали с метил-трет-бутиловым эфиром с получением соединения 37-2 в виде желтого твердого вещества (300 мг; выход более 100%).
Стадия 3: промежуточное соединение 37-2 растворяли в 15 мл метилтетрагидрофурана, затем добавляли 500 мл дигидрата дихлорида олова. Смесь нагревали до 90°С и нагревали с обратным холодильником в течение 5 часов, и взаимодействие завершали с получением соединения 37 (16 мг; выход 5%).
1Н ЯМР (400 МГц, DMSO-d6): 11.60 (s, 1H), 8.30 (s, 1H), 7.33-7.49 (m, 4Н), 6.92 (s, 1H), 6.11 (s, 1H), 3.88 (m, 4Н), 2.08-2.10 (m, 4Н), 1.98 (s, 3Н).
Пример 10: Синтез соединения 38
Стадия 1: 300 мг Вос-пиперазина, 450 мг промежуточного соединения 33-2 и 300 мг карбоната калия взвешивали и добавляли 20 мл ацетонитрила для растворения. Смесь нагревали до 60°С и подвергали взаимодействию в течение 6 часов. Окончание взаимодействия контролировали посредством ЖХ-МС. Добавляли 300 мл воды и 50 мл дихлорметана. Отделенную масляную фазу сушили и упаривали досуха с получением 300 мг промежуточного соединения 38-1, 300 мг.
Стадия 2: 15 мл трифторуксусной кислоты выливали в 300 мг соединения 38-1. Смесь нагревали до 90°С и нагревали с обратным холодильником в течение 4 часов. Окончание взаимодействия контролировали посредством ЖХ-МС, и реакционную смесь упаривали досуха с получением 200 мг соединения 38-2.
Стадия 3: взвешивали 1,10 г дигидрата дихлорида олова и добавляли промежуточное соединение 38-2, 15 мл метилтетрагидрофурана и 0,2 мл воды. Смесь нагревали до 90°С и подвергали взаимодействию в течение 16 часов. После окончания взаимодействия реакционную смесь доводили до значения рН 10, фильтровали, упаривали досуха и сушили с получением 36 мг соединения 38 с выходом 21%.
1Н ЯМР (400 МГц, DMSO): 11.69 (s, 1H), 8.49 (s, 1H), 7.38-7.49 (m, 4Н), 7.10-7.25 (m, 1H), 2.57-2.58 (m, 4Н), 2.26 (s, 2Н), 2.35 (s, 4Н), 1.98 (s, 3Н).
Пример 11: синтез соединений 40, 41, 42, 43, 44, 45, 46, 48
Путь синтеза был следующим:
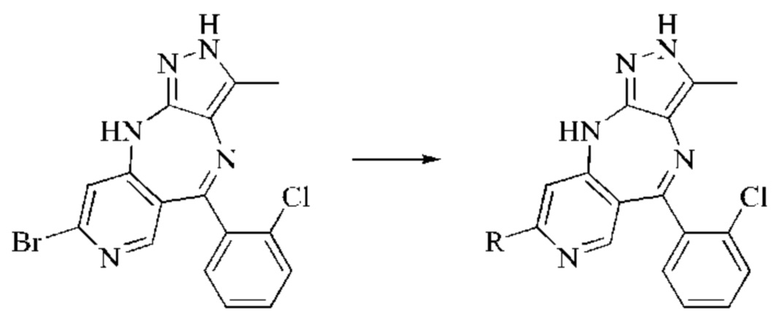
Общий способ синтеза: 8-бром-5-(2-хлорфенил)-3-метил-2,10-дигидропиразоло-[4,3-b]пиридо[4,3-е][1,4]диазепин (1 экв.), полученный на стадии 2 примера 6, растворяли в DMSO (5 мл) и добавляли амин (5 экв.) разной структуры в атмосфере азота. Смесь нагревали до 100°С и подвергали взаимодействию в течение 16 часов. Анализ посредством ТСХ показал окончание взаимодействия. Реакционную смесь выливали в ледяную воду (50 мл), и неочищенный продукт осаждали в виде желтого твердого вещества.
11.1 Синтез соединения 41: структура амина представляла собой  твердый неочищенный продукт очищали с использованием силикагелевой пластины (DCM : МеОН, 30:1) с получением соединения 41 (15 мг; выход: 13,8%).
твердый неочищенный продукт очищали с использованием силикагелевой пластины (DCM : МеОН, 30:1) с получением соединения 41 (15 мг; выход: 13,8%).
1Н ЯМР (400 МГц, CDCl3): 8.85 (s, 1H), 7.28-7.42 (m, 4Н), 5.91 (s, 1H), 5.62 (s, 1H), 3.98-4.02 (d, 2Н), 2.66-2.72 (t, 2Н), 2.29 (s, 3Н), 2.22 (s, 4Н), 1.14-1.16 (m, 6Н).
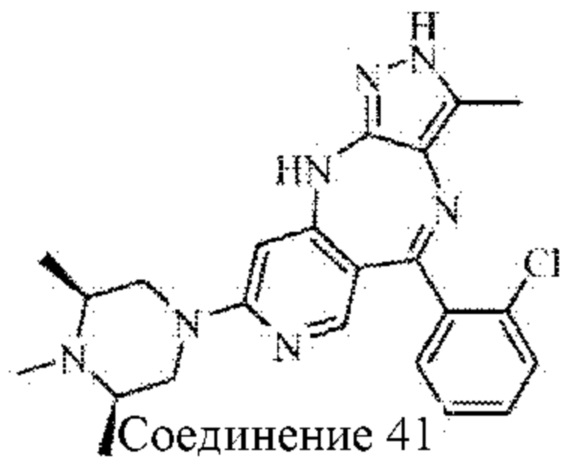
11.2 Синтез соединения 42: структура амина представляла собой  твердый неочищенный продукт растворяли в дихлорметане (20 мл), сушили над сульфатом магния и упаривали досуха с получением соединения 42 (22 мг; выход: 20,4%).
твердый неочищенный продукт растворяли в дихлорметане (20 мл), сушили над сульфатом магния и упаривали досуха с получением соединения 42 (22 мг; выход: 20,4%).
1Н ЯМР (400 МГц, CDCl3): 8.85 (s, 1H), 7.28-7.42 (m, 4Н), 5.91 (s, 1H), 5.62 (s, 1H), 3.98-4.02 (d, 2Н), 2.66-2.72 (t, 2Н), 2.29 (s, 3Н), 2.22 (m, 4Н), 1.14-1.16 (m, 6Н).
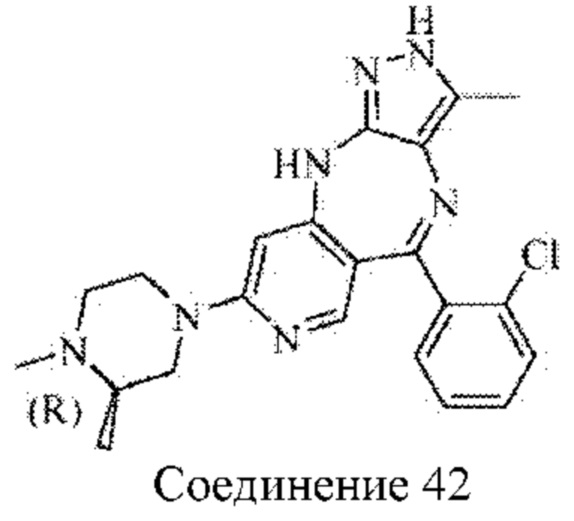
11.3 Синтез соединения 44: структура амина представляла собой  неочищенный продукт сушили с получением соединения 44 (90 мг; выход: 40,35%).
неочищенный продукт сушили с получением соединения 44 (90 мг; выход: 40,35%).
1Н ЯМР (400 МГц, DMSO): 11.59 (s, 1H), 8.26 (s, 1H), 7.32-7.48 (m, 4Н), 6.89 (s, 1H), 5.77 (s, 1H), 1.98 (s, 3Н), 1.62 (m, 1H), 0.33-0.43 (m, 4Н).
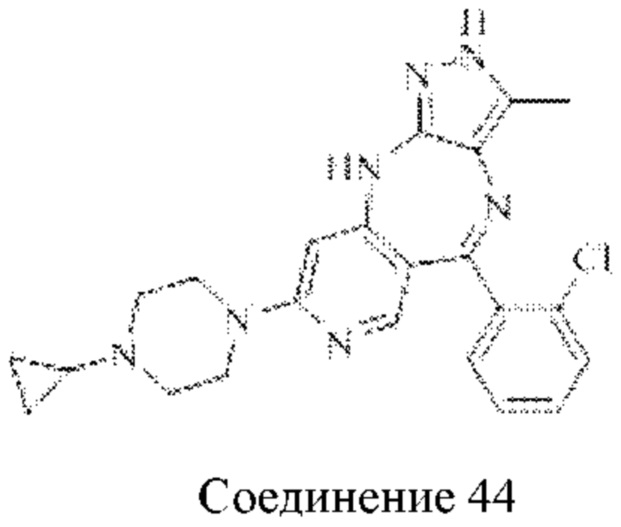
11.4 Синтез соединения 45: структура амина представляла собой  неочищенный продукт растворяли в дихлорметане (10 мл), сушили над сульфатом магния и упаривали досуха с получением желтого твердого вещества, которое промывали смесью дихлорметан : петролейный эфир, 1:1, с получением соединения 45 (42 мг; выход: 20,1%).
неочищенный продукт растворяли в дихлорметане (10 мл), сушили над сульфатом магния и упаривали досуха с получением желтого твердого вещества, которое промывали смесью дихлорметан : петролейный эфир, 1:1, с получением соединения 45 (42 мг; выход: 20,1%).
1Н ЯМР (400 МГц, DMSO): 11.59 (s, 1H), 8.28 (s, 1H), 7.32-7.48 (m, 4Н), 6.86 (s, 1H), 4.62-4.67 (m, 3Н), 3.54-3.71 (dd, 2H), 3.07-3,13 (m, 2H), 1.98 (s, 3H), 1.77-1.84 (m, 2H), 0.82-0.87 (m, 2H).
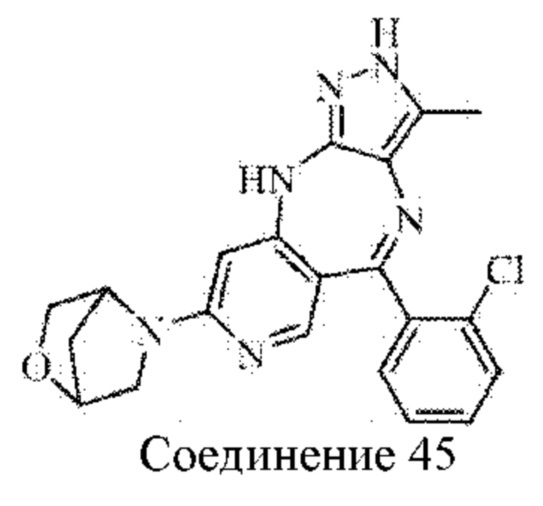
11.5 Синтез соединения 46: структура амина представляла собой  неочищенный продукт сушили с получением соединения 46 (130 мг; выход: 59,9%).
неочищенный продукт сушили с получением соединения 46 (130 мг; выход: 59,9%).
1Н ЯМР (400 МГц, DMSO): 11.55 (s, 1H), 8.28 (s, 1H), 7.32-7.48 (m, 4Н), 6.87 (s, 1H), 5.69 (s, 1H), 3.76-3.80 (m, 2Н), 3.46-3.53 (m, 4Н), 3.17-3,20 (d, 2Н), 2.95 (s, 2Н), 1.97 (s, 3Н).
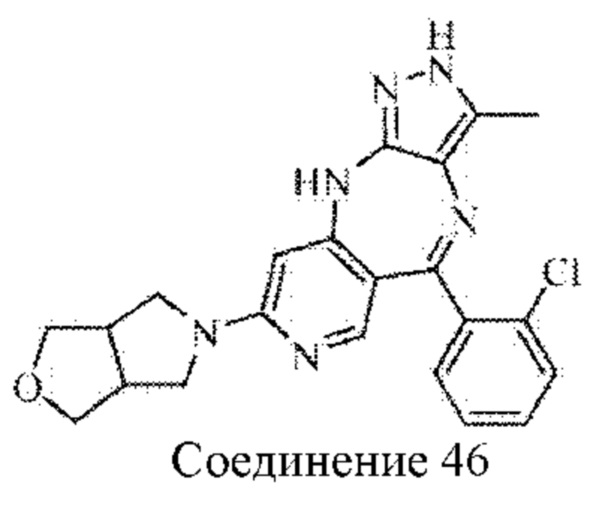
11.6 Синтез соединения 43: структура амина представляла собой  неочищенный продукт сушили с получением соединения 43 (55 мг; выход: 57,9%).
неочищенный продукт сушили с получением соединения 43 (55 мг; выход: 57,9%).
1Н ЯМР (400 МГц, DMSO): 11.59 (s, 1H), 8.30 (s, 1H), 7.33-7.46 (m, 4Н), 6.90 (s, 1H), 5.92 (s, 1H), 3.89 (s, 2Н), 3.65-3.67 (m, 2Н), 3.31-3.36 (m, 2Н), 3.15-3,17 (d, 1H), 2.86 (s, 3Н), 1.97 (s, 3Н).
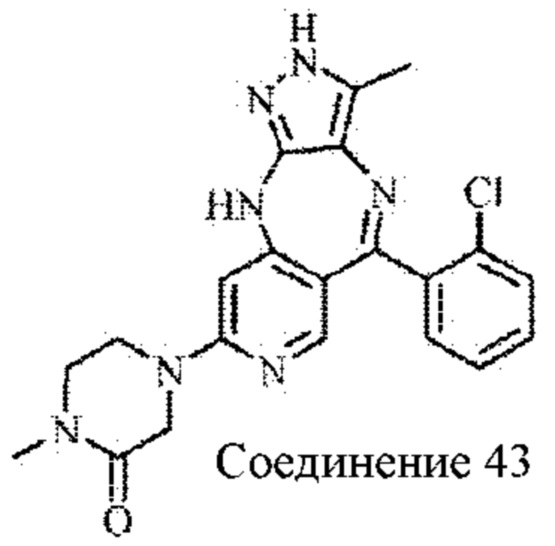
11.7 Синтез соединения 40: структура амина представляла собой  неочищенный продукт сушили с получением соединения 40 (30 мг; выход: 39,9%).
неочищенный продукт сушили с получением соединения 40 (30 мг; выход: 39,9%).
1Н ЯМР (400 МГц, DMSO): 11.57 (s, 1H), 8.24 (s, 1H), 7.33-7.46 (m, 4Н), 6.89 (s, 1H), 5.96 (s, 1H), 3.79-3.93 (m, 4Н), 3.45-3.46 (m, 2Н), 2.75-2.81 (t, 1H), 3.15-3,17 (d, 1H), 1.97 (s, 3Н), 1.08-1.09 (d, 3Н).
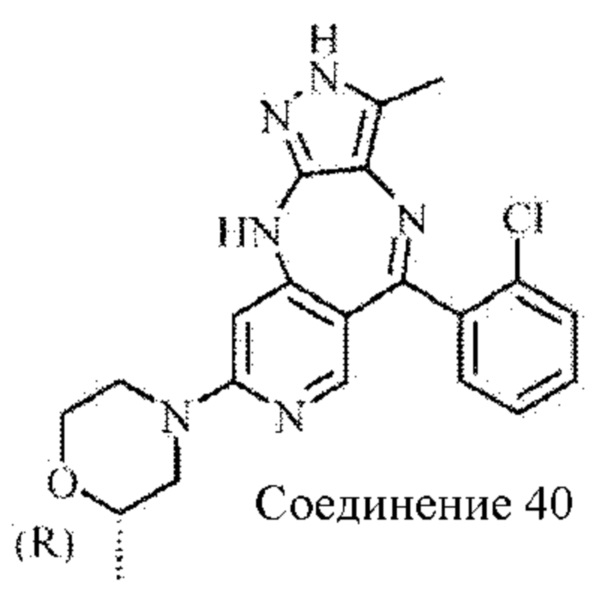
11.8 Синтез соединения 48: структура амина представляла собой  неочищенный продукт сушили с получением соединения 48 (127 мг; выход: 60%).
неочищенный продукт сушили с получением соединения 48 (127 мг; выход: 60%).
1Н ЯМР (400 МГц, DMSO): 11.56 (s, 1H), 8.33 (s, 1H), 7.32-7.43 (m, 4Н), 6.84 (s, 1H), 5.53 (s, 1H), 4.67 (s, 3Н), 4.01 (s, 4Н), 1.97 (s, 3Н).
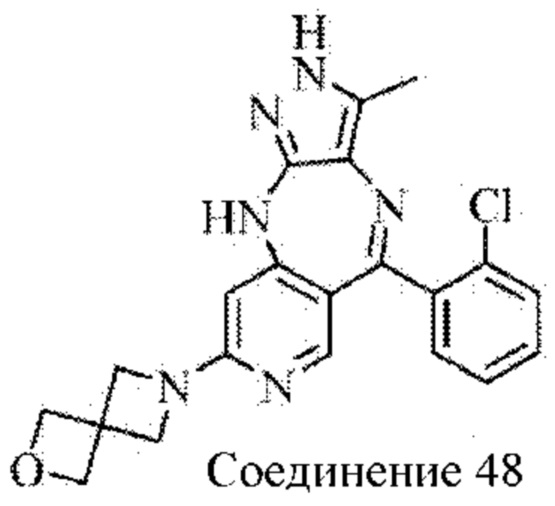
Пример 12: Синтез соединения 47
Стадия 1: Синтез промежуточного соединения 47-1
Промежуточное соединение 1-4 (500 мг; 0,9 ммоль; 1 экв.) добавляли в DMF (20 мл), затем добавляли фосфат калия (573 мг; 2,7 ммоль; 3 экв.), 2-(3,6-дигидро-2Н-пиран-4-ил)-4,4,5,5-тетрагидро-1,3,2-диоксаборолан (246 мг; 1,17 ммоль; 1,2 экв.), ацетат палладия (10 мг; 0,045 ммоль; 0,05 экв.). Реактор трижды продували азотом. Смесь нагревали до 100°С и подвергали взаимодействию в течение 16 часов. Окончание взаимодействия контролировали посредством ТСХ. Реакционную смесь выливали в смесь воды со льдом (50 мл) и экстрагировали этилацетатом (50 мл×3). Органическую фазу отделяли, сушили, упаривали досуха с получением промежуточного соединения 47-1 в виде желтого твердого вещества (500 мг; неочищенный продукт использовали непосредственно на следующей стадии).
Стадия 2: Получение промежуточного соединения 47-2
Промежуточное соединение 47-1 (1,50 г; 2,67 ммоль; 1 экв.) добавляли в 100 мл одногорлую колбу в атмосфере азота, затем добавляли THF (тетрагидрофуран) (5 мл), метанол (5 мл), палладий на угле (0,15 г). По каплям добавляли триэтилсилан (3,1 г; 26,7 ммоль; 10 экв.) в атмосфере азота, и смесь перемешивали при 15°С в течение 5 минут. Полное превращение исходных веществ контролировали посредством ТСХ. После взаимодействия реакционную смесь фильтровали, и фильтрат упаривали досуха. Неочищенный продукт добавляли в МТВЕ (10 мл) и фильтровали с получением соединения 47-2 (0,67 г; выход 44,67%).
Стадия 3: Синтез промежуточного соединения 47-3
Промежуточное соединение 47-2 (0,67 г; 1,2 ммоль; 1 экв.) добавляли в одногорлую колбу на 25 мл, затем добавляли трифторуксусную кислоту (5 мл). Смесь нагревали до 80°С и подвергали взаимодействию в течение 16 часов. Окончание взаимодействия контролировали посредством ТСХ. Реакционную смесь выпаривали досуха с получением промежуточного соединения 47-3 в виде желтого масла (1,0 г; неочищенный продукт использовали непосредственно на следующей стадии).
Стадия 4: Синтез соединения 47
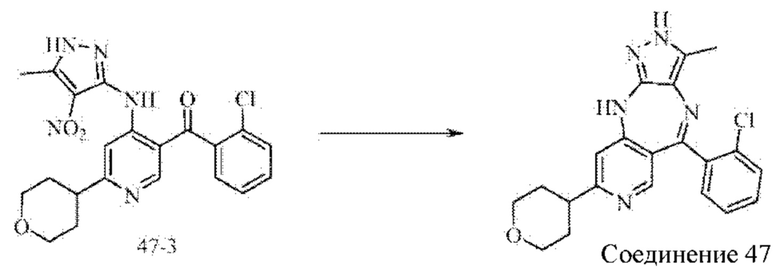
Промежуточное соединение 47-3 (1,0 г; 1,2 ммоль; 1 экв.) добавляли в 25 мл одногорлую колбу, затем добавляли 2-метилтетрагидрофуран (5 мл) и хлорид олова (1,89 г; 8,6 ммоль). Смесь нагревали до 90°С и перемешивали в течение 5 часов. Окончание взаимодействия контролировали посредством ТСХ. Реакционную смесь охлаждали, и значение рН доводили до 8 гидрокарбонатом натрия. Смесь экстрагировали DCM (50 мл), и органическую фазу отделяли, сушили, объединяли и упаривали досуха с получением соединения 47 (36 мг; выход: 6,2% за две стадии) с применением силикагелевой пластины (DCM : MeOH, 30:1).
1Н ЯМР (400 МГц, DMSO-d6): 11.68 (s, 1H), 8.45 (s, 1H), 7.36-7.49 (m, 4Н), 7.11 (s, 1H), 6.41 (s, 1H), 3.86-3.88 (d, 2Н), 3.35-3.38 (m, 2Н), 2.55-2.59 (m, 3Н), 1.97 (s, 3Н), 1.48-1.63 (m, 3Н).
Пример 13: Получение кристаллической формы I по изобретению
Соединение 29 (500,0 мг) формулы (III) добавляли в 100 мл одногорлую колбу, затем добавляли 80,0 мл смеси этанол : 2-метилтетрагидрофуран, 5:1 (об.:об.). Реакционную смесь нагревали до 100°С, чтобы сделать ее прозрачной, медленно порциями добавляли соединение 29 в суммарном количестве 100,00 мг, пока реакционная смесь не становилась прозрачной. Затем раствор в естественных условиях охлаждали до комнатной температуры, перемешивали в течение ночи, фильтровали и сушили с получением 320,00 мг кристаллической формы I.
При использовании Cu-Kα-излучения дифракция рентгеновских лучей на порошке кристаллической формы I, выраженная углами 2θ (°), показала сильные характеристические пики при 7,4±0,2°, 17,9±0,2°, 18,9±0,2°, 19,4±0,2°, 21,5±0,2° и 23,7±0,2°; а также характеристические пики при 14,0±0,2°, 15,0±0,2°, 20,7±0,2° и 25,4±0,2°; и также характеристические пики при 11,7±0,2°, 22,8±0,2° и 27,8±0,2°. Анализ XRPD показан на Фиг. 1.
Эндотерму кристаллической формы I, начиная примерно с 311°С, и пик эндотермы примерно при 312°С измеряли при помощи дифференциальной сканирующей калориметрии. Спектр DSC показан на Фиг. 2.
Пример 14: Получение кристаллической формы I по изобретению
Соединение 29 (500,0 мг) формулы (III) добавляли в одногорлую колбу на 100 мл, затем добавляли 6,0 мл смеси диметилсульфоксид : вода, 2:1 (об.:об.). Реакционную смесь нагревали до 100°С и медленно по каплям добавляли 57 мл смеси диметилсульфоксид : вода, 2:1, до тех пор пока раствор не становился прозрачным. Реакционную смесь в естественных условиях охлаждали до комнатной температуры, перемешивали в течение ночи, фильтровали и сушили с получением 320,00 мг кристаллической формы I.
Пример 15: Получение кристаллической формы I по изобретению
Соединение 29 (500,0 мг) формулы (III) добавляли в одногорлую колбу на 100 мл, добавляли смесь MeOH : DCM, 5:1, (60 мл) и нагревали до полного растворения. Смесь концентрировали при пониженном давлении при 35-40°С на роторном испарителе с получением кристаллической формы I (320,00 мг).
Пример 16: получение кристаллической формы I по изобретению
Соединение 29 (500,0 мг) формулы (III) добавляли в одногорлую колбу на 25 мл, затем добавляли МеОН (1,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 7 суток, фильтровали при пониженном давлении, сушили с получением кристаллической формы I (320,00 мг).
Пример 17: получение кристаллической формы I по изобретению
Стадия 1: Синтез (2-хлорфенил)(4-((1-(4-метоксибензил)-5-метил-4-нитро-1H-пиразол-3-ил)амино)-6-морфолинопиридин-3-ил)метанона (промежуточное соединение III-Е)
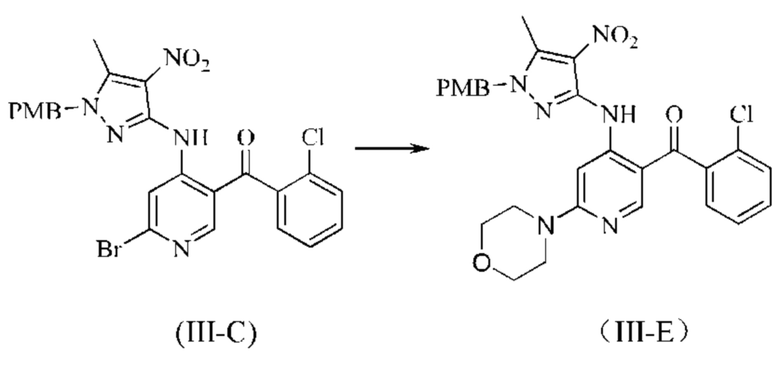
(6-Бром-4-((1-(4-метоксибензил)-5-метил-4-нитро-1H-пиразол-3-ил)амино)-пиридин-3-ил)(2-хлорфенил)метанон (промежуточное соединение III-С) (150,0 г; 269 ммоль) растворяли в DMSO (300 мл). Смесь нагревали до 50°С для растворения твердого вещества, затем по каплям добавляли морфолин (III-D) (70,4 г; 808 ммоль). Затем смесь нагревали до 90°С и подвергали взаимодействию в течение 3 часов. Анализ ТСХ показал окончание взаимодействия. Реакционную смесь выливали в воду (3 л), осаждали большое количество твердого вещества и фильтровали. Осадок на фильтре промывали водой (500 мл), сушили с получением промежуточного соединения III-Е в виде желтого твердого вещества (160,0 г неочищенного продукта).
Стадия 2: синтез (2-хлорфенил)(4-((-5-метил-4-нитро-1H-пиразол-3-ил)амино)-6-морфолинопиридин-3-ил)метанона (промежуточное соединение III-F)
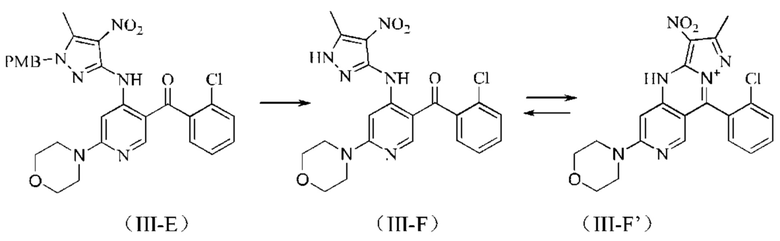
К (2-хлорфенил)(4-((1-(4-метоксибензил)-5-метил-4-нитро-1H-пиразол-3-ил)амино)-6-морфолинопиридин-3-ил)метанону (промежуточное соединение III-Е) (160,0 г; неочищенный продукт) добавляли трифторуксусную кислоту (500 мл). Смесь нагревали до 80°С и подвергали взаимодействию в течение 8 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. Затем реакционную смесь концентрировали, неочищенный продукт растирали с метил-трет-бутиловым эфиром (800 мл), фильтровали с получением промежуточного соединения III-F в виде кирпично-красного твердого вещества (160 г; неочищенный продукт).
1H ЯМР (400 МГц, DMSO-d6) δ м.д.: 13.63 (ушир. s., 1H), 12.39 (s, 1H), 7.95-7.93 (d, J=10,6 Гц, 2Н), 7.62-7.48 (m, 4Н), 3.74-3.70 (m, 4Н), 3.64-3.63 (m, 4Н), 2.58 (s, 3Н).
В способе получения также возможно получение переходного состояния формулы (III-F') из соединения формулы (III-Е), и полученный неочищенный продукт подвергали кислотной обработке (например обрабатывали соляной кислотой) с окончательным получением целевого промежуточного соединения формулы (III-F).
Стадия 3: синтез 4-(5-(2-хлорфенил)-3-метил-2,10-дигидропиразоло[4,3-b]пиридо-[4,3-е][1,4]диазепин-8-ил)морфолина
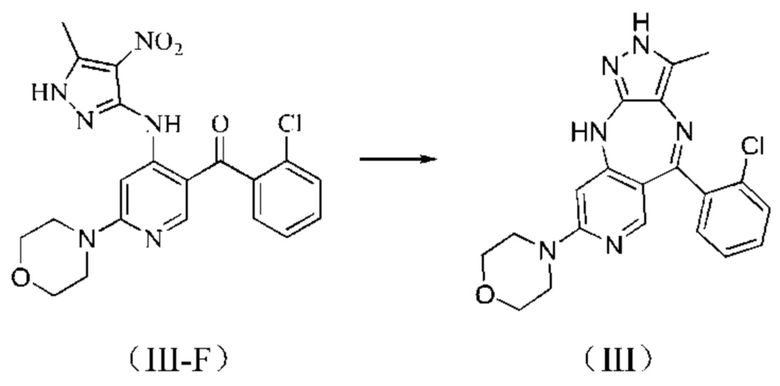
(2-Хлорфенил)(4-((5-метил-4-нитро-1H-пиразол-3-ил)амино)-6-морфолинопиридин-3-ил)метанон (промежуточное соединение III-F) (160 г; неочищенный продукт) растворяли в 2-метилтетрагидрофуране (1,6 л), затем добавляли дигидрат дихлорида олова (320 г; 1420 ммоль). Смесь нагревали до 90°С и подвергали взаимодействию в течение 4 часов. Анализ посредством ЖХ-МС показал окончание взаимодействия. После охлаждения до комнатной температуры (10°С) реакционную смесь выливали в насыщенный раствор гидрокарбоната натрия (4 л), добавляли 2-метилтетрагидрофуран (1 л), фильтровали. Фильтрат отделяли. Водную фазу экстрагировали 2-метилтетрагидрофураном (1 л). Органические фазы объединяли, промывали водой (2 л), затем рассолом (2 л), сушили над безводным сульфатом натрия, концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт растирали с метил-трет-бутиловым эфиром (400 мл) и фильтровали с получением желтого твердого вещества (65,0 г; чистота 95%), очищали посредством хроматографии на силикагеле (дихлорметан : метанол, 20:1) с получением бледно-желтого твердого вещества (55 г). Твердое вещество растворяли в DMSO (примерно 200 мл) и нагревали до 40-50°С, пока твердое вещество не растворялось. Вышеуказанный раствор медленно по каплям добавляли в дистиллированную воду (2 л), затем осаждали большое количество твердого вещества, перемешивали при комнатной температуре в течение ночи, фильтровали посредством всасывания. Осадок на фильтре сушили под вакуумом при 35°С с получением бледно-желтого порошкообразного твердого вещества (53 г; выход 49,9% за три стадии).
1Н ЯМР (400 МГц, DMSO-d6): 11.58 (s, 1H), 8.29 (s, 1H), 7.33-7.47 (m, 4Н), 6.90 (s, 1H), 5.96 (s, 1H), 3.61 (m, 4Н), 3.31 (m, 4Н), 1.98 (s, 3Н).
Кристаллическую форму этого образца определяли посредством дифракции рентгеновских лучей на порошке, и эта кристаллическая форма была такой же, как кристаллическая форма, полученная в способах получения соединений по примерам 13, 14, 15 и 16, то есть кристаллическая форма I.
Настоящее изобретение станет более понятным из следующих примеров биологических экспериментов. Однако специалистам в данной области техники очевидно, что описание примеров экспериментов предназначено только для иллюстрации изобретения и не должно и не будет ограничивать настоящее изобретение.
Пример биологического эксперимента 1: Тестирование ферментной активности соединения по настоящему изобретению
Исследуемые образцы: соединения с 1 по 4 по настоящему изобретению (их порядковые номера и структуры показаны в таблице 1), концентрация разведения: 0,03 мкМ - 3 мкМ, всего 10 градиентов концентраций.
Метод исследования: тестирование ферментативной активности киназы Aurora (включая Aurora А и Aurora В) и VEGFR2 (KDR) проводили с использованием многофункционального микропланшетного ридера.
Методика эксперимента:
1) Тестирование активности в отношении киназы Aurora А:
Белок, киназу Aurora А, и соединения последовательно добавляли в следующую реакционную систему: 8 мМ MOPS (3-(N-морфолино)пропансульфониевая кислота) с рН 7,0; 0,2 мМ EDTA (этилендиаминтетрауксусная кислота); 200 мкМ LRRASLG (Кемптид); 10 мМ ацетата магния и [γ-33Р]-АТФ (аденозинтрифосфат, меченый γ-Р33) (радиоактивность составляла примерно 500 имп/мин/пмоль). Затем в вышеуказанную реакционную систему добавляли АТФ, чтобы начать реакцию, и инкубировали при комнатной температуре в течение 40 минут. Затем реакцию останавливали путем добавления 3% раствора фосфорной кислоты. Отбирали из реакционной смеси 10 мкл и затем по каплям добавляли на фильтровальную мембрану Р30 filtermat, трижды промывали в 75 мМ растворе фосфорной кислоты в течение 5 минут и затем однократно промывали метанолом. После сушки фильтровальной мембраны подсчет проводили с помощью сцинтилляционного счетчика с жидкокристаллическим индикатором.
2) Тестироание активности в отношении киназы Aurora В:
Белок, киназу Aurora В, и соединения последовательно добавляли в следующую реакционную систему: 8 мМ MOPS с рН 7,0; 0,2 мМ EDTA; 200 мкМ AKRRRLSSLRA; 10 мМ ацетата магния и [γ-33Р]-АТФ (радиоактивность составляла примерно 500 имп/мин/пмоль). Затем в вышеуказанную реакционную систему добавляли АТФ, чтобы начать реакцию, и инкубировали при комнатной температуре в течение 40 минут. Затем реакцию останавливали путем добавления 3% раствора фосфорной кислоты. Отбирали из реакционной смеси 10 мкл, и затем по каплям добавляли на фильтровальную мембрану Р30 filtermat, трижды промывали в 75 мМ растворе фосфорной кислоты в течение 5 минут и затем однократно промывали метанолом. После сушки фильтра подсчет проводили с помощью сцинтилляционного счетчика с жидкокристаллическим индикатором.
3) Тестирование активности в отношении KDR киназы:
Белок, KDR киназу, и соединения последовательно добавляли в следующую реакционную систему: 8 мМ MOPS с рН 7,0; 0,2 мМ EDTA; 0,33 мг/мл основного белка миелина; 10 мМ ацетата магния и [γ-33Р]-АТФ (радиоактивность составляла примерно 500 имп/мин/пмоль). Затем в вышеуказанную реакционную систему добавляли АТФ, чтобы начать реакцию, и инкубировали при комнатной температуре в течение 40 минут. Затем реакцию останавливали путем добавления 3% раствора фосфорной кислоты. Отбирали из реакционной смеси 10 мкл и затем по каплям добавляли на фильтровальную мембрану Р30 filtermat, трижды промывали в 75 мМ растворе фосфорной кислоты в течение 5 минут и затем однократно промывали метанолом. После сушки фильтра подсчет проводили с помощью сцинтилляционного счетчика с жидкокристаллическим индикатором. Результаты тестирования показаны в таблице 2.
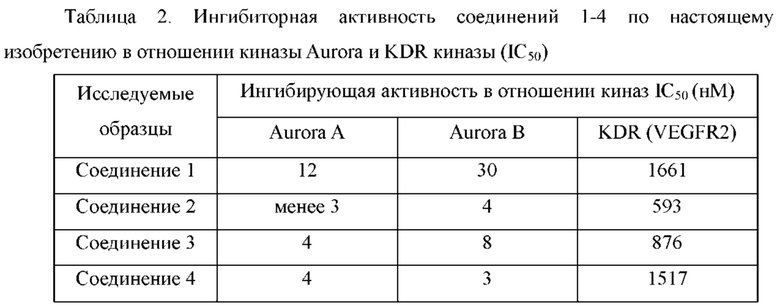
Как можно видеть из результатов эксперимента, показанных в таблице 2, соединения по настоящему изобретению обладают хорошей ингибиторной активностью в отношении мультикиназ, что указывает на то, что соединения по настоящему изобретению обладают хорошим потенциалом для клинического применения в лечении заболеваний, опосредованных аномальной экспрессией киназы Aurora (включая Aurora А и Aurora В) и VEGFR2 (KDR).
Пример биологического эксперимента 2: Тестирование энзиматической активности соединений по настоящему изобретению
Исследуемые образцы: соединения по настоящему изобретению (их порядковые номера и структуры показаны в таблице 1), концентрация разведения: 0,03 мкМ - 3 мкМ, всего 10 градиентов концентраций.
Контрольное лекарственное средство: соединение 1-2, раскрытое в WO 2013123840 A1 
Метод тестирования: тестирования ферментативной активности киназы Aurora (включая Aurora А и Aurora В) и VEGFR2 (KDR) проводили с использованием многофункционального микропланшетного ридера.
Методика эксперимента:
1) Подготовка планшета для соединений
а) 96-луночные планшеты, 10 дозовых групп, 3-кратные серийные разведения, в каждую лунку добавляли DMSO, максимальная концентрация составляла 500 мкМ (50-кратное разведение соединения).
б) 384-луночные планшеты с разведением 1×киназным буфером (50 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота), рН 7,5; 0,0015% Brij-35 (лауриловый эфир полиоксиэтилена); 2 мМ DTT (дитиотреитол)), где каждая лунка содержит 5×соединение, разведенное в 5 мкл 10% DMSO. Каждая лунка отрицательного контроля содержит 5 мкл 1×киназного буфера, содержащего 10% DMSO.
2) Методика тестирования
Aurora A, Aurora В и KDR растворяли в 1×киназном буфере и готовили в виде 2,5× раствора фермента. После того как соединение в различных концентрациях взаимодействовало с 2,5× раствором фермента при комнатной температуре в течение 10 минут, добавляли меченный FAM (5,6-карбоксифлуоресцеин) пептидный субстрат и АТФ для инициирования реакции. После инкубирования в течение 40 минут добавляли 25 мкл терминирующего раствора (100 мМ HEPES, рН 7,5; 0,015% Brij-35; 0,2% реагент покрытия #3; 50 мМ EDTA), чтобы остановить реакцию, и окончательные данные считывали с помощью Caliper. Результаты тестирования показаны в таблице 3.
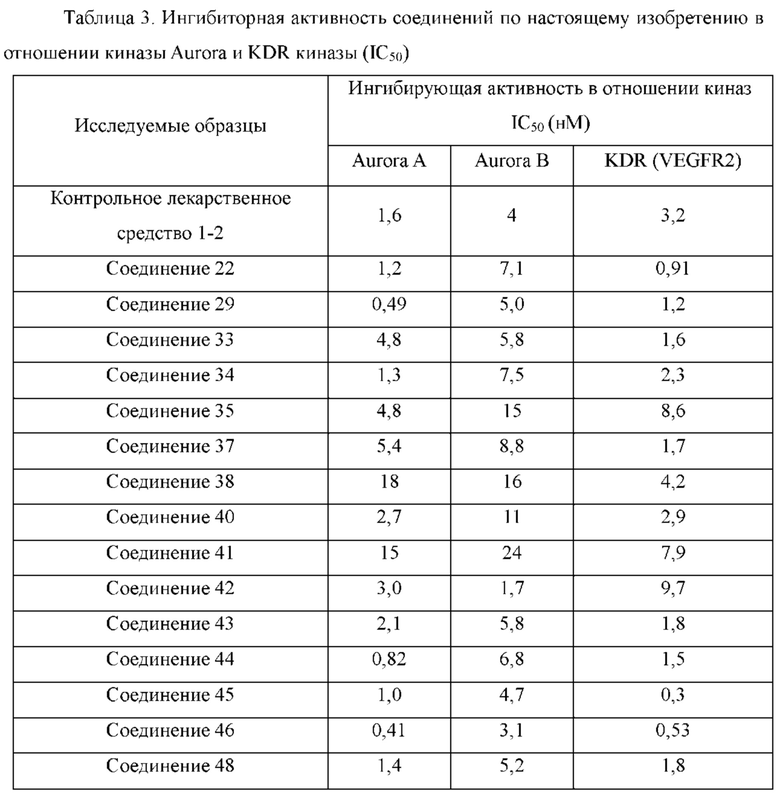
Как можно видеть из результатов эксперимента, приведенных в таблице 3, соединения по настоящему изобретению обладают хорошей ингибиторной активностью в отношении мультикиназ, что указывает на то, что соединения по настоящему изобретению обладают хорошим потенциалом для клинического применения в лечении заболеваний, опосредованных аномальной экспрессией киназы Aurora (включая Aurora А и Aurora В) и VEGFR2 (KDR).
Пример биологического эксперимента 3: Тестирование цитологической активности соединения по настоящему изобретению
Тестируемый образец: соединения по настоящему изобретению (их порядковые номера и структуры показаны в таблице 1).
Контрольное лекарственное средство: соединение 1-2, раскрытое в WO 2013123840 A1 
Методика тестирования: влияние соединений на клеточную пролиферацию разных клеточных линий исследовали с помощью метода Cell Titer-Glo.
Методика эксперимента:
Каждую линию клеток инокулировали в 96-луночный планшет за сутки вперед, и после инкубирования в течение ночи добавляли лекарственные средства в разных концентрациях до конечной концентрации 0-10000 нМ, 3-5-кратное разведение, суммарно для 10 точек концентраций. После инкубирования в течение 72 часов в смесь добавляли реагент Cell titer-Glo, уравновешенный при комнатной температуре, и встряхивали для инкубирования в течение 10 минут, и затем оставляли выстаиваться при комнатной температуре в течение 2 минут для стабилизации светового сигнала. Многофункциональный микропланшетный ридер использовали для считывания данных с каждой лунки и анализировали их. Результаты исследования показаны в таблицах 4 и 5:
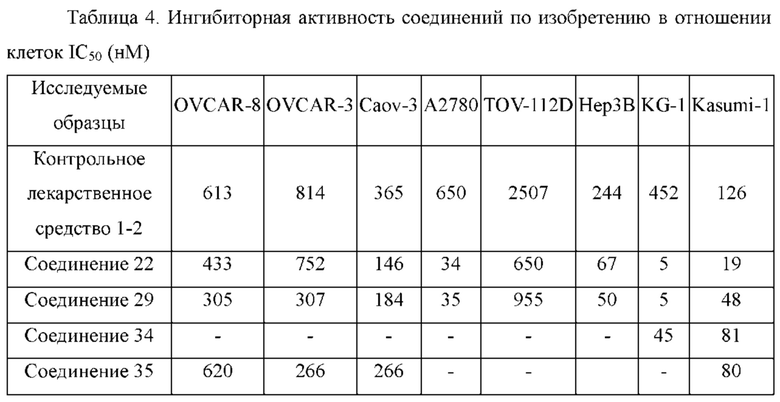
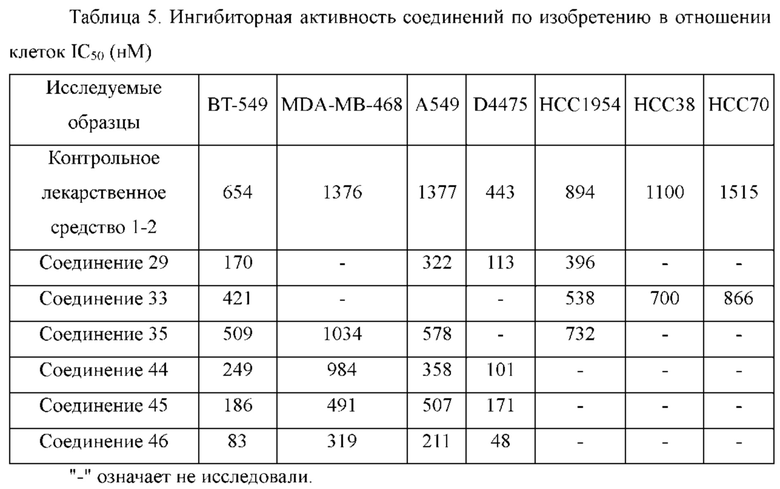
Как можно видеть из результатов экспериментов в таблицах 4 и 5, соединения по настоящему изобретению обладают хорошей ингибиторной активностью в отношении различных раковых клеток и могут быть использованы для лечения соответствующих раковых заболеваний.
Пример биологического эксперимента 4: определение активности кристаллической формы I соединения формулы (III) (соединение 29) в отношении ферментов
Тестируемый образец: кристаллическая форма I, полученная в примере 13, концентрация разведения: 0,03 мкМ-3 мкМ, всего 10 градиентов концентраций.
Метод исследования: исследование ферментативной активности киназ, показанных в таблице 1, проводили с использованием многофункционального микропланшетного ридера.
Методика эксперимента:
1) Подготовка планшета для соединений
а) 96-луночные планшеты, 10 дозовых групп, 3-кратные серийные разведения, в каждую лунку добавляют DMSO вплоть до концентрации 500 мкМ (50x соединение).
б) 384-луночные планшеты с разведением 1× киназным буфером (50 мМ HEPES, рН 7,5; 0,0015% Brij-35; 2 мМ DTT), причем каждая лунка содержит 5× соединение, разведенное в 5 мкл 10% DMSO. Каждая лунка с отрицательным контролем содержит 5 мкл 1× киназного буфера, содержащего 10% DMSO.
2) Методика тестирования
Киназы, показанные в таблице 6, растворяли в 1× киназном буфере и готовили в виде 2,5× раствора фермента соответственно. После того как кристаллическую форму I и 2,5× раствор фермента подвергали взаимодействию при комнатной температуре в течение 10 минут, добавляли FAM-меченый пептидный субстрат и АТФ для инициирования взаимодействия. После инкубирования в течение 40 минут добавляли 25 мкл терминирующего раствора (100 мМ HEPES, рН 7,5; 0,015% Brij-35; 0,2% реагент покрытия #3; 50 мМ EDTA) для остановки взаимодействия. Окончательные данные считывали с помощью Caliper. Результаты исследования показаны в таблице 6.

Согласно результатам экспериментов в таблице 6, кристаллическая форма I соединения формулы (III) по настоящему изобретению обладает хорошей ингибиторной активностью в отношении различных киназ; это указывает на то, что соединения по настоящему изобретению обладают хорошим потенциалом для клинического применения в лечении заболеваний, опосредованных аномальной экспрессией различных киназ, таких как Aurora, VEGFR2 (KDR), FGFR ((рецептор фактора роста фибробластов), FLT (fms-подобная тирозинкиназа) и JAK (янус-киназа).
Пример биологического эксперимента 5: Фармакодинамическое исследование in vivo соединения по настоящему изобретению на модели подкожного ксенотрансплантата опухоли, образованной клетками линии Kasumi-1 острого миелоидного лейкоза человека
Тестируемый образец: кристаллическая форма I, полученная в примере 17 (кристаллическая форма I соединения 29)
Животные, клетки, реагенты и инструменты: клетки линии Kasumi-1, полученные из АТСС.
Мыши линии СВ17 SCID, 6-8 недель, самки, полученные от Shanghai Lingchang Biotechnology Co., Ltd.
Методика эксперимента:
1. Конструирование и распределение по группам мышей, несущих опухоль
Клетки линии Kasumi-1 культивировали in vitro в монослое, и условия культивирования были следующими: среда RPMI 1640 с добавлением 10% инактивированной нагреванием фетальной бычьей сыворотки и 1% пенициллин-стрептомицин двойного антитела, 37°С и 5% CO2. Пересев производили от двух до трех раз в неделю. Когда клетки находились в экспоненциальной фазе роста, клетки собирали, подсчитывали и инокулировали.
0,2 мл клеточной суспензии, содержащей примерно 1×107 клеток Kasumi-1 (клетки, суспендированные в смеси 1:1 основной среды RPMI 1640 и питательного геля), подкожно инокулировали в правую нижнюю часть спины самок мышей СВ17 SCID. Групповое введение начинали, когда средний объем опухоли достигал 100-150 мм3. Метод группировки: животных взвешивали перед введением и измеряли объем опухоли. Распределение по группам проводили в соответствии с объемом опухоли, по 8 мышей на группу.
2. Режим дозирования
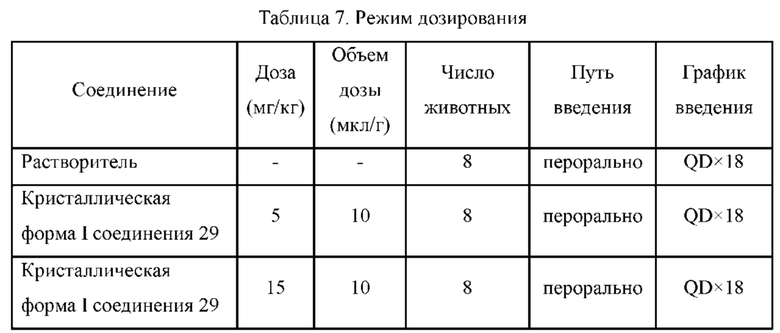
3. Показатели экспериментального наблюдения
Здоровье и смерть животных контролировали каждые сутки. Массу тела измеряли два раза в неделю, образцы собирали после последней дозы, и определяли массу опухоли. Эффективность в отношении массы опухоли оценивали по TGI%, ингибированию роста опухоли (TGI)%=(TWc-TWT)/TWc×100%, TWc: масса опухоли в контрольной группе, TWT: масса опухоли в группе лечения. Согласно рекомендациям NIH (Национальный институт здоровья), лекарственное средство считается эффективным, если TGI составляет не менее 58%.
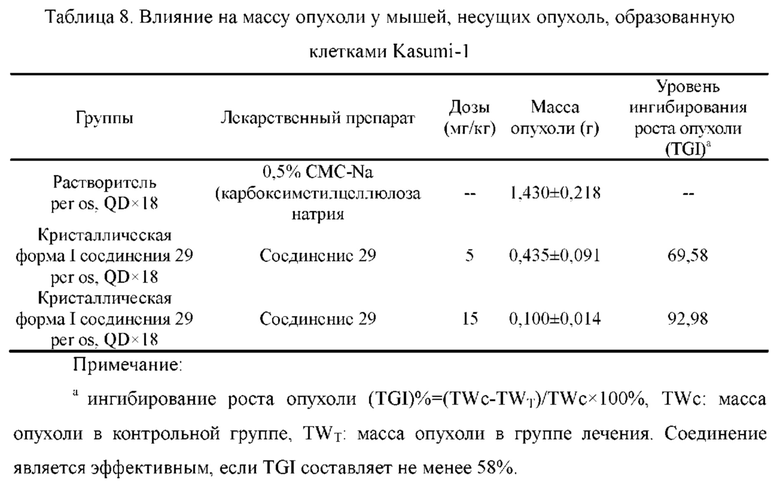

Согласно результатам экспериментов в таблице 8, можно видеть, что соединение 29 (кристаллическая форма I) обладает значительным ингибиторным действием на модель ксенотрансплантата опухоли, образованной клетками линии Kasumi-1; это указывает на то, что соединение по настоящему изобретению обладает хорошим потенциалом для клинического применения при остром лейкозе.
Пример 6: ФК (фармакокинетическая) оценка соединения по настоящему изобретению на собаках породы бигль
Тестируемый образец: кристаллическая форма I, полученная в примере 17 (кристаллическая форма I соединения 29)
Методика эксперимента:
1. Введение и отбор образцов крови
1) Введение животным: все животные не получали пищу в течение более 12 часов перед введением, и их кормили через 4 часа после введения. Воду не ограничивали ни до, ни после введения во время эксперимента. Собаки породы бигль получали внутривенно (в/в) 1 мг/кг кристаллической формы I соединения 29 (рекомендация: 10% DMA + 20% (30% Solutol) + 70% физиологического раствора) в однократной дозе, и кровь отбирали в 0 часов перед введением и через 0,083; 0,25; 0,50; 1,0; 2,0; 4,0; 6,0; 8,0; 12 и 24 часа после введения. Собаки породы бигль получали перорально 2 мг/кг кристаллической формы I соединения 29 (рекомендация: 10% DMA + 20% (30% Solutol) + 70% физиологического раствора) в однократной дозе, и кровь отбирали в 0 часов перед введением и через 0,25; 0,50; 1,0; 2,0; 4,0; 6,0; 8,0; 12 и 24 часа после введения, и 200 мкл крови отбирали через малую подкожную вену ноги и помещали в аналитическую пробирку с сухой EDTA-K2.
2) Получение плазмы: образец цельной крови разделяли посредством низкоскоростного центрифугирования (1800 g; 5 минут; 4°С) (цельную кровь собирали и помещали на ледяную баню, и отделение плазмы следует закончить в пределах 30 минут) с получением плазмы, и отделенную плазму хранили в холодильнике при -20°С для анализа.
2. Метод анализа образца
Исследуемый образец (-80°С) брали из холодильника, размораживали в естественных условиях при комнатной температуре и перемешивали в течение 5 минут; 20 мкл образца плазмы аккуратно аспирировали в 1,5 мл пробирку для центрифугирования; добавляли 200 мкл рабочего раствора внутреннего стандарта в концентрации 50 нг/мл и тщательно перемешивали; после перемешивания в течение 5 минут смесь центрифугировали в течение 5 минут (12000 об/мин); 50 мкл надосадочной жидкости аккуратно аспирировали в 96-луночные планшеты, предварительно наполненные водой 150 мкл/лунка; перемешивали в течение 5 минут, определение посредством ЖХ-МС/МС проводили в объеме вводимой пробы 15 мкл.
3. Метод обработки данных
Концентрацию тестируемого образца выводили с использованием Analyst 1.6.1 от компании АВ SCIEX. Microsoft Excel используют для расчета среднего значения, стандартного отклонения, коэффициента вариаций и других параметров (данные, выводимые непосредственно Analyst 1.6.1, не рассчитывают), ФК параметры рассчитывали, используя программу NCA Pharsight Phoenix 6.1.
Результаты эксперимента:
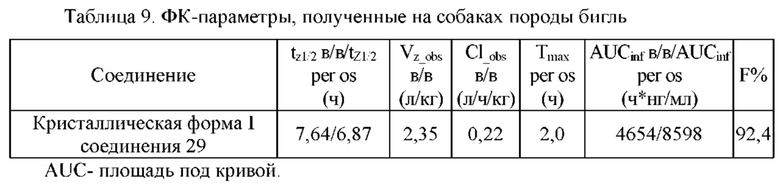
Согласно результатам экспериментов в таблице 9 можно видеть, что соединение 29 (кристаллическая форма I) обладает хорошими фармакокинетическими свойствами у собак породы бигль, демонстрирует очень хорошую возможность использования его в качестве лекарственного средства и обладает очень большим потенциалом для клинического применения.
Вышеизложенное представляет собой лишь предпочтительные воплощения по настоящему изобретению и не предназначено для ограничения настоящего изобретения. Предполагается, что любые модификации, эквивалентные замены и усовершенствования и так далее, сделанные в пределах сущности и объема изобретения, включены в объем изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| Новый ингибитор на основе производного хинолина | 2019 |
|
RU2802283C2 |
| ИНГИБИТОРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2019 |
|
RU2806033C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОНАФТАЛИНА И ТЕТРАГИДРОИЗОХИНОЛИНА В КАЧЕСТВЕ РАЗРУШИТЕЛЕЙ ЭСТРОГЕНОВОГО РЕЦЕПТОРА | 2017 |
|
RU2797244C2 |
| ПРОИЗВОДНЫЕ ТИЕНОПИРРОЛА ДЛЯ ПРИМЕНЕНИЯ ДЛЯ НАЦЕЛИВАНИЯ НА БЕЛКИ, КОМПОЗИЦИИ С УКАЗАННЫМИ ПРОИЗВОДНЫМИ, СПОСОБЫ И ПРИМЕНЕНИЯ | 2017 |
|
RU2771166C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ АКТИВАЦИИ PD1/PD-L1 | 2018 |
|
RU2783211C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА ЭСТРОГЕНОВЫХ РЕЦЕПТОРОВ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ ПРИМЕНЕНИЯ | 2018 |
|
RU2797808C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2811612C2 |
| АЗОТСОДЕРЖАЩЕЕ ГЕТЕРОЦИКЛИЧЕСКОЕ АРОМАТИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2729999C1 |
| ПОЛИЦИКЛИЧЕСКИЕ АГЕНТЫ ДЛЯ ЛЕЧЕНИЯ РЕСПИРАТОРНО-СИНЦИТИАЛЬНЫХ ВИРУСНЫХ ИНФЕКЦИЙ | 2007 |
|
RU2486185C2 |
| МОДУЛЯТОРЫ СЕРИН-ТРЕОНИНПРОТЕИНКИНАЗ И PARP | 2013 |
|
RU2681209C2 |
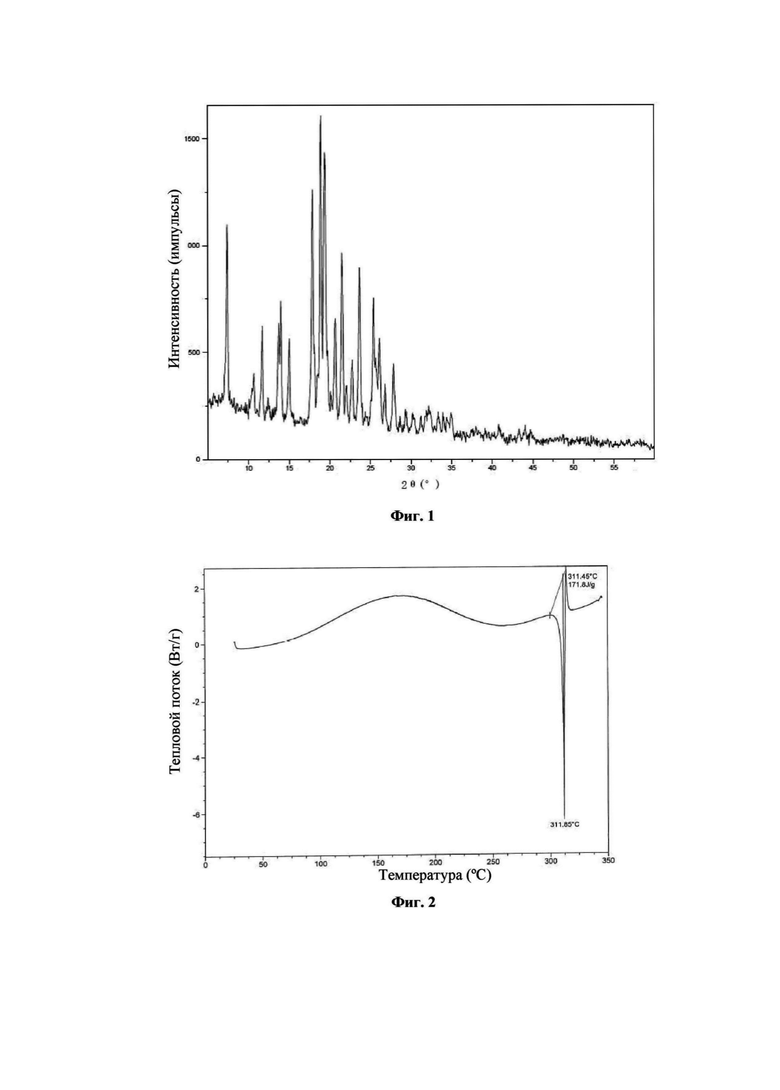
Настоящее изобретение относится к соединению, представленному формулой (I), или его фармацевтически приемлемой соли и стереоизомеру, где R1, R2, X, Y, Р, W и Ar являются такими, как определено в формуле изобретения. Соединение формулы (I) по настоящему изобретению можно использовать в получении лекарственного средства для лечения раковых заболеваний, опосредованных аномальностями мультикиназ. Также предложена кристаллическая форма I соединения 4-(5-(2-хлорфенил)-3-метил-2,10-дигидропиразоло[4,3-b]пиридо[4,3-е]-[1,4]диазепин-8-ил)морфолина, имеющего формулу (III), картина дифракции рентгеновских лучей которого на порошке кристаллической формы I имеет характеристические пики при 7,4±0,2°; 17,9±0,2°; 18,9±0,2°; 19,4±0,2°; 21,5±0,2° и 23,7±0,2°, и способ её получения. Кроме того, изобретение относится к способу получения соединения формулы (III) и промежуточным соединениям для его получения, фармацевтическим композициям, содержащим соединение формулы (I) или кристаллическую форму соединения формулы (III), и способу лечения раковых заболеваний, опосредованных аномальностями мультикиназ. 9 н. и 9 з.п. ф-лы, 2 ил., 9 табл., 17 пр.
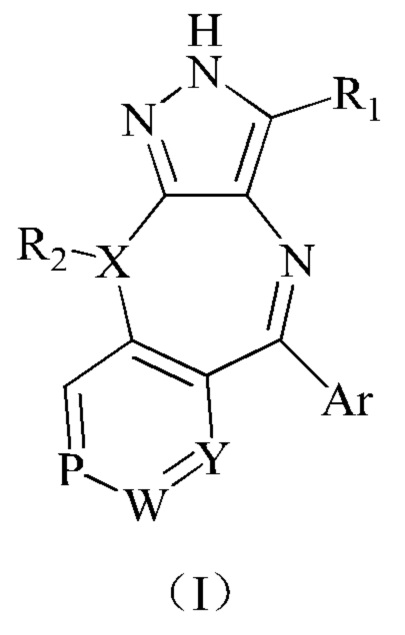
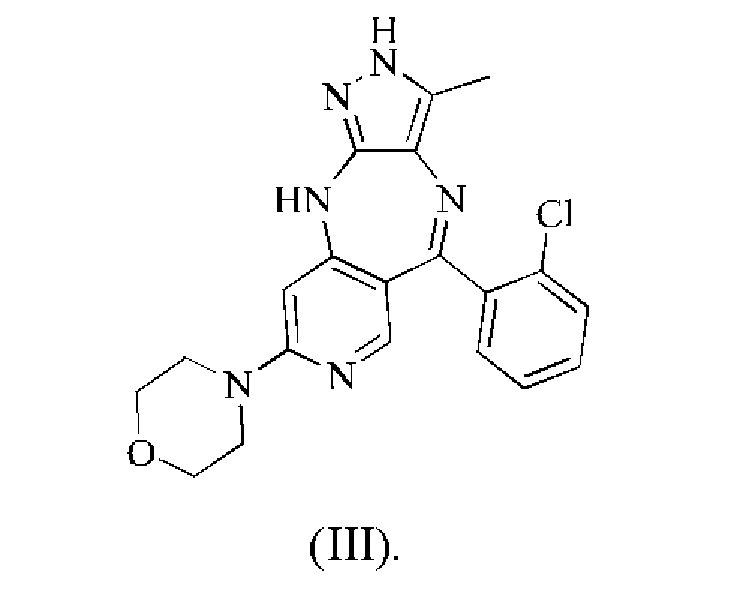
1. Соединение формулы (I), его фармацевтически приемлемые соли или стереоизомеры
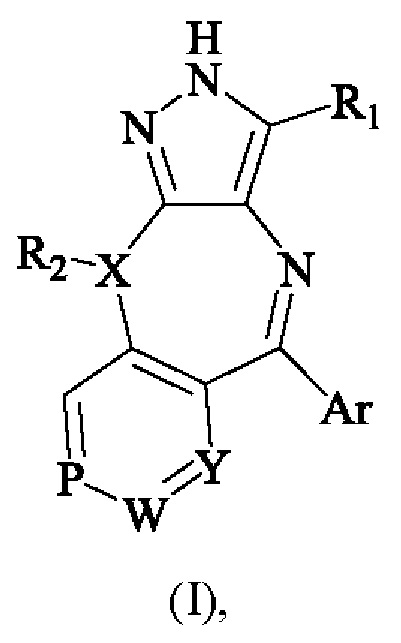
где
X представляет собой N;
R1 выбран из водорода или С1-6алкила;
R2 выбран из водорода или С1-6алкила;
Y выбран из CR3 или N;
Р выбран из CR4 или N;
W выбран из CR5 или N;
R3, R4 и R5 независимо выбраны из группы, состоящей из водорода, гидрокси, амино, карбокси, циано, нитро, галогена, С1-6алкила, С1-6алкокси, С3-6циклоалкилокси, окса-С5-8циклоалкилокси, галогенированного С1-6алкокси, С2-8алкенила, С2-8алкинила, С3-6циклоалкиламино, С1-6алкилсульфонила, С3-8циклоалкилсульфонила, С1-6алкилкарбонила, С3-6циклоалкилкарбонила, -NR11-(CH2)n-N(R7)(R8), С1-6алкилтио-(СН2)n-, -(СН2)n-(3-14)-членного циклоалкила, -(СН2)n-(6-14)-членного арила, -(СН2)n-(5-14)-членного гетероциклила и -(СН2)n-(5-14)-членного гетероарила; где n равен 0-6; образующий кольцо атом S в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до S(O) или S(O)2, образующий кольцо атом С в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до С(О); и циклоалкил, арил, гетероарил или гетероциклил возможно могут быть замещены одним или более независимыми С1-3алкилами или С3-6циклоалкилами;
R7 и R8 независимо выбраны из водорода или С1-6алкила; и
Р, W, и Y одновременно не являются N,
когда Y представляет собой CR3, Р представляет собой CR4, и W представляет собой N, R4 не может являться С1-6алкилом;
когда Y представляет собой CR3, Р представляет собой CR4, и W представляет собой CR5, один из R4 и R5 должен представлять собой Н; и
когда R4 представляет собой Н, R5 выбран из группы, состоящей из гидрокси, карбокси, С1-6алкила, С3-6циклоалкилокси, окса-С5-8циклоалкилокси, галогенированного С1-6алкокси, С2-8алкенила, С2-8алкинила, С3-6циклоалкиламино, С3-8циклоалкилсульфонила, C1-6алкилкарбонила, С3-6циклоалкилкарбонила, -NR11-(CH2)n-N(R7)(R8), С1-6алкилтио-(СН2)n-, -(СН2)n-(3-14)-членного циклоалкила, -(СН2)n-(6-14)-членного арила, -(СН2)n-(5-14)-членного гетероциклила и -(СН2)n-(5-14)-членного гетероарила; где n равен 0-6; образующий кольцо атом S в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до S(O) или S(O)2, образующий кольцо атом С в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до С(О); и циклоалкил, арил, гетероарил или гетероциклил возможно могут быть замещены одним или более независимыми С1-3алкилами или С3-6циклоалкилами;
когда R5 представляет собой Н, R4 выбран из группы, состоящей из гидрокси, амино, карбокси, циано, нитро, галогена, С1-6алкила, С1-6алкокси, С3-6циклоалкилокси, окса-С5-8циклоалкилокси, галогенированного С1-6алкокси, С2-8алкенила, С2-8алкинила, С3-6циклоалкиламино, С1-6алкилсульфонила, С3-8циклоалкилсульфонила, С1-6алкилкарбонила, С3-6циклоалкилкарбонила, -NR11-(CH2)n-N(R7)(R8), С1-6алкилтио-(СН2)n-, -(СН2)n-(3-14)-членного циклоалкила, -(СН2)n-(6-14)-членного арила, -(СН2)n-(5-14)-членного гетероциклила и -(СН2)n-(5-14)-членного гетероарила; где n равен 0-6; образующий кольцо атом S в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до S(O) или S(O)2, образующий кольцо атом С в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до С(О); и циклоалкил, арил, гетероарил или гетероциклил возможно могут быть замещены одним или более независимыми С1-3алкилами или С3-6циклоалкилами;
Ar выбран из 6-14-членного арила или 5-14-членного гетероарила, и Ar возможно может быть замещен 1-3 R6;
каждый R6 независимо выбран из группы, состоящей из водорода, гидрокси, амино, карбокси, циано, нитро, галогена, С1-6алкила, С1-6алкокси, галогенированного С1-6алкокси, галогенированного С1-6алкила, С2-8алкенила, С2-8алкинила, -NR11-(CH2)n-N(R9)(R10), аминоС1-6алкила, С1-6алкилсульфонила, С1-6алкилкарбонила и С1-6алкилтио;
R9 и R10 независимо выбраны из водорода или С1-6алкила; и
R11 выбран из группы, состоящей из водорода, С1-6алкила и С3-6циклоалкила.
2. Соединение, его фармацевтически приемлемые соли или стереоизомеры по п. 1, где
X представляет собой N;
R1 выбран из С1-3алкила;
R2 выбран из группы, состоящей из водорода, метила и этила;
Y выбран из CR3 или N;
Р выбран из CR4 или N;
W выбран из CR5 или N;
R3, R4 и R5 независимо выбраны из группы, состоящей из водорода, гидрокси, амино, карбокси, циано, нитро, галогена, С1-6алкила, С1-6алкокси, С3-6циклоалкилокси, окса-С5-8циклоалкилокси, галогенированного С1-6алкокси, С2-8алкенила, С2-8алкинила, С3-6циклоалкиламино, С1-6алкилсульфонила, С3-8циклоалкилсульфонила, С1-6алкилкарбонила, С3-6циклоалкилкарбонила, -NR11-(CH2)n-N(R7)(R8), С1-6алкилтио-(СН2)n-, -(СН2)n-(3-14)-членного циклоалкила, -(СН2)n-(6-14)-членного арила, -(СН2)n-(5-14)-членного гетероциклила и -(СН2)n-(5-14)-членного гетероарила; где n равен 0-6; образующий кольцо атом S в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до S(O) или S(O)2, и образующий кольцо атом С в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до С(О); циклоалкил, арил, гетероарил или гетероциклил возможно могут быть замещены одним или более независимыми С1-3алкилами или С3-6циклоалкилами;
R7 и R8 независимо выбраны из водорода или С1-6алкила; и
Р, W и Y одновременно не являются N,
когда Y представляет собой CR3, Р представляет собой CR4, и W представляет собой N, R4 не может являться С1-6алкилом,
когда Y представляет собой CR3, Р представляет собой CR4, и W представляет собой CR5, один из R4 и R5 должен представлять собой Н; и
когда R4 представляет собой Н, R5 выбран из группы, состоящей из гидрокси, карбокси, С1-6алкила, С3-6циклоалкилокси, окса-С5-8циклоалкилокси, галогенированного С1-6алкокси, С2-8алкенила, С2-8алкинила, С3-6циклоалкиламино, С3-8циклоалкилсульфонила, C1-6алкилкарбонила, С3-6циклоалкилкарбонила, -NR11-(CH2)n-N(R7)(R8), С1-6алкилтио-(СН2)n-, -(СН2)n-(3-14)-членного циклоалкила, -(СН2)n-(6-14)-членного арила, -(СН2)n-(5-14)-членного гетероциклила и -(СН2)n-(5-14)-членного гетероарила; где n равен 0-6; образующий кольцо атом S в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до S(O) или S(O)2, образующий кольцо атом С в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до С(О); и циклоалкил, арил, гетероарил или гетероциклил возможно могут быть замещены одним или более независимыми С1-3алкилами или С3-6циклоалкилами;
когда R5 представляет собой Н, R4 выбран из группы, состоящей из гидрокси, амино, карбокси, циано, нитро, галогена, С1-6алкила, С1-6алкокси, С3-6циклоалкилокси, окса-С5-8циклоалкилокси, галогенированного С1-6алкокси, С2-8алкенила, С2-8алкинила, С3-6циклоалкиламино, С1-6алкилсульфонила, С3-8циклоалкилсульфонила, С1-6алкилкарбонила, С3-6циклоалкилкарбонила, -NR11-(CH2)n-N(R7)(R8), С1-6алкилтио-(СН2)n-, -(СН2)n-(3-14)-членного циклоалкила, -(СН2)n-(6-14)-членного арила, -(СН2)n-(5-14)-членного гетероциклила и -(СН2)n-(5-14)-членного гетероарила; где n равен 0-6; образующий кольцо атом S в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до S(O) или S(O)2, образующий кольцо атом С в циклоалкиле, ариле, гетероциклиле или гетероариле возможно может быть окислен до С(О); и циклоалкил, арил, гетероарил или гетероциклил возможно могут быть замещены одним или более независимыми С1-3алкилами или С3-6циклоалкилами;
R11 выбран из группы, состоящей из водорода, С1-6алкила и С3-6циклоалкила;
Ar выбран из 6-14-членного арила или 5-10-членного гетероарила; где Ar возможно может быть замещен 1-3 R6; и
каждый R6 независимо выбран из группы, состоящей из водорода, амино, циано, галогена, С1-4алкила, трифторметила и метилсульфонила.
3. Соединение, его фармацевтически приемлемые соли или стереоизомеры по п. 2, где соединение представлено формулой (II):
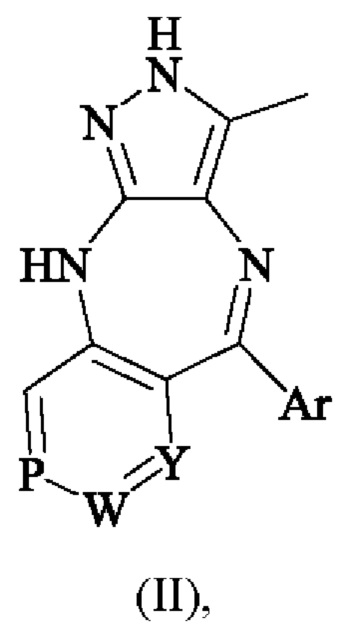
где
Ar выбран из 5-6-членного арила или 5-6-членного гетероарила, где Ar возможно может быть замещен 1-3 R6;
каждый R6 независимо выбран из группы, состоящей из водорода, амино, циано, галогена, С1-4алкила, трифторметила и метилсульфонила;
Y выбран из CR3 или N;
Р выбран из CR4 или N;
W выбран из CR5 или N;
R3, R4 и R5 независимо выбраны из группы, состоящей из водорода, гидрокси, амино, карбокси, циано, нитро, галогена, С1-4алкила, С1-4алкокси, С3-6циклоалкилокси, окса-С5-8циклоалкилокси, галогенированного С1-4алкокси, С2-6алкенила, С2-6алкинила, С3-6циклоалкиламино, С1-4алкилсульфонила, С3-8циклоалкилсульфонила, С1-4алкилкарбонила, С3-6циклоалкилкарбонила, -NR11-(CH2)n-N(R7)(R8), -(СН2)n-(3-14)-членного циклоалкила, -(СН2)n-(5-11)-членного гетероциклила и -(СН2)n-(5-10)-членного гетероарила; где n равен 0-6; образующий кольцо атом S в циклоалкиле, гетероциклиле или гетероариле возможно может быть окислен до S(O) или S(O)2, и образующий кольцо атом С в циклоалкиле, гетероциклиле или гетероариле возможно может быть окислен до С(О); и циклоалкил, гетероарил и гетероциклил возможно могут быть замещены одним или более независимыми С1-3алкилами или С3-6циклоалкилами;
R7 и R8 независимо выбраны из группы, состоящей из водорода, метила, этила и изопропила; и
Р, W и Y одновременно не являются N, и по меньшей мере один из Р, W и Y представляет собой N;
когда Y представляет собой CR3, Р представляет собой CR4, и W представляет собой N, R4 не может являться С1-6алкилом; и
R11 выбран из группы, состоящей из водорода, С1-6алкила и С3-6циклоалкила.
4. Соединение, его фармацевтически приемлемые соли или стереоизомеры по любому из пп. 1-3, где
Ar может быть выбран из группы, состоящей из:
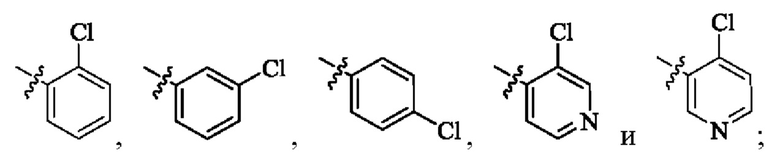
Y выбран из CR3 или N;
Р выбран из CR4 или N;
W выбран из CR5 или N;
R3, R4 и R5 независимо выбраны из группы, состоящей из:
водорода, метила, этила, изопропила, 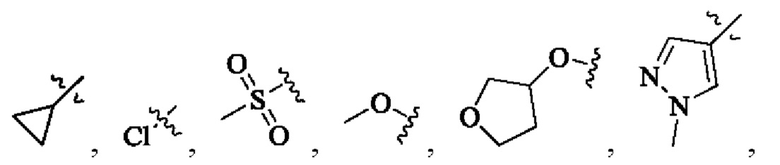
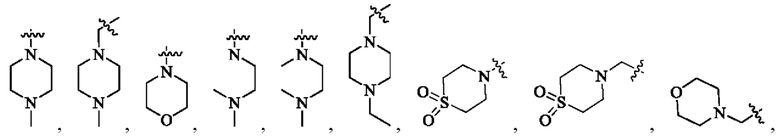
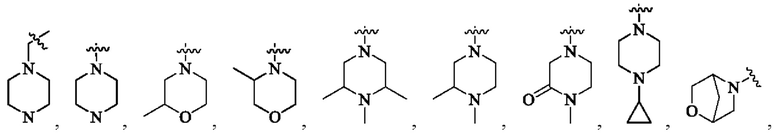
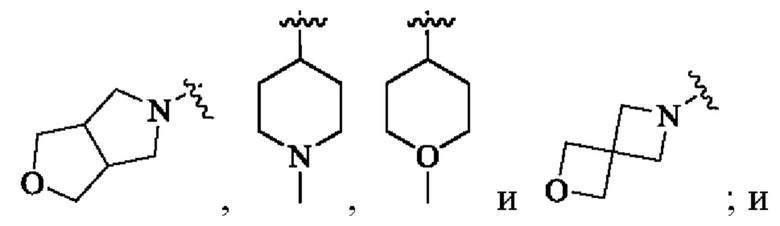
Р, W и Y одновременно не являются N, и по меньшей мере один из Р, W и Y представляет собой N;
когда Y представляет собой CR3, Р представляет собой CR4, и W представляет собой N, R4 не может являться С1-6алкилом.
5. Соединение, его фармацевтически приемлемые соли или стереоизомеры по п. 3 или 4, где
Y представляет собой CR3;
Р представляет собой CR4;
W представляет собой N;
R4 не может являться С1-6алкилом.
6. Соединение, его фармацевтически приемлемые соли или стереоизомеры по п. 3, где
Ar выбран из фенила или 5-6-членного гетероарила, Ar возможно может быть замещен 1-3 R6, каждый R6 независимо выбран из группы, состоящей из водорода, амино, циано, галогена, С1-4алкила, трифторметила и метилсульфонила;
Y представляет собой CR3;
Р представляет собой CR4;
W представляет собой N;
R3 выбран из водорода или С1-4алкила;
R4 выбран из группы, состоящей из водорода, гидрокси, амино, карбокси, циано, нитро, галогена, С1-4алкокси, С3-6циклоалкилокси, окса-С5-8циклоалкилокси, галогенированного С1-4алкокси, С3-6циклоалкиламино, С1-4алкилсульфонила, С3-6циклоалкилсульфонила, C1-4алкилкарбонила, С3-6циклоалкилкарбонила, -NR11-(CH2)n-N(R7)(R8), -(СН2)n-С3-10циклоалкила, -(СН2)n-(5-11)-членного гетероциклила и -(СН2)n-(5-10)-членного гетероарила; где n равен 0-6; образующий кольцо атом S в циклоалкиле, гетероциклиле или гетероариле возможно может быть окислен до S(O) или S(O)2, и образующий кольцо атом С в циклоалкиле, гетероциклиле или гетероариле возможно может быть окислен до С(О); и циклоалкил, гетероарил, гетероциклил возможно могут быть замещены одним или более независимыми С1-3алкилами или С3-6циклоалкилами.
7. Соединение, его фармацевтически приемлемые соли или стереоизомеры по п. 6, где Ar выбран из фенила или пиридила; Ar возможно может быть замещен 1-3 R6, где R6 каждый независимо выбран из группы, состоящей из водорода, амино, циано, галогена, С1-4алкила, трифторметила и метилсульфонила;
Y представляет собой CR3;
Р представляет собой CR4;
W представляет собой N;
R3 выбран из водорода или С1-4алкила;
R4 выбран из группы, состоящей из водорода, гидрокси, амино, карбокси, циано, нитро, галогена, С1-4алкокси, С3-6циклоалкилокси, галогенированного С1-4алкокси, С3-6циклоалкиламино, С1-4алкилсульфонила, С1-4алкилкарбонила, С3-6циклоалкилкарбонила, -NR11-(CH2)n-N(R7)(R8), -(СН2)n-С3-6циклоалкила, -(СН2)n-(5-6)-членного моногетероциклила, -(СН2)n-(7-11)-членного конденсированного гетероциклила, -(СН2)n-(5-6)-членного моногетероарила и -(СН2)n-(8-10)-членного конденсированного гетероарила, где n равен 0-6, образующий кольцо атом S в циклоалкиле, гетероциклиле или гетероариле возможно может быть окислен до S(O) или S(O)2, и образующий кольцо атом С в циклоалкиле, гетероциклиле или гетероариле возможно может быть окислен до С(О); и циклоалкил, гетероарил, гетероциклил возможно могут быть замещены одним или более независимыми С1-3залкилами или С3-6циклоалкилами.
8. Соединение, его фармацевтически приемлемые соли или стереоизомеры по любому из пп. 1-7, где соединение выбрано из:
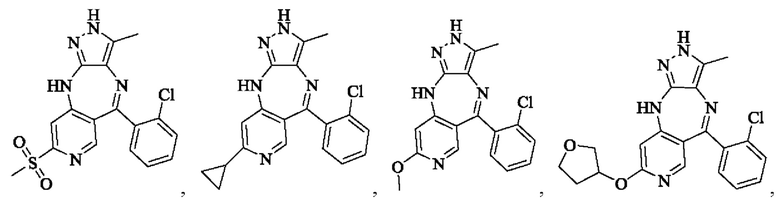
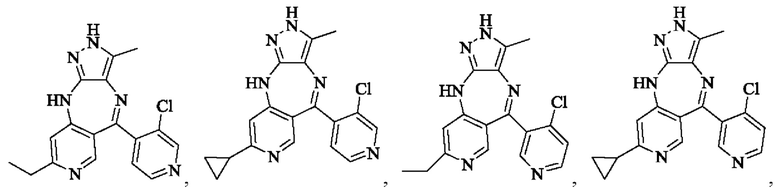
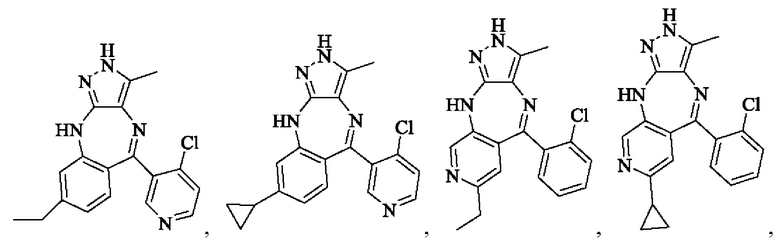
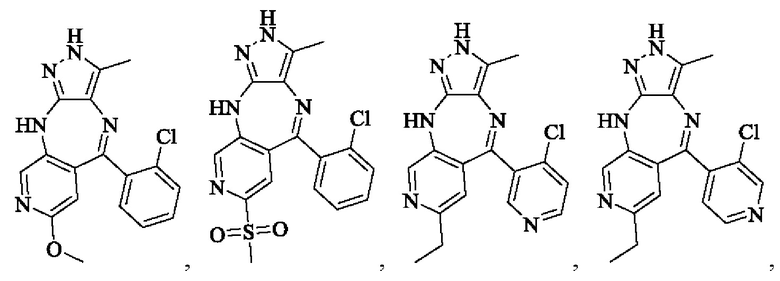
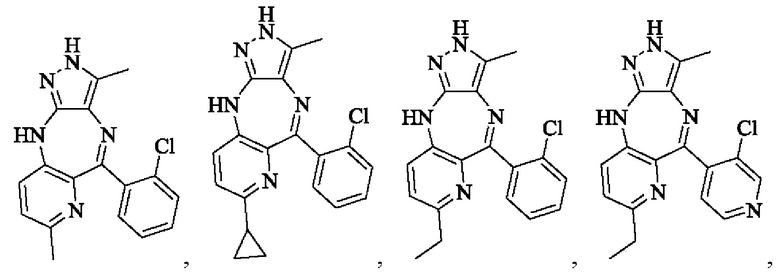
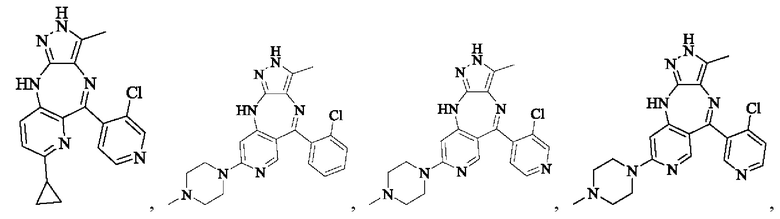
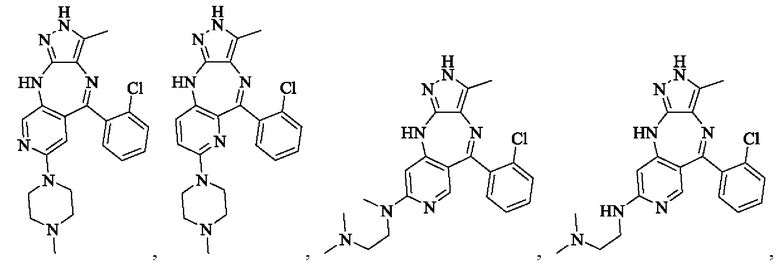
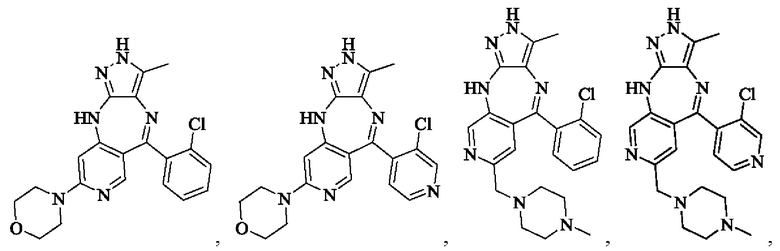
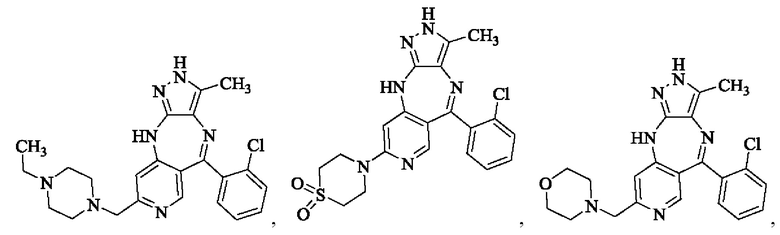
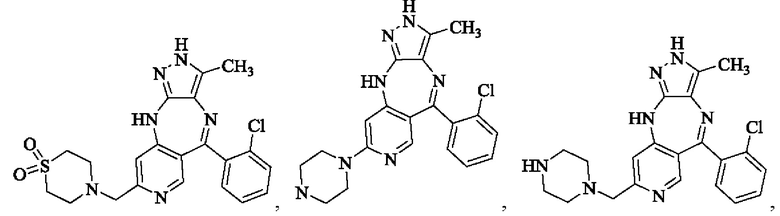
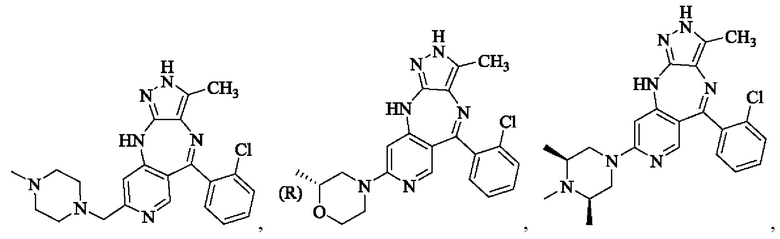
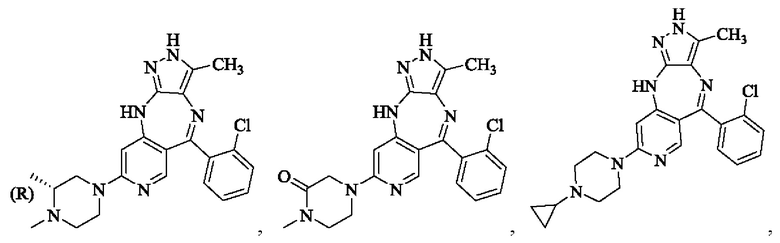
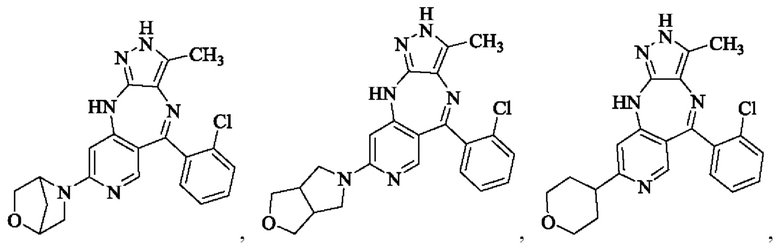
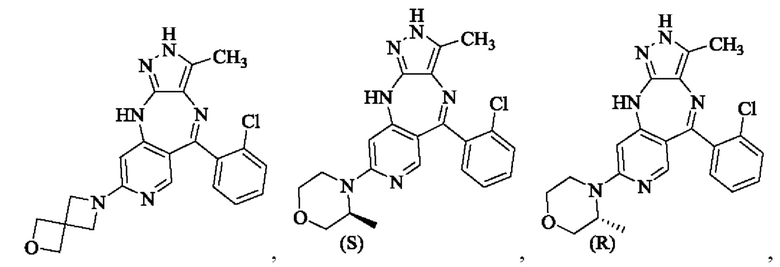
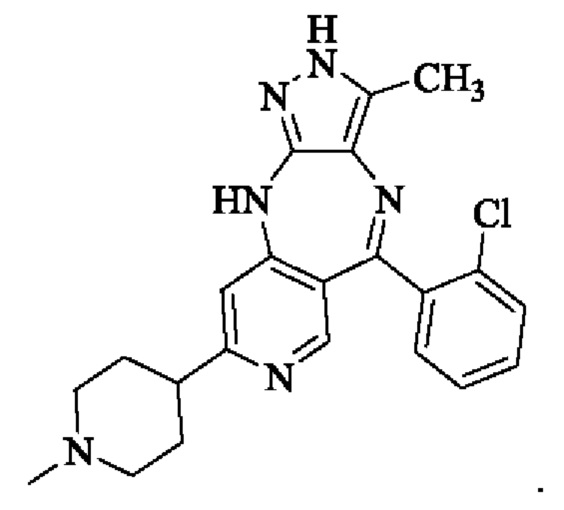
9. Кристаллическая форма I соединения формулы (III), 4-(5-(2-хлорфенил)-3-метил-2,10-дигидропиразоло[4,3-b]пиридо[4,3-e][1,4]диазепин-8-ил)-морфолина, которая имеет характеристические пики при 7,4±0,2°; 17,9±0,2°; 18,9±0,2°; 19,4±0,2°; 21,5±0,2° и 23,7±0,2° на картине дифракции рентгеновских лучей на порошке; для дифракции рентгеновских лучей на порошке используют Cu-Kα-излучение, и характеристический пик выражен углом 2θ;
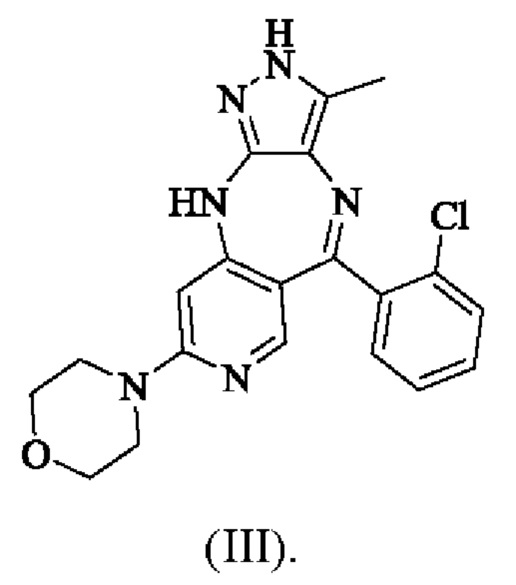
10. Кристаллическая форма I по п. 9, где кристаллическая форма I также имеет характеристические пики при 14,0±0,2°; 15,0±0,2°; 20,7±0,2°; 25,4±0,2° 11,7±0,2°; 22,8±0,2°; 27,8±0,2°.
11. Способ получения кристаллической формы I по п. 9, включающий:
растворение соединения формулы (III) в отдельном или смешанном растворителе при нагревании и
охлаждение для осаждения кристаллической формы I;
или
суспендирование соединения формулы (III) в отдельном или смешанном растворителе, перемешивание и фильтрование с получением кристаллической формы I;
или
растворение соединения формулы (III) в отдельном или смешанном растворителе и концентрирование под вакуумом с получением кристаллической формы I;
или
промывку соединения формулы (III) подходящим количеством отдельного или смешанного растворителя, перемешивание, фильтрование и сушку с получением кристаллической формы I;
где отдельный или смешанный растворитель представляет собой один или более чем один растворитель, выбранный из группы, состоящей из метанола, этанола, тетрагидрофурана, 2-метилтетрагидрофурана, дихлорметана, дихлорэтана, этилацетата, ацетонитрила, диметилсульфоксида, смеси диметилсульфоксид/вода, метанол/тетрагидрофуран, метанол/2-метилтетрагидрофуран, метанол/дихлорметан, этанол/2-метилтетрагидрофуран, дихлорметан/вода.
12. Способ получения соединения формулы (III), включающий:
1) взаимодействие соединения формулы (III-А) с соединением формулы (III-В) с получением соединения формулы (III-C);
2) взаимодействие соединения формулы (III-C) с соединением формулы (III-D) с получением соединения формулы (III-Е);
3) снятие защиты с соединения формулы (III-E) с получением соединения формулы (III-F) или (III-F') и
4) получение соединения формулы (III) из соединения формулы (III-F) или (III-F');
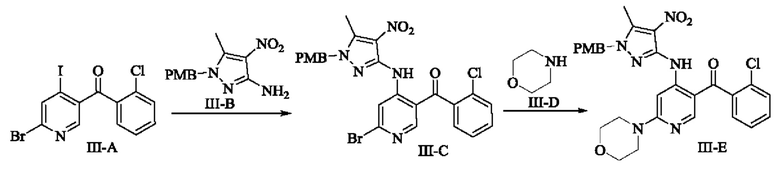
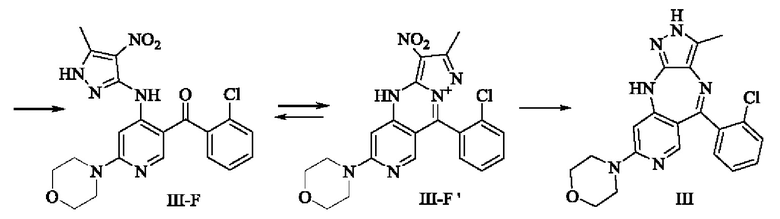
13. Промежуточное соединение для получения соединения формулы (III), представленное следующей структурной формулой:
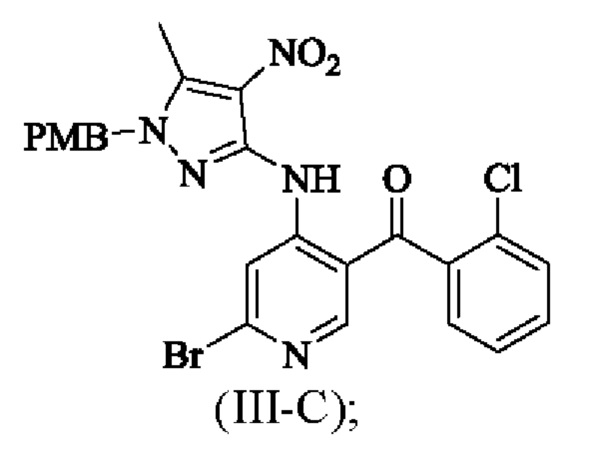
или
или 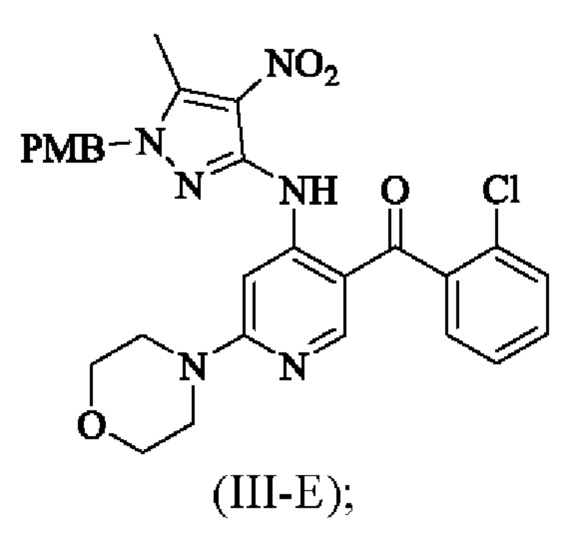
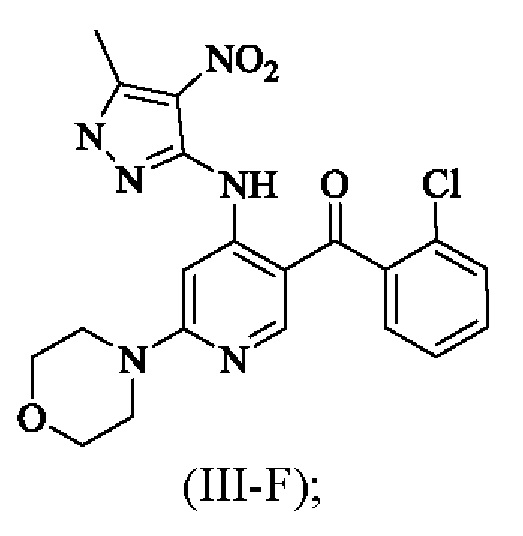
или
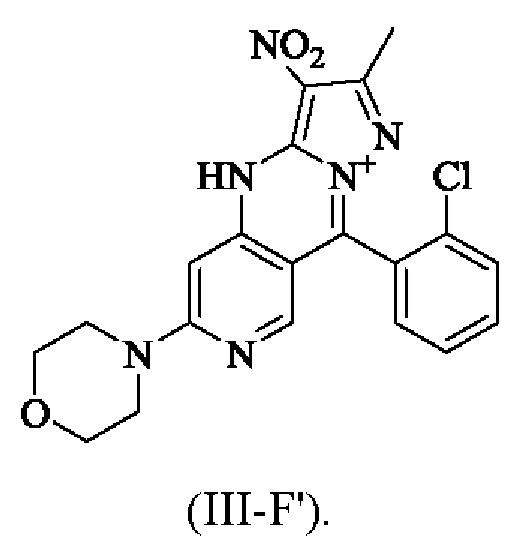
14. Фармацевтическая композиция для лечения рака, опосредованного аномальностью мультикиназ, содержащая соединение, его фармацевтически приемлемые соли или стереоизомеры по любому из пп. 1-8, и/или кристаллическую форму I соединения формулы (III) по п. 9, где фармацевтическая композиция содержит один или более чем один фармацевтически приемлемый носитель.
15. Фармацевтическая композиция по п. 14, дополнительно содержащая один или более чем один второй терапевтически активный агент, где вторые терапевтически активные агенты представляют собой антиметаболиты, ингибиторы фактора роста, ингибиторы митоза, противоопухолевые гормоны, алкилирующие агенты, металлы, ингибиторы топоизомеразы, гормоны, иммуномодуляторы, гены-супрессоры опухолей, противораковые вакцины, иммунные контрольные точки или антитела для иммунотерапии опухолей.
16. Фармацевтическая композиция, содержащая соединение, его фармацевтически приемлемые соли или стереоизомеры по любому из пп. 1-8, и/или кристаллическую форму I соединения формулы (III) по п. 9, и один или более чем один второй терапевтически активный агент, где вторые терапевтически активные агенты представляют собой антиметаболиты, ингибиторы фактора роста, ингибиторы митоза, противоопухолевые гормоны, алкилирующие агенты, металлы, ингибиторы топоизомеразы, гормоны, иммуномодуляторы, гены-супрессоры опухолей, противораковые вакцины, иммунные контрольные точки или антитела для иммунотерапии опухолей.
17. Применение соединений, их фармацевтически приемлемых солей или стереоизомеров по любому из пп. 1-8, кристаллической формы I соединения формулы (III) по п. 9, или фармацевтической композиции по любому из пп. 14-15 или 16 в изготовлении лекарственного средства для лечения рака, опосредованного аномальностью мультикиназ, где рак включает рак легкого, плоскоклеточную карциному, рак мочевого пузыря, рак желудка, рак яичника, рак брюшины, рак молочной железы, протоковую карциному молочной железы, рак головы и шеи, карциному эндометрия, рак тела матки, рак прямой кишки, рак печени, рак почки, опухоль почечной лоханки, эзофагеальную карциному эзофагеальную аденокарциному, глиому, рак предстательной железы, рак щитовидной железы, рак женской репродуктивной системы, карциному in situ, лимфому, нейрофиброматоз, рак кости, рак кожи, рак головного мозга, рак толстой кишки, рак яичка, гастроинтестинальную стромальную опухоль, рак полости рта, фарингеальный рак, множественную миелому, лейкоз, неходжкинскую лимфому, хориоаденому толстой кишки, меланому, цитому и саркому
18. Способ лечения заболевания, включающий введение пациенту нуждающемуся в этом, терапевтически эффективного количества соединений, их фармацевтически приемлемых солей или стереоизомеров по любому из пп. 1-8, кристаллической формы I соединения формулы (III) по п. 9, или фармацевтической композиции по любому из пп. 14-15 или 16, где заболевание включает опосредованные мультикиназами раковые заболевания и опосредованные мультикиназами раковые заболевания включают рак легкого, плоскоклеточную карциному, рак мочевого пузыря, рак желудка, рак яичника, рак брюшины, рак молочной железы, протоковую карциному молочной железы, рак головы и шеи, карциному эндометрия, рак тела матки, рак прямой кишки, рак печени, рак почки, опухоль почечной лоханки, эзофагеальную карциному, эзофагеальную аденокарциному, глиому рак предстательной железы, рак щитовидной железы, рак женской репродуктивной системы, карциному in situ, лимфому нейрофиброматоз, рак кости, рак кожи, рак головного мозга, рак толстой кишки, рак яичка, гастроинтестинальную стромальную опухоль, рак полости рта, фарингеальный рак, множественную миелому, лейкоз, неходжкинскую лимфому, хориоаденому толстой кишки, меланому цитому и саркому.
| ПИРАЗОЛБЕНЗОДИАЗЕПИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ CDK2, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ СОДЕРЖАЩАЯ | 2000 |
|
RU2249593C2 |
| Барражирующий боеприпас | 2023 |
|
RU2818171C1 |

