Настоящее изобретение относится к способу извлечения гидроксида лития высокой чистоты из литийсодержащих источников, которые также содержат ионы фтора. Такие источники могут быть геогенными, как например, литиевый минерал лепидолит, или антропогенными, как например, отходы ионно-литиевых батарей, содержащие по меньшей мере один переходный металл, выбранный из группы, состоящей из никеля, марганца и кобальта.
Для извлечения лития минерал лепидолит, как правило, прокаливают с известняком, из растворов материала, содержащего гидроксид лития и фторид лития, большая часть фторида лития может быть удалена после концентрирования и фильтрации. Полученный фильтрат все еще может содержать небольшое количество фтора, определяемое равновесием раствора.
Подобная ситуация может возникнуть при переработке ионно-литиевых батарей, где в процессе переработки также могут быть получены растворы, содержащие литий и фторид. Способ очистки LiOH согласно настоящему изобретению в частности относится к отделению гидроксида лития от нежелательных примесей, таких как фторидные соли, путем экстракции лития из сырого препарата гидроксида лития жидкостью, содержащей или состоящей из определенных органических растворителей, таких как метанол.
Было описано несколько способов, ведущих от отходов материалов литиевых батарей, таких как целые батареи, разряженные и измельченные, или далее разобранные и/или механически разделенные, к материалу, предпочтительно состоящему из одной или нескольких солей лития, предполагающих, что такие соли могут в дальнейшем использоваться для получения новых ионно-литиевых батарей или электродных материалов из них. В WO 2018223193 описан способ, в котором литий осаждают в виде Li2CO3 после предварительного разделения Ni, Cu и Mn путем ионного обмена и Со путем осаждения. В CN 107017443 А предлагается аналогичный способ, необязательно с прямым отверждением продукта гидроксида лития. В KR 101998691 B раскрыт способ получения предшественника лития, такого как LiOH, из материала электрода ионно-литиевой батареи.
Проблемой, возникающей при извлечении ценных компонентов из аккумуляторного лома, является присутствие примесей, особенно фторидов, происходящих из электролита или связующего материала.
Согласно настоящему изобретению обнаружено, что очистка сырого гидроксида лития, содержащего LiF и/или соли амфотерных элементов, может быть эффективно осуществлена путем растворения такого материала в низшем спирте или в смеси растворителей на основе низшего спирта.
Таким образом, способ согласно настоящему изобретению относится к очистке сырого гидроксида лития, как правило, получаемого из горнодобывающих растворов или из материала, содержащего отходы ионно-литиевых батарей или их частей, где способ содержит стадии
(A) предоставление неочищенного гидроксида лития в виде твердого вещества, которое содержит фторид и
(B) объединение неочищенного гидроксида лития, предоставленного на стадии (А) с низшим спиртом.
Неочищенный гидроксид лития может быть предоставлен на стадии (А) в виде однофазного твердого вещества или в виде смеси твердых фаз. Хотя возможно присутствие жидкости, в предпочтительный способ обеспечивает неочищенный гидроксид лития в виде сухого твердого вещества в отсутствие жидкой фазы. Поскольку на этой стадии нет необходимости в очистке, неочищенный гидроксид лития можно высушить в сушилке, например, в лопастной сушилке, распылительной сушилке, центробежной кольцевой сушилке, сушилке с псевдоожиженным слоем, стояке или вращающейся печи. В зависимости от температуры сушки получают моногидрат гидроксида лития или ангидрат гидроксида лития или любую их смесь и, таким образом, предоставляют на стадии (А) согласно настоящему изобретению. Тем не менее, неочищенный твердый гидроксид лития может все еще содержать остатки растворителя, например, воду в количестве, превышающем кристаллическую воду, полученную из моногидрата гидроксида лития, как правило, до около 5% от общей массы твердого вещества.
Неочищенный гидроксид лития, предоставленный на стадии (А), содержит фторид в качестве примеси, как правило, причем содержит моногидрат гидроксида лития, содержащий от 100 частей на миллион до 1.29% кальция, от 0.1 до 1.29% фтора, от 0.1 до 1.29% натрия, или ангидрат гидроксид лития, содержащий от 175 частей на миллион до 2.26% кальция, от 0.175 до 2.26% фтора, от 0.175 до 2.26% натрия, w где все количества приведены по массе сухого твердого вещества, в важном варианте осуществления количество фторида составляет 500 частей на миллион или более по отношению к общей массе неочищенного твердого гидроксида лития.
Указанный неочищенный гидроксид лития, предоставляемый на стадии (А), часто содержит одну или несколько дополнительных примесей из группы щелочных солей, солей алюминия, солей цинка, сумма щелочных металлов, алюминия и цинка, как правило, составляет от 100 до 500 частей на миллион или более, например, от 500 до 5000 частей на миллион, по отношению к общей массе неочищенного твердого гидроксида лития.
В типичном способе согласно настоящему изобретению 90 мас.% или более неочищенного твердого гидроксида лития, предоставляемого на стадии (А), состоит из ангидрата гидроксида лития или моногидрата гидроксида лития или их смесей.
Низший спирт обычно выбирается из С1-С4 спиртов или представляет собой смесь таких спиртов, такую как метанола и/или этанола.
Низший спирт, добавляемый на стадии (В) согласно настоящему изобретению, как правило, представляет собой технический продукт, который может содержать до около 6 мас.% воды, как правило, продукт низшего спирта или смесь растворителей на основе такого спирта состоит из 93 мас.% или более низшего спирта. Таким образом, продукт низшего спирта, как правило, содержит низший спирт с чистотой по меньшей мере 93 мас.%, предпочтительно по меньшей мере 95 мас.%, более предпочтительно по меньшей мере 98 мас.% и особенно по меньшей мере 99 мас.%, при этом остатки в указанном продукте составляют в основном другие спирты и/или воду, в то время как другие примеси, такие как неспиртовые органические растворители, могут присутствовать в количестве до 1 мас.% от указанного продукта низшего спирта или смеси растворителей на основе такого спирта. Низший спирт предпочтительно представляет собой метанол, этанол или их смесь.
Существует линейное влияние применяемого соотношения спирт:LiOH на стадии (В) на конечное содержание фторида. Это связано с достаточно низкой, но существующей растворимостью LiF в низшем спирте. Таким образом, количество низшего спирта, применяемого на стадии (В), должно быть достаточным для растворения неочищенного гидроксида лития, чтобы обеспечить хороший общий выход лития, но должно быть ограничено, чтобы избежать растворения фторида и эффектов сильного разбавления. В типичном способе объединяют, например, около от 5 до 15, особенно около от 7 до 13 молярных частей низшего спирта, такого как метанол, на стадии (В) с 1 молярной частью лития, предоставленного на стадии (А).
В типичном способе объединенный гидроксид лития и низший спирт со стадии (В) последовательно смешивают с получением суспензии частиц в растворе гидроксида лития.
Полученный раствор, содержащий растворенный LiOH, может быть затем отделен от нерастворенных примесей с помощью разделения твердой и жидкой фаз (стадия С), такого как фильтрация, осаждение, декантация, центрифугирование и т.д. В предпочтительном способе раствор гидроксида лития, полученный на стадии (В), затем подвергают
(C) разделению твердой и жидкой фаз с получением прозрачного раствора,
(D) необязательно с последующим добавлением воды, и
(F) выделению твердого гидроксида лития в виде ангидрата или моногидрата, или в виде смеси ангидрата и моногидрата, или в виде смеси моногидрата с водой и/или низшим спиртом, или в виде смеси ангидрата с низшим спиртом.
Дополнительную очистку можно проводить с помощью дополнительной стадии водной кристаллизации, которую проводят перед стадией (А), например, путем кристаллизации выщелачивающего раствора, содержащего гидроксид лития, или после стадии (В), например, после удаления спирта и добавления воды с последующим удалением твердых веществ путем разделения твердой и жидкой фаз (стадия С).
Очищенный таким образом твердый гидроксид лития может быть извлечен из раствора с использованием известных способов, таких как кристаллизация, кристаллизация с погружением путем добавления менее полярных растворителей, например, высших спиртов, простых эфиров или углеводородов, сушка и т.д.
В конкретном варианте указанного предпочтительного способа раствор гидроксида лития, полученный на стадии (В), в метаноле затем подвергают
(C) разделению твердой и жидкой фаз с удалением нерастворенных примесей, таких как LiF, и получением прозрачного раствора,
(D) необязательно с последующим добавлением воды,
(E) удалению растворителя для немедленного получения очищенного гидроксида лития (Е1), или путем кристаллизации (Е2), и
в случае, если проводят (Е2), (F) отделению кристаллов от жидкости и
(G) сушке кристаллов. В таком способе твердый LiOH получают на стадии (Е1) или (F) и (G) в виде ангидрата, или в виде моногидрата, или в виде смеси ангидрата и моногидрата, или в виде смеси моногидрата с водой и/или низшим спиртом, или в виде смесь ангидрата с низшим спиртом.
Используемый низший спирт преимущественно извлекают путем дистилляции или выпаривания и возвращают в процесс, например, путем сбора растворителя со стадии (Е1) или маточного раствора, отделенного на стадии (F), отделения воды от низшего спирта, если она присутствует, и повторного использования на стадии (В). Для того, чтобы избежать более высокого содержания алкоголята или спирта (как правило, метилата или метанола), в ходе выпаривания спирта можно добавлять воду, предпочтительно в конце выпаривания (Е1) или, альтернативно, во время кристаллизации (Е2).
Согласно настоящему изобретению было обнаружено, что способ согласно настоящему изобретению особенно эффективен, и может быть достигнуто особенно низкое содержание фторида, когда стадии процесса (А), (В) и (С) проводят с использованием лишь небольшого количества воды или без воды, таким образом, неочищенный твердый гидроксид лития, предоставляемый на стадии (А) согласно настоящему изобретению, преимущественно содержит менее одного моля воды на моль лития, т.е. меньше количества воды, присутствующего в моногидрате гидроксида лития. Это может быть достигнуто путем предоставления неочищенного материала для стадии (А) в виде смеси моногидрата и ангидрата гидроксида лития, например, с вычисленным содержанием 90%, или 80%, или особенно 50% или менее всего LiOH на стадии (А), присутствующего в виде моногидрата гидроксида лития помимо ангидрата гидроксида лития (чистый LiOH*H2O содержит 16,55 мас% лития и 42,9 мас.% воды, а ангидрат содержит 28,99 мас.% лития). Следовательно, количество воды, присутствующей в метаноле, добавляемом на стадии (В), должно поддерживаться на как можно более низком уровне, как правило, должно быть ниже количества, способного превратить весь ангидрат, предоставленный на стадии (А), в моногидрат, например, путем использования интенсивно высушенного метанола.
В этом варианте способа воду используют перед стадией (С) только в количествах, меньших стехиометрического, или воду вообще не используют. Таким образом, на стадии (D) предпочтительно добавляют воду, особенно в количествах, снижающих образование алкоголята во время последующих стадий сушки до менее чем 5 мол. %, предпочтительно менее 1 мол. % от общего количества лития в сухом продукте. В примере такого предпочтительного способа на стадии (D) добавляют около 1 молярной части воды или более на каждый моль лития, предоставленного на стадии (А), обычно от 1 до 5 моль, предпочтительно от 1 до 3 моль, в большинстве случаев предпочтительно от 1 до 2 моль.
Следовательно, в предпочтительном способе неочищенный твердый гидроксид лития, предоставленный на стадии (А), содержит 50 мол. % или более гидроксида лития в форме ангидрата, и воду добавляют (на стадии D) после стадии (С) в количество от 1 до 5 моль, предпочтительно от 1 до 3 моль, наиболее предпочтительно от 1 до 2 моль на 1 молярную часть лития.
Как правило, твердый продукт гидроксида лития, полученный в соответствии с настоящим изобретением (стадии Е, F и/или G), содержит определенное количество остаточных углеродных соединений (общий углерод [ТС] или, в частности, неочищаемый органический углерод [NPOC], как описано ниже). Остаточное содержание, особенно NPOC, как правило, составляет от 1 до 5000 частей на миллион, особенно от 1 до 3000 частей на миллион от общей массы твердого продукта, полученного в описанном выше способе. Такой остаточный NPOC определяют, как описано ниже. Примерами NPOC являются остатки низшего спирта, особенно метанола, используемого на данной стадии (В), или элементарный углерод (графит, например, определяется как NPOC), небольшие количества алкоголята лития, такого как LiOCH3, также могут присутствовать, но их предпочтительно избегать из-за проблем с обращением и хранением, особенно когда количество алкоголята в продукте выше 5 мол. % лития или даже 1 мол. %.
Полученный таким образом твердый гидроксид лития, как правило, содержит по меньшей мере 50 мас.% LiOH, часто в присутствии воды, например, в виде моногидрата гидроксида лития.
Предпочтительным является продукт в виде твердого гидроксида лития, содержащий от 1 до 100 частей на миллион фторида и от 1 до 1000 частей на миллион, особенно от 5 до 500 частей на миллион указанного низшего спирта, такого как этанол или особенно метанол, в каждом случае относительно общей массы твердого продукта.
Неочищенный гидроксид лития, предоставленный на стадии (А), например, с уровнями примесей, как описано выше, как правило, получают из отходов литий-ионных батарей.
В основном все растворы, содержащие ионы лития и фторида в растворенной форме, могут быть использованы в качестве источника настоящего неочищенного твердого гидроксида лития согласно настоящему изобретению (А), который является получаемым после удаления по меньшей мере части растворителя, в типичном способе неочищенный твердый гидроксид лития согласно настоящему изобретению, предоставленный на стадии (А), получают сушкой и/или кристаллизацией, необязательно после отделения твердых веществ от раствора. Неочищенный гидроксид лития, предоставленный на стадии (А), может быть получен выщелачиванием материала, содержащего гидроксид лития, он также может быть получен химическим или электрохимическим преобразованием подходящего исходного материала. Таким образом, исходным материалом может быть фторид-содержащая литиевая соль, отличная от гидроксида (например, бикарбонат/карбонат, хлорид, нитрат или сульфат). Из таких солей необработанный гидроксид лития может быть получен, например, реакцией с гидроксидом щелочноземельного металла, или электролизом, или электродиализом, или прокаливанием (нитрат лития) и последующим гидролизом.
Для получения неочищенного гидроксида лития материал, полученный из отходов аккумуляторов, как правило, затем подвергают стадиям восстановления, водному выщелачиванию, необязательно в присутствии гидроксида щелочноземельного металла (предпочтительно гидроксида кальция), отделению твердых веществ и сушке и/или кристаллизации.
Кроме того, настоящее изобретение относится к способу извлечения лития из материала, содержащего отходы ионно-литиевых батарей или их части, где способ содержит перед стадиями (А) и (В) согласно настоящему изобретению, стадии:
(a) предоставления восстановленного материала в виде частиц, содержащего соединение переходного металла и/или переходный металл, где переходный металл выбирают из группы, состоящей из Mn, Ni и Со, причем этот материал в виде частиц дополнительно содержит соль лития и фторидную соль, и где материал в виде частиц необязательно содержит кальций,
(b) обработки материала, предоставленного на стадии (а), полярным растворителем; а также
(c) отделения твердых веществ от жидкости, необязательно с последующей промывкой твердого остатка.
Как правило, указанный материал оксида переходного металла, содержащий литий, получают после механического удаления корпуса, проводки или схемы, таким образом, как правило, он состоит в основном из материала элемента. Из соображений безопасности такие ионно-литиевые батареи разряжаются полностью, например, путем погружения в сухую токопроводящую ванну, такую как металлическая крошка, в противном случае могут возникнуть короткие замыкания, представляющие опасность пожара и взрыва. Такие ионно-литиевые батареи могут быть разобраны, пробиты, измельчены, например, в молотковой мельнице, или измельчены, например, в промышленном измельчителе.
Может быть предпочтительно по меньшей мере частично удалить электролиты перед тем, как подвергнуть материал предварительной стадии (i), особенно электролиты, которые содержат органический растворитель или смесь органических растворителей, например, путем сушки, например, при температурах в диапазоне от 50 до 250°С при атмосферном давлении или ниже. Материал оксида переходного металла, содержащий литий предпочтительно не подвергают воздействию более высоких температур (в частности, не до 400°С или более) в окислительных условиях перед тем, как подвергнуть его стадии (а) согласно настоящему изобретению.
В одном варианте осуществления настоящего изобретения указанный материал оксида переходного металла, содержащий литий, получают из аккумуляторных отходов. В предпочтительном варианте осуществления настоящего изобретения указанный материал оксида переходного металла, содержащий литий, получают из механически обработанных аккумуляторных отходов, например, из аккумуляторных отходов, обработанных в молотковой дробилке или в промышленном измельчителе.
Материал в виде частиц, полученный на стадии (а), как правило, представляет собой материал, полученный из отходов ионно-литиевых батарей после проведения предварительной стадия нагревания в инертных или восстанавливающих условиях до температуры в диапазоне от 80 до 900°С, где восстановление, как правило, проводится после разрядки ионно-литиевой батареи, разборки и/или измельчения. Восстановление целесообразно проводить в условиях, включающих присутствие углерода и восстановительного газа, выбранного из водорода и монооксида углерода. Температура во время такой стадии восстановления предпочтительно находится в интервале от 200 до 800°С, более предпочтительно от 350 до 500°С, особенно от 350 до 450°С. В важном варианте осуществления восстановление проводят в присутствии 35% или более по объему водорода, предпочтительно от 50 до 100 об.% (нормальные условия) водорода, остальное, если присутствует, представляет собой неокисляющий газ. В указанном варианте осуществления настоящего изобретения эта стадия восстановления имеет продолжительность в диапазоне от 10 минут до 30 часов, предпочтительно от 20 минут до 8 часов, более предпочтительно от 30 минут до 4 часов. Особый технический интерес представляет продолжительность стадии восстановления от 20 до 90 минут, особенно от 30 до 60 минут, предпочтительно в присутствии водорода. Концентрация водорода в восстановительной атмосфере и время реакции зависят друг от друга. Обычно низкая концентрация водорода требует более длительного времени восстановления и наоборот.
В другом важном варианте осуществления восстановление проводят путем нагревания в исходном присутствии кислорода и углерода в закрытой печи с образованием моноксида углерода. Используемые ионно-литиевые батареи и, следовательно, материал в виде частиц, полученный на стадии (а), как правило, содержат углерод, например, в форме графита.
В одном варианте осуществления восстанавливающие условия, связанные с концентрацией водорода и/или монооксида углерода, а также температурой и продолжительностью, выбирают таким образом, чтобы по меньшей мере часть полученного материала оксида переходного металла, содержащего литий, содержит пара-, антиферро-, ферро- и/или ферримагнитные компоненты. Предпочтительным является образование ферро- или ферримагнитных компонентов в результате, по меньшей мере, частичного восстановления материала оксида переходного металла, содержащего литий. Степень восстановления может варьироваться в диапазоне от 1 до 100% по отношению к никелю, содержащемуся в материале оксида переходного металла, содержащем литий, предпочтительным является диапазон от 80 до 100%.
Материал в виде частиц, предоставленный на настоящей стадии (а), таким образом, содержит соединение переходного металла и/или переходный металл, где переходный металл выбран из группы, состоящей из Mn, Ni и/или Со, и где дополнительно по меньшей мере часть указанного Ni и/или Со, если они присутствуют, находятся в степени окисления ниже +2, и по меньшей мере часть указанного Mn, если он присутствует, представляет собой оксид марганца (II), никель и/или кобальт, они, как правило, по меньшей мере частично присутствуют в их металлическом состоянии.
В дальнейшем материал в виде частиц этого варианта осуществления, полученный на стадии (а), а также материал, подвергнутый стадии предварительного восстановления, альтернативно будут обобщены как материал оксида переходного металла, содержащий литий. Как правило, такой материал оксида переходного металла, содержащий литий, происходит из ионно-литиевой батареи и содержит фтор предпочтительно в диапазоне от 1% до 8 мас.% и/или фосфор в диапазоне от 0,2% до 2 мас.% относительно массы материала оксида переходного металла, содержащего литий.
В результате восстановительной обработки материалов виде частиц перед вышеуказанной стадией (а) по меньшей мере часть указанного Ni и/или Со, если они присутствуют, находятся в степени окисления ниже +2, и по меньшей мере часть указанного Mn, если он присутствует, представляет собой оксид марганца (II).
Соединение переходного металла и/или переходный металл Ni и/или Со в степени окисления ниже+2, содержащиеся в материале в виде частиц, полученном на стадии (а), в основном содержит Ni и/или Со в металлическом состоянии, как определено порошково-рентгеновской дифрактометрией (Cu-k-альфа-1 излучение). Соль лития и фторидная соль, содержащиеся в материале в виде частиц, полученном на стадии (а), как правило, содержат одну или несколько солей, выбранных из LiOH, LiF, Li2O, Li2CO3, LiHCO3, алюминатов лития, солей фосфата лития, смешанных оксидов Li и один или более из Ni, Со, Mn, Fe, Al, Cu и/или фторидов Ni, Со, Mn, Fe, Al, Cu.
Поскольку сильное нагревание, особенно в окислительных условиях, но в меньшей степени также в восстановительной атмосфере, приводит к увеличению образования нерастворимых веществ (таких как LiMnO2), предпочтительно в общем не подвергать материал оксида переходного металла, содержащего литий, воздействию температур 500°С и более. Таким образом, материал в виде частиц, полученный на стадии (а), предпочтительно не подвергают воздействию температур 400°С или выше в окислительных условиях перед тем, как подвергнуть его стадии (а) согласно настоящему изобретению.
В одном варианте осуществления настоящего изобретения материал, предоставленный на стадии (а), подвергают сухому разделению твердых фаз перед стадией (b) для отделения компонентов, содержащих переходный металл.
В типичном варианте осуществления настоящего изобретения указанный материал оксида переходного металла, содержащий литий, предоставленный на стадии (А), содержит в интервале от 20 частей на миллион до 10 мас.%, особенно от 20 частей на миллион до 3 мас.% меди, в виде металла или в форме одного или нескольких его соединений.
В типичном варианте осуществления настоящего изобретения указанный материал оксида переходного металла, содержащий литий, предоставленный на стадии (А), содержит в интервале от 100 частей на миллион до 15 мас.% алюминия, в виде металла или в форме одного или нескольких его соединений.
В типичном варианте осуществления настоящего изобретения указанный материал оксида переходного металла, содержащий литий, предоставленный на стадии (А), содержит в интервале от 100 частей на миллион до 5 мас.% железа, в виде металла или сплава или в форме одного или нескольких его соединений.
В типичном варианте осуществления настоящего изобретения указанный материал оксида переходного металла, содержащий литий, предоставленный на стадии (А), содержит в интервале от 20 частей на миллион до 2 мас.% цинка, в виде металла или сплава или в форме одного или нескольких его соединений.
В типичном варианте осуществления настоящего изобретения указанный материал оксида переходного металла, содержащий литий, предоставленный на стадии (А), содержит в интервале от 20 частей на миллион до 2 мас.% циркония, в виде металла или сплава или в форме одного или нескольких его соединений.
В типичном варианте осуществления настоящего изобретения указанный материал оксида переходного металла, содержащий литий, предоставленный на стадии (А), содержит в интервале от 1% до 8%, особенно от 2% до 8 мас.% фтора, вычисленного как сумма органического фтора, например, связанного в полимеры, и неорганического фторида в виде одного или нескольких его неорганических фторидов.
В типичном варианте осуществления настоящего изобретения указанный материал оксида переходного металла, содержащий литий, предоставленный на стадии (А), содержит в интервале от 0.2% до 2 мас.% фосфора. Фосфор может встречаться в одном или нескольких неорганических соединениях.
Каждое из приведенных выше процентных значений относится к массе сухого материала (т.е. материала оксида переходного металла, содержащего литий, как предоставлено на стадии (а)) настоящего изобретения.
Указанный материал оксида переходного металла, содержащий литий, содержит никель и кобальт. Примеры материалов оксида переходного металла, содержащих литий, могут быть основаны на литированном оксиде никеля-кобальта-марганца («NCM») или на литерованном оксиде никеля-кобальта-алюминия («NCA») или их смесях.
Предварительная обработка отходов аккумуляторных батарей и/или стадии (а)-(с) могут быть проведены в соответствии с JP 2012229481. Таким образом, отработавшие аккумуляторные батареи можно обрабатывать в одну или несколько стадий для связывания фтора, содержащегося в проводящей электролитной соли, обычно LiPF6 и связующего полимера, обычно поливинилиденфторида (PVDF). Это может быть достигнуто путем обработки отработавших батарей водным раствором гидроксида кальция (гашеной извести) для гидролиза проводящей соли и осаждения фторида в виде фторида кальция. В JP 2012-229481 описан способ извлечения металлов из отработавших ионно-литиевых батарей, включающий стадию предварительного погружения с последующим высокотемпературным окислением, восстановительный обжиг, водную обработку с фильтрацией, последующую конверсию гидроксида лития путем карбонизации и извлечение лития в виде карбоната. Способ согласно настоящему изобретению не требует такой карбонизации.
Материал, предоставленный на стадии (а), как правило, измельчают перед стадией (b) для деагломерации различных твердых частиц друг от друга в случаях, когда они каким-то образом агломерированы, например, остаточными связующими полимерами. Измельчение можно выполнять в сухом или влажном состоянии. Предпочтительно измельчение проводят в водной среде, которую также используют на последующей стадии (b).
Материал в виде частиц, предоставленный на стадии (а), как получено из отходов ионно-литиевых батарей, в особенности материалы их элементов, преимущественно получают в форме сухого порошка, влажного порошка или суспензии частиц в жидкости; содержащиеся частицы, как правило, имеют средний диаметр частиц D50 в диапазоне от 1 мкм до 2 мм при обнаружении в соответствии с ISO 13320 EN:2009-10.
Полярный растворитель, используемый на стадии (b) и, необязательно, для промывки твердого остатка на стадии (с), как правило, выбирают из воды, спиртов, кетонов, сложных эфиров, органических карбонатов, простых полиэфиров, нитрилов и их смесей, способных растворять гидроксид кальция так же хорошо, как вода или даже лучше. Примерами таких растворителей являются полиолы, подобные гликолю, глицерину или полиэтиленгликолю, и их смеси. Полярный растворитель может представлять собой протонный растворитель, такой как вода, спирты и их смеси. Водная среда в основном представляет собой воду или смеси воды с одним или несколькими спиртами.
Полярный растворитель предпочтительно представляет собой воду или водный раствор; водный раствор обычно содержит катионы металлов, упомянутых выше, а также другие металлы, как правило, содержащиеся в материалах отходов аккумуляторных батарей или литиевых минералах, таких как Al, Zn, щелочь и т.д., в виде гидроксидов и/или солей.
Стадию (b) предпочтительно проводят в присутствии гидроксида щелочноземельного металла. Гидроксид щелочноземельного металла обычно выбирают из гидроксидов Mg, Са, Sr и Ва; предпочтительными являются гидроксид кальция, гидроксид бария и их смеси; наиболее предпочтительным является гидроксид кальция. Гидроксид щелочноземельного металла, используемый на стадии (b) настоящего изобретения, может быть использован как таковой или может быть добавлен в виде оксида или смеси оксида и гидроксида для образования гидроксида щелочноземельного металла при контакте с полярным растворителем, выбранным из протонных растворителей, как отмечено выше. Обработку, как правило, проводят путем объединения некоторого количества гидроксида щелочноземельного металла (АЕН) с материалом в виде частиц (РМ), что соответствует по меньшей мере 5% и, как правило, не более 100% его массы, например 50-1000 г АЕН на 1 кг РМ, предпочтительно 100-1000 г АЕН, более предпочтительно 200-1000 г АЕН на 1 кг РМ. Количество полярного растворителя, как правило, выбирают таким образом, чтобы обеспечить смешиваемость компонентов, например, использование полярного растворителя в количестве, превышающем совокупное массовое количество РМ и ЕН в 2-20 раз, предпочтительно в около 3-10 раз.
Стадия (b) в первую очередь обеспечивает суспензию материала в виде частиц в полярном растворителе; ее предпочтительно проводят с нагревом, как правило, при температурах от 60 до 200°С, предпочтительно от 70 до 150°С. Если температура кипения полярного растворителя превышена, обработку проводят под давлением, чтобы растворитель или по меньшей мере его часть оставались в жидком состоянии. Особое техническое значение имеет температурный диапазон, близкий к точке кипения воды, т.е. около от 70 до 150°С, при котором обработка может быть достигнута с использованием водной жидкости или воды при нормальном или слегка повышенном давлении (например, до 5 бар). Альтернативно, стадию (b) согласно настоящему изобретению проводят при более высоких температурах и давлениях, например, от 150 до 300°С и от 1,5 до 100 бар.
В одном варианте осуществления настоящего изобретения стадию (b) проводят в сосуде, защищенном от воздействия сильных оснований, например, из стальных сплавов с высоким содержанием молибдена и меди, сплавов на основе никеля, дуплексной нержавеющей стали или стали, футерованной стеклом или покрытой титаном. Другими примерами являются полимерные вкладыши и полимерные сосуды из устойчивых к действию оснований полимеров, например, полиэтилена, такого как HDPE и UHMPE, фторированного полиэтилена, перфторалкоксиалканов («PFA»), политетрафторэтилена («PTFE»), PVdF и FEP. FEP означает фторированный этиленпропиленовый полимер, сополимер тетрафторэтилена и гексафторпропилена.
Обработку, как правило, проводят с использованием смесительного устройства, например, мешалки, с приложением мощности, как правило, до 10 Вт на кг суспензии, например, от 0,5 до 10 Вт/кг, или циклическим перекачиванием для достижения хорошего перемешивания и избегания осаждения нерастворимых компонентов. Сдвиг можно дополнительно улучшить, используя перегородки. Кроме того, суспензия, полученная на стадии (b), может быть предпочтительно подвергнута измельчению, например, в шаровой мельнице или шаровой мельнице с мешалкой; такая обработка измельчением может привести к лучшему доступу полярного растворителя к материалу в виде частиц оксида переходного металла, содержащему литий. Применяемые режущие и фрезерные устройства, как правило, являются достаточно коррозионностойкими, они могут быть изготовлены из материалов и покрытий, аналогичных описанным выше для сосуда.
В одном варианте осуществления настоящего изобретения стадия (b) имеет продолжительность в диапазоне от 20 минут до 24 часов, предпочтительно от 1 до 10 часов.
В одном варианте осуществления стадию (b) проводят по меньшей мере дважды для достижения оптимального извлечения гидроксида лития или соли лития. Между каждой обработкой проводится разделение твердой и жидкой фаз. Полученные растворы солей лития могут быть объединены или обработаны отдельно для извлечения твердых солей лития.
В одном варианте осуществления настоящего изобретения полярный растворитель на стадии (b) согласно настоящему изобретению представляет собой водную среду, и соотношение водной среды и материала, предоставленного на стадии (а), находится в диапазоне от 1:1 до 99:1, предпочтительно от 5:1 до 20:1 по массе.
В конце стадии (b) давление может быть сброшено, если применимо. Получают водный раствор, содержащий LiOH.
В важном варианте осуществления изобретения перед стадиями (А) и (В) согласно настоящему изобретению стадии (а)-(с) проводят следующим образом:
(а) предоставление материала в виде частиц, содержащего соединение переходного металла и/или переходный металл, где переходный металл выбран из группы, состоящей из Mn, Ni и Со, и где дополнительно по меньшей мере часть указанного Ni и/или Со, если они присутствуют, находятся в степени окисления ниже +2, и по меньшей мере часть указанного Mn, если он присутствует, представляет собой оксид марганца (II), где материал в виде частиц дополнительно содержит соль лития и фторидную соль, и где материал в виде частиц необязательно содержит кальций при условии, что соотношение элементов кальция и фтора составляет 1,7 или менее или равно нулю,
(b) обработка материала, предоставленного на стадии (а), полярным растворителем и гидроксидом щелочноземельного металла, и
(c) отделение твердых веществ от жидкости, необязательно с последующей промывкой твердого остатка полярным растворителем, таким как вода.
Неочищенный твердый гидроксид лития, полученный на стадии (А) согласно настоящему изобретению, может быть получен после удаления полярного растворителя или частей этого растворителя из жидкости, полученной на стадии (с), или после дополнительной обработки, как описано ниже (необязательные стадии d, е, e1, е2, е3).
В одном варианте осуществления настоящего изобретения твердый материал, полученный на стадии (с), подвергают дополнительной стадии (d) разделения твердой фазы и твердой фазы. Материал целесообразно измельчать перед стадией (d), чтобы отделить друг от друга различные твердые частицы в случаях, когда они каким-то образом агломерированы, например, остаточными связующими полимерами. Такое измельчение предпочтительно проводят в шаровых мельницах или шаровых мельницах с мешалкой.
В одном варианте осуществления настоящего изобретения стадия (d) представляет собой влажное разделение твердых фаз с использованием водной среды, предпочтительно воды, в качестве жидкости. Соотношение жидкой среды к твердому материалу, полученному на стадии (b), находится в диапазоне от 1:1 до 99:1, предпочтительно от 2:1 до 9:1 по массе.
В результате влажного разделения твердых фаз на стадии (d) получают две суспензии, одна из которых содержит твердый материал, содержащий целевой переходный металл, а другая содержит другие компоненты, такие как углеродсодержащие материалы и полимеры, и, если применимо, также некоторые неорганические соединения. Посредством подходящего выбора и, при необходимости, сочетания стадий разделения твердых фаз по меньшей мере 60% никеля и, если присутствует, Со получают и концентрируют в одной фракции. Предпочтительно отделяют по меньшей мере от 80 до 99% никеля и, если он присутствует, Со.
В одном варианте осуществления настоящего изобретения жидкая фаза суспензии, подаваемой на стадию (d), содержит растворенный литий. В этом случае одну или другую или обе суспензии, полученные в результате разделения твердой и твердой фаз на стадии (d), подвергают разделению твердой и жидкой фазы для извлечения раствора лития. Затем литиевый раствор дополнительно обрабатывают на стадии (е).
На стадии (е) раствор, полученный на любой из предыдущих стадий, который содержит литий, обрабатывают для извлечения лития в виде гидроксида или солей в виде твердых материалов.
В одном варианте осуществления настоящего изобретения соли Li, особенно LiOH, выделяют путем выпаривания воды, содержащейся в растворе (e1). Для получения желаемой литиевой соли высокой чистоты это выпаривание можно проводить в две последовательные стадии (е2 и е3) с разделением твердой и жидкой фаз между стадиями е2 и е3 для удаления нерастворенных твердых веществ. На стадии е3 раствор либо сушат, либо путем кристаллизации или кристаллизации с погружением получают гидроксид лития; последнее путем добавления высшего спирта или другого компонента с низкой полярностью, но достаточной смешиваемостью с водой, например, простые эфиры и т.д. В предпочтительном варианте стадии (е), (e1), (е2) и (е3) проводят в инертной атмосфере, например, в атмосфере азота или аргона.
Две последовательные стадии могут содержать:
Во-первых, раствор, содержащий Li, полученный на любой из предыдущих стадий, концентрируют до уровня чуть ниже предела растворимости LiOH, как правило, концентрация Li будет по меньшей мере на 5% меньше, чем соответствующая концентрация насыщения LiOH. Эта стадия сопровождается образованием твердого вещества (например, кристаллизацией) примесей, растворимость которых ниже, чем у LiOH; потенциальными примесями могут быть Са(ОН)2, CaF2, СаСО3, LiF, но без ограничения. Их отделяют путем разделения твердой и жидкой фаз, например, фильтрацией или центрифугированием, или своего рода осаждением и декантацией. Поскольку количество твердых веществ, подлежащих отделению, является небольшим, предпочтительно применять глубинные фильтры. В случае, когда LiF осаждается во время этой первой стадии концентрирования (е2), предпочтительно повторно подавать твердый материал на стадию (b).
Во-вторых, фильтрат, полученный в результате разделения твердой и жидкой фаз после одной из стадий концентрирования, т.е. концентрированный раствор LiOH, используют на следующей стадии выпаривания. LiOH высокого качества можно получить выпариванием оставшейся воды и образованием твердого LiOH. В случае кристаллизации кристаллы отделяют от оставшегося маточного раствора снова путем разделения твердой и жидкой фаз и, при необходимости, промывают.
Для всех вышеупомянутых стадий отверждения предпочтительна последующая сушка твердых веществ для получения неочищенного твердого гидроксида лития для стадии (А) согласно настоящему изобретению. Сушка при температуре ниже 60°С или при более высоких температурах в условиях высокой влажности приводит к моногидрату LiOH; в противном случае получают по меньшей мере частично свободный от воды LiOH.
В случае, если фильтрат, полученный после стадии (с), сушат путем полного выпаривания полярного растворителя, например, воды, в соответствии с описанной стадией (el), можно получить неочищенное твердое вещество LiOH, пригодное для стадии (А) согласно настоящему изобретению, которое может иметь чистоту в диапазоне от 90 до 99 мас.%, как правило, чистоту около 98 мас.% в высушенном виде. Наряду с примесями на основе углерода (как правило, <0,5 мас.%) оно может содержать примесный спектр, характерный для вышеописанного способа.
Что касается полученного таким образом моногидрата LiOH, характерными примесями, как правило, являются кальций, фтор и натрий. Их типичные количества в этом моногидрате LiOH составляют:
Са: 100 частей на миллион - 1.29 мас.%
F: 0.1 - 1.29 мас.%
Na: 0.1-1.29 мас.%
Кроме того, в зависимости от состава исходного материала возможно присутствие значительных количеств цинка, алюминия, калия и хлора. В этих случаях их характерные количества в описанном выше моногидрате LiOH, получаемом после стадии (e1), часто находятся в следующих диапазонах:
Zn: 20 частей на миллион - 1.29 мас.%
Al: 50 частей на миллион - 1.29 мас.%
К: 0.1 - 1.29 мас.%
Cl: 0.1 - 1.29 мас.%
В зависимости от условий сушки вместо моногидрата получают безводный LiOH. При этом указанные выше характерные количества примесей, относящиеся к моногидрату, являются более концентрированными, соответственно, в 1,75 раза (соответствует молярной массе моногидрата, поделенной на молярную массу ангидрата) для 100% безводного LiOH.
Все применяемые стадии, включая стадии (b), (с), (d) и (е), предпочтительно проводят в инертной атмосфере, например, в азоте, аргоне или в воздухе, не содержащем СO2.
Типичный вариант осуществления можно описать следующим образом:
Остаток, полученный из отходов аккумуляторных батарей, содержащий литий, фторид и металлический кобальт и/или никель, предоставляют (стадия а) и перемешивают с водой, к которой добавлен гидроксид кальция в количестве, достаточном для связывания всего фторида, содержащегося в материале (стадия b). Суспензию фильтруют (стадия с) для отделения нерастворенных твердых веществ (таких как металлический кобальт и/или никель, фторид кальция, углерод).
Раствор для выщелачивания содержит большую часть лития, содержащегося в исходном материале, растворенного в виде LiOH (основная литиевая соль). Теперь этот раствор для выщелачивания концентрируют путем выпаривания воды до концентрации LiOH, достаточно низкой, чтобы LiOH не выпадал в осадок (стадия е2, т.е. менее 12 г LiOH/100 г воды), и любой оставшийся менее растворимый осаждающийся материал отделяют.
Следы катионов металлов в растворе, полученном, например, на стадии b, с и/или е2, могут быть удалены различными способами в соответствии с уровнем техники. Типичные способы
i) Осаждение и последующее отделение на стадии с или е2 нерастворимых сульфидов металлов при добавлении источника сульфидов, например, Na2S, NaHS, Li2S, LiHS или H2S, из которых последние два являются предпочтительными, или смесей по меньшей мере двух из указанных источников сульфида,
ii) кристаллизация в водном растворе после любой из стадий с или е2 либо в форме одностадийной кристаллизации, либо в форме фракционированной кристаллизации с двумя или более стадиями или непрерывно. Кристаллизация предпочтительна в тех случаях, когда присутствуют примеси с высокой растворимостью, например, Na или К, происходящие из NaOH, NaCl, КОН или KCl; помимо этих катионов даже содержание хлорида может быть значительно уменьшено путем кристаллизации в водном растворе,
iii) экстракция растворителем после любой из стадий с или е2.
Наконец, любой осадок отделяют путем разделения твердой и жидкой фаз, например, фильтрацией.
Жидкость, содержащую гидроксид лития, полученную на любой из предыдущих стадий с или е2 с любой из стадий очистки (i)-(iii) или без нее, окончательно превращают в неочищенный твердый гидроксид лития либо путем выпаривания жидкости до сухости, либо путем кристаллизации или кристаллизации с погружением.
Твердый остаток неочищенного гидроксида лития, предоставленного таким образом на стадии А, затем обрабатывают низшим спиртом, таким как метанол (стадия В), в количестве, достаточном для растворения любого LiOH, содержащегося в твердом остатке; в типичном способе согласно настоящему изобретению концентрация LiOH в полученном таким образом метанольном растворе составляет 9 г LiOH в 100 г раствора или менее.
Этот раствор фильтруют от любого нерастворимого остатка, например, остаточных солей на основе фторида, алюмината или цинката (стадия С).
Метанольный фильтрат LiOH затем концентрируют для отверждения LiOH. Полученный LiOH можно высушить для полного испарения метанола. Его можно перекристаллизовать из воды с образованием гидратированного гидроксида лития, такого как LiOH*H2O, или кристаллизацию проводят непосредственно в смеси метанол/вода. В одном варианте осуществления твердый гидроксид лития получают кристаллизацией с погружением из метанольного раствора путем добавления высших спиртов, простых эфиров и/или углеводородов. Полученный гидроксид лития имеет высокую чистоту, особенно в отношении примесей ионов фтора, а также в отношении солей амфотерных элементов, таких как Al, Zn.
Например, остаток, полученный из отходов аккумуляторных батарей, содержащих фторид, как предоставлено на стадии а, суспендируют в воде и перемешивают с гидроксидом кальция (или оксидом кальция, который образует гидроксид кальция в воде) при содержании твердого вещества от 5 до 30 мас.% и молярном соотношении Ca:Li в диапазоне около от 1:3 до 1:1 в течение периода времени, как правило, в диапазоне от 1 часа до 8 часов, например, при кипячении с обратным холодильником (стадия b, как правило, при нормальном давлении). После охлаждения до комнатной температуры суспензию фильтруют (стадия с). Выщелачивающий раствор концентрируют путем выпаривания воды до концентрации LiOH немного ниже 12 г LiOH/100 г воды (стадия е2). Необязательно добавляют Li2S и/или H2S. Осадки окончательно отделяют фильтрованием. Оставшуюся жидкость выпаривают досуха (стадия е3). Полученный таким образом остаток (стадия А) обрабатывают метанолом (стадия В) с получением концентрации LiOH в метаноле до 9 мас.%. Этот раствор фильтруют от любого нерастворимого остатка (стадия С). Метанольный фильтрат LiOH концентрируют для кристаллизации LiOH высокой чистоты.
В другом варианте осуществления суспензия, полученная при выщелачивании в воде на стадии b в предыдущем примере, может быть обработана сульфидами; затем ее можно выпарить досуха (стадия е3) и впоследствии повторно растворить в метаноле (стадия В), избегая при этом нескольких стадий фильтрации. Метанольную суспензию затем фильтруют (стадия С) и из фильтрата выделяют чистый LiOH с низким содержанием фтора.
В контексте настоящего изобретения содержание примесей часто относится к ангидрату гидроксида лития или моногидрату гидроксида лития. Примесь 10 частей на миллион в моногидрате гидроксида лития соответствует примеси 17,5 частей на миллион в ангидрате гидроксида лития. Такой же уровень примесей может быть связан с содержанием лития, что помогает в случае смеси ангидрата/моногидрата; 10 частей на миллион примеси в моногидрате гидроксида лития, а также 17,5 частей на миллион примеси в ангидрате гидроксида лития соответствуют 60,4 частей на миллион примеси относительно содержания Li.
Описание способов:
Измерения гранулометрического состава, включая определение D50, выполняются в соответствии со стандартом ISO 13320 EN:2009-10.
Элементный анализ лития, кальция и марганца (выполненный среди прочего для определения содержания Li, Са, Mn в материале в виде частиц, предоставленном на стадии (а) согласно настоящему изобретению):
Реагенты: деионизированная вода, соляная кислота (36%), смесь K2CO3-Na2CO3 (сухая), Na2B4O7 (сухая), соляная кислота 50 об.% (смесь деионизированной воды и соляной кислоты (36%) 1:1); все реагенты имеют марку р.а.
Элементный анализ фтора и фторида проводят в соответствии со стандартизированными методами: DIN EN 14582:2016-12 в части подготовки образца для определения общего содержания фтора (образцы отходов); способ обнаружения представляет собой измерение с помощью ионоселективного электрода. DIN 38405-D4-2:1985-07 (образцы воды; расщепление неорганических твердых веществ с последующей дистилляцией с кислотой и определением фторида с использованием ионселективного электрода).
Определение содержания углерода и спирта
Углерод: помимо органического углерода образцы могут содержать диоксид углерода или ионы угольной кислоты. В зависимости от представляющего интерес соединения применяются следующие способы.
Общий углерод (ТС): Типичный анализ общего углерода измеряет как присутствующий общий органический углерод (ТОС), так и дополнительный общий неорганический углерод (TIC), последний представляет собой количество неорганического углерода, такого как углерод в карбонатных минералах. Это проводят путем сжигания в атмосфере, содержащей кислород. Описание этого способа является частью стандарта DIN EN 15936:2012-11 (способ А).
ТОС можно определить косвенно (а) и напрямую (b):
a) Вычитание неорганического углерода из общего углерода (ТС) дает ТОС и описано в DIN EN 15936:2012-11 (Способ А).
b) Другой распространенный вариант анализа ТОС включает сначала удаление части TIC, а затем измерение остаточного углерода. Этот способ включает очистку подкисленного образца азотом или воздухом, не содержащим углерод, перед измерением, и поэтому его более точно называют не очищаемым органическим углеродом (NPOC), поскольку летучие органические вещества также удаляются. NPOC определяют путем продувки подкисленного образца не содержащим углерода воздухом или азотом перед измерением с последующим сжиганием в кислородсодержащей атмосфере (DIN EN 15936:2012-11, В).
Низшие спирты, такие как метанол или этанол, идентифицируют и определяют количественно методом GC-MS (газовая хроматография + масс-спектрометрия) в свободном пространстве (в соответствии с ISO 17943:2016) после отделения низшего спирта от матрицы LiOH в подходящем растворителе, таком как вода или водный раствор соляной кислоты.
Другие металлические примеси и фосфор определяют аналогично элементным анализом с использованием ICP-OES (индуктивно-связанная плазма - оптическая эмиссионная спектроскопия) или ICP-MS (индуктивно-связанная плазма масс-спектрометрия). Общий углерод определяется с помощью детектора теплопроводности после сжигания.
Фазовые составы твердых веществ [включая идентификацию оксида марганца (II), а также никеля и кобальта в степени окисления ниже +2 (как правило, металлические) в материале в виде частиц, предоставляемом на стадии (а) согласно настоящему изобретению], определяют с помощью порошковой рентгеновской дифракции (PXRD). Способ проводят следующим образом:
Образец измельчают до тонкоизмельченного порошка и засыпают в держатель образца.
Используются два устройства, каждое из которых использует свой конкретный источник излучения:
(1) Измерение с использованием Cu-излучения: используется прибор Bruker D8 Advance Series 2 с автодозатором; первичная сторона: Cu-анод, апертура угла раскрытия луча 0,1° с ASS; вторичная сторона: апертура рассеянного луча 8 мм с никелем 0,5 мм, Soller 4°, Lynx-Eye (апертура).
(2) Измерение с использованием излучения Мо: Используемый прибор представляет собой Bruker D8 Discover А25 с автоматическим блоком отбора проб; первичная сторона: Mo-анод с монохроматором Йоханссона (Mo-K-alphal) с осевым цоколем 2,5°; вторичная сторона: ASS, Soller 2,5°, детектор Lynx-Eye ХЕ (апертура 3,77°).
Эталоны используются для идентификации совпадений с полученным образцом отражения. Все соответствующие фазы хорошо известны в литературе; для расчета теоретической картины дифракции используются следующие эталоны (см. положение и интенсивность отражений в Табл. 1 ниже):
CoxNi1-x, пространственная группа Fm-3m, х=0.5: Taylor et al., J. Inst. Met. (1950) 77, 585-594. x=0: Buschow et al., J. Magn. Magn. Mater. 1983, 38, 1-22.
a) Co, пространственная группа Р63/mmc, Buschow et al., J. Magn. Magn. Mater. 1983, 38, 1-22.
b) Li2CO3, пространственная группа C2/c, J. Alloys Compd. (2011), 509, 7915-7921
c) LiAlO2, пространственная группа R-3m, Marezio et al., J. Chem. Phys. (1966) 44, 3143-3145.
d) MnO, пространственная группа Fm-3m, Locmelis et al., Z. Anorg. Allg. Chem. 1999, 625, 1573.
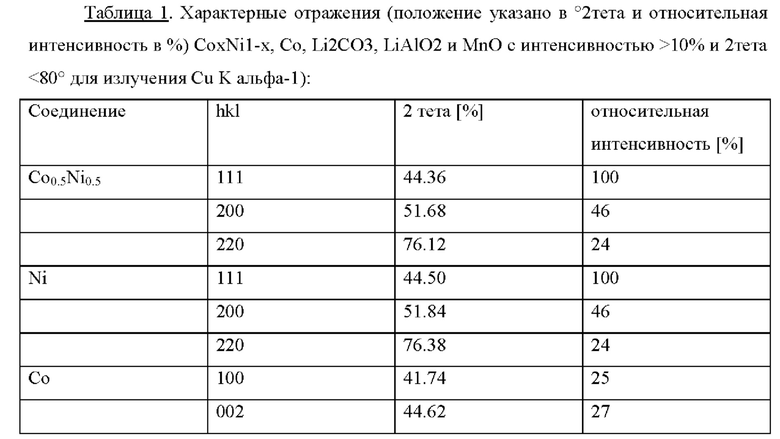
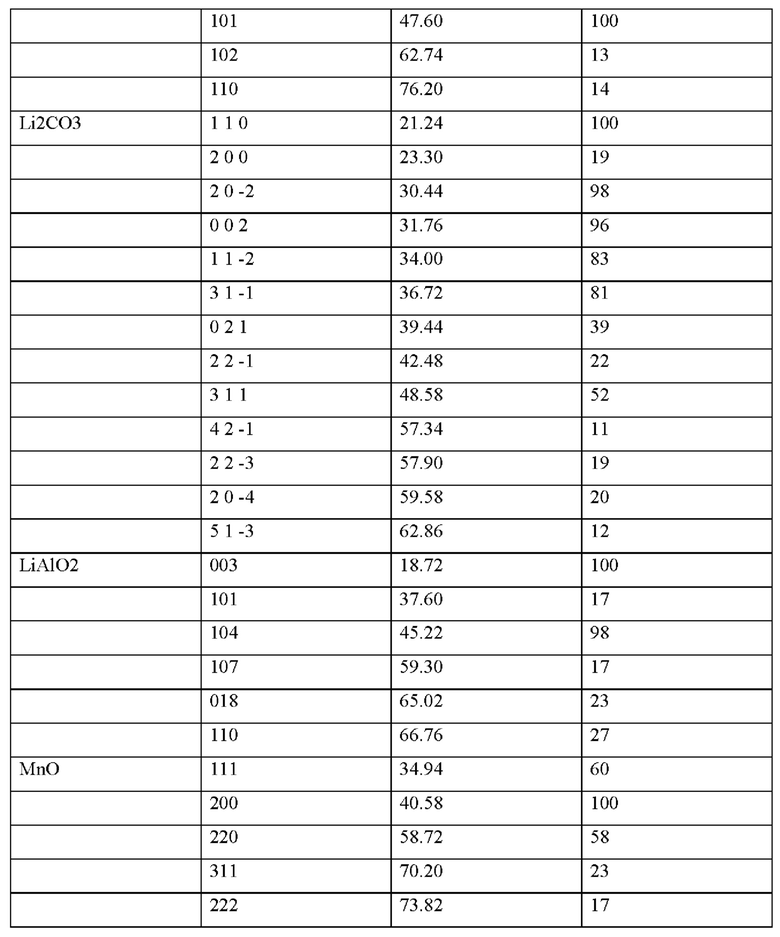
В случае перекрытия характеристических отражений с отражениями различных кристаллических фаз (особенно графита, составляющего наибольшую часть образца), выполняется дополнительное измерение с использованием альтернативного источника излучения (например, Мо К-альфа вместо Cu К-альфа).
Аббревиатуры:
Стадии А и В обозначают обязательные стадии способа согласно настоящему изобретению, другие описанные стадии являются необязательными и могут быть полезными в описанном контексте, например, стадии, обозначенные заглавными буквами (в основном ниже по ходу потока), и стадии, обозначенные строчными буквами (в основном выше по ходу потока).
В контексте настоящего изобретения нормальное давление означает 1 атм или 1013 мбар. «Нормальные условия» означают нормальное давление и 20°С. N1 нормальный литр, литр при нормальных условиях (1 атм, 20°С). PFA означает перфторалкоксиполимер; МеОН означает метанол; EtOH обозначает этанол.
Проценты или части на миллион относятся к % или частям на миллион по массе, «в вакууме» обозначает пониженное давление в диапазоне от 20 до 50 мбар, каждое, если специально не указано иное. Выражения мас.% (% по массе) и мас.% могут использоваться взаимозаменяемо. «Частей на миллион» обозначает массовые части на миллион; конц. обозначает концентрацию. Везде, где упоминается, термины «комнатная температура» и «температура окружающей среды» обозначают температуру от 18 до 25°С. XRD означает порошковое рентгеновское исследование (излучение, как указано, как правило, Cu k-alphal излучение 154 pm или Mo k-alphal 71 pm).
Настоящее изобретение далее иллюстрируется следующими примерами; Содержание LiOH рассчитывали на основании аналитически определенного содержания Li; содержание воды по массе может быть аппроксимировано вычитанием расчетного содержания LiOH из 100%.
Пример 1. Образец синтетического эдукта
Количество 200 г имитации лома отработавших батарей, содержащее
78,8 г отработанного катодного активного материала, содержащего никель, кобальт и марганец в аналогичных молярных количествах, приблизительная формула Li(Ni0.34Co0.33Mn0.33)O2,
62,2 органического углерода в виде графита и сажи,
47,0 г смеси органических электролитов (содержащей LiPF6),
7,4 г поливинилиденфторида в качестве связующего,
2,4 г алюминиевого порошка,
0,2 г порошка железа,
2.0 г металлической меди
помещают в кварцевую круглодонную колбу на 500 мл и присоединяют к роторному испарителю таким образом, чтобы колба погружалась в печь. В течение 4,5 ч вращающуюся колбу нагревают до 800°С в течение 2 ч в потоке аргона (20 л/ч) и выдерживают при этой температуре 1 ч в потоке сухого воздуха (20 л/ч) перед охлаждение до температуры окружающей среды. Получают 173,3 г термообработанного материала, включающего фазовый состав сплава Ni/Co, оксида железа и марганца, Li2CO3, LiF и графита.
Пример 1а. Обеспечение восстановленной массы отходов ионно-литиевых батарей Около 1 т механически обработанного аккумуляторного лома, содержащего отработанный катодный активный материал, содержащий никель, кобальт и марганец, органический углерод в виде графита и сажи и остаточный электролит, а также другие примеси, среди прочего, включающие соединения фтора, фосфор и кальций, обрабатывают с получением восстановленной массы в соответствии с процессом, описанным в Jia Li et al., Journal of Hazardous Materials 302 (2016) 97-104, за исключением того, что атмосфера внутри системы обжига представляет собой воздух, кислород которого реагирует с углеродом в аккумуляторном ломе с образованием монооксид углерода, температура обработки 800°С.
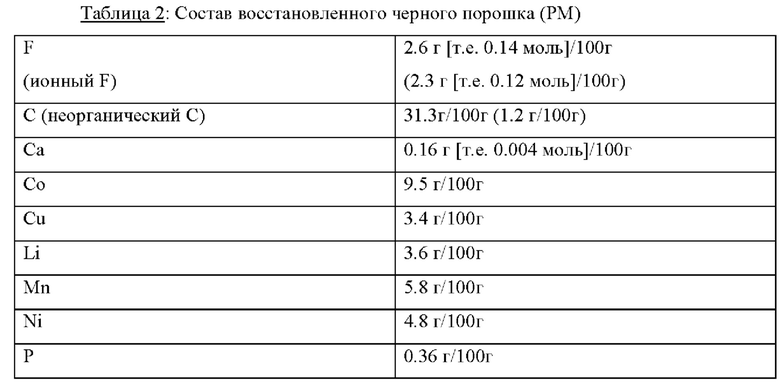
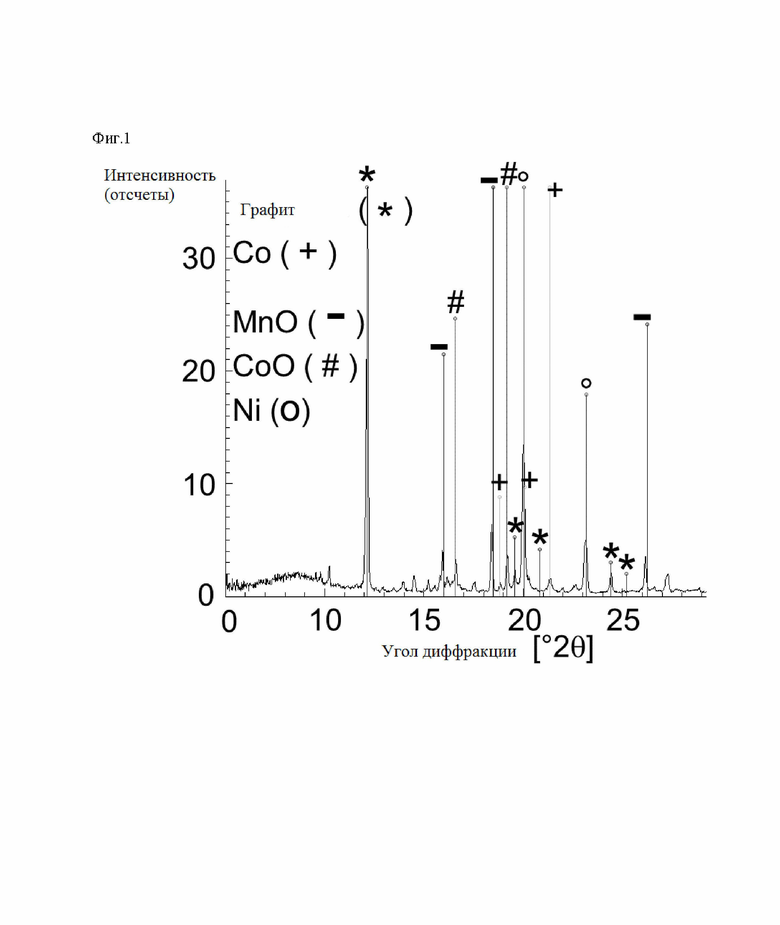
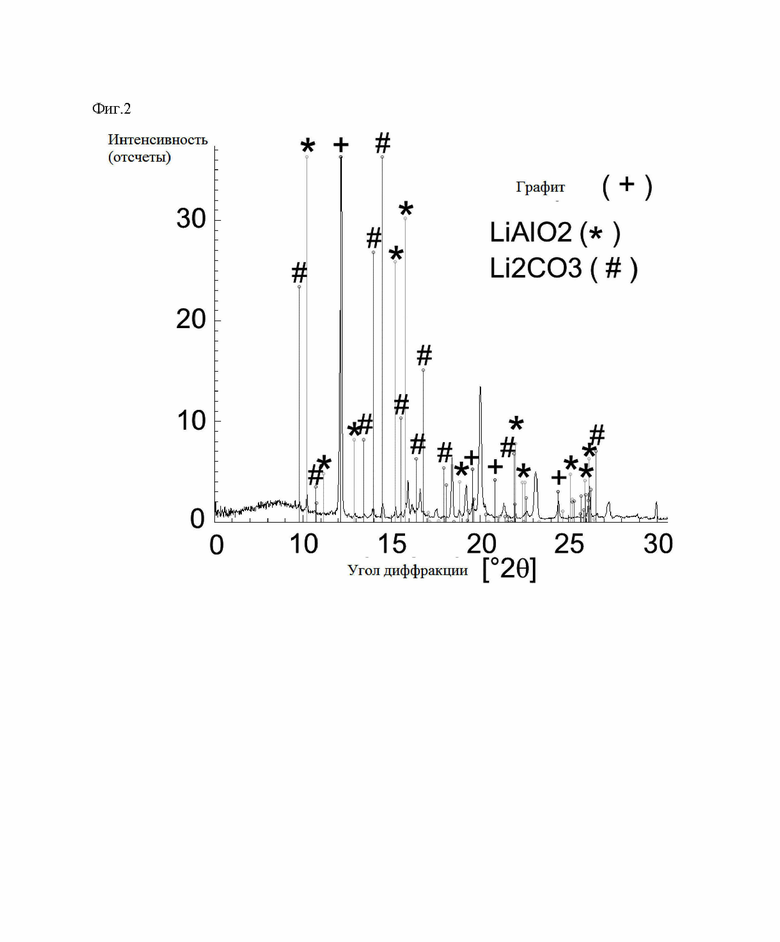
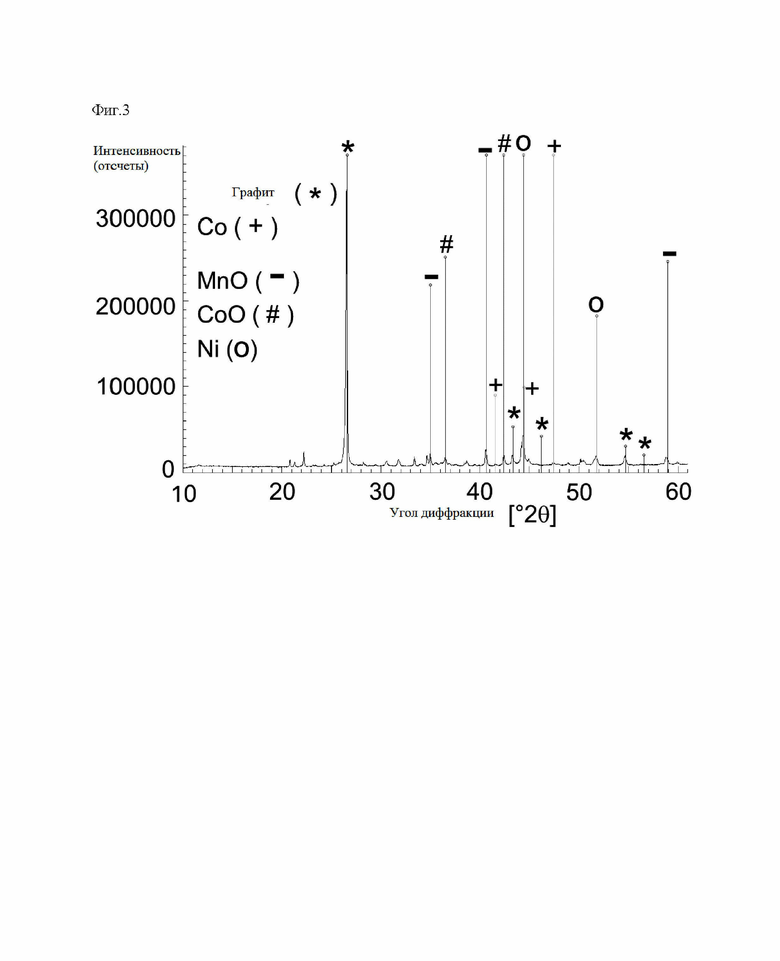
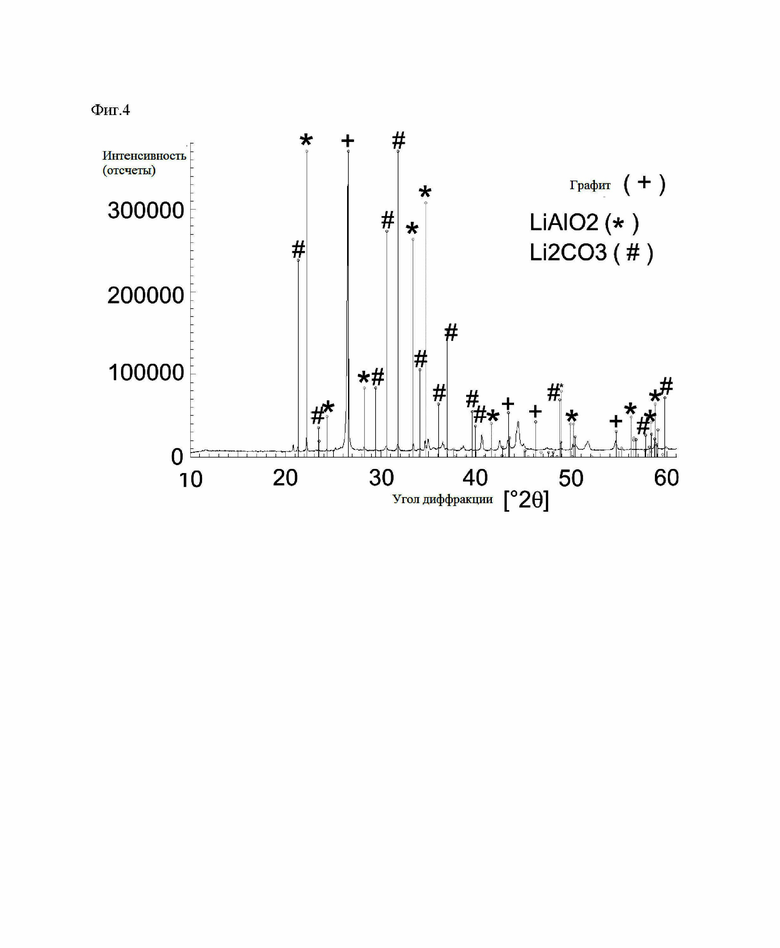
После реакции и охлаждения до температуры окружающей среды термообработанный материал извлекают из печи, подвергают механической обработке для получения материала в виде частиц и анализируют с помощью порошковой рентгеновской дифракции (фиг. 1 и 2: излучение Мо-Ка, фиг. 3 и 4: излучение CuKa), элементный анализ (табл.2) и гранулометрический состав (табл.3).
Содержание Li составляет 3,6 мас.%, что служит эталоном для всех дальнейших примеров выщелачивания (см. ниже). Фтор в основном представлен в виде неорганического фторида (88%). Размеры частиц значительно меньше 1 мм; Определено, что D50 составляет 17,36 мкм.
Сравнивая полученную рентгенограмму XRD с рассчитанными эталонными картинами Ni (которая идентична таковой для CoxNi1-x, х=0-0,6), Со, Li2CO3 и LiAlO2 (см. эталонные картины в табл.1), можно сделать вывод, что Ni присутствует исключительно в виде металлической фазы, либо в виде чистого Ni, либо в виде сплава в сочетании с Со. Для ясности этот результат подтверждается применением двух разных источников излучения. Присутствие металлического никеля подтверждается качественным наблюдением, что весь образец проявляет типичное ферромагнитное поведение при контакте с материалом с постоянными магнитами. Соли лития, Li2CO3, а также LiAlO2 четко идентифицируются по их характерной дифракционной картине.
Состав полученного черного порошка (РМ) представлен в табл. 2.

Пример 2. Неочищенный гидроксид лития в воде может быть получен из материала, представленного в примере 1 или примере 1а, в соответствии со способом, предложенным в KR101998691.
Пример 2а. Выщелачивание с помощью Са(ОН)2
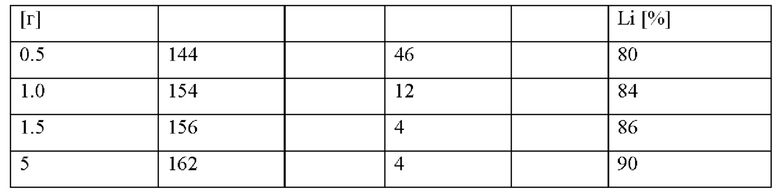
Количество 5 г вышеупомянутого восстановленного материала аккумуляторных батарей (полученного, как показано в примере 1а) помещают в колбу из PFA и смешивают с 5, 1,5, 1,0 и 0,5 г твердого Са(ОН)2, соответственно. При перемешивании добавляют 200 г воды и всю смесь кипятят с обратным холодильником в течение 4 часов.
Через 4 часа твердое содержимое отфильтровывают, отбирают образцы фильтрата и анализируют на содержание Li, F, карбоната, ОН и Са. Результаты собраны в табл. 4 ниже.

Пример 3. Твердый LiOH из выщелоченного фильтрата лития и последующая перекристаллизация в метаноле
Концентрированный водный раствор: Фильтрат, полученный в результате процесса в соответствии с примером 2а, дополнительно обрабатывают в соответствии с описанными выше стадиями (е2) и (е3) с получением твердого LiOH в виде моногидрата: 1 л фильтрата, содержащего 0,21 мас.% лития, концентрируют выпариванием (40°С, 42 мбар) в 11 раз, что сопровождается осаждением тонкораспределенных белых кристаллов. Этот осадок отфильтровывают.
Получение неочищенного твердого LiOH: Оставшийся раствор сушат при 40°С и постоянном потоке азота в течение 24 часов. РСА показывает полученный моногидрат LiOH с незначительными примесями Li2CO3. Последнее связано с контактом с воздухом практически на всех стадиях процесса. Показан элементный анализ, включая все проанализированные примеси, относящиеся к LiOH*H2O. Эта фракция обозначена как неочищенный LiOH*H2O.
Объединение с низшим спиртом:
3 г этого неочищенного LiOH*H2O растворяют в 20 г метанола при комнатной температуре. Оставшиеся нерастворимые твердые вещества отфильтровывают и к прозрачному раствору добавляют 4,5 г воды. Затем жидкую фазу медленно испаряют и получают белые кристаллы LiOH*H2O. Результаты анализа этого очищенного продукта показаны в (правая колонка), оба относятся к LiOH*H2O.
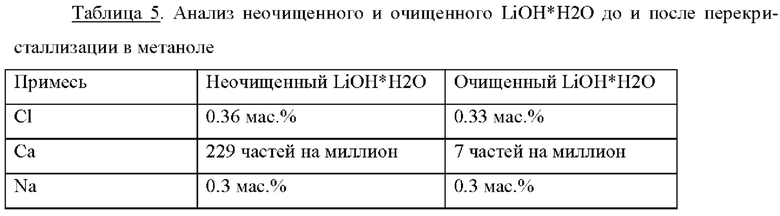

Результаты показывают, что примеси фтора, а также амфотерные элементы Al и Zn эффективно удаляются.
Пример 4. Твердый LiOH из выщелоченного фильтрата лития путем фракционной кристаллизации и последующей перекристаллизации в метаноле
Пример 3 повторяют, применяя на первой стадии фракционированную кристаллизацию: 1 л фильтрата, содержащего 0,21 мас.% лития, концентрируют выпариванием (40°С, 42 мбар) в 11 раз, что сопровождается осаждением тонкораспределенных белых кристаллов. Этот осадок отфильтровывают, а оставшийся раствор ступенчато сушат при 40°С и 42 мбар. После дальнейшего концентрирования в 2 раза первую твердую фракцию отфильтровывают. Оставшуюся смесь снова концентрируют в 2 раза и отделяют вторую фракцию. В Табл. 6 и Табл. 7 показан элементный анализ обеих фракций, включая все проанализированные примеси, относящиеся к LiOH*H2O. Эти фракции помечены как 1я или 2я фракция твердого LiOH*H2O соответственно (средние столбцы).
Затем обе фракции перекристаллизовывают в метаноле: каждый раз по 2 г твердого LiOH*H2O 1й или 2й фракции растворяют в 13 г метанола. Оставшееся нерастворимое твердое вещество отфильтровывают и к прозрачному фильтрату добавляют 3 г воды. Затем жидкую фазу медленно испаряют и получают белые кристаллы LiOH*H2O. Результаты анализа этого очищенного продукта показаны в Табл. 6 и Табл. 7 (правые столбцы).
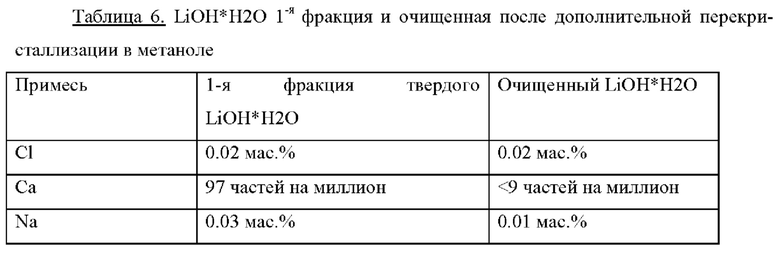

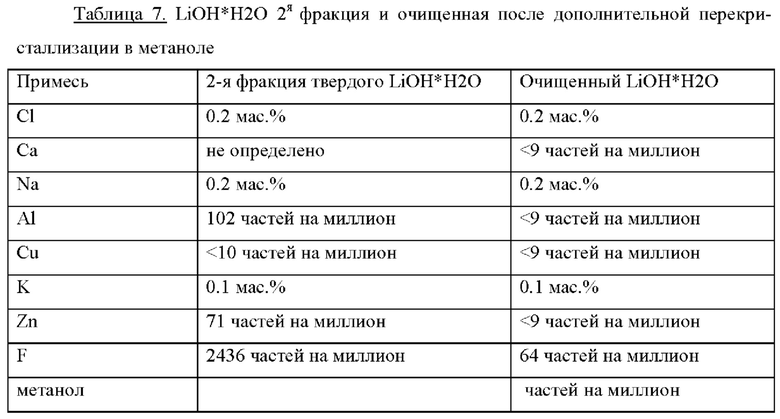
Комбинация фракционированной кристаллизации и перекристаллизации в метаноле согласно настоящему изобретению дает гидроксид лития высокой чистоты.
Пример 5. Перекристаллизация в МеОН, начиная с LiOH, в основном в виде моногидрата (LiOH*H2O)
Около 24 г неочищенного твердого гидроксида лития, полученного в соответствии с примером 3 (в основном включающего моногидрат, содержащий 18,2 мас.% Li, что соответствует 62,8 мас.% LiOH, около 37,2 мас.% воды; c(F)=0,32 мас.%) добавляют к метанолу (m(МеОН), как указано в Табл. 8 ниже). Раствор перемешивают в течение 16 ч при температуре окружающей среды. Получают мутный раствор, который затем фильтруют. Фильтрат сушат при 40°С в вакууме в течение 12 часов. Количество полученного продукта [m(продукт)/г], выход лития и оставшейся фторидной примеси приведены в таблице ниже.
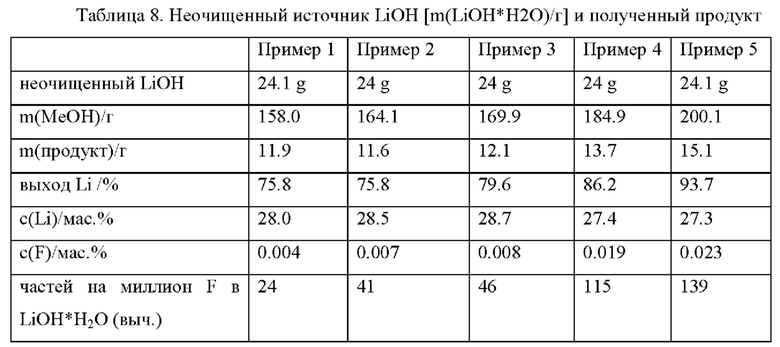
Применяемое соотношение MeOH/LiOH оказывает линейное влияние на конечное содержание фтора. Это связано с достаточно низкой, но существующей растворимостью LiF в МеОН. Поэтому нежелательно применять большие количества МеОН. С другой стороны, повышенное содержание МеОН благотворно влияет на общий выход лития.
Пример б. Перекристаллизация в МеОН, начиная с LiOH, в основном содержащего ангидрат
Последовательность стадий способа: 1) Растворение твердого LiOH в МеОН, 2) Фильтрация для удаления LiF, 3) Добавление воды, 4) Сушка 9,9 г неочищенного твердого гидроксида лития, полученного по аналогии с примером 3, но высушенного при 110°С (в основном содержащего безводный LiOH (c(Li)=23,9 мас.%; c(F)=0,3 мас.%; NPOC 1739 мас. частей на миллион, содержание воды около 17.6 мас.%) добавляли к 115 г метанола. Раствор перемешивают 2 ч при температуре окружающей среды. Получают мутный раствор, который затем фильтруют. Медленно добавляют 8,75 г воды. Затем фильтрат сушат при 70°С и 200 мбар в течение 45 мин, охлаждают до 45°С при 100 мбар и окончательно сушат в течение 3,5 ч при 40°С и 40 мбар. Получают 11,6 г продукта (выход лития=96,1%. Сравнение содержания NPOC и МеОН (3050 мас. частей на миллион) дает оценку остаточного углерода, который не относится к метанолу. Для снижения содержания МеОН продукт повторно растворяют в воде и снова сушат в течение 1 ч при 70°С. Анализ неочищенного твердого исходного материала и высушенных твердых продуктов приведен в Табл. 9. Предполагая постоянное содержание углерода, которое не присутствует в виде метанола после этой перекристаллизации в воде, уменьшение содержания NPOC вычисляют как потерю метанола. Эта оценка приводит к 913 частей на миллион метанола в продукте после повторного растворения и повторной сушки, что соответствует 521 частей на миллион метанола в идеальном LiOH*H2O.
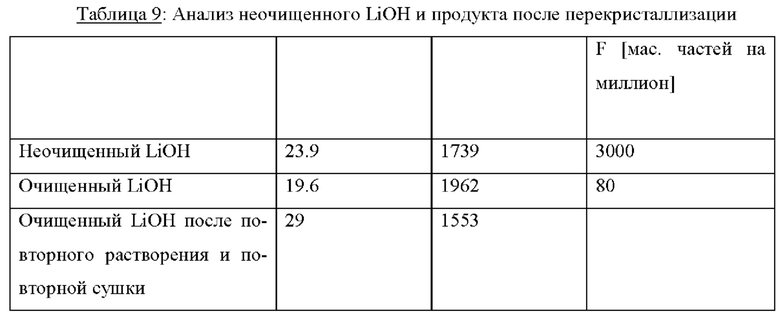
Этот пример представляет собой наиболее предпочтительный способ обработки, поскольку очень эффективное истощение F и хороший выход достигаются за счет поддержания содержания воды на как можно более низком уровне перед отделением нежелательной примеси LiF.
Пример 7. Перекристаллизация из МеОН, начиная с ангидрата LiOH, без добавления воды
22,8 г неочищенного твердого гидроксида лития, полученного по аналогии с примером 6 с высушиванием при 110°С (в основном включающего безводный LiOH (c(Li)=27,8 мас.%, что соответствует 95,9 мас.% LiOH и 4,1 мас.% воды); c(F)=0,37 мас.%) добавляют к 296 г метанола. Раствор перемешивают 2,5 ч при температуре окружающей среды. Получают мутный раствор, который затем фильтруют. Затем фильтрат сушат при 40°С в вакууме в течение 12 ч. Получают 25,5 г продукта (выход лития=90,0%). Анализ высушенного твердого продукта приведен в Табл. 10.
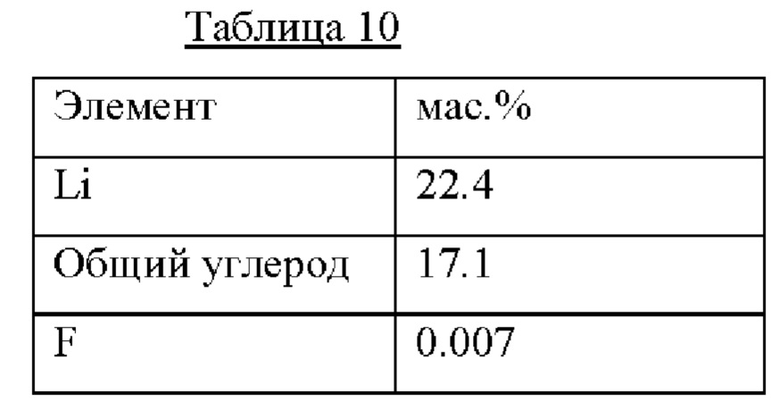
Этот пример менее предпочтителен, поскольку продукт содержит метоксид лития, который самовоспламеняется. Тем не менее, удаление F столь же эффективно, как и в примере 6, поскольку содержание воды, опять же, является настолько низким, насколько это возможно перед отделением нежелательной примеси LiF.
Пример 8. Перекристаллизация в МеОН, предварительное добавление воды
Последовательность стадий процесса: 1) Растворение твердого LiOH в МеОН, 2) Добавление воды, 3) Фильтрация для удаления LiF, 4) Сушка.
9,95 г неочищенного твердого гидроксида лития, полученного аналогично примеру 3 (c(Li)=23,9 мас.%, что соответствует 82,4 мас.% LiOH и 17,6 мас.% воды; c(F)=0,3 мас.%) добавляют к 116 г метанола. Раствор перемешивают в течение 2 ч при температуре окружающей среды. К мутному раствору медленно добавляют 8,55 г воды, затем фильтруют. Фильтрат сушат при 70°С и 200 мбар в течение 1 ч, охлаждают до 45°С при 100 мбар и окончательно сушат в течение 4 ч при 45°С и 45 мбар. Получают 10,8 г продукта, содержащего 21,3 мас.% Li и 0,012 мас.% F (выход лития=96,7%).
Этот пример менее предпочтителен, чем пример 6, поскольку воду добавляют перед отделением нежелательного LiF. Эта дополнительная вода способна растворять LiF. Следовательно, истощение F не так эффективно, как описано для предыдущих примеров.
Пример 9. Крупномасштабная перекристаллизация, начиная с LiOH, в основном содержащего ангидрат
6,4 кг концентрированного водного раствора гидроксида лития (6,2 мас.%), полученного, как описано в примере 3, сушат в смесительно-сушильной машине объемом 0,77 л. Реактор нагревают до 120-135°С, температура продукта 100-105°С при 950 мбар. Раствор добавляют пошагово по 100 мл. Реактор продувают азотом со скоростью 10 нл/ч в течение всего процесса. Температуру стенки повышают до 140-155°С через 15 ч и, наконец, доводят до 130-140°С в течение 1 ч. Затем продукт охлаждают до комнатной температуры. В ходе процесса реактор заполняется на 45-80% от его максимальной вместимости. Получают 191 г неочищенного твердого LiOH, в основном состоящего из ангидрата гидроксида лития (c(Li)=27,8 мас.%, что соответствует 95,9 мас.% LiOH и 4,1 мас.% воды, c(F)=0,33 мас.%).
60 г этого свободнотекучего кристаллического порошка растворяют в 800 г метанола и перемешивают в течение 16 часов. Смесь фильтруют и к фильтрату добавляют 100 мл метанола. Фильтрат сушат при 70°С и 200 мбар в течение 1 ч, охлаждают до 45°С при 100 мбар и окончательно сушат в течение 4 ч при 45°С и 45 мбар. Анализ высушенного твердого продукта приведен в Табл. 11.
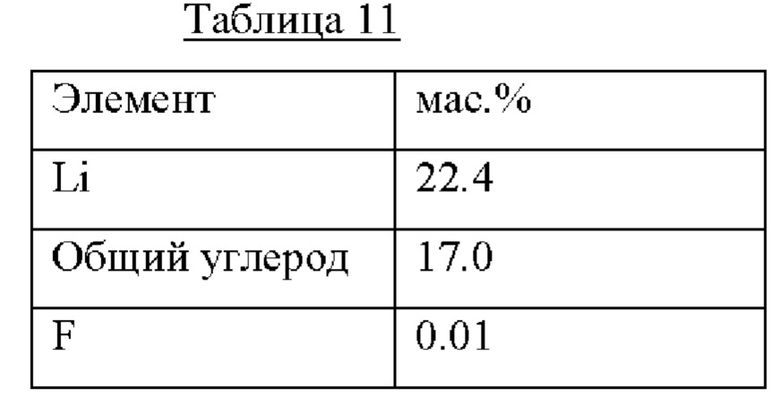
Сравнительный пример А. Кристаллизация с погружением с использованием МеОН
Кристаллизацию с погружением с метанолом проводят для удаления примесей из раствора. Твердый неочищенный LiOH*H2O, рециклированный из черной массы, как описано выше в примере 3, обрабатывают следующим образом: 3 г этого неочищенного LiOH*H2O растворяют в воде (20,48 г; c(Li)=15,21 мас.%), перемешивали в течение около 3 ч при температуре окружающей среды и по каплям добавляют метанол до тех пор, пока не станет видимым образование твердого вещества. В сумме добавляют 13,58 г метанола. Затем эту суспензию фильтруют и полностью сушат прозрачный фильтрат. Высушенный твердый остаток анализируют с помощью элементного анализа, показывающего содержание лития 25,7 мас.% и содержание фторида 0,20 мас.%, что соответствует стехиометрически идеальному LiOH*H2O, содержащему 1288 частей на миллион фторида.
По сравнению с вышеописанными примерами, этот сравнительный пример с применением кристаллизации с погружением явно подчеркивает менее эффективное удаление фторида, что не приводит к предпочтительному содержанию фторида, которое можно получить в соответствии с настоящим изобретением. Это соответствует приведенным выше примерам, показывающим, что повышенное содержание воды приводит к повышенному количеству LiF.
Сравнительный пример В: Кристаллизация с погружением с использованием этанола
Кристаллизацию с погружением проводят с использованием этанола в качестве антирастворителя для кристаллизации LiOH, как это предложено, например, ME Taboada et al., Chem.Eng.Res.and Design 85, 1325 (2007). Концентрированный, почти насыщенный раствор LiOH, как описано выше в примере 3 (источник), содержит:
Литий=2,9 мас.% (соответствует 10,0 мас.% LiOH)
Фторид=0,026 мас.% (соответствует 0,15 мас.% примеси фторида в LiOH*H2O).
i) В первом эксперименте 40 г этанола добавляют в 100 г этого раствора LiOH в виде 70% (мас.%) этанола/воды. После добавления 35 г EtOH наблюдают белые кристаллы LiOH.
ii) Во втором эксперименте 40 г жидкости, состоящей из 90 мас.% этанола и 10 мас.% воды 100 добавляют в 100 г этого раствора LiOH. После добавления 19 г EtOH наблюдают белые кристаллы LiOH.
Каждую из смесей (i) и (ii) перемешивают в течение 15 мин. После этого выпавшую в осадок соль отфильтровывают, сушат (3 ч при 40°С при 30 мбар) и анализируют содержание в ней фторида и лития. Рассчитано содержание фторида относительно чистого LiOH*H2O (чистый LiOH*H2O содержит 16,55 мас.% Li). Результаты суммированы в следующей табл.
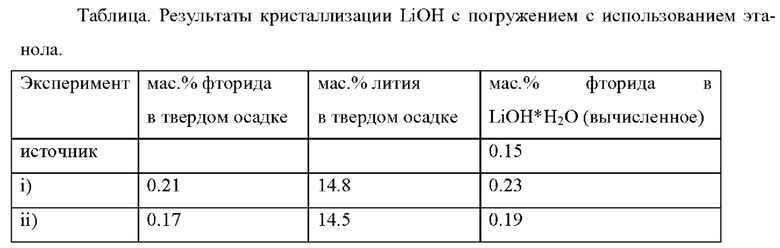
Кристаллизация с погружением с этанолом приводит скорее к небольшому обогащению фторидом осажденного LiOH, а не к его сокращению.
Краткое описание чертеже
Фиг. 1. Рентгеновская порошковая дифрактограмма (Мо Ка) восстановленной массы из отходов ионно-литиевых батарей после термической/восстановительной обработки, как получено в примере 1а и использовано в примере 2а, включая ссылку на дифракто-граммы графита, кобальта, марганца-II-оксида, оксида кобальта и никеля.
Фиг. 2. Рентгеновская порошковая дифрактограмма (Мо Ка) восстановленной массы из отходов ионно-литиевых батарей после термической/восстановительной обработки, как получено в примере 1а и использовано в примере 2а, включая ссылку на дифракто-граммы графита, алюмината лития и карбоната лития.
Фиг.3. Рентгеновская порошковая дифрактограмма (Cu Ка) восстановленной массы из отходов ионно-литиевых батарей после термической/восстановительной обработки, как получено в примере 1а и использовано в примере 2а, включая ссылку на дифракто-граммы графита, кобальта, марганца-II-оксида, оксида кобальта и никеля.
Фиг. 4. Рентгеновская порошковая дифрактограмма (Cu Ка) восстановленной массы из отходов ионно-литиевых батарей после термической/восстановительной обработки, как получено в примере 1а и использовано в примере 2а, включая ссылку на дифракто-граммы графита, алюмината лития и карбоната лития.
Проект, ведущий к этой заявке, получил финансирование от Bundesministerium für Wirtschaft und Energie (Германия; FKZ: 16BZF101A); заявитель несет ответственность за раскрытие информации в настоящем документе.
| название | год | авторы | номер документа |
|---|---|---|---|
| РЕЦИКЛИЗАЦИЯ БАТАРЕИ ПОСРЕДСТВОМ ОБРАБОТКИ ВЫЩЕЛАЧИВАЮЩИМ АГЕНТОМ С МЕТАЛЛИЧЕСКИМ НИКЕЛЕМ | 2018 |
|
RU2794298C2 |
| ФОСФАТ ЛИТИЯ-ЖЕЛЕЗА СО СТРУКТУРОЙ ОЛИВИНА И СПОСОБ ЕГО АНАЛИЗА | 2009 |
|
RU2484009C2 |
| КАТОД НА ОСНОВЕ ДВУХ ВИДОВ СОЕДИНЕНИЙ И ВКЛЮЧАЮЩАЯ ЕГО ЛИТИЕВАЯ ВТОРИЧНАЯ БАТАРЕЯ | 2010 |
|
RU2501125C1 |
| КАТОД НА ОСНОВЕ ДВУХ ВИДОВ СОЕДИНЕНИЙ И ВКЛЮЧАЮЩАЯ ЕГО ЛИТИЕВАЯ ВТОРИЧНАЯ БАТАРЕЯ | 2010 |
|
RU2501124C1 |
| ПРОЦЕСС И СПОСОБ ОЧИСТКИ КАРБОНАТА ЛИТИЯ, ИСХОДЯ ИЗ РАСТВОРА ХЛОРИДА ЛИТИЯ С ПРИМЕСЯМИ | 2020 |
|
RU2795224C1 |
| ФОСФАТ ЛИТИЯ-ЖЕЛЕЗА, ИМЕЮЩИЙ ОЛИВИНОВУЮ СТРУКТУРУ, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2009 |
|
RU2488550C2 |
| СПОСОБ ПОЛУЧЕНИЯ МОНОГИДРАТА ГИДРОКСИДА ЛИТИЯ ВЫСОКОЙ СТЕПЕНИ ЧИСТОТЫ ИЗ МАТЕРИАЛОВ, СОДЕРЖАЩИХ КАРБОНАТ ЛИТИЯ | 2001 |
|
RU2196735C1 |
| ЛИТИЕВАЯ ВТОРИЧНАЯ БАТАРЕЯ С ЭЛЕКТРОЛИТОМ, СОДЕРЖАЩИМ СОЕДИНЕНИЯ АММОНИЯ | 2006 |
|
RU2335044C1 |
| АКТИВНЫЙ КАТОДНЫЙ МАТЕРИАЛ, ОБЕСПЕЧИВАЮЩИЙ УЛУЧШЕННУЮ ЭФФЕКТИВНОСТЬ И ПЛОТНОСТЬ ЭНЕРГИИ ЭЛЕКТРОДА | 2009 |
|
RU2467434C1 |
| СПОСОБ ИЗВЛЕЧЕНИЯ ЛИТИЯ И ПЕРЕХОДНОГО МЕТАЛЛА С ПРИМЕНЕНИЕМ НАГРЕВАНИЯ | 2019 |
|
RU2790318C2 |
Изобретение относится к извлечению гидроксида лития из отходов ионно-литиевых батарей или их частей. Неочищенный гидроксид лития в количестве 1 молярная часть в виде твердого вещества, которое содержит фторид лития, объединяют с низшим спиртом, взятым в количестве от 5 до 15 молярных частей. Разделяют твердую и жидкую фазы с получением прозрачного раствора, добавляют воду. Выделяют твердый гидроксид лития в виде ангидрата или моногидрата, или в виде смеси ангидрата и моногидрата, или в виде смеси моногидрата с водой и/или низшим спиртом, или в виде смеси моногидрата с низшим спиртом, или в виде смеси ангидрата и моногидрата с низшим спиртом. Способ позволяет эффективно очистить гидроксид лития, содержащего LiF и/или соли амфотерных элементов. 13 з.п. ф-лы, 4 ил., 12 табл., 9 пр.
1. Способ очистки гидроксида лития из материала, содержащего отходы ионно-литиевых батарей или их части, при этом способ включает стадии
(A) предоставление неочищенного гидроксида лития в виде твердого вещества, которое содержит фторид лития,
(B) объединение неочищенного гидроксида лития, предоставленного на стадии (A), с низшим спиртом,
(C) разделение твердой и жидкой фаз с получением прозрачного раствора,
(D) последующее добавление воды,
(F) выделение твердого гидроксида лития в виде ангидрата или моногидрата, или в виде смеси ангидрата и моногидрата, или в виде смеси моногидрата с водой и/или низшим спиртом, или в виде смеси моногидрата с низшим спиртом, или в виде смеси ангидрата и моногидрата с низшим спиртом,
причем от 5 до 15 молярных частей низшего спирта на стадии (B) объединяют с 1 молярной частью лития, предоставленного на стадии (A).
2. Способ по п. 1, в котором низший спирт представляет собой C1-C4 спирт или смесь таких спиртов.
3. Способ по п. 1, в котором низший спирт представляет собой метанол или этанол.
4. Способ по п. 1, в котором объединенный гидроксид лития и низший спирт со стадии (В) смешивают с получением суспензии частиц в растворе гидроксида лития.
5. Способ по п. 1, в котором неочищенный гидроксид лития, предоставленный на стадии (A), содержит моногидрат гидроксида лития, содержащий от 100 частей на миллион до 1,29 мас.% кальция, от 0,1 мас.% до 1,29 мас.% фторида, от 0,1 мас.% до 1,29 мас.% натрия, или ангидрат гидроксида лития, содержащий от 175 частей на миллион до 2,26 % кальция, от 0,175 мас.% до 2,26 мас.% фторида, от 0,175 мас.% до 2,26 мас.% натрия, где все количества приведены по массе сухого твердого вещества.
6. Способ по п. 1, в котором неочищенный гидроксид лития, предоставленный на стадии (A), дополнительно содержит одну или более дополнительных примесей из группы солей кальция, солей алюминия, солей цинка, где сумма кальция, алюминия и цинка, как правило, составляет 100 частей на миллион или более таких примесей относительно общей массы неочищенного твердого гидроксида лития.
7. Способ по п. 1, в котором 90 мас.% неочищенного твердого гидроксида лития, предоставленного на стадии (A), состоит из ангидрата гидроксида лития или моногидрата гидроксида лития или их смесей.
8. Способ по п. 1, в котором неочищенный гидроксид лития, предоставленный на стадии (A), содержит фторид в качестве примеси в количестве 500 частей на миллион или более относительно общей массы неочищенного твердого гидроксида лития.
9. Способ по п. 1, в котором процесс дополнительно включает стадию (E) сушки, следующую за стадией (D).
10. Способ по п. 1, в котором комбинация неочищенного твердого гидроксида лития с низшим спиртом, таким как метанол или этанол, на стадии (В) содержит менее одной молярной части воды на одну молярную часть лития, и воду добавляют на стадии (D) в количестве около 1 молярная часть или более на одну молярную часть лития.
11. Способ по п. 1, в котором гидроксид лития дополнительно очищают посредством стадии водной кристаллизации, которую проводят перед стадией (А) или после стадии (В) с последующим удалением твердых веществ путем разделения твердой и жидкой фаз (стадия C).
12. Способ по п. 1, в котором гидроксид лития дополнительно очищают путем добавления ионов сульфида или ионов сульфида водорода к водному раствору неочищенного гидроксида лития перед стадией (A).
13. Способ по п. 1, в котором неочищенный гидроксид лития, предоставленный на стадии (A), получают из отходов ионно-литиевых батарей после проведения последовательно стадий восстановления, водного выщелачивания, при необходимости в присутствии гидроксида щелочноземельного металла, такого как гидроксид кальция, отделения твердых веществ и сушки.
14. Способ по любому из пп. 1-13, в котором используемый низший спирт извлекают путем дистилляции или выпаривания и рециркулируют в процесс.
| TABODA M.E | |||
| et al | |||
| Process design for drowning-out crystallization of lithium hydroxide monohydrate., Chemical Engineering Research and Design, Elsevier, Amsterdam, vol | |||
| Устройство для выпрямления опрокинувшихся на бок и затонувших у берега судов | 1922 |
|
SU85A1 |
| Способ приготовления жидкого горючего | 1919 |
|
SU1325A1 |
| Индикатор качества уплотнения бетонной смеси | 1975 |
|
SU613198A1 |
| JP 2012229481 A, 22.11.2012 | |||
| WO 2008022414 A1, 28.02.2008 | |||
| СПОСОБ ОЧИСТКИ РАСТВОРОВ СОЕДИНЕНИЙ ЛИТИЯ ОТ КАТИОНОВ ЩЕЛОЧНЫХ И ЩЕЛОЧНОЗЕМЕЛЬНЫХ МЕТАЛЛОВ | 1995 |
|
RU2092449C1 |
| СПОСОБ ОЧИСТКИ ГИДРОКСИДА ЛИТИЯ | 2004 |
|
RU2267461C2 |

