ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новому агонисту β-рецептора тиреоидных гормонов, который может применяться для лечения ожирения, гиперлипидемии, гиперхолестеринемии, диабета, заболеваний печени (жирового гепатоза, НАСГ, НАЖБП и т.п.), сердечно-сосудистых заболеваний (атеросклероза и т.п.) и заболеваний щитовидной железы (гипотиреоза, рака щитовидной железы и т.п.).
УРОВЕНЬ ТЕХНИКИ
Тиреоидный гормон - это гормон, секретируемый щитовидной железой и оказывающий действие почти на все клетки человеческого организма. Тиреоидные гормоны включают тироксин (Т4) и трийодтиронин (Т3). Т4 может подвергаться дейодинизации в Т3 с помощью специфической дейодиназы для придания эффективности. Т3 имеет быстрое и сильное действие, и его продолжительность короче, чем у Т4, в то время как Т4 имеет медленное и слабое действие и большую продолжительность. Хотя специфическая дейодиназа присутствует во всех тканях, больше всего ее содержится в печени и почках.
Тиреоидные гормоны необходимы для нормального роста и развития человеческого организма. Недостаточная или избыточная секреция тиреоидных гормонов может вызвать заболевания. Недостаточность тиреоидных гормонов будет влиять на физическое и умственное развитие, что может привести к кретинизму. Взрослые с недостаточностью тиреоидных гормонов могут страдать от микседемы. В случае гипертиреоза возникает повышенная возбудимость, нетерпение, тремор, увеличение частоты сердечных сокращений, увеличение сердечного выброса и другие явления. Тиреоидные гормоны могут способствовать окислению веществ, увеличивать потребление кислорода, повышать скорость базального метаболизма и увеличивать тепловыделение.
В норме центральная нервная система контролирует высвобождение тиреотропин-рилизинг гормона (ТРГ) из гипоталамуса, который регулирует секрецию тиреотропного гормона (ТТГ) аденогипофизом, а ТТГ стимулирует клетки щитовидной железы к секреции Т4 и Т3. При повышении концентрации Т4 и Т3 в крови синтез и высвобождение ТТГ в аденогипофизе ингибируется отрицательной обратной связью, а чувствительность аденогипофиза к ТРГ снижается, тем самым снижая секрецию ТТГ, так что секреция тиреоидных гормонов не слишком высока. Однако при снижении концентраций Т4 и Т3 в крови влияние отрицательной обратной связи на аденогипофиз снижается. Повышение секреции ТТГ вызывает повышение секреции Т4 и Т3. Иными словами, регуляторная ось «гипоталамус - аденогипофиз - щитовидная железа» может поддерживать относительно постоянную секрецию тиреоидных гормонов.
Биологическая активность тиреоидных гормонов опосредуется рецепторами тиреоидных гормонов (TR), которые принадлежат к надсемейству ядерных рецепторов. TR имеет лиганд-связывающий домен, ДНК-связывающий домен и аминоконцевой домен. TR имеет четыре подтипа: TRα1, TRα2, TRβ1 и TRβ2, соответственно. TRα1 в основном содержится в сердце, а TRβ1 в печени. Экспрессия мРНК TRβ2 в основном ограничена аденогипофизом и гипоталамусом. Тиреоидные гормоны связываются с TRα1, TRβ1 и TRβ2, вызывая соответствующие физиологические эффекты. Тиреоидные гормоны не связываются с TRα2.
Терапевтические эффекты, такие как лечение ожирения, могут быть достигнуты за счет наиболее эффективного использования таких благоприятных действий тиреоидных гормонов, как увеличение скорости метаболизма, потребления кислорода и тепловыделения. Гипертиреоз часто приводит к потреблению пищи, но также к общему увеличению скорости базального метаболизма (BMR). Гипертиреоз часто сопровождается потерей массы тела примерно на 15%, в то время как гипотиреоз часто сопровождается увеличением массы тела на 25%-30%. При применении Т3 для лечения гипотиреоза у большинства пациентов наблюдается увеличение массы тела.
Кроме того, тиреоидные гормоны могут также снижать уровень липопротеинов низкой плотности (ЛПНП) в сыворотке (Journal of Molecular and Celluar Cardiology 37(2004): 1137-1146). Существующие исследования показали, что гипертиреоз значительно снижает общий холестерин в сыворотке, что в основном связано с тем, что тиреоидные гормоны повышают экспрессию рецепторов ЛПНП в печени, тем самым способствуя процессу метаболизма холестерина в желчные кислоты; гипотиреоз связан с гиперхолестеринемией. Таким образом, тиреоидные гормоны могут снижать заболеваемость атеросклерозом и другими сердечнососудистыми заболеваниями.
При лечении заболеваний тиреоидными гормонами из-за индивидуальных особенностей часто имеют место побочные действия чрезмерных физиологических доз, включая сердечные заболевания (в основном относится к тахикардии), мышечную слабость, чрезмерную потерю массы тела и т.д., а длительное применение тиреоидных гормонов может привести к потере костной массы. Таким образом, крайне необходимо разработать новые лекарственные средства посредством структурной модификации тиреоидных гормонов для сохранения его благоприятного действия и уменьшения его побочных действий для лечения связанных с ним заболеваний, таких как ожирение, гиперлипидемия, гиперхолестеринемия, диабет, заболевания печени (жировой гепатоз, НАС Г, НАЖБП и т.п.), сердечно-сосудистые заболевания (атеросклероз и т.п.), заболевания щитовидной железы (гипотиреоз, рак щитовидной железы и т.п.) и другие связанные заболевания.
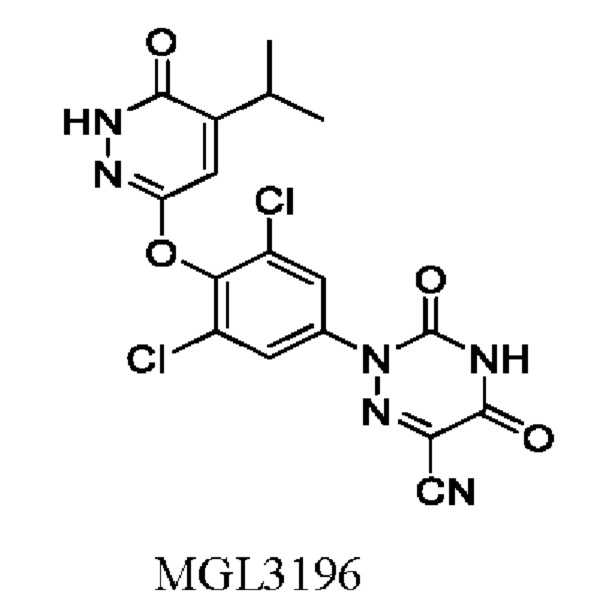
Пиридазиноновый аналог тиреоидных гормонов, представленный структурой MGL3196, был запатентован компанией Madrigal Pharmaceuticals (CN101228135B) и в настоящее время проходит III фазу клинических исследований для лечения НАСГ и НАЖБП. В связи с его низкой активностью и плохой проникающей способностью требуется пероральная доза от 80 мг до 100 мг в сутки. Эта доза значительно выше, чем у других продуктов, направленных против той же мишени.
Соединение VK2809, запатентованное компанией Viking Therapeutics (CN1882327C), проходит фазу 2b клинического исследования для лечения НАСГ. Согласно клиническим данным I фазы соединение имело проблемы с безопасностью и относительно узкое терапевтическое окно. Наблюдалось повышение уровня печеночных ферментов, что является признаком поражения печени. В то же время, в доклинических токсикологических исследованиях было обнаружено поражение хряща (J. Med. Chem. 2014, 57, 3912-3923). III фаза клинического исследования эпротирома, запатентованного компанией Bristol-Myers Squibb Со (CN1216857C), была досрочно прекращена. Согласно сообщенным клиническим данным, также наблюдалось повышение уровня печеночных ферментов. В доклинических токсикологических исследованиях также было обнаружено поражение хряща. Что же касается других патентов, таких как относящиеся к производным пиридина (CN102459185) и производным индола (WO2002051805), в них сообщаются только данные об активности без проведения дальнейших исследований. Ни один из продуктов не стал предметом клинических исследований.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Для решения описанных существующих проблем в настоящей заявке предложен новый агонист β-рецептора тиреоидных гормонов, обладающий лучшей активностью, селективностью или без опасностью.
Один из аспектов настоящей заявки заключается в обеспечении нового агониста β-рецептора тиреоидных гормонов формулы I, его фармацевтически приемлемой сопи или его пролекарства:
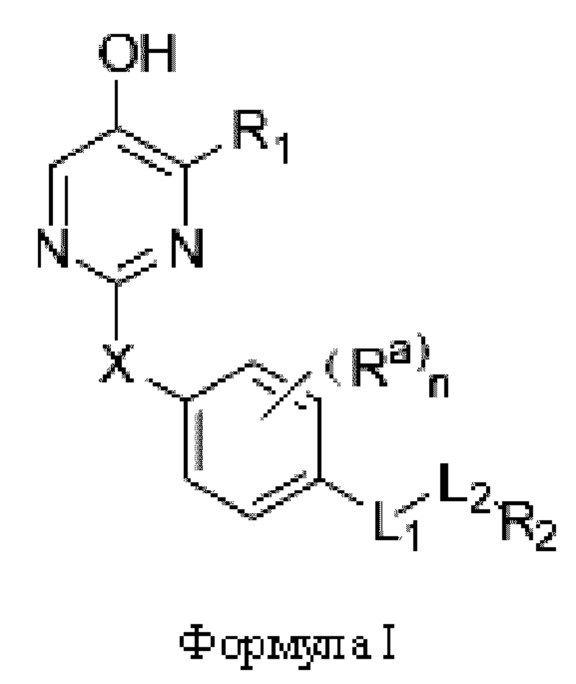
где
R1 представляет собой водород, необязательно замещенный алкил, необязательно замещенный циклоалкил, необязательно замещенный арил, необязательно замещенный гетероциклил, необязательно замещенный гетероарил, необязательно замещенный амино, необязательно замещенный карбамоил или -COR10;
X представляет собой необязательно замещенный метилен, -О-, -S- или -SO2-;
Ra выбран из водорода, галогена, С1-6 линейного и разветвленного акила или циклоалкила; или два соседних Ra связаны с образованием карбоциклического кольца или гетероциклического кольца,
L1 представляет собой одинарную связь, метилен, -СН=СН-, -О-, -СО-, -NR3-, -NR3CO-, -CONR3-, -CH2NR3- или -S-,
L2 представляет собой одинарную связь или - (CR4R5)p;
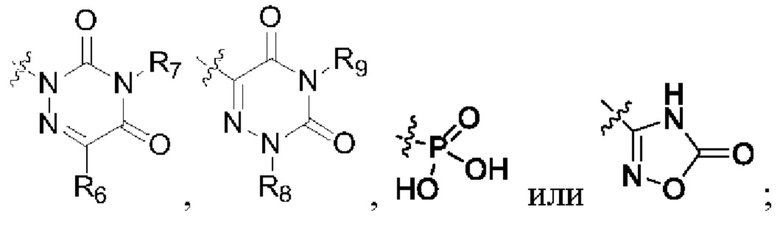
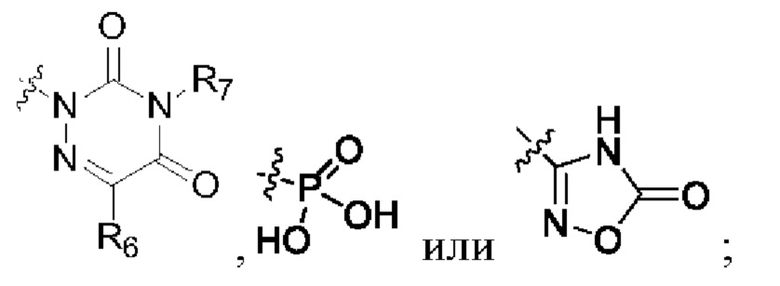
R2 представляет собой карбоксил или группу, представленную следующей формулой:
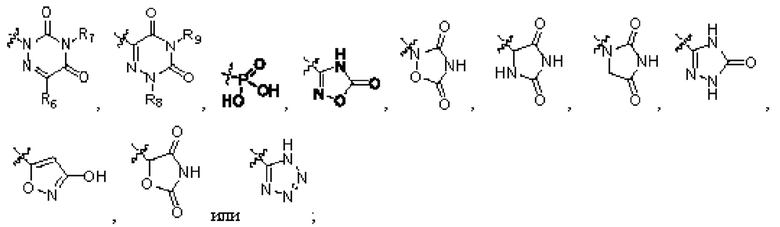
R3 представляет собой водород или необязательно замещенный алкил;
каждый из R4 и R5 независимо выбран из водорода, галогена или необязательно замещенного алкила, или R4 и R5 связаны с образованием циклоалкила;
R6 представляет собой водород, циано, амино, СООН, С1-6 алкил, С3-6 галогеналкил, С3-6 циклоалкил или С3-6 галогенциклоалкил;
R8 представляет собой водород, циано, СООН, С1-6 алкил, С3-6 галогеналкил, С3-6 циклоалкил или С3-6 галогенциклоалкил;
R7 и R9 представляют собой водород, С1-3 алкил или С1-3 галогеналкил;
R10 представляет собой необязательно замещенный алкил, амино, гидроксил, необязательно замещенный циклоалкил, необязательно замещенный арил, необязательно замещенный гетероциклил или необязательно замещенный гетероарил;
n составляет 0, 1, 2, 3 или 4; и
р составляет 0, 1 или 2.
В некоторых предпочтительных вариантах осуществления R1 представляет собой водород или -COR10, или алкил, циклоалкил, циклоалкилалкил, арил, арилалкил, гетероциклил, гетероарил, амино или карбамоил, необязательно замещенный водородом, дейтерием, тритием, C1-6 алкилом, гидроксилом, галогеном или CN. В некоторых предпочтительных вариантах осуществления R1 представляет собой -COR10 или С1-10 алкил, С3-10 циклоалкил, С3-10 циклоалкил С1-6 алкил, С5-10 арил, С5-10 арил С1-6 алкил, 5-10-членный гетероциклил, 5-10-членный гетероарил, амино или карбамоил, необязательно замещенный водородом, дейтерием, тритием, C1-6 алкилом, гидроксилом, галогеном или CN. В некоторых предпочтительных вариантах осуществления R1 представляет собой -COR10 или C1-8 алкил, С3-8 циклоалкил, С3-8 циклоалкил С1-6 алкил, С5-10 арил, С5-10 арил С1-6 алкил, 5-10-членный гетероциклил или 5-10-членный гетероарил, необязательно замещенный водородом, дейтерием, тритием, C1-6 алкилом, гидроксилом, галогеном или CN.
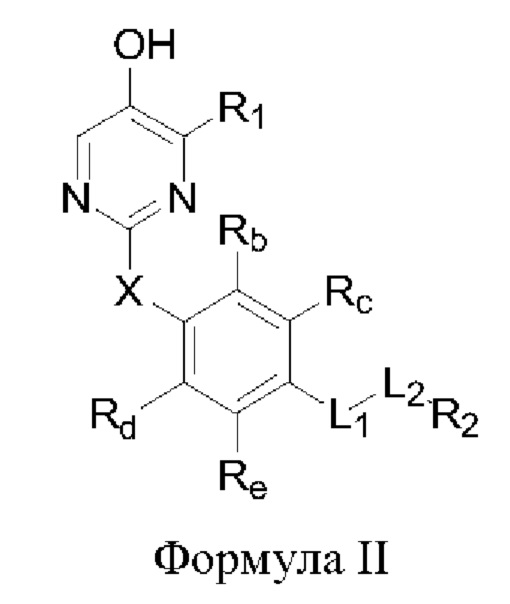
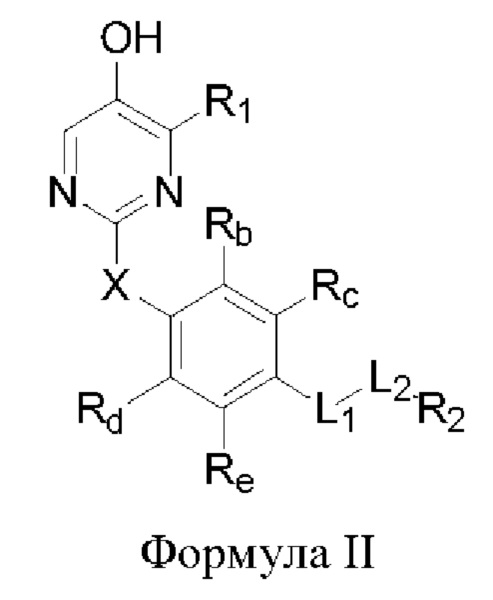
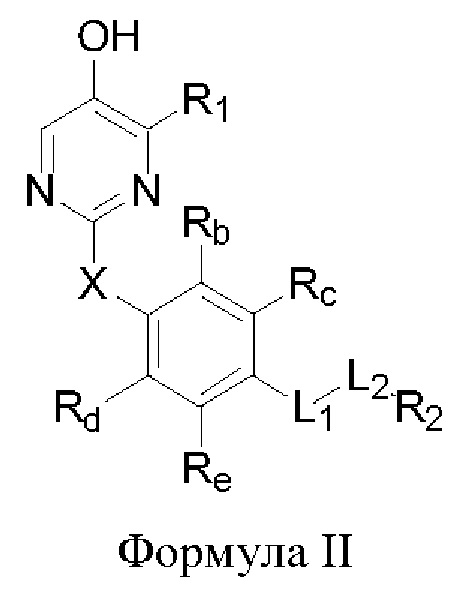
В некоторых аспектах соединение формулы I, предложенное в настоящей заявке, представлено формулой II:
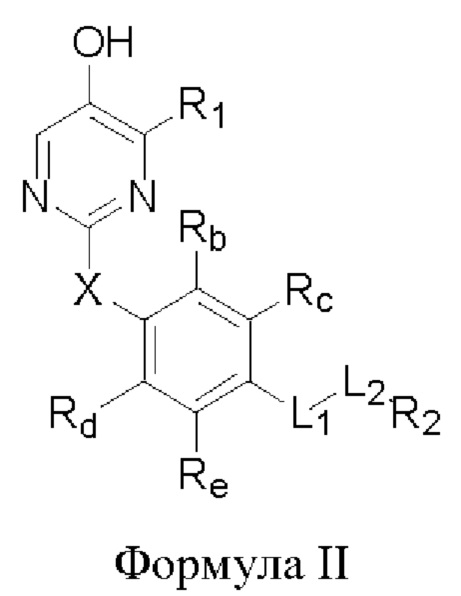
где Rb, Rc, Rd и Re представляют собой водород, дейтерий, галоген, С1-6 линейный или разветвленный алкил или циклоалкил; или Rb и Rc связаны с образованием 5- или 6-членного циклоалкила или 5- или 6-членного неароматического гетероциклического кольца, содержащего 1 или 2 гетероатома, выбранных из атома азота, атома кислорода и атома серы; или Rd и Re связаны с образованием 5- или 6-членного циклоалкила или 5- или 6-членного неароматического гетероциклического кольца, содержащего 1 или 2 гетероатома, выбранных из атома азота, атома кислорода и атома серы.
Другие заместители имеют определения, указанные выше в формуле I.
В некоторых аспектах соединение формулы II, предложенное в настоящей заявке, представляет собой:
где R1 представляет собой необязательно замещенный С1-6 в линейный или разветвленный алкил или С3-8 циклоалкил;
X представляет собой О, S или -CH2-;
Rb, Rc, Rd и Re представляют собой водород, дейтерий, галоген, C1-6 линейный или разветвленный алкил или циклоалкил; или Rb и Rc связаны с образованием 5- или 6-членного циклоалкила или 5- или 6-членного неароматического гетероциклического кольца, содержащего 1 или 2 гетероатома, выбранных из атома азота, атома кислорода и атома серы; или Rd и Re связаны с образованием 5- или 6-членного циклоалкила или 5- или 6-членного неароматического гетероциклического кольца, содержащего 1 или 2 гетероатома, выбранных из атома азота, атома кислорода и атома серы;
L1 представляет собой одинарную связь, -NR3-, -О или -S-;
L2 представляет собой одинарную связь или -СН2-;
R2 представляет собой группу, представленную следующей формулой: 
R3 представляет собой водород или необязательно замещенный С1-6 алкил;
R6 представляет собой водород, циано, амино, СООН, C1-6 алкил, C1-6 галогеналкил, С3-6 циклоалкил или С3-6 галогенциклоалкил;
R8 представляет собой водород, циано, СООН, C1-6 алкил, C1-6 галогеналкил, С3-6 циклоалкил или С3-6 галогенциклоалкил; и
R7 и R9 представляют собой водород, С1-3 алкил или С1-3 галогеналкил.
В некоторых аспектах соединение формулы II, предложенное в настоящей заявке, представляет собой:
где R1 представляет собой необязательно замещенный С1-6 линейный или разветвленный алкил;
Rb, Rc, Rd и Re представляют собой водород, дейтерий, галоген, С1-6 линейный или разветвленный алкил или циклоалкил; или Rb и Rc связаны с образованием 5- или 6-членного циклоалкила или 5- или 6-членного неароматического гетероциклического кольца, содержащего 1 или 2 гетероатома, выбранных из атома азота, атома кислорода и атома серы; или Rd и Re связаны с образованием 5- или 6-членного циклоалкила или 5- или 6-членного неароматического гетероциклического кольца, содержащего 1 или 2 гетероатома, выбранных из атома азота, атома кислорода и атома серы;
X представляет собой О, S или -СН2-;
L1 представляет собой одинарную связь, -О-, -S- или -NH-;
L2 представляет собой одинарную связь;
R2 представляет собой группу, представленную следующей формулой: 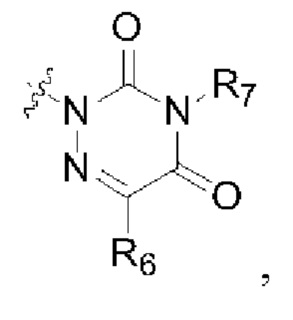
R6 представляет собой водород, циано, С1-6 алкил или C1-6 галогеналкил;
R8 представляет собой водород, циано, С1-6 алкил или С1-6 галогеналкил; и
R7 и R9 представляют собой водород, С1-3 алкил или С1-3 галогеналкил.
В некоторых аспектах соединение формулы II, предложенное в настоящей заявке, представляет собой:
где R1 представляет собой С1-6 линейный или разветвленный алкил, бензил или С5-6 циклоалкилметилен, необязательно замещенный водородом, дейтерием, тритием, С1-6 алкилом, гидроксилом, галогеном или CN, и более предпочтительно изопропил или бензил;
Rb и Rd представляют собой галоген, и Rc и Re представляют собой водород, и Rb и Rd более предпочтительно представляют собой хлор;
X представляет собой О, S или -СН2-;
L1 представляет собой одинарную связь, -О, -S- или -NH-;
L2 представляет собой одинарную связь или -СН2-;
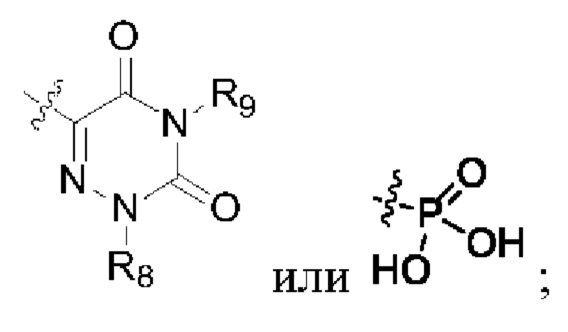
R2 представляет собой группу, представленную следующей формулой: 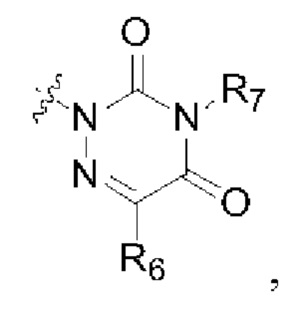
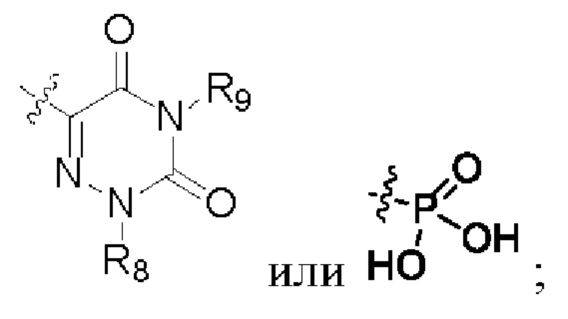
R6, R7, R8 и R9 представляют собой водород, или C1-6 алкил, или С3-8 циклоалкил.
В некоторых аспектах соединение формулы I, предложенное в настоящей заявке, представлено формулой III:
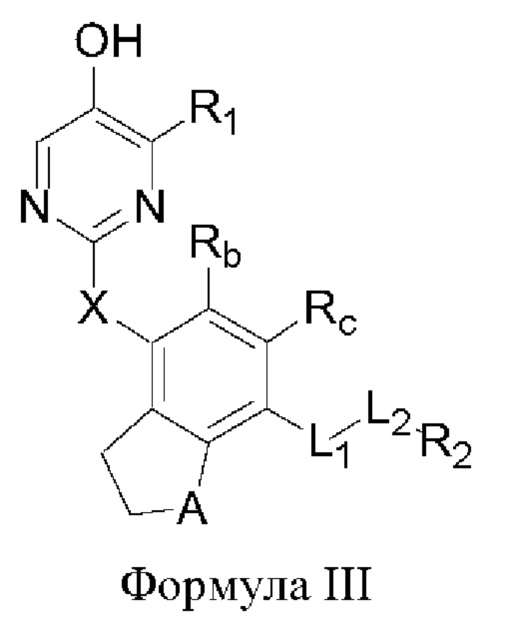
где
Rb и Rc представляют собой водород, дейтерий, галоген, С1-6 линейный или разветвленный алкил или циклоалкил; и
А представляет собой О или метилен.
Другие заместители имеют определения, указанные в формуле I.
В других предпочтительных вариантах осуществления в соединении формулы I, предложенном в настоящей заявке, R1 выбран из:
1) необязательно замещенного C1-6 линейного и разветвленного алкила;
2) необязательно замещенного С3-8 циклоалкила;
3) необязательно замещенного С3-8 неароматического гетероциклила, содержащего от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы;
4) необязательно замещенного фенила; или
5) необязательно замещенного С5-6 гетероарила, содержащего от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы.
В других предпочтительных вариантах осуществления в соединении формулы I, предложенном в настоящей заявке, R1 выбран из -(CR11R12)mR13; R11 и R12 выбраны из водорода, дейтерия, галогена, гидроксила, амино, карбоксила или необязательно замещенного С1-4 алкила; и R13 выбран из:
1) водорода или дейтерия;
2) галогена;
3) гидроксила;
4) амино;
5) карбоксила;
6) необязательно замещенного С1-4 алкила или С1-4 алкокси;
7) необязательно замещенного С3-8 циклоалкила;
8) необязательно замещенного С3-8 неароматического гетероциклила, содержащего от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы;
9) необязательно замещенного фенила; или
10) необязательно замещенного С5-6 гетероарила, содержащего от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы; и
m составляет 0, 1, 2 или 3.
В других предпочтительных вариантах осуществления в соединении формулы I, предложенном в настоящей заявке, R1 выбран из -COR10, где R10 выбран из:
1) амино;
2) гидроксила;
3) необязательно замещенного С1-4 алкила или С1-4 алкокси;
4) необязательно замещенного С3-8 циклоалкила;
5) необязательно замещенного С3-8 неароматического гетероциклила, содержащего от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы;
6) необязательно замещенного фенила; или
7) необязательно замещенного С5-6 гетероарила, содержащего от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы.
В других предпочтительных вариантах осуществления в соединении формулы I R1 представляет собой водород, С1-10 алкил (предпочтительно С1-5 алкил), С3-10 циклоалкил (предпочтительно С3-8 циклоалкил), С3-10 циклоалкил С1-6 алкил (предпочтительно С3-8 циклоалкил С1-4 алкил), С5-10 арил (предпочтительно С5-8 арил), С5-10 арил С1-6 алкил (предпочтительно С5-8 арил С1-4 алкил), 5-10-членный гетероциклил, содержащий от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы, 5-10-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы, амино или -COR10, и указанный С1-10 алкил (предпочтительно С1-5 алкил), С3-10 циклоалкил (предпочтительно С3-8 циклоалкил), С3-10 циклоалкил С-16 алкил (предпочтительно С3-8 циклоалкилС1-4 алкил), С5-10 арил (предпочтительно С5-8 арил), С5-10 арил С1-6 алкил (предпочтительно С5-8 арил С1-4 алкил), 5-10-членный гетероциклил, содержащий от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы, 5-10-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы, или амино является незамещенным или может быть замещен дейтерием, тритием, C1-6 алкилом, гидроксилом, галогеном или CN;
X представляет собой метилен, -О-, -S- или -SO2-;
Ra представляет собой водород, дейтерий, галоген, С1-6 линейный или разветвленный алкил или циклоалкил; или два соседних Ra связаны с образованием 5-10-членного карбоциклического кольца или 5-10-членного гетероциклического кольца, содержащего от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы;
L1 представляет собой одинарную связь, метилен, -О-, -СО-, -NR3-, -NR3CO-, -CONR3-, -CH2NR3- или -S-;
L2 представляет собой одинарную связь или С1-6 алкил (предпочтительно С1-4 алкил);
R2 представляет собой карбоксил или группу, представленную следующей формулой:
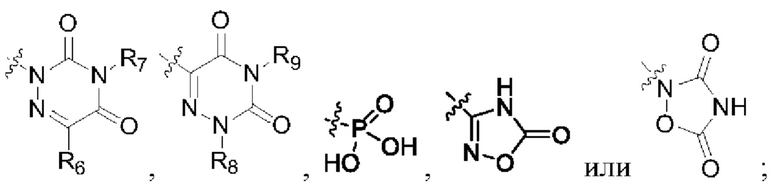
R3 представляет собой водород или С1-6 алкил;
R6 представляет собой водород, циано, амино, СООН, С1-6 алкил или С1-6 галогеналкил;
R8 представляет собой водород, циано, СООН, C1-6 алкил или С1-6 галогеналкил;
R7 и R9 представляют собой водород, С1-3 алкил или С1-3 галогеналкил;
R10 представляет собой С3-10 циклоалкил (предпочтительно С3-8 циклоалкил), С5-10 арил (предпочтительно С5-8 арил), 5-10-членный гетероциклил, содержащий от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы, или 5-10-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы; и
n составляет 0, 1, 2, 3 или 4.
В других предпочтительных вариантах осуществления в соединении формулы I R1 представляет собой C1-8 алкил (предпочтительно С1-5 алкил), С3-8 циклоалкил (предпочтительно С3-6 циклоалкил), С3-8 циклоалкил С1-5 алкил (предпочтительно С3-6 циклоалкил С1-3 алкил), С5-8 арил (предпочтительно С5-6 арил), С5-8 арил С1-5 алкил (предпочтительно С5-6 арил-С1-3 алкил), 5-8-членный гетероциклил, содержащий от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы, 5-8-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы, амино или -COR10, и указанный С1-8 алкил (предпочтительно С1-5 алкил), С3-8 циклоалкил (предпочтительно С3-6 циклоалкил), С3-8 циклоалкил С1-5 алкил (предпочтительно С3-6 циклоалкил С1-3 алкил), С5-8 арил (предпочтительно С5-6 арил), С5-8 арил С1-5 алкил (предпочтительно С5-6 арил C1-3 алкил), 5-8-членный гетероциклил, содержащий от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы, 5-8-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы, или амино является незамещенным или может быть замещен дейтерием, тритием, С1-6 алкилом, гидроксилом, галогеном или CN;
X представляет собой метилен, -О-, -S- или -SO2-;
Ra представляет собой галоген или С1-4 линейный или разветвленный алкил; или два соседних Ra связаны с образованием 5-7-членного карбоциклического кольца или 5-7-членного гетероциклического кольца, содержащего от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы;
L1 представляет собой одинарную связь, -О-, -NR3-, -NR3CO-, -CONR3-, -CH2NR3- или -S-;
L2 представляет собой одинарную связь или С1-5 алкил (предпочтительно С1-3 алкил);
R2 представляет собой карбоксил или группу, представленную следующей формулой:
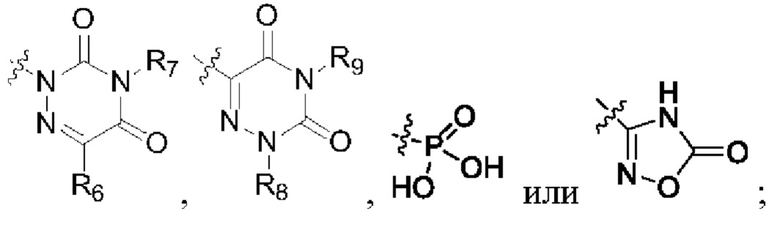
R3 представляет собой водород или С1-3 алкил;
R6 представляет собой водород, циано, СООН или С1-4 алкил;
R8 представляет собой водород или C1-4 алкил;
R7 и R9 представляют собой водород или С1-3 алкил;
R10 представляет собой С5-8 арил (предпочтительно С5-6 арил) или 5-8-членный гетероарил (предпочтительно 5-6-членный гетероарил), содержащий от 1 до 3 гетероатомов, выбранных из атома азота, атома кислорода и атома серы; и
n составляет 1, 2 или 3.
В других предпочтительных вариантах осуществления в соединении формулы I R1 представляет собой метил, этил, пропил, бутил, пентил, циклопропан, циклобутан, циклопентан, циклогексан, циклопропанметил, циклобутанметил, циклопентанметил, циклогексанметил, фенил, бензил или -COR10, и указанный метил, этил, пропил, бутил, пентил, циклопропан, циклобутан, циклопентан, циклогексан, циклопропанметил, циклобутанметил, циклопентанметил, циклогексанметил, фенил или бензил является незамещенным или может быть замещен дейтерием, С1-3 алкилом, гидроксилом, галогеном или CN;
X представляет собой метилен, -О-, -S- или -SO2-;
Ra представляет собой галоген; или два соседних Ra связаны с образованием 5-членного карбоциклического кольца или 5-членного гетероциклического кольца, содержащего от 1 до 2 гетероатомов, выбранных из атома азота, атома кислорода и атома серы;
L1 представляет собой одинарную связь, -О-, -NH-, -NHCO-, -CONH-, -CH2NH- или -S-;
L2 представляет собой одинарную связь, метил, этил или пропил;
R2 представляет собой карбоксил или группу, представленную следующей формулой:
R6 представляет собой водород, циано, СООН, метил, этил или пропил;
R8 представляет собой водород, метил, этил или пропил;
R7 и R9 представляют собой водород или метил;
R10 представляет собой фенил; и n составляет 2 или 3.
В других предпочтительных вариантах осуществления в соединении формулы I R1 представляет собой метил, этил, пропил, бутил, пентил, циклопропан, циклобутан, циклопентан, циклогексан, циклопропанметил, циклобутанметил, циклопентанметил, циклогексанметил, фенил или бензил, и указанный метил, этил, пропил, бутил, пентил, циклопропан, циклобутан, циклопентан, циклогексан, циклопропанметил, циклобутанметил, циклопентанметил, циклогексанметил, фенил или бензил является незамещенным или может быть замещен дейтерием, С1-3 алкилом, гидроксилом, F, Cl, Br или CN;
X представляет собой метилен, -О- или -S-;
Ra представляет собой F, О или Br; или два соседних Ra связаны с образованием 5-членного карбоциклического кольца или 5-членного гетероциклического кольца, содержащего от 1 до 2 гетероатомов, выбранных из атома азота, атома кислорода и атома серы;
L1 представляет собой одинарную связь, -О-, -NH- или -NHCO-;
L2 представляет собой одинарную связь, метил, этил или пропил;
R2 представляет собой карбоксил или группу, представленную следующей формулой:
R6 представляет собой водород, циано или метил;
R7 представляет собой водород; и
n составляет 2 или 3.
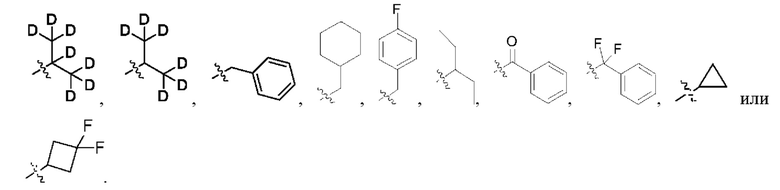
В некоторых конкретных вариантах осуществления в соединении формулы I, предложенном в настоящей заявке, R1 выбран из: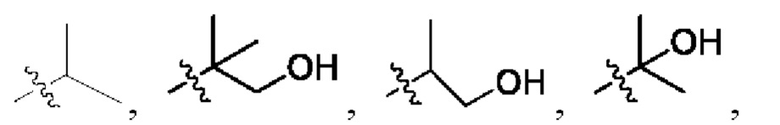
В некоторых конкретных вариантах осуществления в соединении формулы I, предложенном в настоящей заявке, Rb, Rc, Rd и Re выбраны из водорода, дейтерия или галогена.
В некоторых конкретных вариантах осуществления в соединении формулы I, предложенном в настоящей заявке, L1 выбран из одинарной связи, -О-, -NH-, -NHCO- или -NHCH2-.
В некоторых конкретных вариантах осуществления в соединении формулы I, предложенном в настоящей заявке, L2 выбран из одинарной связи или метилена (-СН2-).
В некоторых конкретных вариантах осуществления в соединении формулы I, предложенном в настоящей заявке, R2 выбран из: карбоксила, 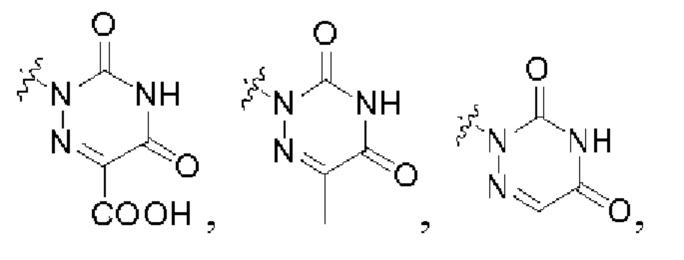
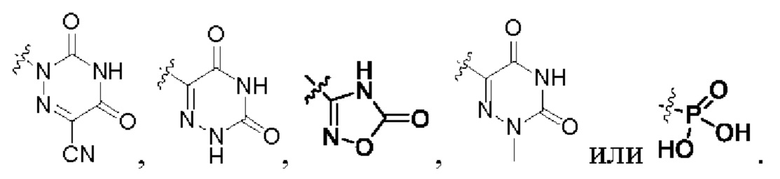
В некоторых конкретных вариантах осуществления соединение формулы I, предложенное в настоящей заявке, выбрано из:
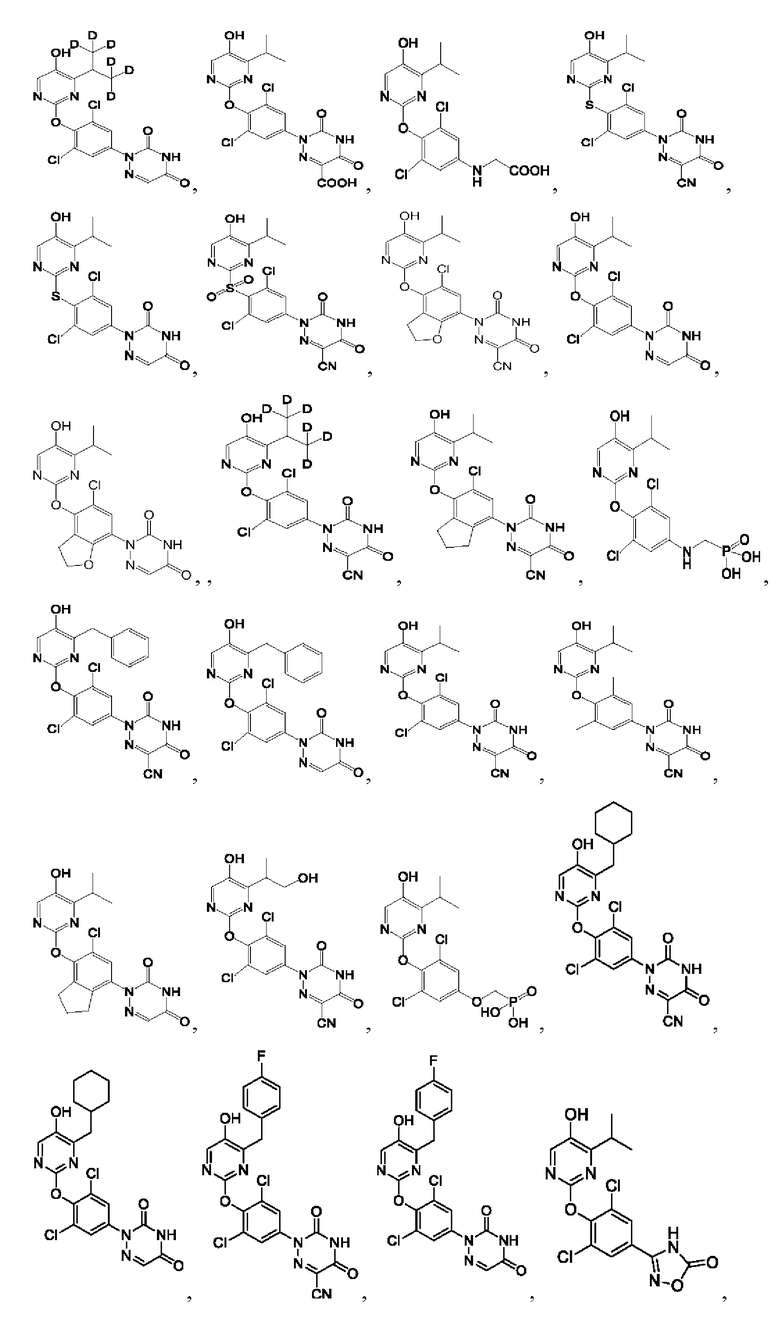
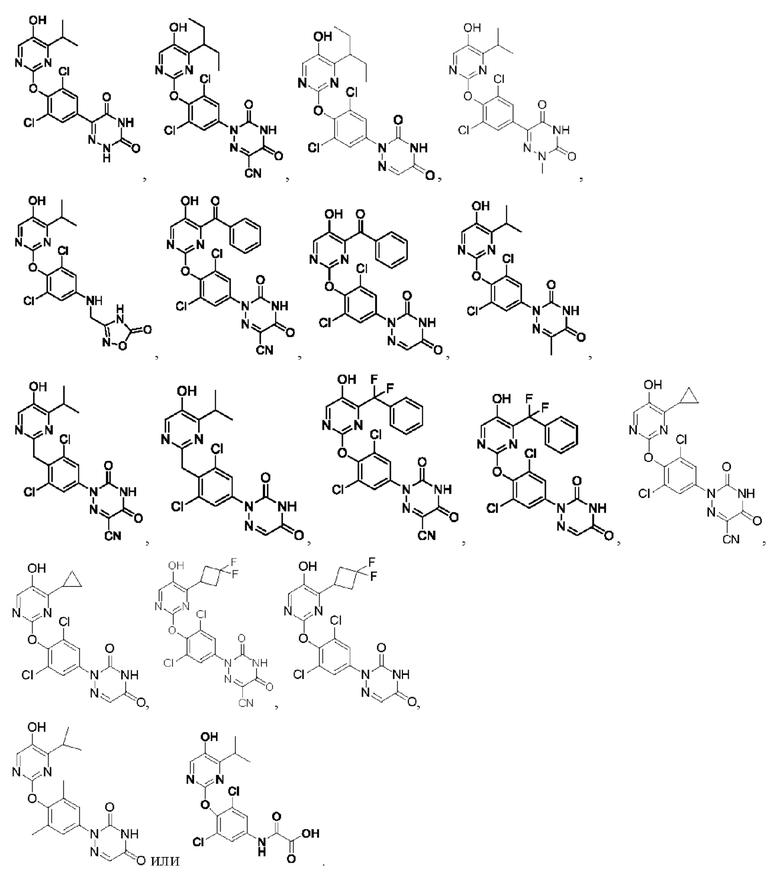
Другой аспект настоящей заявки заключается в обеспечении фармацевтической композиции, содержащей соединение формулы I согласно настоящей заявке, его фармацевтически приемлемую соль или его пролекарство и один или более фармацевтически приемлемых носителей.
Другой аспект настоящей заявки заключается в обеспечении применения соединения согласно настоящему изобретению, его фармацевтически приемлемой соли или его пролекарства для предотвращения или лечения заболевания, связанного с действием агониста THR-β (такого как ожирение, гиперлипидемия, гиперхолестеринемия, диабет, стеатогепатит, неалкогольный стеатогепатит, неалкогольная жировая болезнь печени, атеросклероз, рак щитовидной железы, гипотиреоз). В качестве альтернативы, в настоящей заявке предложено указанное выше соединение, его фармацевтически приемлемая соль или его пролекарство для предотвращения или лечения заболевания, связанного с действием агониста β-рецептора. В качестве альтернативы, в настоящей заявке предложен способ предотвращения или лечения заболевания, связанного с агонизмом THR-β, включающий введение указанного выше соединения, его фармацевтически приемлемой соли или пролекарства нуждающемуся в этом субъекту. Предпочтительно заболевание, связанное с действием агониста β-рецептора, включает, не ограничиваясь перечисленным: гиперхолестеринемию, гиперлипидемию, гипертриглицеридемию, семейную гиперхолестеринемию, дислипидемию, рак щитовидной железы, гипотиреоз, первопричинный гипотиреоз, атеросклероз, метаболический синдром, ожирение, диабет, сердечно-сосудистые заболевания, ишемическая болезнь сердца, инфаркт миокарда, желудочковую недостаточность, сердечную недостаточность, жировой гепатоз, цирроз печени, неалкогольный стеатогепатит (НАСГ), неалкогольную жировую болезнь печени (НАЖБП), депрессию, деменцию, остеопороз, алопецию, заболевания ногтей, кожные заболевания, заболевания почек, хроническую почечную недостаточность и/или рак, и т.п., в особенности гиперхолестеринемию, гиперлипидемию, гипертриглицеридемию, семейную гиперхолестеринемию, дислипидемию, атеросклероз, гипотиреоз и/или первопричинный гипотиреоз.
Определение
Если ниже не приведено других определений, все технические термины и научные термины, используемые в настоящем документе, имеют те же значения, которые обычно понимаются специалистами в данной области техники. Технологии, упомянутые в настоящем документе, относится к технологиям, обычно понимаемым в данной области техники, включающим технические изменения, очевидные для специалистов в данной области техники, или эквивалентные технические замены. Хотя считается, что следующие термины хорошо понятны специалистам в данной области техники, тем не менее, ниже приведены определения для лучшего пояснения настоящей заявки.
В контексте настоящего документа термины «содержащий», «включающий», «имеющий» или «задействующий» и другие их варианты в настоящем документе являются включительными или неисчерпывающими, и не исключают другие, не перечисленные элементы или стадии способа.
В контексте настоящего документа термин «водород» и водород в каждой группе охватывает его природный изотоп, такой как протий (Р), дейтерий (D) или тритий (Т).
«Алкил» представляет собой органическую группу с линейной или разветвленной цепью, содержащую только атомы углерода и атомы водорода. Примеры алкила включают линейный или разветвленный С1-10, предпочтительно C1-6 и более предпочтительно С1-4 алкил, такой как C1, С2, С3, С4, С5, С6, С7, C8, С9 или С10 алкил, и в особенности такой как метил, этил, пропил, изопропил, бутил, изобутил, 1-метилпропил, пентил, гексил и т.п.
«Галоген» включает атом фтора, атом хлора, атом брома и атом иода.
«Циклоалкил» включает моноциклическое, бициклическое или трициклическое неароматическое С3-14, предпочтительно С3-10 и более предпочтительно С6-10 карбоциклическое кольцо, которое необязательно является частично или полностью насыщенным.
«Гетероциклил» включает моноциклическое, бициклическое или трициклическое неароматическое карбоциклическое кольцо, или циклоалканы, содержащие один или более (например, от 1 до 5, от 1 до 4, от 1 до 3 или от 1 до 2) гетероатомов, выбранных из атома фосфора, атома азота, атома кислорода и атома серы (в особенности атома азота, атома кислорода и атома серы). В качестве примера, «гетероциклил» включает 5-12-членное моноциклическое или бициклическое неароматическое карбоциклическое кольцо, или циклоалканы, содержащие от 1 до 4 гетероатомов, выбранных из атома азота, атома кислорода и атома серы, которые необязательно являются частично или полностью насыщенными.
«Арил» относится к полностью углеродной моноциклической или полициклической с конденсированными кольцами ароматической группе с сопряженной системой π-электронов. Например, в контексте настоящего документа термин «С6-14 арил» относится к ароматической группе, содержащей от 6 до 14 атомов углерода, такой как фенил или нафтил. Арил необязательно замещен одним или более (например, 1-3) подходящими заместителями (такими как галоген, -ОН, -CN, -NO2, С1-6 алкил и т.п.).
«Гетероарил» представляет собой ароматическую циклическую группу, содержащую по меньшей мере один гетероатом (азот, кислород или серу) и атом углерода, и включает 5- или 6-членное моноциклическое соединение, 8-10-членную бициклическую группу, в которой конденсированы одинаковые или разные моноциклические гетероароматические кольца, и 8-10-членную бициклическую группу, в которой моноциклическое гетероароматическое кольцо конденсировано с бензолом. Конкретные примеры гетероарила включают фурил, тиенил, пирролил, имидазолил, пиразолил, триазолил, тетразолил, оксазолил, тиазолил, изоксазолил, изотиазолил, оксадиазолил, тиадиазолил, фуразанил, пиридил, пиразинил, пиримидинил, пиридазинил, триазинил, индолил, индазолил, бензимидазолил, пуринил, хинолил, изохинолил, нафтиридинил, хиноксалил, хиназолинил, циннолинил, бензофуранил, бензотиенил, бензоксазолил, бензотиазолил, бензизоксазолил, бензизотиазолил и т.п.
В контексте настоящего документа термин «замещение» относится к тому, что один или более (например, один, два, три или четыре) атомов водорода на указанном атоме замещены вариантами из указанных групп, при условии, что нормальная валентность указанного атома в текущей ситуации не превышена и в результате замещения образуется стабильное соединение. Комбинация заместителей и/или переменных допускается только в том случае, если такая комбинация образует стабильное соединение.
Если заместитель описан как «необязательно замещенный», заместитель может быть (1) незамещенным или (2) замещенным. Если атом углерода заместителя описан как необязательно замещенный одним или более из списка заместителей, один или более атомов водорода при указанном атоме углерода (в случае существования какого-либо атома водорода) могут быть необязательно замещены отдельно и/или совместно независимо выбранными заместителями. Если атом азота заместителя описан как необязательно замещенный одним или более из списка заместителей, один или более атомов водорода при указанном атоме азота (в случае существования какого-либо атома водорода) могут быть необязательно замещены соответственно независимо выбранными заместителями.
«Необязательно замещенный» может относиться к замещению 1-5, предпочтительно 1-3, заместителями, и указанные заместители включают (1) алкил, замещенный 1-3 заместителями, выбранными из галогена, гидроксила, карбоксила, амино, арила, гетероарила, циклоалкила и гетероциклила, (2) карбоциклил, замещенный 1-3 заместителями, выбранными из алкила, галогена, гидроксила, карбоксила, галогеналкила, алкокси, галогеналкокси, алканоила и циано, (3) гетероциклил, замещенный 1-3 заместителями, выбранными из алкила, галогена, гидроксила, карбоксила, галогеналкила, алкокси, галогеналкокси, алканоила и циано, (4) арил, замещенный 1-3 заместителями, выбранными из алкила, галогена, гидроксила, карбоксила, галогеналкила, алкокси, галогеналкокси, алканоила и циано, (5) гетероарил, замещенный 1-3 заместителями, выбранными из алкила, галогена, гидроксила, карбоксила, галогеналкила, алкокси, галогеналкокси, алканоила и циано, (6) гидроксила, (7) алкокси, (8) галогена, (9) амино, необязательно замещенный 1 или 2 алкильными группами, и (10) окси.
Настоящая заявка также охватывает все фармацевтически приемлемые и изотопно-меченые соединения, которые являются такими же, как соединение согласно настоящей заявке, за исключением того, что один или более атомов замещены атомами с тем же атомным числом, что и преобладающее в природе, но другой атомной массой или массовым числом. Примеры изотопов, подходящих для включения в состав соединения согласно настоящей заявке, включают (не ограничиваясь перечисленным) изотопы водорода (такие как дейтерий (2Н) и тритий (3Н)); изотопы углерода (такие как 11С, 13С и 14С); изотопы хлора (такие как 36Cl); изотопы фтора (такие как 18F); изотопы иода (такие как 123I и 125I); изотопы азота (такие как 13N и 15N); изотопы кислорода (такие как 15O,17O и 18O); изотопы фосфора (такие как 32Р); и изотопы серы (такие как 35S). Некоторые изотопно-меченые соединения согласно настоящей заявке (такие как соединения, которые легированы радиоизотопами) могут быть использованы в исследованиях распределения лекарственного средства и/или субстрата в ткани (таких как анализ). Радиоизотопы тритий (3Н) и углерод-14 (14С) особенно подходят для этой цели благодаря простоте легирования и детектирования. Замена изотопами с позитронной эмиссией (такими как 11C, 18F, 15O и 13N) может быть использована для изучения степени связывания субстрата с рецептором методом позитронно-эмиссионной томографии (ПЭТ).
Структура, описанная в настоящем документе, также охватывает все изомерные (например, энантиомерные, диастереомерные и геометрические (или конформационные)) формы структуры, такие как R- и S-конфигурация каждого центра асимметрии, (Z)- и (Е)-изомер расположения относительно двойной связи и (Z)- и (Е)-конформационный изомер. Таким образом, отдельные стереохимические изомеры и энантиотопные, диастереомерные и геометрические (или конформационные) смеси этих соединений входят в объем настоящей заявки. Если не указано иное, все таутомерные формы соединения согласно настоящей заявке входят в объем настоящей заявки. Кроме того, если не указано иное, структура, описанная в настоящем документе, также охватывает все соединения, которые различаются только наличием одного или более изотопно-обогащенных атомов.
Настоящим документом охватываются все возможные кристаллические формы или полиморфные вещества соединения согласно настоящей заявке, которые могут представлять собой одну кристаллическую форму или смесь более чем одного полиморфного вещества в любой пропорции.
Следует также иметь в виду, что некоторые соединения согласно настоящей заявке могут существовать в свободной форме для лечения или, при необходимости, существовать в форме фармацевтически приемлемых производных. В настоящей заявке фармацевтически приемлемые производные включают, не ограничиваясь перечисленным, фармацевтически приемлемую соль, сольват, N-оксид, метаболит или пролекарство, и после введения производных нуждающимся в этом пациентам прямо или косвенно обеспечивается соединение согласно настоящей заявке или его метаболит или остаток. Следовательно, когда в настоящем документе упоминается «соединение согласно настоящей заявке», оно также охватывает вышеуказанные различные производные формы соединения.
Фармацевтически приемлемая соль соединения согласно настоящей заявке включает его кислотно-аддитивную соль и основно-аддитивную соль, при этом типы соли как-либо конкретно не ограничены, при условии, что соль является физиологически приемлемой. Примеры подходящей фармацевтически приемлемой кислотно-аддитивной соли включают, не ограничиваясь перечисленным, гидрохлорид, гидробромид, сульфат, нитрат, фосфат, ацетат, трифторацетат, тартрат, фумарат, оксалат, малеат, цитрат, сукцинат, метансульфонат, бензолсульфонат, малат, аспартат, глюцептат, глюконат, оротат, пальмитат и другие подобные соли. Примеры подходящей фармацевтически приемлемой основно-аддитивной соли включают, не ограничиваясь перечисленным, натриевую соль, калийную соль, аммониевую соль, кальциевую соль, магниевую соль, алюминиевую соль, соль железа, гистидиновую соль, аргининовую соль, холиновую соль и другие подобные соли.
Соединение согласно настоящей заявке может существовать в форме сольвата (предпочтительно гидрата), при этом соединение согласно настоящей заявке содержит полярный растворитель в качестве структурного элемента кристаллической решетки соединения, такой как, вода, метанол или, в особенности, этанол. Количество полярного растворителя, в особенности воды, может находиться в стехиометрическом или нестехиометрическом соотношении.
Специалистам в данной области техники может быть понятно, что, поскольку азот должен иметь доступную неподеленную пару электронов для окисления до оксида, не все азотсодержащие гетероциклы могут образовывать N-оксид. Специалистам в данной области техники может быть понятно, какой азотсодержащий гетероцикл будет способен образовывать N-оксид. Специалистам в данной области техники также может быть понятно, что третичный амин может образовывать N-оксид. Способ синтеза N-оксида гетероцикла и третичного амина хорошо известен специалистам в данной области техники и включает окисление гетероцикла и третичного амина пероксикислотой, такой как перуксусная кислота и м-хлорпероксибензойная кислота (МСРВА), пероксидом водорода, алкилпероксидом водорода, таким как трет-бутилгидропероксид, перборатом натрия и диоксираном, таким как диметилдиоксиран.
В объем настоящей заявки также входит метаболит соединения согласно настоящей заявке, такой как вещество, образованное in vivo при введении соединения согласно настоящей заявке. Такой продукт может быть получен, например, окислением, восстановлением, гидролизом, амидированием, дезамидированием, этерификацией, ферментолизом и тому подобным, вводимого соединения. Следовательно, настоящая заявка охватывает метаболит соединения согласно настоящей заявке и охватывает соединение, полученное способом приведения соединения согласно настоящей заявке в контакт с млекопитающим в течение времени, достаточного для получения метаболита.
Настоящая заявка также охватывает пролекарство соединения согласно настоящей заявке в объеме настоящей заявки, которое может быть превращено в соединение согласно настоящей заявке с желаемой активностью, например, путем гидролитического расщепления, когда некоторые производные соединения согласно настоящей заявке с небольшой или отсутствующей фармакологической активностью вводят в организм или применяют к нему наружно. Как правило, такое пролекарство может представлять собой функциональное производное соединения, которое легко превращается в соединение с желаемой терапевтической активностью in vivo.
«Фармацевтически приемлемый носитель» в настоящей заявке относится к фармакологически и фармацевтически приемлемой добавке, вводимой вместе с активным ингредиентом, и может быть использовано вспомогательное вещество, разрыхлитель, адгезив, смазывающее вещество, покрывающий агент, краситель, разбавитель, основный агент, изотонический агент и т.п.
Лекарственные формы включают, не ограничиваясь перечисленным, таблетку, капсулу, пастилку, леденец, порошок, спрей, крем, мазь, каплю, суппозиторий, гель, пасту, лосьон, водную суспензию, инъекционный раствор, эликсир и сироп.
Примеры лекарственных форм, подходящих для перорального введения, включают таблетку, капсулу, порошок, мелкую гранулу, гранулу, жидкость, сироп и т.п. Примеры лекарственных форм, подходящих для неперорального введения, включают инъекцию, каплю, суппозиторий и т.п.
В тексте, если не указано иное, термины в единственном числе включают объекты во множественном числе, и наоборот.
Если не указано иное, термин «субъект» может использоваться взаимозаменяемо с терминами «индивидуум» и «пациент» и включает позвоночного, такого как птицы, рыбы и млекопитающие, включая, не ограничиваясь перечисленным, мышей, крыс, морских свинок, собак, свиней, овец, крупный рогатый скот, кур, кроликов, обезьян (таких как макаки-резусы), людей и т.п.
В тексте, если не указано иное, все числа, используемые в настоящем документе для обозначения количеств ингредиентов, измеренных значений или условий реакций, следует понимать как во всех случаях модифицированные термином «приблизительно» для обозначения возможных погрешностей измерения. Например, при использовании вместе с процентом термин «приблизительно» может относиться к ±1%.
Соединение формулы I согласно настоящей заявке проявляет действие агониста β-рецептора тиреоидных гормонов и может представлять собой лекарственное средство для предотвращения или лечения заболевания, регулируемого β-рецептором, например, применяться для предотвращения, уменьшения и/или лечения следующих заболеваний: гиперхолестеринемия, гиперлипидемия, гипертриглицеридемия, семейная гиперхолестеринемия, дислипидемия, рак щитовидной железы, гипотиреоз, первопричинный гипотиреоз, атеросклероз, метаболический синдром, ожирение, диабет, сердечно-сосудистые заболевания, ишемическая болезнь сердца, инфаркт миокарда, желудочковая недостаточность, сердечная недостаточность, жировой гепатоз, цирроз печени, неалкогольный стеатогепатит (НАСГ), неалкогольная жировая болезнь печени (НАЖБП), депрессия, деменция, остеопороз, алопеция, заболевания ногтей, кожные заболевания, заболевания почек, хроническая почечная недостаточность и/или рак, и т.п., в особенности гиперхолестеринемия, гиперлипидемия, гипертриглицеридемия, семейная гиперхолестеринемия, дислипидемия, атеросклероз, гипотиреоз и/или первопричинный гипотиреоз, и т.п.
ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На ФИГ. 1 показаны результаты оценки фиброза.
На ФИГ. 2 показаны результаты оценки по шкале NAS.
ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ
Для того, чтобы сделать объекты и технические решения согласно настоящей заявке более ясными, настоящая заявка дополнительно проиллюстрирована ниже со ссылкой на конкретные варианты осуществления. Следует иметь в виду, что эти варианты осуществления использованы только для иллюстрации настоящей заявки и не имеют ограничительного характера в отношении объема настоящей заявки. Кроме того, конкретные экспериментальные методы, не упомянутые в следующих вариантах осуществления, проводят в соответствии с обычными экспериментальными методами.
Пример 1. Синтез ключевого промежуточного продукта KH01
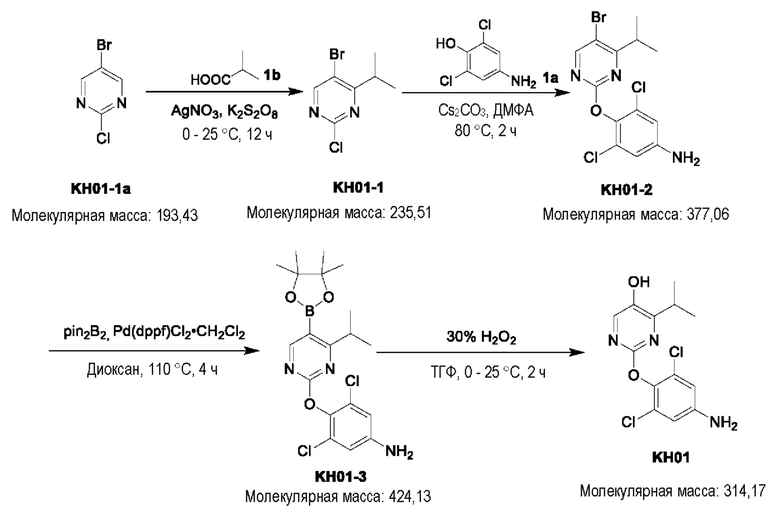
Соединение KH01-1: исходные вещества 2-хлор-5-бромпиримидин (KH01-1a) (100 г, 516 ммоль, 1,00 экв.), изомасляную кислоту (1b) (36,4 г, 413 ммоль, 38,3 мл, 0,80 экв.), персульфат калия (111 г, 413 ммоль, 82,8 мл, 0,80 экв.) и нитрат серебра (17,5 г, 103 ммоль, 0,20 экв.) добавляли в круглодонную колбу при 0°С, затем добавляли 1 л дихлорметана и 1 л воды. После равномерного перемешивания некоторые твердые вещества не растворились, и затем смесь перемешивали при комнатной температуре (25°С) в течение 12 часов под защитой азота. Контроль с помощью ТСХ (тонкослойная хроматография) показал, что сырье полностью прореагировало и образовалось новое пятно. Реакционную смесь фильтровали и дважды промывали фильтровальный осадок дихлорметаном (2×1 л). Фильтрат собирали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (100-200 меш) (петролейный эфир/этил ацетат = 100:1) с получением 54,0 г вещества KH01-1 в виде масла, выход: 44,3%. ЖХ-МС:МС (ESI) m/z=236,9 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3): δ 8,58 (s, 1H), 3,48 -3,41 (m, 1H), 1,29 (d, J=6,8 Гц, 6H).
Соединение KH01-2: реакционную смесь KH01-1 (15,0 г, 63,6 ммоль, 1,00 экв.), 1a (11,3 г, 63,6 ммоль, 1,00 экв.) и карбоната цезия (62,2 г, 191 ммоль, 3,00 экв.) в N,N-диметилформамиде (150 мл) трижды подвергали замене азотом при перемешивании, затем нагревали при внешней температуре 80°С в течение 2 часов под защитой азота. Реакцию контролировали с помощью ТСХ до ее завершения. Реакционную смесь выливали в 100 мл воды и перемешивали до образования прозрачного раствора. Реакционную смесь экстрагировали этилацетатом (2 × 100 мл). Этилацетатные слои объединяли, промывали насыщенным солевым раствором (2 × 100 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали и очищали остаток с помощью колоночной хроматографии на силикагеле (100-200 меш) (петролейный эфир/этил ацетат = 1:2) с получением 13,0 г KH01-2 в виде твердого желтого вещества, выход: 54,1%. ЖХ-МС: МС (ESI) m/z=378,0 [М+Н]+. 1H ЯМР (400 МГц, CDCl3): δ 8,46 (s, 1H), 6,68 (s, 2H), 3,78 (s, 2H), 3,43 - 3,36 (m, 1H), 1,22 (d, J=6,8 Гц, 6H).
Соединение KH01-3: реакционную смесь KH01-2 (10,0 г, 26,5 ммоль, 1,00 экв.), бис(пинаколато)дибора (pin2B2) (13,4 г, 53,0 ммоль, 2,00 экв.), N,N-бис(дифенилфосфино)ферроцен-палладий(II)дихлорида, комплекса с дихлорметаном (1,08 г, 1,33 ммоль, 0,05 экв.) и ацетата калия (5,21 г, 53,0 ммоль, 2,00 экв.) в диоксане (100 мл) трижды подвергали замене азотом, затем нагревали при внешней температуре 110°С в течение 4 часов под защитой азота. ТСХ показала, что сырье полностью прореагировало. К реакционной смеси добавляли 200 мл воды и экстрагировали этилацетатом (3 × 200 мл). Этилацетатные слои объединяли, промывали насыщенным солевым раствором (300 мл × 2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением 12,0 г неочищенного продукта в виде коричневого масла, которое сразу же использовали на следующей стадии без дополнительной очистки. ЖХ-МС: МС (ESI) m/z=425,2 [М+Н]+.
Соединение KH01: неочищенный KH01-3 (12,0 г, 28,2 ммоль, 1,00 экв.), полученный выше, и 30,0% пероксид водорода (6,74 г, 59,4 ммоль, 5,71 мл, 2,10 экв.) растворяли в тетрагидрофуране (120 мл) при внешней температуре 0°С. Смесь перемешивали при комнатной температуре (25°С) в течение 2 часов под защитой азота. ТСХ показала, что реакция была завершена. Реакционную смесь гасили 50 мл 2 М раствора сульфита натрия, затем экстрагировали дихлорметаном (3×5 мл). Объединенные органические слои сушили над безводным сульфатом натрия, концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (100-200 меш) (петролейный эфир/этил ацетат = 1:2) с получением 4,80 г твердого желтого вещества, выход: 53,4%. ЖХ-МС: МС (ESI) m/z=314,1 [М+Н]+. 1Н ЯМР (400 МГц, ДМСО-d6): δ 9,85 (br s, 1H), 7,95 (s, 1H), 6,66 (s, 2Н), 5,52 (s, 2Н), 3,31 -3,18(m, 1H), 1,10 (d, J=6,8 Гц, 6Н).
Пример 2. Синтез соединения KH02
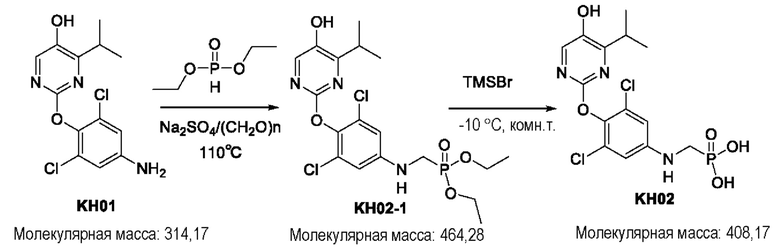
Соединение KH02-1: к смеси KH01 (0,152 г, 0,485 ммоль), диэтилфосфита (0,104 г, 0,754 ммоль), параформальдегида (0,095 г, 1,055 ммоль) и сульфата натрия (0,156 г, 1,098 ммоль) в одногорлой колбе добавляли толуол (8 мл) под защитой N2. Частично растворенную реакционную смесь нагревали при внешней температуре 110°С течение 3 часов. ТСХ показала, что реакция была завершена и образовалось новое пятно с повышенной полярностью. К реакционной смеси добавляли 100 мл воды и экстрагировали этилацетатом (3 × 50 мл). Органические фазы собирали, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этил ацетат = 1:2) с получением KH02-1 в виде бесцветного масла. (142 мг, 63%). ЖХ-МС (ESI, m/z): 465,3 [М+Н]+.
Соединение KH02: соединение KH02-1 (0,142 г, 0,306 ммоль) добавляли в одногорлую колбу и растворяли в дихлорметане (10 мл) под защитой N2. При внешней температуре -10°С к реакционной системе медленно по каплям добавляли триметилбромсилан (3,2 мл) и оставляли реагировать при этой температуре в течение 30 минут, после чего температуру медленно повышали до комнатной температуры для проведения реакции в течение ночи. ЖХ-МС показала, что сырье полностью прореагировало. Реакционную смесь сразу же концентрировали с помощью ротационного испарения. Остаток очищали с помощью препаративной ВЭЖХ с получением после лиофилизации соединения KH02 в виде бледно-желтого твердого вещества (55 мг, 44%). 1Н ЯМР (400 МГц, ДМСО): δ 9,87 (s, 1H), 7,95 (s, 1Н), 6,82 (s, 2Н), 4,61 - 5,22 (m, 3Н), 3,27 (m, 3Н), 1,12 (d, J=4 МГц, 6Н). ЖХ-МС (ESI, m/z): 408,0 [М+1]+.
Пример 3. Синтез соединения KH03
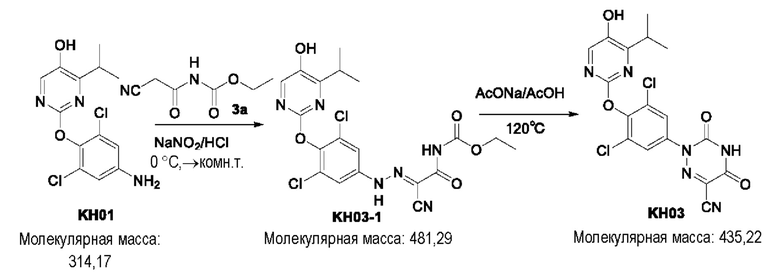
Соединение KH03-1: соединение KH01 (0,265 г, 0,846 ммоль) добавляли к 11,2 мл воды, добавляли 5,6 мл концентрированной соляной кислоты при внешней температуре 0°С, затем навеску нитрита натрия (0,072 г, 1,043 ммоль) растворяли в 0,8 мл воды, медленно по каплям добавляли к реакционному раствору и перемешивали при 0°С в течение 1,5 часов с получением смеси. Дополнительно взвешивали соединение 3а (0,148 г, 0,948 ммоль) и растворяли в 19,4 мл воды, добавляли 5,6 мл пиридина при 0°С, перемешивали в течение 1,5 часов при этой температуре и затем быстро добавляли к вышеупомянутому реакционному раствору при 0°С. На данном этапе образовалось оранжевое твердое вещество, и затем реакционный раствор медленно нагревали до комнатной температуры (25°С) и оставляли непрерывно реагировать в течение ночи. После того, как контроль с помощью ТСХ показал, что реакция была завершена, твердое вещество сразу же фильтровали на фильтровальной воронке и трижды промывали 50 мл воды и РЕ (петролейный эфир), соответственно. Было собрано и получено оранжевое твердое вещество KH03-1 (380 мг, 93,8%). ЖХ-МС (ESI, m/z): 482,3 [М+1]+.
Соединение KH03: соединение KH03-1 (0,380 г, 0,931 ммоль) и ацетат натрия (0,650 г, 7,926 ммоль) добавляли в одногорлую колбу и растворяли в уксусной кислоте (10 мл) под защитой N2. Реакцию проводили в течение 3 часов при внешней температуре 120°С. После того, как контроль с помощью ТСХ показал, что сырье полностью прореагировало, реакцию останавливали. Реакционный раствор охлаждали до 0°С и добавляли 100 мл воды, затем осаждалось большое количество твердого вещества, которое сразу же фильтровали через фильтровальную воронку и промывали твердое вещество водой и РЕ, соответственно (3 × 50 мл). Было собрано и получено оранжевое твердое вещество KH03 (230 мг, 66,9%). 1H ЯМР (400 МГц, ДМСО) δ 10,09 (s, 1H), 8,01 (s, 1H), 7,75 (s, 2Н), 3,27 - 3,28 (m, 1H), 1,13 (d, J=4 МГц, 6Н). ЖХ-МС (ESI, m/z): 435,2 [М+1]+.
Пример 4. Синтез соединения KH04
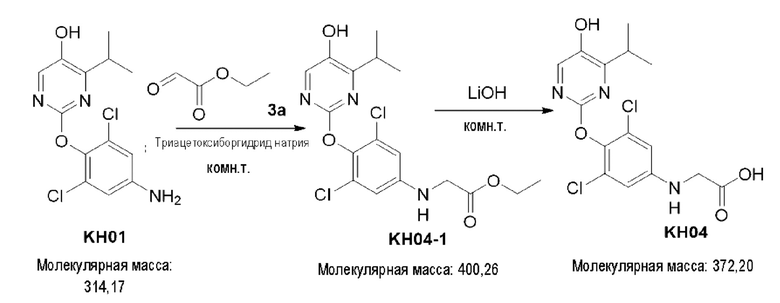
Соединение KH04-1: соединение KH01 (0,0512 г, 0,1635 ммоль), этилглиоксилат (0,0274 г, 0,268 ммоль) и триацетоксиборгидрид натрия (0,1023 г, 0,483 ммоль) взвешивали и добавляли в одногорлую колбу, а затем добавляли 1,2-дихлорэтан (3 мл) для растворения. Реакцию проводили при внешней температуре 75°С в течение 3 часов, контроль с помощью ТСХ показал, что сырье полностью прореагировало и образовалось новое пятно повышенной полярности. Затем реакцию останавливали. К реакционному раствору добавляли 50 мл дихлорметана и 100 мл воды. После перемешивания в течение 10 минут органические фазы разделяли, водные фазы экстрагировали дихлорметаном (3×50 мл), затем органические фазы объединяли, сушили с помощью безводного сульфата натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматопластины (петролейный эфир/этил ацетат = 1:2) с получением бесцветного маслянистого вещества KH04-1 (50 мг, 78,5%). ЖХ-МС (ESI, m/z): 401,3 [М+1]+.
Соединение KH04: соединение KH04-1 (50 мг, 0,125 ммоль) и гидроксид лития (35 мг, 1,458 ммоль) добавляли в одногорлую колбу и добавляли тетрагидрофуран/метанол/воду (4:1:1, 6 мл) для растворения. Реакцию проводили при комнатной температуре в течение ночи. После того, как контроль с помощью ТСХ показал, что сырье полностью прореагировало, реакцию останавливали. Добавляли 20 мл воды для разбавления реакционного раствора, органический растворитель удаляли при пониженном давлении и доводили рН реакционной смеси до 3-4 при 0°С. К реакционному раствору добавляли дихлорметан (50 мл × 3) для экстракции, собирали органические фазы, сушили с помощью безводного Na2SO4, фильтровали и концентрировали для удаления растворителя. Остатки очищали с помощью хроматопластины (дихлорметан/метанол = 5:1). Целевой продукт собирали и лиофилизировали с получением белого твердого вещества KH04 (24 мг, 51,6%). 1H ЯМР (400 МГц, ДМСО) δ 10,48 (s, 1H), 8,00 (s, 1H), 6,65 (s, 2Н), 5,98 (s, 1H), 4,21 - 4,22 (m, 1H), 3,49 (s, 1H), 3,34 - 3,19 (m, 2Н), 1,12 (d, J=4 МГц, 6Н). ЖХ-МС (ESI, m/z): 372,1 [М+1]+.
Пример 5. Синтез соединения KH05
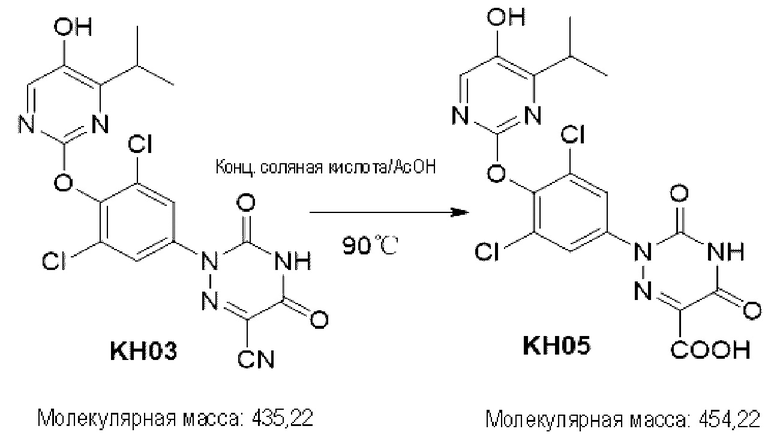
100 мг соединения KH03 растворяли в 5 мл уксусной кислоты, добавляли 1 мл концентрированной соляной кислоты и перемешивали при внешней температуре 90°С в течение 4 часов. После того, как контроль с помощью ТСХ показал, что сырье полностью прореагировало, реакционный раствор подвергали декомпрессии, сушили с помощью ротационного испарения и доводили рН до 9-10 путем добавления насыщенного раствора карбоната натрия. После того, как реакционный раствор экстрагировали с помощью 50 мл этилацетата, органические фазы отбрасывали, водные фазы доводили до рН=3-4 и экстрагировали этилацетатом (50 мл × 3), а затем органические фазы объединяли, сушили с помощью безводного сульфата натрия и концентрировали с получением 70 мг белого твердого вещества. Выход составил 67,1%. ЖХ-МС (ESI, m/z): 455,3 [М+1]+.
Пример 6. Синтез соединения KH06
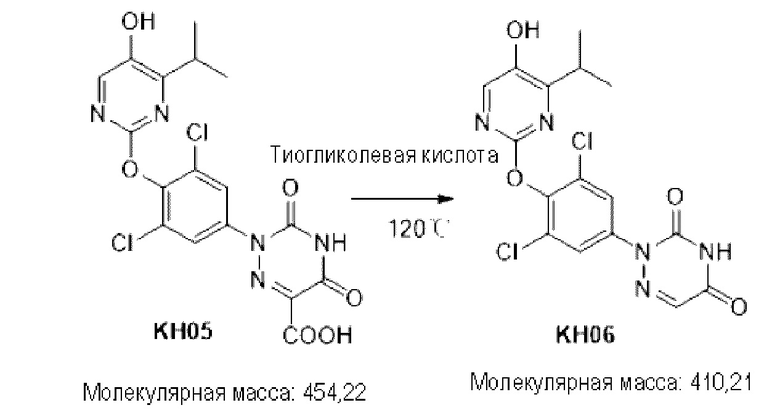
KH03 (60 мг) растворяли в 4 мл тиогликолевой кислоты и перемешивали при внешней температуре 120°С под защитой азота в течение 6 часов, контроль с помощью ТСХ показал, что образовалось пятно низкой полярности. В реакционный раствор добавляли насыщенный тиосульфат натрия для остановки реакции и использовали эти л ацетат для экстракции (25 мл × 3), органические фазы объединяли, сушили с помощью безводного сульфата натрия, концентрировали и очищали с помощью хроматопластины (дихлорметан/метанол=10:1). Целевой продукт собирали и лиофилизировали с получением приблизительно 1,5 мг белого твердого вещества KH06, выход: 27,8%. ЖХ-МС (ESI, m/z): 410,1 [М+1]+. 1H ЯМР (400 МГц, ДМСО) δ: 12,49 (s, 1H), 10,08 (s, 1H), 8,01 (s, 2Н), 7,71 (d, J=4 МГц, 1H), 3,27-3,33 (m, 1Н), 1,12 (d, J=4МГц, 6Н).
Пример 7. Синтез ключевого промежуточного продукта KH07-10
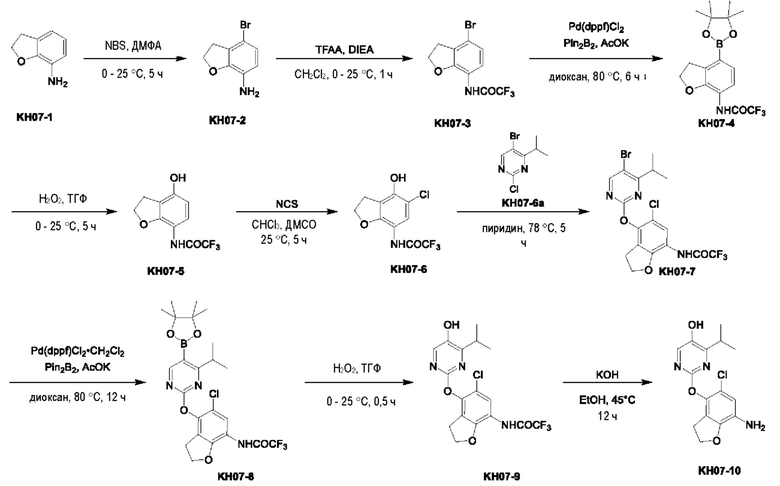
Соединение KH07-2: сырье KH07-1 (25,0 г, 184 ммоль, 1 экв.) растворяли в ДМФА (200 мл), медленно добавляли NBS (32,9 г, 184 ммоль, 1 экв.) при 0°С и после добавления перемешивали при внешней температуре 25°С в течение 5 часов. Контроль с помощью ЖХ-МС показал, что сырье полностью прореагировало и образовалось новое пятно (RT=0,483). Контроль с помощью ТСХ (петролейный эфир/этил ацетат=3/1) показал, что образовались два новых пятна. Реакционный раствор разбавляли водой (250 мл), а затем добавляли этилацетат (250 мл × 2) для экстракции, органические фазы объединяли, промывали насыщенным солевым раствором (250 мл × 2), сушили с помощью безводного сульфата натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 3/1) с получением желтого твердого вещества KH07-2 (29,1 г, 135 ммоль, выход: 73,5%). МС (ESI) m/z: 216,1 [М+Н]+. 1H ЯМР (ДМСО-d6, 400 МГц): 5 6,75 - 6,69 (m, 1H), 6,43 (d, J=8,4 Гц, 1H), 4,76 (s, 2H), 4,58 - 4,49 (m, 2H), 3,15 - 3,08 (m, 2H).
Соединение KH07-3: соединение KH07-2 (28,0 г, 130 ммоль, 1 экв.) и TFAA (32,9 г, 156 ммоль, 21,8 мл, 1,2 экв.) растворяли в дихлорметане (280 мл), медленно по каплям добавляли DIEA (33,8 г, 261 ммоль, 45,5 мл, 2 экв.) при внешней температуре 0°С, и после добавления перемешивали смесь при внешней температуре 25°С в течение 1 часа. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 5/1) показал, что сырье полностью прореагировало. Реакционный раствор выливали в воду (280 мл) и экстрагировали дихлорметаном (300 мл × 3), органические слои объединяли, промывали насыщенным солевым раствором (280 мл), сушили с помощью безводного сульфата натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 3/1) с получением желтого твердого вещества KH07-3 (27,8 г, выход: 68,5%). МС (ESI) m/z: 309,9 [М+Н]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 11,26 - 10,86 (m, 1Н), 7,15 - 7,10 (m, 1H), 7,09 - 7,03 (m, 1Н), 4,71 - 4,62 (m, 1H), 4,66 (t, J=8,8 Гц, 1H), 3,25 (t, J=8,8 Гц, 2Н).
Соединение KH07-4: соединение KH07-3 (27,0 г, 87,0 ммоль, 1 экв.), Pd(dppf)Cl2•CH2Cl2 (3,56 г, 4,35 ммоль, 0,05 экв.), Pin2B2 (55,2 г, 217 ммоль, 2,5 экв.) и ацетат калия (25,6 г, 261 ммоль, 3 экв.) добавляли в диоксан (270 мл) и перемешивали при внешней температуре 80°С в течение 6 часов под защитой азота. После того, как контроль с помощью ЖХ-МС показал, что сырье полностью прореагировало, реакционную смесь выливали в воду (500 мл) и экстрагировали этилацетатом (500 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (500 мл × 3), сушили с помощью безводного сульфата натрия, фильтровали и концентрировали с получением коричневого твердого вещества KH07-4 (33,0 г, неочищенный продукт), которое использовали на следующей стадии без очистки. МС (ESI) m/z: 358,1 [М+Н]+.
Соединение KH07-5: соединение KH07-4 (32,0 г, 89,6 ммоль, 1 экв.) растворяли в тетрагидрофуране (300 мл), медленно по каплям добавляли Н2О2 (30,4 г, 268 ммоль, 25,8 мл, чистота 30,0%, 3 экв.) при внешней температуре 0°С, и после добавления смесь перемешивали при внешней температуре 25°С в течение 5 часов. После того, как контроль с помощью ЖХ-МС показал, что сырье полностью прореагировало, реакционный раствор медленно выливали в насыщенный сульфит натрия (400 мл) для прекращения реакции, и затем экстрагировали этилацетатом (200 мл × 2), органические фазы объединяли, промывали насыщенным солевым раствором (100 мл × 2), сушили с помощью безводного сульфата натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 20/1 до 5/1) с получением белого твердого вещества KH07-5 (20,0 г, 80,9 ммоль, выход: 90,3%). МС (ESI) m/z: 248,1 [М+Н]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 10,6 (s, 1H), 9,63 (s, 1H), 6,87 (d, J=8,4 Гц, 1H), 6,31 (d, J=8,8 Гц, 1H), 4,55 (t, J=8,8 Гц, 2H), 3,09 (t, J=8,8 Гц, 2H).
Соединение KH07-6: соединение KH07-5 (10,0 г, 40,4 ммоль, 1 экв.) и NCS (6,48 г, 48,5 ммоль, 1,20 экв.) добавляли в смешанный растворитель из хлороформа (100 мл) и ДМСО (25,0 мл) и перемешивали при внешней температуре 25°С в течение 5 часов. После того, как контроль с помощью ЖХ-МС показал, что сырье полностью прореагировало, реакционный раствор сразу же концентрировали и очищали с помощью препаративной ВЭЖХ с получением белого твердого вещества KH07-6 (4,50 г, 15,9 ммоль, выход: 39,5%). МС (ESI) m/z: 282,0 [М+Н]+. 1H ЯМР (CDCl3, 400 МГц): δ 8,00 - 7,88 (m, 1H), 7,76 (br s, 1H), 5,57 (br s, 1H), 4,65 (t, J=8,8 Гц, 2Н), 3,24 - 3,18 (m, 2Н).
Соединение KH07-7: соединение KH07-6 (2,50 г, 8,88 ммоль, 1 экв.) и KH07-6а добавляли к 25 мл пиридина и перемешивали при внешней температуре 78°С в течение 5 часов. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 5/1) показал, что сырье полностью прореагировало. Реакционный раствор выливали в 60 мл воды и экстрагировали этилацетатом (100 мл × 3), органические фазы объединяли, промывали насыщенным солевым раствором (100 мл × 3); сушили с помощью безводного сульфата натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 5/1 до 3/1) с получением желтого твердого вещества KH07-7 (1,20 г, 2,50 ммоль, выход: 28,1%). МС (ESI) m/z: 482,0 [М+Н]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 11,20 -11,11 (m, 1H), 8,73 (s, 1Н), 7,47 - 7,35 (m, 1Н), 4,68 - 4,61 (m, 2Н), 3,39 - 3,34 (m, 1H), 3,15 - 3,06 (m, 2Н), 1,15 (d, J=6,8 Гц, 6Н).
Соединение KH07-8: соединение KH07-7 (1,05 г, 2,18 ммоль, 1 экв.), Pin2B2 (1,39 г, 5,46 ммоль, 2,5 экв.), АсОК (643 мг, 6,55 ммоль, 3 экв.) и Pd(dppf)Cl2•CH2Cl2 (107 мг, 131 ммоль, 0,06 экв.) добавляли в 10 мл диоксана, трижды при перемешивании подвергали замене азотом, после чего смесь непрерывно перемешивали при внешней температуре 80°С в течение 12 часов под защитой азота. Контроль с помощью ЖХ-МС показал, что некоторые сырьевые вещества оставались, и образовалось приблизительно 39,8% продукта. Реакционный раствор сразу же концентрировали, и остатки добавляли в 100 мл воды, перемешивали, экстрагировали этилацетатом (100 мл × 2), промывали насыщенным солевым раствором (100 мл × 2), сушили с помощью безводного сульфата натрия, фильтровали и концентрировали с получением коричневого твердого вещества (1,00 г, неочищенный продукт). МС (ESI) m/z: 528,1 [М+Н]+.
Соединение KH07-9: методика была такой же, как и в случае синтеза соединения KH07-5, и реакционный раствор очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 1/0 до 3/1) с получением белого твердого вещества KH07-9 (700 мг). МС (ESI) m/z: 418,0 [М+Н]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 11,12 (s, 1H), 10,00 (s, 1H), 8,00 (s, 1Н), 7,36 (s, 1Н), 4,62 (t, J=8,8 Гц, 2Н), 4,34 (t, J=5,2 Гц, 1Н), 3,30 - 3,26 (m, 1H), 3,02 (t, J=8,8 Гц, 2Н), 1,17- 1,10 (m, 1H), 1,14 (d, J=6,8 Гц, 6Н).
Соединение KH07-10: соединение KH07-9 (700 мг, 1,68 ммоль, 1 экв.) и гидроксид калия (376 мг, 6,70 ммоль, 4 экв.) добавляли в смешанный раствор этанола (4 мл) и воды (3 мл) и перемешивали при внешней температуре 45°С в течение 12 часов. Контроль с помощью ЖХ-МС показал, что сырье полностью прореагировало. Реакционный раствор доводили приблизительно до рН 7 с помощью 1 н. соляной кислоты, затем добавляли 50 мл воды, перемешивали в течение 10 минут, а затем экстрагировали этилацетатом (50 мл × два раза), органические фазы объединяли, промывали насыщенным солевым раствором (100 мл × 2), сушили с помощью безводного сульфата натрия, фильтровали и очищали с помощью препаративной ВЭЖХ с получением бледно-желтого твердого вещества KH07-10 (501,4 мг, выход: 93,0%, чистота 99,6%). МС (ESI) m/z: 322,2 [М+Н]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 9,81 (s, 1H), 7,95 (s, 1H), 6,71 - 6,35 (m, 1H), 5,75 (s, 1H), 4,78 (s, 2Н), 4,50 (br t, J=8,8 Гц, 2Н), 3,31 -3,21 (m, 1H), 2,90 (brt, J=8,8 Гц, 2Н), 1,12 (d, J=6,8 Гц, 6Н).
Пример 8. Синтез соединения KH07
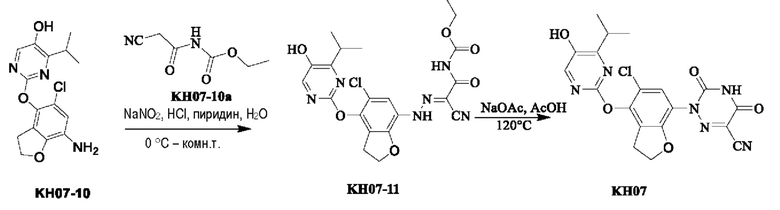
Соединение KH07-11: реакционный раствор А: соединение KH07-10 (202,5 мг, 0,637 ммоль) добавляли к 10 мл воды и добавляли 5,6 мл концентрированной соляной кислоты при 0°С. Навеску нитрита натрия (58,3 мг, 0,803 ммоль) растворяли в 1 мл воды, медленно по каплям добавляли к реакционному раствору и перемешивали при 0°С в течение 1,5 часов с получением раствора нитрита натрия; Реакционный раствор В: соединение KH07-10а (108,8 мг, 0,7 ммоль) добавляли к 20 мл воды, добавляли 5,6 мл пиридина при 0°С, а затем перемешивали при этой температуре в течение 1,5 часов. Затем реакционный раствор А быстро выливали в реакционный раствор В при 0°С с образованием оранжевого твердого вещества, и температуру медленно повышали до комнатной температуры для продолжения реакции в течение ночи. После того, как контроль с помощью ТСХ показал, что реакция была завершена, твердое вещество сразу же фильтровали и промывали водой и петролейным эфиром (25 мл × 3) соответственно. Было получено оранжевое твердое вещество KH07-11 (280 мг, выход: 89,9%, неочищенный продукт), которое сразу же использовали на следующей стадии без очистки. МС (ESI) m/z: 289,2 [М+Н]+.
Соединение KH07: соединение KH07-11 (280 мг, 0,573 ммоль) и ацетат натрия (485,4 мг, 5,73 ммоль) добавляли в одногорлую колбу и растворяли в уксусной кислоте (10 мл) под защитой N2. Реакцию проводили в течение 3 часов при внешней температуре 120°С. После того, как контроль с помощью ТСХ показал, что сырье полностью прореагировало, реакцию останавливали. Реакционный раствор охлаждали до 0°С и после добавления 50 мл воды осаждалось большое количество твердого вещества, которое сразу же фильтровали и промывали водой (20 мл × 3) и петролейным эфиром (20 мл × 3), соответственно. Оранжевое твердое вещество (254 мг, неочищенный продукт) собирали, и 50 мг неочищенного продукта очищали с помощью препаративной ВЭЖХ с получением грязно-белого твердого соединения KH07 (13,5 мг, 27,0%). МС (ESI) m/z: 443,0 [М+Н]+. 1H ЯМР (ДМСО-d6, 400 МГц): 10,06 (s, 1H), 7,40 (s, 1H), 4,65 (t, 2Н, J=8,0 Гц), 3,21-3,32 (m, 1H), 3,05 (t, 2Н, J=8,0 Гц), 1,16 (d, 6Н, J=8,0 Гц).
Пример 9. Синтез соединения KH08
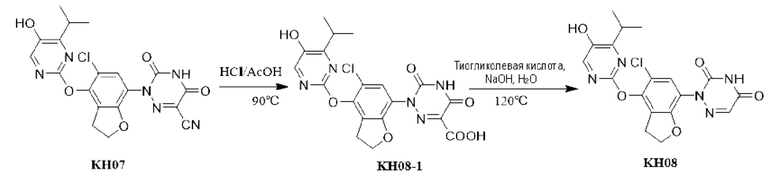
Соединение KH08-1: неочищенное соединение KH07 (200 мг, 0,452 ммоль) добавляли в одногорлую колбу, растворяли в уксусной кислоте (7,5 мл), а затем к реакционной смеси по каплям добавляли концентрированную соляную кислоту (2,5 мл). После проведения реакции в течение 4 часов при 90°С контроль с помощью ТСХ показал, что сырье полностью прореагировало, образовалось новое пятно повышенной полярности и реакцию останавливали. Реакционный раствор сразу же сушили с помощью ротационного испарения и доводили рН реакционной смеси до 9-10 насыщенным раствором карбоната натрия, затем дважды экстрагировали реакционный раствор этилацетатом (20 мл × 2). Водные фазы собирали, и рН водных фаз доводили до 3-4, затем водные фазы экстрагировали этилацетатом (20 мл × 3); органические фазы собирали, сушили с помощью безводного сульфата натрия и фильтровали. Органические фазы концентрировали с получением желтого твердого вещества KH08-1 (186,5 мг, 89,4%), которое сразу же использовали на следующей стадии без очистки.
Соединение KH08: соединение KH08-1 (150 мг, 0,325 ммоль, 1 экв.) и гидроксид натрия (52 мг, 1,3 ммоль, 4 экв.) добавляли в одногорлую колбу и растворяли в воде (20 мл), затем к реакционному раствору добавляли тиогликолевую кислоту (0,6 г, 6,5 ммоль, 20 экв.) и проводили реакцию при 120°С в течение 3 часов. После того, как контроль с помощью ТСХ показал, что сырье полностью прореагировало, образовалось пятно пониженной полярности и реакцию останавливали. Добавляли насыщенный раствор карбоната натрия для доведения рН реакционной системы до нейтрального, затем реакционную систему экстрагировали этилацетатом (20 мл × 3), сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью препаративной ВЭЖХ с получением белого твердого вещества KH08 (36,8 мг, 27,1%). МС (ESI) m/z: 418,2 [М+Н]+.
Пример 10. Синтез ключевого промежуточного продукта KH09-6
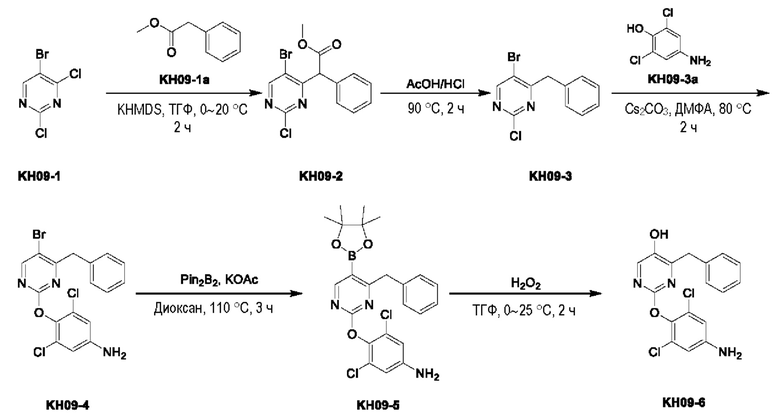
Соединение KH09-2: неочищенные KH09-1 (50,0 г, 219 ммоль, 28,0 мл, 1,00 экв.) и KH09-1а (32,9 г, 219 ммоль, 30,8 мл, 1,00 экв.) растворяли в тетрагидрофуране (200 мл), по каплям добавляли KHMDS (1,00 М, 230 мл, 1,05 экв.) при 0°С, после добавления перемешивали при этой температуре в течение 10 минут, а затем перемешивали при 25°С в течение 2 часов. После того, как контроль с помощью ТСХ (петролейный эфир/этилацетат = 10:1) показал, что сырье полностью прореагировало, реакционный раствор медленно выливали в ледяную воду и экстрагировали дихлорметаном (500 мл × 3), органические фазы объединяли, промывали насыщенным солевым раствором (300 мл), сушили с помощью безводного сульфата натрия и концентрировали с получением желтого маслянистого вещества KH09-2 (75,0 г, неочищенный продукт), которое сразу же использовали на следующей стадии без очистки. 1H ЯМР (ДМСО-d6, 400 МГц): δ 8,97 (s, 1H), 7,37 - 7,28 (m, 5Н), 7,26 - 7,20 (m, 1H), 5,64 (s, 1H), 3,68 - 3,64 (m, 3Н).
Соединение KH09-3: KH09-2 (75,0 г, 219 ммоль, 1,00 экв.) растворяли в растворе хлористого водорода в уксусной кислоте (100 мл) и перемешивали при внешней температуре 90°С в течение 2 часов. После того, как контроль с помощью ЖХ-МС показал, что сырье полностью прореагировало, реакционный раствор сразу же сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 100/1 до 20/1) с получением грязно-белого твердого вещества KH09-3 (23,0 г, 81,1 ммоль, выход: 36,9%). МС (ESI) m/z: 282,9 [М+Н]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 8,93 (s, 1H), 7,37-7,28 (m, 2Н), 7,28-7,21 (m, 3Н), 4,23 (s, 2Н).
Соединение KH09-4: соединение KH09-3 (10,0 г, 35,2 ммоль, 1,00 экв.) и соединение KH09-3а (6,28 г, 35,2 ммоль, 1,00 экв.) растворяли в 100 мл ДМФА, затем добавляли карбонат цезия (34,4 г, 105 ммоль, 3,00 экв.) и перемешивали при внешней температуре 80°С в течение 2 часов. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 100:1) показал, что сырье полностью прореагировало. Реакционный раствор фильтровали и промывали остатки на фильтре этилацетатом (20 мл). Фильтрат собирали, добавляли 50 мл воды и перемешивали в течение 5 минут, затем органические фазы разделяли и водные фазы экстрагировали этилацетатом (20 мл × 2). Органические фазы объединяли, промывали насыщенным солевым раствором (100 мл × 2), сушили с помощью безводного сульфата натрия, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 100/1 до 20/1) с получением белого твердого вещества KH09-4 (12,0 г, 28,2 ммоль, выход: 80,0%). 1H ЯМР (ДМСО-d6, 400 МГц): δ 8,75 (s, 1Н), 7,41-7,12 (m, 5Н), 6,68 (s, 2Н), 5,64 (s, 2Н), 4,14 (s, 2Н).
Соединение KH09-5: соединение KH09-4 (5,00 г, 11,7 ммоль, 1,00 экв.), Pin2B2(5,97 г, 23,5 ммоль, 2,00 экв.), Pd(dppf)Cl2⋅CH2Cl2 (480 мг, 588 мкмоль, 0,05 экв.) и ацетат калия (2,31 г, 23,5 ммоль, 2,00 экв.) добавляли в 50 мл диоксана, воздух в реакционном сосуде трижды заменяли азотом, затем реакционный раствор перемешивали при внешней температуре 110°С в течение 4 часов под защитой азота. После того, как контроль с помощью ТСХ показал, что сырье полностью прореагировало, реакционный раствор фильтровали, а фильтрат собирали, сразу же сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 100:1 до 50:1) с получением коричневого маслянистого вещества (5,00 г, неочищенный продукт, присутствовал продукт бороновой кислоты).
Соединение KH09-6: соединение KH09-5 (5,00 г, 10,5 ммоль, 1,00 экв.) растворяли в тетрагидрофуране, добавляли H2O2 (2,46 г, 21,7 ммоль, 2,08 мл, чистота 30%, 2,05 экв.) при 0°С и перемешивали в течение 30 минут, затем непрерывно добавляли H2O2 (4,80 г, 42,3 ммоль, 4,07 мл, чистота 30%, 4,00 экв.) и перемешивали при комнатной температуре в течение 2 часов. После того, как контроль с помощью ТСХ показал, что сырье полностью прореагировало, реакционный раствор выливали в 50 мл насыщенного раствора сульфита натрия и перемешивали в течение 15 минут; и экстрагировали дихлорметаном (200 мл × 3), органические фазы объединяли, сушили с помощью безводного сульфата натрия, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 100:1 до 10:1) с получением бледно-желтого твердого вещества KH09-6 (1,01 г, 2,74 ммоль, выход: 25,8%). МС (ESI) m/z: 362,0 [М+Н]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 10,03 (s, 1Н), 8,01 (s, 1H), 7,38-7,09 (m, 5Н), 6,66 (s, 2Н), 5,54 (s, 2Н), 3,97 (s, 2Н), 1,98 (s, 1Н).
Пример 11. Синтез соединения KH09
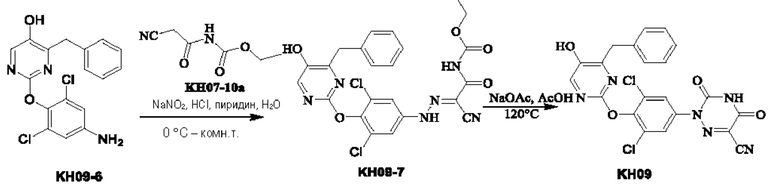
Соединение KH09-7: методика была такой же, как и в случае синтеза KH07-11, и было получено оранжевое твердое вещество KH09-7 (323 мг, неочищенный продукт), которое сразу же использовали на следующей стадии без очистки.
Соединение KH09: методика была такой же, как и в случае синтеза KH07, и было получено оранжевое твердое вещество KH09 (432 мг, неочищенный продукт). 50 мг твердого вещества отбирали и очищали с помощью препаративной ВЭЖХ с получением 6,12 мг белого твердого вещества, выход: 7,65%. МС (ESI) m/z: 482,9 [М+Н]+. 1Н ЯМР(ДМСО-d6, 400 МГц): δ 13,05-13,10 (m, 1Н), 10,26 (s, 1Н), 8,07 (s, 1H), 7,75 (s, 2Н), 7,18-7,30(m, 5Н), 4,00 (s, 1H).
Пример 12. Синтез соединения KH10
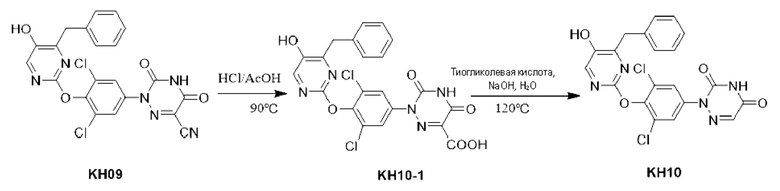
Соединение KH10-1: методика была такой же, как и в случае синтеза KH08-1, и было получено желтое твердое вещество (255,7 мг, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 502,0 [М+1]+.
Соединение KH10: методика была такой же, как и в случае синтеза KH08, и было получено белое твердое вещество KH10 (51 мг, выход: 20,0%). МС (ESI) m/z: 458,0 [М+1]+. 1Н ЯМР (ДМСО-d6, 400 МГц): 12,49(s, 1H), 10,25(s, 1H), 8,01(s, 1H), 7,77(s, 2Н), 7,72(d, 1H, m=4,0 Гц), 7,17-7,30(m, 5Н), 4,0(s, 2Н).
Пример 13. Синтез ключевого промежуточного продукта KH11-3
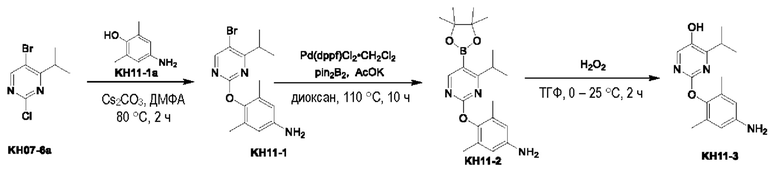
Соединение KH11-1: методика была такой же, как и в случае синтеза KH09-4, и было получено оранжевое твердое вещество KH11-1 (9,00 г, 26,7 ммоль, выход: 73,4%) путем очистки с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 1/0 до 1/2). МС (ESI) m/z: 338,1 [M+H]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 8,42 (s, 1Н), 6,43 (s, 2Н), 3,52 (br s, 2Н), 3,46-3,33 (m, 1Н), 2,04 (s, 6H), 1,25 (d, J=6,8 Гц, 6H).
Соединение KH11-2: соединение KH11-1 (5,00 г, 14,8 ммоль, 1,00 экв.), pin2B2 (7,55 г, 29,7 ммоль, 2,00 экв.), Pd(dppf)Cl2⋅CH2Cl2 (607 мг, 743 мкмоль, 0,05 экв.) и ацетат калия (2,92 г, 29,7 ммоль, 2,00 экв.) добавляли в 50 мл диоксана, воздух в реакционном сосуде трижды заменяли азотом, затем реакционный раствор перемешивали при внешней температуре 110°С в течение 10 часов под защитой азота. После того, как контроль с помощью ТСХ показал, что сырье полностью прореагировало, реакционный раствор сразу же сушили с помощью ротационного испарения, добавляли 50 мл воды и 50 мл этилацетата и перемешивали в течение 10 минут, органические фазы отделяли и водные фазы экстрагировали этилацетатом (50 мл × 2). Органические фазы объединяли, сушили с помощью безводного сульфата натрия, фильтровали и концентрировали с получением черного маслянистого вещества KH11-2 (6,00 г, неочищенный продукт), которое сразу же использовали на следующей стадии без очистки. МС (ESI) m/z: 384,5 [М+Н]+.
Соединение KH11-3: методика была такой же, как и в случае синтеза соединения KH09-6, и было получено грязно-белое твердое вещество (1,11 г, 2,79 ммоль, выход: 17,8%, соль с TFA) путем очистки с помощью препаративной ВЭЖХ (0,1% TFA). МС (ESI) m/z: 274,2 [М+Н]+. 1Н ЯМР (MeOD, 400 МГц): δ 7,93 (s, 1H), 7,12 (s, 2Н), 3,40 - 3,33 (m, 1H), 2,13 (s, 6Н), 1,11 (d, J=6,8 Гц, 6Н).
Пример 14. Синтез соединения KH11
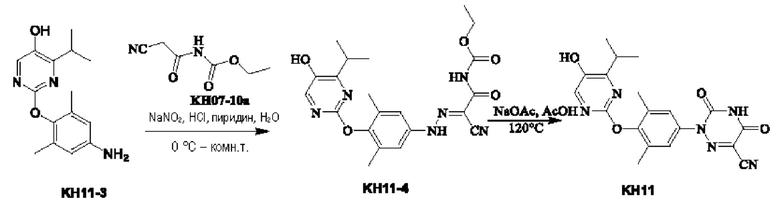
Соединение KH11-4: методика была такой же, как и в случае синтеза KH07-11, и было получено оранжевое твердое вещество KH11-4 (368,4 мг, неочищенный продукт), которое сразу же использовали на следующей стадии без очистки. МС (ESI) m/z: 441,3[М+Н]+.
Соединение KH11: методика была такой же, как и в случае синтеза KH07, и было получено оранжевое твердое вещество KH11 (342,3 мг, неочищенный продукт). 50 мг твердого вещества отбирали и очищали с помощью препаративной ВЭЖХ с получением 11,2 мг белого твердого вещества, выход: 23,2%. МС (EST) m/z: 395,2 [М+Н]+.
Пример 15. Синтез соединения KH12
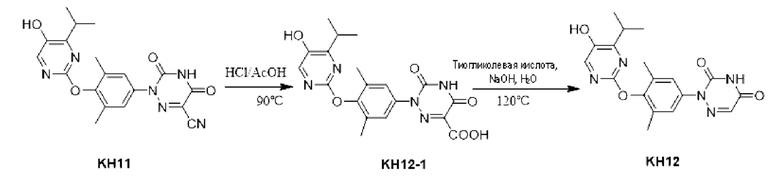
Соединение KH12-1: методика была такой же, как и в случае синтеза KH08-1, и было получено желтое твердое вещество (286 мг, неочищенный продукт), которое сразу же использовали на следующей стадии без очистки. МС (EST) m/z: 414,2 [М+Н]+.
Соединение KH10: методика была такой же, как и в случае синтеза KH08, и было получено белое твердое вещество KH10 (38,4 мг, выход: 15,2%). МС (EST) m/z: 370,1 [М+Н]+.
Вариант осуществления 16. Синтез ключевого промежуточного продукта KH13-7
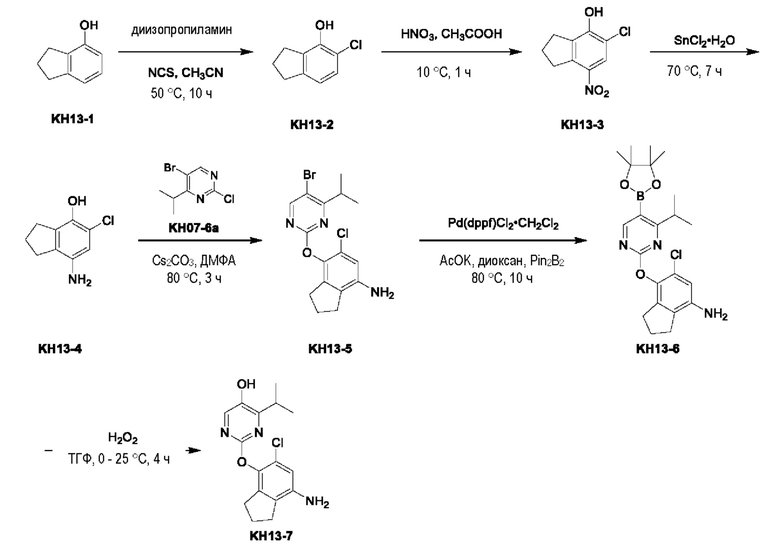
Соединение KH13-2: N,N-диизопропиламин (1,89 г, 18,6 ммоль, 2,63 мл, 0,1 экв.) и неочищенный KH13-1 (25,0 г, 186 ммоль, 1 экв.) растворяли в ацетонитриле (250 мл), а затем к смеси добавляли NCS (26,1 г, 195 ммоль, 1,05 экв.). Смесь перемешивали при 50°С в течение 10 часов. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 10/1) показал, что реакция была завершена. Реакционный раствор сушили с помощью ротационного испарения и разбавляли 100 мл воды, и экстрагировали этилацетатом (100 мл × 3), органические фазы объединяли, сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 1/0 до 20/1) с получением желтого твердого вещества KH13-2 (5,00 г, 29,6 ммоль, выход 15,9%). 1H ЯМР (ДМСО-d6, 400 МГц): δ 9,24 (s, 1H), 7,07 (d, J=8,0 Гц, 1H), 6,69 (d, J=8,0 Гц, 1 H), 2,85 - 2,77 (m, 4Н), 2,05 - 1,96 (m, 2Н).
Соединение KH13-3: соединение KH13-2 (3,70 г, 21,9 ммоль, 1 экв.) растворяли в этаноле (37 мл), добавляли азотную кислоту (2,42 г, 23,0 ммоль, 1,73 мл, чистота 60,0%, 1,05 экв.) при 10°С и непрерывно перемешивали при этой температуре в течение 1 часа. Контроль с помощью ТСХ показал, что сырье полностью исчезло. Смесь разбавляли 100 мл воды, а затем экстрагировали этилацетатом (50 мл × 3), органические фазы собирали, промывали насыщенным солевым раствором (100 мл), сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 1/0 до 10/1) с получением желтого твердого вещества KH13-3 (3,5,0 г, 16,3 ммоль, выход 74,6%). 1Н ЯМР (ДМСО-d6, 400 МГц): δ 10,96 (br s, 1H), 7,98 (s, 1H), 3,25 (t, J=7,6 Гц, 2Н), 2,89 (t, J=7,6 Гц, 2Н), 2,50 (td, J=1,6, 3,5 Гц, 1Н), 2,06 (quin, J=7,6 Гц, 2H).
Соединение KH13-4: соединение KH13-3 (3,50 г, 16,3 ммоль, 1 экв.) и SnCl2•2H2O (18,4 г, 81,9 ммоль, 5 экв.) растворяли в метаноле (10 мл) и перемешивали при 70°С в течение 7 часов. После того, как контроль с помощью ЖХ-МС показал, что сырье полностью исчезло, реакционный растворитель сразу же сушили с помощью ротационного испарения, остатки растворяли с помощью этилацетата, а затем добавляли насыщенный раствор бикарбоната натрия, после чего было получено твердое вещество. Реакционный раствор фильтровали для удаления твердых веществ и собирали фильтрат, органические фазы отделяли, промывали насыщенным солевым раствором (100 мл), сушили с помощью безводного сульфата натрия, а затем фильтровали и сушили с помощью ротационного испарения. Остатки сразу же использовали на следующей стадии без очистки с получением желтого твердого вещества KH13-4 (2,70 г, 14,7 ммоль, выход: 89,7%). 1H ЯМР (ДМСО-d6, 400 МГц): δ 8,12 (s, 1H), 6,39 (s, 1H), 4,48 (s, 2Н), 2,75 (t, J=7,6 Гц, 2Н), 2,59 (t, J=7,6 Гц, 2Н), 2,04 - 1,89 (m, 2Н).
Соединение KH13-5: соединение KH13-4 (2,50 г, 13,6 ммоль, 1 экв.), соединение KH07-6а (3,21 г, 13,6 ммоль, 1 экв.) и карбонат цезия (13,3 г, 40,8 ммоль, 3 экв.) добавляли в ДМФА (30 мл) и перемешивали при 80°С в течение 3 часов. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 3/1) показал, что сырье полностью исчезло. После разбавления реакционного раствора 100 мл воды реакционный раствор экстрагировали этилацетатом (50 мл × 3), а затем органические фазы объединяли и промывали насыщенным солевым раствором, сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 1/0 до 3/1) с получением желтого твердого вещества KH13-5 (3,70 г, 9,57 ммоль, выход: 70,3%, чистота: 99,0%). МС (ESI) m/z: 383,8 [М+Н]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 8,54-8,35 (m, 1H), 6,70-6,51 (m, 1H), 3,59 (s, 2Н), 3,41 (spt, J=6,8 Гц, 1H), 2,76 (td, J=7,6, 17,5 Гц, 4Н), 2,19 - 2,06 (m, 2Н), 1,25 (d, J=6,8 Гц, 6Н).
Соединение KH13-6: методика была такой же, как и в случае синтеза соединения KH11-2, и было получено коричневое твердое вещество KH13-6 (4,30 г, неочищенный продукт). МС (ESI)m/z: 430,3 [М+Н]+.
Соединение KH13-7: методика была такой же, как и в случае синтеза KH11-3, и было получено бледно-желтое твердое вещество KH13-7 (1,20 г, 3,72 ммоль, выход: 38,0%) путем очистки с помощью колоночной хроматографии на силикагеле (SiO2 петролейный эфир/этилацетат = от 1/0 до 1/1). МС (ESI) m/z: 320,0 [М+Н]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 9,74 (s, 1H), 7,92 (s, 1H), 6,50 (s, 1H), 5,22 - 4,80 (m, 2Н), 3,27 (spt, J=6,8 Гц 1Н), 2,64 (br t, J=7,6 Гц, 2Н), 2,59-2,51 (m, 2Н), 1,95 (br dd, J=7,6, 15,1 Гц 2Н), 1,11 (d, J=6,8 Гц, 6Н).
Пример 17. Синтез соединения KH13
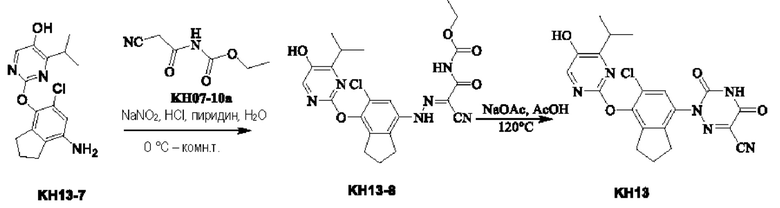
Соединение KH13-8: методика была такой же, как и в случае синтеза KH07-11, и было получено оранжевое твердое вещество (431,3 мг, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 487,2[М+Н]+.
Соединение KH13: методика была такой же, как и в случае синтеза KH07, и было получено оранжевое твердое вещество KH13 (354,2 мг, неочищенный продукт). 120 мг твердого вещества отбирали и очищали с помощью препаративной ВЭЖХ с получением 43 мг белого твердого вещества. МС (ESI) m/z: 441,2 [М+Н]+. 1H ЯМР (ДМСО-d6, 400 МГц): 13,07 (s, 1H), 10,06 (s, 1Н), 7,98 (s, 1H), 7,38 (s, 1H), 3,25 - 3,34 (m, 1H), 2,84 (t, 2Н, J=8,0 Гц), 2,68 (t, 2Н, J=8,0 Гц), 2,01 (t, 2Н, J=8,0 Гц), 1,15 (d, 6Н, J=4,0 Гц).
Пример 18. Синтез соединения KH14
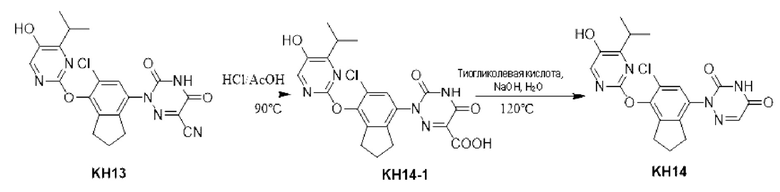
Соединение KH14-1: методика была такой же, как и в случае синтеза KH08-1, и было получено желтое твердое вещество (251,4 мг, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 460,1 [М+Н]+.
Соединение KH14: методика была такой же, как и в случае синтеза KH08, и было получено белое твердое вещество KH14 (6 мг, выход: 11,1%). МС (ESI) m/z: 416,2 [М+Н]+. 1H ЯМР (ДМСО-d6 400 МГц): 12,40 (s, 1H), 9,98 (s, 1H), 7,99 (s, 1H), 7,63 (s, 1H), 7,46 (s, 1H), 3,27-3,30 (m, 1H), 2,83 (t, 2H, J=8,0 Гц), 2,68 (t, 2Н, J=8,0 Гц), 2,0 (t, 2H, J=8,0 Гц), 1,15 (d, 6H, J=4,0 Гц).
Пример 19. Синтез ключевого промежуточного продукта KH15-8
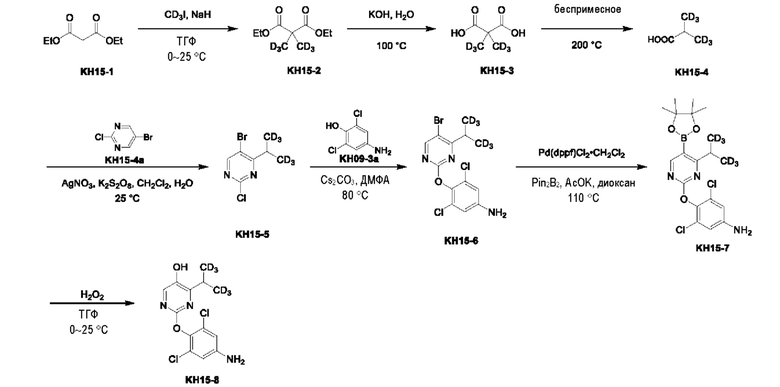
Соединение KH15-2: гидрид натрия (7,49 г, 187 ммоль, чистота 60,0%, 3,00 экв.) растворяли в тетрагидрофуране (100 мл) и к вышеуказанной реакционной системе добавляли KH15-1 (10,0 г, 62,4 ммоль, 9,43 мл, 1,00 экв.) и CD3I (19,0 г) при 0°С. Смесь непрерывно перемешивали при 0°С в течение 0,5 часа, а затем медленно нагревали до 25°С и перемешивали в течение 11,5 часов. Контроль с помощью ТСХ показал, что сырье полностью прореагировало. Реакционный раствор выливали в 200 мл ледяной воды и экстрагировали этилацетатом (100 мл × 3), собирали органические фазы, сушили с помощью безводного сульфата натрия, фильтровали и сушили с помощью ротационного испарения с получением желтого маслянистого вещества KH15-2 (8,35 г, 42,9 ммоль, выход: 68,8%). Твердое вещество сразу же подвергали взаимодействию на следующей стадии без очистки.
Соединение KH15-3: соединение KH15-2 (8,95 г, 46,0 ммоль, 1,00 экв.) растворяли в воде (20 мл) и к вышеуказанному реакционному раствору добавляли гидроксид натрия (6,27 г, 111 ммоль, 2,42 экв.). После добавления проводили реакцию в течение 3 часов при 100°С. Контроль с помощью ТСХ показал, что образовалось новое пятно. рН доводили до 2 с помощью 6 и. соляной кислоты при 0°С, а затем реакционный раствор экстрагировали дихлорметаном (100 мл × 2), органические фазы объединяли, промывали насыщенным солевым раствором (100 мл), сушили с помощью безводного сульфата натрия, фильтровали и сушили с помощью ротационного испарения с получением бесцветного масла KH15-3 (5,02 г, неочищенный продукт), которое сразу же использовали на следующей стадии без очистки.
Соединение KH15-4: соединение KH15-3 (5,00 г, 36,1 ммоль, 1,00 экв.) нагревали до 200°С и перемешивали в течение 0,5 часа. После того, как контроль с помощью ТСХ показал, что образовалось новое соединение, неочищенный продукт перегоняли (140°С, 1 атм) с получением желтого масла KH15-4 (2,52 г, 26,7 ммоль, выход: 73,9%).
Соединение KH15-5: соединение KH15-4 (2,50 г, 28,3 ммоль, 1,00 экв.), соединение KH15-4а (5,49 г, 28,3 ммоль, 1,00 экв.), нитрат серебра (1,64 г, 9,65 ммоль, 0,34 экв.) и K2S2O8 (11,5 г, 42,5 ммоль, 8,52 мл, 1,50 экв.) добавляли в дихлорметан (50 мл) и воду (50 мл) и перемешивали при внешней температуре 25°С в течение 12 часов под защитой азота. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 20/1) показал, что образовалось новое пятно. Реакционный раствор гасили раствором сульфита натрия и экстрагировали дихлорметаном (100 мл×2), органические фазы объединяли, промывали насыщенным солевым раствором (50 мл), сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 1/0 до 10/1) с получением неочищенного продукта, который затем очищали с помощью препаративной ВЭЖХ с получением желтого маслянистого вещества KH15-5 (2,60 г, 10,7 ммоль, выход: 37,9%). МС (ESI) m/z: 243,1 [М+Н]+.
Соединение KH15-6: соединение KH15-5 (2,60 г, 10,7 ммоль, 1,00 экв.), соединение KH09-3а (1,92 г, 10,7 ммоль, 1,00 экв.) и карбонат цезия (10,5 г, 32,29 ммоль, 3,00 экв.) добавляли в ДМ ФА (26 мл) и перемешивали при внешней температуре 80°С в течение 10 часов под защитой азота. Контроль с помощью ЖХ-МС показал, что сырье уже полностью прореагировало. К реакционному раствору добавляли этилацетат (100 мл) и воду (100 мл) для экстракции и разделения, органические фазы собирали, промывали насыщенным солевым раствором (100 мл), сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 1/0 до 5/1) с получением желтого маслянистого вещества KH15-6 (2,70 г, 7,05 ммоль, выход 65,4%). МС (ESI) m/z: 384,1 [М+Н]+.
Соединение KH15-7: методика была такой же, как и в случае соединения KH11-2, и остаток очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 1/0 до 5/1) с получением белого твердого вещества KH15-7 (3,00 г, 6,97 ммоль, выход: 98,9%). МС (ESI) m/z=429,9 [М+Н]+.
Соединение KH15-8: соединение KH15-7 (3,00 г, 6,97 ммоль, 1,00 экв.) растворяли в тетрагидрофуране (30 мл) и в реакционную смесь добавляли пероксид водорода (1,66 г, 14,6 ммоль, 1,41 мл, чистота 30,0%, 2,10 экв.) при 0°С, и после добавления смесь перемешивали при комнатной температуре в течение 8 часов. Контроль с помощью ЖХ-МС показал, что сырье полностью прореагировало, и с помощью МС был обнаружен новый продукт. Реакцию останавливали с помощью насыщенного раствора сульфита натрия (50 мл), экстрагировали дихлорметаном (50 мл × 3), органические фазы объединяли, сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 10/1 до 1/1) с получением бледно-желтого твердого вещества KH15-8 (1,20 г, 3,73 ммоль, выход 53,5%). МС (ESI) m/z: 320,1 [М+Н]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 9,91-9,78 (m, 1H), 8,01-7,90 (m, 1H), 6,70-6,56 (m, 2Н), 5,56-5,42 (m, 2Н), 3,25-3,19 (m, 1H).
Пример 20. Синтез соединения KH15
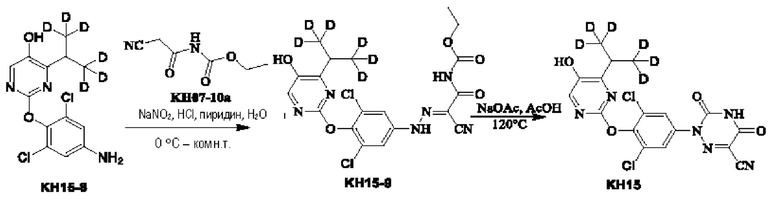
Соединение KH15-9: методика была такой же, как и в случае синтеза KH07-11, и было получено оранжевое твердое вещество (430 мг, неочищенный продукт), которое сразу же подвергали взаимодействию наследующей стадии без очистки. МС (ESI) m/z: 487,2 [М+Н]+.
Соединение KH15: методика была такой же, как и в случае синтеза KH07, и было получено оранжевое твердое вещество KH15 (320 мг, неочищенный продукт). 100 мг твердого вещества отбирали и очищали с помощью препаративной ВЭЖХ с получением 6 мг белого твердого вещества. МС (ESI) m/z: 441,2 [М+Н]+.
Пример 21. Синтез соединения KH16
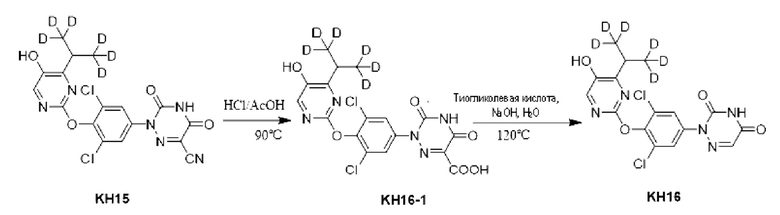
Соединение KH16-1: методика была такой же, как и в случае синтеза KH08-1, и было получено желтое твердое вещество (200 мг, неочищенный продукт), которое сразу же подвергали взаимодействию наследующей стадии без очистки. МС (ESI) m/z: 460,1 [М+Н]+.
Соединение KH16: методика была такой же, как и в случае синтеза KH08, и было получено белое твердое вещество KH16 (11 мг, выход: 21,2%). МС (ESI) m/z: 416,2 [М+Н]+.
Пример 22. Синтез ключевого промежуточного продукта KH17-5
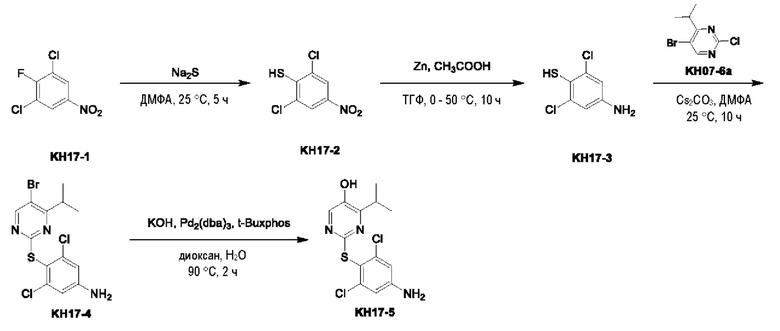
Соединение KH17-2: соединение KH17-1 (50,0 г, 238 ммоль, 1 экв.) и сульфид натрия (27,8 г, 357 ммоль, 14,9 мл, 1,5 экв.) добавляли в ДМФА (500 мл) и перемешивали в течение 5 часов при 25°С. Контроль с помощью ЖХ-МС показал, что соединение KH17-1 уже исчезло. Реакционный раствор выливали в 500 мл ледяной воды и доводили рН до 5 с помощью соляной кислоты (2 н.). Реакционный раствор экстрагировали этилацетатом (200 мл × 2), органические фазы объединяли, промывали насыщенным солевым раствором (300 мл), сушили с помощью безводного сульфата натрия, фильтровали и сушили с помощью ротационного испарения. Остатки сразу же использовали на следующей стадии без очистки с получением красного твердого вещества KH17-2 (43,0 г, неочищенный продукт). МС (ESI) m/z: 224,1 [М+Н]+.
Соединение KH17-3: соединение KH17-2 (40,0 г, 178 ммоль, 1 экв.) и Zn (58,3 г, 892 ммоль, 5 экв.) добавляли к тетрагидрофурану (1,5 л), по каплям добавляли уксусную кислоту (21,4 г, 357 ммоль, 20,4 мл, 2 экв.) при 0°С и перемешивали смесь при 50°С в течение 10 часов. Контроль с помощью ЖХ-МС показал, что сырье полностью исчезло. Реакционный раствор сразу же фильтровали. Фильтрат собирали и сушили с помощью ротационного испарения с получением остатка. Остатки перетирали в твердый порошок с метил-трет-бутиловым эфиром с получением белого твердого вещества KH17-3 (220 г, 108 ммоль, выход: 60,8%, чистота: 95,8%). МС (ESI)m/z: 194,0 [М+Н]+.
Соединение KH17-4: соединение KH17-3 (16,0 г, 82,4 ммоль, 1 экв.), соединение KH07-6а (16,1 г, 57,7 ммоль, 0,7 экв.) и карбонат цезия (40,2 г, 123 ммоль, 1,5 экв.) добавляли в ДМФА (200 мл) и перемешивали в течение 10 часов при 25°С. Контроль с помощью ЖХ-МС показал, что сырье уже полностью прореагировало. Реакционный раствор добавляли к 200 мл воды и экстрагировали этилацетатом (200 мл × 2), собирали органические фазы, промывали насыщенным солевым раствором (200 мл), сушили с помощью безводного сульфата натрия, фильтровали и сушили с помощью ротационного испарения с получением остатков. Остаток перетирали в твердый порошок с метил-трет-бутиловым эфиром с получением желтого твердого вещества KH17-4 (2,60 г, 10,7 ммоль, выход: 37,9%). МС (ESI) m/z: 394,0 [М+Н]+.
Соединение KH17-5: после растворения соединения KH17-4 (5,00 г, 12,7 ммоль, 1 экв.) в воде (25,0 мл) к смеси добавляли диоксан (100 мл), гидроксид калия (2,85 г, 50,8 ммоль, 4 экв.), Pd2(dba)3 (1,16 г, 1,27 ммоль, 0,1 экв.) и t-Buxphos (540 мг, 1,27 ммоль, 0,1 экв.) и перемешивали при 90°С в течение 2 часов под защитой азота. После того, как контроль с помощью ЖХ-МС показал, что сырье уже полностью исчезло, реакционный раствор доводили до рН 5, используя 1М разбавленную соляную кислоту, затем добавляли 100 мл воды и экстрагировали этилацетатом (100 мл × 2), органические фазы собирали, промывали насыщенным солевым раствором (100 мл), сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 1/0 до 3/1) с получением грязно-белого твердого вещества KH17-5 (1,20 г, 3,48 ммоль, выход: 27,3%). МС (ESI) m/z: 330,0 [М+Н]+. 1Н ЯМР (ДМСО-d6 400 МГц): δ 9,99 (br s, 1Н), 8,10 - 7,90 (m, 1H), 6,83 - 6,64 (m, 2H), 6,14 - 5,88 (m, 2H), 3,22 (spt, J=6,8 Гц, 1H), 1,04 (d, J=6,8 Гц, 6H).
Пример 23. Синтез соединения KH17
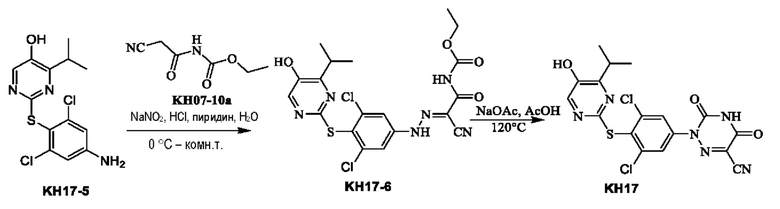
Соединение KH17-6: методика была такой же, как и в случае синтеза KH07-11, и было получено оранжевое твердое вещество (390 мг, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 497,1[М+Н]+.
Соединение KH17: методика была такой же, как и в случае синтеза KH07, и было получено оранжевое твердое вещество KH17 (140 мг, неочищенный продукт). 40 мг твердого вещества отбирали и очищали с помощью препаративной ВЭЖХ с получением 5 мг бледно-желтого твердого вещества, выход: приблизительно 12,5%. МС (ESI)m/z: 451,1 [М+Н]+.
Пример 24. Синтез соединения KH18
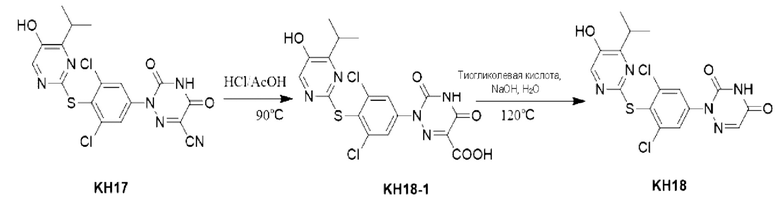
Соединение KH18-1: методика была такой же, как и в случае синтеза KH08-1, и было получено коричневое твердое вещество (102,4 мг, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 470,1 [М+Н]+.
Соединение KH18: методика была такой же, как и в случае синтеза KH08, и было получено белое твердое вещество KH18 (18 мг, выход: 19,8%). МС (ESI) m/z: 426,0 [М+Н]+. 1Н ЯМР (ДМСО-d6, 400 МГц): 12,50 (s, 1Н), 10,21 (s, 1H), 8,05 (s, 1H), 7,94 (s, 2Н), 7,73 (s, 1Н), 3,20-3,27 (m, 1H), 1,04 (d, 6Н, J=4,0 Гц).
Пример 25. Синтез соединения KHE001
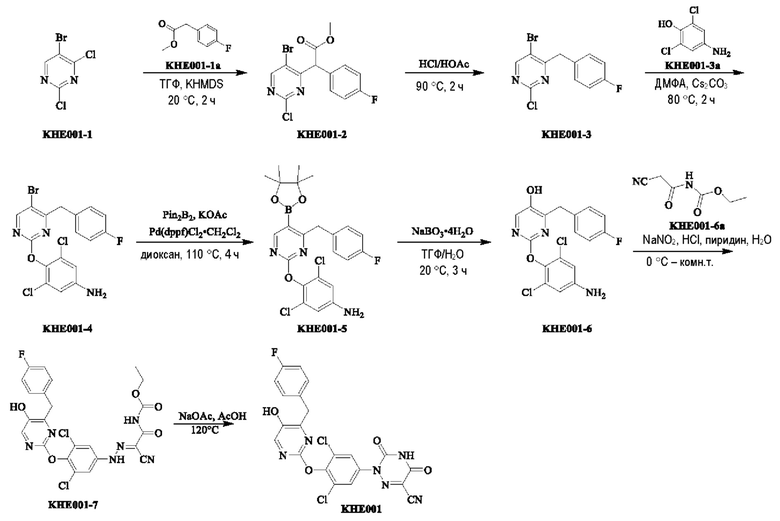
Соединение KHE001-2: соединения KHE001-1 (20,0 г, 87,8 ммоль, 11,2 мл, 1,00 экв.) и KHE001-1а (14,8 г, 87,8 ммоль, 1,00 экв.) растворяли в толуоле (8 мл) и медленно по каплям добавляли бис(триметилсилил)амид калия (KHMDS) (1 М, 92,2 мл, 1,05 экв.) при 0°С. После перемешивания смеси при 20°С в течение 2 часов контроль с помощью ТСХ (петролейный эфир/этилацетат = 10/1) показал, что оставшееся количество сырья составляло менее 5% и образовалось новое пятно повышенной полярности. Реакцию останавливали в ледяной воде (500 мл), и затем подвергали реакционную смесь экстракции этилацетатом (50 мл × 3). Органические фазы собирали, промывали насыщенным солевым раствором (100 мл), сушили с помощью безводного сульфата натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 30/1 до 5/1) с получением желтого маслянистого вещества KHE001-2 (17,1 г, выход: 54,2%).
Соединение KHE001-3: соединение KHE001-2 (17,0 г, 47,3 ммоль, 1,00 экв.) растворяли в соляной кислоте (20 мл, чистота 36%) и уксусной кислоте (80 мл) и перемешивали при 90°С в течение 2 часов. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 10/1) показал, что сырье полностью прореагировало и образовалось новое пятно (Rf=0,60). Реакционную смесь сразу же сушили с помощью ротационного испарения при пониженном давлении с получением неочищенного продукта. Неочищенный продукт растворяли в этилацетате и промывали насыщенным раствором бикарбоната натрия (50 мл × 2). Органические фазы собирали, сушили с помощью безводного сульфата натрия, фильтровали, концентрировали для удаления растворителя и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = 10/1) с получением белого твердого вещества KHE001-3 (9,70 г, выход: 68,0%). 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,62 (s, 1H), 7,31 (dd, J=5,6, 8,8 Гц, 2Н), 7,04 - 6,97 (m, 2Н), 4,22 (s, 2Н).
Соединение KHE001-4: соединения KHE001-3 (9,00 г, 29,9 ммоль, 1,00 экв.), KHE001-3а (6,38 г, 35,8 ммоль, 1,20 экв.) и карбонат цезия (29,2 г, 89,5 ммоль, 3,00 экв.) растворяли в диметилформамиде (90 мл). Смесь перемешивали в течение 2 часов при 80°С под защитой азота. Контроль с помощью ЖХ-МС показал, что сырье KHE001-3 полностью прореагировало, а сигнал целевого продукта является основным пиком. К реакционному раствору для экстракции и разделения добавляли этилацетат (300 мл) и воду (100 мл). Органические фазы собирали, промывали насыщенным солевым раствором, сушили с помощью безводного сульфата натрия и концентрировали для удаления растворителя с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 30/1 до 5/1) с получением желтого твердого вещества KHE001-4 (4,50 г, выход: 34,0%). МС (ESI) m/z: 443,8 [М+Н]+.
Соединение KHE001-5: соединение KHE001-4 (4,50 г, 10,1 ммоль, 1,0 экв.), Pin2B2 (5,16 г, 20,3 ммоль, 2,00 экв.), Pd(dppf)Cl2•CH2Cl2 (415 мг, 508 мкмоль, 0,05 экв.) и ацетат калия (1,99 г, 20,3 ммоль, 2,00 экв.) растворяли в диоксане (45 мл). Реакционная система находилась под защитой азота и перемешивалась в течение 4 часов при 110°С. онтроль с помощью ЖХ-МС показал, что сырье KHE001-4 полностью прореагировало, а сигнал целевого продукта является основным пиком. Реакционную систему сразу же сушили с помощью ротационного испарения с получением коричневого маслянистого вещества KHE001-5 (14,0 г, смесь), которое сразу же использовали на следующей стадии без очистки. МС (ESI) m/z: 490,1 [М+Н]+.
Соединение KHE001-6: соединение KHE001-5 (14,0 г, 28,6 ммоль, 1,00 экв.) растворяли в тетрагидрофуране (70 мл) и воде (70 мл), и к реакционной смеси добавляли перборат натрия (NaBO3⋅4H2O) (13,2 г, 85,7 ммоль, 3,00 экв.). Смесь перемешивали при 20°С в течение 3 часов, и контроль с помощью ТСХ (петролейный эфир/этилацетат = 2/1, Rf=0,15) показал, что реакция уже была завершена. Реакционную смесь разбавляли водой (150 мл) и перемешивали при 25°С в течение 10 минут. Реакционный раствор фильтровали, и фильтровальный осадок промывали этилацетатом (20 мл × 2). Объединенные фильтраты экстрагировали этилацетатом (100 мл × 2). Органические фазы собирали и промывали насыщенным солевым раствором (300 мл × 2), сушили с помощью безводного сульфата натрия, концентрировали для удаления растворителя с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 0/1) с получением коричневого твердого вещества KHE001-6 (1,11 г, выход: 28,8%, чистота: 94,2%). МС (ESI) m/z: 380,0 [M+H]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 10,10-10,05 (m, 1H), 8,02 (s, 1H), 7,30-7,20 (m, 2Н), 7,14-7,02 (m, 2Н), 6,66 (s, 2Н), 5,54 (br s, 2Н), 3,99-3,91 (m, 2Н).
Соединение KHE001-7: реакционный раствор А: соединение KHE001-6 (0,5014 г, 1,322 ммоль) добавляли к 26 мл воды и добавляли 14 мл концентрированной соляной кислоты при 0°С. Нитрит натрия (0,1218 г, 1,765 ммоль) растворяли в 4 мл воды и медленно по каплям добавляли к реакционному раствору, и перемешивали при 0°С в течение 1,5 часов, чтобы он растворился. Реакционный раствор В: соединение KHE001-6а (0,2381 г, 1,526 ммоль) добавляли к 40 мл воды, добавляли 14 мл пиридина при 0°С и непрерывно перемешивали при этой температуре в течение 1,5 часов. Затем реакционный раствор А быстро выливали в реакционный раствор В при 0°С с образованием оранжевого твердого вещества, и температуру медленно повышали до комнатной температуры для продолжения реакции в течение ночи. После того, как контроль с помощью ТСХ показал, что реакция была завершена, твердое вещество фильтровали и трижды промывали 50 мл воды и петролейным эфиром, соответственно. Было получено оранжевое твердое вещество KHE001-7 (600 мг, выход: 82,9%, неочищенный продукт), которое сразу же использовали на следующей стадии без очистки. МС (ESI) m/z: 547,1 [М+Н]+.
Соединение KHE001: соединение KHE001-7 (600 г, 1,097 ммоль) и ацетат натрия (0,9035 г, 11,018 ммоль) добавляли в одногорлую бутыль и растворяли в уксусной кислоте (12 мл) под защитой N2. Реакцию проводили в течение 3 часов при температуре 120°С. После того, как контроль с помощью ТСХ показал, что сырье полностью прореагировало, реакцию останавливали. Реакционный раствор охлаждали до 0°С и добавляли 100 мл воды, затем осаждалось большое количество твердого вещества, которое сразу же фильтровали, и промывали твердое вещество трижды 50 мл воды и петролейным эфиром, соответственно. Оранжевое твердое вещество (560 мг, неочищенный продукт) собирали, и 100 мг неочищенного продукта очищали с помощью препаративной ВЭЖХ с получением грязно-белого твердого соединения KHE001 (21,8 мг, 23,8%). МС (ESI, m/z): 500,9 [М+1]+. 1Н ЯМР (ДМСО-d6, 400 МГц): 3 13,25 (s, 1H), 10,28 (s, 1Н), 8,08 (s, 2Н), 7,74 (s, 2Н), 7,22-7,24 (m, 2Н), 7,07-7,11 (m, 2Н), 3,39 (s, 2Н).
Пример 26. Синтез соединения KHE002
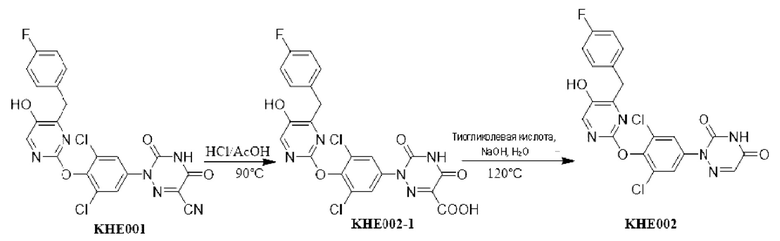
Соединение KHE002-1: неочищенное соединение KHE001 (460 мг, 0,918 ммоль) добавляли в одногорлую колбу, растворяли в уксусной кислоте (15 мл), а затем к реакционной смеси по каплям добавляли концентрированную соляную кислоту (5 мл). После проведения реакции в течение 4 часов при 90°С контроль с помощью ТСХ показал, что сырье полностью прореагировало, образовалось новое пятно повышенной полярности и реакцию останавливали. Реакционный раствор сразу же сушили с помощью ротационного испарения и доводили рН реакционной смеси до 9-10 насыщенным раствором карбоната натрия, затем экстрагировали реакционный раствор этилацетатом (20 мл × 2). Водные фазы собирали, и рН водных фаз доводили до 3-4, затем водные фазы экстрагировали этилацетатом (20 мл × 3). Органические фазы собирали, сушили с помощью безводного сульфата натрия и фильтровали. Органические фазы концентрировали с получением желтого твердого вещества KHE002-1 (300 мг, 62,8%), которое сразу же использовали на следующей стадии без очистки.
Соединение KHE002: соединение KHE002-1 (300 мг, 0,577 ммоль) и гидроксид натрия (0,0985 г, 2,462 ммоль) добавляли в одногорлую колбу и растворяли в воде (40 мл), а затем к реакционному раствору добавляли тиогликолевую кислоту (1,0831 г, 11,773 ммоль) и проводили реакцию при 120°С в течение 3 часов. После того, как контроль с помощью ТСХ показал, что сырье полностью прореагировало, образовалось пятно пониженной полярности и реакцию останавливали. Добавляли насыщенный раствор карбоната натрия для доведения рН реакционной системы до нейтрального, затем реакционную систему экстрагировали этилацетатом (20 мл × 3), сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью препаративной ВЭЖХ с получением белого твердого вещества KHE002 (46,7 мг, 17,0%). МС (ESI, m/z): 476,3[М+1]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 12,49 (s, 1H), 10,25 (s, 1H), 8,08 (s, 1H), 7,69 (s, 2Н), 7,60 (s, 1H), 7,23-7,24 (m, 2Н), 7,07-7,12 (m, 1H), 3,98 (m, 2Н).
Пример 27. Синтез соединения KHE003
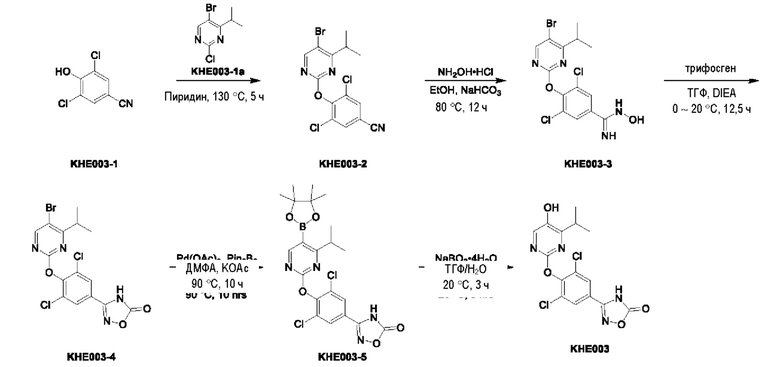
Соединение KHE003-2: KHE003-1а (10,0 г, 42,5 ммоль, 1,60 экв.) и соединение KHE003-1 (5,00 г, 26,6 ммоль, 1,00 экв.) растворяли в пиридине (50 мл) и перемешивали при 130°С в течение 5 часов. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 10/1, Rf=0,52) показал, что реакция была завершена. Реакционную смесь охлаждали до 20°С, разбавляли водой (500 мл) и экстрагировали этилацетатом (200 мл × 2). Объединенные органические фазы промывали насыщенным солевым раствором (300 мл × 2), сушили с помощью безводного сульфата натрия, фильтровали и концентрировали для удаления растворителя с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 5/1) с получением желтого маслянистого вещества KHE003-2 (2,40 г, выход: 23,2%). 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,51 - 8,47 (m, 1Н), 7,73 - 7,70 (m, 2Н), 3,41 (spt, J=6,8 Гц, 1H), 1,19 (d, J=6,8 Гц, 6Н).
Соединение KHE003-3: гидрохлорид гидроксиламина (377 мг, 5,43 ммоль, 1,50 экв.), соединение KHE003-2 (1,40 г, 3,62 ммоль, 1,00 экв.) и бикарбонат натрия (304 мг, 3,62 ммоль, 1,00 экв.) добавляли к этанолу (20 мл) и перемешивали при 80°С в течение 12 часов. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 2/1, Rf=0,11) показал, что реакция была завершена. Реакционную смесь сразу же сушили с помощью ротационного испарения с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 1/0 до 20/1) с получением желтого маслянистого вещества KHE003-3 (1,00 г, выход: 65,8%).
Соединение KHE003-4: трифосген (1,61 г, 5,43 ммоль, 1,20 экв.), соединение KHE017-3 (1,90 г, 4,52 ммоль, 1,00 экв.) и DIE А (2,92 г, 22,6 ммоль, 3,94 мл, 5,00 экв.) растворяли в тетрагидрофуране (20 мл) под защитой азота при 0°С и непрерывно перемешивали при 0°С в течение 0,5 часа, затем нагревали до 20°С и перемешивали в течение 12 часов. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 1/1, Rf=0,22) показал, что реакция была завершена. Реакционный раствор разбавляли водой (500 мл), а затем к реакционному раствору для экстракции и разделения добавляли этилацетат (300 мл) и воду (150 мл). Органические фазы собирали, промывали насыщенным солевым раствором, сушили с помощью безводного сульфата натрия и концентрировали для удаления растворителя с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 0/1) с получением желтого твердого вещества KHE003-4 (1,30 г, выход: 64,4%). 1H ЯМР (ДМСО-d6, 400 МГц): δ 13,02 (br s, 1H), 8,79 (s, 1H), 8,05 (s, 2H), 3,42-3,40 (m, 1H), 1,13 (d, J=6,8 Гц, 6H).
Соединение KHE003-5: ацетат палладия (60,4 мг, 269 мкмоль, 0,10 экв.), соединение KHE003-4 (1,20 г, 2,69 ммоль, 1 экв.), Pin2B2 (6,83 г, 26,9 ммоль, 10,0 экв.) и ацетат калия (792 мг, 8,07 ммоль, 3,00 экв.) добавляли в ДМФА (10 мл) под защитой азота и перемешивали при 90°С в течение 10 часов. Контроль с помощью ЖХ-МС показал, что реакция была завершена. После охлаждения до 20°С к реакционному раствору медленно добавляли воду (100 мл) и экстрагировали этилацетатом (30 мл × 2). Органические фазы собирали, промывали насыщенным солевым раствором (100 мл × 2), сушили с помощью безводного сульфата натрия и фильтровали. Органический растворитель сушили с помощью ротационного испарения с получением желтого маслянистого вещества KHE003-5 (2,00 г, смесь), которое использовали на следующей стадии без очистки. МС (ESI) m/z=493,0 [М+1]+.
Соединение KHE003: соединение KHE003-5 (2,00 г, 4,06 ммоль, 1,00 экв.) растворяли в тетрагидрофуране (10 мл) и воде (10 мл), и затем к реакционной смеси добавляли перборат натрия (NaBO3⋅4Н2О) (1,87 г, 12,2 ммоль, 3,00 экв.). Реакционную смесь перемешивали при 20°С в течение 3 часов, и контроль с помощью ЖХ-МС показал, что реакция была завершена. Реакционный раствор экстрагировали и разделяли с помощью этилацетата (20 мл) и воды (100 мл). Органические фазы собирали, промывали насыщенным солевым раствором, сушили с помощью безводного сульфата натрия и фильтровали. Затем органические фазы сушили с помощью ротационного испарения с получением остатков. Остатки очищали с помощью препаративной ВЭЖХ с получением желтого твердого вещества KHE003 (154 мг, выход: 14,9% и чистота 97,0%). МС (ESI) m/z: 383,1 [М+1]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 10,12 (br s, 1H), 8,00 (s, 1H), 7,97 (s, 2H), 3,27 (br s, 1H), 1,10 (d, J=6,8 Гц, 6H).
Пример 28. Синтез соединения KHE004
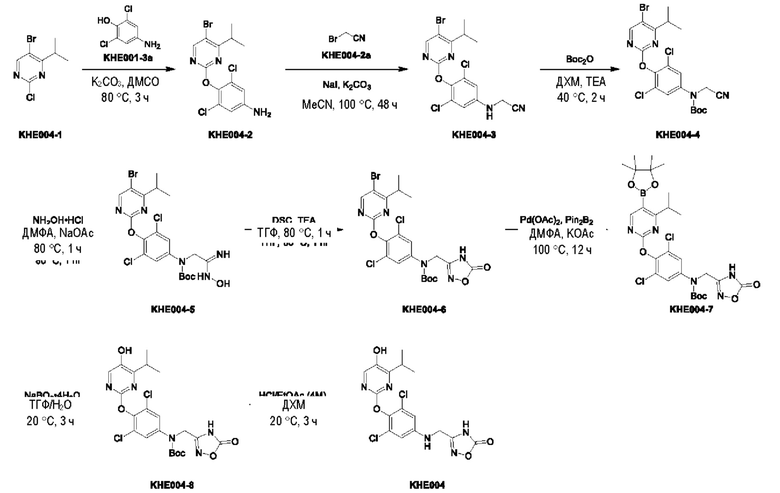
Соединение KHE004-2: KHE001-3а (7,56 г, 42,5 ммоль, 1,00 экв.), соединение KHE004-1 (10,0 г, 42,5 ммоль, 1,00 экв.) и карбонат калия (11,7 г, 84,9 ммоль, 2,00 экв.) добавляли к ДМФА (100 мл) и перемешивали при 80°С в течение 3 часов под защитой азота. Контроль с помощью ЖХ-МС показал, что реакция была завершена. После охлаждения до 20°С реакционный раствор разбавляли водой (200 мл) и экстрагировали этилацетатом (100 мл × 2). Органические фазы собирали, промывали насыщенным солевым раствором, сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 1/0 до 2/1) с получением желтого твердого вещества KHE004-2 (12,0 г, выход: 75,0%). МС (ESI) m/z=378,0 [М+1]+.
Соединение KHE004-3: соединение KHE004-2 (5,00 г, 13,3 ммоль) и бромацетонитрил (KHE004-2а) (11,9 г, 99,5 ммоль) растворяли в MeCN (50 мл), а затем добавляли иодид натрия (5,96 г, 39,8 ммоль) и карбонат калия (5,50 г, 39,8 ммоль). Смесь нагревали до 100°С в закрытой пробирке на 100 мл и перемешивали в течение 48 часов. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 5/1, Rf=0,22) показал, что реакция была завершена. Реакционный раствор сразу же сушили с помощью ротационного испарения, а остатки очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 2/1) с получением желтого твердого вещества KHE004-3 (4,00 г, выход: 72,5%). 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,72 (s, 1H), 6,92 (s, 2Н), 6,77 (br t, J=6,8 Гц, 1H), 4,41 - 4,34 (m, 1H), 4,38 (d, J=6,6 Гц, 2Н), 3,39 - 3,34 (m, 1H), 3,39 - 3,34 (m, 1H), 1,14 (d, J=6,8 Гц, 6Н).
Соединение KHE004-4: соединение KHE004-3 (4,00 г, 9,61 ммоль) и триэтиламин (1,02 г, 10,1 ммоль, 1,40 мл) растворяли в дихлорметане (40 мл), а затем добавляли Вос2О (2,20 г, 10,1 ммоль, 2,32 мл). Смесь перемешивали при 40°С в течение 2 часов. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 5/1, Rf=0,62) показал, что реакция была завершена. Реакционный раствор охлаждали до 20°С, медленно добавляли воду (200 мл) и экстрагировали дихлорметаном (40 мл × 2). Органические фазы собирали и промывали насыщенным солевым раствором (200 мл × 2), сушили с помощью безводного сульфата натрия, фильтровали и сушили с помощью ротационного испарения с получением желтого маслянистого вещества KHE004-4 (4,80 г, выход: 96,7%). 1Н ЯМР (ДМСО-d6, 400МГц): δ 8,78 (s, 1H), 7,65 (s, 2Н), 4,81 (s, 2Н), 3,38 - 3,33 (m, 1H), 1,46- 1,41 (m, 10Н), 1,10 (d, J=6,8 Гц, 6Н).
Соединение KHE004-5: соединение KHE004-4 (4,80 г, 9,30 ммоль) и ацетат натрия (6,10 г, 74,4 ммоль) растворяли в ДМФА (40 мл) и добавляли гидрохлорид гидроксиламина (5,17 г, 74,4 ммоль). Смесь перемешивали при 80°С в течение 1 часа. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 1/1, Rf=0,39) показал, что реакция была завершена. После охлаждения до комнатной температуры реакционный раствор экстрагировали этилацетатом (100 мл) и водой (250 мл). Органические фазы собирали, сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 0/1) с получением желтого твердого вещества KHE004-5 (4,60 г, выход: 90,1%). 1H ЯМР (ДМСО-d6, 400 МГц): δ 8,78 (s, 1Н), 7,65 (s, 2Н), 4,81 (s, 2Н), 3,38 - 3,33 (m, 1H), 1,46 - 1,41 (m, 9Н), 1,10 (d, J=6,8 Гц, 6Н).
Соединение KHE004-6: N,N-дисукцинимидилкарбонат (DSC) (2,67 г, 10,4 ммоль), соединение KHE004-5 (4,40 г, 8,01 ммоль) и триэтиламин (3,05 г, 30,1 ммоль, 4,19 мл) растворяли в ДМФА (10 мл) и перемешивали при 80°С в течение 1 часа под защитой азота. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 2/1, Rf=0,07) показал, что реакция была завершена. Сигнал KHE004-6 контролировали с помощью ЖХ-МС и разделяли реакционный раствор с помощью этилацетата (50 мл) и воды (50 мл). Органические фазы собирали, промывали насыщенным солевым раствором, сушили с помощью безводного сульфата натрия, фильтровали и сушили с помощью ротационного испарения с получением желтого твердого вещества KHE004-6 (4,20 г, смесь), которое сразу же использовали для следующей реакции. МС (ESI) m/z: 576,0 [М+1]+.
Соединение KHE004-7: ацетат палладия (39,0 мг, 174 мкмоль, 0,10 экв.), соединение KHE004-6 (1,00 г, 1,74 ммоль, 1,00 экв.), ацетат калия (512 мг, 5,22 ммоль, 3,00 экв.) и Pim2B2 (4,41 г, 17,4 ммоль, 10,0 экв.) растворяли в ДМФА (15 мл) и перемешивали при 100°С в течение 12 часов под защитой азота. Целевой продукт KHE004-7 контролировали с помощью ЖХ-МС. Реакционный раствор экстрагировали и разделяли с помощью этилацетата (20 мл) и воды (20 мл). Органические фазы собирали, сушили с помощью безводного сульфата натрия, фильтровали и сушили с помощью ротационного испарения с получением желтого твердого вещества KHE004-7 (4,00 г, неочищенный продукт). Неочищенный продукт сразу же использовали для следующей реакции. МС (ESI) m/z: 622,3 [М+1]+.
Соединение KHE004-8: соединение KHE004-7 (4,00 г, 6,43 ммоль, 1,00 экв.) растворяли в тетрагидрофуране (20 мл) и воде (20 мл), затем к реакционной смеси добавляли перборат натрия (NaBO3⋅4H2O) (2,97 г, 19,3 ммоль, 3,00 экв.) и перемешивали при 20°С в течение 3 часов. Контроль с помощью ЖХ-МС показал, что реакция была завершена. Реакционный раствор экстрагировали и разделяли с помощью этилацетата (20 мл) и воды (100 мл). Органические фазы промывали насыщенным солевым раствором, сушили с помощью безводного сульфата натрия, фильтровали и сушили с помощью ротационного испарения с получением желтого твердого вещества KHE004-8 (2,00 г, неочищенный продукт). Неочищенный продукт сразу же использовали для следующей реакции. МС (ESI) m/z: 514,0 [М+1]+.
Соединение KHE004: KHE004-8 (2,00 г, 3,90 ммоль, 1,00 экв.) растворяли в дихлорметане (20 мл), к реакционному раствору добавляли раствор хлористого водорода в этилацетате (4 М, 10 мл, 10,3 экв.) и перемешивали при 20°С в течение 3 часов. Контроль с помощью ЖХ-МС показал, что реакция была завершена. Реакционный раствор сразу же сушили с помощью ротационного испарения и очищали с помощью препаративной ВЭЖХ с получением коричневого твердого вещества KHE004 (19,5 мг, выход: 1,16%, чистота 96,3%). МС (ESI) m/z: 412,0 [М+1]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 12,42 (br s, 1H), 9,89 (br s, 1H), 7,95 (s, 1H), 6,79 (s, 1H), 6,68 - 6,55 (m, 1H), 4,29 (s, 2H), 3,32 - 3,23 (m, 2H), 1,11 (s, 6H).
Пример 29. Синтез соединения KHE005
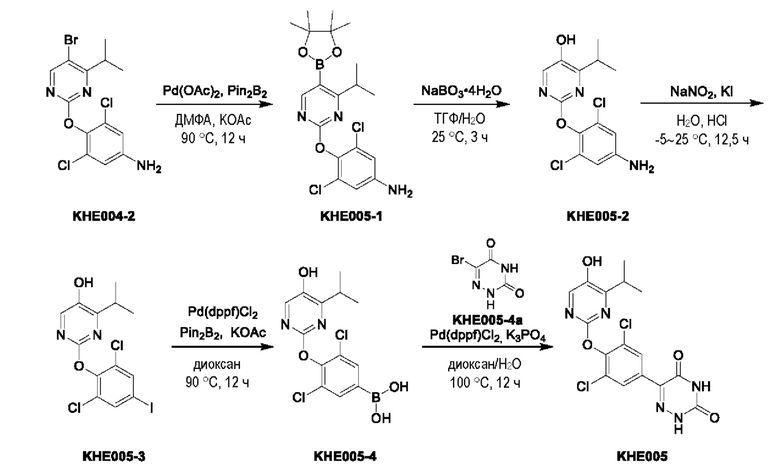
Соединение KHE005-1: ацетат палладия (238 мг, 1,06 ммоль, 0,10 экв.), соединение KHE004-2 (4,00 г, 10,6 ммоль, 1,00 экв.), Pin2B2 (27,0 г, 106 ммоль, 10,0 экв.) и ацетат калия (3,12 г, 31,8 ммоль, 3,00 экв.) растворяли в ДМФА (100 мл) и перемешивали при 90°С в течение 12 часов под защитой азота. Контроль с помощью ЖХ-МС показал, что реакция была завершена, и целевой пик был легко различим. Реакционный раствор экстрагировали и разделяли с помощью этилацетата (50 мл) и воды (50 мл). Органические фазы собирали, сушили с помощью безводного сульфата натрия, фильтровали и сушили с помощью ротационного испарения с получением желтого твердого вещества KHE005-1 (4,00 г, 9,43 ммоль, неочищенный продукт). Неочищенный продукт сразу же использовали для следующей реакции. МС (ESI) m/z: 424,1 [М+Н]+.
Соединение KHE005-2: соединение KHE005-1 (4,00 г, 9,43 ммоль, 1,00 экв.) растворяли в тетрагидрофуране (10 мл) и воде (10 мл), а затем к реакционной смеси добавляли перборат натрия (NaBO3⋅4H2O) (4,35 г, 28,3 ммоль, 3,00 экв.) и перемешивали смесь при 20°С в течение 3 часов. Контроль с помощью ЖХ-МС показал, что реакция была завершена, и целевой пик был легко различим. Реакционный раствор экстрагировали и разделяли с помощью этилацетата (20 мл) и воды (20 мл). Органические фазы собирали, промывали насыщенным солевым раствором, сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 20/1 до 5/1) с получением фиолетового маслянистого вещества KHE005-2 (1,40 г, выход: 42,0%). МС (ESI) m/z: 314,0 [М+Н]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 10,04 (s, 1 Н), 7,97-7,99 (m, 3 Н), 3,27 (d, J=6,8 Гц, 1 H), 1,11 (s, 3 Н), 1,09 (s, 3 Н).
Соединение KHE005-3: соединение KHE005-2 (800 мг, 2,55 ммоль, 1,00 экв.) добавляли к концентрированной соляной кислоте (12,0 М, 20 мл, 94,3 экв.) и охлаждали реакционную смесь до -5°С. Нитрит натрия (1,05 г, 15,3 ммоль, 6,00 экв.) растворяли в воде (4,00 мл), затем медленно по каплям добавляли к вышеуказанному раствору и энергично перемешивали, и температуру реакции поддерживали между -5°С и 0°С. После реакции в течение 0,5 часа иодид калия (6,34 г, 38,2 ммоль, 15,0 экв.) растворяли в воде (6,00 мл), а затем медленно по каплям добавляли к вышеуказанному раствору и энергично перемешивали, и температуру реакции непрерывно поддерживали между -5°С и 0°С. После добавления реакционный раствор нагревали до 25°С и перемешивали в течение 12 часов под защитой азота. Контроль с помощью ЖХ-МС показал, что реакция была завершена. Реакционный раствор экстрагировали и разделяли с помощью этилацетата (20 мл) и воды (100 мл). Органические фазы собирали, промывали насыщенным раствором сульфита натрия, сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 20/1 до 0/1) с получением желтого твердого вещества KHE005-3 (546 мг, выход: 50,5%). МС (ESI) m/z: 424,9 [М+Н]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 10,05 (br s, 1 Н) 7,98 (s, 2 Н) 7,98 (s, 1 Н) 3,27 (d, J=6,8 Гц, 1 Н) 1,11 (s, 3 Н) 1,10 (s, 3 Н).
Соединение KHE005-4: под защитой азота Pd(dppf)Cl2 (93 мг, 0,127 ммоль, 0,10 экв.), соединение KHE005-3 (540 мг, 1,27 ммоль, 1,00 экв.), Pin2B2 (3,227 г, 12,7 ммоль, 10,0 экв.) и ацетат калия (374 мг, 3,81 ммоль, 3,00 экв.) растворяли в диоксане (7 мл) и перемешивали при 90°С в течение 12 часов. Контроль с помощью ЖХ-МС показал, что реакция была завершена. Реакционный раствор сразу же сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, дихлорметан/метанол = 20/1) с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением красного твердого продукта бороновой кислоты KHE005-4 (400 мг, выход: 92,0%). МС (ESI) m/z: 343,0 [М+Н]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 10,05 (s, 1 Н), 8,47 (s, 1 Н), 7,96 (s, 1 Н), 7,86 (s, 1 Н), 3,27 (d, J=6,8 Гц, 1 Н), 1,10 (s, 3 H), 1,08 (s, 3 Н).
Соединение KHE005: под защитой азота Pd(dppf)Cl2 (51,2 мг, 0,07 ммоль, 0,06 экв.), соединение KHE005-4 (400 мг, 1,17 ммоль, 1,00 экв.), KHE005-4а (224 мг, 1,17 ммоль, 1,00 экв.) и фосфат калия (495 мг, 2,33 ммоль, 2,00 экв.) добавляли к системе из диоксана (2,5 мл) и воды (2,5 мл) и перемешивали при 100°С в течение 12 часов. Контроль с помощью ЖХ-МС показал, что реакция была завершена. Реакционный раствор экстрагировали и разделяли с помощью этилацетата (10 мл) и воды (80 мл). Органические фазы собирали, промывали насыщенным солевым раствором, сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, дихлорметан/метанол = 20/1) с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ с получением желтого твердого вещества KHE005-5 (101 мг, выход: 21,0%, чистота 99,5%). МС (ESI) m/z: 410,1 [М+1]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 12,69 (d, J=1,2 Гц, 1H), 12,23 (s, 1H), 10,07 (br s, 1H), 8,04-7,97 (m, 3Н), 3,36-3,21 (m, 1H), 1,12 (d, J=6,8 Гц, 6H).
Пример 30. Синтез соединения KHE006
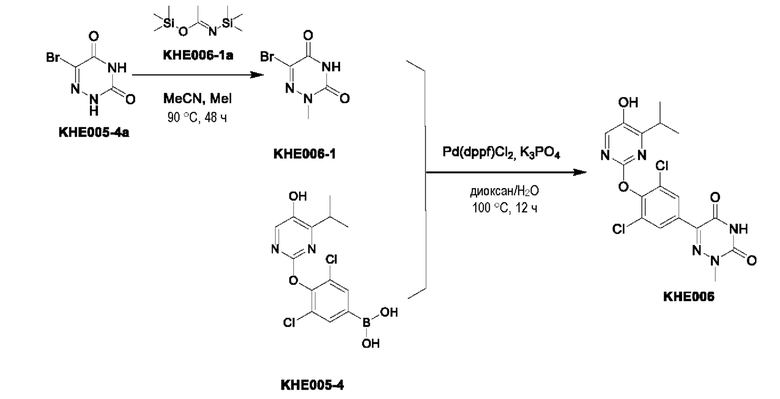
Соединение KHE006-1: соединение KHE005-4а (3,00 г, 15,6 ммоль, 1,00 экв.) растворяли в ацетонитриле (45 мл), затем к реакционной смеси добавляли KHE006-1а (8,14 г, 40,0 ммоль, 9,89 мл, 2,56 экв.) и перемешивали при 82°С в течение 3 часов. Затем к вышеуказанной реакционной системе добавляли метилиодид (2,71 г, 19,1 ммоль, 1,19 мл, 1,22 экв.), перемешивали в течение 24 часов, а затем к реакционной смеси непрерывно добавляли метилиодид (1,11 г, 7,81 ммоль, 486 мкл, 0,50 экв.) и перемешивали в течение 24 часов. ТСХ (петролейный эфир/этилацетат = 2/1, Rf=0,55) показала, что реакция была завершена. Реакционный раствор экстрагировали этилацетатом (50 мл) и водой (50 мл). Органические фазы собирали, сушили с помощью безводного сульфата натрия и фильтровали. Фильтрат концентрировали и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 20/1 до 0/1) с получением желтого твердого вещества KHE006-1 (2,00 г, выход: 62,1%). 1Н ЯМР (ДМСО-d6, 400 МГц): δ 3,44 (s, 3 Н), 12,49 (br s, 1 Н).
Соединение KHE006: под защитой азота Pd(dppf)Cl2 (89,6 мг, 0,122 ммоль, 0,06 экв.), KHE005-4 (700 мг, 2,04 ммоль, 1,00 экв.), KHE006-1 (631 мг, 3,06 ммоль, 1,50 экв.) и фосфат калия (866 мг, 4,08 ммоль, 2,00 экв.) растворяли в системе из диоксана (2,5 мл) и воды (2,5 мл) и перемешивали при 100°С в течение 12 часов. Контроль с помощью ЖХ-МС показал, что реакция была завершена. Реакционный раствор экстрагировали и разделяли с помощью этилацетата (10 мл) и воды (80 мл). Органические фазы собирали, промывали насыщенным солевым раствором, сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью препаративной ВЭЖХ с получением бледно-желтого твердого вещества KHE006 (10,4 мг, выход: 1,11%, чистота 91,9%). МС (ESI) m/z: 424,1 [М+1]+.
Пример 31. Синтез соединения KHE007
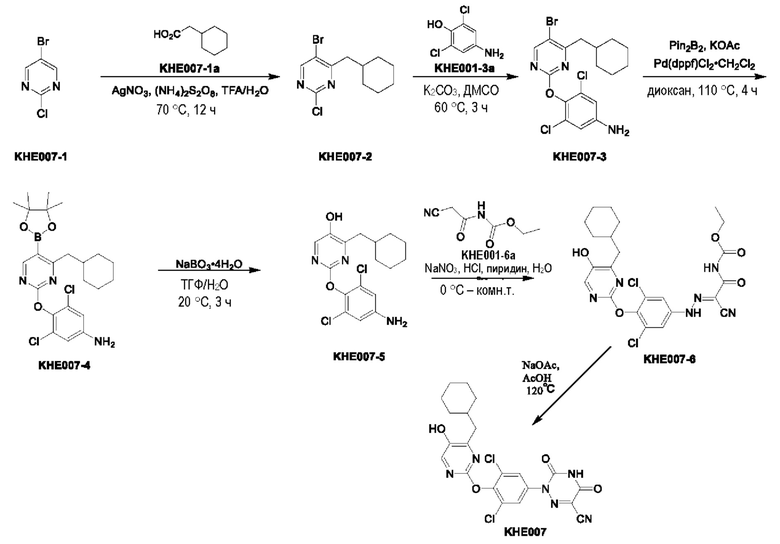
Соединение KHE007-2: в воду (300 мл) добавляли соединение KHE007-1 (30,0 г, 155 ммоль, 1,00 экв.), к вышеуказанной реакционной системе добавляли KHE007-1а (26,5 г, 186 ммоль, 26,2 мл, 1,20 экв.), нитрат серебра (5,27 г, 31,0 ммоль, 0,20 экв.) и трифторуксусную кислоту (8,84 г, 77,6 ммоль, 5,74 мл, 0,50 экв.). Смесь нагревали до 70°С и перемешивали, а затем медленно добавляли персульфат аммония (NH4)2S2O8 (70,8 г, 310 ммоль, 2,00 экв.). После добавления реакцию продолжали в течение 12 часов. ТСХ (петролейный эфир/этилацетат = 1/0, Rf=0,12) показала, что реакция была завершена. Реакционный раствор охлаждали до 20°С и экстрагировали этилацетатом (20 мл) и водой (100 мл). Органические фазы собирали, промывали насыщенным раствором хлорида натрия, сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 1/0 до 10/1) с получением бесцветного маслянистого вещества KHE007-2 (24,4 г, 84,3 ммоль, выход: 54,3%). МС (ESI) m/z: 291,2 [М+Н]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 8,88 (s, 1H), 2,74 (d, J=7,2 Гц, 2Н), 1,81 (dt, J=3,6, 7,2 Гц, 1H), 1,69-1,54 (m, 5Н), 1,27-1,12 (m, 3Н), 1,09-0,96 (m, 2Н).
Соединение KHE007-3: к ДМСО (200 мл) добавляли соединение KHE007-2 (24,4 г, 84,3 ммоль, 1,00 экв.) и K2CO3 (23,3 г, 169 ммоль, 2,00 экв.), и в это время к вышеуказанной реакционной системе добавляли KHE001-3а (15,0 г, 84,3 ммоль, 1,00 экв.). Реакционный раствор нагревали до 60°С и перемешивали в течение 3 часов под защитой азота. ТСХ (петролейный эфир/этилацетат = 5/1, Rf=0,34) показала, что реакция была завершена. Реакционный раствор экстрагировали и разделяли с помощью этилацетата (20 мл) и воды (100 мл). Органические фазы собирали, промывали насыщенным солевым раствором, сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 1/1) с получением бесцветного маслянистого вещества KHE007-3 (28,0 г, 64,9 ммоль, выход: 77,1%). МС (ESI) m/z: 432,1 [М+Н]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,71 (s, 1H), 6,68 (s, 2Н), 5,62 (s, 2Н), 2,68-2,63 (m, 2Н), 1,83-1,69 (m, 1H), 1,66-1,51 (m, 5Н), 1,15-1,05 (m, 3Н), 1,02-0,88 (m, 2Н).
Соединение KHE007-4: соединение KHE007-3 (2,00 г, 4,64 ммоль, 1,00 экв.), ацетат калия (911 мг, 9,28 ммоль, 2,00 экв.) и Pin2B2 (2,36 г, 9,28 ммоль, 2,00 экв.) добавляли в диоксан (10 мл), а затем к вышеуказанной реакционной системе добавляли Pd(dppf)Cl2⋅CH2Cl2 (189 мг, 0,232 ммоль, 0,05 экв.). Реакционный раствор нагревали до 110°С и перемешивали в течение 4 часов под защитой азота. Контроль с помощью ЖХ-МС показал, что реакция была завершена. Реакционный раствор экстрагировали этилацетатом (20 мл) и водой (100 мл). Органические фазы собирали, промывали насыщенным солевым раствором, сушили с помощью безводного сульфата натрия, фильтровали и сушили с помощью ротационного испарения с получением коричневого твердого вещества KHE026-4 (4,00 г, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 478,1 [М+Н]+.
Соединение KHE007-5: неочищенный продукт соединения KHE007-4 (4,00 г, 8,36 ммоль, 1,00 экв.) растворяли в тетрагидрофуране (20 мл) и воде (20 мл), а затем к вышеуказанному раствору добавляли перборат натрия NaBO3⋅4Н2О (3,86 г, 25,1 ммоль, 3,00 экв.). Смесь выдерживали в течение 3 часов при 20°С. ТСХ (петролейный эфир/этилацетат = 2/1, Rf=0,15) показала, что реакция была завершена. Реакционный раствор фильтровали, фильтровальный осадок промывали этилацетатом, и фильтрат собирали и экстрагировали этилацетатом (50 мл × 2). Органические фазы собирали, промывали насыщенным солевым раствором, сушили с помощью безводного сульфата натрия, фильтровали, сушили с помощью ротационного испарения и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 0/1) с получением бледно-желтого маслянистого вещества (1,40 г), которое очищали с помощью препаративной ВЭЖХ с получением серого твердого вещества KHE007-5 (1,23 г, 3,27 ммоль, выход: 72,0%, чистота: 97,8%). МС (ESI) m/z: 368,1 [М+Н]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 9,74 (s, 1H), 8,00 (s, 1H), 6,65 (s, 2Н), 5,51 (s, 2Н), 2,94 (s, 1Н), 1,62-1,41 (m, 4Н), 0,68 (t, J=7,2 Гц, 6Н).
Соединение KHE007-6: реакционный раствор А: соединение KHE007-5 (0,5038 г, 1,369 ммоль) растворяли в воде (26 мл), добавляли 14 мл концентрированной соляной кислоты при 0°С, растворяли навеску нитрита натрия (0,1234 г, 1,788 ммоль) в 4 мл воды, медленно по каплям добавляли к реакционному раствору и поддерживали температуру реакции при 0-5°С. После добавления реакционный раствор непрерывно перемешивали при 0°С в течение 1,5 часов с получением раствора. Реакционный раствор В: соединение KHE001-6а (0,2381 г, 1,526 ммоль) добавляли к 40 мл воды, добавляли 14 мл пиридина при 0°С и перемешивали в течение 1,5 часов при этой температуре. Затем реакционный раствор А быстро выливали в реакционный раствор В при 0°С с образованием оранжевого твердого вещества, и температуру медленно повышали до комнатной температуры для продолжения реакции в течение ночи. После того, как контроль с помощью ТСХ показал, что реакция была завершена, твердое вещество сразу же отфильтровывали и трижды промывали 50 мл воды и петролейным эфиром, соответственно. Твердое вещество собирали с получением оранжевого твердого вещества KHE007-6 (700 мг, 95,6%, неочищенный продукт). Неочищенный продукт сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 535,1 [М+Н]+.
Соединение KHE007: соединение KHE007-6 (700 мг, 1,310 ммоль) и NaOAc (1,075 г, 13,110 ммоль) добавляли в одногорлую колбу и растворяли в уксусной кислоте (12 мл) под защитой азота. Реакцию проводили в течение 3 часов при температуре 120°С. После того, как контроль с помощью ТСХ показал, что сырье полностью прореагировало, реакцию останавливали. Реакционный раствор охлаждали до 0°С и добавляли 100 мл воды, затем осаждалось большое количество твердого вещества, которое сразу же фильтровали и промывали трижды 50 мл воды и петролейным эфиром, соответственно. Собирали и получали оранжевое твердое вещество (620 мг, неочищенный продукт), и 120 мг неочищенного продукта очищали с помощью препаративной ВЭЖХ с получением бледно-желтого твердого соединения KHE007 (26,9 мг, 22,4%). МС (ESI) m/z: 489,3 [M+1]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 13,25 (s, 1Н), 10,02 (s, 1Н), 8,03 (s, 2Н), 7,75 (s, 1H), 2,49 - 2,50 (m, 2Н), 1,55-1,71 (m, 6Н), 1,04 - 1,13 (m, 4Н), 0,92 - 0,97 (m, 2Н).
Пример 32. Синтез соединения KHE008
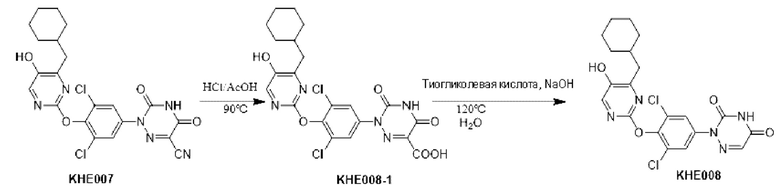
Соединение KHE008-1: соединение KHE007 (503,8 мг, 1,030 ммоль) добавляли в одногорлую колбу, растворяли в уксусной кислоте (15 мл), а затем по каплям добавляли к реакционной смеси концентрированную соляную кислоту (5 мл). После добавления реакцию проводили в течение 4 часов при 90°С, контроль с помощью ТСХ показал, что сырье полностью прореагировало, образовалось новое пятно повышенной полярности и реакцию останавливали. Реакционный раствор сразу же сушили с помощью ротационного испарения и доводили рН до 9-10 насыщенным раствором карбоната натрия. Реакционный раствор экстрагировали этилацетатом (20 мл × 2), собирали водные фазы и доводили рН водных фаз до 3-4, и экстрагировали этилацетатом (20 мл × 2), а затем собирали органические фазы, сушили с помощью безводного сульфата натрия и фильтровали. Органические фазы концентрировали для удаления растворителя и получения желтого твердого вещества KHE008-1 (260 мг, 49,7%), которое сразу же использовали на следующей стадии без очистки.
Соединение KHE008: соединение KHE008-1 (260 мг, 0,581 ммоль) и гидр оксид натрия (0,0891 г, 2,227 ммоль) добавляли в одногорлую колбу, растворяли в воде (40 мл), а затем к реакционной смеси добавляли тиогликолевую кислоту (1,0125 г, 10,331 ммоль). Реакцию проводили в течение 3 часов при 120°С. После того, как контроль с помощью ТСХ показал, что сырье полностью прореагировало, образовалось пятно пониженной полярности и реакцию останавливали. Добавляли насыщенный раствор карбоната натрия для доведения рН реакционной системы до нейтрального, затем реакционную систему экстрагировали этилацетатом (20 мл × 2), сушили с помощью безводного сульфата натрия, фильтровали, концентрировали и очищали с помощью препаративной ВЭЖХ с получением белого твердого вещества KHE008 (96,9 мг, 40,9%). МС (ESI) m/z: 464,3[М+1]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 12,46 (s, 1Н), 9,99 (s, 1H), 8,03 (s, 2Н), 7,77 (s, 1H), 7,69 (s, 1H), 2,49 - 2,50 (m, 2Н), 1,60 - 1,71 (m, 1H), 1,56 - 1,59 (m, 5H), 1,10 - 1,13 (m, 3H), 0,92 - 0,97 (m, 2H).
Пример 33. Синтез соединения KHE009
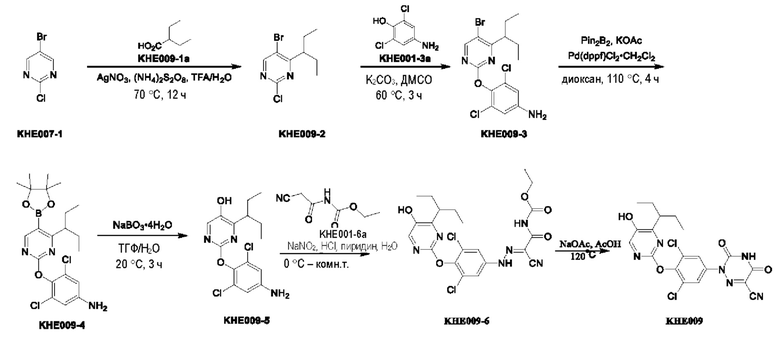
Соединение KHE009-2: методика была такой же, как и в случае синтеза соединения KHE007-2, и неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 1/0 до 10/1) с получением желтого масла KHE009-2 (45,5 г, 173 ммоль, выход: 66,8%). МС (ESI) m/z: 263,0[М+1]+. 1Н ЯМР (CDCl3, 400 МГц): δ 8,93 (s, 1H), 3,15 - 3,05 (m, 1H), 1,73 - 1,59 (m, 4Н), 0,74 (t, J=7,6 Гц, 6Н).
Соединение KHE009-3: методика была такой же, как и в случае синтеза соединения KHE007-3, и неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 0/1) с получением белого твердого вещества KHE009-3 (11,8 г, 29,1 ммоль, выход: 38,4%). МС (ESI) m/z: 404,0[М+1]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,77 (s, 1H), 6,68 (s, 2Н), 5,61 (s, 2Н), 3,04 (t, J=7,0 Гц, 1H), 1,54 (quin, J=7,2 Гц, 4Н), 0,69 (t, J=7,2 Гц, 6Н).
Соединение KHE009-4: методика была такой же, как и в случае синтеза соединения KHE007-4; было получено коричневое твердое вещество KHE009-4 (22,0 г, неочищенный продукт). Неочищенный продукт сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 452,2 [М+Н]+.
Соединение KHE009-5: методика была такой же, как и в случае синтеза соединения KHE007-5; было получено белое твердое вещество KHE009-5 (4,44 г, 12,5 ммоль, выход: 48,7%, чистота 96,5%). МС (ESI) m/z: 342,0 [М+Н]+.
1Н ЯМР (ДМСО-d6, 400МГц): δ 9,74 (s, 1H), 8,00 (s, 1Н), 6,65 (s, 2Н), 5,51 (s, 2Н), 2,82 - 3,01 (m, 1H), 1,62 - 1,41 (m, 4Н), 0,68 (t, J=7,2 Гц, 6Н).
Соединение KHE009-6: методика была такой же, как и в случае синтеза соединения KHE007-6, и было получено оранжевое твердое вещество KHE009-6 (650 мг, 86,7%, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 509,2 [М+Н]+.
Соединение KHE009: методика была такой же, как и в случае синтеза соединения KHE007, и было получено оранжевое твердое вещество (570 мг, неочищенный продукт). 100 мг неочищенного продукта очищали с помощью препаративной ВЭЖХ с получением бледно-желтого твердого вещества KHE009 (18 мг, выход: 18,2%). МС (ESI)m/z: 463,0 [М+1]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 13,26 (s, 1Н), 10,00 (s, 1Н), 8,06 (s, 1Н), 7,75 (s, 2Н), 2,98 - 3,02 (m, 1Н), 1,52-1,55 (m, 4Н), 1,24 (s, 1Н), 0,68 - 0,72 (m, 6Н).
Пример 34. Синтез соединения KHE010
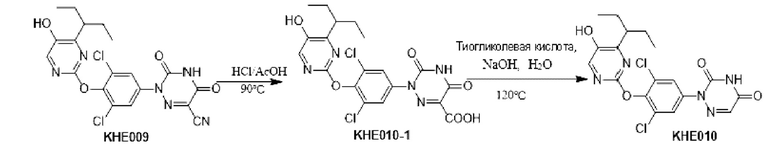
Соединение KHE010-1: методика была такой же, как и в случае синтеза соединения KHE008-1, и было получено желтое твердое вещество KH010-1 (280 мг, 57,4%), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 482,0 [М+1]+.
Соединение KHE010: методика была такой же, как и в случае синтеза соединения KHE008, и после очистки с помощью препаративной ВЭЖХ было получено белое твердое вещество KHE010 (113,6 мг, 44,7%). МС (ESI) m/z: 438,2 [М+1]+. 1Н ЯМР (ДМСО-d6, 400 МГц): 12,47(s, 1Н), 9,98(s, 1Н), S,06(s, 1Н), 7,78(s, 2Н), 7,70(s, 1Н), 2,94-3,01 (m, 1Н), 1,48-1,61 (m, 4Н), 0,70(t, 6Н, J=8,0 Гц).
Пример 35. Синтез соединения KHE011
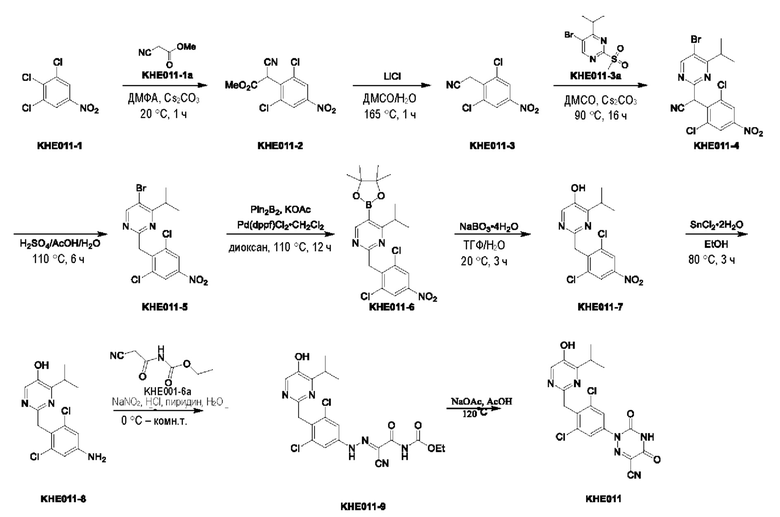
Соединение KHE011-2: соединение KHE011-1 (50,0 г, 221 ммоль, 1,00 экв.), KHE011-1a (30,0 г, 265 ммоль, 28,3 мл, 1,20 экв.) и CS2CO3 (144 г, 442 ммоль, 2,00 экв.) добавляли в ДМФА (250 мл) и перемешивали при 20°С в течение 1 часа под защитой азота.
Контроль с помощью ТСХ (петролейный эфир/этилацетат = 10/1) показал, что сырье полностью прореагировало и образовалось новое пятно (Rf=20). Реакционный раствор выливали в 1 н. HCl (1 л). Желтое твердое вещество осаждали и отфильтровывали. Фильтровальный осадок промывали водой (200 мл × 3), а затем собирали фильтровальный осадок, растворяли в этилацетате (1 л), сушили с помощью безводного сульфата натрия и фильтровали. Фильтрат концентрировали с получением желтого твердого соединения KHE011-2 (60,0 г, 198 ммоль, выход: 89,7%), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 289,0 [М+1]+.
Соединение KHE011-3: соединение KHE001-2 (60,0 г, 198 ммоль, 1,00 экв.) и LiCl (12,6 г, 297 ммоль, 1,50 экв.) добавляли в ДМСО (150 мл) и воду (50 мл), трижды подвергали замене азотом и подвергали взаимодействию при 165°С в течение 1 часа под защитой азота. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 5/1) показал, что сырье полностью прореагировало и образовалось новое пятно (Rf=0,50). Реакционный раствор выливали в ледяную воду (1 л) и экстрагировали этилацетатом (500 мл × 3). Органические фазы собирали, промывали насыщенным солевым раствором (200 мл × 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали фильтрат. Концентрированный фильтрат очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 30/1 до 5/1) с получением желтого твердого вещества KHE011-3 (22,1 г, 95,7 ммоль, выход: 48,3%). МС (ESI)m/z: 231,0 [М+1]+.
Соединение KHE011-4: соединение KHE011-3 (7,50 г, 32,5 ммоль, 1,00 экв.), KHE011-3а (9,06 г, 32,5 ммоль, 1,00 экв.) и Cs2CO3 (21,2 г, 64,9 ммоль, 2,00 экв.) добавляли в ДМСО (70 мл) и перемешивали при 90°С в течение 16 часов. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 5/1) показал, что сырье полностью прореагировало и образовалось новое соединение с низкой полярностью (Rf=0,70). Реакционный раствор выливали в насыщенный солевой раствор (500 мл) и экстрагировали этилацетатом (500 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (200 мл × 3), сушили с помощью безводного сульфата натрия, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 1/1) с получением темно-серого твердого вещества KHE011-4 (3,95 г, 9,18 ммоль, выход: 28,3%). МС (ESI) m/z: 429,0 [М+1]+. 1Н ЯМР (CDCl3, 400 МГц): δ 8,66 (s, 1H), 8,29 (s, 2Н), 6,39 (s, 1H), 3,43 (d, J=6,7 Гц, 1H), 1,24- 1,13 (m, 6Н).
Соединение KHE011-5: соединение KHE011-4 (2,90 г, 6,74 ммоль, 1,0 экв.) добавляли к системе из воды (12 мл), уксусной кислоты (12 мл) и концентрированной серной кислоты (12 мл) и перемешивали при 110°С в течение 6 часов. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 5/1) показал, что сырье полностью прореагировало и образовалось новое пятно (Rf=0,47). Реакционный раствор разбавляли водой (200 мл) и экстрагировали этилацетатом (80 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (200 мл × 3), сушили с помощью безводного сульфата натрия, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 1/1) с получением желтого твердого вещества KHE011-5 (2,50 г, 6,17 ммоль, выход: 91,5%). МС (ESI) m/z: 404,0 [М+1]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 8,79 (s, 1H), 8,35 (s, 2Н), 4,62 (s, 2Н), 3,38 - 3,33 (m, 1H), 1,11 (d, J=6,8 Гц, 6Н).
Соединение KHE011-6: методика была такой же, как и в случае синтеза соединения KHE007-4; было получено коричневое маслянистое вещество KHE011-6 (4,50 г, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI)m/z: 452,2 [М+1]+.
Соединение KHE011-7: методика была такой же, как и в случае синтеза соединения KHE007-5; после очистки с помощью препаративной ВЭЖХ было получено серое твердое вещество KHE011-7 (680 мг, 1,94 ммоль, выход: 31,5%, чистота 97,8%). МС (ESI) m/z: 342,0 [M+1]+.
Соединение KHE011-8: соединение KHE011-7 (680 мг, 1,95 ммоль, чистота 97,9%, 1,00 же.) растворяли в этаноле (10 мл), добавляли двухлористое олово (SnCl2⋅2H2O) (1,32 г, 5,84 ммоль, 3,00 же.) и перемешивали при 80°С в течение 3 часов. Контроль с помощью ЖХ-МС показал, что сырье полностью прореагировало. Реакционный раствор добавляли к системе из этилацетата (30 мл) и воды (80 мл) частями для разделения органических фаз. Водные фазы экстрагировали этилацетатом (30 мл × 2). Органические фазы объединяли, промывали насыщенным солевым раствором (100 мл × 2), сушили с помощью безводного сульфата натрия, концентрировали и очищали с помощью препаративной ВЭЖХ с получением желтого твердого вещества KHE011-8 (254 мг, 788 мкмоль, выход: 40,5%, чистота 96,8%). МС (ESI) m/z: 312,0 [М+1]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 10,19 (s, 1Н), 8,08 (s, 1Н), 6,80 - 6,68 (m, 2Н), 4,24 (s, 2Н), 3,31 - 3,23 (m, 1Н), 2,08 - 2,06 (m, 1Н), 1,10 (d, J=6, 8 Гц, 6Н).
Соединение KHE011-9: методика была такой же, как и в случае синтеза соединения KHE007-6, и было получено желтое твердое вещество KHE011-9 (350 мг, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI)m/z: 479,0 [М+1]+.
Соединение KHE011: методика была такой же, как и в случае синтеза соединения KHE007, и после очистки с помощью препаративной ВЭЖХ было получено желтое твердое вещество KHE011 (194 мг, 433 мкмоль, выход: 63,9%, чистота 96,6%). МС (ESI) m/z: 433,0 [М+1]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 13,21 (s, 1Н), 10,06 (s, 1Н), 8,07 (s, 1Н), 7,63 (s, 2Н), 4,46 (s, 2Н), 3,30 - 3,26 (m, 1Н), 1,11 (d, J=6,8 Гц, 6Н).
Пример 36. Синтез соединения KHE012
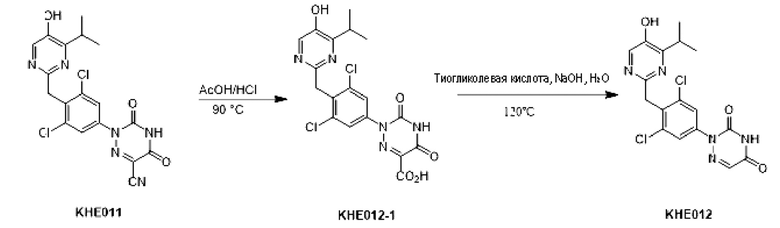
Соединение KHE012-1: методика была такой же, как и в случае синтеза соединения KHE008-1, и было получено желтое твердое вещество KHE012-1 (120 мг, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 451,9 [М+1]+.
Соединение KHE012: методика была такой же, как и в случае синтеза соединения KHE008, и после очистки с помощью препаративной ВЭЖХ было получено бледно-желтое твердое вещество KHE012 (58,0 мг, выход: 51,2%, чистота: 97,9%). МС (ESI)m/z: 408,2 [М+1]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 12,43 (s, 1H), 10,05 (m, 1H), 8,07 (s, 1H), 7,69 (d, J=2,0 Гц, 1H), 7,66 (s, 2Н), 4,44 (s, 2Н), 3,35 - 3,21 (m, 1H), 1,11 (d, J=6,8 Гц, 6Н).
Пример 37. Синтез соединения KHE013
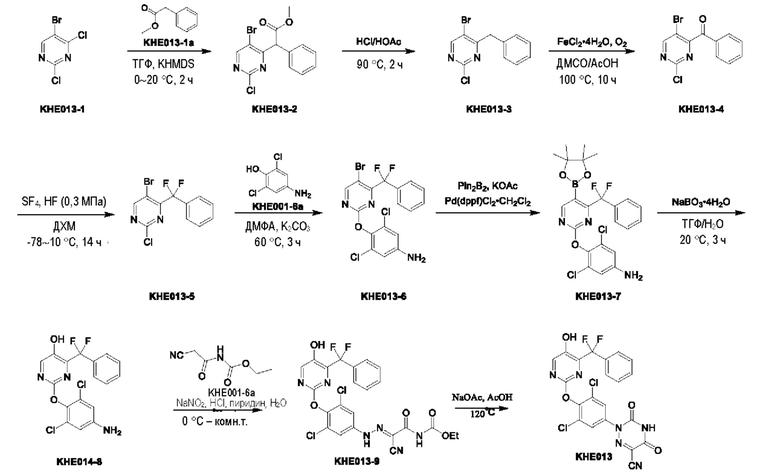
Соединение KHE013-2: KHE013-1 (250 г, 1,10 моль, 140 мл, 1,00 экв.) и KHE013-1а (165 г, 1,10 моль, 154 мл, 1,00 экв.) растворяли в тетрагидрофуране (2 л) и медленно по каплям добавляли KHMDS (1,0 М, 1,15 л, 1,05 экв.) при внешней температуре 0°С. После добавления реакционный раствор нагревали до 20°С и перемешивали в течение 2 часов. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 20/1) показал, что сырье полностью прореагировало и образовалось новое пятно (Rf=0,33). Реакционный раствор медленно выливали в насыщенный раствор хлорида аммония (1 л), а затем добавляли этилацетат для экстракции (600 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили с помощью безводного сульфата натрия и концентрировали с получением коричневого твердого вещества KHE013-2 (360 г, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 343,0 [М+Н]+.
Соединение KHE013-3: KHE013-2 (330 г, 966 ммоль, 1,00 экв.) добавляли к системе растворителя из концентрированной соляной кислоты (165 мл, чистота 37%) и уксусной кислоты (660 мл) и перемешивали в течение 2 часов при внешней температуре 90°С. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 20/1) показал, что сырье полностью прореагировало и образовалось новое соединение (Rf=0,48). Растворитель удаляли с помощью выпаривания при пониженном давлении, и остаточное маслянистое вещество сразу же очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 2/1) с получением белого твердого вещества KHE013-3 (117 г, 412 ммоль, выход: 37,6%). 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,93 (br d, J=6,0 Гц, 1H), 7,37 - 7,21 (m, 5Н), 4,23 (br d, J=2,4 Гц, 2H).
Соединение KHE013-4: KHE013-3 (30,0 г, 106 ммоль, 1,00 экв.) растворяли в смешанном растворе из ДМСО (300 мл) и уксусной кислоты (150 мл), добавляли FeCl2⋅4H2O (2,10 г, 10,6 ммоль, 0,10 экв.) и перемешивали при внешней температуре 100°С в течение 10 часов в токе кислорода. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 20/1) показал, что сырье полностью прореагировало и образовалось новое пятно (Rf=0,24). Реакционный раствор медленно выливали в этилацетат (200 мл) и воду (800 мл), этилацетатные слои разделяли и водные слои экстрагировали этилацетатом (150 мл × 2). Органические слои объединяли, промывали насыщенным солевым раствором (500 мл × 2), сушили с помощью безводного сульфата натрия, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 2/1) с получением желтого твердого вещества KHE013-4 (16,0 г, 53,8 ммоль, выход: 50,8%).
Соединение KHE013-5: SF4 (14,2 г, 131 ммоль, 3,00 экв.) и HF (8,74 г, 437 ммоль, 7,95 мл, 10,0 экв.) добавляли к раствору KHE013-4 (13,0 г, 43,7 ммоль, 1,00 экв.) в дихлорметане (50 мл) при внешней температуре -78°С. После добавления реакционный раствор перемешивали при внешней температуре 10°С и 0,30 МПа в течение 14 часов. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 5/1) показал, что реакция была завершена и образовалось новое соединение (Rf=0,70). Реакционный раствор выливали в этилацетат (100 мл) и доводили рН до 7-8 насыщенным раствором бикарбоната натрия (300 мл). Органические слои разделяли, и водные слои экстрагировали этилацетатом (100 мл × 3). Органические слои объединяли, промывали насыщенным солевым раствором (200 мл × 2), сушили с помощью безводного сульфата натрия, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 1/0 до 1/1) с получением бесцветного маслянистого вещества KHE013-5 (10,0 г, 31,3 ммоль, выход: 71,6%). МС (ESI) m/z: 320,8 [М+Н]+. 1Н ЯМР: (CDCl3, 400 МГц): δ 8,78 (s, 1H), 7,63 (dd, J=1,2, 7,2 Гц, 2Н), 7,54 - 7,41 (m, 3Н). 19F ЯМР: (CDCl3, 400 МГц): δ -96,31.
Соединение KHE013-6: методика была такой же, как и в случае соединения KHE007-3, и реакционный раствор очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 2/1) с получением желтого твердого вещества KHE013-6 (10,2 г, 22,1 ммоль, выход: 88,4%). МС (ESI) m/z: 460,0 [М+Н]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 9,03 (s, 1Н),7,64 - 7,44 (m, 5Н), 6,69 (s,2H),5,70 (s,2H).
Соединение KHE013-7: методика была такой же, как и в случае синтеза соединения KHE007-4; было получено коричневое маслянистое вещество KHE013-7 (25,0 г, неочищенный продукт). Неочищенный продукт сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 508,2 [М+Н]+.
Соединение KHE013-8: методика была такой же, как и в случае синтеза соединения KHE007-5; после очистки с помощью препаративной ВЭЖХ было получено грязно-белое твердое вещество KHE013-8 (3,28 г, 8,09 ммоль, выход: 33,3%, чистота 98,2%). МС (ESI) m/z: 380,0 [М+Н]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 10,65 (s, 1Н), 8,29 (s, 1Н), 7,57 - 7,43 (m, 5Н), 6,67 (s,2H). 19F ЯМР (ДМСО-d6, 400 МГц): δ -95,89.
Соединение KHE013-9: методика была такой же, как и в случае синтеза соединения KHE007-6, и было получено желтое твердое вещество KHE013-9 (512 мг, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 565,0 [М+Н]+.
Соединение KHE013: методика была такой же, как и в случае синтеза соединения KHE007, и после очистки с помощью препаративной ВЭЖХ было получено грязно-белое твердое вещество KHE013 (134 мг, выход: 65,2%, чистота 98,1%). МС (ESI) m/z: 519,1 [М+Н]+.
Пример 38. Синтез соединения KHE014
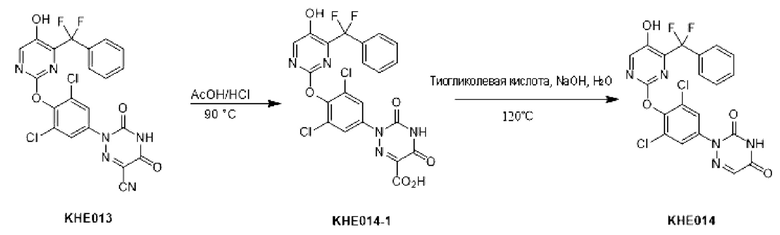
Соединение KHE014-1: методика была такой же, как и в случае синтеза соединения KHE008-1, и было получено желтое твердое вещество KHE014-1 (63 мг, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 538,0 [М+1]+.
Соединение KHE014: методика была такой же, как и в случае синтеза соединения KHE008, и после очистки с помощью препаративной ВЭЖХ было получено бледно-желтое твердое вещество KHE014 (24,0 мг, выход: 23,2% за две стадии, и чистота: 96,8%). МС (ESI) m/z: 494,1 [М+1]+.
Пример 39. Синтез соединения KHE015
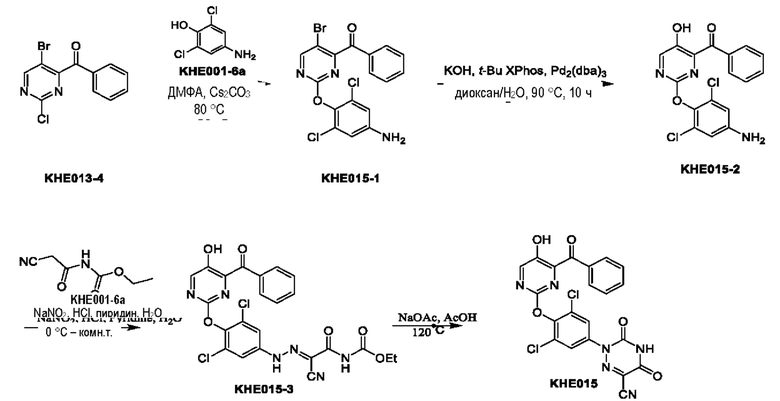
Соединение KHE015-1: методика была такой же, как и в случае синтеза соединения KHE007-3, и было получено желтое твердое вещество KHE015-1 (6,00 г, 13,7 ммоль, выход: 56,5%) после очистки с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 1/1). 1Н ЯМР (ДМСО-d6, 400 МГц): δ 9,09 (s, 1H), 7,85 - 7,75 (m, 3Н), 7,62 (t, J=7,7 Гц, 2Н), 6,66 (s, 2Н), 5,66 (br s, 2Н).
Соединение KHE015-2: соединение KHE015-1 (6,00 г, 13,7 ммоль, 1,00 экв.), гидроксид калия (3,07 г, 54,7 ммоль, 4,00 экв.) и t-Bu Xphos (580 мг, 1,37 ммоль, 0,10 экв.) добавляли к системе из диоксана (60 мл) и воды (15 мл), после чего добавляли Pd2(dba)3 (1,25 г, 1,37 ммоль, 0,10 экв.). После добавления раствор помещали при внешней температуре 90°С и перемешивали в течение 10 часов. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 3/1) показал, что реакция была завершена и образовалось новое пятно (Rf=0,22). Реакционный раствор выливали в этилацетат (50 мл) и воду (200 мл) и перемешивали в течение 10 минут. Органические фазы разделяли, и водные фазы экстрагировали этилацетатом (50 мл × 2). Органические фазы объединяли, промывали насыщенным солевым раствором (100 мл × 2), сушили с помощью безводного сульфата натрия, фильтровали, концентрировали и очищали с помощью препаративной ВЭЖХ с получением желтого твердого вещества KHE015-2 (2,45 г, 6,46 мкмоль, выход: 47,3%, чистота 99,2%). МС (ESI) m/z: 376,0 [М+Н]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 10,62 (s, 1Н), 8,44 (s, 1H), 7,83 - 7,77 (m, 2H), 7,72 (s, 1H), 7,56 (s, 2H), 6,64 (s, 2H), 5,84 - 5,29 (m, 2H).
Соединение KHE015-3: методика была такой же, как и в случае синтеза соединения KHE007-6, и было получено бледно-желтое твердое вещество KHE015-3 (625,7 мг, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (EST) m/z: 543,0 [М+Н]+.
Соединение KHE015: методика была такой же, как и в случае синтеза соединения KHE007, и после очистки с помощью препаративной ВЭЖХ было получено грязно-белое твердое вещество KHE015 (278,4 г, выход за две стадии: 31,1%, чистота 95,9%). 1Н ЯМР (ДМСО-d6, 400 МГц): 10,87(s, 1Н), 8,49(s, 1Н), 7,82(s, 1Н), 7,80(d, 1Н, J=4,0 Гц),7,77(s, 2Н), 7,71-7,74(m, 1Н), 7,55-7,59(m, 2Н).
Пример 40. Синтез соединения KHE016
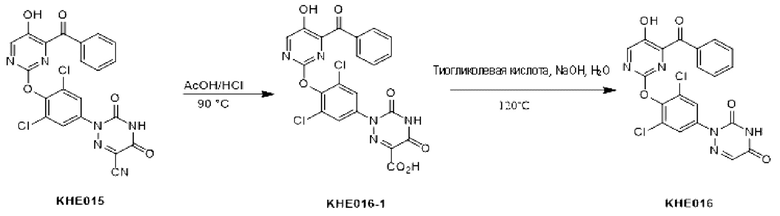
Соединение KHE016-1: методика была такой же, как и в случае синтеза соединения KHE008-1, и было получено желтое твердое вещество KHE016-1 (74,5 мг, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (EST) m/z: 516,0 [M+1]+.
Соединение KHE016: методика была такой же, как и в случае синтеза соединения KHE008, и после очистки с помощью препаративной ВЭЖХ было получено грязно-белое твердое вещество KHE016 (31,2 мг, выход: 26,3% за две стадии, и чистота: 97,2%). МС (EST) m/z: 472,0 [M+1]+.
Пример 41. Синтез соединения KHE017
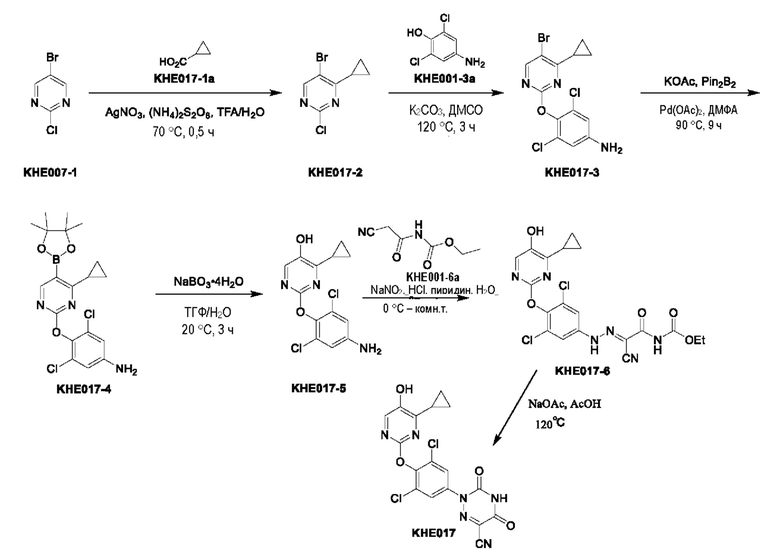
Соединение KHE017-2: методика была такой же, как и в случае соединения KHE007-2, и реакционный раствор очищали с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 1/0 до 5/1) с получением бесцветного маслянистого вещества KHE017-2 (9,50 г, 40,7 ммоль, выход: 78,7%). 1H ЯМР (CDCl3, 400 МГц): δ 8,46 - 8,39 (m, 1Н), 2,39 (tt, J=4,8, 8,0 Гц, 1Н), 1,25 - 1,20 (m, 1Н), 1,25 - 1,20 (m, 1H), 1,18 - 1,11 (m, 2Н).
Соединение KHE017-3: методика была такой же, как и в случае соединения KHE007-3, и было получено серое твердое вещество KHE017-3 (13,0 мг, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 375,9 [М+Н]+.
Соединение KHE017-4: методика была такой же, как и в случае соединения KHE007-4, и было получено желтое твердое вещество KHE017-4 (25,0 мг, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 422,3 [М+Н]+.
Соединение KHE017-5: методика была такой же, как и в случае соединения KHE007-5, и после очистки с помощью препаративной ВЭЖХ было получено желтое твердое вещество KHE017-5 (439 мг, 1,40 ммоль, выход: 13,1%, чистота 99,4%). МС (ESI) m/z: 312,0 [М+Н]+.
1H ЯМР (ДМСО-d6, 400 МГц): δ 9,89 (s, 1H), 7,89 (s, 1H), 6,64 (s, 1H), 6,70 - 6,60 (s, 1H), 5,52 (s, 2H), 2,39 - 2,25 (m, 1H), 1,06 - 0,93 (m, 2H), 0,88 - 0,76 (m, 2H).
Соединение KHE017-6: методика была такой же, как и в случае соединения KHE007-6, и было получено приблизительно 451,6 мг оранжевого твердого вещества KHE017-6, которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 479,1 [М+Н]+.
Соединение KHE017: методика была такой же, как и в случае соединения KHE007, и было взято 200 мг с получением приблизительно 56,2 мг белого твердого вещества KHE017 путем очистки с помощью препаративной ВЭЖХ. МС (ESI) m/z: 433,0 [М+Н]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 13,27 (s, 1H), 10,12 (s, 1H), 7,94 (s, 1H), 7,74 (s, 2Н), 2,28 (m, 1H), 1,04 (m, 2Н), 0,86 (m, 2Н).
Пример 42. Синтез соединения KHE018
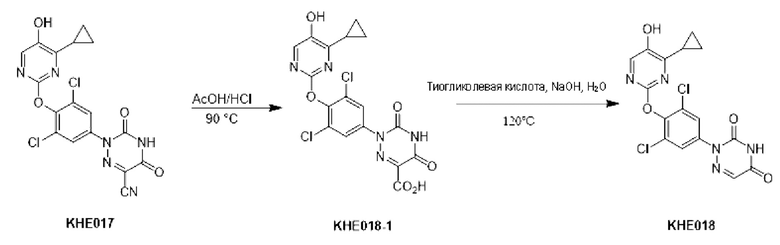
Соединение KHE018-1: методика была такой же, как и в случае синтеза соединения KHE008-1, и было получено желт о-коричневое твердое вещество KHE018-1 (205,7 мг, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 452,0 [М+Н]+.
Соединение KHE018: методика была такой же, как и в случае синтеза соединения KHE008, и после очистки с помощью препаративной ВЭЖХ было получено грязно-белое твердое вещество KHE018 (20 мг, чистота 96,81%). МС (ESI) m/z: 472,0 [М+Н]+. 1Н ЯМР (ДМСО-d6, 400 МГц): 12,46(s, 1H), 10,09(s, 1H), 8,25(s, 1H), 7,75(s, 2Н), 7,68(s, 1H), 2,32-2,39(m, 1H), 1,02-1,07 (m, 2Н), 0,85-0,90 (m, 2Н).
Пример 43. Синтез соединения KHE019
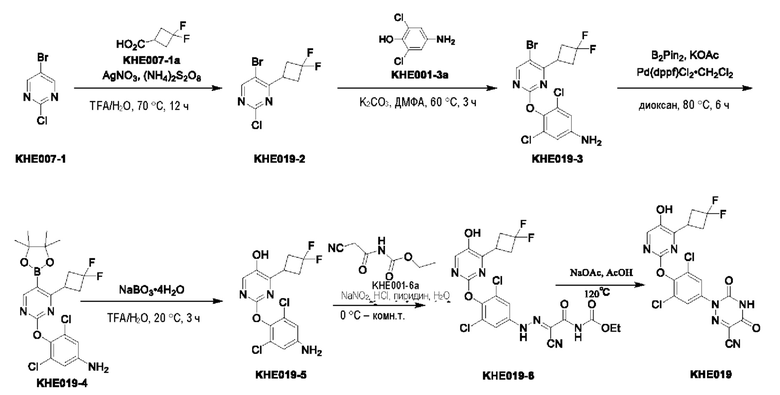
Соединение KHE019-2: методика была такой же, как и в случае KHE007-2, и было получено желтое твердое вещество KHE019-2 (9,00 г, 31,7 ммоль, выход: 68,2%) после очистки с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 10/1). МС (ESI) m/z: 283,0 [М+Н]+. 1H ЯМР (CDCl3, 400 МГц): δ 8,62 (s, 1H), 3,88 -3,60 (m, 1H), 3,11 - 2,90 (m, 4Н).
Соединение KHE019-3: методика была такой же, как и в случае KHE007-3, и было получено желтое твердое вещество KHE019-3 (5,20 г, 12,2 ммоль, выход: 86,7%) после очистки с помощью колоночной хроматографии на силикагеле (SiO2, петролейный эфир/этилацетат = от 50/1 до 10/1). МС (ESI) m/z: 424,0[М+Н]+. 1Н ЯМР (CDCl3, 400 МГц): δ 8,50 (s, 1H), 6,69 (s, 2Н), 4,06 - 3,75 (m, 2Н), 3,75 - 3,64 (m, 1H), 2,91 (td, J=8,4, 16,4 Гц, 4Н).
Соединение KHE019-4: методика была такой же, как и в случае KHE007-4, и было получено коричневое маслянистое вещество KHE019-4 (5,80 г, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 472,1 [М+Н]+.
Соединение KHE019-5: методика была такой же, как и в случае KHE007-5, и после очистки с помощью препаративной ВЭЖХ было получено белое твердое вещество KHE019-5 (1,1 г, 2,96 ммоль, выход: 24,0%, чистота 97,4%). МС (ESI) m/z: 362,0 [М+Н]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 10,13 (s, 1H), 8,01 (s, 1H), 6,67 (s, 2Н), 5,55 (s, 2Н), 3,62 (dquin, J=2,8, 8,8 Гц, 1H), 2,91 - 2,73 (m, 4Н). 19F ЯМР (ДМСО-d6, 400 МГц): δ -80,48 (d, J=12,8 Гц, 1F), -95,69 (d, J=12,8 Гц, 1F).
Соединение KHE019-6: методика была такой же, как и в случае соединения KHE007-6, и было получено приблизительно 324 мг оранжевого твердого вещества KHE019-6, которое фазу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 529,0 [М+Н]+.
Соединение KHE019: методика была такой же, как и в случае соединения KHE007, и было взято 120 мг с получением приблизительно 43,7 мг белого твердого вещества KHE019 путем очистки с помощью препаративной ВЭЖХ. МС (ESI) m/z: 483,0 [М+Н]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 13,25 (s, 1H), 10,377 (s, 1Н), 8,073 (s, 1Н), 7,779 (s, 2Н), 3,653-3,675 (m, 1Н), 2,836 -2,948 (m,4Н).
Пример 44. Синтез соединения KHE020
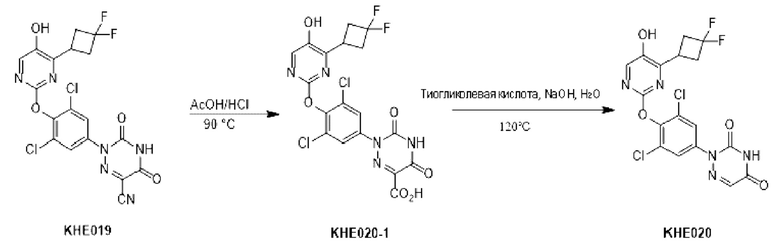
Соединение KHE020-1: методика была такой же, как и в случае синтеза соединения KHE008-1, и было получено коричневое твердое вещество KHE020-1 (157 мг, неочищенный продукт), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z: 502,0 [М+Н]+.
Соединение KHE020: методика была такой же, как и в случае синтеза соединения KHE008, и после очистки с помощью препаративной ВЭЖХ было получено грязно-белое твердое вещество KHE020 (15,4 мг, чистота 97,23%). МС (ESI) m/z: 458,0 [М+Н]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 12,480 (s, 1Н), 10,341 (s, 1Н), 8,073 (s, 1Н), 7,722 (s, 2Н), 7,718 (s, 1Н), 3,343-3,665 (m, 1H), 2,841-2,946 (m, 4Н).
Пример 45. Синтез соединения KHE021
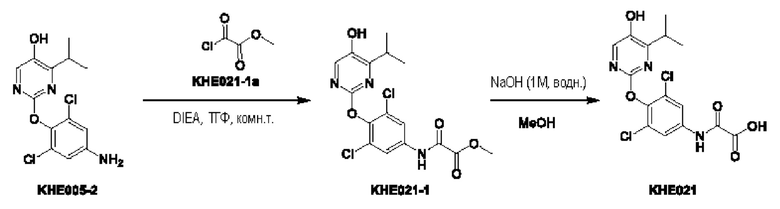
Соединение KHE021-1: соединение KHE005-2 (0,1095 г, 3,498 ммоль, 1,00 экв.) растворяли в ТГФ (10 мл), затем к реакционной системе добавляли KHE021-1а (0,4736 г, 3,882 ммоль, 12 экв.) и DIEA (0,9965 г, 7,665 ммоль, 24 экв.) и перемешивали при комнатной температуре в течение ночи. Контроль с помощью ТСХ показал, что образовалось новое пятно. Реакционный раствор разбавляли водой и экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, сушили с помощью безводного сульфата натрия, фильтровали и сушили с помощью ротационного испарения с получением остатков. Остатки очищали с помощью пластины для ТСХ с получением желтого твердого вещества KHE021-1 (80 мг, выход: 57,3%), отбирали образцы и использовали ЖХ-МС для определения сигнала продукта. МС (ESI)m/z: 400,0 [М+Н]+.
Соединение KHE021: соединение KHE021-1 (80 мг, 0,2 ммоль) растворяли в метаноле (6 мл) и добавляли раствор гидроксида натрия (1 мл, 1 М, водн.). Реакционный раствор перемешивали в течение ночи при комнатной температуре. Контроль с помощью ТСХ показал, что реакция была завершена. Реакционный раствор разбавляли 10 мл воды, и органический растворитель в реакционной смеси сразу же испаряли с помощью ротационного испарения. рН доводили до 3-4, а затем реакционный раствор экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, сушили с помощью безводного сульфата натрия и сушили с помощью ротационного испарения с получением остатков. Остатки очищали с помощью хроматопластины с получением белого твердого соединения KHE021 (12 мг, выход: 15,5%). МС (ESI) m/z: 386,0 [М+Н]+. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 9,87 (s, 1H), 7,94 (s, 1H), 6,65 (s, 2Н), 5,53 (s, 2Н), 3,23 - 3,28 (m, 1H), 1,11 (d, J=6,8 Гц, 6Н).
Пример 46. Синтез соединения KHE022
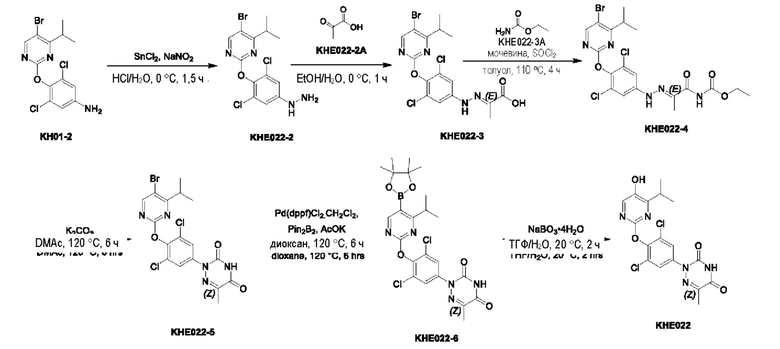
Соединение KHE022-2: KH01-2 (120 г, 318 ммоль, 1,0 экв.) добавляли к раствору соляной кислоты (12 М, 358 мл, 13,5 экв.), перемешивали в течение 30 минут и охлаждали до 0°С. Нитрит натрия (24,1 г, 350 ммоль, 1,1 экв.) растворяли в 50 мл воды, медленно по каплям добавляли к вышеуказанному раствору и после добавления непрерывно перемешивали при 0°С в течение 1 часа. Контроль с помощью ТСХ (петролейный эфир/этилацетат = 5/1) показал, что сырье полностью прореагировало и образовалось новое пятно (Rf=0,80). Двухлористое олово (215 г, 954 ммоль, 3,0 экв.) растворяли в соляной кислоте (12 М, 493 мл, 18,6 экв.), по каплям добавляли к вышеупомянутому реакционному раствору при 0°С и после добавления непрерывно перемешивали при этой температуре в течение 1 часа. Контроль с помощью ЖХ-МС показал, что сырье полностью прореагировало и образовался новый целевой продукт. Реакционный раствор фильтровали, и фильтровальный остаток собирали с получением неочищенного продукта в виде желтого твердого вещества KHE022-2 (80,0 г, 204 ммоль, выход: 64,1%) без дополнительной очистки. МС (ESI) m/z=392,9 [М+1]+.
Соединение KHE022-3: соединение KHE022-2 (80,0 г, 204 ммоль, 1,0 экв.) и KHE022-2А (32,3 г, 367 ммоль, 25,8 мл, 1,8 экв.) добавляли к 480 мл этанола и 1,5 л воды и перемешивали при 0°С в течение 1 часа. Контроль с помощью ЖХ-МС показал, что KHE022-2 полностью прореагировало и образовался целевой продукт. Реакционный раствор дважды экстрагировали этилацетатом (2 л × 2); этилацетатные слои объединяли и промывали 500 мл насыщенного солевого раствора, сушили с помощью безводного сульфата натрия, фильтровали и концентрировали с получением желтого твердого вещества KHE022-3 (26,5 г, 57,34 ммоль, выход: 28,1%), которое сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z=462,9 [М+1]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 1H ЯМР: ДМСО-d6, 400 МГц δ 12,16 (s, 1H), 10,04 (s, 1H), 8,75 (s, 1H), 7,56 (s, 2Н), 2,06 (s, 3Н), 1,24 - 1,14 (m, 6Н).
Соединение KHE022-4: соединение KHE022-3 (10,0 г, 21,64 ммоль, 1 экв.) добавляли к 2,0 л толуола, добавляли тионилхлорид (7,72 г, 64,92 ммоль, 4,71 мл, 3,0 экв.) при комнатной температуре, затем нагревали до 110°С и перемешивали в течение 2 часов. Реакционный раствор сушили с помощью ротационного испарения для удаления избытка тионилхлорида, затем добавляли 2,0 л толуола для растворения, а затем добавляли мочевину (5,78 г, 64,9 ммоль, 3,0 экв.) и KHE022-3А (2,89 г, 32,4 ммоль, 1,5 экв.), и перемешивали при 110°С в течение 2 часов. Контроль с помощью ЖХ-МС показал, что сырье полностью прореагировало и образовался целевой продукт. Реакционный раствор сушили с помощью ротационного испарения, добавляли 1 л воды и 1 л этилацетата и перемешивали при комнатной температуре в течение 30 минут. После этого органические фазы разделяли и водные фазы однократно экстрагировали 1 л этилацетата. Органические фазы объединяли, сушили с помощью безводного сульфата натрия, фильтровали и концентрировали. Остатки очищали с помощью обращенно-фазовой флэш-хроматографии (0,1% TFA) с получением желтого твердого вещества KHE022-4 (1,00 г, 1,45 ммоль, выход: 3,35%, чистота 71,4%). МС (ESI) m/z=534,0 [М+1]+.
Соединение KHE022-5: KHE022-4 (1,00 г, 1,88 ммоль, 1,0 экв.) растворяли в N,N-диметилацетамиде (15 мл), добавляли карбонат калия (777 мг, 5,63 ммоль, 3,0 экв.) и перемешивали при 120°С в течение 6 часов. Контроль с помощью ЖХ-МС показал, что сырье полностью прореагировало и образовался целевой продукт. К реакционному раствору добавляли 30 мл воды и экстрагировали этилацетатом (30 мл × 2). Органические фазы объединяли, сушили с помощью безводного сульфата натрия, фильтровали и концентрировали. Остатки очищали с помощью ВЭЖХ с получением желтого твердого вещества KHE022-5 (400 мг, 821 мкмоль, выход: 43,7%). МС (ESI) m/z=487,9 [М+1]+.
Соединение KHE022-6: KHE022-5 (400 мг, 821 мкмоль, 1,0 экв.) и Pin2B2 (521 мг, 2,05 ммоль, 2,5 экв.) растворяли в диоксане (4,0 мл), добавляли ацетат калия (241 мг, 2,46 ммоль, 3,0 экв.) и Pd(dppf)Cl2•CH2Cl2 (33,5 мг, 41,0 мкмоль, 0,05 экв.), и перемешивали при 120°С в течение 6 часов. Контроль с помощью ЖХ-МС показал, что сырье полностью прореагировало. Реакционный раствор фильтровали. Фильтрат собирали, добавляли 5 мл этилацетата, а затем поочередно промывали 5 мл воды и 5 мл насыщенного солевого раствора. Органические фазы собирали, сушили с помощью безводного сульфата натрия, фильтровали и концентрировали. Остатки очищали с помощью препаративной ВЭЖХ с получением желтого твердого вещества KHE022-6 (100 мг, 187 мкмоль, выход: 22,8%). МС (ESI) m/z=534,1 [М+1]+.
Соединение KHE022: KHE022-6 (90,0 мг, 168 мкмоль, 1 экв.) добавляли в смешанный раствор тетрагидрофурана (1,0 мл) и воды (1,0 мл), добавляли перборат натрия (NaBO3•4H2O) (150 мг, 673,91 мкмоль, 4,0 экв.) и перемешивали при комнатной температуре в течение 2 часов. Контроль с помощью ЖХ-МС показал, что сырье полностью прореагировало и образовался целевой продукт. К реакционному раствору добавляли 5 мл воды и экстрагировали этилацетатом (5,0 мл × 2). Органические фазы объединяли, сушили с помощью безводного сульфата натрия, фильтровали и концентрировали. Остатки очищали с помощью препаративной ВЭЖХ с получением желтого твердого вещества KHE022 (18,0 мг, 42,17 мкмоль, выход: 25,0%, чистота 99,4%). МС (ESI) m/z=424,0 [M+1]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 12,40 (s 1H), 10,07 (s, 1H), 8,01 (s, 1H), 7,78 (s, 2Н), 3,32 - 3,26 (m, 1H), 2,17 (s, 3Н), 1,16 - 1,10 (m, 6Н).
Пример 47. Синтез соединения KHE023
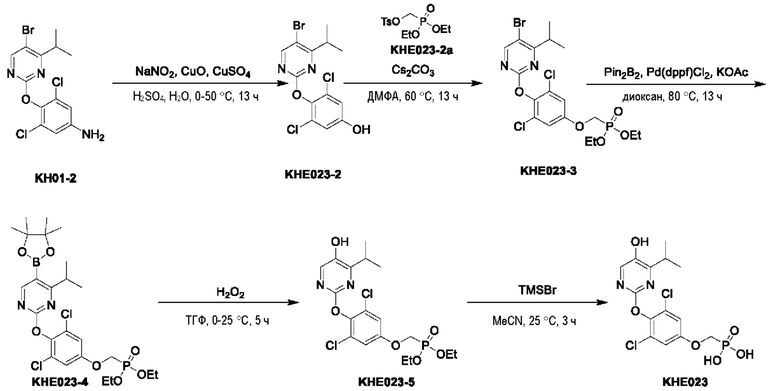
Соединение KHE023-2: KH01-2 (40,0 г, 106 ммоль, 1,00 экв.) добавляли к концентрированной серной кислоте (60,0 мл) и воде (25,0 мл), по каплям добавляли раствор нитрита натрия (7,32 г, 106 ммоль, 1,00 экв., растворенный в 150 мл воды) при 0°С и после добавления непрерывно перемешивали при этой температуре в течение 1 часа. Раствор сульфата меди (253 г, 1,59 моль, 244 мл, 15,0 экв., растворенный в 150 мл воды) по каплям добавляли к вышеупомянутому реакционному раствору, и в это время добавляли монооксид меди (8,44 г, 106,08 ммоль, 1,34 мл, 1,00 экв.). Реакционный раствор нагревали до 50°С и перемешивали в течение 2 часов. Контроль с помощью ЖХ-МС показал, что образовался целевой продукт. Реакционный раствор фильтровали. К фильтрату добавляли 800 мл этилацетата и промывали 200 мл насыщенного солевого раствора. Органические фазы собирали, сушили с помощью безводного сульфата натрия, фильтровали, концентрировали и очищали с помощью обращенно-фазовой ВЭЖХ с получением желтого твердого вещества KHE023-2 (7,60 г, 20,0 ммоль, выход: 18,96%). МС (ESI) m/z=378,9 [M+1]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 10,36 (s, 1H), 8,73 (s, 1H), 6,96 (s, 2Н), 3,40 - 3,36 (m, 1H), 1,12 (d, J=6,8 Гц, 6Н).
Соединение KHE023-3: KHE023-2 (4,50 г, 11,9 ммоль, 1,00 экв.) растворяли в ДМФА (50,0 мл), добавляли KHE023-2а (4,60 г, 14,2 ммоль, 3,68 мл, 1,20 экв.) и карбонат цезия (4,65 г, 14,2 ммоль, 1,20 экв.) и перемешивали при 60°С в течение 13 часов. Контроль с помощью ЖХ-МС показал, что образовался целевой продукт. Реагент выливали в 50 мл воды и экстрагировали этилацетатом (50 мл × 2). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл × 2), сушили с помощью безводного сульфата натрия, фильтровали и концентрировали. Остатки очищали с помощью колоночной хроматографии (петролейный эфир/этилацетат = от 50/1 до 2/1) с получением желтого маслянистого вещества KHE023-3 (3,80 г, 7,19 ммоль, выход: 60,4%). МС (ESI) m/z=529,0 [М+1]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 8,73 (s, 1H), 7,36 (s, 2Н), 4,57 (d, J=9,6 Гц, 2Н), 4,18 - 4,05 (m, 4Н), 3,37 (m, 1H), 1,25 (t, J=7,2 Гц, 6Н), 1,12 (d, J=6,8 Гц, 6Н).
Соединение KHE023-4: KHE023-3 (3,50 г, 6,63 ммоль, 1,00 экв.) растворяли в диоксане (85,0 мл), добавляли ацетат калия (1,30 г, 13,2 ммоль, 2,00 экв.), Pin2B2 (3,37 г, 13,2 ммоль, 2,00 экв.) и Pd(dppf)Cl3 (484 мг, 662 мкмоль, 0,10 экв.) и перемешивали при 80°С в течение 13 часов под защитой азота. Контроль с помощью ЖХ-МС показал, что образовался целевой продукт. Реакционный раствор выливали в 10 мл воды и экстрагировали этилацетатом (10 мл × 2). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл), сушили с помощью безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта желтого маслянистого вещества KHE023-4 (2,00 г), который сразу же подвергали взаимодействию на следующей стадии без очистки. МС (ESI) m/z=575,1 [М+1]+.
Соединение KHE023-5: KHE023-4 (2,00 г, 3,48 ммоль, 1,00 экв.) растворяли в тетрагидрофуране (40,0 мл), добавляли пероксид водорода (3,94 г, 34,7 ммоль, 3,34 мл, чистота 30,0%, 10,0 экв.) при 0°С, а затем нагревали до комнатной температуры и перемешивали в течение 5 часов. Контроль с помощью ЖХ-МС показал, что сырье полностью прореагировало и образовался целевой продукт. Реакционный раствор выливали в насыщенный сульфит натрия (50 мл), перемешивали в течение 10 минут, а затем экстрагировали этилацетатом (50 мл × 2). Органические фазы собирали, промывали насыщенным солевым раствором (50 мл), сушили с помощью безводного сульфата натрия, фильтровали и концентрировали. Остатки очищали с помощью препаративной ВЭЖХ с получением коричневого маслянистого вещества KHE023-5 (0,80 г, 1,72 ммоль, выход: 49,4%). МС (ESI) m/z=465,2 [M+1]+. 1H ЯМР (ДМСО-d-6, 400 МГц): δ 10,0 (br s, 1H), 7,98 (s, 1H), 7,32 (s, 2Н), 4,57 (d, J=9,6 Гц, 2Н), 4,13 (quin, J=7,2 Гц, 4Н), 3,31 - 3,25 (m, 1H), 1,27 (t, J=7,2 Гц, 6Н), 1,11 (d, J=6,8 Гц, 6Н).
Соединение KHE023: KHE023-5 (0,65 г, 1,40 ммоль, 1,00 экв.) растворяли в 20 мл ацетонитрила, добавляли триметилбромсилан (2,6 г, 16,8 ммоль, 2,2 мл, 12,0 экв.) и перемешивали при комнатной температуре в течение 3 часов. Контроль с помощью ЖХ-МС показал, что сырье исчезло и образовался целевой продукт. Реагент выливали в 20 мл метанола, перемешивали в течение 30 минут и сушили с помощью ротационного испарения при пониженном давлении. Остатки очищали с помощью препаративной ВЭЖХ с получением белого твердого вещества KHE023 (0,5 г, 1,22 ммоль, выход: 87,4%). МС (ESI) m/z=409,0 [М+Н]+. 1H ЯМР (ДМСО-d6, 400 МГц): δ 9,96 (br s, 1Н), 7,97 (s, 1H), 7,25 (s, 2H), 4,21 (d, J=10,0 Гц, 2H), 3,30 (br d, J=6,8 Гц, 1H), 1,11 (d, J=6,8 Гц, 6H).
Пример 48. Исследование связывания TRα или TRβ in vitro
In vitro анализ агонистического действия соединения на TRα или TRβ проводили с помощью исследования рекрутинга пептидов на основе анализа по методике времяразрешенного флуоресцентного индуктивно-резонансного переноса энергии. В эксперименте использовали европий-анти-GST антитело (Cisbio, 61GSTKLB), коактивированный пептид биотин-SRC2-2 (Sangon Biotech), стрептавидин-d2 (Cisbio, 610SADAB), RXRα (Pharmaron) и TRα-LBD (Invitrogen, PV4762) или TRβ-LBD (Invitrogen, PV4764) с GST-меткой. Европий-анти-GST антитело опосредованно метило TRα-LBD или TRβ-LBD путем связывания с GST-меткой. Стрептавидин-d2 (Cisbio, 610SADAB) опосредованно метил коактивированный пептид SRC2-2 путем связывания с биотиновой меткой. В случае существования RXRα, TRα-LBD или TRβ-LBD может образовывать гетеродимер TRα-LBD/RXRα или TRβ-LBD/RXRα с RXRα, соответственно. Агонист связывался с TRα-LBD/RXRα или TRβ-LBD/RXRα и приводил к конформациоиному изменению TRα-LBD или TRβ-LBD, таким образом улучшая способность гетеродимера к рекрутингу коактивированного пептида SRC2-2. При этом по мере уменьшения расстояния между (12-меченым коактивированным пептидом SRC2-2 и европий-анти-GST антителом сигнал TR-FRET увеличивался.
В зависимости от эффекта соединения на активность TRα или TRβ в различных концентрациях можно было оценить агонизм соединения.
Этапы работы:
a. Готовили 6 мМ раствор положительного эталонного соединения (MGL-3196) и подлежащих тестированию соединений (100Х) в диметилсульфоксиде (ДМСО), и 100% ДМСО использовали в качестве отрицательного контроля
b. 6 мМ (100Х) раствор положительного эталонного соединения (MGL-3196) или подлежащего тестированию соединения последовательно разбавляли 100% диметилсульфоксидом в соотношении 1:3 для получения в общей сложности 10 концентраций, и переносили в 96-луночный планшет
c. 4Х раствор соединения, подвергнутый разбавлению с градиентом концентрации, получали с помощью 1X рабочего буфера (50 мМ HEPES (рН 7,0), 50 мМ KF, 1 мМ DTT, 0,05% NP-40, 0,2% BSA).
d. 5 мкл 4Х раствора соединения, подвергнутого разбавлению с градиентом концентрации, добавляли в 384-луночный экспериментальный планшет.
e. 4Х TRαLBD и 4Х RXRα получали с помощью 1X рабочего буфера (50 мМ HEPES (рН 7,0), 50 мМ KF, 1 мМ DTT, 0,05% NP-40, 0,2% BSA).
f. 5 мкл 4X TRαLBD и 4X RXRα добавляли в 384-луночный экспериментальный планшет.
g. раствор смеси 2Х биотина-SRC2-2, 2Х европий-анти-GST и 2Х стрептавидина-d2 получали с 1X рабочим буфером (50 мМ HEPES (рН 7,0), 50 мМ KF, 1 мМ DTT, 0,05% NP-40, 0,2% BSA).
h. 10 мкл 2Х раствора смеси (со стадии g) добавляли в 384-луночный экспериментальный планшет.
i. Инкубацию проводили при комнатной температуре в темноте в течение 4 часов.
j. Значения сигнала флуоресценции при длинах волн 665 нм и 615 нм в каждой лунке 384-луночного экспериментального планшета регистрировали с помощью считывателя для микропланшетов Envision 2104 (PerkinElmer) и рассчитывали отношение сигналов флуоресценции при 665 нм/615 нм.
Анализ данных:
Рассчитывали относительное отношение каждой лунки: (отношение 665 нм/615 нм -отношение холостой пробы), а процент активности (% активности) рассчитывали следующим образом:
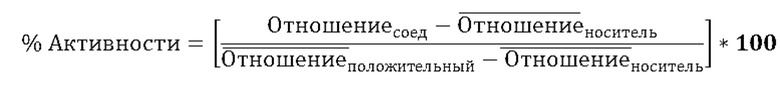
где:
Отношениеположитепьный представляло собой относительное отношение положительного контроля для всего планшета;
Отношениеноситепь представляло собой относительное отношение отрицательного контроля для всего планшета; и
Отношениесоед представляло собой относительное отношение соединения для всего планшета.
ЕС50 рассчитывали с помощью аппроксимации значений % активности и логарифма концентраций соединений методом нелинейной регрессии с помощью Graphpad 8.0.
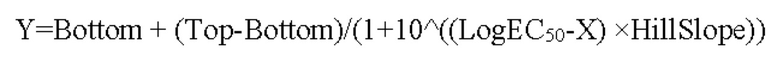
X: Логарифм концентрации соединения; Y: % активности.
Конкретные экспериментальные данные приведены в таблице 1.
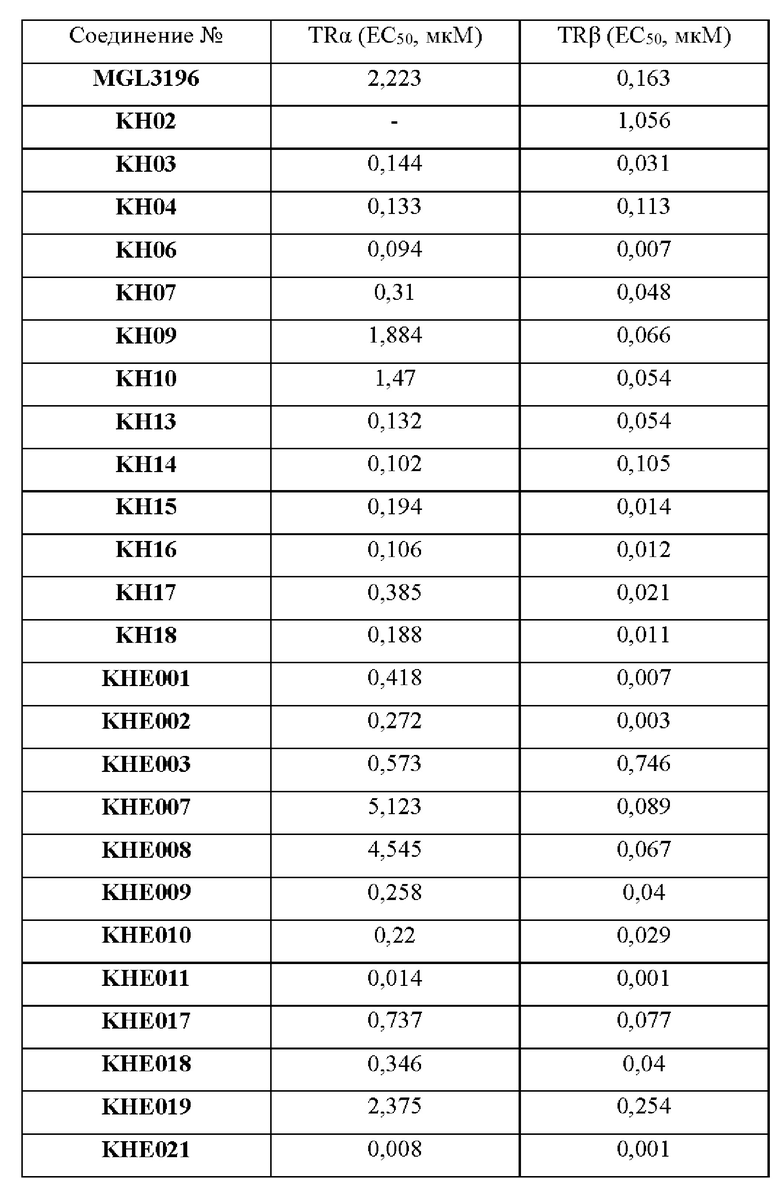
Соединение положительного контроля MGL3196 получали способом, описанным в CN101228135B.
Пример 49. Эксперимент по трансфекции клеток TRα или TRβ in vitro
Этот эксперимент был разработан для оценки агонистического действия соединения на TRα или TRβ. Кодирующие последовательности TRα-LBD или TRβ-LBD и RXRα-LBD вставляли в плазмиду pBIND (Promega, Е1581). Два вектора экспрессии и репортерные векторы (pGL4.35, несущий стабильно интегрированный репортерный ген люциферазы, управляемый промотором GAL4) (Promega, E3370) коэкспрессировали в клетках-хозяевах. Когда агонист связывается с соответствующим химерным рецептором, химерный рецептор связывается с сайтом связывания GAL4 на векторе репортерного гена и стимулирует экспрессию репортерного гена. Агонистическая активность соединения в отношении TRα или TRβ может быть оценена по интенсивности сигнала люминесценции
Подробные этапы:
Приготовление рабочего раствора
a) Готовили 30 мМ раствор эталонного соединения (MGL-3196) или подлежащего тестированию соединения в диметилсульфоксиде (ДМСО).
b) Все соединения подвергали 3-кратному градиентному разбавлению ДМСО, начиная с начальной концентрации 30 мМ и получив в общей сложности 10 градиентов концентрации.
c) Готовили положительный контроль 50 мкМ Т3 (трийодтиронин, полученный путем растворения в ДМСО) и отрицательный контроль (100% ДМСО).
d) Планшет с соединением закрывали и встряхивали в течение 5 минут.
Получение суспензии клеток
a) Все клетки культивировали в соответствии со стандартом АТСС, и анализ в HEK293T проводили во время их экспоненциальной фазы роста
b) Осторожно отбрасывали супернатант культуральной среды. Дважды промывали клетки с помощью PBS.
c) Для расщепления клеток добавляли трипсиновый расщепляющий раствор TrypLE™ (Gibco), и расщепление прекращали с помощью полной культуральной среды (Gibco).
d) Клетки собирали и подсчитывали. Эксперимент можно было проводить только тогда, когда жизнеспособность клеток составляла более 90%
e) 2,5×106 клеток HEK293T инокулировали в каждую чашку для культивирования клеток размером 60 мм, соответственно.
1) Культуральную чашку, инокупированную клетками, культивировали в инкубаторе с 5% СО2 при 37°С в течение ночи.
Трансфекция клеток
a) Реагент для трансфекции Fugene6 (Promega, Е2691) помещали при комнатной температуре.
b) 30 мкл реагента Fugene6 добавляли к культуральной среде Opti-MEM™ (Gibco, 11058-021), избегая контакта со стенкой пробирки.
c) Смесь равномерно перемешивали с помощью пипетки и оставляли отстояться при комнатной температуре в течение 5 минут.
d) К разбавленному реагенту для трансфекции добавляли 6 мкг плазмид (плазмиду pBIND (Pharmaron), в которую были вставлены кодирующие последовательности TRα-LBD или TRβ-LBD и RXRα-LBD, и плазмиду репортерного гена pGL4.35 (Promega, Е1370)). Смесь равномерно перемешивали с помощью пипетки и оставляли отстояться в течение 20 минут при комнатной температуре
e) Реагент для трансфекции, смешанный с плазмидной ДНК, добавляли к чашке для культивирования клеток размером 60 мм, инокулированной клетками.
f) Культуральную чашку культивировали в инкубаторе с 5% СО2 при 37°С в течение 5 часов.
Обработка соединениями
a) Разбавленные растворы соединений, положительный контроль и отрицательный контроль переносили в 384-луночный планшет для культивирования клеток (PerkinElmer, 6007680-50) с помощью Echo550 (Labcyte, 550).
b) Клетки инокулировали в 384-луночный планшет для культивирования клеток в количестве 15000 клеток на лунку и добавляли 25 мкл культуральной среды, содержащей 5% фетальной бычьей сыворотки (Gibco, 16000-044).
c) Клетки культивировали в инкубаторе с 5% СО2 при 37°С в течение ночи.
Детекция соединения
a) Детектирующий реагент Steady-Glo™ (Promega, Е2520) помещали при комнатной температуре.
b) 384-луночный планшет для культивирования клеток помещали при комнатной температуре.
c) 25 мкл детектирующего реагента Steady-Glo™ добавляли в каждую лунку планшета для культивирования клеток.
d) Культуральный планшет помещали на осциллятор для встряхивания в темноте в течение 5 минут.
Значение люминесценции детектировали с помощью Envision 2104 (PerkinElmer, Envision HTS).
Анализ данных:
Рассчитывали сигнал флуоресценции в RLU (Сигнал) для каждой лунки, а затем процент активности (% Активности), как показано ниже
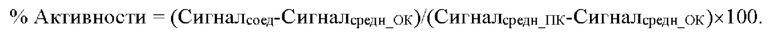
где:
Сигналсредн_ПК представлял собой средний сигнал флуоресценции в RLU положительного контроля для всего планшета;
Сигналсредн_ОК представлял собой средний сигнал флуоресценции в RLU отрицательного контроля для всего планшета; и
Сигналсоед представлял собой средний сигнал флуоресценции в RLU соединения для всего планшета.
ЕС50 рассчитывали с помощью аппроксимации значений % активности и логарифма концентраций соединений методом нелинейной регрессии с помощью Graphpad 8.0.
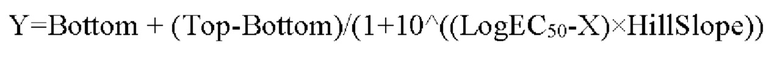
X: Логарифм концентрации соединения; Y: % активности.
Конкретные экспериментальные данные приведены ниже в таблице 2.
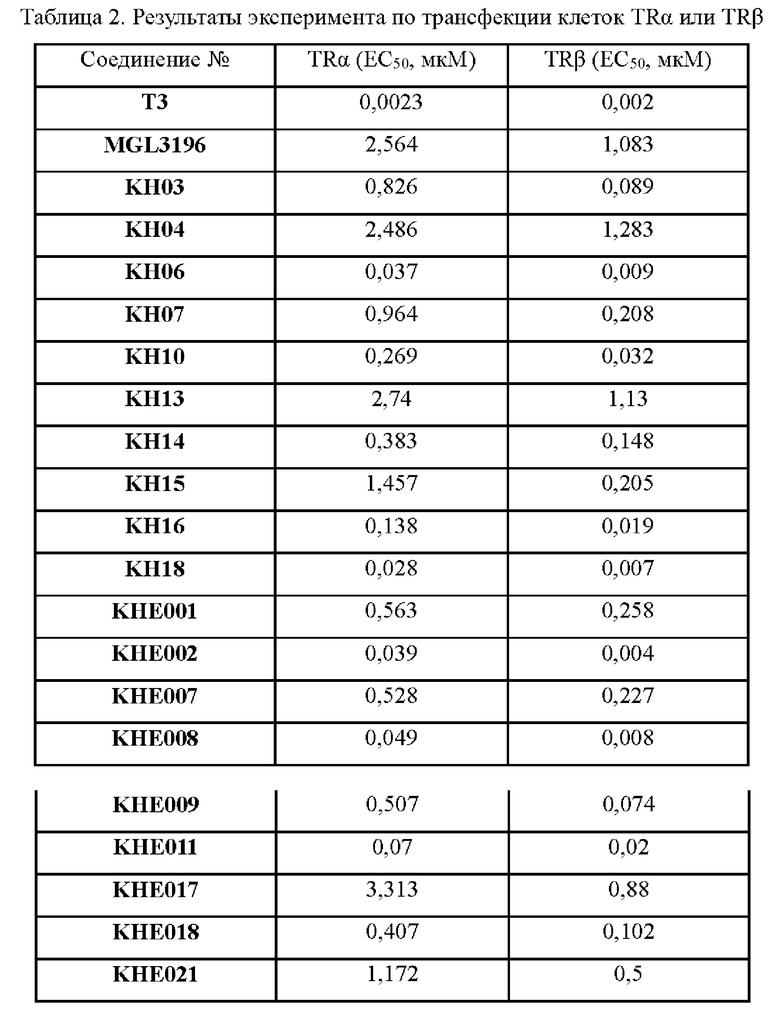
Пример 50. Выявление гепатотоксичности in vitro
Информация о первичных гепатоцитах
Эксперимент:
Этап 1: готовили 200 мМ исходный раствор тестируемого вещества в ДМСО, а затем подвергали 3-кратному последовательному градиентному разбавлению с получением 7 концентраций, затем 1,5 мкл каждого из растворов в 8 концентрациях добавляли в 498,5 мкл инкубационной культуральной среды (имеющей состав, указанной в таблице 5, сразу же равномерно перемешивали) для получения рабочего раствора, и инкубационную культуральную среду предварительно нагревали до 37°С перед приготовлением. ДМСО использовали в качестве контроля растворителем. Содержание ДМСО в рабочем растворе и контроле растворителем составляло 0,3 об.%. Концентрации соединений были следующими:
Этап 2: инокуляция планшета и культивирование клеток Второй этап: инокуляция планшета и культивирование клеток
1) Брали пробирку с гепатоцитами донора, используемого в эксперименте, консервированными при ультранизкой температуре, и перед реанимацией проверяли, что гепатоциты по-прежнему заморожены при низкой температуре. Гепатоциты быстро помещали на водяную баню при 37°С и осторожно встряхивали до тех пор, пока все кристаллы льда не растворились, и переносили в бокс биологической безопасности после распыления 70 об.% этанола.
2) Содержимое пробирки с гепатоцитами выливали в центрифужную пробирку на 50 мл, содержащую 50 мл реанимационной культуральной среды (имеющей состав, указанной в таблице 3, сразу же равномерно перемешивали), и центрифугировали при 80 g в течение 8 минут. После центрифугирования реанимационную культуральную среду аспирировали и добавляли инокуляционную культуральную среду (имеющей состав, указанной в таблице 4, сразу же равномерно перемешивали). Клетки подсчитывали с помощью окрашивания AO/PI (акридиновым оранжевым и иодидом пропидия) с получением клеточной суспензии с плотностью клеток 0,2 × 106 клеток на миллилитр.
3) Вышеуказанную клеточную суспензию инокулировали в 96-луночный планшет, покрытый коллагеном I, по 100 мкл на лунку. Культуральный планшет помещали в инкубатор с 5% CO2 с относительной влажностью 95% для культивирования в течение 4-6 часов.
4) После инкубации в течение 4-6 часов наблюдали состояние клеток под микроскопом. Культуральный планшет осторожно встряхивали, инокуляционную культуральную среду аспирировали, и в каждую лунку добавляли 100 мкл инкубационной культуральной среды (имеющей состав, указанной в таблице 5). Тест на токсичность можно было проводить после культивирования в инкубаторе в течение 18-20 часов.
5) Перед введением под микроскопом наблюдали морфологию клеток. Культуральную среду в культуральном планшете аспирировали, и в каждую лунку добавляли 100 мкл контроля растворителем (ДМСО) или рабочего раствора тестируемого вещества. В каждом случае тестировали три параллельных образца.
6) Свежеприготовленный рабочий раствор или контроль растворителем использовали для замены раствора каждые 24 часа после введения дозы.
После действия рабочего раствора в течение 48 часов под микроскопом наблюдали морфологию клеток для последующего использования, этап 3: выявление цитотоксичности
1) Реагент CellTiter-Glo (Promega, артикульный номер G9243), хранящийся при -20°С, расплавляли на водяной бане при 37°С.
2) После инкубации планшета для культивирования клеток, полученного выше, в течение 48 часов, 50 мкл раствора CellTiter-Glo сразу же добавляли в каждую экспериментальную лунку.
3) Планшет для культивирования клеток перемешивали на вортексе при 400 об/мин в течение 10 минут и инкубировали при комнатной температуре для стабилизации сигнала люминесценции.
Через 10 минут из каждой лунки аспирировали 100 мкл реакционного раствора и переносили в новый белый непрозрачный планшет с плоским дном (96-луночный планшет Corning, кат. №3917).
Значение хемилюминесценции каждой лунки считывали с помощью считывателя для микропланшетов (значение люминесценции белого непрозрачного планшета с плоским дном регистрировали как «значение люминесценцииХолостого определения»; и значение люминесценции контроля растворителем регистрировали как «значение люминесценциирастаорителя»).
этап 4: обработка данных
Жизнеспособность клеток (%)=[(значениелюминесценцииподлежащего тестированию соединения -значение люминесценциихолостого определения)/(значение люминесценциирастворителя - значение люминесценциихолостого определения)] × 100%
Строили график зависимости жизнеспособности клеток (%) от концентрации соединения и рассчитывали IC50 соединения путем аппроксимации жизнеспособности клеток (%) и концентрации соединения с помощью GraphPad Prism 8.0.2.
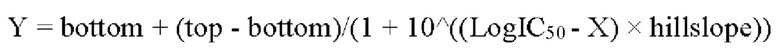
X представлял собой концентрацию соединения; a Y представлял собой жизнеспособность клеток (%).
Конкретные экспериментальные данные приведены в таблице 6.




Пример 51. ФК эксперимент на яванских макаках
Целью данного эксперимента являлась оценка фармакокинетического (ФК) поведения тестируемого соединения после однократного внутривенного и внутрижелудочного введения, а также изучение биодоступности после внутрижелудочного введения, предоставление экспериментальных данных на животных для клинических исследований.
Получение растворов для внутривенного введения и внутрижелудочного введения: соответствующее количество подлежащего тестированию соединения точно взвешивали и смешивали с подходящим растворителем (5 об.% ДМСО + 10 об.% полиэтиленгликоля - 15-гидроксистеарата + 85 об.% физиологического раствора). После вортексной или ультразвуковой обработки получали бесцветный и прозрачный раствор (раствор для внутривенного введения) или однородную суспензию. Раствор, вводимый путем внутривенной инъекции, необходимо было фильтровать через мембрану с фильтром с размером пор 0,22 мкм.
План эксперимента: перед введением первой дозы яванские макаки были разделены на 2 группы по массе тела, по 3 самца яванских макаков в каждой группе, где животным в 1 группе вводили тестируемое соединение (1 мг/кг) путем однократной внутривенной инъекции (в/в); и животным во 2 группе вводили подлежащее тестированию соединение (5 мг/кг) путем однократного внутрижелудочного введения (п/о). Животных взвешивали перед введением и рассчитывали дозировки на основании массы животных.
Взятие образцов: брали образец цельной крови (около 0,2 мл) в определенное время путем венепункции верхней конечности (или других подходящих мест забора крови), и фактическое время забора крови записывали в протоколы испытаний. Допустимая погрешность для одной временной точки сбора данных в течение 1 часа после введения составляет ±1 минуту, а допустимая погрешность для других временных точек составляет ±5% от теоретического времени. Все образцы крови немедленно переносили в маркированную коммерческую центрифужную пробирку, содержащую К2-ЭДТА. После взятия образца крови образец крови центрифугировали при 4°С и 3200 g в течение 10 минут, супернатантную плазму аспирировали, быстро помещали в сухой лед и хранили в холодильнике при -70±10°С для анализа методом ЖХ-МС/МС. Время взятия в обеих группах составляло 0,083 часа, 0,25 часа, 0,5 часа, 1 час, 2 часа, 4 часа, 8 часов и 24 часа после введения.
Обработка данных: площадь под кривой (AUC(o-t) и AUC(o-∞)), период полувыведения (Т1/2), максимальную концентрацию (Cmax), время достижения максимальной концентрации в плазме (Tmax) и т.д. рассчитывали с помощью модуля некомпартментного анализа в Phoenix WinNonlin 7.0.
Биодоступность (F)=площадь под кривой AUC(o-t) в случае п/о введения × дозировкав/в/(площадь под кривой AUC(o-t) в случае в/в введения × дозировкап/о) × 100%
Конкретные данные приведены в таблице 7.
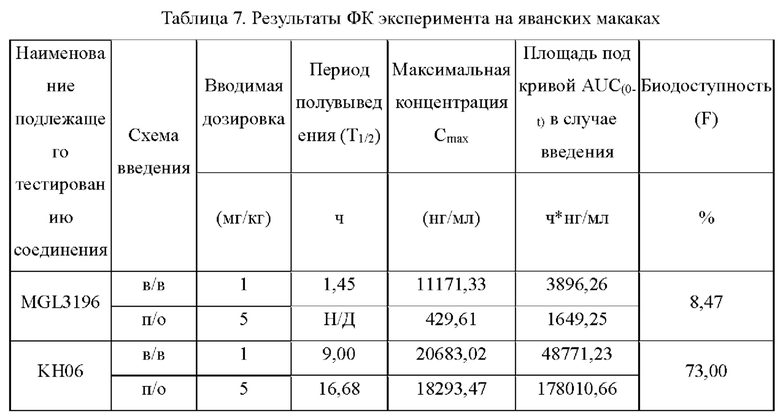
Пример 52. Фармакологический эксперимент in vivo
На ранних стадиях этого эксперимента мыши с ДНО (диета-индуцированное ожирение) получали корм с высоким содержанием жиров, а затем внутрибрюшинную инъекцию CCl4 во время кормления кормом с высоким содержанием жиров для индукции модели НАСГ. В этой модели оценивали эффективность тестируемого соединения против НАСГ
Информация о животных: использовали самцов мышей C57BL/6J (в возрасте 18 недель), включая 32 мыши с ДИО + 8 нормальных мышей, причем масса мышей с ДИО составляла >38 г.
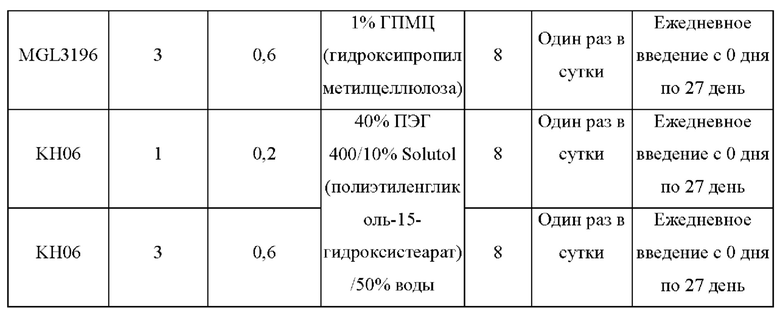
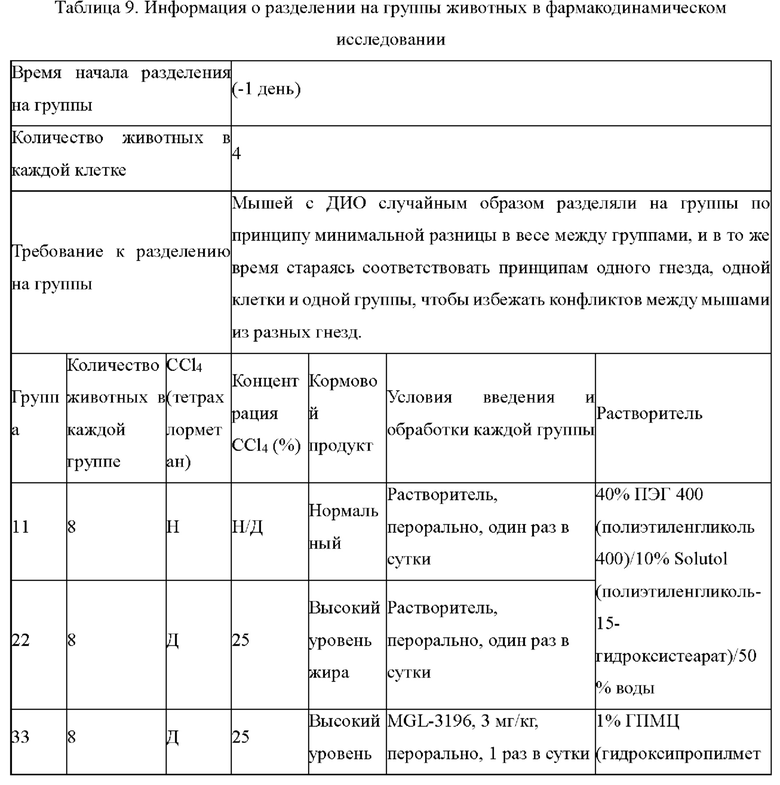
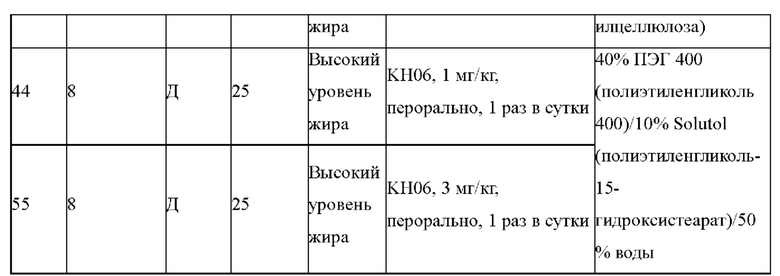
Индукция с помощью CCl4: готовили 25% (объемное соотношение) раствор CCl4 путем помещения 1 части CCl4 и 3 частей оливкового масла в стеклянную бутылку. Раствор хорошо перемешивали и использовали сразу после приготовления. Восьми мышам в первой группе внутрибрюшинно инъецировали физиологический раствор в качестве нормального контроля. Мышам во 2 и 5 группах внутрибрюшинно инъецировали 25% раствор CCl4 два раза в неделю. 25% (объемное соотношение) CCl4 вводили по массе тела, 0,5 мл/кг. Интервал между инъекцией CCl4 и временем введения в этот день должен составлять более 4 часов
Гистопатологический анализ
Все образцы печени обезвоживали с помощью обезвоживающего инструмента (Leica HistoCore Pearl-0348), а затем заливали с помощью аппарата для заливки парафином (HistoCore Arcadia). Залитый образец печени затем нарезали с помощью аппарата Lecia RM2235.
Оценка по шкале NAS (шкала активности НАЖБП)
Оценки по шкале NAS проводили на срезах, окрашенных НЕ (гематоксилин-эозином), и они представляли собой сумму стеатоза, баллонной дистрофии и дольчатого воспаления. Срезы были оценены патологоанатомом в соответствии со стандартами, представленными ниже в таблице 10.


Критерии оценки очагов
(1) Баллонная дегенерация гепатоцитов: в гепатоцитах наблюдалось патологическое изменение, схожее с вакуолью. Вследствие вакуолеподобного изменения размер гепатоцитов был увеличен, а ядра гепатоцитов концентрировались или отклонялись.
(2) Инфильтрация воспалительных клеток: большое количество агрегированных воспалительных клеток, в основном нейтрофилов и макрофагов, было обнаружено вокруг области воротной вены, области брюшной вены или вокруг долей печени.
(3) Стеатоз: в гепатоцитах разных размеров наблюдались регулярные круглые вакуоли, а ядра гепатоцитов располагались по краям.
Процент фиброза
Все срезы, окрашенные сириусом красным, сканировали с помощью сканера Leica Aperio АТ2 Brightfield, а затем рассчитывали процент площади положительной окраски сириусом красным с помощью системы HALO AI для оценки процента площади сириуса красного от общей просканированной площади печени.
Результаты представлены на ФИГ. 1 и ФИГ. 2.
На ФИГ. 1 показаны результаты оценки фиброза, где эффективность KH06 была дозозависимой, и все дозы обеспечили эффект снижения доли фиброза. Эффективность KH06 в дозировке 1 мг/кг была лучше, чем у MGL3196 в дозировке 3 мг/кг.
На ФИГ. 2 показаны результаты оценки по шкале NAS, где ордината оценки по шкале NAS представляла собой сумму баллов стеатоза, баллонной дистрофии и дольчатого воспаления. По сравнению с модельной группой, эффект KH06 был дозозависимым, и у всех животных было достигнуто значительное снижение оценки по шкале. При этом KH06 в дозе 1 мг/кг был столь же эффективен, что и MGL3196 в дозе 3 мг/кг.
В целях описания и раскрытия, все патенты, патентные заявки и другие публикации в явном виде включены в настоящий документ посредством ссылки. Эти публикации приведены лишь потому, что их раскрытие предшествует дате подачи настоящей заявки. Все заявления о датах всех этих документов, или выражения содержания этих документов основаны на информации, имеющейся в распоряжении заявителя, и не являются каким-либо признанием правильности дат этих документов или содержания этих документов. Кроме того, в любой стране любая ссылка на эти публикации в настоящем документе не означает признания того, что данная публикация стала частью общедоступных сведений в данной области техники.
Специалистам в данной области техники будет ясно, что объем настоящей заявки не ограничивается различными конкретными вариантами реализации и примерами, описанными выше. Однако могут быть сделаны различные модификации, замены или перегруппировки, без отклонения от сущности настоящей заявки, все из которых входят в объем охраны настоящей заявки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2625309C2 |
| НОВЫЙ ИНГИБИТОР ЦИКЛИНЗАВИСИМОЙ КИНАЗЫ CDK9 | 2018 |
|
RU2738654C1 |
| МОДУЛЯТОРЫ КАЛЬПАИНА И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2773288C2 |
| МОДУЛЯТОРЫ АКТИВНОСТИ НЕС1 И СПОСОБЫ ДЛЯ НИХ | 2011 |
|
RU2576036C2 |
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ НЕЙРОПРОТЕКТОРНЫМ ДЕЙСТВИЕМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2802457C1 |
| ЗАМЕЩЁННЫЕ ИНДОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ПОНИЖАЮЩИХ РЕГУЛЯТОРОВ РЕЦЕПТОРОВ ЭСТРОГЕНА | 2017 |
|
RU2722441C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ С КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ, ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, СПОСОБ ПОЛУЧЕНИЯ, КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2732576C2 |
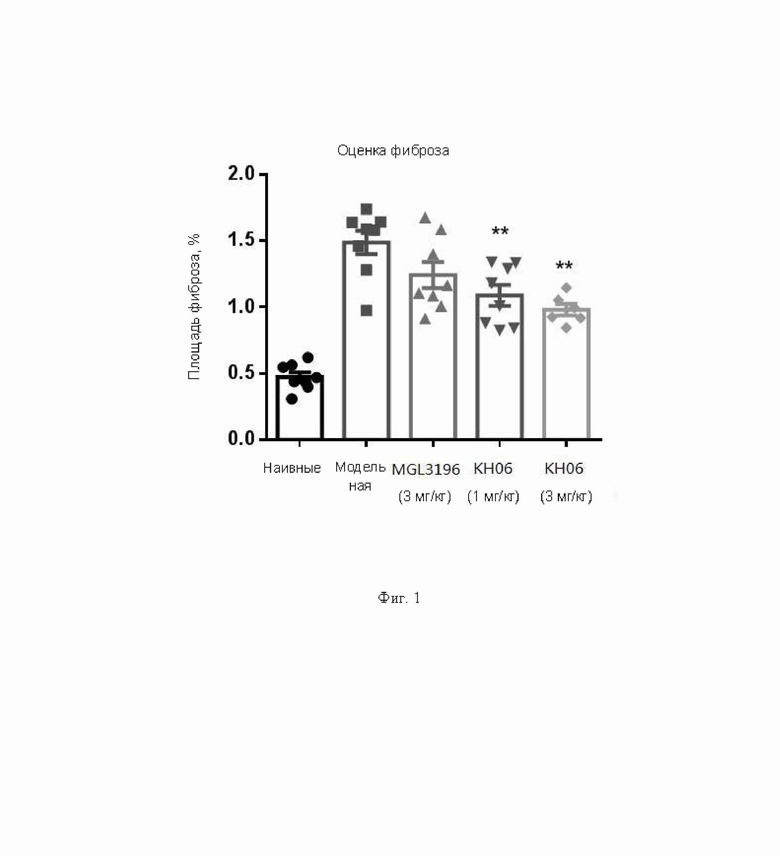
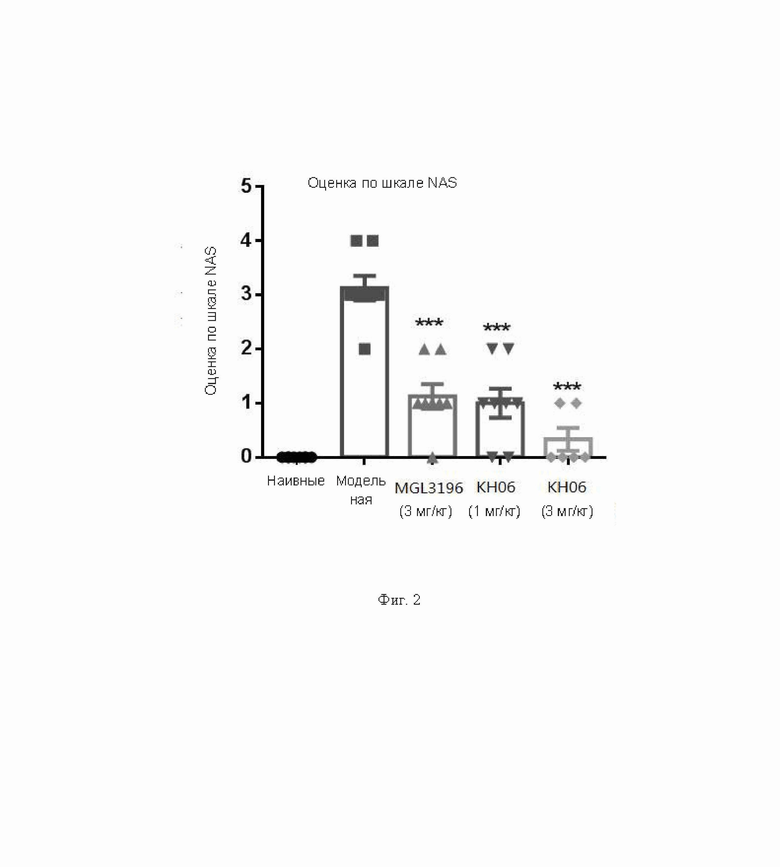
Изобретение относится к новым агонистам β-рецептора тиреоидных гормонов формулы (I) и их применению для предотвращения или лечения заболеваний, связанных с действием агониста β-рецептора, где указанное заболевание включает, например, ожирение, гиперлипидемию, гиперхолестеринемию, диабет, заболевание печени, в частности жировой гепатоз, НАСГ, НАЖБП, сердечно-сосудистые заболевания, в частности атеросклероз, заболевания щитовидной железы, в частности гипотиреоз или рак щитовидной железы. Соединения обладают лучшей активностью, селективностью или безопасностью. 3 н. и 13 з.п. ф-лы, 2 ил., 10 табл., 52 пр.
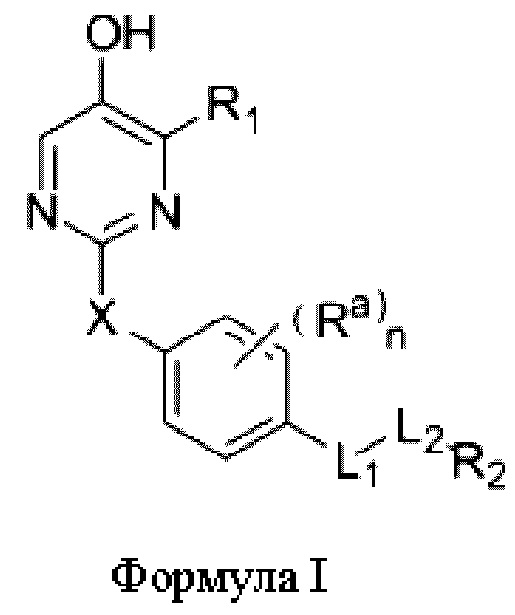
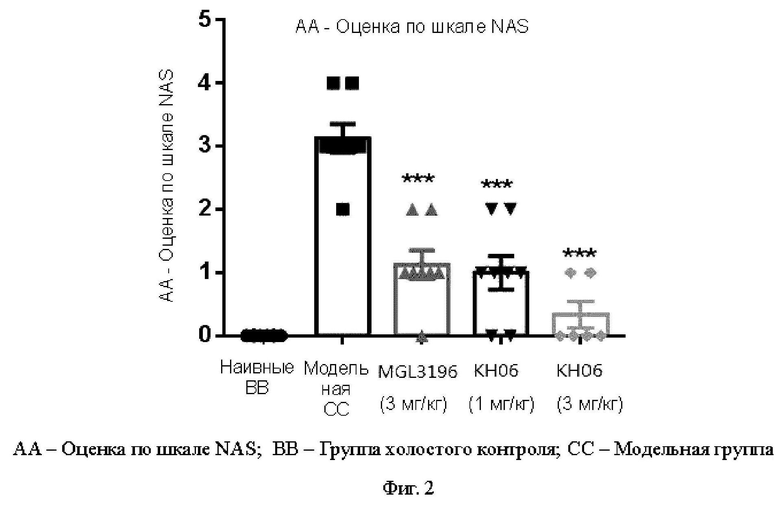
1. Соединение формулы I или его фармацевтически приемлемая соль:
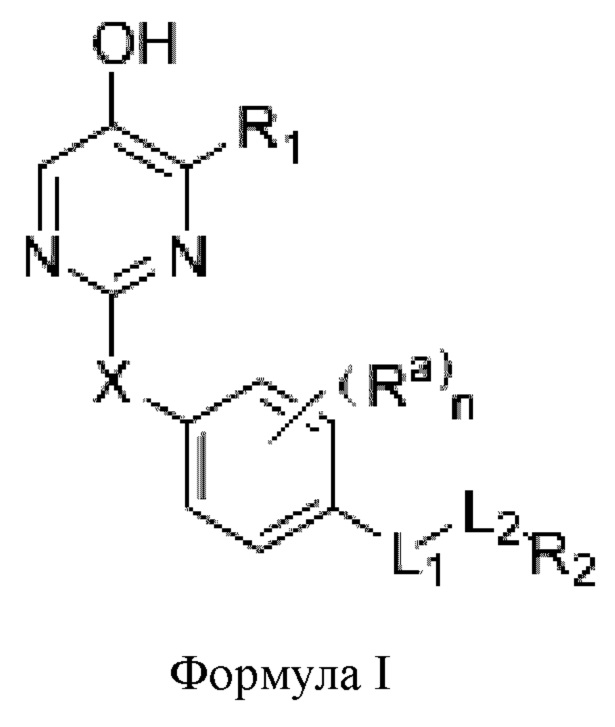
где
R1 представляет собой C1-6 линейный или разветвленный алкил, С3-6циклоалкил, С3-6циклоалкилС1-6алкил, С5-10арилС1-6алкил или -COR10, где указанный C1-6алкил, С3-6циклоалкил, С3-6циклоалкилС1-6алкил, С5-10арилС1-6алкил является незамещенным или замещен дейтерием, C1-6 алкилом или галогеном;
X представляет собой метилен, -О- или -S-;
Ra выбран из водорода, галогена или C1-6 линейного и разветвленного алкила; или два соседних Ra связаны с образованием 5-членного карбоциклического кольца или 5-членного гетероциклического кольца, содержащего 1 гетероатом кислорода;
L1 представляет собой одинарную связь, метилен, -О-, -NR3-, -NR3CO-;
L2 представляет собой одинарную связь или - (CR4R5)p;
R2 представляет собой карбоксил или группу, представленную следующей формулой: 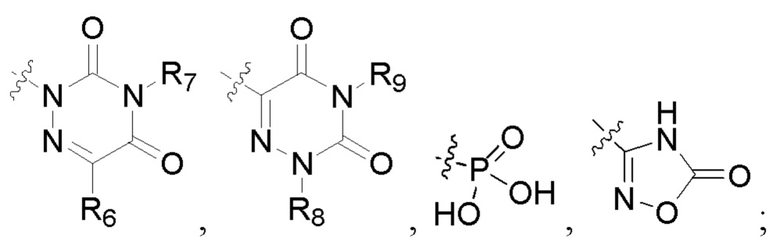
R3 представляет собой водород;
каждый из R4 и R5 независимо выбран из водорода;
R6 представляет собой водород, циано, СООН или C1-6 алкил;
R8 представляет собой водород, C1-6 алкил;
R7 и R9 представляют собой водород;
R10 представляет собой фенил; n составляет 2 или 3; и
р составляет 0 или 1.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где соединение формулы I представлено формулой II:
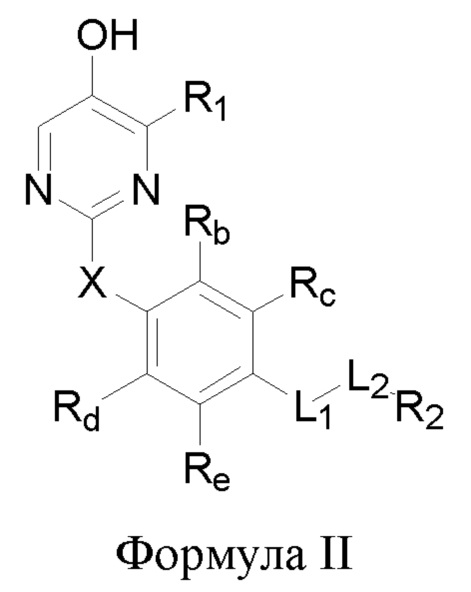
где Rb, Rc, Rd и Re представляют собой водород, галоген или C1-6 линейный или разветвленный алкил; или Rb и Rc связаны с образованием 5-членного циклоалкила или 5-членного неароматического гетероциклического кольца, содержащего 1 гетероатом кислорода; или Rd и Re связаны с образованием 5-членного циклоалкила или 5-членного неароматического гетероциклического кольца, содержащего 1 гетероатом кислорода.
3. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, где соединение формулы I представлено формулой III:
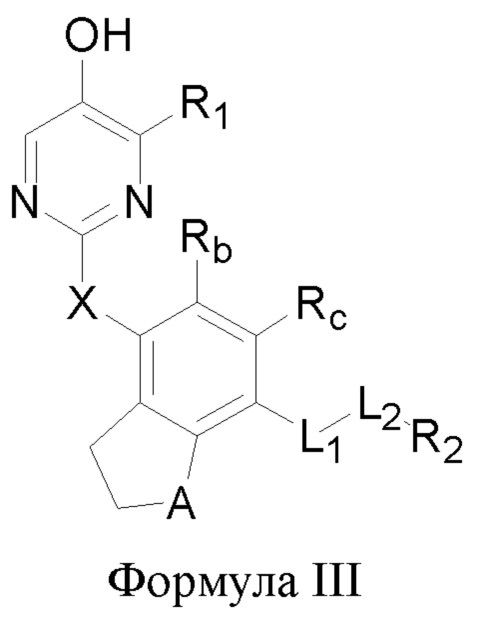
где
Rb и Rc представляют собой водород, галоген или C1-6 линейный или разветвленный алкил; и
А представляет собой О или метилен.
4. Соединение или его фармацевтически приемлемая соль по любому из предыдущих пунктов, где R1 выбран из:
1) C1-6 линейного или разветвленного алкила;
2) С3-6 циклоалкила или С3-6циклоалкилС1-6алкила;
3) С5-10арилС1-6алкила;
где C1-6 линейный или разветвленный алкил, С3-6 циклоалкил, С3-6циклоалкилС1-6алкил или С5-10арилС1-6алкил является незамещенным или замещен дейтерием, С1-6 алкилом или галогеном.
5. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-4, где R1 выбран из -(CR11R12)mR13; R11 и R12 выбраны из водорода, галогена или С1-4 алкила, где С1-4 алкил является незамещенным или замещен дейтерием; и R13 выбран из:
1) C1-4 алкила, где С1-4алкил является незамещенным или замещен дейтерием;
2) С3-6 циклоалкила, где С3-6циклоалкил является незамещенным или замещен галогеном; или
3) фенила, где фенил является незамещенным или замещен галогеном; и
m составляет 0 или 1.
6. Соединение или его фармацевтически приемлемая соль по любому из предыдущих пунктов, где R1 выбран из: 
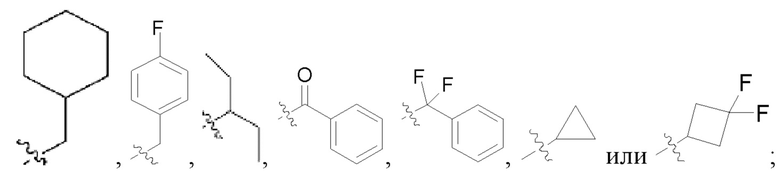
предпочтительно Rb, Rc, Rd и Re выбраны из водорода или галогена;
предпочтительно L1 выбран из одинарной связи, -О-, -NH- или -NHCO-;
предпочтительно L2 выбран из одинарной связи или метилена; и
предпочтительно R2 выбран из: 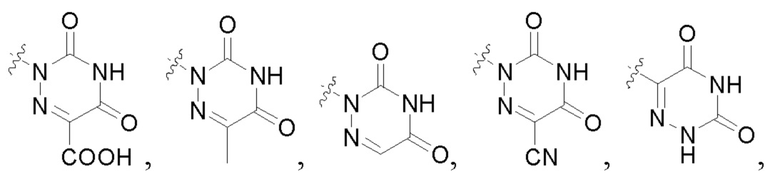
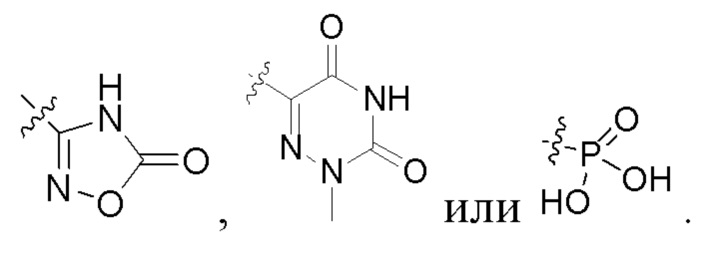
7. Соединение или его фармацевтически приемлемая соль по п. 2, где R1 представляет собой C1-6 линейный или разветвленный алкил или С3-6 циклоалкил или С3-6циклоалкилС1-6алкил, где C1-6 линейный или разветвленный алкил, С3-6 циклоалкил или С3-6циклоалкилС1-6 алкил является незамещенным или замещен дейтерием, C1-6 алкилом или галогеном;
X представляет собой О, S или -СН2-;
Rb, Rc, Rd и Re представляют собой водород, галоген или C1-6 линейный или разветвленный алкил; или Rb и Rc связаны с образованием 5-членного циклоалкила или 5-членного неароматического гетероциклического кольца, содержащего 1 гетероатом кислорода; или Rd и Re связаны с образованием 5-членного циклоалкила или 5-членного неароматического гетероциклического кольца, содержащего 1 гетероатом кислорода;
L1 представляет собой одинарную связь, -NR3-, -О-;
L2 представляет собой одинарную связь или -СН2-;
R2 выбран из группы, представленной следующей формулой: 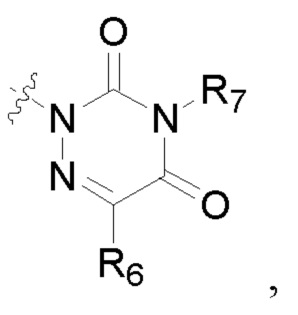
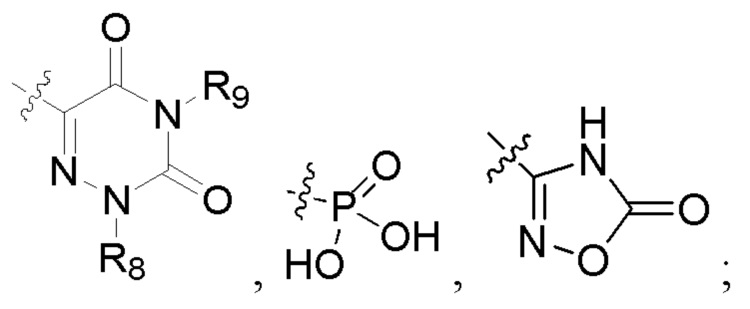
R3 представляет собой водород;
R6 представляет собой водород, циано, СООН или C1-6 алкил;
R8 представляет собой водород или C1-6 алкил; и
R7 и R9 представляют собой водород.
8. Соединение или его фармацевтически приемлемая соль по п. 2 или 7, где R1 представляет собой C1-6 линейный или разветвленный алкил, где указанный C1-6 линейный или разветвленный алкил является незамещенным или замещен дейтерием, C1-6 алкилом или галогеном;
Rb, Rc, Rd и Re представляют собой водород, галоген или C1-6 линейный или разветвленный алкил; или Rb и Rc связаны с образованием 5-членного циклоалкила или 5-членного неароматического гетероциклического кольца, содержащего 1 гетероатом кислорода; или Rd и Re связаны с образованием 5-членного циклоалкила или 5-членного неароматического гетероциклического кольца, содержащего 1 гетероатом кислорода;
X представляет собой О или -СН2-;
L1 представляет собой одинарную связь, -О- или -NH-;
L2 представляет собой одинарную связь;
R2 выбран из группы, представленной следующей формулой: 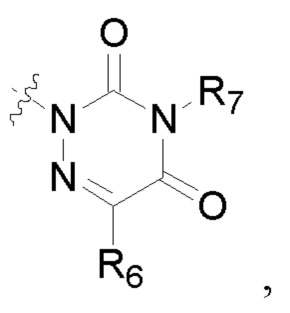
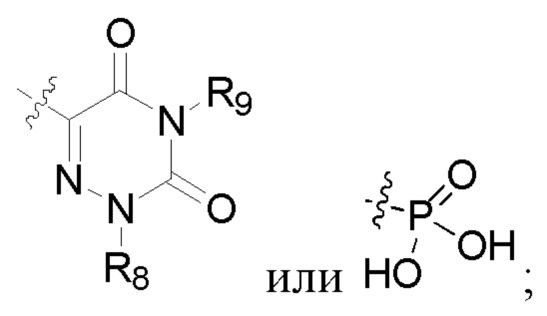
R6 представляет собой водород, циано, C1-6 алкил; R8 представляет собой водород или C1-6 алкил; и
R7 и R9 представляют собой водород.
9. Соединение или его фармацевтически приемлемая соль по п. 2, где R1 представляет собой C1-6 линейный или разветвленный алкил, бензил или С5-6 циклоалкилметилен, где C1-6 линейный или разветвленный алкил, бензил или С5-6 циклоалкилметилен является незамещенным или замещен дейтерием, C1-6 алкилом, или галогеном, предпочтительно R1 представляет собой изопропил или бензил;
Rb и Rd представляют собой галоген, Rc и Re представляют собой водород, и Rb и Rd более предпочтительно представляют собой хлор;
X представляет собой О, S или -СН2-;
L1 представляет собой одинарную связь, -О или -NH-;
L2 представляет собой одинарную связь или -СН2-;
R2 выбран из группы, представленной следующей формулой: 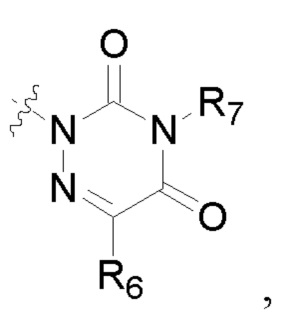
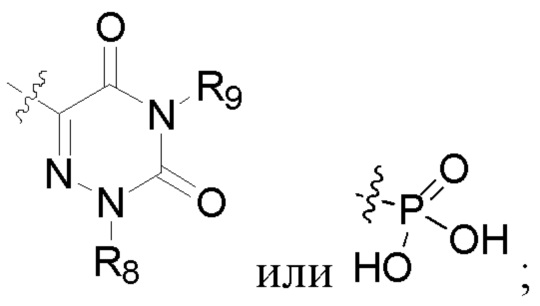 и
и
R6, R8 представляет собой водород или C1-6 алкил; R7 и R9 представляют собой водород.
10. Соединение или его фармацевтически приемлемая соль по п. 7, где R1 представляет собой C1-6 линейный или разветвленный алкил или С5-6 циклоалкилметилен, где C1-6 линейный или разветвленный алкил или С5-6 циклоалкилметилен является незамещенным или замещен дейтерием, C1-6 алкилом или галогеном; предпочтительно R1 представляет собой изопропил;
Rb и Rd представляют собой галоген, Rc и Re представляют собой водород, и Rb и Rd дополнительно предпочтительно представляют собой хлор;
X представляет собой О, S или -СН2-;
L1 представляет собой одинарную связь, -О или -NH-;
L2 представляет собой одинарную связь или -СН2-;
R2 выбран из группы, представленной следующими формулами: 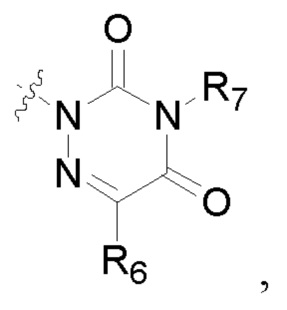
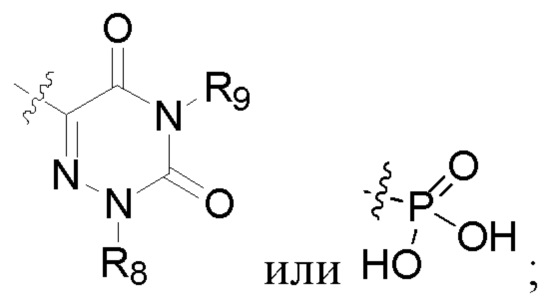 и
и
R6, R8 представляет собой водород или C1-6 алкил;
R7 и R9 представляют собой водород.
11. Соединение или его фармацевтически приемлемая соль по п. 8, где R1 представляет собой C1-6 линейный или разветвленный алкил, где C1-6 линейный или разветвленный алкил является незамещенным или замещен дейтерием, C1-6 алкилом или галогеном; предпочтительно, R1 представляет собой изопропил;
Rb и Rd представляют собой галоген, Rc и Re представляют собой водород, и Rb и Rd более предпочтительно представляют собой хлор;
X представляет собой О, S или -СН2-;
L1 представляет собой одинарную связь, -О или -NH-;
L2 представляет собой одинарную связь;
R2 выбран из группы, представленной следующими формулами: 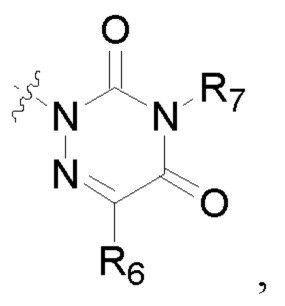
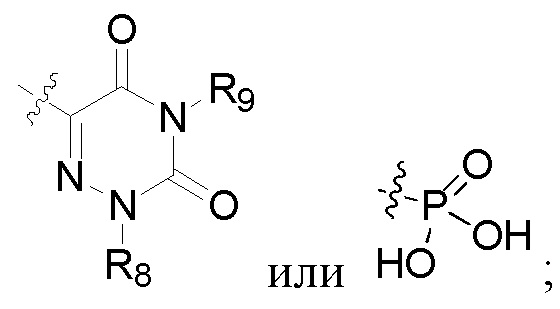 и
и
R6, R8 представляет собой водород или C1-6 алкил;
R7 и R9 представляют собой водород.
12. Соединение или его фармацевтически приемлемая соль по п. 1, где R1 представляет собой метил, этил, пропил, бутил, пентил, циклопропил, циклобутил, циклопентил, циклогексил, циклопропилметил, циклобутилметил, цикломентилметил, циклогексилметил, бензил или -COR10, и указанный метил, этил, пропил, бутил, пентил, циклопропил, циклобутил, циклопентил, циклогексил, циклопропилметил, циклобутилметил, циклопентилметил, циклогексилметил или бензил является незамещенным или может быть замещен дейтерием, С1-3 алкилом или галогеном;
X представляет собой метилен, -О- или -S-;
Ra представляет собой галоген; или два соседних Ra связаны с образованием 5-членного карбоциклического кольца или 5-членного гетероциклического кольца, содержащего 1 гетероатом кислорода;
L1 представляет собой одинарную связь, -О-, -NH-, -NHCO;
L2 представляет собой одинарную связь, метилен;
R2 представляет собой карбоксил или группу, представленную следующей формулой: 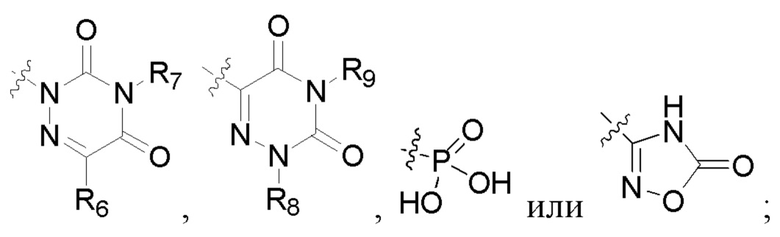
R5 представляет собой водород, циано, СООН, метил, этил или пропил;
R8 представляет собой водород, метил, этил или пропил;
R7 и R9 представляют собой водород;
R10 представляет собой фенил; и
n составляет 2 или 3.
13. Соединение или его фармацевтически приемлемая соль по п. 12, где R1 представляет собой метил, этил, пропил, бутил, пентил, циклопропил, циклобутил, циклопентил, циклогексил, циклопропилметил, циклобутилметил, циклопентилметил, циклогексилметил или бензил, и указанный метил, этил, пропил, бутил, пентил, циклопропил, циклобутил, циклопентил, циклогексил, циклопропилметил, циклобутилметил, циклопентилметил, циклогексилметил или бензил является незамещенным или может быть замещен дейтерием, С1-3 алкилом, F, Cl или Br;
X представляет собой метилен, -О- или -S-;
Ra представляет собой F, Cl или Br; или два соседних Ra связаны с образованием 5-членного карбоциклического кольца или 5-членного гетероциклического кольца, содержащего 1 гетероатом кислорода;
L1 представляет собой одинарную связь, -О-, -NH- или -NHCO-;
L2 представляет собой одинарную связь, метилен;
R2 представляет собой карбоксил или группу, представленную следующей формулой: 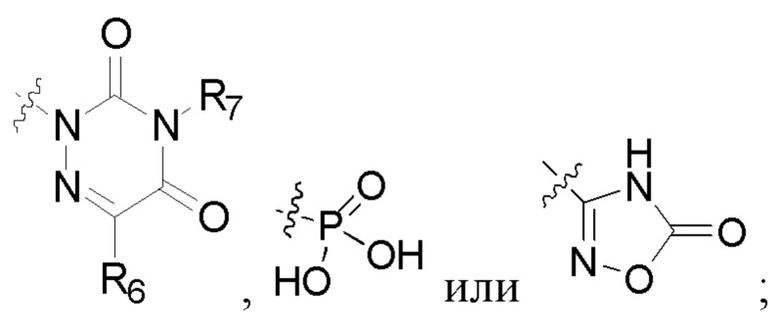
R6 представляет собой водород, циано или метил;
R7 представляет собой водород; и
n составляет 2 или 3.
14. Соединение следующей формулы или его фармацевтически приемлемая соль:
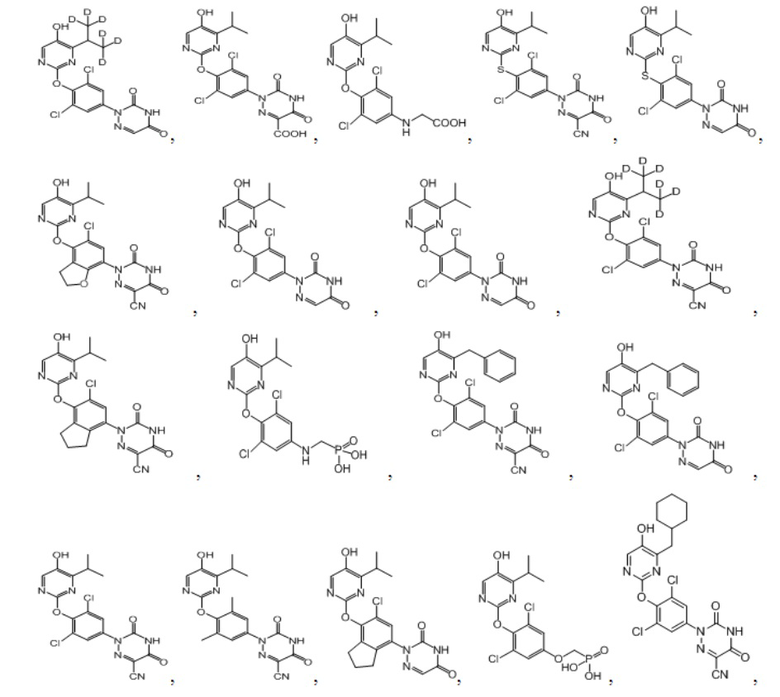
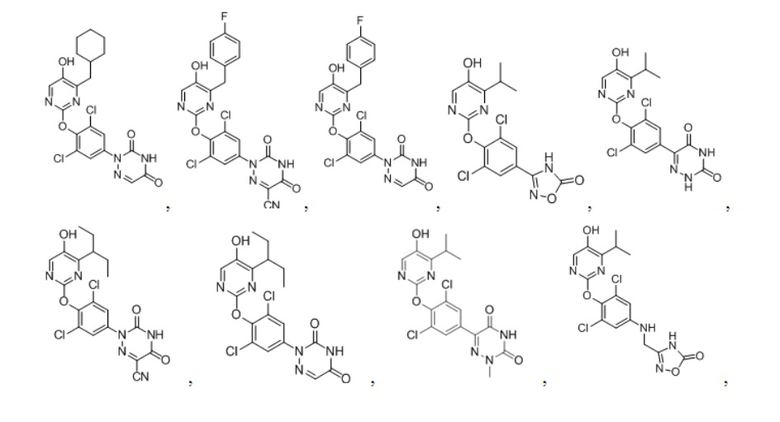
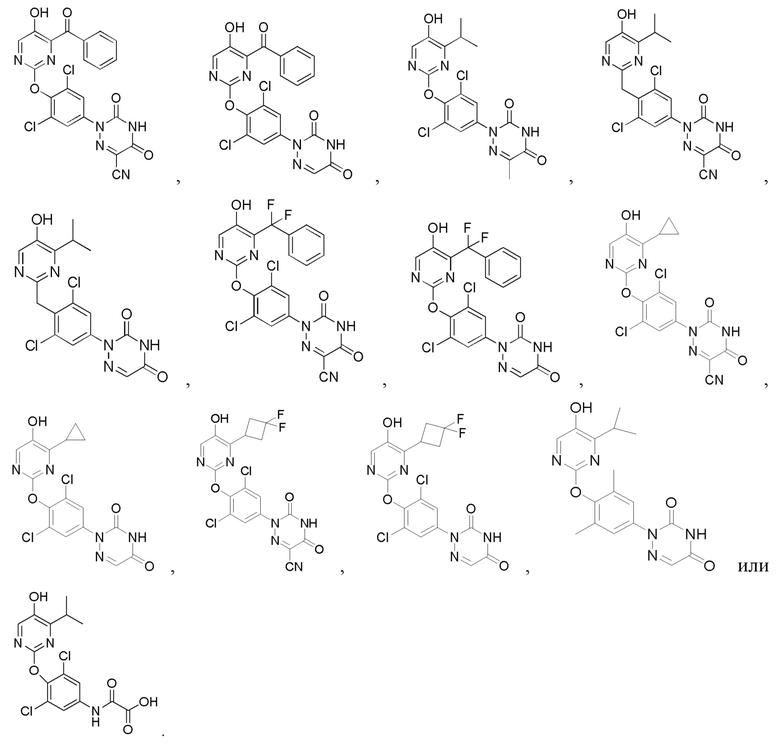
15. Фармацевтическая композиция для предотвращения или лечения заболевания, связанного с действием агониста β-рецептора тиреоидных гормонов, содержащая соединение или его фармацевтически приемлемую соль по любому из пп. 1-14 и один или более фармацевтически приемлемых носителей.
16. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-14 или фармацевтическая композиция по п. 15 для предотвращения или лечения заболевания, связанного с действием агониста β-рецептора тиреоидных гормонов, где предпочтительно заболевание, связанное с действием агониста β-рецептора тиреоидных гормонов, представляет собой заболевание или расстройство, представляющее собой ожирение, гиперлипидемию, гиперхолестеринемию, гипертриглицеридемию, дислипидемию, рак щитовидной железы, метаболический синдром, сердечно-сосудистое заболевание, ишемическую болезнь сердца, инфаркт миокарда, желудочковую недостаточность, сердечную недостаточность, жировой гепатоз, цирроз, диабет, стеатогепатит, неалкогольный стеатогепатит, неалкогольную жировую болезнь печени, атеросклероз или гипотиреоз.
| НОВЫЙ АГОНИСТ БЕТА РЕЦЕПТОРА ТИРЕОИДНОГО ГОРМОНА | 2010 |
|
RU2527948C2 |
| WO 2020073974 A1, 16.04.2020 | |||
| WO 2020077123 A1, 16.04.2020 | |||
| CN 110627773 A, 31.12.2019 | |||
| CN 109574995 A, 05.04.2019 | |||
| JP 2012106996 A, 07.06.2012 | |||
| JP 2011088840 A, 06.05.2011 | |||
| WO 2013097773 A1, 04.07.2013 | |||
| WO 2010003624 A2, 14.01.2010. | |||

