ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к ряду соединений-производных, имеющих структуру с гетероароматическими кольцами, и к их применению в качестве ингибиторов RET-киназ и, в частности, к соединениям формулы (I) или их фармацевтически приемлемым солям.
УРОВЕНЬ ТЕХНИКИ
В 1985 году ученые трансфицировали клетки NIH3T3 с помощью высокомолекулярной ДНК Т-клеточной лимфомы человека и установили, что новый ген RET вызывает трансформацию. Данный ген активировался перестройкой ДНК, в результате которой два несвязанных сегмента ДНК человека рекомбинировались с образованием новой единицы транскрипции. Позже, проведя исследование, RET локализовали на хромосоме 10qll.2, на которой он кодирует рецепторную тирозинкиназу. RET представляет собой однопроходный трансмембранный белок с типичным внутриклеточным тирозинкиназным доменом. Хотя «классическая» активация рецепторных тирозинкиназ (RTK) происходит благодаря лиганд-рецепторным взаимодействиям, активация RET требует взаимодействий между своими лигандами (лиганды семейства нейротрофических факторов глиальной клеточной линии, GFL) и корецептором (рецептор-сх семейства GFL). Связывание комплексов GFL-GFRcx с внеклеточным доменом Ret вызывает фосфорилирование внутриклеточного тирозинкиназного домена и, тем самым, активацию нескольких путей, включающих киназы МАРК, PI3K, JAK-STAT, РКА и PKC.
Б нормальных физиологических условиях ген RET связан с развитием нервной системы в почках и желудочно-кишечном тракте; однако мутации RET ведут к независимой от лигандов конститутивной аномальной активации RET-киназы и, таким образом, к онкогенезу. Существует два основных механизма активации RET-киназ: 1. точечные мутации в гене RET и 2. перестройка гена RET. Нонсенс-мутации RET могут возникать во внеклеточных остатках Cys, создавая аномальную активацию киназ. Мутации также могут происходить во внутриклеточном домене киназной активности и могут стимулировать независимую от лигандов активацию RET-киназ. Точечные мутации в RET очень распространены при медуллярной карциноме щитовидной железы (МТС): они происходят приблизительно в 50% случаев спорадической МТС и почти при всех наследственных МТС.Ген RET перестраивается в новый ген, продукт-слияния, посредством своего разрушения и слияния с другими генами, вызывая независимую от регуляции лигандами активацию тирозинкиназы RET, приводя к дальнейшему аутофосфорилированию. Вследствие этого усиливается функция передачи сигналов, и стимулируется активация киназ, что вызывает онкогенез. Слияния RET обнаружены приблизительно в 20% случаев папиллярного рака щитовидной железы (РТС), 1-2% случаев немелкоклеточного рака легкого (NSCLC) и других раковых заболеваний, таких как рак толстой кишки и рак молочной железы. Приведенные выше данные показывают, что дисрегуляция сигнального пути RET является ключевым действующим фактором множества неопластических заболеваний.
В настоящее время различные мультикиназные ингибиторы, в контексте активности и переносимости, обладают ингибирующей активностью в отношении RET, но не имеют специфической ингибирующей активности, и оказывается высокой частота проявления токсичности 3-4 степени у пациентов, получавших лечение TKI RET в течение длительного периода времени, вследствие их ингибирующего воздействия на VEGFR-киназу. Поэтому существует медицинская потребность в разработке специфических к RET ингибиторов с выраженной активностью и высокой селективностью, которые, как предполагается, должны стать новыми лекарственными препаратами для лечения различных раковых заболеваний, таких как рак щитовидной железы и немелкоклеточный рак легкого.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение предназначено для предоставления класса соединений с гетероароматическими кольцами в качестве ингибиторов RET-киназ.
Цели настоящего изобретения могут достигаться путем получения:
соединения формулы (I) или его фармацевтически приемлемых соли, сложного эфира, стереоизомера, сольвата, оксида или пролекарства,
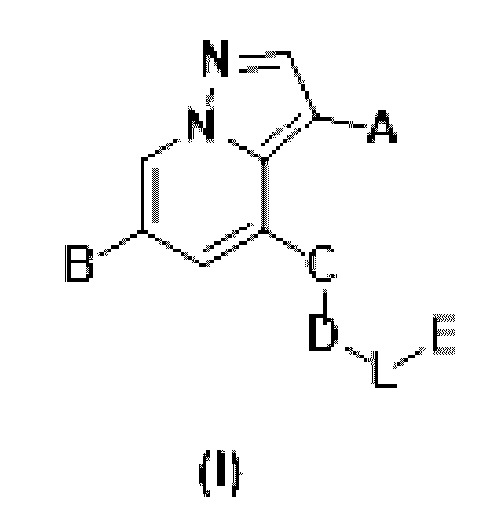
где:
А выбран из Н, -CN, галогена, -С=ONH2, -С=С-CN и 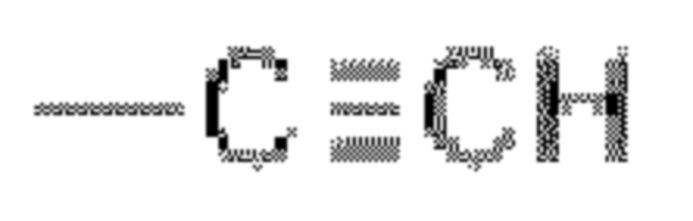 .
.
Б выбран из следующих групп, незамещенных или замещенных одним или более идентичными или различными заместителями: С1-С6 алкилом, С1-С6 алкиламином, С2-С6 алкинилом, С2-С6 алкенилом, HetAr1 и HetCyc1; заместителями, независимо выбранными из галогена, гидрокси, -CN, =O [карбонила), C1-С6 алкила, дейтерированного C1-С6 алкила, C1-С6 алкокси, гидрокси C1-С6 алкила, галоген C1-С6 алкила, циано С1-Сб алкила, (C1-С6 алкокси) C1-С6 алкила, С3-С6 циклоалкила и (С1-С6 алкокси SO6) C1-С6 алкила;
HetAr1 представляет собой от 5- до 6-членное гетероароматическое кольцо, содержащее 1-3 гетероатома, независимо выбранные из N, S и О;
HetCyc1 представляет собой от 4- до S-членный гетероцикл, содержащий 1-3 гетероатома, выбранные из N и О, от 8- до 10-членное спирокольцо, содержащее 1-3 гетероатома, выбранные из N и О, или от 7- до 11-членный конденсированный гетероцикл, содержащий 1-3 гетероатома, выбранные из N и О;
С представляет собой от 5- до 6-членное гетероароматическое кольцо, содержащее 1-3 гетероатома, независимо выбранные из N, S и О, причем гетероароматическое кольцо не замещено или необязательно замещено одним или более идентичными или различными заместителями, выбранными из галогена, гидрокси, -CN, нитро, C1-С3 алкила и галоген C1-С3 алкила;
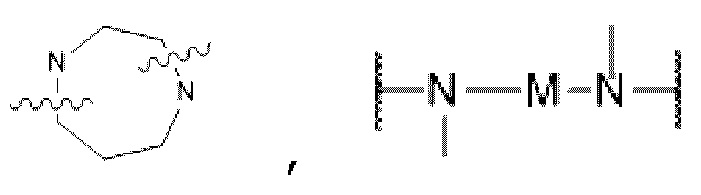
D представляет собой C1-С6 алкил, содержащий 1-3 гетероатома, выбранные из II и О, от 4- до S-членный гетероцикл, содержащий 1-3 гетероатома, выбранные из N и О; от 7- до 8-членное мостиковое кольцо, содержащее 1-3 гетероатома, выбранные из Н и О, от 7- до 11-членное спирокольцо, содержащее 1-3 гетероатома, выбранные из N и О, от 7- до 10-членный конденсированный гетероцикл, содержащий 1-3 гетероатома, выбранные из N и О, 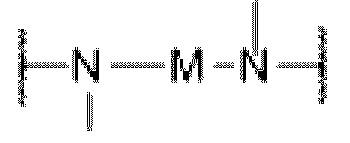 или
или 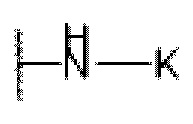 , причем М выбран из C1-С3 алкила и С3-С8 циклоалкила; К представляет собой от 4- до 8-членный гетероцикл, содержащий 1-3 гетероатома, выбранные из N и О;
, причем М выбран из C1-С3 алкила и С3-С8 циклоалкила; К представляет собой от 4- до 8-членный гетероцикл, содержащий 1-3 гетероатома, выбранные из N и О;
L представляет собой 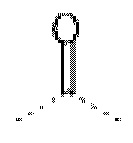 , -С(=O)-C1-С3 алкил или C1-С3 алкил;
, -С(=O)-C1-С3 алкил или C1-С3 алкил;
Е представляет собой HetAr2, незамещенный или замещенный одним или более идентичными или различными заместителями; заместители независимо выбраны из галогена, C1-С6 алкила, дейтерированного C1-С6 алкила, C1-С6 алкокси, дейтерированного C1-С6 алкокси, гидрокси C1-С6 алкила, C1-С6 галогеналкила, циано C1-С6 алкила, (C1-С6 алкокси) C1-С6 алкила, С3-С6 циклоалкила и (C1-С6 алкокси SO2) C1-С6 алкила;
HetAr2 представляет собой от 5- до 6-членное гетероароматическое кольцо, содержащее 1-3 гетероатома кольца, независимо выбранные из N, 5 и О.
В некоторых вариантах осуществления в соединении формулы (I) или его фармацевтически приемлемых соли, сложного эфира, стереоизомера, сольвата, оксида или пролекарства, А выбран из -CN, -C=C-CN и 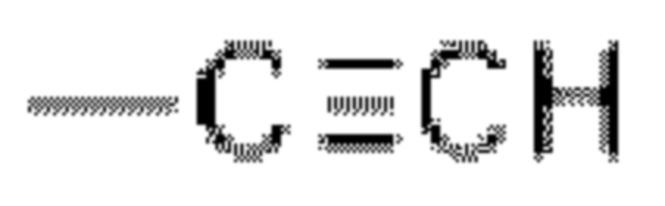 в конкретном варианте осуществления А представляет собой -CN.
в конкретном варианте осуществления А представляет собой -CN.
В некоторых вариантах осуществления в соединении формулы (I) или его фармацевтически приемлемых соли, сложном эфире, стереоизомере, сольвате, оксиде или пролекарстве, В выбран из следующих групп, незамещенных или замещенных одним или двумя идентичными или различными заместителями: 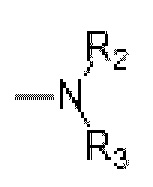 ,
, 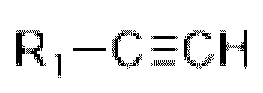 и HetCyc1; R1 выбран из Н, C1-С6 алкила, дейтерированного C1-С6 алкила и C1-С6 гидроксиалкила; Rz или R3 независимо выбран из Н, C1-С6 алкила, дейтерированного C1-С6 алкила и гидрокси C1-С6 алкила; заместителями, независимо выбранными из галогена, гидрокси, -CN,=O (карбонила), C1-С3 алкила, дейтерированного C1-С3 алкила, C1-С3 алкокси, гидрокси C1-С3 алкила, C1-С3 фторалкила, циано C1-С3 алкила, (C1-С3 алкокси) C1-С3 алкила, С3-С6 циклоалкила и (C1-С3 алкокси SO2) C1-С3 алкила;
и HetCyc1; R1 выбран из Н, C1-С6 алкила, дейтерированного C1-С6 алкила и C1-С6 гидроксиалкила; Rz или R3 независимо выбран из Н, C1-С6 алкила, дейтерированного C1-С6 алкила и гидрокси C1-С6 алкила; заместителями, независимо выбранными из галогена, гидрокси, -CN,=O (карбонила), C1-С3 алкила, дейтерированного C1-С3 алкила, C1-С3 алкокси, гидрокси C1-С3 алкила, C1-С3 фторалкила, циано C1-С3 алкила, (C1-С3 алкокси) C1-С3 алкила, С3-С6 циклоалкила и (C1-С3 алкокси SO2) C1-С3 алкила;
HetCyc1 представляет собой от 4- до 8-членный гетероцикл, содержащий 1-2 гетероатома, выбранные из N и О, от 7- до 11-членное спирокольцо, содержащее 1-2 гетероатома, выбранные из N и О, или от 8- до 10-членный конденсированный гетероцикл, содержащий 1-2 гетероатома, выбранные из N и О.
В некоторых вариантах осуществления в соединении формулы (I) или его фармацевтически приемлемых соли, сложном эфире, стереоизомере, сольвате, оксиде или пролекарстве, В выбран из следующих групп, незамещенных или замещенных одним или двумя идентичными или различными заместителями: 
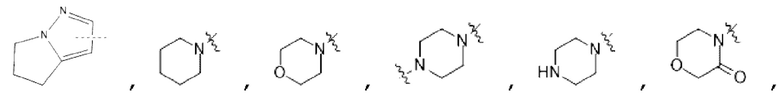
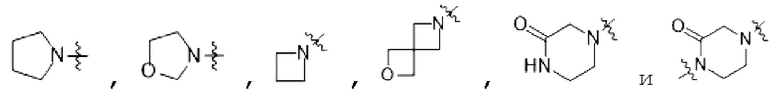 ; R1 выбран из С1-С4 алкила и гидрокси С1-С4 алкила; R2 или R3 независимо выбран из Н, С1-С4 алкила, дейтерированного С1-С4 алкила и гидрокси С1-С4 алкила; заместители независимо выбраны из гидрокси, циано, галогена, C1-С3 алкила, дейтерированного C1-С3 алкила, C1-С3 алкокси и С3-С6 циклоалкила.
; R1 выбран из С1-С4 алкила и гидрокси С1-С4 алкила; R2 или R3 независимо выбран из Н, С1-С4 алкила, дейтерированного С1-С4 алкила и гидрокси С1-С4 алкила; заместители независимо выбраны из гидрокси, циано, галогена, C1-С3 алкила, дейтерированного C1-С3 алкила, C1-С3 алкокси и С3-С6 циклоалкила.
В некоторых вариантах осуществления в соединении формулы (I) или его фармацевтически приемлемых соли, сложном эфире, стереоизомере, сольвате, оксиде или пролекарстве, В представляет собой 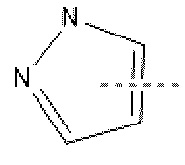 , незамещенный или замещенный одним или двумя идентичными или различными заместителями; заместители независимо выбраны из галогена, гидрокси, -CN, =O (карбонила), C1-С3 алкила, дейтерированного C1-С3 алкила, C1-С3 алкокси, гидрокси C1-С3 алкила, C1-С3 фторалкила, циано C1-С3 алкила, (C1-С3 алкокси) C1-С3 алкила, С3-С6 циклоалкила и (C1-С3 алкокси SO2) C1-С3 алкил C1-С3 алкила; С представляет собой от 5- до 6-членное гетероароматическое кольцо, содержащее 1-2 гетероатома кольца, выбранные из N и S; D представляет собой
, незамещенный или замещенный одним или двумя идентичными или различными заместителями; заместители независимо выбраны из галогена, гидрокси, -CN, =O (карбонила), C1-С3 алкила, дейтерированного C1-С3 алкила, C1-С3 алкокси, гидрокси C1-С3 алкила, C1-С3 фторалкила, циано C1-С3 алкила, (C1-С3 алкокси) C1-С3 алкила, С3-С6 циклоалкила и (C1-С3 алкокси SO2) C1-С3 алкил C1-С3 алкила; С представляет собой от 5- до 6-членное гетероароматическое кольцо, содержащее 1-2 гетероатома кольца, выбранные из N и S; D представляет собой  ,
,  или
или 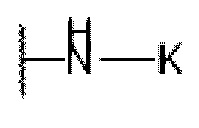 ; где М выбран из С3-С6 циклоалкила; К представляет собой от 4- до 8-членный гетероцикл, содержащий 1-3 гетероатома, выбранные из N и О; L представляет собой -СН2-; Е представляет собой
; где М выбран из С3-С6 циклоалкила; К представляет собой от 4- до 8-членный гетероцикл, содержащий 1-3 гетероатома, выбранные из N и О; L представляет собой -СН2-; Е представляет собой 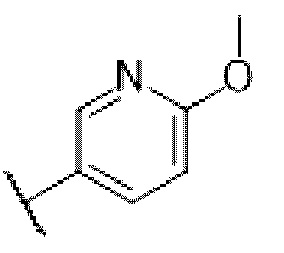 .
.
В некоторых вариантах осуществления в соединении формулы (I) или его фармацевтически приемлемых соли, сложном эфире, стереоизомере, сольвате, оксиде или пролекарстве, С представляет собой следующую группу, незамещенную или замещенную одним или двумя идентичными или различными заместителями: 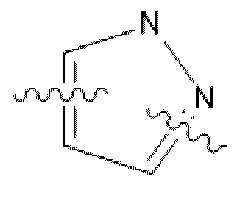 ,
, 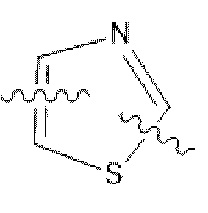 или
или 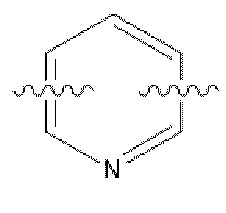 ; заместители независимо выбраны из фтора, хлора и брома.
; заместители независимо выбраны из фтора, хлора и брома.
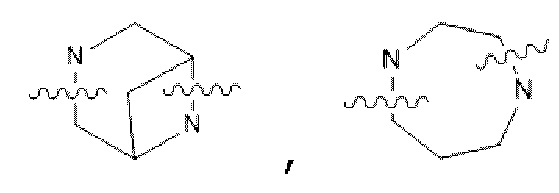
В некоторых вариантах осуществления в соединении формулы (I) или его фармацевтически приемлемых соли, сложном эфире, стереоизомере, сольвате, оксиде или пролекарстве, D представляет собой 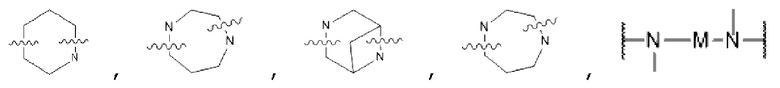 или
или 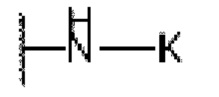 ; где М выбран из C1-С3 алкана и С3-С6 циклоалкила; К представляет собой от 4- до 8-членный гетероцикл, содержащий 1-3 гетероатома, выбранные из N и О; предпочтительнее D представляет собой -N(СН3)CH2CH2N(СН3)-,
; где М выбран из C1-С3 алкана и С3-С6 циклоалкила; К представляет собой от 4- до 8-членный гетероцикл, содержащий 1-3 гетероатома, выбранные из N и О; предпочтительнее D представляет собой -N(СН3)CH2CH2N(СН3)-, 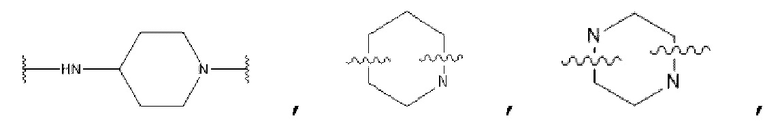
 или
или 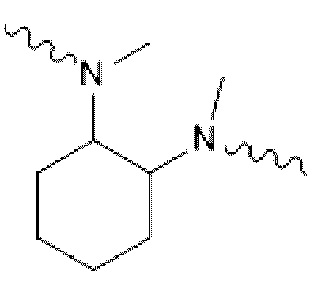
В некоторых вариантах осуществления в соединении формулы (I) или его фармацевтически приемлемых соли, сложном эфире, стереоизомере, сольвате, оксиде или пролекарстве, L представляет собой 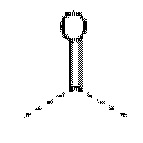 или -СН2-.
или -СН2-.
В некоторых вариантах осуществления в соединении формулы (I) или его фармацевтически приемлемых соли, сложном эфире, стереоизомере, сольвате или пролекарстве, Е представляет собой 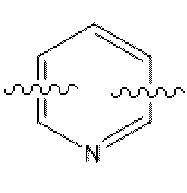 , незамещенный или замещенный одним или двумя идентичными или различными заместителями; заместители независимо выбраны из C1-С3 алкокси и дейтерированного C1-С3 алкокси.
, незамещенный или замещенный одним или двумя идентичными или различными заместителями; заместители независимо выбраны из C1-С3 алкокси и дейтерированного C1-С3 алкокси.
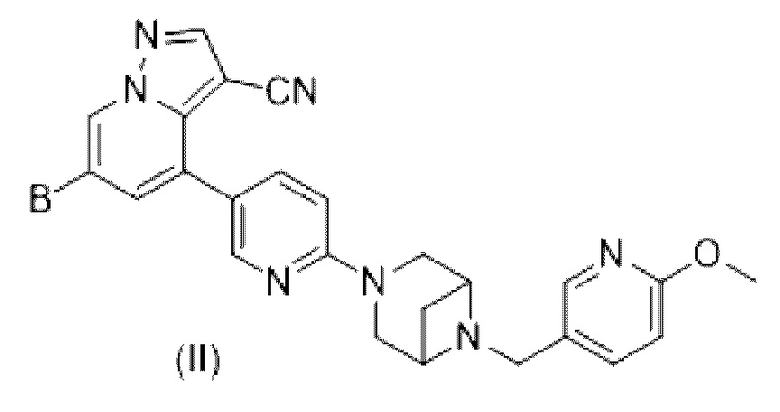
В некоторых вариантах осуществления в настоящем изобретении также предлагается соединение формулы (II) или его фармацевтически приемлемые соль, сложный эфир, стереоизомер, сольват, оксид или пролекарство, в которой
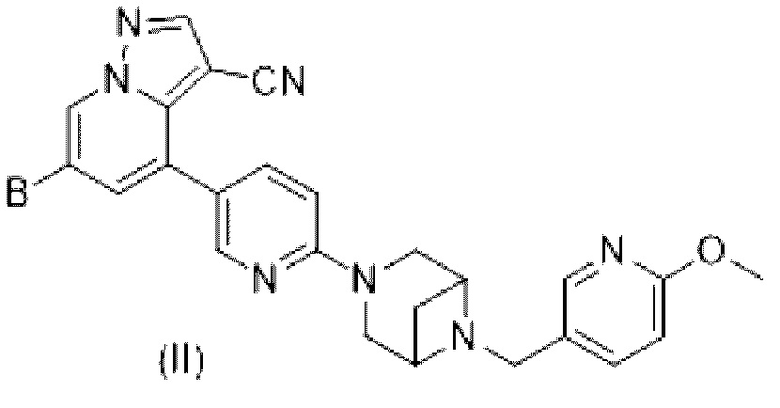
В выбран из HetCyc1, незамещенного или замещенного одним или двумя идентичными или различными заместителями; HetCyc1 представляет собой от 4- до 5-членный гетероцикл, содержащий 1 атом N или  ; заместители независимо выбраны из Н, галогена, гидрокси, -CN, карбонила, C1-С3 алкила, дейтерированного C1-С3 алкила, C1-С3 алкокси, гидрокси C1-С3 алкила, C1-С3 фторалкила и циано C1-С3 алкила; в некоторых вариантах осуществления заместители В в формуле (II) независимо выбраны из галогена, гидрокси, -CN, карбонила, метила, этила, дейтерированного метила и метокси.
; заместители независимо выбраны из Н, галогена, гидрокси, -CN, карбонила, C1-С3 алкила, дейтерированного C1-С3 алкила, C1-С3 алкокси, гидрокси C1-С3 алкила, C1-С3 фторалкила и циано C1-С3 алкила; в некоторых вариантах осуществления заместители В в формуле (II) независимо выбраны из галогена, гидрокси, -CN, карбонила, метила, этила, дейтерированного метила и метокси.
В некоторых вариантах осуществления в соединении формулы (II) или его фармацевтически приемлемых соли, сложном эфире, стереоизомере, сольвате или пролекарстве, в соответствии с настоящим изобретением
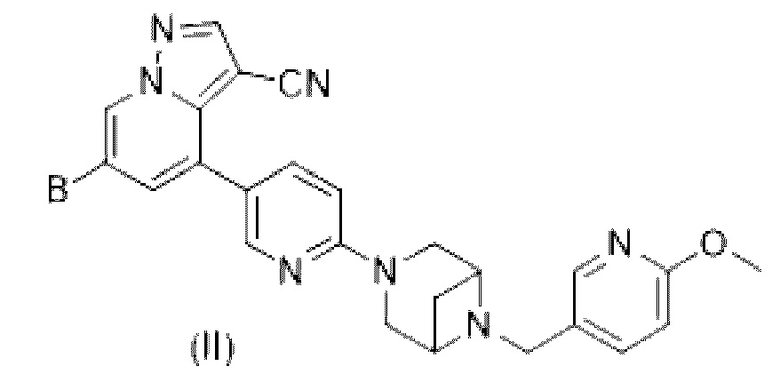
В выбран из замещенного или незамещенного HetCyc2, или В представляет собой замещенное или незамещенное от 7- до 8-членное мостиковое кольцо, содержащее 1-3 гетероатома, выбранные из N, S и О; HetCycz представляет собой от 4- до 6-членный гетероцикл, содержащий атом Р, или от 4- до 6-членный гетероцикл, содержащий атом Р и атом Н; заместители независимо выбраны из Н, галогена, гидрокси, -СН, карбонила, C1-С3 алкила, дейтерированного C1-С3 алкила, C1-С3 алкокси, гидрокси C1-С3 алкила, C1-С3 фторалкила и циано C1-С3 алкила; в некоторых вариантах осуществления В представляет собой замещенный или незамещенный HetCyc2, или В представляет собой замещенное или незамещенное от 7- до 8-членное мостиковое кольцо, содержащее 1-2 гетероатома, включающих Н; HetCyc2 представляет собой от 4- до 6-членный гетероцикл, содержащий атом Р и атом N; в некоторых более конкретных вариантах осуществления В представляет собой замещенный или незамещенный  или
или  ; причем R4 выбран из Н, галогена, гидрокси, -CN, карбонила, C1-С3 алкила, дейтерированного C1-С3 алкила, C1-С3 алкокси, гидрокси C1-С3 алкила, C1-С3 фторалкила и циано C1-С3 алкила; предпочтительно R4 выбран из Н, галогена, гидрокси, -СН, карбонила, метила, этила, дейтерированного метила, метокси или трифторметила; в некоторых вариантах осуществления заместители независимо выбраны из галогена, гидрокси, -СН, карбонила, метила, этила, дейтерированного метила, метокси и трифторметила.
; причем R4 выбран из Н, галогена, гидрокси, -CN, карбонила, C1-С3 алкила, дейтерированного C1-С3 алкила, C1-С3 алкокси, гидрокси C1-С3 алкила, C1-С3 фторалкила и циано C1-С3 алкила; предпочтительно R4 выбран из Н, галогена, гидрокси, -СН, карбонила, метила, этила, дейтерированного метила, метокси или трифторметила; в некоторых вариантах осуществления заместители независимо выбраны из галогена, гидрокси, -СН, карбонила, метила, этила, дейтерированного метила, метокси и трифторметила.
В некоторых вариантах осуществления в соединении формулы (II) или его фармацевтически приемлемых соли, сложном эфире, стереоизомере, сольвате, оксиде или пролекарстве, в соответствии с настоящим изобретением
В выбран из HetCyc1, незамещенного или замещенного одним или двумя идентичными или различными заместителями; HetCyc1 представляет собой 6-членный гетероцикл, содержащий 1-2 атома N; заместители независимо выбраны из Н, C1-С3 алкила, дейтерированного C1-С3 алкила и C1-С3 фторалкила; в некоторых более предпочтительных вариантах осуществления HetCyc1 представляет собой  , незамещенный или замещенный заместителями; заместители В независимо выбраны из галогена, метила, этила, дейтерированного метила и -CF3.
, незамещенный или замещенный заместителями; заместители В независимо выбраны из галогена, метила, этила, дейтерированного метила и -CF3.
Описанный в настоящем изобретении оксид могут получать путем окисления в любом положении, чувствительном для окисления; в некоторых конкретных вариантах осуществления оксид образован путем окисления по атому N из от 4- до 8-членного гетероцикла или мостикового кольца, содержащего гетероатом N; например, в некоторых вариантах осуществления оксид образован путем окисления по атому азота из 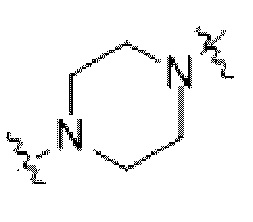 и
и 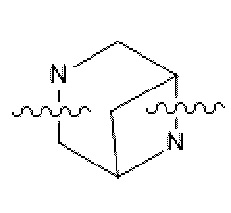 .
.
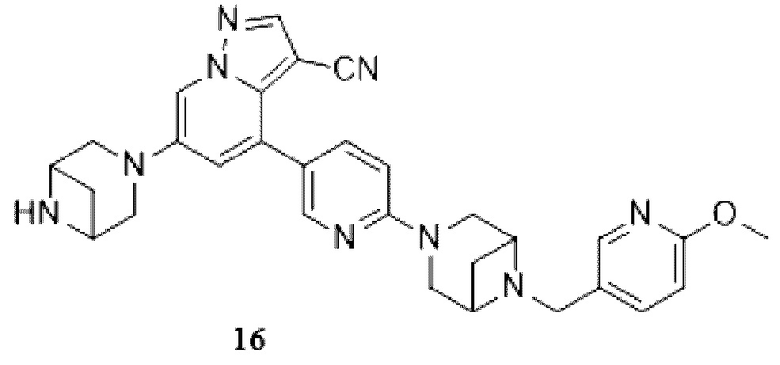
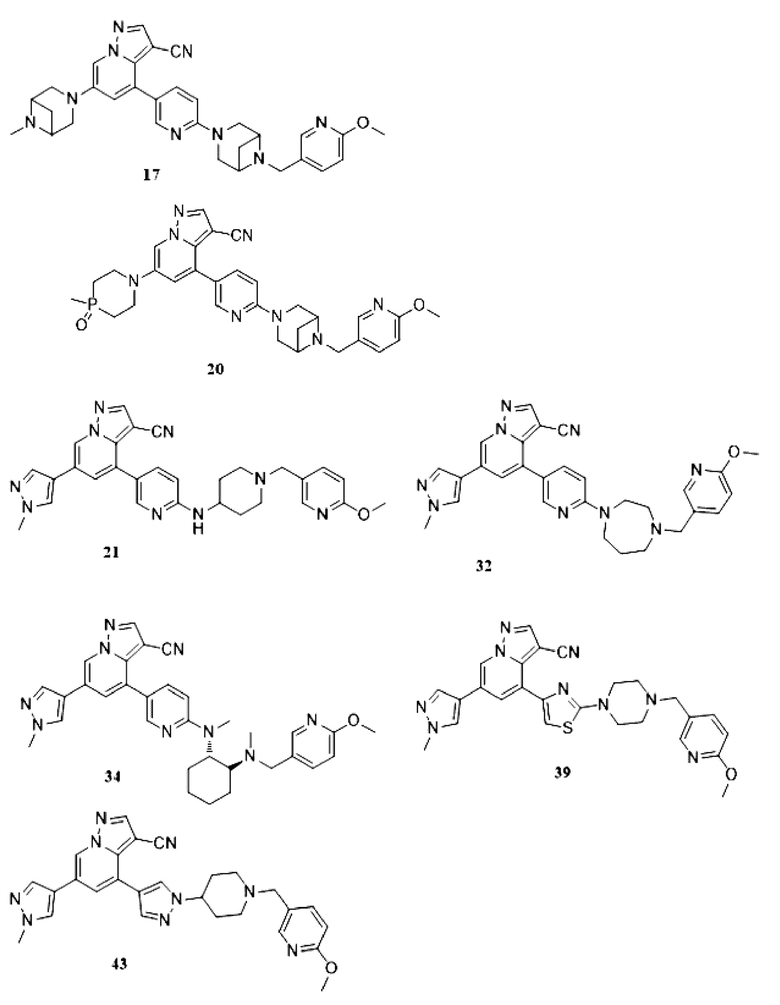
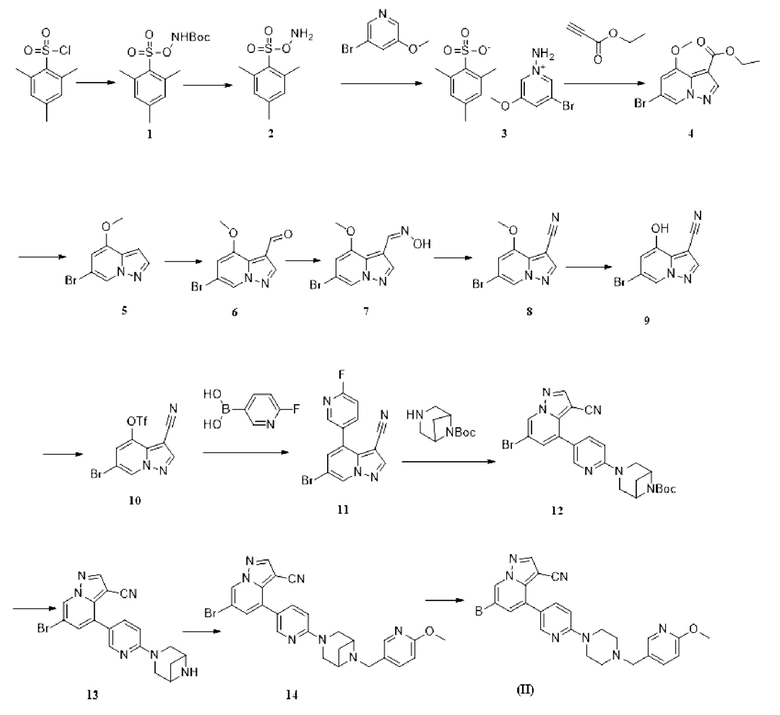
Некоторые конкретные соединения из представленных соединений или их фармацевтически приемлемых солей по настоящему изобретению выбраны из:
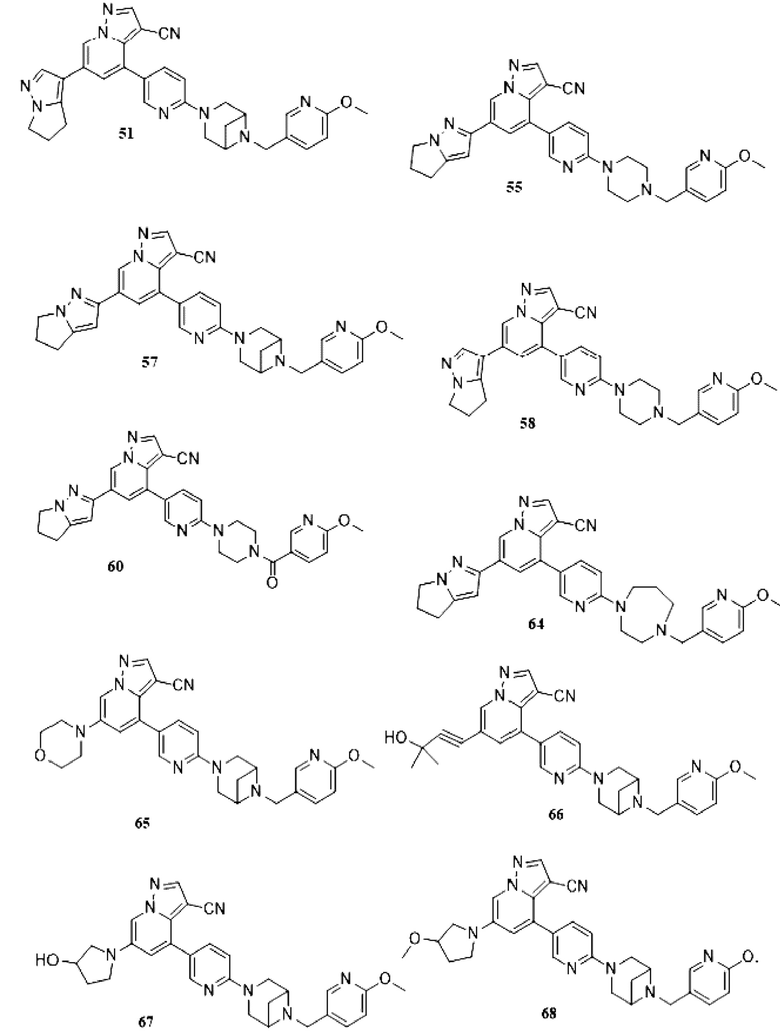
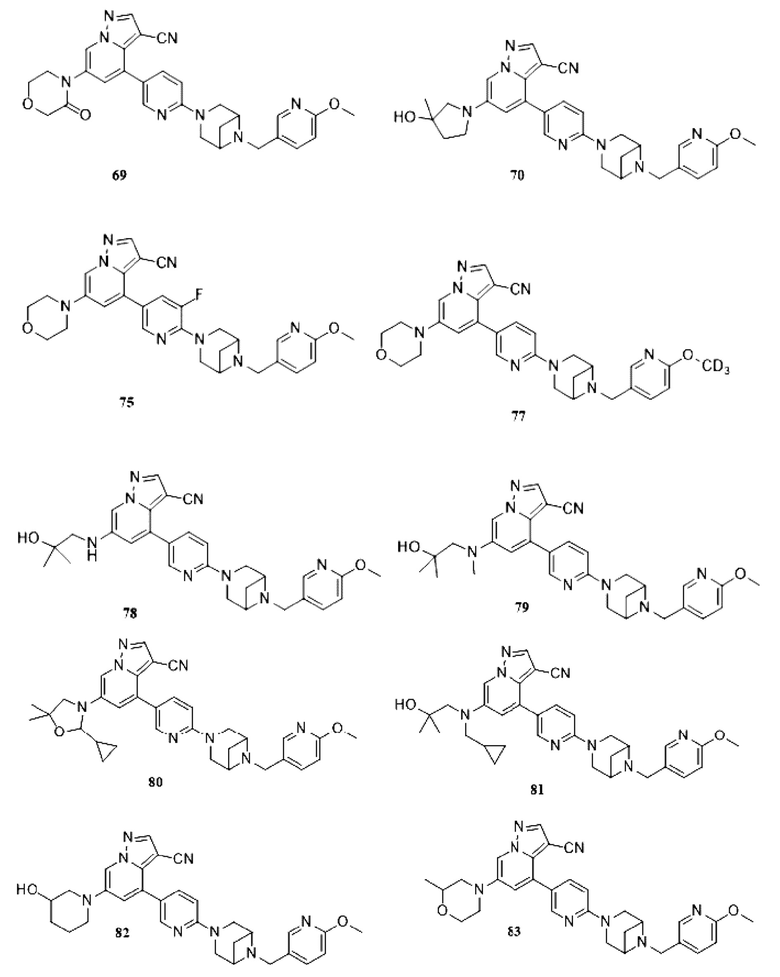
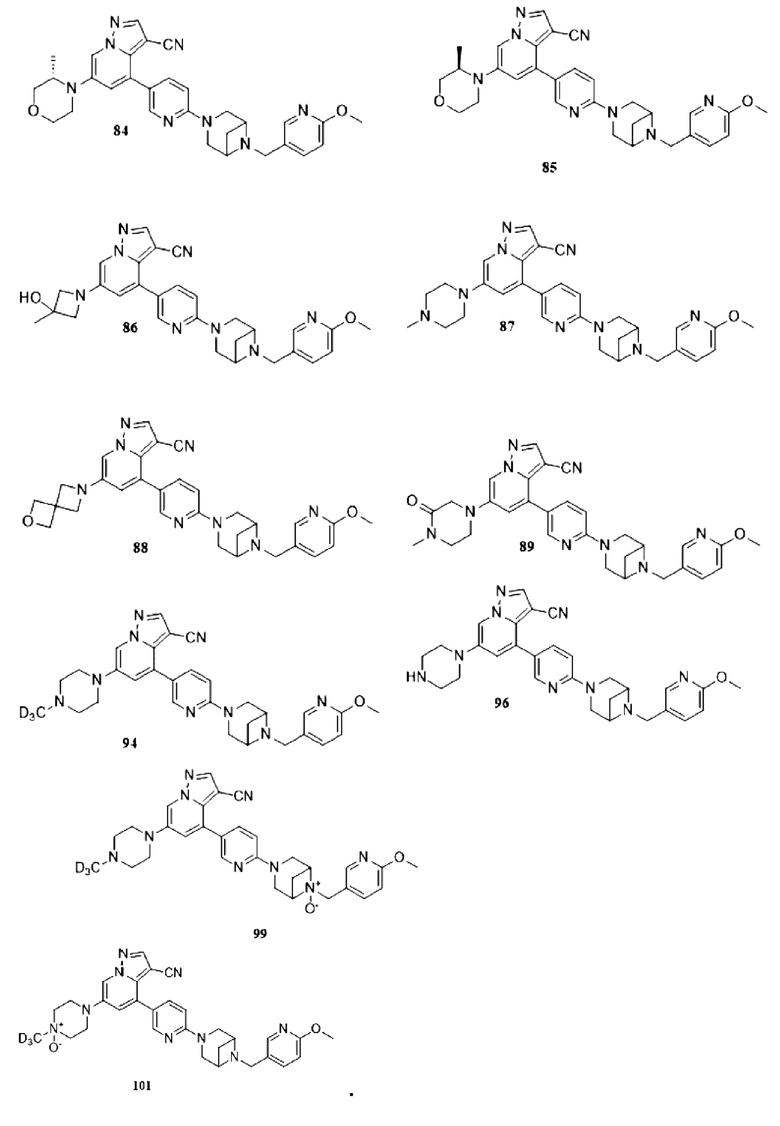
В настоящем изобретении также предлагается схема получения соединения общей формулы (I):
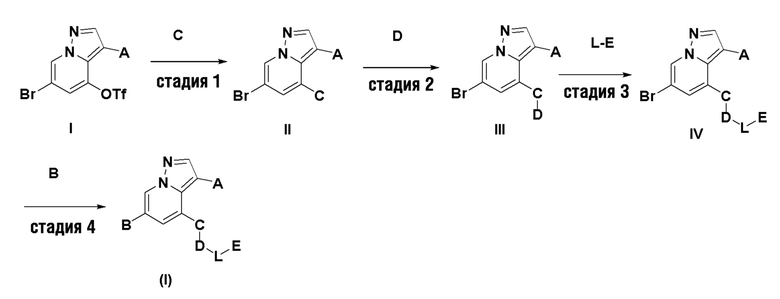
где А, В, С, D, L и Е определены, как указано выше.
В некоторых вариантах осуществления описанная выше схема содержит следующие конкретные стадии:
стадия 1: проведение реакции сочетания соединения I с реактивом С на основе борной кислоты в растворителе диоксане с получением II;
стадия 2: проведение реакции нуклеофильного замещения промежуточного соединения II с аминным соединением с получением ш;
стадия 3: проведение реакции восстановительного аминирования или реакции ацилирования промежуточного соединения III с L-E в растворителе 1,2-дихлорэтане с получением промежуточного соединения IV;
стадия 4: проведение реакции сочетания C-N в растворителях диоксане и N, ЛЬдиметилформамиде с получением конечного продукта (I).
В настоящем изобретении также предлагается способ получения соединения формулы (II) или его фармацевтически приемлемых соли, сложного эфира, стереоизомера, сольвата или пролекарства:
где В определен, как указано выше.
Оксид по настоящему изобретению могут получать путем окисления соединения IV перед получением соединения формулы (I) или путем окисления после получения соединения формулы (I). Окисление могут выполнять с использованием традиционных в данной области способов; например, в некоторых конкретных вариантах осуществления, для проведения окисления применяются обычные окислители, такие как м-хлорпероксибензойная кислота (m-СРВА), с получением оксидов соответствующих соединений.
Соли, которые может образовывать соединение по настоящему изобретению, также находятся в пределах объема настоящего изобретения. Если не указано иное, подразумевается, что соединение по настоящему изобретению включает в себя его соли. Например, соединение формулы (I) вступает в реакцию с определенным количеством, например, эквивалентным количеством, кислоты или основания, и продукт выделяют высаливанием из реакционной среды или путем лиофилизации воднопо раствора.
Соединение по настоящему изобретению содержит основные фрагменты, включающие, но ограничивающиеся ими, аминные или пиридиновые или имидазольные кольца, и они могут образовывать соли с органическими или неорганическими кислотами. Неограничивающий пример фармацевтически приемлемой соли соединения формулы (I) включает моногидрохлоридные,
дигидрохлоридные, трифторацетатные и дифторацетатные соли.
Содержание «по массе» соединения по настоящему изобретению, которое получено путем приготовления, разделения и последующей очистки, равно или больше 90%, например равно или больше 95%, или равно или больше 99% («очень чистое» соединение), как указано в данном текстовом описании. В данном случае такие «очень чистые» соединения по настоящему изобретению также составляют часть настоящего изобретения.
В данном документе также предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемые соль, сложный эфир, стереоизомер, сольват или пролекарство и фармацевтически приемлемый носитель.
В данном документе также предложен способ ингибирования пролиферации клеток in vitro или in vivo, который включает приведение клетки в контакт с эффективным количеством соединения формулы (I), как определено в данном документе, или его фармацевтически приемлемых соли или сольвата или
фармацевтической композиции.
В данном документе также предложен способ лечения RET-ассоциированного заболевания или нарушения у нуждающегося в таком лечении пациента, который включает введение пациенту терапевтически эффективного количества соединения формулы (I), как определено в данном документе, или его фармацевтически приемлемых соли или сольвата или фармацевтической композиции.
В данном документе также предложено применение соединения или его фармацевтически приемлемых соли, сложного эфира, стереоизомера, сольвата или пролекарства, описанных в настоящем изобретении, в качестве ингибитора RET-киназы.
В данном документе также предложено применение соединения или его фармацевтически приемлемых соли, сложного эфира, стереоизомера, сольвата или пролекарства, описанных в настоящем изобретении, в производстве лекарственного средства для лечения RET-ассоциированного заболевания.
В некоторых примерах по настоящему изобретению RET-ассоциированное заболевание или нарушение представляет собой рак.
В данном документе также предложен способ лечения рака или ингибирования раковых метастазов, ассоциированных с конкретным раковым заболеванием, у нуждающегося в таком лечении пациента, который включает введение пациенту терапевтически эффективного количества соединения формулы (I), как определено в данном документе, или его фармацевтически приемлемых соли или сольвата или фармацевтической композиции.
В данном документе также предложено соединение формулы (I), как определено в данном документе, или его фармацевтически приемлемые соль или сольват или фармацевтическая композиция для применения в терапии.
В данном документе также предложено соединение формулы (I), как определено в данном документе, или его фармацевтически приемлемые соль или сольват или фармацевтическая композиция для применения в лечении рака и/или ингибирования раковых метастазов, ассоциированных с конкретным раковым заболеванием.
В данном документе также предложено соединение формулы (I) или его фармацевтически приемлемые соль или сольват для применения в ингибировании активности RET-киназы.
В данном документе также предложено соединение формулы (I), как определено в данном документе, или его фармацевтически приемлемые соль или сольват или фармацевтическая композиция для применения в лечении RET-ассоциированного заболевания или нарушения.
В данном документе также предложено соединение формулы (I), как определено в данном документе, или его фармацевтически приемлемые соль или сольват для производства лекарственного средства для лечения рака и/или для ингибирования раковых метастазов, ассоциированных с конкретным раковым заболеванием.
В данном документе также предложено соединение формулы (I), как определено в данном документе, или его фармацевтически приемлемые соль или сольват для производства лекарственного средства для ингибирования активности RET-киназы.
В данном документе также предложено соединение формулы (I), как определено в данном документе, или его фармацевтически приемлемые соль или сольват для производства лекарственного средства для лечения RET-ассоциированного заболевания или нарушения.
В данном документе также предложен способ лечения рака у нуждающегося пациента, который включает: (а) определение связи рака с дисрегуляцией экспрессии или активности или уровня гена RET, RET-киназы или любого из них (например, имеется RET-ассоциированный рак); и (b) введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемых соли или сольвата или
фармацевтической композиции, если установлено, что рак связан с дисрегуляцией экспрессии или активности или уровня гена RET, RET-киназы или любого из них (например, имеет RET-ассоциированный рак).
В данном документе также предложена фармацевтическая комбинация для лечения рака (например, RET-ассоциированного рака, такого как RET-ассоциированный рак с одной или более мутациями, вызывающими резистентность к ингибиторам RET) у нуждающегося в этом пациента, которая содержит: (а) соединение формулы (I) или его фармацевтически приемлемые соль или сольват, (b) дополнительный терапевтический агент и (с) необязательно по меньшей мере один фармацевтически приемлемый носитель, причем соединение формулы (I) или его фармацевтически приемлемые соль или сольват и дополнительный терапевтический агент готовят в виде отдельной композиции или дозы для одновременного, отдельного или последовательного применения в лечении рака, причем количество соединения формулы (I) или его фармацевтически приемлемых соли или сольвата и количество дополнительного терапевтического агента вместе эффективны в лечении рака. В данном документе также предложена фармацевтическая композиция, содержащая такую комбинацию. В данном документе также предложено применение такой комбинации для производства лекарственного средства для лечения рака. В данном документе также предложена коммерческая упаковка или продукт, содержащие такую комбинацию в комбинированном препарате для одновременного, отдельного или последовательного применения, и способ лечения рака у нуждающегося пациента.
В данном документе также предложен способ реверсии или предотвращения приобретенной резистентности к противораковому лекарственному средству, который включает введение пациенту с риском развития или возникновения приобретенной резистентности к противораковому лекарственному средству терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемых соли или сольвата. В некоторых вариантах осуществления дозу противоракового лекарственного средства вводят пациенту (например, практически одновременно с введением пациенту дозы соединения формулы (I) или его фармацевтически приемлемых соли или сольвата).
В данном документе также предложен способ задержки и/или предотвращения развития резистентности ракового заболевания к противораковому лекарственному средству у индивида, который включает введение индивиду эффективного количества соединения формулы (I) или его фармацевтически приемлемых соли или сольвата до, во время или после введения эффективного количества противоракового лекарственного средства.
В данном документе также предложен способ лечения индивида с раковым заболеванием и повышенной вероятностью развития резистентности к противораковому лекарственному средству, который включает введение индивиду (а) эффективного количества соединения формулы (I) до, во время или после введения (b) эффективного количества противоракового лекарственного средства.
В данном документе также предложен способ лечения индивида с RET-ассоциированным раком и одной или более вызывающими резистентность к ингибиторам RET мутациями, которые увеличивают резистентность ракового заболевания к первому ингибитору RET (например, замена в аминокислотном положении 804, такая как V804M, V804L или V804E), способ включает введение соединение формулы (I) или его фармацевтически приемлемых соли или сольвата до, во время или после введения другого противоракового лекарственного средства (например, второго ингибитора RET-киназы).
В данном документе также предложен способ лечения субъекта с RET-ассоциированным раком, который включает введение соединения формулы (I) или его фармацевтически приемлемых соли или сольвата до, во время или после введения другого противоракового лекарственного средства (например, первого ингибитора RET-киназы).
В некоторых вариантах осуществления любого способа или применения, описанных в данном документе, рак (например, RET-ассоциированный рак) представляет собой гематологическую онкологию. В некоторых вариантах осуществления любого способа или применения, описанных в данном документе, рак (например, RET-ассоциированный рак) представляет собой солидную опухоль. В некоторых вариантах осуществления любого способа или применения, описанных в данном документе, рак (например, RET-ассоциированный рак) представляет собой рак легкого (например, мелкоклеточный рак легкого или немелкоклеточный рак легкого), рак щитовидной железы (например, папиллярный рак щитовидной железы, медуллярный рак щитовидной железы, дифференцированный рак щитовидной железы, рецидивирующий рак щитовидной железы или рефрактерный дифференцированный рак щитовидной железы), аденому щитовидной железы, опухоли эндокринных желез, аденокарциному легкого, бронхиолярную карциному легкого, множественную эндокринную неоплазию типа 2А или 2 В (MEN2A или MEN2B), феохромоцитому, гиперплазию паращитовидных желез, рак груди, рак молочной железы, карциному молочной железы, новообразование молочной железы, колоректальный рак (например, метастатический колоректальный рак), папиллярную почечно-клеточную карциному, ганглиоцитому слизистой оболочки желудочно-кишечного тракта, воспалительные миофибробластические опухоли или рак шейки матки.
В некоторых вариантах осуществления пациент является человеком.
Соединение формулы (I) и его фармацевтически приемлемые соль и сольват также подходят для применения в лечении RET-ассоциированного рака.
В данном документе также предложен способ лечения пациента, у которого диагностирован или определен RET-ассоциированный рак (например, любой из примеров RET-ассоциированных раковых заболеваний, раскрытых в данном документе), который включает введение пациенту терапевтически эффективного количества соединения формулы (I), как определено в данном документе, или его фармацевтически приемлемых соли или сольвата или фармацевтической композиции.
В данном документе также предложено применение соединения или его фармацевтически приемлемых соли, сложного эфира, стереоизомера, сольвата или пролекарства в производстве лекарственного средства ингибитора киназ семейства FGFR; семейство FGFR, описанное в данном документе, включает, но не ограничивается ими, FGFR1, FGFR1 V561M, FGFR2, FGFR2 V564F, FGFR2 N549H, FGFR2 V564I, FGFR2 K641R, FGFR3, FGFR3 V555M, FGFR3 K650E и FGFR4; в некоторых более конкретных воплощениях семейство FGFR включает FGFR2 V564F, FGFR2 N549H, FGFR2 V564I и FGFR3 V555M.
Термины настоящего изобретения определены следующим образом, если не указано иное:
Термин «галоген» относится к -F (иногда упоминаемый в данном документе как «фтор»), -Cl, -Br и -I.
Термины «C1-С3 алкил», «С1-С6 алкил», «С2-С6 алкил» и «С3-С6 алкил» относятся к моновалентным гидрокарбильным группам с насыщенной, линейной или разветвленной цепью, имеющим от одного до трех, от одного до шести, от двух до шести или от трех до шести атомов углерода, соответственно. Примеры включают, в том числе, метил, этил, 1-пропил, изопропил, 1-бутил, изобутил, втор-бутил, трет-бутил, 2-метил-2-пропил, пентил, неопентил и гексил.
Термин «C1-С6 алкокси» относится к моновалентной алкокси группе с насыщенной, линейной или разветвленной цепью, имеющей от одного до шести атомов углерода, в которой связь находится на атоме кислорода. Примеры таких групп включают метокси, этокси, пропокси, изопропокси, бутокси и трет-бутокси.
Термины «(С1-С6 алкокси) С1-С6 алкил-» и «(С1-С6 алкокси) С2-С6 алкил-» относятся к моновалентным группам с насыщенной, линейной или разветвленной цепью, имеющим от одного до шести атомов углерода или от двух до шести атомов углерода, соответственно, один из которых замещен группой (С1-С6 алкокси), как определено в данном документе. Примеры включают метоксиметил (СН3ОСН2-) и метоксиэтил (СН3ОСН2СН2-).
Термины «гидрокси С1-С6 алкил-» и «гидрокси С2-С6 алкил-» относятся к моновалентным алкильным группам с насыщенной, линейной или разветвленной цепью, имеющим от одного до шести атомов углерода или от двух до шести атомов углерода, соответственно, один из которых замещен гидроксигруппой.
Термины «дейтерированный C1-С6 алкил-», «галоген C1-С6 алкил-» и «циано C1-С6 алкил-» относятся к моновалентным алкильным группам с насыщенной, линейной или разветвленной цепью, имеющим от одного до шести атомов углерода, один из которых замещен дейтерием, галогеном или цианогруппой, соответственно.
Термин «С3-С6 циклоалкил» относится к циклопропилу, циклобутилу, циклопентилу или циклогексилу.
Термин «алкенил» относится к линейноцепочечному, разветвленноцепочечному или циклическому неароматическому гидрокарбилу, содержащему по меньшей мере одну углерод-углеродную двойную связь. Таким образом, «С2-С6 алкенил» относится к алкенилу, содержащему от 2 до 6 атомов углерода. Примеры алкенильных групп включают, но не ограничиваются ими, этенил, пропенил, бутенил, 2-метилбутенил, циклогексинил и тому подобные.
Термин «алкинил» относится к линейноцепочечному, разветвленноцепочечному или циклическому неароматическому гидрокарбилу, содержащему по меньшей мере одну углерод-углеродную тройную связь. Таким образом, «С2-С6 алкинил» относится к алкинилу, содержащему от 2 до 6 атомов углерода. Примеры алкинильных групп включают, но не ограничиваются ими, этинил, пропинил, бутинил, 3-метилбутинил и тому подобные.
Термин «гетероцикл» относится к моноциклическому или бициклическому неароматическому гетероциклу, содержащему, в дополнение к атомам углерода, от 1 до 4 гетероатомов, выбранных из группы, состоящей из атома кислорода, атома серы и атома азота, и следующие гетероциклы могут перечисляться в качестве конкретных примеров: от 4- до 7-членные моноциклические неароматические гетероциклы, содержащие, в дополнение к атомам углерода, от 1 до 2 гетероатомов, выбранных из группы, состоящей из атома кислорода, атома серы и атома азота, такие как азетидин, пирролидин, пиразолидин, пиперидин, оксетан, тетрагидрофуран, тетрагидропиран, тетрагидротиофен,
дигидроимидазол, имидазолидин, тетрагидропиразин, пиперазин и морфолин; от 6- до 8-членные бициклические неароматические гетероциклы, содержащие, в дополнение к атомам углерода, 1-4 гетероатома, выбранные из группы, состоящей из атома кислорода, атома серы и атома азота, такие как азабицикло[3.1.0]гексан.
Термин «гетероароматическое кольцо» представляет собой стабильное моноциклическое кольцо, содержащее до 3-10 атомов в кольце, или бициклическое углеродное кольцо, содержащее до 3-10 атомов в каждом кольце, в котором по меньшей мере одно кольцо является ароматическим и содержит 1-4 гетероатома, выбранные из О, N и S. Гетероарильные группы в рамках данного определения включают, но не ограничиваются ими, акридинил, карбазолил, циннолинил, хиноксалинил, пиразолил, индолил, бензотриазолил, фуранил, тиенил, бензотиенил, бензофуранил, хинолинил, изохинолинил, оксазолил, изоксазолил, индолил, пиразинил, пиридазинил, пиридинил, пиримидинил и пирролил.
Термин «спирокольцо» относится к группе из двух колец, соединенных спиросоединением атома углерода, в которой каждое кольцо содержит от 4 до 6 атомов кольца (один атом углерода кольца является общим для двух колец).
Термин «гетероспирокольцо» относится к группе из двух колец, соединенных спиросоединением атома углерода, содержащей один или более идентичных или различных гетероатомов, выбранных из атома азота и атома кислорода, в которой каждое кольцо содержит от 4 до 6 атомов кольца (один атом углерода кольца является общим для двух колец).
Термин «конденсированный гетероцикл» относится к циклическому углеводороду, в котором 2-3 кольца содержат два смежных (орто) атома и в котором по меньшей мере одно кольцо представляет собой ароматическое кольцо, содержащее от 1 до 3 гетероатомов, выбранных из О, N и S, и в некоторых вариантах осуществления к циклическому углеводороду, в котором 2 кольца содержат два смежных (орто) атома и в котором одно кольцо представляет собой ароматическое кольцо, содержащее от 1 до 3 гетероатомов, выбранных из О, N и S, а другое кольцо представляет собой насыщенный гетероцикл.
Термин «мостиковое кольцо» относится к полициклическим углеводородам, в которых два или более атомов углерода (атомы углерода в начале мостика) являются общими, которые классифицируются как бициклические углеводороды, трициклические углеводороды, тетрациклические углеводороды и тому подобные, в соответствии с количеством составляющих колец.
Использование термина «лечить» или «лечение», упоминаемого во всем данном документе, является обычным, например это обозначает оказание помощи человеку или уход за ним с целью противодействия, облегчения, уменьшения или улучшения состояния болезни или нарушения, такого как рак.
Термин «индивид» или «пациент» включает организмы, такие как человеческие организмы и не являющиеся человеком животные, у которых возможно развитие пролиферативного нарушения клеток или которые могут получить пользу от введения соединения по настоящему изобретению. Предпочтительные человеческие организмы включают в себя пациентов-людей, болеющих пролиферативным заболеванием клеток или ассоциированным с ним патологическим состоянием или чувствительных к нему, как описано в данном документе. Термин «не являющиеся человеком животные» включает в себя позвоночных, например млекопитающих, таких как не являющиеся человеком приматы, овцы, коровы, собаки, кошки и грызуны (например, мыши), а также немлекопитающие, например, куры, амфибии, рептилии и тому подобные.
Термин «пролиферация клеток» включает в себя нежелательную или неконтролируемую пролиферацию клеток. Соединение по настоящему изобретению могут применять для предотвращения, ингибирования, блокирования, снижения, контроля и так далее пролиферации клеток и/или деления клеток, и/или для инициирования апоптоза. Способ включает введение нуждающемуся в этом индивиду (включая млекопитающих, включающих людей) определенного количества соединения по настоящему изобретению или его фармацевтически приемлемых соли, изомера, полиморфа, метаболита, гидрата или сольвата, эффективного для лечения или профилактики такого нарушения.
По сравнению с известным уровнем техники настоящее изобретение обладает описанными ниже преимуществами.
Соединение ингибитора RET-киназы по настоящему изобретению обладает биологической активностью на уровне ферментов и клеток, превосходящей активность имеющегося на рынке лекарственного вещества селперкатиниб (LOXO-292), и имеет меньшую
кардиотоксичность. Соединение по настоящему изобретению предоставляет больше возможностей для создания новых противоопухолевых лекарственных веществ и перспективно для применения в лекарственных средствах.
ПОДРОБНОЕ ОПИСАНИЕ
Следующие репрезентативные примеры предназначены для того, чтобы помочь проиллюстрировать настоящее изобретение и не ограничивают объем настоящего изобретения или не должны быть истолкованы как его ограничивающие. При практических реализациях, за исключением представленных и описанных в данном документе, полное содержание документа настоящей заявки, включая примеры в сочетании с цитируемой в данном документе научной литературой и патентами, а также различные модификации и многочисленные дальнейшие вариации, вытекающие из них, будут очевидны специалистам в данной области. Следует также понимать, что цитирование этих ссылок является полезным при изложении раскрытия в настоящем документе. Следующие примеры содержат важную дополнительную информацию, примеры и руководства, адаптируемые к различным вариантам настоящего изобретения и тому подобному.
Пример 1
Схема синтеза выглядит следующим образом:
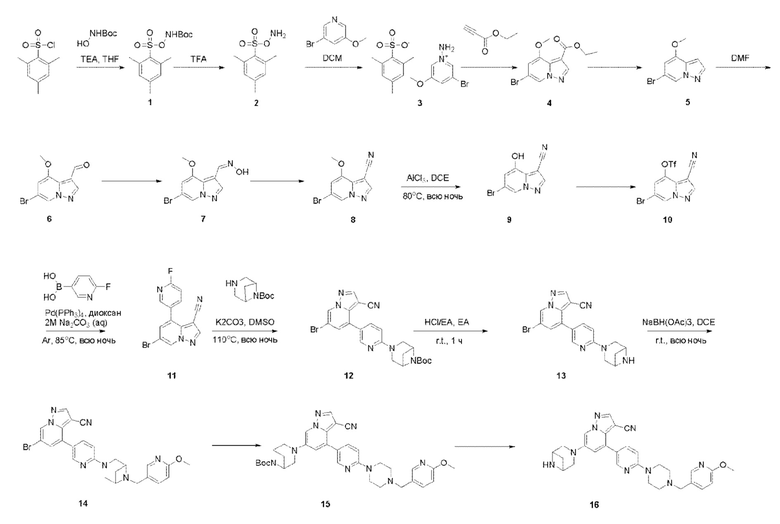
Соединение 1
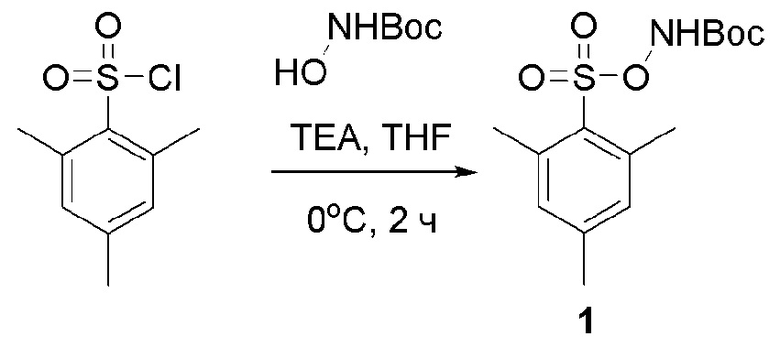
Готовили трехгорлую колбу объемом 2 л, низкотемпературный термометр и устройство для защиты аргоном. В трехгорлую колбу последовательно добавляли 2,4,б-триметилбензолсульфонилхлорид (80 г, 367 ммоль), трет-бутил N-гидроксикарбамат (62,4 г, 468 ммоль) и THF (1,2 л). Температуру снижали до 0°С, и при этой температуре медленно по каплям добавляли TEA (64 мл). Смесь перемешивали при комнатной температуре в течение еще 1 ч, и контроль ТСХ показал отсутствие остатков исходного вещества. Растворитель отгоняли при пониженном давлении. Для растворения остатка добавляли ЕА (1,2 л), раствор промывали Н2О (1,2 л × 3) и раствором 10% NaHCO3 (1,2 л), высушивали над безводным Na2SO4 и растворяли под пониженным давлением с получением продукта 1 в виде бледно-желтого твердого вещества (110 г, 95% выход).
Соединение 2
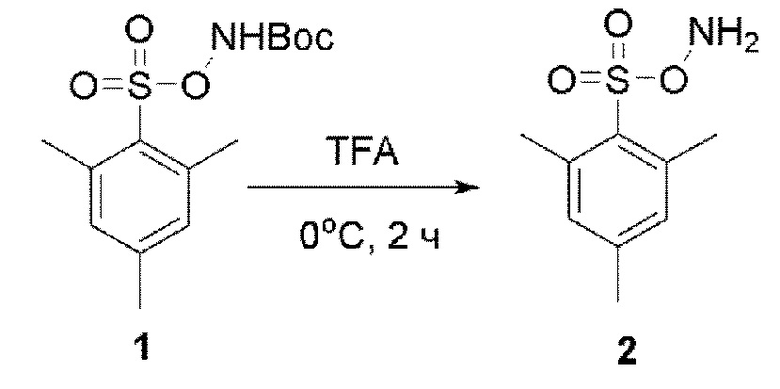
Готовили трехгорлую колбу объемом 500 мл,
низкотемпературный термометр и устройство для защиты аргоном. В трехгорлую колбу добавляли TFA (150 мл). Температуру снижали до 0°С, и при этой температуре порциями добавляли 1 (110 г, 348,8 ммоль). Смесь перемешивали при комнатной температуре в течение еще 3 ч, и контроль ТСХ показал отсутствие остатков исходного вещества. Реакционную смесь медленно выливали в ледяную воду при энергичном перемешивании. После 10 минут перемешивания осаждалось белое твердое вещество. Смесь фильтровали, и отфильтрованный осадок промывали ледяной водой и сушили с получением продукта 2 в виде белого твердого вещества (60,1 г, выход 80%).
Соединение 3
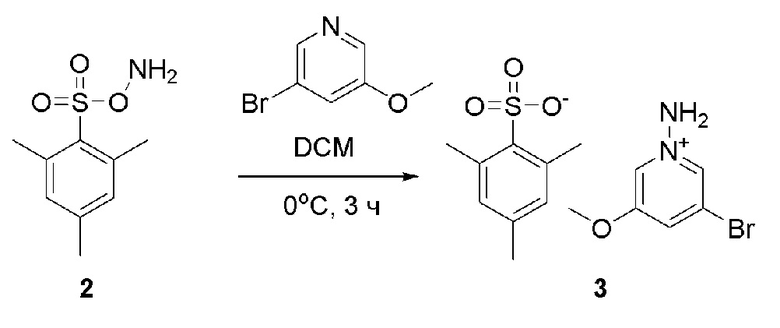
Готовили трехгорлую колбу объемом 500 мл, низкотемпературный термометр и устройство для защиты аргоном. В трехгорлую колбу последовательно добавляли 2 (60 г, 278,5 ммоль), 3-бром-5-метоксипиридин (52,37 г, 278,6 ммоль) и DCM (1 л). Температуру снижали до 0°С, и после 1 ч перемешивания добавляли дополнительно 3-бром-5-метоксипиридин (523 мг, 2,8 ммоль). Смесь перемешивали при 0°С в течение еще 2 ч. При этой температуре добавляли РЕ (1 л). После 10 минут перемешивания осаждалось белое твердое вещество. Смесь фильтровали, и фильтрат концентрировали. Для растворения остатка добавляли DCM (300 мл). После снижения температуры до 0°С добавляли РЕ (300 мл). Смесь перемешивали в течение еще 10 мин, и осаждалось белое твердое вещество. Смесь фильтровали полученные твердые вещества объединяли и сушили с получением продукта 3 в виде белого твердого вещества (87,6 г, выход 78%).
Соединение 4
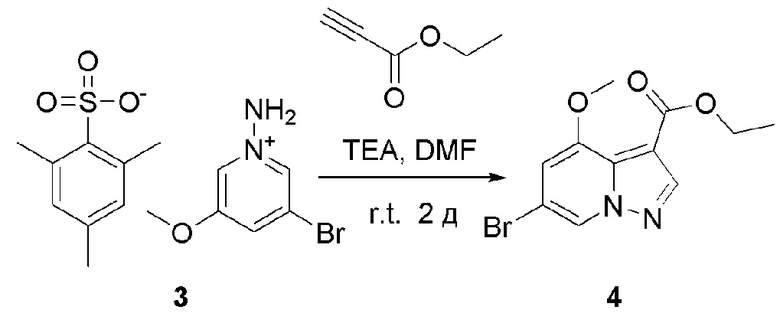
В одногорлую колбу объемом 500 мл последовательно добавляли соединение 3 (87,6 г, 217,2 ммоль), этилпропиолат (42,6 г, 434,4 ммоль) и DMF (200 мл), и при комнатной температуре медленно добавляли по каплям TEA (60,4 мл, 434,4 ммоль). После завершения добавления смесь перемешивали при комнатной температуре в течение еще 2 ч, и контроль ТСХ показал отсутствие остатков исходного вещества. Смесь разбавляли и гасили добавлением H2O (600 мл), и экстрагировали ЕА (200 мл × 3). Экстракты объединяли, концентрировали и очищали колоночной хроматографией с получением продукта 4 в виде оранжевого твердого вещества (48,7 г, выход 75%).
Соединение 5

В одногорлую грушевидную колбу объемом 1 л добавляли 40% НВг (400 мл), и порциями при перемешивании добавляли соединение 4 (48,7 г, 162,8 ммоль). Смесь подогревали до 100°С и перемешивали в течение еще 1 ч, и контроль ТСХ показал отсутствие остатков исходного вещества. Смесь охлаждали до комнатной температуры, затем выливали на колотый лед, и смесь перемешивали. Значение рН корректировали до>8 с помощью 2 М раствора NaOH, и осаждалось твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 5 в виде розового твердого вещества (34,38 г, выход 93%).
Соединение 6
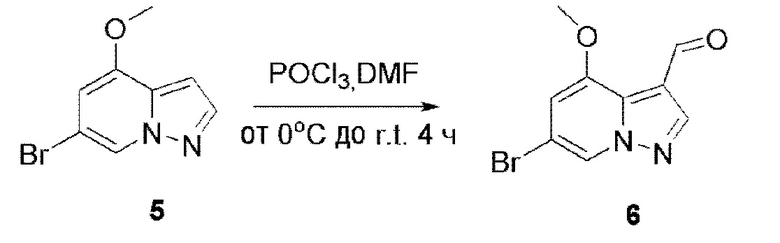
В одногорлую колбу объемом 500 мл последовательно добавляли соединение 5 (34,38 г, 151,4 ммоль) и DMF (200 мл), и при температуре 0°С медленно по каплям добавляли PDCl3 (76 мл). После завершения добавления смесь подогревали до комнатной температуры и перемешивали еще 4 ч, и контроль ТСХ показал отсутствие остатков исходного вещества. Смесь выливали в 200 мл H2O для разбавления, и значение рН корректировали до >8 с помощью 3 М раствора NaOH. Смесь перемешивали при комнатной температуре в течение 20 мин, и осаждалось твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 6 в виде серого твердого вещества (34,7 г, выход 90%).
Соединение 7
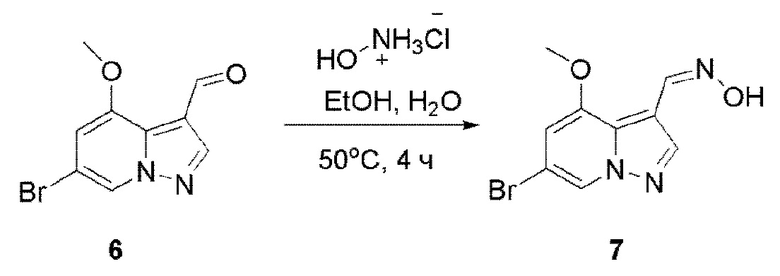
В одногорлую колбу объемом 500 мл последовательно добавляли соединение 6 (34,38 г, 151,4 ммоль), гидрохлорид гидроксиламина (13,7 г, 196,82 ммоль), EtOH (250 мл) и H2O (80 мл). Смесь перемешивали при температуре 50°С в течение еще 4 ч, и контроль ТСХ показал отсутствие остатков исходного вещества. EtOH отгоняли при пониженном давлении. Добавляли 250 мл H2O. Значение рН корректировали до >9 с помощью насыщенного раствора NaHCO3, и осаждалось твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 7 в виде серого твердого вещества (38 г, выход 93%).
Соединение 8
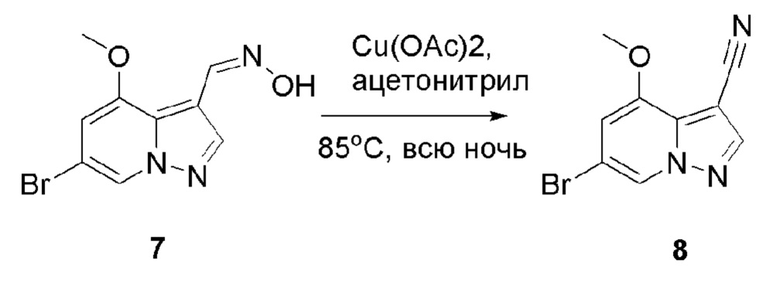
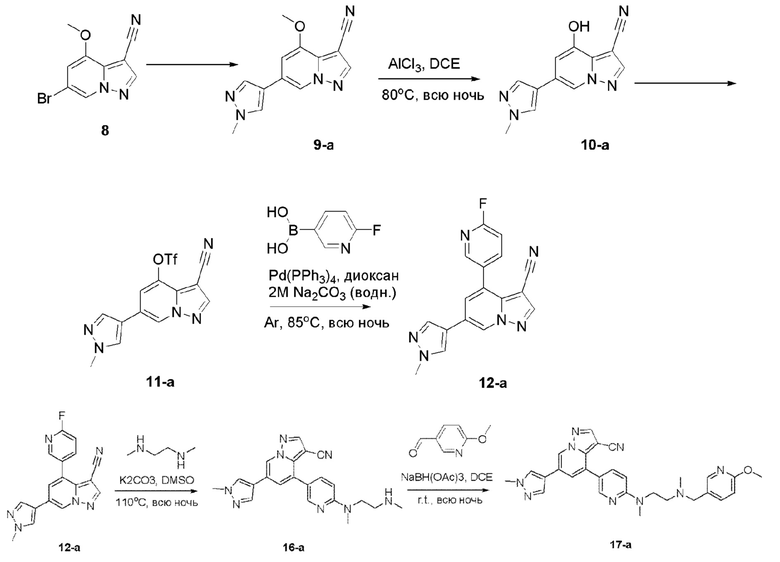
В одногорлую колбу объемом 500 мл последовательно добавляли соединение 7 (38 г, 140,8 ммоль), Cu(ОАс)2 (25,6 г, 140,8 ммоль) и ацетонитрил (200 мл). Смесь реагировала при температуре 85°С в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества. Смесь охлаждали до комнатной температуры. Значение рН корректировали до 8 с помощью NH3⋅H2O, и осаждалось твердое вещество. Смесь фильтровали, и отфильтрованный осадок растирали и промывали метил трет-бутиловым эфиром. Твердое вещество собирали фильтрацией и сушили с получением продукта 8 в виде коричнево-серого твердого вещества (19,4 г, выход 55%).
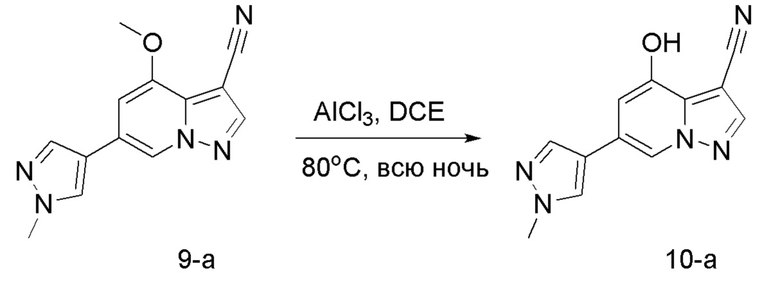
Соединение 9
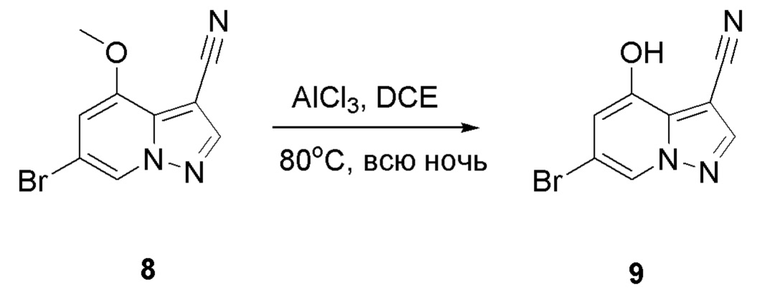
В герметизируемую пробирку объемом 45 мл последовательно добавляли соединение 8 (5 г, 19,8 ммоль) и 1,2-дихлорэтан (100 мл), и при комнатной температуре порциями добавляли AlCl3 (9,34 мг, 70 ммоль). Смесь перемешивали при температуре 80°С в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 8. Смесь охлаждали до комнатной температуры, гасили добавлением Na2SO4⋅10Н2О, перемешивали в течение 1 ч, фильтровали, промывали МеОН, концентрировали для удаления растворителя и очищали колоночной хроматографией с получением продукта 9 в виде серого твердого вещества (3 г, выход 55%).
Соединение 10
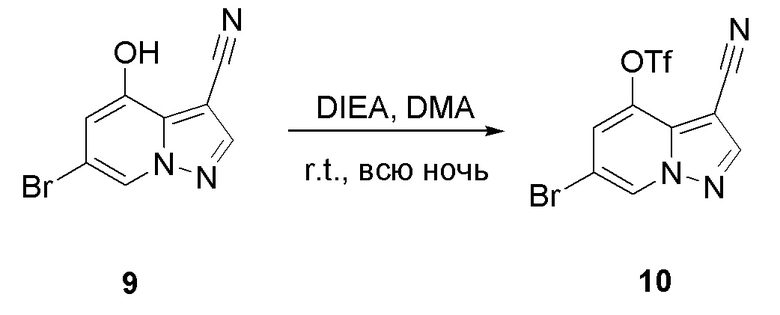
В одногорлую колбу объемом 50 мл последовательно добавляли соединение 9 (3 г, 12,6 ммоль), N-фенилбис(трифторметансульфонил)имид (4,5 г, 12,6 ммоль), DIEA (3,26 г, 25,2 ммоль) и DMA (20 мл). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал завершение реакции. Реакционную смесь выливали при перемешивании в 60 мл H2O, и осаждалось коричневое твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 10 в виде коричневого твердого вещества (4,4 г, выход 95%).
Соединение 11
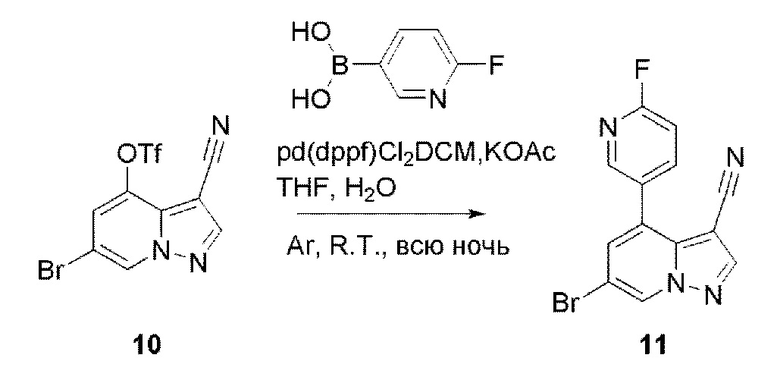
В герметизируемую пробирку объемом 12 мл последовательно добавляли соединение 10 (4,4 г, 11,9 ммоль), комплекс Pd(dppf)Cl2 с дихлорметаном (490 мг, 0,6 ммоль), 2-фторпиридин-5-бороновую кислоту (1,68 г, 11,9 ммоль), KOAc (2,92 г, 29,75 ммоль) и диоксан (50 мл). Смесь перемешивали при 85°С в течение ночи, и контроль ТСХ показал завершение реакции. Смесь охлаждали до комнатной температуры, и добавляли 100 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось коричневато-желтое твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 11 (3 г, выход 80%).
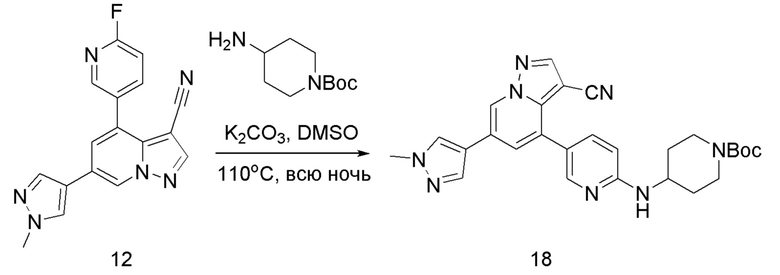
Соединение 12
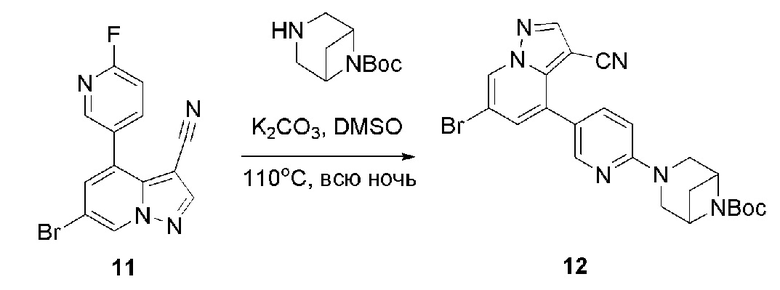
В герметизируемую пробирку объемом 45 мл последовательно добавляли соединение 11 (3 г, 9,46 ммоль), 6-(трет-бутоксикарбонил)-3,6-диазабицикло[3.1.1]гептан (2,25 г, 11,3 ммоль), K2CO3 (3,9 г, 28,35 ммоль) и DMSO (20 мл). Смесь перемешивали при температуре 110°С в течение ночи, и контроль ТСХ показал завершение реакции.Смесь охлаждали до комнатной температуры, и добавляли 50 мл воды.Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 12 (3,2 г, выход 69%).
Соединение 13
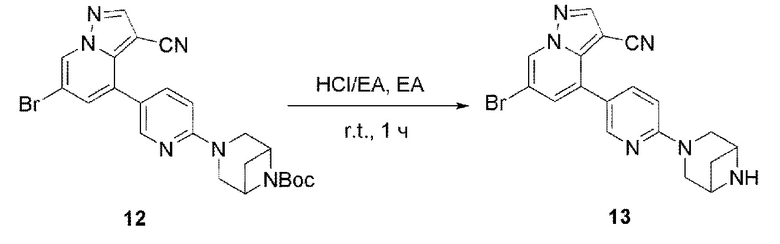
В одногорлую колбу объемом 25 мл добавляли 3,5 М раствор HCl в ЕА (10 мл) и медленно по каплям добавляли раствор соединения 12 (94,5 мг, 0,2 ммоль) в ЕА. После завершения добавления смесь перемешивали при комнатной температуре еще 1 ч, концентрировали для удаления растворителя, нейтрализовали добавлением раствора NH3 в метаноле, концентрировали и очищали колоночной хроматографией с получением продукта 13 в виде коричневато-желтого твердого вещества (68 мг, выход 90%).
Соединение 14
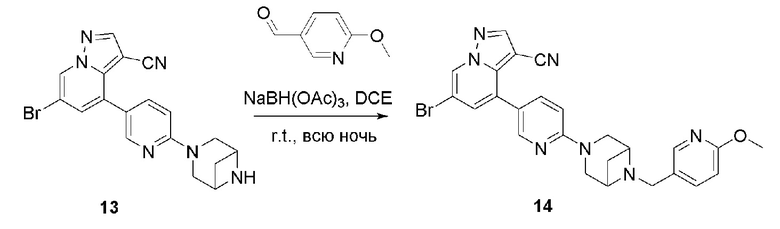
В одногорлую колбу объемом 50 мл последовательно добавляли соединение 13 (68 мг, 0,18 ммоль), 6-метокси-3-пиридинкарбоксальдегид (30,2 мг, 0,22 ммоль) и DCM (10 мл). После 10 мин перемешивания, при комнатной температуре порциями добавляли NaBH(OAc)3 (190,8 мг, 0,9 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 13. Смесь гасили добавлением аммиачной воды. Водную фазу отделяли и экстрагировали DCM (5 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением продукта 14 в виде желтого твердого вещества (55 мг, выход 61%).
Соединение 15
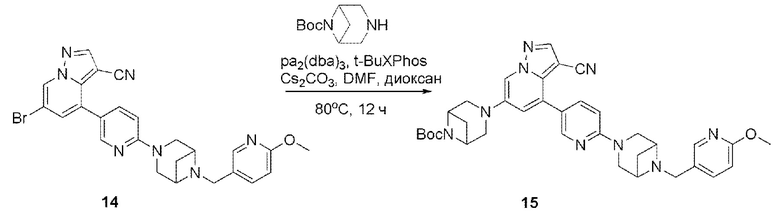
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 14 (500 мг, 0,97 ммоль), Pd2(dba)3 (106,4 мг, 0,12 ммоль), t-BuXPhos (152,9 мг, 0,36 ммоль), 6-(трет-бутоксикарбонил)-3,6-диазабицикло[3.1.1]гептан (384,12 мг, 1,94 ммоль), Cs2CO3 (632,1 мг, 1,94 ммоль), диоксан (6 мл) и DMF (3 мл). Смесь перемешивали при температуре 80°С в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 14. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 15 (450 г, выход 73%).
1Н ЯМР (400 МГц, CDCl3) δ 8,43 (д, J=2,2 Гц, 1Н), 8,17 (с, 1Н), 8,11 (д, J=2,1 Гц, 1Н), 7,95 (д, J=2,1 Гц, 1Н), 7,83 (дд, J=8,8, 2,5 Гц, 1Н), 7,64 (дд, J=8,5, 2,4 Гц, 1Н), 7,10 (д, J=2,1 Гц, 1Н), 6,72 (дд, J=8,6, 6,0 Гц, 2Н), 4,00 (д, J=5,9 Гц, 2Н), 3,92 (д, J=7,4 Гц, 3Н), 3,85 (д, J=12,0 Гц, 2Н), 3,77 (т, J=9, 9 Гц, 2Н), 3,60 (д, J=13,1 Гц, 8Н), 2,86 (дт, J=8,8, 6,2 Гц, 1Н), 2,70 (дд, J=14,2, 6,2 Гц, 1Н), 1,69 (дд, J=20,8, 8,8 Гц, 2Н), 1,42 (с, 9Н).
Соединение 16
В одногорлую грушевидную колбу объемом 25 мл добавляли раствор хлористоводородной кислоты в этилацетате (10 мл) и медленно добавляли соединение 15 (450 мг, 0,71 ммоль). Смесь перемешивали при комнатной температуре в течение 3 ч, и контроль ТСХ показал отсутствие остатков исходного вещества. Смесь концентрировали, корректировали рН до 9 аммиачной водой и экстрагировали DCM (3 мл × 3). Органические фазы объединяли, концентрировали и очищали колоночной хроматографией с получением целевого продукта 16 в виде желтого твердого вещества (350 мг, выход 92%).
1Н ЯМР (400 МГц, CDCl3) δ 8,43 (д, J=2,2 Гц, 1Н), 8,17 (с, 1Н), 8,11 (д, J=2,1 Гц, 1Н), 7,95 (д, J=2,1 Гц, 1Н), 7,83 (дд, J=8,8, 2,5 Гц, 1Н), 7,64 (дд, J=8,5, 2,4 Гц, 1Н), 7,10 (д, J=2,1 Гц, 1Н), 6,72 (дд, J=8,6, 6,0 Гц, 2Н), 4,00 (д, J=5,9 Гц, 2Н), 3,92 (д, J=7,4 Гц, 3Н), 3,85 (д, J=12,0 Гц, 2Н), 3,77 (т, J=9,9 Гц, 2Н), 3,60 (д, J=13,1 Гц, 8Н), 2,86 (дт, J=8,8, 6,2 Гц, 1Н), 2,70 (дд, J=14,2, 6,2 Гц, 1Н), 1,69 (дд, J=20,8, 8,8 Гц, 2Н).
Пример 2
Соединение 17
В одногорлую грушевидную колбу объемом 25 мл последовательно добавляли соединение 16 (200 мг, 0,37 ммоль), параформальдегид (333 мг, 3,7 ммоль) и DCM (15 мл). Смесь перемешивали при комнатной температуре в течение 30 мин, и добавляли борогидрид ацетат натрия (392,2 мг, 1,85 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества. Корректировали рН до 9 аммиачной водой, и смесь экстрагировали DCM (3 мл × 3). Органические фазы объединяли, концентрировали и очищали колоночной хроматографией с получением целевого продукта 17 в виде желтого твердого вещества (150 мг, выход 74,2%).
1Н ЯМР (400 МГц, CDCl3) δ 8,45 (д, J=2,2 Гц, 1Н), 8,18 (с, 1Н), 8,12 (д, J=2,0 Гц, 1Н), 7,98 (д, J=2,1 Гц, 1Н), 7,84 (дд, J=8,8, 2,5 Гц, 1Н), 7,67 (дд, J=8,5, 2,4 Гц, 1Н), 7,13 (д, J=2,1 Гц, 1Н), 6,77-6,68 (м, 2Н), 4,74 (с, 3Н), 3,94 (с, 3Н), 3,90-3,78 (м, 6Н), 3,69-3,55 (м, 6Н), 3,41 (д, J=10,7 Гц, 2Н), 2,83-2,67 (м, 2Н), 1,70 (дт, J=14,4, 7,2 Гц, 2Н).
Пример 3
Соединение 18
Готовили трехгорлую колбу объемом 250 мл, устройство для защиты аргоном и низкотемпературный термометр. В колбу добавляли 100 мл THF и метилфосфонилдихлорид (5 г, 37,6 ммоль). Температуру снижали до -78°С, и в течение 30 мин медленно по каплям добавляли винилмагния бромид (38 мл, 38 ммоль). Смесь нагревали до 0°С и перемешивали в течение 1 ч. По каплям добавляли раствор бензиламина (4,8 г, 44,8 ммолв) в метаноле. Смесь нагревали до температуры 68°С и выдерживали с обратным холодильником в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества. Смесь очищали колоночной хроматографией с получением целевого продукта 18 в виде белого твердого вещества (3,1 г, выход 36,9%).
Соединение 19
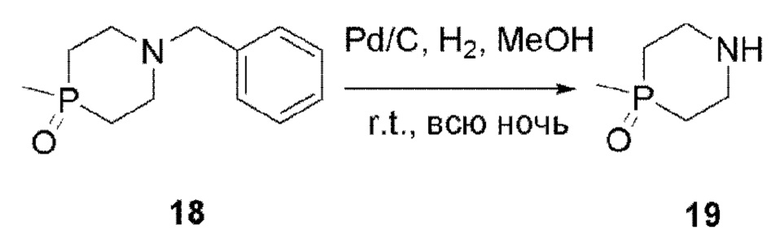
В одногорлую колбу объемом 100 мл последователвно добавляли соединение 18 (3 г, 13,4 ммоль), палладий на углероде (500 мг) и метанол (30 мл). Смесь перемешивали при комнатной температуре в течение ночи в атмосфере Н2, и контроль ТСХ показал отсутствие остатков исходного вещества. Смесь фильтровали для удаления палладия на углероде и концентрировали с получением целевого продукта 19 (1,5 г, выход 83,85%). Продукт непосредственно использовали на следующей стадии без очистки.
Соединение 20
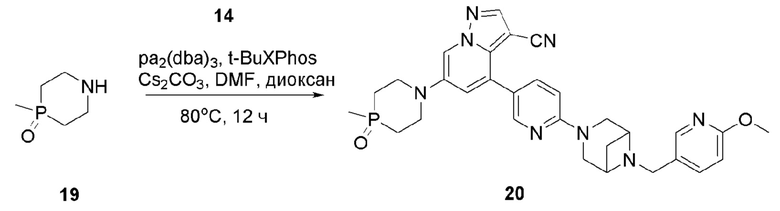
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 14 (300 мг, 0,58 ммолв), Pd2(dba)3 (64,1 мг, 0,07 ммоль), t-BuXPhos (89,2 мг, 0,21 ммоль), соединение 19 (231,6 мг, 1,74 ммоль), Cs2CO3 (378 мг, 1,16 ммоль), диоксан (6 мл) и DMF (3 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 14. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 20 (230 мг, выход 70%).
1Н ЯМР (400 МГц, CDCl3) δ 8,45 (д, J=2,2 Гц, 1Н), 8,18 (с, 1Н), 8,12 (д, J=2,0 Гц, 1Н), 7,98 (д, J=2,1 Гц, 1Н), 7,84 (дд, J=8,8, 2,5 Гц, 1Н), 7,67 (дд, J=8,5, 2,4 Гц, 1Н), 7,13 (д, J=2,1 Гц, 1Н), 6,77-6,68 (м, 2Н), δ 3,92 (с, 3Н), 3,84 (м, 4Н), 3,62 (м, 4Н), 3,51 (м, 5Н), 2,75 (м, 1Н), 2,08 (м, 4Н), 1,64 (м, 3Н).
Пример 4
Схема синтеза выглядит следующим образом:
Соединение 8 синтезировали как в примере 1.
Соединение 9-а

В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 8 (1 г, 3,97 ммоль), Pd(PPh3)4 (229,3 мг, 0,2 ммоль), 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (991 мг, 4,76 ммоль), 2 М K2CO3 (1,26 г, 11,9 ммоль) и 1,4-диоксан (8 мл). Смесь перемешивали при температуре 85°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 8. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 9-а (850 мг, выход 85%).
Соединение 10-а
В герметизируемую пробирку объемом 45 мл последовательно добавляли соединение 9-а (850 мг, 3,36 ммоль) и 1,2-дихлорэтан (15 мл), и порциями при комнатной температуре - AlCl3 (1,57 г, 11,76 ммоль). Смесь перемешивали при температуре 80°С в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 9-а. Смесь охлаждали до комнатной температуры, гасили добавлением Na2SO4⋅10Н2О, перемешивали в течение 1 ч, фильтровали, промывали МеОН, концентрировали для удаления растворителя и очищали колоночной хроматографией с получением продукта 10-а в виде серого твердого вещества (450 мг, выход 56%).
Соединение 11-а
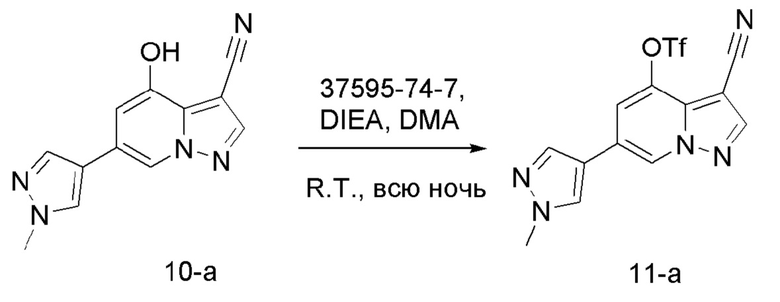
В одногорлую колбу объемом 50 мл последовательно добавляли соединение 10-а (450 мг, 1,88 ммоль), N-фенилбис(трифторметансульфонил)имид (806 г, 2,26 ммоль), DIEA (486 г, 3,76 ммоль) и DMA (5 мл). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 10-а. Реакционную смесь выливали при перемешивании в 10 мл H2O, и осаждалось коричневое твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 11-а в виде коричневого твердого вещества (628 мг, выход 90%).
Соединение 12-а
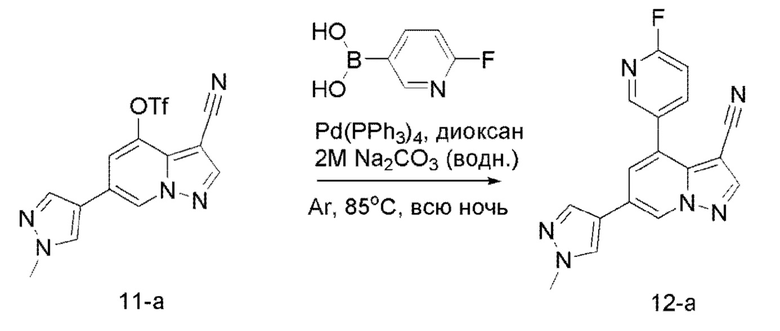
В герметизируемую пробирку объемом 12 мл последовательно добавляли соединение 11-а (628 мг, 1,69 ммоль), Pd(PPh3)4 (97,6 мг, 0,08 ммоль), 2-фторпиридин-5-бороновую кислоту (238,3 мг, 1,69 ммоль), 2 М Na2CO3 (358,3 мг, 3,38 ммоль) и 1,4-диоксан (5 мл). Смесь перемешивали при температуре 85°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 11-а. Смесв охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесв перемешивали в течение 10 мин, и осаждалось коричневато-желтое твердое вещество. Твердое вещество собирали филвтрацией и сушили с получением продукта 12-а (430 мг, выход 80%).
Соединение 16-а
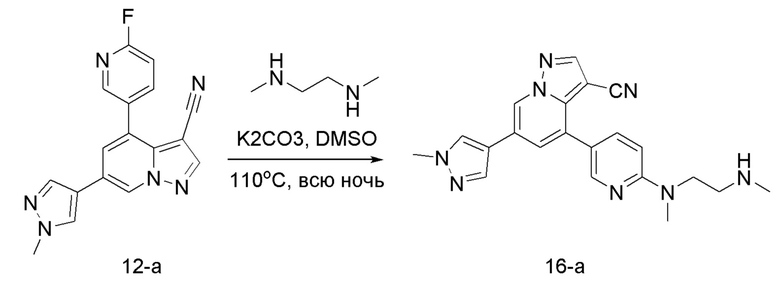
В герметизируемую пробирку объемом 12 мл последовательно добавляли соединение 12-а (70 мг, 0,22 ммоль), N,N'-диметилэтилендиамин (23,2 мг, 0,26 ммоль), K2CO3 (61 мг, 0,44 ммоль) и DMSO (1 мл). Смесь перемешивали при температуре 120°С в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 12-а. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 16-а (51 мг, выход 60%).
ЖХ-МС [М+Н]+ 387,2.
Соединение 17-а
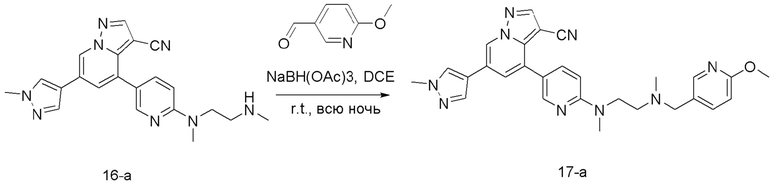
В одногорлую колбу объемом 50 мл последовательно добавляли соединение 16-а (51 мг, 0,13 ммоль), 6-метокси-3-пиридинкарбоксальдегид (26,7 мг, 0,19 ммоль) и DCM (15 мл). После 10 мин перемешивания, при комнатной температуре порциями добавляли NaBH(OAc)3 (137,8 мг, 0,65 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 16-а. Смесь гасили добавлением аммиачной воды. Водную фазу отделяли и экстрагировали DCM (5 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением продукта 17-а в виде желтого твердого вещества (30 мг, выход 46%).
1Н ЯМР (400 МГц, CDCl3) δ 8,62 (с, 1Н), 8,31 (с, 1Н), 8,25 (с, 1Н), 8,02 (с, 1Н), 7,80 (с, 1Н), 7,70 (с, 1Н), 7,68 (с, 1Н), 7,54 (д, J=6,6 Гц, 1Н), 7,39 (с, 1Н), 6,64 (дд, J=36,1, 8,7 Гц, 2Н), 3,99 (с, 3Н), 3,89 (с, 3Н), 3,79 (т, J=6,7 Гц, 2Н), 3,53 (с, 2Н), 3,11 (с, 3Н), 2,67 (т, J=6,6 Гц, 2Н), 2,30 (с, 3Н). ЖХ-МС [М+Н]+ 507,6.
Пример 5
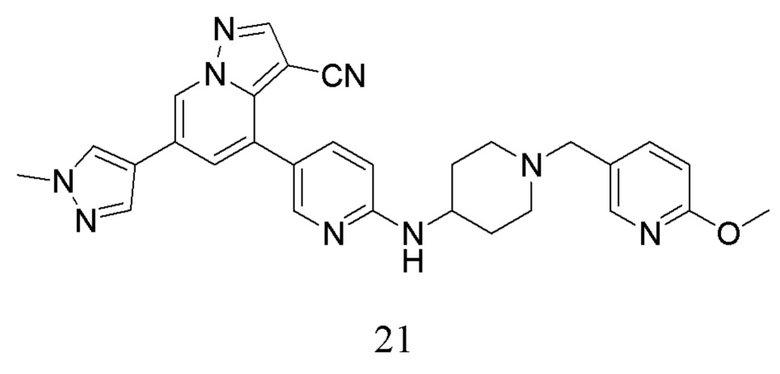
Схема синтеза выглядит следующим образом:
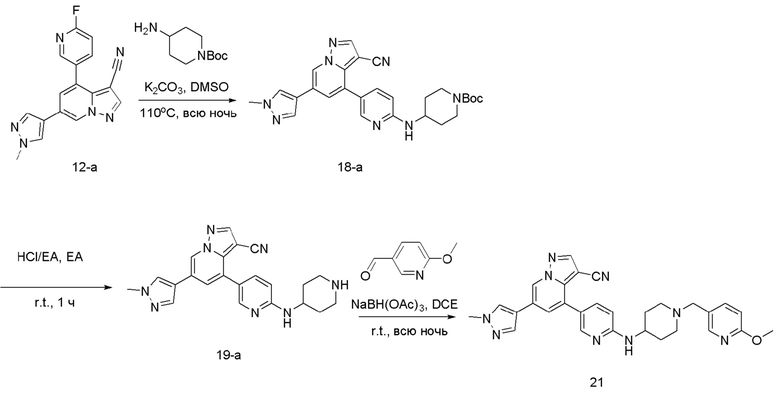
Соединение 18-а
В герметизируемую пробирку объемом 12 мл последовательно добавляли соединение 12-а (80 мг, 0,25 ммоль), 1-Вос-4-аминопиридин (60,5 мг, 0,3 ммоль), K2CO3 (69 мг, 0,5 ммоль) и DMSO (1 мл). Смесь перемешивали при температуре 120°С в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 12-а. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 18-а (70 мг, выход 56%).
ЖХ-МС [М+Н]+ 499,25.
Соединение 19-а
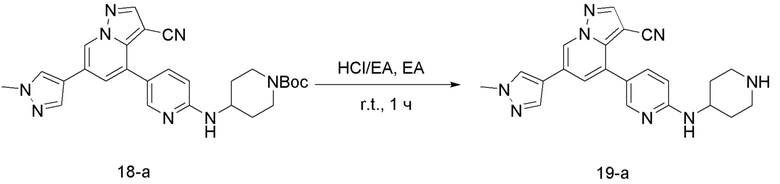
В одногорлую колбу объемом 50 мл добавляли 3,5 М раствор HCl в ЕА (10 мл) и медленно по каплям добавляли раствор соединения 18-а (70 мг, 0,14 ммоль) в ЕА. После завершения добавления смесь перемешивали при комнатной температуре еще 3 ч, концентрировали для удаления растворителя, нейтрализовали добавлением по каплям раствора NH3 в метаноле и концентрировали с получением продукта 19-а в виде желтого твердого вещества (55 мг, выход 98%).
ЖХ-МС [М+Н]+ 399,25.
Соединение 21
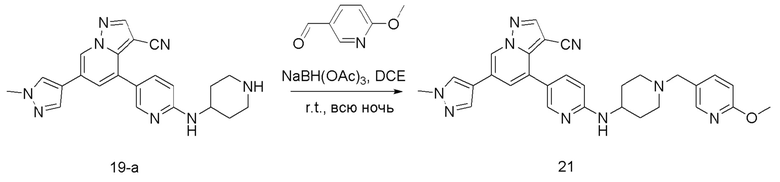
В одногорлую колбу объемом 50 мл последовательно добавляли соединение 19-а (55 мг, 0,138 ммоль), 6-метокси-3-пиридинкарбоксальдегид (22,7 мг, 0,166 ммоль) и DCM (15 мл). После 10 мин перемешивания, при комнатной температуре порциями добавляли NaBH(OAc)3 (146,28 мг, 0,69 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 19-а. Смесь гасили добавлением аммиачной воды. Водную фазу отделяли и экстрагировали DCM (5 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением продукта 21 в виде желтого твердого вещества (30 мг, выход 41,8%).
1Н ЯМР (400 МГц, CDCl3) δ 8,63 (с, 1Н), 8,29-8,20 (м, 2Н), 8,06 (с, 1Н), 7,79 (с, 1Н), 7,64 (ддд, J=19,4, 7,1, 2,8 Гц, 3Н), 7,38 (с, 1Н), 6,74 (д, J=8,4 Гц, 1Н), 6,52 (д, J=8,6 Гц, 1Н), 4,78 (д, J=7,7 Гц, 1Н), 4,00 (с, 3Н), 3,94 (с, 3Н), 3,77 (с, 1Н), 3,54 (с, 2Н), 2,90 (с, 2Н), 2,29 (д, J=10,2 Гц, 2Н), 2,10 (д, J=15,2 Гц, 2Н), 1,61 (д, J=10,2 Гц, 2Н). ЖХ-МС [М+Н]+ 519,6.
Пример 6
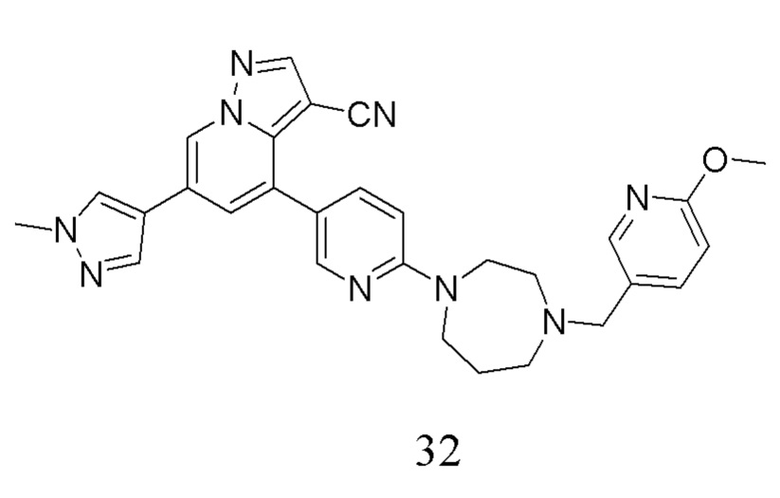
Схема синтеза выглядит следующим образом:
Соединение 30
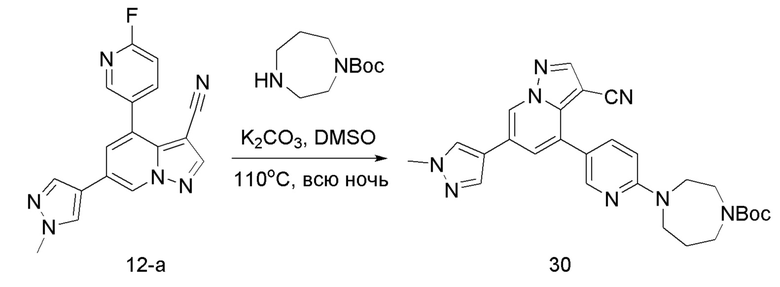
В герметизируемую пробирку объемом 12 мл последовательно добавляли соединение 12-а (70 мг, 0,22 ммоль), трет-бутил 1,4-диазепан-1-карбоксилат (52,1 мг, 0,26 ммоль), K2CO3 (60,8 мг, 0,44 ммоль) и DMSO (3 мл).Смесь перемешивали при температуре 120°С в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 12-а. Смесь охлаждали до комнатной температуры, и добавляли 5 мл воды.Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 30 (55 мг, выход 50%).
ЖХ-МС [М+Н]+ 499,25.
Соединение 31
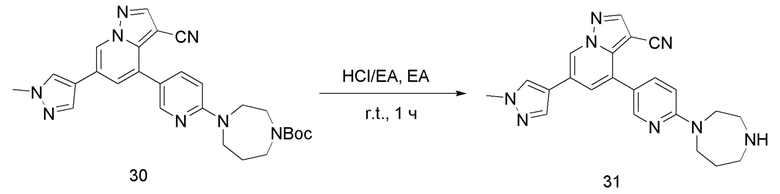
В одногорлую колбу объемом 50 мл добавляли 3,5 М раствор HCl в ЕА (5 мл) и медленно по каплям добавляли раствор соединения 30 (55 мг, 0,11 ммоль) в ЕА. После завершения добавления смесь перемешивали при комнатной температуре еще 3 ч, концентрировали для удаления растворителя, нейтрализовали добавлением по каплям раствора NH3 в метаноле и концентрировали с получением продукта 31 в виде желтого твердого вещества (44,6 мг, выход 100%).
ЖХ-МС [М+Н]+ 399,25.
Соединение 32
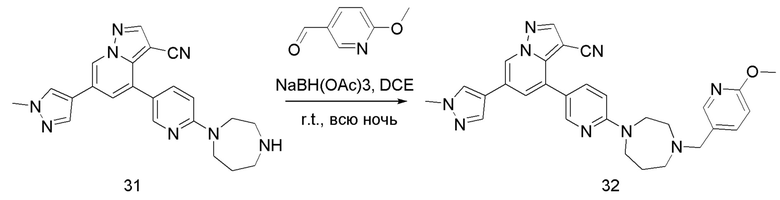
В одногорлую колбу объемом 50 мл последовательно добавляли соединение 31 (44,6 мг, 0,11 ммоль), 6-метокси-3-пиридинкарбоксальдегид (18,1 мг, 0,13 ммоль) и DCM (5 мл). После 10 мин перемешивания, при комнатной температуре порциями добавляли NaBH(OAc)3 (93,3 мг, 0,44 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 31. Смесь гасили добавлением аммиачной воды. Водную фазу отделяли и экстрагировали DCM (5 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением продукта 32 в виде желтого твердого вещества (32 мг, выход 55%).
1Н ЯМР (400 МГц, CDCl3) δ 8,62 (д, J=1,4 Гц, 1Н), 8,34 (д, J=2,4 Гц, 1Н), 8,25 (с, 1Н), 8,05 (д, J=1,8 Гц, 1Н), 7,79 (с, 1Н), 7,71 (дд, J=8,9, 2,5 Гц, 1Н), 7,68 (с, 1Н), 7,63-7,57 (м, 1Н), 7,39 (д, J=1,4 Гц, 1Н), 6,72 (д, J=8,5 Гц, 1Н), 6,63 (д, J=8,9 Гц, 1Н), 3,99 (с, 3Н), 3,93 (с, 3Н), 3,85 (м, 2Н), 3,73 (т, J=6,1 Гц, 2Н), 3,58 (с, 2Н), 2,79 (с, 2Н), 2,64 (д, J=5,1 Гц, 2Н), 1,98 (с, 2Н), 1,68 (с, 2Н). ЖХ-МС [М+Н]+ 519,6.
Пример 7
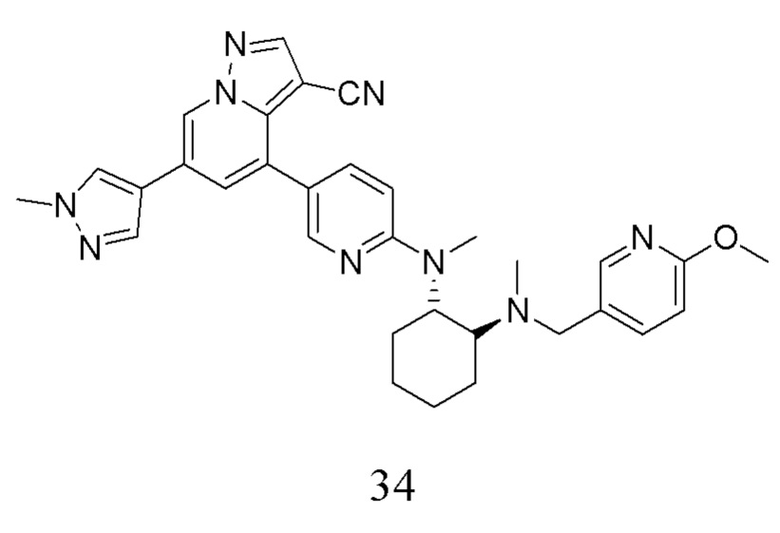
Схема синтеза выглядит следующим образом:
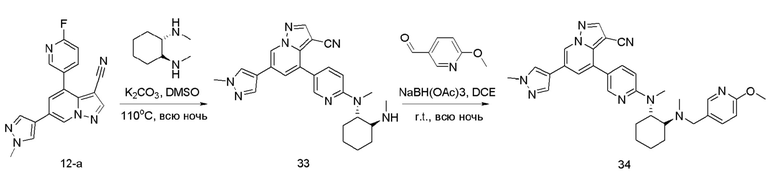
Соединение 33
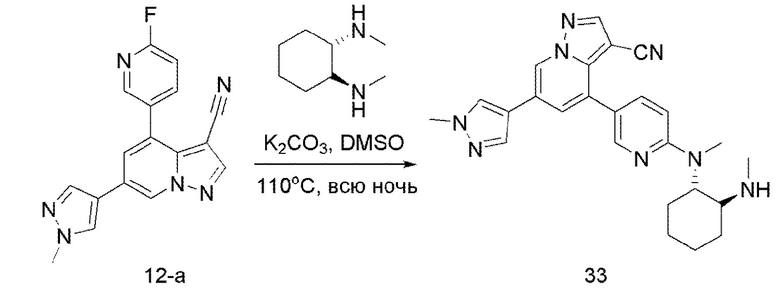
В герметизируемую пробирку объемом 12 мл последовательно добавляли соединение 12-а (70 мг, 0,22 ммоль), (IS,2S)-(+)-N,N'-диметил-1,2-циклогександиамин (31,3 мг, 0,22 ммоль), К?С03 (60,8 мг, 0,44 ммоль) и DMSO (3 мл).Смесь перемешивали при температуре 120°С в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 12-а. Смесь охлаждали до комнатной температуры, и добавляли 5 мл воды.Смесь перемешивали в течение 10 мин, и осаждалосв желтое твердое вещество. Твердое вещество очищали колоночной хроматографией с получением продукта 33 (43,6 мг, выход 45%).
ЖХ-МС [М+Н]+ 441,24.
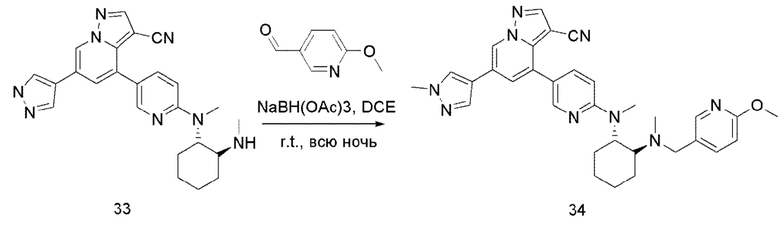
В одногорлую колбу объемом 50 мл последовательно добавляли соединение 33 (43,6 мг, 0,1 ммоль), 6-метокси-3-пиридинкарбоксальдегид (16,7 мг, 0,12 ммоль) и DCM (5 мл). После 10 мин перемешивания, при комнатной температуре порциями добавляли NaBH(ОАс)3 (84,8 мг, 0,4 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 33. Смесь гасили добавлением аммиачной воды. Водную фазу отделяли и экстрагировали DCM (5 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением продукта 34 в виде желтого твердого вещества (30 мг, выход 53%).
1Н ЯМР (400 МГц, CDCl3) δ 8,59 (с, 1Н), 8,24 (с, 1Н), 8,21 (с, 1Н), 7,84 (с, 1Н), 7,78 (с, 1Н), 7,73 (д, J=9,2 Гц, 1Н), 7,67 (с, 1Н), 7,25-7,21 (м, 1Н), 7,15 (с, 1Н), 6,64 (д, J=8,6 Гц, 1Н), 6,58 (д, J=8,2 Гц, 1Н), 3,99 (с, 3Н), 3,83 (с, 3Н), 3,73 (д, J=12,9 Гц, 1Н), 3,25 (д, J=13,0 Гц, 1Н), 2,89 (с, 3Н), 2,70 (с, 1Н), 2,10 (с, 3Н), 2,04 (с, 1Н), 1,92-1,76 (м, 3Н), 1,44 (дд, J=38,9, 14,7 Гц, 3Н). ЖХ-МС [М+Н]+ 561,7.
Пример 8
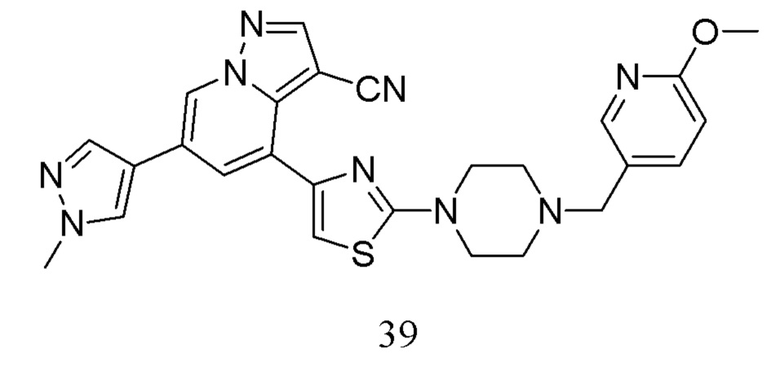
Схема синтеза выглядит следующим образом:
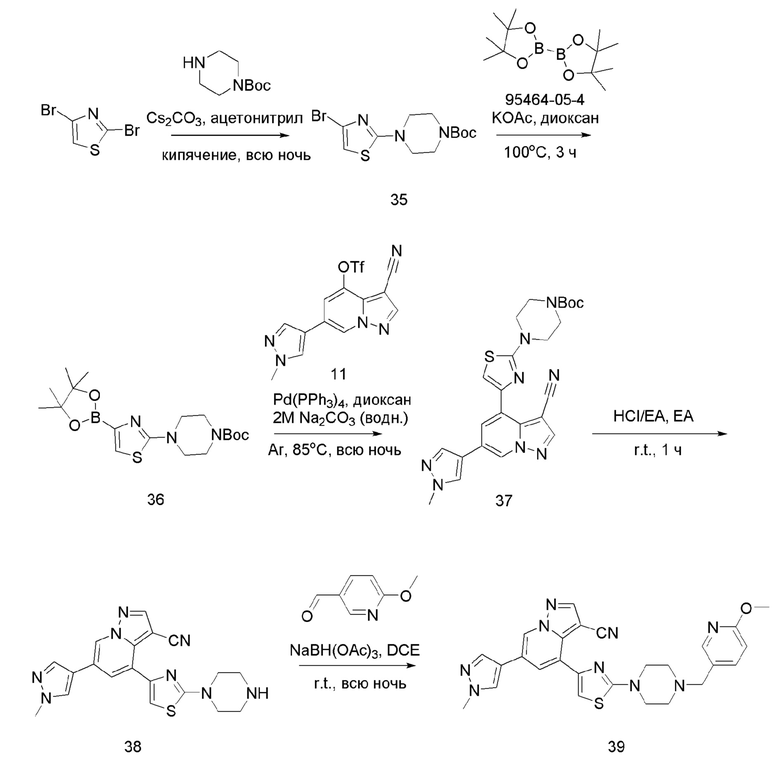
Соединение 35
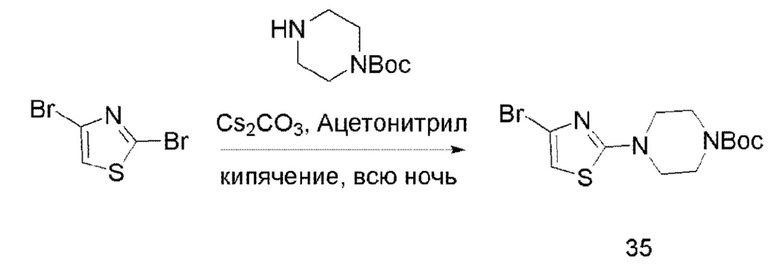
В одногорлую колбу объемом 50 мл последовательно добавляли соединения 2,4-дибромтиазол (1,5 г, 6,17 ммоль), 1-N-BOC-пиперазин (1,7 г, 9,26 ммоль), Cs2CO3 (4,0 г, 12,3 ммоль) и ацетонитрил (20 мл). Смесь нагревали с обратным холодильником в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества. Смесь разбавляли добавлением H2O (50 мл) и экстрагировали ЕА (20 мл × 3). Органические фазы объединяли, концентрировали и очищали колоночной хроматографией с получением продукта 35 в виде белого твердого вещества (1,6 г, выход 74,5%).
ЖХ-МС [М+Н]+ 348,03.
Соединение 36
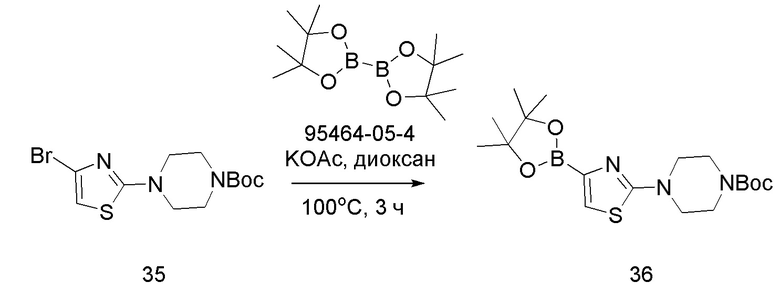
В герметизируемую пробирку объемом 45 мл последовательно добавляли соединение 35 (1,2 г, 3,45 ммоль), бис(пинаколято)диборон (918,7 мг, 3,62 ммоль), Pd(dppf)Cl2⋅DCM (140,9 мг, 0,17 ммоль), KOAc (1,02 г, 10,35 ммоль) и 1,4-диоксан (15 мл).Смесь перемешивали при температуре 100°С в течение 3 ч в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 35. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось красновато-коричневое твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 36 (1,26 г, выход 92,38%). ЖХ-МС [М+Н]+ 396,21.
Соединение 37

В герметизируемую пробирку объемом 45 мл последовательно добавляли соединение 36 (1,25 г, 3,18 ммоль), соединение 11-а (1,0 г, 2,69 ммоль), Pd(PPh3)4 (155 мг, 0,13 ммоль), 4 мл 2 М Na2CO3 (855,4 г, 8,07 ммоль) и 1,4-диоксан (20 мл). Смесь перемешивали при температуре 85°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 11-а. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 37 в виде серого твердого вещества (1,2 г, выход 91%).
ЖХ-МС [М+Н]+ 491,19.
Соединение 38
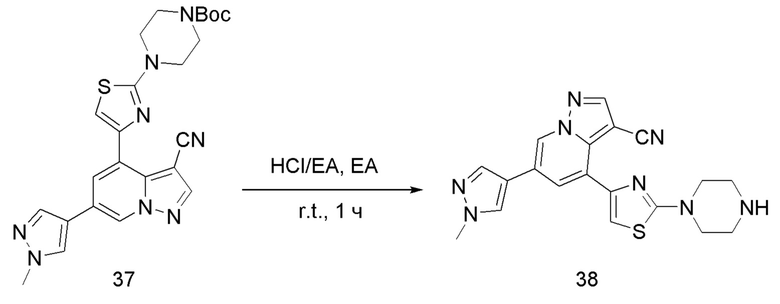
В одногорлую колбу объемом 25 мл добавляли 3,5 М раствор HCl в ЕА (10 мл) и медленно по каплям добавляли раствор соединения 37 (1,2 г, 2,45 ммоль) в ЕА. После завершения добавления смесь перемешивали при комнатной температуре еще 3 ч, концентрировали для удаления растворителя, нейтрализовали добавлением по каплям раствора NH3 в метаноле, концентрировали и очищали колоночной хроматографией с получением продукта 38 в виде коричневато-желтого твердого вещества (680 мг, выход 71,1%).
ЖХ-МС [М+Н]+ 492,19.
Соединение 39
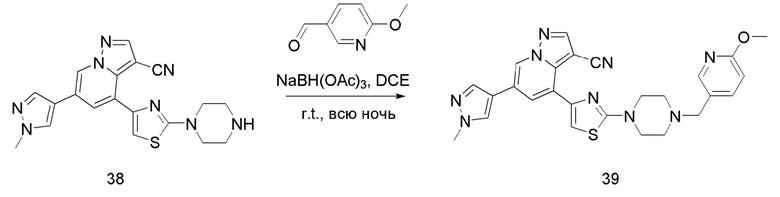
В одногорлую колбу объемом 50 мл последовательно добавляли соединение 38 (200 мг, 0,51 ммоль), 6-метокси-3-пиридинкарбоксальдегид (70,2 мг, 0,51 ммоль) и DCM (10 мл). После 10 мин перемешивания, при комнатной температуре порциями добавляли NaBH(OAc)3 (324,3 мг, 1,53 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 38. Смесь гасили добавлением аммиачной воды. Водную фазу отделяли и экстрагировали DCM (5 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением продукта 39 в виде желтого твердого вещества (170 мг, выход 65%).
1Н ЯМР (400 МГц, CDCl3) δ 8,61 (д, J=1,4 Гц, 1Н), 8,26 (с, 1Н), 8,07 (д, J=2,0 Гц, 1Н), 7,79 (с, 1Н), 7,76 (д, J=1,4 Гц, 1Н), 7,69 (с, 1Н), 7,60 (дд, J=8,5, 2,2 Гц, 1Н), 6,95 (с, 1Н), 6,74 (д, J=8,5 Гц, 1Н), 3,98 (с, 3Н), 3,94 (с, 3Н), 3,66-3,57 (м, 4Н), 3,51 (с, 2Н), 2,65-2,57 (м, 4Н). ЖХ-МС [М+Н]+ 511,6.
Пример 9
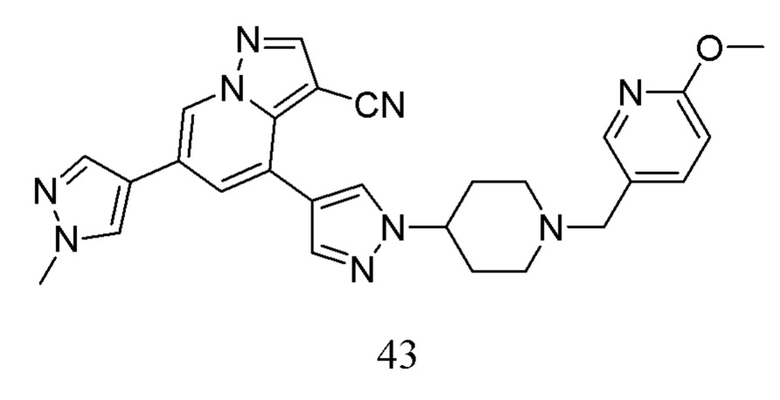
Схема синтеза выглядит следующим образом:
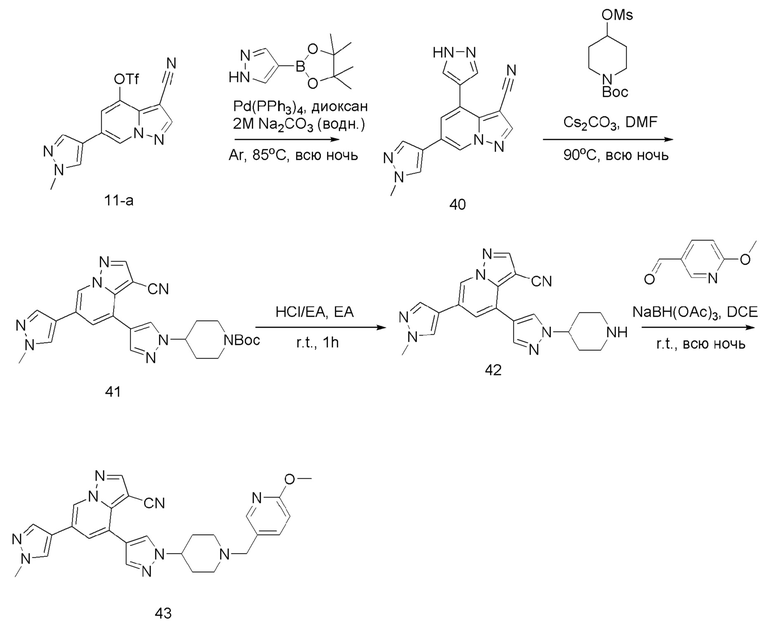
Соединение 40
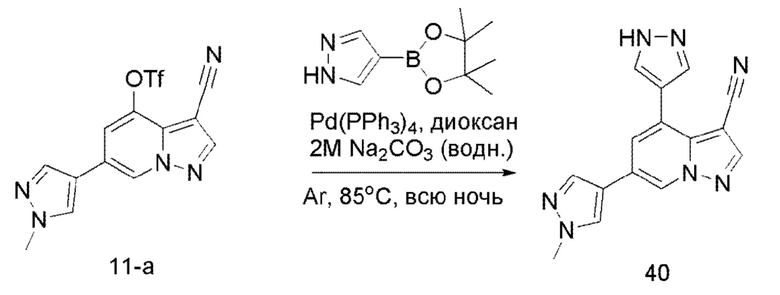
В герметизируемую пробирку объемом 45 мл последовательно добавляли соединение 11-а (200 мг, 0,54 ммоль), сложный эфир 4-пиразобороновой кислоты (104,5 мг, 0,54 ммоль), Pd(PPh3)4 (31,2 мг, 0,07 ммоль), 0,54 мл 2 М Na2CO3 (114,5 мг, 1,08 ммоль) и 1,4-диоксан (2 мл). Смесь перемешивали при температуре 85°С в течение ночи в атмосфере Ar, и контролв ТСХ показал отсутствие остатков исходного вещества 11-а. Смесв охлаждали до комнатной температуры, и добавляли 5 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 40 в виде серого твердого вещества (115,7 мг, выход 74%).
ЖХ-МС [М+Н]+ 290,11.
Соединение 41
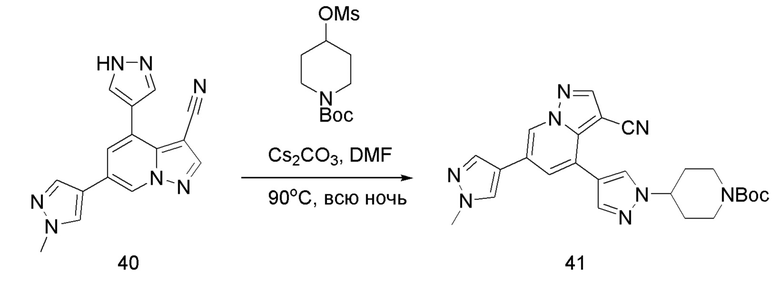
В одногорлую колбу объемом 50 мл последовательно добавляли соединение 40 (115,7 мг, 0,4 ммоль), 1-Вос-4-метансульфонилоксипиперидин (167,6 мг, 0,6 ммоль), Cs2CO3 (260,7 мг, 0,8 ммоль) и DMF (2 мл). Смесь нагревали с обратным холодильником в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 40. Смесь разбавляли добавлением H2O (6 мл) и экстрагировали ЕА (2 мл × 3). Органические фазы объединяли, концентрировали и очищали колоночной хроматографией с получением продукта 41 в виде желтого твердого вещества (94,5 мг, выход 50%).
ЖХ-МС [М+Н]+ 473,23.
Соединение 42
В одногорлую колбу объемом 25 мл добавляли 3,5 М раствор HCl в ЕА (10 мл) и медленно по каплям добавляли раствор соединения 41 (94,5 мг, 0,2 ммоль) в ЕА. После завершения добавления смесь перемешивали при комнатной температуре еще 3 ч, концентрировали для удаления растворителя, нейтрализовали добавлением по каплям раствора NH3 в метаноле, концентрировали и очищали колоночной хроматографией с получением продукта 42 в виде коричневато-желтого твердого вещества (68 мг, выход 90%).
Соединение 43
В одногорлую колбу объемом 50 мл последовательно добавляли соединение 42 (68 мг, 0,18 ммоль), 6-метокси-3-пиридинкарбоксальдегид (30,2 мг, 0,22 ммоль) и DCM (10 мл). После 10 мин перемешивания, при комнатной температуре порциями добавляли NaBH(OAc)3 (190,8 мг, 0,9 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 42. Смесь гасили добавлением аммиачной воды. Водную фазу отделяли и экстрагировали DCM (5 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением продукта 43 в виде желтого твердого вещества (55 мг, выход 61%).
1Н ЯМР (400 МГц, CDCl3) δ 8,59 (с, 1Н), 8,25 (с, 1Н), 8,06 (с, 1Н), 7,95 (с, 1Н), 7,79 (с, 2Н), 7,70 (с, 1Н), 7,62 (дд, J=8,4, 1,9 Гц, 1Н), 7,47 (с, 1Н), 6,73 (д, J=8,4 Гц, 1Н), 4,24 (д, J=10,7 Гц, 1Н), 3,99 (с, 3Н), 3,93 (с, 3Н), 3,50 (с, 2Н), 2,19 (дт, J=22,9, 10,5 Гц, 8Н). ЖХ-МС [М+Н]+ 493,56.
Пример 10
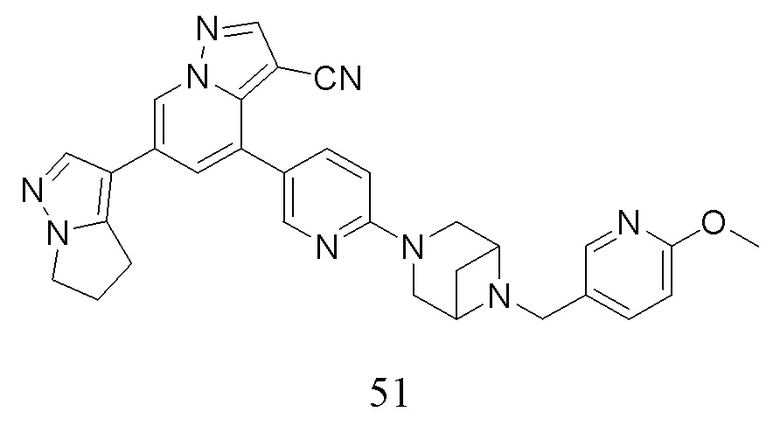
Схема синтеза выглядит следующим образом:
Соединение 44
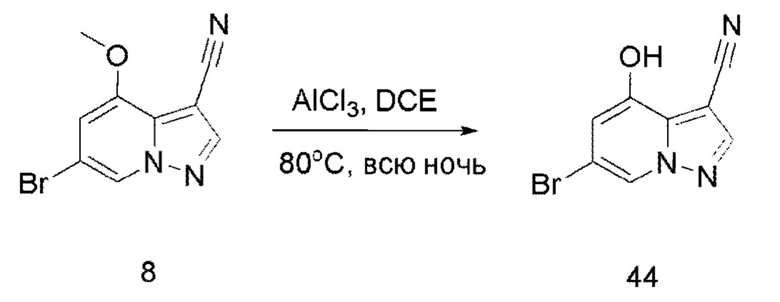
В герметизируемую пробирку объемом 45 мл последовательно добавляли соединение 8 (5 г, 19,8 ммоль) и 1,2-дихлорэтан (100 мл), и при комнатной температуре порциями добавляли AlCl3 (9,34 мг, 70 ммоль). Смесь перемешивали при температуре 80°С в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 8. Смесь охлаждали до комнатной температуры, гасили добавлением Na2SO4⋅10Н2О, перемешивали в течение 1 ч, фильтровали, промывали МеОН, концентрировали для удаления растворителя и очищали колоночной хроматографией с получением продукта 44 в виде серого твердого вещества (3 г, выход 55%).
Соединение 45
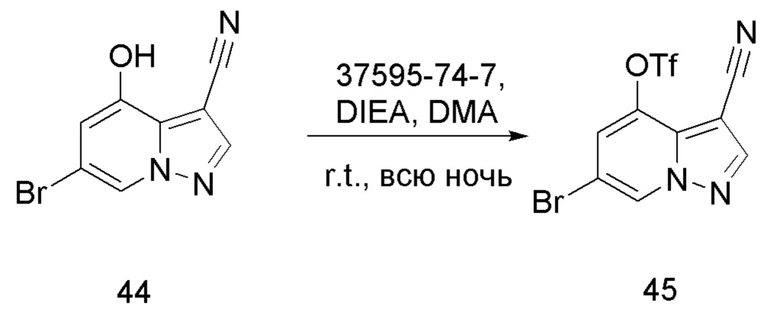
В одногорлую колбу объемом 50 мл последовательно добавляли соединение 44 (3 г, 12,6 ммоль), N-фенилбис(трифторметансульфонил)имид (4,5 г, 12,6 ммоль), DIEA (3,26 г, 25,2 ммоль) и DMA (20 мл). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 44. Реакционную смесь выливали при перемешивании в 60 мл H2O, и осаждалось коричневое твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 45 в виде коричневого твердого вещества (4,4 г, выход 95%).
Соединение 46

В герметизируемую пробирку объемом 12 мл последовательно добавляли соединение 45 (4,4 г, 11,9 ммоль), комплекс Pd(dppf)Cl2 с дихлорметаном (490 мг, 0,6 ммоль), 2-фторпиридин-5-бороновую кислоту (1,68 г, 11,9 ммоль), KOAc (2,92 г, 29,75 ммоль) и 1,4-диоксан (50 мл). Смесь перемешивали при температуре 85°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 45. Смесь охлаждали до комнатной температуры, и добавляли 100 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось коричневато-желтое твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 46 (3 г, выход 80%).
Соединение 47
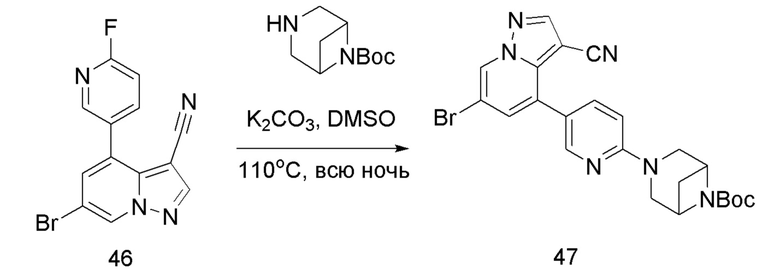
В герметизируемую пробирку объемом 45 мл последовательно добавляли соединение 46 (3 г, 9,46 ммоль), 6-(трет-бутоксикарбонил)-3,6-диазабицикло[3.1.1]гептан (2,25 г, 11,3 ммоль), K2CO3 (3,9 г, 28,35 ммоль) и DMSO (20 мл).Смесь перемешивали при температуре 110°С в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 46. Смесь охлаждали до комнатной температуры, и добавляли 50 мл воды.Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 47 (3,2 г, выход 69%).
Соединение 48
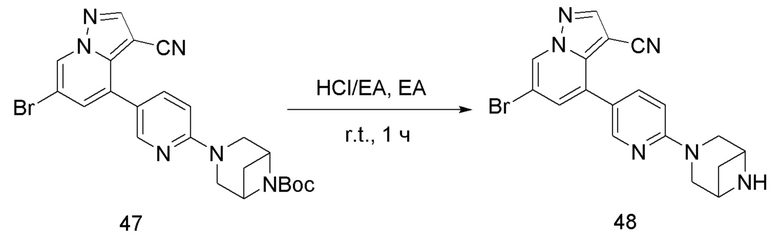
В одногорлую колбу объемом 25 мл добавляли 3,5 М раствор HCl в ЕА (10 мл) и медленно по каплям добавляли раствор соединения 47 (94,5 мг, 0,2 ммоль) в ЕА. После завершения добавления смесь перемешивали при комнатной температуре еще 1 ч, концентрировали для удаления растворителя, нейтрализовали добавлением по каплям раствора NH3 в метаноле, концентрировали и очищали колоночной хроматографией с получением продукта 48 в виде коричневато-желтого твердого вещества (68 мг, выход 90%).
Соединение 49
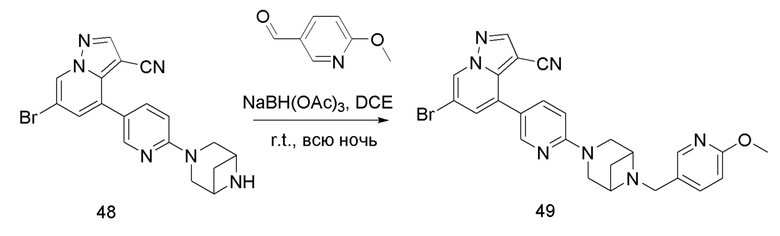
В одногорлую колбу объемом 50 мл последовательно добавляли соединение 48 (68 мг, 0,18 ммоль), 6-метокси-3-пиридинкарбоксальдегид (30,2 мг, 0,22 ммоль) и DCM (10 мл). После 10 мин перемешивания, при комнатной температуре порциями добавляли NaBH(OAc)3 (190,8 мг, 0,9 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 48. Смесь гасили добавлением аммиачной воды. Водную фазу отделяли и экстрагировали DCM (5 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением продукта 49 в виде желтого твердого вещества (55 мг, выход 61%).
Соединение 50
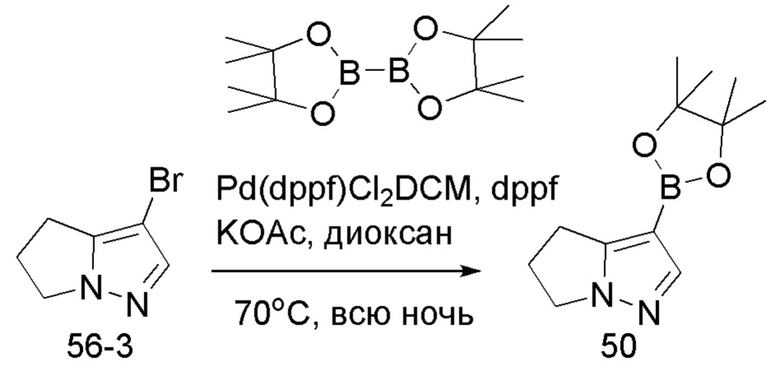
В герметизируемую пробирку объемом 25 мл последовательно добавляли 3-бром-5,б-дигидро-4Я-пирроло[1,2-В]пиразол (1,45 г, 7,8 ммоль), бис(пинаколято)диборон (2,1 г, 8,2 ммоль), Pd(dppf)Cl2 DCM (318,2 мг, 0,4 ммоль), 1,1'-
бис(дифенилфосфино)ферроцен (221,8 мг, 0,4 ммоль), KOAc (2,3 г, 23,4 ммоль) и 1,4-диоксан (40 мл). Смесь перемешивали при температуре 70°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 56-3. Смесь охлаждали до комнатной температуры, и добавляли 60 мл воды. Смесь перемешивали в течение 10 мин, и экстрагировали DCM (20 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением продукта в виде красновато-коричневой жидкости (1,5 г, выход 82%), который непосредственно использовали на следующей стадии без очистки.
ЖХ-МС [М+Н]+ 235,15.
Соединение 51
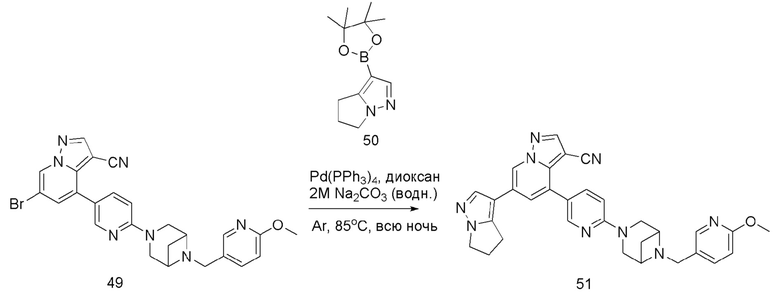
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (55 мг, 0,11 ммоль), Pd(PPh3)4 (6,36 мг, 0,005 ммоль), соединение 50 (25,74 мг, 0,11 ммоль), 2 М Na2CO3 (23,3 мг, 0,22 ммоль) и 1,4-диоксан (5 мл). Смесь перемешивали при температуре 85°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 51 (47,8 мг, выход 80%).
1Н ЯМР (400 МГц, CDCl3) δ 8,56 (д, J=1,3 Гц, 1Н), 8,36 (д, J=2,3 Гц, 1Н), 8,25 (с, 1Н), 8,08 (д, J=1,9 Гц, 1Н), 8,04 (д, J=1,9 Гц, 1Н), 8,02 (с, 1Н), 7,82 (с, 1Н), 7,75 (дд, J=8,8, 2,5 Гц, 1Н), 7,63 (дд, J=8,4, 2,2 Гц, 1Н), 7,40 (д, J=1,4 Гц, 1Н), 4,23 (т, J=7,3 Гц, 2Н), 3,92 (с, 3Н), 3,84 (д, J=11,9 Гц, 2Н), 3,77 (д, J=5,6 Гц, 2Н), 3,62 (с, 2Н), 3,58 (с, 2Н), 2,97 (т, J=7,3 Гц, 2Н), 2,67 (дд, J=14,3, 7,2 Гц, 3Н), 1,67 (д, J=8,6 Гц, 1Н). ЖХ-МС [М+Н]+ 544,6.
Пример 11
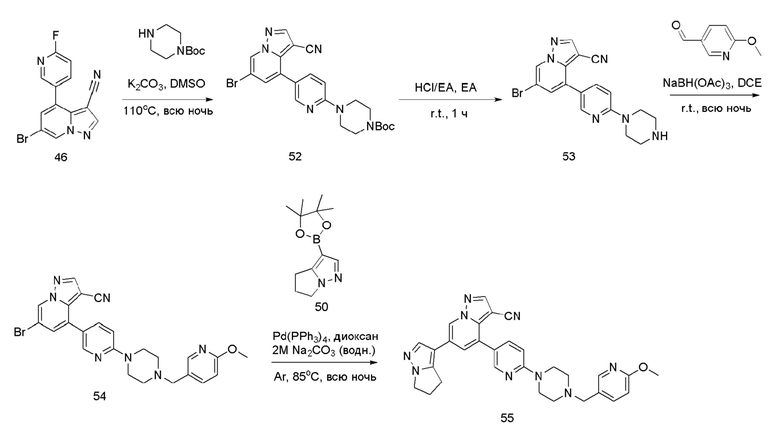
Соединение 52
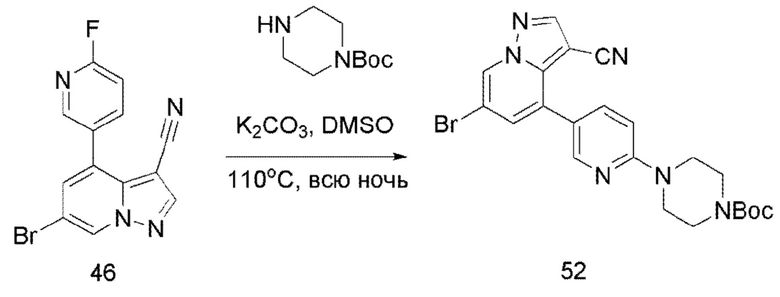
В герметизируемую пробирку объемом 45 мл последовательно добавляли соединение 46 (500 мг, 1,58 ммоль), N-BOC-пиперазин (352,2 мг, 1,89 ммоль), K2CO3 (436,1 мг, 3,16 ммоль) и DMSO (20 мл). Смесь перемешивали при температуре 120°С в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 46. Смесь охлаждали до комнатной температуры, и добавляли 50 мл воды. Смесв перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 52 (611 мг, выход 80%).
Соединение 53
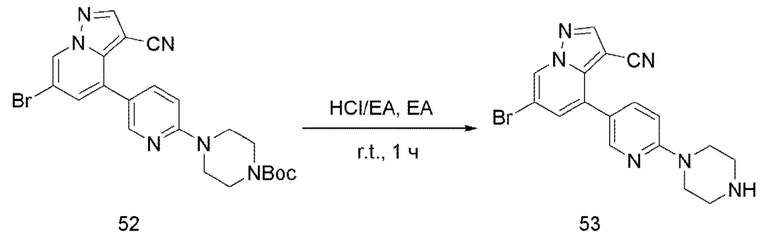
В одногорлую колбу объемом 25 мл добавляли 3,5 М раствор HCl в ЕА (10 мл) и медленно по каплям добавляли раствор соединения 52 (611 мг, 1,26 ммоль) в ЕА. После завершения добавления смесь перемешивали при комнатной температуре еще в течение 1 ч, концентрировали для удаления растворителя, нейтрализовали добавлением по каплям раствора NH3 в метаноле, концентрировали и очищали колоночной хроматографией с получением продукта 53 в виде коричневато-желтого твердого вещества (473,2 мг, выход 98%).
Соединение 54
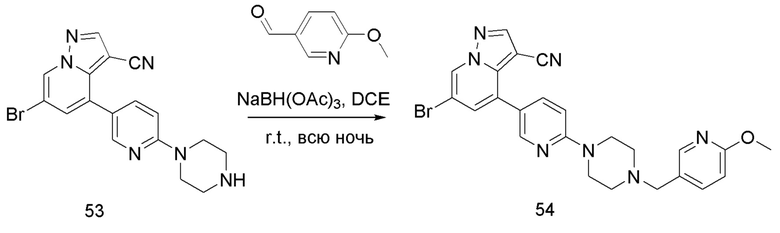
В одногорлую колбу объемом 50 мл последовательно добавляли соединение 53 (473,2 мг, 1,23 ммоль), 6-метокси-3-пиридинкарбоксальдегид (203,2 мг, 1,48 ммоль) и DCM (10 мл). После 10 мин перемешивания, при комнатной температуре порциями добавляли NaBH(OAc)3 (1,3 г, 6,15 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 53. Смесь гасили добавлением аммиачной воды. Водную фазу отделяли и экстрагировали DCM (5 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением продукта 54 в виде желтого твердого вещества (55 мг, выход 61%).
Соединение 55
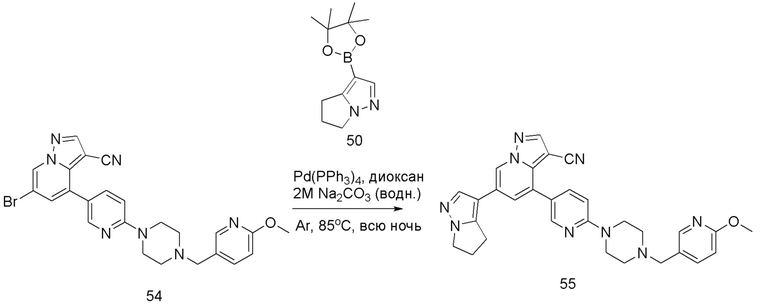
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 54 (50 мг, 0,1 ммоль), Pd(PPh3)4 (6,36 мг, 0,005 ммоль), соединение 50 (23,41 мг, 0,1 ммоль), 2 М Na2CO3 (21,2 мг, 0,2 ммоль) и 1,4-диоксан (5 мл). Смесь перемешивали при температуре 85°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 54. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 55 (60,4 мг, выход 88%).
1Н ЯМР (400 МГц, CDCl3) δ 8,56 (д, J=1,3 Гц, 1Н), 8,36 (д, J=2,3 Гц, 1Н), 8,25 (с, 1Н), 8,08 (д, J=1,9 Гц, 1Н), 8,04 (д, J=1,9 Гц, 1Н), 8,02 (с, 1Н), 7,82 (с, 1Н), 7,75 (дд, J=8,8, 2,5 Гц, 1Н), 7,63 (дд, J=8,4, 2,2 Гц, 1Н), 7,40 (д, J=1,4 Гц, 1Н), 4,23 (т, J=7,3 Гц, 2Н), 3,95 (с, 3Н), 3,93 (с, 3Н), 3,70-3,59 (м, 4Н), 3,50 (с, 2Н), 3,15-3,08 (м, 2Н), 2,79-2,69 (м, 2Н), 2,60-2,51 (м, 4Н). ЖХ-МС [М+Н]+ 531,6.
Пример 12
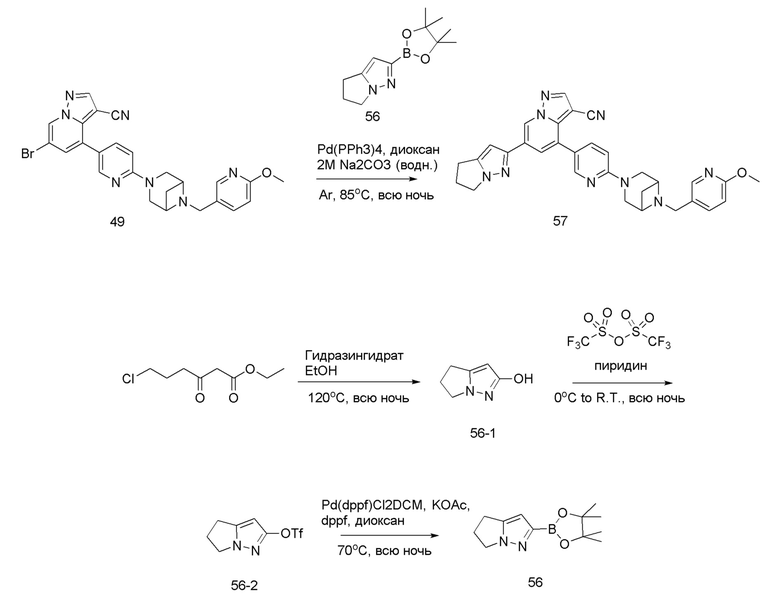
Соединение 56-1
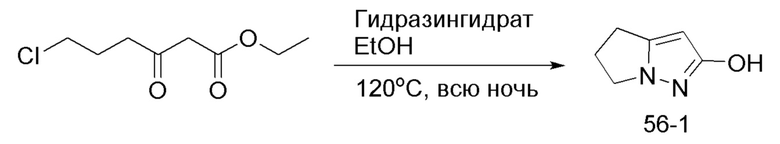
В герметизируемую пробирку объемом 100 мл последовательно добавляли этил 6-хлор-3-оксогексаноат (10 г, 51,9 ммоль), гидрат гидразина (33,3 мл) и EtOH (20 мл). Смесь перемешивали при температуре 120°С в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества. Смесь охлаждали до комнатной температуры, концентрировали и экстрагировали ЕА (20 мл × 3). Органические фазы объединяли, концентрировали и очищали колоночной хроматографией с получением продукта в виде желтого твердого вещества (3,4 мг, выход 53%).
ЖХ-МС [М+Н]+ 125,06.
Соединение 56-2
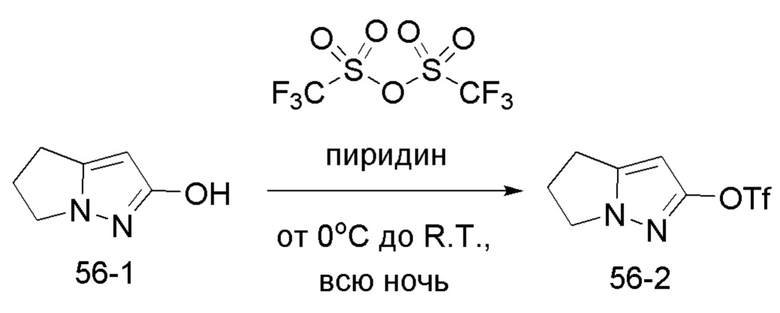
В трехгорлую колбу объемом 100 мл последовательно добавляли соединение 56-1 (3,4 г, 27,4 ммоль) и пиридин (55 мл). Температуру снижали до 0°С, и при этой температуре медленно по каплям добавляли ангидрид трифторметансульфоновой кислоты (8,1 г, 28,8 ммоль). После завершения добавления смесь подогревали до комнатной температуры и перемешивали в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 56-1. Смесь разбавляли вливанием в 20 мл 2 М HCl и экстрагировали ЕА (10 мл × 3). Органические фазы объединяли, последовательно промывали насыщенным солевым раствором и насыщенным раствором карбоната натрия, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением продукта в виде бесцветного масла (1,7 г, выход 24%).
ЖХ-МС [М+Н]+ 257,01.
Соединение 56
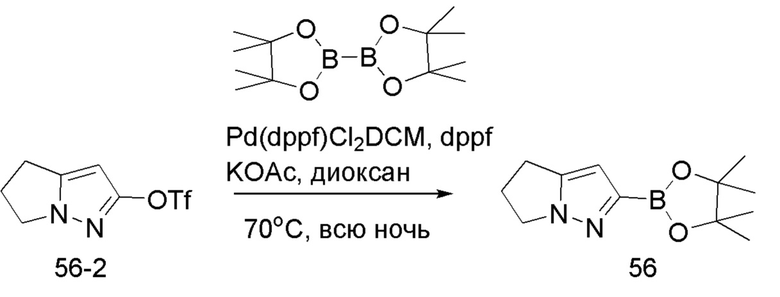
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 56-2 (2 г, 7,8 ммоль), бис(пинаколято)диборон (2,1 г, 8,2 ммоль), Pd(dppf)Cl2 DCM (318,2 мг, 0,4 ммоль), 1,1'-бис(дифенилфосфино)ферроцен (221,8 мг, 0,4 ммоль), KOAc (2,3 г, 23,4 ммоль) и 1,4-диоксан (40 мл). Смесь перемешивали при температуре 70°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 56-2. Смесь охлаждали до комнатной температуры, и добавляли 60 мл воды. Смесь перемешивали в течение 10 мин, и экстрагировали DCM (20 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением продукта в виде красновато-коричневой жидкости (1,7 г, выход 93%), который непосредственно использовали на следующей стадии без очистки.
ЖХ-МС [М+Н]+ 235,15.
Соединение 57
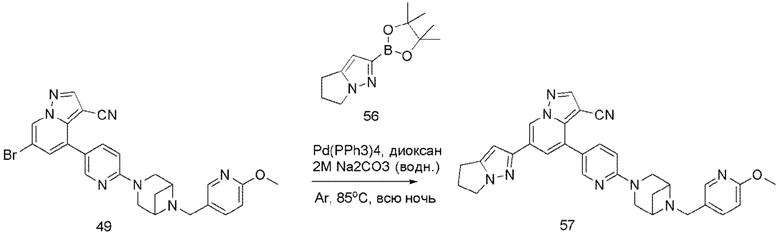
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (50 мг, 0,1 ммоль), Pd(PPh3)4 (6,36 мг, 0,005 ммоль), соединение 56 (23,41 мг, 0,1 ммоль), 2 М Na2CO3 (21,2 мг, 0,2 ммоль) и 1,4-диоксан (5 мл). Смесь перемешивали при температуре 85°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 57 (46,3 мг, выход 88%).
1Н ЯМР (400 МГц, CDCl3) δ 8,90 (с, 1Н), 8,47 (д, J=2,3 Гц, 1Н), 8,28 (с, 1Н), 8,12 (д, J=1,8 Гц, 1Н), 7,82 (дд, J=9,9, 3,4 Гц, 2Н), 7,63 (дд, J=8,5, 2,2 Гц, 1Н), 6,70 (т, J=8,9 Гц, 2Н), 6,31 (с, 1Н), 4,22 (т, J=7,2 Гц, 2Н), 3,92 (с, 3Н), 3,84 (д, J=11,9 Гц, 2Н), 3,77 (д, J=5,6 Гц, 2Н), 3,62 (с, 2Н), 3,58 (с, 2Н), 2,97 (т, J=7,3 Гц, 2Н), 2,67 (дд, J=14,3, 7,2 Гц, 3Н), 1,67 (д, J=8,6 Гц, 1Н). ЖХ-МС [М+Н]+ 543,6.
Пример 13
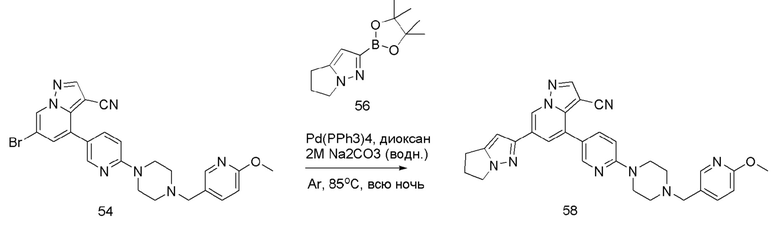
Соединение 58
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 54 (50 мг, 0,1 ммоль), Pd(PPh3)4 (6,36 мг, 0,005 ммоль), соединение 56 (23,41 мг, 0,1 ммоль), 2 М Na2CO3 (21,2 мг, 0,2 ммоль) и 1,4-диоксан (5 мл). Смесь перемешивали при температуре 85°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 54. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесв перемешивали в течение 10 мин, и осаждалосв желтое твердое вещество. Твердое вещество собирали филвтрацией, сушили и очищали колоночной хроматографией с получением продукта 58 (46,3 мг, выход 88%).
1Н ЯМР (400 МГц, CDCl3) δ 8,88 (д, J=1,3 Гц, 1Н), 8,39 (д, J=2,3 Гц, 1Н), 8,26 (с, 1Н), 8,08 (д, J=2,0 Гц, 1Н), 7,77 (д, J=1,3 Гц, 1Н), 7,74 (дд, J=8,9, 2,5 Гц, 1Н), 7,62 (дд, J=8,5, 2,3 Гц, 1Н), 6,76 (д, J=2,7 Гц, 1Н), 6,74 (д, J=2,2 Гц, 1Н), 6,29 (с, 1Н), 4,20 (т, J=7,2 Гц, 2Н), 3,94 (с, 3Н), 3,75-3,59 (м, 4Н), 3,50 (с, 2Н), 2,96 (т, J=7,3 Гц, 2Н), 2,73-2,59 (м, 2Н), 2,60-2,48 (м, 4Н). ЖХ-МС [М+Н]+ 531,6.
Пример 14
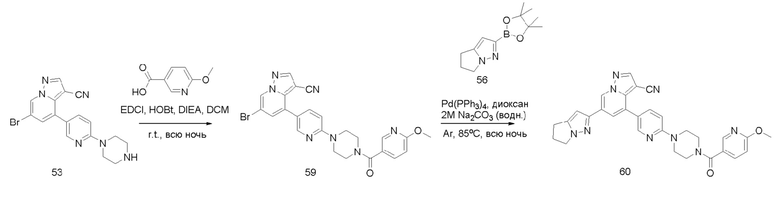
Соединение 59
В одногорлую колбу объемом 25 мл последовательно добавляли соединение 53 (50 мг, 0,13 ммоль), 6-метоксиникотиновую кислоту (19,9 мг, 0,13 ммоль), EDCI (37,38 мг, 0,2 ммоль), HOBt (35,13 мг, 0,26 ммоль), DIEA (50,4 мг, 0,39 ммоль) и DCM (8 мл). Смесь перемешивали при комнатной температуре в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 53. Добавляли 10 мл воды, и смесь перемешивали в течение 10 мин. Органическую фазу отделяли, концентрировали и очищали колоночной хроматографией с получением продукта 59 (55,25 мг, выход 82%).
ЖХ-МС [М+Н]+ 518,09.
Соединение 60
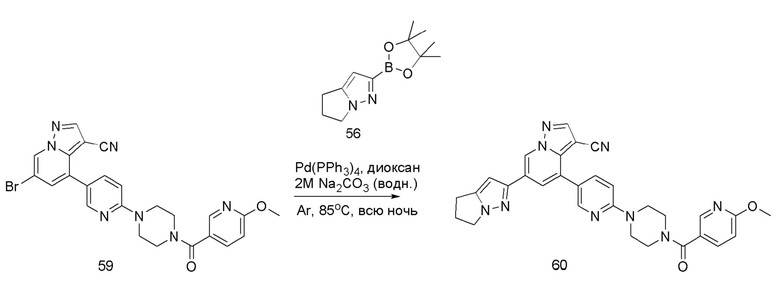
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 59 (55,2 мг, 0,1 ммоль), Pd(PPh3)4 (6,36 мг, 0,005 ммоль), соединение 56 (23,41 мг, 0,1 ммоль), 2 М Na2CO3 (21,2 мг, 0,2 ммоль) и 1,4-диоксан (5 мл). Смесь перемешивали при температуре 85°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 59. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 60 (40 мг, выход 73%).
1Н ЯМР (400 МГц, CDCl3) δ 8,90 (с, 1Н), 8,42 (д, J=2,0 Гц, 1Н), 8,33 (с, 1Н), 8,27 (с, 1Н), 7,87-7,77 (м, 2Н), 7,73 (дд, J=8,6, 2,2 Гц, 1Н), 6,81 (дд, J=8,6, 5,7 Гц, 2Н), 6,31 (с, 1Н), 4,21 (т, J=7,2 Гц, 2Н), 3,99 (д, J=4,7 Гц, 3Н), 3,73 (с, 8Н), 3,00-2,91 (м, 2Н), 2,72-2,57 (м, 3Н). ЖХ-МС [М+Н]+ 545,6.
Пример 15
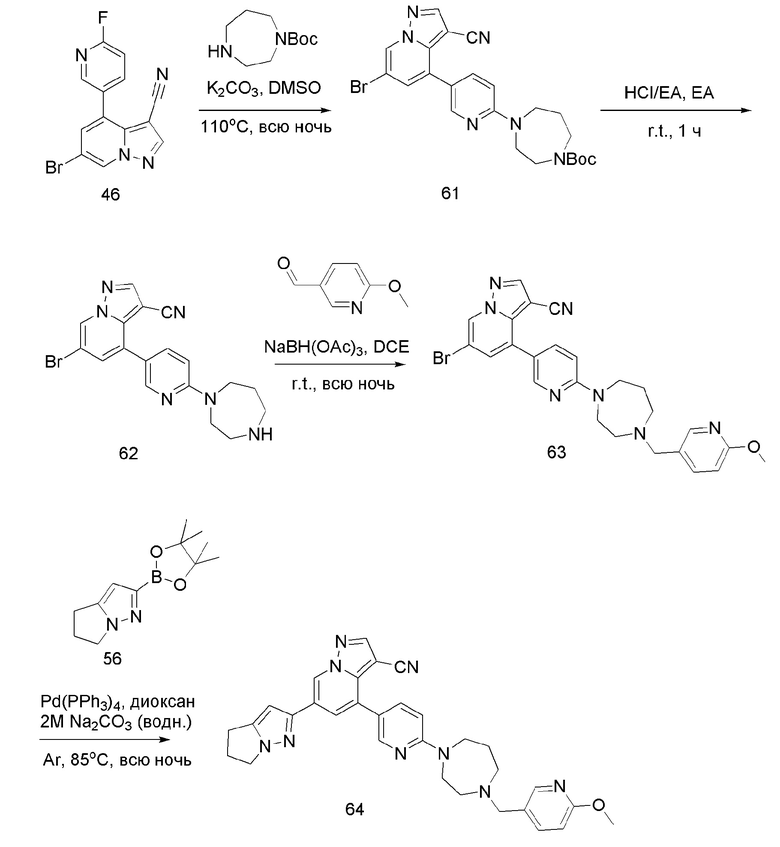
Соединение 61

В герметизируемую пробирку объемом 45 мл последовательно добавляли соединение 46 (200 мг, 0,63 ммоль), трет-бутил 1,4-диазепан-1-карбоксилат (151,57 мг, 0,76 ммоль), K2CO3 (173,88 мг, 1,26 ммоль) и DMSO (10 мл). Смесь перемешивали при температуре 120°С в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 46. Смесь охлаждали до комнатной температуры, и добавляли 50 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 61 (250 мг, выход 80%).
ЖХ-МС [М+Н]+ 497,12.
Соединение 62
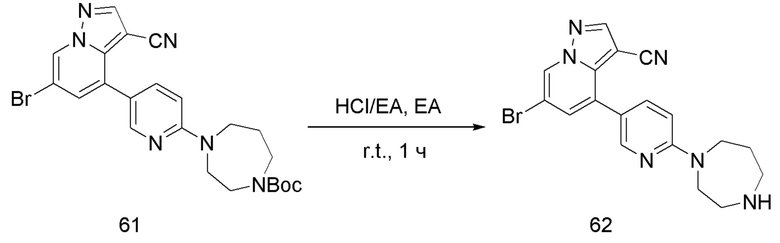
В одногорлую колбу объемом 25 мл добавляли 3,5 М раствор HCl в ЕА (10 мл) и медленно по каплям добавляли раствор соединения 61 (250 мг, 0,5 ммоль) в ЕА. После завершения добавления смесь перемешивали при комнатной температуре еще 1 ч, концентрировали для удаления растворителя, нейтрализовали добавлением по каплям раствора NH3 в метаноле, концентрировали и очищали колоночной хроматографией с получением продукта 62 в виде коричневато-желтого твердого вещества (188,7 мг, выход 95%).
ЖХ-МС [М+Н]+ 397,07.
Соединение 63
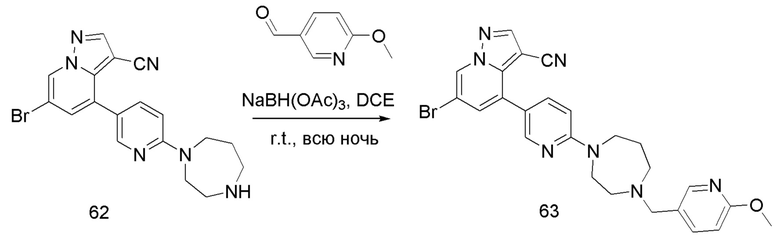
В одногорлую колбу объемом 50 мл последовательно добавляли соединение 62 (188,7 мг, 0,47 ммоль), 6-метокси-3-пиридинкарбоксальдегид (78,2 мг, 0,57 ммоль) и DCM (10 мл). После 10 мин перемешивания, при комнатной температуре порциями добавляли NaBH(OAc)3 (498,2 мг, 2,35 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 62. Смесь гасили добавлением аммиачной воды. Водную фазу отделяли и экстрагировали DCM (5 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением продукта 63 в виде желтого твердого вещества (158 мг, выход 65%).
ЖХ-МС [М+Н]+ 518,12.
Соединение 64
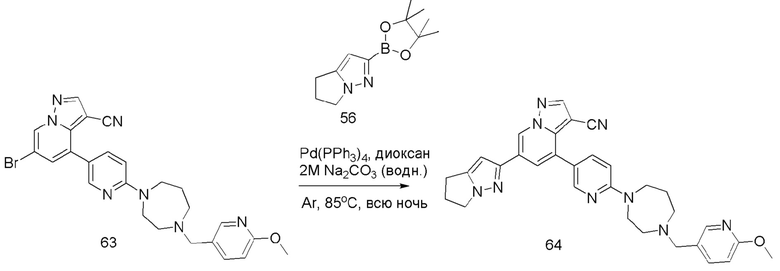
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 63 (52 мг, 0,1 ммоль), Pd(PPh3)4 (6,36 мг, 0,005 ммоль), соединение 56 (23,41 мг, 0,1 ммоль), 2 М Na2CO3 (21,2 мг, 0,2 ммоль) и 1,4-диоксан (5 мл). Смесь перемешивали при температуре 85°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 63. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 64 (39,8 мг, выход 73%).
1Н ЯМР (400 МГц, CDCl3) δ 8,88 (д, J=1,3 Гц, 1Н), 8,37 (д, J=2,4 Гц, 1Н), 8,26 (с, 1Н), 8,05 (д, J=2,0 Гц, 1Н), 7,77 (д, J=1,3 Гц, 1Н), 7,72 (дд, J=8,8, 2,6 Гц, 1Н), 7,61 (д, J=6,9 Гц, 1Н), 6,72 (д, J=8,5 Гц, 1Н), 6,61 (д, J=9,0 Гц, 1Н), 6,31 (с, 1Н), 4,25-4,17 (т, 2Н), 3,93 (с, 3Н), 3,86 (м, 2Н), 3,72 (т, J=6,1 Гц, 2Н), 3,58 (с, 2Н), 3,02-2,92 (м, 2Н), 2,79 (м, 2Н), 2,71, 2,63 (м, 4Н), 1,99 (м, 2Н). ЖХ-МС [М+Н]+ 545,6.
Пример 16
Соединение 65
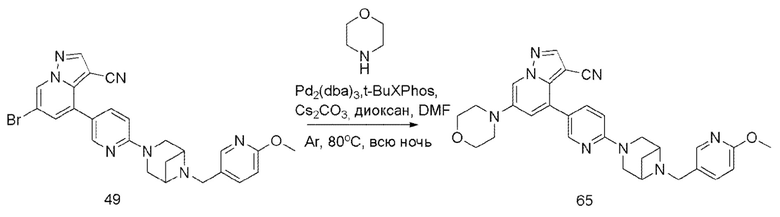
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (52 мг, 0,1 ммоль), Pd2(dba)3 (5,5 мг, 0,006 ммоль), t-BuXPhos (7,6 мг, 0,018 ммоль), морфолин (26 мг, 0,3 ммоль), Cs2CO3 (65,2 мг, 0,2 ммоль), 1,4-диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесв перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 65 (47 мг, выход 90%).
1Н ЯМР (400 МГц, CDCl3) δ 8,39 (д, J=2,3 Гц, 1Н), 8,20 (с, 1Н), 8,10 (д, J=2,0 Гц, 1Н), 8,02 (д, J=1,9 Гц, 1Н), 7,79 (дд, J=8,8, 2,5 Гц, 1Н), 7,65 (дд, J=8,5, 2,2 Гц, 1Н), 7,17 (д, J=1,9 Гц, 1Н), 6,71 (дд, J=13,3, 8,7 Гц, 2Н), 3,91 (м, J=8,4 Гц, 6Н), 3,82 (м, J=19,6, 8,9 Гц, 4Н), 3,59 (м, 4Н), 3,22-3,09 (м, 4Н), 2,71 (м, J=7,0 Гц, 1Н), 1,66 (д, J=8,7 Гц, 2Н). ЖХ-МС [М+Н]+ 522,6.
Пример 17
Соединение 66

В герметизируемую пробирку объемом 25 мл последователвно добавляли соединение 49 (52 мг, 0,1 ммоль), Pd2(PPh3)2 (5,5 мг, 0,005 ммоль), Cul (19 мг, 0,1 ммоль), PPh3 (52 мг, 0,2 ммоль), 2-метилбут-3-ин-2-ол (25 мг, 0,3 ммоль) и TEA (2 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 65 (46,7 мг, выход 90%).
1Н ЯМР (400 МГц, CDCl3) δ 8,61 (д, J=1,3 Гц, 1Н), 8,40 (д, J=2,3 Гц, 1Н), 8,31 (с, 1Н), 8,11 (д, J=2,0 Гц, 1Н), 7,76 (дд, J=8,8, 2,5 Гц, 1Н), 7,67 (д, J=8,3 Гц, 1Н), 7,30 (д, J=1,3 Гц, 1Н), 6,71 (дд, J=13,8, 8,6 Гц, 2Н), 3,92 (с, 3Н), 3,89-3,74 (м, 4Н), 3,61 (м, 4Н), 2,73 (м, 1Н), 2,22 (м, 1Н), 1,66 (с, 6Н). ЖХ-МС [М+Н]+ 519,6.
Пример 18
Соединение 67
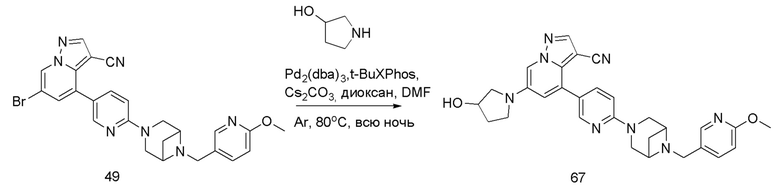
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (52 мг, 0,1 ммоль), Pd2(dba)3 (5,5 мг, 0,006 ммоль), t-BuXPhos (7,6 мг, 0,018 ммоль), 3-гидроксипирролидин (26 мг, 0,3 ммоль), Cs2CO3 (65, 2 мг, 0,2 ммоль), 1,4-диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 67 (47 мг, выход 90%).
1Н ЯМР (400 МГц, CDCl3) δ 8,40 (д, J=2,3 Гц, 1Н), 8,14 (с, 1Н), 8,10 (д, J=1,9 Гц, 1Н), 7,82 (д, J=2,5 Гц, 1Н), 7,79 (д, J=1,8 Гц, 1Н), 7,65 (дд, J=8,5, 2,3 Гц, 1Н), 6,93 (д, J=2,0 Гц, 1Н), 6,71 (т, J=9,2 Гц, 2Н), 4,69 (м, 1Н), 3,92 (с, 3Н), 3,88-3,75 (м, 4Н), 3,67-3,50 (м, 6Н), 3,40 (тд, J=8,7, 3,1 Гц, 1Н), 3,31 (д, J=10,0 Гц, 1Н), 2,70 (м, 1Н), 2,25 (м, 1Н), 2,15 (м, 1Н), 1,67 (д, J=8,6 Гц, 2Н). ЖХ-МС [М+Н]+ 522,6.
Пример 19
Соединение 68
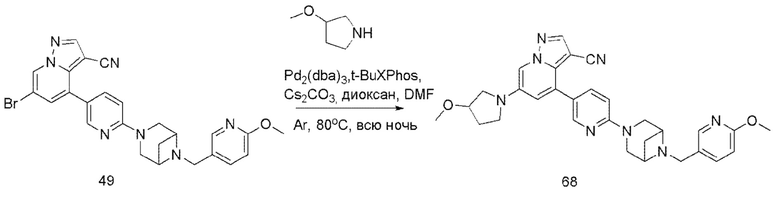
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (52 мг, 0,1 ммоль), Pd2(dba)3 (5,5 мг, 0,006 ммоль), t-BuXPhos (7,6 мг, 0,018 ммоль), 3-метоксипирролидин (26 мг, 0,3 ммоль), Cs2CO3 (65,2 мг, 0,2 ммоль), 1,4-диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 68 (48,3 мг, выход 90%).
1Н ЯМР (400 МГц, CDCl3) δ 8,40 (д, J=2,3 Гц, 1Н), 8,14 (с, 1Н), 8,10 (д, J=2,0 Гц, 1Н), 7,82 (д, J=2,5 Гц, 1Н), 7,80 (т, J=2,5 Гц, 1Н), 7,66 (д, J=8,3 Гц, 1Н), 6,94 (д, J=2,0 Гц, 1Н), 6,76-6,65 (м, 2Н), 4,20-4,10 (м, 1Н), 3,92 (с, 3Н), 3,83 (м, 4Н), 3,65-3,60 (м, 4Н), 3,55-3,42 (м, 2Н), 3,39 (с, 3Н), 3,37-3,34 (м, 1Н), 2,74-2,68 (м, 1Н), 2,29-2,09 (м, 2Н), 1,67 (д, J=8,6 Гц, 2Н). ЖХ-МС [М+Н]+ 536,6.
Пример 20
Соединение 69
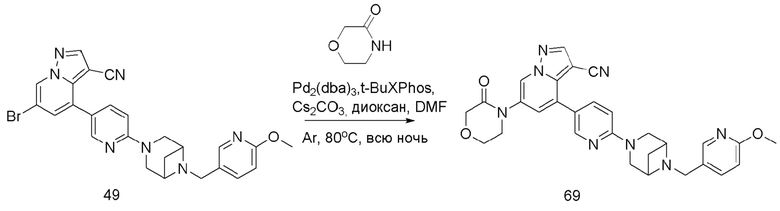
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (52 мг, 0,1 ммоль), Pd2(dba)3 (5,5 мг, 0,006 ммоль), t-BuXPhos (7,6 мг, 0,018 ммоль), морфолин-3-он (30 мг, 0,3 ммоль), Cs2CO3 (65,2 мг, 0,2 ммоль), 1,4-диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 69 (48,3 мг, выход 90%).
1Н ЯМР (400 МГц, CDCl3) δ 8,39 (д, J=2,3 Гц, 1Н), 8,20 (с, 1Н), 8,10 (д, J=2,0 Гц, 1Н), 8,02 (д, J=1,9 Гц, 1Н), 7,79 (дд, J=8,8, 2,5 Гц, 1Н), 7,65 (дд, J=8,5, 2,2 Гц, 1Н), 7,17 (д, J=1,9 Гц, 1Н), 6,71 (дд, J=13,3, 8,7 Гц, 2Н), 3,91 (м, J=8,4 Гц, 6Н), 3,82 (м, J=19,6, 8,9 Гц, 4Н), 3,59 (м, 2Н), 3,22-3,09 (м, 2Н), 2,92 (с, 2Н) 2,71 (м, J=7,0 Гц, 1Н), 1,66 (д, J=8,7 Гц, 2Н). ЖХ-МС [М+Н]+ 537,58.
Пример 21
Соединение 70
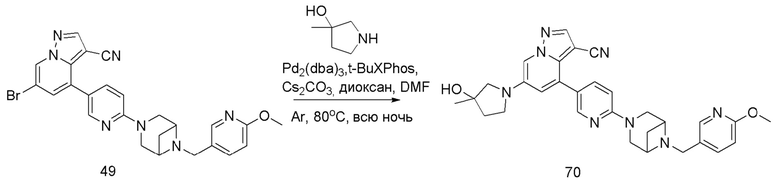
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (52 мг, 0,1 ммоль), Pd2(dba)3 (5,5 мг, 0,006 ммоль), t-BuXPhos (7,6 мг, 0,018 ммоль), 3-гидрокси-3-метилпиррол (30 мг, 0,3 ммоль), Cs2CO3 (65,2 мг, 0,2 ммоль), 1,4-диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 70 (48,3 мг, выход 90%).
1Н ЯМР (400 МГц, CDCl3) δ 8,39 (д, J=2,4 Гц, 1Н), 8,14 (с, 1Н), 8,11 (д, J=2,2 Гц, 1Н), 7,80 (дд, J=8,8, 2,3 Гц, 1Н), 7,76 (д, J=1,3 Гц, 1Н), 7,65 (дд, J=8,5, 2,4 Гц, 1Н), 6,90 (д, J=1,7 Гц, 1Н), 6,72 (дд, J=11,9, 8,7 Гц, 2Н), 3,93 (с, 3Н), 3,85 (д, J=11,7 Гц, 2Н), 3,80 (д, J=5,8 Гц, 2Н), 3,65-3,60 (м, 2Н), 3,59 (с, 3Н), 3,42 (тд, J=8,5, 3,0 Гц, 1Н), 3,38-3,30 (м, 2Н), 2,71 (дд, J=14,1, 6,3 Гц, 1Н), 2,22-2,05 (м, 3Н), 1,67 (д, J=8,7 Гц, ОН), 1,56 (с, 1Н). ЖХ-МС [М+Н]+ 536,6.
Пример 22
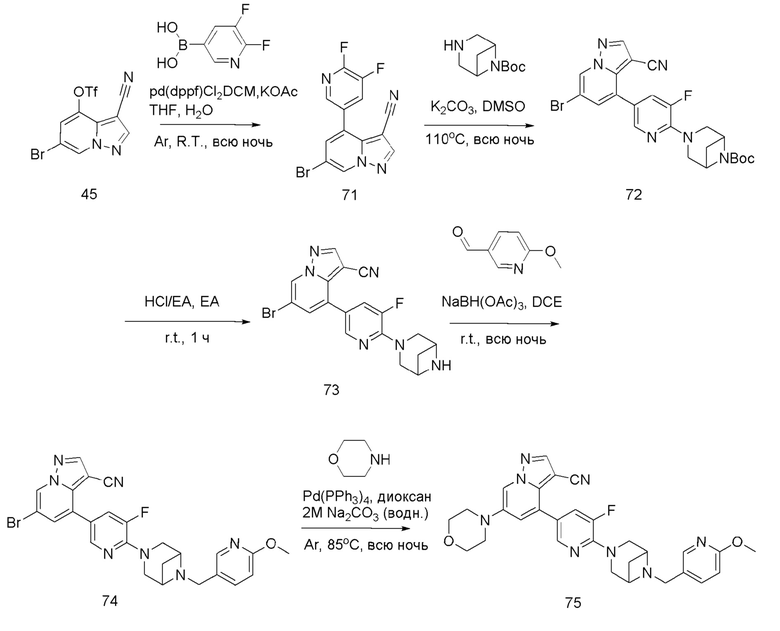
Соединение 71
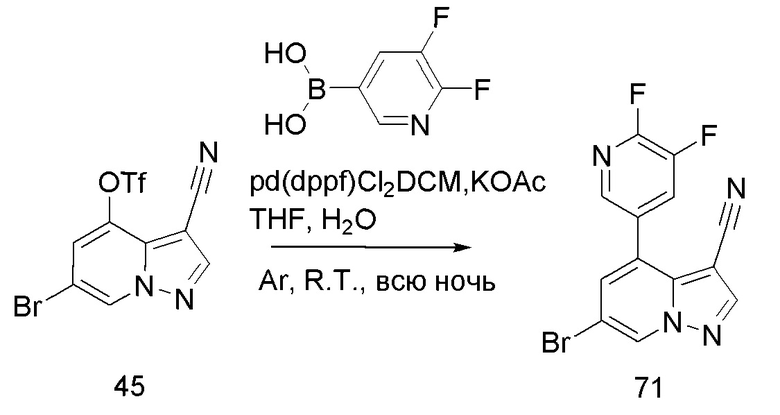
В герметизируемую пробирку объемом 12 мл последовательно добавляли соединение 45 (500 мг, 1,35 ммоль), комплекс Pd(dppf)Cl2 с дихлорметаном (57 мг, 0,07 ммоль), 2,3-дифторпиридин-5-бороновую кислоту (178,7 г, 1,13 ммоль), KOAc (265 мг, 2,7 ммоль) THF (10 мл) и H2O (2 мл). Смесь перемешивали при температуре 85°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 45. Смесь охлаждали до комнатной температуры, и добавляли 20 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось коричневато-желтое твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 71 (93,8 мг, выход 80%).
ЖХ-МС [М+Н]+ 334,97.
Соединение 72
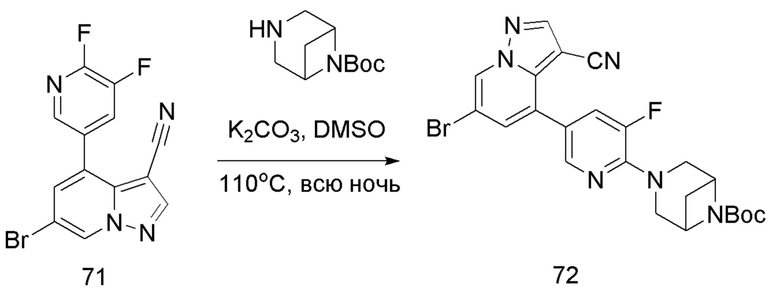
В герметизируемую пробирку объемом 45 мл последовательно добавляли соединение 71 (93,8 мг, 1,08 ммоль), 6-(трет-бутоксикарбонил)-3,6-диазабицикло[3.1.1]гептан (257 мг, 1,3 ммоль), K2CO3 (298,5 мг, 2,16 ммоль) и DMSO (2 мл). Смесь перемешивали при температуре 110°С в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 71. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением продукта 72 (382,5 мг, выход 69%).
ЖХ-МС [М+Н]+ 513,1.
Соединение 73
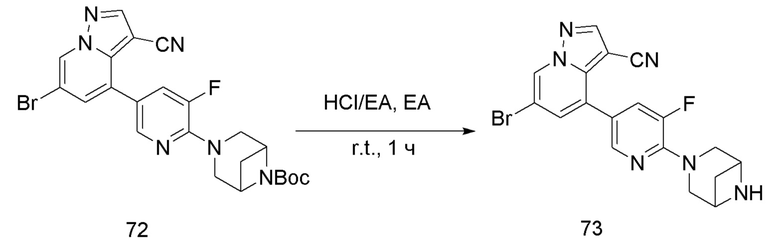
В одногорлую колбу объемом 25 мл добавляли 3,5 М раствор HCl в ЕА (10 мл) и медленно по каплям добавляли раствор соединения 72 (382,5 мг, 0,74 ммоль) в ЕА. После завершения добавления смесь перемешивали при комнатной температуре еще 1 ч, концентрировали для удаления растворителя, нейтрализовали добавлением по каплям раствора NH3 в метаноле, концентрировали и очищали колоночной хроматографией с получением продукта 73 в виде коричневато-желтого твердого вещества (277 мг, выход 90%).
ЖХ-МС [М+Н]+ 413,04.
Соединение 74
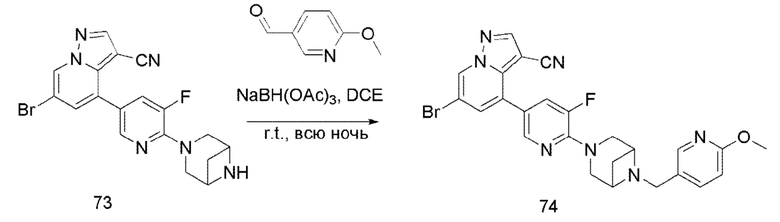
В одногорлую колбу объемом 50 мл последовательно добавляли соединение 73 (100 мг, 0,24 ммолв), 6-метокси-3-пиридинкарбоксальдегид (39,8 мг, 0,29 ммоль) и DCM (10 мл). После 10 мин перемешивания, при комнатной температуре порциями добавляли NaBH(OAc)3 (254,4 мг, 1,2 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 73. Смесь гасили добавлением аммиачной воды. Водную фазу отделяли и экстрагировали DCM (5 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением продукта 74 в виде желтого твердого вещества (77 мг, выход 60%).
ЖХ-МС [М+Н]+ 534,1.
Соединение 75
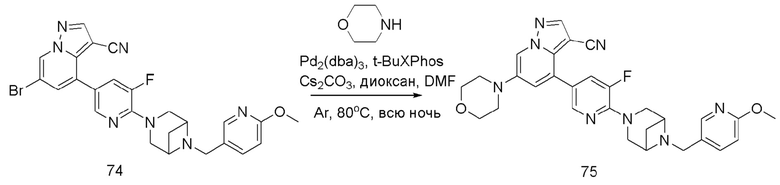
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 74 (77 мг, 0,14 ммоль), Pd2(dba)3 (7,9 мг, 0,009 ммоль), t-BuXPhos (11,5 мг, 0,027 ммоль), морфолин (36,6 мг, 0,42 ммоль), Cs2CO3 (91,2 мг, 0,28 ммоль), 1,4-диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 74. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 75 (68 мг, выход 90%).
1Н ЯМР (400 МГц, CDCl3) δ 8,21 (с, 1Н), 8,18 (с, 1Н), 8,15 (с, 1Н), 8,04 (д, J=1,9 Гц, 1Н), 7,50 (с, 1Н), 7,46 (с, 1Н), 7,19 (д, J=1,9 Гц, 1Н), 6,76 (д, J=8,6 Гц, 1Н), 3,95-3,88 (м, 10Н), 3,78 (с, 2Н), 3,23-3,11 (м, 5Н), 2,39-2,18 (м, 2Н), 2,01 (с, 2Н). ЖХ-МС [М+Н]+ 540,6.
Пример 23
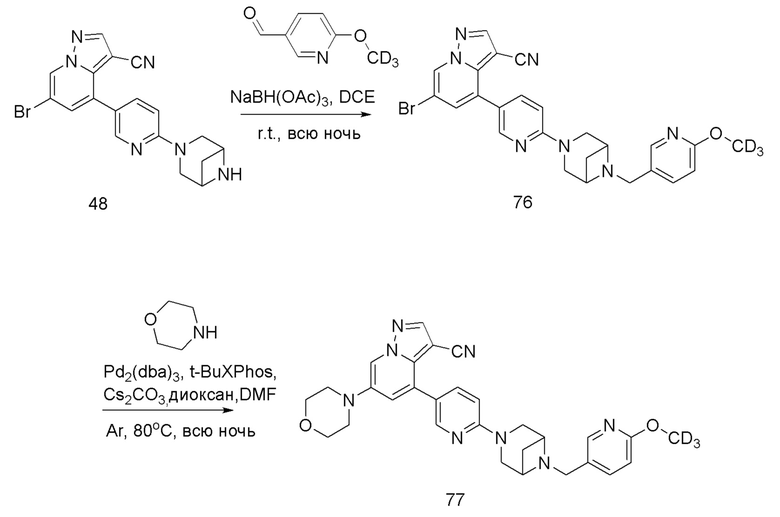
Соединение 76
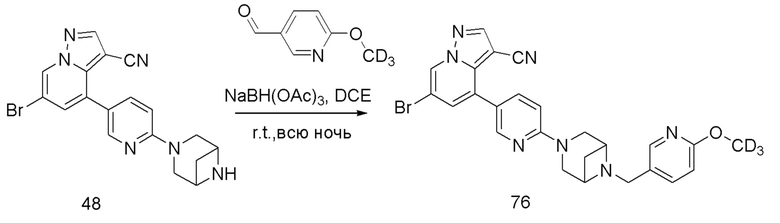
В одногорлую колбу объемом 50 мл последовательно добавляли соединение 48 (60 мг, 0,15 ммоль), 6-дейтерометокси-3-пиридинкарбоксальдегид (25,5 мг, 0,18 ммоль) и DCM (10 мл). После 10 мин перемешивания, при комнатной температуре порциями добавляли NaBH(OAc)3 (159 мг, 0,75 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 48. Смесь гасили добавлением аммиачной воды. Водную фазу отделяли и экстрагировали DCM (5 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением продукта 76 в виде желтого твердого вещества (56 мг, выход 71,8%).
Соединение 77
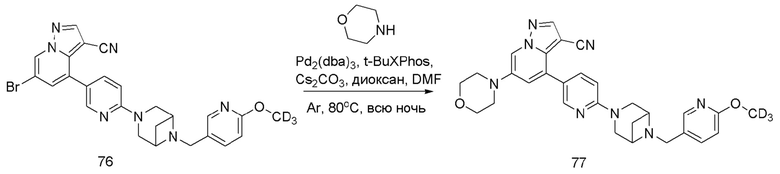
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 76 (56 мг, 0,11 ммоль), Pd2 (dba) 3 (6 мг, 0,007 ммоль), t-BuXPhos (8,4 мг, 0,02 ммоль), морфолин (26 мг, 0,33 ммоль), Cs2CO3 (71,6 мг, 0,22 ммоль), 1,4-диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 76. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 77 (53,7 мг, выход 93%).
1Н ЯМР (400 МГц, CDCl3) δ 8,39 (с, 1Н), 8,20 (с, 1Н), 8,10 (с, 1Н), 8,02 (с, 1Н), 7,79 (д, J=8,4 Гц, 1Н), 7,17 (с, 1Н), 6,99 (д, J=8,9 Гц, 1Н), 6,71 (дд, J=15,3, 8,5 Гц, 2Н), 3,91 (д, J=4,7 Гц, 4Н), 3,86-3,81 (м, 2Н), 3,63 (с, 2Н), 3,20-3,12 (м, 4Н), 2,78-2,74 (м, 1Н), 2,35 (м, 1Н), 2,24 (дд, J=20,5, 6,6 Гц, 2Н), 2,04-2,00 (м, 2Н). ЖХ-МС [М+Н]+ 525,6.
Пример 24
Соединение 78
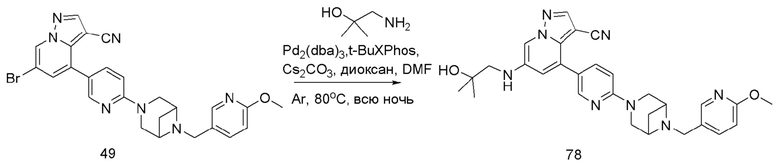
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (52 мг, 0,1 ммоль), Pd2(dba)3 (5,5 мг, 0,006 ммоль), t-BuXPhos (8,4 мг, 0,02 ммоль), 1-амино-2-метил-2-пропанол (26,7 мг, 0,3 ммоль), Cs2CO3 (65 мг, 0,2 ммоль), 1,4-диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 78 (42,5 мг, выход 81%).
1Н ЯМР (400 МГц, CDCl3) δ 8,40 (д, J=2,3 Гц, 1Н), 8,15 (с, 1Н), 8,11 (с, 1Н), 7,93 (д, J=1,4 Гц, 1Н), 7,82 (дд, J=8,7, 2,3 Гц, 1Н), 7,74 (д, J=8,2 Гц, 1Н), 7,08 (д, J=1,5 Гц, 1Н), 6,72 (дд, J=16,0, 8,7 Гц, 2Н), 3,97 (д, J=21,1 Гц, 4Н), 3,91 (с, 3Н), 3,76-3,73 (м, 4Н), 3,33 (дд, J=43,0, 8,5 Гц, 2Н), 2,92-2,86 (м, 2Н), 1,76-1,72 (м, 1Н), 1,47 (с, 3Н), 1,27 (с, 3Н). ЖХ-МС [М+Н]+ 526,6.
Пример 25
Соединение 79
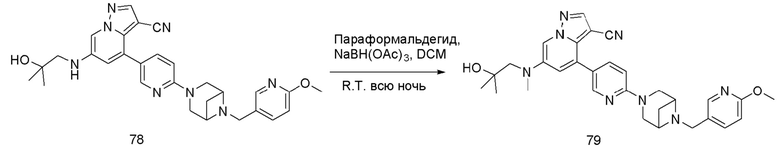
В одногорлую колбу объемом 25 мл последовательно добавляли соединение 78 (50 мг, 0,09 ммоль), параформальдегид (50 мг) и DCM (10 мл). После 10 мин перемешивания, при комнатной температуре порциями добавляли NaBH(OAc)3 (95,4 мг, 0,45 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 78. Смесь гасили добавлением аммиачной воды. Водную фазу отделяли и экстрагировали DCM (5 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением продукта 79 в виде желтого твердого вещества (33,9 мг, выход 70%).
1Н ЯМР (400 МГц, CDCl3) δ 8,40 (д, J=2,3 Гц, 1Н), 8,15 (с, 1Н), 8,11 (с, 1Н), 7,93 (д, J=1,4 Гц, 1Н), 7,82 (дд, J=8,7, 2,3 Гц, 1Н), 7,74 (д, J=8,2 Гц, 1Н), 7,08 (д, J=1,5 Гц, 1Н), 6,72 (дд, J=16,0, 8,7 Гц, 2Н), 3,97 (д, J=21,1 Гц, 4Н), 3,91 (с, 3Н), 3,84 (с, 3Н), 3,76-3,73 (м, 4Н), 3,33 (дд, J=43,0, 8,5 Гц, 2Н), 2,92-2,86 (м, 2Н), 1,76-1,72 (м, 1Н), 1,47 (с, 3Н), 1,27 (с, 3Н). ЖХ-МС [М+Н]+ 539,6.
Пример 26
Соединение 80
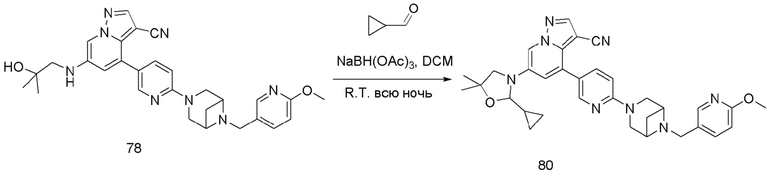
В одногорлую колбу объемом 25 мл последовательно добавляли соединение 78 (50 мг, 0,09 ммоль), циклопропанкарбоксальдегид (0,5 мл) и DCM (10 мл). После 10 мин перемешивания, при комнатной температуре порциями добавляли NaBH(ОАс)3 (95,4 мг, 0,45 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 78. Смесь гасили добавлением аммиачной воды. Водную фазу отделяли и экстрагировали DCM (5 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением продукта 80 в виде желтого твердого вещества (30 мг, выход 58%).
1Н ЯМР (400 МГц, CDCl3) δ 8,40 (д, J=2,3 Гц, 1Н), 8,15 (с, 1Н), 8,11 (с, 1Н), 7,93 (д, J=1,4 Гц, 1Н), 7,82 (дд, J=8,7, 2,3 Гц, 1Н), 7,74 (д, J=8,2 Гц, 1Н), 7,08 (д, J=1,5 Гц, 1Н), 6,72 (дд, J=16,0, 8,7 Гц, 2Н), 4,84 (д, J=5,7 Гц, 1Н), 3,97 (д, J=21,1 Гц, 4Н), 3,91 (с, 3Н), 3,76-3,73 (м, 4Н), 3,33 (дд, J=43,0, 8,5 Гц, 2Н), 2,92-2,86 (м, 2Н), 2,05 (с, 1Н), 1,76-1,72 (м, 1Н), 1,47 (с, 3Н), 1,27 (д, J=4,1 Гц, 3Н), 1,24 (с, 3Н), 1,18-1,11 (м, 1Н), 0,7-0,50 (м, 5Н). ЖХ-МС [М+Н]+ 576,7.
Пример 27
Соединение 81
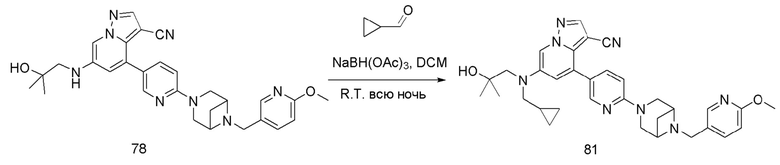
В одногорлую колбу объемом 25 мл последовательно добавляли соединение 78 (50 мг, 0,09 ммоль), циклопропанкарбоксальдегид (0,5 мл) и DCM (10 мл). После 10 мин перемешивания, при комнатной температуре порциями добавляли NaBH(OAc)3 (95,4 мг, 0,45 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 78. Смесь гасили добавлением аммиачной воды. Водную фазу отделяли и экстрагировали DCM (5 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением продукта 81 в виде желтого твердого вещества (28 мг, выход 54%).
1Н ЯМР (400 МГц, CDCl3) δ 8,40 (д, J=2,3 Гц, 1Н), 8,15 (с, 1Н), 8,11 (с, 1Н), 7,93 (д, J=1,4 Гц, 1Н), 7,82 (дд, J=8,7, 2,3 Гц, 1Н), 7,74 (д, J=8,2 Гц, 1Н), 7,08 (д, J=1,5 Гц, 1Н), 6,72 (дд, J=16,0, 8,7 Гц, 2Н), 3,97 (д, J=21,1 Гц, 4Н), 3,91 (с, 3Н), 3,84 (д, J=5,7 Гц, 2Н), 3,76-3,73 (м, 4Н), 3,33 (дд, J=43,0, 8,5 Гц, 2Н), 2,92-2,86 (м, 2Н), 1,76-1,72 (м, 1Н), 1,47 (с, 3Н), 1,27 (с, 3Н), 1,18-1,11 (м, 1Н), 0,65-0,50 (м, 4Н). ЖХ-МС [М+Н]+ 579,7.
Пример 28
Соединение 82
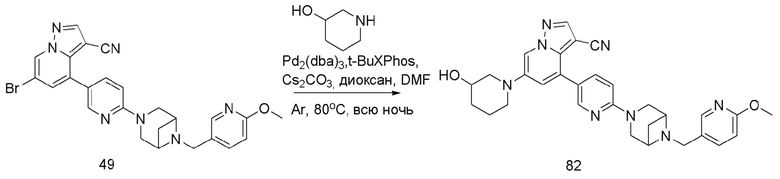
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (52 мг, 0,1 ммоль), Pd2(dba)3 (5,5 мг, 0,006 ммоль), t-BuXPhos (8,4 мг, 0,02 ммоль), 3-гидроксипиперидин (30,3 мг, 0,3 ммоль), Cs2CO3 (65 мг, 0,2 ммоль), 1,4-диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 82 (40,2 мг, выход 75%).
1Н ЯМР (400 МГц, CDCl3) δ 8,40 (с, 1Н), 8,19 (с, 1Н), 8,11 (с, 1Н), 8,05 (с, 1Н), 7,78 (д, J=10,9 Гц, 1Н), 7,29 (с, 1Н), 7,19 (с, 1Н), 6,71 (дд, J=17,2, 8,7 Гц, 2Н), 4,03-3,99 (м, 1Н), 3,92 (с, 3Н), 3,88-3,82 (м, 3Н), 3,65-3,60 (м, 3Н), 3,33 (д, J=9,2 Гц, 1Н), 3,16-3,01 (м, 3Н), 2,83-2,70 (м, 2Н), 1,99-1,94 (м, 4Н). ЖХ-МС [М+Н]+ 536,6.
Пример 29
Соединение 83
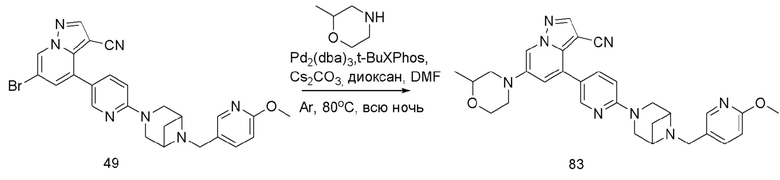
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (52 мг, 0,1 ммолв), Pd2(dba)3 (5,5 мг, 0,006 ммоль), t-BuXPhos (8,4 мг, 0,02 ммоль), 2-метилморфолин (30,3 мг, 0,3 ммоль), Cs2CO3 (65 мг, 0,2 ммоль), 1,4-диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контролв ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 83 (38 мг, выход 70,8%).
1Н ЯМР (400 МГц, CDCl3) δ 8,40 (с, 1Н), 8,20 (с, 1Н), 8,11 (с, 1Н), 8,01 (с, 1Н), 7,80 (д, J=8,6 Гц, 1Н), 7,69 (с, 1Н), 7,18 (с, 1Н), 6,75-6,63 (м, 2Н), 4,08-4,05 (м, 1Н), 3,92 (с, 3Н), 3,88-3,84 (м, 5Н), 3,64-3,58 (м, 4Н), 3,38 (дд, J=23,9, 11,6 Гц, 2Н), 2,93-2,90 (м, 1Н), 2,76-2,72 (м, 1Н), 2,59-2,51 (м, 1Н), 1,41 (д, J=19,1 Гц, 2Н), 1,31-1,24 (м, 3Н). ЖХ-МС [М+Н]+ 536,6.
Пример 30
Соединение 84
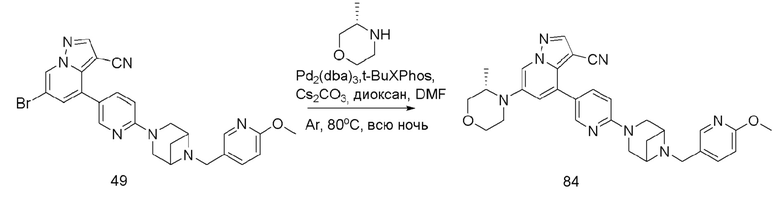
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (52 мг, 0,1 ммолв), Pd2(dba)3 (5,5 мг, 0,006 ммоль), t-BuXPhos (8,4 мг, 0,02 ммоль), 3-(S)-3-метилморфолин (30,3 мг, 0,3 ммоль), Cs2CO3 (65 мг, 0,2 ммоль), 1,4-диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 84 (30 мг, выход 56%).
1Н ЯМР (400 МГц, CDCl3) δ 8,40 (д, J=2,2 Гц, 1Н), 8,20 (с, 1Н), 8,11 (с, 1Н), 8,02 (д, J=1,8 Гц, 1Н), 7,81 (дд, J=8,8, 2,5 Гц, 1Н), 7,70 (с, 1Н), 7,18 (д, J=1,9 Гц, 1Н), 6,72 (дд, J=14,1, 8,7 Гц, 2Н), 4,02 (д, J=11,2 Гц, 1Н), 3,92 (с, 3Н), 3,90-3,85 (м, 5Н), 3,81-3,75 (м, 2Н), 3,75-3,63 (м, 6Н), 3,27-3,16 (м, 1Н), 3,03 (д, J=11,8 Гц, 1Н), 2,79-2,75 (м, 1Н), 1,14 (д, J=6,5 Гц, 3Н). ЖХ-МС [М+Н]+ 536,6.
Пример 31
Соединение 85
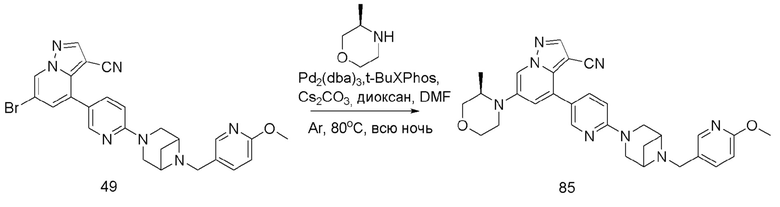
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (52 мг, 0,1 ммоль), Pd2(dba)3 (5,5 мг, 0,006 ммоль), t-BuXPhos (8,4 мг, 0,02 ммоль), (R)-3-метилморфолин (30,3 мг, 0,3 ммоль), Cs2CO3 (65 мг, 0,2 ммоль), 1,4-диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесв охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 85 (32 мг, выход 56%).
1Н ЯМР (400 МГц, CDCl3) δ 8,41 (д, 3=2,1 Гц, 1Н), 8,20 (с, 1Н), 8,13 (с, 1Н), 8,03 (д, J=1,9 Гц, 1Н), 7,82 (дд, J=8,7, 2,2 Гц, 1Н), 7,34 (д, J=7,1 Гц, 1Н), 7,18 (д, J=1,8 Гц, 1Н), 6,74 (дд, J=21,1, 8,7 Гц, 2Н), 4,02 (д, J=11,8 Гц, 2Н), 3,93 (с, 3Н), 3,92-3,89 (м, 2Н), 3,85-3,56 (м, 6Н), 3,38-2,81 (м, 4Н), 2,50-2,39 (м,3Н),1,14 (д, J=6,5 Гц, 3Н). ЖХ-МС [М+Н]+ 536,6.
Пример 32
Соединение 86
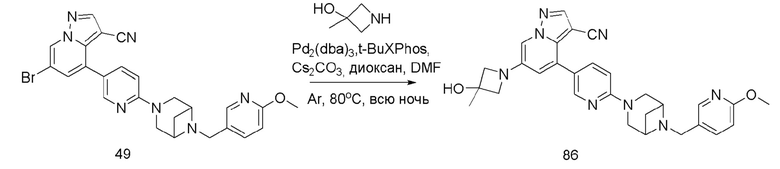
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (52 мг, 0,1 ммоль), Pd2(dba)3 (5,5 мг, 0,006 ммоль), t-BuXPhos (8,4 мг, 0,02 ммоль), 3-метил-3-азетидинол (26 мг, 0,3 ммоль), Cs2CO3 (65 мг, 0,2 ммоль), 1,4-диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 86 (34 мг, выход 65%).
1Н ЯМР (400 МГц, CDCl3) δ 8,37 (д, J=2,3 Гц, 1Н), 8,15 (с, 1Н), 8,10 (д, J=2,0 Гц, 1Н), 7,78 (дд, J=8,8, 2,5 Гц, 1Н), 7,72 (д, J=1,9 Гц, 1Н), 7,65 (дд, J=8,5, 2,2 Гц, 1Н), 6,77-6,66 (м, 3Н), 4,12-3,98 (м, 1Н), 3,92 (с, 3Н), 3,91 (с, 2Н), 3,87-3,74 (м, 6Н), 3,62-3,58 (м, 4Н), 2,73-2,67 (м, 1Н), 1,67 (с, 3Н). ЖХ-МС [М+Н]+ 522,6.
Пример 33
Соединение 87
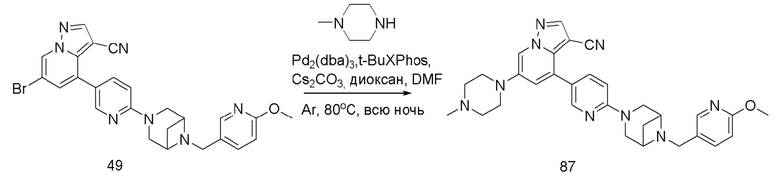
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (52 мг, 0,1 ммоль), Pd2(dba)3 (5,5 мг, 0,006 ммоль), t-BuXPhos (8,4 мг, 0,02 ммоль), JV-метилпиперазин (30 мг, 0,3 ммоль), Cs2CO3 (65 мг, 0,2 ммоль), 1,4-диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 87 (34 мг, выход 63,5%).
1Н ЯМР (400 МГц, CDCl3) δ 8,40 (д, J=2,2 Гц, 1Н), 8,21 (с, 1Н), 8,12 (д, J=2,0 Гц, 1Н), 8,04 (д, J=2,0 Гц, 1Н), 7,80 (дд, J=8,8, 2,5 Гц, 1Н), 7,66 (дд, J=8,5, 2,4 Гц, 1Н), 7,20 (д, J=2,0 Гц, 1Н), 6,72 (дд, J=14,9, 8,6 Гц, 2Н), 3,94 (с, 3Н), 3,85 (д, J=11,9 Гц, 2Н), 3,81 (д, J=5,8 Гц, 2Н), 3,65-3,61 (м, 2Н), 3,60 (с, 2Н), 3,29-3,16 (м, 4Н), 2,77-2,68 (м, 1Н), 2,68-2,63 (м, 4Н), 2,41 (с, 3Н), 1,68 (д, J=8,7 Гц, 1Н). ЖХ-МС [М+Н]+ 535,6.
Пример 34
Соединение 88
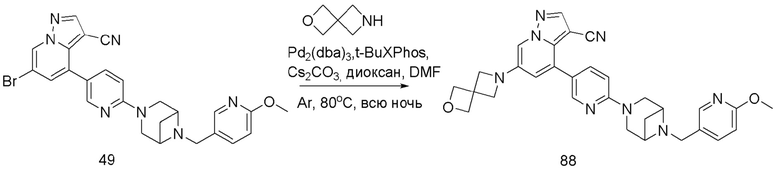
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (52 мг, 0,1 ммоль), Pd2(dba)3 (5,5 мг, 0,006 ммоль), t-BuXPhos (8,4 мг, 0,02 ммоль), 2-окса-6-аза-спиро[3,3]гептан (29,7 мг, 0,3 ммоль), Cs2CO3 (65 мг, 0,2 ммоль), 1,4-диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 88 (35,3 мг, выход 66%).
1Н ЯМР (400 МГц, CDCl3) δ 8,37 (д, J=2,2 Гц, 1Н), 8,16 (с, 1Н), 8,10 (д, J=1,9 Гц, 1Н), 7,78 (дд, J=8,8, 2,5 Гц, 1Н), 7,71 (д, J=1,9 Гц, 1Н), 7,66 (д, J=6,9 Гц, 1Н), 6,76-6,66 (м, 3Н), 4,88 (с, 4Н), 4,10 (с, 4Н), 3,92 (с, 3Н), 3,83-3,80 (м, 4Н), 3,69-3,60 (м, 2Н), 3,59 (с, 2Н), 2,76-2,72 (м, 1Н), 1,96-1,89 (м, 1Н). ЖХ-МС [М+Н]+ 534,6.
Пример 35
Соединение 89
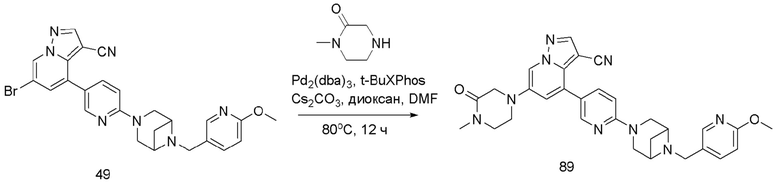
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (52 мг, 0,1 ммоль), Pd2(dba)3 (5,5 мг, 0,006 ммоль), t-BuXPhos (8,4 мг, 0,02 ммоль), 1-метилпиперазин-2-он (34,2 мг, 0,3 ммоль), Cs2CO3 (65 мг, 0,2 ммоль), 1,4-диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 89 (50 мг, выход 90%).
1Н ЯМР (400 МГц, CDCl3) δ 8,39 (д, J=2,3 Гц, 1Н), 8,21 (с, 1Н), 8,10 (д, J=2,0 Гц, 1Н), 8,03 (д, J=2,0 Гц, 1Н), 7,78 (дд, J=8,8, 2,5 Гц, 1Н), 7,66 (дд, J=8,5, 2,2 Гц, 1Н), 7,14 (д, J=2,0 Гц, 1Н), 6,71 (дд, J=11,8, 8,7 Гц, 2Н), 3,92 (с, 3Н), 3,87 (с, 2Н), 3,85-3,75 (м, 4Н), 3,75-3,60 (м, 4Н), 3,57-3,47 (м, 4Н), 3,07 (с, 3Н), 2,72 (д, J=7,1 Гц, 1Н), 2,07-2,01 (м, 1Н). ЖХ-МС [М+Н]+ 549,6.
Пример 36
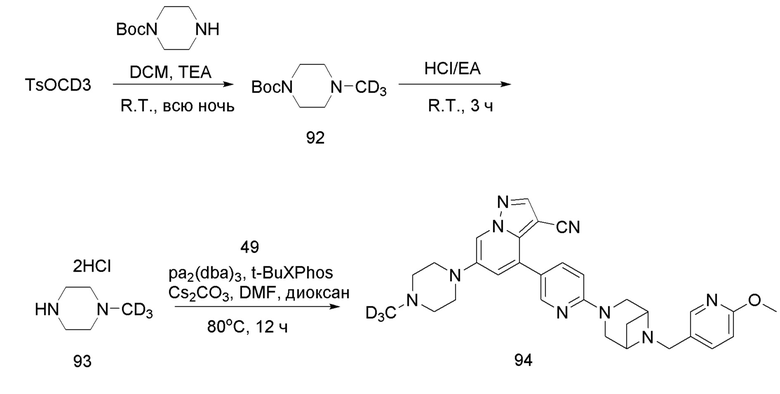
Соединение 92

В одногорлую грушевидную колбу объемом 25 мл последовательно добавляли метил Б3-п-бензолсульфонат (500 мг, 2,64 ммоль), 1-трет-бутоксикарбонилпиперазин (327,8 мг, 1,76 ммоль), триэтиламин (534 мг, 5,28 ммоль) и 1,4-диоксан (10 мл). Смесь перемешивали при комнатной температуре в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и водную фазу отделяли и экстрагировали DCM (5 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта в виде бесцветной жидкости, которую очищали колоночной хроматографией получением продукта 92 в виде бесцветной жидкости (340 мг, выход 95%).
ЖХ-МС [М+Н]+ 204,17.
Соединение 93
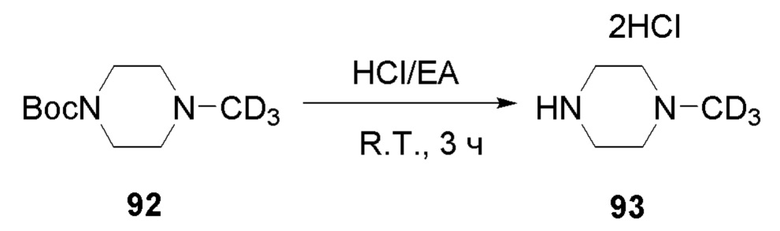
В одногорлую грушевидную колбу объемом 25 мл добавляли раствор хлористоводородной кислоты в этилацетате (50 мл) и медленно по каплям добавляли соединение 92 (340 мг, 1,68 ммоль). Смесь перемешивали при комнатной температуре в течение 3 ч, и контроль ТСХ показал отсутствие остатков исходного вещества. Осаждалось белое твердое вещество. Твердое вещество собирали филвтрацией и сушили с получением целевого продукта 93 в виде белого твердого вещества (296 мг, выход 100%).
Соединение 94
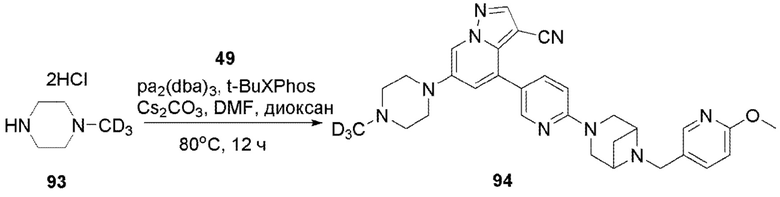
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (52 мг, 0,1 ммоль), Pd2(dba)3 (5,5 мг, 0,006 ммоль), t-BuXPhos (8,4 мг, 0,02 ммоль), соединение 93 (52,8 мг, 0,3 ммоль), Cs2CO3 (65 мг, 0,2 ммоль), диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 94 (42 мг, выход 78%).
1Н ЯМР (400 МГц, CDCl3) δ 8,40 (д, J=2,2 Гц, 1Н), 8,21 (с, 1Н), 8,12 (д, J=2,0 Гц, 1Н), 8,04 (д, J=2,0 Гц, 1Н), 7,80 (дд, J=8,8, 2,5 Гц, 1Н), 7,66 (дд, J=8,5, 2,4 Гц, 1Н), 7,20 (д, J=2,0 Гц, 1Н), 6,72 (дд, J=14,9, 8,6 Гц, 2Н), 3,94 (с, 3Н), 3,85 (д, J=11,9 Гц, 2Н), 3,81 (д, J=5,8 Гц, 2Н), 3,65-3,61 (м, 2Н), 3,60 (с, 2Н), 3,29-3,16 (м, 4Н), 2,77-2,68 (м, 1Н), 2,68-2,63 (м, 4Н), 1,68 (д, J=8,7 Гц, 1Н). ЖХ-МС [М+Н]+ 539,6.
Пример 37
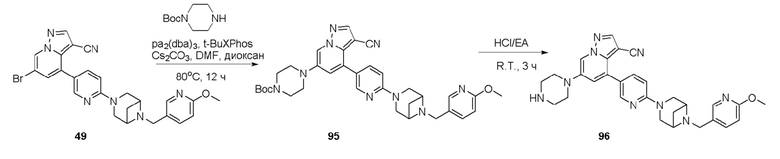
Соединение 95

В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 49 (52 мг, 0,1 ммолв), Pd2(dba)3 (5,5 мг, 0,006 ммоль), t-BuXPhos (8,4 мг, 0,02 ммоль), 1-трет-бутоксикарбонилпиперазин (55,9 мг, 0,3 ммоль), Cs2CO3 (65 мг, 0,2 ммоль), диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 49. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесв перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 95 (53 мг, выход 88%).
ЖХ-МС [М+Н]+ 622,32.
Соединение 96
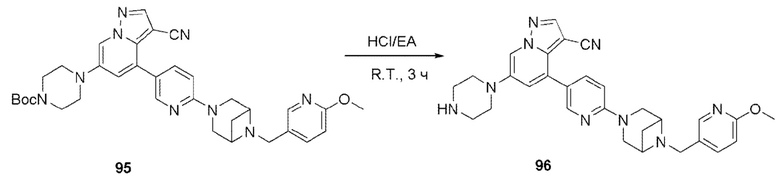
В одногорлую грушевидную колбу объемом 25 мл добавляли раствор хлористоводородной кислоты в этилацетате (10 мл) и медленно по каплям добавляли соединение 95 (53 мг, 0,09 ммоль). Смесь перемешивали при комнатной температуре в течение 3 ч, и контроль ТСХ показал отсутствие остатков исходного вещества. Смесь концентрировали, корректировали рН до 9 аммиачной водой и экстрагировали DCM (3 мл × 3). Органические фазы объединяли, концентрировали и очищали колоночной хроматографией с получением целевого продукта 96 в виде желтого твердого вещества (36 мг, выход 78%).
1Н ЯМР (400 МГц, CDCl3) δ 8,40 (д, J=2,2 Гц, 1Н), 8,21 (с, 1Н), 8,12 (д, J=2,0 Гц, 1Н), 8,04 (д, J=2,0 Гц, 1Н), 7,80 (дд, J=8,8, 2,5 Гц, 1Н), 7,66 (дд, J=8,5, 2,4 Гц, 1Н), 7,20 (д, J=2,0 Гц, 1Н), 6,72 (дд, J=14,9, 8,6 Гц, 2Н), 3,94 (с, 3Н), 3,85 (д, J=11,9 Гц, 2Н), 3,81 (д, J=5,8 Гц, 2Н), 3,65-3,61 (м, 2Н), 3,60 (с, 2Н), 3,29-3,16 (м, 4Н), 2,77-2,68 (м, 1Н), 2,68-2,63 (м, 4Н), 1,68 (д, J=8,7 Гц, 1Н). ЖХ-МС [М+Н]+ 522,6.
Пример 38
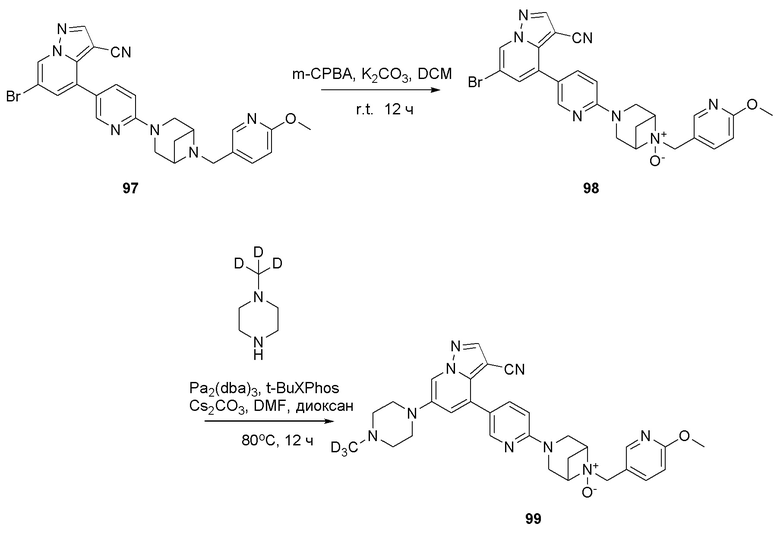
Соединение 98
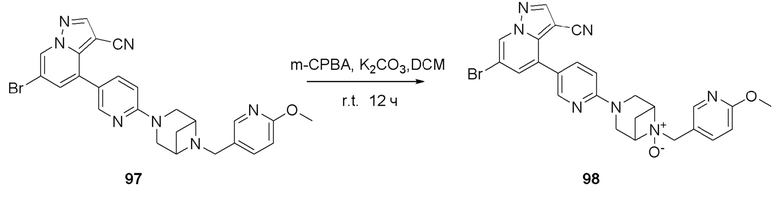
В одногорлую колбу объемом 25 мл последовательно добавляли соединение 97 (500 мг, 0,97 ммоль), K2CO3 (402 мг, 2,91 ммоль) и DCM (50 мл). Порциями при температуре 0°С добавляли m-СРВА (200 мг, 1,16 ммоль). Смесь подогревали до комнатной температуры и перемешивали в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 97. Добавляли 50 мл воды, и смесь перемешивали в течение 10 мин. Органическую фазу отделяли, последовательно промывали H2O и насыщенным раствором NaCl, сушили над безводным Na2SO4, концентрировали и очищали колоночной хроматографией с получением продукта 98 в виде бледно-желтого твердого вещества (439 мг, выход 85%).
Соединение 99
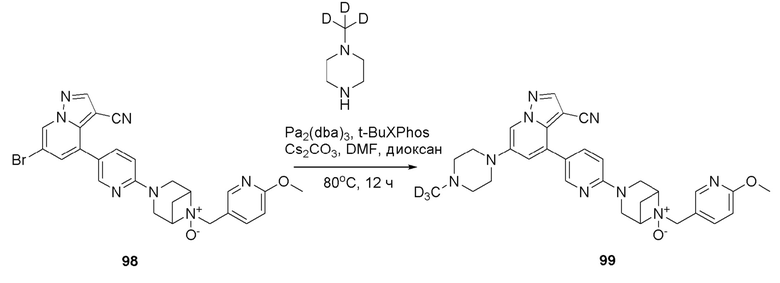
В герметизируемую пробирку объемом 25 мл последовательно добавляли соединение 98 (100 мг, 0,19 ммоль), Pd2(dba)3 (10,4 мг, 0,01 ммоль), t-BuXPhos (12,7 мг, 0,03 ммоль), дейтерированный метилпиперазин (23,7 мг, 0,23 ммоль), Cs2CO3 (187 мг, 0,57 ммоль), диоксан (3 мл) и DMF (1 мл). Смесь перемешивали при температуре 80°С в течение ночи в атмосфере Ar, и контроль ТСХ показал отсутствие остатков исходного вещества 98. Смесь охлаждали до комнатной температуры, и добавляли 10 мл воды. Смесь перемешивали в течение 10 мин, и осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией, сушили и очищали колоночной хроматографией с получением продукта 99 (79 мг, выход 75%).
1Н ЯМР (400 МГц, CDCl3) δ 8,34-8,28 (м, 1Н), 8,17 (д, J=2,0 Гц, 1Н), 8,12 (д, J=2,1 Гц, 1Н), 8,01 (с, 1Н), 7,72-7,63 (м, 2Н), 7,16 (д, J=1,5 Гц, 1Н), 6,76 (дд, J=8,5, 3,2 Гц, 1Н), 6,56 (дд, J=41,0, 8,8 Гц, 1Н), 4,07-3,88 (м, 6Н), 3,84-3,57 (м, 5Н), 3,49 (д, J=4,3 Гц, 1Н), 3,23 (т, 4Н), 2,66 (т, 4Н), 2,49 (д, J=7,0 Гц, 1Н).
Пример 39
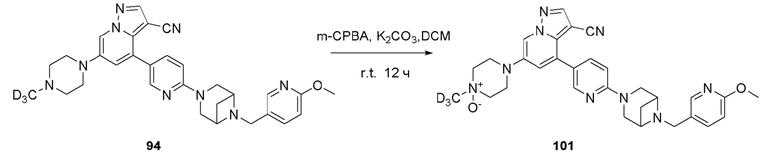
Соединение 101
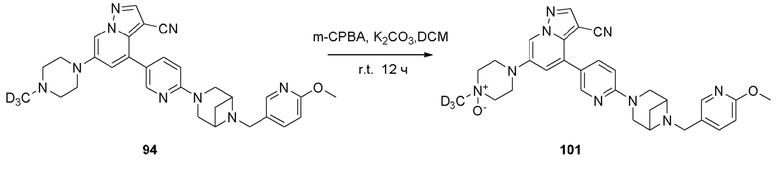
В одногорлую колбу объемом 25 мл последовательно добавляли соединение 94 (100 мг, 0,18 ммоль), K2CO3 (51,2 мг, 0,36 ммоль) и DCM (20 мл). Порциями при температуре 0°С добавляли m-СРВА (37 мг, 0,22 ммоль). Смесь подогревали до комнатной температуры и перемешивали в течение ночи, и контроль ТСХ показал отсутствие остатков исходного вещества 94. Добавляли 20 мл воды, и смесь перемешивали в течение 10 мин. Органическую фазу отделяли, последовательно промывали H2O и насыщенным раствором NaCl, сушили над безводным Na2SO4, концентрировали и очищали колоночной хроматографией с получением продукта 101 в виде бледно-желтого твердого вещества (78 мг, выход 78%).
1Н ЯМР (400 МГц, DMS0) δ 8,55 (с, 1Н), 8,49-8,34 (м, 2Н), 8,07 (с, 1Н), 7,85 (дд, J=8,7, 2,2 Гц, 1Н), 7,68 (дд, J=8,5, 2,1 Гц, 1Н), 7,55 (с, 1Н), 6,78 (т, J=8,2 Гц, 2Н), 3,82 (с, 3Н), 3,73 (д, J=11,9 Гц, 2Н), 3,68 (д, J=5,6 Гц, 2Н), 3,64-3,51 (м, 7Н), 3,50 (с, 2Н), 3,00 (с, 2Н), 2,51 (с, 2Н), 1,59 (д, J=8,3 Гц, 1Н).
Эксперимент 1 по определению активности. Ингибирование полученными соединениями активности киназ семейства RET
Анализировали соединения на наличие активности IC50 в отношении RET-киназ дикого типа (RET WT), с точечными мутациями RET (V804M), RET (М918Т), слитыми мутациями CCDC6-RET и киназы KDR (VEGFR2).Указанные выше киназы приобретали в компаниях Thermo Fisher Scientific и ProQinase GmbH.
Метод анализа активности указанных выше киназ основан на гомогенной флуоресценции с временным разрешением (HTRF), и этим методом определяли ингибирующую активноств соединений. Готовили 8 мкл реакционного раствора, содержащего 1х ферментного буфера (Cisbio, HTRF KinEASE™-TK), 5 мМ MgCl2, 1 мМ MnCl2, 1 мМ DTT, 1 мкМ субстрата-биотина ТК (Cisbio, HTRF KinEASE™-TK), 10 мкМ АТФ (1 мкМ для RET WT, CCDC6-RET; 20 мкМ для KDR), градиентные концентрации соединений и следующие концентрации соответствующих киназ: 0,03 нг/мкл RET WT, 0,2 нг/мкл RET (V804M), 0,04 нг/мкл RET (M918T), 0,12 нг/мкл CCDC6-RET и 0,02 нг/мкл KDR. Киназы и соединения предварительно инкубировали в течение 5 мин, и затем добавляли АТФ и субстрат для начала реакции. Все реакции, катализируемые ферментами, проводили при 25°С в течение 60 мин. После завершения катализируемых ферментами реакций в реакционные смеси добавляли 4 мкл антитела-криптата ТК и 4 мкл стрептавидина-XL665 (концентрация в реакционной смеси составляла 62,5 нМ), и смеси инкубировали при температуре 25°С еще 60 мин. После завершения инкубации значения флуоресценции HTRF определяли на приборе CLARIOstar (BMG LABTECH), и рассчитывали 1С50 с использованием программного обеспечения GraphPad Prism 5.0.
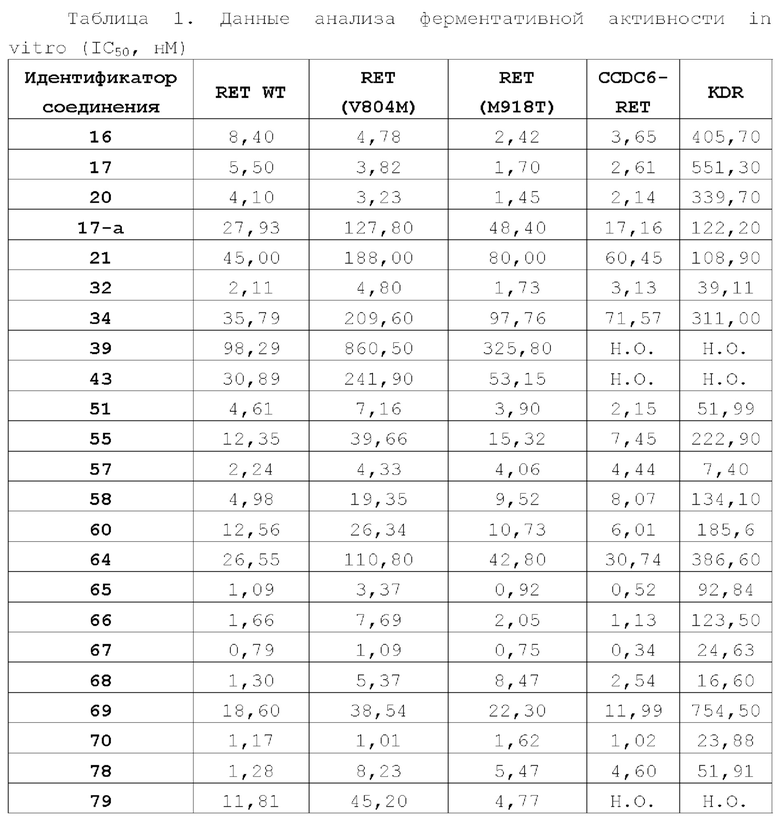
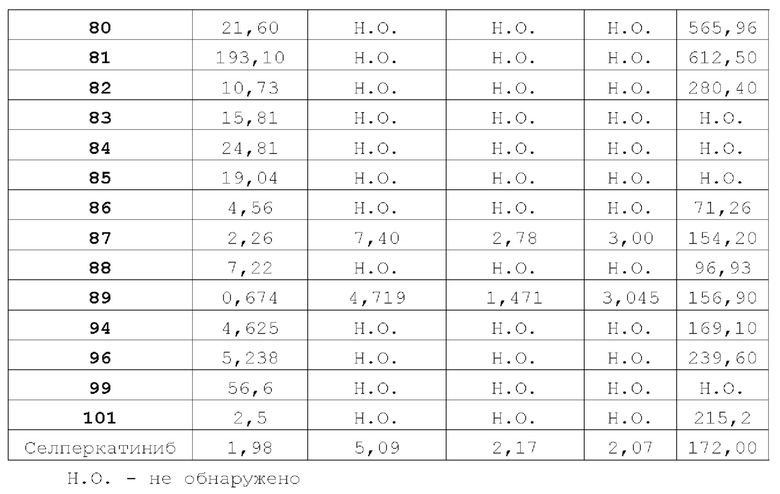
Эксперимент 2 по определению активности. Отчет об исследовании метаболической стабильности полученных соединений в микросомах печени человека
Скорость и степень метаболизма соединений в микросомах печени под действием NADPH-восстанавливающих коферментов исследовали методом жидкостной хроматографии-масс-спектрометрии (ЖХ-МС/МС). Добавляли 445 мкл 0,562 мг/мл рабочего раствора микросом печени человека в соответствующий 96-луночный планшет и предварительно инкубировали на водяной бане при температуре 37°С в течение 10,0 мин. Затем инициировали реакцию путем добавления 5,00 мкл 100 мкМ соединения и 50,0 мкл 10,0 мМ NADPH. Через 0 мин, 2 мин, 5 мин, 10 мин, 20 мин, 30 мин и 60 мин добавляли 50,0 мкл реакционной смеси к 400 мкл ледяного ацетонитрила, содержащего 50,0 нг/мл внутреннего стандарта (толбутамид) для прекращения реакции. Для образца без кофактора (NCF) 445 мкл рабочего раствора микросом каждого вида хорошо перемешивали с 50,0 мкл 10 мМ МдС12 и, наконец, добавляли 5,00 мкл рабочего раствора исследуемого образца. Смесь хорошо перемешивали и инкубировали при 37°С в течение 10 мин. По истечении времени добавляли 50,0 мкл реакционной смеси к 400 мкл ледяного холодного 102 для прекращения реакции. Смесь хорошо перемешивали вихревым перемешиванием и центрифугировали при 1700х g при температуре 4°С в течение 15 мин. Отбирали 150 мкл супернатанта, разбавляли его добавлением 150 мкл сверхчистой воды и подвергали анализу ЖХ-МС/МС. Результаты приведены в таблице 2 и таблице 3.
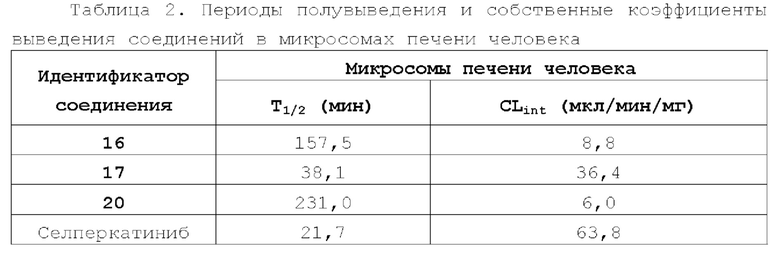
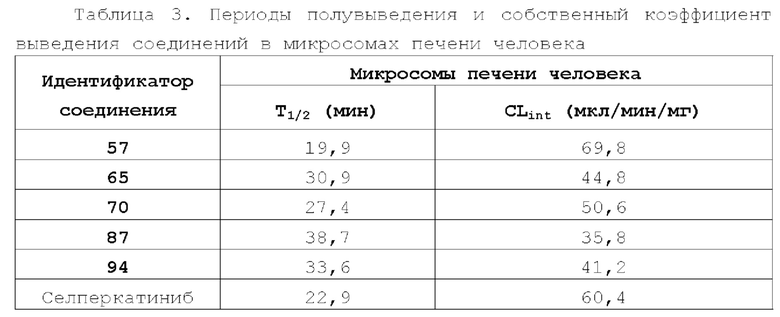
Эксперимент 3 по определению активности. Ингибирование полученными соединениями клеток Ba/F3 KIF5B-RET
Клетки Ва/F3 KIF5B-RET, Ва/F3KIF5B-RET-V804M, Ва/F3 RET-М918Т и Ва/F3 KIF5B-RET-G81OR, выращиваемые в log-фазе, собирали и подсчитывали с использованием счетного планшета. Жизнеспособность клеток определяли методом исключения с трипановым синим, и жизнеспособность клеток сохранялась на уровне более 90%. Концентрацию клеток корректировали, и в 96-луночный планшет добавляли по 90 мкл суспензии клеток. Клетки в 96-луночном планшете инкубировали в течение ночи при температуре 37°С с 5% CO2 и влажностью 95%. Добавляли по 10 мкл/лунку соответствующие градиентные концентрации растворов лекарственных веществ (максимальная концентрация составляла 1000 нМ) к 96-луночному планшету, инокулированному клетками. На одну концентрацию лекарственного вещества отводили по три лунки, и конечная концентрация DMSO составляла 0,1%. Клетки в 96-луночном планшете, обрабатываемые лекарственными веществами, инкубировали в течение еще 72 ч при температуре 37°С с 5% CO2 и влажностью 95%. После завершения обработки лекарственными веществами, в каждую лунку добавляли по 100 мкл реагента CellTiter-Glo. Планшет с клетками встряхивали на орбитальном шейкере в течение 5 мин для лизиса клеток и потом выдерживали при комнатной температуре в течение 20 мин для стабилизации сигналов люминесценции, а затем считывали значения люминесценции.Данные анализировали с использованием программного обеспечения GraphPad Prism 5.0. По данным подбирали кривые «доза-ответ» с использованием нелинейной регрессии в виде S-кривой, и по полученным кривым рассчитывали значения IC50. Результаты приведены в таблице 4.
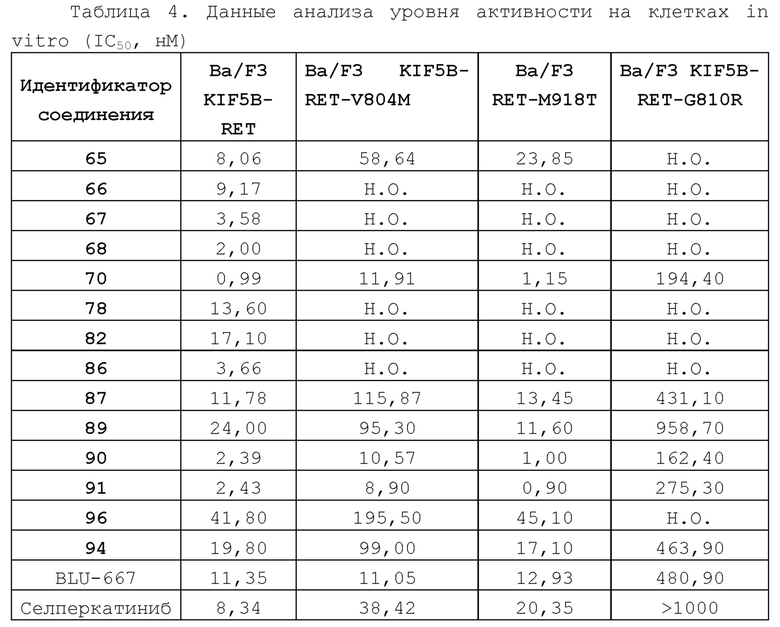
Структурная формула BLU-667 показана ниже:
Структурная формула селперкатиниба (Loxo-292) показана ниже:
Эксперимент 4 по определению активности. Ингибирование полученными соединениями калиевых каналов hERG
Все конечные концентрации исследуемых соединений готовили на день эксперимента, и затем растворяли их во внеклеточной жидкости. Внеклеточная жидкость (мМ) имела состав: NaCl, 137; KCl, 4; CaCl2, 1,8; MgCl2, 1; НЕ PES, 10; глюкоза 10; рН 7,4 (титрование NaOH). Все растворы исследуемых и контрольных соединений содержали 0,3% DMSO. Клетки HEK293, стабильно экспрессирующие ионный канал hERG, переносили в перфузионную камеру и проводили перфузию с использованием внеклеточной жидкости. Внутриклеточную жидкость хранили небольшими партиями в морозильной камере при температуре -80°С и размораживали в день эксперимента. Внутриклеточная жидкость (мМ) имела состав: К аспартат, 130; MgCl2, 5; EGTA 5; НЕ PES, 10; Трис-АТФ 4; рН 7,2 (титрование КОН). Электроды изготовляли путем вытягивания с помощью РС-10 (Narishige, Япония). Проводили фиксацию потенциала целых клеток методом пэтч-кламп.Шум фильтровали на одной пятой частоты дискретизации.
Клетки фиксировали при напряжении -80 мВ, затем деполяризовали до 40 мВ с помощью квадратной волны длительностью 4 секунды и гиперполяризовали до -40 мВ с помощью квадратной волны длительностью 2 секунды для образования следового тока hERG. Эту процедуру повторяли каждые 20 секунд. Следовой ток hERG был чистым следовым током hERG. Определяли максимальное значение тока, индуцированное второй квадратной волной, и после того, как он становился стабильным, проводили перфузию с использованием исследуемых соединений. После стабилизации реакции рассчитывали интенсивность блокирования. Значение 1С50 для ингибирования канала hERG соединениями рассчитывали из значения интенсивности блокирования. Результаты приведены в таблице 5.
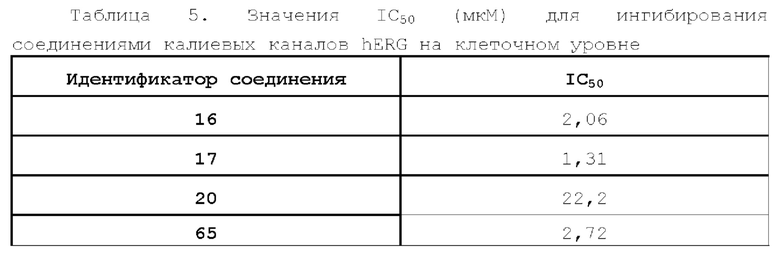
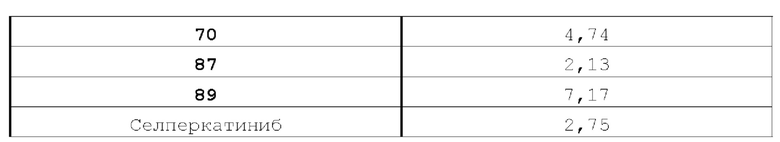
Эксперимент 5 по определению активности.
Фармакокинетический анализ на крысах
Крысам-самцам породы SD (весом 220±20 г) вводили соединение 94 и Loxo-292 в дозе 1,0 мг/кг путем инъекции в хвостовую вену и в дозе 5,0 мг/кг перорально. Каждая группа состояла из 3 животных. Растворителем для введения был раствор 5% DMSO+5% полиэтоксилированное касторовое масло (Cremophor EL) в нормальном солевом растворе. Крысы голодали в течение 12 часов до введения препарата, и через 4 часа после введения им предоставляли ad libitum (неограниченный) доступ к пище и воде. Кровь отбирали из глазницы в количестве около 0,2 мл до введения и через 5 мин, 15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 10 ч и 24 ч после введения, помещали ее в пробирки ЕР с антикоагулянтом EDTA-K2 в ледяную ванну и центрифугировали при температуре 4°С при 8000 об/мин в течение 5 мин для отделения плазмы, которую до анализа хранили при температуре -20°С. Концентрацию соединения в плазме крови количественно определяли с помощью жидкостной хроматографии - тандемной масс-спектрометрии (ЖХ-МС/МС). Фармакокинетические параметры рассчитывали по результатам анализа образцов с использованием WinNonlin 5.2.
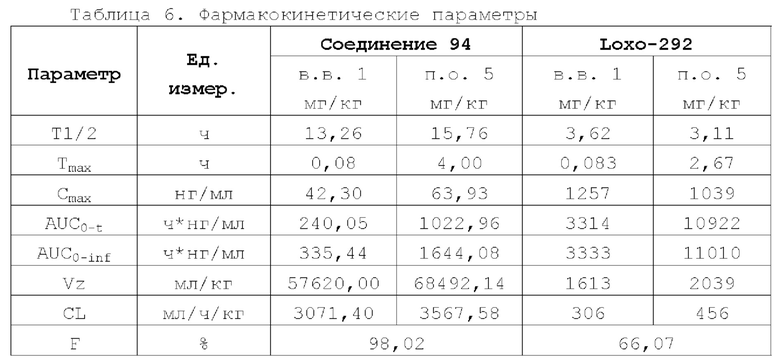
Как видно из данных, приведенных в таблице 6, после перорального введения соединение 94 дольше выводилось из организма крыс и имело более высокую биодоступность, чем Loxo-292.
Эксперимент 6 по определению активности. Анализ распределения в тканях на мышах
Мышам-самцам породы NVSG (весом 20-25 г) перорально вводили соединение 94 и Loxo-292 в дозе 30 мг/кг. Образцы плазмы и тканей печени, мозга, легких и тому подобного у животных собирают через 4 ч после введения. Отбор плазмы крови: цельную кровь собирали в объеме около 80-100 мкл в пробирки ЕР, содержащие EDTA, и центрифугировали при 4000 g в течение 5 мин, затем собирали верхнюю плазму и хранили при температуре -80°С. Отбор тканей: после эвтаназии животных ткани собирали и быстро замораживали в жидком азоте, на грамм ткани добавляли 5 мл раствора для гомогенизации (50% ацетонитрил). Ткани превращали в гомогенаты в тканевом гомогенизаторе и хранили при температуре -80°С. Концентрацию соединения в плазме крови и образцах тканей количественно определяли с помощью жидкостной хроматографии тандемной масс-спектрометрии (ЖХ-МС/МС).
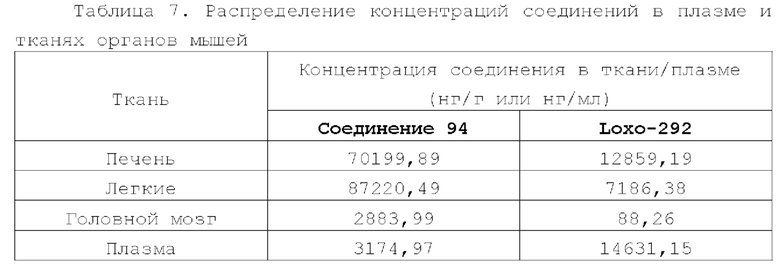
Как видно из данных таблицы 7, после перорального введения соединение 94 давало более высокое распределение концентрации в тканях целевых органов мышей, что является более благоприятным для оказания лекарственных эффектов в целевых органах.
Эксперимент 7 определения активности. Анализ на мышиной модели с подкожным трансплантатом опухоли BA/F3 KIF5B-RET
Клетки ВА/F3 KIF5B-RET культивировали в среде RPMI-1640 с добавлением 10% FBS в инкубаторе с 5% CO2, при температуре 37°С.Каждой из мышей NVSG подкожно в правое плечо вводили около 1 × 106 клеток в объеме 100 мкл путем подкожной инъекции. Когда объем привитых опухолей достиг около 100 мм3, мышей с привитыми опухолями соответствующего размера отбирали и случайным образом объединяли в группы в зависимости от массы тела и размера привитой опухоли. Препараты вводили путем внутрижелудочного введения в объеме 10 мкл/г массы тела. Вещество Loxo-292 вводили в дозе 30 мг/кг дважды в сутки (дважды/сут); соединение 94 вводили в дозе 30 мг/кг дважды в сутки (дважды/сут) и в дозах 30 мг/кг и 60 мг/кг один раз в день (раз/сут); контрольная группа, которой вводился растворитель, получала раствор 2% DMSO+2% полиэтоксилированного касторового масла в нормальном физиологическом растворе. Лекарственные вещества вводили в течение 15 дней подряд, а объем привитых опухолей измеряли дважды в неделю. День 0 относится к первому дню распределения по группам и введения.
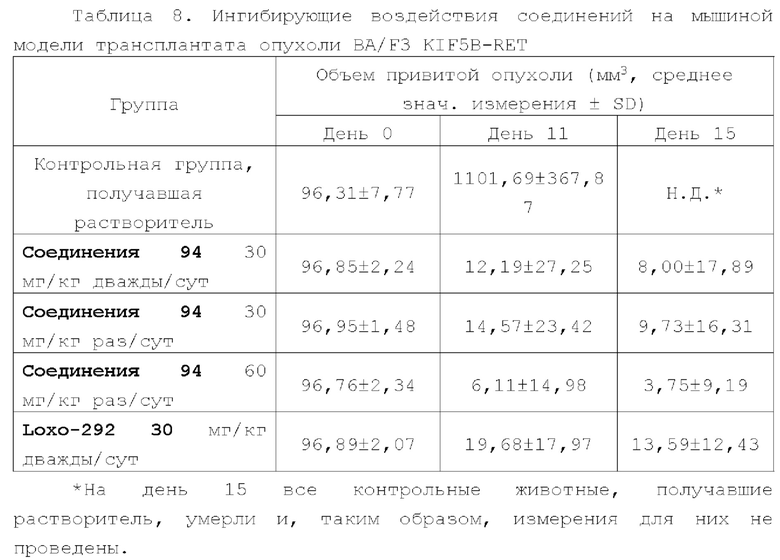
Как видно из данных, приведенных в таблице 8, после 15 дней введения у всех групп, получавших дозы соединения 94, привитые опухоли у мышей были меньше, чем у групп, получавших Loxo-292, что свидетельствует о превосходящей ингибирующей активности против опухолей; кроме того, соединение 94 продемонстрировало хорошую ингибирующую активность против опухолей даже при введении один раз в сутки.
Эксперимент 8 по определению активности. Ингибирование полученными соединениями активности киназ семейства FGFR
Полученные соединения анализировали на наличие активности в виде IC50 в отношении семейства киназ FGFR. Соответствующие киназы приобретали в компаниях Сагпа и Signalchem.
Метод анализа активности соответствующих киназ основан на гомогенной флуоресценции с временным разрешением (HTRF), и этим методом определяли ингибирующую активность соединений. Готовили 5 мкл реакционной смеси, содержащей 1х ферментного буфера (Cisbio, HTRF KinEASE™-TK), 5 мМ MgCl2, 1 мМ DTT, 1 мкМ субстрата-биотина ТК (Cisbio, HTRF KinEASE™-TK), соответственные концентрации АТФ и соответствующих киназ, и градиентные концентрации изучаемых соединений. Концентрации соответствующих киназ и соответственные концентрации АТФ показаны ниже в таблице 9:
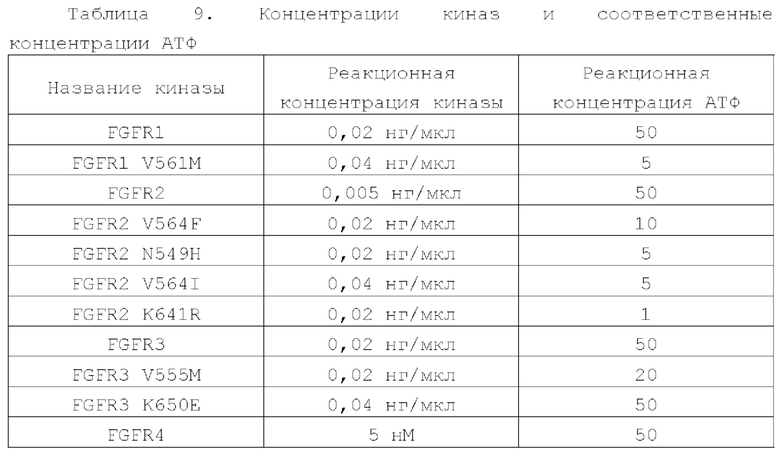
Киназы и соединения предварительно инкубировали в течение 10 мин, и затем добавляли АТФ и субстрат для начала реакции. Все реакции, катализируемые ферментами, проводили при 25°С в течение 40 мин. После завершения катализируемых ферментами реакций в реакционные смеси добавляли 5 мкл антитела-криптата ТК и стрептавидина-ХЬб65 (концентрация в реакционной смеси стрептавидина-XL665 составляла 62,5 нМ), и смеси инкубировали при температуре 25°С еще 60 мин. После завершения инкубации сигналы флуоресценции при 615 нм (криптат) и 665 нм (XL665) считывали на приборе Biotek, и рассчитывали IC50 с использованием программного обеспечения GraphPad Prism 5.0.

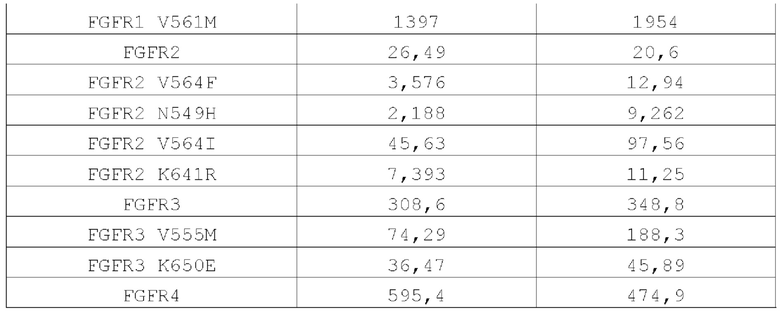
Данные приведенные выше в таблице 10 указывают на то, что соединение 94 обладает ингибирующим воздействием на активность семейства киназ FGFR, и что соединение 94 обладает лучшей ингибирующей активностью в отношении киназ FGFR2 V564F, FGFR2 N549H, FGFR2 V564I и FGFR3 V555M, чем вещество LOXO-292 (IC50 имело коэффициент 2 или даже меньше).
| название | год | авторы | номер документа |
|---|---|---|---|
| КЛАСС БИФУНКЦИОНАЛЬНЫХ ХИМЕРНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ДЛЯ НАПРАВЛЕННОГО РАЗРУШЕНИЯ АНДРОГЕННЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2825000C2 |
| ИНГИБИТОР НЕКРОЗА КЛЕТОК, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2019 |
|
RU2793918C2 |
| Соединение диарилтиогидантоина в качестве антагониста андрогенового рецептора | 2018 |
|
RU2804108C9 |
| ИНГИБИТОР FAK И КОМБИНАЦИИ ЛЕКАРСТВЕННЫХ СРЕДСТВ С НИМ | 2019 |
|
RU2820555C2 |
| НОВЫЙ АГОНИСТ β-РЕЦЕПТОРА ТИРЕОИДНЫХ ГОРМОНОВ | 2021 |
|
RU2839610C1 |
| НОВЫЙ ИНГИБИТОР AURORA КИНАЗЫ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2835080C1 |
| ИНГИБИТОРЫ СЕТР | 2006 |
|
RU2513107C2 |
| ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ 2Н-ПИРАЗОЛА | 2016 |
|
RU2722363C2 |
| МОДУЛЯТОРЫ СВЯЗАННЫХ С MAS РЕЦЕПТОРОВ G-БЕЛКА X4 И СВЯЗАННЫЕ С НИМИ ПРОДУКТЫ И СПОСОБЫ | 2020 |
|
RU2815715C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛОПИРИМИДИНЫ И ЗАМЕЩЕННЫЕ ПУРИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ УБИКВИТИН-СПЕЦИФИЧЕСКОЙ ПРОЦЕССИРУЮЩЕЙ ПРОТЕАЗЫ 1 (USP1) | 2019 |
|
RU2833222C2 |
Группа изобретений относится к органической химии и включает соединения 94, 99, 101, фармацевтическую композицию на их основе и их применение. Технический результат – соединения 94, 99, 101, обладающие активностью ингибитора RET-киназ. 5 н. и 4 з.п. ф-лы, 10 табл., 39 пр.
1. Соединение или его фармацевтически приемлемая соль, выбранное из:
 .
.
2. Фармацевтическая композиция, обладающая активностью ингибитора RET-киназ, содержащая терапевтически эффективное количество соединения или его фармацевтически приемлемой соли по п. 1 и фармацевтически приемлемый носитель.
3. Применение соединения или его фармацевтически приемлемой соли по п. 1 в качестве ингибитора RET-киназ.
4. Применение соединения или его фармацевтически приемлемой соли по п. 1 в производстве лекарственного средства для лечения RET-ассоциированного заболевания.
5. Применение по п. 4, где RET-ассоциированное заболевание представляет собой рак.
6. Применение по п. 5, где рак выбран из рака легкого, рака щитовидной железы, рака молочной железы или колоректального рака.
7. Применение соединения или его фармацевтически приемлемой соли по п. 1 в производстве лекарственного средства ингибитора киназ семейства FGFR.
8. Применение по п. 7, где семейство FGFR включает FGFR1, FGFR1 V561M, FGFR2, FGFR2 V564F, FGFR2 N549H, FGFR2 V564I, FGFR2 K641R, FGFR3, FGFR3 V555M, FGFR3 K650E и FGFR4.
9. Применение по п. 8, где семейство FGFR включает FGFR2 V564F, FGFR2 N549H, FGFR2 V564I и FGFR3 V555M.
| Машина для укладки пачек в ящики с определенным числом пачек в каждом ряду | 1932 |
|
SU35049A1 |
| CN 111285886 A, 16.06.2020 | |||
| CN 110964008 A, 07.04.2020 | |||
| CN 111269229 A, 12.06.2020 | |||
| ИНГИБИТОР RET | 2013 |
|
RU2648818C2 |

